Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ АМИНОЦИКЛОБУТАНА, СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ СРЕДСТВ
Вид РИД
Изобретение
Настоящее изобретение касается производных аминоциклобутана, а также способа их получения и их применения для лечения человека.
Глутаматные NMDA-рецепторы (N-метил-D-аспартатные) являются ионотропными рецепторами, в основном проницаемыми для ионов Са++. Физиологически их активация вызывает открытие ионного канала и продукцию входящего тока, который только медленно инактивируется. Стимуляция этого рецептора требует одновременного присутствия глутамата (эндогенного агониста) и глицина или D-серина (эндогенных со-агонистов), а также деполяризации плазматической мембраны, инициируемой не-NMDA токами. NMDA-рецепторы широко распространены по всей центральной нервной системе, а также присутствуют на периферии. Они обнаружены в нейронах, астроцитах и олигодендроцитах (Karadottir et al., 2005, Nature, 438, 1162-1166). На уровне нейронов они находятся, в основном, в пост-синапсе, но также и в экстра-синаптических областях вдоль аксонов. NMDA-рецепторы играют ключевую роль в коммуникации и нейронной пластичности, а также в эксайтотоксичности.
Физиологическая активность NMDA-рецепторов имеет важное значение для нормального функционирования нейронов (Chen and Lipton, 2006, J. Neurochem., 97, 1611-1626). С другой стороны, сверхактивация этих рецепторов вовлечена как в острые нервные расстройства, например инсульты или черепные травмы, так и в состояния хронического стресса, например нейродегенеративные расстройства. Она также является одной из основных причин, вызывающих сильное возбуждение, приводящее к эпилептическим припадкам. Существуют многочисленные патологии, которые считают связанными с гиперактивностью NMDA-рецепторов и, следовательно, потенциально чувствительными к антагонистам NMDA. В качестве примеров можно привести следующее: эпилепсия, нейродегенеративные расстройства, такие как болезнь Хантингтона, болезнь Паркинсона, болезнь Альцгеймера, инсульт, боковой амилотрофический склероз или рассеянный склероз, СПИД-деменции, тревога, депрессия и болевые синдромы.
В настоящем изобретении заявитель обращает особое внимание на антидепрессантные и анальгетические свойства антагонистов NMDA-рецепторов формулы (1).
В контексте настоящего изобретения термин «хроническая боль» означает болевые симптомы, которые прогрессируют в течение периода времени более трех месяцев, но тяжесть которых может изменяться с течением времени. С другой стороны, термин «острая боль» означает боль, которая длится менее трех месяцев.
В рамках настоящего изобретения боль определена как ненормальный, неприятный, даже мучительный, чувствительный и эмоциональный опыт, который воспринимается и является интегрированным на самом высоком уровне коры головного мозга, которая придает ему эмоциональную и аффективную природу. Под "аналгезией" подразумевают уменьшение интенсивности боли, которую чувствуют в ответ на болезненный раздражитель. Под "анальгетическим лекарственным средством" (или "анальгетиком») подразумевают лекарственное средство, которое снимает или подавляет боль, не приводя к потере чувствительности или сознания.
Боль различной этиологии требует различных терапевтических стратегий. В общем, есть несколько категорий боли в зависимости от вовлеченных механизмов:
- боль из-за чрезмерной ноцицепции результате повреждений или возбуждения (например воспаления) периферических или висцеральных тканей;
- невропатическая (или нейрогенная) боль связана с повреждением, либо с дисфункцией или нарушением соматосенсорной системы; она отличается от ноцицептивной боли тем, что имеет другую симптоматику;
- психогенная (или идиопатическая) боль, которая существует в отсутствие повреждений. Физиологические механизмы этого типа боли четко не определены. Она, как правило, устойчива к анальгетикам.
Тем не менее, некоторые типы боли имеют характеристики, которые являются общими для нескольких типов боли. Например, это имеет место при боли нижней части спины или раковой боли, которая присутствует в виде боли, вызванной чрезмерной ноцицепцией, или в виде невропатической боли или, в большинстве случаев, в виде комбинации того и другого.
Депрессия определяется в психиатрии как расстройство настроения. Она характеризуется потерей мотивации, связанной или не связанной с различными симптомами, такими как безысходность, низкая самооценка, беспокойство, боль и, в крайних случаях, галлюцинации. Она часто является многофакторной и, в целом, имеет несколько причин.
Сообщается, что около 7% европейцев страдают от депрессии и треть из них устойчива к клинически используемым антидепрессантам. Затраты на депрессию в 15-44-летней возрастной группе общества являются одними из самых высоких из всех известных патологий. Одна из задач настоящего изобретения заключается в раскрытии новых антагонистов NMDA, которые обладают полезными свойствами для такого назначения, для которого существующие способы лечения не являются полностью удовлетворительными.
Было показано на мышах, что постоянное введение антидепрессантов, которые имеют разные механизмы действия (ингибиторы моноаминоксидазы, три цикл и ки, ингибиторы обратного захвата серотонина (ИОЗС) или смешанные ингибиторы обратного захвата серотонина и норадреналина), изменяет распределение и плотность NMDA-рецепторов. У крыс однократное внутрибрюшинное введение кетамина, антагониста NMDA-рецепторов, уменьшает время неподвижности в тесте принудительного плавания, признанной доклинической модели для определения молекул с антидепрессантной активностью. Кроме того, недавние исследования показывают, что кетамин имеет свойства антидепрессанта у людей. Таким образом, введение одной субанестетической дозы кетамина внутривенно пациентам с резистентной депрессией значительно улучшает их состояние, и это всего через 2 часа после инъекции. Достигнутые антидепрессантные эффекты продолжались более недели (Zarate et al., 2006, Arch. Gen. Psychiatry, 63, 856-864). Быстрота этого действия контрастирует со временем, необходимым для ответа, которое требуется для обычных антидепрессантов, другими словами, трицикликов первого поколения и ИОЗС или ИОЗН, которые требуют нескольких недель лечения для достижения любого положительного эффекта. Таким образом, представляется, что антагонисты NMDA-рецепторов и, в частности кетамин, эффективны в лечении депрессии, особенно в лечении депрессии, устойчивой к существующим лекарствам.
Терапевтические требования лечения боли значительны. На самом деле, неисчислимое количество людей страдает от острой боли и более одной пятой взрослых в Европе и в Соединенных Штатах страдает от хронической боли (Johannes et al., 2010, J. Pain, 11, 1230-1239). Задачей настоящего изобретения является раскрытие выгодных обезболивающих свойств, которыми обладают соединения формулы (I), и терапевтических перспектив, которые они открывают в лечении острой и хронической боли.
Многие исследования на животных и людях показали, что антагонисты NMDA-рецепторов, такие как кетамин, могут облегчить боль многих типов этиологии, такую как, например, невропатическая, послеоперационная или раковая боль (Cohen et al., 2011, Adv. Psychosom. Med., 30, 139-161). Таким образом, кетамин, вводимый внутривенно, уменьшает невропатическую боль у пациентов, резистентных к лечению обычными антидепрессантами. Он также улучшает аллодинию и повышенную чувствительность к боли у пациентов с КРБС (комплексным региональным болевым синдромом) (Finch et al., 2009, Pain, 146, 18-25). Периоперационное введение низкой дозы кетамина в качестве адъюванта снижает потребность в анальгетиках и ограничивает острую толерантность к морфину после операции (Elia et Tramer, 2005, Pain, 113, 61-70). В качестве профилактического лечения кетамин и декстрометорпан (другой антагонист NMDA) улучшают контроль послеоперационной боли (Muir, 2006, Current Opinion in Pharmacology, 6, 53-60). Также оказывается, что кетамин предотвращает возникновение хронической послеоперационной боли (Wilder-Smith et al., 2002, Pain, 97, 189-194). Результаты, полученные с другими антагонистами NMDA, такими как амантадин или МК-81, при невропатической боли, тем не менее, не являются окончательными (Muir, 2006, см. выше).
Открытие каналов NMDA приводит к увеличению внутриклеточного кальция, который активирует, в частности, NO-синтетазу и циклооксигеназу II типа, что приводит к синтезу простагландинов (ПГ). Ингибируя ПГ, особенно ПГЕ2, антагонисты NMDA, таким образом, имеют прямое влияние на регулирование воспалительных состояний (Beloeil et al., 2009, Anesth. Analg., 109, 943-950). Эта дополнительная противовоспалительная активность антагонистов NMDA может быть выгодной в лечении острой или хронической боли воспалительного происхождения. Аналогичным образом, NMDA-рецепторы экспрессируются в хондроцитах и способствуют механической функции клеток (Salter et al., 2004, Biorheology, 41, 273-281). В частности, оказывается, что они вовлечены в их пролиферацию и воспаление, ведущее к разрушению суставного хряща (Piepoli et al., 2009, Osteoarthritis and Cartilage, 17, 1076-1083). Поскольку суставной хрящ не регенерируется у взрослых, использование антагониста NMDA, следовательно, представляется особенно выгодным для предотвращения или замедления разрушения суставного хряща, которое сопровождает некоторые патологические состояния, такие как, например, воспалительный моноартрит, ревматоидный артрит, септический артрит, остеоартрит, ревматоидный артрит, подагра, спондилоартрит, острый внесуставной ревматизм.
Тем не менее, клиническое применение антагонистов NMDA у людей ограничено их нежелательными эффектами, в частности, на центральную нервную систему, и в особенности при повторном лечении. Среди побочных эффектов антагонистов NMDA можно привести, например, галлюцинации, спутанность сознания, расстройства личности, кошмары, тревожное возбуждение, отсутствие концентрации, изменения настроения, судороги, торможение, сонливость, тошноту (Aarts et Tymianski, 2003, Biochem. Pharmacol., 66, 877-886). Эти побочные эффекты связаны с тем, что антагонисты NMDA блокируют не только чрезмерную активацию системы глутамат/NMDA, но и нарушают ее нормальное физиологическое функционирование. Поэтому представляется существенным на практике улучшить соотношение риск-польза клинически доступных антагонистов NMDA.
Когда тип боли, подлежащий лечению, является подходящим, например в случае артрита, соотношение риск-польза антагонистов NMDA может быть улучшено путем ограничения его действия на центральную нервную систему, например путем местного введения. Концентрация соединения в ткани-мишени, следовательно, является более высокой, чем его концентрация в крови, тем самым снижая риск токсичности. Поэтому исследовали эпидуральное и местное введение некоторых антагонистов NMDA. Было показано, что вводимый местно кетамин является эффективным при лечении невропатической боли, которая не проходила с помощью обычных лекарств. Также были изучены различные комбинации антагониста NMDA с одним или более других анальгетических средств при местном введении. Например, кетамин или другие антагонисты NMDA комбинировали с антидепрессантами или гипотензивными средствами (US 6387957), антиэпилептическими средствами (WO 03/061656, WO 98/07447, WO 99/12537, US 20040204366, WO 2010036937), адренэргическими антагонистами (US 20040101582) или опиоидами (WO 2000003716).
Учитывая важную роль, которую играют NMDA-рецепторы в ряде психиатрических и неврологических расстройств, они были предметом интенсивных исследований, и было описано множество антагонистов/блокаторов/модуляторов. Они могут быть классифицированы в широком смысле на три основные группы в зависимости от их сайта действия на NMDA-рецепторе. Таким образом, они включают в себя:
1) Конкурентные антагонисты, нацеленные либо на сайт связывания глутамата, например, селфотел, перзинфотел и пролекарства (WO 2009029618), либо на сайты связывания глицина, например гавестинел, GV-196771 (Wallace et al., 2002, Neurology, 59, 1694-1700), и хинолины, раскрытые в заявке на патент WO 2010037533. Эта категория также включает частичные агонисты глициновых сайтов, такие как D-циклосерин (US 2011160260).
2) Неконкурентные (или аллостерические) антагонисты, которые действуют на многие модуляторные сайты регуляции рецепторов, такие как, например, полиаминовые и фенилэтаноламиновые сайты. Соединения, принадлежащие к этому семейству, в настоящее время наиболее клинически изучены. Один из главных претендентов является ифенпродил (23210-56-2), и его более селективные производные к NMDA-рецептору, такие как, например, траксодопил, RGH-896, MK-0657, EVT-101 и EVT-103 в настоящее время проходят клинические испытания (Mony et al., 2009, Br. J. Pharmacol., 157, 1301-1317).
3) Неконкурентные антагонисты, блокаторы пор каналов. Это семейство препаратов, которые имели наибольший клинический успех, поскольку кетамин (Ketalar®, обезболивающее/анальгетик), декстрометорфан (Atuxane®, противокашлевое), мемантин (Ebixa®, против болезни Альцгеймера), амантадин (Mantadix®, противовирусное, затем противопаркинсоническое), фелбамат (Taloxa®, противосудорожное) являются коммерчески доступными. Фенциклидин (Sernyl®), разработанный как обезболивающее, был отозван с рынка, а дизоцилпин (МК-801) не является коммерчески доступным в качестве лекарства.
Соединения согласно настоящему изобретению относятся к этому последнему семейству неконкурентных антагонистов, которые блокируют каналы NMDA-рецепторов. Основным преимуществом соединений этого типа является то, что они не блокируют канал за исключением случаев, когда он открыт; поэтому они являются тем более эффективными, чем более чрезмерна активность NMDA-рецепторов. Также можно легко увидеть, что биофизические характеристики блокатора/антагониста, который влияет на частоту и продолжительность открытия канала, будут играть решающую роль в его фармакологической активности и соотношении риск-польза. Было клинически изучено несколько соединений этого типа, таких как, например, CNS-5161 (160754-76-7), нерамексан (219810-59-0), димирацетам (126100-97-8), V-3381 (1104525-45-2), NEU-2000 (640290-67-1). Другие находятся в доклинической стадии, среди которых в качестве примеров можно привести оксазолидины, заявленные в заявке на патент WO 2009092324, инданы (WO 2009069610), диарилэтиламины (WO 2010074647), арилциклогексиламины (WO 2010142890), аналоги кетамина и фенциклидина (Zarantonello et al., 2011, Bioorg. Med. Chem. Lett., 21, 2059-2063).
Настоящее изобретение касается производных общей формулы (1):
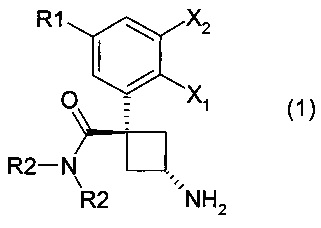
где:
- X1 представляет собой атом водорода или атом фтора;
- Х2 представляет собой атом водорода или атом фтора, или атом хлора;
- R1 представляет собой атом водорода или атом фтора, или атом хлора, или метильную группу, или метоксигруппу, или цианогруппу;
- R2 представляют собой, независимо или оба, метильную группу или этильную группу.
Предпочтительно соединения общей формулы (1) согласно изобретению являются такими, где:
- X1 представляет собой атом водорода или атом фтора;
- Х2 представляет собой атом водорода или атом фтора, или атом хлора;
- R1 представляет собой атом водорода или атом фтора, или атом хлора, или метильную группу, или метоксигруппу, или цианогруппу;
- R2 представляет собой этильную группу.
Соединения согласно изобретению могут существовать в виде чистых диастереоизомеров или в виде смесей диастереоизомеров. Более конкретно, настоящее изобретение относится к чистым диастереоизомерам, где 1-карбоксамидная группа и 3-аминогруппа находятся с противоположных сторон от плоскости, определяемой циклобутаном. Это стереохимическое положение указанных заместителей называется в настоящем изобретении "транс". Таким образом, изобретение касается чистых транс-диастереоизомеров следующих продуктов:
- транс-3-амино-N,N-диэтил-1-фенилциклобутанкарбоксамида,
- транс-3-амино-N,N-диметил-1-фенилциклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(2-фторфенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(3-метоксифенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(3-фторфенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(3-хлорфенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(3-метилфенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(3-цианофенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(2-фтор-3-хлорфенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(2,5-дифторфенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(3,5-дифторфенил)-циклобутанкарбоксамида,
- транс-3-амино-N,N-диэтил-1-(3,5-дихлорфенил)-циклобутанкарбоксамида,
а также их фармацевтически приемлемых солей.
Термин «чистые диастереоизомеры" означает, что «транс» диастереоизомер соединения общей формулы (1) содержит менее 5% «цис» диастереоизомера, то есть соединения, в котором 1-карбоксамидная группа и 3-аминогруппа находятся в одном полупространстве от плоскости, определяемой циклобутаном.
Термин "диастереоизомеры" означает в контексте настоящего изобретения стереоизомеры, которые не являются зеркальным отражением друг друга.
Термин "стереомеры" означает в контексте настоящего изобретения изомеры идентичного строения, но которые отличаются по расположению атомов в пространстве.
Наиболее близкий аналог представлен производными, описанными в заявке на патент WO 2003063797 и имеющими следующую формулу:
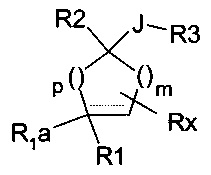
где:
m и p независимо равны 0, 1, 2 или 3;
пунктирная линия означает двойную связь, когда R1a отсутствует;
R1 может представлять собой группу NR6R7, где R6 и R7 могут представлять собой атом водорода;
R1a может представлять собой атом водорода;
R2 может представлять собой замещенную или незамещенную арильную группу;
J может представлять собой связь;
R3 может представлять собой группу -C(Z1)-R5, где R5 может представлять собой группу NR6aR7a;
R6a и R7a могут представлять собой замещенную или незамещенную алкильную группу, и Z1 возможно представляет собой карбонильную группу (С=O);
Rx может представлять собой одну или несколько замещенных или незамещенных групп, присоединенных ко всем доступным атомам углерода в кольце, а также атом водорода.
Следовательно, эта заявка на патент охватывает значительное количество соединений, большая часть которых относится к типу циклобутана (m=0 и p=1). В этой заявке на патент в качестве примеров представлено только четыре соединения из указанной группы соединений. Это касается соединений следующей формулы:
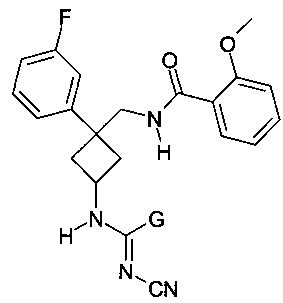
где G представляет собой группы NH2 или N(CH3)2, или NH(CH2CH3), или NH(CH2CHCH2).
Заявлено, что соединения этой заявки на патент являются ингибиторами тока, продуцируемого потенциал-зависимыми калиевыми каналами типа Kv1, в частности тока, продуцируемого изоформой Kv1.5.
Указано, что они полезны в широком диапазоне показаний, которые не включают лечение депрессии или боли.
Важно отметить, что соединения согласно настоящему изобретению не взаимодействуют с калиевыми каналами и, в частности, с каналами типа Kv1.5. Кроме того, обнаружено, что активность соединения согласно изобретению в качестве антагониста NMDA является очень чувствительной к структурным изменениям соединений формулы (1). Таким образом, активность антагониста NMDA подавляется, когда:
1) 1-карбоксамидная группа восстановлена в 1-аминометильную группу, как у циклобутановых соединений заявки на патент WO 2003/063797;
2) аминогруппа в положении 3 в циклобутане отличается от первичной аминогруппы (NH2). В заявке на патент WO 2003063797 3-аминогруппа замещена группой C(G)=NCN;
3) отсутствует «цис» стереохимия между 1-арильной группой и 3-аминогруппой. Фактически, когда 1-арильная группа и 3-аминогруппа находятся в «транс» положениях, соответствующие соединения не имеют сродства к NMDA-рецептору.
Уровень техники также представлен производными, описанными в заявке на патент WO 99/52848 и имеющими следующую формулу:
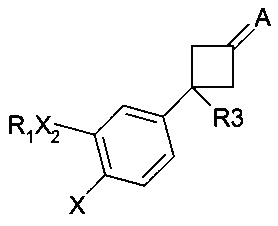
где:
X не является атомом водорода;
А может представлять собой группу NR7, где R7 не является атомом водорода;
R3 может представлять собой группу C(O)NR8R10, где R8 и R10 могут представлять собой С1-С4 алкильные цепи. Заявлено, что эти соединения являются селективными ингибиторами фосфодиэстераз 4 типа, полезными для лечения воспалительных и аутоиммунных заболеваний. Следовательно, соединения согласно настоящему изобретению отличаются от соединений, описанных в заявке WO 99/52848, как в смысле их химической структуры, так и в смысле их фармакологической активности.
Уровень техники также представлен производными, описанными в заявке на патент WO 2010/112597 и имеющими следующую формулу:
где:
 может представлять собой одинарную связь;
может представлять собой одинарную связь;
Ar представляет собой замещенную или незамещенную фенильную группу или пиридин-3-ил, замещенный одним или более атомами галогена или алкильными группами, или алкоксидными группами, или цианогруппой;
R1 и R2 могут, независимо или оба, представлять собой С1-С6 алкильную группу.
В отличие от соединений заявки на патент WO 2010/112597 соединения согласно настоящему изобретению не имеют сродства к сайтам обратного захвата серотонина и норадреналина. Соединения формулы (1), следовательно, отличаются от описанных в заявке WO 2010/112597, не только с точки зрения их химической структуры, но также с точки зрения их фармакологической активности.
Уровень техники, наконец, также представлен соединениями, описанными в заявке на патент WO 2000/051607 и имеющими следующую формулу:
где R12 и R13 представляют собой С1-С6 алкильную группу или С2-С6 алкенильную группу, или С2-С6 алкинильную группу, замещенную или незамещенную.
Указанные производные являются модуляторами хемокинов, полезными для профилактики или лечения некоторых воспалительных или иммунных заболеваний. Здесь снова соединения согласно настоящему изобретению, таким образом, отличаются от описанных в заявке WO 2000/051607, с точки зрения их химической структуры и их фармакологической активности.
Настоящее изобретение также охватывает соли производных общей формулы (1) с фармацевтически приемлемыми органическими или минеральными кислотами. В настоящем изобретении термин "фармацевтически приемлемый" относится к молекулярным фрагментам и композициям, которые не имеют негативного или аллергического эффекта, или любой нежелательной реакции при введении человеку.
При использовании в данном описании термин "фармацевтически приемлемый эксципиент" включает любые разбавители, адъюванты или эксципиенты, такие как консерванты, наполнители, дезинтегрирующие агенты, смачивающие агенты, эмульгаторы, диспергирующие агенты, антибактериальные или противогрибковые агенты, или даже агенты, которые помогают замедлить поглощение и резорбцию в кишечнике и пищеварительном тракте. Использование этих сред или носителей хорошо известно специалистам в данной области. Термин "фармацевтически приемлемые соли" соединения относится к солям, определенным здесь, которые обладают фармакологической активностью исходного соединения. Такие соли включают кислотно-аддитивные соли, образованные с минеральными кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и тому подобное, или образованные с органическими кислотами, такими как уксусная кислота, бензолсульфоновая кислота, бензойная кислота, камфорсульфоновая кислота, лимонная кислота, этансульфоновая кислота, фумаровая кислота, глюкогептоновая кислота, глюконовая кислота, глутаминовая кислота, гликолевая кислота, гидроксинафтоевая кислота, 2-гидроксиэтансульфоновая кислота, молочная кислота, малеиновая кислота, яблочная кислота, миндальная кислота, метансульфоновая кислота, муконовая кислота, 2-нафталинсульфоновая кислота, пропионовая кислота, салициловая кислота, янтарная кислота, дибензоил-L-винная кислота, винная кислота, п-толуолсульфоновая кислота, триметилуксусная кислота, трифторуксусная кислота и т.п.
Фармацевтически приемлемые соли также включают аддитивные формы с растворителем (сольваты) или кристаллические формы (полиморфы), такие как определено в данном описании, тех же кислотно-аддитивных солей.
Настоящее изобретение также охватывает соединения формулы (1) и их фармацевтически приемлемые соли для применения в качестве лекарственного средства.
Настоящее изобретение относится к соединениям формулы (1) и их фармацевтически приемлемым солям для применения в качестве антагонистов NMDA-рецепторов.
Настоящее изобретение также относится к соединениям формулы (1) и их фармацевтически приемлемым солям для применения в качестве лекарственного средства для лечения и/или профилактики депрессии.
Настоящее изобретение также относится к соединениям формулы (1) и их фармацевтически приемлемым солям для применения в качестве лекарственного средства для лечения боли, особенно боли, вызванной чрезмерной ноцицепцией, невропатической боли и боли смешанного типа.
Среди типов боли, потенциально чувствительных к действию соединений общей формулы (1), можно привести, в частности, в качестве не ограничивающих примеров:
- периферийную или центральную невропатическую боль в результате нервных поражений травматического происхождения (например инсульт), метаболического происхождения (например сахарный диабет), инфекционного происхождения (например ВИЧ, опоясывающий лишай, герпес), невралгию тройничного нерва, боль из-за химиотерапии и/или лучевой терапии;
- воспалительную боль, например, ревматоидный артрит, септический артрит, остеоартрит, полиартрит, подагра, спондилоартрит, острый ревматизм, внесуставную висцеральную боль, например синдром раздраженного кишечника, болезнь Крона;
- боль из-за чрезмерной ноцицепции, такую как посттравматическая боль, послеоперационная боль, ожоги, скручивания/растяжения, почечная или печеночная колика, боль в суставах, артрит, спондилоартропатии;
- боль смешанного типа, такую как боль, вызванная раком, боль в спине и пояснице, или другие типы боли, которые трудно классифицировать, например головные боли, фибромиалгии, боли, связанные с сосудистыми/ишемическими проблемами, такими как стенокардия, болезнь Рейно.
Настоящее изобретение также относится к соединениям формулы (1) и их фармацевтически приемлемым солям для применения в качестве лекарственного средства для лечения и/или профилактики воспаления суставов. Среди видов воспаления, потенциально чувствительных к действию соединения общей формулы (1), можно привести, в частности, в качестве не ограничивающих примеров: воспалительный моноартрит, ревматоидный артрит, септический артрит, остеоартрит ревматоидный полиартрит, подагра, спондилоартрит, острый внесуставной ревматизм.
Настоящее изобретение, кроме того, относится к фармацевтической композиции, характеризующейся тем, что она содержит по меньшей мере одно соединение общей формулы (1) или одну из его фармацевтически приемлемых солей в качестве активного ингредиента.
Настоящее изобретение также относится к фармацевтической композиции для применения в качестве лекарственного средства для лечения и/или профилактики депрессии.
Настоящее изобретение также относится к фармацевтической композиции для применения в качестве лекарственного средства для лечения боли, особенно боли, вызванной чрезмерной ноцицепцией, невропатической боли и боли смешанного типа.
Фармацевтические композиции в соответствии с настоящим изобретением могут быть приготовлены для введения человеку. Эти композиции получают таким образом, что они могут быть введены перорально, сублингвально, подкожно, внутримышечно, внутривенно, локально, трансдермально или ректально. В этом случае активный ингредиент может быть введен в виде единичных форм введения, смешанных с обычными фармацевтическими носителями для человека. Подходящие единичные формы введения включают формы для перорального введения, такие как таблетки, капсулы, порошки, гранулы и пероральные растворы или суспензии, сублингвальные и буккальные формы введения, подкожные, местные, внутривенные, внутримышечные, интраназальные или внутриглазные формы введения и ректальные формы введения.
Предпочтительно фармацевтическая композиция согласно настоящему изобретению приготовлена для введения пероральным или местным путем. Местное введение является предпочтительным способом для лечения определенных типов боли, например, такой как суставная боль.
Термин «местное введение» относится к местному введению на кожу или слизистую оболочку.
Подходящие композиции для выбранной формы введения известны специалистам в данной области и описаны, например, в издании Remington, The Science и Practice of Pharmacy, 19th edition, 1995, Mack Publishing Company.
Когда готовят твердую композицию в форме таблеток, активный ингредиент смешивают с фармацевтическим носителем, таким как желатин, крахмал, лактоза, стеарат магния, тальк, гуммиарабик, диоксид кремния или тому подобное. Таблетки могут быть покрыты сахарозой или другими подходящими материалами, или они могут быть обработаны таким образом, что они будут иметь пролонгированное или замедленное действие, и будут высвобождать заданное количество активного ингредиента в непрерывном режиме.
Композицию в форме капсулы получают путем смешивания активного ингредиента с разбавителем и выливания полученной смеси в мягкие или твердые капсулы.
Композиция в форме сиропа или эликсира может содержать активный ингредиент вместе с подсластителем и антисептиком, а также ароматизатором и подходящим красителем.
Порошки или гранулы, диспергируемые в воде, могут содержать активный ингредиент в смеси с диспергирующими агентами или смачивающими агентами, или суспендирующими агентами, а также с корригентами аромата или подсластителями.
Для ректального введения используют суппозитории, которые получают со связующими агентами, которые растворяются при ректальной температуре, например с маслом какао или полиэтиленгликолями.
Для парентерального (внутривенного, внутримышечного, внутрикожного, подкожного), интраназального или внутриглазного введения используют водные суспензии, изотонические солевые растворы или стерильные и инъекционные растворы, которые содержат диспергирующие агенты и/или фармакологически приемлемые смачивающие агенты.
Активные ингредиенты могут быть также приготовлены в виде микрокапсул, возможно с одной или более добавок-носителей, если это необходимо.
Местное введение фармацевтической композиции можно осуществить путем нанесения раствора, дисперсии, геля, лосьона, молочка, мази, мазеобразного крема, капель или другого носителя, используемого для местного нанесения и хорошо известного специалисту в данной области техники. Один возможный способ представляет собой введение фармацевтической композиции посредством аэрозоля, распыляющего мелкие капли жидкости для распределения по всей поверхности, подлежащей обработке, или наоборот для ограничения распределения по точно определенной зоне, подлежащей обработке, или в твердой форме, такой как карандаш. Другим примером является пластырь или лента, которая обеспечивает непрерывное высвобождение местной композиции. Пластырь может быть резервуаром или пористой мембраной, или твердой матрицей, хорошо известной специалисту в данной области техники. Также можно использовать другие способы введения, такие как ионофорез или электропорация.
Композиции, описанные в данном изобретении, могут также включать ингредиенты или соединения, обычно смешиваемые в таких местных композициях, например, композиции могут также включать дополнительные ингредиенты, такие как носители, увлажнители, масла, жиры, воски, поверхностно-активные вещества, загустители, антиоксиданты, стабилизаторы вязкости, хелатирующие агенты, буферы, консерванты, отдушки, красители, влагоудерживающие средства, смягчающие, диспергирующие агенты, солнцезащитные кремы с соединениями, блокирующими излучение, и особенно с УФ-блокаторами, антибактериальные, противогрибковые, дезинфицирующие средства, витамины, антибиотики или другие средства против акне, а также другие адаптированные вещества, не имеющие вредного негативного влияния на активность местной композиции. Например, можно использовать дополнительные ингредиенты, такие как кислый фосфат натрия, экстракт гамамелиса, глицерин, абрикосовое масло, кукурузное масло. В дополнение к соединениям, описанным выше, композиции согласно настоящему изобретению могут необязательно содержать другие ингредиенты. Например, можно добавлять триэтаноламин в качестве агента, образующего сетчатую структуру. Также можно добавлять консервант, такой как бутилированный гидрокситолуол. Также можно добавлять другие агенты, снижающие раздражение, включая, но не ограничиваясь глицерином. Композиции для местного введения могут содержать обычные смягчающие вещества и эмульгаторы, включая альгинаты, стеарат глицерина, ПЭГ-100-стеарат, кетиловый спирт, пропилпарабен, бутилпарабен, сорбитолы, этоксилированный ангидросорбитолмоностеарат (Tween), белый вазелин (вазелин), триэтаноламин, масло эму, алоэ вера, ланолин, масло какао и другие экстракты.
Описанные композиции можно наносить на область кожи пациента, подлежащего лечению. Частота нанесения будет зависеть от обстоятельств и пациента. Например, композиции можно наносить один раз в день, два раза в день или даже чаще.
Дозы соединения общей формулы (1) или одной из его фармацевтически приемлемых солей в композиции по изобретению можно регулировать, чтобы получить количество вещества, которое является эффективным для достижения требуемого терапевтического ответа для композиции, которая является специфичной для способа введения. Эффективная доза соединения согласно изобретению варьирует в зависимости от многих параметров, таких как, например, выбранный способ введения, вес, возраст, пол, вид заболевания, чувствительность пациента, подлежащего лечению. Следовательно, оптимальная дозировка может быть установлена специалистами в данной области в зависимости от параметров, которые специалист считает релевантными. Хотя эффективные дозы могут варьировать в широких пропорциях, суточные дозы можно масштабировать в диапазоне от 1 мг до 1000 мг за 24 ч на взрослого человека среднего веса 70 кг на одну или более разделенных доз.
Наконец, изобретение включает способ синтеза соединений общей формулы (1), а также промежуточных соединений формулы (С) и (D).
Соединения общей формулы (1) могут быть получены способом, описанным ниже на схеме реакции.
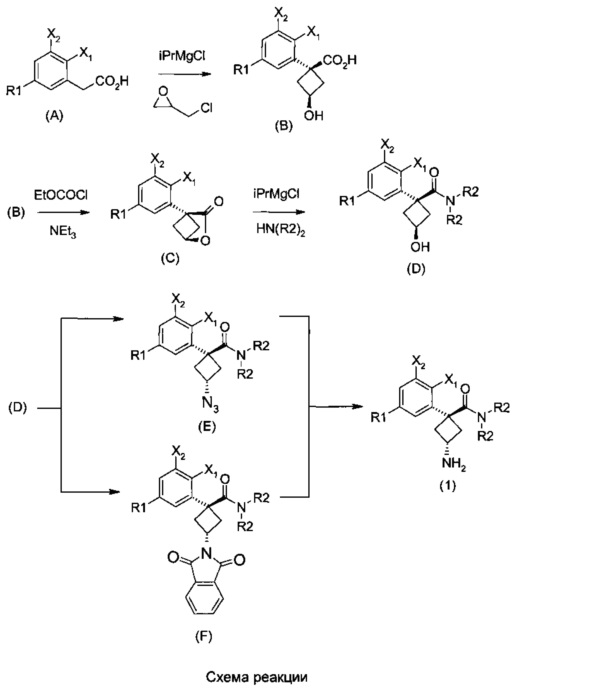
При получении соединений согласно изобретению в качестве исходного материала используют производные бензолуксусной кислоты формулы (А), которые имеются в продаже, такие как бензолуксусная кислота (RN 103-82-2); 2-фторбензолуксусная кислота (RN 451-82-1); 3-фторбензолуксусная кислота (RN 331-25-9); 3-хлорбензолуксусная кислота (RN 1878-65-5); 3-метилбензолуксусная кислота (RN 621-36-3); 3-цианобензолуксусная кислота (RN 1878-71-3); 3-метоксибензолуксусная кислота (RN 1798-09-0); 2,5-дифторбензолуксусная кислота (RN 85068-27-5); 3,5-дифторбензолуксусная кислота (RN 105184-38-1); 3,5-дихлорбензолуксусная кислота (RN 51719-65-4); 2-фтор-3-хлорбензолуксусная кислота (RN 261762-96-3). Производные формулы (А) конденсируют с эпихлоргидрином в соответствии со способом, который адаптирован, исходя из способа, описанного в заявке на патент WO 2007/038452, для получения производных формулы (В), где группы спирта и карбоновой кислоты имеют «цис» стереохимию. Указанный патент не описывает промежуточных продуктов формулы (В). Лактоны формулы (С) затем получают из производных формулы (В) с помощью обычного способа активации кислотной группы, например такого, где используют алкилхлорформиат, как описано в заявке WO 2008/092955. Открытие лактона формулы (С) затем предпочтительно проводят с использованием магниевой соли соответствующего вторичного амина согласно Williams et al. (Tetrahedron Lett., 1995, 36, 5461-5464) с получением соответствующего карбоксамида формулы (D). Введение первичной аминогруппы в положении 3 циклобутана с инверсией стереохимии может быть достигнуто через промежуточный азид формулы (Е) в соответствии с Soltani Rad et al. (Tetrahedron Lett., 2007, 48, 3445-3449). Восстановление азидогруппы в соответствующий первичный амин затем проводят или путем каталитического гидрирования, или путем реакции Штаудингера. Альтернативно превращение соединения формулы (D) в амин формулы (1) может быть осуществлено через промежуточный фталимид формулы (F) в соответствии с обычным способом Габриэля (см., например, WO 2006081179).
Следующие примеры иллюстрируют изобретение без ограничения. В примерах ниже:
i. различные кристаллические формы могут привести к различным температурам плавления; температуры плавления, приведенные в этой заявке, являются температурами плавления продуктов, полученных способами, которые описаны и не корректированы;
ii. структура продуктов, полученных согласно изобретению, подтверждена с помощью спектров ядерного магнитного резонанса (ЯМР) и масс-спектрометрией; чистота конечного продукта проверена с помощью ТСХ и процентного анализа;
iii. спектры ЯМР записаны в указанном растворителе: химические сдвиги (δ) выражены в частях на миллион (ppm) относительно тетраметилсилана; мультиплетность сигналов обозначена: s - синглет; d - дублет; t - триплет; q - квадруплет; qu - квинтуплет, m - мультиплет, l - большой;
iv. различные символы для единиц измерения имеют обычный смысл: мкг (микрограмм); мг (миллиграмм); г (грамм); мл (миллилитр); мВ (милливольт); °С (градус Цельсия); ммоль (миллимоль), нмоль (наномоль); см (сантиметр); нм (нанометр); мин (минута); мс (миллисекунда), Гц (герц);
v. аббревиатуры имеют следующие значения: т.пл. (температура плавления); т.кип. (температура кипения);
vi. термин "температура окружающей среды" относится к температуре между 20°С и 25°С.
Пример 1: транс-3-амино-N,N-диэтил-1-фенилциклобутанкарбоксамид (1а1)
Стадия 1: цис-1-фенил-3-гидрокси-циклобутанкарбоновая кислота (В1)
Помещают 2,2 экв изопропилмагнийхлорида в трехгорлую колбу и охлаждают реакционную среду до 0°С. Добавляют 1 экв фенилуксусной кислоты, разведенной в ТГФ; температура должна быть между 40 и 50°С. Охлаждают среду до 20°С и добавляют 1,8 экв эпихлоргидрина; температура должна быть между 20 и 25°С, и перемешивают при этой температуре в течение 45 мин. Затем добавляют 2 экв изопропилмагнийхлорида (2М в ТГФ) и перемешивают при комнатной температуре в течение 2 ч. Затем нагревают реакционную среду до 60°С в течение 19 ч. Дают среде остыть, затем подкисляют раствором HCl (1Н) до pH 1. Добавляют дихлорметан (ДХМ) и экстрагируют. Декантируют, органическую фазу сушат над MgSO4, затем выпаривают ДХМ при пониженном давлении. Остаток очищают флэш-хроматографией со следующим элюентом: ДХМ, затем ДХМ/метанол 70:30. Продукт, указанный в заголовке, получают в виде бледно-желтого твердого вещества (выход = 70%).
С11Н12О3 (молекулярная масса (MB) = 192).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.50 (m, 2Н), 2.74 (t, 2Н, J=9.4 Гц), 3.32 (s, 1Н), 3.85 (qu, 1Н, J=7.2 Гц), 7.22-7.38 (m, 5Н), 12.21 (s, 1Н).
SM-ESI (масс-спектрометрия с ионизацией электрораспылением): 193.1 (МН+).
Стадия 2: 4-фенил-2-оксабицикло[2.1.1]гексан-3-он (С1)
Помещают 1 экв соединения В1) в колбу, разбавляют в ТГФ и 1,03 экв триэтиламина. Перемешивают при комнатной температуре до полного растворения, затем охлаждают реакционную среду до 0°С. Добавляют 1 экв этилхлорформиата и перемешивают при этой температуре в течение 1 ч, затем доводят до комнатной температуры и перемешивают в течение 20 ч. Выпаривают ТГФ при пониженном давлении, отбирают остаток этилацетатом (AcOEt). Декантируют, сушат ацетат над MgSO4, затем выпаривают при пониженном давлении. Остаток очищают флэш-хроматографией со следующим элюентом: гептан, затем гептан/этилацетат 60:40. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 87%).
С11Н10О2 (MB = 174).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 2.71 (m, 2Н), 2.89 (m, 2Н), 4.97 (s, 1Н), 7.31-7.42 (m, 5Н).
SM-ESI: 175 (МН+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-фенилциклобутанкарбоксамид (D1a)
Помещают 1 экв соединения (С1), 2 экв диэтиламина и ТГФ в трехгорлую колбу. Охлаждают реакционную среду до -20°С, затем добавляют по каплям 3 экв изопропилмагнийхлорида (2М в ТГФ), поддерживая температуру ниже -5°С. Смесь перемешивают в течение 2 ч при температуре между -10 и -20°С. Гидролизуют реакционную среду насыщенным раствором NaCl, затем добавляют раствор HCl (1Н) и экстрагируют этилацетатом. Органическую фазу сушат над MgSO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией со следующей смесью в качестве элюента: ДХМ/метанол 85:15. Продукт, указанный в заголовке, получают в виде бледно-желтого твердого вещества (выход = 99%).
C15H21NO2 (MB = 247).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.63 (t, 3Н, J=7.2 Гц), 1.08 (t, 3Н, J=7.2 Гц), 2.72 (m, 2Н), 2.82 (m, 2Н), 2.90 (q, 2Н, J=7.2 Гц), 3.21 (q, 2Н, J=7.2 Гц), 4.36 (qu, 1Н, J=7.4 Гц), 7.21-7.36 (m, 5Н). Сигнал, соответствующий Н в ОН, в спектре не виден.
SM-ESI: 248 (МН+).
Стадия 4: транс-3-азидо-N,N-диэтил-1-фенилциклобутанкарбоксамид (Е1а)
Помещают 1 экв соединения (D1a), 1,5 экв N-(п-толуолсульфонил)имидазола, 2 экв триэтиламина, 0,025 экв иодида тетрабутиламмония, 3 экв азида натрия и ДМФ в колбу. Перемешивают и нагревают реакционную среду при температуре 160°С в течение 4 ч. Выливают реакционную среду на ледяную воду и экстрагируют этиловым эфиром. Органическую фазу сушат над MgSO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией со следующей смесью в качестве элюента: гептан/AcOEt 70:30. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход=65%).
C15H20N4O (MB = 272).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.52 (t, 3Н, J=7.2 Гц), 1.11 (t, 3Н, J=7.2 Гц), 2.47 (m, 2Н), 2.89 (q, 2Н, J=7.2 Гц), 3.14 (m, 2Н), 3.34 (q, 2Н, J=7.2 Гц), 3.96 (qu, 1Н, J=7.8 Гц), 7.23 (m, 3Н), 7.35 (m, 2Н).
SM-ESI: 273 (М+Н+).
Стадия 5: транс-3-амино-N,N-диэтил-1-фенилциклобутанкарбоксамид (1а1)
Растворяют 1 экв соединения (Е1а) в метаноле в колбе. Раствор дегазируют в течение 30 мин азотом, затем добавляют Pd/C (20 масс. %). Систему продувают (цикл: вакуум/газообразный Н2) и гидрируют реакционную среду в течение 3 ч при комнатной температуре при перемешивании. Катализатор отфильтровывают и выпаривают растворитель. Осадок очищают флэш-хроматографией со следующей смесью в качестве элюента: ДХМ/метанол/NH4OH: 90:9:1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 70%).
C15H22N2O (MB = 246).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.50 (t, 3Н, J=7.2 Гц), 1.10 (t, 3Н, J=7.2 Гц), 2.11 (m, 2Н), 2.92 (q, 2Н, J=6.8 Гц), 3.12 (m, 2Н), 3.32 (q, 2Н, J=6.8 Гц), 3.46 (qu, 1Н, J=8.0 Гц), 7.18-7.35 (m, 5Н). Сигнал, соответствующий Н в NH2, в спектре не виден.
SM-ESI: 247 (МН+).
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 185°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.42 (t, 3Н, J=7.0 Гц), 1.02 (t, 3Н, J=7.0 Гц), 2.56 (m, 2Н), 2.85-2.96 (m, 4Н), 3,25 (q, 2Н, J=6.8 Гц), 3.54 (qu, 1Н, J=8.4 Гц), 6.03 (s, 2Н), 7.26 (m, 3Н), 7.39 (t, 2Н, J=7.6 Гц), 8.00 (s, 2Н). Сигнал, соответствующий Н в NH2, в спектре не виден.
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 12.02, 12.15, 36.93, 39.19, 40.07, 41.19, 46.61, 124.87, 126.51, 128.71, 136.02, 142.69, 167.19, 171.10.
% Теоретический: С 62.97, Н 7.23, N 7.73.
% Обнаруженный: С 63.00, Н 7.17, N 7.78.
Пример 2: транс-3-амино-N,N-диметил-1-фенилциклобутанкарбоксамид (1а2)
Стадия 3: цис-3-тдрокси-N,N-диметил-1-фенилциклобутанкарбоксамид (D1b)
Идентична стадии 3, описанной в Примере 1, при использовании диметиламина вместо диэтиламина. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 89%).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 2.65 (m, 2Н), 2.55 (s, 3Н), 2.95 (s, 3Н), 2.80 (m, 2H), 4.27 (qu, 1H, J=7.8 Гц), 7.19-7.35 (m, 5H). Сигнал, соответствующий Н в ОН, в спектре не виден.
Стадия 4: транс-3-азидо-N,N-диметил-1-фенилциклобутанкарбоксамид (E1b)
Идентична стадии 4, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бежевого твердого вещества (выход = 95%).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 2.50 (m, 2Н), 2.54 (s, 3Н), 2.96 (s, 3Н), 3.18 (m, 2Н), 3.97 (qu, 1Н, J=7.8 Гц), 7.24 (m, 3Н), 7.36 (m, 2Н).
Стадия 5: транс-3-амино-N,N-диметил-1-фенилциклобутанкарбоксамид (1а2)
Идентична стадии 5, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 84%).
C13H18N2O (MB = 218).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 2.13 (m, 2Н), 2.55 (s, 3Н), 2.95 (s, 3Н), 3.15 (m, 2Н), 3.47 (qu, 1Н, J=7.8 Гц), 7.19-7.35 (m, 5Н). Сигнал, соответствующий в NH2, в спектре не виден.
SM-ESI: 219 (МН+).
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 163°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.51 (m, 5Н), 2.86 (s, 3Н), 2.98 (m, 2Н), 3.36 (s, 1Н), 3.53 (qu, 1Н, J=8.4 Гц), 6.03 (s, 2Н), 7.26 (m, 3Н), 7.39 (t, 2Н, J=7.6 Гц), 8.05 (s, 3Н).
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 35.80, 37.20, 37.34, 39.91, 46.54, 124.94, 126.59, 128.72, 136.00, 142.43, 167.15, 171.68.
% Теоретический: С 61.07, Н 6.63, N 8.38.
% Обнаруженный: С 60.73, Н 6.43, N 8.15.
Пример 3: транс-3-амино-N,N-диэтил-1-(2-фторфенил)-циклобутанкарбоксамид (1b)
Стадия 1: цис-3-гидрокси-1-(2-фторфенил)-циклобутанкарбоновая кислота (В2)
Идентична стадии 1, описанной в Примере 1, при использовании 2-фторфенилуксусной кислоты в качестве исходного продукта. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 49%).
C11H11FO3 (MB = 210).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 2.80 (m, 2Н), 2.97 (m, 2Н), 4.29 (qu, 1Н, J=6.4 Гц), 7.04-7.23 (m, 4Н). Сигналы, соответствующие Н в ОН в спирте и кислоте, в спектре не видны.
SM-ESI: 211 (МН+).
Стадия 2: 4-(2-фторфенил)-2-оксабицикло[2.1.1]гексан-3-он (С2)
Идентична стадии 2, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 81%).
C11H9FO2 (MB = 192).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 2.75 (m, 2Н), 2.99 (m, 2Н), 5.01 (s, 1Н), 7.07-7.42 (m, 4Н).
SM-ESI: = 193 (МН+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-(2-фторфенил)-циклобутанкарбоксамид (D2a)
Идентична стадии 3 Примера 1. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 85%).
C15H20NO2F (MB = 265).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.47 (t, 3Н, J=6.8 Гц), 1.10 (t, 3Н, J=6.8 Гц), 2.77-2.89 (m, 4Н), 2.95 (m, 2Н), 3.31 (m, 2Н), 4.32 (qu, 1Н, J=6.8 Гц), 7.04 (t, 1Н, J=7.8 Гц), 7.15 (t, 1Н, J=7.8 Гц), 7.26 (m, 1Н), 7.37 (t, 1Н, J=7.8 Гц). Сигнал, соответствующий Н в ОН, в спектре не виден.
SM-ESI: 266 (МН+).
Стадия 4: транс-3-азидо-N,N-диэтил-1-(2-фторфенил)-циклобутанкарбоксамид (Е2а)
Идентична стадии 4, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 75%).
C15H19N4OF (MB = 290).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.42 (t, 3Н, J=7.0 Гц), 1.10 (t, 3Н, J=7.0 Гц), 2.55 (m, 2Н), 2.98 (q, 2Н, J=7.0 Гц), 3.19 (m, 2Н), 3.31 (q, 2Н, J=7.0 Гц), 4.02 (qu, 1Н, J=8.0 Гц), 7.03 (m, 1Н), 7.14-7.29 (m, 3Н).
SM-ESI: 291 (МН+).
Стадия 5: транс-3-амино-N,N-диэтил-1-(2-фторфенил)-циклобутанкарбоксамид (1b)
Идентична стадии 5, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 90%).
C15H21N2OF (MB = 264).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.42 (t, 3Н, J=6.8 Гц), 1.10 (t, 3Н, J=6.8 Гц), 2.19 (m, 2Н), 3.00 (q, 2H, J=6.8 Гц), 3.17 (m, 2H), 3.31 (q, 2H, J=6.8 Гц), 3.53 (qu, 1H, J=8.0 Гц), 7.00 (m, 1H), 7.11-7.31 (m, 3Н). Сигнал, соответствующий Н в NH2, в спектре не виден.
SM-ESI: 265 (МН+).
Малеат соединения, указанного в заголовке.
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 193°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.01 (t, 3Н, J=6.8 Гц), 0.77 (t, 3Н, J=6.8 Гц), 2.36 (m, 2Н), 2.72 (m, 4Н), 2.97 (q, 2Н, J=6.8 Гц), 3.22 (s, 1Н), 3.38 (qu, 1Н, J=8.0 Гц), 5.81 (s, 2Н), 6.94 (m, 1Н), 7.04-7.14 (m, 2Н), 7.33 (m, 1Н), 7.75 (s, 3Н).
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 12.00, 12.20, 36.14, 39.97, 40.60, 41.19, 43.60, 115.71 (d, 2JC-F = 21 Гц), 124.64 (d, 4JC-F = 4 Гц), 128.00 (d, 3JC-F = 5 Гц), 128.80 (d, 3FC-F = 8 Гц), 130.07 (d, 2JC-F = 13 Гц), 136.04, 158.52, 160.96, 167.14, 169.93.
% Теоретический: С 59.99, H 6.62, N 7.36.
% Обнаруженный: С 60.15, Н 6.48, N 7.20.
Пример 4: транс-3-аминс-N,N-диэтил-1-(3-фторфенил)-циклобутанкарбоксамид (1с)
Стадия 1: цис-3-гидрокси-1-(3-фторфенил)-циклобутанкарбоновая кислота (В3)
Идентична стадии 1 Примера 1 при использовании 3-фторфенилуксусной кислоты в качестве исходной кислоты. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 52%).
C11H11FO3 (MB = 210).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.50 (m, 2Н), 2.75 (m, 2Н), 3.86 (qu, 1Н, J=7.2 Гц), 5.18 (s, 1Н), 7.07-7.21 (m, 3Н), 7.40 (m, 1Н), 12.40 (s, 1Н).
SM-ESI: 211 (МН+).
Стадия 2: 4-(3-фторфенил)-2-оксабицикло[2.1.1]гексан-3-он (С3)
Идентична стадии 2, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 91%).
C11H9O2F (MB = 192).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.83 (s, 4Н), 5.09 (s, 1Н), 7.15-7.22 (m, 3Н), 7.38-7.47 (m, 1Н).
SM-ESI: 193 (МН+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-(3-фторфенил)-циклобутанкарбоксамид (D3a)
Идентична стадии 3 Примера 1. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 92%).
C15H20NO2F (MB = 265).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.62 (t, 3Н, J=7.2 Гц), 0.97 (t, 3Н, J=7.2 Гц), 2.50 (m, 2Н), 2.66 (m, 2Н), 2.86 (q, 2Н, J=7.2 Гц), 3.19 (q, 2Н, J=7.2 Гц), 4.05 (m, 1Н), 5.12 (d, 1Н, J=6.8 Гц), 7.04-7.15 (m, 3Н), 7.39 (m, 1Н).
SM-ESI: 266 (МН+).
Стадия 4: транс-3-азидо-N,N-диэтил-1-(3-фторфенил)-циклобутанкарбоксамид (Е3а)
Идентична стадии 4, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 72%).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.59 (t, 3Н, J=7.2 Гц), 1.00 (t, 3Н, J=7.2 Гц), 2.40 (m, 2Н), 2.86 (q, 2Н, J=7.2 Гц), 3.04 (m, 2Н), 3.24 (q, 2Н, J=7.2 Гц), 4.07 (m, 1Н), 7.04-7.15 (m, 3Н), 7.39 (m, 1Н).
Стадия 5: транс-3-амино-N,N-диэтил-1-(3-фторфенил)-циклобутанкарбоксамид (1с)
Идентична стадии 5, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 93%).
C15H21N2OF (MB = 264).
SM-ESI: 265 (МН+).
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 174°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.49 (t, 3Н, J=7.0 Гц), 1.02 (t, 3Н, J=7.0 Гц), 2.56 (m, 2Н), 2.91 (m, 4Н), 3.26 (q, 2Н, J=7.0 Гц), 3.35 (s, 1Н), 3.53 (qu, 1Н, J=8.4 Гц), 6.03 (s, 2Н), 7.01 (d, 1Н, J=8.0 Гц), 7.09-7.20 (m, 2Н), 7.42 (m, 1Н), 7.99 (s, 3Н).
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 12.10, 36.93, 39.23, 39.91, 41.18, 46.40, 111.03 (d, 2JC-F = 22 Гц), 113.39 (d, 2JC-F = 21 Гц), 121.11 (d, 4JC-F = 2 Гц), 130.77 (d, 3JC-F = 9 Гц), 136.02, 145.55 (d, 3JC-F = 7 Гц), 161.21, 163.64, 167.14, 170.60.
% Теоретический: С 59.99, Н 6.62, N 7.36.
% Обнаруженный: С 59.11, Н 6.40, N 7.07.
Пример 5: транс-3-амино-N,N-диэтил-1-(3-метоксифенил)-циклобутанкарбоксамид (1d)
Стадия 1: цис-3-гидрокси-1-(3-метоксифенил)-циклобутанкарбоновая кислота (В4)
Идентична стадии 1 Примера 1 при использовании 3-метоксифенилуксусной кислоты вместо фенилуксусной кислоты. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 50%).
C12H14O4 (MB = 222).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.50 (m, 2Н), 2.73 (m, 2Н), 3.75 (s, 3Н), 3.86 (qu, 1Н, J=7.2 Гц), 5.14 (s, 1Н), 6.82 (dd, 1Н, J=8.0 Гц и J=2.0 Гц), 6.87 (s, 1Н), 6.93 (d, 1Н, J=8.0 Гц), 7.26 (t, 1Н, J=8.0 Гц), 12.23 (s, 1Н).
SM-ESI: 222.
Стадия 2: 4-(3-метоксифенил)-2-оксабицикло[2.1.1]гексан-3-он (С4)
Идентична стадии 2, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 85%).
C12H12O2 (MB = 188).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.80 (m, 4Н), 3.76 (s, 3Н), 5.06 (s, 1Н), 6.87-6.91 (m, 3Н), 7.30 (t, 1Н, J=8.0 Гц).
SM-ESI: 189 (МН+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-(3-метоксифенил)-циклобутанкарбоксамид (D4a)
Идентична стадии 3, описанной для Примера 1. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 92%).
C16H23NO3 (MB = 277).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.62 (t, 3Н, J=6.8 Гц), 0.97 (t, 3Н, J=6.8 Гц), 2.50 (m, 2Н), 2.64 (m, 2Н), 2.86 (q, 2Н, J=6.8 Гц), 3.19 (q, 2Н, J=6.8 Гц), 3.73 (s, 3Н), 4.06 (se, 1Н, J=7.6 Гц), 5.08 (d, 1Н, J=7.2 Гц), 6.81 (m, 2Н), 6.89 (d, 1Н, J=7.6 Гц), 7.27 (m, 1Н).
SM-ESI: 278 (МН+).
Стадия 4: транс-3-азидо-N,N-диэтил-1-(3-метоксифенил)-циклобутанкарбоксамид (Е4а)
Идентична стадии 4, описанной для Примера 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 82%).
C16H22N4O2 (MB = 302).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.59 (t, 3Н, J=6.8 Гц), 1.00 (t, 3Н, J=6.8 Гц), 2.40 (m, 2Н), 2.86 (q, 2Н, J=6.8 Гц), 3.05 (m, 2Н), 3.24 (q, 2H, J=6.8 Гц), 3.74 (s, 3Н), 3.95 (qu, 1H, J=7.6 Гц), 6.76 (m, 1H), 6.83 (m, 2H), 7.30 (m, 1H).
SM-ESI: 303 (MH+).
Стадия 5: транс-3-амино-N,N-диэтил-1-(3-метоксифенил)-циклобутанкарбоксамид (1d)
Идентична стадии 5, описанной для Примера 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 88%).
C16H24N2O2 (MB = 276).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.50 (t, 3Н, J=7.0 Гц), 1.00 (t, 3Н, J=7.0 Гц), 2.02 (m, 2Н), 2.83 (m, 4Н), 3.10 (qu, 1Н, J=8.0 Гц), 3.23 (q, 2Н, J=7.2 Гц), 3.33 (s, 2Н), 3.73 (s, 3Н), 6.73-6.79 (m, 3Н), 7.25 (t, 1Н, J=8.0 Гц).
SM-ESI: 277 (МН+).
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 156°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.47 (t, 3Н, J=6.8 Гц), 1.02 (t, 3Н, J=6.8 Гц), 2.55 (m, 2Н), 2.89 (m, 4Н), 3.26 (q, 2Н, J=6.8 Гц), 3.35 (s, 1Н), 3.52 (qu, 1Н, J=8.4 Гц), 3.75 (s, 3Н), 6.03 (s, 2Н), 6.78-6.86 (m, 3Н), 7.31 (t, 1Н, J=8.0 Гц), 7.98 (s, 3Н).
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 12.11, 36.98, 39.23, 39.99, 41.23, 46.57, 55.03, 111.05, 111.54, 117.12, 129.89, 136.03, 144.22, 159.54, 167.12, 171.02.
% Теоретический: С 61.21, Н 7.19, N 7.14.
% Обнаруженный: С 61.38, Н 7.09, N 6.98.
Пример 6: транс-3-амино-N,N-диэтил-1-(3-хлорфенил)-циклобутанкарбоксамид (1е)
Стадия 1: цис-3-гидрокси-1-(3-хлорфенил)-циклобутанкарбоновая кислота (В5)
Идентична стадии 1, описанной в Примере 1, при использовании 3-хлорфенилуксусной кислоты в качестве исходной кислоты. Соединение, указанное в заголовке, получают в виде белого твердого вещества (выход = 52%).
C11H11O3Cl (MB = 226.5).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.50 (m, 2Н), 2.75 (m, 2Н), 3.86 (qu, 1Н, J=7.2 Гц), 5.19 (s, 1Н), 7.31-7.40 (m, 4Н), 12.44 (s, 1Н).
Стадия 2: 4-(3-хлорфенил)-2-оксабицикло[2.1.1]гексан-3-он (С5)
Идентична стадии 2, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 78%).
C11H9O2Cl (MB = 208).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.84 (m, 4Н), 5.09 (s, 1Н), 7.29-7.45 (m, 4Н).
SM-ESI: 209 (МН+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-(3-хлорфенил)-циклобутанкарбоксамид (D5a)
Идентична стадии 3, описанной в Примере 1. Соединение, указанное в заголовке, получают в виде белого твердого вещества (выход = 99%).
C15H20NO2Cl (MB = 281.5).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.63 (t, 3Н, J=6.8 Гц), 0.96 (t, 3Н, J=6.8 Гц), 2.51 (m, 2Н), 2.66 (m, 2Н), 2.86 (q, 2Н, J=6.8 Гц), 3.19 (q, 2Н, J=6.8 Гц), 4.05 (qu, 1Н, J=7.6 Гц), 5.13 (s, 1Н), 7.29-7.41 (m, 4Н).
SM-ESI: 282.1 (МН+).
Стадия 4: транс-3-(диоксоизоиндолин-2-ил)-N,N-диэтил-1-(3-хлорфенил)-циклобутанкарбоксамид (F5a).
В колбу в атмосфере азота добавляют 1 экв соединения (D5A), 1,1 экв трифенилфосфина, 1,05 экв фталимида и ТГФ. Затем добавляют 1,2 экв диизопропилдиазодикарбоксилата (ДИАД) по каплям и перемешивают при комнатной температуре в течение 16 ч. Добавляют воду и экстрагируют ДХМ. Органическую фазу сушат над Na2SO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией со следующей смесью в качестве элюента: гептан/AcOEt: 80:20. Продукт, указанный в заголовке, получают с выходом 77%.
C23H23N2O3Cl (MB = 410.5).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.67 (t, 3Н, J=7.2 Гц), 1.18 (t, 3Н, J=7.2 Гц), 2.91 (q, 2Н, J=7.2 Гц), 3.11 (m, 2Н), 3.35 (m, 2Н), 3.42 (q, 2Н, J=7.2 Гц), 4.79 (qu, 1Н, J=8.8 Гц), 7.23 (m, 1Н), 7.32 (m, 2Н), 7.41 (s, 1Н), 7.73 (m, 2Н), 7.83 (m, 2Н).
SM-ESI: 411.1 (МН+).
Стадия 5: транс-3-амино-N,N-диэтил-1-(3-хлорфенил)-циклобутанкарбоксамид (1е)
Помещают производное (F5A) в растворе в этаноламине в колбу. Нагревают реакционную среду при температуре 60°С в течение 1 ч 30 мин. Добавляют смесь льда и воды, перемешивают в течение 15 мин и экстрагируют этилацетатом. Промывают органическую фазу насыщенным раствором NaCl и декантируют. Органическую фазу сушат над MgSO4, фильтруют и концентрируют. Остаток очищают флэш-хроматографией со следующей смесью в качестве элюента: ДХМ/метанол/NH4OH: 90:9:1. Продукт, указанный в заголовке, получают с выходом 40%.
C15H21N2OCl (MB = 280.5).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.58 (t, 3Н, J=7.2 Гц), 1.10 (t, 3Н, J=7.2 Гц), 2.08 (m, 2Н), 2.91 (q, 2Н, J=7.2 Гц), 3.11 (m, 2Н), 3.34 (q, 2Н, J=7.2 Гц), 3.46 (qu, 1Н, J=8,0 Гц), 7.12 (dd, 1Н, J=7.6 Гц и J=1.2 Гц), 7.19 (m, 2Н), 7.26 (m, 1Н). Сигнал, соответствующий Н в NH2, в спектре не виден.
SM-ESI: 281.1 (МН+).
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 167°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.50 (t, 3Н, J=6.8 Гц), 1.02 (t, 3Н, J=6.8 Гц), 2.56 (m, 2Н), 2.86-2.95 (m, 4Н), 3.26 (m, 2Н), 3.34 (s, 1Н), 3.54 (qu, 1Н, J=8.0 Гц), 6.03 (s, 2Н), 7.15 (d, 1Н, J=7.6 Гц), 7.34-7.44 (m, 3Н), 8.00 (s, 3Н).
13С-ЯМР (ДМСО d6. 100 МГц) δ (ppm): 12.09, 12.12, 36.88, 39.19, 39.90, 41.14, 46.37, 123.76, 124.94, 126.61, 130.65, 133.51, 136.00, 145.09, 167.14, 170.54.
% Теоретический: С 57.50, Н 6.35, N 7.06.
% Обнаруженный: С 57.36, Н 6.26, N 6.68.
Пример 7: транс-3-амино-N,N-диэтил-1-(3-метилфенил)-циклобутанкарбоксамид (1f)
Стадия 1: цис-3-гидрокси-1-(3-метилфенил)-циклобутанкарбоновая кислота (В6)
Идентична стадии 1 Примера 1 при использовании 3-метилфенилуксусной кислоты вместо фенилуксусной кислоты. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 40%).
C12H14O3 (MB = 206).
1Н-ЯМР (CDCl3. 400 МГц) δ (ppm): 2.35 (s, 3Н), 2.73 (m, 2Н), 2.94 (m, 2Н), 4.21 (qu, 1Н, J=6.4 Гц), 7.08 (d, 1Н, J=7.6 Гц), 7.16 (s, 2Н), 7.24 (m, 1Н). Сигналы, соответствующие Н в ОН в спирте и кислоте, в спектре не видны.
SM-ESI: 205.
Стадия 2: 4-(3-метилфенил)-2-оксабицикло[2.1.1]гексан-3-он (С6)
Идентична стадии 2, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 74%).
C12H12O2 (MB = 188).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.37 (s, 3Н), 2.70 (m, 2H), 2.87 (m, 2H), 4.96 (s, 1H), 7.09-7.30 (m, 4H).
SM-ESI: 189 (MH+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-(3-метилфенил)-циклобутанкарбоксамид (D6a)
Идентична стадии 3, описанной в Примере 1. Продукт, указанный в заголовке, получают с выходом 77%.
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.65 (t, 3Н, J=7.2 Гц), 1.08 (t, 3Н, J=7.2 Гц), 2.69 (s, 3Н), 2.73 (m, 2Н), 2.81 (m, 2Н), 2.90 (q, 2Н, J=7.2 Гц), 3.31 (q, 2Н, J=7.2 Гц), 4.35 (qu, 1Н, J=7.4 Гц), 7.04 (m, 1Н), 7.11 (m, 2Н), 7.23 (m, 1Н). Сигнал, соответствующий Н в ОН, в спектре не виден.
Стадия 4: транс-3-азидо-N,N-диэтил-1-(3-метилфенил)-циклобутанкарбоксамид (Е6а)
Идентична стадии 4, описанной в Примере 1. Продукт, указанный в заголовке, получают с выходом 70%.
1Н-ЯМР (CDCl3. 400 МГц) δ (ppm): 0.54 (t, 3Н, J=7.2 Гц), 1.11 (t, 3Н, J=7.2 Гц), 2.34 (s, 3Н), 2.47 (m, 2Н), 2.89 (q, 2Н, J=7.2 Гц), 3.12 (m, 2Н), 3.34 (q, 2Н, J=7.2 Гц), 3.95 (qu, 1Н, J=7.8 Гц), 7.04 (m, 3Н), 7.23 (m, 1Н).
Стадия 5: транс-3-амино-N,N-диэтил-1-(3-метилфенил)-циклобутанкарбоксамид (1f)
Идентична стадии 5, описанной в Примере 1. Продукт, указанный в заголовке, получают с выходом 57%.
C16H24N2O (MB = 260).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0,52 (t, 3Н, J=7,2 Гц), 1,10 (t, 3Н, J=7,2 Гц), 2,11 (m, 2Н), 2,33 (s, 3Н), 2,92 (q, 2Н, J=7,2 Гц), 3,10 (m, 2Н), 3,33 (q, 2Н, J=7,2 Гц), 3,44 (qu, 1Н, J=8,0 Гц), 7,03 (m, 3Н), 7,21 (m, 1Н). Сигнал, соответствующий Н в NH2, в спектре не виден.
SM-ESI: 261 (МН+).
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 173°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.45 (t, 3Н, J=6.8 Гц), 1.02 (t, 3Н, J=6.8 Гц), 2.31 (s, 3Н), 2.52 (m, 2Н), 2.89 (m, 4Н), 3.25 (q, 2Н, J=6.8 Гц), 3.52 (qu, 1Н, J=8.4 Гц), 6.02 (s, 2Н), 7.05 (m, 3Н), 7.27 (t, 1Н, J=7.6 Гц), 8.00 (s, 2Н). Сигнал, соответствующий Н в NH2, в спектре не виден.
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 12.07, 12.13, 21.05, 36.97, 39.15, 40.09, 41.18, 46.56, 48.53, 121.99, 125.43, 127.15, 128.63, 136.07, 137.88, 142.66, 167.21, 171.19.
% Теоретический: С 63.81, Н 7.50, N 7.44.
% Обнаруженный: С 63.93, Н 7.45, N 7.27.
Пример 8: транс-3-амино-N,N-диэтил-1-(2-фтор-3-хлорфенил)-циклобутанкарбоксамид (1g)
Стадия 1: цис-3-гидрокси-1-(2-фтор-3-хлорфенил)-циклобутанкарбоновая кислота (В7)
Идентична стадии 1 Примера 1 при использовании 2-фтор-3-хлорфенилуксусной кислоты вместо фенилуксусной кислоты. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 30%).
C11H10FClO3 (MB = 244.5).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.58 (m, 2Н), 2.75 (m, 2Н), 3.93 (qu, 1Н, J=7.6 Гц), 5.33 (s, 1Н), 7.22 (m, 1Н), 7.52 (m, 2Н), 12.56 (m, 1Н).
SM-ESI: 243,0.
Стадия 2: 4-(2-фтор-3-хлорфенил)-2-оксабицикло[2.1.1]гексан-3-он (С7)
Идентична стадии 2, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 66%).
C11H8O2ClF (MB = 226.5).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.89 (s, 4Н), 5.16 (s, 1Н), 7.22-7.29 (m, 2Н), 7.61 (m, 1Н).
SM-ESI: 227 (МН+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-(2-фтор-3-хлорфенил)-циклобутанкарбоксамид (D7a)
Идентична стадии 3, описанной в Примере 1. Продукт, указанный в заголовке, получают с выходом 87%.
C15H19NO2ClF (MB = 299.5).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.35 (t, 3Н, J=7.0 Гц), 0.96 (t, 3Н, J=7.0 Гц), 2.57 (m, 2Н), 2.67 (m, 2Н), 2.87 (q, 2Н, J=7.0 Гц), 3.16 (q, 2Н, J=7.0 Гц), 4.00 (se, 1Н, J=8.0 Гц), 5.02 (d, 1Н, J=7.2 Гц), 7.27 (t, 1Н, J=8.0 Гц), 7.50 (t, 1Н, J=8.0 Гц), 7.65 (t, 1Н, J=8.0 Гц).
SM-ESI: 300 (МН+).
Стадия 4: транс-3-(диоксоизоиндолин-2-ил)-N,N-диэтил-1-(2-фтор-3-хлорфенил)-циклобутанкарбоксамид (F7a)
Идентична стадии 4, описанной для Примера 6. Продукт, указанный в заголовке, получают с выходом 45%.
C23H22FClN2O3 (MB = 428.5).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.29 (t, 3Н, J=6.8 Гц), 1.04 (t, 3Н, J=6.8 Гц), 2.94-3.04 (m, 4Н), 3.22-3.28 (m, 4Н), 4.61 (qu, 1Н, J=8.8 Гц), 7.35 (t, 1Н, J=8.0 Гц), 7.54 (t, 1Н, J=8.4 Гц), 7.62 (t, 1Н, J=7.2 Гц), 7.83 (s, 4Н).
SM-ESI: 429 (МН+).
Стадия 5: транс-3-амино-N,N-диэтил-1-(2-фтор-3-хлорфенил)-циклобутанкарбоксамид (1g)
Идентична стадии 5, описанной для Примера 6. Продукт, указанный в заголовке, получают с выходом 93%.
C15H20FClN2O (MB = 298.5).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.27 (t, 3Н, J=6.8 Гц), 0.97 (t, 3Н, J=6.8 Гц), 2.08 (m, 2Н), 2.88-2.94 (m, 4Н), 3.17-3.25 (m, 3Н), 7.26 (t, 1Н, J=8.0 Гц), 7.44-7.49 (m, 2Н). Сигнал, соответствующий Н в NH2, в спектре не виден.
SM-ESI: 299 (МН+).
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 179°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.26 (t, 3Н, J=6.8 Гц), 0.99 (t, 3Н, J=6.8 Гц), 2.60 (m, 2Н), 2.91-3.00 (m, 4Н), 3.20 (q, 2Н, J=6.8 Гц), 3.34 (s, 1Н), 3.61 (qu, 1Н, J=8.4 Гц), 6.02 (s, 2Н), 7.32 (t, 1Н, J=8.0 Гц), 7.52-7.57 (m, 2Н), 7.97 (s, 3Н).
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 12.15, 12.21, 36.19, 40.08, 40.56, 41.16, 43.81, 120.16, 125.59, 127.11, 129.12, 132.00, 136.11, 153.74, 156.22, 167.19, 169.48.
% Теоретический: С 55.01, Н 5.83, N 6.75.
% Обнаруженный: С 54.73, Н 5.98, N 6.46.
Пример 9: транс-3-амино-N,N-диэтил-1-(2,5-дифторфенил)-циклобутанкарбоксамид (1h)
Стадия 1: цис-3-гидрокси-1-(2,5-дифторфенил)-циклобутанкарбоновая кислота (В8)
Идентична стадии 1, описанной в Примере 1, при использовании 2,5-дифторфенилуксусной кислоты в качестве исходной кислоты. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 69%).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.55 (m, 2Н), 2.71 (m, 2Н), 3.94 (qu, 1Н, J=7.2 Гц), 5.32 (s, 1Н), 7.12-7.23 (m, 2Н), 7.41 (m, 1Н), 12.48 (s, 1Н).
Стадия 2: 4-(2,5-дифторфенил)-2-оксабицикло[2.1.1]гексан-3-он (С8)
Идентична стадии 2, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 91%).
C11H8F2O2 (MB = 210).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 2.77 (m, 2Н), 2.98 (m, 2Н), 5.01 (s, 1Н), 6.93-7.08 (m, 3Н).
SM-ESI: 228 (M+NH4+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-(2,5-дифторфенил)-циклобутанкарбоксамид (D8a)
Идентична стадии 3 Примера 1. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 100%).
C15H19NO2F2 (MB = 283).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.56 (t, 3Н, J=6.8 Гц), 1.09 (t, 3Н, J=6.8 Гц), 2.82 (m, 5Н), 2.95 (q, 2Н, J=6.8 Гц), 3.31 (q, 2Н, J=6.8 Гц), 4.32 (qu, 1Н, J=7.2 Гц), 6.91-7.11 (m, 3Н).
SM-ESI: 284 (МН+).
Стадия 4: транс-3-азидо-N,N-диэтил-1-(2,5-дифторфенил)-циклобутанкарбоксамид (Е8а)
Идентична стадии 4, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 76%).
C15H18N4OF2 (MB = 308).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.51 (t, 3Н, J=7.2 Гц), 1,10 (t, 3Н, J=7,2 Гц), 2.51 (m, 2Н), 2.98 (q, 2Н, J=7.2 Гц), 3.19 (m, 2Н), 3.32 (q, 2Н, J=7.2 Гц), 4.02 (qu, 1Н, J=8.0 Гц), 6.90-7.04 (m, 3Н).
SM-ESI: 309 (МН+).
Стадия 5: транс-3-амино-N,N-диэтил-1-(2,5-дифторфенил)-циклобутанкарбоксамид (1h)
Помещают 1 экв соединения (Е8а) в колбу и растворяют в 20 объемах ТГФ. Перемешивают в атмосфере азота, затем добавляют 1 объем воды и 1,5 экв трифенилфосфина. Продолжают перемешивание в течение ночи. Выпаривают ТГФ при пониженном давлении, отбирают полученный остаток водой и экстрагируют дважды ДХМ. Сушат органическую фазу над MgSO4, фильтруют, затем выпаривают растворитель при пониженном давлении. Полученное масло очищают флэш-хроматографией со следующей смесью в качестве элюента: ДХМ/метанол/NН4ОН 95:4.5:0.5. Продукт, указанный в заголовке, получают в виде бесцветного масла с выходом 97%.
C15H20N2OF2 (MB = 282).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0,51 (t, 3Н, J=6,8 Гц), 1,10 (t, 3Н, J=6,8 Гц), 2,15 (m, 2Н), 2,99 (q, 2Н, J=6,8 Гц), 3,16 (m, 2Н), 3,32 (q, 2Н, J=6,8 Гц), 3,52 (qu, 1Н, J=8,0 Гц), 6,85-7,02 (m, 3Н). Сигнал, соответствующий Н в NH2, в спектре не виден.
SM-ESI: 283 (МН+).
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 184°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.31 (t, 3Н, J=6.8 Гц), 0.99 (t, 3Н, J=6.8 Гц), 2.58 (m, 2Н), 2.93-2.97 (m, 4Н), 3.20 (q, 2Н, J=6.8 Гц), 3.34 (s, 1Н), 3.59 (qu, 1Н, J=8.4 Гц), 6.02 (s, 2Н), 7.13-7.30 (m, 2Н), 7.49-7.53 (m, 1Н), 7.97 (s, 3Н).
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 12.04, 12.15, 36.08, 40.00, 40.61, 41.20, 43.48, 114.90, 117.2, 124.37, 132.1, 136.00, 154.6, 157.05, 157.13, 159.51, 167.12, 169.41.
% Теоретический: С 57.28, Н 6.07, N 7.03.
% Обнаруженный С: 57.21, Н 6.01, N 6.66.
Пример 10: транс-3-амино-N,N-диэтил-1-(3,5-дихлорфенил)-циклобутанкарбоксамид (1i)
Стадия 1: цис-3-гидрокси-1-(3,5-дихлорфенил)-циклобутанкарбоновая кислота (В9)
Идентична стадии 1, описанной в Примере 1, при заблаговременном синтезе 3,5-дихлорфенилуксусной кислоты, которую используют в качестве исходной кислоты. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 50%).
C11H10Cl2O3 (MB = 261).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 2.53 (m, 2Н), 2.77 (m, 2Н), 3.87 (qu, 1Н, J=7.4 Гц), 5.23 (s, 1Н), 7.38 (m, 2Н), 7.51 (m, 1Н), 12.62 (s, 1Н).
SM-ESI: 259.
Стадия 2: 4-(3,5-дихлорфенил)-2-оксабицикло[2.1.1]гексан-3-он (С9)
Идентична стадии 2, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 87%).
C11H8Cl2O2 (MB = 243).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 2.72 (m, 2Н), 2.88 (m, 2Н), 4.99 (s, 1Н), 7.21 (m, 2Н), 7.34 (m, 1Н).
SM-ESI: 244 (МН+).
Стадия 3: цис-3-гидрокси-N,N-диэтил-1-(3,5-дихлорфенил)-циклобутанкарбоксамид (D9a)
Идентична стадии 3 Примера 1. Продукт, указанный в заголовке, получают в виде белого твердого вещества (выход = 100%).
C15H19Cl2O2N (MB = 316).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.77 (t, 3Н, J=7.2 Гц), 1.09 (t, 3Н, J=7.2 Гц), 2.58 (s, 1Н), 2.75 (m, 4Н), 2.88 (q, 2Н, J=7.2 Гц), 3.32 (q, 2Н, J=7.2 Гц), 4.34 (qu, 1Н, J=7.6 Гц), 7.20-7.27 (m, 3Н).
SM-ESI: 316.
Стадия 4: транс-3-азидо-N,N-диэтил-1-(3,5-дихлорфенил)-циклобутанкарбоксамид (Е9а)
Идентична стадии 4, описанной в Примере 1. Продукт, указанный в заголовке, получают в виде бесцветного масла (выход = 79%).
C15H18N4OCl2 (MB = 341).
1Н-ЯМР (CDCl3, 400 МГц) δ (ppm): 0.67 (t, 3Н, J=7.2 Гц), 1.12 (t, 3Н, J=7.2 Гц), 2.40 (m, 2Н), 2.87 (q, 2Н, J=7.2 Гц), 3.15 (m, 2Н), 3.36 (q, 2Н, J=7.2 Гц), 3.99 (qu, 1Н, J=7.6 Гц), 7.13 (m, 2Н), 7.25 (m, 1Н).
SM-ESI: 341.
Стадия 5: транс-3-амино-N,N-диэтил-1-(3,5-дихлорфенил>-циклобутанкарбоксамид (1i)
Идентична стадии 5 Примера 9. Продукт, указанный в заголовке, получают в виде бесцветного масла с выходом 78%.
C15H20N2OClF (MB = 283).
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.58 (t, 3Н, J=7.2 Гц), 1.00 (t, 3Н, J=7.2 Гц), 1.90 (s, 2Н), 2.07 (m, 2Н), 2.85 (m, 4Н), 3.11 (qu, 1Н, J=8.0 Гц), 3.25 (q, 2Н, J=7.2 Гц), 7.23 (m, 2Н), 7.48 (m, 1Н).
SM-ESI: 283.
Малеат соединения, указанного в заголовке
Получение соли предыдущего соединения с использованием малеиновой кислоты приводит к получению малеата соединения, указанного в заголовке, в форме белого порошка.
Т.пл.: 180°С.
1Н-ЯМР (ДМСО d6, 400 МГц) δ (ppm): 0.57 (t, 3Н, J=6.8 Гц), 1.02 (t, 3Н, J=6.8 Гц), 2.58 (m, 2Н), 2.85-2.96 (m, 4Н), 3.28 (q, 2Н, J=6.8 Гц), 3.54 (qu, 1Н, J=8.4 Гц), 6.02 (s, 2Н), 7.28 (s, 2Н), 7.56 (s, 1Н), 7.99 (s, 3Н).
13С-ЯМР (ДМСО d6, 100 МГц) δ (ppm): 11.96, 12.19, 36.83, 39.20, 41.10, 46.2, 124.03, 126.38, 134.50, 136.02, 146.66, 167.12, 170,04.
% Теоретический: С 52.91, Н 5.61, N 6.50.
% Обнаруженный: С 53.01, Н 5.53, N 6.11.
Следующие примеры позволяют лучше понять изобретение, но не ограничивают его объем никоим образом.
Соединения общей формулы (1), а также их фармацевтически приемлемые соли обладают замечательными фармакологическими свойствами: в целом, они являются более сильными блокаторами NMDA-каналов, чем кетамин, но в то же время имеют меньше нежелательных эффектов на центральную нервную систему, чем кетамин.
Было исследовано влияние соединения согласно изобретению на ингибирование тока NMDA на лягушке (Xenopus Laevis, обыкновенной шпорцевой лягушке), экспрессирующей рекомбинантные человеческие NMDA-рецепторы, состоящие из NR1 и NR2B субъединиц. Токи, продуцируемые путем стимуляции этих рецепторов с помощью эндогенных агонистов, были изучены в соответствии с методом двухэлектродной фиксации потенциала, описанным в работе Planells-Cases et al., 2002, J. Pharmacol. Exp. Ther., 302, 163-173.
Протокол: у взрослых лягушек удаляли хирургическим путем овоциты, их ферментативно дефолликулировали и хранили при 17°С в растворе, содержащем: 96 мМ NaCl, 2 мМ KCl, 1 мМ MgCl2, 1,8 мМ CaCl2 и 5 мМ HEPES при pH 7,5 (NaOH) и 50 мг/л гентамицина (Heusler et al., 2005, Neuropharmacology, 49, 963-976). Комплементарную ДНК (кДНК), кодирующую NR1 субъединицы, клонировали с помощью ПЦР с использованием праймеров для стартовых и стоп-кодонов в опубликованной последовательности (номер доступа в Банке генов М_007327). кДНК, кодирующая субъединицу NR2B, была синтезирована компанией Eurogentec (Серен, Бельгия) в соответствии с опубликованной последовательностью (номер доступа в Банке генов NM_000834). NR1 и NR2B кДНК затем субклонировали в носитель высокой экспрессии pGEMHE для in vitro транскрипции кДНК. кРНК, кодирующую NR1 и NR2B, получали в соответствии с методом, описанным в работе Heusler et al. (приведена выше). Аликвоты раствора кРНК инъецировали в овоциты (20-500 пг/овоцит для NR1 и 40-1000 пг/овоцит для NR2B). В каждый овоцит вводили 100 нл раствора, содержащего: 4 мМ Na + ВАРТА (pH 7,2), для того, чтобы блокировать все остаточные токи хлора. После стабилизации NMDA токи активировали перфузией глутамата и глицина, каждого при концентрации 10 мкМ. Соединения, подлежащие тестированию, затем перфузировали в растворе Рингера Ва++ при увеличении концентраций в присутствии глутамата и глицина (было протестировано от 4 до 5 концентраций на овоцит). Полученные показатели концентрация-ответ анализировали для каждого овоцита путем нелинейной регрессии и рассчитывали значение pIC50. pIC50 означает отрицательный логарифм концентрации тестированного соединения, необходимой для уменьшения амплитуды тока NMDA наполовину.
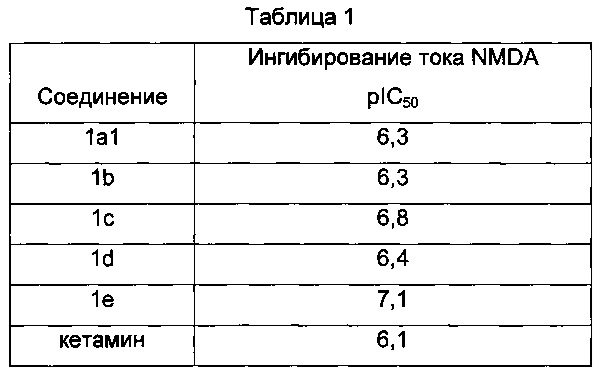
Результаты: ниже в таблице 1 приведены значения рЮ50 для определенных соединений согласно настоящему изобретению. Обнаружено, что при условиях тестирования соединения (1А1), (1b), (1с), (1d) и (1е) блокируют ток NMDA зависящим от концентрации образом и являются более сильными, чем кетамин, который является клинически используемым антагонистом NMDA.
Учитывая низкую биодоступность кетамина, вводимого пероральным путем, был выбран внутрибрюшинный (ip) способ введения в качестве единственного способа введения для экспериментов in vivo. Обезболивающее действие соединений формулы (1) и кетамина, выбранного в качестве контрольного соединения, определяли на классической модели острой воспалительной боли внутрикожной инъекцией формальдегида (Bardin et al., 2001, Eur. J. Pharmacol., 421, 109-114).
Протокол: самцов крыс (Sprague-Dawley Iffa Credo, Франция) помещали для наблюдения в коробки из плексигласа выше зеркала, расположенного под углом, чтобы облегчить наблюдение их задних лап. Через 30 минут акклиматизации животные получали инъекции формальдегида, разведенного до 2,5%, на подошвенную поверхность правой задней лапы. Инъекции формальдегида вызывают поведенческие реакции, которые происходят в две фазы:
- ранняя фаза, через 0-5 минут после инъекции формальдегида, соответствующая стимулированию рецепторов, специализирующихся на передаче ноцицептивных стимулов;
- поздняя фаза, которая имеет место через 20-30 минут после инъекции. Эта фаза соответствует стимуляции рецепторов медиаторами воспаления и/или сверхвозбуждению рога спинного мозга, которые индуцированы в ходе первой фазы. Поэтому эта поздняя фаза запускает центральную сенситизацию системы болевой нейротрансмиссии, в которой важную роль играет система глутамат/NMDA. Следовательно, боль во второй фазе является более типичным образцом невропатической боли, чем боль, которая происходит во время первой фазы. По этой причине в данной заявке рассматриваются только результаты, полученные в этой поздней фазе.
В качестве поведенческого параметра для количественного определения боли был выбран параметр «лизание лапы», в которую была сделана инъекция, и в качестве периодов наблюдений были выбраны периоды, соответствующие поздней фазе (другими словами, через 22,5-27,5 мин после инъекции формальдегида). Во время этого пятиминутного периода животных наблюдали каждые 30 секунд для того, чтобы отметить, лижет ли животное «инъецированную» лапу, или нет; таким образом, максимальная оценка равна 10. Продукты согласно изобретению или носитель вводили внутрибрюшинно за 15 мин до инъекции формальдегида.
Результаты: в этом тесте соединения формулы (1а1) и (1е), которые являются представителями соединений согласно изобретению, имели замечательный обезболивающий эффект (Таблица 2). Таким образом, минимальная значимая доза (МЗД, т.е. доза, необходимая для существенного снижения лизания «инъецированной» лапы) для соединений формулы (1а1) и (1е) меньше, чем минимальная значимая доза для кетамина. Другое преимущество соединений формулы (1а1) и (1е) по сравнению с кетамином относится к полноте обезболивающего эффекта. Фактически было отмечено, что при дозе 40 мг/кг соединения (1а1) и (1е) полностью устранили лизание лапы, тогда как кетамин снизил лизание лапы только на 74%. Поэтому соединения (1а1) и (1е) являются более сильными и эффективными, чем кетамин.

Таким образом, обезболивающий эффект соединений формулы (1а1) и (1е), которые являются представителями соединений формулы (1), является более высоким, чем обезболивающий эффект кетамина на модели острой воспалительной боли у крыс.
Также показано, что соединения согласно изобретению имеют антидепрессантную активность in vivo. Антидепрессантную активность соединений формулы (1) и кетамина определяли на модели принудительного плавания у крыс, которая широко используется, поскольку является прогностической в отношении антидепрессантной активности у человека.
Протокол: самцов крыс (Sprague-Dawley Iffa Credo, Франция) помещают в цилиндр (высотой 45 см и диаметром 20 см), заполненный водой с температурой 25°С ± 0,5°С до высоты 17 см. Эта высота позволяет крысам плавать или держаться на поверхности без помощи лап, касаясь основания цилиндра. За 24 часа до дня тестирования крыс помещают в цилиндр на 15 мин, после этого периода времени они больше не пытаются сбежать и остаются неподвижными на поверхности. В день тестирования соединение, подлежащее тестированию, или носитель внутрибрюшинно вводят животному, которое помещают в цилиндр на 30 мин позже. Продолжительность неподвижности (определяется, когда крысы просто держатся на поверхности и только делают небольшие движения, чтобы оставаться на поверхности) измеряют с точностью до 0,1 сек в течение 5 минут.
Результаты: В тесте принудительного плавания соединения формул (1с) и 1(е), которые являются представителями серии, значительно сокращают время неподвижности животного. При сравнении показателей ED50, то есть доз, которые уменьшают время неподвижности наполовину по сравнению с контрольными животными, обнаружено, что эти показатели для соединений (1с) и 1(е) меньше, чем для кетамина, см. Таблицу 3. Аналогично, полнота эффекта антинеподвижности, наблюдаемого при дозе 20 мг/кг, является большей для соединений (1с) и 1(е), чем для кетамина.

Таким образом, соединения (1с) и 1(e), которые являются представителями соединений формулы (1), являются более сильными и эффективными в прогностическом тесте антидепрессантной активности.
Мы уже подчеркивали важность нормализации функции NMDA-рецепторов, другими словами, важность блокирования их чрезмерной активности без вмешательства или с возможно меньшим вмешательством в их нормальное физиологическое функционирование. В качестве показателя взаимосвязи продуктов согласно изобретению и нормального функционирования NMDA-рецепторов выбран тест преимпульсного ингибирования рефлекса испуга (PPI). Этот тест представляет собой измерение способности организма к фильтрации несущественной информации. Неконкурентные и конкурентные антагонисты, а также блокаторы каналов снижают PPI у крыс (Depoortere et al., 1999, Behav. Pharmacol., 10, 51-62), причем такое снижение считается прогностическим для психотомиметических эффектов NMDA антагонистов у человека.
Протокол: самцов крыс (Sprague-Dawley Iffa Credo, Les Oncins, Франция) помещали в цилиндры высотой 18,4 см с диаметром 8,8 см на опоре, ниже которой установлен пьезоэлектрический акселерометр, действующий в качестве детектора реакции испуга. Эта конструкция вложена в коробку с громкоговорителем, прикрепленного к потолку, для обеспечения звуковых импульсов и предварительных импульсов, и акустически изолирована (SR LAB, San Diego Instruments, Сан-Диего, США). Все события контролировали посредством программного обеспечения. Сначала животных подвергали 13-ти минутному предварительному тесту, чтобы приучить их к процедуре и исключить крыс, которые не отвечают ряду минимальных критериев реакции. Обеспечивали три типа звуковых раздражителей (белый шум); 1) импульс 118 дБ (Р, длительность 40 мс); 2) предварительный импульс 78 дБ (длительность 20 мс), за которым следует импульс 118 дБ (pP); и 3) отсутствие предварительного импульса или импульса (NP). Интервал между началом предварительного импульса и началом импульса составляет 100 мс с фоновым шумом 70 дБ. Реакцию испуга регистрировали в течение 100 мс, через 100 мс после начала стимула (pP или NP) с помощью цифровой/аналоговой платы сбора данных (12 бит). Сеанс начинали с 5-ти минутного периода без стимула, после чего животных подвергали 10 импульсам Р (с промежутками в среднем по 15 сек для стабилизации реакции испуга). Реакции, записанные с этими 10 Р не используют для расчетов. После этого в полуслучайном порядке производили 10 Р, 10 pP и 3 NP со средним интервалом в 15 сек между ними. В конце этого предварительного теста крысам делали внутрибрюшинную инъекцию соединения, подлежащего тестированию, или физиологического раствора в качестве контроля, и возвращали в их клетки. Сеанс фактического тестирования (аналогичный по каждому пункту предварительного теста) осуществляли через 60 минут. Процент преимпульсного ингибирования вычисляли с использованием данных этого сеанса тестирования по формуле:
(средняя амплитуда Р - средняя амплитуда pP)
× 100/(средняя амплитуда Р).
Результаты: исходя из фиг. 1 видно, что соединение (1а1) не нарушает ингибирование рефлекса испуга, вызванного предварительным импульсом (PPI), за исключением дозы 20 мг/кг. Тем не менее, удивительно, что снижение преимпульсного ингибирования (PPI) намного менее выражено, чем это наблюдалось для кетамина. Фактически, в дозе 20 мг/кг ip кетамин приводит к полному исчезновению PPI, тогда как соединение (1а1) только вызывает 30%-ное снижение. Снижение PPI, тем не менее, остается умеренным даже в дозе 40 мг/кг. Следовательно, соединение (1а1) имеет явно меньше выраженную тенденцию вызывать побочные эффекты центрального происхождения, чем кетамин.
Таким образом, соединения согласно изобретению обладают анальгетической и антидепрессантной активностью, которая выше, чем у кетамина, на животной модели, описанной выше. Удивительно, что соединения согласно изобретению вызывают лишь очень умеренные центральные эффекты. Поэтому из этих экспериментов следует, что соотношение риск/польза у соединений согласно изобретению явно более благоприятно, чем у кетамина. Следовательно, соединения согласно настоящему изобретению, а также фармацевтические композиции, содержащие соединение общей формулы (1) или одну из его фармацевтически приемлемых солей в качестве активного ингредиента, являются потенциально полезными в качестве лекарственных средств, в частности, в лечении определенных заболеваний, таких как, например, депрессия и боль, особенно острая или хроническая боль, в областях, в которых терапевтические потребности не полностью удовлетворены и для которых поэтому очень желательно открытие новых способов лечения.
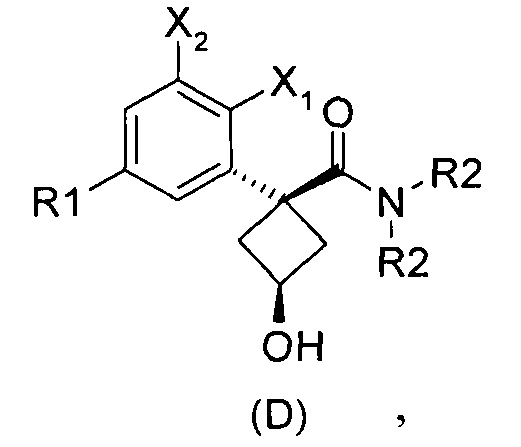
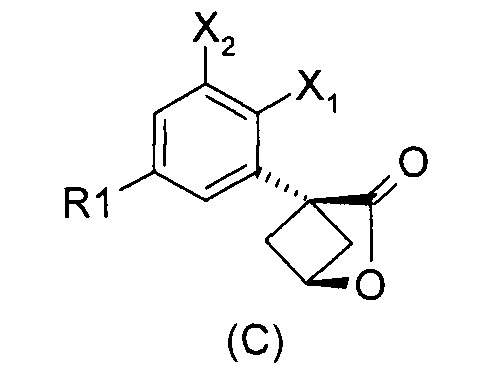
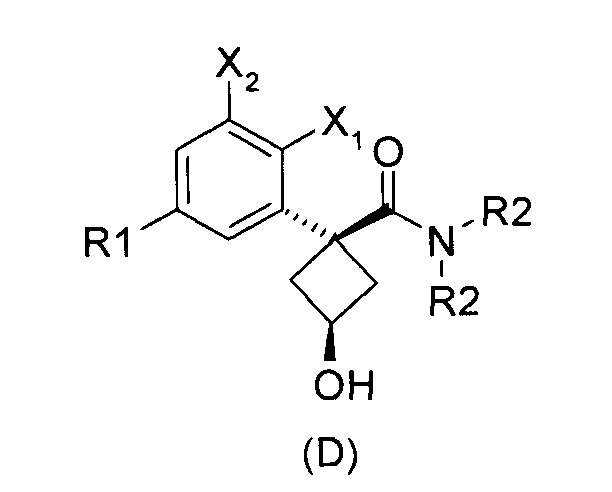
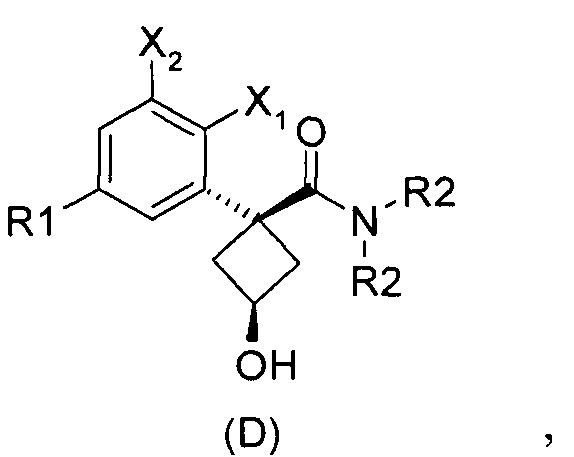
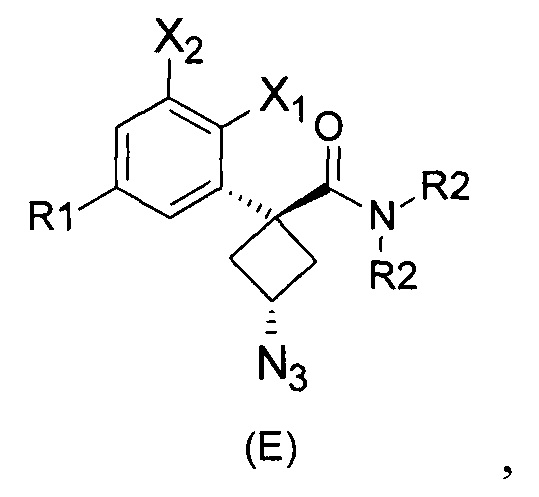
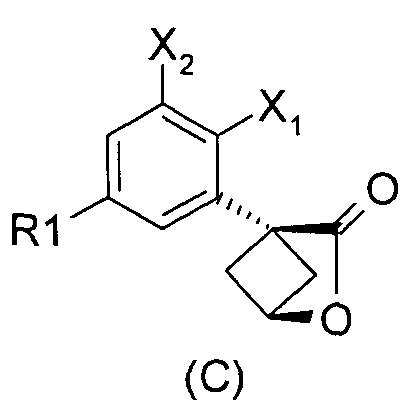
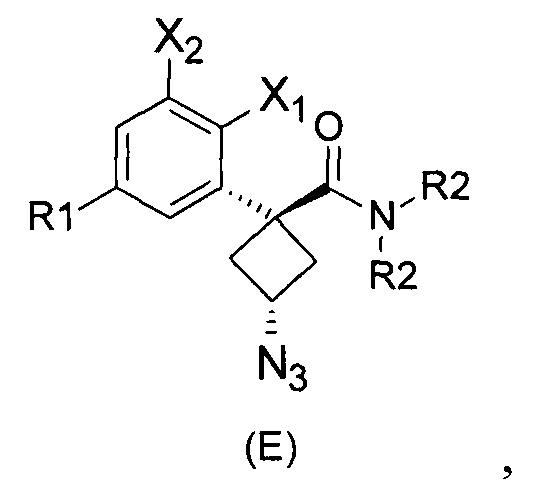
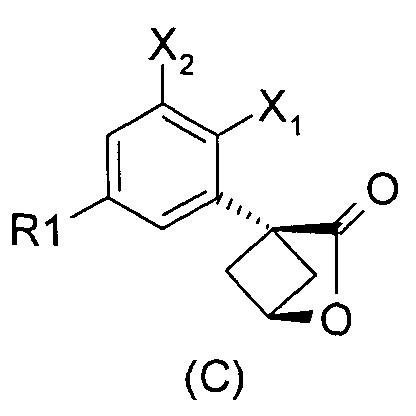
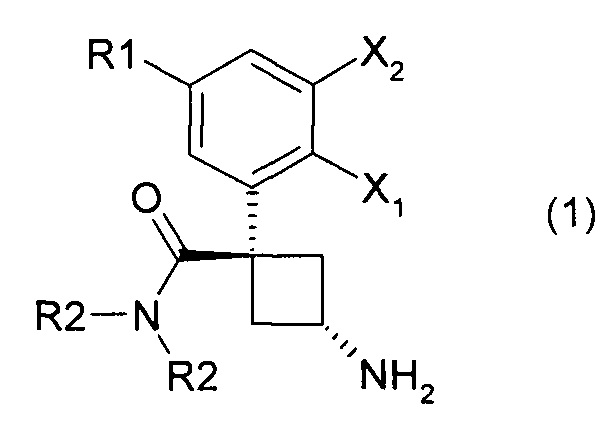
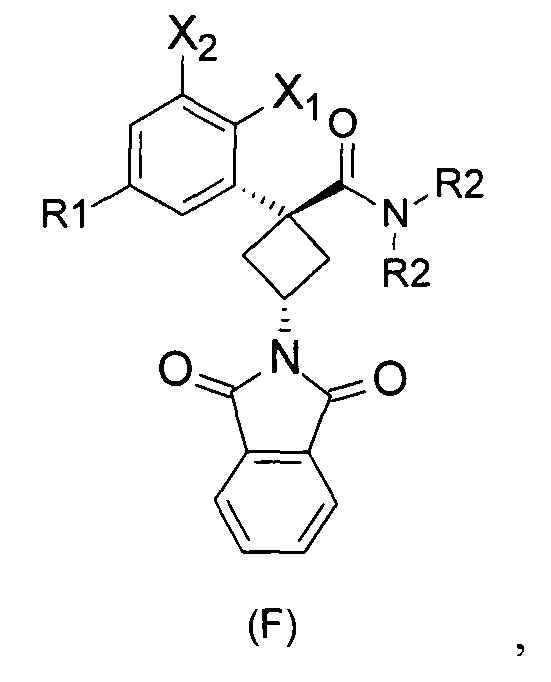
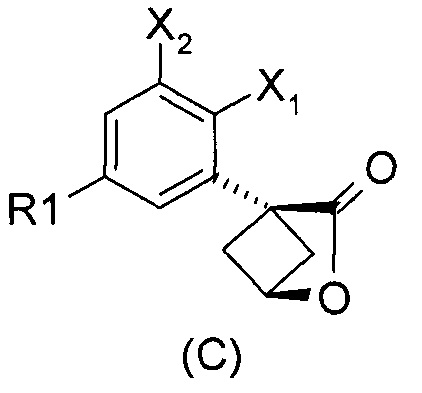
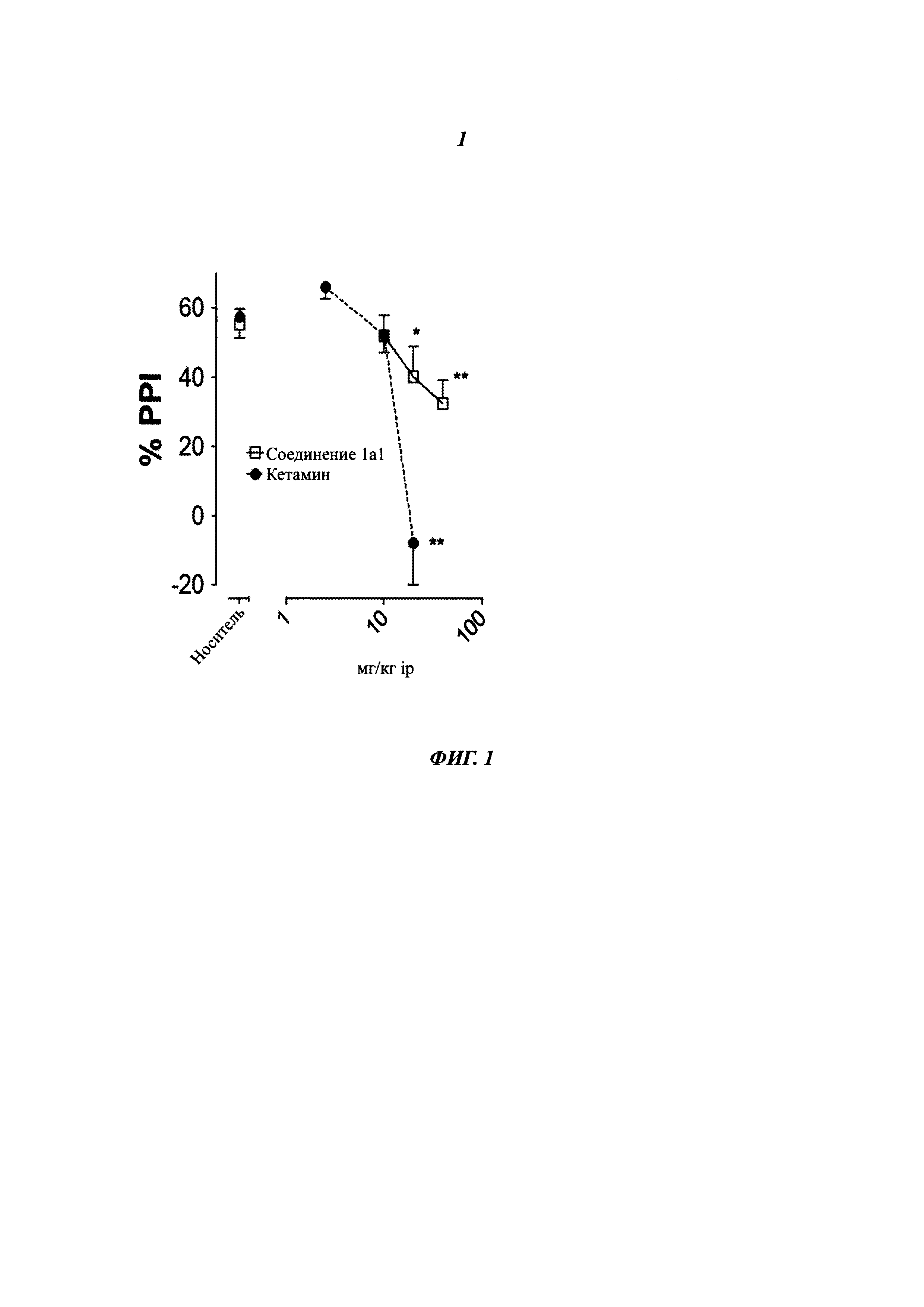
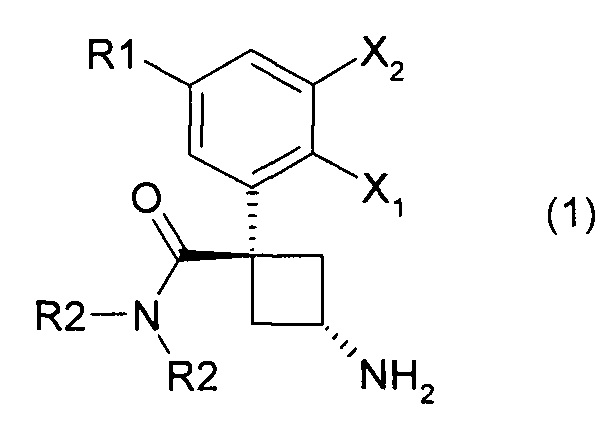
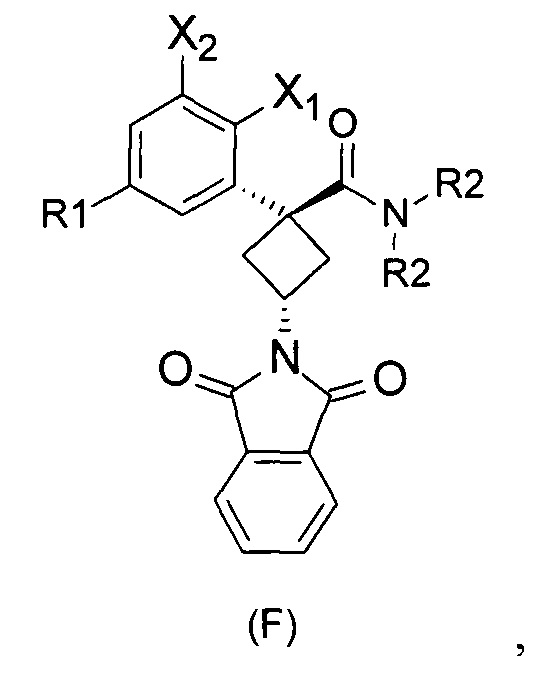
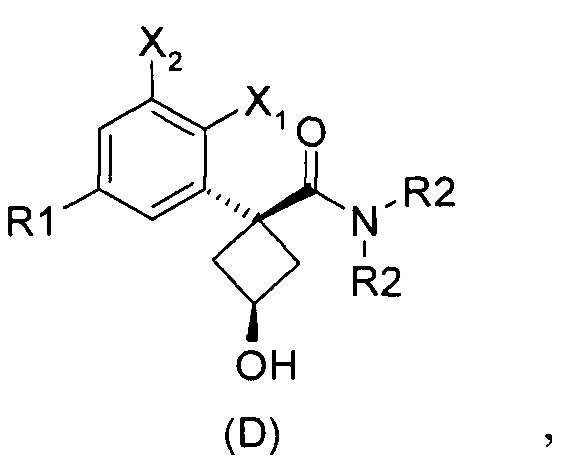
Меквитазин для лечения или предотвращения патологий, в которых задействованы н4 рецепторы гистамина
Способ генерирования активных антител к антигену устойчивости, антитела, полученные этим способом, и их применения
Способ синтеза (1s,2r)-милнаципрана
Производные бензотиазинов, их получение и применение в качестве лекарств
Комбинация антагониста с-мет и аминогетероарила для лечения рака
Новые (поли)аминоалкиламиноалкиламидные, алкил-мочевинные или алкил-сульфонамидные производные эпиподофиллотоксина, способ их получения и их применение в терапии в качестве противораковых средств
Фармацевтическая композиция, содержащая эфир дгк, для парентерального введения
Композиция, содержащая комбинацию экстракта бузины и штамма l. paracasel, l. casei, l. bulgaricus или s. thermophilics
Производные хромонов, способ их получения и их терапевтические применения
Новые антитела, ингибирующие димеризацию с-мет, и их применения
Применение 3-(r)-[3-(2-метоксифенилтио)-2-(s)-метилпропил]амино-3,4-дигидро-2н-1,5-бензоксатиепина для лечения рака и, в частности, для профилактики и/или лечения метастазов рака
Способ получения n-[3-[(2-метоксифенил)сульфанил]-2-метилпропил]-3,4-дигидро-2н-1,5-бензоксатиепин-3-амина
Применение производных пиридин-2-ил-метиламина для получения лекарственного средства, предназначенного для лечения симптомов хронической боли невропатического или психогенного происхождения

