Результат интеллектуальной деятельности: КОМПОЗИЦИИ ИНСУЛИНОВ ДЛИТЕЛЬНОГО ДЕЙСТВИЯ
Вид РИД
Изобретение
Заявка относится к водной фармацевтической композиции, включающей в себя 200–1000 Ед/мл [эквимолярно 200–1000 МЕ инсулина человека] инсулина гларгина, при условии, что концентрация инсулина гларгина в указанной композиции не равна 684 Ед/мл, и к ее применению.
Инсулин гларгин представляет собой 31B-32B-Di-Arg инсулина человека, аналог инсулина человека, с дополнительной заменой аспарагина в положении A21 на глицин.
Лантус® представляет собой продукт инсулина, содержащий инсулин гларгин, обеспечивающий запас базального инсулина в течение 24 часов после подкожной инъекции однократной дозы.
Глюкодинамический эффект Лантуса® отличается от других представленных на рынке продуктов инсулина благодаря отложенной и прогнозируемой абсорбции инсулина гларгина из области подкожной инъекции, что дает в результате сглаженную концентрацию в течение 24 часов и профиль действия без определенного пика. Лантус® был создан в соответствии с медицинской необходимостью в продукте инсулина длительного действия, который может быть введен в виде однократной дневной инъекции для предоставления нормального или близкого к нормальному контроля глюкозы крови с профилем базального инсулина, который является настолько плавным, насколько возможно, на протяжении 24-часового периода времени. Такое средство обеспечивает хороший контроль глюкозы крови на протяжении всего дня, и в то же время минимизирует склонность к возникновению гипогликемии, наблюдаемой в случае других средств инсулина с более определенным “пиковым” действием.
Значительное число пациентов, в частности пациентов с повышенной устойчивостью к инсулину вследствие ожирения, применяют большие дозы для контроля глюкозы крови. Например, доза 100 Ед требует инъекции 1 мл Лантуса® U100, которая может вызвать некоторый дискомфорт; каждый мл Лантуса® U100 содержит 100 Ед (3,6378 мг) инсулина гларгина. Для уменьшения объема инъекции была разработана композиция, содержащая 300 Ед инсулина гларгина в мл. Хотя изобретение не ограничивается композицией инсулина гларгина U300, клинические исследования, описанные здесь, осуществляли с композицией инсулина гларгина U300; каждый мл инсулина гларгина U300 содержит 300 Ед (10,9134 мг) инсулина гларгина. Указанное средство позволило бы пациентам вводить то же число единиц инсулина гларгина в одной третьей объема инъекции.
Ожидали, что оба средства инсулина гларгина, U100 и U300, обеспечат одинаковое воздействие инсулина и одинаковую эффективность, т.е. временные профили.
ПОДРОБНОЕ ОПИСАНИЕ
Воздействие и активность инсулина гларгина U300, исследуемого (T) лекарственного средства, проверяли на здоровых субъектах, не страдающих диабетом, в эугликемических клэмп-тестах на эквивалентность воздействия и активности с Лантусом U100, утвержденным эталонным (R) медицинским средствам. Для оценки долговременности действия инсулина гларгина после подкожного введения выбрали 30 часов. Воздействие оценивали исходя из профилей “концентрация инсулина гларгина – время после подкожного введения”, тогда как активность одновременно оценивали в виде утилизации глюкозы на единицу инсулина.
Дизайн эксперимента с репликами позволил ограничить число субъектов для оценки биоэквивалентности и вариабельности, как рекомендовано в руководстве FDA “Guidance for Industry, Statistical Approaches to Establishing Bioequivalence”.
Предполагали, что соответствующее клиническое исследование установит эквивалентность воздействия и активности.
Для данного исследования выбрали дозу 0,4 Ед/кг; она соответствует средней дозе базального инсулина у пациентов. У здоровых субъектов, не страдающих диабетом, указанная доза вызывает значительное увеличение концентрации инсулина в плазме крови и эффект длительного снижения уровня глюкозы, который может быть измерен количественно в условиях эугликемического клэмп-теста.
Дизайн эксперимента с репликами, одобренный в методических указаниях, требует двух повторных инъекций однократных доз каждого из двух IP (R: Лантус® U100, T: инсулин гларгин U300) в заранее заданных перекрестных последовательностях с четырьмя периодами (RTTR или TRRT), как распределено в плане рандомизации. Указанные инъекции осуществляли в Периоды (P) 1-4 в четыре разных дня. В результате каждый субъект получил две повторные однократные подкожные дозы 0,4 Ед/кг Лантуса® U100 (R) и инсулина гларгина U300 (T), которые вводили поочередно в две противоположные стороны околопупочной области.
Период вымывания от 4 до 18 дней отделял каждый день дозирования. Продолжительность периода вымывания изменялась индивидуально, что позволяло участнику исследования и исследователю согласовывать свои потребности. По опыту, 4 дня составляют минимальный период восстановления, обеспечивая проведение 1 клэмп-теста в неделю для участника, тогда как 18 дней представляют собой перерыв в 3 недели между днями проведения клэмп-теста, обеспечивая субъектам больше свободы для выполнения обязанностей, не связанных с исследованием.
Перед визитами проведения клэмп-теста, во время SCR (скрининговый визит), субъектов проверяли на соответствие требованиям исследования, и в EOS (окончание исследования) визит субъекты проходили заключительное обследование для подтверждения нормального состоянии здоровья. Скрининг и P1 разделяли не более чем 21 день, тогда как визиты EOS имели место не ранее чем в такой же день недели, как День 1 в P4, на следующей неделе, т.е. после дополнительных 4 дней, и не позднее, чем две недели после Дня 2 P4, т.е. после дополнительных 14 дней.
Данное исследование представляло собой однодозовое исследование всего с 4 повторными введениями. Эффект IP составлял по длительности приблизительно 24 часа, поэтому субъекты должны были оставаться в исследовательском центре в течение 2 дней. Субъекты получали лечение 4 раза.
Главная цель исследования заключалась в оценке средней биоэквивалентности (ABE) Лантуса® U100 (коммерческое средство) и инсулина гларгина U300 по биодоступности (воздействии) и биоэффективности (активности) с применением эугликемического клэмп-метода.
Дополнительная цель исследования заключалась в оценке безопасности и переносимости инсулина гларгина U300.
Как упоминалось выше, предполагали, что оба средства инсулина гларгина U100 и U300 обеспечивают одинаковое воздействие инсулина и одинаковую эффективность. Однако, как не удивительно, показали, что воздействие и эффективность инсулина не являются одинаковыми. Инсулин гларгин U100 и инсулин гларгин U300 не являются эквивалентными по биодоступности (воздействию) и биоэффективности (активности). Воздействие и активность после введения инсулина гларгина U300 были приблизительно на 40% меньше по сравнению с воздействием и активностью после введения такого же количества (0,4 Ед/кг) инсулина гларгина U100.
Однако инсулин гларгин U300 продемонстрировал еще более плавный профиль PK (воздействия) и PD (активности), чем инсулин гларгин U100, который был бы желателен для базального инсулина. Указанные удивительные и неожиданные различия в воздействии и активности между средствами инсулина гларгина U100 и инсулина гларгина U300 после одинаковой п/к дозы у здоровых субъектов показаны по существу на фигурах ниже. Следует отметить, что в то же время глюкоза крови оставалась постоянной.
Эффект снижения глюкозы крови инсулина гларгина дополнительно оценивали у здоровых нормогликемических собак породы бигль. При увеличении концентрации инсулина гларгина среднее время действия возрастало от 6,8 часа (U100) до 7,69 часа (U300), соответственно.
При увеличении концентрации гларгина от 100 до 300 Ед/мл у собаки профиль “время снижения глюкозы крови - действие” изменялся по направлению к более плавной и пролонгированной активности. Настоящие данные, полученные на собаках, соответствуют данным, полученным в экспериментах с участием людей, показывающим, что более высокие концентрации инсулина гларгина положительно коррелируют с профилем и с большей продолжительностью действия.
Кроме того, исследовали с помощью микроскопии осадки средств инсулина гларгина с концентрациями 100 Ед/мл, 300 Ед/мл, 500 Ед/мл 700 Ед/мл и 1000 Ед/мл. Указанные исследования выявили различия в свойствах осадков, приводящих к образованию заметно больших частиц при увеличении концентраций.
Кроме того, влияние средств с более высокими концентрациями инсулина гларгина с учетом свойств растворения исследуют с применением тестовой системы in-vitro. Для проведения указанного, исследования осадков осуществляют с применением фосфатного буфера со значением pH 7,4, воспроизводя условия in-vivo.
Супернатант осажденного инсулина исследуют с применением метода ВЭЖХ для определения содержания инсулина гларгина.
Международный патент WO 2008/013938 A2 раскрывает лекарственное средство на водной основе, содержащее инсулин гларгин в концентрации 684 Ед/мл.
Хотя изобретение не ограничивается средством инсулина гларгина U300 и является эффективным с другими более концентрированными средствами инсулина гларгина, как уже указано подробно в описании, клинические исследования, описанные здесь, осуществляли со средством инсулина гларгина U300.
Средство инсулина гларгина U300, 1 мл, содержит 10,913 мг 21A-Gly-30Ba-L-Arg-30Bb-L-Arg инсулина человека [эквимолярно 300 МЕ инсулина человека], 90 мкг цинка, 2,7 мг m-крезола, 20 мг 85% глицерина, HCl и NaOH до значения pH 4,0; удельная плотность 1,006 г/мл
Однако, возможны вариации в отношении типа эксципиентов и их концентраций.
Лекарственное средство содержит 200–1000 Ед/мл инсулина гларгина [эквимолярно 200–1000 МЕ инсулина человека], где концентрация указанной композиции не равна 684 Ед/мл, предпочтительно составляет 250-500 Ед/мл инсулина гларгина [эквимолярно 250–500 МЕ инсулина человека], более предпочтительно 270–330 Ед/мл инсулина гларгина [эквимолярно 270–330 МЕ инсулина человека], и еще более предпочтительно 300 Ед/мл инсулина гларгина [эквимолярно 300 МЕ инсулина человека].
В лекарственное средство могут быть добавлены сурфактанты, например, в числе прочего, неионные сурфактанты. Особенно предпочтительны фармацевтически общепринятые сурфактанты, такие как, например:
частичные сложные эфиры и сложные эфиры жирных кислот, и простые эфиры многоатомных спиртов, таких как глицерин, сорбит и т.д. (Span®, Tween®, в частности Tween® 20 и Tween® 80, Myrj®, Brij®), Cremophor® или полоксамеры. Сурфактанты представлены в лекарственном средстве в концентрации 5-200 мкг/мл, предпочтительно 5–120 мкг/мл и особенно предпочтительно 20–75 мкг/мл.
Средство может дополнительно содержать консерванты (например, фенол, m-крезол, p-крезол, парабены), изотонические агенты (например, маннит, сорбит, лактоза, декстроза, трегалоза, хлорид натрия, глицерин), буферные вещества, соли, кислоты и щелочные металлы, а также другие эксципиенты. Указанные вещества могут быть представлены в каждом случае индивидуально или альтернативно в виде смесей.
Глицерин, декстроза, лактоза, сорбит и маннит могут быть представлены в лекарственном средстве в концентрации 100–250 мМ, NaCl в концентрации до 150 мМ, буферные вещества, такие как, например, фосфат, ацетат, цитрат, аргинин, глицилглицин или TRIS (т.е. 2-амино-2-гидроксиметил-1,3-пропандиол) буфер и соответствующие соли представлены в концентрации 5–250 мМ, предпочтительно 10–100 мМ. Дополнительные эксципиенты могут представлять собой, в числе прочего, соли или аргинин.
Концентрация цинка в средстве находится в диапазоне концентрации, который достигается присутствием 0-1000 мкг/мл, предпочтительно 20–400 мкг/мл цинка, наиболее предпочтительно 90 мкг/мл. Однако цинк может быть представлен в форме хлорида цинка, но соль не ограничивается хлоридом цинка.
В лекарственном средстве глицерин и/или маннит может быть представлен в концентрации 100–250 ммоль/л, и/или NaCl предпочтительно представлен в концентрации до 150 ммоль/л.
В лекарственном средстве буферное вещество может быть представлено в концентрации 5–250 ммоль/л.
Другим объектом изобретения является лекарственное средство инсулина, который содержит дополнительные добавки, такие как, например, соли, которые задерживают высвобождение инсулина. Смеси указанных инсулинов с отсроченным высвобождением и средств, описанных выше, включены в настоящее описание.
Другой объект изобретения относится к способу получения указанных лекарственных средств. Для получения средств ингредиенты растворяют в воде и регулируют значение pH с помощью HCl и/или NaOH. Аналогичным образом, другой объект изобретения относится к применению таких средств для лечения диабета.
Другой объект изобретения относится к применению или к добавлению сурфактантов в качестве стабилизатора во время процесса получения инсулина, аналогов инсулина или производных инсулина или их средств.
Изобретение дополнительно относится к средству, как описано выше, который дополнительно включает в себя также глюкагоноподобный пептид-1 (GLP1) или его аналог или их производные, или эксендин-3 или эксендин-4 или аналог или их производные, предпочтительно эксендин-4.
Изобретение дополнительно относится к средству, как описано выше, в котором аналог эксендина-4 выбран из группы, включающей в себя
H-desPro36-эксендин-4-Lys6-NH2,
H-des(Pro36,37)-эксендин-4-Lys4-NH2 и
H-des(Pro36,37)-эксендин-4-Lys5-NH2,
или их фармакологически переносимая соль.
Изобретение дополнительно относится к средству, как описано выше, в котором аналог эксендина-4 выбран из группы, включающей в себя
desPro36 [Asp28]эксендин-4 (1-39),
desPro36 [IsoAsp28]эксендин-4 (1-39),
desPro36 [Met(O)14, Asp28]эксендин-4 (1-39),
desPro36 [Met(O)14, IsoAsp28]эксендин-4 (1-39),
desPro36 [Trp(O2)25, Asp28]эксендин-2 (1-39),
desPro36 [Trp(O2)25, IsoAsp28]эксендин-2 (1-39),
desPro36 [Met(O)14Trp(O2)25, Asp28]эксендин-4 (1-39) и
desPro36 [Met(O)14Trp(O2)25, IsoAsp28]эксендин-4 (1-39),
или их фармакологически переносимую соль.
Изобретение дополнительно относится к средству, как описано в предыдущем параграфе, в котором пептид -Lys6-NH2 прикреплен к С-концам аналогов эксендина-4.
Изобретение дополнительно относится к средству, как описано выше, в котором аналог эксендина-4 выбран из группы, включающей в себя
H-(Lys)6- desPro36 [Asp28]эксендин-4(1-39)-Lys6-NH2
des Asp28Pro36, Pro37, Pro38 эксендин-4(1-39)-NH2,
H-(Lys)6- desPro36, Pro37, Pro38 [Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5 des Pro36, Pro37, Pro38 [Asp28]эксендин-4(1-39)-NH2,
des Pro36, Pro37, Pro38 [Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36, Pro37, Pro38 [Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-des Pro36, Pro37, Pro38 [Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36 [Trp(O2)25, Asp28]эксендин-4(1-39)-Lys6-NH2,
H-des Asp28 Pro36, Pro37, Pro38 [Trp(O2)25]эксендин-4(1-39)-NH2,
H-(Lys)6-des Pro36, Pro37, Pro38 [Trp(O2)25, Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-desPro36, Pro37, Pro38 [Trp(O2)25, Asp28]эксендин-4(1-39)-NH2,
des Pro36, Pro37, Pro38 [Trp(O2)25, Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36, Pro37, Pro38 [Trp(O2)25, Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5-des Pro36, Pro37, Pro38 [Trp(O2)25, Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6- des Pro36 [Met(O)14, Asp28]эксендин-4(1-39)-Lys6-NH2,
des Met(O)14Asp28 Pro 36, Pro37, Pro38 эксендин-4(1-39)-NH2,
H-(Lys)6-des Pro36, Pro37, Pro38 [Met(O)14, Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-des Pro36, Pro37, Pro38 [Met(O)14, Asp28] эксендин-4(1-39)-NH2,
des Pro36, Pro37, Pro38 [Met(O)14, Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36, Pro37, Pro38 [Met(O)14, Asp28]эксендин-4(1-39)-Lys6-NH2,
H-Asn-(Glu)5 des Pro36, Pro37, Pro38 [Met(O)14, Asp28] эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36 [Met(O)14, Trp(O2)25, Asp28]эксендин-4(1-39)-Lys6-NH2,
des Asp28 Pro36, Pro37, Pro38 [Met(O)14, Trp(O2)25]эксендин-4(1-39)-NH2,
H-(Lys)6-des Pro36, Pro37, Pro38 [Met(O)14, Trp(O2)25, Asp28]эксендин-4(1-39)-NH2,
H-Asn-(Glu)5-des Pro36, Pro37, Pro38 [Met(O)14, Asp28] эксендин-4(1-39)-NH2,
des Pro36, Pro37, Pro38 [Met(O)14, Trp(O2)25, Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-(Lys)6-des Pro36, Pro37, Pro38 [Met(O)14, Trp(O2)25, Asp28]эксендин-4(1-39)-(Lys)6-NH2,
H-Asn-(Glu)5- des Pro36, Pro37, Pro38 [Met(O)14, Trp(O2)25, Asp28] эксендин-4(1-39)-(Lys)6-NH2,
или их фармакологически переносимую соль.
Изобретение дополнительно относится к средству как описано выше, который дополнительно включает в себя Arg34, Lys26 (Nε(γ-глутамил(Nα-гексадеканоил))) GLP-1 (7-37) [лираглутид] или их фармакологически переносимую соль.
В одном воплощении настоящее изобретение относится к лекарственному средству на водной основе, содержащей инсулин гларгин в диапазоне 200–1000 Ед/мл [эквимолярно 200-1000 МЕ инсулина человека], предпочтительно 200 Ед/мл - 650 Ед/мл, еще предпочтительнее 700 Ед/мл - 1000 Ед/мл, более предпочтительно 270–330 Ед/мл и наиболее предпочтительно в концентрации 300 Ед/мл, при условии, что концентрация инсулина гларгина в указанной композиции не равна 684 Ед/мл.
Кроме того, средство также может включать в себя аналог эксендина-4, как, например, ликсисенатид, эксенатид и лираглутид. Указанные аналоги эксендина-4 представлены в средстве в диапазоне от 0,1 мкг до 10 мкг на Ед. инсулина гларгина, предпочтительно от 0,2 до 1мкг на Ед. инсулина гларгина и еще более предпочтительно от 0,25 мкг до 0,7 мкг на Ед. инсулина гларгина. Предпочтительным является ликсисенатид.
Кроме того, лекарственное средство на водной основе может содержать один или более эксципиентов, выбранных из группы, включающей в себя цинк, m-крезол, глицерин, полисорбат 20 и натрий. В частности лекарственное средство на водной основе может содержать 90 мкг/мл цинка, 2,7 мг/мл m-крезола и 20 мг/мл глицерина 85%. Дополнительно лекарственное средство на водной основе может содержать 20 мкг/мл полисорбата 20.
Значение pH лекарственного средства на водной основе составляет от 3,4 до 4,6, предпочтительно от 4 до 4,5.
Настоящее изобретение относится к способу лечения диабета I типа и II типа, включающему в себя введение водной фармацевтической композиции настоящего изобретения пациенту с диабетом. Из числа различных раскрытых диапазонов концентраций предпочтительной является концентрация 300 Ед/мл, и предпочтительным аналогом инсулина является инсулин гларгин. Дополнительно лекарственное средство на водной основе также может содержать цинк, m-крезол, глицерин, полисорбат 20 и натрий и их смеси в диапазонах, раскрытых в описании в отношении лекарственного средства на водной основе настоящего изобретения. В предпочтительном воплощении лекарственное средство на водной основе также включает в себя от 0,1 мкг до 10 мкг ликсисенатида на Ед. инсулина гларгина.
Инсулин вводят предпочтительно один раз в день, но также он может быть введен два раза в день при необходимости. Необходимые дозы являются функцией потребностей конкретного пациента, определяемых достижением нормальных или приемлемых уровней глюкозы крови.
Настоящее изобретение также относится к способу увеличения продолжительности действия инсулина гларгина в лечении диабета I типа и диабета II типа у пациента, включающему в себя введение указанному пациенту лекарственного средства на водной основе настоящего изобретения. Из числа различных раскрытых диапазонов концентраций предпочтительной является концентрация 300 Ед/мл. Дополнительно лекарственное средство на водной основе также может содержать цинк, m-крезол, глицерин, полисорбат 20 и натрий и их смеси в диапазонах, раскрытых в описании в отношении лекарственного средства на водной основе настоящего изобретения. В предпочтительном воплощении лекарственное средство на водной основе также включает в себя от 0,1 мкг до 10 мкг ликсисенатида на Ед. инсулина гларгина.
Настоящее изобретение также относится к способу уменьшения частоты возникновения гипогликемии в лечении диабета I типа и диабета II типа у пациента инсулином гларгином, включающему в себя введение указанному пациенту лекарственного средства настоящего на водной основе изобретения. Из числа различных диапазонов концентраций является концентрация 300 Ед/мл. Дополнительно лекарственное средство на водной основе также может содержать цинк, m-крезол, глицерин, полисорбат 20 и натрий и их смеси в диапазонах, раскрытых здесь в отношении лекарственного средства на водной основе настоящего изобретения. В предпочтительном воплощении лекарственное средство на водной основе также содержит от 0,1 мкг до 10 мкг ликсисенатида на Ед. инсулина гларгина.
Настоящее изобретение также относится к способу предоставления базального инсулина безпикового пролонгированного действия в лечении диабета I типа и диабета II типа у пациента инсулином гларгином, включающему в себя введение указанному пациенту лекарственного средства на водной основе настоящего изобретения. Из числа различных раскрытых диапазонов концентраций предпочтительной является концентрация 300 Ед/мл. Дополнительно лекарственное средство на водной основе также может содержать цинк, m-крезол, глицерин, полисорбат 20 и натрий и их смеси в диапазонах, раскрытых здесь в отношении лекарственного средства на водной основе настоящего изобретения. В предпочтительном воплощении лекарственное средство на водной основе также включает в себя от 0,1 мкг до 10 мкг ликсисенатида на Ед. инсулина гларгина.
Применение средства на водной основе в соответствии с любым из указанных выше пунктов в лечении диабета I типа и диабета II типа.
Дополнительно заявка описывается с помощью нескольких примеров, которые не являются ограничивающими.
Пример 1. Описание протокола
Данное исследование являлось одноцентровым рандомизированным контролируемым слепым с четырьмя периодами, с 2-мя видами лечения, с 2-мя последовательностями перекрестным исследованием на здоровых субъектах с шестью визитами:
|
Субъекты получали однократные подкожные дозы 0,4 Ед/кг инсулина гларгина U100 и инсулина гларгина U300, попеременно инъецируемые в две противоположные стороны околопупочной области (слева, справа, слева, справа) в четыре разных дня. Исследуемое средство вводили с повтором лечения R и T в 2 последовательностях, RTTR или TRRT в P1-P4. Период вымывания от 4 до 18 дней отделял каждый день введения доз.
|
P1 должен иметь место не позднее чем 3-21 день после SCR. Визит EOS должен происходить между 4 и 14 днями после P4.
Во время P1-P4, субъектов подсоединяли к Биостатору для измерения глюкозы крови и регулирования скорости инфузии глюкозы. Осуществляли мониторинг уровней глюкозы крови и скорости инфузии глюкозы (GIR) в течение 90 минут (исходный период) до подкожной инъекции исследуемого средства и в течение 30 часов после введения исследуемого средства. Инициировали инфузию 20% раствора глюкозы для поддержания уровней глюкозы крови на 5% ниже индивидуального уровня глюкозы натощак, определяемого как среднее значение 3 показателей глюкозы крови натощак, измеренных за 60, 30 и 5 минут до введения исследуемого средства. Получали профили GIR. Образцы крови получали в заранее установленные моменты времени во время периода эугликемического клэмп-теста для определения сывороточных концентраций инсулина гларгина. За исключением водопроводной воды, субъекты находились в состоянии натощак во время периода поддержания фиксированной концентрации глюкозы в условиях клэмп-теста.
Предполагалось, что продолжительность настоящего исследования для субъекта составит 13 недель между SCR и визитом EOS.
Протокол исследования был предоставлен для рассмотрения и письменного утверждения независимым комитетам по этике и/или институтским наблюдательным советам. Протокол соответствовал рекомендациям 18-й Всемирной медицинской ассамблеи (Хельсинки,1964) и всех действующих изменений и дополнений. Протокол также соответствовал законам и положениям, а также существующим методическим рекомендациям Германии, где проводилось исследование. Информированное согласие получали до проведения каких-либо процедур, связанных с исследованием.
Пример 2: Отбор субъектов
Двадцать четыре (24) здоровых субъекта были запланированы для проведения лечения, чтобы получить 20 субъектов, завершивших исследование.
Субъекты, отвечавшие всем нижеследующим критериям, рассматривались для включения в исследование:
Демография
 Субъекты любого пола в возрасте от 18 до 50 лет;
Субъекты любого пола в возрасте от 18 до 50 лет;
Вес тела от 50 кг до 110 кг и индекс массы тела от 18 до 28 кг/м²;
Состояние здоровья
Признанные здоровыми после тщательной клинической оценки (подробный анамнез и полное физическое обследование);
Не курящие в течение по меньшей мере 3 месяцев;
Электрокардиограмма в 12 отведениях и основные показатели жизнедеятельности, если только Исследователь рассматривает отклонение как клинически несущественное
Нормальные основные показатели жизнедеятельности после 5 минут отдыха в положении лежа на спине:
95 мм рт.ст. ≤ систолическое давление крови ≤140 мм рт.ст.;
45 мм рт.ст. ≤ диастолическое артериальное давление ≤90 мм рт. ст.;
40 уд/мин ≤ пульс ≤100 уд/мин;
Нормальная ЭКГ в 12 отведениях; 120 мс <PR<220 мс, QRS<120 мс, QTc≤430 мс
(для женщин: QTc≤450 мс);
Лабораторные параметры в пределах диапазона нормальных значений, если только Исследователь рассматривает отклонение как клинически несущественное для здоровых субъектов; однако сывороточный креатинин и ферменты печени (AST, ALT) должны быть строго ниже верхнего предела нормальных лабораторных значений;
Нормальный метаболический контроль, определяемый как глюкоза сыворотки крови натощак (≤100 мг/дл) и гликозилированный гемоглобин (HbA1c≤6,1%);
Субъекты не должны принимать лекарство, отпускаемое по рецепту, на регулярной основе, в течение по меньшей мере четырех (4) недель до участия в исследовании;
Ограничения для женщин-субъектов
Женщины-субъекты детородного возраста (определяемые как женщины в пременопаузе и не стерилизованные хирургически или в постменопаузе менее чем 2 года) и сексуально активные должны использовать адекватные методы контроля рождаемости. Адекватные методы контроля рождаемости определяют как высокоэффективный метод контрацепции (индекс Перла <1%) такой как импланты, инъекции, комбинированные противозачаточные таблетки или гормональные MED (внутриматочные средства). Женщины в постменопаузе для целей настоящего клинического испытания включают в себя: женщин с аменореей в течение 2 или более лет или хирургически стерилизованных женщин;
Женщины-субъекты должны иметь отрицательный результат теста по моче на хорионический бета-гонадотропин (beta-HCG) во время скрининга перед исследованием и до первого клэмп-теста;
Административные положения
Должны дать письменное информированное согласие до проведения какой-либо процедуры исследования;
Должны покрываться системой медицинского страхования и/или в соответствии с рекомендациями действующего федерального закона применительно к биомедицинским исследованиям;
- Не должны находиться под административным или судебным надзором.
Субъекты, соответствующие любому из перечисленного ниже, не включались в исследование:
Анамнез и клинический статус
Присутствие в анамнезе или наличие клинически значимого сердечно-сосудистого, легочного, желудочно-кишечного, печеночного, почечного, метаболического, гематологического, неврологического, психического, системного, глазного или инфекционного заболевания; любое острое инфекционное заболевание или признаки острого заболевания;
Наличие или присутствие в анамнезе лекарственной аллергии, или аллергического заболевания, которое диагносцировал и лечил врач;
Чрезмерное потребление напитков, содержащих ксантиновые основания (>4 чашки или стакана/день);
Противопоказания на основании (в соответствии с диапазонами нормальных значений – если значение находится вне диапазона нормальных значений, субъект может быть включен в исследование, если исследователь расценивает данное аномальное значение как клинически несущественное):
- медицинского/хирургического анамнеза и физического обследования
- лабораторных тестов (гематологический анализ, биохимический анализ крови, и анализа мочи с помощью тестовой полоски)
- стандартной электрокардиограммы в 12 отведениях
- кровяного давления и частоты сердечных сокращений
Текущее лечение прописанными по рецепту лекарствами или любое регулярно проводимое лечение прописанными по рецепту лекарствами в течение 4 недель до участия в исследовании.
Симптомы клинически значимого заболевания в течение 3 месяцев до исследования, или любое заболевание главных внутренних органов в течение 4 недель до исследования, которые, по мнению Исследователя, могут препятствовать целям исследования.
Наличие или осложнение заболеваний или других состояний, которые, как известно, могут препятствовать абсорбции, распределению, метаболизму или выведению лекарств.
Лекарственная или алкогольная зависимость в анамнезе
Гиперчувствительность к исследуемому средству или к лекарствам со сходной химической структурой в анамнезе
Прогрессирующее смертельное заболевание
Заранее запланированное хирургическое вмешательство во время исследования
Сдача крови более чем 500 мл в течение предыдущих 3 месяцев
Не допускалось участие субъекта в исследовании более одного раза.
Общие условия
Субъект, который, по мнению Исследователя, вероятно не будет соблюдать требования терапии во время исследования, или неконтактен ввиду языковой проблемы или слабого умственного развития или вследствие психического состояния, которое не позволяет субъекту понимать сущность, объем и возможные последствия исследования
Субъект в период исключения предыдущего исследования, согласно применяемым положениям;
Субъект является исследователем или младшим исследователем, научным сотрудником, фармацевтом, координатором исследования, другим сотрудником, непосредственно участвующим в осуществлении протокола;
Получение исследуемого лекарства в течение предыдущих 30 дней до SCR.
Биологический статус
Положительная реакция в любом из следующих тестов: HBs-антиген, анти-HCV антитела, анти-HIV1 антитела, anti-HIV2 антитела;
Положительные результаты анализа мочи с целью выявления запрещенных средств при SCR (амфетамины/метамфетамины, барбитураты, бензодиазепины, каннабиноиды, кокаин, опиаты);
Положительный результат пробы на алкоголь в выдыхаемом воздухе проба на алкоголь в выдыхаемом воздухе
Пример 3: Лечение
Сведения о средствах исследования
|
Расчет дозы для средства Лантус®/инсулин гларгин
Чтобы рассчитать количество инсулина гларгина, данное каждому субъекту (0,4 Ед/кг), вес тела (в кг) определяли до одного десятичного знака после запятой и рассчитанное количество инсулина округляли с увеличением или с уменьшением до целых чисел, как показано в следующих примерах: субъект с весом тела 75,3 кг получал 30 Ед инсулина (75,3×0,4=30,12 которые округляют с понижением до 30); субъект с весом тела 74,4 кг получал 30 Ед инсулина (74,4×0,4=29,76, которые округляют с повышением до 30). Вес тела, записанный в День 1 Периода 1, использовали для расчета дозы исследуемого средства во время Периодов 2, 3 и 4, если вес тела изменялся не более чем на 2 кг, по сравнению с Периодом 1.
Количество в единицах было одинаковым для инсулина гларгина U100 и инсулина гларгина U300. Указанная удельная плотность является одинаковой для обоих лекарственных продуктов. Однако, учитывая, что концентрация инсулина гларгина в три раза выше в инсулине гларгине U300 по сравнению с инсулином гларгином U100, объем, который нужно ввести, и, следовательно, вес составлял 1/3 для инсулина гларгина U300. Шприцы, в которых предоставляли индивидуальную дозу, готовили по весу. Вес нетто регистрировался только в исходной документации Исследователя.
Расчет и приготовление дозы инсулинов
|
Раствор глюкозы: проводили инфузию 20% раствора глюкозы с помощью Биостатора, чтобы сохранить индивидуальный уровень глюкозы у субъектов на определенном заранее заданном уровне. Через вторую инфузионную помпу (часть Биостатора) вводили 0,9% раствор хлорида натрия, чтобы иметь в распоряжении проходимый катетер. Если количество необходимого 20% раствора глюкозы превышало инфузионную пропускную способность Биостатора, вторую помпу включали для инфузии глюкозы.
Гепарин: проводили инфузию 10000 МЕ гепарина в 100 мл 0,9% раствора хлорида натрия в катетер с двойным просветом со скоростью приблизительно 2 мл/час, чтобы сохранить его в доступном состоянии для измерения глюкозы крови Биостатором.
Описание методов маскирования:
Исследование являлось простым слепым исследованием. Различные объемы инъекции препятствовали маскированию лечения. Инъекцию выполнял медицинский работник, не участвовавший иным образом в исследовании. Исследователь имел доступ к рандомизационному коду.
Способ распределения субъектов в группу лечения
Исследуемое средство вводили только субъектам, включенным в данное исследование после процедур, изложенных в протоколе клинического исследования.
Создавали схему рандомизации, которая связывала рандомизационные номера, статифицированные по полу, с последовательностями лечения двух средств Лантус®, которые вводили в виде инъекций в P1-P4.
Утром в День 1 Периода 1, как только Исследователь подтверждал, что субъекты удовлетворяют критериям, указанным в протоколе, подходящие субъекты были рандомизированы в исследовательском центре. Рандомизационный номер назначали номеру субъекта последовательно в порядке, в котором подтверждали соответствие требованиям субъектов перед P1. Первый субъект для квалификации в гендерную страту после SCR получал первый рандомизационный номер для соответствующей гендерной страты. Следующий субъект, который квалифицировался в пределах страты, получал следующий рандомизационный номер в пределах страты.
Рандомизационный номер использовали в качестве номера набора для лечения для назначения набора для лечения субъекту. Каждому субъекту давали исследуемое средство, несущий номер набора для лечения, к которому он был отнесен. Набор для лечения, содержащий IP, нес на контейнере-коробке общую информацию, номер набора для лечения, номер периода, поле для записи номера субъекта и дополнительные предписания в соответствии с требованиями местных нормативных документов.
Субъекты, которые навсегда выбывали из исследования, сохраняли номер субъекта и рандомизационный номер, если уже получили их.
Упаковка и маркирование
Исследуемое средство был упакован Sanofi-Aventis Германия GmbH, Франкфурт-на-Майне, Германия, согласно плану рандомизации. Картриджи, содержащие исследуемое средство и картонные коробки, в которые они были упакованы, имели маркировки с номером исследования, рандоминизационным номером, номером партии, условиями хранения, спонсором и номером P.
Запасы исследуемого средства получали в одной поставке. Все контейнеры имели маркировки одинакового формата. Кроме того, в комплект поставки входил 1 комплект маркировок для шприцов. Исследуемое средство и резервное лекарство хранили в разных холодильниках.
Перед введением исследуемого средства фармацевт или лицо, назначенное им, готовил шприцы с соответствующим исследуемым средством и маркировал шприц с указанием номера субъекта, рандоминизационного номера и соответствующего периода согласно контейнерам с исследуемым средством.
Содержание маркировок соответствовало местным регулирующим инструкциям и требованиям.
Условия хранения
Исследуемое средство хранили в защищенном от света месте при температуре от +2°C до +8°C. Исследуемое средство предохраняли от замораживания. Во время приготовления средство не нужно защищать от света.
Резервные образцы (300 картриджей Лантус® U100 и 300 картриджей инсулина гларгина U300) хранили в таких же защищенных условиях на уровне исследовательского центра.
Пример 4. Оценка исследуемого продукта
Активность или фармакодинамика
Способом действия является стимуляция инсулиновых рецепторов инсулином гларгином. Последующий периферический захват глюкозы и супрессия продукции эндогенной глюкозы составляют глюкодинамические эффекты, вызывающие уменьшение концентрации глюкозы в крови. Конечная утилизация глюкозы лучше всего характеризуется показателем количества глюкозы, требуемым для сохранения концентрации глюкозы в крови на постоянном уровне.
Эугликемический клэмп-тест применяли для оценки количества глюкозы, необходимой, чтобы поддержать концентрации глюкозы крови на 5% ниже исходного уровня после инъекции инсулина гларгина.
Методы клинической оценки
Определение глюкозы крови в режиме реального времени осуществляли с помощью Биостатора (Life Sciences instruments, Elkhart, IN, USA) с использованием глюкозооксидазного метода.
Глюкозу крови вне исследовательского центра определяли с помощью анализатора глюкозы Super GL также с применением глюкозооксидазного метода.
Фармакологические переменные/конечные показатели
Количество глюкозы, утилизируемой на единицу (дозы) подкожно введенного инсулина является оценкой измерения глюкодинамического эффекта.
Постоянно регистрируемая скорость инфузии глюкозы (GIR) является отражением профиля действия введенного инсулина.
Первичная переменная/основной конечный показатель
Первичная фармакодинамическая переменная представляет собой площадь под кривой “скорость инфузии глюкозы – время на протяжении 24 часов” [GIR-AUC0-24h (мг⋅кг-1)].
Вторичная переменная/конечный показатель
Вторичная фармакодинамическая переменная представляет собой время до 50% GIR-AUC0-24h
[T50% - GIR-AUC(0-24h) (h)].
Фармакокинетика
Время забора образцов
Образцы крови для оценки сывороточных концентраций инсулина гларгина и C-пептида брали за 1 час, 30 мин и непосредственно перед подкожной инъекцией исследуемого средства, в дальнейшем через 30 мин, 1 час, 2 часа и затем каждые два часа вплоть до 24 часов, и через 30 часов после инъекции.
Нумерация проб инсулина гларгина представляла собой P00, P01, P02, P03, P04 и т.д., нумерация проб C-пептида представляла собой C00, C01, C02, C03, C04 и т.д. (см. также диаграмму исследования).
Число фармакокинетических образцов
Минимально брали 18 образцов за каждый визит проведения клэмп-теста (P1-P4). Всего у субъекта брали 72 образца.
Процедура обработки PK
Точное время сбора образцов должно быть зарегистрировано в CRF. Должны быть использованы специальные процедуры хранения и перевозки фармакокинетических образцов (инсулин гларгин, C-пептид).
Биоаналитический метод
Биоанализ проводили, используя в качестве основы требования Надлежащей лабораторной практики (GLP), применимые к данному типу исследования, установленные в Принципах OECD Надлежащей лабораторной практики (в редакции 1997), ENV/MC/CHEM (98)17 и в правилах GLP в отношении определенной страны.
Поскольку не имеется резервных образцов, приоритет отдается определению инсулина гларгина.
Инсулин гларгин
Концентрации инсулина гларгина в сыворотке крови определяли с помощью радиоиммунологического анализа (RIA) инсулина человека (набор Insulin RIA kit, ADALTIS, Италия) калиброванного для инсулина гларгина. Набор REF 10624.
Нижний предел количественного определения (LLOQ) для данного теста составлял 4,92 мкЕд/мл.
C-пептид
Концентрации C-пептида в сыворотке определяли c помощью радиоиммунологического анализа (RIA) для C-пептида (набор C-peptide RIA, ADALTIS, Италия). Набор REF C-peptide 10282.
Нижний предел количественного определения (LLOQ) составлял 0,090 нмоль/л.
Краткое изложение биоаналитического метода
|
Фармакокинетические переменные/конечные показатели
Кривая концентрация инсулин гларгин - время представляла собой измерение системного воздействия инсулина подкожной инъекции IP.
Первичная переменная/конечный показатель
Первичная фармакокинетическая переменная представляла собой площадь под кривой “концентрация в сыворотке крови инсулина гларгина – время” [INS-AUC0-24h (мкЕд⋅час⋅мл-1)].
Вторичная переменная/конечный показатель
Вторичная фармакокинетическая переменная представляла собой время до 50% INS-AUC0-24h [T50%-INS-AUC(0-24h) (h)].
Отобранный объем крови
|
Меры по сохранению маскирования испытания
Исследование являлось простым слепым исследованием. Биоаналитические определения выполняли после клинического завершения. Код лечения был известен для описания какого-либо серьезного нежелательного явления (SAE), непредвиденного и обоснованно связанного с применением IP, по мнению Исследователя и/или Спонсора.
Пример 5. Процедуры исследования
Расписание визитов
Процедуры скрининга
Медицинскую карту каждого из потенциальных субъектов проверяли перед началом исследования, чтобы установить соответствие требованиям для участия в исследовании. Субъекты находились в состоянии натощак (за исключением воды) в течение 10 часов до скринингового обследования в SCR.
Оценку проводили по следующим пунктам/обследованиям:
Возраст и раса
Физическое обследование (включая сердечно-сосудистую систему, грудь и легкие, щитовидную железу, живот, нервную систему, кожу и слизистые, и костно-мышечную систему)
Значимый медицинский и хирургический анамнез (документируют только сведения, относящиеся к исследованию)
Антропометрические характеристики: рост и вес, расчет BMI [вес в кг⋅(рост в м)-2]
Давление крови и пульс (после 5 мин в положении лежа и 3 мин в положении стоя)
Центральная температура (тимпаническая)
Стандартная ЭКГ в 12 отведениях
Гематологический статус, биохимический анализ крови и анализ мочи (с помощью тестовой полоски)
Коагуляционный статус (INR, aPPT)
Анализ мочи с целью выявления запрещенных средств
Анализ на алкоголь (алкогольно-респираторная трубка)
Нормальный метаболический контроль, определяемый как глюкоза крови натощак (≤100 мг.дл-1) и гликозилированный гемоглобин (HbA1c≤6,1 %)
Тест на гепатиты B/C и HIV
Если субъект не проходил скрининг, все данные, полученные при SCR, включая лабораторные результаты скрининговых тестов, имелись в наличии медицинской карте субъекта.
Описание по типу визита
Период(ы)
Каждый период исследования (с P1 по P4) продолжался 2 дня, День 1 и День 2. День 1 являлся днем начала эугликемического клэмп-теста и введения исследуемого средства. День 2 являлся днем окончания эугликемического клэмп-теста, который продолжался 30 часов после введения исследуемого средства. Между периодами исследования (P1-P4) имелся период вымывания продолжительностью 4-18 дней. Не разрешалась физическая активность (например, горный велосипед, садоводство с тяжелыми нагрузками и т.д.) в течение 2 дней до введения каждого исследуемого средства. Потребление алкогольных напитков, грейпфрутового сока и стимулирующих напитков, содержащих производные ксантина (чай, шоколад, кофе, Кока-Кола™-подобные напитки, и т.д.), и потребление грейпфрута не разрешалось в течение 24 часов перед началом и до завершения эугликемического клэмп-теста. Субъекты находились в состоянии натощак (за исключением воды) в течение 10 часов до Дня 1 каждого периода исследования (P1-P4) и оставались в состоянии натощак (за исключением воды) до окончания эугликемического клэмп-теста. Субъекты должны были оставаться в клинике в течение приблизительно 32 часов во время каждого визита проведения клэмп-теста.
Утром в День 1 Периода 1 субъекту назначали 9-цифровой номер субъекта, начиная с 276001001. Следующий субъект, который соответствовал требованиям для участия в SCR, получал номер субъекта 276001002, и т.д. Первый субъект получал рандомизационный номер 101. Следующий субъект, который соответствовал требованиям, получал рандомизационный номер 102.
Субъектов просили подтвердить, что они не имели клинически значимых изменений в своем физическом состоянии и соблюдали общие ограничения и ограничения в питании, которые указаны в протоколе, после предыдущих периодов. При нарушении критериев исследования субъекты исключаются из участия в исследовании. В зависимости от вида нарушения субъект может быть исключен только на определенный период, что позволяет повторно назначить день исследования. Любые нарушения протокола обсуждаются со Спонсором в индивидуальном порядке заблаговременно.
Любые изменения в состоянии здоровья субъектов со времени последнего периода описывали в медицинских картах субъектов (первичная документация) и в CRF.
Давление крови, пульс и центральную температуру (тимпаническую) записывали в положении лежа на спине, после по меньшей мере 5 минут отдыха, утром в День 1, до и после завершения процедур клэмп-теста через 30 часов после введения каждого исследуемого средства (День 2). Вес тела, анализ на алкоголь и RBC, Hb, HcT (только перед периодом клэмп-теста P3 и P4) оценивали только перед началом клэмп-теста утром в День 1.
В День 1 каждого периода субъектов принимали в клинике в 6:30 утра. После прохождения описанных выше обследований у субъектов готовили по три системы для внутривенного вливания. Вену тыльной стороны руки или вену внешней стороны запястья левой руки катетеризировали ретроградным способом и подсоединяли к Биостатору (Life Sciences instruments, Elkhart, IN, USA) для постоянного получения артериализованной венозной крови для определения глюкозы крови. Для достижения артериализации левую руку помещали в устройство “Hot-Box” с температурой 55°C. Вторую систему для внутривенного вливания соединяли с латеральной подкожной веной левой руки и использовали для сбора образцов для определения сывороточного инсулина гларгина и референсного определения глюкозы крови. Третью вену катетеризировали на противоположном предплечье, чтобы обеспечить инфузию 20% раствора глюкозы и 0,9% физиологического раствора с помощью Биостатора.
Биостатор определял уровни глюкозы крови и регулировал скорость инфузии глюкозы для поддержания уровней глюкозы на 5% ниже индивидуального уровня глюкозы крови в состоянии натощак, определяемого как среднее значение 3 показателей глюкозы крови натощак, измеренных за 60, 30 и 5 минут до введения исследуемого средства. Дополнительные образцы крови по 0,3 мл для определения глюкозы крови брали за 60, 30, и 5 минут до введения исследуемого лекарства, чтобы проверить в сравнении с лабораторным эталоном на основании глюкозооксидазного метода.
Приблизительно в 09:00 утра вводили инъекцию либо инсулина гларгина U100 (коммерческое средство), либо инсулина гларгина U300 в околопупочную область, 5 см в сторону от пупка (слева, справа, слева, справа), используя стандартную технику кожной складки. Применяли инсулиновые шприцы U100 (Производитель: Beckton & Dickinson) объемом 0,5 мл с иглой 0,30 мм × 8 мм (30G).
Исследуемое средство маркировали с нанесением соответствующего номера набора для лечения, номера субъекта (который должен быть задокументирован на контейнере-коробке после рандомизации), и номера периода (см. Раздел 8.5 Упаковка и маркировка).
После введения исследуемого средства начинали инфузию 20% раствора глюкозы с переменной скоростью, если уровень глюкозы опускался ниже, чем на 5% от индивидуального уровня в состоянии натощак, для поддержания указанного уровня. Продолжительность периода клэмп-теста составляла 30 часов. Скорость подачи глюкозы регулировалась Биостатором в ответ на изменения глюкозы крови с интервалами в 1 минуту, с использованием заранее заданного алгоритма. Значения глюкозы крови, полученные с помощью Биостатора, проверяли в сравнении с лабораторным эталоном, на основе глюкозооксидазного метода с 30 минутными интервалами в течение всего клэмп-теста. При необходимости Биостатор калибровали повторно в соответствии с результатами лабораторного эталонного метода. Субъекты оставались в положении лежа на спине во время периода проведения клэмп-теста.
Образцы крови для определения сывороточных концентраций инсулина гларгина и C-пептида брали за 1 ч 30 мин и непосредственно перед введением средства, и в дальнейшем через 30 мин, 1 час, 2 часа и затем каждые два часа вплоть до 24 часов, и 30 часов после введения исследуемого средства.
В День 2 каждого периода исследования (P1-P4), субъектов кормили после завершения эугликемического клэмп-теста. Записывали давление крови, пульс и температуру центральной части тела (тимпаническую) и брали образец крови для определения глюкозы крови. Субъектов выписывали из клиники после того, Исследователь убеждался в безопасности состояния их здоровья.
Места инъекций осматривали во время всего периода проведения клэмп-теста. Любые изменения в состоянии здоровья субъектов записывали в медицинские карты субъектов (первичная документация) и в CRF.
Гематологическая безопасность
В P3 исследовали RBC, Hb и Hct на подверженность анемии в P 4. В случае положительного результата интервал между P3 и P4 увеличивали до максимально разрешенных 18 дней и осуществляли дополнительную оценку RBC, Hb и Hct перед P4.
Процедуры выписки
Субъекты возвращались для визита EOS между 4 и 14 днями после P4. Субъекты были в состоянии натощак (за исключением воды) в течение 10 часов. Все изменения в состоянии здоровья субъектов со времени последнего периода описывали в медицинских картах субъектов (первичная документация) и в CRF.
Оценивали субъектов по следующим пунктам /обследованиям:
Физическое обследование (включая сердечно-сосудистую систему, грудь и легкие, щитовидную железу, живот, нервную систему, кожу и слизистые, и костно-мышечную систему)
Вес
Давление крови и пульс (после 5 мин в положении лежа на спине)
Центральная температура (тимпаническая)
Стандартная ЭКГ в 12 отведениях
Гематологический статус, биохимический анализ крови, и анализ мочи (с помощью тестовой полоски)
Тест β-HCG по моче (только для женщин)
Субъектов выписывали в День 2 каждого периода, после полной проверки Исследователем доступных данных по безопасности из числа имеющихся.
Схема сбора биологических образцов
Кровь
SCR (Скрининг):
Гематология, биохимический анализ крови, HbA1c, серология (тест на гепатит B/C, тест на HIV): собирали приблизительно 20 мл крови.
P1-P4 (День 1 и 2):
Глюкоза крови
Биостатор автоматически измеряет глюкозу крови с интервалами в одну минуту в течение полного периода клэмп-теста, включая период перед ведением исследуемого средства. Объем крови, требуемый Биостатором, составлял 2 мл⋅ч-1. Расчетный объем крови 252 мл был необходим для считывания показаний глюкозы с помощью Биостатора на протяжении четырех периодов. Образцы крови (0,3 мл) для проверки значений глюкозы крови из Биостатора собирали за 60, 30, 5 и 0 минут до введения дозы и с 30-минутными интервалами после введения дозы до конца клэмп-теста (30 часов). В течение четырех периодов собрали планируемый объем крови 41 мл.
Сывороточные концентрации инсулина гларгина и C-пептида
Образцы венозной крови (3,5 мл) брали за 1 час, 30 мин и непосредственно перед введением дозы, через 30 мин, 1 час, 2 часа и затем каждые два часа вплоть до 24 часов, и через 30 часов после введения дозы. Расчетный объем крови 252 мл был собран на протяжении четырех периодов. Приоритет отдавали определению инсулина гларгина. Резервные образцы использовали только для определения концентрации C-пептида.
RBC, Hb, Hct
Венозную кровь собирали до начала клэмп-теста Периода 3 и 4.
Собрали приблизительно 4 мл крови в течение двух периодов.
Визит окончания исследования (EOS):
Гематологический анализ, биохимический анализ крови: собирали приблизительно 12 мл крови.
Тест β-HCG в моче (только для женщин)
Общий объем крови SCR-EOS:
Всего в течение полного исследования собрано приблизительно 585 мл крови каждого субъекта.
Моча
Качественный анализ мочи с целью выявления запрещенных средств проводили в SCR и EOS. Анализ мочи с целью выявления запрещенных средств включает в себя определение амфетаминов/метамфетаминов, барбитуратов, бензодиазепинов, каннабиноидов, кокаина, опиатов. Качественный анализ мочи для оценки безопасности с помощью тест-полосок проводили в SCR и EOS. Анализ мочи на безопасность включает в себя определение: значения pH, белка, глюкозы, крови, эритроцитов, лейкоцитов, билирубина, уробилиногена, кетонов, удельной плотности и нитритов.
Схема измерения других переменных исследования
Физическое обследование осуществляли в SCR и EOS.
Центральную температуру (тимпаническую) измеряли в SCR, P1-P4 до и после периода клэмп-теста, и в EOS.
Давление крови и пульс измеряли после 5 минут отдыха в положении лежа, и также после 3 минут в положении стоя во время SCR и EOS. В P1-P4 давление крови и пульс записывали после по меньшей мере 5 минут в положении лежа на спине, до начала процедур кэмп-теста утром в День 1, и после завершения процедур клэмп-теста, через 30 часов после введения каждого исследуемого средства (День 2).
Электрокардиограмму (стандартную в 12 отведениях) записывали во время SCR и EOS.
Вес тела и рост измеряли в SCR. Вес тела записывали утром в День 1 P1-P4 (до введения исследуемого средства) и в EOS.
Анализ на алкоголь (этанол, алкогольно-респираторная трубка) проводили в SCR и в EOS, и утром в День 1 P1-P4 (до введения исследуемого средства).
Ограничение (ограничения) исследования
С вечера Дня 1 (P1-P4) и на протяжении всех Периодов (дней выполнения клэмп-тестов), субъекты воздерживались от употребления алкоголя, чая, кофе, напитков, содержащих цитрусовые или колу, и от курения. Употребление цитрусовых фруктов также запрещалось во время всего исследования. Субъектов просили вести обычной образ жизни на протяжении всего срока проведения испытания, до заключительного контроля, без интенсивной физической активности.
Описание первичных данных
Все оценки, перечисленные ниже, которые описывают в CRF, были подтверждены соответствующим образом подписанной идентифицирующей первичной документацией, относящейся к:
идентификации субъекта, анамнезу;
клиническому обследованию, основным показателям жизнедеятельности, весу тела и росту;
лабораторным исследованиям, ЭКГ;
фармакокинетическим точкам времени;
дате и времени визитов и оценок;
датам и времени введения, и месту инъекции;
AEs;
продолжительности клэмп-теста (время начала и окончания)
другое
CRF рассматривалась в качестве первичной документации для других пунктов.
Пример 6. Статистические заключения
Данный Пример предоставляет информацию для плана статистического анализа исследования. План статистического анализа составлялся до включения субъектов в исследование.
Определение объема выборки
Основным параметром являлась INS-AUC(0-24h), для которой, соответственно, выполняли расчет объема выборки.
С целью расчета объема выборки рассматривали несколько внутрисубъектных SDwithin log-преобразованной INS-AUC(0-24h) между 0,125 и 0,225. Метод расчета объема выборки для подхода с использованием средней биоэквивалентности применяли для 4-периодного, с 2 видами лечения, с 2 последовательностями перекрестного дизайна. Если 90% CI для отношения параметров средства находились полностью в пределах [0,80-1,25], то делали заключение о средней биоэквивалентности параметра.
Исследование HOE901/1022 является основой для предположений об изменчивости. Исходя из статистического анализа исследования HOE901/1022, значение 0,175 можно ожидать для внутрисубъектного стандартного отклонения (SDwithin) на шкале значений, преобразованных по натуральному log.
В таблице ниже представлено число субъектов необходимое, чтобы продемонстрировать среднюю биоэквивалентность отношения вычисленных средних геометрических значений (исследуемое средство в сопоставлении с эталонным средством), используя референсный интервал биоэквавалентности: [0,80-1,25], принимая истинное отношение между 0,85 и 1,15 со статистической мощностью 90%.
|
Для указанного дизайна необходимо 20 субъектов (по 10 субъектов на последовательность), чтобы показать эквивалентность двух средств инсулина гларгина, с мощностью 90%, предполагая истинным отношение 0,9, если истинное SDwithin на log-шкале составляет 0,175.
Ряд из 24 рандомизированных субъектов учитывает возможные случаи досрочного исключения из исследования.
Описание субъектов
Распределение субъектов
Подробное заключение по учету субъектов, включая число субъектов, включенных в исследование, рандомизированных, получивших лечение (т.е. получивших любое количество исследуемого средства), завершивших (т.е. субъекты, которые закончили все периоды лечения исследования), исключенных из исследования, вместе с основными причинами исключения, было составлено для каждой последовательности и для всех субъектов в совокупности.
Распределение субъектов в заключительном визите представлено в списке, включая группу последовательности, статус распределения в конце исследования с датой последнего введения исследуемого лекарства, дату заключительного визита, причину прекращения. Все досрочные исключения из исследования, имевшие место до или после начала первого введения исследуемого лекарства, полностью документируют в основной части отчета о клиническом исследовании (CSR).
Отклонения от протокола
Перед закрытием базы данных проверяли соблюдение протокола в отношении критериев включения и исключения, соблюдения терапии, запрещенных терапий и своевременности проведения и наличия запланированных обследований. Отклонения от протокола устанавливались командой исследования перед закрытием базы данных и перечислялись в Отчете о проверке данных, включая отсутствующие данные и исключения IP, и классифицированные как небольшие или значительные отклонения.
Перечислены индивидуальные отклонения при включении и критерии исключения, которые сообщались Исследователем.
Другие отклонения перечислены и/или описаны в основной части CSR.
Анализ популяции
Популяция, подлежащая анализу
Субъекты, исключенные из анализа популяции, перечислены с указанием последовательности лечения и причины исключения. Любую существенную информацию полностью документировали в CSR.
в случае если субъекты получали лечение, которое отличалось от назначенного согласно схеме рандомизации, анализы проводили согласно полученному лечению, а не в соответствии с лечением при рандомизации.
Популяция фармакокинетического исследования
Все субъекты без каких-либо серьезных отклонений от протокола, касавшихся введения исследуемого лекарства, и для которых имелись параметры PK, были включены в популяцию фармакокинетического исследования. Для субъектов с неполными профилями PK в некоторые, но не во все дни исследования, параметры полных профилей включали в анализ.
Популяция фармакодинамического исследования
Все субъекты без каких-либо серьезных отклонений от протокола, касавшихся введения исследуемого лекарства, и для которых имелись параметры PD, были включены в популяцию фармакодинамического исследования. Для субъектов с неполными GIR - профилями в некоторые, но не во все дни исследования, параметры полных профилей включали в анализ.
Выборка для оценки безопасности
Оценка безопасности основана на субъектах, которые получили дозу исследуемого лекарства (популяция субъектов, получавших лечение), независимо от количества принятого лекарства, включая субъектов, преждевременно выбывших из исследования.
Демографические и другие исходные характеристики
Демографические характеристики субъектов, анамнезы и диагнозы
Собирали следующие данные: пол, возраст при скрининге, рост, вес, и раса. Индекс массы тела (BMI) субъекта рассчитывали из данных веса тела и роста:
BMI = вес тела [кг]⋅(рост [м])-2
Все переменные, имеющие отношение к демографическим исходным характеристикам перечислены индивидуально и обобщены.
Отклонения от критериев включения, связанные с анамнезами и диагнозами, перечислены и описаны индивидуально.
Исходные фармакодинамические параметры
Исходные уровни крови суммировали в виде последовательности.
Исходные параметры безопасности
В качестве исходного значения для параметров безопасности принимали последнее запланированное значение перед введением исследуемого лекарства вне зависимости от того применяется оно для переменной в пределах периода или в пределах исследования. Если исходное, до введения дозы значение проверяли повторно перед введением дозы, перепроверенное значение рассматривали в качестве исходного и использовали в статистике.
Продолжительность воздействия исследуемого
лечения и соблюдение терапии
Подробности дозирования исследуемого средства и дополнительную информацию описывали по отдельным субъектам и обобщали в соответствующем случае.
Предшествовавшее/сопутствующее лекарственное
средство/терапия
Предшествующие и сопутствующие лекарственные средства/терапии (при наличии) кодировали согласно Перечню лекарств Всемирной Организации Здравоохранения (WHO-DRL) и описывали по отдельным субъектам.
Анализ фармакологических переменных
Описание фармакодинамической переменной (переменных)
Для сопоставимости субъектов при дозировании инсулина в зависимости от веса тела, все значения GIR делили на вес тела субъектов в кг для анализа. Таким образом, GIR в дальнейшем всегда относится к стандартизированной по весу тела скорости инфузии глюкозы.
Первичная переменная PD представляла собой:
Площадь под кривой “стандартизированная по весу тела скорость инфузии глюкозы – время”
[GIR-AUC(0-24h) (мг⋅кг-1)]
Вторичная переменная PD представляла собой:
Время (ч) до 50% GIR-AUC(0-24h) [T50%-GIR-AUC(0-24h) (ч)]
Получали следующие дополнительные переменные PD:
Площадь под кривой “стандартизированная по весу тела скорость инфузии глюкозы – время до конца клэмп-теста” [GIR-AUC(0-end) (мг⋅кг-1)]
Дробные площади под кривой “стандартизированная по весу тела скорость инфузии глюкозы – время” [GIR-AUC(4-20h), GIR-AUC(0-12h), GIR-AUC(12-24h) (мг⋅кг-1)]
Максимальная стандартизированная по весу тела скорость инфузии глюкозы [GIRmax (мг⋅кг-1⋅мин-1)]
Время до GIRmax [GIR-tmax (ч)]
Чтобы предоставить значимые и достоверные данные, значение GIRmax и, соответственно, время до GIRmax получали из сглаженной кривой GIR для каждого субъекта.
Первичный анализ
Чтобы оценить относительную биоэффективность (активность) для GIR-AUC(0-24h) (мг⋅кг-1), непреобразованный параметр анализировали с помощью линейной модели со смешанными эффектами.
Смешанная модель включает в себя фиксированные показатели для последовательности, периода, средства, и случайные показатели для субъекта внутри последовательности, со специфичными для средства межсубъектными и внутрисубъектными дисперсиями и дисперсией показателя среди субъектов в зависимости от средства. Затем получали точечную оценку и 90% доверительный интервал для отношения средств (T/R) на основе теоремы Филлера [Fieller, 1954].
Делали заключение об эквивалентной биоэффективности (активности), если доверительный интервал для отношения средств находился в пределах [0,80-1,25].
Проверяли предположение о распределении переменной.
Вторичный анализ/анализ вторичных переменных
GIR-профили индивидуальных и средних значений, стандартизированных по весу тела, как и кумулятивные профили средних значений, выраженных в процентах, в динамике по времени наносили на график.
Параметры PD перечисляли по отдельным субъектам, и производили описательную статистику.
Отношение средств (T/R) с доверительными пределами получали для дробных GIR-AUC (мг⋅кг-1) и максимальной стандартизованной скорости инфузии глюкозы [GIRmax (мг⋅кг-1⋅мин-1)], используя соответствующую линейную модель со смешанными эффектами, как описано для первичного анализа.
Время до 50%-GIR-AUC (ч) и время до GIRmax [GIR-tmax (ч)] анализировали с помощью непараметрических методов.
Выполнение клэмп-теста
Индивидуальные профили концентрации глюкозы крови наносили на график.
Анализ данных о безопасности
Все обобщенные результаты данных по безопасности были основаны на выборке для оценки безопасности.
Индивидуальная фаза применения исследуемого средства для анализа данных о безопасности начиналась вместе с первым введением исследуемого средства и заканчивалась вместе с визитом EOS.
Нежелательные явления
Все AE кодировали с помощью MedDRA (текущая версия).
Определения
Появившиеся за время лечения AE
Все AE классифицировали следующим образом:
Возникшие во время применения средства AE (TEAE): AE, которые возникли впервые во время периода применения исследуемого средства или усугублялись во время периода применения исследуемого средства, если присутствовали ранее;
Возникшие не во время применения средства AE (NTEAE): AE, которые возникли вне периода применения исследуемого средства без ухудшения во время периода применения исследуемого средства;
Отнесение к средствам
В целях анализа каждое TEAE относили к последнему средству, полученному перед появлением и/или ухудшением AE. Если TEAE развивалось во время приема одного средства и ухудшалось под действием второго средства, его расценивали как TEAE для обоих средств.
Отсутствующая информация
В случае отсутствующей или противоречивой информации, AE считали как TEAE, если нельзя ясно определить, что оно не является TEAE (например, с помощью частичных данных или другой информации).
Если дата начала AE является неполной или не представлена, считали, что оно имело место после первого введения исследуемого средства, за исключением случая, если неполные данные указывали, что AE начиналось до лечения.
Возникшие во время применения средства нежелательные явления
Все AE перечислены по отдельным субъектам. Они суммированы по средствам, включая обзор по классу системы органов.
Смертельные случаи, серьезные и другие значительные нежелательные явления
При наличии таких случаев, смерть, серьезные AE, и другие важные AE перечислены по отдельным субъектам и подробно описаны в отчете об исследовании.
Нежелательные явления, приводящие к прекращению лечения
AE, приводящие к прекращению лечения, перечислены по отдельным субъектам и подробно описаны в отчете об исследовании.
Клинические лабораторные оценки
Потенциально клинически значимые отклонения от нормы (PCSA) и показатели за пределами стандартного диапазона определены в Плане статистического анализа исследования. Описания потенциально клинически значимых отклонений от нормы (PCSA) и определений за пределами стандартных значений производили по параметру.
Индивидуальные данные перечислены по субъектам и по визитам, как и дополнительная информация.
Субъектов со значениями за пределами стандартных диапазонов и субъектов с PCSA анализировали по средству, и в целом для оценки окончания исследования. Перечислены субъекты с PCSA после включения.
Основные показатели жизнедеятельности
Потенциально клинически значимые отклонения от нормы (PCSA) и показатели за пределами стандартного диапазона определены в Плане статистического анализа исследования. Определения PCSA и определения за пределами стандартного диапазона описаны по параметру.
Субъектов с PCSA анализировали по средству и, в целом, для оценки окончания исследования. Перечислены субъекты с PCSA после включения.
Необработанные значения и полученные параметры суммированы по средствам, и в целом для оценки окончания исследования. Индивидуальные данные перечислены по субъектам и по визитам с пометками об отклонениях от нормальных значений, как и дополнительная информация.
ЭКГ
Потенциально клинически значимые отклонения от нормы (PCSA) и показатели за пределами стандартного диапазона определены в Плане статистического анализа исследования. Определения PCSA и определения за пределами стандартного диапазона описаны по параметру.
Субъектов с PCSA в конце исследования анализировали в целом. Перечислены субъекты с PCSA после включения.
Необработанные значения и полученные параметры, полученные в SCR и в EOS, обобщали в целом. Индивидуальные данные описывали по субъектам и по визиту с метками об отклонениях от нормальных значений, а также с дополнительной информацией.
Анализ фармакокинетических данных
Фармакокинетические параметры
Для получения параметров PK применяли фактические значения относительного времени
Первичная переменная представляла собой
INS-AUC(0-24h). (мкЕд⋅ч⋅мл-1)
Вторичная переменная PK представляла собой
Время (h) до 50% INS-AUC(0-24h) [T50% - INS-AUC(0-24h) (ч)]
Получали следующие дополнительные переменные PK:
Дробные INS-AUC [INS-AUC(4-20h), INS-AUC(0-12h), INS-AUC(12-24h) (мкЕд⋅ч⋅мл-1)]
INS-AUC до конца клэмп-теста [INS-AUC(0-end) (мкЕд⋅ч⋅мл-1)]
Максимальная сывороточная концентрация инсулина [INS-Cmax (мкЕд⋅мл-1)]
Время до INS-Cmax [INS-Tmax (ч)]
Статистический анализ
Описательные анализы
Описательная статистика данных по концентрации представлена в соответствии с моментами времени, указанными в протоколе.
Профили концентрации инсулина в сыворотке крови отдельных субъектов и средних значений наносили на график.
Концентрации инсулина в сыворотке крови приведены по отдельным субъектам и выполнена описательная статистика по временным точкам.
Описательную статистику параметров PK производили по средствам.
Профили С-пептида наносили на график и характеризовали описательно.
Первичный анализ
Чтобы оценить относительную биоэквивалентность для INS-AUC(0-24h), log-преобразованный параметр анализировали с помощью линейной модели со смешанными эффектами.
Смешанная модель включает в себя фиксированные показатели для последовательности, периода, средства, и случайные показатели для субъекта внутри последовательности, со специфичными для средства межсубъектными и внутрисубъектными дисперсиями и дисперсией показателя среди субъектов в зависимости от средства.
Для INS-AUC(0-24h) точечную оценку и 90% доверительные интервалы для отношения средств (T/R) получали путем вычисления оценок и 90% доверительных интервалов для различия между средними значениями средств в рамках модели смешанных эффектов, и с последующим преобразованием в шкалу отношений с помощью антилогарифмического преобразования.
Делали вывод об эквивалентной биодоступности, если доверительный интервал для отношения средств находился в пределах [0,80-1,25].
Анализ вторичных и дополнительных параметров PK
Время до 50%-INS-AUC (ч) и время до максимальной концентрации [INS-Tmax (ч)] анализировали с помощью непараметрических методов.
Log-преобразованные дробные INS-AUC и INS-AUC(0-end) (мкЕд⋅ч⋅мл-1) и максимальную сывороточную концентрацию инсулина гларгина [INS-Cmax (мкЕд⋅мл-1)] анализировали с помощью соответствующей линейной модели со смешанными эффектами, как описано для первичного анализа. Точечные оценки и доверительные интервалы представлены в отчете.
C-пептид
Когда возможно, профили C-пептида представляли графически и характеризовали описательно.
АНАЛИЗ PK/PD
Анализы PK/PD выполняли в порядке исследования в соответствующем случае.
Пример 6: Результаты исследования
Распределение субъектов
Всего 35 субъектов, 11 женщин и 24 мужчин, участвовали в скрининге, из которых 24 здоровых подходящих субъектов были включены в исследование, рандомизированы, и получили по меньшей мере одну дозу исследуемого средства. Из 24 рандомизированных субъектов 1 субъект выбыл из исследования по собственному желанию после получения дозы первого периода лечения. Двадцать три (23) субъекта завершили исследование согласно протоколу и были включены в фармакодинамический (PD) и фармакокинетический (PK) анализы. Все субъекты, получавшие лечение, были включены в оценку безопасности.
Значительных отклонений от протокола не имелось.
Демографические характеристики
Собирали следующие данные: пол, возраст при скрининге, рост, вес, и раса. Индексы массы тела (BMI) каждого субъекта рассчитывали из данных веса тела и роста: BMI = вес тела [кг]⋅(рост [м])-2.
|
Выполнение клэмп-теста
Две группы лечения, Лантус U100 и Лантус U300, были одинаковыми в отношении исходных концентраций глюкозы натощак у субъектов, которые служили для определения клэмп-уровня глюкозы субъектов. Продолжительность клэмп-теста после введения дозы составляла 30 часов и была одинаковой во всех периодах лечения.
Первичные конечные показатели
Эквивалентность по биодоступности (воздействие) для Лантус U100 и Лантус U300 не была установлена. Эквивалентность по биоэффективности (активность) для Лантуса U100 и Лантуса U300 не была установлена.
Первичные переменные
Площадь под кривой сывороточная концентрация инсулина гларгина - время от 0 до 24 часов
(INS-AUC(0-24h)) не являлась эквивалентной для Лантуса U100 и Лантуса U300. Воздействие было меньше приблизительно на 40% в случае U300. Площадь под кривой GIR как функция от времени от 0 до 24 часов (GIR-AUC(0-24h)) не была эквивалентна для Лантуса U100 и Лантуса U300. Активность была меньше приблизительно на 40% в случае U300.
Вторичные переменные
Время до 50% INS-AUC(0-24h) (ч) было одинаковым для Лантус U100 и Лантус U300. Время до 50% GIR-AUC(0-24h) (ч) было больше на 0,545 (ч) (0,158–1,030) для Лантуса U300, что является статистически значимым различием.
Безопасность
Не сообщалось о серьезных нежелательных явлениях (AEs). Пять (5) субъектов за время лечения (исследуемое средство и эталонное средство) сообщили всего о 14 TEAE, из которых все имели интенсивность от слабой до умеренной, и разрешались без последствий. Наиболее часто описываемым явлением было головная боль (4 субъекта за время лечения) с последующей тошнотой, рвотой и лихорадкой (по 1 субъекту, каждый на U100), и процедурная боль (1 субъект на U300). Следует отметить, что головная боль является часто встречающимся наблюдением при клэмп-исследованиях и связана с инфузией гиперосмотических растворов глюкозы. Однако, связь с исследуемыми продуктами не может быть исключена. Не сообщали о реакциях в месте инъекции.
Выводы
Инсулин гларгин U100 и инсулин гларгин U300 не являются эквивалентными по биодоступности (воздействию) и биоэффективности (активности). Воздействие и активность после введения инсулина гларгина U300 являлись приблизительно на 40% меньше по сравнению с воздействием и активностью после введения такого же количества (0,4 Ед/кг) инсулина гларгина U100.
Однако инсулин гларгин U300 показал даже более пологий профиль PK (воздействие) и профиль PD (активность), чем инсулин гларгин U100, какой был бы желателен для базального инсулина. Указанные удивительные и неожиданные различия в воздействии и активности средств инсулина гларгина U100 и инсулина гларгина U300 после одинаковой п/к инъекции у здоровых субъектов представлено по существу на фигурах ниже. Следует отметить, что в то же время концентрация глюкозы в крови являлась постоянной.
Введение инсулина гларгина U300 не сопровождалось проблемами с безопасностью или переносимостью.
Пример 7: Обоснование проведения исследования по сравнению глюкодинамической активности и воздействия трех различных подкожных доз инсулина гларгина U300
Результаты исследования с участием здоровых субъектов (смотреть примеры 1-6) показали отсутствие эквивалентности воздействия и эффективности Лантус® U100 и инсулина гларгина U300. субъекты получали одинаковую дозу инсулина гларгина (0,4 Ед/кг) в U100 и U300, но поступление одинакового по числу единиц количества из U300 вызывало приблизительно на 40% меньшее воздействие и эффект, чем поступление из U100. Однако инсулин гларгин U300 показал даже более пологий фармакодинамический профиль, чем Лантус® U100, который был бы желателен для базального инсулина.
В новом исследовании, описываемом в последующих примерах, соответственно сравнивают глюкодинамическую активность и воздействие трех различных подкожных доз инсулина гларгина U300 со стандартной дозой Лантус® U100 в качестве средств сравнения в условиях эугликемического клэмп-теста у пациентов с диабетом 1 типа. Целью данного исследования является провести аппроксимацию дозы U300, которая соответствует по эффективности 0,4 Ед/кг Лантуса® U100, как определили с помощью параметров утилизации глюкозы крови, предоставляемых клэмп-методом.
Воздействие инсулина гларгина оценивают с помощью профилей “концентрация-время” после подкожного введения, и активность оценивают как утилизацию глюкозы на единицу инсулина.
Исследование разработано, чтобы оценить метаболический эффект и воздействие различных доз инсулина гларгина U300 в сравнении со стандартной дозой Лантуса® U100 в условиях эугликемического клэмп-теста у субъектов с диабетом 1 типа. В исследовании сравнивают 4 вида лечения (R, T1, T2 и T3), 4 периода лечения (TP1-4) и 4 последовательности. Проводится один скрининговый визит (D-28-D-3), 4 визита лечения (D1-D2 в TP1-TP4), и один визит окончания лечения (между D5 и D14 после введения последней дозы) с заключительной оценкой параметров безопасности.
Субъекты получали каждый вид лечения R, T1, T2 и T3 однократно в перекрестном, двойном слепом и рандомизированном методе согласно плану опыта по схеме латинского квадрата. Указанный дизайн считается подходящим для оценки фармакологического эффекта и воздействия различных доз инсулина гларгина U300 по сравнению с Лантусом® U100.
Доза Лантуса® U100 0,4 Ед/кг, выбранная для исследования, хорошо характеризуется в обеспечении эугликемии у пациентов с диабетом 1 типа и полностью изучена в других исследованиях с применением клэмп-метода с участием пациентов с диабетом 1 типа.
Проверяют три различные дозы инсулина гларгина U300, 0,4, 0,6 и 0,9 Ед/кг. Указанный диапазон доз позволяет провести интерполирование приблизительной дозы, равноэффективной 0,4 Ед/кг Лантус® U100. Дозу 0,4 Ед/кг инсулина гларгина U300 уже проверяли на здоровых добровольцах (см. примеры 1-6) и обнаружили, что она является менее активной чем доза 0,4 Ед/кг Лантуса® U100 в пределах 30 часов, заранее установленного окончания периода наблюдения. Биоактивность 0,4 Ед/кг инсулина гларгина U300, которую определяли по общей утилизации глюкозы, являлась на 39,4% ниже, чем биоактивность эталонного лекарства (0,4 Ед/кг Лантус® U100). Предполагали, соответственно, что более высокая доза инсулина гларгина U300, например, 0,6 Ед/кг инсулина гларгина U300, даст в результате приблизительно эквивалентную глюкодинамическую активность, по сравнению с 0,4 Ед/кг Лантуса® U100. Кроме того, пропорциональное повышение дозы позволяет изучить профили воздействия и эффекта в отношении пропорциональности доз.
Исследование с участием пациентов с диабетом 1 типа позволяет избежать смешанного влияния эндогенного инсулина и дает возможность лучше оценить воздействие и продолжительность действия. Кроме того, отсутствие теста, специфичного к инсулину гларгину, вынуждает применять тест, который показывает весь эндогенный инсулин. Таким образом, любой дополнительный источник инсулина, другой, чем экзогенный инсулин гларгин будет являться причиной ошибочных очень высоких концентраций инсулина.
Данное исследование имеет перекрестный дизайн; по практическим и этическим причинам не более чем 3 дозы U300 сравнивают с Лантусом® U100. Оценка глюкодинамической активности продуктов инсулина длительного действия требует условий эугликемического клэмп-теста вплоть до 36 часов в связи с увеличенной продолжительностью действия.
Активный фармацевтический ингредиент, инсулин гларгин, является одинаковым в обоих средствах, U100 и U300. Дозы, используемые в исследовании, находятся в диапазоне доз регулярного использования. Хотя совокупный риск гипогликемии не исключен полностью, он контролируется с помощью эугликемического клэмп-метода.
Фармакодинамика
Фармакодинамическую активность инсулина гларгина оценивают у пациентов с диабетом 1 типа с помощью эугликемического клэмп-метода, который является общепризнанным стандартным методом оценки эффекта экзогенных введенных продуктов инсулина на утилизацию глюкозы крови.
Параметрами, специфичными для оценки утилизации глюкозы в условиях эугликемического клэмп-теста, являются стандартизированная по весу тела скорость инфузии глюкозы (GIR),общее количество утилизированной глюкозы, GIR-AUC0-36, и время достижения установленного процента GIR-AUC0-36, такое как время до 50% GIR-AUC0-36.
Дополнительными параметрами являлись максимальная сглаженная стандартизированная по весу тела GIR, GIRmax, и время до GIRmax, GIR-Tmax.
Продолжительность действия инсулина гларгина получают из времени от введения дозы и до достижения заранее заданного отклонения выше эугликемического (клэмп) уровня глюкозы.
Мониторинг глюкозы выполняют в течение 36 часов ввиду большой продолжительности действия инсулина гларгина после подкожного введения.
Фармакокинетика
Благодаря основному свойству замедленного высвобождения инсулина гларгина, в профиле концентрации отсутствуют выраженные пики. Следовательно, время до 50% INS-AUC (T50% INS-AUC0-36) рассчитывают как оценку “время-положение” профиля воздействия инсулина гларгина, и INS-Cmax и INS-Tmax будут служить в качестве дополнительных количественных показателей.
Главные цели исследования
Главной целью исследования является оценка отношений метаболических эффектов трех различных доз инсулина гларгина U300 в сопоставлении с 0,4 Ед/кг Лантуса® U100.
Дополнительные цели исследования
Дополнительными задачами исследования являются оценить отношения воздействий трех различных доз инсулина гларгина U300 в сравнении с 0,4 Ед/кг Лантуса® U100, сравнить продолжительность действия различных доз инсулина гларгина U300 в сравнении с 0,4 Ед/кг Лантуса® U100, изучить зависимость “доза – ответ” и “доза – воздействие” для инсулина гларгина U300, и оценить безопасность и переносимость инсулина гларгина U300 у субъектов с диабетом 1 типа.
Пример 8: Дизайн исследования, описание протокола
Фаза I, одноцентровое, двойное слепое, рандомизированное, перекрестное (4 вида лечения, 4 периода лечения и 4 последовательности; “латинский квадрат”), с активным контролем, с периодом вымывания между периодами лечения (5-18 дней, предпочтительно 7 дней) с участием субъектов мужчин и женщин с диабетом 1 типа, получавших однократные дозы инсулина гларгина
0,4 Ед/кг Лантус® U100 (= Референсное средство R)
0,4 Ед/кг Инсулин гларгин U300 (= Тестируемое средство T1)
0,6 Ед/кг Инсулин гларгин U300 (= Тестируемое средство T2)
0,9 Ед/кг Инсулин гларгин U300 (= Тестируемое средство T3)
Четыре вида лечения R и T1-3 дают перекрестно в четырех периодах лечения (TP1-TP4) с четырьмя последовательностями,
R-T1-T2-T3
T3-R-T1-T2
T2-T3-R-T1
T1-T2-T3-R
назначаемыми субъектам в случайном порядке (соотношение 1:1:1:1).
Продолжительность участия в исследовании
Общая продолжительность исследования для одного субъекта: приблизительно 4-11 недель (продолжительность min-max, в зависимости от периода вымывания, исключая скрининг)
Продолжительность каждой части исследования для одного субъекта:
- Скрининг: от 3 до 28 дней (D-28-D-3)
- Период лечения 1-4: 2 дня (1 ночь в клинике)
- Вымывание: 5-18 дней (предпочтительно 7 дней между последовательными введениями доз)
- Визит окончания исследования: 1 день между D5 и D14 после последнего введения исследуемого лекарства
Пример 9: Отбор субъектов
Запланированное число субъектов: включают в исследование по меньшей мере 24 субъекта, чтобы получить 20 подлежащих оценке субъектов.
Критерии включения
Демографические данные
I 01. Субъекты, мужчины или женщины, в возрасте от 18 до 65 лет, включительно, с диабетом 1 типа на протяжении более 1 года, в соответствии с определениями Американской ассоциации диабетологов (American Diabetic Association. Report of the Expert Committee on the Diagnosis and Classificatijn of Diabetes Mellitus. Diabetes Care 1988; 21: 5-19).
I 02. Общая доза инсулина <1,2 Ед/кг/день.
I 03. Вес тела от 50,0 кг до 95,0 кг включительно, если мужчина, от 50,0 кг до 85,0 кг включительно, если женщина, индекс массы тела от 18,0 до 30,0 кг/м2 включительно.
Состояние здоровья
I 04. Отрицательный результат теста на C-пептид в сыворотке крови в состоянии натощак (<0,3 нмоль/л)
I 05. Гликозилированный гемоглобин (HbA1c) ≤9,0%
I 06. Стабильная схема приема инсулина в течение по меньшей мере 2 месяцев до исследования (что касается безопасности субъекта и научной целостности исследования)
I 07. Нормальные показатели в медицинской истории и при физическом обследовании (сердечно-сосудистая система, грудь и легкие, щитовидная железа, живот, нервная система, кожа и слизистые, и костно-мышечная система), кроме случаев, когда исследователь рассматривает какое-либо отклонение как клинически незначимое и не влияющее на проведение исследования (с учетом безопасности субъекта и научной целостности исследования)
I 08. Нормальные основные показатели жизнедеятельности после 10 минут отдыха в положении лежа на спине: 95 мм рт. ст.< систолическое давление крови <140 мм рт. ст.; 45 мм рт. ст.< диастолическое давление крови <90 мм рт. ст.; 40 ударов/мин <пульс<100 ударов/мин
I 09. Нормальная стандартная ЭКГ в 12 отведениях после 10 минут отдыха в положении лежа на спине; 120 мс <PQ<220 мс, QRS<120 мс, QTc≤440 мс если мужчина, ≤450 мс если женщина
I 10. Лабораторные параметры в пределах диапазона нормальных значений (или определенных скрининговых границы исследовательского центра), если только Исследователь считает отклонение от нормы несущественными для пациентов с диабетом; однако значение креатинина в сыворотке крови должно быть строго ниже верхней границы лабораторной нормы; значения печеночных ферментов (AST, ALT) и билирубина (за исключением субъекта с документально подтвержденным синдромом Жильбера) должны быть не выше 1,5 ULN
Только женщины-субъекты
I 11. Женщины репродуктивного возраста (менопауза менее двух лет или хирургически стерилизованные менее 3 месяцев) должны иметь отрицательный результат теста на беременность β-HCG по сыворотке крови при скрининге и отрицательный результат теста на беременность β-HCG по моче в День 1 во время TP1-TP4, и обязаны использовать высокоэффективный метод контрацепции, который определяется как метод с низким показателем неэффективности (т.е. менее 1% в год) согласно Примечание к руководству по безопасности неклинических исследований для проведения клинических испытаний с участием людей для фармацевтов (CPMP/ICH/286/95, поправки). Во время вхождения в исследование женщины-субъекты репродуктивного возраста должны применять два независимых метода контрацепции, например диафрагма и презерватив, покрытый спермицидом. Применение презерватива и спермицидных кремов не является достаточно надежным. Для женщин в постменопаузе менее двух лет и не стерилизованных хирургически более 3 месяцев назад, должен быть определен гормональный статус (FSH>30 МЕ/л, эстрадиол <20 рг/мл)
Критерии исключения
Анамнез и клинический статус
E 01. Присутствие в анамнезе или наличие клинически значимого сердечно-сосудистого, легочного, желудочно-кишечного, печеночного, почечного, метаболического (кроме диабета 1 типа), гематологического, неврологического, психического, системного (поражающего организм в целом), офтальмологического, гинекологического (если женщина), или инфекционного заболевания; любое острое инфекционное заболевание или признаки острого заболевания
E 02. Более одного эпизода тяжелой гипогликемии с пароксизмом, комой или потребность в помощи другого человека в течение последних 6 месяцев
E 03. Частые тяжелые головные боли и/или мигрень, повторяющаяся тошнота и/или рвота (чаще, чем два раза в месяц)
E 04. Кровопотеря (>300 мл) в течение 3 месяцев перед включением
E 05. Симптоматическая гипотензия (любое снижение давления крови), или бессимптомная ортостатическая гипотензия, определяемое по снижению SBP равному или большему чем 20 мм рт. ст. в течение трех минут при переходе из положения лежа в положение стоя
E 06. Наличие или присутствие в анамнезе лекарственной аллергии или клинически значимого аллергического заболевания по оценке Исследователя
E 07. Вероятность необходимости лечения во время периода исследования лекарствами, не разрешенными протоколом клинического исследования
E 08. Участие в испытании какого-либо исследуемого лекарства в течение предыдущих трех месяцев
E 09. Симптомы клинически значимого заболевания в течение 3 месяцев перед началом исследования, которые, по мнению Исследователя, могут препятствовать целям исследования
E 10. Лекарственная или алкогольная зависимость (потребление алкоголя >40 грамм/день)
E 11. Курение 5 или более сигарет в день, невозможность отказаться от курения во время исследования
E 12. Чрезмерное потребление напитков, содержащих ксантиновые основания (> 4 чашек или стаканов/день)
E 13. Если женщина, беременность (определяемая по положительному результату теста на β-HCG), кормление грудью.
Мешающее вещество
E 14. Любое лекарство (включая St John's Wort) в пределах 14 дней до включения, или в пределах 5-кратного периода полувыведения или фармакодинамического периода полувыведения указанного лекарства, любое продолжительное и регулярное применение какого-либо лекарства, другого, чем инсулины в течение последнего месяца перед началом исследования за исключением гормонов щитовидной железы, липидопонижающих и противогипертонических лекарств и, если женщина, за исключением гормональной контрацепции или менопаузальной гормонозаместительной терапии; любая вакцинация в течение последних 28 дней
Общие условия
E 15. Субъект, который, по мнению Исследователя, не сможет соблюдать требования во время исследования, или не способен к взаимодействию по причине языковой проблемы или недостаточного умственного развития
E 16. Субъект в период исключения предыдущего исследования согласно применяемым положениям
E 17. Субъект, с которым нельзя связаться в экстренных случаях
E 18. Субъект является исследователем или субисследователем, научным сотрудником, провизором, координатором исследования или другим сотрудником, непосредственно участвующим в осуществлении протокола
Биологический статус
E 19. Положительная реакция в любом из следующих тестов: поверхностный антиген вируса гепатита B (HBs Ag), антитела к ядерному антигену вируса гепатита В (anti-HBc Ab), если соединение обладает возможными иммунными активностями, антитела против вируса гепатита C (anti-HCV2), антитела 1 и 2 против вируса иммунодефицита человека (anti-HIV1 и anti HIV2 Ab)
E 20. Положительные результаты анализа мочи с целью выявления запрещенных средств (амфетамины/метамфетамины, барбитураты, бензодиазепины, каннабиноиды, кокаин, опиаты)
E 21. Положительный результат теста на алкоголь
Условия, характерные для исследования
E 22. Известная гиперчувствительность к инсулину гларгину и к эксципиентам.
E 23. Присутствие в анамнезе или наличие тромбоза глубоких вен ног или часто встречающийся тромбоз глубоких вен ног у родственников первой линии (родители, родные братья и сестры или дети).
Пример 10: Лечение
Исследуемый продукт
β Инсулин гларгин
Применяют два различных средства инсулина гларгина:
- Лантус® U100 раствор для инъекций, содержащий 100 Ед/мл инсулина гларгина (рыночный продукт)
- Инсулин гларгин U300 раствор для инъекций, содержащий 300 Ед/мл инсулина гларгина
β Доза:
- Лантус® U100: 0,4 Ед/кг (= Референсный препарат R)
- Инсулин гларгин U300: 0,4, 0,6 и 0,9 Ед/кг (= Тестируемое средство T1-T3)
Контейнер: стеклянные картриджи объемом 3 мл
Способ употребления: подкожно горизонтально 5 см справа и слева от пупка
Условия: натощак
Продолжительность лечения: 1 день в каждый период, разовая доза
Начало: 09:00 в День 1 (D1) в Периоды лечения 1-4 (TP1-4)
Предоставляется дополнительное лечение для 100% включенных субъектов
|
Дозировка
Данное исследование является однодозовым исследованием всего с 4 введениями исследуемого средства. Субъектов рандомизируют по различным последовательностям эталонного и исследуемого лечения, так что каждый субъект получает эталонное лечение (R) и каждое из исследуемых видов лечения (T1-3) однократно.
Инъекции вводят слева или справа от пупка, причем обе области используют для отдельных инъекций. Период вымывания 5-18 дней отделяет последующие дни введения, предпочтительным является период 7 дней (7 дней между последовательными введениями доз). Продолжительность периода вымывания варьирует по отдельным субъектам, что позволяет участнику и исследователю согласовать свои потребности. По опыту, 5 дней составляют минимальный период для восстановления, позволяющий 1 клэмп-тест в неделю для одного участника, тогда как 18 дней представляют собой перерыв в 3 недели между днями введения дозы, обеспечивая субъектам свободу для выполнения не связанных с исследованием обязательств, при необходимости.
Введение IP производят натощак, субъект остается в состоянии натощак на протяжении всего периода проведения клэмп-теста.
Концентрация глюкозы крови находится в диапазоне 5,5 ммоль/л (100 мг/дл) ±20% без какой-либо инфузии глюкозы в течение последнего часа перед введением дозы во время преклэмп-теста. Если глюкоза крови остается стабильной в течение по меньшей мере 1 часа без какой-либо инфузии глюкозы, вводят IP. Введение IP производят не ранее 09:00 часов утра и позже 14:00 часов в День 1 Периодов лечения 1-4. Если глюкоза крови не стабилизируется до 14:00 часов, дозирование не производят. Визит оканчивается, и субъекту назначают следующий визит для введения дозы через 1-7 дней.
На каждого субъекта и на каждое введение дозы используют новый картридж.
Введение IP производится лицом, которое иным образом не вовлечено в исследование или в часть команды исследования в CRO. Указанное лицо получает случайный код, чтобы подготовить введение IP в соответствии открытым списком случайных номеров и соответственно вводит дозы субъектам. Приготовление и введение доз контролируется и проверяется вторым независимым лицом. Соответствующие документы о приготовлении доз и последовательности лечения хранятся строго конфиденциально и не раскрываются какому-либо другому лицу.
Расчет дозы IP (инсулин гларгин)
Для расчета количества инсулина гларгина, получаемого каждым субъектом, определяют вес тела (в кг) до десятичного знака и рассчитанное количество инсулина округляют с увеличением или с уменьшением до целых чисел, как показано в следующих примерах для дозы 0,6 Ед/кг инсулина гларгина:
субъект с весом тела 75,3 кг получает 45 Ед инсулина (75,3×0,6=45,18 которые округляют с уменьшением до 45);
субъект с весом тела 74,4 кг получает 45 Ед инсулина (74,4×0,6=44,64, которые округляют с увеличением до 45).
Вес тела, записанный во время TP1 D1, используют для расчета дозы исследуемого средства для всех периодов лечения. Дозу исследуемого средства не меняют, вес субъекта меняется менее чем или на 2 кг между TP1 и одним из последующих TP. Если вес тела субъекта изменяется более чем на 2 кг между TP1 и одним из последующих TP, дозу исследуемого средства рассчитывают повторно исходя из веса в D1 соответствующего периода лечения.
Шприцы и иглы
Используют только шприцы с прикрепленными иглами, предназначенные для точного введения небольших количеств инъекционного раствора (например, Becton Dickinson, Ref 305502, размеры: 1 мл 27G 3/8 0,40×10). Шприцы предоставляются Исследователем.
Другие продукты
Другие продукты, использованные во время процедуры клэмп-теста, описаны в таблице 5.
|
Раствор глюкозы, хлорид натрия, гепарин и инсулин глулизин предоставляется Исследователем.
Раствор глюкозы: проводят инфузию 20% раствора глюкозы с помощью Биостатора™, чтобы сохранить концентрацию глюкозы в крови субъектов на определенном целевом уровне. Вторая инфузионная помпа (часть Биостатора™) доставляет 0,9% раствор хлорида натрия для поддержания проходимости катетера. Если необходимое количество 20% раствора глюкозы превышает инфузионную емкость Биостатора™, включают вторую помпу для инфузии глюкозы.
Гепарин: проводят инфузию раствор гепарина в низкой дозе (10000 Единиц гепарина/100 мл физиологического раствора) через катетер с двойным просветом. Раствор гепарина забирается вместе с кровью, используемой Биостатором™ для измерения глюкозы крови в другом просвете катетера, и применяется для предотвращения свертывания крови в системе.
Инсулин глулизин: 15 Ед Апидра® [100 Ед/мл] добавляют к 49 мл физиологического раствора, к которым добавляют 1 мл собственной крови субъекта для предотвращения адгезии, получая раствор с концентрацией 0,3 Ед/мл, инфузию которого проводят с индивидуальной скоростью для достижения состояния эугликемии.
Описание методов маскирования
Субъекты получают четыре различных лечения S (R, T1, T2 и T3) в соответствии с рандомизированным, слепым и перекрестным дизайном. Чтобы сохранить маскирование исследования, третье частично немаскированное лицо привлекают для распределения и введения IP. Указанное лицо не участвует иным образом в исследовании и/или не является частью команды исследования в CRO, не раскрывает информацию кому-либо и гарантирует сохранение условия маскирования исследования. Он/она получает случайный код и соответственно вводит дозы субъектам. Приготовление и введение доз IP контролируется и проверяется вторым независимым лицом, которое также имеет доступ к случайному коду, но в равной мере связано условиями конфиденциальности.
Метод назначения субъектов в группу лечения
IP вводят согласно протоколу клинического исследования только субъектам, которые дали письменное информированное согласие.
Субъектам, которые удовлетворяют всем критериям включения/исключения присваивают только перед введением исследуемого продукта в День 1 Периода лечения 1:
возрастающий номер субъекта в соответствии с хронологическим порядком включения утром в D1 в Период лечения 1. Девятизначный номер субъекта состоит из 3 компонентов (например, 276 001 001, 276 001 002, 276 001 003, и т.д.), из которых первые 3 цифры (276) являются номером страны, средние 3 цифры представляют собой номер центра проведения исследования и последние 3 цифры представляют собой номер субъекта в возрастающем порядке в пределах центра проведения исследования. Номер субъекта остается неизменным и позволяет идентифицировать субъекта на протяжении всего исследования.
Номер лечения в заранее запланированном порядке, следуя рандомизационному списку, при этом следующий подходящий субъект всегда получает следующий номер лечения в соответствии со списком рандомизации
Введение IP происходит в соответствии с рандомизированной последовательностью лечения.
Субъекты, досрочно исключенные из исследования, сохраняют свой номер субъекта и свой номер лечения, если они уже назначены. Запасные субъекты имеют другой идентификационный номер (т.е. 500 + номер субъекта, исключенного из исследования). Каждый субъект получает такую же последовательность лечения, как субъект, и субъект, досрочно выбывший из исследования.
Субъектам, не прошедшим скрининг, назначают другой номер, например, 901, 902 (который должен быть зарегистрирован в CRF только в случае AE, происходящем в период скрининга после подписания информированного согласия).
Замечания. Рандомизация субъекта происходит после подтверждения Исследователем пригодности субъекта для данного исследования. Исходные параметры представляют собой параметры, последние по времени перед введением дозы.
Упаковка и маркировка
Раствор инсулина гларгина U300 предоставляется Sanofi-Aventis в перестраиваемых контейнерах для картриджей по 3 мл.
Соответствующее число IP упаковывают под ответственность Sanofi-Fventis согласно Надлежащей производственной практике и местным нормативным требованиям, и предоставляется CRO.
Содержимое маркировки соответствует местным нормативным правилам и требованиям.
Лантус® U100 является коммерчески доступным и заказывается CRO.
Условия хранения
Весь IP хранится в соответствующем закрытом помещении под ответственностью Исследователя, и должен быть доступен только утвержденному персоналу.
IP хранили при температуре +2°C до +8°C, в защищенном от света месте и не допуская замораживания.
Доступ к коду рандомизации во время исследования
Чтобы сохранить маскирование, третье частично выведенное из слепого метода лицо является ответственным за распределение и введение IP. Указанное лицо не вовлечено иным образом в исследование и/или часть команды исследования в CRO, не раскрывает информацию кому-либо и гарантирует сохранение условия маскирования исследования. Он/она получает случайный код и соответственно вводит дозы субъектам. Приготовление IP и введение дозы контролируется и проверяется вторым независимым лицом, которое также имеет доступ к случайному коду, но в равной мере связано условиями конфиденциальности.
В случае Нежелательного Явления, код не раскрывают, кроме как в обстоятельствах, когда знание исследуемого продукта является необходимым для лечения субъекта. Для каждого субъекта, материал, раскрывающий код, который содержит наименование лечения, предоставляется в виде конверта. Он хранится в недоступном месте в исследовательском центре в течение Клинического Испытания. Спонсор извлекает весь материал, раскрывающий код (открытый или опечатанный) после окончания Клинического Испытания.
Если маскирование исследования нарушено, Исследователь документирует дату открытия и причину раскрытия кода в первичных данных.
Исследователь, фармаколог исследовательского центра или другой персонал, обеспечивающий хранение и распределение IP, несет ответственность за безопасное хранение IP, применяемого в исследовании, как указано Спонсором, и в соответствии с соответствующими нормативными требованиями.
Весь IP распределяют в соответствии с протоколом клинического испытания, и Исследователь несет ответственность за обеспечение точной записи выданного и возвращенного IP.
Сопутствующее лечение
Применение сопутствующей лекарственной терапии не разрешается во время исследования, как указано в критерии исключения No. E14, за исключением лекарств, упомянутых дополнительно, и прекращается в течение указанного интервала времени (смотреть E14) перед включением субъекта в День 1 Периода лечения 1.
Чтобы предотвратить воздействие стандартного инсулинового лечения субъектов на результаты измерения клэмп-теста, субъекты должны воздерживаться от использования базальных инсулинов и перейти на
продукты инсулина средней и короткой продолжительности действия за 48 часов до введения дозы в D1 с TP1 до TP4, если принимают пролонгированные продукты инсулина, т.е. Лантус® (инсулин гларгин), Левемир® (детемир) или инсулины ультраленте,
инсулины короткого действия за 24 часа до введения дозы в D1 с TP1 до TP4, если принимают продукты инсулина средней продолжительности действия, т.е. NPH-инсулин
Последняя подкожная инъекция инсулина короткого действия происходит не позднее, чем за 9 часов до введения исследуемого лекарства. Субъекты, принимающие помповую терапию, прекращают инфузию инсулина утром в День 1, по меньшей мере за 6 часов до каждого введения IP (приблизительно в 03:00 часа, принимая начало введения IP в 09:00).
Для симптоматических нежелательных явлений, которые не угрожают безопасности субъекта (например, головная боль) сопутствующая лекарственная терапия откладывается до нежелательных явлений высокой интенсивности или средней интенсивности, которые продолжаются в течение длительного времени. В частности, запрещено применение ацетаминофена /парацетамола, если имеется известный риск гепатотоксичности, или наблюдаются только нарушения ферментов печени.
Однако если по какой-либо причине требуется специальное лечение, в соответствующей регистрационной карте должна остаться точная запись, включая название лекарства (международное непатентованное название), дневную дозировку и длительность применения. Спонсор должен быть информирован в течение 48 часов с помощью e-мейла или факса, за исключением случаев лечения головной боли.
Лечение возможных аллергических реакций проводят в соответствии с рекомендациями, которые опубликованы в другом месте (Samspon HA, Munoz-Furlong A, Campbell RL et al. Second symposium on the definition and management of anaphylaxis: summary report - Second National Institute of Allergy and Infectious Disease/Food Allergy and Anaphylaxis Network symposMEm. Journal of Allergy and Clinical ImMunology 2006; 117 (2):391-397). В зависимости от тяжести аллергической реакции может быть рассмотрено лечение антигистаминными средствами, кортикостероидами и эпинефрином.
Учет средства и соблюдение терапии
Соблюдение терапии IP:
- Ведение IP осуществляется под непосредственным медицинским наблюдением, и соответствующая запись выполняется лицом, ответственным за приготовление и введение IP или его/ее представителем; любая информация о последовательности лечения или дозе не раскрывается и документы хранятся в закрытом помещении без доступа других лиц, участвующих в исследовании
- Введение IP подтверждается измеряемыми результатами теста на определение лекарственного средства
Учет IP:
- Лицо, ответственное за приготовление и введение IP или его/ее представитель подсчитывает число картриджей, оставшихся в возвращенных упаковках, затем заполняет в форму в журнале лечения
- Исследователь записывает информацию о дне и времени введения дозы на соответствующей странице (страницах) индивидуальной регистрационной карты (CRF)
- Команда Монитора, ответственная за исследование, затем проверяет данные в CRF путем их сравнения с IP и соответствующими формами учета средства после закрытия базы данных (для предотвращения раскрытия рандомизационного кода исследования)
Использованные картриджи хранятся Исследователем вплоть до полного документально подтвержденного согласования, выполняемого Спонсором в конце исследования после закрытия базы данных.
Пример 11: Оценка исследуемого продукта
Настоящее исследование разработано, чтобы оценить метаболический эффект и отношения воздействий трех различных доз инсулина гларгина U300 в сравнении с 0,4 Ед/кг Лантуса® U100, сравнить продолжительность действия различных доз инсулина гларгина U300 против 0,4 Ед/кг Лантуса® U100, изучить зависимость “доза-ответ” и “доза-воздействие” для инсулина гларгина U300, и оценить безопасность и переносимость инсулина гларгина U300 в условиях эугликемического клэмп-теста у субъектов с диабетом 1 типа.
Фармакодинамика
Эугликемический клэмп-тест
Фармакодинамический эффект инсулина гларгина, по преимуществу, общую утилизацию глюкозы и продолжительность действия инсулина оценивают с помощью эугликемического клэмп-метода.
Во время эугликемического клэмп-теста, концентрацию глюкозы в артеризованной венозной крови, которая отражает обеспечение глюкозой для общей утилизации всеми тканями, и скорость инфузии глюкозы (GIR), необходимую для поддержания концентрации глюкозы крови субъекта на заданном уровне (клэмп-уровень) постоянно измеряют и записывают с помощью устройства Биостатор™ (система постоянного мониторинга глюкозы, Life Sciences Instruments, Elkhart, IN, USA).
Требуемое количество глюкозы (GIR-AUC) является показателем захвата глюкозы в тканях (утилизация глюкозы или глюкозопонижающая активность) опосредованного избытком экзогенного инсулина. Биостатор™ уровни глюкозы крови с интервалами в 1 мин и регулирует скорость инфузии глюкозы в ответ на изменения уровня глюкозы крови, используя заранее заданный алгоритм.
Порядок проведения клэмп-теста
Чтобы предотвратить противодействие стандартного лечения инсулином субъектов на результаты измерения клэмп-теста, субъекты должны воздерживаться от использования базальных инсулинов и перейти на
продукты инсулина средней и короткой продолжительности действия за 48 часов до введения дозы в D1 с TP1 до TP4, если принимают пролонгированные продукты инсулина, т.е. Лантус® (инсулин гларгин), Левемир® (детемир) или инсулины ультраленте,
инсулины короткого действия за 24 часа до введения дозы в D1 с TP1 до TP4, если принимают продукты инсулина средней продолжительности действия, т.е. NPH-инсулин
Последняя подкожная инъекция инсулина короткого действия происходит не позднее, чем за 9 часов до введения IP. Субъекты, принимающие помповую терапию, прекращают инфузию инсулина утром в День 1, по меньшей мере за 6 часов до каждого введения IP (приблизительно в 03:00 часа, считая, что время начала введения IP 09:00).
Во время периодов лечения 1-4 (TP1–TP4), субъектов принимают в клинике утром в D1 в состоянии натощак с вечера, в течение по меньшей мере 10 часов.
Утром в День 1 начинают процедуру преклэмп-теста и субъектов соединяют с Биостатором™. Концентрацию глюкозы крови доводят до 4,4–6,6 ммоль/л (80-120 мг/дл) и удерживают в указанных пределах с помощью в/в болюс-введений быстродействующего аналога инсулина (например, инсулин глулизин) и последующих индивидуальных инфузий глюкозы по мере необходимости.
За 60 мин до введения исследуемого средства уровень глюкозы крови доводят до 5,5 ммоль/л (100 мг/дл) ±20% (эугликемический клэмп-уровень) без какой-либо инфузии глюкозы в течение часа перед введением дозы. Инфузию инсулина глулизина прекращают непосредственно перед введением исследуемого средства.
Если глюкоза крови остается стабильной в течение по меньшей мере 1 часа в диапазоне 5,5 ммоль/л (100 мг/дл) ±20% без инфузии глюкозы, вводят IP (= T0 в D1 в TP1-TP4, приблизительно 09:00). Субъекты получают эталонный или исследуемое средство (R, T1-3, смотреть таблицу 4), как установлено при рандомизации. Инъекции вводят слева или справа от пупка.
Введение IP происходит не ранее 09:00 часов утра и не позднее 14:00 часов в День 1 Периодов лечения 1-4. Если глюкоза крови не стабилизируется во время преклэмп-теста до 14:00 часов, дозирование не осуществляют. Визит заканчивают, и субъекту назначают новый визит для введения дозы спустя 1-7 дней.
Введение IP производят в условиях натощак; субъект остается в состоянии натощак в течение всего периода проведения клэмп-теста.
Уровень глюкозы крови во время эугликемического клэмп-теста постоянного поддерживается с помощью в/в инфузии раствора глюкозы до окончания клэмп-теста.
Целью добавления какого-либо базального инсулина является дополнение или даже замена секреции эндогенного инсулина между приемами пищи. У субъектов без секреции эндогенного инсулина, которые приглашены для участия в исследовании, экзогенный инсулин должен обеспечивать только количество инсулина, требуемое, чтобы упорядочить продукцию глюкозы в печени. При идеальном подборе нет потребности в дополнительной глюкозе, чтобы компенсировать избыточный инсулин. Результирующая скорость инфузии глюкозы приближается к нулю. Как только действие инсулина заканчивается, концентрация глюкозы крови возрастает. Время до начала роста и до момента времени, когда концентрации глюкозы крови превышают заранее заданные границы, считывается Биостатором™.
Выбранные дозы Лантуса® U100 и инсулина гларгина U300 превышают среднюю базальную потребность, что в свою очередь вызывает некоторую потребность в глюкозе, которая отражена в значительной по размеру GIR, вплоть 36 часов.
Соответствующий параметр, указывающий на выполнение клэмп-теста, т.е. на точность сохранения значений глюкозы крови на исходном клэмп-уровне, представляет собой вариабельность глюкозы крови на протяжении периода клэмп-теста. Показателем вариабельности глюкозы крови является коэффициент вариации (CV%) для отдельного клэмп-теста.
Низкий коэффициент вариации глюкозы крови является предварительным условием правильной оценки эффекта инсулина в условиях клэмп-теста.
Период проведения клэмп-теста не превышает 36 часов после инъекции исследуемого средства, заранее заданного времени окончания клэмп-теста.
Субъекты остаются в состоянии натощак на протяжении всего периода проведения глюкозного клэмп-теста (преклэмп и клэмп) при доступе к воде ad libitum.
В случае если глюкоза крови превышает 11,1 ммоль/л (200 мг/дл) перед окончанием клэмп-теста в течение 30 минут после прекращения инфузии глюкозы, и исследователь подтверждает, что исключены возможные ошибки, приводящие к некорректным результатам глюкозы крови выше 11,1 ммоль/л (200 мг/дл), вводят инсулин глулизин, применяемый перед введением IP во время клэмп-теста, для продления периода наблюдения до 36 часов. В таком случае должен быть информирован спонсор.
Субъектов отсоединяют от установки для клэмп-теста, если значение глюкозы крови находится в изогликемическом диапазоне.
Участники возобновляют прием своих ранее принимаемых средств в день выписки из исследовательского центра в TP1-TP4, т.е. в День 2.
Эффект IP продолжается приблизительно 24–36 часов, и поэтому участники остаются в исследовательском центре в течение 2 дней.
Период вымывания от 5 до 18 дней отделяет дни последовательных периодов клэмп-тестов, предпочтительным является период 7 дней (7 дней между последовательным введением доз). Продолжительность периода вымывания варьирует по отдельным субъектам, что позволяет участнику и исследователю согласовать свои потребности. По опыту, 5 дней составляют минимальный период для восстановления, позволяющий 1 клэмп-тест в неделю для одного участника, тогда как 18 дней представляют собой перерыв в 3 недели между днями введения дозы, обеспечивая субъектам свободу для выполнения не связанных с исследованием обязательств, при необходимости.
Скрининг и D1 TP1 разделены не более чем 28 днями, тогда как EOS имеет место не ранее D5 или не позже D14 после введения последней дозы, соответственно.
Время отбора фармакодинамических образцов
Артериализованную венозную кровь постоянно получают со скоростью 2 мл/ч для определения концентрации глюкозы в артериальной крови каждую минуту во время преклэмп-теста (до введения IP) и периода проведения клэмп-теста (до 36 часов после введения IP).
Образцы артериализованной венозной крови (0,2 мл) для сопутствующей колибровки Биостатора™, которая является техническим требованием, собирают по меньшей мере с 30 минутными интервалами после подсоединения к Биостатору™ вплоть до 36 часов после введения лекарства.
Число фармакодинамических образцов
Глюкозу крови постоянно измеряют во время процедуры проведения клэмп-теста. Кроме того, собирают по меньшей мере 74 образца на каждого субъекта и период лечения для калибровки Биостатор™ после введения IP. Всего собирают 74*4*24 образцов или 7104 образцов (см. таблицу ниже).
|
Процедура проведения фармакодинамических анализов
|
Фармакодинамические параметры
Рассчитывают площадь под кривой стандартизированной по весу тела GIR в пределах 36 часов (GIR AUC0-36) и время до 50% общей GIR-AUC в пределах 36 часов (T50%-GIR-AUC0-36).
Продолжительность контроля глюкозы крови считают как время в эугликемическом состоянии от введения дозы до отклонения выше клэмп уровня глюкозы (100 мг/дл). Время контролирования глюкозы крови в заранее заданных пределах считают от момента введения дозы до достижения заданных пороговых точек, например уровней глюкозы в крови 110, 130 и 150 мг/дл.
Кроме того, оценивают максимальную сглаженную скорректированную по весу тела GIR (GIRmax) и время до GIRmax, GIR-Tmax.
Другие дополнительные параметры получают в соответствующих случаях.
Безопасность
Исходные демографические характеристики
Исходные демографические характеристики включают в себя:
Возраст (годы)
Вес тела (кг)
Рост (см)
Индекс массы тела (BMI) (кг/м2)
Оценка безопасности на исходном уровне и во время исследования
Физическое обследование при скрининге: сердечно-сосудистая система, грудь и легкие, щитовидная железа, живот, нервная система, кожа и мышцы, и костно-мышечная система и соответствующий медицинский и хирургический анамнез, диабетический анамнез (диагноз диабета, начало лечения инсулином, поздние осложнения); документируют только сведения, относящиеся к исследованию
Курение в предшествующее и настоящее время
Физическое обследование до приема лекарства и во время исследования: сердечно-сосудистая система, живот и легкие; документируют только сведения, относящиеся к исследованию
Температура тела (ушная)
Основные показатели жизнедеятельности: пульс, частота дыхания и систолическое и диастолическое давление крови, измеренное через 10 минут отдыха в положении лежа, пульс и систолическое и диастолическое давление крови, также измеренное через 3 минуты, в положении стоя (за исключением незапланированных измерений во время подключения к Биостатору™)
Лабораторные тесты (в состоянии натощак для образцов крови):
Гематологический анализ: число эритроцитов (RBC), гематокрит (Hct), гемаглобин (Hb), число лейкоцитов (WBC) с дифференциальным подсчетом (нейтрофилов, эозинофилов, базофилов, моноцитов и лимфоцитов), тромбоциты, INR и aPTT
Биохимический анализ:
- Электролиты: натрий, калий, бикарбонат, хлорид, кальций
- Оценка функции печени: AST, ALT, щелочная фосфатаза, гамма-глутамилтрансфераза (γGT), общий и конъюгированный билирубин
- Оценка функции почек: креатинин, мочевина
- Метаболизм: глюкоза, альбумин, общие белки, общий холестерин, триглицериды, HbA1c (при скрининге, D1 TP1 и EOS), LDH,амилаза, липаза, C-пептид (только скрининг)
- Возможная мышечная токсичность: креатининфосфокиназа (CPK)
- Серология: антиген гепатита B (HBs Ag), антитела к ядерному антигену вируса гепатита В (anti-HBc Ab), антитела к вирусу гепатита C (anti-HCV2), антитела анти-HIV1 и анти-HIV2
Архивный образец крови: образец крови объемом 5 мл собирают в сухую вакуумную пробирку без антикоагулянта, центрифугируют при 1500 g в течение 10 минут при 4°C; затем переносят сыворотку в три пробирки для хранения, которые немедленно закрывают и замораживают в вертикальном положении при -20°C. Данный образец используют, если возникает любой неожиданный вопрос безопасности, чтобы гарантировать, что исходное значение до введения лекарства доступно для ранее не оцениваемых параметров (например, серология). Если данный образец не используют, Исследователь уничтожает его после разрешения Спонсора
Анализ мочи: белки, глюкоза, кровь, кетоновые тела, pH
- Качественный анализ: анализ с помощью тест-полоски выполняют на свежесобранном образце для качественного определения с помощью индикаторной полоски;
- Количественный анализ: количественное измерение глюкозы, белка, числа эритроцитов и лейкоцитов требуется в случае, когда анализ образца мочи с помощью тест-полоски является положительным по какому-либо из выше перечисленных параметров (например, чтобы подтвердить положительный результат какого-либо параметра, положительного на на тест-полоске, количественным измерением).
анализ мочи на запрещенные средства: амфетамины/метамфетамины, барбитураты, бензодиазепины, каннабиноиды, кокаин, опиаты
Проба на алкоголь в выдыхаемом воздухе
Беременность/гормональный тест (если женщина):
- β-HCG в крови при скрининге
- β-HCG в моче в TP1-TP4, День 1
- FSH/эстрадиол, если менопауза менее 2 лет, только при скрининге
Нежелательные явления: самостоятельно сообщаемые субъектом или наблюдаемые Исследователем
ЭКГ-телеметрия (одно отведение)
ЭКГ в 12 отведениях (автоматическое)
Антитела против инсулина
Образцы крови для лабораторных тестов берут в состоянии натощак.
Методика ЭКГ
ЭКГ-телеметрия
ЭКГ-телеметрия постоянно контролируется медицинским персоналом. Все аритмические эпизоды документируются с помощью распечатки и вносятся в CRF субъекта. Указанная документация диагноз эпизода, время появления, и продолжительность, и подписывается Исследователем или его представителем. Записи ЭКГ-телеметрии хранятся для возможного повторного анализа, учитывающего воздействие исследуемого продукта.
ЭКГ в 12 отведениях
ЭКГ в 12 отведениях записывают по меньшей мере после 10 минут в положении лежа на спине, используя электрокардиографическое устройство (MAC 5500™). Электроды помещают в одно и то же место для каждой записи ЭКГ на протяжении всего исследования (места присоединения отведений отмечают несмываемой ручкой).
ЭКГ всегда записывают перед отбором проб для PK (если имеется). Пробы для PK забирают в максимально короткий срок (в течение 15 минут) после ЭКГ.
Каждая ЭКГ состоит из 10-секундной записи одновременно в 12 отведениях, приводящей к:
- распечатке отдельной ЭКГ в 12 отведениях (25 мм/s, 10мм/mV) с HR, PR, QRS, QT, QT c автоматической коррекцией оценки, включая дату, время, инициалы и номер субъекта, подпись исследователя, и по меньшей мере 3 комплекса для каждого отведения. Медицинское заключение исследователя и автоматические показания записывают в CRF. Указанная распечатка сохраняется на уровне исследовательского центра.
- хранение информации в цифровой форме, которое позволяет последующее дополнительное рассмотрение ЭКГ в центральном учреждении: каждый цифровой файл идентифицируется с помощью теоретического времени (дата и время DxxTxxHxx), реальной даты и реального времени (время записи), кода спонсора исследования, номера субъекта (т.е., 3 цифры) и номера исследовательского центра и страны, если применимо.
Цифровая запись, хранение и передача данных (когда требуется) соответствуют всем существующим нормативным требованиям (т.е., FDA 21 CFR, часть 11).
Если проверка основных показателей жизнедеятельности, ЭКГ и взятие образцов крови запланированы на то же самое время, как и введение исследуемого продукта и/или питание, они выполняются до введения исследуемого продукта и/или до питания. Всегда, когда совпадают измерения основных показателей жизнедеятельности, ЭКГ, и забор образов крови для PK, PD, или оценки безопасности, соблюдается следующий порядок: ЭКГ, основные показатели жизнедеятельности, забор образцов для PD, PK и оценки безопасности; чтобы не нарушать точного времени забора образцов для PK (относится к графику для обеспечения допустимого отклонения от интервала времени для забора образцов PK), другие оценки выполняют ранее запланированного времени. Расписание оценок, производимых в ходе исследования, адаптируют к дизайну исследования
Местная переносимость в месте инъекции
Изменения в месте инъекции (такие как покраснение кожи, отек, папулы, уплотнение, пузырьки, буллезное поражение) классифицируются преимущественно согласно Общей шкале раздражения. Местная реакция в участке инъекции с оценкой ≥3 согласно шкала оценки документируется дополнительно в качестве нежелательного явления.
Субъектов просят описать ощущения в месте инъекции.
Фармакокинетика
Для оценки фармакокинетики инсулина гларгина получают площадь под кривой концентрации инсулина (INS-AUC) вплоть до 36 часов, INS-AUC0-36 и время до 50% INS-AUC0-36. Дополнительно получают максимальную концентрацию инсулина INS-Cmax, и время до Cmax (INS-Tmax).
Время забора образцов
Кровь собирают для определения концентраций инсулина гларгина в моменты времени 0H, 1H, 2H, 4H, 6H, 8H, 12H, 16H, 20H, 24H, 28H, 32H и 36H после инъекции исследуемого средства.
Число фармакокинетических образцов
|
Процедура проведения фармакокинетических анализов
Точное время введения IP и сбора образцов должно быть записано в CRF.
Фармакокинетические параметры
Рассчитывают следующие фармакокинетические параметры с помощью некомпартментных методов для концентраций инсулина гларгина после однократной дозы. Параметры включают в себя, но не ограничены следующим.
|
Объем собранных образцов крови
|
Меры по сохранению маскирования испытания
Чтобы сохранить маскирование исследования, третье частично немаскированное лицо привлекают для распределения и введения IP. Указанное лицо не участвует иным образом в исследовании и/или не является частью команды исследования в CRO или спонсором. Он/она получает случайный код, предоставляемый Sanofi-Aventis, и не раскрывает случайный код или другую информацию какому-либо другому лицу. По соображениям безопасности рандомизационный код лечения раскрыт для сообщения врачебной комиссии о подозреваемой непредвиденной серьезной нежелательной реакции (SUSAR),обоснованно связанной с применением IP по решению исследователя и/или спонсора.
Безопасность субъекта
Исследователь является главным лицом, ответственным за все клинически важные решения в случае возникновения проблем безопасности.
Если считают необходимым, мнение специалиста должно быть своевременно рассмотрено (например, острая почечная недостаточность, судороги, кожная сыпь, отек Квинке, остановка сердца, электрокардиографические изменения, и т.д.).
Пример 12: Процедуры исследования
План проведения визита
Процедуры скрининга
Процедуры скрининга осуществляют от 28 дней до 3 дней до включения для определения соответствия требованиям для участия субъекта в исследовании. Субъект получает от Исследователя информацию о целях и процедурах исследования. Субъект подписывает информированное согласие до какого-либо действия, связанного с исследованием. С этого времени начинают запись о нежелательных явлениях.
До скрининга субъекты находились в состоянии натощак (за исключением воды) в течение 10 часов (за исключением небольшого количества углеводов, как противодействие гипогликемии, если необходимо).
Скрининговый визит включает в себя следующие обследования:
1. Демографические данные (возраст, пол, раса, курение в предшествующее и настоящее время, рост, вес тела, BMI)
2. Физическое обследование (сердечно-сосудистая система, грудь и легкие, щитовидная железа, живот, нервная система, кожа и слизистые и костно-мышечная система) и соответствующий медицинский и хирургический анамнез, диабетический анамнез (диагноз диабета, начало лечения инсулином, поздние осложнения); документируют только сведения, имеющие отношение к исследованию
3. Важное предыдущее и все сопутствующее лечение, средняя схема приема инсулина в последние 2 месяца до включения в исследование
4. ЭКГ (стандартное в 12 отведениях) и основные показатели жизнедеятельности (пульс, систолическое и диастолическое давление крови, измеренное после 10 минут отдыха в положении лежа, и после 3 минут в положении стоя), и центральная температура тела (ауральная).
5. Лабораторные тесты, включая гематологический анализ, HbA1c, C-пептид, биохимический анализ крови, серологический анализ, общий анализ мочи, анализ мочи с целью выявления запрещенных средств, проба на алкоголь в выдыхаемом воздухе, определение в крови β-HCG и FSH/эстрадиола (только женщины, если применимо).
Разрешен один повторный тест в течение недели, при этом результат последнего теста является окончательным.
Субъекты, которые отвечают всем критериям включения, и не отвечают критериям исключения, допускаются к визиту включения.
В случае непрохождения скрининга основные результаты скринингового осмотра записывают в первичных документах.
Процедуры включения (День 1 Периода лечения 1)
Субъектов, которые соответствуют критериям включения в исследование, принимают в клинике в состоянии натощак утром D1 в TP1 приблизительно в 07:00.
Осмотр при включении проводится в первый день введения дозы (D1, TP1) и включает в себя следующие обследования:
Физическое обследование с дополненным медицинским анамнезом (AEs), предшествующая/сопутствующая лекарственная терапия и центральная температура тела
Вес тела, BMI (рост, измеренный при скрининге)
ЭКГ (стандартное в 12 отведениях), Основные показатели жизнедеятельности (пульс, частота дыхания, систолическое и диастолическое давление крови, измеренное после 10 минут отдыха в положении лежа на спине, и после 3 минут в положении стоя)
Лабораторные тесты вместе с гематологическим анализом, биохимическим анализом крови, общим анализом мочи, анализом мочи с целью выявления запрещенных средств, пробой на алкоголь в выдыхаемом воздухе, тесном на β-HCG в моче (только женщины, если применимо).
Каждый субъект получает идентификационный номер, возрастающий в соответствии с хронологическим порядком включения его/ее в исследование.
Рандомизация происходит в D1/TP1 после подтверждения Исследователем соответствия субъектов критериям отбора. Если более чем один субъект рандомизируется в одно и то же время, то субъекты рандомизируются последовательно в соответствии с хронологическим порядком включения утром в День 1/ TP1, т.е. субъект с наименьшим номером субъекта получает следующий имеющийся рандомизационный номер.
Результаты лабораторных тестов D1/TP1 представляют собой исходные значения и рассматриваются как подтверждающие, за исключением теста β-HCG в моче (на основе образца, полученного во время скринингового визита), результат которого должен быть отрицательным.
Если субъекта в результате включают в исследование, берут образец крови для архивирования и для определения антитела против инсулина (только в D1/TP1).
Описание по типу визита
Периоды лечения 1-4 (D1-D2)
Чтобы предотвратить воздействие стандартного инсулинового лечения субъектов на результаты измерения клэмп-теста, субъекты должны воздерживаться от использования базальных инсулинов и перейти на
продукты инсулина средней и короткой продолжительности действия за 48 часов до введения дозы в D1 с TP1 до TP4, если принимают пролонгированные продукты инсулина, т.е. Лантус® (инсулин гларгин), Левемир® (детемир) или инсулины ультраленте,
инсулины короткого действия за 24 часа до введения дозы в D1 с TP1 до TP4, если принимают продукты инсулина средней продолжительности действия, т.е. NPH-инсулин
Последняя подкожная инъекция инсулина короткого действия происходит не позднее, чем за 9 часов до введения исследуемого лекарства. Субъекты, принимающие помповую терапию, прекращают инфузию инсулина утром в День 1, по меньшей мере за 6 часов до каждого введения IP (приблизительно в 03:00 часа, принимая начало введения IP в 09:00).
После поступления в клинику субъектов просят гарантировать, что у них не произошло клинически значимых изменений в состоянии здоровья со времени предыдущего визита, что они соблюдали общие ограничения и ограничения в питании, которые определены в протоколе, и что они изменили инсулиновую терапию, если требовалось. При нарушении критериев исследования субъект исключается из дальнейшего участия в исследовании. В зависимости от вида нарушения субъект может быть исключен только из конкретного дня исследования, что позволяет однократное повторное назначение дня исследования, или субъект может быть полностью исключен из исследования.
Любые изменения в состоянии здоровья и сопутствующая лекарственная терапия субъектов со времени последнего визита описывается в медицинских картах субъектов (первичная документация) и в CRF.
Утром, незадолго до введения исследуемого средства (D1 каждого TP) записывают вес тела, основные показатели жизнедеятельности, ЭКГ в 12 отведениях, мониторинг ЭКГ и центральную температуру тела, выполняют общий анализ мочи и анализ мочи с целью выявления запрещенных средств и анализ на алкоголь.
Количество инсулина гларгина, необходимое для инъекции, рассчитывают в зависимости от веса тела субъекта.
Гематологические показатели анализируют на наличие анемии в День 1 периода лечения 3. В случае положительного результата интервал вымывания между Периодами лечения 3 и 4 увеличивают до максимально разрешенных 18 дней или начало TP4 откладывают до нормализации гематологических параметров. Дополнительную гематологическую оценку проводят в День 1 периода лечения 4.
Субъекты остаются в состоянии натощак (за исключением воды) до окончания эугликемического клэмп-теста.
Затем субъектов готовят к началу преклэмп-процедуры, при этом три системы для внутривенного вливания подсоединяют к устройству для автоматического определения глюкозы крови (Биостатор™) и субъекты остаются в положении полулежа на протяжении всего периода отбора образцов. Около 07:30 вену тыльной стороны руки или вену внешней стороны запястья левой руки катетеризируют и подсоединяют к Биостатору™ для постоянного получения артериализованной венозной крови для определения концентрации глюкозы крови. Левую руку помещают в нагретый контейнер (“Hot-Box”), который нагревает воздух до температуры 55°C, обеспечивая артериализацию венозной крови. Вторую систему для внутривенного вливания соединяют с латеральной подкожной веной левой руки и используют для сбора образцов для определения сывороточного инсулина и референсного определения глюкозы крови. Третью вену катетеризируют на противоположном предплечье, чтобы обеспечить инфузию 0,9% физиологического раствора и 20% раствора глюкозы с помощью насоса в Биостаторе™ или инсулина глулизина с помощью внешнего насоса.
От времени введения сосудистых катетеров до 60 мин перед введением исследуемого средства, приблизительно в 09:00 в D1, уровень глюкозы крови поддерживается в пределах 4,4–6,6 ммоль/л (80-120 мг/дл, преклэмп). В зависимости от уровня глюкозы крови, предоставляют дополнительную внутривенную болюсную инъекцию инсулина глулизина, чтобы сохранить уровень глюкозы крови в пределах заданного диапазона. За 1 час до введения исследуемого средства внутривенные болюсные инъекции прекращают до окончания клэмп-теста.
Дополнительные образцы крови для определения глюкозы получают по меньшей мере с 30 мин интервалами, чтобы проверить в сравнении с лабораторным стандартом на основе глюкозооксидазного метода. При необходимости Биостатор™ повторно калибруют в соответствии с результатами лабораторного эталонного метода.
Скорости инфузии глюкозы регулируют для отдельных субъектов. В то время, как глюкоза крови сохраняется на заранее заданном уровне, инсулин и скорость инфузии глюкозы уменьшаются до минимальных значений во время вводной фазы клэмп-периода. Инфузию раствора инсулина глулизина осуществляют с помощью высокоточной инфузионной помпы (Terumo Spritzenpumpe TE 311™), 20% раствор глюкозы вводят с помощью высокоточной инфузионной помпы (Terumo Infusionspumpe TE 171™).
Клэмп-уровень устанавливают за 60 мин до введения исследуемого средства, чтобы поддержать глюкозу в крови на уровне 5,5 ммоль/л (100 мг/дл) до окончания периода клэмп-теста. Преклэмп фазу продлевают, и введение IP откладывается до 14:00 часов, если заданный уровень глюкозы не достигнут во время фазы введения (преклэмп). Если заданный уровень глюкозы не может быть установлен в срок не позднее 14:00 часов, визит заканчивается и субъекту может быть назначен новый визит для введения дозы спустя 1-7 дней.
Инфузию инсулина глулизина прекращают непосредственно перед введением исследуемого средства. Первую пробу для оценки PK инсулина собирают немедленно, следующим шагом. В 09:00 вводят средства исследования (таблица 4), либо
эталонное средство (R, 0,4 Ед/кг Лантус® U100)
либо исследуемое средство (T1-3) в один участок околопупочной области
согласно плану рандомизации, используя стандартный метод кожной складки.
Во время клэмп-теста ЭКГ в 12 отведениях снимают через 2 часа и через 12 часов после инъекции IP и в конце клэмп-теста.
Исследуемое средство вводится предпочтительно одним и тем же лицом на протяжении всего исследования. Окончание инъекции задает момент времени ноль (T0), который определяет время старта последующего периода проведения клэмп-теста и отбора PK-образцов.
Каждый период наблюдения клэмп-теста продолжается 36 часов и соответственно заканчивается приблизительно в 21:00 в D2, заранее определенное окончание клэмп-теста. Затем субъектов отключают от установки для проведения эугликемического клэмп-теста, если глюкоза крови находится в пределах изогликемического диапазона, субъекты получают питание и свое обычное лечение инсулином.
В случае если глюкоза крови превышает 11,1 ммоль/л (200 мг/дл) во время периода клэмп-теста в течение 30 минут после прекращения инфузии глюкозы, и исследователь подтверждает, что исключены возможные ошибки, приводящие к некорректным результатам глюкозы крови выше 11,1 ммоль/л (200 мг/дл), вводят быстродействующий аналог инсулина (например, инсулин глулизин), применяемый перед введением IP во время клэмп-теста, для продления периода клэмп-теста до 36 часов для сбора фармакокинетических образцов крови. В таком случае должен быть информирован спонсор. Затем субъектов отключают от установки для проведения эугликемического клэмп-теста, если глюкоза крови находится в пределах изогликемического диапазона, субъекты получают питание и свое обычное лечение инсулином.
Реакцию в месте инъекции оценивают через 15 минут и через час после инъекции средства исследования и документируют как AE если наблюдают оценку >3 по шкале раздражения.
Перед выпиской, предоставляют питание ad libitum и субъекты получают свое обычное лечение инсулином.
Основные показатели жизнедеятельности (пульс; систолическое и диастолическое давление крови, измеренное после 10 минут отдыха в положении лежа на спине и после 3 минут отдыха в положении стоя) повторяют, и измеряют глюкозу крови (значение глюкозы крови должно быть выше 80 мг/дл). Субъектов выписывают из клиники в D2 в TP1-TP4 после подтверждения их хорошего самочувствия Исследователем.
Визит окончания исследования
Субъекты возвращаются для проведения визита окончания исследования (EOS) между D5 и D14 после введения последней дозы в TP4. Субъекты находились в состоянии натощак (за исключением воды) в течение 10 часов. EOS включает в себя следующие оценки:
Физическое обследование (вес, температура тела) с дополненной медицинской историей
ЭКГ, измерение основных показателей жизнедеятельности
Лабораторные тесты, включая гематологический анализ, HbA1c, биохимический анализ крови, общий анализ мочи, и, если женщина, определение в крови β-HCG
Любое AE, имевшее место, или сопутствующая лекарственная терапия, принятая после TP4
Образец крови для определения антител против инсулина
Исследователь подтверждает на основании доступных клинических результатов, что субъект может быть выписан из исследовательского центра без опасности для здоровья.
Ограничение (ограничения) исследования
Субъекты прекращают свое обычное лечение инсулином в Дни со -2 до -1, в зависимости от типа применяемого инсулина (пролонгированного, NPH, средней продолжительности действия). Соответственно, уровни глюкозы крови контролируются исключительно многократными подкожными инъекциями обычного инсулина короткого действия.
Обычное лечение инсулином возобновляют после выписки из клиники в День 2 в TP1-TP4.
Субъекты не должны получать сопутствующей лекарственной терапии, которая может влиять на метаболический контроль или чувствительность субъектов к инсулину, на протяжении всего исследования и за две недели до начала исследования.
Запрещено употребление алкогольных напитков, грейпфрутового сока и стимулирующих напитков, содержащих производные ксантина (чай, кофе, напитки, подобные кока-кола, шоколад), за 24 часа до введения каждого исследуемого средства и до окончания клэмп-теста.
Апельсиновый сок или аналогичные углеводы дают в качестве корректирующих мер при гипогликемии во время клэмп-теста, в случае неадекватного противодействия с помощью внутривенной инфузии глюкозы, когда субъект подключен к Биостатору™.
Не разрешается напряженная физическая активность на протяжении 2 дней перед каждым введением исследуемого средства.
Субъектов, которые выкуривают 5 или меньше сигарет в день, включают в исследование, и субъекты могут курить во время исследования, за исключением D1 и D2 в TP1-TP4.
В день скрининга, субъекты приходят в клинику в состоянии натощак с вечера по меньшей мере в течение 10 часов (за исключением небольшого количества углеводов, в качестве меры противодействия гипогликемии, в случае необходимости).
Утром в День 1 в TP1-TP4, субъекты поступают в клинику в состоянии натощак с вечер, по меньшей мере в течение 10 часов, и остаются в состоянии натощак до окончания периода проведения клэмп-теста в День 2. Питание ad libitum предоставляют после окончания клэмп-теста.
Обеспечение жидкостью составляет по меньшей мере 2500 мл для каждого 36-часового периода.
Определение первичных данных
Все оценки, перечисленные ниже, которые описываются в CRF, подтверждаются надлежащим образом подписанной идентифицированной первичной документацией, к которой относится:
идентификация субъекта
медицинская история (в случае аллергической реакции)
клинический осмотр, основные показатели жизнедеятельности, вес тела и рост, температура тела;
лабораторные обследования, ЭКГ
фармакокинетические временные точки
даты и время визитов и обследования
Нежелательные явления
Введение IP
Предшествовавшая/сопутствующая лекарственная терапия
Начало/конец процедуры клэмп-теста, данные клэмп-теста
Пример 13: Статистические оценки
Определение объема выборки
Главной целью исследования является оценить относительный метаболический эффект инсулина гларгина, получаемого в виде одной дозы U100 (R) и трех различных доз U300 (T1-T3).
На основе данных исследования PKD10086, можно предположить значение SDwithin для GIR-AUCend of clamp на преобразованной по натуральному логарифму шкале приблизительно 0,375.
С целью расчета объема выборки использовали значения внутрисубъектных SD от 0,325 до 0,425.
В таблице 11 представлена максимальная погрешность (в терминах 90% доверительного интервала) для парного отношения исправленных геометрических средних значений средств, которые будут получены с 90 % достоверностью, при обще количестве субъектов N от 16 до 24, принимая истинным внутрисубъектное SD значений от 0,325 до 0,425 для log GIR-AUC0-36.
|
В случае 20 субъектов, если истинное внутрисубъектное SD для GIR-AUC0-36 составляет 0,375, отношение параметров средств будет оцениваться с максимальной погрешностью 19,9% (т.е. 90% CI будет равен 0,80, и 1/0,80=1,25 умножить на измеренное отношение), с достоверностью 90%.
В исследование включают 24 субъекта, чтобы получить 20 субъектов, завершивших исследование.
Описание субъектов
Распределение субъектов
Создается подробное заключение по учету субъектов, включая число субъектов, включенных в исследование, рандомизированных, получивших лечение (т.е. получивших любое количество исследуемого средства), завершивших (т.е. субъекты, которые закончили все периоды лечения исследования), исключенных из исследования, вместе с основными причинами исключения.
Распределение субъектов в заключительном визите представлено в списке, включая группу последовательности, статус распределения в конце исследования с датой последнего введения исследуемого лекарства, дату заключительного визита, причину прекращения. Все досрочные исключения из исследования, имевшие место до или после начала первого введения исследуемого лекарства, полностью документируют в основной части отчета о клиническом исследовании (CSR).
Отклонения от протокола
Перед закрытием базы данных исследования, проверяют отклонения от протокола клинических испытаний в отношении критериев, задаваемых для популяций, и других критериев исследования, включающих в себя:
Критерии включения и исключения;
Соблюдение терапии;
Соблюдение протокола клинического исследования в отношении запрещенных лекарств;
Соблюдение протокола клинического исследования в отношении интервалов между визитами и общей продолжительности лечения; и
Выполнение запланированной оценки активности и безопасности, и т.д.
Описываемые отклонения включают в себя, но не ограничиваются:
Субъекты без какой-либо оценки (любых переменных) после рандомизации;
Субъекты не получили лечения;
Субъект без какой-либо оценки первичной переменной (если применимо);
Субъекты вошли в исследование, хотя не соответствовали критериям включения;
Субъекты соответствовали критериям прекращения участия во время исследования, но не были исключены;
Субъекты получили неправильное лечение или ненадлежащую дозу;
Субъекты получили неразрешенную сопутствующую лекарственную терапию.
Важные отклонения перечислены и суммированы.
Анализ популяции
Все исключения из анализа популяций (фармакодинамического, фармакокиненического и/или безопасности) полностью задокументированы в CSR.
Субъекты, исключенные из какого-либо анализа популяции, перечислены с указанием последовательности лечения и причины исключения. Любая существенная информация полностью задокументирована в CSR. Частоты встречаемости субъектов, в целом и по лечению, для анализа популяций представлены в виде таблиц.
Для случая, когда субъекты получали терапию, которые отличалась от терапии, назначенной по плану рандомизации, анализы проводят в соответствии с полученной терапией, а не с назначенной в соответствии с рандомизацией терапией.
Популяция для фармакодинамической оценки
Все субъекты без значительных отклонений, связанных с введением исследуемого средства, и PD-параметры которых доступны, включены в популяцию для фармакодинамической оценки. Для субъектов с недостаточными PD-профилями в один, но не в оба периода лечения, параметры надлежащих профилей включены в анализ.
Для субъектов, которые получают (по соображениям безопасности) инсулин глулизин на протяжении периода наблюдения, 36 часов после введения дозы IP, используют только фармакодинамические данные до времени введения инсулина глулизина.
Исключения из фармакодинамического анализа
Все исключения из фармакодинамического анализа перечислены вместе с указанием причины. Решения об исключениях принимают и документируют на основе проверки данных перед закрытием базы данных и раскрытием рандомизационного кода.
Выборка для оценки безопасности
Все субъекты, которые получили любое лечение сравнительного исследования, независимо от количества проведенной терапии, включены в выборку для оценки безопасности.
Популяции для фармакокинетической оценки
Все субъекты без значительных отклонений, связанных с введением исследуемого средства, и PK-параметры инсулина которых доступны, включены в популяцию для оценки фармакокинетики. Для субъектов с недостаточными PK-профилями инсулина в один, но не во все Периоды лечения, параметры надлежащих профилей включены в анализ.
На биоаналитическое определение инсулина гларгина влияют другие инсулины, подобные инсулину глулизину. Поэтому из оценки исключаются фармакокинетические данные по определению инсулина гларгина у тех субъектов, которыеполучили (по соображениям безопасности) инсулин глулизин в пределах 36 часов периода наблюдения клэмп-теста после введения IP.
Демографические и другие исходные характеристики
Демографические характеристики субъектов, медицинская история и диагнозы
Собирают следующие данные: пол, возраст, рост, вес, и раса. Исходный индекс массы тела (BMI) субъекта рассчитывают из значений веса тела и роста, полученных до введения дозы:
BMI = вес тела [кг]/(рост [м])²
Все переменные, касающиеся демографических и других исходных характеристик, перечислены по отдельным субъектам и суммированы в выборке для оценки безопасности.
Отклонения от критериев включения, связанные с медицинской историей и диагнозами, перечислены и описаны по отдельным субъектам.
Исходные значения параметров безопасности
Для переменных безопасности, последнее запланированное значение перед введением исследуемого средства в пределах периода или в пределах исследования, применимое для переменной, используется в качестве исходного значения. Если исходное значение проверяют повторно перед введением дозы, измеренное повторно значение рассматривают как исходное и применяют в статистике.
Продолжительность исследуемого лечения и соблюдение терапии
Подробности дозирования исследуемого средства и дополнительная информация перечислены по отдельным субъектам и суммированы в соответствующем случае.
Индивидуальные общие дозы инсулина гларгина суммированы по лечению.
Предшествовавшая/сопутствующая лекарственная терапия/терапии
Предшествовавшая и сопутствующая лекарственная терапия/терапии (при наличии) кодируют согласно Рекомендательному списку лекарственных средсв Всемирной организации здравоохранения (WHO-DRL, последняя действующая версия на момент закрытия базы данных) и перечислены по отдельным субъектам.
Сопутствующее введение инсулина (подкожное) перечислено отдельно.
Инфузия инсулина или болюс, полученные в любое время в течение процедуры клэмп-теста, перечисляются или наносятся на график в динамике по времени в индивидуальном порядке. Инфузия инсулина или болюс, полученные после ведения дозы во время процедуры клэмп-теста, перечисляются в индивидуальном порядке.
Анализ фармакодинамических переменных
Все фармакодинамические анализы включают данные, полученные для фармакодинамической популяции. Не проводят корректировку уровня значимости для множественных анализов.
Для оценки фармакодинамики инсулина гларгина во время процедуры клэмп-теста постоянно записывают концентрацию глюкозы крови и скорость инфузии глюкозы (GIR).
Статистические анализы сравнивают исследуемые средства (T1-T3) с эталонным средством (R)
Описание фармакологических переменных
Для достижения сопоставимости при анализе доз инсулина, скорректированных по весу тела субъектов, все значения GIR делят на вес тела субъектов в кг. Таким образом, в дальнейшем, если не указано иначе, GIR всегда относится к стандартизированной по весу тела скорости инфузии глюкозы.
Первичная PD переменная
Следующую PD переменную считают первичной.
Площадь под кривой “стандартизированная по весу тела скорость инфузии глюкозы – время”
[GIR-AUC0-36 (мг/кг)]
GIR-AUC0-36 рассчитывают по правилу треугольников для кусочно-постоянной функции со шкалой времени в минутах.
Вторичные переменные PD
Следующие PD переменные получают и считают вторичными:
Время (ч) до 50% GIR-AUC0-36 [T50%-GIR-AUC0-36 (ч)]
Максимальная сглаженная стандартизированная по весу тела скорость инфузии глюкозы [GIRmax (мг*мин/кг)]
Время после введения дозы до первого достижения
GIRmax [GIR-Tmax (ч)]
Продолжительность эугликемии (время до повышения сглаженного профиля глюкозы крови выше клэмп-уровня) рассчитывают как время от введения дозы до последнего значения сглаженной кривой концентрации глюкозы крови, которое равно или меньше 105 мг/дл.
Продолжительность контролирования глюкозы в заранее заданных пределах определяют как время от введения дозы до достижения последнего значения сглаженной кривой концентрации глюкозы крови, которое равно или ниже
- 110 мг/дл
- 130 мг/дл
- 150 мг/дл
Сглаживание
Максимальное значение необработанной стандартизированной по весу тела GIR подвершается “шуму” во время регулирования GIR. Таким образом, расчет GIRmax и времени до GIRmax, основан на способе сглаживания LOESS (локально взвешенное сглаживание диаграмм рассеяния) для необработанных данных стандартизированной по весу тела GIR. Благодаря ожидаемой морфологии GIR-профилей, которые известны для Лантуса®, используют фактор сглаживания 6% (SAS®, PROC LOESS, factor 0.06).
Уровни глюкозы крови достаточно подвержены “шуму”. Поэтому оценка продолжительности эугликемии и длительности контроля глюкозы крови основана на методе сглаживания LOESS (локально взвешенная регрессия в сглаживании диаграмм разброса) необработанных уровней глюкозы крови. Благодаря ожидаемой морфологии, используют фактор сглаживания 6% (SAS®, PROC LOESS, factor 0.06).
В случае неадекватного сглаживания применяют другой фактор сглаживания для дополнительного анализа.
Дополнительные переменные PD
Получают дополнительные параметры в виде:
Время до окончания инфузии глюкозы, последний момент времени после введения дозы, когда значение GIR выше нуля
Если считают необходимым, получают дополнительные переменные PD для интерпретации результатов.
Первичный анализ PD
Перед проведением описанного ниже анализа, GIR-AUC0-36 log-преобразуют (натуральный log).
Log-преобразованную GIR-AUC0-36 анализируют с помощью линейной модели со смешанными эффектами с фиксированными показателями для последовательности, периода и средства
log(параметр) = последовательность + период + средство + ошибка
и с неструктурированной матрицей R (i, i) дисперсий и ковариаций средства для субъекта в блоках последовательности, используя SAS PROC MIXED.
Получают 90% доверительный интервал (CI) для отношения геометрических средних средств (T1/R, T2/R, T3/R) путем вычисления оценки и 90% CI для различия между средними значениями для средств в рамках линейной модели со смешанными эффектами, и затем переводят в отношение геометрических средних с помощью антилогарифмического преобразования. Эквивалентность подтверждают, если 90% CI для отношения полностью находится в пределах референтного интервала биоэквивалентности 0,80-1,25.
Списки индивидуальных отношений (исследуемые виды лечения и эталонное лечение) предоставляются с соответствующей описательной статистикой.
Вторичный анализ/анализ вторичных переменных
Описательное представление профилей GIR
Индивидуальная стандартизированная по весу тела GIR (мг*мин/кг) наносится на графики для неисправленных, сглаженных и кумулятивных неисправленных значений.
Стандартизированные по весу тела GIR-профили для средних значений и медиан, а также кумулятивные профили для медиан, выраженных в процентах, в динамике по времени наносят на графики по средствам.
Кумулятивные графики охватывают время от введения дозы до окончания клэмп-теста.
Описательное представление производных параметров PD
Параметры PD перечислены по отдельным субъектам, и описательная статистика произведена по лечению.
Отношения средств для вторичных параметров PD
Отношение средств (T1/R, T2/R, T3/R) с границами доверительного интервала получают для максимальной стандартизированной скорости инфузии глюкозы [GIRmax (мг*мин/кг)] с использованием соответствующей линейной модели со смешанными эффектами, как описано выше для первичного анализа. Исследовательские сравнения между средствами основаны на обычно применяемом критерии биоэквивалентности (границы доверительного интервала 90% 0,80–1,25).
Распределение значений GIR-Tmax представляют с помощью гистограмм для каждого средства. Дополнительно предоставляют гистограмму различий в GIR-Tmax для исследуемых средств и эталонного средства.
Различия между средствами для вторичных параметров PD
T50%-GIR-AUC0-36 (ч) анализируют непараметрически на основе критерия Ходжа-Лемана для парных сравнений средств. Получают CI для парных различий по средствам (T1-R, T2-R, T3-R) для медиан. Распределение значений T50%-GIR-AUC0-36 представляют с помощью гистограмм для каждого лечения. Дополнительно предоставляют гистограмму различий в T50%-GIR-AUC0-36 между лечениями (T1-R, T2-R, T3-R).
Распределение значений GIR-Tmax представляют с помощью гистограмм для каждого лечения. Кроме того, предоставляют гистограмму различий в GIR-Tmax между исследуемыми видами лечения и эталонным лечением.
Продолжительность эукликемии и контроль глюкозы крови представлены с помощью гистограмм. Сравнения лечений выполняли с помощью непараметрических методов.
Выполнение клэмп-теста
Профили концентрации глюкозы крови по субъектам наносят на график.
Продолжительность клэмп-теста получают для каждого клэмп-теста, как время между введением дозы и окончанием клэмп-теста, измеренное в часах.
Индивидуальную вариабельность глюкозы крови для каждого клэмп-теста устанавливают как коэффициент вариации (CV%) значений глюкозы крови между индивидуальным началом и индивидуальным окончанием клэмп-теста (или первым введением инсулина глузилина во время клэмп-теста). Индивидуальный средний уровень глюкозы крови для каждого клэмп-теста получают как среднее арифметическое значений глюкозы крови между индивидуальным началом и индивидуальным окончанием клэмп-теста (или первым введением инсулина глузилина во время клэмп-теста).
Параметры перечислены по отдельным субъектам и суммированы с помощью описательной статистики в пределах лечения.
Анализ данных безопасности
Оценка безопасности основана на рассмотрении индивидуальных значений (потенциально клинически значимые отклонения от нормы), описательной статистике (сводные таблицы, графики) и при необходимости на статистическом анализе (соответствующие оценки, доверительные интервалы). Применяют критерии "Потенциально клинически значимые отклонения от нормы" (PCSA) в соответствии со стандартными критериями Sanofi-Aventis. Критерии документируют в Плане статистического анализа исследования. Анализ безопасности проводят согласно стандартам, относящимся к анализу и предоставлению отчетов результатов по безопасности на основании клинических испытаний.
Все анализы безопасности включают в себя указанные выборки для оценки безопасности.
Для всех данных по безопасности период наблюдения делится на сегменты трех различных типов:
период перед применением исследуемого средства определяют как время от момента подписания субъектом информированного согласия и до первого введения исследуемого средства.
период применения исследуемого средства определяют как время от (первого) введения исследуемого средства вплоть до 72 часов спустя.
период после применения исследуемого средства определяют как время после периода применения исследуемого средства либо до (первого) введения исследуемого средства в следующий период, либо до окончания периода наблюдения.
Нежелательные явления
Все AE кодируют, используя MedDRA (последняя действующая версия на момент закрытия базы данных).
Предоставляют следующие списки всех нежелательных явлений:
Список всех нежелательных явлений (по субъектам)
Список замечаний, связанных с нежелательными явлениями
Определения
Для данных по безопасности период наблюдения делят на сегменты трех различных типов:
период перед применением исследуемого средства определяют как время от момента подписания субъектом информированного согласия и до первого введения исследуемого средства сравнения.
период применения исследуемого средства в каждый период определяют как время от (первого) введения исследуемого средства вплоть до 72 часов спустя.
период после применения исследуемого средства определяют как время после периода применения исследуемого средства либо до (первого) введения исследуемого средства в следующий период, либо до окончания периода наблюдения.
Возникшие после начала лечения нежелательные явления
Все AE классифицируют следующим образом:
Возникшие после начала лечения нежелательные явления (TEAE) представляют собой любые AE с появлением (включая ухудшение) во время периода применения исследуемого средства
Нежелательные явления, возникшие не во время применения средства (NTEAE), представляют собой любые AE, не классифицируемые как TEAE:
- Долечебные AE, определяемые как AE, которые развились (или усугубились) в период перед применением исследуемого средства, до получения первой дозы исследуемого средства
- Постлечебные AE, определяемые как AE, которые развились в период после применения исследуемого средства без ухудшения в фазу применения исследуемого средства.
Связь с принимаемыми средствами
В целях анализа каждое TEAE соотносят с последним средством, полученным перед возникновением (или усугублением) AE. Если TEAE развивается во время применения одного средства и усугубляется под действием другого, оба лечения считают, что оно появилось за время лечения обеими средствами.
Отсутствующая информация
В случае отсутствующей или противоречивой информации, AE считают как TEAE, если только очевидно может быть исключено, что оно не является TEAE (например, с помощью частичных данных или другой информации).
Если данные о начале AE являются неполными или отсутствуют, считают, что оно имело место после первого введения исследуемого средства за исключением случая, когда неполные данные показывают, что AE началось перед лечением.
Возникшие после начала лечения нежелательные явления
Возникшие после начала лечения нежелательные явления перечислены и суммированы по лечению:
Обзор TEAE (число и процент субъектов по меньшей мере с одним TEAE, тяжелым TEAE, TEAE, приводящим к прекращению лечения, смерти (при наличии))
Заключение обо всех возникших после начала лечения нежелательных явлениях по первичному классу системы органов и предпочтительному термину (число и процент субъектов по меньшей мере с одним TEAE) (“текстовая таблица”)
- Таблица без указания числа нежелательных явлений (для основной части отчета о клиническом исследовании)
- Таблица с указанием числа явлений (для приложения отчета о клиническом исследовании)
- Таблица с указанием числа субъектов на каждом средстве (U100, U300) и субъектов overall (для приложения отчета о клиническом исследовании)
Список субъектов, представляющий возникшие после начала лечения нежелательные явления по лечению, классу систем органов и предпочтительному термину
Смертельные случаи, серьезные и другие
важные нежелательные явления
В случае каких-либо явлений, смертельные случаи, серьезные AE и другие важные AE перечислены по отдельным субъектам и подробно описаны в отчете об исследовании.
Нежелательные явления, приводящие к прекращению лечения
В случае каких-либо явлений создают списки по отдельным субъектам для всех нежелательных явлений, приводящих к прекращению лечения.
Клинические лабораторные оценки
Гематологические и биохимические данные
Лабораторные параметры безопасности измеряют в D1 периода лечения 1 и в EOS. По графику указанные параметры безопасности оценивают во время периода применения исследуемого средства (за исключением гематологического анализа в TP3 и TP4).
Значения, которые используют в качестве исходных показателей (гематологических и биохимических) представляют собой значения, полученные в D1 перед использованием средства в первый период лечения. Если любой из запланированных тестов исходного уровня проводят повторно для любого субъекта, последние перепроверенные значения рассматривают в качестве исходных, при условии что они выполнены до первого введения IP.
Предоставляют следующие таблицы и списки:
Описательная статистика для необработанных данных и изменений от исходного уровня (включая % изменения креатинина)
Предоставляется специальный список отдельных данных субъектов с PCSA после включения, сортированных по функции и времени измерения
Все отдельные данные, включая повторно проверенные значения запланированных гематологических и биохимических анализов, перечислены по биологической функции и времени измерения. Результаты незапланированных лабораторных тестов, если имеются, включены в указанный список. В указанных списках отмечены отдельные данные, которые меньше или больше нижних или верхних лабораторных пределов и/или достигают абсолютного предела критериев PCSA, если определены
Список результатов оценки функции печени субъектов, которые перенесли по меньшей мере одно из следующего:
- по меньшей мере один случай ALT>3ULN и по меньшей мере один случай общего билирубина >2 ULN во время исследования, при этом по меньшей мере одно из указанного произошло после первой дозы
- конъюгированный билирубин >35% общего билирубина и общий билирубин >1,5 ULN будут установлены в одной и той же пробе после получения первой дозы, независимо от определения во время фазы применения исследуемого средства.
Предоставляется список, относящийся к увеличению ALT≥2 ULN, включая в частности информацию о приеме лекарства, медицинский и хирургический анамнез, алкогольные привычки, инициирующие факторы, подробности осложнения вместе со значениями ALT, сопутствующие факторы и симптомы.
Предоставляется список результатов определений, выходящих за пределы стандартного диапазона.
В списках субъектов с PCSA данные о функции печени, CPK и эозинофилы выражают в виде кратного для соответствующего ULN.
Результаты анализа мочи
Все результаты качественного анализа мочи (тест-полоска), включая повторно проверенные значения, перечислены.
Основные показатели жизнедеятельности
Давление крови и пульс
Пульс и систолическое и диастолическое давление крови (SBP и DBP) измеряют после 10 минут отдыха в положении лежа и также после 3 минут в положении стоя, если субъекты не подключены к Биостатору™.
Значения, которые используют в качестве в качестве исходных, представляют собой значение оценки перед введением дозы в D1 каждого периода лечения. Если любой из запланированных тестов исходного уровня повторяют для какого-либо субъекта, последние перепроверенные значения рассматривают в качестве исходных, при условии, что они были выполнены перед введением IP.
Для пульса и давления крови ортостатические различия рассчитывают как изменение при переходе из положения лежа в положение стоя.
Для всех параметров анализ данных “по применению средства” (“On-Treatment”) выполняют, включая все незапланированные значения и повторно проверенные значения.
Предоставляют следующие таблицы и списки:
Сводные таблицы числа субъектов с PCSA предоставляются в виде таблиц числа случаев PCSA после включения, независимо от нормального или не соответствующего норме статуса на этапе включения
Для пульса и давления крови (в положениях лежа на спине и стоя) необработанные данные и изменения от исходных значений (только для положения лежа на спине) суммируют с помощью описательной статистики, для типа измерения (положения) каждого параметра и временной точки, на основе запланированных измерений перед введением дозы и оцененных исходных значений.
Все индивидуальные данные, включая незапланированные и перепроверенные значения, перечислены (в положении супинации, в положении стоя, ортостатическая разница). В списках, отмечают значения, достигающие пределов критериев PCSA, если они определены
Предоставляется список данных по субъектам с PCSA после включения
Комментарии, касающиеся показателей жизнедеятельности, также перечислены в приложении, если есть в наличии.
Вес тела, индекс массы тела и температура тела
Значения, которые используют в качестве исходных значений для веса тела и BMI, являются значениями, полученными в D1 TP1.
Значения, которые используют в качестве исходных значений для температуры тела, являются значениями, полученными в D1 каждого TP.
Перечислены данные по субъектам, включая метки (только вес) для значений, которые достигают пределов критериев PCSA.
ЭКГ
Пульс, PQ-, QRS-, и QT-интервалы и скорректированные QT (QTc) на основании автоматической регистрации показаний анализируют как неисправленное значение параметра и как изменение от исходного уровня.
Значения, которые используют в качестве исходных, представляют собой значения до приема дозы в День 1 каждого периода. Если любой из запланированных тестов исходного уровня повторяют для субъекта, повторно проверенные значения рассматривают в качестве исходных в том случае, если они были получены перед введением средства периода.
Для всех параметров, анализ данных по применению исследуемого средства выполняют, используя все обследования после включения, выполненные во время периода применения исследуемого средства, включая повторно проверенные значения. Производят подсчет пациентов с PCSA после этапа включения в сводных таблицах независимо от нормального или измененного статуса на исходном уровне, по группе лечения.
Неисправленные данные для всех параметров и изменение от исходного значения суммируют в описательной статистике по параметру, лечению, и времени измерения.
Индивидуальные данные, включая повторно измеренные значения, перечислены, отсортированы по лечению, субъекту, визиту и времени измерения. В списках выделяют значения, достигающие пределов критериев PCSA.
Предоставляют список индивидуальных значений субъектов с PCSA после включения, отсортированных по типу измерения и по субъекту, периоду, и времени измерения.
Кроме того, предоставляется также отдельный список сердечных профилей для субъектов с пролонгированным QTc (>450 мс для мужчин и >470 мс для женщин) или с изменениями от исходного уровня QTc >60 мс (для мужчин и женщин) и список субъектов по меньшей мере с одним нарушением в качественной оценке (т.е., не соответствующая норме ЭКГ) после получения 1ой дозы.
Другие параметры, связанные с безопасностью
Физическое обследование
Предоставляется список комментариев, относящихся к физическому обследованию, если они есть в наличии.
Местная переносимость в месте инъекции
Распределение частот по лечению предоставляется для уровней местной переносимости в месте инъекции. Данные перечислены по субъектам. В пределах каждого критерия и лечения считают субъектов по наиболее тяжелому результату их реакции на инъекцию.
Аллергические реакции
Списки аллергических реакций
Любые случаи аллергической реакции документируют как нежелательные явления с подробной дополнительной информацией. Все случаи подробно описывают в отчете о клиническом исследовании.
Случаи по пациентам и все дополнительные данные перечислены.
Аллергический анамнез и семейный анамнез
Аллергический анамнез и семейный анамнез документируется для субъектов с проявлением возможной аллергической реакции. Все подробности аллергического анамнеза и аллергического семейного анамнеза перечислены по отдельным субъектам.
Антитела против инсулина
Предоставляется сводная таблица с указанием числа субъектов, имевших положительные результаты теста на антитела против инсулина во время исследования и во время обследований после завершения исследования. Предоставляется список по отдельным субъектам.
Анализ фармакокинетических данных
Фармакокинетические параметры
Список PK-параметров показан выше. Дополнительно получают T50%-AUC0-36 для инсулина в контексте статистического анализа.
Статистический анализ
Фармакокинетические параметры инсулина гларгина перечислены и суммированы с применением по меньшей мере арифметического и геометрического среднего, стандартного отклонения (SD), стандартной ошибки среднего (SEM), коэффициента вариации (CV%), минимального значения, медианы и максимального значения для каждого лечения.
Все фармакокинетические анализы включают данные соответствующих популяций для фармакокинетического анализа, которые определены выше. Корректировку уровня значимости критерия для множественных анализов не проводят.
Статистические анализы сравнивают исследуемые средства (T1-T3) с эталонным средством(R).
Анализ отношений средств
Анализ выполняют для AUC0-36 инсулина гларгина. Перед выполнением всех описанных ниже анализов, значения AUC0-36 логарифмически преобразуют (натуральный логарифм).
Log-преобразованные параметры анализируют с помощью линейной модели с фиксированными параметрами последовательности, периода и вида лечения
log(параметр) = последовательность + период + лечение + ошибка,
и с неструктурированной матрицей R лечения (i, i) вариациями и ковариациями для субъекта внутри блоков последовательностей, используя SAS PROC MIXED.
Оценку и 90% доверительный интервал (CI) для отношения геометрических средних средств (T1/R, T2/R, T3/R) получают путем вычисления оценки и 90% CI для различия между средними значениями параметров в рамках линейной модели со смешанными эффектами, и затем переводят в отношение геометрических средних с помощью антилогарифмической трансформации. Делают заключение о биоэквивалентности, если 90% CI для указанного отношения полностью находится в референсном интервале эквивалентности 0,80–1,25.
Списки отдельных отношений средств (T1/R, T2/R, T3/R) предоставляют с соответствующей описательной статистикой.
T50%-AUC0-36 для инсулина
Распределение значений T50%-AUC0-36 для инсулина представляют с помощью гистограмм для каждого средства. Кроме того, предоставляют гистограмму различий в T50%-AUC0-36 между средствами (T1-R, T2-R, T3-R).
T50%-AUC0-36 (ч) анализируют с помощью непараметрических методов.
Зависимость “доза-воздействие” для инсулина гларгина U300
Описательные анализы зависимости “доза-воздействие”
Зависимость “доза воздействие” для инсулина гларгина U300 описывают графически с помощью
графики по субъектам “воздействие, деленное на общую дозу для каждого субъекта”
графики по субъектам “воздействие, деленное на дозу, на кг веса тела”
графики по субъектам “нормализованное по дозе воздействие, деленное на дозу, на кг веса тела (корректировка дозы на 0,6 Ед/кг)”
Если считают необходимым для интерпретации результатов, включают дополнительные описательные анализы.
Статистический анализ зависимости “доза-воздействие ”
Для AUC инсулина гларгина, рассчитанного для исследуемых средств T1-T3, зависимость “доза-воздействие” оценивают, используя эмпирическую степенную модель (PK-параметр = a* дозаb), наряду с “оценочной” интерпретацией, в соответствии с рекомендациями в Gough et al. (Gough K, Hutchison M, Keene O et al. Assessment of dose proportionality: report from the pharmaceutical industry. Drug Information Journal 1995; 29: 1039-1048).
Эмпирическая степенная модель предоставляет явную и интерпретируемую оценку степени непропорциональности, которая может быть использована как для подтверждения пропорциональности, так и для оценки фармакокинетической и клинической значимости каких-либо отклонений. Анализ исследований пропорциональности доз, однако, требует вычисления оценки, а не проверки достоверности, чтобы могла быть определена фармакокинетическая и клиническая значимость какой-либо непропорциональности.
Степенная модель отображается на log-преобразованный шкале с применением случайных коэффициентов степенной модели для дозы (в Ед/кг вес тела):
log(параметр) = (log(альфа) + альфа[i]) + (бета+бета[i]) * log(доза)
где log(альфа) и бета представляют собой популяционное постоянное слагаемое и наклон соответственно, и альфа[i] и бета[i] представляют собой случайные отклонения от альфа и бета, соответственно, для i-того субъекта.
Оценки для бета с 90% доверительными интервалами получают с помощью оцениваемых обобщенных наименьших квадратов в методе SAS®/PROC MIXED, с использованием оценок метода ограниченного максимального правдоподобия (REML) для параметров ковариации. Затем применяют оценки и 90% доверительные интервалы для бета, чтобы получить оценки и 90% доверительные интервалы для увеличения параметра PK, ассоциированного с r-кратным увеличением дозы (r=1,5 и r=2,25 [т.е. высокая доза/низкая доза]), путем потенцирования r до степени бета-оценки и границ доверительного интервала.
В случае неадекватности модели для анализа применяют модель смешанных эффектов (которую использовали для анализа отношений средств). Оценки с 90% CI для увеличения параметра, ассоциированного с парным увеличением доз получают, вычисляя вначале оценки с CI для парных различий между дозами в рамках модели смешанных эффектов, и затем преобразуют в отношения с помощью антилогарифмического преобразования.
Анализ PK/PD
В соответствующем случае создают графические изображения (диаграммы рассеяния) для изучения взаимодействия PK/PD.
Пример 14: Результаты исследования
Распределение субъектов
Всего 24 субъекта с диабетом 1 типа были включены в исследование, рандомизированы и получили по меньшей мере одну дозу исследуемого средства. Из 24 рандомизированных субъектов 2 субъекта выбыли из исследования по собственному желанию. Двадцать два (22) субъекта завершили исследование согласно протоколу и были включены в фармакодинамический (PD) и фармакокинетический (PK) анализы. Все 24 субъекта, получивших средство, были включены в оценку безопасности.
Значительных отклонений от протокола не имелось.
Демографические характеристики
Собирали следующие данные (таблица 12): пол, возраст при скрининге, рост, вес и раса. Индексы массы тела (BMI) субъектов рассчитывали из данных веса тела и роста: BMI = вес тела [кг]⋅(рост [м])-2.
|
Выполнение клэмп-теста
В четырех периодах лечения для каждого субъекта R (Лантус U100), T1 (0,4 Ед/кг HOE901-U 300), T2 (0,6 Ед/кг HOE901-U 300) и T3 (0,9 Ед/кг HOE901-U 300), исходные концентрации глюкозы крови отдельных участников до введения средства инсулина являлись сходными, определяя уровень клэмп-теста 100 мг/дл. Продолжительность периода наблюдения в клэмп-тестах после введения дозы составляла 36 часов и являлась одинаковой во всех периодах лечения.
Первичные конечные показатели
Эквивалентность биодоступности (воздействия) и биоэффективности (активности) R и T не была установлена.
Первичные переменные
Площадь под кривой концентрация инсулина гларгина в сыворотке крови – время от 0 до 36 часов (INS-AUC(0-36h)) не была эквивалентна для R и T1 и T2 и была приблизительно эквивалентна в случае T3. Установили, что воздействие, по сравнению с R было меньше на 37% в случае T1, меньше на 43 % в случае T2 и являлось сходным в случае T3.
Площадь под кривой GIR в зависимости от времени от 0 до 36 часов (GIR-AUC(0-36h)) не была эквивалентна для R и T1 и T2 и являлась приблизительно эквивалентной в случае T3. Установили, что потребление экзогенной глюкозы, требуемой для сохранения контроля уровня глюкозы в крови, было меньше на 88% в случае T1, на 67% в случае T2, в то время, как являлось сходным в случае T3.
Вторичные переменные
Время до 50% в INS-AUC(0-36h) (ч) в случае R составляло приблизительно 14 часов, и, таким образом, было меньше, по сравнению с 16 ч, 16 ч и 19 ч в случае T1, T2 и T3, соответственно.
Время до 50% в GIR-AUC(0-36h) (ч) в случае R составляло приблизительно 12 часов, и, таким образом, было меньше по сравнению с 17 ч, 18 ч и 20 ч в случае T1, T2 и T3, соответственно.
Безопасность
Не сообщалось о серьезных нежелательных явлениях (AE) или отменах средства по причине AE. Двое субъектов на R, 2 в T1 и 4 в T3 сообщали всего о 8 TEAE, все из которых имели интенсивность от слабой до умеренной, и разрешались без последствий. Наиболее часто описываемым нежелательным явлением была головная боль. Примечательно, что головная боль часто наблюдается при клэмп-исследованиях и имеет отношение к инфузии гиперосмолярных растворов глюкозы. Однако связь с исследуемыми продуктами не может быть исключена. Не сообщалось о реакциях в месте инъекции в случае T1, T2 и T3, тогда как у 2 субъектов, получавших R, развилась едва заметная эритема в месте инъекции.
Выводы
Одинаковые дозы R и T U 300 не являются эквивалентными по биодоступности (воздействию) и биоэффективности (активности) после введения разовой дозы. Воздействие и активность, наблюдаемые после введения T1 (0,4 Ед/кг) и T2 (0,6 Ед/кг), были меньше, по сравнению с воздействием и активностью после введения R (0,4 Ед/кг). R и T3 являлись, по существу, эквивалентными в отношении воздействия и потребления экзогенной глюкозы.
Однако T1, T2 и T3 показали еще более пологие PK (воздействие) и PD (активность) профили даже с меньшим отклонением от средних значений, чем R, т.е., профиль, который был бы желателен при подаче базального инсулина. Данный факт очевиден, в частности, при сравнении R и T3, которые обеспечивают номинальное эквивалентное общее воздействие и общее потребление глюкозы, несмотря на различные профили.
Указанные удивительные и неожиданные различия в воздействии и активности между средствами R (Лантус U100) и T (HOE901-U300) у субъектов с диабетом 1типа показаны по существу на фигурах ниже.
Кроме того, введение T (HOE901-U300) происходило без проблем, связанных с безопасностью и переносимостью.
Пример 15. Обоснование целесообразности исследования по сравнению глюкодинамической активности и воздействия двух различных подкожных доз (HOE901-U300) с Лантусом U100 у пациентов с диабетом 1 типа
Результаты исследования с участием здоровых субъектов и субъектов с диабетом 1 типа (смотреть предыдущие примеры) показали, что воздействие и эффективность Лантуса® U100 и инсулина гларгина U300 не эквивалентны. Субъекты получали одинаковую дозу инсулина гларгина (0,4 Ед/кг) в U100 и U300, но применение одинакового по единицам количества U300 вызывало меньшее воздействие из расчета меньшего потребления экзогенной глюкозы для сохранения контроля глюкозы крови, чем применение U100. Хотя Лантус U100 имеет профиль воздействия и фармакодинамический профиль без выраженных отклонений от средних значений, однако HOE901-U300 показал даже меньшее отклонение в профиле воздействия и в фармакодинамическом профиле, которое можно было бы желать при обеспечении базальным инсулином, даже с еще большей продолжительностью действия.
Чтобы оценить фармакокинетический и фармакодинамический профиль в условиях стационарного состояния, в новом исследовании, описанном в следующих примерах, сравнивают соответственно две различных подкожных дозы инсулина гларгина U300 в сопоставлении со стандартной дозой Лантуса® U100, в качестве средства сравнения, с конечным установлением эугликемических клэмп-условий у пациентов с диабетом 1 типа. Настоящее исследование имеет своей целью оценить дозу U300, которая является равной по эффективности 0,4 Ед/кг Лантуса® U100, как определено с помощью параметров контроля глюкозы крови и утилизации глюкозы крови, предусмотренных клэмп-методом.
Воздействие инсулина гларгина оценивают на основании профилей “концентрация–время” после повторного подкожного введения в стационарном состоянии, и активности, в виде утилизации глюкозы на единицу инсулина в стационарном состоянии.
Исследование включает в себя два перекрестных лечения (R и T1, и R и T2) в 2 параллельных группах, с 2 периодами лечения (TP1, TP2) и с 2 последовательностями, каждое. Проводят один скрининговый визит (D-21-D-3), визиты лечения (D1-D10 в TP1 и TP2 с введением дозы вечером), с периодами пребывания в клинике (D1-D4 утро и D8 утро до вечера D10 для клэмп-обследований) и один визит окончания исследования (между D7 и D10, после последней дозы) с заключительной оценкой параметров безопасности.
Доза 0,4 Ед/кг Лантуса® U100, выбранная для исследования, хорошо характеризуется в обеспечении контроля эугикемического уровня глюкозы крови у пациентов с диабетом 1 типа и полностью изучена в других клэмп-исследованиях с участием пациентов с диабетом 1 типа.
Тестируют две различные дозы инсулина гларгина U300 0,4 и 0,6 Ед/кг. Указанный диапазон доз позволяет провести интерполирование приблизительной дозы, равноэффективной 0,4 Ед/кг Лантуса® U100. Дозу 0,4 Ед/кг инсулина гларгина U300 уже изучали на здоровых добровольцах и субъектах с сахарным диабетом (смотреть предыдущие примеры) и обнаружили, что она являлась менее активной, чем 0,4 Ед/кг Лантуса® U100 в пределах 30 и 36 часов, соответственно, заранее заданных окончаний периодов наблюдения. Контроль глюкозы крови с помощью 0,4 Ед/кг инсулина гларгина U300 требовал меньшей утилизации общей глюкозы, чем контроль с помощью эталонного средства (0,4 Ед/кг Лантус® U100). Предполагают, что соответственно более высокая доза инсулина гларгина U300, например, 0,6 Ед/кг инсулина гларгина U300 приведет даже к более строгому контролю глюкозы крови при меньшей общей утилизации глюкозы. Кроме того, пропорциональное увеличение дозы позволяет изучить воздействие и эффект профилей ”доза-пропорциональность”.
Исследование с участием пациентов с диабетом 1 типа позволяет избежать смешанного влияния эндогенного инсулина и дает возможность лучше оценить воздействие и продолжительность действия.
Данное исследование имеет перекрестный дизайн; исходя из результата предыдущих исследований, не более чем две дозы HOE901-U300 сравнивают с Лантус® U100. Оценка глюкодинамической активности продуктов инсулина длительного действия требует условий эугликемического клэмп-теста после 24 часов, заранее заданного интервала наблюдения после инъекции, в связи с увеличенной продолжительностью действия.
Активный фармацевтический ингредиент, инсулин гларгин, является одинаковым в обоих средствах, U100 и U300. Дозы, используемые в исследовании, находятся в диапазоне доз регулярного использования. Хотя совокупный риск гипогликемии не исключен полностью, он контролируется с помощью метода эугликемического клэмп-теста.
Фармакодинамика
Фармакодинамическую активность инсулина гларгина оценивают с помощью метода эугликемического клэмп-теста у пациентов с диабетом 1 типа, который является установленной стандартной процедурой для оценки эффекта экзогенных вводимых продуктов инсулина на утилизацию глюкозы.
Параметры, специфичные для оценки утилизации глюкозы в условиях эугикемического клэмп-теста являются скорость инфузии глюкозы, стандартизированная по весу тела (GIR), общая глюкоза, утилизированная на протяжении 24 и 36 часов, соответственно, GIR-AUC0-24 и GIR-AUC0-36, и время до достижения указанного процента в GIR-AUC0-24 и GIR-AUC0-36,такое как время до 50% в GIR-AUC0-36.
Дополнительные параметры представляют собой миксимальную сглаженную стандартизированную по весу тела GIR, GIRmax, и время до GIRmax, GIR-Tmax.
Продолжительность действия инсулина гларгина получают из времени между введением дозы и предварительно заданными отклонениями выше эугликемического (клэмп) уровня.
Мониторинг глюкозы проводят в течение 36 часов вследствие большей продолжительности действия инсулина гларгина после подкожного введения
Фармакокинетика
Благодаря основному свойству замедленного высвобождения инсулина гларгина в профиле концентрации отсутствуют выраженные пики. Следовательно, время до 50% в INS-AUC (например, T50% INS-AUC0-36) рассчитывают как оценку “время-положение” профиля воздействия инсулина гларгина, и INS-Cmax и INS-Tmax будут служить в качестве дополнительных количественных показателей.
Главные цели исследования
Главной целью исследования является оценка контроля глюкозы крови и потребление требуемой экзогенной глюкозы двух различных доз инсулина гларгина U300 в сопоставлении с 0,4 Ед/кг Лантуса® U100 в стационарном состоянии.
Дополнительные цели исследования
Дополнительными целями исследования является оценка в стационарном состоянии скорости воздействия двух различных доз инсулина гларгина U300 в сопоставлении с 0,4 Ед/кг Лантуса® U100, сравнение продолжительности действия двух различных доз инсулина гларгина U300 в сопоставлении с 0,4 Ед/кг Лантуса® U100, изучение зависимостей “доза-ответ”и “доза-воздействие” инсулина гларгина U300, и оценка безопасности и переносимости инсулина гларгина U300 у субъектов с диабетом 1 типа.
Пример 16: Изменение свойств растворимости кислых средств инсулинов длительного действия в высоких концентрациях
Влияние более высоких концентраций средств инсулина гларгина в отношении свойств растворимости исследуют с применением тестовой системы in-vitro. Для этого исследования осадков выполняют с использованием фосфатного буфера со значением рН 7,4, который имитирует условия in-vivo.
Супернатант осажденного инсулина исследуют с помощью метода ВЭЖХ для определения содержания инсулина гларгина.
Подробное описание исследований:
Приготовление буферного раствора для осаждения:
Растворяют 19,32 мг дигидрофосфата натрия моногидрата (M: 137,98 г/ моль) в 1 мл воды. Применяют 0,1 M гидроксид натрия или 0,1 M соляную кислоту для доведения значения pH до 7,4.
Выполнение исследований осадков:
Растворы лекарственного продукта инсулина гларгина с концентрациями до 1000 Ед/мл и содержащие одинаковое общее количество инсулина гларгина и буфер, помещали в пластиковые пробирки и слегка встряхивали. После осаждения инсулина гларгина дисперсионные растворы центрифугируют при низких скоростях вращения в течение заранее заданного периода времени. Заданный объем эксципиентов растворения забирают и заменяют свежим буферным эксципиентом.
Определение содержания инсулина:
Содержание инсулина гларгина в образцах супернатанта определяют количественно против соответствующего образца инсулина для сравнения с помощью ВЭЖХ с обратной фазой, используя систему с двумя мобильными фазами, содержащими буфер дигидрофосфата натрия в воде, хлорид натрия (NaCl) и различные количества ацетонитрила.
В качестве стационарной фазы используют октадодецил-колонку, определение проволят при длине волны 215 нм.
Профиль высвобождения инсулина гларгина из более концентрированных растворов (например, U500 и U1000) является более пологим и пролонгированным по сравнению с Лантусом U100.
Пример 17: Микроскопическое исследование осадков
Осадки средств инсулина гларгина с концентрациями100 Ед/мл, 300 Ед/мл, 500 Ед/мл 700 Ед/мл и 1000 Ед/мл исследовали с помощью микроскопии. Указанные средства (с одинаковым количеством 60 Ед инсулина гларгина) осаждали в 200 мкл фосфатного буфера, pH 7,4 и исследовали с помощью оптического микроскопа в проходящем свете (Olympus Model BX61) с увеличением 100x, в дальнейшем показаны изображения, также представляющие максимальные диаметры. Указанные исследования выявили различия в характеристиках осадков, приводящих к образованию частиц заметно большего размера при увеличении концентраций. Результаты представлены на фигуре 8.
Пример 18: Эффект инсулина гларгина по снижению глюкозы крови у собак
Эффект инсулина гларгина по снижению глюкозы крови оценивали у здоровых, нормогликемических собаках породы бигль. Собаки получали разовые подкожные инъекции 0,3 МЕ/кг. Глюкозу в венозной крови определяли перед первой инъекцией и после, вплоть до 24 часов.
Животные были взяты из когорты ~30 здоровых, нормогликемических, самцов собак породы бигль, полученных из компании Harlan. Собак содержали в питомниковых группах в стандартизированных условиях. За день до начала исследования собак распределяли случайным образом по камерам для исследования. Они находились в состоянии натощак в течение 18 часов до начала и в течение эксперимента со свободным доступом к водопроводной воде. В настоящем исследовании вес тела собак составлял от 13 до 27 кг. После каждого эксперимента собакам давали время на восстановление в течение по меньшей мере двух недель.
Животных рандомизировали в группы по n=6. В нулевой момент времени животные получали разовые дозы тестируемого соединения. Инсулин гларгин вводили в виде разовой подкожной инъекции дозы 0,3 МЕ/кг.
Отбор проб крови осуществляли последовательно с помощью пунктирования вены предплечья (Vena cephalica) перед введением лекарства (0 ч) и после, вплоть до 24 часов. Глюкозу в крови определяли ферментативным методом (набор Gluco-quant® Glucose/HK на приборе Roche/Hitachi 912).
Воздействие на уровень глюкозы в крови после подкожной инъекции различных концентраций средств инсулина гларгина, 100 и 300 единиц/мл, тестировали на здоровых, нормогликемических собаках породы бигль.
При увеличении концентрации инсулина гларгина среднее время действия возрастало от 6,8 часов (U100) до 7,69 часов (U300), соответственно.
При увеличении концентрации гларгина от 100 до 300 Ед/мл профиль “время снижения глюкозы крови - действие” у собаки изменялся в сторону более пологого и пролонгированного действия.
Настоящие данные, полученные в исследованиях на собаках, соответствуют данным, полученным в исследованиях с участием людей, показывающим, что более высокие концентрации инсулина гларгина положительно коррелируют с профилем и большей продолжительностью действия.
ФИГУРЫ
Фигуры, приведенные ниже, по существу показывают удивительные и неожиданные различия в действии (PK) и активности (PD) средств Лантус U100 и Лантус U300 (средства инсулин гларгин U100 и инсулин гларгин U300) после одинаковой п/к дозы введенной здоровым субъектам, в то время как глюкоза крови (PD) оставалась постоянной.
Краткое описание чертежей
Фиг. 1: Скорость инфузии глюкозы (GIR) Лантус U100
Фиг. 2: Скорость инфузии глюкозы (GIR) Лантус U300
Фиг. 3: Сывороточные концентрации инсулина, Лантус U100 (слева) и U300 (справа)
Фиг. 4: Глюкоза крови (1/2)
Фиг. 5: Глюкоза крови (2/2)
Фиг. 6: Результаты рандомизированного, с 4-мя последовательностями, перекрестного, двойного слепого, “доза–ответ” исследования 0,4, 0,6 и 0,9 Ед/кг HOE-901-U300 (инсулин гларгин U300) в сравнении с 0,4 Ед/кг Лантус® U100 (инсулин гларгин U100) у пациентов с диабетом 1 типа с применением эугликемического клэмп-метода. Верхняя панель: инсулин гларгин концентрация (мЕд/л), средняя панель: глюкоза крови (BG, мг/дл), нижняя панель: скорость инфузии глюкозы (GIR, мг⋅кг-1⋅мин-1). На кривых представлены средние значения, сглаженные с помощью метода LOWESS, всех экспериментальных точек всех субъектов (средние показатели популяции); LOWESS является методом анализа данных для получения “сглаженного” набора значений из временного ряда, который содержит “помехи”, или из диаграммы рассеяния, отражающей взаимосвязь 2 переменных с “помехами”.
Фиг. 7: Скорость инфузии глюкозы (GIR, мг.кг-1.мин-1). На кривых представлены средние значения, сглаженные с помощью метода LOWESS, всех экспериментальных точек всех субъектов (средние показатели популяции); LOWESS является методом анализа данных для получения “сглаженного” набора значений из временного ряда, который содержит “помехи”, или из диаграммы рассеяния, отражающей взаимосвязь 2 переменных с “помехами”.
Обозначение: профили с 1 по 3 (сверху вниз)
Результаты рандомизированного двойного слепого с параллельными группами “доза-ответ” исследования 0,4, 0,6 и 1,2 Ед/кг Лантуса® U100 (инсулин гларгин U100) у пациентов с диабетом 1 типа с применением эугликемического клэмп-метода.
Обозначение: профили с 4 по 7 (сверху вниз)
Результаты рандомизированного, с 4-мя последовательностями, перекрестного, двойного слепого, “доза-ответ” исследования 0,4, 0,6 и 0,9 Ед/кг HOE-901-U300 (инсулин гларгин U300) в сравнении с 0,4 Ед/кг Лантуса® U100 (инсулин гларгин U100) у пациентов с диабетом 1 типа с применением эугликемического клэмп-метода.
Фиг. 8: Изображения, полученные с помощью световой микроскопии, осадков средств инсулина гларгина
С увеличением концентрации:
A: 100 Ед/мл,
B: 300 Ед/мл,
C: 500 Ед/мл,
D: 700 Ед/мл и
E: 1000 Ед/мл,
С увеличением 100x и включая максимальные диаметры.
Все осадки получали при использовании 60U инсулина гларгина
Фиг. 9: Профиль время – действие инсулина гларгина U-100 в сравнении с U-300 у нормогликемических собак
Список сокращений
|
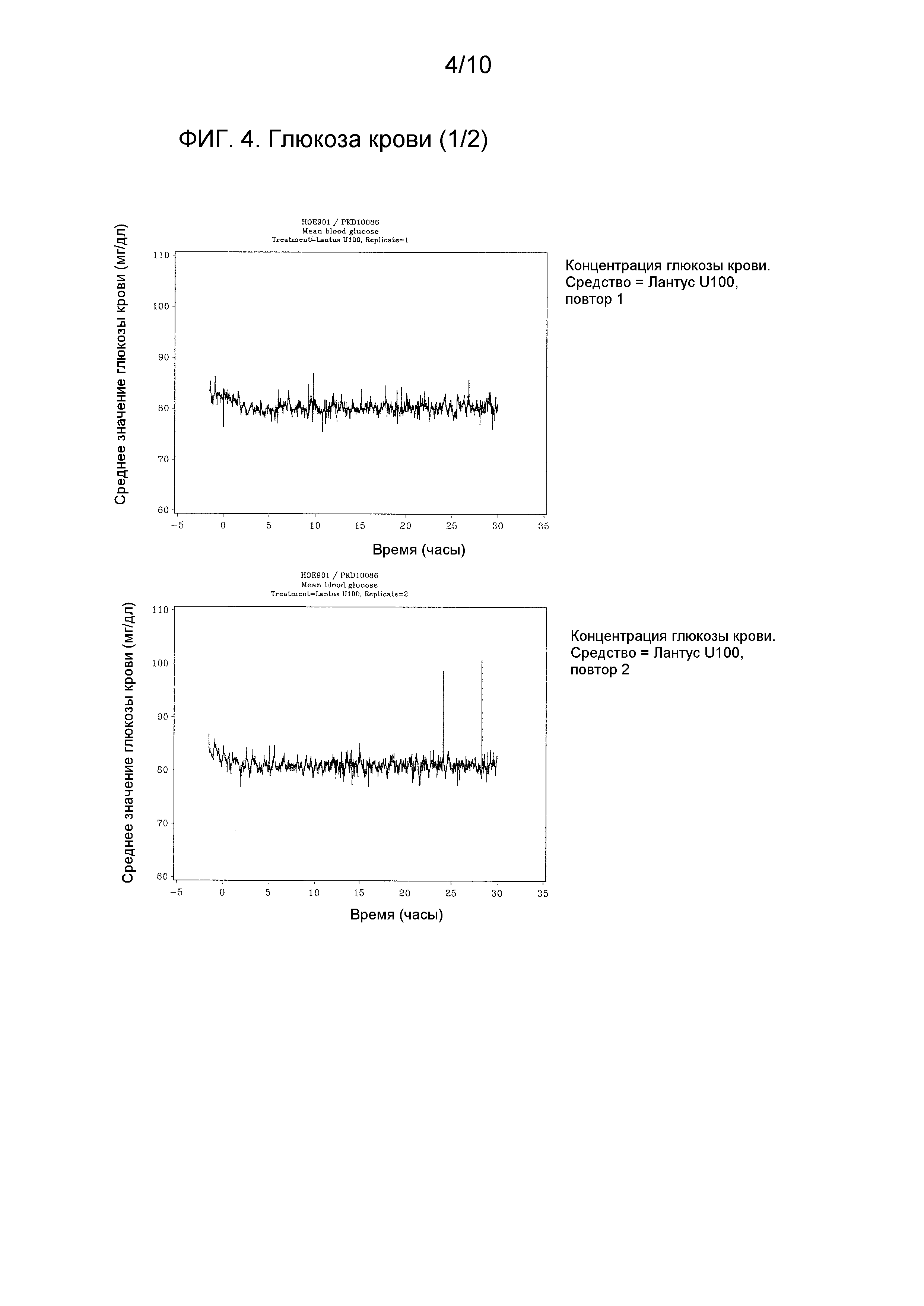
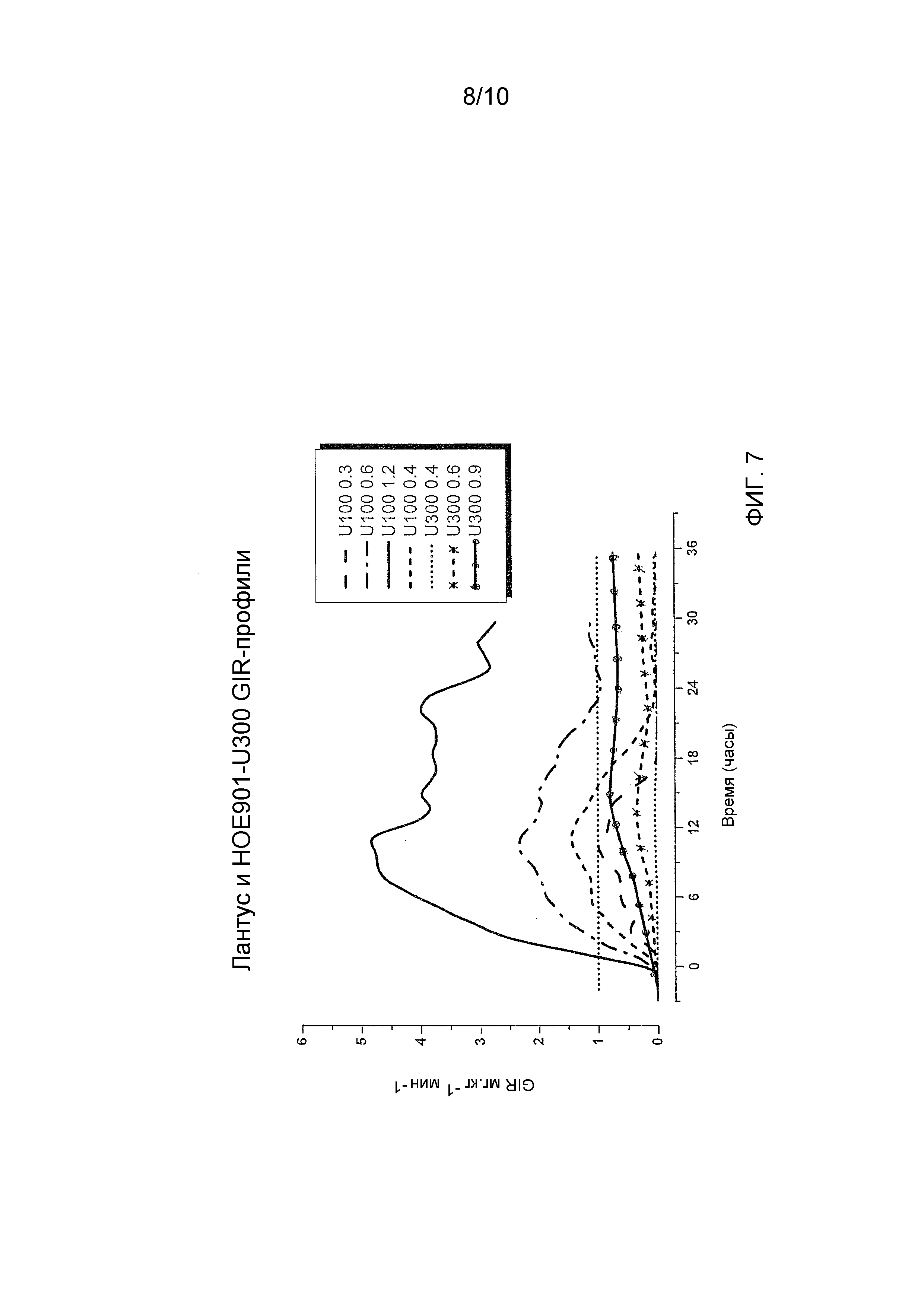
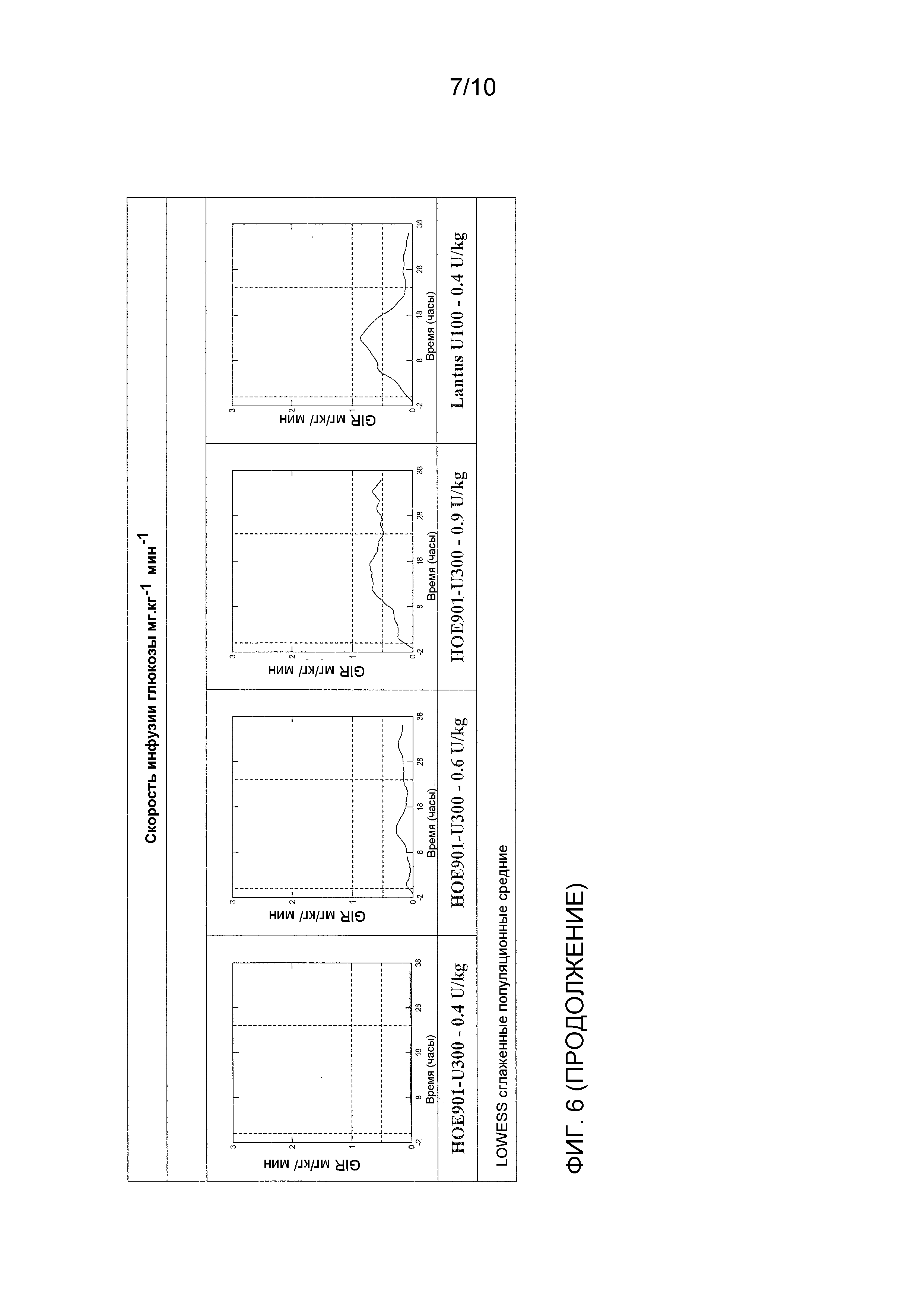
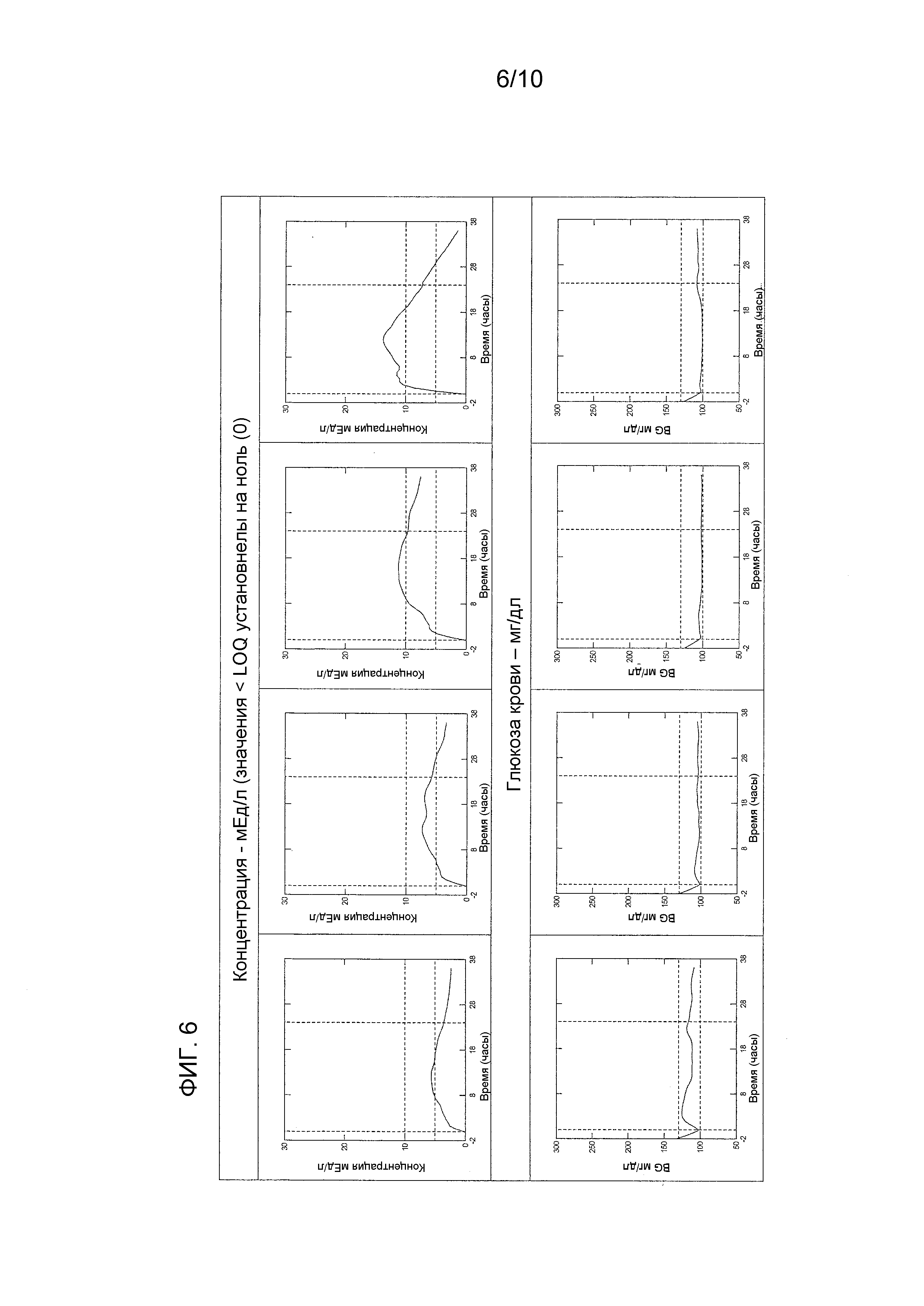
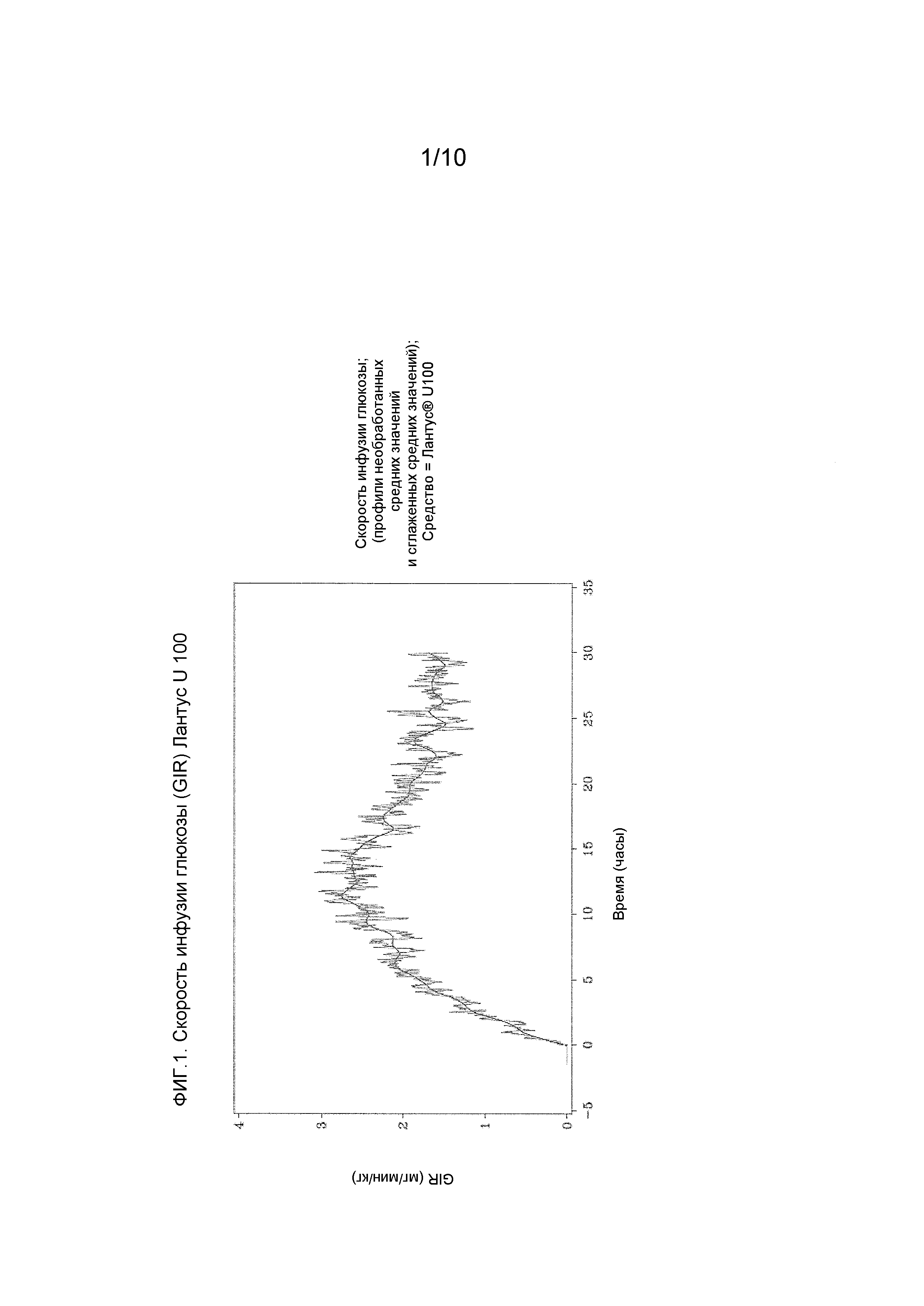
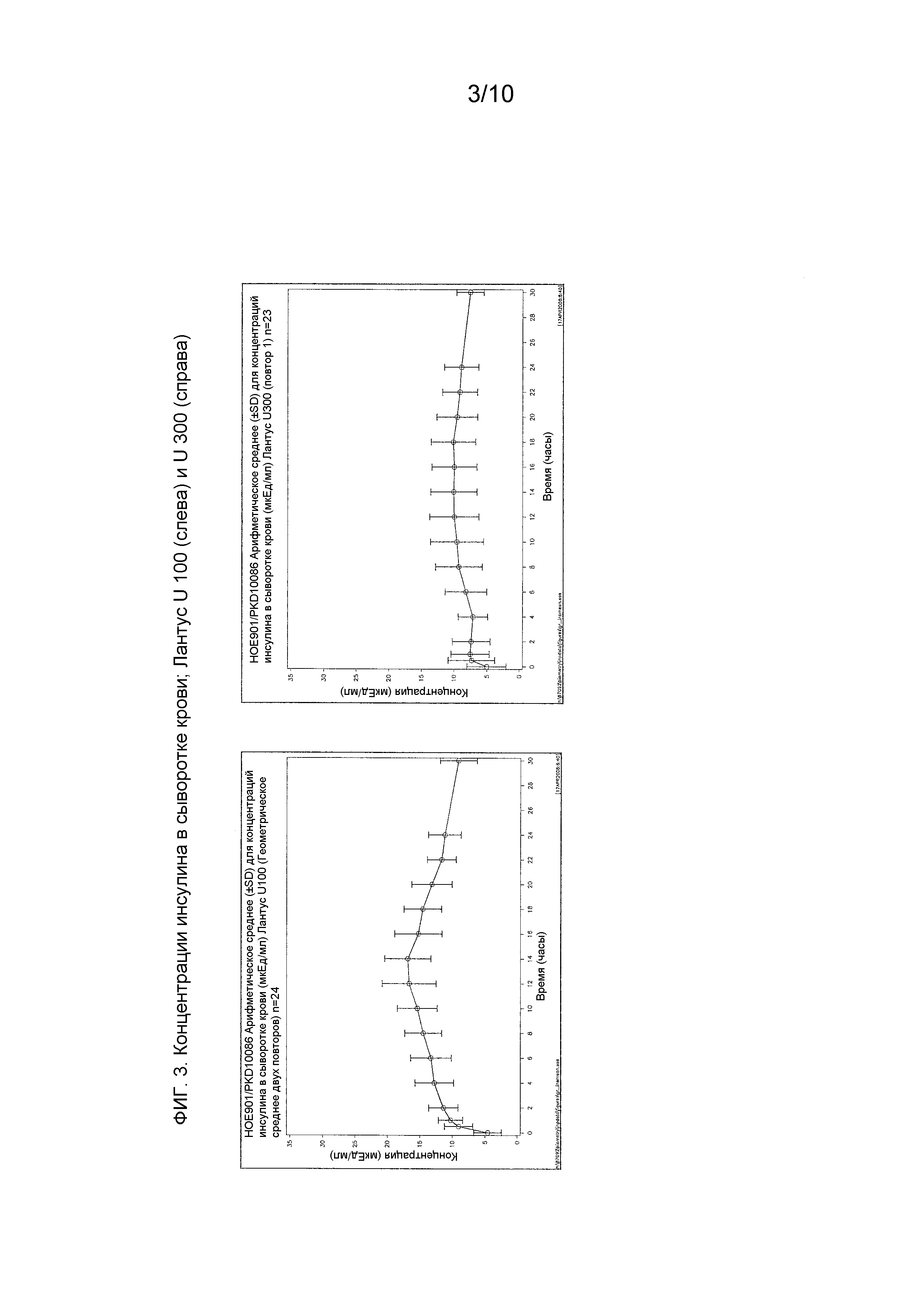

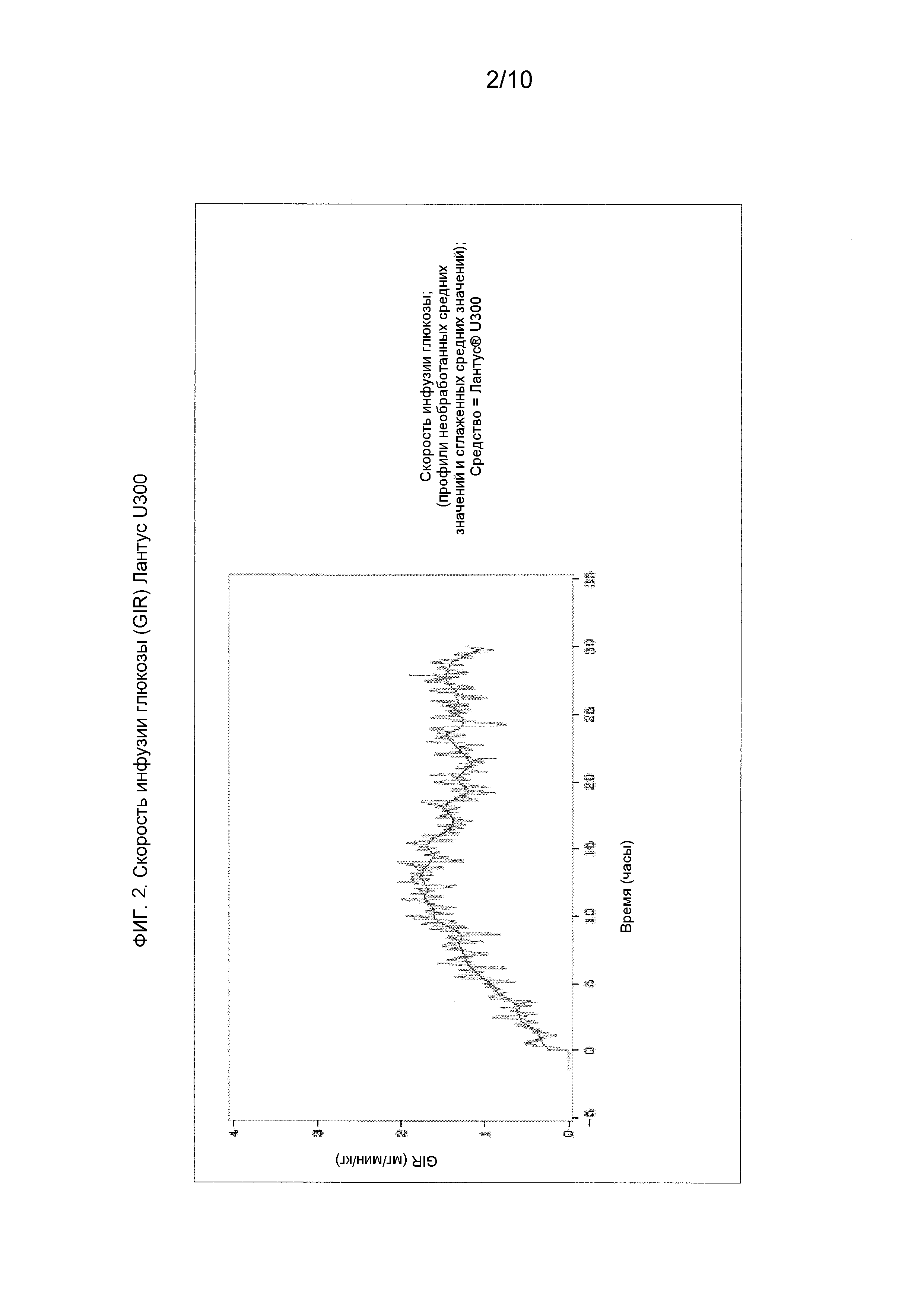
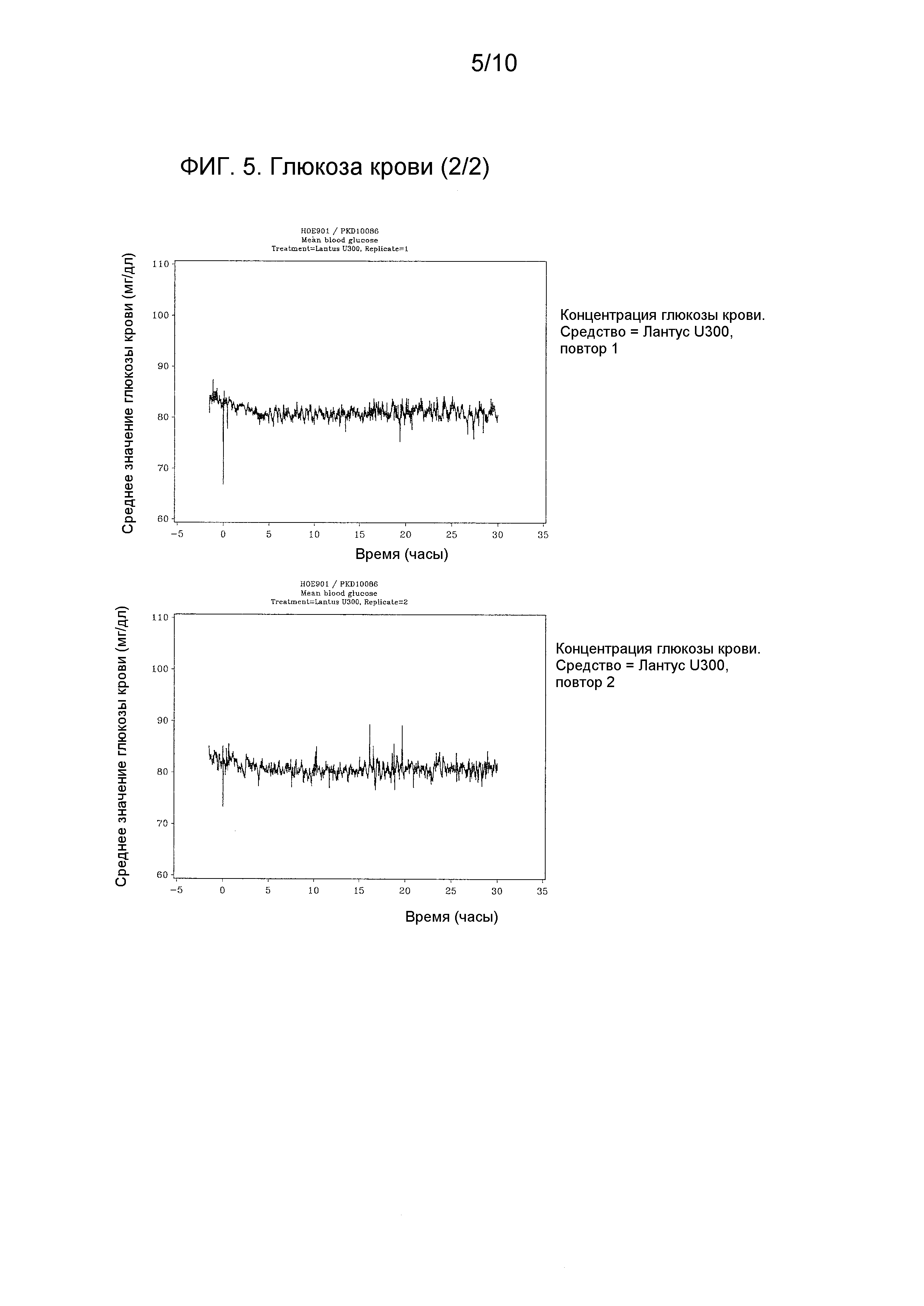
Производные 1-аминоалкилциклогексана для лечения воспалительных заболеваний кожи
Новая комбинация активных ингредиентов, содержащая нестероидное противовоспалительное лекарственное средство и производное колхикозида
Способ получения активированных сложных эфиров
Замещенные тетрагидропиранспиропирролидинон и тетрагидропиранспиропиперидинон, фармацевтическая композиция на их основе и применение в лечебных целях
Лечение дерматологических аллергических состояний
Способ получения [4-(2- хлор-4- метокси-5- метилфенил)-5- метилтиазоло-2- ил] [2-циклопропил-1- (3- фтор-4- метилфенил) - этил ]- амина
Новые производные инсулина с чрезвычайно замедленным профилем время/действие
Производные 1н-пиразоло[4,3-c]изохинолинов, способ их получения и применение в терапии
Комбинация инсулина и агониста glp-1
Фармацевтическая композиция, включающая агонист glp-1, инсулин и метионин
Новая комбинация активных ингредиентов, содержащая нестероидное противовоспалительное лекарственное средство и производное колхикозида
Способ получения активированных сложных эфиров
Замещенные тетрагидропиранспиропирролидинон и тетрагидропиранспиропиперидинон, фармацевтическая композиция на их основе и применение в лечебных целях
Способы и применения, включающие гемсвязывающий белок 1
Лечение дерматологических аллергических состояний
Способ получения [4-(2- хлор-4- метокси-5- метилфенил)-5- метилтиазоло-2- ил] [2-циклопропил-1- (3- фтор-4- метилфенил) - этил ]- амина
Новые производные инсулина с чрезвычайно замедленным профилем время/действие
Производные 1н-пиразоло[4,3-c]изохинолинов, способ их получения и применение в терапии
Комбинация инсулина и агониста glp-1
Фармацевтическая композиция, включающая агонист glp-1, инсулин и метионин

