Результат интеллектуальной деятельности: Замещенные 4-нитропиразолин-5-оны, способ их получения и их применение в качестве фунгицидных средств
Вид РИД
Изобретение
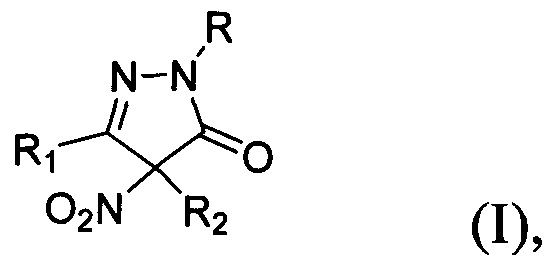
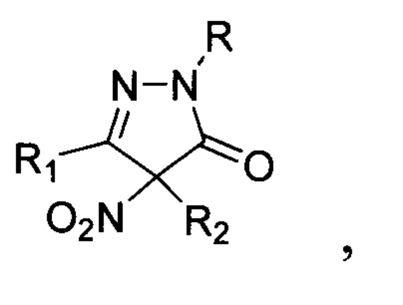
Настоящее изобретение относится к области органической химии, а именно, к замещенным 4-нитропиразолин-5-онам общей формулы:
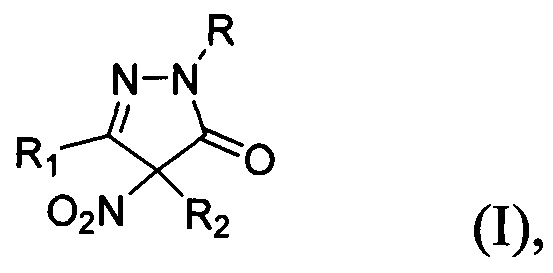
где R=Н, фенил; R1 = низший алкил, фенил; R2 = алкил С1-С6, бензил, аллил, CH2CH2CN либо R1+R2=(СН2)4, к способу их получения и применению этих соединений в качестве фунгицидных средств. Соединения общей формулы I могут найти применение в сельском хозяйстве в качестве фунгицидных средств в составе композиций для борьбы с грибковыми заболеваниями растений.
Производные пиразолонов широко известны как противовоспалительные препараты, анальгетики и антипиретики, давно применяемые в медицине. Следует отметить, что фунгицидная активность была ранее описана лишь для немногих пиразолонов, как правило, отличающихся сложной структурой и содержащих дополнительные карбоциклические и гетероциклические циклы. Так в литературе описаны фенилгидразоны пиразолин-5-онов с тиазольным фрагментом общей формулы:
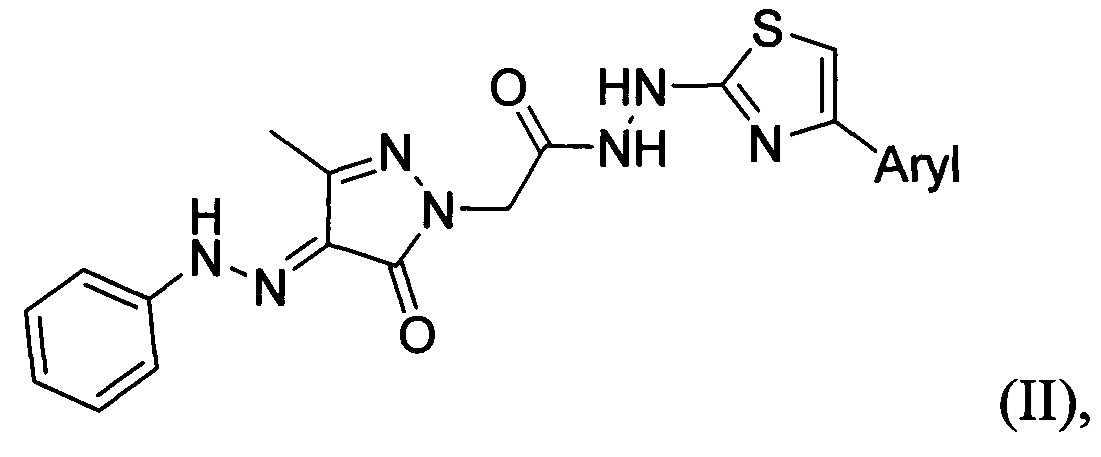
которые обладают фунгицидной активностью по отношению к грибкам Aspergillus niger и Candida albicans [М. R. D. Reddy, A. R. G. Prasad, Y. N. Spoorthy, L. R. K. R. Ravindranath, Synthesis, Characterization and Antimicrobial Activity of Certain Novel Aryl Hydrazone Pyrazoline-5-Ones Containing Thiazole Moiety, Advanced Pharmaceutical Bulletin,, 2013, 3, 153. doi: 10.5681/apb.2013.026].
Известны N1-никотиноил-3-метил-4-(арилазо)-1,2-пиразолин-5-оны общей формулы:
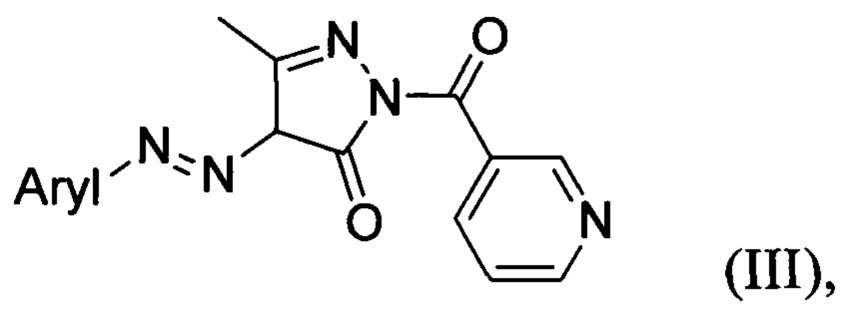
обладающие фунгицидной активностью по отношению к Candida albicans, Candida neoformans, Sporothrix schenckii, Trichophyton mentagrophytes и Aspergillus fumigatus [S.S. Nayal, C.P. Singh, Synthesis and antifungal activity of N1-nicotinoyl-3-methyl-4-(substituted azo)-1,2-pyrazoline-5-one, Asian J. Chem. 1999, 11, 207-212].
Известны также 1-[(2-бензоиламино)бензоил]-3-метилпиразолин-5-оны общей формулы:
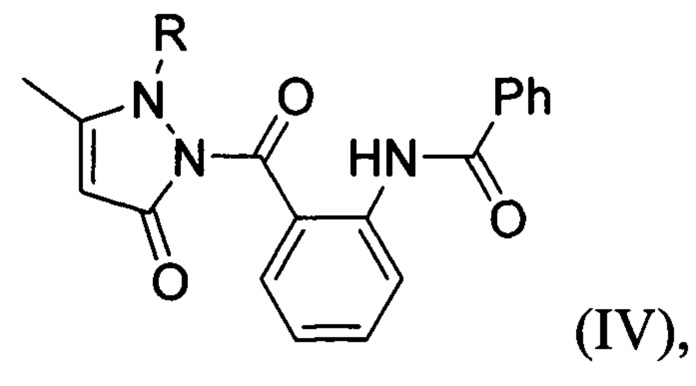 где R=Н, Ph,
где R=Н, Ph,
обладающие фунгицидной активностью по отношению к Aspergillus niger и Aspergillus flavus, которая сопоставима с активностью салициловой кислоты [М. Kidwai, R. Mohan, Ecofriendly Synthesis of Antifungal Azoles, J. Korean Chem. Soc. 2004, 48, 177-181. DOI: 10.5012/jkcs.2004.48.2.177]. В патенте [US 5028717A, Herbicidal and fungicidal agents based on substituted pyrazolin-5-one derivatives] описаны пиразолоны общей формулы:
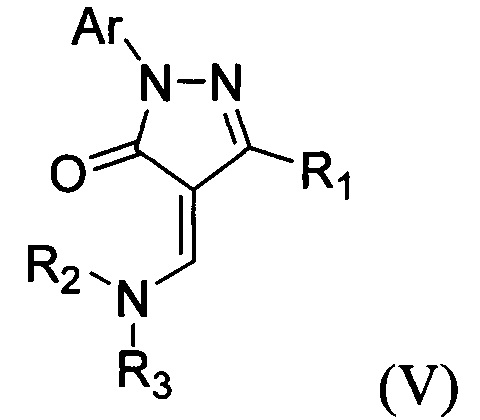
в качестве гербицидов и фунгицидов.
В патентах [US 4666933A, (O-substituted oximino)-pyrazolin-5-one pesticides; ЕР 0166171 А1, Substituted pyrazolin-5-one] описаны пиразолоны общей формулы:
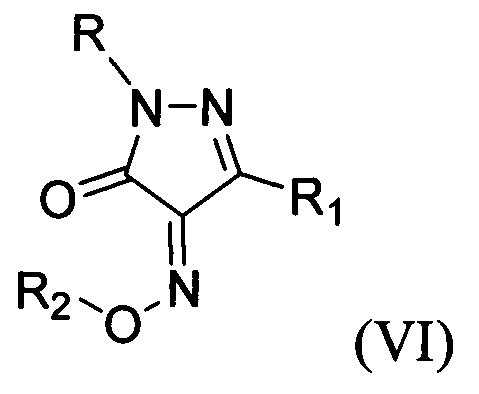
в качестве фунгицидов.
В заявке [WO 9626191 A1, Fungicidal cyclic amides], посвященной циклическим амидам с фунгицидной активностью, в формуле изобретения фигурируют структуры с нитрогруппой формулы:
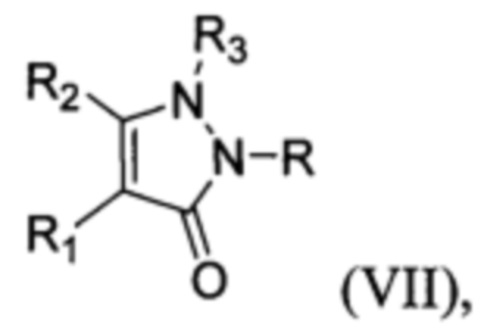 где R = алкил и др., R1 = нитро и др., R2 и R3 = арил и др. [WO 9626191 А1, Fungicidal cyclic amides].
где R = алкил и др., R1 = нитро и др., R2 и R3 = арил и др. [WO 9626191 А1, Fungicidal cyclic amides].
Однако в патенте не приводятся данные о фунгицидной активности соединений общей формулы VII, имеющих при R1 = нитрогруппу.
В ряде публикаций сообщается о фунгицидной активности других полизамещенных пиразолин-5-онов сложного строения [(1) WO 9636229 A1, WO 9636615 A1, Fungicidal cyclic amides; (2) WO 9931070 A1, Substituted phenylpyrazolones, method and intermediate products for the production thereof and their utilization for combating parasitic fungi and animal parasites; (3) EP 0967212 A1, Pyrazoline compounds and use as plant disease control agent].
Отличием предлагаемых в настоящем изобретении структур от известных пиразолин-5-онов, проявляющих фунгицидную активность, является наличие двух заместителей при С-4 пиразолин-5-она, одним из которых является нитрогруппа, а вторым - углеводородный заместитель согласно общей формуле I.
Недостатками этих известных соединений является трудоемкость синтеза и необходимость использования дорогостоящих реагентов.
Известный способ получения нитропиразолонов состоит в обработке соответствующих исходных пиразолин-5-онов азотной кислотой, смесью азотной и серной кислоты или смесью азотной кислоты и трифторуксусного ангидрида [WO 9626191 A1, Fungicidal cyclic amides]. Недостатком этого способа является жесткие сильнокислые условия синтеза, а также низкая селективность. Например, при наличии в молекуле фенильных заместителей они также подвергаются нитрованию [(1) K.-А. Kovar, W. Rohlfes, Н. Auterhoff, Nitrierungsprodukte des Propyphenazons und deren Farbreaktionen mit Aceton und Kalilauge, Arch. Pharm. 1981, 314, 532-541. DOI: 10.1002/ardp.19813140611. (2) A. G. Burton, M. Dereli, A. R. Katritzky, H. O. Tarhan, The kinetics and mechanism of the electrophilic substitution of heteroaromatic compounds. Part XXXV. The Nitration of Phenylpyrazolones, J. Chem. Soc, Perkin Trans. 2, 1974, 382-388.], поэтому известный способ непригоден для получения предлагаемых соединений.
Технической задачей настоящего изобретения является расширение ассортимента фунгицидных средств на основе замещенных 4-нитропиразолин-5-онов, а также разработка способа их получения. Поставленная техническая задача достигается замещенными 4-нитропиразолин-5-онами общей формулы:
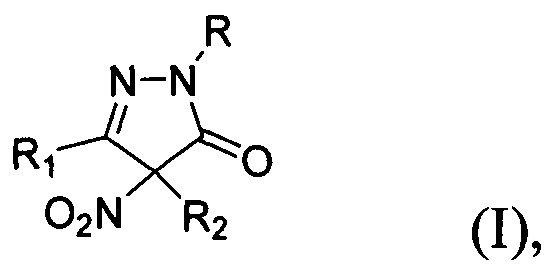
где R=Н, фенил; R1 = низший алкил, фенил; R2 = алкил C1-С6, бензил, аллил, CH2CH2CN либо R1+R2=(СН2)4, обладающими фунгицидной активностью. Предлагаемые в настоящем изобретении 4-нитропиразолин-5-оны общей формулы I принципиально отличаются от всех ранее описанных фунгицидов, содержащих пиразолоновый цикл, наличием двух заместителей в положении 4 пиразолона, одним из которых обязательно является нитрогруппа. Предлагаемые фунгициды являются новыми неописанными в литературе. Соединение общей формулы I, где R = фенил, a R1 и R2=Me, (3,4-диметил-4-нитро-1-фенилпиразолин-5-она (Ia) участвовало в скрининге биологической активности (CAS 957359-02-3) (данные из базы данных PubChem, SID 22412834, https://pubchem.ncbi.nlm.nih.gov/bioassay/804#sid=22412834§ion=Top), однако фунгицидной активности обнаружено не было. Кроме того, не приводится способ его получения.
Предлагаемые соединения представляют интерес в качестве фунгицидных средств для борьбы с возбудителями болезней растений.
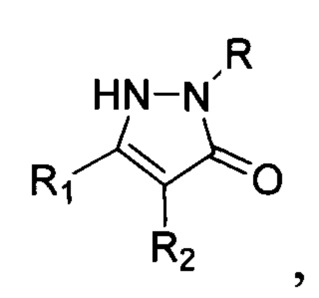
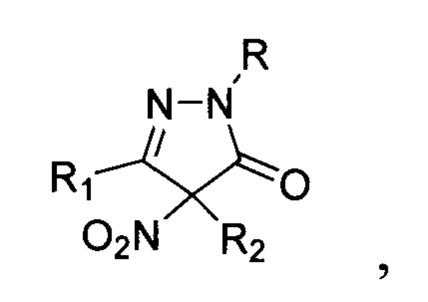
Предложен также неописанный в литературе способ получения замещенных 4-нитропиразолин-5-онов общей формулы:
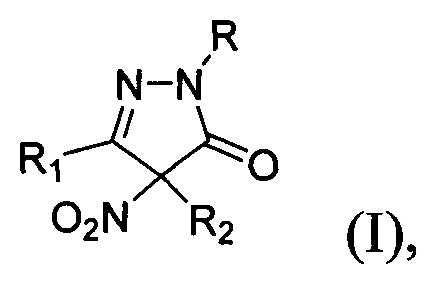
где R=H, фенил; R1 = низший алкил, фенил; R2 = алкил С1-С6, бензил, аллил, CH2CH2CN либо R1+R2=(СН2)4, заключающийся в том, что соответствующие замещенные пиразолин-5-оны общей формулы:
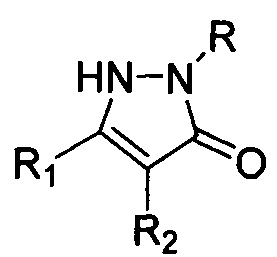 , где R, R1 и R2 имеют вышеуказанные значения, подвергают взаимодействию с нитритом натрия (NaNO2) и нитратом железа (Fe(NO3)3) в среде органического растворителя, и процесс проводят при мольном соотношении пиразолин-5-он : NaNO2 : Fe(NO3)3=1:1-2:2-4. Процесс протекает по следующей схеме:
, где R, R1 и R2 имеют вышеуказанные значения, подвергают взаимодействию с нитритом натрия (NaNO2) и нитратом железа (Fe(NO3)3) в среде органического растворителя, и процесс проводят при мольном соотношении пиразолин-5-он : NaNO2 : Fe(NO3)3=1:1-2:2-4. Процесс протекает по следующей схеме:
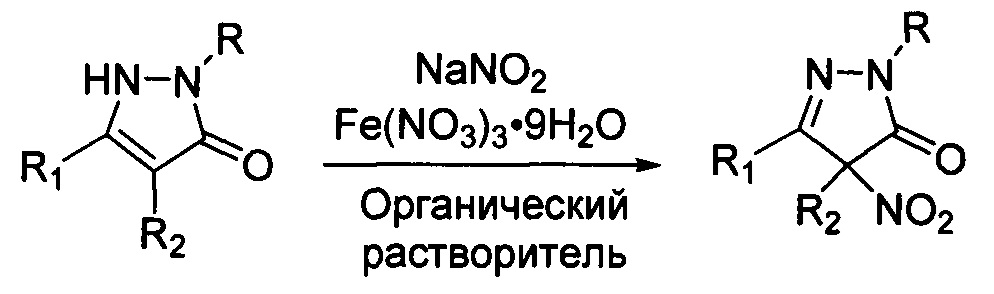
В приведенной схеме R, R1, и R2 имеют вышеуказанные значения общей формулы I.
Процесс проводят при комнатной температуре, а в качестве органического растворителя используют, например, ацетонитрил (MeCN).
Процесс можно проводить путем прибавления к исходным соответствующим замещенным пиразолин-5-онам смеси NaNO2 и Fe(NO3)3 в органическом растворителе либо путем прибавления Fe(NO3)3 к смеси соответствующего исходного замещенного пиразолин-5-она, NaNO2 и органического растворителя.
Изобретение иллюстрируется примерами получения предлагаемых соединений общей формулы I, и результатами их испытаний в качестве фунгицидов против возбудителей болезней растений.
Пример 1. Получение 3,4-диметил-4-нитро-1-фенилпиразолин-5-она (Ia). К смеси NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд. Через 5 минут в перемешиваемую реакционную смесь вносили 3,4-диметил-1-фенилпиразолин-5-он (1 ммоль, 188 мг) в течение 5-15 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли CH2Cl2 (10 мл) и 3% водным раствором HCl (30 мл), взбалтывали и отделяли органический слой. Водный слой экстрагировали CH2Cl2 (2×10 мл), после чего все органические экстракты объединяли, промывали водой (2×20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием CH2Cl2 в качестве элюента. Получали 3,4-диметил-4-нитро-1-фенилпиразолин-5-он в виде вязкой массы, кристаллизующейся при стоянии с выходом 58% (0.58 ммоль, 135 мг). Тпл=40-41°С; 1Н ЯМР (300.13 МГц, CDCl3): δ=7.94-7.84 (m, 2Н), 7.51-7.39 (m, 2Н), 7.32-7.22 (m, 1Н), 2.22 (s, 3Н), 1.95 (s, 3Н); 13C ЯМР (75.47 МГц, CDCl3): δ=164.6, 154.7, 137.0, 129.2, 126.2, 119.0, 91.8, 17.8, 13.3; ИК (тонкий слой): νmax=1731, 1597, 1558, 1501, 1383, 1368, 1294, 1147, 759, 691 см-1; масс-спектр высокого разрешения (Электроспрей): m/z=256.0697, расч. Для C11H11N3O3+Na+: 256.0693.
Пример 2. Получение 3а-нитро-2-фенил-2,3а,4,5,6,7-гексагидро-3Н-индазол-3-она (Ib).
К смеси NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд. Через 5 минут в перемешиваемую реакционную смесь вносили 2-фенил-1,2,4,5,6,7-гексагидро-3Н-индазол-3-он (1 ммоль, 214 мг) в течение 5-15 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли CH2Cl2 (10 мл) и 3% водным раствором HCl (30 мл), взбалтывали и отделяли органический слой. Водный слой экстрагировали CH2Cl2 (2×10 мл), после чего все органические экстракты объединяли, промывали водой (2×20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием CH2Cl2 в качестве элюента. 3а-Нитро-2-фенил-2,3а,4,5,6,7-гексагидро-3Н-индазол-3-он был получен в виде коричневатого порошка с выходом 66% (171 мг, 0.66 ммоль). Тпл=86-87°С; 1Н ЯМР (300.13 МГц, CDCl3): δ=7.89 (d, J=8.1 Hz, 2Н), 7.50-7.38 (m, 2Н), 7.31-7.21 (m, 1Н), 3.26-3.12 (m, 1H), 2.97-2.83 (m, 1H), 2.59-2.42 (m, 1H), 2.30-2.10 (m, 1H), 2.04-1.74 (m, 2H), 1.70-1.48 (m, 2H); 13C ЯМР (75.47 МГц, CDCl3): δ=164.0, 157.5, 137.1, 129.1, 126.1, 119.0, 91.3, 33.9, 27.54, 27.47, 21.0; ИК (KBr): νmax=1726, 1557, 1500, 1385 см-1; Элементный анализ, рассчитано (%) для C13H13N3O3: С 60.23, Н 5.05, N 16.21; найдено: С 60.27, Н 5.18, N 16.08.
Пример 3. Получение 4-бензил-3-метил-4-нитро-1Н-пиразолин-5-она (Ic)
К смеси 4-бензил-3-метил-пиразолин-5-она (1 ммоль, 188 мг), NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли CH2Cl2 (10 мл) и 3% водным раствором HCl (30 мл), взбалтывали и отделяли органический слой. Водный слой экстрагировали CH2Cl2 (2×10 мл), после чего все органические экстракты объединяли, промывали водой (2×20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 4-бензил-3-метил-4-нитро-1Н-пиразолин-5-он в виде желтоватого порошка с выходом 83% (0.83 ммоль, 194 мг). Тпл=84-85°C с разл.; 1Н ЯМР (300.13 МГц, CDCl3): δ=8.53 (bs, 1H), 7.35-7.26 (m, 3Н), 7.23-7.14 (m, 2Н), 3.79 (d, J=13.5 Hz, 1Н), 3.62 (d, J=13.5 Hz, 1H), 2.16 (s, 3Н); 13C ЯМР (75.47 МГц, CDCl3): δ=167.8, 153.8, 129.9, 129.8, 129.2, 128.6, 93.2, 37.7, 14.4; ИК (KBr): νmax=3238, 1733, 1564, 1352, 1312, 1271, 773, 757, 711, 699, 604 см-1; масс-спектр высокого разрешения (Электроспрей): m/z=256.0698, расч. Для C11H11N3O3+Na+: 256.0693. Элементный анализ, рассчитано (%) для C11H11N3O3: С 56.65, Н 4.75, N 18.02; найдено: С 56.47, Н 4.58, N 17.77.
Пример 4. Получение 4-метил-4-нитро-3-фенил-1Н-пиразолин-5-она (Id).
К смеси NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд. Через 5 минут в перемешиваемую реакционную смесь вносили 4-метил-3-фенилпиразолин-5-он (1 ммоль, 174 мг) в течение 5-15 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли EtOAc (10 мл) и раствором, приготовленным из воды (14 мл), концентрированной хлороводородной кислоты (2 мл) и насыщенного раствора NaCl (20 мл). После взбалтывания полученной смеси органический слой отделяли, а водный слой экстрагировали EtOAc (2×10 мл). Все органические экстракты объединяли, промывали насыщенным водным раствором NaCl (20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. 4-Метил-4-нитро-3-фенил-1Н-пиразолин-5-он (Id) был получен в виде белого порошка с выходом 77% (169 мг, 0.77 ммоль). Тпл=138-140°С; 1Н ЯМР (300.13 МГц, CDCl3): δ=9.14 (bs, 1Н), 7.70-7.55 (m, 2H), 7.54-7.37 (m, 3Н), 2.07 (s, 3Н); 13C ЯМР (75.47 МГц, CDCl3): δ=169.3, 154.7, 131.6, 129.5, 128.2, 126.0, 88.9, 19.3; ИК (KBr): νmax=3242, 1727, 1550, 1385, 769, 758, 693, 651 см-1; Элементный анализ, рассчитано (%) для C10H9N3O3: С 54.79, Н 4.14, N 19.17; найдено: С 54.78, Н 4.12, N 19.16.
Пример 5. Получение 4-аллил-3-метил-4-нитро-1Н-пиразолин-5-она (Ie)
К смеси 4-аллил-5-метил-1,2-дигидро-3Н-пиразол-3-она (1 ммоль, 138 мг), NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли EtOAc (10 мл) и раствором, приготовленным из воды (14 мл), концентрированной хлороводородной кислоты (2 мл) и насыщенного раствора NaCl (20 мл). После взбалтывания полученной смеси органический слой отделяли, а водный слой экстрагировали EtOAc (2×10 мл). Все органические экстракты объединяли, промывали насыщенным водным раствором NaCl (20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 4-аллил-3-метил-4-нитро-1Н-пиразолин-5-он в виде слегка коричневатой вязкой массы с выходом 84% (0.84 ммоль, 154 мг). 1Н ЯМР (300.13 МГц, CDCl3): δ=8.92 (bs, 1Н), 5.59-5.20 (m, 3Н), 3.18 (dd, J=13.7, 6.8 Hz, 1H), 3.04 (dd, J=13.7, 7.0 Hz, 1H), 2.10 (s, 3Н); 13C ЯМР (75.47 МГц, CDCl3): δ=167.7, 154.0, 126.2, 123.4, 91.8, 36.1, 14.0; ИК (KBr): νmax=3528, 1730, 1557, 1422, 1404, 1384, 1350, 1305, 1264, 942, 818, 761, 746, 694, 611, 548 cm-1; Элементный анализ, рассчитано (%) для C7H9N3O3: С 45.90, Н 4.95, N 22.94; найдено: С 45.98, Н 4.90, N 22.98.
Пример 6. Получение 4-этил-3-метил-4-нитро-1Н-пиразолин-5-она (If)
К смеси 4-этил-5-метил-1,2-дигидро-3Н-пиразол-3-она (1 ммоль, 126 мг), NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли EtOAc (10 мл) и раствором, приготовленным из воды (14 мл), концентрированной хлороводородной кислоты (2 мл) и насыщенного раствора NaCl (20 мл). После взбалтывания полученной смеси органический слой отделяли, а водный слой экстрагировали EtOAc (2×10 мл). Все органические экстракты объединяли, промывали насыщенным водным раствором NaCl (20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 4-этил-3-метил-4-нитро-1Н-пиразолин-5-он в виде желтоватого порошка с выходом 74% (0.74 ммоль, 93 мг). Тпл=64-66°С; 1Н ЯМР (300.13 МГц, CDCl3): δ=9.01 (bs, 1Н), 2.51 (dq, J=14.9, 7.5 Hz, 1H), 2.28 (dq, J=14.9, 7.5 Hz, 1H), 2.09 (s, 3H), 0.90 (t, J=7.5 Hz, 3Н); 13C ЯМР (75.47 МГц, CDCl3): δ=168.0, 154.2, 93.5, 25.1, 13.7, 6.8; ИК (KBr): νmax=3323, 3259, 1742, 1717, 1561, 1546, 1358, 821, 663, 582 cm-1; масс-спектр высокого разрешения (электроспрей): m/z=194.0530, calcd. for C6H9N3O3+Na+: 194.0536.
Пример 7. Получение 4-бутил-3-метил-4-нитро-1Н-пиразолин-5-она (Ig)
К смеси 4-бутил-5-метил-1,2-дигидро-3Н-пиразол-3-она (1 ммоль, 154 мг), NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли CH2Cl2 (10 мл) и 3% водным раствором HCl (30 мл), взбалтывали и отделяли органический слой. Водный слой экстрагировали CH2Cl2 (2×10 мл), после чего все органические экстракты объединяли, промывали водой (2×20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 4-бутил-3-метил-4-нитро-1Н-пиразолин-5-он в виде желтоватой вязкой массы с выходом 80% (0.80 ммоль, 123 мг). 1Н ЯМР (300.13 МГц, CDCl3): δ=8.70 (bs, 1Н), 2.55-2.38 (m, 1Н), 2.31-2.15 (m, 1Н), 2.08 (s, 3Н), 1.47-1.30 (m, 2Н), 1.30-1.01 (m, 2Н), 0.92 (t, J=7.3 Hz, 3Н); 13С ЯМР (75.47 МГц, CDCl3): δ=168.0, 154.4, 92.9, 31.3, 24.4, 22.5, 13.75, 13.71; ИК (KBr): νmax=3279, 2963, 2935, 2875, 1739, 1556, 1432, 1383, 1352, 1251, 819, 760 cm-1; масс-спектр высокого разрешения (электроспрей): m/z=222.0842, calcd. for C8H13N3O3+Na+: 222.0849.
Пример 8. Получение 4-гексил-3-метил-4-нитро-1Н-пиразолин-5-она (Ih)
К смеси 4-гексил-5-метил-1,2-дигидро-3Н-пиразол-3-она (1 ммоль, 182 мг), NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли CH2Cl2 (10 мл) и 3% водным раствором HCl (30 мл), взбалтывали и отделяли органический слой. Водный слой экстрагировали CH2Cl2 (2×10 мл), после чего все органические экстракты объединяли, промывали водой (2×20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 4-гексил-3-метил-4-нитро-1Н-пиразолин-5-он в виде коричневатой вязкой массы с выходом 70% (0.70 ммоль, 159 мг). 1Н ЯМР (300.13 МГц, CDCl3): δ=8.98 (bs, 1Н), 2.45 (td, J=12.8, 4.8 Hz, 1Н), 2.21 (td, J=12.8, 4.8 Hz, 1H), 2.08 (s, 3H), 1.46-1.02 (m, 8H), 0.87 (t, J=6.4 Hz, 3H); 13C ЯМР (75.47 МГц, CDCl3): δ=168.2, 154.5, 93.0, 31.5, 31.3, 29.0, 22.5, 22.2, 14.1, 13.7; ИК (тонкий слой): νmax=3278, 2958, 2931, 2861, 1737, 1557, 1381, 1349, 819, 760 cm-1; Элементный анализ, рассчитано (%) для C10H17N3O3: С 52.85, Н 7.54, N 18.49; найдено: С 53.13, Н 7.78, N 18.06.
Пример 9. Получение 4-метил-4-нитро-3-пропил-1Н-пиразолин-5-она (Ii)
К смеси 4-метил-5-пропил-1,2-дигидро-3Н-пиразол-3-она (1 ммоль, 140 мг), NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли CH2Cl2 (10 мл) и 3% водным раствором HCl (30 мл), взбалтывали и отделяли органический слой. Водный слой экстрагировали CH2Cl2 (2×10 мл), после чего все органические экстракты объединяли, промывали водой (2×20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 4-метил-4-нитро-3-пропил-1Н-пиразолин-5-он в виде желтоватого порошка с выходом 84% (0.84 ммоль, 118 мг). Тпл=67-69°С разл.; 1Н ЯМР (300.13 МГц, CDCl3): δ=9.05 (bs, 1Н), 2.38-2.25 (m, 2Н), 1.84 (s, 3Н), 1.76-1.57 (m, 2Н), 0.99 (t, J=7.3 Hz, 3Н); 13С ЯМР (75.47 МГц, CDCl3): δ=169.1, 158.5, 89.6, 29.5, 18.4, 17.5, 13.7; ИК (KBr): νmax=3300, 1736, 1711, 1568, 1560, 1384, 667 cm-1; масс-спектр высокого разрешения (электроспрей): m/z=208.0696, calcd. for C7H11N3O3+Na+: 208.0693.
Пример 10. Получение 3-(3-Метил-4-нитро-5-оксо-4,5-дигидро-1Н-пиразол-4-ил)пропионтрила (Ij)
К смеси 3-(5-метил-3-оксо-2,3-дигидро-1Н-пиразол-4-ил)пропионитрила (1 ммоль, 196 мг), NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли EtOAc (10 мл) и раствором, приготовленным из воды (14 мл), концентрированной хлороводородной кислоты (2 мл) и насыщенного раствора NaCl (20 мл). После взбалтывания полученной смеси органический слой отделяли, а водный слой экстрагировали EtOAc (2×10 мл). Все органические экстракты объединяли, промывали насыщенным водным раствором NaCl (20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 3-(3-Метил-4-нитро-5-оксо-4,5-дигидро-1Н-пиразол-4-ил)пропионтрил в виде желтоватого порошка с выходом 80% (0.80 ммоль, 157 мг). Тпл=105-107°С; 1Н ЯМР (300.13 МГц, DMSO-d6): δ=12.10 (bs, 1Н), 2.78-2.65 (m, 2H), 2.65-54 (m, 2H), 2.07 (s, 3Н); 13C ЯМР (75.47 МГц, DMSO-d6): δ=167.1, 153.0, 118.5, 91.5, 25.7, 13.4, 10.6; ИК (KBr): νmax=3249, 2263, 1735, 1556, 1345, 1256, 826, 674, 604, 552 cm-1; Элементный анализ, рассчитано (%) для C7H8N4O3: С 42.86, Н 4.11, N 28.56; найдено: С 42.93, Н 4.08, N 28.39.
Пример 11. Получение 4-изопропил-3-метил-4-нитро-1Н-пиразолин-5-она (Ik)
К смеси 4-изопропил-3-метил-пиразолин-5-она (1 ммоль, 140 мг), NaNO2 (2 ммоль, 138 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (4 ммоль, 1616 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли CH2Cl2 (10 мл) и 3% водным раствором HCl (30 мл), взбалтывали и отделяли органический слой. Водный слой экстрагировали CH2Cl2 (2×10 мл), после чего все органические экстракты объединяли, промывали водой (2×20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 4-изопропил-3-метил-4-нитро-1Н-пиразолин-5-он в виде желтоватого порошка с выходом 74% (0.74 ммоль, 137 мг). Тпл=80-81°C с разл.; 1Н ЯМР (300.13 МГц, CDCl3): δ=8.74 (bs, 1Н), 3.01-2.85 (m, 1Н), 2.13 (s, 3Н), 1.14 (d, J=6.8 Hz, 3Н), 1.01 (d, J=7.1 Hz, 3Н); 13C ЯМР (75.47 МГц, CDCl3): δ=167.3, 153.8, 95.9, 32.8, 16.2, 14.9, 14.5; ИК (KBr): νmax=3285, 1726, 1555, 1471, 1436, 1397, 1377, 1356, 1342, 1265, 822, 757, 727, 680, 565 см-1. Элементный анализ, рассчитано (%) для C7H11N3O3: С 45.40, Н 5.99, N 22.69; найдено: С 45.64, Н 6.16, N 22.60.
Пример 12. Получение 3а-нитро-2,3а,4,5,6,7-гексагидро-3Н-индазол-3-она (Il)
К смеси 1,2,4,5,6,7-гексагидро-3Н-индазол-3-она (1 ммоль, 138 мг), NaNO2 (1 ммоль, 69 мг) и MeCN (5 мл) при перемешивании при комнатной температуре прибавляли Fe(NO3)3⋅9H2O (2 ммоль, 808 мг) в течение 5-10 секунд, перемешивание смеси продолжали 20 минут. Реакционную смесь разбавляли CH2Cl2 (10 мл) и 3% водным раствором HCl (30 мл), взбалтывали и отделяли органический слой. Водный слой экстрагировали CH2Cl2 (2×10 мл), после чего все органические экстракты объединяли, промывали водой (2×20 мл), осушали MgSO4. Растворитель удаляли в вакууме водоструйного насоса на ротационном испарителе. Продукт выделяли колоночной хроматографией на силикагеле с использованием смеси EtOAc/CH2Cl2=1/20 в качестве элюента. Получали 3а-нитро-2,3а,4,5,6,7-гексагидро-3Н-индазол-3-он в виде белого порошка с выходом 61% (0.61 ммоль, 84 мг). Тпл=127-128°С разл.; 1Н ЯМР (300.13 МГц, CDCl3): δ=9.21 (bs, 1H), 3.18-3.01 (m, 1H), 2.85-2.68 (m, 1H), 2.50-2.29 (m, 1H), 2.21-2.06 (m, 1H), 1.97-1.82 (m, 1H), 1.81-1.64 (m, 1H), 1.62-1.38 (m, 2H); 13C ЯМР (75.47 МГц, CDCl3): δ=168.1, 158.1, 89.1, 33.6, 27.5, 21.0; ИК (KBr): νmax=3336, 1721, 1557, 1351, 1339, 1325, 829 cm-1; Элементный анализ, рассчитано (%) для C7H9N3O3: С 45.90, Н 4.95, N 22.94; найдено: С 45.87, Н 4.83, N 22.87.
Испытания на фунгицидную активность соединений общей формулы I проводили in vitro [Методические рекомендации по испытанию химических веществ на фунгицидную активность. Черкассы: НПО «Защита растений», ВНИИ ХСЗР, 1990-68 с]. Для этого готовили растворы веществ в ацетоне с концентрацией 3 мг/мл. Полученные растворы добавляли в разогретый до 50°С сахарозно-картофельный агар в количестве 0,9 мл на 90 мл агара. Таким образом, концентрация испытуемого вещества в среде составляла 30 мг/л. Приготовленные таким образом среды разливали по 15 мл в чашки Петри с внутренним диаметром 9 см. В качестве эталонов использовали высокоэффективные фунгициды триадимефон формулы:

Поверхность охлажденной до комнатной температуры среды инокулировали кусочками мицелия трехдневной культуры грибов и выдерживали при 25°С в течение 72 часов. Затем измеряли диаметр колоний микроорганизмов и вычисляли подавление роста мицелия в процентах к необработанному контролю по Эбботу.
Для биологических испытаний были использованы фитопатогенные грибы различных таксономических классов: Venturia inaequalis (V. i.) (возбудитель парши яблони, класс аскомицеты), Rhizoctonia solani (R. s.) (возбудитель черной парши картофеля, класс базидомицеты), Fusarium oxysporum (F. о.) (возбудитель трахеомикозного увядания различных культур, класс дейтеромицеты), Fusarium moniliforme (F. m.) (возбудитель фузариоза зерновых, «болезни дурных побегов» риса, класс аскомицеты), Helminthosporium sativum (Н. s.) (возбудитель корневой гнили, пятнистости листьев, «черного зародыша» и других болезней зерновых, класс дейтеромицеты), Sclerotinia sclerotiorum (S. s.) (возбудитель белой гнили различных культур, класс аскомицеты). Результаты приведены в таблице. Таблица. Результаты испытаний замещенных 4-нитропиразолин-5-онов на фунгицидную активность в концентрации 30 мг/л.
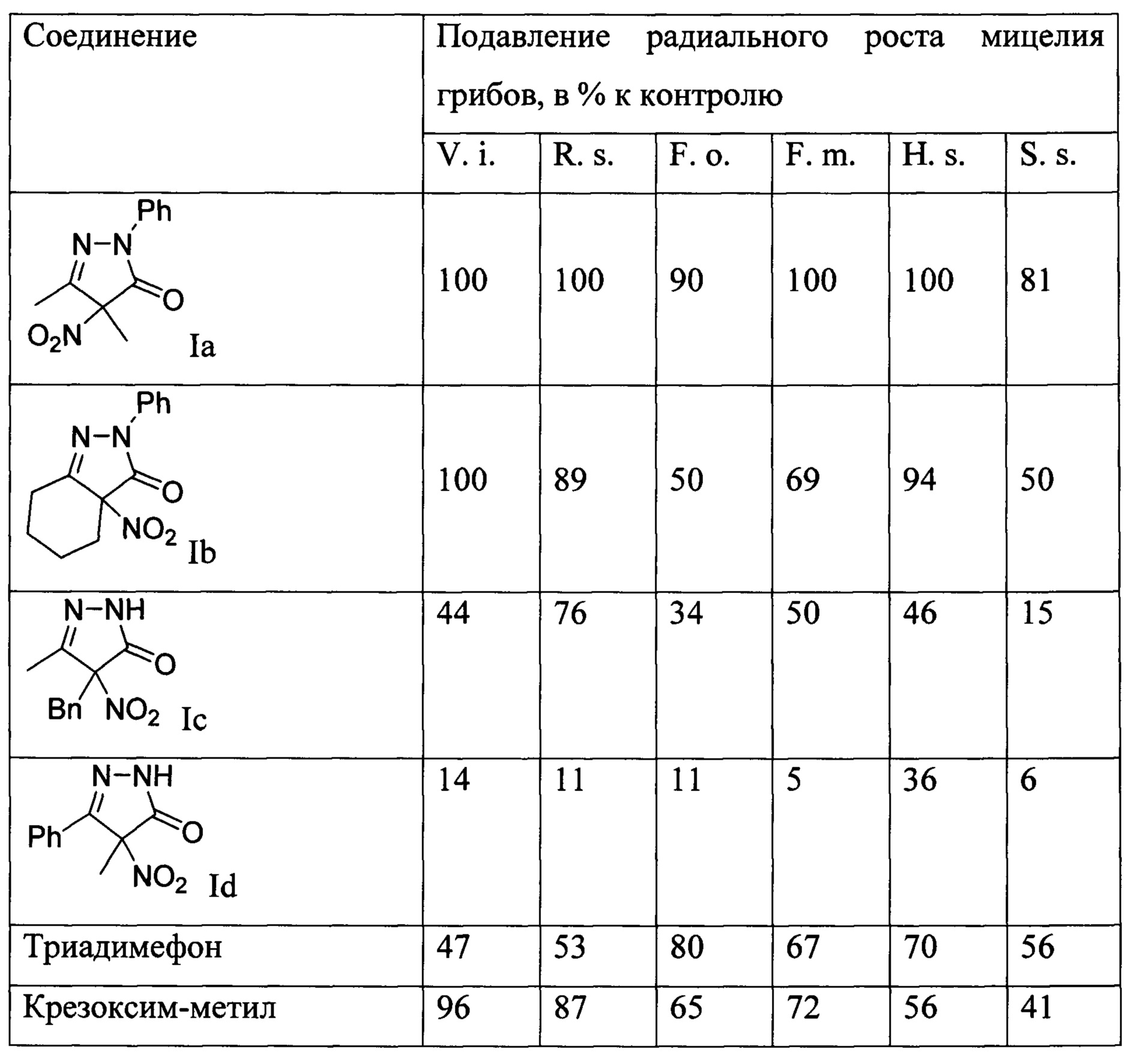
Приведены данные испытаний фунгицидной активности соединений Ia-Id. Как видно из таблицы, соединения общей формулы I проявляют фунгицидную активность по отношению к фитопатогенным грибам. Соединение Ia в концентрации 30 мг/л превосходит по активности оба эталона в отношении практически всех использованных культур фитопатогенных грибков.
Техническим результатом изобретения является создание новых фунгицидных средств на основе замещенных 4-нитропиразолин-5-онов общей формулы I, проявляющих высокую фунгицидную активность по сравнению с известными триадимефоном и крезоксим-метилом. По результатам фунгицидных испытаний in vitro наиболее активным является 3,4-диметил-4-нитро-1-фенилпиразолин-5-она (Ia), превосходящий эталонные триадимефон и крезоксим-метил по отношению практически ко всем из испытанных видов грибов-патогенов.
Применение соединений общей формулы I в композициях позволит более эффективно бороться с грибковыми заболеваниями сельскохозяйственных культур по сравнению с такими широко используемыми фунгицидами как триадимефон и крезоксим-метил.
3,3`-бис(фтординитрометил-onn-азоксифуразанил)фуроксан и способ его получения
Способ получения солей нитрония
Способ получения динитрометил-onn-азоксисоединений
Способ получения 6-метилено-16α,17α-циклогексанопрегн-4-ен-3,20-диона
Способ получения 6-метилено-16α,17α-циклогексанопрегн-4-ен-3,20-диона
Катализатор для гидроаминирования жидких ацетиленовых углеводородов и способ гидроаминирования жидких ацетиленовых углеводородов с использованием этого катализатора
Ионные жидкости с силоксановым фрагментом в составе катиона в качестве теплоносителей
Способ переработки лигнина в жидкие углеводороды
Способ получения полимерного материала, содержащего неорганические нано- или микрочастицы
Адсорбент для улавливания, концентрирования и хранения диоксида углерода
Способ получения ацилоксизамещенных барбитуровых кислот
N-замещенные 3-алкилсульфанил-5-(1,2,4-триазол-1-илметил)-1,2,4-триазолы, способ их получения, фунгицидные и рострегуляторные композиции на их основе

