Результат интеллектуальной деятельности: ФЕНИЛЬНЫЕ ПРОИЗВОДНЫЕ В КАЧЕСТВЕ АГОНИСТОВ КАННАБИНОИДНОГО РЕЦЕПТОРА 2
Вид РИД
Изобретение
Настоящее изобретение относится к органическим соединениям, полезным для лечения и/или профилактики у млекопитающих, и, в частности, к соединениям, которые являются селективными агонистами каннабиноидных рецепторов 2.
Настоящее изобретение относится, в частности к соединению формулы (I)
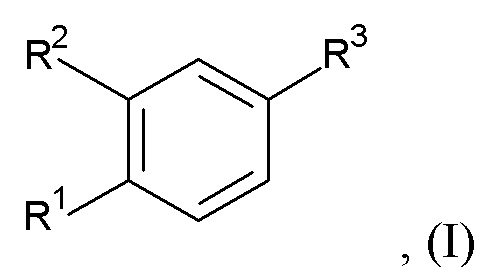
где R1 представляет собой циклопропил, алкил или галоазетидинил;
R2 представляет собой циклопропилметокси, алкокси, галоалкокси, галопиридинил, алкилпиразолил или галопирролидинил;
при условии, что по меньшей мере один из R1 и R2 представляет собой циклопропил или циклопропилметокси;
R3 представляет собой -C(O)-NH-C(R4R5)-R6, -C(O)-R7 или R8;
R4 и R5 независимо выбраны из водорода, алкила, циклоалкила, циклоалкилалкила, алкилсульфонилалкила и алкилоксетанила;
или R4 и R5 вместе с атомом углерода, к которому они присоединены, образуют оксетанил или диоксотиетанил;
R6 представляет собой аминокарбонил, 5-метил-1,2,4-оксадиазол-3-ил, гидроксиалкил, тиазолил, алкоксикарбонил, карбокси, дифторазетидинилкарбонил, 5-амино-1,2,4-оксадиазол-3-ил, алкиламинокарбонил или аминокарбонилалкил;
R7 представляет собой (аминокарбонил)(дифтор)пирролидинил или (аминокарбонил)азаспиро[2.4]гептил; и
R8 представляет собой 3-алкил-1,2,4-оксадиазол-5-ил или 5-алкил-1,2,4-оксадиазол-3-ил;
или его фармацевтически приемлемой соли или эфиру.
Соединение формулы (I) особенно полезно при лечении или профилактике, например, боли, атеросклероза, связанной с возрастом дегенерации желтого пятна, диабетической ретинопатии, глаукомы, окклюзии вен сетчатки, ретинопатии недоношенных, глазного ишемического синдрома, географической атрофии, сахарного диабета, воспаления, воспалительного заболевания кишечника, ишемически-реперфузионного повреждения, острой печеночной недостаточности, фиброза печени, фиброза легких, фиброза почек, системного фиброза, острого отторжения аллотрансплантата, хронической нефропатии аллотрансплантата, диабетической нефропатии, гломерулонефропатии, кардиомиопатии, сердечной недостаточности, ишемии миокарда, инфаркта миокарда, системного склероза, тепловых травм, жжения, гипертрофических рубцов, келоидных рубцов, гингивита с лихорадкой, цирроза или опухоли печени, регулирования костной массы, нейродегенерации, латерального амиотрофического склероза, инсульта, транзиторных ишемических атак или увеита.
Соединение формулы (I) является особенно полезным для лечения или профилактики диабетической ретинопатии, окклюзии вен сетчатки или увеита.
Каннабиноидные рецепторы являются классом рецепторов клеточной мембраны, относящимся к суперсемейству G-белок сопряженных рецепторов. В настоящее время известно два подтипа, названных Каннабиноидный Рецептор 1 (CB1) и Каннабиноидный Рецептор 2 (CB2). CB1 рецептор в основном экспрессируется в центральной нервной системе (например, в миндалине мозжечка, гиппокампе) и в меньшем количестве в периферической нервной системе. CB2, который кодируется геном CNR2, в основном экспрессируется на периферии, на клетках иммунной системы, таких как макрофаги и T-клетки (Ashton, J. C. et al. Curr Neuropharmacol 2007, 5(2), 73-80; Miller, A. M. et al. Br J Pharmacol 2008, 153(2), 299-308; Centonze, D., et al. Curr Pharm Des 2008, 14(23), 2370-42), и в желудочно-кишечном тракте (Wright, K. L. et al. Br J Pharmacol 2008, 153(2), 263-70). CB2 рецепторы также широко распространены в головном мозге, где их обнаружили в основном в микроглие, а не в нейронах (Cabral, G. A. et al. Br J Pharmacol 2008, 153(2): 240-51).
Интерес к агонистам рецепторов CB2 неуклонно растет в течение последнего десятилетия (в настоящее время 30-40 патентных заявок в год) в связи с тем, что некоторые из ранних соединений, как было показано, оказывают благотворное воздействие на доклинических моделях некоторых заболеваний человека, включая хроническую боль (Beltramo, M. Mini Rev Med Chem 2009, 9(1), 11-25), атеросклероз (Mach, F. et al. J Neuroendocrinol 2008, 20 Suppl 1, 53-7), регулирование костной массы (Bab, I. et al. Br J Pharmacol 2008, 153(2), 182-8), нейровоспаление (Cabral, G. A. et al. J Leukoc Biol 2005, 78(6), 1192-7), ишемические/реперфузионные повреждения (Pacher, P. et al. Br J Pharmacol 2008, 153(2), 252-62), системный фиброз (Akhmetshina, A. et al. Arthritis Rheum 2009, 60(4), 1129-36; Garcia-Gonzalez, E. et al. Rheumatology (Oxford) 2009, 48(9), 1050-6), фиброз печени (Julien, B. et al. Gastroenterology 2005, 128(3), 742-55; Munoz-Luque, J. et al. J Pharmacol Exp Ther 2008, 324(2), 475-83).
Ишемическое/реперфузионное (E/R) повреждение является основной причиной повреждения тканей, происходящего в условиях, таких как инсульт, инфаркт миокарда, искусственное кровообращение и другие сосудистые операции и трансплантация органов, а также основным механизмом повреждения органов-мишеней, осложняющим течение циркуляторного шока различной этиологии . Все эти состояния характеризуются нарушением нормального кровоснабжения, приводящего к недостаточной оксигенации тканей. Ре-оксигенация, например, реперфузия, является основным лечением для восстановления нормальной оксигенации тканей. Однако отсутствие кислорода и питательных веществ из крови создает условия, при которых восстановление циркуляции приводит к дальнейшему повреждению тканей. Ущерб от реперфузионного повреждения является отчасти результатом воспалительной реакции поврежденных тканей. Белые клетки крови, собирающиеся в области по вновь возвращающейся крови, высвобождают множество воспалительных факторов, таких как интерлейкины, а также свободных радикалов в ответ на повреждение ткани. Восстановленный кровоток вновь вводит кислород в клетки, что повреждает клеточные белки, ДНК и плазматическую мембрану.
Дистанционное ишемическое прекондиционирование (RIPC) отражает стратегию освоения эндогенных защитных возможностей организма от повреждений в результате ишемии и реперфузии. Она описывает интересный феномен, при котором временная несмертельная ишемия и реперфузия одного органа или ткани придает устойчивость к последующему эпизоду "летального" ишемического реперфузионного повреждения в удаленном органе или ткани. Реальный механизм, посредством которого временная ишемия и реперфузия органа или ткани обеспечивает защиту в настоящее время неизвестно, хотя существует несколько гипотез.
Гуморальная гипотеза предполагает, что эндогенное вещество (например, аденозин, брадикинин, опиоиды, CGRP, эндоканнабиноиды, ангиотензин I или какой-либо другой еще не идентифицированный гуморальный фактор), генерируемое в удаленном органе или ткани, попадает в кровь и активирует свой соответствующий рецептор в целевой ткани и тем самым мобилизует различные внутриклеточные пути кардиопротекции, причастные к ишемическому прекондиционированию.
Последние данные показывают, что эндоканнабиноиды и их рецепторы, в частности CB2, могут быть вовлечены в прекондиционирование и способствуют предотвращению реперфузионного повреждения посредством подавления воспалительного ответа (Pacher, P. et al. Br J Pharmacol 2008, 153(2), 252-62). В частности, недавние исследования с использованием агонистов CB2 продемонстрировали эффективность этой концепции для уменьшения I/R повреждений в сердце (Defer, N. et al. Faseb J 2009, 23(7), 2120-30), мозге (Zhang, M. et al. J Cereb Blood Flow Metab 2007, 27(7), 1387-96), печени (Batkai, S. et al. Faseb J 2007, 21(8), 1788-800) и почках (Feizi, A. et al. Exp Toxicol Pathol 2008, 60(4-5), 405-10).
Более того, за последние несколько лет, все большее количество литературы показывает, что CB2 также может представлять интерес при суб-хронических и хронических условиях. Специфическая положительная регуляция CB1 и CB2, как было показано, ассоциирована, в животных моделях хронических заболеваний, связанных с фиброзом (Garcia-Gonzalez, E. et al. Rheumatology (Oxford) 2009, 48(9), 1050-6; Yang, Y. Y. et al. Liver Int 2009, 29(5), 678-85), с соответствующей экспрессией CB2 в миофибробластах, клетках, ответственных за прогрессирование фиброза.
Активация CB2 рецепторов селективными агонистами CB2, по сути, как было показано, оказывает анти-фиброзный эффект при диффузной системной склеродермии (Garcia-Gonzalez, E. et al. Rheumatology (Oxford) 2009, 48(9), 1050-6) и CB2 рецепторы рассматривали как критическую цель при экспериментальном кожном фиброзе (Akhmetshina, A. et al. Arthritis Rheum 2009, 60(4), 1129-36) и в патофизиологии печени, в том числе фиброгенезе, связанном с хроническими заболеваниями печени (Lotersztajn, S. et al. Gastroenterol Clin Biol 2007, 31(3), 255-8; Mallat, A. et al. Expert Opin Ther Targets 2007, 11(3), 403-9; Lotersztajn, S. et al. Br J Pharmacol 2008, 153(2), 286-9).
Соединения по изобретению связываются с и модулируют CB2 рецепторы и обладают сниженной активностью в отношении рецепторов CB1.
В настоящем описании термин “алкил”, самостоятельно или в комбинации, означает прямоцепочечную или с разветвленной цепью алкильную группу с 1-8 атомами углерода, в частности, прямоцепочечную или с разветвленной цепью алкильную группу с 1-6 атомами углерода и, более конкретно, прямоцепочечную или с разветвленной цепью алкильную группу с 1-4 атомами углерода. Примерами прямоцепочечных или с разветвленной цепью C1-C8 алкильных групп являются метил, этил, пропил, изопропил, бутил, изобутил, трет-бутил, изомерные пентилы, изомерные гексилы, изомерные гептилы и изомерные октилы, в частности, метил, этил, пропил, бутил и пентил. Конкретным примером алкила является метил, более конкретно метил, этил, пропил, изопропил, изобутил, трет-бутил и изопентил. Конкретными примерами алкила являются метил, этил, пропил, изопропил, бутил и трет-бутил.
Термин “циклоалкил”, самостоятельно или в комбинации, означает циклоалкильное кольцо из 3-8 атомов углерода и, в частности, циклоалкильное кольцо из 3-6 атомов углерода. Примерами циклоалкила являются циклопропил, циклобутил, циклопентил и циклогексил, циклогептил и циклооктил. Конкретным примером "циклоалкила" является циклопропил.
Термин “алкокси”, самостоятельно или в комбинации, означает группу формулы алкил-O-, в которой термин "алкил" обладает данным ранее значением, такую как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, втор-бутокси и трет-бутокси. Конкртеными “алкокси” являются метокси, этокси и изопропокси.
Термин “окси”, самостоятельно или в комбинации, обозначает группу -O-.
Термины “галоген” или “гало”, самостоятельно или в комбинации, означают фтор, хлор, бром или йод и, в частности, фтор, хлор или бром, более конкретно фтор и хлор. Термин "галоген", в сочетании с другой группой, обозначает замещение указанной группы по меньшей мере одним атомом галогена, в частности, замещенные одним-пятью атомами галогена, в частности, одним-четырьмя галогенами, т.е. одним, двумя, тремя или четырьмя галогенами. Конкретным “галогеном” является фтор в заместителях R1 - R3.
Термин "галоалкокси", самостоятельно или в комбинации, обозначает алкокси группу, замещенную по меньшей мере одним атомом галогена, в частности, замещенную от одного до пяти атомами галогена, в частности одним-тремя галогенами, в частности одним-тремя фторами. Конкретные «галоалкокси» представляют собой фторэтокси, дифторэтокси и трифторэтокси.
Термины “гидроксил” и “гидрокси”, самостоятельно или в комбинации, означают группу -OH.
Термин “карбонил”, самостоятельно или в комбинации, означает группу -C(O)-.
Термин “амино”, самостоятельно или в комбинации, означает группу первичного амина (-NH2), аминогруппу вторичного амина (-NH-) или аминогруппу третичного амина (-N-).
Термин “сульфонил”, самостоятельно или в комбинации, означает -S(O)2- группу.
Термин " фармацевтически приемлемые соли" относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот, которые не являются биологически или иным образом нежелательными. Соли образуются с неорганическими кислотами, такими как соляная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота, в частности соляная кислота, и органических кислот, таких как уксусная кислота, пропионовая кислота, гликолевая кислота, пировиноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота, салициловая кислота, N- ацетилцистеин. Кроме того, эти соли могут быть получены посредством добавления неорганического основания или органического основания к свободной кислоте. Соли, полученные из неорганических оснований включают, без ограничения, соли натрия, калия, лития, аммония, кальция, магния. Соли, полученные из органических оснований, включают, без ограничения, соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, лизин, аргинин, N- этилпиперидин, пиперидин, полиаминовые смолы. Соединение формулы (I) может также присутствовать в виде цвиттерионов. Особенно предпочтительными фармацевтически приемлемыми солями соединений формулы (I) являются соли соляной кислоты, бромистоводородной кислоты, серной кислоты, фосфорной кислоты и метансульфоновой кислоты.
"Фармацевтически приемлемые эфиры" означает, что соединения общей формулы (I) могут быть дериватизированы по функциональным группам с получением производных, которые способны превратиться обратно в исходные соединения в условиях in vivo. Примеры таких соединений включают физиологически приемлемые и метаболически лабильные эфирные производные, такие как метоксиметиловые эфиры, метилтиометиловые эфиры и пивалоилоксиметиловые эфиры. Дополнительно, любые физиологически приемлемые эквиваленты соединений общей формулы (I), аналогичные метаболически лабильным эфирам, которые способны превращаться в исходные соединения общей формулы (I) in vivo, включены в объем настоящего изобретения.
Если один из исходных материалов или соединений формулы (I) содержат одну или более функциональных групп, которые не являются стабильными или являются реактивными в условиях реакции одной или нескольких реакционных стадий, соответствующие защитные группы (как описано, например, в “Protective Groups in Organic Chemistry” by T. W. Greene and P. G. M. Wuts, 3rd Ed., 1999, Wiley, New York) могут быть введены перед критической стадией, используя способы, хорошо известные в данной области. Такие защитные группы могут быть удалены на более поздней стадии синтеза с использованием стандартных способов, описанных в литературе. Примерами защитных групп являются трет-бутоксикарбонил (Boc), 9-флуоренилметил карбамат (Fmoc), 2-триметилсилилэтил карбамат (Теос), карбобензилокси (Cbz) и п-метоксибензилоксикарбонил (Moz).
Соединение формулы (I) может содержать несколько асимметричных центров и может присутствовать в форме оптически чистых энантиомеров, смесей энантиомеров, таких как, например, рацематы, смесей диастереоизомеров, диастереоизомерных рацематов или смесей диастереоизомерных рацематов.
Термин "асимметричный атом углерода" означает атом углерода с четырьмя различными заместителями. Согласно правилам Кана - Ингольда - Прелога асимметричный атом углерода может быть "R" или "S" конфигурации.
Настоящее изобретение таким образом относится в частности к:
Соединение формулы (I), в котором R1 представляет собой циклопропил;
Соединение формулы (I), в котором R2 представляет собой циклопропилметокси, алкокси, галоалкокси или галопирролидинил;
Соединение формулы (I), в котором R2 представляет собой циклопропилметокси, пропилокси, фторэтокси, трифторэтокси или дифторпирролидинил;
Соединение формулы (I), в котором R4 и R5 независимо выбраны из водорода, алкила, циклоалкила и циклоалкилалкила;
Соединение формулы (I), в котором R4 и R5 независимо выбраны из водорода, метила, бутила, циклопропила и циклопропилметила;
Соединение формулы (I), в котором R6 представляет собой аминокарбонил, 5-метил-1,2,4-оксадиазол-3-ил, гидроксиалкил или алкиламинокарбонил;
Соединение формулы (I), в котором R6 представляет собой аминокарбонил, 5-метил-1,2,4-оксадиазол-3-ил, гидроксиметил или метиламинокарбонил;
Соединение формулы (I), в котором R7 представляет собой (аминокарбонил)(дифтор)пирролидинил; и
Соединение формулы (I), в котором R8 представляет собой 3-трет-бутил-1,2,4-оксадиазол-5-ил, 5-трет-бутил-1,2,4-оксадиазол-3-ил или 5-метил-1,2,4-оксадиазол-3-ил.
Настоящее изобретение, кроме того, относится к соединению формулы (I), выбранному из следующих соединений:
(R)-N-(1-амино-4-метил-1-оксопентан-2-ил)-3-(циклопропилметокси)-4-метилбензамид;
3-(циклопропилметокси)-4-метил-N-[2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид;
4-циклопропил-3-(циклопропилметокси)-N-[2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид;
N2-[4-циклопропил-3-(циклопропилметокси)бензоил]-L-лейцинамид;
4-циклопропил-3-(циклопропилметокси)-N-(1-гидрокси-2-метилпропан-2-ил)бензамид;
4-циклопропил-3-(циклопропилметокси)-N-[2-(1,3-тиазол-2-ил)пропан-2-ил]бензамид;
этил 2-[4-циклопропил-3-(циклопропилметокси)бензамидо]-2-этилбутаноат;
2-[4-циклопропил-3-(циклопропилметокси)бензамидо]-2-этилбутановая кислота;
4-циклопропил-3-(циклопропилметокси)-N-[3-(3,3-дифторазетидин-1-карбонил)пентан-3-ил]бензамид;
3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)-N-[2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид;
N-[2-(5-амино-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензамид;
N2-[3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензоил]-N-метил-L-лейцинамид;
3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)-N-[(2S)-1-гидрокси-4-метилпентан-2-ил]бензамид;
3-трет-бутил-5-[4-циклопропил-3-(циклопропилметокси)фенил]-1,2,4-оксадиазол;
N-[3-(2-амино-2-оксоэтил)оксетан-3-ил]-4-циклопропил-3-(циклопропилметокси)бензамид;
N-[3-(2-амино-2-оксоэтил)-1,1-диоксотиетан-3-ил]-4-циклопропил-3-(циклопропилметокси)бензамид ;
1-[4-циклопропил-3-(циклопропилметокси)бензоил]-4,4-дифтор-L-пролинамид;
N-(3-карбамоилпентан-3-ил)-4-циклопропил-3-(циклопропилметокси)бензамид;
N2-[4-циклопропил-3-(циклопропилметокси)бензоил]-N-метил-L-лейцинамид;
4-циклопропил-3-(циклопропилметокси)-N-[(2S)-1-(метансульфонил)-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид;
4-циклопропил-3-(циклопропилметокси)-N-[(2R)-1-(метансульфонил)-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид;
5-[4-циклопропил-3-(циклопропилметокси)бензоил]-5-азаспиро[2.4]гептан-6-карбоксамид;
5-трет-бутил-3-[4-циклопропил-3-(2,2,2-трифторэтокси)фенил]-1,2,4-оксадиазол;
5-трет-бутил-3-[4-циклопропил-3-(2,2-дифторэтокси)фенил]-1,2,4-оксадиазол;
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид;
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид;
N-[3-(2-амино-2-оксоэтил)-1,1-диоксо-тиетан-3-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид;
4-циклопропил-N-[(2R)-1-(метансульфонил)-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид;
4-циклопропил-N-[(2S)-1-(метансульфонил)-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид;
5-трет-бутил-3-[4-циклопропил-3-(2-фторэтокси)фенил]-1,2,4-оксадиазол;
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2-дифторэтокси)бензамид;
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2-дифторэтокси)бензамид;
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2-фторэтокси)бензамид;
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2-фторэтокси)бензамид;
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид;
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид;
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-[(пропан-2-ил)окси]бензамид;
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-[(пропан-2-ил)окси]бензамид;
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-(2-фторэтокси)бензамид;
1-[4-циклопропил-3-(2-фторэтокси)бензоил]-4,4-дифтор-L-пролинамид;
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-(2,2,2-трифторэтокси)бензамид;
1-[4-циклопропил-3-(2,2,2-трифторэтокси)бензоил]-4,4-дифтор-L-пролинамид;
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-[(пропан-2-ил)окси]бензамид;
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-[(пропан-2-ил)окси]бензамид;
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2-фторэтокси)бензамид;
3-трет-бутил-5-{4-циклопропил-3-[(пропан-2-ил)окси]фенил}-1,2,4-оксадиазол;
3-трет-бутил-5-[4-циклопропил-3-(3,3-дифторпирролидин-1-ил)фенил]-1,2,4-оксадиазол;
1-{4-циклопропил-3-[(пропан-2-ил)окси]бензоил}-4,4-дифтор-L-пролинамид;
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-[(пропан-2-ил)окси]бензамид;
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(6-фторпиридин-3-ил)бензамид;
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид;
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(6-фторпиридин-3-ил)бензамид;
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(6-фторпиридин-3-ил)бензамид;
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2-фторэтокси)бензамид;
1-[4-циклопропил-3-(1-метил-1H-пиразол-5-ил)бензоил]-4,4-дифтор-L-пролинамид;
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-(1-метил-1H-пиразол-5-ил)бензамид;
1-[4-циклопропил-3-(6-фторпиридин-3-ил)бензоил]-4,4-дифтор-L-пролинамид;
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-(6-фторпиридин-3-ил)бензамид;
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(1-метил-1H-пиразол-5-ил)бензамид;
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(6-фторпиридин-3-ил)бензамид;
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензамид;
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензамид;
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(3,3-дифторпирролидин-1-ил)бензамид;
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(3,3-дифторпирролидин-1-ил)бензамид;
1-[4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензоил]-4,4-дифтор-L-пролинамид;
4-циклопропил-3-(3,3-дифторпирролидин-1-ил)-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]бензамид;
4-циклопропил-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]-3-(2,2,2-трифторэтокси)бензамид;
4-циклопропил-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]-3-[(пропан-2-ил)окси]бензамид;
N-[3-амино-1-(3-метилоксетан-3-ил)-3-оксопропил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид; и
4-циклопропил-3-(2-фторэтокси)-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]бензамид.
Настоящее изобретение также относится к соединению формулы (I), выбранному из следующих соединений:
N2-[4-циклопропил-3-(циклопропилметокси)бензоил]-L-лейцинамид;
4-циклопропил-3-(циклопропилметокси)-N-(1-гидрокси-2-метилпропан-2-ил)бензамид;
3-трет-бутил-5-[4-циклопропил-3-(циклопропилметокси)фенил]-1,2,4-оксадиазол;
N-[3-(2-амино-2-оксоэтил)оксетан-3-ил]-4-циклопропил-3-(циклопропилметокси)бензамид;
N-[3-(2-амино-2-оксоэтил)-1,1-диоксотиетан-3-ил]-4-циклопропил-3-(циклопропилметокси)бензамид;
1-[4-циклопропил-3-(циклопропилметокси)бензоил]-4,4-дифтор-L-пролинамид;
5-трет-бутил-3-[4-циклопропил-3-(2-фторэтокси)фенил]-1,2,4-оксадиазол;
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид;
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-[(пропан-2-ил)окси]бензамид;
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-[(пропан-2-ил)окси]бензамид; и
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-[(пропан-2-ил)окси]бензамид.
Получение соединений формулы I согласно настоящему изобретению может проводиться в соответствии с последовательными или конвергентными путями синтеза. Синтез соединений согласно настоящему изобретению показан на следующей схеме. Навыки, необходимые для осуществления реакций и очистки полученных соединений известны квалифицированным специалистам. Заместители и индикаторы, используемые в данных ниже описаниях способов получения, прежде всего, имеют значения данные здесь, если не указано иного. Более детально, соединения формулы I могут быть получены способами, описанными ниже, способами, приведенными в примерах или аналогичными способами. Подходящие реакционные условия для индивидуальных реакций известны квалифицированным специалистам. Более подробно, соединения формулы (I) могут быть получены способами, данными ниже, способами, приведенными в примерах или аналогичными способами. Подходящие условия реакции для отдельных реакционных стадий известны специалисту в данной области техники. Кроме того, для условий реакций, описанных в литературе, затрагивающих описанные реакции см., например: Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2nd Edition, Richard C. Larock. John Wiley & Sons, New York, NY. 1999). Авторами было обнаружено, что удобно проводить реакции в присутствии или в отсутствие растворителя. Нет особых ограничений в отношении природы используемого растворителя, при условии, что он не оказывает вредного влияния на реакцию или участвующие в ней реагенты, и что он может растворить реагенты, по меньшей мере, в некоторой степени. Описанные реакции могут протекать в широком диапазоне температур, и точная температура реакции не является критической для изобретения. Удобно проводить описанные реакции в температурном диапазоне между -78°C до температуры кипения. Время, необходимое для реакции, также может широко варьироваться в зависимости от многих факторов, особенно от реакционной температуры и природы реагентов. Тем не менее, период времени от 0,5 ч до нескольких суток, обычно будет достаточным для получения описанных промежуточных соединений и соединений. Последовательности реакций не ограничены показанными на схемах, однако, зависимость от исходного материала и их соответствующая химическая активность последовательности реакционных стадий может быть изменена без ограничений. Исходные материалы являются как коммерчески доступными, так и могут быть получены согласно способам, аналогичным описанным здесь, способами, описанными в ссылках, приведенных в описании или в примерах, или способами известными в уровне техники
Соединения настоящего изобретения могут быть получены, например, с помощью общих методик синтеза, описанных ниже.
На следующих схемах и описании R1-R8 обладают значениями, как определено выше, если не указано иного.
Следуя методике в соответствии со схемой 1, соединение AA может быть использовано в качестве исходного вещества (R = H, метил, этил, изопропил, трет-бутил или другая подходящая защитная группа, описанная, например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition). AA является либо коммерчески доступным, описанным в литературе, может быть синтезирован квалифицированным специалистом в уровне техники или как описано в экспериментальном разделе.
Схема 1
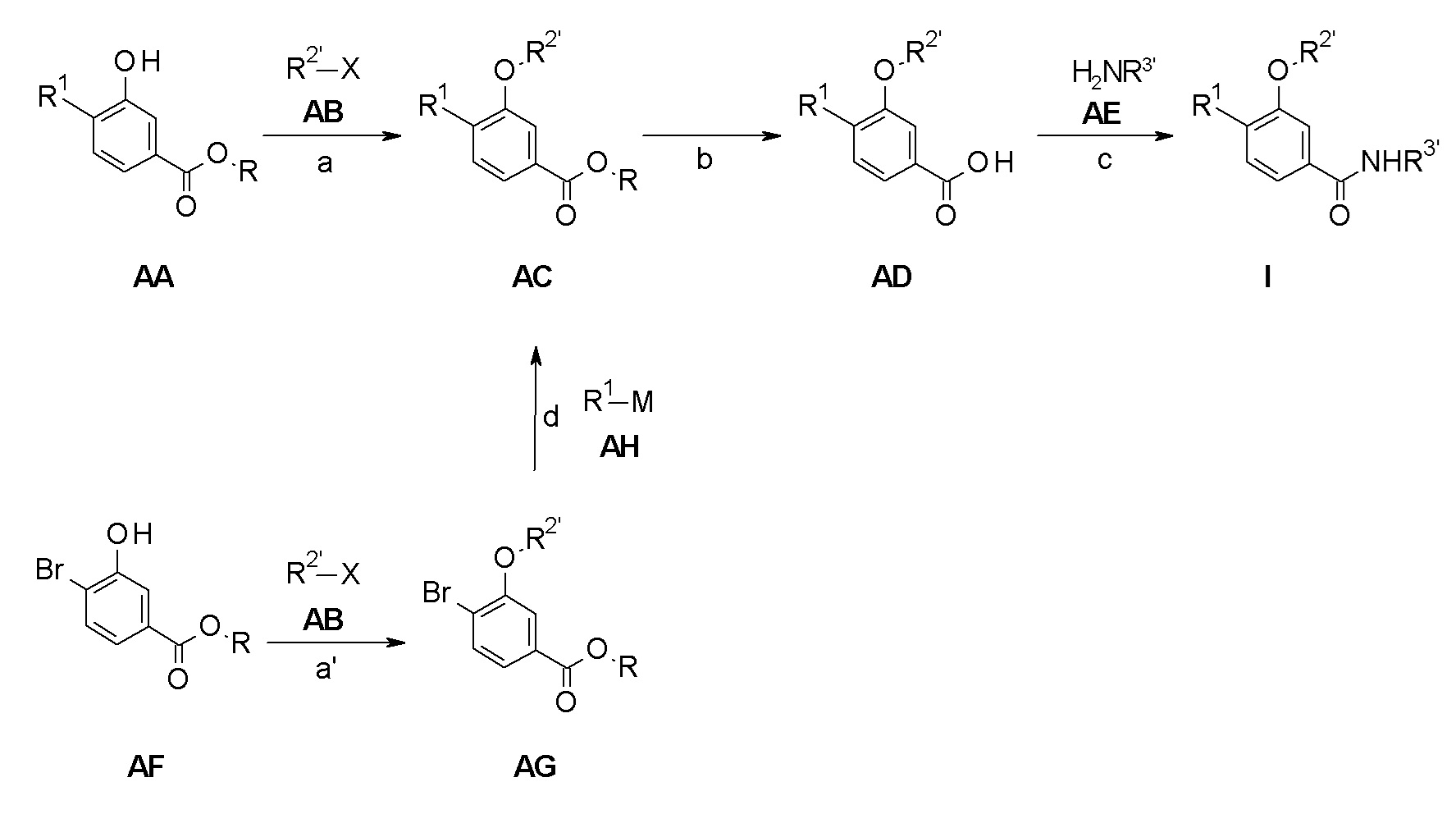
Соединение AC может быть получено из AA посредством взаимодействия с подходящим образом замещенным алкокси или галоалкокси производным R2'-X AB (R2' = циклопропилметил, алкил, галоалкил; X = Cl, Br или другая подходящая уходящая группа) в присутствии основания, например, карбоната калия, в растворителе, таком как ДМФ, при температурах, предпочтительно в диапазоне от комнатной температуры до 50°C (стадия a).
Сапонификация эфира общей формулы AC (R ≠ H) посредством способов, хорошо известных квалифицированным в уровне техники специалистам - с применением, например, водных LiOH, NaOH или KOH в тетрагидрофуране / этаноле или другом подходящем растворителе при температурах между 0°C и температурой кипения используемого растворителя - приводит к кислоте общей формулы AD (стадия b).
Соединение I может быть получено из кислоты AD и соответствующего амина NH2-R3' AE (NH2-R3' представляет собой NH2-C(R4R5)-R6 или H-R7) посредством подходящих реакций образования амидной связи (стадия c). Эти реакции известны из уровня техники. Например, связывающие реагенты, такие как N,N'-карбонил-диимидазол (CDI), N,N'-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), 1-[бис(диметиламино)-метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфат (HATU), 1-гидрокси-1,2,3-бензотриазол (HOBT), O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония тетрафторборат (TBTU), и O-бензотриазол-N,N,N',N'-тетраметил-урония-гексафтор-фосфат (HBTU) могут быть использованы для осуществления такого превращения. Обычным способом является применение, например, HBTU и основания, например, N-метилморфолина в инертном растворителе, таком как например диметилформамид при комнатной температуре. Амины AE являются либо коммерчески доступными, описанными в литературе, могут быть синтезированы квалифицированным специалистом в уровне техники, или как описано в экспериментальном разделе.
Альтернативно, соединение AF может быть использовано в качестве исходного вещества (R = H, метил, этил, изопропил, трет-бутил или другая подходящая защитная группа, описанная, например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition). AF является либо коммерчески доступным, описанным в литературе, может быть синтезирован квалифицированным специалистом в уровне техники или как описано в экспериментальном разделе.
Соединение AG может быть получено из AF посредством взаимодействия с подходящим образом замещенным алкокси или галоалкокси производным R2'-X AB (R2' = циклопропилметил, алкил, галоалкил; X = Cl, Br или другая подходящая уходящая группа) в присутствии основания, например, карбоната калия, в растворителе, таком как ДМФ, при температурах, предпочтительно в диапазоне от комнатной температуры до 50°C (стадия a').
Конверсия соединения AG в соединение AC может быть проведена посредством связывания подходящим образом замещенных циклоалкилметаллов R1-M AH (например, трифторборат [BF3]-K+, бороновая кислота B(OH)2 или бороновой кислоты пинаколовый эфир) в присутствии подходящего катализатора, в частности палладиевый катализатор и более конкретно палладия(II)ацетат/ трифенилфосфин или бутил-1-адамантилфосфиновые смеси, и основания такого как карбонат цезия в смеси инертных растворителей, например, толуол/вода, предпочтительно при температуре кипения смеси растворителя (стадия d). Альтернативно, соединение AG может быть превращено в аминопроизводные AC посредством обработки амином R1-M AH (M представляет собой H) с использованием способов, хорошо известных в уровне техники (стадия d), например, используя инициируемую палладием реакцию аминирования с помощью палладия(II)ацетат/ 2-(дициклогексилфосфино)бифенил в качестве системы катализаторов в присутствии основания, такого как карбонат калия, в диоксане при кипении с обратным холодильником.
Если одно из исходных веществ, соединений формулы AA, AB, AE, AF или AH, содержит одну или более функциональных групп, которые нестабильны или реактивны в реакционных условиях одной или более стадий взаимодействия, подходящие защитные группы (P) (как описано , например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition) могут быть введены перед критическими стадиями с использованием способов, хорошо известных в уровне техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных способов, известных в уровне техники.
Если одно или более соединений формулы AA - AH содержит хиральные центры, фенилы формулы I могут быть получены в виде смесей диастереомеров или энантиомеров, которые могут быть разделены хорошо известными в уровне техники способами, например (хиральной) ВЭЖХ или кристаллизацией. Рацемические соединения могут быть, например, разделены на свои антиподы через диастереомерные соли посредством кристаллизации или посредством разделения антиподов с помощью специальных хроматографических способов с использованием либо хирального абсорбента, либо хирального элюента.
Следуя методике в соответствии со схемой 2, соединение BA может быть использовано в качестве исходного вещества (Y = Br, I; R = H, метил, этил, изопропил, трет-бутил или другая подходящая защитная группа, описанная например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition). BA является либо коммерчески доступным, описанным в литературе, могут быть синтезированы квалифицированным специалистом в уровне техники или как описано в экспериментальном разделе.
Схема 2
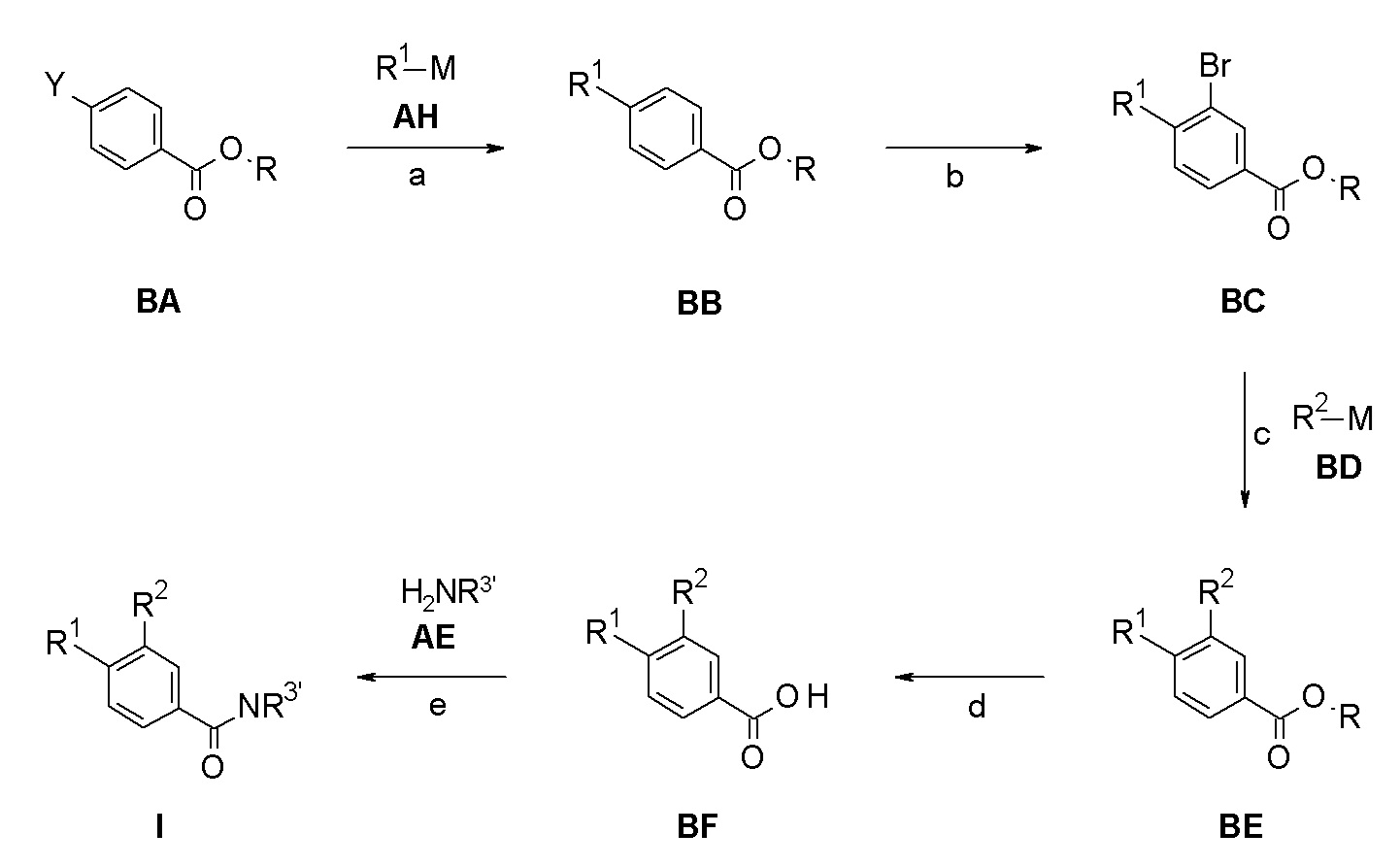
Конверсия соединения BA в соединение BB может быть проведена посредством связывания подходящим образом замещенных циклоалкилметаллов R1-M AH (например, трифторборат [BF3]-K+, бороновая кислота B(OH)2 или бороновой кислоты пинаколовый эфир) или амин R1-M AH (M = H) как описано в стадии d на Схеме 1 (стадия a).
Бромирование фенила BB в соответствии с методиками, хорошо известными квалифицированным специалистам, например, обработкой с помощью N-бромсукцинимида в присутствии трифторуксусной кислоты при температурах около 50°C, дает бромин BC (стадия b).
Соединение BE может быть получено из BC посредством связывания с подходящим образом замещенными арил или гетероарилметаллами R2-M формулы BD (стадия c), например, с органотрифторборатной калиевой солью в присутствии палладиевого катализатора, такого как палладия(II)ацетат/ бутил-1-адамантилфосфин и основания, такого как карбонат цезия, в инертном растворителе, таком как тоуол, при температурах между 50°C и температурой кипения растворителя, или арибороновой кислотой или эфиром арилбороновой кислоты в присутствии подходящего катализатора, в частности палладиевый катализатор и более конкретно палладия(II)ацетат/ трифенилфосфиновая смесь или палладия(II)хлорид-dppf (1,1'-бис(дифенилфосфино)ферроцен) комплексы, и основания, такого как триэтиламин, карбонат натрия или фосфат калия, в инертном растворителе, таком как диметилформамид, толуол, тетрагидрофуран, ацетонитрил или диметоксиэтан. Возможно, соединение BD (M = H) также может быть амином, который связывают с BC способами, хорошо известными квалифицированным специалистам (стадия c), например, с использованием палладиевого катализатора, такого как трис(дибензилиденацетон)дипалладий / диметилбисдифенил-фосфиноксантен, и основания, такого как карбонат цезия, в растворителе, таком как 1,4-диоксан, предпочтительно при температуре кипения растворителя.
Сапонификация эфира общей формулы BE (R ≠ H) посредством способов, хорошо известных квалифицированным в уровне техники специалистам - с применением, например, водных LiOH, NaOH или KOH в тетрагидрофуране / этаноле или другом подходящем растворителе, при температурах между 0°C и температурой кипения используемого растворителя - приводит к кислоте общей формулы BF (стадия d).
Соединение I может быть получено из кислоты BF и соответствующего амина NH2-R3' AE (NH2-R3' представляет собой NH2-C(R4R5)-R6 или H-R7) посредством подходящих реакций образования амидной связи (стадия e). Эти реакции известны из уровня техники. Например, связывающие реагенты, такие как N,N'-карбонил-диимидазол (CDI), N,N'-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), 1-[бис(диметиламино)-метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфат (HATU), 1-гидрокси-1,2,3-бензотриазол (HOBT), O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония тетрафторборат (TBTU), и O-бензотриазол-N,N,N',N'-тетраметил-урония-гексафтор-фосфат (HBTU) могут быть использованы для осуществления такого превращения. Обычным способом является применение, например HBTU и основания, например N-метилморфолина в инертном растворителе, например, диметилформамиде при комнатной температуре. Амины AE являются либо коммерчески доступными, описаны в литературе, могут быть синтезированы квалифицированным специалистом в уровне техники или как описано в экспериментальном разделе.
Если одно из исходных веществ, соединений формулы BA, AH, BD или AE содержит одну или более функциональных групп, которые нестабильны или реактивны в реакционных условиях одной или более стадий взаимодействия, подходящие защитные группы (P) (как описано, например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition) могут быть введены перед критическими стадиями с использованием способов, хорошо известных в уровне техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных способов, известных в уровне техники.
Если одно или более соединений формулы BA - BF, AH или AE содержит хиральные центры, фенилы формулы I могут быть получены в виде смесей диастереомеров или энантиомеров, которые могут быть разделены хорошо известными в уровне техники способами, например, (хиральной) ВЭЖХ или кристаллизацией. Рацемические соединения могут быть, например, разделены на свои антиподы через диастереомерные соли посредством кристаллизации или посредством разделения антиподов с помощью специальных хроматографических способов с использованием либо хирального абсорбента, либо хирального элюента.
Следуя методике в соответствии со схемой 3, соединение CA (R8' = алкил) может быть использовано в качестве исходного вещества. CA является либо коммерчески доступным, описанным в литературе, может быть синтезировано квалифицированным специалистом в уровне техники или как описано в экспериментальном разделе.
Схема 3
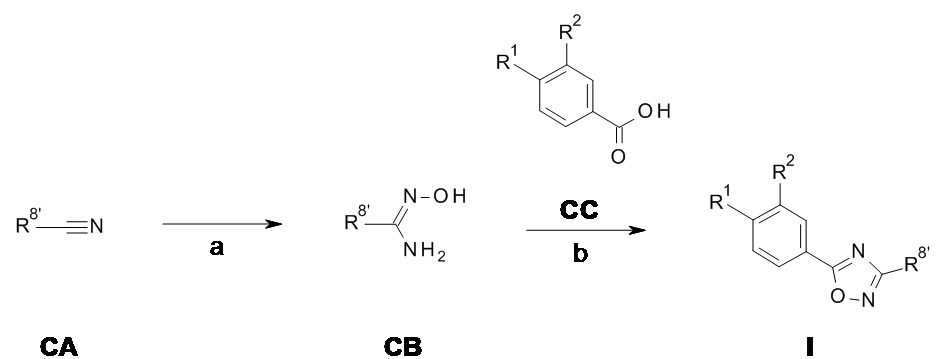
Соединение CB может быть получено посредством взаимодействия нитрила CA с гидроксиламином с использованием способов, хорошо известных квалифицированным в области техники специалистам (стадия a), например через реакцию с гидроксиламина гидрохлоридом в присутствии основания, такого как карбонат калия, в растворителе, таком как этанол, при температурах между 0°C и температурой кипения растворителя, предпочтительно при комнатной температуре.
Конденсация кислоты CB (идентичной соединению AD на Схеме 1 или соединению BF на Схеме 2) с амидом гидроксиимида CB, например, в присутствии карбонилдиимидазола в растворителе, таком как N,N-диметилформамид при температурах приблизительно 100°C, дает соединение I (стадия b).
Если исходное вещество, соединение формулы CC содержит одну или более функциональных групп, которые нестабильны или реактивны в реакционных условиях одной или более стадий взаимодействия, подходящие защитные группы (P) (как описано , например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition) могут быть введены перед критическими стадиями с использованием способов, хорошо известных в уровне техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных способов, известных в уровне техники.
Если одно или более соединений формулы CA - CC содержит хиральные центры, фенилы формулы I могут быть получены в виде смесей диастереомеров или энантиомеров, которые могут быть разделены хорошо известными в уровне техники способами, например (хиральной) ВЭЖХ или кристаллизацией. Рацемические соединения могут быть, например, разделены на свои антиподы через диастереомерные соли посредством кристаллизации или посредством разделения антиподов с помощью специальных хроматографических способов с использованием либо хирального абсорбента, либо хирального элюента.
Следуя методике в соответствии со схемой 4, соединение DA может быть использовано в качестве исходного вещества (R = метили или другая подходящая защитная группа, описанная, например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition). DA является либо коммерчески доступным, описанным в литературе, может быть синтезирован квалифицированным специалистом в уровне техники или как описано в экспериментальном разделе.
Схема 4
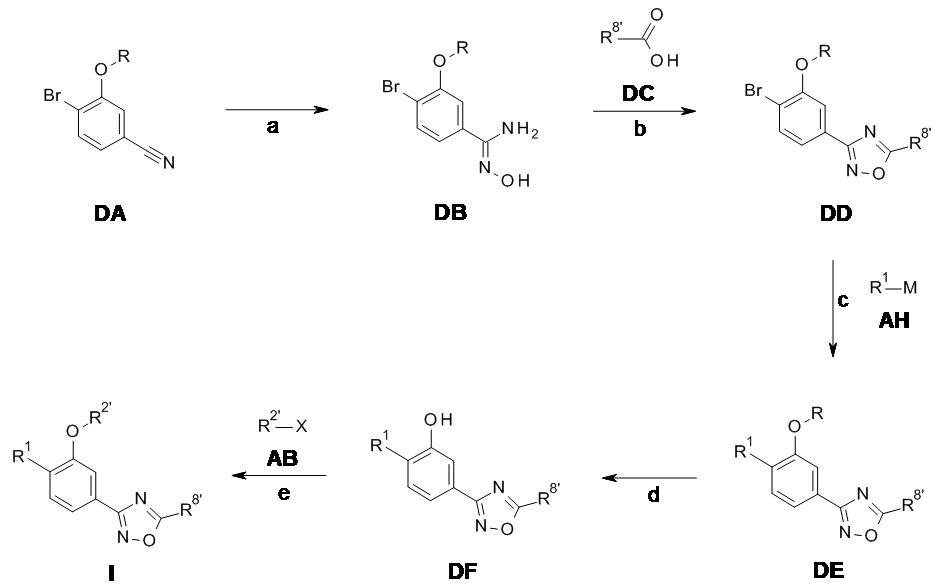
Соединение DB может быть получено из DA посредством обработки гидроксиламина гидрохлоридом в присутствии основания, такого как триэтиламин в растворителе, таком как этанол, аналогично методике, описанной на стадии а Схемы 3 (стадия a).
Циклизация соединения DB в соединение DD может быть проведена способами амидного связывания, хорошо известными квалифицированному специалисту, с использованием подходящим образом замещенной коммерчески доступной карбоновой кислотой DC (R8' = алкил), с последующим нагреванием для циклизации в оксадиазольное кольцо в растворителе с высокой температурой кипения растворителя, например, ДМФ, например, по аналогии со способом, описанным на стадии b Схемы 3 (стадия b).
Конверсия соединения DD в соединение DE может быть проведена посредством связывания подходящим образом замещенных циклоалкилметаллов R1-M AH (например, трифторборат [BF3]-K+, бороновая кислота B(OH)2 или бороновой кислоты пинаколовый эфир) в присутствии подходящего катализатора, в частности палладиевый катализатор и более конкретно палладия(II)ацетат/ трифенилфосфин или бутил-1-адамантилфосфиновые смеси и основания такого как карбонат цезия в смеси инертных растворителей, например, толуол/вода, предпочтительно при температуре кипения смеси растворителя (стадия c). Альтернативно, соединение DD может быть превращено в аминопроизводные DE посредством обработки амином R1-M AH (M представляет собой H) с использованием способов, хорошо известных в уровне техники (стадия c), например, используя инициируемую палладием реакцию аминирования с помощью палладия(II)ацетат/ 2-(дициклогексилфосфино)бифенил в качестве системы катализаторов в присутствии основания, такого как карбонат калия в диоксане при кипении с обратным холодильником.
Соединение DE может быть превращено в соответствующее фенольное соединение DF с использованием способов депротекции, известных квалифицированным специалистам, например, сильные кислоты Льюиса (например, BBr3) в подходящем растворителе, таком как дихлорметан при комнатной температуре для R эквивалента метила (стадия d).
Соединение I может быть получено из DF посредством взаимодействия с подходящим образом замещенным алкокси или галоалкоксипроизводным R2'-X AB (R2' = циклопропилметил, алкил, галоалкил; X = Cl, Br или другая подходящая уходящая группа) в присутствии основания, например, карбоната калия, в растворителе, таком как ДМФ, при температурах в диапазоне от предпочтительно комнатной температуре до 50°C (стадия a).
Если одно из исходных веществ, соединений формулы DC, AH или AB содержит одну или более функциональных групп, которые нестабильны или реактивны в реакционных условиях одной или более стадий взаимодействия, подходящие защитные группы (P) (как описано , например, в T.W. Greene et al., Protective Groups in Organic Chemistry, John Wiley and Sons Inc. New York 1999, 3rd edition) могут быть введены перед критическими стадиями с использованием способов, хорошо известных в уровне техники. Такие защитные группы могут быть удалены на последующих стадиях синтеза с использованием стандартных способов, известных в уровне техники.
Если одно или более соединений формулы DC - DF, AH или AB содержит хиральные центры, фенилы формулы I могут быть получены в виде смесей диастереомеров или энантиомеров, которые могут быть разделены хорошо известными в уровне техники способами, например, (хиральной) ВЭЖХ или кристаллизацией. Рацемические соединения могут быть, например, разделены на свои антиподы через диастереомерные соли посредством кристаллизации или посредством разделения антиподов с помощью специальных хроматографических способов с использованием либо хирального абсорбента, либо хирального элюента.
Настоящее изобретение таким образом относится к способу получения соединение формулы (I), содержащему следующие стадии:
(a) Взаимодействие соединение формулы (A)
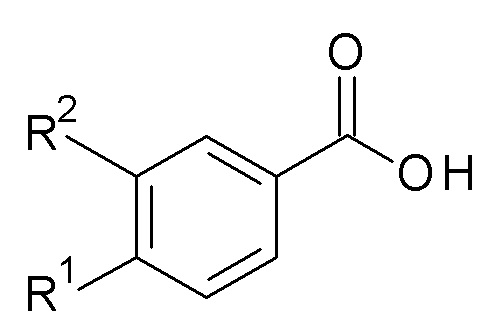 (A)
(A)
в присутствии H2N-C(R4R5)-R6, связывающего агента и основания, где R2 представляет собой циклопропилметокси, алкокси или галоалкокси;
(b) Взаимодействие соединение формулы (A) как определено выше в присутствии H-R7, связывающего агента и основания, где R2 представляет собой циклопропилметокси, алкокси или галоалкокси;
(c) Взаимодействие соединение формулы (A) как определено выше в присутствии соединение формулы (B)
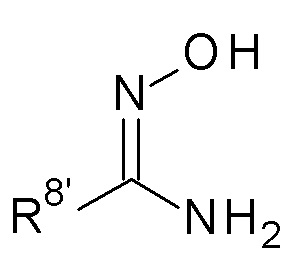 (B)
(B)
и карбонилдиимидазола, где R8' представляет собой метил или трет-бутил; и или
(d) Взаимодействие соединение формулы (C)
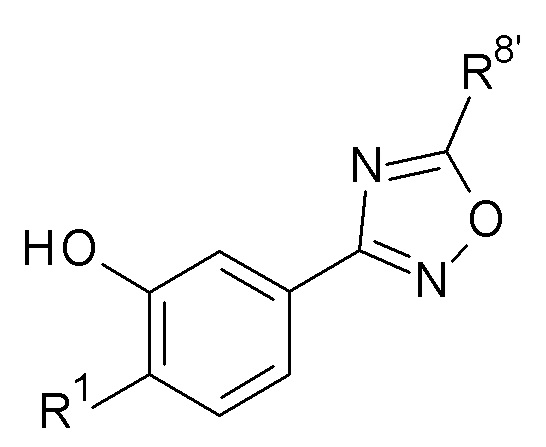 (C)
(C)
в присутствии R2'-X, где R2' представляет собой циклопропилметил, алкил или галоалкил, R8' представляет собой метил или трет-бутил и X представляет собой уходящую группу.
На стадиях (a) и (b), связывающим реагентом является, например, N,N'-карбонил-диимидазол (CDI), N,N'-дициклогексилкарбодиимид (DCC), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (EDCI), 1-[бис(диметиламино)-метилен]-1H-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфат (HATU), 1-гидрокси-1,2,3-бензотриазол (HOBT), O-бензотриазол-1-ил-N,N,N',N'-тетраметилурония тетрафторборат (TBTU) или O-бензотриазол-N,N,N',N'-тетраметил-урония-гексафтор-фосфат (HBTU). Основанием является, например, N-метилморфолина. Обычным способом является применение, например HBTU и основания, например N-метилморфолина, в инертном растворителе таком как, например, диметилформамид при комнатной температуре.
На стадии (d), уходящая группа представляет собой, например, хлор или бром.
Настоящее изобретение также относится к соединению формулы (I) полученному способам по настоящему изобретению.
Настоящее изобретение также относится, в частности, к:
Соединение формулы (I) для применения в качестве терапевтически активного вещества;
Фармацевтическая композиция, содержащая соединение формулы (I) и терапевтически инертный носитель;
Применение соединения формулы (I) для лечения или профилактики боли, атеросклероза, связанной с возрастом дегенерации желтого пятна, диабетической ретинопатии, глаукомы, окклюзии вен сетчатки, ретинопатии недоношенных, глазного ишемического синдрома, географической атрофии, сахарного диабета, воспаления, воспалительного заболевания кишечника, ишемически-реперфузионного повреждения, острой печеночной недостаточности, фиброза печени, фиброза легких, фиброза почек, системного фиброза, острого отторжения аллотрансплантата, хронической нефропатии аллотрансплантата, диабетической нефропатии, гломерулонефропатии, кардиомиопатии, сердечной недостаточности, ишемии миокарда, инфаркта миокарда, системного склероза, тепловых травм, жжения, гипертрофических рубцов, келоидных рубцов, гингивита с лихорадкой, цирроза или опухоли печени, регулирования костной массы, нейродегенерации, латерального амиотрофического склероза, инсульта, транзиторных ишемических атак или увеита;
Применение соединения формулы (I) для получения лекарственного средства для лечения или профилактики боли, атеросклероза, связанной с возрастом дегенерации желтого пятна, диабетической ретинопатии, глаукомы, окклюзии вен сетчатки, ретинопатии недоношенных, глазного ишемического синдрома, географической атрофии, сахарного диабета, воспаления, воспалительного заболевания кишечника, ишемически-реперфузионного повреждения, острой печеночной недостаточности, фиброза печени, фиброза легких, фиброза почек, системного фиброза, острого отторжения аллотрансплантата, хронической нефропатии аллотрансплантата, диабетической нефропатии, гломерулонефропатии, кардиомиопатии, сердечной недостаточности, ишемии миокарда, инфаркта миокарда, системного склероза, тепловых травм, жжения, гипертрофических рубцов, келоидных рубцов, гингивита с лихорадкой, цирроза или опухоли печени, регулирования костной массы, нейродегенерации, латерального амиотрофического склероза, инсульта, транзиторных ишемических атак или увеита;
Соединение формулы (I) для применения для лечения или профилактики боли, атеросклероза, связанной с возрастом дегенерации желтого пятна, диабетической ретинопатии, глаукомы, окклюзии вен сетчатки, ретинопатии недоношенных, глазного ишемического синдрома, географической атрофии, сахарного диабета, воспаления, воспалительного заболевания кишечника, ишемически-реперфузионного повреждения, острой печеночной недостаточности, фиброза печени, фиброза легких, фиброза почек, системного фиброза, острого отторжения аллотрансплантата, хронической нефропатии аллотрансплантата, диабетической нефропатии, гломерулонефропатии, кардиомиопатии, сердечной недостаточности, ишемии миокарда, инфаркта миокарда, системного склероза, тепловых травм, жжения, гипертрофических рубцов, келоидных рубцов, гингивита с лихорадкой, цирроза или опухоли печени, регулирования костной массы, нейродегенерации, латерального амиотрофического склероза, инсульта, транзиторных ишемических атак или увеита; и
Способ лечения или профилактики боли, атеросклероза, связанной с возрастом дегенерации желтого пятна, диабетической ретинопатии, глаукомы, окклюзии вен сетчатки, ретинопатии недоношенных, глазного ишемического синдрома, географической атрофии, сахарного диабета, воспаления, воспалительного заболевания кишечника, ишемически-реперфузионного повреждения, острой печеночной недостаточности, фиброза печени , фиброза легких, фиброза почек, системного фиброза, острого отторжения аллотрансплантата, хронической нефропатии аллотрансплантата, диабетической нефропатии, гломерулонефропатии, кардиомиопатии, сердечной недостаточности, ишемии миокарда, инфаркта миокарда, системного склероза, тепловых травм, жжения, гипертрофических рубцов, келоидных рубцов, гингивита с лихорадкой, цирроза или опухоли печени, регулирования костной массы, нейродегенерации, латерального амиотрофического склероза, инсульта, транзиторных ишемических атак или увеита, который включает введение эффективного количества соединения формулы (I) нуждающемуся в этом пациенту.
Настоящее изобретение, в частности, относится к соединению формулы (I) для лечения или профилактики ишемии, реперфузионного повреждения, фиброза печени или фиброза почек, в частности, ишемии или реперфузионного повреждения.
Настоящее изобретение, кроме того, в частности относится к соединению формулы (I) для лечения или профилактики диабетической ретинопатии, окклюзии вен сетчатки или увеитов.
Настоящее изобретение кроме того относится к соединению формулы (I), полученному способом по настоящему изобретению.
В другом воплощении настоящего изобретения предложены фармацевтическая композиция или лекарственное средство, содержащие соединение по настоящему изобретению и терапевтически инертный носитель, разбавитель или эксципиент, а также способ применения соединений настоящего изобретения для получения такой композиции и лекарственного средства. В одном примере, соединение формулы (I) может быть приготовлено путем смешивания при комнатной температуре, соответствующем рН и при желаемой степени чистоты, с физиологически приемлемыми носителями, например, носителями, которые нетоксичны для реципиентов в используемых дозах и концентрациях, в галенову форму введения. РН композиции зависит в основном от конкретного применения и концентрации соединения, но предпочтительно может варьироваться от примерно 3 до примерно 8. В одном примере соединение формулы (I) готовится в ацетатном буфере при рН 5. В другом воплощении соединение формулы (I) является стерильным. Соединение может храниться, например, в виде твердой или аморфной композиции, в виде лиофилизированного препарата или в виде водного раствора.
Композиции изготавливаются, дозируются и вводятся в соответствии с надлежащей медицинской практикой. Факторы, рассматриваемые в данном контексте, включают конкретное расстройство, подлежащее лечению, конкретное млекопитающее, подлежащее лечению, клиническое состояние конкретного пациента, причину расстройства, место доставки агента, способ введения, схему введения и другие факторы, известные практикующим врачам.
Соединения по изобретению можно вводить любыми подходящими путями, в том числе перорально, местно (в том числе трансбуккально и подъязычно), ректально, вагинально, трансдермально, парентерально, подкожно, внутрибрюшинно, внутрилегочно, внутрикожно, интратекально и эпидурально и интраназально, и, при желании для местного лечения, введение в очаг поражения. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное, внутрибрюшинное или подкожное введение. Соединения по изобретению могут быть введены в частности интраветриально.
Соединения по настоящему изобретению можно вводить в любой удобной форме для введения, например, таблетки, порошки, капсулы, растворы, дисперсии, суспензии, сиропы, спреи, суппозитории, гели, эмульсии, пластыри и т.п. Такие композиции могут содержать обычные для фармацевтических препаратов компоненты, например, разбавители, носители, модификаторы рН, подсластители, наполнители и другие активные агенты.
Типичный препарат готовят путем смешивания соединения по настоящему изобретению и носителя или эксципиента. Подходящие носители и эксципиенты хорошо известны специалистам в данной области техники и подробно описаны, например, в Ansel, Howard C., et al., Ansel's Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; and Rowe, Raymond C. Handbook of Pharmaceutical Excipients. Chicago, Pharmaceutical Press, 2005. Композиции могут также включать один или более буферов, стабилизирующих агентов, поверхностно-активные вещества, смачивающие агенты, смазывающие агенты, эмульгаторы, суспендирующие агенты, консерванты, антиоксиданты, кроющие агенты, глиданты, технологические добавки, красители, подсластители, отдушки, ароматизаторы, разбавители и другие известные добавки для обеспечения элегантной презентации препарата (например, соединения по настоящему изобретению или его фармацевтической композиции) или помощи в изготовлении фармацевтического продукта (например, лекарственного средства).
Настоящее изобретение далее иллюстрировано примерами, которые не являются ограничивающими.
Примеры
Аббревиатуры
MS = масс-спектрометрия; EI = электронная ионизация; ESI = электроспрей; CAN = регистрационный номер CAS; CDI = 1,1'-карбонилдиимидазол; DCM = дихлорметан; DIEA = N-этил-N-изопропилпропан-2-амин; DBU = 1,8-Диазабицикло[5.4.0]ундец-7-ен; ДМФ = диметилформамид; DMSO = диметилсульфоксид; EtOAc = этилацетат; ВЭЖХ = ЖХ = высокоэффективная жидкостная хроматография; iPrOAc = изопропилацетат; TBME = метил трет-бутиловый эфир; TBTU = O-(бензотриазол-1-ил)-N,N,N',N'-тетраметил-урония-тетрафторборат; THF = тетрагидрофуран; ТСХ = тонкослойная хроматография.
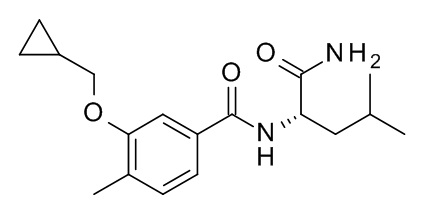
Пример 1
N-[(2S)-1-амино-4-метил-1-оксопентан-2-ил]-3-(циклопропилметокси)-4-метилбензамид
a) Метил 3-(циклопропилметокси)-4-метилбензоат
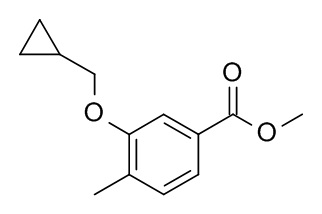
Метил 3-гидрокси-4-метилбензоат (CAN 3556-86-3; 1 г, 6.02 ммоль) растворили в ДМФ (10 мл). (Бромометил)циклопропан (CAN 7051-34-5, 894 мг, 579 мкл, 6.62 ммоль) и карбонат калия (1.66 г, 12.0 ммоль) добавили. Реакционную смесь перемешивали в течение 20 ч, влили в 25 мл 1 M HCl и экстрагировали iPrOAc (2×25 мл). Органические слои промыли льдом/солевым раствором (2×20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 1.1 г светло-желтого масла. Неочищенное вещество очистили с помощью флеш-хроматографии (20 г силикагель, 0 - 10% гептан/iPrOAc) с получением 880 мг (3.99 ммоль, 66%) соединения, указанного в заголовке, в виде бесцветного масла. MS: m/e = 221.3 [M+H]+.
b) 3-(циклопропилметокси)-4-метилбензойная кислота
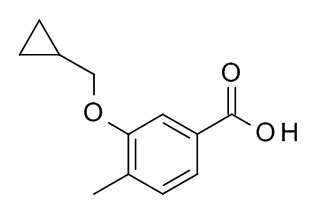
Метил 3-(циклопропилметокси)-4-метилбензоат (880 мг, 4 ммоль) растворили в ТГФ (8.8 мл) и воде (4.4 мл). Моногидрат гидроксида лития (201 мг, 4.79 ммоль) добавили. Реакционную смесь перемешивали в течение 60 ч при комнатной температуре, влили в 1 M HCl (100 мл) и экстрагировали i/ProAc) (200 мл). Органический слой промыли с помощью льда/воды/насыщ. NaCl (3×100 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 830 мг (4 ммоль, колич.) соединения, указанного в заголовке, в виде бесцветного осадка. MS = 204.9 [M-H]-.
c) N-[(2S)-1-амино-4-метил-1-оксопентан-2-ил]-3-(циклопропилметокси)-4-метилбензамид
Смесь 3-(циклопропилметокси)-4-метилбензойной кислоты (20 мг, 97.0 мкмоль), (R)-2-амино-4-метилпентанамида гидрохлорида (CAN 80970-09-8; 17.8 мг, 107 мкмоль), 2-(3H-[1,2,3]триазоло[4,5-b]пиридин-3-ил)-1,1,3,3-тетраметилизоурония гексафторфосфата (V) (73.7 мг, 194 мкмоль) и N-этил-N-изопропилпропан-2-амина (37.6 мг, 50.8 мкл, 291 мкмоль) в ДМФ (235 мкл) перемешивали в течение 18 ч при комнатной температуре. Реакционную смесь влили в 1 M HCl/лед/воду (1×20 мл), экстрагировали iPrOAc (2×25 мл) и промыли льдом/водой (2×25 мл) до pH 6. Органические слои высушили над Na2SO4 и эвапорировали при пониженном давлении. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 2.0 мм, гептан/iPrOAc 1:2), элюировали с помощью iPrOAc, отфильтровали и эвапорировали с получением 21 мг соединения, указанного в заголовке. MS: 319.1 [M+H]+.
Пример 2
3-(циклопропилметокси)-4-метил-N-[2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид
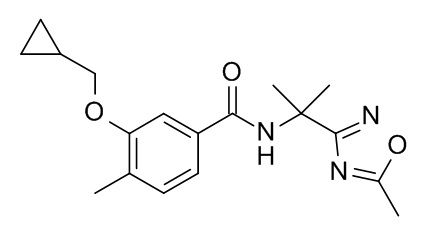
Смесь 3-(циклопропилметокси)-4-метилбензойной кислоты (Пример 1b; 20 мг, 97.0 мкмоль), 2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (CAN 1240526-27-5; 17.2 мг, 97.0 мкмоль), TBTU (46.7 мг, 145 мкмоль) и N,N-диизопропилэтиламин (62.7 мг, 83.0 мкл, 485 мкмоль) в ДМФ (647 мкл) перемешивали в атмосфере аргона в течение 18 ч при комнатной температуре. Реакционную смесь влили в 30 мл льда/воды, экстрагировали iPrOAc (2×40 мл) и промыли 30 мл смеси вода со льдом/солевой раствор. Органические слои объединили, высушили над Na2SO4 и сконцентрировали под вакуумом с получением 45 мг светло-коричневого масла. Неочищенный материал очистили с помощью препаративной ТСХ (силикагель, 2.0 мм, iPrOAc) и элюировали с помощью iPrOAc/DCM 1:1 с получением 28 мг соединения, указанного в заголовке, в виде белого осадка. MS: 330.1 [M+H]+.
Пример 3
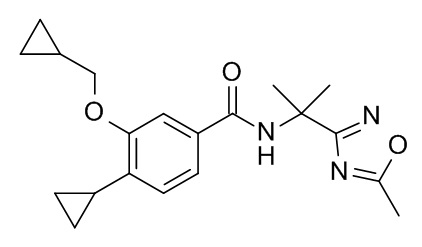
4-циклопропил-3-(циклопропилметокси)-N-[2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид
a) 4-бромо-3-циклопропилметокси-бензойной кислоты этиловый эфир
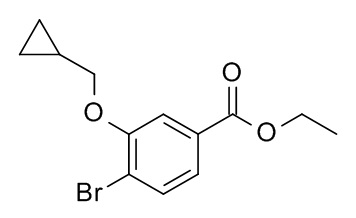
Смесь этил 4-бромо-3-гидроксибензоата (CAN 33141-66-1; 4.85 г, 19.8 ммоль) (бромометил)циклопропана (CAN 7051-34-5, 3.21 г, 2.27 мл, 23.7 ммоль) и карбоната калия (6.56 г, 47.5 ммоль) в N,N-диметилформамиде (50 мл) нагрели до 50°C в течение 19 ч. Реакционную смесь влили в H2O (200 мл) и экстрагировали iPrOAc (2×200 мл). Органические слои промыли льдом/насыщ. NaCl (2×150 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 6.35 г светло-желтой жидкости. 500 мг очистили с помощью флеш-хроматографии с получением 293 мг соединения, указанного в заголовке, в виде бесцветной жидкости. MS: 301.0 [M+H]+.
b) Этил 4-циклопропил-3-(циклопропилметокси)бензоат
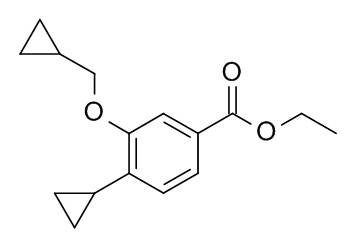
Палладия(II) ацетат (7.5 мг, 33.4 мкмоль), бутил-1-адамантилфосфин (18.0 мг, 50.1 мкмоль), калия циклопропилтрифторборат (CAN 1065010-87-8; 250 мг, 1.69 ммоль) и карбоната цезия (1.63 г, 5.01 ммоль) объединили с получением белого осадка. К этому осадку раствор 4-бромо-3-циклопропилметокси-бензойной кислоты этилового эфира (500 мг, 1.67 ммоль) в толуоле (12.6 мл) и воде (1.4 мл) (откачали и продули аргоном) добавили через крышку с диафрагмой. Реакционную смесь нагревали до 120°C в течение 20 ч. После охлаждения до комнатной температуры неочищенную смесь разбавили H2O (10 мл). Реакционную смесь влили в 100 мл льда/солевого раствора и экстрагировали iPrOAc (2×200 мл). Объединенные органические слои промыли льдом/солевым раствором (100 мл), высушили над Na2SO4 и сконцентрировали под вакуумом. Неочищенный продукт очистили флеш-хроматографией с градиентом гептан/iPrOAc с получением 283 мг соединения, указанного в заголовке. MS: m/e = 261.3 [M+H]+.
c) 4-циклопропил-3-(циклопропилметокси)бензойная кислота
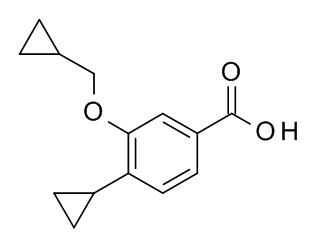
Этил 4-циклопропил-3-(циклопропилметокси)бензоат (311 мг, 1.19 ммоль) и гидрат гидроксида лития (60.2 мг, 1.43 ммоль) объединили с ТГФ (2.5 мл) и водой (625 мкл) с получением желтого раствора, который перемешивали в течение 24 ч при комнатной температуре. Гидрат гидроксида лития (60.2 мг, 1.43 ммоль) добавили и перемешивание продолжили в течение 24 ч. Реакционную смесь влили в лед/воду/1N NaOH (20 мл) и экстрагировали с помощью TBME (2×30 мл). Объединенные экстракты промыли водой со льдом (20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 49 мг желтого масла. Водный слой подкислили 1 н HCl (3 мл). Образовавшийся преципитат затем отфильтровали с получением 166 мг светло-коричневого осадка. Водный слой снова экстрагировали EtOAc (2×30 мл). Органические слои промыли водой со льдом (20 мл), объединили, высушили над Na2SO4 и сконцентрировали под вакуумом с получением 20 мг соединения, указанного в заголовке, в виде желтого осадка. MS(ESI): m/e = 231.3 [M-H]-.
d) 4-циклопропил-3-(циклопропилметокси)-N-[2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид
Смесь 4-циклопропил-3-(циклопропилметокси)бензойной кислоты (10 мг, 43.1 мкмоль), 2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (CAN 1240526-27-5; 8.41 мг, 47.4 мкмоль), TBTU (20.7 мг, 64.6 мкмоль) и N,N-диизопропилэтиламина (27.8 мг, 36.8 мкл, 215 мкмоль) в ДМФ (287 мкл) перемешивали в атмосфере аргона в течение 18 ч при комнатной температуре. Реакционную смесь влили в 30 мл воды со льдом и экстрагировали iPrOAc (2×40 мл). Объединенные экстракты промыли 30 мл воды/льда/солевого раствора, высушили над Na2SO4 и сконцентрировали под вакуумом с получением 45 мг коричневого масла. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 2 мм, iPrOAc) и элюировали в ДХМ/iPrOAc 1:1 с получением 6 мг соединения, указанного в заголовке, в виде светло-желтого осадка. MS: 356.1 [M+H]+.
Пример 4
N2-[4-циклопропил-3-(циклопропилметокси)бензоил]-L-лейцинамид
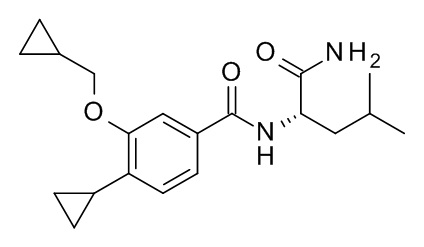
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту и (S)-2-амино-4-метилпентанамида гидрохлорид (CAN 10466-61-2) в качестве исходных веществ, и сразу очистили с помощью препаративной ВЭЖХ без какой-либо обработки. MS (ESI, m/z): 345.1 [M+H]+.
Пример 5
4-циклопропил-3-(циклопропилметокси)-N-(1-гидрокси-2-метилпропан-2-ил)бензамид
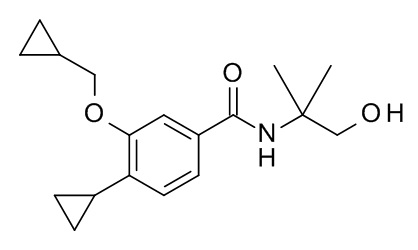
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту и 2-амино-2-метилпропан-1-ол (CAN 124-68-5) в качестве исходных веществ, и сразу очистили с помощью препаративной ВЭЖХ без какой-либо обработки. MS (ESI, m/z): 304.1 [M+H]+.
Пример 6
4-циклопропил-3-(циклопропилметокси)-N-[2-(1,3-тиазол-2-ил)пропан-2-ил]бензамид
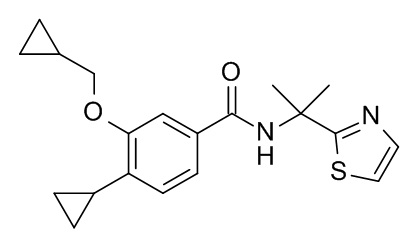
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту и 2-(тиазол-2-ил)пропан-2-амин (CAN 1082393-38-1) в качестве исходных веществ. Реакционную смесь влили в 20 мл воды со льдом, экстрагировали с помощью iPrOAc (2×30 мл) и промыли 20 мл смеси лед/вода/солевой раствор. Органические слои объединили, высушили над Na2SO4 и сконцентрировали под вакуумом с получением 29 мг светло-желтого осадка. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 2 мм, гептан/iPrOAc, 1:1) и элюировали в ДХМ/iPrOAc 1:1 с получением 15 мг соединения, указанного в заголовке, в виде белого осадка. MS (ESI, m/z): 357.1 [M+H]+.
Пример 7
Этил 2-[4-циклопропил-3-(циклопропилметокси)бензамидо]-2-этилбутаноат
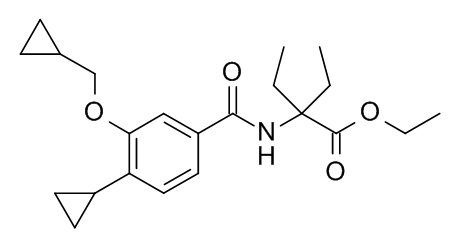
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту и этил 2-амино-2-этилбутаноата гидрохлорид (CAN 1135219-29-2) в качестве исходных веществ. Реакционную смесь перемешивали в течение 4 дней при комнатной температуре, влили в 1 M HCl/смеси лед/вода/солевой раствор (25 мл) и экстрагировали с помощью EtOAc (2×30 мл). Органические слои объединили и промыли водой со льдом/солевым раствором (25 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 122 мг желтого осадка. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 2×2.0 мм, гептан/EtOAc 4:1) и элюировали в ДХМ/EtOAc 1:1 с получением 30 мг соединения, указанного в заголовке, в виде белого осадка. MS: 374.3 [M+H]+.
Пример 8
2-[4-циклопропил-3-(циклопропилметокси)бензамидо]-2-этилбутановая кислота
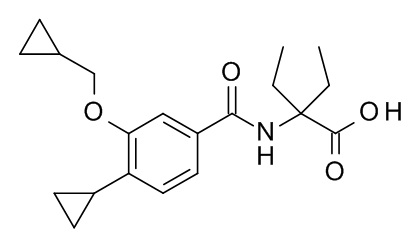
Смесь этил 2-[4-циклопропил-3-(циклопропилметокси)бензамидо]-2-этилбутаноата (Пример 7; 25 мг, 66.9 мкмоль) и гидроксида натрия (268 мкл, 268 мкмоль) в ТГФ (266 мкл) и MeOH (266 мкл) перемешивали при 100°C в течение 40 ч. Реакционную смесь влили в смесь лед/вода/солевой раствор/1 н HCl (25 мл) и экстрагировали с помощью EtOAc (2×30 мл). Объединенные экстракты промыли водой со льдом/солевым раствором (25 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 22 мг соединения, указанного в заголовке, в виде светло-желтого осадка. MS: 344.3 [M-H]-.
Пример 9
4-циклопропил-3-(циклопропилметокси)-N-[3-(3,3-дифторазетидин-1-карбонил)пентан-3-ил]бензамид
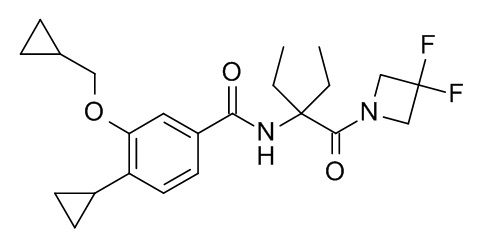
Смесь 2-(4-циклопропил-3-(циклопропилметокси)бензамидо)-2-этилбутановой кислоты (Пример 8; 12 мг, 34.7 мкмоль), 3,3-дифторазетидин гидрохлорида (CAN 288315-03-7; 5.4 мг, 41.7 мкмоль), 1-гидроксибензотриазол гидрата (10.6 мг, 69.5 мкмоль) и DIEA (18 мг, 23.8 мкл, 139 мкмоль) в ДМФ (120 мкл) перемешивали в течение 20 ч при комнатной температуре. Реакционную смесь влили в воду со льдом/солевой раствор/1 мл 1 н HCl (20 мл) и экстрагировали с помощью EtOAc (2×30 мл). Объединенные экстракты промыли водой со льдом/солевым раствором (20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 22 мг светло-желтого осадка. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 1 мм, гептан/EtOAc 1:1) и элюировали в ДХМ/EtOAc 1:1 с получением 7 мг соединения, указанного в заголовке, в виде белого осадка. 421.2 [M+H]+.
Пример 10
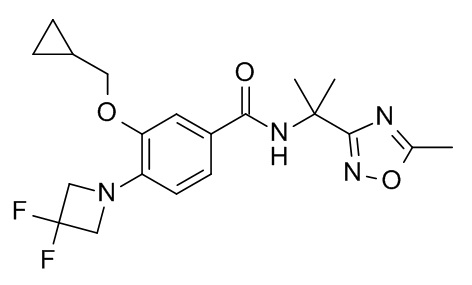
3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)-N-[2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид
a) 4-бромо-3-циклопропилметокси-бензойной кислоты метиловый эфир
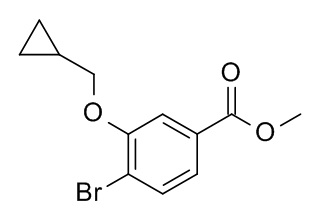
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3a, используя метил 4-бромо-3-гидроксибензоат (CAN 106291-80-9) и (бромометил)циклопропан (CAN 7051-34-5). MS: 285.0 [M+H]+.
b) 3-циклопропилметокси-4-(3,3-дифтор-азетидин-1-ил)-бензойной кислоты метиловый эфир
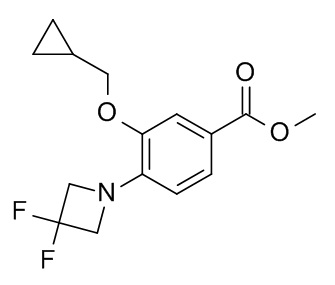
Метил 4-бромо-3-(циклопропилметокси)бензоат (500 мг, 1.75 ммоль) растворили в толуоле (28 мл). 3,3-Дифторазетидина гидрохлорид (CAN 288315-03-7; 250 мг, 1.93 ммоль), карбонат цезия (1.43 г, 4.38 ммоль), рацемический 2,2'-бис(дифенилфосфино)-1,1'-бинафтил (76.4 мг, 123 мкмоль) и палладия (II) ацетат (19.7 мг, 87.7 мкмоль) добавили в атмосфере аргона. Получившуюся реакционную смесь нагревали до 110°C в течение 16 ч. После охлаждения до комнатной температуры добавили EtOAc (40 мл). Смесь влили в воду со льдом /1 н HCl/солевой раствор (80 мл) и экстрагировали с помощью EtOAc. Органические слои снова промыли солевым раствором, высушили над Na2SO4, отфильтровали и сконцентрировали под вакуумом. Неочищенное вещество очистили с помощью флеш хроматографии flashmaster (силикагель, 50 г, градиент EtOAc в гептане).
c) 3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензойная кислота
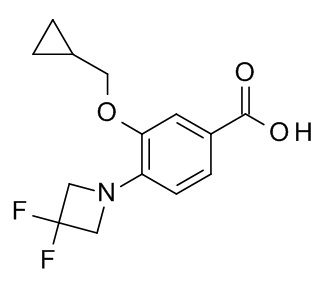
Метил 3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензоат (356 мг, 1.2 ммоль) объединили с тетрагидрофураном (21 мл) и водой (7 мл) с получением бесцветного раствора. Моногидрат гидроксида лития (151 мг, 3.59 ммоль) добавили и получившуюся реакционную смесь перемешивали при кипении с обратным холодильником в течение 24 ч. После охлаждения до комнатной температуры воду (10 мл) добавили. Реакционную смесь подкислили с помощью 1 н HCl (pH=2) и экстрагировали с помощью TBME (100 мл). Водный слой снова экстрагировали с помощью TBME. Объединенные органические фазы высушили над Na2SO4 и сконцентрировали под вакуумом с получением 320 мг соединения, указанного в заголовке, в виде серо-белого осадка. MS: 284.3 [M+H]+.
d) 3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)-N-[2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид
Смесь 3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензойной кислоты (50 мг, 177 мкмоль), DIEA (114 мг, 154 мкл, 883 мкмоль), TBTU (62.3 мг, 194 мкмоль) и 2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина (CAN 1153831-97-0; 27.4 мг, 194 мкмоль) в ДМФ (2 мл) перемешивали при комнатной температуре в течение ночи. После концентрирования под вакуумом (глубокий вакуум, 40°C, 30 мин) остаток растворили в EtOAc (3 мл). 2 н NaOH добавили. Смесь перемешивали в течение 1 минуты и влили в 10 г колонку Varian chemElut. Через 10 минут колонку промыли EtOAc (40 мл) и раствор сконцентрировали под вакуумом. Неочищенное вещество очистили с помощью флеш-хроматографии (силикагель, 10 г, градиент EtOAc в гептане) с получением соединения, указанного в заголовке. MS: 407.18 [M+H]+.
Пример 11
N-[2-(5-амино-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензамид
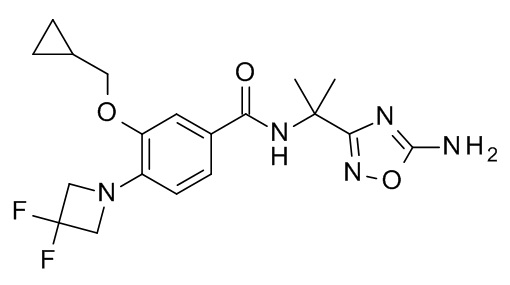
Соединение, указанное в заголовке, синтезировали по аналогии с примером 10d, используя 3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензойную кислоту (Пример 10c) и 1-(5-амино-[1,2,4]оксадиазол-3-ил)-1-метил-этил-аммония хлорид (CAN 1415899-80-7) в качестве исходных веществ. Неочищенную реакционную смесь сконцентрировали под вакуумом. Остаток перемешивали с EtOAc (3 мл). 2 н NaOH добавили. Метанол (1 мл) добавили к слою EtOAc для растворения осадка после разделения. Органическую фазу высушили над Na2SO4, отфильтровали и сконцентрировали под вакуумом. Неочищенный продукт перемешивали с EtOAc при кипении с обратным холодильником и медленно охладили до комнатной температуры. Преципитированное соединение, указанное в заголовке, собрали посредством фильтрации. MS: 408.18 [M+H]+.
Пример 12
N2-[3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензоил]-N-метил-L-лейцинамид
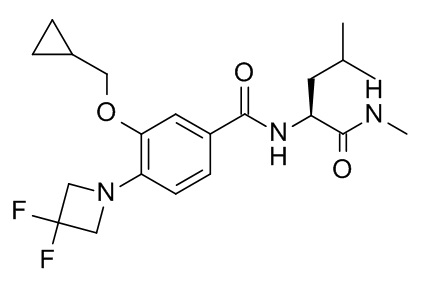
Соединение, указанное в заголовке, синтезировали по аналогии с примером 10d, используя 3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензойную кислоту (Пример 10c) и (S)-2-амино-N,4-диметилпентанамида гидрохлорид (CAN 99145-71-8) в качестве исходных веществ. Неочищенный продукт сконцентрировали под вакуумом (глубокий вакуум, 40°C). Остаток растворили в EtOAc (3 мл). 2 н NaOH добавили. Смесь перемешивали в течение 1 минуты и влили в 10 г колонку Varian chemElut. Через 10 минут колонку промыли EtOAc (40 мл). Неочищенную смесь сконцентрировали под вакуумом и очистили с помощью флеш-хроматографии (силикагель, 10 г, градиент EtOAc в гептане) с получением 35 мг соединения, указанного в заголовке, в виде белого осадка. MS: 410.22 [M+H]+.
Пример 13
3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)-N-[(2S)-1-гидрокси-4-метилпентан-2-ил]бензамид
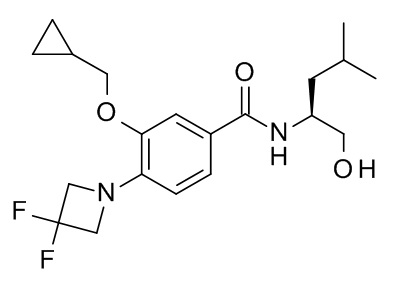
Соединение, указанное в заголовке, синтезировали по аналогии с примером 10d, используя 3-(циклопропилметокси)-4-(3,3-дифторазетидин-1-ил)бензойную кислоту (Пример 10c) и (S)-2-амино-4-метилпентан-1-ол (CAN 7533-40-6) в качестве исходных веществ. Неочищенную смесь сконцентрировали под вакуумом (глубокий вакуум, 40°C). Остаток растворили в EtOAc (3 мл). 2 н NaOH добавили. Раствор перемешивали 1 минуту и влили в 10 г колонку Varian chemElut. Через 10 минут колонку промыли EtOAc (40 мл). Растворитель эвапорировали под вакуумом и неочищенный материал очистили с помощью флеш-хроматографии (силикагель, 10 г, градиент EtOAc в гептане) с получением 37 мг соединения, указанного в заголовке, в виде белого осадка. MS: 383.21 [M+H]+.
Пример 14
3-трет-бутил-5-[4-циклопропил-3-(циклопропилметокси)фенил]-1,2,4-оксадиазол
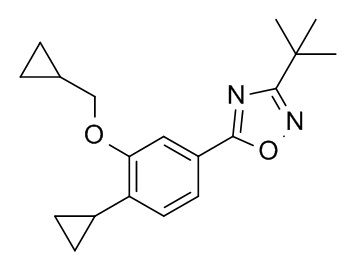
К раствору 4-циклопропил-3-(циклопропилметокси)бензойной кислоты (Пример 3c; 15 мг, 64.6 мкмоль) в безводном ДМФ (0.643 мл) добавили CDI (15.7 мг, 96.9 мкмоль). Смесь перемешивали в течение 30 мин при комнатной температуре. (E)-N'-гидроксипивалимидамид (CAN 1240301-71-6; 11.3 мг, 96.9 мкмоль) добавили и перемешивание при комнатной температуре продолжили в течение 1 ч. Температуру повысили до 100°C. Через 72 ч смесь охладили до комнатной температуры и сразу очистили с помощью препаративной ВЭЖХ без какой-либо обработки с получением 13 мг соединения, указанного в заголовке. MS (ESI) m/e = 313.5 [M+H]+.
Пример 15
N-[3-(2-амино-2-оксоэтил)оксетан-3-ил]-4-циклопропил-3-(циклопропилметокси)бензамид
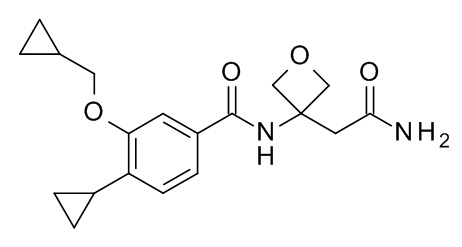
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту (Пример 3c) и 2-(3-амино-оксетан-3-ил)-ацетамид (CAN 1417638-25-5) в качестве исходных веществ в присутствии DIEA в ТГФ. Реакционную смесь влили в воду со льдом/1 н HCl (20 мл) и экстрагировали с помощью EtOAc (2×40 мл). Объединенные органические слои промыли водой со льдом (20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 22 мг белого осадка. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 1.0 мм, гептан/EtOAc 1:1) и элюировали в CH2Cl2/EtOAc 1:1 с получением 10 мг соединения, указанного в заголовке, в виде белого осадка. MS(ESI): m/e = 345.2 [M+H]+.
Пример 16
N-[3-(2-амино-2-оксоэтил)-1,1-диоксотиетан-3-ил]-4-циклопропил-3-(циклопропилметокси)бензамид
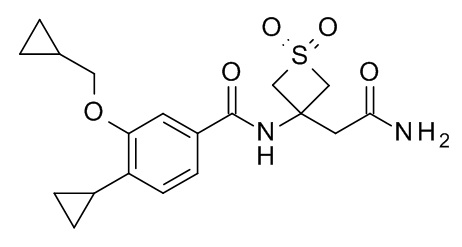
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту (Пример 3c) и 2-(3-амино-1,1-диоксо-тиетан-3-ил)ацетамид (CAN 1613239-56-7) в качестве исходных веществ. Реакционную смесь перемешивали в течение 1 дня при комнатной температуре. Реакционную смесь влили в воду со льдом/1M HCl (20 мл) и экстрагировали с помощью EtOAc (2×30 мл). Объединенные экстракты промыли водой со льдом/солевым раствором (20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 39 мг серо-белого осадка. Неочищенное вещество очистили с помощью препаративной ВЭЖХ с получением 18 мг соединения, указанного в заголовке. MS (ESI) m/e = 393.7 [M+H]+.
Пример 17
1-[4-циклопропил-3-(циклопропилметокси)бензоил]-4,4-дифтор-L-пролинамид
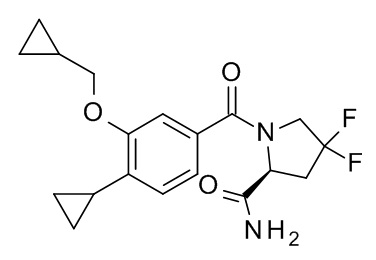
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту (Пример 3c) и (2S)-4,4-дифторпролинамид (CAN 719267-96-6) в качестве исходных веществ в присутствии DIEA в ТГФ. Реакционную смесь перемешивали в течение 1 дня при комнатной температуре. Реакционную смесь влили в смесь вода со льдом/1 н HCl (20 мл) и экстрагировали с помощью EtOAc (2×40 мл). Объединенные экстракты промыли водой со льдом (20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 11 мг бесцветного масла. Неочищенное вещество очистили посредством ТСХ (силикагель, гептан/EtOAc 1:1) и элюировали в CH2Cl2/EtOAc 1:1 с получением 4 мг соединения, указанного в заголовке, в виде бесцветного масла. MS(ESI): m/e = 365.3 [M+H]+.
Пример 18
N-(3-Карбамоилпентан-3-ил)-4-циклопропил-3-(циклопропилметокси)бензамид
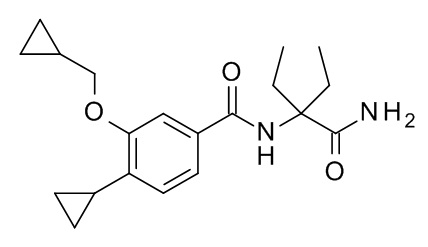
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту (Пример 3c) и 2-амино-2-этилбутирамида гидрохлорид (CAN 17704-75-5) в качестве исходных веществ в присутствии DIEA в ТГФ. Реакционную смесь перемешивали в течение 2 дней при комнатной температуре, влили в смесь вода со льдом/1 н HCl (20 мл) и экстрагировали с помощью EtOAc (2×40 мл). Объединенные экстракты промыли водой со льдом (20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением 24 мг белого осадка. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 1.0 мм гептан/EtOAc 1:1 и элюировали в CH2Cl2/EtOAc 1:1 с получением 11 мг соединения, указанного в заголовке, в виде белого осадка. MS(ESI): m/e = 345.7 [M+H]+.
Пример 19
N2-[4-циклопропил-3-(циклопропилметокси)бензоил]-N-метил-L-лейцинамид
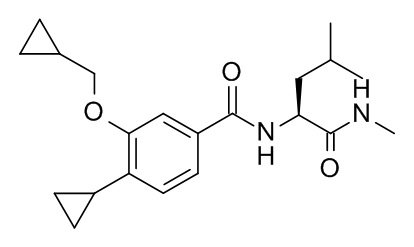
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту (Пример 3c) и (S)-2-амино-N,4-диметилпентанамида гидрохлорид (CAN 99145-71-8) в качестве исходных веществ в присутствии DIEA в ТГФ. Реакционную смесь перемешивали в течение 1 дня при комнатной температуре, влили в смесь вода со льдом/1 н HCl (20 мл) и экстрагировали с помощью EtOAc (2×40 мл). Объединенные экстракты промыли водой со льдом (20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом. Неочищенное вещество очистили посредством ВЭЖХ с получением 11 мг соединения, указанного в заголовке, в виде белого осадка. MS(ESI): m/e = 359.2 [M+H]+.
Пример 20
4-циклопропил-3-(циклопропилметокси)-N-[(2S)-1-(метансульфонил)-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид
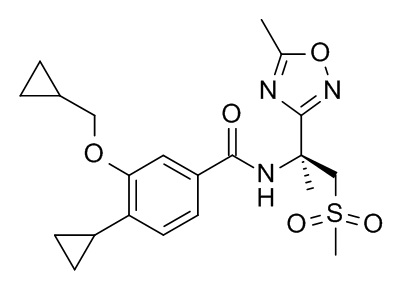
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту (Пример 3c) и (S)-2-(5-метил-1,2,4-оксадиазол-3-ил)-1-(метилсульфонил)пропан-2-амин (CAN 1613239-21-6) в качестве исходных веществ в присутствии DIEA в диоксане. Реакционную смесь перемешивали в течение 1 дня при комнатной температуре, влили в лед /0.1 н HCl (25 мл) и экстрагировали с помощью EtOAc (2×25 мл). Объединенные экстракты промыли смесью вода со льдом/солевой раствор (25 мл) до pH 6, высушили над Na2SO4 и сконцентрировали при пониженном давлении с получением 34 мг оранжевой жидкости. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 2.0 мм, гептан/AcOEt 1:2) и элюировали с помощью EtOAc с получением 6 мг соединения, указанного в заголовке. MS (ESI) m/e = 434.3 [M+H]+.
Пример 21
4-циклопропил-3-(циклопропилметокси)-N-[(2R)-1-(метансульфонил)-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]бензамид
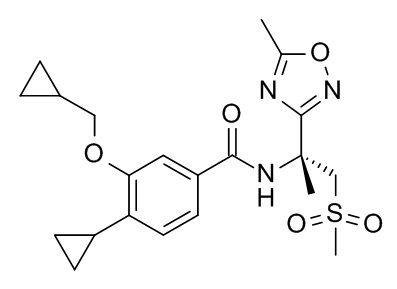
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту (Пример 3c) и (R)-2-(5-метил-1,2,4-оксадиазол-3-ил)-1-(метилсульфонил)пропан-2-амин (CAN 1613239-20-5) в качестве исходных веществ в присутствии DIEA в диоксане. Реакционную смесь перемешивали в течение 1 дня при комнатной температуре, влили в лед /0.1 н HCl (25 мл) и экстрагировали с помощью EtOAc (2×25 мл). Объединенные экстракты промыли водой со льдом/солевым раствором (25 мл) до pH 6, высушили над Na2SO4 и сконцентрировали при пониженном давлении. Неочищенное вещество очистили с помощью препаративной ТСХ (силикагель, 2.0 мм, гептан/AcOEt 1:2) и элюировали с помощью EtOAc с получением 10 мг соединения, указанного в заголовке. MS (ESI) m/e = 434.3 [M+H]+.
Пример 22
5-[4-циклопропил-3-(циклопропилметокси)бензоил]-5-азаспиро[2.4]гептан-6-карбоксамид
Соединение, указанное в заголовке, синтезировали по аналогии с примером 3d, используя 4-циклопропил-3-(циклопропилметокси)бензойную кислоту (Пример 3c) и 5-азаспиро[2.4]гептан-6-карбоксамида гидрохлорид (CAN 1613115-26-6) в качестве исходных веществ в присутствии DIEA в диоксане. Реакционную смесь перемешивали в течение 4 дней при комнатной температуре, влили в лед/0.1 н HCl (25 мл) и экстрагировали с помощью EtOAc (2×25 мл). Объединенные экстракты промыли водой со льдом/солевым раствором (25 мл) до pH 6, высушили над Na2SO4 и сконцентрировали при пониженном давлении. Неочищенный продукт очистили посредством препаративной ВЭЖХ с получением 5 мг соединения, указанного в заголовке. MS (ESI) m/e = 355.3 [M+H]+.
Пример 23
5-трет-бутил-3-[4-циклопропил-3-(2,2,2-трифторэтокси)фенил]-1,2,4-оксадиазол
a) (Z)-4-бромо-N'-гидрокси-3-метоксибензимидамид
К раствору 4-бромо-3-метоксибензонитрила (CAN 120315-65-3; 700 мг, 3.3 ммоль) в EtOH (16.5 мл) добавили гидроксиламина гидрохлорид (344 мг, 4.95 ммоль) и триэтиламин (920 мкл, 6.6 ммоль). Реакционную смесь перемешивали при 60°C в течение ночи. ДХМ (60 мл) добавили и смесь промыли насыщенным водным раствором NaHCO3. Водную фазу снова экстрагировали с помощью этилацетата. Объединенные органические слои высушили над Na2SO4 и эвапорировали досуха. Неочищенный продукт сразу использовали на следующей реакционной стадии без дополнительной очистки. MS (ESI) m/e = 247.1 [M+H]+.
b) 3-(4-бромо-3-метоксифенил)-5-трет-бутил-1,2,4-оксадиазол
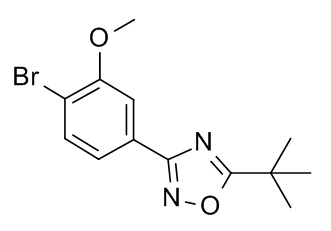
К раствору (Z)-4-бромо-N'-гидрокси-3-метоксибензимидамида (875 мг, 3.39 ммоль) в безводном ДМФ (22.6 мл) добавили пивалоилхлорид (543 мкл, 4.41 ммоль) и триэтиламин (946 мкл, 6.78 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Температуру повысили до 110°C и перемешивание продолжили в течение ночи. Смесь сконцентрировали при пониженном давлении. Этилацетат и водный насыщенный раствор NaHCO3 добавили и слои разделили. Органический слой высушили над Na2SO4 и эвапорировали досуха. Колоночная хроматография на силикагеле с использованием MPLC ISCO с градиентом гептан/этилацетата дала соединение, указанное в заголовке. MS (ESI) m/e = 311.1 [M+H]+.
c) 5-трет-бутил-3-(4-циклопропил-3-метоксифенил)-1,2,4-оксадиазол
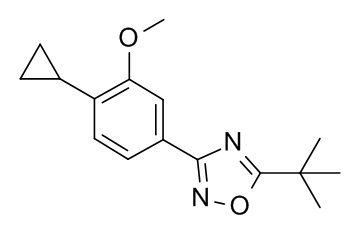
К раствору 3-(4-бромо-3-метоксифенил)-5-трет-бутил-1,2,4-оксадиазола (380 мг, 1.22 ммоль) в толуоле/воде (5.4 мл / 0.7 мл) добавили калия циклопропилтрифторборат (217 мг, 1.47 ммоль), палладия(II) ацетат (11 мг, 0.048 ммоль), карбонат кальция (995 мг, 3.05 ммоль) и бутилди1-адамантилфосфин (26 мг, 0.073 ммоль) в атмосфере аргона. Реакционную смесь перемешивали при 120°C в течение ночи. Этилацетат и / водный насыщенный раствор NaHCO3 добавили. Слои разделили. Органический слой высушили над Na2SO4 и эвапорировали досуха. Неочищенный продукт очистили посредством колоночной хроматографии на силикагеле с использованием MPLC ISCO с помощью градиента гептан/этилацетат с получением соединения, указанного в заголовке. MS (ESI) m/e = 273.2 [M+H]+.
d) 5-(5-трет-бутил-1,2,4-оксадиазол-3-ил)-2-циклопропилфенол
К раствору 5-трет-бутил-3-(4-циклопропил-3-метоксифенил)-1,2,4-оксадиазол (300 мг, 1.1 ммоль) в безводном CH2Cl2 (4.5 мл) в атмосфере аргона добавили 1.0 M раствор BBr3 в CH2Cl2 (1.65 мл, 1.65 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 12 ч, погасили посредством добавления воды и перемешивали в течение 10 мин. Водный насыщенный раствор NH4Cl добавили и слои разделили. Водную фазу снова экстрагировали с помощью CH2Cl2. Органические фазы объединили, высушили над MgSO4 и эвапорировали досуха с получением 255 мг соединения, указанного в заголовке. MS (ESI) m/e = 259.2 [M+H]+.
e) 5-трет-бутил-3-[4-циклопропил-3-(2,2,2-трифторэтокси)фенил]-1,2,4-оксадиазол
К раствору 5-(5-трет-бутил-1,2,4-оксадиазол-3-ил)-2-циклопропилфенола (44 мг, 0.17 ммоль) в безводном ДМФ (1.1 мл) добавили карбонат кальция (166 мг, 0.511 ммоль) и 2,2,2-трифторэтила трифторметансульфонат (CAN 6226-25-1; 35 мкл, 0.256 ммоль). Смесь перемешивали в течение 4 ч при комнатной температуре. Растворитель частично удалили при пониженном давлении. Воду и этилацетат добавили, слои разделили и органический слой высушили над MgSO4. Эвапорация растворителя сопровождалась колоночной хроматографией на силикагеле с помощью градиента гептан/этилацетат с получением соединения, указанного в заголовке, в виде бесцветного вязкого масла. MS (ESI) m/e = 341.2 [M+H]+.
Пример 24
5-трет-бутил-3-[4-циклопропил-3-(2,2-дифторэтокси)фенил]-1,2,4-оксадиазол
Соединение, указанное в заголовке, синтезировали по аналогии с примером 23e, используя 5-(5-трет-бутил-1,2,4-оксадиазол-3-ил)-2-циклопропилфенол (Пример 23d) и 2,2-дифторэтил трифторметансульфонат (CAN 74427-22-8) в качестве исходных веществ с получением бесцветного вязкого масла. MS (ESI) m/e = 323.3 [M+H]+.
Пример 25
(-)-4-циклопропил-N-[1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид
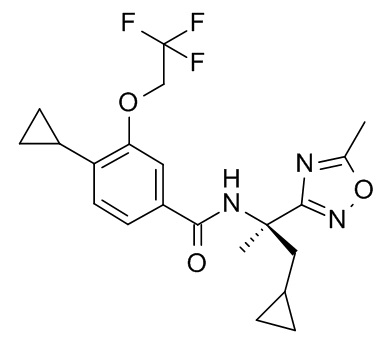
a) Этил 4-бромо-3-(2,2,2-трифторэтокси)бензоат
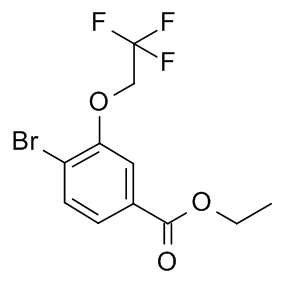
К раствору этил 4-бромо-3-гидроксибензоата (CAN 33141-66-1; 2.09 г, 8.53 ммоль) в ДМФ (57 мл) добавили карбонат кальция (8.34 г, 25.6 ммоль) и 2,2,2-трифторэтил трифторметансульфонат (CAN 6226-25-1; 1.28 мл, 9.38 ммоль). Реакционную смесь перемешивали в течение 8 ч при комнатной температуре. Растворитель удалили при пониженном давлении. Остаток растворили в ДХМ и промыли водным насыщенным раствором NaHCO3. Слои разделили, органический слой высушили над MgSO4 и продули досуха. Неочищенный продукт очистили посредством колоночной хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением 2.7 г соединения, указанного в заголовке. MS m/e = 326 [M]+.
b) Этил 4-циклопропил-3-(2,2,2-трифторэтокси)бензоат
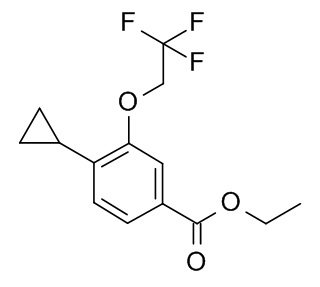
К раствору этил 4-бромо-3-(2,2,2-трифторэтокси)бензоата (2.46 г, 7.52 ммоль) в толуоле/воде (33 мл / 4.4 мл) добавили калия циклопропилтрифторборат (1.34 г, 9.02 ммоль), палладия(II) ацетат (67.5 мг, 0.301 ммоль), карбонат кальция (6.13 г, 18.8 ммоль) и бутилди1-адамантилфосфин (162 мг, 0.451 ммоль) в атмосфере аргона. Смесь перемешивали при 120°C в течение 12 ч. Этилацетат и насыщенный водный раствор NaHCO3 добавили. Слои разделили. Органический слой высушили над Na2SO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии на силикагеле с гептан/этилацетатом с получением соединения, указанного в заголовке. MS (ESI) m/e = 289.2 [M+H]+.
c) 4-циклопропил-3-(2,2,2-трифторэтокси)бензойная кислота
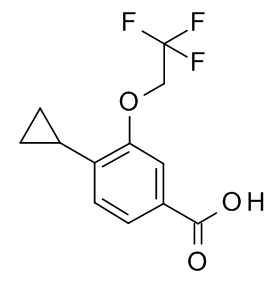
К раствору этил 4-циклопропил-3-(2,2,2-трифторэтокси)бензоата (1.635 г, 5.67 ммоль) в диоксане/воде 1/1 (38 мл) добавили LiOH×H2O (476 мг, 11.3 ммоль). Смесь перемешивали при комнатной температуре в течение 12 ч. 1M водный раствор HCl и этилацетат/этанол (3/1) добавили. Слои разделили. Органический слой высушили над MgSO4 и эвапорировали. Остаток очистили посредством колоночной хроматографии на силикагеле с использованием ЖХСД ISCO с помощью градиента гептан/этилацетат с получением соединения, указанного в заголовке. MS (ESI) m/e = 259.1 [M-H]-.
d) (-)-4-циклопропил-N-[1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид
К раствору 4-циклопропил-3-(2,2,2-трифторэтокси)бензойной кислоты (100 мг, 0.384 ммоль) в ДМФ (2.5 мл) добавили DIEA (134 мкл, 0.769 ммоль) и TBTU (148 мг, 0.461 ммоль). Смесь перемешивали в течение 5 мин при комнатной температуре. 1,2,4-Оксадиазол-3-метанамин, α-(циклопропилметил)-α,5-диметил-, гидрохлорид (CAN 1415900-39-8; 92 мг, 0.423 ммоль добавили и перемешивание продолжили в течение 3 ч. Растворитель удалили при пониженном давлении. Остаток растворили в этилацетате и промыли насыщенным водным раствором NaHCO3. Слои разделили. Органический слой высушили над Na2SO4 и эвапорировали досуха. Остаток очистили посредством колоночной хроматографии на силикагеле с помощью градиента гептан/этилацетат. Энантиомеры разделили посредством хиральной препаративной ВЭЖХ с получением соединения, указанного в заголовке. MS (ESI) m/e = 424.3 [M+H]+.
Пример 26
(+)-4-циклопропил-N-[1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид
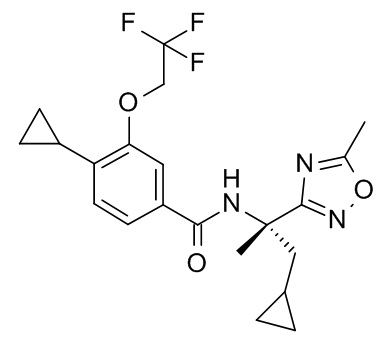
Соединение, указанное в заголовке, синтезировали по аналогии с примером 25d, используя 4-циклопропил-3-(2,2,2-трифторэтокси)бензойную кислоту (Пример 25c) и 1,2,4-оксадиазол-3-метанамин, α-(циклопропилметил)-α,5-диметил-, гидрохлорид (CAN 1415900-39-8) в качестве исходных веществ. Энантиомеры разделили посредством хиральной препаративной ВЭЖХ с получением соединения, указанного в заголовке. MS (ESI) m/e = 424.3 [M+H]+.
Пример 27
N-[3-(2-амино-2-оксоэтил)-1,1-диоксотиетан-3-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид
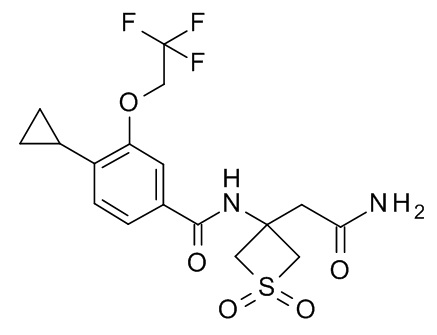
Соединение, указанное в заголовке, синтезировали по аналогии с примером 25d, используя 4-циклопропил-3-(2,2,2-трифторэтокси)бензойную кислоту (Пример 25c) и 2-(3-амино-1,1-диоксо-тиетан-3-ил)ацетамид (CAN 1613239-56-7) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением соединения, указанного в заголовке. MS (ESI) m/e = 421.2 [M+H]+.
Пример 28
4-циклопропил-N-[(2R)-1-(метансульфонил)-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид
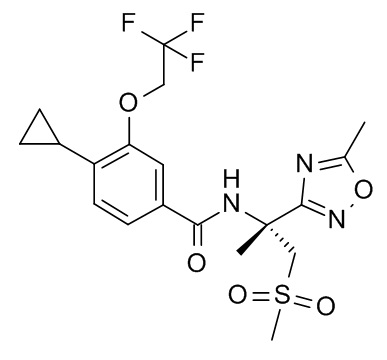
Соединение, указанное в заголовке, синтезировали по аналогии с примером 25d, используя 4-циклопропил-3-(2,2,2-трифторэтокси)бензойную кислоту (Пример 25c) и (R)-2-(5-метил-1,2,4-оксадиазол-3-ил)-1-(метилсульфонил)пропан-2-амин (CAN 1613239-20-5) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением соединения, указанного в заголовке. MS (ESI) m/e = 462.2 [M+H]+.
Пример 29
4-циклопропил-N-[(2S)-1-(метансульфонил)-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2,2-трифторэтокси)бензамид
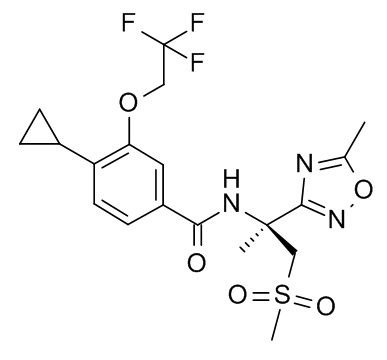
Соединение, указанное в заголовке, синтезировали по аналогии с примером 25d, используя 4-циклопропил-3-(2,2,2-трифторэтокси)бензойную кислоту (Пример 25c) и (S)-2-(5-метил-1,2,4-оксадиазол-3-ил)-1-(метилсульфонил)пропан-2-амин (CAN 1613239-21-6) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением соединения, указанного в заголовке. MS (ESI) m/e = 462.2 [M+H]+.
Пример 30
5-трет-бутил-3-[4-циклопропил-3-(2-фторэтокси)фенил]-1,2,4-оксадиазол
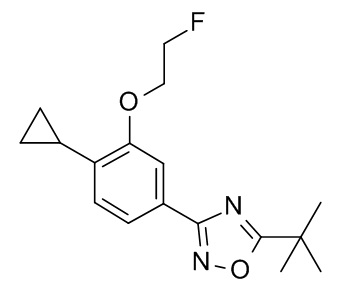
Соединение, указанное в заголовке, синтезировали по аналогии с примером 23e, используя 5-(5-трет-бутил-1,2,4-оксадиазол-3-ил)-2-циклопропилфенол (Пример 23d) и 1-фтор-2-йодэтан (CAN 762-51-6) в качестве исходных веществ. MS (ESI) m/e = 305.2 [M+H]+.
Пример 31
(-)-4-циклопропил-N-[1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2-дифторэтокси)бензамид
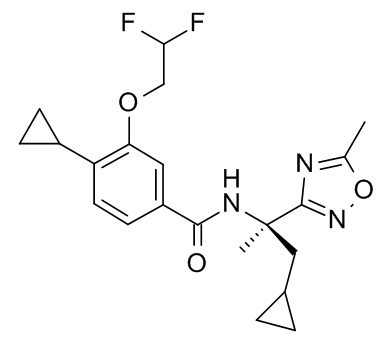
Соединение, указанное в заголовке, синтезировали по аналогии с примером 25d, используя 4-циклопропил-3-(2,2-дифторэтокси)бензойную кислоту, полученную ранее по аналогии с примером 25 с использованием этил 4-бромо-3-гидроксибензоата (CAN 33141-66-1) и 2,2-дифторэтил трифторметансульфоната (CAN 74427-22-8) на стадии a, и 1,2,4-оксадиазол-3-метанамина,α-(циклопропилметил)-α,5-диметил-, гидрохлорида (CAN 1415900-39-8) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением смеси энантиомеров. Разделение энантиомеров посредством хиральной препаративной ВЭЖХ дало соединение, указанное в заголовке. MS (ESI) m/e = 406.2 [M+H]+.
Пример 32
(+)-4-циклопропил-N-[1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2,2-дифторэтокси)бензамид
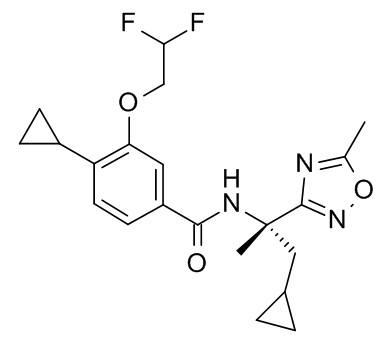
Соединение, указанное в заголовке, синтезировали по аналогии с примером 31, используя 4-циклопропил-3-(2,2-дифторэтокси)бензойную кислоту и 1,2,4-оксадиазол-3-метанамин, α-(циклопропилметил)-α,5-диметил-, гидрохлорид (CAN 1415900-39-8) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением смеси энантиомеров. Разделение энантиомеров посредством хиральной препаративной ВЭЖХ дало соединение, указанное в заголовке. MS (ESI) m/e = 406.2 [M+H]+.
Пример 33
(-)-4-циклопропил-N-[(1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2-фторэтокси)бензамид
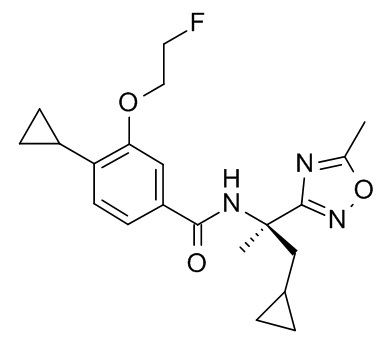
Соединение, указанное в заголовке, синтезировали по аналогии с примером 25d, используя 4-циклопропил-3-(2-фторэтокси)бензойную кислоту, полученную ранее по аналогии с примером 25 с использованием этил 4-бромо-3-гидроксибензоата (CAN 33141-66-1) и 1-фтор-2-йодэтана (CAN 762-51-6) на стадии a, и 1,2,4-оксадиазол-3-метанамина, α-(циклопропилметил)-α,5-диметил-, гидрохлорида (CAN 1415900-39-8) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением смеси энантиомеров. Разделение энантиомеров посредством хиральной препаративной ВЭЖХ дало соединение, указанное в заголовке. MS (ESI) m/e = 388.2 [M+H]+.
Пример 34
(+)-4-циклопропил-N-[1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(2-фторэтокси)бензамид
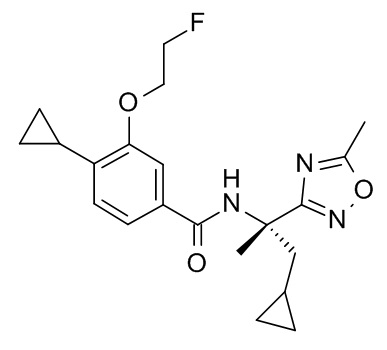
Соединение, указанное в заголовке, синтезировали по аналогии с примером 33, используя 4-циклопропил-3-(2-фторэтокси)бензойную кислоту и 1,2,4-оксадиазол-3-метанамин, α-(циклопропилметил)-α,5-диметил-, гидрохлорид (CAN 1415900-39-8) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением смеси энантиомеров. Разделение энантиомеров посредством хиральной препаративной ВЭЖХ дало соединение, указанное в заголовке. MS (ESI) m/e = 388.2 [M+H]+.
Пример 35
(-)-N-[4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид
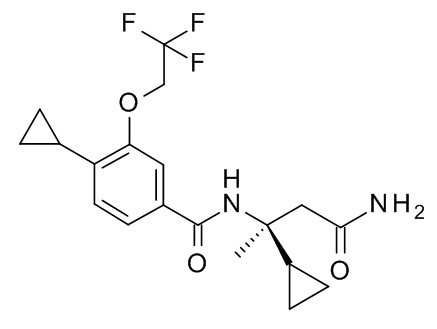
Соединение, указанное в заголовке, синтезировали по аналогии с примером 25d, используя 4-циклопропил-3-(2,2,2-трифторэтокси)бензойную кислоту (Пример 25c) и 3-амино-3-циклопропил-бутанамид (CAN 1534510-01-4) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением смеси энантиомеров. Разделение энантиомеров посредством хиральной препаративной ВЭЖХ дало соединение, указанное в заголовке. MS (ESI) m/e = 385.2 [M+H]+.
Пример 36
(+)-N-[4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид
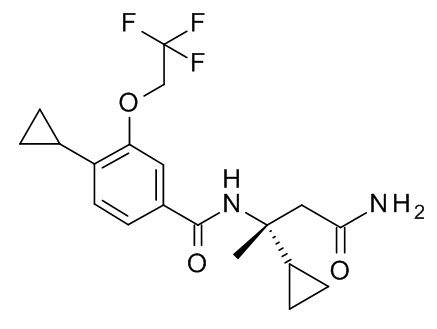
Соединение, указанное в заголовке, синтезировали по аналогии с примером 25d, используя 4-циклопропил-3-(2,2,2-трифторэтокси)бензойную кислоту (Пример 25c) и 3-амино-3-циклопропил-бутанамид (CAN 1534510-01-4) в качестве исходных веществ. Неочищенный продукт очистили с помощью флеш-хроматографии на силикагеле с использованием градиента гептан/этилацетат с получением смеси энантиомеров. Разделение энантиомеров посредством хиральной препаративной ВЭЖХ дало соединение, указанное в заголовке. MS (ESI) m/e = 385.2 [M+H]+.
Пример 37
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-[(пропан-2-ил)окси]бензамид
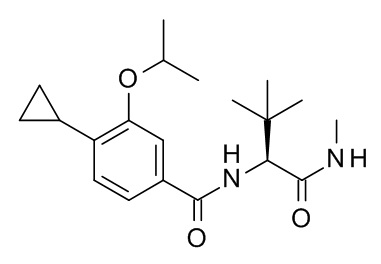
a) Метил 4-бромо-3-(пропан-2-илокси)бензоат
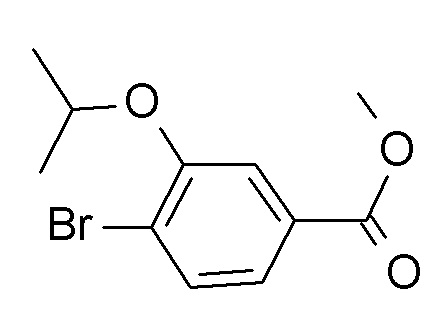
К перемешанному раствору метил 4-бромо-3-гидроксибензоата (CAN 106291-80-9; 4 г, 17.3 ммоль) в ТГФ (100 мл), пропан-2-ол (2 мл, 25.96 ммоль), трифенилфосфин (6.83 г, 25.96 ммоль) и диизопропилазодикарбоксилат (DIAD; 5.14 мл, 25.96 ммоль) добавили при 25°C. Реакционную смесь перемешивали при 25°C в течение 15 h. Летучие компоненты реакционной смеси удалили при пониженном давлении с получением неочищенного продукта, который очистили посредством колоночной хроматографии с использованием 10% этилацетата в гексане в качестве элюента с получением продукта, указанного в заголовке (4 г, 85%) в виде светло-красной жидкости.
b) Метил 4-циклопропил-3-(пропан-2-илокси)бензоат
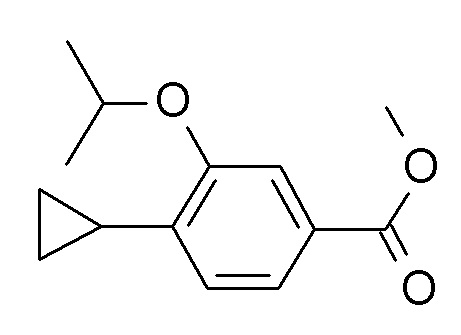
Метил 4-бромо-3-(пропан-2-илокси)бензоат (3 г, 10.98 ммоль), циклопропилбороновую кислоту (1.2 г, 14.27 ммоль) и K3PO4 (5.83 г, 27.45 ммоль) растворили в смеси толуол-вода (60 мл / 2.5 мл) и смесь дегазировали азотом в течение 30 мин. Палладия(II)ацетат (250 мг, 1.09 ммоль) и трициклогексилфосфин (308 мг, 1.09 ммоль) добавили. Смесь дегазировали аргоном в течение 20 мин и затем нагревали до 100°C в течение 15 ч. Реакционную смесь разбавили водой (50 мл) и экстрагировали с помощью этилацетата (3×100 мл). Объединенные органические слои промыли солевым раствором и высушили над безводным Na2SO4. Растворитель удалили при пониженном давлении с получением неочищенного продукта, который очистили посредством колоночной хроматографии combiflash с использованием 15% этилацетата в гексане в качестве элюента с получением соединения, указанного в заголовке (2 г, 78%) в виде светло-желтой жидкости.
c) 4-циклопропил-3-(пропан-2-илокси)бензойная кислота
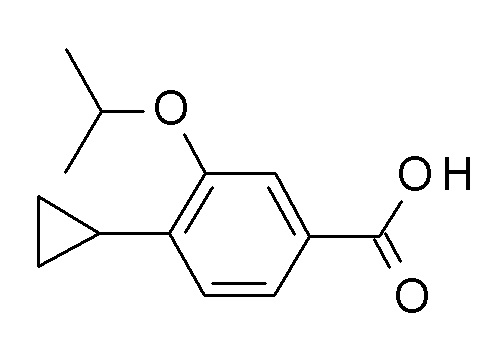
К перемешанному раствору метил 4-бромо-3-(пропан-2-илокси)бензоата (2.1 г, 8.97 ммоль) в диоксане/воде 1/1 (60 мл) добавили LiOH×H2O (753 мг, 17.94 ммоль) при 25°C. Реакционную смесь перемешивали при 25°C в течение 15 ч и затем довели до pH 2-3 посредством добавления 1M водного раствора HCl. Экстрагирование этилацетатом (3×50 мл) сопровождалось впоследствии промывкой объединенных органических слоев солевым раствором и высушиванием над Na2SO4. Удаление растворителя при пониженном давлении дало неочищенный продукт, который очистили посредством колоночной хроматографии combiflash с использованием 40% этилацетата в гексане в качестве элюента с получением соединения, указанного в заголовке (1.4 г, 71%) в виде серо-белого осадка. MS (ESI) m/e = 219.0 [M-H]-.
d) 4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-[(пропан-2-ил)окси]бензамид
К перемешанному раствору 4-циклопропил-3-(пропан-2-илокси)бензойной кислоты (100 мг, 0.45 ммоль) в ДМФ (2.5 мл) добавили DIEA (0.3 мл, 1.81 ммоль) и 2-хлор-1-метилпиридиния йодид (290 мг, 1.13 ммоль). Смесь перемешивали в течение 1.5 ч при 25°C. (2S)-2-амино-N,3,3-триметилбутанамид (CAN 89226-12-0; 78.7мг, 0.55ммоль) добавили и перемешивание продолжили при 25°C в течение 16 ч. Реакционную смесь разбавили водой (20 мл) и экстрагировали с помощью этилацетата (2×50 мл). Объединенные органические слои промыли солевым раствором (50 мл), высушили над безводным Na2SO4 и сконцентрировали под вакуумом с получением неочищенного продукта, который очистили с помощью препаративной ВЭЖХ с получением продукта, указанного в заголовке (13.5 мг, 9%), в виде серо-белого осадка. MS (ESI) m/e = 347.1 [M+H]+.
Пример 38
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-[(пропан-2-ил)окси]бензамид или энантиомер
a) 2-циклопропил-N-метокси-N-метилацетамид
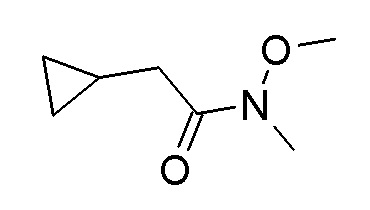
К перемешанному раствору циклопропил-уксусной кислоты (40 г, 400 ммоль) в ДХМ (400 мл) добавили CDI (70 г 431.6 ммоль) по порциям и реакционную смесь перемешивали в течение 2 ч при 25°C. O,N-диметил-гидроксиламина гидрохлорид (39.76 г, 407.6 ммоль) добавили за одну порцию. Реакционную смесь перемешивали в течение 12 ч при 25°C, влили в ледяную воду (300 мл) и экстрагировали с помощью ДХМ (2×200 мл). Объединенный ДХМ слой промыли солевым раствором (200 мл), высушили над безводным Na2SO4 и сконцентрировали при пониженном давлении. Неочищенный продукт очистили посредством колоночной хроматографии combiflash с получением соединения, указанного в заголовке (45 г, 79%), в виде бесцветной жидкости.
b) 1-Циклопропилпропан-2-он
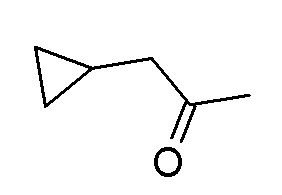
К перемешанному раствору 2-циклопропил-N-метокси-N-метилацетамида (25 г, 174.9 ммоль) в безводном диэтиловом эфире (125 мл) добавили метиллитий (1.6 M раствор в эфире; 120 мл, 192.3 ммоль) при -15°C в течение 30 мин. Реакционную смесь перемешивали в течение 1.5 ч при 0°C, погасили насыщенным водным раствором NH4Cl (100 мл) и экстрагировали с помощью диэтилового эфира (2×300 мл). Объединенный эфирный слой промыли солевым раствором (200 мл), высушили над безводным Na2SO4 и сконцентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке (52 г) в виде светло-желтой жидкости, которую использовали на следующей стадии без дополнительной очистки.
c) 2-амино-3-циклопропил-2-метилпропаннитрил
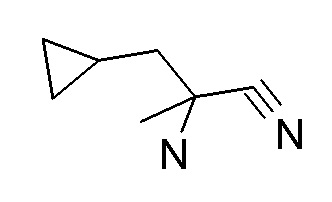
К перемешанному раствору 1-циклопропилпропан-2-она (36 г, 367 ммоль) в этаноле (360 мл) добавили NH4OH (25% в воде; 360 мл) при 25°C и хлорид аммония (20 г, 374 ммоль). Реакционную смесь перемешивали при 25°C в течение 1 ч. Цианид калия (37 г, 572 ммоль) добавили по порциям и перемешивание продолжили в течение 12 ч. Смесь сконцентрировали при пониженном давлении, разбавили водой (500 мл) и экстрагировали с помощью этилацетата (3×200 мл). Объединенный органический слой промыли насыщенным раствором сульфата железа (3×300 мл) и солевым раствором (200 мл), высушили над безводным Na2SO4 и сконцентрировали при пониженном давлении с получением неочищенного соединения, указанного в заголовке (25 г), в виде светло-желтого масла, которое использовали не следующей стадии без дополнительной очистки. MS m/e = 123 [M-H]-.
d) Бензил N-(1-циано-2-циклопропил-1-метилэтил)карбамат
К перемешанному раствору 2-амино-3-циклопропил-2-метилпропаннитрила (24 г, 194 ммоль) в безводном ТГФ (570 мл) добавили DIEA (70 мл, 426 ммоль) и бензилкарбонохлоридат (50% в толуоле; 79.2 мл, 232 ммоль) при 25°C. Смесь перемешивали при 45°C в течение 18 ч и сконцентрировали при пониженном давлении. Остаток растворили в этилацетате (300 мл) и промыли 1 M водным раствором NaHCO3 (250 мл). Слои разделили. Водный слой экстрагировали с помощью этилацетата (2×200 мл). Объединенный органический слой промыли солевым раствором (200 мл), высушили над безводным Na2SO4 и эвапорировали досуха. Неочищенный продукт очистили посредством колоночной хроматографии combiflash с элюцией 10% этилацетатом в гексане с получением продукта, указанного в заголовке (42 г, 44%) в виде бесцветного масла. MS m/e = 258.9 [M]+.
e) Бензил N-[2-циклопропил-1-(N-гидроксикарбамимидоил)-1-метилэтил]карбамат
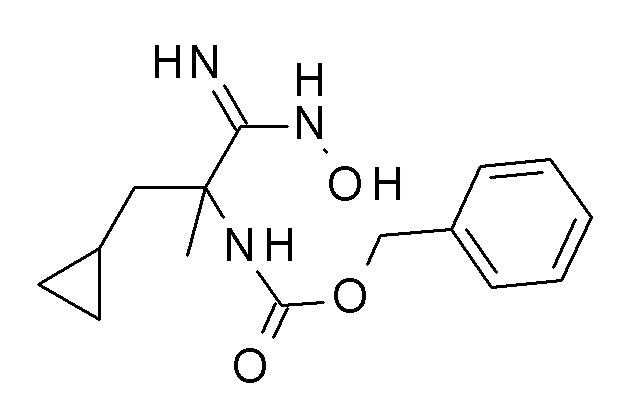
К перемешанному раствору бензил N-(1-циано-2-циклопропил-1-метилэтил)карбамата (42 г, 162.8 ммоль) в этаноле (520 мл) добавили триэтиламин (25 мл, 179.1 ммоль) и гидроксиламина гидрохлорид (11.3 г, 162.5 ммоль). Реакционную смесь перемешивали при 60°C в течение 18 ч. Летучие компоненты удалили при пониженном давлении. Остаток разбавили этилацетатом (300 мл) и водным раствором NaHCO3 (200 мл). Слои разделили. Водный слой экстрагировали с помощью этилацетата (2×200 мл). Объединенный органический слой высушили над безводным Na2SO4 и эвапорировали досуха. Неочищенный продукт очистили посредством колоночной хроматографии combiflash с элюцией 15-20% этилацетатом в гексане с получением продукта, указанного в заголовке (40 г, 84%) в виде белого осадка. MS (ESI) m/e = 292.2 [M+H]+.
f) Бензил N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]карбамат
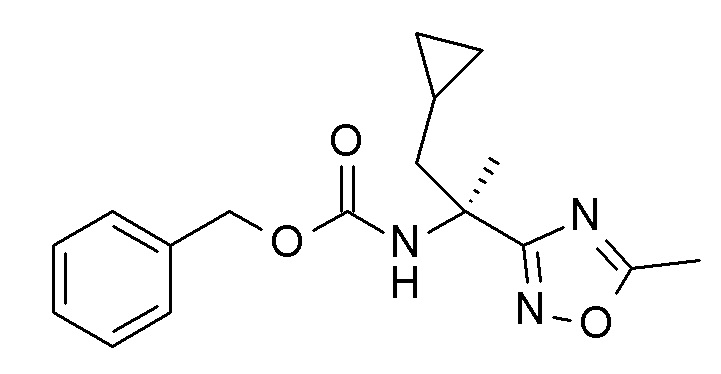
К перемешанному раствору бензил N-[2-циклопропил-1-(N-гидроксикарбамимидоил)-1-метилэтил]карбамата (26 г, 89.3 ммоль) в изопропиловом спирте (466 мл) добавили 1,1 диметокси-N,N-диметилэтанамин (47.6 г, 357.4 ммоль). Реакционную смесь перемешивали в течение 17 ч при 25°C. После охлаждения до 0°C 4 M соляную кислоту в диоксане (112 мл, 447 ммоль) добавили по каплям. Перемешивание продолжили в течение 2 ч при 0°C. Этилацетат (300 мл) добавили и смесь промыли водным раствором 2M карбоната натрия (500 мл). Водный слой экстрагировали с помощью этилацетата (2×300 мл). Объединенный органический слой промыли солевым раствором (200 мл), высушили над безводным Na2SO4 и эвапорировали досуха. Неочищенный продукт очистили посредством колоночной хроматографии combiflash с элюцией 20-30% этилацетатом в гексане с получением рацемического бензил N-[1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]карбамата (25 г, 85%) в виде бесцветного вязкого масла. Хиральным разделением получили соединение, указанное в заголовке (11.5 г, 46%), в виде бесцветного вязкого масла. MS (ESI) m/e = 315.9 [M+H]+.
g) (2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амин
К перемешанному раствору бензил N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]карбамата (11.5 г, 36.5 ммоль) в безводном ДХМ (250 мл) добавили BCl3 (1 M раствор в ДХМ; 186 мл) при 0°C в атмосфере азота. Реакционную смесь перемешивали при 25°C в течение 1.5 ч. Раствор погасили метанолом (30 мл) и H2O (10 мл). Растворитель удалили при пониженном давлении. Остаток растворили в воде (100 мл), подщелачили насыщенным раствором бикарбонат натрия и экстрагировали с помощью ДХМ (2×200 мл). Объединенные экстракты промыли солевым раствором (100 мл), высушили над безводным Na2SO4 и эвапорировали досуха с получением соединения, указанного в заголовке (5.4 г, 82%) в виде светло-коричневой жидкости.
h) (2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорид
К перемешанному раствору (2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина (5.4 г, 29.8 ммоль) в метаноле (50 мл) добавили 4 M раствор соляной кислоты в диоксане (37 мл, 149 ммоль) по каплям при 0°C. Раствор перемешивали в течение 2 ч при 25°C. Летучие компоненты удалили при пониженном давлении. Совместная дистилляция с толуолом (2 x) сопровождалась последующей лиофилизацией с получением продукта, указанного в заголовке (6.2 г, 96%), в виде серо-белого осадка.
i) 4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-[(пропан-2-ил)окси]бензамид или энантиомер
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(пропан-2-илокси)бензойной кислоты (Пример 37c; 110 мг, 0.5 ммоль) и (2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (109 мг, 0.5 ммоль) в виде серо-белого осадка (46 мг, 24%). MS (ESI) m/e = 384.1 [M+H]+.
Пример 39
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-(2-фторэтокси)бензамид
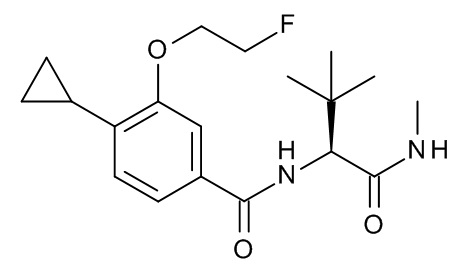
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(2-фторэтокси)бензойной кислоты (Пример 33; 100 мг, 0.45 ммоль) и (2S)-2-амино-N,3,3-триметилбутанамида (CAN 89226-12-0; 66.5 мг, 0.46 ммоль) в виде серо-белого осадка (110 мг, 70%). MS (ESI) m/e = 350.9 [M+H]+.
Пример 40
1-[4-циклопропил-3-(2-фторэтокси)бензоил]-4,4-дифтор-L-пролинамид
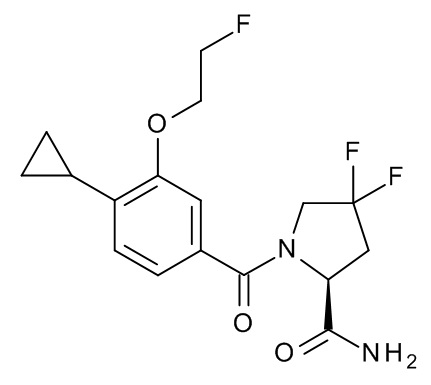
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(2-фторэтокси)бензойной кислоты (Пример 33; 100 мг, 0.45 ммоль) и (2S)-4,4-дифторпирролидин-2-карбоксамида гидрохлорид (CAN 426844-51-1; 86 мг, 0.46 ммоль) в виде серо-белого осадка (63 мг, 46%). MS (ESI) m/e = 356.9 [M+H]+.
Пример 41
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-(2,2,2-трифторэтокси)бензамид
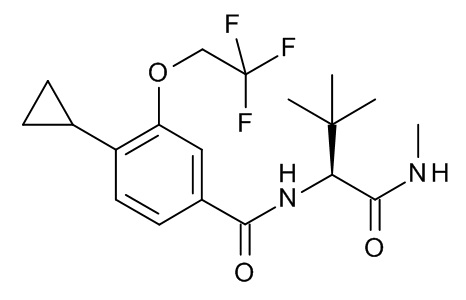
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(2,2,2-трифторэтокси)бензойной кислоты (Пример 25c; 100 мг, 0.38 ммоль) и (2S)-2-амино-N,3,3-триметилбутанамида (CAN 89226-12-0; 67 г, 0.46 ммоль) в виде серо-белого осадка (49 мг, 70%). MS (ESI) m/e = 386.8 [M+H]+.
Пример 42
1-[4-циклопропил-3-(2,2,2-трифторэтокси)бензоил]-4,4-дифтор-L-пролинамид
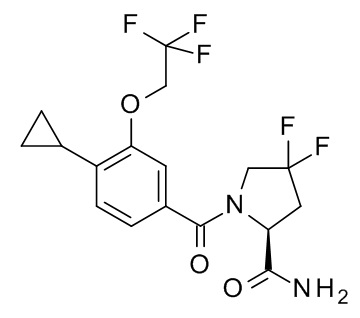
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(2,2,2-трифторэтокси)бензойной кислоты (Пример 25c; 100 мг, 0.38 ммоль) и (2S)-4,4-дифторпирролидин-2-карбоксамида гидрохлорида (CAN 426844-51-1; 86.1 мг, 0.46 ммоль) в виде серо-белого осадка (55 мг, 37%). MS (ESI) m/e = 393.1 [M+H]+.
Пример 43
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-[(пропан-2-ил)окси]бензамид
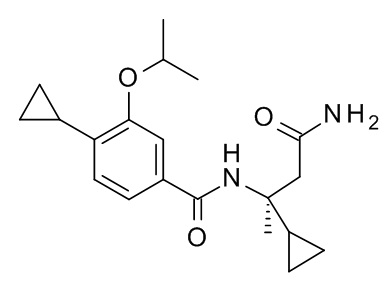
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(пропан-2-илокси)бензойной кислоты (Пример 37c; 100 мг, 0.45 ммоль) и (3S)-3-амино-3-циклопропилбутанамида гидрохлорида (97 мг, 0.54 ммоль) в виде серо-белого осадка (56 мг, 36%). (3S)-3-амино-3-циклопропилбутанамид гидрохлорид получили по аналогии с 3-циклопропил-3-[(2-метилпропан-2-сульфинил)амино]бутановой кислотой (CAN 1534510-01-4) с использованием в качестве исходного вещества (R)-2-метилпропан-2-сульфинамида (CAN 196929-78-9) и 1-циклопропил-этанона (CAN 765-43-5). MS (ESI) m/e = 345.0 [M+H]+.
Пример 44
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-[(пропан-2-ил)окси]бензамид
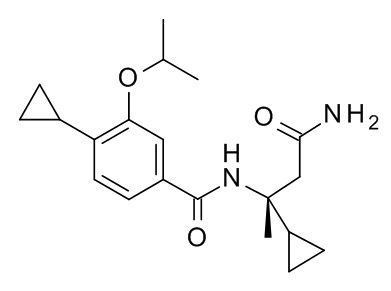
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(пропан-2-илокси)бензойной кислоты (Пример 37c; 100 мг, 0.45 ммоль) и (3R)-3-амино-3-циклопропилбутанамида гидрохлорида (97 мг, 0.54 ммоль) в виде серо-белого осадка (45 мг, 29%). (3R)-3-амино-3-циклопропилбутанамид гидрохлорид получили по аналогии с 3-циклопропил-3-[(2-метилпропан-2-сульфинил)амино]бутановой кислотой (CAN 1534510-01-4) с использованием в качестве исходного вещества (S)-2-метилпропан-2-сульфинамида (CAN 343338-28-3) и 1-циклопропил-этанона (CAN 765-43-5). MS (ESI) m/e = 345.0 [M+H]+.
Пример 45
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2-фторэтокси)бензамид
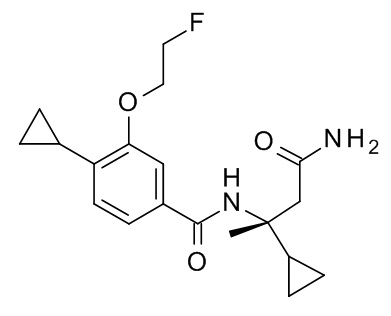
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(2-фторэтокси)бензойной кислоты (Пример 33; 100 мг, 0.45 ммоль) и (3R)-3-амино-3-циклопропилбутанамида гидрохлорида (Пример 44; 96 мг, 0.54 ммоль) в виде серо-белого осадка (60 мг, 39%). MS (ESI) m/e = 348.8 [M+H]+.
Пример 46
3-трет-бутил-5-{4-циклопропил-3-[(пропан-2-ил)окси]фенил}-1,2,4-оксадиазол
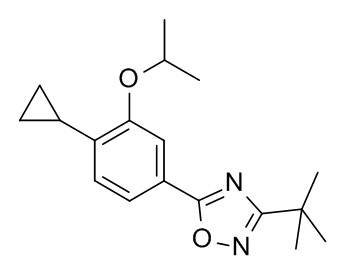
К перемешанному раствору 4-циклопропил-3-(пропан-2-илокси)бензойной кислоты (Пример 37c; 50 мг, 0.22 ммоль) в безводном ДМФ (3 мл) добавили N,N'-дициклогексилкарбодиимид (54 мг, 0.33 ммоль). Смесь перемешивали в течение 30 мин при 25°C. (E)-N'-гидрокси-2,2-диметилпропимидамид (39 мг, 0.33 ммоль) добавили и перемешивание продолжили в течение 1 ч при 25°C. Температуру повышали до 100°C в течение 72 ч. После охлаждения до комнатной температуры концентрированную неочищенную смесь очистили с помощью препаративной ВЭЖХ с получением соединения, указанного в заголовке (34 мг, 51%), в виде бесцветной жидкости. MS (ESI) m/e = 331.2 [M+H]+.
Пример 47
3-трет-бутил-5-[4-циклопропил-3-(3,3-дифторпирролидин-1-ил)фенил]-1,2,4-оксадиазол
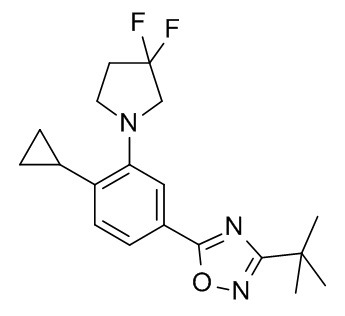
a) Метил 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензоат
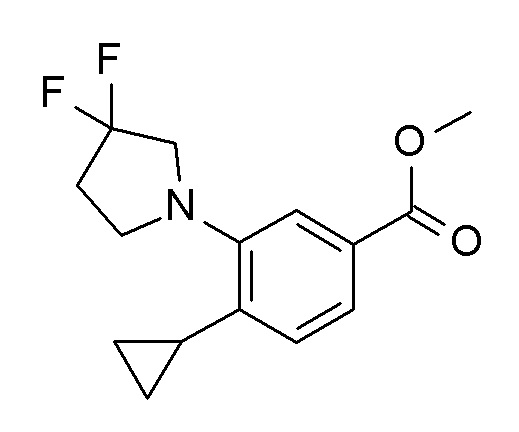
К перемешанному раствору метил 3-бромо-4-циклопропилбензоата (CAN 1131615-05-8; 1 г, 3.92 ммоль) в диоксане (10 мл) добавили 3,3-дифторпирролидина гидрохлорид (1.1 г, 7.84 ммоль) и трет-бутоксид натрия (1.88 г, 19.6 ммоль). Смесь дегазировали азотом в течение 10 мин. Ru-Phos (220 мг, 0.47 ммоль) и Brett-Phos палладоцикл (188 мг, 0.23 ммоль) добавили. Суспензию дегазировали 5 мин., перемешивали при 100°C в течение 45 ч и отфильтровали через слой целита. Сконцентрированный фильтрат очистили препаративной ТСХ с получением продукта, указанного в заголовке (200 мг, 19%) в виде серо-белого осадка. MS (ESI) m/e = 282.2 [M+H]+.
b) 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензойная кислота
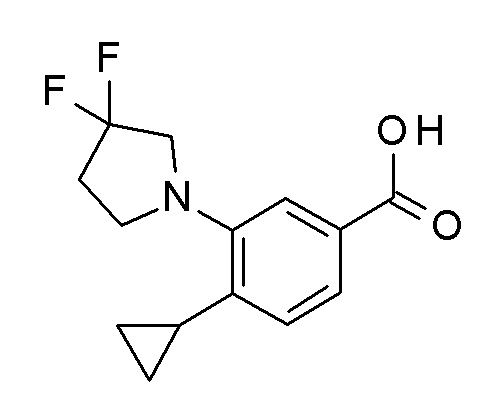
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 37c, с использованием в качестве исходного вещества метил 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензоата (200 мг, 0.71 ммоль) с использованием LiOH×H2O (60 мг, 1.42 ммоль) в виде серо-белого осадка (150 мг, 79%). MS (ESI) m/e = 268.1 [M+H]+.
c) 3-трет-бутил-5-[4-циклопропил-3-(3,3-дифторпирролидин-1-ил)фенил]-1,2,4-оксадиазол
Соединение, указанное в заголовке, синтезировали по аналогии с примером 46 из 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензойной кислоты (50 мг, 0.18 ммоль) и (E)-N'-гидрокси-2,2-диметилпропимидамида (32 мг, 0.28 ммоль) в виде бесцветной жидкости (23 мг, 35%). MS (ESI) m/e = 348.3 [M+H]+.
Пример 48
1-{4-циклопропил-3-[(пропан-2-ил)окси]бензоил}-4,4-дифтор-L-пролинамид
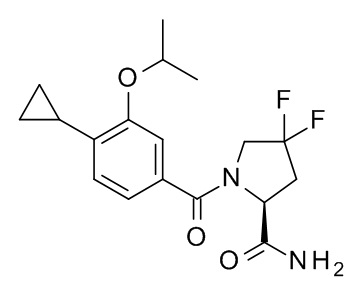
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(пропан-2-илокси)бензойной кислоты (Пример 37c; 100 мг, 0.45 ммоль) и (2S)-4,4-дифторпирролидин-2-карбоксамида гидрохлорида (CAN 426844-51-1; 102 мг, 0.54 ммоль) в виде серо-белого осадка (18 мг, 11%). MS (ESI) m/e = 353.1 [M+H]+.
Пример 49
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-[(пропан-2-ил)окси]бензамид
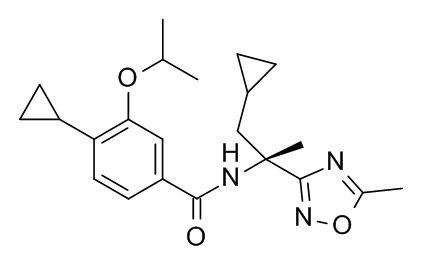
a) Бензил N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]карбамат
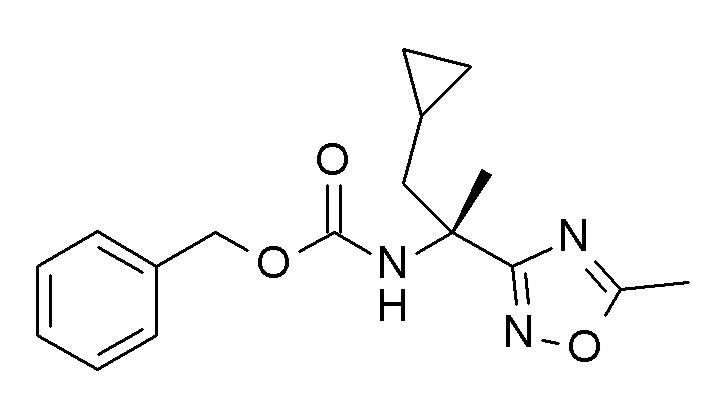
К перемешанному раствору бензил N-[2-циклопропил-1-(N-гидроксикарбамимидоил)-1-метилэтил]карбамата (26 г, 89.3 ммоль) в изопропиловом спирте (466 мл) добавили 1,1-диемтокси-N,N-диметилэтанамин (47.6 г, 357.4 ммоль). Реакционную смесь перемешивали в течение 17 ч при 25°C. После охлаждения до 0°C 4 M соляную кислоту в диоксане (112 мл, 447 ммоль) добавили по каплям. Перемешивание продолжили в течение 2 ч при 0°C. Этилацетат (300 мл) добавили и смесь промыли водным 2M раствором карбоната натрия (500 мл). Водный слой экстрагировали с помощью этилацетата (2×300 мл). Объединенный органический слой промыли солевым раствором (200 мл), высушили над безводным Na2SO4 и эвапорировали досуха. Неочищенный продукт очистили посредством колоночной хроматографии combiflash с элюцией 20-30% этилацетатом в гексане с получением рацемического бензил N-[1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]карбамата (25 г, 85%) в виде бесцветного вязкого масла. Хиральное разделение дало соединение, указанное в заголовке (10.5 г, 42%), в виде бесцветного вязкого масла. MS (ESI) m/e = 316.1 [M+H]+.
b) (2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амин
Соединение, указанное в заголовке (5.7 г, 99%), синтезировали по аналогии с примером 38g с использованием в качестве исходного вещества бензил N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]карбамата в виде светло-коричневой жидкости.
c) (2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорид
Соединение, указанное в заголовке (6.3 г, 92%) синтезировали по аналогии с примером 38h с использованием в качестве исходного вещества (2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина в виде серо-белого осадка.
d) 4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-[(пропан-2-ил)окси]бензамид
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(пропан-2-илокси)бензойной кислоты (Пример 37c; 110 мг, 0.5 ммоль) и (2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (109 мг, 0.5 ммоль) в виде серо-белого осадка (31 мг, 16%). MS (ESI) m/e = 383.9 [M+H]+.
Пример 50
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(6-фторпиридин-3-ил)бензамид
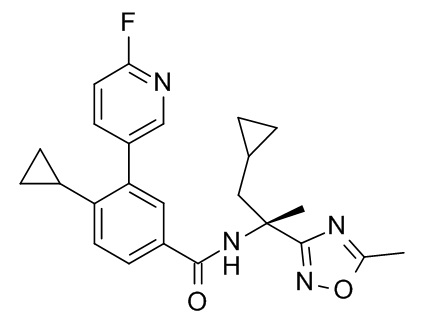
a) Метил 4-циклопропил-3-(6-фторпиридин-3-ил)бензоат
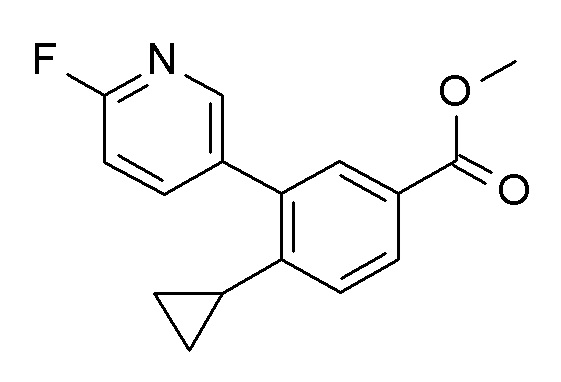
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37b с использованием в качестве исходного вещества метил 3-бромо-4-циклопропилбензоата (CAN 1131615-05-8; 2.0 г, 7.84 ммоль) и (6-фторпиридин-3-ил)бороновой кислоты (2.8 г, 19.6 ммоль) в виде белого осадка (1.4 г, 66%). MS m/e = 271 [M]+.
b) 4-циклопропил-3-(6-фторпиридин-3-ил)бензойная кислота
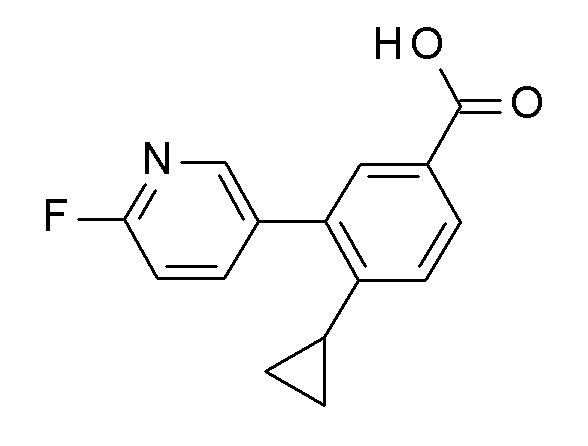
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 37c, с использованием в качестве исходного вещества метил 4-циклопропил-3-(6-фторпиридин-3-ил)бензоата (1.4 г, 5.2 ммоль) с использованием LiOH×H2O (433 мг, 10.3 ммоль) в виде белого осадка (1.3 г, 98%). MS (ESI) m/e = 257.9 [M+H]+.
c) 4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(6-фторпиридин-3-ил)бензамид
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(6-фторпиридин-3-ил)бензойной кислоты (110 мг, 0.43 ммоль) и (2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (Пример 49c; 93 мг, 0.43 ммоль) в виде серо-белого осадка (62 мг, 34%). MS (ESI) m/e = 421.0 [M+H]+.
Пример 51
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид
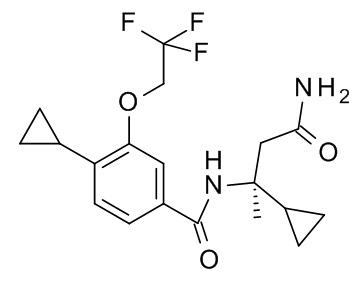
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(2,2,2-трифторэтокси)бензойной кислоты (Пример 25c; 100 мг, 0.38 ммоль) и (3S)-3-амино-3-циклопропилбутанамида гидрохлорида (Пример 43; 82 мг, 0.46 ммоль) в виде серо-белого осадка (50 мг, 34%). MS (ESI) m/e = 384.8 [M+H]+.
Пример 52
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(6-фторпиридин-3-ил)бензамид
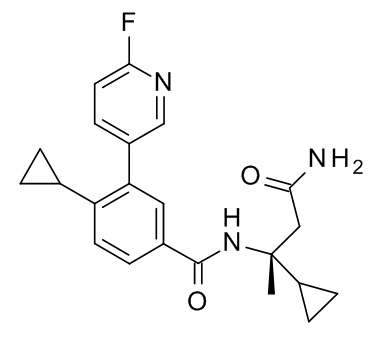
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(6-фторпиридин-3-ил)бензойной кислоты (Пример 50b; 80 мг, 0.31 ммоль) и (3R)-3-амино-3-циклопропилбутанамида гидрохлорида (Пример 44; 67 мг, 0.37 ммоль) в виде серо-белого осадка (65 мг, 55%). MS (ESI) m/e = 381.9 [M+H]+.
Пример 53
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(6-фторпиридин-3-ил)бензамид
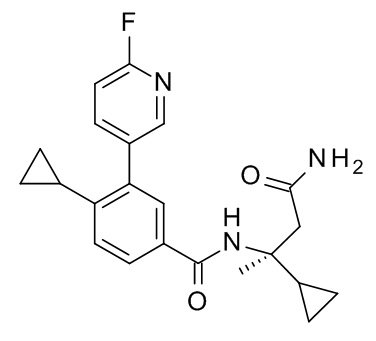
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(6-фторпиридин-3-ил)бензойной кислоты (Пример 50b; 80 мг, 0.31 ммоль) и (3S)-3-амино-3-циклопропилбутанамида гидрохлорида (Пример 43; 67 мг, 0.37 ммоль) в виде серо-белого осадка (68 мг, 57%). MS (ESI) m/e = 381.8 [M+H]+.
Пример 54
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(2-фторэтокси)бензамид
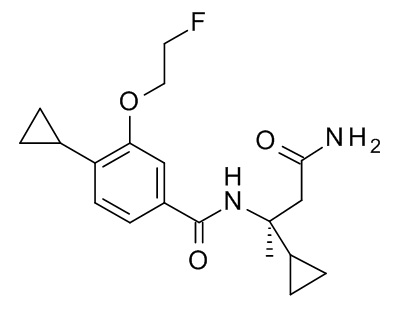
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(2-фторэтокси)бензойной кислоты (Пример 33; 100 мг, 0.45 ммоль) и (3S)-3-амино-3-циклопропилбутанамида гидрохлорида (Пример 43; 96 мг, 0.53 ммоль) в виде серо-белого осадка (75 мг, 42%). MS (ESI) m/e = 349.2 [M+H]+.
Пример 55
1-[4-циклопропил-3-(1-метил-1H-пиразол-5-ил)бензоил]-4,4-дифтор-L-пролинамид
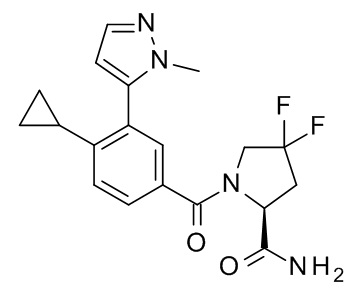
a) Метил 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензоат
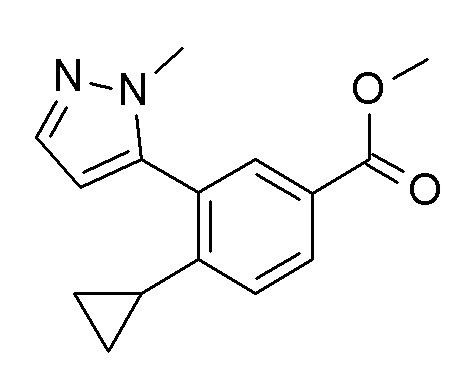
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37b с использованием в качестве исходного вещества метил 3-бромо-4-циклопропилбензоата (CAN 1131615-05-8; 2 г, 7.84 ммоль) и 1-метил-5-(тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразола (2.4 г, 11.8 ммоль) в виде светло-коричневого масла (1.4 г, 70%). MS m/e = 256.5 [M]+.
b) 4-циклопропил-3-(1-метил-1H-пиразол-5-ил)бензойная кислота
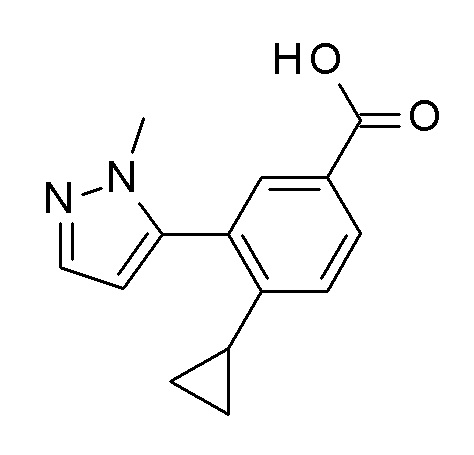
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 37c, с использованием в качестве исходного вещества метил 4-циклопропил-3-(1-метил-1H-пиразол-5-ил)бензоата (2 г, 7.8 ммоль) с использованием LiOH×H2O (655 мг, 15.6 ммоль) в виде серо-белого осадка (1.2 г, 63%). MS (ESI) m/e = 241.0 [M-H]-.
c) 1-[4-циклопропил-3-(1-метил-1H-пиразол-5-ил)бензоил]-4,4-дифтор-L-пролинамид
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(1-метил-1H-пиразол-5-ил)бензойной кислоты (80 мг, 0.33 ммоль) и (2S)-4,4-дифторпирролидин-2-карбоксамида гидрохлорида (CAN 426844-51-1; 57 мг, 0.4 ммоль) в виде серо-белого осадка (50 мг, 40%). MS (ESI) m/e = 375.1 [M+H]+.
Пример 56
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-(1-метил-1H-пиразол-5-ил)бензамид
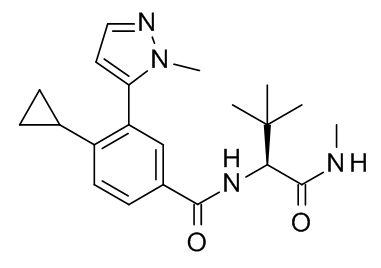
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(1-метил-1H-пиразол-5-ил)бензойной кислоты (Пример 55b; 80 мг, 0.33 ммоль) и (2S)-2-амино-N,3,3-триметилбутанамида (CAN 89226-12-0; 57 г, 0.39 ммоль) в виде серо-белого осадка (72 мг, 59%). MS (ESI) m/e = 369.2 [M+H]+.
Пример 57
1-[4-циклопропил-3-(6-фторпиридин-3-ил)бензоил]-4,4-дифтор-L-пролинамид
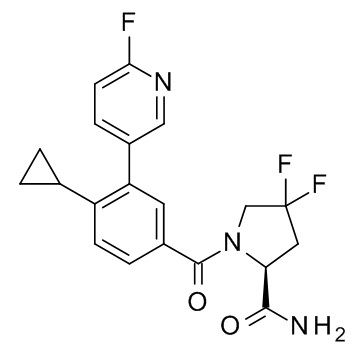
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(6-фторпиридин-3-ил)бензойной кислоты (Пример 50b; 100 мг, 0.39 ммоль) и (2S)-4,4-дифторпирролидин-2-карбоксамида гидрохлорида (CAN 426844-51-1; 87 мг, 0.46 ммоль) в виде серо-белого осадка (65 мг, 43%). MS (ESI) m/e = 390.1 [M+H]+.
Пример 58
4-циклопропил-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]-3-(6-фторпиридин-3-ил)бензамид
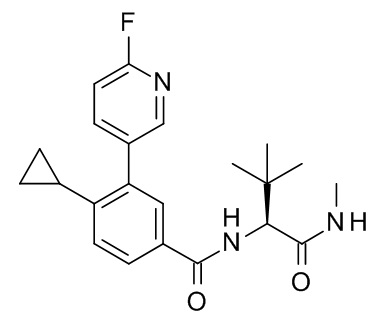
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(6-фторпиридин-3-ил)бензойной кислоты (Пример 50b; 100 мг, 0.39 ммоль) и (2S)-2-амино-N,3,3-триметилбутанамида (CAN 89226-12-0; 67 мг, 0.46 ммоль) в виде серо-белого осадка (70 мг, 47%). MS (ESI) m/e = 384.2 [M+H]+.
Пример 59
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(1-метил-1H-пиразол-5-ил)бензамид
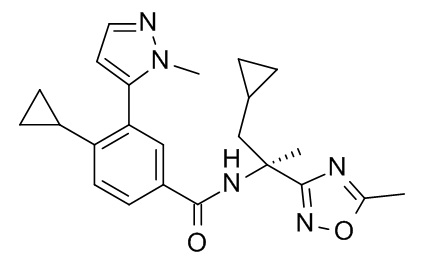
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(1-метил-1H-пиразол-5-ил)бензойной кислоты (Пример 55b; 110 мг, 0.45 ммоль) и (2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (Пример 38h; 99 мг, 0.45 ммоль) в виде серо-белого осадка (73 мг, 40%). MS (ESI) m/e = 404.2 [M-H]-.
Пример 60
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(6-фторпиридин-3-ил)бензамид
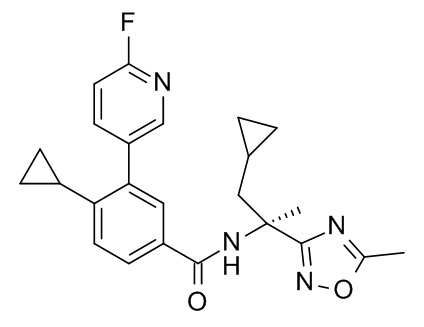
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(6-фторпиридин-3-ил)бензойной кислоты (Пример 50b; 110 мг, 0.43 ммоль) и (2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (Пример 38h; 93 мг, 0.43 ммоль) в виде серо-белого осадка (49 мг, 27%). MS (ESI) m/e = 420.9 [M+H]+.
Пример 61
N-[(2R)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензамид
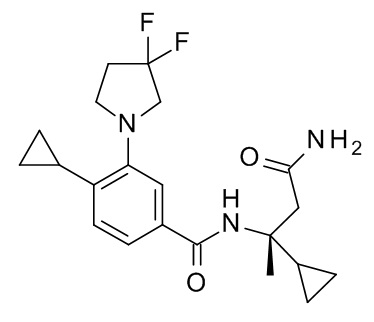
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензойной кислоты (65 мг, 0.24 ммоль) и (3R)-3-амино-3-циклопропилбутанамида гидрохлорида (Пример 44; 52 мг, 0.29 ммоль) в виде серо-белого осадка (19 мг, 20%). MS (ESI) m/e = 392.1 [M+H]+.
Пример 62
N-[(2S)-4-амино-2-циклопропил-4-оксобутан-2-ил]-4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензамид
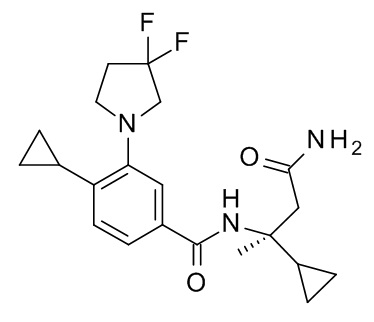
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензойной кислоты (60 мг, 0.22 ммоль) и (3S)-3-амино-3-циклопропилбутанамида гидрохлорида (Пример 43; 40 мг, 0.22 ммоль) в виде серо-белого осадка (24 мг, 27%). MS (ESI) m/e = 391.7 [M+H]+.
Пример 63
4-циклопропил-N-[(2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(3,3-дифторпирролидин-1-ил)бензамид
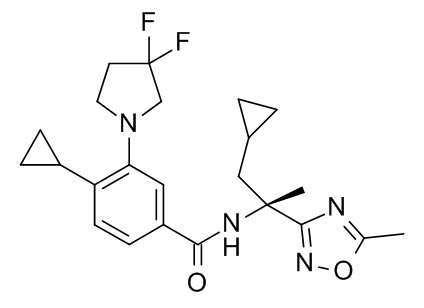
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензойной кислоты (60 мг, 0.22 ммоль) и (2R)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (Пример 49c; 49 мг, 0.22 ммоль) в виде серо-белого осадка (35 мг, 36%). MS (ESI) m/e = 428.8 [M-H]-.
Пример 64
4-циклопропил-N-[(2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-ил]-3-(3,3-дифторпирролидин-1-ил)бензамид
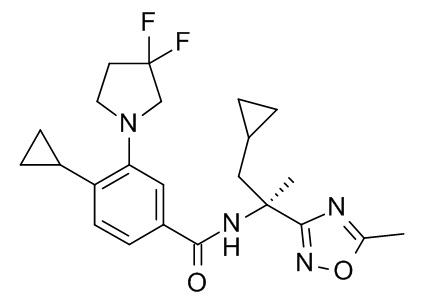
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензойной кислоты (65 мг, 0.24 ммоль) и (2S)-1-циклопропил-2-(5-метил-1,2,4-оксадиазол-3-ил)пропан-2-амина гидрохлорида (Пример 38h; 63 мг, 0.3 ммоль) в виде серо-белого осадка (44 мг, 42%). MS (ESI) m/e = 431.2 [M+H]+.
Пример 65
1-[4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензоил]-4,4-дифтор-L-пролинамид
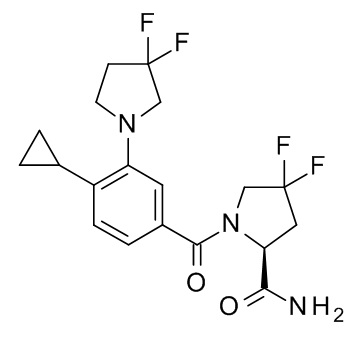
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензойной кислоты (50 мг, 0.18 ммоль) и (2S)-4,4-дифторпирролидин-2-карбоксамида гидрохлорида (CAN 426844-51-1; 42мг, 0.22 ммоль) в виде серо-белого осадка (50 мг, 67%). MS (ESI) m/e = 400.1 [M+H]+.
Пример 66
4-циклопропил-3-(3,3-дифторпирролидин-1-ил)-N-[(2S)-3,3-диметил-1-(метиламино)-1-оксобутан-2-ил]бензамид
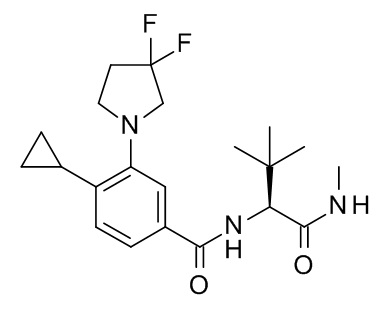
Соединение, указанное в заголовке, синтезировали по аналогии с примером 37d из 4-циклопропил-3-(3,3-дифторпирролидин-1-ил)бензойной кислоты (50 мг, 0.18 ммоль) и (2S)-2-амино-N,3,3-триметилбутанамида (CAN 89226-12-0; 32 мг, 0.22 ммоль) в виде серо-белого осадка (20 мг, 28%). MS (ESI) m/e = 394.3 [M+H]+.
Пример 67
4-циклопропил-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]-3-(2,2,2-трифторэтокси)бензамид
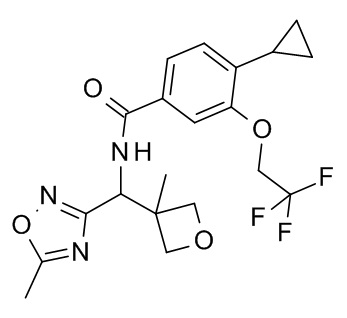
a) 2-метил-пропан-2-сульфиновой кислоты 1-(3-метил-оксетан-3-ил)-мет-(E)-илиденамид
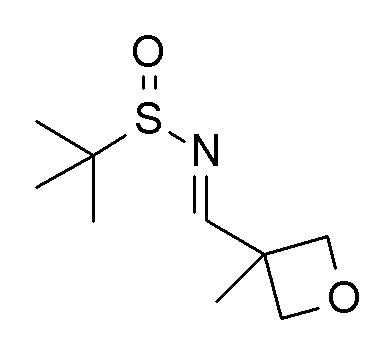
Раствор 3-метил-оксетан-3-карбальдегида (15 г, 149.8 ммоль), 2-метилпропан-2-сульфинамида (18 г, 149.8 ммоль) и Ti(OEt)4 (63 мл, 299.6 ммоль) в ТГФ (300 мл) кипятили с обратным холодильником в течение 16 ч. Реакционную смесь охладили до 25°C, погасили солевым раствором (800 мл), и экстрагировали с помощью EtOAc (3×300 мл). Объединенный органический слой промыли солевым раствором (400 мл), высушили над безводным Na2SO4, отфильтровали, и эвапорировали под вакуумом. Неочищенный продукт очистили посредством колоночной хроматографии combiflash с использованием 30% EtOAc в гексане в качестве элюентов с получением продукта, указанного в заголовке (20 г, 66%), в виде светло-желтого масла. MS (ESI) m/e = 203.9 [M+H]+.
b) 2-метил-пропан-2-сульфиновой кислоты [циано-(3-метил-оксетан-3-ил)-метил]-амид
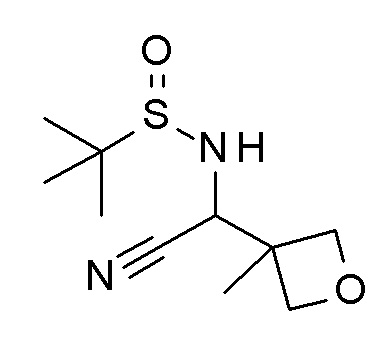
К перемешанному раствору 2-метил-пропан-2-сульфиновой кислоты 1-(3-метил-оксетан-3-ил)-мет-(E)-илиданеамида (4 г, 19.68 ммоль) в ТГФ (100 мл) добавили фторид цезия (3.6 г, 23.6 ммоль) и триметилсилилцианид (3.1 мл, 23.6 ммоль) при 25°C в атмосфере азота. Реакционную смесь перемешивали в течение 4 ч при 25°C. Летучие компоненты удалили под вакуумом. Неочищенную смесь разбавили этилацетатом (50 мл) и водой (50 мл). Слои разделили. Водный слой экстрагировали с помощью EtOAc (2×50 мл). Объединенный органический слой промыли солевым раствором (20 мл), высушили над Na2SO4, отфильтровали и эвапорировали. Неочищенный продукт очистили посредством колоночной хроматографии combiflash с получением продукта, указанного в заголовке, в виде желтого осадка (3.5 г, 78%). MS (ESI) m/e = 231.0 [M+H]+.
c) 2-амино-2-(3-метилоксетан-3-ил)ацетонитрил
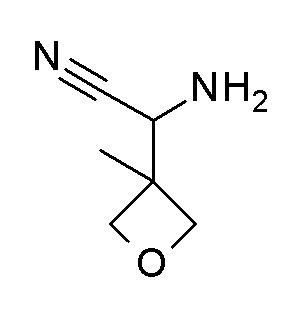
К охлажденному на льду перемешанному раствору 2-метил-пропан-2-сульфиновой кислоты [циано-(3-метил-оксетан-3-ил)-метил]-амида (1.5 г, 6.51 ммоль) в метаноле (35 мл) добавили раствор 4 M HCl в диоксане (2.4 мл). Реакционную смесь перемешивали в течение 2 ч при 0°C. Триэтиламин добавили (1.8 мл, 13.03 ммоль) при 0°C. Летучие компоненты удалили при пониженном давлении с получением неочищенного соединения, указанного в заголовке (800 мг), которое использовали на следующей стадии без дополнительной очистки.
d) N-[циано-(3-метил-оксетан-3-ил)-метил]-4-циклопропил-3-(2,2,2-трифтор-этокси)-бензамид
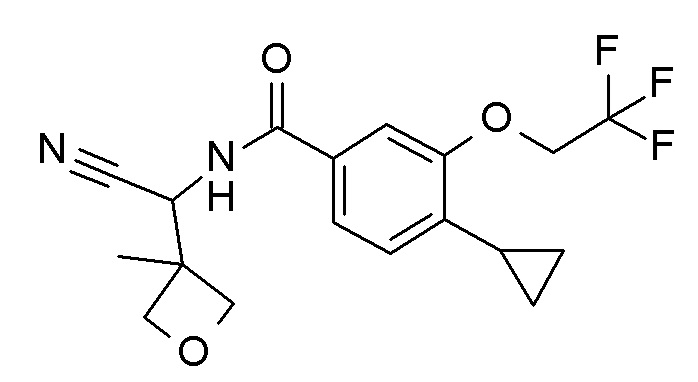
К перемешанному раствору 4-циклопропил-3-(2,2,2-трифтор-этокси)-бензойной кислоты (Пример 25c; 134 мг, 0.51 ммоль) в ДМФ (1.0 мл) добавили DIEA (0.33 мл, 2.1 ммоль) и 2-хлор-1-метилпиридиния йодид (336 мг, 1.3 ммоль). Смесь перемешивали при 25°C в течение 1.5 ч. Неочищенный 2-амино-2-(3-метилоксетан-3-ил)ацетонитрил (130 мг, 1.03 ммоль) в ДМФ (2.0 мл) добавили и перемешивание продолжили в течение 15 ч при 25°C. Реакционную смесь разбавили водой (10 мл) и экстрагировали с помощью EtOAc (3×20 мл). Объединенные органические слои промыли водой (10 мл), насыщенным водным раствором NaHCO3 (10 мл) и солевым раствором (10 мл), высушили над безводным Na2SO4 и сконцентрировали под вакуумом. Неочищенный продукт очистили посредством колоночной хроматографии combiflash с получением соединения, указанного в заголовке, в виде серо-белого осадка (150 мг, 40%). MS (ESI) m/e = 368.9 [M+H]+.
e) 4-циклопропил-N-[(N-гидроксикарбамимидоил)-(3-метил-оксетан-3-ил)-метил]-3-(2,2,2-трифтор-этокси)-бензамид
К перемешанному раствору N-[циано-(3-метил-оксетан-3-ил)-метил]-4-циклопропил-3-(2,2,2-трифтор-этокси)-бензамида (150 мг, 0.407 ммоль) в этаноле (2 мл) добавили триэтиламин (61 мл, 0.45 ммоль) и гидроксиламина гидрохлорид (28 мг, 0.407 ммоль). Реакционную смесь перемешивали при 25°C в течение 18 ч. Растворитель удалили при пониженном давлении. Остаток растворили 10% метанолом в ДХМ (20 мл). Добавили насыщенный водный раствор NaHCO3 (20 мл). Органический слой разделили и водный слой экстрагировали с помощью 10% метанола в ДХМ (2×20 мл). Объединенные органические слои высушили над безводным Na2SO4 и сконцентрировали под вакуумом. Неочищенный продукт очистили с помощью колоночной хроматографии combiflash с использованием 90% EtOAc в гексане в качестве элюентов с получением соединения, указанного в заголовке, в виде серо-белого вязкого осадка (100 мг, 61%). MS (ESI) m/e = 402.1 [M+H]+.
f) 4-циклопропил-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]-3-(2,2,2-трифторэтокси)бензамид
К перемешанному раствору 4-циклопропил-N-[(N-гидроксикарбамимидоил)-(3-метил-оксетан-3-ил)-метил]-3-(2,2,2-трифтор-этокси)-бензамида (100 мг, 0.25 ммоль) в изопропиловом спирте (2.0 мл) добавили (1,1-диметокси-этил)-диметил-амин (265 мг, 1.99 ммоль) при 25°C. Реакционную смесь перемешивали при 25°C в течение 17 ч. Воду (20 мл) и 10% метанол в ДХМ (20 мл) добавили. Органический слой разделили и водный слой экстрагировали с помощью 10% метанолом в ДХМ (3×20 мл). Объединенные органические слои промыли солевым раствором (10 мл), высушили над Na2SO4 и сконцентрировали под вакуумом с получением неочищенного продукта, который очистили с помощью колоночной хроматографии combiflash с использованием EtOAc в качестве элюента с последующей отмывкой пентаном и в заключение высушили лиофилизацией с получением продукта, указанного в заголовке (20.0 мг, 19%), в виде серо-белого осадка. MS (ESI) m/e = 426.0 [M+H]+.
Пример 68
4-циклопропил-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]-3-[(пропан-2-ил)окси]бензамид
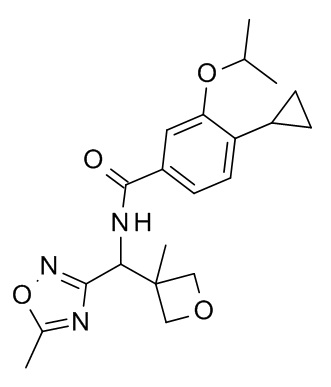
a) N-[циано-(3-метил-оксетан-3-ил)-метил]-4-циклопропил-3-изопропокси-бензамид
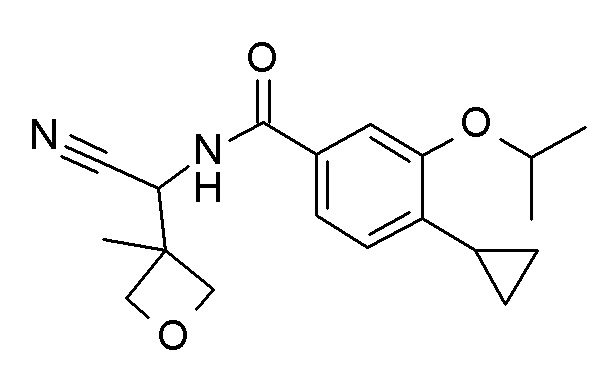
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 67d из 4-циклопропил-3-(пропан-2-илокси)бензойной кислоты (Пример 37c; 174 мг, 0.79 ммоль) и неочищенного 2-амино-2-(3-метилоксетан-3-ил)ацетонитрила (Пример 67c; 200 мг, 1.58 ммоль) в виде серо-белого осадка (250 мг, 48%). MS (ESI) m/e = 329.0 [M+H]+.
b) 4-циклопропил-N-[(N-гидроксикарбамимидоил)-(3-метил-оксетан-3-ил)-метил]-3-изопропокси-бензамид
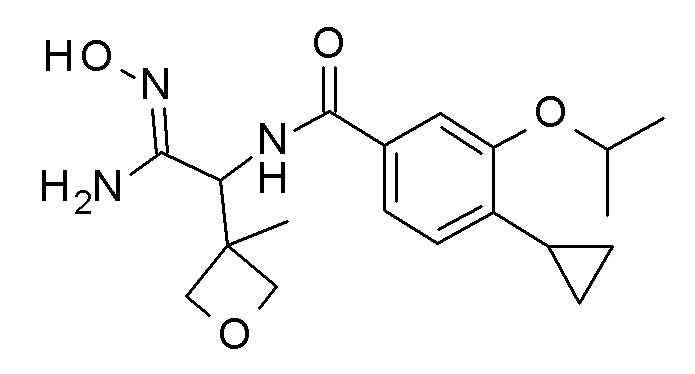
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 67e, из N-[циано-(3-метил-оксетан-3-ил)-метил]-4-циклопропил-3-изопропокси-бензамида (250 мг, 0.76 ммоль) в виде серо-белого осадка (200 мг, 84%). MS (ESI) m/e = 362.3 [M+H]+.
c) 4-циклопропил-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]-3-[(пропан-2-ил)окси]бензамид
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 67f из 4-циклопропил-N-[(N-гидроксикарбамимидоил)-(3-метил-оксетан-3-ил)-метил]-3-изопропокси-бензамида (200 мг, 0.55 ммоль) в виде серо-белого осадка (48 мг, 23%). MS (ESI) m/e = 386.1 [M+H]+.
Пример 69
N-[3-амино-1-(3-метилоксетан-3-ил)-3-оксопропил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид
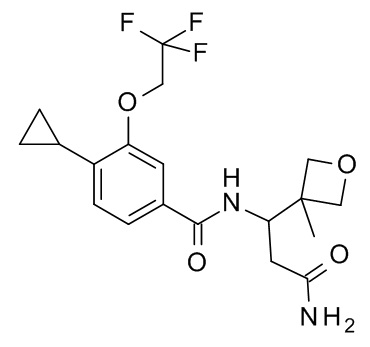
a) 3-метил-оксетан-3-карбоновой кислоты бензиловый эфир
К раствору 3-метил-оксетан-3-карбоновой кислоты (5 г, 43.1 ммоль) в CH3CN (100 мл) добавили DBU (7.1 мл, 47.4 ммоль) и бензилбромид (5.5 мл, 45.6 ммоль). Реакционную смесь перемешивали в течение 18 ч при 25°C. Растворитель удалили при пониженном давлении. Этилацетат (100 мл) и 1 н HCl (20 мл) добавили. Органический слой разделили, высушили над сульфатом натрия и сконцентрировали под вакуумом. Неочищенный продукт очистили с помощью колоночной хроматографии combiflash с использованием 10% EtOAc в гексане в качестве элюентов с получением соединения, указанного в заголовке, в виде бесцветной жидкости (5.4 г. 61%). MS (ESI) m/e = 386.1 [M+NH4]+.
b) 3-(3-метил-оксетан-3-ил)-3-оксо-пропионитрил
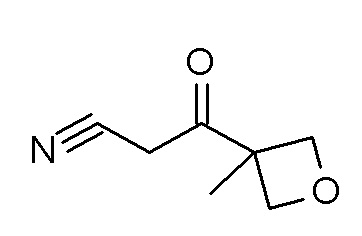
К охлажденному на льду раствору трет-бутоксида калия (2.97 г, 26.47 ммоль) в ТГФ (40 мл) добавили CH3CN (1.08 г, 26.47 ммоль). Смесь перемешивали в течение 10 мин при 0°C. Раствор 3-метил-оксетан-3-карбоновой кислоты бензилового эфира (5.2 г, 25.21 ммоль) в ТГФ (10 мл) добавили. Реакционную смесь нагревали до 25°C и перемешивали в течение 3 ч. После охлаждения до 0°C 2 н водный раствор HCl 20 мл) добавили и смесь экстрагировали с помощью CH2Cl2 (2×100 мл). Объединенный органический слой промыли солевым раствором (10 мл), высушили над сульфатом натрия и сконцентрировали при пониженном давлении. Неочищенное вещество очистили с помощью колоночной хроматографии combiflash с использованием 10% EtOAc в гексане в качестве элюентов с получением соединения, указанного в заголовке, в виде бледно-желтого масла (1.7 г, 47%). MS (ESI) m/e = 138.1 [M-H]-.
c) (Z)-3-[(бензо[1,3]диоксол-5-илметил)-амино]-3-(3-метил-оксетан-3-ил)-акрилонитрил
Смесь 3-(3-метил-оксетан-3-ил)-3-оксо-пропионитрила (1.7 г, 12.21 ммоль), 2H-1,3-бензодиоксол-5-илметанамина (1.84 г, 12.26 ммоль) и изопропоксида титана (IV) (4.56 мл, 15.27 ммоль) в ТГФ (15 мл) перемешивали при 25°C в течение 1 ч. Воду (50 мл) и EtOAc (100 мл) добавили. Смесь отфильтровали через слой целита и промыли с помощью EtOAc (50 мл). Органический слой разделили, промыли солевым раствором (20 мл), высушили над Na2SO4 и сконцентрировали под вакуумом. Неочищенный продукт очистили с помощью колоночной хроматографии combiflash с получением продукта, указанного в заголовке, в виде светло-желтого осадка (1.9 г, 57%). MS (ESI) m/e = 271.2 [M-H]-.
d) 3-[(бензо[1,3]диоксол-5-илметил)-амино]-3-(3-метил-оксетан-3-ил)-пропионитрил
NaBH3CN (650 мг, 10.47 ммоль) добавили в раствор (Z)-3-[(бензо[1,3]диоксол-5-илметил)-амино]-3-(3-метил-оксетан-3-ил)-акрилонитрила (1.9 г, 6.98 ммоль) в AcOH (15 мл). Реакционную смесь перемешивали при 25°C в течение 3 ч. Смесь разбавили водой (50 мл) и экстрагировали с помощью EtOAc (3×50 мл). Объединенный органический слой промыли насыщенным водным NaHCO3 и солевым раствором (10 мл), высушили над Na2SO4 и сконцентрировали под вакуумом. Неочищенный продукт очистили с помощью колоночной хроматографии combiflash с использованием 30% EtOAc в гексане в качестве элюентов с получением соединения, указанного в заголовке, в виде бесцветного смолистого осадка (1.5 г, 78%). MS (ESI) m/e = 275.0 [M+H]+.
e) 3-[(бензо[1,3]диоксол-5-илметил)-амино]-3-(3-метил-оксетан-3-ил)-пропионамид
К раствору 3-[(бензо[1,3]диоксол-5-илметил)-амино]-3-(3-метил-оксетан-3-ил)-пропионитрила (4.2 г, 15.3 ммоль) в ДМСО (50 мл) добавили K2CO3 (3.59 г, 26.1 ммоль) и по каплям 30% H2O2 (31 мл). Реакционную смесь перемешивали при 25°C в течение 17 ч. Воду (200 мл) добавили и суспензию экстрагировали с помощью CH2Cl2 (2×100 мл). Объединенные экстракты промыли водой (50 мл) и солевым раствором (50 мл), высушили над Na2SO4, и сконцентрировали под вакуумом. Неочищенный продукт очистили с помощью колоночной хроматографии combiflash с использованием 5% MeOH в ДХМ в качестве элюентов с получением соединения, указанного в заголовке, в виде серо-белой смолистой жидкости (2.35 г, 52%). MS (ESI) m/e = 293.0 [M+H]+.
f) 3-амино-3-(3-метил-оксетан-3-ил)-пропионамид
Раствор 3-[(бензо[1,3]диоксол-5-илметил)-амино]-3-(3-метил-оксетан-3-ил)-пропионамида (1 г, 3.42 ммоль) в MeOH (70 мл) продули аргоном в течение 30 мин. 20% Pd(OH)2/C (2 г) добавили. Смесь продули азотом в течение 30 мин., перемешивали в течение 18 ч при 25°C под давлением баллонного H2 и отфильтровали через слой целита. Слой целита промыли 10% MeOH в CH2Cl2 (2×20 мл). Объединенные фильтраты сконцентрировали под вакуумом. Неочищенный продукт тритурировали эфиром (2×20 мл) с получением неочищенного соединения, указанного в заголовке, в виде бесцветной жидкости (550 мг), которую использовали на следующей стадии без дополнительной очистки.
g) N-[3-амино-1-(3-метилоксетан-3-ил)-3-оксопропил]-4-циклопропил-3-(2,2,2-трифторэтокси)бензамид
К раствору 4-циклопропил-3-(2,2,2-трифторэтокси)бензойной кислоты (Пример 25c; 50 мг, 0.192 ммоль) в ДМФ (2 мл) добавили DIEA (0.13 мл, 0.78 ммоль) и 2-хлор-1-метилпиридиния йодид (122.8 мг, 0.48 ммоль). Реакционную смесь перемешивали в течение 1.5 ч при 25°C. Неочищенный (3-амино-3-(3-метилоксетан-3-ил)пропанамид (45.6 мг, 0.288 ммоль) в ДМФ (1.0 мл) добавили. Перемешивание продолжили в течение 16 ч при 25°C. Смесь разбавили водой (20 мл) и экстрагировали с помощью этилацетата (2×50 мл). Объединенный органический слой промыли солевым раствором (20 мл), высушили над безводным Na2SO4 и сконцентрировали при пониженном давлении. Воду (10 мл) добавили. Суспензию отфильтровали. Полученный серо-белый осадок очистили с помощью колоночной хроматографии combiflash с использованием EtOAc в качестве элюента с получением продукта, указанного в заголовке (23.5 мг, 31%) в виде серо-белого осадка. MS (ESI) m/e = 400.9 [M+H]+.
Пример 70
4-циклопропил-3-(2-фторэтокси)-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]бензамид
a) N-[циано-(3-метил-оксетан-3-ил)-метил]-4-циклопропил-3-(2-фтор-этокси)-бензамид
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 67d из 4-циклопропил-3-(2-фторэтокси)бензойной кислоты (Пример 33; 240 мг, 1.07 ммоль) и неочищенного 2-амино-2-(3-метилоксетан-3-ил)ацетонитрила (Пример 67c; 270 мг, 1.07 ммоль) в виде серо-белого осадка (250 мг, 35%). MS (ESI) m/e = 333.1 [M+H]+.
b) 4-циклопропил-3-(2-фтор-этокси)-N-[(N-гидроксикарбамимидоил)-(3-метил-оксетан-3-ил)-метил]-бензамид
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 67e, из N-[циано-(3-метил-оксетан-3-ил)-метил]-4-циклопропил-3-(2-фтор-этокси)-бензамида (250 мг, 0.75 ммоль) в виде серо-белого осадка (200 мг, 73%). MS (ESI) m/e = 365.9 [M+H]+.
c) 4-циклопропил-3-(2-фторэтокси)-N-[(5-метил-1,2,4-оксадиазол-3-ил)(3-метилоксетан-3-ил)метил]бензамид
Соединение, указанное в заголовке, синтезировали по аналогии со способом, описанным в примере 67f из 4-циклопропил-3-(2-фтор-этокси)-N-[(N-гидроксикарбамимидоил)-(3-метил-оксетан-3-ил)-метил]-бензамида (200 мг, 0.55 ммоль), и (1,1-диметокси-этил)-диметил-амина (583 мг, 4.38 ммоль) в виде серо-белого осадка (168 мг, 79%). MS (ESI) m/e = 390.0 [M+H]+.
Пример 71
Фармакологические тесты
Следующие тесты проводились для определения активности соединений формулы I:
Анализ связывания радиолиганда
Аффинность соединений настоящего изобретения в отношении каннабиноидных рецепторов CB1 определяли с использованием рекомендуемого количества препаратов мембран (PerkinElmer) клеток почек эмбриона человека (HEK), экспрессирующих рецепторы человека CNR1 или CNR2 в сочетании с 1.5 или 2.6 нМ [3H]-CP-55,940 (Perkin Elmer) в качестве радиолиганда, соответственно. Связывание проводили в связывающем буфере (50 мМ Tris, 5 мМ MgCl2, 2.5 нМ ЭДТА и 0.5% (мас./об.) свободного от жирных кислот БСА, pH 7.4 для CB1 рецепторов и 50 мМ Tris, 5 mM MgCl2, 2.5 мМ ЭГТА и 0.1% (мас./об.) свободного от жирных кислот БСА, pH 7.4 для CB2 рецепторов) в общем объеме 0.2 мл в течение 1 ч при 30°C со встряхиванием. Реакцию остановили быстрой фильтрацией через микрофильтровальные планшеты покрытые 0.5% полиэтиленимином (UniFilter GF/B фильтровальный планшет; Packard). Связавшуюся радиоактивность анализировали для Ki с использованием нелинейного регрессионного анализа (Activity Base, ID Business Solution, Limited), с определением Kd значений для [3H]CP55,940 из экспериментов по насыщению. Соединения формулы (I) показали прекрасную аффинность в отношении CB2 рецепторов с аффинностью ниже 10 мкМ, более конкретно, от 1 нМ до 3 мкМ и наиболее конкретно от 1 нМ до 100 нМ.
цАМФ анализ
Клетки CHO, экспрессирующие CB1 или CB2 рецепторы человека засеяли за 17-24 часов до эксперимента в размере 50.000 клеток на лунку в черные 96 луночные планшеты с плоским прозрачным дном (Corning Costar #3904) в среде DMEM (Invitrogen No. 31331), поддержанной 1x HT, с 10 % фетальной телячьей сывороткой и инкубировали при 5% CO2 и 37°C в инкубаторе с контролем влажности. Среду роста заменили на бикарбонатный буфер Кребса-Рингера с 1 мМ IBMX и инкубировали при 30°C в течение 30 мин. Соединения добавили до конечного анализируемого объема 100 мкл и инкубировали в течение 30 мин при 30°C. Используя cAMP-Nano-TRF набор для детекции (Roche Diagnostics) анализ остановили добавлением 50 мкл лизирующего реагента (Tris, NaCl, 1.5% Triton X100, 2.5% NP40, 10% NaN3) и 50 мкл раствора для детекции (20 мкМ mAb Alexa700-cAMP 1:1, и 48 мкМ Ruthenium-2-AHA-cAMP) и встряхивали в течение 2 ч при комнатной температуре. Разрешенный во времени перенос энергии измеряли с помощью TRF-ридера (Evotec Technologies GmbH), оснащенного ND:YAG лазером в качестве источника возбуждения. Планшет измеряли дважды при волне возбуждения при 355 нм и и эмиссии с задержкой в 100 нс и промежутком 100 нс, общим временем экспозиции 10 с при 730 (ширина полосы 30 нм) или 645 нм (ширина полосы 75 нм), соответственно. FRET сигнал рассчитывался следующим образом: FRET = T730-Alexa730-P(T645-B645) с P = Ru730-B730/Ru645-B645, где T730 представляет собой тестируемую лунку, измеренную при 730 нМ, T645 представляет собой тестируемую лунку, измеренную при 645 нм, B730 и B645 представляют собой контрольные буферы при 730 нм и 645 нм, соответственно. содержание цАМФ определяли по функции стандартной кривой, охватывающей от 10 мкМ до 0.13 нМ сАМФ.
Значения EC50 определяли с использованием анализа Activity Base (ID Business Solution, Limited). Значения EC50 для широкого диапазона агонистов каннабиноида согласно данному анализу согласовывались со значениями, опубликованными в научной литературе.
Соединения по настоящему изобретению являются агонистами рецепторов CB2 со значением EC50 ниже 0?5 мкМ и по меньшей мере 10-кратной селективностью по отношению к CB1 в соответствующем анализе. Конкретные соединения изобретения являются агонистами рецепторов CB2 со значением EC50 ниже 0,05 мкМ и по меньшей мере 500-кратной селективностью по отношению к CB1 в соответствующем анализе.
Например, следующие соединения показали следующие значения EC50 человека в функциональном цАМФ анализе, описанном выше:
|
Пример А
Покрытые оболочкой таблетки, содержащие следующие ингредиенты могут быть изготовлены обычным способом:
|
Активный ингредиент просеивают и смешивают с микрокристаллической целлюлозой и смесь гранулируют с раствором поливинилпирролидона в воде. Затем гранулят смешивают с карбоксиметилкрахмалом натрия и стеаратом магния и прессуют с получением ядер массой 120 или 350 мг соответственно. Ядра лакируют с помощью водного раствора/суспензии упомянутого выше пленочной оболочки.
Пример B
Капсулы, содержащие следующие ингредиенты, могут быть изготовлены обычным способом:
|
Компоненты просеивают и смешивают и ими наполняют капсулы размера 2.
Пример C
Растворы для инъекций могут обладать следующим составом:
|
Активный ингредиент растворили в смеси полиэтиленгликоля 400 и воды для инъекций (часть). РН довели до 5,0 посредством добавления уксусной кислоты. Объем довели до 1,0 мл посредством добавления остаточного количества воды. Раствор отфильтровали, разлили в пузырьки с помощью соответствующего оборудования и стерилизовали.
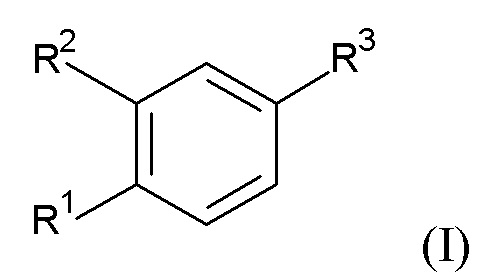
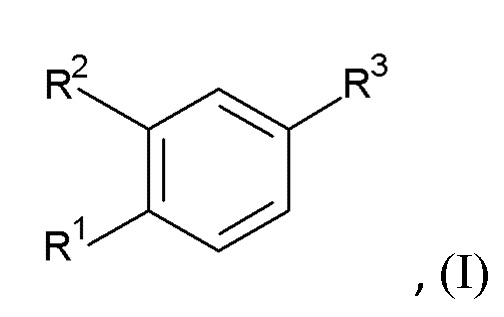
Препарат антитела
Производные гетероарилпирролидинил- и пиперидинилкетона
Производные изоксазоло-пиридина
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Ингибиторы jnk
Бензопирановые и бензоксепиновые ингибиторы рi3k и их применение
Арилциклогексилэфиры дигидротетраазабензоазуленов для применения в качестве антагонистов рецептора вазопрессина v1a
Пуриновые соединения, ингибирующие рi3к, и способы применения
Алкилциклогексиловые эфиры дигидротетраазабензоазуленов
Способ синтеза производных амино-метилтетралина
Спиро-5,6-дигидро-4н-2,3,5,10в-тетрааза-бензо[е]азулены
2-амино-5,5-дифтор-5,6-дигидро-4н-оксазины в качестве ингибиторов васе 1 и(или) васе 2
2,5,6,7-тетрагидро-[1,4]оксазепин-3-иламины или 2,3,6,7-тетрагидро-[1,4]оксазепин-5-иламины
1,4 тиазепины/сульфоны в качестве ингибиторов васе1 и(или) васе2
Новые производные пиразина
Новые производные тетразолона
Производные индол-3-карбонил-спиро-пиперидина в качестве антагонистов рецепторов v1a
2,2,2-трифторэтил-тиадиазины
Производные [1,2,3]триазоло[4,5-d]пиримидина с аффинностью к каннабиноидным рецепторам типа 2

