Результат интеллектуальной деятельности: 1,4 ТИАЗЕПИНЫ/СУЛЬФОНЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ВАСЕ1 И(ИЛИ) ВАСЕ2
Вид РИД
Изобретение
Болезнь Альцгеймера (БА) представляет собой нейродегенеративное расстройство центральной нервной системы и главную причину прогрессирующей деменции у пожилого населения. Ее клиническими симптомами являются ухудшение памяти, познавательной способности, временной и пространственной ориентации, способности связывать понятия и аргументации, а также тяжелые эмоциональные расстройства. В настоящее время нет доступных способов лечения, которые могут предупреждать данное заболевание или его развитие, или стабильно обращать его клинические симптомы. БА стала важной проблемой со здоровьем во всех обществах с высокой продолжительностью жизни и также значительным экономическим бременем для их систем здравоохранения.
БА характеризуется 2 главными патологиями в центральной нервной системе (ЦНС): появлением амилоидных бляшек и нейрофибриллярных клубков (Hardy et al., The amyloid hypothesis of Alzheimer′s disease: progress and problems on the road to therapeutics, Science. 2002 Jul 19; 297(5580):353-6, Selkoe, Cell biology of the amyloid beta-protein precursor and the mechanism of Alzheimer′s disease, Annu Rev Cell Biol. 1994; 10: 373-403). Обе патологии также обычно наблюдаются у пациентов с синдромом Дауна (трисомия 21-ой хромосомы), у которых также развиваются симптомы, подобные БА, в начале жизни. Нейрофибриллярные клубки представляют собой внутриклеточные агрегаты ассоциированного с микротрубочками белка тау (МАРТ). Амилоидные бляшки появляются во внеклеточном пространстве; их основными компонентами являются Aβ-пептиды. Последние представляют собой группу протеолитических фрагментов, происходящих из β-амилоидного белка-предшественника (АРР) посредством ряда стадий протеолитического расщепления. Было идентифицировано несколько форм АРР, из которых наиболее представленными являются белки из 695, 751 и 770 аминокислот в длину. Они все происходят от одного гена посредством дифференциального сплайсинга. Aβ-пептиды происходят из того же самого домена АРР, но отличаются в их N- и C-концах, главные виды имеют 40 и 42 аминокислоты в длину. Существуют несколько доказательств, которые явно свидетельствуют о том, что агрегированные Aβ-пептиды являются существенными молекулами в патогенезе БА: 1) амилоидные бляшки, образованные Aβ-пептидами, неизменно являются частью патологии БА; 2) Aβ-пептиды являются токсичными для нейронов; 3) при семейной болезни Альцгеймера (СБА) мутации в генах заболевания АРР, PSN1, PSN2 приводят к повышенным уровням Aβ-пептидов и раннему амилоидозу мозга; 4) у трансгенных мышей с экспрессией генов СБА, развивается патология, которая имеет много сходств с человеческим заболеванием. Aβ-пептиды образуются из АРР посредством последовательного действия 2 протеолитических ферментов, именуемых β- и γ-секретазой. β-Секретаза сначала осуществляет расщепление во внеклеточном домене АРР приблизительно в 28 аминокислотных остатках снаружи от трансмембранного домена (ТМ) с образованием C-концевого фрагмента АРР, содержащего ТМ- и цитоплазматический домен (CTFβ). CTFβ является субстратом для γ-секретазы, которая осуществляет расщепление в нескольких смежных положениях в пределах ТМ с образованием Aβ-пептидов и цитоплазматического фрагмента. γ-Секретаза представляет собой комплекс из по меньшей мере 4 разных белков, ее каталитическая субъединица с большой вероятностью представляет собой белок пресенилин (PSEN1, PSEN2). β-Секретаза (BACE1, Asp2; BASE обозначает фермент, расщепляющий АРР в β-сайте) представляет собой аспартилпротеазу, которая заякорена в мембране трансмембранным доменом (Vassar et al., Beta-secretase cleavage of Alzheimer′s amyloid precursor protein by the transmembrane aspartic protease BACE, Science. 1999 Oct 22; 286(5440): 735). Она экспрессируется во многих тканях человеческого организма, но ее уровень является особенно высоким в ЦНС. Генетическое удаление гена BACE1 у мышей ясно показало, что его активность является существенной для процессинга АРР, что приводит к образованию Aβ-пептидов, в отсутствие BACE1 Aβ-пептиды не образуются (Luo et al., Mice deficient in BACE1, the Alzheimer′s beta-secretase, have normal phenotype and abolished beta-amyloid generation, Nat Neurosci. 2001 Mar; 4(3):231-2, Roberds et al., BACE knockout mice are healthy despite lacking the primary beta-secretase activity in brain: implications for Alzheimer′s disease therapeutics, Hum Mol Genet. 2001 Jun 1; 10(12):1317-24). Мыши, которые были генетически модифицированы для экспрессии человеческого гена АРР и которые формируют обширные амилоидные бляшки и патологии, подобные болезни Альцгеймера, на протяжении старения, не могли это делать при уменьшении активности β-секретазы путем генетического удаления одного из аллелей BACE1 (McConlogue et al., Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP Transgenic Mice. J Biol Chem. 2007 Sep 7; 282(36): 26326). Таким образом, предполагается, что ингибиторы активности BACE1 могут быть полезными агентами для терапевтического вмешательства в болезнь Альцгеймера (БА).
Диабет типа 2 (ДТ2) вызван резистентностью к инсулину и неадекватной секрецией инсулина из β-клеток поджелудочной железы, приводящей к плохому контролю за уровнем глюкозы в крови и к гипергликемии (М Prentki & CJ Nolan, "Islet beta-cell failure in type 2 diabetes." J. Clin. Investig. 2006, 116(7), 1802-1812). Пациенты с ДТ2 имеют повышенный риск заболевания микро- и макрососудов и ряд связанных с этим осложнений, включающих диабетическую нефропатию, ретинопатию и седечно-сосудистое заболевание. В 2000 году оценочно 171 миллион человек имели указанное заболевание с ожиданием того, что это число удвоится к 2030 году (S Wild, G Roglic, A Green, R. Sicree & Н King, "Global prevalence of diabetes", Diabetes Care 2004, 27(5), 1047-1053), делая данное заболевание важной проблемой для здравоохранения. Увеличение распространенности ДТ2 ассоциировано со все более сидячим образом жизни и с потреблением мировым населением высокоэнергетической пищи (P Zimmet, KGMM Alberti & J Shaw, "Global and societal implications of the diabetes epidemic" Nature 2001, 414, 782-787).
Недостаточность работы β-клеток и последующее резкое снижение секреции инсулина и гипергликемия маркируют начало ДТ2. Большинство современных способов лечения не предупреждает потерю массы β-клеток, характеризующую выраженный ДТ2. Однако недавние разработки аналогов GLP-1 (глюкагон-подобный пептид-1), гастрина и других агентов показывают, что можно добиться сохранения и пролиферации β-клеток, что приводит к улучшенной толерантности к глюкозе и более медленному развитию до выраженного ДТ2 (LL Baggio & DJ Drucker, "Therapeutic approaches to preserve islet mass in type 2 diabetes", Annu. Rev. Med. 2006, 57, 265-281).
Tmem27 был идентифицирован в качестве белка, стимулирующего пролиферацию бета-клеток (P Akpinar, S Kuwajima, J Krutzfeldt, M Stoffel, "Tmem27: A cleaved and shed plasma membrane protein that stimulates pancreatic β cell proliferation", Cell Metab. 2005, 2, 385-397) и секрецию инсулина (К Fukui, Q Yang, Y Cao, N Takahashi et al., "The HNF-1 target Collectrin controls insulin exocytosis by SNARE complex formation", Cell Metab. 2005, 2, 373-384). Tmem27 представляет собой 42 кДа мембранный гликопротеин, который конститутивно отделяется от поверхности β-клеток, что происходит в результате деградации полноразмерного клеточного Tmem27. Сверхэкспрессия Tmem27 у трансгенной мыши увеличивает массу β-клеток и улучшает толерантность к глюкозе у модели диабета с индуцированным диетой ожирением (DIO). Кроме того, нокаут Tmem27 посредством миРНК (малая интерферирующая РНК) в анализе пролиферации β-клеток грызунов (например с использованием клеток INS1e) уменьшает скорость пролиферации, указывая на роль Tmem27 в контроле массы β-клеток.
В том же самом анализе пролиферации ингибиторы BACE2 также увеличивают пролиферацию. Однако ингибирование BACE2 в сочетании с нокдауном Tmem27 посредством миРНК приводит к низким скоростям пролиферации. Следовательно, делается заключение о том, что BACE2 представляет собой протеазу, ответственную за деградацию Tmem27. Кроме того, in vitro, BACE2 расщепляет пептид, основанный на последовательности Tmem27. Близкородственная протеаза BACE1 не расщепляет этот пептид, и одно селективное ингибирование BACE1 не усиливает пролиферацию β-клеток.
Близким гомологом BACE2 является мембраносвязанная аспартилпротеаза, и она солокализуется с Tmem27 в человеческих β-клетках поджелудочной железы (G Finzi, F Franzi, С Placidi, F Acquati et al., "BACE2 is stored in secretory granules of mouse and rat pancreatic beta cells", Ultrastruct Pathol. 2008, 32(6), 246-251). Также известно, что она способна деградировать АРР (I Hussain, D Powell, D Howlett, G Chapman et al., "ASP1 (BACE2) cleaves the amyloid precursor protein at the β-secretase site" Mol Cell Neurosci. 2000, 16, 609-619), IL-1R2 (рецептор 2 интерлейкина 1) (P Kuhn, E Marjaux, A Imhof, В De Strooper et al., "Regulated intramembrane proteolysis of the interleukin-1 receptor II by alpha-, beta-, and gamma-secretase" J. Biol. Chem. 2007, 282(16), 11982-11995) и ACE2 (ангиотензинпревращающий фермент 2). Способность деградировать ACE2 указывает на возможную роль BACE2 в контроле гипертензии.
Ингибирование BACE2, следовательно, предложено в качестве лечения ДТ2 с потенциалом сохранения и восстановления массы β-клеток и стимуляции секреции инсулина у преддиабетических и диабетических пациентов. Следовательно, целью настоящего изобретения является предложение селективных ингибиторов BACE2. Такие соединения являются полезными в качестве терапевтически активных веществ, особенно при лечении и/или предупреждении заболеваний, которые ассоциированы с ингибированием BACE2.
Кроме того, настоящими соединениями ингибируется образование или образование и отложение β-амилоидных пептидов в, на или около нервной ткани (например мозга), т.е. осуществляется ингибирование образования Aβ из АРР или фрагмента АРР.
Согласно настоящему изобретению предложены новые соединения формулы I, их изготовление, лекарственные средства на основе соединения согласно изобретению и их получение, а также применение соединений формулы I в контроле или предупреждении таких заболеваний, как болезнь Альцгеймера и диабет типа 2.
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к 1,4-тиазепинам и 1,4-сульфонам, имеющим свойства ингибирования BACE1 и/или BACE2, к их изготовлению, содержащим их фармацевтическим композициям, и к их применению в качестве терапевтически активных веществ.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединениям формулы I,
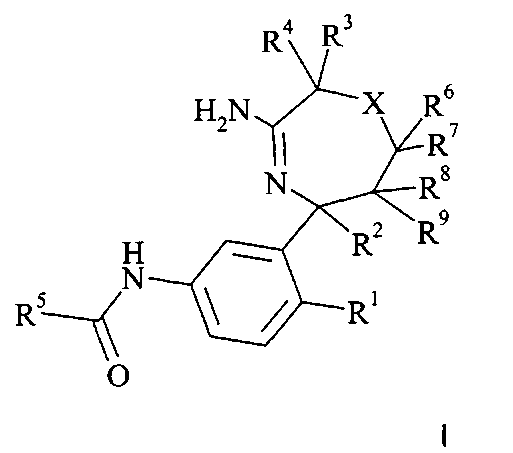
где заместители и переменные являются такими, как описано ниже и в формуле изобретения, или к их фармацевтически приемлемым солям.
Настоящие соединения имеют ингибирующую активность в отношении Asp2 (β-секретаза, BACE1 или мемапсин-2) и, следовательно, могут использоваться в терапевтическом и/или профилактическом лечении заболеваний и расстройств, характеризующихся повышенными уровнями β-амилоидов и/или β-амилоидных олигомеров, и/или β-амилоидных бляшек и дополнительными отложениями, или болезни Альцгеймера. И/или настоящие соединения имеют ингибирующую активность в отношении BACE2 и, следовательно, могут использоваться в терапевтическом и/или профилактическом лечении таких заболеваний и расстройств, как диабет типа 2 и другие метаболические расстройства.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Согласно настоящему изобретению предложены соединения формулы I и их фармацевтически приемлемые соли, получение вышеупомянутых соединений, содержащие их лекарственные средства, и их изготовление, а также применение вышеупомянутых соединений в терапевтическом и/или профилактическом лечении заболеваний и расстройств, которые ассоциированы с ингибированием активности BACE1 и/или BACE2, таких как болезнь Альцгеймера и диабет типа 2. Кроме того, образование или образование и отложение β-амилоидных бляшек в, на или около нервной ткани (например мозга) ингибируется настоящими соединениями путем ингибирования образования Aβ из АРР или фрагмента АРР.
Следующие определения общих терминов, используемых в настоящем описании, применяются независимо от того, появляются ли рассматриваемые термины одни или в комбинации с другими группами.
Если не утверждается иное, следующие термины, используемые в данной заявке, включая описание и формулу изобретения, имеют приведенные ниже определения. Следует отметить, что формы единственного числа в том виде, в котором они используются в описании и в приложенной формуле изобретения, включают объекты во множественном числе, если контекст явно не диктует иного.
Термин «C1-6-алкил», один или в комбинации с другими группами, обозначает углеводородный радикал, который может быть линейным или разветвленным, с одиночным или многократным ветвлением, когда алкильная группа, в общем, содержит от 1 до 6 атомов углерода, например, метил (Me), этил (Et), пропил, изопропил (изо-пропил), н-бутил, изо-бутил (изобутил), 2-бутил (втор-бутил), трет-бутил, изопентил, 2-этил-пропил, 1,2-диметил-пропил и тому подобное. Конкретными «С1-6-алкилами» являются группы с 1-5 атомами углерода. Конкретными группами являются метил, этил и трет-бутил. Наиболее конкретно - метил.
Термин «галоген-С1-6-алкил», один или в комбинации с другими группами, относится к С1-6-алкилу, как здесь определено, который замещен одним или многими атомами галогена, предпочтительно 1-5 атомами галогена, более предпочтительно 1-3 атомами галогена, наиболее предпочтительно 1 атомом галогена или 3 атомами галогена. Конкретным галогеном является фтор. Конкретным «галоген-С1-6-алкилом» является фтор-С1-6-алкил. Примерами являются дифторметил, хлорметил, фторметил и тому подобное. Конкретными примерами являются трифторметил и дифторметил.
Термин «галоген», один или в комбинации с другими группами, обозначает хлор (Cl), йод (I), фтор (F) и бром (Br). Конкретным «галогеном» является Cl и F. Конкретно - F.
Термин «гетероарил», один или в комбинации с другими группами, относится к ароматической карбоциклической группе, имеющей одно 4-8-членное кольцо или многочисленные конденсированные кольца, содержащие от 6 до 14, в частности от 6 до 10 атомов кольца, и содержащие 1, 2 или 3 гетероатома, индивидуально выбранных из N, O и S, в частности, N и O, причем в данной группе по меньшей мере одно гетероциклическое кольцо является ароматическим. Примеры «гетероарила» включают бензофурил, бензоимидазолил, 1Н-бензоимидазолил, бензооксазинил, бензоксазолил, бензотиазинил, бензотиазолил, бензотиенил, бензотриазолил, фурил, имидазолил, индазолил, 1Н-индазолил, индолил, изохинолинил, изотиазолил, изоксазолил, оксазолил, пиразинил, пиразолил (пиразил), 1Н-пиразолил, пиразоло[1,5-a]пиридинил, пиридазинил, пиридинил, пиримидинил, пирролил, хинолинил, тетразолил, тиазолил, тиенил, триазолил, 6,7-дигидро-5Н-[1]пиридинил и тому подобное. Конкретным «гетероарилом» является пиридинил и 2Н-пиразолил. Конкретными примерами являются пиридин-2-ил и 2Н-пиразол-3-ил.
Термин «C1-6-алкокси», один или в комбинации с другими группами, обозначает -O-C1-6-алкильный радикал, который может быть линейным или разветвленным, с одиночным или многократным ветвлением, где алкильная группа, в общем, содержит от 1 до 6 атомов углерода, например, метокси (OMe, MeO), этокси (OEt), пропокси, изопропокси (изо-пропокси), н-бутокси, изо-бутокси, 2-бутокси (втор-бутокси), трет-бутокси, изопентилокси и тому подобное. Конкретными «С1-6-алкокси» являются группы с 1-4 атомами углерода. Конкретными являются метокси и этокси.
Термин «гетероциклил», один или в комбинации с другими группами, относится к 4-8-членному кольцу, содержащему 1, 2 или 3 гетероатома кольца, индивидуально выбранных из N, О или S. Предпочтительными являются 1 или 2 гетероатома кольца. Конкретными примерами являются 4-6-членные «гетероциклилы», более конкретно - 5-6-членные «гетероциклилы», причем каждый из них содержит 1 или 2 гетероатома кольца, выбранных из N, O или S, в частности О. Конкретным примером является 5-членный гетероцикл, содержащий один O. Примеры «гетероциклила» включают азепанил, азетидил, диазепанил, морфолинил, оксазепанил, оксазолидил, окситанил, пиперазинил, пиперидил, пирролидинил, тетрагидрофурил, тетрагидропиридил, тетрагидропирил, тетрагидротиенил, тиазолидил, тиоморфолинил и тому подобное. Конкретным примером является тетрагидрофурил.
Термин «фармацевтически приемлемые соли» относится к солям, которые являются подходящими для применения в контакте с тканями людей и животных. Примерами подходящих солей с неорганическими и органическими кислотами являются соли с уксусной кислотой, лимонной кислотой, муравьиной кислотой, фумаровой кислотой, соляной кислотой, молочной кислотой, малеиновой кислотой, яблочной кислотой, метансульфоновой кислотой, азотной кислотой, фосфорной кислотой, пара-толуолсульфоновой кислотой, янтарной кислотой, серной кислотой, винной кислотой, трифторуксусной кислотой и тому подобными, но не ограничиваются ими. Предпочтительными являются соли с муравьиной кислотой, трифторуксусной кислотой и соляной кислотой.
Термины «фармацевтически приемлемый носитель» и «фармацевтически приемлемое вспомогательное вещество» относятся к носителям и вспомогательным веществам, таким как разбавители и эксципиенты, которые являются совместимыми с другими ингредиентами композиции.
Термин «фармацевтическая композиция» охватывает продукт, содержащий определенные ингредиенты в заданных количествах или пропорциях, а также любой продукт, который образуется, прямо или опосредованно, в результате объединения определенных ингредиентов в определенных количествах. Предпочтительно она охватывает продукт, содержащий один или более чем один активный ингредиент и возможный носитель, содержащий инертные ингредиенты, а также любой продукт, который образуется, прямо или опосредованно, в результате объединения, комплексообразования или агрегации двух или более чем двух ингредиентов, или в результате диссоциации одного или более чем одного ингредиента, или в результате других типов реакций или взаимодействий одного или более чем одного ингредиента.
Термин «ингибитор» обозначает соединение, которое конкурирует с, уменьшает или предотвращает связывание конкретного лиганда с конкретным рецептором, или которое уменьшает или предотвращает ингибирование функции конкретного белка.
Термин «полумаксимальная ингибирующая концентрация» (ИК50) обозначает концентрацию конкретного соединения, требующуюся для получения 50%-ного ингибирования биологического процесса in vitro. Значения ИК50 можно логарифмически преобразовать в значения рИК50 (-log ИК50), в которых более высокие значения указывают экспоненциально большую эффективность. Значение ИК50 не является абсолютным значением, но зависит от экспериментальных условий, например, использованных концентраций. Значение ИК50 можно превращать в абсолютную константу ингибирования (Ki) с использованием уравнения Ченга-Прусова (Biochem. Pharmacol. (1973) 22:3099). Термин «константа ингибирования» (Ki) обозначает абсолютную аффинность связывания конкретного ингибитора с рецептором. Ее измеряют с использованием конкурентных анализов связывания, и она равна концентрации, при которой конкретный ингибитор занял бы 50% рецепторов, если бы отсутствовал конкурирующий лиганд (например, радиоактивный лиганд). Значения Ki можно логарифмически преобразовать в значения pKi (-log Ki), в которых более высокие значения указывают экспоненциально большую эффективность.
«Терапевтически эффективное количество» означает количество соединения, которое, при введении субъекту для лечения болезненного состояния, является достаточным для осуществления такого лечения в отношении болезненного состояния. «Терапевтически эффективное количество» будет варьировать, в зависимости от соединения, болезненного состояния, которое лечат, тяжести заболевания, которое лечат, возраста и относительного состояния здоровья субъекта, пути и формы введения, решения лечащего врача или ветеринара и других факторов.
Термин «как здесь определено» и «как здесь описано», при отнесении к переменной, включает, посредством ссылки, широкое определение переменной, а также предпочтительное, более предпочтительное и наиболее предпочтительные определения, если таковые имеются.
Термины «осуществление обработки», «приведение в контакт» и «осуществление взаимодействия», при отнесении к химической реакции, означают добавление или смешивание двух или более чем двух реактивов при подходящих условиях с получением указанного и/или желательного продукта. Следует понимать, что реакция, которая дает указанный и/или желательный продукт, не обязательно может происходить в результате объединения двух реактивов, которые были исходно добавлены, т.е. может быть одно или более чем одно промежуточное соединение, которое образуется в смеси, что в конечном счете приводит к образованию указанного и/или желательного продукта.
Термин «защитная группа» обозначает группу, которая селективно блокирует реакционноспособный сайт в мультифункциональном соединении, так что можно селективно проводить химическую реакцию в другом незащищенном реакционноспособном сайте, в значении, традиционно ассоциированном с ней в синтетической химии. Защитные группы можно удалять в подходящий момент времени. Типичными защитными группами являются амино-защитные группы, карбокси-защитные группы или гидрокси-защитные группы. Термин «амино-защитная группа» обозначает группы, предназначенные для защиты аминогруппы, и они включают бензил, бензилоксикарбонил (карбобензилокси, CBZ), 9-флуоренилметилоксикарбонил (FMOC), пара-метоксибензилоксикарбонил, пара-нитробензилоксикарбонил, трет-бутоксикарбонил (ВОС) и трифторацетил. Другие примеры данных групп находятся в Т.W. Greene and P.G.M. Wuts, "Protective Groups in Organic Synthesis", 2nd ed., John Wiley & Sons, Inc., New York, NY, 1991, chapter 7; E. Haslam, "Protective Groups in Organic Chemistry", J.G.W. McOmie, Ed., Plenum Press, New York, NY, 1973, Chapter 5 и T.W. Greene, "Protective Groups in Organic Synthesis", John Wiley and Sons, New York, NY, 1981. Термин «защищенная аминогруппа» относится к аминогруппе, замещенной амино-защитными группами. Конкретными амино-защитными группами являются трет-бутоксикарбонильная группа, бис(диметоксифенил)-фенилметильная и диметокситритильная.
Термин «уходящая группа» обозначает группу со значением, традиционно ассоциированным с ней в синтетической органической химии, т.е. атом или группу, вытесняемую при условиях реакции замещения. Примеры уходящих групп включают галоген, в частности бром, алкан- или ариленсульфонилокси, как, например, метансульфонилокси, этансульфонилокси, тиометил, бензолсульфонилокси, тозилокси и тиенилокси, дигалофосфиноилокси, возможно замещенный бензилокси, изопропилокси и ацилокси.
Термин «ароматический» обозначает традиционную идею ароматичности, как определено в литературе, в частности в IUPAC - Compendium of Chemical Terminology, 2nd, A.D. McNaught & A. Wilkinson (Eds). Blackwell Scientific Publications, Oxford (1997).
Термин «фармацевтически приемлемый эксципиент» обозначает любой ингредиент, не имеющий терапевтической активности и не являющийся токсичным, такой как разрыхлители, связующие вещества, наполнители, растворители, буферы, агенты, регулирующие тоничность, стабилизаторы, антиоксиданты, поверхностно-активные вещества или смазки, используемые при приготовлении фармацевтических продуктов.
Всякий раз, когда в химической структуре присутствует хиральный углерод, подразумевается, что структурой охватываются все стереоизомеры, ассоциированные с данным хиральным углеродом.
Согласно изобретению также предложены фармацевтические композиции, способы применил и способы получения вышеупомянутых соединений.
Все отдельные воплощения могут быть объединены.
Определенным воплощением является соединение формулы I,
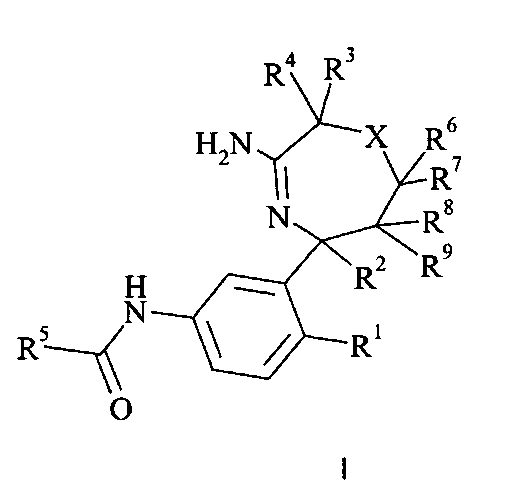
где
R1 выбран из группы, состоящей из
i) водорода,
ii) галогена и
iii) C1-6-алкила;
R2 выбран из группы, состоящей из
i) водорода,
ii) C1-6-алкила и
iii) галоген-C1-6-алкила;
R3 выбран из группы, состоящей из
i) водорода и
ii) C1-6-алкила;
R4 выбран из группы, состоящей из
i) водорода и
ii) C1-6-алкила;
R5 представляет собой гетероарил, не замещенный или замещенный одним или двумя заместителями, индивидуально выбранными из группы, состоящей из
i) C1-6-алкила,
ii) галогена,
iii) C1-6-алкокси и
iv) галоген-C1-6-алкила,
R6 представляет собой водород;
R7 представляет собой водород;
R8 представляет собой водород;
R9 представляет собой водород;
или R6 и R8 вместе образуют 5-6-членный гетероциклил,
X выбран из группы, состоящей из
i) -S и
ii) -SO2;
или его фармацевтически приемлемые соли.
Определенным воплощением изобретения является соединение формулы I′,
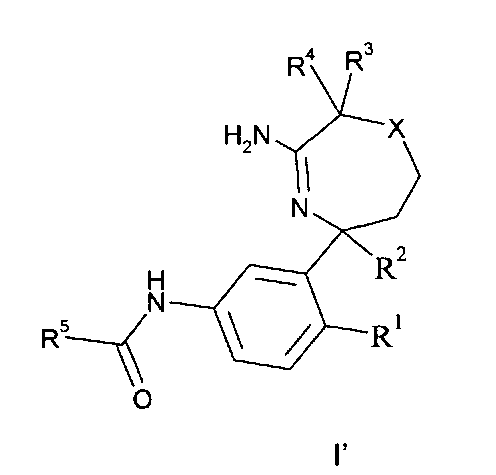
где
R1 выбран из группы, состоящей из
i) водорода,
ii) галогена и
iii) C1-6-алкила;
R2 выбран из группы, состоящей из
i) водорода,
ii) C1-6-алкила и
iii) галоген-C1-6-алкила;
R3 выбран из группы, состоящей из
i) водорода и
ii) C1-6-алкила;
R4 выбран из группы, состоящей из
i) водорода и
ii) C1-6-алкила;
R5 представляет собой гетероарил, не замещенный или замещенный одним или двумя заместителями, индивидуально выбранными из группы, состоящей из
i) C1-6-алкила,
ii) галогена,
iii) C1-6-алкокси и
iv) галоген-C1-6-алкила,
X выбран из группы, состоящей из
i) -S и
ii) -SO2;
или его фармацевтически приемлемые соли.
Определенное воплощение изобретения относится к соединению формулы I′a, как здесь описано,
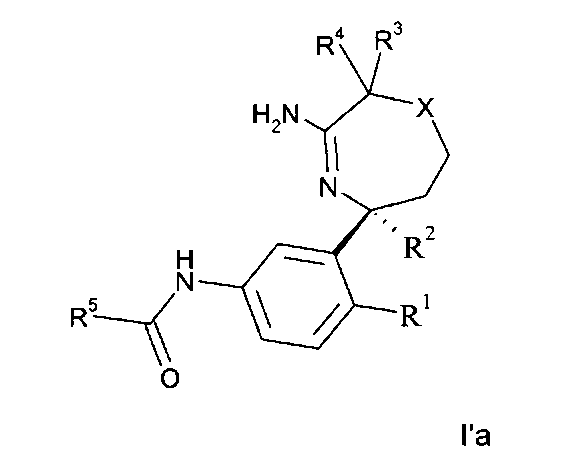
где
R1 выбран из группы, состоящей из
i) водорода,
ii) галогена и
iii) C1-6-алкила;
R2 выбран из группы, состоящей из
i) водорода,
ii) C1-6-алкила и
iii) галоген-C1-6-алкила;
R3 выбран из группы, состоящей из
i) водорода и
ii) C1-6-алкила;
R4 выбран из группы, состоящей из
i) водорода и
ii) C1-6-алкила;
R5 представляет собой гетероарил, не замещенный или замещенный одним или двумя заместителями, индивидуально выбранными из группы, состоящей из
i) C1-6-алкила,
ii) галогена,
iii) C1-6-алкокси и
iv) галоген-C1-6-алкила,
X выбран из группы, состоящей из
i) -S и
ii) -SO2;
или его фармацевтически приемлемым солям.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R1 представляет собой галоген.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R1 представляет собой F.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R2 представляет собой C1-6-алкил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R2 представляет собой Me.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R2 представляет собой галоген-C1-6-алкил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R2 представляет собой -CHF2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R3 представляет собой C1-6-алкил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R3 представляет собой Me.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R4 представляет собой C1-6-алкил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R4 представляет собой Me.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R4 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 представляет собой Me.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R6 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R7 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R8 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R9 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R6 и R8 вместе образуют 5-6-членный гетероциклил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R6 и R8 образуют тетрагидрофурил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где X представляет собой SO2, R1 представляет собой галоген, R2 представляет собой C1-6-алкил, R3 представляет собой водород, и R4 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где X представляет собой SO2, R1 представляет собой F, R2 представляет собой Me, R3 представляет собой водород, и R4 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где X представляет собой SO2, R1 представляет собой галоген, R2 представляет собой C1-6-алкил, R3 представляет собой водород, и R4 представляет собой C1-6-алкил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где X представляет собой SO2, R1 представляет собой F, R2 представляет собой Me, R3 представляет собой водород, и R4 представляет собой Me.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где X представляет собой SO2, R5 представляет собой гетероарил, замещенный одним галогеном, выбранным из хлора и фтора.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R1 представляет собой галоген, R2 представляет собой C1-6-алкил, R3 представляет собой водород, и R4 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R1 представляет собой F, R2 представляет собой Me, R3 представляет собой водород, и R4 представляет собой водород.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R1 представляет собой галоген, R2 представляет собой C1-6-алкил, R3 представляет собой водород, и R4 представляет собой C1-6-алкил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R1 представляет собой F, R2 представляет собой Me, R3 представляет собой водород, и R4 представляет собой Me.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 представляет собой гетероарил, замещенный одним галогеном, выбранным из хлора и фтора.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 выбран из
i) хлор-пиридинила,
ii) фтор-пиридинила и
iii) 2H-пиразолила.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 представляет собой хлор-пиридинил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 представляет собой фтор-пиридинил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 представляет собой 2Н-пиразол-3-ил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 представляет собой 5-хлор-пиридин-2-ил или 5-фтор-пиридин-2-ил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 представляет собой 5-хлор-пиридин-2-ил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где R5 представляет собой 5-фтор-пиридин-2-ил.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где X представляет собой S.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, где X представляет собой -SO2.
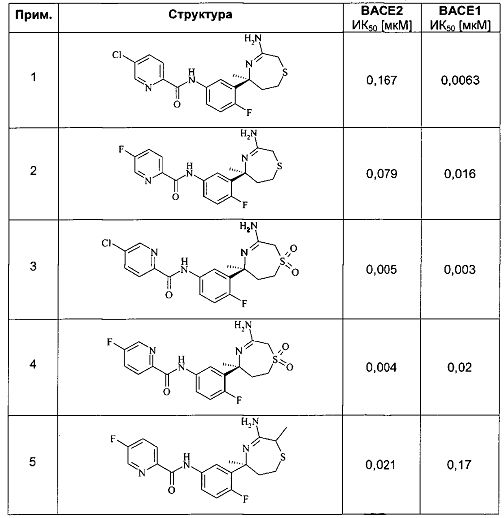
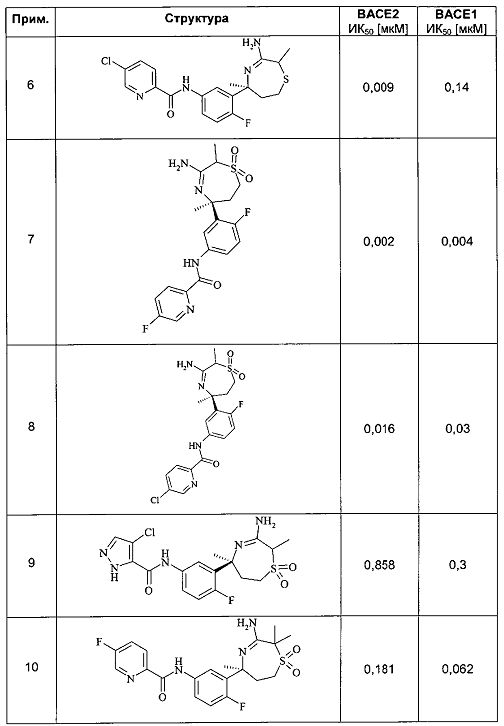
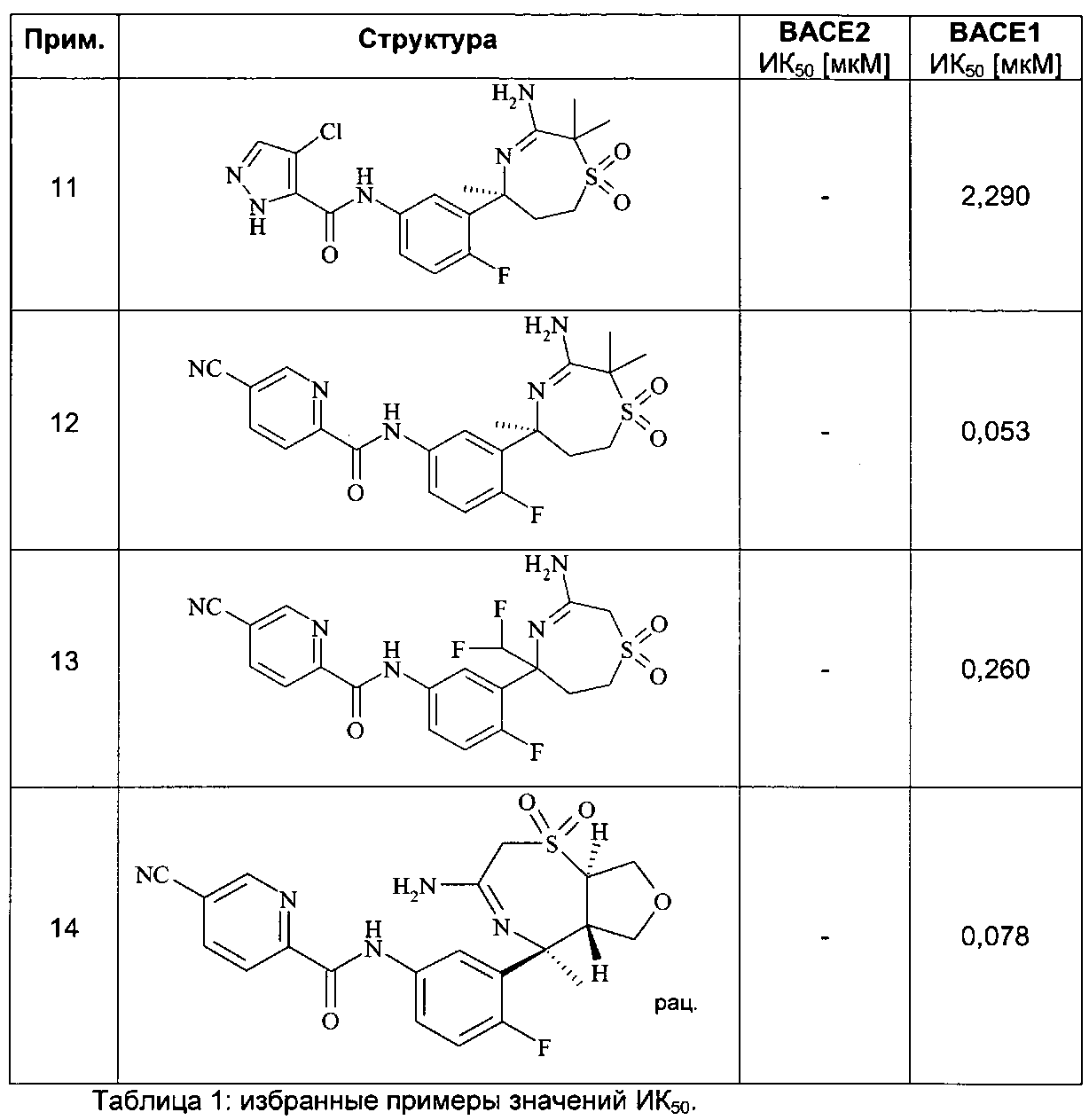
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, выбранному из группы, состоящей из следующих:
[3-((S)-3-амино-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 4-хлор-2Н-пиразол-3-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 4-хлор-2Н-пиразол-3-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-циано-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-циано-пиридин-2-карбоновой кислоты и
[3-((3aR,8S,8aS)-rel-6-амино-8-метил-4,4-диоксо-3,3a,4,5,8,8a-гексагидро-1Н-2-окса-4λ6-тиа-7-аза-азулен-8-ил)-4-фтор-фенил]-амид 5-циано-пиридин-2-карбоновой кислоты
или их фармацевтически приемлемые соли.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, выбранному из группы, состоящей из следующих:
[3-((S)-3-амино-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 4-хлор-2Н-пиразол-3-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 4-хлор-2Н-пиразол-3-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты и
[3-((S)-3-амино-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты
или их фармацевтически приемлемые соли.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, выбранному из группы, состоящей из следующих:
[3-((S)-3-амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты,
[3-((S)-3-амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты и
[3-((S)-3-амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, которое представляет собой [3-((S)-3-амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, которое представляет собой [3-((S)-3-амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, которое представляет собой [3-((S)-3-амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты.
Определенное воплощение изобретения относится к способу получения соединения формулы I, как здесь определено, включающему проведение взаимодействия соединения формулы A9 с соединением формулы А10,
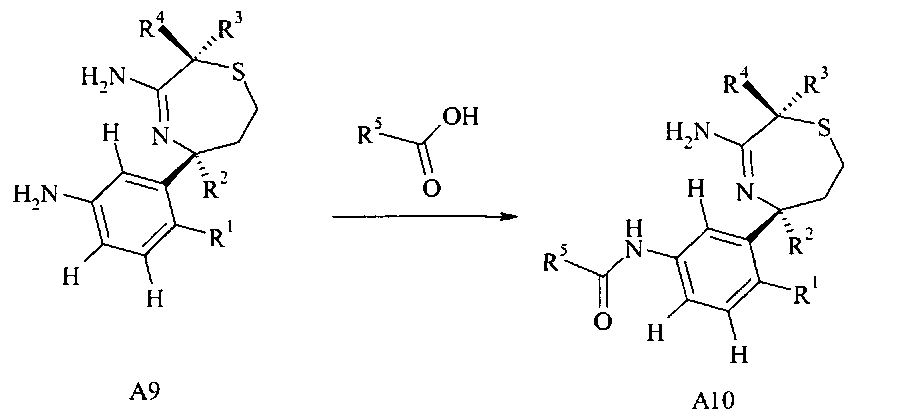
возможно, соединение формулы А10 может далее реагировать с пероксидом до соединения формулы А11,
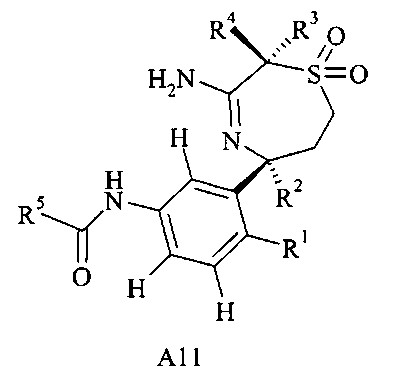
где R1, R2, R3, R4, R5 являются такими, как здесь определено.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, во всех случаях, когда оно получено способом, как определено выше.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве терапевтически активного вещества.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве ингибитора активности BACE1 и/или BACE2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве ингибитора активности BACE1.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве ингибитора активности BACE2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве ингибитора активности BACE1 и BACE2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения заболеваний и расстройств, характеризуемых повышенными уровнями β-амилоида и/или β-амилоидных олигомеров, и/или β-амилоидных бляшек и дополнительных отложений, или болезни Альцгеймера.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения болезни Альцгеймера.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения диабета или диабета типа 2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения диабета.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в качестве терапевтически активного вещества для терапевтического и/или профилактического лечения болезни Альцгеймера, диабета или диабета типа 2.
Определенное воплощение изобретения относится к фармацевтической композиции, содержащей соединение формулы I, как здесь описано, и фармацевтически приемлемый носитель, и/или фармацевтически приемлемое вспомогательное вещество.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для применения при ингибировании активности BACE1 и/или BACE2.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для применения при ингибировании активности BACE1.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для применения при ингибировании активности BACE2.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для применения при ингибировании активности BACE1 и BACE2.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для терапевтического и/или профилактического лечения заболеваний и расстройств, характеризуемых повышенными уровнями β-амилоида и/или β-амилоидных олигомеров, и/или β-амилоидных бляшек и дополнительных отложений, или болезни Альцгеймера.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для терапевтического и/или профилактического лечения болезни Альцгеймера.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для терапевтического и/или профилактического лечения диабета или диабета типа 2.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для терапевтического и/или профилактического лечения диабета.
Определенное воплощение изобретения относится к применению соединения формулы I, как здесь описано, для изготовления лекарственного средства для терапевтического и/или профилактического лечения болезни Альцгеймера, диабета или диабета типа 2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения при ингибировании активности BACE1 и/или BACE2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения при ингибировании активности BACE1.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения при ингибировании активности BACE2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения при ингибировании активности BACE1 и BACE2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в терапевтическом и/или профилактическом лечении заболеваний и расстройств, характеризующихся повышенными уровнями β-амилоида и/или β-амилоидных олигомеров, и/или β-амилоидных бляшек и дополнительных отложений, или болезни Альцгеймера.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в терапевтическом и/или профилактическом лечении болезни Альцгеймера.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в терапевтическом и/или профилактическом лечении диабета или диабета типа 2.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в терапевтическом и/или профилактическом лечении диабета.
Определенное воплощение изобретения относится к соединению формулы I, как здесь описано, для применения в терапевтическом и/или профилактическом лечении болезни Альцгеймера, диабета или диабета типа 2.
Определенное воплощение изобретения относится к способу для применения при ингибировании активности BACE1 и/или BACE2, конкретно для терапевтического и/или профилактического лечения заболеваний и расстройств, характеризующихся повышенными уровнями β-амилоида и/или β-амилоидных олигомеров, и/или β-амилоидных бляшек и дополнительных отложений, болезни Альцгеймера, диабета или диабета типа 2, причем данный способ включает введение соединения формулы I, как здесь описано, человеку или животному.
Определенное воплощение изобретения относится к способу для применения в терапевтическом и/или профилактическом лечении болезни Альцгеймера, диабета или диабета типа 2, причем данный способ включает введение соединения формулы I, как здесь описано, человеку или животному.
Кроме того, изобретение включает все оптические изомеры, т.е. диастереоизомеры, диастереомерные смеси, рацемические смеси, все их соответствующие энантиомеры и/или таутомеры, а также их сольваты соединений формулы I.
Специалист в данной области поймет, что соединения формулы I или I′ могут существовать в таутомерных формах, например, I′ может существовать в следующих таутомерных формах:
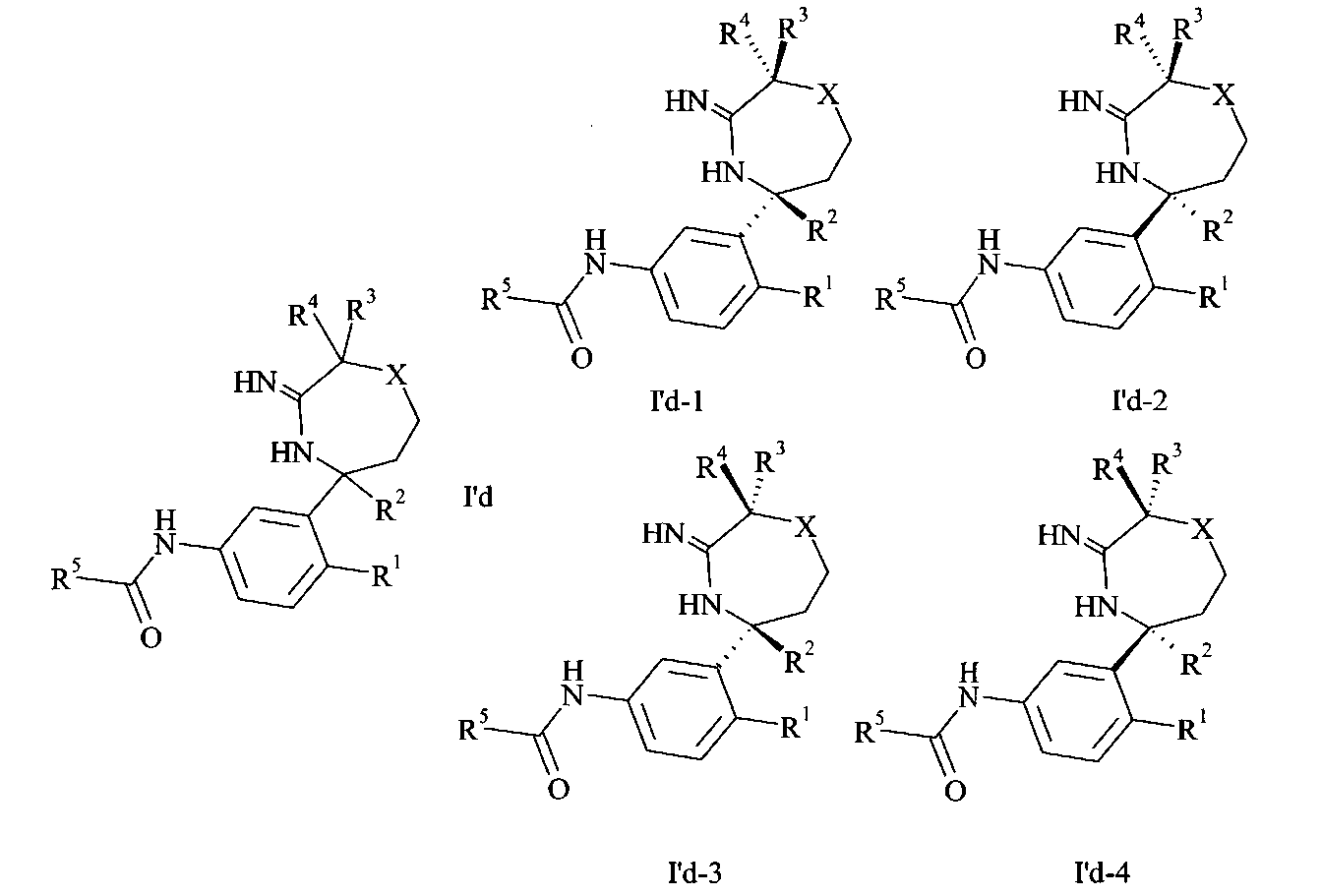
В настоящем изобретении охвачены все таутомерные формы.
Соединения формулы I могут содержать один или более чем один асимметрический центр и, следовательно, могут встречаться в виде рацематов, рацемических смесей, одиночных энантиомеров, диастереомерных смесей и индивидуальных диастереомеров. Могут присутствовать дополнительные асимметрические центры, в зависимости от природы разных заместителей на молекуле. Каждый такой асимметрический центр будет независимо давать два оптических изомера, и подразумевается, что в данное изобретение включены все возможные оптические изомеры и диастереомеры в смесях и в виде чистых или частично очищенных соединений. Подразумевается, что настоящее изобретение охватывает все такие изомерные формы данных соединений. Независимый синтез данных диастереомеров или их хроматографические разделения могут достигаться, как известно в данной области, посредством подходящей модификации раскрытой здесь методологии. Их абсолютную стереохимию можно определять рентгеновской кристаллографией кристаллических продуктов или кристаллических промежуточных соединений, которые, при необходимости, дериватизированы реактивом, содержащим асимметрический центр известной абсолютной конфигурации. Если это желательно, рацемические смеси соединений могут быть разделены, так что выделяются индивидуальные энантиомеры. Разделение можно проводить способами, хорошо известными в данной области, такими как связывание рацемической смеси соединений с энантиомерно чистым соединением с образованием диастереомерной смеси, с последующим разделением индивидуальных диастереомеров стандартными методами, такими как фракционная кристаллизация или хроматография. Предпочтительными примерами изомеров соединения формулы I′ являются соединение формулы I′a или соединение формулы I′b, в частности, I′b, где остатки имеют значение, как описано в любом из воплощений.
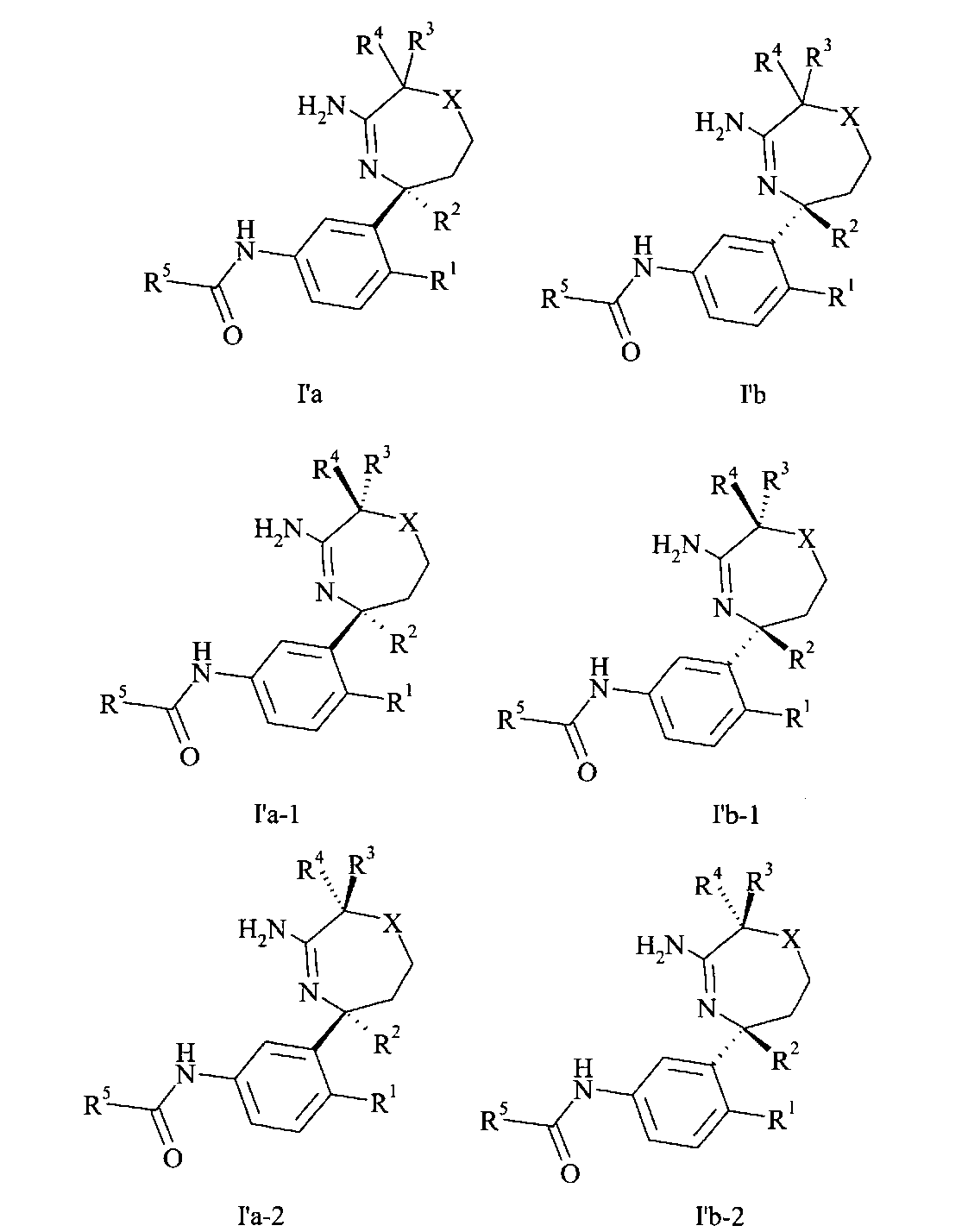
В воплощениях, где предложены оптически чистые энантиомеры, термин «оптически чистый энантиомер» означает то, что соединение содержит больше 90% желательного изомера по массе, предпочтительно больше 95% желательного изомера по массе, или более предпочтительно более 99% желательного изомера по массе, причем указанные проценты по массе основаны на общей массе изомера(ров) соединения. Хирально чистые или хирально обогащенные соединения могут быть получены хирально селективным синтезом или разделением энантиомеров. Разделение энантиомеров может проводиться на конечном продукте или, в качестве альтернативы, на подходящем промежуточном соединении.
Соединения формулы I можно получать согласно следующим схемам. Исходное вещество имеется в продаже или может быть получено согласно известным методам. Любые определенные ранее остатки и переменные будут продолжать иметь определенное ранее значение, если не указано иное.
Соединения формулы I можно получать посредством целого ряда путей синтеза, например, как проиллюстрировано в схемах, приведенных ниже. Получение соединений формулы I по настоящему изобретению можно осуществлять в последовательных или конвергентных синтетических путях. Синтез соединений по изобретению показан на следующих схемах. Навыки, требующиеся для проведения реакции и очистки образующихся продуктов, известны специалистам в данной области. Заместители и индексы, используемые в следующем описании способов, имеют значение, данное здесь ранее, если не указано противоположное.
Более подробно, соединения формулы I можно изготовлять способами, приведенными ниже, способами, приведенными в примерах, или аналогичными способами. Подходящие условия реакции для отдельных стадий реакции известны специалисту в данной области. Последовательность реакций не ограничивается последовательностью, продемонстрированной на схемах, описанных ниже, однако, в зависимости от исходных веществ и их соответствующей реакционной способности, последовательность стадий реакции можно свободно изменять. Исходные вещества либо имеются в продаже, либо могут быть получены способами, аналогичными способам, приведенным ниже, способами, описанными в ссылках, процитированных в описании или в примерах, или способами, известными в данной области.
Некоторые типичные методики для получения соединений формулы I проиллюстрированы на Схемах A, Б и В.
Сульфинилимины общей формулы A2 можно получать по аналогии с Т.Р. Tang & J.A. Ellman, J. Org. Chem. 1999, 64, 12, путем конденсации арилкетона и сульфинамида, например, алкилсульфинамида, наиболее предпочтительно (R)-(+)-трет-бутилсульфинамида, в присутствии кислоты Льюиса, такой как, например, алкоксид титана(IV), более предпочтительно этоксид титана(IV), в растворителе, таком как эфир, например, диэтиловый эфир или более предпочтительно THF (тетрагидрофуран).
Превращение сульфинилимина A2 до сложного эфира сульфинамида A3 идет стереоселективно посредством хиральной направляющей группы, как описано Tang & Ellman. Сульфинилимин A2 может реагировать с енолатом титана, полученным, например, из алкилацетата, предпочтительно этилацетата, LDA и хлортриизопропоксититана при низкой темпертуре, предпочтительно при -78°C в растворителе, таком как эфир, например, диэтиловый эфир, или более предпочтительно THF. В качестве альтернативы, сложный эфир сульфинамида A3 может быть получен из сульфинилимина A2 реакцией Реформатского бромацетилового сложноэфирного производного и цинковой пыли, возможно в присутствии хлорида меди(I) в растворителе, таком как эфир, например, диэтиловый эфир, или более предпочтительно THF, при температурах от 0 до 70°C, предпочтительно при 23°C.
Сложный эфир сульфинамида A3 может быть восстановлен до спирта A4 путем восстановления этилэфира гидридом щелочного металла, предпочтительно боргидридом лития или лития алюминия гидридом, в растворителе, таком как эфир, например, диэтиловый эфир, или более предпочтительно THF.
Тиоацетат А5 может быть получен из спирта A4 посредством протокола Митсунобу с использованием тиоуксусной кислоты, трифенилфосфина и диазокарбоксилата, предпочтительно DCAD или DEAD, в растворителе, таком как дихлорметан.
Получение нитрила сульфинамида A6 можно осуществлять из тиоацетата А5 путем расщепления тиоацетата в присутствии алкилирующего реактива, такого как галоидные аналоги ацетонитрила, с неорганическим основанием, предпочтительно K2CO3, в растворителе, таком как метанол.
Гидролиз хиральной направляющей группы в нитриле сульфинамида A6 с получением аминонитрила A7 можно осуществлять неорганической кислотой, например, серной кислотой, или предпочтительно соляной кислотой, в растворителе, таком как эфир, например, диэтиловый эфир, или более предпочтительно - 1,4-диоксан.
Аминотиазепин А8 можно получать реакцией аминонитрила A7 и триметилалюминия в растворителе, таком как ксилол, предпочтительно толуол.
Восстановление нитрогруппы в аминотиазепине А8 до анилина A9 можно осуществлять восстановлением металлом, таким как железо или олово, более предпочтительно хлоридом олова в спирте, более предпочтительно в водном этаноле при повышенной температуре, более предпочтительно при 80°C.
Связывание амида анилина A9 и карбоновой кислоты с получением амида А10 можно осуществлять с карбодиимидом, например, DCC или EDCI, или триазином, таким как DMTMM, в растворителе, таком как дихлорметан или метанол соответственно.
Получение сульфона А11 можно осуществлять из амида А10 посредством обработки пероксидом, предпочтительно мета-хлорпербензойной кислотой, в растворителе, таком как дихлорметан.
Схема A
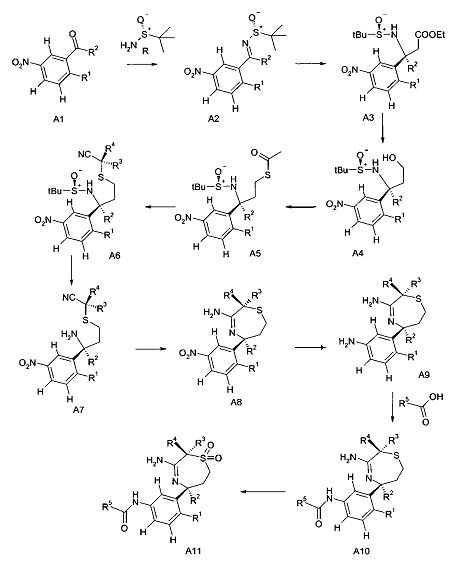
Схема Б
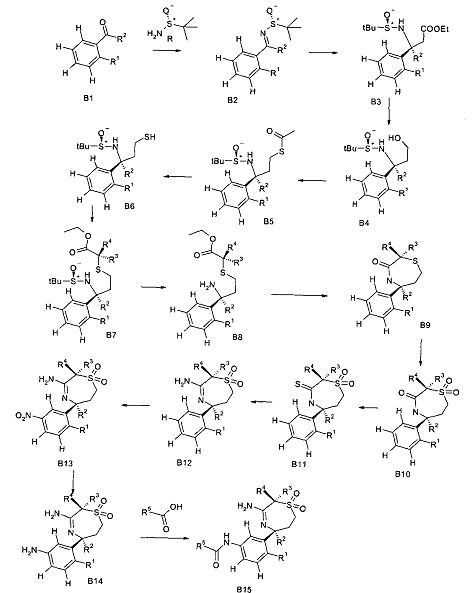
Сульфинилимины общей формулы В2 можно получать по аналогии с Т.Р. Tang & J.A. Ellman, J. Org. Chem. 1999, 64, 12, путем конденсации арилкетона и сульфинамида, например, алкилсульфинамида, наиболее предпочтительно (R)-(+)-трет-бутилсульфинамида, в присутствии кислоты Льюиса, такой как, например, алкоксид титана (IV), более предпочтительно этоксид титана (IV), в растворителе, таком как эфир, например, диэтиловый эфир или более предпочтительно THF.
Превращение сульфинилимина B2 до сложного эфира сульфинамида B3 идет стереоселективно посредством хиральной направляющей группы, как описано Tang & Ellman. Сульфинилимин B2 можно подвергать взаимодействию с енолатом титана, полученным, например, из алкилацетата, предпочтительно этилацетата, LDA и хлортриизопропоксититана при низкой температуре, предпочтительно при -78°C в растворителе, таком как эфир, например, диэтиловый эфир, или более предпочтительно THF. В качестве альтернативы, сложный эфир сульфинамида B3 можно получать из сульфинилимина B2 реакцией Реформатского бромуксусного сложноэфирного производного и цинковой пыли, возможно в присутствии хлорида меди(1), в растворителе, таком как эфир, например, диэтиловый эфир или более предпочтительно THF, при температурах от 0 до 70°C, предпочтительно при 23°C.
Сложный эфир сульфинамида B3 можно восстанавливать до спирта B4 путем восстановления этилэфира гидридом щелочного металла, предпочтительно боргидридом лития или лития алюминия гидридом, в растворителе, таком как эфир, например, диэтиловый эфир, или более предпочтительно THF.
Тиоацетат В5 можно получать из спирта B4 посредством протокола Митсунобу с использованием тиоуксусной кислоты, трифенилфосфина и диазокарбоксилата, предпочтительно DCAD или DEAD в растворителе, таком как дихлорметан.
Тиол B6 можно получать из тиоацетата В5 путем расщепления ацетата с использованием неорганического основания, такого как карбонат калия, в метаноле в качестве растворителя.
Тиол B6 можно алкилировать до сложного эфира В7 с использованием алкилирующего реактива, такого как α-галоидные аналоги сложных эфиров, с георганическим основанием, таким как карбонат калия в ацетонитриле в качестве растворителя.
Гидролиз хиральной направляющей группы в сложном эфире B7 с получением сложного эфира амина В8 можно осуществлять неорганической кислотой, например серной кислотой или предпочтительно соляной кислотой, в растворителе, таком как этанол, при температурах от 0 до 23°C.
Сложный эфир амина В8 можно циклизировать до лактама В9 с использованием основания, такого как бис[бис(триметилсилил)амино]олово(II) в эфирном растворителе, предпочтительно в THF, при температуре от 23°C до 50°C.
Тиоэфир B9 можно окислять до сульфона В10 обработкой окисляющим реактивом, предпочтительно мета-хлорпербензойной кислотой, в дихлорметане в качестве растворителя при комнатной температуре.
Тиолактам В11 можно получать из сульфона В10 с использованием реактива Лоуссона в эфирном растворителе, таком как диоксан или предпочтительно THF, с обратным холодильником.
1,1-диоксо-[1,4]тиазепин В12 можно получать из тиолактама В11 путем обработки хлоридом ртути(II) и раствором аммиака в метаноле в растворителе, таком как THF, при температуре 120°C в микроволновом резонаторе.
Введение нитрогруппы в 1,1-диоксо-[1,4]тиазепин В12 с получением В13 лучше всего было проводить согласно стандартной методике, включающей серную кислоту и азотную кислоту, при низкой температуре, предпочтительно при 0°C.
Восстановление нитрогруппы в В12 до анилина В14 можно осуществлять посредством восстановления металлом, таким как железо или олово, более предпочтительно хлоридом олова в спирте, более предпочтительно в водном этаноле, при повышенной температуре, более предпочтительно при 80°C.
Связывание амида анилина В14 и карбоновой кислотой с получением амида В15 (аналогичен А11) можно осуществлять с карбодиимидом, например, DCC или EDCI, или триазином, таким как DMTMM, в растворителе, таком как дихлорметан или метанол соответственно.
Схема В
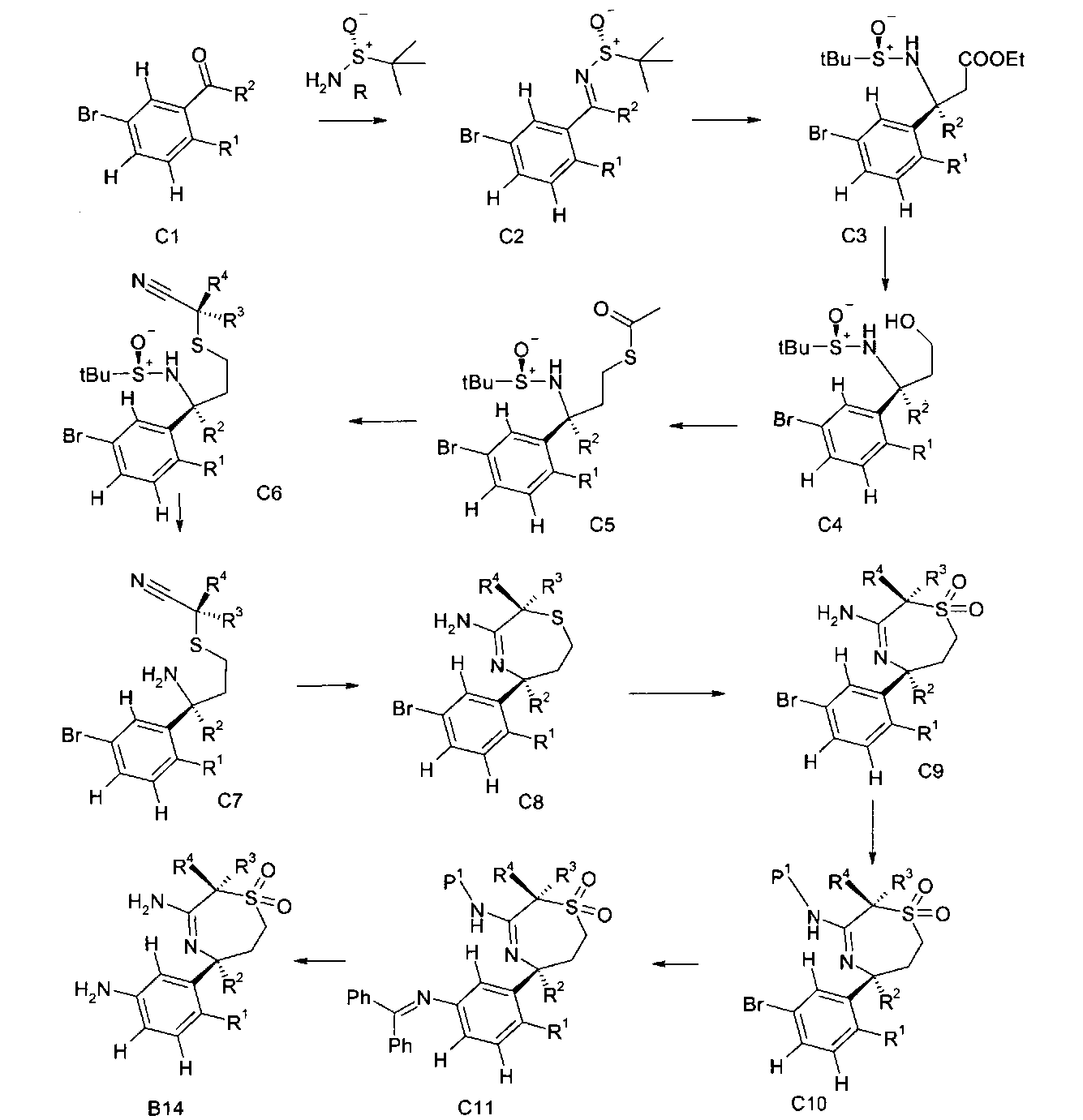
Сульфинилимины общей формулы С2 можно получать по аналогии с Т.Р. Tang & J.A. Ellman, J. Org. Chem. 1999, 64, 12, путем конденсации арилкетона и сульфинамида, например, алкилсульфинамида, наиболее предпочтительно (R)-(+)-трет-бутилсульфинамида, в присутствии кислоты Льюиса, такой как, например, алкоксид титана(IV), более предпочтительно этоксид титана(IV), в растворителе, таком как эфир, например, диэтиловый эфир или более предпочтительно THF.
Превращение сульфинилимина С2 до сложного эфира сульфинамида С3 идет стереоселективно посредством хиральной направляющей группы, как описано Tang & Ellman. Сульфинилимин C2 можно подвергать взаимодействию с енолатом титана, полученным, например, из алкилацетата, предпочтительно этилацетата, LDA и хлортриизопропоксититана при низкой температуре, предпочтительно при -78°C в растворителе, таком как эфир, например, диэтиловый эфир, или более предпочтительно THF. В качестве альтернативы, сложный эфир сульфинамида С3 можно получать из сульфинилимина С2 реакцией Реформатского бромуксусного сложноэфирного производного и цинковой пыли, возможно в присутствии хлорида меди(I), в растворителе, таком как эфир, например, диэтиловый эфир или более предпочтительно THF, при температурах от 0 до 70°C, предпочтительно при 23°C.
Сложный эфир сульфинамида С3 можно восстанавливать до спирта C4 путем восстановления этилэфира гидридом щелочного металла, предпочтительно боргидридом лития или лития алюминия гидридом, в растворителе, таком как эфир, например, диэтиловый эфир, или более предпочтительно THF.
Тиоацетат С5 можно получать из спирта C4 посредством протокола Митсунобу с использованием тиоуксусной кислоты, трифенилфосфина и диазокарбоксилата, предпочтительно DCAD или DEAD в растворителе, таком как дихлорметан.
Получение нитрила сульфинамида C6 можно осуществлять из тиоацетата С5 путем расщепления тиоацетата в присутствии алкилирующего реактива, такого как галоидные аналоги ацетонитрила, с неорганическим основанием, предпочтительно K2CO3, в растворителе, таком как метанол.
Гидролиз хиральной направляющей группы в нитриле C6 с получением аминонитрила С7 можно осуществлять неорганической кислотой, например, серной кислотой, или предпочтительно соляной кислотой, в растворителе, таком как диоксан, THF, этилацетат или метанол, при температурах от 0 до 23°C.
Аминонитрил C7 можно циклизировать до амидина С8 с использованием кислоты Льюиса, такой как триметилалюминий, в инертном растворителе, предпочтительно в толуоле, при температурах от 23°C до 100°C, предпочтительно при 60°C.
Тиоэфир С8 можно окислять до сульфона C9 путем обработки окисляющим реактивом, предпочтительно мета-хлорпербензойной кислотой, в дихлорметане в качестве растворителя при комнатной температуре. В качестве альтернативы, окисление можно проводить с использованием пероксимоносульфата калия (оксон) в растворителе, таком как метанол, при температуре окружающей среды.
Защиту аминогруппы в соединениях формулы С9 с получением бромистых арилов формулы C10 можно осуществлять с триарилметилхлоридами, такими как трифенилметилхлорид (Tr-Cl), пара-метоксифенилдифенилметилхлорид (MMTr-Cl), ди(пара-метоксифенил)фенилметилхлорид (DMTr-Cl) или три(пара-метоксифенил)метилхлорид (TMTr-Cl), предпочтительно с DMTr-Cl при основных условиях, например, в присутствии амина, такого как триэтиламин или диизопропилэтиламин, в хлорирующем растворителе, таком как дихлорметан или хлороформ, при температурах от 0°C до температуры окружающей среды.
Бромистые арилы формулы C10 можно подвергать взаимодействию с эквивалентами аммиака, такими как бензофенона имин, в присутствии подходящего катализатора на основе переходного металла, такого как бис(дибензилиденацетон)палладий(0) ((dba)2Pd) или трис(дибензилиденацетон)дипалладий(0) ((dba)3Pd2)) и подходящего лиганда, такого как рацемический 2,2′-бис(дифенилфосфино)-1,1′-бинафтил (рацемический BINAP), 2-дициклогексилфосфино-2′,4′,6′-триизопропилбифенил (X-PHOS) или 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенил (t-Bu X-PHOS), в присутствии основания, такого как трет-бутоксид натрия, фосфат калия или карбонат цезия, в подходящем растворителе, таком как толуол или 1,4-диоксан, в инертной атмосфере, такой как атмосфера азота или аргона, при температурах от 80 до 110°C с получением соединений формулы С11.
Снятия защиты обеих аминогрупп в соединениях формулы С11 можно добиться посредством однореакторной методики, сначала подвергая их взаимодействию с сильной органической кислотой, такой как трифторуксусная кислота, в хлорирующих растворителях, таких как дихлорметан или хлороформ, при безводных условиях при температурах от 0°C до температуры окружающей среды с расщеплением P1-группы. Последующее добавление воды или водной соляной кислоты для расщепления бензофенонимина и реакция при температуре окружающей среды дают диамины формулы B14.
Схема Г
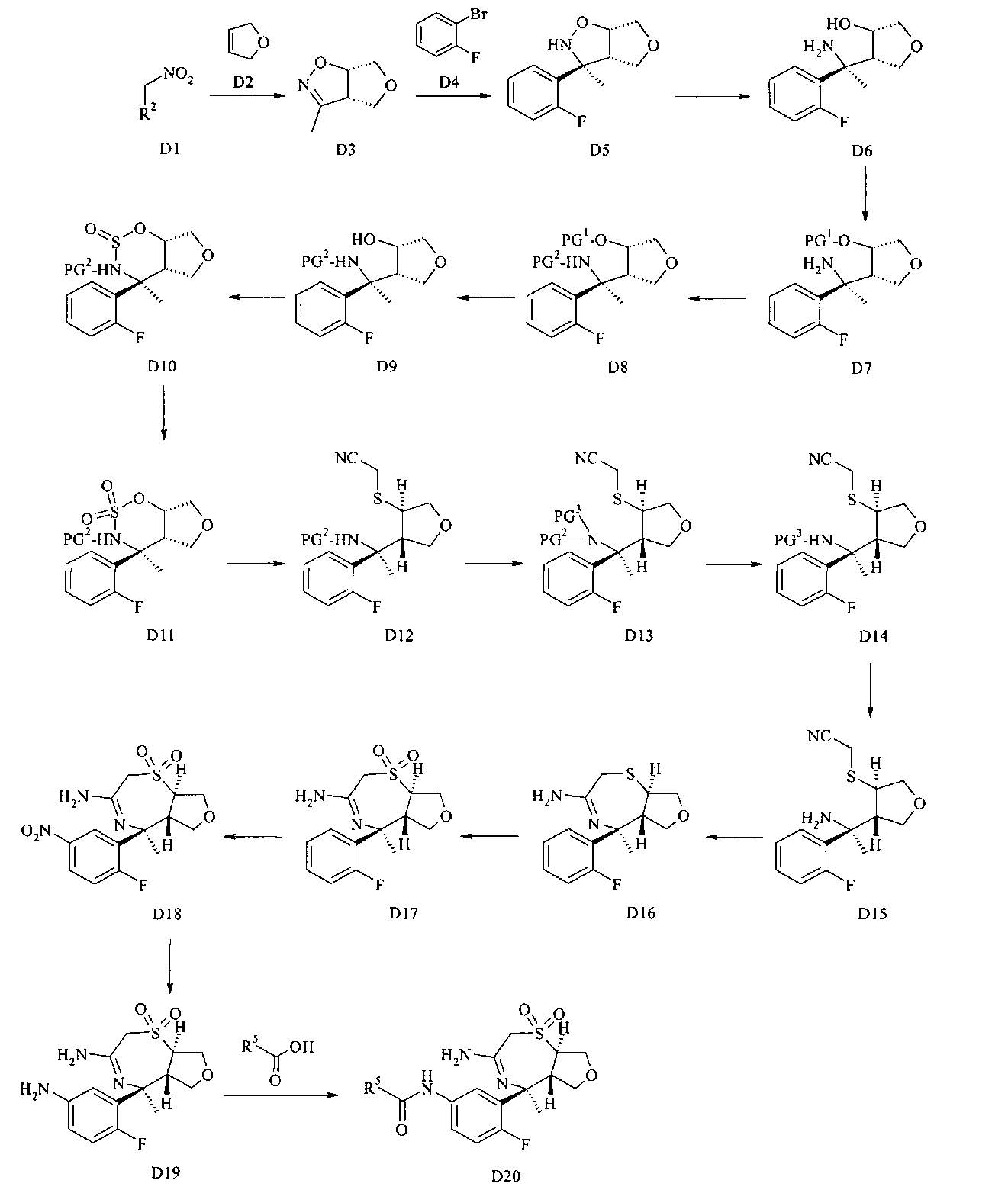
Вещества с конденсированным ядром общей формулы D20 можно получать, как описано на схеме Г. Исходное вещество имеется в продаже или может быть получено согласно известным способам. Любые ранее определенные остатки и переменные будут продолжать иметь ранее определенное значение, если не указано иное.
Нитросоединение D1 подвергают взаимодействию с олефином D2 в присутствии активирующего реактива, такого как, например, изоцианат, в частности, фенилизоцианат, и каталитического количества основания, в частности алкиламина, более конкретно Et3N, в растворителе, таком как бензол или толуол, в частности бензол, или алкиловый эфир, в частности диэтиловый эфир, или хлорированный растворитель, в частности дихлорметан, с получением дигидроизоксазола D3.
Арилирование дигидроизоксазола D3 арилбромидом D4 с получением изоксазолидина D5 проводится путем взаимодействия арилгалогенида, в частности арилбромида, с алкиллитиевым реактивом, в частности н-BuLi, с получением ариллитиевых соединений, которые могут быть подвергнуты взаимодействию с дигидроизоксазолом D3 в присутствии основания Льюиса, предпочтительно трифторидэтерата бора, в смеси растворителей, состоящей из эфира, в частности THF, и толуола, при температуре от -100°C до -20°C, в частности при -78°C.
Разделение рацемического изоксазолидина D5 с получением хирального изоксазолидина может осуществляться хиральной высокоэффективной жидкостной хроматографией (ВЭЖХ) с использованием колонки Chiralpack AD в смеси н-гептана и этанола в качестве элюента.
Гидрогенолиз изоксазолидина D5 до аминоспирта D6 может лучше всего осуществляться путем гидрогенолиза с переносом, используя Pd катализатор, в частности Pd на углероде, и источник водорода, например, соль муравьиной кислоты, в частности формиат аммония в протонном растворителе, таком как спирт, в частности этанол.
Аминоспирт D6 можно селективно защищать по кислороду O-силированием с образованием O-силированного аминоспирта D7 посредством хлорсилана, в частности трет-бутилхлордиметилсилана (PG1 представляет собой трет-BuMe2Si), в хлорированном растворителе, таком как дихлорметан, в присутствии триалкиламинового основания, в частности триэтиламина, и пиридинового катализатора, в частности 4-диметиламинопиридина, при температуре от 0°C до 23°C.
O-силированный аминоспирт D7 можно восстановительно аминировать до O-силированного N-бензилированного аминоспирта D8 посредством альдегида, в частности пара-метоксибензальдегида (PG2 представляет собой РМВ) или 2,4-диметоксибензальдегида (PG2 представляет собой DMB), используя восстанавливающий агент, в частности цианоборгидрид натрия или триацетоксиборгидрид натрия, в хлорированном растворителе, в частности 1,2-дихлорэтане или дихлорметане, в присутствии слабой органической кислоты, в частности уксусной кислоты, при температуре от 0°C до 60°C, предпочтительно при 23°C.
O-силированный N-бензилированный аминоспирт D8 может подвергаться десилированию с образованием N-бензилированного аминоспирта D9 путем его взаимодействия с источником фтора, в частности с тетрабутиламмония фторидом (TBAF), в растворителе, таком как THF, при температуре от 0°C до 50°C, предпочтительно при 23°C.
N-бензилированный аминоспирт D9 можно подвергать взаимодействию с тионилхлоридом с образованием циклического сульфамидита D10 в присутствии аминного основания, в частности пиридина, в хлорированном растворителе, в частности в дихлорметане, начиная с низкой температуры, такой как -78°C, и осуществляя нагревание вплоть до 0°C или до температуры окружающей среды.
Циклический сульфамидит D10 можно окислять до циклического сульфамидата D11 посредством перйодата щелочного металла, такого как перйодат натрия или калия, в присутствии соли рутения, такой как хлорид рутения(III), в смеси растворителей, состоящей из воды, ацетонитрила и этилацетата или тетрахлорметана, при температурах от 0°C до 50°C, предпочтительно при 23°C.
Циклический сульфамидат D11 можно регио- и стереоселективно открывать серным нуклеофилом, таким как меркаптоацетонитрил, и затем гидролизовать в кислых условиях до N-бензилированного аминонитрила D12. Открытие кольца идет в присутствии аминного основания, такого как 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU) или 1,1,3,3-тетраметилгуанидин (TMG), в полярном апротонном растворителе, таком как N,N-диметилформамид, при температурах от 23°C до 80°C, предпочтительно при 60°C. После удаления всех летучих веществ, остающихся со стадии открытия кольца, под вакуумом посредством выпаривания, неочищенную реакционную смесь подвергают кислотному гидролизу в смеси неорганической кислоты, в частности 20%-ной водной серной кислоты, и растворителя, такого как диэтиловый эфир или дихлорметан, при температурах от 0°C до 50°C, предпочтительно при 23°C.
С N-бензилированного аминонитрила D12 снимают защиту с получением аминонитрила D15 в трехэтапном протоколе: во-первых, N-бензилированный аминонитрил D12 подвергают взаимодействию с органическим ангидридом, в частности с трифторуксусным ангидридом, в присутствии аминного основания, в частности триэтиламина или диизопропилэтиламина, в хлорированном растворителе, таком как дихлорметан, при температурах от 0°C до 40°C, предпочтительно при 23°C, с получением N-бензилированного N-трифторацетилированного аминонитрила D13. Во-вторых, N-бензилированный N-трифторацетилированный аминонитрил D13 подвергают дебензилированию с получением N-трифторацетилированного аминонитрила D14 посредством чистой реакции с сильной органической кислотой, в частности с трифторуксусной кислотой, при температурах от 0°C до 50°C, предпочтительно при 23°C. В третьих, N-трифторацетилированный аминонитрил D14 деацилируют с получением аминонитрила D15 путем обработки восстанавливающим агентом, таким как боргидрид натрия, в спиртовом растворителе, в частности в метаноле или этаноле, при температурах от 0°C до 60°C, предпочтительно при 23°C.
Аминонитрил D15 можно циклизировать до амидина D16 с использованием кислоты Льюиса, такой как триметилалюминий в инертном растворителе, предпочтительно в толуоле, при температурах от 23°C до 100°C, предпочтительно при 60°C.
Тиоэфир D16 можно окислять до сульфона D17 посредством обработки окисляющим реактивом, предпочтительно мета-хлорпербензойной кислотой, в дихлорметане в качестве растворителя при комнатной температуре. В качестве альтернативы, окисление можно проводить с использованием пероксимонофосфата калия (оксон) в растворителе, таком как метанол, при температуре окружающей среды.
Нитрирование амидина D17 с получением нитроамидина D18 следует согласно стандартной методике с участием чистой серной кислоты и дымящей азотной кислоты без применения растворителя при температурах от 0°C до 23°C.
Восстановление нитрогруппы в промежуточном соединении D18 с получением анлилина D19 можно осуществлять гидрогенизацией с использованием катализатора, такого как Pd на углероде, в протонных растворителях, таких как спирты, в частности этанол или метанол.
Селективное связывание амида анилина D19 и карбоновой кислоты с получением амида D20 можно осуществлять с 4-(4,6-диметокси[1.3.5]триазин-2-ил)-4-метилморфолиния хлоридом (DMTMM) гидратом в растворителе, таком как спирт, в частности метанол.
Соответствующие фармацевтически приемлемые соли с кислотами можно получать стандартными методами, известными специалисту в данной области, например, путем растворения соединения формулы I в подходящем растворителе, таком как, например, диоксан или THF, и добавления подходящего количества соответствующей кислоты. Продукты обычно можно выделять фильтрованием или хроматографией. Превращение соединения формулы I в фармацевтически приемлемую соль с основанием можно проводить посредством обработки такого соединения таким основанием. Одним возможным методом получения такой соли является, например, добавление 1/n эквивалентов основной соли, такой как, например, М(ОН)n, где М представляет собой металл или катион аммония, и n равно числу гидроксидных анионов, в раствор соединения в подходящем растворителе (например, этанол, смесь этанол-вода, смесь тетрагидрофуран-вода) и удаление растворителя выпариванием или лиофилизацией. Конкретными солями являются гидрохлорид, формиат и трифторацетат.
Поскольку их получение не описано в примерах, соединения формулы I, а также все промежуточные продукты могут быть получены согласно аналогичным методам или согласно изложенным здесь методам. Исходные вещества имеются в продаже, известны в данной области или могут быть получены методами, известными в данной области, или по аналогии с ними.
Будет понятно, что в данном изобретении соединения общей формулы I могут быть дериватизированы по функциональным группам для получения производных, которые способны к обратному превращению до родительского соединения in vivo.
Фармакологические тесты
Соединения формулы I и их фармацевтически приемлемые соли обладают ценными фармакологическими свойствами. Обнаружили, что соединения по настоящему изобретению ассоциированы с ингибированием активности BACE1 и/или BACE2. Соединения исследовали согласно тесту, приведенному ниже.
Клеточный анализ снижения уровня Aβ
Для оценки эффективности соединений в клеточном анализе использовали человеческие клетки НЕК293 (клетки человеческой эмбриональной почки), которые стабильно трансфицированы вектором, экспрессирующим кДНК человеческого гена АРР дикого типа (АРР695). Клетки высевали в 96-луночные планшеты для микротитрования в среду для культуры клеток (Iscove, плюс 10% (об./об.) фетальной коровьей сыворотки, глутамин, пенициллин/стрептомицин) приблизительно до 80%-ной конфлюентности, и добавляли соединения в 10х концентрации в 1/10 объема среды без FCS (фетальная коровья сыворотка), содержащей 8% DMSO (диметилсульфоксид) (конечную концентрацию DMSO поддерживали на уровне 0,8% об./об.). После 18-20 ч инкубации при 37°C и 5% CO2 в увлажненном инкубаторе супернатант культуры отбирали для определения концентраций Aβ40. 96-луночные планшеты ELISA (твердофазный иммуноферментный анализ) (например, Nunc MaxiSorb) покрывали моноклональным антителом, которое специфично распознает C-концевой конец Aβ40 (Brockhaus et al., NeuroReport 9, 1481-1486; 1998). После блокирования сайтов неспецифичного связывания, например, 1% BSA (бычий сывороточный альбумин) и промывки добавляли супернатанты культуры в подходящих разведениях совместно с детектирующим антителом против Aβ, связанным с пероксидазой хрена (например, антитело 4G8, Senetek, Maryland Heights, MO), и инкубировали в течение 5-7 ч. Затем лунки планшетов микротитрования обильно промывали физиологическим раствором, буферизованным Tris, содержащим 0,05% Tween 20, и анализ проявляли с использованием тетраметилбензидина/H2O2 в буфере на основе лимонной кислоты. После остановки реакции одним объемом 1 н. H2SO4, реакцию измеряли на планшет-ридере для ELISA при длине волны 450 нм. Концентрации Aβ в супернатантах культуры рассчитывали из стандартной кривой, полученной с известными количествами чистого пептида Aβ.
Анализ ингибирования ВАСЕ путем измерения клеточного расщепления ТМЕМ27
В данном анализе используется принцип ингибирования расщепления человеческого ТМЕМ27 эндогенным клеточным BACE2 в линии крысиных клеток Insle и отделения от поверхности клетки в культуральную среду, с последующей детекцией в анализе ELISA. Ингибирование BACE2 предотвращает расщепление и отделение дозозависимым образом.
Стабильная линия клеток «INS-TMEM27» представляет собой линию клеток, полученную от INS1e, с индуцибельной экспрессией (с использованием системы TetOn) полноразмерного hTMEM27, зависимой от доксициклина. На протяжении всего эксперимента клетки культивируют в RPMI1640 + Glutamax (Invitrogen), пенициллин/стрептомицин, 10% фетальная коровья сыворотка, 100 мМ пируват, 5 мМ бета-меркаптоэтанол, 100 микрограммов/мл G418 и 100 микрограммов/мл гигромицина и выращивают в неприкрепленной культуре при 37°C в стандартном инкубаторе для клеточной культуры с CO2.
Клетки INS-TMEM27 высевают в 96-луночные планшеты. После 2 суток в культуре добавляют ингибитор BACE2 в ряде концентраций, как требуется анализом, и еще через два часа добавляют доксициклин до конечной концентрации 500 нг/мл. Клетки инкубируют в течение еще 46 часов, и супернатант отбирают для детекции отделенного ТМЕМ27.
Для детекции ТМЕМ27 в культуральной среде используют анализ ELISA (с использованием пары мышиных антител против человеческого ТМЕМ27, полученных против внеклеточного домена ТМЕМ27). EC50 (эффективная концентрация) для ингибирования BACE2 рассчитывают с использованием данных ELISA для каждой концентрации ингибитора с использованием программы для аппроксимации с помощью стандартной кривой, такой как XLFit для таблиц Excel.
Фармацевтические композиции
Соединения формулы I и фармацевтически приемлемые соли можно использовать в качестве терапевтически активных веществ, например, в форме фармацевтических препаратов. Фармацевтические препараты можно вводить перорально, например, в форме таблеток, покрытых таблеток, драже, твердых и мягких желатиновых капсул, растворов, эмульсий или суспензий. Однако введение также можно осуществлять ректально, например, в форме суппозиториев, или парентерально, например, в форме инъецируемых растворов.
Для получения фармацевтических препаратов соединения формулы I и их фармацевтически приемлемые соли можно перерабатывать с фармацевтически инертными неорганическими или органическими носителями. Например, в качестве таких носителей для таблеток, покрытых таблеток, драже и твердых желатиновых капсул можно использовать лактозу, кукурузный крахмал или их производные, тальк, стеариновую кислоту или их соли и тому подобное. Подходящими носителями для мягких желатиновых капсул являются, например, растительные масла, воска, жиры, полутвердые и жидкие полиолы и тому подобное. В зависимости от природы активного вещества, в случае мягких желатиновых капсул, однако, обычно не требуются носители. Подходящими носителями для получения растворов и сиропов являются, например, вода, полиолы, глицерин, растительное масло и тому подобное. Подходящими носителями для суппозиториев являются, например, природные или отвержденные масла, воска, жиры, полутвердые и жидкие полиолы и тому подобное.
Кроме того, фармацевтические препараты могут содержать фармацевтически приемлемые вспомогательные вещества, такие как консерванты, солюбилизаторы, стабилизаторы, увлажнители, эмульгаторы, подсластители, красители, ароматизаторы, соли для изменения осмотического давления, буферы, маскирующие агенты или антиоксиданты. Кроме того, они также могут содержать другие терапевтически полезные вещества.
Согласно настоящему изобретению также предложены лекарственные средства, содержащие соединение формулы I или его фармацевтически приемлемую соль и терапевтически инертный носитель, а также способ их получения, который включает объединение одного или более чем одного соединения формулы I и/или его фармацевтически приемлемых солей и, если желательно, одного или более чем одного другого терапевтически полезного вещества в медицинскую форму введения совместно с одним или более чем одним терапевтически инертным носителем.
Дозировка может варьировать в широких пределах и, естественно, должна корректироваться для индивидуальных требований в каждом конкретном случае. В случае перорального введения дозировка для взрослых соединения общей формулы I или соответствующего количества его фармацевтически приемлемой соли может варьировать от примерно 0,01 мг до примерно 1000 мг в сутки. Суточную дозировку можно вводить в виде одной дозы или в раздельных дозах и, кроме того, верхний предел также может быть превышен, когда имеются показания для этого.
Следующие примеры иллюстрируют настоящее изобретение без его ограничения, просто служа в качестве его образца. Фармацевтические препараты для удобства содержат примерно 1-500 мг, предпочтительно 1-100 мг соединения формулы I. Примерами композиций согласно изобретению являются следующие:
Пример А
Таблетки следующего состава изготовляют обычным способом:
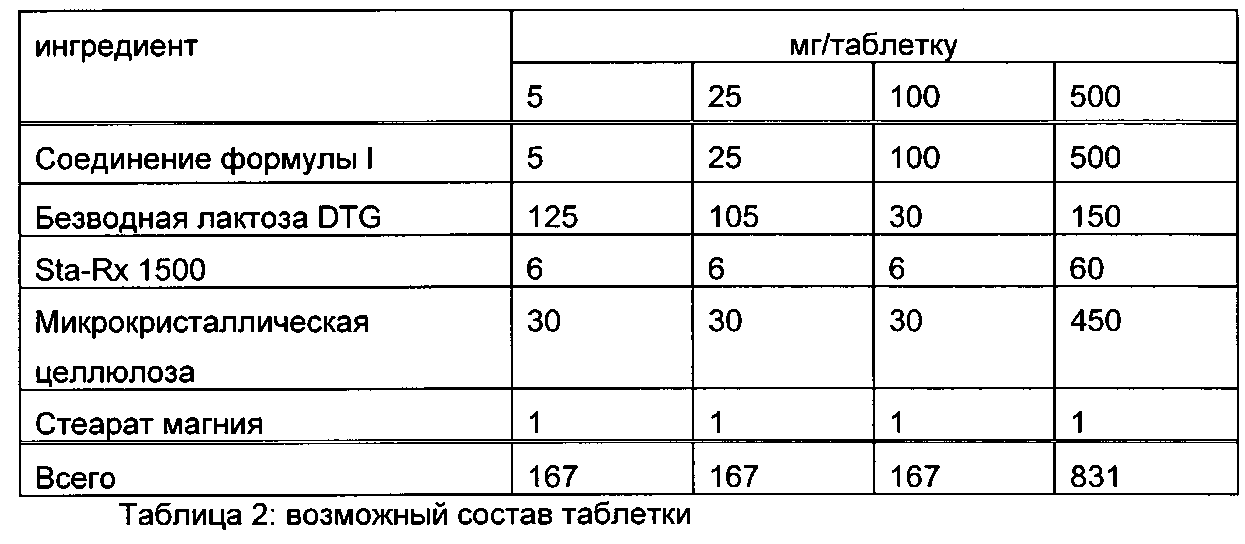
Методика изготовления
1. Смешать ингредиенты 1, 2, 3 и 4 и гранулят с очищенной водой.
2. Высушить гранулы при 50°C.
3. Пропустить гранулы через подходящее размалывающее оборудование.
4. Добавить ингредиент 5 и перемешивать в течение трех минут; прессовать на подходящем прессе.
Пример Б-1
Капсулы следующего состава изготовляют так:

Методика изготовления
1. Смешать ингредиенты 1, 2 и 3 в подходящем миксере в течение 30 минут.
2. Добавить ингредиенты 4 и 5 и перемешивать в течение 3 минут.
3. Заполнить в подходящую капсулу.
Соединение формулы I, лактозу и кукурузный крахмал сперва смешивают в миксере и затем в измельчающей установке. Смесь возвращают в миксер; добавляют в нее тальк и тщательно перемешивают. Установка заполняет смесью подходящие капсулы, например, твердые желатиновые капсулы.
Пример Б-2
Мягкие желатиновые капсулы следующего состава изготовляют так:


Методика изготовления
Соединение формулы I растворяют в теплом расплаве других ингредиентов, и смесью заполняют мягкие желатиновые капсулы подходящего размера. Заполненные мягкие желатиновые капсулы обрабатывают согласно обычным методикам.
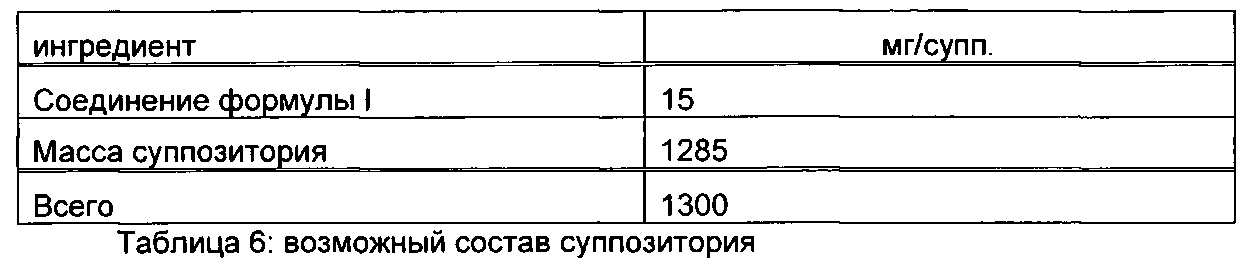
Пример В
Суппозитории следующего состава изготовляют так:
Методика изготовления
Массу суппозитория расплавляют в стеклянном или стальном сосуде, тщательно перемешивают и охлаждают до 45°C. Вслед за этим, в нее добавляют соединение формулы I в форме мелкого порошка и производят перемешивание до его полного диспергирования. Смесь вливают в формы для суппозитория подходящего размера, оставляют охлаждаться; затем суппозитории удаляют из форм и индивидуально упаковывают в восковую бумагу или в металлическую фольгу.
Пример Г
Изготовляют инъекционные растворы следующего состава:
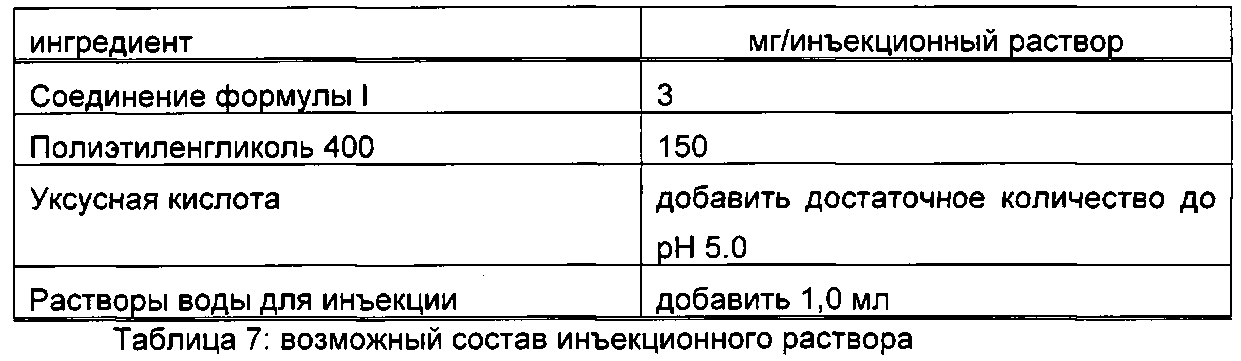
Методика изготовления
Соединение формулы I растворяют в смеси полиэтиленгликоля 400 и воды для инъекции (часть). pH доводят до 5,0 уксусной кислотой. Объем доводят до 1,0 мл добавлением остаточного количества воды. Раствор фильтруют, наполняют им флаконы с использованием допустимого избытка и стерилизуют.
Пример Д
Изготовляют пакетики со следующим составом:
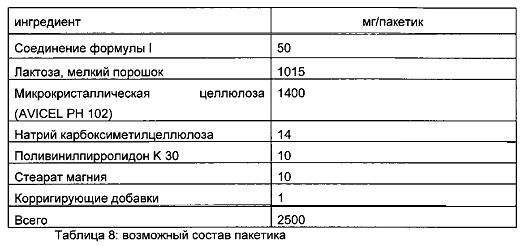
Методика изготовления
Соединение формулы I смешивают с лактозой, микрокристаллической целлюлозой и натрий карбоксиметилцеллюлозой и гранулируют со смесью поливинилпирролидона в воде. Гранулят смешивают со стеаратом магния и корригирующими добавками и заполняют им пакетики.
Экспериментальный раздел
Следующие примеры приведены для иллюстрации изобретения. Они не должны рассматриваться как ограничивающие объем изобретения, но просто как его образец.
Общее:
МС: масс-спектры (MS) измеряли методом распыления положительных или отрицательных ионов (ISP или ISN) на Perkin-Elmer SCI EX API 300, либо методом электронного удара (EI, 70 эВ) на спектрометре Finnigan MAT SSQ 7000.
Сокращения:
DCAD - ди-пара-хлорбензилазодикарбоксилат; DCC - N,N′-дициклогексил-карбодиимид, DCE - 1,2-дихлорэтан, DCM - дихлорметан, DIPEA - диизопропилэтиламин, DMAc - диметилацетамид, DMAP - 4-диметиламинопиридин, DMF - N,N-диметилформамид, DMSO - диметилсульфоксид, EDCI - N-(3-диметиламинопропил)-N-этил-карбодиимида гидрохлорид, HATU - 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиния-3-оксида гексафторфосфат, DMTMM - 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид; HCl - соляная кислота, ВЭЖХ - высокоэффективная жидкостная хроматография, LDA - лития диизопропиламид, MS - масс-спектр, ЯМР - ядерный магнитный резонанс, TEA - триэтиламин, TBME - трет-бутилметиловый эфир и THF - тетрагидрофуран.
Следующие примеры приведены для иллюстрации изобретения. Их не следует рассматривать как ограничивающие объем изобретения, но просто как его образец.
Синтез промежуточных соединений - сульфинилиминов A2
Общая методика
К раствору (R)-(+)-трет-бутилсульфинамида (66 ммоль) в THF (350 мл) последовательно добавляли кетон A1 (72,6 ммоль) и этоксид титана(IV) (132 ммоль), и раствор перемешивали при температуре флегмообразования в течение 5 ч. Смесь охлаждали до 22°C, обрабатывали рассолом (400 мл), суспензию перемешивали в течение 10 мин и фильтровали через дикалит. Слои разделяли, водный слой экстрагировали этилацетатом, объединенные органические слои промывали водой, сушили и концентрировали в вакууме. Остаток очищали хроматографией на диоксиде кремния, используя циклогексан/этилацетат, с получением чистого сульфинилимина A2.
Промежуточное соединение А2А
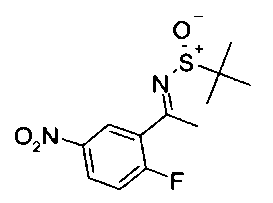
Начиная с 1-(2-фтор-5-нитро-фенил)-этанона, получали продукт - [1-(2-фтор-5-нитро-фенил)-эт-(Е)-илиден]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде бледно-коричневого масла. MS (ISP): m/z равно 287,1 [М+Н]+.
Синтез промежуточных соединений - сложных эфиров сульфинамида A3
Общая методика (через реакцию Реформатского)
В сухой установке нагревали суспензию свежеактивированного цинкового порошка (1,63 г, 24,9 ммоль) в сухом THF (70 мл) в инертной атмосфере до температуры флегмообразования. Добавляли по каплям раствор сульфинилимина A2 (24,9 ммоль) и бромацетата (24,9 ммоль) в сухом THF (15 мл) в течение периода 15 мин, и суспензию нагревали до температуры флегмообразования в течение 5 ч. Охлажденную смесь распределяли между водным насыщенным NH4Cl и этилацетатом, органический слой сушили и упаривали. Неочищенное вещество очищали флэш-хроматографией, используя гептан/этилацетат, с получением сложного эфира сульфинамида A3.
Промежуточные соединения A3A
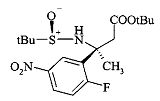
Начиная с [1-(2-фтор-5-нитро-фенил)-эт-(Е)-илиден]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - сложный трет-бутиловый эфир (S)-3-(2-фтор-5-нитро-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-масляной кислоты в виде бледно-коричневого масла. MS (ISP): m/z равно 403,2 [М+Н]+.
Синтез промежуточных соединений - спиртов сульфинамида А4
Общая методика
Раствор сложного эфира сульфинамида A3 (12,7 ммоль) в сухом THF (50 мл) обрабатывали при 0°C боргидридом лития (25,3 ммоль), и перемешивание продолжали при 0°C в течение 4 ч. Реакционную смесь гасили добавлением уксусной кислоты (2 мл) и воды (50 мл), экстрагировали этилацетатом, и органический слой сушили и упаривали. Остаток очищали хроматографией на диоксиде кремния, используя смесь н-гептана и этилацетата, с получением чистого промежуточного соединения - спирта сульфинамида А4.
Промежуточное соединение А4А
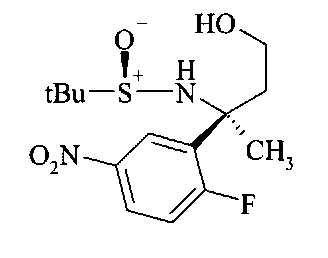
Начиная со сложного трет-бутилового эфира (S)-3-(2-фтор-5-нитро-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-масляной кислоты, получали продукт - [(S)-1-(2-фтор-5-нитро-фенил)-3-гидроски-1-метил-пропил]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде желтого масла. MS (ISP): m/z равно 333,3 [М+Н]+.
Синтез промежуточного соединения - тиоацетата сульфинамида А5 Общая методика
Раствор трифенилфосфина (4,73 г, 18,1 ммоль) в 50 мл сухого DCM обрабатывали DCAD (6,63 г, 18,1 ммоль) при 0°C под аргоном, и образующуюся реакционную смесь перемешивали при 0°C в течение 20 мин. В предыдущую реакционную смесь добавляли раствор тиоуксусной кислоты (1,37 мг, 18,1 ммоль) и спирта А4 (3,0 г, 9,03 ммоль) в 10 мл сухого DCM. Реакционную смесь перемешивали в течение 20 мин при 0°C, давали ей нагреться до комнатной температуры и перемешивали в течение 18 ч при данной температуре. Белый осадок, который образовался по ходу реакции, отфильтровывали. Фильтрат разбавляли дополнительным количеством дихлорметана и экстрагировали 1 М водным раствором карбоната натрия. Органический слой сушили над сульфатом натрия и упаривали досуха. Остаток очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 3,2 г чистого продукта.
Промежуточное соединение А5А
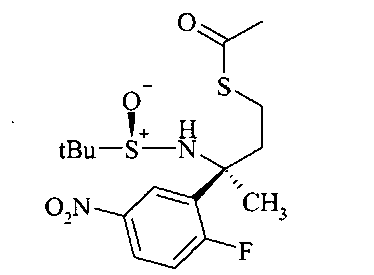
Начиная с [(S)-1-(2-фтор-5-нитро-фенил)-3-гидроски-1-метил-пропил]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - сложный S-[(S)-3-(2-фтор-5-нитро-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-бутиловый эфир] тиоуксусной кислоты в виде желтого масла. MS (ISP): m/z равно 391,3 [М+Н]+.
Синтез промежуточного соединения - нитрила сульфинамида A6
Общая методика
Раствор тиоацетата А5 (1,3 г, 3,33 ммоль) в 19 мл метанола обрабатывали под аргоном бромацетонитрилом (2,23 г, 16,6 ммоль) и карбонатом калия (460 мг, 3,33 ммоль). Образующуюся реакционную смесь перемешивали при комнатной температуре в течение одного часа. Реакционную среду выливали в делительную воронку, заполненную этилацетатом, и экстрагировали водой, органический слой сушили над сульфатом натрия и упаривали досуха. Неочищенный продукт очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 1,05 г нитрила сульфонамида A6.
Промежуточное соединение А6А
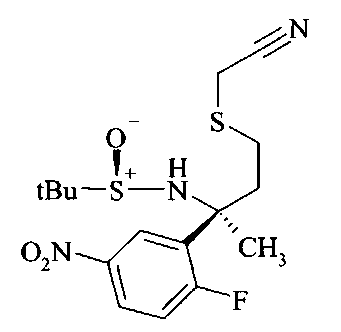
Начиная со сложного S-[(S)-3-(2-фтор-5-нитро-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-бутилового эфира] тиоуксусной кислоты, получали продукт - [(S)-3-цианометилсульфанил-1-(2-фтор-5-нитро-фенил)-1-метил-пропил]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде желтого масла. MS (ISP): m/z равно 388,1 [М+Н]+.
Промежуточное соединение А6В
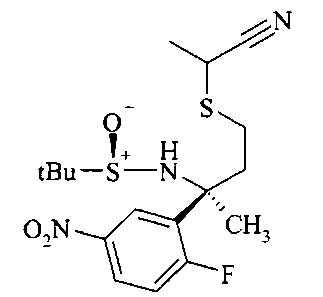
Начиная со сложного S-[(S)-3-(2-фтор-5-нитро-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-бутилового эфира]тиоуксусной кислоты, получали продукт - [(S)-3-(циано-метил-метилсульфанил)-1-(2-фтор-5-нитро-фенил)-1-метил-пропил]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде смеси эпимеров в виде желтого масла. MS (ISP): m/z равно 402,1 [М+Н]+.
Синтез промежуточного соединения - аминонитрила A7
Общая методика
Раствор нитрила сульфонамида (1 г, 2,49 ммоль) в 20 мл MeOH обрабатывали раствором 4 М соляной кислоты в диоксане (1,56 мл, 6,23 ммоль) при 0°C, и реакционную смесь перемешивали при 0°C в течение 30 мин до полного превращения до желательного продукта. Реакционную смесь разбавляли этилацетатом и экстрагировали 2 М водным раствором карбоната натрия. Органический слой сушили над сульфатом натрия и выпаривали досуха под вакуумом. Неочищенный продукт очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 502 мг аминонитрила А7.
Промежуточное соединение А7А
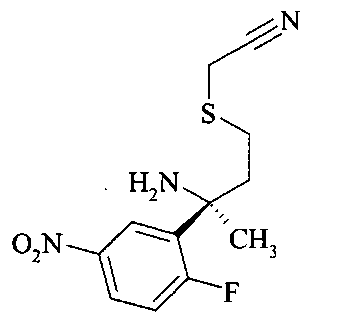
Начиная с [(S)-3-цианометилсул ьфанил-1-(2-фтор-5-нитро-фенил)-1-метил-пропил]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт -[(S)-3-амино-3-(2-фтор-5-нитро-фенил)-бутилсульфанил]-ацетонитрил в виде желтого масла. MS (ISP): m/z равно 284,3 [М+Н]+.
Промежуточное соединение А7В
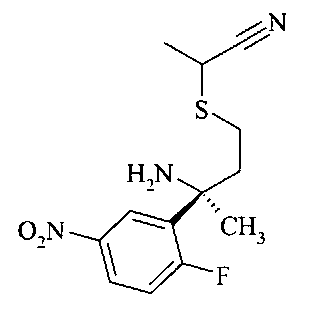
Начиная с [(S)-3-(циано-метил-метилсульфанил)-1-(2-фтор-5-нитро-фенил)-1-метил-пропил]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - 2-[(S)-3-амино-3-(2-фтор-5-нитро-фенил)-бутилсульфанил]-пропионитрил в виде желтого масла. MS (ISP): m/z равно 298,0 [М+Н]+.
Синтез промежуточного соединения - 1,4-тиазепина А8
Общая методика
К раствору аминонитрила A6 (0,715 г, 2,51 ммоль) в 25 мл сухого толуола при 0°C медленно добавляли 2,0 М раствор триметилалюминия в толуоле (1,26 мл, 2,51 ммоль). Образующуюся реакционную смесь перемешивали в течение 30 мин при 0°C и, наконец, перемешивали при 60°C в течение периода двух часов. Реакционную смесь тщательно гасили добавлением воды при 0°C и перемешивали в течение периода 15 мин, образующийся осадок фильтровали через целит. Фильтрат разбавляли этилацетатом, экстрагировали 2 М водным раствором карбоната натрия, и органический слой сушили над сульфатом натрия и упаривали досуха. Неочищенный продукт очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 575 мг желтого масла. Выход: 81%.
Промежуточное соединение А8А
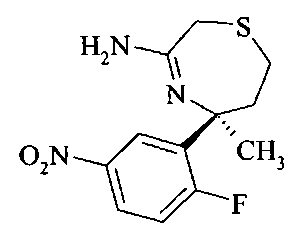
Начиная с [(S)-3-амино-3-(2-фтор-5-нитро-фенил)-бутилсульфанил]-ацетонитрила, получали продукт (S)-5-(2-фтор-5-нитро-фенил)-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламин в виде желтого масла. MS (ISP): m/z равно 284,3 [М+Н]+.
Промежуточное соединение А8В
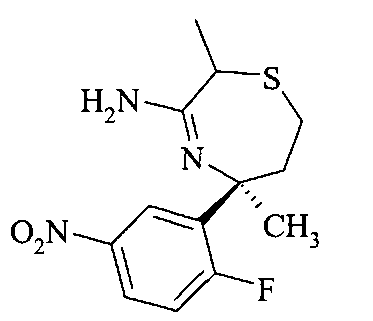
Начиная с 2-[(S)-3-амино-3-(2-фтор-5-нитро-фенил)-бутилсульфанил]-пропионитрила, получали продукт - (S)-5-(2-фтор-5-нитро-фенил)-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламин в виде желтого масла. MS (ISP): m/z равно 298,3 [М+Н]+.
Синтез промежуточного соединения - анилина 9
Общая методика
К раствору нитробензола А7 (140 мг, 0,47 ммоль) в 4,0 мл EtOH добавляли SnCl2·2H2O (321 мг, 1,42 ммоль) (мгновенно формировался осадок, который растворялся при нагревании). Реакционную смесь перемешивали при 80°C в течение 1,5 ч и контролировали посредством ТСХ (тонкослойная хроматография) Si-NH2 (CH2Cl2/MeOH/NH4OH 80:18:2), которая показывала полное превращение. Реакционную смесь вливали в водный раствор 1 н. NaOH, добавляли этилацетат, и смесь перемешивали в течение 10 мин. Осадок фильтровали через целит, и две фазы в фильтрате разделяли. Органическую фазу сушили над Na2SO4, фильтровали и упаривали досуха. Остаток очищали хроматографией на аминомодифицированном диоксиде кремния со смесью CH2Cl2 и МеОН с получением чистого анилина.
Промежуточное соединение А9А
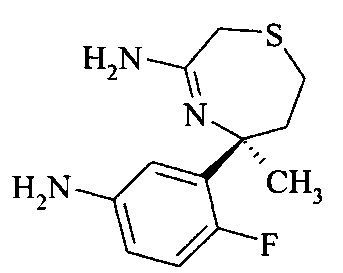
Начиная с (S)-5-(2-фтор-5-нитро-фенил)-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламина, получали продукт - (S)-5-(5-амино-2-фтор-фенил)-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламин в виде бесцветного масла. MS (ISP): m/z равно 254,3 [М+Н]+.
Промежуточное соединение А9 В
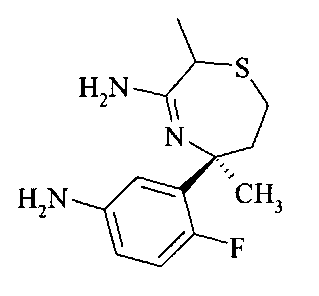
Начиная с (S)-5-(2-фтор-5-нитро-фенил)-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламина, получали продукт - (S)-5-(5-амино-2-фтор-фенил)-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламин в виде бесцветного масла. MS (ISP): m/z равно 268,1 [М+Н]+.
Синтез промежуточного соединения - амида А10
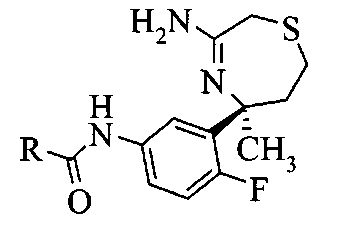
Общая методика
К раствору кислоты (0,16 ммоль) в МеОН (1 мл) при 22°C добавляли 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метил-морфолиния хлорид (0,19 ммоль), и перемешивание продолжали при 0°C в течение 30 мин. К смеси добавляли раствор анилина А9 (0,15 ммоль) в МеОН (2 мл), и перемешивание продолжали при 0°C в течение 2 ч. Смесь разбавляли насыщенным водным Na2CO3, МеОН выпаривали, и водный раствор экстрагировали этилацетатом. Органический слой сушили, упаривали, и остаток очищали посредством хроматографии на силикагеле, используя смесь дихлорметана и раствора 3% триэтиламина в метаноле.
Синтез промежуточного соединения - амида А11
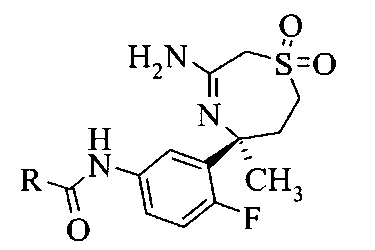
Общая методика
Раствор амида А10 (0,05 г, 0,133 ммоль) в 1,5 мл сухом CH2Cl2 обрабатывали мета-СРВА (0,065 г, 0,266 ммоль) при 0°C, и образующуюся реакционную смесь перемешивали в течение периода 1 ч при комнатной температуре. Реакционную смесь разбавляли дихлорметаном и экстрагировали 2 М водным раствором карбоната натрия. Органический слой сушили над сульфатом натрия и выпаривали досуха. Неочищенный продукт очищали хроматографией на силикагеле с использованием смеси дихлорметана и раствора 3% триэтиламина в метаноле.
Синтез промежуточных соединений - сульфинилиминов B2
Общая методика
К раствору (R)-(+)-трет-бутилсульфинамида (66 ммоль) в THF (350 мл) последовательно добавляли кетон В1 (72,6 ммоль) и этоксид титана(IV) (132 ммоль), и раствор перемешивали при температуре флегмообразования в течение 5 ч. Смесь охлаждали до 22°C, обрабатывали рассолом (400 мл), суспензию перемешивали в течение 10 мин и фильтровали через дикалит. Слои разделяли, водный слой экстрагировали этилацетатом, объединенные органические слои промывали водой, сушили и концентрировали в вакууме. Остаток очищали хроматографией на диоксиде кремния, используя циклогексан/этилацетат, с получением чистого сульфинилимина B2.
Промежуточное соединение В2А
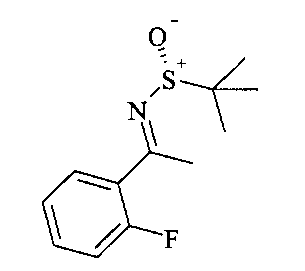
Начиная с 1-(2-фтор-фенил)-этанона, получали продукт - [1-(2-фтор-фенил)-эт-(Е)-илиден]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде бледно-коричневого масла. MS (ISP): m/z равно 242,2 [М+Н]+.
Синтез промежуточных соединений - сложных эфиров сульфинамида B3
Общая методика (через реакцию Реформатского)
В сухой установке нагревали суспензию свежеактивированного цинкового порошка (1,63 г, 24,9 ммоль) в сухом THF (70 мл) в инертной атмосфере до температуры флегмообразования. Добавляли по каплям раствор сульфинилимина B2 (24,9 ммоль) и бромацетата (24,9 ммоль) в сухом THF (15 мл) в течение периода 15 мин, и суспензию нагревали до температуры флегмообразования в течение 5 ч. Охлажденную смесь распределяли между водным насыщенным NH4Cl и этилацетатом, органический слой сушили и упаривали. Неочищенное вещество очищали флэш-хроматографией, используя гептан/этилацетат, с получением сложного эфира сульфинамида B3.
Промежуточные соединения В3А
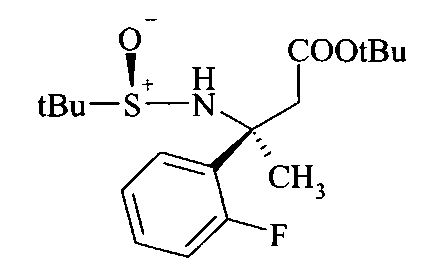
Начиная с [1-(2-фтор-фенил)-эт-(Е)-илиден]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - сложный трет-бутиловый эфир (S)-3-(2-фтор-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-масляной кислоты в виде бледно-коричневого масла. МС (ISP): m/z равно 358,1 [М+Н]+.
Синтез промежуточных соединений - спиртов сульфинамида В4
Общая методика
Раствор сложного эфира сульфинамида B3 (12,7 ммоль) в сухом THF (50 мл) обрабатывали при 0°C боргидридом лития (25,3 ммоль), и перемешивание продолжали при 0°C в течение 4 ч. Реакционную смесь гасили добавлением уксусной кислоты (2 мл) и воды (50 мл), экстрагировали этилацетатом, и органический слой сушили и упаривали. Остаток очищали хроматографией на диоксиде кремния, используя смесь н-гептана и этилацетата, с получением чистого промежуточного соединения - спирта сульфинамида B4.
Промежуточное соединение В4А
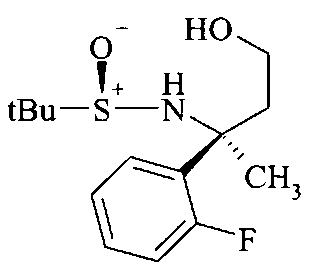
Начиная со сложного трет-бутилового эфира (S)-3-(2-фтор-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-масляной кислоты, получали продукт - [(S)-1-(2-фтор-фенил)-3-гидрокси-1-метил-пропил]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде желтого масла. MS (ISP): m/z равно 288,2 [М+Н]+.
Синтез промежуточного соединения - тиоацетата сульфинамида В5
Общая методика
Раствор трифенилфосфина (4,73 г, 18,1 ммоль) в 50 мл сухого DCM обрабатывали DCAD (6,63 г, 18,1 ммоль) при 0°C под аргоном, и образующуюся реакционную смесь перемешивали при 0°C в течение 20 мин. Предыдущую реакционную смесь добавляли в раствор тиоуксусной кислоты (1,37 мг, 18,1 ммоль) и спирта В4 (3,0 г, 9,03 ммоль) в 10 мл сухого DCM. Реакционную смесь перемешивали в течение 20 мин при 0°C, оставляли нагреваться до комнатной температуры и перемешивали в течение 18 ч при данной температуре. Белый осадок, который формировался по ходу реакции, отфильтровывали. Фильтрат разбавляли дополнительным количеством DCM и экстрагировали 1 М водным раствором Na2CO3. Органический слой сушили над сульфатом натрия и упаривали досуха. Остаток очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 3,2 г тиоацетата сульфонамида В5.
Промежуточное соединение В5А
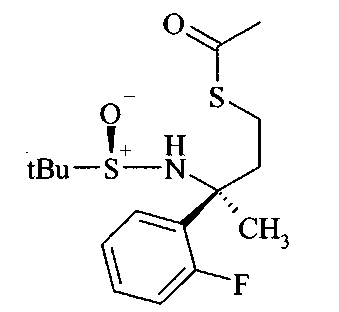
Начиная с [(3)-1-(2-фтор-фенил)-3-гидрокси-1-метил-пропил]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - сложный S-[(S)-3-(2-фтор-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-бутиловый] эфир тиоуксусной кислоты в виде желтого масла. MS (ISP): m/z равно 346,2 [М+Н]+.
Синтез промежуточного соединения - тиола сульфинамида B6
Общая методика
Раствор тиоацетата сульфонамида В5 (1,25 г, 3,62 ммоль) в 25 мл МеОН обрабатывали K2CO3 (600 мг, 4,34 ммоль), реакционную смесь перемешивали при комнатной температуре в течение периода один час. Реакционную смесь разбавляли этилацетатом и экстрагировали насыщенным раствором хлорида аммония. Органический слой сушили над сульфатом натрия и упаривали досуха в вакууме. Остаток очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 810 мг тиола сульфонамида B6.
Промежуточное соединение В6А
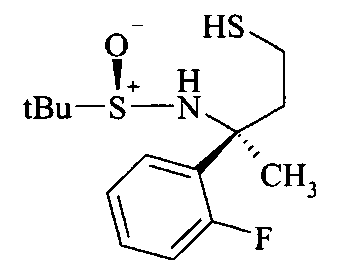
Начиная со сложного 5-[(S)-3-(2-фтор-фенил)-(R)-3-(2-метил-пропан-2-сульфиниламино)-бутилового] эфира тиоуксусной кислоты, получали продукт - [(S)-1-(2-фтор-фенил)-3-меркапто-1-метил-пропил]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде желтого масла. MS (ISP): m/z равно 304,4 [М+Н]+.
Синтез промежуточного соединения - сложного эфира сульфинамида B7
Общая методика
Раствор тиола сульфинамида B6 (1,8 г, 5,93 ммоль) в 30 мл CH3CN обрабатывали бромзамещенным сложным эфиром (1,45 г, 7,41 ммоль) и карбонатом калия (1,23 г, 8,9 ммоль), реакционную смесь перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли этилацетатом и экстрагировали водой, органический слой сушили над сульфатом натрия и упаривали досуха. Остаток очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 1,68 г сложного эфира сульфинамида В7 в виде бесцветного масла.
Промежуточное соединение В7А
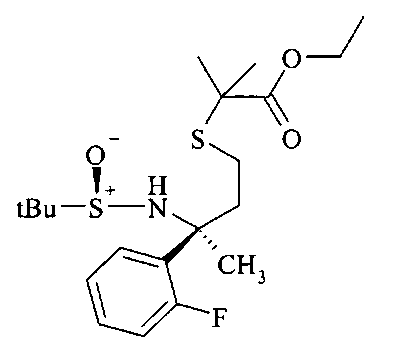
Начиная с [(S)-1-(2-фтор-фенил)-3-меркапто-1-метил-пропил]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - сложный этиловый эфир (R)-2-[(S)-3-(2-фтор-фенил)-3-(2-метил-пропан-2-сульфиниламино)-бутилсульфанил]-2-метил-пропионовой кислоты в виде желтого масла. MS (ISP): m/z равно 418,4 [М+Н]+.
Синтез промежуточного соединения - сложного эфира амина В8
Общая методика
Раствор сложного эфира сульфонамида B7 (1,47 г, 3,52 ммоль) в 15 мл МеОН при 0°C обрабатывали 4 М раствором соляной кислоты в диоксане (4,4 мл, 17,6 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение периода один час. Реакционную смесь разбавляли этилацетатом и экстрагировали 2 М водным раствором карбоната натрия. Органический слой сушили над сульфатом натрия и упаривали досуха под вакуумом. Остаток очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 1,05 г сложного эфира амина В8.
Промежуточное соединение В8А
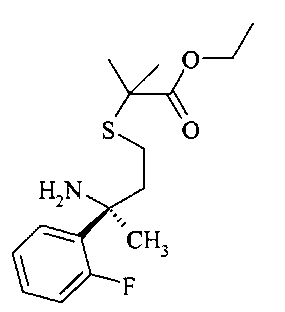
Начиная со сложного этилового эфира (R)-2-[(S)-3-(2-фтор-фенил)-3-(2-метил-пропан-2-сульфиниламино)-бутилсульфанил]-2-метил-пропионовой кислоты, получали продукт - сложный этиловый эфир 2-[(S)-3-амино-3-(2-фтор-фенил)-бутилсульфанил]-2-метил-пропионовой кислоты в виде желтого масла. MS (ISP): m/z равно 314,3 [М+Н]+.
Синтез промежуточного соединения - лактама B9
Общая методика
Раствор сложного эфира амина В8 (1,05 г, 3,35 ммоль) в 20 мл сухого THF под аргоном обрабатывали бис[бис(триметилсилил)амино]оловом(II) (1,49 мл, 3,85 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 50°C в течение периода трех часов, и реакционную среду вливали в смесь воды и этилацетата, которую перемешивали в течение периода 15 мин. Образующуюся смесь фильтровали через Celite® для удаления осадка, органический слой собирали, сушили над сульфатом натрия и упаривали досуха под вакуумом. Остаток очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 480 мг лактама В9.
Промежуточное соединение В9А
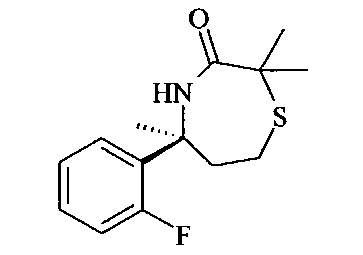
Начиная со сложного этилового эфира 2-[(S)-3-амино-3-(2-фтор-фенил)-бутилсульфанил]-2-метил-пропионовой кислоты, получали продукт - (S)-5-(2-фтор-фенил)-2,2,5-триметил-[1,4]тиазепан-3-он в виде желтого масла. MS (ISP): m/z равно 268,1 [М+Н]+.
Синтез промежуточного соединения - сульфона В10
Общая методика
Раствор лактама В9 (480 мг, 1,8 ммоль) в 15 мл CH2Cl2 обрабатывали мета-СРВА (929 мг, 3,77 ммоль) при комнатной температуре, реакционную смесь перемешивали в течение периода 14 часов при комнатной температуре. Реакционную смесь разбавляли дихлорметаном и экстрагировали 2 М водным раствором карбоната натрия, органический слой сушили над сульфатом натрия и упаривали досуха под вакуумом. Неочищенный продукт очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 525 мг сульфона В10.
Промежуточное соединение В10А
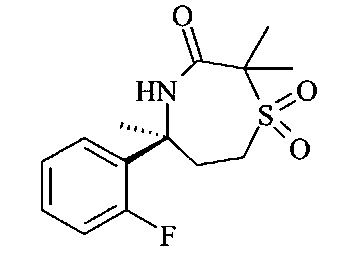
Начиная с (S)-5-(2-фтор-фенил)-2,2,5-триметил-[1,4]тиазепан-3-она, получали продукт - (S)-5-(2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-1λ6-[1,4]тиазепан-3-он в виде бесцветного масла. MS (ISP): m/z равно 300,1 [М+Н]+.
Синтез промежуточного соединения - тиоамида В11
Общая методика
Раствор сульфона В10 (525 мг, 1,75 ммоль) в 10 мл сухого THF обрабатывали реактивом Лоуссона (922 мг, 2,28 ммоль) при комнатной температуре, и реакционную смесь перемешивали при 85°C в течение периода 2 часов. Реакционную среду разбавляли этилацетатом, экстрагировали водой. Органический слой сушили над сульфатом натрия и упаривали досуха под вакуумом. Неочищенный продукт очищали хроматографией на силикагеле, используя смесь н-гептана и этилацетата, с получением 415 мг тиоамида В11.
Промежуточное соединение В11А
Начиная с (S)-5-(2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-1λ6-[1,4]тиазепан-3-она, получали продукт - (S)-5-(2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепан-3-тион в виде желтого масла. MS (ISP): m/z равно 316,1 [М+Н]+.
Синтез промежуточного соединения - 1,1-диоксо-[1,4] тиазепина В12
Общая методика
Раствор тиоамида В11 (415 мг, 1,32 ммоль) в 5 мл THF обрабатывали хлоридом ртути(II) (357 мг, 1,32 ммоль) и раствором 7 н. аммиака в МеОН (752 мкл, 5,26 ммоль). Реакционную смесь перемешивали при 120°C в течение 30 мин в микроволновом резонаторе. Реакционную смесь фильтровали на целите для удаления соли ртути, которая осаждалась в виде черного порошка, фильтрат разбавляли этилацетатом и экстрагировали 2 М водным раствором карбоната натрия. Органический слой сушили над сульфатом натрия и упаривали досуха под вакуумом. Неочищенный продукт очищали хроматографией на силикагеле, используя смесь дихлорметана и метанола, с получением 1,1-диоксо-[1,4]тиазепина В12.
Промежуточное соединение В12А
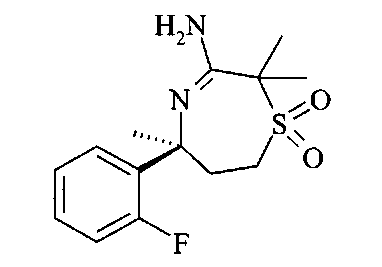
Начиная с (S)-5-(2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепан-3-тиона, получали продукт - (S)-5-(2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-иламин в виде бесцветного масла. MS (ISP): m/z равно 299,1 [М+Н]+.
Синтез промежуточного нитросоединения В13
Общая методика
1,1-диоксо-[1,4]тиазепин В12 (300 мг, 1,01 ммоль) растворяли в H2SO4 97% (для полного растворения была необходимой обработка ультразвуком, при полном растворении появлялось сильное темно-красное окрашивание), и реакционную смесь охлаждали до 0°C, с последующим медленным добавлением 100% азотной кислоты (45 мкл, 1,01 ммоль). Образующуюся реакционную смесь перемешивали в течение периода 15 минут при 0°C. Реакционную среду медленно вливали в смесь этилацетата и воды/льда, образующуюся смесь перемешивали в течение 10 мин. Предыдущую смесь охлаждали до 0°C, и добавляли карбонат калия, пока pH не достигал 10-11. Разделяли две фазы, и органический слой сушили над сульфатом натрия и упаривали досуха под вакуумом. Неочищенный продукт очищали хроматографией на силикагеле, используя смесь дихлорметана и раствора 10% триэтиламина в метаноле, с получением 185 мг нитросоединения В13.
Промежуточное соединение В13А
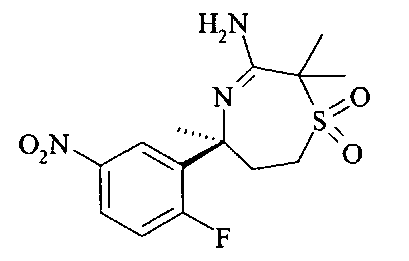
Начиная с (S)-5-(2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-иламина, получали продукт - (S)-5-(2-фтор-5-нитро-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-иламин в виде бесцветного масла. MS (ISP): m/z равно 344,1 [М+Н]+.
Синтез промежуточного соединения - анилина В14
Общая методика
К раствору нитробензола В13 (140 мг, 0,47 ммоль) в 4,0 мл EtOH добавляли SnCl2·2H2O (321 мг, 1,42 ммоль) (мгновенно формировался осадок, который растворялся при нагревании). Реакционную смесь перемешивали при 80°C в течение 1,5 ч и контролировали посредством ТСХ Si-NH2 (CH2Cl2/MeOH/NH4OH 80:18:2), которая показывала полное превращение. Реакционную смесь вливали в водный раствор 1 н. NaOH, добавляли этилацетат, и смесь перемешивали в течение 10 мин. Осадок фильтровали через Celite®, две фазы в фильтрате разделяли. Органическую фазу сушили над Na2SO4, фильтровали и упаривали досуха. Остаток очищали хроматографией на аминомодифицированном диоксиде кремния со смесью CH2Cl2 и МеОН с получением чистого анилина В14.
Промежуточное соединение В14А
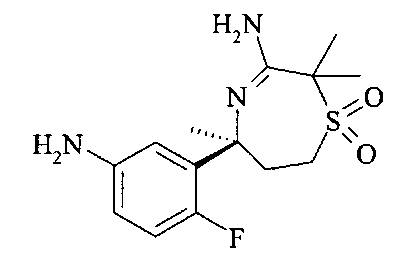
Начиная с (S)-5-(2-фтор-5-нитро-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-3-иламина, получали продукт - (S)-5-(5-амино-2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-иламин в виде бесцветного масла. MS (ISP): m/z равно 314,1 [М+Н]+.
Промежуточное соединение В14А
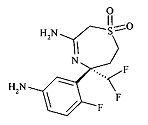
Раствор {(RS)-5-[5-(бензгидрилиден-амино)-2-фтор-фенил]-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-ил}-[бис-(4-метокси-фенил)-фенил-метил]-амина (207 мг, 263 мкмоль) в дихлорметане (10 мл) обрабатывали трифторуксусной кислотой (1,01 мл, 13,1 ммоль). Раствор оранжевого цвета перемешивали при комнатной температуре в течение 1 часа. Для расщепления промежуточного соединения - бензофенонимина добавляли соляную кислоту (1 М, 2,63 мл) и диоксан (10 мл). Смесь перемешивали при 23°C в течение 4 часов. Для обработки реакционную смесь вливали в водный раствор карбоната натрия (1 М), с последующей экстракцией дихлорметаном. Органический слой промывали рассолом, сушили над сульфатом натрия и упаривали. Неочищенный продукт очищали хроматографией на силикагеле с использованием смеси 110:10:1 дихлорметана, метанола и гидроксида аммония в качестве элюента. (RS)-5-(5-амино-2-фтор-фенил)-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-иламин (72 мг, выход 85%) получали в виде светло-коричневой пены. MS (ISP): m/z равно 322,0 [М+Н]+.
Синтез промежуточного соединения - амида В15
Общая методика
В раствор кислоты (0,16 ммоль) в МеОН (1 мл) добавляли 4-(4,6-диметокси-1,3,5-триазин-2ил)-4-метил-морфолиния хлорид (0,19 ммоль) при 22°C, и перемешивание продолжали при 0°C в течение 30 мин. Смесь добавляли в раствор анилина В15 (0,15 ммоль) в МеОН (2 мл), и перемешивание продолжали при 0°C в течение 2 ч. Смесь разбавляли насыщенным водным Na2CO3, MeOH выпаривали, и водный раствор экстрагировали этилацетатом. Органический слой сушили, упаривали, и остаток очищали хроматографией на силикагеле, используя смесь дихлорметана и раствора 3% триэтиламина в метаноле.
Промежуточное соединение С2А
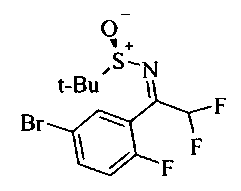
Аналогично способу, описанному для получения А2А, начиная с 1-(5-бром-2-фторфенил)-2,2-дифторэтанона (WO 2011009943; CAS 1262858-97-8), получали продукт - [1-(5-бром-2-фтор-фенил)-2,2-дифтор-эт-(Е)-илиден]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде желтого масла. MS (EI): m/z равно 298 [M-t-Bu+H]+ и 300 [M-t-Bu+2+H]+.
Промежуточное соединение С3А
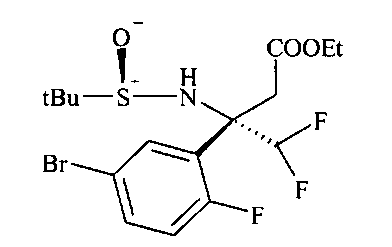
Аналогично способу, описанному для получения А3А, начиная с [1-(5-бром-2-фтор-фенил)-2,2-дифтор-эт-(Е)-илиден]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - сложный этиловый эфир (S)-3-(5-бром-2-фтор-фенил)-4,4-дифтор-3-(R)-2-метил-пропан-2-сульфиниламино)-масляной кислоты (содержащий 30% нежелательного (R)-диастереомера, который удаляли препаративной хиральной ВЭЖХ на колонке Reprosil Chiral NR с 5% EtOH в н-гептане в качестве элюента) в виде бесцветного масла. MS (ISP): m/z равно 444,0 [М+Н]+ и 446,0 [М+2+Н]+.
Промежуточное соединение С4А
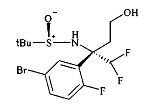
Аналогично способу, описанному для получения А4А, начиная со сложного этилового эфира (S)-3-(5-бром-2-фтор-фенил)-4,4-дифтор-3-(R)-2-метил-пропан-2-сульфиниламино)-масляной кислоты, получали продукт - [(S)-1-(5-бром-2-фтор-фенил)-1-дифторметил-3-гидрокси-пропил]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде бесцветного масла. MS (ISN): m/z равно 399,9 [M-H]- и 401,9 [М+2-Н]-.
Промежуточное соединение С5А
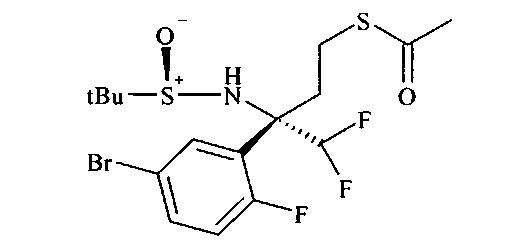
Аналогично способу, описанному для получения А5А, начиная с [(S)-1-(5-бром-2-фтор-фенил)-1-дифторметил-3-гидрокси-пропил]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - сложный S-[(S)-3-(5-бром-2-фтор-фенил)-4,4-дифтор-3-((R)-2-метил-пропан-2-сульфиниламино)-бутиловый] эфир тиоуксусной кислоты в виде светло-желтого масла. MS (ISP): m/z равно 460,2 [М+Н]+ и 462,1 [М+2+Н]+.
Промежуточное соединение С6А
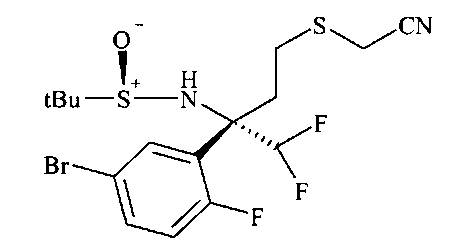
Аналогично способу, описанному для получения А6А, начиная со сложного 8-[(S)-3-(5-бром-2-фтор-фенил)-4,4-дифтор-3-((R)-2-метил-пропан-2-сульфиниламино)-бутилового] эфира тиоуксусной кислоты, получали продукт - [(S)-1-(5-бром-2-фтор-фенил)-3-цианометилсульфанил-1-дифторметил-пропил]-амид (R)-2-метил-пропан-2-сульфиновой кислоты в виде бесцветного масла. MS (ISP): m/z равно 454,9 [М+Н]+ и 457,1 [М+2+Н]+.
Промежуточное соединение С7А
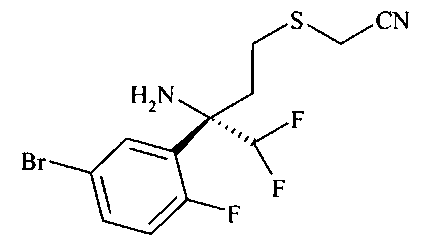
Аналогично способу, описанному для получения А7А, начиная с [(S)-1-(5-бром-2-фтор-фенил)-3-цианометилсульфанил-1-дифторметил-пропил]-амида (R)-2-метил-пропан-2-сульфиновой кислоты, получали продукт - [(S)-3-амино-3-(5-бром-2-фтор-фенил)-4,4-дифтор-бутилсульфанил]-ацетонитрил в виде бесцветного масла. MS (ISP): m/z равно 352,9 [М+Н]+ и 354,9 [М+2+Н]+.
Промежуточное соединение С8А
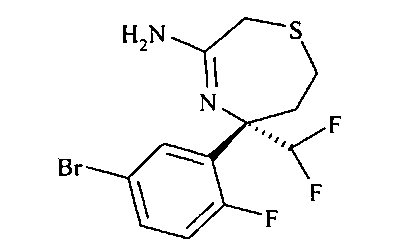
Аналогично способу, описанному для получения А8А, начиная с [(S)-3-амино-3-(5-бром-2-фтор-фенил)-4,4-дифтор-бутилсульфанил]-ацетонитрила, получали продукт - (S)-5-(5-бром-2-фтор-фенил)-5-дифторметил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламин в виде светло-желтого твердого вещества. MS (ISP): m/z равно 353,0 [М+Н]+ и 355,0 [М+2+Н]+.
Промежуточное соединение С9А
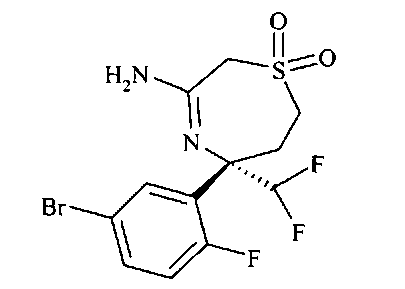
Раствор (S)-5-(5-бром-2-фтор-фенил)-5-дифторметил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламина (364 мг, 1,03 ммоль) в метаноле (20 мл) обрабатывали в двух порциях пероксимоносульфатом калия (оксон) (1,27 г, 2,06 ммоль) при 23°C. Белую суспензию перемешивали при 23°C в течение 5 часов. Для обработки реакционную смесь гасили при 0°C водой (10 мл) при энергичном перемешивании, затем обрабатывали разбавленным раствором гидросульфита натрия, насыщенным раствором натрия и дихлорметаном. Энергичное перемешивание продолжали в течение 10 мин. Органический слой отделяли, промывали водой, сушили над сульфатом натрия и упаривали с получением бесцветного масла. Неочищенный продукт очищали хроматографией на силикагеле, используя градиент дихлорметана и метанола от 100:0 до 90:10 в качестве элюента. (S)-5-(5-бром-2-фтор-фенил)-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-иламин (322 мг, выход 81%) получали в виде белого твердого вещества. MS (ISP): m/z равно 384,9 [М+Н]+ и равно 386,9 [М+2+Н]+.
Промежуточное соединение С10А
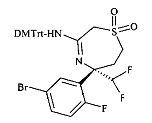
Раствор (S)-5-(5-бром-2-фтор-фенил)-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-3-иламина (318 мг, 826 мкмоль) и триэтиламина (167 мг, 1,65 ммоль) в дихлорметане (15 мл) обрабатывали 4,4′-диметокситритилхлоридом (336 мг, 991 мкмоль). Зеленую реакционную смесь перемешивали при комнатной температуре в течение 6 часов. Затем реакционную смесь упаривали, и неочищенный продукт очищали хроматографией на силикагеле, используя градиент гептана и этилацетата от 100:0 до 50:50 в качестве элюента. Получали [бис-(4-метокси-фенил)-фенил-метил]-[(S)-5-(5-бром-2-фтор-фенил)-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-ил]-амин (365 мг, выход 63%) в виде серой пены. MS (ISP): m/z равно 687,0 [М+Н]+ и 689,3 [М+2+Н]+.
Промежуточное соединение С11А
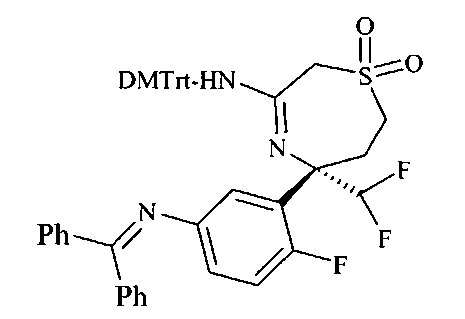
В пробирке в атмосфере аргона раствор [бис-(4-метокси-фенил)-фенил-метил]-[(S)-5-(5-бром-2-фтор-фенил)-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-3-ил]-амина (356 мг, 518 мкмоль) в толуоле (5 мл) последовательно обрабатывали трет-бутоксидом натрия (149 мг, 1,55 ммоль), 2-ди-трет-бутилфосфино-2′,4′,6′-триизопропилбифенилом (33 мг, 77,7 мкмоль), аддуктом трис(дибензилиденацетон)дипалладия(0) и хлороформа (26,8 мг, 26 мкмоль) и бензофенонимином (188 мг, 1,04 ммоль). Пробирку запечатывали, и смесь нагревали при 105°C при перемешивании в течение 4 часов. Для обработки коричневый раствор экстрагировали этилацетатом и водой. Органический слой промывали рассолом, сушили над сульфатом натрия и упаривали при пониженном давлении. Неочищенный продукт очищали хроматографией на силикагеле, используя градиент гептана и этилацетата от 100:0 до 50:50 в качестве элюента. {(S)-5-[5-(Бензгидрил иден-амино)-2-фтор-фенил]-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-3-ил}-[бис-(4-метокси-фенил)-фенил-метил]-амин (207 мг, выход 51%) получали в виде светло-желтой пены. MS (ISN): m/z равно 786,5 [M-H]+.
Промежуточное соединение D3
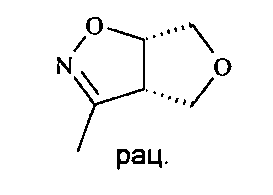
Раствор 2,5-дигидрофурана (5 г, 69,9 ммоль) в эфире (90 мл) обрабатывали по каплям раствором нитроэтана (5,46 г, 71,3 ммоль) в эфире (15 мл) при комнатной температуре. Затем добавляли триэтиламин (70,7 мг, 699 мкмоль), с последующим добавлением по каплям фенилизоцианата (17,3 г, 143 ммоль). Реакционную смесь перемешивали в течение 3 суток при комнатной температуре. Для обработки белый осадок фильтровали и промывали эфиром. Фильтрат упаривали, и неочищенный продукт очищали хроматографией с использованием градиента гептана и этилацетата от 100:0 до 50:50 в качестве элюента. После второй хроматографии получали (3aS,6aS)-rel-3-метил-3a,4,6,6a-тетрагидрофуро[3,4-d]изоксазол (выход 58%) в виде желтого масла.
Промежуточное соединение D5
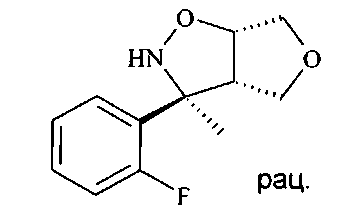
Раствор 1-бром-2-фторбензола (31,1 г, 178 ммоль) в смеси толуола (750 мл) и тетрагидрофурана (75 мл) охлаждали до -100°C. В пределах 10 минут добавляли по каплям раствор н-бутиллития (1,6 М в гексане; 100 мл, 160 ммоль) с такой скоростью, что могла поддерживаться температура от -95 до 102°C. После 10 минут перемешивания при -100°C добавляли смесь (3aS,6aS)-rel-3-метил-3a,4,6,6а-тетрагидрофуро[3,4-d]изоксазола (11,3 г, 88,9 ммоль) и бора трифториддиэтилэтерата (25,2 г, 178 ммоль) в смеси толуола (75 мл) и тетрагидрофурана (14 мл) в пределах 3-5 минут, поддерживая температуру ниже -94°C. Реакционную смесь перемешивали еще 10 минут при температуре от -95 до -102°C. Для обработки добавляли насыщенный раствор хлорида аммония (60 мл), воду (100 мл) и этилацетат (200 мл), и оставляли реакционную смесь нагреваться до 0°C. Слои разделяли, и водный слой экстрагировали еще один раз этилацетатом. Объединенные органические слои промывали рассолом, сушили над сульфатом натрия и упаривали при пониженном давлении. Неочищенный продукт очищали хроматографией с использованием градиента гептана и этилацетата от 100:0 до 0:100 в качестве элюента. Получали (3S,3aS,6aS)-rel-3-(2-фтор-фенил)-3-метил-гексагидро-фуро[3,4-d]изоксазол (17 г, выход 81%) в виде беловатого твердого вещества. MS (ISP): m/z равно 224,1 [М+Н]+.
Промежуточное соединение D6
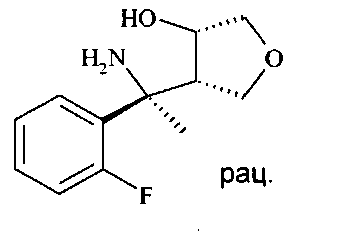
Раствор (3S,3aS,6aS)-rel-3-(2-фтор-фенил)-3-метил-гексагидро-фуро[3,4-d]изоксазола (16,72 г, 72,9 ммоль) в этаноле (300 мл) обрабатывали формиатом аммония (36,8 г, 583 ммоль) и палладием (5% на углероде; 7,76 г). Реакционную смесь перемешивали при комнатной температуре в течение 18 часов, затем фильтровали, и фильтрат упаривали при пониженном давлении. Неочищенный продукт измельчали с диизопропиловым эфиром (50 мл) и фильтровали. Фильтрат упаривали при пониженном давлении и получали (3S,4S)-4-[(S)-rel-1-амино-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ол (8,69 г, выход 48%) в виде густого бесцветного масла. MS (ISP): m/z равно 226,2 [М+Н]+. Твердое вещество, полученное после фильтрования, rel-(3S,4S)-4-[(S)-1-амино-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ол (8,21 г, выход 42%), обрабатывали насыщенным раствором гидрокарбоната натрия (100 мл), дихлорметаном (100 мл) и перемешивали в течение 1 часа. Водный слой отделяли и экстрагировали дихлорметаном, затем объединенные органические слои сушили над сульфатом натрия и упаривали. Получали дополнительное количество rel-(3S,4S)-4-[(S)-1-амино-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ола (5,6 г, выход 31%) в виде густого бесцветного масла. MS (ISP): m/z равно 226,2 [М+Н]+.
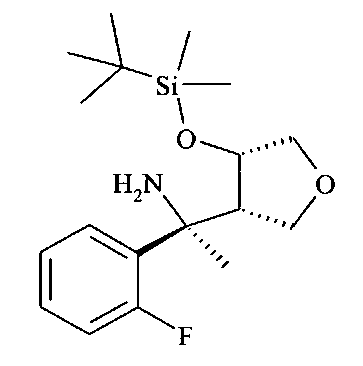
Промежуточное соединение D7
В раствор rel-(3S,4S)-4-[(S)-1-амино-1-(2-фторфенил)этил]тетрагидрофуран-3-ола (5,6 г, 24,9 ммоль) в дихлорметане (100 мл) добавляли триэтиламин (8,05 г, 79,6 ммоль) при 0°C, затем 4-диметиламинопиридин (1,52 г, 12,4 ммоль), с последующим добавлением трет-бутилдиметилхлорсилана (7,49 г, 49,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Для обработки реакционную смесь экстрагировали насыщенным раствором гидрокарбоната натрия и рассолом. Органический слой сушили над сульфатом натрия и упаривали. Неочищенное вещество очищали флэш-хроматографией на силикагеле, используя градиент гептана и этилацетата от 100:0 до 50:50 в качестве элюента. Получали rel-(S)-1-[(35,45)-4-(ттрет-бутил-диметил-силанокси)-тетрагидро-фуран-3-ил]-1-(2-фтор-фенил)-этиламин (8,13 г, выход 96%) в виде бесцветного масла. MS (ISP): m/z равно 340,1 [М+Н]+.
Промежуточное соединение D9
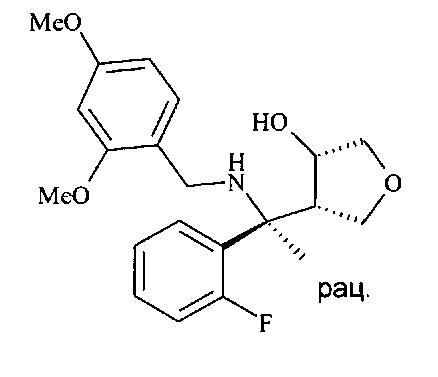
Раствор rel-(S)-1-[(3S,4S)-4-(трет-бутил-диметил-силанокси)-тетрагидро-фуран-3-ил]-1-(2-фтор-фенил)-этиламина (8,13 г, 23,9 ммоль) в 1,2-дихлорэтане (80 мл) обрабатывали в атмосфере аргона 2,4-диметоксибензальдегидом (3,98 г, 23,9 ммоль), триацетоксиборгидридом натрия (10,2 г, 47,9 ммоль) и уксусной кислотой (1,37 мл, 23,9 ммоль). Реакционную смесь перемешивали в течение ночи. Для завершения реакции добавляли 2,4-диметоксибензальдегид (1,99 г, 12,0 ммоль) и триацетоксиборгидрид натрия (5,08 г, 23,9 ммоль), и смесь перемешивали в течение ночи. Затем реакционную смесь охлаждали до 0°C и добавляли тетрабутиламмония фторид тригидрат (7,56 г, 23,9 ммоль). Давали смеси нагреться до комнатной температуры при перемешивании в течение 4 часов. Для завершения реакции вновь добавляли тетрабутиламмония фторид тригидрат (2,27 г, 7,18 ммоль), и перемешивание продолжали в течение 4 суток. Для обработки реакционную смесь дважды экстрагировали смесью насыщенного раствора гидрокарбоната натрия и дихлорметана. Объединенные органические слои сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле, используя градиент гептана и этилацетата от 100:0 до 50:50 в качестве элюента. Получали rel-(3S,4S)-4-[(S)-1-(2,4-диметокси-бензиламино)-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ол (6,8 г, выход 76%) в виде бесцветного аморфного вещества.
Промежуточное соединение D10
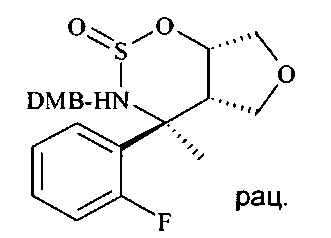
В сухой атмосфере аргона раствор rel-(3S,4S)-4-[(S)-1-(2,4-диметокси-бензиламино)-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-ола (6,8 г, 18,1 ммоль) в пиридине (7,32 мл, 90,5 ммоль) и дихлорметане (120 мл) охлаждали до -77°C. Бесцветный раствор обрабатывали по каплям хлористым тионилом (2,15 г, 18,1 ммоль) в течение примерно 10 мин, в то время как температура увеличивалась до -73°C. После удаления охлаждающей бани давали реакционной смеси достигнуть комнатной температуры. Для обработки реакционную смесь разбавляли дихлорметаном (150 мл) и последовательно промывали соляной кислотой (1 н.) и насыщенным раствором гидрокарбоната натрия. Органический слой сушили над сульфатом натрия и упаривали. (3aS,7S,7aS)-rel-6-(2,4-диметокси-бензил)-7-(2-фтор-фенил)-7-метил-гексагидро-2,4-диокса-5-тиа-6-аза-инден 5-оксид (7,24 г, выход 95%) получали в виде белого твердого вещества. MS (ISP): m/z равно 422,0 [М+Н]+.
Промежуточное соединение D11
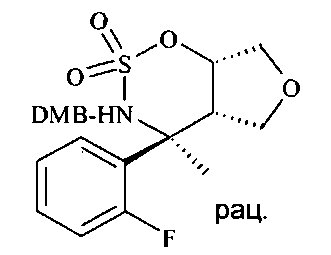
Раствор (3aS,7S,7aS)-rel-6-(2,4-диметокси-бензил)-7-(2-фтор-фенил)-7-метил-гексагидро-2,4-диокса-5-тиа-6-аза-инден-5-оксида (7,24 г, 17,2 ммоль) и перйодата натрия (4,04 г, 18,9 ммоль) в смеси этилацетата (60 мл), ацетонитрила (60 мл) и холодной воды (99,6 мл) обрабатывали хлоридом рутения(III) (35,6 мг, 172 мкмоль). Реакционную смесь перемешивали при 23°C в течение 30 минут. Для обработки реакционную смесь экстрагировали насыщенным раствором гидрокарбоната натрия, водный слой повторно экстрагировали этилацетатом, и объединенные органические слои сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле, используя градиент гептана и этилацетата от 100:0 до 0:100 в качестве элюента. (3aS,7S,7aS)-rel-6-(2,4-диметокси-бензил)-7-(2-фтор-фенил)-7-метил-гексагидро-2,4-диокса-5-тиа-6-аза-инден 5,5-диоксид (4,1 г, выход 55%) получали в виде светло-желтого твердого вещества. MS (ISP): m/z равно 460,2 [M+Na]+.
Промежуточное соединение D12
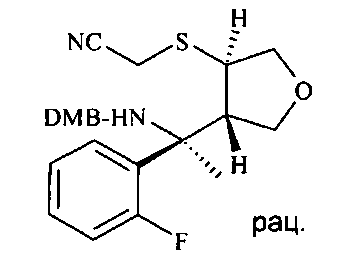
2-Меркаптоацетонитрил (1,03 г, 14,1 ммоль) добавляли по каплям в раствор (3aS,7S,7aS)-rel-6-(2,4-диметокси-бензил)-7-(2-фтор-фенил)-7-метил-гексагидро-2,4-диокса-5-тиа-6-аза-инден 5,5-диоксида (4,1 г, 9,37 ммоль) и 1,1,3,3-тетраметилгуанидина (1,62 г, 14,1 ммоль) в N,N-диметилформамиде (55 мл) при комнатной температуре. Затем реакционную смесь перемешивали при 60°C в течение 12 часов. Для обработки растворитель выпаривали при пониженном давлении, и остаток растворяли в дихлорметане (50 мл). После добавления серной кислоты (20%; 50 мл, 185 ммоль) и перемешивания в течение ночи смесь выливали на насыщенный раствор гидрокарбоната натрия, с последующей экстракцией (2х) дихлорметаном. Объединенные органические слои сушили над сульфатом натрия и упаривали. Остаток очищали флэш-хроматографией на силикагеле, используя градиент гептана и этилацетата от 100:0 до 0:100 в качестве элюента. {Rel-(3R,4S)-4-[(S)-1-(2,4-диметокси-бензиламино)-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-илсульфанил}-ацетонитрил (3,92 г, выход 97%) получали в виде светло-коричневого масла. MS (ISP): m/z равно 431,2 [М+Н]+.
Промежуточное соединение D13
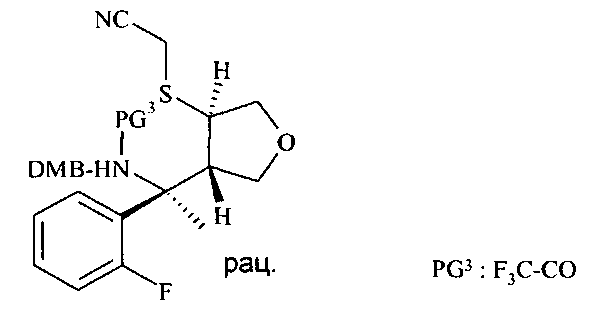
Раствор {rel-(3R,4S)-4-[(S)-1-(2,4-диметокси-бензиламино)-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-илсульфанил}-ацетонитрила (1,75 г, 4,06 ммоль) и триэтиламина (1,13 мл, 8,13 ммоль) в дихлорметане (8,75 мл) обрабатывали по каплям при 0°C трифторуксусным ангидридом (1,28 г, 861 мкл, 6,1 ммоль). Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Для обработки реакционную смесь разбавляли этилацетатом и экстрагировали водой. Органический слой сушили над сульфатом натрия и упаривали при пониженном давлении. N-[rel-(S)-1-((3S,4R)-4-цианометилсульфанил-тетрагидро-фуран-3-ил)-1-(2-фтор-фенил)-этил]-N-(2,4-диметокси-бензил)-2,2,2-трифтор-ацетамид (1,65 г, выход 77%) получали в виде светло-желтой пены. MS (ISP): m/z равно 544,4 [M+NH4]+.
Промежуточное соединение D14
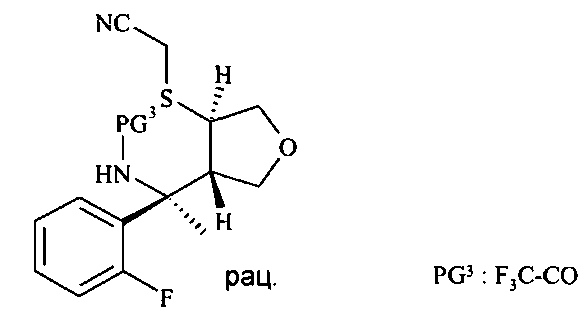
Раствор N-[rel-(S)-1-((3S,4R)-4-цианометилсульфанил-тетрагидро-фуран-3-ил)-1-(2-фтор-фенил)-этил]-N-(2,4-диметокси-бензил)-2,2,2-трифтор-ацетамида (1,65 г, 3,13 ммоль) в трифторуксусной кислоте (14,5 мл, 188 ммоль) перемешивали при комнатной температуре в течение ночи. Реакционную смесь экстрагировали смесью насыщенного раствора гидрокарбоната натрия и этилацетата/тетрагидрофурана. Объединенные органические слои сушили над сульфатом натрия и упаривали при пониженном давлении. Неочищенный N-[rel-(S)-1-((3S,4R)-4-цианометилсульфанил-тетрагидро-фуран-3-ил)-1-(2-фтор-фенил)-этил]-2,2,2-трифтор-ацеамид получали в виде белого твердого вещества и задействовали на следующей стадии без дальнейшей очистки. MS (ISP): m/z равно 375,3 [М+Н]+.
Промежуточное соединение D15
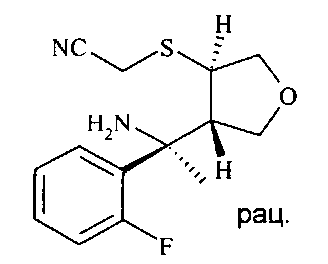
Раствор неочищенного N-[rel-(S)-1-((3S,4R)-4-цианометилсульфанил-тетрагидро-фуран-3-ил)-1-(2-фтор-фенил)-этил]-2,2,2-трифтор-ацетамида (2 г, 3,19 ммоль) в этаноле (60 мл) подвергали взаимодействию с боргидридом натрия (482 мг, 12,8 ммоль) при 0°C. Реакционную смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Для обработки реакционную смесь экстрагировали смесью насыщенного раствора гидрокарбоната натрия и этилацетата. Объединенные органические слои сушили над сульфатом натрия и упаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, используя градиент гептана и этилацетата от 100:0 до 0:100 в качестве элюента. {Rel-(3R,4S)-4-[(S)-1-амино-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-илсульфанил}-ацетонитрил (510 мг, выход 57%) получали в виде светло-желтого масла. MS (ISP): m/z равно 281,0 [М+Н]+.
Промежуточное соединение D16
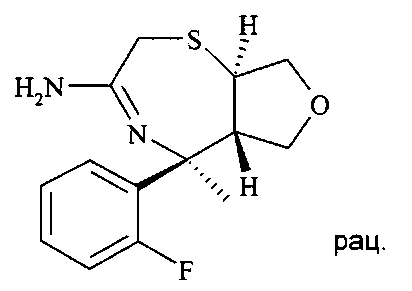
Раствор {rel-(3R,4S)-4-[(S)-1-амино-1-(2-фтор-фенил)-этил]-тетрагидро-фуран-3-илсульфанил}-ацетонитрила (460 мг, 1,64 ммоль) в толуоле (6 мл) обрабатывали по каплям триметилалюминием (902 мкл, 1,8 ммоль) при комнатной температуре. Реакционную смесь перемешивали при 60°C в течение 2 часов, затем экстрагировали смесью насыщенного раствора гидрокарбоната натрия и этилацетата. Объединенные органические слои сушили над сульфатом натрия и упаривали при пониженном давлении. Остаток очищали флэш-хроматографией на силикагеле, используя смесь этилацетата, метанола, гидроксида аммония 100:10:1 в качестве элюента. (3aR,8S,8aS)-rel-8-(2-фтор-фенил)-8-метил-1,3,3a,5,8,8а-гексагидро-2-окса-4-тиа-7-аза-азулен-6-иламин (200 мг, выход 43%) получали в виде светло-желтого масла. MS (ISP): m/z равно 281,0 [М+Н]+.
Промежуточное соединение D17
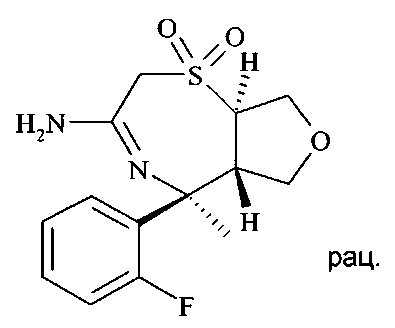
3-Хлорпербензойную кислоту (251 мг, 1,02 ммоль) добавляли при 0°C в раствор (3aR,8S,8aS)-rel-8-(2-фтор-фенил)-8-метил-1,3,3а,5,8,8а-гексагидро-2-окса-4-тиа-7-аза-азулен-6-иламина (130 мг, 464 мкмоль) в дихлорметане (20 мл). Реакционную смесь перемешивали при 0°C в течение 2 часов, затем экстрагировали смесью насыщенного раствора гидрокарбоната натрия и этилацетата. Объединенные органические слои сушили над сульфатом натрия и упаривали при пониженном давлении. (3aR,8S,8aS)-rel-8-(2-фтор-фенил)-8-метил-4,4-диоксо-3,3a,4,5,8,8а-гексагидро-1H-2-окса-4λ6-тиа-7-аза-азулен-6-иламин (110 мг, выход 76%) получали в виде светло-коричневого твердого вещества. MS (ISP): m/z равно 281,0[М+Н]+.
Промежуточное соединение D18
Раствор (3aR,8S,8aS)-rel-8-(2-фтор-фенил)-8-метил-4,4-диоксо-3,3a,4,5,8,8а-гексагидро-1Н-2-окса-4λ6-тиа-7-аза-азулен-6-иламина (110 мг, 176 мкмоль) в трифторуксусной кислоте (1,33 г, 11,7 ммоль) охлаждали до 0-5°C. Медленно добавляли серную кислоту (149 мг, 81,2 мкл, 1,52 ммоль), затем азотную кислоту (12,1 мг, 8,01 мкл, 192 мкмоль). Реакционную смесь перемешивали при 0°C в течение 1 часа. Для того чтобы завершить реакцию, добавляли еще один эквивалент азотной кислоты (12,1 мг, 8,01 мкл, 192 мкмоль) и перемешивание продолжали при 0°C в течение 2 часов. Для обработки в реакционную смесь добавляли ледяную воду, и образующуюся суспензию доводили до pH ~12 добавлением раствора гидроксида натрия (32%). Смесь дважды экстрагировали этилацетатом. Объединенные органические слои промывали водой, сушили над сульфатом натрия и упаривали. Неочищенный продукт (3aR,8S,8aS)-rel-8-(2-фтор-5-нитро-фенил)-8-метил-4,4-диоксо-3,3a,4,5,8,8а-гексагидро-1Н-2-окса-4λ6-тиа-7-аза-азулен-6-иламин получали в виде коричневого твердого вещества и задействовали на следующей стадии без очистки. MS (ISP): m/z равно 358,3 [М+Н]+.
Промежуточное соединение D19
Неочищенный (3aR,8S,8aS)-rel-8-(2-фтор-5-нитро-фенил)-8-метил-4,4-диоксо-3,3a,4,5,8,8а-гексагидро-1H-2-окса-4λ6-тиа-7-аза-азулен-6-иламин (73 мг, 204 мкмоль) гидрогенизировали при атмосферном давлении при комнатной температуре в этаноле (5 мл), используя палладий на углероде (5%; 39 мг, 18,3 мкмоль) в качестве катализатора. Через 3 часа катализатор отфильтровывали, и фильтрат упаривали при пониженном давлении. Для того чтобы завершить реакцию, остаток гидрогенизировали в смеси этанола (5 мл) и тетрагидрофурана (5 мл) при вышеупомянутых условиях на протяжении 20 часов. Затем катализатор отфильтровывали, и фильтрат упаривали при пониженном давлении. Неочищенный продукт (3aR,8S,8aS)-rel-8-(5-амино-2-фтор-фенил)-8-метил-4,4-диоксо-3,3a,4,5,8,8а-гексагидро-1H-2-окса-4λ6-тиа-7-аза-азулен-6-иламин (70 мг) получали в виде коричневого полутвердого вещества и задействовали на следующей стадии без очистки. MS (ISP): m/z равно 328,3 [М+Н]+.
Пример 1
[3-((S)-3-Амино-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты
Соединение получали с использованием методики синтеза амида А9 из (S)-5-(5-амино-2-фтор-фенил)-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламина (промежуточное соединение А8А). Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 393,3 [(М+Н)+].
Пример 2
[3-((S)-3-Амино-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 1, из (S)-5-(5-амино-2-фтор-фенил)-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламина (промежуточное соединение А8А). Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 377,1 [(М+Н)+].
Пример 3
[3-((S)-3-Амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты
Соединение получали с использованием методики синтеза сульфона А10 из [3-((S)-3-амино-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амида 5-хлор-пиридин-2-карбоновой кислоты. Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 425,1 [(М+Н)+].
Пример 4
[3-((S)-3-Амино-5-метил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 3, из [3-((S)-3-амино-5-метил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амида 5-фтор-пиридин-2-карбоновой кислоты. Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 409,1 [(М+Н)+].
Пример 5
[3-((S)-3-Амино-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 1, из (S)-5-(5-амино-2-фтор-фенил)-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламина (промежуточное соединение А8В). Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 391,1 [(М+Н)+].
Пример 6
[3-((S)-3-Амино-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 1, из (S)-5-(5-амино-2-фтор-фенил)-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламина (промежуточное соединение А8В). Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 407,0 [(М+Н)+].
Пример 7
[3-((S)-3-Амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 3, из [3-((S)-3-амино-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амида 5-фтор-пиридин-2-карбоновой кислоты. Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 423,0 [(М+Н)+].
Пример 8
[3-((S)-3-Амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-хлор-пиридин-2-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 3, из [3-((S)-3-амино-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амида 5-хлор-пиридин-2-карбоновой кислоты. Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 439,1 [(М+Н)+].
Пример 9
[3-((S)-3-Амино-2,5-диметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 4-хлор-2Н-пиразол-3-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 3, из (S)-5-(5-амино-2-фтор-фенил)-2,5-диметил-2,5,6,7-тетрагидро-[1,4]тиазепин-3-иламина (промежуточное соединение А9В). Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 428,0 [(М+Н)+].
Пример 10
[3-((S)-3-Амино-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-фтор-пиридин-2-карбоновой кислоты
Соединение получали с использованием методики синтеза амида В15 из (S)-5-(5-амино-2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-иламина (промежуточное соединение В14). Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 437,1 [(М+Н)+].
Пример 11
[3-((S)-3-Амино-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 4-хлор-2Н-пиразол-3-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 10, из (S)-5-(5-амино-2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1H-1λ6-[1,4]тиазепин-3-иламина (промежуточное соединение В14). Соединение, указанное в заголовке, получали в виде белого твердого вещества. MS (ISP): m/z равно 442,4 [(М+Н)+].
Пример 12
[3-((3)-3-Амино-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-циано-пиридин-2-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 10, из (S)-5-(5-амино-2-фтор-фенил)-2,2,5-триметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-3-иламина (промежуточное соединение В14). Соединение, указанное в заголовке, получали в виде светло-желтого масла. МС (ISP): m/z равно 444,3 [(М+Н)+].
Пример 13
[3-((3)-3-амино-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-5-ил)-4-фтор-фенил]-амид 5-циано-пиридин-2-карбоновой кислоты
Соединение получали аналогично способу, описанному для примера 10, из (S)-5-(5-амино-2-фтор-фенил)-5-дифторметил-1,1-диоксо-2,5,6,7-тетрагидро-1Н-1λ6-[1,4]тиазепин-3-иламина (промежуточное соединение В14В). Соединение, указанное в заголовке, получали в виде беловатого твердого вещества. MS (ISP): m/z равно 452,0 [(М+Н)+].
Пример 14
[3-((3aR,8S,8aS)-Rel-6-амино-8-метил-4,4-диоксо-3,3a,4,5,8,8а-гексагидро-1Н-2-окса-4λ6-тиа-7-аза-азулен-8-ил)-4-фтор-фенил]-амид 5-циано-пиридин-2-карбоновой кислоты
Раствор 5-цианопиридин-2-карбоновой кислоты (15,8 мг, 107 мкмоль) в метаноле (1 мл) охлаждали до 0°C. После добавления 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метил-морфолиния хлорида (29,6 мг, 107 мкмоль) смесь перемешивали при 0°C в течение 30 минут. Добавляли неочищенный (3aR,8S,8aS)-rel-8-(5-амино-2-фтор-фенил)-8-метил-4,4-диоксо-3,3a,4,5,8,8а-гексагидро-1Н-2-окса-4λ6-тиа-7-аза-азулен-6-иламин (70 мг, 107 мкмоль), и оставляли смесь нагреваться до комнатной температуры. После перемешивания в течение 21 часа светло-коричневый раствор декантировали от образовавшегося липкого твердого вещества и упаривали при пониженном давлении. Остаточное аморфное вещество очищали препаративной ВЭЖХ, используя градиент воды и метанола от 95:5 до 0:100 (+0,05% муравьиная кислота) в качестве элюента. [3-((3aR,8S,8aS)-Rel-6-амино-8-метил-4,4-диоксо-3,3a,4,5,8,8а-гексагидро-1Н-2-окса-4λ6-тиа-7-аза-азулен-8-ил)-4-фтор-фенил]-амид 5-циано-пиридин-2-карбоновой кислоты (17 мг) получали в виде светло-коричневого аморфного вещества. MS (ISP): m/z равно 458,4 [(М+Н)+].
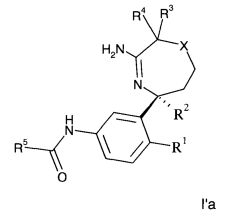
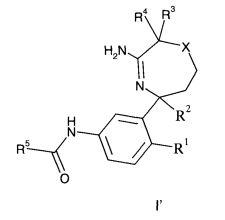
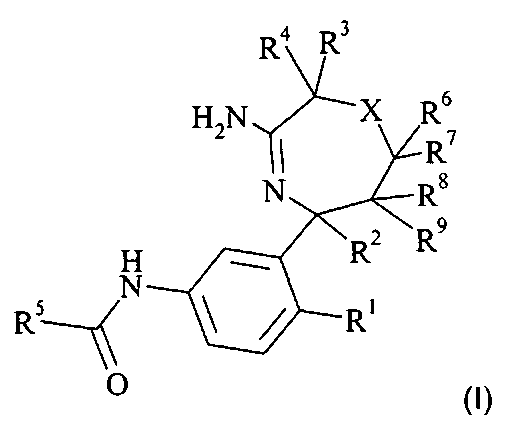
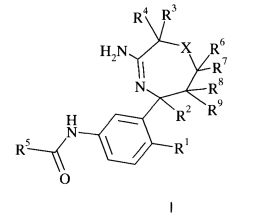
Препарат антитела
Производные гетероарилпирролидинил- и пиперидинилкетона
Производные изоксазоло-пиридина
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Ингибиторы jnk
Бензопирановые и бензоксепиновые ингибиторы рi3k и их применение
Арилциклогексилэфиры дигидротетраазабензоазуленов для применения в качестве антагонистов рецептора вазопрессина v1a
Пуриновые соединения, ингибирующие рi3к, и способы применения
Алкилциклогексиловые эфиры дигидротетраазабензоазуленов
Способ синтеза производных амино-метилтетралина
Аналитический инструмент с тестовой лентой, содержащий двигатель постоянного тока и редуктор
Препарат антитела
Производные гетероарилпирролидинил- и пиперидинилкетона
Производные изоксазоло-пиридина
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Ингибиторы jnk
Бензопирановые и бензоксепиновые ингибиторы рi3k и их применение
Арилциклогексилэфиры дигидротетраазабензоазуленов для применения в качестве антагонистов рецептора вазопрессина v1a
Пуриновые соединения, ингибирующие рi3к, и способы применения
Алкилциклогексиловые эфиры дигидротетраазабензоазуленов

