Результат интеллектуальной деятельности: СПОСОБ ПОИСКА МОЛЕКУЛЯРНЫХ МАРКЕРОВ ПАТОЛОГИЧЕСКОГО ПРОЦЕССА ДЛЯ ДИФФЕРЕНЦИАЛЬНОЙ ДИАГНОСТИКИ, МОНИТОРИНГА И ТАРГЕТНОЙ ТЕРАПИИ
Вид РИД
Изобретение
Изобретение относится к области биотехнологии и таргетной терапии, касается, способа поиска набора генов и мРНК-транскриптов, специфичных для развития определенного патологического процесса, и может быть использовано в медицине для идентификации молекулярно-генетических маркеров с целью разработки средств дифференциальной диагностики, мониторинга течения заболевания и таргетной терапии.
Современный подход к диагностике, мониторингу и лечению патологических состояний заключается в идентификации уникальных молекулярно-генетических или белковых маркеров, характеризующих развитие и течение заболевания, а также являющихся потенциальными мишенями для фармакотерапии. Финальным этапом поисковых работ, направленных на выявление таких маркеров, является анализ большого количества данных, получаемых с помощью высокопроизводительных технологий (биочипы, секвенирование РНК, полногеномное секвенирование), который, по сути, представляет собой процедуру отбора признаков (feature selection) [Bermingham, Mairead Lesley; Pong-Wong, Ricardo; Spiliopoulou, Athina; Hay-ward, Caroline; Rudan, Igor; Campbell, Harry; Wright, Alan F.; Wilson, Jim; ward, Caroline; Rudan, Igor; Campbell, Harry; Wright, Alan F.; Wilson, Jim; Agakov, Felix; Navarro, Pau; Haley, Chris. Application of high-dimensional feature selection: evaluation for genomic prediction in man//Scientific Reports. 2015. Vol. 5, 10312.]. Отбор признаков заключается в выделении элементов, значимых для построения математической модели или интерпретации результатов.
Можно выделить три основные группы методов для отбора признаков (молекулярно-генетических маркеров):
1. Группа методов традиционной статистики, основанных на оригинальном и модифицированных Т-тестах, использующих для ранжирования признаков уровень значимости. Серьезным недостатком таких методов является то, что наличие статистически значимых различий экспрессии какого-либо гена в двух сравниваемых группах не гарантирует того, что это различие достаточно велико, чтобы иметь биологическое значение. Большинство известных методов этой группы пытаются устранить описанный недостаток за счет введения порогового значения t-статистики [Tan Q., Thomassen М., Kruse Т.А. Feature selection for predicting tumor metastases in microarray experiments using paired design//Cancer. - 2007. V. 3. - P. 213-218.; Tusher V.G., Tibshirani R., Chu G. Significance analysis of microarrays applied to the ionizing radiation response // Proc. Natl. Acad. Sci. U.S.A. - 2001. V. 98. No. 9. - P. 5116-5121.] или кратности изменения экспрессии генов [Zhongbo Cao; Yan Wang; Ying Sun; Wei Du; Yanchun Liang Effective and stable feature selection method based on filter for gene signature identification in paired microarray data//2013. IEEE International conference on bioinformatics and biomedicine. - P. 189-192.].
2. Группа методов логистической регрессии, позволяющих спрогнозировать вероятность возникновения некоторого события путем «подгонки» данных к логистической кривой. Методы широко применяются при дизайне исследования по типу «связанных пар» [Sen Liang, AnjunMa, Sen-Yang, Yan Wang, QinMa A review of matched-pairs feature selection for gene expression data analysis//Comput. Struct. Biotechnol. J. - 2018.V. 16. - P. 88-97.]. Общим недостатком этих методов является чувствительность к коллинеарности предикторов - наличие взаимосвязи между анализируемыми параметрами приводит к смещенным оценкам и большим ошибкам [N.Е. Breslow, N.Е. Day, K.Т. Halvorsen, R.L. Prentice, С. Sabai Estimation of multiple relative risk functions in matched case-control studies// Am. J. Epidemiology. - 1978. V. 108. No. 4. - P. 299-307.].
3. Группа методов, решающих проблему классификации с помощью бустинга. Стратегия бустинга основана на комбинации множества «слабых» классификаторов, в совокупности формирующих мощный классификатор [Friedman J., Hastie Т., Tibshiranti R. Additive logistic regression: a statistic view of boosting (with discussion and a rejoinder by authors)//Ann. Statist. - 2000. V. 28. - P. 337-407.; Adewale A.J., Dinu I., Yasui Y. Boosting for correlated binary classification//Journal of Computational and Graphical Statistics. - 2010. V. 19. No. 1. - P. 140-153.]. Количественной мерой, используемой для отбора признаков при бустинге, является показатель значимости для классификации (feature importance), характеризующий «вклад» признака в решение задачи классификации. По причине простоты, универсальности, гибкости и высокой обобщающей способности в течение последних десяти лет бустинг остается одним из наиболее популярных методов машинного обучения. Разновидность бустинга - бустинг над решающими деревьями - считается одним из наиболее эффективных методов с точки зрения качества классификации, поскольку демонстрирует практически неограниченное уменьшение частоты ошибок по мере наращивания композиции [G. James, D. Witten, Т. Hastie, R. Tibshirani An introduction to statistical learning: with applications in R. Springer Verlag, New York, 2013. - 426 р.]. К недостаткам методов этой группы можно отнести плохую имплементацию результатов в медицинскую практику: анализ экспрессии сотен генов и транскриптов чрезвычайно дорогостоящий, а критерии отбора значимых параметров не удовлетворяют требованиям доказательной медицины.
Общим недостатком известных методов идентификации маркеров патологического состояния является то, что они позволяют определить молекулы, количество которых изменяется при развитии заболевания, но не гарантируют того, что эти же молекулы не изменяются при другой патологии. Полученные таким образом маркеры зачастую невозможно применить для дифференциальной диагностики заболеваний, обладающих схожей симптоматикой. Так, нарушение экспрессии регулятора клеточного цикла ТР53 (Р53) наблюдается в 50% случаев онкологических заболеваний различного генеза [Ozaki Т., Nakagawara A. Role of р53 in cells death and human cancers//Cancers (Basel). - 2011. V. 3. No. - P. 994-1013.]. Экспрессия лимфотоксина альфа (LT-alpha, TNF-beta) повышается при реализации процессов врожденного иммунного ответа, аутоиммунных и онкологических заболеваниях [G. Seleznik, J .Zoller, T. O'Connor, R. Graf, M. Heikenwalder The role of lymphotoxin signaling in the development of autoimmune pancreatitis and associated secondary extra-pancreatic patholo-gies//Cytokine growth factor rev. - 2014. V. 25. No. 2. - P. 125-137.]. Изменение экспрессии трансдукторного протеина кальмодулина выявлено в случае прионной энцефалопатии, гипоксии мозга, некоторых нейроэндокринных заболеваниях, отравлении тяжелыми металлами, а также при развитии злокачественных новообразований [Vallilobo A. The multifunctional role of phosphor-calmodulin in pathophysiological processes//Biochem. J. - 2018. V. 475. No. 24. - P. 4011-4023.].
Пока не разработаны устоявшиеся алгоритмы и подходы по интеграции массивов различных типов NGS-данных с целью их биологического осмысления, что сильно затрудняет клиническое применение результатов проведенных геномных и транскриптомных анализов.
Задачей изобретения является создание способа поиска молекулярных маркеров патологического процесса, позволяющего идентифицировать набор маркеров в транскриптоме биологического образца, уникальных для исследуемой патологии.
Техническим результатом является оптимизация способов дифференциальной диагностики, мониторинга и специфической терапии.
Изучение транскриптома становится все более востребованным в современной биомедицине. Анализ экспрессии генов, регулирующих базовые клеточные процессы, позволяет выявлять молекулярные механизмы развития патологического процесса, оценивать вероятность развития заболевания и/или его осложнений. Выраженное изменение экспрессии определенного набора генов может служить своеобразным молекулярным паспортом патологии, а выявление ключевых транскриптов-участников патогенеза открывает возможности их использования в качестве маркеров заболевания с целью диагностики, мониторинга, а также в качестве потенциальной терапевтической мишени [Binjie Li, Qiyi Zeng Personalized identification of differentially expressed pathways in pediatric sepsis//Mol. med. rep. - 2017. V. 16. No. 4 - P. 5085-5090.; M. Omar, F. Klawonn. S. Brand, M. Stiesch, Ch. Krettek J. Eberhard Transcriptome-Wide high-density microarray analysis reveals differential gene transcription in periprosthetic tissue from hips with chronic periprosthetic joint infection vs aseptic loosening//J. Anthroplasty. 2017. V. 32. No. l. - P. 234-240; A. Ruffo-Campos, I. Riquelme, P. Brebi-Mieville Tools for sequence-based miRNA target prediction: What to choose?// Int. J. Mol. Sci. - 2016. V. 17(12). - P. 1987; Sano D., M. Tazawa, M. Inaba, S. Kadova, R. Watanabe, T. Miura, K. Kitajima, S. Okabe Selection of cellular genetic markers for the detection of infectious poliovirus//J. Appl. Microbiol. - 2018. V. 124(4). - P. 1001-1007.]. Средства специфической диагностики, мониторинга и лечения обладают высокой эффективностью и, при условии внедрения в медицинскую практику, увеличивают качество и продолжительность жизни населения.
Изучение транскриптома осуществляют с помощью высокопроизводительных технологий (ДНК-биочипы, секвенирование РНК). Данные об экспрессии генов (суммарной экспрессии всех сплайсированных изоформ мРНК каждого гена) и транскриптов (экспрессии индивидуальных сплайсированных изоформ мРНК каждого гена) могут быть получены с помощью различных технологий. Наиболее распространены два подхода - метод биочипов (microarray) и метод RNASeq, представляющий собой секвенирование следующего поколения (NGS) и основанный на прямом прочтении последовательности нуклеотидов в молекулах дезоксирибону-клеиновых кислот. Набор генов и транскриптов, составляющих исследуемые при патологии сигнальные пути, определяются индивидуально для каждого заболевания исходя из данных научной литературы. Анализ полученных результатов требует привлечения сложного статистического аппарата. Большое количество и возможная тесная взаимосвязь анализируемых параметров, каждый из которых обладает различной информативностью для решения текущей исследовательской задачи, приводит к необходимости применять методы прикладной статистики в сочетании с алгоритмами машинного обучения.
Технический результат достигается способом поиска молекулярных маркеров патологического процесса, включающим следующие этапы: а. получение транскриптомных данных об уровнях экспрессии генов и транскриптов в образцах, полученных от пациентов с исследуемой патологией (патология исследования), пациентов с заболеванием иной этиологии со схожей клинической картиной (патология сравнения) и клинически здоровых доноров сопоставимого возраста и пола (норма); б. парная классификация образцов для групп сравнения норма - патология исследования, норма - патология сравнения, патология сравнения - патология исследования с выделением для каждой пары групп значимых для классификации прессии каждого гена и транскрипта в паре групп норма - патология исследования, норма - патология сравнения, патология сравнения - патология исследования с определением кратности изменения экспрессии и уровня статистической значимости; г. выбор для каждой пары групп норма - патология исследования, норма - патология сравнения, патология сравнения - патология исследования кандидатных маркеров - генов и транскриптов, отвечающих следующим критериям: статистически значимое различие транскриптомных данных об уровнях экспрессии в двух группах; высокие показатели кратности изменения экспрессии и/или значимости для классификации; д. идентификация молекулярно-генетических маркеров исследуемой патологии, отвечающих следующим критериям: принадлежность к кандидатным маркерам для групп сравнения норма - патология исследования и патология исследования - патология сравнения; отсутствие среди кандидатных маркеров для групп норма - патология сравнения.
Способ осуществляется следующим образом.
Идентификацию молекулярно-генетических маркеров патологического процесса осуществляют в четыре этапа:
а. Получение транскриптомных данных об уровнях экспрессии генов и транскриптов в образцах, полученных от пациентов с исследуемой патологией (патология исследования), пациентов с заболеванием иной этиологии со схожей клинической картиной (патология сравнения) и клинически здоровых доноров сопоставимого возраста и пола (норма).
б. Парная классификация образцов для групп сравнения норма -патология исследования, норма - патология сравнения, патология сравнения - патология исследования с выделением для каждой пары групп значимых для классификации генов и транскриптов с определением их значимости. Классификацию проводят с использованием алгоритмов построения модели, в частности, градиентного бустинга над решающими деревьями с применением кросс-валидации [Adewale A.J., Dinu I, Yasui Y. Boosting for correlated binary classification // Journal of Computational and Graphical Statistics.- 2010. V. 19. No. 1. - P. 140-153.]. Классификатор считают эффективным при значении медианы показателя аккуратности (accuracy) и медианы показателя площади под кривой ошибок (area under receiver operating characteristic curve (ROC), AUC) не менее 0,75. В случае построения эффективного классификатора для улучшения качества классификации проводят отбор признаков и повторное моделирование [Pirooznia М. и др. A comparative study of different machine learning methods on microarray gene expression data // BMC Genomics. - 2008. V. 9. Suppl. 1.: S. 13.]. В результате повторного моделирования получают показатели значимости для каждого прошедшего отбор гена и транскрипта.
в. Сравнение транскриптомных данных об уровнях экспрессии каждого гена и транскрипта в паре групп норма - патология исследования, норма - патология сравнения, патология сравнения - патология исследования с определением кратности изменения экспрессии и уровня статистической значимости. Сравнение проводят с использованием методов традиционной статистики, в частности, Т-теста с поправкой на ожидаемую долю ложных отклонений (false discovery rate, FDR) [Benjamini Y., Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing // Journal of the Royal Statistical Society. Series В (Methodological). - 1995. V. 57. No. 1. - P. 289-300.]. Рассчитывают кратность изменения экспрессии (в абсолютных значениях, процентах, логарифме отношений величин экспрессии и пр.) и уровень статистической значимости. Пороговую отметку для разграничения «высоких» и «низких» значений значимости и кратности изменения экспрессии определяют, исходя из распределения величин, но не менее 70-го процентиля.
г. Выбор для каждой пары групп норма - патология исследования, норма - патология сравнения, патология сравнения - патология исследования кандидатных маркеров - генов и транскриптов, отвечающих следующим критериям: статистически значимое различие транскриптомных данных об уровнях экспрессии в двух группах; высокие показатели кратности изменения экспрессии и/или значимости для классификации.
д. Идентификация молекулярно-генетических маркеров исследуемой патологии, отвечающих следующим критериям: принадлежность к кандидатным маркерам для групп сравнения норма - патология исследования и патология исследования - патология сравнения; отсутствие среди кандидатных маркеров для групп норма - патология сравнения.
Сущность способа поясняется примером.
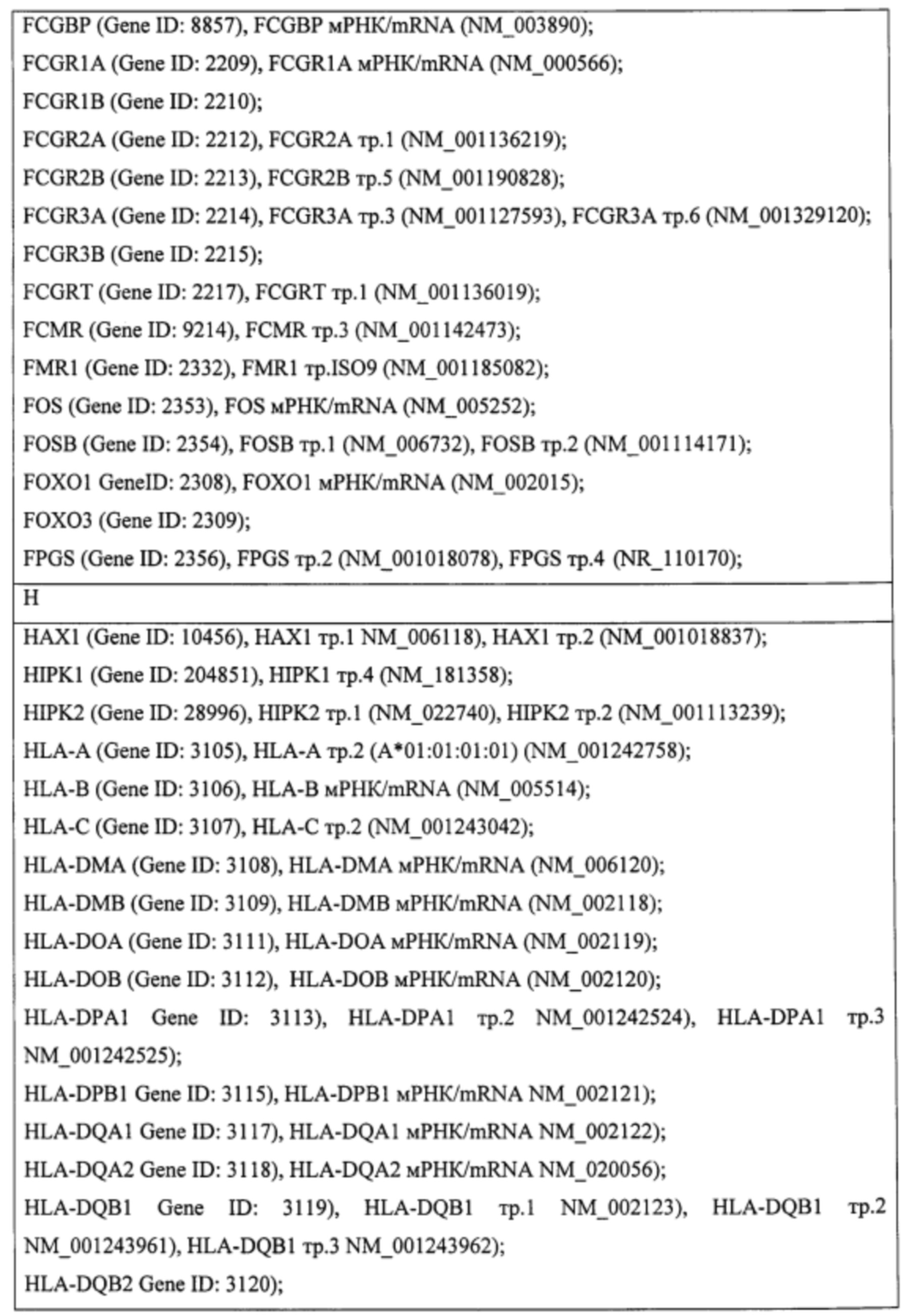
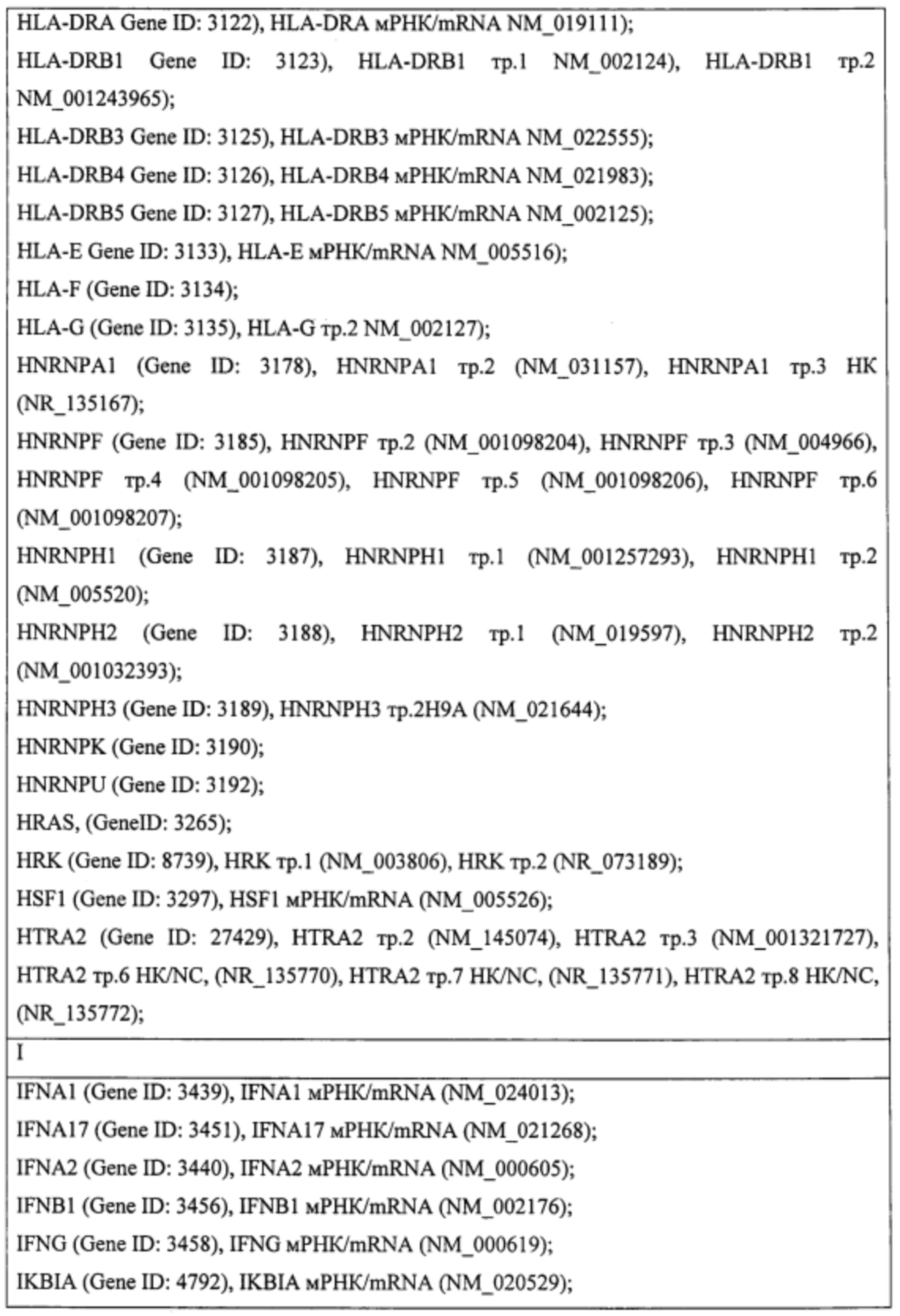
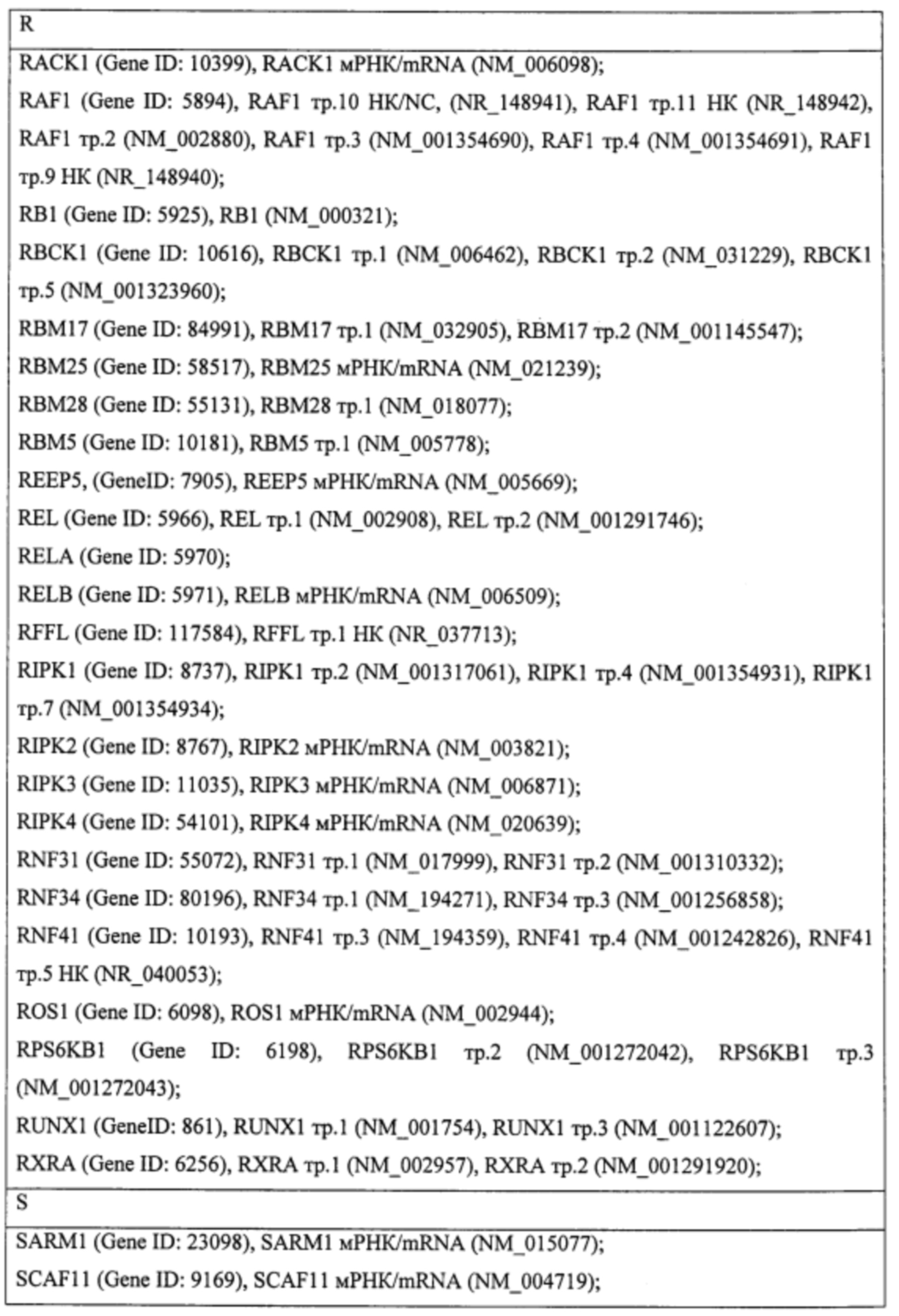
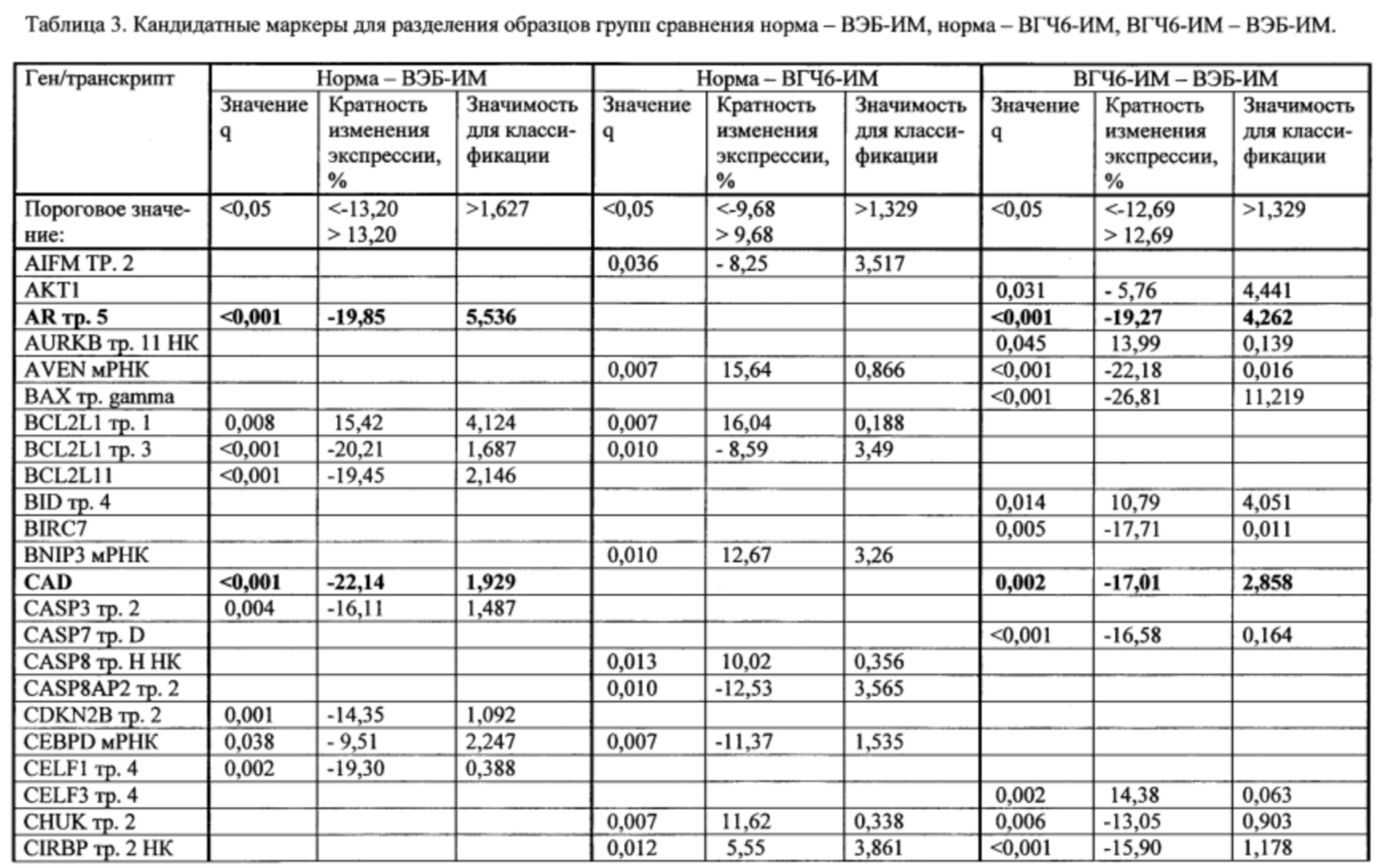
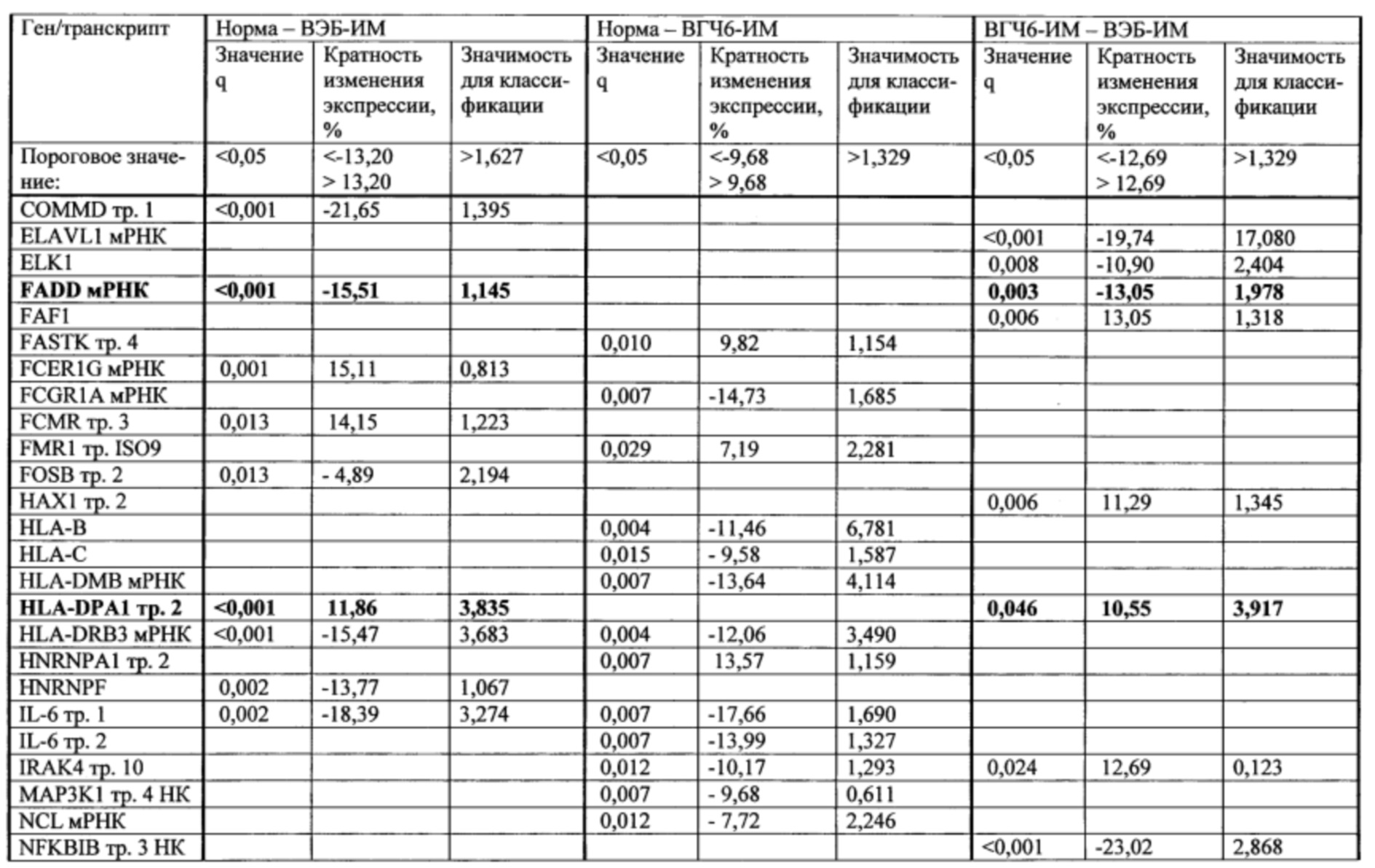
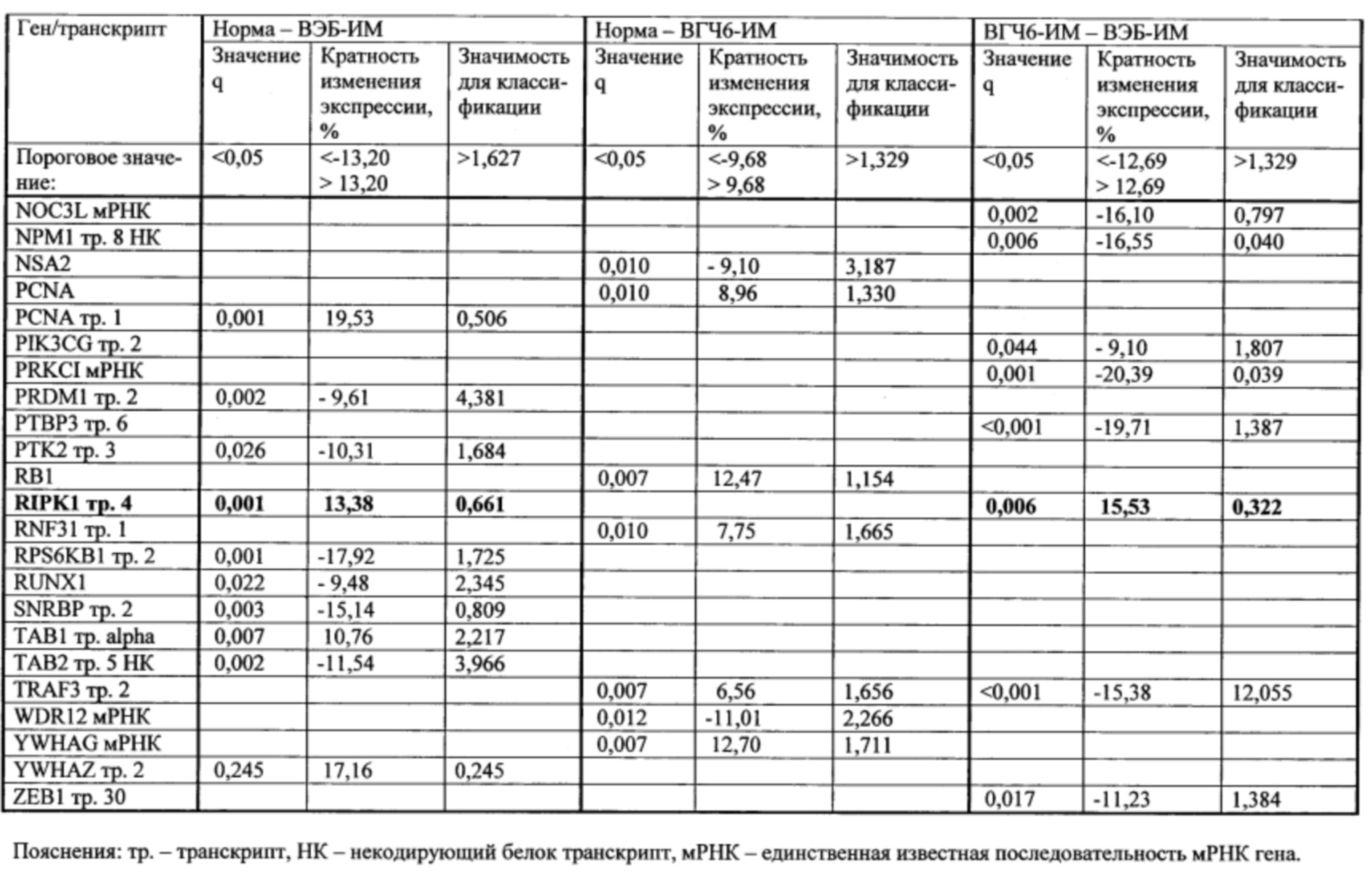
Пример. Идентификация молекулярно-генетических маркеров инфекционного мононуклеоза, ассоциированного с вирусом Эпштейна-Барр. На этапе а получали данные о транскриптоме лейкоцитов периферической крови детей и подростков 7-14 лет с диагнозом «острый инфекционный мононуклеоз (ИМ)», а также практически здоровых доноров сопоставимого пола и возраста (группа норма). На основании этиологической причины заболевания пациенты с ИМ были разделены на две группы: пациенты с ИМ, ассоциированным с вирусом Эпштейна-Барр (ВЭБ-ИМ) (патология исследования), и пациенты с ИМ, ассоциированным с вирусом герпеса человека 6 типа (ВПГЧ6-ИМ) (патология сравнения). ВЭБ-ИМ и ВГЧ6-ИМ обладают схожей симптоматикой, однако молекулярные механизмы, обуславливающие патогенез двух заболеваний, различаются. Из лейкоцитов экстрагировали пул тотальной РНК, который подвергали серии реакций с получением биотин-меченой РНК. Меченую РНК гибридизовали на биочип собственного дизайна для получения амперометрического сигнала, который принимали за уровень экспрессии гена или транскрипта. Всего были получены данные об экспрессии 403 генов и 712 транскриптов генов, участвующих в активации клеток иммунной системы, их пролиферации, дифференцировке и апоптозе. Полный перечень генов и их транскриптов представлен в таблице 1.













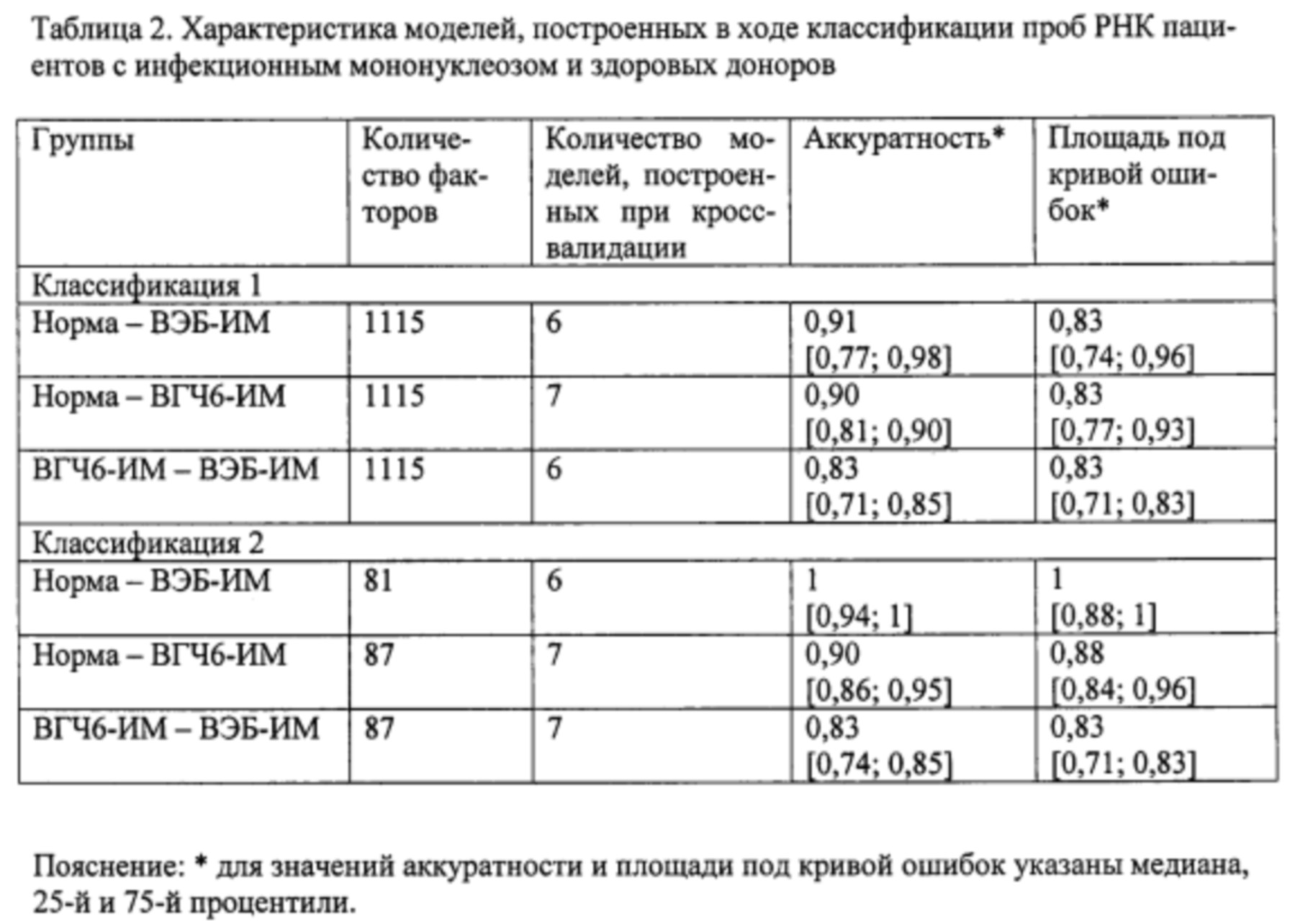
На этапе б. с применением метода градиентного бустинга над решающими деревьями [Adewale A.J., Dinu I., Yasui Y. Boosting for correlated binary classification//Journal of computational and graphical statistics. - 2010. V. 19. №1. - P. 140-153.] и метода кросс-валидации построили две модели классификации образцов пар групп норма - ВЭБ-ИМ, норма - ВГЧ6-ИМ, ВГЧ6-ИМ - ВЭБ-ИМ. Для построения первой модели в качестве факторов использовали все анализируемые гены и транскрипты (1115 наименований). Качество классификации считали удовлетворительным при средних значениях аккуратности и площади под кривой ошибок не менее 0,75. Из всех моделей, построенных в ходе кросс-валидации, отобрали по 20 факторов, обладавших наибольшей значимостью, что составило менее 8% от первоначального количества факторов. Повторяющиеся факторы исключали и итоговый набор использовали для повторной классификации. В результате при повторной классификации количество факторов было сокращено в среднем более чем на 90%, что значительно улучшило качество моделей при повторной классификации. Характеристики построенных моделей представлены в таблице 2. Полученные значения важности факторов для второй классификации рассматривали как показатель значимости генов и транскриптов.
На этапе в. для каждой пары анализируемых групп оценивали изменения уровней экспрессии генов и транскриптов, составивших факторы при второй классификации. Кратность изменения экспрессии рассчитывали по формуле:
[(среднее группы 2*100/среднее группы 1)-100] (%).
Оценку статистической значимости изменения экспрессии проводили с помощью Т-теста с поправкой на ожидаемую долю ложных отклонений [Benjamini Y., Hochberg Y. Controlling the false discovery rate: a practical and powerful approach to multiple testing // Journal of the Royal Statistical Society. Series В (Methodological). - 1995. V. 57. No. l. - P. 289-300.].
На этапе г. для каждой пары анализируемых групп составляли списки кандидатных маркеров - генов и транскриптов, показатели экспрессии которых позволяли различить образцы, принадлежащие к двум группам. В список кандидатных маркеров вошли гены и транскрипты, отвечавшие следующим критериям: уровень эксперссии статистически значимо различался в двух группах; высокие показатели кратности изменения экспрессии и/или значимости для классификации. Для определения порогового значения важности и кратности изменения экспрессии рассчитывали 75-ый процентиль распределения показателей (использовали величины кратности изменения в абсолютных значениях). Перечни кандидатных маркеров для разделения образцов анализируемых групп представлены в таблице 3. Из таблицы 3 видно, что ряд генов и транскриптов: транскрипт 5 AR, ген CAD, мРНК FADD, транскрипт 2 HLA-DPA1 и транскрипт 4 RIPK1, - изменяли свою экспрессию при ВЭБ-ИМ по сравнению с нормой, и при ВЭБ-ИМ по сравнению с ВГЧ6-ИМ. В перечень кандидатных маркеров для разделения образцов между группами норма и ВГЧ6-ИМ перечисленные гены и транскрипты не вошли.
Таким образом, транскрипт 5 AR, ген CAD, мРНК FADD, транскрипт 2 HLA-DPA1 и транскрипт 4 RIPK1 в совокупности формировали уникальный набор молекулярно-генетических маркеров инфекционного мононуклеоза, ассоциированного с вирусом Эпштейна-Барр. В дальнейшем для применения в медицинской практике экспрессия выявленных маркерных транскриптов может оцениваться с помощью более дешевых и простых в реализации методов молекулярной биологии, например, ПЦР в реальном времени.
Способ приготовления лечебно-профилактического препарата из живых штаммов микроорганизмов лактобацилл и бифидобактерий "lb-комплекс плюс"
Биомаркеры риска развития осложнений инфекционного мононуклеоза, ассоциированного с вирусом эпштейна-барр
Способ идентификации инфекционного мононуклеоза, ассоциированного с вирусом герпеса человека 6 типа
Способ прогнозирования риска развития вэб-ассоциированных опухолей у пациентов с острой вэб-инфекцией
Способ дифференциальной диагностики инфекционного мононуклеоза различной этиологии
Питательная среда для консервации и транспортировки клеток для дальнейшего цитологического и иммуноцитохимического исследования
Биомаркеры риска развития осложнений инфекционного мононуклеоза, ассоциированного с вирусом эпштейна-барр
Способ идентификации инфекционного мононуклеоза, ассоциированного с вирусом герпеса человека 6 типа
Способ скрининговой дифференциальной диагностики предраковых заболеваний и рака слизистой оболочки рта (сор)
Способ многоцветной иммуноцитохимической диагностики паранеоплазии шейки матки

