Результат интеллектуальной деятельности: ЛЕЧЕНИЕ САХАРНОГО ДИАБЕТА С ПОМОЩЬЮ СОСТАВОВ ИНСУЛИНОВ ДЛИТЕЛЬНОГО ДЕЙСТВИЯ
Вид РИД
Изобретение
Настоящая заявка относится к водному фармацевтическому составу для применения в лечении сахарного диабета I типа или II типа, где лечение снижает риск развития ночной гипогликемии, при этом указанный состав содержит 200-1000 ЕД/мл [эквимолярно 200-1000 МЕ человеческого инсулина] инсулина гларгина, при условии, что концентрация инсулина гларгина в указанном составе не составляет 684 ЕД/мл.
Инсулин гларгин представляет собой человеческий инсулин 31в-32в-ди-Arg, аналог человеческого инсулина с дополнительной заменой аспарагина в положении А21 на глицин.
В WO 2008/013938 А2 раскрыт водный фармацевтический состав, содержащий инсулин гларгин в концентрации 684 ЕД/мл.
Метформин представляет собой гипогликемическое средство группы бигуанидов, применяемое в лечении инсулинозависимого сахарного диабета (сахарного диабета 2 типа), не поддающегося коррекции модификацией рациона. Метформин улучшает гликемический контроль путем повышения чувствительности к инсулину и снижения всасывания глюкозы в кишечнике. Метформин обычно вводят перорально.
Lantus® представляет собой продукт на основе инсулина, содержащий инсулин гларгин, обеспечивающий 24-часовой запас базального инсулина после подкожной инъекции одной дозы.
По влиянию на глюкодинамику Lantus® отличается от других представленных в настоящее время на рынке продуктов на основе инсулина благодаря отсроченному и прогнозируемому всасыванию инсулина гларгина из места подкожной инъекции, результатом чего является плавный 24-часовой профиль зависимости "время-концентрация" и профиль действия без выраженного пика. Lantus® был разработан, чтобы удовлетворить медицинскую необходимость в продукте на основе инсулина длительного действия, который можно вводить в виде однократной суточной инъекции для достижения нормального или практически нормального контроля уровня глюкозы в крови с как можно более плавным профилем базального инсулина на протяжении 24-часового периода. Такой препарат обеспечивает хороший контроль уровня глюкозы в крови на протяжении всего дня, минимизируя при этом тенденцию к вызыванию гипогликемии, наблюдаемую у других препаратов инсулина с более выраженным "пиковым" эффектом.
Значительное число пациентов, в частности, пациентов с повышенной резистентностью к инсулину, обусловленной ожирением, применяют большие дозы для контроля уровня глюкозы в крови.
Например, для дозы 100 ЕД необходима инъекция 1 мл Lantus® U100, что может причинить некоторое неудобство; каждый мл Lantus® U100 содержит 100 ЕД (3,6378 мг) инсулина гларгина. Для уменьшения объема инъекции был разработан состав, содержащий 300 ЕД инсулина гларгина на мл.
Целью многоцентрового рандомизированного открытого исследования на параллельных группах, описанного в примере 1, было сравнение эффективности и безопасности инсулина гларгина U300 с таковыми Lantus, оба из которых вводят один раз в день подкожно (S.C.) как часть базис-болюсной схемы введения инсулина у пациентов с сахарным диабетом 2 типа. К участию в исследовании допускали пациентов с сахарным диабетом 2 типа и с уровнем гликозилированного гемоглобина A1c (HbA1c) в диапазоне от 7% до 10%, которым инъецировали по меньшей мере 42 ЕД Lantus U100 или эквивалентные количества инсулина нейтрального протамина Хагедорна (NPH) по схеме применения базального инсулина в сочетании с прандиальным. Для этих пациентов на относительно высоких дозах базального инсулина меньший объем инъекции составов инсулина U300 был более благоприятным по сравнению с составами U100.
Каждый мл инсулина гларгина U300 содержит 300 ЕД (10,9134 мг) инсулина гларгина. Данный состав может обеспечить возможность инъекции пациентам того же количества единиц инсулина гларгина в одной трети объема инъекции. Данный состав в данном документе также имеет название HOE901-U300.
В исследовании, описанном в примере 1, пациентов стратифицировали в зависимости от их уровня HbA1c (<8,0%; ≥8,0%). В первичном анализе эффективности исследовали наличие не меньшей эффективности инсулина гларгина U300 по сравнению с Lantus по изменению уровня HbA1c от начала исследования до конечной точки (запланированной на 6 месяце; предел не меньшей эффективности 0,4% единиц HbA1c). HbA1c отражает средний уровень гликемии на протяжении нескольких месяцев и имеет высокую прогностическую ценность в отношении осложнений сахарного диабета. Продолжительность лечения в рамках исследования, составлявшая 6 месяцев, как полагают, является достаточной для достижения условий установившегося состояния с инсулином гларгином U300 после перехода с Lantus или инсулина NPH, что обеспечивает возможность адекватной оценки зависимых от времени изменений уровня HbA1c и сопутствующего риска развития гипогликемии.
Основные вторичные конечные точки включали ночную гипогликемию. Гипогликемия является крайне важным ограничивающим фактором в контроле гликемии при сахарном диабете, как в краткосрочном, так и в долгосрочном периоде. Несмотря на постоянное улучшение контроля гликемии при сахарном диабете, данные популяционного масштаба указывают на то, что гипогликемия остается основной проблемой для людей с сахарным диабетом как 1 типа, так и 2 типа (American diabetes association, workgroup on hypoglycemia: Defining and Reporting Hypoglycemia in Diabetes. Diabetes Care 28(5), 2005, 1245-1249).
В исследовании, описанном в примере 1, было неожиданно обнаружено, что посредством лечения пациентов с сахарным диабетом 2 типа с помощью состава инсулина гларгина U300 риск развития эпизода ночной гипогликемии может быть значительно снижен по сравнению с лечением с помощью Lantus U100. Количество пациентов по меньшей мере с одним случаем ночной тяжелой и/или подтвержденной гипогликемии в промежутке между началом на 9 неделе и 6 месяцем в группе U300 [136/404 (33,7%)] было меньше, чем в группе Lantus [180/400 (45,0%)] (см. таблицу 6). Превосходство U300 по сравнению с Lantus было показано при относительном риске 0,75 (95% CI [0,63, 0,89]) (р=0,0010).
В примере 2 описано клиническое испытание, в котором сравнивали эффективность и безопасность состава инсулина гларгина U300 (HOE901-U300) и Lantus® U100, оба из которых применяли в сочетании с пероральным(и) гипогликемическим(и) лекарственным(и) средством(ами), у пациентов с сахарным диабетом 2 типа.
В примере 2 не меньшая эффективность U300 по сравнению с Lantus была доказана на основании разницы полученного методом наименьших квадратов среднего значения HbA1c по сравнению с Lantus, составлявшей -0,01% (95% CI [-0,139; 0,119]) (таблица 26). Изменение полученных методом наименьших квадратов средних значений прединъекционной концентрации SMPG было сходным в группах U300 (-0,56 ммоль/л) и Lantus (-0,51 ммоль/л) (таблица 28).
В примере 2 подтверждено, что посредством лечения с помощью состава инсулина гларгина U300 риск развития эпизода ночной гипогликемии может быть значительно снижен по сравнению с лечением с помощью Lantus® U100. В другой группе пациентов, а именно у пациентов с сахарным диабетом 2 типа, у которых гипогликемическое(ие) лекарственное(ые) средство (средства) в отдельности не обеспечивали адекватный контроль, было неожиданно обнаружено, что количество пациентов по меньшей мере с одним случаем ночной тяжелой и/или подтвержденной гипогликемии в исследуемом промежутке времени в группе U300 [87/403 (21,6%)] было меньше, чем в группе Lantus [113/405 (27,9%)] (см. таблицу 27). Превосходство U300 по сравнению с Lantus было показано при относительном риске 0,77 (95% CI [0,61, 0,99]) (р=0,0380).
В примере 3 изменяемые интервалы дозирования сравнивают с фиксированными интервалами дозирования, предусматривающими введение состава инсулина гларгина U300 в сочетании с прандиальным инсулином один раз в день. В примере 3 представлено дополнительное исследование из испытания, описанного в примере 1. Отрицательных эффектов в отношении уровня HbA1c (таблица 50) и концентрации глюкозы в плазме крови натощак (таблица 51) не наблюдали. Общая частота возникновения гипогликемии была сходной для обеих схем вне зависимости от разновидности гипогликемии (таблица 53).
В примере 6 изменяемые интервалы дозирования сравнивают с фиксированными интервалами дозирования, предусматривающими введение состава инсулина гларгина U300 в сочетании с пероральным(и) гипогликемическим(и) лекарственным(и) средством(ами) один раз в день. В примере 6 представлено дополнительное исследование из испытания, описанного в примере 2. Отрицательных эффектов в отношении уровня HbA1c (таблица 67) и концентрации глюкозы в плазме крови натощак (таблица 68) не наблюдали. Общая частота возникновения гипогликемии была сходной для обеих схем вне зависимости от разновидности гипогликемии (таблица 70).
Один аспект настоящего изобретения относится к водному фармацевтическому составу для применения в лечении сахарного диабета I типа или II типа, где лечение снижает риск развития ночной гипогликемии, при этом указанный состав содержит 200-1000 ЕД/мл [эквимолярно 200-1000 ME человеческого инсулина] инсулина гларгина, при условии, что концентрация инсулина гларгина в указанном составе не составляет 684 ЕД/мл. Настоящее изобретение относится к водному фармацевтическому составу для применения в снижении риска развития ночной гипогликемии.
Состав по настоящему изобретению может уменьшать частоту возникновения ночной гипогликемии при введении пациенту с сахарным диабетом, как описано в данном документе.
"Уменьшение частоты возникновения ночной гипогликемии" включает уменьшение числа эпизодов ночной гипогликемии и/или тяжести эпизодов ночной гипогликемии. Состав, описанный в данном документе, пригоден для применения в уменьшении частоты возникновения ночной гипогликемии.
Состав по настоящему изобретению может предупреждать ночную гипогликемию при введении пациенту с сахарным диабетом, как описано в данном документе. "Предупреждение ночной гипогликемии" включает уменьшение числа эпизодов ночной гипогликемии и/или тяжести эпизодов ночной гипогликемии. Состав, описанный в данном документе, пригоден для применения в предупреждении ночной гипогликемии.
Состав по настоящему изобретению пригоден для применения в уменьшении числа эпизодов ночной гипогликемии и/или тяжести эпизодов ночной гипогликемии.
Согласно настоящему изобретению гипогликемия представляет собой состояние, при котором у пациента с сахарным диабетом 2 типа концентрация глюкозы в плазме крови составляет ниже 70 мг/дл (или ниже 3,9 ммоль/л), ниже 60 мг/дл (или ниже 3,3 ммоль/л), ниже 54 мг/дл (или ниже 3,0 ммоль/л), ниже 50 мг/дл, ниже 40 мг/дл или ниже 36 мг/дл.
Согласно настоящему изобретению "симптоматическая гипогликемия" или "эпизод симптоматической гипогликемии" представляет собой состояние, связанное с клиническим симптомом, обусловленным гипогликемией, при котором концентрация глюкозы в плазме крови может составлять ниже 70 мг/дл (или ниже 3,9 ммоль/л), ниже 60 мг/дл (или ниже 3,3 ммоль/л), ниже 54 мг/дл (или ниже 3,0 ммоль/л), ниже 50 мг/дл или ниже 40 мг/дл. Клиническим симптомом могут быть, например, потливость, учащенное сердцебиение, чувство голода, беспокойство, тревога, утомляемость, раздражительность, головная боль, снижение концентрации, сонливость, нарушения психики, нарушения зрения, транзиторные сенсорные нарушения, транзиторные двигательные нарушения, спутанность сознания, судороги и кома. В способе по настоящему изобретению можно выбрать один или несколько клинических симптомов симптоматической гипогликемии, указанных в данном документе. Симптоматическая гипогликемия может быть связана с быстрой утилизацией после перорального введения углеводов.
Согласно настоящему изобретению "тяжелая симптоматическая гипогликемия" или "эпизод тяжелой симптоматической гипогликемии" представляет собой состояние с клиническим симптомом, указанным в данном документе, обусловленное гипогликемией, при котором концентрация глюкозы в плазме крови может составлять ниже 70 мг/дл (или ниже 3,9 ммоль/л), ниже 54 мг/дл (или ниже 3,0 ммоль/л) или ниже 36 мг/дл (или ниже 2,0 ммоль/л). Тяжелая симптоматическая гипогликемия может быть связана с острым неврологическим нарушением, обусловленным эпизодом гипогликемии. При тяжелой симптоматической гипогликемии пациент может нуждаться в помощи другого человека для активного введения углеводов, глюкагона или выполнения других реанимационных действий. Эти приступы могут быть связаны со степенью нейрогликопении, достаточной, чтобы вызвать судорожный припадок, потерю сознания или кому. Измерение концентрации глюкозы в плазме крови может быть недоступно во время такого эпизода, но нормализация неврологического статуса, связанная с восстановлением нормальной концентрации глюкозы в плазме крови, считается достаточным доказательством того, что эпизод был вызван низкой концентрацией глюкозы в плазме крови.
Определение тяжелой симптоматической гипогликемии может включать все приступы, при которых неврологическое нарушение является достаточно тяжелым, чтобы препятствовать самостоятельному лечению, и которые тем самым, как считается, подвергают пациентов риску причинения вреда себе или окружающим. Острое неврологическое нарушение может представлять собой по меньшей мере нарушение, выбранное из сонливости, нарушений психики, нарушений зрения, транзиторных сенсорных нарушений, транзиторных двигательных нарушений, спутанности сознания, судорог и комы. "Нуждается в помощи" означает, что пациент(ка) не может сам(а) оказать себе помощь. Помощь пациенту из доброты, когда помощь не является необходимой, не следует считать случаем, когда "нуждаются в помощи".
Тяжелая симптоматическая гипогликемия может быть связана с быстрой утилизацией после перорального введения углеводов, внутривенного введения глюкозы или/и глюкагона.
Согласно настоящему изобретению "документально подтвержденная симптоматическая гипогликемия" или "документально подтвержденный эпизод симптоматической гипогликемии" представляет собой эпизод, во время которого при типичных симптомах гипогликемии измеренная концентрация глюкозы в плазме крови составляет ≤70 мг/дл (≤3,9 ммоль/л) или является меньшей или равной 54 мг/дл (≤3,0 ммоль/л). Клиническими симптомами, которые, как считают, обусловлены приступом гипогликемии, являются, например, повышенная потливость, нервозность, астения/слабость, тремор, головокружение, повышенный аппетит, учащенное сердцебиение, головная боль, нарушение сна, спутанность сознания, судорожные припадки, потеря сознания, кома.
Согласно настоящему изобретению "бессимптомная гипогликемия" или "эпизод бессимптомной гипогликемии" представляет собой эпизод, не сопровождающийся типичными симптомами гипогликемии, но при котором измеренная концентрация глюкозы в плазме крови является меньшей или равной 70 мг/дл (3,9 ммоль/л) или меньшей или равной 54 мг/дл (3,0 ммоль/л).
Согласно настоящему изобретению "вероятная симптоматическая гипогликемия" или "вероятный эпизод симптоматической гипогликемии" представляет собой эпизод, во время которого при симптомах гипогликемии не проводят определение концентрации глюкозы в плазме крови, но который был предположительно обусловлен концентрацией глюкозы в плазме крови, меньшей или равной 70 мг/дл (или меньшей или равной 3,9 ммоль/л) или меньшей или равной 54 мг/дл (или меньшей или равной 3,0 ммоль/л); при этом симптомы лечат с помощью перорального введения углеводов без проведения теста для определения концентрации глюкозы в плазме крови.
Согласно настоящему изобретению "относительная гипогликемия" или "эпизод относительной гипогликемии" представляет собой эпизод, во время которого человек с сахарным диабетом сообщает о каких-либо типичных симптомах гипогликемии и интерпретирует симптомы как указывающие на гипогликемию, но при этом измеренная концентрация глюкозы в плазме крови составляет более 70 мг/дл (или более 3,9 ммоль/л).
Согласно настоящему изобретению "ночной гипогликемией" или "эпизодом ночной гипогликемии" является любая гипогликемия из разновидностей гипогликемии, описанных выше, которая возникает в ночное время. "Ночная гипогликемия" может быть определена по времени суток. В частности, ночной гипогликемией является гипогликемия, возникающая в часы с 00:00 до 05:59 утра. Пациент может бодрствовать или может проснуться из-за эпизода. Пациент также может спать во время эпизода.
Согласно настоящему изобретению "дневной гипогликемией" или "эпизодом дневной гипогликемии" является, в частности, любая гипогликемия из разновидностей гипогликемии, описанных выше, которая возникает между 06:00 утра и 23:59.
Согласно настоящему изобретению ночной гипогликемией может являться симптоматическая гипогликемия, тяжелая симптоматическая гипогликемия, документально подтвержденная симптоматическая гипогликемия, вероятная симптоматическая гипогликемия, относительная симптоматическая гипогликемия или бессимптомная гипогликемия. Предпочтительной является симптоматическая гипогликемия, более предпочтительно тяжелая симптоматическая гипогликемия.
"Снижение риска развития гипогликемии", как используется в данном документе, может включать уменьшение частоты возникновения гипогликемии. Частоту возникновения гипогликемии на пациент-год можно рассчитать для пациента следующим образом: 365,25 × (число приступов гипогликемии)/(число дней лечения) и суммировать по типу эпизода и группе лечения. "Снижение риска развития гипогликемии", как используется в данном документе, может дополнительно включать предупреждение гипогликемии у пациента, когда состав, описанный в данном документе, вводят пациенту с сахарным диабетом, как описано в данном документе. "Снижение риска развития гипогликемии", как используется в данном документе, может дополнительно включать уменьшение числа эпизодов ночной гипогликемии и/или тяжести эпизодов ночной гипогликемии.
В примерах 3 и 6 показано, что периодическое изменение интервалов между инъекциями инсулина гларгина U300 не оказывало отрицательных эффектов в отношении уровня HbA1c (таблицы 50 и 67) и концентрации глюкозы в плазме крови натощак (таблицы 51 и 69). Общая частота возникновения гипогликемии была сходной при введении с изменяемыми интервалами дозирования и при введении с фиксированными интервалами дозирования вне зависимости от разновидности гипогликемии (таблицы 53 и 70).
Один аспект настоящего изобретения относится к водному фармацевтическому составу для введения с изменяемыми временными интервалами. Данный аспект относится к водному фармацевтическому составу для применения в лечении сахарного диабета I типа или II типа, где состав вводят пациенту один раз в день, и где временной интервал после предыдущего введения находится в диапазоне от 24,5 ч. до 28 ч. или в диапазоне от 20 ч. до 23,5 ч. в течение по меньшей мере двух дней в неделю, и где средний временной интервал после предыдущего введения составляет приблизительно 24 ч., при этом указанный состав содержит 200-1000 ЕД/мл [эквимолярно 200-1000 ME человеческого инсулина] инсулина гларгина, при условии, что концентрация инсулина гларгина в указанном составе не составляет 684 ЕД/мл.
Как используется в данном документе, временной интервал после предыдущего введения представляет собой временной интервал между двумя последовательными введениями, в частности, инъекциями.
Предпочтительно, чтобы состав содержал 300 ЕД/мл инсулина гларгина.
По схеме лечения состав можно вводить во временном диапазоне, отсчитываемом от фиксированного времени, например, отсчитываемом от фиксированного времени вечером или утром. Средний временной интервал после предыдущего введения может составлять приблизительно 24 ч. (см. таблицу 46). Интервал "приблизительно 24 ч.", в частности, означает диапазон 24 ч. +/-10 мин., диапазон 24 ч. +/-20 мин. или диапазон 24 ч. +/-30 мин. Средний временной интервал можно рассчитать, например, на неделю, на месяц или на два или три месяца или можно рассчитать на более длительное время.
В таблице 47 описано соблюдение схемы дозирования лекарственного средства в тестовой группе и контрольной группе пациентов из примера 3. Приведен % инъекций у пациентов из разных категорий по интервалу дозирования. В контрольной группе (с фиксированным интервалом дозирования 24 ч.) приблизительно 88% доз U300 инъецировали с интервалом 23-25 ч. после предыдущей инъекции. Приблизительно 12% доз инъецировали с интервалом менее 23 ч. или более 25 ч. С расчетом на одну инъекцию в день пациентам дозировали состав U300 с интервалом менее 23 ч. или более 25 ч. менее одного дня в неделю. В тестовой группе (с изменяемым интервалом дозирования) приблизительно 63% доз U300 инъецировали с интервалом 23-25 ч. после предыдущей инъекции. Приблизительно 37% доз инъецировали с интервалом менее 23 ч. или более 25 ч. С расчетом на одну инъекцию в день пациентам дозировали состав U300 с интервалом менее 23 ч. или более 25 ч. два или три дня в неделю.
Водный состав можно вводить с временным интервалом, указанным в данном документе, в течение по меньшей мере двух дней в неделю, в течение по меньшей мере трех дней в неделю, в течение по меньшей мере четырех дней в неделю или в течение по меньшей мере пяти дней в неделю. Водный состав можно вводить с временным интервалом, указанным в данном документе, в течение не более пяти дней в неделю, в течение не более четырех дней в неделю или в течение не более трех дней в неделю. Более конкретно, водный состав вводят с временным интервалом, указанным в данном документе, в течение двух или трех дней в неделю или в течение двух-трех дней в неделю.
"Периодическое изменение", в частности, означает, что водный состав вводят в течение двух или трех дней в неделю с временным интервалом, указанным в данном документе.
Количество "дней в неделю", как указано в данном документе, можно рассчитать, например, на неделю, на месяц или на два или три месяца или можно рассчитать на более длительное время.
"Изменяемые интервалы между инъекциями" означают, что временной интервал после предыдущей инъекции варьирует в пределах заранее установленного временного диапазона. Временной интервал после предыдущего введения может находиться в диапазоне от 24,5 ч. до 28 ч. или в диапазоне от 20 ч. до 23,5 ч. В частности, временной интервал после предыдущего введения находится в диапазоне от 25 ч. до 28 ч. или в диапазоне от 20 ч. до 23 ч.
Временной интервал после предыдущего введения может также находиться в диапазоне от 25 ч. до 27 ч. или в диапазоне от 21 ч. до 23 ч.
Временной интервал после предыдущего введения может также находиться в диапазоне от 25 ч. до 26,5 ч. или в диапазоне от 21,5 ч. до 23 ч.
В данном аспекте наполнители в составе могут представлять собой наполнители, описанные в данном документе. Пациентом, подлежащим лечению, может быть пациент, описанный в данном документе.
Схему лечения с изменяемыми временными интервалами, описанную в данном документе, можно комбинировать со снижением риска развития ночной гипогликемии, как описано в данном документе.
Состав для применения в лечении сахарного диабета 1 или 2 типа, вводимый с изменяемыми временными интервалами, как описано в данном документе, можно комбинировать с применением в лечении сахарного диабета 1 или 2 типа со снижением риска развития ночной гипогликемии, как описано в данном документе.
Согласно настоящему изобретению нормогликемия может означать концентрацию глюкозы в плазме крови от 70 мг/дл до 140 мг/дл (что соответствует от 3,9 ммоль/л до 7,8 ммоль/л).
Пациентом, подлежащим лечению с помощью состава, описанного в данном документе, может быть пациент с сахарным диабетом I типа или II типа. Пациентом предпочтительно является пациент с сахарным диабетом II типа.
Фармацевтический состав по настоящему изобретению можно вводить в сочетании по меньшей мере с одним гипогликемическим средством. В частности, по меньшей мере одним гипогликемическим средством является метформин или/и его фармацевтически приемлемая соль. "Метформин" является международным непатентованным названием 1,1-диметилбигуанида (номер CAS 657-24-9). Согласно настоящему изобретению термин "метформин" включает любую его фармацевтически приемлемую соль.
Согласно настоящему изобретению метформин можно вводить перорально. Специалист в данной области знает составы метформина, пригодные для лечения сахарного диабета посредством перорального введения. Метформин можно вводить пациенту, нуждающемуся в этом, в количестве, достаточном, чтобы вызвать терапевтический эффект. Метформин можно вводить в дозе по меньшей мере 1,0 г/день или по меньшей мере 1,5 г/день. Для перорального введения метформин может быть составлен в твердой лекарственной форме, такой как таблетка или пилюля. Метформин может быть составлен с подходящими фармацевтически приемлемыми носителями, адъювантами или/и вспомогательными веществами.
Состав по настоящему изобретению и метформин можно вводить посредством разных путей введения. Метформин можно вводить перорально, а состав по настоящему изобретению можно вводить парентерально.
Пациентом, подлежащим лечению с помощью состава по настоящему изобретению, может быть пациент, страдающий сахарным диабетом 2 типа, где лечение по меньшей мере одним гипогликемическим средством в отдельности не обеспечивает адекватный контроль сахарного диабета 2 типа. Гипогликемическим средством может быть метформин, при этом введение не обеспечивает адекватный контроль сахарного диабета 2 типа, например, после лечения в течение по меньшей мере 2 или по меньшей мере 3 месяцев, например, при дозе метформина по меньшей мере 1,0 г/день или по меньшей мере 1,5 г/день.
Согласно настоящему изобретению у пациента не обеспечивается адекватный контроль сахарного диабета 2 типа, если по меньшей мере один физиологический показатель, отражающий концентрацию глюкозы в крови (например, значение HbA1c, прединъекционная концентрация SMPG или/и концентрация глюкозы в плазме крови натощак), превышает нормогликемические значения, описанные в данном документе. В частности, у пациента, у которого не обеспечивается адекватный контроль сахарного диабета 2 типа, может иметься
(i) значение HbA1c в диапазоне от 7% до 10% или даже больше,
(ii) прединъекционная концентрация SMPG, составляющая по меньшей мере 9 ммоль/л, или/и
(iv) концентрация глюкозы в плазме крови натощак, составляющая по меньшей мере 8,0 ммоль/л.
У пациента, подлежащего лечению с помощью состава, описанного в данном документе, значение HbA1c может находиться в диапазоне от 7% до 10% в начале лечения. Более конкретно, у пациента, подлежащего лечению, значение HbA1c может составлять по меньшей мере 8% или значение HbA1c может находиться в диапазоне от 8% до 10% в начале лечения по настоящему изобретению.
Пациентом, подлежащим лечению с помощью состава, описанного в данном документе, может быть взрослый субъект. Возраст пациента может составлять по меньшей мере 50 лет, по меньшей мере 57 лет, по меньшей мере 58 лет, по меньшей мере 59 лет, по меньшей мере 60 лет, по меньшей мере 65 лет, по меньшей мере 70 лет или по меньшей мере 75 лет в начале лечения по настоящему изобретению.
Пациентом, подлежащим лечению с помощью состава, описанного в данном документе, может быть субъект с ожирением в начале лечения по настоящему изобретению. Согласно настоящему изобретению у субъекта с ожирением индекс массы тела (BMI) может составлять по меньшей мере 30 кг/м2, по меньшей мере 31 кг/м2, по меньшей мере 32 кг/м2, по меньшей мере 33 кг/м2, по меньшей мере 34 кг/м2, по меньшей мере 35 кг/м2, по меньшей мере 36 кг/м2, по меньшей мере 37 кг/м2, по меньшей мере 38 кг/м2, по меньшей мере 39 кг/м2 или по меньшей мере 40 кг/м2 в начале лечения. Предпочтительно, чтобы у пациента BMI составлял по меньшей мере 34 кг/м2 или по меньшей мере 36 кг/м2 в начале лечения.
У пациента, подлежащего лечению с помощью состава, описанного в данном документе, может иметься повышенный риск развития гипогликемии, в частности, у пациента с сахарным диабетом 2 типа, у которого был по меньшей мере один эпизод гипогликемии.
Пациент, подлежащий лечению с помощью состава, описанного в данном документе, мог получать инсулин непосредственно перед лечением, описанным в данном документе. В частности, пациент мог получать базальный инсулин, например, в дозе по меньшей мере 32 ЕД/день или по меньшей мере 42 ЕД/день. Согласно настоящему изобретению может учитываться любое предварительное лечение базальным инсулином. В частности, базальный инсулин может быть выбран из инсулина гларгина, детемира, NPH, ленте, ультраленте, новолина, хумалога и их смесей. Смесь может содержать два разных базальных инсулина. Например, можно использовать смесь, содержащую детемир и гларгин, или смесь, содержащую NPH и новолин. Базальный инсулин предпочтительно представляет собой инсулин гларгин или смесь, содержащую инсулин гларгин. Согласно настоящему изобретению "базальный инсулин" включает его пригодные фармацевтически приемлемые соли.
Пациент, подлежащий лечению с помощью состава, описанного в данном документе, мог получать прандиальный инсулин короткого действия непосредственно перед лечением, описанным в данном документе. Прандиальным инсулином короткого действия может быть аналог инсулина, например, инсулин глулизин, инсулин лизпро или инсулин аспарт.
Состав, описанный в данном документе, можно вводить один или два раза в день. В частности, состав, описанный в данном документе, можно вводить один раз в день, например, вечером. Состав, описанный в данном документе, можно вводить один раз в день вечером в заранее установленное время.
Пациент может дополнительно получать прандиальный инсулин короткого действия. Прандиальным инсулином короткого действия может быть аналог инсулина, например, инсулин глулизин, инсулин лизпро или инсулин аспарт.
У пациента, подлежащего лечению с помощью состава по настоящему изобретению, прединъекционная концентрация глюкозы в плазме крови при самоконтроле (SMPG) составляет по меньшей мере 9 ммоль/л, по меньшей мере 10 ммоль/л, по меньшей мере 10,5 ммоль/л или по меньшей мере 11 ммоль/л в начале лечения по настоящему изобретению. Согласно настоящему изобретению концентрация глюкозы в плазме крови при самоконтроле может представлять собой концентрацию SMPG натощак или прединъекционную концентрацию SMPG (например, измеряемую за 30 минут до инъекции состава, описанного в данном документе).
У пациента, подлежащего лечению, концентрация глюкозы в плазме крови натощак может составлять по меньшей мере 7 ммоль/л, по меньшей мере 7,5 ммоль/л, по меньшей мере 8 ммоль/л, по меньшей мере 8,5 ммоль/л или по меньшей мере 9 ммоль/л в начале лечения по настоящему изобретению.
Хотя настоящее изобретение не ограничено составом инсулина гларгина U300, а является эффективным с другими более концентрированными составами инсулина гларгина, как подробно изложено в настоящем описании, клиническое исследование, описанное в данном документе, проводили в отношении состава инсулина гларгина U300.
1 мл состава инсулина гларгина U300 содержит 10,913 мг человеческого инсулина 21A-Gly-30Ba-L-Arg-30Bb-L-Arg [эквимолярно 300 ME человеческого инсулина], 90 мкг цинка, 2,7 мг м-крезола, 20 мг 85% глицерина, HCl и NaOH до рН 4,0; удельная плотность 1,006 г/мл.
Однако возможны изменения типа наполнителей и их концентраций.
Фармацевтический состав содержит 200-1000 ЕД/мл инсулина гларгина [эквимолярно 200-1000 ME человеческого инсулина], где концентрация указанного состава не составляет 684 ЕД/мл, предпочтительно составляет 250-500 ЕД/мл инсулина гларгина [эквимолярно 250-500 ME человеческого инсулина], более предпочтительно 270-330 ЕД/мл инсулина гларгина [эквимолярно 270-330 ME человеческого инсулина] и еще более предпочтительно 300 ЕД/мл инсулина гларгина [эквимолярно 300 ME человеческого инсулина].
В фармацевтический состав могут быть добавлены поверхностно-активные вещества, например, помимо прочего, неионогенные поверхностно-активные вещества. В частности, предпочтительными являются стандартные с фармацевтической точки зрения поверхностно-активные вещества, такие как, например, неполные эфиры и сложные эфиры жирных кислот и эфиры многоатомных спиртов, таких как глицерин, сорбит и т.п. (Span®, Tween®, в частности, Tween® 20 и Tween® 80, Myrj®, Brij®), Cremophor или полоксамеры. Поверхностно-активные вещества присутствуют в фармацевтической композиции в концентрации 5-200 мкг/мл, предпочтительно 5-120 мкг/мл и особенно предпочтительно 20-75 мкг/мл.
Состав может дополнительно содержать консерванты (например, фенол, м-крезол, п-крезол, парабены), изотонические средства (например, маннит, сорбит, лактозу, декстрозу, трегалозу, хлорид натрия, глицерин), буферные вещества, соли, кислоты и щелочи, а также дополнительные наполнители. Эти вещества в каждом случае могут присутствовать отдельно или, альтернативно, в виде смесей.
Глицерин, декстроза, лактоза, сорбит и маннит могут присутствовать в фармацевтическом препарате в концентрации 100-250 мМ, NaCl - в концентрации до 150 мМ. Буферные вещества, такие как, например, фосфатный, ацетатный, цитратный, аргининовый, глицилглициновый или TRIS (т.е. 2-амино-2-гидроксиметил-1,3-пропандиоловый) буфер и соответствующие соли, присутствуют в концентрации 5-250 мМ, предпочтительно 10-100 мМ. Дополнительными наполнителями могут быть, помимо прочего, соли или аргинин.
Концентрация цинка в составе находится в диапазоне концентрации, которая достигается за счет присутствия 0-1000 мкг/мл, предпочтительно 20-400 мкг/мл цинка, наиболее предпочтительно 90 мкг/мл. Однако, цинк может присутствовать в форме хлорида цинка, но соль не ограничена хлоридом цинка.
В фармацевтическом составе глицерин и/или маннит могут присутствовать в концентрации 100-250 ммоль/л, и/или NaCl предпочтительно присутствует в концентрации до 150 ммоль/л.
В фармацевтическом составе буферное вещество может присутствовать в концентрации 5-250 ммоль/л.
Дополнительным объектом настоящего изобретения является фармацевтический состав инсулина для применения, описанного в данном документе, который содержит дополнительные добавки, такие как, например, соли, которые отсрочивают высвобождение инсулина. Смеси таких инсулинов с отсроченным высвобождением с составами, описанными выше, включены в данный документ.
Дополнительным объектом, на который направлено настоящее изобретение, является способ получения таких фармацевтических составов для применения, описанного в данном документе. Для получения составов ингредиенты растворяют в воде, и рН регулируют с помощью HCl и/или NaOH. Также дополнительным объектом, на который направлено настоящее изобретение, является применение таких составов для лечения сахарного диабета.
Дополнительным объектом, на который направлено настоящее изобретение, является применение или добавление поверхностно-активных веществ в качестве стабилизаторов в ходе выполнения способа получения инсулина, аналогов инсулина или производных инсулина или препаратов на их основе.
Настоящее изобретение дополнительно относится к составу, описанному выше, который также дополнительно содержит глюкагоноподобный пептид-1 (GLP1) или его аналог или производное или эксендин-3 или -4 или его аналог или производное, предпочтительно эксендин-4.
Настоящее изобретение дополнительно относится к составу, описанному выше, в котором аналог эксендина-4 выбран из группы, включающей
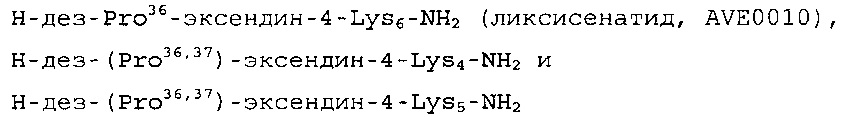
или их фармакологически допустимую соль.
Настоящее изобретение дополнительно относится к составу, описанному выше, в котором аналог эксендина-4 выбран из группы, включающей

или их фармакологически допустимую соль.
Настоящее изобретение дополнительно относится к составу, описанному в предыдущем абзаце, в котором пептид -Lys6-NH2 присоединен к С-концам аналогов эксендина-4.
Настоящее изобретение дополнительно относится к составу, описанному выше, в котором аналог эксендина-4 выбран из группы, включающей
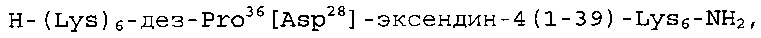


или их фармакологически допустимую соль.
Настоящее изобретение дополнительно относится к составу, описанному выше, который дополнительно содержит Arg34, Lys26-(Nε(γ-глутамил(Nα-гексадеканоил)))-GLP-1 (7-37) [лираглутид] или его фармакологически допустимую соль.
В одном варианте осуществления настоящее изобретение направлено на водный фармацевтический состав для применения, описанного в данном документе, который содержит инсулин гларгин в диапазоне 200-1000 ЕД/мл [эквимолярно 200-1000 МЕ человеческого инсулина], предпочтительно от 200 ЕД/мл до 650 ЕД/мл, еще предпочтительнее от 700 ЕД/мл до 1000 ЕД/мл, более предпочтительно 270-330 ЕД/мл и наиболее предпочтительно в концентрации 300 ЕД/мл при условии, что концентрация инсулина гларгина в указанном составе не составляет 684 ЕД/мл.
Дополнительно, состав также может содержать аналог эксендина-4, такой как, например, ликсисенатид, эксенатид и лираглутид. Эти аналоги эксендина-4 присутствуют в составе в диапазоне от 0,1 мкг до 10 мкг на ЕД инсулина гларгина, предпочтительно от 0,2 до 1 мкг на ЕД инсулина гларгина и более предпочтительно от 0,25 мкг до 0,7 мкг на ЕД инсулина гларгина. Ликсисенатид является предпочтительным.
Дополнительно, водный фармацевтический состав может содержать один или несколько наполнителей, выбранных из группы, включающей цинк, м-крезол, глицерин, полисорбат 20 и натрий. Более конкретно, водный фармацевтический состав может содержать 90 мкг/мл цинка, 2,7 мг/мл м-крезола и 20 мг/мл 85% глицерина. Водный фармацевтический состав необязательно может содержать 20 мкг/мл полисорбата 20.
Значение рН водного фармацевтического состава, описанного в данном документе, может составлять 4,6 или ниже, предпочтительно 4,5 или ниже.
Значение рН водного фармацевтического состава, описанного в данном документе, может также находиться в диапазоне от 3,4 до 4,6, предпочтительно в диапазоне от 4 до 4,5.
Другой аспект настоящего изобретения направлен на способ лечения заболевания или состояния, описанного в данном документе, в частности, на способ лечения сахарного диабета I типа или II типа, включающий введение указанному пациенту водной фармацевтической композиции по настоящему изобретению для пациента с диабетом, где лечение снижает риск развития ночной гипогликемии. Способ предпочтительно относится к лечению сахарного диабета II типа. Среди различных раскрытых диапазонов концентраций предпочтительной является концентрация 300 ЕД/мл. Дополнительно, водный фармацевтический состав также может содержать цинк, м-крезол, глицерин, полисорбат 20 и натрий, а также их смеси в диапазонах, раскрытых в данном документе, в зависимости от водного фармацевтического состава по настоящему изобретению. В предпочтительном варианте осуществления водный фармацевтический состав также содержит от 0,1 мкг до 10 мкг ликсисенатида на ЕД инсулина гларгина. Ночная гипогликемия может представлять собой любую ночную гипогликемию, которая определена в данном документе. Пациентом может быть любой пациент, который определен в данном документе.
Инсулин вводят предпочтительно один раз в день, но его можно вводить два раза в день при необходимости. Требования к дозировке зависят от нужд отдельного пациента, определяемых достижением нормальных или приемлемых уровней глюкозы в крови.
Способ также может представлять собой способ лечения сахарного диабета I типа или II типа у пациента, включающий введение указанному пациенту водной фармацевтической композиции, описанной в данном документе, где состав вводят один раз в день, и где временной интервал после предыдущего введения находится в диапазоне от 24,5 ч. до 28 ч. или в диапазоне от 20 ч. до 23,5 ч. в течение по меньшей мере двух дней в неделю, и где средний временной интервал после предыдущего введения составляет приблизительно 24 ч. Временной интервал может представлять собой временной интервал, который определен в данном документе. Водный состав можно вводить в течение по меньшей мере трех дней в неделю, в течение по меньшей мере четырех дней в неделю или в течение по меньшей мере пяти дней в неделю с временным интервалом, указанным в данном документе. Способ предпочтительно относится к лечению сахарного диабета II типа. Среди различных раскрытых диапазонов концентраций предпочтительной является концентрация 300 ЕД/мл. Дополнительно, водный фармацевтический состав также может содержать цинк, м-крезол, глицерин, полисорбат 20 и натрий, а также их смеси в диапазонах, раскрытых в данном документе, в зависимости от водного фармацевтического состава по настоящему изобретению. В предпочтительном варианте осуществления водный фармацевтический состав также содержит от 0,1 мкг до 10 мкг ликсисенатида на ЕД инсулина гларгина. Пациентом может быть любой пациент, который определен в данном документе.
Способ лечения сахарного диабета 1 или 2 типа, осуществляемого с изменяемыми временными интервалами, как описано в данном документе, можно комбинировать со способом лечения сахарного диабета 1 или 2 типа со снижением риска развития ночной гипогликемии, как описано в данном документе.
Еще один аспект настоящего изобретения направлен на применение водного состава, описанного в данном документе, для изготовления медикамента для лечения заболевания или состояния, описанного в данном документе, в частности, для лечения сахарного диабета I типа или II типа, где лечение снижает риск развития ночной гипогликемии. Применение предпочтительно относится к лечению сахарного диабета II типа. Среди различных раскрытых диапазонов концентраций предпочтительной является концентрация 300 ЕД/мл. Дополнительно, водный фармацевтический состав также может содержать цинк, м-крезол, глицерин, полисорбат 20 и натрий, а также их смеси в диапазонах, раскрытых в данном документе, в зависимости от водного фармацевтического состава по настоящему изобретению. В предпочтительном варианте осуществления водный фармацевтический состав также содержит от 0,1 мкг до 10 мкг ликсисенатида на ЕД инсулина гларгина. Ночная гипогликемия может представлять собой любую ночную гипогликемию, которая определена в данном документе. Пациентом может быть любой пациент, который определен в данном документе.
Инсулин вводят предпочтительно один раз в день, но его можно вводить два раза в день при необходимости. Требования к дозировке зависят от нужд отдельного пациента, определяемых достижением нормальных или приемлемых уровней глюкозы в крови.
Другой аспект относится к применению водного состава, описанного в данном документе, для изготовления медикамента для лечения сахарного диабета I типа или II типа у пациента, включающий введение указанному пациенту водной фармацевтической композиции, описанной в данном документе, где состав вводят один раз в день, и где временной интервал после предыдущего введения находится в диапазоне от 24,5 ч. до 28 ч. или в диапазоне от 20 ч. до 23,5 ч. в течение по меньшей мере двух дней в неделю, и где средний временной интервал после предыдущего введения составляет приблизительно 24 ч. Временной интервал может представлять собой временной интервал, который определен в данном документе. Водный состав можно вводить в течение по меньшей мере трех дней в неделю, в течение по меньшей мере четырех дней в неделю или в течение по меньшей мере пяти дней в неделю с временным интервалом, указанным в данном документе. Способ предпочтительно относится к лечению сахарного диабета II типа. Среди различных раскрытых диапазонов концентраций предпочтительной является концентрация 300 ЕД/мл. Дополнительно, водный фармацевтический состав также может содержать цинк, м-крезол, глицерин, полисорбат 20 и натрий, а также их смеси в диапазонах, раскрытых в данном документе, в зависимости от водного фармацевтического состава по настоящему изобретению. В предпочтительном варианте осуществления водный фармацевтический состав также содержит от 0,1 мкг до 10 мкг ликсисенатида на ЕД инсулина гларгина. Пациентом может быть любой пациент, который определен в данном документе.
Применение для изготовления медикамента для лечения сахарного диабета 1 или 2 типа, осуществляемого с изменяемыми временными интервалами, как описано в данном документе, можно комбинировать с применением для изготовления медикамента для лечения сахарного диабета 1 или 2 типа со снижением риска развития ночной гипогликемии, как описано в данном документе.
Настоящее изобретение относится, помимо прочего, к следующим пунктам.
1. Водный фармацевтический состав для применения в лечении сахарного диабета I типа или II типа, где лечение снижает риск развития ночной гипогликемии, при этом указанный состав содержит 200-1000 ЕД/мл [эквимолярно 200-1000 ME человеческого инсулина] инсулина гларгина, при условии, что концентрация инсулина гларгина в указанном составе не составляет 684 ЕД/мл.
2. Водный состав для применения по пункту 1, содержащий от 200 ЕД/мл до 650 ЕД/мл инсулина гларгина.
3. Водный состав для применения по пункту 1, содержащий от 700 ЕД/мл до 1000 ЕД/мл инсулина гларгина.
4. Водный состав для применения по пункту 2, содержащий 270-330 ЕД/мл инсулина гларгина [эквимолярно 270-330 ME человеческого инсулина].
5. Водный состав для применения по пункту 4, содержащий 300 ЕД/мл инсулина гларгина [эквимолярно 300 ME человеческого инсулина].
6. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где ночная гипогликемия выбрана из симптоматической гипогликемии, тяжелой симптоматической гипогликемии, документально подтвержденной симптоматической гипогликемии, вероятной симптоматической гипогликемии, относительной симптоматической гипогликемии и бессимптомной гипогликемии.
7. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где у пациента, подлежащего лечению, значение HbA1c составляет по меньшей мере 8% в начале лечения.
8. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где возраст пациента, подлежащего лечению, составляет по меньшей мере 60 лет в начале лечения.
9. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где у пациента, подлежащего лечению, BMI составляет по меньшей мере 30 кг/м2 в начале лечения.
10. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где пациент, подлежащий лечению, получал базальный инсулин непосредственно перед лечением.
11. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где пациент, подлежащий лечению, получал прандиальный инсулин короткого действия непосредственно перед лечением.
12. Водный фармацевтический состав для применения по пункту 10 или 11, где у пациента, подлежащего лечению, прединъекционная концентрация SMPG составляет по меньшей мере 9 ммоль/л в начале лечения.
13. Водный фармацевтический состав для применения по пункту 10 или 11, где у пациента, подлежащего лечению, концентрация глюкозы в плазме крови натощак составляет по меньшей мере 8 ммоль/л в начале лечения.
14. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где состав вводится один раз в день вечером в заранее установленное время.
15. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где пациент дополнительно получает прандиальный инсулин короткого действия.
16. Водный фармацевтический состав для применения по любому из предыдущих пунктов, содержащий аналог эксендина-4.
17. Водный состав для применения по пункту 16, где аналог эксендина-4 выбран из группы, включающей ликсисенатид, эксенатид и лираглутид.
18. Водный состав для применения по пункту 17, содержащий от 0,1 мкг до 10 мкг ликсисенатида на ЕД инсулина гларгина.
19. Водный состав для применения по пункту 18, содержащий от 0,2 до 1 мкг ликсисенатида на ЕД инсулина гларгина.
20. Водный состав для применения по пункту 19, содержащий от 0,25 мкг до 0,7 мкг ликсисенатида на ЕД инсулина гларгина.
21. Водный состав для применения по любому из предыдущих пунктов, содержащий один или несколько наполнителей, выбранных из группы, включающей цинк, м-крезол, глицерин, полисорбат 20 и натрий.
22. Водный состав для применения по пункту 21, содержащий 90 мкг/мл цинка, 2,7 мг/мл м-крезола и 20 мг/мл 85% глицерина.
23. Водный состав для применения по пункту 21, содержащий 90 мкг/мл цинка, 2,7 мг/мл м-крезола, 20 мкг/мл полисорбата 20 и 20 мг/мл 85% глицерина.
24. Водный состав для применения по любому из предыдущих пунктов, где значение рН составляет от 3,4 до 4,6.
25. Водный состав для применения по пункту 24, где значение рН составляет 4.
26. Водный состав для применения по пункту 24, где значение рН составляет 4,5.
27. Фармацевтический состав для применения по любому из пунктов 1-26, где сахарный диабет является сахарным диабетом II типа.
28. Фармацевтический состав для применения по пункту 27, где по меньшей мере одно пероральное гипогликемическое средство в отдельности не обеспечивает адекватный контроль сахарного диабета II типа.
29. Фармацевтический состав для применения по пункту 28, где по меньшей мере одним пероральным гипогликемическим средством является метформин.
30. Фармацевтический состав для применения по пункту 29, где лечение с помощью по меньшей мере 1,5 г/день метформина не обеспечивает адекватный контроль сахарного диабета.
31. Водный фармацевтический состав для применения по любому из пунктов 27-30, вводимый в сочетании по меньшей мере с одним пероральным гипогликемическим средством.
32. Водный фармацевтический состав для применения по пункту 31, где по меньшей мере одним гипогликемическим средством является метформин.
33. Водный фармацевтический состав для применения по любому из предыдущих пунктов, где состав вводится один раз в день, и где временной интервал после предыдущего введения находится в диапазоне от 24,5 ч. до 28 ч. или в диапазоне от 20 ч. до 23,5 ч. в течение по меньшей мере двух дней в неделю, и где средний временной интервал после предыдущего введения составляет приблизительно 24 ч.
34. Способ лечения сахарного диабета I типа или II типа у пациента, включающий введение указанному пациенту водной фармацевтической композиции, содержащей инсулин гларгин в концентрации 300 ЕД/мл, где лечение снижает риск развития ночной гипогликемии.
35. Способ по пункту 34, где указанная фармацевтическая композиция дополнительно содержит наполнители, выбранные из группы, включающей цинк, м-крезол, глицерин, полисорбат 20 и натрий.
36. Способ по пункту 34, где указанная фармацевтическая композиция дополнительно содержит от 0,1 мкг до 10 мкг ликсисенатида на ЕД инсулина гларгина.
37. Применение водного состава по любому из предыдущих пунктов для изготовления лекарственного средства для лечения сахарного диабета 1 типа и сахарного диабета 2 типа, где лечение снижает риск развития ночной гипогликемии.
38. Водный фармацевтический состав для применения в лечении сахарного диабета I типа или II типа, где состав вводят пациенту один раз в день, и где временной интервал после предыдущего введения находится в диапазоне от 24,5 ч. до 28 ч. или в диапазоне от 20 ч. до 23,5 ч. в течение по меньшей мере двух дней в неделю, и где средний временной интервал после предыдущего введения составляет приблизительно 24 ч., при этом указанный состав содержит 200-1000 ЕД/мл [эквимолярно 200-1000 ME человеческого инсулина] инсулина гларгина, при условии, что концентрация инсулина гларгина в указанном составе не составляет 684 ЕД/мл.
39. Водный состав по пункту 38, вводимый в течение по меньшей мере трех дней в неделю с временным интервалом, указанным в пункте 38.
40. Водный состав по пункту 38, вводимый в течение по меньшей мере четырех дней в неделю с временным интервалом, указанным в пункте 38.
41. Водный состав по любому из пунктов 38-40, где временной интервал после предыдущего введения находится в диапазоне от 25 ч. до 28 ч. или в диапазоне от 20 ч. до 23 ч.
42. Водный состав по любому из пунктов 38-41, где временной интервал после предыдущего введения находится в диапазоне от 25 ч. до 27 ч. или в диапазоне от 21 ч. до 23 ч.
43. Водный состав по любому из пунктов 38-42, где временной интервал после предыдущего введения находится в диапазоне от 25 ч. до 26,5 ч. или в диапазоне от 21,5 ч. до 23 ч.
44. Водный состав для применения по любому из пунктов 38-43, содержащий инсулин гларгин в количестве, определенном в любом из пунктов 2-5.
45. Водный состав для применения по любому из пунктов 38-44, где пациент определен в любом из пунктов 7-15.
46. Водный состав для применения по любому из пунктов 38-45, дополнительно содержащий аналог эксендина-4, определенный в любом из пунктов 16-20.
47. Водный состав для применения по любому из пунктов 38-46, дополнительно содержащий один или несколько наполнителей, определенных в любом из пунктов 21-23.
48. Водный состав для применения по любому из пунктов 38-47, имеющий значение рН, определенное в любом из пунктов 24-26.
49. Водный состав для применения по любому из пунктов 38-48, где лечение снижает риск развития ночной гипогликемии.
50. Водный состав для применения по пункту 49, где ночная гипогликемия выбрана из симптоматической гипогликемии, тяжелой симптоматической гипогликемии, документально подтвержденной симптоматической гипогликемии, вероятной симптоматической гипогликемии, относительной симптоматической гипогликемии и бессимптомной гипогликемии.
51. Водный состав для применения по любому из пунктов 38-50, где сахарный диабет является сахарным диабетом II типа.
52. Водный состав для применения по пункту 51, где по меньшей мере одно пероральное гипогликемическое средство, определенное в любом из пунктов 28-32, в отдельности не обеспечивает адекватный контроль сахарного диабета II типа.
53. Способ лечения сахарного диабета I типа или II типа у пациента, включающий введение указанному пациенту водной фармацевтической композиции, содержащей инсулин гларгин в концентрации 300 ЕД/мл, где состав вводят один раз в день, и где временной интервал после предыдущего введения находится в диапазоне от 24,5 ч. до 28 ч. или в диапазоне от 20 ч. до 23,5 ч. в течение по меньшей мере двух дней в неделю, и где средний временной интервал после предыдущего введения составляет приблизительно 24 ч.
54. Способ по пункту 53, где указанная фармацевтическая композиция дополнительно содержит наполнители, выбранные из группы, включающей цинк, м-крезол, глицерин, полисорбат 20 и натрий.
55. Способ по пункту 53, где указанная фармацевтическая композиция дополнительно содержит от 0,1 мкг до 10 мкг ликсисенатида на ЕД инсулина гларгина.
56. Применение водного состава по любому из предыдущих пунктов для изготовления лекарственного средства для лечения сахарного диабета 1 типа и сахарного диабета 2 типа, где состав вводят один раз в день, и где временной интервал после предыдущего введения находится в диапазоне от 24,5 ч. до 28 ч. или в диапазоне от 20 ч. до 23,5 ч. в течение по меньшей мере двух дней в неделю, и где средний временной интервал после предыдущего введения составляет приблизительно 24 ч.
57. Изделие, содержащее упаковочный материал, водный состав по любому из предыдущих пунктов и этикетку или упаковочный материал, на которых указано, что состав вводится один раз в день, и при этом временной интервал после предыдущего введения составляет от 21 ч. до 27 ч., и при этом средний временной интервал после предыдущего введения составляет приблизительно 24 ч.
58. Изделие, содержащее упаковочный материал, водный состав по любому из предыдущих пунктов и этикетку или упаковочный материал, на которых указано, что состав можно вводить вместе с другими гипогликемическими лекарственными препаратами.
59. Изделие, содержащее упаковочный материал, водный состав по любому из предыдущих пунктов и этикетку или упаковочный материал, на которых указано, что переход с введения продуктов на основе базального инсулина один раз в день на введение состава один раз в день можно осуществлять с сохранением количества единиц, исходя из дозы применяемого ранее базального инсулина; и при переходе с введения продукта на основе базального инсулина два раза в день на введение состава один раз в день рекомендуемая начальная доза состава составляет 80% от общей суточной дозы базального инсулина, введение которого прекращают.
60. Изделие, содержащее упаковочный материал, водный состав по любому из предыдущих пунктов и этикетку или упаковочный материал, на которых указано, что в случае введения состава вместе с веществом, которое может усиливать эффект состава, заключающийся в снижении уровня глюкозы в крови, выбранным из группы, включающей гипогликемические лекарственные препараты, ингибиторы ангиотензин-превращающего фермента (АСЕ), дизопирамид, фибраты, флуоксетин, ингибиторы моноаминоксидазы (МАО), пентоксифиллин, пропоксифен, салицилаты и сульфаниламидные антибиотики, может потребоваться корректировка дозы состава.
61. Изделие, содержащее упаковочный материал, водный состав по любому из предыдущих пунктов и этикетку или упаковочный материал, на которых указано, что в случае введения состава вместе с веществом, которое может ослаблять эффект состава, заключающийся в снижении уровня глюкозы в крови, выбранным из группы, включающей кортикостероиды, даназол, диазоксид, диуретики, глюкагон, изониазид, эстрогены и прогестогены, производные фенотиазина, соматропин, симпатомиметические лекарственные препараты (например, эпинефрин [адреналин], сальбутамол, тербуталин), гормоны щитовидной железы, атипичные антипсихотические лекарственные препараты (например, клозапин и оланзапин) и ингибиторы протеазы, может потребоваться корректировка дозы состава.
Настоящая заявка описана ниже с помощью следующих графических материалов и примеров, которые, как предполагается, не служат для какого-либо ограничения.
Пояснения
Фигура 1 - основной анализ эффективности - средние значения уровня HbA1c (%) по визитам на протяжении основного 6-месячного периода применения исследуемого препарата - популяция mITT (пример 1). BAS = исходное значение, M6LOCF = последнее значение в основном 6-месячном периоде применения исследуемого препарата (LOCF). LOCF = перенос данных последнего наблюдения вперед.
Фигура 2 - другие вторичные конечные точки эффективности - средние значения средней прединъекционной концентрации SMPG (ммоль/л) по визитам на протяжении основного 6-месячного периода применения исследуемого препарата - популяция mITT (пример 1). BAS = исходное значение, M6LOCF = последнее значение в основном 6-месячном периоде применения исследуемого препарата (LOCF). LOCF = перенос данных последнего наблюдения вперед.
Фигура 3 - другие вторичные конечные точки эффективности - средние значения концентрации SMPG (ммоль/л) в профиле по 8 моментам времени в начале исследования и в конечной точке на 6 месяце - популяция mITT (пример 1). LOCF = перенос данных последнего наблюдения вперед.
Фигура 4 - другие вторичные конечные точки эффективности - средние суточные дозы базального инсулина и прандиального инсулина (ЕД) по визитам на протяжении основного 6-месячного периода применения исследуемого препарата - популяция mITT (пример 1). BAS = исходное значение, M6LOCF = последнее значение в основном 6-месячном периоде применения исследуемого препарата (LOCF). LOCF = перенос данных последнего наблюдения вперед.
Фигура 5 - основной анализ эффективности - средние значения уровня HbA1c (%) по визитам на протяжении основного 6-месячного периода применения исследуемого препарата - популяция mITT (пример 2). BAS = исходное значение, M6LOCF = конечная точка на 6 месяце (LOCF), LOCF = перенос данных последнего наблюдения вперед. Примечание. Для всех пациентов, подвергнутых резервной терапии на протяжении 6-месячного периода, последнее после начала исследования измерение уровня HbA1c перед началом резервной терапии и на протяжении 6-месячного периода применения исследуемого препарата использовали в качестве конечной точки для HbA1c.
Фигура 6 - другие вторичные конечные точки эффективности - средние значения средней прединъекционной концентрации SMPG (ммоль/л) по визитам на протяжении основного 6-месячного периода применения исследуемого препарата - популяция mITT (пример 2). BAS = исходное значение, M6LOCF = конечная точка на 6 месяце (LOCF). SMPG = концентрация глюкозы в плазме крови при самоконтроле. LOCF = перенос данных последнего наблюдения вперед. Примечание. Для всех пациентов, подвергнутых резервной терапии на протяжении 6-месячного периода, последнее после начала исследования измерение средней прединъекционной концентрации SMPG перед началом резервной терапии и на протяжении 6-месячного периода применения исследуемого препарата использовали в качестве конечной точки для средней прединъекционной концентрации SMPG.
Фигура 7 - другие вторичные конечные точки эффективности - средние значения концентрации SMPG (ммоль/л) в профиле по 8 моментам времени в начале исследования и в конечной точке на 6 месяце - популяция mITT (пример 2). LOCF = перенос данных последнего наблюдения вперед. SMPG = концентрация глюкозы в плазме крови при самоконтроле. М6 (LOCF)=LOCF для конечной точки на 6 месяце. Примечание. Для всех пациентов, подвергнутых резервной терапии на протяжении 6-месячного периода, последнее после начала исследования измерение SMPG для профиля по 8 моментам времени перед началом резервной терапии и на протяжении 6-месячного периода применения исследуемого препарата использовали в качестве конечной точки для профиля концентрации SMPG по 8 моментам времени.
Фигура 8 - другие вторичные конечные точки эффективности - средние суточные дозы базального инсулина (ЕД) по визитам на протяжении основного 6-месячного периода применения исследуемого препарата - популяция mITT (пример 2). BAS = исходное значение, M6LOCF = последнее значение в основном 6-месячном периоде применения исследуемого препарата (LOCF). LOCF = перенос данных последнего наблюдения вперед. Примечание. Для всех пациентов, подвергнутых резервной терапии на протяжении 6-месячного периода, последнее после начала исследования измерение дозы инсулина перед началом резервной терапии и на протяжении 6-месячного периода применения исследуемого препарата использовали в качестве конечной точки для дозы инсулина.
Фигура 9 - основной анализ эффективности - средние значения уровня HbA1c (%) по визитам на протяжении 3-месячного периода применения схемы, используемой для сравнения - популяция mITT дополнительного исследования. BASM6 = исходное значение (6 месяц), M9LOCF = последнее значение в 3-месячном периоде применения схемы, используемой для сравнения (LOCF). LOCF = перенос данных последнего наблюдения вперед.
Фигура 10 - средние суточные дозы базального (гларгин) и прандиального инсулина (ЕД) по визитам на протяжении 3-месячного периода применения схемы, используемой для сравнения - популяция mITT дополнительного исследования. BASM6 = исходное значение (6 месяц), M9LOCF = последнее значение в 3-месячном периоде применения схемы, используемой для сравнения (LOCF). LOCF = перенос данных последнего наблюдения вперед.
Фигура 11 - график зависимости средней концентрации глюкозы (мг/дл) от времени суток в течение всего периода лечения популяция CGM.
Фигура 12 - график зависимости средней концентрации глюкозы (мг/дл) от времени суток в течение всего периода применения утренних инъекций - популяция CGM.
Фигура 13 - график зависимости средней концентрации глюкозы (мг/дл) от времени суток в течение всего периода применения вечерних инъекций - популяция CGM.
Фигура 14 - основной анализ эффективности - средние значения уровня HbA1c (%) по визитам на протяжении 3-месячного периода применения схемы, используемой для сравнения - популяция mITT дополнительного исследования. BASM6 = исходное значение (6 месяц), M9LOCF = последнее значение в 3-месячном периоде применения схемы, используемой для сравнения (LOCF). LOCF = перенос данных последнего наблюдения вперед. Примечание. Для всех пациентов, подвергнутых резервной терапии на протяжении 3-месячного периода применения схемы, используемой для сравнения, последнее после начала исследования измерение уровня HbA1c перед началом резервной терапии и на протяжении 3-месячного периода применения исследуемого препарата в дополнительном исследовании использовали в качестве конечной точки для HbA1c.
Фигура 15 - средние суточные дозы базального инсулина (гларгин) (ЕД) по визитам на протяжении 3-месячного периода применения схемы, используемой для сравнения - популяция mITT дополнительного исследования. BASM6 = исходное значение (6 месяц), M9LOCF = последнее значение в 3-месячном периоде применения схемы, используемой для сравнения (LOCF). LOCF = перенос данных последнего наблюдения вперед. Примечание. Для всех пациентов, подвергнутых резервной терапии на протяжении 3-месячного периода применения схемы, используемой для сравнения, последнее после начала исследования измерение дозы инсулина перед началом резервной терапии и на протяжении 3-месячного периода применения исследуемого препарата в дополнительном исследовании использовали в качестве конечной точки для дозы инсулина.
Пример 1. 6-Месячное многоцентровое рандомизированное открытое исследование на параллельных группах, в котором сравнивали эффективность и безопасность нового состава инсулина гларгина и Lantus®, оба из которых применяли в сочетании с прандиальным инсулином у пациентов с сахарным диабетом 2 типа, с 6-месячным продленным периодом исследования безопасности
ОБЩЕЕ ОПИСАНИЕ ИССЛЕДОВАНИЯ
Фаза разработки: фаза 3
Цели
Первичная цель: оценка эффектов HOE901-U300 в отношении гликемического контроля по сравнению с Lantus при введении в качестве базального инсулина по схеме, включающей прандиальный инсулин, с точки зрения изменения уровня HbA1c за период 6 месяцев у пациентов с сахарным диабетом 2 типа.
Основные вторичные цели: сравнение HOE901-U300 и Lantus с точки зрения частоты ночной гипогликемии, изменения прединъекционной концентрации глюкозы в плазме крови и изменения вариабельности прединъекционной концентрации глюкозы в плазме крови.
Дополнительные вторичные цели:
Сравнение HOE901-U300 и Lantus с точки зрения достижения целевых значений HbA1c и контролируемой концентрации глюкозы в плазме крови;
сравнение HOE901-U300 и Lantus с точки зрения удовлетворенности пациентов лечением с помощью опросника удовлетворенности лечением сахарного диабета (статус) (DTSQs) (не представлен в KRM);
оценка безопасности и переносимости HOE901-U300.
Методика. Рандомизацию производили в соотношении 1:1 (HOE901-U300 по отношению к Lantus), а стратификацию осуществляли в зависимости от значений HbA1c при скрининге (<8,0%; ≥8,0%). Объем выборки (400 для HOE901-U300 и 400 для Lantus) выбирали для обеспечения достаточную мощность исследования по первичной конечной точке (изменению уровня HbA1c от начала исследования до конечной точки [6 месяц]), а также для обеспечения возможности сделать выводы по первой основной вторичной конечной точке (частоте ночной гипогликемии).
Число пациентов: Запланировано: 800 (400 на группу лечения) Рандомизировано: 807
Получали лечение: 806
Подвергнуты оценке: эффективности: 804, безопасности: 806
Диагностика и критерии включения. Критерии включения: Пациенты с сахарным диабетом 2 типа, как определено согласно WHO; подписавшие письменное информированное согласие. Ключевые критерии невключения: возраст <18 лет; HbA1c <7,0% или >10% при скрининге; сахарный диабет, отличный от сахарного диабета 2 типа; менее 1 года применения базального инсулина в сочетании с прандиальным и самоконтроля уровня глюкозы в крови; общая суточная доза инсулина гларгина <42 ЕД или эквивалентная доза NPH в течение последних 4 недель перед исследованием (если NPH применяли в качестве базального инсулина перед исследованием).
Исследуемые средства лечения
Экспериментальные лекарственные препараты: изучаемое лекарственное средство: HOE901-U300; контрольное лекарственное средство: Lantus.
Составы. HOE901-U300 (раствор инсулина гларгина, 300 ЕД/мл) представляет собой стерильный апирогенный прозрачный бесцветный раствор в стеклянном картридже, который был укомплектован в шприц-ручку (предварительно наполненную, т.е. одноразовую ручку). Lantus (раствор инсулина гларгина, 100 ЕД/мл) представляет собой стерильный апирогенный прозрачный бесцветный раствор, поставляемый в продаваемой Solostar® (предварительно наполненной, т.е. одноразовой ручке).
Путь введения: подкожная инъекция.
Схема дозирования: инъекция один раз в день вечером. Время инъекции фиксировали в момент рандомизации, и оно должно было сохраняться на протяжении исследования.
HOE901-U300 или Lantus инъецировали один раз в день подкожно вечером, т.е. в любое время непосредственно перед вечерним приемом пищи до отхода ко сну. Время инъекции всегда было одним и тем же в пределах этого временного промежутка и было зафиксировано при рандомизации по усмотрению пациента/исследователя. Пациенты продолжали применять свой прандиальный аналог инсулина.
Начальная доза: пациенты, применявшие Lantus или NPH один раз в день до визита исходного уровня: суточная доза (ЕД) HOE901-U300 или Lantus была равна медианному значению общих суточных доз базального инсулина в последние 3 дня до визита исходного уровня.
Пациенты, применявшие NPH более одного раза в день до визита исходного уровня: суточная доза HOE901-U300 или Lantus (ЕД) должна была быть примерно на 20% меньше медианного значения общих суточных доз инсулина NPH в последние 3 дня до визита исходного уровня.
Дозу базального инсулина корректировали один раз в неделю для достижения концентрации SMPG натощак в целевом диапазоне от 80 до 100 мг/дл (от 4,4 до 5,6 ммоль/л):
на +3 ЕД, если медианное значение концентрации SMPG натощак для последних 3 дней находилось в диапазоне >100 мг/дл и <140 мг/дл (>5,6 и <7,8 ммоль/л);
на +6 ЕД, если медианное значение концентрации SMPG натощак для последних 3 дней составляло ≥140 мг/дл (≥7,8 ммоль/л);
на -3 ЕД, если медианное значение концентрации SMPG натощак для последних 3 дней находилось в диапазоне ≥60 мг/дл и <80 мг/дл (≥3,3 и <4,4 ммоль/л).
Дозы прандиального инсулина должны были быть скорректированы для оптимизации гликемического контроля после оптимизации доз базального инсулина. Дозы болюсного инсулина могли быть уменьшены, тогда как дозы базального инсулина были увеличены.
Неэкспериментальные лекарственные препараты
Пациенты в обеих группах лечения должны были продолжать применять свой прандиальный аналог инсулина в ходе исследования. Пациенты, получавшие сопутствующее лечение метформином, должны были на протяжении исследования продолжать получать постоянную дозу, которую они получали до исследования, за исключением случаев уменьшения дозы или отмены метформина, продиктованных проблемами безопасности.
Продолжительность лечения: до 12 месяцев.
Продолжительность наблюдения: до 54 недель (до 2 недель периода скрининга + 6 месяцев периода анализа эффективности и безопасности + 6 месяцев продленного периода исследования безопасности + 2 дня последующего наблюдения для контроля безопасности).
Периодом анализа эффективности и безопасности является основной 6-месячный период применения исследуемого препарата. Результаты, представленные в настоящем KRM, относятся к этому периоду.
Критерии оценки
Эффективность
Первичная конечная точка эффективности: изменение уровня HbA1c от начала исследования до конечной точки (6 месяц).
Основные вторичные конечные точки: количество пациентов (%) по меньшей мере с одним случаем ночной гипогликемии в промежутке между началом на 9 неделе и конечной точкой (6 месяц), указанной как тяжелая и/или подтвержденной концентрацией глюкозы в плазме крови ≤70 мг/дл (3,9 ммоль/л); изменение прединъекционной концентрации SMPG от начала исследования до конечной точки (6 месяц) и изменение вариабельности прединъекционной концентрации SMPG от начала исследования до конечной точки (6 месяц).
Безопасность. Гипогликемия, частота нежелательных явлений, особенно нежелательных явлений, возникших в ходе лечения (ТЕАЕ), и серьезных нежелательных явлений (SAE), реакций в месте инъекции и реакций гиперчувствительности. Следующая информация не представлена в KRM: физикальное обследование, другая информация, касающаяся безопасности, в том числе данные клинико-лабораторных исследований, основные показатели состояния организма (включая массу тела), ECG в 12 отведениях и наличие антител к инсулину.
Статистические методы. Анализ первичной конечной точки эффективности (изменения уровня HbA1c от начала исследования до конечной точки [6 месяц]) осуществляли с использованием модели ковариационного анализа (ANCOVA) с лечением, типическими группами по уровню HbA1c при скрининге (<8,0 и ≥8,0%) и страной в качестве фиксированных эффектов и с использованием исходного значения уровня HbA1c в качестве ковариаты. Различия между HOE901-U300 и Lantus и двусторонние 95% доверительные интервалы оценивали в рамках ANCOVA.
Подход со ступенчатым закрытым тестированием использовали в отношении первичной конечной точки эффективности для последовательной проверки гипотезы о не меньшей эффективности и превосходстве. На стадии 1 оценивали не меньшую эффективность HOE901-U300 по сравнению с Lantus. Для оценки не меньшей эффективности верхнюю границу двустороннего 95% CI для различия изменения среднего значения уровня HbA1c от начала исследования до конечной точки между HOE901-U300 и Lantus сравнивали с предварительно установленным порогом не меньшей эффективности, составлявшим 0,4% для HbA1c. Не меньшая эффективность будет демонстрироваться, если верхняя граница двустороннего 95% CI для различия между HOE901-U300 и Lantus в популяции mITT будет составлять <0,4%. На стадии 2 оценивали превосходство НОЕ901-U300 по сравнению с Lantus, только если была продемонстрирована не меньшая эффективность. Превосходство HOE901-U300 над Lantus демонстрировалось, если верхняя граница двустороннего 95% CI для различия между HOE901-U300 и Lantus в популяции mITT составляла <0.
Только в случае, если по первичной конечной точке демонстрировалась не меньшая эффективность HOE901-U300 по сравнению с Lantus, осуществляли тестирование в отношении превосходства HOE901-U300 над Lantus по основным вторичным конечным точкам в рамках процедуры иерархического тестирования. Анализы безопасности были описательными, основанными на популяции для оценки безопасности.
Краткое описание
Характеристики популяции
В общей сложности 807 пациентов с сахарным диабетом 2 типа рандомизировали в группы, получающие HOE901-U300 (n=404) или Lantus (n=403); 806 пациентов подвергали воздействию IMP (популяция для оценки безопасности). Популяция mITT (популяция для оценки эффективности) включала 8 04 пациентов.
В целом, в каждой группе лечения досрочно прекращало участие в исследовании сопоставимое число пациентов (НОЕ901-U300: 30/404, 7,4%; Lantus: 31/403, 7,7%).
Демографические данные и исходные параметры были сбалансированы среди групп лечения. Средний возраст в исследуемой популяции составлял 60 лет, у 246/807 (30,4%) составлял ≥65 лет. Среднее значение BMI в начале исследования составляло 36,6 кг/м2. Средняя продолжительность сахарного диабета до начала исследования составляла 15,8 лет, средняя продолжительность предшествующего лечения с помощью базального инсулина составляла 6,6 лет, а медианное значение общей суточной дозы инсулина составляло 1,1 ЕД/кг массы тела. В обеих группах лечения среднее значение уровня HbA1c в начале исследования составляло 8,14%.
Результаты в отношении эффективности
Первичная конечная точка. Изменение полученного методом LS среднего значения уровня HbA1c от начала исследования до конечной точки (6 месяц) было сходным в обеих группах лечения (HOE901-U300: -0,83% (95% CI [-0,946; -0,709]); Lantus: -0,83% (95% CI [-0,944; -0,706]). Не меньшая эффективность HOE901-U300 по сравнению с Lantus была продемонстрирована с разницей полученного методом LS среднего значения уровня HbA1c по сравнению с Lantus, составлявшей -0,00% (95% CI [-0,112; 0,107]), при этом верхняя граница была ниже предварительно установленного порога не меньшей эффективности, составлявшего 0,4%. Превосходство HOE901-U300 по сравнению с Lantus не было продемонстрировано.
1-я основная вторичная конечная точка. Количество пациентов по меньшей мере с одним случаем ночной тяжелой и/или подтвержденной гипогликемии в промежутке между началом на 9 неделе и 6 месяцем в группе HOE901-U300 [136/404 (33,7%)] было меньшим, чем в группе Lantus [180/400 (45,0%)]. Превосходство HOE901-U300 по сравнению с Lantus было показано при относительном риске 0,75 (95% CI [0,63, 0,89]) (р=0,0010).
2-я основная вторичная конечная точка. Изменение полученного методом LS среднего значения прединъекционной концентрации SMPG от начала исследования до конечной точки (6 месяц) было сходным в группах HOE901-U300 (-0,90 ммоль/л) и Lantus (-0,84 ммоль/л). Различие между группами лечения не было статистически значимым (разница между полученными методом LS средними значениями составляла -0,06 (95% CI [-0,383, 0,255], р=0,6921).
3-я основная вторичная конечная точка. Поскольку превосходство HOE901-U300 по сравнению с Lantus не было продемонстрировано по второй основной вторичной конечной точке, то по третьей основной вторичной конечной точке (снижению вариабельности прединъекционной концентрации SMPG на 6 месяце, которое было сходным в обеих группах лечения) не проводили дополнительное тестирование.
Другие вторичные конечные точки эффективности (6 месяц).
Как доля пациентов, у которых был достигнут уровень HbA1c <7%, так и изменение среднего значения концентрации FPG были сходными среди групп лечения. На графическом изображении профилей концентрации SMPG по 8 моментам времени для пациентов, получавших лечение с помощью HOE901-U300 и Lantus, видно значительное снижение концентрации глюкозы в плазме крови в конечной точке (6 месяц) по сравнению с началом исследования. Профили 2 групп лечения практически совпадали как в начале исследования, так и в конечной точке.
Увеличение дозы базального инсулина в группе HOE901-U300 давало в результате среднюю суточную дозу 103 ЕД на 6 месяце по сравнению с группой Lantus, где средняя суточная доза составляла 94 ЕД (средняя доза базального инсулина в начале исследования составляла 70 ЕД в обеих группах лечения). Увеличение суточной дозы прандиального инсулина было сопоставимым среди групп лечения с небольшим повышением в первые две недели. В дальнейшем дозы прандиального инсулина оставались постоянными.
Результаты в отношении безопасности
В целом, в группе HOE901-U300 процентная доля пациентов, сообщавших о гипогликемии, была неизменно меньшей, чем в группе Lantus. Это различие было еще более выраженным на протяжении первых 2 месяцев лечения в рамках исследования, так же как и для эпизодов ночной гипогликемии. На протяжении основного 6-месячного периода применения исследуемого препарата тяжелая гипогликемия была зарегистрирована у 21/404 (5,2%) пациентов, получавших лечение с помощью HOE901-U300, и у 23/402 (5,7%) пациентов, получавших лечение с помощью Lantus.
Процентная доля пациентов с какими-либо ТЕАЕ (HOE901-U300, 222/404 [55,0%]; Lantus: 215/402 [53,5%]) или с серьезными ТЕАЕ (HOE901-U300, 25 [6,2%]; Lantus, 21 [5,2%]) была сходной в обеих группах. У сходной доли пациентов были серьезные кардиальные ТЕАЕ (SOC - нарушения сердечной деятельности) в обеих группах лечения (HOE901-U300: n=5, 1,2%; Lantus: n=7; 1,7%).
Шесть пациентов умерли в ходе исследования, по 3 (0,7%) в каждой группе лечения. Из них 4 пациента умерли в течение первых 6 месяцев, по 2 (0,5%) в каждой группе лечения. Явления со смертельным исходом у троих пациентов в группе HOE901-U300 включали следующие состояния: инфицированный тромбоз и эмболию сердца, бронхогенную карциному с метастазированием и - для третьего пациента - эмболию легочной артерии. Основная причина явлений, приведших к летальному исходу, у троих пациентов в группе Lantus включала хроническую депрессию и интоксикацию лекарственным веществом, у одного пациента имелось несколько состояний, в том числе усугубление хронической сердечной недостаточности (IV класс по NYHA), хроническая почечная недостаточность 4 стадии с острой декомпенсацией, декомпенсированный сахарный диабет и диабетическая нефропатия, вызвавших смертельный исход, а последний пациент страдал от острой остановки кровообращения неизвестной этиологии. Ни один из случаев смерти не считался связанным с исследуемым лекарственным средством.
У сходного числа пациентов в обеих группах лечения были ТЕАЕ, приведшие к окончательному прекращению лечения (НОЕ901-U300: n=6, 1,5%; Lantus: n=7, 1,7%).
Реакции гиперчувствительности на протяжении основного 6-месячного периода применения исследуемого препарата были зарегистрированы в сходном количестве в обеих группах лечения (HOE901-U300: n=3, 0,7%; Lantus: n=2, 0,5%).
Общее количество реакций в месте инъекции на протяжении основного 6-месячного периода применения исследуемого препарата было сходным в обеих группах лечения (HOE901-U300: n=9, 2,2%; Lantus: n=6, 1,5%).
Выводы
В данном исследовании с участием 807 пациентов с T2DM, применявших базальный инсулин в сочетании с прандиальным инсулином, исходные параметры и демографические данные были сбалансированы в группах лечения. Не меньшая эффективность HOE901-U300 по сравнению с Lantus была показана по первичной конечной точке эффективности (изменению уровня HbA1c от начала исследования до конечной точки [месяц 6]). Количество пациентов (%), сообщавших о ночной гипогликемии (тяжелой и/или подтвержденной концентрацией SMPG ≤70 мг/дл [3,9 ммоль/л]) в промежутке между началом на 9 неделе и 6 месяцем, в группе HOE901-U300 было значительно меньшим, чем в группе Lantus (33,7% и 45%, соответственно, RR составлял 0,75, р-значение 0,0010; 1-ая основная вторичная конечная точка эффективности). Для других вторичных конечных точек, таких как прединъекционная концентрация глюкозы в плазме крови, вариабельность прединъекционной концентрации глюкозы в плазме крови, число пациентов, у которых был достигнут целевой уровень HbA1c, изменение среднего значения концентрации FPG и профили концентрации глюкозы в плазме крови по 8 моментам времени были получены сопоставимые результаты среди групп лечения.
Общая частота возникновения гипогликемии (% пациентов по меньшей мере с одним эпизодом) на протяжении основного 6-месячного периода применения исследуемого препарата в группе HOE901-U300 было меньшим, чем в группе Lantus, вне зависимости от разновидности гипогликемии.
HOE901-U300 хорошо переносился на протяжении основного 6-месячного периода применения исследуемого препарата в исследовании, и особых проблем безопасности не наблюдалось.
Краткое описание результатов в отношении эффективности и безопасности из двенадцатимесячного расширенного исследования 1 версии
- HbA1c: на протяжении продленного периода исследования безопасности (от конечной точки основного исследования [6 месяц] до конца лечения [12 месяц]) уровень HbA1c оставался постоянным и был сопоставимым в обеих группах лечения;
- Гипогликемия: в целом, так же как и на протяжении основного 6-месячного периода лечения, на протяжении всего периода применения исследуемого препарата в исследовании гипогликемия в группе HOE901-U300 возникала у сходной или меньшей процентной доли пациентов по сравнению с группой Lantus;
- Безопасность: HOE901-U300 хорошо переносился в ходе исследования, и особых проблем безопасности не наблюдалось; на протяжении всего периода лечения процентная доля пациентов с какими-либо ТЕАЕ без учета конкретных включенных SOC была сходной в обеих группах (289/404 [71,5%] в группе лечения HOE901-U300 и 278/402 [69,2%] в группе лечения Lantus). О серьезных ТЕАЕ сообщало сходное число пациентов (53 [13,1%]) в группе лечения HOE901-U300 и 62 [15,4%] в группе лечения Lantus). На протяжении всего периода применения исследуемого препарата в исследовании у двух (0,5%) пациентов в группе лечения HOE901-U300 и 4 (1,0%) пациентов в группе лечения Lantus было ТЕАЕ, приведшее к смерти.
- Масса тела: в обеих группах лечения на протяжении всего периода применения исследуемого препарата в исследовании отмечалось небольшое увеличение массы тела (1,17 кг для НОЕ901-U300 и 1,40 кг для Lantus).
1. РЕЗУЛЬТАТЫ
1.1 ПАЦИЕНТЫ-УЧАСТНИКИ ИССЛЕДОВАНИЯ
1.1.1 Распределение участников исследования


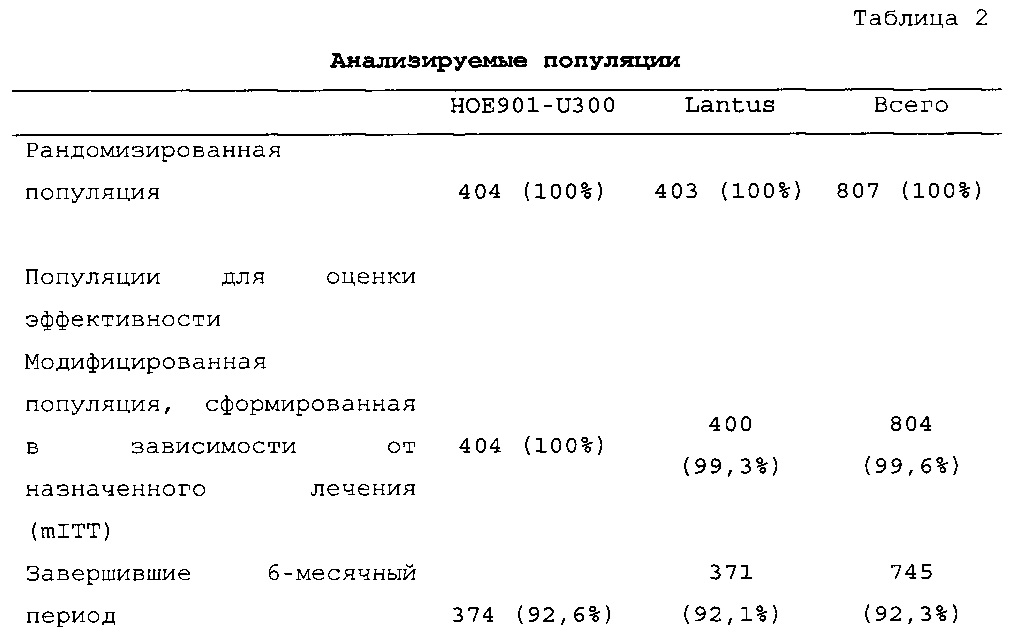

1.1.2 Демографические данные и исходные параметры
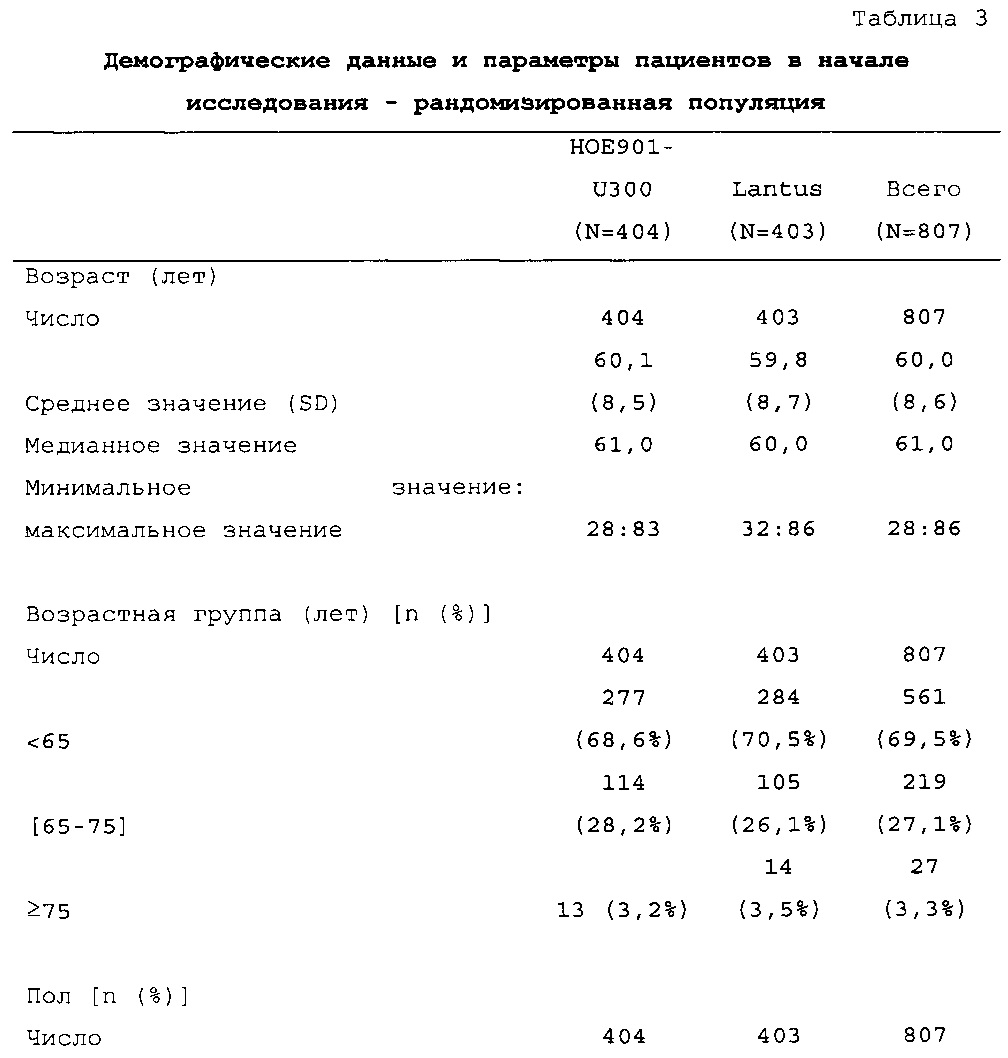
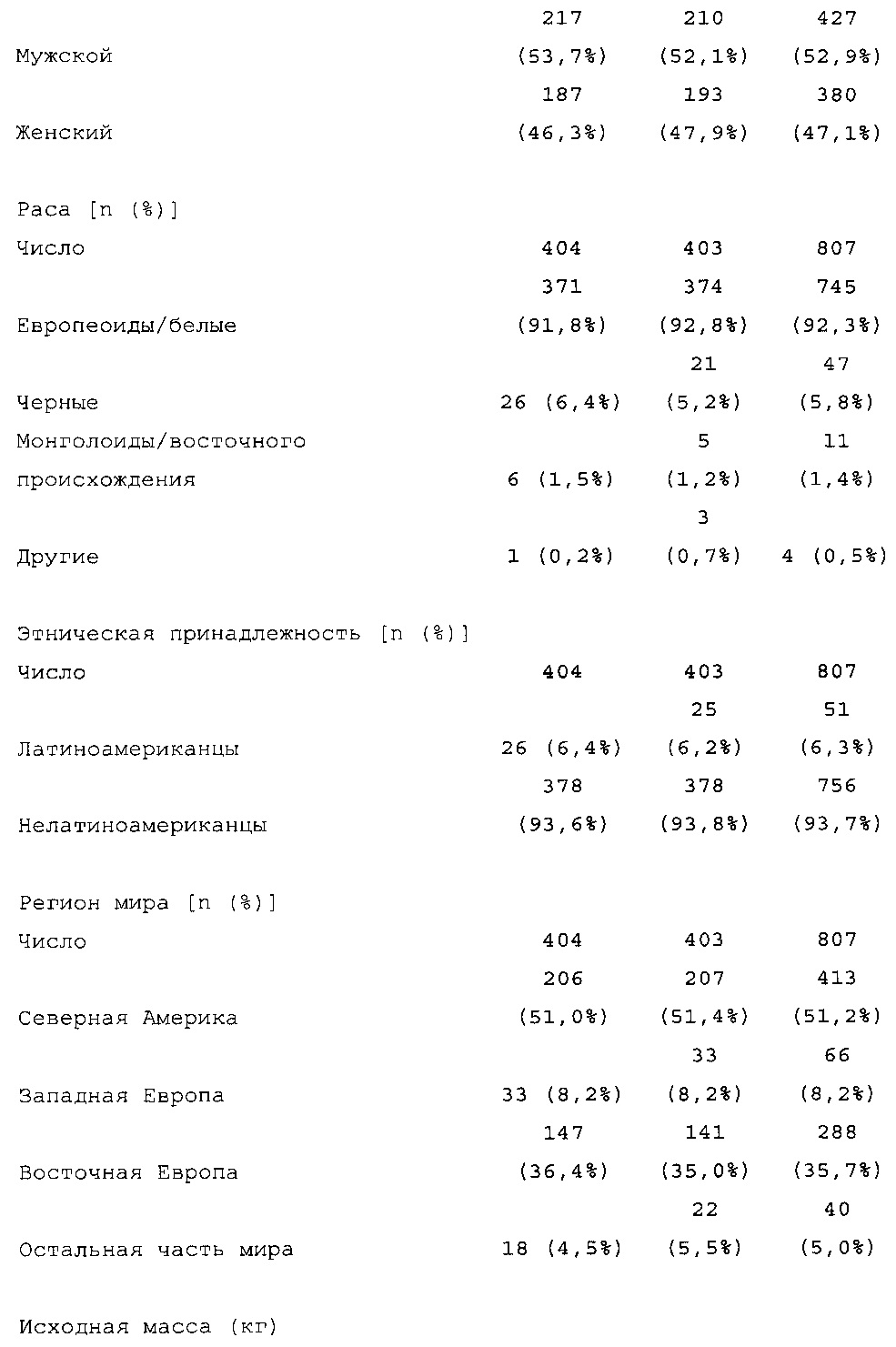
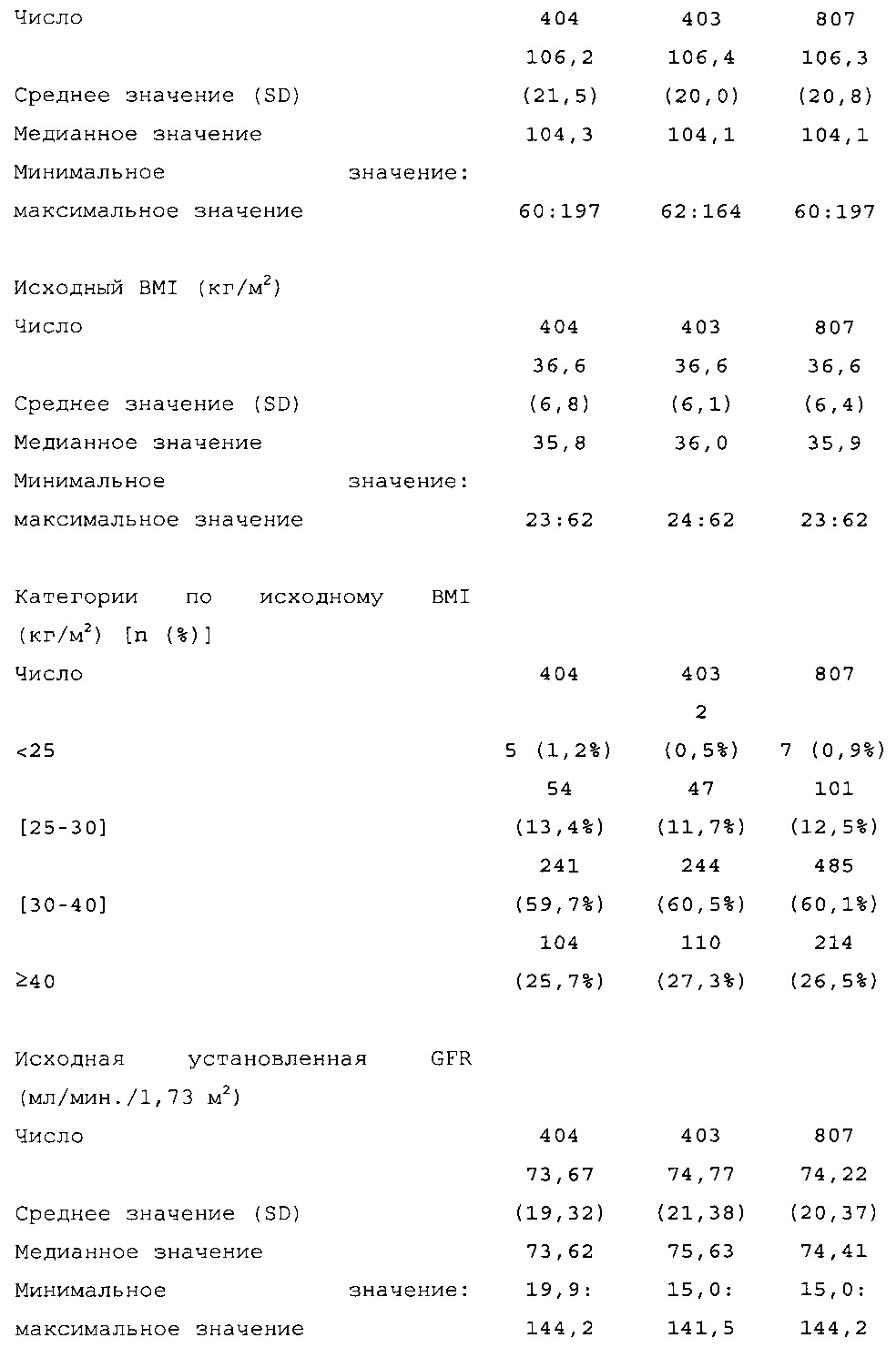
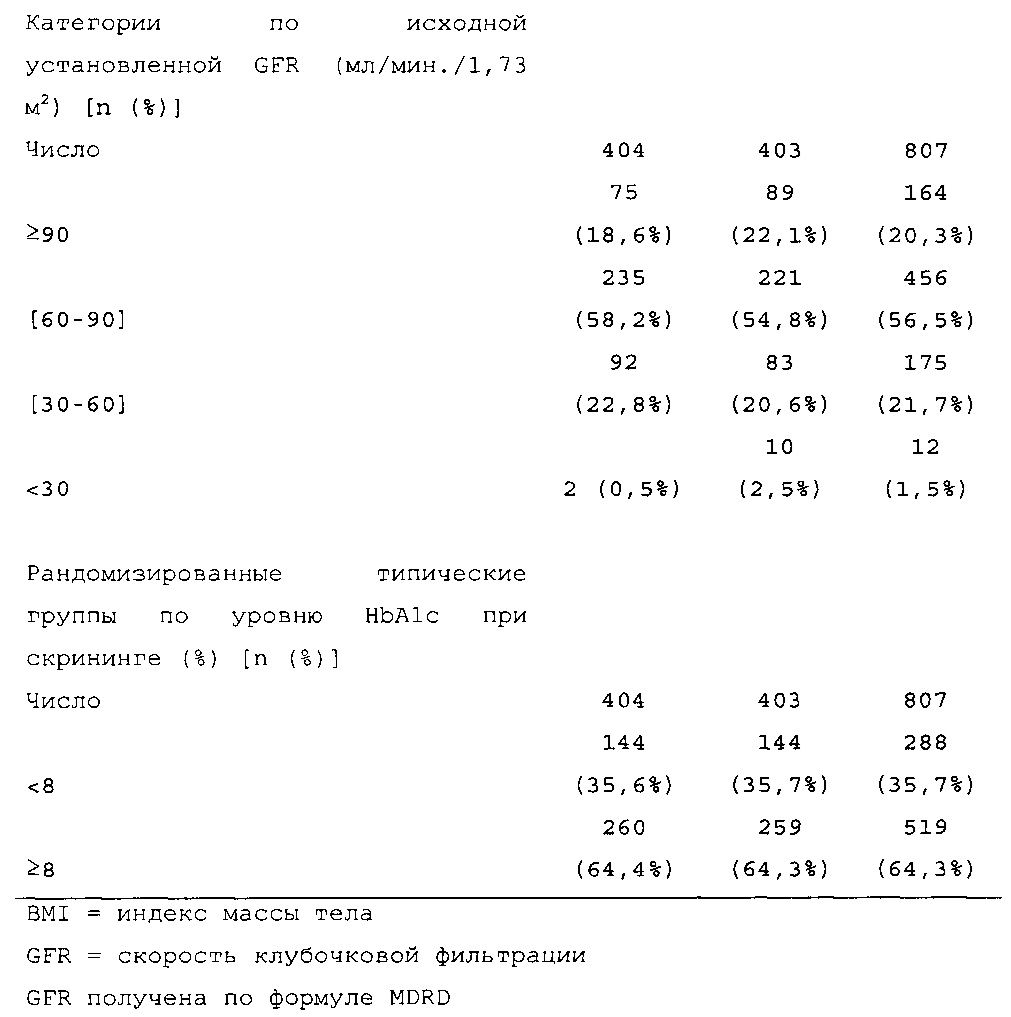

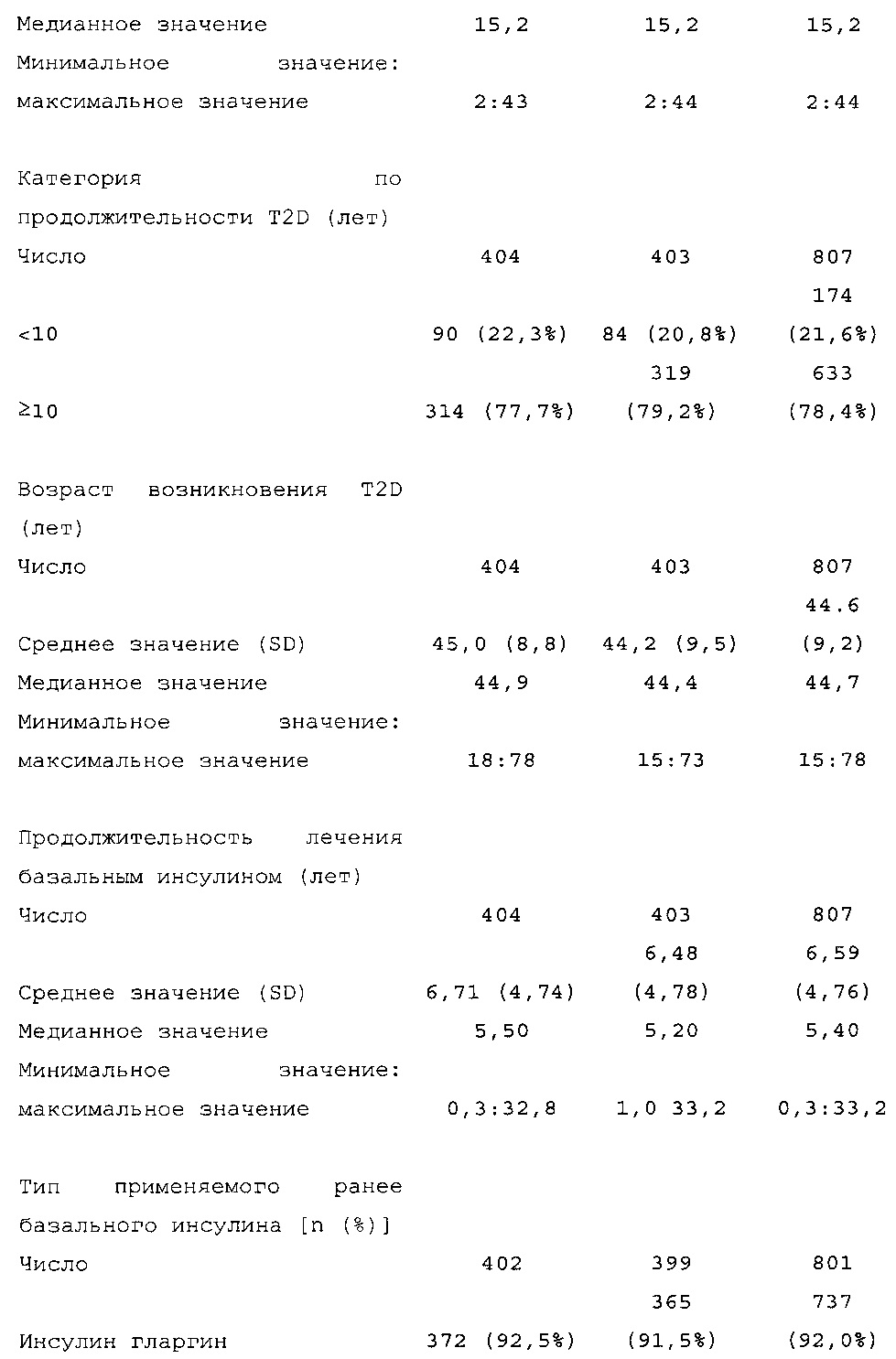

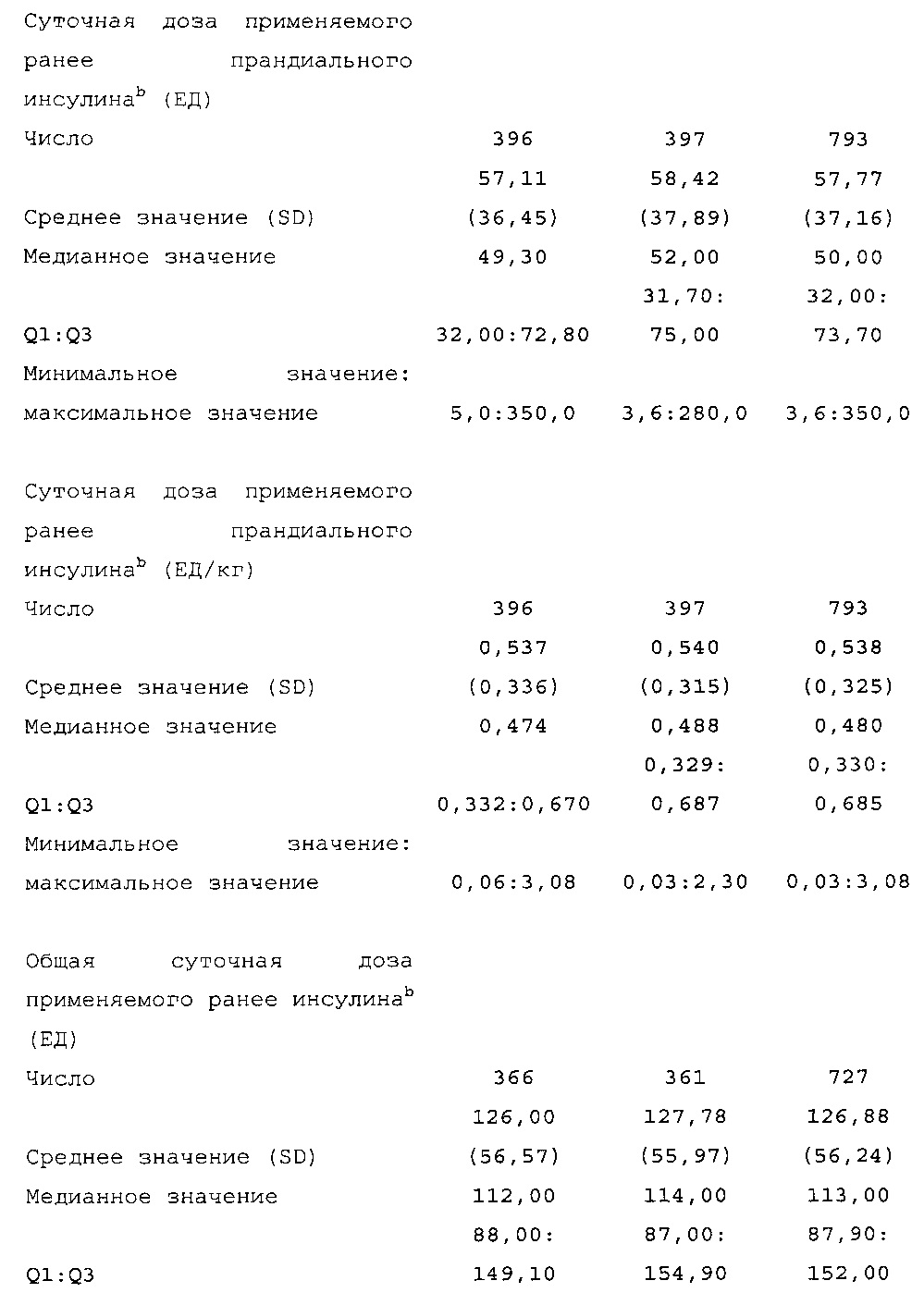
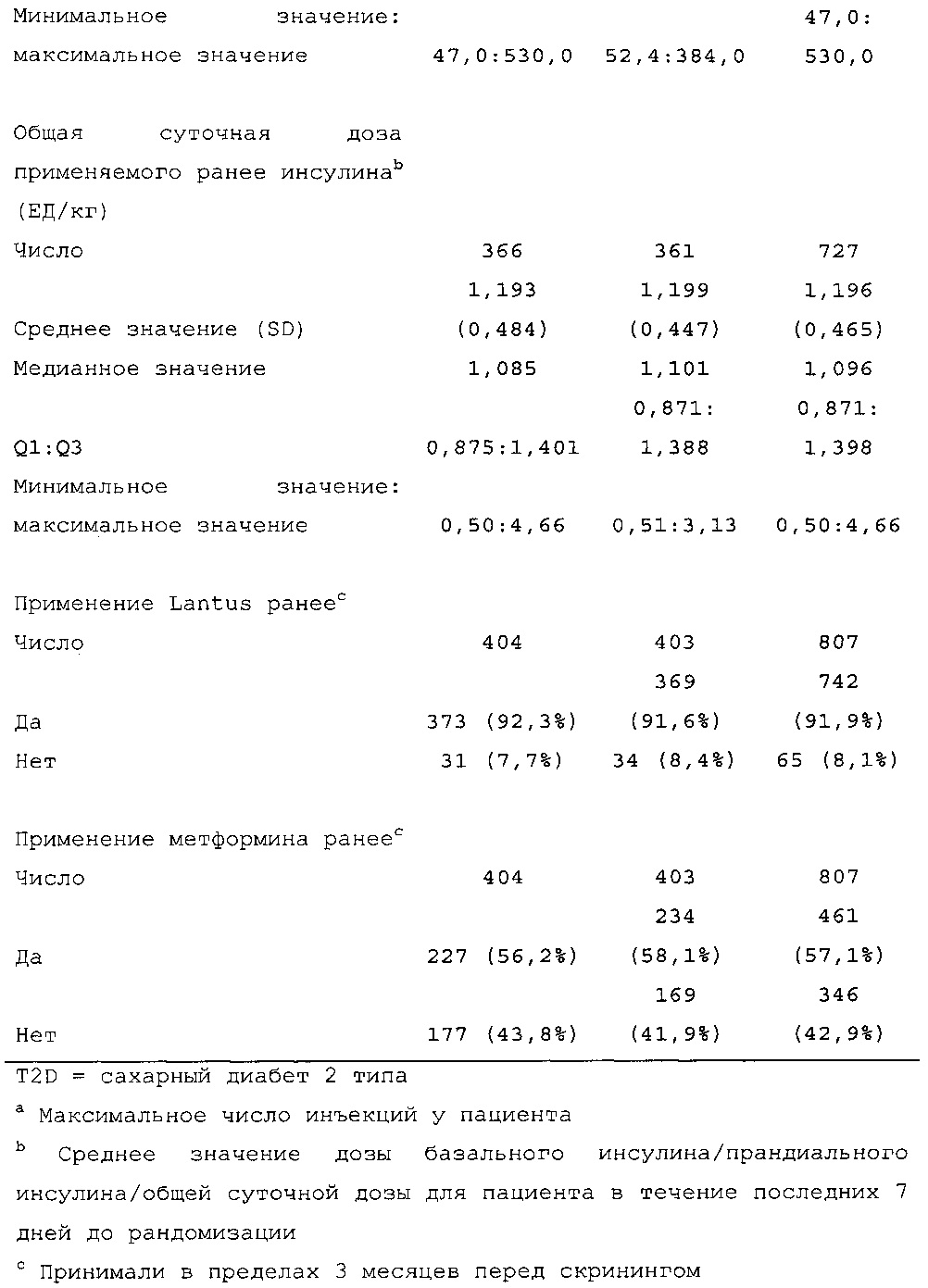
1.2 ОЦЕНКА ЭФФЕКТИВНОСТИ
1.2.1 Первичная конечная точка эффективности
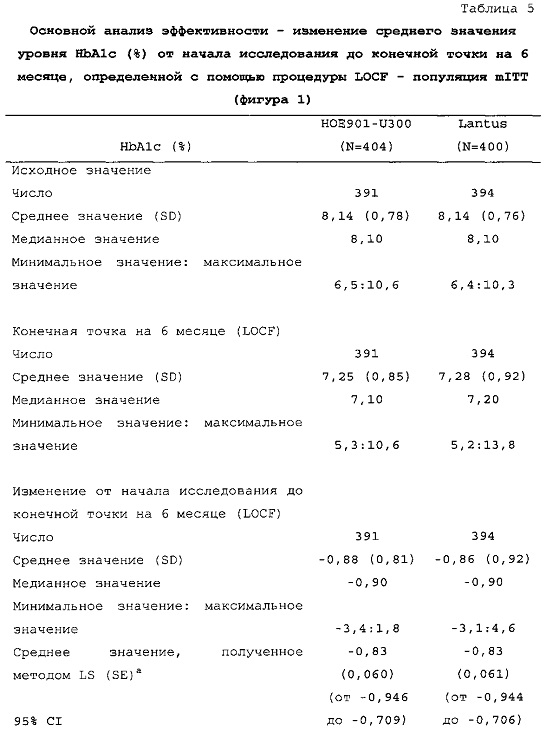

1.2.2 Основные вторичные конечные точки
1.2.2.1 Ночная гипогликемия


1.2.2.2 Прединъекционная концентрация глюкозы в плазме крови - конечная точка на 6 месяце


В V1 (неделя 2) исследователь или член персонала, задействованного в исследовании, предоставлял пациентам глюкометр для измерения концентрации глюкозы в крови и соответствующие расходные материалы (иглы, контрольные растворы, тест-полоски и т.д.), а также дневники для проведения самостоятельного измерения концентрации глюкозы в плазме крови и записи его результатов. Пациентам показывали, как правильно измерять значения концентрации глюкозы в плазме крови с помощью глюкометра для измерения концентрации глюкозы в крови. Исследователь или член персонала, задействованного в исследовании, объяснял необходимость измерения концентрации глюкозы в моменты времени, необходимые для профилей, и корректной записи значений в дневниках пациентов. Обучение повторяли во время визитов исследования так часто, как это было необходимо, и персонал, задействованный в исследовании, проверял дневник пациента во время каждого визита. Значения концентрации глюкозы в крови пациент измерял с помощью предоставленного спонсором прибора для измерения уровня глюкозы в крови. Пациенты документировали свои данные о концентрации SMPG в дневнике.
Пациентам давали указание приносить с собой глюкометры для измерения уровня глюкозы в крови, предоставленные спонсором, во время каждого визита в клинику. Глюкометры для измерения уровня глюкозы в крови должны были быть откалиброваны согласно инструкциям, приведенным в аннотации, и исследовательский центр также должен был регулярно проверять глюкометры с использованием предоставленных контрольных растворов на предмет достоверности данных.
Начиная с V1 (скринингового визита), дневник включает разделы для записи пациентами следующего.
Исследуемое средство лечения и прандиальный инсулин: время и доза инъекций HOE901-U300 или Lantus (инъекций Lantus или NPH на протяжении периода скрининга) и инъекций прандиального/болюсного аналога инсулина.
Концентрация SMPG: время и значение концентрации SMPG.
1. Концентрация SMPG натощак утром (перед завтраком).
2. Концентрация SMPG в пределах 30 минут до инъекции базального инсулина на протяжении 7 дней перед каждым визитом.
3. Концентрация SMPG: профиль по 4 моментам времени и профиль по 8 моментам времени.
4. Концентрация SMPG в связи с эпизодами гипогликемии.
5. Любые другие значения концентрации SMPG (вне зависимости от причины измерения).
Измерения концентрации SMPG запланированы следующим образом:
Концентрация глюкозы в плазме крови натощак (SMPG).
Концентрацию SMPG натощак (перед завтраком) измеряли ежедневно на протяжении всего исследования. На протяжении недели перед 3 визитом (1 день, начало исследования) соблюдение графика измерения концентрации SMPG натощак использовали для оценки соответствия требованиям вступления в период рандомизированного лечения. В ходе исследования, когда повышение дозы было завершено и концентрация SMPG натощак (перед завтраком) была постоянной в целевом диапазоне, необходимо было выполнять по меньшей мере 3 измерения в неделю.
Концентрация глюкозы в плазме крови в пределах 30 минут до инъекции исследуемого лекарственного средства (прединъекционная концентрация SMPG). Прединъекционную концентрацию SMPG определяли в пределах 30 мин. до инъекции IMP (HOE901-U300 или Lantus) в течение по меньшей мере 7 дней перед началом исследования и перед каждым визитом в исследовательский центр на протяжении всего исследования. В дни, когда определяли профили по 4 моментам времени или по 8 моментам времени: если время определения прединъекционной концентрации SMPG совпадало с моментом времени профиля по 4 моментам времени или по 8 моментам времени, пациент должен был в дневнике отнести значение концентрации SMPG к обоим моментам времени (например, прединъекционная концентрация PG; при отходе ко сну).
Профили концентрации SMPG по 4 моментам времени (перед завтраком, перед обедом, перед ужином, при отходе ко сну). На протяжении недели перед визитом исходного уровня и на протяжении первых 12 недель лечения с помощью IMP пациенты определяли профили концентрации SMPG по 4 моментам времени в течение по меньшей мере 3 дней в неделю. Когда цель подбора дозы была достигнута, число профилей концентрации SMPG по 4 моментам времени могло быть уменьшено по решению исследователя, но определение профилей концентрации SMPG по 4 моментам времени в течение по меньшей мере 3 дней в неделю перед каждым визитом было обязательным. Однако рекомендовалось определять профили концентрации SMPG по 4 моментам времени ежедневно на протяжении всего исследования для оптимальной корректировки схемы введения инсулина.
Профили концентрации глюкозы в крови по 8 моментам времени (начиная с измерения в 03:00 ночи; перед завтраком и через 2 часа после завтрака; перед обедом и через 2 часа после обеда; перед ужином и через 2 часа после ужина; при отходе ко сну).
Пациенты определяли профили концентрации SMPG по 8 моментам времени в течение по меньшей мере одного дня из 5 дней перед каждым визитом в исследовательский центр. Однако рекомендовалось определять профили концентрации SMPG по 8 моментам времени по меньшей мере один раз в неделю до 8 недели и один раз в две недели в дальнейшем. Особое внимание следует обратить на то, чтобы было записано значение концентрации SMPG, полученное в 3:00 ночи.
Концентрация SMPG во время приступов симптоматической гипогликемии. В какое бы время пациенты ни ощущали симптомы гипогликемии, при возможности концентрация глюкозы в плазме крови должна была измеряться пациентом (или окружающими в соответствующих случаях). Пациентам должны были дать указание измерять уровни глюкозы в плазме крови перед введением глюкозы или потреблением углеводов в любое время, когда подозревают симптоматическую гипогликемию, кроме случаев, когда из соображений безопасности необходимо немедленное экстренное введение глюкозы/углеводов до подтверждения.
Следующие значения концентрации SMPG необходимо было переносить в eCRF.
На протяжении недели перед визитом исходного уровня и на протяжении первых 12 недель до 8 визита (12 недели):
концентрация SMPG натощак (перед завтраком): ежедневно;
прединъекционная концентрация SMPG (в пределах 30 минут до инъекции базального инсулина): в течение 7 дней перед каждым визитом в исследовательский центр;
профиль концентрации SMPG по 4 моментам времени: в течение 3 разных дней на каждой неделе до 8 визита (12 недели);
профиль концентрации SMPG по 8 моментам времени: 1 профиль в пределах 5 дней перед каждым визитом в исследовательский центр;
концентрация SMPG в связи с эпизодом гипогликемии: во всех случаях документального подтверждения.
Примечание: во время визитов после телефонного разговора в e-CRF вносили следующий минимальный набор данных: концентрацию SMPG натощак (перед завтраком) за последние 3 дня, концентрацию SMPG в связи с эпизодом гипогликемии во всех случаях документального подтверждения. Остальные данные по концентрации SMPG в течение недели перед визитами после телефонного разговора вносили в e-CRF во время следующего визита в исследовательский центр.
После 8 визита (12 недели):
концентрация SMPG натощак (перед завтраком): на протяжении 7 дней перед каждым визитом в исследовательский центр;
прединъекционная концентрация SMPG (в пределах 30 минут до инъекции базального инсулина): в течение 7 дней перед каждым визитом в исследовательский центр;
профиль концентрации SMPG по 4 моментам времени: в течение 3 разных дней в пределах 7 дней перед каждым визитом в исследовательский центр;
профиль концентрации SMPG по 8 моментам времени: в течение одного дня в пределах 5 дней перед каждым визитом в исследовательский центр;
концентрация SMPG в связи с эпизодом гипогликемии: во всех случаях документального подтверждения.
Все значения концентрации глюкозы исследователь использовал для мониторинга гликемии.
1.2.2.3 Вариабельность прединъекционной концентрации SMPG - конечная точка на 6 месяце

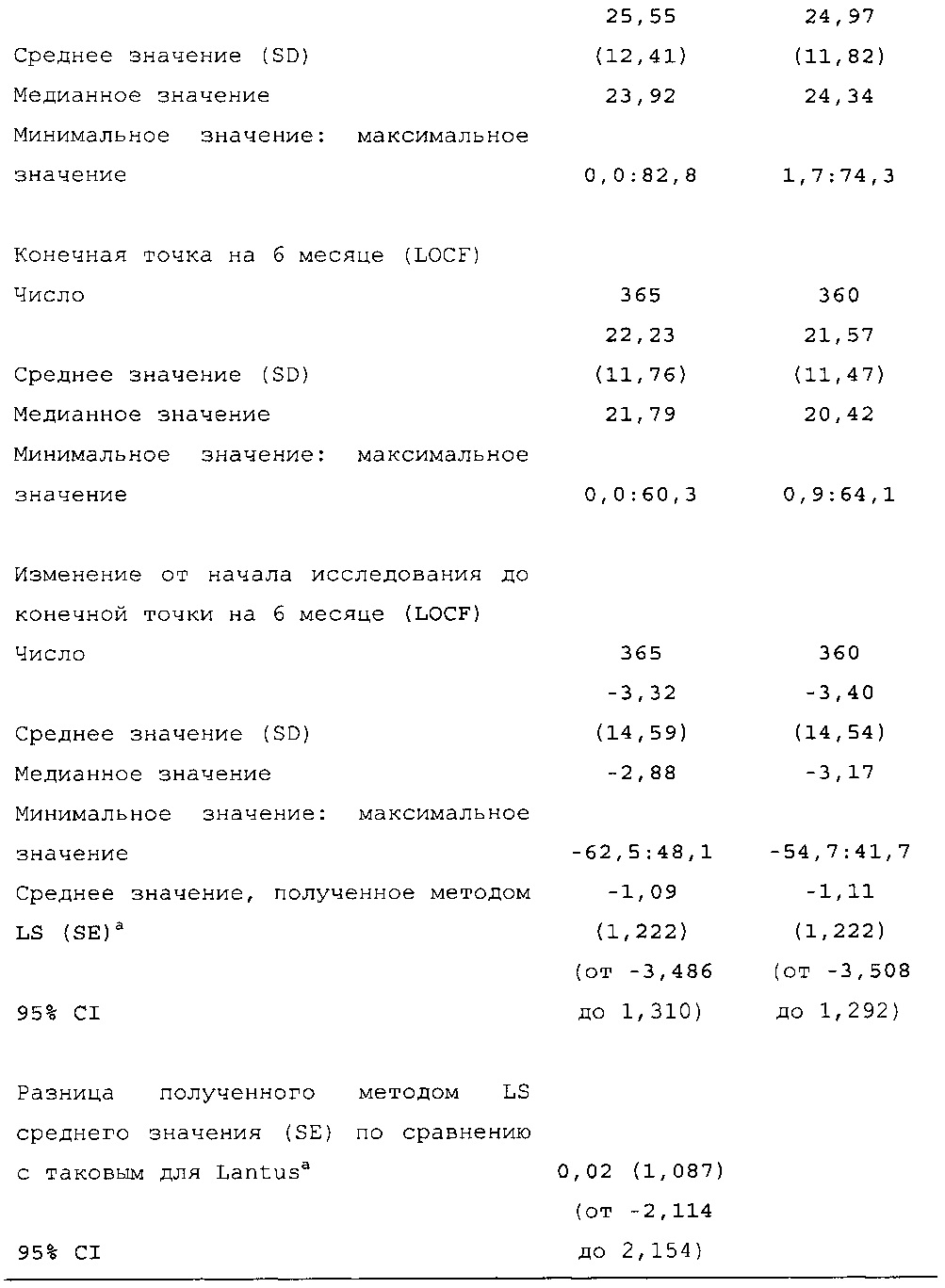

1.2.3 Другие вторичные конечные точки эффективности
1.2.3.1 Процентная доля пациентов с уровнем HbA1c<7% на 6 месяце


1.2.3.2 Изменение концентрации FPG от начала исследования до конечной точки на 6 месяце


1.2.3.3 Профиль концентрации SMPG по восьми моментам времени
На фигуре 3 показаны средние значения концентрации SMPG (ммоль/л) в профиле по 8 моментам времени в начале исследования и в конечной точке на 6 месяце - популяция mITT.
1.2.3.4 Доза базального и прандиального инсулина
На фигуре 4 показаны средние суточные дозы базального инсулина и прандиального инсулина (ЕД) по визитам на протяжении основного 6-месячного периода применения исследуемого препарата - популяция mITT.
1.3 ОЦЕНКА БЕЗОПАСНОСТИ
1.3.1 Длительность проведения лечения
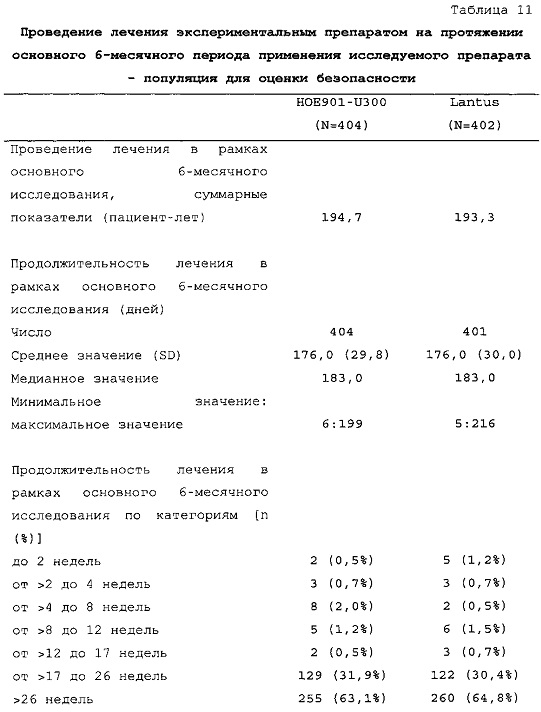

1.3.2 Гипогликемия
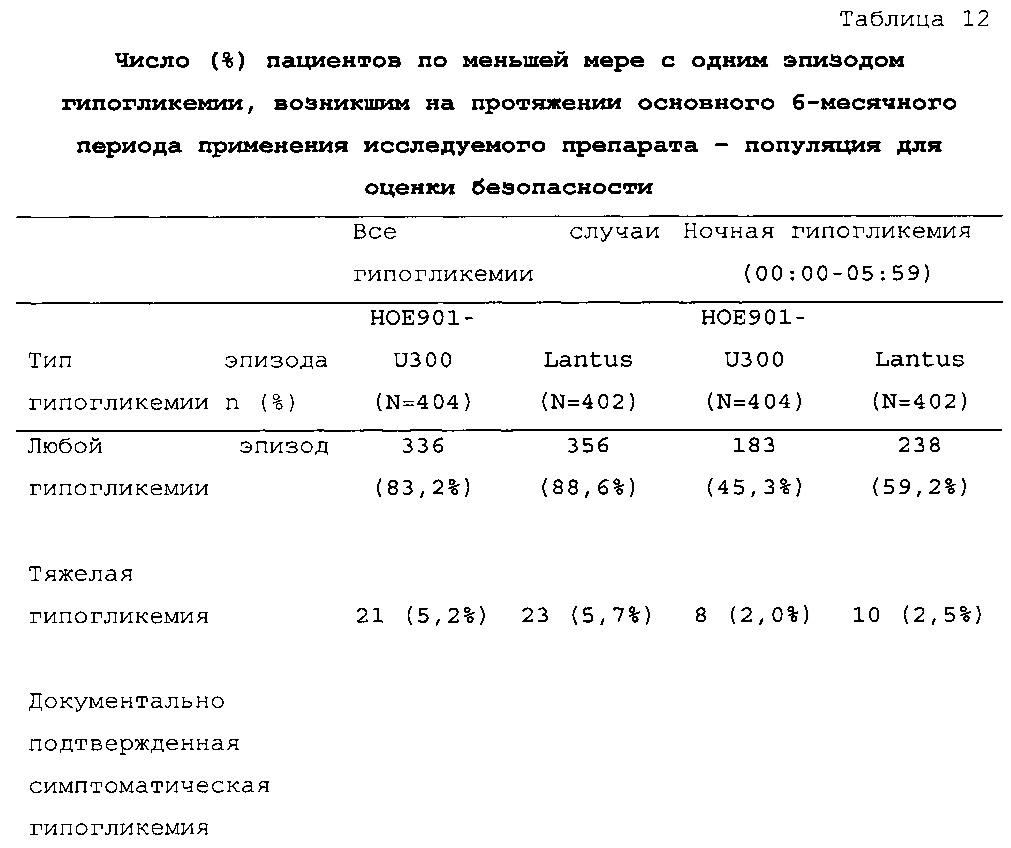
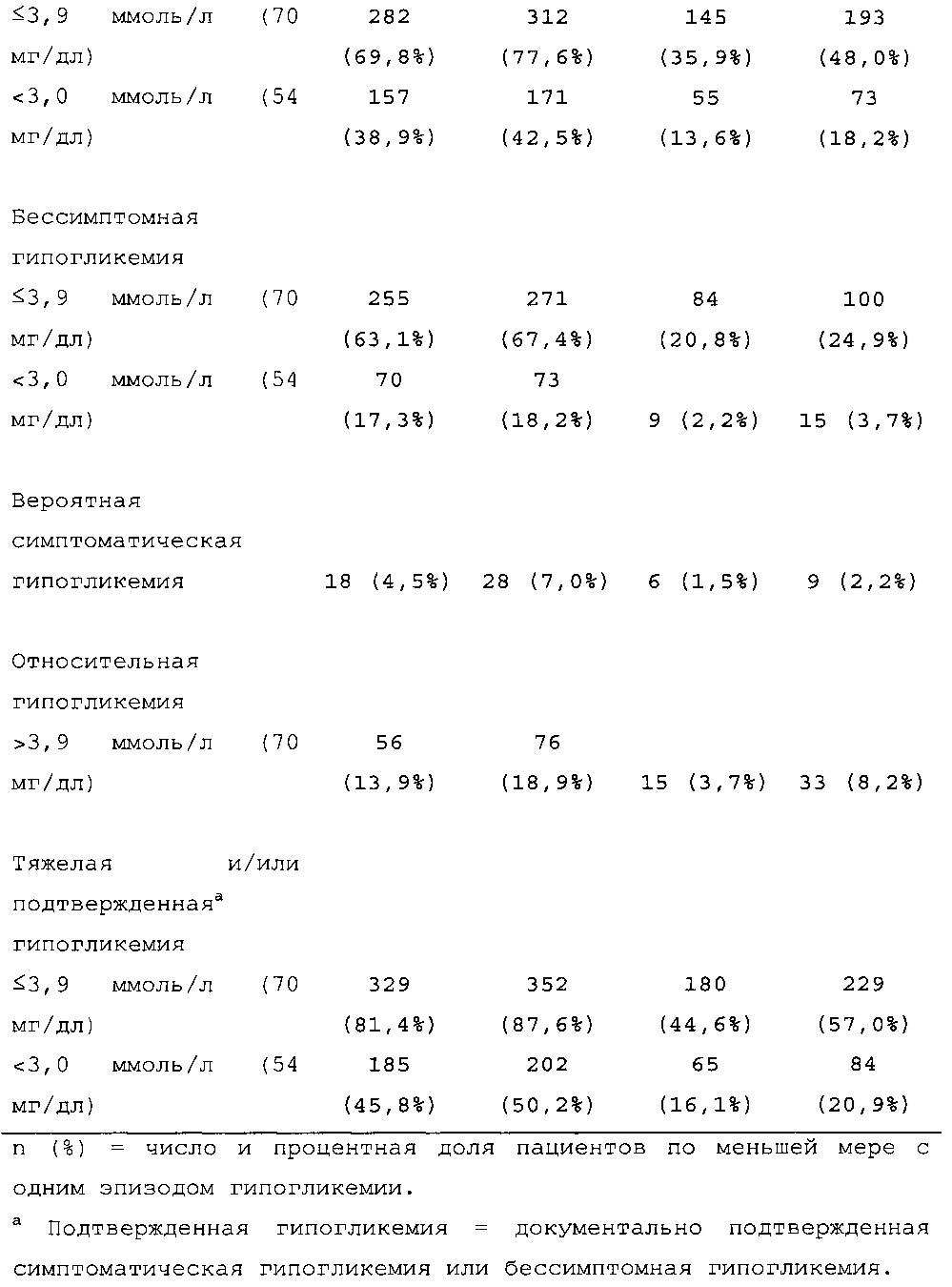
Эпизоды гипогликемии были разделены на категории следующим образом (American Diabetes Association Workgroup on Hypoglycemia. Defining and Reporting Hypoglycemia in Diabetes. Diabetes Care 2005; 28:1245-49).
Тяжелая гипогликемия
Тяжелая гипогликемия представляет собой эпизод, при котором требуется помощь другого человека для активного введения углеводов, глюкагона или выполнения других реанимационных действий.
Эти приступы могут быть связаны со степенью нейрогликопении, достаточной, чтобы вызвать судорожный припадок, потерю сознания или кому. Измерение концентрации глюкозы в плазме крови может быть недоступно во время такого эпизода, но нормализация неврологического статуса, связанная с восстановлением нормальной концентрации глюкозы в плазме крови, считается достаточным доказательством того, что эпизод был вызван низкой концентрацией глюкозы в плазме крови.
Определение тяжелой симптоматической гипогликемии включает все приступы, при которых неврологическое нарушение является достаточно тяжелым, чтобы препятствовать самостоятельному лечению, и которые тем самым, как считается, подвергают пациентов риску причинения вреда себе или окружающим.
Нужно отметить, что "нуждается в помощи" означает, что пациент(ка) не может сам(а) оказать себе помощь. Помощь пациенту из доброты, когда помощь не является необходимой, не следует считать случаем, когда "нуждаются в помощи".
Тяжелая симптоматическая гипогликемия будет расценена как SAE, только если она удовлетворяет критериям SAE. Все эпизоды судорожных припадков, потери сознания или комы должны регистрироваться как SAE.
Документально подтвержденная симптоматическая гипогликемия
Документально подтвержденная симптоматическая гипогликемия представляет собой эпизод, во время которого при типичных симптомах гипогликемии измеренная концентрация глюкозы в плазме крови составляет ≤70 мг/дл (3,9 ммоль/л) (American Diabetes Association Workgroup on Hypoglycemia. Defining and Reporting Hypoglycemia in Diabetes. Diabetes Care 2005; 28:1245-49).
Клиническими симптомами, которые, как считают, обусловлены приступом гипогликемии, являются, например, повышенная потливость, нервозность, астения/слабость, тремор, головокружение, повышенный аппетит, учащенное сердцебиение, головная боль, нарушение сна, спутанность сознания, судорожные припадки, потеря сознания, кома.
Бессимптомная гипогликемия
Бессимптомная гипогликемия представляет собой эпизод, не сопровождающийся типичными симптомами гипогликемии, но при котором измеренная концентрация глюкозы в плазме крови является меньшей или равной 70 мг/дл (3,9 ммоль/л).
Вероятная симптоматическая гипогликемия
Вероятная симптоматическая гипогликемия представляет собой эпизод, во время которого при симптомах гипогликемии не проводят определение концентрации глюкозы в плазме крови, но который был предположительно обусловлен концентрацией глюкозы в плазме крови, меньшей или равной 70 мг/дл (3,9 ммоль/л); при этом симптомы лечат с помощью перорального введения углеводов без проведения теста для определения концентрации глюкозы в плазме крови.
Относительная гипогликемия
Относительная гипогликемия представляет собой эпизод, во время которого человек с сахарным диабетом сообщает о каких-либо типичных симптомах гипогликемии и интерпретирует симптомы как указывающие на гипогликемию, но при этом измеренная концентрация глюкозы в плазме крови составляет более 70 мг/дл (3,9 ммоль/л).
Ночная гипогликемия
Ночной гипогликемией является любая гипогликемия из указанных выше разновидностей, которая возникает в часы между 00:00 и 05:59. Примечание. Относительная ночная гипогликемия не была включена в анализ основной вторичной конечной точки (пациенты по меньшей мере с одним случаем ночной гипогликемии).
Помимо порогового значения, меньшего или равного 70 мг/дл (3,9 ммоль/л), отдельно анализировали приступы гипогликемии при концентрации глюкозы в плазме крови <54 мг/дл (3,0 ммоль/л) (Guideline on clinical investigation of medicinal products in the treatment of diabetes mellitus. Draft. EMA, 20 January 2010).
Классификация гипогликемии была основана на показаниях часов.
Приступы гипогликемии анализировали в зависимости от их суточного распределения (0:00-24:00) и, кроме того, в зависимости от времени суток:
- ночная гипогликемия, определяемая по времени суток: любая гипогликемия из указанных выше разновидностей, которая возникает в часы между 00:00 и 05:59 утра, вне зависимости от того, бодрствовал ли пациент или проснулся из-за эпизода;
- дневная гипогликемия: любая гипогликемия из указанных выше разновидностей, которая возникает между 6:00 утра и 23:59.
Пациентам давали указание измерять уровни глюкозы в плазме крови, взятой путем прокола пальца, перед введением углеводов в любое время, когда подозревают симптоматическую гипогликемию, кроме случаев, когда из соображений безопасности необходимо немедленное экстренное введение глюкозы до подтверждения, в этом случае измерение уровня глюкозы следовало выполнять, как только это становилось безопасным, с соответствующим документированием в дневнике.
Подробная информация о приступах гипогликемии фиксировалась в дневниках пациентов, и пациенты связывались с исследовательскими центрами в максимально короткие сроки после тяжелых явлений, чтобы сообщить подробную информацию и определить любые необходимые меры, которые нужно предпринять.
Все приступы гипогликемии документировали в "специальном бланке для регистрации данных по гипогликемии" в e-CRF. Они включали все эпизоды симптоматической гипогликемии и бессимптомной гипогликемии. Эпизоды гипогликемии, удовлетворявшие критериям SAE, документировали в форме для регистрации данных по SAE в e-CRF.
Частоту возникновения гипогликемии на пациент-год рассчитывали для пациента следующим образом: 365,25 × (число приступов гипогликемии)/(число дней лечения) и суммировали по типу эпизода и группе лечения.
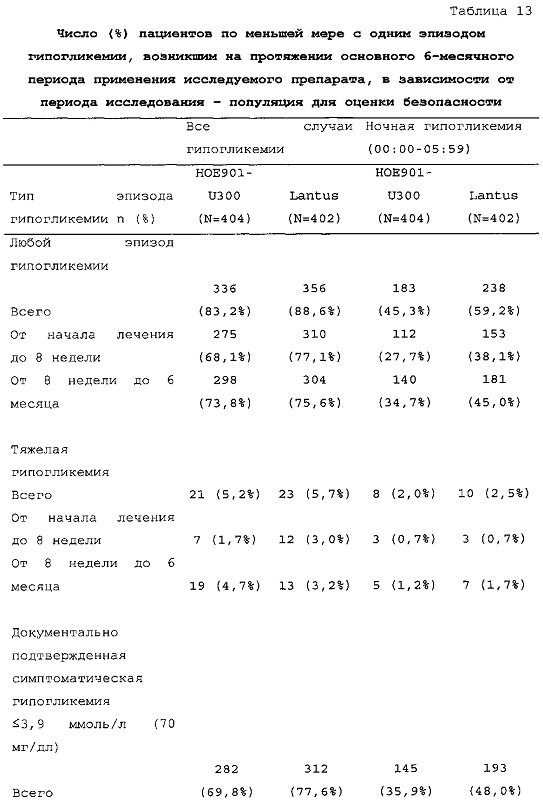
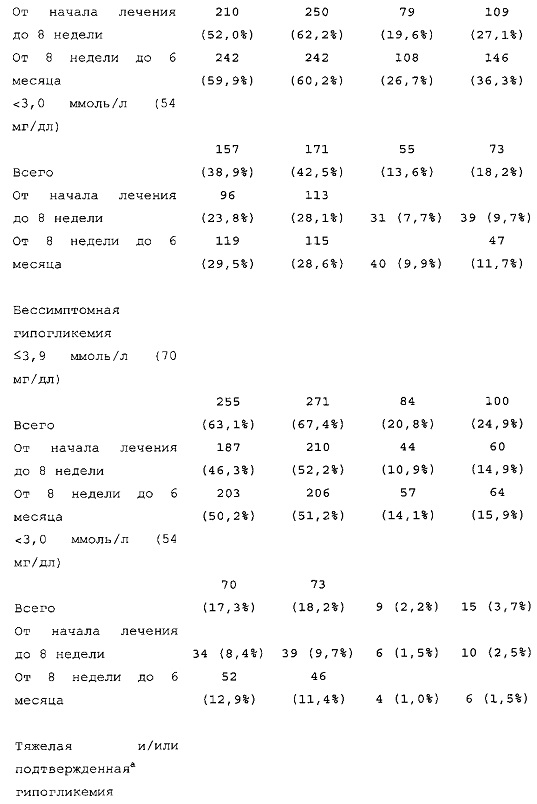

1.3.3 Нежелательные явления, возникшие в ходе лечения

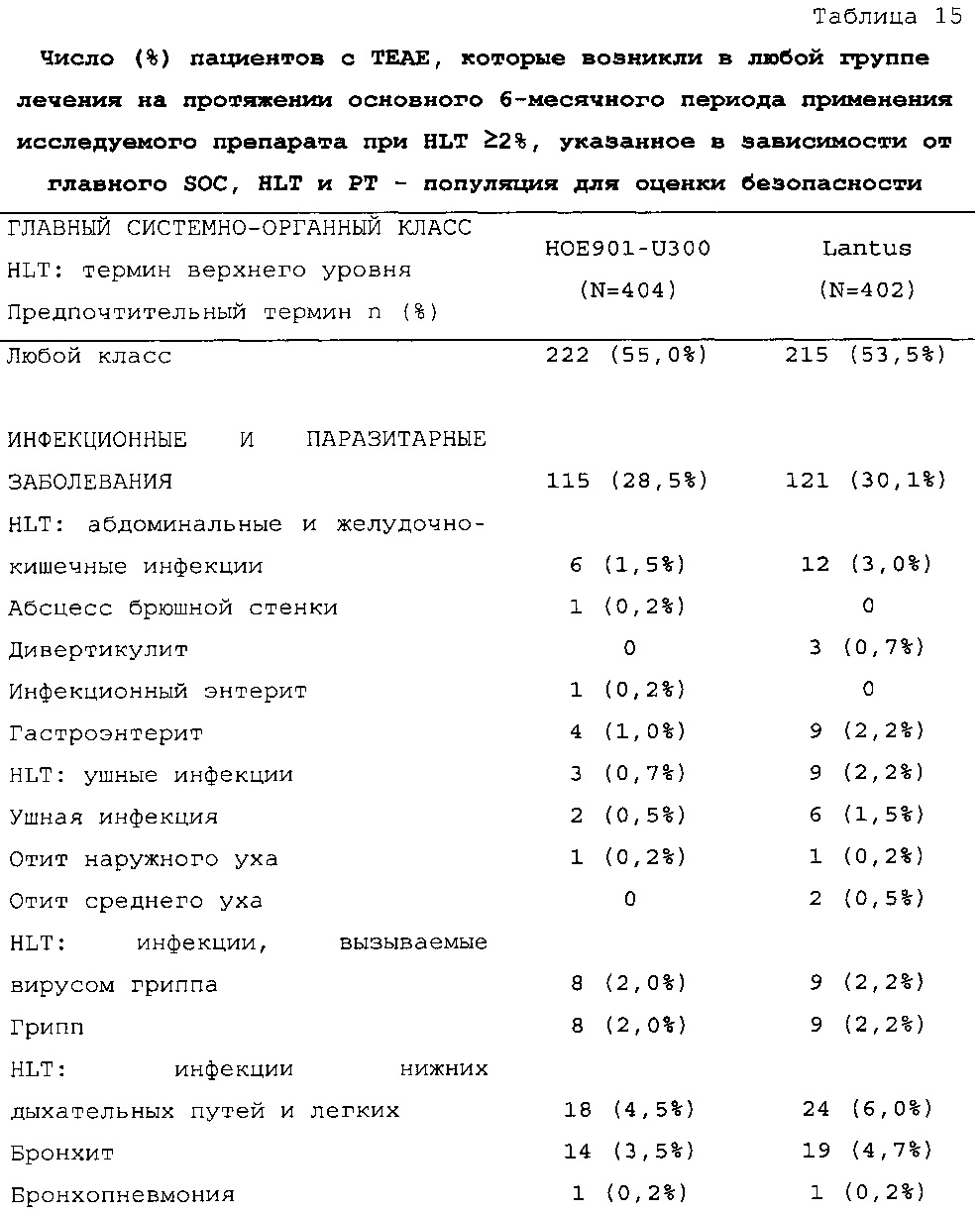
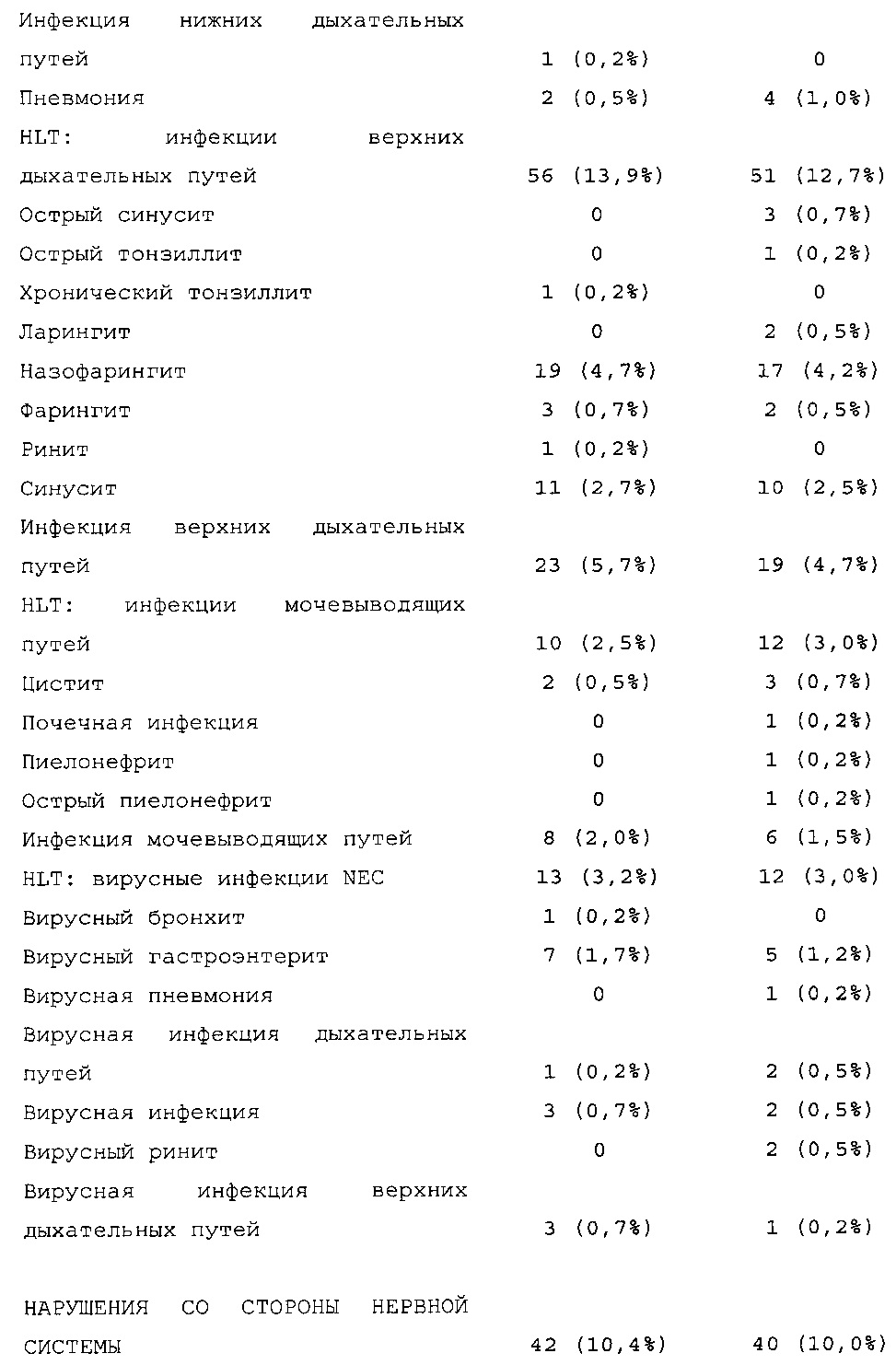
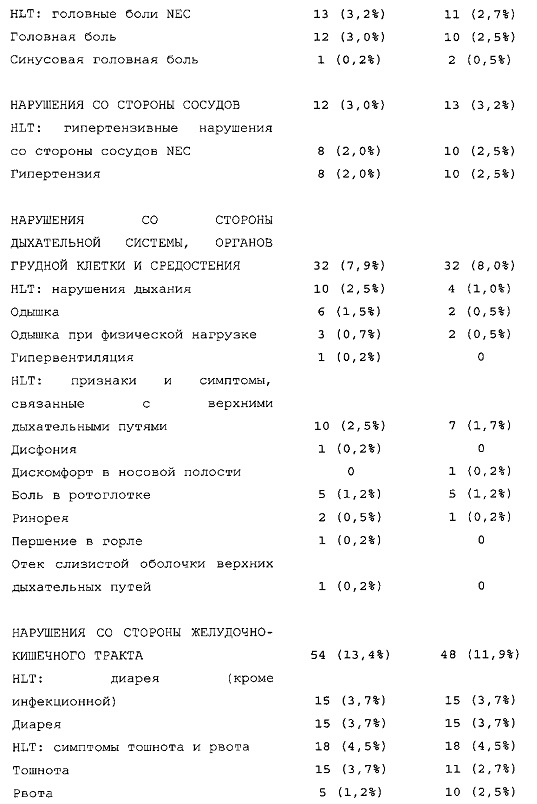
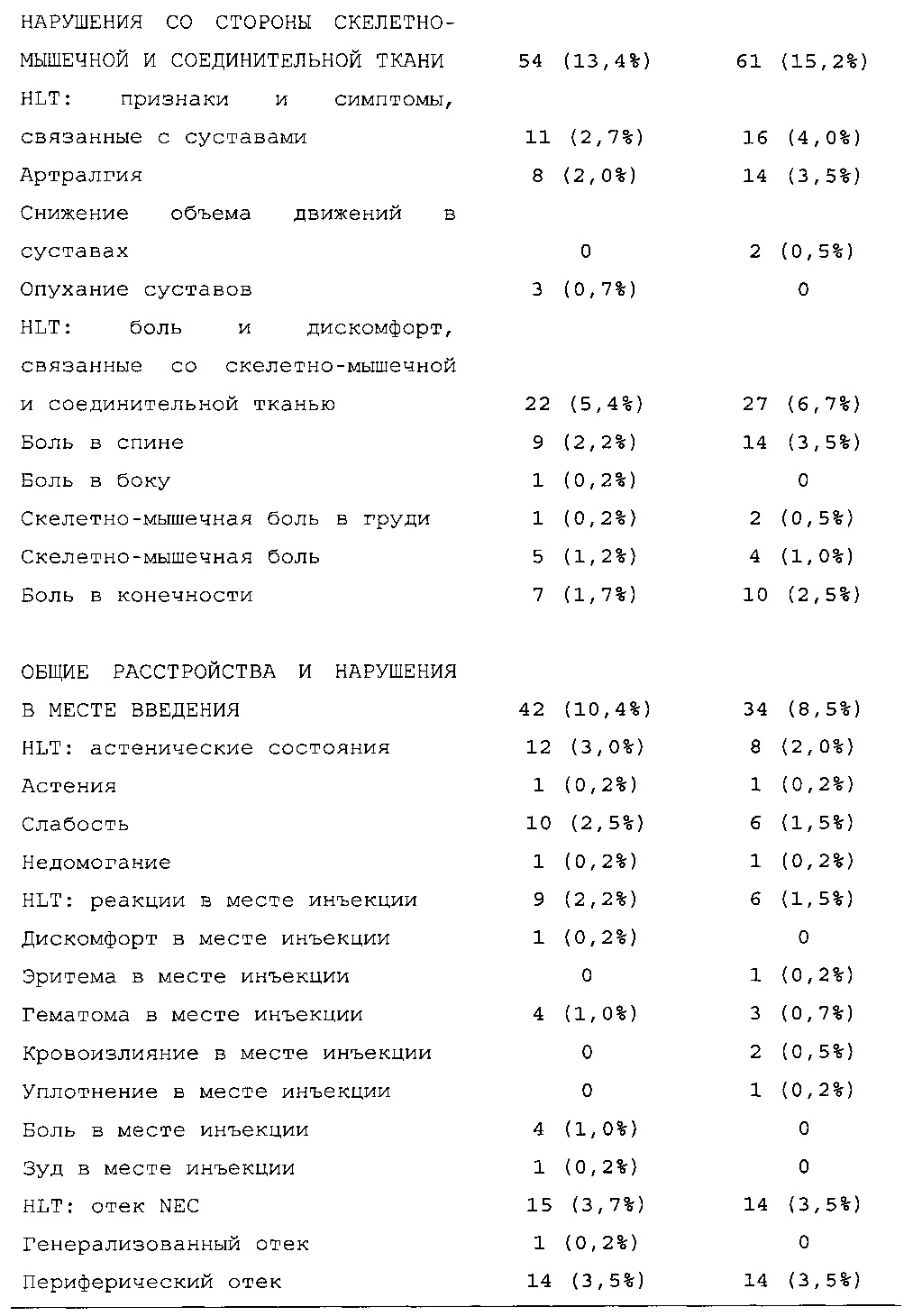

1.3.4 Случаи смерти, серьезные нежелательные явления, возникшие в ходе лечения
1.3.4.1 Случаи смерти


1.3.4.2 Серьезные нежелательные явления
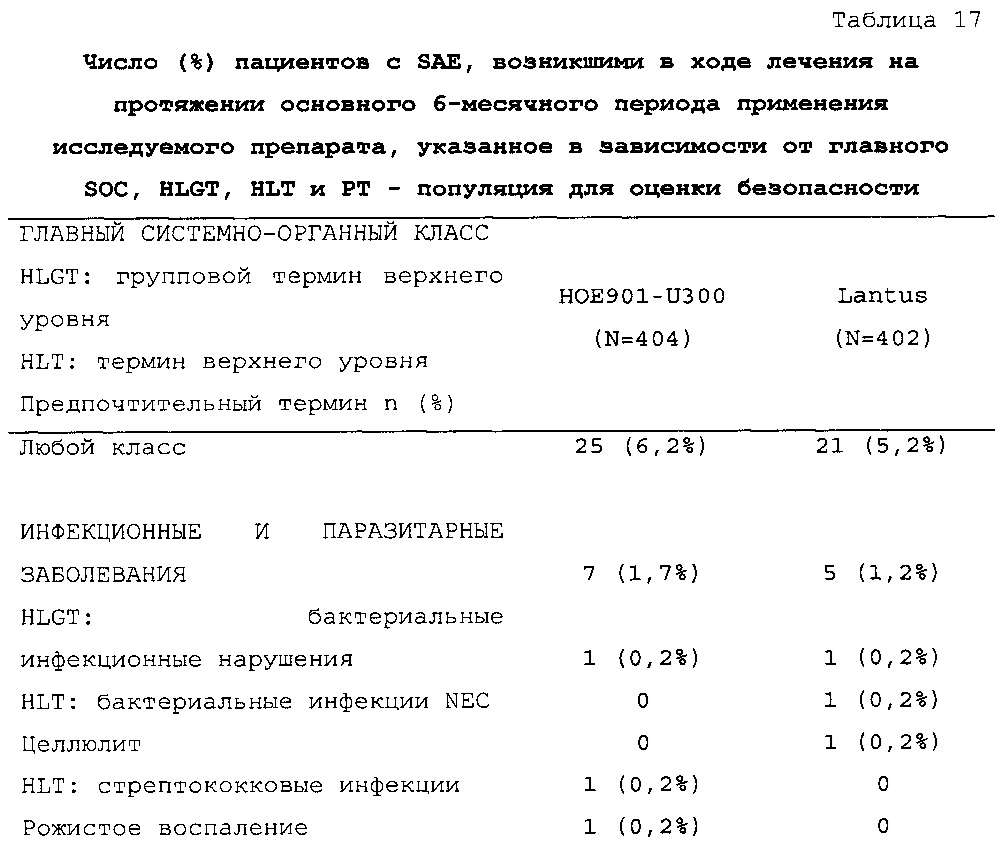


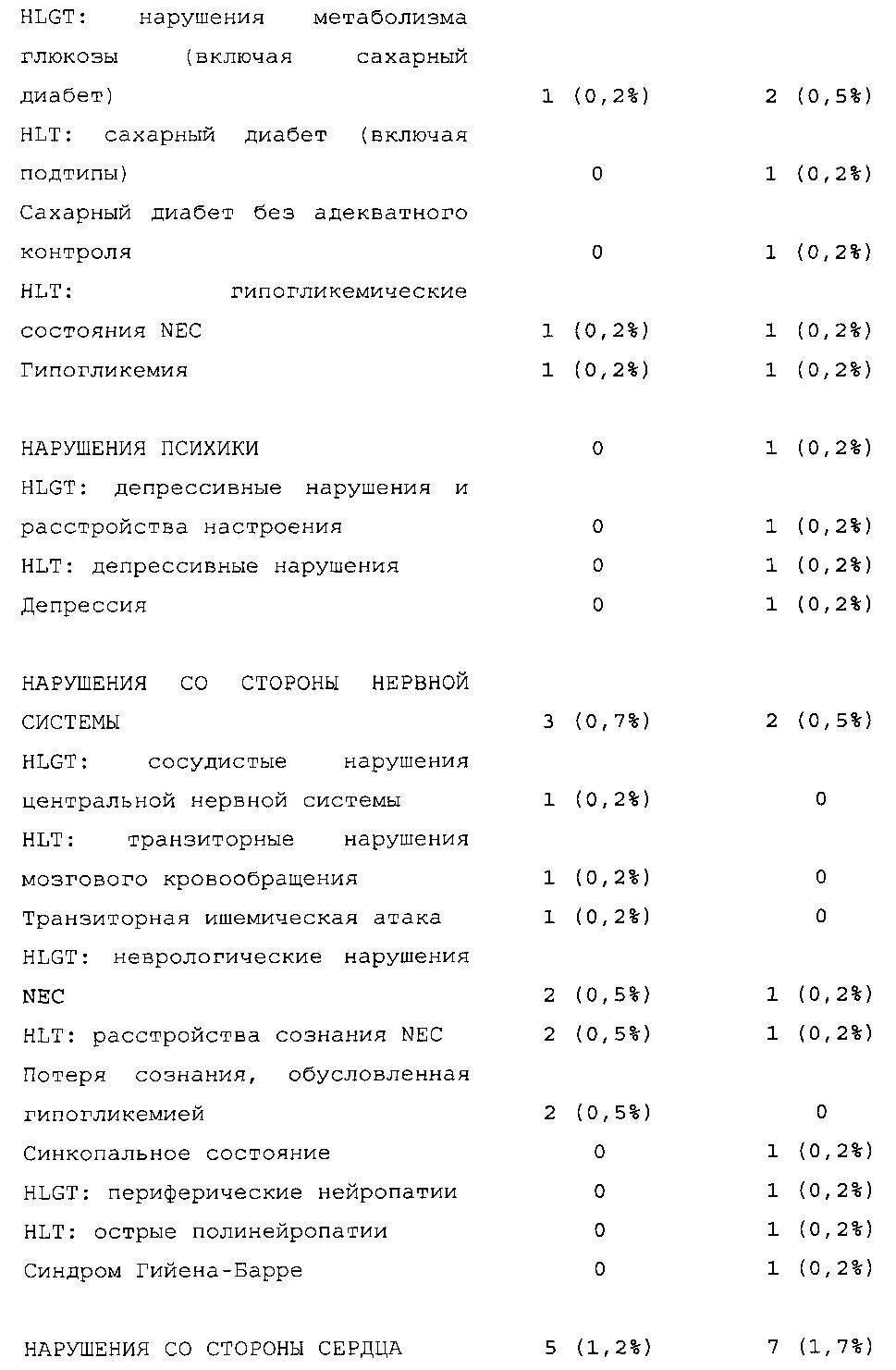
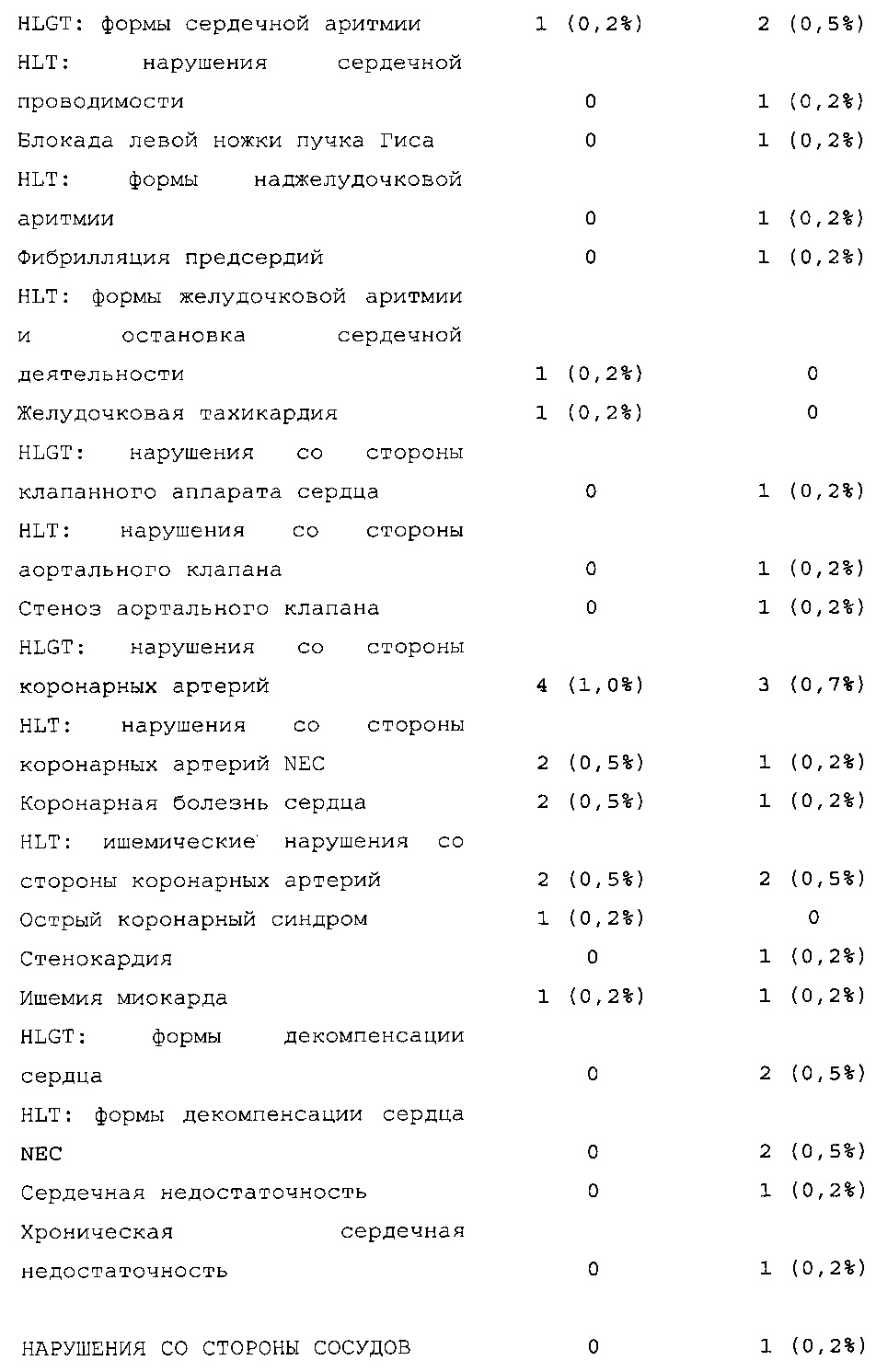



1.3.5 Нежелательные явления, приведшие к прекращению участия
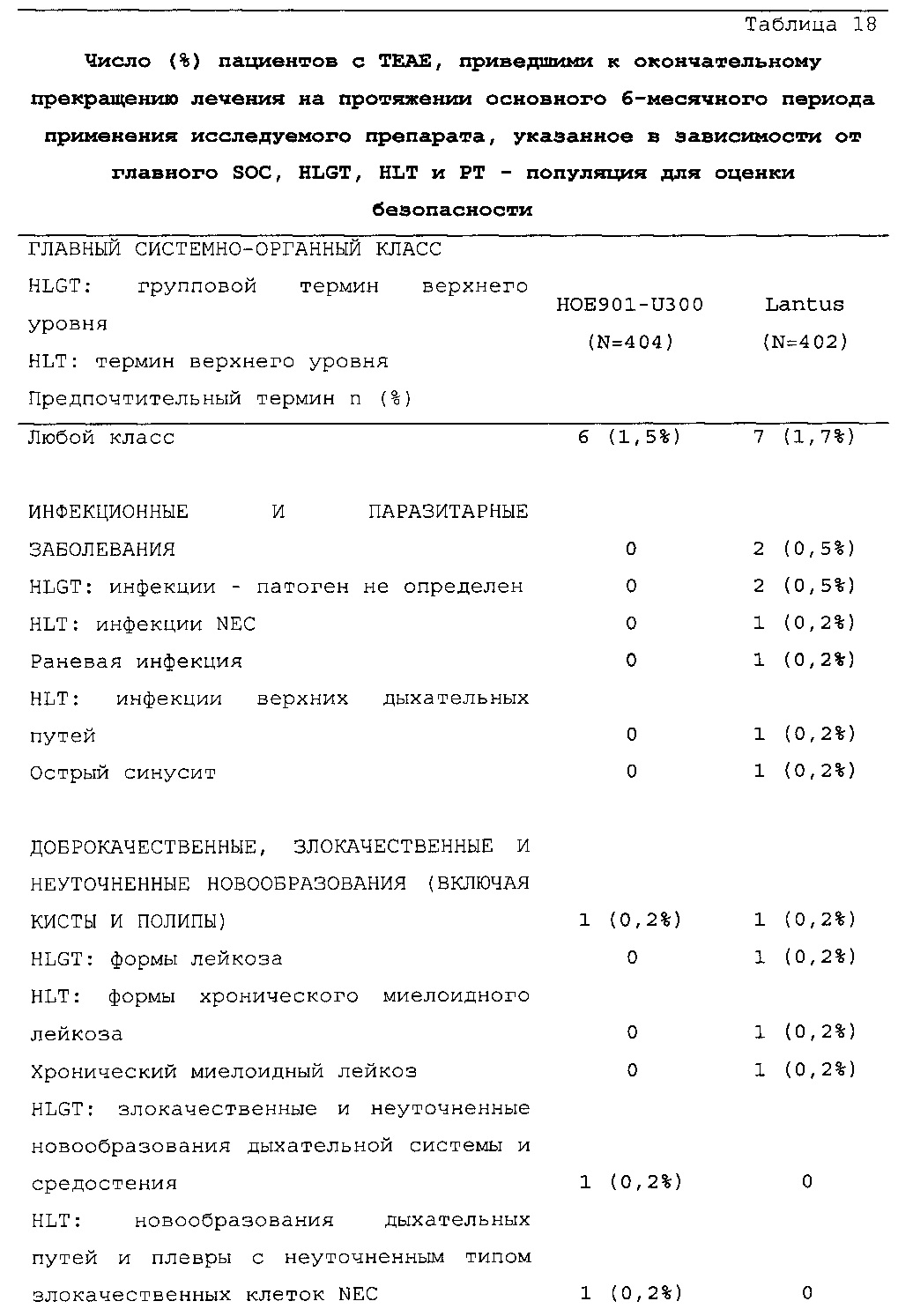
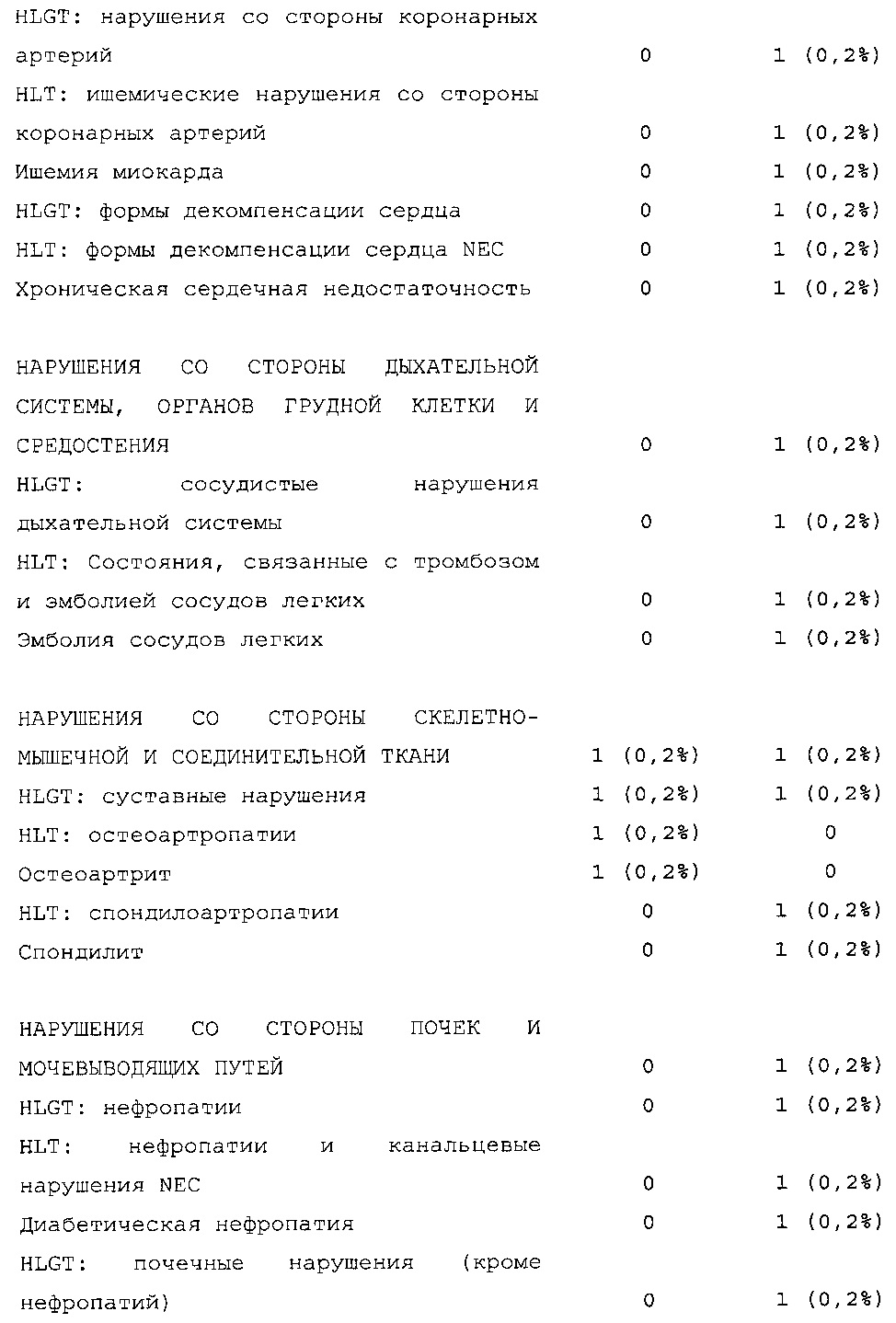
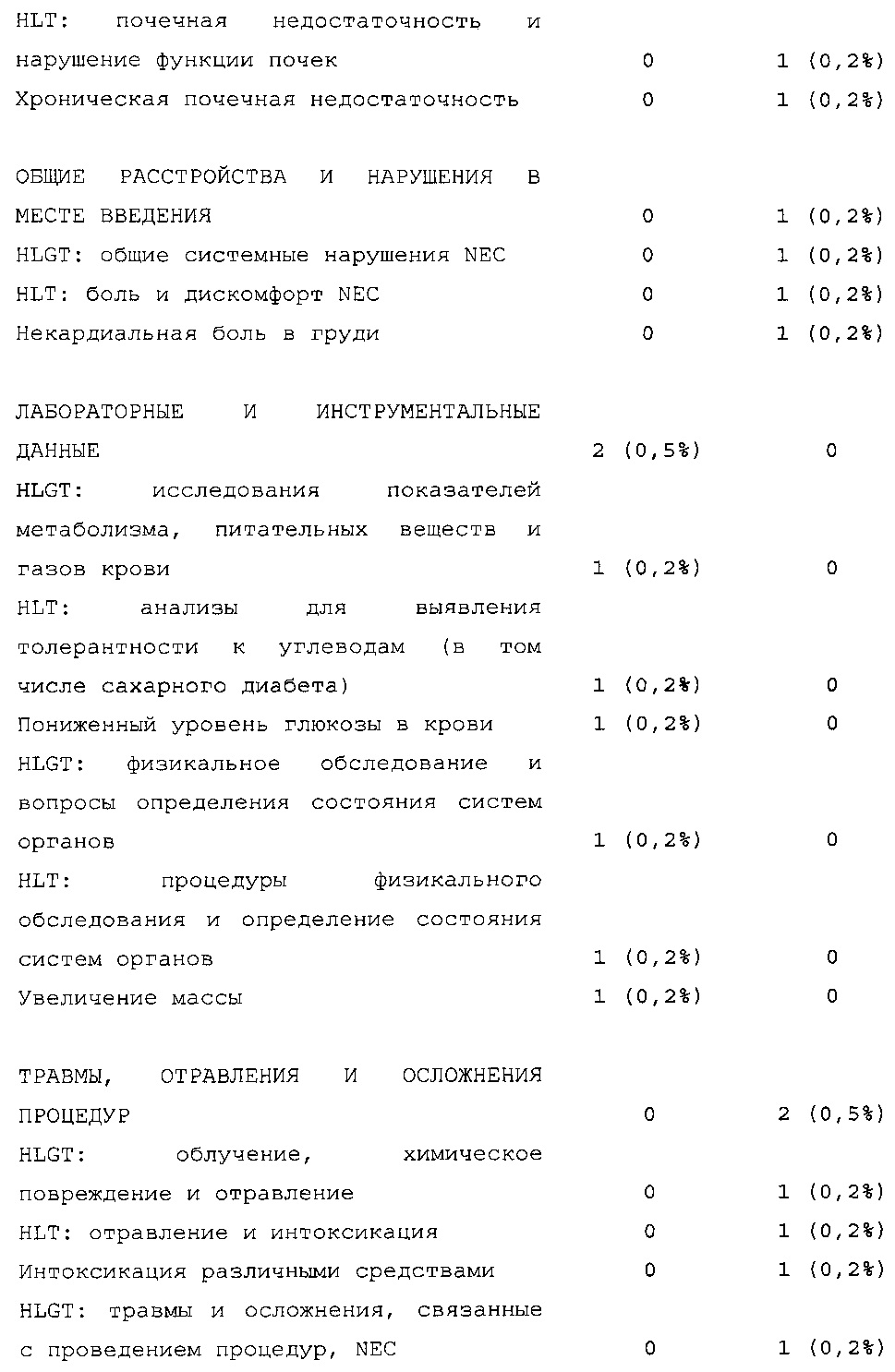

1.3.6 Другие значительные нежелательные явления
1.3.6.1 Реакция гиперчувствительности
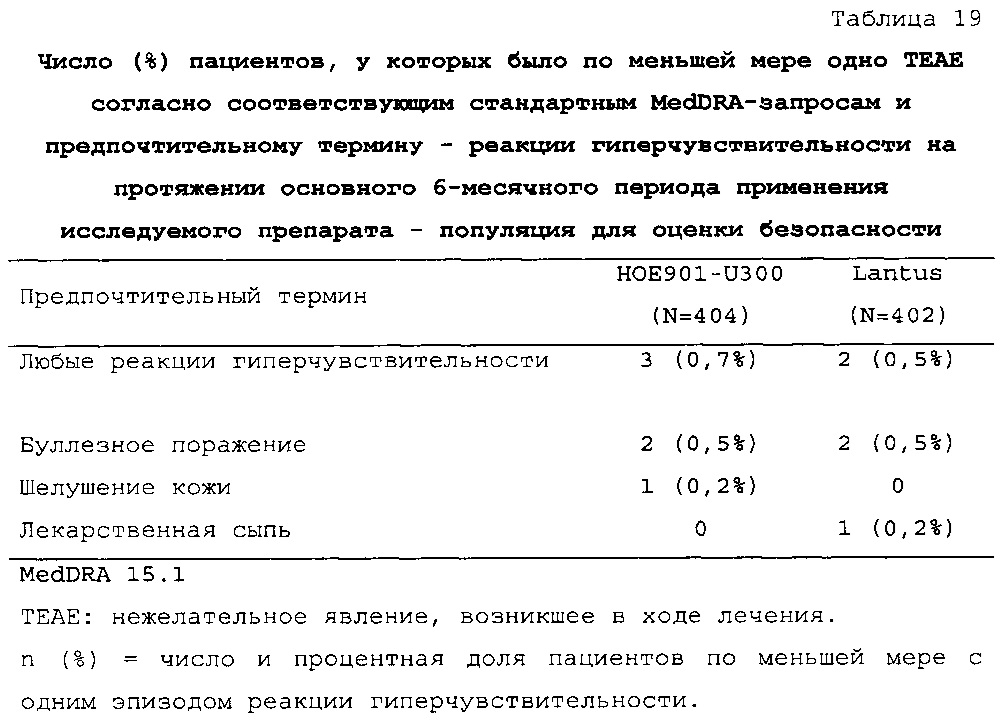
1.3.6.2 Реакции в месте инъекции

Пример 2. 6-месячное многоцентровое рандомизированное открытое исследование на параллельных группах, в котором сравнивали эффективность и безопасность нового состава инсулина гларгина и Lantus®, оба из которых применяли в сочетании с пероральным(и) гипогликемическим(и) средством(ами) у пациентов с сахарным диабетом 2 типа, с 6-месячным продленным периодом исследования безопасности
ОБЩЕЕ ОПИСАНИЕ ИССЛЕДОВАНИЯ
Исследовательский(ие) центр(ы):многоцентровое.
Фаза разработки: 3
Цели
Первичная цель: оценка эффектов HOE901-U300 в отношении гликемического контроля по сравнению с Lantus при введении в качестве базального инсулина по схеме, включающей пероральное(ые) гипогликемическое(ие) средство(а), с точки зрения изменения уровня HbA1c за период 6 месяцев у пациентов с сахарным диабетом 2 типа.
Основные вторичные цели: сравнение HOE901-U300 и Lantus с точки зрения частоты ночной гипогликемии, изменения прединъекционной концентрации глюкозы в плазме крови и изменения вариабельности прединъекционной концентрации глюкозы в плазме крови.
Дополнительные вторичные цели:
сравнение HOE901-U300 и Lantus с точки зрения достижения целевых значений HbA1c и контролируемой концентрации глюкозы в плазме крови;
сравнение HOE901-U300 и Lantus с точки зрения удовлетворенности пациентов лечением с помощью опросника удовлетворенности лечением сахарного диабета (статус) (DTSQs) (не представлен в KRM);
оценка безопасности и переносимости HOE901-U300.
Методика. Рандомизацию производили в соотношении 1:1 (HOE901-U300 по отношению к Lantus), а стратификацию осуществляли в зависимости от значений HbA1c при скрининге (<8,0%; ≥8,0%). Объем выборки (400 для HOE901-U300 и 400 для Lantus) выбирали для обеспечения достаточную мощность исследования по первичной конечной точке (изменению уровня HbA1c от начала исследования до конечной точки [6 месяц]), а также для обеспечения возможности сделать выводы по первой основной вторичной конечной точке (частоте ночной гипогликемии).
Число пациентов
Запланировано: 800 (400 на группу лечения) Рандомизировано: 811 Получали лечение: 809
Подвергнуты оценке: эффективности: 808, безопасности: 809
Диагностика и критерии включения. Критерии включения: пациенты с сахарным диабетом 2 типа, определенным согласно WHO, у которых диагноз был поставлен по меньшей мере за 1 год до момента скринингового визита; подписавшие письменное информированное согласие. Ключевые критерии невключения: возраст <18 лет; HbA1c <7,0% или >10% при скрининге; сахарный диабет, отличный от сахарного диабета 2 типа; менее 6 месяцев лечения базальным инсулином вместе с пероральными гипогликемическими средствами и самоконтроля уровня глюкозы в крови; общая суточная доза инсулина гларгина <42 ЕД или эквивалентная доза NPH в течение последних 4 недель перед исследованием (если NPH применяли в качестве базального инсулина перед исследованием).
Исследуемые средства лечения
Экспериментальные лекарственные препараты: изучаемое лекарственное средство: HOE901-U300; контрольное лекарственное средство: Lantus.
Составы: HOE901-U300 (раствор инсулина гларгина, 300 ЕД/мл) представляет собой стерильный апирогенный прозрачный бесцветный раствор в стеклянном картридже, который был укомплектован в шприц-ручку (предварительно наполненную, т.е. одноразовую ручку). Lantus (раствор инсулина гларгина, 100 ЕД/мл) представляет собой стерильный апирогенный прозрачный бесцветный раствор, поставляемый в продаваемой Solostar® (предварительно наполненной, т.е. одноразовой ручке).
Путь введения: подкожное введение.
Схема дозирования: инъекция один раз в день вечером. Время инъекции фиксировали в момент рандомизации, и оно должно было сохраняться на протяжении исследования.
Начальная доза: пациенты, применявшие Lantus или NPH один раз в день до визита исходного уровня: суточная доза (ЕД) HOE901-U300 или Lantus была равна медианному значению общих суточных доз базального инсулина в последние 3 дня до визита исходного уровня.
Пациенты, применявшие NPH более одного раза в день до визита исходного уровня: суточная доза HOE901-U300 или Lantus (ЕД) должна была быть примерно на 20% меньше медианного значения общих суточных доз инсулина NPH в последние 3 дня до визита исходного уровня.
Дозу базального инсулина корректировали один раз в неделю для достижения концентрации SMPG натощак в целевом диапазоне от 80 до 100 мг/дл (от 4,4 до 5,6 ммоль/л):
на +3 ЕД, если медианное значение концентрации SMPG натощак для последних 3 дней находилось в диапазоне >100 мг/дл и <140 мг/дл (>5,6 и <7,8 ммоль/л);
на +6 ЕД, если медианное значение концентрации SMPG натощак для последних 3 дней составляло ≥140 мг/дл (≥7,8 ммоль/л);
на -3 ЕД, если медианное значение концентрации SMPG натощак для последних 3 дней находилось в диапазоне ≥60 мг/дл и <80 мг/дл (≥3,3 и <4,4 ммоль/л).
Резервное лечение
Если посредством корректировки дозы базального инсулина не удавалось снизить уровень FPG/HbA1c ниже пороговых значений 11,1 ммоль/л (200 мг/дл) для FPG и 8% для HbA1c к 12 неделе или позже и не было обнаружено явной причины недостаточного контроля, следовало рассмотреть возможность интенсификации лечения. Выбор противодиабетического средства лечения для добавления к базовой терапии с помощью базального инсулина и перорального гипогликемического средства основывался на решении исследователя и местных вариантах инструкций по применению лекарственного средства.
Неэкспериментальные лекарственные препараты
Пациенты в обеих группах лечения должны были продолжать применять свою базовую терапию пероральным гипогликемическим средством в постоянной дозе в ходе исследования, за исключением сульфонилмочевины, применение которой было запрещено в пределах 2 месяцев перед скрининговым визитом и в ходе исследования. Средство резервной терапии также считалось неэкспериментальным лекарственным препаратом.
Продолжительность лечения: до 12 месяцев.
Продолжительность наблюдения: до 58 недель (до 2 недель периода скрининга + 6 месяцев периода анализа эффективности и безопасности + 6 месяцев продленного периода исследования безопасности + 4 недели периода последующего наблюдения после лечения).
Периодом анализа эффективности и безопасности является основной 6-месячный период применения исследуемого препарата. Результаты, представленные в настоящем KRM, относятся к этому периоду.
Для всех пациентов, которым потребовалась резервная терапия на протяжении 6-месячного периода лечения, последнее после начала исследования определение эффективности перед началом резервной терапии использовали в качестве конечной точки эффективности. Эти пациенты были исключены из анализов эффективности после начала резервного лечения. Для конечных точек безопасности анализируемым периодом являлся основной 6-месячный период применения исследуемого препарата вне зависимости от применения резервной терапии.
Критерии оценки
Эффективность
Первичная конечная точка эффективности: изменение уровня HbA1c от начала исследования до конечной точки (6 месяц).
Основные вторичные конечные точки: количество пациентов (%) по меньшей мере с одним случаем ночной гипогликемии в промежутке между началом на 9 неделе и конечной точкой (6 месяц), указанной как тяжелая и/или подтвержденной по концентрации глюкозы в плазме крови ≤70 мг/дл (3,9 ммоль/л); изменение прединъекционной концентрации SMPG от начала исследования до конечной точки (6 месяц) и изменение вариабельности прединъекционной концентрации SMPG от начала исследования до конечной точки (6 месяц).
Безопасность: гипогликемия, частота нежелательных явлений, особенно нежелательных явлений, возникших в ходе лечения (ТЕАЕ), и серьезных нежелательных явлений (SAE), ТЕАЕ, приведших к прекращению участия, и ТЕАЕ, приведших к смерти, реакций в месте инъекции и реакций гиперчувствительности. Следующая информация не представлена в данном KRM: физикальное обследование, другая информация, касающаяся безопасности, в том числе данные клинико-лабораторных исследований, основные показатели состояния организма, ECG в 12 отведениях и наличие антител к инсулину.
Следующая информация не представлена в данном KRM: физикальное обследование, другая информация, касающаяся безопасности, в том числе данные клинико-лабораторных исследований, основные показатели состояния организма, ECG в 12 отведениях и наличие антител к инсулину.
Статистические методы. Анализ первичной конечной точки эффективности (изменения уровня HbA1c от начала исследования до конечной точки [6 месяц]) осуществляли с использованием модели ковариационного анализа (ANCOVA) с лечением, типическими группами по уровню HbA1c при скрининге (<8,0 и ≥8,0%) и страной в качестве фиксированных эффектов и с использованием исходного значения уровня HbA1c в качестве ковариаты. Различия между HOE901-U300 и Lantus и двусторонние 95% доверительные интервалы оценивали в рамках ANCOVA.
Подход со ступенчатым закрытым тестированием использовали в отношении первичной конечной точки эффективности для последовательной проверки гипотезы о не меньшей эффективности и превосходстве. На стадии 1 оценивали не меньшую эффективность HOE901-U300 по сравнению с Lantus. Для оценки не меньшей эффективности верхнюю границу двустороннего 95% CI для различия изменения среднего значения уровня HbA1c от начала исследования до конечной точки между HOE901-U300 и Lantus сравнивали с предварительно установленным порогом не меньшей эффективности, составлявшим 0,4% для HbA1c. Не меньшая эффективность будет демонстрироваться, если верхняя граница двустороннего 95% CI для различия между HOE901-U300 и Lantus в популяции mITT будет составлять <0,4%. На стадии 2 оценивали превосходство НОЕ901-U300 по сравнению с Lantus, только если была продемонстрирована не меньшая эффективность. Превосходство HOE901-U300 над Lantus демонстрировалось, если верхняя граница двустороннего 95% CI для различия между HOE901-U300 и Lantus в популяции mITT составляла <0.
Только в случае, если по первичной конечной точке демонстрировалась не меньшая эффективность HOE901-U300 по сравнению с Lantus, осуществляли тестирование в отношении превосходства HOE901-U300 над Lantus по основным вторичным конечным точкам в рамках процедуры иерархического тестирования. Анализы безопасности были описательными, основанными на популяции для оценки безопасности.
Краткое описание
Характеристики популяции
В общей сложности 811 пациентов с сахарным диабетом 2 типа рандомизировали в группы, получающие HOE901-U300 (n=404) или Lantus (n=407); 809 пациентов подвергали воздействию IMP (популяция для оценки безопасности). Популяция mITT (популяция для оценки эффективности) включала 808 пациентов.
В целом, в каждой группе лечения досрочно прекращало лечение в рамках исследования сопоставимое число пациентов (HOE901-U300: 36/404, 8,9%; Lantus: 38/407, 9,3%). В общей сложности 344 (85,1%) пациента в группе HOE901-U300 и 349 (85,7%) в группе Lantus завершили основной 6-месячный период лечения (пациенты, получавшие лекарственное вещество для резервной терапии, были исключены из популяции завершивших лечение).
Демографические данные и исходные параметры были сбалансированы среди групп лечения. Средний возраст в исследуемой популяции составлял 58,2 лет, у 190/811 (23,4%) составлял ≥65 лет. Среднее значение BMI в начале исследования составляло 34,8 кг/м2. В группе HOE901-U300 пациентов с BMI, превышающим 40 кг/м2 (21,5%), было немного больше, чем в группе Lantus (16,7%). Средняя продолжительность сахарного диабета до начала исследования составляла 12,6 лет; средняя продолжительность предшествующего лечения с помощью базального инсулина составляла 3,8 лет. Большинство пациентов принимали инсулин гларгин (78,8% по сравнению с 21,2%, принимавших NPH) в течение 7 дней перед началом лечения в рамках исследования; в группе Lantus инсулин гларгин применяло больше пациентов (82,8%) по сравнению с группой HOE901-U300 (74,9%). Медианное значение суточной дозы базального инсулина в начале исследования составляло 0,614 ЕД/кг массы тела.
Среднее значение уровня HbA1c в начале исследования было сходным в обеих группах лечения (НОЕ901-U300: 8,28% и Lantus: 8,22%; для пациентов, подвергаемых оценке, т.е. тех, у кого производили оценку уровня HbA1c в начале исследования и по меньшей мере один раз после начала исследования).
Результаты в отношении эффективности
Первичная конечная точка. Изменение полученного методом LS среднего значения уровня HbA1c от начала исследования до конечной точки (6 месяц) было сходным в обеих группах лечения (HOE901-U300: -0,57% (95% CI [-0,756; -0,387]); Lantus: -0,56% (95% CI [-0,744; -0,379]). Не меньшая эффективность HOE901-U300 по сравнению с Lantus была продемонстрирована с разницей полученного методом LS среднего значения уровня HbA1c по сравнению с Lantus, составлявшей -0,01% (95% CI [-0,139; 0,119]), при этом верхняя граница была ниже предварительно установленного порога не меньшей эффективности, составлявшего 0,4%. Не меньшая эффективность также демонстрировалась в случае, когда порог не меньшей эффективности составлял 0,3%. Превосходство HOE901-U300 по сравнению с Lantus не было продемонстрировано.
1-я основная вторичная конечная точка. Количество пациентов по меньшей мере с одним случаем ночной тяжелой и/или подтвержденной гипогликемии в промежутке между началом на 9 неделе и 6 месяцем в группе HOE901-U300 [87/403 (21,6%)] было меньшим, чем в группе Lantus [113/405 (27,9%)]. Превосходство HOE901-U300 по сравнению с Lantus было показано при относительном риске 0,77 (95% CI [0,61, 0,99]) (р=0,0380).
2-я основная вторичная конечная точка. Изменение полученного методом LS среднего значения прединъекционной концентрации SMPG от начала исследования до конечной точки (6 месяц) было сходным в группах HOE901-U300 (-0,56 ммоль/л) и Lantus (-0,51 ммоль/л). Различие между группами лечения не было статистически значимым (разница между полученными методом LS средними значениями составляла -0,04 (95% CI [-0,438, 0,350], р=0,8279).
3-я основная вторичная конечная точка. Поскольку превосходство HOE901-U300 по сравнению с Lantus не было продемонстрировано по второй основной вторичной конечной точке, то по третьей основной вторичной конечной точке (снижению вариабельности прединъекционной концентрации SMPG на 6 месяце, которое было количественно большим в группе HOE901-U300 (-2,34) по сравнению с группой Lantus (-0,53)) не проводили дополнительное тестирование.
Другие вторичные конечные точки эффективности (6 месяц). Доля пациентов, у которых был достигнут уровень HbA1c <7%, была сходной в группах лечения (30,6% в HOE901-U300; 30,4% в Lantus). Что касается изменения среднего значения концентрации FPG, для двух групп лечения было показано сходное снижение. На графическом изображении профилей концентрации SMPG по 8 моментам времени в обеих группах лечения показано сопоставимое значительное снижение концентрации глюкозы в плазме крови в конечной точке (6 месяц) по сравнению с началом исследования.
На 6 месяце средняя суточная доза инсулина в группе НОЕ901-U300 составляла 91 ЕД (0,92 ЕД/кг), а в группе Lantus 82 ЕД (0,84 ЕД/кг).
В обеих группах лечения резервную терапию на протяжении основного 6-месячного периода лечения получало сходное число пациентов (5,7% для HOE901-U300, 4,9% для Lantus).
Результаты в отношении безопасности
В целом, в группе HOE901-U300 процентная доля пациентов, сообщавших о гипогликемии, была неизменно меньшей, чем в группе Lantus. Это различие было еще более выраженным на протяжении первых 2 месяцев лечения в рамках исследования, так же как и для эпизодов ночной гипогликемии. На протяжении основного 6-месячного периода применения исследуемого препарата тяжелая гипогликемия была зарегистрирована у 4/403 (1%) пациентов, получавших лечение с помощью HOE901-U300, и у 6/406 (1,5%) пациентов, получавших лечение с помощью Lantus.
Процентная доля пациентов с какими-либо ТЕАЕ (НОЕ901-U300, 236/403 [58,6%]; Lantus: 206/406 [50,7%]) без учета конкретных включенных SOC среди пациентов в группе лечения HOE901-U300 была более высокой, чем в группе Lantus. О серьезных ТЕАЕ сообщало сходное число пациентов в обеих группах лечения (HOE901-U300, 15 [3,7%]; Lantus, 15 [3,7%]).
Два (0,5%) пациента в группе лечения HOE901-U300 и 1 (0,2%) пациент в группе лечения Lantus умерли на протяжении 6-месячного периода применения исследуемого препарата.
Явления со смертельным исходом у двух пациентов в группе HOE901-U300 включали инфаркт миокарда и внезапную сердечную смерть, вызванную коронарной болезнью сердца на поздней стадии. Оба пациента страдали от предсуществующей выраженной сердечнососудистой патологии, и у них имелось несколько факторов риска, вызвавших смертельный исход. У пациента в группе Lantus произошло обострение хронического пиелонефрита со смертельным исходом. Ни один из случаев смерти, произошедших на протяжении 6-месячного периода лечения, не считался связанным с исследуемым лекарственным средством.
У сходного числа пациентов в обеих группах лечения были ТЕАЕ, приведшие к окончательному прекращению лечения (НОЕ901-U300: n=6, 1,5%; Lantus: n=4, 1,0%).
Реакции гиперчувствительности на протяжении основного 6-месячного периода применения исследуемого препарата были зарегистрированы в сходном количестве в обеих группах лечения (HOE901-U300: n=13, 3,2%; Lantus: n=16, 3,9%).
Зарегистрированное общее количество реакций в месте инъекции на протяжении основного 6-месячного периода применения исследуемого препарата было большим в группе лечения Lantus, чем в группе HOE901-U300 (Lantus: n=12, 3,0%; HOE901-U300: n=4, 1,0%).
В обеих группах лечения отсутствовало явное изменение массы тела (0,08 кг для HOE901-U300 и 0,66 кг для Lantus).
Выводы
В данном исследовании с участием 811 пациентов с T2DM, применявших базальный инсулин в сочетании с пероральным(и) гипогликемическим(и) средством(ами), исходные параметры и демографические данные были сбалансированы в группах лечения. Не меньшая эффективность HOE901-U300 по сравнению с Lantus была показана по первичной конечной точке эффективности (изменению уровня HbA1c от начала исследования до конечной точки [месяц 6]). Количество пациентов (%), сообщавших о ночной гипогликемии (тяжелой и/или подтвержденной концентрацией SMPG ≤70 мг/дл [3,9 ммоль/л]) в промежутке между началом на 9 неделе и 6 месяцем в группе HOE901-U300 было значительно меньшим, чем в группе Lantus (21,6% и 27,9%, соответственно, RR составлял 0,77, р-значение 0,0380; 1-я основная вторичная конечная точка эффективности). Для других вторичных конечных точек - прединъекционной концентрации глюкозы в плазме крови, вариабельности прединъекционной концентрации глюкозы в плазме крови, числа пациентов, у которых был достигнут целевой уровень HbA1c, и изменения среднего значения концентрации FPG, изменения концентрации SMPG в профиле по 8 моментам времени и вариабельности средней концентрации глюкозы в плазме крови в течение 24 часов - были получены сопоставимые результаты среди групп лечения.
Общая частота возникновения гипогликемии (% пациентов по меньшей мере с одним эпизодом) на протяжении основного 6-месячного периода применения исследуемого препарата в группе HOE901-U300 было неизменно меньшим, чем в группе Lantus, вне зависимости от разновидности гипогликемии. Эта разница в пользу HOE901-U300 была еще более выраженной для всех разновидностей ночной гипогликемии.
HOE901-U300 хорошо переносился на протяжении основного 6-месячного периода применения исследуемого препарата в исследовании, и особых проблем безопасности не наблюдалось.
Краткое описание результатов в отношении эффективности и безопасности из двенадцатимесячного расширенного исследования 2 версии
- HbA1c: на протяжении продленного периода исследования безопасности (от конечной точки основного исследования [6 месяц] до конца лечения [12 месяц]) уровень HbA1c оставался постоянным и был сопоставимым в обеих группах лечения.
- Резервная терапия: на протяжении всего периода лечения процентная доля пациентов, которым потребовалась резервная терапия, была сходной в обеих группах лечения (8,2% для НОЕ901-U300, 10,1% для Lantus). На протяжении 6-месячного продленного периода исследования безопасности в группе лечения HOE901-U300 резервную терапию начинала меньшая процентная доля пациентов, чем в группе Lantus (2,5% для HOE901-U300, 5,4% для Lantus).
- Гипогликемия: в целом, так же как и на протяжении основного 6-месячного периода лечения, на протяжении всего периода применения исследуемого препарата в исследовании гипогликемия в группе HOE901-U300 возникала у меньшей процентной доли пациентов, чем в группе Lantus, вне зависимости от разновидности гипогликемии.
- Безопасность: HOE901-U300 хорошо переносился в ходе исследования, и особых проблем безопасности не наблюдалось; на протяжении всего периода лечения процентная доля пациентов с какими-либо ТЕАЕ без учета конкретных включенных SOC среди пациентов в группе лечения HOE901-U300 была выше, чем в группе Lantus (278/403 [69,0%] и 244/406 [60,1%], соответственно). О серьезных ТЕАЕ сообщало сходное число пациентов (30 [7,4%]) в обеих группах лечения. На протяжении всего периода применения исследуемого препарата в исследовании у четырех (1,0%) пациентов в группе лечения HOE901-U300 и 2 (0,5%) пациентов в группе лечения Lantus было ТЕАЕ, приведшее к смерти.
- Масса тела: в обеих группах лечения на протяжении всего периода применения исследуемого препарата в исследовании отмечалось небольшое увеличение массы тела (0,41 кг для НОЕ901-U300 и 1,15 кг для Lantus).
2. РЕЗУЛЬТАТЫ
2.1 ПАЦИЕНТЫ-УЧАСТНИКИ ИССЛЕДОВАНИЯ
2.1.1 Распределение участников исследования





2.1.2 Демографические данные и исходные параметры
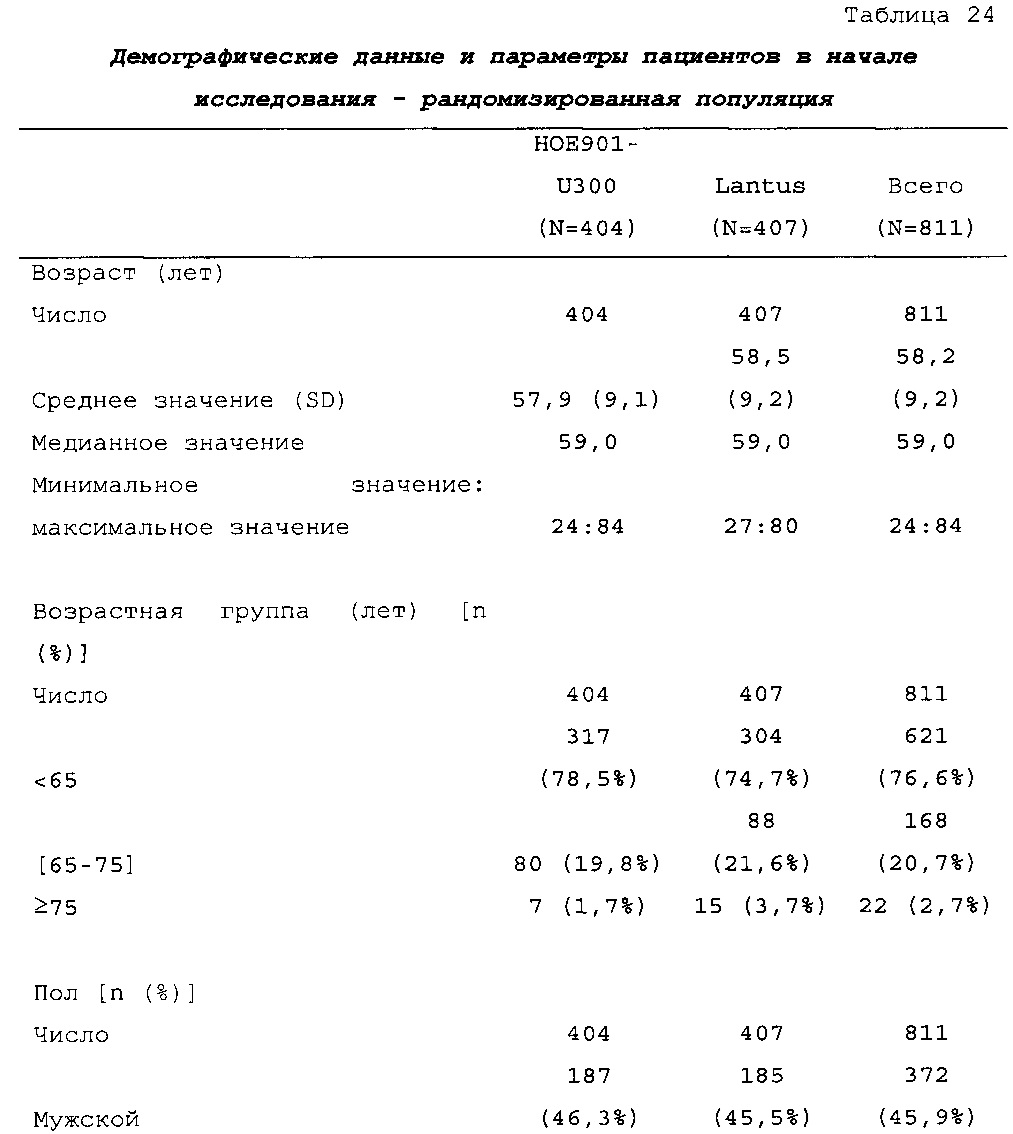
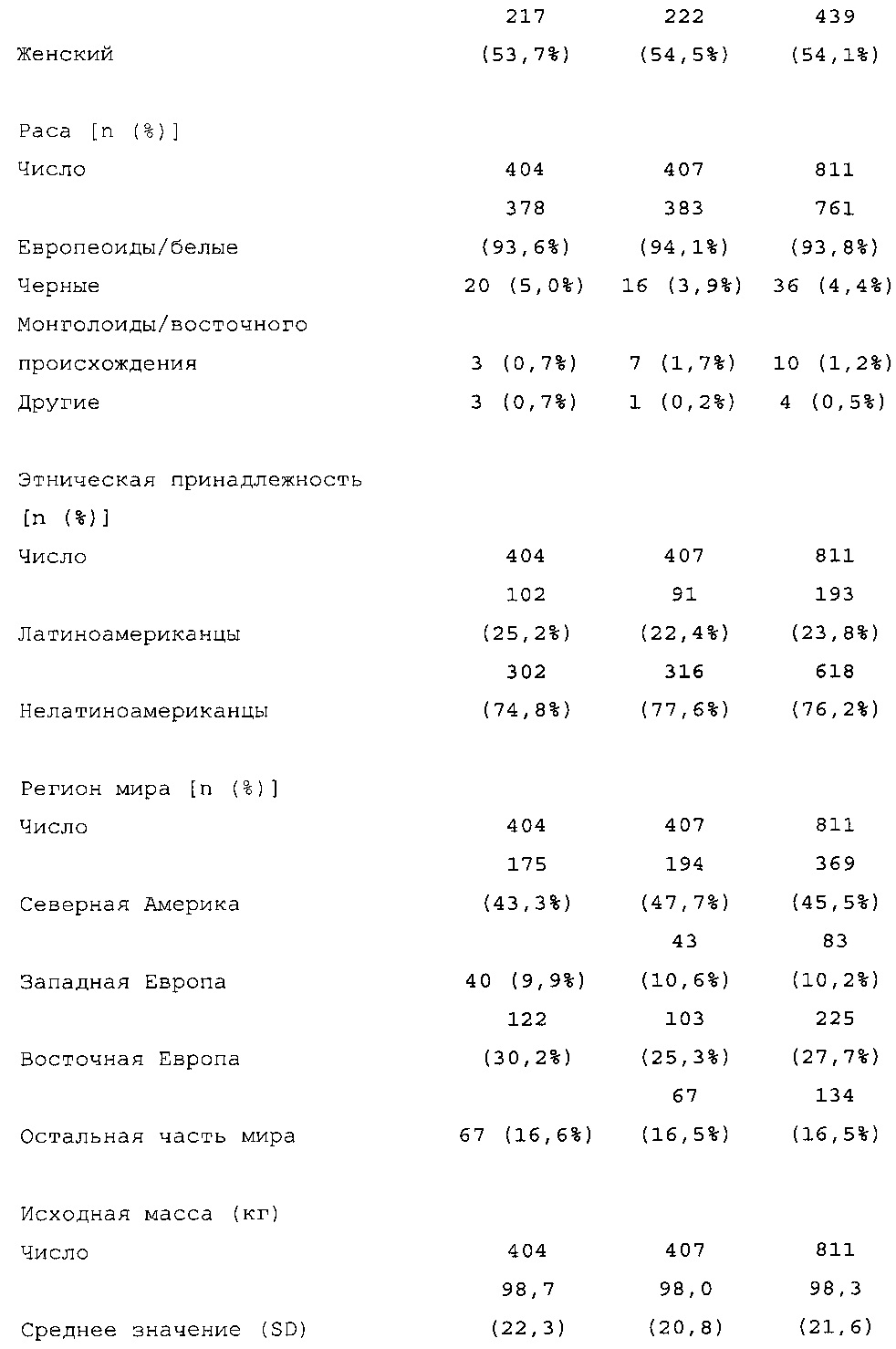
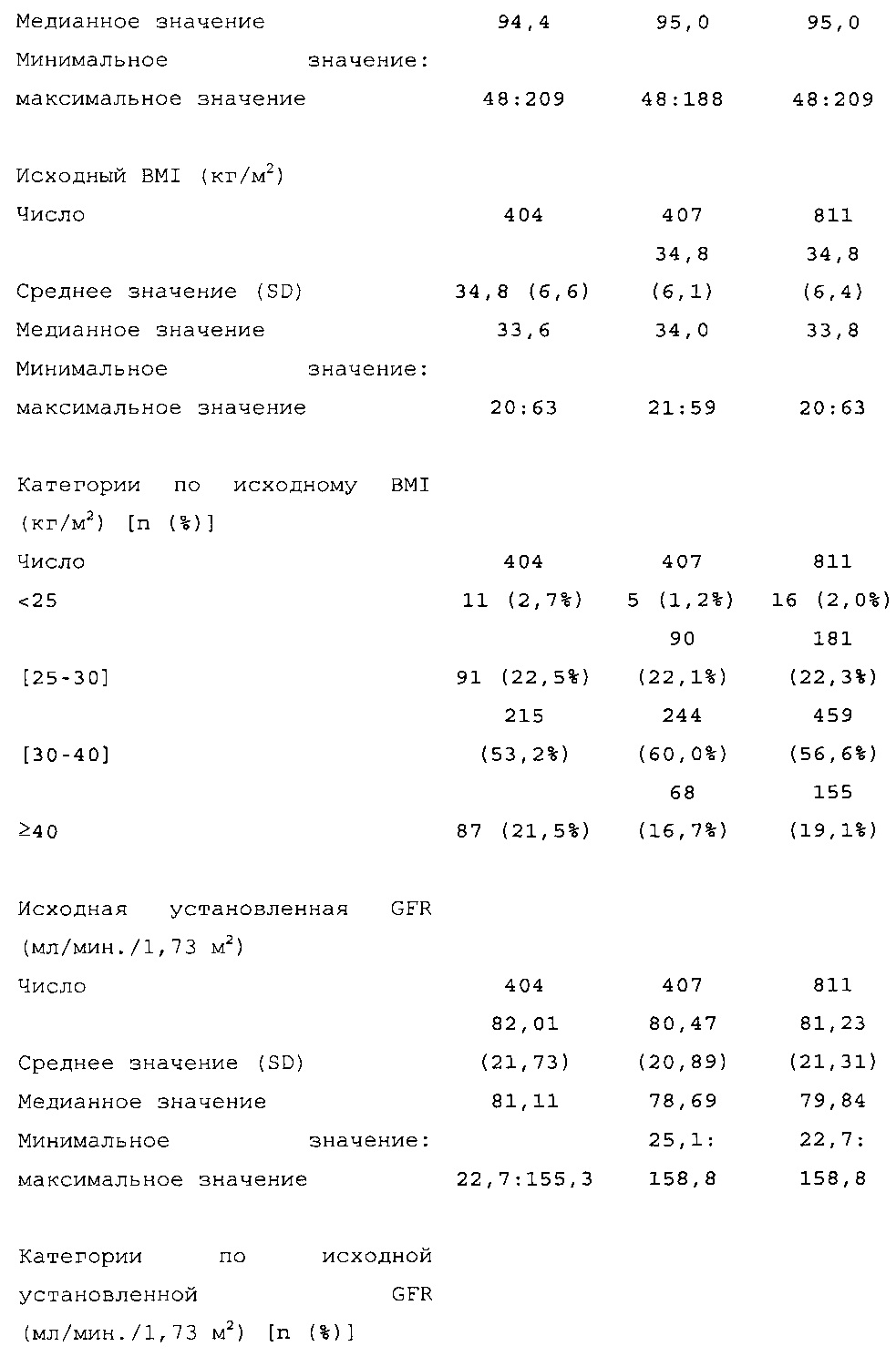

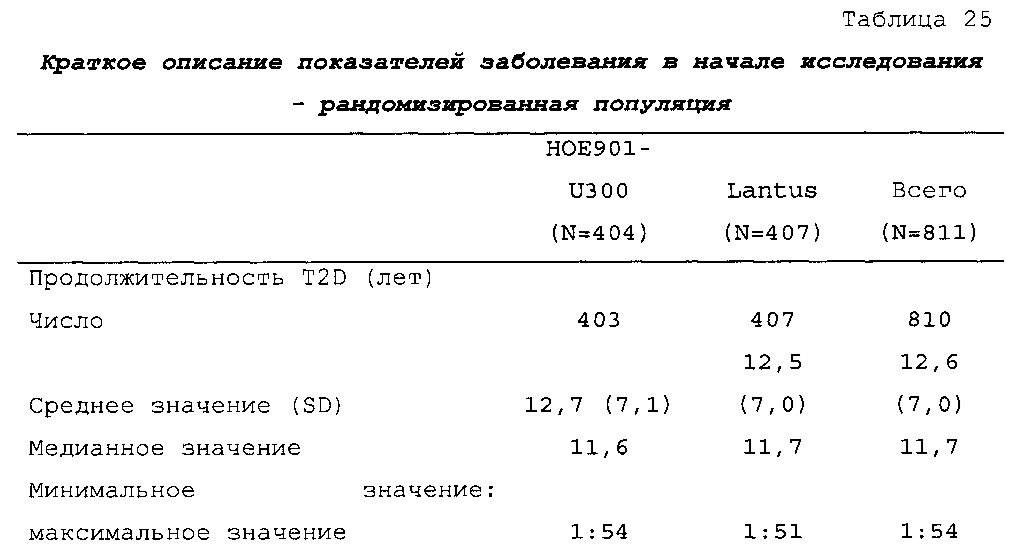
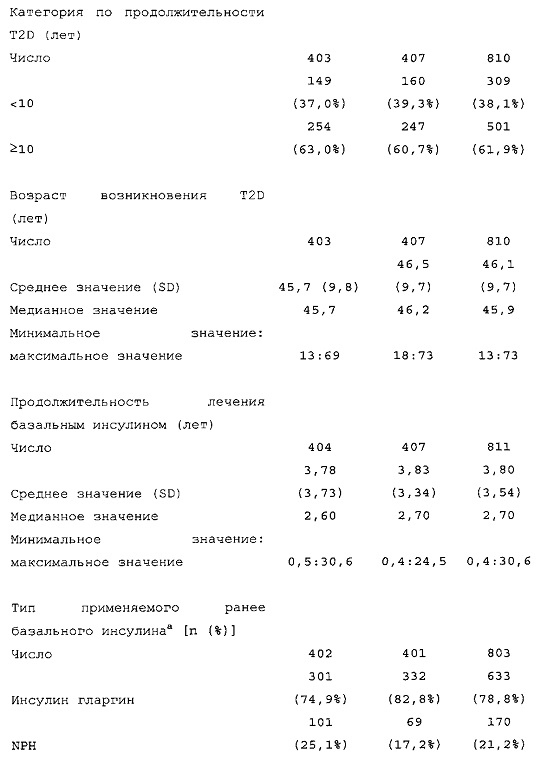
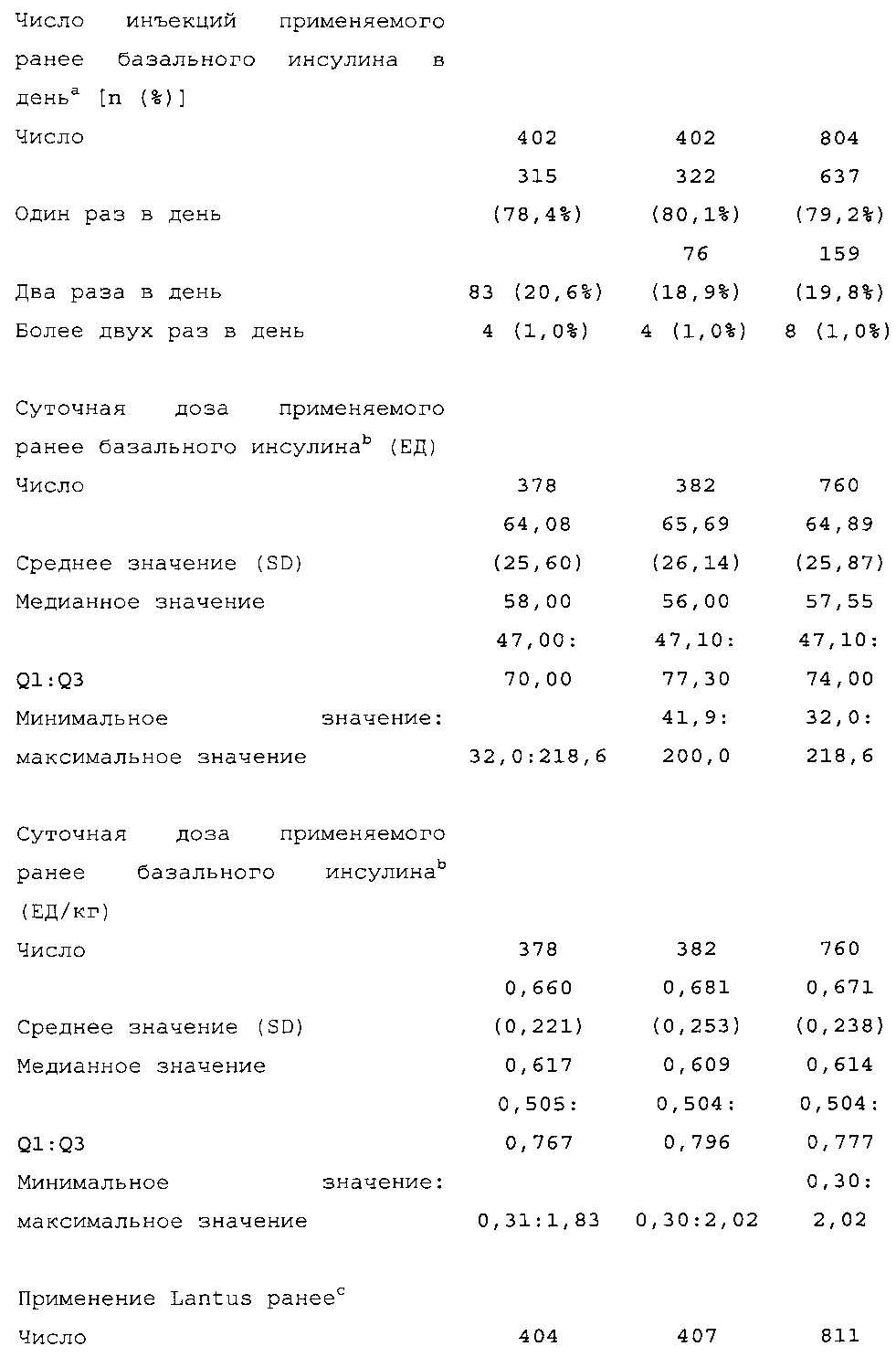

2.2 ОЦЕНКА ЭФФЕКТИВНОСТИ
2.2.1 Первичная конечная точка эффективности
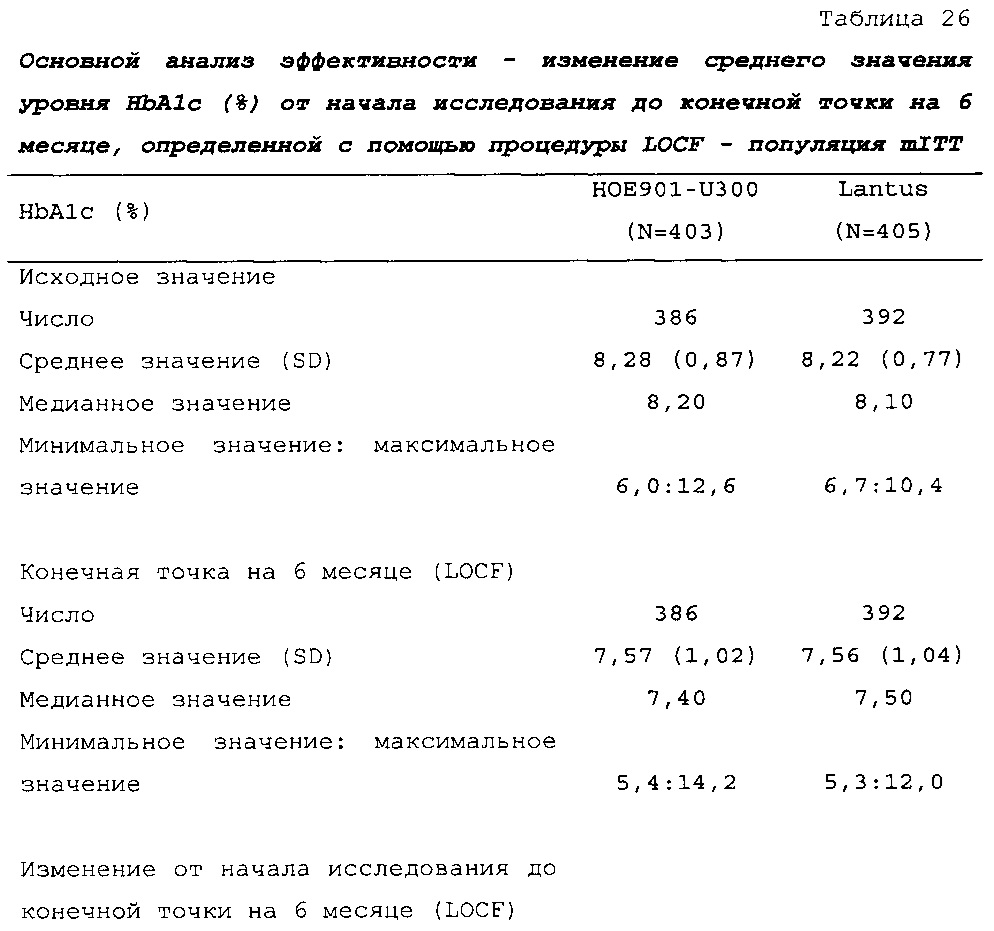

Данные из таблицы 26 обобщены на фигуре 5.
2.2.2 Основные вторичные конечные точки
2.2.2.1 Ночная гипогликемия

2.2.2.2 Прединъекционная концентрация глюкозы в плазме крови - конечная точка на 6 месяце
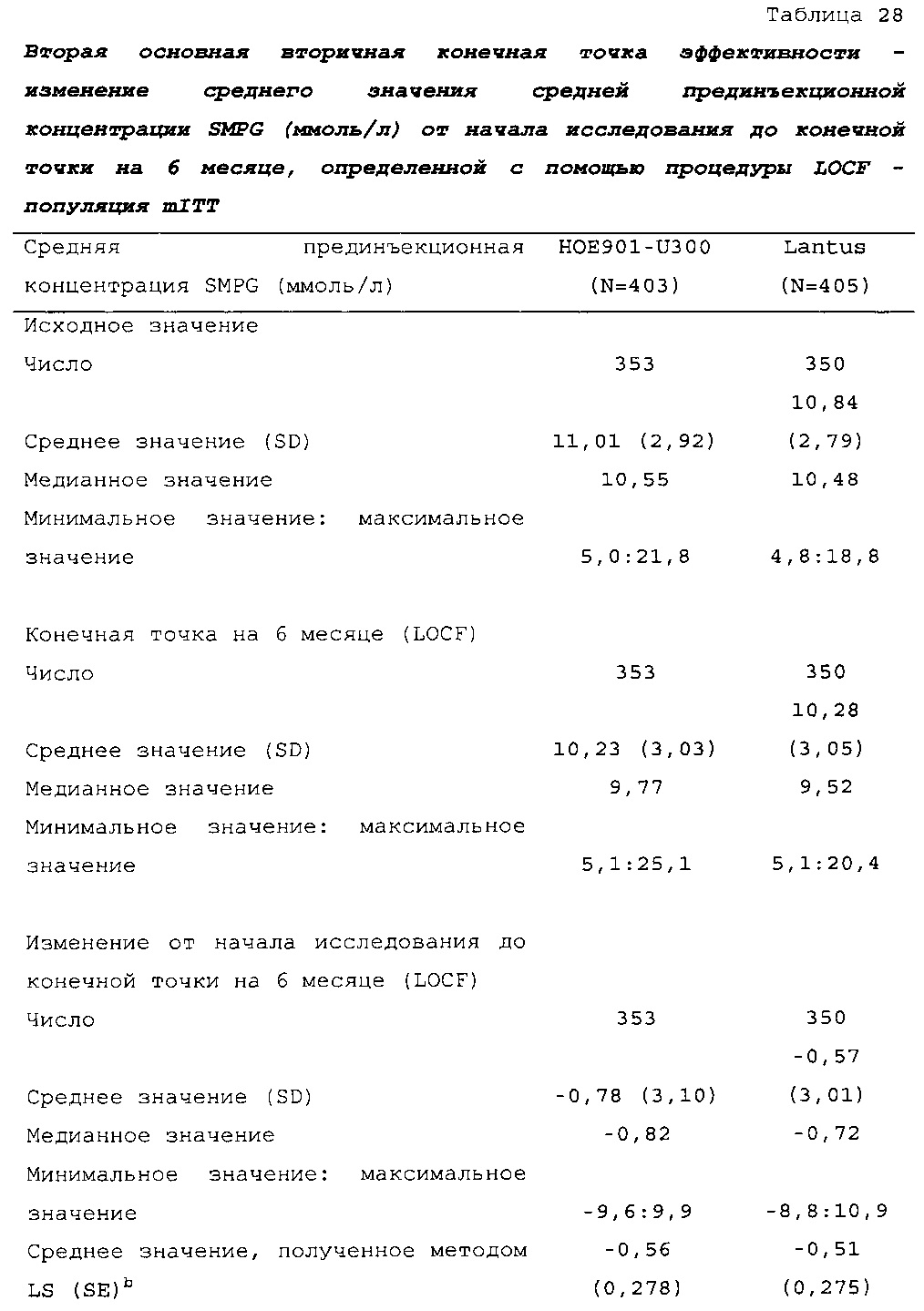

Данные из таблицы 28 обобщены на фигуре 6.
2.2.2.3 Вариабельность прединъекционной концентрации SMPG - конечная точка на 6 месяце
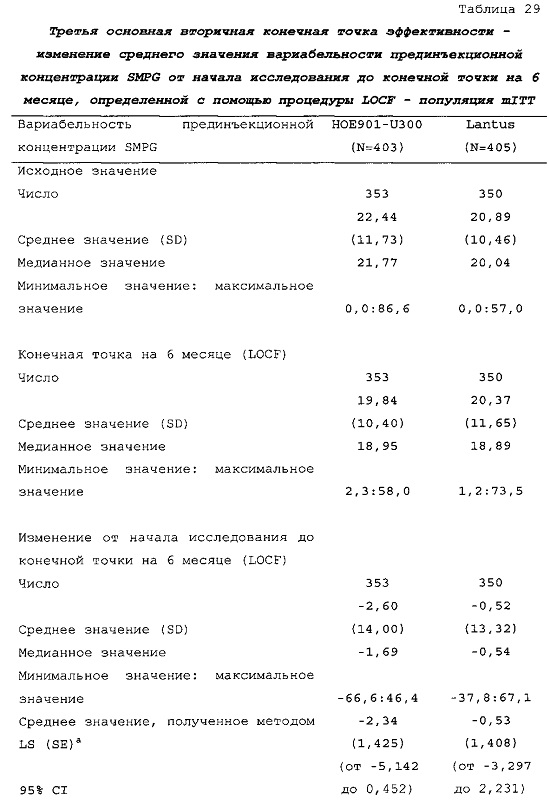

2.2.3 Другие вторичные конечные точки эффективности
2.2.3.1 Процентная доля пациентов с уровнем HbA1c <7% на 6 месяце


2.2.3.2 Изменение концентрации FPG от начала исследования до конечной точки на 6 месяце
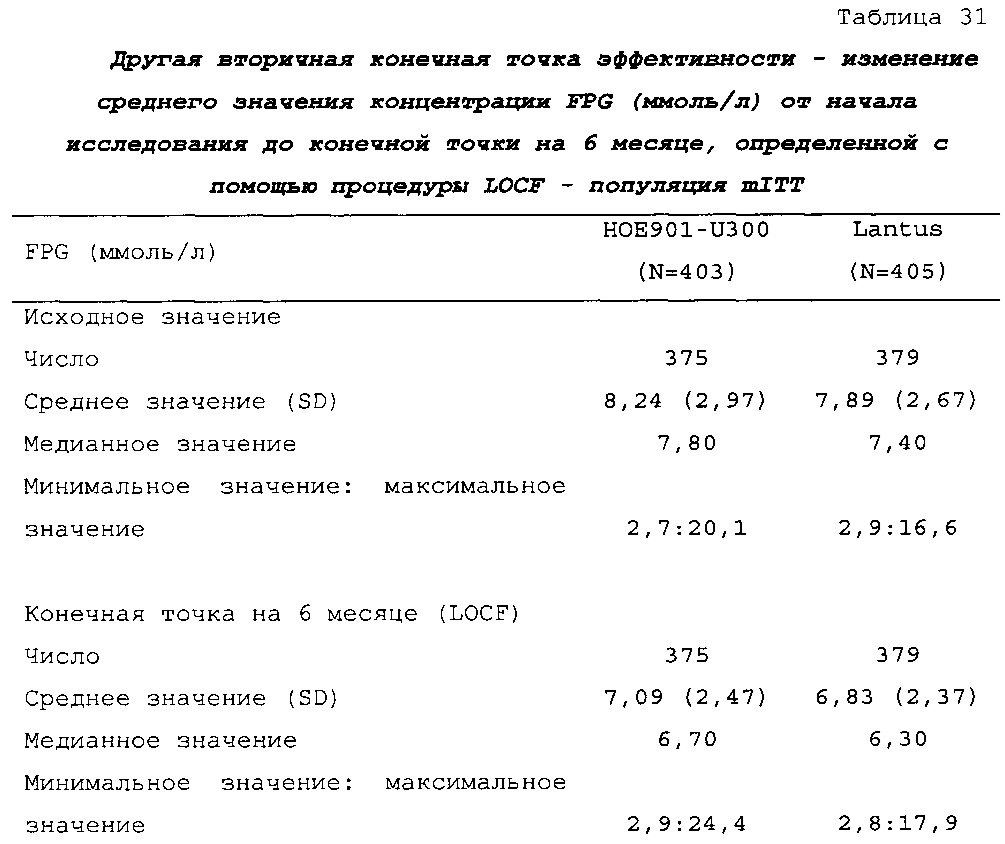

2.2.3.3 Профиль концентрации SMPG по восьми моментам времени
На фигуре 7 показаны средние значения концентрации SMPG (ммоль/л) в профиле по 8 моментам времени в начале исследования и в конечной точке на 6 месяце (популяция mITT).
2.2.3.4 Доза базального инсулина
На фигуре 8 показаны средние суточные дозы базального инсулина (ЕД) по визитам на протяжении основного 6-месячного периода применения исследуемого препарата (популяция mITT).
2.3 ОЦЕНКА БЕЗОПАСНОСТИ
2.3.1 Длительность проведения лечения

2.3.2 Гипогликемия



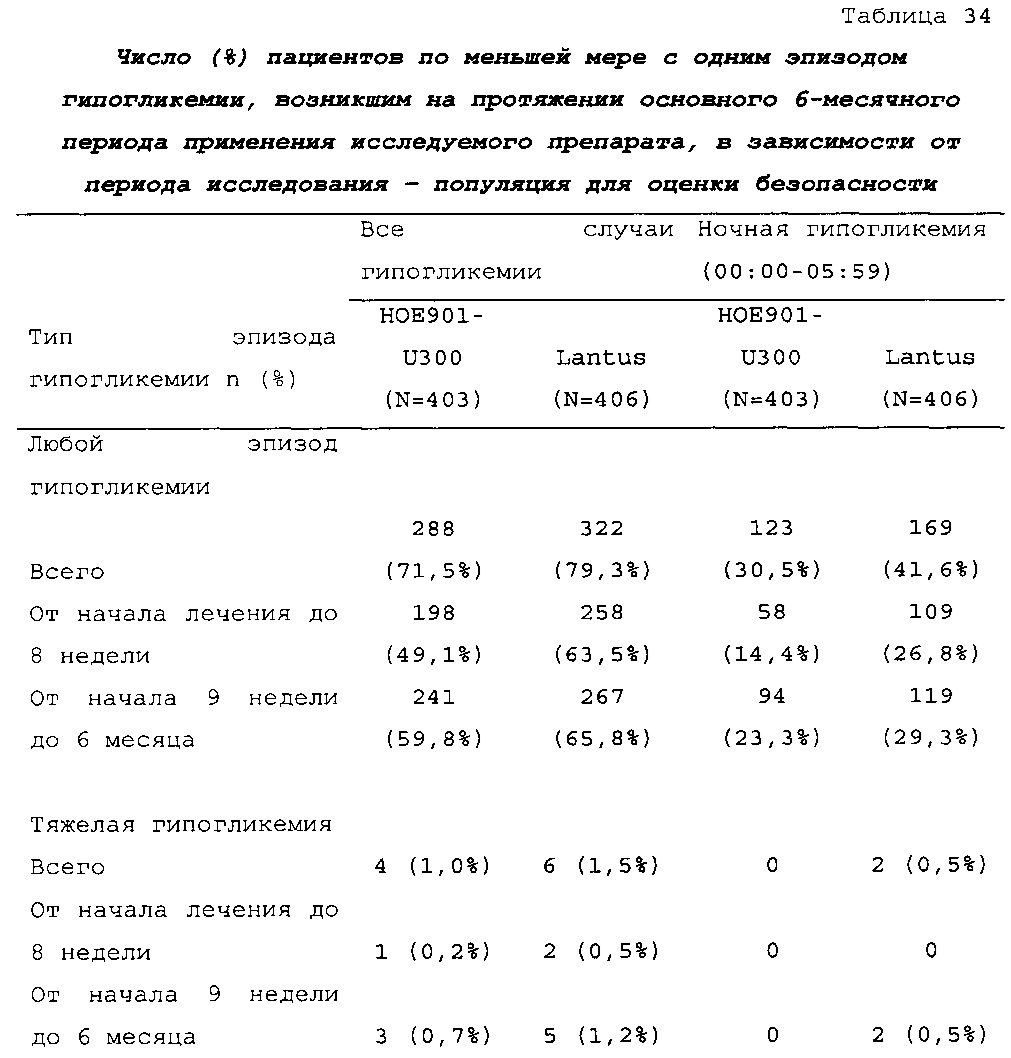
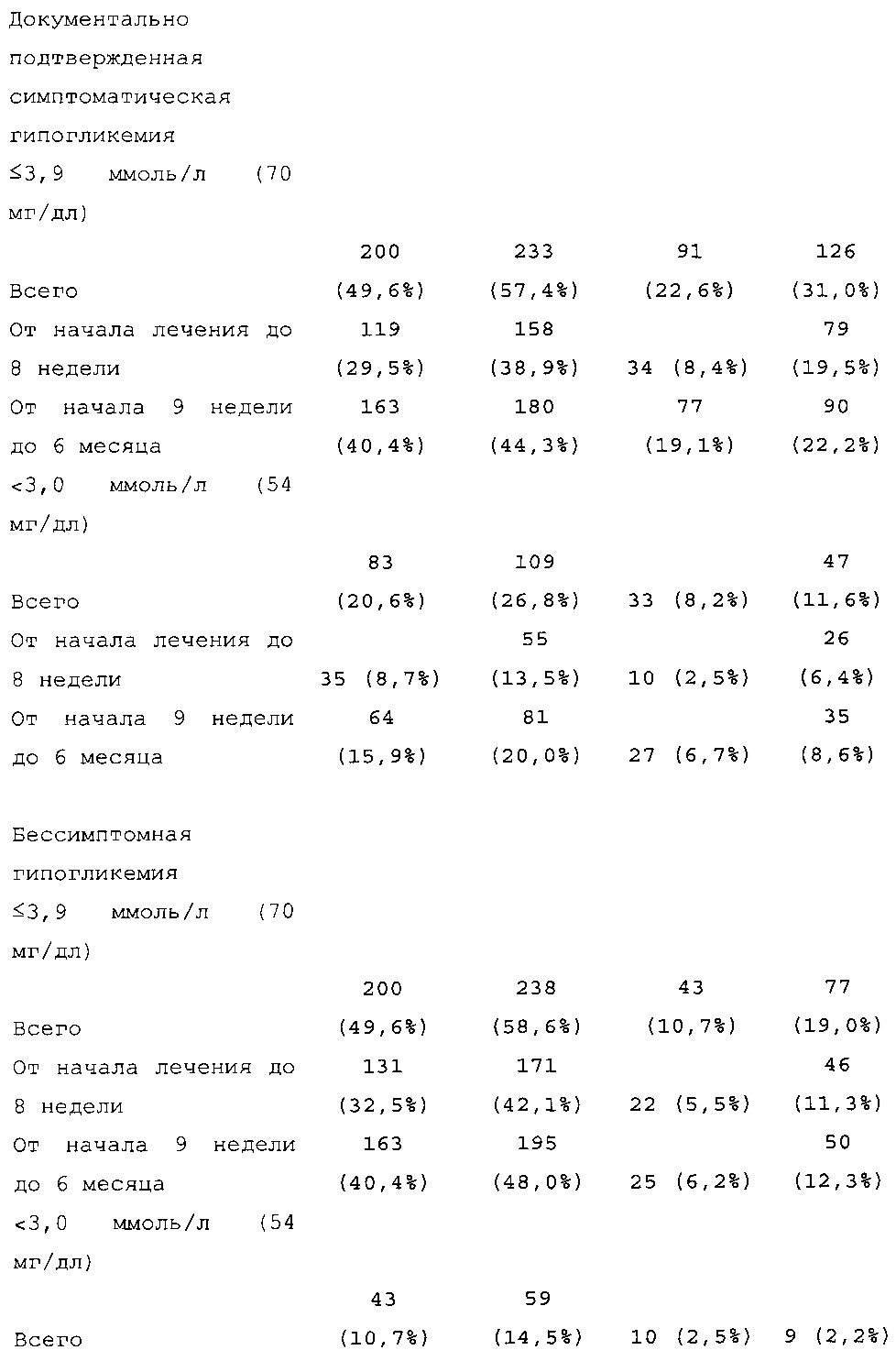
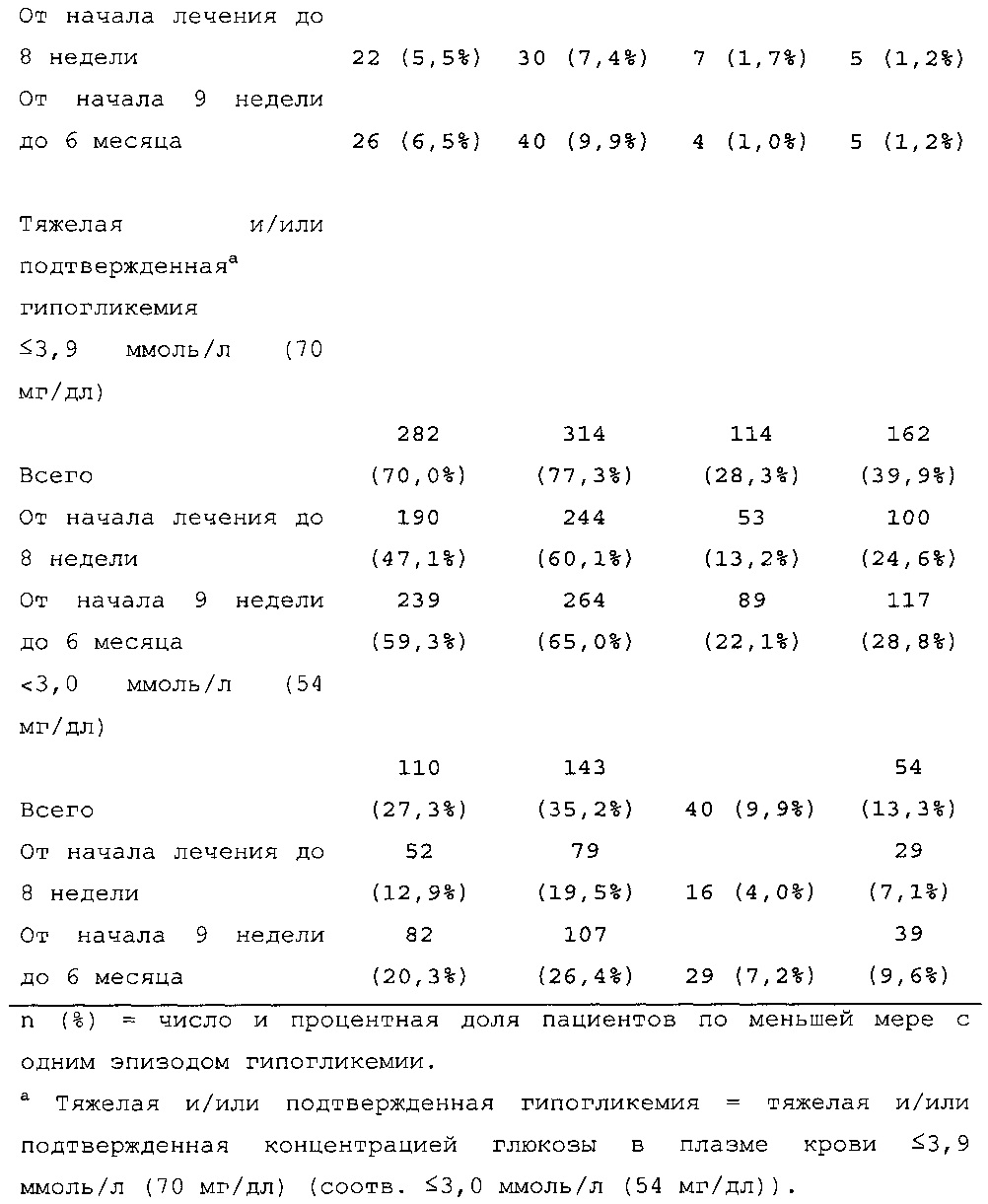
2.3.3 Нежелательные явления, возникшие в ходе лечения
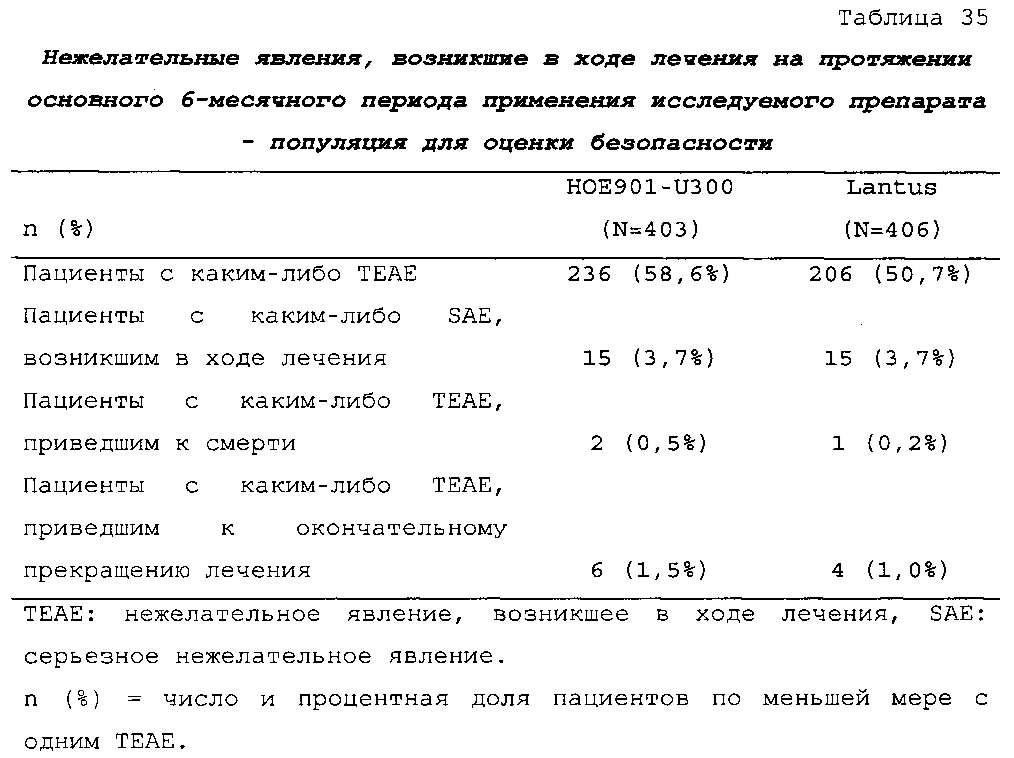



2.3.4 Случаи смерти, серьезные нежелательные явления, возникшие в ходе лечения
2.3.4.1 Случаи смерти


2.3.4.2 Серьезные нежелательные явления
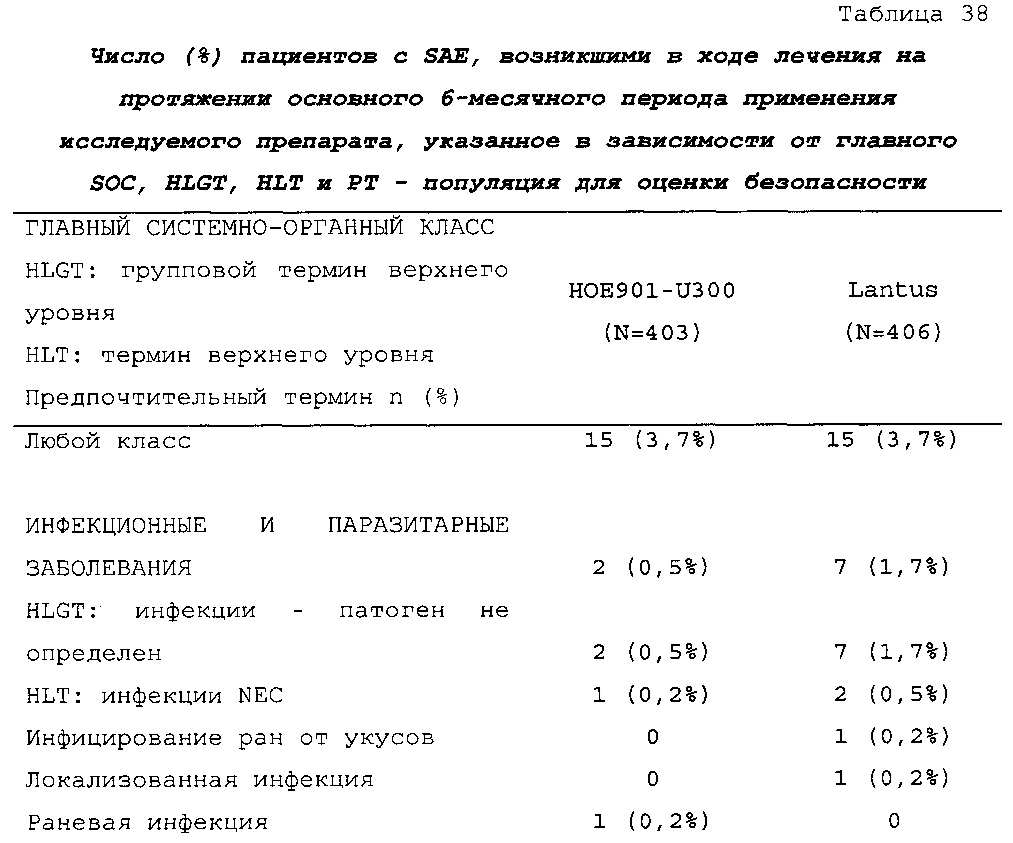
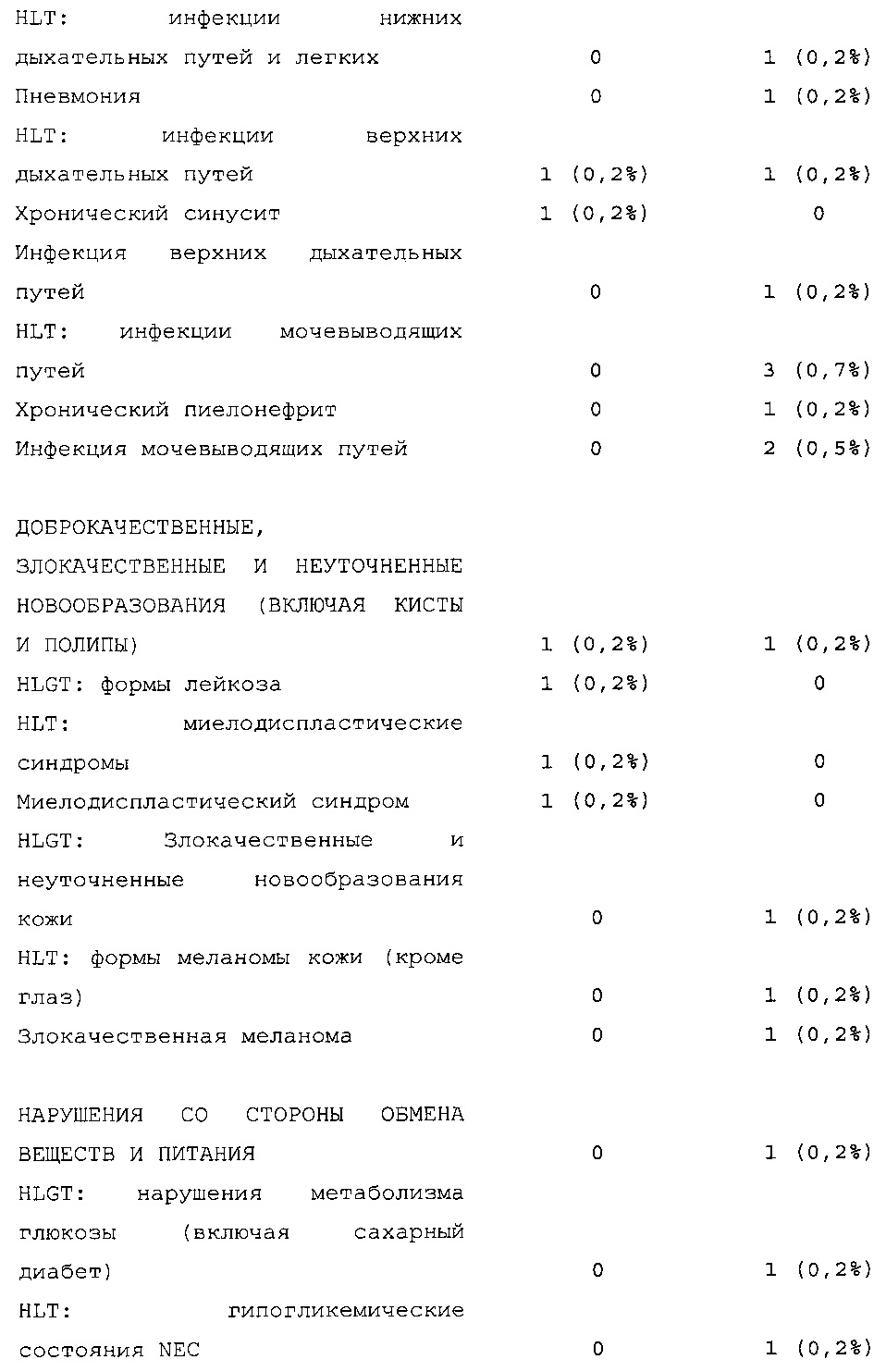
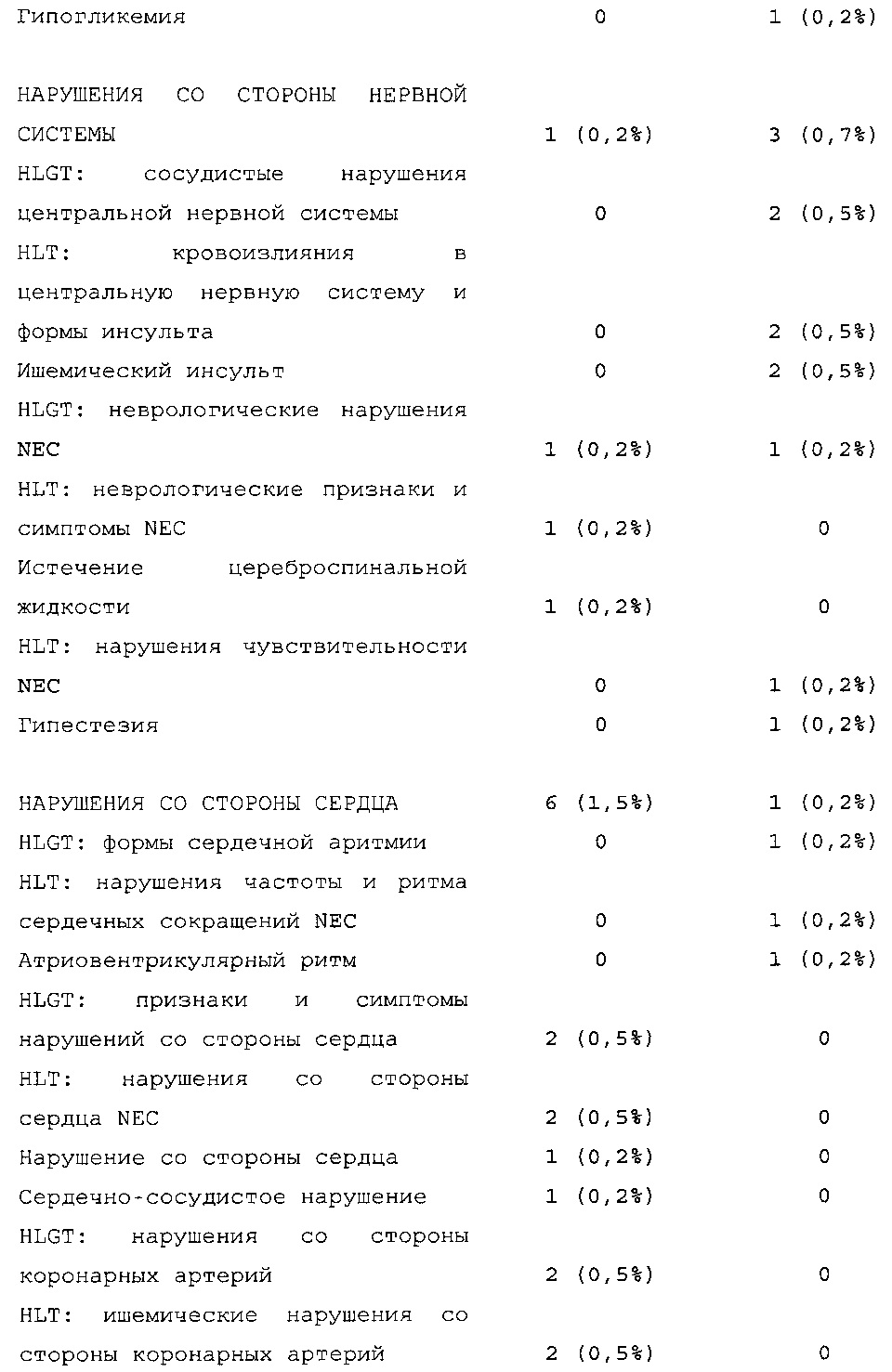
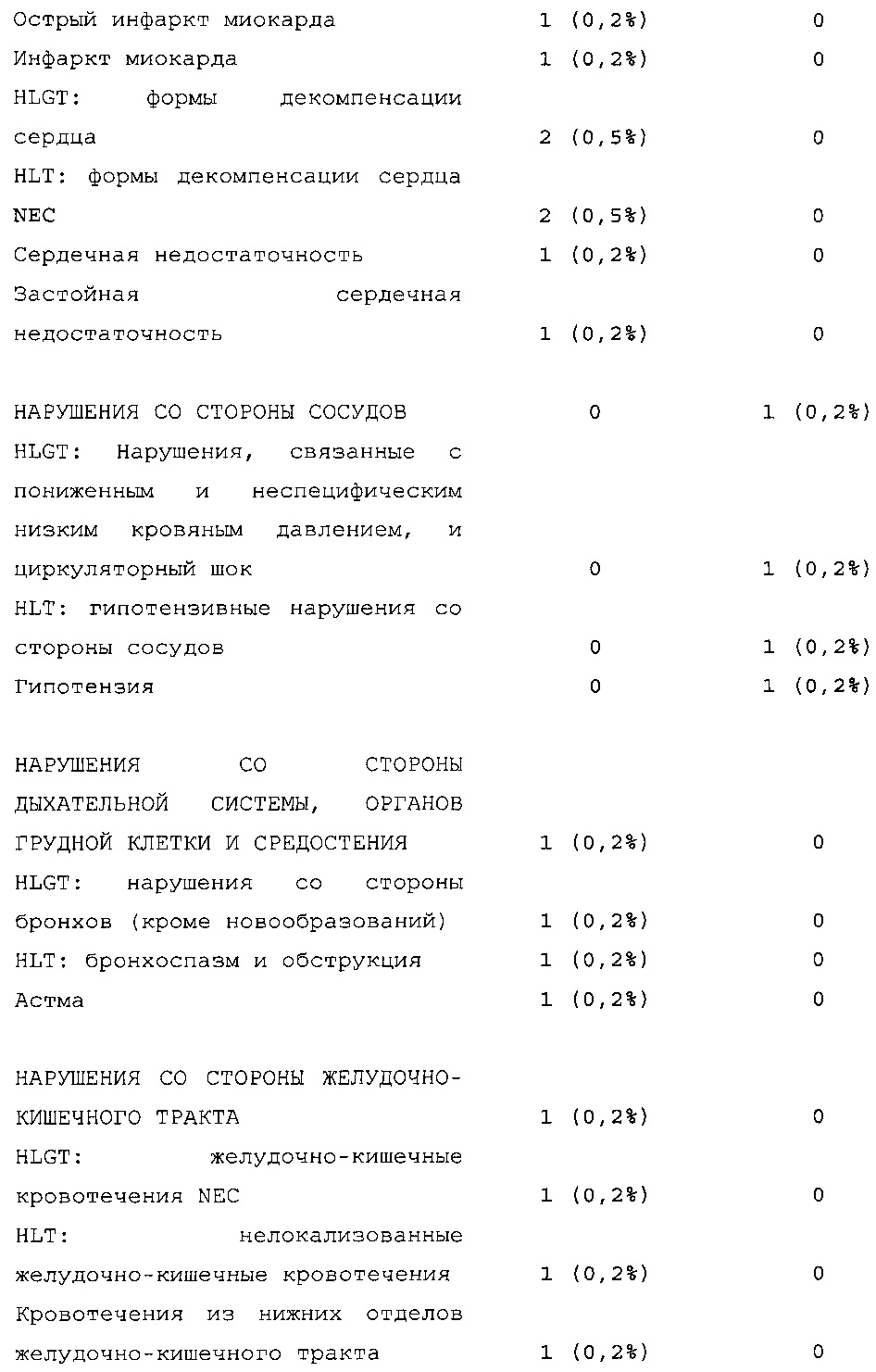


2.3.5 Нежелательные явления, приведшие к прекращению участия




2.3.6 Другие значительные нежелательные явления
2.3.6.1 Реакция гиперчувствительности
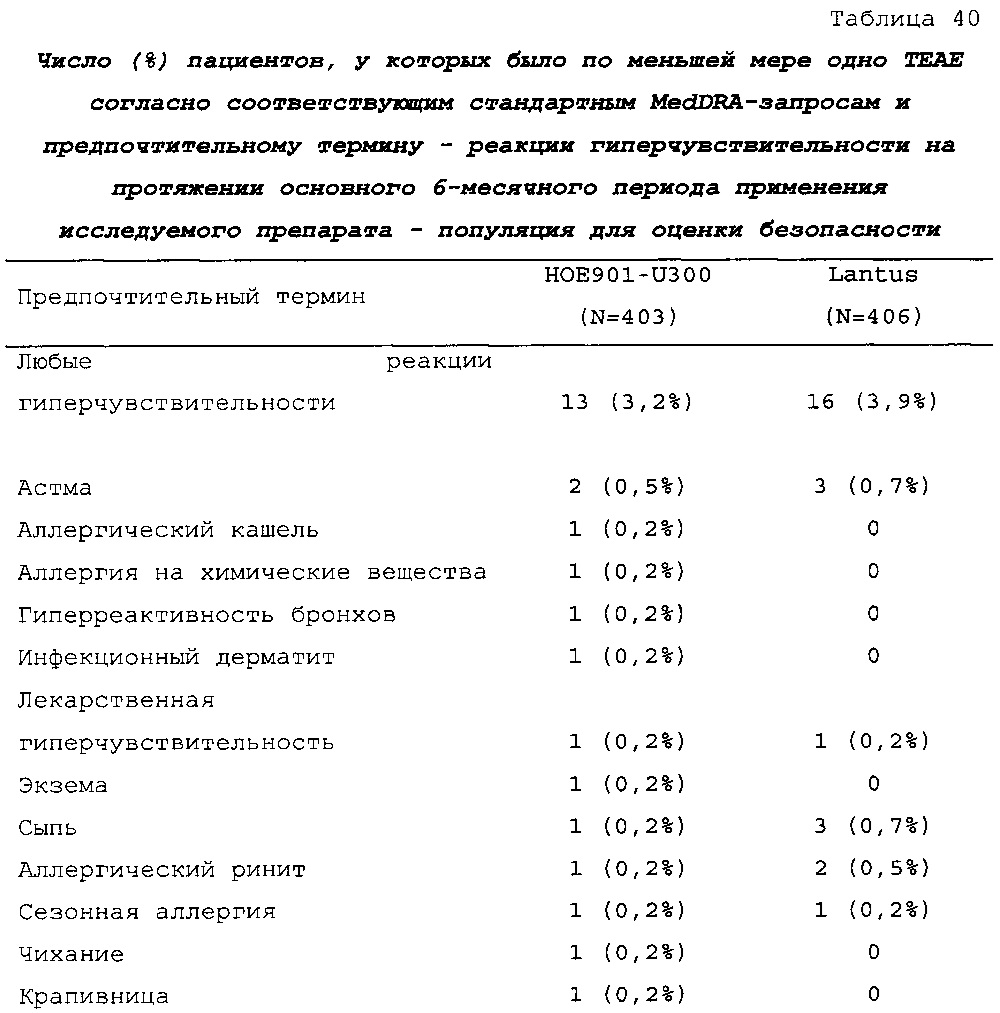

2.3.6.2 Реакции в месте инъекции
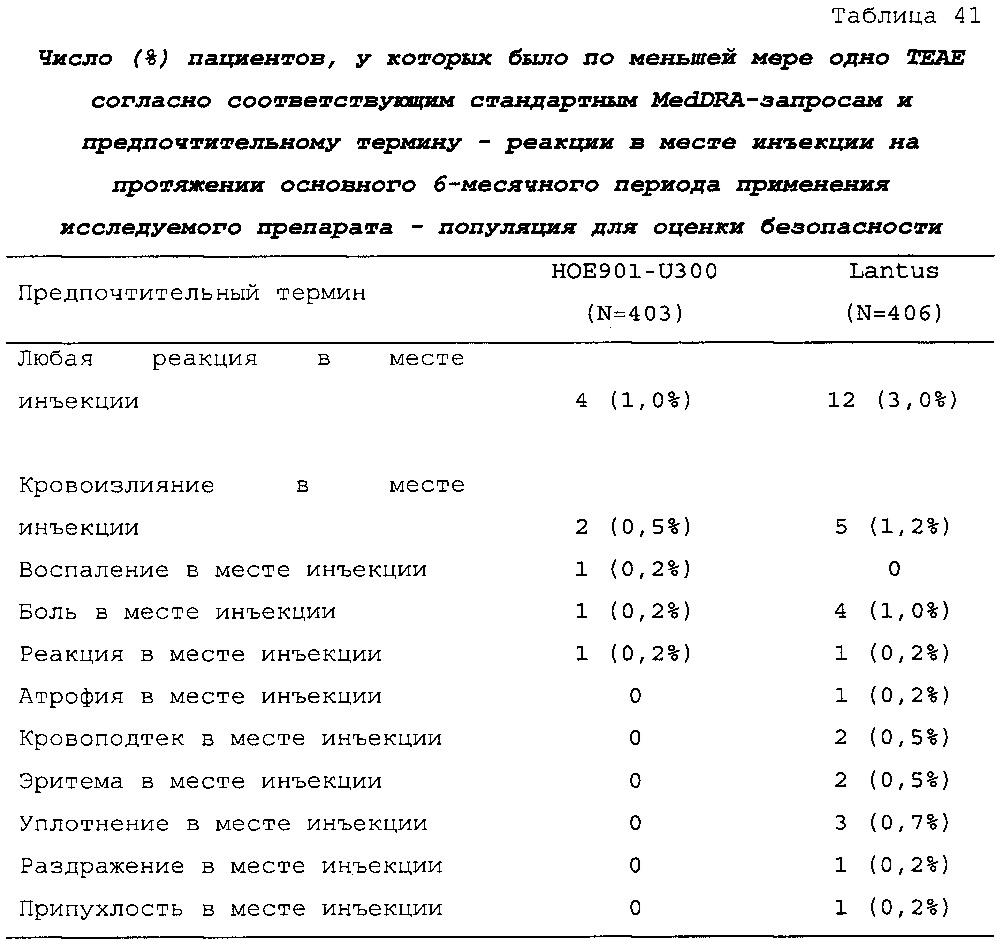

2.3.7 Масса тела
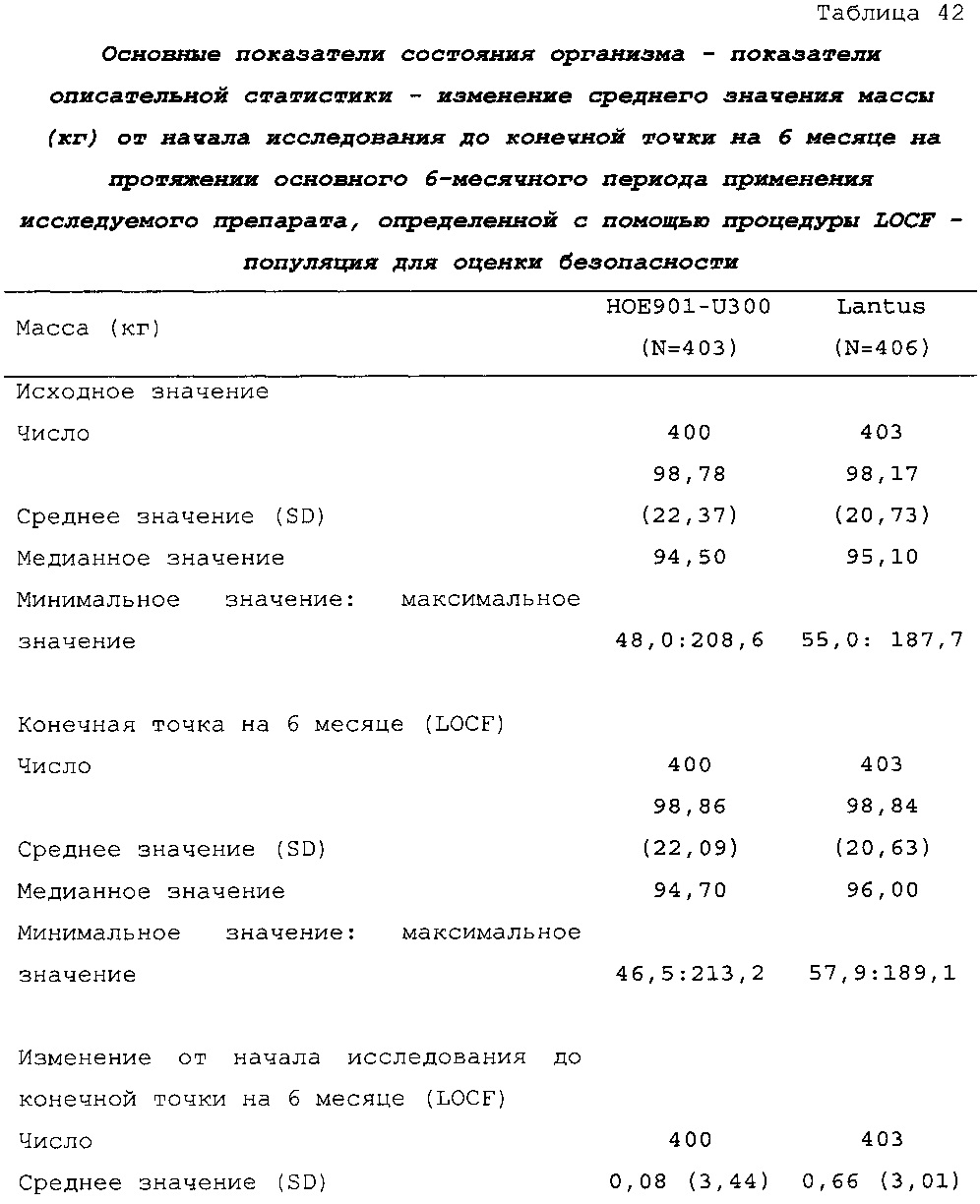

Пример 3. 6-месячное многоцентровое рандомизированное открытое исследование на параллельных группах, в котором сравнивали эффективность и безопасность нового состава инсулина гларгина и Lantus®, оба из которых применяли в сочетании с прандиальным инсулином у пациентов с сахарным диабетом 2 типа, с 6-месячным продленным периодом исследования безопасности дополнительное исследование введения, в котором сравнивали изменяемые интервалы дозирования с фиксированными интервалами дозирования
1 ОБЩЕЕ ОПИСАНИЕ ИССЛЕДОВАНИЯ
Фаза разработки: 3
Цели:
Первичная цель: сравнение эффективности HOE901-U300, инъецируемого один раз в день каждые 24 часа, и HOE901-U300, инъецируемого один раз в день с интервалами 24±3 часа, с точки зрения изменения уровня HbA1c от 6 месяца основного исследования (= начало дополнительного исследования) до 9 месяца основного исследования (= конечная точка дополнительного исследования) у пациентов с сахарным диабетом 2 типа.
Основные вторичные цели: сравнение безопасности двух схем инъекций HOE901-U300 с точки зрения частоты гипогликемии.
Методика. Пациентов, рандомизированных в группу HOE901-U300 и получавших HOE901-U300 на протяжении 6-месячного периода основного исследования, рандомизировали в соотношении 1:1 для введения HOE901-U300 один раз в день каждые 24 часа (фиксированные интервалы дозирования) либо каждые 24±3 часа (изменяемые интервалы дозирования).
Пациенты, применявшие HOE901-U300, завершившие 6-месячный период основного исследования (см. пример 1) и отвечающие критериям отбора для дополнительного исследования, были допущены к дополнительному исследованию. Для первичного анализа, который был описательным, не требовался конкретный объем выборки.
Число пациентов: Запланировано: до 300 (150 в группе предварительного лечения) Рандомизировано: 110
Получали лечение: 110
Подвергнуты оценке: эффективности: 109, безопасности: 110
Диагностика и критерии включения. Критерии включения: завершение 6-месячного периода исследования в основном исследовании, описанного в примере 1 (визит 10), рандомизация и получение лечения с помощью HOE901-U300 на протяжении 6-месячного периода лечения (начало исследования - 6 месяц), полученное подписанное письменное информированное согласие на участие в дополнительном исследовании.
Ключевые критерии невключения: нежелание пациента применять изменяемые интервалы между инъекциями, составляющие 24±3 часа, в течение по меньшей мере двух дней в неделю; невозможность соблюдения изменяемой схемы дозирования по мнению исследователя; состояние здоровья, препятствующее дальнейшему участию пациента в исследовании.
Исследуемые средства лечения
Экспериментальный лекарственный препарат: изучаемое лекарственное средство: HOE901-U300
Состав: HOE901-U300 (раствор инсулина гларгина, 300 ЕД/мл) представляет собой стерильный апирогенный прозрачный бесцветный раствор в стеклянном картридже, укомплектованном в шприц-ручку (предварительно наполненную, одноразовую ручку).
Путь введения: подкожная инъекция.
Исследуемая схема
Изменяемые интервалы дозирования: введение HOE901-U300 один раз в день каждые 24±3 часа.
Время инъекции могло быть изменено в зависимости от потребностей индивидуума в пределах до 3 часов до или после фиксированного в начале основного исследования времени ежедневной инъекции вечером. Максимальные интервалы, т.е. предполагающие ежедневную инъекцию за 3 часа до или спустя 3 часа после фиксированного времени, необходимо было применять в течение по меньшей мере 2 дней в неделю по выбору пациента. Зафиксированное в начале основного исследования время инъекции должно было оставаться опорным временем для варьирования.
Контрольная схема
Фиксированные интервалы дозирования: инъекция HOE901-U300 один раз в день каждые 24 часа.
Пациенты продолжали инъецировать HOE901-U300 один раз в день каждые 24 часа в зафиксированное в начале основного исследования время инъекции.
Доза
Следовало подобрать дозу HOE901-U300, необходимую для достижения или поддержания концентрации глюкозы в плазме крови натощак в целевом диапазоне от 80 до 100 мг/дл (от 4,4 ммоль/л до 5,6 ммоль/л) без гипогликемии. Изменения дозы инсулина основывались на данных измерений концентрации глюкозы в плазме капиллярной крови натощак при самоконтроле (SMPG).
Неэкспериментальные лекарственные препараты
Пациенты в обеих группах лечения должны были продолжать применять свой прандиальный аналог инсулина на протяжении дополнительного исследования.
Пациенты, получавшие сопутствующее лечение метформином, должны были на протяжении дополнительного исследования продолжать получать постоянную дозу, за исключением случаев уменьшения дозы или отмены метформина, продиктованных проблемами безопасности.
Продолжительность лечения. Дополнительное исследование состояло из 3-месячного периода сравнения эффективности и безопасности лечения, начинавшегося по окончании 6-месячного периода основного исследования и заканчивавшегося по окончании 9 месяца основного исследования.
После окончания пациенты, входившие в группу с изменяемой схемой дозирования, могли продолжать придерживаться этой схемы до конца основного исследования (12 месяца). Пациенты, инъецировавшие HOE901-U300 каждые 24 часа, продолжали придерживаться своей схемы лечения до конца исследования.
Продолжительность наблюдения. Периодом анализа эффективности и безопасности являлся 3-месячный период исследования, начинавшийся на 6 месяце основного исследования и заканчивавшийся на 9 месяце основного исследования. Результаты, представленные в настоящем KRM, относятся к этому периоду.
Критерии оценки
Эффективность
Первичная конечная точка эффективности: изменение уровня HbA1c от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц).
Вторичные конечные точки: изменение концентрации FPG (центральная лаборатория) от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц), суточной дозы базального инсулина и прандиального инсулина.
Безопасность: Гипогликемия, частота нежелательных явлений, особенно нежелательных явлений, возникших в ходе лечения (ТЕАЕ), и серьезных нежелательных явлений (SAE), реакций в месте инъекции и реакций гиперчувствительности. Следующая информация не представлена в KRM: другая информация, касающаяся безопасности, в том числе об основных показателях состояния организма и передозировке.
Статистические методы
Для данного 3-месячного дополнительного исследования в качестве начала определен 6 месяц периода основного исследования; конечной точкой являлся 9 месяц основного исследования.
Анализ первичной конечной точки эффективности (изменения уровня HbA1c от начала дополнительного исследования [6 месяц] до конечной точки [9 месяц]) осуществляли с использованием модели ковариационного анализа (ANCOVA) со схемой лечения и страной в качестве фиксированных эффектов и с использованием исходного значения уровня HbA1c в качестве ковариаты. Различия между изменяемой схемой дозирования HOE901-U300 и фиксированной схемой дозирования HOE901-U300 и двусторонние 95% доверительные интервалы оценивали в рамках ANCOVA.
Все непрерывные переменные вторичной эффективности (кроме изменения вариабельности прединъекционной концентрации SMPG) анализировали с использованием модели ANCOVA со схемой лечения и страной в качестве фиксированных эффектов и с использованием соответствующего исходного значения в качестве ковариаты.
Изменение вариабельности прединъекционной концентрации SMPG от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц) анализировали с использованием модели дисперсионного анализа (ANOVA) со схемой лечения и страной в качестве фиксированных эффектов.
Анализы безопасности были описательными, основанными на популяции для оценки безопасности.
Краткое описание
Характеристики популяции
В общей сложности 110 пациентов с сахарным диабетом 2 типа рандомизировали для дополнительного исследования: 56 пациентов для применения HOE901-U300 по схеме с изменяемыми интервалами дозирования и 54 пациента для применения HOE901-U300 по схеме с фиксированными интервалами дозирования; 110 пациентов подвергали воздействию IMP (популяция для оценки безопасности). Популяция mITT дополнительного исследования (популяция для оценки эффективности) включала 109 пациентов.
Один пациент (1/56, 1,8%), рандомизированный для применения HOE901-U300 с изменяемыми интервалами дозирования, досрочно прекратил участие в дополнительном исследовании, а также прекратил участие в продленном периоде основного исследования (фиксированные интервалы дозирования HOE901-U300: 0/54, 0%).
Демографические данные и параметры пациентов в начале дополнительного исследования (6 месяц) были сбалансированы среди обеих групп применения схем. Средний возраст в популяции дополнительного исследования составлял 60 лет; у 35 из 110 (31,8%) пациентов он составлял ≥65 лет.
В ходе дополнительного исследования в среднем 23,0% инъекций у пациентов в группе изменяемых интервалов дозирования были сделаны с предельными интервалами <21,5 часа или >26,5 часа после предыдущей инъекции по сравнению с 3,9% инъекций у пациентов в группе фиксированных интервалов дозирования. В среднем 13,5% инъекций у пациентов в группе изменяемых интервалов дозирования были сделаны с промежуточным интервалом (в промежутке 21,5-23 часов или в промежутке 25-26,5 часа после предыдущей инъекции) по сравнению с 8,2% инъекций у пациентов в группе фиксированных интервалов дозирования. В пределах 23-25 часов после предыдущей инъекции у пациентов в группе изменяемых интервалов дозирования было сделано меньшее число инъекций (63,4%) по сравнению с группой фиксированных интервалов дозирования (88,0%).
В общей сложности 34,5% пациентов в группе изменяемых интервалов дозирования делали менее 20% инъекций за пределами интервала 23-25 часов после предыдущей инъекции, и, следовательно, считалось, что примерно 65% пациентов соблюдали схему с изменяемыми интервалами дозирования. В группе фиксированных интервалов дозирования 78,8% пациентов делали более 80% инъекций в пределах 23-25 часов после предыдущей инъекции и, следовательно, могли считаться соблюдающими схему с фиксированными интервалами дозирования.
Соблюдение обеих схем было сходным, когда интервалы отсчитывали от опорного времени инъекции, запланированного в начале основного исследования.
Результаты в отношении эффективности
Первичная конечная точка. Изменение полученного методом LS среднего значения уровня HbA1c от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц) было сходным в группах изменяемых интервалов дозирования (0,22% [95% CI: от -0,006 до 0,436]) и фиксированных интервалов дозирования (0,14% [95% CI: от -0,099 до 0,380]), при этом разница между полученными методом LS средними значениями составляла 0,07% [95% CI: от -0,169 до 0,318].
Вторичные конечные точки эффективности (9 месяц):
- FPG (изменяемый интервал дозирования - 1,40 ммоль/л [95% CI: от 0,624 до 2,177]; фиксированный интервал дозирования - 1,18 ммоль/л [95% CI: от 0,350 до 2,015]; разница между полученными методом LS средними значениями - 0,22 ммоль/л [95% CI: от -0,634 до 1,070]).
- Прединъекционная концентрация SMPG, вариабельность прединъекционной концентрации SMPG:
изменение интервалов между инъекциями HOE901-U300, дававшее в результате более короткие и более длинные интервалы по сравнению с обычным 24-часовым интервалом, могло влиять на вторичные конечные точки эффективности прединъекционную концентрацию SMPG и вариабельность прединъекционной концентрации SMPG. Следовательно, помимо общего анализа с помощью схем дозирования с интервалами, в котором показано среднее значение средних значений по пациентам за период до 7 дней перед визитом, в CSR была представлена разбивка данных о концентрации SMPG по интервалам между инъекциями.
В обеих группах применения схем дозирования с интервалами средние суточные дозы базального и прандиального инсулина оставались постоянными на протяжении 3-месячного периода применения схемы, используемой для сравнения.
Безопасность
На протяжении 3-месячного периода применения схемы, используемой для сравнения, об эпизодах гипогликемии, как в целом, так и по каждой разновидности гипогликемии, сообщала сходная процентная доля пациентов, применявших HOE901-U300 по схеме с изменяемыми интервалами дозирования и применявших HOE901-U300 по схеме с фиксированными интервалами дозирования.
Процентные доли пациентов с каким-либо ТЕАЕ (изменяемые интервалы дозирования - 15/56 [26,8%]; фиксированные интервалы дозирования - 15/54 [27,8%]) или с серьезным ТЕАЕ (изменяемые интервалы дозирования - 4/56 [7,1%]; фиксированные интервалы дозирования - 5/56 [9,3%]) были сопоставимыми среди различных схем.
ТЕАЕ, приведшие к прекращению лечения, приведшие к смерти или связанные с реакциями в месте инъекции, не наблюдались ни для одной схемы дозирования с интервалами на протяжении 3-месячного периода дополнительного исследования. У одного пациента [1,9%], применявшего схему с фиксированными интервалами дозирования, было ТЕАЕ, связанное с реакцией гиперчувствительности.
Выводы
Большинство пациентов в обеих группах применения схем дозирования с интервалами следовали графику инъекций в зависимости от группы рандомизации и применяли изменяемые интервалы дозирования (НОЕ901-U300 один раз в день каждые 24 часа ± 3 часа в течение по меньшей мере 2 дней в неделю) либо фиксированные интервалы дозирования (HOE901-U300 один раз в день каждые 24 часа). Это обеспечивало возможность сравнения двух схем дозирования с интервалами для анализов эффективности и безопасности.
Анализы эффективности с точки зрения уровня HbA1c и концентрации FPG показали сопоставимые результаты для двух схем дозирования с интервалами.
Общая частота возникновения гипогликемии (% пациентов по меньшей мере с одним эпизодом) на протяжении 3-месячного периода дополнительного исследования была сходной для обеих схем вне зависимости от разновидности гипогликемии.
HOE901-U300 с изменяемыми интервалами дозирования и НОЕ901-U300 с фиксированными интервалами дозирования хорошо переносились на протяжении 3-месячного периода сравнительного дополнительного исследования; особых проблем безопасности не наблюдалось.
В совокупности, согласно результатам дополнительного исследования при периодических изменениях интервалов между инъекциями отрицательные эффекты в отношении основных конечных точек эффективности и безопасности не наблюдались.
2. РЕЗУЛЬТАТЫ
2.1 ПАЦИЕНТЫ-УЧАСТНИКИ ИССЛЕДОВАНИЯ
2.1.1 Распределение участников исследования




2.1.2 Демографические данные и исходные параметры
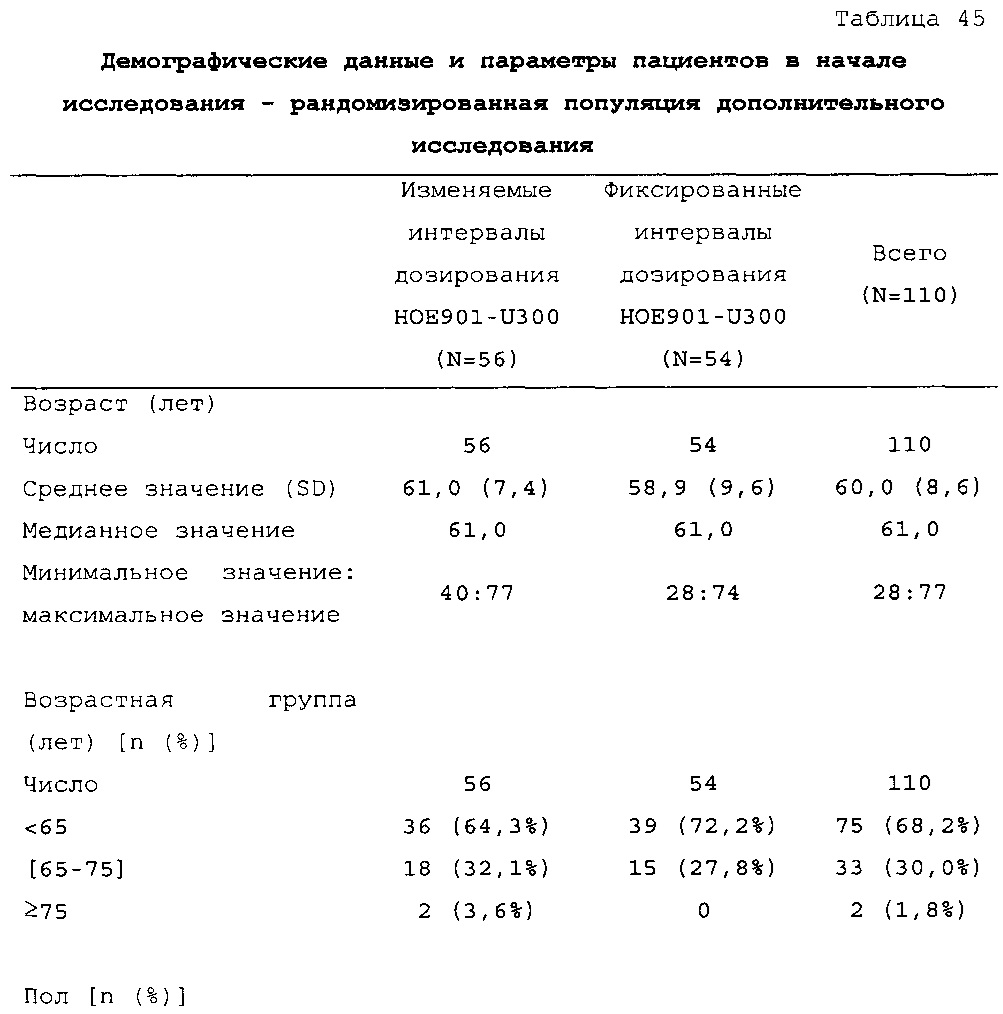
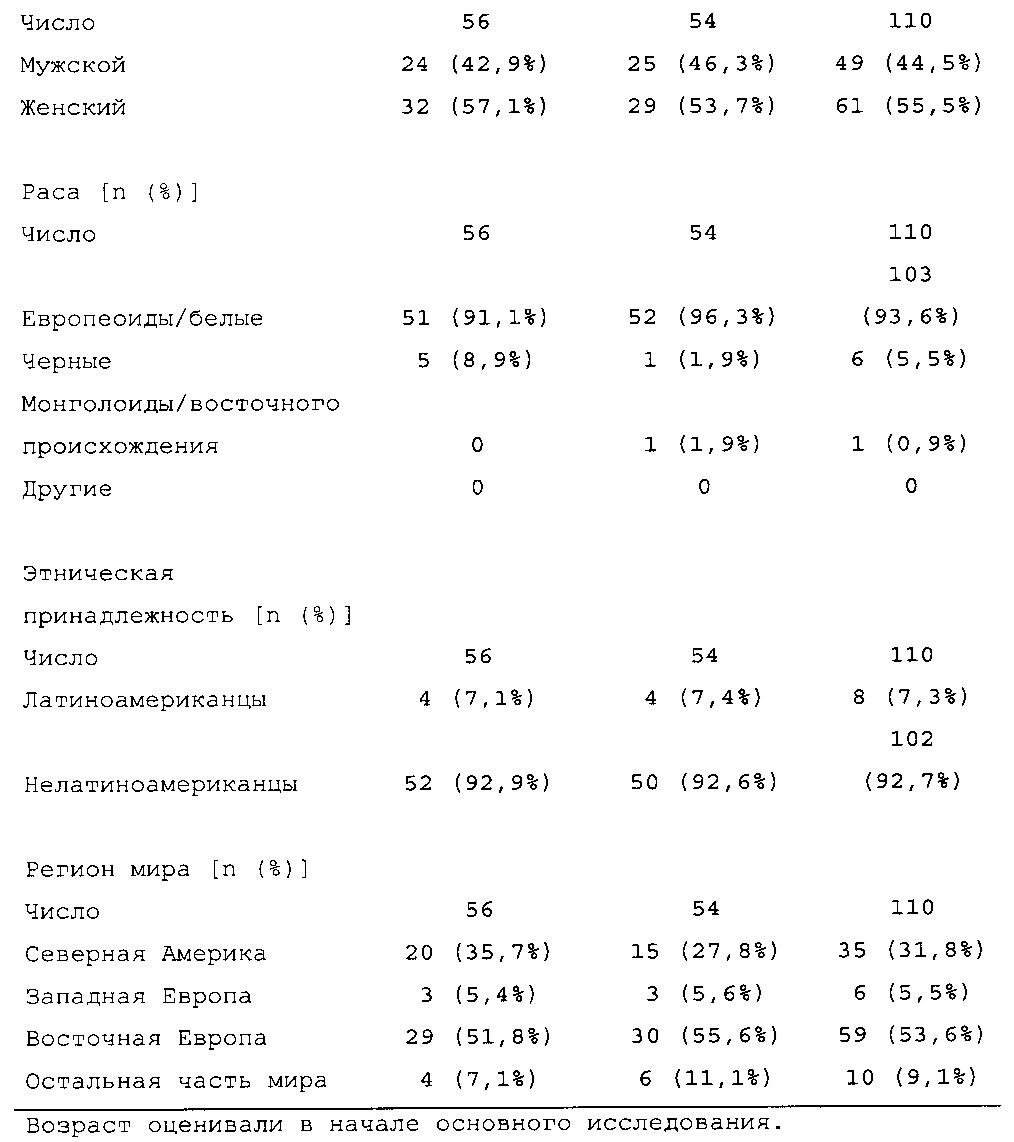
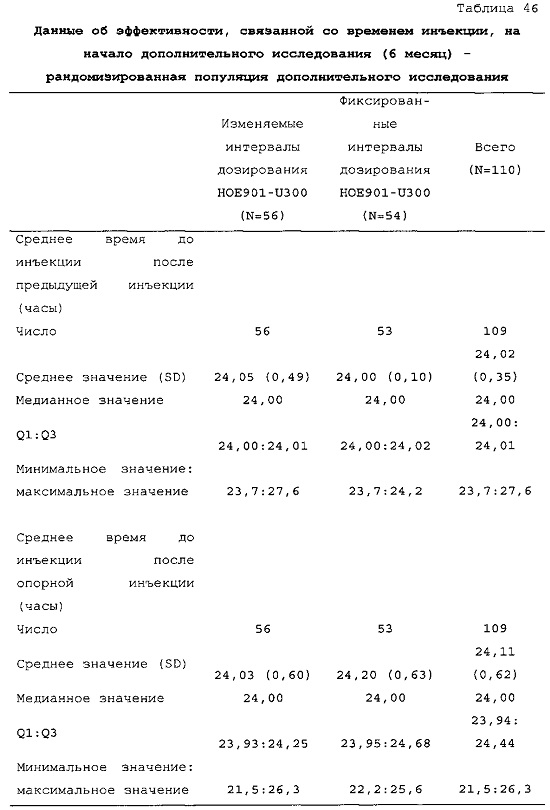

2.1.3 Определение соблюдения режима лечения




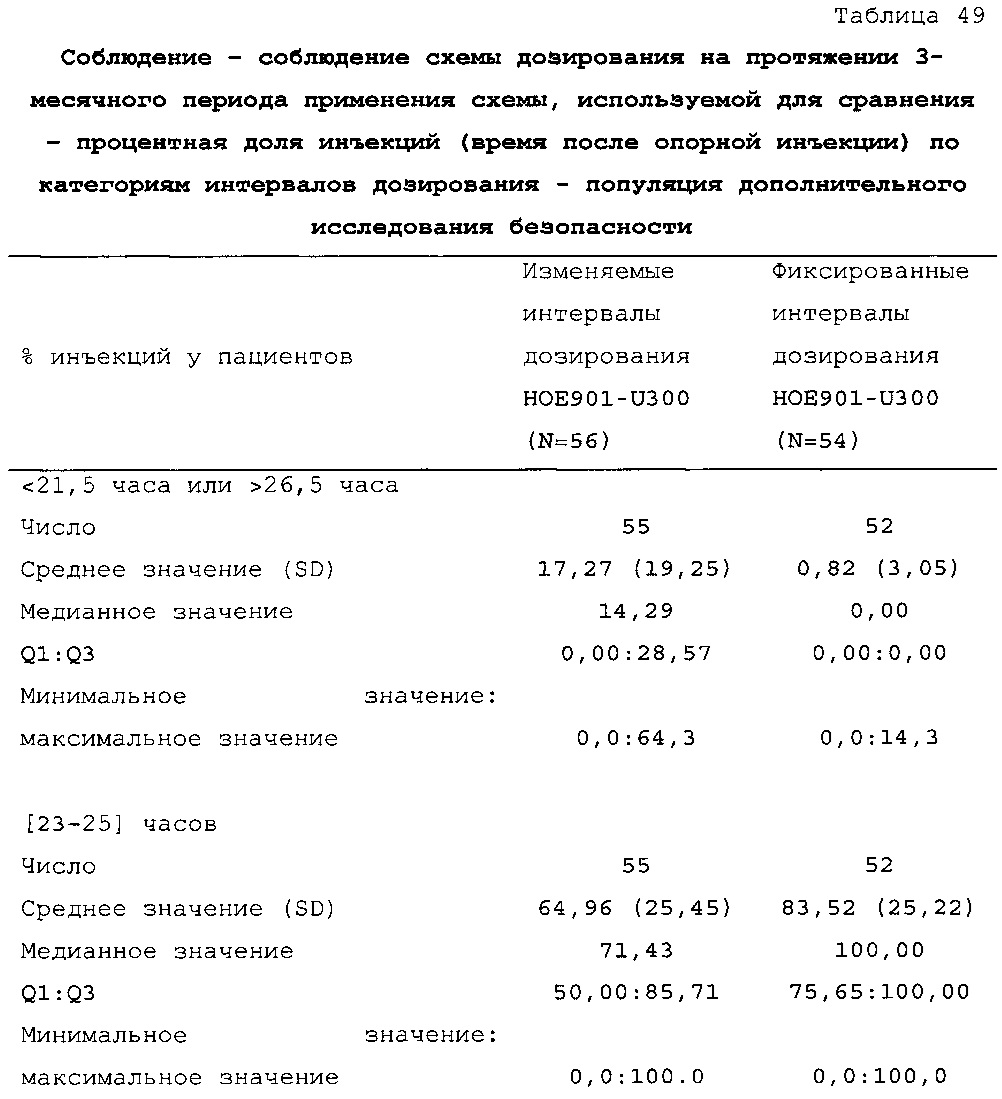

2.2 ОЦЕНКА ЭФФЕКТИВНОСТИ
2.2.1 Первичная конечная точка эффективности



2.2.2 Вторичные конечные точки
2.2.2.1 Концентрация глюкозы в плазме крови натощак


2.2.2.2 Доза базального и прандиального инсулина
Средние суточные дозы базального (гларгин) и прандиального инсулина (ЕД) по визитам на протяжении 3-месячного периода применения схемы, используемой для сравнения, в популяции mITT дополнительного исследования показаны на фигуре 10.
2.3 ОЦЕНКА БЕЗОПАСНОСТИ
2.3.1 Длительность проведения лечения

2.3.2 Гипогликемия
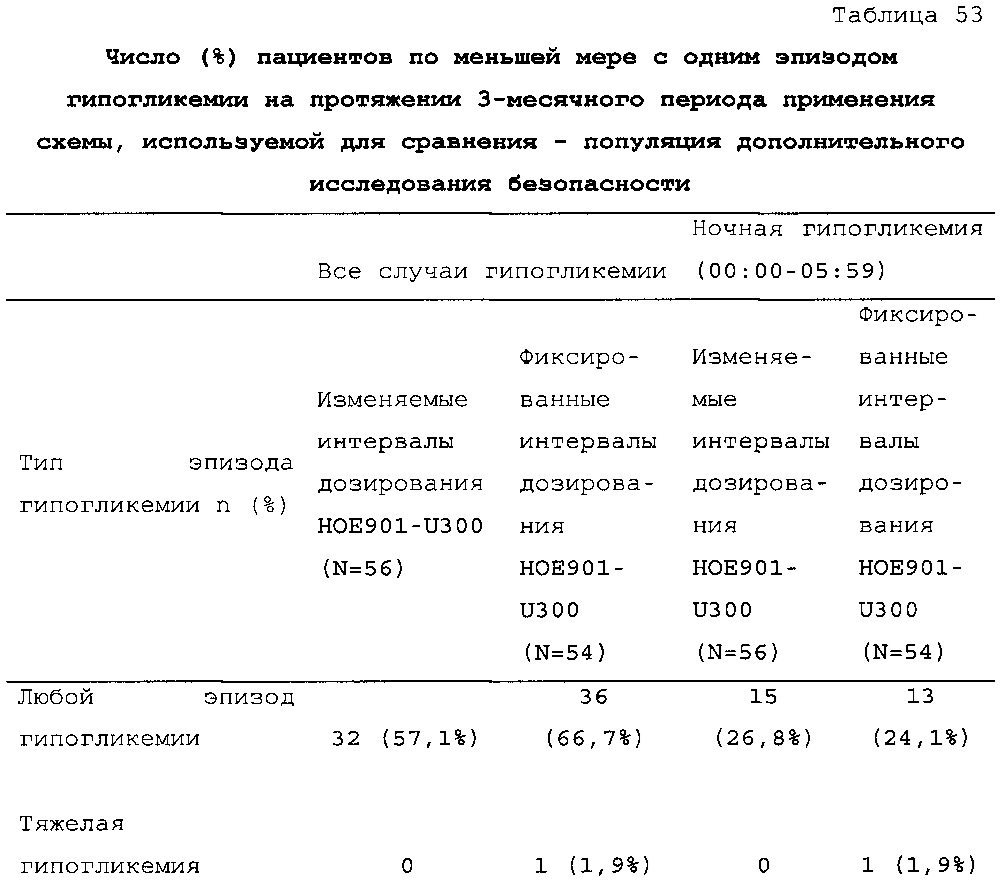
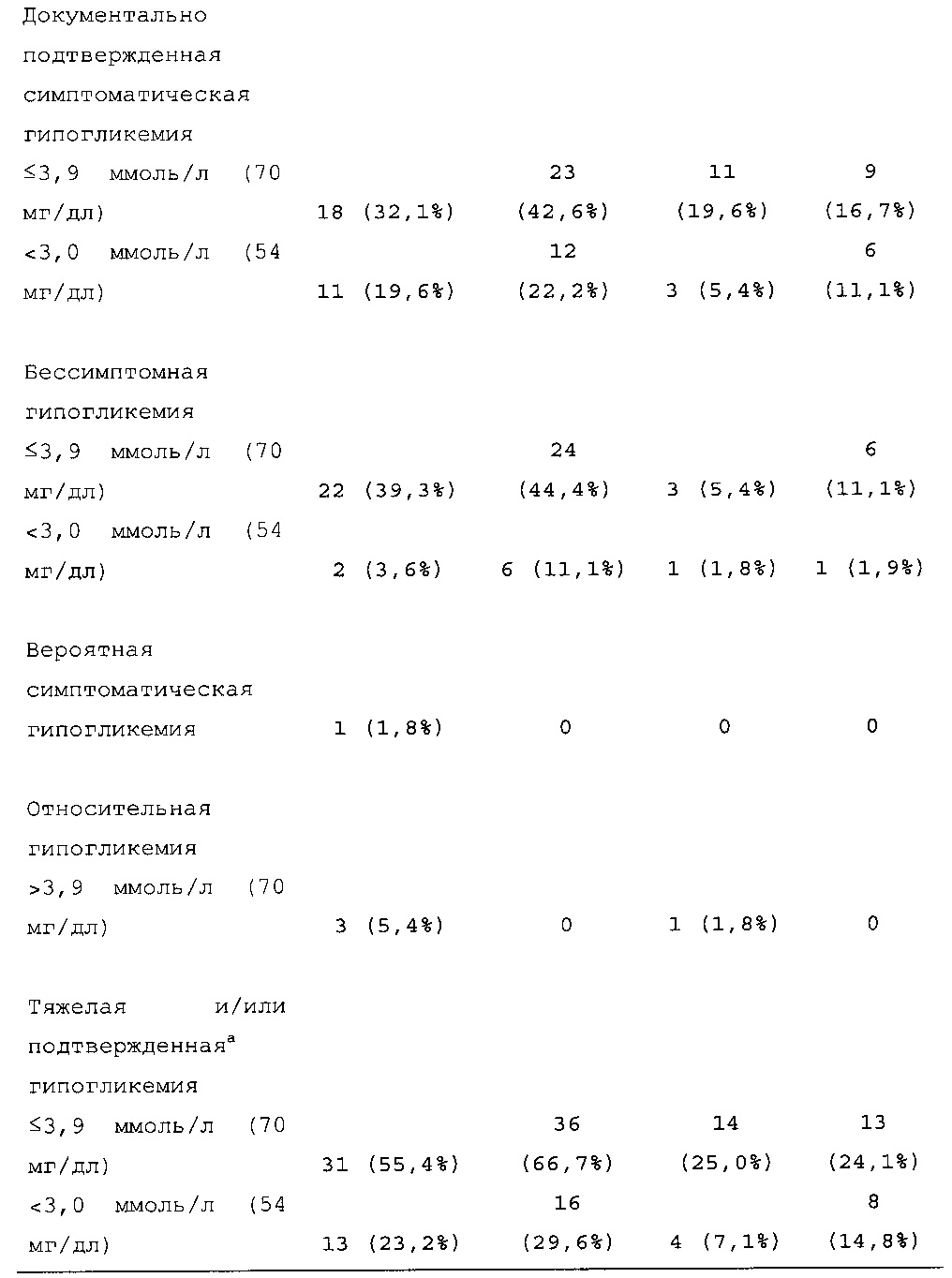

2.3.3 Нежелательные явления, возникшие в ходе лечения
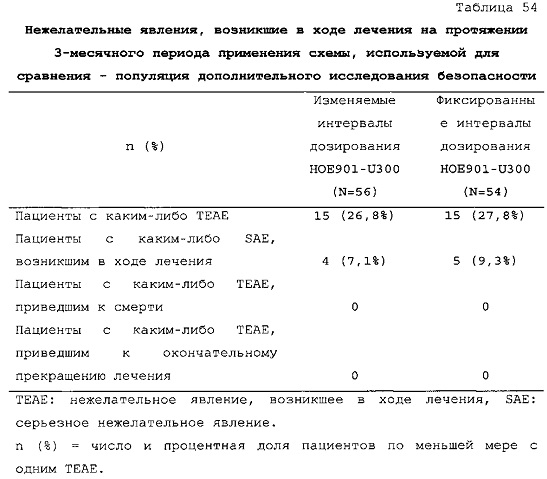







2.3.4 Серьезные нежелательные явления, возникшие в ходе лечения



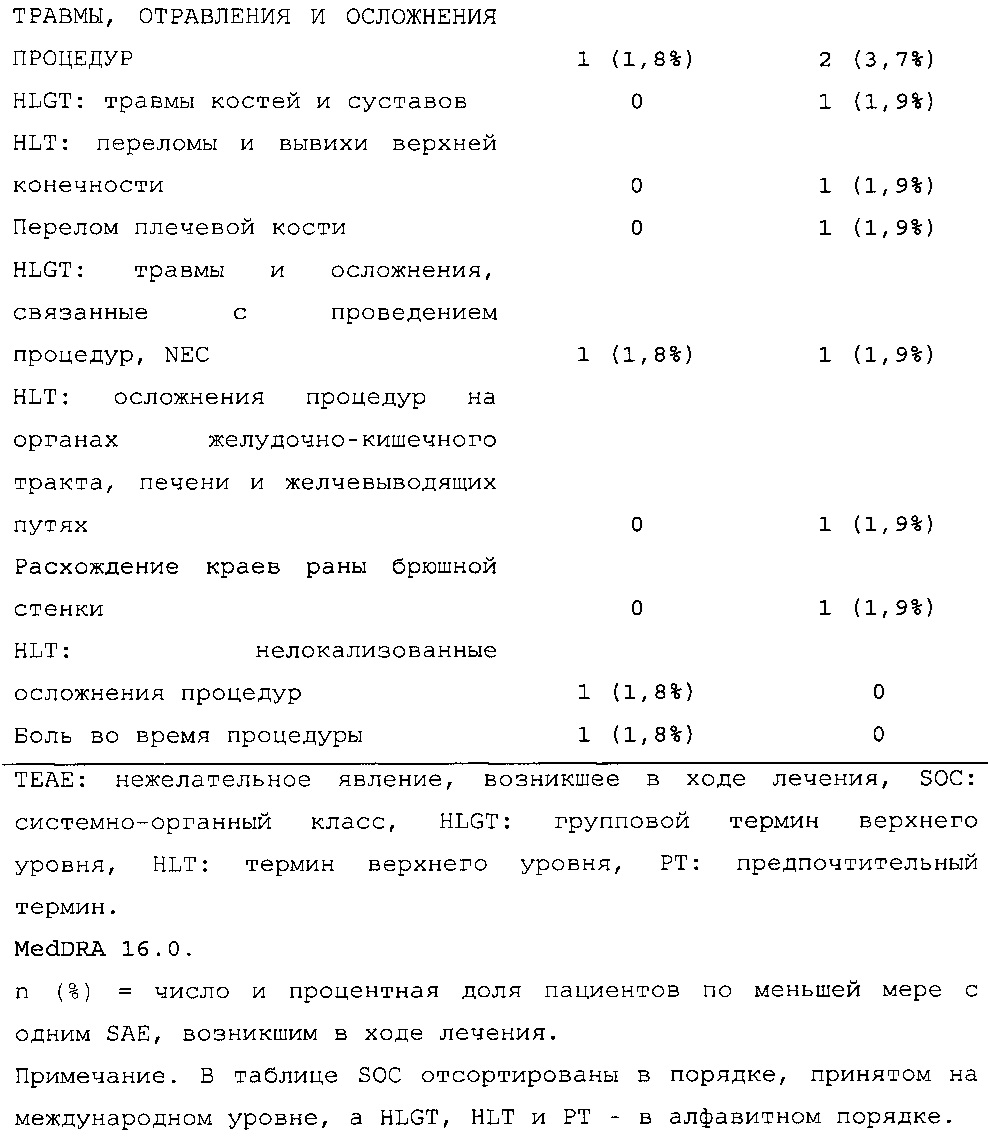
2.3.5 Возникшие в ходе лечения нежелательные явления, приведшие к прекращению участия

2.3.6 Другие значительные нежелательные явления, возникшие в ходе лечения
2.3.6.1 Реакции в месте инъекции

2.3.6.2 Реакции гиперчувствительности

Пример 4. 16-недельное рандомизированное открытое контролируемое исследование, в котором сравнивали эффективность и безопасность нового состава инсулина гларгина и Lantus у пациентов с сахарным диабетом 1 типа
Фаза разработки: 2
Цели
Первичная цель: сравнение контроля концентрации глюкозы в ходе лечения новым составом инсулина гларгина (HOE901-U300) и Lantus у взрослых пациентов с сахарным диабетом 1 типа.
Вторичные цели:
- сравнение HOE901-U300 и Lantus, вводимых утром или вечером, в отношении данных непрерывного контроля концентрации глюкозы (CGM): концентрации глюкозы в течение суток; постоянства концентрации глюкозы в течение суток, измеряемого по скорости изменения на медианной кривой; вариабельности концентрации глюкозы в течение суток, измеряемой по интерквартильному размаху (IQR); среднего значения и вариабельности в профилях концентрации глюкозы;
- сравнение HOE901-U300 и Lantus в отношении уровня гликозилированного гемоглобина A1c (HbA1c), концентрации глюкозы в плазме крови при самостоятельном измерении (концентрации глюкозы в плазме крови натощак перед инъекцией исследуемого лекарственного средства в профилях по 7 моментам времени);
- сравнение количества и частоты приступов гипогликемии, классифицированных согласно определению критериев ADA, как симптоматической, подтвержденной концентрацией глюкозы в плазме крови ≤70 мг/дл, так и выявляемой при CGM;
- оценка безопасности и переносимости HOE901-U300.
Методика. Многоцентровое открытое рандомизированное сравнительное исследование 2 фазы на 4 параллельных группах, в котором сравнивали HOE901-U300 и Lantus у пациентов с сахарным диабетом 1 типа. Пациентов рандомизировали для получения базального инсулина (HOE901-U300 или Lantus) один раз в день с последовательными временами инъекций в ходе периода А исследования и периода В исследования (утром, а затем вечером или вечером, а затем утром) в соотношении 1:1:1:1. Для данного поискового исследования не выполняли формальную оценку объема выборки.
Число пациентов: запланировано 56, рандомизировали 59, получали лечение 59, подвергнуты оценке: эффективности 59, безопасности 59.
Диагностика и критерии включения. Критерии включения: пациенты с сахарным диабетом 1 типа; подписавшие письменное информированное согласие. Ключевые критерии невключения: возраст <18 лет и >70 лет; HbA1c >9% при скрининге; менее 1 года применения базального инсулина в сочетании с прандиальным; пациенты, получавшие >0,5 ЕД/кг массы тела базального инсулина, и пациенты, не применявшие постоянную дозу инсулина (±20% от общей дозы базального инсулина) в течение последних 30 дней перед скрининговым визитом; госпитализация в связи с диабетическим кетоацидозом или тяжелая гипогликемия в анамнезе (потребовавшая помощи третьих лиц) в течение последних 6 месяцев до рандомизации.
Исследуемые средства лечения
Экспериментальные лекарственные препараты: изучаемое лекарственное средство: HOE901-U300; контрольное лекарственное средство: Lantus.
Составы: HOE901-U300 поставляли в виде раствора инсулина гларгина в концентрации 300 ЕД/мл для подкожной (SC) инъекции в картриджах на 3 мл. Lantus поставляли в виде раствора инсулина гларгина в концентрации 100 ЕД/мл для SC инъекции во флаконах на 10 мл.
Путь введения: SC инъекция.
Инъекцию HOE901-U300 делали с помощью коммерчески доступного инсулинового шприца с короткой иглой BD Ultra-Fine™ с шагом шкалы в половину единицы. Инъекцию Lantus необходимо было делать с помощью коммерчески доступных инсулиновых шприцев BD:
для доз инсулина гларгина 1-30 ЕД: инсулиновый шприц с короткой иглой BD Ultra-Fine™ с шагом шкалы в половину единицы [8 мм (5/16'') × 31 G];
для доз инсулина гларгина >30 ЕД: инсулиновый шприц с короткой иглой BD Ultra-Fine™ [8 мм (5/16'') × 31 G] с шагом шкалы в целую единицу.
Схема дозирования: инъекция один раз в день утром или вечером в течение 8 недель в ходе периода А, затем вечером или утром, соответственно, в течение еще 8 недель в ходе периода В согласно рандомизации.
Начальная доза: пациенты, применявшие Lantus, или NPH один раз в день, или инсулин детемир один раз в день перед визитом исходного уровня: суточная доза (ЕД) HOE901-U300 или Lantus была равна суточным дозам базального инсулина в день, предшествующий дню визита исходного уровня. Пациенты, применявшие NPH или инсулин детемир более одного раза в день перед визитом исходного уровня: 80% от общей суточной дозы NPH или инсулина детемира (= общая суточная доза, сниженная на 20%) в день, предшествующий дню визита исходного уровня.
Дозы на протяжении исследования. Дозирование инсулина гларгина, вводимого в виде HOE901-U300 или Lantus, осуществляли, исходя из уровней глюкозы в плазме крови при самостоятельном измерении натощак перед завтраком (целевой диапазон 80-130 мг/дл; 4,4-7,2 ммоль/л), а также принимая в расчет наличие гипогликемии. Минимальный шаг повышения дозы для базального инсулина должен был составлять 1,5 ЕД.
Номер партии: HOE901-U300: С1011129; Lantus: поступал из местных аптек.
Неэкспериментальные лекарственные препараты. Прандиальный (болюсный) аналог инсулина короткого действия (глулизин, аспарт или лизпро).
Пациенты в обеих группах лечения должны были продолжать применять свой прандиальный аналог инсулина в ходе исследования.
Дозы прандиального инсулина необходимо было корректировать, исходя из данных концентрации SMPG, включающих результаты измерения концентрации глюкозы в плазме крови через 2 часа после приема пищи, и содержания углеводов в пище для оптимизации гликемического контроля. Целевой диапазон для концентрации глюкозы в плазме крови через 2 часа после приема пищи составлял <160 мг/дл (8,3 ммоль/л). Дозы болюсного инсулина могли быть уменьшены, тогда как дозы базального инсулина были увеличены.
Продолжительность лечения до 16 недель (8 недель в ходе периода А и 8 недель в ходе периода В).
Продолжительность наблюдения:
до 4 недель скрининга (включая 2 недели периода обучения CGM);
2×8 недель периода сравнения эффективности и безопасности лечения;
4 недели периода последующего наблюдения после лечения для исследования безопасности после окончания исследования или окончательного прекращения лечения в рамках исследования.
В целом, максимальная продолжительность исследования для пациента составляла до 24 недель.
Критерии оценки
Эффективность
Первичная конечная точка эффективности: процент (%) времени нахождения в диапазоне гликемии 80-140 мг/дл (4,4-7,8 ммоль/л) на протяжении 7 и 8 недели в рамках периода лечения А и на протяжении 15 и 16 недели в рамках периода лечения В на основании CGM.
Вторичные конечные точки эффективности: процент времени нахождения выше верхней границы/ниже нижней границы диапазона гликемии (% времени гипергликемии/гипогликемии).
Следующие вторичные конечные точки эффективности не представлены в данном KRM: концентрация глюкозы в течение суток; постоянство концентрации глюкозы в течение суток; вариабельность концентрации глюкозы в течение суток; среднее значение и вариабельность в профилях концентрации глюкозы; среднее время нахождения в диапазоне гликемии в течение последних четырех часов каждого интервала дозирования на протяжении 14 дней применения CGM в последние 2 недели 8-недельного периода лечения; AUC в области гипергликемии (площадь ниже кривой профиля CGM и выше верхней границы диапазона гликемии, деленная на общий период времени) и AUC в области гипогликемии (площадь выше кривой профиля CGM и ниже нижней границы диапазона гликемии, деленная на общий период времени).
Дополнительные вторичные конечные точки эффективности: доза инсулина; не представлены в данном KRM: уровень HbA1c, концентрация глюкозы в плазме крови натощак (FPG), прединъекционная концентрация SMPG, профиль SMPG по 7 моментам времени.
Безопасность
Гипогликемия, частота нежелательных явлений, особенно нежелательных явлений, возникших в ходе лечения (ТЕАЕ), и серьезных нежелательных явлений (SAE), реакций в месте инъекции и реакций гиперчувствительности. Следующая информация не представлена в данном KRM: физикальное обследование, другая информация, касающаяся безопасности, в том числе данные клинико-лабораторных исследований, основные показатели состояния организма и ECG в 12 отведениях.
Статистические методы.
Анализ по первичной конечной точке осуществляли с использованием линейной смешанной модели с лечением (HOE901-U300 или Lantus) и периодом (периодом лечения А или В) в качестве фиксированных эффектов и данных пациентов в качестве случайного эффекта. Будут представлены скорректированные оценки средних значений для каждого вида лечения со стандартными ошибками, скорректированная оценка разницы средних значений между видами лечения со стандартной ошибкой и 95% доверительный интервал для разницы средних значений между видами лечения. Статистическими критериями были двусторонние критерии с 5% номинальным уровнем значимости. Эту же модель использовали для вторичных конечных точек эффективности: % времени гипергликемии/гипогликемии, концентрации глюкозы в течение суток, постоянства концентрации глюкозы в течение суток и вариабельности концентрации глюкозы в течение суток. Другие конечные точки эффективности были описательными. Параметры, связанные с CGM, анализировали на основе популяции CGM, параметры, не связанные с CGM, основывались на популяции mITT.
Анализы безопасности были описательными, основанными на популяции для оценки безопасности.
Краткое описание
Характеристики популяции
В общей сложности 59 пациентов с сахарным диабетом 1 типа рандомизировали в 1 из 4 групп: группа утренней инъекции НОЕ901-U300 в период А с последующей вечерней инъекцией в период В (n=15), группа вечерней, а затем утренней инъекции HOE901-U300 (n=15), группа утренней, а затем вечерней инъекции Lantus (n=15) или группа вечерней, а затем утренней инъекции Lantus (n=14). В общей сложности 5 9 пациентов подвергали воздействию IMP (популяция для оценки безопасности) и включали в популяции mITT и CGM (популяции для оценки эффективности). Один пациент (3,3%) в группе HOE901-U300 и 3 пациента (10,3%) в группе Lantus досрочно прекратили лечение в рамках исследования.
Демографические данные и исходные параметры были сбалансированы среди групп лечения. Средний возраст в исследуемой популяции составлял 44,2 года, у 2 пациентов он составлял 65 лет или более. Все пациенты были европеоидами. Среднее значение BMI в начале исследования составляло 27,3 кг/м2. Средняя продолжительность сахарного диабета до начала исследования составляла 22,1 года. Медианное значение общей суточной дозы инсулина составляло 0,565 ЕД/кг массы тела. Среднее значение уровня HbA1c в начале исследования составляло 7,46%.
Результаты в отношении эффективности
Первичная конечная точка эффективности. На протяжении последних 2 недель каждого 8-недельного периода лечения, когда дозу базального инсулина необходимо было поддерживать как можно более постоянной, наблюдаемая концентрация глюкозы в плазме крови, измеряемая с помощью CGM, находилась в пределах диапазона гликемии на протяжении 31,75% (среднее значение, полученное методом LS) времени в группе HOE901-U300 и на протяжении 30,99% (среднее значение, полученное методом LS) времени в группе Lantus. Разница между полученными методом LS средними значениями составляла 0,75% [95% CI: от -3,614 до 5,124].
Вторичные конечные точки эффективности. На протяжении последних 2 недель каждого 8-недельного периода лечения процент времени нахождения выше верхней границы диапазона гликемии 140 мг/дл (7,8 ммоль/л) был сопоставимым среди групп лечения (среднее значение, полученное методом LS, составляло 58,24% в группе HOE901-U300 и 57,38% в группе Lantus), так же как и процент времени нахождения ниже нижней границы 80 мг/дл (4,4 ммоль/л) при среднем его значении, полученном методом LS, составлявшим 10,01% в группе HOE901-U300 и 11,64% в группе Lantus.
Графическое изображение средней концентрации глюкозы на основе CGM в зависимости от времени суток в течение всего периода лечения (фигура 11) свидетельствует о меньшем отклонении в группе HOE901-U300 по сравнению с группой Lantus. Профиль оказывается более плоским для HOE901-U300, чем для Lantus, в особенности в течение периода утренних инъекций (фигура 12) по сравнению с периодом вечерних инъекций (фигура 13).
В целом, в группах лечения HOE901-U300 и Lantus доза базального инсулина увеличивалась сходным образом преимущественно в первые 6 недель исследования и оставалась относительно постоянной в дальнейшем (в начале исследования среднее значение суточной дозы базального инсулина было сходным в обеих группах лечения: HOE901-U300: 24,9 единицы; Lantus: 25,0 единицы; на 16 неделе - HOE901-U300: 30,11 единицы; Lantus: 28,22 единицы).
Среднее значение суточной дозы прандиального инсулина было более высоким в начале исследования в группе HOE901-U300 (29,92 единицы), чем в группе Lantus (23,69 единицы), но было сопоставимым на 16 неделе (HOE901-U300: 27,34 единицы; Lantus: 26,31 единицы).
Результаты в отношении безопасности
В течение периода применения исследуемого препарата процентные доли пациентов с гипогликемией были в целом сопоставимы по эпизодам гипогликемии в целом и по каждой их разновидности (все случаи гипогликемии) в группе HOE901-U300 и группе Lantus. В сообщениях о гипогликемии наблюдалось неизменное сходство между следующими подгруппами:
группы утренних и вечерних инъекций в группе HOE901-U300;
группы утренних и вечерних инъекций в группе Lantus;
группа утренних инъекций HOE901-U300 и группа утренних инъекций Lantus;
группа вечерних инъекций HOE901-U300 и группа вечерних инъекций Lantus.
Процентные доли пациентов с ночной гипогликемией были неизменно более низкими в группе HOE901-U300, чем в группе Lantus, вне зависимости от того, утренним или вечерним было время инъекции. Благоприятные количественные тенденции в группе HOE901-U300 следует интерпретировать с осторожностью по причине небольшого числа пациентов.
Процентная доля пациентов с каким-либо ТЕАЕ была более высокой в группе HOE901-U300 (24/30 [80,0%]), чем в группе Lantus (19/29 [65,5%]). В группе HOE901-U300 один пациент испытывал тяжелую непроходимость кишечника (не связанную с IMP), а другая пациентка прекратила лечение в связи с беременностью. В ходе исследования не сообщалось о случаях смерти. ТЕАЕ, связанные с реакциями в месте инъекции, наблюдали у 2/30 [6,7%] пациентов из группы HOE901-U300 и у 1/29 [3,4%] пациентов из группы Lantus. Не наблюдалось проблем в отношении ТЕАЕ, связанных с реакцией гиперчувствительности, которые встречались у 4/40 пациентов из группы HOE901-U300 и у 1/30 пациентов из группы Lantus.
Выводы
Наблюдаемая концентрация глюкозы в плазме крови, измеряемая с помощью CGM, находилась в пределах целевого диапазона гликемии (80-140 мг/дл или 4,4-7,8 ммоль/л) в течение сходного процента времени на протяжении последних 2 недель каждого 8-недельного периода лечения в группе HOE901-U300 и в группе Lantus. Примечательно, что данный целевой диапазон был более узким по сравнению с рекомендуемым ADA, составляющим 70-180 мг/дл (3,9-10,0 ммоль/л).
В обеих группах лечения процент времени, проведенного выше верхней границы диапазона гликемии, был более высоким (57%-58%), чем процент времени, проведенного ниже нижней границы целевого диапазона (10-11%).
В целом, процентная доля пациентов по меньшей мере с одним эпизодом вне зависимости от разновидности гипогликемии в ходе исследования была сопоставима в обеих группах лечения (НОЕ901-U300, Lantus) и для обоих времен инъекции (утреннего или вечернего). Благоприятные количественные тенденции в группе HOE901-U300 для ночной гипогликемии следует интерпретировать с осторожностью по причине небольшого числа пациентов.
HOE901-U300 и Lantus, которые вводили утром либо вечером, хорошо переносились в ходе периода исследования, и особых проблем безопасности не наблюдалось.
Пример 5. Экспериментальный новый инсулин U300: контроль концентрации глюкозы и гипогликемия при сахарном диабете 2 типа с применением базального инсулина (II версия)
Цели. Экспериментальный новый инсулин U300, имеющий еще более плоский профиль и подвергающийся более длительным процессам PK/PD, чем инсулин гларгин 100 ЕД/мл (U100), находится на стадии клинических исследований. В исследовании 3 фазы II версии сравнивали эффективность и безопасность U300 и U100 у людей с T2DM, применяющих схему введения базального инсулина в сочетании с OAD.
Способы. В этом многоцентровом открытом 6-месячном исследовании участников рандомизировали (1:1) для применения U300 или U100 один раз в день вечером. Дозу инсулина подбирали до достижения целевой концентрации глюкозы в плазме крови натощак (FPG) 80-100 мг/дл. Первичной конечной точкой являлось изменение уровня HbA1c от начала исследования до 6 месяцев, а 1-й основной вторичной конечной точкой эффективности в иерархическом анализе являлась процентная доля участников исследования с ≥1 эпизодом тяжелой или подтвержденной (≤70 мг/дл) ночной (24:00-05:59 ч.) гипогликемии от 9 недели до 6 месяца.
Результаты. Рандомизировали 811 участников [средний возраст 58,2 (SD 9,2) лет, продолжительность сахарного диабета 12,6 (7,0) лет, BMI 34,8 (6,4) кг/м2, доза базального инсулина 0,67 (0,24) ЕД/кг]. HbA1c в начале исследования был сопоставим среди групп; U300: 8,26% (0,86) по сравнению с U100: 8,22% (0,77). U300 характеризовался не меньшей эффективностью, чем U100, в отношении изменения уровня HbA1c [изменение полученного методом LS среднего значения -0,57% (SE: 0,09) и -0,56% (SE: 0,09), соответственно, к 6 месяцу; разница -0,01 (95% CI: от -0,14% до +0,12%)]. Не наблюдалось значимых различий для FPG, профилей концентрации глюкозы в плазме крови при самоконтроле по 8 моментам времени и прединъекционной концентрации глюкозы в плазме крови. Процентная доля участников с тяжелой или подтвержденной ночной гипогликемией была значительно ниже для U300 по сравнению с U100 от 9 недели до 6 месяца [21,6% по сравнению с 27,9%; относительный риск (RR) 0,77 (95% CI: от 0,61 до 0,99); р=0,038]. В течение 6-месячного периода лечения частота возникновения какой-либо ночной гипогликемии (% участников с ≥1 эпизодом) была ниже для U300 по сравнению с U100 [30,5% по сравнению с 41,6%; RR 0,73 (95% CI: от 0,60 до 0,89)], равно как и частота возникновения любого эпизода гипогликемии в любое время суток (24 ч.) [U300 71,5%; U100 79,3%; RR 0,90 (95% CI: от 0,84 до 0,97)]. О тяжелой гипогликемии в любое время суток сообщали 1,0% участников в группе U300 и 1,5% в группе U100. Между видами лечения не наблюдалось различий относительно серьезных нежелательных явлений.
Выводы: У людей с T2DM, применявших схему введения базального инсулина в сочетании с OAD, U300 хорошо переносился и был таким же эффективным, как и U100, в отношении контроля уровня глюкозы в крови. U300 был связан с 23% снижением частоты возникновения тяжелой или подтвержденной ночной гипогликемии от 9 недели до 6 месяца по сравнению с U100 и с более низкой частотой возникновения любого эпизода ночной гипогликемии и гипогликемии в любое время суток (24 ч.) на протяжении всего 6-месячного исследования.
Пример 6. 6-месячное многоцентровое рандомизированное открытое исследование на параллельных группах, в котором сравнивали эффективность и безопасность нового состава инсулина гларгина и Lantus, оба из которых применяли в сочетании с пероральным(и) гипогликемическим(и) средством(ами) у пациентов с сахарным диабетом 2 типа, с 6-месячным продленным периодом исследования безопасности - дополнительное исследование введения, в котором сравнивали изменяемые интервалы дозирования с фиксированными интервалами дозирования
1 ОБЩЕЕ ОПИСАНИЕ ИССЛЕДОВАНИЯ
Фаза разработки: дополнительное исследование для основного исследования 3 фазы
Цели
Первичная цель: сравнение эффективности HOE901-U300, инъецируемого один раз в день каждые 24 часа, и HOE901-U300, инъецируемого один раз в день с интервалами 24±3 часа, с точки зрения изменения уровня HbA1c от 6 месяца основного исследования (= начало дополнительного исследования) до 9 месяца основного исследования (= конечная точка дополнительного исследования) у пациентов с сахарным диабетом 2 типа.
Основные вторичные цели: сравнение безопасности двух схем инъекций HOE901-U300 с точки зрения частоты гипогликемии.
Методика
Пациентов, рандомизированных в группу HOE901-U300 и получавших HOE901-U300 на протяжении 6-месячного периода основного исследования, рандомизировали в соотношении 1:1 для введения HOE901-U300 один раз в день каждые 24 часа (фиксированные интервалы дозирования) либо каждые 24±3 часа (изменяемые интервалы дозирования).
Пациенты, применявшие HOE901-U300, завершившие 6-месячный период основного исследования и отвечающие критериям отбора для дополнительного исследования, были допущены к дополнительному исследованию. Для первичного анализа, который был описательным, не требовался конкретный объем выборки.
Число пациентов Запланировано: до 300 (150 в группе предварительного лечения). Рандомизировано: 89
Получали лечение: 87
Подвергнуты оценке: эффективности: 86, безопасности: 87
Диагностика и критерии включения
Критерии включения: завершение 6-месячного периода исследования в основном исследовании (визит 10), рандомизация и получение лечения с помощью HOE901-U300 на протяжении 6-месячного периода лечения (начало исследования - 6 месяц), полученное подписанное письменное информированное согласие на участие в дополнительном исследовании.
Ключевые критерии невключения: нежелание пациента применять изменяемые интервалы между инъекциями, составляющие 24±3 часа, в течение по меньшей мере двух дней в неделю; невозможность соблюдения изменяемой схемы дозирования по мнению исследователя; состояние здоровья, препятствующее дальнейшему участию пациента в исследовании.
Исследуемые средства лечения
Экспериментальный лекарственный препарат: изучаемое лекарственное средство: HOE901-U300
Состав: HOE901-U300 (раствор инсулина гларгина, 300 ЕД/мл) представляет собой стерильный апирогенный прозрачный бесцветный раствор в стеклянном картридже, укомплектованном в шприц-ручку (предварительно наполненную, одноразовую ручку).
Путь введения: подкожная инъекция.
Исследуемая схема
Изменяемые интервалы дозирования: введение HOE901-U300 один раз в день каждые 24±3 часа.
Время инъекции могло быть изменено в зависимости от потребностей индивидуума в пределах до 3 часов до или после фиксированного в начале основного исследования опорного времени ежедневной инъекции вечером. Максимальные интервалы, т.е. предполагающие ежедневную инъекцию за 3 часа до или спустя 3 часа после фиксированного опорного времени, необходимо было применять в течение по меньшей мере 2 дней в неделю по выбору пациента. Зафиксированное в начале основного исследования время инъекции должно было оставаться опорным временем для варьирования.
Контрольная схема
Фиксированные интервалы дозирования: инъекция HOE901-U300 один раз в день каждые 24 часа.
Пациенты продолжали инъецировать HOE901-U300 один раз в день каждые 24 часа в фиксированное в начале основного исследования время инъекции.
Доза
Следовало подобрать дозу HOE901-U300, необходимую для достижения или поддержания концентрации глюкозы в плазме крови натощак в целевом диапазоне от 80 до 100 мг/дл (от 4,4 ммоль/л до 5,6 ммоль/л) без гипогликемии. Изменения дозы инсулина основывались на данных измерений концентрации глюкозы в плазме капиллярной крови натощак при самоконтроле (SMPG).
Неэкспериментальные лекарственные препараты
Для обеих схем лечения пациенты должны были продолжать свою базовую терапию пероральным гипогликемическим средством в ходе участия в дополнительном исследовании. На протяжении всего исследования дозы нужно было поддерживать постоянными, если не было особой проблемы безопасности, связанной с этими средствами лечения. В этом исследовании не нужно было применять никаких других сопутствующих противодиабетических средств лечения.
Кратковременное применение (т.е. не более 10 дней) терапии инсулином короткого действия (т.е. в связи с острой болезнью или хирургическим вмешательством) не считалось резервной терапией. Лекарственное вещество для резервной терапии считалось неэкспериментальным лекарственным препаратом.
Продолжительность лечения. Дополнительное исследование состояло из 3-месячного периода сравнения эффективности и безопасности лечения, начинавшегося по окончании 6-месячного периода основного исследования и заканчивавшегося по окончании 9 месяца основного исследования.
После окончания пациенты, входившие в группу с изменяемой схемой дозирования, могли продолжать придерживаться этой схемы до конца основного исследования (12 месяца). Пациенты, инъецировавшие HOE901-U300 каждые 24 часа, продолжали придерживаться своей схемы лечения до конца основного исследования.
Продолжительность наблюдения. Периодом анализа
эффективности и безопасности являлся 3-месячный период исследования, начинавшийся на 6 месяце основного исследования и заканчивавшийся на 9 месяце основного исследования. Результаты, представленные в настоящем KRM, относятся к этому периоду.
Критерии оценки
Первичная конечная точка эффективности: изменение уровня HbA1c от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц).
Вторичные конечные точки эффективности: изменение
концентрации FPG (центральная лаборатория) и изменение суточной дозы базального инсулина от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц).
Безопасность: гипогликемия (в том числе ночная), частота нежелательных явлений, особенно нежелательных явлений, возникших в ходе лечения (ТЕАЕ), и серьезных нежелательных явлений (SAE), ТЕАЕ, приведших к прекращению участия, реакций в месте инъекции и реакций гиперчувствительности. Следующая информация не представлена в KRM: другая информация, касающаяся безопасности, в том числе об основных показателях состояния организма и передозировке.
Статистические методы
Для данного 3-месячного дополнительного исследования в качестве начала определен 6 месяц основного исследования; конечной точкой, определенной с помощью процедуры переноса данных последнего наблюдения вперед (LOCF), являлся 9 месяц основного исследования.
Анализ первичной конечной точки эффективности (изменения уровня HbA1c от начала дополнительного исследования [6 месяц] до конечной точки [9 месяц]) осуществляли с использованием модели ковариационного анализа (ANCOVA) со схемой лечения и страной в качестве фиксированных эффектов и с использованием исходного значения уровня HbA1c в качестве ковариаты. Различия между изменяемой схемой дозирования HOE901-U300 и фиксированной схемой дозирования HOE901-U300 и двусторонние 95% доверительные интервалы оценивали в рамках ANCOVA.
Все непрерывные переменные вторичной эффективности анализировали с использованием модели ANCOVA со схемой лечения и страной в качестве фиксированных эффектов и с использованием соответствующего исходного значения в качестве ковариаты.
Анализы безопасности были описательными, основанными на популяции для оценки безопасности.
Краткое описание
Характеристики популяции
В общей сложности 89 пациентов с сахарным диабетом 2 типа рандомизировали для дополнительного исследования: 45 пациентов для применения HOE901-U300 по схеме с изменяемыми интервалами дозирования и 44 пациента для применения HOE901-U300 по схеме с фиксированными интервалами дозирования; 87 пациентов подвергали воздействию IMP (популяция для оценки безопасности). Популяция mITT дополнительного исследования (популяция для оценки эффективности) включала 86 пациентов.
В общей сложности 40 (88,9%) пациентов, рандомизированных для применения HOE901-U300 по схеме с изменяемыми интервалами дозирования, и 38 (86,4%) пациентов, рандомизированных для применения HOE901-U300 по схеме с фиксированными интервалами дозирования, завершили 3-месячный период применения схемы, используемой для сравнения. 1 пациент (2,2%), рандомизированный для применения HOE901-U300 по схеме с изменяемыми интервалами дозирования, и 2 пациента (4,5%), рандомизированные для применения HOE901-U300 по схеме с фиксированными интервалами дозирования, досрочно прекращали участие в дополнительном исследовании и также прекращали участие в продленном периоде основного исследования.
Демографические данные и параметры пациентов в начале дополнительного исследования (6 месяц) были сбалансированы среди обеих групп применения схем. Средний возраст в популяции дополнительного исследования составлял 57,8 лет; у 16 из 89 (18,0%) пациентов он составлял ≥65 лет. 3 пациента, применяющие схему с изменяемыми интервалами дозирования, и 2 пациента, применяющие схему с фиксированными интервалами дозирования, начинали прием инсулина или стимулятора секреции инсулина в качестве резервной терапии на протяжении периода основного исследования. Ни один пациент ни в одной из групп применения схем не начинал резервную терапию на протяжении 3-месячного периода применения схемы, используемой для сравнения.
Соблюдение схем дозирования с интервалами
Соблюдение схемы дозирования с интервалами оценивали, принимая в расчет временной интервал между 2 последовательными инъекциями и временной интервал между инъекцией и опорным временем инъекции, запланированным в начале основного исследования.
В ходе дополнительного исследования в группе изменяемых интервалов дозирования с предельными интервалами <21,5 часа или >26,5 часа между 2 последовательными инъекциями в среднем было произведено 28,04% инъекций в расчете на одного пациента по сравнению с 2,41% инъекций для пациентов в группе фиксированных интервалов дозирования, при этом в группе фиксированных интервалов дозирования в пределах интервала 23-25 часов между 2 последовательными инъекциями было произведено 88,77% инъекций в расчете на одного пациента по сравнению с 53,09% инъекций в расчете на одного пациента в группе изменяемых интервалов дозирования.
Оценка временных интервалов между фактическим временем инъекции и опорным временем инъекции показала, что процентная доля инъекций в расчете на одного пациента в пределах интервала 23-25 часов в группе фиксированных интервалов дозирования (среднее значение 65,07%) является более высокой по сравнению с группой изменяемых интервалов дозирования (56,38%). В группе изменяемых интервалов дозирования 21,69% инъекций в расчете на одного пациента было произведено с интервалами 21,5-23 часа или с интервалами 25-26,5 часа (25,51% в группе фиксированных интервалов дозирования). Эти данные позволяют предположить, что большинство инъекций было произведено в промежутке до 3 часов до или после опорного времени инъекции, которое должно было быть зафиксировано вечером согласно протоколу.
На основании документально подтвержденных времен инъекции в течение недели, предшествующей визитам в 7,5 месяце и в 9 месяце, было определено, что в общей сложности 47,5% пациентов в группе изменяемых интервалов дозирования производили инъекции так, что 4 или более интервала между ними находились за пределами интервала 21,5-26,5 часа после предыдущей инъекции, и, следовательно, считались соблюдавшими схему с изменяемыми интервалами дозирования по сравнению с 2,6% пациентов, применяющих схему с фиксированными интервалами дозирования. В группе фиксированных интервалов дозирования у 61,5% пациентов все интервалы между последовательными инъекциями находились в пределах 23-25 часов, и, следовательно, пациенты могли считаться соблюдающими схему с фиксированными интервалами дозирования.
Эффективность
Первичная конечная точка эффективности (9 месяц): Изменение полученного методом LS среднего значения уровня HbA1c от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц) было сходным в группах изменяемых интервалов дозирования (-0,12% [95% CI: от -0,422 до 0,183]) и фиксированных интервалов дозирования (-0,25% [95% CI: от -0,574 до 0,072]), при этом разница между полученными методом LS средними значениями составляла 0,13% [95% CI: от -0,152 до 0,415].
Вторичные конечные точки эффективности (9 месяц): изменение полученного методом LS среднего значения концентрации FPG от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц) было сходным в группах изменяемых интервалов дозирования (-0,46 ммоль/л [95% CI: от -1,521 до 0,609]) и фиксированных интервалов дозирования (-0,25 ммоль/л [95% CI: от -1,378 до 0,881]), при этом разница между полученными методом LS средними значениями составляла -0,21 [95% CI: от -1,200 до 0,784].
В обеих схемах дозирования с интервалами средняя суточная доза базального инсулина оставалась постоянной на протяжении 3-месячного периода применения схемы, используемой для сравнения.
Безопасность
На протяжении 3-месячного периода сравнительного дополнительного исследования об эпизодах гипогликемии сообщали 16/44 (36,4%) пациентов, применяющих схему с изменяемыми интервалами дозирования, и 18/43 (41,9%) пациентов, применяющих схему с фиксированными интервалами дозирования. О каждой разновидности эпизодов гипогликемии сообщала сопоставимая процентная доля пациентов для обеих схем. Ни для одной схемы не наблюдались эпизоды тяжелой гипогликемии или тяжелой ночной гипогликемии.
Процентные доли пациентов с каким-либо ТЕАЕ (изменяемые интервалы дозирования - 9/44 [20,5%]; фиксированные интервалы дозирования - 11/43 [25,6%]) или с серьезным ТЕАЕ (изменяемые интервалы дозирования - 2/44 [4,5%]; фиксированные интервалы дозирования - 0/43) были сопоставимыми среди различных схем.
ТЕАЕ, приведшие к прекращению лечения или к смерти или связанные с местной реакцией на инъекцию или с реакцией гиперчувствительности, не наблюдались ни для одной схемы дозирования с интервалами на протяжении 3-месячного периода дополнительного исследования.
Выводы
Оценка продолжительности интервалов между инъекциями и % пациентов, применявших более короткие или более длинные интервалы между инъекциями по сравнению с обычным 2 4-часовым периодом, позволяет предположить, что большинство пациентов в обеих группах применения схем дозирования с интервалами следовали графику инъекций в зависимости от группы рандомизации и применяли изменяемые интервалы дозирования (HOE901-U300 один раз в день каждые 24 часа ± 3 часа в течение по меньшей мере 2 дней в неделю) либо фиксированные интервалы дозирования (HOE901-U3 00 один раз в день каждые 24 часа). Это обеспечивало возможность сравнения двух схем дозирования с интервалами для анализов эффективности и безопасности.
Анализы эффективности с точки зрения изменения уровня HbA1c и концентрации FPG от начала дополнительного исследования (6 месяц) до конечной точки (9 месяц) показали сопоставимые результаты для двух схем дозирования с интервалами.
Общая частота возникновения гипогликемии (% пациентов по меньшей мере с одним эпизодом) на протяжении 3-месячного периода дополнительного исследования была сопоставимой для обеих схем вне зависимости от разновидности гипогликемии.
HOE901-U300, вводимый с изменяемыми либо с фиксированными интервалами дозирования, хорошо переносился на протяжении 3-месячного периода сравнительного дополнительного исследования; особых проблем безопасности не наблюдалось.
В совокупности результаты дополнительного исследования позволяют предположить, что периодическое изменение интервалов между инъекциями в пределах до 3 часов до или после опорного времени для инъекции HOE901-U300 один раз в день не оказывало эффектов в отношении основных конечных точек эффективности (HbA1c) и безопасности, в частности, эпизодов гипогликемии, по сравнению с инъекциями один раз в день с 24-часовыми интервалами.
2. РЕЗУЛЬТАТЫ
2.1 ПАЦИЕНТЫ-УЧАСТНИКИ ИССЛЕДОВАНИЯ
2.1.1 Распределение участников исследования



2.1.2 Демографические данные и исходные параметры
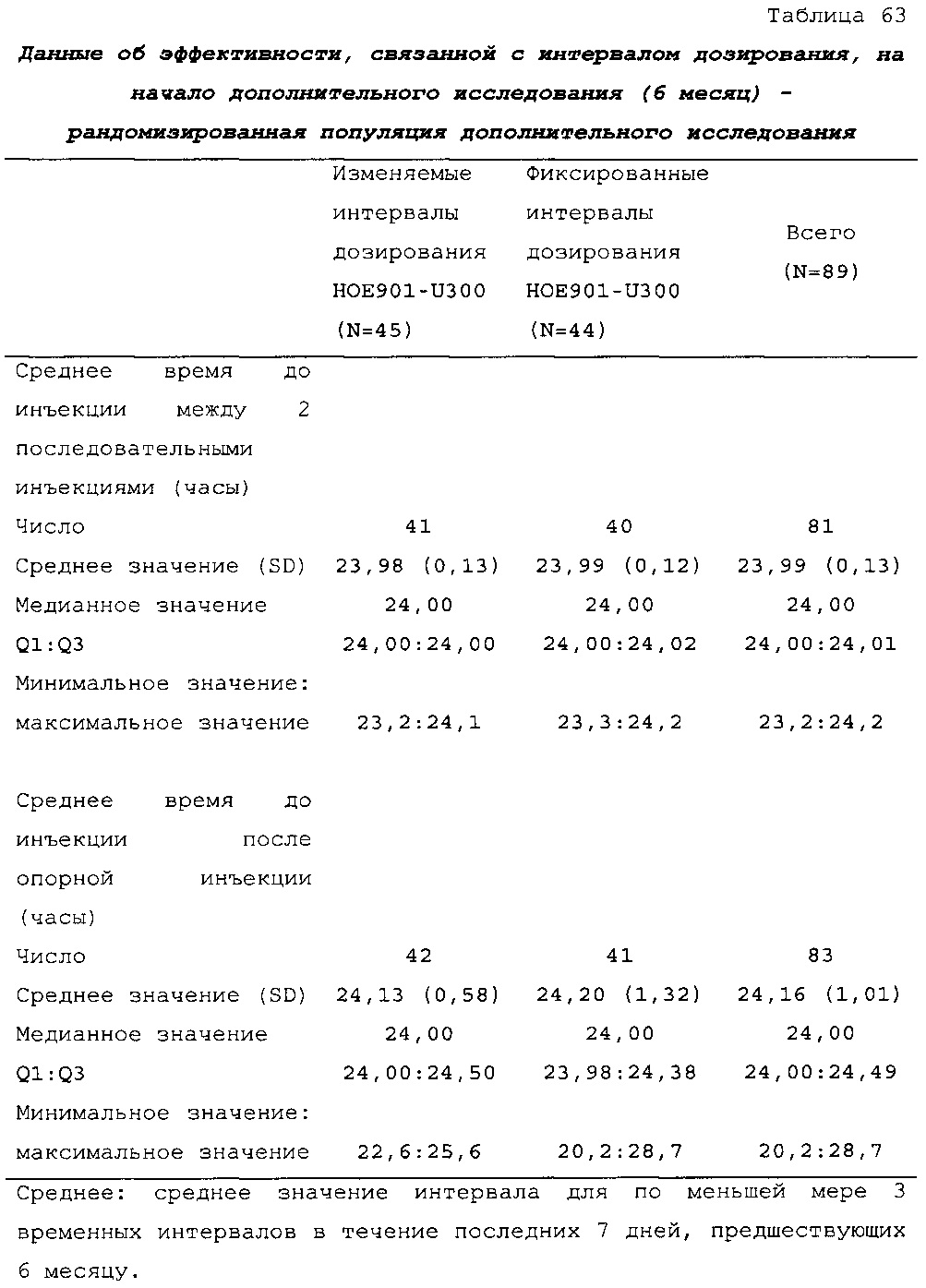
2.1.3 Соблюдение схемы дозирования с интервалами


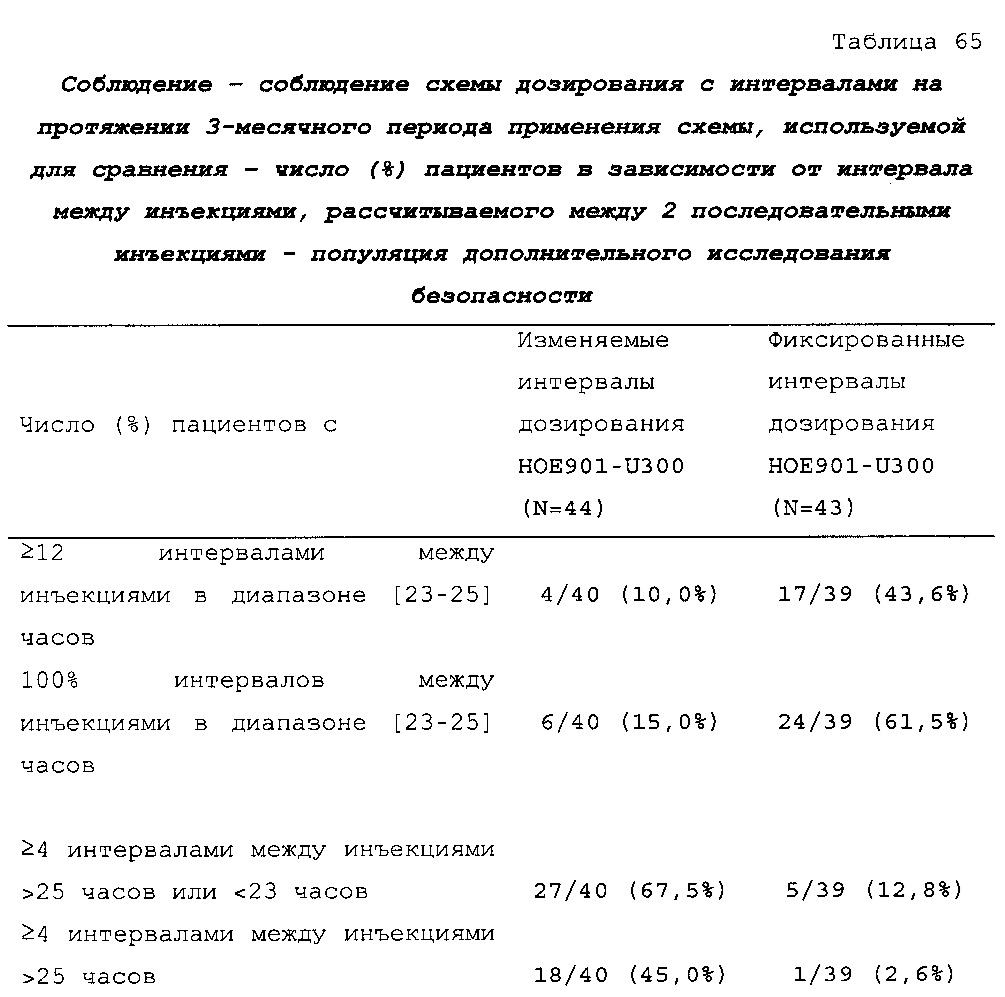


2.2 ОЦЕНКА ЭФФЕКТИВНОСТИ
2.2.1 Первичная конечная точка эффективности
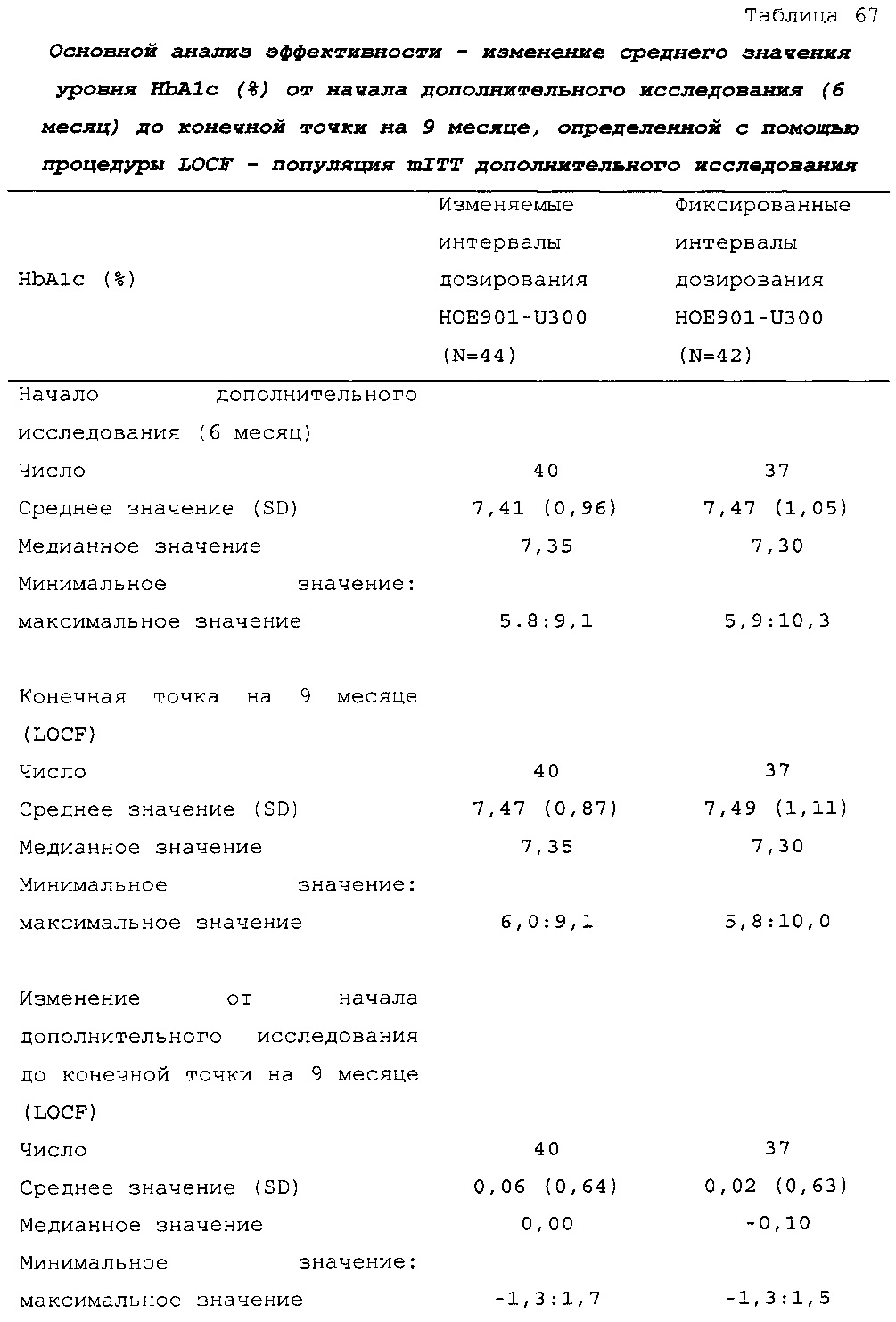

На фигуре 14 показаны средние значения уровня HbA1c (%) по визитам на протяжении 3-месячного периода применения схемы, используемой для сравнения, в популяции mITT дополнительного исследования.
2.2.2 Вторичные конечные точки
2.2.2.1 Концентрация глюкозы в плазме крови натощак
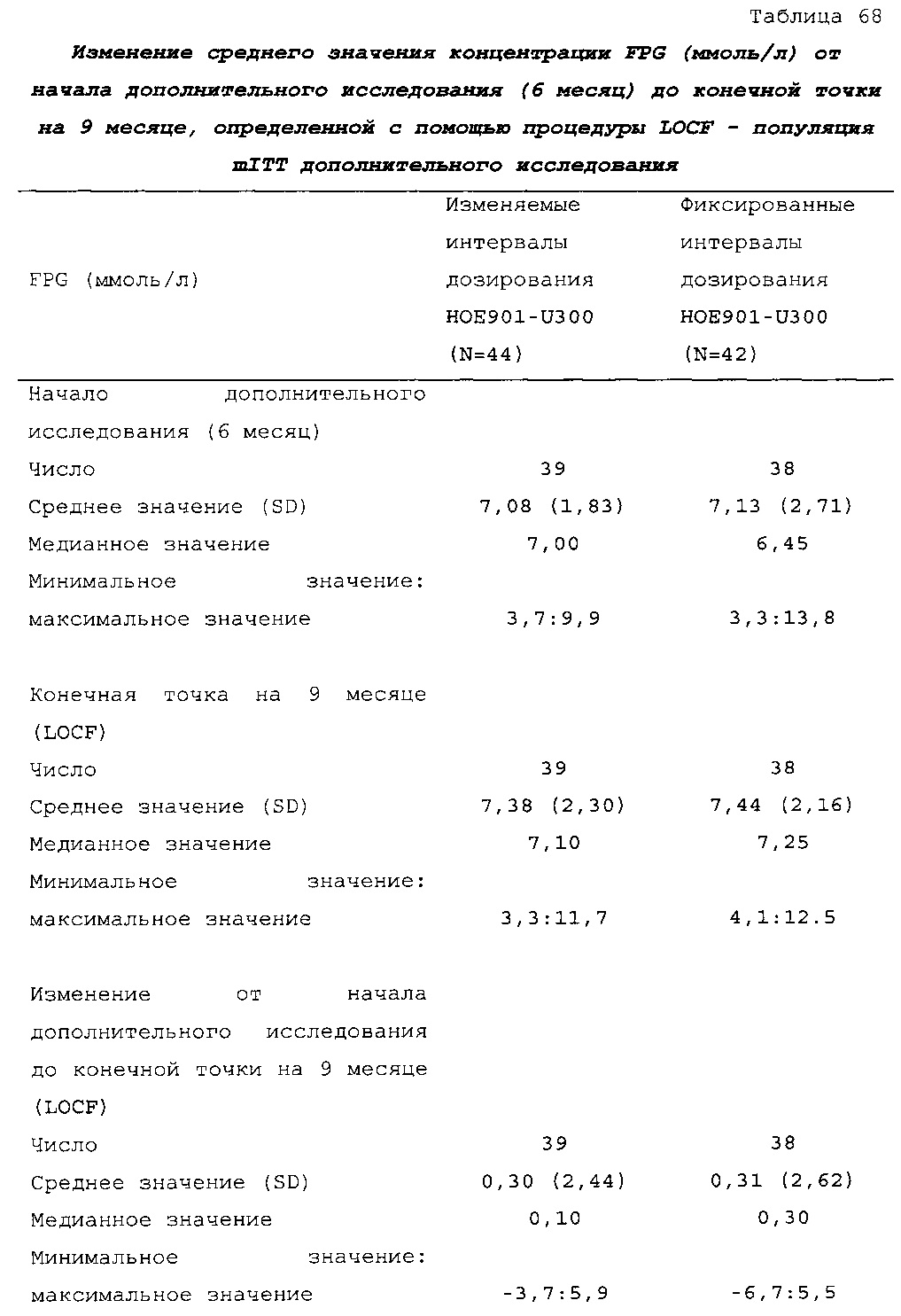

2.2.2.2 Доза базального инсулина
На фигуре 15 показаны средние суточные дозы базального инсулина (гларгин) (ЕД) по визитам на протяжении 3-месячного периода применения схемы, используемой для сравнения, в популяции mITT дополнительного исследования.
2.3 ОЦЕНКА БЕЗОПАСНОСТИ
2.3.1 Длительность проведения лечения


2.3.2 Гипогликемия



2.3.3 Нежелательные явления, возникшие в ходе лечения





2.3.4 Серьезные нежелательные явления, возникшие в ходе лечения


2.3.5 Возникшие в ходе лечения нежелательные явления, приведшие к прекращению участия

2.3.6 Другие значительные нежелательные явления, возникшие в ходе лечения
2.3.6.1 Местные реакции на инъекцию

2.3.6.2 Реакции гиперчувствительности

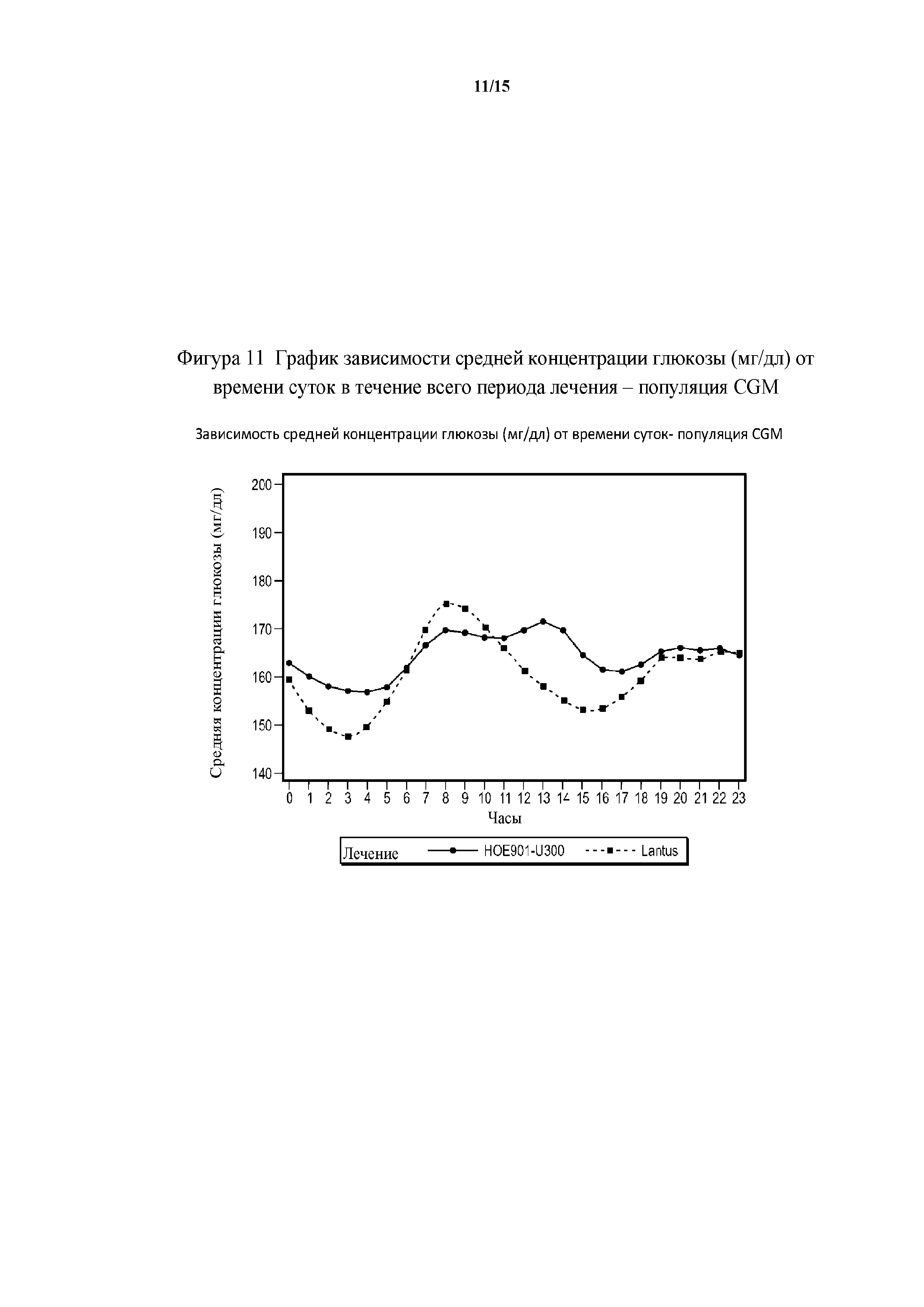
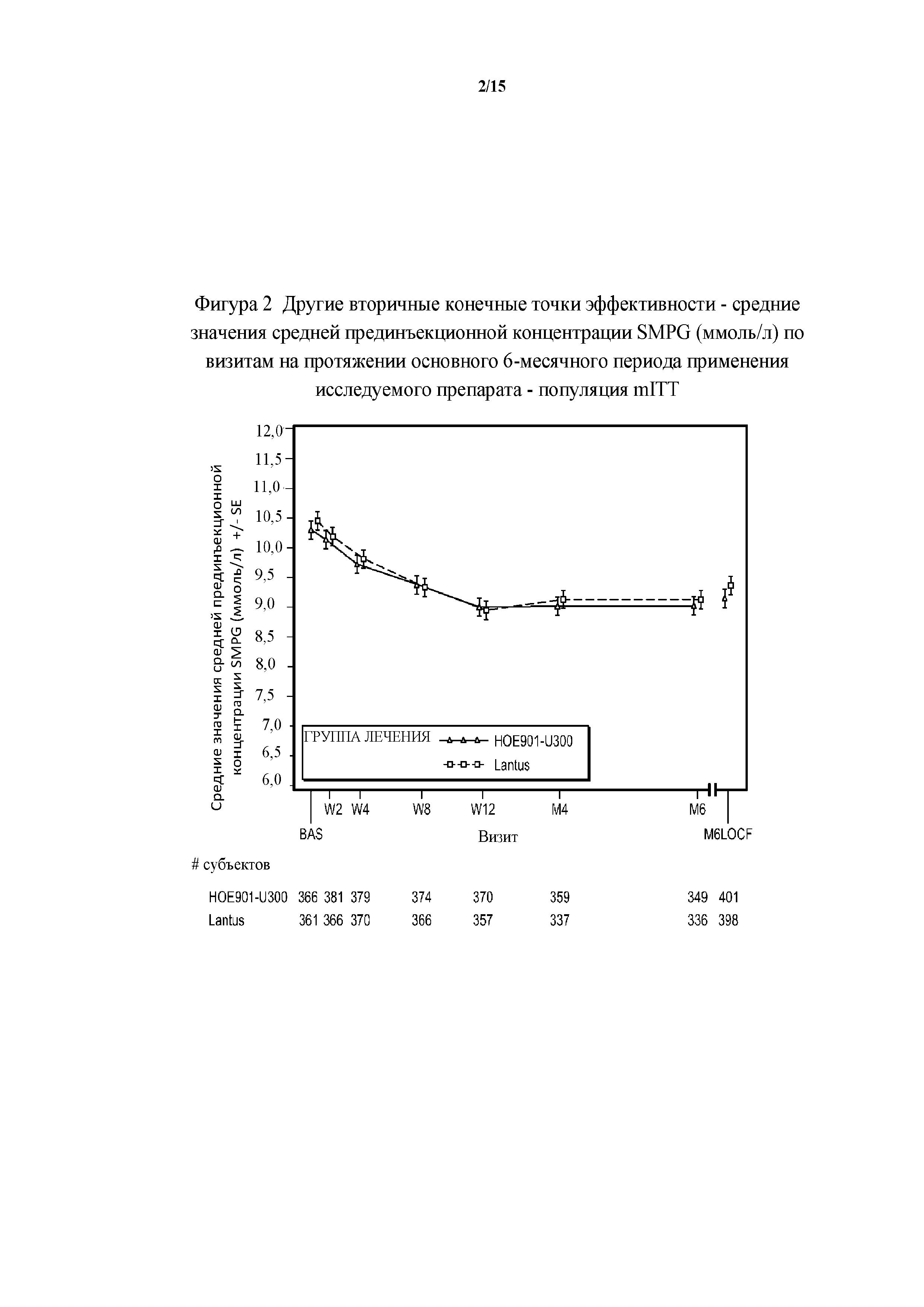
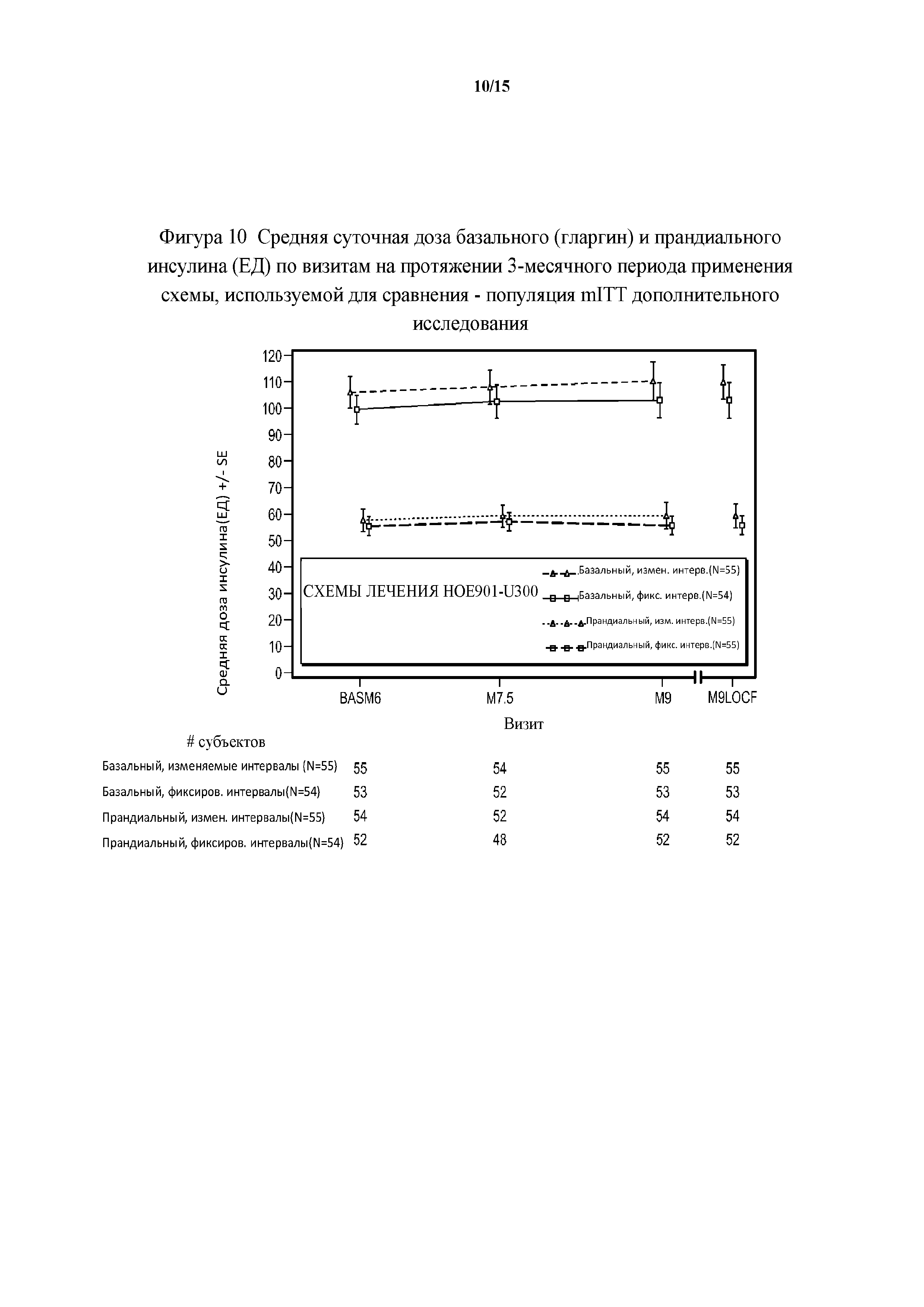
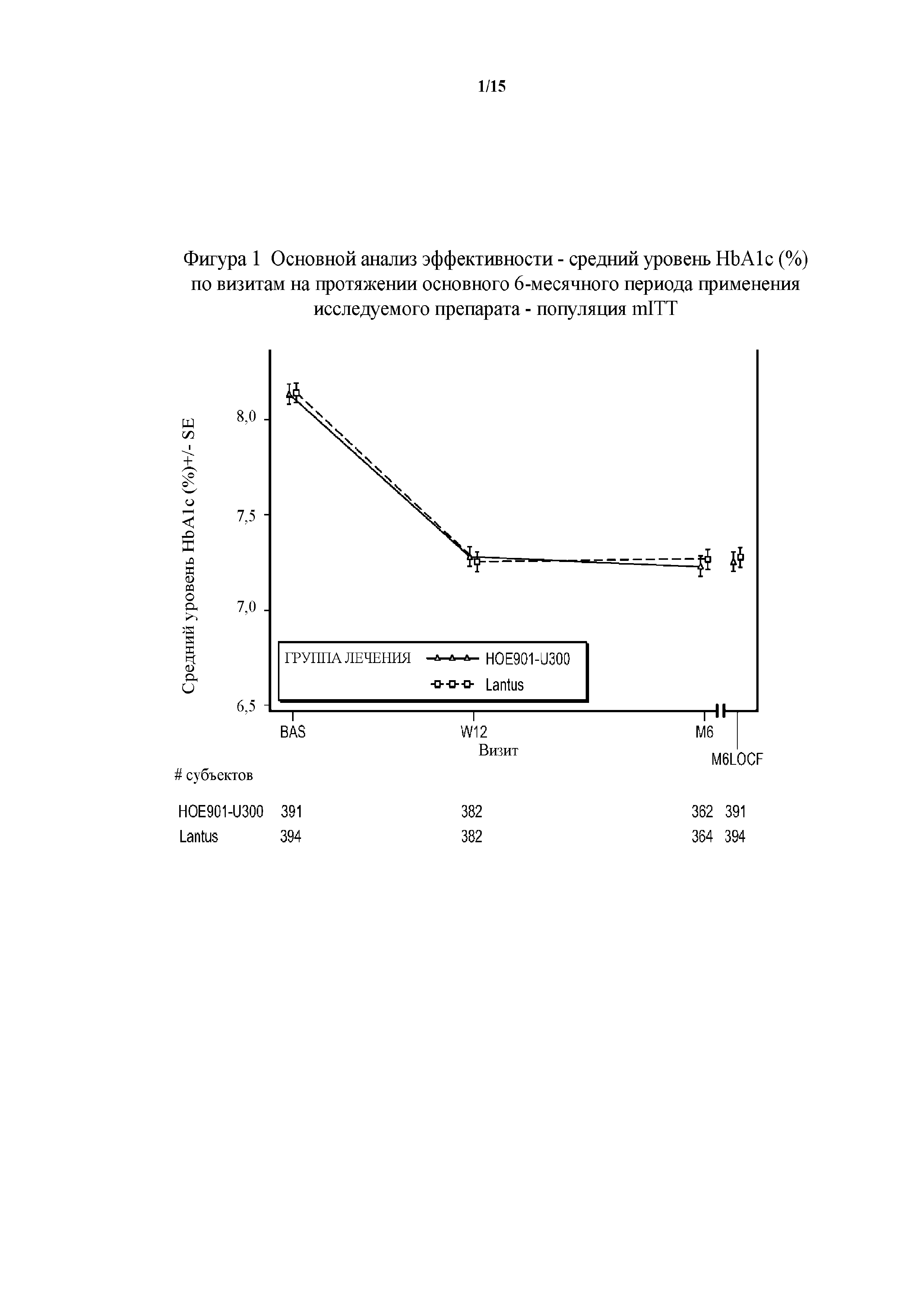
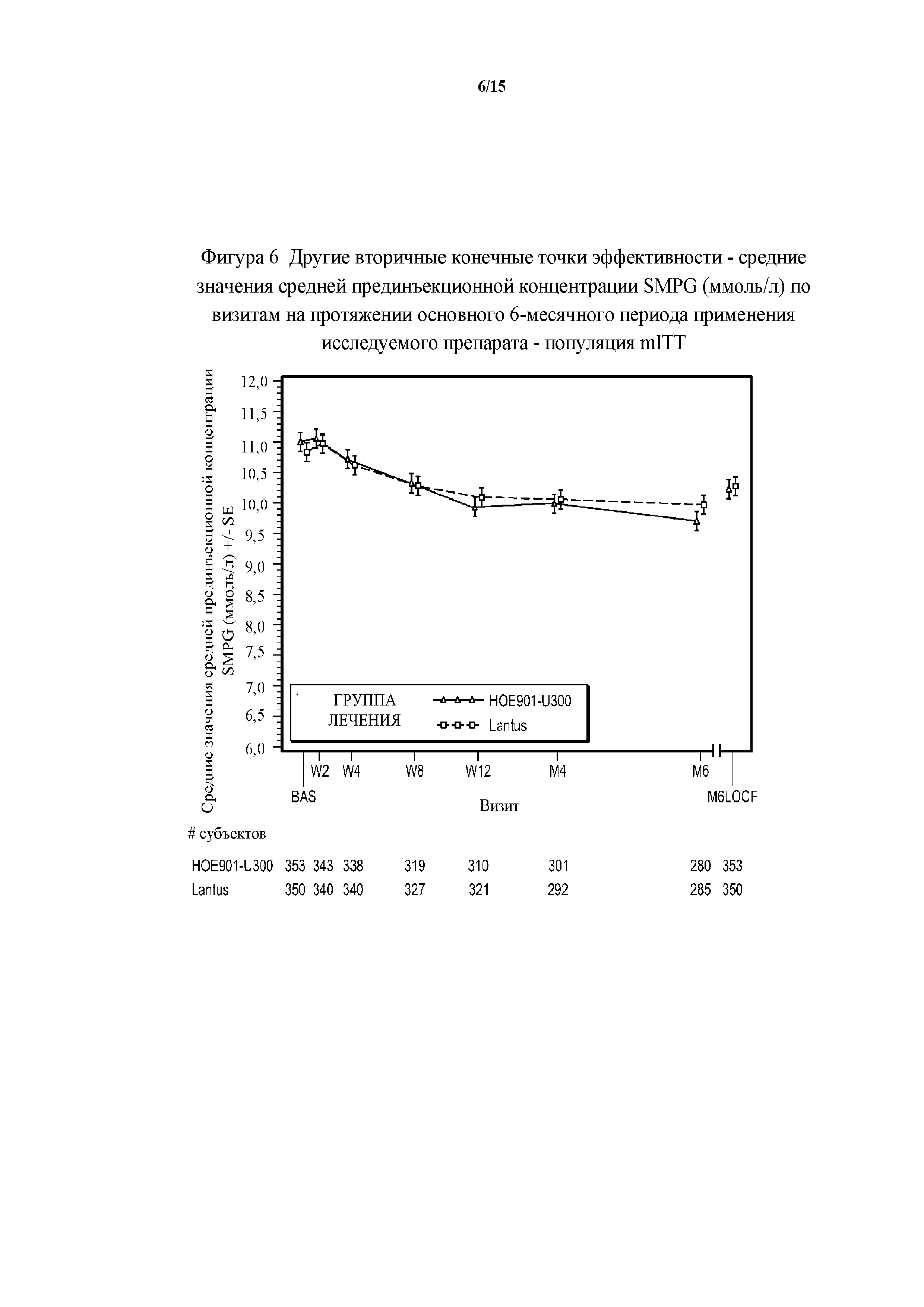
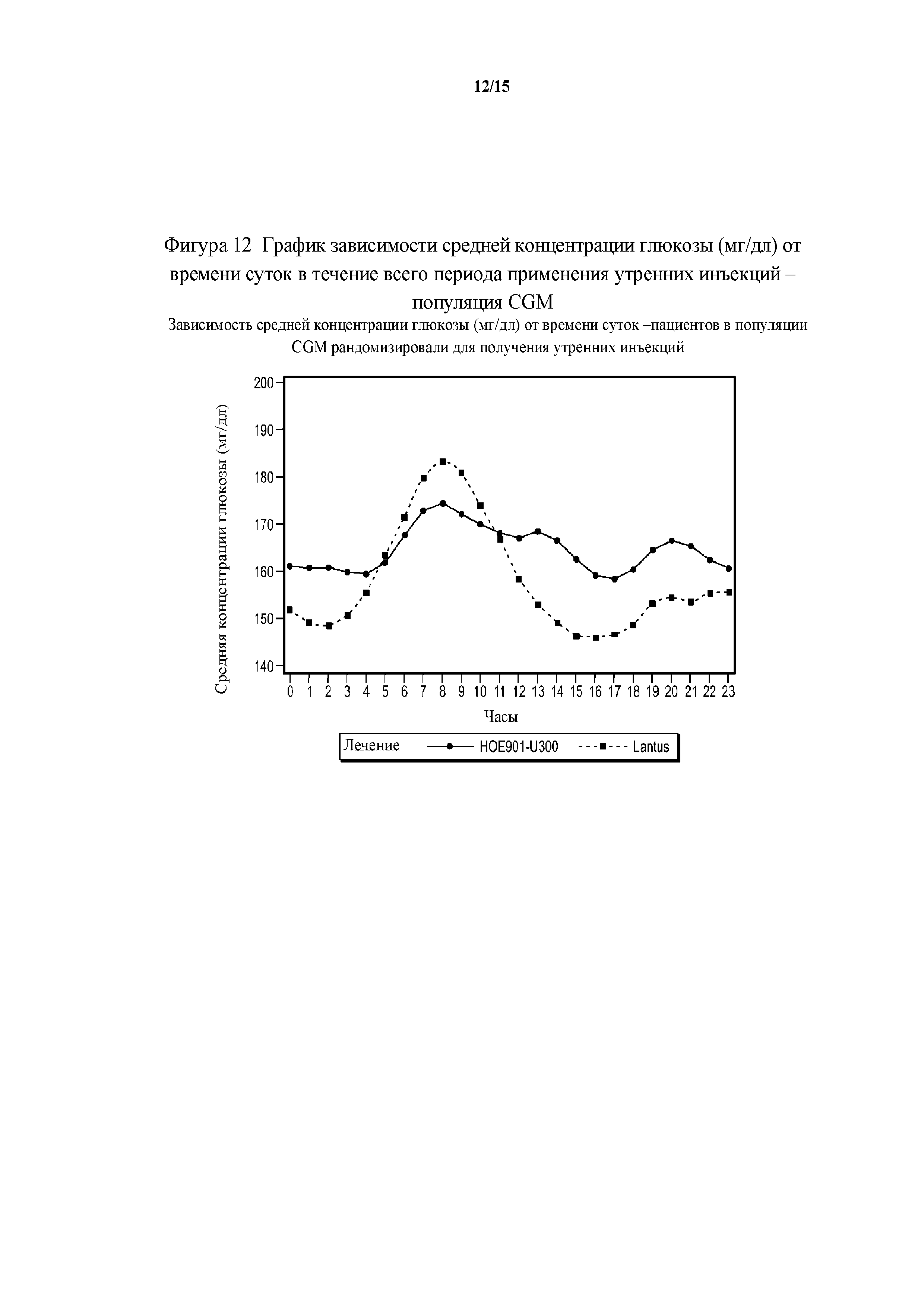
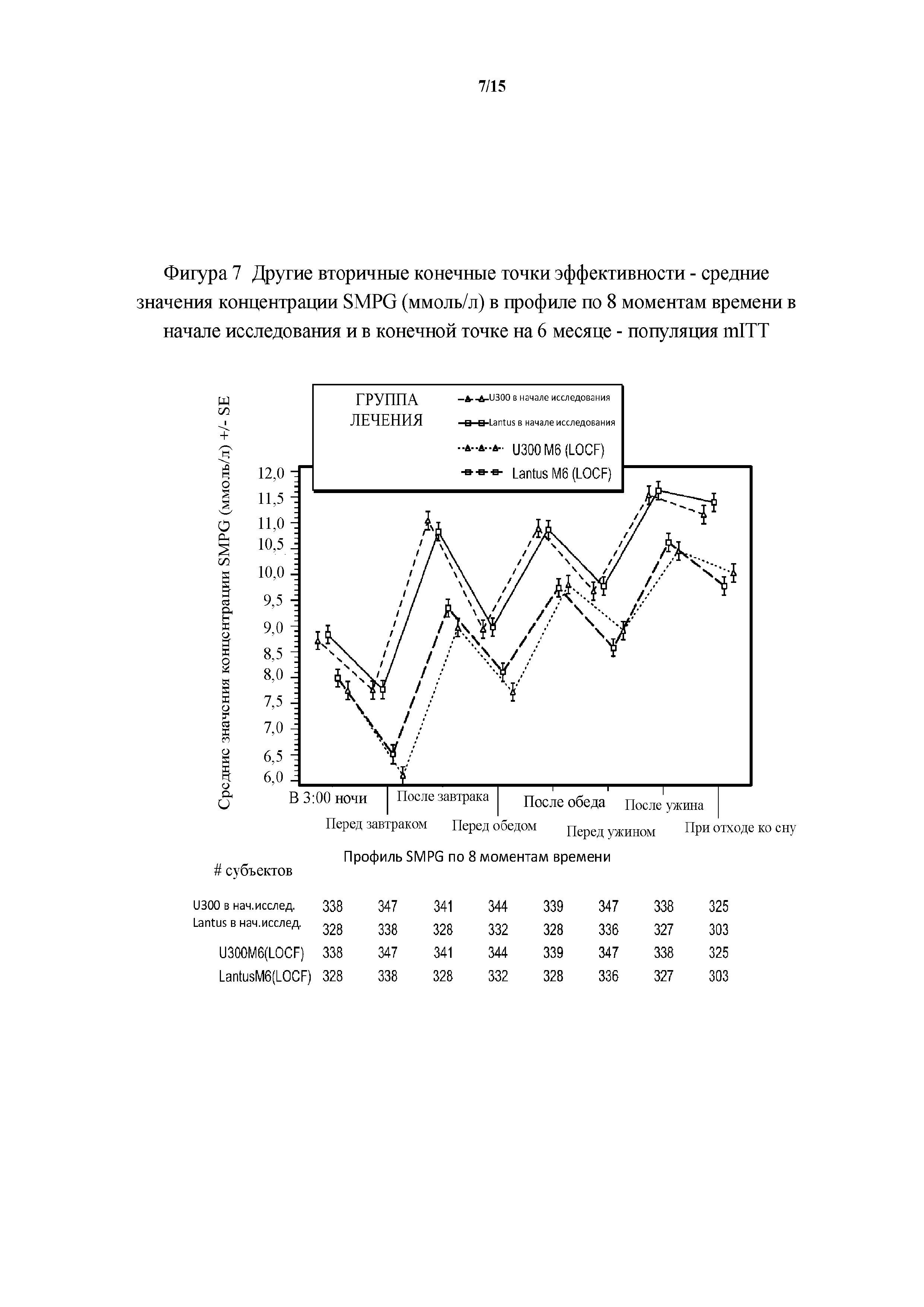
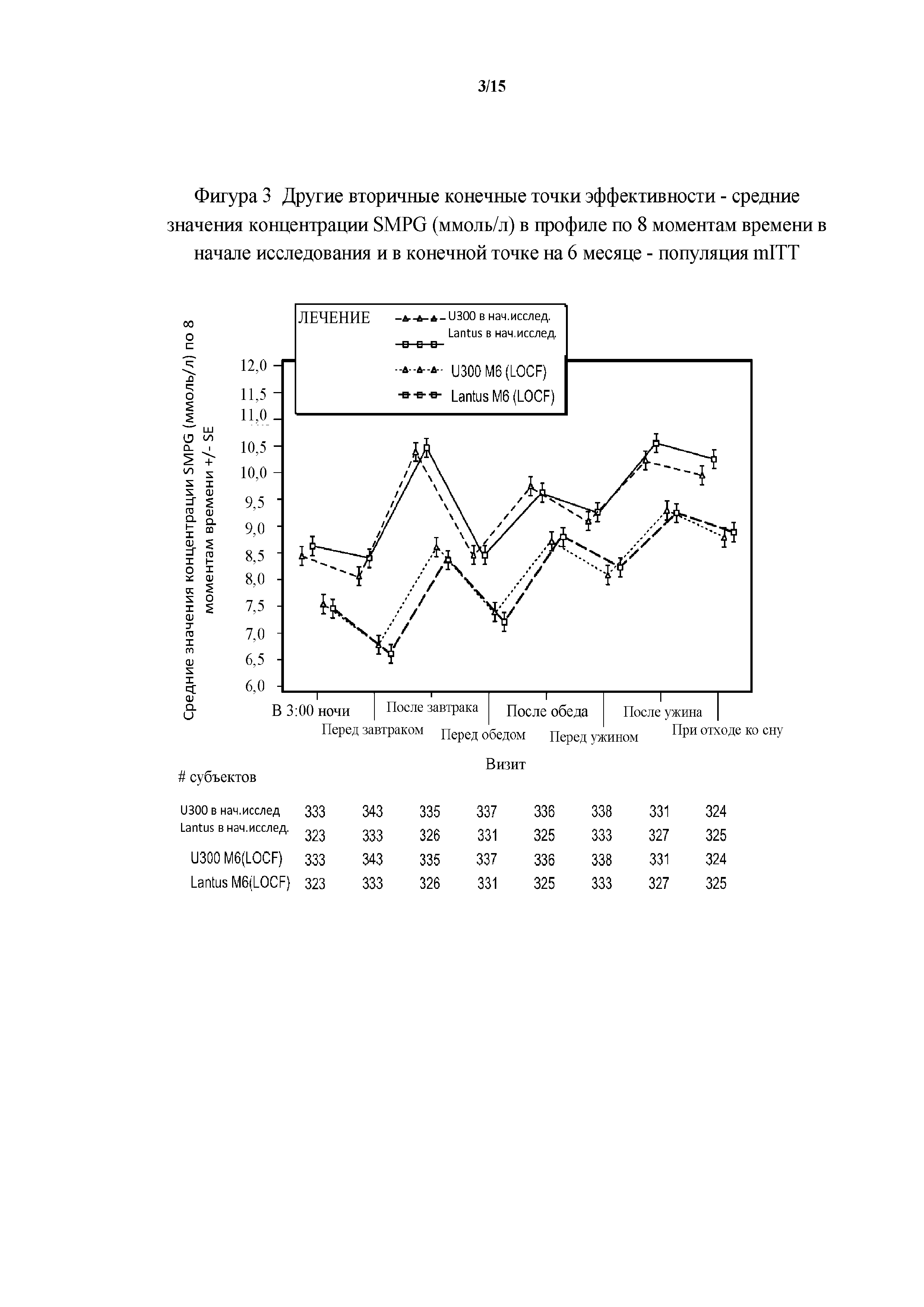
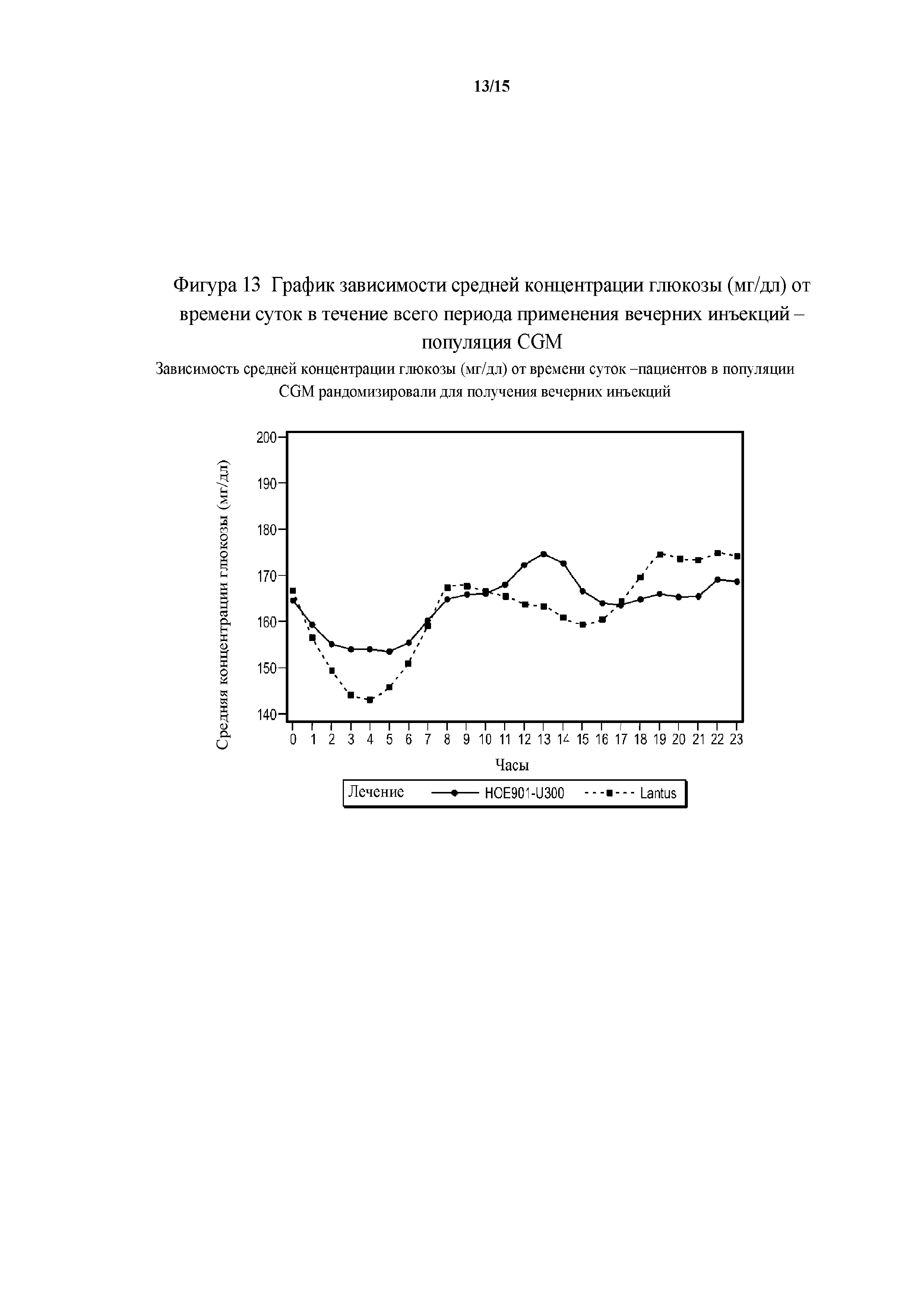
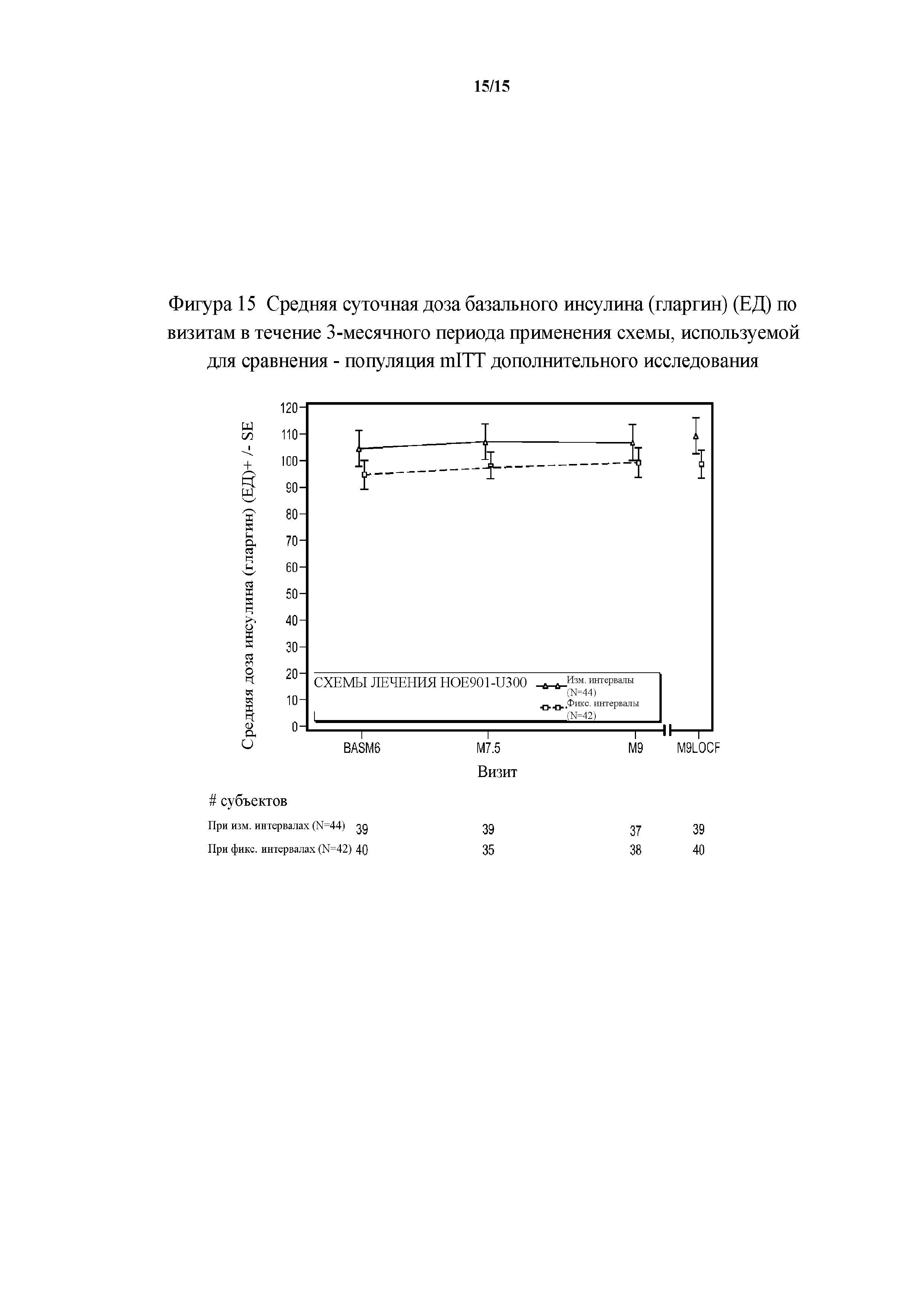
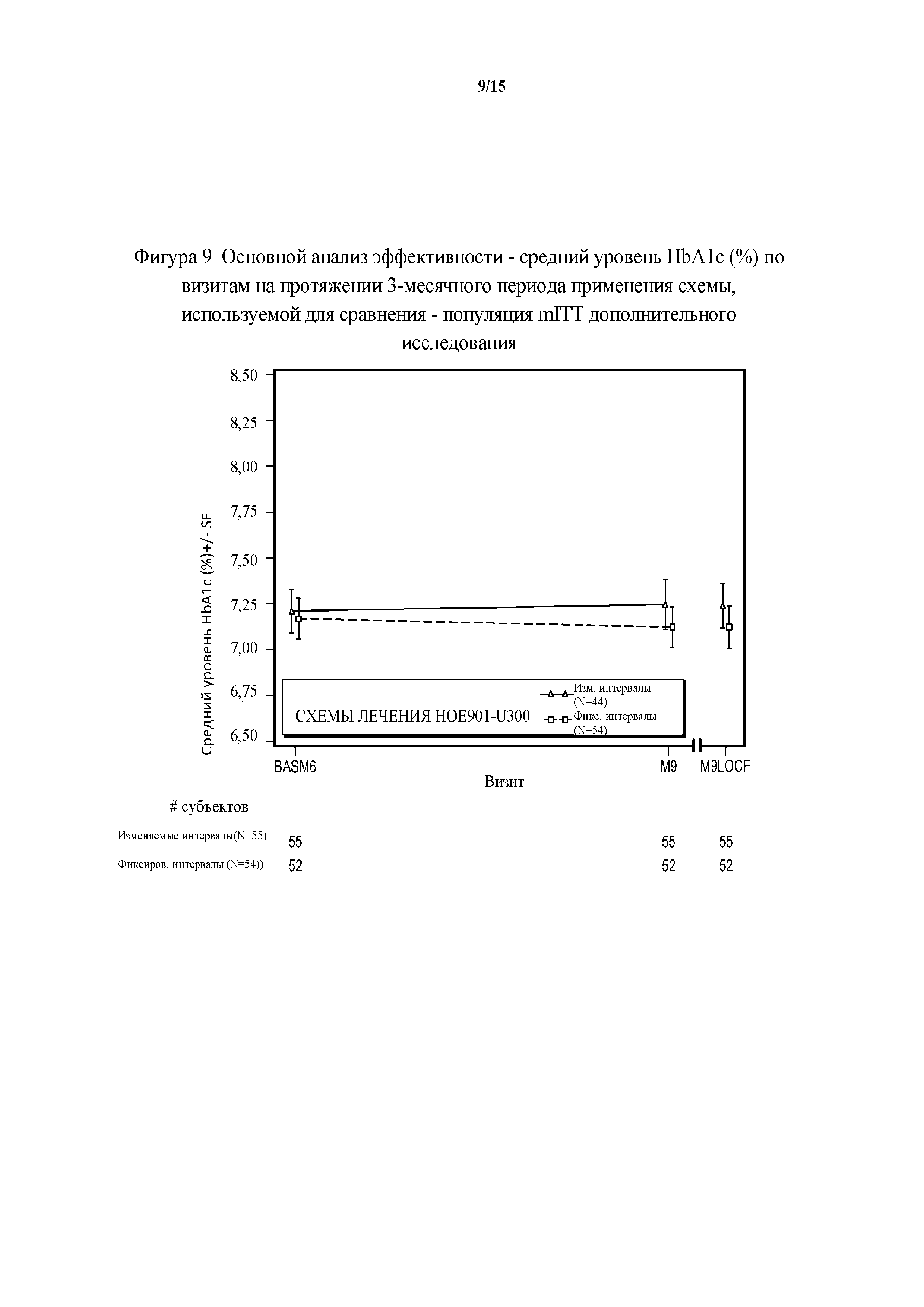
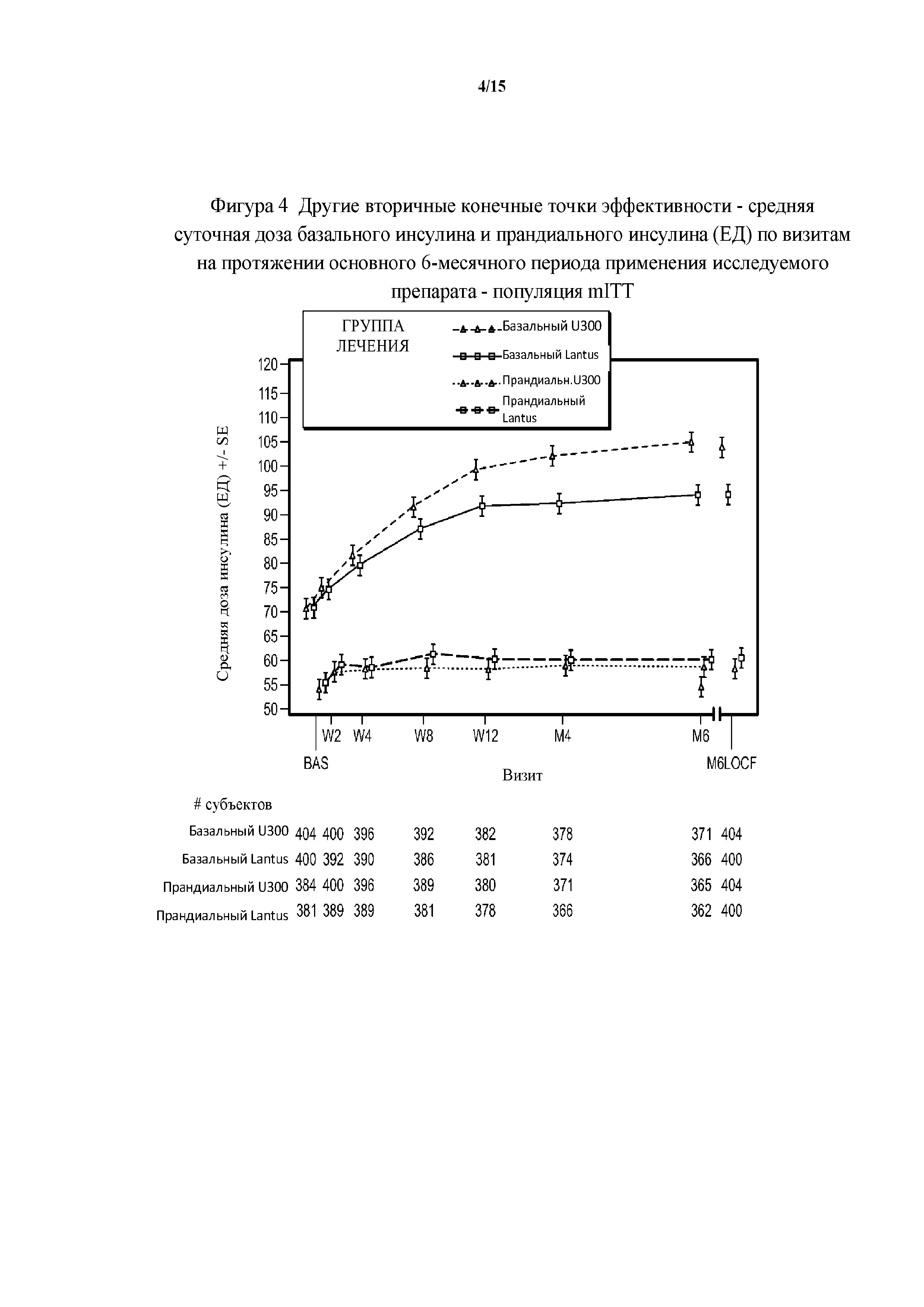
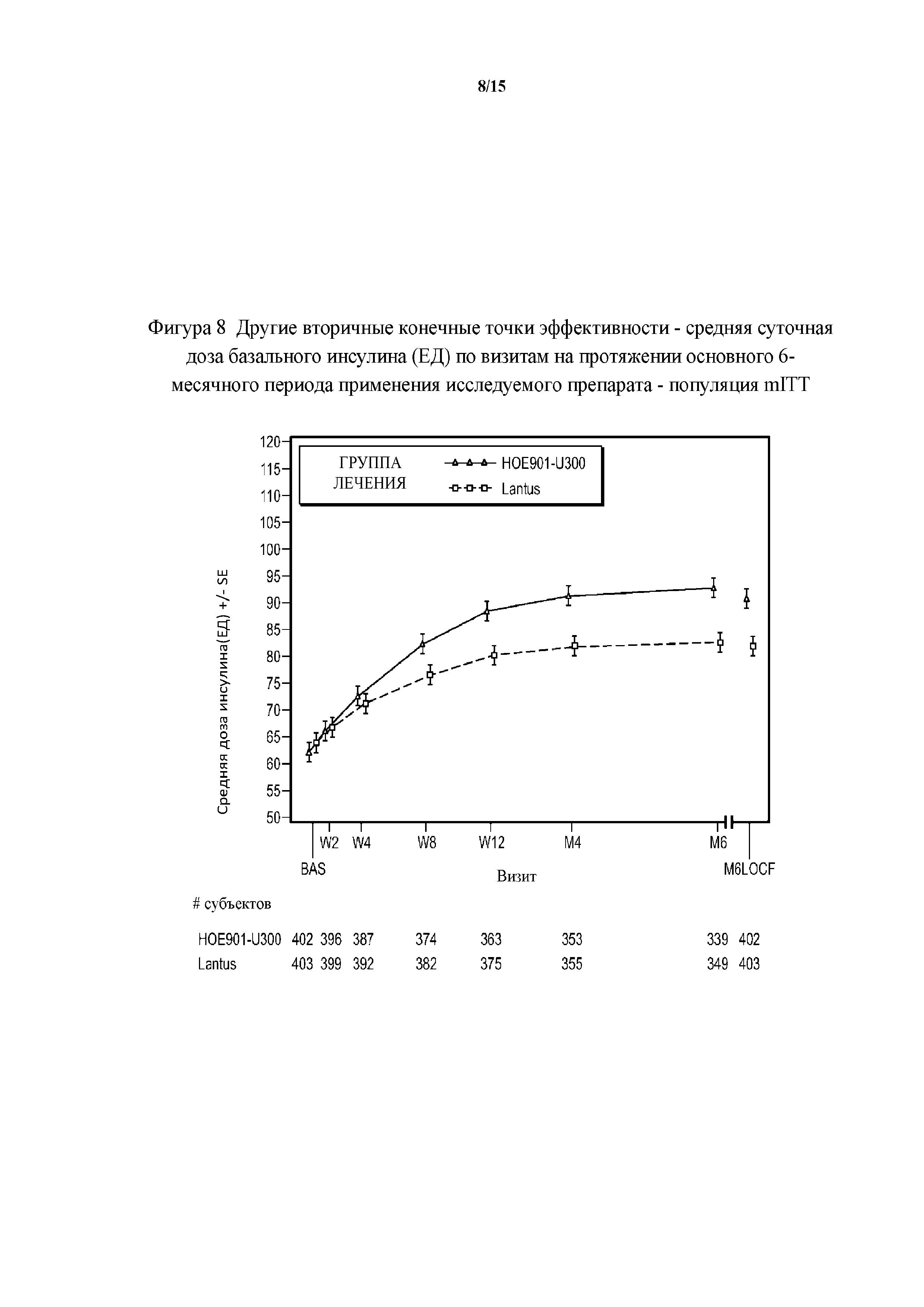
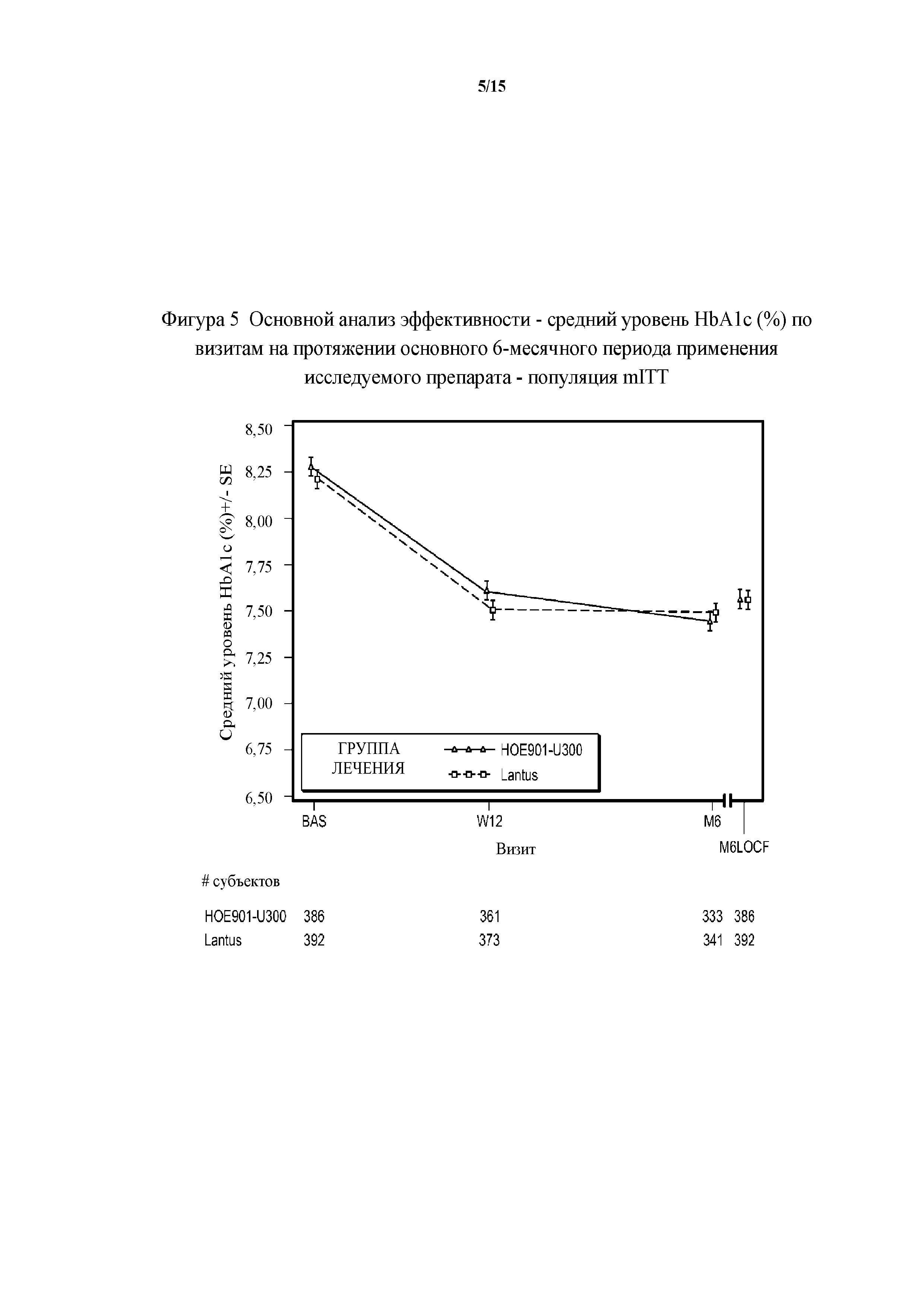
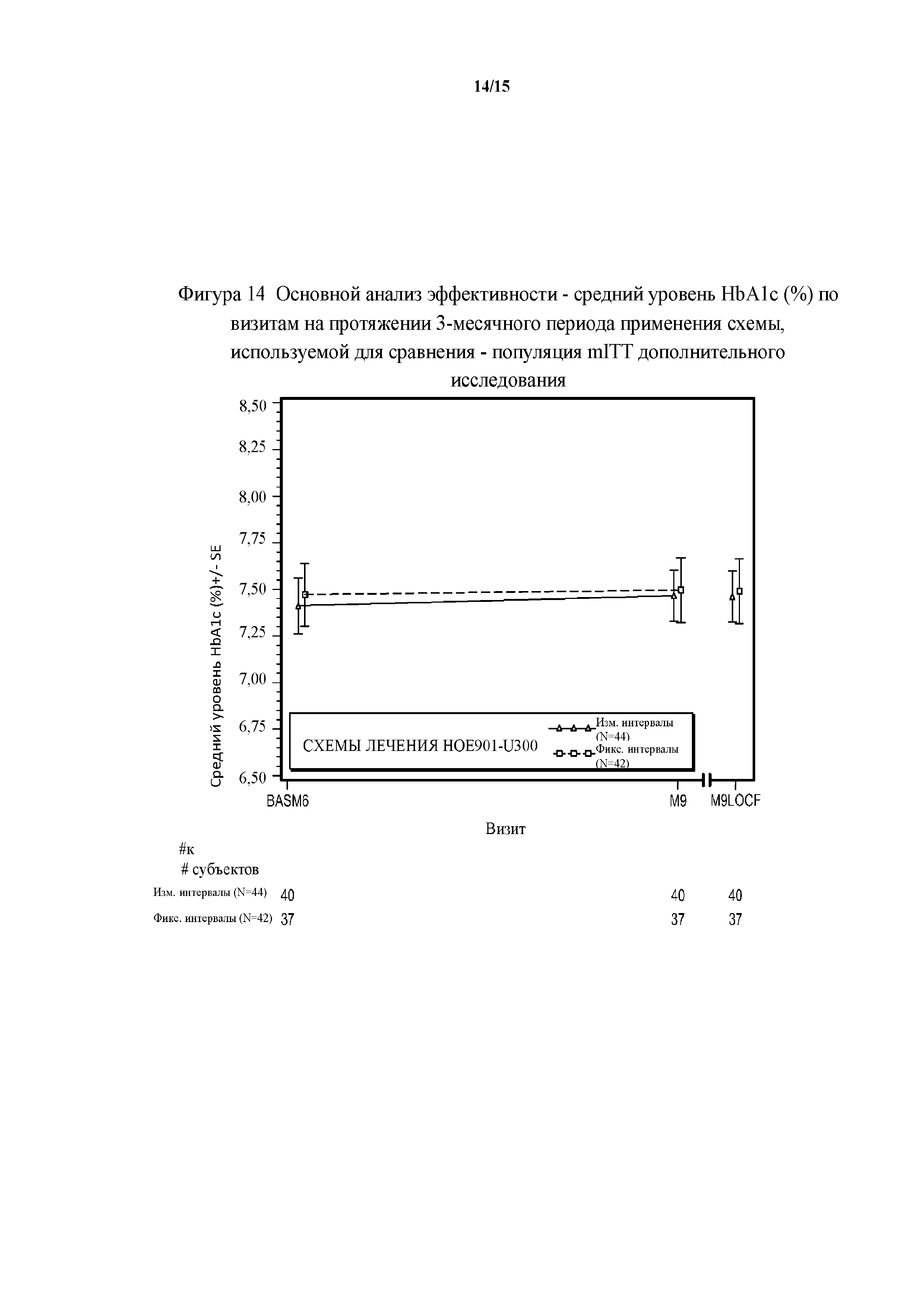
Новая комбинация активных ингредиентов, содержащая нестероидное противовоспалительное лекарственное средство и производное колхикозида
Способ получения активированных сложных эфиров
Замещенные тетрагидропиранспиропирролидинон и тетрагидропиранспиропиперидинон, фармацевтическая композиция на их основе и применение в лечебных целях
Лечение дерматологических аллергических состояний
Способ получения [4-(2- хлор-4- метокси-5- метилфенил)-5- метилтиазоло-2- ил] [2-циклопропил-1- (3- фтор-4- метилфенил) - этил ]- амина
Производные 1н-пиразоло[4,3-c]изохинолинов, способ их получения и применение в терапии
Новые фумаратные соли антагониста гистаминового рецептора н3
Способ получения соединения, применимого в качестве ингибитора tafia
Циклические (аза) индолизинкарбоксамиды, их получение и их примнение в качестве фармацевтических средств
Отамиксабан для лечения инфаркта миокарда без подъема сегмента st у пациентов пожилого возраста и пациентов с нарушенной функцией почек
Фармацевтическая комбинация для применения при гликемическом контроле у пациентов с сахарным диабетом 2 типа

