Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ПИРИМИДИНИЛЦИКЛОПЕНТАНОВЫХ СОЕДИНЕНИЙ
Вид РИД
Изобретение
Родственная заявка
В данной заявке заявлен приоритет Европейской патентной заявки № 131930307, поданной 15 ноября 2013 г., которая полностью включена в данный документ в качестве ссылки.
Область изобретения
Настоящее изобретение относится к способам получения пиримидинилциклопентановых соединений, которые полезны в качестве промежуточных продуктов при получении ингибиторов AKT протеинкиназы с терапевтической активностью в отношении таких заболеваний, как рак.
Уровень техники
В/Akt ферменты протеинкиназы представляют собой группу серин/треонин киназ, которые избыточно экспрессируются в некоторых опухолях человека. В международной патентной заявке WO 2008/006040 и в патенте США № 8063050 обсуждают ряд ингибиторов AKT, в том числе соединения (S)-2-(4-хлорофенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)пропан-1-oна (ипатасертиб, GDC-0068), который в настоящее время исследуется в клинических исследованиях для лечения различных видов рака.
В то время как процессы, описанные в WO 2008/006040 и US 8063050 полезны при обеспечении соединений гидроксилированного циклопента[d] пиримидина в качестве ингибиторов AKT протеинкиназы, необходимы альтернативные или улучшенные процессы, в том числе для крупномасштабного производства этих соединений.
Краткое описание изобретения
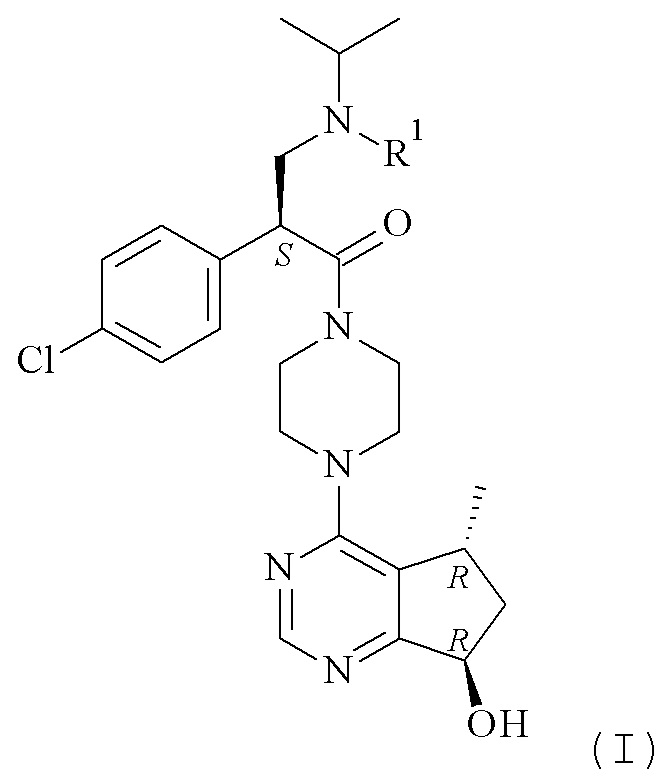
Данное изобретение представляет способы получения соединения формулы (I)
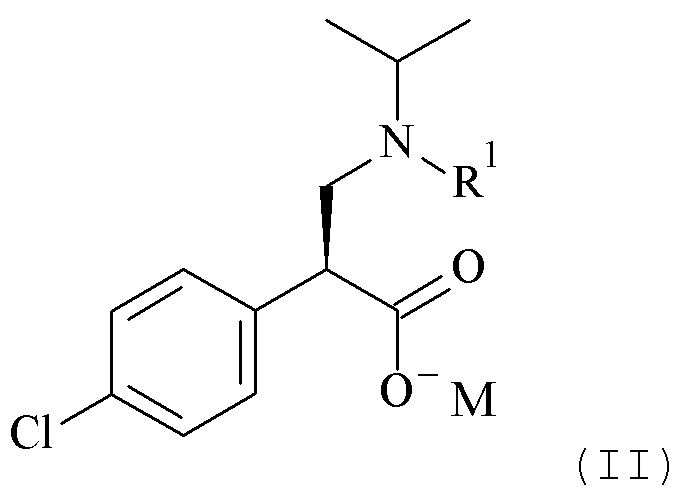
или его солей, включающие реакцию сочетания соединения формулы (II)
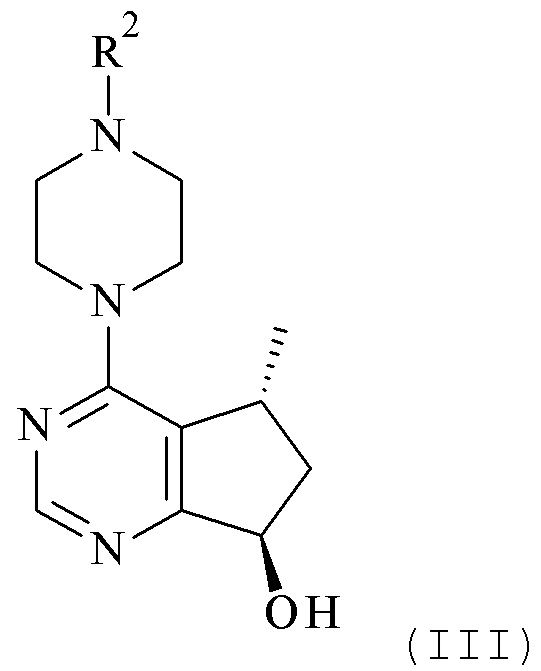
с соединением формулы (III)
где R1, R2 и M являются такими, как описано в данном документе.
ПОДРОБНОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Если иное не определено, то все технические и научные термины, используемые в данном документе, имеют такое же значение, как обычно понимает специалист в данной области техники, к которой относится данное изобретение. Хотя способы и материалы, подобные или эквивалентные описанным в настоящем документе, могут быть использованы на практике или при испытании данного изобретения, подходящие способы и материалы описаны ниже.
Номенклатура, используемая в данной заявке, основана на систематической номенклатуре IUPAC, если не указано иное.
Любая открытая валентность, указанная на атоме углерода, кислорода, серы или атоме азота в структурах в данном документе, указывает на присутствие водорода, если не указано иное.
При указании количества заместителей, термин "один или более" относится к диапазону от одного заместителя до максимально возможного количества заместителей, то есть от замещения одного водорода до замещения всех водородов заместителями.
Термин "необязательный" или "необязательно" означает, что впоследствии описанное событие или обстоятельство может, но не обязательно, иметь место, и что описание включает случаи, когда событие или обстоятельство имеет место, и случаи, в которых это не так.
Термин "фармацевтически приемлемые соли" обозначает соли, которые не являются биологически или иным образом нежелательными. Фармацевтически приемлемые соли включают как кислотные, так и основные аддитивные соли.
Термин "фармацевтически приемлемая кислотно-аддитивная соль" обозначает такие фармацевтически приемлемые соли, образованные с неорганическими кислотами, такими как соляная кислота, бромоводородная кислота, серная кислота, азотная кислота, угольная кислота, фосфорная кислота, и органические кислоты, выбранные из алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоновых, и сульфоновых классов органических кислот, таких как муравьиная кислота, уксусная кислота, пропионовая кислота, гликолевая кислота, глюконовая кислота, молочная кислота, пировиноградная кислота, щавелевая кислота, яблочная кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, аспарагиновая кислота, аскорбиновая кислота, глутаминовая кислота, антраниловая кислота, бензойная кислота, коричная кислота, миндальная кислота, эмбоновая кислота, фенилуксусная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфоновая кислота и салициловая кислота.
Термин "фармацевтически приемлемая основная аддитивная соль" обозначает фармацевтически приемлемые соли, образованные с органическим или неорганическим основанием. Примеры приемлемых неорганических оснований включают соли натрия, калия, аммония, кальция, магния, железа, цинка, меди, марганца, и алюминия. Соли, полученные из фармацевтически приемлемых органических нетоксичных оснований, включают соли первичных, вторичных и третичных аминов, замещенных аминов, включая природные замещенные амины, циклические амины и основные ионообменные смолы, такие, как изопропиламин, триметиламин, диэтиламин, триэтиламин, трипропиламин, этаноламин, 2-диэтиламиноэтанол, триметамин, дициклогексиламин, лизин, аргинин, гистидин, кофеин, прокаин, гидрабамин, холин, бетаин, этилендиамин, глюкозамин, метилглюкамин, теобромин, пурины, пиперизин, пиперидин, N-этилпиперидин и полиаминные смолы.
Стереохимические определения и условные термины, используемые в данном документе, как правило, приведены в соответствии с S. P. Parker, Ed., McGraw-Hill Dictionary of Chemical Terms (1984) McGraw-Hill Book Company, New York; и Eliel, E. and Wilen, S., Stereochemistry of Organic Compounds, John Wiley & Sons, Inc., New York, 1994. При описании оптически активного соединения префиксы D и L, или R и S, используются для обозначения абсолютной конфигурации молекулы относительно ее хирального центра (центров). Заместители, присоединенные к рассматриваемому хиральному центру ранжированы в соответствии с правилом последовательности Кана, Ингольда и Прелога. (Cahn et al. Angew. Chem. Inter. Edit. 1966, 5, 385; errata 511). Префиксы D и L или (+) и (-) используются для обозначения знака вращения плоско-поляризованного света соединением, причем (-) или L обозначают, что соединение левовращающее. Соединение с префиксом (+) или D является правовращающим.
Термин "стереоизомер" означает соединение, которое обладает идентичной молекулярной связностью и несколькими связями, но отличается по расположению его атомов в пространстве.
Термин "хиральный центр" означает атом углерода, связанный с четырьмя неодинаковыми заместителями. Термин "хиральный" означает способность к не совмещению с зеркальным отражением, в то время как термин "ахиральный" относится к вариантам реализации изобретения, которые совмещаются с их зеркальным отражением. Хиральные молекулы являются оптически активными, т.е., они обладают способностью вращать плоскость плоско-поляризованного света.
Соединения в соответствии с данным изобретением могут иметь один или несколько хиральных центров и могут существовать в форме оптически чистых энантиомеров, смесей энантиомеров, таких как, например, рацематы, оптически чистые диастереоизомеры, смеси диастереоизомеров, диастереоизомерные рацематы или смеси диастереоизомерных рацематов. Всякий раз, когда хиральный центр присутствует в химической структуре, предполагается, что все стереоизомеры, связанные с этим хиральным центром, охватываются настоящим изобретением.
Термин "энантиомеры" обозначает два стереоизомерных соединения, которые являются не совмещающимися зеркальными отражениями друг друга.
Термин "диастереомер" обозначает стереоизомеры, с двумя или более центрами хиральности, молекулы которых не являются зеркальными отражениями друг друга. Диастереоизомеры имеют различные физические свойства, например, точки плавления, точки кипения, спектральные свойства и реакционные способности.
Термин "диастереомерный избыток" (de) обозначает диастереомерную чистоту, т.е. (диастереомер А - диастереоизомер B)/(диастереомер А+диастереомер В) (в %площади).
Термин "энантиомерный избыток" (ее) обозначает энантиомерную чистоту, то есть (энантиомер А - энантиомер В)/( энантиомера А+энантиомер B)(в % площади).
Термин "гало" и "галоген" используют в данном документе взаимозаменяемо, и они обозначают фтор, хлор, бром или йод.
Термин "галид" обозначает ион галогена, в частности, фторид, хлорид, бромид или иодид.
Термин "алкил" означает одновалентную линейную или разветвленную насыщенную углеводородную группу, содержащую от 1 до 12 атомов углерода. В конкретных вариантах реализации изобретения, алкил имеет от 1 до 7 атомов углерода, и в более конкретных вариантах реализации изобретения от 1 до 4 атомов углерода. Примеры алкила включают метил, этил, пропил, изопропил, н-бутил, изо-бутил, втор-бутил или трет-бутил.
Термин "алкенил" обозначает одновалентную линейную или разветвленную углеводородную группу, содержащую от 2 до 7 атомов углерода, с по меньшей мере, одной двойной связью. В конкретных вариантах реализации изобретения, алкенил содержит от 2 до 4 атомов углерода, с по меньшей мере одну двойной связью. Примеры алкенила включают этенил, пропенил, проп-2-енил, изопропенил, н-бутенил, и изо-бутенил.
Термин "алкинил" обозначает одновалентную линейную или разветвленную, насыщенную углеводородную группу, содержащую от 2 до 7 атомов углерода, содержащую одну, две или три тройных связи. В конкретных вариантах реализации изобретения алкинил имеет от 2 до 4 атомов углерода, содержит одну или две тройные связи. Примеры алкинила включают этинил, пропинил, и н-бутинил.
Термин "алкокси" обозначает группу формулы -O-R', где R' представляет собой алкильную группу. Примеры алкокси групп включают метокси, этокси, изопропокси и трет-бутокси.
Термин "галоалкил" обозначает алкильную группу, в которой по меньшей мере один из атомов водорода алкильной группы был замещен одинаковыми или различными атомами галогена, в частности атомами фтора. Примеры галоалкила включают монофторо-, дифторо- или трифторо-метил, этил или пропил, например, 3,3,3-трифторопропил, 2-фтороэтил, 2,2,2-трифторэтил, фторометил или трифторометил. Термин "пергалоалкил" обозначает алкильную группу, где все атомы водорода алкильной группы были замещены одинаковыми или различными атомами галогена.
Термин "галоалкокси" означает алкоксигруппу, в которой по меньшей мере один из атомов водорода алкоксигруппы был замещен одинаковыми или различными атомами галогена, в частности атомами фтора. Примеры галоалкоксила включают монофторо-, дифторо- или трифторо-метокси, этокси или пропокси, например 3,3,3-трифторпропокси, 2-фтороэтокси, 2,2,2-трифторэтокси, фторометокси, или трифторометокси. Термин "пергалоалкокси" обозначает алкоксильную группу, где все атомы водорода алкоксигруппы были замещены одинаковыми или разными атомами галогена.
Термин "циклоалкил" обозначает одновалентную насыщенную моноциклическую или бициклическую углеводородную группу, содержащую от 3 до 10 атомов углерода в кольце. В конкретных вариантах реализации изобретения циклоалкил обозначает одновалентную насыщенную моноциклическую углеводородную группу, содержащую от 3 до 8 кольцевых атомов углерода. Термин «бициклический» означает состоящий из двух насыщенных карбоциклов, имеющих один или более атомов углерода в общем. Конкретные циклоалкильные группы являются моноциклическими. Примерами моноциклических циклоалкилов являются циклопропил, циклобутанил, циклопентил, циклогексил или циклогептил. Примерами бициклических циклоалкилов являются бицикло [2.2.1]гептанил, или бицикло [2.2.2]октанил.
Термин "гетероциклоалкил" означает одновалентную насыщенную или частично ненасыщенную моно- или бициклическую кольцевую систему, содержащую от 3 до 9 кольцевых атомов, содержащую 1, 2 или 3 гетероатомов в кольце, выбранных из N, О и S, остальные кольцевые атомы представляют собой углерод. В конкретных вариантах реализации изобретения, гетероциклоалкил представляет собой одновалентную насыщенную моноциклическую кольцевую систему, содержащую от 4 до 7 атомов в кольце, содержащую 1, 2 или 3 гетероатома в кольце, выбранных из N, O и S, остальные кольцевые атомы представляют собой углерод. Примерами моноциклических насыщенных гетероциклоалкилов являются азиридинил, оксиранил, азетидинил, оксетанил, пирролидинил, тетрагидрофуранил, тетрагидро-тиенил, пиразолидинил, имидазолидинил, оксазолидинил, изоксазолидинил, тиазолидинил, пиперидинил, тетрагидропиранил, тетрагидротиопиранил, пиперазинил, морфолинил, тиоморфолинил, 1,1-диоксо-тиоморфолин-4-ил, азепанил, диазепанил, гомопиперазинил, или оксазепанил. Примерами бициклических насыщенных гетероциклоалкилов являются 8-аза-бицикло [3.2.1]октил, хинуклидинил, 8-окса-3-аза-бицикло[3.2.1]октил, 9-аза-бицикло[3.3.1] нонил, 3-окса-9-аза-бицикло[3.3.1] нонил, или 3-тиа-9-аза-бицикло[3.3.1] нонил. Примерами частично ненасыщенных гетероциклоалкилов являются дигидрофурил, имидазолинил, дигидро-оксазолил, тетрагидро-пиридинил или дигидропиранил.
Термин "арил" обозначает одновалентную ароматическую карбоциклическую моно- или бициклическую кольцевую систему, содержащую от 6 до 10 атомов углерода в кольце. Примеры арильных групп включают фенил и нафтил. Конкретный арил представляет собой фенил.
Термин "гетероарил" означает одновалентную ароматическую гетероциклическую моно- или бициклическую кольцевую систему, содержащую от 5 до 12 атомов в кольце, содержащем 1, 2, 3 или 4 гетероатома, выбранных из N, O и S, остальные кольцевые атомы представляют собой углерод. Примеры гетероарильных групп включают пирролил, фуранил, тиенил, имидазолил, оксазолил, тиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиразинил, пиразолил, пиридазинил, пиримидинил, триазинил, азепинил, диазепинил, изоксазолил, бензофуранил, изотиазолил, бензотиенил, индолил, изоиндолил, изобензофуранил, бензимидазолил, бензоксазолил, бензоизоксазолил, бензотиазолил, бензоизотиазолил, бензооксадиазолил, бензотиадиазолил, бензотриазолил, пуринил, хинолинил, изохинолинил, хиназолинил, или хиноксалинил.
"Отходящая группа" относится к части первого реагента в химической реакции, которая смещена от первого реагента в химической реакции. Примеры отходящих групп включают, но не ограничиваются ими, водород, галоген, гидроксильные группы, сульфгидрильные группы, аминогруппы (например, -NRR, где R независимо представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R независимо необязательно замещена), силильные группы (например -SiRRR, где R независимо представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R независимо необязательно замещена), -N(R)OR (где R независимо представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R независимо необязательно замещена), алкокси группы (например -OR, где R независимо представляет собой алкил, алкенил, алкинил, цмклоалкил, фенил или гетероциклил, и R независимо необязательно замещена), тиольные группы (например, -SR, где R независимо представляет собой алкил, алкенил, алкинил, цмклоалкил, фенил или гетероциклил, и R независимо необязательно замещена), сульфонилоксигруппы (например, -OS(O)1-2R, где R независимо представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R независимо необязательно замещена), сульфаматные группы (например, -OS(O)1-2NRR, где R независимо представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R независимо необязательно замещена), карбаматные группы (например -OC(O)2NRR, где R независимо представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил и R независимо необязательно замещена), и карбонатные группы (например -OC(O)2R, где R независимо представляет собой алкил, алкенил, алкинил, циклоалкил, фенил или гетероциклил, и R независимо необязательно замещена). Иллюстративные карбонатные группы включают трет-бутил карбонат. Иллюстративные сульфонилокси группы включают, но не ограничиваются ими, алкилсульфонилокси группы (например, метил сульфонилокси (мезилатная группа) и трифторметилсульфонилокси группу (трифлатная группа)) и арилсульфонилокси группы (например, п-толуолсульфонилокси (тозилатная группа), и п-нитросульфонилокси (нозилатная группа)). Другие примеры отходящих групп включают замещенные и незамещенные аминогруппы, такие как амино, алкиламино, диалкиламино, гидроксиламино, алкоксиламино, N-алкил-N-алкоксиамино, ациламино, сульфониламино, трет-бутилокси и тому подобное.
Термин "защитная группа" обозначает группу, которая селективно блокирует реакционноспособный участок в многофункциональном соединении, так что химическая реакция может быть проведена селективно в другом незащищенном реакционно-способном месте в значении, которое обычно связано с ним в синтетической химии. Защитные группы могут быть удалены в соответствующий момент. Примерами защитных групп являются группы, амино защитные группы, карбокси защитные группы или гидрокси защитные группы.
Термин "амино-защитная группа" обозначает группы, предназначенные для защиты аминогруппы, и включает бензил, бензилоксикарбонил (карбобензилокси, CBZ), Fmoc (9-флуоренилметилоксикарбонил), п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, трет-бутоксикарбонил (ВОС), и трифторацетил. Дополнительные примеры этих групп можно найти в T. W. Greene and P. G. M. Wuts, ʺProtective Groups in Organic Synthesisʺ, 2nd ed., John Wiley & Sons, Inc., New York, NY, 1991, chapter 7; E. Haslam, ʺProtective Groups in Organic Chemistryʺ, J. G. W. McOmie, Ed., Plenum Press, New York, NY, 1973, Chapter 5, and T.W. Greene, ʺProtective Groups in Organic Synthesisʺ, John Wiley and Sons, New York, NY, 1981. Термин "защищенная аминогруппа" относится к аминогруппе, замещенной амино-защитными группами. Конкретным примером аминозащитной группы является трет-бутоксикарбонил (ВОС).
Термин "снятие защиты" обозначает процесс, посредством которого защитную группу удаляют после того, как селективная реакция завершается. Реагенты удаления защитной группы включают кислоты, основания или водород, в частности карбонаты калия или натрия, гидроксид лития в спиртовых растворах, цинк в метаноле, уксусную кислоту, трифторуксусную кислоту, палладиевые катализаторы или трибромид бора. Конкретным реагентом снятия защиты является соляная кислота.
Термин "буфер" обозначает наполнитель, который стабилизирует рН состава. Подходящие буферы хорошо известны в данной области и их можно найти в литературе. Конкретные фармацевтически приемлемые буферы включают гистидиновые буферы, аргининовые буферы, цитратные буферы, сукцинатные буферы, ацетатные буферы и фосфатные буферы. Вне зависимости от используемого буфера, рН может быть отрегулирован кислотой или основанием, известным в данной области техники, таким как, соляная кислота, уксусная кислота, фосфорная кислота, серная кислота и лимонная кислота, гидроксид натрия и гидроксид калия.
Термин "щелочной металл" относится к химическим элементам 1-й группы периодической таблицы элементов, т.е. литию (Li), натрию (Na), калию (K), рубидию (Rb), цезию (Cs) и францию (Fr). Конкретными примерами щелочных металлов являются Li, Na и К, наиболее особенно Na.
Термин "щелочноземельный металл" относится к химическим элементам 2-й группы Периодической таблицы элементов, т.е. бериллию (Be), магнию (Mg), кальцию (Са), стронцию (Sr), барию (Ва), и радию (Ra). Конкретными примерами щелочноземельных металлов являются Mg и Ca.
Термин "переходный металл" означает химические элементы, атомы которых имеют неполную d подоболочку.
Сокращения
Ac ацетил
AcOH уксусная кислота
AN ацетонитрил
BINAP 2,2'-бис(дифенилфосфино)-1,1'-бинафтил
BINAPHANE 1,2-бис[4,5-дигидро-3H-бинафто(1,2-c:2',1'-e)фосфепино]бензол
BIPHEMP (6,6’-диметилбифенил-2,2’-диил)бис(дифенил-фосфин)
BOC трет-бутоксикарбонил
(Boc)2O ди-трет-бутил дикарбонат
CBS катализатор Кори-Бакша-Шибата
CBZ бензилоксикарбонил, карбобензилокси
COD 1,5-циклооктадиен
CPME циклопентил метиловый эфир
de диастереомерный избыток
DIPEA диизопропилэтиламин
DMAP диметиламинопиридин
DMF N,N-диметилформамин
DPEN 1,2-дифенил этилендиамин
ee энантиомерный избыток
Et этил
EtOAc этилацетат
Fmoc 9-флуоренилметилоксикарбонил
(2-Фурил)-MeOBIPHEP (6,6’-диметоксибифенил-2,2ʹ-диил)бис[бис(2-фурил)-фосфин]
HAP опасный загрязнитель воздуха
HBTU N,N,N’,N’-тетраметил-O-(1H-бензотриазол-1-ил)уроний гексафторофосфат
iBu изо-бутил
ICM Международная конференция по гармонизации
IPC контроль в ходе процесса
iPr изопропил
iPr-DUPHOS 1,2-бис(2,5-ди-изопропилфосфолано)бензол
Me метил
MeOBIPHEP (6,6’-диметоксибифенил-2,2’-диил)бис(дифенил-фосфин)
MES 2-(н-морфолино)этансульфоновая кислота
MTBE метил трет-бутиловый эфир
NAD никотинамид аденин динуклеотид
NADP никотинамид аденин динуклеотид фосфат
nBu н-бутил
NEM N-этилморфолин
nPr n-пропил
OAc ацетат
PBS буфер на основе кислого фосфата калия
pCym п-кумол
PDE разрешенное ежедневное воздействие
Ph фенил
pTol п-толил
pTol-Binap 2,2'-бис(ди-п-толилфосфино)-1,1'-бинафтил
S/C молярное соотношение субстрата и катализатора
T3P пропилфосфониевый ангидрид
tBu трет-бутил
t-BuOK трет-бутоксид калия
TEA триэтиламин
TFA трифторацетат
ТГФ тетрагидрофуран
TMBTP 2,2’,5,5’-тетраметил-4,4’-бис(дифенилфосфино)-3,3’-битиофен
TPA три(н-пропил)амин
Xyl 3,5-диметилфенил
3,5-Xyl,4-MeO-MeOBIPHEP (6,6’-диметоксибифенил-2,2’-диил)бис[бис(3,5-диметил-4-метокси-фенил)-фосфин]
3,5-Xyl-BINAP 2,2’-бис[ди(3,5-ксилил)фосфин]-1,1’-бинафтил
3,5-Xyl-MeOBIPHEP (6,6’-диметоксибифенил-2,2’-диил)бис[бис(3,5-диметилфенил)-фосфин]
Настоящее изобретение обеспечивает способы получения соединения формулы (I), или его соли, которые включают реакцию сочетания соединения формулы (II) с соединением формулы (III) где R1, R2 и М являются такими, как описано в данном документе (Схема 1 ниже).
Один дополнительный аспект данного изобретения относится к способу получения соединений формулы (II), включающему асимметричную гидрогенизацию соединения формулы (IV), с помощью катализатора на основе металлокомплекса (C) (Схема 1 ниже).
Одним из аспектов в соответствии с данным изобретением относится к способу получения соединений формулы (III), включающему асимметричное восстановление соединения формулы (V), катализируемое оксидоредуктазой (Схема 1 ниже).
Один дополнительный аспект данного изобретения относится к способу получения соединений формулы (VI) или их фармацевтически приемлемых солей, где с соединения формулы (I) удаляют защитную группу (Схема 1 ниже).
Схема 1
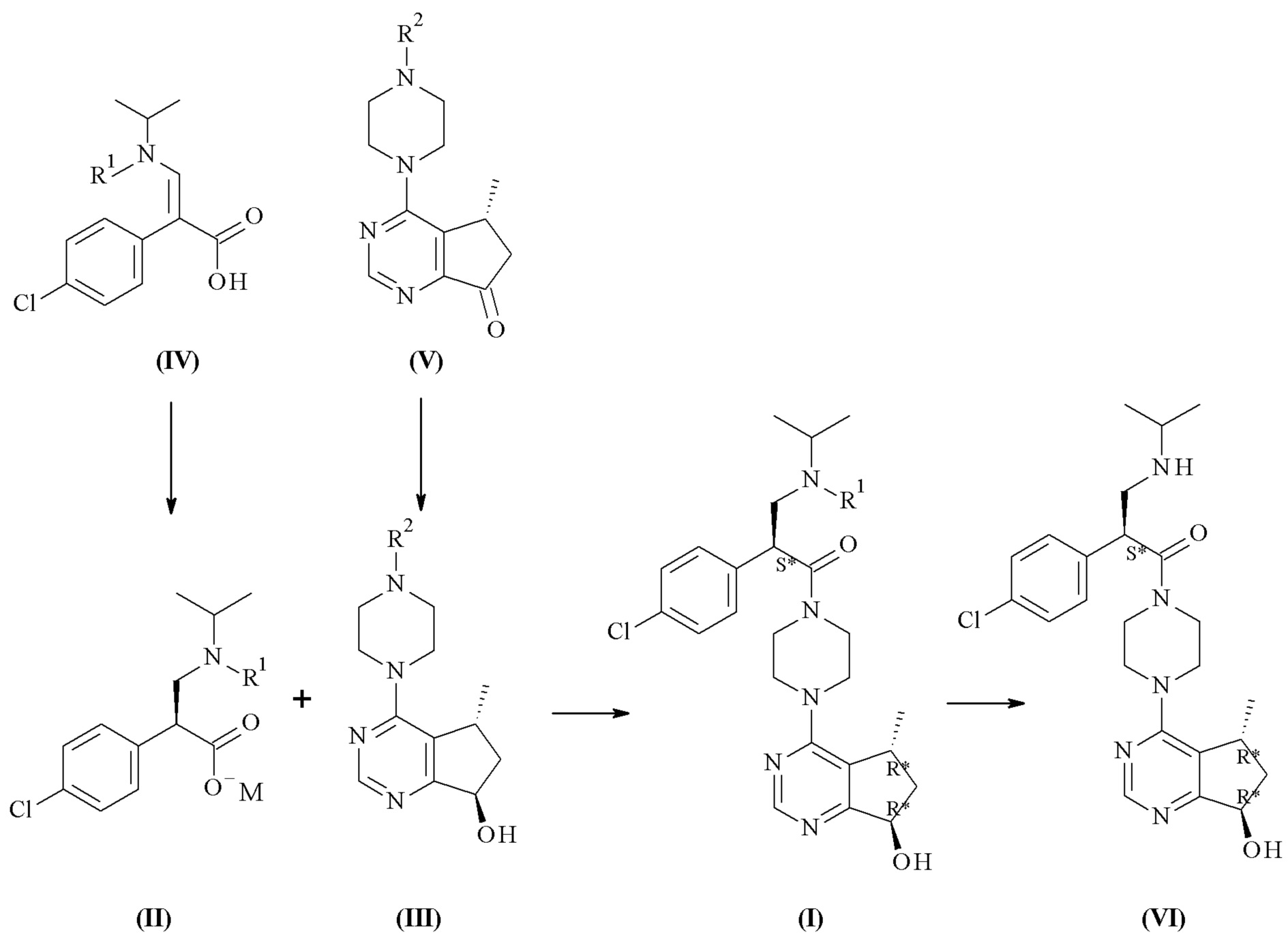
В одном варианте реализации изобретения, R1 представляет собой амино-защитную группу, выбранную из списка бензил, бензилоксикарбонил (карбобензилоки, CBZ), 9-флуоренилметилоксикарбонил (Fmoc), п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, трет-бутоксикарбонил (BOC), и трифтороацетил.
В конкретном варианте реализации изобретения, R1 представляет собой трет-бутоксикарбонил (BOC).
В одном варианте реализации изобретения, R2 представляет собой амино-защитную группу, выбранную из списка бензил, бензилоксикарбонил (карбобензилoкси, CBZ), 9-флуоренилметилоксикарбонил (Fmoc), п-метоксибензилоксикарбонил, п-нитробензилоксикарбонил, трет-бутоксикарбонил (BOC), и трифтороацетил.
В конкретном варианте реализации изобретения, R2 представляет собой трет-бутоксикарбонил (BOC).
В одном варианте реализации изобретения, M представляет собой ион металла, выбранный из списка ион щелочного металла, ион щелочноземельного металла и ион переходного металла.
В конкретном варианте реализации изобретения, M представляет собой ион металла, в частности щелочного металла, ион щелочноземельного металла или ион переходного металла при условии, что он не является K+.
В конкретном варианте реализации изобретения, M представляет собой ион щелочного металла.
В конкретном варианте реализации изобретения, M представляет собой Li+, K+ или Na+.
В конкретном варианте реализации изобретения, M не является K+.
В наиболее конкретном варианте реализации изобретения, M представляет собой Na+.
WO 2008/006040 описывает аминокислоты формулы (II-pa) и способы их получения, где R6 и R9 могут быть различными альтернативами и t равен 0-4.
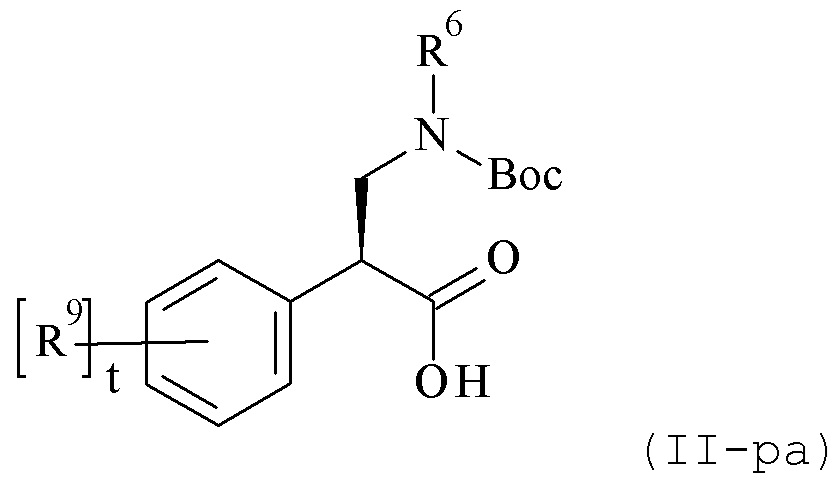
В данном документе описаны способы получения соединений формулы (II-ра), которые либо включают а) не энантиоселективную реакции алкиламина с 2-арилакрилатом с получением рацемической смеси или b) асимметричное добавление алкоксиметаламина к 2-фенилацетату, содержащему соответствующий хиральный вспомогательное вещество. Оба процесса не связаны с асимметричной гидрогенизации, но реакцией присоединения. Следовательно, оба процесса требуют дополнительных стадий для добавления, расщепления и разделения вспомогательного вещества.
Синтез в соответствии со способом а) выше протекает через образование рацемического сложноэфирного промежуточного продукта, который дополнительно гидролизуется до рацемической кислоты, в сочетании с хиральным вспомогательным веществом (как например, только S-энантиомером), чтобы генерировать 50:50 смесь диастереомеров R-аминокислоты/S-вспомогательного вещества и S-аминокислоты/S-вспомогательного вещества. Диастереомеры должны быть разделены с помощью хроматографии. Выход целевого S-S промежуточного продукта составляет только 38%. Дополнительно, S-S промежуточный продукт должен быть гидролизован с получением S-II кислоты (с потерей другого хирального компонента, хирального вспомогательного вещества). Эта процедура занимает много времени и малоэффективна, так как на одной стадии 72% вещества теряется. Таким образом, не-энантиоселективное добавление амина и акрилата демонстрирует внутреннюю проблему отсутствия стереоселективности и, таким образом, обязательное разделение рацемической смеси с помощью, например, хроматографии. Следовательно, выход по меньшей мере 100% ниже по сравнению со стереоселективной последовательностью.
Кроме того, асимметричное добавление к промежуточному продукту, содержащему хиральное вспомогательное вещество (метод b) выше) требует дополнительных стадий для добавления, расщепления и разделения вспомогательного вещества. Предшественник при синтезе целевой кислоты комбинируют с хиральным вспомогательным веществом, и полученный промежуточный продукт сочетают с алкоксиметанамином. Продукт состоит тогда в лучшем случае из слегка обогащенной смеси диастереоизомеров R/S и S/S, если не из смеси 1:1, которая должна быть дополнительно обработана, как указано выше, чтобы выделить (S)-изомер соединения формулы (II-ра) с лучшим умеренным выходом.
Существует, таким образом, неудовлетворенная потребность в улучшенных способах получения соединений формулы (II), которые обеспечивают лучшую стереоселективность, что устраняет необходимость в последующей хиральной хроматографии, требуют меньшего количества реакционных стадий, обеспечивают более высокий выход, и, следовательно, являются более эффективными, экологически чистыми и менее дорогостоящими.
Авторы данного изобретения нашли новый способ получения соединений формулы (II), который включает асимметричную гидрогенизацию соединения формулы (IV), с помощью катализатора на основе металлокомплексов (C).
Этот новый способ получения соединений формулы (II) имеет ряд соответствующих преимуществ по сравнению со способами, известными в данной области техники:
Высокостереоселективная реакция вводится в синтез;
Последующая очистка с использованием хиральной хроматографии пропускается;
Количество стадий реакции уменьшается;
Общий выход улучшается;
В целом реакция является более эффективной, экологически чистой и менее дорогостоящей.
Конкретные катализаторы на основе металлокомплексов в соответствии с данным изобретением, как было установлено, являются гораздо более эффективными и гораздо более активными и селективными, чем другие известные катализаторы в том смысле, что при тех же условиях реакции (то есть, без добавок) молярное соотношение субстрата и катализатора (S/C) вплоть до 10'000 может быть использовано, в то время как другие известные катализаторы должны использоваться при S/C 200-250. Таким образом, использование в 40-50 раз меньшего количества катализатора оказывает существенное влияние на эффективность, затраты и экологичность.
Некоторые известные катализаторы требуют большого количества LiBF4 в качестве добавки (до 5,8% мол на субстрат гидрогенизации, вплоть до 100 мольных эквивалентов по отношению к катализатору) для повышения активности катализаторов. Высокие количества LiBF4 невыгодны для промышленного процесса, ввиду наличия большого количества фторид-ионов (вплоть до 23,2% субстрата гидрогенизации), что представляет собой проблему в отношении коррозии стальных реакторов под давлением при расширении масштабов. С другой стороны, даже с LiBF4 добавкой катализатор не достигает активности наших новых катализаторов (например, вплоть до S/C 10'000).
Гомогенно катализируемые реакции, такие как, например, асимметричное гидрирование, как известно в данной области техники, требуют весьма трудоемкой процедуры обработки, включающей множество циклов экстракции и концентрации растворов. Дополнительно, асимметричные гидрирования, как известно в данной области техники, требуют удаления металлических катализаторов при помощи ловушки (например, тиольных смол) в больших количествах (вплоть до 6 мас % на субстрат гидрогенизации; вплоть до 193 раз по массе катализатора). Такое удаление рутениевых загрязнений с использованием смолистых ловушек является далеко не простым и довольно дорогостоящим. Дополнительно, содержание рутения уменьшается только частично (например, до около 50 ppm), и осуществляется на следующей стадии, тем самым увеличивая потенциал для образования побочных продуктов. Это добавляет материальные и трудовые затраты и открывает дискуссию о потенциальных примесях.
В заключение отметим, что эффективность известных процессов очистки и выделения продукта гидрирования из катализаторов и добавок является низкой.
В противоположность этому, способ в соответствии с данным изобретением обеспечивает соли соединения формулы (II), которые осаждаются непосредственно из смеси гидрогенизации, и которые могут быть легко отфильтрованы. Такое выделение и очистка продукта гидрирования обеспечивает высокие выходы (> 94%) при 100% ее и с содержанием рутения ниже предела обнаружения 5 ppm. Обработка продукта реакции асимметричной гидрогенизацией, как обнаружено авторами данного изобретения, таким образом, значительно проще, дешевле и более полезна, чем традиционные процессы.
Один из аспектов данного изобретения относится к формуле (II)
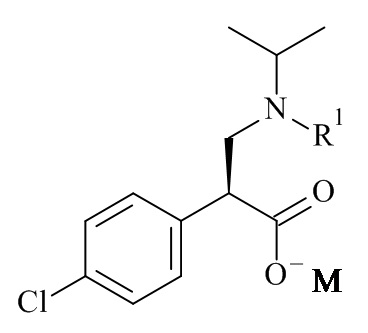 (II)
(II)
где R1 и M являются такими, как определено в данном документе.
Один из аспектов данного изобретения относится к соединению формулы (II), которое представляет собой натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноат.
Один из аспектов данного изобретения относится к способу получения соединений формулы (II)
(II)
включающему асимметричную гидрогенизацию соединения формулы (IV)
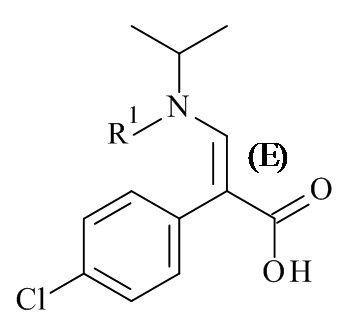 (IV)
(IV)
с использованием катализатора на основе металлокомплексов (C), где R1 и M являются такими, как определено в данном документе.
В одном варианте реализации изобретения, катализатор на основе металлокомплексов (C) представляет собой катализатор на основе комплексов рутения.
В одном варианте реализации изобретения, катализатор на основе комплексов рутения содержит рутений, характеризующийся степенью окисления II.
В одном варианте реализации изобретения, катализатор на основе комплексов рутения содержит хиральный фосфиновый лиганд (D).
В одном варианте реализации изобретения, катализатор на основе комплексов рутения содержит лиганды, в частности нейтральные лиганды (L) и/или анионные лиганды (Z).
Примерами нейтральных лигандов (L) являются олефины, такие как этилен или пропилен, циклооктен, 1,3-гексадиен, норборнадиен, 1,5-циклооктадиен, бензол, гексаметилбензол, 1,3,5-триметилбензол, и п-кумол или также растворители, такие как тетрагидрофуран, диметилформамин, ацетонитрил, бензонитрил, ацетон толуол и метанол.
Примерами анионных лигандов (Z) являются ацетат (CH3COO-), трифтороацетат (CF3COO-), η5-2,4-пентадиенил, η5-2,4-диметил-пентадиенил, и галоген-ионы, такие как фторид, хлорид, бромид или йодид.
Если катализатор на основе комплексов рутения заряжен, он дополнительно содержит не-координирующиеся анионы (Y). Примерами не-координирующихся анионов (Y) являются галоген-ионы, такие как фторид, хлорид, бромид или йодид, BF4-, ClO4-, SbF6-, PF6-, B(фенил)4-, B(3,5-ди-трифторометил-фенил)4-, CF3SO3-, и C6H5SO3-.
Катализатор на основе комплексов рутения может необязательно быть дополнительно скоординирован с кислотой Льюиса, такой как AlCl3.
В одном варианте реализации изобретения, катализатор на основе комплексов рутения выбирают из соединения формулы (C1), (C2) или (C3):
Ru(Z)2D (C1)
[Ru(Z)2-p(D)(Л)m](Y)p (C2)
Ru(E)(Eʹ)(D)(F) (C3)
где:
D представляет собой хиральный фосфиновый лиганд;
L представляет собой нейтральный лиганд, выбранный из C2-7 алкена, циклооктена, 1,3-гексадиена, норборнадиена, 1,5-циклооктадиена, бензола, гексаметилбензола, 1,3,5-триметилбензола, п-кумола, тетрагидрофурана, диметилформамина, ацетонитрила, бензонитрила, ацетона, толуола и метанола;
Z представляет собой анионный лиганд, выбранный из гидрида, фторида, хлорида, бромида, η5-2,4-пентадиенила, η5-2,4-диметил-пентадиенила или группы A-COO-, при условии, что если две группы Z присоединены к атому Ru, они могут быть одинаковыми или различными;
A представляет собой C1-7 алкил, C1-7 галоалкил, арил, или галоарил;
Y представляет собой координирующийся анион, выбранный из фторида, хлорида, бромида, BF4-, ClO4-, SbF6-, PF6-, B(фенил)4-, B(3,5-ди-трифторометил-фенил)4-, CF3SO3-, и C6H5SO3-;
F представляет собой необязательно хиральный диамин;
E и Eʹ оба представляют собой иона галогена, или E представляет собой гидрид и Eʹ представляет собой BH4-;
m представляет собой 1, 2, 3 или 4;
p представляет собой 1 или 2.
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения выбирают из соединения формулы (C1) или (C2), где Z, D, L, Y, m и p являются такими, как описано в данном документе.
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения выбирают из соединения формулы (C1), где Z и D являются такими, как описано в данном документе.
В конкретном варианте реализации изобретения, катализаторы на основе комплексов рутения представляют собой Ru(Z)2D, где Z и D являются такими, как описано в данном документе.
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения выбирают из соединения формулы (C2), где Z, D, L, Y, m и p являются такими, как описано в данном документе.
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения выбирают из соединения формулы (C3), где E, Eʹ, D и F являются такими, как описано в данном документе.
В конкретном варианте реализации изобретения, анионный лиганд (Z) независимо выбирают из хлорида, бромида, йодида, OAc, и TFA.
В конкретном варианте реализации изобретения, анионный лиганд (Z) представляет собой A-COO-.
В конкретном варианте реализации изобретения, A представляет собой -CF3.
В конкретном варианте реализации изобретения, анионный лиганд (Z) представляет собой трифтороацетат (TFA).
В конкретном варианте реализации изобретения, нейтральный лиганд (L) независимо выбирают из бензола (C6H6), п-кумола (pCym), и ацетонитрила (AN).
В конкретном варианте реализации изобретения, нейтральный лиганд (L) представляет собой бензол (C6H6).
В конкретном варианте реализации изобретения, не-координирующийся анион (Y) выбирают из хлорида, бромида, йодида и BF4-.
В конкретном варианте реализации изобретения, не-координирующийся анион (Y) представляет собой BF4-.
В конкретном варианте реализации изобретения, m представляет собой 1 или 4.
В конкретном варианте реализации изобретения, m представляет собой 1.
В конкретном варианте реализации изобретения, m представляет собой 4.
В конкретном варианте реализации изобретения, p представляет собой 1.
В конкретном варианте реализации изобретения, p представляет собой 2.
В конкретном варианте реализации изобретения, E и Eʹ представляют собой оба хлорид;
В конкретном варианте реализации изобретения, хиральный диамин F представляет собой (1S,2S)-1,2-дифенилэтилендиамин (с,S-DPEN).
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения координируется с кислотой Льюиса, в частности с AlCl3.
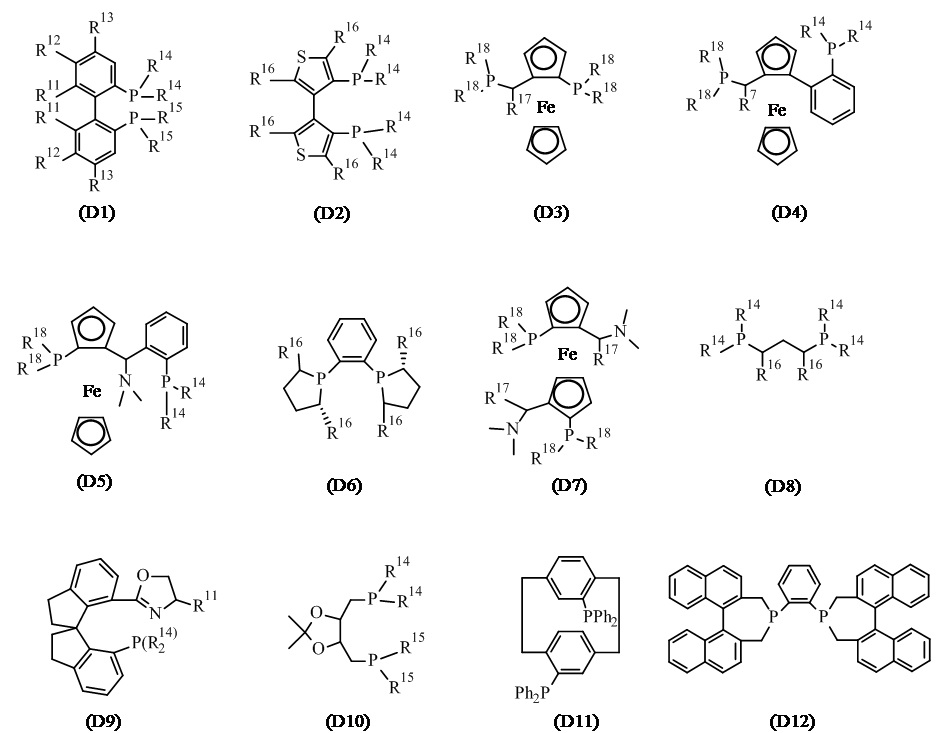
В одном варианте реализации изобретения, хиральный фосфиновый лиганд D выбирают из соединения формулы (D1) - (D12):
где:
R11 представляет собой C1-7 алкил, C1-7 алкокси, бензилокси, гидрокси или C1-7 алкил-C(O)O-;
R12 и R13 каждый независимо представляет собой водород, C1-7 алкил, C1-7 алкокси или ди(C1-7 алкил)амино; или
R11 и R12, присоединенные к той же самой фенильной группе, или R12 и R13 присоединенные к той же самой фенильной группе, взятые вместе, представляют собой-X-(CH2)r-Y-, где X представляет собой -O-, или -C(O)O-, Y представляет собой -O-, -N(нижний алкил)-, или -CF2- и R представляет собой целое число от 1 дo 6; или
два R11 взятые вместе, представляют собой-O-(CH2)s-O- или O-CH(CH3)-(CH2)s-CH(CH3)-O-, где s представляет собой целое число от 1 до 6; или
R11 и R12, или R12 и R13, вместе с атомами углерода, к которым они присоединены, образуют нафтильное, тетрагидронафтильное или дибензофурановое кольцо;
R14 и R15 каждый независимо представляет собой C1-7 алкил, C3-8 циклоалкил, фенил, нафтил или гетероарил, необязательно замещенный 1-7 заместителями, независимо выбранными из группы, состоящей из C1-7 алкил, C1-7 алкокси, ди(C1-7 алкил)амино, морфолинил, фенил, три(C1-7 алкил)силил, C1-7 алкоксикарбонил, гидроксикарбонил, гидроксисульфонил, (CH2)t-OH и (CH2)t-NH2, где t представляет собой целое число от 1 до 6;
R16 представляет собой C1-7 алкил;
R17 представляет собой C1-7 алкил; и
R18 независимо представляет собой арил, гетероарил, C3-8 циклоалкил или C1-7 алкил.
В конкретном варианте реализации изобретения, хиральный фосфиновый лиганд (D) выбирают из соединения формулы (D1), где R11 - R15 являются такими, как описано в данном документе.
В конкретном варианте реализации изобретения, хиральный фосфиновый лиганд (D) выбирают из (R)-3,5-Xyl-BINAP, (R)-BINAP, (S)-2-фурил-MeOBIPHEP, (S)-BINAP, (S)-BINAPHANE,(S)-BIPHEMP, (S)-MeOBIPHEP, (S)-pTol-BINAP), (S)-TMBTP и (с,S)-iPr-DUPHOS.
В конкретном варианте реализации изобретения, хиральный фосфиновый лиганд (D) выбирают из (S)-BIPHEMP, (S)-BINAP, и (S)-MeOBIPHEP.
В конкретном варианте реализации изобретения, хиральный фосфиновый лиганд (D) представляет собой (S)-BINAP.
В конкретном варианте реализации изобретения, хиральный фосфиновый лиганд (D) представляет собой (S)-2,2'-Бис(дифенилфосфино)-1,1'-бинафтил.
В конкретном варианте реализации изобретения, хиральный фосфиновый лиганд (D) представляет собой
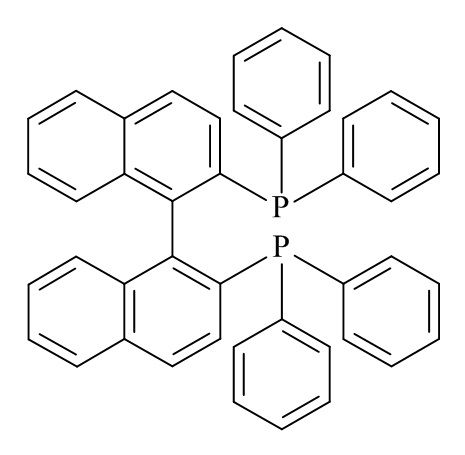 .
.
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения выбирают из группы:
Ru(TFA)2((R)-3,5-Xyl-BINAP),
Ru(OAc)2((S)-2-фурил-MeOBIPHEP),
Ru(OAc)2((S)-BINAP),
[Ru(OAc)2((S)-BINAP)]AlCl3,
Ru(TFA)2((S)-BINAP),
Ru(TFA)2((S)-BINAPHANE),
Ru(TFA)2((S)-BIPHEMP),
Ru(OAc)2((S)-MeOBIPHEP),
Ru(TFA)2((S)-TMBTP),
Ru(TFA)2((с,S)-iPr-DUPHOS),
[Ru((R)-BINAP)(pCym)(AN)](BF4)2,
[RuBr((S)-BINAP)(C6H6)]Br,
[RuCl((S)-BINAP)(C6H6)]BF4,
[RuCl((S)-BINAP)(C6H6)]Cl,
[RuI((S)-BINAP)(C6H6)]I,
[Ru((S)-BINAP)(AN))4](BF4)2, и
RuCl2((S)-pTol-BINAP)(с,S-DPEN).
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения выбирают из группы:
Ru(TFA)2((R)-3,5-Xyl-BINAP),
Ru(OAc)2((S)-2-фурил-MeOBIPHEP),
Ru(OAc)2((S)-BINAP),
[Ru(OAc)2((S)-BINAP)]AlCl3,
Ru(TFA)2((S)-BINAP),
Ru(TFA)2((S)-BINAPHANE),
Ru(TFA)2((S)-BIPHEMP),
Ru(OAc)2((S)-MeOBIPHEP),
Ru(TFA)2((S)-TMBTP),
Ru(TFA)2((с,S)-iPr-DUPHOS),
[Ru((R)-BINAP)(pCym)(AN)](BF4)2,
[RuBr((S)-BINAP)(C6H6)]Br,
[RuCl((S)-BINAP)(C6H6)]BF4,
[RuI((S)-BINAP)(C6H6)]I,
[Ru((S)-BINAP)(AN))4](BF4)2, и
RuCl2((S)-pTol-BINAP)(с,S-DPEN).
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения представляет собой соединение формулы (C1), выбранное из группы:
Ru(TFA)2((R)-3,5-Xyl-BINAP),
Ru(OAc)2((S)-2-фурил-MeOBIPHEP),
Ru(OAc)2((S)-BINAP),
[Ru(OAc)2((S)-BINAP)]AlCl3,
Ru(TFA)2((S)-BINAP),
Ru(TFA)2((S)-BINAPHANE),
Ru(TFA)2((S)-BIPHEMP),
Ru(OAc)2((S)-MeOBIPHEP),
Ru(TFA)2((S)-TMBTP), и
Ru(TFA)2((с,S)-iPr-DUPHOS).
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения представляет собой соединение формулы (C2), выбранное из группы:
[Ru((R)-BINAP)(pCym)(AN)](BF4)2,
[RuBr((S)-BINAP)(C6H6)]Br,
[RuCl((S)-BINAP)(C6H6)]BF4,
[RuCl((S)-BINAP)(C6H6)]Cl,
[RuI((S)-BINAP)(C6H6)]I, и
[Ru((S)-BINAP)(AN)4](BF4)2.
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения представляет собой соединение формулы (C3), в частности RuCl2((S)-pTol-BINAP)(с,S-DPEN)).
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения представляет собой Ru(TFA)2((S)-BINAP).
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения представляет собой [RuCl(S-BINAP)(C6H6)]Cl.
В конкретном варианте реализации изобретения, катализатор на основе комплексов рутения не является [RuCl(S-BINAP)(C6H6)]Cl.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят в растворителе, выбранном из спиртов, углеводородов, хлорированных углеводородов, фторированных и полифторированных алифатических или ароматических углеводородов, надкритического или жидкого диоксида углерода, ТГФ, воды или их смесей.
Конкретные растворители для асимметричной гидрогенизации представляют собой спирты, хлорированные углеводороды и ТГФ.
Конкретные растворители для асимметричной гидрогенизации выбирают из списка MeOH, EtOH, i-PrOH, EtOH/циклопентил метиловый эфир, EtOH/CH2Cl2, EtOH/EtOAc, EtOH/ТГФ, EtOH/H2O, CH2Cl2 и ТГФ.
Наиболее конкретным растворителем для асимметричной гидрогенизации является этанол (EtOH).
Растворители могут быть использованы по отдельности или в виде смеси растворителей, указанных выше.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят при концентрации соединения формулы (IV) 1-50% мас, в частности 5% мас, 10% мас, 20% мас или 30% мас.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят при концентрации 10-25% мас соединения формулы (IV).
Неожиданно было обнаружено, что в особых случаях добавление определенных добавок улучшает асимметричную гидрогенизацию соединения формулы (IV). Существует гипотеза, что активность, а также стабильность рутениевого катализатора существенно улучшена и поэтому уменьшается необходимое количество катализатора. Более низкие используемые количества катализатора приводят к упрощению обработки и снижению стоимости.
В конкретном варианте реализации изобретения, асимметричная гидрогенизация соединения формулы (IV) дополнительно содержит одну или несколько добавок, выбранных из списка LiBF4, LiPF6, LiO3SCF3, NaCl, NaBr, NaI, KCl, KBr, KI, LiCl, LiBr, LiI, HBF4, HCl, HBr, H2SO4, и CH3SO3H.
В конкретном варианте реализации изобретения, асимметричная гидрогенизация соединения формулы (IV) не содержит LiBF4, LiPF6 или LiO3SCF3 в качестве добавки. Ввиду их высоко коррозионного характера, такие фторид-содержащие добавки сложно обрабатывать и поэтому они не являются предпочтительными.
В конкретном варианте реализации изобретения, асимметричная гидрогенизация соединения формулы (IV) дополнительно содержит одну или несколько добавок, выбранных из списка NaCl, NaBr, KCl, KBr, HCl и HBr.
В конкретном варианте реализации изобретения, асимметричная гидрогенизация соединения формулы (IV) дополнительно содержит одну или несколько добавок, выбранных из списка LiBF4, HBF4, HCl, H2SO4, и CH3SO3H.
В конкретном варианте реализации изобретения, асимметричная гидрогенизация соединения формулы (IV) дополнительно содержит одну или несколько добавок, выбранных из списка LiBF4, NaCl, NaBr, LiCl, LiBr, LiI, HBF4, HCl, HBr, H2SO4 и CH3SO3H.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят с водородом в качестве источника водорода.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят при давлении водорода 1-150 бар, в частности 10-30 бар, наиболее особенным образом 17-21 бар.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят при температуре 10-120°C, в частности 20-90°C.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят в течение периода времени 5-30 часов, в частности 6-25 часов, более особенным образом 6-23 часов.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят при соотношении субстрат/катализатор (S/C) 5-100’000, в частности 100-15’000, наиболее особенным образом 100-10’000.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят периодически.
В конкретном варианте реализации изобретения, асимметричную гидрогенизацию соединения формулы (IV) проводят непрерывным образом.
Один из аспектов данного изобретения относится к способу получения соединений формулы (II)
(II)
включающему асимметричную гидрогенизацию соединения формулы (IV)
(IV)
при помощи катализатора на основе металлокомплексов (C), с образованием соли путем добавления к гидрогенизационной смеси спиртового раствора алкоксида металла формулы C1-7 алкил-OM, где R1 и M являются такими, как определено в данном документе.
Один из аспектов данного изобретения относится к способу получения соединений формулы (II), включающему асимметричную гидрогенизацию соединения формулы (IV) при помощи катализатора на основе металлокомплексов (C), с образованием соли путем добавления к гидрогенизационной смеси спиртового раствора алкоксида металла формулы C1-7 алкил-OM, без предварительного выделения или очистки кислотного промежуточного вещества, где R1 и M являются такими, как определено в данном документе.
В конкретном варианте реализации изобретения, алкоксид металла, используемый на стадии образования соли, представляет собой MeOM, EtOM, iPrOM, nPrOM, nBuOM, iBuOM или tBuOM, наиболее особенным образом EtOM.
В конкретном варианте реализации изобретения, спирт, используемый в качестве растворителя на стадии образования соли, представляет собой C1-7 алкил-OH, более особенным образом MeOH, EtOH, iPrOH, nPrOH, nBuOH, iBuOH или tBuOH, наиболее особенным образом EtOH.
Один из аспектов данного изобретения относится к способу получения соединений формулы (II), включающему асимметричную гидрогенизацию соединения формулы (IV) при помощи катализатора на основе металлокомплексов (C), с образованием соли путем добавления к гидрогенизационной смеси этанольного раствора этоксида натрия.
Соединения формулы (IV) могут быть получены в соответствии со способами известными специалистам в данной области техники. Конкретный общий способ получения соединений формулы (IV) приведен на Схеме 2. Более подробное описание отдельных стадий реакции приведено в разделе Примеры ниже.
Схема 2
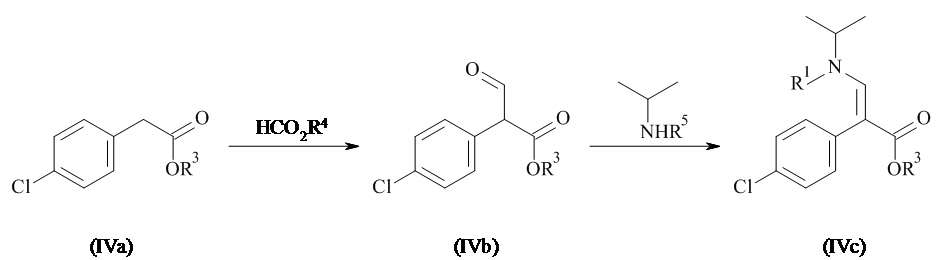
Соединение формулы (IVa), где R3 представляет собой необязательно замещенный C1-7 алкил, в особенности этил, конденсируется в основных условиях с соединением HCO2R4, где R4 является необязательно замещенным C1-7 алкилом, в особенности этилом, с образованием соединения формулы (IVb). Дополнительная конденсация соединений формулы (IVb) с амином HN(изопропил)R5, где R5 представляет собой водород, C1-7 алкил или аминозащитную группу, образует соединения формулы (IVc). Когда R5 представляет собой водород в соединениях формулы (IVc), дополнительная защита амина может быть выполнена с образованием защищенных соединений формулы (IVc) (например, где R5 представляет собой аминозащитную группу, такую как Вос). Гидролиз сложного эфира соединения (IVc) приводит к получению соединения формулы (IV).
Катализаторы на основе комплексов рутения в соответствии с данным изобретением могут в принципе быть получены способом, известным как таковой. Они могут быть выделены или использованы непосредственно (получение in situ), например, в соответствии с B. Heiser et al., Tetrahedron: Asymmetry 1991, 2, 51; или N. Feiken et al., Organometallics 1997, 16, 537; или J.-P. Genet, Acc. Chem. Res. 2003, 36, 908; или K. Mashima et al., J. Org. Chem. 1994, 53, 3064; Angew. Chem. Int. Ed. 1998, 37, 1703-1707; или M.P. Fleming et al., US 6,545,165 B1, и ссылками, приведенными в них; а также O. Briel et al. in Catalysis of Organic Reactions, CRC Press, Boca Raton, 2009 особенно для комплексов Ru на основе ферроцена, описания всех этих документов включены в данный документ полностью для всех целей.
Синтез[Ru(TFA)2((S)-BINAP)] описан в B. Heiser et al, Tetrahedron: Asymmetry 1991, 2, 51.
Катализаторы на основе комплексов рутения могут быть получены in situ, то есть непосредственно перед использованием и без выделения. Раствор, в котором такой катализатор получают, может уже содержать субстрат для энантиоселективной гидрогенизации или раствор может быть смешан с субстратом непосредственно перед тем, как инициируют реакцию гидрогенизации.
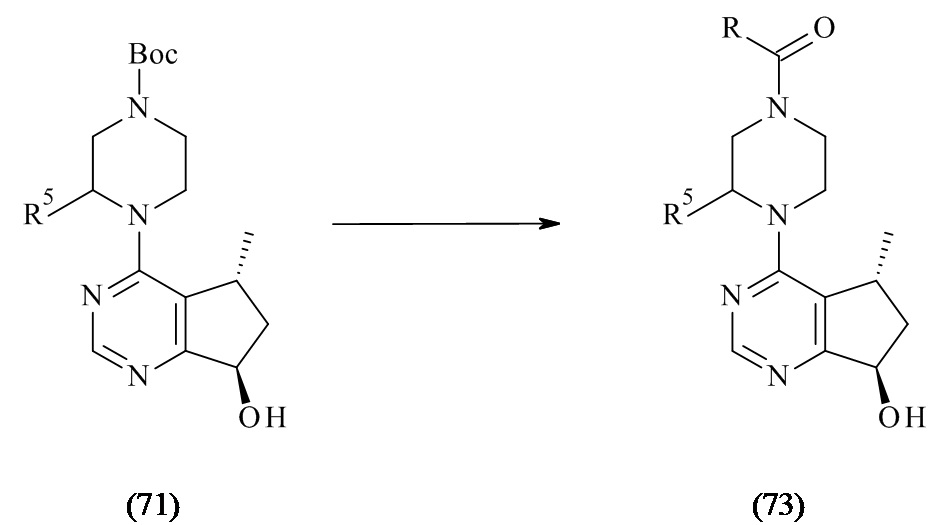
В WO 2008/006040 раскрыты 5-метил-6,7-дигидро-5Н-циклопента[d]пиримидин-7-олы формулы (71) и способы их получения, где R5 могут иметь различные альтернативы.
 (71)
(71)
В частности, в WO 2008/006040 раскрывает асимметричное восстановление 5-метил-5,6-дигидроциклопента[d]пиримидин-7-онов в (R) или (S)-5-метил-6,7-дигидро-5Н циклопента[d]пиримидин-7-олов с применением хирального катализатора в присутствии водорода, катализатора Кори-Бакша-Шибата (CBS), боргидридного восстановителя в присутствии хирального лиганда, или нехирального восстановителя (например, H2, Pd/C).
Способы, известные в данной области техники для получения соединений формулы (III), проявляют присущие им недостатки, они требуют жестких условий реакции (например, высокого давления), использования тяжелых металлов и хиральных вспомогательных веществ, и полученная диастереоселективность только ограниченная (т.е. 88% де), таким образом требуя дополнительных стадий очистки.
Авторами данного изобретения были найдены новые ферментные процессы для получения соединений формулы (III), где R2 является таким, как описано в данном документе.
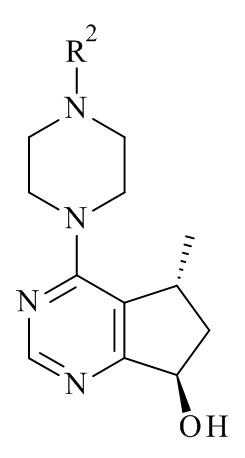 (III)
(III)
Эти новые способы получения соединений формулы (III) в соответствии с настоящим изобретением имеют ряд соответствующих преимуществ по сравнению с процессом, как известно в данной области техники. Преимуществами ферментативного восстановления являются его каталитическая природа, очень высокая диастереоселективность, чтобы избежать потенциальной необходимости последующей пептизации диастереоизомеров, образованных и мягких условиях реакции. Кроме того, никакие тяжелые металлы и хиральные вспомогательные вещества не требуются.
Ферментативное восстановление в соответствии с данным изобретением упрощает технические требования, сокращает перечень и количество ингредиентов и позволяет обеспечить более высокий пространственно-временной выход. Преимущества в соответствии с данным изобретением иллюстрируются как улучшенные технические соответствующие критерии, такие, как увеличение концентрации субстрата (до 25%), увеличение концентрации продукта (до 25%), снижение кофакторной нагрузки (вплоть до 1/3000 соединения формулы (V)) и более простая система кофакторной регенерации с 2-пропанолом в качестве конечного восстановителя. Система кофакторной регенерации с 2-пропанолом в качестве конечного восстановителя избегает второго фермента, снижает вязкость, позволяет избежать непрерывной нейтрализации глюконовой кислоты в качестве окисленного ко-субстрата и позволяет непрерывное удаление ацетона, который образуется.
Один из аспектов в соответствии с данным изобретением относится к способу получения соединений формулы (III)
(III)
включающему асимметричное восстановление соединения формулы (V)
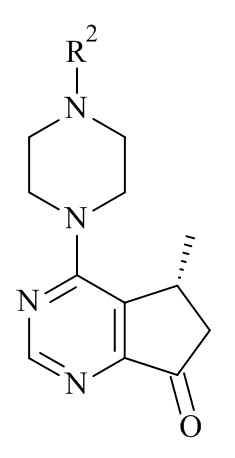 (V)
(V)
катализируемое оксидоредуктазой, где R2 является такой, как определено в данном документе.
В одном аспекте данного изобретения, оксидоредуктаза, которая катализирует асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой кеторедуктазу.
В одном аспекте данного изобретения, оксидоредуктаза катализирует асимметричное восстановление соединения формулы (V) в соединение формулы (III), с диастереоселективностью по меньшей мере 95% диастереомерного избытка (de), в частности, с диастереоселективностью по меньшей мере 98% de, в частности, с диастереоселективность по меньшей мере 99% de.
В одном аспекте данного изобретения, асимметричное восстановление соединения формулы (V) в соединение формулы (III), катализируется оксидоредуктазой в присутствии кофактора.
В одном аспекте данного изобретения, кофактор, который окисляется при асимметрическом восстановлении соединения Формулы (V) в соединение формулы (III), представляет собой NADH или NADPH.
В одном аспекте данного изобретения, кофактор, который окисляется при асимметрическом восстановлении соединения Формулы (V) в соединение формулы (III), регенерируется in situ применением фермента в сочетании с кофактором регенерации (например, на основе глюкозы в качестве конечного восстановителя и глюкоза дегидрогеназы) или субстрат-комбинированной регенерацией (например, с использованием вторичного спирта в качестве ко-субстрата).
В одном аспекте данного изобретения, кофактор, который окисляется при асимметрическом восстановлении соединения формулы (V) в соединение формулы (III), регенерируется in situ применением фермента в сочетании с кофактором регенерации с использованием глюкозы и глюкоза-дегидрогеназы в качестве ко-субстрата.
В одном аспекте данного изобретения, кофактор, который окисляется при асимметрическом восстановлении соединения формулы (V) в соединение формулы (III), регенерируется in situ регенерацией в сочетании с субстратом с использованием вторичного спирта в качестве ко-субстрата.
В одном аспекте данного изобретения, вторичный спирт в качестве ко-субстрата для регенерации в сочетании с субстратом выбирают из 2-пропанола, 2-бутанола, бутан-1,4-диола, 2-пентанола, пентан-1,5-диола, 4-метил-2-пентанола, 2-гексанола, гексан-1,5-диола, 2-гептанола, или 2-октанола, в частности, 2-пропанола.
Особенно полезным является 2-пропанол для регенерации кофактора на том же ферменте, также катализируя целевую реакцию и непрерывное удаление ацетона, который образуется.
В одном аспекте данного изобретения, оксидоредуктаза, которая катализирует асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу.
В одном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу, выбранную из списка:
KRED-NADPH-111 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
KRED-NADPH-112 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
KRED-NADPH-113 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
KRED-NADPH-114 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
KRED-NADPH-115 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
KRED-NADPH-121 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
KRED-NADPH-123 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
KRED-NADPH-145 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
KRED-NADPH-155 (от Codexis Inc., Редвуд-Сити, Калифорния, США),
A231 (от Almac Group Ltd. Крэйгавон, Великобретания), и
KRED-NADPH-136 (от Enzysource, Хангжу, Китай).
Дополнительные подходящие оксидоредутазы, катализирующие асимметричное восстановление соединения формулы (V) в соединение формулы (III) представляют собой диастереоселективную NADPH-зависимую оксидоредуктазу, выбранную из списка:
KRED-X1, генетически сконструированная кеторедуктаза от Lactobacillus kefir, как описано в международной публикации РСТ № WO2010/025238A2, и идентифицированная как SEQ. ID. NO. 34, и
KRED-X2, генетически сконструированная кеторедуктаза от Sporobolomyces salmonicolor как описано в международной публикации РСТ № WO2009/029554A2, и идентифицированная как SEQ. ID. NO. 138.
Дополнительные подходящие оксидоредуктазы, катализирующие асимметричное восстановление соединения формулы (V) в соединение формулы (III) представляют собой варианты KRED-X1, которые коммерчески доступны (от Codexis Inc., Редвуд-Сити, Калифорния, США).
В частности полезной является кеторедуктаза KRED-X1-P1B06, KRED вариант P1B06 от Codexis KRED специальный планшетный продукт KRED-X1-SPECIALTY-PLT.
Дополнительные подходящие оксидоредуктазы, катализирующие асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляют собой варианты KRED-X1, которые коммерчески доступны (от Codexis Inc., Редвуд-Сити, Калифорния, США). В частности полезными являются следующие сконструированные редуктазы от Codexis KRED специальный планшетный продукт ʺKRED-X1.1-B06-SPECIALTY-PLTʺ:
ʺKRED-X1.1-P1F01ʺ (KRED вариант P1F01),
ʺKRED-X1.1-P1H10ʺ (KRED вариант P1H10),
ʺKRED-X1.1-P1G11ʺ (KRED вариант P1G11),
ʺKRED-X1.1-P1C04ʺ (KRED вариант P1C04),
ʺKRED-X1.1-P1C11ʺ (KRED вариант P1C11), и
ʺKRED-X1.1-P1C08ʺ (KRED вариант P1C08).
В частности полезными являются сконструированные редуктазы ʺKRED-X1.1-P1C04ʺ и ʺKRED-X1.1-P1F01ʺ. Наиболее особой кеторедуктазой является сконструированная кеторедуктаза ʺKRED-X1.1-P1F01ʺ.
Международные публикации РСТ № WO2010/025085A2 и WO2009/029554A2 настоящим включены путем ссылки полностью для всех целей, в частности аспектов в данном документе, относящихся к получению и использованию оксидоредуктаз.
Все из указанных выше ферментов могут использовать также кофактор NADH.
В одном конкретном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу выбранную из списка KRED-NADPH-111, KRED-NADPH-112, KRED-NADPH-113, KRED-NADPH-114, KRED-NADPH-115, KRED-NADPH-121, KRED-NADPH-123, KRED-NADPH-145, KRED-NADPH-155, A231, KRED-NADPH-136, KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1F01, KRED-X1.1-P1H10, KRED-X1.1-P1G11, KRED-X1.1-P1C04, KRED-X1.1-P1C11 и KRED-X1.1-P1C08.
В одном конкретном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу выбранную из спискаKRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1F01, KRED-X1.1-P1H10, KRED-X1.1-P1G11, KRED-X1.1-P1C04, KRED-X1.1-P1C11, и KRED-X1.1-P1C08.
В одном конкретном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу выбранную из списка KRED-X1, KRED-X2, KRED-X1-P1B06, KRED-X1.1-P1C04 и KRED-X1.1-P1F01.
В одном конкретном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу, выбранную из списка KRED-X1, KRED-X1-P1B06, KRED-X1.1-P1C04 and KRED-X1.1-P1F01.
В одном конкретном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу, выбранную из списка KRED-X1 и KRED-X2.
В одном конкретном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу, выбранную из списка KRED-X1 и KRED-X1-P1B06.
В одном конкретном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу выбранную из списка KRED-X1.1-P1C04 и KRED-X1.1-P1F01.
В одном конкретном аспекте данного изобретения, оксидоредуктаза, катализирующая асимметричное восстановление соединения формулы (V) в соединение формулы (III), представляет собой диастереоселективную NADPH-зависимую оксидоредуктазу KRED-X1.1-P1F01.
В одном аспекте данного изобретения, асимметричное восстановление соединения формулы (V) в соединение формулы (III) выполняют в водной среде в присутствии одного или нескольких органических сорастворителей.
В одном аспекте данного изобретения, асимметричное восстановление соединения формулы (V) в соединение формулы (III) выполняют в водной среде в присутствии одного или нескольких органических сорастворителей, где органические сорастворители присутствуют в общей концентрации от 1 до 50% об, в частности от 4 до 40% об.
В одном аспекте данного изобретения, сорастворители, присутствующие в асимметричном восстановлении соединения формулы (V) в соединение формулы (III) выбирают из списка глицерин, 2-пропанoл, диэтиловый эфир, трет-бутилметиловый эфир, диизопропиловый эфир, дибутиловый эфир, метил тетрагидрофуран, этилацетат, бутилацетат, толуол, гептан, гексан, циклогексен и их смеси; в частности 2-пропанoл.
2-пропанoл, в частности, полезен как сорастворитель, поскольку может служить конечным восстановителем для субстрат-комбинированной кофакторной регенерации.
В одном аспекте данного изобретения, асимметричное восстановление соединения формулы (V) в соединение формулы (III) выполняют при температуре реакции от 1°C до 50°C, в частности от 20°C до 45°C.
Температуры в верхнем диапазоне увеличивают скорость реакции и способствуют удалению ацетона.
В одном аспекте данного изобретения, асимметричное восстановление соединения формулы (V) в соединение формулы (III) выполняют при pH от 5,5 до 8,5.
В одном аспекте данного изобретения, асимметричное восстановление соединения формулы (V) в соединение формулы (III) выполняют в водном буфере. Подходящие буферы известны специалисту в данной области. Конкретными буферами являются 2-(н-морфолино)этансульфоновая кислота (MES) или кислый фосфат калия (PBS).
Оптимальный pH диапазон и поэтому любые приемлемые буферы зависят от конкретной используемой оксидоредуктазы.
Один аспект данного изобретения относится к асимметричному восстановлению соединения формулы (V) в соединение формулы (III), где соединение формулы (V) изначально присутствует при концентрации 1-25% мас, в частности 10-20% мас.
Один аспект данного изобретения относится к асимметричному восстановлению соединения формулы (V) в соединение формулы (III), где концентрация реакции (общая концентрация кетона формулы (V) и хирального спирта формулы (III) в реакционной смеси) составляет от 1 до 25% мас, в частности от 10 до 20% мас.
Один из аспектов данного изобретения относится к способу получения соединений формулы (III), включающему асимметричное восстановление соединения формулы (V), катализируемому оксидоредуктазой, с последующей обработкой путем экстракции или фильтрованием.
Один аспект данного изобретения относится к асимметричному восстановлению соединения формулы (V) в соединение формулы (III), катализируемому оксидоредуктазой, где продукт традиционным образом обрабатывают путем экстракции или фильтрованием.
Чистота неочищенного продукта может быть дополнительно повышена путем кристаллизации или он может быть использован как есть в последующих последовательностях реакций для получения соединений формулы (I).
Один из аспектов данного изобретения относится к способу получения соединений формулы (III), включающему асимметричное восстановление соединения формулы (V), катализируемому оксидоредуктазой, с последующей обработкой путем экстракции или фильтрованием и дополнительно кристаллизацией.
Один аспект данного изобретения относится к асимметричному восстановлению соединения формулы (V) в соединение формулы (III), где продукт традиционным образом обрабатывают путем экстракции или фильтрованием и дополнительно кристаллизацией.
Один аспект данного изобретения относится к способу получения соединений формулы (IVc):
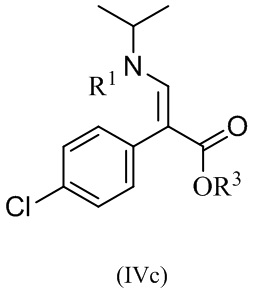 ,
,
или его соли, где R1 и R3 определены в данном документе, включающему приведение в контакт соединение формулы (IVd):
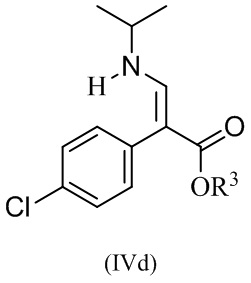 ,
,
или его соли, с R1-X, где X представляет собой отходящую группу, в условиях, достаточных для получения соединения формулы IVc или его соли.
В одном варианте реализации изобретения, способ включает получение этил (E)-3-(трет-бутоксикарбонил(изопропил)-амино)-2-(4-хлорофенил)акрилат, или его соли, где R1 представляет собой BOC защитную группу, R3 представляет собой этил, и где R1-X представляет собой (BOC)2O.
В одном конкретном варианте реализации изобретения, способ включает приведение в контакт соединения формулы IVd или его соли с менее чем около 8 эквивалентами (BOC)2O, в частности менее чем около 4 эквивалентов, более особенным образом около 3 эквивалента в условиях, приводящих к получению соединения формулы IVc или его соли с выходами более чем около 50%, в частности около 75% или больший выход, в смеси полярных растворителей, содержащих DMF.
В одном более конкретном варианте реализации изобретения, условия включают приведение в контакт соединения формулы IVd или его соли с около 3 эквивалентами (BOC)2O, и основной смесью, содержащей около 2 эквивалента каждая, трибутиламина и диметиламинопиридина (DMAP), в смеси полярных растворителей, содержащих DMF. В варианте реализации изобретения, способ дополнительно включает удаление части жидкости из реакционной смеси под вакуумом во время добавления (BOC)2O.
Соединения формулы (V) могут быть получены в соответствии со способами известными специалистам в данной области техники. Конкретный общий способ получения соединений формулы (V) приведен на Схеме 3. Более подробное описание отдельных стадий реакции, приведено в разделе Примеры ниже.
Схема 3
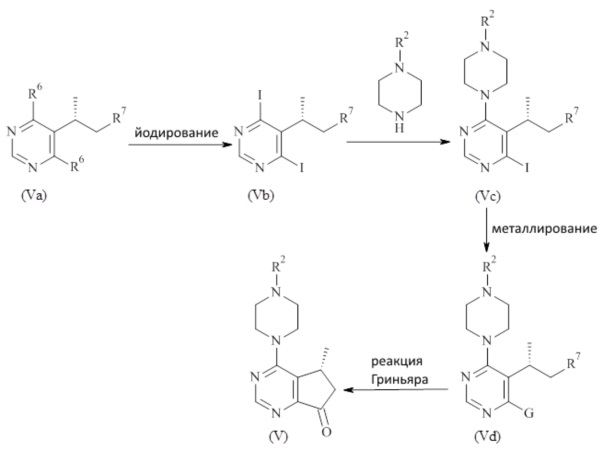
Реакция соединения формулы (Va) с агентом йодирования (например йодидной солью, такой как NaI и необязательно с кислотой) приводит к получению дийодоoпиримидина формулы (Vb), который может дополнительно реагировать с моно-защищенным пиперазином с получением соединения формулы (Vc). Соединение формулы (Vc) метилировано агентом метилирования, таким как реактив Гриньяра (например C1-7 алкилмагний галид, такой как iPrMgCl) с образованием соединения формулы (Vd) которое дополнительно циклизуется с образованием циклопентил кетона формулы (V), где
R2 является таким, как описано в данном документе,
G представляет собой Li или Mg,
R6 представляет собой Cl или OH,
R7 представляет собой -CN, -COORa или -CONRaRb, где Ra и Rb независимо выбраны из списка водород, -OH, C1-7 алкокси, C1-7 алкил, C2-7 алкенил, C2-7 алкинил, C3-8 циклоалкил, фенил или 3-12 членный гетероциклоалкил; или Ra и Rb взятые вместе с атомом азота, к которому они присоединены, образуют 3-7 членный гетероциклоалкил.
WO 2008/006040 описывает способы получения соединений формулы (73), где соединения формулы (71) после снятия защитной группы при помощи кислоты, ацилированы соответствующей аминокислотой, где R и R5 могут иметь различные альтернативы.
Реакции ацилирования, как описано в предшествующем уровне техники, проявляют следующие недостатки:
• Способ, включающий HBTU в качестве агента сочетания, не подходит для крупномасштабного коммерческого способа производства. HBTU повышает серьезные проблемы, связанные с промышленной гигиеной, поскольку случаи анафилаксии и профессиональной контактной аллергии описаны в литературе (Hannu T. et al, Occup Med, 2006, 56 (6), 430-433 and M. A. Aleer et al, Contact Dermatitis, 2010 62, 2 123).
• Способ, включающий дихлорметан в виде растворителя, не подходит для крупномасштабного процесса химического производства, поскольку классифицирован как опасный загрязнитель воздуха (HAP) в США. Дополнительно, дихлорметан оценен Международной конференцией по гармонизации (ICH) как растворитель класса 2 с ограниченным разрешенным ежедневным воздействием (PDE) ввиду присущей ему токсичности.
• Очистка продукта с использованием хроматографии не является приемлемым способом очистки для крупномасштабного низкомолекулярного производства ввиду его очень высокого потребления растворителя и низкой продуктивности.
• В случае коммерческой крупномасштабной реакции способ включает более одного растворителя в смеси, растворители требуют различных точек кипения, которые достаточно различаются друг от друга, для того, чтобы позволить их разделение и повторное использование при помощи перегонки. Способы, включающие на той же стадии четыре растворителя (например с циклопентил метиловым эфиром (CPME)) которые не являются повторно используемыми, поскольку смесь не может быть разделена, не подходят для крупномасштабного производства.
• Обработка продуктов требует многочисленных (например, шести) водных экстракций, все из них с концентрированными органическими солями, приводя к получению значительных количеств загрязненных сточных вод. Такие условия способа приводят к неблагоприятному производственному процессу с точки зрения загрязнения окружающей среды.
Авторами данного изобретения был открыт новый усовершенствованный способ получения соединения формулы (I), включающий сочетание соединения формулы (II), которое является солью, в частности натриевой солью, с соединением формулы (III). Было найдено, что использование соединения формулы (II) в виде соли, в частности натриевой соли, в значительной степени облегчает и упрощает такой процесс, по сравнению с использованием свободной аминокислоты.
Способ получения соединений формулы (I) в соответствии с данным изобретением отличается рядом соответствующих преимуществ по сравнению со способами, описанными в уровне техники, среди прочего, например:
• Обработка соединения формулы (I) значительно улучшена. Используют только три растворителя (изопропанoл, толуол и гептан), которые хорошо разделяются.
• Пропилфосфониевый ангидрид (T3P) является нетоксичным агентом сочетания без аллергических или сенсибилизирующих свойств.
• Побочные продукты реакции водорастворимы и могут поэтому легко могут быть удалены например тройной водной экстракцией.
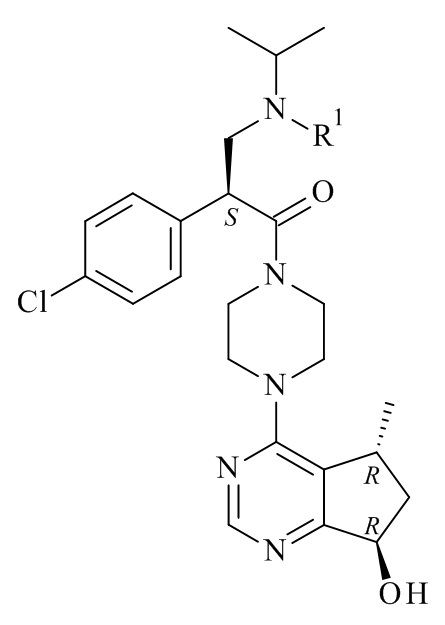
Один из аспектов данного изобретения представляет способ получения соединения формулы (I)
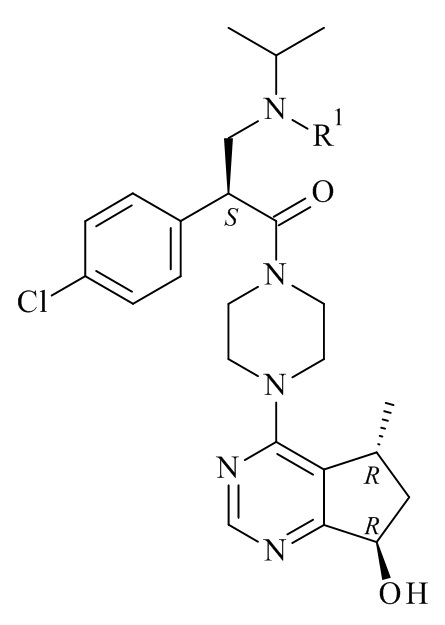 (I)
(I)
или его солей, включающий реакцию сочетания соединения формулы (II)
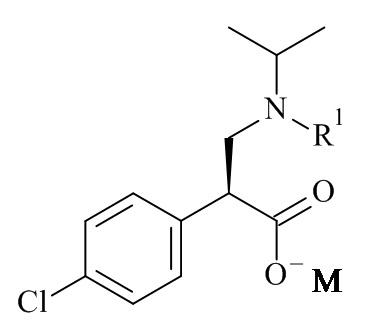 (II)
(II)
с соединением формулы (III)
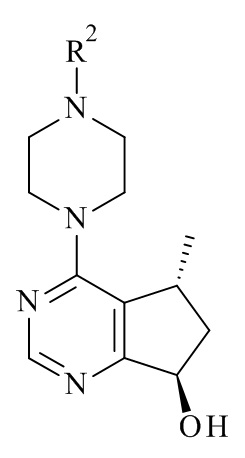 (III)
(III)
где R1, R2 и M являются такими, как определено в данном документе.
Один из аспектов данного изобретения представляет способ получения соединения формулы (I) или его солей, включающий реакцию сочетания соединения формулы (II) с соединением формулы (III) где R1, R2 и M являются такими, как определено в данном документе, включающий следующие стадии реакции:
a) Снятие защиты соединения формулы (III) в растворителе в кислотных условиях;
b) Регулирование до щелочного pH при помощи основания;
c) Добавление раствора, содержащего соединение формулы (II) в растворитель;
d) Добавление раствора, содержащего агент сочетания, в растворитель.
В одном аспекте данного изобретения, снятие защиты на стадии a) выполняют при помощи соляной кислоты, серной кислоты, трифторуксусной кислоты или бромистоводородной кислоты.
В конкретном аспекте данного изобретения, снятие защиты на стадии a) выполняют при помощи соляной кислоты.
В одном аспекте данного изобретения, растворитель, используемый для снятия защиты на стадии a) выбирают из воды, метанола, этанола, н-пропанoла, изопропанола, н-бутанола, и трет-бутанола.
В конкретном аспекте данного изобретения, растворитель, используемый для снятия защиты на стадии a) выбирают из н-пропанoла или изопропанoла.
В одном аспекте данного изобретения, снятие защиты на стадии a) выполняют при температуре от 50 до 100°C, в частности при 80°C.
В одном аспекте данного изобретения, снятие защиты на стадии a) выполняют в течение времени реакции 0.1-24 часа, в частности в течение времени реакции 1-2 часа.
В одном аспекте данного изобретения, основание на стадии b) является жидким основанием, выбранным из N-этилморфолина (NEM), триэтиламина (TEA), три(н-пропил)амина (TPA), диизопропилэтиламина (DIPEA), пиридина и лутидина.
В одном аспекте данного изобретения, основание на стадии b) представляет собой N-этилморфолин (NEM).
В одном аспекте данного изобретения, на стадии b) 4-8 эквивалента основания добавляют относительно формулы (III), в частности 6-7 эквивалента основания, наиболее особенным образом 6,5 эквивалента основания.
В одном аспекте данного изобретения, растворитель, используемый на стадии c) идентичен растворителю, используемому на стадии a).
В одном аспекте данного изобретения, растворитель, используемый на стадии c) выбирают из воды, метанола, этанола, н-пропанoла, изопропанола, н-бутанола, и трет-бутанола.
В конкретном аспекте данного изобретения, растворитель на стадии c) выбирают из н-пропанoла или изопропанола.
В одном аспекте данного изобретения, агент сочетания, используемый на стадии d) представляет собой пропилфосфониевый ангидрид (T3P).
В одном аспекте данного изобретения, растворитель, используемый на стадии d) выбирают из метанола, этанола, н-пропанoла, изопропанола, н-бутанола, трет-бутанола, толуола, ацетонитрила, тетрагидрофурана, N,N-диметилформамина, хлороформа, метиленхлорида, дихлорметана, дихлорэтана, диэтилового эфира, ацетона, метил этил кетона, диметил сульфоксида, N,N-диметил ацетамида, N-метил пирролидинона, диоксана, тетрагидропирана, пиридина, 2-пропанoна, 2-бутанона, этиленгликоль диметилового эфира, этилацетата, бутил ацетата, изопропил ацетата, и смесей вышеуказанных соединений.
В конкретном аспекте данного изобретения, растворитель, используемый на стадии d) выбирают из смеси н-пропанoла и толуола или изопропанола и толуола, наиболее особенным образом смеси н-пропанoла и толуола.
В одном аспекте данного изобретения, реакцию сочетания на стадии d) выполняют при температуре от -10 до 50°C, в частности от 0 до 25°C.
В одном аспекте данного изобретения, реакцию сочетания на стадии d) выполняют в течение времени реакции 0,1-24 часа, в частности в течение времени 1-4 часа.
Один из аспектов данного изобретения относится к реакции сочетания соединения формулы (II) с соединением формулы (III), где после стадии d) продукт обрабатывают водной экстракцией.
В конкретном аспекте данного изобретения, обработка продукта после стадии d) включает от одной до шести экстракций водой, в частности три экстракции водой.
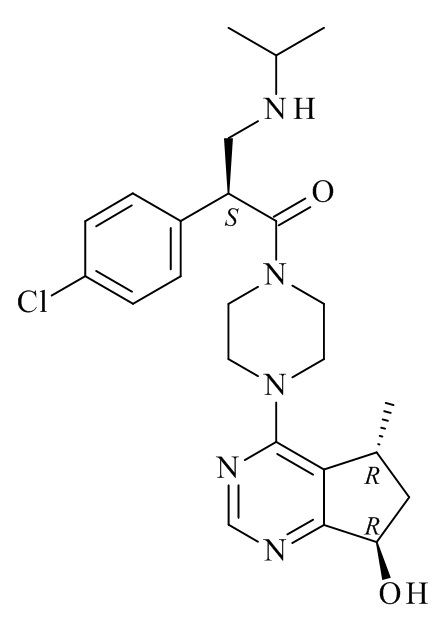
Один из аспектов данного изобретения относится к способу получения соединений формулы (VI)
 (VI)
(VI)
или его фармацевтически приемлемых солей, где с соединения формулы (I) снята защита
 (I)
(I)
где R1 является такой, как определено в данном документе.
Один из аспектов данного изобретения относится к способу получения соединений формулы (VI) или его фармацевтически приемлемых солей, где с соединения формулы (I) снята защита, где R1 является такой, как определено в данном документе, включающему следующие стадии реакции:
i) снятие защиты с соединения формулы (I) в растворителе в кислотных условиях;
ii) регулирование pH при помощи основания в растворителе;
iii) необязательно кристаллизацию соединения формулы (VI).
В одном аспекте данного изобретения, снятие защиты на стадии i) выполняют при помощи соляной кислоты, серной кислоты, трифторуксусной кислоты или бромистоводородной кислоты.
В конкретном аспекте данного изобретения, снятие защиты на стадии i) выполняют при помощи соляной кислоты.
В одном аспекте данного изобретения, растворитель, используемый для снятия защиты на стадии i) выбирают из воды, метанола, этанола, н-пропанoла, изопропанола, и трет-бутанола или их смесей.
В конкретном аспекте данного изобретения, растворитель, используемый для снятия защиты на стадии i) выбирают из н-пропанoа, изопропанола и 1:1 смеси н-пропанoл/вода.
В одном аспекте данного изобретения, снятие защиты на стадии i) выполняют при температуре от 30 до 100°C, в частности при 80°C.
В одном аспекте данного изобретения, снятие защиты на стадии i) выполняют в течение времени реакции 1-24 часа, в частности в течение времени реакции 1-4 часа.
В одном аспекте данного изобретения, основание на стадии ii) представляет собой NaOH в 1:1 смеси н-пропанoл/вода.
В одном аспекте данного изобретения, основание на стадии ii) представляет собой аммиак.
В одном аспекте данного изобретения, растворитель, используемый на стадии ii) идентичен растворителю, используемому на стадии i).
В одном аспекте данного изобретения, растворитель, используемый на стадии ii) выбирают из воды, метанола, этанола, н-пропанoла, изопропанoла, н-бутанола, и трет-бутанола или их смесей.
В конкретном аспекте данного изобретения, растворитель на стадии ii) выбирают из н-пропанола, изопропанола и 1:1 смеси н-пропанoл/вода.
В конкретном аспекте данного изобретения, регулирование pH выполняют путем добавления по каплям раствора аммиака (2-4% мас, в частности 3,8% мас) в изопропаноле или раствора NaOH (5-10M, в частности 7M) в 1:1 смеси н-пропанoл/вода.
В конкретном аспекте данного изобретения, конечное значение pH после регулирования на стадии ii) превышает pH 6, в частности от pH 6 до 7.
В одном аспекте данного изобретения, кристаллизацию на стадии iii) выполняют растворителем, переходя на растворитель кристаллизации, подходящий для кристаллизации соединения формулы (VI).
В конкретном аспекте данного изобретения, растворитель кристаллизации на стадии iii) выбирают из толуола, гептана, тетрагидрофурана, 2-пропанoна, 2-бутанона, этилeн гликоль диметилового эфира, этилацетата, бутил ацетата, изопропил ацетата и их смесей.
В конкретном аспекте данного изобретения, растворитель кристаллизации на стадии iii) представляет собой этилацетат.
Один аспект данного изобретения относится к соединению, которое может быть получено любым способом, как описано в данном документе.
Один аспект данного изобретения относится к фармацевтическим композициям, содержащим соединения, которые могут быть получены любым способом, описанным в данном документе.
Один аспект данного изобретения относится к соединению формулы (VI), как описано в данном документе, содержащему от 1 ppb до 100 ppm соединения формулы (I), где R1 является такой, как определено в данном документе.
Один аспект данного изобретения относится к соединению формулы (VI), как описано в данном документе, содержащему от 1 ppb до 1 ppm соединения формулы (I), где R1 является такой, как определено в данном документе.
Один аспект данного изобретения относится к фармацевтической композиции, содержащей соединения формулы (VI), как описано в данном документе.
Один аспект данного изобретения относится к соединению формулы (I), как описано в данном документе, содержащему от 1 ppb до 100 ppm соединения формулы (II), где R1 и M являются такими, как определено в данном документе.
Один аспект данного изобретения относится к соединению формулы (I), как описано в данном документе, содержащему от 1 ppb до 1 ppm соединения формулы (II), где R1 и M являются такими, как определено в данном документе.
Один аспект данного изобретения относится к соединению формулы (I), как описано в данном документе, содержащему от 1 ppb до 100 ppm соединения формулы (III), где R1 и R2 являются такими, как определено в данном документе.
Один аспект данного изобретения относится к соединению формулы (I), как описано в данном документе, содержащему от 1 ppb до 1 ppm соединения формулы (III), где R1 и R2 являются такими, как определено в данном документе.
Один аспект данного изобретения относится к соединению формулы (I), как описано в данном документе, содержащему от 1 ppb до 100 ppm соединения формулы (II) и от 1 ppb до 100 ppm соединения формулы (III), где R1, R2 и M являются такими, как определено в данном документе.
Один аспект данного изобретения относится к соединению формулы (I), как описано в данном документе, содержащему от 1 ppb до 1 ppm соединения формулы (II) и от 1 ppb до 1 ppm соединения формулы (III), где R1, R2 и M являются такими, как определено в данном документе.
Примеры
Следующие Примеры 1-15 представлены для иллюстрации данного изобретения. Они не должны быть рассмотрены как ограничивающие объем данного изобретения, но являются только репрезентативными.
Пример 1
(E)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)акриловая кислота
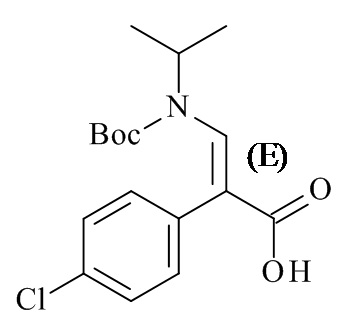
В раствор этилформиата (123,9 л, 1538,9 моль) в MTBE (189 л) добавляли этил 4-хлорофенилацетат (120 кг, 604,1 моль). Смесь перемешивали при 15-30°C в течение 30 минут и затем смесь t-BuOK (136,8 кг, 1219,1 моль) в MTBE (1215,8 л) добавляли, поддерживая внутреннюю температуру ниже 5°C. Смесь перемешивали при 0-10°C в течение 1,5 ч. Реакционную смесь добавляли в водный раствор соляной кислоты (35%, 99,8 л в 560 л H2О), поддерживая внутреннюю температуру ниже 10°C. Смесь перемешивали в течение 30 минут при 0-10°C до наблюдения конечного рН=2. Слои разделяли и органический слой промывали 25% NaCl раствором (496 л).
Смесь охлаждали до -5°C и затем изопропиламин (107,2 л, 1251,9 моль) и AcOH (70,5л, 1233,3моль) медленно добавляли, поддерживая температуру <10°C. Смесь перемешивали в течение 3 ч при 0-10°C и затем органический слой промывали H2O (760 л), 15% водным Na2CО3 (424 л) и затем 25% водным NaCl (650 л). Водный слой отделяли и DMF (443 л) и DMAP (14,4 кг, 117,9 моль) добавляли к органическому раствору. Смесь затем нагревали до 60-65°C с последующим медленным добавлением (Boc)2O (951,6 л, 4142 моль), DMF (228,6 л) и триэтиламина (263,0 л, 1821,8 моль) в течение 24 ч. После перемешивания ~6 ч, смесь охлаждали до комнатной температуры и MTBE (1434 л), воду (1010 л) и добавляли 10% водную лимонную кислоту (938 л). Водный слой отделяли и смесь промывали 25% водным NaCl (984 л). Органический слой затем концентрировали посредством перегонки до минимального рабочего объема (~240 л), поддерживая температуру ниже 50°C. Органический слой затем перемешивали в течение 5 ч при 0-5°C и фильтровали. Осадок на фильтре промывали гептаном (20,6 л) и высушивали с получением (E)-этил 3-((трет-бутоксикарбонил)(изопропил)амино)-2-(4-хлорофенил)акрилата (148,55 кг, 63% выход за три стадии) в виде белого твердого вещества.
(E)-этил 3-((трет-бутоксикарбонил)(изопропил)амино)-2-(4-хлорофенил)акрилат (133,5 кг, 362,9 моль) добавляли в смесь H2O (252 л), NaOH (58,25 кг, 1456 моль) и EtOH (383,5 л) перемешивали при комнатной температуре. Смесь нагревали до 40-45°C в течение 2,5 ч до образования прозрачного раствора. Смесь концентрировали до минимального рабочего объема, поддерживая температуру ниже 50°C. Смесь затем охлаждали до 10-25°C и раствор HCl добавляли (842 л 2N HCl и 11 л 35% HCl) до конечного pH=2~4. Водный слой отделяли и органический слой промывали 25% водным NaCl (810 л). н-гептан добавляли при перегонке с получением суспензии. Продукт собирали и промывали н-гептаном и высушивали при 40-45°C в течение ~10 ч с получением 110,7 кг (90,5% выход) (E)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)акриловой кислоты с 99,9% чистотой по ВЭЖХ. E-конфигурацию подтверждали при помощи рентгеноструктурного анализа.
Пример 1a
(E)-этил 3-((трет-бутоксикарбонил)-(изопропил)амино)-2-(4-хлорофенил)акрилат
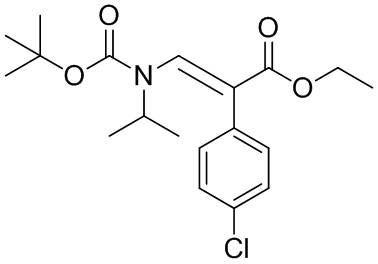
К концентрированному раствору этил 2-(4-хлорофенил)-3-(изопропиламино)акрилата (полученному, как описано выше в Примере 1 из 120 кг этил 2-(4-хлорофенил)ацетата, 0,604 кмоль) добавляли DMF (354 кг) и партию концентрировали до 3 объемов. DMAP (14,0 кг, 114,6 моль) и n-Bu3N (224,21 кг, 1,21 кмоль) добавляли и смесь нагревали до 70-75°C и раствор (BOC)2O (330кг, 1,51 кмоль) в растворе DMF (169кг) добавляли в течение 2 ч при 70-75 °C. После завершения добавления около 200 л DMF удаляли в вакууме в течение 3 ч при менее 75ºC. Добавление (BOC)2O (68,6 кг, 0,314 кмоль) в DMF (32,4 кг) раствор продолжали в течение 0,5 ч при 70-75ºC. После завершения добавления, партию концентрировали при температуре ниже 75ºC и затем охлаждали до около 23.5ºC. MTBE (899,6 кг) загружали и затем смесь охлаждали до около 12,6ºC. Моногидрат лимонной кислоты (197,4 кг) в водном растворе (702 кг) добавляли при 10-20ºC. Слои разделяли и органический слой промывали 5% водный NaCl (582 кг). Слои разрезали и органический слой концентрировали до 240-360 л при ниже 50ºC. После того, как н-гептан (77кг) загружали, смесь концентрировали до 240-360 л при ниже 50ºC. После того, как н-гептан (70кг) загружали, суспензию перемешивали в течение 4 ч при 0-10ºC и продукт собирали центрифугированием. Осадок промывали н-гептаном (28,2кг) и получали (E)-этил 3-((трет-бутоксикарбонил)(изопропил)амино)-2-(4-хлорофенил)акрилат (170,6 кг, 77% выход, 99,8A % ВЭЖХ), который может быть использован так, как описано выше в данном Примере 1 с получением (E)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)акриловой кислоты.
Пример 2
Натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноат
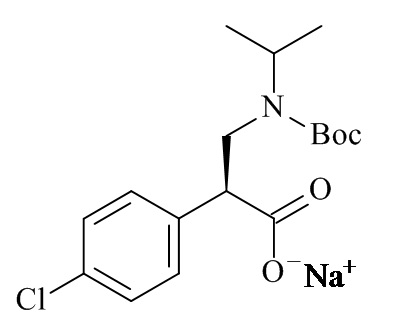
В герметизированном рабочем боксе (O2 содержание ≤ 2 ppm), 50 мл автоклав загружали 6,8 г (20,0 ммоль) (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевой кислоты, 34 мл этанола и 4,81 мг (0,0051 ммоль, S/C 4ʹ000) [Ru(TFA)2((S)-BINAP]. Асимметричная гидрогенизация была проведена в течение 7 ч при 60°C при 18 бар водорода. После охлаждения до комнатной температуры давление сбрасывали из автоклава и пробу желтого реакционного раствора анализировали, которая демонстрировала >99% превращение в (S)-3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовую кислоту с S/R энантиомерным соотношением 99,3-0,7. Гидрогенизационную смесь переносили при помощи 100 мл трет-бутил метилового эфира в 1 л стеклянный реактор под аргоном, содержащий неочищенную реакционную смесь 6 аналогичных гидрогенизационных экспериментов (всего 47,9 г (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевой кислоты), затем этанольный раствор этоксида натрия (52,3 мл, 140 ммоль) добавляли по каплям при 50 °C при перемешивании. Образовывался желтоватый осадок, который перемешивали при той же температуре и затем при комнатной температуре в течение ночи. Осажденный продукт отфильтровывали, промывали смесью трет-бутил метиловый эфир/этанол 4:1 (300 мл) и трет-бутил метиловым эфиром (200 мл), высушивали в вакууме до постоянной массы с получением натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноата с 96,3% выходом (49,03 г) и с S/R энантиомерным соотношением >99,9 - <0,1% в виде белых кристаллов. 1H-ЯМР (D2O): δ 7,32 (д, 2H), 7,22 (д, 2H), 3,65-3,85 две bs, арил-CH и N-CH(CH3)2), 3,55 (м, 2H), 1,29 (с, 9H), 1,00 (д, 3H), 0,80 (bs, 3H).
Энантиомерное соотношение определяли при помощи ВЭЖХ с использованием колонки Chiralpak-AD-3, 150 мм*4,6 мм. Элюенты: A) н-гептан с 0,10% трифторуксусной кислотой, B) этанол, поток: 1,25 мл/мин, 25°C, 5 мкл объем впрыска, 220 нм. Времена удержания: (S)-3-(трет-Бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовая кислота 2,48 мин, (R)-3-(трет-Бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовая кислота 2,77 мин, (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевая кислота 3,16 мин.
Пример 3
Натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноат
В герметизированном рабочем боксе (O2 содержание ≤ 2 ppm), 185 мл автоклав загружали 17,0 г (50,0 ммоль) (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевой кислоты, 70 мл этанола и 4,74 мг (0,005 ммоль, S/C 10ʹ000) [Ru(TFA)2((S)-BINAP]. Асимметричная гидрогенизация была проведена в течение 22 ч при 70°C при 18 бар водорода. После охлаждения до комнатной температуры давление сбрасывали из автоклава и пробу желтого реакционного раствора анализировали, которая демонстрировала >99% превращение в (S)-3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовую кислоту с S/R энантиомерным соотношением 98,2-1,8. Гидрогенизационную смесь переносили при помощи 200 мл трет-бутил метилового эфира в 400 мл стеклянный реактор под аргоном then an этанольный раствор этоксида натрия (18,7 мл, 50 ммоль) добавляли по каплям при 50°C при перемешивании. Образовывался желтоватый осадок, который перемешивали при той же температуре и затем при комнатной температуре в течение в общем 3,5 ч. Осажденный продукт отфильтровывали, промывали смесью трет-бутил метиловый эфир/этанол 4:1 (80 мл) и трет-бутил метиловым эфиром (20 мл), высушивали в вакууме до постоянной массы с получением натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноата с 94,6% выходом (17,35 г) и с S/R энантиомерным соотношением 100-0% в виде белых кристаллов.
Пример 3a
Натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноат
В герметизированном рабочем боксе (O2 содержание ≤ 2 ppm), 185 мл автоклав загружали 10,0 г (29,4 ммоль) (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевой кислоты, 125 мл этанола, 0,118 мл, NaBr раствора в воде (1M) и 5,77 мг (0,006 ммоль, S/C 5ʹ000) [Ru(TFA)2((S)-BINAP]. Асимметричная гидрогенизация была проведена в течение 22 ч при 60°C при 18 бар водорода. После охлаждения до комнатной температуры давление сбрасывали из автоклава и пробу желтого реакционного раствора анализировали, которая демонстрировала >99% превращение в (S)-3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовую кислоту с S/R энантиомерным соотношением 98 к 2. Гидрогенизационную смесь переносили при помощи 10 мл этанола в 0,5 л реактор. Реакционную смесь испаряли при 45°C в вакууме до остаточного объема 65 мл.
70 мл трет-бутил метилового эфира добавляли при 45°C. Затем этанольный раствор этоксида натрия (11,4 г, 35 ммоль) добавляли по каплям при 45°C при перемешивании. Воронку промывали 1,3 г этанола. Образовывался желтоватый осадок, который перемешивали при той же температуре в течение 1 часа и затем при комнатной температуре в течение 1 часа. Осажденный продукт отфильтровывали, промывали смесью трет-бутил метиловый эфир/этанол 1:1 (13,6г) и трет-бутил метиловым эфиром (16г), высушивали в вакууме до постоянной массы с получением натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноата с 89% выходом (9,52 г) и с S/R энантиомерным соотношением 100-0% в виде белых кристаллов.
Пример 3b
Натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноат
В герметизированном рабочем боксе (O2 содержание ≤ 2 ppm), 185 мл автоклав загружали 10,0 г (29,4 ммоль) (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевой кислоты, 120 мл этанола (перегнанного под Ar), 0,118 мл, NaCl раствор в воде (1M) и 5,8 мг (0,006 ммоль, S/C 5ʹ000) [Ru(TFA)2((S)-BINAP]. Асимметричная гидрогенизация была проведена в течение 12 ч при 60°C при 18 бар водорода. После охлаждения до комнатной температуры давление сбрасывали из автоклава и пробу желтого реакционного раствора анализировали, которая демонстрировала >99% превращение в (S)-3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовую кислоту с S/R энантиомерным соотношением 99 к 1. Гидрогенизационную смесь переносили при помощи 10 мл этанола в 0,5 л реактор. Реакционную смесь испаряли при 45°C в вакууме до остаточного объема 65 мл.
70 мл трет-бутил метилового эфира добавляли при 20°C. Затем раствор этоксида натрия (21% (мас./мас.), 9,5 г, 29.4 ммоль) в этаноле добавляли по каплям при 45°C при перемешивании. Воронку промывали 1,3 г этанола. Образовывался осадок, который перемешивали при той же температуре в течение 1 ч и затем при комнатной температуре в течение 1 ч. Осажденный продукт отфильтровывали, промывали трет-бутил метиловый эфир/этанол 1:1 смесью (13,6г) и трет-бутил метиловый эфир (16г), высушивали в вакууме до постоянной массы с получением натрий (S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропаноат с 89% выходом (9,5 г) и с S/R энантиомерным соотношением 100 к 0% в виде белых кристаллов.
Пример 3c
(S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)-пропионовая кислота
В герметизированном рабочем боксе (O2 содержание ≤ 2 ppm), 185 мл автоклав загружали 17,1 г (50,0 ммоль) (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевой кислоты и 75 мл этанола. В отдельной колбе смесь 4,79 мг (0,005 ммоль, S/C 10ʹ000) [Ru(TFA)2((S)-BINAP] и 8.3 мл этанола обрабатывали 1,67 мл, (0.10 ммоль) 60 миллимолярного HCl раствора в воде, полученную в результате суспензию перемешивали в течение 30 минут и затем добавляли в автоклав. После герметизации в автоклаве асимметричная гидрогенизация была проведена в течение 12 ч при 60°C при 18 бар водорода. После охлаждения до комнатной температуры давление сбрасывали из автоклава и пробу желтого реакционного раствора анализировали, которая демонстрировала 99,5% превращение в (S)-3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовую кислоту с S/R энантиомерным соотношением 98,7 к 1,3.
Пример 3d
(S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)-пропионовая кислота
Процедуру Примера 3c повторяли с использованием HBr в качестве добавки. Гидрогенизация продолжалась с 99,8% превращением, желательную (S)-кислоту выделяли с количественным выходом с S/R энантиомерным соотношением 98,7:1,3.
Пример 3e
(S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)-пропионовая кислота
В герметизированном рабочем боксе (O2 содержание ≤ 2 ppm), 185 мл автоклав загружали 17.0 г (50,0 ммоль) (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевой кислоты и 75 мл этанола. В отдельной колбе смесь 9,59 мг (0,010 ммоль, S/C 5ʹ000) [Ru(TFA)2((S)-BINAP] и 9,8 мл этанола обрабатывали 0,20 мл, (0,20 ммоль) 1 молярного HCl раствора в воде, полученную в результате суспензию перемешивали в течение 30 минут и затем добавляли в автоклав. После герметизации в автоклаве асимметричная гидрогенизация была проведена в течение 12 ч при 60°C при 18 бар водорода. После охлаждения до комнатной температуры давление сбрасывали из автоклава и пробу желтого реакционного раствора анализировали которая демонстрировала 99,7% превращение в (S)-3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовую кислоту с S/R энантиомерным соотношением 99,0 к 1,0.
Пример 3f
(S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)-пропионовая кислота
Процедуру Примера 3f повторяли с использованием LiBr в качестве добавки. Гидрогенизация продолжалась с 98,9% превращением, желаемую (S)-кислоту отделяли с количественным выходом с S/R энантиомерным соотношением 98,5:1,5.
Пример 4
(S)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)-пропионовую кислоту
В герметизированном рабочем боксе (O2 содержание ≤ 2 ppm), 35 мл, автоклав, снабженный стеклянной вставкой и магнитной мешалкой загружали 400 мг (1,18 ммоль) (E)-2-(4-хлорофенил)-3-[(2-метилпропан-2-ил)оксикарбонил-пропан-2-иламино]проп-2-еноевой кислоты, 5,92 мг (0,00589 ммоль) [Ru(TFA)2((S)-BINAP)] (S/C 200) и 4 мл этанола. Автоклав герметизировали и вакуумировали 20 бар водорода, асимметричная гидрогенизация была проведена при перемешивании в течение 14 ч при 60°C. После охлаждения до комнатной температуры давление сбрасывали из автоклава, этанольный раствор испаряли в вакууме с получением (S)-3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовой кислотой с количественным выходом и с S/R энантиомерным соотношением 99:1. Превращение составляло >= 99,9%.
Пример 5.1-5.17(S) или (R)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропионовая кислота
Процедуру Примера 4 повторяли с использованием различных хиральных рутениевых катализаторов с получением соответствующих (R) и (S) изомеров 3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовой кислоты. Результаты приведены в Таблице 1, вместе с катализатором, % превращения и S/R энантиомерным соотношением. Масштаб реакции составлял во всех экспериментах (если специально не указано в сноске) 400 мг, температура составляла 60°C, давление водорода составляло 20 бар при S/C соотношении 200, время реакции составляло 14 ч. Реактор имел объем 35 мл, автоклав. Указанное количество добавки приведено относительно количества металлического катализатора.
|
35 мл, автоклав, 1,7 г шкала; S/C 250, 22 ч. b) 1,7 г шкала, S/C 250,14 ч; c) 6,8 г субстрат в 50 мл, автоклав, S/C 1500, 5 ч. d) Катализатор получали in situ путем перемешивания 2,56 мг [Ru(COD)(TFA)2]2 и 2,2 молярных эквивалентов хирального дифосфина в герметизированном рабочем боксе в 3 мл этанола в течение 3 ч при 50°C.
Пример 6.1- 6.8
(S) или (R)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропионовая кислота
Аналогично Примерам 5.1-5.16 следующие гидрогенизации выполняли с использованием различных веществ в качестве добавок с получением (R) и (S) изомеров 3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовой кислоты при чистоте и энантиомерной чистоте, указанных в Таблице 2. Масштаб реакции составлял во всех экспериментах (если специально не указано в сноске) 400 мг, температура составляла 60°C, давление водорода составляло 18-20 бар в течение 4 ч при S/C соотношении 200. Реактор имел объем 35 мл, автоклав. Указанное количество добавки приведено относительно количества металлического катализатора.
Таблица 2
|
Пример 7.1-7.11
(S) или (R)-3-(трет-бутоксикарбонил(изопропил)амино)-2-(4-хлорофенил)пропионовая кислота
Процедуру Примеров 5.1-5.16 повторяли, но условия реакции варьировались в терминах давления водорода, концентрации и растворителя с получением соответствующих (R) и (S) изомеров 3-(трет-бутоксикарбонил-изопропил-амино)-2-(4-хлоро-фенил)-пропионовой кислоты. Рзультаты приведены в Таблице 3, масштаб реакции во всех экспериментах (если специально не указано в сноске) 400 мг в 4 мл, растворителя, температура составляла 60°C при S/C соотношении 200, время реакции составляло 14 ч. Реактор имел объем35 мл, автоклав, катализатор был Ru(TFA)2((S)-BINAP).
|
Пример 8
(R)-трет-бутил 4-(5-метил-7-оксо-5,6-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-кабоксилат
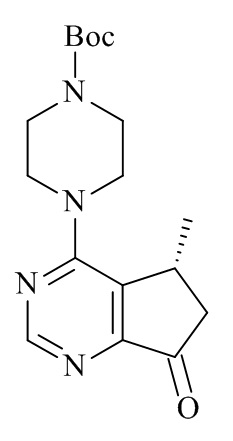
(R)-метил-3-(4,6-дихлоропиримидин-5-ил)бутаноат В смесь (R)-метил 3-(4,6-дигидроxyпиримидин-5-ил)бутаноата (1,00 кг, 4,70 моль), толуола (4,00 л), и 2,6-лутидина (0,550 л, 4,70 моль) медленно добавляли оксихлорид фосфора (0,960 л, 10,6 моль) при 50°C. Смесь перемешивали при 70°C в течение 24 ч. Раствор охлаждали до 0°C. К смеси медленно добавляли 20% водный натрий гидроксид (приблизительно 40,0 моль, 1,60 кг в 8,00 л H2О), поддерживая внутреннюю температуру ниже 30°C, с получением конечного значения pH от 5 до 6. Этилацетат (2,50 л) добавляли, перемешивали в течение 0,5 ч, и затем слои разделяли. Водную фазу экстрагировали этилацетатом (3×1,00 л). Органические слои комбинировали и промывали 1 N соляной кислотой (2×2,50 л), и солевым раствором (2,50 л). Органические слои комбинировали и высушивали над натрий сульфатом и фильтровали через стекловолоконный фильтр. Раствор концентрировали до около 3,00 мл/г, и разбавляли ацетонитрилом до около 7,00 мл/г. Эту последовательность повторяли дважды для удаления остаточного этилацетата и толуола (подтверждено 1H ЯМР анализом). Оставшийся неочищенный раствор использовали непосредственно для следующей стадии без дополнительной очистки или выделения.
(R)-метил 3-(4,6-дийодопиримидин-5-ил)бутаноат
В раствор (R)-метил 3-(4,6-дихлоропиримидин-5-ил)бутаноата (36,0 г, 145 ммоль) в ацетонитриле (540 мл) добавляли натрий йодид (152 г, 1,02 моль). Смесь перемешивали при 25°C в течение 30 минут и затем охлаждали до около 5°C. Mэтансульфоновую кислоту (9,41 мл, 1,00 эквив.) добавляли в течение 5 минут. Смесь перемешивали при около 5°C в течение 3 часов. Реактор охлаждали до около 5°C и добавляли N,N-диизопропилэтиламином (20,3 мл, 116 ммоль). Смесь перемешивали в течение 1 ч при нагревании смеси до 20°C. Насыщенный раствор сульфита натрия добавляли, пока не переставали наблюдать изменение цвета до удаления йода. Воду (540 мл) добавляли и pH регулировали до около 5-7. Двухфазную смесь концентрировали при сниженном давлении при температуре менее чем 40°C для удаления ацетонитрила. Водную суспензию фильтровали с получением 48,8 г (78% выход) практически белого твердого продукта.
(R)-трет-бутил 4-(6-йодо-5-(4-метокси-4-оксобутан-2-ил)пиримидин-4-ил)пиперазин-1-кабоксилат В раствор (R)-метил 3-(4,6-дийодопиримидин-5-ил)бутаноата (212 г, 491 ммоль) и Boc-пиперазина (101 г, 540 ммоль) в метаноле (424 мл) добавляли N,N-диизопропилэтиламин (94,3 мл, 540 ммоль). Смесь нагревали при 60°C в течение 24 часов. Метанол отгоняли при сниженном давлении ниже 40°C. К смеси добавляли 318 мл тетрагидрофурана. Указанный выше процесс обмена растворителей дважды повторяли. К смеси добавляли 424 мл тетрагидрофурана, 212 мл, насыщенного водного хлорида аммония и 21,2 мл воды. Органический слой промывали 212 мл, (1,00 об.) насыщенным водным хлоридом аммония. Данный раствор тетрагидрофурана использовали для следующей стадии без дополнительной очистки (91% масс. аналитический выход).
(R)-3-(4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-6-йодо-пиримидин-5-ил)бутаноевая кислота
В раствор (R)-трет-бутил 4-(6-йодо-5-(4-метокси-4-оксобутан-2-ил)пиримидин-4-ил)пиперазин-1-кабоксилата (219 г, 0,447 моль) в тетрагидрофуране (657 мл) добавляли раствор литий гидроксида моноглицерата (56,2 г, 1,34 моль) в 329 мл воды при 25°C. Смесь перемешивали при 25°C в течение 5 ч. Нижний водный слой отбрасывали. Смесь подкисляли 1 N соляной кислотой при 5°C с получением конечного значения pH от около 1 до 2. Слои разделяли. Верхний слой затем экстрагировали изопропил ацетатом (440 мл, x 3), комбинировали с нижним слоем, и промывали водой (220 мл, x 2). Растворитель отгоняли при сниженном давлении ниже 50°C. Оставшийся изопропил ацетат азеотропно отгоняли гептаном при сниженном давлении ниже 50°C. Продукт постепенно осаждали и фильтровали с получением желтовато-белого порошка (196 г, 84% выход).
(R)-трет-бутил 4-(6-йодо-5-(4-(метокси(метил)амино)-4-оксобутан-2-ил)пиримидин-4-ил)пиперазин-1-кабоксилат В раствор (R)-3-(4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)-6-йодо-пиримидин-5-ил)бутаноевой кислоты (100 г, 210 ммоль) в тетрагидрофуране (700 мл) частями добавляли 1,1ʹ-карбонилимидазола (40,9 г, 252 ммоль). Реакционную смесь перемешивали при 20°C в течение 1 ч и охлаждали до 5°C. N,O-диметилгидроксиамин гидрохлорид (41,0 г, 420 ммоль) добавляли частями с последующим добавлением N-метилфорфолина (6,94 мл, 63,0 ммоль). Смесь перемешивали при 5°C в течение около 1 ч, медленно нагревали до комнатной температуры и перемешивали в течение 24 часов. Насыщенный водный хлорид аммония (500 мл) и воду (150 мл) добавляли с получением четкого разделения фаз. Органический слой промывали насыщенным водным хлоридом аммония (500 мл) и солевым раствором (200 мл). Остаточную воду азеотропно отгоняли до менее чем 500 ppm путем со-испарения с тетрагидрофураном. Продукт, в виде раствора тетрагидрофурана, использовали для следующей стадии без дополнительной очистки или выделения (масс. аналитический выход: >99%.
(R)-трет-бутил 4-(5-метил-7-оксо-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-кабоксилат Раствор (R)-трет-бутил 4-(6-йодо-5-(4-(метокси(метил)амино)-4-оксобутан-2-ил)пиримидин-4-ил)пиперазин-1-кабоксилата (109 г, 210 ммоль) в тетрагидрофуране (600 мл) продували азотом в течение 30 минут. Раствор изопропил магний хлорида (159 мл, 210 ммоль, 1,32 M в тетрагидрофуране) добавляли по каплям при -15°C. Смесь перемешивали при -10°C в течение 1 ч и медленно переносили в холодный 20 мас % водный хлорид аммония (600 мл) при перемешивании, поддерживая внутреннюю температуру ниже 10°C. Органический слой затем промывали насыщенным водным хлоридом аммония (500 мл). Тетрагидрофуран отгоняли при сниженном давлении ниже 40°C. Метил трет-бутиловый эфир (350 мл) медленно добавляли при поддержании внутренней температуры от 35°C до 40 °C, с последующим добавлением гептана (350 мл). Смесь медленно охлаждали до 20°C и продукт постепенно осаждали в течение процесса. Суспензию фильтровали и осадок высушивали при 40°C в вакууме с получением серого твердого осадка (52,3 г, 75% выход за две стадии), 1H ЯМР (300 МГц, CDCl3) δ 8,73 (с. IH), 3,92-3,83 (м. 2H), 3,73-3,49 (м. 7H), 2,96 (дд. J= 16,5, 7,2 Гц, 1H), 2,33 (дд. J=16,5, 1,8 Гц, IH), 1,50 (с. 9H), 1,32 (д. J=6,9 Гц, 3H). HRМС рассч. для C17H25N403 [M+H]+: 333,1921, найдено 333,1924.
Пример 9
Трет-бутил 4-[(5R,7R)-7-гидрокси-5-метил-6,7-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилат
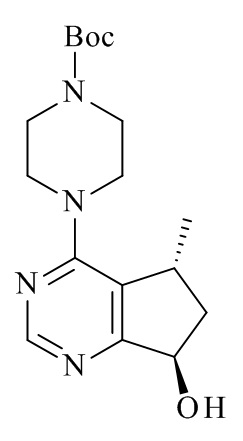
Блекло-желтая суспензия 3 г трет-бутила 4-[(5R)-5-метил-7-оксо-5,6-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилата в 21 мл, водного буфера (100 мM 2-(н-морфолино)этансульфоновой кислоты pH 5,8), 6 мл, 2-пропанoл и 3 мг окисленного кофактора NADP [Roche] образовывалась при энергичном перемешивании. Реакционный раствор нагревали до 40°C и перемешивали в течение 5 минут и затем, восстановление начинали путем добавления 30 мг KRED-X1-P1B06. pH регулировали с 5,6 до 5,8. В течение хода реакции при 40°C в течение 21,5 ч достигая практически полного превращения (IPC: 0.6 площадь % выделенного вещества), значение pH возрастало до 6,4. В реакцию добавляли 30 мл изопропил ацетата и энергично перемешивали в течение 15 минут. Разделение фаз происходило спонтанно. Отделенную водную фазу дважды экстрагировали 50 мл изопропил ацетата, всего 100 мл изопропил ацетата. Соединенные органические фазы высушивали над MgSO4, фильтровали и испаряли в вакууме при 50°C получая 3,07 г (102%) светло-красной пены в виде неочищенного продукта титульного соединения, содержащего около 4% изопропил ацетата. ГХ-EI-МС: 334.2 (M+H)+; хиральная ВЭЖХ: 99,88% (R,R), 0,12% (R,S) [254 нм; Chiralpak IC-3; 150*4,6 мм, 3 мкм, поток 0,8 мл, 30 °C, A: 60% н-гептан, B: 40% EtOH+0,1 DEA, 0-15 мин 100% B, 15-17 мин 100% B, 17,1 мин 40% B]; химическая чистота по ВЭЖХ: 99,2%площади (содержит 0.6%площади выделенного продукта). 1H ЯМР (600 МГц, CDCl3) δ ppm 1,17-1,22 (м. 3 H) 1,45-1,51 (м. 9 H) 2,02 (с.1 H) 2,12-2,24 (м. 2 H) 3,43-3,83 (м. 9 H) 3,85-4,08 (м. 1 H) 5,12 (т. J=7,2 Гц, 1 H) 8,53(с. 1 H) (содержит ~4% изопропилацетат).
Пример 10.1-10.6
Трет-бутил 4-[(5R,7R)-7-гидрокси-5-метил-6,7-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилат
Для Примеров 9.1-9.6, повторяли процедуру Примера 9, но кофакторное соотношение (NADP [Roche]) варьировали, как указано в таблице ниже и применяли различные кеторедуктазные варианты, а именно KRED-X1.
|
Пример 11
Трет-бутил 4-[(5R,7R)-7-гидрокси-5-метил-6,7-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилат
Блекло-желтая суспензия 6 г трет-бутил 4-[(5R)-5-метил-7-оксо-5,6-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилата в 18 мл водного буфера (100 мM 2-(н-морфолино)этансульфоновая кислота pH 5,8), 6 мл, 2-пропанола и 6 мг окисленного кофактора NADP [Roche] образовывалась при интенсивном перемешивании. Реакционный раствор нагревали до 40°C и перемешивали в течение 5 минут и затем начинали реакцию путем добавления 60 мг KRED-X1-P1B06. Значение pH регулировали от 5,5 до 5,8. В течение хода реакции при 40°C на протяжении 2 дней, при достижении практически полного превращения (IPC: 1d 1,3% площади выделенного вещества, 2d 1,2% площади выделенного вещества) значение pH возрастало до 6,0. В реакцию добавляли 30 мл изопропил ацетата и интенсивно перемешивали в течение 15 мин. Разделение фаз происходило спонтанно. Отделенную водную фазу дважды экстрагировали 50 мл изопропил ацетата, всего 100 мл изопропил ацетата. Соединенные органические фазы высушивали над MgSO4, фильтровали и испаряли в вакууме при 50°C с получением 6,02 г (99,7%) светло-красной пены в виде неочищенного продукта титульного соединения, содержащего около 4% изопропил ацетата. ГХ-EI-МС: 334.2 (M+H)+; хиральная ВЭЖХC: 99,88% (R,R), 0,12% (R,S) [254 нм; Chirapakl IC-3; 150*4,6 мм, 3мкм, поток 0,8 мл, 30 °C, A: 60% н-гептан, B: 40% EtOH+0,1 DEA, 0-15 мин 100% B, 15-17 мин 100% B, 17,1 мин 40% B]; химическая чистота по ВЭЖХ: 98,4%площади (содержит 1,3%площади выделенного продукта). 1H ЯМР (600 МГц, CDCl3) δ ppm 1,2 (д, J=7,1 Гц 3 H) 1,49 (с, 9 H) 2,14-2,23 (м, 2 H) 3,46-3,59 (м, 5 H) 3,64 (ддд, J=13,1, 6,9, 3,3 Гц, 2 H) 3,78 (ддд J=13,1, 7,2, 3,3 Гц, 2H) 5,12 (т, J=7,2 Гц, 1 H) 8,53(с, 1 H) (содержит ~4% изопропилацетата).
Пример 12
Трет-бутил 4-[(5R,7R)-7-гидрокси-5-метил-6,7-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-карбоксилат
Блекло-желтая суспензия 3 г трет-бутил 4-[(5R)-5-метил-7-оксо-5,6-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилата в 21 мл водного буфера (100 мM кислого фосфата калия pH 7.2; 2 мM хлорида магния), 6 мл, 2-пропанoла и 3 мг окисленного кофактора NADP [Roche] образовывалась при интенсивном перемешивании. Реакционный раствор нагревали до 40°C и перемешивали в течение 5 мин и затем начинали реакцию путем добавления 30 мг KRED-X1-P1B06. Значение pH регулировали от 7,5 до 7,2. В течение хода реакции при 40°C на протяжении 18,5 ч достигая практически полного превращения (IPC: 0,8% площади выделенного продукта), значение pH уменьшалось до 7,15. В реакцию добавляли 30 мл изопропил ацетата и интенсивно перемешивали в течение 15 мин. Разделение фаз происходило спонтанно. Отделенную водную фазу дважды экстрагировали 50 мл изопропил ацетата, всего 100 мл изопропил ацетата. Соединенные органические фазы высушивали над MgSO4, фильтровали и испаряли в вакууме при 50°C с получением 3,06 г (102%) светло-красной пены в виде неочищенного продукта титульного соединения, содержащего около 4% изопропил ацетата. ГХ-EI-МС: 334.2 (M+H)+; хиральная ВЭЖХ: 99,76% (R,R), 0,24% (R,S) [254 нм; Chirapakl IC-3; 150*4,6 мм, 3мкм, поток 0,8 мл, 30 °C, A: 60% н-гептан, B: 40% EtOH+0,1 DEA, 0-15 мин 100% B, 15-17 мин 100% B, 17,1 мин 40% B]; химическая чистота по ВЭЖХ: 98,9%площади (содержит 0,8% площади выделенного продукта), 1H ЯМР (600 МГц, CDCl3) δ ppm 1,16-1,22 (м. 3 H) 1,45-1,53 (м. 9H) 2,12-2,25 (м. 2 H) 3,42-3,86 (м. 9 H) 4,13 (уш. с. 1 H) 5,12 (т. J=7,2 Гц, 1H) 8,44-8,59 (м. 1 H) (содержит ~4% изопропил ацетата).
Пример 13.1-13.7
Трет-бутил 4-[(5R,7R)-7-гидрокси-5-метил-6,7-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилат
10 мг трет-бутил 4-[(5R)-5-метил-7-оксо-5,6-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилата растворяли в смеси 50 мкл ДМСО и 50 мкл 2-пропанoла добавляли в каждую лунку планшеты с глубокими лунками, содержащей 300 мкл буфера (MES 100 мM, MgCl2 2 мM; pH 5,8) 1 мг NADP и варианты KRED-X1. После перемешивания в течение 1,5 ч при комнатной температуре в каждую лунку добавляли 0.5 мл MeOH и анализировали при помощи ВЭЖХ. Результаты наилучших вариантов приведены в таблице 5.
|
Пример 13a
Трет-бутил 4-[(5R,7R)-7-гидрокси-5-метил-6,7-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-карбоксилат
Суспензия 50 г (150 ммоль) трет-бутил 4-[(5R)-5-метил-7-оксо-5,6-дигидроциклопента[d]-пиримидин-4-ил]пиперазин-1-кабоксилата в 100 мл водного буфера (100 мM кислого фосфата калия pH 7,2), 78 г 2-пропанола и 50 мг NAD (75 мкмоль) образовывалась при интенсивном перемешивании. Восстановление начинали путем добавления 500 мг KRED-X1.1-P1F01. Реакционную смесь продували азотом и нагревали до 40°C в течение 22 часов. После завершения реакции добавляли 174 г изопропилацетата, перемешивали, фазы разделялись и водную фазу удаляли. Водную фазу снова экстрагировали 174 г изопропилацетата. Водную фазу удаляли и органические фазы соединяли и концентрировали при 35°C в вакууме до конечного объема 115 мл. При той же самой температуре добавляли 212 г гептана в течение 1 часа, суспензию выдерживали в течение 1 часа и охлаждали до 10°C в течение 6 часов. Суспензию фильтровали и промывали 68 г гептана. После высушивания осадка на фильтре в течение 4 часов при 50°C и получали 41,1 г (82% выход, чистота 100% площади) белых кристаллов.
Пример 13b
Трет-бутил 4-[(5R,7R)-7-гидрокси-5-метил-6,7-дигидроциклопента[d]пиримидин-4-ил]пиперазин-1-кабоксилат
Суспензия 40 г (150 ммоль) трет-бутил 4-[(5R)-5-метил-7-оксо-5,6-дигидроциклопента[d]-пиримидин-4-ил]пиперазин-1-кабоксилата в 240 мл водного буфера (содержащего 3,3 г KH2PO4 и 8,4 г K2HPO4), 26 г глюкозы и 40 мг NAD образовывалась при интенсивном перемешивании. Реакцию восстановления нагревали до 35°C и начинали путем добавления 400 мг KRED-X1.1-P1F01 и 400 мг GDH-101. В ходе реакции (26 часа) значение pH поддерживали при 7,0 с использованием 58,8 мл водн. KOH (10% (мас./мас.)). После завершения реакции добавляли 290 г изопропилацетата и 117 г NaSCN, перемешивали, фазы разделялись и водную фазу удаляли. Органическую фазу промывали 200 г воды и фильтровали при помощи фильтровальной пластины filtrox, водную фазу промывали 175 г изопропилацетата. Соединенные органические слои концентрировали при 25°C в вакууме до конечного объема 100 мл. При 25°C 383 г гептана добавляли в течение 1 часа. Суспензию охлаждали до 0°C в течение 30 минут и выдерживали в течение 30 минут, суспензию фильтровали и промывали 91 г гептана. После высушивания осадка на фильтре в течение 16 часов при 50°C и получали 30.9 г (76% выход, чистота 100% площади) белых кристаллов.
Пример 14
трет-бутил ((S)-2-(4-хлорофенил)-3-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропил)(изопропил)карбамат
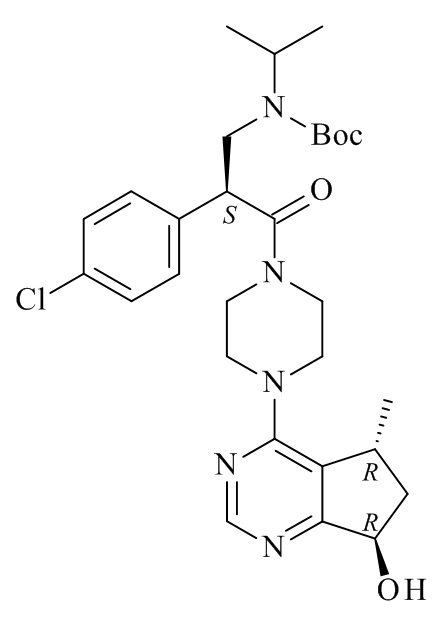
В трехгорлый реактор на 500 мл, оснащенный механической мешалкой, входом для азота, и термометром, загружали трет-бутил 4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-кабоксилат (16.7 г, 52.5 ммоль) и 2-пропанoл (65 мл). Раствор нагревали до 55°C. Затем 20,8% (мас./мас.) HCl в 2-пропанoле (24.6 г, 140 ммоль) добавляли в течение 10 минут при 55°C. Суспензию перемешивали до завершения реакции. Реакционную смесь охлаждали до 10 °C и добавляли 4-метилфорфолин (32,9 г, 325 ммоль). Смесь перемешивали при 15°C в течение 30 минут. Натриевую соль (S)-3-((трет-бутоксикарбонил)(изопропил)амино)-2-(4-хлорофенил)пропаноевой кислоты (19.1г, 52,5 ммоль) и 2-пропанoл (73 г) добавляли и реакционную смесь охлаждали до 5°C. Пропан фосфониевый ангидрид (T3P) (50 мас % (мас./мас.) в толуоле) (35 г, 57,3 ммоль) добавляли при скорости, поддерживая температуру при 5°C.
После завершения реакции, добавляли 20 г воды. Раствор концентрировали путем отгонки при 45°C и 150 мбар до конечного объема 100 мл. Добавляли толуол (260 г). Раствор снова концентрировали путем отгонки при 45°C и 150 мбар до конечного объема 300 мл. Добавляли воду (150 г) и суспензию перемешивали в течение 15 минут. Фазы разделяли в течение 15 минут и водную фазу удаляли. Добавляли воду (100 г) и суспензию перемешивали в течение 15 минут. Фазы разделялись в течение 15 минут и водную фазу удаляли. Снова добавляли воду (100 г) и суспензию перемешивали в течение 15 минут. Фазы разделялись в течение 15 минут и водную фазу удаляли.
Раствор концентрировали путем отгонки при 45°C и 150 мбар до конечного объема 100 мл. н-гептан (34 г) добавляли, раствор охлаждали до 0°C в течение 1 часа, чтобы позволить кристаллизацию трет-бутил ((S)-2-(4-хлорофенил)-3-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропил)(изопропил)карбамата. Добавляли дополнительный н-гептан (170 г). Суспензию выдерживали в течение 2 часов, фильтровали и промывали смесью толуола (6,4г) и н-гептана (29,2 г) с последующим добавлением гептана (каждый 68,4 г). Осадок на фильтре высушивали при ≤ 55°C с получением трет-бутил ((S)-2-(4-хлорофенил)-3-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропил)(изопропил)карбамата в виде грязно-белого твердого вещества, выделяли 23,9 г, 86% выход. (1H ЯМР (600 МГц, CDCl3) δ ppm 0,68 (уш. с., 3 H) 0,94-1,08 (м. 3 H) 1,14 (д, J=7,0 Гц, 3 H) 1,47 (с. 10 H) 2,06-2,27 (м. 2 H) 3,30 (уш.с., 1 H) 3,38-3,53 (м. 5 H) 3,56 -3,73 (м. 4 H) 3,78 (уш. с., 3 H) 4,62 (уш. с., 1 H) 5,10 (т. J=7,1 Гц, 1 H) 7,24 (с. 1 H) 8,49 (с. 1H).
Пример 15
(S)-2-(4-хлорофенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)пропан-1-oн моногидрохлорид
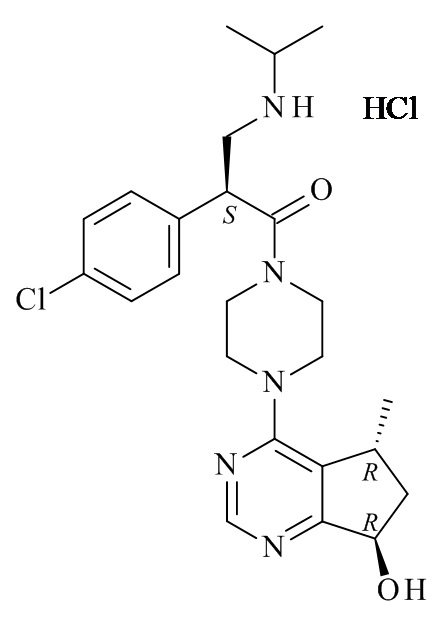
В реактор на 500 мл, оснащенный механической мешалкой, входом для азота, термометром и pH-метром, добавляли трет-бутил ((S)-2-(4-хлорофенил)-3-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропил)(изопропил)карбамат (50 г) и 2-пропанoл (128 г). Раствор нагревали до 50°C. Раствор HCl в 2-пропанoле (21% мас (мас./мас.), 46,7 г) добавляли при 50°C. Раствор выдерживали при 50°C до завершения реакции и смесь охлаждали до 25°C. Раствор аммония в 2-пропанoле (2M, 66,6 г, 1,66 экв.) добавляли в течение около 1 часа до достижения pH 6,7. Суспензию охлаждали до 0°C и фильтровали. Осадок промывали 2-пропанoлом (39 г). Фильтрат концентрировали отгонкой при 50°C и 150 мбар до конечного объема 100 мл. Этилацетат (130 г) добавляли в раствор. В суспензии меняли растворитель при 40°C при постоянном объеме (300 мл), используя этилацетат (670 г). Суспензию охлаждали до 5°C и суспензию фильтровали. Осадок на фильтре промывали EtOAc (105 мл) и высушивали в вакууме при 100°C в течение 16 часов с получением (S)-2-(4-хлорофенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)пропан-1-oна моногидрохлорида в виде грязно-белого твердого вещества: 36,4 г (82% выход). (1H ЯМР (600 МГц, D2O) δ ppm 0.92 (д, J=7,1 Гц, 3 H) 1.23 (т, J=6,4 Гц, 6H) 1,89-2,15 (м, 2 H) 2,85-3,06 (м, 1 H) 3,17-3,59 (м, 10 H) 3,83 (д, J=10,5 Гц, 2 H) 4,33(dd, J=8,5, 4,9 Гц, 1 H) 4,98 (т, J=7,0 Гц, 1 H) 7,23 (д, J=8,5 Гц, 2 H) 7,36 (д, J=8,7 Гц, 2 H) 8,10-8,35 (м, 1 H). ЖХ-МС [M+H]+ 458,2).
Пример 16
(S)-2-(4-хлорофенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)пропан-1-oн моногидрохлорид
В реактор на 500 мл, оснащенный механической мешалкой, входом для азота, трмометром и pH-метром, добавляли трет-бутил ((S)-2-(4-хлорофенил)-3-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-оксопропил)(изопропил)карбамат (50 г) и 1-пропанoл (131 г). Раствор нагревали до 60°C. Раствор HCl в 1-пропанoле (22% мас (мас./мас.), 38.0 г) добавляли при 60°C. Раствор выдерживали при 50°C до завершения реакции и смесь охлаждали до 25°C. Водн. NaOH (28%) (16 г) добавляли до достижения pH 6. Суспензию концентрировали при 60°C в вакууме до достижения финального объема 100 мл. Суспензию охлаждали до 20°C, добавляли 90 г этилацетата и фильтровали при помощи пластины filtrox. Реактор и фильтровальную установку промывали 41 г 1-пропанoл/этилацетат. Раствор фильтровали при 20°C через фильтрующие угольные прокладки, Реактор и фильтр промывали 82г 1-пропанoл/этилацетат. При 60°C раствор концентрировали в вакууме до конечного объема 300 мл. Отгонку продолжали при 60°C и одновременно добавляли 1260 г этилацетата, поддерживая объем постоянным.
Суспензию охлаждали до 5°C и суспензию фильтровали. Осадок на фильтре промывали EtOAc (105 мл) и высушивали в вакууме при 60°C в течение 16 часов с получением (S)-2-(4-хлорофенил)-1-(4-((5R,7R)-7-гидрокси-5-метил-6,7-дигидро-5H-циклопента[d]пиримидин-4-ил)пиперазин-1-ил)-3-(изопропиламино)пропан-1-он моногидрохлорида в виде грязно-белого твердого вещества: 36,5 г (81% выход, 91,4% (мас./мас.) чистота, 99,9% (площадь) анализ).
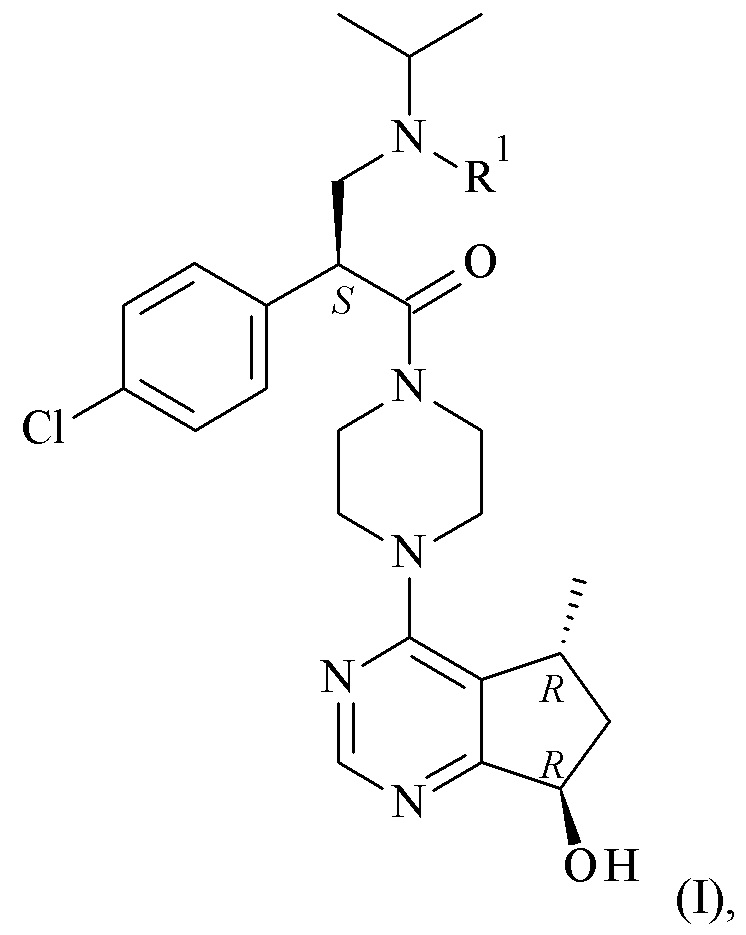
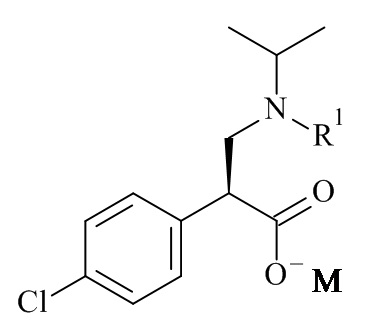
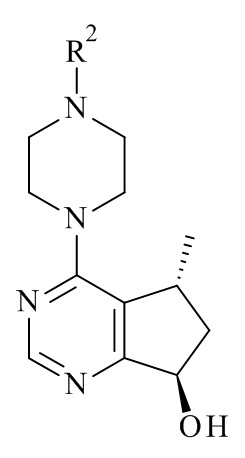
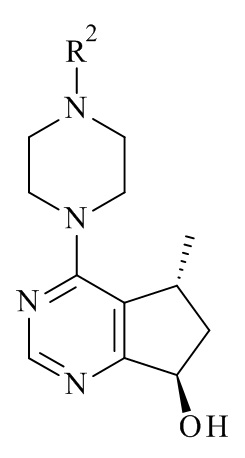
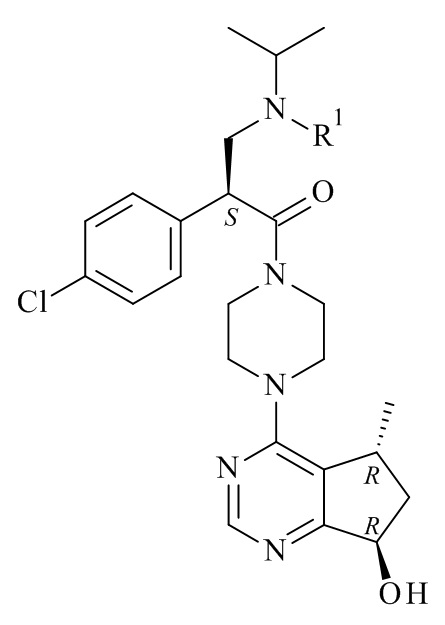
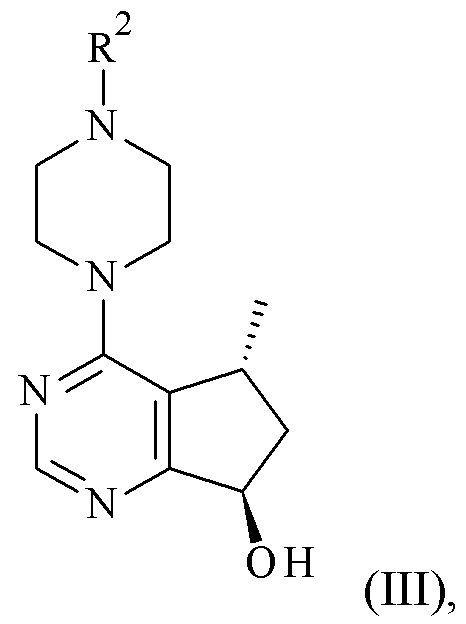
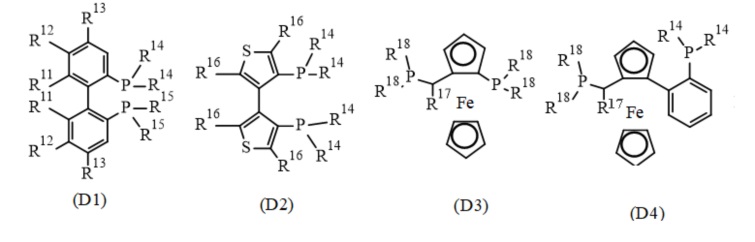
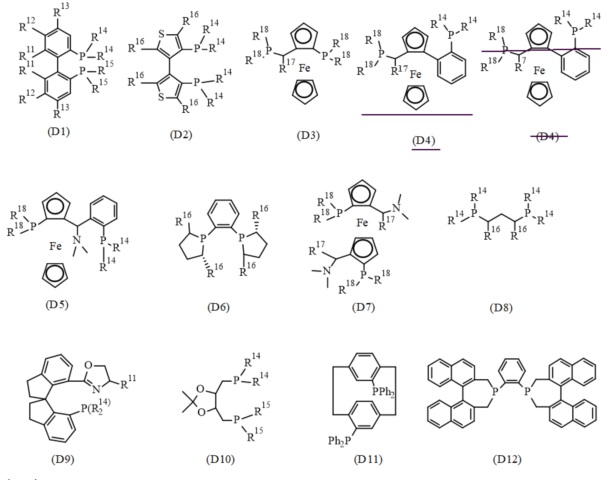
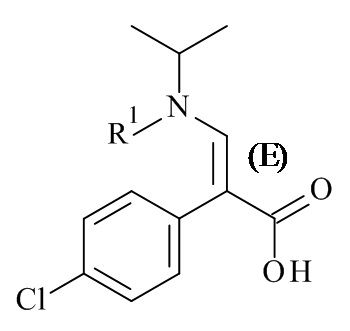
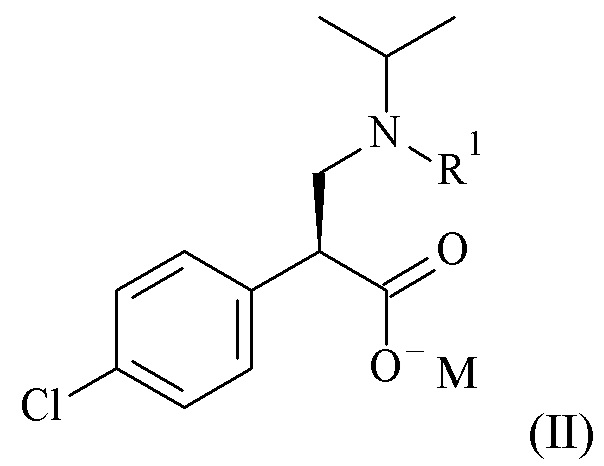
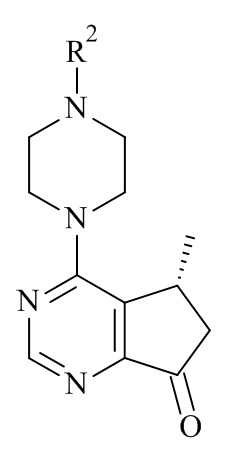
Ингибирование ангиогенеза
Пиримидиновые соединения, композиции и способы применения
Антитела против c3b и способы профилактики и лечения связанных с комплементом нарушений
Осаждение и очистка белков полиэлектролитами
Гуманизированные антитела к фактору d и их применения
Препарат антитела
Антитела против nrr notch1 и способы их применения
Гидроксилированные и метоксилированные циклопента[d]пиримидины в качестве ингибиторов акт протеинкиназ
Производные гетероарилпирролидинил- и пиперидинилкетона
Циклопента(d)пиримидины в качестве ингибиторов протеинкиназ акт
Способ синтеза производных амино-метилтетралина
Способ получения ароматических тиольных производных при гидрировании дисульфидов
Новые соединения и композиции для ингибирования nampt
Способ получения гидроксилированных циклопентапиримидиновых соединений и их солей
Формы и составы пиримидинилциклопентанового соединения, композиции и способы, относящиеся к ним
Способ получения соединения
Способ получения хиральных 2-арилморфолинов
Способ получения лекарственных средств
Способы получения (циклопентил[d]пиримидин-4-ил)пиперазиновых соединений

