Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ХИРАЛЬНЫХ 2-АРИЛМОРФОЛИНОВ
Вид РИД
Изобретение
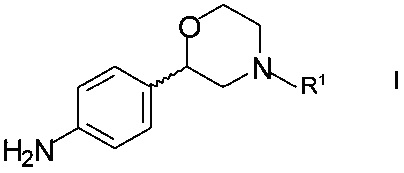
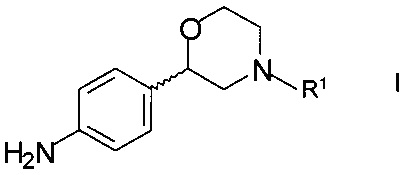
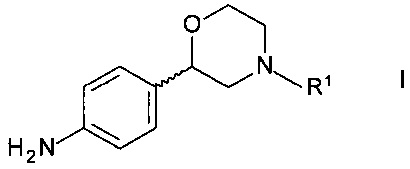
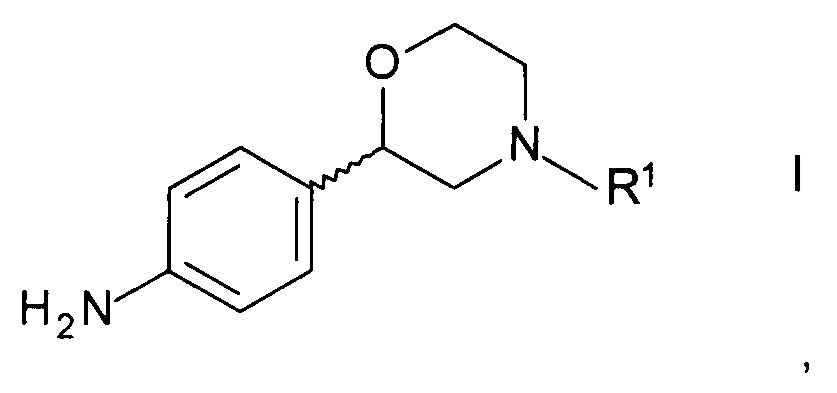
Изобретение относится к новому способу получения хиральных 2-(4-аминофенил)морфолинов формулы
 ,
,
где R1 представляет собой водород или аминозащитную группу.
Хиральные 2-(4-аминофенил)морфолины формулы I являются ключевыми промежуточными продуктами для получения соединений, обладающих сродством к рецепторам, ассоциированным со следовыми аминами (TAAR, англ. trace амин associated receptors), в особенности, TAAR1, как описано, например, в публикациях РСТ патентных документов WO 2012/016879 и WO 2012//126922.
Вследствие этого изобретение также относится к применению способа по настоящему изобретению для получения соединений формулы
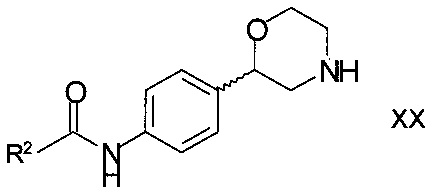 ,
,
где
R2 является арилом или гетероарилом, в котором ароматические кольца необязательно замещены одним или двумя заместителями, выбранными из С1-7-алкила, галогена, CF3, OCF3, OCH2CF3, С1-7-алкокси или циано;
или его фармацевтически приемлемых солей присоединения кислоты, или
для получения соединений формулы
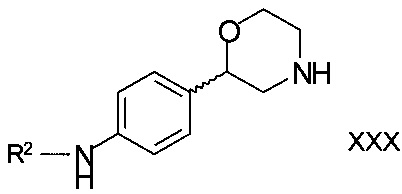 ,
,
где
R2 является арилом или гетероарилом, в котором ароматические кольца необязательно замещены одним или двумя заместителями, выбранными из C1-7-алкила, галогена, CF3, OCF3, OCH2CF3, С1-7-алкокси или циано;
или его фармацевтически приемлемых солей присоединения кислоты.
Целью настоящего изобретения был поиск способа, осуществимого в промышленном масштабе.
Цель может быть достигнута при помощи способа, такого как описан ниже.
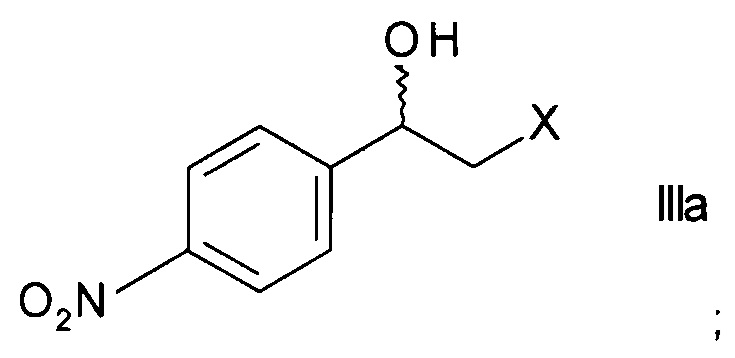
Способ получения хирального 2-(4-аминофенил)морфолина формулы
 ,
,
где R1 представляет собой водород или аминозащитную группу PG (англ. PG - protecting group), включает в себя стадии
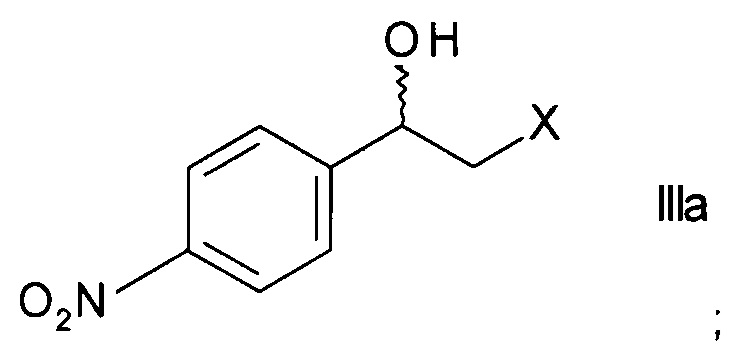
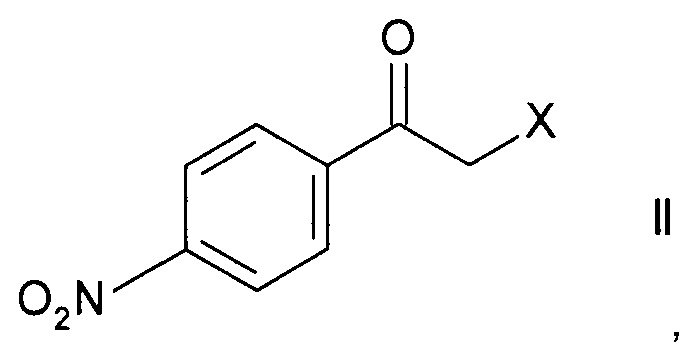
а) ферментативного восстановления кетона формулы
 ,
,
где X представляет собой атом галогена, оксидоредуктазой с образованием хирального спирта формулы
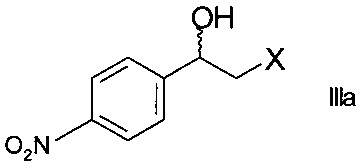 ;
;
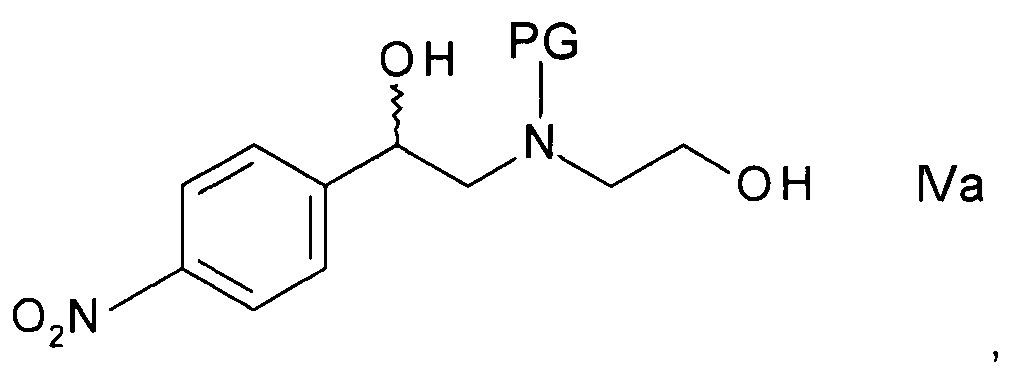
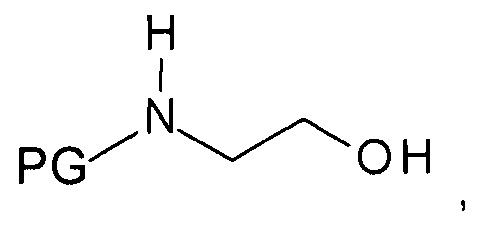
b) образования N-защищенного этаноламина формулы
 ,
,
где PG представляет собой аминозащитную группу;
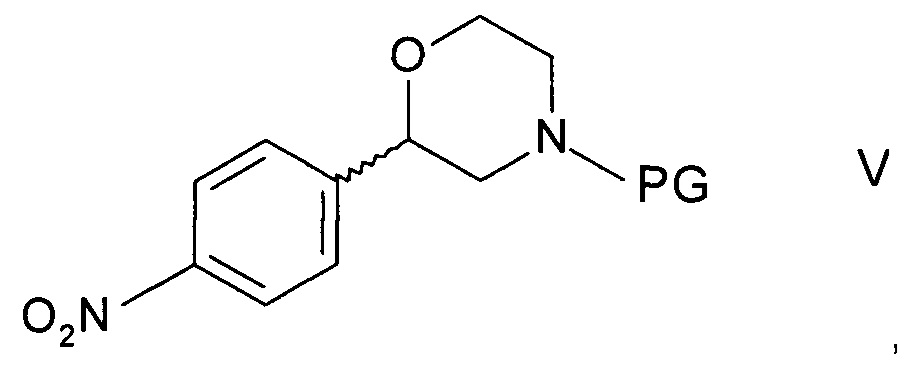
c) циклизации N-защищенного этаноламина формулы IVa с образованием 2-(4-нитрофенил)морфолина формулы
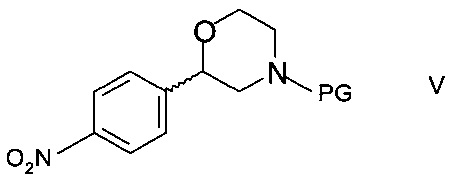 ,
,
где PG такая как определена выше; и
d) восстановления нитрогруппы с образованием хирального 2-(4-аминофенил)морфолина формулы I, и
e) необязательно удаления аминозащитной группы PG.
Следующие определения представлены для иллюстрации и пояснения смысла и содержания различных терминов, используемых в контексте описания изобретения.
Термин "C1-7-алкил" относится к одновалентному насыщенному алифатическому углеводородному радикалу с разветвленной или нормальной неразветвленной цепью, содержащему от одного до шести атомов углерода, предпочтительно, от одного до четырех атомов углерода, более предпочтительно, от одного до двух атомов углерода. Примерами этого термина также являются такие радикалы, как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил или трет-бутил, пентил и его изомеры, гексил и его изомеры, и гептил и его изомеры.
Термин "C1-7-алкокси" относится к С1-7-алкильной группе, такой как определена выше, к которой присоединен атом кислорода.
Термин "галоген" относится к фтору, хлору, брому или йоду, но в особенности, к хлору и брому.
Термин "арил" относится к ароматическому углеродному кольцу, такому как фенильное или нафтильное кольцо, предпочтительно, к фенильному кольцу.
Термин "гетероарил" относится к ароматическому от 5 до 6-членному моноциклическому кольцу или от 9 до 10-членному бициклическому кольцу, содержащему 1, 2 или 3 гетероатома, выбранных из азота, кислорода и/или серы, такому как пиридинил, пиразолил, пиримидинил, бензоимидазолил, хинолинил и изохинолинил.
Термин "фармацевтически приемлемые соли присоединения кислот" охватывают соли с неорганическими и органическими кислотами, такими как соляная кислота, азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, фумаровая кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфокислота, п-толуолсульфокислота и тому подобное.
Термин "аминозащитная группа" относится к заместителю, чувствительному к кислотам или кислотам Льюиса, обычно используемому для сдерживания реакционной способности аминогруппы. Подходящие аминозащитные группы, чувствительные к кислотам или кислотам Льюиса, описаны в работе Green Т., "Protective Groups in Organic Synthesis (Защитные группы в органическом синтезе)", 4th Ed. by Wiley Interscience, 2007, глава 7, с. 696 и далее. Таким образом, подходящие аминозащитные группы PG могут быть выбраны из Boc (трет-бутоксикарбонила), бензила, 4-метоксибензила, бензгидрила, Fmoc (флуоренилметоксикарбонила), Cbz (бензилоксикарбонила), Moz (р-метоксибензилкарбонила), Troc (2,2,2-трихлорэтоксикарбонила), Teoc (2-(триметилсилил)этоксикарбонила), Adoc (адамантоксикарбонила), формила, ацетила или циклобутоксикарбонила. В частности, используют Boc или бензил.
Спиралевидная связь
„ „
„
означает „ „ или „
„ или „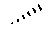 ", указывая тем самым на хиральность молекулы.
", указывая тем самым на хиральность молекулы.
Во всех случаях, когда в химической структуре присутствует хиральный атом углерода, предполагается, что все стереоизомеры, обусловленные таким хиральным атомом углерода, охватываются структурой, причем, как чистые стереоизомеры, так и их смеси.
Стадия а)
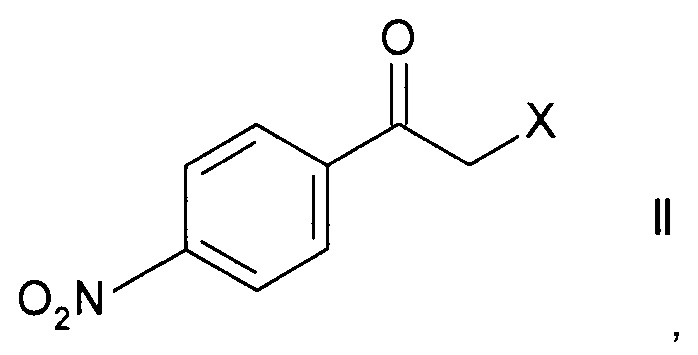
Стадия а) предполагает ферментативное восстановление кетона формулы II.
Кетоны формулы II являются коммерчески доступными соединениями или же могут быть синтезированы в соответствии с методами, известными специалистам в данной области техники.
Наиболее часто используемым кетоном формулы II является 2-бром-1-(4-нитрофенил)этанон.
Асимметрическое восстановление катализируется оксидоредуктазой обычно в присутствии НАДН (никотинамидадениндинуклеотид восстановленный) или НАДФН (англ. nicotinamide adenine dinucleotide phosphate, H form - никотинамидадениндинуклеотидфосфат восстановленный) в качестве кофактора, генерируемого in situ (с лат. - на месте).
Окисленный кофактор, как правило, постоянно регенерируется с помощью вторичного спирта в качестве косубстрата. Типичные косубстраты могут быть выбраны из пропанола-2, бутанола-2, пентан-1,4-диола, пентанола-2, 4-метилпентанола-2, гептанола-2, гексан-1,5-диола, гептанола-2 или октанола-2, предпочтителен пропанол-2. Предпочтительно, кофактор регенерируется при помощи косубстрата тем же ферментом, также катализирующим целевую реакцию. Ацетон, образующийся при использовании пропанола-2 в качестве косубстрата, согласно еще одному предпочтительному варианту осуществления, непрерывно удаляется из реакционной смеси.
Также хорошо известна регенерация кофактора с помощью дополнительного фермента, окисляющего его природный субстрат и обеспечивающего восстановленный кофактор, примерами являются системы дегидрогеназа вторичного спирта/спирт; глюкозодегидрогеназа/глюкоза; формиатдегидрогеназа/муравьиная кислота; глюкозо-6-фосфатдегидрогеназа/глюкозо-6-фосфат; фосфитдегидрогеназа/фосфит; гидрогеназа/молекулярный водород и тому подобное. Кроме того, известны методы электрохимической регенерации и применяются химические методы регенерации кофактора, включающие в себя металлический катализатор и восстановитель.
Предпочтительные микробиологические ферменты класса оксидоредуктаз происходят из дрожжей, бактерий или из клеток млекопитающих.
Оксидоредуктаза может использоваться в форме выделенного фермента (ферментов) или цельных клеток, необязательно в иммобилизованной форме, при помощи любого стандартного метода, описанного в литературе.
Согласно частному варианту осуществления настоящего изобретения, асимметрическое восстановление проводят в водной среде в присутствии органического сорастворителя, который может быть выбран, например, из глицерина, пропанола-2, диэтилового эфира, трет-бутилметилового эфира, диизопропилового эфира, дибутилового эфира, этилацетата, бутилацетата, гептана, гексана или циклогексана либо из их смеси.
Присутствие органического сорастворителя является весьма целесообразным, поскольку при этом может образовываться гомогенная суспензия, позволяющая осуществлять простое отделение требуемого кетона формулы II при помощи фильтрации.
Температуру реакции обычно поддерживают в диапазоне от 1°С до 50°С, предпочтительно, от 20°С до 40°С.
Реакционную концентрацию (концентрацию кетона формулы II и хирального спирта формулы IIIa в реакционной смеси) обычно поддерживают в диапазоне от 1% до 25%, предпочтительно, от 10% до 20%.
По окончании реакции (как правило, при более 90% превращении) продукт обычно обрабатывают при помощи экстракции или, предпочтительно, фильтрации.
В зависимости от кетонного субстрата предпочтительные системы катализатор/кофактор/косубстрат могут меняться.
Как правило, выбирают оксидоредуктазы, способные превращать кетон формулы II в требуемый хиральный спирт формулы IIIa с энантиомерным избытком 98% и выше.
Для получения (S)-2-бром-1-(4-нитрофенил)этанола оказались полезными следующие оксидоредуктазы.
НАДФН-зависимые оксидоредуктазы могут быть выбраны из кеторедуктаз (англ. KRED, кеторедуктаза) KRED-Y1, KRED-NADPH-P1A04 (англ. nicotinamide adenine dinucleotide, Н form - никотинамидадениндинуклеотид восстановленный), KRED-NADPH-P2H07, KRED-NADPH-P1B10, KRED-NADPH-107, KRED-NADPH-135, KRED-NADPH-136, KRED-NADPH-147 или KRED-NADPH-162 С, которые доступны из компании Codexis Inc., Редвуд Сити, Калифорния, США.
Наиболее предпочтительной является НАДФН-зависимая оксидоредуктаза KRED-Y1, рекомбинантная кеторедуктаза из лактобактерии Lactobacillus kefir, такая как раскрыта в Международной публикации РСТ патентного документа No. WO 2008103248 A1, идентифицированная как SEQ. ID. NO. 124, имеющая дополнительное замещение Е145А, из компании Codexis Inc., Редвуд Сити, Калифорния, США.
НАДН-зависимые оксидоредуктазы могут быть выбраны из типов KRED-NADH-110 и KRED-NADH-124, все из компании Codexis Inc., Редвуд Сити, Калифорния, США, из типов А161, А291 и А401 из компании Almac Group Ltd. Крейгавон, Соединенное Королевство, и типа А11 из компании Johnson Matthey, Лондон, Соединенное Королевство, а также из 1.1.200 из компании evocatal GmbH, Монхайм-на-Рейне, Германия, из ES-KRED-120 и из Enzysource, Ханчжлу, Китай. Особенно предпочтительными являются НАДН-зависимая оксидоредуктаза KRED-NADH-110 из компании Codexis Inc., Редвуд Сити, Калифорния, США, и А11 из Johnson Matthey, Лондон, Соединенное Королевство.
Асимметрическое восстановление можно осуществлять либо при помощи регенерации кофакторов с использованием сопряженных ферментативных реакций, основанной на глюкозе в качестве конечного восстановителя, либо при помощи регенерации с использованием сопряженных субстратов с пропанолом-2 в качестве конечного восстановителя. При восстановлении с использованием глюкозы в качестве конечного восстановителя величину pH следует поддерживать при помощи регулируемого добавления основания для нейтрализации образующейся глюконовой кислоты - окисленного побочного продукта регенерации восстановленного кофактора никотинамида при использовании глюкозодегидрогеназы (GDH 105 [Codexis]) в диапазоне от 1/10 до 1/2000 (соотношение фермент/субстрат). Температуру реакции можно поддерживать в диапазоне от 20°С до 40°С. Реакция может протекать как превращение кетона формулы II в хиральный спирт формулы IIIа в суспензии при концентрациях до 25%. Продукт может быть выделен с помощью стандартной процедуры экстракции, например, ТБМЭ (трет-бутилметиловым эфиром) или этилацетатом. Продукт предпочтительно выделяют фильтрацией - если это целесообразно - после предварительного упаривания органического сорастворителя.
Для получения (R)-2-бром-1-(4-нитрофенил)этанола оказались полезными следующие оксидоредуктазы.
НАДФН-зависимая оксидоредуктаза может быть выбрана из типов KRED-NADPH-104, KRED-NADPH-130 or KRED-NADPH-148, все из компании Codexis Inc., Редвуд Сити, Калифорния, США. Особенно предпочтительной является НАДФН-зависимая оксидоредуктаза KRED-NADPH-104 из компании Codexis Inc., Редвуд Сити, Калифорния, США.
НАДН-зависимая оксидоредуктаза может быть выбрана из типов KRED-Y2, KRED-NADH-117, KRED-NADH-126, все из компании Codexis Inc., Редвуд Сити, Калифорния, США, из типа Х1 из Johnson Matthey, Лондон, Соединенное Королевство, и из типа 127 из Enzysource, Ханчжлу, Китай, а также из типа А131 из компании Almac Group Ltd. Craigavon, United Kingdom.
Особенно полезной является НАДН-зависимая оксидоредуктаза KRED-Y2, рекомбинантная кеторедуктаза из Novosphingobium aromaticivorans, такая как описана в Международной публикации РСТ патентного документа No. WO 2011/005527 A2 и идентифицирована как SEQ. ID. NO. 2., из компании Codexis Inc., Редвуд Сити, Калифорния, США.
Асимметрическое восстановление выполняли при помощи регенерации кофакторов с использованием сопряженных ферментативных реакций на основе глюкозы в качестве конечного восстановителя. Во время реакции величину pH поддерживали при помощи контролируемого добавления основания, такого как водный раствор гидроксида натрия, для нейтрализации образующейся глюконовой кислоты - окисленного побочного продукта регенерации восстановленного кофактора никотинамида при использовании глюкозодегидрогеназы (GDH 105 из Codexis). Температуру реакции можно поддерживать в диапазоне от 20°С до 40°С. Реакция может протекать как превращение кетона формулы II в хиральный спирт формулы IIIa в суспензии при концентрациях до 20%. Продукт может быть выделен при помощи стандартной процедуры экстракции, например, ТБМЭ или этилацетатом. Предпочтительным способом выделения продукта является простая фильтрация продукта - если это целесообразно - после отгонки органических сорастворителей.
Стадия b)
Стадия b) подразумевает образование N-защищенного этаноламина формулы IVa.
Согласно частному варианту осуществления, хиральный спирт формулы IIIа, полученный на стадии a), может быть непосредственно, без выделения его из реакционной смеси, использован на данной стадия b).
Как правило, получение N-защищенного этаноламина формулы IVa выполняют либо
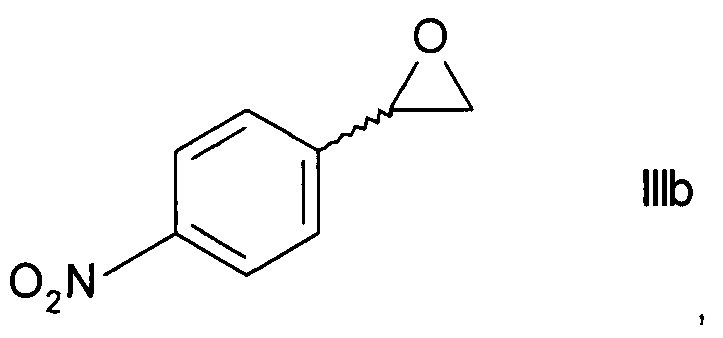
i) в три стадии путем превращения на первой стадии хирального спирта формулы IIIa в присутствии основания в эпоксид формулы
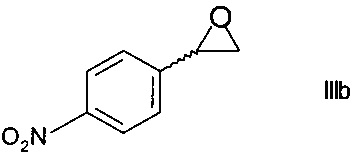 ,
,
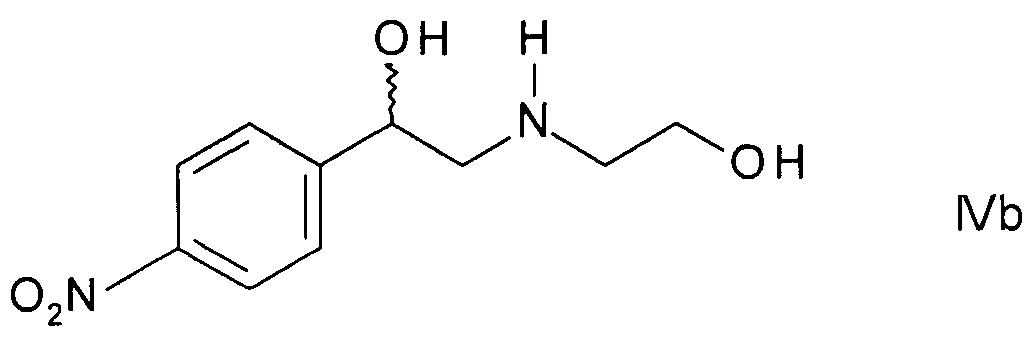
превращения на следующей стадии эпоксида формулы IIIb при воздействии этаноламина в незащищенный этаноламин формулы

и введения на последней стадии аминозащитной группы PG;
ii) в две стадии путем превращения на первой стадии хирального спирта формулы IIIa при воздействии этаноламина в незащищенный этаноламин формулы IVb и введения на следующей стадии аминозащитной группы PG, или
iii) в одну стадию путем реакции хирального спирта формулы IIIa с N-защищенным этаноламином формулы
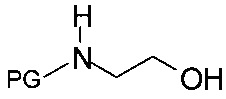 ,
,
где PG представляет собой аминозащитную группу.
Образование эпоксида по способу i) может осуществляться путем обработки хирального спирта формулы IIIa водным основанием, таким как водный раствор гидроксида натрия, в присутствии органического растворителя, такого как тетрагидрофуран, метилтетрагидрофуран, трет-бутилметиловый эфир, циклопентилметиловый эфир, 1,2-диэтоксиэтан, или низшими алифатическими спиртами, такими как этанол. Эпоксид формулы IIIb может быть выделен из органического слоя при помощи отгонки растворителя.
Получение незащищенного этаноламина формулы IVa по способу i) можно осуществлять при помощи обработки эпоксида формулы IIIb этаноламином в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в подходящем органическом растворителе, таком как диэтиловый эфир, тетрагидрофуран, диоксан или трет-бутилметиловый эфир, при температуре от 0°С до 60°С.
Как правило, используют избыток по отношению к стехиометрии от 2 до 30 эквивалентов, предпочтительно, избыток приблизительно 10 эквивалентов этаноламина.
Незащищенный этаноламин формулы IVa может быть выделен из реакционной смеси путем экстракции подходящим растворителем, таким как смесь этилацетата с водой, с последующим концентрированием органической фазы.
Аминозащитную группу PG по способу i) можно вводить при помощи способов, хорошо известных специалистам в данной области. Согласно частному варианту осуществления, выбирают Вос-группу и вводят ее, используя Вос-ангидрид в присутствии подходящего органического растворителя, такого как диэтиловый эфир, тетрагидрофуран, диоксан или трет-бутилметиловый эфир, при температуре от 0°С до 40°С. N-защищенный этаноламин формулы IVa может быть выделен из органического слоя путем отгонки растворителя.
Согласно способу ii), хиральный спирт формулы IIIa обрабатывают этаноламином в присутствии подходящего органического растворителя, такого как диэтиловый эфир, тетрагидрофуран, диоксан или трет-бутилметиловый эфир, при температуре от 0°С до 60°С.
Как правило, используют избыток по отношению к стехиометрии от 2 до 30 эквивалентов, предпочтительно избыток приблизительно 10 эквивалентов этаноламина.
Выделение незащищенного этаноламина формулы IVa из реакционной смеси можно осуществлять при помощи экстракции подходящим растворителем, таким как смесь этилацетата с водой, с последующим концентрированием органической фазы.
Аминозащитную группу PG можно вводить, как описано выше для способа i).
Согласно способу iii), N-защищенный этаноламин формулы IVa также может быть получен обработкой хирального спирта формулы III N-защищенным этаноламином, предпочтительно, бензил-защищенным этаноламином, в присутствии подходящего растворителя, такого как н-пропанол, и органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, при температуре от 40°С до температуры рефлюкса растворителя.
В качестве альтернативы, способ iii) также можно осуществлять, исходя из эпоксида формулы IIIb, используя условия реакции, такие как изложены выше для способа iii).
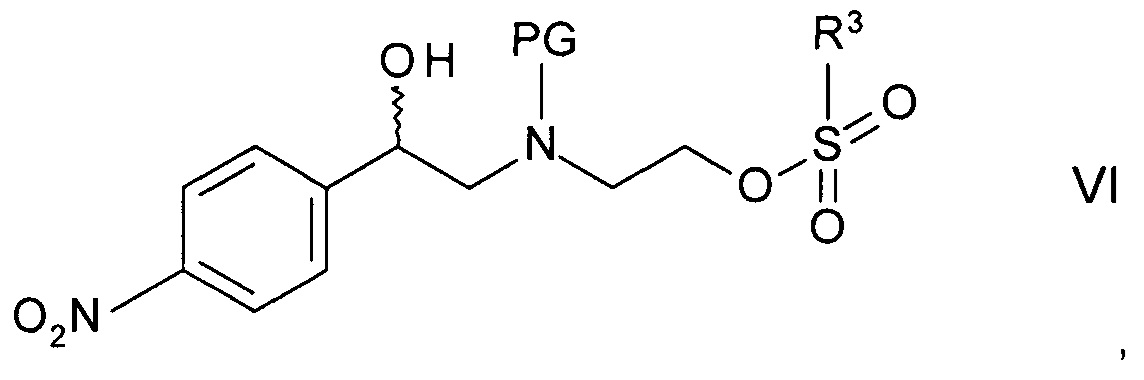
Стадия c)
Стадия c) предполагает циклизацию N-защищенного этаноламина формулы IVa с образованием 2-(4-нитрофенил)морфолина формулы V.
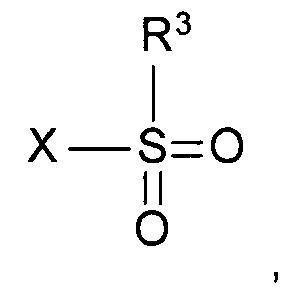
Реакцию, как правило, выполняют постадийно взаимодействием N-защищенного этаноламина формулы IVa с сульфонилгалогенидом формулы
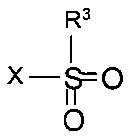 ,
,
где R3 и X такие как определены выше, с образованием промежуточного сульфоната формулы
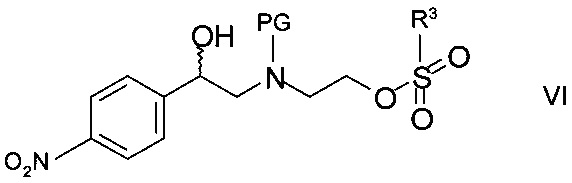 ,
,
где PG такая как определена выше, a R3 представляет собой C1-4-алкил или фенил, необязательно замещенный С1-4-алкильной группой, нитрогруппой или атомом галогена. Подходящим сульфонилгалогенидом является метансульфохлорид (R1 - метил, X - хлор). Реакцию проводят в присутствии органического основания, такого как триэтиламин, N,N-диизопропилэтиламин или N-метилморфолин, в особенности, триэтиламин, и подходящего органического растворителя, такого как диэтиловый эфир, тетрагидрофуран, диоксан или трет-бутилметиловый эфир, более предпочтителен тетрагидрофуран, при температуре от 0°С до 40°С.
Промежуточный сульфонат может быть выделен с помощью методов, известных специалистам в данной области, однако, как правило, реакционную смесь циклизуют напрямую обработкой ненуклеофильным основанием.
Подходящими основаниями являются такие ненуклеофильные основания, как алкоксиды щелочных металлов, такие как трет-бутоксид калия или 2-метил-2-бутоксид калия; реакцию, таким образом, проводят в по существу безводной среде, используя подходящие апротонные органические растворители, такие как диэтиловый эфир, тетрагидрофуран, диоксан или трет-бутилметиловый эфир.
Альтернативой ненуклеофильным основаниям являются катализаторы межфазного переноса, такие как соли четвертичного аммония или фосфония, тетраалкиламмониевые соли, такие как, например, гидросульфат тетрабутиламмония, хлорид бензилтриметиламмония, бромид этилгексадецилдиметиламмония или бромид тетрабутилфосфония. При использовании оснований такого типа, как правило, присутствует водное неорганическое основание, такое как водный раствор гидроксида натрия, калия или лития, и подходящий органический растворитель, такой как диэтиловый эфир, тетрагидрофуран, 2-метилтетрагидрофуран или толуол.
Температуру реакции циклизации выбирают в диапазоне от 0°С до 40°С.
Образовавшийся 2-(4-нитрофенил)морфолин формулы V может быть выделен путем экстракции водой и подходящим органическим растворителем, таким как трет-бутилметиловый эфир, с последующим концентрированием органической фазы.
Стадия d)
Стадия d) предполагает восстановление нитрогруппы с образованием хирального 2-(4-аминофенил)морфолина формулы I, где R1 представляет собой PG.
Восстановление может осуществляться путем гидрирования водородом при атмосферном или повышенном давлении в присутствии металлического катализатора гидрирования, такого как PtO2, Pd/C, Pt/V или катализатор на основе никеля Ренея, в протонных растворителях, таких как метанол, этанол, пропанол-2, вода или их смеси, при температуре от 0°С до 40°С.
Хиральный 2-(4-аминофенил)морфолин формулы I, где R1 представляет собой PG, может быть выделен фильтрацией реакционной смеси с последующим концентрированием фильтрата.
Стадия e)
Стадия e) включает в себя необязательное удаление защитной группы PG.
Способы удаления аминозащитных групп хорошо известны специалистам в данной области техники.
N-защитную группу ВОС можно удалять с помощью водных растворов минеральных кислот, таких как соляная кислота, H2SO4 или H3PO4, или при помощи органических кислот, таких как трифторуксусная кислота, хлоруксусная кислота, дихлоруксусная кислота, уксусная кислота, метансульфокислота или п-толуолсульфокислота, в растворителях, таких как хлористый метилен, хлороформ, тетрагидрофуран, метанол, этанол, пропанол-1, ацетонитрил или вода, при температуре реакции от 0°С до 80°С.
Согласно предпочтительному варианту осуществления, N-защитную группу ВОС можно удалять при помощи трифторуксусной кислоты в водном ацетонитриле при температуре приблизительно 60°С в течение 2 часов или при помощи 25% водного раствора соляной кислоты в пропаноле-1 при температуре приблизительно 60°С в течение 2 часов.
Бензильную защитную группу предпочтительно можно удалять в условиях гидрогенолиза в присутствии металлического катализатора гидрирования, такого как Pd/C.
Согласно еще одному варианту осуществления изобретения и как было указано выше, способ настоящего изобретения может применяться для получения соединений формулы
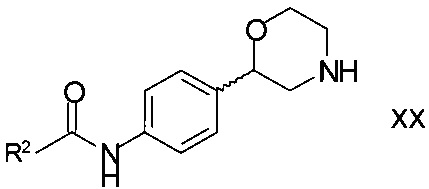 ,
,
где
R2 является арилом или гетероарилом, в котором ароматические кольца необязательно замещены одним или двумя заместителями, выбранными из C1-7-алкила, галогена, CF3, OCF3, OCH2CF3, C1-7-алкокси или циано;
или их фармацевтически приемлемой соли присоединения кислоты или
для получения соединений формулы
 ,
,
где
R2 является арилом или гетероарилом, в котором ароматические кольца необязательно замещены одним или двумя заместителями, выбранными из C1-7-алкила, галогена, CF3, OCF3, OCH2CF3, С1-7-алкокси или циано;
или их фармацевтически приемлемой соли присоединения кислоты.
Соединения формулы XX могут, например, быть получены взаимодействием хирального 2-(4-аминофенил)морфолина формулы
 ,
,
где R1 - аминозащитная группа, со сложным эфиром формулы
R2COOR4,
где R2 такой как определен выше, a R4 является С1-7-алкилом.
Согласно частному варианту осуществления настоящего изобретения, амид может быть образован путем конденсации хирального 2-(4-аминофенил)морфолина формулы I с карбоновой кислотой формулы
R2COOH,
где R2 такой как определен выше,
при использовании в качестве связующего агента пропилфосфинового ангидрида. Оказалось, что подходящим основанием является триэтиламин, а подходящим растворителем - этилацетат. Температуру реакции можно выбрать в диапазоне от 0°С до 50°С.
Согласно еще одному частному варианту осуществления настоящего изобретения, амид может быть образован путем конденсации сложного эфира формулы, такой как была определена выше, с хиральным 2-(4-аминофенил)морфолином формулы I в присутствии подходящего алкоголята щелочного металла, такого как трет-бутилат натрия или калия, и подходящего органического растворителя, такого как эфирные растворители, как, например, тетрагидрофуран, 2-метилтетрагидрофуран, трет-бутилметиловый эфир или циклопентилметиловый эфир. Температуру реакции обычно выбирают в диапазоне от -10°С до 30°С.
На следующей стадии аминозащитная группа может быть удалена при помощи методов, описанных выше для стадии e).
Согласно еще одному варианту осуществления настоящего изобретения, соединения формулы XX также могут быть получены взаимодействием хирального 2-(4-аминофенил)морфолина формулы I, где R1 представляет собой водород, со сложным эфиром формулы
R2COOR4,
где R2 такой как определен выше, a R4 - С1-7-алкил.
R4, в частности, является метилом.
Реакция, как правило, протекает в присутствии гексаметилдисилазана щелочного металла, такого как гексаметилдисилазан лития, натрия или калия, и подходящего органического растворителя, такого как эфирные растворители, как, например, тетрагидрофуран, 2-метилтетрагидрофуран, циклопентилметиловый эфир или трет-бутилметиловый эфир. Реакцию обычно проводят при температуре приблизительно от -50°С до -78°С.
Соединения формулы XXX могут быть получены, например, взаимодействием хирального 2-(4-аминофенил)морфолина формулы
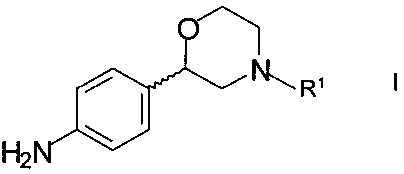 ,
,
где R1 такой как определен выше, с галогенидом формулы
R2X,
где R2 такой как определен выше, а X представляет собой галоген.
X, в частности, представляет собой хлор.
Реакцию, как правило, проводят в присутствии подходящего третичного амина, такого как триэтиламин, N,N-диизопропилэтиламин или тому подобное, и полярного апротонного растворителя, такого как тетрагидрофуран, этилацетат, диметилформамид, или полярного протонного растворителя, такого как алифатические спирты, в частности, третичные спирты, такие как 2-метил-2-бутанол или тому подобное. Реакцию, как правило, осуществляют в условиях кипячения с обратным холодильником.
На следующей стадии аминозащитная группа может быть удалена при помощи методов, описанных выше для стадии e).
ОПИСАНИЕ ПРИМЕРОВ ОСУЩЕСТВЛЕНИЯ ИЗОБРЕТЕНИЯ
Сокращения:
ЦПМЭ - циклопентилметиловый эфир (англ. СРМЕ)
ДИПЭА - диизопропилэтиламин (англ. DIPEA)
EtOH - этанол
IPC - контроль в ходе процесса (англ. in process control)
ВЭЖХ - высокоэффективная жидкостная хроматография (англ. HPLC, high pressure liquid chromatography)
ТЭА - триэтиламин (англ. TEA)
ТФУК - трифторуксусная кислота (англ. TFA)
ТБМЭ - трет-бутилметиловый эфир (англ. ТВМЕ)
ТГФ - тетрагидрофуран (англ. THF)
2-Ме-ТГФ - 2-метилтетрагидрофуран
rt - комнатная температура (англ. room temperature)
Пример 1
(R)-2-Бром-1-(4-нитрофенил)этанол

Субстрат, 100 г 2-бром-1-(4-нитрофенил)-этанона, суспендировали в двухфазной реакционной смеси, содержащей 600 мл водного буферного раствора (2,45 г дигидрофосфата калия (30 мМ), 1,29 г тетрагидрата ацетата магния (10 мМ), 100 г моногидрата D-глюкозы и 100 мг НАД) и 200 мл н-гептана. Температуру повышали при перемешивании до 30°С, и величину pH доводили до 7,2 (15,7 мл 1 N NaOH). Восстановление начиналось при добавлении оксидоредуктазы KRED-Y2 [Codexis] (1,0 г) и фермента регенерации кофактора - глюкозодегидрогеназы (1,0 г GDH 105 [Codexis]), образуя тонкодисперсную суспензию светло-желтого цвета. В течение 18 ч реакционного времени величину pH поддерживали на уровне pH 7,2 добавлением 403 мл 1М NaOH, достигая практически полного превращения (IPC: 0,8% площади II). После охлаждения до комнатной температуры продукт отфильтровывали, дважды промывали 118 мл воды и 118 мл гептана, сушили при движении в вакууме 20 мбар (2000 Па) при температуре 30°С и получали 97,7 г титульного соединения. ГХ-ЭИ-МС (англ. GC-EI-MS, Gas Chromatography-Electron Ionization-Mass Spectrometry - газовая хроматография в сочетании с масс-спектрометрией с ионизацией электронным ударом): 245 (М+Н)+; хиральная ВЭЖХ: э.и. (англ. ее, enantiomeric excess - энантиомерный избыток) 99,9% [268 нм; Chiracel OZ-H; 250*4,6 мм, изократическое элюирование 90% н-гептана, 5% EtOH, 5% н-гептана с 0,4% ТФУК]; 12°С: 1 мл/мин содержание, соответствующее (2,6% (R)-эпоксида IIIb, э.и. 99,9%.
Пример 2
(S)-2-Бром-1-(4-нитрофенил)этанол
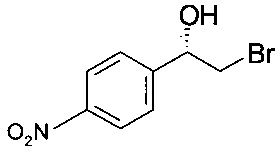
При интенсивном перемешивании 100 г 2-бром-1-(4-нитрофенил)-этанона в 300 мл водного буферного раствора (100 мМ дигидрофосфата калия, pH 7,2; 2 мМ хлорида магния) и 100 мл пропанола-2 образовывалась суспензия светло-желтого цвета. Реакционный раствор нагревали до температуры 30°С и перемешивали в течение 15 минут, при этом фактическая величина pH соответствовала 7,7. После этого запускали реакцию восстановления добавлением окисленного кофактора НАДФ (200 мг [Roche]) и оксидоредуктазы (500 мг KRED-Y1 [Codexis]). В ходе реакции в течение 23 ч величина pH снижалась до pH 6,5, реакция протекала практически до полного превращения (IPC: 1,6% площади II). После охлаждения до комнатной температуры реакционную массу - с учетом трехкратного ополаскивания 4-горлой плоскодонной реакционной колбы 100 мл воды - переносили в круглодонную колбу для отгонки органических растворителей, пропанола-2 и ацетона (образовавшегося), при давлении от 100 до 50 мбар (от 10000 до 5000 Па), температуре 40°С в течение 30 минут. После охлаждения до комнатной температуры продукт отфильтровывали, промывали 200 мл воды и 200 мл гептана, сушили в высоком вакууме и получали 96,6 г титульного соединения. ГХ-ЭИ-МС: 245 (М+Н)+; хиральная ВЭЖХ: э.и. 99,5% [268 нм; Chiracel OZ-H; 250*4,6 мм, изократическое элюирование 90% н-гептана, 5% EtOH, 5% н-гептана с 0,4% ТФУК]; 12°С: 1 мл/мин содержание, соответствующее 1,2% (S)-эпоксида IIIb, э.и. более 99,5%..
Пример 3
Пример 3.1
Получение (S)-2-(4-нитрофенил)оксирана
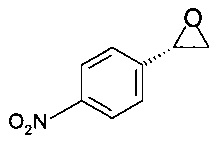
2,46 г (10,0 ммоль) (S)-2-бром-1-(4-нитрофенил)этанола растворяли в 12,0 мл ТГФ, при комнатной температуре добавляли 10,0 мл 2М NaOH, реакционную массу перемешивали при комнатной температуре в течение 1 ч. Мутный раствор темно-коричневого цвета фильтровали через стекловолоконный фильтр, промывали 20 мл ТБМЭ, органический слой отделяли и промывали 20 мл 1М KH2PO4, сушили над Na2SO4, фильтровали, концентрировали в вакууме при 40°С/20 мбар (2000 Па)/1 ч и получали 1,60 г титульного продукта в виде твердого вещества желтого цвета.
МС-ЭРИ- (англ. MS-ESI-, масс-спектроскопия - электрораспылительная ионизация с регистрацией отрицательных ионов): МН- 164,035
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak IA-3. Энантиомерное соотношение: 99,8/0,2% (S/R)
Пример 3.2
Получение (R)-2-(4-нитрофенил)оксирана
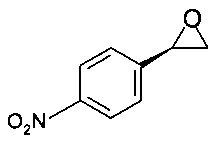
К раствору (R)-2-бром-1-(4-нитрофенил)этанола (2,46 г, 10 ммоль, экв.: 1,00) в ТГФ (10,9 г, 12,3 мл) прибавляли при комнатной температуре NaOH (10,0 мл, 20,0 ммоль, экв.: 2), и смесь перемешивали при комнатной температуре в течение 1 ч.
Смесь фильтровали, и осадок на фильтре промывали 20 мл ТБМЭ. Фильтрат экстрагировали и разделяли, органический слой промывали 20 мл 1М KH2PO4, сушили над Na2SO4, фильтровали, фильтрат концентрировали в вакууме при 40°С/20 мбар (2000 Па)/1 ч и получали 1,5 г титульного продукта в виде твердого вещества желтого цвета.
МС-ЭРИ-: МН- 164,035
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak IA-3. Энантиомерное соотношение: 99,95/0,05 (R/S)
Пример 4
Пример 4.1
Получение (S)-2-(2-гидроксиэтиламино)-1-(4-нитрофенил)этанола
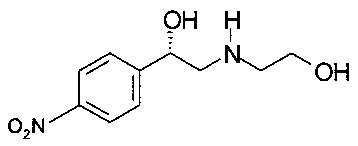
К 124,0 г (2,02 моль) 2-аминоэтанола в течение 30 минут по каплям прибавляли раствор 50,0 г (202 ммоль) (S)-2-бром-1-(4-нитрофенил)этанола в 50 мл ТГФ. Смесь охлаждали на водяной бане до температуры менее 30°С. Смесь перемешивали в течение 16 часов при комнатной температуре. Раствор экстрагировали 500 мл этилацетата и 500 мл воды. Водный слой повторно экстрагировали 250 мл этилацетата. Водный слой насыщали 160 г NaCl и еще раз экстрагировали 500 мл этилацетата. Объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали в вакууме при 20 мбар (2000 Па)/40°С/2 часа и получали 40,05 г технического (S)-2-(2-гидроксиэтиламино)-1-(4-нитрофенил)этанола в виде масла коричневого цвета, которое использовали в Примере 5.1 без дополнительной очистки.
МС-ЭРИ+: МН+ 227,3
Пример 4.2
Получение (R)-2-(2-гидроксиэтиламино)-1-(4-нитрофенил)этанола

Аналогично Примеру 4.1 (R)-2-бром-1-(4-нитрофенил)этанол вступал в реакцию с 2-аминоэтанолом. 110 г титульного продукта получали в виде технического масла коричневого цвета, которое использовали в Примере 5.2 без дополнительной очистки.
МС-ЭРИ+: МН+ 227,3
Пример 5
Пример 5.1
Получение (S)-трет-бутил-2-гидрокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамата
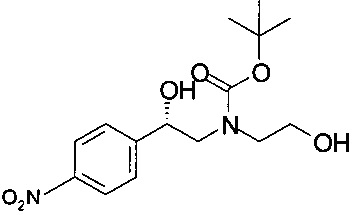
К смеси 45,0 г (199 ммоль) (S)-2-(2-гидроксиэтиламино)-1-(4-нитрофенил)этанола (45 г, 199 ммоль, экв.: 1,00) в ТГФ (399 г, 450 мл, 5,51 моль, экв.: 27,7) прибавляли Вос-ангидрид (43,8 г, 46,6 мл, 201 ммоль, экв.: 1,01). Температура повышалась до 35°С. Через 15 минут снова добавляли Вос-ангидрид (6,95 г, 31,8 ммоль, экв.: 0,16), и реакционную массу перемешивали в течение 30 минут при комнатной температуре. Прибавляли 650 мл ТБМЭ и 650 мл 1М раствора Na2CO3 и перемешивали в течение 10 минут. Органический слой отделяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме. Воду удаляли при помощи азеотропной вакуумной перегонки с 2×100 мл ТБМЭ. Вязкое масло красного цвета сушили при 40°С/12 мбар (1200 Па) в течение 4 часов и получали 75,26 г технического (S)-трет-бутил-2-гидрокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамата в виде масла коричневого цвета, которое использовали в Примерах 6.1 и 7.1 без дополнительной очистки.
МС-ЭРИ-: MHCOO- 371,1
Пример 5.2
Получение (R)-трет-бутил-2-гидрокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамата
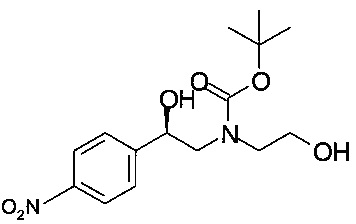
Аналогично Примеру 5.1 (R)-2-(2-гидроксиэтиламино)-1-(4-нитрофенил)этанол вступал в реакцию с Вос-ангидридом, при этом получали 55,6 г титульного продукта, который использовали в Примерах 6.2 и 7.2 без дополнительной очистки.
МС-ЭРИ-: (М+НСОО)- 371,1
Пример 5.3
Получение (S)-2-(бензил(2-гидроксиэтил)амино)-1-(4-нитрофенил)этанола (из (S)-2-бром-1-(4-нитрофенил)этанола)
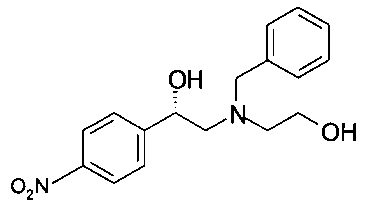
24,6 г (100 ммоль) (S)-2-бром-1-(4-нитрофенил)этанола растворяли в 120 мл пропанола-2, прибавляли 13,9 мл (100 ммоль) триэтиламина и 17,1 мл (120 ммоль) 2-(бензиламино)этанола, и реакционную массу нагревали в сосуде с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры; пропанол-2 удаляли в вакууме при 40°С/от 100 до 50 мбар (от 10000 Па до 5000 Па)/1 ч. Остаток обрабатывали 320 мл 1,75М NH3 в солевом растворе (смесь 130 мл 25% водного раствора аммиака и 870 мл солевого раствора) и дважды экстрагировали 320 мл ТБМЭ. Объединенные органические слои сушили над Na2SO4, фильтровали, фильтрат концентрировали в вакууме при 40°С/20 мбар (2000 Па)/2 ч и получали 30,5 г технического (S)-2-(бензил(2-гидроксиэтил)амино)-1-(4-нитрофенил)этанола в виде масла темно-красного цвета, которое использовали без дополнительной очистки.
МС-ЭРИ+: МН+ 317,15
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak AY-3. Энантиомерное соотношение: 91,6/8,4 (S/R).
Пример 5.4
Получение (S)-2-(бензил(2-гидроксиэтил)амино)-1-(4-нитрофенил)этанола (из (S)-2-(4-нитрофенил)оксирана)
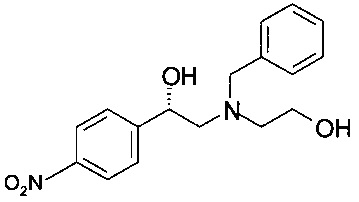
0,16 г (1,0 ммоль) (S)-2-(4-нитрофенил)оксирана растворяли в 0,65 мл пропанола-2, добавляли 0,14 мл (1,0 ммоль) триэтиламина и 0,18 мл (1,2,0 ммоль) 2-(бензиламино)этанола, и реакционную массу нагревали в сосуде с обратным холодильником в течение 16 часов. Реакционную смесь охлаждали до комнатной температуры; пропанол-2 удаляли в вакууме при 40°С/от 100 до 50 мбар (от 10000 Па до 5000 Па)/1 ч. Остаток обрабатывали 3,5 мл 1,75М NH3 в солевом растворе (смесь 130 мл 25% водного раствора аммиака и 870 мл солевого раствора) и дважды экстрагировали 3,5 мл МТБЭ. Объединенные органические слои сушили над Na2SO4, фильтровали, фильтрат концентрировали в вакууме при 40°С/20 мбар (2000 Па)/2 ч и получали 0,34 г технического (S)-2-(бензил(2-гидроксиэтил)амино)-1-(4-нитрофенил)этанола в виде масла темно-красного цвета, которое использовали без дополнительной очистки.
МС-ЭРИ+: МН+ 317,15
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak AY-3. Энантиомерное соотношение: 99,7/0,3 (S/R).
Пример 6
Пример 6.1
Получение (S)-трет-бутил-2-мезилокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамата
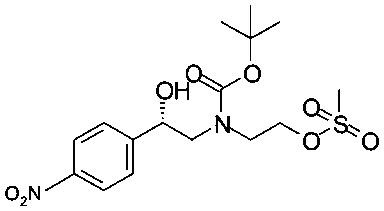
К раствору 0,32 г (1,0 ммоль) (S)-трет-бутил-2-гидрокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамата в 3,3 мл ТГФ прибавляли 0,15 мл (1,1 ммоль) триэтиламина, раствор охлаждали до температуры от 0 до 5°С.
После этого в течение 5 минут прибавляли раствор 1,05 ммоль метансульфохлорида в 82 мкл ТГФ (температура от 0 до 5°С). После перемешивания смеси в течение 15 мин при температуре от 0 до 5°С анализ методом ВЭЖХ показал, что осталось 23% исходного вещества. К белой суспензии медленно прибавляли 42 мкл, 0,30 ммоль, триэтиламина и 20 мкл, 0,25 ммоль, метансульфохлорида. Суспензию перемешивали в течение 15 минут при температуре от 0 до 5°С, фильтровали и промывали предварительно охлажденным (от 0 до 5°С) ТГФ. Маточный раствор, содержащий технический (S)-трет-бутил-2-мезилокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамат в растворе, хранили при температуре -20°С (продукт в виде вещества нестабилен, в растворе стабилен в течение нескольких дней).
МС-ЭРИ-: (М+НСОО)- 449,12
Пример 6.2
Получение (R)-трет-бутил-2-мезилокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамата

Аналогично Примеру 6.1 (R)-трет-бутил-2-гидрокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамат вступал в реакцию с метансульфонилхлоридом. Маточный раствор, содержащий технический (R)-трет-бутил-2-мезилокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамат в растворе, хранили при температуре -20°С.
МС-ЭРИ-: (М+НСОО)- 449,12
Пример 7
Пример 7.1
Получение (S)-трет-бутил-2-(4-нитрофенил)морфолин-4-карбоксилата
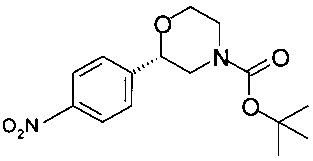
К раствору 18,0 г (55,2 ммоль) (S)-трет-бутил-2-гидрокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамата в 180 мл ТГФ прибавляли 8,50 мл (60,7 ммоль) триэтиламина и охлаждали до температуры от 0 до 5°С. Раствор 4,5 мл (57,9 ммоль) метансульфохлорида в 4,5,0 мл ТГФ прибавляли в течение 15 минут (температура 0-5°С). Смесь перемешивали в течение 15 минут при температуре от 0 до 5°С. После анализа методом ВЭЖХ оставалось 18% исходного вещества. К суспензии медленно прибавляли 2,3 мл (16,5 ммоль) триэтиламина и 0,86 мл (11,0 ммоль) метансульфохлорида. Суспензию перемешивали в течение 15 минут при температуре от 0 до 5°С, суспензию светло-желтого цвета фильтровали и промывали 50 мл предварительно охлажденного ТГФ (от 0 до 5°С). К холодному раствору, содержащему промежуточный (S)-трет-бутил-2-мезилокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамат, прибавляли 27,5 мл (110 ммоль) 4М NaOH и 0,38 г (1,1 ммоль) гидросульфата тетрабутиламмония. Смесь тщательно перемешивали в течение 16 часов при комнатной температуре, затем экстрагировали 140 мл воды и 170 мл ТБМЭ, отделенные органические слои сушили над Na2SO4, фильтровали, концентрировали в вакууме при 40°С/10 мбар (1000 Па)/2 ч. 17,7 г технического продукта обрабатывали 53 мл МеОН при нагревании с обратным холодильником в течение 5 минут, охлаждали в течение 1 ч до комнатной температуры, суспензию перемешивали в течение 16 ч при температуре от 0 до 5°С, фильтровали, и осадок на фильтре промывали 13 мл предварительно охлажденного МеОН, кристаллы сушили при 40°С/10 мбар (1000 Па)/2 ч и получали 12,0 г (S)-трет-бутил-2-(4-нитрофенил)морфолин-4-карбоксилата в виде кристаллов белого цвета.
ГХ-ЭИ-МС: М308+.
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak AD-H. Энантиомерное соотношение: 99,92/0,08% (S/R).
Пример 7.2
Получение (R)-трет-бутил-2-(4-нитрофенил)морфолин-4-карбоксилата
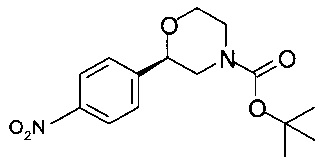
Аналогично Примеру 7.1 циклизовали (R)-трет-бутил-2-гидрокси-2-(4-нитрофенил)этил-(2-гидроксиэтил)карбамат. Получали 44,3 г титульного продукта в виде кристаллов грязно-белого цвета.
ГХ-ЭИ-МС: М308+.
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak AD-H. Энантиомерное соотношение: 99,95/0,05 (R/S).
Пример 7.3
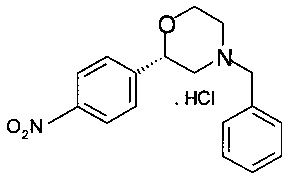
Получение хлоргидрата (S)-4-бензил-2-(4-нитрофенил)морфолина
 via
via 
30,2 г (95,5 ммоль) (S)-2-(бензил(2-гидроксиэтил)амино)-1-(4-нитрофенил)этанола растворяли в 330 мл ТГФ, прибавляли 29,3 мл (210 ммоль) триэтиламина, и раствор охлаждали до температуры 0-5°С. После этого прибавляли по каплям раствор 11,9 мл (153 ммоль) метансульфохлорида в 12 мл ТГФ при температуре от 0 до 5° в течение 20 минут. Суспензию перемешивали в течение 30 мин при температуре от 0 до 5°С, фильтровали и промывали 100 мл предварительно охлажденного ТГФ. К объединенной маточной жидкости (содержащей преимущественно промежуточное мезилокси-производное) прибавляли 95 мл 4М NaOH и 0,65 г (1,91 ммоль) гидросульфата тетрабутиламмония. Реакционную массу перемешивали в течение 2 ч при комнатной температуре, экстрагировали 300 мл воды и 300 мл трет-бутиметилового эфира (ТБМЭ), отделенные органические слои сушили над Na2SO4, фильтровали, фильтрат концентрировали в вакууме при 40°С/10 мбар (1000 Па)/5 ч и получали 35,9 г технического (S)-4-бензил-2-(4-нитрофенил)морфолина в виде масла темно-коричневого цвета. Технический продукт растворяли в 50 мл этилацетата, добавляли 24,0 мл 4М раствора HCl в этаноле (полученного in situ из хлористого ацетила в этаноле). Образовавшуюся суспензию нагревали в сосуде с обратным холодильником в течение 5 мин, добавляли 50 мл этилацетата и снова нагревали в сосуде с обратным холодильником в течение 5 минут. Суспензию охлаждали в течение 1 ч до комнатной температуры, перемешивали в течение 1 ч при комнатной температуре, фильтровали и промывали 25 мл смеси растворителей, содержащей этилацетат и этанол, 4/1. Кристаллы сушили в вакууме при 40°С/10 мбар (1000 Па)/2 ч и получали 11,8 г хлоргидрата (S)-4-бензил-2-(4-нитрофенил)морфолина в виде кристаллов грязно-белого цвета.
МС-ЭРИ+: МН+ 299,1393.
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak AD-3. Энантиомерное соотношение: 93,40/6,60% (S/R).
Пример 8
Пример 8.1
Получение (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата
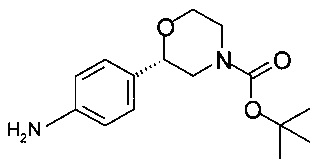
К суспензии 6,0 г (19,5 ммоль) (S)-трет-бутил-2-(4-нитрофенил)морфолин-4-карбоксилата (6,0 г, 44,1 ммоль, экв.: 1,00) в 60 мл МеОН в атмосфере аргона прибавляли 0,23 г Pd/C (10%), и смесь перемешивали с газообразным водородом (1,1 бар - 110000 Па) в течение 2 ч при температуре от 0 до 5°С, затем в течение 16 часов при комнатной температуре. Суспензию фильтровали, фильтрат концентрировали в вакууме при 40°С/10 мбар (1000 Па)/2 ч и получали 5,4 г (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата в виде бесцветной смолы (которая кристаллизуется при стоянии).
ГХ-ЭИ-МС: М278+.
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak IA-3. Энантиомерное соотношение: 99,65/0,35% (S/R).
Пример 8.2
Получение (R)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата
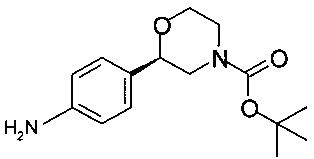
Аналогично Примеру 8.1 восстанавливали (R)-трет-бутил-2-(4-нитрофенил)морфолин-4-карбоксилат и получали 47,8 г титульного продукта в виде масла светло-желтого цвета (которое кристаллизуется при стоянии).
ГХ-ЭИ-МС: М278+
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak IA-3. Энантиомерное соотношение: 99,99/0,01 (R/S).
Пример 9
Пример 9.1
Получение (S)-2-(4-аминофенил)морфолина (из (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата)
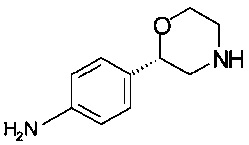
16,7 г (60,0 ммоль) (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата растворяли в 85 мл метанола, прибавляли 47 мл (360 ммоль) 25% соляной кислоты, и реакционную массу нагревали в сосуде с обратным холодильником в течение 1,5 ч, охлаждали до температуры от 0 до 5°С, в течение 5 минут по каплям прибавляли 42 мл (386 ммоль) 9,2М NaOH. Для удаления метанола суспензию концентрировали в вакууме при 40°С/от 150 до 50 мбар (от 15000 Па до 5000 Па), водную суспензию трижды экстрагировали 100 мл этилацетата и трижды - 100 мл ТГФ, объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали в вакууме при 40°С/от 150 до 10 мбар (от 15000 Па до 5000 Па) и получали 10,65 г технического продукта в виде твердого вещества красного цвета, которое кристаллизовали с помощью 100 мл ТБМЭ, нагревали в сосуде с обратным холодильником, отгоняли 70 мл ТБМЭ, суспензию желтого цвета перемешивали в течение 1 ч при комнатной температуре, фильтровали и промывали 10 мл ТБМЭ, кристаллы светло-розового цвета сушили при 40°С/10 мбар (1000 Па)/2 ч и получали 9,24 г (S)-2-(4-аминофенил)морфолина.
ГХ-ЭИ-МС: М178+.
Пример 9.2
Получение (S)-2-(4-аминофенил)морфолина (из хлоргидрата (S)-4-бензил-2-(4-нитрофенил)морфолина)

11,8 г (35,2 ммоль) хлоргидрата (S)-4-бензил-2-(4-нитрофенил)морфолина суспендировали в 118 мл метанола и прибавляли 1,18 г 10% Pd/C, продували сначала аргоном, затем газообразным водородом (1,1 бар - 110000 Па), гидрировали при комнатной температуре в течение 20 часов. Добавляли 12 мл воды и снова гидрировали в течение 4 ч. Суспензию черного цвета нагревали до температуры 60°С в течение 10 минут, фильтровали через стекловолоконный фильтр, промывали 100 мл метанола. Фильтрат концентрировали в вакууме при 40°С/10 мбар (1000 Па)/5 ч и получали 7,50 г технического хлоргидрата (S)-2-(4-аминофенил)морфолина в виде твердого вещества желтого цвета. 5,37 г (25 ммоль) технического продукта экстрагировали 35 мл смеси 1М NaOH/солевой раствор (приготовленной из 500 мл солевого раствора и 500 мл 2М NaOH) и 50 мл смеси ТГФ/ТБМЭ, 1/1. Водный слой повторно экстрагировали 5 раз по 50 мл ТГФ/ТБМЭ, 1/1. Объединенные органические слои сушили над Na2SO4, фильтровали, фильтрат концентрировали в вакууме при 40°С/10 мбар (1000 Па)/5 ч и получали 4,25 г (S)-2-(4-аминофенил)морфолина в виде кристаллов светло-желтого цвета.
ГХ-ЭИ-МС: М178+
Пример 10
Получение (S)-трет-бутил-2-(4-(2-(трифторметил)изоникотинамидо)фенил)морфолин-4-карбоксилата
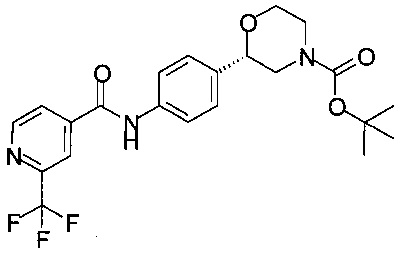
2,78 г (10,0 ммоль) (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата растворяли в 27 мл этилацетата, прибавляли 1,91 г (10,0 ммоль) 2-(трифторметил)изоникотиновой кислоты и 2,80 мл (20,0 ммоль) триэтиламина. При комнатной температуре прибавляли 7,70 мл (13,0 ммоль) 50% раствора ангидрида н-пропилфосфоновой кислоты (циклический тример) в этилацетате (Р3Р®), реакционную массу перемешивали в течение 15 ч при комнатной температуре, экстрагировали 45 мл воды и 45 мл 1М раствора NaHCO3. Органический слой сушили над Na2SO4, фильтровали, концентрировали в вакууме при 40°С и получали 4,57 г в виде пены светло-желтого цвета.
МС-ЭРИ-: (М-Н)- 450,16
Пример 11
Получение хлоргидрата (S)-2-(4-(2-(трифторметил)изоникотинамидо)фенил)морфолина
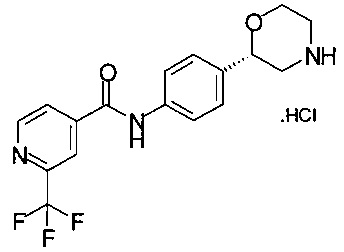
4,01 г (8,88 ммоль) (S)-трет-бутил-2-(4-(2-(трифторметил)изоникотинамидо)фенил)морфолин-4-карбоксилата (4,01 г, 8,88 ммоль, экв.: 1,00) обрабатывали 16,6 мл пропанола-1, прибавляли 3,50 мл 26,6 ммоль 25% соляной кислоты, раствор перемешивали при температуре 60°С в течение 30 минут. Раствор концентрировали в вакууме при 40°C/50 мбар (5000 Па) для отгонки 10 мл смеси растворителей, затем добавляли 10 мл пропанола-1 и снова сгоняли 10 мл смеси растворителей, эту процедуру повторяли три раза. Образовавшуюся суспензию нагревали до температуры 60°С в течение 10 минут, перемешивали в течение 1 ч при комнатной температуре, фильтровали и промывали 5 мл пропанола-1, кристаллы белого цвета сушили при 40°С/10 мбар (1000 Па)/2 ч и получали 3,11 г хлоргидрата (S)-2-(4-(2-трифторметил)изоникотинамидо)фенил)морфолина.
МС-ЭРИ+: (МН)+ 352,12
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak AY-3. Энантиомерное соотношение: 99,50/0,50 (S/R).
Пример 12
Пример 12.1
Получение (R)-трет-бутил-2-(4-(6-хлор-2-(трифторметил)пиримидин-4-иламино)фенил)морфолин-4-карбоксилата
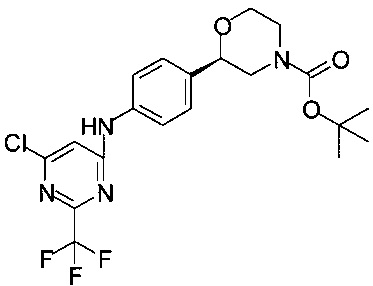
К раствору 2,78 г (10 ммоль) (R)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата в 8,4 мл 2-метил-бутанола-2 прибавляли 2,62 мл (15,0 ммоль) ДИПЭА и 1,58 мл (11,0 ммоль) 4,6-дихлор-2-(трифторметил)пиримидина, и смесь нагревали в сосуде с обратным холодильником в течение 1 ч. Смесь разбавляли 45 мл ТБМЭ и промывали два раза по 45 мл воды. Органический слой сушили над Na2SO4, фильтровали, фильтрат концентрировали в вакууме при 40°С/20 мбар (2000 Па)/1 ч. Получали 5,13 г титульного продукта в виде пены желтого цвета, которую использовали в Примере 12.2 без дополнительной очистки
МС-ЭРИ+: (МН)+ 459,14
Пример 12.2
Получение (R)-трет-бутил-2-(4-(2-(трифторметил)пиримидин-4-иламино)фенил)морфолин-4-карбоксилата
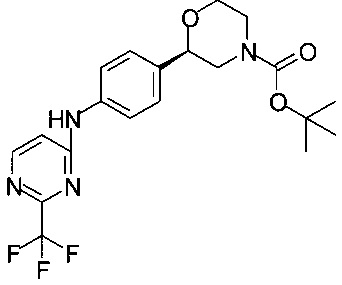
К 37,0 мл раствора 3,7 г (8,06 ммоль) (R)-трет-бутил-2-(4-(6-хлор-2-(трифторметил)пиримидин-4-иламино)фенил)морфолин-4-карбоксилата в пропаноле-2 и 1,35 мл (9,68 ммоль) ТЭА прибавляли 0,19 г 10% Pd/C, и смесь выдерживали в атмосфере Н2 при перемешивании в течение 1 ч.
Суспензию фильтровали, осадок на фильтре промывали пропанолом-2, и фильтрат концентрировали в вакууме при 40°С/20 мбар (2000 Па)/2 ч. Технический продукт растворяли в 35 мл этилацетата и промывали 35 мл 0,25М раствора HCl. Органический слой сушили над Na2SO4, фильтровали, фильтрат концентрировали в вакууме при 40°С/20 мбар (2000 Па)/1 ч. Технический продукт размешивали в 4,5 мл МеОН при комнатной температуре и медленно по каплям прибавляли 1,5 мл воды. Образовавшуюся светлую суспензию перемешивали в течение 16 часов. Суспензию фильтровали, осадок на фильтре промывали 1,5 мл смеси МеОН/вода и сушили в вакууме при 40°С/20 мбар (2000 Па)/3 ч. Получали 2,82 г титульного продукта в форме кристаллов белого цвета, которые использовали в Примере 12.3 без дополнительной очистки.
МС-ЭРИ+: (МН)+ 425,18
Пример 12.3
Получение (R)-трет-бутил-2-(4-(2-(трифторметил)пиримидин-4-иламино)фенил)морфолин-4-карбоксилата
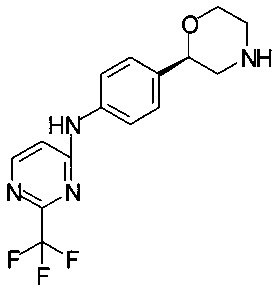
К раствору 2,80 г (6,60 ммоль) (R)-трет-бутил-2-(4-(2-(трифторметил)пиримидин-4-иламино)фенил)морфолин-4-карбоксилата в 28,0 мл МеОН прибавляли 5,15 мл (39,6 ммоль) 25% HCl, и смесь перемешивали при температуре 60°С в течение 1,5 ч. Смесь концентрировали в вакууме при 40°С/от 200 до 20 мбар (от 20000 Па до 2000 Па)/30 мин.
К твердому остатку медленно прибавляли 40 мл Na2CO3 (1М) и 5 мл воды (выделение газа), и смесь экстрагировали 25 мл этилацетата, обработанного 5 мл EtOH. Водный слой повторно экстрагировали 10 мл этилацетата. Объединенные органические слои сушили над Na2SO4, фильтровали, фильтрат концентрировали в вакууме.
Технический продукт перемешивали в 4 мл ТБМЭ, обработанного 200 мкл EtOH, при температуре 56°С в течение 20 мин до образования гомогенной суспензии. Затем смесь охлаждали до комнатной температуры и перемешивали в течение 2 ч, после чего фильтровали, осадок на фильтре промывали 1 мл ТБМЭ и сушили в вакууме при 40°С/20 мбар (2000 Па)/1 ч. Получали 1,9 г титульного продукта в форме кристаллов белого цвета.
МС-ЭРИ+: (МН)+ 325,13
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak IC-3. Энантиомерное соотношение: 99,89/0,11 (R/S).
Пример 13
Получение (S)-трет-бутил-2-(4-(2-(трифторметил)изоникотинамидо)фенил)морфолин-4-карбоксилата
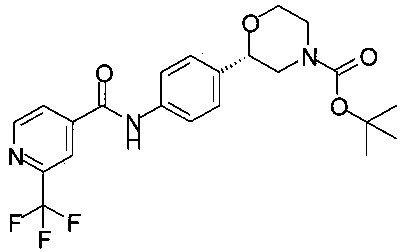
27,8 г (10,0 ммоль) (S)-трет-бутил-2-(4-аминофенил)морфолин-4-карбоксилата и 22,6 г (110 ммоль) метил-2-(трифторметил)изоникотината растворяли в 110 мл ТГФ. Раствор желтого цвета охлаждали до температуры от 0 до 5°С. Прибавляли по каплям в течение 30 минут раствор 22,9 г (200 ммоль) трет-бутоксида калия в 160 мл ТГФ. Раствор темно-желтого цвета перемешивали при температуре от 0 до 5°С в течение 1 ч. В течение 20 минут при температуре от 0 до 5°С прибавляли 140 мл воды, и перемешивали в течение 30 минут при температуре от 0 до 5°С. Реакционную смесь нейтрализовали при температуре от 0 до 5°С в течение 30 минут 97 мл (194 ммоль) водного 2М раствора HCl до pH 7-8. Реакционную массу экстрагировали 200 мл МТБЭ. Органический слой отделяли, сушили над Na2SO4, фильтровали, концентрировали в вакууме и получали 49,8 г технического (S)-трет-бутил-2-(4-(2-(трифторметил)изоникотинамидо)фенил)морфолин-4-карбоксилата в виде пены желтого цвета, содержащей некоторое количество органического растворителя, которую использовали в Примере 14 без дополнительной очистки.
МС-ЭРИ-: (М-Н)- 450,16
Пример 14
Получение хлоргидрата (S)-2-(4-(2-(трифторметил)изоникотинамидо)фенил)морфолина
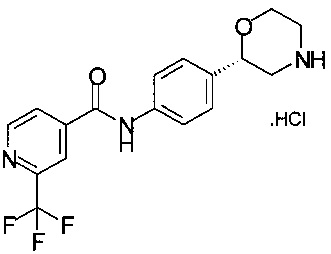
49,8 г (100,0 ммоль) технического (S)-трет-бутил-2-(4-(2-(трифторметил)изоникотинамидо)фенил)морфолин-4-карбоксилата в виде пены желтого цвета (из Примера 13) дважды упаривали со 100 мл пропанола-1 и получали раствор 57,2 г, который разбавляли 170 мл пропанола-1, прибавляли 39,0 мл (300 ммоль) 25% соляной кислоты. Смесь нагревали до температуры от 55 до 60°С в течение 2,5 часов. Суспензию с помощью 50 мл пропанола-1 переносили в круглодонную колбу емкостью 500 мл и концентрировали в вакууме при 40°С/от 60 до 30 мбар (от 6000 Па до 3000 Па). Всего отгоняли 140 мл смеси растворителей. Добавляли 150 мл пропанола-1 и еще раз сгоняли в вакууме. Эту процедуру повторяли трижды. Суспензию разбавляли 150 мл пропанола-1 и нагревали до температуры от 60 до 65°С в течение 10 минут, охлаждали 1 ч до комнатной температуры и перемешивали в течение 18 часов при комнатной температуре, суспензию желтого цвета фильтровали, и осадок на фильтре промывали порциями в общей сложности 50 мл пропанола-1. Кристаллы белого цвета сушили при 40°C/15 мбар (1500 Па) в течение 3 часов и получали 35,1 г хлоргидрата (S)-2-(4-(2-трифторметил)изоникотинамидо)фенил)морфолина.
МС-ЭРИ+: (МН)+ 352,12.
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak AY-3. Энантиомерное соотношение: 99,60/0,40 (S/R).
Пример 15
Получение (S)-2-(4-(2-(хлор)изоникотинамидо)фенил)морфолина
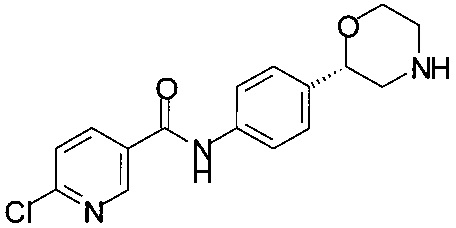
178 мг (1,0 ммоль) (S)-2-(4-аминофенил)морфолина растворяли в 4,0 мл ТГФ и прибавляли 172 мг (1,0 ммоль) метил-6-хлорникотината. Раствор желтого цвета охлаждали до температуры в диапазоне от -70 до -78°С. К желтой суспензии прибавляли 2,0 мл 1М раствора гексаметилдисилазана натрия в ТГФ в течение 30 минут, перемешивали в течение 1 ч при температуре от -70 до -78°С. Добавляли 2,0 мл 1М HCl и отделяли органический слой, водный слой снова экстрагировали 2,0 мл этилацетата, объединенные органические слои сушили над Na2SO4, фильтровали, концентрировали в вакууме при 40°С и получали 290 мг технического продукта в виде твердого вещества светло-желтого цвета. Технический продукт обрабатывали 3,0 мл толуола, нагревали до рефлюкса, затем охлаждали до комнатной температуры, перемешивали в течение 2 ч при комнатной температуре, фильтровали и промывали 1,0 мл толуола, сушили при 40°С/2 ч и получали 270 мг в виде кристаллов белого цвета.
МС-ЭРИ+: (МН)+ 318,10.
Хиральность определяли при помощи хиральной ВЭЖХ с колонкой Chiralpak AY-3. Энантиомерное соотношение: 94,77/5,23 (S/R).



Препарат антитела
Производные гетероарилпирролидинил- и пиперидинилкетона
Производные изоксазоло-пиридина
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Ингибиторы jnk
Бензопирановые и бензоксепиновые ингибиторы рi3k и их применение
Арилциклогексилэфиры дигидротетраазабензоазуленов для применения в качестве антагонистов рецептора вазопрессина v1a
Пуриновые соединения, ингибирующие рi3к, и способы применения
Алкилциклогексиловые эфиры дигидротетраазабензоазуленов
Способ синтеза производных амино-метилтетралина
Способ получения 2-трифторметилизоникотиновой кислоты и ее эфиров
Способ получения лекарственных средств
Способ получения пиримидинилциклопентановых соединений
Способы получения (циклопентил[d]пиримидин-4-ил)пиперазиновых соединений

