Результат интеллектуальной деятельности: Способ преимплантационной генетической диагностики спинальной мышечной атрофии типа 1
Вид РИД
Изобретение
Изобретение относится к преимплантационной генетической диагностике моногенных заболеваний. В настоящее время в мире насчитывается более 350 миллионов людей, страдающих редким заболеванием (по данным RARE Project). Общее количество таких заболеваний по подсчетам Европейской ассоциации по редким заболеваниям (European Organization for Rare Diseases (EURORDIS)) варьируется от 5 до 7 тысяч. При этом около 80% редких заболеваний имеют генетическую причину. Генетическая основа заболевания позволяет с высокой точностью предсказать не только здоровье уже родившегося ребенка, но и оценить риск рождения такого ребенка при анализе генотипов родителей, а также провести генетическую диагностику на самых ранних этапах. Преимплантационная генетическая диагностика (ПГД) моногенного заболевания становится мощным инструментом для профилактики таких заболеваний.
Основной проблемой при генетической диагностике эмбрионов является малое исходное количество биоматериала, так как биоптат содержит от одной до трех клеток. В этом случае для повышения эффективности и точности анализа важно как полностью исключить возможность контаминации, так и нивелировать возможный эффект неравномерной и/или неполной амплификации, а также деградации материала. Это требует разработки тест-системы с особыми характеристиками. При этом тест-система разрабатывается с учетом возможности использовать биоматериал разного типа - тотальную ДНК, выделенную из разных тканей, продукт полногеномной амплификации (whole genome amplification - WGA), а также единичные клетки. Сочетание универсальности по отношению к биоматериалу с поэтапной амплификацией целевых фрагментов позволяет проводить анализ нескольких мутаций для одного образца, в том числе на единичных клетках, а также выявить ситуацию неполной амплификации или деградации образца.
ПГД моногенного заболевания проводится для семей, имеющих подтвержденную молекулярно-генетическую природу заболевания. Обязательно наличие заключения врача-генетика и генетической лаборатории, что выявлена определенная мутация в определенном гене, и именно данная мутация является причиной клинического проявления заболевания. ПГД моногенного заболевания рекомендуется семьям с высоким риском рождения ребенка с тяжелым (неизлечимым) наследственным заболеванием с установленной мутацией, обуславливающей этот риск. ПГД позволяет выбрать из всех эмбрионов, полученных при ЭКО, такие, которые не несут мутацию, являющуюся причиной заболевания.
Настоящее изобретение относится к способу преимплатационной генетической диагностики спинальной мышечной атрофии. Мутации гена SMN1, и прежде всего его делеция связаны с развитием заболевания - спинальная мышечная атрофия (СМА) типа 1, которое характеризуется нарушением функции нервных клеток спинного мозга, приводящее к прогрессивному развитию слабости мышц, их атрофии, и в итоге, обездвиживанию пациента. Частота встречаемости этого заболевания очень высока. Каждый 35 человек является бессимптомным носителем мутации, приводящей к СМА, и больной ребенок рождается, когда встречаются 2 таких мутации, со стороны матери и со стороны отца. Это происходит примерно 1 раз на 6000 рождений - в семьях, где никто, как правило, не слышал про такую болезнь, где не было больных родственников, вредных факторов среды - ничего, что могло бы навести на мысль о высоком риске генетических проблем.
Причина болезни - мутация в гене, кодирующем белок SMN (survival of motor neuron protein, белок, способствующий выживанию моторных нейронов). Дефицит этого белка приводит к гибели моторных нейронов. Область, кодирующая белок SMN расположена на пятой хромосоме в локусе 5q13 и состоит из гена SMN1 и псевдогена SMN2. Псевдоген отличается от гена всего несколькими нуклеотидами в экзонах 7 и 8 и интронах 7 и 8, однако разница в 7 экзоне приводит к тому, что у 80-90% транскриптов SMN2 отсутствует седьмой экзон. Такой транскрипт не дает начала функциональному белку, и, следовательно, псевдоген SMN2 обеспечивает всего около одной десятой общего количества функционального белка в клетке. Иногда в геноме присутствуют несколько идущих подряд копий SMN2. Примерно 95% случаев спинальной мышечной амиотрофии 1-3 типов вызваны делецией только седьмого или седьмого и восьмого экзона гена SMN1. Остальные 5% - точечными мутациями в этом же гене. Если продукт гена SMN1 нефункционален, благодаря SMN2 все еще остается небольшое количество белка. Поэтому большее число копий SMN2 связано с более мягкой формой заболевания.
По степени тяжести и времени появления симптомов развивающиеся в детстве спинальные мышечные амиотрофии делят на три типа.
Первый тип - болезнь Верднига-Гоффмана - наиболее тяжелый вариант заболевания. Симптомы появляются в первые полгода жизни. Иногда болезнь можно заподозрить еще до рождения по слабому шевелению плода. Такие дети никогда не смогут держать голову или сидеть самостоятельно. У них возникают сложности с дыханием и приемом пищи. Прогноз неблагоприятен. Второй тип - промежуточный. Первые симптомы появляются в возрасте от полугода до полутора. Дети могут сидеть самостоятельно, стоять при поддержке и не испытывают сложностей при приеме пищи. Однако они имеют повышенный риск развития инфекций дыхательных путей. Третий тип - болезнь Кюгельбера-Веландера - характеризуется более мягким течением. Первые симптомы возникают после полутора лет. Больные способны самостоятельно сидеть, стоять и даже ходить.
Перечень иллюстраций:
Фиг. 1 Семья А с диагнозом СМА 1.
Фиг. 2 Семья Б, в которой родилось 2 ребенка со СМА и носителями мутации являлись оба супруга.
Из уровня техники известна опубликованная заявка на получение патента на изобретение в Китае CN105112541A1, которая предлагает к защите способ детекции мутации в гене SMN1 с помощью композиции праймеров, специфично амплифицирующиеся с участками экзона 7 гена SMN1, а также анализа сцепленных с мутацией маркеров типа однонуклеотидных полиморфизмов (SNP). Сниженная гетерозиготность и полиморфность косвенных маркеров приводит к снижению вероятности точной диагностики при ПГД, а использование платформы IontorrentProton значительно удорожает анализ, а также делает невозможным тестирование единичных клеток без этапа полногеномной амплификации. Таким образом, предложенный способ детекции отличается высокой стоимостью и продолжительностью проведения.
Задачей являлась разработка недорогого и точного способа преимплантационной генетической диагностики моногенного заболевания спинальная мышечная атрофия.
Техническим результатом стало создание точной диагностики делеции экзона 7 гена SMN1, для чего была разработана система с двойной детекцией - прямой и косвенной. Такая система необходима при работе с малым количеством биоматериала, так как нестабильная амплификация может привести к потере информации или сниженной точности анализа. Прямая диагностика подразумевает анализ непосредственно наличия-отсутствия экзона 7. Для более точных результатов в тест-систему также включен анализ экзона 8 гена SMN1, так как именно его последовательность отличается от гена SMN2 и может выступать маркером количества копий гена SMN1, а также степени конверсии гена SMN1 в SMN2 при наличии таковой. Экзоны 7 и 8 гена SMN1 по последовательности очень схожи с экзонами 7 и 8 гена SMN2, отличаясь лишь 2 нуклеотидами: g.70247773C в 7 экзоне гена SMN1 соответствует g.69372353T в 7 экзоне гена SMN2, g.70248501G в 8 экзоне гена SMN1 соответствует g.69373081A в 8 экзоне гена SMN2. Для различения этих точек была разработана тест-система на основе ПЦР-ПДРФ. Косвенная диагностика заключается в анализе наследования молекулярно-генетических маркеров, сцепленных с мутацией. Для этого на расстоянии не более 1 Mb (что соответствует 1% кроссинговера в среднем) от гена SMN1 были выбраны полиморфные локусы, называемые короткие тандемные повторы (STR - single tandem repeat), с гетерозиготностью не менее 0,75. Для каждого из этих локусов были подобраны праймеры для амплификации по типу полугнездовой ПЦР, позволяющей повысить точность и эффективность анализа. В тест-систему были включены 15 STR локусов: D5S1408, D5S1414, D5S1417, D5S1556, D5S351, D5S435, D5S610, D5S629, D5S637, D5S68.3, D5S68.73, D5S68.77, D5S70.8, D5S70.9, D5S71.2. Праймеры для амплификации представлены на SEQ ID NO 1.
Все 15 STR локусов находятся на 5 хромосоме в районе координат 68096973-71817906 (в соответствии с hg19). Важно отметить, что при подборе праймеров соблюдали ряд особенных требований: длина продукта с внешний праймеров не должна превышать 500 п.н. (для наработки с фрагментов, получаемых при полногеномной амплификации), длина продукта с внутренних праймеров от 120 до 300 п.н., высокая специфичность внешних праймеров, температура отжига не отличается более, чем на 0,5оС.
Отработка тест-системы для ПГД
На этапе отработки тест-системы проводится подбор условий амплификации, оптимальных для работы праймеров, анализ эффективности и специфичности ПЦР-амплификации в обоих раундах, универсальность тест-системы для биообразцов различного типа. При отработке тест-системы были приготовлены стоковые разведения праймеров с концентрацией 100mM, и рабочие разведения комбинаций праймеров (комбинация пар праймеров для 1 и 2 раунда полугнездовой ПЦР) с концентрацией 10mM каждого праймера в растворе. Так как в рамках диагностики клинического материала могут быть использованы различные типы матриц, при отработке тест-системы были использованы две биопсии единичных клеток, находящихся в специальном лизирующем буфере (1×PCR Buffer, 0,1% Tween-20, 0,1% Triton Х-100, 1 мкг Proteinase K), два или три образца продуктов полногеномной амплификации биопсиий эмбриона (WGA), а также тотальной ДНК членов семьи, выделенной из крови, для составления родословной.
В рамках полугнездовой ПЦР (seminested-PCR) амплификация проводится в два этапа. На первом этапе проводится мультиплексная ПЦР со всеми внешними праймерами для всех локусов, входящих в тест-систему, для обогащения образца всеми целевыми фрагментами. Во втором раунде проводится индивидуальная амплификация каждого фрагмента с внутренними праймерами. Полугнездовая ПЦР Для первого раунда были подобраны внешние высокоспецифичные праймеры для амплификации фрагментов от 300 до 500 п.н.
Для второго раунда были подобраны праймеры для амплификации фрагментов длиной не более 350 пар оснований, а также были введены метки для детекции методом фрагментного анализа на приборе ABI3130XL. ПЦР-смесь для первого раунда амплификации содержала 1xPCR Buffer with Mg2+ (Евроген, Россия), 0.1 mM каждого деоксинуклеотида, 0.15 μМ каждого праймера, 2,5 U/μl of HsTaq DNA-polymerase (Евроген, Россия), 6% of DMSO and 1 мкл тотальной ДНК или 2,5 мкл WGA или 5 мкл лизирующего буфера с образцом в качестве матрицы. Первый этап амплификации проводился по следующему протоколу: этап денатурации 94оС в течение 2 минут, 30 циклов с понижением температуры отжига праймеров с 62 до 45оС в каждом, этап достройки всех матриц 72оС 10 минут. Далее продукты 1-ого раунда были разнесены в индивидуальные пробирки с одной парой праймеров на определенный локус.В состав ПЦР смеси для второго раунда входили 1xPCR Buffer with Mg2+ (Евроген, Россия), 0.5xRediLoad™ Loading Buffer (Thermo Fisher Scientific, USA), 0.2 mM каждого деоксинуклеотида, 0.2 μM каждого праймера, 1U/μl of HsTaq DNA-polymerase (Евроген, Россия), 6% of DMSO and 1 μl ПЦР-продукта первого раунда амплификации в качестве матрицы. Второй этап амплификации проводился по следующему протоколу: этап денатурации 95оС в течение 2 минут, 35 циклов: денатурация 95оС 30 секунд, отжиг праймеров - 57оС 30 секунд, синтез матрицы - 72оС 1 минута, этап достройки всех матриц 72оС 5 минут.
Оценку эффективности и специфичности амплификации проводили с помощью электрофореза в 2% агарозном геле. Результат электрофореза в агарозном геле позволяет определить степень разведения продуктов амплификации для нанесения на фрагментный анализ (продукты амплификации с ДНК членов семьи). Фрагментный анализ продуктов амплификации проводили с помощью капиллярного электрофореза на приборе 3130xl Genetic Analyzer (Applied Biosystems, USA). По результатам фрагментного анализа составляется родословная и отмечаются информативные маркеры для каждой семьи, которые в дальнейшем будут использованы в клинической диагностике.
Полимеразная цепная реакция - полиморфизм длин рестрикционных фрагментов (ПЦР-ПДРФ).
Полиморфизм длин рестрикционных фрагментов (Restriction fragment length polymorphism, RFLP) - это способ исследования геномной ДНК, путем разрезания ДНК с помощью эндонуклеаз рестрикции и дальнейшего анализа размеров образующихся фрагментов (рестриктов) путем гель-электрофореза (ДНК электрофореза).
При использовании данного метода наблюдаются фрагменты различной длины в зависимости от различий в последовательности нуклеотидов в сайте рестрикции, что позволяет при помощи ПДРФ идентифицировать некоторые различия в последовательности нуклеотидов ДНК, в случае, когда они располагаются в сайте рестрикции. В виду того, что технологии секвенирования ДНК могут охарактеризовать ДНК очень точно, ПДРФ был разработан как первый и дешевый метод для массового применения.
Для детекции мутации была разработана тест-система на основе ПЦР-ПДРФ, которая позволяет различить экзоны 7 и 8 генов SMN1 и SMN2 и, таким образом, выявить полное отсутствие экзонов 7 и 8 гена SMN1 в образце. Стадия амплификации подробно описана в предыдущем разделе. Были использованы следующие праймеры: g.70247773C (экзон 7) 
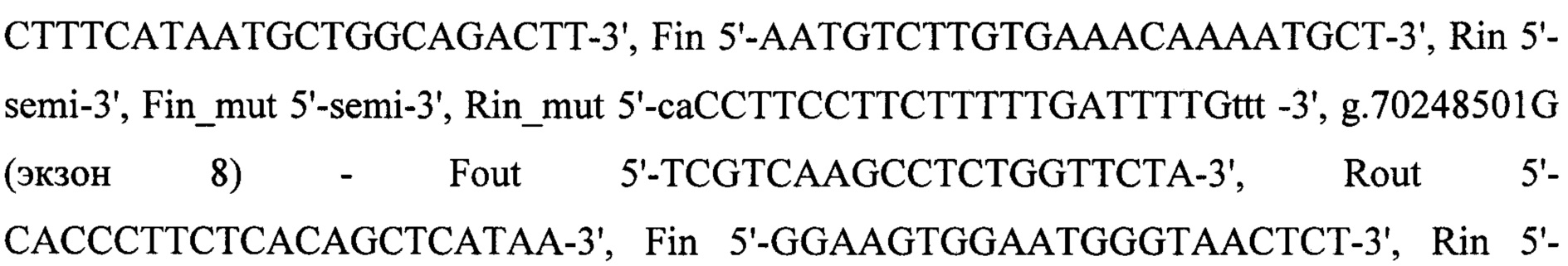
 . Далее продукты амплификации с внутренних праймеров для детекции мутации использовали в реакции рестрикции. Эндонуклеаза Hpy188I разрезает только фрагмент экзона 7 гена SMN1, эндонуклеаза DraI разрезает только фрагмент экзона 7 гена SMN2 при введении мисматча с помощью праймера
. Далее продукты амплификации с внутренних праймеров для детекции мутации использовали в реакции рестрикции. Эндонуклеаза Hpy188I разрезает только фрагмент экзона 7 гена SMN1, эндонуклеаза DraI разрезает только фрагмент экзона 7 гена SMN2 при введении мисматча с помощью праймера 
 . Эндонуклеаза DdeI разрезает только фрагмент экзона 8 гена SMN2, а эндонуклеаза BseNI разрезает фрагмент экзона 8 генов SMN1 и SMN2 на фрагменты разной длины: 113, 74, 34 п.н. для гена SMN1 и 147, 74 п.н. для гена SMN2. Детекция проводилась с помощью электрофореза в 12% полиакриламидном геле.
. Эндонуклеаза DdeI разрезает только фрагмент экзона 8 гена SMN2, а эндонуклеаза BseNI разрезает фрагмент экзона 8 генов SMN1 и SMN2 на фрагменты разной длины: 113, 74, 34 п.н. для гена SMN1 и 147, 74 п.н. для гена SMN2. Детекция проводилась с помощью электрофореза в 12% полиакриламидном геле.
Пример 1.
В ЦГРМ Генетико обратилась семья А, в которой носителями мутации являлись оба супруга. В таком случае риск для потомства составляет 50% (вероятность рождения ребенка с данным заболеванием). Им было рекомендовано проведение ПГД СМА в рамках ЭКО для отбора эмбрионов, имеющих хотя бы одну копию гена SMN1.
Разработка родословной
На первом этапе был получен биоматериал (периферическая кровь) супругов и их погибшего от СМА 1 типа ребенка для детекции мутации и выявления групп сцепления полиморфных маркеров (см. Фиг. 1). Было проанализировано 15 STR локусов. Из них 12 оказались информативными по матери и/или отцу. Таким образом, образцы эмбрионов тестировались только на информативные маркеры. Полученные результаты по информативным маркерам: партнер пациентки - D5S435 126/139, D5S629 145/145, D5S1417 121/119, D5S68.3 301/282, D5S1414 175/161, D5S68.73 305/305, SMN1 ex. 7 -/+, SMN1 ex. 8 -/+, D5S1408 231/227, D5S70.9 130/136, D5S610 167/172, D5S637 137/135, D5S71.2 161/175, D5S351 298/298, пациентка - D5S435 139/135, D5S629 145/152, D5S1417 121/123, D5S68.3 301/292, D5S1414 175/167, D5S68.73 305/307, SMN1 ex. 7 -/+, SMN1 ex. 8 -/+, D5S1408 231/227, D5S70.9 130/136, D5S610 167/172, D5S637 137/140, D5S71.2 161/169, D5S351 302/302, ребенок - D5S435 139/139, D5S629 145/145, D5S1417 121/121, D5S68.3 301/301, D5S1414 175/175, D5S68.73 305/305, SMN1 ex. 7 -/-, SMN1 ex. 8 -/-, D5S1408 231/231, D5S70.9 130/130, D5S610 167/167, D5S637 137/137, D5S71.2 161/161, D5S351 298/302.
ПГД CMA
В цикле ЭКО был получен 1 эмбрион, проведена его биопсия на 5 день развития (в клинике ЭКО), биоптат в буфере для WGA (1xPBS (Invitrogen, США), 1% PVP (Fertipro, Бельгия)) направлен в лабораторию Генетико. Для контроля контаминации на разных этапах работы с образцом в лаборатории разработана система контролей: контроль контаминации буфера для биопсии, контроль контаминации при транспортировке (одна пробирка с буфером не открывается эмбриологом), контроль контаминации каждого образца (проба среды из последней отмывочной капли биопсиийного материала). Все эти контроли вместе с образцами проходят этап полногеномной амплификации, после которого будет заметно малейшее количество ДНК, контаминировавшей контроли. Полногеномную амлификацию проводили с помощью коммерческого набора SurePlex (Illumina, США).
Продукт полногеномной амплификации, а также ДНК всех членов семьи амплифицировали в 1 раунде в мультиплексной ПЦР с праймерами для детекции мутации и праймерами для информативных для семьи А полиморфных маркеров в соответствии с разработанным протоколом для тест-системы. 2 раунд проводили для каждого маркера отдельно в соответствии с разработанным протоколом для тест-системы. Таким образом были выявлены группы сцепления, унаследованные эмбрионом, а также проведен анализ на наличие у него хотя бы одной копии гена SMN1. Полученные результаты: D5S435 126/139, D5S629 145/145, D5S1417 119/121, D5S68.3 282/301, D5S1414 161/175, D5S68.73 305, SMN1 ex. 7 +/-, SMN1 ex. 8 +/-, D5S1408 227/231, D5S70.9 136/130, D5S610 172/167, D5S637 135/137, D5S71.2 175/161, D5S351 298/302.
По результатам прямой и косвенной диагностики у эмбриона обнаружено носительство мутации без риска клинических проявлений заболевания. С согласия родителей для эмбриона провели преимплантационный генетический скрининг хромосомных аномалий, который не выявил нарушений. По результатам всех проведенных анализов эмбрион был рекомендован к переносу. Перенос оказался результативным, беременность закончилась рождением здорового ребенка.
Пример 2.
В ЦГРМ Генетико обратилась семья Б, в которой родилось 2 ребенка со СМА и носителями мутации являлись оба супруга (см. Фиг. 2). В таком случае риск для потомства составляет 50% (вероятность рождения ребенка с данным заболеванием). Им было рекомендовано проведение ПГД СМА в рамках ЭКО для отбора эмбрионов, имеющих хотя бы одну копию гена SMN1. Однако пара забеременела естественным путем, поэтому проводилась инвазивная пренатальная диагностика.
Пренатальная диагностика
В лабораторию поступил плодный материал (ворсины хориона, полученных при аспирации в клинике ЭКО на 10-11 неделе естественной беременности пациентки), а также кровь родителей. Из биоматериалов была выделена ДНК и проведена прямая и косвенная диагностика с помощью тест-системы, разработанной для ПГД СМА: ПЦР в 2 раундах в соответствии с разработанным протоколом для тест-системы. Были выявлены группы сцепления, унаследованные плодом, а также проведен анализ наличия хотя бы одной копии гена SMN1 с помощью ПЦР-ПДРФ в соответствии с разработанным протоколом для тест-системы. В рамках пренатальной диагностики доступно большее количество биоматериала плода, чем при ПГД. Это позволяет использовать для диагностики метод MLPA (коммерческий набор SALSA MLPA Р021 SMA (MRC-Holland, Нидерланды)), который детектирует не только наличие хотя бы одной копии гена, но и позволяет оценить количество копий экзонов 7 и 8 обоих генов SMN1 и SMN2. Такая диагностика подтвердила результат, полученный с помощью разработанной тест-системы и выявила наличие двух копий гена SMN1. Беременность не была прервана, родился здоровый ребенок.
Общество с ограниченной ответственностью «Центр Генетики и Репродуктивной Медицины «ГЕНЕТИКО»
Способ преимплантационной генетической диагностики спинальной мышечной атрофии типа 1
Праймеры для амплификации
SEQ ID NO 1


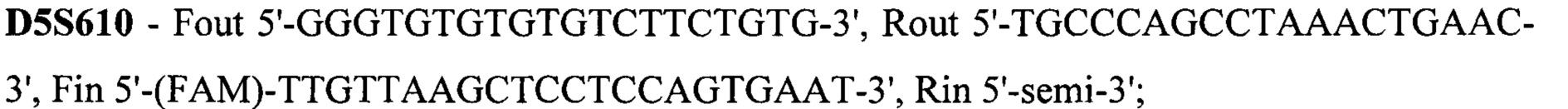
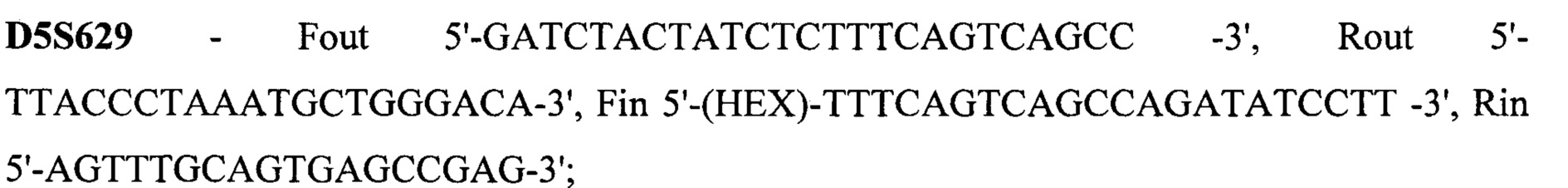

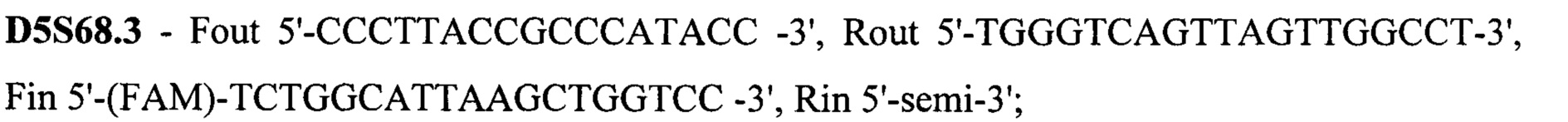
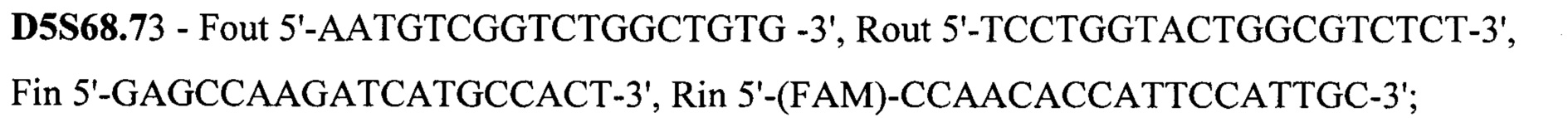
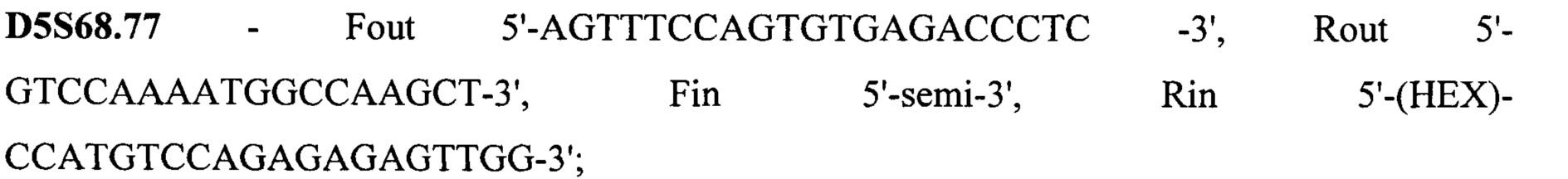


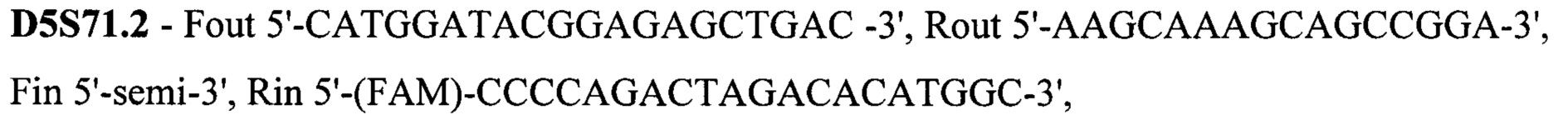

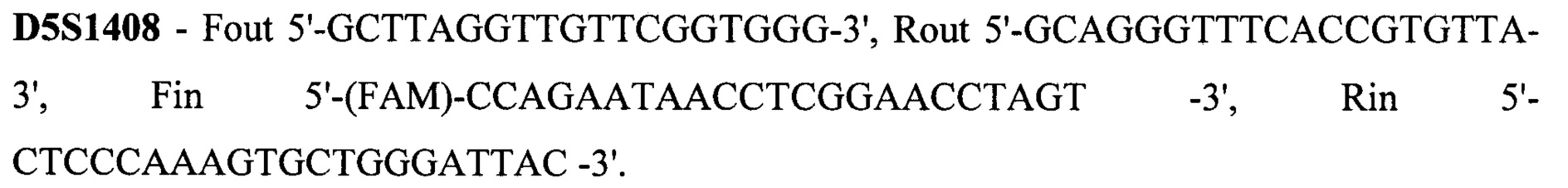
Способ преимплантационной генетической диагностики анемии фанкони
Тест система для пренатального генетического тестирования на hla-совместимость
Способ повышения эффективности вирусной трансдукции
Биокомпозит для обеспечения восстановительных процессов после повреждения у млекопитающего, способ его получения (варианты) и применения
Кодон-оптимизированная кднк, кодирующая дисферлин человека, генно-инженерная конструкция, рекомбинантный аденовирус и фармацевтическая композиция для лечения дисферлинопатий
Ингибиторы калиевых каналов как лекарственные средства при лимфомах и лейкозах
Способ получения фармацевтической композиции для индукции развития кровеносных сосудов в тканях, фармацевтическая композиция, полученная этим способом, и способ лечения ишемии тканей и/или органов человека
Набор олигонуклеотидных зондов, днк-микрочип, способ его получения, комплект для молекулярно-генетического исследования человека и их применение
Кодон-оптимизированные последовательности и фармацевтическая композиция для восстановления кровеносных сосудов
Кодон-оптимизированная рекомбинантная плазмида, способ стимуляции регенерации периферического нерва, способ лечения поврежденного нерва человека
Способ получения суспензии ядросодержащих клеток из пуповинной крови со стандартизированной концентрацией
Способ создания персонализированного ген-активированного имплантата для регенерации костной ткани

