Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ СОЕДИНЕНИЙ 2-АМИНО-1,3-ПРОПАНДИОЛА И ИХ СОЛЕЙ
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к области лекарственных средств. Изобретение относится к способам получения соединений 2-амино-1,3-пропандиола и их гидрохлоридных солей. В частности, настоящее изобретение относится к способам синтеза 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола и его гидрохлоридной соли, т.е. гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола. Этот способ является безопасным, коммерчески осуществимым для крупномасштабного синтеза и имеет улучшенную эффективность наряду со многими другими преимуществами.
Предшествующий уровень техники
Соединения 2-амино-1,3-пропандиола являются полезными лекарственными средствами, которые используются особенно в качестве иммунодепрессантов. Гидрохлорид 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола принадлежит к этому же классу соединений, имеющих общее название гидрохлорид финголимода. Гидрохлорид 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (гидрохлорид финголимода) является иммуномодулятором, одобренным для лечения рассеянного склероза (MS) Управлением по контролю за качеством пищевых продуктов и лекарственных препаратов США (US FDA) в сентябре 2010 года. Гидрохлорид финголимода продается под торговым названием Gilenya®. С химической точки зрения Gilenya® является гидрохлоридом 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (формула 1), имеющим молекулярную массу 380,44 и молекулярную формулу C19H34NO2Cl.
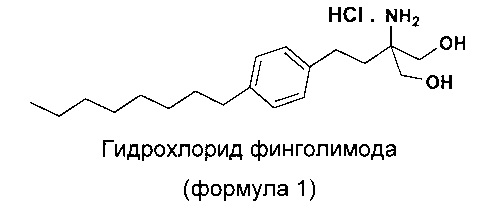
В патенте US 5604229 раскрыто применение соединений 2-амино-1,3-пропандиола в качестве лекарственных средств, в частности иммунодепрессантов. В этом документе раскрыт синтез гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (гидрохлорида финголимода) различными способами, а также его применение в качестве иммунодепрессанта. Однако раскрытые способы синтеза для получения гидрохлорида финголимода включают трудоемкие стадии очистки на хроматографических колонках, а также применение реагентов с высокой реакционной активностью и реакций, которые сложно выполнять безопасно в коммерческих масштабах.
В публикации US 2002/0072635 А1 раскрыт способ получения гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола, включающий восстановление соответствующего замещенного фенола в соответствующий замещенный бензиловый спирт, а затем восстановительное дезоксигенирование указанного замещенного бензилового спирта путем трудоемкой реакции гидрогенизации.
В WO 2010/055027 раскрыты различные соли 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (финголимода), такие как тартрат, лактат, бензоат, сукцинат, малонат, ацетат, пропионат, и их соответствующие полиморфные формы.
В WO 2010/055028 А2 раскрыты гидратные и кристаллические полиморфы гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола, а именно форма-I, форма-II, форма-III.
В WO 2011/009634 А2 раскрыт способ получения фармацевтически приемлемых солей финголимода, таких как аскорбат, сукцинат, оксалат, фосфат, манделат, адипат, и их соответствующие полиморфные формы, который включает способ лиофилизации.
Таким образом, в данной области техники существует необходимость в разработке надежного и коммерчески выполнимого способа получения финголимода и/или его гидрохлоридной соли, который является безопасным, эффективным и не включает каких-либо хроматографических очисток.
Настоящее изобретение направлено на преодоление вышеуказанных недостатков известного уровня техники.
Сущность изобретения
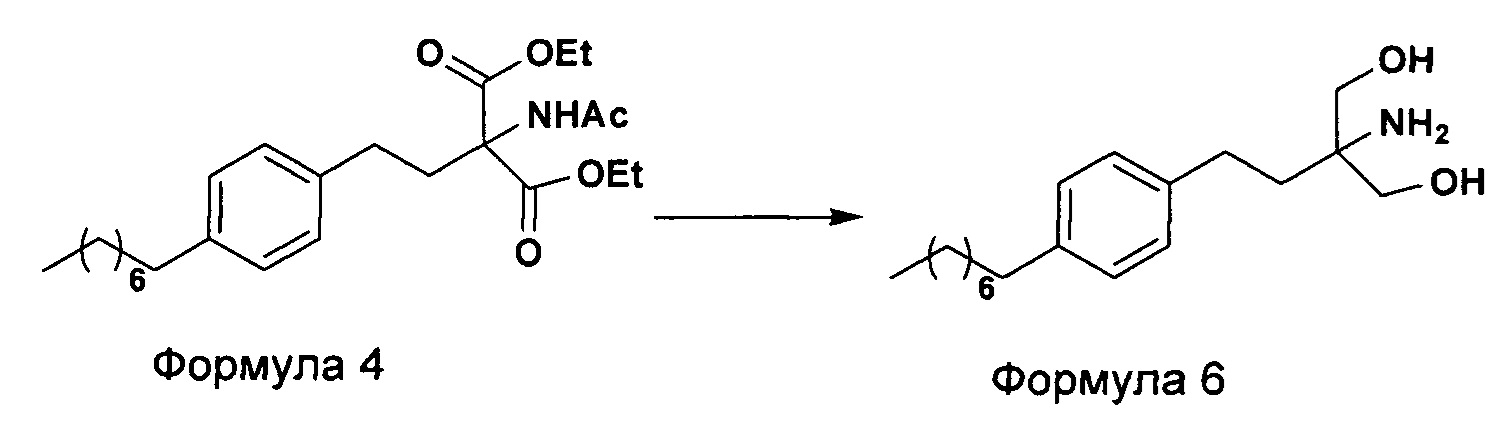
Соответственно, настоящее изобретение относится к способу получения 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6), который включает следующие стадии:
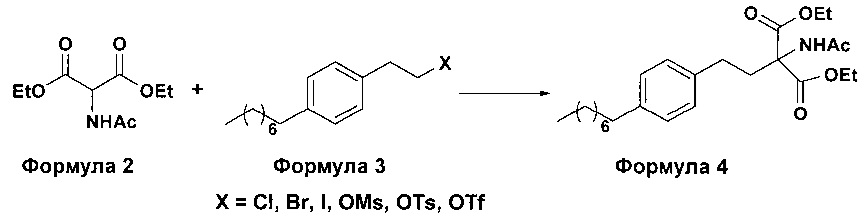
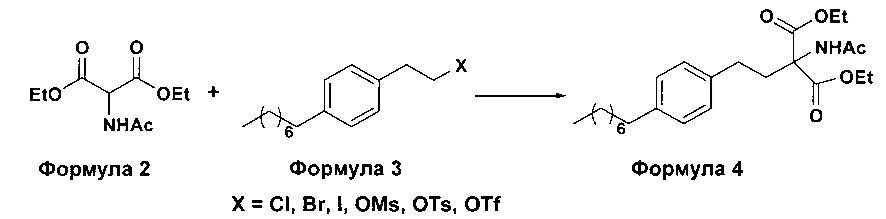
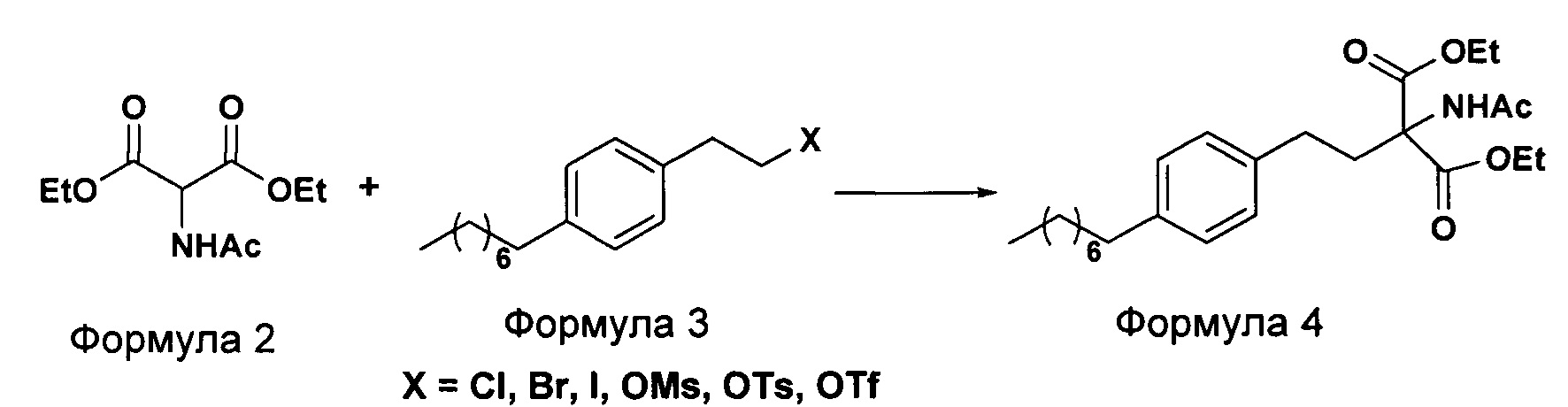
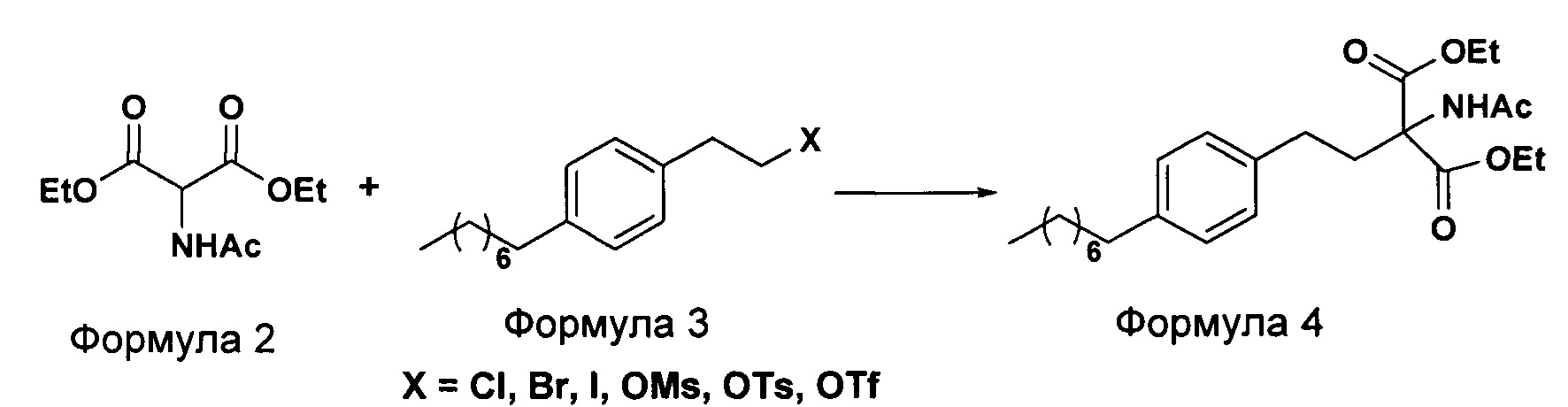
а. осуществление взаимодействия соединения формулы 3 с диэтилацетамидомалонатом (Формула 2) с получением диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4),
и
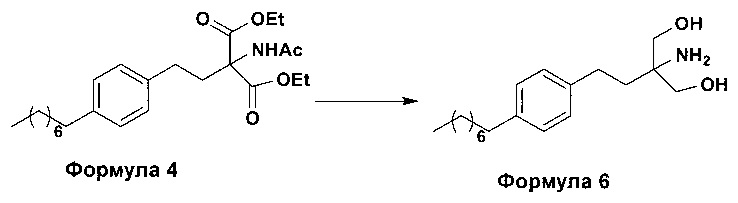
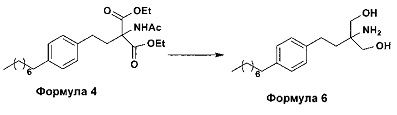
b. превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты, полученного на стадии (а), с получением 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6),
или
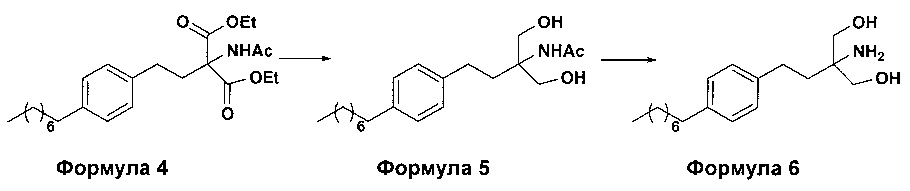
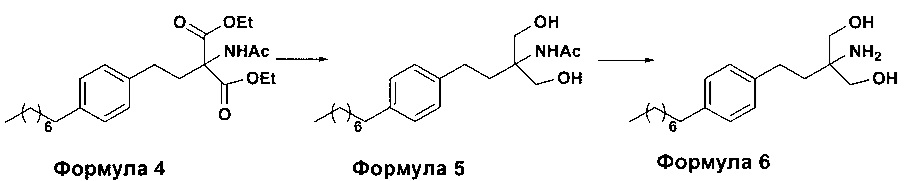
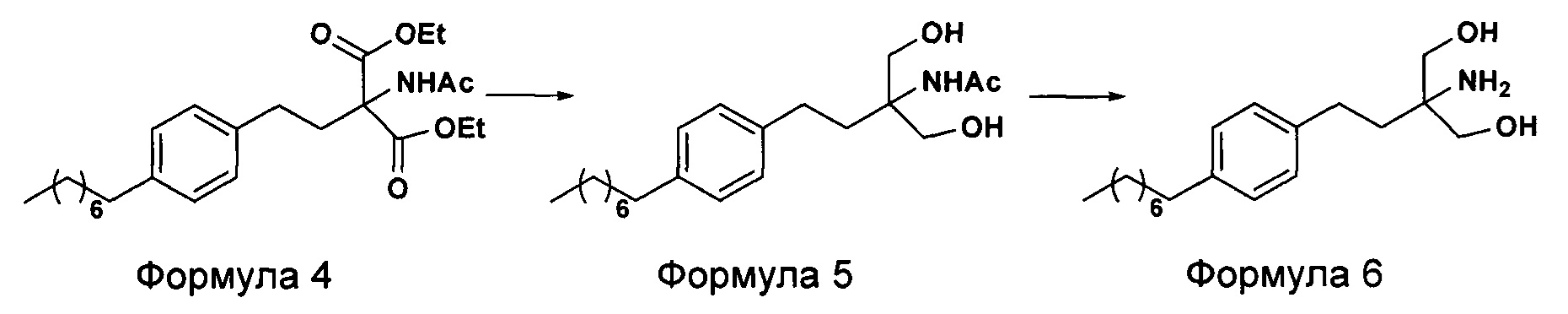
c. превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты, полученного на стадии (а), в N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамид (Формула 5) с последующим гидролизом N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида с получением 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6),
или
d. превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты, полученного на стадии (а), в N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамид (Формула 5) с последующим превращением N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида (Формула 5) в 2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетат (Формула 5а) с последующим гидролизом ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетата (Формула 5а) с получением 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6);
причем полиморфная форма А 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6), полученная указанным способом, имеет значения 2θ XRPD (порошковой рентгеновской дифракции): 3.876, 5.744, 7.739, 11.65, 14.886, 15.356, 16.774, 17.65, 18.008, 18.963, 19.473, 20.845, 21.626, 23.431, 24.643, 27.389, 27.894, 30.566, 31.421, 34.267, 35.01, 35.5, 38.756, 42.214, 43.767, 46.201, 48.026, 50.269 и 52.314; и полиморфная форма Y гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 1), полученная указанным способом, имеет значения 2θ XRPD: 3.549, 5.185, 5.832, 7.052, 8.62, 9.305, 10.625, 12.149, 12.82, 14.163, 14.713, 15.174, 15.61, 16.374, 17.329, 17.749, 18.254, 18.698, 19.255, 19.948, 20.879, 21.389, 22.248, 22.578, 22.838, 23.527, 24.449, 24.953, 25.847, 26.139, 27.127, 28.094, 28.604, 29.47, 29.697, 31.786, 32.24, 33.147, 36.955 и 44.474.
Краткое описание графических материалов
Для более легкого понимания изобретения и его осуществления на практике приведены иллюстративные варианты осуществления изобретения со ссылкой на прилагаемые графические материалы. Графические материалы вместе с подробным описанием ниже включены в описание и составляют часть описания изобретения, а также служат для дополнительной иллюстрации вариантов осуществления и пояснения различных принципов и преимуществ в соответствии с настоящим изобретением.
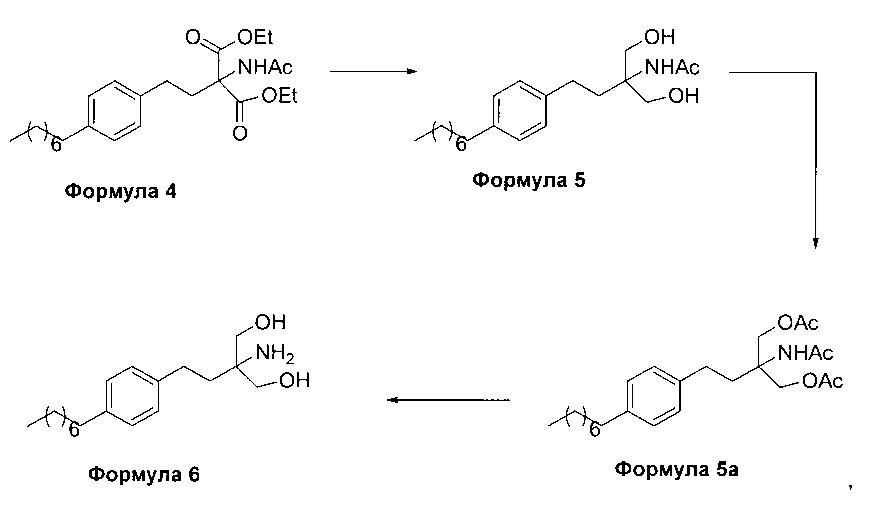
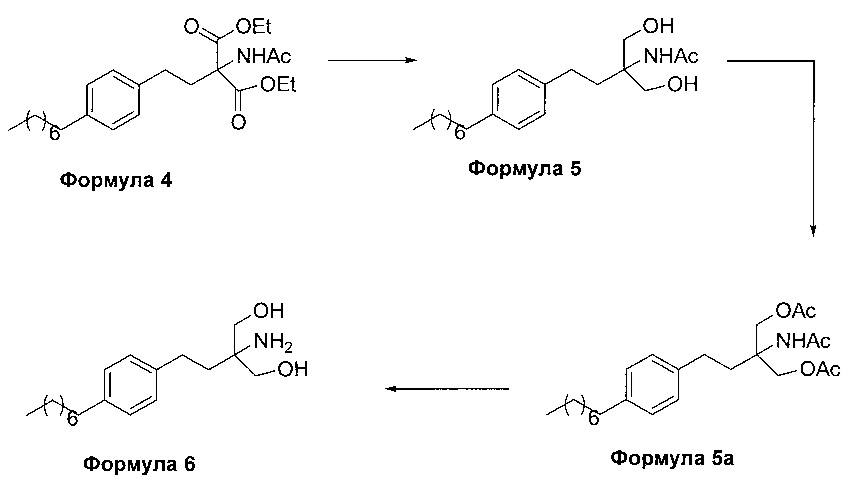
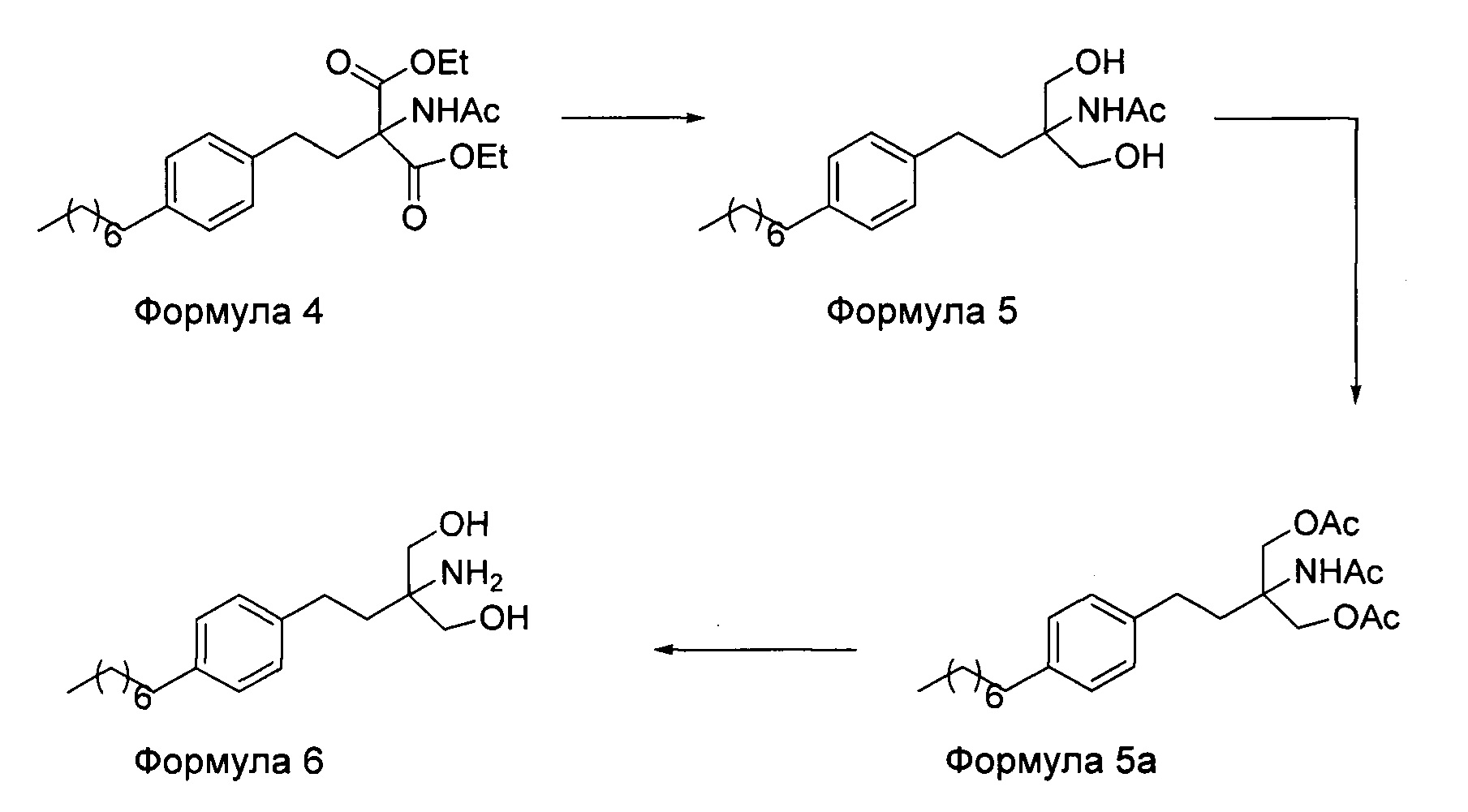
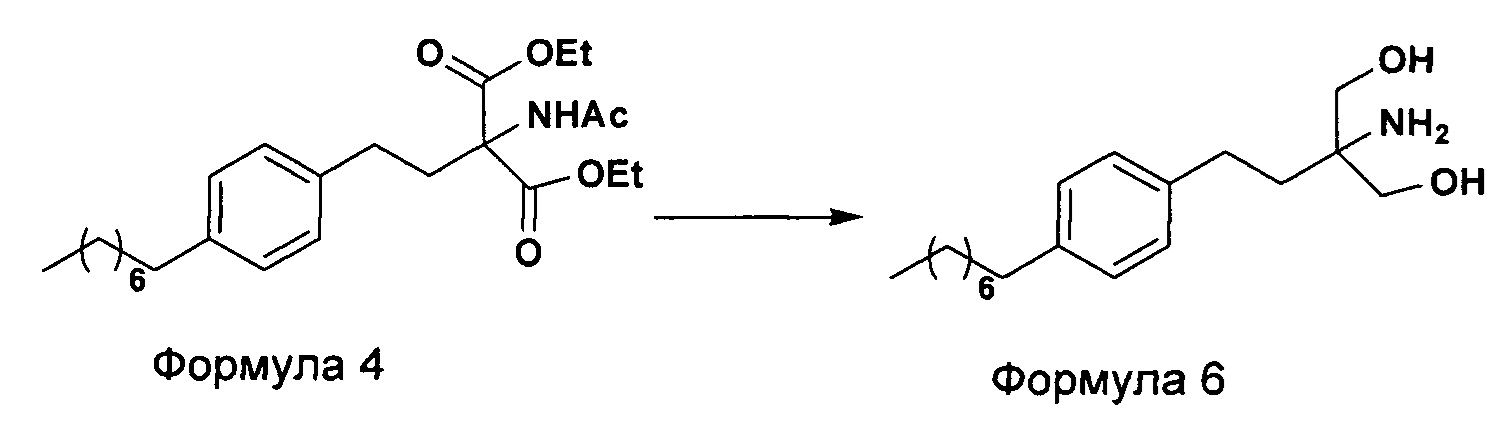
На фиг. 1 представлен один из вариантов синтеза гидрохлорида финголимода [гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола] согласно настоящему изобретению.
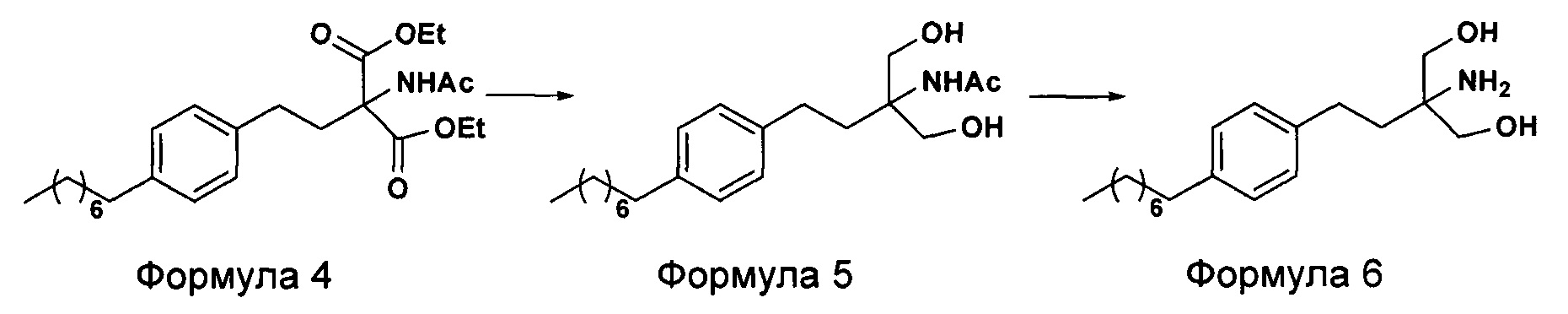
На фиг. 2 представлен другой вариант синтеза гидрохлорида финголимода [гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола] согласно настоящему изобретению.
На фиг. 3 представлена дифракционная рентгенограмма (ДРГ) полиморфной формы А финголимода.
На фиг. 4 представлена ДРГ полиморфной формы Y гидрохлорида финголимода.
Подробное описание изобретения
Настоящее изобретение относится к способу получения 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6), который включает следующие стадии:
а. осуществление взаимодействия соединения формулы 3 с диэтилацетамидомалонатом (Формула 2) с получением диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4),
и
b. превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты, полученного на стадии (а), с получением 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6),
или
c. превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты, полученного на стадии (а), в N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамид (Формула 5) с последующим гидролизом N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида с получением 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6),
или
d. превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты, полученного на стадии (а), в N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамид (Формула 5) с последующим превращением N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида (Формула 5) в 2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетат (Формула 5а) с последующим гидролизом ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетата (Формула 5а) с получением 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6);
В одном из вариантов осуществления настоящего изобретения реакцию стадии (а) описанного выше способа осуществляют в присутствии растворителя, выбранного из группы, включающей толуол, ксилол, гептаны, гексаны, диэтиловый эфир, метил-трет-бутиловый эфир и тетрагидрофуран, или любую их комбинацию; и способ дополнительно включает добавление реагента, выбранного из группы, включающей карбонат щелочного металла и карбонат щелочноземельного металла, или их комбинацию.
В другом варианте осуществления настоящего изобретения объем растворителя составляет от приблизительно 1 до приблизительно 30 объемов; а карбонат щелочного металла и карбонат щелочноземельного металла выбирают из группы, включающей карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, карбонат магния, карбонат кальция и карбонат бария.
В еще одном варианте осуществления настоящего изобретения стадию (а) описанного выше способа осуществляют при температуре в диапазоне от приблизительно 10°С до приблизительно 160°С и в течение периода времени от приблизительно 3 часов до приблизительно 24 часов.
В еще одном варианте осуществления настоящего изобретения превращение стадии (b), или стадии (с), или стадии (d) описанного выше способа осуществляют в присутствии реагента в растворителе, причем растворитель представляет собой С1-С4 низший спирт, и реагент выбирают из группы, включающей борогидрид щелочноземельного металла и алкоксиборогидрид щелочноземельного металла, или их комбинацию.
В еще одном варианте осуществления настоящего изобретения С1-С4 низший спирт выбирают из группы, включающей метанол, этанол, н-пропанол, изопропанол, н-бутанол, 2-бутанол, трет-бутанол, тетрагидрофуран, толуол, воду, диэтиловый эфир, метил-трет-бутиловый эфир или любую их комбинацию, причем их объем составляет от приблизительно 2 до приблизительно 30 объемов; а борогидрид щелочноземельного металла выбирают из группы, включающей боргидрид магния, боргидрид кальция, боргидрид натрия и боргидрид бария; и алкоксиборгидрид щелочноземельного металла выбирают из группы, включающей триацетоксиборгидрид магния, триацетоксиборгидрид кальция и триацетоксиборгидрид бария.
В еще одном варианте осуществления настоящего изобретения реагент представляет собой комбинацию боргидрида щелочного металла и соли, выбранной из группы, включающей сульфат бария, хлорид бария, сульфат магния, ацетат кальция, хлорид кальция, хлорид магния и ацетат магния, или любую смесь этих солей.
В еще одном варианте осуществления настоящего изобретения превращение на стадии (b), или на стадии (с), или на стадии (d) описанного выше способа осуществляют при температуре в интервале от приблизительно -5°С до приблизительно 110°С; при рН в диапазоне от приблизительно 1 до приблизительно 14; и где указанный диапазон рН достигают с помощью раствора, выбранного из раствора кислоты, раствора основания или их комбинации.
В еще одном варианте осуществления настоящего изобретения раствор кислоты выбирают из группы, включающей соляную кислоту и уксусную кислоту, или их комбинацию; раствор основания выбирают из группы, включающей гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид магния, гидроксид кальция и гидроксид бария, или любую их комбинацию.
В еще одном варианте осуществления настоящего изобретения реакция стадий (а), (b), (с) и (d) описанного выше способа возможно включает использование катализатора фазового переноса.
В еще одном варианте осуществления настоящего изобретения катализатор фазового переноса представляет собой галогенид тетраалкиламмония; а указанный галогенид тетраалкиламмония выбирают из группы, включающей бромид тетраметиламмония, бромид тетраэтиламмония, бромид тетрабутиламмония и иодид тетрабутиламмония, или любую их комбинацию.
В еще одном варианте осуществления настоящего изобретения гидролиз осуществляют в присутствии:
a. раствора неорганического основания или
b. соляной кислоты, после чего доводят рН с использованием основания, выбранного из группы, включающей гидроксид натрия, гидроксид лития, гидроксид калия, гидроксид магния.
В еще одном варианте осуществления настоящего изобретения неорганическое основание выбирают из группы, включающей гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид магния, гидроксид кальция и гидроксид бария, или любую их комбинацию.
В еще одном варианте осуществления настоящего изобретения после добавления раствора неорганического основания или соляной кислоты осуществляют кипячение с обратным холодильником в течение периода времени в диапазоне от приблизительно 0,5 часов до приблизительно 24 часов.
В еще одном варианте осуществления настоящего изобретения превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты в 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол и гидролиз N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида в 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол дополнительно включает возможные стадии добавления растворителя, перемешивания, фильтрации и сушки.
В еще одном варианте осуществления настоящего изобретения полученный 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол является кристаллическим по природе и обозначен как полиморф А.
В еще одном варианте осуществления настоящего изобретения списанный выше способ дополнительно включает превращение 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6) в гидрохлорид 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 1).
В еще одном варианте осуществления настоящего изобретения превращение осуществляют путем добавления соляной кислоты к 2-амино-2-(2-(4-октилфенил) этил)-1,3-пропандиолу в присутствии растворителя при температуре от приблизительно -25°С до приблизительно 45°С.
В еще одном варианте осуществления настоящего изобретения соляную кислоту добавляют в изопропаноле; причем растворитель выбирают из группы, включающей толуол, метилацетат, этилацетат, изопропилацетат, бутилацетат, ацетонитрил, метилизобутилкетон и метилэтилкетон, или их комбинацию; и объем растворителя составляет от приблизительно 2 объемов до приблизительно 25 объемов.
В еще одном варианте осуществления настоящего изобретения полученный гидрохлорид 2-амино-2-(2-(4-октилфенил) этил)-1,3-пропандиола является кристаллическим по природе и обозначен как полиморф Y.
В еще одном варианте осуществления настоящего изобретения 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол или гидрохлорид 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола необязательно очищают и сушат; и очистку проводят путем перекристаллизации в присутствии растворителя, выбранного из группы, включающей толуол, метилацетат, этилацетат, изопропилацетат, бутилацетат, ацетонитрил, метилизобутилкетон и метилэтилкетон, или любую их комбинацию.
В еще одном варианте осуществления настоящего изобретения объем растворителя составляет от приблизительно 2 до приблизительно 30 объемов; и температура очистки 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола составляет от приблизительно 10°С до приблизительно 110°С; а температура очистки гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола составляет от приблизительно -25°С до приблизительно 50°С.
В еще одном варианте осуществления настоящего изобретения сушку проводят в вакууме при температуре от приблизительно 20°С до приблизительно 75°С.
В еще одном варианте осуществления настоящего изобретения соединение формулы 3 представляет собой:
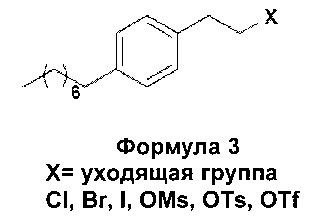
В еще одном варианте осуществления настоящего изобретения соединение формулы 3 выбрано из группы, включающей 1-(2-иодэтил)-4-октилбензол, 1-(2-бромэтил)-4-октилбензол, 1-(2-хлорэтил)-4-октилбензол, 1-(2-этилмезилат)-4-октилбензол, 1-(2-этилтозилат)-4-октилбензол и 1-(2-этилтрифлат)-4-октилбензол, или любую их комбинацию.
Кроме того, настоящее изобретение относится к полиморфной форме А 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6), полученной указанным способом, имеющей значения 2θ XRPD: 3.876, 5.744, 7.739, 11.65, 14.886, 15.356, 16.774, 17.65, 18.008, 18.963, 19.473, 20.845, 21.626, 23.431, 24.643, 27.389, 27.894, 30.566, 31.421, 34.267, 35.01, 35.5, 38.756, 42.214, 43.767, 46.201, 48.026, 50.269 и 52.314.
Кроме того, настоящее изобретение относится к полиморфной форме Y гидрохлорида 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 1), полученной указанным способом, имеющей значения 2θ XRPD: 3.549, 5.185, 5.832, 7.052, 8.62, 9.305, 10.625, 12.149, 12.82, 14.163, 14.713, 15.174, 15.61, 16.374, 17.329, 17.749, 18.254, 18.698, 19.255, 19.948, 20.879, 21.389, 22.248, 22.578, 22.838, 23.527, 24.449, 24.953, 25.847, 26.139, 27.127, 28.094, 28.604, 29.47, 29.697, 31.786, 32.24, 33.147, 36.955 и 44.474.
Настоящее изобретение преодолевает ограничения предшествующего уровня техники с обеспечением надежного способа синтеза финголимода и его гидрохлоридной соли, который является безопасным, коммерчески выполнимым, эффективным и не включает каких-либо хроматографических очисток.
Следовательно, одной из задач настоящего изобретения является обеспечение улучшенных способов крупномасштабного синтеза финголимода и его гидрохлоридной соли.
В одном из вариантов осуществления настоящего изобретения различные химические соединения и промежуточные соединения, раскрытые в данном описании, является следующими:
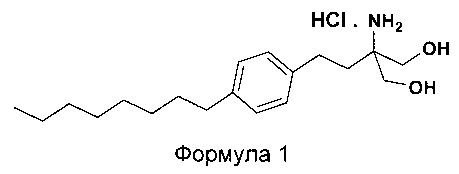
Гидрохлорид 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола
[гидрохлорид финголимода]
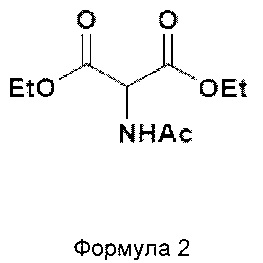
Диэтилацетамидомалонат
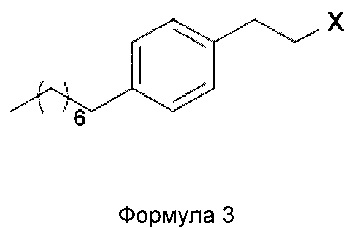
Х= Cl, Br, I, OMs, OTs, OTf
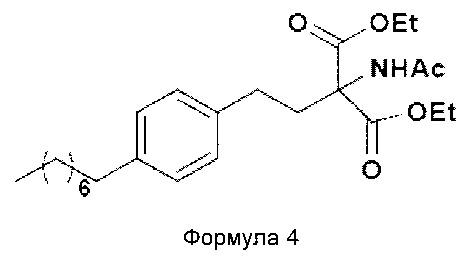
Диэтиловый эфир 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты
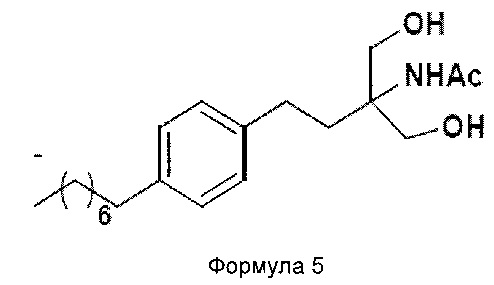
N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамид
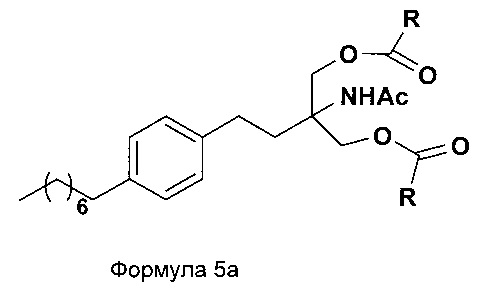
2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диилдиацетат
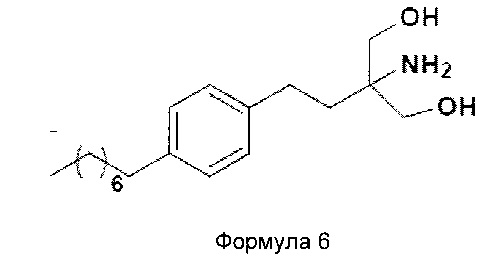
2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол [финголимод]
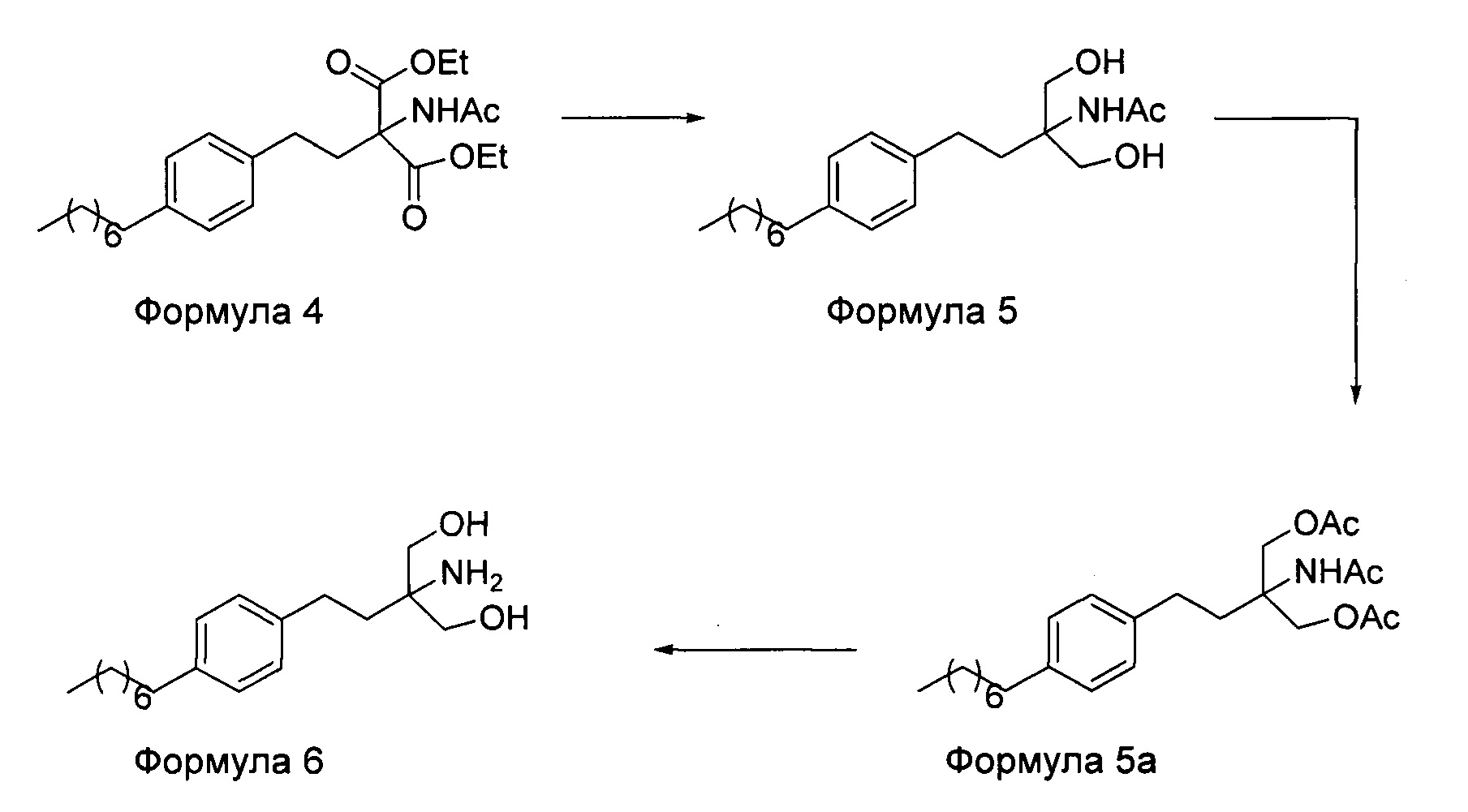
В первом варианте осуществления настоящего изобретения (Схема 1 ниже) гидрохлорид финголимода [гидрохлорид 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола] синтезируют способом, состоящим из следующих стадий:
i. взаимодействие 1-(2-иодэтил)-4-октилбензола (Формула 3) с диэтилацетамидомалонатом (Формула 2) в подходящих условиях с получением соответствующего конденсированного продукта: диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4);
ii. превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4) в 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол (Формула 6) (свободное основание финголимода) в подходящих условиях в присутствии подходящих реагентов;
iii. при необходимости, очистка свободного основания финголимода (Формула 6) и осуществление стадии сушки после очистки;
iv. превращение 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (Формула 6) (свободного основания финголимода) в гидрохлоридную соль (т.е. гидрохлоридную соль финголимода) (Формула 1); и
v. при необходимости, очистка гидрохлоридной соли финголимода и осуществление стадии сушки после очистки.
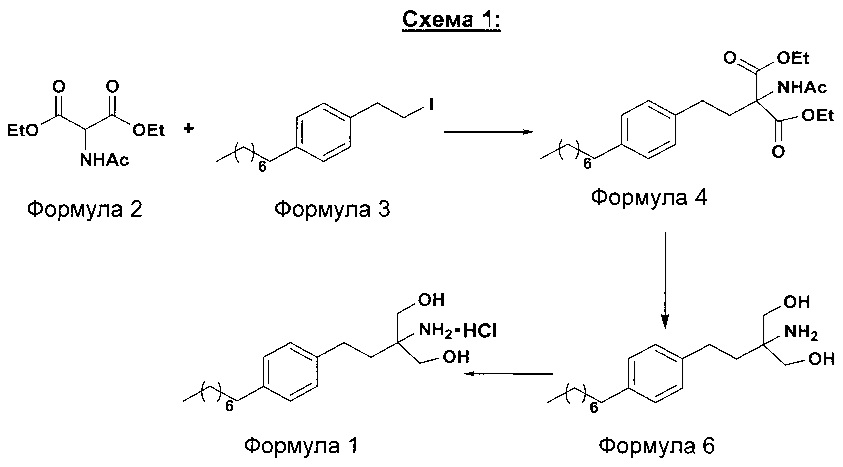
В варианте осуществления изобретения фиг. 1 иллюстрирует описанный выше способ синтеза, который подробно описан ниже:
(А) Взаимодействие 1-(2-иодэтил)-4-октилбензола [Формула 3] с диэтилацетамидомалонатом [Формула 2]:
Осуществляют конденсацию 1-(2-Иодэтил)-4-октилбензола (Формула 3) с диэтилацетамидомалонатом (Формула 2) в органическом растворителе, выбранном из группы, включающей толуол, ксилол, гептаны, гексаны, диэтиловый эфир, метил-трет-бутиловый эфир и тетрагидрофуран, или любую смесь указанных растворителей, при подходящей температуре в присутствии подходящих реагентов с получением диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4). Объемы растворителей составляют приблизительно 1-30 объемов по отношению к объему диэтилацетамидомалоната (Формула 2), и интервал температур составляет приблизительно от 10°С до 160°С.
Используемые реагенты выбирают из группы, включающей карбонаты щелочных металлов и карбонаты щелочноземельных металлов, такие как карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, карбонат магния, карбонат кальция и карбонат бария, или любую их смесь. При необходимости, также используют катализаторы фазового переноса для повышения реакционной способности. Указанный катализатор фазового переноса выбирают из галогенида тетраалкиламмония, такого как бромид тетраметиламмония, бромид тетраэтиламмония, бромид тетрабутиламмония и йодид тетрабутиламмония.
(В) Превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты [Формула 4] в 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол [Формула 6] (свободное основание финголимода):
Диэтиловый эфир 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4) превращают в 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол (Формула 6) (свободное основание финголимода) путем реакции в присутствии подходящих реагентов в подходящем растворителе (растворителях) при подходящей температуре. Растворители выбирают из С1-С4 низших спиртов, таких как метанол, этанол, н-пропанол, изопропанол, н-бутанол, 2-бутанол, трет-бутанол, тетрагидрофуран, толуол, вода, диэтиловый эфир, метил-трет-бутиловый эфир или их смеси. Объемы растворителей составляют приблизительно 2-30 объемов, и температура составляет приблизительно от -5°С до 110°С.
Используемые реагенты выбирают из группы, включающей борогидриды щелочноземельных металлов и алкоксиборгидриды щелочноземельных металлов, такие как борогидрид магния, боргидрид кальция, борогидрид бария, боргидрид натрия, триацетоксиборгидрид магния, триацетоксиборгидрид кальция и триацетоксиборгидрид бария, или любую их смесь. В другом аспекте настоящего изобретения также используют комбинацию боргидрида щелочного металла и соли, выбранной из сульфата бария, хлорида бария, сульфата магния, ацетата кальция, хлорида кальция, хлорида магния и ацетата магния, или любой смеси солей, в качестве альтернативных реагентов для указанного превращения.
В другом аспекте настоящего изобретения рН реакционной смеси доводят до кислого значения путем добавления раствора кислоты, выбранной из соляной кислоты и уксусной кислоты, или их смеси.
В еще одном аспекте настоящего изобретения рН реакционной смеси доводят до щелочного значения с получением свободного основания путем добавления щелочного раствора, выбранного из гидроксида лития, гидроксида натрия, гидроксида калия, гидроксида магния, гидроксида кальция и гидроксида бария, или любых их смесей.
В другом варианте осуществления изобретения осуществляют необязательную стадию, в которой добавляют органический растворитель и реакционную смесь перемешивают до осаждения. Осадок, содержащий свободное основание финголимода в твердой форме, отделяют фильтрованием и дополнительно необязательно сушат. Кроме того, выполняют еще одну необязательную стадию очистки, в которой свободное основание финголимода очищают перекристаллизацией в подходящем растворителе, выбранном из группы, включающей толуол, метилацетат, этилацетат, изопропилацетат, бутилацетат, ацетонитрил, метилизобутилкетон и метилэтилкетон, или любую их смесь. Объемы используемого растворителя составляют приблизительно 2-25 объемов, и диапазон температур составляет приблизительно от 10°С до температуры кипения.
В еще одном аспекте настоящего изобретения свободное основание финголимода (Формула 6) является кристаллическим по природе и обозначено как полиморф А, имеющий следующие значения 2θ XRPD: 3.876, 5.744, 7.739, 11.65, 14.886, 15.356, 16.774, 17.65, 18.008, 18.963, 19.473, 20.845, 21.626, 23.431, 24.643, 27.389, 27.894, 30.566, 31.421, 34.267, 35.01, 35.5, 38.756, 42.214, 43.767, 46.201, 48.026, 50.269, 52.314 (фиг. 3).
(С) Превращение 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола [Формула 6] (свободного основания финголимода) в гидрохлорид 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола [Формула 1]:
Свободное основание финголимода (Формула 6) превращают в его гидрохлоридную соль (Формула 1) путем добавления соляной кислоты в изопропаноле к финголимоду (Формула 6) в присутствии подходящего растворителя, выбранного из группы, включающей толуол, метилацетат, этилацетат, изопропилацетат, бутилацетат, ацетонитрил, метилизобутилкетон и метилэтилкетон, или любую их смесь, при температуре приблизительно от -25°С до 45°С. В предпочтительном варианте осуществления температура составляет приблизительно от -20°С до 25°С, более предпочтительно приблизительно от -10°С до 15°С и наиболее предпочтительно приблизительно от -5°С до 10°С. Объемы используемого растворителя составляют приблизительно 2-25 объемов. Кроме того, осажденный материал, полученный после завершения реакции, выделяют и, при необходимости, сушат в вакууме при температуре от приблизительно 25°С до 75°С.
В еще одном аспекте настоящего изобретения гидрохлорид финголимода (Формула 1) является кристаллическим по природе и обозначен как полиморф Y, имеющий пики на XRPD при 2θ следующие: 3.549, 5.185, 5.832, 7.052, 8.62, 9.305, 10.625, 12.149, 12.82, 14.163, 14.713, 15.174, 15.61, 16.374, 17.329, 17.749, 18.254, 18.698, 19.255, 19.948, 20.879, 21.389, 22.248, 22.578, 22.838, 23.527, 24.449, 24.953, 25.847, 26.139, 27.127, 28.094, 28.604, 29.47, 29.697, 31.786, 32.24, 33.147, 36.955, 44.474 (фиг. 4).
В еще одном аспекте настоящего изобретения осуществляют дополнительную стадию очистки, в которой полученный гидрохлорид финголимода очищают перекристаллизацией в подходящем растворителе, выбранном из группы, включающей толуол, ацетонитрил, метилацетат, этилацетат, изопропилацетат, бутилацетат, метанол, этанол и изопропанол, или любую их смесь, в температурном диапазоне приблизительно от -25 до 50°С. После дополнительной стадии очистки осуществляют сушку очищенного гидрохлорида финголимода в вакууме при температуре приблизительно от 20 до 75°С.
В другом варианте осуществления настоящего изобретения (Схема 2 ниже) гидрохлорид финголимода синтезируют способом, включающим следующие стадии:
i. взаимодействие 1-(2-иодэтил)-4-октилбензола (Формула 3) с диэтилацетамидомалонатом (Формула 2) в подходящих условиях с получением соответствующего конденсированного продукта: диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4);
ii. превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4) в N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамид (Формула 5) в подходящих условиях в присутствии подходящих реагентов;
iii. при необходимости, превращение N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида (Формула 5) в соединение Формулы 5а путем защиты гидроксильных групп;
iv. гидролиз N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида (Формула 5) или соединения формулы 5а в 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол (свободное основание финголимода) (Формула 6) в присутствии основания;
v. при необходимости, очистка свободного основания финголимода (Формула 6) и осуществление стадии сушки после очистки;
vi. превращение 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (свободного основание финголимода) (Формула 6) в его гидрохлоридную соль (т.е. гидрохлоридную соль финголимода) (Формула 1); и
vii. при необходимости, очистка гидрохлоридной соли финголимода и осуществление стадии сушки после очистки.
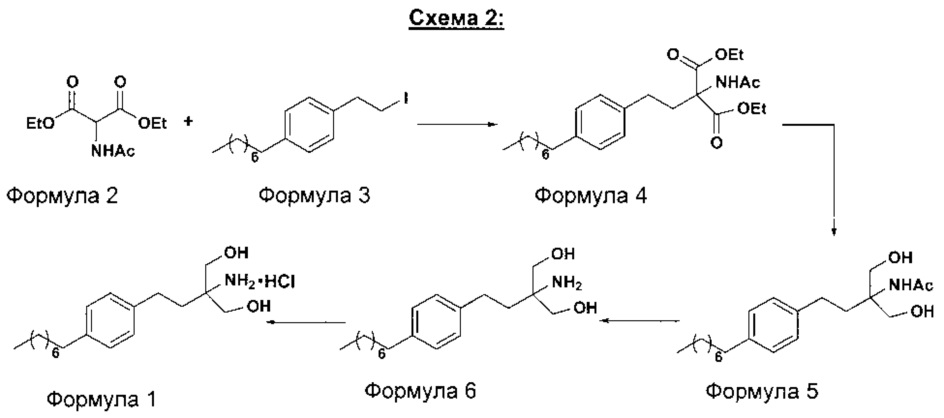
В варианте осуществления изобретения фиг. 2 иллюстрирует описанный выше способ синтеза, который подробно описан ниже:
(А) Взаимодействие 1-(2-иодэтил)-4-октилбензола [Формула 3] с диэтилацетамидомалонатом [Формула 2]:
Осуществляют конденсацию 1-(2-Иодэтил)-4-октилбензола (Формула 3) с диэтилацетамидомалонатом (Формула 2) в органическом растворителе, выбранном из группы, включающей толуол, ксилол, гептаны, гексаны, диэтиловый эфир, метил-трет-бутиловый эфир и тетрагидрофуран, или любую смесь указанных растворителей, при подходящей температуре. Реакцию осуществляют в присутствии подходящих реагентов с получением диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4). Объемы растворителей составляют приблизительно 1-30 объемов, и интервал температур составляет приблизительно от 10°С до 160°С.
Используемые реагенты выбирают из группы, включающей карбонаты щелочных металлов и карбонаты щелочноземельных металлов, такие как карбонат лития, карбонат натрия, карбонат калия, карбонат цезия, карбонат магния, карбонат кальция и карбоната бария, или любую их смесь. При необходимости, также используют катализаторы фазового переноса для повышения реакционной способности. Указанный катализатор фазового переноса выбирают из галогенида тетраалкиламмония, такого как бромид тетрабутиламмония, бромид тетраэтиламмония, йодид тетраэтиламмония и иодид тетрабутиламмония.
(В) Превращение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты [Формула 4] в N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамид [Формула 5]:
Диэтиловый эфир 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4) превращают в N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамид (Формула 5) путем реакции с подходящими реагентами в подходящем растворителе, причем растворители выбирают из С1-С4 низших спиртов, таких как метанол, этанол, н-пропанол, изопропанол, н-бутанол, 2-бутанол, трет-бутанол, тетрагидрофуран, толуол, вода, диэтиловый эфир, метил-трет-бутиловый эфир или любая их смесь, при подходящей температуре. Объемы растворителей составляют приблизительно 2-30 объемов, и температура составляет приблизительно от -5°С до 110°С.
В варианте осуществления используемые реагенты выбирают из группы, включающей борогидриды щелочноземельных металлов и алкоксиборгидриды щелочноземельных металлов, такие как борогидрид магния, боргидрид кальция, борогидрид бария, боргидрид натрия, триацетоксиборгидрид магния, триацетоксиборгидрид кальция и триацетоксиборгидрид бария, или любую их смесь. Интересно, что использование на этой стадии катализатора фазового переноса контролирует образование нежелательных побочных продуктов. Указанный катализатор фазового переноса выбирают из галогенида тетраалкиламмония, такого как бромид тетрабутиламмония, бромид тетраэтиламмония, йодид тетраэтиламмония и иодид тетрабутиламмония.
В другом аспекте настоящего изобретения также используют комбинацию боргидрида щелочного металла и соли, выбранной из сульфата бария, хлорида бария, сульфата магния, ацетата кальция, хлорида кальция, хлорида магния и ацетата магния, или любой смеси солей, в качестве альтернативных реагентов для указанного превращения.
В другом аспекте настоящего изобретения рН реакционной смеси доводят до кислого значения путем добавления раствора кислоты, выбранной из соляной кислоты, уксусной кислоты или их смеси.
В еще одном аспекте настоящего изобретения рН реакционной смеси доводят до щелочного значения с получением свободного основания путем добавления щелочного раствора, выбранного из гидроксида лития, гидроксида натрия, гидроксида калия, гидроксида магния, гидроксида кальция и гидроксида бария, или любых их смесей.
(С) Гидролиз N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида [Формула 5] в 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиол (свободное основание финголимода) [Формула 6]:
Осуществляют гидролиз N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида (Формула 5) с получением 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (свободного основания финголимода) (Формула 6) с использованием раствора неорганического основания, выбранного из группы, включающей гидроксид лития, гидроксид натрия, гидроксид калия, гидроксид магния, гидроксид кальция и гидроксид бария. При необходимости, добавляют также органический растворитель и реакционную смесь перемешивают с получением осадка. Осадок, содержащий материал в твердой форме, отделяют фильтрованием, затем необязательно сушат с получением свободного основания финголимода. Еще в одном аспекте свободное основание финголимода, при необходимости, очищают перекристаллизацией в подходящем растворителе, выбранном из группы, включающей толуол, метилацетат, этилацетат, изопропилацетат, бутилацетат, ацетонитрил, метилизобутилкетон и метилэтилкетон, или любую их смесь. Объемы используемого растворителя составляют приблизительно 2-25 объемов, и диапазон температур составляет приблизительно от 10°С до температуры кипения.
В другим альтернативном варианте осуществления настоящего изобретения проводят гидролиз N-(1,1-бисгидроксиметил-3-(4-оксилфенил)-пропил)-ацетамида (Формула 5) с получением 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола (свободного основания финголимода) (Формула 6) с использованием соляной кислоты, после чего доводят рН гидроксидом натрия.
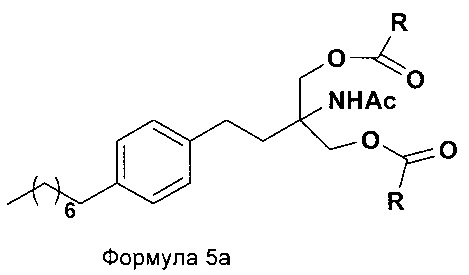
В еще одном варианте осуществления настоящего изобретения соединение формулы 5 превращают в полностью защищенное соединение формулы 5а (2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диилдиацетат), и затем соединение формулы в 5а превращают в свободное основание финголимода (Формула 6) путем одностадийного гидролиза в подходящих условиях. В соединении формулы 5а R выбран из защищенных гидроксильных групп, R представляет собой С1-С4-алкил, арил, такой как ацетил, бензоил и замещенный ароматический фрагмент.
В еще одном аспекте настоящего изобретения свободное основание финголимода (Формула 6) является кристаллическим по природе и обозначено как полиморф А, имеющий следующие значения 2θ XRPD: 3.876, 5.744, 7.739, 11.65, 14.886, 15.356, 16.774, 17.65, 18.008, 18.963, 19.473, 20.845, 21.626, 23.431, 24.643, 27.389, 27.894, 30.566, 31.421, 34.267, 35.01, 35.5, 38.756, 42.214, 43.767, 46.201, 48.026, 50.269, 52.314 (фиг. 3).
(D) Превращение 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола [Формула 6] в гидрохлорид 2-амино-2-(2-(4-октилфенил)этил)-1,3-пропандиола [Формула 1]
Свободное основание финголимода (Формула 6) превращают в его гидрохлоридную соль (Формула 1) путем добавления соляной кислоты в изопропаноле к финголимоду (Формула 6) в присутствии подходящего растворителя, выбранного из группы, включающей толуол, метилацетат, этилацетат, изопропилацетат, бутилацетат, ацетонитрил, метилизобутилкетон и метилэтилкетон, или любую их смесь, при температуре приблизительно от -25°С до 45°С. В предпочтительном варианте осуществления температура составляет приблизительно от -20°С до 25°С, более предпочтительно приблизительно от -10°С до 15°С и наиболее предпочтительно приблизительно от -5°С до 10°С. Объемы используемого растворителя составляют приблизительно 2-25 объемов. Кроме того, осажденный материал (Формула 6), полученный в результате реакции, выделяют и, при необходимости, сушат в вакууме при температуре от приблизительно 25°С до 75°С.
В еще одном аспекте настоящего изобретения гидрохлорид финголимода (Формула 1), полученный описанным выше способом, является кристаллическим по природе и обозначен как полиморф Y, имеющий пики на XRPD при 2θ следующие: 3.549, 5.185, 5.832, 7.052, 8.62, 9.305, 10.625, 12.149, 12.82, 14.163, 14.713, 15.174, 15.61, 16.374, 17.329, 17.749, 18.254, 18.698, 19.255, 19.948, 20.879, 21.389, 22.248, 22.578, 22.838, 23.527, 24.449, 24.953, 25.847, 26.139, 27.127, 28.094, 28.604, 29.47, 29.697, 31.786, 32.24, 33.147, 36.955, 44.474 (фиг. 4).
В еще одном варианте осуществления настоящего изобретения гидрохлорид финголимода, при необходимости, очищают перекристаллизацией в подходящем растворителе в температурном диапазоне приблизительно от -25 до 50°С. Растворитель выбирают из группы, включающей толуол, ацетонитрил, метилацетат, этилацетат, изопропилацетат, бутилацетат, метанол, этанол и изопропанол, или любую их смесь. Кроме того, после необязательной очистки осуществляют сушку в вакууме при температуре приблизительно от 20 до 75°С.
Технология данного изобретения далее подробно описана с помощью следующих примеров. Однако эти примеры не следует рассматривать как ограничивающие объем изобретения.
Примеры
Пример 1
Получение диэтилового эфира 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4)
К суспензии диэтилацетамидомалоната (DEAM) [Формула 2] (28,39 г) и карбоната цезия (189,13 г) в толуоле (300 мл) добавляют тетрабутилбромид (0,468 г) и 1-(2-иодэтил)-4-октилбензол [Формула 3] (50 г) в толуоле (50 мл) в атмосфере азота. Содержимое нагревают до кипения с обратным холодильником и выдерживают в течение приблизительно 3-10 часов. После завершения реакции полученную массу охлаждают до приблизительно 30°С с последующим добавлением воды (300 мл) и разделением образовавшихся слоев. Органический слой промывают водой, затем солевым раствором и сушат над сульфатом натрия. Растворитель выпаривают при пониженном давлении с получением неочищенного соединения Формулы 4 (59 г).
Пример 2
Получение N-[1,1-бисгидроксиметил-3-(4-октилфенил)пропил]ацетамида (Формула 5)
К диэтиловому эфиру 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4) (58 г) в 50% водном IPA (изопропиловый спирт) (600 мл), добавляют тетрабутиламмонийбромид (4,31 г) и боргидрид натрия (25,41 г), и реакционную массу перемешивают в течение приблизительно 8-24 часов при температуре приблизительно от 20°С до 35°С. После завершения реакции рН доводят до приблизительно 6,0±0,5 с использованием 6Н водного раствора соляной кислоты. Реакционную массу фильтруют через воронку Бюхнера, чтобы удалить твердые частицы, и фильтрат концентрируют при пониженном давлении с получением сиропа. В полученный сироп добавляют воду (116 мл), и соединение экстрагируют в этилацетат (2×174 мл). Органический слой промывают водой, затем солевым раствором с последующей сушкой над сульфатом натрия, затем выпаривают при пониженном давлении. Продукт кристаллизуют с использованием петролейного эфира для получения указанного в заголовке соединения формулы 5 (27 г).
Пример 3
Получение N-[1,1-бисгидроксиметил-3-(4-октилфенил)пропил]ацетамида (Формула 5)
К диэтиловому эфиру 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (Формула 4) (58 г) в IPA (928 мл) добавляют ацетат кальция (63,47 г), и реакционную смесь перемешивают при температуре приблизительно 10-15°С в течение приблизительно 30 мин. После этого добавляют боргидрид натрия (25,41 г), и реакционную массу перемешивают в течение приблизительно 8-12 часов при температуре приблизительно 10-15°С. Завершение реакции контролируют с помощью ТСХ (тонкослойной хроматографии). Реакционную массу гасят 1,5Н раствором HCl для достижения нейтрального рН. Реакционную массу фильтруют, чтобы удалить твердые частицы, и фильтрат подвергают выпариванию в вакууме. Затем добавляют воду (116 мл), и соединение экстрагируют в этилацетат (2×174 мл). Органический слой промывают водой (2×174 мл). Органический слой промывают солевым раствором (58 мл) и сушат над сульфатом натрия. Полученный продукт фильтруют и выпаривают при пониженном давлении с образованием сиропа, который представляет собой соединение формулы 5 (38 г), указанное в заголовке.
Пример 4
Получение N-[1,1-бисгидроксиметил-3-(4-октил фенил)пропил]ацетамида (Формула 5)
К диэтиловому эфиру (2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (0,196 г) [Формула 4] в IPA (3,2 мл) и воде (0,8 мл) добавляют BaCl2.2H2O (0,332 г), и реакционную смесь перемешивают в течение 10-15 минут. Реакционную массу охлаждают до приблизительно 0-5°С с использованием ледяной бани. Добавляют порциями NaBH4 (0,085 г) с последующим перемешиванием в течение приблизительно 12-16 часов при приблизительно 10-15°С. Завершение реакции контролируют с помощью ТСХ. Значение рН реакционной массы доводят до нейтрального с помощью 1,5H раствора HCl. Твердое вещество отфильтровывают, и фильтрат подвергают выпариванию при пониженном давлении. К этому добавляют воду (5 мл) и экстрагируют соединение в этилацетат (3×10 мл). Органический слой промывают солевым раствором (2×5 мл) и сушат над Na2SO4, фильтруют и выпаривают при пониженном давлении с получением соединения формулы 5 в виде не совсем белого твердого вещества (0,13 г).
Пример 5
Получение N-[1,1-бисгидроксиметил-3-(4-октил фенил)пропил]ацетамида (Формула 5)
Литий бромид (72,02 г) и NaBH4 (25,41 г) добавляют к IPA (928 мл) и воде (232 мл) при температуре приблизительно 10-15°С. Содержимое перемешивают в течение приблизительно 2 часов при указанной температуре. К указанной смеси добавляют диэтиловый эфир 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (58 г) [Формула 4], и реакционную смесь перемешивают в течение приблизительно 24 часов при температуре приблизительно 10-15°С. Завершение реакции контролируют с помощью ТСХ. Значение рН реакционной массы доводят до нейтрального с помощью 1,5Н раствора HCl. Полученное твердое вещество после завершения реакции отфильтровывают, и фильтрат подвергают выпариванию. К фильтрату добавляют воду, и соединение экстрагируют в этилацетат (2×174 мл). Органический слой промывают водой. Органический слой промывают насыщенным раствором соли и сушат над сульфатом натрия, затем фильтруют и выпаривают при пониженном давлении с получением сиропа, содержащего указанное в заголовке соединение формулы 5 в виде неочищенного сиропа (36 г). Полученное соединение формулы 5 используют без дополнительной очистки.
Пример 6
Получение (2-амино-2[2-(4-октилфенил)этил]пропан-1,3-диола (основания финголимода; Формула 6)
Берут N-[1,1-бисгидроксиметил-3-(4-октилфенил)пропил]ацетамид (38 г) [Формула 5] в IPA (133 мл) и 6М HCl (133 мл), и содержимое нагревают до кипения и кипятят с обратным холодильником в течение приблизительно 2 часов. Реакционную массу охлаждают до приблизительно 40°С, и IPA выпаривают при пониженном давлении. Затем рН реакционной массы доводят до приблизительно 9-10 с помощью 25%-ного раствора NaOH, и продукт экстрагируют в этилацетат (3×114 мл). Органический слой промывают водой (114 мл), затем насыщенным раствором хлорида натрия (2×38 мл), сушат над сульфатом натрия и концентрируют при пониженном давлении. Содержимое охлаждают до приблизительно 0-2°С, твердое вещество перемешивают в течение приблизительно 1 часа и фильтруют. Влажный осадок на фильтре промывают охлажденным этилацетатом (38 мл). Сырой продукт перекристаллизовывают с использованием этилацетата с получением основания финголимода (Формула 6) в чистом виде (21,5 г).
Пример 7
Получение (2-амино-2[2-(4-октилфенил)этил]пропан-1,3-диола (основания финголимода; Формула 6)
N-(1-гидрокси-2 гидроксиметил-3-(4-октилфенил)-пропил)-ацетамид (38 г) [Формула 5] в IPA (114 мл) и 6N HCl (114 мл) объединяют. Содержимое нагревают до приблизительно 80±5°С и перемешивают в течение приблизительно 2 часов. Реакционную смесь охлаждают до приблизительно 10±5°С, и рН доводят до приблизительно 9,5±0,5 с использованием 25% раствора гидроксида натрия. Добавляют 5 объемов воды, и реакционную смесь перемешивают в течение приблизительно 1 часа. Твердое вещество фильтруют, фильтрат перекристаллизовывают в этилацетате. Сырой продукт перекристаллизовывают с использованием этилацетата с получением основания финголимода (Формула 6) в чистом виде (18,2 г), имеющего чистоту 99,5%.
Пример 8
Получение (2-амино-2[2-(4-октилфенил)этил]пропан-1,3-диола (основания финголимода; Формула 6)
К диэтиловому эфиру 2-(ацетиламино)-2-(2-(4-октилфенил)этил)пропандиовой кислоты (0,50 г) [Формула 4] в этаноле (8,5 мл) и воде (2,0 мл) добавляют CaCl2 (0,32 г), и реакционную смесь перемешивают в течение приблизительно 10-15 минут. Реакционную массу охлаждают до приблизительно 10-15°С с использованием ледяной бани. Добавляют порциями NaBH4 (0,21 г), и реакционную смесь перемешивают в течение приблизительно 4-20 часов при приблизительно 10-15°С. Завершение реакции контролируют с помощью ТСХ. К реакционной массе добавляют 6М HCl (1,5 мл), и реакционную массу нагревают до кипения с обратным холодильником. Кипячение с обратным холодильником продолжают в течение приблизительно 2 часов. После исчезновения исходного вещества реакционную смесь охлаждают до приблизительно 10±5°С, и доводят рН до приблизительно 9,5±0,5, используя 25%-ный раствор гидроксида натрия. Добавляют 5 объемов воды с последующим перемешиванием в течение приблизительно 1 часа. Твердые вещества отфильтровывают, и фильтрат перекристаллизовывают в этилацетате с получением свободного основания финголимода.
Пример 9
Получение гидрохлорида финголимода (Формула 1) [как показано на схеме 1 согласно настоящему изобретению]
Объединяют основание финголимода (21 г) [Формула 6] в этилацетате (63 мл) и HCl в IPA (15,75 мл). Содержимое перемешивают в течение приблизительно 1 часа при температуре приблизительно 75±5°С. Затем содержимое охлаждают до приблизительно 25-30°С, и охлаждение продолжают в течение приблизительно 1 ч при 25-30°С. Суспензию дополнительно охлаждают до приблизительно 0-5°С, и охлаждение поддерживают в течение приблизительно 1 часа. Полученное твердое вещество после охлаждения фильтруют и промывают охлажденным этилацетатом (21 мл), и сушат отсасыванием в течение приблизительно 1 часа при пониженном давлении. Полученную смесь дополнительно сушат в течение приблизительно 8-10 ч при температуре приблизительно 20-75°С с получением указанного в заголовке соединения (формула 1) в виде белого твердого вещества (20 г) с хроматографической чистотой 99,7%.
Пример 10
Получение 2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетата [Формула 5а]
Берут N-(1-гидрокси-2-гидроксиметил-3-(4-октилфенил)-пропил)-ацетамид (38 г) [Формула 5] в пиридине (129 мл) и перемешивают в течение приблизительно 10-15 минут. Реакционную массу охлаждают до приблизительно 0-5°С, используя ледяную баню. Добавляют уксусный ангидрид (168 мл) при температуре приблизительно 0-5°С. Завершение реакции контролируют с помощью ТСХ. Значение рН реакционной массы доводят до нейтрального с помощью 5%-ного раствора HCl. Продукт экстрагируют в этилацетат (135 мл). Органический слой промывают приблизительно 100 мл воды, а затем приблизительно 60 мл солевого раствора. После этого органический слой сушат над безводным сульфатом натрия, и затем растворитель полностью отгоняют при пониженном давлении. Продукт перекристаллизовывают в гексане с получением соединения 2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетата (формула 5а) в виде твердого вещества.
Пример 11
Получение 2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетата [Формула 5а]
Пиридин (6,0 мл) добавляют к N-(1-гидрокси-2-гидроксиметил-3-(4-октилфенил)-пропил)-ацетамиду (2 г) [Формула 5] в дихлорметане (12 мл), и смесь перемешивают в течение приблизительно 5 минут. Реакционную массу охлаждают до приблизительно 0-5°С, используя ледяную баню. Добавляют ацетилхлорид (12 мл) при температуре приблизительно 0-5°С. Завершение реакции контролируют с помощью ТСХ. Значение рН реакционной массы доводят до нейтрального с помощью 5%-ного раствора HCl. Органический слой промывают приблизительно 10 мл воды, а затем приблизительно 4 мл солевого раствора. Органический слой сушат над безводным сульфатом натрия, и растворитель полностью отгоняют при пониженном давлении. Продукт перекристаллизовывают в гексане с получением соединения 2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетата (формула 5а) в виде твердого вещества.
Пример 12
Получение (2-амино-2[2-(4-октилфенил)этил]пропан-1,3-диола (основания финголимода) [Формула 6]:
К 2-ацетамидо-2-(4-октилфенэтил)пропан-1,3-диил диацетату (формула 5а) (4 г) в метаноле (12 мл) добавляют раствор LiOH (12 мл), и содержимое нагревают до кипения с обратным холодильником и кипятят в течение приблизительно 2 часов. Реакционную массу охлаждают до приблизительно 40°С, и упаривают метанол при пониженном давлении. Продукт экстрагируют в этилацетат (2×10 мл). Органический слой промывают водой (приблизительно 8 мл), затем насыщенным раствором хлорида натрия (приблизительно 4 мл), сушат над сульфатом натрия и концентрируют при пониженном давлении. Содержимое охлаждают до приблизительно 0-2°С, твердые вещества перемешивают в течение приблизительно 1 часа и фильтруют. Влажный осадок промывают охлажденным этилацетатом (приблизительно 2 мл). Сырой продукт перекристаллизовывают, используя этилацетат, с получением основания финголимода (формула 6) с хроматографической чистотой приблизительно 99,6%.


Хроматографические способы и соединения, очищенные этими способами
Составы антитела
Способ снижения степени гликозилирования белков, способы и белки
Нацеленные/иммуномодулирующие слитые белки и способы их получения
Способ получения деферазирокса
Новый способ получения терифлуномида
Способ обнаружения нейтрализующих антител против рекомбинантного человеческого инсулина в сыворотке крови человека
Способ получения деферазирокса

