Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ ДЕФЕРАЗИРОКСА
Вид РИД
Изобретение
Область техники
Настоящее изобретение раскрывает надежный способ получения кристаллической полиморфной формы I деферазирокса высокого качества. Деферазирокс получают с использованием новой соли соответствующего промежуточного соединения с металлом и соли деферазирокса с металлом, что обеспечивает легкость операций и более высокое качество продукта.
Предшествующий уровень техники
Деферазирокс (1) является хелатором железа, который снижает перегрузку железом у пациентов, получающих переливания крови в течение длительного времени при состояниях, таких как бета-талассемия и хроническая анемия другого типа. Он селективно связывается с ионами Fe3+ в соотношении 2:1. Деферазирокс утвержден FDA и продается под товарным знаком Exjade®. Химически деферазирокс представляет собой 4-[3,5-бис(2-гидроксифенил)-1H-1,2,4-триазол-1-ил]-бензойную кислоту, имеющую молекулярную формулу C21H15N3O4 и молекулярную массу 373,4.
В US 6465504 B1 раскрыты замещенные 3,5-дифенил-1,2,4-триазолы и их применение в качестве фармацевтических хелаторов металлов, в котором салицилоилхлорид (формула 2) подвергают взаимодействию с салициламидом (формула 3) при 170°C с получением 2-(2-гидроксифенил)бенз[e][1,3]оксазин-4-она (формула 4) в виде слегка желтоватых кристаллов, имеющих точку плавления 206-208°C, который затем подвергают взаимодействию с 4-гидразинбензойной кислотой (формула 5) в этаноле при кипячении с обратным холодильником с получением 4-[3,5-бис(2-гидроксифенил)-1H-1,2,4-триазол-1-ил]бензойной кислоты (деферазирокса) в виде бесцветных кристаллов, имеющих точку плавления 264-265°C.

При повторении этого известного способа был получен деферазирокс (1) в виде кристаллической полиморфной формы I. Однако полученный материал не соответствует параметрам качества, таким как хроматографическая чистота, зольность и внешний вид, в соответствии с Руководством Международной конференции по гармонизации (ICH). Следовательно, потребность в более надежном способе очистки этого сырого деферазирокса является неизбежной для соответствия параметрам качества конечного активного фармацевтического агента (API). В известных способах конденсацию салицилоилхлорида с салициламидом проводили при 170°C, что является весьма сложным и опасным в коммерческих масштабах. В этой реакции при такой пожароопасной температуре также образуются побочные продукты, такие как нециклическое производное - соединение 2-гидрокси-N-(2-гидроксибензоил)бензамид (бис-салициламид) формулы 6, которое трудно удалить из нужного продукта. Хотя при реакции в лабораторном масштабе указанная примесь образуется в количестве более 10%, при масштабировании этой реакции образуется примерно >20% этой примеси. Следовательно, контролирование образования этого бис-салициламида до возможно минимального уровня имеет важное значение.

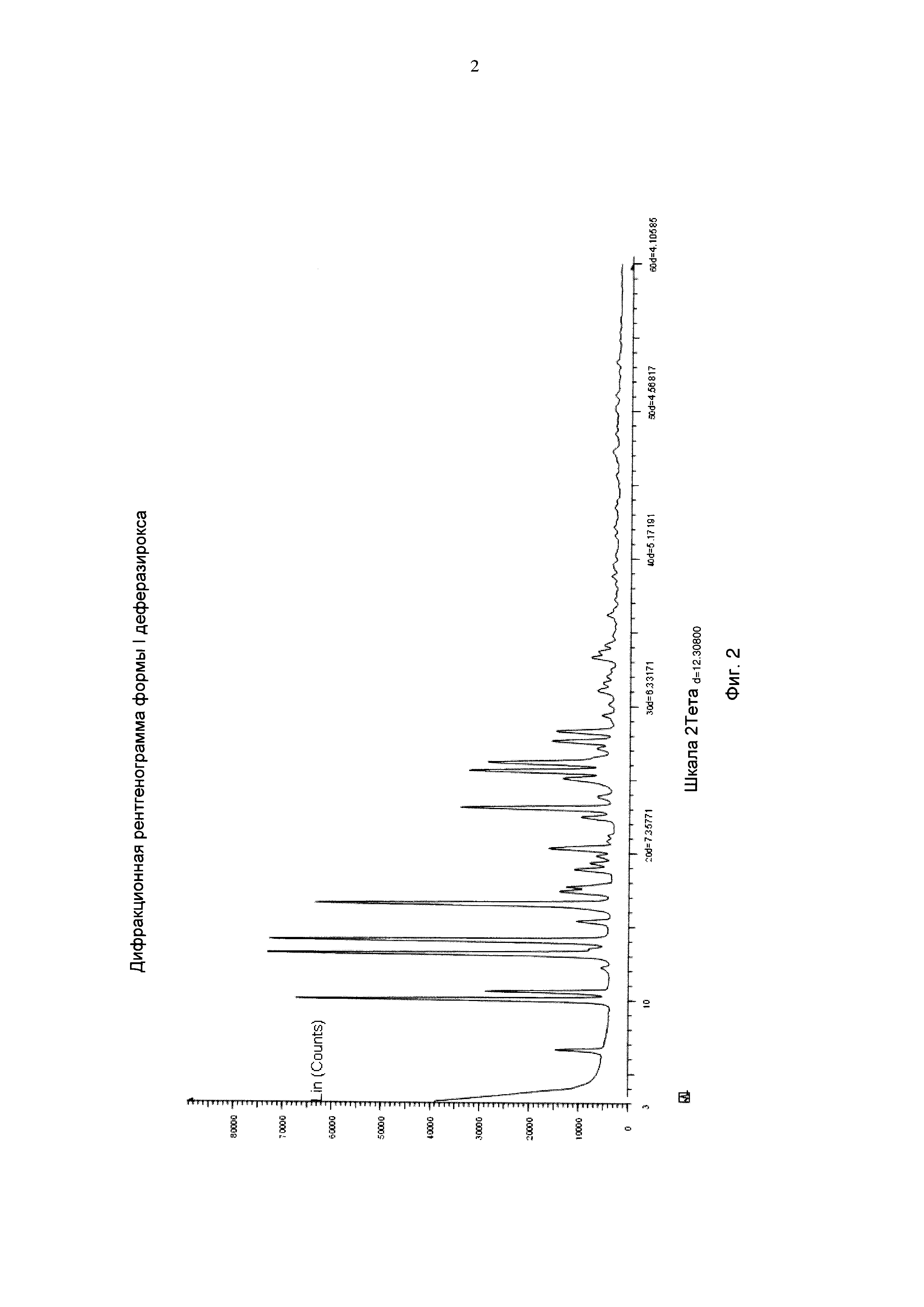
Кристаллическая полиморфная форма I деферазирокса раскрыта в IPCOM000146862D и характеризуется значениями 2θ ДРГ (порошковой рентгеновской дифракции) приблизительно 13.2, 14.1 и 16.6±0.2 градусов. Дополнительно получены характеристические значения 26 формы I: 6.6, 10.0, 10.6, 20.3, 23.1, 25.7, 26.2±0.2 градусов.
В WO 2008/065123 раскрыто применение и способ получения новых полиморфов, обозначенных как форма A, B, C, D и Sb. С другой стороны, в WO 2008/094617 сообщается о полиморфных формах деферазирокса, обозначенных как форма II, форма III, форма IV, и их превращении в наиболее стабильную полиморфную форму I. В Индийский патентной заявке 1924/CHE/2008 сообщается о новых сольватах деферазирокса и их превращении в деферазирокс.
Следовательно, существует большая потребность в коммерчески надежном, включающем простые операции способе получения кристаллической полиморфной формы I деферазирокса, удовлетворяющей параметрам качества API и требованиям ICH.
Сущность изобретения
Таким образом, настоящее изобретение относится к способу получения кристаллической формы деферазирокса, имеющей чистоту по меньшей мере 99%, причем указанный способ включает следующие стадии: a) осуществление взаимодействия салицилоилхлорида формулы 2 с салициламидом формулы 3 в присутствии катализатора с получением 2-(2-гидроксифенил)бенз[е]оксазин-4-она формулы 4, содержащего менее 1% нециклических примесей, b) осуществление взаимодействия салицилоилхлорида формулы 2 с салициламидом формулы 3 в присутствии катализатора и основания металла с получением соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она с металлом формулы 4, содержащей менее 1% нециклических примесей, c) осуществление взаимодействия 2-(2-гидроксифенил)бенз[е]оксазин-4-она со стадии (a) или соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она с металлом со стадии (b) с гидразинбензойной кислотой формулы 5 с получением получистой формы деферазирокса и d) очистку получистой формы деферазирокса в растворителе с получением кристаллической формы деферазирокса, имеющей чистоту по меньшей мере 99%, и 2-(2-гидроксифенил)бенз[е]оксазин-4-она - соединения формулы 4, или его соли с металлом, имеющими менее 1% нециклических примесей.
Краткое описание графических материалов
Для более легкого понимания изобретения и его осуществления на практике приведены иллюстративные варианты осуществления изобретения со ссылкой на прилагаемые графические материалы. Графические материалы вместе с подробным описанием ниже включены в описание и составляют часть описания изобретения, а также служат для дополнительной иллюстрации вариантов осуществления и пояснения различных принципов и преимуществ в соответствии с настоящим изобретением.
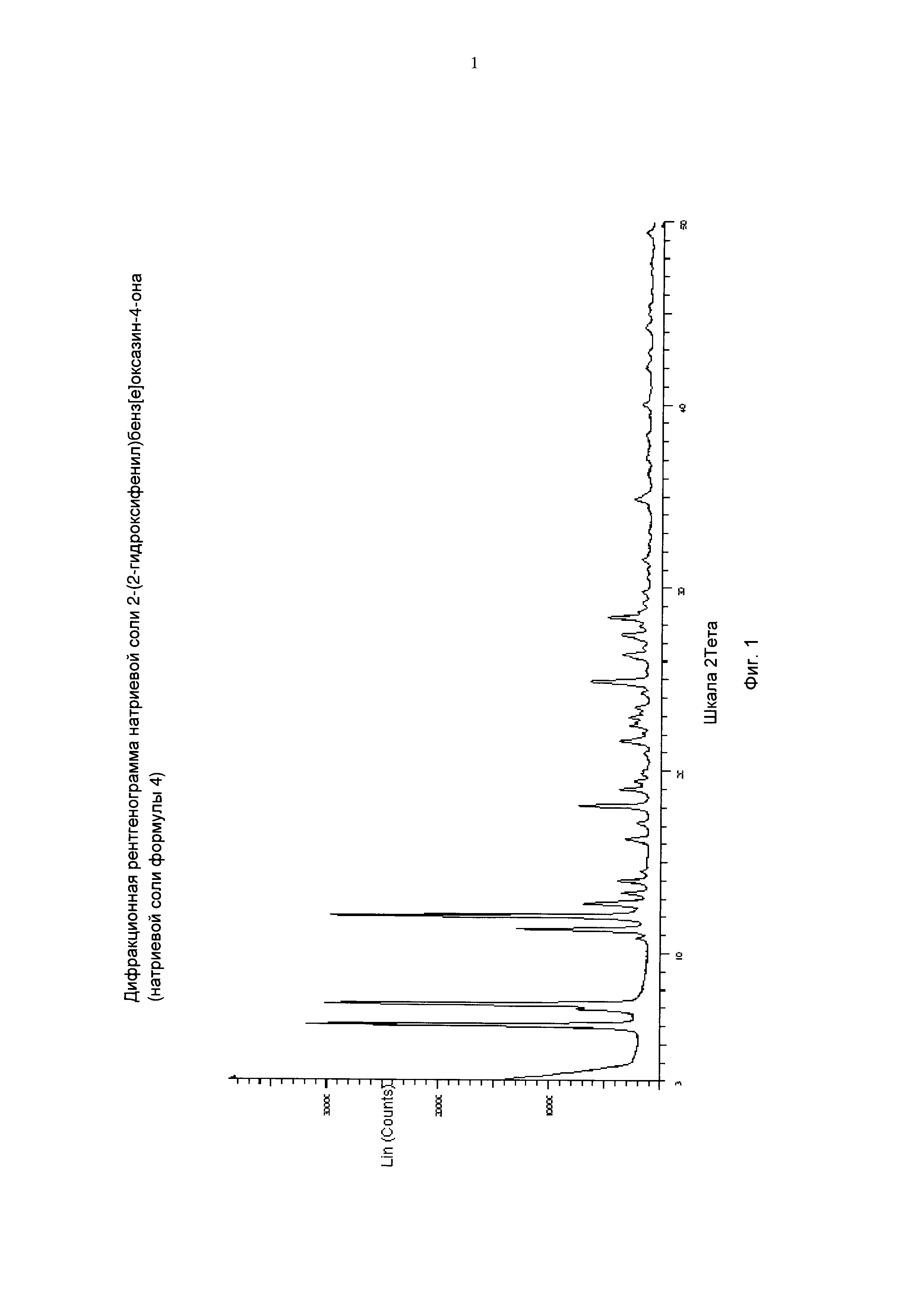
На фиг. 1 представлена дифракционная рентгенограмма (ДРГ) натриевой соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она (натриевой соли формулы 4).
На фиг. 2 представлена ДРГ формы I деферазирокса.
Подробное описание изобретения
Настоящее изобретение относится к способу получения кристаллической полиморфной формы I деферазирокса, который может быть реализован безопасно в коммерческих масштабах. Удивительно, что настоящее изобретение одновременно решает проблемы как технических аспектов способа, так и аспектов качества. Настоящее изобретение также относится к реакции при пониженных температурах, контролируемой в отношении образования примесей, к очищающей фильтрации для удаления неорганических примесей и очистке сырого деферазирокса для удаления нежелательных примесей и цвета.
Настоящее изобретение относится к способу получения кристаллической формы деферазирокса, имеющей чистоту по меньшей мере 99%, причем указанный способ включает следующие операции:
a) осуществление взаимодействия салицилоилхлорида формулы 2 с салициламидом формулы 3 в присутствии катализатора с получением 2-(2-гидроксифенил)бенз[е]оксазин-4-она формулы 4, содержащего менее 1% нециклических примесей;
b) осуществление взаимодействия салицилоилхлорида формулы 2 с салициламидом формулы 3 в присутствии катализатора и основания металла с получением соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она с металлом формулы 4, содержащей менее 1% нециклических примесей;
c) осуществление взаимодействия 2-(2-гидроксифенил)бенз[е]оксазин-4-она со стадии (a) или соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она с металлом со стадии (b) с гидразинбензойной кислотой формулы 5 с получением получистой формы деферазирокса и
d) очистку получистой формы деферазирокса в растворителе с получением кристаллической формы деферазирокса, имеющей чистоту по меньшей мере 99%.
В одном из вариантов осуществления настоящего изобретения катализатор представляет собой катализатор фазового переноса.
В другом варианте осуществления настоящего изобретения катализатор фазового переноса представляет собой галогенид четвертичного аммония, выбранный из группы, включающей галогенид тетраалкиламмония и галогенид бензилтриалкиламмония или их комбинацию.
В еще одном варианте осуществления настоящего изобретения галогенид тетраалкиламмония выбирают из группы, включающей бромид тетрабутиламмония, бромид тетраэтиламмония, хлорид тетрабутиламмония и иодид тетрабутиламмония или любую их комбинацию; и где галогенид бензилтриалкиламмония представляет собой бромид бензилтриалкиламмония.
В еще одном варианте осуществления настоящего изобретения катализатор используют в количестве от приблизительно 0,001 до приблизительно 2 эквивалентов.
В еще одном варианте осуществления настоящего изобретения основание металла выбирают из группы, включающей основание щелочного металла и основание щелочноземельного металла или их комбинацию.
В еще одном варианте осуществления настоящего изобретения металл представляет собой литий, натрий, калий, магний и кальций или любую их комбинацию.
В еще одном варианте осуществления настоящего изобретения основание щелочного металла или щелочноземельного металла выбирают из группы, включающей гидроксид лития, гидроксид натрия, метоксид натрия, гидроксид калия и трет-бутоксид калия или любую их комбинацию.
В еще одном варианте осуществления настоящего изобретения нециклическая примесь представляет собой 2-гидрокси-N-(2-гидроксибензоил)бензамид (бис-салициламид) формулы 6.
В еще одном варианте осуществления настоящего изобретения взаимодействие на стадии (a), т.е. взаимодействие салицилоилхлорида формулы 2 с салициламидом формулы 3 в присутствии катализатора с получением 2-(2-гидроксифенил)бенз[е]оксазин-4-она формулы 4, содержащего менее 1% нециклической примеси, включает следующие операции:
a) добавление салициламида и катализатора к салицилоилхлориду в растворителе с получением твердой массы;
b) нагревание твердой массы до температуры от приблизительно 90°C до приблизительно 130°C в течение периода времени от приблизительно 3 часов до приблизительно 5 часов, затем охлаждение массы до температуры менее приблизительно 40°C с последующим перемешиванием с получением осадка; и
c) промывка осадка растворителем и сушку с получением 2-(2-гидроксифенил)бенз[е]оксазин-4-она формулы 4, содержащего менее 1% нециклической примеси.
В еще одном варианте осуществления настоящего изобретения взаимодействие на стадии (b), т.е. взаимодействие салицилоилхлорида формулы 2 с салициламидом формулы 3 в присутствии катализатора и основания металла с получением соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она с металлом формулы 4, содержащей менее 1% нециклической примеси, включает следующие операции:
a) добавление салициламида и катализатора к салицилоилхлориду в растворителе с получением твердой массы;
b) нагревание твердой массы до температуры от приблизительно 90°C до приблизительно 130°C в течение периода времени от приблизительно 3 часов до приблизительно 5 часов, затем охлаждение массы до температуры менее приблизительно 40°C с последующим добавлением растворителя с получением раствора;
c) фильтрование раствора и добавление основания металла к фильтрату, затем перемешивание с получением осадка; и
d) промывка осадка растворителем и сушку с получением соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она с металлом формулы 4, содержащей менее 1% нециклической примеси.
В еще одном варианте осуществления настоящего изобретения взаимодействие на стадиях (a) и (b) основного способа, как описано выше, осуществляют при температуре от приблизительно 35°C до приблизительно 170°C.
В еще одном варианте осуществления настоящего изобретения взаимодействие на стадии (c), т.е. взаимодействие 2-(2-гидроксифенил)бенз[е]оксазин-4-она со стадии (a) или соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она с металлом со стадии (b) с гидразинбензойной кислотой формулы 5 с получением получистой формы деферазирокса, включает следующие операции:
a) добавление гидразинбензойной кислоты к 2-(2-гидроксифенил)бенз[е]оксазин-4-ону формулы 4 и кипячение реакционной смеси с обратным холодильником в течение периода времени от приблизительно 1 часа до приблизительно 3 часов, затем охлаждение до температуры менее приблизительно 40°C с получением осадка; и
b) промывка осадка растворителем и сушку с получением получистой формы деферазирокса.
В еще одном варианте осуществления настоящего изобретения взаимодействие на стадии (c) основного способа включает следующие операции:
a) добавление гидразинбензойной кислоты к соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она с металлом формулы 4 и кипячение реакционной смеси с обратным холодильником в течение периода времени от приблизительно 1 часа до приблизительно 3 часов, затем охлаждение до температуры менее приблизительно 40°C с получением осадка; и
b) промывка осадка растворителем с получением твердой массы и суспендирование твердой массы в растворителе, затем подкисление до значения pH от приблизительно 3 до приблизительно 5 с получением второго осадка; и
c) фильтрование и сушку осадка с получением получистой формы деферазирокса.
В еще одном варианте осуществления настоящего изобретения подкисление осуществляют кислотами, выбранными из группы, содержащей соляную кислоту и бромисто-водородную кислоту или их комбинацию.
В еще одном варианте осуществления настоящего изобретения очистка на стадии (d), т.е. очистка получистой формы деферазирокса в растворителе с получением кристаллической формы деферазирокса, имеющей чистоту по меньшей мере 99%, включает следующие операции:
a) растворение получистой формы деферазирокса в растворителе при температуре кипения с получением реакционной массы;
b) добавление в реакционную массу угля с последующим перемешиванием в течение периода времени от приблизительно 30 минут до приблизительно 60 минут с получением раствора;
c) фильтрование раствора, затем охлаждение с получением осадка;
d) повторное перемешивание раствора в течение периода времени от приблизительно 30 минут до приблизительно 90 минут с получением твердого вещества; и
e) фильтрование и промывку твердого вещества растворителем, затем сушку с получением кристаллической формы деферазирокса, имеющей чистоту по меньшей мере 99%.
В еще одном варианте осуществления настоящего изобретения очистка на стадии (d) основного способа включает следующие операции:
a) растворение получистой формы деферазирокса в растворителе при температуре кипения с получением реакционной массы;
b) добавление в реакционную массу угля с последующим перемешиванием в течение периода времени от приблизительно 30 минут до приблизительно 60 минут с получением раствора;
c) фильтрование раствора, затем охлаждение с получением осадка;
d) повторное перемешивание раствора в течение периода времени от приблизительно 30 минут до приблизительно 90 минут с получением твердого вещества; и
e) фильтрование и промывку твердого вещества растворителем, затем сушку с получением кристаллической формы деферазирокса, имеющей чистоту по меньшей мере 99%.
В еще одном варианте осуществления настоящего изобретения растворитель выбирают из группы, включающей дихлорметан, дихлорэтан, хлороформ, метанол, этанол, изопропанол, толуол, ксилол, тетрагидрофуран, диметилформамид, этилацетат, изопропилацетат, диэтиловый эфир, диизопропиловый эфир, метилтретбутиловый эфир, петролейный эфир, гексаны, гептаны и воду или любую их комбинацию.
В еще одном варианте осуществления настоящего изобретения кристаллическая форма деферазирокса представляет собой полиморфную форму I деферазирокса, имеющую значения 2θ 6.6, 10.0, 10.6, 20.3, 23.1, 25.7 и 26.2±0.2 градусов.
Настоящее изобретение относится к 2-(2-гидроксифенил)бенз[е]оксазин-4-ону - соединению формулы 4 или к его соли с металлом, которые содержат менее 1% нециклической примеси.
В варианте осуществления настоящего изобретения нециклическая примесь представляет собой 2-гидрокси-N-(2-гидроксибензоил)бензамид (бис-салициламид) формулы 6.
В другом варианте осуществления настоящего изобретения соль с металлом представляет собой натриевую соль 2-(2-гидроксифенил)бенз[е]оксазин-4-она.
В еще одном варианте осуществления настоящего изобретения натриевая соль 2-(2-гидроксифенил)бенз[е]оксазин-4-она характеризуется ДРГ, имеющей значения 29 приблизительно: 6.02, 6.90, 7.18, 10.80, 11.29, 12.02, 12.73, 13.30, 13.98, 14.49, 16.29, 17.13, 18.06, 18.99, 19.42, 19.97, 20.97, 21.63, 22.50, 22.93, 23.48, 24.26, 24.89, 26.38, 26.75, 27.47, 27.95, 28.42, 29.22, 29.80, 31.57, 34.88 и 40.06±0.2 градусов.
В варианте осуществления настоящего изобретения салицилоилхлорид (формула 2) получают из салициловой кислоты путем реакции с хлорангидридом, выбранным из группы, включающей тионилхлорид, оксалоилхлорид, фосфорилхлорид и пентахлорид фосфора или любую их смесь, в подходящем растворителе, выбранным из группы, включающей толуол, ксилол, тетрагидрофуран и диметилформамид или любую их смесь.
В другом варианте осуществления настоящего изобретения полученный салицилоилхлорид (формула 2) подвергают взаимодействию с салициламидом (формула 3) в присутствии стабилизатора реакции, который представляет собой катализатор фазового переноса, с получением 2-(2-гидроксифенил)бенз[е][1,3]оксазин-4-она (формула 4) с максимальным превращением, имеющим минимальное содержание побочного продукта (формула 6) в сырой реакционной смеси.
В одном варианте осуществления катализатор фазового переноса представляет собой галогенид четвертичного аммония, выбранный из группы, включающей галогенид тетраалкиламмония и галогенид бензилтриалкиламмония или их комбинацию. В одном варианте осуществления галогенид тетраалкиламмония выбирают из группы, включающей бромид тетрабутиламмония, бромид тетраэтиламмония, хлорид тетрабутиламмония и иодид тетрабутиламмония или любую их комбинацию. В другом варианте осуществления галогенид бензилтриалкиламмония представляет собой бромид бензилтриалкиламмония.
В одном из вариантов осуществления настоящего изобретения катализатор фазового переноса используют в соотношениях от каталитических до стехиометрических, которые стабилизируют реакцию и контролируют образование бис-салициламида. Взаимодействие осуществляют в подходящем растворителе, выбранном из группы, включающей толуол, ксилолы, ТГФ, ДМФ и дихлорметан или любую их смесь, или возможно взаимодействие осуществляют в чистом виде (без растворителя). Температуру реакции поддерживают в диапазоне 35-170°C, более предпочтительно 65-130°C и наиболее предпочтительно 75-120°C. Кроме того, использование растворителя в этой реакции позволяет промывать осадок для удаления нежелательных примесей. Извлечение продукта формулы 4 является значительно более высоким, чем в известном способе.
В одном из вариантов осуществления настоящего изобретения реакционную смесь, в которой получен продукт формулы 4, оставляют осаждаться, осадок выделяют в виде твердого вещества с лучшим качеством, имеющего содержание побочного продукта формулы 6 приблизительно <1%, более предпочтительно <0,5%, наиболее предпочтительно <0,2%. При желании, промежуточное соединение формулы 4 также выделяют в виде раствора в органических растворителях, выбранных из группы, включающей дихлорметан, дихлорэтан и хлороформ или любую их смесь, и затем переносят в следующую реакцию.
В еще одном варианте осуществления настоящего изобретения промежуточное соединение формулы 4 или натриевую соль промежуточного соединения формулы 4 получают за одну операцию следующим образом: к салициловой кислоте добавляют салициламид и бромид тетрабутиламмония в смеси диметилформамида и толуола, и тионилхлорид, и осуществляют взаимодействие до завершения реакции. После завершения реакции летучие соединения отгоняют из реакционной смеси. Добавляют изопропанол, и твердое вещество отделяют фильтрованием. В случае натриевой соли: после завершения реакции к реакционной смеси добавляют метоксид натрия. Полученную натриевую соль осаждают и выделяют фильтрованием.
В еще одном варианте осуществления настоящего изобретения после завершения реакции между салицилоилхлоридом и салициламидом в присутствии катализатора фазового переноса реакционную смесь растворяют в дихлорметане и добавляют метоксид натрия с получением соответствующей натриевой соли промежуточного соединения 2-(2-гидроксифенил)бенз[е][1,3]оксазин-4-она (формула 4). Полученную таким образом соль оставляют осаждаться, осадок затем выделяют фильтрованием. Обнаружено, что соль является чистой и кристаллической по природе. Полиморф натриевой соли формулы 4, обозначенный как форма X, характеризуется ДРГ, имеющей значения 2θ приблизительно 6.02, 6.90, 7.18, 10.80, 11.29, 12.02, 12.73, 13.30, 13.98, 14.49, 16.29, 17.13, 18.06, 18.99, 19.42, 19.97, 20.97, 21.63, 22.50, 22.93, 23.48, 24.26, 24.89, 26.38, 26.75, 27.47, 27.95, 28.42, 29.22, 29.80, 31.57, 34.88, 40.06±0.2.
В другом варианте осуществления настоящего изобретения для получения деферазирокса 2-(2-гидроксифенил)бенз[е][1,3]оксазин-4-он (формула 4) или соответствующую натриевую соль подвергают взаимодействию с 4-гидразинбензойной кислотой (формула 5) в подходящем растворителе, таком как C1-C4 спирт, выбранный из группы, включающей метанол, этанол, изопропанол, диметилформамид, дихлорметан и вода, или их смесь. Более предпочтительно, когда в качестве растворителей используют C1-C4 спирты или водные спирты, чистота и превращение являются более хорошими. Наиболее предпочтительно в качестве единственного растворителя для этой реакции использовать воду. Объемы используемых растворителей составляют 2-70 объемов, более предпочтительно 10-45 объемов и наиболее предпочтительно 20-35 объемов. Температуру реакции поддерживают в диапазоне 35-120°C, более предпочтительно 50-110°C, наиболее предпочтительно 65-105°C. После завершения реакции массу охлаждают до менее приблизительно 70°C, более предпочтительно до менее 45°C и наиболее предпочтительно до менее 30°C, для полного осаждения. Затем осажденное твердое вещество отделяют. Выделенный влажный осадок необязательно промывают и сушат отсасыванием. Выделенный материал является получистым в отношении хроматографической чистоты, составляющей приблизительно >99% чистоты, и внешний вид является серовато бледно-желтым.
В еще одном варианте осуществления настоящего изобретения реакция между натриевой солью 2-(2-гидроксифенил)бенз[е][1,3]оксазин-4-она и 4-гидразинбензойной кислотой (формула 5) приводит к получению натриевой соли деферазирокса. Затем ее суспендируют в органическом растворителе, таком как изопропанол, и подкисляют водной кислотой, такой как соляная кислота. Полученную смесь оставляют для осаждения и полученный деферазирокс выделяют в виде кристаллической полиморфной формы I.
В другом варианте осуществления настоящего изобретения получистый деферазирокс (возможно, высушенный осадок) очищают с получением по существу чистых бесцветных кристаллов деферазирокса с хроматографической чистотой >99,2%. Используемыми для очистки растворителями являются C1-C4 спирты, выбранные из группы, включающей метанол, этанол и изопропанол, низшие сложные эфиры, выбранные из группы, включающей этилацетат и изопропилацетат, низшие простые эфиры, выбранные из группы, включающей диэтиловый эфир, диизопропиловый эфир и метилтретбутиловый эфир, углеводороды, выбранные из группы, включающей петролейный эфир, гексаны и гептаны, или любая их смесь. Объемы используемых растворителей составляют от 5-75 объемов, более предпочтительно от 10-50 объемов и наиболее предпочтительно от 15-45 объемов. Температура процесса очистки составляет 20-120°C, более предпочтительно 40-90°C и наиболее предпочтительно 50-85°C. При необходимости, осуществляют обработку углем для удаления цветных примесей и фильтрование через целит для удаления нерастворимых частиц. Добавление растворителя в получистый деферазирокс выполняют порциями, либо непрерывно, либо однократно при соответствующей температуре. Очищенный раствор деферазирокса охлаждают для осаждения и дополнительно перемешивают для полного осаждения. Полученный твердый продукт отделяют фильтрованием или центрифугированием. При необходимости влажный осадок промывают и сушат отсасыванием под вакуумом. Выделенное вещество дополнительно сушат в вакууме при соответствующей температуре до содержания остаточных растворителей в соответствии с требованиями согласно ICH. Обнаружено, что конечный деферазирокс является по существу чистым с хроматографической чистотой >99,2%, более предпочтительно >99,5% и наиболее предпочтительно >99,7%. ДРГ выделенного деферазирокса имеет пики, относящиеся к форме I, приблизительно при значениях 2θ: 6.6, 10.0, 10.6, 20.3, 23.1, 25.7, 26.2±0.2 градусов.
Более полное понимание может быть получено со ссылкой на следующие конкретные примеры, которые приведены для целей иллюстрации и не предназначены для ограничения объема изобретения.
Примеры
Для определения качества продукта формулы 4 проводили экспериментальную процедуру, описанную в патенте US 6465504 B1: салицилхлорид получали из салициловой кислоты. К салицилоилхлориду добавляли салициламид, реакционную смесь нагревали выше 170°C и затем выдерживали в течение приблизительно 0,5 ч. Реакционную массу охлаждали до 70°C, добавляли этанол и затем охлаждали до комнатной температуры. Суспензию фильтровали, влажный осадок промывали этанолом и полученный материал сушили. (Извлечение: 80%, чистота: 74%, примесь формулы 6: 16,9%).
Пример 1: Получение салицилоилхлорида (Формула 2)
Тионилхлорид (0,65 л) медленно добавляют к салициловой кислоте (1 кг) в смеси толуол (10 л)/ДМФ (100 мл) и нагревают до приблизительно 70-75°C, одновременно перемешивая. Перемешивание продолжают в течение приблизительно 1 ч и летучие компоненты отгоняют с получением сиропа салицилоилхлорида (Формула 2) (1,1 кг).
Пример 2: Получение салицилоилхлорида (Формула 2)
Оксалоилхлорид (1 л) медленно добавляют к салициловой кислоте (1,2 кг) в смеси тетрагидрофуран (10 л)/ДМФ (100 мл) и нагревают до приблизительно 50-75°C, одновременно перемешивая. Перемешивание продолжают в течение приблизительно 1 ч и летучие компоненты отгоняют с получением сиропа салицилоилхлорида (Формула 2) (1,3 кг).
Пример 3: Типичное получение 2-(2-гидроксифенил)бенз[е]оксазин-4-она (Формула 4) с использованием катализатора фазового переноса
К салицилоилхлориду (Формула 2) (1 кг) в толуоле (10 л) добавляют салициламид (Формула 3) (1 кг) и катализатор фазового переноса (выбранный из перечня, представленного в этом описании) (100 г) и полученную массу нагревают до приблизительно 110°C в течение приблизительно 4 ч. Затем реакционную смесь охлаждают до приблизительно <40°C и продолжают перемешивание до полного осаждения. Затем к суспензии добавляют изопропанол (5 л) и перемешивают в течение приблизительно 1 ч с получением и выделением осажденного твердого вещества путем фильтрации. Полученное твердое вещество промывают изопропанолом (1 л) и материал сушат с получением соединения формулы 4 (1,3 кг), содержащего менее 0,3% примеси (Формула 6).
Пример 4: Получение 2-(2-гидроксифенил)бенз[е]оксазин-4-она (Формула 4)
К салицилоилхлориду (Формула 2) (110 г) в толуоле (1 л) добавляют салициламид (Формула 3) (119 г) и бромид тетрабутиламмония (1 г) и полученную массу нагревают до приблизительно 110°C в течение приблизительно 4 ч. Затем реакционную смесь охлаждают до приблизительно <40°C, и продолжают перемешивание до полного осаждения. Затем к суспензии добавляют изопропанол (500 мл) и перемешивают в течение приблизительно 30 мин с получением и выделением осажденного твердого вещества путем фильтрации. Полученное твердое вещество промывают изопропанолом (100 мл), и материал сушат с получением соединения формулы 4 (130 г), содержащего менее 0,5% примеси (Формула 6).
Пример 5: Получение натриевой соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она (натриевой соли формулы 4)
К салицилоилхлориду (Формула 2) (330 г) в толуоле (1.5 л) добавляют салициламид (формула 3) (359 г) и бромид тетрабутиламмония (3 г), и полученную массу кипятят с обратным холодильником в течение приблизительно 4 ч. Затем реакционную смесь охлаждают до комнатной температуры, добавляют дихлорметан (3 л) для экстрагирования растворимых в дихлорметане материалов, смесь фильтруют через целит для удаления нерастворимого материала. Затем к полученному фильтрату добавляют метоксид натрия (1,1 экв. и перемешивают до полного осаждения при комнатной температуре в атмосфере азота. Осажденное твердое вещество выделяют фильтрацией. Полученное твердое вещество промывают дихлорметаном (300 мл), и материал сушат с получением зеленовато-желтого твердого вещества (350 г) натриевой соли формулы 4.
Пример 6: Получение 4-[3,5-Бис(2-гидроксифенил)-[1,2,4]триазол-1-ил]бензойной кислоты (деферазирокса)
К приблизительно 100 г 2-(2-гидроксифенил)бенз[е]оксазин-4-она (Формула 4) в изопропаноле (1,5 л) добавляют приблизительно 70 г гидразинбензойной кислоты (Формула 5) и кипятят с обратным холодильником в течение приблизительно 2 ч. После завершения реакции реакционную смесь охлаждают до комнатной температуры до полного осаждения. Осажденное твердое вещество выделяют путем фильтрации. Полученное твердое вещество промывают изопропанолом (100 мл) и сушат в вакууме с получением получистого деферазирокса (Формула 1) (125 г) зеленовато-желтого цвета.
Пример 7: Получение 4-[3,5-Бис(2-гидроксифенил)-[1,2,4]триазол-1-ил]бензойной кислоты (деферазирокса) из натриевой соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она
К приблизительно 100 г натриевой соли 2-(2-гидроксифенил)бенз[е]оксазин-4-она в изопропаноле (1,5 л) добавляют приблизительно 70 г гидразинбензойной кислоты (Формула 5) и кипятят с обратным холодильником в течение приблизительно 2 ч. После завершения реакции реакционную смесь охлаждают до комнатной температуры до полного осаждения. Осажденное твердое вещество выделяют путем фильтрации. Полученное твердое вещество промывают изопропанолом (100 мл). Полученное твердое вещество суспендируют в изопропаноле и подкисляют до pH 4 с использованием соляной кислоты. Реакционную смесь перемешивают до полного осаждения с получением осадка, содержащего сырой деферазирокс. Сырой деферазирокс выделяют путем фильтрации, осадок сушат в вакууме с получением получистого деферазирокса (Формула 1) (125 г) зеленовато-желтого цвета.
Пример 8: Получение 4-[3,5-Бис(2-гидроксифенил)-[1,2,4]триазол-1-ил]бензойной кислоты (деферазирокса)
К приблизительно 200 г 2-(2-гидроксифенил)бенз[е]оксазин-4-она (Формула 4) в воде (3 л) добавляют приблизительно 150 г гидразинбензойной кислоты (Формула 5) и нагревают до 90°C в течение 2 ч. Суспензию охлаждают до комнатной температуры для полного осаждения с получением осадка. Затем осадок фильтруют и дважды промывают изопропанолом (250 мл), и сушат отсасыванием с получением деферазирокса (Формула 1) (310 г) бледно-желтого цвета.
Пример 9: Очистка 4-[3,5-Бис(2-гидроксифенил)-[1,2,4]триазол-1-ил]бензойной кислоты (деферазирокса)
Приблизительно 150 г получистой 4-[3,5-Бис(2-гидроксифенил)-[1,2,4]триазол-1-ил]бензойной кислоты (деферазирокса) растворяют в смеси изопропанол (3 л)/этилацетат (1 л) при температуре кипения с получением реакционной массы. В реакционную массу добавляют приблизительно 2 г угля и перемешивают в течение приблизительно 45 мин при кипячении с обратным холодильником. Полученный раствор фильтруют через слой целита, фильтрат охлаждают до комнатной температуры до полного осаждения. Раствор дополнительно перемешивают в течение приблизительно 1 ч с получением твердого вещества, которое выделяют фильтрацией. Полученное твердое вещество промывают изопропанолом (150 мл) и влажный осадок сушат до тех пор, пока не будет достигнуто содержание растворителя в соответствии с требованиями, с получением по существу чистой формы I деферазирокса (120 г) в виде бесцветного кристаллического вещества (чистота: 99,4%).
Пример 10: Очистка 4-[3,5-Бис(2-гидроксифенил)-[1,2,4]триазол-1-ил]бензойной кислоты (деферазирокса)
Приблизительно 275 г получистого деферазирокса бледно-желтого цвета в изопропаноле (2,5 л) и метилтретбутиловом эфире (MTBE) (50 мл) нагревают до кипения с обратным холодильником и добавляют 10 г угля, а затем медленно изопропанол (3 л). Нагревание продолжают до полного растворения деферазирокса. Полученный раствор фильтруют через картридж, затем раствор охлаждают для полного осаждения с получением кристаллического твердого вещества. Затем выделяют кристаллическое твердое вещество, содержащее по существу чистую форму I деферазирокса. Кристаллическое твердое вещество промывают изопропанолом (200 мл) и влажный осадок сушат отсасыванием, а затем дополнительно в вакууме до тех пор, пока не будет достигнуто содержание растворителя в соответствии с требованиями. Конечный материал представляет собой форму I в виде бесцветного кристаллического вещества, имеющего чистоту приблизительно >99,5% (243 г).
Пример 11: Очистка 4-[3,5-Бис(2-гидроксифенил)-[1,2,4]триазол-1-ил]бензойной кислоты (деферазирокса)
Приблизительно 100 г получистой 4-[3,5-Бис(2-гидроксифенил)-[1,2,4]триазол-1-ил]бензойной кислоты (деферазирокса) растворяют в изопропаноле (3 л) при температуре кипения с получением реакционной массы. В реакционную массу добавляют приблизительно 2 г угля и перемешивают в течение приблизительно 45 мин при кипячении с обратным холодильником. Раствор фильтруют через слой целита и растворитель отгоняют до минимального объема. Затем к раствору добавляют этилацетат (6 об.) и растворитель опять отгоняют до минимального объема (2 об.). Суспензию охлаждают до комнатной температуры для полного осаждения. Раствор дополнительно перемешивают в течение приблизительно 1 ч с получением твердого вещества, которое дополнительно выделяют путем фильтрации. Полученное твердое вещество промывают изопропанолом (150 мл) и влажный осадок сушат до тех пор, пока не будет достигнуто содержание растворителя в соответствии с требованиями, с получением по существу чистой формы I деферазирокса (120 г) в виде бесцветного кристаллического вещества (чистота: 99,4%).
Хроматографические способы и соединения, очищенные этими способами
Составы антитела
Способ снижения степени гликозилирования белков, способы и белки
Нацеленные/иммуномодулирующие слитые белки и способы их получения
Способ получения соединений 2-амино-1,3-пропандиола и их солей
Новый способ получения терифлуномида
Способ обнаружения нейтрализующих антител против рекомбинантного человеческого инсулина в сыворотке крови человека
Способ получения соединений 2-амино-1,3-пропандиола и их солей

