Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ОКСОТИОИМИДАЗОЛИДИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ В КАЧЕСТВЕ ИНГИБИТОРОВ АНДРОГЕННОГО РЕЦЕПТОРА
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к производным имидазолина, способам получения, фармацевтическим композициям, включающим их, и к их применению в качестве терапевтического агента, в частности в качестве ингибитора андрогенного рецептора (АР), а также в получении лекарственного препарата для лечения и предотвращения рака предстательной железы и т.п.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Рак предстательной железы является наиболее распространенной формой рака у мужчин на западе и третьей по значимости причиной смертности от рака по причине высокой заболеваемости. На основе глобальных оценок, рак предстательной железы насчитывает, в общем, более 900000 случаев (лишь немногим менее чем рак легкого и карцинома бронхов (1095200)) и около 260000 смертельных исходов (приблизительно 6% от общего количества смертей от рака) у мужчин в 2008 году. На ранней излечимой стадии рак предстательной железы in situ возможно диагностировать с помощью анализа простатоспецифического антигена (ПСА) и лечить путем хирургического удаления или радиотерапии. Большинство пациентов с раком предстательной железы отвечают на вид терапии, называемый андрогенная блокада (АБ) в конкретный период. Механизм АБ заключается в блокировании или снижении активности андрогенного рецептора (АР) путем снижения уровня андрогена, что, в свою очередь, вызывает подавление активации андрогенозависимого сигнального пути. Тем не менее практически у всех пациентов развивается "кастрационно-резистентный рак предстательной железы" (КР РПЖ).
Функциональный сигнальный путь андрогенного рецептора (АР) необходим для развития рака предстательной железы. Сигнальный путь АР отсутствует или ослаблен при синдроме нечувствительности к андрогенам и у пациентов со спинальной и бульбарной мышечной атрофией, что приводит к недоразвитию предстательных желез, в которых не образуются карциномы. Выяснилось, что экспрессия АР и его сигнальный путь остаются неизменными при развитии андрогеночувствительного рака в гормононезависимый рак предстательной железы (ГН РПЖ). Генетические и эпигенетические изменения означают, что опухоли предстательной железы продолжают зависеть от ростового сигнального пути АР, и они продолжают являться мишенями "гормональной" терапии. Развитие новых стратегий и новых лекарственных препаратов, которые более эффективно подавляют сигнальный путь АР, возможно, приведет к значительным клиническим преимуществам.
АР, ген которого расположен на хромосоме Xq11-12, является членом семейства рецепторов стероидных гормонов лиганд-активируемых ядерных транскрипционных факторов. АР включает четыре функциональных района: амино-концевой регуляторный домен (сайт AF-1), ДНК-связывающий домен, состоящий из двух "цинковых пальцев", шарнирного района, включающего сигнал ядерной локализации, а также карбокси-концевого лиганд-связывающего домена (сайт AF-2). АР не связанные с лигандами локализуются в основном в цитоплазме и связаны с белками теплового шока (БТШ) 90, 70, 56 и 23, которые стабилизируют третичную структуру АР в конформации, которая обеспечивает связывание с андрогеном. Андроген связывается с АР, что приводит к диссоциации БТШ и АР, вызывая димеризацию АР и последующее фосфорилирование тирозинкиназами, что в свою очередь приводит к перемещению АР в ядро. При попадании в ядро АР связывается с элементами андрогенного ответа, расположенными в промоторных и энхансерных областях генов-мишеней, что приводит к сопутствующему привлечению дополнительных регуляторных белков и образованию активного транскрипционного комплекса. Дополнительные регуляторные белки образуют мост между АР, комплексом преинициации и РНК полимеразой; коактиваторы способствуют транскрипции путем привлечения белковых комплексов к ДНК, что преобразует структуру хроматина в более транскрипционно-активную форму, а корепрессоры опосредуют конденсацию хроматина и подавляют транскрипцию.
Амплификация гена АР показана у 25% - 30% пациентов с ГН РПЖ, но присутствует с очень низкой частотой (1-2%) у пациентов с первичным раком предстательной железы, что указывает на то, что амплификация гена АР связана с развитием ГН РПЖ. Амплификация гена АР подчеркивает сильное селективное давление на непрерывный сигнальный путь АР, так как опухоли развиваются в среде лишенной андрогена и обеспечивает толчок для развития более эффективного ингибирования сигнала АР. Точечная мутация АР может приводить к изменению специфичности к лиганду таким образом, что АР могут быть активированы неандрогенными лигандами, такими как антиандрогены.
Бикалутамид (торговое название Касодекс) является наиболее часто применяемым антиандрогенным лекарственным препаратом, который ингибирует АР при гормонозависимом раке предстательной железы. Однако бикалутамид не способен эффективно ингибировать активность АР, если рак становится гормоннезависимым. Новый антагонист MDV-3100, разработанный Medivation Inc., способен эффективно подавлять сочетание андрогена и белка АР, блокировать перемещение АР в ядро и привлечение коактиватора комплекса лиганд-рецептор. До настоящего времени никем не было показано, что MDV-3100 мог бы стать агонистом и способствовать развитию рака при опухолях со сверхэкспрессированным АР. 21 мая 2012 года MDV-3100 вступил в стадию предварительной регистрации.
До настоящего времени ряд антагонистов АР раскрыт в некоторых заявках, включая WO 2010092371, WO 2011008543, WO 2012011840 и WO 2012015723 и т.п.
Несмотря на то что ряд антагонистов АР для лечения рака предстательной железы был раскрыт, все еще остается необходимость разработки новых соединений с более высокой эффективностью. После продолжительных усилий, настоящее изобретение обеспечивает соединения формулы (I) и обнаруживает, что соединения, имеющие данную структуру, обладают превосходными эффектами и действиями.
КРАТКОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к соединению формулы (I), таутомеру, мезомеру, рацемату, энантиомеру или диастереомеру, и их смеси, или его фармацевтически приемлемой соли, а также их метаболита, метаболического предшественника или пролекарства.
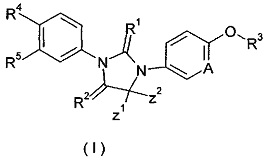
где
А представляет собой CR' или Ν;
R' представляет собой водород, галоген, алкил, циклоалкил или гетероциклил, при этом каждый указанный алкил, циклоалкил и гетероциклил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, гидрокси, алкила, алкокси, карбоксила и эфира карбоновой кислоты,
Ζ1 и Ζ2 каждый независимо представляет собой алкил, или Ζ1 и Ζ2 объединяются вместе с присоединенными атомами углерода с образованием одного циклоалкила или гетероциклила;
R1 и R2 каждый независимо выбирают из группы, включающей S или О;
R3 выбирают из группы, включающей алкил, алкенил, алкинил, циклоалкил, гетероциклил, арил, гетероарил и -S(O)mR6, при этом каждый алкенил, алкинил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, амино, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6, при этом алкил замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, амино, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6, при этом каждый циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, амино, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6,
R4 и R5 каждый независимо выбирают из группы, состоящей из циано, нитро, алкила, галогеналкила, гидрокси, галогена, алкокси и галогеналкокси,
R6 представляет собой водород, алкил, алкенил, алкинил, гидрокси, галоген, алкокси, циклоалкил, гетероциклил, арил или гетероарил, при этом каждый алкил, алкокси, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, гидрокси, амино, оксо, алкила, галогеналкила, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и эфира карбоновой кислоты,
R7 и R8 каждый независимо выбирают из группы, состоящей из водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, при этом каждый алкил, циклоалкил, гетероциклил, арил или гетероарил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, гидрокси, амино, оксо, алкила, галогеналкила, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила, гетероарила, карбоксила и эфира карбоновой кислоты, и
m представляет собой 0, 1 или 2.
В одном из вариантов осуществления изобретения соединение формулы (I) или таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или его фармацевтически приемлемая соль, выбраны из соединения (II) или таутомера, мезомера, рацемата, энантиомера диастереомера, или их смеси, или его фармацевтически приемлемой соли:
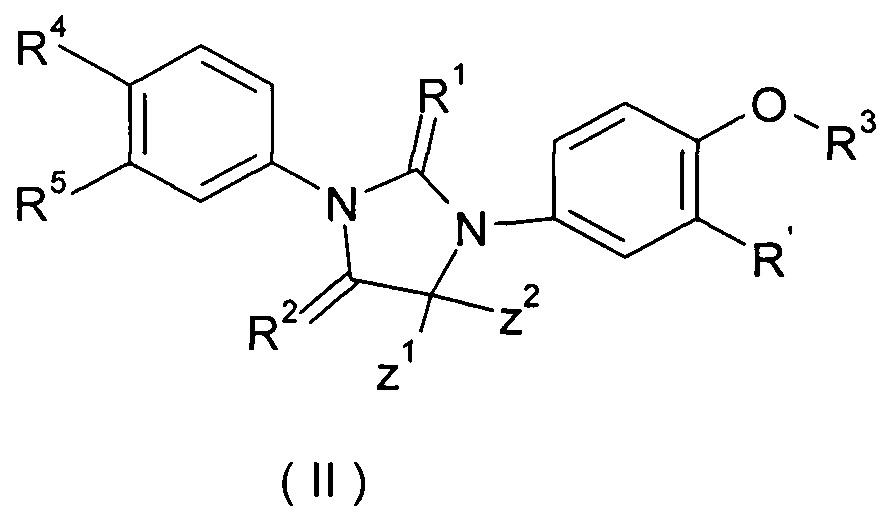
где R' представляет собой водород или галоген, Z1, Z2 и R1 - R5 являются такими, как определено в формуле (I).
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, А представляет собой N.
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, А представляет собой -CR', R' представляет собой галоген.
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, R1 представляет собой S.
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, R2 представляет собой О.
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, Ζ1 и Ζ2 каждый независимо представляет собой метил.
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, R4 представляет собой циано.
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, R5 представляет собой галогеналкил.
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, R5 представляет собой трифторметил.
В другом варианте осуществления изобретения в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, R3 выбирают из группы, состоящей из алкила, циклоалкила, гетероциклила, арила и гетероарила, при этом каждый циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, амино, алкила, -OR6, -C(O)NR7R8, -S(O)mR6, -C(O)R6 и -C(O)OR6, при этом алкил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, амино, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)NR7R8, -S(O)mR6 и -C(O)OR6, при этом каждый циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, амино, алкила, галогеналкила, гидроксиалкила, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)NR7R8, -S(O)mR6 и -C(O)OR6,
R6, R7 и R8 каждый независимо выбирают из группы, состоящей из водорода и алкила, при этом алкил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, гидрокси, амино, оксо, алкила и галогеналкила, и
m представляет собой 2.
В другом варианте осуществления изобретения, в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, R3 представляет собой алкил, при этом алкил замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, амино, циклоалкила, гетероциклила, арила, гетероарила, -OR6, -C(O)NR7R8, -S(O)mR6 и -C(O)OR6;
R6, R7 и R8 каждый независимо выбирают из группы, состоящей из водорода и алкила, при этом алкил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, гидрокси, амино, оксо, алкила и галогеналкила, и
m представляет собой 2.
В другом варианте осуществления изобретения, в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или его фармацевтически приемлемой соли, R3 представляет собой алкил, при этом алкил замещен одной или более гидроксигруппой.
В другом варианте осуществления изобретения, в соединении формулы (I) или таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, или их фармацевтически приемлемой соли, R3 представляет собой гетероциклил, при этом гетероциклил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, амино, алкила, -OR6, -C(O)NR7R8, -S(O)mR6, -C(O)R6 и -C(O)OR6,
R6, R7 и R8 каждый независимо выбирают из группы, состоящей из водорода и алкила, и
m представляет собой 2.
Типичные соединения по настоящему изобретению включают следующие соединения, но не ограничиваются ими:
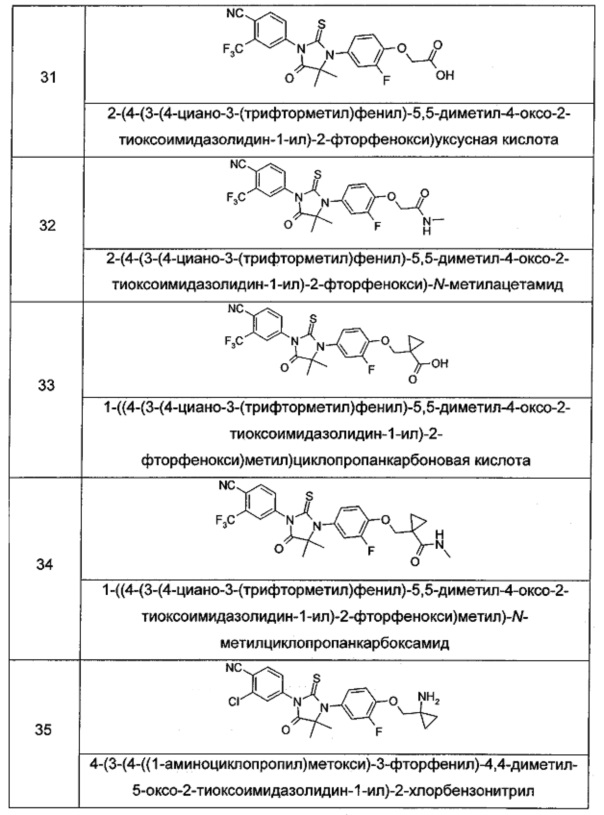
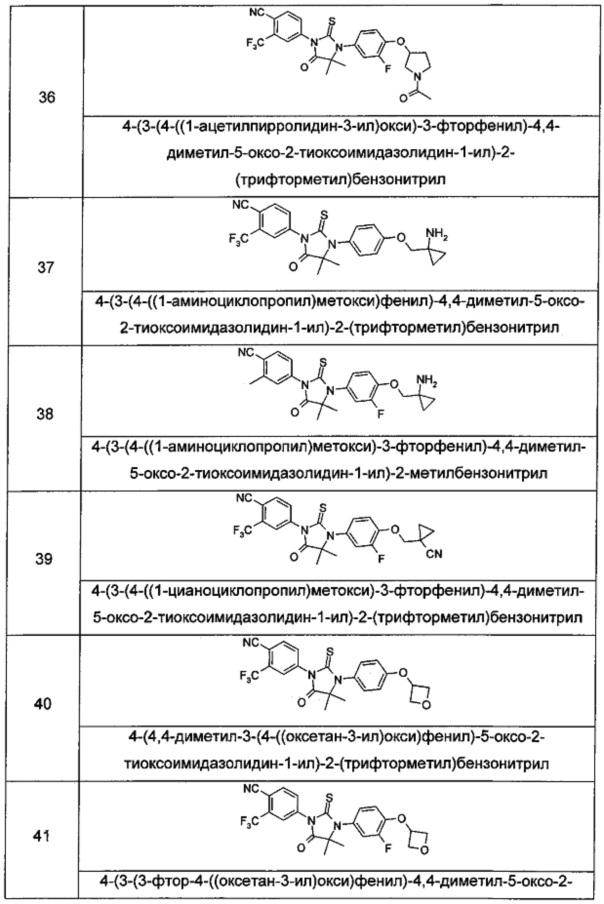
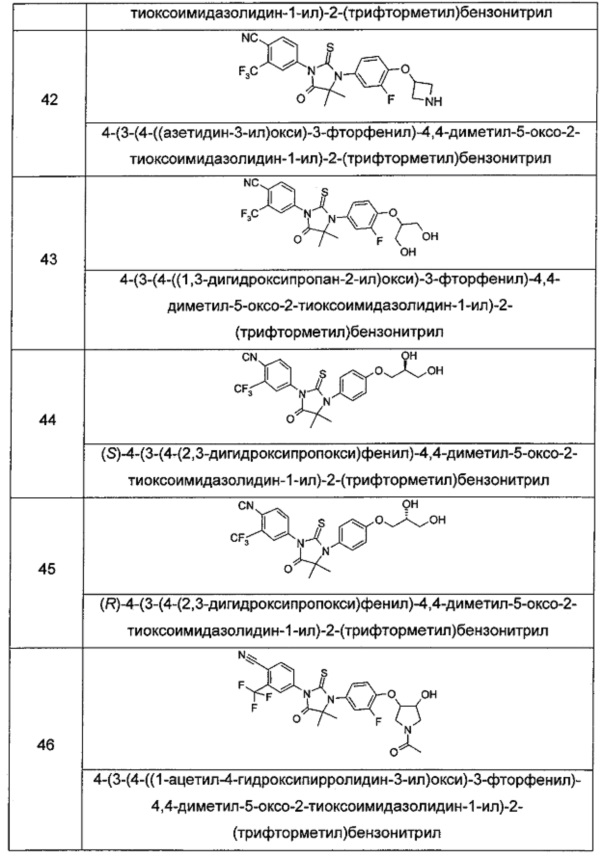
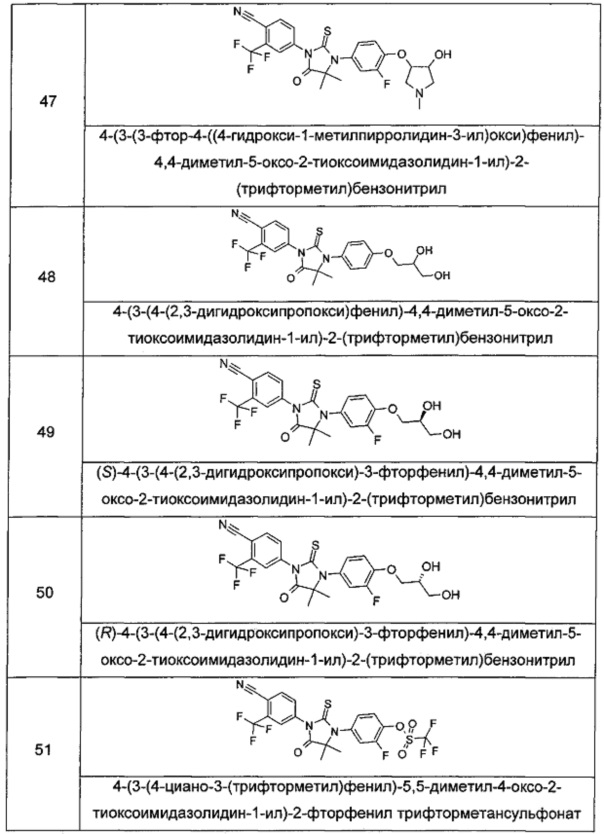
или таутомер, мезомер, рацемат, энантиомер, диастереомер, или их смесь, или их фармацевтически приемлемая соль.
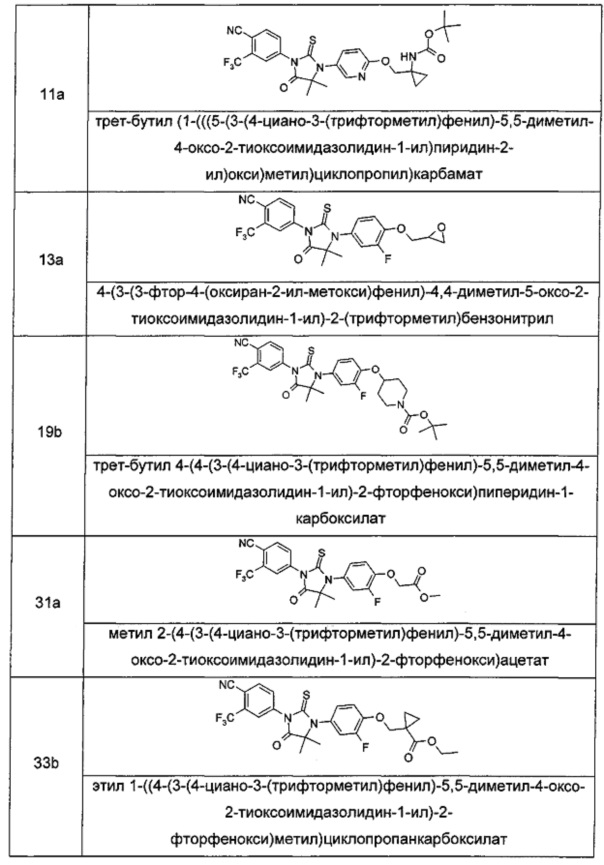
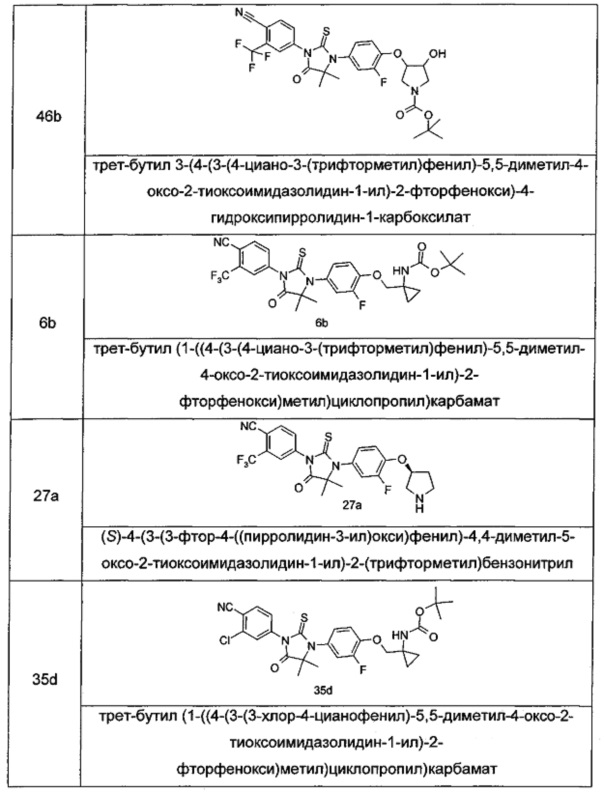
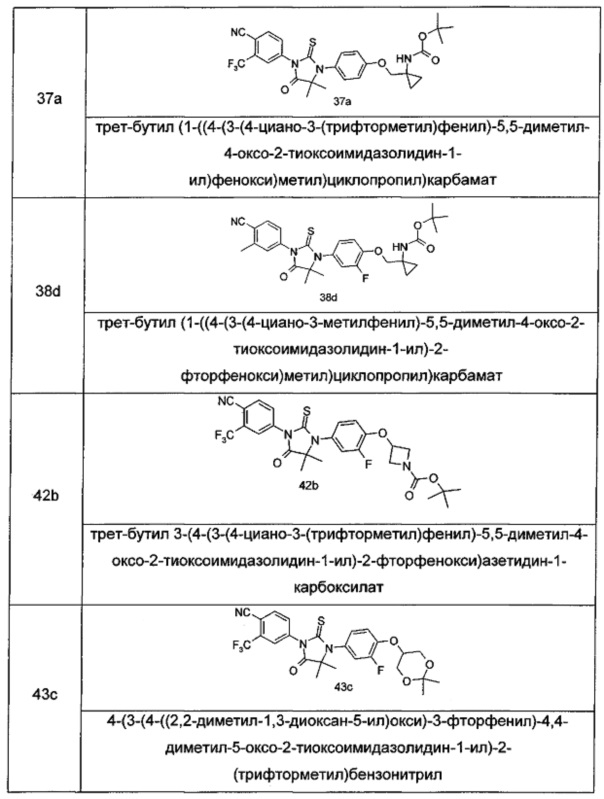
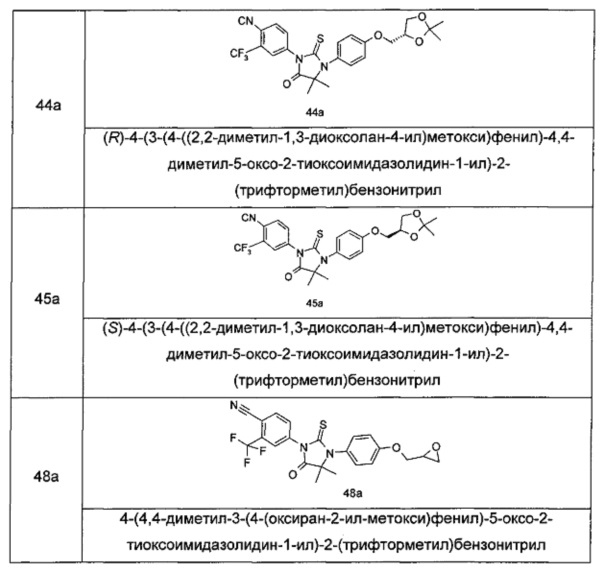
В другом аспекте настоящее изобретение направлено на обеспечение промежуточного продукта для синтеза соединений, описанных выше, например, 11а, 13а, 19b, 31а, 33b, 46b, 6b, 27а, 35d, 37а, 38d, 42b, 43с, 44а, 45а, 48а можно использовать в качестве промежуточного продукта для синтеза соответствующих соединений. Например, 11а можно использовать в качестве промежуточного продукта для синтеза соединения 11; 13а можно использовать в качестве промежуточного продукта для синтеза соединения 13, и так далее.
В другом аспекте настоящее изобретение обеспечивает способ получения соединения формулы (I), или таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или его фармацевтически приемлемой соли, включающий этап:
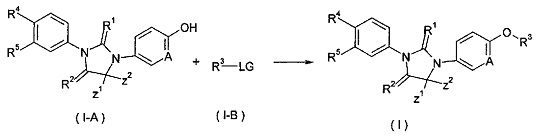
взаимодействия соединения формулы (1-А) с соединением формулы (1-В) в щелочных условиях с получением соединения формулы (I);
где LG представляет собой замещаемую группу, предпочтительно галоген или п-толуолсульфонилокси-; и Α, Ζ1, Ζ2, с R1 по R5 такие, как определено в формуле (I).
В другом аспекте, настоящее изобретение обеспечивает способ получения соединения формулы (I), или таутомера, мезомера, рацемата, энантиомера, диастереомера, или их смеси, или их фармацевтически приемлемой соли, включающий этап:
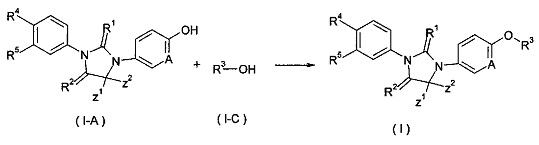
объединения соединения формулы (Ι-Α) с соединением формулы (I-C) в присутствии трифенилфосфина или три-н-бутилфосфина, азодикарбоксилата (предпочтительно 1,1'-(азодикарбонил)дипиперидина или диизопропил азодикарбоксилата) с получением соединения формулы (I);
где Α, Ζ1, Ζ2 и R1-R5 такие, как определено в формуле (I).
В другом аспекте, настоящее изобретение обеспечивает соединение формулы (IIA):
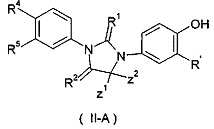
используемое в качестве промежуточного продукта для получения соединения формулы (II), где:
R' представляет собой водород, галоген, алкил, циклоалкил или гетероциклил, где каждый алкил, циклоалкил и гетероциклил необязательно замещен одной или более группой, выбранной из группы, состоящей из галогена, циано, гидрокси, алкила, алкокси, карбоксила и эфира карбоновой кислоты; R' представляет собой предпочтительно водород или галоген;
Z1 и Z2 каждый независимо представляет собой алкил; или Z1 и Z2 объединяются вместе с присоединенными атомами углерода с образованием одного циклоалкила или гетероциклила;
R1 и R2 каждый независимо выбирают из группы, состоящей из S или О; и
R4 и R5 каждый независимо выбирают из группы, состоящей из циано, нитро, алкила, галогеналкила, гидрокси, галогена, алкокси и галогеналкокси.
В другом аспекте настоящее изобретение обеспечивает способ получения соединения формулы (II) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, включающее этап:
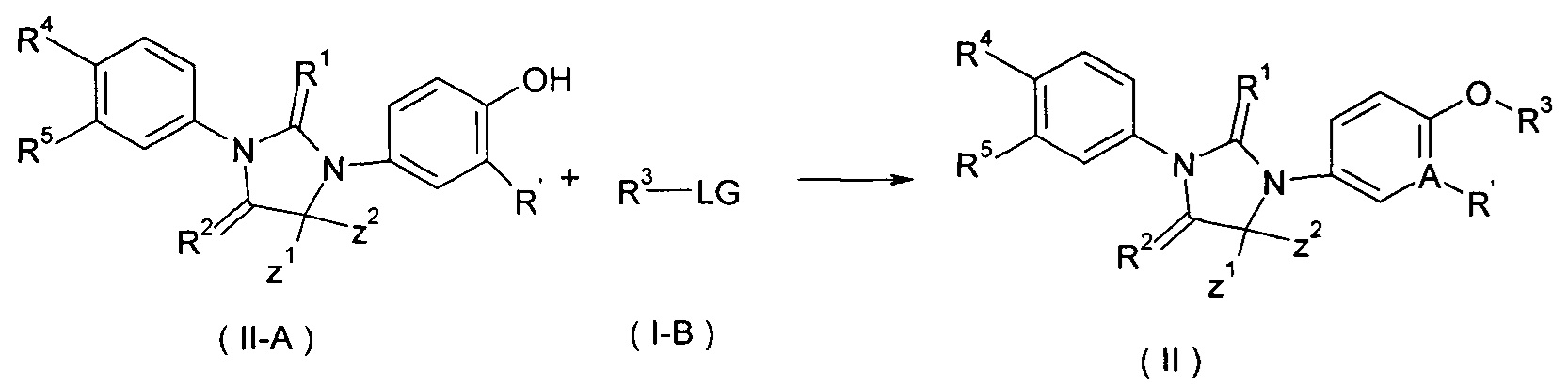
реакция соединения формулы (I-А) с соединением формулы (I-В) в щелочных условиях с получением соединения (II);
где LG представляет собой замещаемую группу, предпочтительно галоген или п-толуолсульфонилокси; и A, Z1, Z2 и R1 - R5 такие, как определено в формуле (I).
В другом аспекте настоящее изобретение обеспечивает способ получения соединения формулы (II) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, включающий этап:
объединения соединения формулы (II-А) и соединения формулы (I-С) в присутствии трифенилфосфина или три-н-бутилфосфина, азодикарбоксилата (предпочтительно 1,1'-(азодикарбонил)дипиперидина или диизопропил азодикарбоксилата) с получением соединения формулы (II);
где Α, Ζ1, Ζ2 и R1-R5 такие, как определено в формуле (I).
Настоящее изобретение также обеспечивает фармацевтическую композицию, включающую терапевтически эффективное количество соединения формулы (I) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, с фармацевтически приемлемым носителем, разбавителем или наполнителем.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции, для получения лекарственного препарата для регулирования активности андрогенного рецептора.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции, в получении лекарственного препарата для подавления активности андрогенного рецептора.
Настоящее изобретение также относится к соединению формулы (I) или таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции, для применения в качестве лекарственного препарата для регулирования активности андрогенного рецептора, предпочтительно для подавления андрогенного рецептора.
Настоящее изобретение также относится к способу регулирования активности андрогенного рецептора, предпочтительно, подавлению активности андрогенного рецептора, включающему этап введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции, для получения лекарственного препарата для лечения или предотвращения заболеваний или нарушений, опосредованных андрогенным рецептором, где нарушения или заболевания, опосредованные андрогенным рецептором, выбраны из группы, состоящей из рака предстательной железы, гиперплазии предстательной железы, гирсутизма, алопеции, нервной анорексии, рака молочной железы, угрей, мужской половой дисфункции, СПИДа и кахексии, предпочтительно рака молочной железы или рака предстательной железы, более предпочтительно рака предстательной железы, и наиболее предпочтительно гормонозависимого рака предстательной железы или гормононезависимого рака предстательной железы.
Настоящее изобретение также относится к способу лечения или предотвращения заболеваний или нарушений, опосредованных андрогенным рецептором, включающий этап введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции, где нарушения или заболевания, опосредованные андрогенным рецептором, выбраны из группы, состоящей из рака предстательной железы, гиперплазии предстательной железы, гирсутизма, алопеции, нервной анорексии, рака молочной железы, угрей, мужской половой дисфункции, СПИДа и кахексии, предпочтительно рака молочной железы или рака предстательной железы, более предпочтительно рака предстательной железы, и наиболее предпочтительно гормонозависимого рака предстательной железы или гормононезависимого рака предстательной железы.
Настоящее изобретение также относится к соединению формулы (I) или таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции, для применения в качестве лекарственного препарата для лечения или предотвращения нарушений или заболеваний, опосредованных андрогенным рецептором, где нарушения или заболевания, опосредованные андрогенным рецептором выбраны из группы, состоящей из рака предстательной железы, гиперплазии предстательной железы, гирсутизма, алопеции, нервной анорексии, рака молочной железы, угрей, мужской половой дисфункции, СПИДа и кахексии, предпочтительно рака молочной железы или рака предстательной железы, более предпочтительно рака предстательной железы, и наиболее предпочтительно гормонозависимого рака предстательной железы или гормононезависимого рака предстательной железы.
Настоящее изобретение также относится к применению соединения формулы (I) или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции, в получении лекарственного средства для мужской контрацепции.
Настоящее изобретение также относится к соединению формулы (I) или таутомеру, мезомеру, рацемату, энантиомеру, диастереомеру или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции, для применения в качестве лекарственного средства для мужской контрацепции.
Настоящее изобретение также относится к способу мужской контрацепции, включающему этап введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I), или таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, или включающей их фармацевтической композиции.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют значения, описанные ниже.
"Алкил" относится к насыщенной алифатической углеводородной группе, включающей прямую цепь С1-С20 и группы с разветвленной цепью. Предпочтительно алкильная группа представляет собой алкил, имеющий от 1 до 10 атомов углерода, более предпочтительно, алкил, имеющий от 1 до 6 атомов углерода. Типичные примеры включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, сек-бутил, н-пентил, 1,1-диметил пропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилфенил, 2-метил-3-этилфенил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилфенил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и изомеры их разветвленной цепи. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, типичные примеры включают, но не ограничиваются ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, сек-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метил пропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, и т.д.. Алкильная группа может быть замещенной или незамещенной. Если она является замещенной, то замещающая(ие) группа(ы) может быть замещена(ы) в любой возможной точке соединения, предпочтительно, замещающая(ие) группа(ы) представляет(ют) собой одну или более группу, независимо выбранные из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксогруппы, -OR6, -NR7R8, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6.
"Циклоалкил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода, более предпочтительно от 3 до 10 атомов углерода, и наиболее предпочтительно от 3 до 6 атомов углерода. Типичные примеры моноциклических циклоалкилов включают, но не ограничиваются ими, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил, и т.д.. Полициклический циклоалкил включает циклоалкил, имеющий спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком.
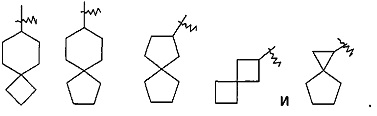
"Спироциклоалкил" относится к от 5- до 20-членной полициклической группе с кольцами, соединенными с помощью одного общего атома углерода (называемого спироатом), где одно или более кольцо может содержать одну или более двойную связь, но ни одно из колец не имеет полностью сопряженную систему пи-электронов. Предпочтительно спироциклоалкил является от 6- до 14-членным, и более предпочтительно от 7- до 10-членным. В соответствии с количеством общих спироатомов спироциклоалкил относится к моноспироциклоалкилу, диспироциклоалкилу или полиспироциклоалкилу, и предпочтительно относится к моноспироциклоалкилу или диспироциклоалкилу, более предпочтительно 4-членному/4-членному, 4-членному/5-членному, 4-членному/6-членному, 5-членному/5-членному, 5-членному/6-членному моноспироциклоалкилу. Типичные примеры спироциклоалкилов включают следующие группы, но не ограничиваются ими:
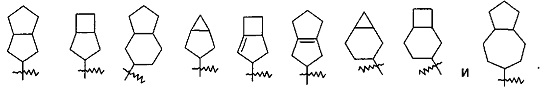
"Конденсированный циклоалкил" относится к от 5- до 20-членной полициклической углеводородной группе, где каждое кольцо в системе делит смежную пару атомов углерода с другим кольцом, где одно или более кольцо может содержать одну или более двойную связь, но ни одно из колец не имеет полностью сопряженную систему пи-электронов. Предпочтительно конденсированная циклоалкильная группа является от 6- до 14-членной, и более предпочтительно от 7- до 10-членной. В соответствии с количеством членных колец, конденсированный алкил относится к бициклическому, трициклическому, тетрациклическому или полициклическому циклоалкилу, и предпочтительно относится к бициклическому или трициклическому конденсированному циклоалкилу, и более предпочтительно 5-членному/5-членному или 5-членному/6-членному бициклическому конденсированному циклоалкилу. Типичные примеры конденсированных циклоалкилов включают следующие группы, но не ограничиваются ими:
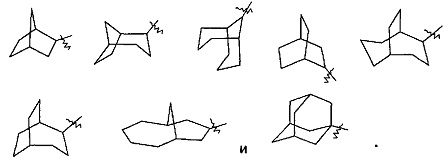
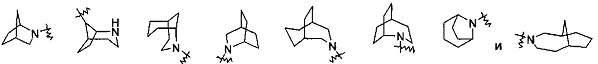
"Мостиковый циклоалкил" относится к от 5- до 20-членной полициклической углеводородной группе, где каждые два кольца в системе делят два несмежных атома углерода. Кольца могут иметь одну или более двойную связь, но ни одно из колец не имеет полностью сопряженную систему пи-электронов. Предпочтительно, мостиковый циклоалкил является от 6-до 14-членным, и более предпочтительно от 7- до 10-членным. В соответствии с количеством членных колец, мостиковый циклоалкил относится к бициклическому, трициклическому, тетрациклическому или полициклическому мостиковому циклоалкилу, и предпочтительно относится к бициклическому, трициклическому или тетрациклическому мостиковому циклоалкилу, и более предпочтительно к бициклическому или трициклическому мостиковому циклоалкилу. Типичные примеры включают следующие группы, но не ограничиваются ими:
Циклоалкил может быть сконденсированным с кольцом арила, гетероарила или гетероциклического алкила, где кольцо, связанное с родительской структурой, представляет собой циклоалкил. Типичные примера включают инданилацетат, тетрагидронафталин, бензоциклогептил и так далее, но не ограничиваются ими. Циклоалкил может быть необязательно замещенным или незамещенным. Если он является замещенным, то замещающая(ие) группа(ы) представляет(ют) собой одну или более группу, независимо выбранные из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксогруппы, -OR6, -NR7R8, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6.
"Гетероциклил" относится к от 3- до 20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более гетероатом, выбранный из группы, включающей N, О и S(O)m (где m представляет собой целое число, выбранное из от 0 до 2) в качестве кольцевых атомов, за исключением -O-O-, -O-S- или -S-S- в кольце, остальные кольцевые атомы представляют собой С. Предпочтительно гетероциклил имеет от 3 до 12 атомов, где от 1 до 4 атомов являются гетероатомами; более предпочтительно от 3 до 10 атомов; и наиболее предпочтительно от 4 до 6 атомов. Типичные примеры моноциклических гетероциклилов включают пирролидил, пиперидил, пиперазинил, морфолинил, сульфоморфолинил, гомопиперазинил, пиранил, тетрагидрофуранил, 1,1-диоксотетрагидрофуранил, оксетанил, азетидинил и так далее, но не ограничиваются ими. Полициклический гетероциклил включает гетероциклил, имеющий спирокольцо, конденсированное кольцо или кольцо с внутренним мостиком.
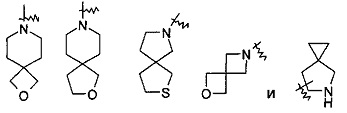
"Спирогетероциклил" относится к от 5- до 20-членному полициклическому гетероциклилу с кольцами, соединенными с помощью одного общего атома углерода (называемого спироатомом), где указанные кольца имеют один или более гетероатом, выбранный из группы, включающей Ν, О и S(O)m (где m представляет собой целое число, выбранное из от 0 до 2) в качестве кольцевых атомов, остальные кольцевые атомы представляют собой С, где одно или более кольцо может содержать одну или более двойную связь, но ни одно из колец не имеет полностью сопряженную систему пи-электронов. Предпочтительно спирогетероциклил является от 6- до 14-членным, и более предпочтительно от 7- до 10-членным. В соответствии с количеством общих спироатомов спирогетероциклил относится к моноспирогетероциклилу диспирогетероциклилу или полиспирогетероциклилу, и предпочтительно относится к моноспирогетероциклилу или диспирогетероциклилу, и более предпочтительно 4-членному/4-членному, 4-членному/5-членному, 4-членному/6-членному, 5-членному/5-членному, 5-членному/6-членному моноспирогетероциклилу. Типичные примеры спирогетероциклилов включают следующие группы, но не ограничиваются ими:
"Конденсированный гетероциклил" относится к от 5- до 20-членной полициклической гетероциклической групп, где каждое кольцо в системе делит смежную пару атомов углерода с другим кольцом, где одно или более кольцо может содержать одну или более двойную связь, но ни одно из колец не имеет полностью сопряженную систему пи-электронов, и где указанные кольца имеют один или более гетероатом, выбранный из группы, включающей N, О и S(O)p (где p представляет собой целое число, выбранное из от 0 до 2) в качестве кольцевых атомов, остальные кольцевые атомы представляют собой С. Предпочтительно, конденсированный гетероциклил является от 6- до 14-членным, и более предпочтительно от 7- до 10-членным. В соответствии с количеством членных колец, конденсированный гетероциклил относится к бициклическому, трициклическому, тетрациклическому или полициклическому гетероциклилу, предпочтительно относится к бициклическому или трициклическому конденсированному гетероциклилу, и более предпочтительно 5-членному/5-членному или 5-членному/6-членному бициклическому конденсированному гетероциклилу. Типичные примеры конденсированных гетероциклилов включают следующие группы, но не ограничиваются ими:
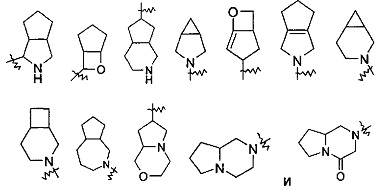
"Мостиковый гетероциклил" относится к от 5- до 14-членной полициклической гетероциклической алкильной группе, где каждые два кольца в системе делят два несмежных атома, кольца могут иметь одну или более двойную связь, но ни одно из колец не имеет полностью сопряженную систему пи-электронов, и кольца имеют один или более гетероатом, выбранный из группы, включающей Ν, О и S(O)m (где m представляет собой целое число, выбранное из от 0 до 2) в качестве кольцевых атомов, остальные кольцевые атомы представляют собой С. Предпочтительно, мостиковый гетероциклил является от 6- до 14-членным, и более предпочтительно от 7- до 10-членным. В соответствии с количеством членных колец, мостиковый гетероциклил относится к бициклическому, трициклическому, тетрациклическому или полициклическому мостиковому гетероциклилу, и предпочтительно относится к бициклическому, трициклическому или тетрациклическому мостиковому гетероциклилу, и более предпочтительно к бициклическому или трициклическому мостиковому гетероциклилу. Типичные примеры включают следующие группы, но не ограничиваются ими:
Циклоалкил может быть сконденсированным с кольцом арила, гетероарила или гетероциклического алкила, где кольцо, связанное с родительской структурой, представляет собой циклоалкил. Типичные примеры включают следующие группы, но не ограничиваются ими:
 , и т.д.
, и т.д.
Гетероциклил может быть необязательно замещенным или незамещенным. Если он является замещенным, то замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более группу, независимо выбранные из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксогруппы, -OR6, -NR7R8, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6.
"Арил" относится к от 6- до 14-членной группе, представляющей полностью углеродное моноциклическое кольцо или полициклическое конденсированное кольцо ("конденсированная" кольцевая система обозначает, что каждое кольцо в системе делит смежную пару углеродных атомов с другим кольцом в системе), и имеет полностью сопряженную систему пи-электронов. Предпочтительно арил является от 6- до 10-членным, более предпочтительно фенилом или нафтилом, и наиболее предпочтительно фенилом. Арил может быть сконденсированным с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с родительской структурой, представляет собой арил. Типичные примеры включают следующие группы, но не ограничиваются ими:
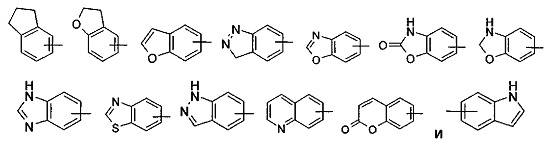
Арильная группа может являться необязательно замещенной или незамещенной. Если она является замещенной, то замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более группу, независимо выбранные из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, -OR6, -NR7R8, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6.
"Гетероарил" относится к арильной системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, включающей О, S и N в качестве кольцевых атомов и имеющей от 5 до 14 членов. Предпочтительно гетероарил является от 5- до 10-членным, более предпочтительно от 5- до 6-членным, и еще более предпочтительно фурилом, тиенилом, пиридилом, пирролилом, N-алкилпирролилом, пиримидинилом, пиразинилом, имидазолилом, тетразолилом и подобным. Гетероарил может быть сконденсирован с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с родительской структурой представляет собой гетероарил. Типичные примеры включаю следующие группы, но не ограничиваются ими:
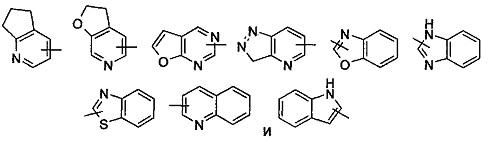
Гетероарильная группа может быть замещенной или незамещенной. Если она является замещенной, то замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более группу, независимо выбранные из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, -OR6, -NR7R8, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6.
"Алкенил" относится к алкилу как определено выше, который имеет по меньшей мере два атома углерода и по меньшей мере одну двойную связь углерод-углерод, предпочтительно С2-6 алкенил, и более предпочтительно С2-4 алкенил, например, винил, 1-пропенил, 2-пропенил, 1-, 2-, или 3-бутенил и т.д. Алкенильная группа может являться замещенной или незамещенной. Если она является замещенной, то замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более группу, независимо выбранные из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксогруппы, -OR6, -NR7R8, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6.
"Алкинил" относится к алкилу как определено выше, который имеет по меньшей мере два атома углерода и по меньшей мере одну тройную связь углерод-углерод, предпочтительно С2-6 алкинил, и более предпочтительно С2-4 алкинил, например, этинил, 1-пропинил, 2-пропинил, 2-, 2- или 3-бутилил и т.д. Алкинильная группа может Если она является замещенной, то замещающая(ие) группа(ы) предпочтительно представляет(ют) собой одну или более группу, независимо выбранные из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксогруппы, -OR6, -NR7R8, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6.
"Алкокси" относится как к группе -О-(алкил), так и к группе -О-(незамещенный циклоалкил), где алкил, циклоалкил являются такими как определено выше. Типичные примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и подобные, но не ограничиваются ими. Алкокси может быть замещенной или незамещенной. Если она является замещенной, то замещающая группа предпочтительно представляет собой одну или более группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилсульфо, алкиламино, галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, -OR6, -NR7R8, -C(O)NR7R8, -S(O)mR6, -C(O)R6, -OC(O)R6, -NR7C(O)R8, -NR7C(O)OR8 и -C(O)OR6.
"Гидроксиалкил" относится к -(алкил)-ОН, где алкил является таким, как определено выше.
"Галогеналкил" относится к алкилу, замещенному одним или более галогеном, где алкил является таким, как определено выше.
"Гидрокси" относится к -ОН группе.
"Галоген" относится к фтор-, хлор-, бром- или йод-.
"Амино" относится к-NH2 группе.
"Циано" относится к -CN группе.
"Нитро" относится к -NO2 группе.
"Оксогруппа" относится к=O группе.
"Карбоксил" относится к -С(O)ОН группе.
"Алкоксикарбонил" относится к группам -С(O)O(алкил) или (циклоалкил), где алкил и циклоалкил являются такими, как определено выше.
"Необязательно" или "необязательный" обозначает, что событие или обстоятельство, описанное далее, может произойти, но не в обязательном порядке, и описание включает случаи, в которых событие или обстоятельство может произойти или не произойти. Например, "гетероциклическая группа необязательно является замещенной алкилом" обозначает, что алкильная группа может присутствовать, но не в обязательном порядке, и описание включает случаи, когда гетероциклическая группа является замещенной алкилом, а также когда гетероциклическая группа не является замещенной алкилом.
"Замещенный" относится к одному или более атому водорода в группе, предпочтительно до 5, более предпочтительно от 1 до 3 атомов водорода, независимо замещенных соответствующим количеством заместителей. Само собой разумеется, что заместители существуют в их единственно возможном химическом положении. Специалист в данной области техники способен определить, возможно, или невозможно замещение без приложения чрезмерных экспериментальных или теоретических усилий. Например, сочетание амино или гидрокси группы, имеющей свободные атомы водорода и углерода, имеющие ненасыщенные связи (такие как олефиновые) может являться нестабильным.
"Фармацевтическая композиция" относится к смеси одного или более соединений, описанных в настоящем изобретении, или их физиологически/фармацевтически приемлемых солей или пролекарств и других химических компонентов, таких как физиологически/фармацевтически приемлемых носителей или наполнителей. Целью фармацевтической композиции является облегчение введения соединения в организм, что способствует абсорбции активного ингредиента и таким образом демонстрирует биологическую активность.
m и R6-R8 являются такими, как определено в соединении формулы (I).
Способ синтеза соединения по настоящему изобретению
Для того чтобы завершить цель изобретения, настоящее изобретение относится к следующему техническому решению, но не ограничивается им:
Способ получения соединения формулы (I) по настоящему изобретению, или таутомера, рацемата, энантиомера, диастереомера или их смеси, или его фармацевтически приемлемой соли, включающий этапы:
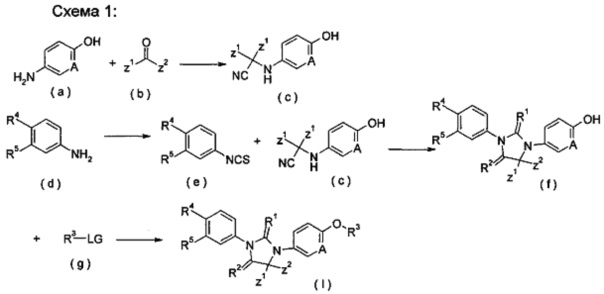
взаимодействие аминосоединения (а) с кетоном (b) и триметилсилилцианидом в растворителе в присутствии триметилсилил трифторметансульфоната с получением цианосоединения (с); реакция фениламинового соединения (d) с тиофосгеном в растворителе с получением изотиоцианатобензольного соединения (е); циклизация
изотиоцианатобензольного соединения (е) с цианосоединением (с) в растворителе и гидролиз полученного продукта в кислотных условиях с получением тиоимидазолидинового соединения (f); затем реакция тиоимидазолидинового соединения (f) с LG-замещенным соединением R3 (g) в растворителе в щелочных условиях с получением соединения формулы (I); где LG представляет собой замещаемую группу, предпочтительно галоген или п-толуолсульфонилокси; и Α, Ζ1, Z2 и R1-R5 являются такими, как определено в формуле (I).
Кислотные условия включают, но не ограничивается, трифторуксусную кислоту, муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту или метансульфоновую кислоту, предпочтительно соляную кислоту.
Щелочные условия включают органические щелочи и неорганические щелочи, где органические щелочи включают, без ограничения, триэтиламин, Ν,Ν-диизопропилэтиламин, Ν,Ν-диметилформамид, н-бутиллитий, трет-бутоксид калия или бромид тетрабутиламмония; и неорганические щелочи включают, но не ограничивается, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, предпочтительно карбонат натрия, карбонат калия или гидроксид калия.
Растворитель включает, но не ограничивается, уксусную кислоту, этанол, тетрагидрофуран, диметилсульфоксид, 1,4-диоксан, н-гексан, ацетон, метанол, воду, ацетонитрил, дихлорметан, метилбензол, Ν,Ν-диметилформамид или Ν,Ν-диметилацетамид.

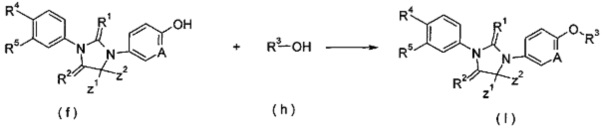
конденсация тиоксоимидазол иди нового соединения (f) с гидрокси-замещенным R3 соединением (h) в растворителе в присутствии трифенилфосфина или три-н-бутилфосфина, производных азодикарбоновой кислоты (предпочтительно 1,1'-(азодикарбонил)дипиперидина или диизопропилазодикарбоксилата) с получением соединения формулы (I), где А, Ζ1, Z2 и R1-R5 являются такими, как определено в формуле (I).
Щелочные условия включают органические щелочи и неорганические щелочи, где органические щелочи включают, но не ограничиваются, триэтиламин, Ν,Ν-диизопропилэтиламин, Ν,Ν-диметилформамид, н-бутиллитий, трет-бутоксид калия или бромид тетрабутиламмония; где неорганические щелочи включают, но не ограничиваются, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, предпочтительно карбонат натрия, карбонат калия или гидроксид калия.
Растворитель включает, но не ограничивается, уксусную кислоту, этанол, тетрагидрофуран, диметилсульфоксид, 1,4-диоксан, н-гексан, ацетон, метанол, воду, ацетонитрил, дихлорметан, метилбензол, Ν,Ν-диметилформамид или Ν,Ν-диметилацетамид.
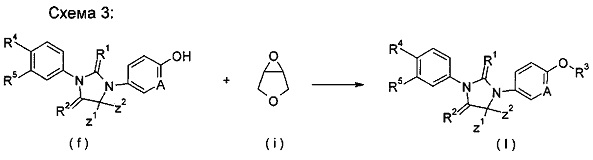
взаимодействие тиоксоимидазолидинового соединения (f) с 3,4-эпокситетрагидрофураном (i) в растворителе в щелочных условиях с получением соединения формулы (I); где Α, Ζ1, Z2, R1, R2, R4 и R5 являются такими, как определено в формуле (I), и R3 представляет собой 4-гидрокситетрагидрофуран.
Щелочные условия включают органические щелочи и неорганические щелочи, где органические щелочи включают, но не ограничиваются ими, триэтиламин, Ν,Ν-диизопропилэтиламин, Ν,Ν-диметилформамид, н-бутиллитий, трет-бутоксид калия или бромид тетрабутиламмония; и неорганические щелочи включают, без ограничения, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, предпочтительно карбонат натрия, карбонат калия или гидроксид калия.
Растворитель включает, но не ограничивается ими, уксусную кислоту, этанол, тетрагидрофуран, диметилсульфоксид, 1,4-диоксан, н-гексан, ацетон, метанол, воду, ацетонитрил, дихлорметан, метилбензол, Ν,Ν-диметилформамид или Ν,Ν-диметилацетамид.
Способ получения соединения формулы (II) по изобретению, или его таутомера, рацемата, энантиомера, диастереомера, или их смеси, или его фармацевтически приемлемой соли, включающий следующие этапы:
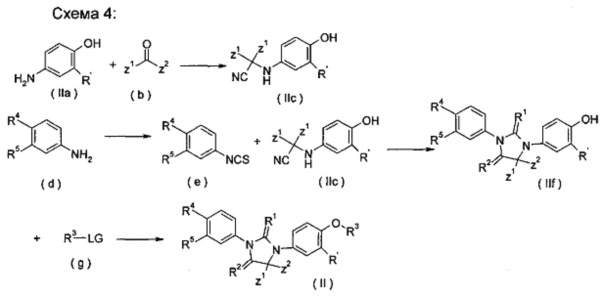
взаимодействие аминосоединения (IIa) с кетоном (b) и триметилсилилцианидом в растворителе в присутствии триметилсилил трифторметансульфоната с получением цианосоединения (IIc); взаимодействие фениламинового соединения (d) с тиофосгеном в растворителе с получением изотиоцианатобензольного соединения (е); циклизация изотиоцианатобензолового соединения (д) с цианосоединением (IIc) в растворителе и гидролиз полученного продукта в кислотных условиях с получением тиоксоимидазолидинового соединения (IIf); затем реакция тиоимидазолидинового соединения (IIf) с LG-замещенным соединением R3 (g) в растворителе в щелочных условиях с получением соединения формулы (II); где LG представляет собой замещаемую группу, предпочтительно галоген или п-толуолсульфонилокси; и Α, Ζ1, Z2, R' и R1-R5 являются такими, как определено в формуле (II).
Кислотные условия включают, но не ограничиваются ими, трифторуксусную кислоту, муравьиную кислоту, уксусную кислоту, соляную кислоту, серную кислоту или метансульфоновую кислоту, предпочтительно, соляную кислоту.
Щелочные условия включают органические щелочи и неорганические щелочи, где органические щелочи включают, но не ограничиваются, триэтиламин, Ν,Ν-диизопропилэтиламин, Ν,Ν-диметилформамид, н-бутиллитий, трет-бутоксид калия или бромид тетрабутиламмония; и неорганические щелочи включают, без ограничения, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, предпочтительно карбонат натрия, карбонат калия или гидроксид калия.
Растворитель включает, но не ограничивается, уксусную кислоту, этанол, тетрагидрофуран, диметилсульфоксид, 1,4-диоксан, н-гексан, ацетон, метанол, воду, ацетонитрил, дихлорметан, метилбензол, Ν,Ν-диметилформамид или Ν,Ν-диметилацетамид.
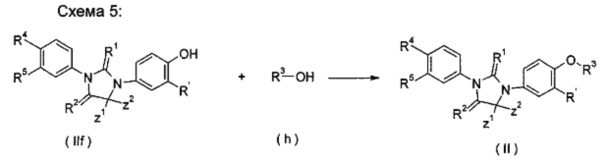
взаимодействие соединения тиоксоимидазолидинового соединения (IIf) с гидрокси-замещенным R3 соединением (h) в растворителе в присутствии трифенилфосфина или три-н-бутилфосфина, производных азодикарбоновой кислоты (предпочтительно 1,1'-(азодикарбонил)дипиперидина или диизопропилазодикарбоксилата) с получением соединения формулы (II), где А, Ζ1, Z2, R' и R1-R5 являются такими, как определено в формуле (II).
Щелочные условия включают органические щелочи и неорганические щелочи, где органические щелочи включают, но не ограничиваются ими, триэтиламин, Ν,Ν-диизопропилэтиламин, Ν,Ν-диметилформамид, н-бутиллитий, трет-бутоксид калия или бромид тетрабутиламмония; и неорганические щелочи включают, но не ограничиваются ими, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, предпочтительно карбонат натрия, карбонат калия или гидроксид калия.
Растворитель включает, но не ограничивается, уксусную кислоту, этанол, тетрагидрофуран, диметилсульфоксид, 1,4-диоксан, н-гексан, ацетон, метанол, воду, ацетонитрил, дихлорметан, метилбензол, Ν,Ν-диметилформамид или Ν,Ν-диметилацетамид.
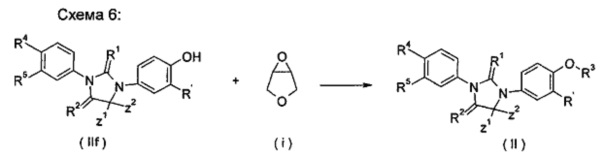
взаимодействие тиоксоимидазолидинового соединения (IIf) с 3,4-эпокситетрагидрофураном (i) в растворителе в щелочных условиях с получением соединения формулы (II); где Α, Ζ1, Z2, R', R1, R2, R4, R5 являются такими, как определено в формуле (II), и R3 представляет собой 4-гидрокситетрагидрофуран.
Щелочные условия включают органические щелочи w-неорганические щелочи, где органические щелочи включают, но не ограничиваются ими, триэтиламин, Ν,Ν-диизопропилэтиламин, Ν,Ν-диметилформамид, н-бутиллитий, трет-бутоксид калия или бромид тетрабутиламмония; и неорганические щелочи включают, но не ограничиваются ими, гидрид натрия, гидроксид натрия, гидроксид калия, карбонат натрия, бикарбонат натрия, карбонат калия, бикарбонат калия или карбонат цезия, предпочтительно карбонат натрия, карбонат калия или гидроксид калия.
Растворитель включает, без ограничения, уксусную кислоту, этанол, тетрагидрофуран, диметилсульфоксид, 1,4-диоксан, н-гексан, ацетон, метанол, воду, ацетонитрил, дихлорметан, метилбензол, Ν,Ν-диметилформамид или Ν,Ν-диметилацетамид.
Предпочтительные варианты осуществления изобретения
Следующие примеры служат для иллюстрации изобретения, но примеры не следует рассматривать как ограничивающие объем изобретения.
Если конкретные условия экспериментального способа не указаны в примерах настоящего изобретения, они в целом соответствуют традиционным условиям или условиям, рекомендованным производителем сырьевого материала или продукта. Реагенты без конкретного указанного источника являются коммерчески доступными, традиционными реагентами.
ПРИМЕРЫ
Структуры соединений определяли с помощью ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). ЯМР осуществляли с помощью аппарата Bruker AVANCE-400. Химические сдвиги ЯМР (δ) приведены в 10-6 (миллионные доли, м.д.). Растворителями являлись дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD) с тетраметилсиланом (ТМС) в качестве внутреннего стандарта.
МС осуществляли с помощью масс-спектрометра FINNIGAN LCQAd (ИЭР) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Высокоэффективную жидкостную хроматографию (ВЭЖХ) осуществляли с помощью спектрометра для жидкостной хроматографии с высоким давлением Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и спектрометра для жидкостной хроматографии с высоким давлением Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
Общий уровень подавления киназы и значения IC50 определяли с помощью NovoStar ELISA (BMG Co., Германия).
Для тонкослойной хроматографии на силикагеле (ТСХ) применяли пластины из силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластин, которые применяли в ТСХ, составлял от 0,15 мм до 0,2 мм, а размер пластин, которые применяли при очистке продукта, составлял от 0,4 мм до 0,5 мм.
При колоночной хроматографии в качестве носителя, как правило, применяли силикагель Yantai Huanghai от 200 до 300 меш.
Известный исходный материал согласно изобретению можно получить с помощью обычных методов синтеза в предшествующем уровне техники, или возможно приобрести в ABCR GmbH & Co. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc. или Dari chemical Company и т.п.
Реагент оксон представляет собой 2KHSO5⋅KHSO4⋅K2SO4.
Если не указано иное, следующие реакции проводили в атмосфере азота или аргона.
Термин "атмосфера азота" или "атмосфера аргона" обозначает, что реакционная колба оснащена 1 л баллоном азота или аргона.
Термин "атмосфера водорода" обозначает, что реакционная колба оснащена 1 л баллоном водорода.
Реакции гидрирования под давлением осуществляли с помощью спектрометра гидрирования Parr 3916EKX и генератора водорода QL-500.
В реакциях гидрирования, реакционную систему, как правило, вакуумировали и заполняли водородом, с трехкратным повторением указанной операции.
Если не указано иное, раствор, применяемый в следующих реакциях, является водным раствором.
Если не указано иное, температура реакции в следующих реакциях являлась комнатной температурой, и диапазон температуры составлял от 20°С до 30°С.
Реакционный процесс контролировали с помощью тонкослойной хроматографии (ТСХ), система проявляющих растворителей включала: А: смесь дихлорметана и метанола, В: н-гексан и этилацетат, С: петролейный эфир и этилацетат, D: ацетон. Отношение объема растворителя регулировали в соответствии с полярностью соединений.
Элюирующая система для очистки соединений путем колоночной хроматографии и тонкослойной хроматографии включала: А: система дихлорметан и метанол, В: система н-гексан и этилацетат, С: н-гексан и ацетон, D: н-гексан, Е: этилацетат. Объем растворителя регулировали в соответствии с полярностью соединений, и иногда также добавляли небольшое количество щелочного реагента, такого как триэтиламин, или кислотного реагента, такого как уксусная кислота
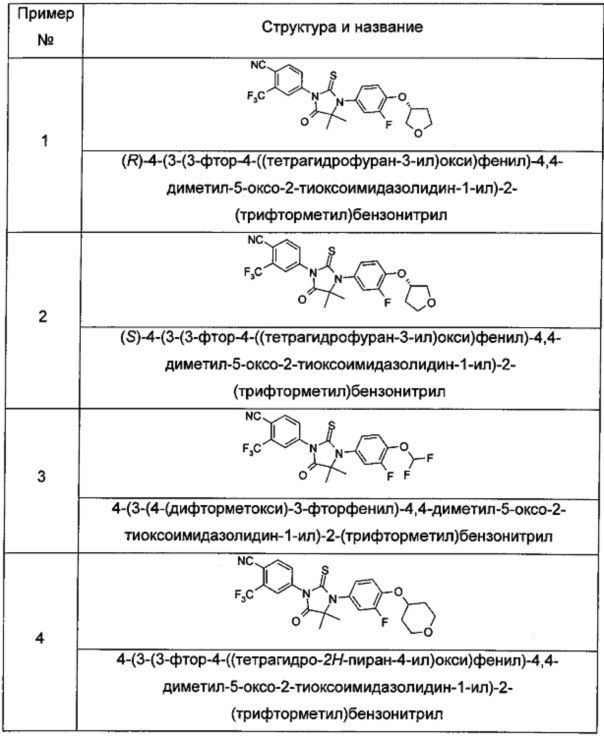
Пример 1
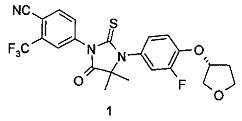
(R)-4-(3-(3-фтор-4-((тетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
Этап 1
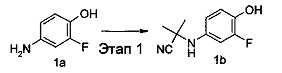
2-((3-Фтор-4-гидроксифенил)амино)-2-метилпропаннитрил 4-Амино-2-фторфенол 1а (6 г, 0,05 моль) растворяли в 90 мл смеси ацетона и дихлорметана (об./об.=1:2), с последующим добавлением триметилсилилцианида (9,4 мл, 0,07 моль) и триметилсилиловый эфир трифторметансульфоновой кислоты (0,4 мл, 2,30 ммоль). Реакционный раствор перемешивали в течение 2,5 часов. Реакционный раствор концентрировали при сниженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, 2-((3-фтор-4-гидроксифенил)амино)-2-метилпропаннитрила 1b (7,02 г, выход 76,6%).
МС m/z (ИЭР): 194,4 [М+1]. ИЭР - ионизация электрораспылением.
Этап 2
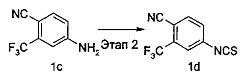
4-Изотиоцианато-2-(трифторметил)бензонитрил 4-Амино-2-(трифторметил)бензонитрил 1 с (10 г, 0,05 моль) растворяли в 60 мл смеси н-гексана и воды (об./об.=1:1), с последующим добавлением тиофосгена (4,6 мл, 0,06 моль) на ледяной бане (от 0 до 5°С). Затем ледяную баню удаляли, реакционный раствор нагревали до комнатной температуры и проводили реакцию в течение 12 часов, затем добавляли тиофосген (3 мл, 0,04 моль). После проведения реакции в течение 24 часов реакцию отстаивали и расслаивали, водную фазу экстрагировали смесью (50 мл) н-гексана и этилацетата (об./об.=10:1). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (50 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении с получением соединения, указанного в названии, 4-изотиоцианато-2-(трифторметил)бензонитрила 1d (10 г, выход 80,8%) в форме светло-коричневого маслянистого вещества.
Этап 3
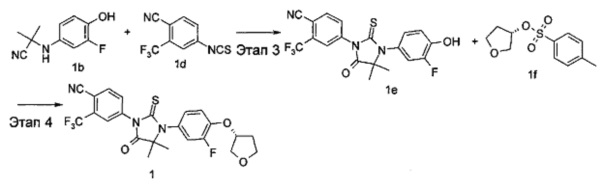
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 2-((3-Фтор-4-гидроксифенил)амино)-2-метилпропаннитрил 1b (3 г, 15 ммоль) и 4-изотиоцианато-2-(трифторметил)бензонитрил 1d (4,20 г, 18 ммоль) растворяли в 50 мл Ν,Ν-диметилацетамида и перемешивали в течение 3 часов.
Реакционный раствор смешивали с 30 мл метанола и 30 мл 2 Μ соляной кислоты, и нагревали до 70°С. После проведения реакции в течение 2 часов реакционный раствор охлаждали до комнатной температуры, смешивали с 50 мл воды и экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения 4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1е (6 г, выход 91,7%) в виде твердого белого вещества.
МС m/z(ИЭР): 424,3 [М+1]
Этап 4
(R)-4-(3-(3-Фтор-4-((тетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (80 мг, 0,19 ммоль) помещали в реакционную колбу с последующим добавлением (S)-тетрагидрофуран-3-ил-4-метилбензолсульфоната 1f (92 мг, 0,38 ммоль, полученного способом, раскрытым в патентной заявке США US 2003/153752 А1), карбоната цезия (186 мг, 0,57 ммоль) и дополнительно 1 мл Ν,Ν-диметилацетамида. Реакционный раствор нагревали до 50°С. После проведения реакции в течение 3 часов реакционный раствор охлаждали до комнатной температуры, смешивали с 15 мл Н2О и экстрагировали этилацетатом (15 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного в названии, (R)-4-(3-(3-Фтор-4-((тетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1 (63 мг, выход 67,6%) в виде белого твердого вещества.
МС m/z (ИЭР): 494,4 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,96-8,00 (m, 2Н), 7,84 (d, 1Н), 7,00-7,09 (m, 3Н), 5,00-5,03 (m, 1Н), 4,12414 (m, 2Н), 4,06-4,08 (m, 1Н), 3,96-4,01 (m, 1Н), 2,23-2,26 (m, 2Н), 1,59 (s, 6Н).
Пример 2
(S)-4-(3-(3-Фтор-4-((тетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
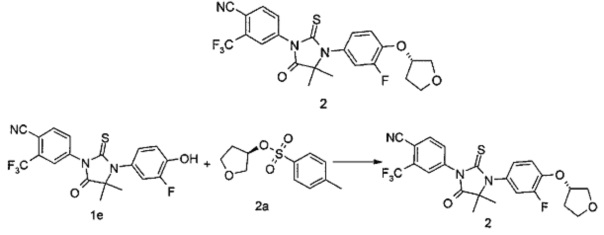
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (80 мг, 0,19 ммоль) помещали в реакционную колбу с последующим добавлением (R)-тетрагидрофуран-3-ил-4-метилбензолсульфоната 2а (92 мг, 0,38 ммоль, полученного хорошо известным способом, описанным в "Journal of Medicinal Chemistry, 2011, 54 (12), 4092-4108"), карбоната цезия (186 мг, 0,57 ммоль) и дополнительно 3 мл Ν,Ν-диметилацетамида. Реакционный раствор нагревали до 60°С. После проведения реакции в течение 2 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 15 мл Н2О и экстрагировали этилацетатом (25 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, (S)-4-(3-(3-Фтор-4-((тетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 2 (73 мг, выход 77,9%) в виде бледно-желтого твердого вещества.
МС m/z (ИЭР): 494,4 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,06 (m, 2Н), 7,84 (q, 1Н), 7,08 (d, 1Н), 7,04-7,03 (m, 2Н), 5,04-5,02 (m, 1Н), 4,07-3,94 (m, 4Н), 2,29-2,24 (m, 2Н), 1,60 (s, 6Н).
Пример 3
4-(3-(4-(Дифторметокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
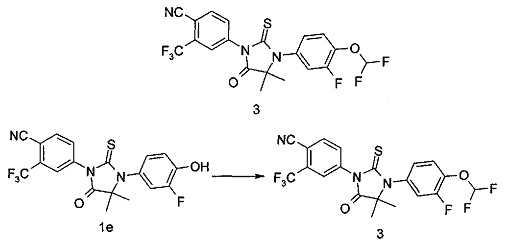
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (300 мг, 0,71 ммоль) растворяли в 10 мл смеси ацетонитрила и воды (об./об.=1:1) с последующим добавлением гидроксида калия (79 мг, 1,42 ммоль) и бромдифторметандиэтилфосфата (0,15 мл, 0,85 ммоль) последовательно на водяной бане (0°С). Реакционный раствор нагревали до комнатной температуры, перемешивали в течение 12 часов, затем смешивали с 20 мл воды и экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой D и тонкослойной хроматографии с элюирующей системой D последовательно с получением соединения, указанного в названии, 4-(3-(4-(дифторметокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 3 (90 мг, выход 26,8%) в виде белого твердого вещества.
MC m/z (ИЭР): 474,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,96-7,99 (m, 2Н), 7,84 (d, 1Н), 7,44 (s, 1Н), 7,13-7,20 (m, 2Н), 6,66 (t, 1Н), 1,62 (s, 6Н).
Пример 4
4-(3-(3-фтор-4-((тетрагидро-2Н-пиран-4-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
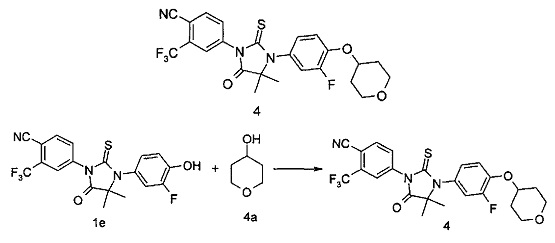
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением тетрагидро-2Н-пиран-4-ола 4а (29 мг, 0,28 ммоль, полученного способом, раскрытым в патентной заявке US 2011/71196 А1), 1,1'-(азодикарбонил)дипиперидина (95 мг, 0,38 ммоль), 10 мл метилбензола и три-н-бутилфосфина (94 мкл, 0,38 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 3 часов. Реакционный раствор концентрировали при сниженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((тетрагидро-2Н-пиран-4-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 4 (82 мг, выход 68,4%) в виде белого твердого вещества.
МС m/z (ИЭР): 508,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,96-8,00 (m, 2Н), 7,84 (d, 1Н), 7,03-7,13 (m, 3Н), 4,58-4,59 (m, 1Н), 4,00-4,03 (m, 2Н), 3,59-3,63 (m, 2Н), 2,05-2,08 (m, 2Н), 1,87-1,91 (m, 2Н), 1,59 (s, 6Н).
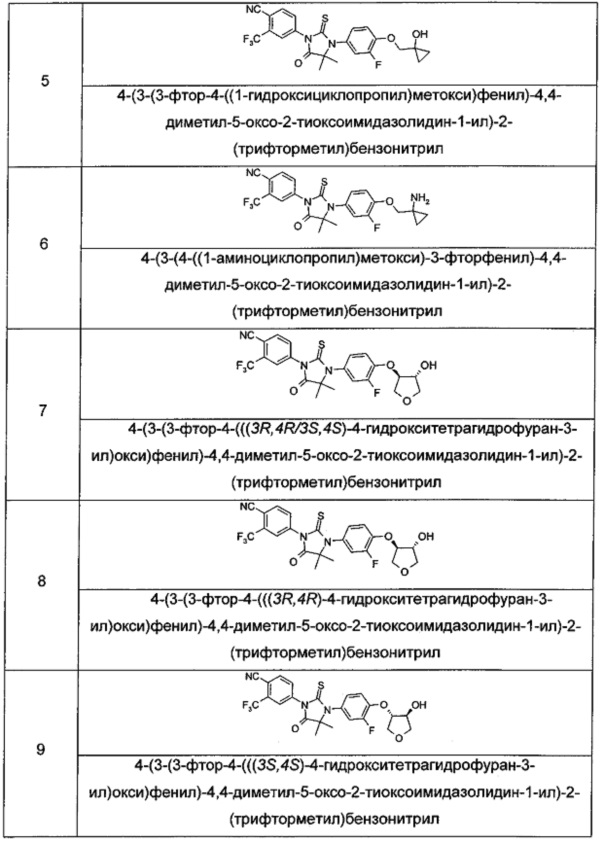
Пример 5
4-(3-(3-Фтор-4-((1-гидроксициклопропил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
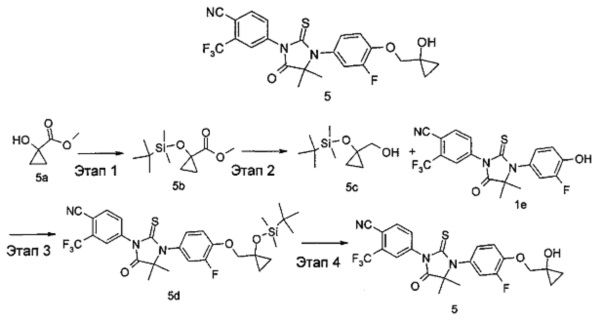
Этап 1
Метил-1-(трет-бутилдиметилсилилокси)циклопропанкарбоксилат
Метил-1-гидроксициклопропанкарбоксилат 5а (350 мг, 3,02 ммоль) растворяли в 30 мл дихлорметана с последующим добавлением трет-бутил диметилсилилхлорида (495 мг, 3,30 ммоль) и имидазола (306 мг, 4,49 ммоль). После проведения реакции в течение 12 часов, реакционную смесь смешивали с 20 мл дихлорметана и экстрагировали насыщенным раствором хлорида натрия (20 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении с получением грубого продукта, указанного в названии, метил-1-(трет-бутилдиметилсилилокси)циклопропанкарбоксилата 5b (600 мг, бесцветное маслянистое вещество), который непосредственно применяли на следующем этапе без дополнительной очистки.
Этап 2
(1-((трет-Бутилдиметилсилил)окси)циклопропил)метанол
Метил-1-(трет-бутилдиметилсилилокси)циклопропанкарбоксилат 5b (600 мг, 2,61 ммоль) растворяли в 30 мл тетрагидрофурана и охлаждали до -40°С на бане с сухим льдом и ацетоном, с последующим добавлением диизобутилалюминийгидрида (7,8 мл, 7,8 ммоль). Реакционный раствор перемешивали в течение 3 часов при -40°С с последующим добавлением 20 мл Н2О. Реакционный раствор нагревали до комнатной температуры и перемешивали в течение 10 минут для погашения реакции. Реакционную смесь фильтровали. Фильтрат экстрагировали этилацетатом (20 мл×2). Органические фазы совмещали, промывали насыщенным раствором хлорида натрия (20 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении с получением грубого продукта, указанного в названии, (1-((трет-бутилдиметилсилил)окси)циклопропил)метанола 5 с (500 мг, прозрачное маслянистое вещество), которое непосредственно применяли на следующем этапе без дополнительной очистки.
Этап 3
4-(3-(4-((1-((трет-Бутилдиметилсилил)окси)циклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
(1-((трет-Бутилдиметилсилил)окси)циклопропил)метанол 5 с (500 мг, 0,23 ммоль) помещали в реакционную колбу с последующим добавлением 4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1е (80 мг, 0,19 ммоль), 1,1'-(азодикарбонил)дипиперидина (77 мг, 0,31 ммоль), 5 мл метилбензола и три-н-бутилфосфина (61 мг, 0,31 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 2 часов. Реакционный раствор растворяли в небольшом количестве метанола, очищали с помощью тонкослойной хроматографии с элюирующей системой В с получением соединения, указанного в названии, 4-(3-(4-((1-((трет-бутилдиметилсилил)окси)циклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 5d (73 мг, выход 63,3%) в виде желтого маслянистого вещества.
Этап 4
4-(3-(3-Фтор-4-((1-гидроксициклопропил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(4-((1-((трет-Бутилдиметилсилил)окси)циклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 5d (73 мг, 0,12 ммоль) растворяли в 7 мл тетрагидрофурана с последующим добавлением фторида тетрабутиламмония (0,13 мл, 0,13 ммоль). Реакционный раствор перемешивали в течение 1 часа. Реакционный раствор концентрировали при сниженном давлении, смешивали с 5 мл Н2О и экстрагировали этилацетатом (20 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А последовательно с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((1-гидроксициклопропил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 5 (50 мг, выход 84,4%) в виде твердого белого вещества.
МС m/z (ИЭР): 494,4 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,83 (m, 1Н), 7,13-7,03 (m, 3Н), 4,15 (s, 2Н), 1,60 (s, 6Н), 1,04-1,00 (m, 2Н), 0,77-0,74 (m, 2Н).
Пример 6
4-(3-(4-((1-Аминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
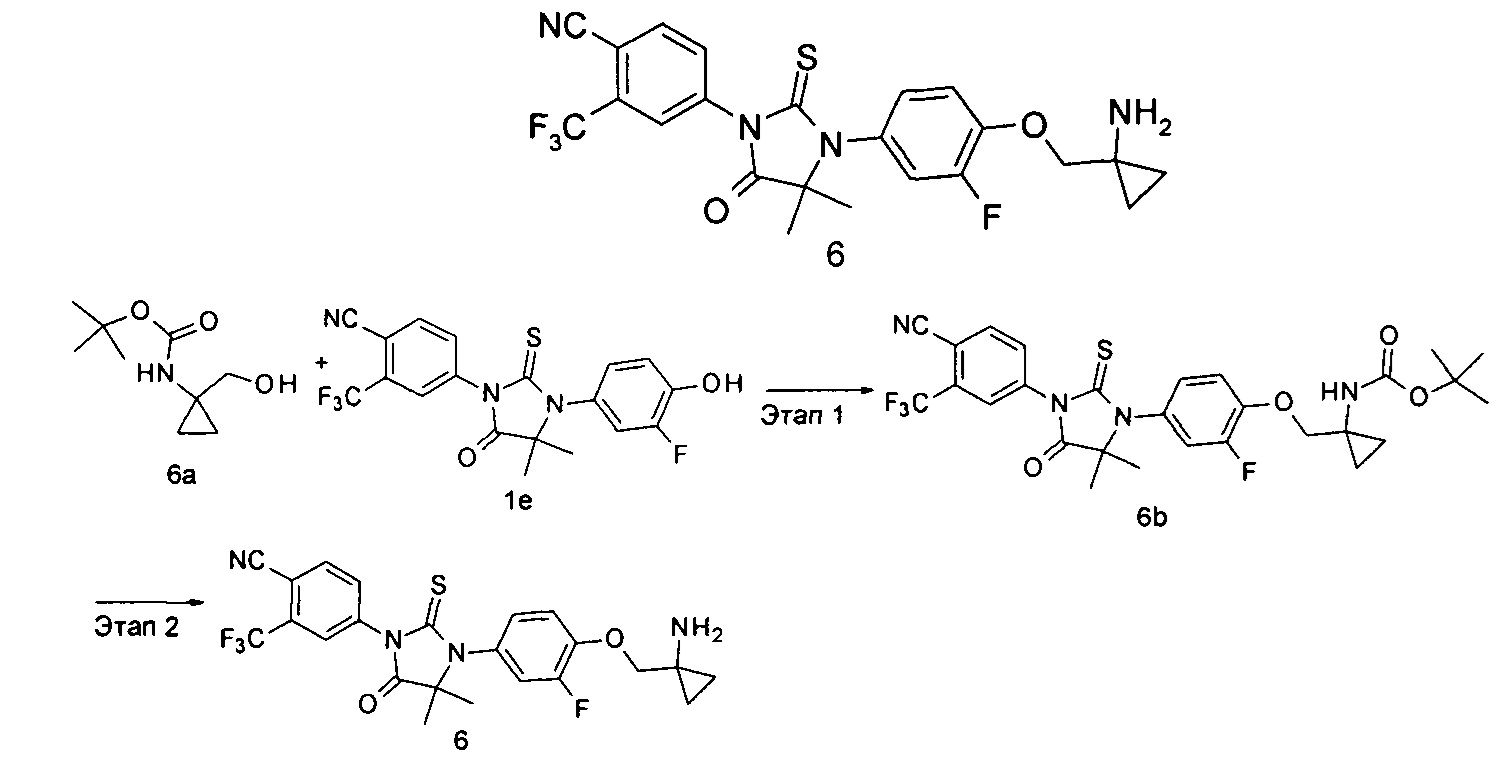
трет-Бутил (1-((4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамат
трет-Бутил (1-(гидроксиметил)циклопропил)карбамат 6а (53 мг, 0,28 ммоль) помещали в реакционную колбу с последующим добавлением 4-(3-(3-фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1е (100 мг, 0,24 ммоль), 1,1'-(азодикарбонил)дипиперидина (95 мг, 0,38 ммоль), 5 мл метилбензола и три-н-бутилфосфина (76 мг, 0,38 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 2 часов. Реакционный раствор растворяли в небольшом количестве метанола и очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, трет-бутил (1-((4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамата 6b (115 мг, выход 82,3%) в виде белого твердого вещества.
Этап 2
4-(3-(4-((1-Аминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
трет-Бутил (1-((4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамат 6b (115 мг, 0,19 ммоль) растворяли в 4 мл раствора 2М хлористого водорода в метаноле. Реакционный раствор перемешивали в течение 4 часов с последующим добавлением 2Ν водного раствора гидроксида натрия для доведения pH до 7. Большую часть метанола выпаривали, и реакционный раствор экстрагировали этилацетатом (30 мл). Органические фазы высушивали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при сниженном давлении с получением продукта, указанного в названии, 4-(3-(4-((1-аминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 6 (97 мг, выход 94,7%) в виде белого твердого вещества.
МС m/z(ИЭР): 493,4 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,41-8,39 (m, 1Н), 8,28-8,27 (m, 1Н), 8,08-8,05 (m, 1Н), 7,40-7,34 (m, 2Н), 7,21-7,19 (m, 1Н), 4,25 (s, 2Н), 1,51 (s, 6Н), 1,14-1,11 (m, 2Н), 1,12-0,99 (m, 2Н).
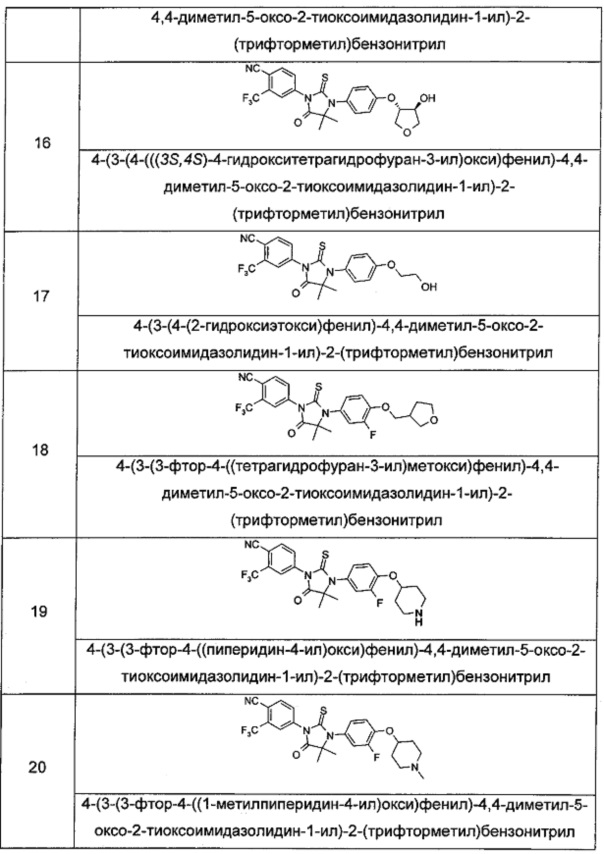
Пример 7
4-(3-(3-Φтοр-4-(((3R,4R/3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
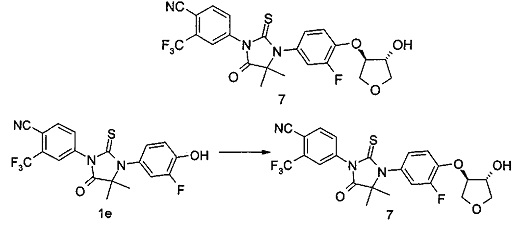
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением 3,4-эпокситетрагидрофурана (24 мг, 0,28 ммоль), карбоната цезия (115 мг, 0,35 ммоль) и 4 мл Ν,Ν-диметилацетамида последовательно. Реакционную смесь нагревали до 120°С. После проведения реакции в течение 1 часа, в реакционный раствор добавляли 3,4-эпокситетрагидрофуран (100 мг, 1,16 ммоль) и перемешивали при 120°С в течение еще 1 часа. Реакционный раствор смешивали с 30 мл Н2О и экстрагировали этилацетатом (30 мл). Органические фазы объединяли, промывали водой (15 мл×3) и насыщенным раствором хлорида натрия (15 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А и последующей элюирующей системой В с получением соединения, указанного в названии, 4-(3-(3-фтор-4-(((3R,4R/3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 7 (40 мг, выход 33,3%) в форме белого твердого вещества.
МС m/z (ИЭР): 510,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,82 (m, 1Н), 7,19-7,15 (t, 1Н), 7,08-7,04 (m, 2Н), 4,81-4,80 (m, 1Н), 4,52-4,50 (m, 1Н), 4,33-4,29 (m, 1Н), 4,14-4,10 (m, 1Н), 4,03-4,00 (m, 1Н), 3,88-3,86 (m, 1Н), 1,60 (s, 6Н).
Примеры 8, 9
4-(3-(3-Фтор-4-(((3R,4R)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(3-Фтор-4-(((3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
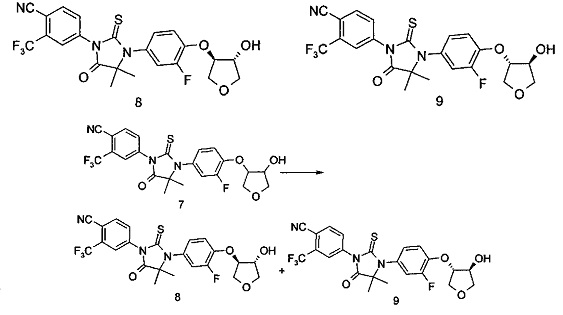
4-(3-(3-Фтор-4-(((3R,4R/3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 7 (320 мг, 0,63 ммоль) разделяли с помощью хиральной ВЭЖХ (условия разделения: хиральная колонка CHIRALCEL IC, подвижная фаза: н-гексан: изопропанол=85:15, скорость потока 15 мл/минута). Соответствующие фракции собирали и выпаривали для удаления растворителя с получением продукта, указанного в названии, 4-(3-(3-фтор-4-(((3R,4R)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 8 (130 мг, 0,26 ммоль) и 4-(3-(3-фтор-4-(((3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 9 (130 мг, 0,26 ммоль).
8: МС m/z (ИЭР): 510,3 [М+1], время удержания=26,958 минут, значение энантиомерного избытка (ее)>99,0%.
9: МС m/z (ИЭР): [М+1], время удержания=32,291 минут, значение энантиомерного избытка (ее)>99,0%.
8: 1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,82 (m, 1Н), 7,19-7,15 (t, 1Η), 7,08-7,04 (m, 2Η), 4,81-4,80 (m, 1Н), 4,52-4,50 (m, 1Н), 4,33-4,29 (m, 1Н), 4,14-4,10 (m, 1Н), 4,03-4,00 (m, 1Н), 3,88-3,86 (m, 1Н), 1,60 (s, 6Н).
9: 1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,82 (m, 1Н), 7,19-7,15 (t, 1Н), 7,08-7,04 (m, 2Н), 4,81-4,80 (m, 1Н), 4,52-4,50 (m, 1Н), 4,33-4,29 (m, 1Н), 4,14-4,10 (m, 1Н), 4,03-4,00 (m, 1Н), 3,88-3,86 (m, 1Н), 1,60 (s, 6Н).
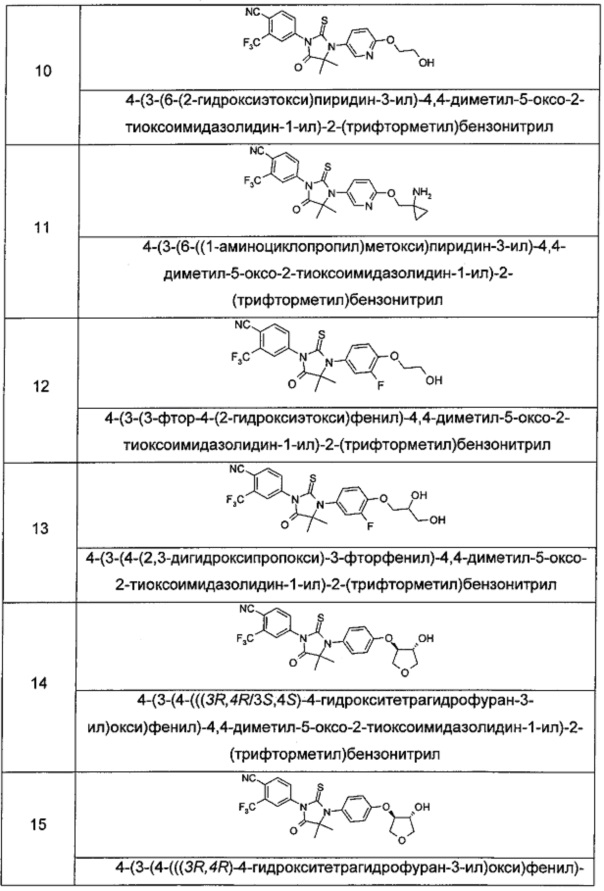
Пример 10
4-(3-(6-(2-Гидроксиэтокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
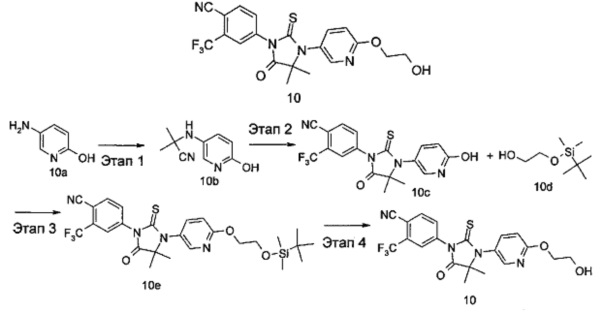
Этап 1
2-((6-Гидроксипиридин-3-ил)амино)-2-метилпропаннитрил 5-Аминопиридин-2-ол 10а (400 мг, 3,63 ммоль) растворяли в 9 мл смеси ацетона и дихлорметана (об./об.=1:2) с последующим добавлением триметилсилилцианида (0,7 мл, 5,40 ммоль) и триметилсилилового эфира трифторметансульфоновой кислоты (33 мкл, 0,18 ммоль). Реакционный раствор перемешивали в течение 12 часов. Полученный раствор концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного названии 2-((6-гидроксипиридин-3-ил)амино)-2-метилпропаннитрила 10b (507 мг, выход 79,5%) в виде коричневого твердого вещества.
МС m/z (ИЭР): 178,2 [М+1]
Этап 2
4-(3-(6-Гидроксипиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
2-((6-Гидроксипиридин-3-ил)амино)-2-метилпропаннитрил 10b (167 мг, 0,94 ммоль) и 4-амино-2-(трифторметил)бензонитрил 1с (175 мг, 0,94 ммоль) растворяли в 5 мл Ν,Ν-диметилацетамида с последующим добавлением тиофосгена (72 мкл, 0,94 ммоль). Реакционный раствор нагревали до 60°С и перемешивали в течение 12 часов с последующим добавлением 4 мл метанола и 2 мл концентрированной соляной кислоты. Реакционный раствор охлаждали до комнатной температуры и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(6-гидроксипиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10с (200 мг, выход 51,9%) в виде серого твердого вещества.
Этап 3
4-(3-(6-(2-((трет-Бутилдиметилсилил)окси)этокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 4-(3-(6-Гидроксипиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10с (55 мг, 0,14 ммоль) помещали в реакционную колбу с последующим добавлением 2-((трет-бутилдиметилсилил)окси)этанола 10d (48 мг, 0,27 ммоль, полученного хорошо известным способом из "Bioorganic & Medicinal chemistry, 2006, 14(7), 2375-2385"), трифенилфосфина (53 мг, 0,20 ммоль), 5 мл дихлорметана и диизопропилазодикарбоксилата (41 мг, 0,20 ммоль), последовательно. Реакционный раствор перемешивали в течение 2 часов. Реакционный раствор концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой В с получением соединения, указанного в названии, 4-(3-(6-(2-((трет-бутилдиметилсилил)окси)этокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10е (28 мг, выход 18,4%), в виде белого твердого вещества.
МС m/z (ИЭР): 565,3 [М+1]
Этап 4
4-(3-(6-(2-Гидроксиэтокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(6-(2-((трет-Бутилдиметилсилил)окси)этокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 10е (28 мг, 0,05 ммоль) растворяли в 3 мл тетрагидрофурана с последующим добавлением фторида тетрабутиламмония (54 мкл, 0,05 ммоль). Реакционный раствор перемешивали в течение 1 часа. Реакционный раствор концентрировали при сниженном давлении, смешивали с этилацетатом (30 мл), промывали насыщенным раствором хлорида натрия (10 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат высушивали при сниженном давлении, и полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(6-(2-гидроксиэтокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10 (9 мг, выход 40,9%), в виде белого твердого вещества.
МС m/z (ИЭР): 451,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,10-8,09 (m, 1Н), 8,01-7,97 (m, 2Н), 7,86-7,83 (m, 1Н), 7,57-7,55 (m, 1Н), 6,99-6,96 (m, 1Н), 4,55-4,53 (m, 2Н), 4,02-4,00 (m, 2Н), 1,60 (s,6H).
Пример 11
4-(3-(6-((1-Аминоциклопропил)метокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
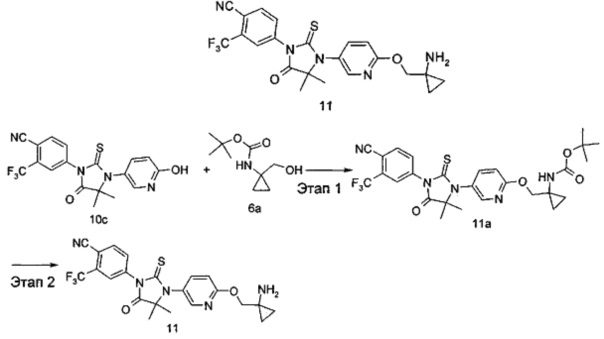
Этап 1
трет-Бутил (1-(((5-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)пиридин-2-ил)окси)метил)циклопропил)карбамат
трет-Бутил (1-(гидроксиметил)циклопропил)карбамат 6а (92 мг, 0,49 ммоль) помещали в реакционную колбу с последующим добавлением 4-(3-(6-гидроксипиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10с (100 мг, 0,24 ммоль), трифенилфосфина (97 мг, 0,37 ммоль), 5 мл дихлорметана и диизопропилазодикарбоксилата (75 мг, 0,37 ммоль) последовательно. Реакционный раствор и перемешивали в течение 2 часов. Реакционный раствор очищали с помощью тонкослойной хроматографии с элюирующей системой А и последующей элюирующей системой В с получением соединения, указанного в названии, трет-бутил (1-(((5-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)пиридин-2-ил)окси)метил)циклопропил)карбамата 11а (40 мг, выход 28,4%) в виде белого твердого вещества.
МС m/z(ИЭP): 576,2 [М+1]
Этап 2
4-(3-(6-((1-Аминоциклопропил)метокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
трет-Бутил (1-(((5-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)пиридин-2-ил)окси)метил)циклопропил)карбамата 11а (40 мг, 0,07 ммоль) растворяли в 5 мл раствора 2М хлористого водорода в метаноле. Реакционный раствор перемешивали в течение 1 часа с последующим добавлением 2Ν водного раствора гидроксида натрия для доведения pH до 7. Большую часть метанола выпаривали, и реакционный раствор экстрагировали этилацетатом (20 мл). Органические фазы высушивали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при сниженном давлении с получением продукта, указанного в названии, 4-(3-(6-((1-аминоциклопропил)метокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 11 (35 мг, выход 98,5%) в виде желтого твердого вещества.
МС m/z (ИЭР): 476,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,19-8,16 (m, 3Н), 8,00-7,98 (m, 1Н), 7,80-7,78 (m, 1Н), 7,10-7,08 (m, 1Н), 4,53 (s, 2Н), 1,57 (s, 6Н), 1,24-1,13 (m, 4Н).
Пример 12
4-(3-(3-Фтор-4-(2-гидроксиэтокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1е (50 мг, 0,12 ммоль) помещали в реакционную колбу с последующим добавлением карбоната калия (33 мг, 0,24 ммоль), 2 мл Ν,Ν-диметилформамида и бромэтанола (30 мл, 0,24 ммоль) последовательно. Реакционную смесь нагревали до 80°С. После проведения реакции в течение 12 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 20 мл воды и экстрагировали этилацетатом (15 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В и последующей элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-(2-гидроксиэтокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 12 (25 мг, выход 45,3%) в виде белого твердого вещества.
МС m/z(ИЭР): 556,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,96-7,99 (m, 2Н), 7,83 (d, 1Н), 7,04-7,15 (m, 3Н), 4,22 (t, 2Н), 4,03 (t, 2Н), 1,59 (s, 6Н).
Пример 13
4-(3-(4-(2,3-Дигидроксипропокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
Этап 1
4-(3-(3-Фтор-4-(оксиран-2-илметокси)фенил)-4,4-диметил-5-оксо-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1е (500 мг, 1,18 ммоль) растворяли в 20 мл ацетонитрила с последующим добавлением эпоксихлорпропана (218 мг, 2, 36 ммоль) и карбоната калия (407 мг, 2, 95 ммоль) последовательно. Реакционный раствор нагревали до 80°С и нагревали с обратным холодильником в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, смешивали с 20 мл воды и экстрагировали этилацетатом (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-(оксиран-2-илметокси)фенил)-4,4-диметил-5-оксо-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 13а (300 мг, выход 53,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 480,2 [М+1]
Этап 2
4-(3-(4-(2,3-Дигидроксипропокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(3-Фтор-4-(оксиран-2-илметокси)фенил)-4,4-диметил-5-оксо-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 13а (100 мг, 0,21 ммоль) растворяли в 10 мл смеси воды и тетрагидрофурана (об/об=1:1) с последующим добавлением 0,2 мл концентрированной серной кислоты. Реакционный раствор нагревали с обратным холодильником в течение 4 часов. Реакционный раствор охлаждали до комнатной температуры, смешивали с 10 мл 1М раствора гидроксида натрия и экстрагировали этилацетатом (30 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(4-(2,3-дигидроксипропокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 13 (40 мг, выход 40,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 498,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,95 (m, 2Н), 7,84-7,82 (m, 1Н), 7,15-7,04 (m, 3Н), 4,22-4,18 (m, 3Н), 3,91-3,79 (m, 2Н), 1,59 (s, 6Н).
Пример 14
4-(3-(4-(((3R,4R/3S,4S)-4-Гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
Этап 1
2-((4-Гидроксифенил)амино)-2-метилпропаннитрил 4-Аминофенол 14а (4 г, 36,65 ммоль) растворяли в 72 мл смеси ацетона и дихлорметана (об/об=1:2) с последующим добавлением триметилсилилцианида (7,4 мл, 55,05 ммоль) и триметилсилилового эфира трифторметансульфоновой кислоты (0,3 мл, 1.84 ммоль). Реакционный раствор перемешивали в течение 12 часов. Полученный в результате раствор концентрировали при сниженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, 2-((4-гидроксифенил)амино)-2-метилпропаннитрила 14b (1,5 г, выход 23,2%), в виде белого твердого вещества.
Этап 2
4-(3-(4-Гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
2-((4-Гидроксифенил)амино)-2-метилпропаннитрила 14b (1,50 г, 8,51 ммоль) и 4-изотиоцианато-2-(трифторметил)бензонитрил 1d (2,90 г, 12,75 ммоль) растворяли в 5 мл Ν,Ν-диметилацетамида. Реакционный раствор нагревали до 60°С. После проведения реакции в течение 1,5 часов, реакционный раствор смешивали с 20 мл метанола и 20 мл концентрированной соляной кислоты, охлаждали до комнатной температуры и экстрагировали этилацетатом (20 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, 4-(3-(4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 14 с (3,20 г, выход 92,9%) в виде бледно-желтого твердого вещества.
МС m/z (ИЭР): 406,2 [М+1]
Этап 3
4-(3-(4-((4-Гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(4-Гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 14с (2,38 г, 5,87 ммоль) помещали в реакционную колбу с последующим добавлением карбоната цезия (2,86 г, 8,81 ммоль) последовательно. Реакционный раствор нагревали до 120°С. После проведения реакции в течение 0,5 часов, реакционный раствор смешивали со 100 мл воды и экстрагировали этилацетатом (100 мл). Органические фазы объединяли, промывали водой (30 мл×3) и насыщенным раствором хлорида натрия (30 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением неочищенного соединения. Неочищенное соединение разделяли с помощью метода ВЭЖХ с получением продукта, указанного в названии, 4-(3-(4-(((3R,4R/3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 14 (350 мг, выход 12,1%) в виде белого твердого вещества.
МС m/z(ИЭР): 492,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,03-8,01 (m, 2Н), 7,90-7,88 (m, 1Н), 7,28-7,26 (m, 2Н), 7,12-7,10 (m, 2Н), 4,81-4,80 (m, 1Н), 4,53-4,52 (m, 1Н), 4,37-4,33 (m, 1Н), 4,14-4,10 (m, 1Н), 4,02-3,99 (m, 1Н), 3,91-3,89 (m, 1Н), 1,62 (s, 6Н).
Примеры 15, 16
4-(3-(4-(((3R,4R)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(4-(((3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
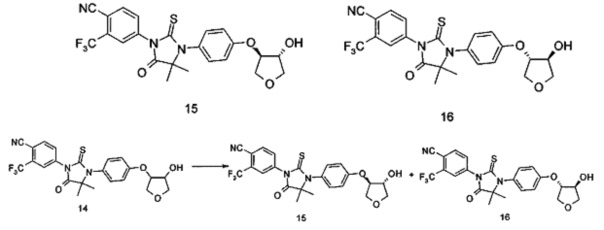
4-(3-(4-(((3R,4R/3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 14 (350 мг, 0,71 ммоль) подвергали хиральному разделению с помощью оборудования для предварительной подготовки и хиральной колонки путем метода ВЭЖХ (условия разделения: хиральная колонка CHIRALCEL IA, подвижная фаза: н-гексан: этанол: дихлормтан=80:10:10, скорость потока 20 мл/минута). Соответствующие фракции собирали и выпаривали для удаления растворителя с получением продукта, указанного в названии, 4-(3-(4-(((3R,4R)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 15 (150 мг, 0,31 ммоль) и 4-(3-(4-(((3S,4S)-4-гидрокситетрагидрофуран-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазол идин-1-ил)-2-(трифторметил)бензонитрила 16 (150 мг, 0,31 ммоль).
15: МС m/z (ИЭР): 492,3 [М+1], время удержания=6,136 минут, значение энантиомерного избытка (ее)>99.0%.
16: МС m/z (ИЭР): 492,3 [М+1], время удержания=7,139 минут, значение энантиомерного избытка (ее)>99.0%.
15: 1Н ЯМР (400 МГц, CDCl3): δ 8,03-8,01 (m, 2Н), 7,90-7,88 (m, 1Н), 7,28-7,26 (m, 2Н), 7,12-7,10 (m, 2Н), 4,81-4,80 (m, 1Н), 4,53-4,52 (m, 1Н), 4,37-4,33 (m, 1Н), 4,14-4,10 (m, 1H), 4,02-3,99 (m, 1H), 3,91-3,89 (m, 1H), 1,62 (s, 6H).
16: 1H ЯМР (400 МГц, CDCl3): δ 8,03-8,01 (m, 2Н), 7,90-7,88 (m, 1Н), 7,28-7,26 (m, 2Н), 7,12-7,10 (m, 2Н), 4,81-4,80 (m, 1Н), 4,53-4,52 (m, 1Н), 4,37-4,33 (m, 1Н), 4,14-4,10 (m, 1Н), 4,02-3,99 (m, 1Н), 3,91-3,89 (m, 1Н), 1,62 (s, 6Н).
Пример 17
4-(3-(4-(2-Гидроксиэтокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
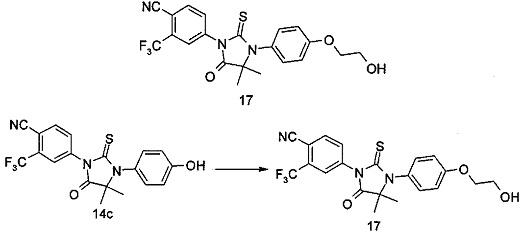
4-(3-(4-Гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 14с (100 мг, 0,25 ммоль) помещали в реакционную колбу с последующим добавлением карбоната калия (69 мг, 0,50 ммоль), 2 мл Ν,Ν-диметилформамида и бромэтанола (62 мг, 0,50 ммоль) последовательно. Реакционный раствор нагревали до 80°С. После проведения реакции в течение 12 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 20 мл воды и экстрагировали этилацетатом (20 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой В с получением соединения, указанного в названии, 4-(3-(4-(2-гидроксиэтокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 17 (40 мг, выход 36,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 450,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,97 (m, 2Н), 7,86-7,84 (m, 1Н), 7,24-7,21 (m, 2Н), 7,09-7,06 (m, 2Н), 4,16-4,14 (m, 2Н), 4,02-4,00 (m, 2Н), 1,58 (s, 6Н).
Пример 18
4-(3-(3-Фтор-4-((тетрагидрофуран-3-ил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
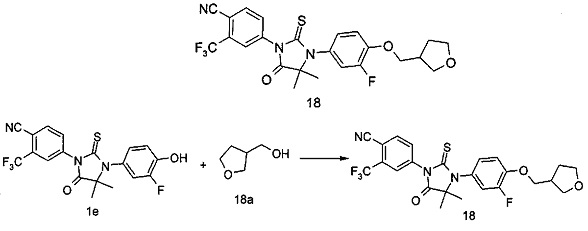
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением (тетрагидрофуран-3-ил)метанола 18а (29 мг, 0,28 ммоль), 1,1'-(азодикарбонил)дипиперидина (95 мг, 0,38 ммоль), 5 мл метилбензола и три-н-бутилфосфина (76 мг, 0,38 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 2 часов. Реакционный раствор растворяли в небольшом количестве метанола, очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((тетрагидрофуран-3-ил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 18 (97 мг, выход 81,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 508,4 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,83 (m, 1Н), 7,11-7,03 (m, 3Н), 4,07-3,99 (m, 2Н), 3,96-3,91 (m, 2Н), 3,84-3,75 (m, 2Н), 2,88-2,80 (m, 1Н), 2,22-2,14 (m, 1Н), 1,80-1,75 (m, 1Н), 1,59 (s, 6Н).
Пример 19
4-(3-(3-Фтор-4-((пиперидин-4-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
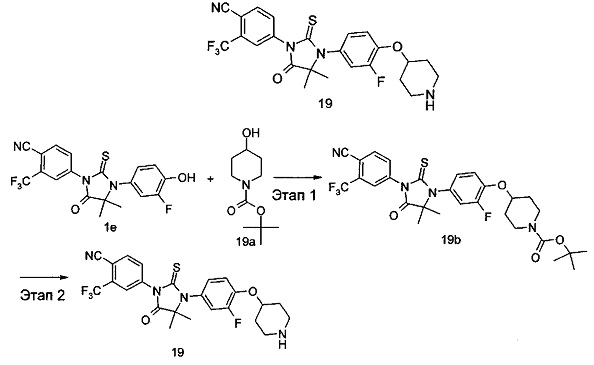
Этап 1
трет-бутил 4-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)пиперидин-1-карбоксилат 4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 1е (200 мг, 0,47 ммоль) помещали в реакционную колбу с последующим добавлением трет-бутил 4-гидроксипиперидин-1-карбоксилата 19а (114 мг, 0,57 ммоль), 1,1'-(азодикарбонил)дипепиридина (191 мг, 0,76 ммоль), 10 мл метилбензола и три-н-бутилфосфина (153 мг, 0,76 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 3 часов. Реакционный раствор концентрировали при сниженном давлении, и полученный остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, трет-бутил 4-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)пиперидин-1-карбоксилата 19b (200 мг, выход 69,8%) в виде белого твердого вещества.
МС m/z (ИЭР): 551,4 [М-56+1]
Этап 2
4-(3-(3-Фтор-4-((пиперидин-4-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
трет-бутил 4-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)пиперидин-1-карбоксилат 19b растворяли в 6 мл раствора 2М хлористого водорода в метаноле. Реакционный раствор перемешивали в течение 12 часов. Реакционный раствор концентрировали при сниженном давлении с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((пиперидин-4-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазол идин-1-ил)-2-(трифторметил)бензонитрила 19 (160 мг, выход 99,4%) в виде белого твердого вещества.
МС m/z (ИЭР): 507,4 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,95-7,99 (m, 2Н), 7,84 (d, 1Н), 7,07-7,11 (m, 3Н), 4,56-4,57 (m, 1Н), 2,99-3,02 (m, 2Н), 2,85-2,88 (m, 2Н), 2,35-2,37 (m, 2Н), 2,05-2,09 (m, 2Н), 1,59 (s, 6Н).
Пример 20
4-(3-(3-Фтор-4-((1-метилпиперидин-4-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
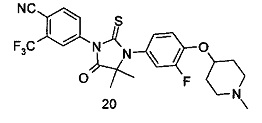
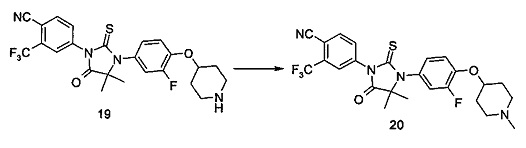
4-(3-(3-фтор-4-((пиперидин-4-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 19 (150 мг, 0,28 ммоль) растворяли в 1,5 мл метанола с последующим добавлением 1 мл 40% раствора формальдегида и 1 мл цианоборогидрида натрия в насыщенном растворе хлорида цинка в метаноле последовательно. Реакционный раствор перемешивали в течение 3 часов, затем смешивали с 15 мл воды и экстрагировали этилацетатом (15 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((1-метилпиперидин-4-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 20 (90 мг, выход 62,6%) в виде белого твердого вещества.
МС m/z (ИЭР): 521,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,95-7,99 (m, 2Н), 7,84 (d, 1Н), 7,07-7,11 (m, 3Н), 4,56-4,57 (m, 1Н), 2,97-2,99 (m, 2Н), 2,81-2,85 (m, 2Н), 2,56 (s, 3Н), 2,34-2,36 (m, 2Н), 2,05-2,09 (m, 2Н), 1,59 (s, 6Н).
Пример 21
(R)-4-(4,4-Диметил-5-оксо-3-(6-((тетрагидрофуран-3-ил)окси)пиридин-3-ил)-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
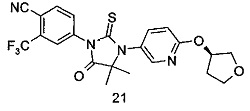
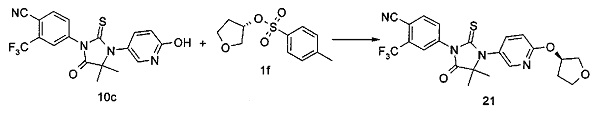
4-(3-(6-Гидроксипиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10с (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением (S)-тетрагидрофуран-3-ил-4-метилбензолсульфоната 1f (116 мг, 0,48 ммоль), карбоната цезия (235 мг, 0,72 ммоль) и 3 мл Ν,Ν-диметилацетамида последовательно. Реакционный раствор нагревали до 60°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, смешивали с 5 мл насыщенного раствора хлорида натрия и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, (R)-4-(4,4-диметил-5-оксо-3-(6-((тетрагидрофуран-3-ил)окси)пиридин-3-ил)-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 21 (46 мг, выход 40,3%), в виде желтого твердого вещества.
МС m/z (ИЭР): 477,1 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,09-8,08 (m, 1Н), 8,01-7,97 (m, 2Н), 7,86-7,84 (m, 1Н), 7,54-7,51 (m, 1Н), 6,92-6,90 (m, 1Н), 5,76-5,60 (m, 1Н), 4,09-3,93 (m, 4Н), 2,32-2,21 (m, 2Н), 1,61 (s, 6Н).
Пример 22
4-(3-(3-Фтор-4-(3-(метилсульфонил)пропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
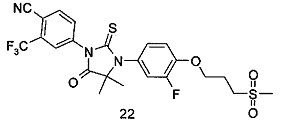
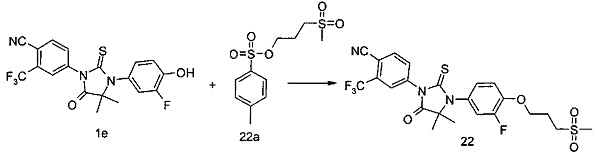
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением 3-(метилсульфонил)пропил-4-метилбензолсульфоната 22а (138 мг, 0,47 ммоль, полученного способом, раскрытым в патентной заявке РСТ "WO 2008/1931 А2"), карбоната цезия (231 мг, 0,71 ммоль) и 2 мл Ν,Ν-диметилацетамида последовательно. Реакционный раствор нагревали до 70°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, смешивали с 155 мл воды и экстрагировали этилацетатом (15 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-(3-(метилсульфонил)пропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 22 (50 мг, выход 63,8%) в виде белого твердого вещества.
МС m/z (ИЭР): 544,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,95-8,00 (m, 2Н), 7,84 (d, 1Н), 7,05-7,10 (m, 3Н), 4,26-4,29 (m, 2Н), 3,29-3,32 (m, 2Н), 2,99 (s, 3Н), 2,41-2,46 (m, 2Н), 1,59 (s, 6Н).
Пример 23
4-(3-(4-((1,1-Диоксидотетрагидро-2Н-тиопиран-4-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
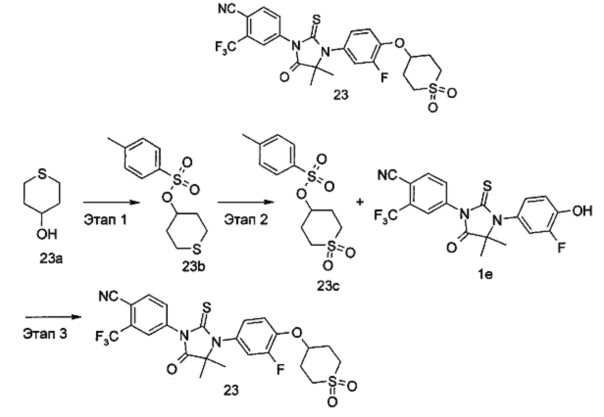
Этап 1
Тетрагидро-2Н-тиопиран-4-ил-4-метилбензолсульфонат Тетрагидро-2Н-тиопиран-4-ол 23а (350 мг, 2,97 ммоль, полученный способом, раскрытым в патентной заявке ЕР 1466898 А1) помещали в реакционную колбу с последующим добавлением триэтиламина (606 мг, 5,94 ммоль), 4-диметиламинопиридина (36 мг, 0,30 ммоль), 20 мл дихлорметана и п-толуолсульфонилхлорида (848 мг, 4,45 ммоль). После проведения реакции в течение 12 часов, реакционный раствор смешивали с 30 мл воды, отстаивали и расслаивали, водную фазу экстрагировали дихлорметаном (10 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, тетрагидро-2Н-тиопиран-4-ил-4-метилбензолсульфоната 23b (556 мг, выход 68,9%), в виде белого твердого вещества.
Этап 2
1,1-Диоксидотетрагидро-2Н-тиопиран-4-ил-4-метилбензолсульфонат
Тетрагидро-2Н-тиопиран-4-ил-4-метилбензолсульфонат 23b (280 мг, 2,57 ммоль) растворяли в 6 мл смеси дихлорметана и метанола (об/об=1:1) с последующим добавлением 0,6 мл воды и реагента Оксон (1,58 г, 2,57 ммоль) последовательно. Реакционный раствор перемешивали в течение 3 часов, концентрировали при сниженном давлении, смешивали с 20 мл воды и 20 мл этилацетата. Реакционный раствор отстаивали и расслаивали, водную фазу экстрагировали этилацетатом (10 мл×2). Органические фазы объединяли, промывали насыщенным раствором сульфата натрия (10 мл×2) и насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, с получением соединения, указанного в названии, тетрагидро-2Н-тиопиран-4-ил-4-метилбензолсульфоната 23 с (279 мг, выход 889,1%), в виде белого твердого вещества.
Этап 3
4-(3-(4-((1,1-Диоксидотетрагидро-2Н-тиопиран-4-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением тетрагидро-2Н-тиопиран-4-ил-4-метилбензолсульфоната 23 с (144 мг, 0,47 ммоль), карбоната цезия (231 мг, 0,71 ммоль) и 1 мл Ν,Ν-диметилацетамида последовательно. Реакционную смесь нагревали до 70°С. После проведения реакции в течение 3 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 15 мл Н2О и экстрагировали этилацетатом (15 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой В и последующей элюирующей системой А с получением соединения, указанного в названии, 4-(3-(4-((1,1-диоксидотетрагидро-2Н-тиопиран-4-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 23 (70 мг, выход 72,4%), в виде белого твердого вещества.
МС m/z (ИЭР): 556,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,95-8,00 (m, 2Н), 7,83 (d, 1Н), 7,06-7,12 (m, 3Н), 4,70-4,71 (m, 1 Η), 3,45-3,49 (m, 2H), 2,99-3,02 (m, 2H), 2,42-2,50 (m, 4H), 1,60 (s, 6H).
Пример 24
(S)-4-(4,4-диметил-5-оксо-3-(6-((тетрагидрофуран-3-ил)окси)пиридин-3-ил-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
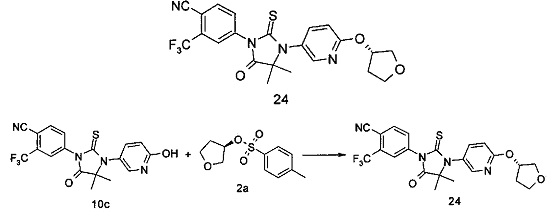
4-(3-(6-Гидроксипиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10с (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением (R)-тетрагидрофуран-3-ил-4-метилбензолсульфоната 2а (116 мг, 0,72 ммоль) и 3 мл Ν,Ν-диметилацетамида последовательно. Реакционный раствор нагревали до 60°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, смешивали с 10 мл насыщенного раствора хлорида натрия и экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, (S)-4-(4,4-диметил-5-оксо-3-(6-((тетрагидрофуран-3-ил)окси)пиридин-3-ил-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 24 (60 мг, выход 52,6%) в виде белого твердого вещества.
МС m/z (ИЭР): 477,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,09-8,08 (m, 1Н), 8,00-7,96 (m, 2Н), 7,86-7,84 (m, 1Н), 7,54-7,51 (m, 1Н), 6,92-6,90 (m, 1Н), 5,63-5,60 (m, 1Н), 4,14-3,91 (m, 4Н), 2,35-2,16 (m, 2Н), 1,60 (s, 6Н).
Пример 25
4-(4,4-Диметил-5-оксо-3-(6-((тетрагидрофуран-3-ил)метокси)пиридин-3-ил)-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
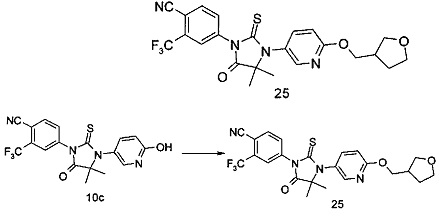
4-(3-(6-Гидроксипиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10 с (80 мг, 0,20 ммоль) помещали в реакционную колбу с последующим добавлением (тетрагидрофуран-3-ил)метанола 18а (24 мг, 0,24 ммоль), 1,1'-(азодикарбонил)дипиперидина (80 мг, 0,32 ммоль), 5 мл метилбензола и три-н-бутилфосфина (64 мг, 0,32 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 2 часов. Реакционный раствор растворяли в небольшом количестве метанола, и очищали с помощью тонкослойной хроматографии с элюирующей системой А с получение соединения, указанного в названии, 4-(4,4-диметил-5-оксо-3-(6-((тетрагидрофуран-3-ил)метокси)пиридин-3-ил)-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 25 (40 мг, выход 41,4%) в виде желтого твердого вещества.
МС m/z (ИЭР): 491,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,10-8,09 (m, 1Н), 8,00-7,97 (m, 2Н), 7,86-7,84 (m, 1Н), 7,54-7,51 (m, 1Н), 6,92-6,90 (m, 1Н), 4,38-4,25 (m, 2Н), 3,96-3,90 (m, 2Н), 3,84-3,70 (m, 2Н), 2,80-2,77 (m, 1Н), 2,25-2,11 (m, 2Н), 1,60 (s, 6Н).
Пример 26
2-(4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)-N-метилбензамид
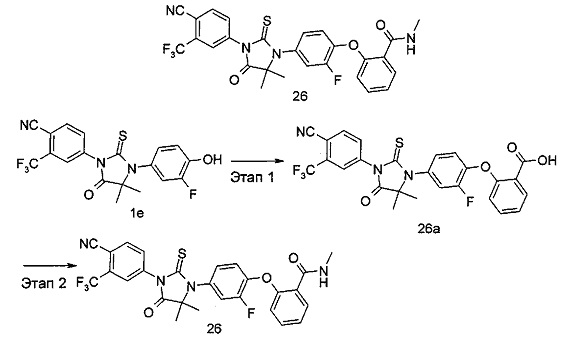
Этап 1
2-(4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)бензойная кислота 4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением 2-йодобензойной кислоты (70 мг, 0,28 ммоль), йодида меди (14 мг, 0,07 ммоль), фенантролина (13 мг, 0,07 ммоль), карбоната цезия (186 мг, 0,57 ммоль) и 4 мл метилбензола последовательно. Реакционный раствор нагревали до 120°С и перемешивали в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, и добавляли 2М соляную кислоту для доведения pH до 2 с последующим добавлением Н2О (20 мл), и экстрагировали этилацетатом (15 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)бензойной кислоты 26а (32 мг, выход 24,9%) в виде желтого твердого вещества.
МС m/z (ИЭР): 544,2 [М+1]
Этап 2
2-(4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)-N-метилбензамид
2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)бензойную кислоту 26а (30 мг, 0,06 ммоль) помещали в реакционную колбу с последующим добавлением 2М раствора метиламина в тетрагидрофуране (0,33 мкл, 0,07 ммоль), 2-(7-азабензатриазол-1-ил)-N,N,N',N'-тетраметилурониум гексафторфосфата (25 мг, 0,07 ммоль) и 2 мл дихлорметана последовательно. Реакционную смесь перемешивали в течение 48 часов. Реакционный раствор концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)-N-метилбензамида 26 (32 мг, выход 32,7%) в виде белого твердого вещества.
МС m/z (ИЭР): 557,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,96-8,01 (m, 2Н), 7,84 (d, 1Н), 7,32-7,41 (m,4Н), 7,06-7,12 (m, 3Н), 6,56-6,57 (m, 1Н), 2,99 (d, 3Н), 1,60 (s, 6Н).
Пример 27
(S)-4-(3-(3-Фтор-4-((1-метилпирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
Этап 1
(S)-4-(3-(3-Фтор-4-((пирролидин-3-ил)окси)фенилI)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (200 мг, 0,47 ммоль) помещали в реакционную колбу с последующим добавлением (R)-пирролидин-3-ола (50 мг, 0,58 ммоль), 1,1'-(азодикарбонил)дипиперидина (192 мг, 0,76 ммоль), 20 мл метилбензола и три-н-бутилфосфина (154 мг, 0,76 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при сниженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного в названии, (S)-4-(3-(3-фтор-4-((пирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 27а (100 мг, выход 43,1%) в виде белого твердого вещества.
Этап 2
(S)-4-(3-(3-Фтор-4-((1-метилпирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
(S)-4-(3-(3-Фтор-4-((пирролидин-3-ил)окси)фенилI)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 27а (100 мг, 0,20 ммоль) растворяли в 1,5 мл метанола с последующим добавлением 1 мл 40% раствора формальдегида и 1 мл 0,3М цианоборгидрида натрия в насыщенном метанольном растворе хлорида цинка последовательно. Реакционный раствор перемешивали в течение 12 часов, затем смешивали с 20 мл воды и экстрагировали этилацетатом (20 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, (S)-4-(3-(3-фтор-4-((1-метилпирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 27 (50 мг, выход 49,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 507,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,95 (m, 2Н), 7,85-7,82 (m, 1Н), 7,08-7,00 (m, 3Н), 4,97-4,93 (m, 1Н), 3,16-3,14 (m, 1Н), 2,90-2,84 (m, 2Н), 2,76-2,75 (m, 1Н), 2,50 (s, 3Н), 2,42-2,38 (m, 1Н), 2,18-2,16 (m, 1Н), 1,59 (s, 6Н).
Пример 28
4-(3-(3-Фтор-4-((пирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
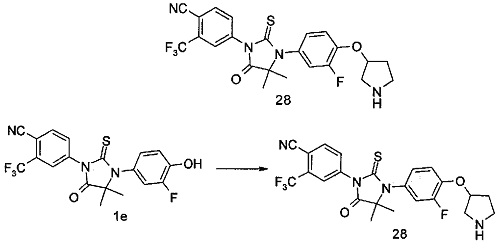
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением пирролидин-3-ола (12 мг, 0,14 ммоль), 1,1'-(азодикарбонил)дипиперидина (47 мг, 0,19 ммоль), 5 мл метилбензола и три-н-бутилфосфина (38 мг, 0,19 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при сниженном давлении и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((пирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 28 (45 мг, выход 77,6%) в виде белого твердого вещества.
МС m/z (ИЭР): 493.2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,82 (m, 1Н), 7,17-7,04 (m, 3Н), 3,54-3,35 (m, 5Н), 2,27-2,18 (m, 2Н), 1,60 (s, 6Н).
Пример 29
4-(3-(3-Фтор-4-((1-метилпирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
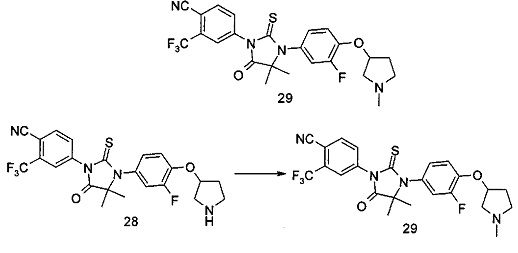
4-(3-(3-фтор-4-((пирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 28 (80 мг, 0,16 ммоль) растворяли в 1,5 мл метанола с последующим добавлением 1 мл 40% раствора формальдегида и 1 мл 0,3М цианоборгидрида натрия в насыщенном метанольном растворе хлорида цинка последовательно. Реакционный раствор перемешивали в течение 12 часов, затем смешивали с 50 мл воды и экстрагировали этилацетатом (50 мл). Органические фазы объединяли, промывали водой (30 мл×3) и насыщенным раствором хлорида натрия (30 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((1-метилпирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 29 (50 мг, выход 60,9%) в виде белого твердого вещества.
МС m/z (ИЭР): 507,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,83 (m, 1Н), 7,09-7,03 (m, 3Н), 3,34-3,33 (m, 1Н), 2,95-2,93 (m, 3Н), 2,59 (s, 3Н), 2,44-2,42 (m, 1Н), 2,17-2,16 (m,2H), 1,59 (s, 6Н).
Пример 30
4-(3-(6-(Дифторметокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
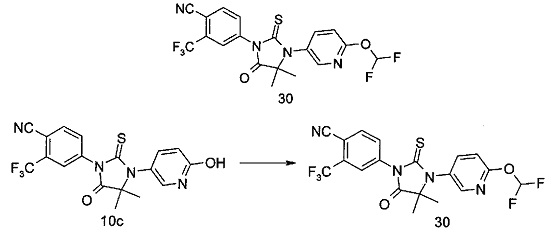
4-(3-(6-Гидроксипиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 10 с (50 мг, 0,12 ммоль) растворяли в 2 мл ацетонитрила с последующим добавлением безводного сульфата натрия (2 мг, 0,01 ммоль) и 2-(фторсульфонил)дифторуксусной кислоты (26 мг, 0,15 ммоль) последовательно. Реакционный раствор перемешивали в течение 12 часов. В реакционный раствор дополнительно добавляли 2-(фторсульфонил)дифторуксусную кислоту (26 мг, 0,15 ммоль) и небольшое количество безводного сульфата натрия, нагревали до 60°С и перемешивали в течение 2 часов. Реакционный раствор смешивали с насыщенным раствором хлорида натрия (5 мл) и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой В и последующей элюирующей системой А с получением соединения, указанного в названии, 4-(3-(6-(дифторметокси)пиридин-3-ил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 30 (20 мг, выход 35,7%) в виде желтого твердого вещества.
МС m/z (ИЭР): 457,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,18-8,17 (m, 1Н), 8,02-7,94 (m, 2Н), 7,87-7,83 (m, 1Н), 7,72-7,69 (m, 1Н), 7,51 (t, 1Н), 7,12-7,09 (m, 1Н), 1,62 (s, 6Н).
Пример 31
2-(4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)уксусная кислота
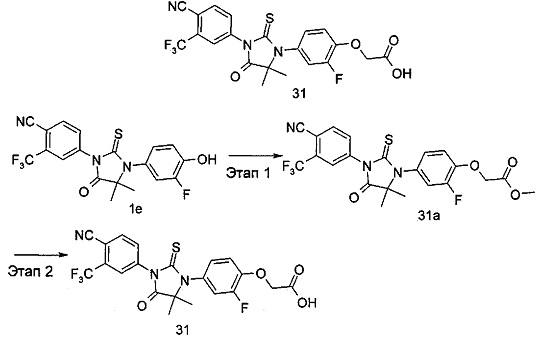
Этап 1
Метил 2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоим идазолидин-1-ил)-2-фторфенокси)ацетат 4-(3-(3-фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением метилгликолята (42 мг, 0,47 ммоль), трифенилфосфина (93 мг, 0,35 ммоль) 5 мл дихлорметана и диизопропилазодикарбоксилата (72 мг, 0,35 ммоль) последовательно. Реакционный раствор перемешивали в течение 1 часа. Реакционный раствор концентрировали при сниженном давлении и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, метил 2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)ацетата 31а (104 мг, выход 89,7%) в идее белого твердого вещества.
МС m/z (ИЭР): 496,2 [М+1]
Этап 2
2-(4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1 -ил)-2-фторфенокси)уксусная кислота
Метил 2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)ацетата 31а (60 мг, 0,12 ммоль) растворяли в 4 мл смеси тетрагидрофурана и метанола (об./об.=1:1) с последующим добавлением 1 мл гидроксида натрия (48 мг, 1,20 ммоль). Реакционный раствор перемешивала в течение 10 минут. Реакционный раствор концентрировали при сниженном давлении с последующим добавлением этилацетата (5 мл), затем по каплям добавляли 2М соляной кислоты для доведения pH до 2-3. Реакционный раствор высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении с получением соединения, указанного в названии, 2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)уксусной кислоты 31 (58 мг, выход 99,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 482,1 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,83 (m, 1Н), 7,14-7,05 (m, 3Н), 4,83 (s, 2Н), 1,60 (s, 6Н).
Пример 32
2-(4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)-N-метилацетамид
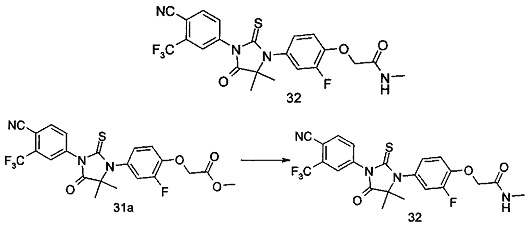
Метил 2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)ацетата 31а (60 мг, 0,12 ммоль) растворяли в 2 мл тетрагидрофурана с последующим добавлением спиртового раствора метиламина (5 мл). После проведения реакции в течение 2 часов, реакционный раствор нагревали до 60°С и перемешивали в течение 12 часов. Реакционный раствор концентрировали при сниженном давлении, и полученный осадок очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 2-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)-N-метилацетамида 32 (30 мг, выход 68,2%) в виде белого твердого вещества.
МС m/z (ИЭР): 495,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,02-7,95 (m, 2Н), 7,85-7,82 (m, 1Н), 7,15-7,08 (m, 3Н), 4,61 (s, 2Н), 2,98 (d, 3Н), 1,60 (s, 6Н).
Пример 33
1-((4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропанкарбоновая кислота
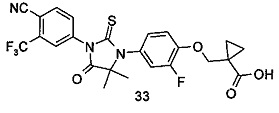
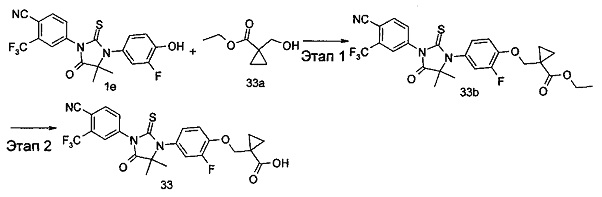
Этап 1
Этил 1-((4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропанкарбоксилат 4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением 1-(гидроксиметил)циклопропанкарбоксилата 33а (20 мг, 0,14 ммоль, полученного способом, раскрытым в патентной заявке US 2012/110702 А1), трифенилфосфина (46 мг, 0,18 ммоль), 8 мл дихлорметана и диизопропилазодикарбоксилата (36 мг, 0,18 ммоль) последовательно на ледяной бане при 0°С. Реакционный раствор перемешивали в течение 1 часа, концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, этил 1-((4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропанкарбоксилата 33b (80 мг, выход >100%) в виде желтого маслянистого вещества.
Этап 2
1-((4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропанкарбоновая кислота
Этил 1-((4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропанкарбоксилата 33b (70 мг, 0,13 ммоль) растворяли в 8 мл смеси тетрагидрофурана и метанола (об./об.=1:1) с последующим добавлением 1 мл гидроксида натрия (51 мг, 1,28 ммоль). Реакционный раствор нагревали до 50°С и перемешивали в течение 1 часа. Реакционный раствор концентрировали при сниженном давлении с последующим добавлением воды (50 мл) и этилацетата (50 мл), затем по каплям добавляли 1М соляной кислоты для доведения pH до 3-4. Реакционный раствор расслаивали, этилацетатную фазу промывали водой (10 мл×3) и насыщенным раствором хлорида натрия (10 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 1-((4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропанкарбоновой кислоты 33 (40 мг, выход 60,6%) в виде белого твердого вещества.
МС m/z (ИЭР): 522,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,96-7,95 (m, 2Н), 7,84-7,83 (m, 1Н), 7,11-7,04 (m, 3Н), 4,26 (s, 2Н), 1,58 (s, 6Н), 1,52-1,51 (m, 2Н), 1,19-1,18 (m, 2Н),
Пример 34
1-((4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)-N-метилциклопропанкарбоксамид
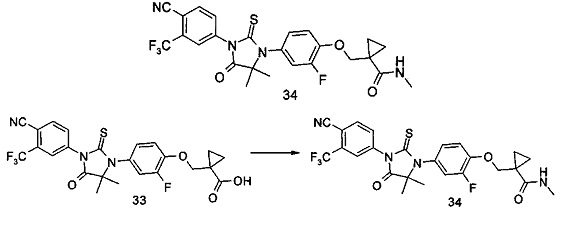
1-((4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропанкарбоновую кислоту 33 (20 мг, 0,04 ммоль) растворяли в 6 мл смеси тетрагидрофурана и дихлорметана (об./об.=2:1) с последующим добавлением 2М раствора метиламина в тетрагидрофуране (0,38 мкл, 0,08 ммоль), 2-(7-азабензатриазол-1-ил)-N,N,N',N'-тетраметилурониум гексафторфосфата (22 мг, 0,06 ммоль) и N,N-диизопропилэтиламина(10 мг, 0,08 ммоль). Реакционный раствор перемешивали в течение 12 часов. Реакционный раствор концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 1-((4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)-N-метилциклопропанкарбоксамида 34 (15 мг, выход 25,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 535,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,83 (m, 1Н), 7,14-7,06 (m, 3Н), 4,15 (s, 2Н), 2,88 (d, 3Н), 1,60 (s, 6Н), 1,42-1,41 (m, 2Н), 0,84-0,83 (m, 2Н).
Пример 35
4-(3-(4-((1-аминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-хлорбензонитрил
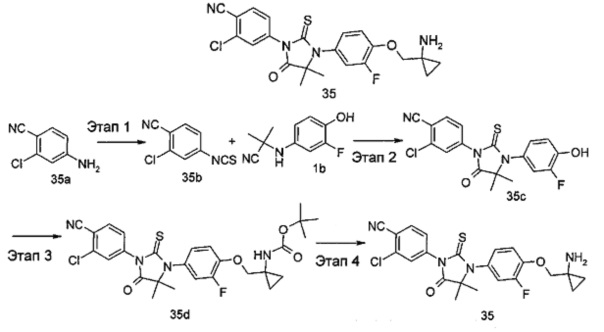
Этап 1
2-Хлор-4-изотиоцианатобензонитрил
4-Амино-2-хлорбензонитрил 35а (12 г, 0,08 моль) растворяли в 30 мл 1,2-дихлорэтана с последующим добавлением тиофосгена (13.60 г, 0,12 моль). Реакционный раствор нагревали до 60°С. После проведения реакции в течение 12 часов, реакционный раствор охлаждали до комнатной температуры, вливали в 100 мл воды, водную фазу экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, 2-хлор-4-изотиоцианатобензонитрила 35b (8,50 г, выход 56,7%) в виде желтого твердого вещества.
Этап 2
2-Хлор-4-(3-(3-фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)бензонитрил
2-Хлор-4-изотиоцианатобензонитрил 35b (2,20 г, 11,30 ммоль) растворяли в 20 мл Ν,Ν-диметилацетамида с последующим добавлением 2-((3-фтор-4-гидроксифенил)амино)-2-метилпропаннитрила 1b (2 г, 10,3 ммоль). Реакционный раствор нагревали до 60°С и перемешивали в течение 12 часов с последующим добавлением 20 мл метанола и 20 мл 2М соляной кислоты. Реакционный раствор нагревали до 75°С и перемешивали в течение 2 часов. Реакционный раствор охлаждали до комнатной температуры, смешивали с Н2О (30 мл) и экстрагировали этилацетатом (30 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×3), высушивали над безводным сульфатом натрия и фильтровали). Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, 2-хлор-4-(3-(3-фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)бензонитрила 35 с (2,20 г, выход 45,9%) в виде белого твердого вещества.
Этап 3
трет-Бутил (1-((4-(3-(3-хлор-4-цианофенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамат
2-Хлор-4-(3-(3-фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)бензонитрил 35 с (100 мг, 0,26 ммоль) растворяли в метилбензоле с последующим добавлением трет-бутил (1-(гидроксиметил)циклопропил)карбамата 6а (49 мг, 0,26 ммоль), 1,1'-(азодикарбонил)дипиперидина (106 мг, 0,42 ммоль) и три-н-бутилфосфина (85 мг, 0,42 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 2 часов. Полученный раствор концентрировали при сниженном давлении. Остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, трет-бутил (1-((4-(3-(3-хлор-4-цианофенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамата 35d (80 мг, выход 55,9%) в виде белого твердого вещества.
Этап 4
4-(3-(4-((1-Аминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-хлорбензонитрил
трет-Бутил (1-((4-(3-(3-хлор-4-цианофенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамат 35d (70 мг, 0,13 ммоль) растворяли в 3 мл раствора 2М раствора хлористого водорода в метаноле и перемешивали в течение 2 часов. Реакционный раствор концентрировали при сниженном давлении, и остаток промывали диэтиловым эфиром (10 мл) и высушивали с получением соединения, указанного в названии, 4-(3-(4-((1-вминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-хлорбензонитрила 35 (60 мг, выход 96,7%) в виде желтого твердого вещества.
МС m/z (ИЭР): 459,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,97-7,95 (m, 1Н), 7,85 (s, 1Н), 7,64-7,62 (m, 1Н), 7,31-7,27 (m, 2Н), 7,19-7,17 (m, 1Н), 4,27 (s, 2Н), 1,54 (s, 6Н), 1,16-1,13 (m, 4Н).
Пример 36
4-(3-(4-((1-Ацетилпирролидин-3-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
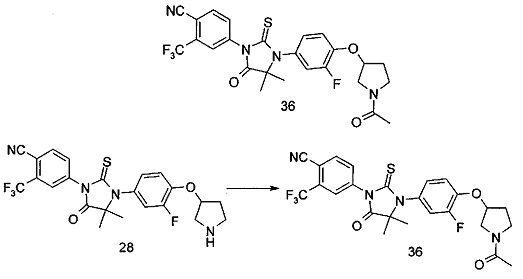
4-(3-(3-Фтор-4-((пирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 28 (50 мг, 0,10 ммоль) растворяли в 5 мл дихлорметана и охлаждали до 0°С с последующим добавлением триэтиламина (20 мг, 0,20 ммоль), 4-диметиламинопиридина (12 мг, 0,10 ммоль) и добавлением по каплям ацетилхлорида (16 мг, 0,20 ммоль). Реакционный раствор перемешивали в течение 30 минут при 0°С. Реакционный раствор концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(4-((1-ацетилпирролидин-3-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 36 (20 мг, выход 37,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 535,2 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,96 (m, 2Н), 7,85-7,83 (m, 1Н), 7,12-7,00 (m, 3Н), 3,75-3,60 (m, 4Н), 2,42-2,27 (m, 3Н), 2,18 (s, 3Н), 1,60 (s, 6Н).
Пример 37
4-(3-(4-((1-Аминоциклопропил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
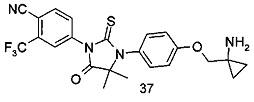
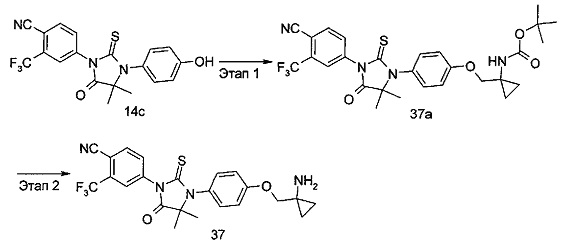
Этап 1
(1-((4-(3-(4-циано-3-(трифторметил(фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)фенокси)метил)циклопропил)карбамат
4-(3-(4-Гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 14 с (100 мг, 0,25 ммоль)помещали в реакционную колбу с последующим добавлением трет-бутил (1-(гидроксиметил)циклопропил)карбамата 6а (93 мг, 0,49 ммоль), трифенилфосфина (97 мг, 0,37 ммоль), 5 мл дихлорметана и диизопропилазодикарбоксилата (75 мг, 0,37 ммоль) последовательно. Реакционный раствор перемешивали в течение 2 часов. Реакционный раствор концентрировали при сниженном давлении и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии (50 мг, выход 35,2%) в виде белого твердого вещества.
МС m/z (ИЭР): 519,3 [М-56+1]
Этап 2
4-(3-(4-((1-Аминоциклопропил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
трет-Бутил (1-((4-(3-(4-циано-3-(трифторметил(фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)фенокси)метил)циклопропил)карбамата 37а (50 мг, 0,09 ммоль) растворяли в 5 мл раствора 2М хлористого водорода в метаноле и перемешивали в течение 2 часов. Реакционный раствор концентрировали при сниженном давлении с получением соединения, указанного в названии, 4-(3-(4-((1-аминоциклопропил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 37 (50 мг, выход>100%) в виде желтого твердого вещества.
МС m/z (ИЭР): 475,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,40-8,38 (m, 1Н), 8,29-8,28 (m, 1Н), 8,09-8,06 (m, 1Н), 7,34-7,31 (m, 2Н), 7,16-7,11 (m, 2Н), 4,15 (s, 2Н), 1,50 (s, 6Н), 1,09-1,06 (m, 2Н), 0,99-0,94 (m, 2Н).
Пример 38
4-(3-(4-((1-Аминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазол идин-1 -ил)-2-метилбензонитрил
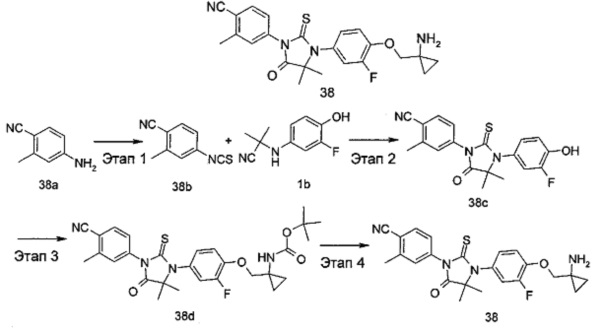
Этап 1
4-Изотиоцианато-2-метилбензонитрил
4-Амино-2-метилбензонитрил 38а (110 мг, 0,83 ммоль) растворяли в 5 мл тетрагидрофурана и охлаждали до 0°С, с последующим добавлением тиофосгена (115 мг, 0,99 ммоль). Реакционную смесь перемешивали в течение 1 часа при 0°С, затем смешивали с водой (20 мл) и экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении с получением соединения, указанного в названии, 4-изотиоцианато-2-метилбензонитрила 38b (130 мг, выход 89,7%) в виде белого твердого вещества.
Этап 2
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-метилбензонитрил
4-Изотиоцианато-2-метилбензонитрил 38b (110 мг, 0,63 ммоль) растворяли в 3 мл Ν,Ν-диметилацетамида с последующим добавлением 2-((3-фтор-4-гидроксифенил)амино)-2-метилпропаннитрила 1b (98 мг, 0,50 ммоль). Реакционный раствор перемешивали в течение 12 часов с последующим добавлением метанола (1,5 мл) и 2М соляной кислоты (1,5 мл). Реакционный раствор нагревали до 80°С. После проведения реакции в течение 2 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 20 мл H2O и экстрагировали этилацетатом (15 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой В с получением соединения, указанного в названии, 4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-метилбензонитрила 38 с (20 мг, выход 10,7%) в виде желтого твердого вещества.
МС m/z (ИЭР): 370,1 [М+1]
Этап 3
трет-Бутил (1-((4-(3-(4-циано-3-метилфенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамат 4-(3-(3-фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-метилбензонитрила 38 с (22 мг, 0,06 ммоль) помещали в реакционную колбу с последующим добавлением трет-бутил (1-(гидроксиметил)циклопропил)карбамата 6а (12 мг, 0,07 ммоль), 1,1'-(азодикарбонил)дипиперидина (24 мг, 0,10 ммоль) и три-н-бутилфосфина (19 мг, 0,10 ммоль) последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 24 часов. Реакционный раствор концентрировали при сниженном давлении, и полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой В с получением соединения, указанного в названии, трет-бутил (1-((4-(3-(4-циано-3-метилфенил)-5,5-димети л-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамата 38d (17 мг, выход 52,9%) в виде белого твердого вещества.
МС m/z (ИЭР): 483,3 [М-56+1]
Этап 4
4-(3-(4-((1-Аминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-метилбензонитрил
трет-Бутил (1-((4-(3-(4-циано-3-метилфенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)метил)циклопропил)карбамата 38d (16 мг, 0,03 ммоль) растворяли в 2 мл 4М раствора хлористого водорода в метаноле. Реакционный раствор перемешивали в течение 1 часа. Реакционный раствор концентрировали при сниженном давлении с получением соединения, указанного в названии, 4-(3-(4-((1-аминоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-метилбензонитрила 38 (14 мг, выход 99,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 439,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,79 (d, 1Н), 7,52 (s, 1Н), 7,42 (d, 1Н), 7,26 (t, 2Н), 7,18 (d, 1Н), 4,28 (s, 2Н), 2,59 (s, 3Н), 1,54 (s, 6Н), 1,12-1,17 (m, 4Н).
Пример 39
4-(3-(4-((1-Цианоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
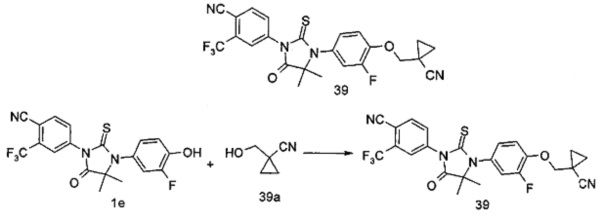
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) помещали в реакционную колбу с последующим добавлением 1 (гидроксиметил)циклопропанкарбонитрила 39а (28 мг, 0,28 ммоль, полученного хорошо известным способом из "Bioorganic and Medicinal Chemistry Letters 2009, 19(6), 1797-1801"), 1,1,1'-(азодикарбонил)дипиперидина (95 мг, 0,38 ммоль), 10 мл метилбензола и три-н-бутилфосфина (76 мг, 0,38 ммоль последовательно. Реакционный раствор нагревали до 50°С и перемешивали в течение 12 часов. Реакционный раствор концентрировали при сниженном давлении, и полученный остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(4-((1-цианоциклопропил)метокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 39 (110 мг, выход 93,2%) в идее белого твердого вещества.
МС m/z (ИЭР): 503,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,95-8,00 (m, 2Н), 7,84 (d, 1Н), 7,04-7,11 (m, 3Н), 4,12 (s, 2Н), 1,59 (s, 6Н), 1,45 (t, 2Н), 1,18 (t, 2Н),
Пример 40
4-(4,4-Диметил-3-(4-((оксетан-3-ил)окси)фенил)-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
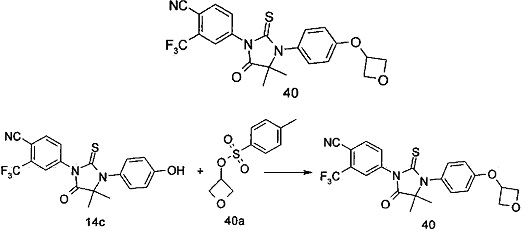
4-(3-(4-Гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 14 с (100 мг, 0,25 ммоль) растворяли в 5 мл Ν,Ν-диметилацетамида с последующим добавлением оксэтан-3-ил-4-метилбензол сульфоната 40а (114 мг, 0,50 ммоль, полученного хорошо известным способом из "Organic Letters, 2008, 10(15), 3259-3262") и карбоната калия (103 мг, 0,75 ммоль). Реакционный раствор нагревали до 80°С. После проведения реакции в течение 12 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 20 мл Н2О и экстрагировали этилацетатом (20 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(4,4-диметил-3-(4-((оксетан-3-ил)окси)фенил)-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 40 (20 мг, выход 17,7%).
МС m/z (ИЭР): 462,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,96 (m, 2Н), 7,86-7,83 (m, 1Н), 7,23-7,20(m, 2Н), 6,86-6,83 (m, 2Н), 5,27-5,24 (m, 1Н), 5,03-4,99 (m, 2Н), 4,83-4,80 (m, 2Н), 1,58 (s, 6Н).
Пример 41
4-(3-(3-Фтор-4-((оксетан-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
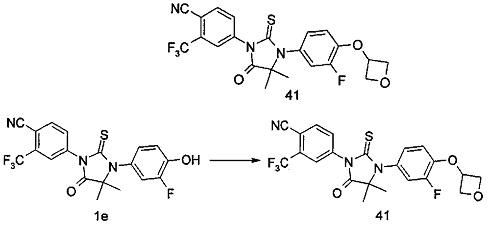
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) растворяли в 5 мл Ν,Ν-диметилацетамида с последующим добавлением оксетан-3-ил-4-метилбензолсульфоната 40а (110 мг, 0,48 ммоль) и карбоната калия (100 мг, 0,72 ммоль). Реакционный раствор нагревали до 80°С. После проведения реакции в течение 4 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 20 мл H2O и экстрагировали этилацетатом (20 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((оксетан-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 41 (30 мг, выход 26,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 480,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,95 (m, 2Н), 7,84-7,82 (m, 1Н), 7,12-7,09(m, 1Н), 7,01-7,00 (m, 1Н), 6,75-6,71 (m, 1Н), 5,32-5,30 (m, 1Н), 5,03-4,99 (m, 2Н), 4,89-4,86 (m, 2Н), 1,59 (s, 6Н).
Пример 42
4-(3-(4-((Азетидин-3-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
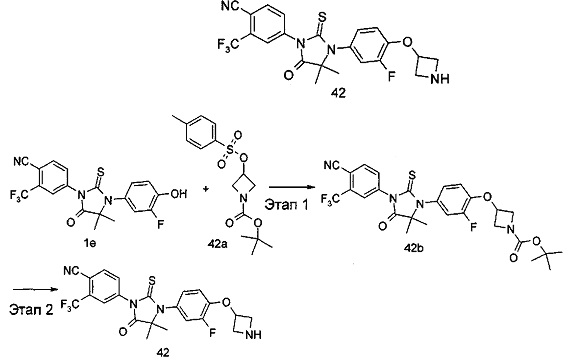
Этап 1
3-(4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазол идин-1 -ил)-2-фторфенокси)азетидин-1-карбоксилат
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (100 мг, 0,24 ммоль) растворяли в 5 мл Ν,Ν-диметилацетамида с последующим добавлением трет-бутил 3-(тозилокси)азетидин-1-карбоксилата 42а (157 мг, 0,48 ммоль, полученного с помощью способа, раскрытого в патентной заявке РСТ WO 2011/103196 А1) и карбоната калия (100 мг, 0,72 ммоль). Реакционный раствор нагревали до 90°С. После проведения реакции в течение 4 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 20 мл H2O и экстрагировали этилацетатом (20 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой Ас получением соединения, указанного в названии, 3-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)азетидин-1-карбоксилата 42b (70 мг, выход 51,4%) в виде белого твердого вещества.
Этап 2
4-(3-(4-((Азетидин-3-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
трет-Бутил 3-(4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)азетидин-1-карбоксилат 42b (70 мг, 0,12 ммоль) растворяли в 4М растворе хлористого водорода в метаноле и перемешивали в течение 12 часов. Реакционный раствор концентрировали при сниженном давлении с получением соединения, указанного в названии, 4-(3-(4-((азетидин-3-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 42 (60 мг, 96,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 479,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 9,22 (S, 1Н), 8,39-8,37 (m, 1Н), 8,26-8,25 (m, 1Н), 8,06-8,03 (m, 1Н), 7,42-7,38 (m, 1Н), 7,18-7,12 (m, 2Н), 5,22-5,19 (m, 1Н), 4,49-4,44 (m, 2Н), 4,10-4,09 (m, 2Н), 1,50 (s, 6Н).
Пример 43
4-(3-(4-((1,3-Дигидроксипропан-2-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
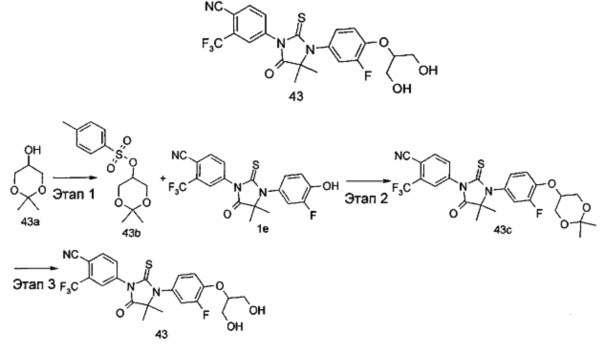
Этап 1
2,2-Диметил-1,3-диоксан-5-ил-4-метилбензолсульфонат
2,2-Диметил-1,3-диоксан-5-ол 43а (200 мг, 1,51 ммоль, полученный способом, раскрытым в патентной заявке US 2007/10542 А1) растворяли в 30 мл дихлорметана с последующим добавлением п-толуолсульфонилхлорида (420 мг, 2,20 ммоль) и триэтиламина (300 мг, 2,97 ммоль). Реакционную смесь перемешивали в течение 3 часов, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой В с получением соединения, указанного в названии, 2,2-диметил-1,3-диоксан-5-ил-4-метилбензолсульфоната 43b (100 мг, выход 23,3%) в виде бесцветного маслянистого вещества.
МС m/z (ИЭР): 287,2 [М+1]
Этап 2
4-(3-(4-((2,2-Диметил-1,3-диоксан-5-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (200 мг, 0,47 ммоль) растворяли в 5 мл Ν,Ν-диметилацетамида с последующим добавлением 2,2-диметил-1,3-диоксан-5-ил-4-метилбензолсульфоната 43b (200 мг, 0,71 ммоль) и карбоната калия (130 мг, 0,94 ммоль). Реакционный раствор нагревали до 60°С. После проведения реакции в течение 6 часов, реакционный раствор охлаждали до комнатной температуры, смешивали с 20 мл воды и экстрагировали этилацетатом (30 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой В с получением соединения, указанного в названии, 4-(3-(4-((2,2-диметил-1,3-диоксан-5-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 43 с (170 мг, выход 67,1%) в виде белого твердого вещества.
МС m/z (ИЭР): 538,3 [М+1]
Этап 3
4-(3-(4-((1,3-Дигидроксипропан-2-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(4-((2,2-Диметил-1,3-диоксан-5-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 43 с (70 мг, 0,12 ммоль) растворяли в 10 мл метанола с последующим добавлением 1М соляной кислоты (1 мл). Реакционный раствор перемешивали в течение 2 часов, смешивали с 10 мл воды и 20 мл насыщенного раствора карбоната калия и экстрагировали этилацетатом (30 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл×3), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(4-((1,3-дигидроксипропан-2-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 43 (40 мг, выход 43,4%) в виде белого твердого вещества.
МС m/z (ИЭР): 498,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,95 (m, 2Н), 7,84-7,82 (m, 1Н), 7,29-7,26 (m, 1Н), 7,11-7,03 (m, 2Н), 4,51-4,47 (m, 1Н), 3,978 (s, 4Н), 1,59 (s, 6Н).
Пример 44
(S)-4-(3-(4-(2,3-Дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1 -ил)-2-(трифторметил)бензонитрил
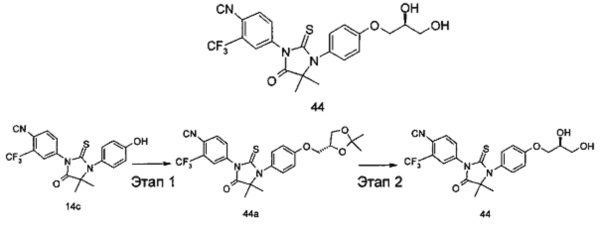
Этап 1
(R)-4-(3-(4-((2,2-Диметил-1,3-диоксолан-4-ил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(4-Гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 14с (2,38 г, 5,87 ммоль) растворяли в 50 мл метилбензола с последующим добавлением (R)-(2,2-диметил-1,3-диоксопентан-4-ил)метанола (819 мг, 6,20 ммоль) и 1,1,1'-(азодикарбонил)дипиперидина (2,5 г, 9,92 ммоль) последовательно. Реактор продували аргоном 3 раза с последующим добавлением три-н-бутилфосфина (2 г, 9,92 ммоль). Реакционный раствор нагревали до 50°С и перемешивали в течение 3 часов. Затем реакционный раствор концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой Ε с получением соединения, указанного в названии, (R)-4-(3-(4-((2,2-диметил-1,3-диоксолан-4-ил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 44а (2,2 г, выход 68,7%) в виде желтого твердого вещества.
МС m/z (ИЭР): 520,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,38 (d, 1Н), 8,29 (s, 1Н), 8,08 (dd, 1Н), 7,29 (d, 2Н), 77,12 (d, 2Н), 4,47-4,41 (m, 1Н), 4,14-4,03 (m, 3Н), 3,81-3,32 (m, 1Н), 1,50 (s, 6Н), 1,37 (s, 3Н), 1,32 (s, 3Н).
Этап 2
(S)-4-(3-(4-(2,3-дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
(R)-4-(3-(4-((2,2-диметил-1,3-диоксолан-4-ил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 44а (2,2 г, 4,20 ммоль) растворяли в 100 мл уксусной кислоты с последующим добавлением 50 мл Н2О. Реакционный раствор нагревали до 70°С и перемешивали в течение 1 часа. Затем реакционный раствор концентрировали при сниженном давлении для удаления остатков уксусной кислоты, смешивали с Н2О (100 мл) и этилацетатом (100 мл), отстаивали и расслаивали. Органические фазы промывали насыщенным раствором бикарбоната натрия (100 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного в названии, (S)-4-(3-(4-(2,3-дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 44 (1,1 г, выход 55,0%) в виде белого твердого вещества.
МС m/z (ИЭР): 480,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,02-8,00 (m, 2Н), 7,89-7,87 (m, 1Н), 7,30-7,25 (m, 2Н), 7,11-7,09 (m, 2Н), 4,20-4,14 (m, 3Н), 3,91-3,80 (m, 2Н), 2,61-2,60 (d, 1Н), 2,02-1,99 (m, 1Η), 1,61 (s, 6Н),
Пример 45
(R)-4-(3-(4-(2,3-дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
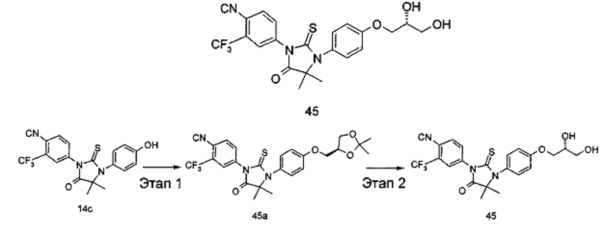
Этап 1
(S)-4-(3-(4-((2,2-Диметил-1,3-диоксолан-4-ил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(4-Гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 14 с (2,5 г, 6,20 ммоль) растворяли в 50 мл метилбензола с последующим добавлением (R)-(2,2-диметил-1,3-диоксопентан-4-ил)метанола (819 мг, 6,20 ммоль) и 1,1,1'-(азодикарбонил)дипиперидина (2,5 г, 9,92 ммоль) последовательно. Реактор продували аргоном 3 раза с последующим добавлением три-н-бутилфосфина (2 г, 9,92 ммоль). Реакционный раствор нагревали до 50°С и перемешивали в течение 3 часов. Затем реакционный раствор концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой Ε с получением соединения, указанного в названии, (S)-4-(3-(4-((2,2-диметил-1,3-диоксолан-4-ил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 45а (2,5 г, выход 78,1%) в виде желтого твердого вещества.
МС m/z (ИЭР): 520,3 [М+1]
Этап 2
(R)-4-(3-(4-(2,3-Дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
(S)-4-(3-(4-((2,2-Диметил-1,3-диоксолан-4-ил)метокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 45а (2,5 г, 4,80 ммоль) растворяли в 100 мл уксусной кислоты с последующим добавлением 50 мл Н2О. Реакционный раствор нагревали до 70°С и перемешивали в течение 1 часа. Затем реакционный раствор концентрировали при сниженном давлении для удаления остатков уксусной кислоты, смешивали с Н2О (100 мл) и этилацетатом (100 мл), отстаивали и расслаивали. Органические фазы промывали насыщенным раствором бикарбоната натрия (100 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного в названии, (R)-4-(3-(4-(2,3-дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 45 (1,32 г, выход 57,3%) в виде белого твердого вещества.
МС m/z (ИЭР): 480,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,97 (m, 2Н), 7,86-7,83 (m, 1Н), 7,24-7,22 (m, 2Н), 7,08-7,06 (m, 2Н), 4,15-4,10 (m, 3Н), 3,88-3,78 (m, 2H), 2,59-2,58 (d, 1H), 2,02-1,99 (m, 1H), 1,59 (s, 6H).
Пример 46
4-(3-(4-((1-Ацетил-4-гидроксипирролидин-3-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
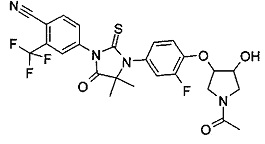
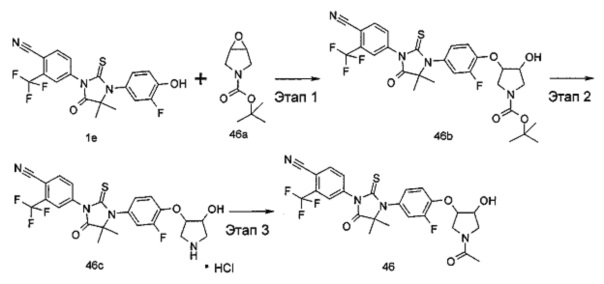
Этап 1
трет-Бутил 3-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)-4-гидроксипирролидин-1-карбоксилат
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (212 мг, 0,50 ммоль) помещали в реакционную колбу с последующим добавлением трет-бутил 6-окса-3-азабицикло[3.1.0]гексан-3-карбоксилата 46а (463 мг, 2,5 ммоль) и 0,5 мл Ν,Ν-диизопропилэтиламина. Реакционный раствор нагревали до 100°С и перемешивали в течение 16 часов. Затем реакционный раствор охлаждали естественным образом до комнатной температуры, смешивай с Н2О (50 мл) и этилацетатом (50 мл) и расслаивали. Органические фазы промывали насыщенным раствором хлорида натрия (50 мл×2), высушивали над безводным сульфатом натрия и фильтровали для удаления осушающего агента. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой С с получением соединения, указанного в названии, трет-бутил 3-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)-4-гидроксипирролидин-1-карбоксилата 46b (280 мг, выход 92,1%) в виде бледно-желтого твердого вещества.
МС m/z (ИЭР): 553,3 [М-56+1]
Этап 2
4-(3-(3-Фтор-4-((4-гидроксипирролидин-3-ил)окси)фенил)-4,4-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил гидрохлорид
трет-Бутил 3-(4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенокси)-4-гидроксипирролидин-1-карбоксилат 46b (280 мг, 0,46 ммоль) растворяли в 50 мл метанола с последующим добавлением раствора соляной кислоты в метаноле (2%, 5 мл). Реакционный раствор перемешивали в течение 4 часов, затем концентрировали при сниженном давлении с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((4-гидроксипирролидин-3-ил)окси)фенил)-4,4-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил гидрохлорида 46 с (230 мг, выход 98,3%) в виде бледно-желтого твердого вещества.
МС m/z (ИЭР): 509,3 [М+1]
1Н ЯМР (400 МГц, ДМСО): δ 9,62 (m, 2Н), 8,40-8,36 (m, 1Н), 8,28-8,27 (m, 1Н), 8,08-8,06 (m, 1Н), 7,57-7,53 (m, 1Н), 7,41-7,37 (m, 1Н), 7,23-7,21 (m, 1Н), 4,99-4,98 (m, 1Н), 4,43-4,42 (m, 1Н), 3,67-3,63 (m, 1Н), 3,36-3,30 (m, 2Н), 2,25-2,16 (m, 1Н), 1,51 (s, 6H).
Этап 3
4-(3-(4-((1-Ацетил-4-гидроксипирролидин-3-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(3-Фтор-4-((4-гидроксипирролидин-3-ил)окси)фенил)-4,4-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил гидрохлорида 46 с (80 мг, 0,16 ммоль), триэтиламин (14 мг, 0,18 ммоль) и 4-диметиламинопиридин (2 мг, 0,02 ммоль) добавляли к 5 мл дихлорметана последовательно и хорошо перемешивали с последующим добавлением ацетилхлорида. Реакционный раствор перемешивали в течение 2 часов, концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой С с получением соединения, указанного в названии, 4-(3-(4-((1-ацетил-4-гидроксипирролидин-3-ил)окси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 46 (15 мг, выход 18,8%) в виде белого твердого вещества.
МС m/z (ИЭР): 551,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,00-7,95 (m, 2Н), 7,84-7,82 (m, 1Н), 7,15-7,06 (m, 3Н), 4,86-4,78 (m, 1Н), 4,63-4,53 (m, 1Н), 3,99-3,89 (m, 2Н), 3,77-3,73 (m, 1Н), 3,69-3,56 (m, 1Н), 2,12 (s, 3Н), 1,60 (s, 6Н).
Пример 47
4-(3-(3-фтор-4-((4-гидрокси-1-метилпирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
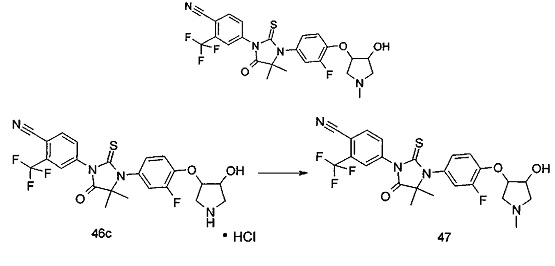
Цианоборгидрид натрия (35 мг, 0,55 ммоль) и хлорид цинка (37 мг, 0,28 ммоль) добавляли к 3 мл метанола и хорошо перемешивали с последующим добавлением 4-(3-(3-фтор-4-((4-гидроксипирролидин-3-ил)окси)фенил)-4,4-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил гидрохлорида 46с (100 мг, 0,18 ммоль) и формальдегида (1 мл). Реакционный раствор перемешивали в течение 16 часов, смешивали с H2O (50 мл) и этилацетатом (50 мл) и расслаивали. Органические фазы промывали насыщенным раствором хлорида натрия (50 мл×2), высушивали над безводным сульфатом натрия и фильтровали для удаления осушающего агента. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью тонкослойной хроматографии с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(3-фтор-4-((4-гидрокси-1-метилпирролидин-3-ил)окси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 47 (35 мг, выход 36,5%) в виде белого твердого вещества.
МС m/z (ИЭР): 523,4 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,96 (m, 2Н), 7,84-7,82 (m, 1Н), 7,25-7,23 (m, 1Н), 7,08-7,03 (m, 2Н), 4,70 (m, 1Н), 4,33 (m, 1Н), 3,42-3,38 (m, 1Н), 2,86-2,79 (m, 2Н), 2,63-2,59 (m, 1Н), 2,45 (s, 3Н), 1,59 (s, 6Н).
Пример 48
4-(3-(4-(2,3-Дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
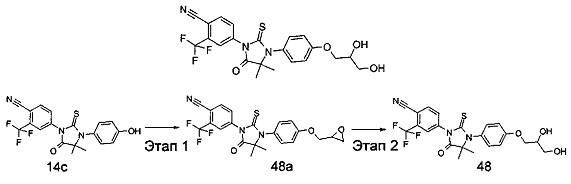
Этап 1
4-(4,4-Диметил-3-(4-(оксиран-2-ил-метокси)фенил)-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(3-(4-Гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 14с (4,5 г, 11,1 моль) растворяли в 50 мл ацетонитрила с последующим добавлением эпоксихлорпропана (2,05 г, 22,2 ммоль) и карбоната калия (3,8 г, 27,8 ммоль) последовательно. Реакционный раствор нагревали до 80°С и нагревали с обратным холодильником в течение 16 часов. Реакционный раствор охлаждали до комнатной температуры, смешивали со 100 мл воды и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой С с получением соединения, указанного в названии, 4-(4,4-диметил-3-(4-(оксиран-2-илметокси)фенил)-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 48а (3,0 г, выход 58,8%) в виде бесцветного маслянистого вещества.
МС m/z (ИЭР): 480,3 [М+1]
Этап 2
4-(3-(4-(2,3-Дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил
4-(4,4-диметил-3-(4-(оксиран-2-илметокси)фенил)-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 48а (2,9 г, 6,3 ммоль) растворяли в 80 мл смеси воды и тетрагидрофурана (об./об.=1:1) с последующим добавлением концентрированной серной кислоты (2,0 мл). Реакционный раствор перемешивали в течение 4 часов, смешивали с соляной кислотой (1М, 50 мл) и экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении, и остаток очищали с помощью колоночной хроматографии на силикагеле с элюирующей системой А с получением соединения, указанного в названии, 4-(3-(4-(2,3-дигидроксипропокси)фенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 48 (1,1 г, выход 36,7 ммоль) в виде белого твердого вещества.
МС m/z (ИЭР): 480,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,97 (m, 2Н), 7,86-7,84 (m, 1Η), 7,27-7,22(m, 2H), 7,08-7,06 (m, 2H), 4,16-4,10 (m, 3Н), 3,87-3,78 (m, 2H), 1,58 (s, 6H).
Примеры 49 и 50
(S)-4-(3-(4-(2,3-Дигидроксипропокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 49
(R)-4-(3-(4-(2,3-Дигидроксипропокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 50
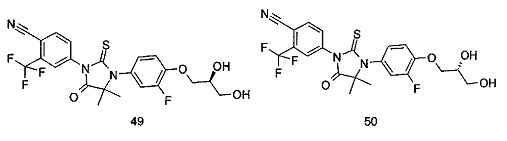
Рацемат 4-(3-(4-(2,3-дигидроксипропокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 13 (1,6 г, белое твердое вещество) хирально разделяли с помощью оборудования для предварительной подготовки и хиральной колонки путем метода ВЭЖХ (условия разделения: хиральная колонка CHIRALPAK AD-Η, размер колонки: 3 см внутренний диаметр × 25 длина, подвижная фаза: н-гексан:изопропанол=80:20 (об/об), скорость потока – 25 мл/минута). Соответствующие фракции собирали и выпаривали для удаления растворителя с получением продукта, указанного в названии, (S)-4-(3-(4-(2,3-дигидроксипропокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 49 (745 мг, выход 46,6%) в виде твердого белого вещества и
(R)-4-(3-(4-(2,3-дигидроксипропокси)-3-фторфенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрила 50 (796 мг, выход 49,8%) в виде твердого белого вещества.
49: МС m/z (ИЭР):498,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,95 (m, 2Н), 7,84-7,82 (m, 1Н), 7,15-7,04 (m, 3Н), 4,22-4,18 (m, 3Н), 3,91-3,79 (m, 2Н), 1,59 (s, 6Н).
50: МС m/z (ИЭР):498,3 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7,99-7,95 (m, 2Н), 7,84-7,82 (m, 1Н), 7,15-7,04 (m, 3Н), 4,22-4,18 (m, 3Н), 3,91-3,79 (m, 2Н), 1,59 (s, 6Н).
Пример 51
4-(3-(4-Циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенил трифторметансульфонат
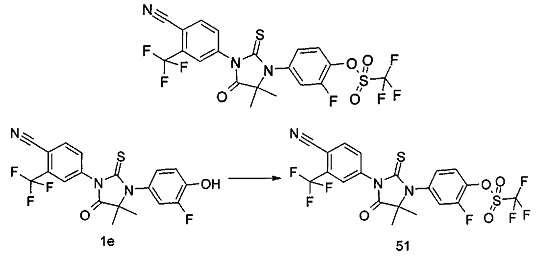
4-(3-(3-Фтор-4-гидроксифенил)-4,4-диметил-5-оксо-2-тиоксоимидазолидин-1-ил)-2-(трифторметил)бензонитрил 1е (200 мг, 0,47 ммоль) растворяли в 4 мл дихлорметана на ледяной бане с последующим добавлением триэтиламина (0,2 мл 1,42 ммоль) и трифторметансульфоновой кислоты (160 мг, 0,57 ммоль). Реактор продували аргоном 3 раза и перемешивали в течение 1 часа с последующим добавлением 20 мл смеси дихлорметана и воды (об./об.-1:1). Реакционный раствор расслаивали, и водную фазу экстрагировали дихлорметаном (10 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при сниженном давлении с получением соединения, указанного в названии, 4-(3-(4-циано-3-(трифторметил)фенил)-5,5-диметил-4-оксо-2-тиоксоимидазолидин-1-ил)-2-фторфенил трифторметансульфоната 51 (250 мг, выход 95,3%) в виде коричневого твердого вещества.
МС m/z (ИЭР):614,1 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 8,01-7,95 (m, 2Н), 7,84-7,83 (m, 1Н), 7,57-7,52(m, 1Н), 7,32-7,29 (m, 1Н), 7,27-7,24 (m, 1Н), 1,59 (s, 6Н)
ПРИМЕРЫ ИСПЫТАНИЙ
Биологический анализ
Изобретение дополнительно проиллюстрировано со ссылкой на следующие примеры испытаний. Следует понимать, что данные примеры предназначены только для демонстрации изобретения без ограничения объема настоящего изобретения.
Экспериментальные методы в следующих примерах, для которых не указаны конкретные условия, проводят при традиционных условиях или при условиях, рекомендованных производителем. Экспериментальные реагенты, для которых не указаны конкретные источники, являются традиционными реагентами, как правило, доступными на рынке.
Пример тестирования 1. Биологическая оценка ингибиторной активности на секрецию ПСА (простатоспецифический антиген) клеток LNcap
Следующий in vitro анализ направлен на определение активности соединений по настоящему изобретению в ингибировании секреции ПСА клетками LNcap.
1. Материалы и приборы
(1) Среда для культивирования RIPM 1640 (Hyclone, SH30809.01B)
(2) Эмбриональная телячья сыворотка, FBS (GIBCO, 10099)
(3) Пенициллин-стрептомицин (GIBCO, 15140-122)
(4) CS-FBS (Эмбриональная телячья сыворотка, очищенная на активированном угле) (SERANA, 1090111)
(5) Клетки линии LNcap (клеточный банк Академии наук Китая, TCHu173)
(6) Тестостерон (Dr.Ehrenstorfer Gmbh, С 17322500, LOT: 10519)
(7) Набор реагентов PSA kit (abnova, KA0208)
(8) Считыватель микропланшетов NOVO Star
(9) Клеточная культура: среда для культивирования RIPM 1640, 10% FBS, 1% пенициллин-стрептомицин
(10) Среда для культивирования с очисткой на активированном угле: RPMI 1640, 10% CS-FBS, 1% Пенициллин-стрептомицин
(11) Среда для культивирования с тестостероном: RIPM 1640, 10% CS-FBS, 1% Пенициллин-стрептомицин, 1 нМ тестостерон.
2. Экспериментальная методика
Клетки линии LNcap культивировали в среде для культивирования клеток, ресуспендировали в среде для культивирования с очисткой на активированном угле и затем сеяли на 96-луночный планшет в объеме 100 мкл/лунка с плотностью посева 5×104 клеток/лунка.
Тестируемые соединения растворяли в ДМСО (диметилсульфоксид) и готовили в виде 10 мМ стоковых растворов. При применении стоковые растворы разводили в 5× ДМСО и затем разводили с трехкратным градиентом с получением 8 градиентных концентраций. Затем, каждую из 8 градиентных концентраций разводили в 20× среде для культивирования с тестостероном (для того, чтобы обеспечить 0,5% концентрацию ДМСО в каждой культуральной системе). Разведенные тестируемые соединения сохраняли для тестирования.
10 мкл разведенных тестируемых соединений добавляли в 96-луночный планшет и тщательно перемешивали. 10 мкл 0,5% ДМСО, разведенного средой для культивирования с очисткой на активированном угле, добавляли в пустую группу, и 10 мкл 0,5% ДМСО, разведенного средой для культивирования с тестостероном, добавляли в контрольную группу. 96-луночный планшет культивировали в инкубаторе при условиях 37°С, 5% СО2.
3. Результаты тестирования
После 4 дней инкубации, собирали 60 мкл культурального супернатанта и измеряли значение оптической плотности OD450 на лунку с помощью считывателя микропланшетов NOVO Star с применением набора реагентов PSA kit (abnova, KA0208).
Значения IC50 (концентрация полумаксимального ингибирования) рассчитывали из данных об уровнях ингибирования тестируемых соединений в различных концентрациях.
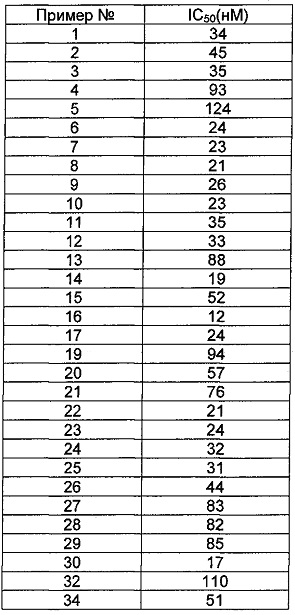
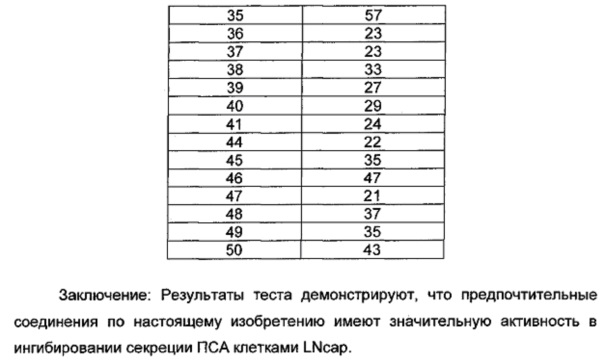
Пример тестирования 2. Биологическая оценка ингибирования пролиферации клеток LNcap
Следующий in vitro анализ направлен на определение активности соединений по настоящему изобретению для ингибирования пролиферации клеток LNcap.
1. Материалы и приборы:
(1) Реагент для подсчета клеток CCK-8 (DOJINDO, CK04)
(2) Считыватель микропланшетов Victor3 (Perkin Elmer, 1420).
2. Экспериментальная методика
См. экспериментальную методику из Примера тестирования 1.
3. Результаты тестирования
После культивирования клеток LNcap в течение 3 дней, половину объема жидкости культуральной среды, тестируемых соединений, пустой группы и контрольной группы обновляли, и культивировали еще 3 дня. Затем 96-луночные планшеты тщательно перемешивали с 10 мкл CCK-8 и культивировали в инкубаторе в течение 2-3 часов при условиях 37°С, 5% СО2. Значение OD на лунку измеряли при 450 нм с помощью считывателя микропланшетов Victor3.
Значения IC50 рассчитывали из данных об уровнях ингибирования тестируемых соединений в различных концентрациях.
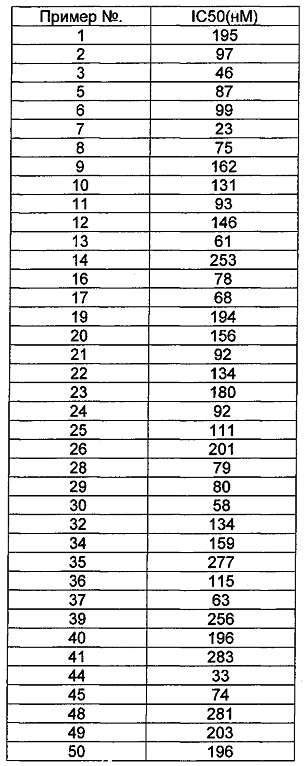

Пример тестирования 3. Биологическая оценка клеток LNcap-AR Следующий in vitro анализ направлен на определение активности высоких концентраций соединений по настоящему изобретению в активировании секреции ПСА клетками LNcap-AR (клетками LNcap со сверхэкспрессией АР (для краткости клетками LNcap-AR), полученных с помощью ретровирусной трансфекции).
1. Клеточная культура
Клетки линии LNcap-AR культивировали в среде для культивирования клеток, ресуспендировали в среде для культивирования с очисткой на активированном угле и затем сеяли на 96-луночный планшет в объеме 100 мкл/лунка с плотностью посева 5×104 клеток/лунка.
Тестируемые соединения растворяли в ДМСО (диметилсульфоксид) и готовили в виде 10 мМ стоковых растворов. При применении стоковые растворы разводили в ДМСО до двух концентраций 2 мМ и 0,6 мМ. Затем, каждую из градиентных концентраций разводили в 20× среде для культивирования, с очисткой на активированном угле (для того, чтобы обеспечить 0,5% концентрацию ДМСО в каждой культуральной системе). Разведенные тестируемые соединения сохраняли для тестирования.
10 мкл разведенных тестируемых соединений добавляли в 96-луночный планшет и тщательно перемешивали. 10 мкл 0,5% ДМСО, разведенного средой для культивирования с очисткой на активированном угле, добавляли в пустую группу.
2. Результаты тестирования
После 4 дней инкубации, собирали 60 мкл культурального супернатанта и измеряли значение оптической плотности OD ПСА на лунку с помощью считывателя микропланшетов NOVO Star с применением набора реагентов PSA kit (abnova, KA0208).
Агонистическую активность тестируемых соединений по настоящему изобретению на секрецию ПСА клетками LNcap-AR оценивали с помощью процентного соотношения значение OD (тестируемое соединение)/ значение OD (пустой контроль). Если значение процентного соотношения выше 100, тестируемые соединения имеют агонистическую активность на секрецию ПСА клетками LNcap-AR по сравнению с пустым контролем. Наоборот, если значение процентного соотношения менее 100, тестируемые соединения не имеют агонистической активности на секрецию ПСА клетками LNcap-AR по сравнению с пустым контролем. Конкретные результаты тестирования приведены ниже:
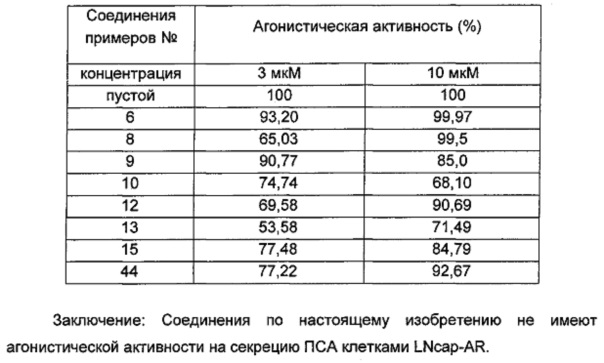
АНАЛИЗ ФАРМАКОКИНЕТИКИ
Пример тестирования 4. Анализ фармакокинетики соединений по настоящему изобретению.
1. Краткое описание
Самцов крысы применяли в качестве экспериментальных животных. Соединения Примера 6, Примера 9, Примера 11, Примера 13, Примера 15, Примера 16, Примера 17, Примера 44 и Примера 45 вводили крысам внутрижелудочно для определения концентрации лекарственного препарата в плазме на различных временных точках с помощью метода ЖХ/МС/МС (жидкостная хроматография с тандемной масс-спектрометрией). Фармакокинетические свойства соединений по настоящему изобретению изучали и оценивали на крысах.
2. Протокол
2.1. Образцы
Соединения Примера 6, Примера 9, Примера 11, Примера 13, Примера 15, Примера 16, Примера 17, Примера 44 и Примера 45
2.2. Экспериментальные животные
27 здоровых взрослых самцов крысы Sprague-Dawley (SD), приобретенных в SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, Certificate No.: SCXK (Шанхай) 2008-0016, разделяли на 9 групп, по 3 крысы в каждой группе.
2.3. Приготовление тестируемых соединений
Соответствующие количества тестируемых соединений взвешивали и смешивали с 0,5% КМЦ-Na (натрий-карбоксиметилцеллюлоза) для приготовления 0,5 мг/мл суспензии с помощью ультразвукового метода.
2.4. Введение
После ночного голодания 27 крыс SD делили на 9 групп, и вводили им внутрижелудочно дозу 5,0 мг/кг, при объеме введения 10 мл/кг.
3. Способ
Соединения Примера 6, Примера 9, Примера 11, Примера 13, Примера 15, Примера 16, Примера 17, Примера 44 и Примера 45 вводили крысам внутрижелудочно. Образцы крови (0,1 мл) отбирали из орбитального синуса до введения и через 0,5 ч, 1,0 ч, 2,0 ч, 3,0 ч, 4,0 ч, 6,0 ч, 8,0 ч, 11,0 ч и 24,0 после введения, хранили в гепаринизированных пробирках и центрифугировали в течение 3 минут при 3,500 об/мин для отделения плазмы крови. Образцы плазмы хранили при -20°С. Крыс кормили через 2 часа после введения.
Концентрацию различных соединений в плазме крыс после внутрижелудочного введения определяли с помощью метода ЖХ/МС/МС. Линейность метода составляет 1,00-2000 нг/мл. Образцы плазмы анализировали после осаждения белка, которое осуществляли путем добавления метанола.
4. Результаты Фармакокинетических Параметров
Фармакокинетические Параметры соединений по настоящему изобретению представляли собой следующие:
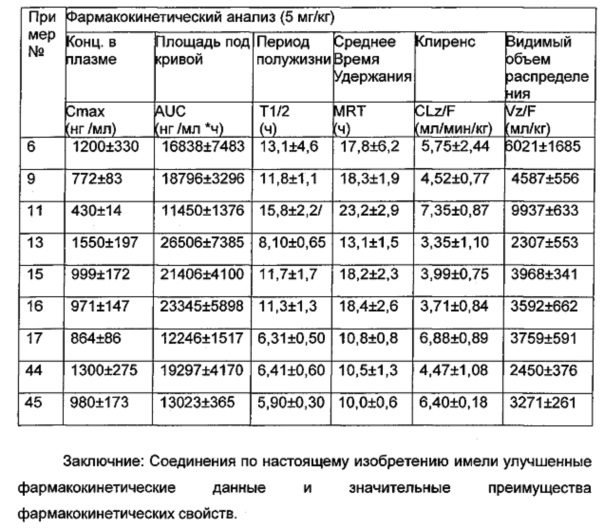
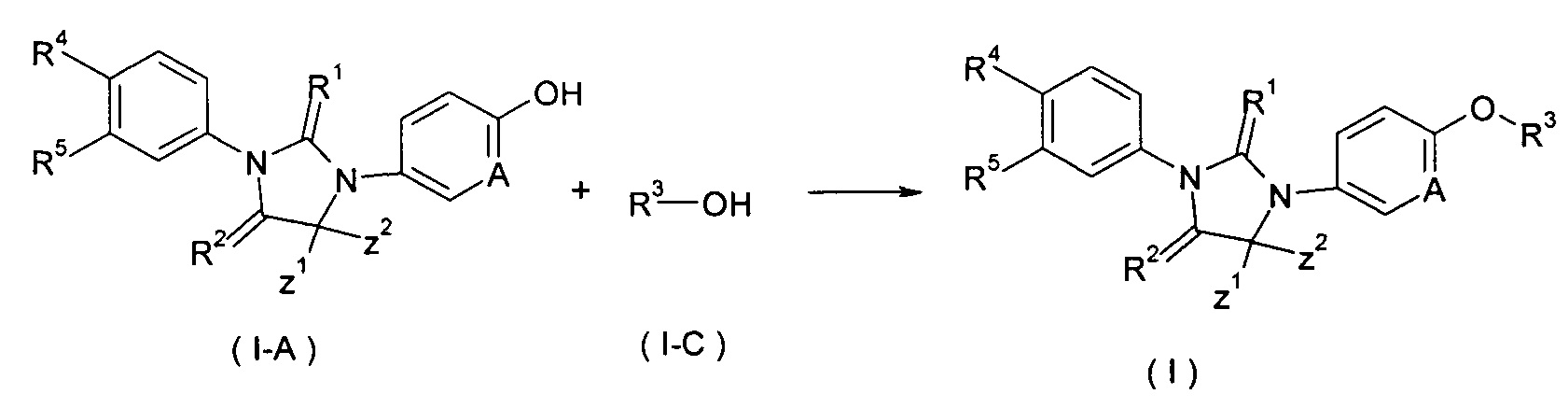
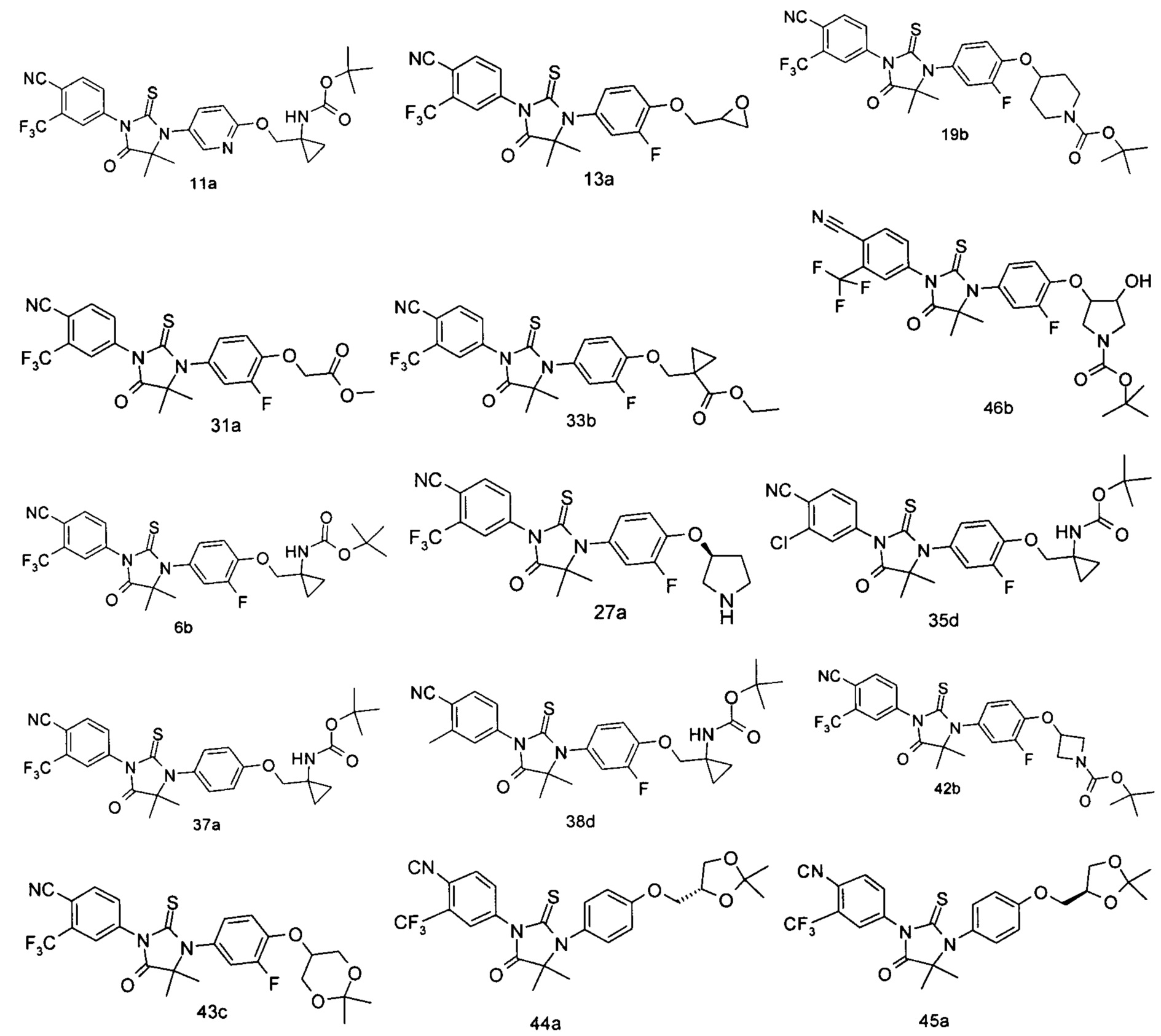
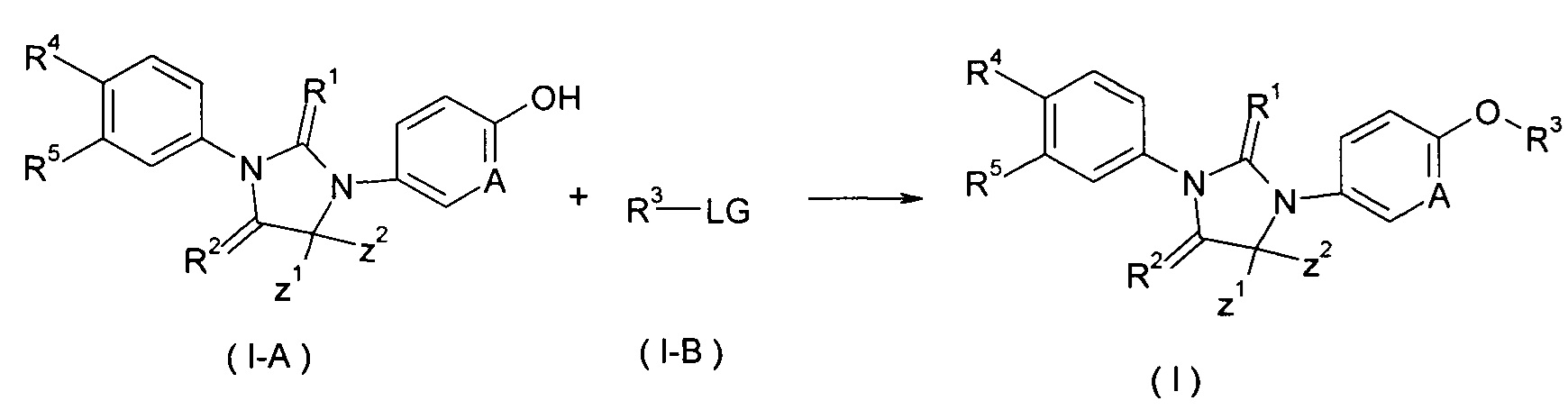
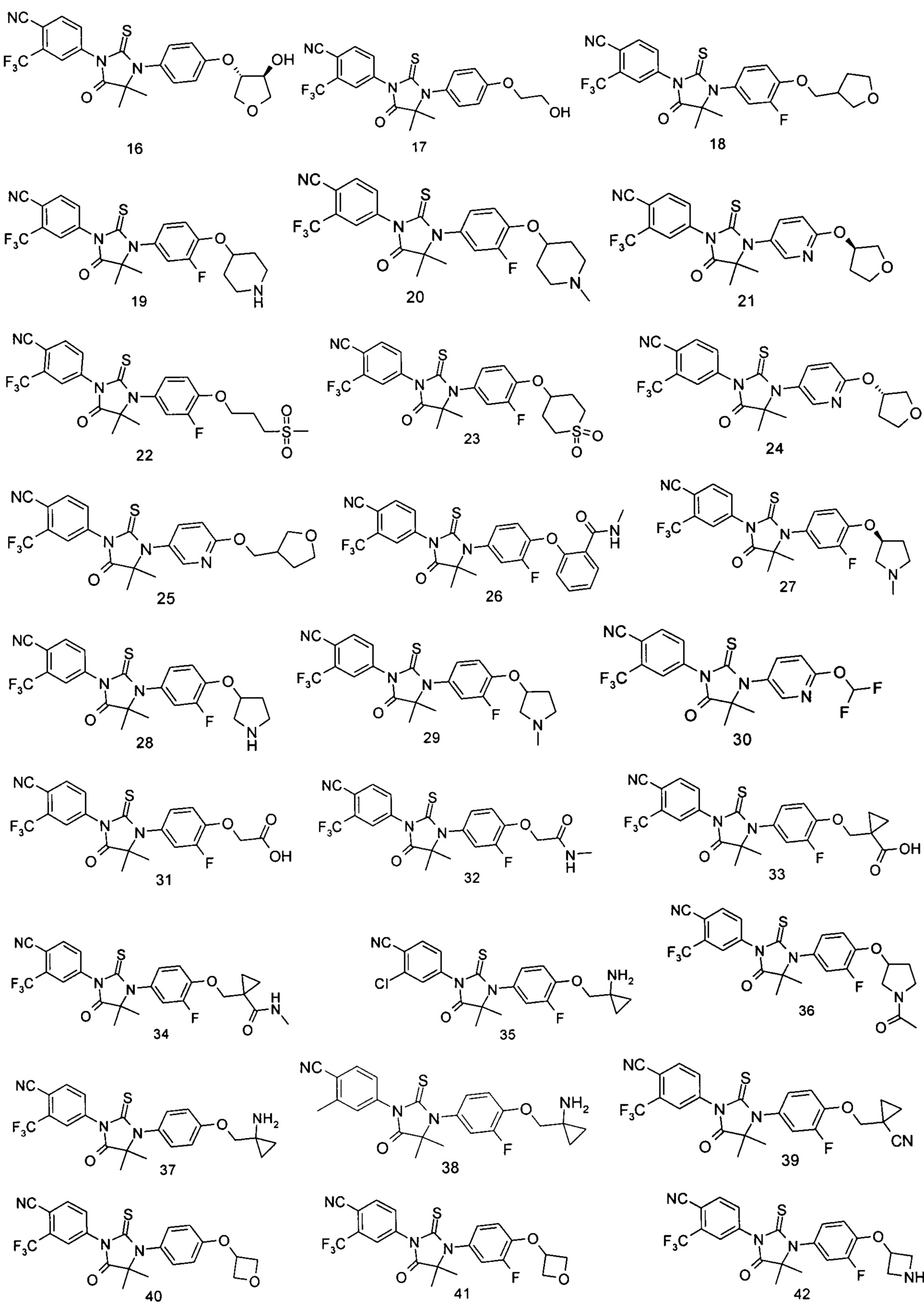
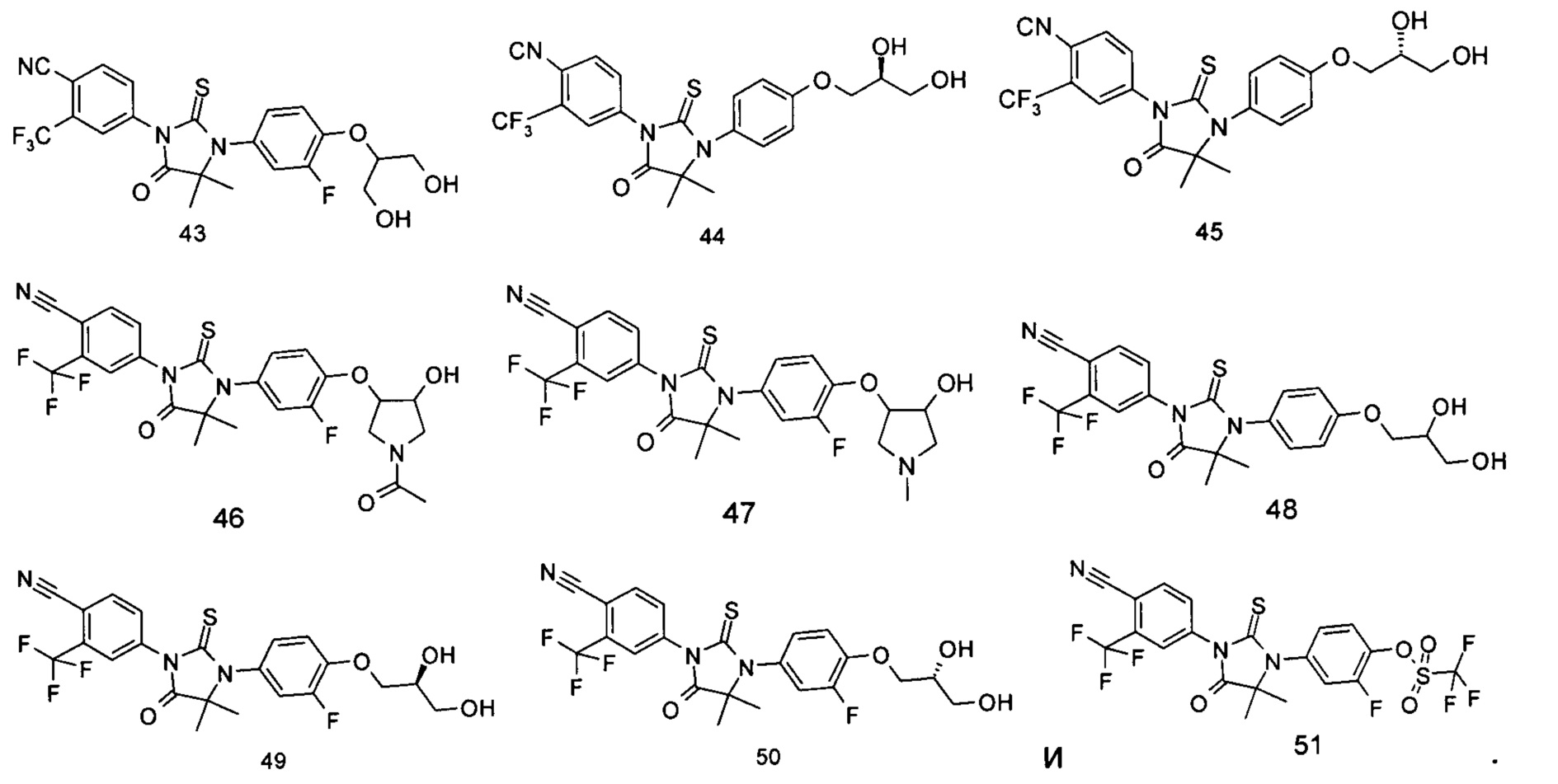
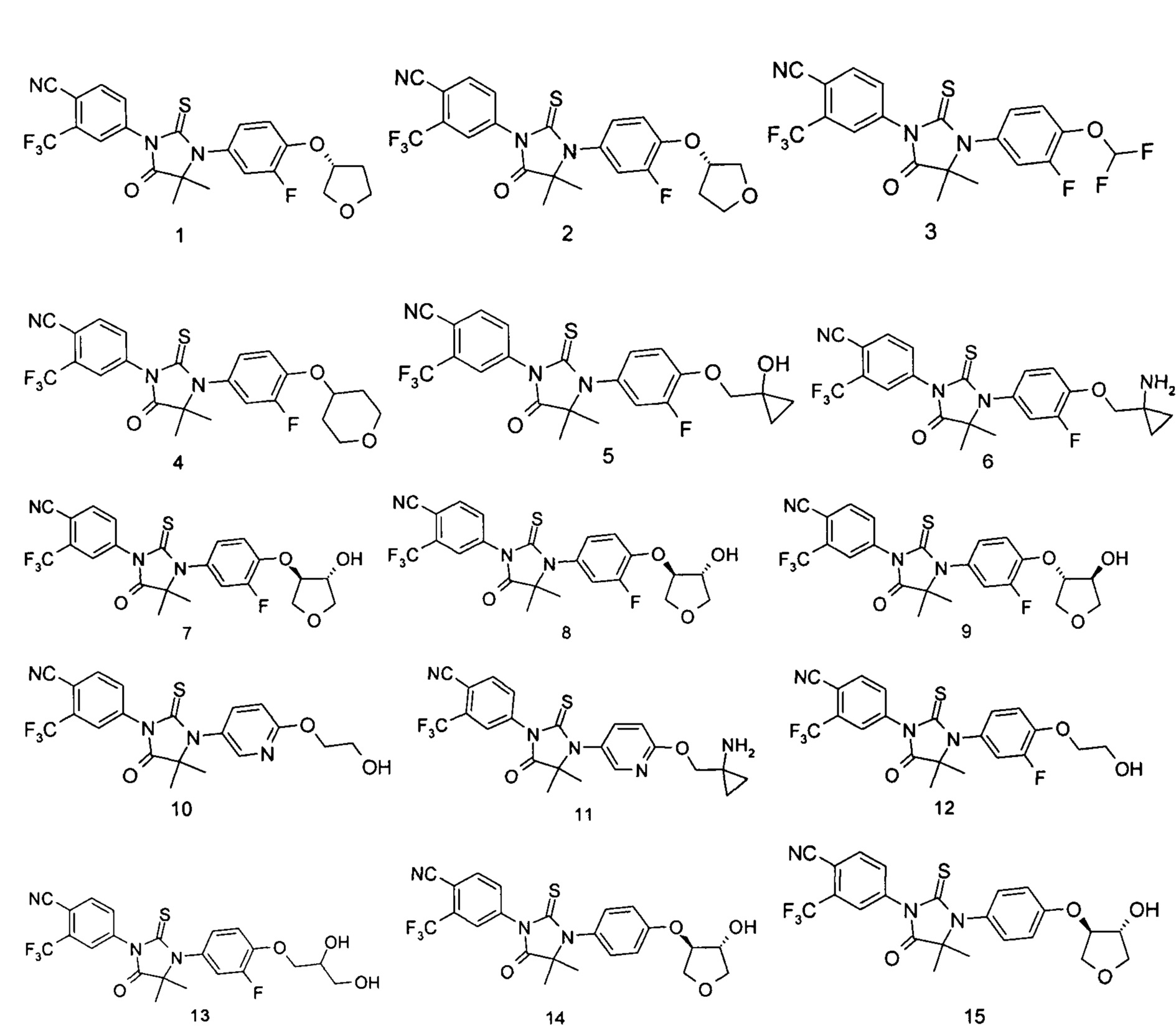
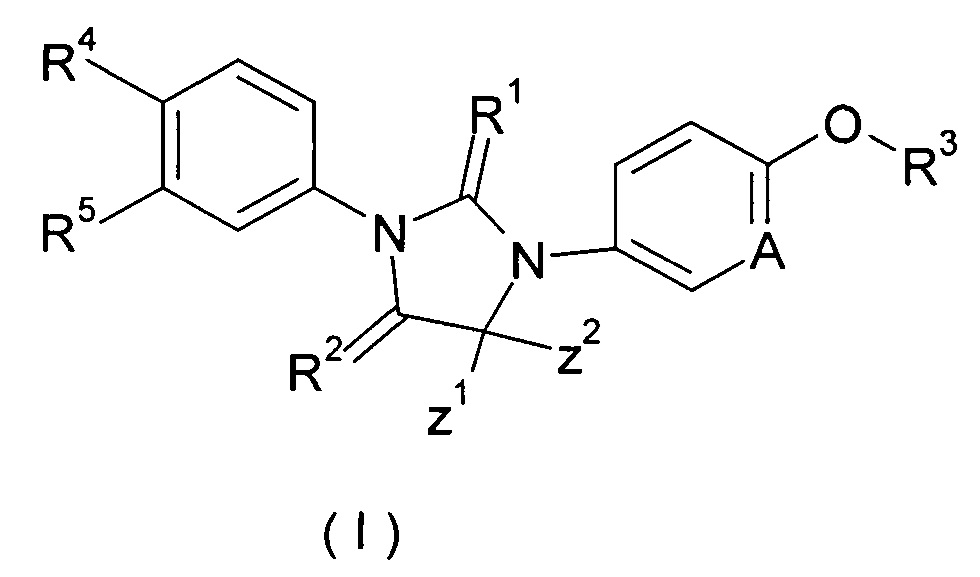
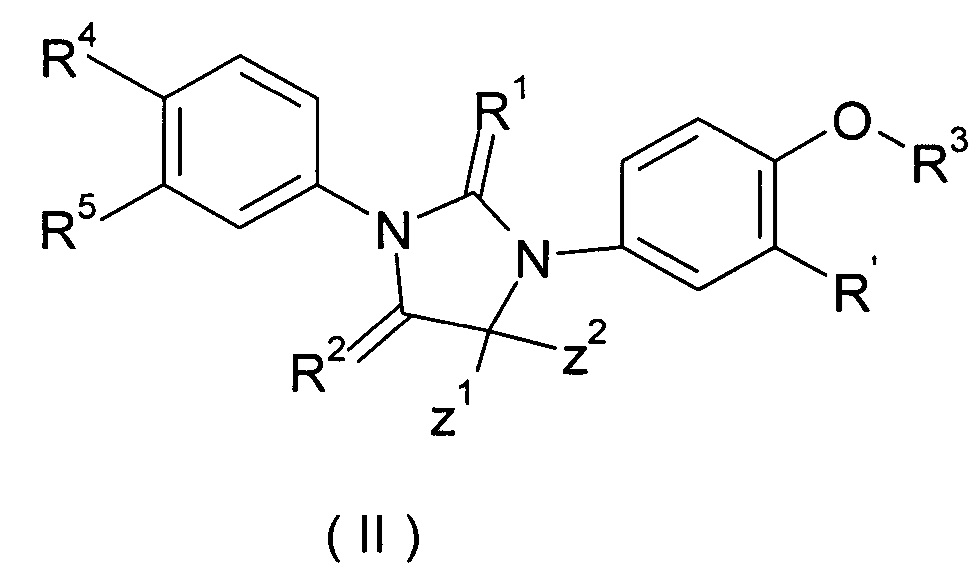
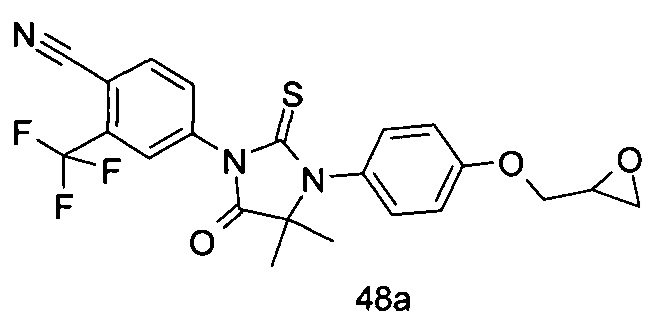
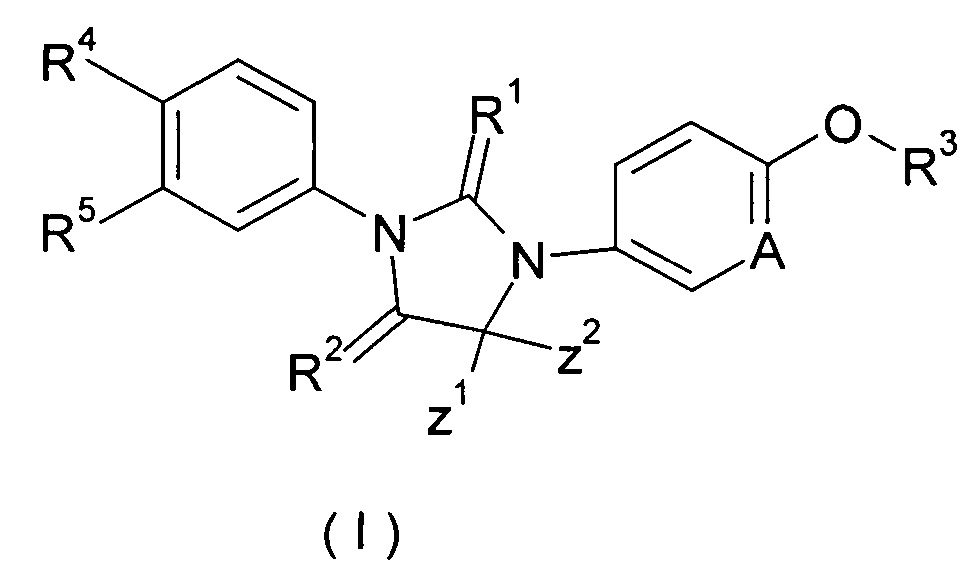
Соединения 2-(2-оксоиндолин-3-илиден)метил-5-(2-гидрокси-3-морфолин-4-илпропил)-6,7 дигидро-1-н-пиррол[3,2-с]пиридин-4(5н)-она и их применение в качестве ингибиторов протеинкиназы
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Производные дициклоазаалкана, способы их получения и их применение в медицине
Бициклозамещенные азопроизводные пиразолона, способ их получения и фармацевтическое применение
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Фармацевтическая композиция для лечения диабета 2 типа
Производные 6-аминохиназолина или 3-цианохинолина, способы их получения и их применение в качестве ингибитора рецепторных тирозинкиназ egfr или her-2
Соединения 2-(2-оксоиндолин-3-илиден)метил-5-(2-гидрокси-3-морфолин-4-илпропил)-6,7 дигидро-1-н-пиррол[3,2-с]пиридин-4(5н)-она и их применение в качестве ингибиторов протеинкиназы
Гетероциклические азотистые производные пиррола, их получение и фармацевтическое применение
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Производные дициклоазаалкана, способы их получения и их применение в медицине
Бициклозамещенные азопроизводные пиразолона, способ их получения и фармацевтическое применение
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Фармацевтическая композиция для лечения диабета 2 типа

