Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ДИЦИКЛОАЗААЛКАНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В МЕДИЦИНЕ
Вид РИД
Изобретение
Область изобретения
Изобретение относится к производным азабициклоалкана, способу их получения содержащим их композициям, и к их применению, в частности их фармацевтическому применению в качестве ингибиторов дипептидилпептидазы IV (DPP-IV).
Предшествующий уровень техники
Диабет относится к заболеванию, происходящему в силу множества причинных факторов, и характеризующемуся повышенными уровнями глюкозы в плазме крови или гипергликимией, наряду с заболеваниями метаболизма сахаров, жиров и белков, к которым приводят дефекты выделения инсулина и/или его действия. Диабет представляет собой древнее заболевание, при нем человеческий организм абсолютно или относительно лишен инсулина, что приводит к повышенным концентрациям глюкозы в крови. Глюкоза в основном выделяется с мочой. Высокие уровни глюкозы в крови могут вызывать несколько проблем, таких как полидипсия, частое мочеиспускание, сниженный аппетит, потеря массы организма, головокружение, усталость, а также другие симптомы.
Дипептидилпептидаза-IV (DPP-IV) представляет собой сериновую пептидазу, которая расщепляет N-концевые дипептиды пептидной цепи, содержащей, предпочтительно, остатки пролина в предпоследнем положении. Хотя ее биологическая роль в системах млекопитающих не была установлена окончательно, предполагают, что DPP-IV играет важную роль в метаболизме нейропептидов, активации Т-клеток, прикреплении раковых клеток к эндотелию и проникновению ВИЧ (вируса иммунодефицита) в лимфоидные клетки (WO 98/19998).
Совсем недавно было описано, что DPP-IV отвечает за ингибирование секреции глюкагон-подобного пептида (GLP)-1. В частности, DPP-IV расщепляет аминоконцевой His-Ala дипептид GLP-1, разрушая активный GLP-1(7-36)NH2 в неактивный GLP-1(9-36)NH2 (Endocrinology, 1999, 140: 5356-5363). В физиологических условиях период полувыведения целого GLP-1 в системе кровообращения мал, неактивный метаболит GLP-1, который разрушается с помощью DPP-IV, может связываться с рецептором GLP-1, вызывая противодействие активному GLP-1, таким образом укорачивается физиологический ответ на GLP-1. Эндогенный, и даже экзогенный, GLP-1 можно полностью защитить с помощью ингибитора DPP-IV от разрушения DPP-IV, а также биологическую активность GLP-1 можно значительно повысить (от 5 до 10 раз). Поскольку GLP-1 представляет собой главный стимулятор выработки инсулина поджелудочной железой и может оказывать непосредственный эффект на потребление глюкозы, инигибитор DPP-IV необходим для лечения инсулиннезависимого сахарного диабета (NIDDM) (US 6110949).
Краткое изложение сущности изобретения
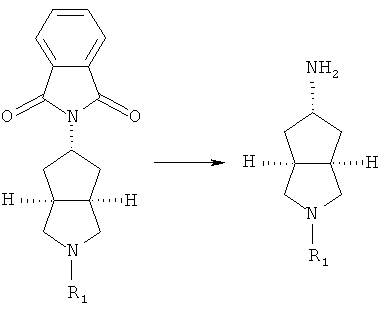
Соответственно настоящее изобретение относится к соединениям, представленным формулой (I), или их фармацевтически приемлемым солям.
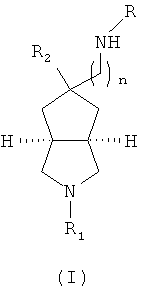
где R выбран из группы, состоящей из алкила, циклоалкила, галогеноалкила, арила, гетероарила, аминокарбонилалкила, амидалкила, аминокарбонилалкила, имеющего гетероцикл и аминоалкил, где гетероцикл представляет собой 5- или 6-членное гетероциклическое кольцо, дополнительно замещенное одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галоалкоксила, гидроксила, амино, алкиламина, амида, аминокарбонила, циано, алкинила, алкоксила, арилоксила, аминоалкила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты и галогена;
R1 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, -C(O)NR3R4, -C(O)R3 и -C(O)OR3, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гидроксила, амино-, алкоксила-, арилоксила- и гетероциклического алкила;
R2 выбран из группы, состоящей из водорода и алкила, где алкил дополнительно замещен одной или более чем одной группой, независимо выбранной из группы, состоящей из циклоалкила и арила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил дополнительно необязательно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероарилоксила, галогена, гидроксила, амино-, циано-, гидроксиалкила-, гетероциклического алкила, гетероциклического алкоксила, трифторметила, карбоновой кислоты и эфира карбоновой кислоты; и
R3 и R4 необязательно соединены вместе с атомом N с образованием 3-8-членного гетероциклического кольца, где 3-8-членное гетероциклическое кольцо дополнительно необязательно содержит один или более чем один гетероатом, выбранный из N, O и S, а 3-8-членное кольцо дополнительно необязательно замещено одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галоалкоксила, гидроксила, амино-, циано-, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты, галогена и -NR3R4; и
n представляет собой целое число от 0 до 4.
Кроме того, настоящее изобретение включает соединения, имеющие формулу (IA), или их фармацевтически приемлемые соли:
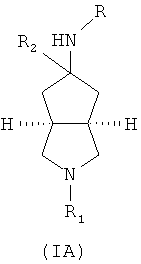
где R выбран из группы, состоящей из алкила, циклоалкила, галогеноалкила, арила, гетероарила, аминокарбонилалкила, амидалкила, аминокарбонилалкила, имеющего гетероцикл и аминоалкил, где гетероцикл представляет собой 5- или 6-членное гетероциклическое кольцо, дополнительно замещенное одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галогеноалкоксила, гидроксила, амино, алкиламина, амида, аминокарбонила, циано, алкинила, алкоксила, арилоксила, аминоалкила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты и галогена;
R1 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, -C(O)NR3R4, -C(O)R3 и -C(O)OR3, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил необязательно дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гидроксила, амино-, алкоксила-, арилоксила- и гетероциклического алкила;
R2 выбран из группы, состоящей из водорода и алкила, где алкил дополнительно замещен одной или более чем одной группой, независимо выбранной из группы, состоящей из циклоалкила и арила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил необязательно дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероарилоксила, галогена, гидроксила, амино-, циано-, гидроксиалкила-, гетероциклического алкила, гетероциклического алкоксила, трифторметила, карбоновой кислоты и эфира карбоновой кислоты; и
R3 и R4 необязательно соединены вместе с атомом N с образованием 3-8-членного гетероциклического кольца, где 3-8-членное гетероциклическое кольцо необязательно дополнительно содержит один или более чем один гетероатом, выбранный из N, O и S, а 3-8-членное кольцо необязательно дополнительно замещено одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галогеноалкоксила, гидроксила, амино-, циано-, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты, галогена и -NR3R4.
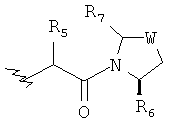
Предпочтительно у соединений, представленных формулой (I), или у их фармацевтически приемлемых солей R имеет следующую формулу:
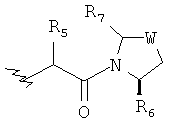
где R5 выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил необязательно дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероарилоксила, галогена, гидроксила, амино, алкиламина, циано, гидроксиалкила, гетероциклического алкила, гетероциклического алкоксила, карбоновой кислоты и эфира карбоновой кислоты;
каждый из R6 и R7 независимо выбран из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галогеноалкоксила, гидроксила, амино, циано, алкинила, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты и галогена; и
W представляет собой C, S или O, где C необязательно дополнительно замещен на R6 или R7.
Кроме того, настоящее изобретение включает соединения,
представленные формулой (IB), или их фармацевтически приемлемые соли:
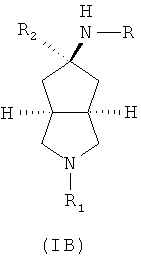
где R имеет следующую формулу:
R1 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, -C(O)NR3R4, -C(O)R3 и -C(O)OR3, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гидроксила, амино, алкоксила, арилоксила и гетероциклического алкила;
R2 выбран из группы, состоящей из водорода и алкила, где алкил дополнительно замещен одной или более чем одной группой, независимо выбранной из группы, состоящей из циклоалкила и арила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил дополнительно замещены одной или более чем одной группой, выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероарилоксила, галогена, гидроксила, амино, циано, гидроксиалкила, гетероциклического алкила, гетероциклического алкоксила, трифторметила, карбоновой кислоты и эфира карбоновой кислоты; и
R3 и R4 связаны вместе атомом N с образованием 3-8-членного гетероциклического кольца, где 3-8-членное гетероциклическое кольцо дополнительно содержит один или более чем один гетероатом, выбранный из N, О и S, а 3-8-членное кольцо дополнительно замещено одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галогеноалкоксила, гидроксила, амино, циано, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты, галогена и -NR3R4;
R5 выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероарилоксила, галогена, гидроксила, амино, алкиламина, циано, гидроксиалкила, гетероциклического алкила, гетероциклического алкоксила, карбоновой кислоты и эфира карбоновой кислоты;
каждый из R6 и R7 независимо выбран из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галогеноалкоксила, гидроксила, амино, циано, алкинила, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты и галогена; и
W представляет собой C, S или O, где C дополнительно замещен R6 или R7.
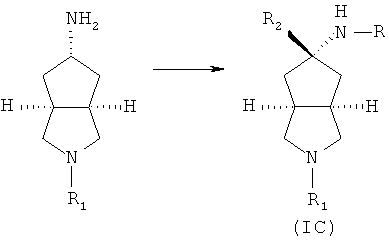
Кроме того, настоящее изобретение включает соединения, представленные формулой (IC), или их фармацевтически приемлемые соли:
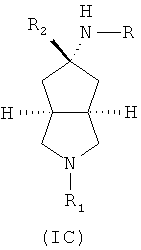
где R имеет следующую формулу:
R1 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, -C(O)NR3R4, -C(O)R3 и -C(O)OR3, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гидроксила, амино, алкоксила, арилоксила и гетероциклического алкила;
R2 выбран из группы, состоящей из водорода или алкила, где алкил дополнительно замещен одной или более чем одной группой, независимо выбранной из группы, состоящей из циклоалкила и арила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероарилоксила, галогена, гидроксила, амино, циано, гидроксиалкила, гетероциклического алкила, гетероциклического алкоксила, трифторметила, карбоновой кислоты и эфира карбоновой кислоты; и
R3 и R4 связаны вместе атомом N с образованием 3-8-членного гетероциклического кольца, где 3-8-членное гетероциклическое кольцо дополнительно содержит один или более чем один гетероатом, выбранный из N, O и S, а 3-8-членное кольцо дополнительно замещено одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галогеноалкоксила, гидроксила, амино, циано, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты, галогена и -NR3R4;
R5 выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероарилоксила, галогена, гидроксила, амино, алкиламина, циано, гидроксиалкила, гетероциклического алкила, гетероциклического алкоксила, карбоновой кислоты и эфира карбоновой кислоты;
каждый из R6 и R7 независимо выбран из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галогеноалкоксила, гидроксила, амино, циано, алкинила, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты и галогена; и
W представляет собой C, S или O, где C дополнительно замещен R6 или R7.
В настоящем изобретении предложены соединения, представленные формулой (I), или их фармацевтически приемлемые соли, где соли получены с помощью взаимодействия соединений, представленных формулами (I), с кислотами, выбранными из группы, состоящей из соляной кислоты, пара-толуолсульфоновой кислоты, винной кислоты, малеиновой кислоты, молочной кислоты, метансульфоновой кислоты, серной кислоты, ортофосфорной кислоты, лимонной кислоты, уксусной кислоты и трифторуксусной кислоты (TFA). Предпочтительными являются пара-толуолсульфоновая кислота, соляная кислота, винная кислота и трифторуксусная кислота.
В наиболее предпочтительном воплощении соединения, представленные формулой (I), или их фармацевтически приемлемые соли включают:
|
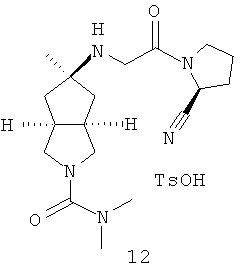
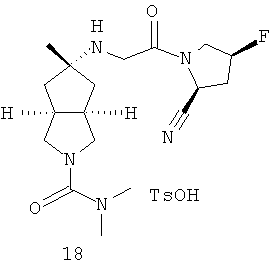
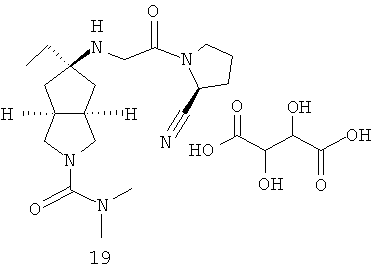
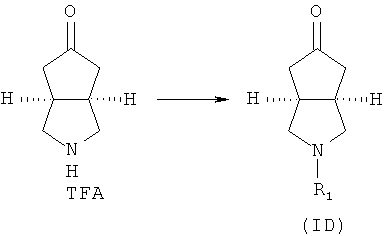
Кроме того, изобретение относится к соединениям, представленным формулой (ID), как промежуточным продуктам в синтезе соединений, имеющих формулу (I):
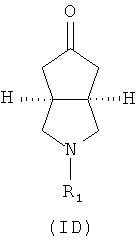
где R1 выбран из группы, состоящей из водорода, алкила, циклоалкила, гетероциклического алкила, арила, гетероарила, -C(O)NR3R4, -C(O)R3 и -C(O)OR3, где алкил, циклоалкил, гетероциклический алкил, арил или гетероарил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, арила, гидроксила, амино-, алкоксила, арилоксила и гетероциклического алкила;
каждый из R3 и R4 независимо выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероарилоксила, галогена, гидроксила, амино, циано, гидроксиалкила, гетероциклического алкила, гетероциклического алкоксила, трифторметила, карбоновой кислоты и эфира карбоновой кислоты; и
R3 и R4 связаны вместе с атомом N с образованием 3-8-членного гетероциклического кольца, где 3-8-членное гетероциклическое кольцо дополнительно содержит один или более чем один гетероатом, выбранный из N, O и S, а 3-8-членное кольцо дополнительно замещено одной или более чем одной группой, выбранной из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галогеноалкоксила, гидроксила, амино, циано, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты, галогена и -NR3R4.
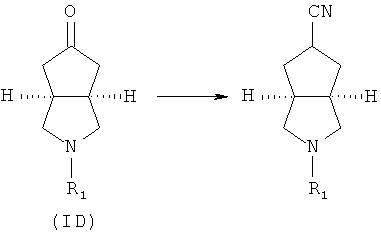
Кроме того, данное описание относится к способу получения соединений, представленных формулой (ID), включающему следующие стадии:
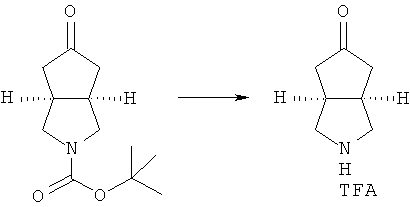
исходное вещество эфир трет-5-оксогексагидроциклопента[c]пиррол-2-карбоновой кислоты приводят во взаимодействие с трифторуксусной кислотой в подходящем растворителе, таком как дихлорметан, при охлаждении в бане с ледяной водой с получением гексагидроциклопента[c]пиррол-5-он трифторацетата; и
гексагидроциклопента[c]пиррол-5-он трифторацетат приводят во взаимодействие с соответствующим ацилхлоридом или эфиром в присутствии основания с получением соединений формулы (ID).
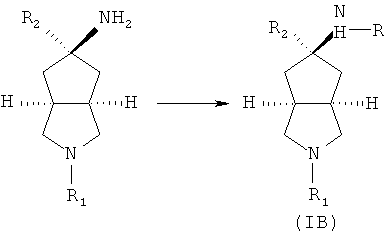
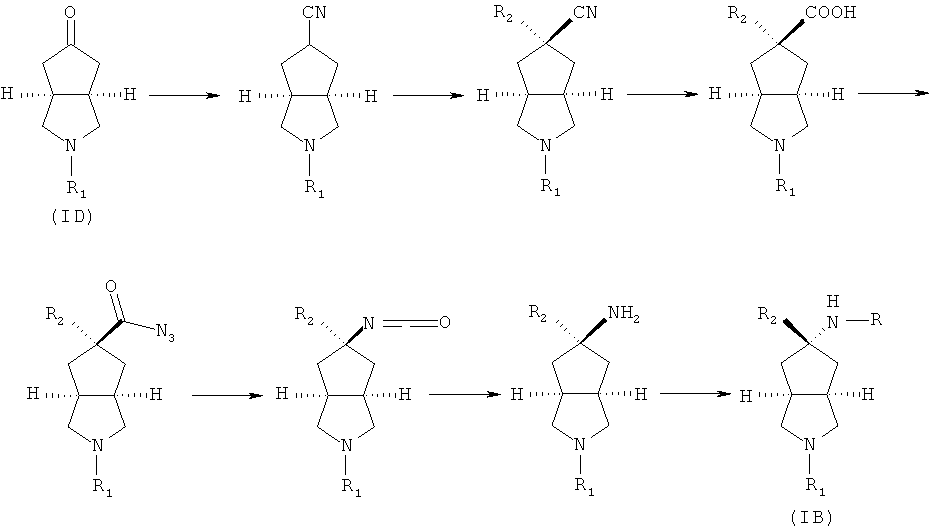
Данное описание относится к способу получения соединений, представленных формулой (IB), включающему следующие стадии:
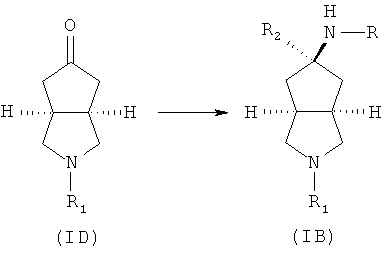
промежуточное соединение формулы (ID) в подходящем растворителе, таком как метанол или этанол, приводят во взаимодействие с амином, замещенным боргидридом натрия, и подходящим основанием, таким как триэтиламин, с получением соединений формулы (IB).
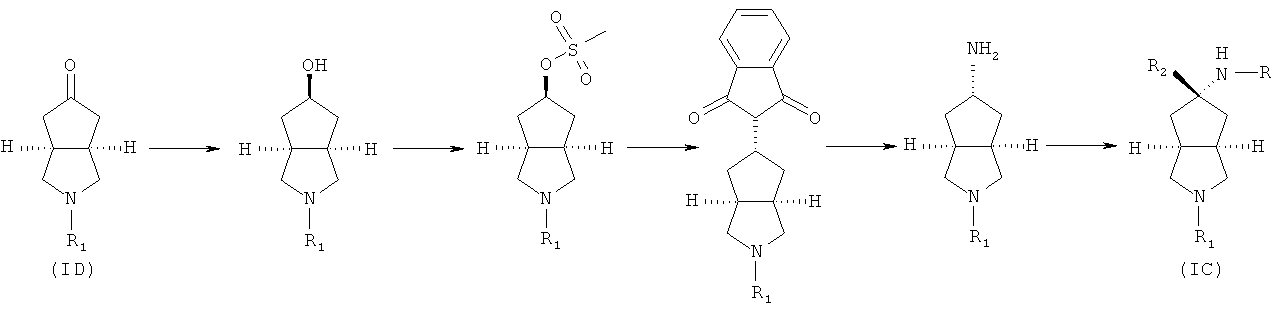
Данное описание относится к способу получения соединений, представленных формулой (IC), включающему следующие стадии:
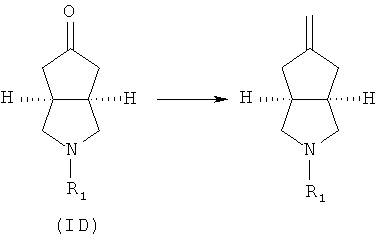
нагревают трет-бутоксид калия и раствор метилтрифенилфосфоний йодида в толуоле; затем добавляют промежуточный продукт формулы (ID) при комнатной температуре с получением азабициклоалкенильного соединения;
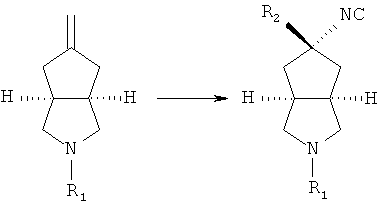
азабициклоалкенильное соединение в подходящем растворителе, таком как дихлорметан, приводят во взаимодействие с триметилсилилцианидом в присутствии перхлората серебра при комнатной температуре с получением азабициклоциано соединения;
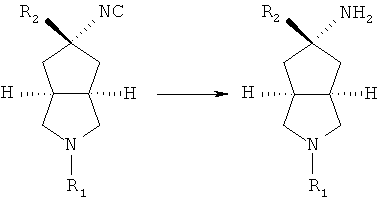
азабициклоциано соединение в подходящем растворителе, таком как этанол, приводят во взаимодействие с подходящей кислотой при комнатной температуре с получением азабициклоамино соединения;
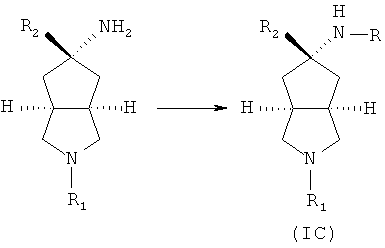
азабициклоамино соединение приводят во взаимодействие с замещенным галогеном соединением в присутствии щелочного растворителя, такого как N,N-диметилформамид, с получением соединения формулы (IC).
Данное описание относится к способу получения соединений, представленных формулой (IB), включающему следующие стадии:
промежуточное соединение формулы (ID) приводят во взаимодействие с тозилметилизоцианидом в подходящем растворителе, таком как этиленгликоль диметиловый эфир, посредством изоцианидной реакции с получением азабициклоциано соединения;
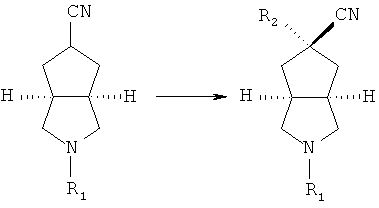
азабициклоциано соединение в подходящем растворителе, таком как тетрагидрофуран, приводят во взаимодействие с галогеносоединением в присутствие гексаметилдисилазида лития с получением R2 замещенного азабициклоциано соединения;
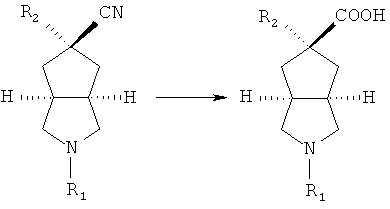
осуществляют гидролиз R2 замещенного азабициклоциано соединения в присутствии кислоты с получением R2 замещенного азабициклокарбоксильного соединения;
или R2 замещенное соединение азабициклоциано приводят во взаимодействие с восстановителем, таким как DIBAL-H, в подходящем растворителе, таком как дихлорметан, в условиях охлаждения в бане с ледяной водой с получением альдегидного соединения; осуществляют взаимодействие альдегидного соединения в смеси растворителей тетрагидрофуран/вода с дигидрофосфатом натрия, хлоритом натрия и 2-метил-2-бутеном в условиях охлаждения в бане с ледяной водой с получением R2 замещенного азабициклокарбоксильного соединения;
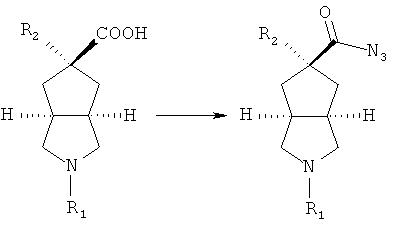
R2 замещенное азабициклокарбоксильное соединение приводят во взаимодействие с этилхлорформатом в присутствии основания, такого как триэтиламин, посредством азидореакции с получением азабициклоазидо соединения;
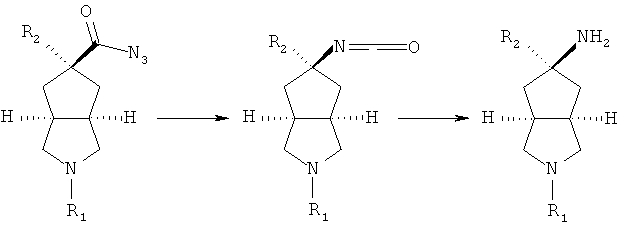
осуществляют нагревание азабициклоазидо соединения в подходящем растворителе, таком как толуол, с последующим помешиванием в кислом растворе; реакционный раствор нейтрализуют до значения pH, демонстрирующего слабощелочное значение, с получением R2 замещенного азабициклоамино соединения;
R2 замещенное азабициклоамино соединение приводят во взаимодействие с галоген-замещенным соединением в присутствии щелочного растворителя, такого как N,N-диметилформамид, с получением соединения формулы (IB).
Данное описание относится к способу получения соединений, представленных формулой (IC), включающему следующие стадии:
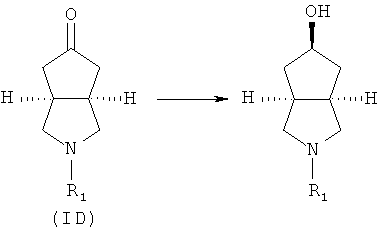
осуществляют взаимодействие промежуточного продукта, имеющего формулу (ID), в подходящем растворителе, таком как тетрагидрофуран, с восстановителем, таким как три-трет-бутоксиалюмогидрид лития, в условиях охлаждения в бане с ледяной водой с получением азабициклогидроксильного соединения;
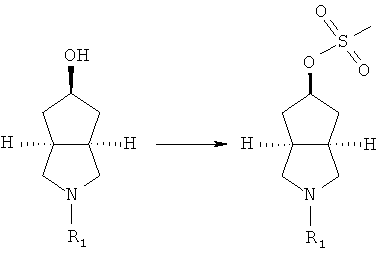
азабициклогидроксильное соединение в подходящем растворителе, таком как дихлорметан, приводят во взаимодействие с щелочным агентом, таким как триэтиламин, и метилсульфонилхлоридом с получением соединения азабициклометилсульфоновой кислоты;
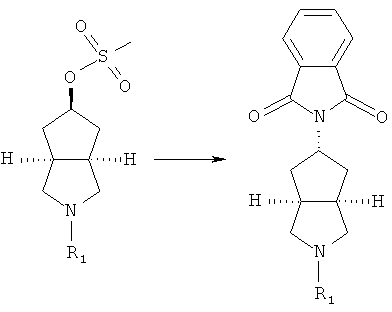
осуществляют нагревание соединения азабициклометилсульфоновой кислоты и фталимида калия в присутствии щелочного агента, такого как N,N-диметилформамид, с получением замещенного фталимидом азабицикло соединения;
осуществляют нагревание замещенного фталимидом азабицикло соединения и гидразина в подходящем растворителе, таком как этанол, с получением азабициклоамино соединения;
осуществляют нагревание азабициклоамино соединения и галогено соединения в подходящем растворителе, таком как дихлорметан, с получением соединения формулы (IC).
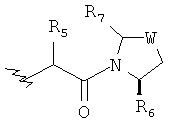
Предпочтительно, в вышеописанном способе получения R имеет следующую формулу:
R5 выбран из группы, состоящей из водорода, алкила, циклоалкила, арила, гетероарила и гетероциклического алкила, где алкил, циклоалкил, арил, гетероарил или гетероциклический алкил дополнительно замещены одной или более чем одной группой, независимо выбранной из группы, состоящей из алкила, циклоалкила, арила, гетероарила, алкоксила, циклоалкоксила, арилоксила, гетероариоксила, галогена, гидроксила, амино, алкиламина, циано, гидроксиалкила, гетероциклического алкила, гетероциклического алкоксила, карбоновой кислоты и эфира карбоновой кислоты;
каждый из R6 и R7 независимо выбран из группы, состоящей из алкила, арила, гетероарила, галогеноалкила, галоалкоксила, гидроксила, амино, циано, алкинила, алкоксила, арилоксила, гидроксиалкила, гетероциклического алкила, карбоновой кислоты, эфира карбоновой кислоты и галогена; и
W представляет собой C, S или O, где C дополнительно замещен R6 или R7.
Очищенные соединения, представленные формулой (IB) и формулой (IC), далее приводят во взаимодействие с кислотами в растворителях метаноле, дихлорметане или этилацетате с получением солей присоединения кислоты.
Кроме того, данное описание относится к фармацевтической композиции, содержащей соединения в соответствии с настоящим описанием или их соли, в эффективной терапевтической дозе, а также фармацевтически приемлемый носитель.
Кроме того, данное описание относится к применению соединений в соответствии с настоящим описанием или их фармацевтически приемлемых солей в изготовлении лекарственного средства в качестве ингибитора дипептидилпептидазы (DPP-IV).
Данное описание также относится к соединениям, представленным формулой (I), или их фармацевтически приемлемым солям, где соединения формулы (I) могут быть представлены в свободной форме или в форме солей присоединения кислоты, которые представляют собой фармацевтически приемлемые нетоксичные соли. Фармацевтически приемлемые соли включают гидрохлорид, пара-толуолсульфонат, тартрат, малеат, лактат, метансульфонат, сульфат, фосфат, цитрат, ацетат и трифторацетат, предпочтительно пара-толуолсульфонат, гидрохлорид, тартрат и трифторацетат.
Подробное описание изобретения
Если не указано иное, то следующие термины, используемые в описании и формуле изобретения, имеют значения, обсуждаемые ниже.
Термин “алкил” относится к насыщенной алифатической углеводородной группе, включающей C1-C20прямоцепочечные и имеющие разветвленную цепь группы. Предпочтительно алкильная группа представляет собой алкил умеренного размера, имеющий от 1 до 10 атомов углерода, например метил, этил, пропил, 2-пропил, н-бутил, изобутил, трет-бутил, пентил и т.п. Более предпочтительно, она представляет собой низший алкил, имеющий от 1 до 4 атомов углерода, например метил, этил, пропил, 2-пропил, н-бутил, изобутил или трет-бутил и т.п. Алкильная группа может быть замещенной или незамещенной. Будучи замещенной, группа(ы) заместителя(ей) предпочтительно представляет собой галогено, гидроксил, низшую алкокси, арил, арилокси, гетероарил, гетероциклический алкил, C(O)R3 и C(O)NR3R4.
Термин “циклоалкил” относится к 3-8 членному состоящему полностью из углерода моноциклическому кольцу, состоящему полностью из углерода 5-членному/6-членному или 6-членному/6-членному конденсированному бициклическому кольцу или мультициклической конденсированной кольцевой (“конденсированная” кольцевая система означает, что каждое кольцо в системе разделяет соседнюю пару атомов углерода с другим кольцом в системе) группе, где одно или более чем одно кольцо может содержать одну или более чем одну двойные связи, но ни одно из колец не имеет полностью конъюгированную систему пи-электронов. Примеры циклоалкильных групп представляют собой циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексадиенил, адамантил, циклогептил, циклогептатриенил и т.п. Циклоалкильная группа может быть замещенной или незамещенной. Будучи замещенной, группа(ы) заместителя(ей) предпочтительно представляет(ют) собой одну или более чем одну, независимо выбранную из группы, состоящей из низшего алкила, тригалогеноалкила, галогено, гидрокси, низшей алкокси, арила (необязательно замещенного одной или более чем одной группой, каждая из которых независимо представляет собой группы галогено, гидрокси, низшего алкила или низшей алкокси), арилокси (необязательно замещенной одной или более чем одной группой, где каждая независимо представляет собой группы галогено, гидрокси, низшего алкила или низшей алкокси), 6-членного гетероарила (имеющего от 1 до 3 атомов азота в кольце, где атомы углерода в кольце необязательно замещены одной или более чем одной группой, каждая из которых независимо представляет собой группу галогено, гидрокси, низшего алкила или низшей алкокси), 5-членного гетероарила (имеющего от 1 до 3 гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где атомы углерода и атомы азота группы необязательно замещены одной или более чем одной группой, каждая из которых независимо представляет собой группы галогено, гидрокси, низшего алкила или низшей алкокси), 5- или 6-членного гетероциклического алкила (имеющего от 1 до 3 гетероатомов, выбранных из группы, состоящей из азота, кислорода и серы, где атомы углерода и азота (если представлены) группы необязательно замещены одной или более чем одной группой, каждая из которых независимо представляет собой группы галогено, гидрокси, низшего алкила или низшей алкокси), меркапто, (низший алкил) тио, арилтио (необязательно замещенная одной или более чем одной группой, каждая из которых независимо представляет собой группы галогено, гидрокси, низшего алкила или низшей алкокси), циано, ацил, тиоацил, О-карбамил, н-карбамил, О-тиокарбамил, н-тиокарбамил, С-амидо, н-амидо, нитро, я-сульфонамидо, S-сульфонамидо, C(O)R3, C(O)NR3R4 и -C(O)OR3.
Термин “алкенил” относится к алкильной группе, как определено выше, имеющий по меньшей мере 2 атома углерода и по меньшей мере одну межуглеродную двойную связь. Типичные примеры включают этенил, 1-пропенил, 2-пропенил, 1-, 2-, 3-бутенил и т.п., но не ограничиваются ими.
Термин “алкинил” относится к алкильной группе, как определено выше, имеющей по меньшей мере 2 атома углерода и по меньшей мере одну межуглеродную тройную связь. Типичные примеры включают этинил, 1-пропинил, 2-пропинил, 1-, 2-, 3-бутинил и т.п., но не ограничиваются ими.
Термин “арил” относится к группам, имеющим по меньшей мере одно ароматическое кольцо, т.е. имеющим конъюгированную систему пи-электронов, включающим состоящую полностью из углерода группу циклического арила, гетероарила и биарила. Указанная арильная группа может быть необязательно замещена одной или более чем одной группой, каждая из которых независимо выбрана из группы, состоящей из галогено, тригалогенометила, гидрокси, SR, нитро, циано, алкоксила и алкила.
Термин “гетероарил” относится к арилу, имеющему от 1 до 3 гетероатомов, выбранному из группы, состоящей из N, О, и S в качестве кольцевых атомов, где оставшиеся кольцевые атомы представляют собой C. Указанное кольцо представляет собой 5- или 6-членное кольцо. Примеры гетероарильных групп включают фурил, тиенил, пиридил, пирролил, н-алкилпирролил, пиримидинил, пиразинил, имидазолил и т.п.
Термин “гетероциклический алкил” относится к группе моноциклического или конденсированного кольца из 5-9 кольцевых атомов, где один или два кольцевых гетероатомы выбраны из группы, состоящей из N, О, и S(O)n (n представляет собой целое число от 0 до 2), оставшиеся кольцевые атомы представляют собой C, дополнительно, кольцо может также иметь одну или более чем одну двойную связь, но не иметь полностью конъюгированной системы пи-электронов. Незамещенный гетероциклический алкил включает пирролидил, пиперидил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.п., но не ограничивается ими. Гетероциклический алкил может быть замещенным или незамещенным. Если он является замещенным, тогда замещающая группа предпочтительно одна или более чем одна, более предпочтительно одна, две или три, кроме того, более предпочтительно одна или две группы, каждая из которых независимо выбрана из группы, состоящей из низшего алкила, тригалогеноалкила, галогено, гидрокси, низшей алкокси, циано и ацила. Предпочтительно гетероциклический алкил необязательно замещен одной или двумя группами, независимо выбранными из группы, состоящей из галогено, низшего алкила, тригалогеноалкила, гидрокси, меркапто, циано, н-амидо и карбокси.
Термин “гидрокси” относится к группе -OH.
Термин “алкоксил” относится к группе -O-(алкил) и -O-(незамещенный циклоалкил). Типичные примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси и т.п., но не ограничиваются ими.
Термин “галогеноалкокси” относится к -O-(галогеноалкилу). Типичные примеры включают трифторметокси, трибромметокси и т.п., но не ограничиваются ими.
Термин “арилоксил” относится к -O-арильной и -O-гетероарильной группе, где арил и гетероарил являются такими, как определено выше. Типичные примеры включают фенокси, пиридинилокси, фуранилокси, тиенилокси, пиримидинилокси, пиразинилокси и т.п. и их производные, но не ограничиваются ими.
Термин “меркапто” относится к группе -SH.
Термин “алкилтио” относится к группе -S-(алкил) и -S-(незамещенный циклоалкил). Типичные примеры включают метилтио, этилтио, пропилтио, бутилтио, циклопропилтио, циклобутилтио, циклопентилтио, циклогексилтио и т.п., но не ограничиваются ими.
Термин “арилтио” относится к -S-арильной и -S-гетероарильной группе, где арил и гетероарил являются такими, как определено выше. Типичные примеры включают, например, фенилтио, пиридинилтио, фуранилтио, тиенилтио, пиримидинилтио и т.п. и их производные, но не ограничиваются ими.
Термин “ацил” относится к группе -C(O)-R”, где R” выбран из группы, состоящей из водорода, низшего алкила, тригалогенометила, незамещенного циклоалкила, арила (необязательно замещенного одним или более чем одним, предпочтительно одним, двумя или тремя заместителями, выбранными из группы, состоящей из групп низшего алкила, тригалогенометила, низшей алкокси и галогено), гетероарила (связанного через атом углерода кольца) (необязательно замещенного одним или более чем одним, предпочтительно одним, двумя или тремя заместителями, выбранными из группы, состоящей из групп низшего алкила, тригалогеноалкила, низшей алкокси и галогено), и гетероалициклической группы (связанной через атом углерода кольца) (необязательно замещенной одним или более чем одним, предпочтительно одним, двумя или тремя заместителями, выбранными из группы, состоящей из групп низшего алкила, тригалогеноалкила, низшей алкокси и галогено). Типичные ацильные группы включают ацетил, трифторацетил, бензоил и т.п., но не ограничиваются ими.
Термин “тиоацил” относится к группе -C(S)-R”, где R” является таким, как определено выше.
Термин “ацетил” относится к группе -C(O)CH3.
Термин “галогено” относится к фтор-, хлор-, бром- или йод-, предпочтительно фтор- или хлор-.
Термин “трифторметил” относится к группе -CF3.
Термин “циано” относится к группе -C≡N.
Термин “амино” относится к группе -NH2.
Термин “карбоновая кислота” относится к группе -COOH.
Термин “эфир карбоновой кислоты” относится к группе -COOR, где R представляет собой алкил или циклоалкил.
Термин “гидроксиалкил” относится к группе -(CH2)OH, где R представляет собой целое число от 1 до 4.
Термин “необязательный” или “необязательно” обозначает, что описанное далее явление или событие может произойти или может не произойти, и что описание включает случаи, когда явление или событие может произойти или может не произойти. Например, “группа гетероцикла, необязательно замещенная алкильной группой” означает, что алкил может присутствовать или может не присутствовать, и что описание включает случаи, когда гетероциклическая группа замещена алкильной группой, и случаи, когда гетероциклическая группа не замещена алкильной группой.
Термин “фармацевтическая композиция” относится к смеси одного или более чем одного из описанных здесь соединений или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами, такими как физиологически/фармацевтически приемлемыми носителями и эксципиентами. Задача фармацевтической композиции заключается в том, чтобы облегчить введение в организм соединения.
Способ синтеза раскрытого соединения
Для полного раскрытия объекта описания в последнем применяется следующее техническое решение:
Схема I
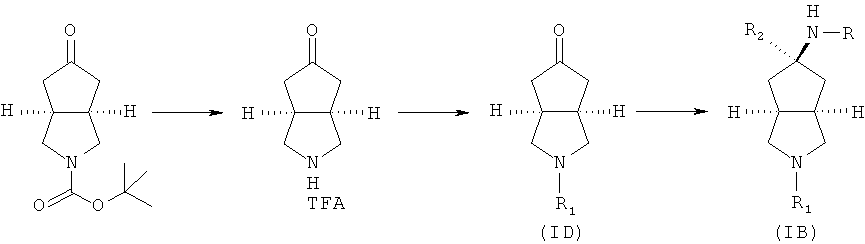
трет-бутил-5-оксо-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты эфир приводят во взаимодействие с трифторуксусной кислотой в подходящем растворителе, таком как дихлорметан, при охлаждении в бане с ледяной водой, с получением гексагидро-циклопента[c]пиррол-5-он трифторацетата; гексагидро-циклопента[c]пиррол-5-он трифторацетат приводят во взаимодействие с хлорангидридом или сложным эфиром в присутствии основания с получением соединения формулы (ID); промежуточное соединение формулы (ID) в подходящем растворителе, таком как метанол или этанол, приводят во взаимодействие с соответствующими аминами, замещенным боргидридом натрия и подходящим основанием, таким как триэтиламин, с получением соединения формулы (IB) при комнатной температуре;
Схема II
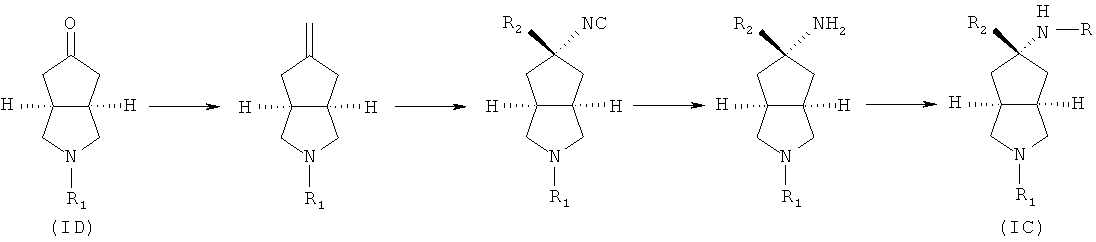
нагревают калий трет-бутоксид и раствор метилтрифенилфосфония йодида в толуоле, а затем добавляют промежуточное соединение формулы (ID) при комнатной температуре с получением азабициклоалкенильного соединения; азабициклоалкенильное соединение в подходящем растворителе, таком как дихлорметан, приводят во взаимодействие с триметилсилилцианидом в присутствии перхлората серебра при комнатной температуре с получением азабициклоциано соединения; азабициклоциано соединение в подходящем растворителе, таком как этанол, приводят во взаимодействие с подходящей кислотой, такой как соляная кислота, при комнатной температуре с получением азабициклоамино соединения; азабициклоамино соединение приводят во взаимодействие с галоген-замещенным соединением в присутствии щелочного растворителя, такого как N,N-диметилформамид, с получением соединения формулы (IC).
Схема III
промежуточное соединение формулы (ID) приводят во взаимодействие с тозилметилизоцианидом в подходящем растворителе, таком как этиленгликоль диметиловый эфир, путем изоцианидного взаимодействия с получением азабициклоциано соединения; азабициклоциано соединение в подходящем растворителе, таком как тетрагидрофуран, приводят во взаимодействие с галогеносоединением в присутствии гексаметилдисилазида лития с получением R2 замещенного азабициклоциано соединения; R2 замещенное азабициклоциано соединение гидролизуют в присутствии кислоты с получением R2 замещенного азабициклокарбоксильного соединения; или R2 замещенное азабициклоциано соединение приводят во взаимодействие с восстановителем, таким как DIBAL-H, в подходящем растворителе, таком как дихлорметан, при охлаждении в бане с ледяной водой, с получением соединения альдегида; соединение альдегида в смеси растворителей тетрагидрофуран/вода приводят во взаимодействие с дигидрофосфатом натрия, хлоритом натрия и 2-метил-2-бутеном при охлаждении в бане с ледяной водой, с получением R2 замещенного азабициклокарбоксильного соединения; R2 замещенное азабициклокарбоксильное соединение приводят во взаимодействие с этилхлорформиатом в присутствии основания, такого как триэтиламин, путем азидореакции с получением азабициклоазидо соединения; нагревают азабициклоазидо соединение в подходящем растворителе, таком как толуол, а затем перемешивание в кислом растворе; реакционный раствор нейтрализуют до слабощелочного pH с получением R2 замещенного азабициклоамино соединения; R2 замещенное азабициклоамино соединение приводят во взаимодействие с галоген-замещенным соединением в присутствии щелочного растворителя, такого как N,N-диметилформамид, с получением соединения формулы (IB).
Схема IV
промежуточное соединение формулы (ID) приводят во взаимодействие с восстановителем, таким как литий три-трет-бутоксиалюмогидрид, в подходящем растворителе, таком как тетрагидрофуран, при охлаждении в бане с ледяной водой с получением азабициклогидроксильного соединения; азабициклогидроксильное соединение приводят во взаимодействие с щелочным реагентом, таким как триэиламин и метилсульфонилхлорид, в подходящем растворителе, таком как дихлорметан, с получением соединения азабициклометилсульфоновой кислоты; нагревают соединение азабициклометилсульфоновой кислоты и фталимид-калий в присутствии щелочного агента, такого как N,N-диметилформамид, с получением фталимид-замещенного азабицикло соединения; нагревают фталимид-замещенное азабицикло соединение и в подходящем растворителе, таком как этанол, с получением азабициклоамино соединения; нагревают азабициклоамино соединение и галогено-соединение в подходящем растворителе, таком как дихлорметан, с получением соединения формулы (IC).
Очищенные соединения, представленные формулой (IB) и формулой (IC), дополнительно приводят во взаимодействие с кислотами в растворителе метаноле, дихлорметане или этилацетате, с получением солей присоединения кислоты.
Кроме того, данное описание относится к фармацевтической композиции, содержащей соединения в соответствии с данным описанием или их соли в эффективной терапевтической дозе, и фармацевтически приемлемый носитель; и к применению соединений в соответствии с данным описанием или фармацевтически приемлемых солей в изготовлении лекарственного средства в качестве ингибитора дипептидилпептидазы (DPP-IV). Другими словами, в этом описании также предложена фармацевтическая композиция, содержащая вышеупомянутые соединения в эффективной терапевтической дозе, а также их применение в изготовлении лекарственного средства в качестве ингибитора дипептидилпептидазы (DPP-IV).
Конкретные способы воплощений
Следующие примеры служат для иллюстрации описания, но примеры не следует рассматривать, как ограничивающие объем изобретения.
Примеры
Структуры всех соединений идентифицировали с помощью ядерного магнитного резонанса (1H ЯМР) и масс-спектрометрии (MS). Химические сдвиги 1H ЯМР (δ) регистрировали в миллионных долях млн-1 (10-6). ЯМР осуществляли на спектрометре AVANCE-400. Приемлемые растворители представляли собой дейтерированный хлороформ (CDCl3), дейтерированный диметилсульфоксид (DMSO-d6) и дейтерированный метанол (CD3OD) с триметилсиланом (TMS) в качестве внутреннего стандарта, а химические сдвиги регистрировали в млн-1 (10-6).
MS определяли с помощью масс-спектрометра FINNIGAN LCQ Ad (ESI (ионизация распылением электронов)).
Среднее скорости ингибирования киназы и IC50 (средняя ингибирующая концентрация) определяли с помощью NovoStar ELIASA (BMG Co. German).
Тонкослойный силикагель представлял собой пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254.
В качестве носителя для колоночной хроматографии, как правило, применяли силикагель Yantai Huanghai 200-300 меш.
DMSO-D6: дейтерированный диметилсульфоксид.
CDCl3: дейтерированный хлороформ.
CD3OD: дейтерированный метанол.
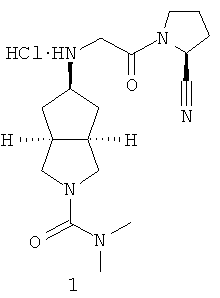
Пример 1
цис-5-[2-((S)-2-Цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамид гидрохлорид
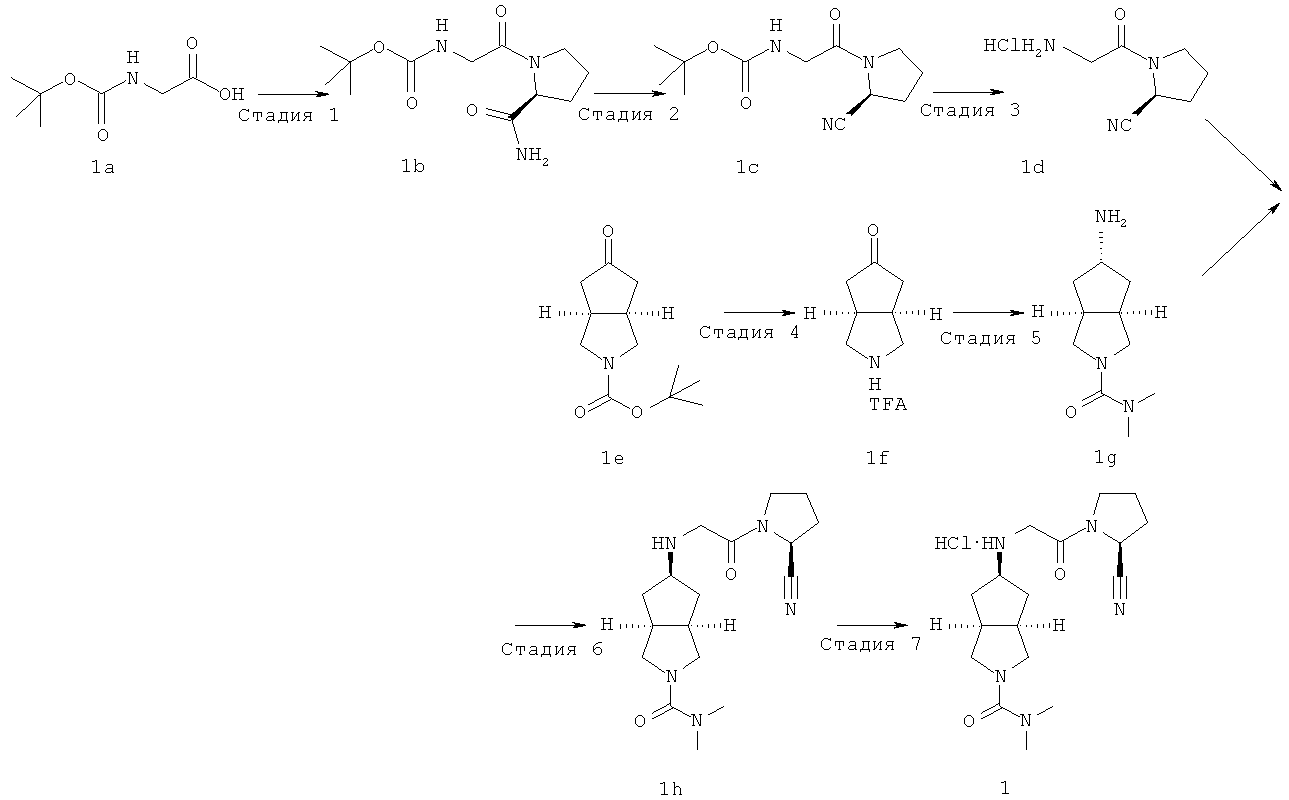
Стадия 1
Получение (S)-[2-(2-карбамоилпирролидин-1-ил)-2-оксоэтил]карбаминовой кислоты трет-бутилового эфира 1b
N-трет-бутилоксикарбонилглицин 1a (5 г, 28,56 ммоль) и L-пролинамид (3,25 г, 28,50 ммоль) растворяли в 75 мл N,N-диметилформамида и смесь охлаждали до 0°C. Затем добавляли 1-гидроксибензотриазол (11,8 г, 87,3 ммоль), N-этил-N'-(диметиламинопропил)карбодиимид (11,3 г, 59 ммоль) и триэтиламин (12,1 мл, 87,3 ммоль) и перемешивали. Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. За реакцией наблюдали с помощью тонкослойной хроматографии (ТСХ) до исчезновения исходных веществ. N,N-диметилформамид выпаривали при температуре ниже 50°C. Полученную смесь экстрагировали этилацетатом (200 мл × 3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Остаток перекристаллизовали из этилацетата с получением указанного в заголовке соединения (S)-[2-(2-карбамоилпирролидин-1-ил)-2-оксоэтил]карбаминовой кислоты трет-бутилового эфира 1b (7,42 г, выход 95,8%) в виде белого порошка.
MS m/z (ESI): 272,1 [M+1]
Стадия 2
Получение (S)-[2-(2-цианопирролидин-1-ил)-2-оксоэтил]карбаминовой кислоты трет-бутилового эфира 1c
В сухую трехгорлую колбу последовательно добавляли 286 мл пиридина, (S)-[2-(2-карбамоилпирролидин-1-ил)-2-оксоэтил]карбаминовой кислоты трет-бутилового эфира 1b (13,5 г, 49,8 ммоль) и имидазол (7,11 г, 104,6 ммоль) в атмосфере азота. Затем реакционную систему охлаждали до -35°C с последующим добавлением в нее по капле при перемешивании оксихлорида фосфора (19 мл, 204,2 ммоль). Реакционную смесь перемешивали в течение 1 часа при той же температуре. После нагревания до комнатной температуры реакционную смесь перемешивали в течение еще 30 минут. Смесь упаривали для удаления пиридина и получающуюся в результате смесь разводили этилацетатом с последующим добавлением воды. Смесь экстрагировали этилацетатом (200 мл×3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (S)-[2-(2-цианопирролидин-1-ил)-2-оксоэтил]карбаминовой кислоты трет-бутилового эфира 1с (10,7 г, выход 84,9%) в виде белого порошка.
MS m/z (ESI): 254,3 [M+1]
Стадия 3
Получение (S)-1-(2-аминоацетил)пирролидин-2-карбонитрил гидрохлорида 1d
(S)-[2-(2-Цианопирролидин-1-ил)-2-оксоэтил]карбаминовой кислоты трет-бутиловый эфир 1с (13,7 г, 54,2 ммоль) растворяли в 140 мл эфира и 40 мл воды и перемешивали. После охлаждения на ледяной водяной бане по каплям добавляли 37% соляную кислоту (90 мл) и реакционную смесь перемешивали в течение 1 часа при той же температуре. Смесь концентрировали в условиях пониженного давления и остаток, разведенный простым эфиром, фильтровали на фильтрующей центрифуге с получением указанного в заголовке соединения (S)-1-(2-аминоацетил)-пирролидин-2-карбонитрил гидрохлорида 1d (10 г, выход 98%) в виде белого порошка.
MS m/z (ESI): 154,4 [M+1]
Стадия 4
Получение гексагидроциклопента[c]пиррол-5-он трифторацетата 1f
Трет-бутил-5-оксогексагидроциклопента[c]пиррол-2-карбоксилат 1e (0,32 г, 1,42 ммоль) растворяли в 10 мл дихлорметана и перемешивали. При охлаждении на ледяной водяной бане по каплям добавляли трифторуксусную кислоту (3,27 мл, 42,7 ммоль) и реакционной смеси давали возможность прореагировать в течение 30 минут при 0°C. Смесь концентрировали в условиях пониженного давления с получением неочищенного указанного в заголовке соединения гексагидроциклопента[c]пиррол-5-он трифторацетата 1f, которое непосредственно применяли на следующей стадии.
MS m/z (ESI): 126,4 [M+1]
Стадия 5
Получение N,N-диметилил-5-оксо-гексагидроциклопента[c]пиррол-2-карбоксамида 1g
Гексагидроциклопента[c]пиррол-5-он трифторацетат 1f, полученный на предыдущей стадии, растворяли в 15 мл ацетонитрила и перемешивали. При охлаждении на ледяной водяной бане добавляли карбонат калия (0,24 г, 1,71 ммоль) с последующим добавлением диметилкарбамидхлорида (0,14 мл, 1,56 ммоль). Реакционную смесь нагревали до комнатной температуры и давали возможность взаимодействовать в течение 2 часов. Смесь концентрировали в условиях пониженного давления и разбавляли 50 мл воды. Смесь экстрагировали этилацетатом (50 мл × 3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения N,N-диметил-5-оксогексагидроциклопента[c]пиррол-2-карбоксамида 1g (0,19 г, выход 68,3%) в виде светло-желтого масла.
MS m/z (ESI): 197,4 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 3,56 (m, 2H), 2,85 (m, 2H), 2,7 (s, 6H), 2,81 (m, 2H), 2,5 (m, 2H), 2,01 (m, 2H).
Стадия 6
Получение цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамида 1h
(S)-1-(2-Аминоацетил)-пирролидин-2-карбонитрил гидрохлорид 1d (0,36 г, 1,91 ммоль) растворяли в 20 мл метанола и перемешивали с последующим добавлением N,N-диметил-5-оксогексагидроциклопента[c]пиррол-2-карбоксимида 1g (0,25 г, 1,28 ммоль) и триацетоксиборогидрида натрия (1,22 г, 5,74 ммоль). Реакционной смеси давали возможность взаимодействовать в течение 3 часов при комнатной температуре. Смесь концентрировали и разбавляли 20 мл насыщенного водного карбоната натрия. Затем смесь экстрагировали дихлорметаном (20 мл × 10). Объединенные органические экстракты промывали 10 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамида 1h (0,3 мг, выход 53%) в виде белого порошка.
MS m/z (ESI): 334,5 [M+1]
Стадия 7
Цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамид гидрохлорид
Цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамид 1h (200 мг, 0,687 ммоль) растворяли в 10 мл дихлорметана. Во время охлаждения на ледяной водяной бане добавляли 2 мл соляной кислоты в простом эфире (0,5 н.). Смесь концентрировали в условиях пониженного давления и разбавляли 10 мл простого эфира. Полученную смесь фильтровали на фильтрующей центрифуге с получением указанного в заголовке соединения цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамид гидрохлорида 1 (180 мг, 80%) в виде белого порошка.
1H ЯМР (CD3OD, 400 МГц) δ 4,82 (dd, 1H, J1=4 Гц, J2=5,2 Гц), 4,02 (dd, 2H, J1=J2=16,4 Гц), 3,62-3,25 (m, 7H), 2,76 (s, 6H), 2,51-1,49 (m, 10H).
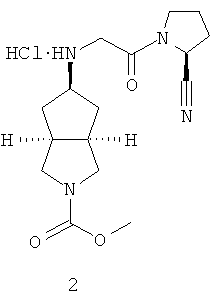
Пример 2
Цис-метил-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]гексагидроциклопента[c]пиррол-2-карбоксамид гидрохлорид
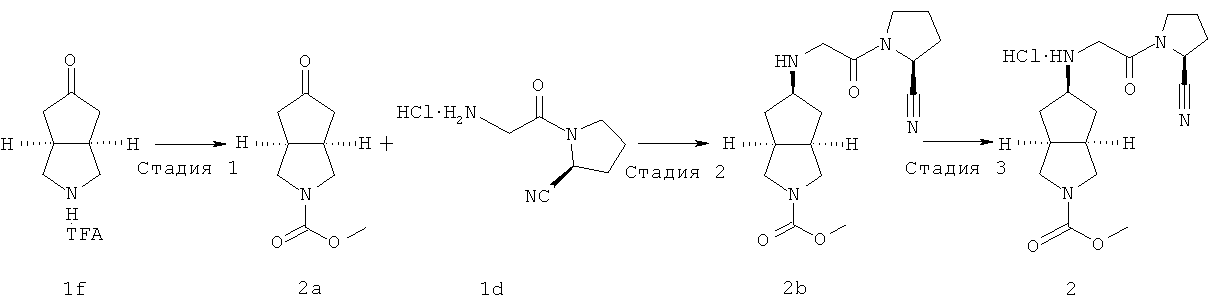
Стадия 1
Получение метил-5-оксогексагидроциклопента[c]пиррол-2-карбоксамида 2a
Гексагидроциклопента[c]пиррол-5-он трифторацетат 1f (0,559 г, 2,34 ммоль) растворяли в 20 мл ацетонитрила. При охлаждении на ледяной водяной бане последовательно добавляли карбонат калия (0,646 г, 4,68 ммоль) и метил-2-хлорацетат (0,22 мл, 2,8 ммоль). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Смесь концентрировали в условиях пониженного давления и разбавляли 50 мл воды. Смесь экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты последовательно промывали 50 мл насыщенного солевого раствора и 50 мл воды, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения метил-5-оксогексагидроциклопента[c]пиррол-2-карбоксамида 2a (0,25 г, выход 58,4%) в виде бесцветного масла.
MS m/z (ESI): 184 [M+1]
Стадия 2
Получение цис-метил-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]гексагидроциклопента[c]пиррол-2-карбоксамида 2b
(S)-1-(2-Аминоацетил)-пирролидин-2-карбонитрил гидрохлорид 1d (0,43 г, 2,29 ммоль) растворяли в 20 мл метанола и перемешивали с последующим добавлением метил-5-оксогексагидроциклопента[c]пиррол-2-карбоксамида 2a (0,28 г, 1,53 ммоль) и триацетоксиборгидрида натрия (1,46 г, 6,88 ммоль). Реакционной смеси давали возможность взаимодействовать в течение 3 часов при комнатной температуре. Смесь концентрировали и разбавляли 20 мл насыщенного водного карбоната натрия. Смесь экстрагировали дихлорметаном (20 мл × 3). Объединенные органические экстракты промывали 10 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-метил-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-гексагидроциклопента[c]пиррол-2-карбоксамида 2b (0,22 г, выход 41%) в виде белого порошка.
MS m/z (ESI): 357 [M+1]
Стадия 3
Цис-метил-5-[2-(2-цианопирролидин-1-ил)-2-оксоэтиламино]гексагидроциклопента[c]пиррол-2-карбоксамид гидрохлорид 2
Цис-метил-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]гексагидроциклопента[c]пиррол-2-карбоксамид 2b растворяли в 10 мл простого эфира. При охлаждении на ледяной водяной бане добавляли 2 мл соляной кислоты в простом эфире (0,5 н.). Полученную смесь фильтровали на фильтрующей центрифуге с получением указанного в заголовке соединения цис-метил-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]гексагидроциклопента[c]пиррол-2-карбоксамид гидрохлорида 2 (200 мг) в виде белого порошка.
1H ЯМР (CD3OD, 400 МГц) δ 4,71 (m, 1H), 3,93 (m, 2H), 3,59-3,28 (m, 10H), 2,64 (m, 2H), 2,34 (m, 2H), 2,17 (m, 2H), 2,08 (m, 2H).
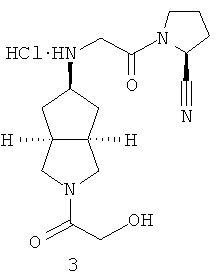
Пример 3
Цис-(S)-1-{2-[2-(2-гидроксиацетил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорид
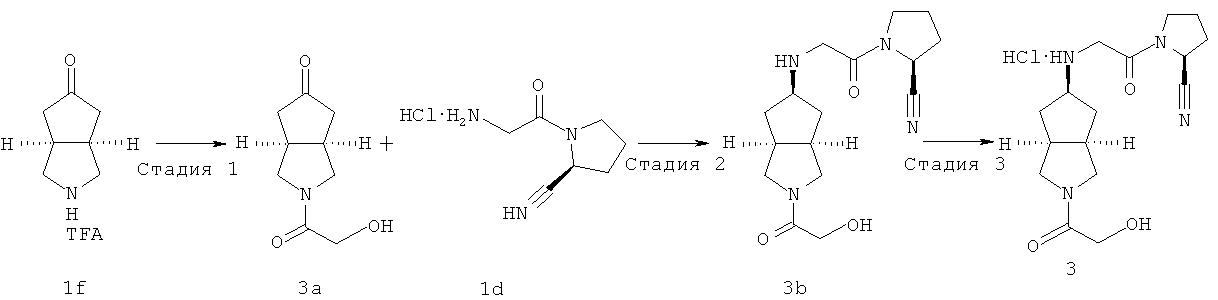
Стадия 1
Получение 2-(2-гидроксиацетил)гексагидроциклопента[c]пиррол-5-она 3а Гексагидроциклопента[c]пиррол-5-он трифторацетат 1f (764,8 мг, 3,2 ммоль) и 2-гидроксилэтановую кислоту (267,5 мг, 3,52 ммоль) растворяли в 10 мл ацетонитрила. При охлаждении на ледяной водяной бане добавляли гидроксиуксусную кислоту (1,3 г, 9,6 ммоль), 1-этил-3-диметиламинопропилкарбодиимид гидрохлорид (1,23 г, 6,4 ммоль) и триэтиламин (1,3 мл, 9,6 ммоль). Ледяную водяную баню удаляли, а реакционной смеси давали возможность взаимодействовать в течение ночи при 25°С. Смесь концентрировали и разбавляли 20 мл этилацетата. Смесь фильтровали в условиях пониженного давления и фильтрат промывали 20 мл воды. Органическую фазу сушили над безводным сульфатом магния, фильтровали в условиях пониженного давления и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 2-(2-гидроксиацетил)гексагидроциклопента[c]пиррол-5-она 3a (0,375 г, выход 64%) в виде бесцветного масла.
MS m/z (ESI): 184 [M+1]
Стадия 2
Получение цис-(S)-1-{2-[2-(2-гидроксиацетил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрила 3b
2-(2-Гидроксиацетил)гексагидроциклопента[c]пиррол-5-он 3a (0,375 г, 2,05 ммоль) и (S)-1-(2-аминоацетил)пирролидин-2-карбонитрил гидрохлорид 1d (0,78 г, 4,1 ммоль) растворяли в 5 мл метанола и 10 мл тетрагидрофурана. После того как смеси давали возможность взаимодействовать при комнатной температуре в течение 30 минут, добавляли триацетоксиборгидрид натрия (0,87 г, 4,1 ммоль). Затем смеси давали возможность прореагировать в течение ночи при комнатной температуре. Смесь концентрировали в условиях пониженного давления и разбавляли 50 мл метанола с последующим добавлением карбоната калия (2 г, 7 ммоль). После перемешивания в течение 30 минут смесь фильтровали и фильтрат концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-(S)-1-{2-[2-(2-гидроксиацетил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрила 3b, которое непосредственно использовали на следующей стадии.
MS m/z (ESI): 357 [M+1]
Стадия 3
Получение цис-(S)-1-{2-[2-(2-гидроксиацетил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорида 3
Цис-(S)-1-{2-[2-(2-гидроксиацетил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил 3b растворяли в 10 мл простого эфира. При охлаждении на ледяной водяной бане добавляли 2 мл соляной кислоты в простом эфире (0,5 н.). Полученную смесь фильтровали с помощью фильтрующей центрифуги с получением указанного в заголовке соединения цис-(S)-1-{2-[2-(2-гидроксиацетил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорида 3 (100 мг) в виде белого порошка.
Пример 4
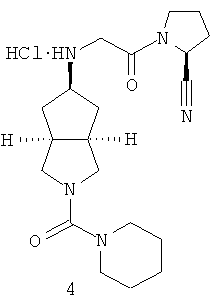
Цис-(S)-1-{2-[2-(пиперидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорид
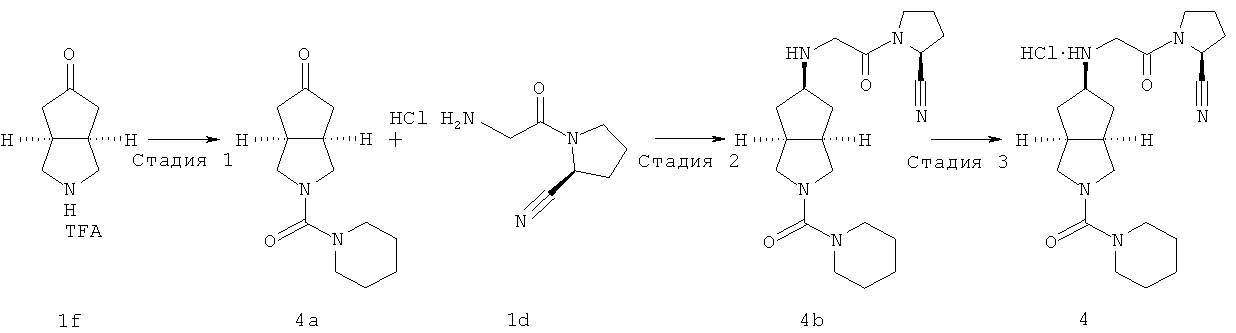
Стадия 1
Получение 2-(пиперидин-1-карбонил)гексагидроциклопента[c]пиррол-5-она 4a
Гексагидроциклопента[c]пиррол-5-он трифторацетат 1f (478 мг, 2 ммоль) растворяли в 20 мл дихлорметана и перемешивали, а затем добавляли йод[3-(1-пиперидинформил)имидазол-1-метил] (0,96 г, 3 ммоль) и триэтиламин (0,84 мл, 6 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре. Реакцию гасили 20 мл воды и смесь экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты последовательно промывали 50 мл 10% раствора лимонной кислоты и 50 мл насыщенного рассола, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 2-(пиперидин-1-карбонил)гексагидроциклопента[c]пиррол-5-она 4a (0,41 г, выход 87%) в виде бесцветного масла.
MS m/z (ESI): 237 [M+1]
Стадия 2
Получение цис-(S)-1-{2-[2-(пиперидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}-пирролидин-2-карбонитрила 4b
2-(Пиперидин-1-карбонил)гексагидроциклопента[c]пиррол-5-он 4а (0,41 г, 1,74 ммоль) и (S)-1-(2-аминоацетил)пирролидин-2-карбонитрил гидрохлорид 1d (0,5 г, 2,6 ммоль) растворяли в 50 мл тетрагидрофурана с последующим добавлением сульфата натрия (5 г) и 0,05 мл уксусной кислоты. После того как смесь перемешивали при комнатной температуре в течение 30 минут, добавляли триацетоксиборгидрид натрия (1,1 г, 5,2 ммоль). Реакционной смеси давали возможность прореагировать при комнатной температуре в течение 3 часов и ее концентрировали в условиях пониженного давления. Смесь разводили 50 мл насыщенного водного карбоната натрия и экстрагировали этилацетатом (50 мл × 3). Объединенные органические экстракты последовательно отмывали 50 мл насыщенного солевого раствора и 50 мл воды, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-(S)-1-{2-[2-(пиперидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрила 4b, которое непосредственно использовали на следующей стадии.
MS m/z (ESI): 410 [M+1]
Стадия 3
Получение цис-(S)-1-{2-[2-(пиперидин-1-карбониил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорида 4
цис-(S)-1-{2-[2-(пиперидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил 4b растворяли в 10 мл простого эфира. При охлаждении на ледяной водяной бане добавляли 2 мл соляной кислоты в простом эфире (0,5 н.). Полученную смесь фильтровали с помощью фильтрующей центрифуги с получением указанного в заголовке соединения цис-(S)-1-{2-[2-(пиперидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорида 4 (0,16 г) в виде белого осадка.
1H ЯМР (CD3OD, 400 МГц) δ 4,83 (dd, 1H, J1=3,0 Гц, J2=5,8 Гц), 4,09 (dd, 2H, J1=J2=13,1 Гц), 3,70-3,30 (m, 10H), 2,72 (m, 2H), 2,47 (m, 2H), 2,31-2,00 (m, 5H), 1,66-1,52 (m, 8H).
Пример 5
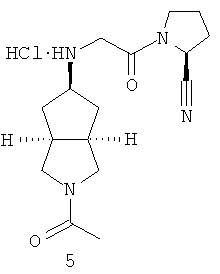
Цис-(S)-1-[2-(2-ацетилоктагидроциклопента[c]пиррол-5-иламино)ацетил]пирролидин-2-карбонитрил гидрохлорид
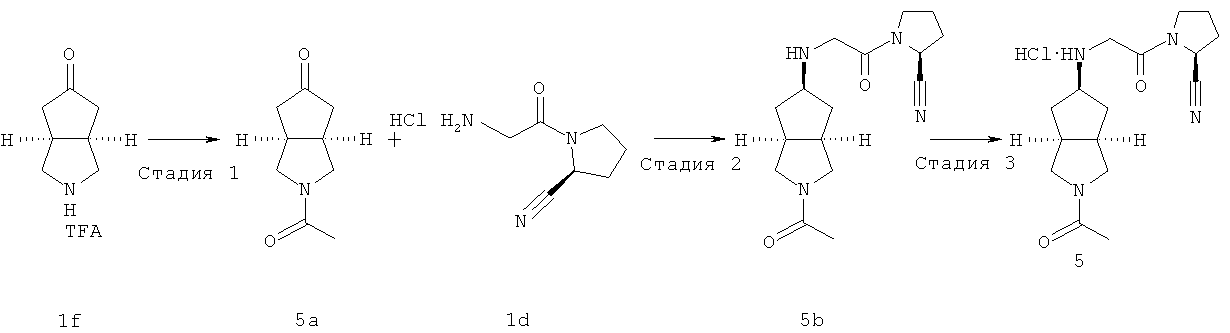
Стадия 1
Получение 2-ацетилгексагидроциклопента[c]пиррол-5-она 5а Гексагидроциклопента[c]пиррол-5-он трифторацетат 1f (717 мг, 3 ммоль) растворяли в 20 мл ацетонитрила с последующим добавлением ди-трет-бутил бикарбоната (0,42 мл, 4,5 ммоль) и триэтиламина (0,98 мл, 9 ммоль) при охлаждении на ледяной водяной бане. Реакционную смесь перемешивали в течение ночи при той же температуре. Смесь концентрировали и разбавляли 50 мл воды. Смесь экстрагировали этилацетатом (50 мл × 3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 2-ацетилгексагидроциклопента[c]пиррол-5-она 5a (0,36 г, выход 72%) в виде бесцветного масла.
MS m/z (ESI): 168,4 [M+1]
Стадия 2
Получение цис-(S)-1-[2-(2-ацетилоктагидроциклопента[c]пиррол-5-иламино)ацетил]пирролидин-2-карбонитрила 5b
2-Ацетилгексагидроциклопента[c]пиррол-5-он 5a (0,36 г, 2,15 ммоль) и (S)-1-(2-аминоацетил)пирролидин-2-карбонитрил гидрохлорид 1d (0,614 г, 3,23 ммоль) растворяли в 50 мл тетрагидрофурана с последующим добавлением 5 г сульфата натрия и 0,05 мл уксусной кислоты. После того как смесь перемешивали при комнатной температуре в течение 30 минут, добавляли триацетоксиборгидрид натрия (1,37 г, 6,46 ммоль) и смеси давали возможность прореагировать в течение следующих 3 часов. Смесь концентрировали в условиях пониженного давления и разбавляли 50 мл насыщенного водного карбоната натрия. Реакционную смесь экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты последовательно промывали 50 мл насыщенного солевого раствора и 50 мл воды, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-(S)-1-[2-(2-ацетилоктагидроциклопента[c]пиррол-5-иламино)ацетил]пирролидин-2-карбонитрила 5b, которое непосредственно использовали на следующей стадии.
MS m/z (ESI): 305,5 [M+1]
Стадия 3
Получение цис-(S)-1-[2-(2-ацетилоктагидроциклопента[c]пиррол-5-иламино)ацетил]пирролидин-2-карбонитрил гидрохлорида 5
Цис-(S)-1-[2-(2-ацетилоктагидроциклопента[c]пиррол-5-иламино)ацетил]пирролидин-2-карбонитрил 5b, полученный на предыдущей стадии, растворяли в 20 мл простого эфира. При охлаждении на ледяной водяной бане добавляли 4 мл соляной кислоты в простом эфире (0,5 н.). Полученную смесь фильтровали с помощью фильтрующей центрифуги с получением указанного в заголовке соединения цис-(S)-1-[2-(2-ацетилоктагидроциклопента[c]пиррол-5-иламино)ацетил]пирролидин-2-карбонитрил гидрохлорида 5 (0,23 г) в виде белого порошка.
1H ЯМР (CD3OD, 400 МГц) δ 4,71 (m, 1H), 3,92 (m, 2H), 3,69-3,37 (m, 7H), 2,69 (m, 2H), 2,33 (m, 2H), 2,13 (m, 2H), 2,04-2,00 (m, 5H), 1,48 (m, 2H).
Пример 6
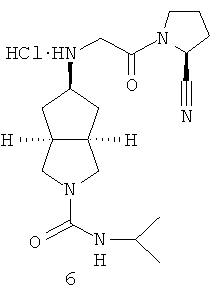
цис-5-[2-((S)-2-Цианопирролидин-1-ил)-2-оксоэтиламино]гексагидроциклопента[c]пиррол-2-карбоновой кислоты изопропиламидогидрохлорид
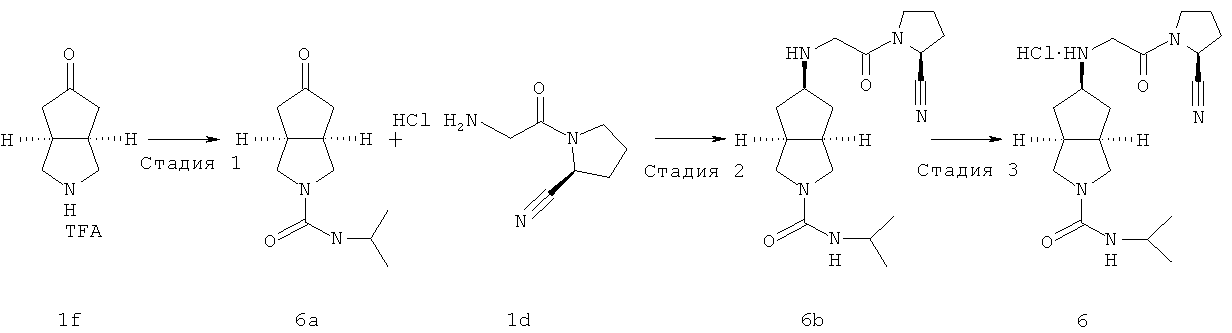
Стадия 1
Получение N-изопропил-5-оксогексагидроциклопента[c]пиррол-2-карбоксамида 6a
Гексагидроциклопента[c]пиррол-5-он трифторацетат 1f (717 мг, 3 ммоль) растворяли в 20 мл дихлорметана и смешивали при охлаждении на ледяной водяной бане с последующим добавлением изоцианата (9 мл, 9 ммоль) и триэтиламина (1,7 мл, 12 ммоль). Реакционной смеси давали возможность прореагировать в течение ночи при комнатной температуре и ее разбавляли 50 мл воды. Смесь экстрагировали дихлорметаном (50 мл × 3). Объединенные органические экстракты последовательно промывали 10% раствором лимонной кислоты (50 мл) и 50 мл насыщенного солевого раствора, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением N-изопропил-5-оксогексагидроциклопента[c]пиррол-2-карбоновой кислоты изопропиламида 6а (0,3 г, выход 47,6%) в виде бесцветного масла.
MS m/z (ESI): 211 [M+1]
Стадия 2
Получение цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-гексагидроциклопента[c]пиррол-2-карбоновой кислоты изопропиламида 6b
N-Изопропил-5-оксогексагидроциклопента[c]пиррол-2-карбоновой кислоты изопропиламид 6a (0,3 г, 1,43 ммоль) и (S)-1-(2-аминоацетил)-пирролидин-2-карбонитрил гидрохлорид 1d (0,407 г, 2,14 ммоль) растворяли в 50 мл тетрагидрофурана с последующим добавлением 5 г сульфата натрия и 0,05 мл уксусной кислоты. После того, как реакционную смесь перемешивали при комнатной температуре в течение 30 минут, добавляли триацетоксиборгидрид натрия (0,9 г, 4,3 ммоль) и смесь перемешивали в течение 3 часов. Смесь концентрировали в условиях пониженного давления и разбавляли 50 мл насыщенного водного карбоната натрия. Реакционную смесь экстрагировали этилацетатом (50 мл × 3). Объединенные органические экстракты последовательно промывали 50 мл насыщенного солевого раствора и 50 мл воды, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-гексагидроциклопента[c]пиррол-2-карбоновой кислоты изопропиламида 6b, которое непосредственно использовали на следующей стадии.
MS m/z (ESI): 384 [M+1]
Стадия 3
Получение цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-гексагидроциклопента[c]пиррол-2-карбоновой кислоты изопропиламида гидрохлорида 6
цис-5-[2-((S)-2-Цианопирролидин-1-ил)-2-оксоэтиламино]гексагидроциклопента[c]пиррол-2-карбоновой кислоты изопропиламид 6b, полученный на предыдущей стадии, растворяли в 10 мл простого эфира. При охлаждении на ледяной водяной бане добавляли 2 мл соляной кислоты в простом эфире (0,5 н.). Полученную смесь фильтровали на фильтрующей центрифуге с получением указанного в заголовке соединения цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]гексагидроциклопента[c]пиррол-2-карбоновой кислоты изопропиламида гидрохлорида 6 (80 мг) в виде белого порошка.
1H ЯМР (CD3OD, 400 МГц) δ 4,70 (m, 1H), 3,92 (m, 2H), 3,76-3,32 (m, 8H), 2,63-1,41 (m, 10H), 1,01 (d, 6H, J=6 Гц).
Пример 7
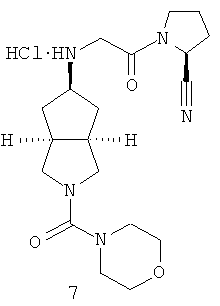
Цис-(S)-1-{2-[2-(морфолин-4-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорид
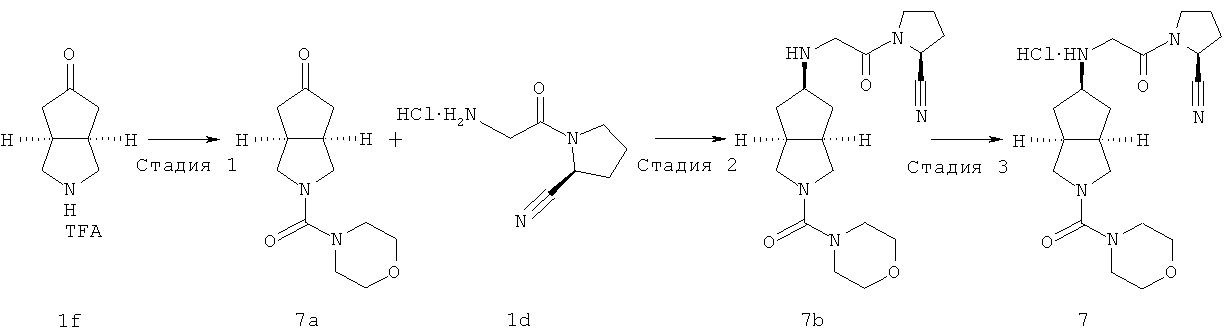
Стадия 1
Получение 2-(морфолин-4-карбонил)гексагидроциклопента[c]пиррол-5-она 7a
Гексагидроциклопента[c]пиррол-5-он трифторацетат 1f (574 мг, 2,4 ммоль) растворяли в 20 мл ацетонитрила и перемешивали с последующим добавлением карбоната калия (0,397 г, 2,88 ммоль), а также морфолин-4-карбонил хлорида (0,323 мл, 2,64 ммоль) при охлаждении на ледяной водяной бане. Реакционной смеси давали возможность прореагировать в течение ночи при одинаковой температуре. Смесь концентрировали в условиях пониженного давления и разбавляли 50 мл воды. Смесь экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силакагеле с получением указанного в заголовке соединения 2-(морфолин-4-карбонил)гексагидроциклопента[c]пиррол-5-она 7а (0,572 г, выход 77,3%) в виде бесцветного масла.
MS m/z (ESI): 239 [M+1]
Стадия 2
Получение цис-(S)-1-{2-[2-(морфолин-4-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрила 7b
2-(Морфолин-4-карбонил)гексагидроциклопента[c]пиррол-5-он 7а (0,64 г, 2,69 ммоль) и (S)-1-(2-аминоацетил)пирролидин-2-карбонитрил гидрохлорид 1d (0,764 г, 4,03 ммоль) растворяли в 50 мл тетрагидрофурана с последующим добавлением 5 г сульфата натрия и 0,05 мл уксусной кислоты. После того как смесь перемешивали при комнатной температуре в течение 30 минут, к ней добавляли триацетоксиборгидрид натрия (1,71 г, 8,07 ммоль) и смеси давали возможность прореагировать в течение следующих 3 часов. Смесь концентрировали в условиях пониженного давления и разбавляли 50 мл насыщенного водного карбоната натрия. Реакционную смесь экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты последовательно промывали 50 мл насыщенного солевого раствора и 50 мл воды, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-(S)-1-{2-[2-(морфолин-4-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрила 7b, которое непосредственно использовали на следующей стадии.
MS m/z (ESI): 376,7 [M+1]
Стадия 3
Получение цис-1-{2-[(S)-2-(морфолин-4-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрила гидрохлорида 7
Цис-1-{2-[(S)-2-(морфолин-4-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил 7b, полученный на предыдущей стадии, растворяли в 10 мл простого эфира. При охлаждении на ледяной водяной бане добавляли 2 мл соляной кислоты в простом эфире (0,5 н.). Полученную смесь фильтровали с помощью фильтрующей центрифуги с получением указанного в заголовке соединения цис-1-{2-[(S)-2-(морфолин-4-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорида 7 (30 мг, выход 3%) в виде белого порошка.
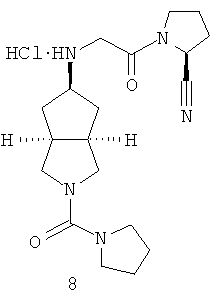
Пример 8
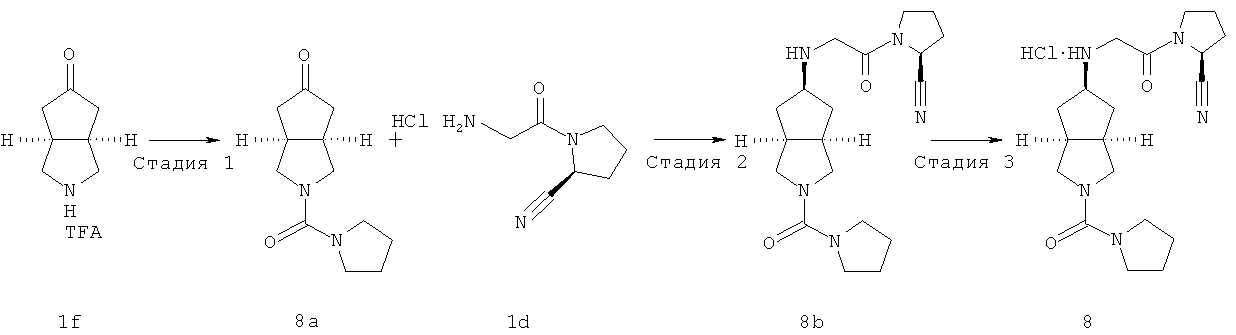
Цис-(S)-1-{2-[2-(пирролидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорид
Стадия 1
Получение 2-(пирролидин-1-карбонил)гексагидроциклопента[c]пиррол-5-она 8a
Гексагидроциклопента[c]пиррол-5-он трифторацетат 1f (478 мг, 2 ммоль) растворяли в 20 мл дихлорметана с последующим добавлением пирролидин-1-карбонилхлорида (0,276 мл, 2,5 ммоль) и триэтиламина (0,84 мл, 6 ммоль). Реакционной смеси давали возможность прореагировать в течение ночи при комнатной температуре. Смесь доводили до значения pH 4 раствором 10% лимонной кислоты. Смесь экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом магния, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 2-(пирролидин-1-карбонил)гексагидроциклопента[c]пиррол-5-она 8а (0,26 г, выход 58,5%) в виде бесцветного масла.
MS m/z (ESI): 223 [M+1]
Стадия 2
Получение цис-(S)-1-{2-[2-(пирролидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрила 8b
2-(Пирролидин-1-карбонил)гексагидроциклопента[c]пиррол-5-он 8a (0,26 г, 1,17 ммоль) и (S)-1-(2-аминоацетил)пирролидин-2-карбонитрил гидрохлорид 1d (0,33 г, 1,75 ммоль) растворяли в 50 мл тетрагидрофурана с последующим добавлением 5 г сульфата натрия и 0,05 мл уксусной кислоты. После взаимодействия смеси при комнатной температуре в течение 30 минут добавляли триацетоксиборгидрид натрия (0,75 г, 3,5 ммоль) и реакционной смеси давали возможность прореагировать в течение 3 часов. Смесь концентрировали в условиях пониженного давления и разбавляли 50 мл насыщенного водного карбоната натрия. Реакционную смесь экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора и 50 мл воды, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-(S)-1-{2-[2-(пирролидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрила 8b, который непосредственно использовали на следующей стадии.
MS m/z (ESI): 396 [M+1]
Стадия 3
Получение цис-(S)-1-{2-[2-(пирролидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорида
Цис-(S)-1-{2-[2-(пирролидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил 8b, полученный на предыдущей стадии, растворяли в 10 мл простого эфира. При охлаждении на ледяной водяной бане добавляли 2 мл соляной кислоты в простом эфире (0,5 н.). Полученную смесь фильтровали с помощью фильтрующей центрифуги с получением указанного в заголовке соединения цис-(S)-1-{2-[2-(пирролидин-1-карбонил)октагидроциклопента[c]пиррол-5-иламино]ацетил}пирролидин-2-карбонитрил гидрохлорида 8 (90 мг) в виде белого порошка.
1H ЯМР (CD3OD, 400 МГц) δ 4,72 (m, 1H), 4,09 (m, 2H), 3,43-3,30 (m, 11H), 2,62 (m, 2H), 2,35 (m, 2H), 2,18 (m, 2H), 2,08 (m, 2H), 1,77 (m, 4H).
Пример 9
Цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметил-гексагидроциклопента[c]пиррол-2-карбоксамид трифторацетат
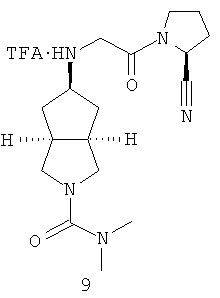
5-[2-((S)-2-Цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоновой кислоты диметиламид 1h, полученный в примере 1, растворяли в 10 мл дихлорметана с последующим добавлением 2 мл трифторуксусной кислоты при охлаждении на ледяной водяной бане. Реакционную смесь перемешивали в течение 30 минут. Полученную смесь фильтровали с помощью фильтрующей центрифуги с получением указанного в заголовке соединения цис-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамида трифторацетата 9 (201 мг) в виде белого порошка.
1H ЯМР (CDCl3, 400 МГц) δ 4,74 (t, 1H, J=5,2 Гц), 3,98 (d, 1H, J=15,6 Гц), 3,79 (d, 1H, J=15,6 Гц), 3,57-3,25 (m, 7H), 2,75 (s, 6H), 2,55 (m, 2H), 2,33 (m, 2H), 2,20-2,08 (m, 4H), 1,74 (m, 2H).
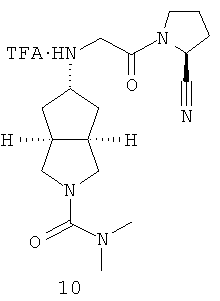
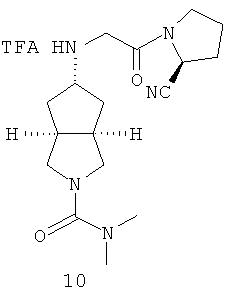
Пример 10
Транс-5-[2-((S)-2-цианопирролидин-1-ил)-2-оксоэтиламино]-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамид трифтоацетат
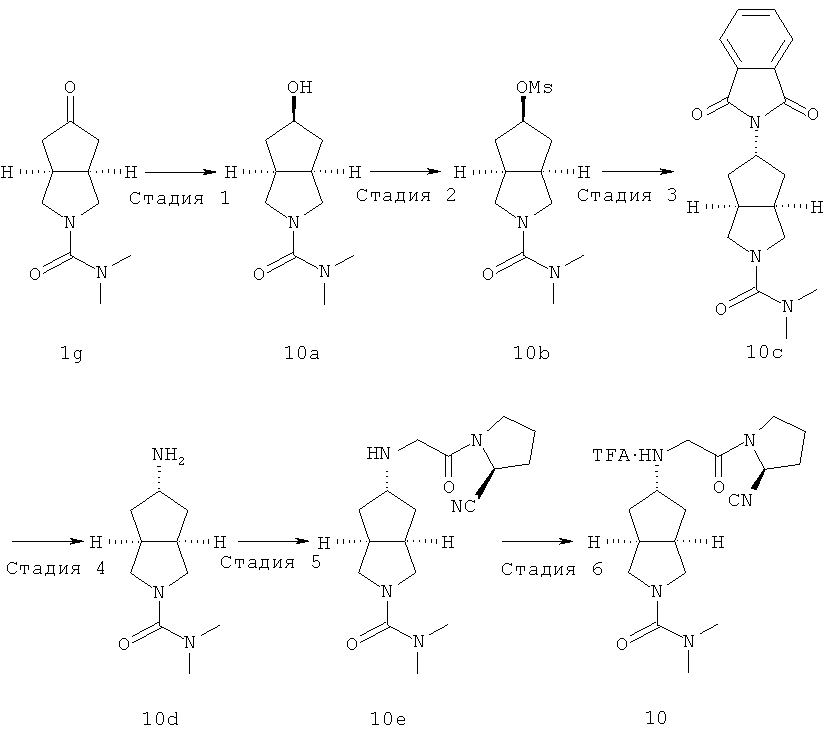
Стадия 1
Получение цис-5-гидроксигексагидроциклопента[c]пиррол-2-карбоксамида N,N-диметиламида 10а
В сухой трехгорлой колбе N,N-диметил-5-оксогексагидроциклопента[c]пиррол-2-карбоксамид 1 г (1,58 г, 8,06 ммоль) растворяли в 30 мл тетрагидрофурана и перемешивали в атмосфере азота. Смесь охлаждали до -25°C с последующим добавлением по капле раствора три-трет-бутоксиалюмогидрида лития (2,45 г, 9,6 ммоль) в 30 мл тетрагидрофурана. После взаимодействия реакционной смеси в течение 2,5 часов при той же температуре реакцию гасили водой. Смесь разбавляли 20 мл насыщенного водного хлорида аммония и нагревали до комнатной температуры. Слои разделяли и затем водный слой экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-5-гидроксигексагидроциклопента[c]пиррол-2-карбоновой кислоты N,N-диметиламида 10a (1,27 г, выход 80%) в виде бесцветного масла.
MS m/z (ESI): 199 [M+1]
Стадия 2
Получение 2-диметилкарбамоилоктагидроциклопента[c]пиррол-5-илового эфира цис-метансульфоновой кислоты 10b
В сухой одногорлой колбе цис-5-гидроксигексагидроциклопента[c]пиррол-2-карбоновой кислоты диметиламид 10a (1,69 г, 8,5 ммоль) растворяли в 30 мл дихлорметана и перемешивали в атмосфере азота. При охлаждении в бане лед-соль -5~0°C последовательно добавляли триэтиламин (1,66 мл, 14,45 ммоль) и метенсульфонилхлорид (2,2 г, 21,74 ммоль). Реакционную смесь перемешивали в течение 30 минут и нагревали до комнатной температуры. После того как реакционной смеси давали возможность прореагировать в течение 2 часов, смесь концентрировали в условиях пониженного давления и разбавляли 20 мл воды. Реакционную смесь экстрагировали этилацетатом (50 мл × 6). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали в условиях пониженного давления. Полученный остаток очищали с помощью колоночной хроматографии на силикагеле с получением указанного в заголовке соединения цис-метансульфоновой кислоты 2-диметилкарбамоилоктагидроциклопента[c]пиррол-5-илового эфира 10b (1,94 г, выход 83%) в виде белого твердого осадка.
MS m/z (ESI):277 [M+1]
Стадия 3
Получение транс-5-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 10c
В сухую одногорлую колбу при перемешивании в атмосфере азота добавляли метансульфоновой кислоты 2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-ильный сложный эфир 10b (1 г, 3,6 ммоль) в 20 мл N,N-диметилформамида и соль фталимид калия (993 мг, 5,4 ммоль). Реакционную смесь нагревали до 70°C и взаимодействие осуществляли в течение 3 часов. Смесь концентрировали при пониженном давлении для удаления N,N-диметилформамида и остаток разбавляли 20 мл воды. Смесь экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты промывали 50 мл насыщенного солевого раствора, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения транс-5-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 10c (1,06 г, выход 90%) в виде белого твердого вещества, которое непосредственно использовали на следующей стадии.
MS m/z (ESI): 328 [M+1]
Стадия 4
Получение транс-5-амино-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты N,N-диметиламида 10d
В одногорлой колбе транс-5-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 10c растворяли в 20 мл этанола (95%) при перемешивании, а затем добавляли гидразин (490 мг, 15,3 ммоль). Реакционную смесь кипятили с обратным холодильником в течение 8 часов и затем охлаждали до комнатной температуры. Смесь фильтровали и фильтрат концентрировали при пониженном давлении с получением белого твердого вещества. Получающееся в результате твердое вещество растворяли в 25 мл метанола, фильтровали и затем фильтрат концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на оксиде алюминия с получением указанного в заголовке соединения транс-5-амино-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты N,N-диметиламида 10d (290 мг, выход 48%) в виде бесцветного масла.
MS m/z (ESI): 198 [M+1]
Стадия 5
Получение транс-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-N,N-диметил-гексагидро-циклопента[c]пиррол-2-карбоксамида 10e
В сухой одногорлой колбе трансе-(2-хлор-этил)-пиррол-2-циано (334 мг, 1,94 ммоль) и раствор транс-5-амино-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты N,N-диметиламида 10d (290 мг, 1,46 ммоль) растворяли в 20 мл дихлорметана. Реакционную смесь кипятили с обратным холодильником в течение 48 часов. Смесь концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения транс-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-N,N-диметил-гексагидро-циклопента[c]пиррол-2-карбоксамида 10e, которое непосредственно использовали на следующей стадии.
Стадия 6
Получение транс-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-N,N-диметил-гексагидро-циклопента[c]пиррол-2-карбоксамида трифторацетата 10
транс-5-[2-((S)-2-Циано-пирролидин-1-ил)-2-оксо-этиламино]-N,N-диметил-гексагидро-циклопента[c]пиррол-2-карбоксамид 10е, полученный на вышеприведенной стадии, при перемешивании растворяли в 10 мл дихлорметана, а затем добавляли 2 мл трифторуксусной кислоты. Реакционную смесь перемешивали в течение 30 минут с получением указанного в заголовке соединения транс-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-N,N-диметил-гексагидро-циклопента[c]пиррол-2-карбоксамида трифторацетата 10 (201 мг) в виде белого твердого вещества.
MS m/z (ESI): 334 [M+1]
1H ЯМР (CDCl3, 400 МГц) δ 4,65 (m, 1H), 3,93 (d, 1H, J=15,2 Гц), 3,74 (d, 1H, J=15,2 Гц), 3,69-3,19 (m, 7H), 2,77 (s, 6H), 2,18-1,96 (m, 10H).
Пример 11
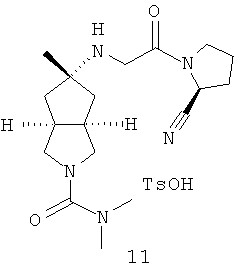
5-[2-((S)-2-Циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид пара-толуолсульфонат
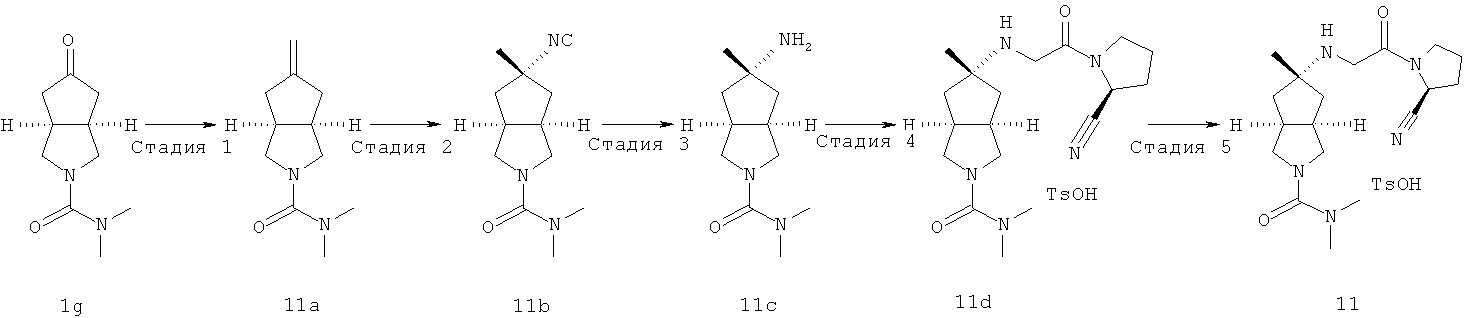
Получение 5-метилен-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
Калия трет-бутоксид (7,17 г, 0,064 моль) и метилтрифенилфосфония йодид (25,8 г, 0,064 моль) растворяли в 150 мл толуола в атмосфере азота. Реакционную смесь кипятили с обратным холодильником в течение 3 часов и охлаждали до комнатной температуры. Добавляли раствор [N,N-диметил-5-оксо-гексагидро-циклопента[c]пиррол-2-карбоксилата 1g (5,0 г, 0,0255 моль) в толуоле. Реакционную смесь перемешивали в течение 30 минут. За реакцией следили при помощи TCX до исчезновения исходных веществ. Смесь разбавляли 30 мл воды и 30 мл насыщенного солевого раствора и экстрагировали этилацетатом (200 мл×4). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-метилен-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 11a (4,0 г, выход 80%) в виде светло-желтого масла.
MS m/z (ESI): 195,2 [M+1]
Стадия 2
Получение 5-изоциано-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Метилен-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 11a (1,6 г, 8,23 ммоль) при перемешивании растворяли в 30 мл дихлорметана, а затем в атмосфере азота добавляли триметилсилилцианид (4,08 г, 41,2 ммоль) и перхлорат серебра (5,12 г, 24,7 ммоль). Реакционную смесь перемешивали в течение ночи при комнатной температуре и затем часть реакционной смеси обрабатывали насыщенным водным карбонатом натрия. За реакцией следили при помощи TCX, которая продемонстрировала, что остается большая часть исходных веществ. Затем смесь обрабатывали 20 мл насыщенного водного карбоната натрия при охлаждении в бане с ледяной водой, и в реакции высвобождалось тепло. Через 10 минут смесь фильтровали для удаления остатка. Отделенную водную фазу экстрагировали этилацетатом (50 мл×4). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением 5-изоциано-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 11b (0,33 г, выход 18,1%) в виде светло-желтого масла.
ГХ-МС: 221,1 [M+]
1H ЯМР (CDCl3, 400 МГц) δ 3,31 (m, 4H), 2,97 (m, 2H), 2,84 (s, 6H), 2,32 (m, 2H), 1,52 (s, 3H), 1,46 (m, 2H).
Стадия 3
Получение 5-амино-5-метил-гексагидро-циклопента[c]пиррол-2-
карбоновой кислоты диметиламида
К раствору 5-изоциано-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 11b (0,388 г, 1,75 ммоль) в 15 мл этанола при охлаждении в бане с ледяной водой добавляли 0,38 мл соляной кислоты (6 н.). Реакционную смесь нагревали до комнатной температуры и перемешивали в течение 1,5 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь гасили 20 мл насыщенного водного карбоната натрия. Смесь экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты выпаривали досуха. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-амино-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 11с (0,28 г, выход 75,7%) в виде желтого масла.
MS m/z (ESI): 212,2 [M+1]
Стадия 4
Получение 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Амино-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 11c (200 мг, 0,92 ммоль) растворяли в 8 мл N,N-диметилформамида, а затем добавляли 1-(2-хлор-ацетил)-пирролидин-2-циано (175 мг, 1,02 ммоль). Реакционную смесь оставляли для протекания реакции в течение ночи при комнатной температуре. Смесь концентрировали при пониженном давлении при 40~50°C для удаления N,N-диметилформамида. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 11d (0,135 г, выход 42,3%) в виде бесцветного масла. И выделяли 5-амино-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 11c (130 мг).
MS m/z (ESI): 348,2 [M+1]
Стадия 5
Получение 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида пара-толуолсульфоната
5-[2-((S)-2-Циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 11d (20 мг, 0,057 ммоль) и пара-толуолсульфоновой кислоты моногидрат (12 мг, 0,063 ммоль) растворяли в 2 мл дихлорметана при перемешивании при охлаждении в бане с ледяной водой. Реакционную смесь перемешивали в течение 10 минут при той же самой температуре, а баню лед-вода удаляли. Смесь концентрировали при пониженном давлении с получением масла. Смесь обрабатывали 5 мл этилацетата при перемешивании с получением белого осадка. Смесь фильтровали при пониженном давлении с получением указанного в заголовке соединения 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[с]пиррол-2-карбоновой кислоты диметиламида пара-толуолсульфоната 11 (0,026 г, выход 86,8%) в виде белого твердого вещества.
MS m/z (ESI): 348,2 [M+1]
1H ЯМР (CDCl3, 400 МГц) δ 7,68 (d, 2H), 7,17 (d, 2H), 3,72 (m, 1H), 3,68 (m, 1H), 3,58 (m, 1H), 3,30 (m, 4H), 2,98 (s, 2H), 2,81 (s, 6H), 2,55 (m, 2H), 2,22 (m, 4H), 2,05 (m, 2H), 1,62 (m, 2H), 1,56 (s, 3H).
Пример 12
5-[2-((S)-2-Циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида пара-толуолсульфонат
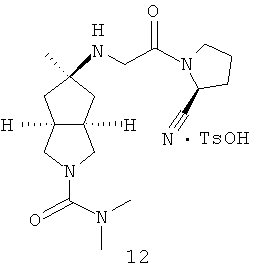
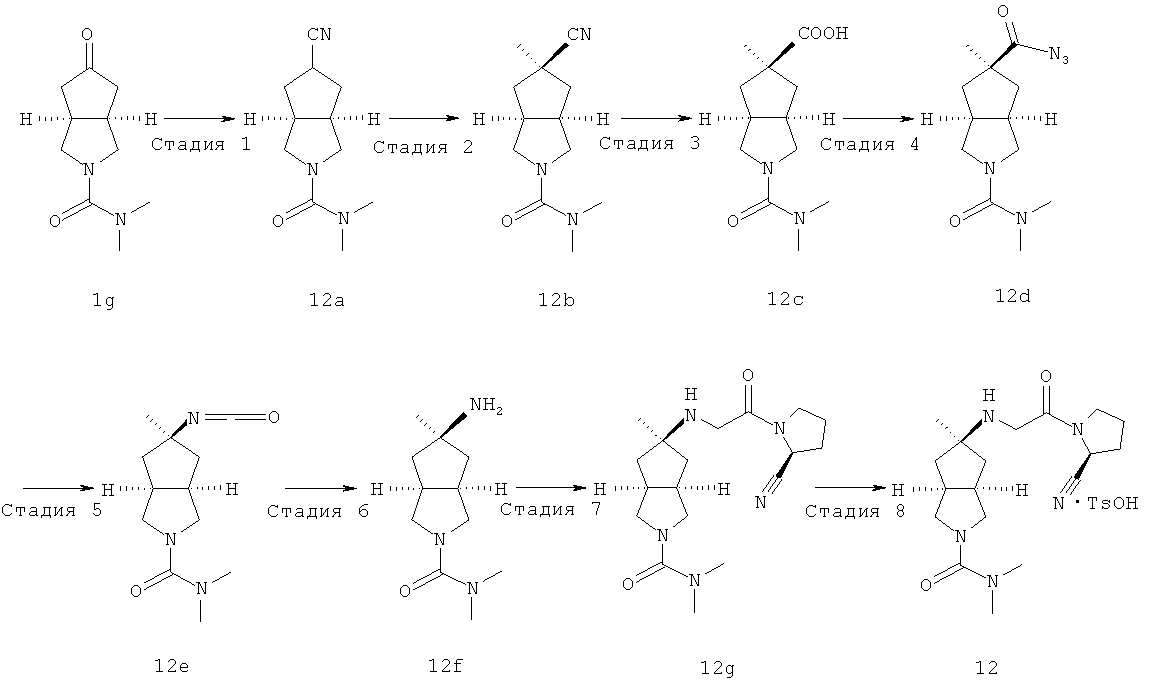
Стадия 1
Получение 5-циано-гексагидро-циклопента[c]пиррол-2-карбоновой
кислоты диметиламида
N,N-диметил-5-оксо-гексагидро-циклопента[c]пиррол-2-карбоксамид 1g (12,9 г, 0,066 моль) и 4-толуолсульфонилметил изоцианид (14,2 г, 0,0727 моль) растворяли в 240 мл 1,2-диметоксиэтана при перемешивании при охлаждении в бане с ледяной водой, а затем по каплям добавляли раствор трет-бутоксида калия (14,8 г, 0,132 моль) в трет-бутаноле. После завершения добавления баню лед-вода удаляли. Реакционную смесь нагревали до комнатной температуры, и реакция протекала в течение ночи. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь разбавляли 100 мл воды и 50 мл насыщенного солевого раствора и экстрагировали этилацетатом (200 мл × 3). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 12а (7,0 г, выход 51%) в виде светло-желтого масла.
MS m/z (ESI): 208,1 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 3,5-3,0 (m, 4H), 2,75 (s, 6H), 2,6 (m, 1H), 2,1-1,5 (m, 6H).
Стадия 2
5-Циано-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид
5-Циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12a (4,2 г, 20,2 ммоль) и йодметан (11,5 г, 80,8 ммоль) растворяли в 100 мл тетрагидрофурана, а затем по каплям при комнатной температуре в атмосфере азота добавляли гексаметилдисилазид лития (80,8 мл, 80,8 ммоль). Реакционной смеси давали возможность прореагировать в течение 2 часов до тех пор, пока MS не продемонстрировала исчезновение исходных веществ. Смесь разбавляли 100 мл воды и экстрагировали этилацетатом (200 мл×3). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением масла. ТСХ продемонстрировала, что существуют две соседние точки, и затем масло очищали путем колоночной хроматографии на силикагеле с получением соответствующего нижней точке соединения 5-циано-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 12b (2,411 г) в виде светло-желтого масла.
MS m/z (ESI): 222,2 [M+1]
1H ЯМР (CDCl3, 400 МГц) δ 3,44 (m, 2H), 3,3 (m, 2H), 2,85 (s, 6H), 2,77 (m, 2H), 2,06 (m, 2H), 2,00 (m, 2H), 1,36 (s, 3H).
Стадия 3
Получение 2-диметилкарбамоил-5-метил-октагидро-циклопента[c]пиррол-5-карбоновой кислоты
5-Циано-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12b (6,99 г, 31,6 ммоль) растворяли в 90 мл соляной кислоты (36%). Реакционную смесь нагревали при 50°C в масляной бане и перемешивали в течение 48 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Реакционную смесь разбавляли 100 мл воды. При охлаждении в бане с ледяной водой смесь доводили до pH 6 карбонатом калия и последовательно экстрагировали этилацетатом (1500 мл × 3) и дихлорметаном (150 мл × 3). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 2-диметилкарбамоил-5-метил-октагидро-циклопента[c]пиррол-5-карбоновой кислоты 12c (7,0 г, выход 92%) в виде желтой жидкости, которую непосредственно использовали на следующей стадии.
MS m/z (ESI): 241,2 [M+1]
Стадия 4
Получение 2-диметилкарбамоил-5-метилгексагидроциклопента[c]пиррол-5-карбонилазида
При охлаждении в бане с ледяной водой 2-диметилкарбамоил-5-метил-октагидро-циклопента[c]пиррол-5-карбоновую кислоту 12c (1,0 г, 4,2 ммоль) растворяли в 25 мл ацетона при перемешивании, а затем по каплям добавляли раствор триэтиламина (463,5 мг, 4,58 ммоль) и этилхлорформиата (497 мг, 4,58 ммоль) в 10 мл ацетона при -5°C. Реакционной смеси давали возможность прореагировать в течение 15 минут при -5°C, а затем добавляли раствор азида натрия (546 мг, 8,4 ммоль) в 10 мл воды. Затем реакционную смесь перемешивали в течение еще 30 минут при -5°C, и гасили 25 мл воды, и экстрагировали этилацетатом (50 мл × 3). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного соединения 2-диметилкарбамоил-5-метилгексагидроциклопента[c]пиррол-5-карбонилазида 12d (700 мг) в виде желтого масла.
Стадия 5 - Стадия 6
2-Диметилкарбамоил-5-метилгексагидроциклопента[c]пиррол-5-карбонилазид 12d (700 мг) добавляли к 20 мл толуола и реакционную смесь кипятили с обратным холодильником в течение 2 часов. Толуол упаривали, что приводило в результате к образованию (3aS,5r,6aR)5-изоцианато-5-метил-N,N,5-триметил-гексагидро-циклопента[c]пиррол-2(1H)-2-карбоксамида 12e. При охлаждении в бане с ледяной водой к раствору 8 мл соляной кислоты (8 н.) добавляли вышеупомянутую смесь. Реакционную смесь перемешивали в течение 30 минут и за реакцией следили при помощи ТСХ до исчезновения исходных веществ. При охлаждении в бане с ледяной водой смесь доводили до pH>12 при помощи 3 н. водного раствора гидроксида натрия и экстрагировали этилацетатом (15 мл × 3). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-амино-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 12f (500 мг) в виде светло-желтого масла.
MS m/z (ESI): 212,2 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 3,5-3,0 (m, 4H), 2,72 (s, 6H), 1,7-1,3 (m, 6H), 1,04 (s, 3H).
Стадия 7
Получение 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Амино-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12f (332 мг, 1,57 ммоль) растворяли в 10 мл смеси растворителей N,N-диметилформамид/дихлорметан (1:1), а затем добавляли 1-(2-хлор-ацетил)-пирролидин-2-циано (217 мг, 1,26 ммоль). Реакционной смеси давали возможность прореагировать в течение ночи при комнатной температуре и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 12g (240 мг, выход 55%) в виде бесцветного масла.
MS m/z (ESI): 348,2 [M+1]
Стадия 8
Получение 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида пара-толуолсульфоната
5-[2-((S)-2-Циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12g (150 мг, 0,43 ммоль) и пара-толуолсульфоновой кислоты моногидрат (82 мг, 0,43 ммоль) растворяли в 4 мл дихлорметана при перемешивании. Реакционную смесь перемешивали в течение 10 минут и концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида пара-толуолсульфоната 12 (0,022 г, выход 95,3%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 348,2 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 4,82 (m, 1H), 3,97 (s, 2H), 3,79 (m, 2H), 3,49 (m, 2H), 3,21 (m, 4H), 2,75 (s, 6H), 2,62 (m, 2H), 2,19 (m, 2H), 1,92-1,61 (m, 3H), 2,06 (m, 3H), 1,2 (s, 3H).
Пример 13
5-[2-((2S,4S)-2-Циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-N,N,5-триметил-гексагидро-циклопента[c]пиррол-2-карбоксамид тартрат
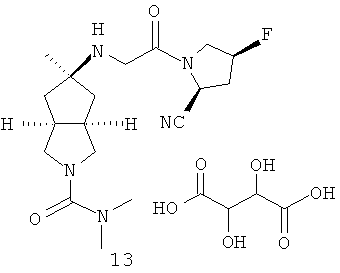
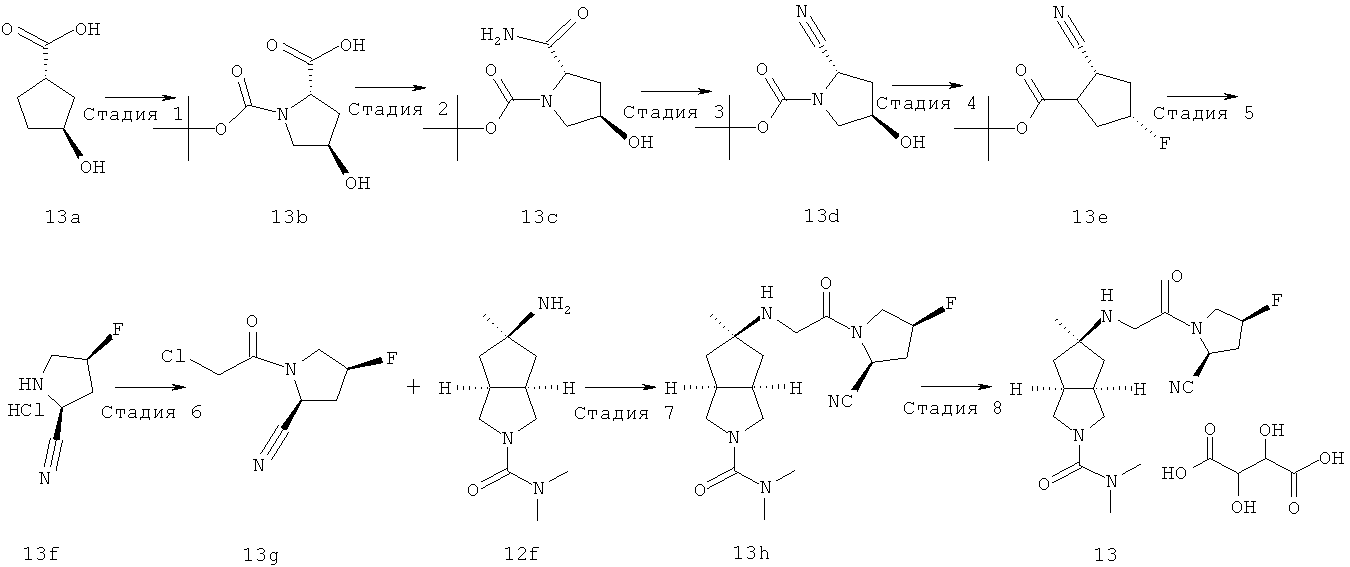
Стадия 1
Получение (2S,4R)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-2-карбоновой кислоты
L-Гидроксипролин 13a (60 г, 0,458 моль) добавляли к раствору 750 мл смеси растворителей тетрагидрофуран/вода (2:1), а затем 252 мл водного гидроксида натрия (10%) и раствор ди-трет-бутилдикарбоната (136 г, 0,624 моль) в 750 мл смеси растворителей тетрагидрофуран/вода (2:1). Реакционной смеси давали возможность прореагировать в течение ночи при комнатной температуре и разбавляли 500 мл этилацетата. Слои разделяли, а затем органический слой удаляли, и водный слой доводили до pH 2 концентрированной соляной кислотой. Смесь экстрагировали 1,5 л этилацетата. Объединенные органические экстракты промывали насыщенным солевым раствором, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,4R)-1-(трет-бутоксикарбонил)-4-гидроксипирролидин-2-карбоновой кислоты 13b (86,4 г, выход 80%) в виде бесцветного масла.
Стадия 2
Получение (2S,4R)-трет-бутил-2-карбамоил-4-гидроксипирролидин-1-карбоксилата
(2S,4R)-1-(трет-Бутоксикарбонил)-4-гидроксипирролидин-2-карбоновую кислоту 13b (86,4 г, 0,374 моль) растворяли в 1,2 л тетрагидрофурана, а затем добавляли триэтиламин (41 г, 0,411 моль) в атмосфере аргона. Реакционную смесь охлаждали до -15°C, добавляли этилхлорформиат (43,84 г, 0,411 моль). После перемешивания смеси в течение 10 минут добавляли водный раствор аммиак (236,8 мл). Реакционную смесь медленно нагревали до 5°C в течение 2 часов, а затем добавляли хлорид аммония (32 г). Реакционную смесь перемешивали в течение 30 минут и разделяли. Отделенную органическую фазу сушили над безводным сульфатом натрия и водную фазу экстрагировали этилацетатом (100 мл × 2). Объединенные органические экстракты концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,4R)-трет-бутил 2-карбамоил-4-гидроксипирролидин-1-карбоксилата 13c (74 г, выход 86%) в виде белого твердого вещества.
Стадия 3
Получение (2S,4R)-трет-бутил-2-циано-4-гидроксипирролидин-1-карбоксилата
(2S,4R)-трет-Бутил-2-карбамоил-4-гидроксипирролидин-1-карбоксилат 13с (74 г, 0,3217 моль) растворяли в 740 мл пиридина при перемешивании в атмосфере аргона. Смесь охлаждали до -20°C, а затем по каплям добавляли трифторацетангидрид (169 г, 0,804 моль). После завершения добавления реакционную смесь нагревали до комнатной температуры. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь гасили водой и разбавляли 0,8 л этилацетата. Отделенную органическую фазу промывали 500 мл насыщенного солевого раствора и нейтрализовали 400 мл концентрированной соляной кислоты до слабой кислотности. Получающуюся в результате смесь последовательно промывали 300 мл водного гидроксида натрия (2 M) и 500 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения (2S,4R)-трет-бутил-2-циано-4-гидроксипирролидин-1-карбоксилата 13d (49,7 г, выход 73%) в виде коричневого масла.
Стадия 4
Получение (2S,4R)-трет-бутил-2-циано-4-фторпирролидин-1-карбоксилата
(2S,4R)-трет-Бутил-2-циано-4-гидроксипирролидин-1-карбоксилат 13d (49,7 г, 0,2344 моль) растворяли в 1130 мл дихлорметана при перемешивании в атмосфере аргона. Смесь охлаждали до -30°C, а затем добавляли трифторид диэтиламиносеры (56,7 г, 0,3516 моль). После перемешивания в течение 45 минут реакционную смесь нагревали до -5°C. Затем смеси давали прореагировать в течение ночи при комнатной температуре. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Реакционную смесь нейтрализовали насыщенным водным раствором карбоната натрия до pH>7 с такой скоростью, что температуру реакционной смеси поддерживали ниже 20°C. Затем добавляли ледяную воду и 500 мл дихлорметана. Отделенную органическую фазу последовательно промывали 500 мл насыщенного водного гидросульфата натрия и 500 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, поддерживая температуру ниже 38°C, с получением указанного в заголовке соединения (2S,4S)-трет-бутил 2-циано-4-фторпирролидин-1-карбоксилата 13e (50 г, выход 100%) в виде светло-желтого твердого вещества.
Стадия 5
Получение (2S,4S)-4-фторпирролидин-2-карбонитрил
(2S,4S)-трет-Бутил-2-циано-4-фторпирролидин-1-карбоксилат 13e (1 г, 4,6 ммоль) растворяли в 2 мл этилацетата при перемешивании в атмосфере аргона. Смесь охлаждали до 15°C, а затем добавляли раствор хлорида водорода в 2,5 мл 1,4-диоксана (3 M). Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. Затем смеси давали возможность прореагировать в течение еще 5 часов при 25°C. За реакцией следили при помощи ТСХ, указывающей на присутствие исходных веществ. Реакционную смесь фильтровали и фильтрат перемешивали при комнатной температуре в течение 2 часов, что приводило в результате к образованию белого осадка. Смесь вновь фильтровали и фильтрат перемешивали в течение еще 2 часов, затем фильтровали. Осадки на фильтрах объединяли с получением указанного в заголовке соединения (2S,4S)-4-фторпирролидин-2-карбонитрила 13f (15,6 г, выход 76,6%) в виде белого твердого вещества.
Стадия 6
Получение (2S,4S)-1-(2-хлорацетил)-4-фторпирролидин-2-карбонитрила
Хлорацетилхлорид (11,13 г, 98,5 ммоль) растворяли в 120 мл дихлорметана при перемешивании в атмосфере аргона. После охлаждения до 0°C (2S,4S)-4-фторпирролидин-2-карбонитрил 13f (11,4 г, 75,7 ммоль) растворяли в 400 мл дихлорметана, а затем добавляли триэтиламин (16,1 г, 158,97 ммоль). К раствору хлорацетилхлорида в дихлорметане добавляли вышеупомянутую смесь в течение 30 минут. Реакционной смеси давали возможность прореагировать при 0°C в течение 2 часов и разбавляли 200 мл ледяной воды и 150 мл дихлорметана. Отделенную органическую фазу нейтрализовали насыщенным бисульфатом натрия, последовательно промывали 300 мл воды и 300 мл насыщенного солевого раствора, сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (2S,4S)-1-(2-хлорацетил)-4-фторпирролидин-2-карбонитрила 13д (8 г, выход 60%) в виде белого кристаллического вещества.
Ссылка: WO 2003002553.
Стадия 7
Получение 5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-N,N,5-триметил-гексагидро-циклопента[c]пиррол-2-карбоксамида
5-Амино-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12f (130 мг, 0,616 ммоль), (2S,4S)-1-(2-хлорацетил)-4-фторпирролидин-2-карбонитрил 13g (117,4 мг, 0,616 ммоль) и карбонат калия (85 мг, 0,616 ммоль) при перемешивании в атмосфере азота растворяли в смеси растворителей 2 мл дихлорметана и 2 мл N,N-диметилформамида. Реакционной смеси давали возможность прореагировать в течение ночи при комнатной температуре. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали при пониженном давлении для удаления N,N-диметилформамида и дихлорметана. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением 5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-N,N,5-триметил-гексагидро-циклопента[c]пиррол-2(1H)-карбоксамида 13h (0,13 г, выход 58%) в виде бесцветного масла.
Стадия 8
Получение 5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-N,N,5-триметил-гексагидро-циклопента[c]пиррол-2(1H)-карбоксамид тартрата
5-[2-((2S,4S)-2-Циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-N,N,5-триметил-гексагидро-циклопента[c]пиррол-2(1H)-карбоксамид 13 (0,16 г, 0,44 ммоль) при перемешивании растворяли в 5 мл дихлорметана, а затем по каплям добавляли раствор 5 мл винной кислоты (65,6 мг, 0,44 ммоль) в ацетоне. Реакционной смеси давали возможность прореагировать в течение 30 минут, приводя в результате к образованию белого осадка. Смесь фильтровали и осадок на фильтре промывали ацетоном с получением указанного в заголовке соединения 5-[2-((25,45)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-N,N,5-триметил-гексагидро-циклопента[c]пиррол-2-карбоксамид тартрата 13 (0,18 г, выход 82%) в виде белого твердого вещества.
1H ЯМР (DMSO-D6, 400 МГц) δ 5,76 (m, 1H), 5,46 (m, 1H), 5,0 (m, 1H), 4,08-4,05 (m, 4H), 3,97 (m, 2H), 3,69 (m, 2H), 2,73 (s, 6H), 2,61 (m, 2H), 1,87 (m, 3H), 1,57 (m, 3H), 1,18 (s, 3H), 1,9 (m, 2H).
Пример 14
5-Бензил-5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид тартрат
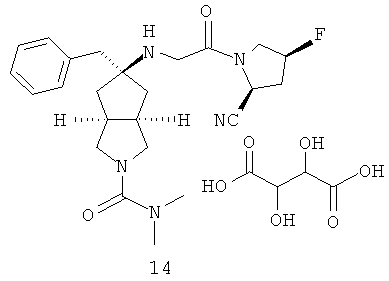
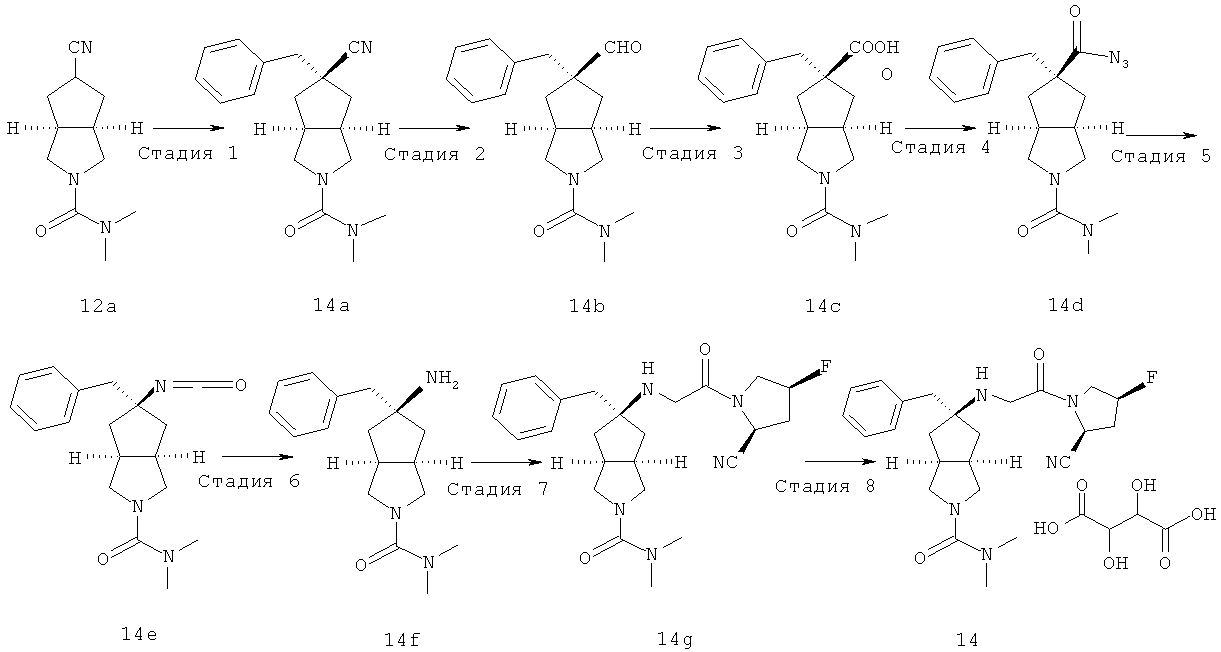
Стадия 1
Получение 5-бензил-5-циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12a (4,0 г, 19,3 ммоль) растворяли в 60 мл тетрагидрофурана при перемешивании, а затем добавляли бензилхлорид (5,4 г, 42,5 ммоль) и гексаметилдисилазид лития (42,5 мл, 42,5 ммоль) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 4 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ и гасили 50 мл воды. Смесь экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-бензил-5-циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 14a (2,6 г, выход 46%) в виде светло-желтого твердого вещества.
Стадия 2
Получение 5-бензил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Бензил-5-циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 14a (2,0 г, 6,7 ммоль) растворяли в 100 мл дихлорметана при перемешивании. После охлаждения до 0°C по каплям добавляли диизобутилалюминия гидрид (20,2 мл, 20,2 ммоль). За реакцией следили при помощи ТСХ до исчезновения исходных веществ и гасили водой. Смесь экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-бензил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 14b (729 мг, выход 36%) в виде бесцветного масла.
Стадия 3
Получение 5-бензил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновой кислоты
5-Бензил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 14b (0,729 г, 2,43 ммоль) растворяли в смеси растворителей 28 мл тетрагидрофурана и 14 мл воды в атмосфере азота, а затем при охлаждении до 0°C добавляли натрия дигидрофосфата дигидрат (1,14 г, 7,29 ммоль), хлорит натрия (0,66 г, 7,29 ммоль) и 2-метил-2-бутен (0,513 г, 7,32 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-бензил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновой кислоты 14c (0,76 г, выход 98%) в виде бесцветного масла.
MS m/z (ESI): 317,3 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 7,5-7,0 (m, 5H), 3,24 (m, 2H), 3,1 (m, 2H), 2,76 (s, 2H), 2,7 (s, 6H), 2,68 (m, 2H), 1,99-1,55 (m, 4H).
Стадия 4
Получение 5-бензил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазида
5-Бензил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновую кислоту 14 с (0,86 г, 2,72 ммоль) растворяли в 30 мл ацетона при охлаждении в бане с ледяной водой. Когда смесь охлаждали до -5°C по каплям последовательно добавляли раствор триэтиламина (0,303 г, 2,99 ммоль) и этилхлорформиата (0,325 г, 2,99 ммоль) в 15 мл ацетона. Реакционную смесь перемешивали в течение 15 минут, а затем по каплям добавляли раствор азида натрия (0,353 г, 5,44 ммоль) в 15 мл воды. Реакционную смесь перемешивали в течение еще 30 минут. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали при пониженном давлении и экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-бензил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазида 14d (0,9 г, выход 97%) в виде светло-желтого масла.
Стадия 5
Получение 5-бензил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Бензил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазид 14d (1,0 г, 2,72 ммоль) растворяли в 20 мл толуола при перемешивании. Реакционную смесь кипятили с обратным холодильником в течение 1,5 часов и растворитель упаривали с получением неочищенного указанного в заголовке соединения 5-бензил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 14e, который непосредственно использовали на следующей стадии.
Стадия 6
Получение 5-амино-5-бензил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
К раствору 12 мл соляной кислоты (8 н.) по каплям добавляли 5-бензил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 14e, полученный на вышеприведенной стадии при комнатной температуре. Реакционную смесь перемешивали в течение 30 минут и доводили до pH 9 при помощи 4 н. водного раствора гидроксида натрия. Смесь экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-амино-5-бензил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 14f (0,55 г, выход 70%) в виде бесцветного масла.
Стадия 7
Получение 5-бензил-5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Амино-5-бензил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 14f (0,1 г, 0,35 ммоль) растворяли в 4 мл смеси растворителей дихлорметана / N,N-диметилформамида (V/V=1/1), а затем добавляли (2S,4S)-1-(2-хлорацетил)-4-фторпирролидин-2-карбонитрил 13g (66,5 мг, 0,35 ммоль) и карбонат калия (49 мг, 0,35 ммоль). Реакционной смеси давали возможность прореагировать при 40°C в течение 12 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали и получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-бензил-5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 14g (87 мг, выход 56%) в виде белого твердого вещества.
MS m/z (ESI): 442,2 [M+1]
Стадия 8
Получение 5-бензил-5-[(2S,4S)-2-(2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата
5-Бензил-5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 14g (87 мг, 0,197 ммоль) растворяли в 3 мл дихлорметана при перемешивании, а затем по каплям добавляли раствор 3 мл винной кислоты в ацетоне. Реакционной смеси давали возможность прореагировать при комнатной температуре в течение 30 минут и концентрировали. Получающийся в результате остаток перекристаллизовывали из смеси растворителей этилацетата/ацетона с получением указанного в заголовке соединения 5-бензил-5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата 14 (80 мг, выход 68%) в виде белого твердого вещества.
1H ЯМР (DMSO-D6, 400 МГц) δ 7,32-7,2 (m, 5H), 5,55 (d, 1H), 5,41 (d, 1H), 4,97 (m, 1H), 4,31 (s, 2H), 4,08 (m, 1H), 4,0-3,5 (m, 6H), 2,81 (s, 2H), 2,78 (s, 6H), 2,5 (m, 2H), 2,0 (m, 3H), 1,6-1,3 (m, 6H).
Пример 15
5-Циклогексилметил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрат
Стадия 1
Получение 5-циано-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12а (3,66 г, 17,6 ммоль) растворяли в 150 мл тетрагидрофурана при перемешивании, а затем по каплям добавляли циклогексилметил бромид (6,2 г, 35,2 ммоль) и гексаметилдисилазид лития (35,2 мл, 32,5 ммоль). Реакционной смеси давали возможность взаимодействовать при комнатной температуре в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Затем добавляли 150 мл водного насыщенного раствора хлорида аммония. Смесь экстрагировали этилацетатом (60 мл × 3). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циано-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 15а (2,2 г, выход 41,5%) в виде желтого твердого вещества.
MS m/z (ESI): 304,5 [M+1]
Стадия 2
Получение 5-циклогексилметил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
После охлаждения до 0°C при помощи бани лед-вода 5-циано-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 15а (1,2 г, 3,95 ммоль) растворяли в 30 мл дихлорметана при перемешивании, а затем по каплям добавляли диизобутилалюминия гидрид (11,8 мл, 11,8 ммоль). Реакционной смеси давали возможность прореагировать в течение 1 часа. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Затем добавляли раствор насыщенного водного тартрата калия и натрия. Смесь перемешивали до прояснения раствора и экстрагировали этилацетатом (100 мл × 3). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циклогексилметил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 15b (0,4 г, выход 33%) в виде светло-желтого масла.
MS m/z (ESI): 307,4 [M+1]
Стадия 3
Получение 5-циклогексилметил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновой кислоты
5-Циклогексилметил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 15b (0,713 г, 2,33 ммоль) растворяли в смеси растворителей 60 мл тетрагидрофурана и 30 мл воды. После охлаждения до 0°C добавляли натрия дигидрофосфата дигидрат (1,09 г, 6,99 ммоль), хлорит натрия (0,79 г, 6,99 ммоль) и 2-метил-2-бутен (0,62 мл, 7,0 ммоль). Реакционной смеси давали возможность взаимодействовать при 0°C в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали для удаления тетрагидрофурана и экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циклогексилметил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновой кислоты 15c (0,75 г, выход 100%) в виде желтого твердого вещества.
MS m/z (ESI): 323,3 [M+1]
Стадия 4
Получение 5-циклогексилметил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазида
При охлаждении в бане с ледяной водой 5-циклогексилметил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновую кислоту 15c (0,75 г, 2,33 ммоль) растворяли в 20 мл ацетона при перемешивании, а затем по каплям добавляли триэтиламин (0,36 мл, 2,56 ммоль) и раствор 2 мл этилхлорформиата (0,25 мл, 2,56 ммоль) в ацетоне. Реакционную смесь перемешивали в течение 15 минут и добавляли раствор 2 мл азида натрия (0,303 г, 4,66 ммоль) в воде. Реакционной смеси давали возможность прореагировать в течение еще 30 минут при 0~-5°C. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали для удаления ацетона и разбавляли 10 мл воды. Смесь экстрагировали этилацетатом (10 мл × 5). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-циклогексилметил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазида 15d (0,79 г) в виде желтого масла, которое непосредственно использовали на следующей стадии.
Стадия 5
Получение 5-циклогексилметил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Циклогексилметил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазид 15d (0,79 г, 2,27 ммоль) растворяли в 20 мл толуола и реакционную смесь кипятили с обратным холодильником в течение 1 часа. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали с получением указанного в заголовке соединения 5-циклогексилметил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 15e (0,65 г) в виде серого твердого вещества, которое непосредственно использовали на следующей стадии.
MS m/z (ESI): 320,4 [M+1]
Стадия 6
Получение 5-амино-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
При охлаждении в бане с ледяной водой к раствору 10 мл соляной кислоты (8 н.) по каплям добавляли 5-циклогексилметил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 15e (0,65 г, 2,03 ммоль). Затем баню лед-вода удаляли. Реакционной смеси давали возможность прореагировать при 50°C в течение 20 минут. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь доводили до pH>8 концентрированным аммиаком и экстрагировали этилацетатом (30 мл × 5). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением 5-амино-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 15f (0,5 г, выход 84%) в виде серого масла.
MS m/z (ESI): 294,3 [M+1]
Стадия 7
Получение 5-(циклогексилметил)-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Амино-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 15f (0,115 г, 0,39 ммоль) растворяли в 10 мл смеси растворителей дихлорметан/N,N-диметилформамид (V/V=1/1) при перемешивании, а затем добавляли 1-(2-хлор-ацетил)-пирролидин-2-циано (54 мг, 0,31 ммоль). Реакционной смеси давали возможность взаимодействовать при 50°C в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали при пониженном давлении для удаления дихлорметана и N,N-диметилформамида. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-(циклогексилметил)-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 15g (113 мг, выход 67%) в виде бесцветного масла.
MS m/z (ESI): 430,5 [M+1]
Стадия 8
Получение 5-(циклогексилметил)-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата
5-(Циклогексилметил)-5-[2-((S)-2-Циано-пирролидин-1-ил)-2-оксо-этиламино]-5-циклогексилметил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 15g (113 мг, 0,263 ммоль) растворяли в 10 мл этилацетата, а затем по каплям добавляли раствор 2 мл винной кислоты в ацетоне. Реакционную смесь перемешивали в течение 30 минут и фильтровали с получением указанного в заголовке соединения 5-(циклогексилметил)-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата 15 (40 мг) в виде белого твердого вещества.
1H ЯМР (DMSO-D6, 400 МГц) δ 5,2 (m, 1H), 4,77 (s, 2H), 4,18 (m, 3H), 4,02 (m, 1H), 3,63 (m, 2H), 3,32 (m, 2H), 2,73 (s, 6H), 2,58 (m, 2H), 2,16-1,87 (m, 9H), 1,59-1,19 (m, 8H).
Пример 16
5-Циклопентил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрат
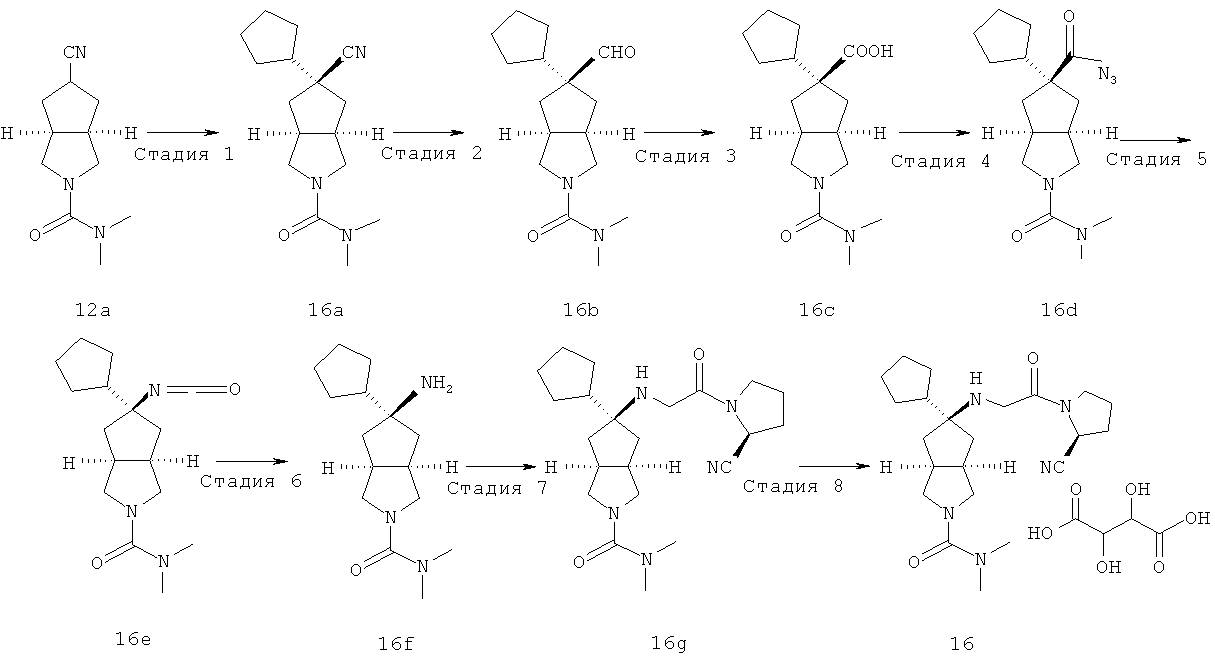
Стадия 1
Получение 5-циано-5-циклопентил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12а растворяли в 30 мл N,N-диметилформамида, а затем добавляли йодоциклопентан (5,4 г, 27,5 ммоль) и гексаметилдисилазид лития (27,5 мл, 27,5 ммоль) в атмосфере азота. Реакционную смесь перемешивали при комнатной температуре в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь разбавляли 10 мл воды и концентрировали для удаления N,N-диметилформамида и экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циано-5-циклопентил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 16a (2,6 г, выход 42%) в виде светло-желтого твердого вещества.
MS m/z (ESI): 276,2 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 3,35-3,0 (m, 4H), 2,24 (s, 6H), 1,34-2,5 (m, 15H).
Стадия 2
Получение 5-циклопентил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Циано-5-циклопентил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 16a (0,817 г, 2,97 ммоль) растворяли в 40 мл дихлорметана при перемешивании при 0°C, а затем по каплям добавляли диизобутилалюминия гидрид (8,9 мл, 8,9 ммоль). Реакционную смесь перемешивали в течение 45 минут. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали для удаления дихлорметана, и остаток разбавляли 100 мл насыщенного водного тартрата калия и натрия. Смесь перемешивали до просветления раствора и экстрагировали этилацетатом (100 мл×4). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циклопентил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 16b (0,266 г, выход 32%) в виде светло-желтого масла.
MS m/z (ESI): 279,3 [M+1]
Стадия 3
Получение 5-циклопентил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновой кислоты
5-Циклопентил-5-формил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 16b (0,266 г, 0,955 ммоль) растворяли в смеси растворителей 20 мл тетрагидрофурана и 10 мл воды. Затем при 0°С добавляли натрия дигидрофосфата дигидрат (0,448 г, 2,87 ммоль), хлорит натрия (0,26 г, 2,87 ммоль) и 2-метил-2-бутен (0,24 мл, 2,88 ммоль). Реакционную смесь перемешивали при 0°C в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали для удаления тетрагидрофурана и экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циклопентил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновой кислоты 16c (0,28 г, выход 99,6%) в виде желтого твердого вещества.
MS m/z (ESI): 295,5 [M+1]
Стадия 4
Получение 5-циклопентил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазида
5-Циклопентил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбоновую кислоту 16c (0,28 г, 6,95 ммоль) растворяли в 20 мл ацетона при охлаждении в бане с ледяной водой, а затем добавляли триэтиламин (0,15 мл, 1,05 ммоль) и раствор 2 мл этилхлорформиата (0,1 мл, 1,05 ммоль) в ацетоне при 0°C~-5°C. После перемешивания реакционной смеси в течение 15 минут по каплям добавляли раствор азида натрия (0,124 г, 1,9 ммоль) в воде. Реакционную смесь перемешивали в течение еще 30 минут. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали и остаток разбавляли 10 мл воды. Смесь экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-циклопентил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазида 16d (0,287 г, выход 95%) в виде желтого масла, которое непосредственно использовали на следующей стадии.
Стадия 5
Получение 5-циклопентил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Циклопентил-2-диметилкарбамоил-октагидро-циклопента[c]пиррол-5-карбонилазид 16d (0,287 г, 0,9 ммоль) растворяли в 10 мл толуола при перемешивании. Реакционную смесь кипятили с обратным холодильником в течение 1 часа. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали с получением указанного в заголовке соединения 5-циклопентил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 16e, который непосредственно использовали на следующей стадии.
MS m/z (ESI): 292,3 [M+1]
Стадия 6
Получение 5-амино-5-циклопентил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
При охлаждении в бане с ледяной водой к раствору 10 мл соляной кислоты (8 н.) по каплям добавляли 5-циклопентил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 16e, полученный на вышеприведенной стадии. Затем баню лед-вода удаляли и реакционной смеси давали возможность прореагировать при 50°C в течение 15 минут. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь доводили до pH>8 при помощи концентрированного аммиака и экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-амино-5-циклопентил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 16f (0,18 г, выход 81,8%) в виде желтого масла.
MS m/z (ESI): 266,2 [M+1]
Стадия 7
Получение 5-циклопентил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Амино-5-циклопентил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 16f (0,108 г, 0,407 ммоль) и 1-(2-хлор-ацетил)-пирролидин-2-циано (70 мг, 0,407 ммоль) растворяли в 3 мл N,N-диметилформамида при перемешивании, а затем добавляли карбонат калия (57 мг, 0,407 ммоль) в атмосфере азота. Реакционной смеси давали возможность прореагировать в течение ночи при 80°C при нагревании в масляной бане. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали и получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циклопентил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 16g (100 мг, выход 61,3%) в виде бесцветного масла.
MS m/z (ESI): 402,3 [M+1]
Стадия 8
Получение 5-циклопентил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата
5-Циклопентил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 16g (102 мг, 0,254 ммоль) растворяли в 2 мл этилацетата, а затем добавляли раствор 3 мл винной кислоты в ацетоне. Реакционную смесь перемешивали в течение 30 минут, что приводит в результате к образованию белого осадка, а затем добавляли н-гексан и вновь перемешивали. Смесь фильтровали с получением указанного в заголовке соединения 5-циклопентил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата 16 (88 мг, выход 65%) в виде белого твердого вещества.
Пример 17
5-Бензил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрат
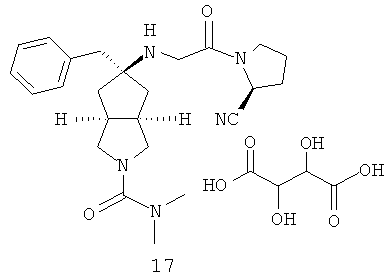
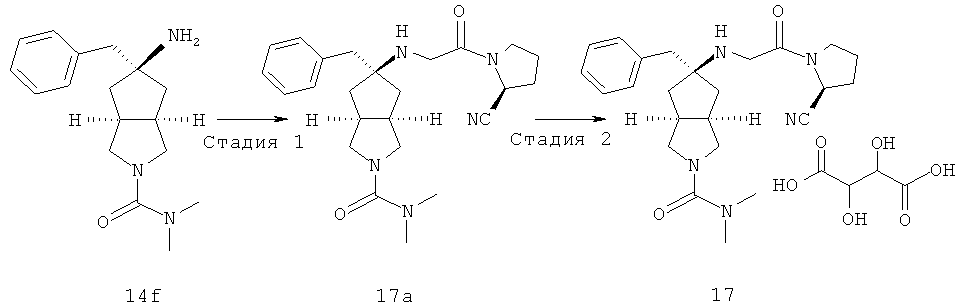
Стадия 1
Получение 5-бензил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Амино-5-бензил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 14f (0,1 г, 0,35 ммоль), 1-(2-хлорацетил)-пирролидин-2-циано (120,4 мг, 0,7 ммоль) и карбонат калия (49 мг, 0,35 ммоль) растворяли в 3 мл N,N-диметилформамида в атмосфере азота. Реакционной смеси давали возможность прореагировать в течение ночи при 80°C при нагревании в масляной бане. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали, и получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-бензил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 17a (56 мг, выход 40%) в виде бесцветного масла.
MS m/z (ESI): 424,3 [M+1]
Стадия 2
Получение 5-бензил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата
5-Бензил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 17a (56 мг, 0,132 ммоль) растворяли в 1 мл этилацетата при перемешивании, а затем добавляли раствор 1 мл винной кислоты в ацетоне. Реакционную смесь перемешивали в течение 30 минут, приводя в результате к образованию белого осадка, а затем добавляли н-гексан и вновь перемешивали. Смесь фильтровали с получением указанного в заголовке соединения 5-бензил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата 17 (50 мг, выход 67%) в виде белого твердого вещества.
1H ЯМР (DMSO-D6, 400 МГц) δ 7,32-7,19 (m, 5H), 4,78 (m, 1H), 4,23 (s, 2H), 3,8-3,0 (m, 8H), 2,75 (s, 2H), 2,73 (s, 6H), 2,18 (m, 2H), 2,16 (m, 2H), 1,87 (m, 3H), 1,35 (m, 3H).
Пример 18
5-[2-((2S,4S)-2-Циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида пара-толуолсульфонат
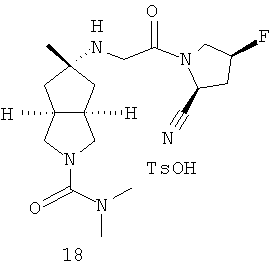
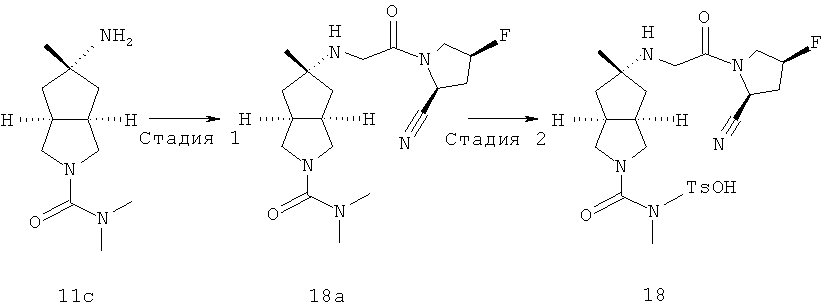
Стадия 1
Получение 5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Амино-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 11с (1,3 г, 7,58 ммоль), (25,45)-1-(2-хлорацетил)-4-фторпирролидин-2-карбонитрил 13g (1,74 г, 9,1 ммоль), карбонат калия (1,26 г, 9,1 ммоль), 30 мл N,N-диметилформамида и 18 мл дихлорметана добавляли в колбу в атмосфере азота. Реакционной смеси давали возможность взаимодействовать в течение ночи при 30°C при нагревании в масляной бане. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали для удаления N,N-диметилформамида. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 18a (1,39 г, выход 50%) в виде белого твердого вещества.
MS m/z (ESI): 348,2 [M+1]
Стадия 2
Получение 5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида пара-толуолсульфоната
5-[2-(2-Циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 18a (0,9 г, 2,47 ммоль) растворяли в 5 мл дихлорметана при перемешивании при комнатной температуре. К раствору пара-толуолсульфоновой кислоты моногидрата (469 мг, 2,47 ммоль) в 3 мл ацетона добавляли вышеупомянутый раствор, что приводило в результате к образованию белого осадка. Реакционную смесь перемешивали в течение 30 минут, а затем добавляли 1 мл н-гексана. Смесь фильтровали с получением указанного в заголовке соединения 5-[2-((2S,4S)-2-циано-4-фтор-пирролидин-1-ил)-2-оксо-этиламино]-5-метил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида пара-толуолсульфоната 18 (1,3 г, выход 93%) в виде белого твердого вещества.
MS m/z (ESI): 366,1 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 7,74 (d, 2H), 7,29 (d, 2H), 5,57-5,44 (m, 1H), 5,07 (m, 1H), 4,19-4,07 (m, 4H), 3,49 (m, 2H), 3,35 (m, 2H), 3,0 (m, 2H), 2,9 (s, 6H), 2,6 (m, 2H), 2,39 (m, 5H), 1,57 (m, 2H), 1,34 (s, 3H).
Пример 19
5-Этил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрат
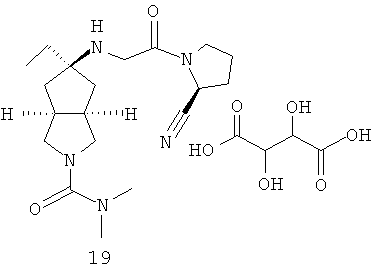
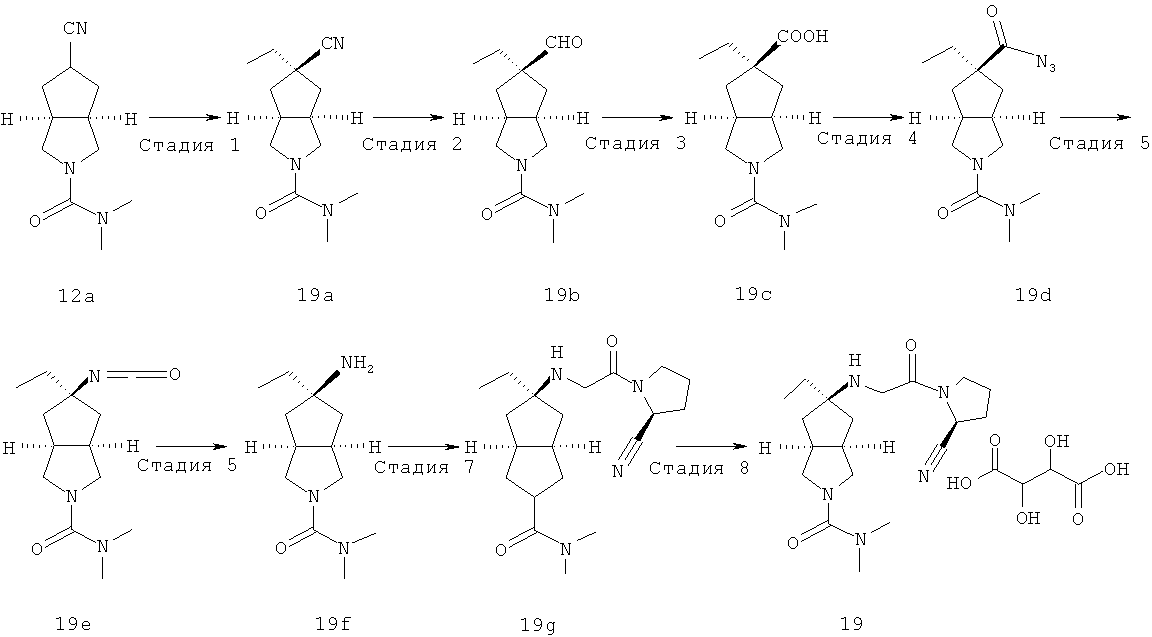
Стадия 1
5-Циано-5-этил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид
5-Циано-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 12a (3,0 г, 14,5 ммоль) растворяли в 60 мл тетрагидрофурана при перемешивании, а затем добавляли этилйодид (4,52 г, 29 ммоль) и гексаметилдисилазид лития (29 мл, 29 ммоль) в атмосфере азота. Реакционной смеси давали возможность взаимодействовать при комнатной температуре в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь разбавляли 20 мл воды и экстрагировали этилацетатом (200 мл×3). Объединенные органические экстракты сушили над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением масла. ТСХ продемонстрировала, что существуют две соседние точки, и затем масло очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-циано-5-этил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 19а (1,64 г) в виде светло-желтого твердого вещества.
MS m/z (ESI): 236,3 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 3,5-3,0 (m, 4H), 2,7 (s, 6H), 1,9-1,2 (m, 8H), 1,19 (m, 3H).
Стадия 2
Получение 5-этил-5-формил-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамида
5-Циано-5-этил-гексагидро-циклопента[фиррол-2-карбоновой кислоты диметиламид 19a (1,54 г, 6,55 ммоль) растворяли в 60 мл дихлорметана. По каплям при 0°C добавляли диизобутилалюмогидрид (19,6 мл, 19,6 ммоль). Реакционную смесь перемешивали при 0°C в течение 30 минут. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь гасили 1,5 мл воды, сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-этил-5-формил-N,N-диметилгексагидроциклопента[c]пиррол-2-карбоксамид 19b (0,4 г, выход 26%) в виде светло-желтого масла.
MS m/z (ESI): 239,1 [M+1]
Стадия 3
Получение 5-этил-2-диметилкарбамоил-гексагидро-циклопента[c]пиррол-5-карбоновой кислоты
5-Этил-5-формил-N,N-диметилгексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 19b (0,4 г, 1,68 ммоль) растворяли в смеси растворителей 18 мл тетрагидрофурана и 9 мл воды, а затем при 0°C добавляли натрий дигидрофосфата дигидрат (787 мг, 5,04 ммоль), хлорит натрия (0,454 г, 5,04 ммоль) и 2-метил-2-бутен (354 мг, 5,06 ммоль). Реакционной смеси давали возможность прореагировать при 0°C в течение 2 часов в атмосфере азота. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь экстрагировали этилацетатом. Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-этил-2-диметилкарбамоил-гексагидро-циклопента[c]пиррол-5-карбоновой кислоты 19c (0,426 г, выход 100%) в виде желтого твердого вещества, которое непосредственно использовали на следующей стадии.
MS m/z (ESI): 255,2 [M+1]
Стадия 4
Получение 5-этил-2-диметилкарбамоил-гексагидро-циклопента[c]пиррол-5-карбонилазида
5-Этил-2-диметилкарбамоил-гексагидро-циклопента[c]пиррол-5-карбоновую кислоту 19c (0,56 г, 2,2 ммоль) растворяли в 60 мл ацетона при охлаждении в бане с ледяной водой. После охлаждения до -5°C по каплям добавляли триэтиламин (0,245 мг, 2,43 ммоль) и раствор 2 мл этилхлорформиата (263 мг, 2,43 ммоль) в ацетоне. После того как реакционную смесь перемешивали в течение 15 минут, добавляли раствор азида натрия (0,286 г, 4,4 ммоль) в 2 мл воды. Реакционную смесь перемешивали при -5°C~0°C в течение еще 30 минут. За реакцией следили при помощи ТСХ до исчезновения исходных веществ.
Смесь разбавляли 10 мл воды, экстрагировали этилацетатом (10 мл×5). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением указанного в заголовке соединения 5-этил-2-диметилкарбамоил-гексагидро-циклопента[c]пиррол-5-карбонилазида 19d (0,5 г) в виде светло-желтого масла, которое непосредственно использовали на следующей стадии.
Стадия 5
Получение 5-этил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Этил-2-диметилкарбамоил-гексагидро-циклопента[c]пиррол-5-карбонилазид 19d (0,5 г, 1,86 ммоль) растворяли в 30 мл толуола. Реакционную смесь кипятили с обратным холодильником в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали с получением указанного в заголовке соединения 5-этил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 19e, которое непосредственно использовали на следующей стадии.
Стадия 6
Получение 5-амино-5-этил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
К раствору 12 мл соляной кислоты (8 н.) добавляли 5-этил-5-изоцианато-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 19e, полученный на вышеприведенной стадии при комнатной температуре. Реакционную смесь перемешивали в течение 30 минут и доводили до pH 9-10 при помощи 8 н. водного раствора гидроксида натрия. Отделенный водный слой экстрагировали дихлорметаном (30 мл×5). Объединенные органические экстракты сушили над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-амино-5-этил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида 19f (0,3 г, выход 71%) в виде бесцветного масла.
MS m/z (ESI): 226,2 [M+1]
Стадия 7
Получение 5-этил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида
5-Амино-5-этил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 19f (0,202 г, 0,897 ммоль), 1-(2-хлор-ацетил)-пирролидин-2-циано (185 мг, 1,07 ммоль), карбонат калия (148 мг, 1,07 ммоль) и смеси растворителей 9 мл N,N-диметилформамид/дихлорметан (V/V=1/1) добавляли в колбу в атмосфере азота. Реакционной смеси давали возможность взаимодействовать при 60°C в течение 2 часов. За реакцией следили при помощи ТСХ до исчезновения исходных веществ. Смесь концентрировали при пониженном давлении для удаления дихлорметана и N,N-диметилформамида. Получающийся в результате остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-этил-гексагидро-циклопента[с]пиррол-2-карбоновой кислоты диметиламида 19g (120 мг, выход 40%) в виде светло-желтого масла.
MS m/z (ESI): 362,2 [M+1]
Стадия 8
Получение 5-этил-5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата
5-[2-((S)-2-Циано-пирролидин-1-ил)-2-оксо-этиламино]-5-этил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламид 19g (120 мг, 0,33 ммоль) растворяли в 1 мл этилацетата, а затем по каплям добавляли раствор 2 мл винной кислоты (50 мг, 0,33 ммоль) в ацетоне. Реакционную смесь перемешивали в течение 30 минут и фильтровали с получением указанного в заголовке соединения 5-[2-((S)-2-циано-пирролидин-1-ил)-2-оксо-этиламино]-5-этил-гексагидро-циклопента[c]пиррол-2-карбоновой кислоты диметиламида тартрата 19 (160 мг, выход 94%) в виде белого твердого вещества.
MS m/z (ESI): 362,2 [M+1]
1H ЯМР (DMSO-D6, 400 МГц) δ 4,79 (m, 1H), 4,11 (s, 2H), 3,65 (m, 2H), 3,49 (m, 2H), 3,22 (m, 4H), 2,75 (s, 6H), 2,55 (m, 2H), 2,19 (m, 2H), 2,09 (m, 2H), 1,99 (m, 2H), 1,51 (m,4H), 0,86 (t, 3G).
Примеры тестов:
Биологические анализы
Анализ ингибирующей активности DPP-IV/DPP-VIII/DPP-IX
Следующие способы могут быть использованы для измерения активностей соединений в соответствии с настоящим описанием, ингибирующих ферментативную активность DPP-IV/DPP-VIII/DPP-IX. Соединения в соответствии с настоящим описанием тестируют в отношении их способности ингибировать ферментативную активность очищенного DPP-IV/DPP-VIII/DPP-IX. Скорость ингибирования или среднюю ингибирующую концентрацию IC50 (концентрацию тестируемого соединения, при которой ингибируется 50% ферментативной активности) для каждого соединения определяют путем инкубации фиксированных количеств смешанного субстрата фермента с отличающимися концентрациями тестируемых соединений.
Анализ ингибирующей активности DPP-IV
Материалы и способы:
Материалы:
|
Процедура:
Анализ осуществляли следующим образом:
|
Определение степени ингибирования:
[1-(S-B)/(N-B)]·100%
S: образец
B: бланк-контроль
N: отрицательный контроль
Анализ ингибирующей активности DPP-VIII Материалы и способы:
Материалы:
|
Процедура:
Анализ выполняли следующим образом:
|
Определение степени ингибирования:
[1-(S-B)/(N-B)]·100%
S: образец
B: бланк-контроль
N: отрицательный контроль
Анализ ингибирующей активности DPP-IX
Материалы и способы:
Материалы:
|
Процедура:
Анализ осуществляли следующим образом:
|
Определение степени ингибирования:
[1-(S-B)/(N-B)]·100%
S: образец
B: бланк-контроль
N: отрицательный контроль
IC50 в отношении DPP-IV/DPP-VIII/DPP-IX для тестируемых соединений продемонстрирована в Таблице 1:
|
Предварительная оценка гипогликемических действий ингибиторов DPP-IV
Задача:
Для обнаружения действий ингибиторов DPP-IV соединений Примера 1 и Примера 2 в отношении толерантности к перорально вводимой глюкозе у нормальных мышей ICR оценивали гипогликемические действия in vivo.
Тестируемые животные:
|
Лекарства:
|
Способ приготовления: Соединения точно взвешивали и затем растворяли в дважды дистиллированной воде. Готовили соответственно суспензии 0,5, 0,15 и 0,05 мг/мл (Примечание: Хотя инструкция к продукту указывает на то, что тестируемые соединения растворимы в воде, в эксперименте он был плохо растворим в воде, т.е. в низких концентрациях он мог быть растворен, но в концентрации 0,5 мг/мл имелись видимые не вооруженным глазом частицы. Попытались использовать 1% CMC (карбоксиметилцеллюлоза) для суспендирования соединений, но этот раствор оказался не лучше, чем дважды дистиллированная вода.)
|
Способ приготовления: Соединения точно взвешивали и затем растворяли в дважды дистиллированной воде и полностью смешивали для приготовления раствора 1,5 мг/мл и затем разбавляли соответственно до 0,5, 0,15 и 0,05 мг/мл прозрачного раствора.
|
Способ:
1. Действия соединений на уровни глюкозы в крови у нормальных мышей ICR
Нормальных самцов мышей ICR случайным образом группировали по массе, 6 мышей в каждой группе. Группы включали бланк-контрольную группу, а также обрабатываемые различными дозами группы в соответствии со следующим:
Тест 1:
Бланк-контроль: дважды дистиллированная вода через зонд.
|
Тест 2:
Бланк-контроль: дважды дистиллированная вода через зонд.
|
Животных в каждой группе не кормили в течение 6 часов, и затем предварительно обрабатывали соответственно тестируемыми соединениями или дважды дистиллированной водой через зонд во время разового введения. Через 30 минут животным вводили дозу глюкозы 2,5 г/кг через зонд. До и после введения глюкозы в момент времени 30, 60 и 120 минут кровь отбирали для измерения уровней глюкозы в сыворотке крови.
2. Определение уровня глюкозы в сыворотке крови:
Уровень глюкозы в сыворотке крови измеряют при помощи набора. Готовили 250 мкл рабочего раствора фермента и затем в раствор добавляли 5 мкл сыворотки крови. Пробирку с бланк-контролем (добавляли 5 мкл дважды дистиллированной воды) и пробирку со стандартом (добавляли 5 мкл раствора стандарта глюкозы) готовили одновременно и встряхивали в водяной бане при 37°C вода в течение 20 минут. Пробирку с бланк-контролем использовали для установки на приборе нулевого значения, колориметрический анализ осуществляли при OD505 нм.
Концентрация глюкозы в сыворотке крови (BG, ммоль/л)=ODпробирка с образцом/ODстандартная пробирка×5,55
Обработка данных и статистический анализ:
1. Среднее ± СО (стандартное отклонение) и t-тест Стьюдента использовали в статистическом анализе.
2. Рассчитывали процентное уменьшение уровня глюкозы в крови через 30 минут после приема сахара, а также площадь под кривой (AUC).
Результаты:
Тест 1:
Самцов мышей ICR не кормили в течение 6 часов, и затем обрабатывали дважды дистиллированной водой, различными дозами тестируемых соединений Примера 1 и Примера 2 через зонд. Через 30 минут после введения проводили тест толерантности к перорально вводимой глюкозе. Результаты продемонстрировали, что уровень глюкозы в крови в контрольной группе значительно увеличивался после введения 2,5 г/кг глюкозы через зонд и достигал максимального значения через 30 минут. При низкой, средней и высокой дозах соединения Примера 1 уровень глюкозы в крови был значительно ниже по сравнению с контрольной группой через 30 минут, и процентное содержание глюкозы в крови уменьшалось соответственно на 19,16%, 22,85% и 31,85%. При каждой дозе соединения Примера 2 уровень глюкозы в крови был значительно ниже по сравнению с контрольной группой через 30 минут после введения глюкозы (P<0,01). По сравнению с контрольной группой процентное содержание глюкозы в крови уменьшалось на 25,54%, 25,92% и 26,93%.
Тест 2:
Самцов мышей ICR не кормили в течение 6 часов, и затем обрабатывали дважды дистиллированной водой, различными дозами тестируемых соединений Примера 1 и Примера 2 через зонд. Через 30 минут после введения проводили тест толерантности к перорально вводимой глюкозе. Результаты продемонстрировали, что уровень глюкозы в крови в контрольной группе значительно увеличивался после введения 2,5 г/кг глюкозы через зонд и достигал максимального значения через 30 минут. При каждой дозе SHR1039 (т.е. соединения Примера 1) уровень глюкозы в крови был значительно ниже по сравнению с контрольной группой через 30 минут после введения глюкозы (P<0,01), и процентное содержание глюкозы в крови уменьшалось соответственно на 26,10%, 30,24 и 32,05%. При низкой, средней и высокой дозах SHR1040 (т.е. соединения Примера 2) уровень глюкозы в крови был значительно ниже по сравнению с контрольной группой через 30 минут (P<0,01) и процентное содержание глюкозы в крови уменьшалось на 24,51%, 26,96% и 27,75%.
Заключение:
Два экспериментальных результата в этом отчете демонстрируют, что тестируемые соединения Примера 1 и Примера 2 оказывают значительное гипогликемическое действие в тесте толерантности к глюкозе, перорально вводимой нормальным мышам ICR. Кроме того, тестируемое соединение Примера 1 демонстрирует более хорошую связь доза-эффект.
Действия ингибиторов DPP-IV на толерантность к глюкозе, перорально вводимой мышам KKAy
Задача:
Для обнаружения ингибирующих действий в отношении DPP-IV соединений Примера 1 и Примера 2 в тесте толерантности к перорально вводимой глюкозе у мышей KKAy, страдающих от сахарного диабета II типа, осуществляли предварительную оценку их гипогликемического действия in vivo.
Тестируемые животные:
|
Лекарства:
Название: соединения Примера 1 и Примера 2
Способ приготовления: Соединения точно взвешивали, затем растворяли в дважды дистиллированной воде и полностью смешивали с получением суспензии 3 мг/мл, затем разбавляли соответственно до 1, 0,3 и 0,1 мг/мл прозрачного раствора.
|
Способы:
Действия соединений на уровень глюкозы в крови мышей KKAy
Нормальных мышей KKAy не кормили в течение 6 часов, и затем случайным образом группировали в соответствии с массами организма и уровнем глюкозы в крови натощак, 5 мышей в каждой группе. Группы включали бланк-контрольную группу, а также обрабатываемые различными дозами группы в соответствии со следующим:
|
Животных в каждой группе не кормили в течение 6 часов и затем предварительно обрабатывали соответственно соединениями или дважды дистиллированной водой через зонд во время разового введения. Через 30 минут животным вводили 2,5 г/кг (самки мышей KKAy) или 1,5 г/кг (самцы мышей KKAy) глюкозу через зонд. После введения глюкозы через 0, 30, 60 и 120 минут уровни глюкозы в сыворотке крови измеряли при помощи глюкометра.
Обработка данных и статистический анализ:
3. Среднее ± СО и t-тест Стьюдента использовали в статистическом анализе.
4. Рассчитывали процентное уменьшение уровня глюкозы в крови через 30 минут после приема сахара, а также площадь под кривой (AUC).
Результаты:
1. Соединение Примера 1: Тест 1, 2
Самок мышей KKAy не кормили в течение 6 часов и затем обрабатывали дважды дистиллированной водой, различными дозами тестируемых соединений Примера 1 через зонд. Через 30 минут после введения проводили тест толерантности к перорально вводимой глюкозе. Результаты продемонстрировали, что уровень глюкозы в крови в контрольной группе значительно увеличивался после введения 1,5 г/кг глюкозы через зонд и достигал максимального значения через 30 минут. В группах, которым вводили дозы 10 мг/кг и 30 мг/кг соединения Примера 1, уровни глюкозы в крови были ниже, чем в контрольной группе через 30 минут после введения глюкозы. По сравнению с контрольной группой процентное содержание глюкозы в крови уменьшалось соответственно на 16,22% и 17,15%.
Самок мышей KKAy не кормили в течение 6 часов, и затем обрабатывали дважды дистиллированной водой, различными дозами тестируемых соединений Примера 1 через зонд. Через 30 минут после введения проводили тест толерантности к перорально вводимой глюкозе. Результаты продемонстрировали, что уровень глюкозы в крови в контрольной группе значительно увеличивался после введения 2,5 г/кг глюкозы через зонд и достигал максимального значения через 30 минут. В группах, которым вводили дозы 3 мг/кг и 10 мг/кг соединения Примера 1, уровни глюкозы в крови были значительно ниже, чем в контрольной группе через 30 минут после введения глюкозы. Процентное содержание глюкозы в крови уменьшалось соответственно на 40,63% и 24,68%.
2. Соединение Примера 2: Тест 3, 4
Самцов мышей KKAy не кормили в течение 6 часов, и затем обрабатывали дважды дистиллированной водой, различными дозами тестируемых соединений Примера 2 через зонд. Через 30 минут после введения проводили тест толерантности к перорально вводимой глюкозе. Результаты продемонстрировали, что уровень глюкозы в крови в контрольной группе значительно увеличивался после введения 1,5 г/кг глюкозы через зонд и достигал максимального значения через 30 минут. В группах, которым вводили дозы 10 мг/кг и 30 мг/кг соединения Примера 2, уровни глюкозы в крови были ниже, чем в контрольной группе через 30 минут после введения глюкозы. По сравнению с контрольной группой процентное содержание глюкозы в крови уменьшалось соответственно на 13,79% и 12,23%.
Самок мышей KKAy не кормили в течение 6 часов и затем обрабатывали дважды дистиллированной водой, различными дозами тестируемых соединений Примера 2 через зонд. Через 30 минут после введения проводили тест толерантности к перорально вводимой глюкозе. Результаты продемонстрировали, что уровень глюкозы в крови в контрольной группе значительно увеличивался после введения 2,5 г/кг глюкозы через зонд и достигал максимального значения через 30 минут. В группе, которой вводили дозу 10 мг/кг соединения Примера 2, уровни глюкозы в крови были ниже, чем в контрольной группе через 30 минут после введения глюкозы (P=0,075, anova). Процентное содержание глюкозы в крови уменьшалось на 21,55%. Тем не менее, ввиду значительных индивидуальных различий у мышей результаты не отличаются значимо.
Заключение:
Тестируемые соединения Примера 1 и Примера 2 обладают некоторыми гипогликемическими действиями в тесте толерантности к перорально вводимой глюкозе у мышей KKAy, страдающих от сахарного диабета II типа.
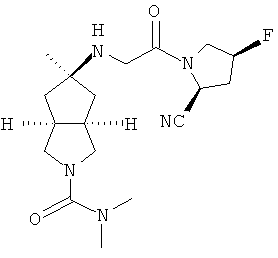
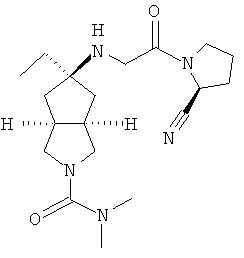
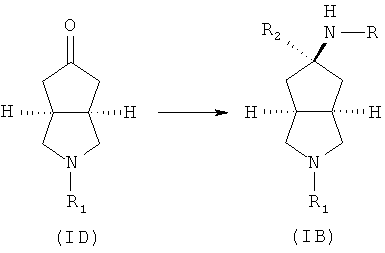
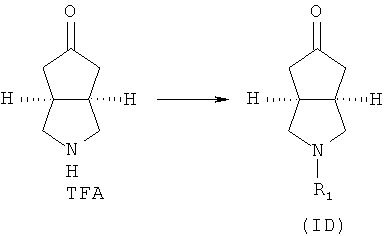
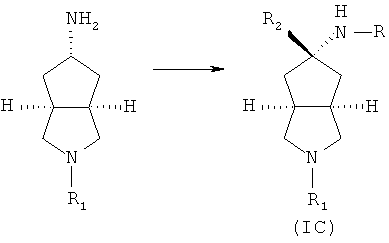
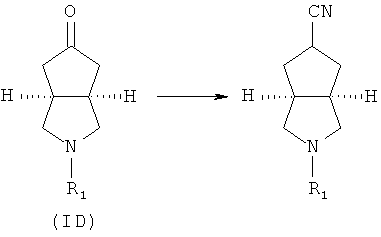
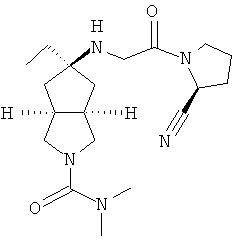
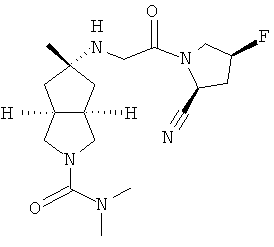
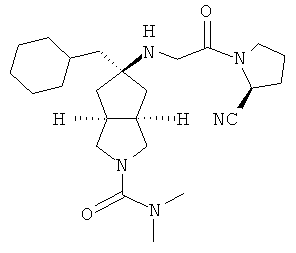
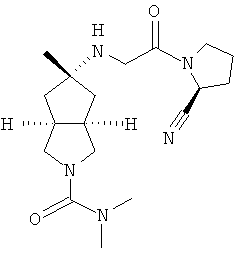
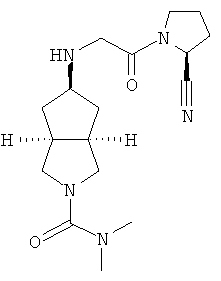
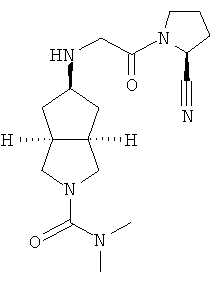
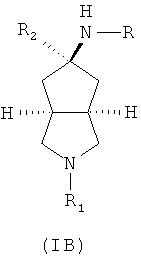
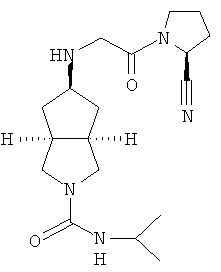
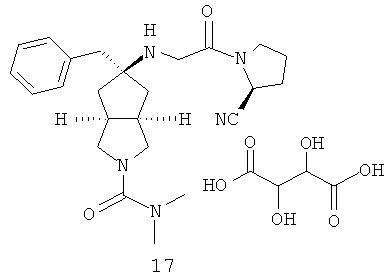
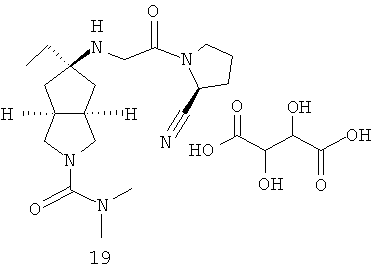
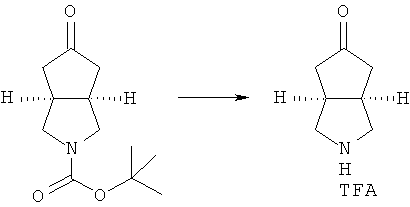
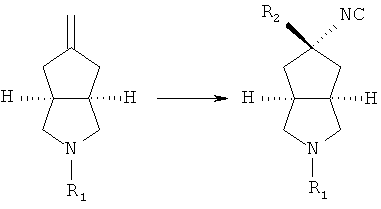
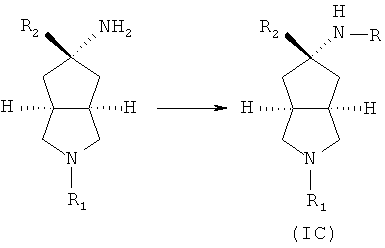
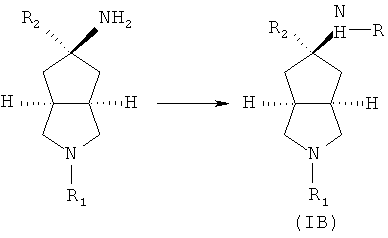
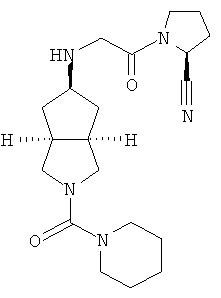
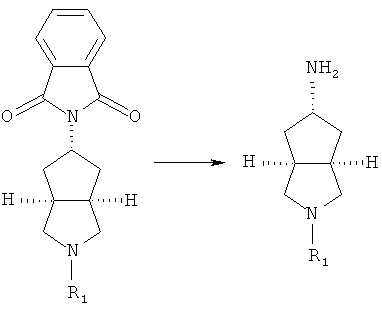
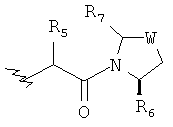
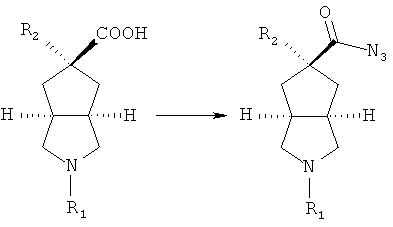
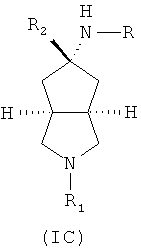
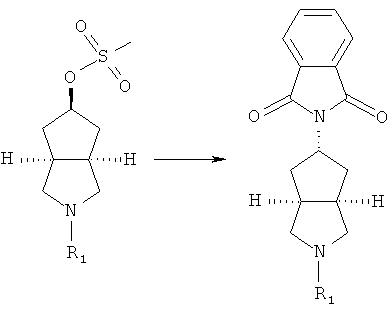
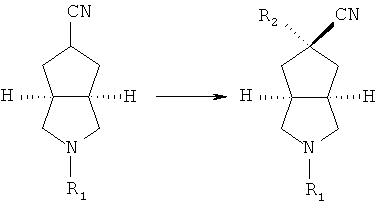
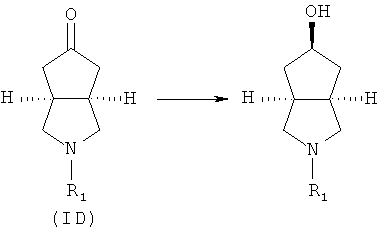
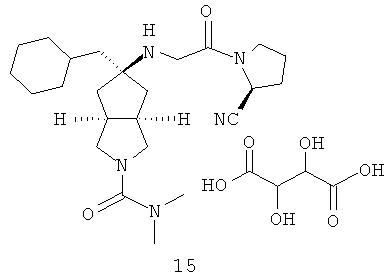
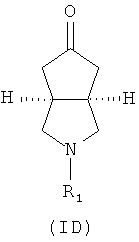
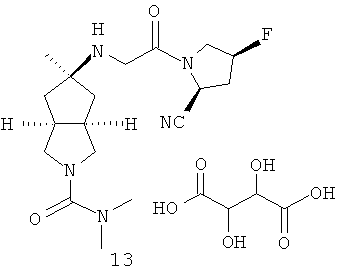
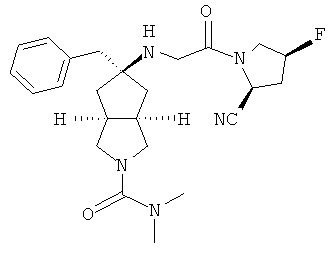
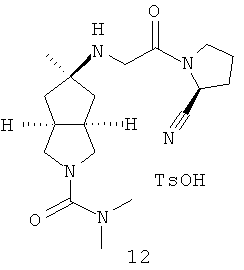
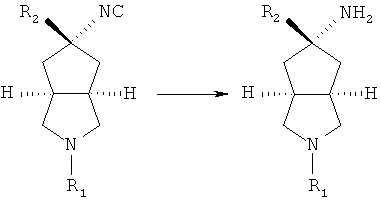
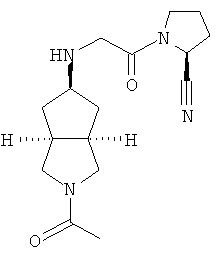
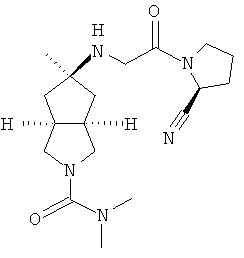
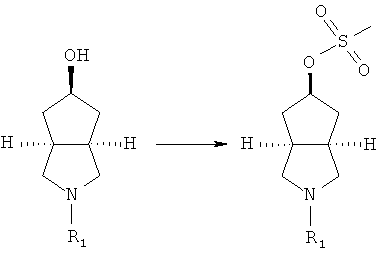
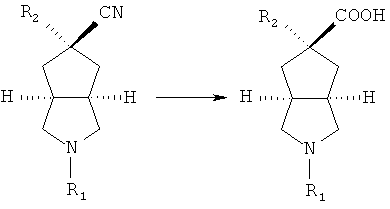
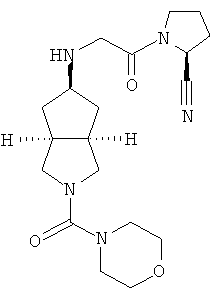
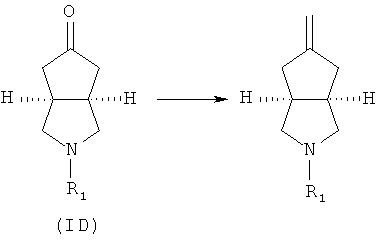
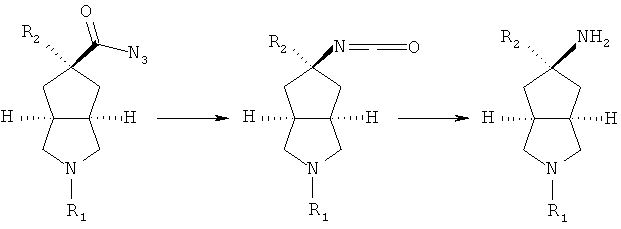
Соединения 2-(2-оксоиндолин-3-илиден)метил-5-(2-гидрокси-3-морфолин-4-илпропил)-6,7 дигидро-1-н-пиррол[3,2-с]пиридин-4(5н)-она и их применение в качестве ингибиторов протеинкиназы
Гетероциклические азотистые производные пиррола, их получение и фармацевтическое применение
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Бициклозамещенные азопроизводные пиразолона, способ их получения и фармацевтическое применение
Пептидное производное - миметик эритропоэтина и его фармацевтические соли, их получение и применение
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Производные 6-аминохиназолина или 3-цианохинолина, способы их получения и их применение в качестве ингибитора рецепторных тирозинкиназ egfr или her-2
Фармацевтически приемлемая соль (е)-n-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2r)-1-метилпирролидин-2-ил]проп-2-енамида, способ ее получения и применение при лечении рака
Содержащие фосфор соединения в качестве ингибиторов протеинкина3
Соединения 2-(2-оксоиндолин-3-илиден)метил-5-(2-гидрокси-3-морфолин-4-илпропил)-6,7 дигидро-1-н-пиррол[3,2-с]пиридин-4(5н)-она и их применение в качестве ингибиторов протеинкиназы
Гетероциклические азотистые производные пиррола, их получение и фармацевтическое применение
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Бициклозамещенные азопроизводные пиразолона, способ их получения и фармацевтическое применение
Пептидное производное - миметик эритропоэтина и его фармацевтические соли, их получение и применение
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Производные 6-аминохиназолина или 3-цианохинолина, способы их получения и их применение в качестве ингибитора рецепторных тирозинкиназ egfr или her-2
Фармацевтически приемлемая соль (е)-n-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2r)-1-метилпирролидин-2-ил]проп-2-енамида, способ ее получения и применение при лечении рака
Содержащие фосфор соединения в качестве ингибиторов протеинкина3

