Результат интеллектуальной деятельности: СОЛИ N-[4-(1-ЦИАНОЦИКЛОПЕНТИЛ)ФЕНИЛ]-2-(4-ПИРИДИЛМЕТИЛ)АМИНО-3-ПИРИДИНКАРБОКСАМИДА
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтически приемлемым солям N-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амидо-3-пиридинкарбоксамида.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Опухолевый ангиогенез играет критическую роль в росте и метастазах злокачественной опухоли. Когда опухоли перерастают 1 мм3, ангиогенез или древовидное разветвление кровеносных сосудов путем почкования от существующих кровеносных сосудов необходимо для обеспечения достаточного количества крови для выживания опухолевых клеток. Скорость роста опухолей и их склонность к метастазам связаны с уровнем факторов неоваскуляризации и количеством образующихся микрососудов. С тех пор как гипотеза "антиангиогенной терапии" была выдвинута Folkman в ранние 1970-е годы, люди совершили значительный прогресс в данной области, и ингибирование ангиогенеза опухолей повсеместно принято в качестве новой противораковой стратегии.
Тирозинкиназный эндотелиальный фактор роста сосудов (VEGF) и его рецептор (VEGFR) играют очень важные роли в ангиогенезе опухолей, и оба они являются важными мишенями при блокировании ангиогенеза опухолей. Эндотелиальный фактор роста сосудов (VEGF) является первоочередным фактором in vivo, стимулирующим ангиогенез. Связывание VEGF с рецептором эндотелиального фактора роста сосудов (VEGFR) в эндотелиальных клетках приводит к различным реакциям ангиогенеза, таким как пролиферация клеток, метастазы клеток, увеличение проницаемости кровеносных сосудов и движение предшественников эндотелиальных клеток из костного мозга. Семейство VEGFR включает VEGFR1 (Flt-1), VEGFR2 (KDR/Flk-1) и VEGFR3 (Flt-4). Стимуляция ангиогенеза, главным образом, опосредована связанным VEGF и VEGFR2 (KDR/Flk-1). Различные опухоли человека проявляют высокие уровни VEGFR. В настоящее время более 40 лекарств, способных к ингибированию ангиогенеза, находится в клинических испытаниях, как, например, моноклональные антитела к VEGF и его рецептору (VEGFR) и низкомолекулярные ингибиторы VEGFR тирозинкиназы. Моноклональное антитело к VEGF Авастин, которое разрабатывалось фирмой Genetech в течение более десяти лет, было одобрено к продаже в 2004 году. Эффективность Авастина в комбинации с другими лекарствами при раке ободочной кишки, раке легких и раке молочной железы доказала, что механизм действия Авастина в качестве лекарства анти-VEGF осуществим. Авастин также внес выдающиеся вклады в механизм антиангиогенеза как противораковой мишени.
Наиболее примечательные лекарства из низкомолекулярных ингибиторов VEGFR в недавние годы включают ингибитор VEGFR Ваталаниб (РТК787) для лечения рака ободочной кишки, разработанный Novartis/Schering, и ингибитор двойной мишени VEGFR и рецептора эпидермального фактора роста (EGFR) Зактима (ZD-6474) для лечения рецидивирующего/резистентного немелкоклеточного рака легкого, разработанный Astrazeneca. Ингибиторы VEGF постепенно становятся новыми не цитотоксическими противораковыми лекарствами с хорошими перспективами применения. По сравнению с традиционными цитотоксическими лекарствами, которые ингибируют рост опухолей, лекарства, направленные на ангиогенез, более специфичны и менее токсичны, а также помогают преодолению лекарственной устойчивости опухолей и могут применяться для лечения различных опухолей.
N-[4-(1-цианоцикпопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамид (здесь далее называемый "соединением A") является ингибитором тирозинкиназы нового поколения, и данное соединение имеет формулу (I):
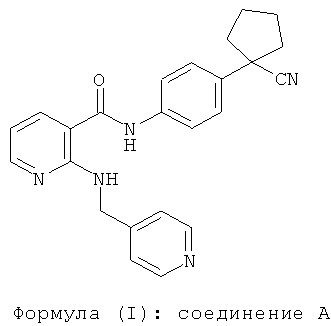
Вышеупомянутое соединение описано в китайской заявке на патент №02138671.4, содержание которой полностью включено в данную заявку посредством ссылки. Обнаружено, что соединение A обладает очень сильным селективным ингибированием в отношении VEGFR-2 в тестах рецепторов тирозинкиназы на ферментативном уровне в различных лабораториях, IC50 которых составляла примерно 1 нМ. Кроме того, оно обладает некоторой активностью селективного ингибирования в отношении киназ Ret, VEGFR-1, PDGFR-P, c-kit, cSRC и т.д. При исследовании фармакодинамики на опухоли человека, трансплантированной бестимусным мышам, обнаружено, что эффективность соединения A на раке ободочной кишки Ls174t, трансплантированном бестимусным мышам, была значительно лучшей, чем РТК787; и эффективность соединения A улучшалась, когда его использовали в комбинации с оксалиплатином, причем его токсичность не повышалась. При использовании отдельно или в комбинации эффективность соединения A была лучше, чем РТК787. Также обнаружено, что эффективность соединения A на немелкоклеточном раке легкого А549, трансплантированном бестимусным мышам, была значительно лучшей, чем РТК787, максимальная эффективность которого была эквивалентна ZD6474 при обычной дозе. В аспекте токсичности соединение A было хорошо переносимым бестимусными мышами при максимальной дозе 400 мг/кг.
Однако во время исследования этого лекарства было обнаружено, что N-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамид был неудовлетворительным в некоторых аспектах, например, в аспектах стабильности и биодоступности.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Посредством длительных усилий авторы изобретения обнаружили, что проблемы, такие как стабильность и биодоступность, были бы решены за счет превращения
N-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида в его соответствующие фармацевтически приемлемые соли.
В одном аспекте настоящее изобретение относится к фармацевтически приемлемым солям N-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида, где эти фармацевтически приемлемые соли представляют собой общепринятые неорганические соли или органические соли в данной области техники. Кроме того, неорганическая соль предпочтительно выбрана из группы, состоящей из соли гидрохлорид, соли гидробромид, соли сульфат, соли нитрат и соли фосфат, а органическая соль предпочтительно выбрана из группы, состоящей из соли мезилат, соли малеат, соли тартрат, соли сукцинат, соли ацетат, соли трифторацетат, соли фумарат, соли цитрат, соли бензолсульфонат, соли бензоат, соли нафталинсульфонат, соли лактат и соли малат. Особенно предпочтительными фармацевтически приемлемыми солями являются соль мезилат и соль гидробромид, которые обладают большими преимуществами в отношении стабильности, характера и биодоступности, чем другие соли.
В другом аспекте настоящее изобретение относится к способу получения фармацевтически приемлемых солей N-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида, который является общепринятым способом солеобразования в данной области техники.
В третьем аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество фармацевтически приемлемых солей N-[4-(1-цианоцикпопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида, которая может дополнительно содержать один или более чем один фармацевтически приемлемый носитель.
В четвертом аспекте настоящее изобретение относится к применению фармацевтически приемлемых солей N-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида при получении противоопухолевых лекарств.
КРАТКОЕ ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
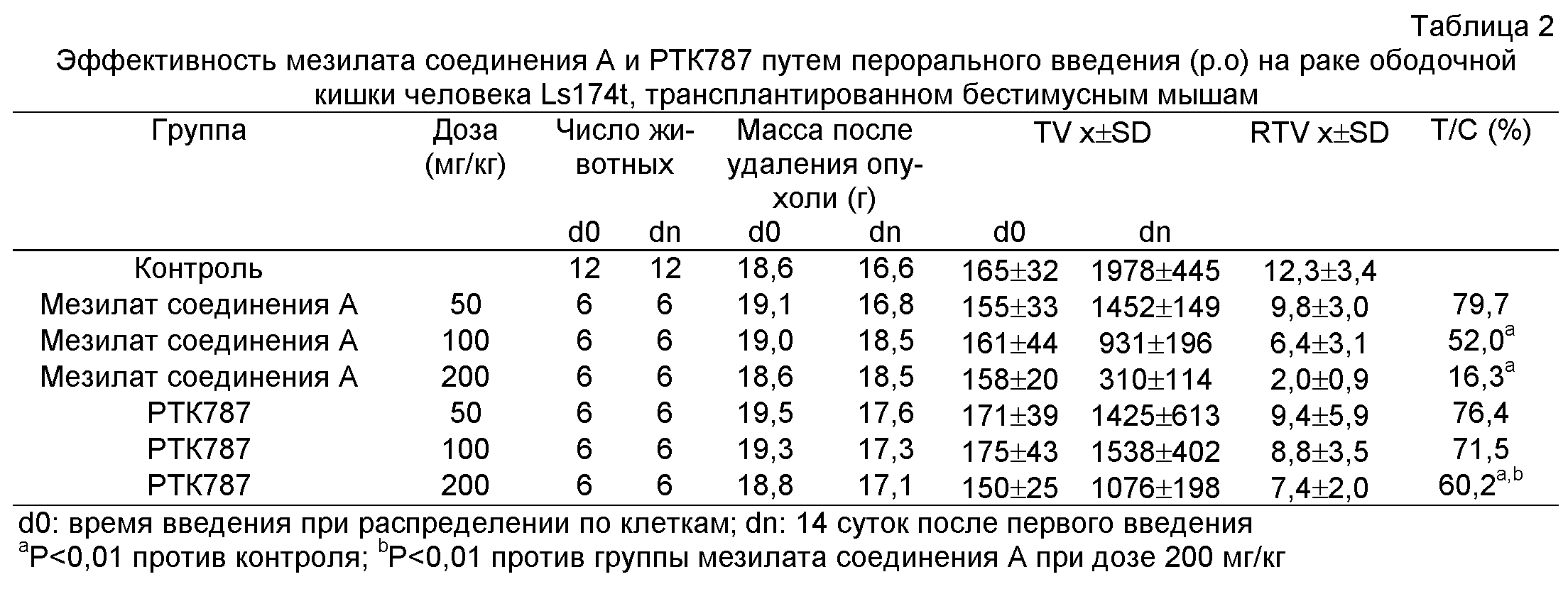
Фиг.1: Эффективность мезилата соединения A и РТК787 на раке ободочной кишки человека Ls174t, трансплантированном бестимусным мышам.
Фиг.2: Эффективность мезилата соединения A и РТК787 на раке ободочной кишки человека HT-29, трансплантированном бестимусным мышам.
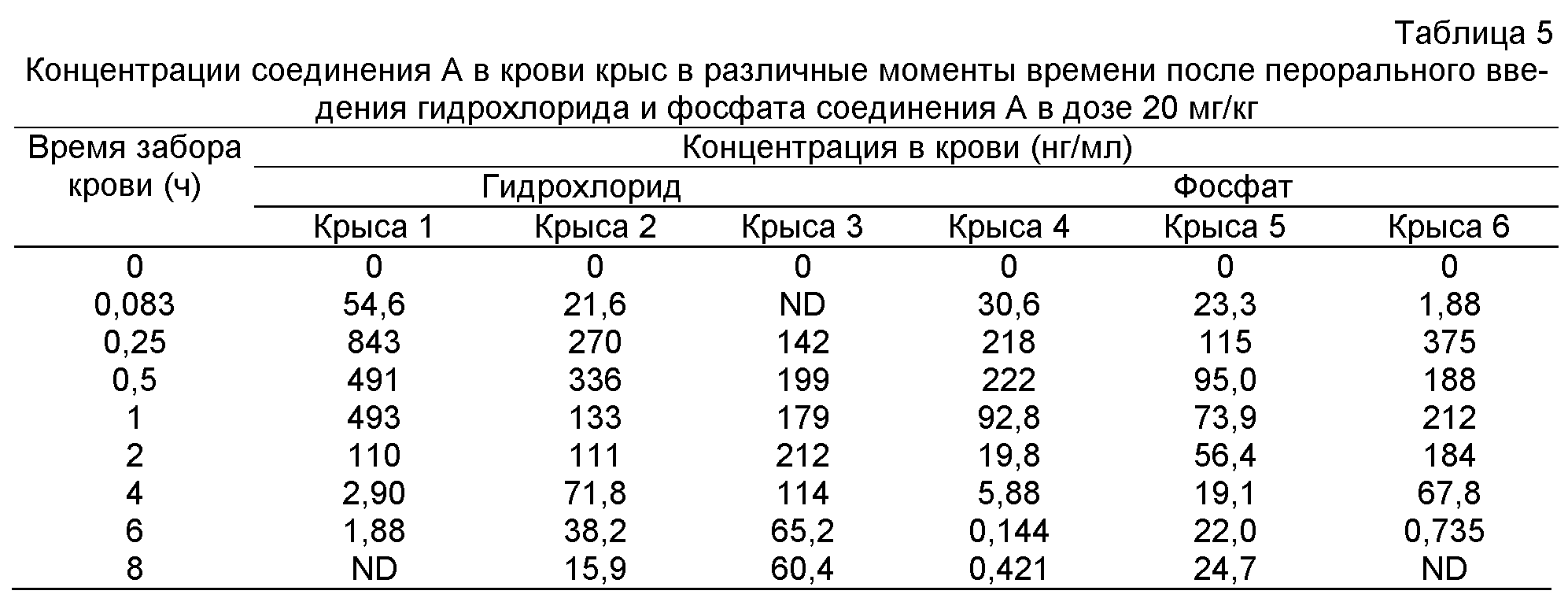
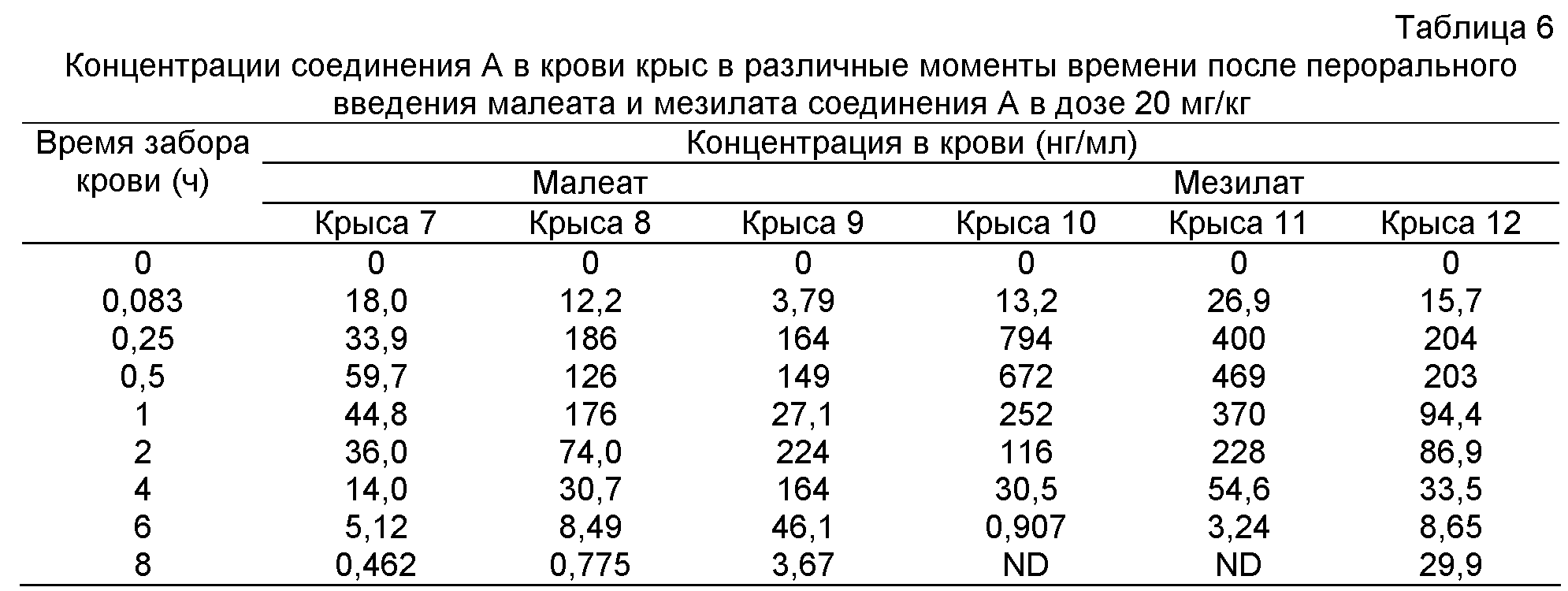
Фиг.3: Кривая концентрация лекарства - время гидрохлорида (крыса 1-3), фосфата (крыса 4-6), малеата (крыса 7-9) и мезилата (крыса 10-12) соединения A, вводимого крысам перорально 20 мг/кг.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
1. ПОЛУЧЕНИЕ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ СОЕДИНЕНИЯ A
Пример 1 для получения. Получение гидрохлорида соединения A:
5,049 г соединения A (12,7 ммоль) суспендировали в 120 мл этанола, добавляли по каплям 23,89 мл стандартного раствора соляной кислоты (0,5322 моль/л), и смесь нагревали до образования флегмы до получения прозрачного раствора (если существует какое-либо нерастворимое вещество, можно осуществить горячее фильтрование). После охлаждения до комнатной температуры (23°C) кристаллы выпадали в осадок из раствора. Полученную в результате смесь фильтровали, и фильтрационный кек промывали этанолом (20 мл × 2), переносили в вакуумную сушильную печь (CaCl2) и фильтровали с помощью наноса в течение 5 часов при 80°C с получением 3,619 г (65,7%) гидрохлорида соединения A. Диапазон плавления: 200-202,5°C, содержание воды 5,1% и остаток растворителя 0,025%.
Пример 2 для получения. Получение сульфата соединения A:
3,092 г соединения A (7,778 ммоль) суспендировали в 120 мл этанола, добавляли по каплям 14,89 мл (7,793 ммоль) стандартного раствора серной кислоты (0,5234 моль/л), и смесь нагревали до образования флегмы до получения прозрачного раствора (если существует какое-либо нерастворимое вещество, можно осуществить горячее фильтрование). Смесь концентрировали до 100 мл при пониженном давлении. После охлаждения до комнатной температуры (23°C) кристаллы выпадали в осадок из раствора. Полученную в результате смесь фильтровали, и фильтрационный кек промывали этанолом (8 мл × 2), переносили в вакуумную сушильную печь (CaCl2) и фильтровали с помощью наноса в течение 5 часов при 80°C с получением 2,662 г (выход 57,7% на основе содержания свободного основания) сульфата соединения A. Диапазон плавления: 199,5-230°C (не полностью плавится).
Пример 3 для получения. Получение фосфата соединения A:
Смесь 1,910 г соединения A (4,805 ммоль), 225 мл этанола и 9,29 мл (4,803 ммоль) стандартного раствора фосфорной кислоты (0,5008 моль/л) нагревали до образования флегмы. Через 4 часа твердые вещества полностью растворялись. Затем смеси давали охладиться до комнатной температуры (25°C), и кристаллы выпадали в осадок из раствора. Полученную в результате смесь фильтровали, и фильтрационный кек промывали этанолом (5 мл × 2), переносили в вакуумную сушильную печь (CaCl2) и фильтровали с помощью наноса в течение 6 часов при 80°C с получением 1,150 г (выход 46,1% на основе содержания свободного основания) фосфата соединения A. Диапазон плавления: 205-258°C (не полностью плавится).
Пример 4 для получения. Получение мезилата соединения A:
170 г (0,428 моль) соединения A, 42,5 г (0,442 моль) метансульфоновой кислоты и 2,55 л 95% водного раствора изопропанола добавляли в 5 л реакционную колбу. Смесь перемешивали и нагревали до полного растворения при защите азота и в темноте. Получили светло-желтый прозрачный раствор, и его фильтровали горячим. После охлаждения до комнатной температуры кристаллы выпадали в осадок из раствора. Полученные в результате осадки собирали фильтрованием и промывали изопропанолом, высушивали в вакууме с получением 180,2 г (0,365 моль) белых игольчатых кристаллов. Выход: 85,4%.
180,2 г мезилата соединения A в 2,52 л 95% водного раствора изопропанола добавляли в 5 л реакционную колбу. Смесь перемешивали и нагревали до полного растворения при азотной защите и в темноте и фильтровали горячей. После охлаждения до комнатной температуры кристаллы выпадали в осадок из раствора. Полученные в результате осадки собирали фильтрованием и промывали изопропанолом, высушивали в вакууме с получением 161,5 г белых игольчатых кристаллов. Выход: 85,4%. Диапазон плавления: 193,5-195°C.
Пример 5 для получения. Получение цитрата соединения A:
2,886 г свободного основания соединения A, 0,522 г лимонной кислоты и 80 мл этанола смешивали и нагревали почти до кипения до получения бесцветного прозрачного раствора. После охлаждения до комнатной температуры белые кристаллы осаждали и фильтровали. Фильтрационный кек промывали этанолом (3 мл × 2), фильтровали в вакуумной сушильной печи в течение 6 часов при 80°C с получением 2,283 г игольчатых кристаллов. Выход: 79%. Диапазон плавления: 160,5-162,0°C.
Пример 6 для получения. Получение малеата соединения A:
2,508 г свободного основания соединения A, 0,351 г малеиновой кислоты и 110 мл этанола смешивали и нагревали до образования флегмы до получения прозрачного светло-желтого раствора. Этот раствор кипятили и добавляли активированный углерод. Зону хлопьевидных нерастворимых веществ удаляли горячим фильтрованием. Фильтрат концентрировали примерно до 90 мл и охлаждали до комнатной температуры. Светло-желтые кристаллические твердые вещества осаждали и фильтровали. Фильтрационный кек промывали небольшим количеством этанола, фильтровали в вакуумной сушильной печи в течение 6 часов при 80°C с получением 1,009 г светло-желтых игольчатых кристаллов. Выход: 40%. Диапазон плавления: 115-160°C.
Пример 7 для получения. Получение сукцината соединения A:
2,827 г свободного основания соединения A, 0,401 г янтарной кислоты и 70 мл этанола смешивали и нагревали до образования флегмы. Твердые вещества полностью растворялись. Этот раствор кипятили и добавляли активированный углерод. Зону хлопьевидных нерастворимых веществ удаляли горячим фильтрованием. Фильтрат концентрировали примерно до 25 мл и охлаждали до комнатной температуры. Белые кристаллические твердые вещества осаждали и фильтровали. Фильтрационный кек промывали небольшим количеством этанола, фильтровали в вакуумной сушильной печи в течение 6 часов при 80°C с получением 1,009 г светло-желтых игольчатых кристаллов. Выход: 77%. Диапазон плавления: 117-161,5°C.


Вывод: В соответствии с результатом экспериментов по стабильности стабильность гидрохлорида и мезилата наиболее удовлетворительна. В частности, мезилат наиболее стабилен.
3. ИССЛЕДОВАНИЕ ФАРМАКОЛОГИЧЕСКОЙ АКТИВНОСТИ ФАРМАЦЕВТИЧЕСКИ ПРИЕМЛЕМЫХ СОЛЕЙ СОЕДИНЕНИЯ А
Пример 1: Ингибирование мезилата соединения A на рецепторном белке тирозинкиназе
(1) Метод
Метод ELISA (I Posner et al., J. Biol. Chem., Oct, 1992, Vol.267, Issue 29, 20638-20647): Планшет ферментативной метки покрывали субстратом ферментативной реакции Poly(Glu, Tyr)4:1, затем добавляли фермент, образец и АТФ. Фосфорилирование субстрата определяли моноклональным антителом против фосфорилирования тирозина (PY99). Затем добавляли IgG коза против мыши, меченый HRP, и степень фосфорилирования субстрата определяли с помощью окрашивания OPD. В то же время ставили контрольную группу без тирозинкиназы и контрольные планшеты соответствующей концентрации ДМСО. Для остановки реакции добавляли 2 М H2SO4 в количестве 50 мкл/лунка. Данные считывали с помощью прибора для микропланшетов с ферментативной меткой с регулируемой длиной волны VERSAmax (Sunnyvale, CA, U.S.A.) с последующей визуализацией реакции, затем наблюдали значение OD при 490 нм.

Определяли относительный коэффициент ингибирования лекарств на белке тирозинкиназе.
Ингибиторную концентрацию 50% IC50 вычисляли методом LOGIT в соответствии с коэффициентами ингибирования различных концентраций. Каждый вышеупомянутый эксперимент повторяли 3 раза, и среднее значение IC50 3 повторов экспериментов принимали за конечный показатель ингибирующей способности.
(2) Результаты
Результаты ингибирования мезилата соединения A и положительного контрольного соединения РТК787 на 8 типах тирозинкиназ суммированы в таблице 1. Эти результаты показывают, что мезилат соединения A обладает значимым ингибированием в отношении киназной активности KDR, Flt1, PDGFR (3, c-Kit и c-Src на молекулярном уровне, IC50 которого составляет 2,43 нМ, 70,08 нМ, 537,31 нМ, 420,31 нМ и 348,53 нМ, соответственно. Напротив, IC50 положительного контрольного соединения РТК787 на KDR, Flt1, PDGFRp и c-Kit составляет 33,30 нМ, 84,69 нМ, 416,51 нМ и 606,11 нМ, соответственно. Эти результаты также показывают, что мезилат соединения A обладает сильным ингибированием в отношении киназной активности рецепторов эндотелиального фактора роста сосудов 1 и 2 (Flt1/VEGFR1 и KDR/VEGFR2). Его ингибирование на киназе KDR значимо сильнее, чем на киназе Flt1, и IC50 на киназе KDR в 13,7 раз ниже, чем для контрольного соединения. То есть ингибирование мезилата соединения A на KDR сильнее, чем для РТК787. В то же время, мезилат соединения A также обладает значительным ингибированием в отношении третьих рецепторных тирозинкиназ, таких как рецептор β тромбоцитарного фактора роста (PDGFR(3) и рецептор фактора роста стволовых клеток (c-Kit), которое является более слабым, чем ингибирование в отношении рецептора эндотелиального фактора роста сосудов. Когда концентрация повышается до 104 нМ, положительное соединение РТК787 не обладает ингибированием в отношении не рецепторной тирозинкиназы c-Src, тогда как IC50 ингибирования мезилата соединения A на c-Src составляет 348,53 нМ. Однако, когда концентрация повышается до 104 нМ, мезилат соединения A не обладает ингибированием в отношении киназной активности киназ из других семейств, таких как рецептор эпидермального фактора роста EGFR1 и ErbB2, и рецептор фактора роста фибробластов FGFR1. Кроме того, результаты показывают, что ингибирование мезилата соединения A в отношении киназной активности KDR сильнее, чем для положительного соединения РТК787, на молекулярном уровне, хотя их ингибирование в отношении тирозинкиназы Flt1, PDGFR, c-Kit является по существу одинаковым при силе ингибирования, находящейся на уровне одного диапазона. В отношении селективности, мезилат соединения A обладает более широким диапазоном, чем РТК787, и также обладает ингибированием в отношении киназной активности не рецепторной тирозинкиназы c-Src. Как вывод, мезилат соединения A является ингибитором тирозинкиназы, обладающим значимо селективным ингибированием в отношении KDR, вместе с ингибированием в отношении киназ Flt1, PDGFR, c-Kit, c-Src и т.д.
|
Пример 2: Эффективность мезилата соединения A на раке ободочной кишки человека Ls174t, трансплантированном бестимусным мышам
(1) Подопытные животные
Бестимусные мыши BALB/cA, самки, 5-6-недельного возраста, доступные от компании с ограниченной ответственностью подопытных животных Shanghai Slaccas. Сертификационный номер: SCXK (hu) 2004-0005. Условия выращивания: квалификация SPF.
(2) Экспериментальные методы
После адаптации в течение недели подопытных животных инокулировали подкожно опухолевыми тканями рака ободочной кишки человека Ls174t. Когда опухоли вырастали до 100-300 мм3, животных делили случайным образом на несколько групп на сутки 0 (d0). Дозы мезилата соединения A составляли 50 мг/кг, 100 мг/кг и 200 мг/кг соответственно. РТК787 вводили в таких же дозах. Как мезилат соединения A, так и РТК787 вводили перорально (с помощью зонда) один раз в сутки от суток 0 (d0) до суток 13 (d13), всего 14 раз. Объемы опухолей и массы мышей измеряли 2-3 раза каждую неделю, и данные записывали. Уравнение для вычисления объема опухолей (V) приведено ниже:
V=1/2×a×b2
где а и b представляют собой длину и ширину, соответственно.
(3) Результаты
Экспериментами по энзимологии и на клеточном уровне доказано, что ведущей мишенью действия мезилата соединения A является VEGFR2/KDR (IC50=2,43±1,30 нМ). В качестве положительного соединения в эксперименте было выбрано РТК787 (IC50 на KDR равна 33,30±14,45 нМ) фирмы Novartis, соединение, имеющее подобную мишень действия, клинические испытания которого были проведены ранее. В соответствии с предварительными испытаниями мезилата соединения A и контрольного соединения РТК787 было выбрано три дозы 50, 100, 200 мг/кг, и оценку и сравнение эффективности проводили, используя те же дозы и режим дозировки. Результаты приведены в таблице 2. Эти результаты показывают, что мезилат соединения A дозозависимо ингибировал рост рака ободочной кишки человека Ls174t, и его Т/С% составляло 16,3% при дозе 200 мг/кг. РТК787 также ингибировал рост Ls174t при дозе 200 мг/кг, однако его Т/С% составляло только 60,2%, что указывает на то, что эффективность РТК787 значительно ниже по сравнению с мезилатом соединения A. В статье J.M. Wood et al., Cancer Research 60, 2178-2189, April 15, 2000, описано, что лучшее Т/С% могло достигать 40% при введении РТК787 в дозе 75 мг/кг, но экспериментальные результаты авторов изобретения показали, что РТК787, вводимый в дозе 100 мг/кг, не обладает значимым эффектом, и Т/С% составляло только 71,5%. При сравнении отмечено, что в эксперименте J.M. Wood et al. исходный объем опухолей в момент введения лекарств составлял 25-100 мм3, что по меньшей мере в 1,5-6 раз меньше, чем в эксперименте авторов изобретения, и введение длилось в течение 28 суток или более, что было дольше, чем в эксперименте авторов изобретения. Кроме того, Т/С%, описанное J.M. Wood et al., было лучшим среди других в их эксперименте, но не конечным Т/С% по окончании эксперимента. Напротив, лучшее Т/С% в эксперименте авторов изобретения оказывалось на 10е сутки в течение введения, и в это время Т/С% составляло 60,7% и 45,8% для дозы РТК787 100 мг/кг и 200 мг/кг соответственно, что было близко к значениям в эксперименте J.M. Wood et al. Кроме того, следует подчеркнуть, что существуют различные факторы, влияющие на эффективность эксперимента, и сравнение следует проводить в той же системе. Хотя эффективность РТК787 в настоящем эксперименте была не идентична литературным данным, сравнение между эффективностью РТК787 и мезилата соединения A не было подвергнуто воздействию. На основании таблицы 2 можно было вычислить, что ED50 мезилата соединения A на раке ободочной кишки Ls174t составляла 97,2 мг/кг, тогда как ED50 РТК787 составляла 458,7 мг/кг, что указывает на то, что эффективность мезилата соединения A на раке ободочной кишки Ls174t была значительно лучше, чем РТК787.
На основании этого можно заключить, что, когда мезилат соединения A и РТК787 оба вводят в дозе 400 мг/кг, их эффективность значимо не увеличивается, хотя они могут быть переносимы мышами, то есть отсутствует явное отношение доза - эффект. Этот результат был подобен другому ингибитору ангиогенеза SU11248. Поэтому в последующих экспериментах для оценки эффективности были выбраны дозы 200, 100, 50 мг/кг мезилата соединения A.
Согласно схеме эксперимента два соединения последовательно вводили мышам, несущим опухоли, в течение 14 суток соответственно. Результаты показывают, что оба эти соединения были хорошо переносимыми, и у мышей отсутствовала явная потеря массы. Токсичность этих двух соединений незначительно различалась в этой схеме эксперимента.
Пример 3: Эффективность мезилата соединения A на раке ободочной кишки человека НТ-29, трансплантированном бестимусным мышам
(1) Подопытные животные
Бестимусные мыши BALB/cA, самки, 5-6-недельного возраста, приобретенные от компании с ограниченной ответственностью подопытных животных Shanghai Slaccas. Сертификационный номер: SCXK (Ни) 2004-0005. Условия выращивания: квалификация SPF.
(2) Экспериментальные методы
После адаптации в течение недели подопытных животных инокулировали подкожно опухолевыми тканями рака ободочной кишки человека НТ-29. Когда опухоли вырастали до 100-300 мм3, животных делили случайным образом на несколько групп на сутки 0 (d0). Дозы мезилата соединения A составляли 50 мг/кг, 100 мг/кг, 200 мг/кг, соответственно, а доза РТК787 составляла 200 мг/кг. Как мезилат соединения A, так и РТК787 вводили перорально (с помощью зонда) один раз в сутки от суток 0 (d0) до суток 20 (d20), всего 21 раз. Объемы опухолей и массы мышей измеряли 2-3 раза каждую неделю, и данные записывали. Уравнение для вычисления объема опухолей (V) приведено ниже:
V=1/2×a×b2
где а и b представляют собой длину и ширину, соответственно.
(1) Результаты (см. таблицу 3):
Результаты показывают, что мезилат соединения A значимо ингибировал рост рака ободочной кишки человека НТ-29 с очевидной дозозависимостью. Эффективность РТК787 была также хорошей, но ниже эффективности мезилата соединения A. Т/С% мезилата соединения A и РТК787 при дозе 200 мг/кг составляло 25,5% и 56,5%, соответственно, и различались значимо (Р<0.01). Это указывает на то, что эффективность мезилата соединения A была значительно лучшей, чем РТК787.
Кроме того, оба эти соединения были хорошо переносимыми, и их токсичность эквивалента.

Пример 4: Исследование биодоступности соединения A посредством перорального введения
(1) Подопытные животные
Самцов крыс линии Спраг-Доули (SD) (масса: примерно 250 г, сертификат годности подопытных животных: 0006473) покупали от компании с ограниченной ответственностью подопытных животных Shanghai Slaccas (сертификационный номер: SCXK (hu) 2003-0003). В первую очередь проверяли релевантные квалификации и состояние здоровья крыс SD, и пригодных крыс помещали в комнаты асептической категории для крыс в Институте фармакологии, Шанхай.
(2) Экспериментальные приборы
Система анализа жидкостной хроматографии - масс-спектрометрии (ЖХ/МС/МС) включает двухканальный насос серии Agilent1100, подключенный деаэратор, автоматический дозатор, нагреватели колонок и тройной квадрупольный масс-спектрометр TSQ Quantum от фирмы Thermo Finnigan Company. Рабочими программными обеспечениями системы являются Xcalibur и Chemstation (США). Другие экспериментальные приборы включают: аппарат для азотной сушки Techne (Германия); морозильник сверхнизких температур на -80°C SANYO (Япония); мини-качалку Vibrax VXR (Германия); турбинную мешалку MS1 (Германия); магнитную мешалку стабильной температуры с таймером 92-2 (Шанхай); электронные аналитические весы с двойной шкалой METTLERAE240 (0,01 мг/41 г, 0,1 мг/205 г) (Германия) и флакон-диспенсер EPPENDORF (Германия).
(3) Экспериментальные методы
I. Условия анализа ЖХ/МС/МС
Условия анализа жидкостной хроматографии
Хроматографическая колонка: колонка Agilent Zorbax SB-C18 (50 мм × 2,1 мм ID);
Температура колонки: 25°C;
Подвижная фаза: A: H2O-CH3CN (2:98, об/об), В: H2O-CH3CN (10:90, об/об)
А: 25% + В : 75%, элюирование постоянным градиентом;
Скорость тока: 0,25 мл/мин;
Объем впрыска: 10 мкл;
Время анализа: 3 минуты.
II. Эксперименты с крысами
Световой цикл переключали на 12/12 часов день/ночь в комнатах асептической категории для крыс. Влажность и температура составляли 40-60% и 20-24°C, соответственно. Каждые 4 крысы содержали в стальных клетках для крыс размером 36×24×19 см3. Крысы имели свободный доступ к воде, и их кормили специальным кормом для крыс регулярно один раз в сутки. Только после одной недели адаптации крыс можно было использовать для эксперимента по фармакокинетике на животных. Трем крысам линии Спраг-Доули перорально вводили соединение A в дозе 20 мг/кг.
24 мг порошка соединения A точно взвешивали, растворяли в 4 мл воды, помещали в ступку и измельчали, а затем смывали 8 мл воды в 15 мл пробирку с получением суспензии 2 мг/мл для экспериментов на животных.
Образцы крови собирали в 0 часов перед введением и через 0,083, 0,25, 0,5, 1,0, 2, 4, 6, 8 часов после введения. Образцы крови крыс 250-300 мкл собирали в каждый момент времени из заднего венозного синуса глаз после респирационной анестезии эфиром (степень анестезии в высокой степени контролировали). Образцы крови собирали в пробирки, содержащие предварительно добавленный гепарин, а затем центрифугировали для получения плазмы. Полученную плазму делили на 2 части (50 мкл для каждой части) и хранили при -70°C до анализа. Концентрации соединения A в образцах крови в различные моменты времени анализировали, используя метод ЖХ/МС/МС. Эвтаназию проводили на использованных крысах газом CO2.
Фармакокинетические параметры эксперимента на животных каждой группы вычисляли, используя программное обеспечение InnaPhase Kinetica™ (США).
III. Результаты экспериментов
|
*Cmax: максимальная концентрация лекарства в плазме после экстраваскулярного введения; Tmax: время экстраваскулярного введения; AUC0-8ч: площадь под кривой концентрация лекарства в плазме - время (от 0 до 8 часов); Т1/2: время полувыведения; Kel: константа скорости элиминации; MRT: среднее время пребывания in vivo отдельной молекулы; CL: плазматический клиренс; Vd: кажущийся объем распределения на основании концентрации в плазме.
Пример 5: Сравнение биодоступности четырех фармацевтически приемлемых солей соединения A посредством перорального введения
(1) Подопытные животные
Самцов крыс линии Спраг-Доули (SD) (масса: примерно 250 г, сертификат годности подопытных животных: 0006473) покупали от компании с ограниченной ответственностью подопытных животных Shanghai Slaccas (сертификационный номер: SCXK (hu) 2003-0003). В первую очередь проверяли релевантные квалификации и состояние здоровья крыс SD, и пригодных крыс помещали в комнаты асептической категории для крыс в Институте фармакологии, Шанхай.
(2) Экспериментальные приборы
Система анализа жидкостной хроматографии - масс-спектрометрии (ЖХ/МС/МС) включает двухканальный насос серии Agilent1100, подключенный деаэратор, автоматический дозатор, нагреватели колонок и тройной квадрупольный масс-спектрометр TSQ Quantum от фирмы Thermo Finnigan Company. Рабочими программными обеспечениями системы являются Xcalibur и Chemstation (США). Другие экспериментальные приборы включают: аппарат для азотной сушки Techne (Германия); морозильник сверхнизких температур на -80°C SANYO (Япония); мини-качалку Vibrax VXR (Германия); турбинную мешалку MS1 (Германия); магнитную мешалку стабильной температуры с таймером 92-2 (Шанхай); электронные аналитические весы с двойной шкалой METTLERAE240 (0,01 мг/41 г, 0,1 мг/205 г) (Германия) и флакон-диспенсер EPPENDORF (Германия).
(3) Экспериментальные методы
I. Условия анализа ЖХ/МС/МС
Условия анализа жидкостной хроматографии
Хроматографическая колонка: колонка Agilent Zorbax SB-C18 (50 мм × 2,1 мм ID);
Температура колонки: 25°C;
Подвижная фаза: A: H2O-CH3CN (2:98, об/об), В: H2O-CH3CN (10:90, об/об) А: 25% + В : 75%, элюирование постоянным градиентом;
Скорость тока: 0,25 мл/мин;
Объем впрыска: 10 мкл;
Время анализа: 3 минуты.
II. Эксперименты с крысами
Световой цикл переключали на 12/12 часов день/ночь в комнатах асептической категории для крыс. Влажность и температура составляли 40-60% и 20-24°C, соответственно. Каждые 4 крысы содержали в стальных клетках для крыс размером 36×24×19 см3. Крысы имели свободный доступ к воде, и их кормили специальным кормом для крыс регулярно один раз в сутки. Только после одной недели адаптации крыс использовали для проведения исследований фармакокинетики. 12 крыс линии Спраг-Доули делили на 4 группы, 3 на каждую группу. Четырем группам вводили перорально гидрохлорид, фосфат, малеат и мезилат соединения A в дозе 20 мг/кг, соответственно.
24 мг порошка гидрохлорида, фосфата, малеата и мезилата соединения A, соответственно, точно взвешивали, растворяли в 4 мл воды, помещали в ступки и измельчали, а затем смывали 8 мл воды в 15 мл пробирку с получением суспензии 2 мг/мл для экспериментов на животных.
Образцы крови собирали в 0 часов перед введением и через 0,083, 0,25, 0,5, 1,0, 2, 4, 6, 8 часов после введения. Образцы крови крыс 250-300 мкл собирали в каждый момент времени из заднего венозного синуса глаз после респирационной анестезии эфиром (степень анестезии в высокой степени контролировали). Образцы крови собирали в пробирки, содержащие предварительно добавленный гепарин, а затем центрифугировали для получения плазмы. Полученную плазму делили на 2 части (50 мкл для каждой части) и хранили при -70°C до анализа. Концентрации соединения A в образцах крови в различные моменты времени анализировали, используя метод ЖХ/МС/МС. После экспериментов эвтаназию проводили газом СО2.
Фармакокинетические параметры эксперимента на животных каждой группы вычисляли, используя программное обеспечение InnaPhase Kinetica™ (США).
III. Результаты экспериментов на животных
Концентрации в крови гидрохлорида, фосфата, малеата и мезилата соединения A, вводимых крысам перорально в дозе 20 мг/кг, в различные моменты времени приведены в таблицах 5 и 6, соответственно. Соответствующие кривые концентрация лекарства в плазме - время представлены на фиг.3, и Фармакокинетические параметры приведены в таблицах 7 и 8.
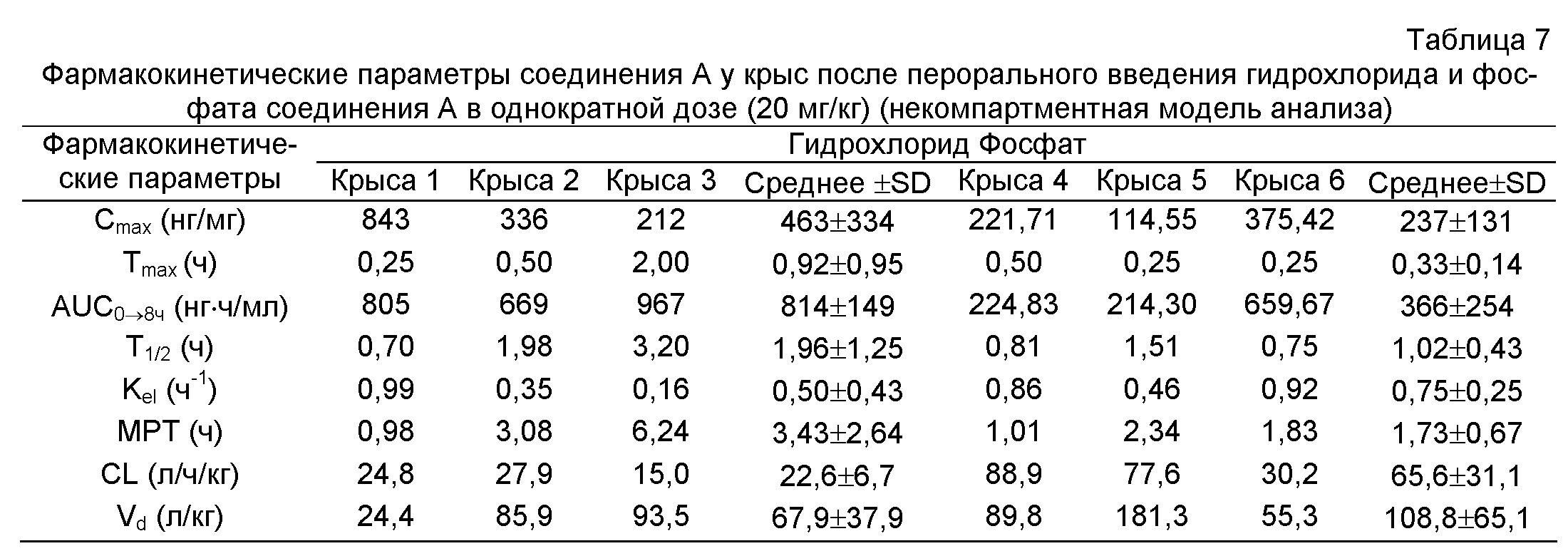
Cmax: максимальная концентрация лекарства в плазме после экстраваскулярного введения; Tmax: время, необходимое для достижения максимальная концентрация лекарства в плазме после экстраваскулярного введения; AUC0→8 ч: площадь под кривой концентрация лекарства в плазме - время (0-8 ч); AUMC0→8 ч площадь под кривой первый момент - время (0-8 часов); T1/2: время полувыведения; Kel: константа скорости элиминации; MRT: среднее время пребывания in vivo отдельной молекулы; CL: плазматический клиренс; Vd: кажущийся объем распределения на основе концентрации в плазме.
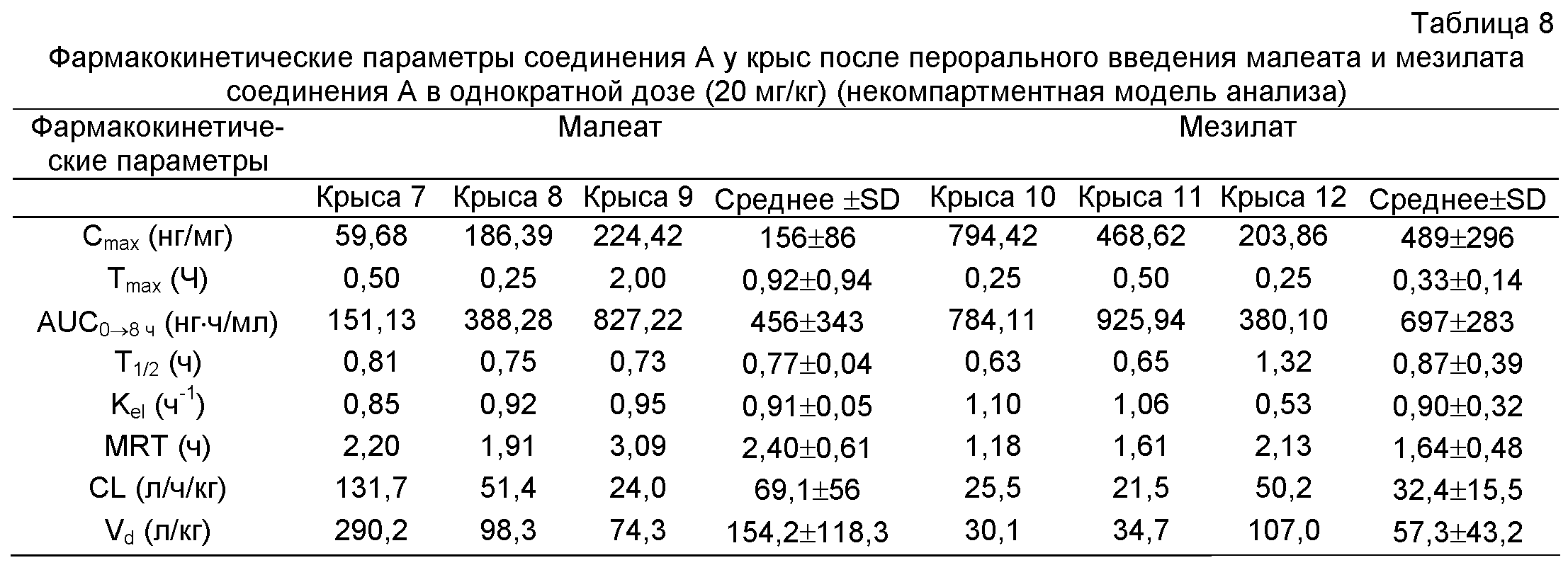
Cmax: максимальная концентрация лекарства в плазме после экстраваскулярного введения; Tmax: время, необходимое для достижения максимальная концентрация лекарства в плазме после экстраваскулярного введения; AUC0→8 ч - площадь под кривой концентрация лекарства в плазме - время (0-8 ч); AUMC0→8 ч: площадь под кривой первый момент - время (0-8 часов); Т1/2: время полувыведения; Kel: константа скорости элиминации; MRT: среднее время пребывания in vivo отдельной молекулы; CL: плазматический клиренс; Vd: кажущийся объем распределения на основе концентрации в плазме.
|
Cmax: максимальная концентрация лекарства в плазме после экстраваскулярного введения; Tmax: время, необходимое для достижения максимальная концентрация лекарства в плазме после экстраваскулярного введения; AUC0→8 ч: площадь под кривой концентрация лекарства в плазме - время (0-8 ч); AUC0→8 ч моль дозы (нг·ч/мл): площадь под кривой концентрация лекарства в плазме - время (0-8 ч) при дозе 1 ммоль/кг; Относительная F: относительная биодоступность.
Вывод: по сравнению с биодоступностью соединения A, определенной в примере 4, было обнаружено, что соли соединения A в настоящем изобретении значительно улучшали биодоступность соединения A, особенно гидрохлорид и мезилат соединения A.
|
|
Способ получения: Фармацевтически приемлемые соли соединения A просеивали через сито 100-200 меш и смешивали с крахмалом. Добавляли 2% крахмальную суспензию, и смесь гранулировали, высушивали и смешивали со 5 стеаратом магния. Полученную в результате смесь прессовали и тестировали. Пригодные таблетки упаковывали.
|
Способ получения: Смесь гранулировали, инкапсулировали, тестировали и упаковывали общепринятым способом.
![СОЛИ N-[4-(1-ЦИАНОЦИКЛОПЕНТИЛ)ФЕНИЛ]-2-(4-ПИРИДИЛМЕТИЛ)АМИНО-3-ПИРИДИНКАРБОКСАМИДА](https://fips.edrid.ru/images/rid/82/96/fb/29af2d006e30932633377425d6929c94.png)
![СОЛИ N-[4-(1-ЦИАНОЦИКЛОПЕНТИЛ)ФЕНИЛ]-2-(4-ПИРИДИЛМЕТИЛ)АМИНО-3-ПИРИДИНКАРБОКСАМИДА](https://fips.edrid.ru/images/rid/82/96/fb/65ed78f68cc53cd7c87cb4ce56c99573.png)
![СОЛИ N-[4-(1-ЦИАНОЦИКЛОПЕНТИЛ)ФЕНИЛ]-2-(4-ПИРИДИЛМЕТИЛ)АМИНО-3-ПИРИДИНКАРБОКСАМИДА](https://fips.edrid.ru/images/rid/82/96/fb/d46632054335ee661a3e59520b6c0ba2.png)
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Соли метил(r)-7-[3-амино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-a]пиразин-1-карбоксилата
Фармацевтическая композиция для лечения диабета 2 типа
Производные 6-аминохиназолина или 3-цианохинолина, способы их получения и их применение в качестве ингибитора рецепторных тирозинкиназ egfr или her-2
Соли бициклозамещенных производных азопиразолона, способ их получения и применение
Твердая дисперсия толваптана и способ ее получения
Промежуточное соединение для синтеза каспофунгина и способ его получения
Производные оксотиоимидазолидина, способы их получения и их применение в медицине в качестве ингибиторов андрогенного рецептора
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Соли метил(r)-7-[3-амино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-a]пиразин-1-карбоксилата
Фармацевтическая композиция для лечения диабета 2 типа
Производные 6-аминохиназолина или 3-цианохинолина, способы их получения и их применение в качестве ингибитора рецепторных тирозинкиназ egfr или her-2
Соли бициклозамещенных производных азопиразолона, способ их получения и применение
Твердая дисперсия толваптана и способ ее получения
Промежуточное соединение для синтеза каспофунгина и способ его получения
Производные оксотиоимидазолидина, способы их получения и их применение в медицине в качестве ингибиторов андрогенного рецептора

