Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ 6-АМИНОХИНАЗОЛИНА ИЛИ 3-ЦИАНОХИНОЛИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЕ В КАЧЕСТВЕ ИНГИБИТОРА РЕЦЕПТОРНЫХ ТИРОЗИНКИНАЗ EGFR ИЛИ HER-2
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к новым производным 6-аминохиназолина или 3-цианохинолина, к способам их получения, к фармацевтическим композициям, содержащим такие производные, и к применению таких производных в качестве терапевтических агентов, в частности в качестве ингибиторов протеинкиназ.
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Преобразование сигнала является фундаментальным механизмом, посредством которого внеклеточные стимулы передаются внутрь клеток. Эти сигналы регулируют широкое разнообразие физических ответов в клетке, включающих пролиферацию, дифференциацию и апоптоз. Многие из этих процессов преобразования сигнала используют обратимый процесс фосфорилирования белков, в который вовлечены специфичные протеинкиназы и фосфатазы.
Существует два класса протеинкиназ (РК): протеин-тирозинкиназы (РТК) и серин-треонинкиназы (STK). РТК могут фосфорилировать остаток тирозина на белке. STK могут фосфорилировать остаток серина и/или треонина. Тирозинкиназы можно разделить на киназы либо рецепторного типа (рецепторная тирозинкиназа, RTK), либо нерецепторного типа (нерецепторная тирозинкиназа). В настоящее время в геноме человека идентифицировано примерно 90 тирозинкиназ, из которых примерно 60 принадлежит к рецепторному типу и примерно 30 принадлежит к нерецепторному типу.
Семейство рецепторных тирозинкиназ (RTK) включает: (1) семейство EGF рецепторных тирозинкиназ, таких как EGFR, HER-2, HER-3 и HER-4; (2) инсулиновое семейство рецепторных тирозинкиназ, таких как рецептор инсулина (IR) и рецептор инсулиноподобного фактора роста-I (IGF-IR) и рецептор, подобный рецептору инсулина (IRR); (3) семейство класса III рецепторных тирозинкиназ, таких как рецепторные тирозинкиназы тромбоцитарного фактора роста (PDGF), рецепторная тирозинкиназа фактора стволовых клеток SCF RTK (общеизвестная как c-Kit), рецепторная тирозинкиназа fms-подобная тирозинкиназа 3 (Flt3) и рецепторная тирозинкиназа колониестимулирующего фактора роста 1 (CSF-1R) и тому подобное. Другие тирозинкиназы, рецептор фактора роста гепатоцитов (HGFR) c-Met и фактора роста эндотелия сосудов (VEGFR), принадлежат к семейству RTK. Они играют критическую роль в контроле клеточного роста и дифференциации и являются ключевыми медиаторами клеточных сигналов, ведущих к продуцированию цитокинов, таких как факторы роста (Schlessinger and Ullrich, Neuron 1992, 9, 383).
EGFR (ErbB, HER) играет критическую роль в регуляции клеточной пролиферации и роста. Эти RTK состоят из внеклеточного гликозилированного лиганд-связывающего домена, трансмембранного домена и внутриклеточного цитоплазматического каталитического домена. Ферментативную активность рецепторных тирозинкиназ можно стимулировать путем лиганд-опосредованной гомодимеризации или гетеродимеризации. Димеризация приводит в результате к фосфорилированию остатков тирозина на рецепторах в каталитическом домене, продуцируя будущий сайт связывания. За этим следует активация внутриклеточных биохимических путей передачи сигнала, таких как пути, в которые вовлечена протеинкиназа, ассоциированная с микротрубочками (MAP киназа), и фосфатидилинозит-3-киназа (Р13 киназа). Показано, что активация этих биохимических путей приводит к клеточной пролиферации и к ингибированию апоптоза. Идентифицировано, что такие мутированные или гиперэкспрессируемые формы тирозинкиназ, таких как EGFR, HER-2, присутствуют в большой доле распространенных раков человека, таких как рак молочной железы, рак простаты, немелкоклеточный рак легкого, рак пищевода, рак яичника и рак поджелудочной железы и тому подобное. Распространенность и релевантность тирозинкиназ подтверждена при онкогенезе и раковом росте.
Что касается семейства класса III рецепторных тирозинкиназ, группа рецептора тромбоцитарного фактора роста (PDGFR), которая включает c-Kit и Fms-подобную тирозинкиназу 3 (FLT-3), имеет такую же структуру и способ активации, как для семейства EGFR. Они передают сигналы посредством димеризации, последовательно регулируют физические ответы, заключающиеся в клеточной пролиферации, дифференциации, подвижности, и рост кровеносных сосудов. Таким образом, члены этого семейства тесно связаны с инициацией и развитием рака. Паттерн экспрессии с-Kit исследован, например, в группе различных первичных солидных опухолей. Высокая экспрессия c-Kit может быть обнаружена, среди прочего, в мелкоклеточной бронхиальной карциноме, тестикулярных внутриэпителиальных новообразованиях, меланомах, карциномах молочной железы, необластомах, в частности в желудочно-кишечных стромальных опухолях (GIST) [см. Weber et al., J. Clin. Oncol. 22(148), 9642 (2004)]. Большинство (50-80%) случаев GIST происходит посредством мутаций гена с-Kit. Мутации могут сделать c-Kit имеющим постоянную активацию рецепторных тирозинкиназ, приводящую к высокой скорости деления клеток и, возможно, геномной нестабильности. Таким образом, индуцируется рак.
Другим важным членом рецепторных тирозинкиназ является рецептор фактора роста эндотелия сосудов (VEGFR). VEGFR тесно вовлечен в ангиогенез. VEGF может активировать родственные биохимические пути передачи сигнала для стимуляции ангиогенеза посредством связывания с VEGFR. Недавние данные указывают на то, что VEGF может индуцировать пролиферацию и миграцию эндотелиальных клеток, что впоследствии приводит к образованию капиллярных трубок, которые способствуют образованию сверхпроницаемой, незрелой сосудистой сети, которая питает рост рака. В дополнение к их ангиогенной активности, VEGFR и VEGF могут непосредственно стимулировать опухолевый рост посредством эффектов, способствующих выживанию опухолевых клеток. Наблюдали, что VEGFR высоко экспрессируется в ряде солидных злокачественных опухолей, таких как карциномы легкого, карциномы молочной железы, карцинома яичника, рак поджелудочной железы и меланома. Таким образом, развитие опухолей можно ингибировать посредством ингибирования активации VEGFR. Это является полезным при лечении рака.
Что касается одного члена RTK, рецептора фактора роста гепатоцитов (HGF) (с-Met или HGFR), показано, что при многих раках человека он вовлечен в онкогенез, инвазию опухоли и метастаз, а также в усиленную клеточную подвижность (см. Ма, Р.С. et al. (2003b). Cancer Metastasis Rev, 22, 309-25; Maulik, G. et al. (2002b). Cytokine Growth Factor Rev, 13, 41-59).
Что касается других членов РТК, нерецепторные тирозинкиназы (сокращенно "NRTK" или "СТК") представляют собой протеин-тирозинкиназы в цитоплазме. По сравнению с RTK, у СТК отсутствует внеклеточный функциональный домен и трансмембранный домен. Активация тирозинкиназы СТК также тесно вовлечена в рак. Более подробное описание СТК приведено в статье Bolen, 1993, Oncogen 8: 2025-2031.
Двумя основными характеристиками рака являются геномная нестабильность и неконтролируемые биохимические пути передачи сигнала для регуляции клеточного цикла и пролиферации. Геномная нестабильность приводит к изменению или утрате биологической функции ключевых регуляторных белков, затем к вмешательству в биохимические пути преобразования сигнала или их повреждению, и аберрантные биохимические пути преобразования сигнала неспособны регулировать и контролировать нормальное протекание клеточного цикла и апоптоз, в то время как раковая клетка может продолжить жить и пролиферировать в состоянии генетического повреждения. Что касается основы достижения этих регуляторных процессов, РК, включающие обсуждаемые выше RTK и цитоплазматические РТК (СТК) тесно вовлечены в онкогенез и раковый рост и становятся важной мишенью для лечения рака.
Ожидают синтезировать новые соединения, обладающие противоопухолевыми активностями в отношении клеточной пролиферации. Ожидают, что эти соединения ингибируют одну или более чем одну RTK, СТК или STK, и полезны для лечения или ослабления опосредованных ангиогенезом, опосредованным RTK, СТК или STK, физиологических расстройств с гиперпролиферацией клеток.
Вплоть до настоящего времени имеется серия литературных данных об ингибиторах протеинкиназ, таких как WO 00/18761 A1, WO 2003089439 A1, WO 2005028443 A1, WO 2007055514 A1. В них раскрыты производные хинолина или хиназолина, их применение и получение. В статье Hwei-Ru Tsou et al. in J.Med.Chem. 48, 1107-1131 (2005) также раскрыты производные хинолина в качестве ингибиторов протеинкиназ.
Хотя некоторые ингибиторы протеинкиназ для лечения раков раскрыты, все еще существует необходимость в разработке новых соединений, которые обладают лучшим лечебным эффектом и фармакокинетическим всасыванием. После продолжительных усилий автор изобретения разработал новые соединения формулы (I) в настоящем изобретении и раскрыл, что эти соединения показали лучшую эффективность и функцию.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
В целях преодоления недостатков предшествующего уровня техники настоящее изобретение направлено на разработку новых производных 6-аминохиназолина и 3-цианохинолина формулы (I), а также их таутомеров, энантиомеров, диастереомеров, рацематов и фармацевтически приемлемых солей и метаболитов, предшественников или пролекарств,
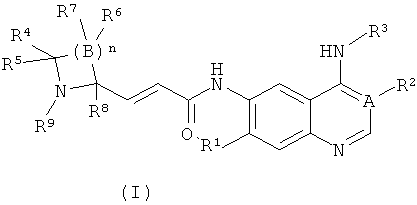
где:
А выбран из группы, состоящей из атома углерода или атома азота;
когда А представляет собой атом углерода, R1 выбран из группы, состоящей из атома водорода или алкоксила; где алкоксил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкоксила; R2 представляет собой циано;
когда А представляет собой атом азота, R1 выбран из группы, состоящей из атома водорода или алкоксила; где алкоксил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкоксила; R2 отсутствует;
R3 представляет собой радикал, имеющий приведенную ниже формулу:
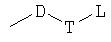 или
или  ;
;
где:
D выбран из группы, состоящей из арила или гетероарила, где арил или гетероарил каждый независимо необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена, алкила или трифторметила;
Т выбран из группы, состоящей из -(CH2)r-, -O(СН2)r-, -NH(CH2)r- или -S(O)r(CH2)r-;
L выбран из группы, состоящей из арила или гетероарила, где арил или гетероарил каждый независимо необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкила;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкоксила, гидроксила, гидроксиалкила, атома галогена, карбонила, амино, циано, нитро, карбокси или эфира карбоновой кислоты;
В выбран из группы, состоящей из атома углерода, атома кислорода или S(O)r;
когда В представляет собой атом углерода, R6 и R7 каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкоксила, гидроксила, гидроксиалкила, атома галогена, карбонила, амино, циано, нитро, карбокси или эфира карбоновой кислоты;
когда В представляет собой атом кислорода или S(O)r, R6 и R7 отсутствуют;
R8 выбран из группы, состоящей из атома водорода или алкила;
R9 выбран из группы, состоящей из атома водорода, алкила, арила, карбокси или эфира карбоновой кислоты;
r равно 0, 1 или 2; и
n равно 1, 2, 3, 4 или 5.
Предпочтительны соединения формулы (I) или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли, где А представляет собой атом углерода, R1 представляет собой алкоксил; R2 представляет собой циано.
Предпочтительны соединения формулы (I) или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли, где А представляет собой атом азота, R1 представляет собой атом водорода; R2 отсутствует.
Предпочтительны соединения формулы (I) или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли, где n равно 2.
Предпочтительно соединения формулы (I) или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли включают соединения формулы (II) или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли:
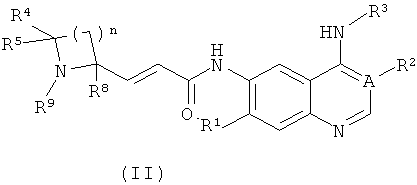
где:
А выбран из группы, состоящей из атома углерода или атома азота;
когда А представляет собой атом углерода, R1 выбран из группы, состоящей из атома водорода или алкоксила; где алкоксил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкоксила; R2 представляет собой циано;
когда А представляет собой атом азота, R1 выбран из группы, состоящей из атома водорода или алкоксила; где алкоксил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкоксила; R2 отсутствует;
R3 представляет собой радикал, имеющий приведенную ниже формулу:
или ;
где:
D выбран из группы, состоящей из арила или гетероарила, где арил или гетероарил каждый независимо необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена, алкила и трифторметила;
Т выбран из группы, состоящей из -(CH2)r-, -O(СН2)r-, -NH(CH2)r- или -S(O)r(CH2)r-;
L выбран из группы, состоящей из арила или гетероарила, где арил или гетероарил каждый независимо необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкила;
R4 и R5 каждый независимо выбран из группы, состоящей из атома водорода, алкила, алкоксила, гидроксила, гидроксиалкила, атома галогена, карбонила, амино, циано, нитро, карбокси или эфира карбоновой кислоты;
R8 выбран из группы, состоящей из атома водорода или алкила;
R9 выбран из группы, состоящей из атома водорода, алкила, арила, карбокси или сложного эфира карбоновой кислоты;
r равно 0, 1 или 2; и
n равно 1, 2, 3, 4 или 5.
Предпочтительны соединения формулы (II) или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли, где А представляет собой атом углерода, R1 представляет собой алкоксил; R2 представляет собой циано.
Предпочтительны соединения формулы (II) или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли, где А представляет собой атом азота, R1 представляет собой атом водорода; R2 отсутствует.
Предпочтительны соединения формулы (II) или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли, где n равно 2.
Соединения по настоящему изобретению включают, но не ограничены ими, приведенные ниже.
|
или их таутомеры, рацематы, энантиомеры, диастереомеры и их смеси, а также их фармацевтически приемлемые соли.
В другом аспекте данное изобретение относится к соединениям, имеющим приведенную ниже формулу (IA), в качестве промежуточных соединений при синтезе соединений формулы (I):
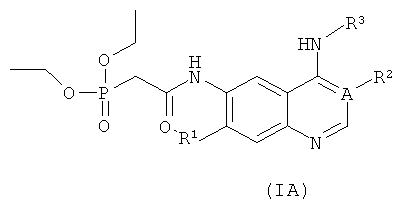
где:
А выбран из группы, состоящей из атома углерода или атома азота;
когда А представляет собой атом углерода, R1 выбран из группы, состоящей из атома водорода или алкоксила; где алкоксил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкоксила; R2 представляет собой циано;
когда А представляет собой атом азота, R1 выбран из группы, состоящей из атома водорода или алкоксила; где алкоксил необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкоксила; R2 отсутствует;
R3 представляет собой радикал, имеющий приведенную ниже формулу:
или ;
D выбран из группы, состоящей из арила или гетероарила, где арил или гетероарил каждый независимо необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена, алкила или трифторметила;
Т выбран из группы, состоящей из -(СН2)r-, -O(СН2)r-, -NH(СН2)r- или -S(O)r(CH2)r-;
L выбран из группы, состоящей из арила или гетероарила, где арил или гетероарил каждый независимо необязательно дополнительно замещен одной или более чем одной группой, выбранной из группы, состоящей из атома галогена или алкила;
r равно 0, 1 или 2.
В другом аспекте данное изобретение относится к способу получения соединения формулы (IA), включающему приведенные ниже стадии:

преобразование соединений формулы (IA_1) в соединения формулы (IA); где А, R1, R2 и R3 определены, как в формуле (IA).
В другом аспекте данное изобретение относится к способу получения соединений формулы (I) или их фармацевтически приемлемых солей, включающему приведенные ниже стадии:
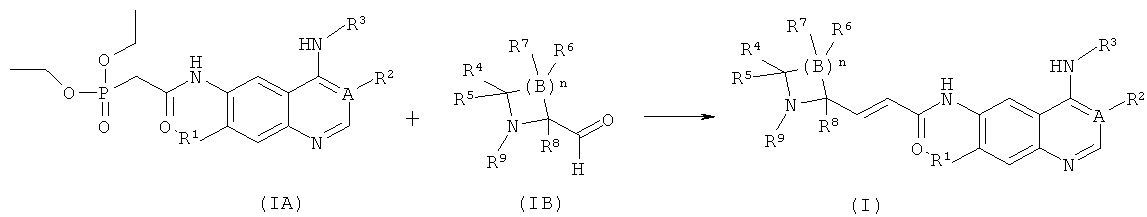
взаимодействие фосфатов соединений формулы (IA) с соединениями формулы (IB) с получением соединений формулы (I); где А, В, n, R1-R9 определены, как в формуле (I).
В другом аспекте данное изобретение относится к способу получения соединений формулы (II) или их фармацевтически приемлемых солей, включающему приведенные ниже стадии:
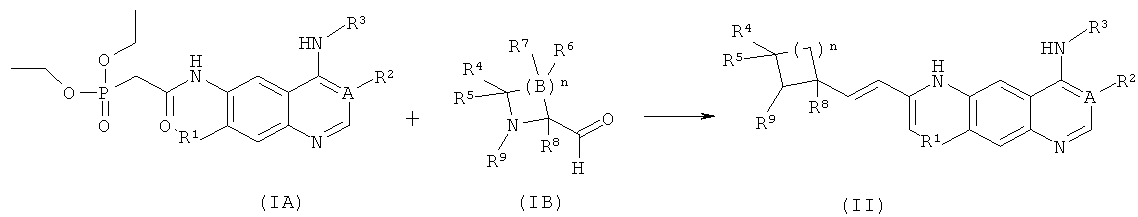
взаимодействие соединений формулы (IA) с соединениями формулы (IIB) с получением соединений формулы (II); где A, n, R1-R5, R8 и R9 определены, как в формуле (II).
Данное изобретение относится к соединениям формулы (I) или их таутомерам, рацематам, энантиомерам, диастереомерам и их смесям, а. также к их фармацевтически приемлемым солям для применения в качестве ингибиторов рецепторных тирозинкиназ, ингибирующих VEGFR, EGFR, HER-2, HER-3, HER-4, c-Met, Jak3 или их смесей.
Данное изобретение относится к применению соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереомеров и их смесей, а также их фармацевтически приемлемых солей при получении ингибиторов рецепторной тирозинкиназы, выбранной из группы, состоящей из VEGFR, EGFR, HER-2, HER-3, HER-4, c-Met, Jak3 или их смесей.
Данное изобретение относится к применению соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереомеров и их смесей, а также их фармацевтически приемлемых солей для получения лекарственного средства для лечения заболеваний, связанных с протеинкиназами, где протеинкиназы выбраны из группы, состоящей из рецепторной тирозинкиназы, нерецепторной тирозинкиназы или серин-треонинкиназ; где рецепторная тирозинкиназа выбрана из группы, состоящей из VEGFR, EGFR, HER-2, HER-3, HER-4, c-Met, Jak3 или их смесей.
Еще в одном другом аспекте данное изобретение относится к соединениям формулы (I) или их таутомерам, рацематам, энантиомерам, диастереомерам и их смесям, а также их фармацевтически приемлемым солям для применения в качестве лекарственного средства для лечения заболеваний, связанных с протеинкиназами, где протеинкиназы выбраны из группы, состоящей из рецепторной тирозинкиназы, нерецепторной тирозинкиназы или серин-треонинкиназ; где рецепторная тирозинкиназа выбрана из группы, состоящей из VEGFR, EGFR, HER-2, HER-3, HER-4, c-Met, Jak3 или их смесей.
Еще в одном другом аспекте данное изобретение относится к применению соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереомеров и их смесей, а также их фармацевтически приемлемых солей для получения лекарственного средства для лечения рака, где рак выбран из группы, состоящей из рака легкого, рака молочной железы, чешуйчато-клеточного рака или рака желудка.
Еще в одном другом аспекте данное изобретение относится к соединениям формулы (I) или их таутомерам, рацематам, энантиомерам, диастереомерам и их смесям, а также их фармацевтически приемлемым солям для применения в качестве лекарственного средства для лечения рака.
Еще в одном другом аспекте настоящее изобретение относится к фармацевтической композиции, содержащей терапевтически эффективное количество соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереомеров и их смесей, а также их фармацевтически приемлемых солей или их пролекарств и фармацевтически приемлемые носители или эксципиенты. Настоящее изобретение также относится к применению фармацевтической композиции для получения лекарственного средства для лечения заболеваний, связанных с протеинкиназами, где протеинкиназы выбраны из группы, состоящей из рецепторных тирозинкиназ, выбранных из группы, состоящей из VEGFR, EGFR, HER-2, HER-3, HER-4, c-Met, Jak3 или их смесей. Далее настоящее изобретение относится к применению фармацевтической композиции для получения лекарственного средства для лечения рака, где рак выбран из группы, состоящей из рака легкого, рака молочной железы, чешуйчато-клеточного рака или рака желудка.
В другом аспекте данное изобретение относится к способу получения фармацевтической композиции, включающему стадию объединения соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереомеров и их смесей, а также их фармацевтически приемлемых солей или их пролекарств с фармацевтически приемлемыми носителями или разбавителями.
В другом аспекте настоящее изобретение относится к способу регулирования каталитической активности протеинкиназ, включающему приведение в контакт протеинкиназ с соединениями формулы (I) или их таутомерами, рацематами, энантиомерами, диастереомерами и их смесями, а также их фармацевтически приемлемыми солями; где протеинкиназы представляют собой рецепторные тирозинкиназы, выбранные из группы, состоящей из VEGFR, EGFR, HER-2, HER-3, HER-4, c-Met, Jak3 или их смесей.
В другом аспекте настоящее изобретение относится к способу лечения рака, включающему введение отдельно или совместное введение с другими лекарствами субъекту, нуждающемуся в этом, терапевтически эффективного количества соединений формулы (I) или их таутомеров, рацематов, энантиомеров, диастереомеров и их смесей, а также их фармацевтически приемлемых солей, где совместно вводимыми лекарствами являются противоопухолевые лекарства, выбранные из группы, состоящей из трастузумаба, герцептина, цетуксимаба, лапатиниба, нератиниба, летрозола, капецитабина, топотекана, доцетаксела и т.д.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, приведенные ниже термины, используемые в описании и формуле изобретения, имеют значения, обсуждаемые ниже.
"Алкил" относится к насыщенной алифатической углеводородной группе, включающей C1-C20 прямоцепочечные и разветвленные группы. Предпочтительно алкильная группа представляет собой алкил, имеющий от 1 до 12 атомов углерода. Репрезентативные примеры включают, но не ограничены ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексил и их изомеры разветвленной цепи. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода. Репрезентативные примеры включают, но не ограничены ими, метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.д. Алкильная группа может быть замещенной или незамещенной. Когда она замещена, группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, карбокси или эфира карбоновой кислоты.
"Циклоалкил" относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе и имеет от 3 до 20 атомов углерода. Предпочтительно циклоалкильная группа представляет собой циклоалкил, имеющий от 3 до 12 атомов углерода, циклоалкильная группа представляет собой циклоалкил, имеющий от 3 до 10 атомов углерода. Репрезентативные примеры моноциклического алкила включают, но не ограничены ими, циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, циклогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.д. Полициклический циклоалкил включает циклоалкил, имеющий спиро-кольцо, конденсированное кольцо и кольцо с внутренним мостиком.
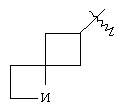
"Спиро-циклоалкил" относится к 5-20-членной полициклической углеводородной группе с кольцами, соединенными через один общий атом углерода (называемый спиро-атомом), где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы. Предпочтительно спиро-циклоалкил является 6-14-членным, более предпочтительно является 7-10-членным. В соответствии с числом общих спиро-атомов спиро-циклоалкил делят на моноциклическое спиро-кольцо, бициклическое спиро-кольцо или полициклическое спиро-кольцо, предпочтительно оно относится к моноциклическому спиро-кольцу или бициклическому спиро-кольцу. Более предпочтительно спиро-циклоалкил представляет собой 4-членное/4-членное, 4-членное/5-членное, 4-членное/6-членное, 5-членное/5-членное или 5-членное/б-членное моноциклическое спиро-кольцо. Репрезентативные примеры спиро-циклоалкила включают, но не ограничены ими, приведенные ниже группы:
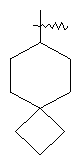
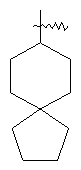
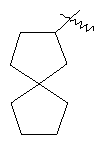
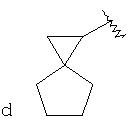 .
.
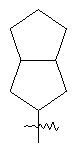
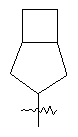
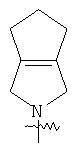
"Конденсированный циклоалкил" относится к 5-20-членной полициклической углеводородной группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы. Предпочтительно конденсированная циклоалкильная группа представляет собой 6-14-членную, более предпочтительно 7-10-членную. В соответствии с числом колец-членов конденсированный циклоалкил делят на конденсированное бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или полициклическое кольцо, предпочтительно он относится к конденсированному бициклическому или трициклическому кольцу. Более предпочтительно конденсированный циклоалкил представляет собой 5-членное/5-членное или 5-членное/6-членное конденсированное бициклическое кольцо. Репрезентативные примеры конденсированного циклоалкила включают, но не ограничены ими, приведенные ниже группы:
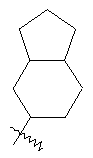
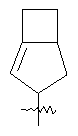
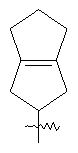
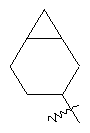
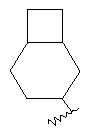 и
и 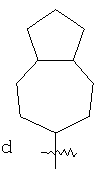 .
.
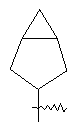
"Циклоалкил с внутренним мостиком" относится к 5-20-членной полициклической углеводородной группе, где каждые два кольца в системе имеют два общих разъединенных атома углерода. Эти кольца могут иметь одну или более чем одну двойную связь, но не имеют полностью сопряженной пи-электронной системы. Предпочтительно циклоалкил с внутренним мостиком является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов циклоалкил с внутренним мостиком делят на бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или полициклическое кольцо с внутренним мостиком, предпочтительно он относится к бициклическому, трициклическому или тетрациклическому циклоалкилу с внутренним мостиком, более предпочтительно относится к бициклическому или трициклическому циклоалкилу с внутренним мостиком. Репрезентативные примеры циклоалкила с внутренним мостиком включают, но не ограничены ими, приведенные ниже группы:
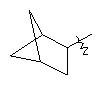
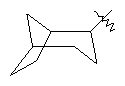
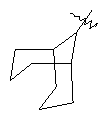
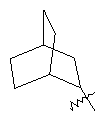
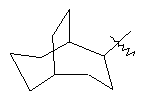
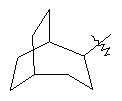
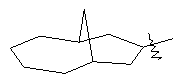
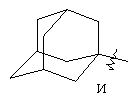
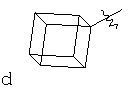 .
.
Циклоалкил может быть конденсирован с арилом, гетероарилом или гетероциклическим алкилом, где кольцом, соединенным с исходной структурой, является циклоалкил. Репрезентативные примеры циклоалкила с внутренним мостиком включают, но не ограничены ими, инданилуксусную кислоту, тетрагидронафталин, 6,7,8,9-тетрагидро-5Н-бензо[7]анилин и т.д. Циклоалкил может быть замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, карбокси или эфира карбоновой кислоты.
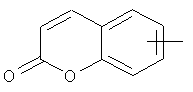
"Арил" относится к 6-14-членной полностью углеродной моноциклической кольцевой или к полициклической конденсированной кольцевой ("конденсированная" кольцевая система означает, что каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом в системе) группе и имеет полностью сопряженную пи-электронную систему. Предпочтительно арил является 6-10-членным, таким как фенил и нафтил. Арил может быть конденсирован с гетероарилом, гетероциклическим алкилом или циклоалкилом, где кольцом, соединенным с исходной структурой, является арил. Репрезентативные примеры арила включают, но не ограничены ими, приведенные ниже группы:
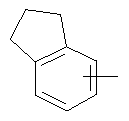
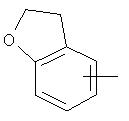
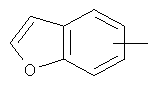
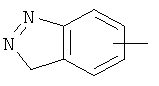
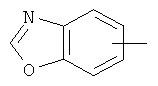
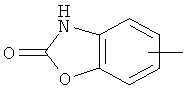
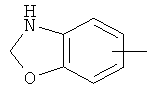
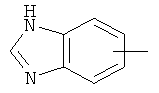
 и
и 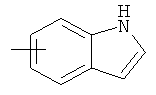 .
.
Арил может быть замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, карбокси или эфира карбоновой кислоты.
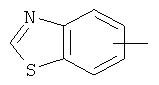
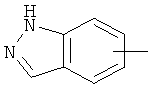
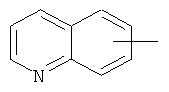
"Гетероарил" относится к 5-14-членному арилу, имеющему от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N, в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С. Предпочтительно кольцо представляет собой 6- или 10-членное кольцо. Предпочтительно гетероарил представляет собой 5- или 6-членное кольцо. Примерами гетероарильных групп являются фуран, тиофен, пиридин, пиррол, N-алкилпиррол, пиримидин, пиразин, имидазол, тетразолил и т.д. Гетероарил может быть конденсирован с арилом, гетероциклическим алкилом или циклоалкилом, где кольцом, соединенным с исходной структурой, является гетероарил. Репрезентативные примеры гетероарила включают, но не ограничены ими, приведенные ниже группы:
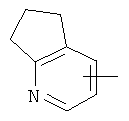

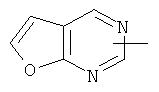
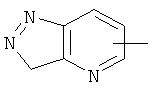
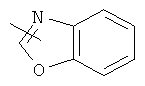
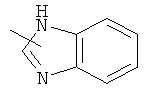
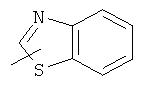
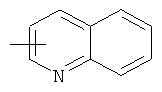 и
и 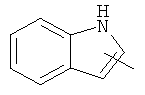 .
.
Гетероарил может быть замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, карбокси или эфира карбоновой кислоты.
"Гетероциклический алкил" относится к 3-20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более чем один гетероатом, выбранный из группы, состоящей из N, О или S(O)p (где n равно 0, 1 или 2), в качестве кольцевых атомов, но исключая -O-O-, -O-S- или -S-S- в кольце, где остальными кольцевыми атомами являются атомы С. Предпочтительно гетероциклический алкил является 3-12-членным, имеющим от 1 до 4 указанных гетероатомов, более предпочтительно 3-10-членным. Репрезентативные примеры моноциклического гетероциклического алкила включают, но не ограничены ими, пирролидил, пиперидил, пиперазинил, морфолинил, сульфоморфолинил, гомопиперазинил и т.д. Пол и циклически и гетероциклический алкил включает гетероциклический алкил, имеющий спиро-кольцо, конденсированное кольцо и кольцо с внутренним мостиком.
"Спиро-гетероциклический алкил" относится к 5-20-членной полициклической гетероциклической алкильной группе с кольцами, соединенными через один общий атом углерода (называемый спиро-атомом), где кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О или S(O)p (где p равно 0, 1 или 2) в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы. Предпочтительно спиро-гетероциклический алкил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом общих атомов спиро-гетероциклический алкил делят на моноциклический спиро-гетероциклический алкил, бициклический спиро-гетероциклический алкил или полициклический спиро-гетероциклический алкил, предпочтительно он относится к моноциклическому спиро-гетероциклическому алкилу или бициклическому спиро-гетероциклическому алкилу. Более предпочтительно спиро-гетероциклический алкил представляет собой 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноциклический спиро-гетероциклический алкил. Репрезентативные примеры спиро-гетероциклического алкила включают, но не ограничены ими, приведенные ниже группы:
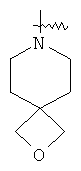
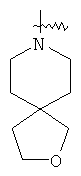
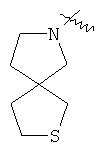
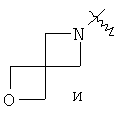
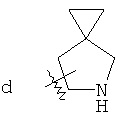 .
.
"Конденсированный гетероциклический алкил" относится к 5-20-членной полициклической гетероциклической алкильной группе, где каждое кольцо в системе имеет общую пару соседних атомов углерода с другим кольцом, где одно или более чем одно кольцо может содержать одну или более чем одну двойную связь, но ни одно из колец не имеет полностью сопряженной пи-электронной системы, и где кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О или S(O)p (где p равно 0, 1 или 2) в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С. Предпочтительно конденсированный гетероциклический алкил является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов конденсированный гетероциклический алкил делят на конденсированное бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или полициклическое кольцо, предпочтительно он относится к конденсированному бициклическому или трициклическому кольцу. Более предпочтительно конденсированный гетероциклический алкил представляет собой 5-членное/5-членное или 5-членное/6-членное конденсированное бициклическое кольцо. Репрезентативные примеры конденсированного гетероциклического алкила включают, но не ограничены ими, приведенные ниже группы:
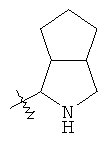
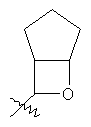
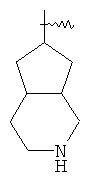
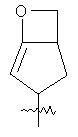
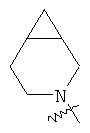
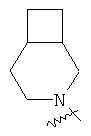
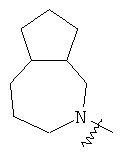
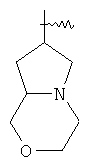
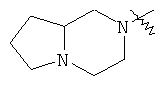 и
и 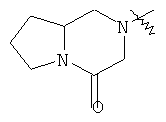 .
.
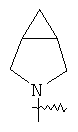
"Гетероциклический алкил с внутренним мостиком" относится к 5-14-членной полициклической гетероциклической алкильной группе, где каждые два кольца в системе имеют два общих разъединенных атома углерода, кольца могут иметь одну или более чем одну двойную связь, но не имеют полностью сопряженной пи-электронной системы, кольца имеют один или более чем один гетероатом, выбранный из группы, состоящей из N, О или S(O)p (где p равно 0, 1 или 2) в качестве кольцевых атомов, где остальными кольцевыми атомами являются атомы С. Предпочтительно гетероциклический алкил с внутренним мостиком является 6-14-членным, более предпочтительно 7-10-членным. В соответствии с числом колец-членов гетероциклический алкил с внутренним мостиком делят на бициклическое кольцо, трициклическое кольцо, тетрациклическое кольцо или полициклическое кольцо с внутренним мостиком, предпочтительно он относится к бициклическому, трициклическому или тетрациклическому гетероциклическому алкилу с внутренним мостиком, более предпочтительно относится к бициклическому или трициклическому гетероциклическому алкилу с внутренним мостиком. Репрезентативные примеры гетероциклического алкила с внутренним мостиком включают, но не ограничены ими, приведенные ниже группы:
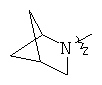
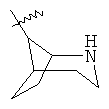
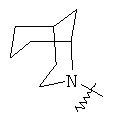
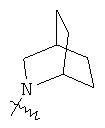
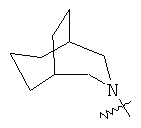
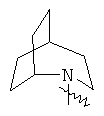
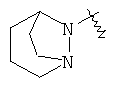
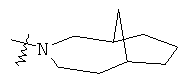 .
.
Гетероциклический алкил может быть конденсирован с арилом, гетероциклическим алкилом или циклоалкилом, где кольцом, соединенным с исходной структурой, является гетероциклический алкил. Репрезентативные примеры гетероциклического алкила включают, но не ограничены ими, приведенные ниже группы:
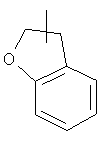
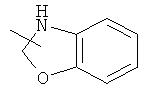
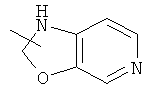 и
и 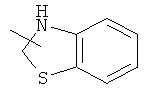 .
.
Гетероциклический алкил может быть замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляет собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, карбокси или эфира карбоновой кислоты.
"Алкоксил" относится как к группе -О-(алкил), так и к группе -O-(незамещенный циклоалкил), где алкил определен выше. Репрезентативные примеры включают, но не ограничены ими, метоксил, этоксил, пропоксил, бутоксил, циклопропоксил, циклобутоксил, циклопентилоксил, циклогексилоксил и тому подобное. Алкоксил может быть замещенным или незамещенным. Когда он замещен, группа(ы) заместителя предпочтительно представляют собой одну или более чем одну группу, независимо выбранную из группы, состоящей из алкила, алкенила, алкинила, алкоксила, алкилсульфо, алкиламино, атома галогена, тиола, гидроксила, нитро, циано, циклоалкила, гетероциклического алкила, арила, гетероарила, циклоалкоксила, гетероциклического алкоксила, циклоалкилтио, гетероциклического алкилтио, карбонила, карбокси или эфира карбоновой кислоты.
"Гидроксил" относится к -ОН группе.
"Гидроксиалкил" относится к -алкил-ОН, где алкил определен выше.
"Галогено" относится к фторо, хлоро, бромо или йодо, предпочтительно фторо или хлоро.
"Карбонил" относится к -С(=O)-.
"Нитро" относится к -NO2.
"Циано" относится к -CN.
"Амино" относится к -NH2.
"Карбокси" относится к -С(=O)ОН.
"Эфир карбоновой кислоты" относится к -С(=O)O-алкилу.
"Необязательный" или "необязательно" означает, что описанное впоследствии событие или обстоятельство может произойти или не произойти, и что описание включает случаи, где событие или обстоятельство может произойти или не произойти. Например, "гетероциклическая группа необязательно дополнительно замещена алкильной группой" означает, что этот алкил может присутствовать или отсутствовать, и описание включает ситуации, где гетероциклическая группа замещена алкильной группой, и ситуации, где гетероциклическая группа не замещена алкильной группой.
"Фармацевтическая композиция" относится к смеси одного или более чем одного из соединений, описанных в данной заявке, или их физиологически/фармацевтически приемлемых солей или пролекарств с другими химическими компонентами, такими как физиологически/фармацевтически приемлемые носители и эксципиенты. Целью фармацевтической композиции является облегчение введения соединения в организм, где польза состоит в более эффективном приеме активного ингредиента.
СПОСОБ СИНТЕЗА СОЕДИНЕНИЯ ПО ИЗОБРЕТЕНИЮ
Чтобы осуществить цель изобретения, в изобретении применено приведенное ниже техническое решение:
Способ получения соединения формулы (I) или его фармацевтически приемлемых солей в соответствии с данным изобретением, включающий приведенные ниже стадии:
Взаимодействие соединения формулы (IA-1) с диэтилфосфоноуксусной кислотой с получением соединения формулы (IA) в присутствии агента конденсации; в бане с сухим льдом, взаимодействие соединения формулы (IA) с бис(триметилсилил)амидом лития, нагревание реакционного раствора до комнатной температуры и взаимодействие его с соединением формулы (IB) посредством реакции Виттига с получением соединения формулы (I);
где А, В, n и R1-R9 являются такими, как определено в формуле (I). Способ получения соединения формулы (II) или его фармацевтически приемлемых солей в соответствии с данным изобретением, включающий приведенные ниже стадии:
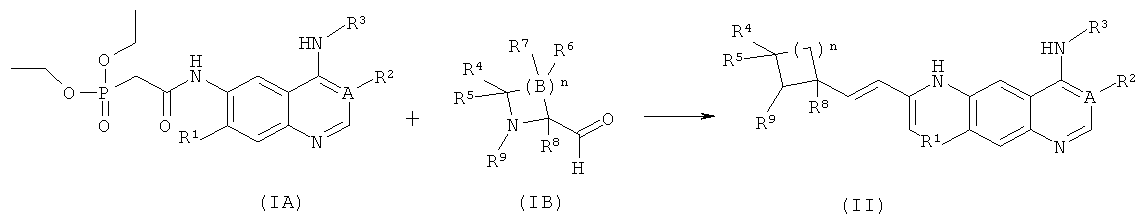
Взаимодействие соединений формулы (IA) с бис(триметилсилил)амидом лития в бане с сухим льдом, затем нагревание реакционного раствора до комнатной температуры и взаимодействие его с соединением формулы (ИВ) посредством реакции Виттига с получением соединения формулы (II);
где А, n, R1~R5 и R8-R9 являются такими, как определено в формуле (II).
КОНКРЕТНЫЕ СПОСОБЫ ОСУЩЕСТВЛЕНИЯ
Далее настоящее изобретение описано приведенными ниже Примерами, которые не предназначены для ограничения объема изобретения.
ПРИМЕРЫ
Структуры всех соединений были идентифицированы с помощью ядерного магнитного резонанса (1Н ЯМР) и/или масс-спектрометрии (МС). Химические сдвиги 1H ЯМР записаны в млн-1 (10-6). 1H ЯМР проводили на спектрометре Bruker AVANCE-400. Соответствующие растворители включали дейтерированный метанол (CD3OD), дейтерированный хлороформ (CDCl3) и дейтерированный диметилсульфоксид (ДМСО-d6) с тетраметилсиланом (ТМС) в качестве внутреннего стандарта.
МС определяли на масс-спектрометре FINNIGAN LCQ Ad (ИЭР) (Thermo, модель: Finnigan LCQ advantage MAX).
ВЭЖХ определяли на спектрометре жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire C18 150×4,6 мм) и на спектрометре жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini C18 150×4,6 мм).
IC50 определяли на NovoStar ELISA (BMG Co. German).
В качестве тонкослойного силикагеля использовали пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Размер пластин, используемых в ТСХ, составлял от 0,15 мм до 0,2 мм, а размер пластин, используемых для очистки продукта, составлял от 0,4 мм до 0,5 мм.
При колоночной хроматографии в качестве носителя, как правило, использовали силикагель Yantai Huanghai с размером ячеек от 200 до 300 меш.
При колоночной хроматографии на щелочном глиноземе в качестве носителя, как правило, использовали щелочной глинозем GuoYao FCP от 200 до 300 меш.
Исходные вещества по настоящему изобретению известны или приобретены у фирм ABCR GmbH & Со. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc, Darui Finechemical Co., Ltd и т.д., либо их можно получить общепринятыми способами синтеза предшествующего уровня техники.
Если не указано иное, нижеописанные реакционные смеси помещали в атмосферу азота или в атмосферу аргона.
Термин "атмосфера аргона" или "атмосфера азота" относится к тому, что реакционная колба оборудована баллоном, заполненным примерно 1 л азота.
Термин "атмосфера водорода" относится к тому, что реакционная колба оборудована баллоном, заполненным примерно 1 л водорода.
Реакции гидрогенизации под давлением проводили с помощью спектрометра гидрогенизации Parr 3916EKX и генератора водорода QL-500 или спектрометра гидрогенизации HC2-SS.
При реакциях гидрогенизации реакционную систему обычно помещали в вакуум и заполняли водородом, повторяя вышеуказанную операцию три раза.
Если не указано иное, раствор, используемый в примерах, относится к водному раствору.
Если не указано иное, температурой реакций является комнатная температура.
Комнатная температура представляет собой самую высокую температуру окружающей среды, которая составляет 20ºС-30ºС.
За ходом реакций примеров следили с помощью тонкослойной хроматографии (ТСХ). Хроматографическая система растворителей включала систему дихлорметана и метанола, систему гексана и этилацетата, систему петролейного эфира и этилацетата и ацетон. Соотношение объемов растворителей регулировали в соответствии с полярностью соединений.
Система элюирования колоночной хроматографии и хроматографическая система растворителей тонкослойной хроматографии включала: А: систему дихлорметана и метанола, В: систему гексана и этилацетата, С: систему дихлорметана и ацетона. Соотношение объемов растворителей регулировали в соответствии с полярностью соединений, и иногда также добавляли основной агент, такой как триэтиламин, или кислотный агент, такой как уксусная кислота.
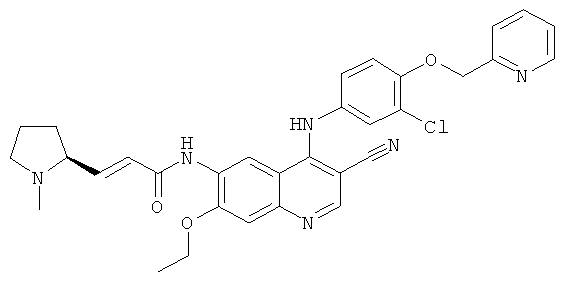
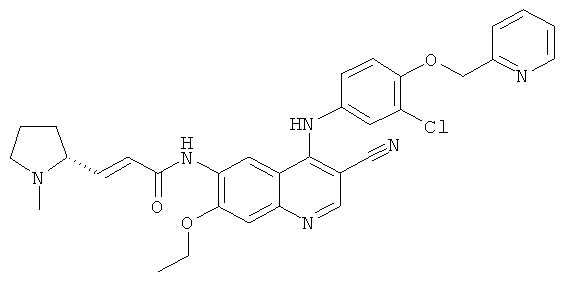
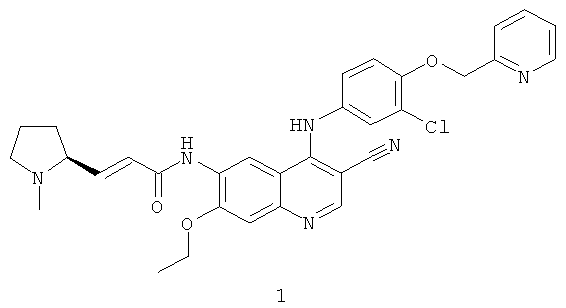
Пример 1
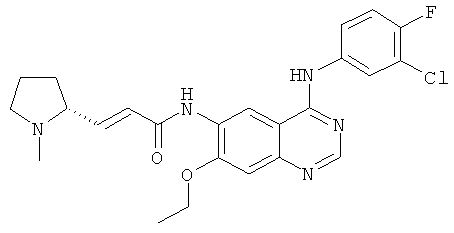
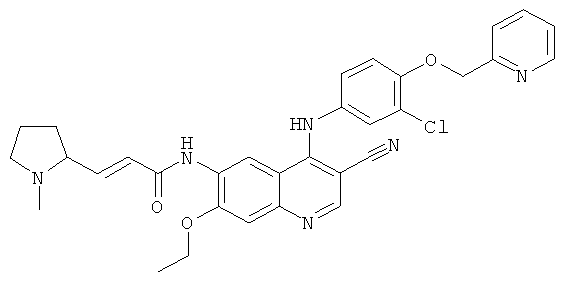
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2S)-1-метилпирролидин-2-ил]проп-2-енамид
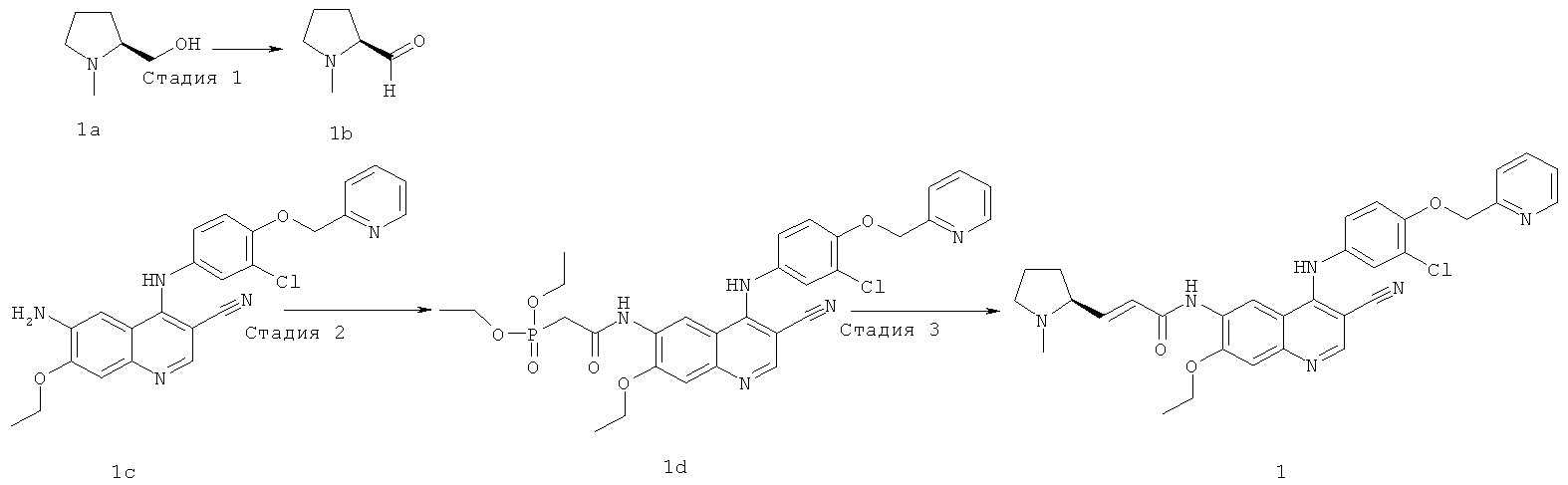
Стадия 1
(2S)-1-Метилпирролидин-2-карбальдегид
Оксалилхлорид (1,1 мл, 13,02 ммоль) растворяли в диметилсульфоксиде (1,9 мл, 26,04 ммоль) в бане с сухим льдом. Через 30 минут добавляли по каплям раствор [(25)-1-метилпирролидин-2-ил]метанола 1а (1 г, 8,68 ммоль) в дихлорметане (25 мл). Затем смесь перемешивали при -30ºС в течение 45 минут с последующим добавлением по каплям триэтиламина (6,15 г, 60,77 ммоль). Смесь подогревали до комнатной температуры и перемешивали в течение 12 часов. К реакционной смеси добавляли 250 мл дихлорметана, последовательно промывали насыщенным бикарбонатом натрия (100 мл), насыщенным хлоридом аммония (100 мл) и насыщенным рассолом (100 мл). Объединенные органические экстракты высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на щелочном глиноземе с системой элюирования А с получением соединения, указанного в заголовке, (2S)-1-метилпирролидин-2-карбальдегида 1b (308 мг, выход 31,4%) в виде светло-желтого масла.
Стадия 2
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорилацетамид
N,N'-Карбонилдиимидазол (487 мг, 3 ммоль) растворяли в 4 мл тетрагидрофурана. Смесь нагревали до 40ºС в масляной бане, к смеси добавляли по каплям раствор диэтилфосфоноуксусной кислоты (588 мг, 3 ммоль) в тетрагидрофуране (4 мл) и перемешивали в течение 30 минут до следующей стадии.
6-Амино-4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-7-этоксихинолин-3-карбонитрил 1с (446 мг, 1 ммоль, полученный хорошо известным способом: заявка на патент WO 2005028443) растворяли в 4 мл тетрагидрофурана при 40ºС с последующим добавлением по каплям полученного выше реакционного раствора. После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении и экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорилацетамида 1d (624 мг, выход 99,9%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 624 [М+1]
Стадия 3
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(25)-1-метилпирролидин-2-ил]проп-2-енамид
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорилацетамид 1d (50 мг, 0,08 ммоль) растворяли в 2 мл тетрагидрофурана при -78ºС с последующим добавлением по каплям раствора бис(триметилсилил)амида лития (1 М) в толуоле (80 мкл, 0,08 ммоль). После того, как смесь перемешивали в течение 45 минут, добавляли (2S)-1-метилпирролидин-2-карбальдегид 1b (20 мг, 0,17 ммоль). После перемешивания еще в течение 1 часа реакционную смесь подогревали до комнатной температуры и перемешивали в течение 12 часов. К реакционной смеси добавляли 1 мл воды и 1 мл метанола, затем органические экстракты экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2S)-1-метилпирролидин-2-ил]проп-2-енамида 1 (25 мг, выход 53,5%) в виде желтого твердого вещества.
МС m/z (ИЭР): 583 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.63 (s, 2Н), 8.95 (s, 1H), 8.60 (d, 1H), 8.48 (s, 1H), 7.89 (t, 1H), 7.59 (d, 1H), 7.37 (m, 3Н), 7.27-7.20 (m, 2Н), 6.80-6.60 (m, 2Н), 5.29 (s, 2Н), 4.34 (dd, 2Н), 2.33-2.24 (m, 3Н), 2.23-2.15 (m, 2Н), 1.99-1.88 (m, 3Н), 1.80-1.78 (m, 2Н), 1.49 (t, 3H).
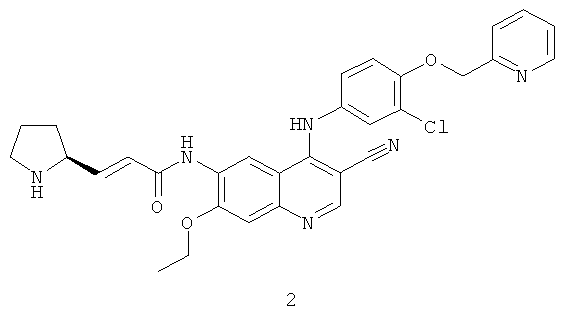
Пример 2
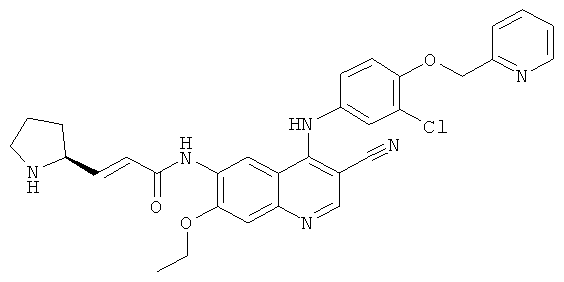
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2S)-пирролидин-2-ил]проп-2-енамид
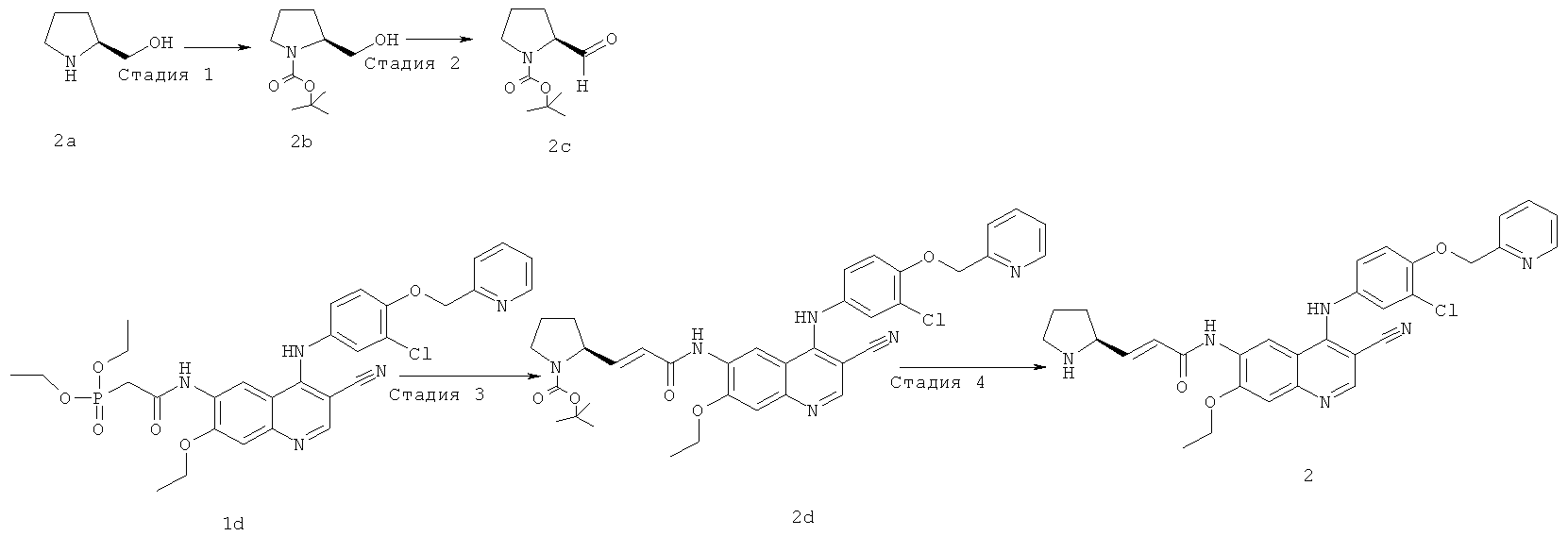
Стадия 1
Трет-бутил-(2S)-2-(гидроксиметил)пирролидин-1-карбоксилат
[(2S)-Пирролидин-2-ил]метанол 2а (5,06 г, 0,05 ммоль) и триэтиламин (10,12 г, 0,10 ммоль) растворяли в 100 мл дихлорметана в бане лед-вода. К реакционной смеси добавляли ди-трет-бутилпирокарбонат (16,37 г, 0,08 ммоль) порциями и перемешивали при комнатной температуре в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении, экстрагировали этилацетатом (50 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, трет-бутил-(2S)-2-(гидроксиметил)пирролидин-1-карбоксилата 2b (10 г, выход 99,9%) в виде светло-желтого масла.
Стадия 2
Трет-бутил-(2S)-2-формилпирролидин-1-карбоксилат
Оксалилхлорид (3,2 мл, 0,04 моль) и диметилсульфоксид (4,3 мл, 0,06 моль) растворяли в 100 мл дихлорметана в бане с сухим льдом. После перемешивания в течение 30 минут добавляли по каплям раствор трет-бутил-(2S)-2-(гидроксиметил)пирролидин-1-карбоксилат 2b (2 г, 0,01 моль) в 20 мл дихлорметана. Реакционную смесь перемешивали в течение 45 минут и добавляли по каплям триэтиламин (7,08 г, 0,07 моль). После перемешивания еще в течение 1 часа при 0ºС к реакционной смеси добавляли 500 мл дихлорметана. Объединенные органические слои промывали насыщенным рассолом (100 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, трет-бутил-(2S)-2-формилпирролидин-1-карбоксилата 2 с (1,10 г, выход 55,4%) в виде светло-желтого масла.
Стадия 3
Трет-бутил-(2S)-2-[(Е)-3-[[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]амино]-3-оксопроп-1-енил]пирролидин-1-карбоксилат
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорилацетамид 1d (156 мг, 0,25 ммоль) растворяли в 3 мл тетрагидрофурана в бане с сухим льдом с последующим добавлением по каплям раствора бис(триметилсилил)амида лития (1 М) в толуоле (375 мкл, 0,38 ммоль). После того, как реакционную смесь перемешивали в течение 45 минут, добавляли раствор трет-бутил-(2S)-2-формилпирролидин-1-карбоксилата 2с (100 мг, 0,50 ммоль) в 2 мл тетрагидрофурана. Реакционную смесь перемешивали еще в течение 1 часа, затем подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, трет-бутил-(2S)-2-[(Е)-3-[[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]амино]-3-оксопроп-1-енил]пирролидин-1-карбоксилата 2d (161 мг, выход 96,2%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 669 [М+1]
Стадия 4
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2S)-пирролидин-2-ил]проп-2-енамид
Трет-бутил-(2S)-2-[(Е)-3-[[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]амино]-3-оксопроп-1-енил]пирролидин-1-карбоксилат 2d (161 мг, 0,24 ммоль) растворяли в растворе хлорида водорода (2 М) в 25 мл 1,4-диоксана. После перемешивания в течение 12 часов реакционную смесь концентрировали при пониженном давлении и экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2S)-пирролидин-2-ил]проп-2-енамида 2 (20 мг, выход 14,6%) в виде желтого твердого вещества.
МС m/z (ИЭР): 569,4 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 10.01 (s, 1Н), 9.76 (s, 1Н), 9.71 (s, 2H), 9.40 (s, 1H), 8.92 (s, 1Н), 8.61 (s, 1Н), 8.60 (s, 1Н), 7.90 (t, 1Н), 7.60 (d, 1Н), 7.58-7.41 (s, 2H), 7.39-7.38 (m, 2H), 6.95 (dd, 1Н), 6.79 (d, 1Н), 5.29 (s, 1Н), 4.35 (t, 2H), 4.21-4.20 (m, 1Н), 3.23-3.22 (m, 3Н), 2.21-2.20 (m, 1Н), 2.039-1.94 (m, 1H), 1.84-1.76 (m, 1H),1.49 (t, 3H).
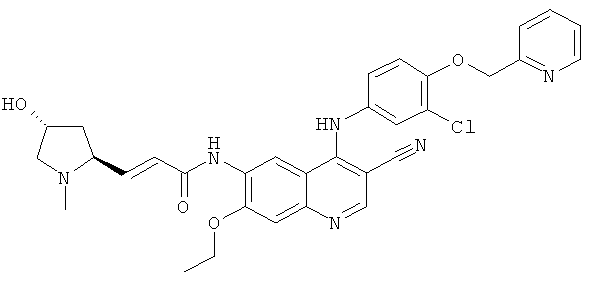
Пример 3
(Е)-N-[4-[[3-Хпор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2S,4R)-4-гидрокси-1-метил-пирролидин-2-ил]проп-2-енамид
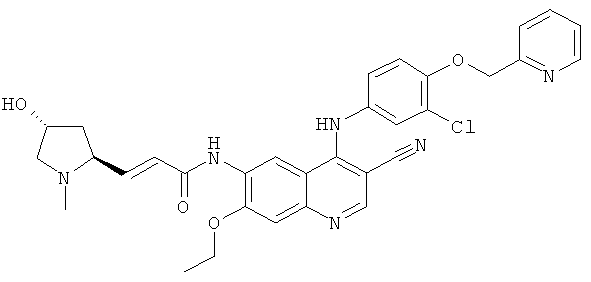
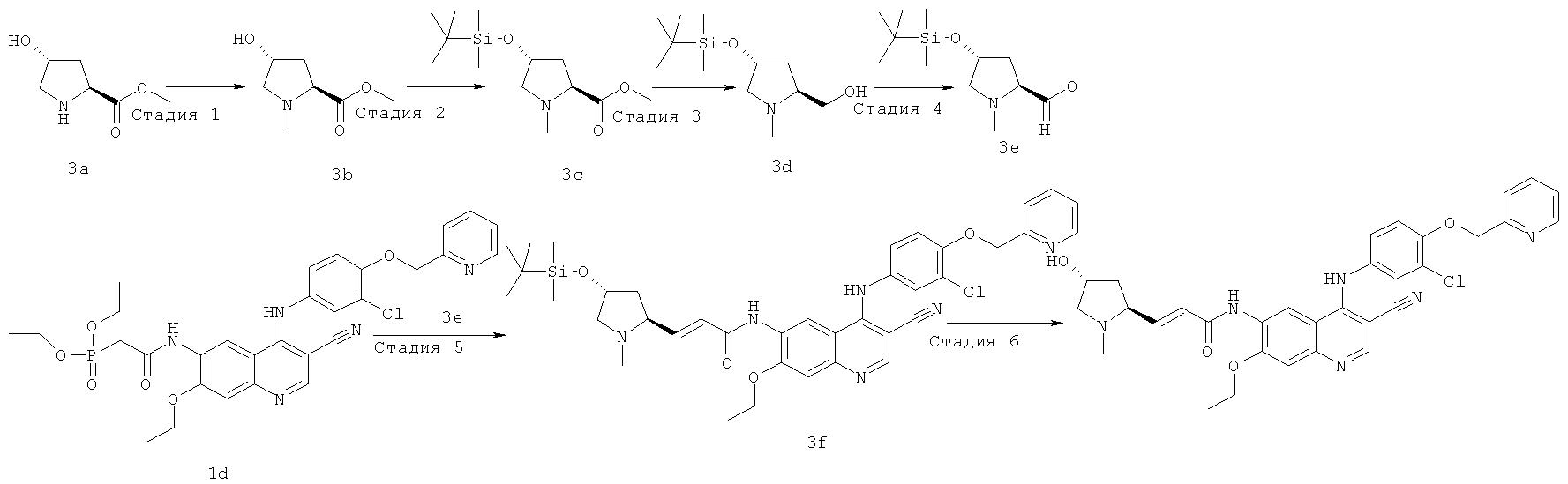
Стадия 1
Метил-(2S,4R)-4-гидрокси-1-метилпирролидин-2-карбоксилат
Метил-(2S,4R)-4-гидроксипирролидин-2-карбоксилат 3а (5,53 г, 38 ммоль) растворяли в 80 мл метанола в бане лед-вода с последующим добавлением 40% раствора формальдегида (31 мл, 380 ммоль) и цианоборгидрида натрия (12 г, 190 ммоль) порциями. Реакционную смесь перемешивали в течение 0,5 часа, затем подогревали до комнатной температуры и перемешивали в течение 3 часов. Смесь гасили 40 мл воды, концентрировали при пониженном давлении и экстрагировали дихлорметаном (80 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, сырого продукта метил-(2S,4R)-4-гидрокси-1-метилпирролидин-2-карбоксилата 3b в виде бесцветного масла, который использовали непосредственно на следующей стадии.
Стадия 2
Метил-(2S,4R)-4-(трет-бутил(диметил)силил)окси-1-метилпирролидин-2-карбоксилат
Метил-(2S,4R)-4-гидрокси-1-метилпирролидин-2-карбоксилат 3b (6 г, 37 ммоль) растворяли в 100 мл дихлорметана с последующим добавлением имидазола (7,70 г, 113 ммоль) и трет-бутилдиметилхлорсилана (6,80 г, 45 ммоль) последовательно. После перемешивания в течение 12 часов реакционную смесь разбавляли 100 мл дихлорметана, последовательно промывали водой (50 мл) и насыщенным рассолом (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, сырого продукта метил-(2S,4R)-4-(трет-бутил(диметил)силил)окси-1-метилпирролидин-2-карбоксилата 3с в виде бесцветного масла, который использовали непосредственно на следующей стадии без очистки.
МС m/z (ИЭР): 274 [М+1]
Стадия 3
[(2S,4R)-4-(Трет-бутил(диметил)силил)окси-1-метилпирролидин-2-ил]метанол
Метил-(2S,4R)-4-(трет-бутил(диметил)силил)окси-1-метилпирролидин-2-карбоксилат 3с (2,50 г, 9,10 ммоль) растворяли в 50 мл дихлорметана в бане лед-вода. Диизобутилалюмогидрид (18 мл, 18 ммоль) медленно добавляли по каплям, и смесь перемешивали в течение 6 часов. Реакционную смесь гасили 1 мл метанола, разбавляли 200 мл дихлорметана, добавляли безводный сульфат натрия и перемешивали в течение 30 минут. Реакционную смесь фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на щелочном глиноземе с системой элюирования А с получением соединения, указанного в заголовке, [(2S,4R)-4-(трет-бутил(диметил)силил)окси-1-метилпирролидин-2-ил]метанола 3d (570 мг, выход 50,0%) в виде желтого масла.
МС m/z (ИЭР): 246 [М+1]
Стадия 4
(2S,4R)-4-(Трет-бутил(диметил)силил)окси-1-метилпирролидин-2-карбальдегид
Диметилсульфоксид (174 мкл, 2,45 ммоль) растворяли в 20 мл дихлорметана в бане с сухим льдом. После того, как температура системы стабилизировалась, медленно добавляли по каплям оксалилхлорид (156 мкл, 1,80 ммоль). После перемешивания в течение 30 минут добавляли по каплям раствор [(2S,4R)-4-(трет-бутил(диметил)силил)окси-1-метилпирролидин-2-ил]метанола 3d (300 мг, 1,20 ммоль) в 2 мл дихлорметана. После перемешивания в течение 45 минут к реакционной смеси добавляли триэтиламин (510 мкл, 3,67 ммоль) и перемешивали в течение 10 минут, затем подогревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь разбавляли 100 мл дихлорметана, последовательно промывали насыщенным бикарбонатом натрия (20 мл), насыщенным хлоридом аммония (20 мл) и насыщенным рассолом (20 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, сырого продукта (2S,4R)-4-(трет-бутил(диметил)силил)окси-1-метилпирролидин-2-карбальдегида 3е (320 мг) в виде желтого масла, который использовали непосредственно на следующей стадии.
Стадия 5
(Е)-3-[(2S,4R)-4-(Трет-бутил(диметил)силил)окси-1-метилпирролидин-2-ил]-М-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]проп-2-енамид
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорилацетамид 1d (418 мг, 0,67 ммоль) растворяли в 2,5 мл тетрагидрофурана в бане с сухим льдом с последующим добавлением по каплям раствора бис(триметилсилил)амида лития (1 М) в толуоле (1 мл, 1 ммоль). После того, как смесь перемешивали в течение 45 минут, добавляли раствор (2S,4R)-4-(трет-бутил(диметил)силил)окси-1-метилпирролидин-2-карбальдегида 3е (326 мг, 1,34 ммоль) в 2,5 мл тетрагидрофурана и перемешивали еще в течение 1 часа, затем подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (E)-3-[(2S,4R)-4-(трет-бутил(диметил)силил)окси-1-метилпирролидин-2-ил]-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]проп-2-енамида 3f (292 мг, выход 61,2%) в виде желтого твердого вещества.
МС m/z (ИЭР): 713 [М+1]
Стадия 6
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(25,4Я)-4-гидрокси-1-метилпирролидин-2-ил]проп-2-енамид
(Е)-3-[(2S,4R)-4-(Трет-бутил(диметил)силил)окси-1-метилпирролидин-2-ил]-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]проп-2-енамид 3f (50 мг, 0,07 ммоль) и тетрабутиламмония фторид (51 мг, 0,21 ммоль) растворяли в 5 мл тетрагидрофурана, и смесь перемешивали в течение 12 часов. К реакционной смеси добавляли 1 мл воды, концентрировали при пониженном давлении и экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2S,4R)-4-гидрокси-1-метилпирролидин-2-ил]проп-2-енамида 3 (17 мг, выход 40,4%) в виде желтого твердого вещества.
МС m/z (ИЭР): 599,4 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.63 (s, 1Н), 9.52 (s, 1H), 8.97 (s, 1H), 8.61-8.60 (m, 1H), 8.48 (s, 1H), 7.904-7.862 (m, 1H), 7.60 (d, 1H), 7.41-7.36 (m, 3Н), 7.28-7.20 (m, 2H), 6.76 (dd, 1H), 6.61 (d, 1H), 5.29 (s, 2H), 4.82 (s, 1H), 4.35-4.29 (m, 2H), 4.21 (d, 1H), 3.42-3.38 (m, 2H), 3.36-3.33 (m, 3Н), 2.93 (d, 1H), 2.41-2.37 (m, 1H), 2.20-2.18 (m, 1H), 1.49 (t, 3Н).
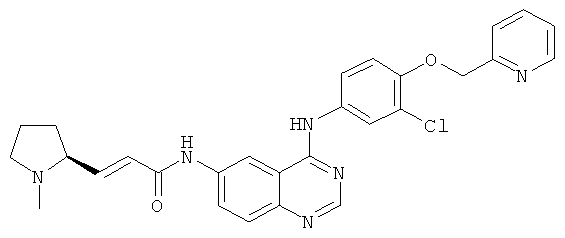
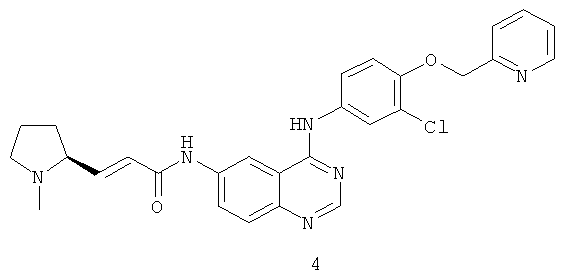
Пример 4
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]хиназолин-6-ил]-3-[(25)-1-метилпирролидин-2-ил]проп-2-енамид
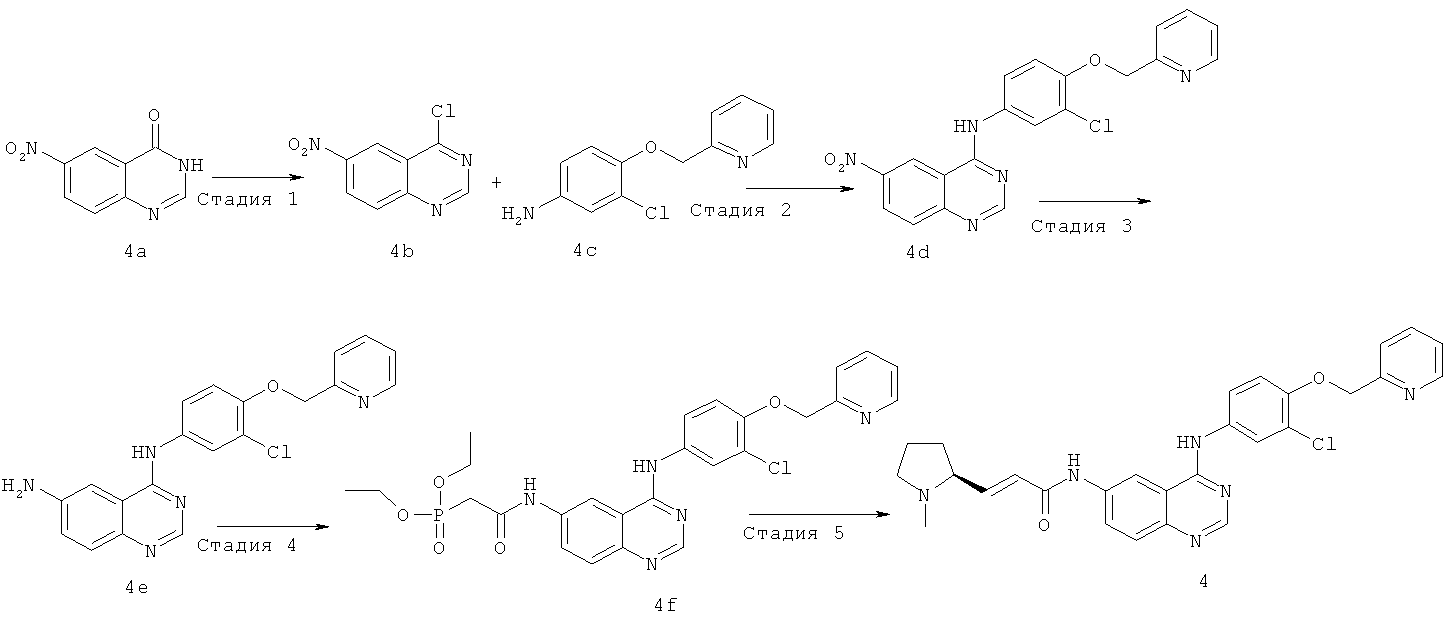
Стадия 1
4-Хлор-6-нитрохиназолин
6-Нитро-3Н-хиназолин-4-он 4a (18,88 г, 99,40 ммоль) растворяли в хлориде фосфора (31,03 г, 149 ммоль). Смесь подогревали до 160ºС и перемешивали в течение 3 часов. К реакционной смеси в горячем виде добавляли 250 мл н-гексана, перемешивали, и из раствора осаждалось много твердого вещества, которое фильтровали, фильтрационный кек промывали н-гексаном, высушивали в вакууме с получением соединения, указанного в заголовке, 4-хлор-6-нитрохиназолина 4b (18,14 г, выход 87,2%) в виде желтого твердого вещества.
Стадия 2
N-[3-Хлор-4-(2-пиридилметокси)фенил]-6-нитрохиназолин-4-амин Сырой продукт 4-хлор-6-нитрохиназолина 4b (6,06 г, 28,90 ммоль) растворяли в 100 мл изопропанола с последующим добавлением 3-хлор-4-(2-пиридилметокси)анилина 4c (7,47 г, 31,8 ммоль). Реакционную смесь нагревали до образования флегмы в течение 5 часов. Смесь охлаждали до комнатной температуры, твердое вещество осаждали, фильтровали и фильтрационный кек последовательно промывали этилацетатом, насыщенным рассолом (50 мл) и водой (150 мл), высушивали в вакууме с получением соединения, указанного в заголовке, N-[3-хлор-4-(2-пиридилметокси)фенил]-6-нитрохиназолин-4-амина 4d (8,38 г, выход 74,8%) в виде желтого твердого вещества.
МС m/z (ИЭР): 319 [М+1]
Стадия 3
N4-[3-Хлор-4-(2-пиридилметокси)фенил]хиназолин-4,6-диамин
N-[3-Хлор-4-(2-пиридилметокси)фенил]-6-нитрохиназолин-4-амин 4d (4,07 г, 10 ммоль) и концентрированную соляную кислоту (2 мл, 24 ммоль) растворяли в 130 мл смеси растворителей 95% этанола и воды (об/об=10/3) с последующим добавлением порошка железа (11,17 г, 200 ммоль). Реакционную смесь нагревали до образования флегмы в течение 2 часов, фильтровали в горячем виде и фильтрат концентрировали при пониженном давлении для удаления этанола. Полученный в результате остаток доводили до рН>7 гидроксидом аммония, фильтровали и фильтрационный кек высушивали в вакууме, очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, N4-[3-хлор-4-(2-пиридилметокси)фенил]хиназолин-4,6-диамина 4е (2,04 г, выход 54,1%) в виде белого твердого вещества.
МС m/z (ИЭР): 378 [М+1]
Стадия 4
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]хиназолин-6-ил]-2-диэтоксифосфорилацетамид
Диэтилфосфоноуксусную кислоту (1,04 г, 5,30 ммоль) растворяли в 10 мл дихлорметана в бане лед-вода с последующим добавлением оксалилхлорида (1,34 г, 10 ммоль) и 1 капли N,N-диметилформамида. После перемешивания в течение 1 часа смесь подогревали до комнатной температуры и перемешивали еще в течение 1 часа, концентрировали при пониженном давлении и добавляли 10 мл тетрагидрофурана для следующей стадии.
N4-[3-Хлор-4-(2-пиридилметокси)фенил]хиназолин-4,6-диамин 4е (1 г, 2,65 ммоль) растворяли в N,N-диизопропилэтиламине (1,03 г, 7,94 ммоль) с последующим добавлением по каплям полученного выше реакционного раствора. Смесь подогревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]хиназолин-6-ил]-2-диэтоксифосфорилацетамида 4f (671 мг, выход 45,7%) в виде белого твердого вещества.
МС m/z (ИЭР): 556 [М+1]
Стадия 5
(E)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]хиназолин-6-ил]-3-[(25)-1-метилпирролидин-2-ил]проп-2-енамид
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]хиназолин-6-ил]-2-диэтоксифосфорилацетамид 4f (277 мг, 0,50 ммоль) растворяли в 2,5 мл тетрагидрофурана в бане с сухим льдом с последующим добавлением по каплям раствора бис(триметилсилил)амида лития (1 М) в толуоле (750 мкл, 0,75 ммоль). После перемешивания в течение 45 минут к реакционной смеси добавляли (2S)-1-метилпирролидин-2-карбальдегид 1b (113 мг, 2 ммоль) и перемешивали в течение 1 часа, затем подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]хиназолин-6-ил]-3-[(25)-1-метилпирролидин-2-ил]проп-2-енамида 4 (85 мг, выход 33,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 515,3 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 10.47 (s, 1Н), 9.82 (s, 1H), 8.79 (s,1H), 8.61 (d, 1H), 8.52 (s, 1H), 8.00 (s, 1H), 7.91-7.89 (т, 2Н), 7.78-7.69 (т, 2Н), 7.61 (d, 1H), 7.38 (d, 1H), 7.28 (d, 1H), 6.78-6.72 (т, 1H), 6.47 (d, 1H), 5.30 (s, 2Н), 3.14-3.00 (т, 2Н), 3.001 (s, 1H), 2.31 (т, 4Н), 2.10-2.07 (т, 1H), 1.81 (т, 2Н), 1.63 (т, 1H)
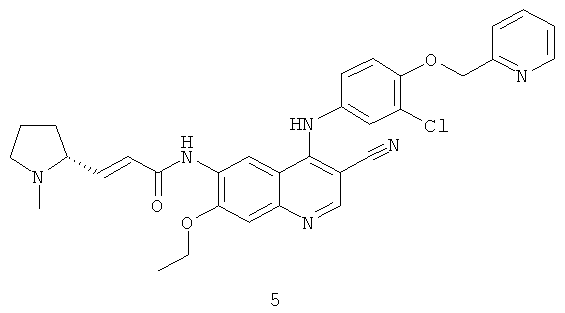
Пример 5
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2Д)-1-метилпирролидин-2-ил]проп-2-енамид
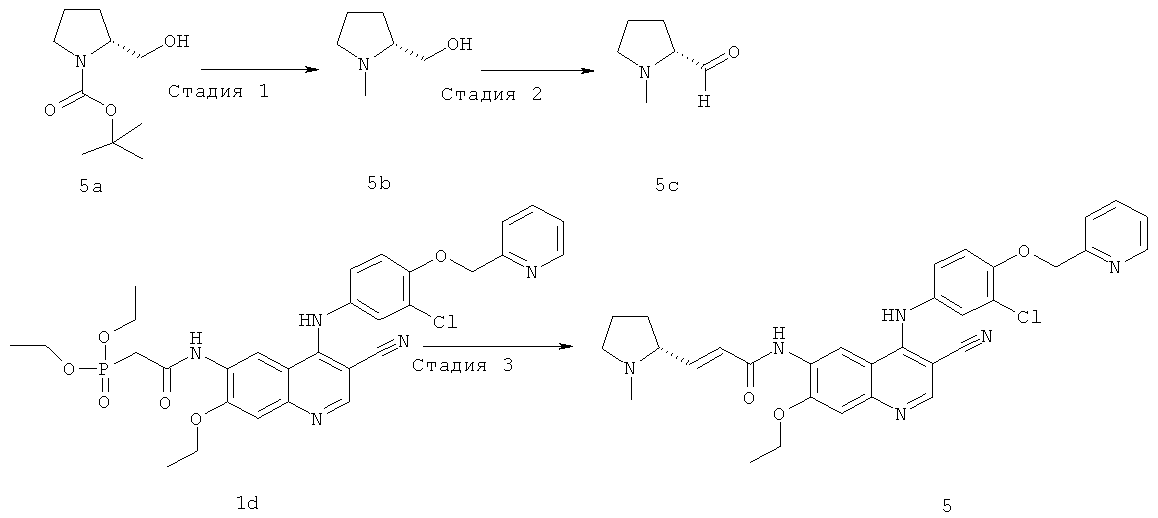
Стадия 1
[(2R)-1-Метилпирролидин-2-ил]метанол
Алюмогидрид лития (230 мг, 6 ммоль) и N-трет-бутоксикарбонил-L-пролинол 5а (400 мг, 2 ммоль) растворяли в 10 мл сухого тетрагидрофурана в бане лед-вода порциями. После того, как видимый газ больше не выделялся, реакционную смесь нагревали до образования флегмы в течение 2 часов. К реакционной смеси добавляли по каплям 5 мл метанола в бане лед-вода с последующим добавлением 5 мл воды, высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, [(2R)-1-метилпирролидин-2-ил]метанола 5b (221 мг, выход 77,0%) в виде бесцветного масла.
МС m/z (ИЭР): 116 [М+1]
Стадия 2
(2R)-1-Метилпирролидин-2-карбальдегид
Диметилсульфоксид (820 мкл, 11,46 ммоль) растворяли в 5 мл дихлорметана в бане с сухим льдом с последующим медленным добавлением по каплям оксалилхлорида (968 мг, 7,64 ммоль). После перемешивания в течение 45 минут к этому раствору добавляли раствор [(2R)-1-метилпирролидин-2-ил]метанола 5b (220 мг, 1,91 ммоль) в 2 мл дихлорметана. Реакционную смесь перемешивали в течение 45 минут и добавляли триэтиламин (1,9 мл, 13,37 ммоль). Реакционную смесь перемешивали в течение 10 минут, затем подогревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь последовательно промывали водой (20 мл) и насыщенным рассолом (10 мл). Объединенные органические экстракты высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на щелочном глиноземе с системой элюирования А с получением соединения, указанного в заголовке, (2R)-1-метилпирролидин-2-карбальдегида 5 с (300 мг) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии без очистки.
Стадия 3
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорилацетамид 1d (250 мг, 0,40 ммоль) растворяли в 10 мл сухого тетрагидрофурана в бане с сухим льдом с последующим добавлением по каплям раствора бис(триметилсилил)амида лития (1 М) в толуоле (440 мкл, 0,44 ммоль). Реакционную смесь перемешивали в течение 30 минут, добавляли по каплям раствор (2R)-1-метилпирролидин-2-карбальдегида 5 с (90 мг, 0,80 ммоль) в 5 мл тетрагидрофурана и перемешивали в течение 30 минут, затем подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида 5 (46 мг, выход 19,7%) в виде желтого твердого вещества.
MC m/z (ИЭР): 583,4 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): S 9.16 (s, 1Н), 8.63 (d, 1Н), 8.56 (s, 1H), 8.26 (s, 1H), 7.83-7.80 (dd, 1H), 7.76-7.50 (m, 2H), 7.57-7.56 (m,1H), 7.40 (s, 1H), 7.38(s, 1H), 7.19 (d, 1H), 7.06-7.03 (m, 2H), 6.34-6.31 (d, 1H), 5.35 (s, 2H), 4.39 (m, 2H), 4.27-4.26 (m, 1H), 3.32 (m, 1H), 3.10 (m, 1H), 2.73 (s, ЗН), 2.37-2.36 (m, 2H), 2.07-2.01 (m, 2H), 1.64 (t, 3H)
Пример 6
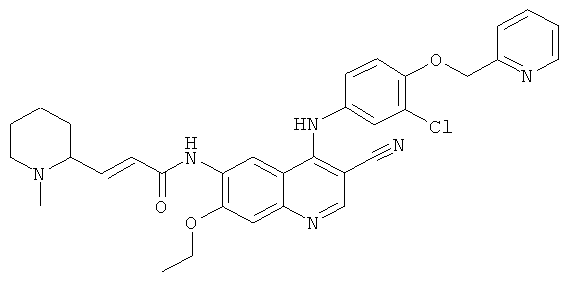
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-(1-метил-2-пиперидил)проп-2-енамид
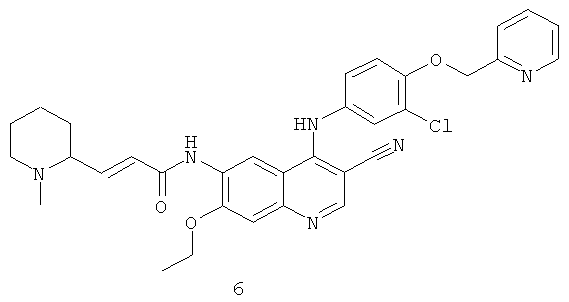
Стадия 1
1-Метилпиперидин-2-карбальдегид
Диметилсульфоксид (3,3 мл, 46 ммоль) растворяли в 15 мл дихлорметана в бане с сухим льдом с последующим медленным добавлением по каплям оксалилхлорида (2,6 мл, 31 ммоль). После перемешивания в течение 45 минут к этому раствору добавляли по каплям раствор (1-метил-2-пиперидил)метанола 6а (1 г, 7,74 ммоль) в 5 мл дихлорметана. Реакционную смесь перемешивали в течение 45 минут, затем добавляли триэтиламин (7,2 мл, 52 ммоль) и перемешивали в течение 10 минут, затем подогревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь последовательно промывали водой (20 мл) и насыщенным рассолом (20 мл). Объединенные органические экстракты высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, затем полученный в результате остаток очищали колоночной хроматографией на щелочном глиноземе с системой элюирования А с получением соединения, указанного в заголовке, 1-метилпиперидин-2-карбальдегида 6b (300 мг, выход 31,0%) в виде коричневого масла, которое использовали непосредственно на следующей стадии без очистки.
Стадия 2
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-(1-метил-2-пиперидил)проп-2-енамид
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорилацетамид 1d (300 мг, 0,48 ммоль) растворяли в 10 мл тетрагидрофурана в бане с сухим льдом с последующим добавлением по каплям раствора бис(триметилсилил)амида лития (1 М) в толуоле (530 мкл, 0,53 ммоль). После перемешивания в течение 30 минут добавляли по каплям раствор 1-метилпиперидин-2-карбальдегида 6b (120 мг, 0,96 ммоль) в 5 мл тетрагидрофурана. Реакционную смесь перемешивали еще в течение 30 минут, затем подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-(1-метил-2-пиперидил)проп-2-енамида 6 (14 мг, выход 4,9%) в виде желтого твердого вещества.
МС m/z (ИЭР): 597,3 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.09 (s, 1H), 8.63 (d, 1H), 8.51 (s, 1H), 7.83-7.79 (т, 2Н), 7.58-7.56 (т, 1H), 7.30-7.27 (т, ЗН), 7.14-7.12 (т, 2Н), 7.04 (d, 1H), 6.69-6.66 (т, 1H), 5.32 (s, 2Н), 4.32-4.29 (т, 2Н), 4.27-4.24 (т, 2Н), 3.60-3.40 (т, 2Н), 2.71 (s, 3Н), 2.05-1.72(m, 6H),1.62(t, 3H)
Пример 7
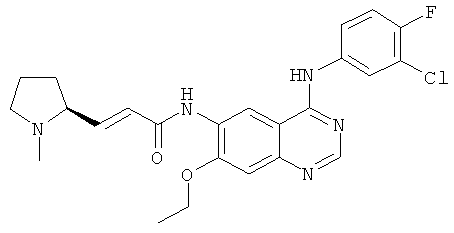
(Е)-N-[4-[(3-Хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-3-[(2S)-1-метилпирролидин-2-ил]проп-2-енамид
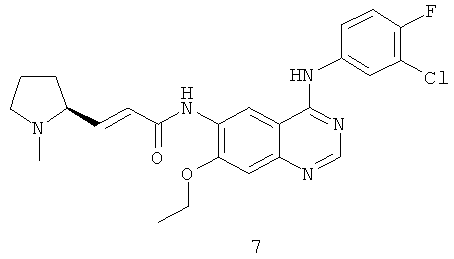
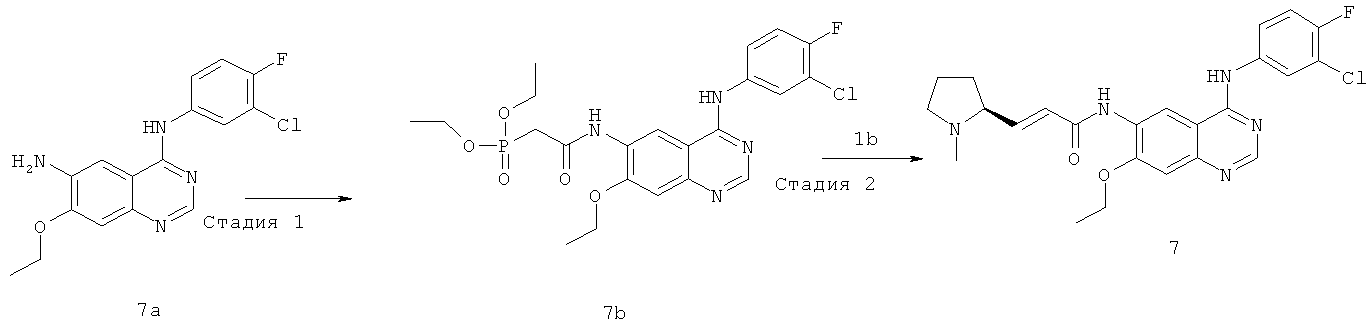
Стадия 1
N-[4-[(3-Хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-2-диэтоксифосфорилацетамид
N,N'-Карбонилдиимидазол (292 мг, 1,80 ммоль) растворяли в 4 мл тетрагидрофурана. Смесь нагревали до 50ºС в масляной бане, добавляли по каплям раствор диэтилфосфоноуксусной кислоты (353 мг, 1,8 ммоль) в 3 мл тетрагидрофурана и перемешивали в течение 1,5 часов до следующей стадии.
N4-(3-Хлор-4-фторфенил)-7-этоксихиназолин-4,6-диамин 7a (200 мг, 0,60 ммоль, полученный хорошо известным способом: заявка на патент WO 2005028443) растворяли в 10 мл тетрагидрофурана с последующим добавлением по каплям полученного выше реакционного раствора при 50ºС. После перемешивания в течение 3 часов при 40ºС реакционную смесь концентрировали при пониженном давлении и экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (50 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, N-[4-[(3-хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-2-диэтоксифосфорилацетамида 7b (100 мг, выход 33,3%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 511,1 [М+1]
Стадия 2
(Е)-N-[4-[(3-Хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-3-[(28)-1-метилпирролидин-2-ил]проп-2-енамид
N-[4-[(3-Хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-2-диэтоксифосфорилацетамид 7b (100 мг, 0,20 ммоль) растворяли в 10 мл тетрагидрофурана. Смесь охлаждали до -78ºС в бане с сухим льдом с последующим добавлением по каплям раствора бис(триметилсилил)амида лития (1 М) в толуоле (400 мкл, 0,40 ммоль) и смесь перемешивали в течение 45 минут, добавляли к ней (2S)-1-метилпирролидин-2-карбальдегид 1b (100 мг, 0,85 ммоль). После перемешивания еще в течение 1 часа реакционную смесь подогревали до комнатной температуры и перемешивали в течение 12 часов. К реакционной смеси добавляли воду (1 мл) и метанол (1 мл), экстрагировали дихлорметаном (100 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и фильтрат концентрировали при пониженном давлении, затем полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[(3-хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-3-[(28)-1-метилпирролидин-2-ил]проп-2-енамида 7 (60 мг, выход 65,2%) в виде желтого твердого вещества.
МС m/z (ИЭР): 470,2 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.78 (s, 1Н), 9.53 (s, 1Н), 8.91 (s, 1H), 8.52 (s, 1H), 8.13-8.15 (m, 1H), 7.79-7.81 (m, 1H), 7.39-7.43 (m, 1Н), 7.26 (s, 1H), 6.67-6.69 (m, 2H), 4.26-4.31 (m, 2Н), 4.09-4.10 (m, 1Н), 3.17-3.15 (m, 2Н), 3.08-3.04 (m, 1Н), 2.77-2.79 (m, 1Н), 2.87-2.82 (m, 1Н), 2.23 (s,3H), 1.74-1.76 (m,1H), 1.47 (m, 3Н).
Пример 8
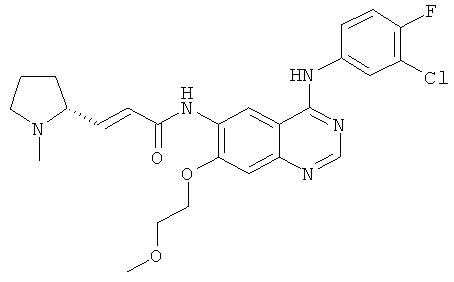
(Е)-N-[4-[(3-Хлор-4-фторфенил)амино]-7-(2-метоксиэтокси)хиназолин-6-ил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид
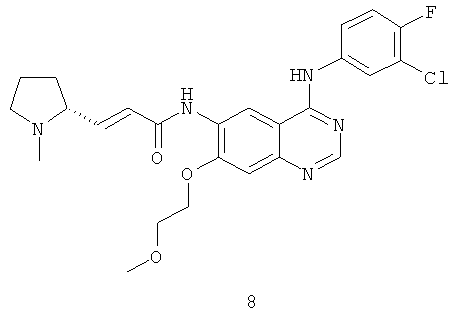
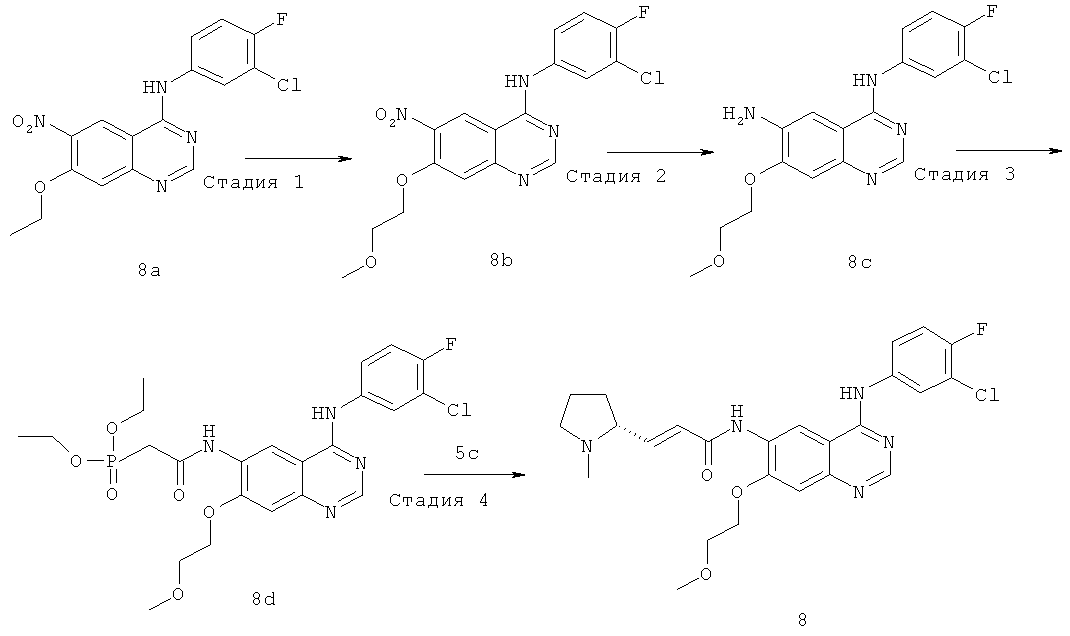
Стадия 1
N-(3-Хлор-4-фторфенил)-7-(2-метоксиэтокси)-6-нитрохиназолин-4-амин 2-Метоксиэтанол (152 мг, 2 ммоль) растворяли в 30 мл диметилсульфоксида в бане лед-вода, с последующим добавлением 60% гидрида натрия (80 мг, 2 ммоль). Смесь подогревали до 40ºС и перемешивали в течение 2 часов, затем добавляли N-(3-хлор-4-фторфенил)-7-фтор-6-нитрохиназолин-4-амин 8a (336 мг, 1 ммоль). Реакционную смесь перемешивали в течение 4 часов при 40ºС, затем подогревали до 50ºС и перемешивали в течение 12 часов. К реакционной смеси добавляли воду (20 мл), фильтровали и фильтрационный кек промывали водой (50 мл), высушивали в вакууме с получением соединения, указанного в заголовке, N-(3-хлор-4-фторфенил)-7-(2-метоксиэтокси)-6-нитрохиназолин-4-амина 8b (392 мг, выход 100%) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии.
МС m/z (ИЭР): 393,0 [М+1]
Стадия 2
N4-(3-Хлор-4-фторфенил)-7-(2-метоксиэтокси)хиназолин~4,6-диамин
N-(3-Хлор-4-фторфенил)-7-(2-метоксиэтокси)-6-нитрохиназолин-4-амин 8b (392 мг, 1 ммоль) и порошок железа (392 мг, 7 ммоль) растворяли в 20 мл уксусной кислоты. Реакционную смесь нагревали до образования флегмы в течение 4 часов, концентрировали при пониженном давлении. К остатку добавляли 100 мл насыщенного бикарбоната натрия, экстрагировали дихлорметаном (100 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (50 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, N4-(3-хлор-4-фторфенил)-7-(2-метоксиэтокси)хиназолин-4,6-диамина 8 с (200 мг, выход 55,2%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 363,1 [М+1]
Стадия 3
N-[4-[(3-Хлор-4-фторфенил)амино]-7-(2-метоксиэтокси)хиназолин-6-ил]-2-диэтоксифосфорилацетамид
N,N'-Карбонилдиимидазол (292 мг, 1,80 ммоль) растворяли в 4 мл тетрагидрофурана. Смесь нагревали до 50ºС в масляной бане, добавляли по каплям раствор диэтилфосфоноуксусной кислоты (353 мг, 1,8 ммоль) в 3 мл тетрагидрофурана и перемешивали в течение 1,5 часов до следующей стадии.
N4-(3-Хлор-4-фторфенил)-7-(2-метоксиэтокси)хиназолин-4,6-диамин 8 с (200 мг, 0,55 ммоль) растворяли в 10 мл тетрагидрофурана с последующим добавлением по каплям полученного выше реакционного раствора при 50ºС. После перемешивания в течение 3 часов при 40ºС реакционную смесь концентрировали при пониженном давлении и экстрагировали дихлорметаном (50 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (50 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, N-[4-[(3-хлор-4-фторфенил)амино]-7-(2-метоксиэтокси)хиназолин-6-ил]-2-диэтоксифосфорилацетамида 8d (150 мг, выход 50,5%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 541,2 [М+1]
Стадия 4
(Е)-N-[4-[(3-Хлор-4-фторфенил)амино]-7-(2-метоксиэтокси)хиназолин-6-ил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид
N-[4-[(3-Хлор-4-фторфенил)амино]-7-(2-метоксиэтокси)хиназолин-6-ил]-2-диэтоксифосфорилацетамид 8d (200 мг, 0,37 ммоль) растворяли в 10 мл тетрагидрофурана. Смесь охлаждали до -78ºС в бане с сухим льдом. В атмосфере аргона добавляли по каплям раствор бис(триметилсилил)амида лития (1 М) в толуоле (740 мкл, 0,74 ммоль). После перемешивания в течение 30 минут к реакционному раствору добавляли (2R)-1-метилпирролидин-2-карбальдегид 5 с (84 мг, 0,74 ммоль). Реакционную смесь перемешивали еще в течение 1 часа, затем подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали, добавляли к ней 10 мл воды, экстрагировали дихлорметаном (25 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[(3-хлор-4-фторфенил)амино]-7-(2-метоксиэтокси)хиназолин-6-ил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида 8 (100 мг, выход 54,2%) в виде желтого твердого вещества.
МС m/z (ИЭР): 500,2 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.82 (s, 1Н), 9.58 (s, 1H), 8.89 (s, 1H), 8.53 (s, 1H), 8.12-8.13 (m, 1H), 7.79-7.81 (m, 1H), 7.40-7.44 (m, 1H), 7.32 (s, 1H), 6.57-6.75 (m, 2H), 4.36-4.37 (m, 2H), 3.80-3.81 (m, 2H), 3.35-3.32 (m, 4H), 3.15-3.13 (m, 1H), 2.5(s, 3H), 2.40-2.31 (m, 2H), 2.08 (m, 1H), 1.90-1.81(m, 1H), 1.70-1.64 (m, 1H)
Пример 9
(Е)-N-[4-[(3-Хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид
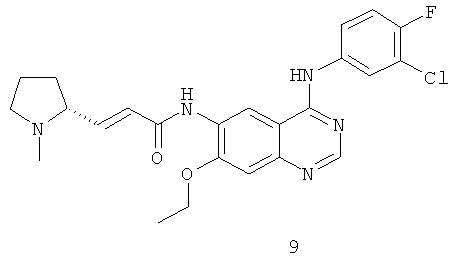
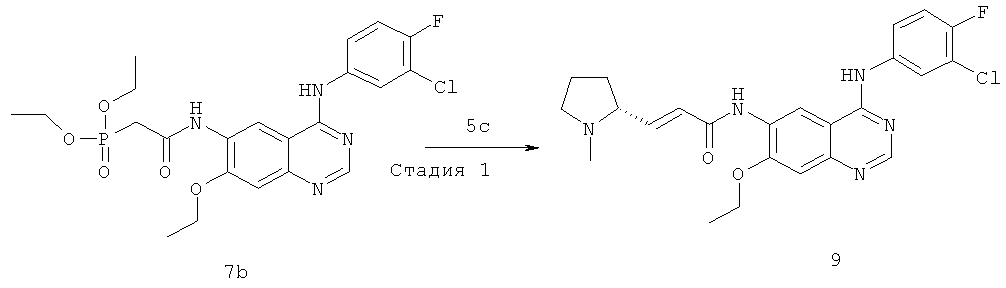
Стадия 1
(Е)-N-[4-[(3-Хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамид
N-[4-[(3-Хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-2-диэтоксифосфорилацетамид 7b (300 мг, 0,59 ммоль) растворяли в 10 мл тетра гидрофура на. Смесь охлаждали до -78ºС в бане с сухим льдом. В атмосфере аргона добавляли по каплям раствор бис(триметилсилил)амида лития (1 М) в толуоле (1,2 мл, 1,18 ммоль). После того, как смесь перемешивали в течение 30 минут, добавляли (2R)-1-метилпирролидин-2-карбальдегид 5 с (133 мг, 1,18 ммоль) и смесь перемешивали еще в течение 1 часа, затем подогревали до комнатной температуры и перемешивали в течение 12 часов. Реакционную смесь концентрировали, добавляли 10 мл воды, экстрагировали дихлорметаном (25 мл×3). Объединенные органические экстракты промывали насыщенным рассолом (30 мл×2), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (E)-N-[4-[(3-хлор-4-фторфенил)амино]-7-этоксихиназолин-6-ил]-3-[(2R)-1-метилпирролидин-2-ил]проп-2-енамида 9 (130 мг, выход 47,3%) в виде желтого твердого вещества.
МС m/z (ИЭР): 470,2 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 9.79 (s, 1H), 9.53 (s, 1H), 8.93 (s, 1H), 8.53 (s, 1H), 8.12-8.15 (m, 1H), 7.79-7.83 (m, 1H), 7.40-7.45 (m, 1H), 7.27 (s, 1H), 6.67-6.73 (m, 1H), 6.56-6.60 (m, 1H), 4.27-4.32 (m, 2Н), 4.09-4.10 (m, 1H), 3.17 (m, 2Н), 3.04 (m, 1H), 2.77-2.79 (m, 1H), 2.18-2.16 (m, 1H), 2.21 (s, 3H), 1.74-1.76 (m, 1H), 1.47 (t, 3H)
Пример 10
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-(1-метилпирролидин-2-ил)проп-2-енамид
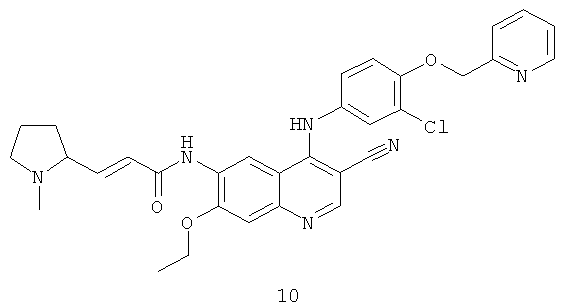
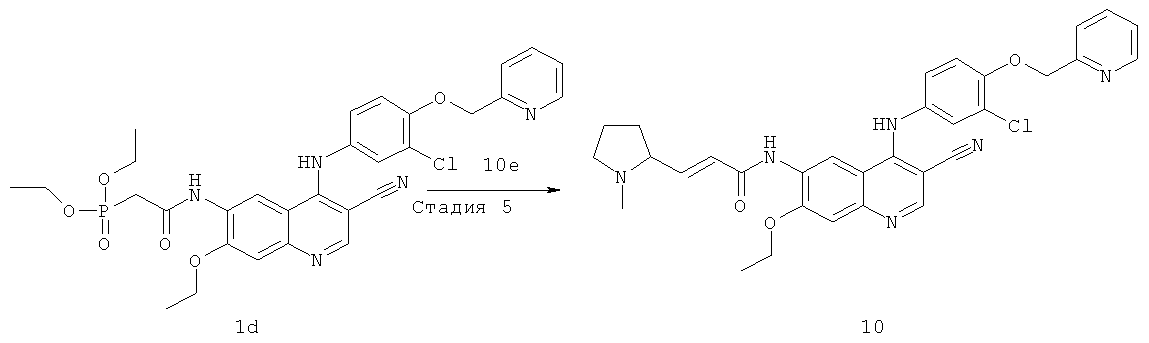
Стадия 1
Метилпирролидин-2-карбоксилат
7 мл тионилхлорида растворяли в 50 мл метанола в бане лед-вода с последующим добавлением пирролидин-2-карбоновой кислоты 10а (5 г, 43,40 ммоль). Смесь подогревали до комнатной температуры и перемешивали в течение 24 часов. Смесь концентрировали при пониженном давлении с получением сырого продукта метилпирролидин-2-карбоксилата 10b (10 г) в виде белого твердого вещества, который использовали непосредственно на следующей стадии без очистки.
MC m/z (ИЭР): 130,1 [М+1]
Стадия 2
Метил-1-метилпирролидин-2-карбоксилат
Сырой продукт метилпирролидин-2-карбоксилат 10b (5 г) растворяли в 100 мл метанола. Раствор охлаждали до 0-5ºС в бане лед-вода с последующим добавлением 13 мл 40% формальдегида. Реакционную смесь подогревали до комнатной температуры и перемешивали в течение 2 часов, затем охлаждали до 0-5ºС в бане лед-вода и добавляли порциями цианоборгидрид натрия (5,45 г, 87,20 ммоль). Реакционную смесь подогревали до комнатной температуры и перемешивали в течение 24 часов. Смесь концентрировали при пониженном давлении и добавляли 5 мл воды, экстрагировали дихлорметаном (5 мл×3). Объединенные органические экстракты высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением сырого продукта метил-1-метилпирролидин-2-карбоксилата 10 с (4,7 г, выход 70,1) в виде коричневого масла.
МС m/z (ИЭР): 144,1 [М+1]
Стадия 3
(1-Метилпирролидин-2-ил)метанол
Диизобутилалюмогидрид (60 мл, 66 ммоль) добавляли по каплям к раствору метил-1-метилпирролидин-2-карбоксилата 10 с (4,7 г, 33 ммоль) в 50 мл дихлорметана. Реакционную смесь перемешивали в течение 6 часов в бане лед-вода с последующим добавлением 10 мл метанола. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (1-метилпирролидин-2-ил)метанола 10d (1,8 г, выход 47,4%) в виде коричневого масла.
Стадия 4
1-Метилпирролидин-2-карбальдегид
Диметилсульфоксид (2,2 мл, 31,20 ммоль) растворяли в 20 мл дихлорметана в бане сухой лед - ацетон с последующим добавлением оксалилхлорида (2 мл, 23,40 ммоль). После перемешивания в течение 45 минут при -18ºС добавляли (1-метилпирролидин-2-ил)метанола 10d (1,8 г, 15,60 ммоль). После перемешивания еще в течение 45 минут добавляли триэтиламин (6,5 мл, 46,80 ммоль). Реакционную смесь подогревали до комнатной температуры и перемешивали в течение 1 часа. Реакционную смесь промывали насыщенным рассолом (50 мл), высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на щелочном глиноземе с системой элюирования А с получением 1-метилпирролидин-2-карбальдегида 10е (1 г, выход 56,8%) в виде коричневого масла.
Стадия 5
(Е)-N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-(1-метилпирролидин-2-ил)проп-2-енамид
N-[4-[[3-Хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-2-диэтоксифосфорилацетамид 1d (3 г, 4,40 ммоль) растворяли в 30 мл тетрагидрофурана в бане с сухим льдом с последующим добавлением по каплям раствора бис(триметилсилил)амида лития (1 М) в толуоле (9,6 мл, 8,80 ммоль). После того, как смесь перемешивали в течение 30 минут, добавляли по каплям раствор 1-метилпирролидин-2-карбальдегида 10е (1 г, 8,80 ммоль) в 5 мл тетрагидрофурана. Реакционную смесь перемешивали еще в течение 30 минут, затем подогревали до комнатной температуры и перемешивали в течение 24 часов. Реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюирования А с получением соединения, указанного в заголовке, (Е)-N-[4-[[3-хлор-4-(2-пиридилметокси)фенил]амино]-3-циано-7-этокси-6-хинолил]-3-(1-метилпирролидин-2-ил)проп-2-енамида 10 (500 мг, выход 20,8%) в виде желтого твердого вещества.
МС m/z (ИЭР): 583,2 [М+1]
1H ЯМР (400 МГц, ДМСО-d6): δ 11.59 (s, 1Н), 11.28 (s, 1H), 9.19 (s,1H), 9.05 (s, 1H), 8.71 (d, 1H), 8.09-8.07 (m, 1H), 7.74-7.68 (m, ЗН), 7.56-7.55 (m, 1H), 7.45-7.37 (m, 2H), 7.04-7.00 (m, 1H), 6.88-6.84 (m, 1H), 5.43 (s, 2H), 4.38 (dd, 2H), 4.10 (m, 2H), 3.63-3.60 (m, 1H), 3.13-3.08 (m, 1H), 2.73-2.72 (m, ЗН), 2.31-2.29 (m, 1H), 2.08-2.02 (m, 2H), 1.53(t, 3H).
ТЕСТОВЫЕ ПРИМЕРЫ
БИОЛОГИЧЕСКИЕ АНАЛИЗЫ
ПРИМЕР 1: Анализ ингибирования пролиферации клеток EGFR
Приведенный ниже анализ in vitro предназначен для определения активности соединений по изобретению по ингибированию пролиферации клеток эпидермоидной карциномы человека А431, которые обладают высокой экспрессией EGFR.
Приведенный ниже анализ in vitro предназначен для определения активности тестируемых соединений по ингибированию пролиферации раковых клеток, которые обладают высокой экспрессией EGFR. Эта активность представлена в виде значения 1Сбо. Общие методы этого анализа являются следующими: раковые клетки А431, обладающие высокой экспрессией EGFR (Институт биохимии и клеточной биологии), отбирали и высевали в 96-луночный планшет для клеточных культур при подходящей концентрации (например, 5000 клеток/мл среды). Затем клетки инкубировали в инкубаторе с диоксидом углерода (CO2) до достижения ими 85% конфлюентности. Затем клеточную культуральную среду заменяли свежей средой с тестируемыми соединениями, добавленными в нее в серийных разведениях (обычно от 6 до 7 концентраций). Затем клетки помещали обратно в инкубатор и культивировали непрерывно. Спустя 72 часа активность тестируемых соединений по ингибированию клеточной пролиферации определяли путем использования метода сульфородамина В (SRB). Значение IC50 на тестируемых клетках вычисляют по данным уровней ингибирования серийных концентраций тестируемых соединений.
Активность соединений по изобретению.
Биологическую активность соединений по изобретению тестировали путем использования анализа, описанного выше. Значения IC50 измерены и представлены в приведенной ниже таблице.
|
Вывод: соединения по настоящему изобретению обладают очевидной активностью по ингибированию пролиферации клеток А431.
ПРИМЕР 2: Анализ активности киназы EGFR
Анализ активности киназы EGFR in vitro тестировали с помощью приведенного ниже анализа.
Приведенный ниже анализ использовали для определения активности соединений по изобретению по ингибированию активности киназы EGFR. Половинную максимальную ингибирующую концентрацию IC50 (концентрацию тестируемого соединения, проявляющую 50% ингибирование активности фермента) каждого соединения определяли путем инкубации нескольких различных концентраций тестируемых соединений со специфичным ферментом и субстратом. Киназа EGFR, используемая в данном анализе, представляет собой рекомбинантный белок человеческого происхождения (Cell signaling technology, #7908), который подвергали взаимодействию с пептидным субстратом и различными концентрациями тестируемых соединений в буферном растворе, содержащем 60 мМ HEPES (рН 7,5), 5 мМ MgCl2, 5 мМ MnCl2, 3 мкМ Na3VO4, 1,25 M ДТТ (1000 х) и 20 мкМ АТФ при 25ºС в течение 45 минут. Активность киназы EGFR определяли путем использования метода времяпролетной флуоресценции.
Активность соединений по изобретению.
Биологическую активность соединений по изобретению тестировали путем использования вышеописанного анализа. Значения IC50 измерены и представлены в приведенной ниже таблице.
|
Вывод: соединения по настоящему изобретению обладают очевидной активностью по ингибированию активности киназы EGFR.
ФАРМАКОКИНЕТИЧЕСКИЙ АНАЛИЗ
Тестовый Пример 1: фармакокинетический анализ соединений Примера 1 и Примера 5 по настоящему изобретению
1. Краткое описание
Соединения Примера 1 и Примера 5 по настоящему изобретению вводили внутрижелудочно крысам, чтобы определить концентрацию лекарства в плазме в различные моменты времени методом ЖХ/МС/МС. Фармакокинетические свойства соединений по настоящему изобретению исследовали и оценивали на крысах.
2. Протокол
2.1 Образцы
Соединения Примера 1 и Примера 5
2.2 Подопытные животные
8 здоровых взрослых крыс SD, половину самцов и половину самок, покупали в фирме SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., CO, номер лицензии: SCXK (Shanghai) 2003-0002
2.3 Приготовление тестируемых соединений
Точное количество соединений взвешивали и однородно измельчали с 0,5% натриевой соли карбоксиметилцеллюлозы, суспендировали с 1% Твин 80 до 2,5 мг/мл суспензии перед использованием.
2.4 Введение
8 здоровым взрослым крысам SD, половине самцов и половине самок, после голодания в течение ночи вводили соединения внутрижелудочно в дозе 25,0 мг/кг, в объеме 10 мл/кг.
2.5 Сбор образцов
8 здоровых взрослых крыс SD, половину самцов и половину самок, делили на 2 группы. После голодания в течение ночи крысам вводили соединения внутрижелудочно в дозе 25,0 мг/кг. Образцы крови (0,2 мл) брали из орбитального синуса перед введением и через 0,5, 1,0, 2,0, 3,0, 4,0, 5,0, 7,0, 9,0, 12,0, 24,0 и 30,0 часов после введения, хранили их в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин. Образцы плазмы хранили при -20ºС до анализа. Крыс кормили через 2 часа после введения.
3. Работа
50 мкл плазмы крыс, взятой в различные моменты времени после введения, смешивали с 50 мкл раствора стандартных серий до получения концентрации в плазме 50,0,100, 200, 500, 1000, 2000, 5000 нг/мл. Добавляли 150 мкл метанола, а затем смесь перемешивали в течение 3 минут, используя вортекс, и центрифугировали в течение 10 минут при 13500 об/мин. 10 мкл супернатанта анализировали с помощью ЖХ-МС/МС. Средние фармакокинетические параметры вычисляли, используя программное обеспечение DAS 2.0.
4. Результаты фармакокинетических параметров
Фармакокинетические параметры соединений по настоящему изобретению показаны ниже.
|
Вывод: соединения по настоящему изобретению обладают хорошим всасыванием в фармакокинетике и очевидно улучшенными фармакокинетическими характеристиками.
Терапевтические эффекты против ксенотрансплантатов рака легкого человека Calu-3 у бестимусных мышей
1. Краткое описание
Оценивали терапевтический эффект соединения Примера 1 против ксенотрансплантатов рака легкого человека Calu-3 у бестимусных мышей. Соединение Примера 1 заметно ингибировал рост рака легкого человека Calu-3, и тестируемые мыши были хорошо толерантными.
2. Цель
Оценивали и сравнивали терапевтические эффекты соединений Примера 1 и Примера 5 против ксенотрансплантатов рака легкого человека Calu-3 у бестимусных мышей.
3. Тестируемые лекарства
Название лекарств и партии лекарств: соединения Примера 1 и Примера 5
Способ приготовления: Соединения Примера 1 и Примера 5 готовили до соответствующей концентрации путем использования дистиллированной воды, содержащей 0,1% Твин-80.
4. Подопытные животные
Бестимусных мышей BALB/cA в возрасте от 6 до 7 недель, $, покупали у фирмы Slaccas Experimental Animal LTD., CO.
Номер сертификата: SCXK 2007 - 0005. Окружающая среда: уровень SPF (свободный от специфичных патогенов).
5. Экспериментальный протокол
Бестимусным мышам инокулировали подкожно клетки рака легкого человека Calu-3. После того, как опухоли вырастали до 150-250 мм3, мышей случайным образом делили на группы (d0).
Объем опухолей и массу мышей измеряли и регистрировали 2-3 раза в неделю.
Формула вычисления объема опухоли (V): V=1/2×a×b2, а: длина опухоли, b: ширина опухоли.
6. Краткое описание
Соединение Примера 1 заметно ингибировало рост рака легкого человека Calu-3. Низкая доза (100 мг/кг) соединения Примера 1 уменьшала объем опухоли на 2/6, высокая доза (200 мг/кг) соединения Примера 1 уменьшала объем опухоли на 1/6 и полностью разрушала другую 1/6 часть опухоли. Низкая доза (100 мг/кг) соединения Примера 5 уменьшала объем опухоли на 3/6, высокая доза (200 мг/кг) соединения Примера 5 уменьшала объем опухоли на 4/6. Кроме того, мыши обладали хорошей толерантностью к соединениям Примера 1 и Примера 5 в соответствии с настоящим протоколом введения.
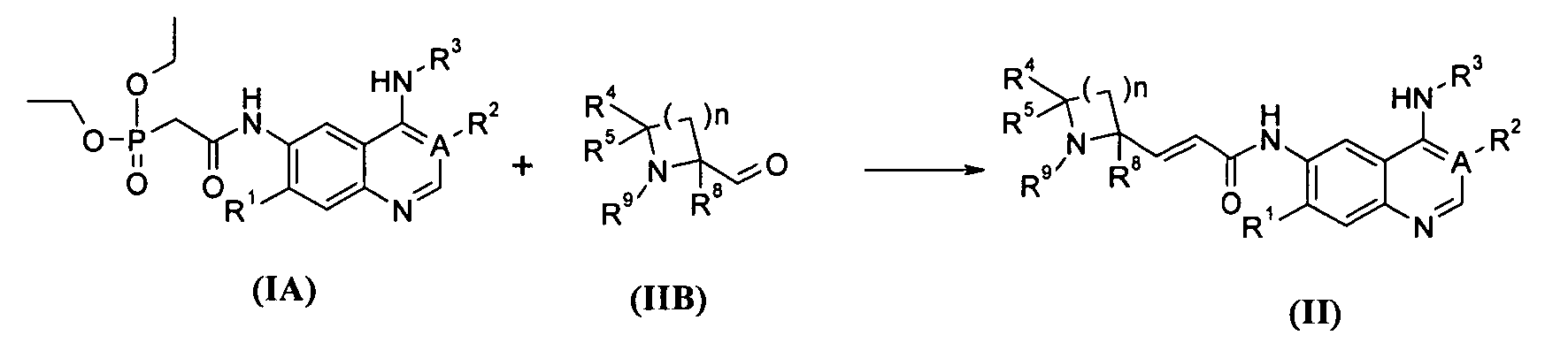
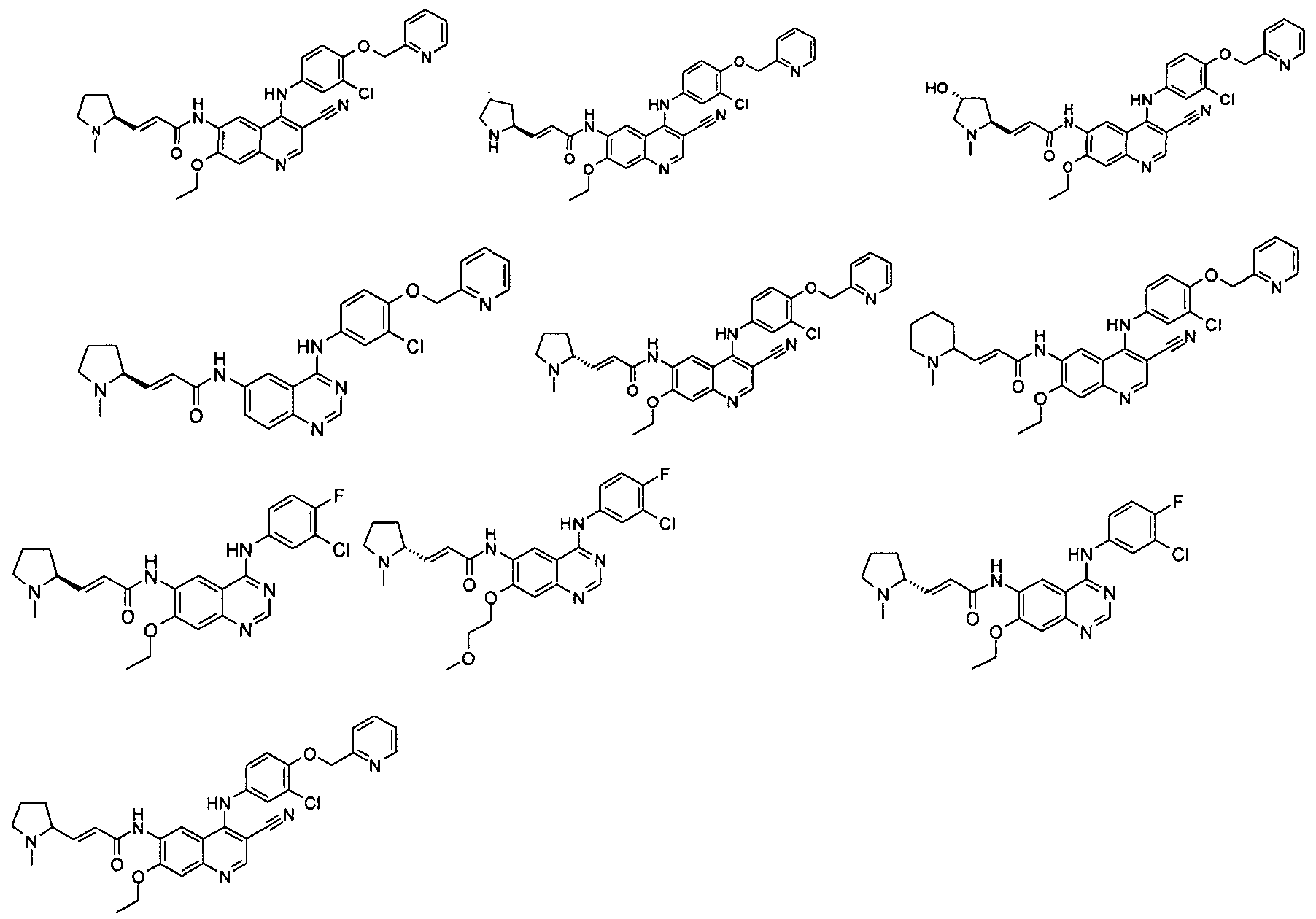
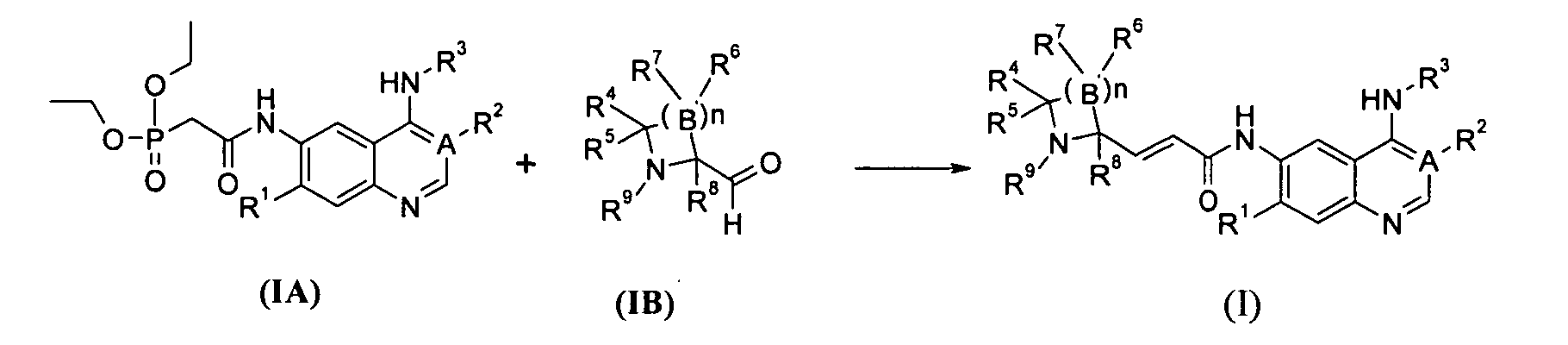
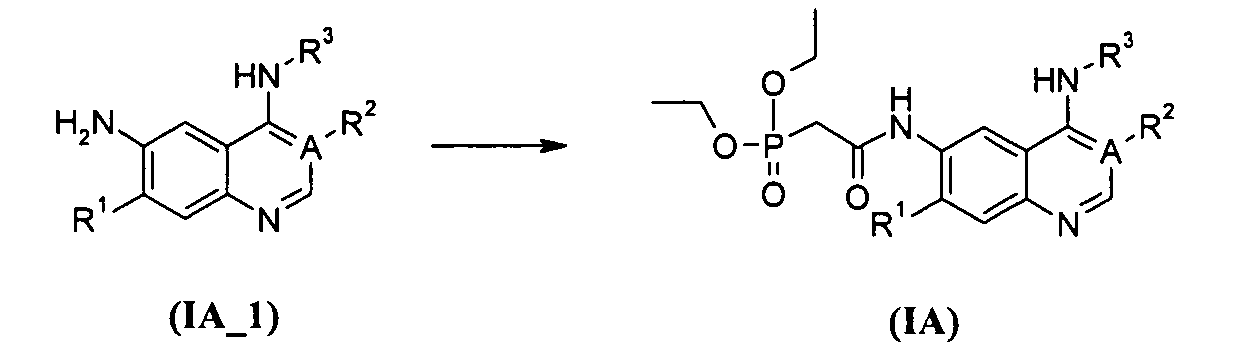
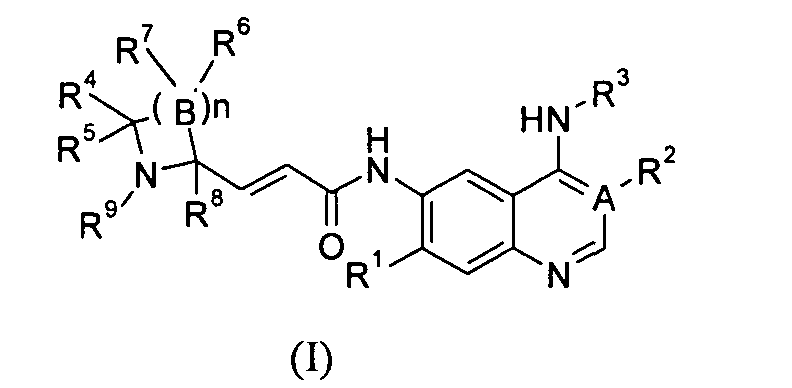
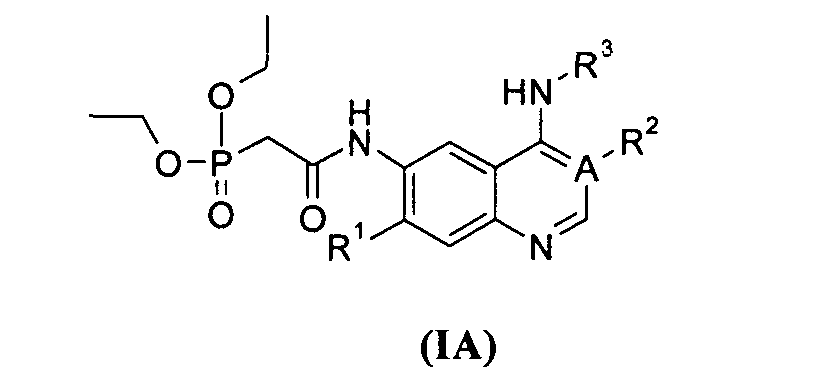
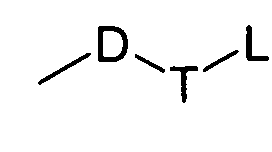
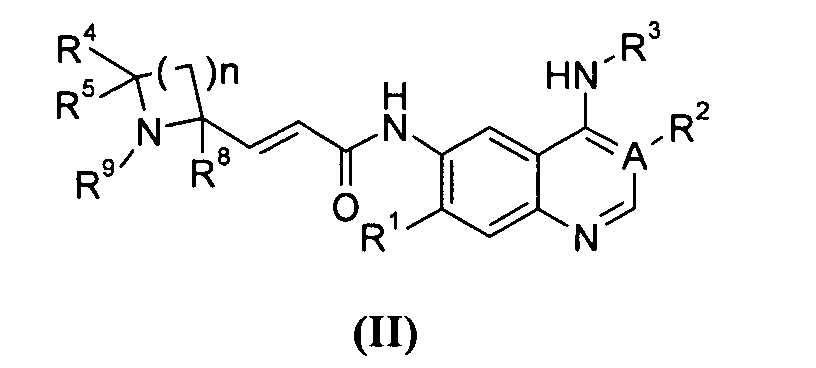
Соединения 2-(2-оксоиндолин-3-илиден)метил-5-(2-гидрокси-3-морфолин-4-илпропил)-6,7 дигидро-1-н-пиррол[3,2-с]пиридин-4(5н)-она и их применение в качестве ингибиторов протеинкиназы
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Производные дициклоазаалкана, способы их получения и их применение в медицине
Бициклозамещенные азопроизводные пиразолона, способ их получения и фармацевтическое применение
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Способ поискового вызова, устройство и система для соты с множеством несущих
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Соли метил(r)-7-[3-амино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-a]пиразин-1-карбоксилата
Соединения 2-(2-оксоиндолин-3-илиден)метил-5-(2-гидрокси-3-морфолин-4-илпропил)-6,7 дигидро-1-н-пиррол[3,2-с]пиридин-4(5н)-она и их применение в качестве ингибиторов протеинкиназы
Гетероциклические азотистые производные пиррола, их получение и фармацевтическое применение
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Производные дициклоазаалкана, способы их получения и их применение в медицине
Бициклозамещенные азопроизводные пиразолона, способ их получения и фармацевтическое применение
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Соли метил(r)-7-[3-амино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-a]пиразин-1-карбоксилата

