Результат интеллектуальной деятельности: СОЕДИНЕНИЯ ДЛЯ ЛЕЧЕНИЯ РАКА
Вид РИД
Изобретение
Область техники, к которой относится изобретение
Настоящее изобретение касается новых соединений, обладающих противораковой активностью, способов получения таких соединений и их применения для лечения рака, лечения лекарственно-устойчивых опухолей, лекарственно-устойчивого рака, метастатического рака, метастатической меланомы, лекарственно-устойчивой меланомы, рака предстательной железы и лекарственно-устойчивого рака предстательной железы.
Уровень техники
Рак является второй по частоте причиной смерти в Соединенных Штатах, уступая только сердечным заболеваниям. В Соединенных Штатах рак является причиной каждой четвертой смерти. Уровень относительной пятилетней выживаемости для всех пациентов, у которых был диагностирован рак, в 1996-2003 годах составлял 66%, повысившись со значения 50%, зафиксированного в 1975-1977 годах (Cancer Facts & Figures American Cancer Society: Atlanta, GA (2008)). Такое повышение выживаемости отражает прогресс в диагностике на ранних стадиях и улучшение в методиках лечения. Разработка высокоэффективных противораковых средств, имеющих низкую токсичность, является приоритетной задачей в исследовании рака.
Микротрубочки представляют собой цитоскелетные волокна, состоящие из гетеродимеров α,β-тубулина, и участвуют в широком наборе клеточных функций, включая поддержание формы, везикулярный транспорт, моторику клеток и деление. Тубулин представляет собой главный структурный компонент микротрубочек и хорошо известную мишень для многих высокоуспешных противораковых лекарственных средств. Соединения, способные нарушать равновесие между микротрубочками и тубулином в клетках эффективны в лечении рака. Противораковые лекарства, способные нарушать равновесие между микротрубочками и тубулином в клетках, такие как таксол и винбластин, широко применяются в химиотерапии рака. Существует три основных класса противоопухолевых средств. Средства, стабилизирующие микротрубочки, которые связываются с полностью сформированными микротрубочками и предотвращают деполимеризацию субъединиц тубулина, представлены таксанами и эпотилонами. Двумя другими классами средств являются средства, дестабилизирующие микротрубочки, которые связываются с димерами тубулина и ингибируют их полимеризацию в микротрубочки. Алкалоиды барвинка, такие как винбластин, связываются с сайтом барвинка и являются представителями одного из таких классов. Колхицин и вещества, связывающиеся с сайтом колхицина, взаимодействуют с другим сайтом тубулина и представляют собой третий класс антимитотических средств.
И таксаны, и алкалоиды барвинка широко применяются для лечения раковых заболеваний у человека, в то время как ни одно из веществ, связывающихся с сайтом колхицина, к настоящему моменту не было одобрено для применения в химиотерапии рака. Однако, средства, связывающиеся с сайтом колхицина, такие как комбретастатин А-4 (СА-4) и АВТ-751 (фиг.19), находятся на стадии клинических испытаний в качестве потенциальных новых средств для химиотерапии (Luo, Y.; Hradil, V.Р.; Frost, D.J.; Rosenberg, S.H.; Gordon, G.В.; Morgan, S.J.; Gagne, G.D.; Cox, B.F.; Tahir, S.K.; Fox, G.В., АВТ-751, "a novel tubulin-binding agent, decreases tumor perfusion и disrupts tumor vasculature". Anticancer Drugs 2009, 20, (6), 483-92.; Mauer, A.M.; Cohen, E.E.; Ma, P.C; Kozloff, M.F.; Schwartzberg, L.; Coates, A.I.; Qian, J.; Hagey, A.E.; Gordon, G.В., "A phase II study of АВТ-751 in patients with advanced non-small cell lung cancer". J Thorac Oncol 2008, 3, (6), 631-6.; Rustin, G.J.; Shreeves, G.; Nathan, P.D.; Gaya, A.; Ganesan, T.S.; Wang, D.; Boxall, J.; Poupard, L.; Chaplin, D.J.; Stratford, M.R.; Balkissoon, J.; Zweifel, M., "A Phase Ib trial of CA4P (combretastatin A-4 phosphate), carboplatin, and paclitaxel in patients with advanced cancer". Br J Cancer 2010, 102, (9), 1355-60.).
К сожалению, противораковые лекарственные средства, взаимодействующие с микротрубочками, при клиническом применении имеют две главные проблемы: резистентность и нейротоксичность. Их эффективность ограничивается обычным механизмом множественной лекарственной резистентности (MDR), а именно выведением лекарства посредством белков-транспортеров АТФ-связывающей кассеты (АВС) (Green, H.; Rosenberg, P.; Soderkvist, P.; Horvath, G.; Peterson, C, "beta-Tubulin mutations in ovarian cancer using single strand conformation analysis-risk of false positive results from paraffin embedded tissues". Cancer letters 2006, 236, (1), 148-54.; Wang, Y.; Cabral, F., "Paclitaxel resistance in cells with reduced beta-tubulin". Biochimica et Biophysica Acta, Molecular Cell Research 2005, 1744, (2), 245-255.; Leslie, E.M.; Deeley, R.G.; Cole, S.P.C, "Multidrug resistance proteins: role of P-glycoprotein, MRP1, MRP2, and BCRP (ABCG2) in tissue defense". Toxicology and Applied Pharmacology 2005, 204, (3), 216-237.).
Р-гликопротеины (Р-gp, кодируются геном MDR1) являются важными представителями суперсемейства АВС. P-gp предотвращают внутриклеточное накопление многих противораковых лекарств путем усиления их выведения из раковых клеток, а также участвуя в их выведении через печень, почки или кишечник. Успех попыток одновременного введения еще и P-gp модуляторов или ингибиторов для усиления клеточной доступности посредством блокировки действия P-gp был очень ограничен (Gottesman, M.M.; Pastan, I., "The multidrug transporter, a double-edged sword". J Biol Chew 1988, 263, (25), 12163-6.; Fisher, G.A.; Sikic, B.I., "Clinical studies with modulators of multidrug resistance". Hematology/oncology clinics of North America 1995, 9, (2), 363-82).
Другой важной проблемой для таксанов, так же как для многих биологически активных природных продуктов, является их липофильность и нерастворимость в водных системах. Это приводит к применению в клинических препаратах эмульгаторов, таких как Cremophor EL и Tween 80. Был описан ряд биологических эффектов, вызываемых применением этих носителей в лекарственных препаратах, включая острые аллергические реакции и периферические нейропатии (Hennenfent, К.L.; Govindan, R., "Novel formulations of taxanes: a review. Old wine in a new bottle?" Ann Oncol 2006, 17, (5), 735-49.; ten Tije, A.J.; Verweij, J.; Loos, W.J.; Sparreboom, A., "Pharmacological effects of formulation vehicles: implications for cancer chemotherapy". Clin Pharmacokinet 2003, 42, (7), 665-85.).
По сравнению с соединениями, связывающимися с сайтами паклитаксела или алкалоидов барвинка, колхицин-связывающиеся средства обычно имеют относительно простую структуру. Тем самым они дают больше возможностей для достижения пероральной биодоступности посредством оптимизации структуры для улучшения растворимости и фармакокинетических параметров. Кроме того, как оказалось, эффективность многих из этих лекарственных средств не страдает от множественной лекарственной резистентности, обеспечиваемой P-gp. По этой причине такие новые соединения, нацеленные на колхицин-связывающие сайты, имеют большой потенциал в качестве терапевтических средств, в особенности благодаря более высокой растворимости в воде и способности избегать множественной лекарственной резистентности, обеспечиваемой P-gp.
Рак предстательной железы является одним из наиболее часто диагностируемых некожных видов рака среди мужчин в США и является второй по частоте причиной смерти от рака. Ожидаемая в этом году заболеваемость составляет свыше 180000, а смертность - почти 29000. Пациенты с поздними стадиями рака предстательной железы проходят антиандрогенную терапию (ADT), обычно с применением агонистов гормона, высвобождающего лютеинизирующий гормон (LHRH), или двусторонней орхиэктомии. Антиандрогенная терапия не только снижает уровень тестостерона, но также понижает уровень эстрогена, поскольку эстроген получается при ароматизации тестостерона, уровень которого снижается при ADT. Дефицит эстрогена, вызванный антиандрогенной терапией, вызывает заметные побочные эффекты, которые включают приливы, гинекомастию и масталгию, разрежение костей, снижение качества и прочности костной ткани, остеопороз и угрожающие жизни трещины, неблагоприятные изменения липидов, повышенный риск сердечно-сосудистых заболеваний и инфаркта миокарда, депрессию и другие изменения настроения. Считается, что многие из побочных эффектов от дефицита эстрогена при ADT вызываются ERα.
Лейпролид ацетат (Lupron®) представляет собой синтетический нонапептидный аналог природного гонадотропин-рилизинг гормона (GnRH или LH-RH). Лейпролид ацетат представляет собой суперагонист LH-RH, который в конечном счете подавляет выработку LH в гипофизе. Лейпролид ацетат работает как мощный ингибитор секреции гонадотропина, подавляя стероидогенез в яичниках и яичках. У человека введение лейпролида ацетата приводит к повышению уровня в крови лютеинизирующего гормона (LH) и фолликулостимулирующего гормона (FSH), в результате временно повышается уровень гонадных стероидов (тестостерона и дигидротестостерона у мужчин, и эстрона и эстрадиола у женщин предклимактерического возраста). Однако продолжительный прием лейпролида ацетата приводит к понижению уровня LH и FSH. У мужчин уровень тестостерона понижается до уровня кастратов (ниже 50 нг/дл). У женщин предклимактерического возраста уровень эстрогена понижается до уровня, характерного для постклимактерического возраста. Тестостерон является известным стимулом для раковых клеток в предстательной железе. Подавление секреции тестостерона или ингибирование действия тестостерона является, таким образом, необходимым компонентом терапии рака предстательной железы. Лейпролид ацетат может применяться для подавления LH, и, как следствие, снижения уровня тестостерона в плазме крови до уровня кастратов с целью лечения рака предстательной железы.
Злокачественная меланома представляет собой наиболее опасную форму рака кожи, на долю которой приходится около 75% смертей от рака кожи. Заболеваемость меланомой в Западной популяции постепенно повышается. Число заболевших удвоилось за последние 20 лет. Около 160000 новых случаев меланомы диагностируется в мире ежегодно, и она чаще встречается у мужчин и представителей европеоидной расы. Согласно отчету ВОЗ, в год в мире фиксируется около 48000 смертей от меланомы.
В настоящее время не существует эффективного метода лечения метастатической меланомы. Она крайне устойчива к известным в настоящее время методам химиотерапии, радиотерапии и иммунотерапии. Метастатическая меланома характеризуется очень плохим прогнозом, при этом медиана выживаемости составляет 6 месяцев, а уровень выживаемости в течение 5 лет составляет менее 5%. За последние 30 лет единственным одобренным FDA средством лечения метастатической меланомы является дакарбазин (DTIC). Однако он обеспечивает полную ремиссию всего лишь для менее 5% пациентов. В последние годы предпринимались большие усилия для борьбы с метастатической меланомой. Ни комбинирование DTIC с другими средствами химиотерапии (например, цисплатином, винбластином и кармустином), ни добавление интерферона-α2b к DTIC не дали повышения выживаемости по сравнению с лечением только DTIC. Совсем недавние клинические испытания с использованием антител и вакцин для лечения метастатической меланомы также не показали удовлетворительной эффективности.
Клетки меланомы имеют низкий уровень спонтанного апоптоза in vivo no сравнению с опухолевыми клетками других типов, и они относительно устойчивы к индуцируемому лекарствами апоптозу in vitro. Природная роль меланоцитов состоит в защите внутренних органов от УФ-излучения, потенциально способного повредить ДНК. Поэтому неудивительно, что клетки меланомы могут иметь специальные системы починки повреждений ДНК и повышенные характеристики выживаемости. Кроме того, недавние исследования показали, что при развитии меланома приобретает комплексные генетические изменения, приводящие к гиперактивации выкачивающих насосов, ферментов детоксикации и мультифакторному изменению путей выживания и апоптоза. Предполагается, что все вышеперечисленное обуславливает множественный лекарственно-устойчивый фенотип меланомы. Принимая во внимание рост случаев заболевания данной болезнью и высокую устойчивость к существующим терапевтическим средствам, разработка более эффективных лекарств для лечения распространенной меланомы и других типов рака, способных преодолевать множественную лекарственную резистентность, обеспечит значительные преимущества пациентам, страдающим от рака.
Краткое описание изобретения
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы (Ia):
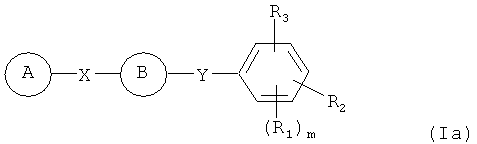 ,
,
где
А представляет собой замещенную или незамещенную моноциклическую, конденсированную или полициклическую арильную или (гетеро)циклическую систему; замещенный или незамещенный, насыщенный или ненасыщенный N-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный S-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный O-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный циклический углеводород; или замещенный или незамещенный, насыщенный или ненасыщенный смешанный гетероцикл;
В представляет собой
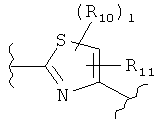 (тиазол),
(тиазол), 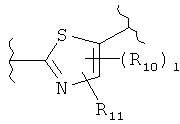 (тиазол),
(тиазол),  (тиазолидин),
(тиазолидин),
 (оксазол),
(оксазол),  (оксазолин),
(оксазолин),  (оксазолидин),
(оксазолидин),  (бензол),
(бензол),
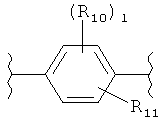 (бензол),
(бензол), 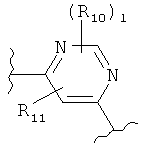 (пиримидин),
(пиримидин), 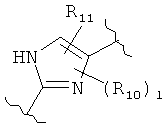 (имидазол),
(имидазол),
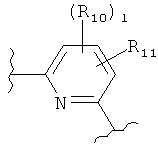 (пиридин),
(пиридин), 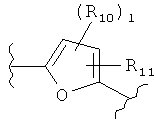 (фуран),
(фуран), 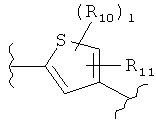 (тиофен),
(тиофен), 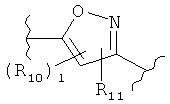 (изоксазол),
(изоксазол),
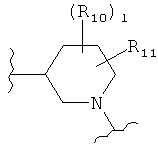 ,
,  (пиперидин),
(пиперидин),  (пиразол),
(пиразол),  (индол), или
(индол), или
 (изохинолин);
(изохинолин);
R1, R2 и R3 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Х представляет собой связь, NH, C1-C5 углеводород, О или S;
Y представляет собой связь, -С=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN; -C=N-CN, -СН=СН-, -С=С(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(С=О)-О, -O-(С=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
где указанные циклы А и В необязательно замещены 1-5 заместителями, которые независимо представляют собой O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2;
m представляет собой целое число от 1 до 3; и
где если В представляет собой бензольное кольцо, тиофеновый цикл, фурановый цикл или индольный цикл, то Х не является связью или СН2, и А не является индолом;
если В представляет собой индол, то Х не является О; и
если В представляет собой тиазольный цикл, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы II:
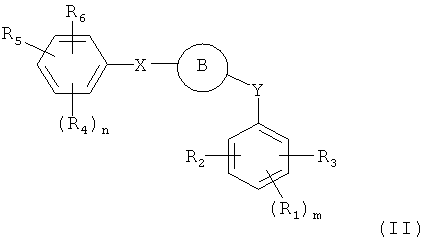 ,
,
где
В представляет собой
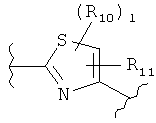 (тиазол),
(тиазол), 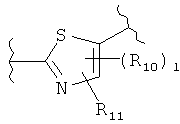 (тиазол),
(тиазол), 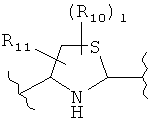 (тиазолидин),
(тиазолидин),
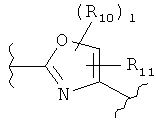 (оксазол),
(оксазол), 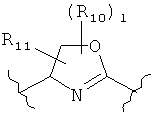 (оксазолин),
(оксазолин), 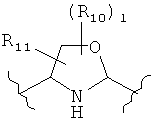 (оксазолидин),
(оксазолидин), 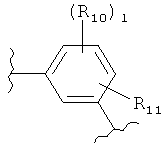 (бензол),
(бензол),
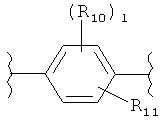 (бензол),
(бензол), 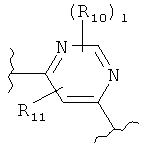 (пиримидин),
(пиримидин), 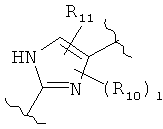 (имидазол),
(имидазол),
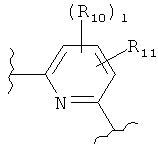 (пиридин),
(пиридин), 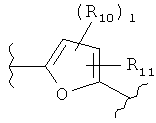 (фуран),
(фуран), 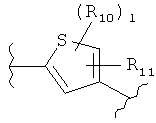 (тиофен),
(тиофен), 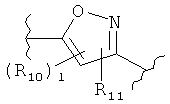 (изоксазол),
(изоксазол),
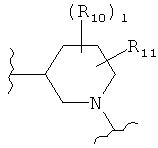 ,
, 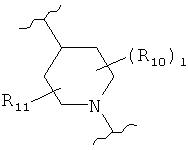 (пиперидин),
(пиперидин), 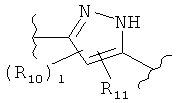 (пиразол),
(пиразол), 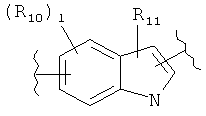 (индол), или
(индол), или
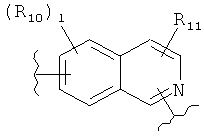 (изохинолин);
(изохинолин);
R1, R2, R3, R4, R5 и R6 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Х представляет собой связь, NH, С1-С5 углеводород, О или S;
Y представляет собой связь, -С=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, -C=N-CN, -CH=CH-, С=СН(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2;
n представляет собой целое число от 1 до 3; и
m представляет собой целое число от 1 до 3;
где если В представляет собой индол, то Х не является О; и
если В представляет собой тиазольный цикл, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы (V):
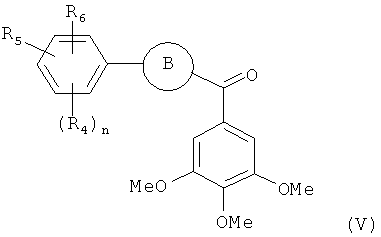
где
В представляет собой
(тиазол), (тиазолидин), (оксазолидин),
(бензол), (бензол), (пиримидин), (пиридин),
(фуран), (тиофен), (изоксазол), ,
(пиперидин), (пиразол), (индол), или
(изохинолин);
R4, R5 и R6 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2; и
n представляет собой целое число от 1 до 3;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы (XI):
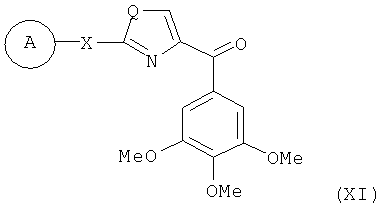 ,
,
где
Х представляет собой связь, NH или S;
Q представляет собой О, NH или S;
А представляет собой замещенную или незамещенную моноциклическую, конденсированную или полициклическую арильную или (гетеро)циклическую систему; замещенный или незамещенный, насыщенный или ненасыщенный N-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный S-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный O-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный циклический углеводород; или замещенный или незамещенный, насыщенный или ненасыщенный смешанный гетероцикл; где указанный цикл А необязательно замещен 1-5 заместителями, которые независимо представляют собой O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2; и
i представляет собой целое число от 0 до 5;
где если Q представляет собой S, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы (VIII):
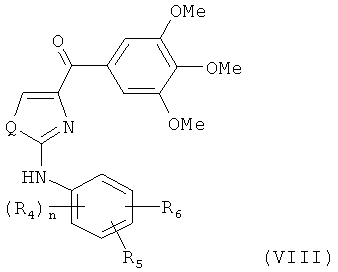 ,
,
где
R4, R5 и R6 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, С1-С5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Q представляет собой S, О или NH;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 3;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы [XI(b)]:
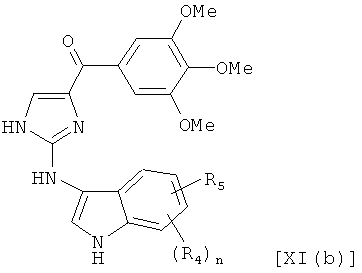 ,
,
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, С1-С5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы [XI(с)]:
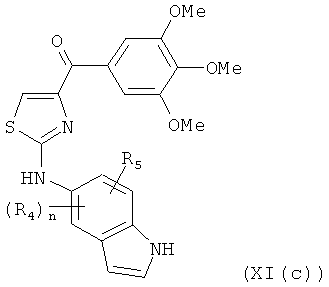 ,
,
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, С1-С5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы [XI(е)]:
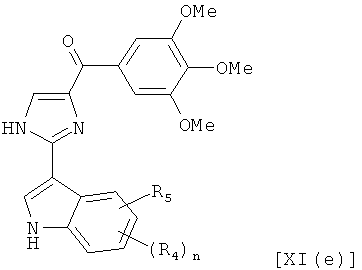 ,
,
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы (XVI):
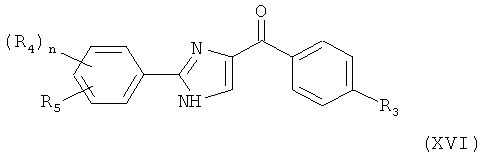 ,
,
где R4 и R5 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкил, COOH, С(O)O-алкил или С(O)Н;
R3 представляет собой I, Br, Cl, F;
i представляет собой целое число от 0 до 5; и
n равен 1-4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы IX:
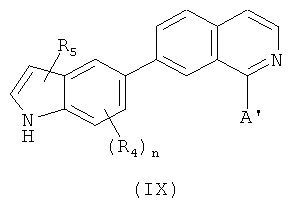 ,
,
R4 и R5 независимо выбраны из атома водорода, O-алкила, O-галогеналкила, F, Cl, Br, I, галогеналкила, CF3, CN, -CH2CN, NH2, гидроксила, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейного или разветвленного алкила, галогеналкила, алкиламино-группы, аминоалкила, -OCH2Ph, -NHCO-алкила, COOH, -C(O)Ph, C(O)O-алкила, С(O)Н, -(O)NH2 или NO2;
А' представляет собой замещенную или незамещенную моноциклическую, конденсированную или полициклическую арильную или (гетеро)циклическую систему, включая насыщенные и ненасыщенные N-гетероциклы, насыщенные и ненасыщенные S-гетероциклы, и насыщенные и ненасыщенные O-гетероциклы, насыщенный или ненасыщенный циклический углеводород, насыщенный или ненасыщенный смешанный гетероцикл или алифатический линейный или разветвленный C1-С30 углеводород; где указанный цикл А необязательно замещен 1-5 одинаковыми или разными заместителями, включая O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 3;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой соединения (55):
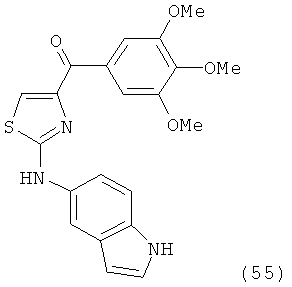 .
.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой соединения (17ya):
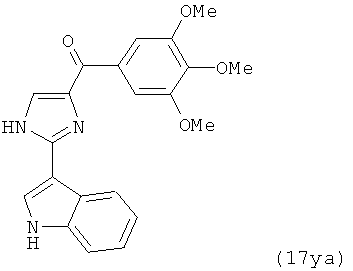 .
.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой соединения 12da:
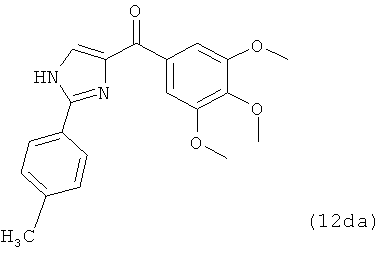 .
.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой соединения (12fa):
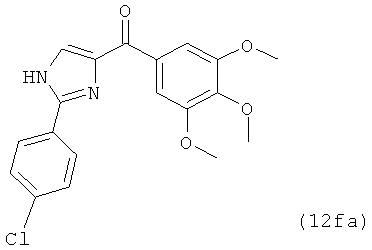 .
.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой соединения (12cb):
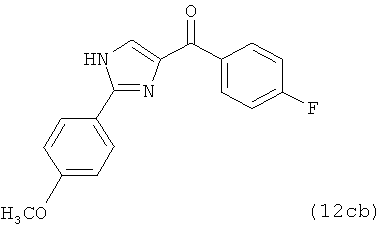 .
.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой соединения (12fb):
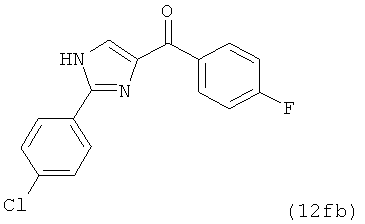 .
.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой соединения (6b):
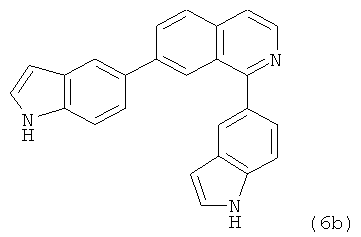 .
.
В одном варианте осуществления настоящее изобретение касается фармацевтической композиции, содержащей соединение по настоящему изобретению и фармацевтически приемлемый носитель.
В одном варианте осуществления настоящее изобретение касается способа (а) лечения, подавления, уменьшения тяжести, снижения риска, или ингибирования рака; (b) лечения лекарственно-устойчивой опухоли или опухолей; и (с) разрушения раковой клетки, включающего введение соединения по настоящему изобретению. В другом варианте осуществления рак выбран из группы, состоящей из рака предстательной железы, рака груди, рака яичника, рака кожи, меланомы, рака легких, рака толстой кишки, лейкемии, рака почки, рака ЦНС и их комбинаций.
Краткое описание чертежей
Объект настоящего изобретения предметно описан и однозначно сформулирован в завершающей части настоящего описания. Однако настоящее изобретение, в плане организации и способа осуществления, вместе с задачами, отличительными особенностями и преимуществами, наилучшим образом можно понять из приведенного далее подробного описания в совокупности с прилагающимися чертежами, в которых:
На фиг.1 изображен синтез различных В-циклических промежуточных соединений на основе оксазола. Реагенты и условия: (а) МеОН, CH3COCl, 83%; (b) Этиловый эфир бензимидной кислоты, CH2Cl2, Et3N, 96%; (с) LiOH, МеОН, H2O, 65%; (d) EDCI, HOBt, NMM, CH3OCH3NH·HCl, 61%; (e) бромид 3,4,5-триметоксифенилмагния, ТГФ, 48%-71%; (f) CBrCl3, DBU, CH2Cl2, 56%.
На фиг.2 изображен синтез различных В-циклических промежуточных соединений. Реагенты и условия: (a) EDCI, HOBt, NMM, CH3OCH3NH·HCl, CH2Cl2, 51-95%; (b) бромид 3,4,5-триметоксифенилмагния, ТГФ, 48-78%; (с) LAH, -78°С, ТГФ, 85%; (d) Реагент Десс-Мартина, CH2Cl2, 81%; (e) EDCI, HOBt, NMM, 3,4,5-триметоксибензойная кислота, CH2Cl2, 58%.
На фиг.3 изображена схема синтеза соединений по настоящему изобретению. Реагенты и условия: (а) МеОН/рН=6.4 фосфатный буфер, комнатная температура; (b) EDCI, HOBt, NMM, HNCH3OCH3; (с) CBrCl3, DBU, CH2Cl2; (d) бромид 3,4,5-триметоксифенилмагния, ТГФ; (е) иодид изопропилтрифенилфосфония, н-BuLi, ТГФ; (f) LAH, ТГФ; (g) Для 2е-цис и 2е-транс, NH2OH·HCl, C2H5OH, H2O, NaOH; Для 2g и 2h, NH2OMe-HCl, пиридин; (h) TsCl, NaH, основный Al2O3; (i) NH2NH2•xH2O, CH2Cl2, i-BuOH; (j) диэтил цианометилфосфонат, н-BuLi, ТГФ; (k) бис-триметилсилилкарбодиимид, TiCl4, CH2Cl2; (l) EDCI, HOBt, Et3N, 3,4,5-триметоксианилин, CH2Cl2.
На фиг.4 изображена схема синтеза соединений по настоящему изобретению. Реагенты и условия: (а) бром, EtOH; (b) бензотиоамид, EtOH, кипячение; (с) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (d) CBrCl3, DBU, CH2Cl2; (е) LAH, ТГФ; (f) 5-(бромметил)-1,2,3-триметоксибензол, Ph3P, ТГФ; (g) н-BuLi, ТГФ; (h) (l) HCl, H2O; (2) NaNO2, H2O, 0°С; (i) ксантат этил калия; (j) KOH/EtOH; (k) H2O, HCl; (l) 5-иод-1,2,3-триметоксибензол, CuI, t-BuONa; (m) 2 экв или 1 экв м-СРВА, CH2Cl2; (n) 3,4,5-триметоксианилин, NEt3, ДМФА.
На фиг.5 изображена схема синтеза соединений по настоящему изобретению. Реагенты и условия: (а) L-цистеин, EtOH, 65°С; (b) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (с) TBDMSCl, имидазол, ТГФ; (d) 3,4,5-триметоксифенилбромид, BuLi, ТГФ; (е) TBAF, ТГФ; (f) SOCl2, Et2O; (g) NH3, МеОН; (h) POCl3; (i) PhSO2Cl, Bu4NHSO4, толуол, 50% NaOH; (j) 1 н. NaOH, EtOH, кипячение; (k) Boc2O, 1 н. NaOH, 1,4-диоксан; (l) CBrCl3, DBU, CH2Cl2; (m) 4 н. HCl в 1,4-диоксане; (n) NaH, ДМФА, MeI; (о) НСНО, NaBH3CN, Et3N.
На фиг.6 изображена схема синтеза соединений по настоящему изобретению. Реагенты и условия: (а) EtOH, 65°С; (b) NaOH, C2H5OH, кипячение; (с) EDCI, HOBt, NMM, HNCH3OCH3, CH2Cl2; (d) 3,4,5-триметоксифенилбромид, BuLi, ТГФ; (е) 2 н. HCl в 1,4-диоксане.
На фиг.7 изображена схема синтеза Арил-Бензоил-Имидазольных (ABI) соединений по настоящему изобретению. Реагенты и условия: (a) t-BuOH, I2, этилендиамин, К2СО3, кипячение; (b) PhI(ОАс)2, К2СО3, ДМСО; (с) DBU, CBrCl3, ДМФА; (d) NaH, PhSO2Cl, ТГФ, 0°С - комнатная температура; (е) t-BuLi, замещенный бензоилхлорид, ТГФ, -78°С; (f) Bu4NF, ТГФ, комнатная температура.
На фиг.8 изображена схема синтеза Арил-Бензоил-Имидазольных (ABI) соединений по настоящему изобретению. Реагенты и условия: (a) NH4OH, щавелевый альдегид, этанол, комнатная температура; (b) NaH, PhSO2Cl, ТГФ, 0°С - комнатная температура; (с) t-BuLi, замещенный бензоилхлорид, ТГФ, -78°С; (d) Bu4NF, ТГФ, комнатная температура; (е) BBr3, CH2Cl2; (f) конц-HCl, АсОН, кипячение.
На фиг.9 изображена схема синтеза Арил-Бензоил-Имидазольных (ABI) соединений по настоящему изобретению. Реагенты и условия: (a) NaH, замещенный бензоилхлорид, ТГФ.
На фиг.10 изображена схема синтеза соединений 12dc, 12fc, 12daa, 12dab, 12cba. (а) AlCl3, ТГФ, кипячение; (b) NaH, CH3I для 12dab и 12cba и BnBr для 12daa, ТГФ, кипячение.
На фиг.11 изображена схема синтеза соединений 11gaa, 12la. (a) NH4OH, этанол, глиоксаль, комнатная температура; (b) NaH, замещенный PhSO2Cl, ТГФ, 0°С - комнатная температура; (с) t-BuLi (1.7 М в пентане), замещенный бензоилхлорид, ТГФ, -78°С; (d) Bu4NF, комнатная температура.
На фиг.12 изображена схема синтеза соединений 15хаа и 12ха. (а) 1. КОН, этанол; 2. PhSO2Cl, ацетон; (b) NH4OH, глиоксаль, этанол, комнатная температура; (с) NaH, PhSO2Cl, ТГФ, 0°С - комнатная температура; (d) t-BuLi (1.7 М в пентане), бензоилхлорид, ТГФ, -78°С; (е) NaOH, этанол, Н2О, кипячение.
На фиг.13 изображена схема синтеза соединения 17ya. (а) 1. КОН, этанол, 2. PhSO2Cl, ацетон, комнатная температура; (b) NH4OH, глиоксаль, этанол, комнатная температура; (с) NaH, PhSO2Cl, ТГФ, 0°С - комнатная температура; (d) t-BuLi (1.7 М в пентане), бензоилхлорид, ТГФ, -78°С; (е) NaOH, этанол, H2O, кипячение.
На фиг.14 изображена схема синтеза соединения 12fa. (a) NH4OH, щавелевый альдегид, этанол, комнатная температура; (b) NaH, PhSO2Cl, ТГФ, 0°С - комнатная температура; (с) t-BuLi, 3,4,5-триметоксибензоилхлорид, ТГФ, -78°С; (d) Bu4NF, ТГФ, комнатная температура.
На фиг.15 изображена схема синтеза соединения 55.
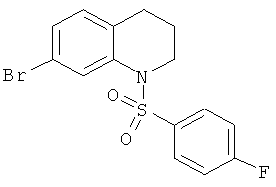
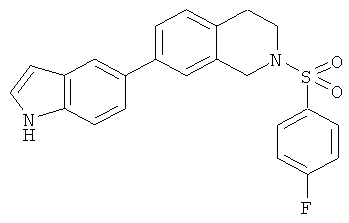
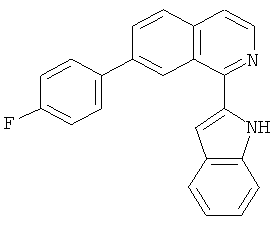
Фиг.16. Схема синтеза соединений на основе изохинолина и хинолина. На фиг.16А изображена схема синтеза производных изохинолина. Реагенты и условия: а) арилбороновая кислота (1 экв.), Pd(PPh3)4 (0.01 экв.), К2СО3, H2O, ДМФА, 5 ч; b) арилбороновая кислота (2.4 экв.), Pd(PPh3)4 (0.04 экв.), К2СО3, H2O, ДМФА, 16 ч; с) арилбороновая кислота (1.2 экв.), Pd(PPh3)4 (0.04 экв.), К2СО3, H2O, ДМФА, 16 ч. На фиг.16В изображена схема синтеза соединений 41 и 44. Реагенты и условия: а) п-фторбензолсульфонил хлорид, пиридин, пиридин, 80°С, 3 ч; b) 5-индолбороновая кислота (1.2 экв.), Pd(PPh3)4 (0.02 экв.), К2СО3, H2O, ДМФА, 16 ч. На фиг.16С изображена схема синтеза изохинолинового производного 6d. На фиг.16D изображена схема синтеза изохинолинового производного 6с. На фиг.16Е изображена схема синтеза изохинолинового производного 6b.
На фиг.17 изображена стандартная кривая растворимости для ABI соединения 12ga (растворено в ацетонитриле). По оси x приведено количество соединения, а по оси y площадь пика m/z.
На фиг.18 приведена измеренная растворимость в воде для анти-тубулиновых соединений 1h, 1с, 66а, 2r-HCl, 5а и 5с.
На фиг.19 приведены структуры ингибиторов тубулина, связывающихся по сайту колхицина.
На фиг.20 отражена способность анти-тубулиновых соединений 1h, 1c, 2j, 66а и 5а ингибировать полимеризацию тубулина in vitro.
На фиг.21 изображены кривые зависимости доза-ответ для 2-арил-4-бензоил-имидазольных соединений (ABI) в сравнении с другими противораковыми лекарствами и соединениями на линии клеток множественно-лекарственно-резистентной меланомы (MDR клетки) и соответствующей чувствительной материнской линии клеток (нормальные клетки меланомы). Большое расстояние между двумя кривыми для паклитаксела, винбластина и колхицина указывает, что они являлись субстратами для Р-гликопротеина (P-gp). Перекрывание двух кривых каждого ABI соединения указывает, что данные ABI соединения не являлись субстратами для P-gp и преодолевали множественную лекарственную резистентность.
На фиг.22 представлено влияние ABI соединений на полимеризацию тубулина in vitro. Тубулин (0.4 мг/анализ) подвергали воздействию 10 мкМ ABI соединения (контрольный образец с носителем, 5% ДМСО). Регистрировали поглощение при 340 нм при 37°С каждую минуту в течение 15 минут и показывали, что ABI соединения 12da, 12cb и 12db ингибировали полимеризацию тубулина in vitro.
На фиг.23 представлены данные анализа формирования колоний B16-F1 меланомы в мягком агаре, показывающие, что ABI соединения ингибировали формирование колоний зависимым от концентрации образом. На фиг.23А приведена репрезентативная картина контрольного образца и каждого протестированного соединения (12cb, 12da и 12fb) при 100 нМ. Диаметр каждой лунки составлял 35 мм. На фиг.23В приведены количественные результаты анализа для каждого протестированных соединений (12cb, 12da и 12fb). Значение Р вычисляли сравнением с контрольным образцом с использованием t критерия Стьюдента и программы GraphPad Prism. Колонки, среднее из трех повторов; столбики, SD.
На фиг.24 приведены результаты in vivo исследования ABI соединений. На фиг.24А приведены данные по in vivo активности соединения 12cb в отношении опухолей B16-F1 меланомы у C57/BL мышей. На фиг.24В приведены данные по in vivo активности соединения 12fb в отношении B16-F1 меланомы у C57/BL/6 мышей и SHO безтимусных (голых) мышей. Результаты показали, что 12fb ингибирует рост опухоли меланомы дозо-зависимым образом. C57BL/6 мыши с аллографтами B16-F1 меланомы (n=5 в группе). Каждая мышь получала 0.5×106 клеток подкожной инъекцией в бок. Ежедневное введение 30 мкл внутрибрюшинно начинали, когда размер опухоли достигал ~100 мм3. На фиг.24С приведены данные по in vivo активности 12fb в отношении ксенотрансплантата А375 меланомы человека. SHO безтимусные мыши несли ксенотрансплантаты А375 меланомы человека (n=5 в группе). Каждая мышь получала 2.5×106 клеток подкожной инъекцией в бок. Ежедневное введение 30 мкл внутрибрюшинно начинали, когда размер опухоли достигал ~150 мм3. Контроль, раствор чистого носителя; точки, средние; гистограмма, SD. DTIC, (5-(3,3,-диметил-1-триазенил)-имидазол-4-карбоксамид, дакарбазин.
На фиг.25 приведены результаты исследования конкурентного связывания колхицина. На фиг.25А приведены результаты сцинтилляционного анализа сближения при конкурентном связывании 3H-меченого колхицина, которые показывают, что 12cb конкурентно связывается с сайтом связывания колхицина в тубулине. На фиг.25В изображены репрезентативные диаграммы анализа клеточного цикла с использованием поточной цитометрии, показывающие, что ABI соединения (показаны примеры для 12da и 12fb) останавливают А375 клетки в фазе G2/M после 24 часов инкубирования. Действие и активность близки к колхицину. На фиг.25С графически показаны количественные результаты анализа клеточного цикла. Все протестированные соединения (показаны примеры для 12cb, 12da и 12fb) останавливают А375 клетки в фазе G2/M дозо-зависимым образом. ABI 12da показало более высокую активность, чем колхицин. На фиг.25D показаны результаты анализа клеточного цикла с использованием поточной цитометрии А375 клеток после инкубирования с 12cb, 12da и 12fb в различных концентрациях в течение 24 часов. Колхицин останавливал большинство клеток в фазе G2/M начиная с 50 нМ. 12cb, 12da и 12fb также останавливали большинство клеток в фазе G2/M начиная с 200, 50 и 200 нМ, соответственно
На фиг.26 показано влияние 17ya (верх) и 55 (низ) на полимеризацию тубулина. Тубулин (0.4 мг) обрабатывали испытуемыми соединениями (1 и 5 мкМ). Поглощение при 340 нм отслеживали каждую минуту в течение 15 минут. В качестве положительного контроля применяли 5 мкМ колхицин.
На фиг.27 показано ингибирование опухоли соединением 17ya на ксенографической модели таксол-устойчивого рака предстательной железы (PC-3_TxR) (верх). Животные продолжали набирать вес (низ), несмотря на уменьшение опухоли, демонстрируя отсутствие токсичности у 17ya.
На фиг.28 показано, что соединения 1h, 2k и 2l ингибируют полимеризацию тубулина посредством связывания с сайтом связывания колхицина в тубулине. (А) Структуры 1h (-H), 2k (-F) и 2l (-ОН). (B) Влияние соединений на полимеризацию тубулина. Тубулин (0.4 мг) обрабатывали соединениями 1h, 2k и 2l (10 мкМ). Поглощение при 340 нм отслеживали каждую минуту в течение 15 минут. (С) Способность 1h конкурировать за сайты связывания колхицина, винбластина и паклитаксела в тубулине, с применением масс-спектрального анализа конкурентного связывания (n=3); гистограмма, SD.
На фиг.29 показано, что соединения 1h, 2k и 2l останавливают клетки в фазе G2/M и индуцируют апоптоз. (А) Репрезентативные диаграммы анализа клеточного цикла после обработки соединениями в течение 24 часов на клетках РС-3 и A375. (В) Изменения доли G2/M, вызванные соединениями 1h, 2k и 2l в клетках PC-3 и А375 после 24-часовой обработки. (С) Способность 1h, 2k и 2l усиливать формирование комплекса ДНК-гистон за 24 часа (n=3); гистограмма, SD. В качестве положительного контроля применяли колхицин и винбластин.
На фиг.30 представлены результаты исследования фармакокинетики соединений 1h, 2k и 2l при внутрибрюшинном введении мышам и крысам. (А) Кривая зависимости концентрации от времени для SMART соединений (замещенные метоксибензоил-арил-тиазольные соединения, аббревиатура от Substituted Methoxybenzoyl Aryl Thiazole) в ICR мышах (n=3); гистограмма, SD. SMART соединения вводили внутривенной инъекцией в хвостовую вену в дозировке 15 мг/кг. (B) Кривая зависимости концентрации от времени для соединений 1h и 2k в SD крысах (n=4); гистограмма, SD. Spague-Dawley крысам вводили 2.5 мг/кг внутривенно в виде готовой формы в ДМСО/PEG300 (1/4).
На фиг.31 показана in vivo противораковая эффективность (введение внутрибрюшинно) и нейротоксичность SMART соединений в мышах. (А) Эффективность SMART соединений в отношении ксенографтов PC-3 опухоли предстательной железы на безтимусных мышах (n=6-8). (B) Эффективность винбластина в отношении ксенографтов PC-3 опухоли предстательной железы на безтимусных мышах (n=8). Применялось в качестве положительного контроля. (C) in vivo эффективность 1h и 2k в отношении безтимусных мышей с ксенографтами А375 меланомы (n=10). Безтимусным мышам инокулировали 2.5×106 клеток PC-3 или А375 и проводили внутрибрюшинное введение ежедневно (SMART соединения) и один раз в 2 дня (винбластин) после формирования опухоли (150-200 мм3). Каждая точка соответствует среднему объему опухоли для животных в каждой группе. (D) in vivo нейротоксичность (испытание на вращающемся стержне) 1h в ICR мышах (n=7 или 8). 1h (5 и 15 мг/кг), винбластин (0.5 мг/кг) и носитель ежедневно вводили внутрибрюшинно, и винбластин использовался в качестве положительного контроля. Введение останавливали на 31-й день. *,p<0.05. Гистограмма, SE.
На фиг.32 представлено молекулярное моделирование ABI соединений, мишенью которых является сайт связывания колхицина в тубулине. На фиг.32А и 32В представлены молекулярные модели соединений 12cb и 11cb, соответственно.
На фиг.33 представлены микроскопические изображения иммуно-флуоресцентно меченых микротрубочек в клетках меланомы WM-164, которые показывают, что расположение микротрубочек значительно изменилось после обработки соединениями в течение 18 часов. Это служит визуальным доказательством того, что ABI соединения взаимодействуют с тубулином и нарушают формирование функциональных микротрубочек.
На фиг.34 показана эффективность и переносимость 6b и 6с на ксенографических моделях после введения внутрибрюшинной инъекцией. А. РС-3 ксенографты обрабатывали носителем (раз в день), 6b (40 мг/кг, раз в день) или 6с (40 мг/кг, раз в день) в течение 3 недель. Носитель для введения представлял собой 20% Captex200 в Tween80. Значения объема опухоли (мм3) откладывали на графике относительно времени и представляли собой средние значения ±SD для восьми животных. Объем опухоли представлен на левом графике, а вес тела - на правом графике. В. Размер печени (г) каждой безтимусной мыши измеряли после 3 недель дозирования. С. Число белых кровяных телец подсчитывали в цельной крови, взятой у животных после 3 недель дозирования.
Следует понимать, что для простоты и ясности представления элементы на фигурах необязательно изображены в правильном масштабе. Например, размеры некоторых элементов для ясности могут быть увеличены относительно других элементов. Кроме того, там, где это уместно, цифровые обозначения на фигурах могут повторяться для обозначения соответствующих или аналогичных элементов.
Подробное описание изобретения
В одном варианте осуществления настоящее изобретение касается соединения формулы (I)
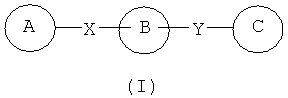 ,
,
где
А и С каждый независимо представляют собой замещенные или незамещенные моноциклические, конденсированные или полициклические арильные или (гетеро)циклические системы; замещенные или незамещенные, насыщенные или ненасыщенные N-гетероциклы; замещенные или незамещенные, насыщенные или ненасыщенные S-гетероциклы; замещенные или незамещенные, насыщенные или ненасыщенные O-гетероциклы; замещенные или незамещенные, насыщенные или ненасыщенные циклические углеводороды; или замещенные или незамещенные, насыщенные или ненасыщенные смешанные гетероциклы;
В представляет собой
(тиазол), (тиазол), (тиазолидин),
(оксазол), (оксазолин), (оксазолидин), (бензол),
(бензол), (пиримидин), (имидазол),
(пиридин), (фуран), (тиофен), (изоксазол),
, (пиперидин), (пиразол), (индол), или
(изохинолин);
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Х представляет собой связь, NH, C1-C5 углеводород, О или S;
Y представляет собой связь, -С=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, C=N-CN, -CH=CH-, -С=С(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
где указанные циклы А и С необязательно замещены 1-5 заместителями, которые независимо представляют собой O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2;
где если В представляет собой бензольное кольцо, тиофеновый цикл, фурановый цикл или индольный цикл, то Х не является связью или СН2, и А не является индолом;
если В представляет собой индол, то Х не является О; и
если В представляет собой тиазольный цикл, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
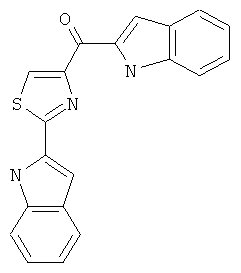
В одном варианте осуществления А в соединении Формулы I представляет собой индолил. В другом варианте осуществления А представляет собой 2-индолил. В другом варианте осуществления А представляет собой фенил. В другом варианте осуществления А представляет собой пиридил. В другом варианте осуществления А представляет собой нафтил. В другом варианте осуществления А представляет собой изохинолин. В другом варианте осуществления С в соединении Формулы I представляет собой индолил. В другом варианте осуществления С представляет собой 2-индолил. В другом варианте осуществления С представляет собой 5-индолил. В другом варианте осуществления В в соединении Формулы I представляет собой тиазол. В другом варианте осуществления В в соединении Формулы I представляет собой тиазол; Y представляет собой СО и Х представляет собой связь. Неограничивающие примеры соединения формулы I выбраны из: (2-(1Н-Индол-2-ил)тиазол-4-ил)(1Н-индол-2-ил)метанона (8), (2-(1Н-индол-2-ил)тиазол-4-ил)(1Н-индол-5-ил)метанона (21).
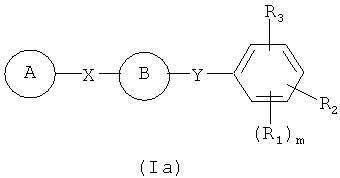
В одном варианте осуществления настоящее изобретение касается соединения формулы (Ia)
 ,
,
где
А представляет собой замещенную или незамещенную моноциклическую, конденсированную или полициклическую арильную или (гетеро)циклическую систему; замещенный или незамещенный, насыщенный или ненасыщенный N-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный S-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный O-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный циклический углеводород; или замещенный или незамещенный, насыщенный или ненасыщенный смешанный гетероцикл;
В представляет собой
(тиазол), (тиазол), (тиазолидин),
(оксазол), (оксазолин), (оксазолидин), (бензол),
(бензол), (пиримидин), (имидазол),
(пиридин), (фуран), (тиофен), (изоксазол),
, (пиперидин), (пиразол), (индол), или
(изохинолин);
R1, R2 и R3 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Х представляет собой связь, NH, C1-C5 углеводород, О или S;
Y представляет собой связь, -С=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, -C=N-CN, -CH=CH-, -С=С(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
где указанный цикл А необязательно замещен 1-5 заместителями, которые независимо представляют собой O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2;
m представляет собой целое число от 1 до 3;
где
если В представляет собой бензольное кольцо, тиофеновый цикл, фурановый цикл или индольный цикл, то Х не является связью или СН2, и А не является индолом;
если В представляет собой индол, то Х не является О; и
если В представляет собой тиазольный цикл, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
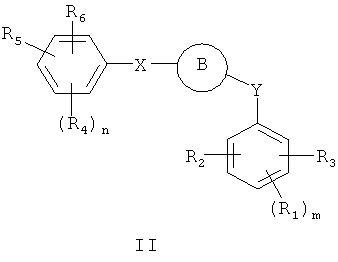
В одном варианте осуществления настоящее изобретение касается соединения формулы (II):
 ,
,
где
В представляет собой
(тиазол), (тиазол), (тиазолидин),
(оксазол), (оксазолин), (оксазолидин), (бензол),
(бензол), (пиримидин), (имидазол),
(пиридин), (фуран), (тиофен), (изоксазол),
, (пиперидин), (пиразол), (индол), или
(изохинолин);
R1, R2, R3, R4, R5 и R6 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-С5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Х представляет собой связь, NH, C1-C5 углеводород, О или S;
Y представляет собой связь, -С=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, -C=N-CN, -CH=CH-, С=СН(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2;
n представляет собой целое число от 1 до 3; и
m представляет собой целое число от 1 до 3;
где
если В представляет собой индол, то Х не является О; и
если В представляет собой тиазольный цикл, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
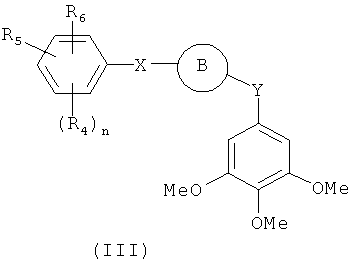
В одном варианте осуществления настоящее изобретение касается соединения формулы (III)
 ,
,
где
В представляет собой
(тиазол), (тиазол), (тиазолидин),
(оксазол), (оксазолин), (оксазолидин), (бензол),
(бензол), (пиримидин), (имидазол),
(пиридин), (фуран), (тиофен), (изоксазол),
, (пиперидин), (пиразол), (индол), или
(изохинолин);
R4, R5 и R6 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2; и
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Х представляет собой связь, NH, C1-C5 углеводород, О или S;
Y представляет собой связь, -С=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, -C=N-CN, -CH=CH-, С=СН(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2; и
n представляет собой целое число от 1 до 3; где
если В представляет собой индол, то Х не является О; и
если В представляет собой тиазольный цикл, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
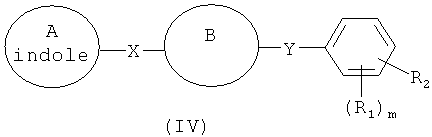
В одном варианте осуществления настоящее изобретение касается соединения формулы (IV)
 ,
,
где цикл А представляет собой индолил;
В представляет собой
(тиазол), (тиазол), (тиазолидин),
(оксазол), (оксазолин), (оксазолидин), (бензол),
(бензол), (пиримидин), (имидазол),
(пиридин), (фуран), (тиофен), (изоксазол),
, (пиперидин), (пиразол), (индол), или
(изохинолин);
R1 и R2 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Х представляет собой связь, NH, C1-С5 углеводород, О или S;
Y представляет собой связь, С=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, -C=N-CN, -CH=CH-, С=СН(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
где указанный А необязательно замещен O-алкилом, O-галогеналкилом, F, Cl, Br, I, галогеналкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейным или разветвленным алкилом, галогеналкилом, алкиламиногруппой, аминоалкилом, -OCH2Ph, -NHCO-алкилом, СООН, -C(O)Ph, C(O)O-алкилом, С(O)Н, -C(O)NH2 или NO2; и
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2; и
m представляет собой целое число от 1 до 4;
где
если В представляет собой бензольное кольцо, тиофеновый цикл, фурановый цикл или индольный цикл, то Х не является связью или СН2;
если В представляет собой тиазольный цикл, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В другом варианте осуществления индолил цикла А в формуле IV присоединен по одному из своих положений 1-7 к Х или напрямую к В, если Х представляет собой связь (т.е. отсутствует).
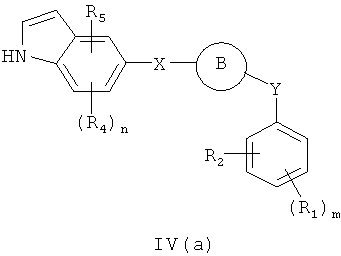
В одном варианте осуществления настоящее изобретение касается соединения формулы IV(a)
В представляет собой
(тиазол), (тиазол), (тиазолидин),
(оксазол), (оксазолин), (оксазолидин), (бензол),
(бензол), (пиримидин), (имидазол),
(пиридин), (фуран), (тиофен), (изоксазол),
, (пиперидин), (пиразол), (индол), или
(изохинолин);
R1, R2, R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2; и
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Х представляет собой связь, NH, C1-C5 углеводород, О или S;
Y представляет собой связь или С=О, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, -C=N-CN, -СН=СН-, C=СН(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(С=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
i представляет собой целое число от 0 до 5;
l представляет собой целое число от 1 до 2;
n представляет собой целое число от 1 до 2; и
m представляет собой целое число от 1 до 4;
где
если В представляет собой бензольное кольцо, тиофеновый цикл, фурановый цикл или индольный цикл, то Х не является связью или СН2;
если В представляет собой тиазольный цикл, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
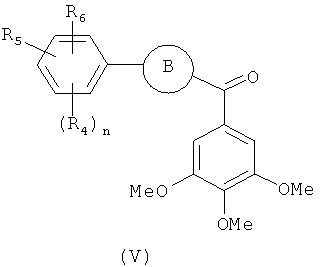
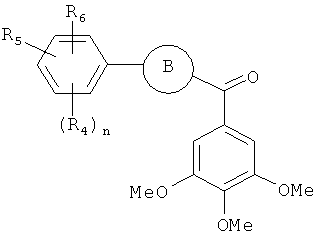
В одном варианте осуществления настоящее изобретение касается соединения формулы (V)
В представляет собой
(тиазол), (тиазол), (тиазолидин),
(оксазол), (оксазолин), (оксазолидин), (бензол),
(бензол), (пиримидин), (имидазол),
(пиридин), (фуран), (тиофен), (изоксазол),
, (пиперидин), (пиразол), (индол), или
(изохинолин);
R4, R5 и R6 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 1 до 5;
l представляет собой целое число от 1 до 2; и
n представляет собой целое число от 1 до 3;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В другом варианте осуществления В в формуле V не является тиазолом . В другом варианте осуществления В в формуле V не является оксазолом. В другом варианте осуществления В в формуле V не является оксазолином. В другом варианте осуществления В в формуле V не является имидазолом. В другом варианте осуществления В в формуле V не является тиазолом, оксазолом, оксазолином или имидазолом.
В одном варианте осуществления настоящее изобретение касается следующих соединений:
|
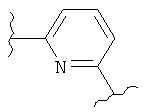
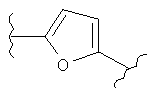
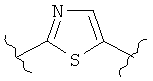
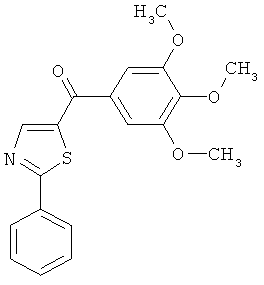
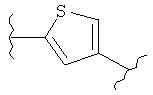
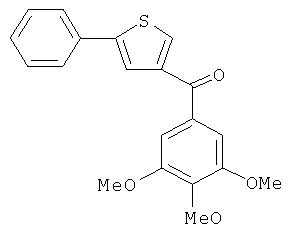
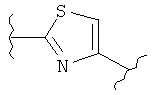
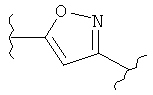
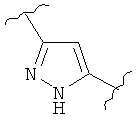
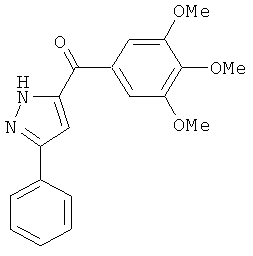
|
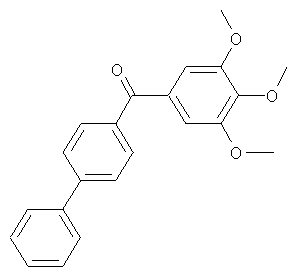
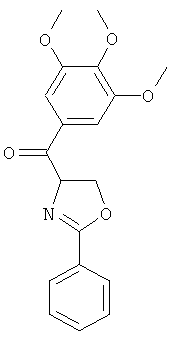
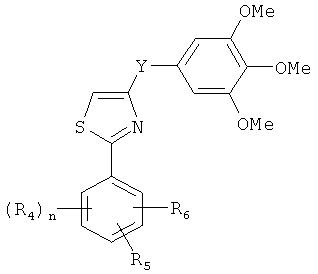
В одном варианте осуществления настоящее изобретение касается соединения формулы (VI)
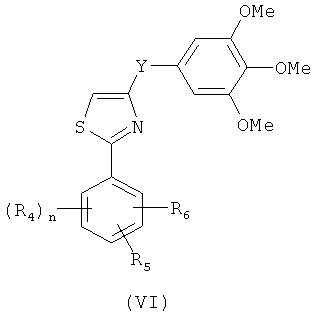 ,
,
где
R4, R5 и R6 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2; и
Y представляет собой связь или С=O, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, -C=N-CN, -CH-CH-, С=СН(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(C=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
n представляет собой целое число от 1 до 3; и
i представляет собой целое число от 1 до 5;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
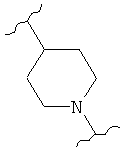
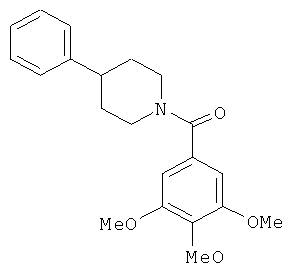
В одном варианте осуществления настоящее изобретение касается следующих соединений:
|
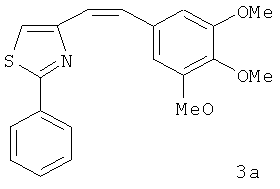
В одном варианте осуществления настоящее изобретение касается соединения 3а:
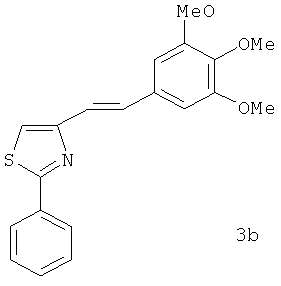
В одном варианте осуществления настоящее изобретение касается соединения 3b:
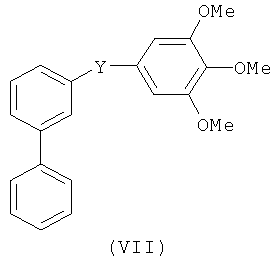
В одном варианте осуществления настоящее изобретение касается соединения формулы (VII)
где
Y представляет собой связь или С=О, -C=S, -C=N-NH2, -C=N-OH, -CH-OH, -C=CH-CN, -C=N-CN, -СН=СН-, С=СН(СН3)2, -C=N-OMe, -(C=O)-NH, -NH-(C=O), -(C=O)-O, -O-(С=O), -(CH2)1-5-(C=O), (C=O)-(CH2)1-5, -(SO2)-NH-, -NH-(SO2)-, SO2, SO или S;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается следующих соединений:
|
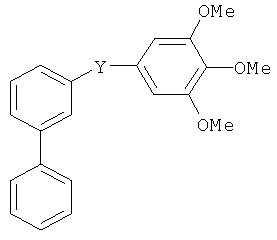
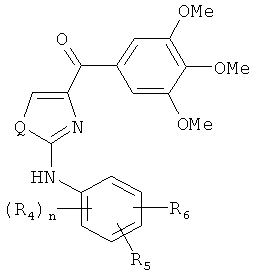
В одном варианте осуществления настоящее изобретение касается соединения формулы (VIII)
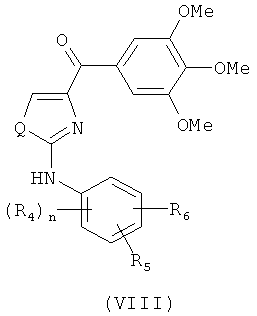 ,
,
где
R4, R5 и R6 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
Q представляет собой S, О или NH;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 3;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается следующих соединений:
|
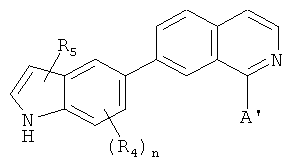
В одном варианте осуществления настоящее изобретение касается соединения формулы (IX)
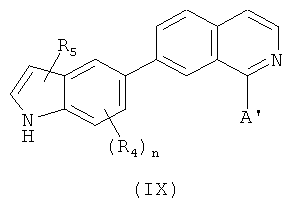 ,
,
где
R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, С1-С5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -(O)NH2 или NO2;
А' представляет собой галоген; замещенную или незамещенную моноциклическую, конденсированную или полициклическую арильную или (гетеро)циклическую систему; замещенный или незамещенный, насыщенный или ненасыщенный N-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный S-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный O-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный циклический углеводород; или замещенный или незамещенный, насыщенный или ненасыщенный смешанный гетероцикл; где указанный цикл А' необязательно замещен 1-5 заместителями, которые независимо представляют собой O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 1 до 5; и
n представляет собой целое число от 1 до 3;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления соединение формулы IX представлено структурами следующих соединений:
|
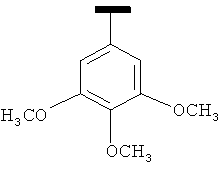
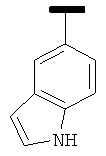
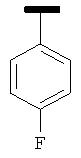
В одном варианте осуществления А' в формуле IX представляет собой фенил. В другом варианте осуществления А' в формуле IX представляет собой замещенный фенил. В другом варианте осуществления А' в формуле IX представляет собой галоген. В другом варианте осуществления заместитель в А' представляет собой галоген. В другом варианте осуществления заместитель представляет собой 4-F. В другом варианте осуществления заместитель представляет собой 3,4,5-(ОСН3)3. В другом варианте осуществления А' в формуле IX представляет собой замещенный или незамещенный 5-индолил. В другом варианте осуществления А' в формуле IX представляет собой замещенный или незамещенный 2-индолил. В другом варианте осуществления А' в формуле IX представляет собой замещенный или незамещенный 3-индолил. В другом варианте осуществления соединения формулы IX изображены на фиг.16A.
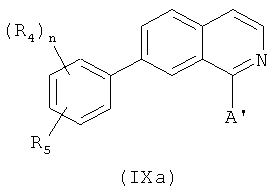
В одном варианте осуществления настоящее изобретение касается соединения формулы (IXa)
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -(O)NH2 или NO2;
А' представляет собой галоген; замещенную или незамещенную моноциклическую, конденсированную или полициклическую арильную или (гетеро)циклическую систему; замещенный или незамещенный, насыщенный или ненасыщенный N-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный S-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный O-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный циклический углеводород; или замещенный или незамещенный, насыщенный или ненасыщенный смешанный гетероцикл; где указанный цикл А' необязательно замещен 1-5 заместителями, которые независимо представляют собой O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 1 до 5; и
n представляет собой целое число от 1 до 3;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления А' в формуле IXa представляет собой фенил. В другом варианте осуществления А' в формуле IXa представляет собой замещенный фенил. В другом варианте осуществления А' в формуле IXa представляет собой галоген. В другом варианте осуществления заместитель в А' представляет собой галоген. В другом варианте осуществления этот заместитель представляет собой 4-F. В другом варианте осуществления этот заместитель представляет собой 3,4,5-(ОСН3)3. В другом варианте осуществления А' в формуле IXa представляет собой замещенный или незамещенный 5-индолил. В другом варианте осуществления А' в формуле IXa представляет собой замещенный или незамещенный 2-индолил. В другом варианте осуществления А' в формуле IXa представляет собой замещенный или незамещенный 3-индолил.
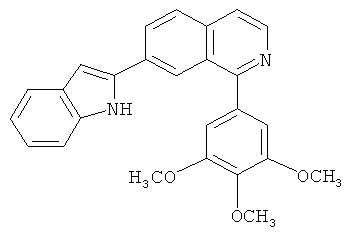
В другом варианте осуществления соединение формулы IXa представляет собой 1-хлор-7-(4-фторфенил)изохинолин. В другом варианте осуществления соединение формулы IXa представляет собой 7-(4-фторфенил)-1-(1Н-индол-5-ил)изохинолин. В другом варианте осуществления соединение формулы IXa представляет собой 7-(4-фторфенил)-1-(3,4,5-триметоксифенил)изохинолин. В другом варианте осуществления соединение формулы IXa представляет собой 1,7-бис(4-фторфенил)изохинолин (40). В другом варианте осуществления соединение формулы IXa представляет собой 1,7-бис(3,4,5-триметоксифенил)изохинолин. В другом варианте осуществления соединение формулы IXa представляет собой 1-(4-фторфенил)-7-(3,4,5-триметоксифенил)изохинолин. В другом варианте осуществления соединение формулы IXa представляет собой 1-(1Н-индол-5-ил)-7-(3,4,5-триметоксифенил)изохинолин. В другом варианте осуществления соединение формулы IXa представляет собой 1-хлор-7-(3,4,5-триметоксифенил)изохинолин.
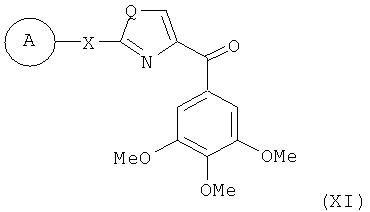
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы XI:
 ,
,
где
Х представляет собой связь, NH или S;
Q представляет собой О, NH или S; и
А представляет собой замещенную или незамещенную моноциклическую, конденсированную или полициклическую арильную или (гетеро)циклическую систему; замещенный или незамещенный, насыщенный или ненасыщенный N-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный S-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный O-гетероцикл; замещенный или незамещенный, насыщенный или ненасыщенный циклический углеводород; или замещенный или незамещенный, насыщенный или ненасыщенный смешанный гетероцикл; где указанный цикл А необязательно замещен 1-5 заместителями, которые независимо представляют собой O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2; и
i представляет собой целое число от 0 до 5;
где если Q представляет собой S, то Х не является связью;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления А в соединении формулы XI представляет собой Ph. В другом варианте осуществления А в соединении формулы XI представляет собой замещенный Ph. В другом варианте осуществления заместитель представляет собой 4-F. В другом варианте осуществления заместитель представляет собой 4-Ме. В другом варианте осуществления Q в соединении формулы XI представляет собой S. В другом варианте осуществления Х в соединении формулы XI представляет собой NH. Неограничивающие примеры соединений формулы XI выбраны из следующих: (2-(фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5а), (2-(п-толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5b), (2-(п-фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5с), (2-(фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон гидрохлорид (5На), (2-(п-толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон гидрохлорид (5Hb), (2-(п-фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон гидрохлорид (5Нс).
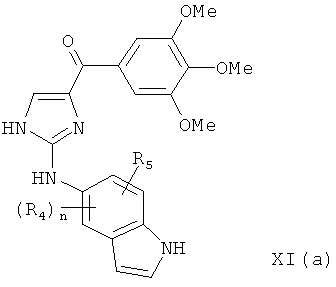
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы XI(а):
 ,
,
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
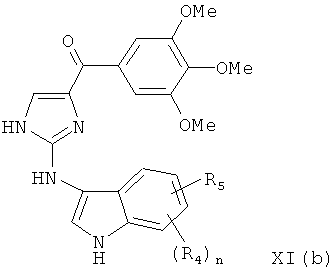
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы XI(b):
 ,
,
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
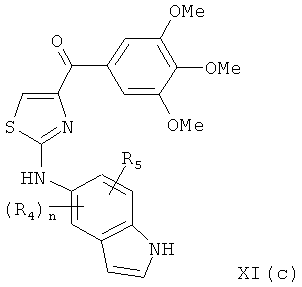
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы XI(с):
 .
.
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы XI(d):
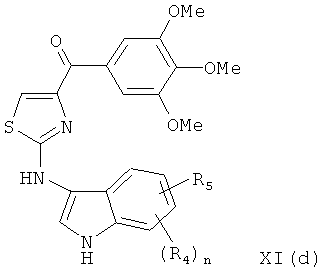 ,
,
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(СН2)iNHCH3, -(CH2)iNH2-(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, С(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения, представленного структурой формулы XI(е):
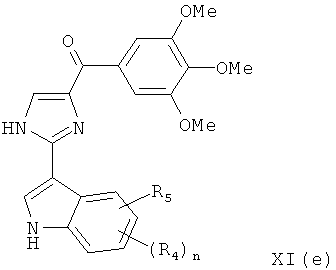
где R4 и R5 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, C1-C5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В другом варианте осуществления соединение формулы XI представлено структурой соединения 55:
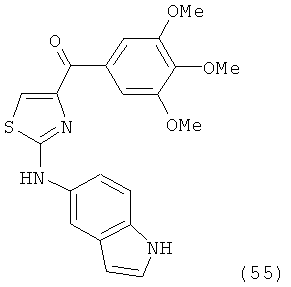 .
.
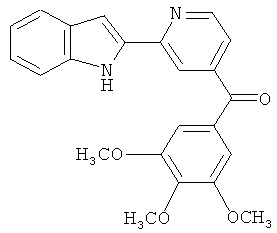
В другом варианте осуществления соединение формулы XI представлено структурой соединения 17ya:
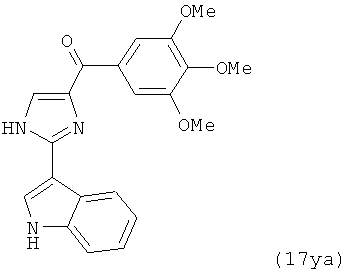 .
.
В одном варианте осуществления в настоящем изобретении описано соединение, представленное следующими структурами:
|
|
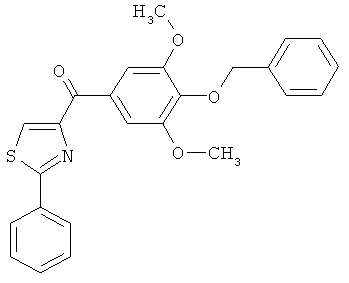
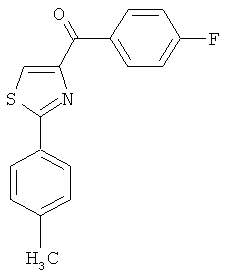
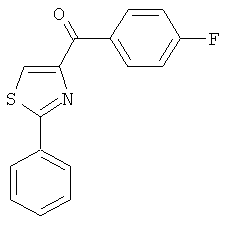
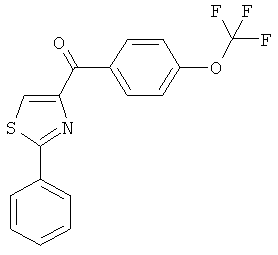
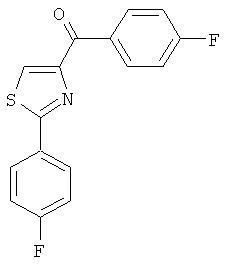
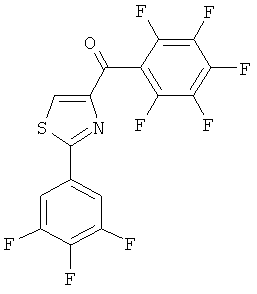
|
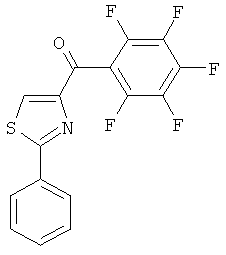
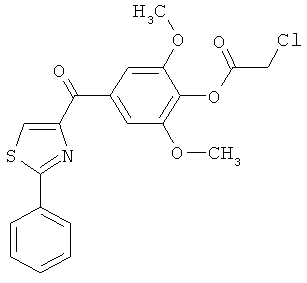
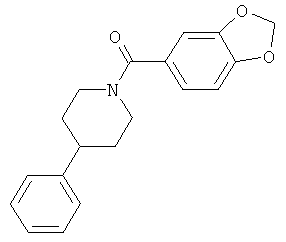
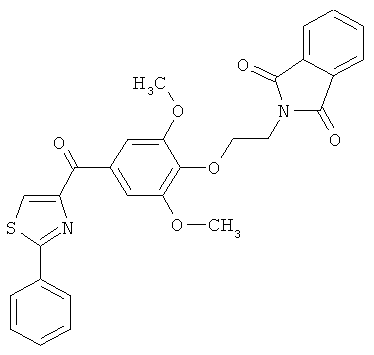
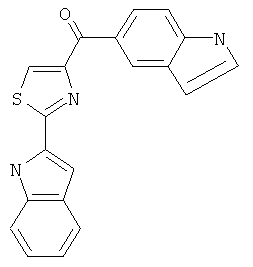
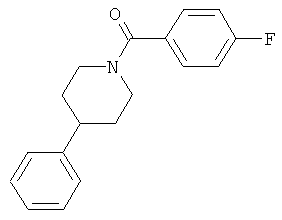
|
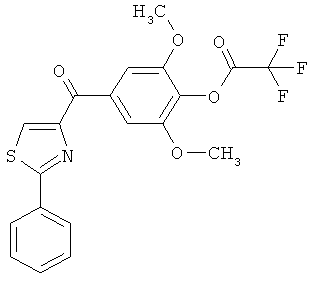
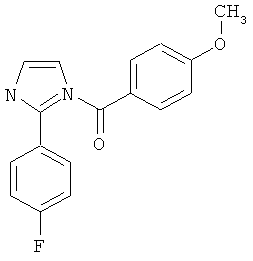
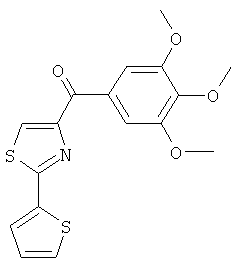
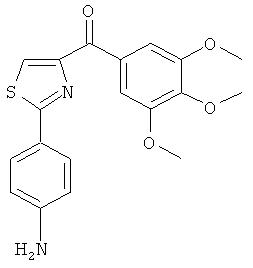
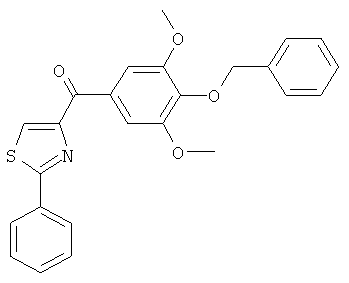
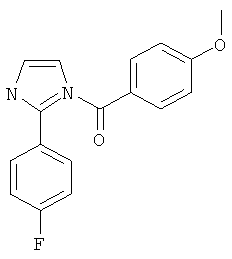
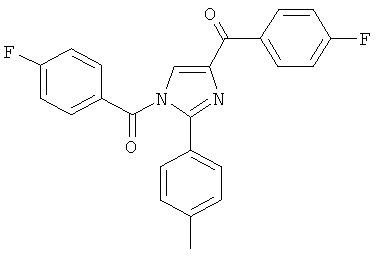
|
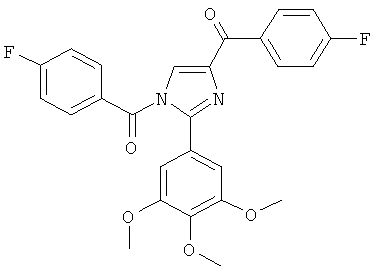
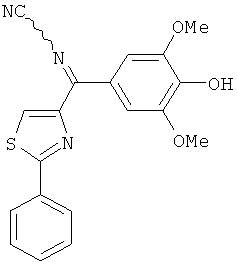
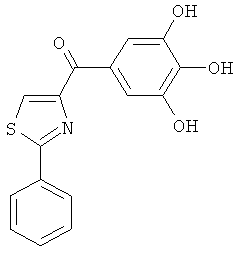
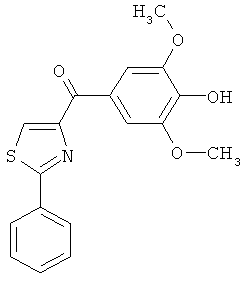
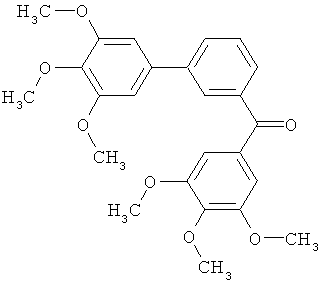
|
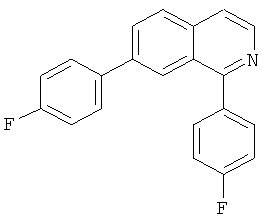
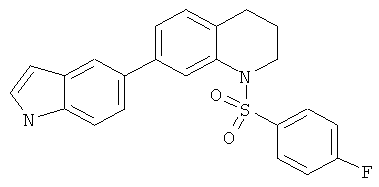
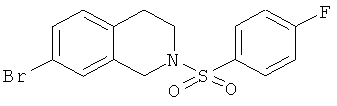
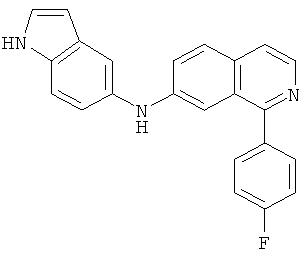
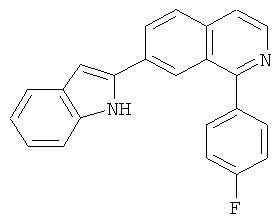
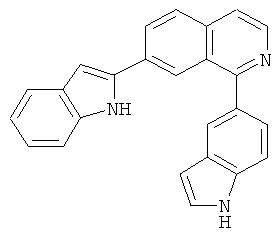
|
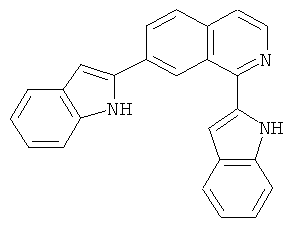
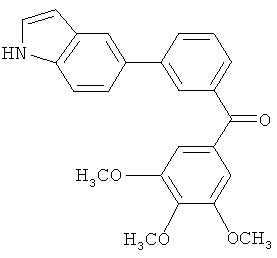
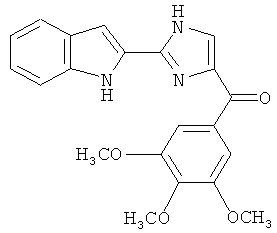
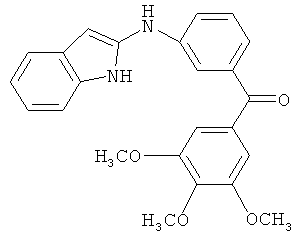
Само собой разумеется, что в представленных в настоящем изобретении соединениях, в которых атом азота имеет меньше 3 связей, остальные валентности атома азота заполнены связями с атомами водорода.
В одном варианте осуществления группы А, А' и/или С в формуле I, I(а), IV, IX, IX(а) и XI независимо представляют собой замещенный и незамещенный фуранил, индолил, пиридинил, фенил, бифенил, трифенил, дифенилметан, адамантанил, флуоренил и другие гетероциклические аналоги, такие как те, что перечислены выше (например, пирролил, пиразолил, имидазолил, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил, пирролизинил, индолил, изохинолинил, хинолинил, изохинолинил, бензимидазолил, индазолил, хинолизинил, циннолинил, хиналолинил, фталазинил, нафтиридинил, хиноксалинил, оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, диоксанил, фуранил, пирилий, бензофуранил, бензодиоксолил, тиранил, тиетанил, тетрагидротиофенил, дитиоланил, тетрагидротиопиранил, тиофенил, тиепинил, тианафтенил, оксатиоланил, морфолинил, тиоксанил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазолил).
В одном варианте осуществления наиболее предпочтительными группами А, А' и/или С является замещенный или незамещенный фенил. В одном варианте осуществления наиболее предпочтительными группами А' А' и/или С является замещенный или незамещенный изохинолинил. В одном варианте осуществления группы А, А' и/или С включают замещенные и незамещенные индолильные группы; наиболее предпочтительно, замещенный и незамещенный 3-индолил и 5-индолил.
В одном варианте осуществления группы А, А' и/или С в формуле I, I(а), IV, IX, IX(а) и XI могут быть замещенными или незамещенными. Так, несмотря на то, что примеры групп, приведенные в предыдущем абзаце, являются незамещенными, квалифицированным специалистам в данной области будет понятно, что указанные группы могут быть замещены одним, двумя или более, тремя или более, и даже вплоть до пяти, заместителями (отличными от водорода).
В одном варианте осуществления наиболее предпочтительные группы А, А' и/или С замещены 3,4,5-триметоксифенилом. В другом варианте осуществления группы А, А' и/или С замещены алкокси-группой. В другом варианте осуществления группы А, А' и/или С замещены метокси-группой. В другом варианте осуществления группы А, А' и/или С замещены алкилом. В другом варианте осуществления группы А, А' и/или С замещены метилом. В другом варианте осуществления группы А, А' и/или С замещены галогеном. В другом варианте осуществления группы А, А' и/или С замещены атомом F. В другом варианте осуществления группы А, А' и/или С замещены атомом Cl. В другом варианте осуществления циклы А, А' и/или С замещены атомом Br.
Заместители в указанных группах А, А' и/или С в формуле I, I(а), IV, IX, IX(а) и XI независимо выбраны из группы, состоящей из атома водорода (например, отсутствие заместителя в конкретном положении), гидроксила, алифатического линейного или разветвленного C1-С10 углеводорода, алкокси-группы, галоген-алкокси-группы, арилокси-группы, нитро-группы, циано-группы, алкил-CN, галогена (например, F, Cl, Br, I), галогеналкила, дигалогеналкила, тригалогеналкила, СООН, C(O)Ph, С(O)-алкила, С(O)O-алкила, С(O)Н, C(O)NH2, -ОС(O)CF3, OCH2Ph, амино-группы, аминоалкила, алкиламино-группы, мезиламино-группы, диалкиламино-группы, ариламино-группы, амидной группы, NHC(O)-алкила, мочевины, алкил-мочевины, алкиламидо-группы (например, ацетамидной), галогеналкиламидо-группы, ариламидо-группы, арила и C5-C7 циклоалкила, арилалкила и их комбинаций. Единичные заместители могут присутствовать в орто-, мета- или параположениях. Если имеется два или более заместителей, один из них предпочтительно, хотя и не обязательно, находится в параположении.
В одном варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V выбрана из замещенного или незамещенного тиазола, тиазолидина, оксазола, оксазолина, оксазолидина, бензола, пиримидина, имидазола, пиридина, фурана, тиофена, изоксазола, пиперидина, пиразола, индола и изохинолина, где указанный цикл В связан по любым двум положениям цикла с Х и Y, или напрямую с фенильным, индолильным циклом А и/или С.
В одном варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V является незамещенной. В другом варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V представляет собой:
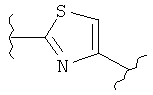 (тиазол),
(тиазол), 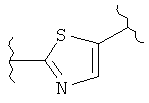 (тиазол),
(тиазол), 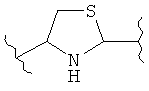 (тиазолидин),
(тиазолидин), 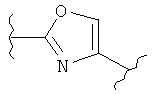 (оксазол),
(оксазол),
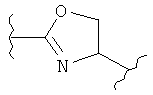 (оксазолин),
(оксазолин), 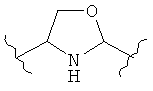 (оксазолидин),
(оксазолидин), 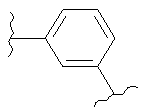 (бензол),
(бензол), 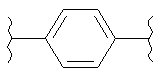 (бензол),
(бензол),
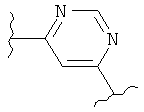 (пиримидин),
(пиримидин),  (имидазол),
(имидазол), 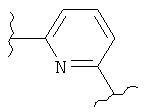 (пиридин),
(пиридин), 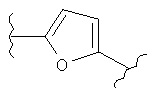 (фуран),
(фуран),
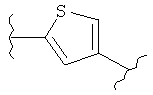 (тиофен),
(тиофен), 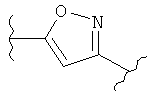 (изоксазол),
(изоксазол), 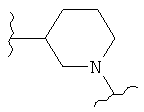 ,
, 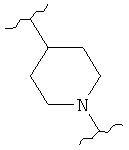 (пиперидин),
(пиперидин),
 (пиразол),
(пиразол),  (индол), или
(индол), или  (изохинолин);
(изохинолин);
В другом варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V является замещенной. В другом варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V представляет собой:
(тиазол), (тиазол), (тиазолидин),
(оксазол), (оксазолин), (оксазолидин), (бензол),
(бензол), (пиримидин), (имидазол),
(пиридин), (фуран), (тиофен), (изоксазол),
, (пиперидин), (пиразол), (индол), или
(изохинолин);
где R10 и R11 независимо представляют собой атом водорода, O-алкил, O-галогеналкил, F, Cl, Br, I, галогеналкил, CF3, CN, -CH2CN, NH2, гидроксил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, С1-С5 линейный или разветвленный алкил, галогеналкил, алкиламино, аминоалкил, -OCH2Ph, -NHCO-алкил, СООН, -C(O)Ph, C(O)O-алкил, С(O)Н, -C(O)NH2 или NO2.
В другом варианте осуществления группа В представляет собой  (тиазол). В другом варианте осуществления группа В представляет собой
(тиазол). В другом варианте осуществления группа В представляет собой 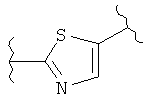 (тиазол). В другом варианте осуществления группа В представляет собой
(тиазол). В другом варианте осуществления группа В представляет собой 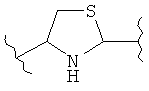 (тиазолидин). В другом варианте осуществления группа В представляет собой
(тиазолидин). В другом варианте осуществления группа В представляет собой 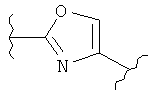 (оксазол). В другом варианте осуществления группа В представляет собой
(оксазол). В другом варианте осуществления группа В представляет собой 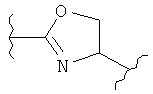 (оксазолин). В другом варианте осуществления группа В представляет собой
(оксазолин). В другом варианте осуществления группа В представляет собой 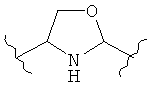 (оксазолидин). В другом варианте осуществления группа В представляет собой
(оксазолидин). В другом варианте осуществления группа В представляет собой 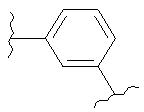 (бензол). В другом варианте осуществления группа В представляет собой
(бензол). В другом варианте осуществления группа В представляет собой 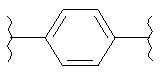 (бензол). В другом варианте осуществления группа В представляет собой
(бензол). В другом варианте осуществления группа В представляет собой 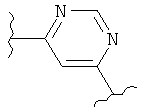 (пиримидин). В другом варианте осуществления группа В представляет собой
(пиримидин). В другом варианте осуществления группа В представляет собой 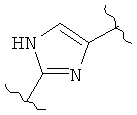 (имидазол). В другом варианте осуществления группа В представляет собой
(имидазол). В другом варианте осуществления группа В представляет собой 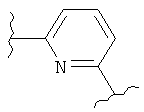 (пиридин). В другом варианте осуществления группа В представляет собой
(пиридин). В другом варианте осуществления группа В представляет собой 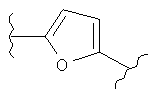 (фуран). В другом варианте осуществления группа В представляет собой
(фуран). В другом варианте осуществления группа В представляет собой 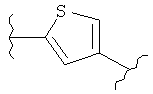 (тиофен). В другом варианте осуществления группа В представляет собой
(тиофен). В другом варианте осуществления группа В представляет собой 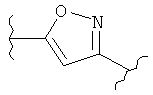 (изоксазол). В другом варианте осуществления группа В представляет собой
(изоксазол). В другом варианте осуществления группа В представляет собой 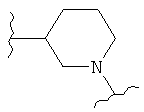 (пиперидин). В другом варианте осуществления группа В представляет собой
(пиперидин). В другом варианте осуществления группа В представляет собой 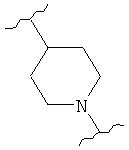 (пиперидин). В другом варианте осуществления группа В представляет собой
(пиперидин). В другом варианте осуществления группа В представляет собой 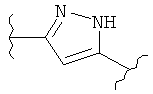 (пиразол). В другом варианте осуществления группа В представляет собой
(пиразол). В другом варианте осуществления группа В представляет собой  (индол). В другом варианте осуществления группа В представляет собой
(индол). В другом варианте осуществления группа В представляет собой  (изохинолин).
(изохинолин).
В одном варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V замещена заместителями R10 и R11. В другом варианте осуществления R10 и R11 оба представляют собой атомы водорода. В другом варианте осуществления R10 и R11 независимо представляют собой O-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой O-галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой F. В другом варианте осуществления R10 и R11 независимо представляют собой Cl. В другом варианте осуществления R10 и R11 независимо представляют собой Br. В другом варианте осуществления R10 и R11 независимо представляют собой I. В другом варианте осуществления R10 и R11 независимо представляют собой галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой CF3. В другом варианте осуществления R10 и R11 независимо представляют собой CN. В другом варианте осуществления R10 и R11 независимо представляют собой -CH2CN. В другом варианте осуществления R10 и R11 независимо представляют собой NH2. В другом варианте осуществления R10 и R11 независимо представляют собой гидроксил. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iNHCH3. В другом варианте осуществления R10 и R11 независимо представляют собой -(CH2)iNH2. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iN(СН3)2. В другом варианте осуществления R10 и R11 независимо представляют собой -ОС(O)CF3. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный алкил. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейную или разветвленную алкиламино-группу. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный аминоалкил. В другом варианте осуществления R10 и R11 независимо представляют собой -OCH2Ph. В другом варианте осуществления R10 и R11 независимо представляют собой -NHCO-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой СООН. В другом варианте осуществления R10 и R11 независимо представляют собой -C(O)Ph. В другом варианте осуществления R10 и R11 независимо представляют собой С(O)O-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой С(O)Н. В другом варианте осуществления R10 и R11 независимо представляют собой -C(O)NH2. В другом варианте осуществления R10 и R11 независимо представляют собой NO2.
В другом варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V представляет собой (тиазол), где R10 и R11 независимо представляют собой Н, и l равно 1. В другом варианте осуществления R10 и R11 независимо представляют собой O-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой O-галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой F. В другом варианте осуществления R10 и R11 независимо представляют собой Cl. В другом варианте осуществления R10 и R11 независимо представляют собой Br. В другом варианте осуществления R10 и R11 независимо представляют собой I. В другом варианте осуществления R10 и R11 независимо представляют собой галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой CF3. В другом варианте осуществления R10 и R11 независимо представляют собой CN. В другом варианте осуществления R10 и R11 независимо представляют собой -CH2CN. В другом варианте осуществления R10 и R11 независимо представляют собой NH2. В другом варианте осуществления R10 и R11 независимо представляют собой гидроксил. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iNHCH3. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iNH2. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iN(СН3)2. В другом варианте осуществления R10 и R11 независимо представляют собой -ОС(O)CF3. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный алкил. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой С1-С5 линейную или разветвленную алкиламино-группу. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный аминоалкил. В другом варианте осуществления R10 и R11 независимо представляют собой -OCH2Ph. В другом варианте осуществления R10 и R11 независимо представляют собой -NHCO-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой СООН. В другом варианте осуществления R10 и R11 независимо представляют собой -C(O)Ph. В другом варианте осуществления R10 и R11 независимо представляют собой С(O)O-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой С(O)Н. В другом варианте осуществления R10 и R11 независимо представляют собой -C(O)NH2. В другом варианте осуществления R10 и R11 независимо представляют собой NO2.
В другом варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V представляет собой (имидазол), где R10 и R11 независимо представляют собой Н, и l равно 1. В другом варианте осуществления R10 и R11 независимо представляют собой O-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой O-галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой F. В другом варианте осуществления R10 и R11 независимо представляют собой Cl. В другом варианте осуществления R10 и R11 независимо представляют собой Br. В другом варианте осуществления R10 и R11 независимо представляют собой I. В другом варианте осуществления R10 и R11 независимо представляют собой галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой CF3. В другом варианте осуществления R10 и R11 независимо представляют собой CN. В другом варианте осуществления R10 и R11 независимо представляют собой -CH2CN. В другом варианте осуществления R10 и R11 независимо представляют собой NH2. В другом варианте осуществления R10 и R11 независимо представляют собой гидроксил. В другом варианте осуществления R10 и R11 независимо представляют собой -(CH2)iNHCH3. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iNH2. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iN(СН3)2. В другом варианте осуществления R10 и R11 независимо представляют собой -ОС(O)CF3. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный алкил. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейную или разветвленную алкиламино-группу. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный аминоалкил. В другом варианте осуществления R10 и R11 независимо представляют собой -OCH2Ph. В другом варианте осуществления R10 и R11 независимо представляют собой -NHCO-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой СООН. В другом варианте осуществления R10 и R11 независимо представляют собой -C(O)Ph. В другом варианте осуществления R10 и R11 независимо представляют собой С(O)O-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой С(O)Н. В другом варианте осуществления R10 и R11 независимо представляют собой -C(O)NH2. В другом варианте осуществления R10 и R11 независимо представляют собой NO2.
В другом варианте осуществления группа В в формуле I, I(а), II, III, IV, IVa и V представляет собой (изохинолин), где R10 и R11 независимо представляют собой Н, и l равно 1. В другом варианте осуществления R10 и R11 независимо представляют собой O-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой O-галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой F. В другом варианте осуществления R10 и R11 независимо представляют собой Cl. В другом варианте осуществления R10 и R11 независимо представляют собой Br. В другом варианте осуществления R10 и R11 независимо представляют собой I. В другом варианте осуществления R10 и R11 независимо представляют собой галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой CF3. В другом варианте осуществления R10 и R11 независимо представляют собой CN. В другом варианте осуществления R10 и R11 независимо представляют собой -CH2CN. В другом варианте осуществления R10 и R11 независимо представляют собой NH2. В другом варианте осуществления R10 и R11 независимо представляют собой гидроксил. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iNHCH3. В другом варианте осуществления R10 и R11 независимо представляют собой -(CH2)iNH2. В другом варианте осуществления R10 и R11 независимо представляют собой -(СН2)iN(СН3)2. В другом варианте осуществления R10 и R11 независимо представляют собой -ОС(O)CF3. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный алкил. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный галогеналкил. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейную или разветвленную алкиламино-группу. В другом варианте осуществления R10 и R11 независимо представляют собой C1-C5 линейный или разветвленный аминоалкил. В другом варианте осуществления R10 и R11 независимо представляют собой -OCH2Ph. В другом варианте осуществления R10 и R11 независимо представляют собой -NHCO-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой СООН. В другом варианте осуществления R10 и R11 независимо представляют собой -C(O)Ph. В другом варианте осуществления R10 и R11 независимо представляют собой С(O)O-алкил. В другом варианте осуществления R10 и R11 независимо представляют собой С(O)Н. В другом варианте осуществления R10 и R11 независимо представляют собой -C(O)NH2. В другом варианте осуществления R10 и R11 независимо представляют собой NO2.
В одном варианте осуществления мостик Х в формуле I, Ia, II, III, IV, IVa и XI представляет собой связь. В другом варианте осуществления мостик Х представляет собой NH. В другом варианте осуществления мостик Х представляет собой C1-C5 углеводород. В другом варианте осуществления мостик Х представляет собой СН2. В другом варианте осуществления мостик Х представляет собой -СН2-СН2-. В другом варианте осуществления мостик Х представляет собой О. В другом варианте осуществления мостик Х представляет собой S.
В одном варианте осуществления мостик Y в формуле I, Ia, II, III, IV, IVa, VI и VII представляет собой С=O. В другом варианте осуществления мостик Y представляет собой C=S. В другом варианте осуществления мостик Y представляет собой C=N(NH2)-. В другом варианте осуществления мостик Y представляет собой -C=NOH. В другом варианте осуществления мостик Y представляет собой -СН-ОН. В другом варианте осуществления мостик Y представляет собой -C=CH-(CN). В другом варианте осуществления мостик Y представляет собой -C=N(CN). В другом варианте осуществления мостик Y представляет собой -C=CH(CH3)2. В другом варианте осуществления мостик Y представляет собой -C=N-OMe. В другом варианте осуществления мостик Y представляет собой -(C=O)NH-. В другом варианте осуществления мостик Y представляет собой -NH(C=O)-. В другом варианте осуществления мостик Y представляет собой -(С=O)-O. В другом варианте осуществления мостик Y представляет собой -O-(С=O). В другом варианте осуществления мостик Y представляет собой -(CH2)1-5-(C=O). В другом варианте осуществления мостик Y представляет собой -(C=O)-(CH2)1-5. В другом варианте осуществления мостик Y представляет собой S. В другом варианте осуществления мостик Y представляет собой SO. В другом варианте осуществления мостик Y представляет собой SO2. В другом варианте осуществления мостик Y представляет собой -СН=СН-. В другом варианте осуществления мостик Y представляет собой -(SO2)-NH-. В другом варианте осуществления мостик Y представляет собой -NH-(SO2)-.
В одном варианте осуществления R1, R2, R3, R4, R5 и R6 в формуле Ia, II, III, IV, IV(a), V, VI, VIII, IX, IX(а), XI(а), XI(b), XI(c), XI(d) и XI(е) независимо представляют собой атом водорода. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой O-алкил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой O-галогеналкил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой F. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой Cl. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой Br. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой I. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой галогеналкил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой CF3. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой CN. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -CH2CN. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой NH2. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой гидроксил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -(СН2)iNHCH3. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -(CH2)iNH2. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -(СН2)iN(СН3)2. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -ОС(O)CF3. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой C1-C5 линейный или разветвленный алкил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой галогеналкил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой алкиламино. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой аминоалкил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -OCH2Ph. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -NHCO-алкил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой СООН. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -C(O)Ph. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой С(O)O-алкил. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой С(O)Н. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой -C(O)NH2. В другом варианте осуществления R1, R2, R3, R4, R5 и R6 независимо представляют собой NO2.
В одном варианте осуществления настоящее изобретение касается соединения формулы XII:
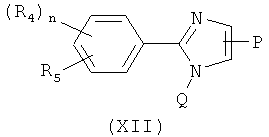 .
.
где
Р и Q независимо представляют собой Н или
 ;
;
W представляет собой С=O, C=S, SO2 или S=O;
где по меньшей мере один из Q или Р не является атомом водорода;
R1 и R4 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2; С(O)O-алкил или С(O)Н; где по меньшей мере один из R1 и R4 не является атомом водорода;
R2 и R5 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
m представляет собой целое число от 1 до 4;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения формулы XIII:
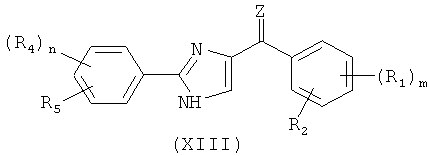 ,
,
где
Z представляет собой О или S;
R1 и R4 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2; СООН, С(O)O-алкил или С(O)Н; где по меньшей мере один из R1 и R4 не является атомом водорода;
R2 и R5 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2; OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
m представляет собой целое число от 1 до 4;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления настоящее изобретение касается соединения формулы XIV:
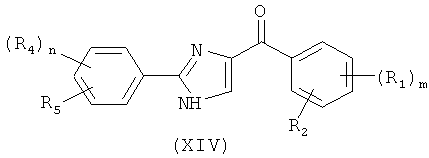 ,
,
где R1 и R4 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н; где по меньшей мере один из R1 и R4 не является атомом водорода;
R2 и R5 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
m представляет собой целое число от 1 до 4;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R1 в соединении формулы XII, XIII и XIV представляет собой ОСН3. В другом варианте осуществления R1 в соединении формулы XII, XIII и XIV представляет собой 4-F. В другом варианте осуществления R1 в соединении формулы XII, XIII и XIV представляет собой ОСН3, и m равно 3. В другом варианте осуществления R4 в соединении формулы XII, XIII и XIV представляет собой 4-F. В другом варианте осуществления R4 в соединении формулы XII, XIII и XIV представляет собой ОСН3. В другом варианте осуществления R4 в соединении формулы XIV представляет собой СН3. В другом варианте осуществления R4 в соединении формулы XII, XIII и XIV представляет собой 4-Cl. В другом варианте осуществления R4 в соединении формулы XII, XIII и XIV представляет собой 4-N(Me)2. В другом варианте осуществления R4 в соединении формулы XII, XIII и XIV представляет собой OBn. В другом варианте осуществления R4 в соединении формулы XII, XIII и XIV представляет собой 4-Br. В другом варианте осуществления R4 в соединении формулы XII, XIII и XIV представляет собой 4-CF3. Неограничивающие примеры соединений формулы XIV выбраны из следующих: (2-фенил-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12аа), (4-фторфенил)(2-фенил-1Н-имидазол-4-ил)метанон (12af), (2-(4-фторфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ba), (2-(4-метоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12са), (4-фторфенил)(2-(4-метоксифенил)-1Н-имидазол-4-ил)метанон (12cb), (2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12da), (4-фторфенил)(2-(п-толил)-1Н-имидазол-4-ил)метанон (12db), (4-гидрокси-3,5-диметоксифенил)(2-(п-толил)-1Н-имидазол-4-ил)метанон (12dc), (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12fa), (2-(4-хлорфенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12fb), (2-(4-хлорфенил)-1Н-имидазол-4-ил)(4-гидрокси-3,5-диметоксифенил)метанон (12fc), (2-(4-(диметиламино)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ga); (2-(4-(диметиламино)фенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12gb), (2-(3,4-диметоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ha), (2-(4-(бензилокси)фенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12jb), (2-(4-бромфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12la), (2-(4-(Трифторметил)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ра).
В одном варианте осуществления настоящее изобретение касается соединения формулы XIVa:
где R1 и R4 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н; где по меньшей мере один из R1 и R4 не является атомом водорода;
R2 и R5 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
R9 представляет собой Н, линейный или разветвленный, замещенный или незамещенный алкил, замещенный или незамещенный арил, CH2Ph, замещенный бензил, галогеналкил, аминоалкил, OCH2Ph, замещенный или незамещенный SO2-арил, замещенный или незамещенный -(С=O)-арил или ОН;
где заместители независимо выбраны из группы, состоящей из атома водорода (например отсутствие заместителя в определенном положении), гидроксила, алифатического линейного или разветвленного C1-C10 углеводорода, алкокси-группы, галогеналкокси-группы, арилокси-группы, нитро-группы, циано-группы, алкил-CN, галогена (например F, Cl, Br, I), галогеналкила, дигалогеналкила, тригалогеналкила, СООН, C(O)Ph, С(O)-алкила, С(O)O-алкила, С(O)Н, C(O)NH2, -ОС(O)CF3, OCH2Ph, амино-группы, аминоалкила, алкиламино-группы, мезиламино-группы, диалкиламино-группы, ариламино-группы, амидной группы, NHC(O)-алкила, мочевины, алкил-мочевины, алкиламидо-группы (например, ацетамид), галогеналкиламидо-группы, ариламидо-группы, арила и C5-C7 циклоалкила, арилалкила и их комбинаций;
m представляет собой целое число от 1 до 4;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R9 в соединении формулы XIVa представляет собой СН3. В другом варианте осуществления R9 в соединении формулы XIVa представляет собой CH2Ph. В другом варианте осуществления R9 в соединении формулы XIVa представляет собой (SO2)Ph. В другом варианте осуществления R9 в соединении формулы XIVa представляет собой (SO2)-Ph-ОСН3. В другом варианте осуществления R9 в соединении формулы XIVa представляет собой Н. В другом варианте осуществления R4 в соединении формулы XIVa представляет собой Н. В другом варианте осуществления R4 в соединении формулы XIVa представляет собой СН3. В другом варианте осуществления R4 в соединении формулы XIVa представляет собой ОСН3. В другом варианте осуществления R4 в соединении формулы XIVa представляет собой ОН. В другом варианте осуществления R4 в соединении формулы XIVa представляет собой 4-Cl. В другом варианте осуществления R4 в соединении формулы XIVa представляет собой 4-N(Me)2. В другом варианте осуществления R4 в соединении формулы XIVa представляет собой OBn. В другом варианте осуществления R1 в соединении формулы XIVa представляет собой ОСН3; m равно 3 и R2 представляет собой Н. В другом варианте осуществления R1 в соединении формулы XIVa представляет собой F; m равно 1, и R2 представляет собой Н. Неограничивающие примеры соединений формулы XIVa выбраны из следующих: (4-фторфенил)(2-фенил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон (11af), (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон (11cb), (4-фторфенил)(1-(фенилсульфонил)-2-(п-толил)-1Н-имидазол-4-ил)метанон (11db), (2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11fb), (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ga), (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11gb), (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ha), (2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11jb), (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12gba), (1-бензил-2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12daa); (1-метил-2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12dab); (4-фторфенил)(2-(4-метоксифенил)-1-метил-1Н-имидазол-4-ил)метанон (12cba).
В одном варианте осуществления настоящее изобретение касается соединения формулы XV:
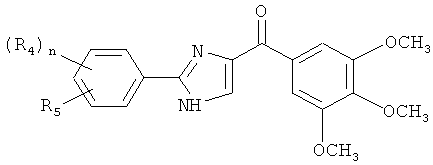
где R4 и R5 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R4 в соединении формулы XV представляет собой Н. В другом варианте осуществления R4 в соединении формулы XV представляет собой F. В другом варианте осуществления R4 в соединении формулы XV представляет собой Cl. В другом варианте осуществления R4 в соединении формулы XV представляет собой Br. В другом варианте осуществления R4 в соединении формулы XV представляет собой I. В другом варианте осуществления R4 в соединении формулы XV представляет собой N(Me)2. В другом варианте осуществления R4 в соединении формулы XV представляет собой OBn. В другом варианте осуществления R4 в соединении формулы XV представляет собой ОСН3. В другом варианте осуществления R4 в соединении формулы XV представляет собой СН3. В другом варианте осуществления R4 в соединении формулы XV представляет собой CF3. Неограничивающие примеры соединений формулы XV выбраны из следующих: (2-фенил-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12аа), (2-(4-фторфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ba), (2-(4-метоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12са), (2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12da), (3,4,5-триметоксифенил)(2-(3,4,5-триметоксифенил)-1Н-имидазол-4-ил)метанон (12еа), (2-(4-хлорфенил)-1Н-имидазо-4-ил)(3,4,5-триметоксифенил)метанон (12fa), (2-(4-(диметиламино)фенил)-1Н-имидазо-4-ил)(3,4,5-триметоксифенил)метанон (12ga), (2-(3,4-диметоксифенил)-1Н-имидазо-4-ил)(3,4,5-триметоксифенил)метанон (12ha), (2-(2-(трифторметил)фенил)-1Н-имидазо-4-ил)(3,4,5-триметоксифенил)метанон (12ia), (2-(4-(бензилокси)фенил)-1Н-имидазо-4-ил)(3,4,5-триметоксифенил)метанон (12ja), (2-(4-гидроксифенил)-1Н-имидазо-4-ил)(3,4,5-триметоксифенил)метанон (12ka), (2-(4-бромфенил)-1Н-имидазо-4-ил)(3,4,5-триметоксифенил)метанон (12la), (2-(4-(трифторметил)фенил)-1Н-имидазо-4-ил)(3,4,5-триметоксифенил)метанон (12ра).
В одном варианте осуществления настоящее изобретение касается соединения формулы XVI:
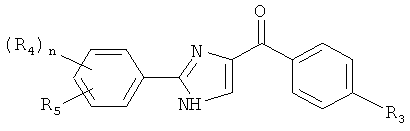
где R4 и R5 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкил, COOH, С(O)O-алкил или С(O)Н;
R3 представляет собой I, Br, Cl, F;
i представляет собой целое число от 0 до 5; и
n представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R3 в соединении формулы XVI представляет собой галоген. В другом варианте осуществления R3 представляет собой F. В другом варианте осуществления R3 представляет собой Cl. В другом варианте осуществления R3 представляет собой Br. В другом варианте осуществления R3 представляет собой I. В другом варианте осуществления R4 представляет собой Н. В другом варианте осуществления R4 представляет собой ОСН3. В другом варианте осуществления R4 представляет собой ОСН3; n равно 3 и R5 представляет собой Н. В другом варианте осуществления R4 представляет собой СН3. В другом варианте осуществления R4 представляет собой F. В другом варианте осуществления R4 представляет собой Cl. В другом варианте осуществления R4 представляет собой Br. В другом варианте осуществления R4 представляет собой I. В другом варианте осуществления R4 представляет собой N(Me)2. В другом варианте осуществления R4 представляет собой OBn. В другом варианте осуществления R3 представляет собой F; R5 представляет собой атом водорода; n равно 1 и R4 представляет собой 4-Cl. В другом варианте осуществления R3 представляет собой F; R5 представляет собой атом водорода; n равно 1 и R4 представляет собой 4-ОСН3. В другом варианте осуществления R3 представляет собой F; R5 представляет собой атом водорода; n равно 1 и R4 представляет собой 4-СН3. В другом варианте осуществления R3 представляет собой F; R5 представляет собой атом водорода; n равно 1, и R4 представляет собой 4-N(Me)2. В другом варианте осуществления R3 представляет собой F; R5 представляет собой атом водорода; n равно 1 и R4 представляет собой 4-OBn. Неограничивающие примеры соединений формулы XVI выбраны из следующих: (4-фторфенил)(2-фенил-1Н-имидазол-4-ил)метанон (12af), (4-фторфенил)(2-(4-метоксифенил)-1Н-имидазол-4-ил)метанон (12cb), (4-фторфенил)(2-(п-толил)-1Н-имидазол-4-ил)метанон (12db), 4-фторфенил)(2-(3,4,5-триметоксифенил)-1Н-имидазол-4-ил)метанон (12eb), (2-(4-хлорфенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12fb), (2-(4-(диметиламино)фенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12gb), (2-(4-(бензилокси)фенил)-1H-имидазол-4-ил)(4-фторфенил)метанон (12jb).
В одном варианте осуществления настоящее изобретение касается соединения формулы XVII:
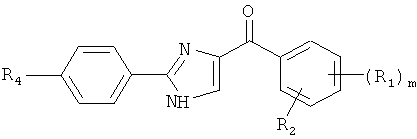
где R4 представляет собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
где R1 и R2 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
и
m представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R4 в соединении формулы XVII представляет собой галоген. В другом варианте осуществления R4 представляет собой F. В другом варианте осуществления R4 представляет собой Cl. В другом варианте осуществления R4 представляет собой Br. В другом варианте осуществления R4 представляет собой I. В другом варианте осуществления R4 представляет собой ОСН3. В другом варианте осуществления R4 представляет собой СН3. В другом варианте осуществления R4 представляет собой N(Me)2. В другом варианте осуществления R4 представляет собой CF3. В другом варианте осуществления R4 представляет собой ОН. В другом варианте осуществления R4 представляет собой OBn. В другом варианте осуществления R1 в соединении формулы XVII представляет собой галоген. В другом варианте осуществления R1 в соединении формулы XVII представляет собой F. В другом варианте осуществления R1 в соединении формулы XVII представляет собой Cl. В другом варианте осуществления R1 в соединении формулы XVII представляет собой Br. В другом варианте осуществления R1 в соединении формулы XVII представляет собой I. В другом варианте осуществления R1 в соединении формулы XVII представляет собой ОСН3. В другом варианте осуществления R1 в соединении формулы XVII представляет собой ОСН3, m равно 3, и R2 представляет собой Н. В другом варианте осуществления R1 в соединении формулы XVII представляет собой F, m равно 1, и R2 представляет собой Н. В другом варианте осуществления R4 представляет собой F; R2 представляет собой атом водорода; n равно 3, и R1 представляет собой ОСН3. В другом варианте осуществления R4 представляет собой ОСН3; R2 представляет собой атом водорода; n равно 3, и R1 представляет собой ОСН3. В другом варианте осуществления R4 представляет собой СН3; R2 представляет собой атом водорода; n равно 3, и R1 представляет собой ОСН3. В другом варианте осуществления R4 представляет собой Cl; R2 представляет собой атом водорода; n равно 3, и R1 представляет собой ОСН3. В другом варианте осуществления R4 представляет собой N(Me)2; R2 представляет собой атом водорода; n равно 3, и R1 представляет собой ОСН3. В одном варианте осуществления R4 в соединении формулы XVII представляет собой галоген, R1 представляет собой Н, и R2 представляет собой галоген. В одном варианте осуществления R4 в соединении формулы XVII представляет собой галоген, R1 представляет собой галоген и R2 представляет собой Н. В одном варианте осуществления R4 в соединении формулы XVII представляет собой алкокси-группу, R1 представляет собой галоген и R2 представляет собой Н. В одном варианте осуществления R4 в соединении формулы XVII представляет собой метокси-группу, R1 представляет собой галоген, и R2 представляет собой Н. Неограничивающие примеры соединений формулы XVII выбраны из следующих: (2-(4-фторфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ba), (2-(4-метоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12са), (4-фторфенил)(2-(4-метоксифенил)-1Н-имидазол-4-ил)метанон (12cb), (2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12da), (4-фторфенил)(2-(п-толил)-1Н-имидазол-4-ил)метанон (12db), (4-гидрокси-3,5-диметоксифенил)(2-(п-толил)-1Н-имидазол-4-ил)метанон (12dc), (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12fa), (2-(4-хлорфенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12fb), (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-тригидроксифенил)метанон (13fa), (2-(4-(диметиламино)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ga), (2-(4-(диметиламино)фенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12gb), (2-(4-(бензилокси)фенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12jb), (2-(4-гидроксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ka), (2-(4-бромфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12la), (2-(4-(трифторметил)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ра).
В другом варианте осуществления соединение формулы XVII представлено структурой формулы 12fb:
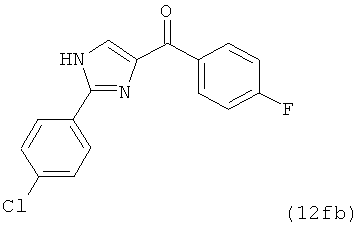 .
.
В другом варианте осуществления соединение формулы XVII представлено структурой формулы 12cb:
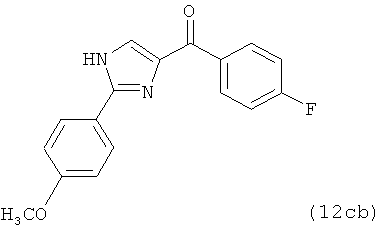 .
.
В одном варианте осуществления настоящее изобретение касается соединения формулы XVIII:
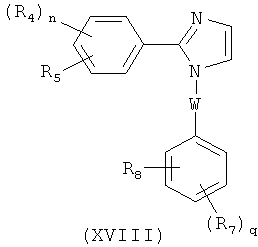 ,
,
где W представляет собой С=O, C=S, SO2 или S=O;
R4 и R7 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкил, COOH, С(O)O-алкил или С(O)Н;
R5 и R8 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(СН2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, COOH, С(O)O-алкил или С(O)Н;
n представляет собой целое число от 1 до 4;
i представляет собой целое число от 0 до 5; и
q представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления W в соединении формулы XVIII представляет собой С=O. В другом варианте осуществления W в соединении формулы XVIII представляет собой SO2. В другом варианте осуществления R4 в соединении формулы XVIII представляет собой Н. В другом варианте осуществления R4 в соединении формулы XVIII представляет собой NO2. В другом варианте осуществления R4 в соединении формулы XVIII представляет собой OBn. В другом варианте осуществления R7 в соединении формулы XVIII представляет собой Н. В другом варианте осуществления R7 в соединении формулы XVIII представляет собой ОСН3. В другом варианте осуществления R7 в соединении формулы XVIII представляет собой ОСН3, и q равно 3. Неограничивающие примеры соединений формулы XVII выбраны из следующих: (4-метоксифенил)(2-фенил-1Н-имидазол-1-ил)метанон (12aba), (2-фенил-1Н-имидазол-1-ил)(3,4,5-триметоксифенил)метанон (12ааа), 2-фенил-1-(фенилсульфонил)-1Н-имидазол (10а), 2-(4-нитрофенил)-1-(фенилсульфонил)-1Н-имидазол (10x), 2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1Н-имидазол (10j).
В одном варианте осуществления настоящее изобретение касается соединения формулы XIX:
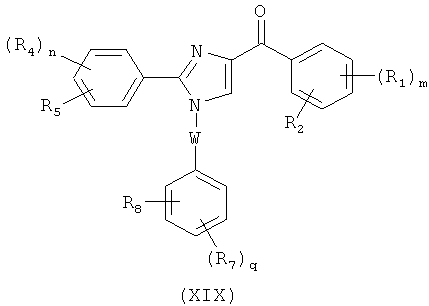 ,
,
где
W представляет собой С=O, C=S, SO2, S=O;
R1, R4 и R7 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
R2, R5 и R8 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
m представляет собой целое число от 1 до 4;
n представляет собой целое число от 1 до 4;
i представляет собой целое число от 0 до 5; и
q представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R1, R4 и R7 в формуле XIX независимо представляют собой Н. В другом варианте осуществления R1, R4 и R7 в формуле XIX независимо представляют собой O-алкил. В другом варианте осуществления R1, R4 и R7 в формуле XIX независимо представляют собой галоген. В другом варианте осуществления R1, R4 и R7 в формуле XIX независимо представляют собой CN. В другом варианте осуществления R1, R4 и R7 в формуле XIX независимо представляют собой ОН. В другом варианте осуществления R1, R4 и R7 в формуле XIX независимо представляют собой алкил. В другом варианте осуществления R1, R4 и R7 в формуле XIX независимо представляют собой OCH2Ph. В одном варианте осуществления R2, R5 и R8 в формуле XIX независимо представляют собой Н. В другом варианте осуществления R2, R5 и R8 в формуле XIX независимо представляют собой O-алкил. В другом варианте осуществления R2, R5 и R8 в формуле XIX независимо представляют собой галоген. В другом варианте осуществления R2, R5 и R8 в формуле XIX независимо представляют собой CN. В другом варианте осуществления R2, R5 и R8 в формуле XIX независимо представляют собой ОН. В другом варианте осуществления R2, R5 и R8 в формуле XIX независимо представляют собой алкил. В другом варианте осуществления R2, R5 и R8 в формуле XIX независимо представляют собой OCH2Ph. В другом варианте осуществления R5, R2 и R8 в формуле XIX представляют собой Н, R4 представляет собой 4-N(Me)2, R1 представляет собой ОСН3, m равно 3, и R7 представляет собой ОСН3. В другом варианте осуществления R5, R2, R7 и R8 в формуле XIX представляют собой Н, R4 представляет собой 4-Br, R1 представляет собой ОСН3, и m равно 3. В другом варианте осуществления W представляет собой SO2. В другом варианте осуществления W представляет собой С=O. В другом варианте осуществления W представляет собой C=S. В другом варианте осуществления W представляет собой S=O. Неограничивающие примеры соединений формулы XIX выбраны из следующих: (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11gaa); (2-(4-бромфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11la), (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон (11cb), (2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11fb), (4-фторфенил)(2-фенил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон (11af), (4-фторфенил)(1-(фенилсульфонил)-2-(п-толил)-1Н-имидазол-4-ил)метанон (11db), (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ga), (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11gb), (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ha), (2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11jb), (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12gba).
В другом варианте осуществления соединение формулы XIX представлено структурой формулы 11cb:
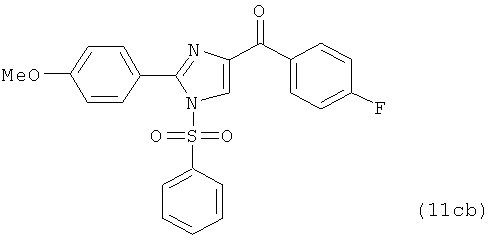 .
.
В другом варианте осуществления соединение формулы XIX представлено структурой формулы 11fb:
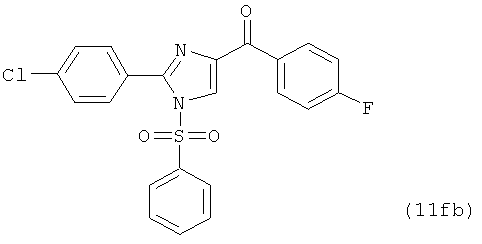 .
.
В одном варианте осуществления настоящее изобретение касается соединения формулы XX:
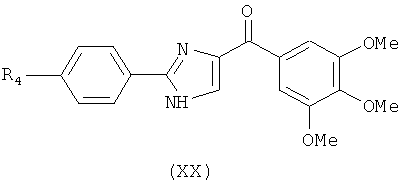 ,
,
где
R4 представляет собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, OH, CN, NO2, -NHCO-алкил, COOH, С(O)O-алкил или С(O)Н; и i представляет собой целое число от 0 до 5;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R4 в соединении формулы XX представляет собой Н. В другом варианте осуществления R4 в соединении формулы XX представляет собой галоген. В другом варианте осуществления R4 представляет собой F. В другом варианте осуществления R4 представляет собой Cl. В другом варианте осуществления R4 представляет собой Br. В другом варианте осуществления R4 представляет собой I. В другом варианте осуществления R4 представляет собой алкил. В другом варианте осуществления R4 представляет собой метил. Неограничивающие примеры соединений формулы XX выбраны из следующих: (2-фенил-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12аа), (2-(4-фторфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ba), (2-(4-метоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12са), (2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12da), (3,4,5-триметоксифенил)(2-(3,4,5-триметоксифенил)-1Н-имидазол-4-ил)метанон (12еа), (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12fa), (2-(4-(диметиламино)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ga), (2-(3,4-диметоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ha), (2-(2-(трифторметил)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ia), (2-(4-(бензилокси)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ja), (2-(4-гидроксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ka), (2-(4-бромфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12la), (2-(4-(трифторметил)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ра).
В другом варианте осуществления соединение формулы XX представлено структурой формулы 12da:
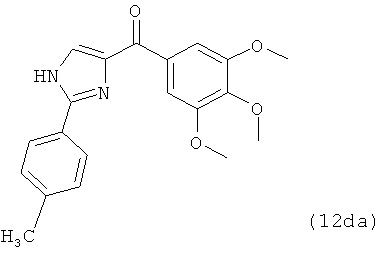 .
.
В другом варианте осуществления соединение формулы XX представлено структурой формулы 12fa:
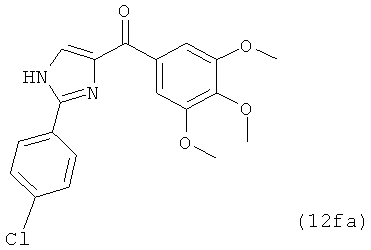 .
.
В одном варианте осуществления настоящее изобретение касается соединения формулы XXI:
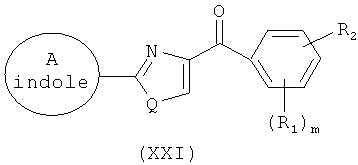 ,
,
где
А представляет собой индолил;
Q представляет собой NH, О или S;
R1 и R2 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н; и
i представляет собой целое число от 0 до 5;
где указанный А необязательно замещен замещенным или незамещенным O-алкилом, O-галогеналкилом, F, Cl, Br, I, галогеналкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, замещенным или незамещенным -SO2-арилом, замещенным или незамещенным C1-C5 линейным или разветвленным алкилом, замещенным или незамещенным галогеналкилом, замещенной или незамещенной алкиламино-группой, замещенным или незамещенным аминоалкилом, -OCH2Ph, замещенным или незамещенным -NHCO-алкилом, СООН, замещенным или незамещенным -C(O)Ph, замещенным или незамещенным С(O)O-алкилом, С(O)Н, -C(O)NH2, NO2 или их комбинацией;
i представляет собой целое число от 0 до 5; и
m представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R1 в соединении формулы XXI представляет собой ОСН3; m равно 3, и R2 представляет собой атом водорода. В другом варианте осуществления R1 представляет собой F; m равно 1, и R2 представляет собой атом водорода. В одном варианте осуществления Q в формуле XXI представляет собой О. В другом варианте осуществления Q в формуле XXI представляет собой NH. В другом варианте осуществления Q в формуле XXI представляет собой S.
В одном варианте осуществления цикл А в соединении формулы XXI представляет собой замещенный 5-индолил. В другом варианте осуществления заместитель представляет собой -(С=O)-арил. В другом варианте осуществления арил представляет собой 3,4,5-(ОСН3)3-Ph.
В другом варианте осуществления цикл А в соединении формулы XXI представляет собой 3-индолил. В другом варианте осуществления цикл А в соединении формулы XXI представляет собой 5-индолил. В другом варианте осуществления цикл А в соединении формулы XXI представляет собой 2-индолил. Неограничивающие примеры соединений формулы XXI выбраны из следующих: (5-(4-(3,4,5-триметоксибензоил)-1Н-имидазол-2-ил)-1Н-индол-2-ил)(3,4,5-триметоксифенил)метанон (15хаа); (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-2-(3,4,5-триметоксибензоил)-1Н-индол-5-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (16хаа); 2-(1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (17ya); (2-(1Н-индол-2-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (62а); и (2-(1Н-индол-5-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (66а).
В одном варианте осуществления настоящее изобретение касается соединения формулы XXIa:
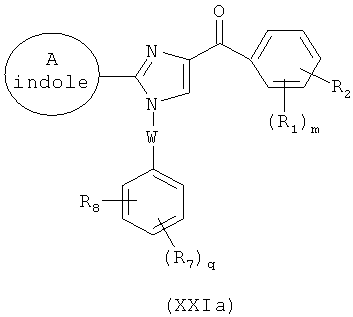 ,
,
где
W представляет собой С=O, OS, SO2, S=O;
А представляет собой индолил;
R1 и R2 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
R7 и R8 независимо представляют собой Н, O-алкил, I, Br, Cl, F, алкил, галогеналкил, аминоалкил, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, OCH2Ph, ОН, CN, NO2, -NHCO-алкил, СООН, С(O)O-алкил или С(O)Н;
где указанный А необязательно замещен замещенным или незамещенным O-алкилом, O-галогеналкилом, F, Cl, Br, I, галогеналкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(СН2)iNHCH3, -(CH2)iNH2, -(СН2)iN(СН3)2, -ОС(O)CF3, замещенным или незамещенным -SO2-арилом, замещенным или незамещенным C1-C5 линейным или разветвленным алкилом, замещенным или незамещенным галогеналкилом, замещенной или незамещенной алкиламино-группой, замещенным или незамещенным аминоалкилом, -OCH2Ph, замещенным или незамещенным -NHCO-алкилом, СООН, замещенным или незамещенным -C(O)Ph, замещенным или незамещенным С(O)O-алкилом, С(O)Н, -C(O)NH2, NO2 или их комбинацией;
i представляет собой целое число от 0 до 5; и
m представляет собой целое число от 1 до 4;
q представляет собой целое число от 1 до 4;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления R1 в соединении формулы XXIa представляет собой ОСН3; m равно 3, и R2 представляет собой атом водорода. В другом варианте осуществления R1 представляет собой F; m равно 1, и R2 представляет собой атом водорода. В другом варианте осуществления цикл А в соединении формулы XXIa представляет собой замещенный 5-индолил. В другом варианте осуществления цикл А в соединении формулы XXIa представляет собой 3-индолил. Неограничивающие примеры соединений формулы XXIa выбраны из следующих: (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-2-(3,4,5-триметоксибензоил)-1Н-индол-5-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (16хаа); (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (17yaa).
В одном варианте осуществления настоящее изобретение касается соединения формулы XXII:
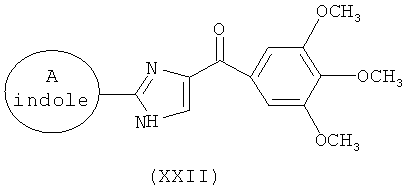 ,
,
где
А представляет собой индолил;
где указанный А необязательно замещен замещенным или незамещенным O-алкилом, O-галогеналкилом, F, Cl, Br, I, галогеналкилом, CF3, CN, -CH2CN, NH2, гидроксилом, -(CH2)iNHCH3, -(CH2)iNH2, -(CH2)iN(CH3)2, -ОС(O)CF3, замещенным или незамещенным -SO2-арилом, замещенным или незамещенным C1-С5 линейным или разветвленным алкилом, замещенным или незамещенным галогеналкилом, замещенной или незамещенной алкиламино-группой, замещенным или незамещенным аминоалкилом, -OCH2Ph, замещенным или незамещенным -NHCO-алкилом, СООН, замещенным или незамещенным -C(O)Ph, замещенным или незамещенным С(O)O-алкилом, С(O)Н, -C(O)NH2, NO2 или их комбинацией;
i представляет собой целое число от 0 до 5;
или его фармацевтически приемлемая соль, гидрат, полиморф, метаболит, таутомер или изомер.
В одном варианте осуществления цикл А в соединении формулы XXII представляет собой замещенный 5-индолил. В другом варианте осуществления заместитель представляет собой -(С=O)-арил. В другом варианте осуществления арил представляет собой 3,4,5-(ОСН3)3-Ph.
В другом варианте осуществления цикл А в соединении формулы XXII представляет собой 3-индолил. Неограничивающие примеры соединений формулы XXII выбраны из следующих: (5-(4-(3,4,5-триметоксибензоил)-1Н-имидазол-2-ил)-1Н-индол-2-ил)(3,4,5-триметоксифенил)метанон (15хаа); 2-(1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (17ya).
В другом варианте осуществления соединение формулы XXI или XXII представлено структурой формулы 17ya:
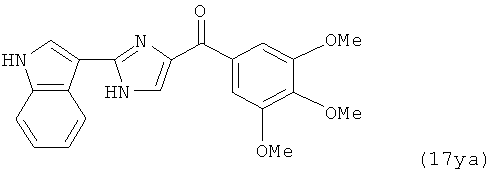 .
.
В одном варианте осуществления Q в соединении формулы XII представляет собой Н, и Р представляет собой . В другом варианте осуществления Р в соединении формулы XII представляет собой Н, и Q представляет собой . В другом варианте осуществления Р в соединении формулы XII представляет собой , и Q представляет собой SO2-Ph. В одном варианте осуществления Q в соединении формулы XII представляет собой Н, и Р представляет собой , где W представляет собой С=O. В другом варианте осуществления W в соединении формулы XII, XVIII, XIX, или XXIa представляет собой С=O. В другом варианте осуществления W в соединении формулы XII, XVIII, XIX, или XXIa представляет собой SO2. В другом варианте осуществления W в соединении формулы XII, XVIII, XIX, или XXIa представляет собой C=S. В другом варианте осуществления W в соединении формулы XII, XVIII, XIX, или XXIa представляет собой S=O.
В одном варианте осуществления Z в соединении формулы XIII представляет собой кислород. В другом варианте осуществления Z в соединении формулы XIII представляет собой серу.
В одном варианте осуществления R5 в соединении формулы XII-XVI, XVIII или XIX представляет собой атом водорода, n равно 1 и R4 находится в параположении.
В одном варианте осуществления R4 в соединении формулы XII-XX представляет собой алкил. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой Н. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой метил (СН3). В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой O-алкил. В другом варианте осуществления R4 в соединении формулы XII-XIX представляет собой ОСН3. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой I. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой Br. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой F. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой Cl. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой N(Me)2. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой OBn. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой ОН. В другом варианте осуществления R4 в соединении формулы XII-XX представляет собой CF3.
В одном варианте осуществления R2 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой атом водорода; R1 представляет собой ОСН3, и m равно 3. В другом варианте осуществления R2 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой атом водорода; m равно 1, и R1 находится в параположении. В другом варианте осуществления R2 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой атом водорода; m равно 1, и R1 представляет собой I. В другом варианте осуществления R2 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой атом водорода; m равно 1, и R1 представляет собой Br. В другом варианте осуществления R2 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой атом водорода; m равно 1, и R1 представляет собой F. В другом варианте осуществления R2 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой атом водорода; m равно 1, и R1 представляет собой Cl. В другом варианте осуществления R1 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой I. В другом варианте осуществления R1 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой Br. В другом варианте осуществления R1 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой Cl. В другом варианте осуществления R1 в соединении формулы XII, XIII, XIV, XIVa, XVII, XIX, XXI или XXIa представляет собой F.
В одном варианте осуществления Q в соединении формулы XII представляет собой Н, и Р представляет собой , где W представляет собой С=О. Неограничивающие примеры соединений формулы XII-XVII и XX-XXII выбраны из следующих: (2-фенил-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12аа); (4-метоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ab); (3-метоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ас); (3,5-диметоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ad); (3,4-диметоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ае); (4-фторфенил)(2-фенил-1Н-имидазол-4-ил)метанон (12af); (3-фторфенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ag); (2-фенил-1Н-имидазол-4-ил)(п-толил)метанон (12ah); (2-фенил-1Н-имидазол-4-ил)(м-толил)метанон (12ai); (2-(4-фторфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ba); (2-(4-метоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12са); (4-фторфенил)(2-(4-метоксифенил)-1Н-имидазол-4-ил)метанон (12cb); (2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12da); (4-фторфенил)(2-(п-толил)-1Н-имидазол-4-ил)метанон (12db); (4-фторфенил)(2-(п-толил)-1Н-имидазол-4-ил)метанон гидрохлорид (12db-HCl); (4-гидрокси-3,5-диметоксифенил)(2-(п-толил)-1Н-имидазол-4-ил)метанон (12dc); (3,4,5-триметоксифенил)(2-(3,4,5-триметоксифенил)-1Н-имидазол-4-ил)метанон (12еа); (4-фторфенил)(2-(3,4,5-триметоксифенил)-1Н-имидазол-4-ил)метанон (12eb); (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12fa); (2-(4-хлорфенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12fb); (2-(4-хлорфенил)-1Н-имидазол-4-ил)(4-гидрокси-3,5-диметоксифенил)метанон (12fc); (2-(4-(диметиламино)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ga); (2-(4-(диметиламино)фенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12gb); (2-(3,4-диметоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ha); (2-(3,4-диметоксифенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12hb); (2-(2-(трифторметил)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ia); (4-фторфенил)(2-(2-(трифторметил)фенил)-1Н-имидазол-4-ил)метанон (12ib); (2-(4-(бензилокси)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ja); (2-(4-(бензилокси)фенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12jb); (2-(4-гидроксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ka); (2-(4-(гидроксифенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (12kb); (2-(4-бромфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12la); (2-(4-(трифторметил)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12ра); (3,4,5-тригидроксифенил)(2-(3,4,5-тригидроксифенил)-1Н-имидазол-4-ил)метанон (13еа); (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-тригидроксифенил)метанон (13fa); и 2-(3,4-дигидроксифенил)-1Н-имидазол-4-ил)(3,4,5-тригидроксифенил)метанон (13ha).
В одном варианте осуществления Р в соединении формулы XII представляет собой , и Q представляет собой SO2-Ph. Неограничивающие примеры соединений формулы XII, где Р в соединении формулы XII представляет собой , и Q представляет собой SO2-Ph, выбраны из следующих: (4-метоксифенил)(2-фенил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон (11ab); (3-метоксифенил)(2-фенил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон (11ас); (2-фенил-1-(фенилсульфонил)-1Н-имидазол-4-ил)(п-толил)метанон (11ah); (4-фторфенил)(2-фенил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон (11af); (3-фторфенил)(2-фенил-1-(фенилсульфонил)-1H-имидазол-4-ил)метанон (11ag); (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон (11cb); (1-(фенилсульфонил)-2-(п-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11da); (4-фторфенил)(1-(фенилсульфонил)-2-(п-толил)-1Н-имидазол-4-ил)метанон (11db); (1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ea); (4-фторфенил)(1-(фенилсульфонил)-2-(3,4,5-триметоксифенил)-1Н-имидазол-4-ил)метанон (11eb); (2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11fb); (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ga); (2-(4-(диметиламино)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11gb); (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ha); (2-(3,4-диметоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11hb); (1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11ia); (1-(фенилсульфонил)-2-(2-(трифторметил)фенил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11ib); и (2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанон (11jb); (2-(4-бромфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11la); (1-(фенилсульфонил)-2-(4-(трифторметил)фенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11pa).
В одном варианте осуществления R4 и R5 в соединениях формулы XIII-XVI представляют собой атомы водорода. Неограничивающие примеры соединений формулы XIII-XVI, где R4 и R5 представляют собой атомы водорода, выбраны из следующих: (2-фенил-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12аа); (4-метоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ab); (3-метоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ас); (3,5-диметоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ad); (3,4-диметоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ае); (4-фторфенил)(2-фенил-1Н-имидазол-4-ил)метанон (12af); (3-фторфенил)(2-фенил-1Н-имидазол-4-ил)метанон (12ag); (2-фенил-1Н-имидазол-4-ил)(п-толил)метанон (12ah); и (2-фенил-1Н-имидазол-4-ил)(м-толил)метанон (12ai).
В одном варианте осуществления Р в соединении формулы XII представляет собой Н, и Q представляет собой . В другом варианте осуществления W представляет собой С=O. В другом варианте осуществления W в соединении формулы XVIII представляет собой С=O. Неограничивающие примеры соединений формулы XVIII, где W представляет собой C=O, выбраны из (4-метоксифенил)(2-фенил-1Н-имидазол-1-ил)метанон (12aba) и (2-фенил-1Н-имидазол-1-ил)(3,4,5-триметоксифенил)метанон (12ааа).
В другом варианте осуществления W в соединении формулы XVIII представляет собой SO2. Неограничивающие примеры в соединении формулы XVIII, где W представляет собой SO2, выбраны из следующих: 2-фенил-1-(фенилсульфонил)-1Н-имидазол (10а); 2-(4-нитрофенил)-1-(фенилсульфонил)-1Н-имидазол (10х) и 2-(4-(бензилокси)фенил)-1-(фенилсульфонил)-1Н-имидазол (10j).
При использовании в настоящем тексте "моноциклические, конденсированные или полициклические арильные или (гетеро)циклические системы" могут представлять собой любой такой цикл, включая (но не ограничиваясь только ими) фенил, бифенил, трифенил, нафтил, циклоалкил, циклоалкенил, циклодиенил, флуорен, адамантан и т.д.
"Насыщенные или ненасыщенные N-гетероциклы" могут представлять собой любой такой N-содержащий гетероцикл, включая (но не ограничиваясь только ими) аза- и диаза-циклоалкилы, такие как азиридинил, азетидинил, диазатидинил, пирролидинил, пиперидинил, пиперазинил, и азоканил, пирролил, пиразолил, имидазолил, пиридинил, пиримидинил, пиразинил, пиридазинил, триазинил, тетразинил, пирролизинил, индолил, хинолинил, изохинолинил, бензимидазолил, индазолил, хинолизинил, циннолинил, хинололинил, фталазинил, нафтиридинил, хиноксалинил и т.п.
"Насыщенные или ненасыщенные O-гетероциклы" могут представлять собой любой такой O-содержащий гетероцикл, включая (но не ограничиваясь только ими) оксиранил, оксетанил, тетрагидрофуранил, тетрагидропиранил, диоксанил, фуранил, пирилий, бензофуранил, бензодиоксолил и т.п.
"Насыщенные или ненасыщенные S-гетероциклы" могут представлять собой любой такой S-содержащий гетероцикл, включая (но не ограничиваясь только ими) тиранил, тиетанил, тетрагидротиофенил, дитиоланил, тетрагидротиопиранил, тиофенил, тиепинил, тианафтенил и т.п.
"Насыщенные или ненасыщенные смешанные гетероциклы" могут представлять собой любой гетероцикл, содержащий два или более S-, N- или O-гетероатомов, включая (но не ограничиваясь только ими) оксатиоланил, морфолинил, тиоксанил, тиазолил, изотиазолил, тиадиазолил, оксазолил, изоксазолил, оксадиазолил и т.п.
При использовании в настоящем тексте термин "алифатический линейный или разветвленный углеводород" относится как к алкиленовым группам, которые содержат от одного до максимального указанного количества атомов углерода, так и к алкенильным группам и алкинильным группам, которые содержат от двух до максимального указанного количества атомов углерода, где атомы углерода могут формировать как линейную цепочку, так и разветвленную цепочку. Если не указано иное, углеводород может содержать до примерно 30 атомов углерода, или до примерно 20 атомов углерода, или до примерно 10 атомов углерода. Алкенильные и алкинильные группы могут быть мононенасыщенными или полиненасыщенными. В другом варианте осуществления алкил содержит C1-С6 атомов углерода. В другом варианте осуществления алкил содержит C1-C8 атомов углерода. В другом варианте осуществления алкил содержит C1-С10 атомов углерода. В другом варианте осуществления алкил содержит C1-C12 атомов углерода. В другом варианте осуществления алкил содержит C1-C5 атомов углерода.
При использовании в настоящем тексте термин "алкил" может означать любую линейную или разветвленную алкильную группу, содержащую до примерно 30 атомов углерода, если не указано иное. В другом варианте осуществления алкил содержит C1-С6 атомов углерода. В другом варианте осуществления алкил содержит C1-C8 атомов углерода. В другом варианте осуществления алкил содержит C1-C10 атомов углерода. В другом варианте осуществления алкил содержит C1-C12 атомов углерода. В другом варианте осуществления алкил содержит C1-C20 атомов углерода. В другом варианте осуществления циклическая алкильная группа содержит 3-8 атомов углерода. В другом варианте осуществления разветвленный алкил представляет собой алкил, замещенный алкильными боковыми цепями, содержащими 1-5 атомов углерода.
Алкильная группа может быть отдельным заместителем или она может быть компонентом большего заместителя, такого как алкокси-группа, галогеналкил, арилалкил, алкиламино-группа, диалкиламино-группа, алкиламидо-группа, алкилмочевина и т.п. Предпочтительными алкильными группами являются метил, этил и пропил, и, таким образом, галогенметил, дигалогенметил, тригалогенметил, галогенэтил, дигалогенэтил тригалогенэтил, галогенпропил, дигалогенпропил, тригалогенпропил, метокси, этокси, пропокси, арилметил, арилэтил, арилпропил, метиламино, этиламино, пропиламино, диметиламино, диэтиламино, метиламидо, ацетамидо, пропиламидо, галогенметиламидо, галогенэтиламидо, галогенпропиламидо, метил-мочевина, этилмочевина, пропилмочевина и т.п.
При использовании в настоящем тексте термин "арил" относится к любому ароматическому кольцу, непосредственно связанному с другой группой. Арильная группа может быть отдельным заместителем или арильная группа может быть компонентом большего заместителя, такого как арилалкил, ариламино, ариламидо и т.п. Примеры арильных групп включают (но не ограничиваются только ими) фенил, толил, ксилил, фуранил, нафтил, пиридинил, пиримидинил, пиридазинил, пиразинил, триазинил, тиазолил, оксазолил, изооксазолил, пиразолил, имидазолил, тиофенил, пирролил, фенилметил, фенилэтил, фениламино, фениламидо и т.п.
При использовании в настоящем тексте термин "аминоалкил" относится к аминогруппе, замещенной алкильной группой, определение которой дано выше. Термин «аминоалкил» относится к моноалкиламину, диалкиламину и триалкиламину. Неограничивающими примерами аминоалкильных групп являются -N(Me)2, -NHMe, -NH3.
В другом варианте осуществления "галогеналкильная" группа означает алкильную группу, определение которой дано выше, которая имеет в качестве заместителей один или более атомов галогенов, например F, Cl, Br или I. Неограничивающими примерами галогеналкильных групп являются CF3, CF2CF3, CH2CF3.
В одном варианте осуществления в настоящем изобретении описано соединение по настоящему изобретению или его изомер, метаболит, фармацевтически приемлемая соль, фармацевтический продукт, таутомер, гидрат, N-оксид, полиморф или кристаллическая форма, или их комбинации. В одном варианте осуществления в настоящем изобретении описан изомер соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описан метаболит соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описана фармацевтически приемлемая соль соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описан фармацевтический продукт соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описан таутомер соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описан гидрат соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описан N-оксид соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описан полиморф соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описана кристаллическая форма соединения по настоящему изобретению. В другом варианте осуществления в настоящем изобретении описана композиция, содержащая соединение по настоящему изобретению, или, в другом варианте осуществления, - комбинация изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа или кристаллической формы соединения по настоящему изобретению.
В одном варианте осуществления термин "изомер" включает (но не ограничивается только ими) оптические изомеры и аналоги, структурные изомеры и аналоги, конформационные изомеры и аналоги, и т.п.
В одном варианте осуществления соединения по настоящему изобретению представляют собой чистые (E)-изомеры. В другом варианте осуществления соединения по настоящему изобретению представляют собой чистые (Z)-изомеры. В другом варианте осуществления соединения по настоящему изобретению представляют собой смеси (Е) и (Z) изомеров. В одном варианте осуществления соединения по настоящему изобретению представляют собой чистые (R)-изомеры. В другом варианте осуществления соединения по настоящему изобретению представляют собой чистые (S)-изомеры. В другом варианте осуществления соединения по настоящему изобретению представляют собой смеси (R) и (S) изомеров.
Соединения по настоящему изобретению могут также присутствовать в форме рацемической смеси, содержащей практически эквивалентные количества стереоизомеров. В другом варианте осуществления соединения по настоящему изобретению могут быть получены или другим путем выделены, с использованием известных методик, с получением стереоизомера, практически не содержащего иного стереоизомера (т.е. практически в чистом виде). Под термином «практически чистый» понимается, что стереоизомер имеет чистоту по меньшей мере примерно 95%, более предпочтительно по меньшей мере примерно 98%, наиболее предпочтительно по меньшей мере примерно 99%.
Соединения по настоящему изобретению могут также находиться в форме гидрата, это означает, что соединение дополнительно включает стехиометрическое или нестехиометрическое количество воды, связанной нековалентными межмолекулярными силами.
Соединения по настоящему изобретению могут существовать в форме одного или более возможных таутомеров, и, в зависимости от конкретных условий, может быть возможным разделить некоторые или все таутомеры на индивидуальные и различающиеся вещества. Следует понимать, что все возможные таутомеры, включая все дополнительные енольные и кето-таутомеры и/или изомеры, входят в объем настоящего изобретения. Например, включены следующие таутомеры (не ограничиваясь только ими):
Таутомерия имидазольного цикла
Настоящее изобретение включает "фармацевтически приемлемые соли" соединений по настоящему изобретению, которые могут быть получены реакцией соединения по настоящему изобретению с кислотой или основанием. Некоторые соединения, в частности содержащие кислые или основные группы, могут также присутствовать в форме соли, предпочтительно фармацевтически приемлемой соли. Термин "фармацевтически приемлемая соль" относится к солям, которые сохраняют биологическую эффективность и свойства свободных оснований или свободных кислот и которые не являются нежелательными в биологическом или другом отношении. Соли формируются с неорганическими кислотами, такими как хлористоводородная кислота, бромистоводородная кислота, серная кислота, азотная кислота, фосфорная кислота и т.п., и органическими кислотами, такими как уксусная кислота, пропионовая кислота, гликолевая кислота, виноградная кислота, щавелевая кислота, малеиновая кислота, малоновая кислота, янтарная кислота, фумаровая кислота, винная кислота, лимонная кислота, бензойная кислота, коричная кислота, миндальная кислота, метансульфоновая кислота, этансульфоновая кислота, п-толуолсульфокислота, салициловая кислота, N-ацетилцистеин и т.п. Другие соли известны квалифицированным специалистам в данной области и могут быть легко адаптированы для применения согласно настоящему изобретению.
Подходящие фармацевтически приемлемые соли аминных соединений по настоящему изобретению можно получить с использованием неорганической кислоты или органической кислоты. В одном варианте осуществления примеры неорганических солей аминов представляют собой бисульфаты, бораты, бромиды, хлориды, гемисульфаты, гидроброматы, гидрохлораты, 2-гидроксиэтилсульфонаты (гидроксиэтансульфонаты), иодаты, иодиды, изотионаты, нитраты, персульфаты, фосфаты, сульфаты, сульфаматы, сульфанилаты, сульфоновые кислоты (алкилсульфонаты, арилсульфонаты, галоген-замещенные алкилсульфонаты, галоген-замещенные арилсульфонаты), сульфонаты и тиоцианаты.
В одном варианте осуществления примеры органических солей аминов могут быть выбраны из классов алифатических, циклоалифатических, ароматических, аралифатических, гетероциклических, карбоксильных и сульфоновых органических кислот, примерами которых являются ацетаты, аргинины, аспартаты, аскорбаты, адипаты, антранилаты, альгенаты, алканкарбоксилаты, замещенные алканкарбоксилаты, альгинаты, бензолсульфонаты, бензоаты, бисульфаты, бутираты, бикарбонаты, битартраты, цитраты, камфораты, камфорсульфонаты, циклогексилсульфаматы, циклопентанпропионаты, кальций-эдетаты, камсилаты, карбонаты, клавуланаты, циннаматы, дикарбоксилаты, диглюконаты, додецилсульфонаты, дигидрохлориды, деканоаты, энантуаты, этансульфонаты, эдетаты, эдизилаты, эстолаты, эзилаты, фумараты, формиаты, фториды, галактуронаты, глюконаты, глутаматы, гликоляты, глюкораты, глюкогептаноаты, глицерофосфаты, глуцептаты, гликолиларсанилаты, глутараты, глютаматы, гептаноаты, гексаноаты, гидроксималеаты, гидроксикарбоновые кислоты, гексилрезорцинаты, гидроксибензоаты, гидроксинафтоаты, гидрофлуораты, лактаты, лактобионаты, лаураты, малаты, малеаты, метиленбис(бета-оксинафтоаты), малонаты, манделаты, мезилаты, метансульфонаты, метилбромиды, метилнитраты, метилсульфонаты, монокалий-малеаты, мукаты, монокарбоксилаты, нафталинсульфонаты, 2-нафталинсульфонаты, никотинаты, нитраты, напсилаты, N-метилглюкамины, оксалаты, октаноаты, олеаты, памоаты, фенилацетаты, пикраты, фенилбензоаты, пивалаты, пропионаты, фталаты, фенилацетаты, пектинаты, фенилпропионаты, палмитаты, пантотенаты, полигалактураты, пируваты, хинаты, салицилаты, сукцинаты, стеараты, сульфанилаты, субацетаты, тартраты, теофиллинацетаты, п-толуолсульфонаты (тозилаты), трифторацетаты, терефталаты, таннаты, теоклаты, тригалогенацетаты, триэтиодид, трикарбоксилаты, ундеканоаты и валераты.
В одном варианте осуществления примеры неорганических солей с карбоновыми кислотами или гидроксильными соединениями могут быть выбраны из солей аммония, щелочных металлов, включая литий, натрий, калий, цезий; щелочноземельных металлов, включая кальций, магний, алюминий; цинка, бария, холинов, четвертичных аммониевых соединений.
В другом варианте осуществления примеры органических солей с карбоновыми кислотами или гидроксильными соединениями могут быть выбраны из солей аргинина, органических аминов, включая алифатические органические амины, алициклические органические амины, ароматические органические амины, бензатины, трет-бутиламины, бенетамины (N-бензилфенетиламин), дициклогексиламины, диметиламины, диэтаноламины, этаноламины, этилендиамины, гидрабамины, имидазолы, лизины, метиламины, мегламины, N-метил-D-глюкамины, N,N'-дибензилэтилендиамины, никотинамиды, органические амины, орнитины, пиридины, пиколины, пиперазины, прокаин, трис(гидроксиметил)метиламины, триэтиламины, триэтаноламины, триметиламины, трометамины и мочевины.
В одном варианте осуществления соли можно получить общеизвестными способами, такими как реакция продукта в форме свободного основания или свободной кислоты с одним или более эквивалентами подходящей кислоты или основания в растворителе или среде, в которой соль нерастворима, или в растворителе, таком как вода, который удаляют в вакууме или на лиофильной сушилке, или обменом ионов в существующей соли на другой ион, или с помощью подходящей ионообменной смолы.
В некоторых вариантах осуществления в настоящем изобретении описан способ получения соединений по настоящему изобретению. В одном варианте осуществления арил-имидазол получают реакцией соответствующим образом замещенного бензальдегида с этилендиамином с получением имидазолинового цикла, после чего имидазолин окисляют с помощью окислительного агента в соответствующий имидазол. В другом варианте осуществления окислительный агент представляет собой диацетоксииодбензол, бромтрихлорметан и 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU), систему углерод-O2 или систему палладий-углерод. В другом варианте осуществления арил-имидазол получают реакцией соответствующим образом замещенного бензальдегида с этилендиамином в присутствии йода и карбоната калия с получением имидазолинового цикла, после чего имидазолиновый цикл окисляют до соответствующего имидазола с использованием в качестве катализатора диацетоксииодбензола, бромтрихлорметана и 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU), системы углерод-O2 или системы палладий-углерод. В другом варианте осуществления арил-имидазол получают реакцией соответствующим образом замещенного бензальдегида с этилендиамином в присутствии йода и карбоната калия с получением имидазолинового цикла, после чего имидазолиновый цикл окисляют до соответствующего имидазола с использованием в качестве катализатора диацетоксииодбензола. В другом варианте осуществления арил-имидазол получают реакцией соответствующим образом замещенного бензальдегида с этилендиамином в присутствии йода и карбоната калия с получением имидазолинового цикла, после чего имидазолиновый цикл окисляют до соответствующего имидазола с использованием в качестве катализатора бромтрихлорметана и 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU). В одном варианте осуществления арил-имидазол получают реакцией соответствующего бензальдегида в этаноле со щавелевым ангидридом и гидроксидом аммония с получением имидазольного цикла.
В одном варианте осуществления арил-бензоил-имидазольное соединение по настоящему изобретению получают путем защиты арил-имидазола с последующим сочетанием с подходящим образом замещенным бензоилхлоридом, после чего защитную группу удаляют. В другом варианте осуществления защитная группа представляет собой фенилсульфонильную группу, фталамид, дитрет-бутил-дикарбонат (Boc), флуоренилметилоксикарбонил (Fmoc), бензилоксикарбонил (Cbz) или монометокситритил (ММТ). В другом варианте осуществления арил-имидазол защищают фенилсульфонильной группой, получая N-сульфонил-защищенный арил-имидазол. В другом варианте осуществления защищенное арил-имидазольное соединение получают реакцией арил-имидазола с фенилсульфонилхлоридом и гидридом натрия в ТГФ. В другом варианте осуществления защищенный арил-имидазол получают согласно фиг.7 и 8.
В одном варианте осуществления защищенный арил-имидазол вводят в реакцию с соответствующим замещенным бензоилхлоридом, получая защищенный арил-бензоил имидазол. В другом варианте осуществления арил-имидазол вводят в реакцию с соответствующим замещенным бензоилхлоридом в присутствии трет-бутиллития, получая арил-фенилсульфонил (2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон. В другом варианте осуществления (2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанон получают согласно фиг.7 и 8, стадии е и с, соответственно.
В одном варианте осуществления арил-бензоил-имидазол получают удалением защитной группы с арил-бензоил-имидазола. В другом варианте осуществления удаление защитной группы зависит от применяемой защитной группы и ее можно удалить в условиях, известных в данной области техники. В другом варианте осуществления фенилсульфонильную защитную группу удаляют с помощью тетрабутиламмоний фторида в ТГФ. В другом варианте осуществления фенилсульфонил удаляют согласно фиг.7 и 8.
В одном варианте осуществления соединения формулы I, Ia, II, III, V и XI получают согласно фиг.1. В другом варианте осуществления соединения формулы I, Ia, II, III, V, VI, VII и XI получают согласно фиг.2. В другом варианте осуществления соединения формулы I, Ia, II, III, V и VI получают согласно фиг.3. В другом варианте осуществления соединения формулы I, Ia, II, III, V и VI получают согласно фиг.4. В другом варианте осуществления соединения формулы I, Ia, II, III, IV, IVa, V, VI и XI получают согласно фиг.5. В другом варианте осуществления соединения формулы I, Ia, II, III, VIII и XI получают согласно фиг.6.
В одном варианте осуществления соединения формулы XII и XVIII получают согласно фиг.9. В другом варианте осуществления соединения формулы XII, XIII, XIV, XIVa, XV, XVI, XVII, XIX и XX получают согласно фиг.10. В другом варианте осуществления соединения формулы XIVa и XIX получают согласно фиг.11. В другом варианте осуществления соединения формулы I, Ia, IV, IVa, XI, XXI, XXIa и XXII получают согласно фиг.12. В другом варианте осуществления соединения формулы I, Ia, IV, IVa, XI, XIb, XXI, XXIa и XXII получают согласно фиг.13. В другом варианте осуществления соединения формулы I, Ia, II, III, V, XI, XII, XIII, XIV, XV, XVII, XIX и XX получают согласно фиг.14. В другом варианте осуществления соединения формулы I, Ia, II, IV, IVa, XI и XIc получают согласно фиг.15.
В одном варианте осуществления соединения формулы IX и IXa получают согласно фиг.16.
Фармацевтическая композиция
Другой аспект настоящего изобретения касается фармацевтической композиции, содержащей фармацевтически приемлемый носитель и соединение согласно различным аспектам настоящего изобретения. Фармацевтическая композиция может содержать одно или более из описанных выше соединений по настоящему изобретению. В типичном случае фармацевтическая композиция по настоящему изобретению содержит соединение по настоящему изобретению или его фармацевтически приемлемую соль, а также фармацевтически приемлемый носитель. Термин "фармацевтически приемлемый носитель" относится к любым подходящим вспомогательным веществам, носителям, эксципиентам или стабилизаторам, и они могут находиться в твердой или жидкой форме, такой как таблетки, капсулы, порошки, растворы, суспензии или эмульсии.
В типичном случае композиция содержит от около 0.01 до 99%, предпочтительно от около 20 до 75% действующего вещества (веществ), вместе с вспомогательными веществами, носителями и/или эксципиентами. Поскольку индивидуальные потребности могут различаться, определение оптимальных диапазонов эффективных количеств каждого компонента находится в рамках компетенции квалифицированных специалистов в данной области. Типичные дозировки содержат от около 0.01 до около 100 мг/кг веса тела. Предпочтительные дозировки содержат от около 0.1 до около 100 мг/кг веса тела. Наиболее предпочтительные дозировки содержат от около 1 до около 100 мг/кг веса тела. Режим приема соединений по настоящему изобретению также может быть легко определен квалифицированными специалистам в данной области. То есть частоту введения и размер дозы можно определить рутинной оптимизацией, предпочтительно с минимизацией любых побочных эффектов.
Твердые дозированные лекарственные формы могут быть любого общепринятого типа. Твердая лекарственная форма может представлять собой капсулу и т.п., например обычные желатиновые капсулы, содержащие соединения по настоящему изобретению, и носитель, например, лубриканты и инертные наполнители, такие как лактоза, сахароза или кукурузный крахмал. В другом варианте осуществления данные соединения смешивают с обычными основами для таблеток, такими как лактоза, сахароза или кукурузный крахмал, в комбинации со связующими веществами, такими как смола акации, кукурузный крахмал или желатин, разрыхлителями, такими как кукурузный крахмал, картофельный крахмал или альгиновая кислота, и лубрикантом, таким как стеариновая кислота или стеарат магния.
Таблетки, капсулы и т.д. могут также содержать связующее вещество, такое как трагакантовая камедь, смола акации, кукурузный крахмал или желатин; наполнители, такие как дикальция фосфат; разрыхлители, такие как кукурузный крахмал, картофельный крахмал, альгиновая кислота; лубрикант, такой как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Когда дозированная лекарственная форма представляет собой капсулу, она может содержать, помимо веществ описанного выше типа, жидкий носитель, такой как жирное масло.
Различные другие вещества могут присутствовать в качестве покрытия или для модификации физической формы дозированной лекарственной формы. Например, таблетки могут быть покрыты шеллаком, сахаром, или обоими перечисленными веществами. Сироп может содержать, помимо действующего вещества, сахарозу в качестве подсластителя, метил- и пропил-парабены в качестве консервантов, краситель, ароматизатор, такой как вишневый или апельсиновый ароматизатор.
Для перорального применения указанные действующие вещества можно смешивать с эксципиентами и применять в форме таблеток, капсул, эликсиров, суспензий, сиропов и т.п. Такие композиции и препараты должны содержать по меньшей мере 0.1% действующего вещества. Процентное содержание соединения в описанных композициях может, разумеется, варьироваться и обычно составляет от около 2% до около 60% от веса готовой формы. Количество действующего вещества в таких фармацевтически пригодных композициях таково, что позволяет обеспечить необходимую дозировку. Предпочтительные композиции по настоящему изобретению получают таким образом, чтобы дозированная форма для перорального применения содержала от около 1 мг до 800 мг действующего вещества.
Активные соединения по настоящему изобретению можно вводить перорально, например, с инертным разбавителем или с усваиваемым съедобным носителем, или их можно заключать в твердые или мягкие капсулы, или их можно прессовать в таблетки, или их можно вводить непосредственно в пищу.
Фармацевтические формы, пригодные для инъекционного применения, включают стерильные водные растворы или дисперсии, и стерильные порошки для приготовления стерильных инъецируемых растворов или дисперсий непосредственно перед приемом. Во всех случаях лекарственная форма должна быть стерильной и должна быть жидкой до такой степени, чтобы ее можно было легко вводить шприцом. Она должна быть стабильной в условиях производства и хранения, и должна быть защищена от загрязняющего действия микроорганизмов, таких как бактерий и грибы. Носитель может представлять собой растворитель или диспергирующую среду, содержащую, например, воду, этанол, полиол (например, глицерин, пропиленгликоль и жидкий полиэтиленгликоль), их подходящие смеси и растительные масла.
Соединения или фармацевтические композиции по настоящему изобретению можно также вводить инъекционно в виде растворов или суспензий данных веществ в физиологически приемлемом разбавителе с фармацевтическим вспомогательным веществом, носителем или эксципиентом. Такие вспомогательные вещества, носители и/или эксципиенты включают (но не ограничиваются только ими) стерильные жидкости, такие как вода и масла, с добавлением или без добавления поверхностно-активных веществ и других фармацевтически и физиологически приемлемых компонентов. Примерами масел являются масла нефтяного, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло или минеральное масло. В целом, вода, физиологический раствор, водный раствор декстрозы и раствор родственных Сахаров, гликоли, такие как пропиленгликоль или полиэтиленгликоль, являются предпочтительными жидкими носителями, в частности для инъецируемых растворов.
Указанные действующие вещества можно также вводить парэнтерально. Растворы или суспензии указанных действующих веществ можно готовить в воде, с добавлением подходящих поверхностно-активных веществ, таких как гидроксипропилцеллюлоза. Дисперсии также можно готовить в глицерине, жидких полиэтиленгликолях и их смесях в маслах. Примерами масел являются масла нефтяного, животного, растительного или синтетического происхождения, например, арахисовое масло, соевое масло или минеральное масло. В целом, вода, физиологический раствор, водный раствор декстрозы и раствор родственных сахаров, гликоли, такие как пропиленгликоль или полиэтиленгликоль, являются предпочтительными жидкими носителями, в частности для инъецируемых растворов. В обычных условиях хранения и использования, данные препараты содержат консерванты для предотвращения роста микроорганизмов.
Для использования в качестве аэрозолей соединения по настоящему изобретению в растворе или суспензии можно упаковывать в находящийся под давлением аэрозольный контейнер вместе с подходящими пропеллантами, например углеводородными пропеллантами, такими как пропан, бутан или изобутан, вместе со вспомогательными веществами. Вещества по настоящему изобретению можно также вводить в виде формы, не находящейся под давлением, такой как небулайзер или аэрозольный ингалятор.
В одном варианте осуществления соединения по настоящему изобретению вводят в комбинации с противораковым средством. В одном варианте осуществления противораковое средство представляет собой моноклональное антитело. В некоторых вариантах осуществления моноклональные антитела применяются для диагностики, мониторинга или лечения рака. В одном варианте осуществления моноклональные антитела реагируют со специфическими антигенами на раковых клетках. В одном варианте осуществления моноклональное антитело выступает в роли антагониста рецептора раковой клетки. В одном варианте осуществления моноклональные антитела усиливают иммунный ответ пациента. В одном варианте осуществления моноклональные антитела работают против факторов роста клеток, тем самым блокируя рост раковых клеток. В одном варианте осуществления противораковые моноклональные антитела конъюгированы или связаны с противораковыми лекарствами, радиоизотопами, другими модификаторами биологического ответа, другими токсинами или с их комбинацией. В одном варианте осуществления противораковые моноклональные антитела конъюгированы или связаны с соединением по настоящему изобретению, описанным выше в настоящем тексте.
Другой аспект настоящего изобретения касается способа лечения рака, который включает выбор субъекта, нуждающегося в лечении рака, и введение данному субъекту фармацевтической композиции, содержащей соединение по первому аспекту настоящего изобретения и фармацевтически приемлемый носитель, в условиях, эффективных для лечения рака.
При введении соединений по настоящему изобретению, их можно вводить системно или, альтернативно, их можно вводить непосредственно в конкретное место, в котором имеются раковые или прераковые клетки. Таким образом, введение можно осуществлять любым образом, эффективным в смысле доставки соединений или фармацевтических композиций в раковые клетки или прераковые клетки. Примеры способов введения включают (но не ограничиваются только ими) введение соединений или композиций перорально, наружно, чрезкожно, парэнтерально, подкожно, внутривенно, внутримышечно, внутрибрюшинно, закапыванием в нос, внутриполостным или внутрипузырным закапыванием, внутриглазным образом, внутриартериально, внутриочагово или нанесением на слизистые оболочки, такие как оболочки носа, горла и бронхиальных каналов.
Биологическая активность
В одном варианте осуществления в настоящем изобретении описаны соединения и композиции, включая любой вариант осуществления, описанный в настоящем тексте, для использования в любом из способов по настоящему изобретению. В одном варианте осуществления использование соединения по настоящему изобретению или содержащей его композиции, применяется для ингибирования, подавления, усиления или стимулирования желаемого ответа субъекта, как будет понятно квалифицированному специалисту в данной области. В другом варианте осуществления композиции могут дополнительно содержать дополнительные активные ингредиенты, активность которых полезна для конкретной области применения, в рамках которой осуществляется ввод соединения по настоящему изобретению.
В одном варианте осуществления настоящее изобретение касается способа лечения, подавления, уменьшения тяжести, снижения риска развития или ингибирования рака, включающего введение соединения по настоящему изобретению субъекту, страдающему раком, в условиях, эффективных для лечения рака.
Лекарственная резистентность является главной причиной неудач химиотерапии рака. Главный вклад во множественную лекарственную резистентность вносит сверхвыработка Р-гликопротеина (P-gp). Этот белок является клинически важным белком-транспортером, принадлежащим к семейству АТФ-связывающей кассеты клеточных мембранных транспортеров. Он может выкачивать из клеток опухолей субстраты, включая противораковые лекарства, по АТФ-зависимому механизму.
В одном варианте осуществления в настоящем изобретении описаны способы: а) лечения, подавления, уменьшения тяжести, снижения риска или ингибирования лекарственно-устойчивый опухоли; b) лечения, подавления, уменьшения тяжести, снижения риска или ингибирования метастатического рака; с) лечения, подавления, уменьшения тяжести, снижения риска или ингибирования лекарственно-устойчивого рака; d) лечения, подавления, уменьшения тяжести, снижения риска или ингибирования лекарственно-устойчивого рака, где рак представляет собой меланому; е) способ лечения, подавления, уменьшения тяжести, снижения риска или ингибирования лекарственно-устойчивого рака, где рак представляет собой рак предстательной железы; f) способ лечения, подавления, уменьшения тяжести, снижения риска или ингибирования метастатической меланомы; g) способ лечения, подавления, уменьшения тяжести, снижения риска или ингибирования рака предстательной железы; h) лечения, подавления, уменьшения тяжести, снижения риска или ингибирования рака у субъекта, где субъект до этого подвергался химиотерапии, радиотерапии или биологической терапии; включающий стадию введения указанному субъекту соединения по настоящему изобретению и/или изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа или кристаллической формы указанного соединения, или любой комбинации перечисленного.
Соединения по настоящему изобретению могут применяться для лечения, подавления, уменьшения тяжести, снижения риска или ингибирования рака, метастатического рака, лекарственно-устойчивой опухоли, лекарственно-устойчивого рака и различных форм рака. В предпочтительном варианте осуществления рак представляет собой рак предстательной железы, рак груди, рак яичника, рак кожи (например, меланому), рак легких, рак толстой кишки, лейкемию, лимфому, рак головы и шеи, рак поджелудочной железы, рак пищевода, рак почки или рак ЦНС (например, глиома, глиобластома). Лечение перечисленных разных видов рака подтверждается приведенными в настоящем тексте примерами. Кроме того, основываясь на том, что предполагаемым механизмом их действия является ингибирование тубулина, авторы полагают, что другие формы рака также могут лечиться или предотвращаться путем введения пациенту соединений или композиций по настоящему изобретению. Предпочтительные соединения по настоящему изобретению селективно разрушают раковые клетки, вызывая разложение раковых клеток, но, предпочтительно, не нормальных клеток. Важно, что вред для нормальных клеток минимизирован, поскольку раковые клетки подвержены разрушению при намного меньших концентрациях соединений по настоящему изобретению.
В некоторых вариантах осуществления в настоящем изобретении описано применение описанного в настоящем изобретении соединения, или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, полиморфа, кристаллической формы, N-оксида, гидрата или любой их комбинации для лечения, подавления, уменьшения тяжести, снижения риска или ингибирования рака у субъекта. В другом варианте осуществления рак представляет собой карциному коры надпочечников, рак анального канала, рак мочевого пузыря, опухоль мозга, рак ствола мозга, рак груди, глиому, астроцитому мозжечка, астроцитому головного мозга, эпендимому, медуллобластому, супратенториальную примитивную нейроэктодермальную опухоль, опухоли шишковидного тела, гипоталамическую глиому, рак груди, карциноидную опухоль, карциному, рак шейки матки, рак толстой кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак желчных протоков, опухоли семейства саркомы Юинга (примитивная эктодермальная опухоль), опухоль внечерепных стволовых клеток, рак глаза, внутриглазную меланому, рак желчного пузыря, рак желудка, герминогенные опухоли, внегонадную гестационную трофобластическую болезнь, рак головы и шеи, гипофарингеальный рак, карциному островных клеток, рак гортани, лейкемию, острый лимфобластный лейкоз, рак ротовой полости, рак печени, рак легких, не-мелкоклеточный рак легких, мелкоклеточную лимфому, вызванную СПИДом лимфому, лимфому центральной нервной системы (первичную), лимфому Т-клеток кожи, болезнь Ходжкина, не-Ходжкинскую лимфому, злокачественную мезотелиому, меланому, карциному из клеток Меркеля, метастатическую плоскоклеточную карциному, множественную миелому, новообразования плазматических клеток, грибовидный микоз, миелодиспластический синдром, миелопролиферативные нарушения, рак носоглотки, нейробластому, рак ротоглотки, остеобластическую саркому, рак яичника, рак эпителия яичника, герминогенную опухоль яичника, пограничную опухоль яичника, рак поджелудочной железы, экзокринный рак поджелудочной железы, карциному островных клеток, рак околоносовой полости и носовой полости, рак паращитовидной железы, рак полового члена, феохромоцитому, рак гипофиза, опухоль плазматических клеток, рак предстательной железы, рабдомиосаркому, рак прямой кишки, рак почки, рак клеток почечного эпителия, рак слюнных желез, синдром Сезари, рак кожи, лимфому Т-клеток кожи, рак кожи, саркому Капоши, рак кожи, меланому, рак тонкого кишечника, саркому мягких тканей, рак яичка, злокачественную тимому, рак щитовидной железы, рак уретры, рак матки, саркому, необычный детский рак, рак вагины, рак вульвы, опухоль Вильмса или любые их комбинации. В другом варианте осуществления субъект ранее проходил химиотерапию, радиотерапию или биологическую терапию.
В некоторых вариантах осуществления в настоящем изобретении описано применение описанного в настоящем изобретении соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, полиморфа, кристаллической формы, N-оксида, гидрата или любой их комбинации, для лечения, подавления, уменьшения тяжести, снижения риска, или ингибирования метастатического рака у субъекта. В другом варианте осуществления рак представляет собой карциному коры надпочечников, рак анального канала, рак мочевого пузыря, опухоль мозга, рак ствола мозга, рак груди, глиому, астроцитому мозжечка, астроцитому головного мозга, эпендимому, медуллобластому, супратенториальную примитивную нейроэктодермальную опухоль, опухоли шишковидного тела, гипоталамическую глиому, рак груди, карциноидную опухоль, карциному, рак шейки матки, рак толстой кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак желчных протоков, опухоли семейства саркомы Юинга (примитивная эктодермальная опухоль), опухоль внечерепных стволовых клеток, рак глаза, внутриглазную меланому, рак желчного пузыря, рак желудка, герминогенные опухоли, внегонадную гестационную трофобластическую болезнь, рак головы и шеи, гипофарингеальный рак, карциному островных клеток, рак гортани, лейкемию, острый лимфобластный лейкоз, рак ротовой полости, рак печени, рак легких, не-мелкоклеточный рак легких, мелкоклеточную лимфому, вызванную СПИДом лимфому, лимфому центральной нервной системы (первичную), лимфому Т-клеток кожи, болезнь Ходжкина, не-Ходжкинскую лимфому, злокачественную мезотелиому, меланому, карциному из клеток Меркеля, метастатическую плоскоклеточную карциному, множественную миелому, новообразования плазматических клеток, грибовидный микоз, миелодиспластический синдром, миелопролиферативные нарушения, рак носоглотки, нейробластому, рак ротоглотки, остеобластическую саркому, рак яичника, рак эпителия яичника, герминогенную опухоль яичника, пограничную опухоль яичника, рак поджелудочной железы, экзокринный рак поджелудочной железы, карциному островных клеток, рак околоносовой полости и носовой полости, рак паращитовидной железы, рак полового члена, феохромоцитому, рак гипофиза, опухоль плазматических клеток, рак предстательной железы, рабдомиосаркому, рак прямой кишки, рак почки, рак клеток почечного эпителия, рак слюнных желез, синдром Сезари, рак кожи, лимфому Т-клеток кожи, рак кожи, саркому Капоши, рак кожи, меланому, рак тонкого кишечника, саркому мягких тканей, рак яичка, злокачественную тимому, рак щитовидной железы, рак уретры, рак матки, саркому, необычный детский рак, рак вагины, рак вульвы, опухоль Вильмса или любые их комбинации.
В некоторых вариантах осуществления в настоящем изобретении описано применение описанного в настоящем изобретении соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, полиморфа, кристаллической формы, N-оксида, гидрата или любой их комбинации, для лечения, подавления, уменьшения тяжести, снижения риска, или ингибирования лекарственно-устойчивого рака или резистентного рака у субъекта. В другом варианте осуществления рак представляет собой карциному коры надпочечников, рак анального канала, рак мочевого пузыря, опухоль мозга, рак ствола мозга, рак груди, глиому, астроцитому мозжечка, астроцитому головного мозга, эпендимому, медуллобластому, супратенториальную примитивную нейроэктодермальную опухоль, опухоли шишковидного тела, гипоталамическую глиому, рак груди, карциноидную опухоль, карциному, рак шейки матки, рак толстой кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак желчных протоков, опухоли семейства саркомы Юинга (примитивная эктодермальная опухоль), опухоль внечерепных стволовых клеток, рак глаза, внутриглазную меланому, рак желчного пузыря, рак желудка, герминогенные опухоли, внегонадную гестационную трофобластическую болезнь, рак головы и шеи, гипофарингеальный рак, карциному островных клеток, рак гортани, лейкемию, острый лимфобластный лейкоз, рак ротовой полости, рак печени, рак легких, не-мелкоклеточный рак легких, мелкоклеточную лимфому, вызванную СПИДом лимфому, лимфому центральной нервной системы (первичную), лимфому Т-клеток кожи, болезнь Ходжкина, не-Ходжкинскую лимфому, злокачественную мезотелиому, меланому, карциному из клеток Меркеля, метастатическую плоскоклеточную карциному, множественную миелому, новообразования плазматических клеток, грибовидный микоз, миелодиспластический синдром, миелопролиферативные нарушения, рак носоглотки, нейробластому, рак ротоглотки, остеобластическую саркому, рак яичника, рак эпителия яичника, герминогенную опухоль яичника, пограничную опухоль яичника, рак поджелудочной железы, экзокринный рак поджелудочной железы, карциному островных клеток, рак околоносовой полости и носовой полости, рак паращитовидной железы, рак полового члена, феохромоцитому, рак гипофиза, опухоль плазматических клеток, рак предстательной железы, рабдомиосаркому, рак прямой кишки, рак почки, рак клеток почечного эпителия, рак слюнных желез, синдром Сезари, рак кожи, лимфому Т-клеток кожи, рак кожи, саркому Капоши, рак кожи, меланому, рак тонкого кишечника, саркому мягких тканей, рак яичка, злокачественную тимому, рак щитовидной железы, рак уретры, рак матки, саркому, необычный детский рак, рак вагины, рак вульвы, опухоль Вильмса или любые их комбинации.
В одном варианте осуществления "метастатический рак" означает рак, распространяющийся (дающий метастазы) из места изначального появления в другие области тела. Практически все виды рака потенциально могут давать метастазы. Развитие метастазов зависит от комплексного взаимодействия многих факторов раковых клеток, включая типа рака, степень зрелости (дифференциации) опухолевых клеток, локализации и длительности течения ракового заболевания, а также от других, полностью не изученных, факторов. Метастазы распространяются тремя способами - локальным распространением из опухоли в окружающие ткани, через кровеносную систему в удаленные области или через лимфатическую систему в соседние или удаленные лимфоузлы. Каждый вид рака может иметь типичный способ распространения. Опухоль называют по месту ее первоначального возникновения (например, рак груди, распространившийся в мозг, называют раком груди, давшим метастазы в мозг).
В одном варианте осуществления термин "лекарственно-устойчивый рак" относится к раковым клеткам, обладающим устойчивостью к химиотерапии. Раковые клетки могут приобретать устойчивость к химиотерапии по нескольким механизмам, включая мутацию и сверхвыработку мишени для лекарства, инактивацию лекарства или вывод лекарства из клетки. Опухоли, дающие рецидив после изначального ответа на химиотерапию, могут быть устойчивыми к множеству лекарств (их называют множественно-лекарственно устойчивыми). В общепринятом взгляде на лекарственную устойчивость, одна или несколько клеток в популяции опухоли претерпевают генетические изменения, обуславливающие устойчивость к лекарственному средству. Соответственно, причинами лекарственной устойчивости, среди прочего, являются: а) некоторые клетки, не погибшие при химиотерапии, мутируют (изменяются) и становятся резистентными к лекарственному средству. При их размножении формируются клетки, которые могут быть более устойчивыми, чем клетки, сензитивные к химиотерапии; b) Амплицикация генов. Раковые клетки могут продуцировать сотни копий отдельного гена. Данный ген вызывает сверхпроизводство белка, что делает противораковое лекарство неэффективным; с) раковые клетки могут выводить лекарственное средство из клетки сразу после его поступления в клетку, используя молекулу, называемую p-гликопротеином; d) раковые клетки могут прекращать доставку лекарственного средства в клетку, поскольку перестает работать белок, транспортирующий лекарственное средство через клеточную стенку; е) раковые клетки могут учиться восстанавливать повреждения ДНК, вызываемые некоторыми противораковыми лекарствами; f) в раковых клетках могут развиваться механизмы, инактивирующие лекарственное средство. Большой вклад во множественную лекарственную устойчивость вносит сверхвыработка Р-гликопротеина (Р-gp). Данный белок представляет собой клинически важный транспортерный белок, принадлежащий семейству клеточных мембранных транспортеров АТФ-связывающей кассеты. Он может выводить из опухолевых клеток субстраты, включая противораковые лекарства, по АТФ-зависимому механизму. Таким образом, резистентность к применяемым в химиотерапии противораковым средствам является главной причиной неудач лечения при злокачественных нарушениях, провоцируя развитие резистентности опухолей. Лекарственная устойчивость является главной причиной неудач в химиотерапии рака.
В одном варианте осуществления "резистентный рак" означает лекарственно-устойчивый рак, как описано выше в тексте. В другом варианте осуществления термин "резистентный рак" относится к раковым клеткам, приобретающим устойчивость к любому виду лечения, такому как химиотерапия, радиотерапия или биологическая терапия.
В одном варианте осуществления настоящее изобретение касается лечения подавления, уменьшения тяжести, снижения риска, или ингибирования рака у субъекта, где субъект ранее проходил химиотерапию, радиотерапию или биологическую терапию.
В одном варианте осуществления термин "химиотерапия" означает химическое лечение рака, такое как применение лекарственных средств, напрямую убивающих раковые клетки. Такие лекарственные средства называют «противораковыми» лекарствами или «противоопухолевыми» лекарствами. В современной терапии применяется свыше 100 лекарственных средств для лечения рака. Химиотерапия применяется для контроля роста опухоли, когда излечение невозможно; для уменьшения размеров опухолей перед хирургической операцией или радиотерапией; для облегчения симптомов (таких как боль); и для уничтожения микроскопических раковых клеток, которые могут оставаться после удаления опухоли хирургическим путем (называют вспомогательной терапией). Вспомогательную терапию применяют для предотвращения возможного рецидива рака
В одном варианте осуществления термин "радиотерапия" означает применение высокоэнергетического рентгеновского излучения и сходного излучения (такого как электронное) для лечения заболевания. Множество людей, страдающих от рака, проходят радиотерапию как часть курса лечения. Ее можно применять как в форме внешней радиотерапии, используя источник рентгеновского излучения вне тела, так и в форме внутренней радиотерапии изнутри тела. Радиотерапия работает путем разрушения раковых клеток в обрабатываемой области. Хотя нормальные клетки тоже могут повреждаться при радиотерапии, они обычно обладают способностью к самовосстановлению. Радиотерапевтическое лечение может излечить некоторые виды рака, а также снизить вероятность рецидива рака после операции. Ее можно применять для облегчения симптомов рака.
В одном варианте осуществления термин "биологическая терапия" относится к субстанциям, которые естественным образом имеются в теле, используемым для разрушения раковых клеток. Существует несколько типов лечения, включая: моноклональные антитела, ингибиторы роста раковых опухолей, вакцины и генную терапию. Биологическая терапия также известна как иммунотерапия.
В одном варианте осуществления в настоящем изобретении описан способ лечения субъекта, страдающего от рака предстательной железы, метастатического рака предстательной железы, резистентного рака предстательной железы или лекарственно-устойчивого рака предстательной железы, включающий стадию введения описанного соединения по настоящему изобретению или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинации, или содержащей их композиции в количестве, эффективном для лечения рака предстательной железы у субъекта. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В одном варианте осуществления в настоящем изобретении описан способ подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования рака предстательной железы, метастатического рака предстательной железы, резистентного рака предстательной железы или лекарственно-устойчивого рака предстательной железы у субъекта, включающий введение указанному субъекту соединения по настоящему изобретению и/или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинация или содержащей их композиции. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В одном варианте осуществления в настоящем изобретении описан способ лечения пациента, страдающего от рака груди, метастатического рака груди, резистентного рака груди или лекарственно-устойчивого рака груди, включающий введение указанному субъекту соединения по настоящему изобретению или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинация или содержащей их композиции. В другом варианте осуществления субъект представляет собой мужчину или женщину. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В одном варианте осуществления в настоящем изобретении описан способ подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования рака груди, метастатического рака груди, резистентного рака груди или лекарственно-устойчивого рака груди у субъекта, включающий стадию введения указанному субъекту соединения по настоящему изобретению или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинация или содержащей их композиции. В другом варианте осуществления субъект представляет собой мужчину или женщину. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинации для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования рака яичника, метастатического рака яичника, резистентного рака яичника или лекарственно-устойчивого рака яичника у субъекта. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В одном варианте осуществления в настоящем изобретении описан способ лечения, подавления, уменьшения тяжести, снижения риска или ингибирования меланомы, метастатической меланомы, резистентной меланомы или лекарственно-устойчивой меланома у субъекта, включающий введение указанному субъекту соединения по настоящему изобретению и/или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинации. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинации, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития, или ингибирования рака легких, метастатического рака легких, резистентного рака легких или лекарственно-устойчивого рака легких. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинации, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования немелкоклеточного рака легких, метастатического мелкоклеточного рака легких, резистентного мелкоклеточного рака легких или лекарственно-устойчивого мелкоклеточного рака легких. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинации, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования рака толстой кишки, метастатического рака толстой кишки, резистентного рака толстой кишки или лекарственно-устойчивого рака толстой кишки. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинации, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования лейкемии, метастатической лейкемии, резистентной лейкемии или лекарственно-устойчивой лейкемии. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы или любой их комбинации, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования лимфомы, метастатической лимфомы, лимфомы или лекарственно-устойчивой лимфомы. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллического вещества или любой их комбинации, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития, или ингибирования рака головы и шеи, метастатического рака головы и рака шеи, или лекарственно-устойчивого рака головы и шеи. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы и любых их комбинаций, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования рака поджелудочной железы, метастатического рака поджелудочной железы, резистентного рака поджелудочной железы или лекарственно-устойчивого рака поджелудочной железы. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы и любых их комбинаций, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования рака пищевода, метастатического рака пищевода, резистентного рака пищевода или лекарственно-устойчивого рака пищевода. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы и любых их комбинаций, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования рака почки, метастатического рака почки, резистентного рака почки или лекарственно-устойчивого рака почки. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, гидрата, N-оксида, полиморфа, кристаллической формы и любых их комбинаций, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования рака ЦНС, метастатического рака ЦНС, резистентного рака ЦНС или лекарственно-устойчивого рака ЦНС. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В некоторых вариантах осуществления в настоящем изобретении описано применение описанного в настоящем тексте соединения или его изомера, метаболита, фармацевтически приемлемой соли, фармацевтического продукта, таутомера, полиморфа, кристаллической формы N-оксида, гидрата и любых их комбинаций, для лечения, подавления, уменьшения тяжести, снижения риска, замедления развития или ингибирования лекарственно-устойчивой раковой опухоли или опухоли у субъекта. В другом варианте осуществления рак представляет собой карциному коры надпочечников, рак анального канала, рак мочевого пузыря, опухоль мозга, рак ствола мозга, рак груди, глиому, астроцитому мозжечка, астроцитому головного мозга, эпендимому, медуллобластому, супратенториальную примитивную нейроэктодермальную опухоль, опухоли шишковидного тела, гипоталамическую глиому, рак груди, карциноидную опухоль, карциному, рак шейки матки, рак толстой кишки, рак центральной нервной системы (ЦНС), рак эндометрия, рак пищевода, рак желчных протоков, опухоли семейства саркомы Юинга (примитивная эктодермальная опухоль), опухоль внечерепных стволовых клеток, рак глаза, внутриглазную меланому, рак желчного пузыря, рак желудка, герминогенные опухоли, внегонадную гестационную трофобластическую болезнь, рак головы и шеи, гипофарингеальный рак, карциному островных клеток, рак гортани, лейкемию, острый лимфобластный лейкоз, рак ротовой полости, рак печени, рак легких, не-мелкоклеточный рак легких, мелкоклеточную лимфому, вызванную СПИДом лимфому, лимфому центральной нервной системы (первичную), лимфому Т-клеток кожи, болезнь Ходжкина, не-Ходжкинскую лимфому, злокачественную мезотелиому, меланому, карциному из клеток Меркеля, метастатическую плоскоклеточную карциному, множественную миелому, новообразования плазматических клеток, грибовидный микоз, миелодиспластический синдром, миелопролиферативные нарушения, рак носоглотки, нейробластому, рак ротоглотки, остеобластическую саркому, рак яичника, рак эпителия яичника, герминогенную опухоль яичника, пограничную опухоль яичника, рак поджелудочной железы, экзокринный рак поджелудочной железы, карциному островных клеток, рак околоносовой полости и носовой полости, рак паращитовидной железы, рак полового члена, феохромоцитому, рак гипофиза, опухоль плазматических клеток, рак предстательной железы, рабдомиосаркому, рак прямой кишки, рак почки, рак клеток почечного эпителия, рак слюнных желез, синдром Сезари, рак кожи, лимфому Т-клеток кожи, рак кожи, саркому Капоши, рак кожи, меланому, рак тонкого кишечника, саркому мягких тканей, рак яичка, злокачественную тимому, рак щитовидной железы, рак уретры, рак матки, саркому, необычный детский рак, рак вагины, рак вульвы, опухоль Вильмса или любые их комбинации. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления указанная опухоль представляет собой опухоль при раке предстательной железы. В другом варианте осуществления указанная опухоль представляет собой опухоль при раке яичника. В другом варианте осуществления указанная опухоль представляет собой меланому. В другом варианте осуществления указанная опухоль представляет собой мультилекарственно-устойчивую опухоль при меланоме.
В одном варианте осуществления настоящее изобретение касается способа разрушения раковой клетки, включающего: получение соединения по настоящему изобретению и введение раковой клетки в контакт с соединением в условиях, в которых эффективно происходит разрушение контактирующей раковой клетки. В различных вариантах разрушения раковых клеток, клетки, подвергающиеся разрушению, могут находиться in vivo или ex vivo (т.е., в культуре). В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
В другом варианте осуществления рак выбран из группы, состоящей из рака предстательной железы, рака груди, рака яичника, рака кожи, меланомы, рака легких, рака толстой кишки, лейкемии, рака почки, рака ЦНС и их комбинации.
Другой аспект настоящего изобретения касается способа лечения или профилактики ракового состояния, который включает: получение соединения по настоящему изобретению и последующее введение эффективного количества соединения пациенту способом, обеспечивающим эффективное лечение или профилактику ракового состояния.
В одном варианте осуществления подвергающийся лечению пациент характеризуется наличием преракового состояния, и введение соединения эффективно предотвращает развитие преракового состояния в раковое состояние. Это может происходить путем разрушения прераковой клетки до или во время ее дальнейшего перехода в раковое состояние.
В другом варианте осуществления подвергающийся лечению пациент характеризуется наличием ракового состояния, и введение соединения эффективно вызывает регрессию ракового состояния или ингибирует развитие ракового состояния, т.е. полностью останавливает его развитие или снижает скорость его развития. Это предпочтительно происходит за счет разрушения раковых клеток, независимо от их локализации в теле пациента. То есть независимо от того, находятся ли раковые клетки в месте первичной опухоли или раковые клетки дали метастазы и создали вторичные опухоли в теле пациента.
При использовании в настоящем тексте термины «субъект» или «пациент» относятся к любому пациенту-млекопитающему, включая (но не ограничиваясь только ими) людей и других приматов, собак, кошек, лошадей, коров, овец, свиней, крыс, мышей и других грызунов. В одном варианте осуществления субъектом является самец. В другом варианте осуществления субъектом является самка. В некоторых вариантах осуществления описанные в настоящем тексте способы могут быть эффективны для лечения либо самцов, либо самок.
При введении соединений по настоящему изобретению их можно вводить системно или, альтернативно, их можно вводить непосредственно в конкретное место, в котором имеются раковые или прераковые клетки. Таким образом, введение можно осуществлять любым образом, эффективным в смысле доставки соединений или фармацевтических композиций в раковые клетки или прераковые клетки. Примеры способов введения включают (но не ограничиваются только ими) введение соединений или композиций перорально, наружно, чрезкожно, парэнтерально, подкожно, внутривенно, внутримышечно, внутрибрюшинно, закапыванием в нос, внутриполостным или внутрипузырным закапыванием, внутриглазным образом, внутриартериально, внутриочагово или нанесением на слизистые оболочки, такие как оболочки носа, горла и бронхиальных каналов.
Соединения по настоящему изобретению могут применяться в лечении или профилактике различных форм рака, в частности рака предстательной железы, рака груди, рака яичника, рака кожи (например, меланомы), рака легких, рака толстой кишки, лейкемии, рака почки и рака ЦНС (например, глиомы, глиобластомы). Лечение перечисленных разных видов рака подтверждается приведенными в настоящем тексте примерами. Кроме того, основываясь на том, что предполагаемым механизмом их действия является ингибирование тубулина, авторы полагают, что другие формы рака также могут лечиться или предотвращаться путем введения пациенту соединений или композиций по настоящему изобретению. Предпочтительные соединения по настоящему изобретению селективно разрушают раковые клетки, вызывая разложение раковых клеток, но, предпочтительно, не нормальных клеток. Важно, что вред для нормальных клеток минимизирован, поскольку раковые клетки подвержены разрушению при намного меньших концентрациях соединений по настоящему изобретению.
Соединения по настоящему изобретению могут применяться для лечения, уменьшения тяжести заболевания, снижения риска или ингибирования рака, метастатического рака, резистентного рака или лекарственно-устойчивого рака. В другом варианте осуществления рак представляет собой рак предстательной железы, рак груди, рак яичника, рака кожи (например, меланомы), рака легких, рака толстой кишки, лейкемии, лимфомы, рака головы и шеи, рака поджелудочной железы, рака пищевода, рака почки или рака ЦНС. Лечение перечисленных разных видов рака подтверждается приведенными в настоящем тексте примерами. Кроме того, основываясь на том что предполагаемым механизмом их действия является ингибирование тубулина, авторы полагают, что другие формы рака также могут лечиться или предотвращаться путем введения пациенту соединений или композиций по настоящему изобретению. Предпочтительные соединения по настоящему изобретению селективно разрушают раковые клетки, вызывая разложение раковых клеток, но, предпочтительно, не нормальных клеток. Важно, что вред для нормальных клеток минимизирован, поскольку раковые клетки подвержены разрушению при намного меньших концентрациях соединений по настоящему изобретению. В другом варианте осуществления указанное соединение представляет собой соединение 12db. В другом варианте осуществления указанное соединение представляет собой соединение 11cb. В другом варианте осуществления указанное соединение представляет собой соединение 11fb. В другом варианте осуществления указанное соединение представляет собой соединение 12da. В другом варианте осуществления указанное соединение представляет собой соединение 12fa. В другом варианте осуществления указанное соединение представляет собой соединение 12fb. В другом варианте осуществления указанное соединение представляет собой соединение 12cb. В другом варианте осуществления указанное соединение представляет собой соединение 55. В другом варианте осуществления указанное соединение представляет собой соединение 6b. В другом варианте осуществления указанное соединение представляет собой соединение 17ya.
При использовании в настоящем тексте термины «субъект» или «пациент» относятся к любому пациенту-млекопитающему, включая (но не ограничиваясь только ими) людей и других приматов, собак, кошек, лошадей, коров, овец, свиней, крыс, мышей и других грызунов. В некоторых вариантах осуществления описанные в настоящем тексте способы могут быть эффективны для лечения либо самцов, либо самок.
В одном варианте осуществления соединение по настоящему изобретению вводят в комбинации с противораковым средством, вводя указанные соединения как описано в настоящем тексте, отдельно или в комбинации с другими средствами.
Когда соединения или фармацевтические композиции по настоящему изобретению вводят для лечения, подавления, уменьшения тяжести заболевания, снижения риска или ингибирования ракового состояния, фармацевтическая композиция может также содержать (или вводиться совместно с) другие терапевтические средства, или применяться совместно с режимом лечения, известным в настоящее время или разработанным в дальнейшем, различных типов рака. Примеры других терапевтических средств или режимов лечения включают (но не ограничиваются только ими) радиационную терапию, иммунотерапию, химиотерапию, хирургическое вмешательство и их комбинации.
Описанные далее примеры представлены для более полной иллюстрации предпочтительных вариантов осуществления настоящего изобретения. Они никоим образом не должны рассматриваться как ограничивающие объем настоящего изобретения.
Примеры
Представленные ниже примеры имеют исключительно иллюстративную функцию и никоим образом не предназначены для ограничения объема настоящего изобретения.
Материалы и Методы
Общие. Все реактивы приобретались в Sigma-Aldrich Chemical Co., Fisher Scientific (Pittsburgh, PA), AK Scientific (Mountain View, CA), Oakwood Products (West Columbia, SC) и т.д., и использовались без дополнительной очистки. Реакции, чувствительные к влаге, проводили в атмосфере аргона. АВТ-751 получали способом, описанным Yoshino с соавторами26. Рутинную тонкослойную хроматографию (ТСХ) проводили на пластинах с алюминиевой подложкой Uniplates (Analtech, Newark, DE). Температуры плавления измеряли на приборе для определения температур плавления Fisher-Johns (нескорректированы). ЯМР-спектры записывали на спектрометре Bruker AX 300 (Billerica, MA) или спектрометре Varian Inova-500 (Vernon Hills, Illinois). Значения химических сдвигов приведены в миллионных долях (м.д.) относительно TMS в CDCl3. Macc-спектральные данные получали на приборе Bruker ESQUIRE электроспрей/ионная ловушка в режиме регистрации положительно и отрицательно заряженных ионов. Элементный анализ проводились в Atlantic Microlab Inc.
Исследования клеточных культур и цитотоксичности для рака предстательной железы и меланомы. Все клеточные линии получали от АТСС (American Type Culture Collection, Manassas, VA, USA), а все культуры клеток были приобретены в Cellgro Mediatech (Herndon, VA, USA). Авторы анализировали антипролиферативную активность полученных анти-тубулиновых соединений на четырех клеточных линиях рака предстательной железы человека (LNCaP, DU 145, PC-3 и РРС-1) и двух клеточных линиях меланомы человека (А375 и WM-164). Клеточная линия рака яичника человека OVCAR-8 и его резистентная клеточная линия, отличающаяся сверхвыработкой P-gp (NCI/ADR-RES) использовались в качестве MDR моделей. Обе клеточные линии рака яичника были получены от National Cancer Institutes (NCI). Все клеточные линии были проанализированы и проверены на аутентичность либо в АТСС либо в NCI. Все клеточные линии рака предстательной железы и рака яичника выращивали в RPMI 1640, с добавлением 10% эмбриональной бычьей сыворотки (FBS). Клетки меланомы выращивали в DMEM, с добавлением 5% FBS, 1% смеси антибиотика/противогрибкового средства (Sigma-Aldrich, Inc., St. Louis, МО, USA) и бычьего инсулина (5 мкг/мл; Sigma-Aldrich). Цитотоксический потенциал анти-тубулиновых соединений определяли анализом с сульфородамином В (SRB) через 96 часов после обработки.
Растворимость в воде. Растворимость лекарственных средств определяли на Multiscreen Solubility Filter Plate (Millipore Corporate, Billerica, MA), соединенном с LC-MS/MS. Вкратце, 198 мкл фосфатно-солевого буферного раствора (PBS) (рН 7.4) помещали в 96-луночный планшет, и диспергировали и смешивали с 2 мкл 10 мМ раствора испытуемых соединений (в ДМСО) при аккуратном встряхивании (200-300 об/мин) в течение 1.5 часов при комнатной температуре (N=3). Планшет центрифугировали при 800 g в течение 5 минут, и фильтрат использовали для определения его концентрации и растворимости испытуемого соединения методом LC-MS/MS, как описано ниже.
Исследование фармакокинетики. Самок крыс Sprague-Dawley (n=3 или 4; 254±4 г) покупали в Harlan Inc. (Indianapolis, IN). Торакальные катетеры для яремной вены крыс покупали в Braintree Scientific Inc. (Braintree, MA). После прибытия, животные акклиматизировались в течение 3 дней в комнате с контролируемой температурой (20-22°С) с 12-часовым циклом темнота/свет перед какими либо экспериментами. Соединение 1h вводили внутривенно (i.v.) через катетеры в яремной вене в дозировке 2.5 мг/кг (в ДМСО/PEG300, 2/8), а 5На и 5Нс - в дозировке 5 мг/кг (в ДМСО/PEG300, 1/9). Равные объемы гепаринизированного физраствора инъецировали для замены забираемой крови, и отбирали образцы крови (250 мкл) через катетеры в яремной вене через 10, 20, 30 минут, и 1, 2, 4, 8, 12, 24 часа. Соединения 1h, 5На и 5Нс принудительно вводили перорально (р.о.) в дозировке 10 мг/кг (в Tween80/ДМСО/Н2О, 2/1/7). Все образцы крови (250 мкл) после перорального введения отбирали через катетеры в яремной вене через 30, 60, 90 минут, 120 минут, 150 минут, 180 минут, 210 минут, 240 минут, и через 8, 12, 24 часа. Гепаринизированные шприцы и виалы готовили до забора крови. Образцы плазмы готовили центрифугированием образцов крови при 8000 g в течение 5 минут. Все образцы плазмы сразу замораживали и хранили при -80°С до момента анализа.
Аналиты экстрагировали из 100 мкл плазмы 200 микролитрами ацетонитрила, содержащего 200 нМ внутреннего стандарта ((3,5-диметоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон). Образцы тщательно перемешивали, центрифугировали, и органический экстракт переносили в автосэмплер для LC-MS/MS анализа. Для наблюдения наиболее чувствительных сигналов применяли режим мониторинга нескольких реакций (multiple reaction monitoring (MRM)), сканируя m/z 356 → 188 (соединение 1h), m/z 371 → 203 (соединение 5На), m/z 389 → 221 (соединение 5Нс) и m/z 309 → 171 (внутренний стандарт). Фармакокинетические параметры определяли с применением анализа без учета компартментов (WinNonlin, Pharsight Corporation, Mountain View, CA).
Метод анализа. Раствор образца (10 мкл) вводили в ВЭЖХ систему Agilent (Agilent 1100 Series Agilent 1100 Chemstation, Agilent Technology Co, Ltd). Все аналиты разделяли на narrow-bore C18 колонке (Alltech Alltima HP, 2.1×100 мм, 3 мкм. Fisher, Fair Lawn, NJ). Применяли два градиентных режима. Градиентный режим применяли для достижения разделения аналитов в смесях подвижной фазы A [ACN/H2O (5/95, об/об), содержащей 0.1% муравьиной кислоты] и подвижной фазы В [ACN/H2O (95%/5%, об/об) содержащей 0.1% муравьиной кислоты] при скорости потока 300 мкл/мин. Подвижная фаза А использовалась в количестве 15% с 0 до 1 мин, затем линейный градиент до 100% подвижной фазы В в течение 6 мин, 100% подвижной фазы В выдерживали в течение 0.5 мин, затем быстрый переход на 15% подвижной фазы А. Подвижная фаза А подавалась еще 12 минут до конца анализа.
In Vitro анализ полимеризации тубулина. Тубулин бычьего мозга (0.4 мг, >97% чистота) (Cytoskeleton, Denver, CO) смешивали с 10 мкМ анализируемого соединения и инкубировали в 100 мкл общего тубулинового буфера (80 мМ PIPES, 2.0 мМ MgCl2, 0.5 мМ EGTA и 1 мМ GTP) при рН 6.9. Поглощение при длине волны 340 нм измеряли каждую минуту в течение 20 минут на микропланшетном ридере SYNERGY 4 (Bio-Tek Instruments, Winooski, VT). Температура в спектрофотометре поддерживалась равной 37°С для полимеризации тубулина.
Использовали трехквадрупольный масс-спектрометр API Qtrap 4000™ (Applied Biosystems/MDS SCIEX, Concord, Ontario, Canada), работающий с источником TurboIonSpray. Напряжение на игле-распылителе составляло 5 кВ в режиме регистрации положительных ионов. Газовую завесу выставляли на 10, Газ 1 и газ 2 выставляли на 50. Газ активированной столкновениями диссоциации (CAD) выставляли на значение "medium", а температуру нагревателя образцов - на 500°С. Регистрацию данных и количественный обсчет значений проводили с применением программного обеспечения Analyst(TM) software, Ver. 1.4.1 (Applied Biosystems). Чистоту конечных соединений проверяли методом обращенно-фазной ВЭЖХ на ВЭЖХ-системе Waters 2695, снабженной детектором с фотодиодной матрицей. Два метода обращенно-фазной ВЭЖХ применяли на Supelco Ascentis™ 5 мкм С-18 колонке (250 × 4.6 мм) при комнатной температуре и скорости потока 0.7 мл/мин. ВЭЖХ1: Градиент: Растворитель А (вода) и Растворитель В (метанол): 0-20 мин 40-100% В (линейный градиент), 20-27 мин 100% В. ВЭЖХ2: Градиент: Растворитель А (вода) и Растворитель В (метанол): 0-15 мин 40-100% В (линейный градиент), 15-25 мин 100% В. УФ-детектирование при 254 нм.
Соединения по настоящему изобретению получали согласно фиг.1-17.
Пример 1
Синтез соединений с разными вариантами цикла В
Соединения с разными вариантами цикла В синтезировали согласно фиг.1 и 2.
Оксазольный цикл В:
Синтез (2-фенил-оксазол-4-ил)-(3,4,5-триметокси-фенил)-метанона (36а) (фиг.1):
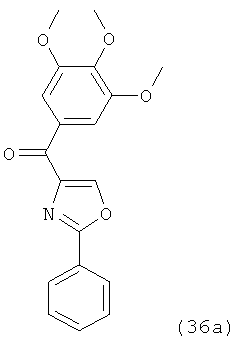
Метиловый эфир (2R)-2-фенил-4,5-дигидро-оксазол-4-карбоновой кислоты (32а). Ацетилхлорид (6.8 мл) прикалывали к охлаждаемому льдом метанолу (30 мл). После добавления L-серина (0.48 ммоль) реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Упаривание растворителя дало белое твердое вещество - гидрохлорид метилового эфира (2R)-3-гидрокси-2-метил-пропионовой кислоты, который использовали без очистки в следующей стадии. Триэтиламин (11 мл, 72.3 ммоль) медленно добавляли к раствору гидрохлорида этилбензимидата (11.6 г, 62.8 ммоль) в CH2Cl2 (150 мл). Реакционную смесь перемешивали при комнатной температуре 30 мин и порциями добавляли гидрохлорид метилового эфира (2R)-3-гидрокси-2-метил-пропионовой кислоты (13.5 г, 79.6 ммоль). Полученную смесь перемешивали в течение 48 часов и упаривали при пониженном давлении. Соединение 32а выделяли методом колоночной флэш-хроматографии в виде желтого масла (12.3 г, 95.9%). 1Н-ЯМР (CDCl3) [δ] 7.99-7.38 (м, 5Н), 4.97 (дд, 1Н, J=7.8 Гц, J=10.5 Гц), 4.70 (т, 1H, J=8.7 Гц), 4.62 (дд, 1Н, J=8.7 Гц, J=10.5 Гц), 3.82 (с, 3Н); MS (ESI) m/z 206.1 (M+H)+.
(2R)-2-фенил-4,5-дигидро-оксазол-4-карбоновая кислота (33a). К охлаждаемому льдом раствору 32а в МеОН/Н2О при перемешивании добавляли LiOH (2.5 экв). Смесь оставляли нагреваться до комнатной температуры в течение 1 часа, упаривали в вакууме, и полученное белое твердое вещество растворяли в H2O и подкисляли добавлением 1 н. раствора HCl до рН 2.0, экстрагировали MgSO4, фильтровали и упаривали в вакууме, получая кислоту 33а в виде белого твердого вещества (95.8%). 1Н-ЯМР (CDCl3) [δ] 7.98 (д, 2Н), 7.57-7.42 (м, 3Н), 5.04 (дд, 1Н, J=7.8 Гц, J=10.8 Гц), 4.80 (т, 1Н, J=8.7 Гц), 4.70 (дд, 1Н, J=9.0 Гц, J=10.8 Гц); MS (ESI) m/z 191.9 (M+H)+, 189.7 (М-Н)-, 145.8 (М-СООН)-.
Метокси-метил-амид (2R)-2-фенил-4,5-дигидро-оксазол-4-карбоновой кислоты (34а). К смеси 33а (5 ммоль), EDCI (6 ммоль), HOBt (5 ммоль) и Et3N (5 ммоль) в CH2Cl2 (50 мл) добавляли HNCH3OCH3 (5 ммоль) и продолжали перемешивать при комнатной температуре 6-8 часов. Реакционную смесь разбавляли добавлением CH2Cl2 (100 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая неочищенный продукт 34а, который очищали методом колоночной хроматографии и получали в виде белого твердого вещества (61.0%). 1Н-ЯМР (CDCl3) [δ] 7.98-7.36 (м, 5Н), 7.57-7.42 (м, 3Н), 5.35 (ушир, т, 1Н), 4.81 (ушир, т, 1Н), 4.52 (дд, 1Н, J=8.7 Гц, J=10.2 Гц), 3.90 (с, 3Н), 3.27 (с, 3Н); MS (ESI) m/z 257.0 (М+Н)+.
(2R)-(2-фенил-4,5-дигидро-оксазол-4-ил)-(3,4,5-триметокси-фенил)-метанон (35а). К раствору н-BuLi (1.6 М, 0.713 мл) в 8 мл ТГФ добавляли раствор 3,4,5-триметоксибромбензола (1.09 ммоль) в 3 мл ТГФ при -78°С. Смесь перемешивали в течение 2 часов и добавляли раствор амида Вайнреба 34а (1.14 ммоль) в 3 мл ТГФ. Температуру повышали до комнатной и перемешивали в течение ночи. Реакционную смесь гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 35а в виде белого твердого вещества (47.9%). 1Н-ЯМР (CDCl3) [δ] 7.97-7.94 (м, 2Н), 7.62 (с, 2Н), 7.54-7.37 (м, 3Н), 5.61 (кв, 1Н, J=7.5 Гц, 9.9 Гц), 5.12 (т, 1Н, J=7.5 Гц), 4.57 (кв, 1Н, J=7.8 Гц, 9.9 Гц), 3.96 (с, 6Н), 3.95 (с, 3Н); MS (ESI) m/z 364.1 (M+Na)+, 340.1 (М-Н)-.
(2-фенил-оксазол-4-ил)-(3,4,5-триметокси-фенил)-метанон (36а). Смесь 35а (1.48 ммоль), CBrCl3 (2.59 ммоль) и DBU (2.97 ммоль) в CH2Cl2 (20 мл) перемешивали в течение ночи. Реакционную смесь адсорбировали на силикагеле и очищали методом колоночной хроматографии, получая чистый целевой продукт 36а (61.6%). 1Н-ЯМР (CDCl3) [δ] 8.37 (с, 1Н), 8.14-8.12 (м, 2Н), 7.74 (с, 2Н), 7.52-7.49 (м, 3Н), 3.97 (с, 9Н); MS (ESI) m/z 362.1 (M+Na)+.
Бензольный, пиримидиновый, пиридиновый, фурановый, тиофеновый, тиазольный, пиразольный и пиперидиновый варианты цикла В (фиг.2): Варианты цикла В (1a-1d, 1k) получали из их соответствующих кислот (37а-37d, 37k). Соединение 1f с тиофеном в положении цикла В невозможно выделить из смеси 1f и побочного продукта сочетания реагента Гриньяра 3,4,5,3',4',5'-гексаметоксибифенила с помощью флэш-колонки. Поэтому для получения 1f был использован альтернативный метод: Амид Вайнреба 38f превращали в соответствующий альдегид, который затем вводили в реакцию с 3,4,5-триметоксифенилмагния бромидом, получая спирт 40f, который можно легко отделить от 3,4,5,3',4',5'-гексаметоксибифенила при помощи колоночной флэш-хроматографии. Окисление пиридиний дихроматом (PDC) или ДМСО не привело к получению 1f из вторичного амина 40f с хорошим выходом. Но использование в качестве окислителя периодинана Десс-Мартина позволило успешно получить целевое кетоновое соединение 1f. 1e и 1i получали из спиртов 40е и 40i аналогичным образом. Соединение 1g получали реакцией сочетания из пиперидина 41g и 3,4,5-триметоксибензойной кислоты.
Бензольный цикл В:
Синтез Бифенил-3-ил(3,4,5-триметоксифенил)метанона (1а) (фиг.2)
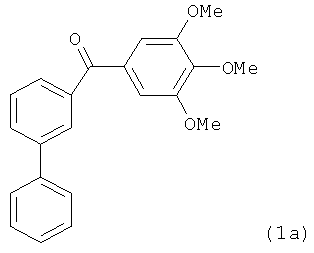
N-Метокси-N-метилбифенил-3-карбоксамид (38а). К смеси 37а (5 ммоль), EDCI (6 ммоль), HOBt (5 ммоль) и NMM (11 ммоль) в CH2Cl2 (50 мл) добавляли HNCH3OCH3 гидрохлорид (5 ммоль) и продолжали перемешивать при комнатной температуре 2 часа. Реакционную смесь разбавляли добавлением CH2Cl2 (100 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая бесцветное масло, которое использовали в следующей стадии (58.4%). MS (ESI) m/z 264.0 (M+Na)+.
Бифенил-3-ил(3,4,5-триметоксифенил)метанон (1а). К раствору 38а (фиг.2) (0.174 г, 0.72 ммоль) в 5 мл ТГФ добавляли тетрагидрофурановый раствор 3,4,5-триметоксифенилмагний бромида (0.5 н., 1.08 ммоль) при 0°С. Смесь перемешивали в течение 30 мин, гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 1а в виде белого твердого вещества (43.8%). 1Н-ЯМР (CDCl3) [δ] 8.02 (т, 1Н), 7.84-7.74 (м, 2Н), 7.64-7.38 (м, 6Н), 7.11 (с, 2Н), 3.95 (с, 3Н), 3.88 (с, 6Н); MS (ESI) m/z 371.1 (M+Na)+.
Пиримидиновый цикл В:
Синтез (6-фенилпиримидин-4-ил)(3,4,5-триметоксифенил)метанона (1b) (фиг.2)
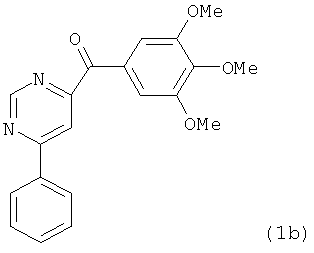
N-Метокси-N-метил-6-фенилпиримидин-4-карбоксамид (38b). К смеси 37b (5 ммоль), EDCI (6 ммоль), HOBt (5 ммоль) и NMM (11 ммоль) в CH2Cl2 (50 мл) добавляли HNCH3OCH3 гидрохлорид (5 ммоль) и продолжали перемешивать при комнатной температуре в течение ночи. Реакционную смесь разбавляли добавлением CH2Cl2 (100 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 38b в виде желтого твердого вещества (62.3%). 1Н-ЯМР (CDCl3) [δ] 9.28 (с, 1Н), 8.14-8.06 (м, 2Н), 7.96 (ушир, с, 1Н), 7.54-7.50 (м, 3Н), 5.35 (ушир, т, 1Н), 4.81 (ушир, т, 1Н), 4.52 (дд, 1Н, J=8.7 Гц, J=10.2 Гц), 3.79 (с, 3Н), 3.42 (с, 3Н); MS (ESI) m/z 266.0 (M+Na)+.
(6-фенилпиримидин-4-ил)(3,4,5-триметоксифенил)метанон (1b). К раствору 38b (0.243 г, 1 ммоль) в 5 мл ТГФ добавляли тетрагидрофурановый раствор 3,4,5-триметоксифенилмагния бромида (0.5 н., 5.6 мл, 1.4 ммоль) при 0°С. Смесь перемешивали в течение 30 мин, гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 1b (52.3%). 1Н-ЯМР (CDCl3) [δ] 9.40 (д, 1Н, J=1.5 Гц), 8.29 (д, 1Н, J=1.5 Гц), 8.22-8.18, 7.57-7.54 (м, 5Н), 7.46 (с, 2Н), 3.96 (с, 3Н), 3.91 (с, 6Н); MS (ESI) m/z 351.1 (M+H)+.
Пиридиновый цикл В:
Синтез (6-фенилпиридин-2-ил)(3,4,5-триметоксифенил)метанона (1с) (фиг.2)
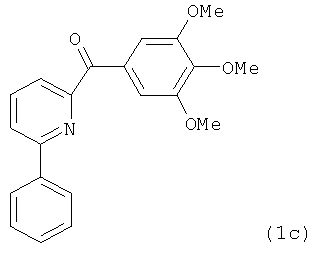
N-Метокси-N-метил-6-фенилпиколинамид (38с). К смеси 37с (1.77 ммоль), EDCI (2.12 ммоль), HOBt (1.86 ммоль) и NMM (3.54 ммоль) в CH2Cl2 (20 мл) добавляли HNCH3OCH3 гидрохлорид (1.86 ммоль) и продолжали перемешивать при комнатной температуре в течение ночи. Реакционную смесь разбавляли добавлением CH2Cl2 (40 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 38с в виде бесцветного масла (51.2%). 1Н-ЯМР (CDCl3) [δ] 8.02 (д, 1Н, J=7.0 Гц), 7.86-7.81 (м, 2Н), 7.55 (ушир, 1Н), 7.48 (т, 2Н), 7.44-7.41 (м, 1Н), 3.82 (с, 3Н), 3.44 (с, ушир., 3Н); MS (ESI) m/z 265.0 (M+Na)+.
(6-фенилпиридин-2-ил)(3,4,5-триметоксифенил)метанон (1с). К раствору 38с (0.210 г, 0.86 ммоль) в 5 мл ТГФ добавляли тетрагидрофурановый раствор 3,4,5-триметоксифенилмагний бромида (0.5 н., 3.5 мл, 1.73 ммоль) при 0°С. Смесь перемешивали в течение 30 мин, гасили добавлением воды, экстрагировали этилацетатом и сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистый 1с в виде белых игольчатых кристаллов (78%). 1Н ЯМР (CDCl3) [δ] 8.10 (д, ушир., 2Н), 8.02-8.00 (м, 1Н), 7.97-7.96 (м, 2Н), 7.66 (с, 2Н), 7.49-7.43 (м, 3Н), 3.97 (с, 3Н), 3.89 (с, 6Н); MS (ESI) m/z 372.6 (M+Na)+.
Фурановый цикл В:
Синтез (5-фенилфуран-2-ил)(3,4,5-триметоксифенил)метанона (1d) (фиг.2)
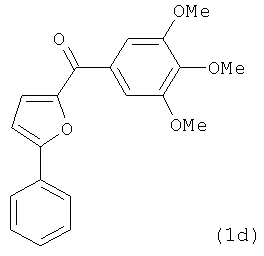
N-Метокси-N-метил-5-фенилфуран-2-карбоксамид (38d). К смеси 37d (10 ммоль), EDCI (12 ммоль), HOBt (11 ммоль) и NMM (21 ммоль) в CH2Cl2 (200 мл) добавляли HNCH3OCH3 гидрохлорид (10.5 ммоль) и продолжали перемешивать при комнатной температуре в течение ночи. Реакционную смесь разбавляли добавлением CH2Cl2 (200 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 38d. (95.2%). 1Н-ЯМР (CDCl3) [δ] 7.82 (д, 1Н, J=7.0 Гц), 7.46-7.43 (т, 2Н), 7.37-7.34 (м, 1Н), 7.25 (д, 1Н, J=4.0 Гц), 6.78 (д, 1Н, J=4.0 Гц), 3.86 (с, 3Н), 3.41 (с, 3Н); MS (ESI) m/z 254.1 (M+Na)+.
(5-фенилфуран-2-ил)(3,4,5-триметоксифенил)метанон (1d). К раствору 38d (0.231 г, 1 ммоль) в 5 мл ТГФ добавляли тетрагидрофурановый раствор 3,4,5-триметоксифенилмагний бромида (0.5 н., 4.0 мл, 2 ммоль) при 0°С. Смесь перемешивали в течение 30 мин, гасили добавлением воды, экстрагировали этилацетатом и сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 1d в виде белых кристаллов (35.5%). 1Н-ЯМР (CDCl3) [δ] 7.85-7.82 (м, 1Н), 7.48-7.36 (м, 4Н), 7.35 (с, 2Н), 7.25 (д, 1Н, J=4.0 Гц), 6.86 (д, 1Н, J=4.2 Гц), 3.96 (с, 3Н), 3.95 (с, 6Н); MS (ESI) m/z 339.1 (М+Н)+.
Тиазольный цикл В:
Синтез (2-фенилтиазол-5-ил)(3,4,5-триметоксифенил)метанона (1е) (фиг.2)
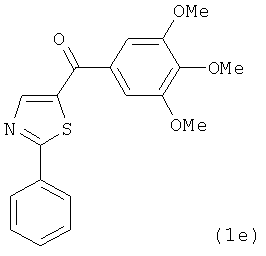
(2-фенилтиазол-5-ил)(3,4,5-триметоксифенил)метанол (40е). К раствору 2-фенилтиазол-5-карбальдегида 38е (0.567 г, 3 ммоль) в 15 мл ТГФ добавляли тетрагидрофурановый раствор 3,4,5-триметоксифенилмагний бромида (0.5 н., 6.5 мл, 3.25 ммоль) при 0°С. Смесь перемешивали в течение 30 мин и гасили насыщенным раствором NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 40е (72.9%). 1Н ЯМР (CDCl3) [δ] 7.90 (м, 2Н), 7.64 (с, 1Н), 7.41 (м, 3Н), 6.69 (с, ушир., 2Н), 6.04 (с, 1Н), 3.86 (с, 6Н), 3.85 (с, 3Н), 1.57 (д, 1Н, J=5.5 Гц); MS (ESI) m/z 358.1 (M+Na)+.
(2-фенилтиазол-5-ил)(3,4,5-триметоксифенил)метанон (1e). К раствору 40е (0.357 г, 1 ммоль) в 40 мл безводного CH2Cl2 добавляли реагент Десс-Мартина (0.848 г, 2 ммоль). Смесь перемешивали в течение 30 мин, гасили насыщенным раствором Na2S2O3, экстрагировали этилацетатом и сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 1е (80.1%). 1Н-ЯМР (CDCl3) [δ] 8.33 (с, 1Н), 8.04 (м, 2Н), 7.51 (м, 3Н), 7.18 (с, 2Н), 3.96 (с, 3Н), 3.93 (с, 6Н); MS (ESI) m/z 378.1 (M+H)+.
Тиофеновый цикл В:
Синтез (5-фенилтиофен-3-ил)(3,4,5-триметоксифенил)метанон (1f) (фиг.2)
N-Метокси-N-метил-5-фенилтиофен-3-карбоксамид (38f). К смеси 37f (2.5 ммоль), EDCI (2.9 ммоль), HOBt (2.6 ммоль) и NMM (5.3 ммоль) в CH2Cl2 (30 мл) добавляли HNCH3OCH3 гидрохлорид (2.6 ммоль) и продолжали перемешивать при комнатной температуре в течение ночи. Реакционную смесь разбавляли добавлением CH2Cl2 (20 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 38f (90.8%). 1Н-ЯМР (CDCl3) [δ] 8.28 (д, 1Н, J=1.5 Гц), 7.69 (д, 1Н, J=1.5 Гц), 7.64 (д, 2Н, J=7.0 Гц), 7.44 (т, 2Н, J=7.0 Гц), 7.35-7.32 (м, 1Н), 6.78 (д, 1Н, J=4.0 Гц), 3.86 (с, 3Н), 3.41 (с, 3Н); MS (ESI) m/z 270.0 (M+Na)+.
(5-фенилтиофен-3-ил)(3,4,5-триметоксифенил)метанол (40f). При -78°С к раствору 38f (2.5 ммоль) в 5 мл ТГФ в атмосфере аргона добавляли раствор LiAlH4 в ТГФ (1 н., 1.42 мл) и продолжали перемешивать 1 час при -20°С. Реакционную смесь помещали в ледяную баню и гасили 20%-ным раствором H2SO4, экстрагировали этилацетатом и сушили над MgSO4. Растворитель удаляли при пониженном давлении и очищали методом колоночной хроматографии, получая 5-фенилтиофен-3-карбальдегид (не изображен) (84.8%). 1Н-ЯМР (CDCl3) [δ] 9.98 (с, 1Н), 8.04 (д, 1H, J=1.5 Гц), 7.86 (ушир, 1Н), 7.61-7.58 (ушир, 2Н), 7.47-7.33 (м, 3Н), 7.35-7.32 (м, 1Н), 6.78 (д, 1Н, J=4.0 Гц); MS (ESI) m/z 210.9 (M+Na)+. К раствору 5-фенилтиофен-3-карбальдегида (0.195 г, 1.04 ммоль) в 5 мл ТГФ добавляли тетрагидрофурановый раствор 3,4,5-триметоксифенилмагний бромида (0.5 н., 2.3 мл, 1.14 ммоль) при 0°С. Смесь перемешивали в течение 30 мин, гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 40f. (70.5%). 1Н-ЯМР (CDCl3) [δ] 7.55-7.52 (м, 2Н), 7.40-7.35 (м, 3Н), 7.30 (ушир, 1Н), 7.20 (ушир, 1Н), 6.72 (с, 2Н), 6.01 (д, 1Н, J=3.9 Гц), 3.86 (с, 6Н), 3.85 (с, 3Н), 2.42 (д, 1Н, J=3.9 Гц); MS (ESI) m/z 339.1 (M-OH)-.
(5-фенилтиофен-3-ил)(3,4,5-триметоксифенил)метанон (1f). К раствору 40f (0.260 г, 0.73 ммоль) в 20 мл безводного CH2Cl2 добавляли реагент Десс-Мартина (0.465 г, 1.36 ммоль). Смесь перемешивали в течение 30 мин, гасили насыщенным раствором Na2S2O3, экстрагировали этилацетатом и сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 1f в виде светло-желтых кристаллов (60.9%). 1Н-ЯМР (CDCl3) [δ] 7.97 (д, 1Н, J=1.5 Гц), 7.82 (д, 1Н, J=1.5 Гц), 7.59-7.57 (м, 2Н), 7.45-7.34 (м, 3Н), 7.19 (с, 2Н), 3.95 (с, 3Н), 3.93 (с, 6Н); MS (ESI) m/z 355.1 (М+Н)+.
Пиперидиновый цикл В:
Синтез (4-фенилпиперидин-1-ил)(3,4,5-триметоксифенил)метанон (1g) (фиг.2)
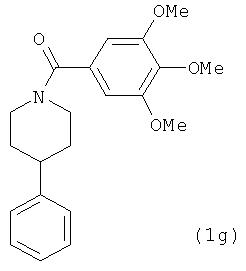
(4-фенилпиперидин-1-ил)(3,4,5-триметоксифенил)метанон (1g). К смеси 4-фенилпиперидина 41g (5 ммоль), EDCI (6 ммоль), HOBt (5.5 ммоль) и NMM (6 ммоль) в CH2Cl2 (50 мл) добавляли 3,4,5-триметоксибензойную кислоту (5.3 ммоль) и продолжали перемешивать при комнатной температуре в течение ночи. Реакционную смесь разбавляли добавлением CH2Cl2 (100 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 1g. (57.9%). 1Н-ЯМР (CDCl3) [δ] 7.35-7.21 (м, 5Н), 6.66 (с, 2Н), 4.84 (ушир, 1Н), 3.95 (ушир, 1Н), 3.88 (с, 6Н), 3.86 (с, 3Н), 3.20-2.87 (ушир, 2Н), 2.85-2.74 (тт, 1Н, J=3.6 Гц, J=15.6 Гц) 1.92 (ушир, 2Н), 1.70 (ушир, 2Н); MS (ESI) m/z 378.1 (M+Na)+.
Изоксазольный цикл В:
Синтез (5-фенилизоксазол-3-ил)(3,4,5-триметоксифенил)метанон (1i) (фиг.2)
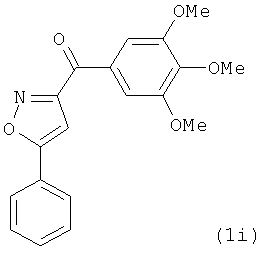
(5-фенилизоксазол-3-ил)(3,4,5-триметоксифенил)метанол (40i). К раствору 5-фенилизоксазол-3-карбальдегида 38i (0.365 г, 2.1 ммоль) в 15 мл ТГФ добавляли тетрагидрофурановый раствор 3,4,5-триметоксифенилмагний бромида (0.5 н., 5.5 мл, 2.74 ммоль) при 0°С. Смесь перемешивали в течение 30 мин, гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 40i в виде белого твердого вещества. (48.8%). 1Н-ЯМР (CDCl3) [δ] 7.78-7.77 (м, 2Н), 7.48-7.46 (м, 3Н), 6.74 (с, 2Н), 6.45 (с, 1Н), 5.98 (д, 1Н, J=3.5 Гц), 3.89 (с, 6Н), 3.86 (с, 3Н), 2.77 (д, 1Н, J=3.5 Гц); MS (ESI) m/z 364.1 (M+Na)+.
(5-фенилизоксазол-3-ил)(3,4,5-триметоксифенил)метанон (1i). К раствору 40i (0.110 г, 0.73 ммоль) в 8 мл безводного CH2Cl2 добавляли реагент Десс-Мартина (0.274 г, 0.645 ммоль). Смесь перемешивали в течение 30 мин, гасили насыщенным раствором Na2S2O3, экстрагировали этилацетатом и сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 1i (70.1%). 1Н-ЯМР (CDCl3) [δ] 7.87-7.85 (м, 2Н), 7.72 (с, 2Н), 7.53-7.49 (м, 3Н), 7.05 (с, 1Н), 7.82 (д, 1Н, J=1.5 Гц), 3.97 (с, 3Н), 3.96 (с, 6Н); MS (ESI) m/z 362.1 (M+H)+.
Пиразольный цикл В:
Синтез (3-фенил-1Н-пиразол-5-ил)(3,4,5-триметоксифенил)метанона (1k) (фиг.2)
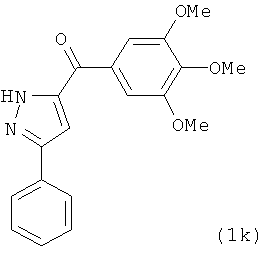
(3-фенил-1Н-пиразол-5-ил)(3,4,5-триметоксифенил)метанон (1k) получали тем же способом, который применялся для получения соединения 1с, из 3-фенил-1Н-пиразол-5-карбоновой кислоты. 1Н-ЯМР (500 МГц, CDCl3) [δ] 10.97 (ушир, 1Н), 7.77 (с, ушир, 2Н), 7.48-7.38 (м, 5Н), 7.14 (с, ушир., 1Н), 3.96 (с, 3Н), 3.94 (с, 6Н); MS (ESI) m/z 361.1 (M+Na)+, 337.0 (M-H)-.
Пример 2
Синтез соединений по настоящему изобретению, имеющих различные линкеры y
Соединения по настоящему изобретению имеют различные линкеры Y. Такие соединения с различными линкерами Y синтезировали согласно фиг.3 и 4.
Соединение 1h синтезировали из 2-фенил-4,5-дигидро-тиазол-4-карбоновой кислоты 42а в три стадии, как описано ранее (Lu, Y.; Wang, Z.; Li, С.М.; Chen, J.; Dalton, J.Т.; Li, W.; Miller, D.D., Synthesis, in vitro structure-activity relationship, and in vivo studies of 2-arylthiazolidine-4-carboxylic acid amides as anticancer agents. Bioorg Med Chem 2010, 18, (2), 477-95, включено в полном объеме в настоящий текст посредством ссылки). 1h превращали в изомеры оксима 2е-цис,транс и 2f-цис,транс реакцией с гидроксиламинами, NH2OH или NH2OCH3. Отнесения были сделаны на основе химических и спектральных данных, как описано ниже. Модифицированная перегруппировка Бекмана позволила легко получить перегруппированные амиды 2g и 2h из двух геометрических стереоизомеров 2е-цис и 2е-транс реакцией с тозилхлоридом и последующей колонкой на основном оксиде алюминия. Гидразидные производные 2d-цис и 2d-транс получали смешиванием 1h с гидразингидратом в этаноле и кипячением в течение 24 часов. Акрилонитрилы 2с-транс,цис получали по реакции Виттига 1h с диэтил цианометилфосфонатом. Цианоимин 2j получали по методике, описанной Cuccia (Cuccia, S.J.; Fleming, L.В.; France, D.J., A novel and efficient synthesis of 4-phenyl-2-chloropyrimidines from acetophenone cyanoimines. Synthetic Communications 2002, 32, (19), 3011-3018, включено в полном объеме в настоящий текст посредством ссылки). Карбонильную группу в соединении 1h также восстанавливали до вторичного спирта 2b или превращали в алкен (2а), как показано на фиг.3.
Попытки удалить карбонильную группу между циклами В и С в 1h привели к образованию соединения 2i, как показано на фиг.4. Введение цис- и транс-двойных связей в положение карбонила дало соединения (3а и 3b), которые синтезировали по реакции Виттига с 2-фенилтиазол-4-карбальдегидом. Сульфидное соединение 4а, сульфон 4b и сульфоксид 4с получали, используя 3-аминобифенил в качестве исходного соединения, сначала проводя реакцию Зандмейера, получая карбонодитиоат 52а, после чего проводили CuI-катализируемую реакцию сочетания и окисление с помощью m-СРВА. Соединение 4d с сульфонамидным линкером получали по реакции 3-бифенилсульфонилхлорида с 3,4,5-триметоксианилином в присутствии NEt3 в ДМФА.
Синтез (2-фенил-тиазол-4-ил)-(3,4,5-триметокси-фенил)-метанона (1h) (фиг.3)
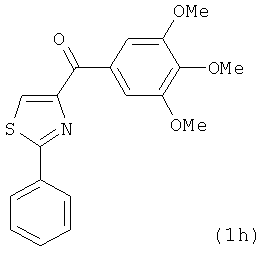
(2-фенил-тиазол-4-ил)-(3,4,5-триметокси-фенил)-метанон (1h). Смесь 2-фенил-4,5-дигидротиазол-4-карбоновой кислоты (5 ммоль), EDCI (6 ммоль) и HOBt (5 ммоль) в CH2Cl2 (50 мл) перемешивали в течение 10 мин. К полученному раствору добавляли NMM (5 ммоль) и HNCH3OCH3 (5 ммоль), и продолжали перемешивать при комнатной температуре 6-8 часов. Реакционную смесь разбавляли добавлением CH2Cl2 (100 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая метоксиметиламид 2-фенил-4,5-дигидротиазол-4-карбоновой кислоты. Раствор метоксиметиламида 2-фенил-4,5-дигидротиазол-4-карбоновой кислоты (1 экв) в CH2Cl2 охлаждали до 0°С, и добавляли перегнанный DBU (2 экв). Затем шприцом добавляли бромтрихлорметан (1.7 экв) по каплям в течение 10 мин. Реакционные смеси оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. После промывки насыщенным водным раствором NH4Cl (2×50 мл), водную фазу экстрагировали этилацетатом (3×50 мл). Объединенные органические слои сушили над MgSO4, фильтровали и упаривали в вакууме. Остаток очищали методом флэш-хроматографии, получая метоксиметиламид 2-фенил-тиазол-4-карбоновой кислоты (73.6%). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.01 (с, 1Н), 7.99-7.96 (м, 2Н), 7.47-7.44 (м, 3Н), 3.88 (с, 3Н), 3.49 (с, 3Н). MS (ESI) m/z 271.0 (M+Na)+. К раствору 3,4,5-триметоксифенилмагния бромида (0.5 н., 3 мл) в 2 мл ТГФ добавляли раствор метоксиметиламида 2-фенил-тиазол-4-карбоновой кислоты (1 ммоль) в 3 мл ТГФ при 0°С. Смеси перемешивали 30 мин до исчезновения амидов по данным ТСХ. Реакционную смесь гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 1h. Выход: 27.3%. 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.29 (с, 1Н), 8.03 (кв, 2Н), 7.80 (с, 2Н), 7.49-7.47 (м, 3Н), 3.96 (с, 6Н), 3.97 (с, 3Н). MS (ESI) m/z 378.1 (M+Na)+.
Синтез 4-(2-метил-1-(3,4,5-триметоксифенил)проп-1-енил)-2-фенилтиазола (2а) (фиг.3)
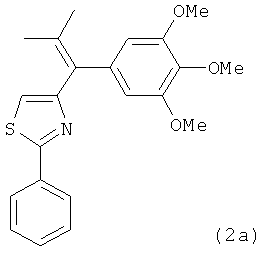
4-(2-метил-1-(3,4,5-триметоксифенил)проп-1-енил)-2-фенилтиазол (2а) (фиг.3). При -78°С к раствору 223 мг иодида изопропилтрифенилфосфония (0.52 ммоль) в 5 мл ТГФ прикалывали 0.4 мл 1.6 н. раствора н-BuLi в гексане в атмосфере аргона. Смесь перемешивали при 0°С 40 минут. Раствор 140 мг (0.39 ммоль) 1h в 5 мл ТГФ прикапывали при 0°С, и полученную смесь перемешивали в течение 1 часа при комнатной температуре. В реакционную смесь добавляли насыщенный раствор NH4Cl. После обычной обработки, колоночная хроматография (силикагель, петролейный эфир/этилацетат) дала соединение 2а (86 мг, 57.3%). 1Н-ЯМР (300 МГц, CDCl3) [δ] 7.98-7.97 (м, 2Н), 7.45-7.40 (м, 3Н), 6.77 (с, 1Н), 6.48 (с, 2Н), 3.86 (с, 3Н), 3.82 (с, 6Н), 2.15 (с, 3Н), 1.81 (с, 3Н). MS (ESI) m/z 404.1 (M+Na)+.
Синтез (2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанола (2b) (фиг.3)
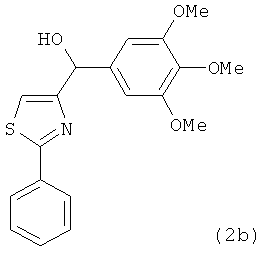
2-фенил-4,5-дигидротиазол-4-карбоновая кислота (42а). Бензонитрил (40 ммоль) объединяли с L-цистеином (45 ммоль) в 100 мл смеси 1:1 МеОН/рН 6.4 фосфатный буферный раствор. Реакционную смесь перемешивали при 40°С 3 дня. Осадок отделяли фильтрованием и МеОН удаляли на роторном испарителе. К оставшемуся раствору добавляли 1М HCl при 0°С, доводя значение рН до 2. Отфильтровывали выпавший осадок, получая белое твердое вещество - 2-фенил-4,5-дигидротиазол-4-карбоновую кислоту 42а, которую напрямую использовали в следующей стадии без дополнительной очистки.
2-фенилтиазол-4-карбальдегид (42b). При -78°С к раствору метоксиметиламида 2-фенил-тиазол-4-карбоновой кислоты (1 экв) в ТГФ добавляли LiAlH4 (1 экв, 1 н. раствор в ТГФ) и перемешивали 1 час при -20°С. Реакционную смесь помещали в ледяную баню и гасили 20%-ным раствором H2SO4, экстрагировали этилацетатом и сушили над MgSO4. Растворитель удаляли при пониженном давлении и очищали методом колоночной хроматографии, получая 42b (45.8%). 1Н-ЯМР (300 МГц, CDCl3) [δ] 10.1 (с, 1Н), 8.17 (с, 1Н), 8.02-8.00 (м, 2Н), 7.50-7.48 (м, 3Н). MS (ESI) m/z 244.1 (M+Na+MeOH)+.
(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанол (2b) (фиг.3). При 0°С к раствору 104 мг 42b (0.55 ммоль, 1 экв.) в 6 мл ТГФ добавляли 3,4,5-триметоксифенилмагния бромид (0.5 н. раствор в ТГФ, 2.9 мл). Смеси перемешивали 30 мин до исчезновения альдегида по данным ТСХ. Реакционную смесь гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение (2b). 1Н-ЯМР (300 МГц, CDCl3) [δ] 7.95-7.92 (м, 2Н), 7.44-7.43 (м, 4Н), 6.97 (с, 1Н), 6.76 (с, 2Н), 5.93 (д, 1Н, J=3.6 Гц), 3.86 (с, 9Н). MS (ESI) m/z 402.1 (M+Na)+.
Синтез (Z)-3-(2-фенилтиазол-4-ил)-3-(3,4,5-триметоксифенил)акрилонитрила (2с-транс) и (E)-3-(2-фенилтиазол-4-ил)-3-(3,4,5-триметоксифенил)акрилонитрила (2с-цис) (фиг.3)
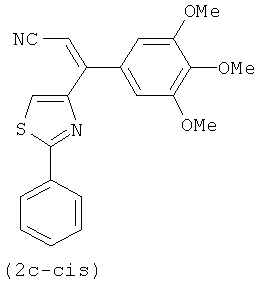
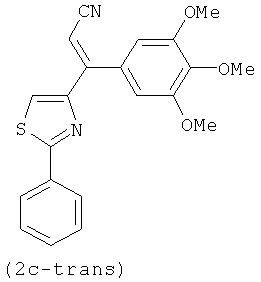
(Z)-3-(2-фенилтиазол-4-ил)-3-(3,4,5-триметоксифенил)акрилонитрил (2с-транс). К смеси 0.4 мл 2.5 н. раствора н-BuLi в гексане и 10 мл ТГФ прикалывали раствор 177 мг (1 ммоль) диэтилцианометилфосфоната в 5 мл ТГФ при 0°С под аргоном. Ледяную баню убирали, и смесь перемешивали при 25°С 40 минут. Прикалывали раствор 200 мг (0.56 ммоль) 1h в 10 мл ТГФ при 0°С, и полученную смесь перемешивали в течение 1 часа при комнатной температуре. В реакционную смесь добавляли насыщенный раствор NH4Cl. После обычной обработки, колоночная хроматография (силикагель, петролейный эфир/этилацетат) дала соединения 2с-транс (83 мг) и 2с-цис (76 мг). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.01-7.99 (м, 2Н), 7.44-7.40 (м, 3Н), 7.21 (с, 1Н), 6.74 (с, 2Н), 6.67 (с, 1Н), 3.93 (с, 3Н), 3.89 (с, 6Н). MS (ESI) m/z 401.1 (M+Na)+.
(Е)-3-(2-фенилтиазол-4-ил)-3-(3,4,5-триметоксифенил)акрилонитрил (2с-цис). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.07-8.05 (м, 2Н), 7.49-7.46 (м, 4Н), 6.66 (с, 2Н), 5.64 (с, 1Н), 3.91 (с, 3Н), 3.86 (с, 6Н). MS (ESI) m/z 401.1 (M+Na)+.
Синтез (Z)-4-(гидразоно(3,4,5-триметоксифенил)метил)-2-фенилтиазола (2d-цис) и (E)-4-(гидразоно(3,4,5-триметоксифенил)метил)-2-фенилтиазола (2d-транс) (фиг.3)
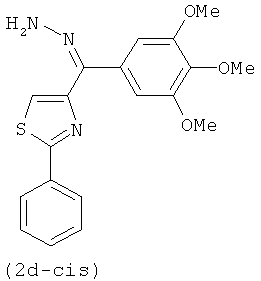
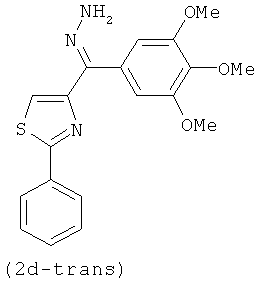
(E)-4-(гидразоно(3,4,5-триметоксифенил)метил)-2-фенилтиазол (2d-цис). К смеси 1h (230 мг, 0.65 ммоль) в 3 мл CH2Cl2 и 3 мл этанола добавляли гидразин гидрат (2 мл). Затем реакционную смесь кипятили в течение ночи. По окончании реакции остаток адсорбировали на силикагеле и очищали методом колоночной хроматографии, получая соединения 2d-цис (80 мг) и 2d-транс (56 мг). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.01-7.98 (м, 2Н), 7.49-7.46 (м, 5Н), 7.33 (с, 1Н), 6.82 (с, 2Н), 3.87 (с, 3Н), 3.85 (с, 6Н). MS (ESI) m/z 370.1 (M+H)+.
(E)-4-(гидразоно(3,4,5-триметоксифенил)метил)-2-фенилтиазол (2d-транс). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.04-8.01 (м, 2Н), 7.44-7.40 (м, 3Н), 6.95 (с, 1Н), 6.65 (с, 2Н), 5.62 (с, 2Н), 3.93 (с, 3Н), 3.87 (с, 6Н). MS (ESI) m/z 370.1 (M+H)+.
Синтез (Z)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон оксим (2е-цис) и (E)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон оксим (2е-транс) (фиг.3)
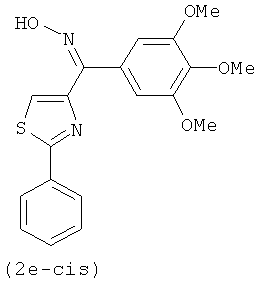
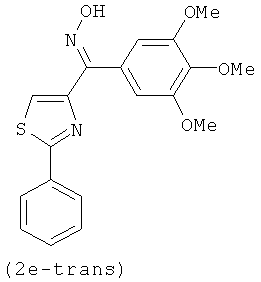
(Z)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон оксим (2е-цис). К суспензии 1h (210 мг, 0.59 ммоль) в 10 мл этанола добавляли водный раствор (2 мл) гидроксиламина гидрохлорида (127 мг, 1.83 ммоль). Затем прикалывали в реакционную смесь 2 мл 1 н. раствора NaOH, и полученную смесь перемешивали 3 часа при 55°С. По окончании реакции, остаток адсорбировали на силикагеле и очищали методом колоночной хроматографии, получая соединения 2е-цис (85 мг) и 2е-транс (50 мг). 1Н-ЯМР (300 МГц, ДМСО-d6) [δ] 11.95 (с, 1Н), 8.35 (с, 1Н), 7.91-7.89 (м, 2Н), 7.50-7.44 (ушир, 3Н), 6.85 (с, 2Н), 3.73 (с, 6Н), 3.70 (с, 3Н). MS (ESI) m/z 393.1 (M+Na)+; 368.9 (M-Н)-.
(Е)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон оксим 2е-транс). 1Н-ЯМР (300 МГц, ДМСО-d6) [δ] 11.49 (с, 1Н), 7.92-7.89 (м, 2Н), 7.64 (с, 1Н), 7.51-7.49 (м, 3Н), 7.34 (с, 1Н), 6.75 (с, 2Н), 3.75 (с, 6Н), 3.72 (с, 3Н). MS (ESI) m/z 393.1 (M+Na)+; 368.9 (M-H)-.
Синтез (Z)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон O-метилоксим (2f-цис) и (E)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон O-метилоксим (2f-транс) (фиг.3)
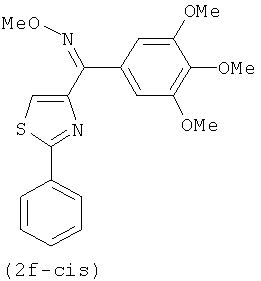
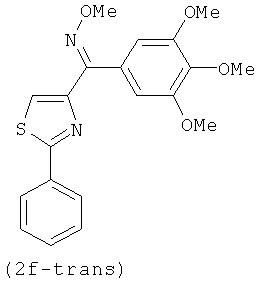
(Z)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанон O-метилоксим (2f-цис). К суспензии 1h (110 мг, 0.59 ммоль) в 10 мл пиридина добавляли O-метилгидроксиламин гидрохлорид (52 мг, 0.63 ммоль), и полученную смесь перемешивали при 60°С в течение ночи. Реакционную смесь гасили добавлением 1 н. раствора HCl, экстрагировали этилацетатом и сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистые соединения 2f-цис (41 мг) и 2f-транс (33 мг). 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.13 (с, 1Н), 7.96-7.94 (м, 2Н), 7.45-7.44 (м, 3Н), 6.94 (с, 2Н), 4.13 (с, 3Н), 3.91 (с, 6Н), 3.88 (с, 3Н). MS (ESI) m/z 407.2 (M+Na)+.
(Е)-(2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метанона O-метилоксим (2f-транс). 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.00-7.98 (м, 2Н), 7.44-7.43 (м, 3Н), 7.28 (с, 1Н), 6.70 (с, 2Н), 4.08 (с, 3Н), 3.91 (с, 6Н), 3.85 (с, 3Н). MS (ESI) m/z 407.0 (M+Na)+.
Синтез 2-фенил-N-(3,4,5-триметоксифенил)тиазол-4-карбоксамида (2g) (фиг.3)
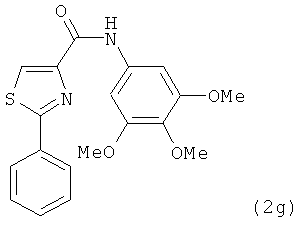
2-фенил-N-(3,4,5-триметоксифенил)тиазол-4-карбоксамид (2g). К раствору 2е-цис (21 мг, 0.06 ммоль) в 5 мл CH2Cl2 добавляли п-толуолсульфонилхлорид (23 мг, 0.12 ммоль) и NaH (5 мг, 60% в легком минеральном масле). Затем реакционную смесь перемешивали в течение 20 мин. По окончании реакции, остаток адсорбировали на силикагеле и очищали колоночной хроматографией на Al2O3, получая соединение 2g (15 мг). 1Н-ЯМР (300 МГц, CDCl3) [δ] 9.22 (с, 1Н), 8.19 (с, 1Н), 8.02-7.99 (м, 2Н), 7.52-7.50 (м, 3Н), 7.07 (с, 2Н), 3.92 (с, 6Н), 3.85 (с, 3Н). MS (ESI) m/z 371.1 (M+H)+.
Синтез 3,4,5-триметокси-N-(2-фенилтиазол-4-ил)бензамида (2h) (фиг.3)
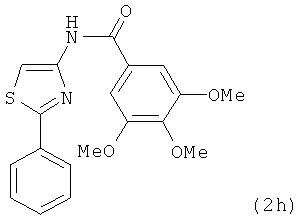
3,4,5-триметокси-N-(2-фенилтиазол-4-ил)бензамид (2h). К раствору 2е-транс (26 мг, 0.07 ммоль) в 5 мл CH2Cl2 добавляли п-толуолсульфонилхлорид (27 мг, 0.14 ммоль) и NaH (5 мг, 60% в легком минеральном масле). Затем реакционную смесь перемешивали в течение 20 мин. По окончании реакции остаток адсорбировали на силикагеле и очищали методом колоночной хроматографии на Al2O3, получая соединение 2h (15 мг). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.88 (с, 1Н), 7.94-7.91 (м, 2Н), 7.83 (с, 1Н), 7.48-7.46 (м, 3Н), 7.18 (с, 2Н), 3.97 (с, 6Н), 3.94 (с, 3Н). MS (ESI) m/z 393.1 (M+Na)+.
Синтез N-((2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метилен)цианамида (2j) (фиг.3)
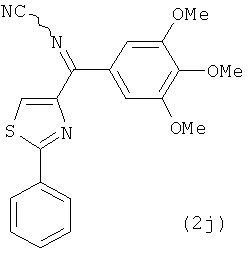
N-((2-фенилтиазол-4-ил)(3,4,5-триметоксифенил)метилен)цианамид (2j).
100 мг 1h (0.28 ммоль, 1 экв.) растворяли в 10 мл метиленхлорида. Тетрахлорид титана в метиленхлориде (1.0 н, 0.7 мл, 2.5 экв.) прикалывали при 0°С и перемешивали в течение 30 мин. Бис-триметилсилилкарбодиимид (2.4 экв.) в 2 мл метиленхлорида добавляли, и реакцию перемешивали в течение ночи в условиях защиты от воздуха и влаги. В реакционную смесь добавляли смесь лед-вода, после чего проводили экстракцию метиленхлоридом. Органическую фазу сушили над сульфатом магния, фильтровали через целит, получая неочищенные ацетофенон цианоимины, которые очищали методом флэш-хроматографии в качестве изомеров, в соотношении 3:7. 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.72 (ушир, 0.3Н), 8.63 (с, 0.7Н), 8.09-8.07 (м, 1.4Н), 7.99 (ушир, 0.6Н), 7.58-7.56 (ушир, 3Н), 7.26 (с, 1.4Н), 7.18 (с, 0.6Н), 3.84, 3.83 (с, с, 6Н), 3.82 (с, 3H). MS (ESI) m/z 402.1 (M+Na)+.
Синтез N-((4-гидрокси-3,5-диметоксифенил)(2-фенилтиазол-4-ил)метилен)цианамида (32).
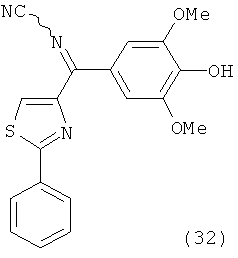
N-((4-гидрокси-3,5-диметоксифенил)(2-фенилтиазол-4-ил)метилен)цианамид (32) получали в качестве побочного продукта при синтезе 2j. 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.23 (с, 1Н), 8.02 (м, 2Н), 7.92 (с, 2Н), 7.55 (м, 3Н), 6.02 (с, 1Н), 3.99 (с, 6Н). MS (ESI) m/z 364.1 (M+H)+
Синтез (Z)-2-фенил-4-(3,4,5-триметоксистирил)тиазол (3а) и (E)-2-фенил-4-(3,4,5-триметоксистирил)тиазол (3b) (фиг.4)
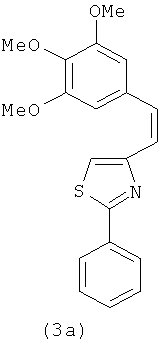
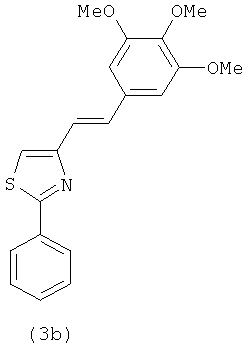
(Е)-2-фенил-4-(3,4,5-триметоксистирил)тиазол (3а). Трифенилфосфин (3.41 г, 13 ммоль) добавляли к раствору 5-(бромметил)-1,2,3-триметоксибензола (2.61 г, 10 ммоль) в безводном ТГФ (30 мл). Смесь кипятили при перемешивании 6 часов. Образовавшееся белое твердое вещество отфильтровывали и промывали смесью эфир/гексан, получая продукт - 3,4,5-триметоксибензилтрифенилфосфония бромид с выходом 96.4%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.77-7.73, 7.65-7.61 (м, 15Н), 6.44 (д, 2H, J=1.5 Гц), 5.37 (д, 2H, J=14 Гц), 3.76 (с, 3H), 3.51 (д, 6H); MS (ESI) m/z 443.1 (М-Br]+. При -78°С н-BuLi (0.42 мл, 2.5 н. раствор в гексане) добавляли к раствору 3,4,5-триметоксибензилтрифенилфосфония бромида (500 мг, 0.96 ммоль) в 10 мл ТГФ. После перемешивания при комнатной температуре в течение 2 часов добавляли альдегид 42b (109 мг, 0.58 ммоль) в 3 мл ТГФ и перемешивали в течение 30 мин. В реакционную смесь добавляли насыщенный раствор NH4Cl. После обычной обработки, колоночная хроматография (силикагель, петролейный эфир/этилацетат) дала соединения 3а (57 мг) и 3b (99 мг). 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.90-7.89 (м, 2Н), 7.42-7.40 (м, 3Н), 7.07 (с, 1Н), 6.71 (с, 2Н), 6.66 (с, 1Н), 3.87 (с, 6Н), 3.75 (с, 3Н); MS (ESI) m/z 376.1 (M+Na)+.
(Е)-2-фенил-4-(3,4,5-триметоксистирил)тиазол (3b). 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.03-8.01 (м, 2H), 7.52 (д, 1H, J=16 Гц), 7.47-7.44 (м, 3H), 7.16 (с, 1H), 7.05 (д, 1H, J=16 Гц), 6.79 (с, 2H), 3.92 (с, 6H), 3.88 (с, 3H). MS (ESI) m/z 354.1 (M+H)+.
Синтез бифенил-3-ил(3,4,5-триметоксифенил)сульфана (4а), 3-(3,4,5-триметоксифенилсульфонил)бифенила (4b) и 3-(3,4,5-триметоксифенилсульфинил)бифенил (4с) (фиг.4)
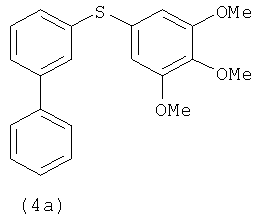
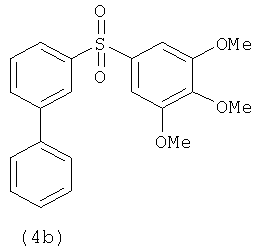
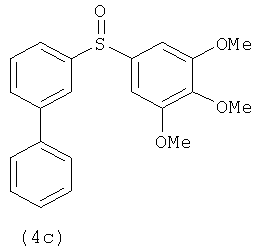
S-Бифенил-3-ил O-этилкарбонодитиоат (52а). К раствору 1 экв. бифенил-3-амина (1 г, 5.92 ммоль) в воде (7.3 мл) при 0°С добавляли концентрированную соляную кислоту (1 мл). Медленно добавляли холодный раствор 1.1 экв. нитрита натрия (450 мг, 6.5 ммоль) в воде (3 мл) и перемешивали в течение 15 мин. Холодный раствор соли диазония медленно добавляли к раствору 1.3 экв. этилксантата калия (1.16 г, 1.3 ммоль) в воде (1.3 мл) при 45°С. Реакционную смесь перемешивали еще 30 мин при 45°С и затем охлаждали до комнатной температуры. Реакционную смесь экстрагировали диэтиловым эфиром (3×50 мл). Объединенные органические экстракты промывали 1 н. раствором NaOH (100 мл), водой (3×50 мл), насыщенным раствором соли (50 мл), сушили над MgSO4, фильтровали и упаривали при пониженном давлении. Полученный неочищенный ксантат 52а использовали напрямую в следующей стадии без дополнительной очистки. MS (ESI) m/z 275.0 (М+Н)+.
Бифенил-3-ил(3,4,5-триметоксифенил)сульфан (4а). К раствору 52а (1.1 г, неочищенное вещество) в этаноле (8 мл) добавляли гидроксид калия (2.1 г, 12 мл) и кипятили в течение ночи. Раствор охлаждали до комнатной температуры и упаривали этанол при пониженном давлении. Остаток растворяли в воде и промывали диэтиловым эфиром (10 мл). Водный слой подкисляли добавлением 2 н. раствора HCl и экстрагировали диэтиловым эфиром (3×50 мл). Органические экстракты промывали водой (50 мл), насыщенным раствором соли (50 мл), сушили над MgSO4, фильтровали и упаривали при пониженном давлении, получая 0.85 г (77.3%) неочищенного бифенил-3-тиольного продукта (в сумме за 3 стадии). В круглодонную колбу, снабженную магнитной мешалкой, помещали 0.1 г (1.04 ммоль) трет-бутоксида натрия и 83 мг иодида меди (0.43 ммоль). После того, как реакционный сосуд был герметично закрыт, через септу шприцем вводили 0.13 г (0.71 ммоль) 4-метоксибензолтиола и 0.19 г (0.65 ммоль) 5-иод-1,2,3-триметоксибензола в 3.0 мл толуола. Реакционную смесь нагревали в течение ночи при 110°С. Очистку проводили методом флэш-хроматографии, и получали аморфное твердое вещество (40% выход). 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.54-7.52 (м, 3Н), 7.44-7.41 (м, 3Н), 7.37-7.33 (м, 2Н), 7.23 (с, ушир, 1Н), 6.69 (с, 2Н), 3.86 (с, 3Н), 3.80 (с, 6Н). MS (ESI) m/z 353.2 (M+H)+.
3-(3,4,5-триметоксифенилсульфонил)бифенил (4b). К раствору 60 мг (0.17 ммоль) соединения 4а в 5 мл дихлорметана очень медленно добавляли 2 экв. m-СРВА в течение 3 часов. Формирование сульфоксида отслеживали методом тонкослойной хроматографии. Очистку проводили методом колоночной флэш-хроматографии, и получали аморфный порошок (4b) (73% выход). 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.14 (ушир, 1Н), 7.89 (д, 1Н), 7.78 (д, 1Н), 7.59-7.56 (м, 3Н), 7.49-7.39 (м, 3Н), 7.19 (с, 2Н), 3.89 (с, 6Н), 3.87 (с, 3Н). MS (ESI) m/z 385.0 (M+Na)+.
3-(3,4,5-триметоксифенилсульфинил)бифенил (4с). При 0°С к раствору 500 мг (1.42 ммоль) соединения (4а) в 5 мл дихлорметана очень медленно добавляли 1 экв. м-СРВА в течение 3 часов. Формирование сульфоксида отслеживали методом тонкослойной хроматографии. Очистку проводили методом колоночной флэш-хроматографии, и получали аморфный порошок (4с) (87% выход). 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.92 (ушир, 1Н), 7.71 (д, 2Н), 7.62-7.60 (м, 3Н), 7.58-7.40 (м, 4Н), 6.94 (с, 2Н), 3.79 (с, 3Н), 3.74 (с, 6Н). MS (ESI) m/z 369.1 (М+H)+.
Синтез N-(3,4,5-триметоксифенил)бифенил-3-сульфонамида (4d) (фиг.4)
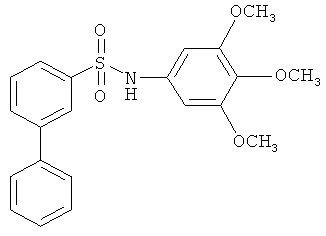
N-(3,4,5-триметоксифенил)бифенил-3-сульфонамид (4d). Смесь 65 мг бифенил-3-сульфонилхлорида (0.25 ммоль), 44 мг 3,4,5-триметоксианилина (0.24 ммоль) и 0.3 ммоль триэтиламина в 5 мл ДМФА перемешивали в течение ночи. Реакционную смесь обрабатывали водой и экстрагировали этилацетатом. После обычной обработки, методом колоночной хроматографии (силикагель, петролейный эфир/этилацетат) выделяли 88 мг соединения (4d) (91.7%). 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.96 (т, 1Н, J=1.8 Гц), 7.81-7.74 (м, 2Н), 7.57-7.40 (м, 6Н), 6.33 (с, 2Н), 3.86 (с, 3Н), 3.80 (с, 6Н). MS (ESI) mJz 422.1 (M+Na)+.
2-фенил-4-(3,4,5-триметоксифенил)тиазол (2i) (фиг.4)
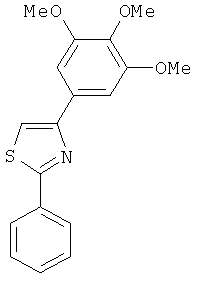
2-фенил-4-(3,4,5-триметоксифенил)тиазол (2i). Бром (160 мг, 1 ммоль) прикалывали к перемешиваемому раствору 1-(3,4,5-триметоксифенил)этанона (210 мг, 1 ммоль) в этаноле (30 мл), и полученный раствор перемешивали при 0°С 1 час, после чего выливали в воду, получая осадок. Осадок перекристаллизовывали из этанола, получая бромацетофенон (70%), который напрямую использовали в следующей стадии. Смесь бромацетофенона (288 мг, 1 ммоль) и бензотиоамида (137 мг, 1 ммоль) в этаноле кипятили 1 час. Реакционную смесь упаривали в вакууме и очищали методом колоночной флэш-хроматографии, получая 2i (167 мг, 51.1%). 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.05-8.03 (м, 2Н), 7.48-7.44 (м, 3Н), 7.41 (с, 1Н), 7.22 (с, 2Н), 3.97 (с, 6Н), 3.89 (с, 3Н). MS (ESI) m/z 350.1 (M+Na)+.
Пример 3
Синтез метоксибензоилтиазольных соединений, имеющих различные циклы "а" и/или замещенный цикл "а"
Соединения по настоящему изобретению содержат различные замещенные или незамещенные циклы А, такие как бензил или индолил. Такие соединения синтезировали согласно фиг.5 и 6.
Гидроксил и аминометил вводили в параположение фенильного А-цикла, а также фенил заменяли на 5-индолильный и 2-индолильный циклы. Амиды Вайнреба 57а, 61а, 65а и 67а получали методом, проиллюстрированным на фиг.5, используя арилнитрилы в качестве исходных соединений. 2-Циано-индол 60а получали по стандартной методике (Pletnev, A.A.; Tian, Q.; Larock, R.С, Carbopalladation of nitriles: synthesis of 2,3-diarylindenones and polycyclic aromatic ketones by the Pd-catalyzed annulation of alkynes and bicyclic alkenes by 2-iodoarenenitriles. J Org Chem 2002, 67, (26), 9276-87, включено в настоящий текст в полном объеме посредством ссылки). В синтезах использовали защиту гидроксильной (TBDMSCl), индолильной (PhSO2Cl) и амино-групп (Вос2О). Снятие TBDMS и окисление тиазолина (58а) в тиазол (21) осуществляли в одну стадию, используя раствор TBAF/ТГФ. Данное окисление тиазолина в тиазол происходит спонтанно в реакции тиазолинового амида Вайнреба и реактива Гриньяра. То же самое наблюдалось при получении индольных соединений 62а и 66а.
Соединение 62а выделяли в виде чистого тиазольного соединения после реакции с 3,4,5-триметоксифениллитием без необходимости дальнейшей очистки. Соединение 66а получали снятием фенилсульфонильных защитных групп в горячем растворе NaOH в этаноле. Пара-ОН и NH2 в цикле А в соединениях 2l и 2r получали аналогичной реакцией Гриньяра из амидов Вайнреба 58а и 68а. Соединение 2r далее превращали в гидрохлорид (2r-HCl) и гидрохлорид монометиламина 2s-HCl в условиях NaH/MeI, и в диметиламин 2u в условиях HCHO/NaBH3CN.
Замещенный цикл А:
Синтез (2-(4-Гидроксифенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (2l) (фиг.5)
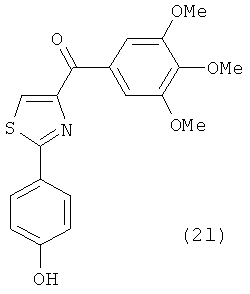
(R)-2-(4-Гидроксифенил)-N-метокси-N-метил-4,5-дигидротиазол-4-карбоксамид (57а) синтезировали способом, описанным для 38d. Количественный выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.56 (д, 2Н, J=8.5 Гц), 6.84 (ушир, 1Н), 6.73 (д, 2Н, J=8.5 Гц), 5.64 (т, ушир, 1Н), 3.87 (с, 3Н), 3.30 (с, 3Н). MS (ESI) m/z 289.0 (M+Na)+, 264.9 (M-H)-.
(R)-(2-(4-(трет-бутилдиметилсилилокси)фенил)-4,5-дигидротиазол-4-ил)(3,4,5-триметоксифенил)метанон (58а) синтезировали способом, описанным для (35а) - см. пример 1. 67.0% выход. 1Н-ЯМР (300 МГц, CDCl3) [δ] 7.73 (д, 2Н, J=8.7 Гц), 7.61 (с, 2Н), 6.83 (д, 2Н, J=8.7 Гц), 5.95 (дд, 1Н, J=8.1 Гц, 9.0 Гц), 4.09, (дд, 1Н, J=7.8 Гц, 11.1 Гц), 3.95 (с, 3Н), 3.94 (с, 6Н), 3.55 (дд, 1Н, J=9.3 Гц, 11.1 Гц), 0.97 (с, 9Н), 0.19 (с, 6Н). MS (ESI) m/z 510.4 (M+Na)+, 486.0 (M-H)-.
(2-(4-Гидроксифенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (2l). При 0°С, к раствору 58а (0.2 ммоль) в 5 мл CH2Cl2 добавляли раствор фторида тетрабутиламмония в ТГФ (1 н. раствор, 0.6 ммоль) и перемешивали при комнатной температуре примерно 14 часов, до окончания реакции по данным ТСХ-мониторинга. 67.0% выход. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 10.1 (с, 1Н), 8.51 (с, 1Н), 7.85 (д, 2Н, J=8.50 Гц), 7.62 (с, 2Н), 6.91 (д, 2Н, J=8.5 Гц), 3.86 (с, 6Н), 3.79 (с, 3Н). MS (ESI) m/z 394.1 (M+Na)+, 369.9 (M-Н)-.
(2-(4-(Аминометил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон гидрохлорид (2r или 2r-HCl) (фиг.5)
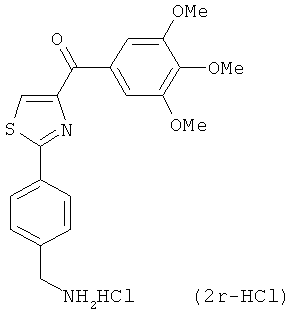
(R)-трет-бутил 4-(4-(метокси(метил)карбамоил)-4,5-дигидротиазол-2-ил)бензилкарбамат (67а). 4-(Аминометил)бензонитрил (25.09 г, 0.149 моль) и L-цистеин (18.1 г, 0.149 моль) суспендировали в 500 мл МеОН и рН 6.4 буферном растворе (1:1), и перемешивали 3 дня при комнатной температуре. В смесь добавляли триэтиламин (30 мл) и Boc2O (68 г, 0.31 моль) и перемешивали в течение 2 часов. Растворители удаляли, и после фильтрования получали белое твердое вещество (R)-2-(4-((трет-бутоксикарбониламино)метил)фенил)-4,5-дигидротиазол-4-карбоновую кислоту (38.4 г, 76.8%). Соединение 67а получали из этой кислоты по той же методике, которая применялась для получения 38d. Выход: 84.4%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.75-7.77 (д, 2H, J=7.5 Гц), 7.27-7.26 (д, 2H, J=7.5 Гц), 7.23 (с, 1Н), 5.62 (ушир, 1Н), 4.87 (ушир, 1Н), 4.30 (ушир, 2Н), 3.86 (с, 3Н), 3.78 (т, J=10.0 Гц, 1Н), 3.48-3.4 (м, 1Н), 3.25 (с, 3Н), 1.42 (с, 9H). MS (ESI) m/z 402.1 (M+Na)+, 378.0 (M-H)-.
Трет-бутил 4-(4-(3,4,5-триметоксибензоил)тиазол-2-ил)бензилкарбамат (68а). Смесь 67а (2.5 ммоль), CBrCl3 (3.2 ммоль) и DBU (5.0 ммоль) в CH2Cl2 (20 мл) перемешивали в течение ночи. Реакционную смесь адсорбировали на силикагеле и очищали методом колоночной хроматографии, получая промежуточный тиазольный амид Вайнреба. К раствору (3,4,5-триметоксифенил)магния бромида (0.5 М, 5.5 мл) в ТГФ добавляли раствор промежуточного тиазольного амида Вайнреба (1.83 ммоль) в 10 мл ТГФ при 0°С и перемешивали в течение 30 мин. Реакционную смесь гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение в виде светло-желтого твердого вещества (32.3%). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.27 (с, 1Н), 7.98 (д, 2Н, J=8.1 Гц), 7.78 (с, 2Н), 7.39 (д, 2Н, J=8.1 Гц), 7.27-7.26 (д, 2Н, J=7.5 Гц), 7.23 (с, 1Н), 4.93 (ушир, 1Н), 4.37 (ушир, д, 1Н), 3.96 (с, 3Н), 3.95 (с, 6Н), 1.47 (с, 9Н); MS (ESI) m/z 507.1 (M+Na)+.
(2-(4-(Аминометил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (2r или 2r-HCl). При 0°С к раствору 68а (200 мг) в 10 мл CH2Cl2 добавляли раствор HCl в 1,4-диоксане (4 н. раствор, 2 мл) и перемешивали при комнатной температуре 4 часа. Осадок (2r) отфильтровывали и промывали диэтиловым эфиром. Выход: 81.3%. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 8.68 (с, 1Н), 8.38 (ушир, 3Н), 8.10 (д, 2Н, J=8.4 Гц), 7.66 (д, 2Н, J=8.4 Гц), 7.62 (с, 2Н), 4.11 (с, 2Н), 3.87 (с, 6Н), 3.80 (с, 3Н). MS (ESI) m/z 385.1 (M+H)+.
(2-(4-((Диметиламино)метил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон гидрохлорид (2u или 2u-HCl) (фиг.5)
Трет-бутилметил(4-(4-(3,4,5-триметоксибензоил)тиазол-2-ил)бензил)карбамат (71а). При 0°С к раствору соединения 68а (100 мг, 0.2 ммоль) в 5 мл ДМФА добавляли гидрид натрия (10 мг, 0.2 ммоль), затем в реакционную смесь добавляли иодметан (77 мг, 0.4 ммоль) и перемешивали при комнатной температуре в течение ночи. Смесь гасили добавлением насыщенного раствора NaHCO3, экстрагировали этилацетатом и сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 71а. Выход: 61.3%. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 8.30 (с, 1Н), 8.02 (д, 2Н, J=8.0 Гц), 7.82 (с, 2Н), 7.36 (ушир, 2Н), 4.50 (с, 2Н), 4.00 (с, 3Н), 3.98 (с, 6Н), 2.90 (д, ушир., 3Н), 1.50 (с, 9Н). MS (ESI) m/z 521.2 (M+Na)+, 496.9 (M-H)-.
(2-(4-((Метиламино)метил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (2s или 2s-HCl). При 0°С, к раствору 71а (60 мг) в 5 мл CH2Cl2 добавляли раствор HCl в 1,4-диоксане (4 н. раствор, 2 мл) и перемешивали при комнатной температуре в течение ночи. Осадок (2s-HCl) отфильтровывали и промывали диэтиловым эфиром. Выход: 81.3%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 10.0 (с, 1Н), 8.29 (с, 1Н), 8.05 (д, 2Н, J=6.0 Гц), 7.74 (с, 2Н), 7.72 (д, 2Н, J=6.0 Гц), 4.15 (с, 2Н), 3.99 (с, 3Н), 3.96 (с, 6Н), 2.61 (с, 3H). MS (ESI) m/z 399.1 (M+H)+.
(2-(4-((Диметиламино)метил)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (2u или 2u-HCl). К раствору 2r (53 мг, 0.14 ммоль) в 5 мл CH2Cl2 добавляли раствор формальдегида (37%-ный в H2O, 340 мг, 4.2 ммоль), и цианоборгидрид натрия (34 мг, 0.55 ммоль), реакционную смесь адсорбировали на силикагеле и выделяли чистое свободное основание колоночной флэш-хроматографией (41 мг, 70.9%). При 0°С к раствору свободного основания (41 мг) в 5 мл CH2Cl2 добавляли раствор HCl в 1,4-диоксане (4 н. раствор, 2 мл) и перемешивали при комнатной температуре в течение ночи. Осадок (2u) отфильтровывали и промывали диэтиловым эфиром. Выход: 71.3%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 13.0 (с, 1Н), 8.34 (с, 1Н), 8.13 (д, 2Н, J=7.0 Гц), 7.82 (д, 2Н, J=7.5 Гц), 7.75 (с, 2Н), 4.24 (с, 2Н), 3.99 (с, 3Н), 3.97 (с, 6Н), 2.83 (с, 6Н). MS (ESI) m/z 413.1 (М+Н)+.
2-(4-(4-(3,4,5-триметоксибензоил)тиазол-2-ил)фенил)ацетонитрил (2n)
2-(4-(4-(3,4,5-триметоксибензоил)тиазол-2-ил)фенил)ацетонитрил (2n) получали по той же методике, которая применялась для получения соединения 1h, из терефталонитрила и цистеина. 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.30 (с, 1Н), 8.04 (д, 2Н), 7.76 (с, 2Н), 7.46 (д, 2Н), 3.97 (с, 3Н), 3.95 (с, 6Н), 3.83 (с, 2Н).
Синтез (2-(4-(диметиламино)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (2о)
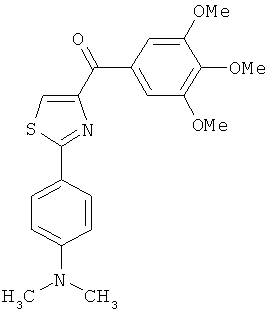
(2-(4-(Диметиламино)фенил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (2о) получали по той же методике, которая применялась для получения соединения 1h, из 4-(диметиламино)бензонитрила и цистеина. 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.12 (с, 1Н), 7.88 (д, 2Н), 7.80 (с, 2Н), 6.73 (д, 2Н), 3.96 (с, 3Н), 3.95 (с, 6Н), 3.05 (с, 6Н); MS (ESI) m/z 421.1 (M+Na)+.
Индолильный цикл А:
Синтез (2-(1Н-индол-2-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (62а) (фиг.5)
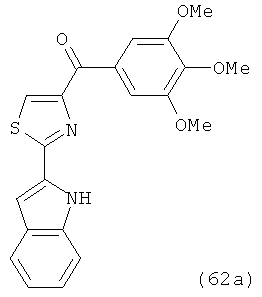
1Н-Индол-2-карбонитрил (60а). К охлажденному раствору индол-2-карбоновой кислоты (2.0 г, 12.4 ммоль) в 60 мл безводного Et2O добавляли 1.9 мл SOCl2 (26 ммоль). После перемешивания в течение 40 мин при комнатной температуре эфир удаляли при пониженном давлении при температуре не выше 35°С. Полученный ацилхлорид растворяли в 40 мл безводного Et2O, и полученный раствор немедленно добавляли к перемешиваемому раствору жидкого аммиака в 80 мл Et2O. Реакционную смесь перемешивали при комнатной температуре 24 часа. Затем упаривали растворитель при пониженном давлении, и белый индол-2-карбоксамид кристаллизовали из 50%-ного водного EtOH и сушили на воздухе, после чего растворяли в POCl3 и кипятили в течение 5 мин. Охлажденный раствор выливали в измельченный лед и добавляли водный раствор NH4OH до основного рН. Водный раствор экстрагировали эфиром, экстракты сушили над Na2SO4 и упаривали. Получали коричневый индол-2-карбонитрил 60а (63.3% общий выход из индол-2-карбоновой кислоты). 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.56 (ушир, с, 1H), 7.68 (д, 1Н, J=8.0 Гц), 7.43-7.34 (м, 2Н), 7.24-7.21 (м, 2Н). MS (ESI) m/z 144.0 (M+H)+, 140.8 (М-Н)-.
(R)-2-(1H-индол-2-ил)-N-метокси-N-метил-4,5-дигидротиазол-4-карбоксамид (61а) синтезировали по той же методике, которая применялась для получения 38d. 67.1% выход. 1Н-ЯМР (300 МГц, CDCl3) [δ] 9.06 (с, ушир., 1Н), 7.64 (д, 2Н, J=8.1 Гц), 7.36-7.24 (м, 2Н), 7.12 (дт, 1Н, J=8.1 Гц, 1.2 Гц), 6.95 (д, 1Н, J=1.8 Гц), 5.60 (т, ушир., 1Н, J=8.7 Гц), 3.86 (с, 3Н), 3.78 (т, 1Н, J=10.2 Гц), 3.58 (дд, 1H, J=9.0 Гц, 10.2 Гц), 3.30 (с, 3Н). MS (ESI) m/z 312.1 (M+Na)+, 287.9 (M-H)-.
(2-(1Н-индол-2-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (62а) синтезировали из 61а по той же методике, которая применялась для получения 35а. 45.8% выход. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 9.26 (с, 1Н), 8.11 (с, 1Н), 7.66 (д, 1Н, J=8.0 Гц), 7.46 (с, 2Н), 7.42 (д, 1Н, J=8.0 Гц), 7.29 (т, 1Н, J=7.5 Гц), 7.16 (т, 1Н, J=7.5 Гц), 7.10 (с, 1Н), 3.97 (с, 3Н), 3.93 (с, 6Н). MS (ESI) m/z 417.1 (M+Na)+, 392.9 (M-H)-.
Синтез (2-(1Н-индол-5-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанона (66а) (фиг.5)
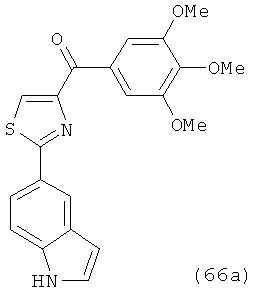
(R)-2-(1-(фенилсульфонил)-1Н-индол-5-ил)-4,5-дигидротиазол-4-карбоновая кислота (64а). (R)-2-(1Н-иидол-5-ил)-4,5-дигидротиазол-4-карбоновую кислоту 63а синтезировали способом, описанным для 42а, из 1H-индол-5-карбонитрила и использовали без дополнительной очистки. К интенсивно перемешиваемому раствору 63а (1 ммоль) и гидросульфата тетрабутиламмония (0.15 ммоль) в толуоле (10 мл) при 0°С добавляли 50%-ный водный раствор гидроксида натрия (10 мл) и сульфонилхлорид (2 ммоль). Полученный раствор перемешивали при комнатной температуре 6 часов. Затем добавляли 1 н. раствор HCl, подкисляя смесь до рН=2 и экстрагировали CH2Cl2, органический слой отделяли и сушили (MgSO4); затем упаривали досуха, получая 64а, который использовали в последующих стадиях без дополнительной очистки.
(R)-N-метокси-N-метил-2-(1-(фенилсульфонил)-1H-индол-5-ил)-4,5-дигидротиазол-4-карбоксамид (65а) получали из 64а по той же методике, которая применялась для получения 38d. 57.1% выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.92 (м, 2Н), 7.77 (м, 3Н), 7.51 (д, 1Н, J=3.0 Гц). 7.46 (т, 1Н), 7.35 (т, 1Н), 6.61 (д, 1Н), 5.58 (ушир, т, 1Н) 3.82 (с, 3Н), 3.73 (т, 1Н), 3.43 (м, 1Н), 3.21 (с, 3Н). MS (ESI) m/z 452.1 (M+Na)+.
(2-(1Н-индол-5-ил)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (66а). К раствору н-BuLi (1.6 М, 1.7 мл) в 8 мл ТГФ добавляли раствор 3,4,5-триметоксибромбензола (2.47 ммоль) в 3 мл ТГФ при -78°С. Смесь перемешивали в течение 2 часов и добавляли раствор амида Вайнреба 65а (1.24 ммоль) в 3 мл ТГФ. Температуру повышали до комнатной и перемешивали в течение ночи. Реакционную смесь гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который кипятили в 1 н. растворе NaOH в 5 мл этанола, получая соединение 66а со снятой защитной группой, и очищали методом колоночной хроматографии, получая чистое соединение в виде светло-желтого твердого вещества (36.3%). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.36 (ушир, с, 1Н), 8.31 (с, 1Н), 8.21 (с, 1Н), 7.92, 7.89 (дд, 1Н, J=1.8, 2.7 Гц), 7.46 (д, 1Н), 7.62 (с, 2Н, J=8.7 Гц), 7.29 (т, 1Н, J=2.7 Гц), 6.64 (ушир, 1Н), 3.97 (с, 6Н), 3.97 (с, 3Н); MS (ESI) m/z 417.1 (M+Na)+, 392.9 (M-H)-.
Синтез (2-(1Н-Индол-2-ил)тиазол-4-ил)(1Н-индол-2-ил)метанона (8).
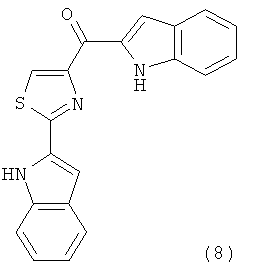
(2-(1Н-Индол-2-ил)тиазол-4-ил)(1Н-индол-2-ил)метанон (8) получали способом, аналогичным описанному для соединения 1h, из 2-(1Н-индол-2-ил)-4,5-дигидротиазол-4-карбоновой кислоты и цистеина. 1Н-ЯМР (500 МГц, CDCl3) [δ] 9.39 (с, 1Н), 8.54 (с, 1Н), 8.46 (с, 1Н), 8.06 (с, 1Н), 8.03 (дд, 1Н), 7.66 (д, 1Н), 7.51 (д, 1Н), 7.41 (д, 1Н), 7.33 (т, 1Н), 7.29 (д, 1Н), 7.15 (т, 1Н), 7.09 (д, 1Н), 6.72 (с, 1Н). MS (ESI) m/z 366.1 (М+Na)+, 341.9 (М-Н)-.
Синтез (2-(1Н-индол-2-ил)тиазол-4-ил)(1Н-индол-5-ил)метанона (2l).
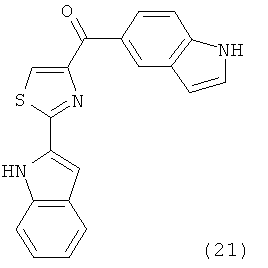
(2-(1Н-индол-2-ил)тиазол-4-ил)(1Н-индол-5-ил)метанон (2l) получали способом, аналогичным описанному для соединения 1h, из 2-(1Н-индол-2-ил)-4,5-дигидротиазол-4-карбоновой кислоты и цистеина. 1Н-ЯМР (500 МГц, CDCl3) [δ] 9.60 (с, 1Н), 9.26 (с, 1Н), 8.31 (с, 1Н), 8.03 (с, 1Н), 7.83 (дд, 1Н), 7.69 (д, 1Н), 7.53-7.49 (м, 2Н), 7.41 (т, 1Н), 7.33 (т, 1Н), 7.21-7.18 (м, 2Н), 7.13 (с, 1Н). MS (ESI) m/z 366.1 (M+Na)+, 341.9 (M-H)-.
Пример 4
Синтез соединений по настоящему изобретению, содержащих азотный линкер (x=nh)
Для улучшения биодоступности был введен NH линкер между фенильным циклом А и тиазольным циклом В. Эту новую серию соединений синтезировали как показано на фиг.6. Реакцией этилового эфира 3-бром-2-оксопропановой кислоты и арилтиомочевины в этаноле при 65°С получали 2-(ариламино)-тиазол-4-карбоновые кислоты 73а-с с высокими выходами. Полученные кислоты превращали в амиды Вайнреба 74а-с, которые после реакции с 3,4,5-триметоксифениллитием дали свободные основания 5а-с с анилиновым линкером, которые можно превратить в гидрохлориды 5На-с.
Синтез (2-(фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метаноновых производных (5а-с) и их гидрохлоридов (фиг.6)
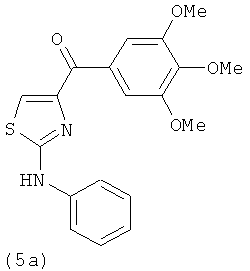
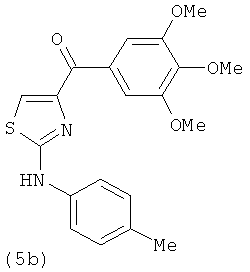
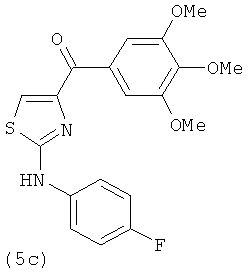
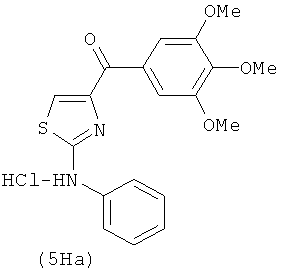
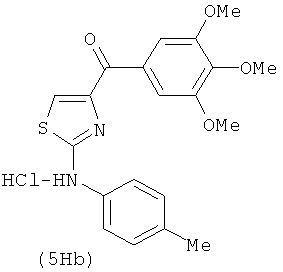
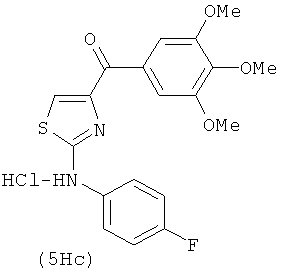
Общая методика синтеза 2-(ариламино)тиазол-4-карбоновых кислот (37а-с). N-арилтиомочевину (0.01 моль) и этилбромпируват (0.011 моль) растворяли в 3 мл этанола и кипятили 2 часа. Реакционную смесь охлаждали, кристаллический этил 2-(замещенный фениламино)тиазол-4-карбоксилат отфильтровывали и промывали этанолом. Кипячением смеси этиловых эфиров с раствором NaOH в этаноле получали конечные соединения 73а-с, которые напрямую использовали в следующих стадиях.
N-Метокси-N-метил-2-(ариламино)тиазол-4-карбоксамиды (74а-с) синтезировали способом, аналогичным описанному для 38d (см. пример 1, фиг.2).
N-Метокси-N-метил-2-(фениламино)тиазол-4-карбоксамид (74а). 90.2% выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.39 (с, 2Н), 7.38 (ушир, 1Н), 7.36-7.33 (м, ушир., 4Н), 7.09 (т, ушир., 1Н), 3.77 (с, 3Н), 3.43 (с, 3Н), 2.33 (с, 3Н). MS (ESI) m/z 286.0 (M+Na)+.
N-Метокси-N-метил-2-(п-толиламино)тиазол-4-карбоксамид (74b). 93.3% выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.35 (с, 1Н), 7.31 (ушир, 1Н), 7.22 (д, 2Н), 7.16 (д, 2Н), 3.76 (с, 3Н), 3.42 (с, 3Н), 2.33 (с, 3Н). MS (ESI) m/z 278.0 (M+H)+.
2-(4-Фторфениламино)-N-метокси-N-метилтиазол-4-карбоксамид (74c). 89.7% выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.36 (с, 1Н), 7.36-7.31 (м, 2Н), 7.07-7.04 (м, 6Н), 3.76 (с, 3Н), 3.42 (с, 3Н). MS (ESI) m/z 282.0 (M+Na)+, 280.8 (M-H)-.
Общая методика синтеза (2-(ариламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанонов (5а-с). При -78°С к раствору 5-бром-1,2,3-триметоксибензола (1.235 г, 5.0 ммоль) в 30 мл ТГФ добавляли н-BuLi в гексане (2.5 н. раствор, 2.4 мл, 6 ммоль) в атмосфере аргона и перемешивали в течение 10 мин. К полученному литиевому реагенту добавляли амид Вайнреба 74а-с (1 ммоль) в 10 мл ТГФ и перемешивали при комнатной температуре 2 часа. Реакционную смесь гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение (5а-с).
(2-(Фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5а). 33.3% выход. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 10.4 (с, 1Н), 7.85 (с, 1Н), 7.68 (д, 2Н, J=8.0 Гц), 7.31 (т, 2Н, J=8.0 Гц), 6.98 (т, 1Н, J=8.0 Гц), 3.83 (с, 6Н), 3.78 (с, 3Н). MS (ESI) m/z 393.1 (М+Н)+, 368.9 (M-H)-.
(2-(п-толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5b). 40.6% выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.48 (с, 1Н), 7.47 (с, 2Н), 7.30 (ушир, 1Н), 7.27 (д, 2Н, J=8.5 Гц), 7.17 (д, 2Н, J=8.5 Гц), 3.93 (с, 3Н). 3.90 (с, 6Н), 2.34 (с, 3Н). MS (ESI) m/z 385.1 (M+H)+, 382.9 (М-Н)-.
(2-(п-фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон (5с). 39.6% выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.52 (ушир, 1Н), 7.49 (с, 1Н), 7.45 (с, 2Н), 7.40-7.37 (кв, 2Н, J=4.5 Гц), 7.08-7.04 (т, 2Н, J=8.0 Гц), 3.93 (с, 3Н), 3.89 (с, 6Н). MS (ESI) m/z 389.3 (M+H)+, 386.9 (М-Н)-.
Общая методика синтеза гидрохлоридов (5На-с). При 0°С к раствору соединения 5а-с (0.1 ммоль) в 5 мл CH2Cl2 добавляли раствор HCl в 1,4-диоксане (4 н. раствор, 2 мл) и перемешивали при комнатной температуре в течение ночи. Осадки 5На-с собирали и промывали диэтиловым эфиром.
(2-(фениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (5На). 91.6% выход. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 12.9 (ушир, 1Н), 7.49-7.46 (м, 2Н), 7.42-7.40 (м, 2Н), 7.37-7.34 (м, ушир., 2Н), 7.11 (с, 2Н), 3.94 (с, 3Н), 3.92 (с, 6Н). MS (ESI) m/z 389.1 (M+H)+.
(2-(п-толиламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанона гидрохлорид (5Hb). 39.6% выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.30-7.25 (м, ушир., 5Н), 7.12 (с, 2Н), 3.94 (с, 3Н), 3.92 (с, 6Н), 2.38 (с, 3Н). MS (ESI) m/z 389.1 (М+Н)+.
(2-(п-фторфениламино)тиазол-4-ил)(3,4,5-триметоксифенил)метанон гидрохлорид (5Нс). 89.3% выход. 1Н-ЯМР (500 МГц, CDCl3) [δ] 10.55 (с, 1Н), 7.85 (с, 1Н), 7.72-7.69 (кв, 2Н, J=4.5 Гц), 7.50 (с, 2Н), 7.18-7.15 (т, 2Н, J=8.5 Гц), 4.30 (ушир, 1Н), 3.82 (с, 6Н), 3.78 (с, 3Н). MS (ESI) m/z 389.3 (M+H)+.
Пример 5
Синтез отдельных арил-бензоил-имидазольных соединений
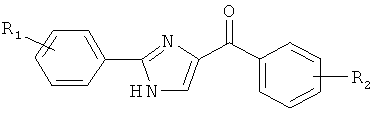
Получение 2-арил-4,5-дигидро-1Н-имидазолов 14b, 14с, 14х (фиг.7).
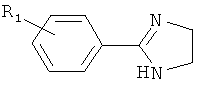
К раствору соответствующего бензальдегида 8(b, с, х) (60 ммоль) в изо-BuOH (300 мл) добавляли этилендиамин (66 ммоль) и перемешивали в течение 30 мин при комнатной температуре. В реакционную смесь последовательно добавляли карбонат калия (75 ммоль) и йод (180 ммоль), после чего смесь перемешивали при 70°С 3 часа. Добавляли сульфит натрия (Na2SO3), и смеси экстрагировали хлороформом. Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (хлороформ:метанол 20:1), получая белое твердое вещество. Выход: 50-60%.
Получение 2-арил-1Н-имидазолов (9a-j, p, х; фиг.7 и 8).
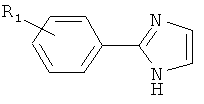
Метод А (применим только для 9b, 9х фиг.7): К раствору 2-арил-4,5-дигидро-1Н-имидазола 14b, x (35 ммоль) в ДМСО (100 мл) добавляли карбонат калия (38.5 ммоль) и диацетоксииодбензол (38.5 ммоль). Реакционную смесь перемешивали в течение ночи в темноте. Добавляли воду, после чего проводили экстракцию дихлорметаном. Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 3:2), получая белое твердое вещество. Выход: 30%-50%.
Метод В (применим только для 9с; фиг.7): К раствору 2-арил-4,5-дигидро-1Н-имидазола 14с (50 ммоль) в ДМФА (70 мл) добавляли DBU (55 ммоль) и CBrCl3 (55 ммоль). Реакционную смесь перемешивали в течение ночи и добавляли насыщенный водный раствор NaHCO3, после чего проводили экстракцию дихлорметаном. Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (хлороформ:метанол 50:1), получая белое твердое вещество. Выход: 7%.
Метод С (применим только для 9а, 9d-j, 9p; фиг.8): К раствору соответствующего бензальдегида (8а, 8d-j, 8p) (100 ммоль) в этаноле (350 мл) при 0°С добавляли 40%-ный раствор щавелевого альдегида в воде (12.8 мл, 110 ммоль) и 29%-ный раствор гидроксида аммония в воде (1000 ммоль, 140 мл). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь упаривали, и остаток очищали методом колоночной флэш-хроматографии, используя дихлорметан в качестве элюента, получая указанное в заголовке соединение в виде желтого порошка. Выход: 20%-40%.
Получение 2-арил-1-(фенилсульфонил)-1Н-имидазолов (10а-j, р, x; фиг.7 и 8).
К раствору 2-арил-1H-имидазола 9a-j, р, х (20 ммоль) в безводном ТГФ (200 мл) при 0°С добавляли гидрид натрия (60%-ная дисперсия в минеральном масле, 1.2 г, 30 ммоль) и перемешивали в течение 30 мин. Добавляли бензолсульфонилхлорид (2.82 мл, 22 ммоль), и реакционную смесь перемешивали в течение ночи. После добавления 100 мл насыщенного водного раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (500 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 2:1), получая опалесцирующее твердое вещество. Выход: 50%-70%.
Получение арил (2-арил-l-(фенилсульфонил)-1Н-имидазол-4-ил)метанонов (11aa-ai, ba, ca, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa; фиг.7 и 8).
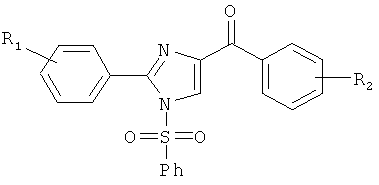
К раствору 2-арил-1-(фенилсульфонил)-1Н-имидазола (6.0 ммоль) 10a-j, p, x в безводном ТГФ (30 мл) при -78°С добавляли 1.7М раствор трет-бутиллития в пентане (5.3 мл, 9.0 ммоль) и перемешивали в течение 10 мин. Добавляли соответствующий замещенный бензоилхлорид (7.2 ммоль) при -78°С и перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 100 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (200 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 4:1), получая белое твердое вещество. Выход: 15%-40%.
Общая методика получения арил (2-арил-1Н-имидазол-4-ил)метанонов (12aa-ai, ba, ca, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa; фиг.7 и 8).
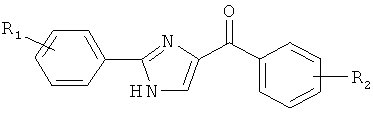
К раствору арил (2-арил-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанонов (2.0 ммоль) 11aa-ai, ba, ca, cb, da, db, ea, eb, fa, fb, ga, gb, ha, hb, ia, ib, ja, jb, pa в ТГФ (20.0 мл) добавляли 1.0М раствор фторида тетрабутиламмония (4.0 ммоль) и раствор перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 50 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (100 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 3:1) или перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 80-95%.
Получение (2-(4-гидроксифенил)-1H-имидазол-4-ил)(арил)метанонов (12ka, 12kb; фиг.8).
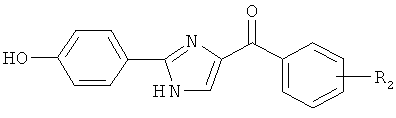
К раствору (2-(4-(бензилокси)фенил)-1Н-имидазол-4-ил)(арил)метанона 12ja или 12jb (1 ммоль) в АсОН (20 мл) добавляли концентрированную HCl (2 мл) и кипятили в течение ночи. После удаления растворителя, остаток перекристаллизовывали из дихлорметана, получая указанное в заголовке соединение в виде желтого твердого вещества. Выход: 70-85%.
Получение (2-арил-1H-имидазол-4-ил)(3,4,5-тригидроксифенил)метанонов 13еа, 13fa, 13ha (фиг.8).
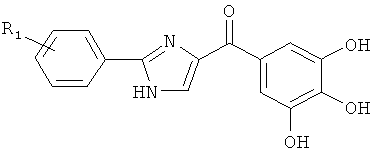
К раствору арил (2-арил-1Н-имидазол-4-ил)метанона 12еа, 12fa или 12ha (0.5 ммоль) в CH2Cl2 (6.0 мл) добавляли 1.0 М раствор BBr3 (2 ммоль) в CH2Cl2 и перемешивали в течение 1 часа при комнатной температуре. Добавляли воду для разрушения избытка BBr3. Выпавший твердый осадок отфильтровывали и перекристаллизовывали из МеОН, получая желтое твердое вещество. Выход: 60-80%.
Получение арил (2-арил-1Н-имидазол-4-ил)метанон-HCl соли (12db-HCl).
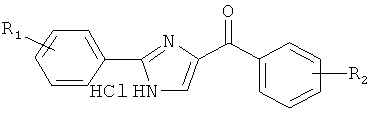
К раствору 12db (0.5 ммоль) в метаноле (20 мл) добавляли 2М раствор хлороводорода (5 ммоль) в этиловом эфире и перемешивали в течение ночи при комнатной температуре. Реакционную смесь упаривали и остаток промывали CH2Cl2, получая указанное в заголовке соединение. Выход: 95%.
Получение арил (2-фенил-1Н-имидазол-1-ил)метанона 12aba, 12ааа; фиг.9).
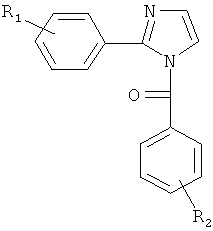
К раствору 2-фенил-1Н-имидазола 9а (10 ммоль) в ТГФ (20 мл) добавляли NaH (15 ммоль) и замещенный бензоилхлорид (12 ммоль) при 0°С. Реакционную смесь перемешивали в течение ночи и разбавляли насыщенным водным раствором NaHCO3, после чего проводили экстракцию этилацетатом. Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (хлороформ), получая белое твердое вещество. Выход: 12-16%.
Получение 1-замещенного-(2-фенил-1Н-имидазол-1-ил)-арил-метанона (12dc, 12fc, 12daa, 12dab, 12cba, 11gaa, 12la; фиг.10-11).
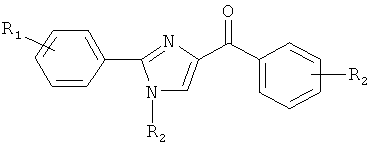
Синтез 12dc, 12fc и 12daa, 12dab и 12cba суммарно описан на фиг.10. Соединения 12da, 12cb и 12fa синтезированы по методике, описанной выше и на фиг.7 и 8. Обработка 12da и 12fa хлоридом алюминия дала пара-деметилированные 12dc, 12fc, 3,5-диметокси-группы остались незатронутыми. Соединение 12daa получали бензилированием положения N-1 в 12da. Метилирование положения N-1 в 12da и 12cb дало соединения 12dab и 12cba, соответственно.
Синтез 12dc, 12fc, 12daa, 12dab, 12cba: Метод D. (для 12dc и 12fc) (фиг.10):
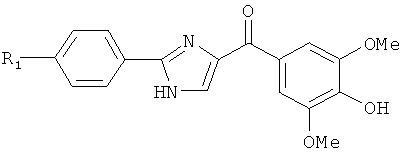
R1=СН3 (12dc)
R1=Cl (12fc)
К раствору 12da и 12fa (200 мг) в ТГФ (20 мл) добавляли хлорид алюминия (10 экв). Реакционную смесь перемешивали в течение ночи. Добавляли воду, после чего проводили экстракцию этилацетатом. Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 1:1) получая бело-желтое твердое вещество. Выход: 60-80%.
Синтез 12daa, 12dab, 12cba, Метод Е: (фиг.10):
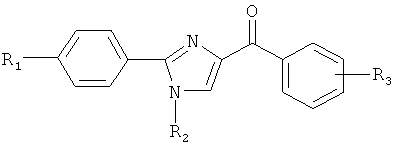
R1=Me; R2=Bn; R3=3,4,5-(ОМе)3 (12daa)
R1=Me; R2=СН3; R3=3,4,5-(ОМе)3 (12dab)
R1=OMe; R2=СН3; R3=F (12cba)
К раствору 12da и 12cb (100 мг) в ТГФ (10 мл) на ледяной бане добавляли гидрид натрия (1.2 экв), затем добавляли метилиодид (для 12dab, 12cba) или бензилбромид (для 12daa) (2 экв). Полученную реакционную смесь перемешивали в течение 5 часов при кипячении. После добавления 50 мл насыщенного водного раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (100 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 2:1), получая белое твердое вещество. Выход: 50-98%.
Синтез 11gaa и 12la (фиг.11)
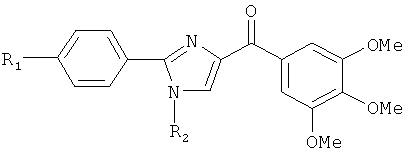
R1=N(Me)2; R2=(4-OMe)PhSO2 (11gaa)
R1=Br; R2=H (12la)
Замещенные бензальдегиды 8(l, g) превращали в соединения 9(l, g) в присутствии гидроксида аммония и глиоксаля, формируя имидазольный фрагмент. Имидазольные циклы в соединениях 9(l, g) защищали соответствующей фенилсульфонильной группой, после чего проводили сочетание в 3,4,5-триметоксибензоилхлоридом, получая соединение 11(la, gaa). Обработка 11la фторидом трет-бутиламмония для удаления защитной группы дала 12la.
Структурная характеризация (1-Бензил-2-(n-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12daa) (фиг.11).
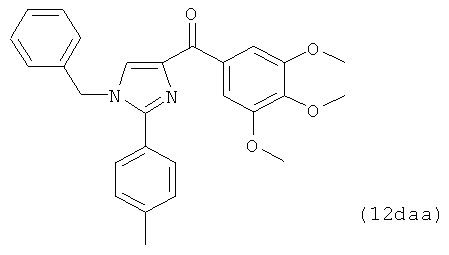
Выход: 92.8%; т.пл. 135-137°С. 1Н-ЯМР (CDCl3, 500 МГц) [δ] 7.81 (с, 1Н), 7.80 (д, J=6.5 Гц, 2Н), 7.58 (д, J=8.0 Гц, 2Н), 7.41-7.45 (м, 3Н), 7.31-7.33 (м, 2Н), 7.20 (д, J=7.0 Гц, 2Н), 5.33 (с, 2Н), 3.99 (с, 3Н), 3.98 (с, 6Н), 2.47 (с, 3Н). MS (ESI) вычислено для C27H26N2O4 442.2, найдено 443.1 [M+Na]+. ВЭЖХ1: tR 4.28 мин, чистота >99%.
Определение структуры (2-(4-(диметиламино)фенил)-1-((4-метоксифенил)сульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанона (12gba).
Выход: 34.1%; т.пл. 147-149°С. 1Н-ЯМР (CDCl3, 500 МГц) [δ] 8.07 (кв, J=8.5 Гц, 5.5 Гц, 2Н), 7.78 (д, J=9.0 Гц, 2Н), 7.41 (д, J=8.5 Гц, 2Н), 7.39 (с, 1Н), 7.23 (т, J=8.5 Гц, 2Н), 6.91 (д, J=9.0 Гц, 2Н), 6.68 (д, J=9.0 Гц, 2Н), 3.89 (с, 3Н), 3.08 (с, 3Н). MS (ESI) вычислено для C25H22FN3O4S 479.1, найдено 502.1 [M+Na]+. ВЭЖХ2: tR 18.6 мин, чистота 96.9%.
Синтез (2-(4-бромфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12la) (фиг.11)
Синтез 9l, 9g: К раствору подходящего бензальдегида (8l и 8g, 100 ммоль) в этаноле (400 мл) при 0°С добавляли 40%-ный раствор щавелевого альдегида (глиоксаля) в воде (1.1 экв) и 29%-ный раствор гидроксида аммония в воде (10 экв). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь упаривали и остаток очищали методом колоночной флэш-хроматографии с дихлорметаном в качестве элюента, получая указанное в заголовке соединение в виде желтого порошка. Выход: 10%-30%.
Синтез 10la, 10gb: К раствору имидазолов (9l, 9g) (10 ммоль) в безводном ТГФ (200 мл) при 0°С добавляли гидрид натрия (60%-ная дисперсия в минеральном масле, 1.2 экв) и перемешивали в течение 20 мин. Добавляли 4-метоксибензолсульфонил хлорид (для 10gb) или бензолсульфонил хлорид (для остальных) (1.2 экв), и реакционную смесь перемешивали в течение ночи. После добавления 200 мл насыщенного водного раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (600 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 2:1, получая опалесцирующее твердое вещество. Выход: 40-95%.
Синтез 11la, 11gaa: К раствору 2-арил-1-(фенилсульфонил)-1Н-имидазола (10la, 10gb) (5.0 ммоль) в безводном ТГФ (30 мл) при -78°С добавляли 1.7 М раствор трет-бутиллития в пентане (1.2 экв) и перемешивали в течение 10 мин. Добавляли 3,4,5-триметоксибензоил хлорид (1.2 экв) при -78°С и перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 100 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (300 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 3:1), получая белое твердое вещество. Выход: 5-45%.
Синтез 12la: К раствору арил (2-арил-l-(фенилсульфонил)-1Н-имидазол-4-ил)метанона (11la), 2.0 ммоль) в ТГФ (25.0 мл) добавляли 1.0 М раствор фторида тетрабутиламмония (2 экв) и перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 60 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (150 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 4:1) или перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 80-98%.
Синтез (4-фторфенил)(2-(4-метоксифенил)-1Н-имидазол-4-ил)метанона (12cb) (фиг.7).
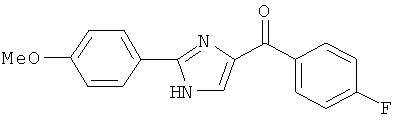
К раствору (4-фторфенил)(2-(4-метоксифенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)метанона (11cb, 872 мг, 2.0 ммоль) в ТГФ (20.0 мл) добавляли 1.0 М раствор фторида тетрабутиламмония (4.0 мл, 4.0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 50 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (100 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 90%; т.пл. 245-247°С.
Синтез (2-(n-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12da) (фиг.8).
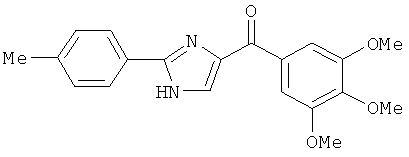
К раствору (1-(фенилсульфонил)-2-(n-толил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11da, 492 мг, 1.0 ммоль) в ТГФ (15.0 мл) добавляли 1.0 М раствор фторида тетрабутиламмония (2.0 мл, 2.0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 30 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (80 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 88.5%.
Синтез (2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (12fa) (фиг.8 и 14).
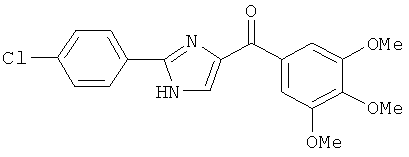
2-(4-хлорфенил)-1Н-имидазол (9f): К раствору 4-хлорбензальдегида (8f) (100 ммоль) в этаноле (350 мл) при 0°C добавляли 40%-ный раствор щавелевого альдегида в воде (12.8 мл, 110 ммоль) и 29%-ный раствор гидроксида аммония в воде (1000 ммоль, 140 мл). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь упаривали и остаток очищали методом колоночной флэш-хроматографии, с дихлорметаном в качестве элюента, получая указанное в заголовке соединение в виде желтого порошка. Выход: 19.8%. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 13.60 (ушир, 1Н), 7.94 (д, J=8.5 Гц, 2Н), 7.51 (д, J=8.0 Гц, 2Н), 7.27 (с, 1Н), 7.03 (с, 1Н). MS (ESI): вычислено для C9H7ClN2, 178.0, найдено 178.9 [M+H]+.
2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол (10f): К раствору 2-(4-хлорфенил)-1Н-имидазола (9f) (20 ммоль) в безводном ТГФ (200 мл) при 0°С добавляли гидрид натрия (60%-ная дисперсия в минеральном масле, 1.2 г, 30 ммоль) и перемешивали в течение 30 мин. Добавляли бензолсульфонил хлорид (2.82 мл, 22 ммоль), и реакционную смесь перемешивали в течение ночи. После добавления 100 мл насыщенного водного раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (500 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 2:1), получая опалесцирующее твердое вещество. Выход: 54.9%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 7.65 (д, J=2.0 Гц, 1Н), 7.58 (т, J=7.5 Гц, 1Н), 7.43 (д, J=8.5 Гц, 2Н), 7.38 (т, J=8.0 Гц, 2Н), 7.34-7.36 (м, 4Н), 7.12 (д, J=1.5 Гц, 1Н). MS (ESI): вычислено для C15H11ClN2O2S, 318.0, найдено 341.0 [М+Na]+.
(2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (11fa): К раствору 2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазола (10f) (6.0 ммоль) в безводном ТГФ (30 мл) при -78°С добавляли 1.7 М раствор трет-бутиллития в пентане (5.3 мл, 9.0 ммоль) и перемешивали в течение 10 мин. Добавляли 3,4,5-триметоксибензоилхлорид (7.2 ммоль) при -78°С и перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 100 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (200 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 4:1), получая белое твердое вещество. Выход: 36.8%; 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.05 (д, J=7.5 Гц, 2Н), 7.77 (т, J=7.5 Гц, 1Н), 7.62 (т, J=8.0 Гц, 2Н), 7.48 (с, 1Н), 7.44 (д, J=9.0 Гц, 2Н), 7.39 (д, J=8.5 Гц, 2Н), 7.37 (с, 2Н). MS (ESI): вычислено для C25H21ClN2O6S, 512.1, найдено 513.1 [М+Н]+.
(2-(4-хлорфенил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (12fa): К раствору (2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (11fa) (2.0 ммоль) в ТГФ (20.0 мл) добавляли 1.0 М раствор фторида тетрабутиламмония (4.0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 50 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (100 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 3:1) или перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 80-95%. Выход: 36.9%; т.пл. 193-195°С. 1Н-ЯМР (500 МГц, CDCl3) [δ] 10.75 (ушир, 1Н), 7.96 (д, J=8.5 Гц, 2Н), 7.83 (с, 1Н), 7.47 (д, J=9.0 Гц, 2Н), 7.23 (с, 2Н), 3.97 (с, 3Н), 3.94 (с, 6Н), 2.43 (с, 3Н). MS (ESI): вычислено для C19H17ClN2O4, 372.1, найдено 395.1 [M+Na]+, 370.9 [М-Н]-. ВЭЖХ градиент: растворитель А (вода) и растворитель В (метанол): 0-15 мин 40-100% В (линейный градиент), 15-25 мин 100% В: tR 16.36 мин, чистота >99%.
Синтез (2-(4-хлорфенил)-1Н-имидазол-4-ил)(4-фторфенил)метанона (12fb) (фиг.8).
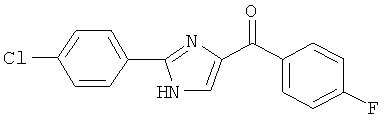
К раствору (2-(4-хлорфенил)-1-(фенилсульфонил)-1Н-имидазол-4-ил)(4-фторфенил)метанона (11fb, 440 мг, 1.0 ммоль) в ТГФ (12.0 мл) добавляли 1.0 М раствор фторида тетрабутиламмония (2.0 мл, 2.0 ммоль) и перемешивали в течение ночи. Реакционную смесь разбавляли добавлением 20 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (60 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 83.7%.
Физикохимические характеристики Арил-Бензоил-Имидазольных соединений и интермедиатов
|
|
|
|
|
|
|
|
|
|
Пример 6
Синтез некоторых индолил-бензоил-имидазольных соединений
Синтез 15хаа схематически изображен на фиг.12. Данный путь синтеза изначально был разработан для синтеза 12ха, но неселективность бензоилирования по положениям индол-2 и имидазол-4 привела к получению 15хаа, который представляет собой близкородственный, но более объемный аналог 11хаа. Индол-5-карбоксальдегид 8х защищали фенилсульфонильной группой по индольному NH, получая интермедиат 8ха. 8ха вводили в реакцию с глиоксалем и гидроксидом аммония, получая 2-арил-имидазол 9ха. После защиты имидазольного NH фенилсульфонильной группой получали интермедиат 10хаа, который вводили в реакцию сочетания с 3,4,5-триметоксибензоил хлоридом, получая 16хаа. После удаления защитной группы с 16хаа получали 15хаа.
Синтез 1-(фенилсульфонил)-1Н-индол-5-карбальдегида (8ха). К раствору индол-3-карбоксальдегида (100 ммоль) в этаноле (500 мл) при комнатной температуре добавляли гидроксид калия (110 экв), смесь перемешивали до полной стабилизации. Этанол полностью удаляли в вакууме и добавляли ацетон (250 мл), после чего добавляли бензолсульфонил хлорид (110 экв). Выпавший осадок отфильтровывали, и фильтрат упаривали и перекристаллизовывали из метанола, получая белое твердое вещество. Выход: 32.6%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 10.17 (с, 1Н), 8.25-8.39 (м, 2Н), 7.97-8.09 (м, 3Н), 7.69 (т, J=7.33 Гц, 1Н), 7.59 (т, J=7.5 Гц, 2Н), 7.39-7.54 (м, 2Н). MS (ESI) вычислено для C15H11NO3S 285.1, найдено 286.0 [М+Н]+.
Синтез (5-(4-(3,4,5-триметоксибензоил)-1Н-имидазол-2-ил)-1Н-индол-2-ил)(3,4,5-триметоксифенил)метанона (15хаа): К раствору (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-2-(3,4,5-триметоксибензоил)-1Н-индол-5-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (16хаа) (1 ммоль) в этаноле (20 мл) добавляли гидроксид натрия (10 экв) и перемешивали в течение ночи в темноте. Реакционную смесь разбавляли добавлением 50 мл воды и экстрагировали этилацетатом (250 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 3:1) или перекристаллизовывали из воды и метанола, получая белое твердое вещество. Выход: 30-95%.
5-(1Н-имидазол-2-ил)-1-(фенилсульфонил)-1Н-индол (9ха). Выход: 12.0%. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 8.33 (д, J=2.9 Гц, 2Н), 8.13 (д, J=7.8 Гц, 2Н), 7.98-8.04 (м, 1Н), 7.62-7.67 (м, 1Н), 7.55 (д, J=7.82 Гц, 2Н), 7.22-7.34 (м, 4Н). MS (ESI) вычислено для C17H13N3O2S 323.1, найдено 324.0 [М+Н]+.
1-(фенилсульфонил)-5-(1-(фенилсульфонил)-1Н-имидазол-2-ил)-1Н-индол (10хаа). Выход: 23.6%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.01 (д, J=8.5 Гц, 1Н), 7.95 (д, J=7.5 Гц, 2Н), 7.73 (д, J=1.0 Гц, 1Н), 7.70 (д, J=4.0 Гц, 1Н), 7.63-7.66 (м, 2Н), 7.52-7.56 (м, 3Н), 7.31-7.34 (м, 3Н), 7.22 (т, J=8.5 Гц, 2Н), 7.17 (с, 1Н), 6.14 (д, J=3.5 Гц, 1Н). MS (ESI) вычислено для C23H17N3O4S2 463.1, найдено 464.0 [М+Н]+.
(1-(фенилсульфонил)-2-(1-(фенилсульфонил)-2-(3,4,5-триметоксибензоил)-1Н-индол-5-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанон (16хаа). Выход: 15.9%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.18-8.25 (м, 3Н), 8.04 (д, J=8.1 Гц, 2Н), 7.70-7.78 (м, 2Н), 7.61-7.69 (м, 3Н), 7.55 (т, J=7.7 Гц, 3Н), 7.50 (с, 1Н), 7.38 (с, 2Н), 7.34 (с, 2Н), 6.94 (с, 1Н), 3.99-4.06 (м, 12Н), 3.94-3.99 (м, 6Н). MS (ESI) вычислено для C43H37N3O12S2 851.2, найдено 852.1 [М+Н]+.
(5-(4-(3,4,5-триметоксибензоил)-1Н-имидазол-2-ил)-1Н-индол-2-ил)(3,4,5-триметоксифенил)метанон (15хаа). Выход: 45.9%; т.пл. 239-241°С. 1Н-ЯМР (500 МГц, CDCl3) [δ] 10.45 (с, 1Н), 9.44 (с, 1Н), 8.41 (с, 1Н), 8.04 (д, J=8.5 Гц, 1Н), 7.86 (с, 1Н), 7.61 (д, J=8.5 Гц, 1Н), 7.29 (с, 2Н), 7.26 (с, 2Н), 3.99 (с, 3Н), 3.95-3.97 (м, 15Н). MS (ESI) вычислено для C31H29N3O8 571.2, найдено 572.2 [М+Н]+. ВЭЖХ2: tR 4.09 мин, чистота 96.3%.
Пример 7
Синтез (2-(1h-индол-3-ил)-1h-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17ya) (фиг.13)
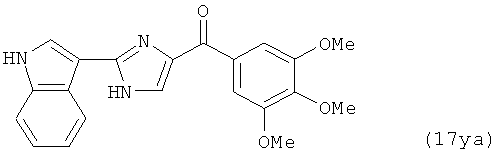
Синтез 1-(фенилсульфонил)-1Н-индол-3-карбоксальдегида (8ya): К раствору индол-3-карбоксальдегида (8y) (100 ммоль) в этаноле (500 мл) при комнатной температуре добавляли гидроксид калия (1.1 экв). Смесь перемешивали до полной стабилизации. Этанол полностью удаляли в вакууме, и остаток растворяли в ацетоне (250 мл), после чего добавляли бензолсульфонил хлорид (1.1 экв, 110 ммоль). Реакционную смесь перемешивали в течение получаса. Выпавший осадок отфильтровывали, фильтрат упаривали и перекристаллизовывали из метанола, получая белое твердое вещество. Выход: 33%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 10.17 (с, 1Н), 8.25-8.39 (м, 2Н), 7.97-8.09 (м, 3Н), 7.69 (т, 7=7.33 Гц, 1Н), 7.59 (т, 7=7.5 Гц, 2Н), 7.39-7.54 (м, 2H). MS (ESI) вычислено для C15H11NO3S 285.1, найдено 286.0 [М+Н]+.
Синтез 3-(1Н-имидазол-2-ил)-1-(фенилсульфонил)-1Н-индола (9ya): К раствору 1-(фенилсульфонил)-1Н-индол-3-карбоксальдегида (8ya) (100 ммоль) в этаноле (400 мл) при 0°С добавляли 40%-ный раствор щавелевого альдегида (глиоксаля) в воде (1.1 экв, 110 ммоль) и 29%-ный раствор гидроксида аммония в воде (10 экв, 1000 ммоль). После перемешивания в течение 2-3 дней при комнатной температуре, реакционную смесь гасили водой и экстрагировали дихлорметаном. Органический слой удаляли в вакууме, и остаток очищали методом колоночной флэш-хроматографии, используя смесь гексан/этилацетат (4:1-2:1) в качестве элюента, получая указанное в заголовке соединение в виде желтого порошка. Выход: 12%. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 8.33 (д, J=2.9 Гц, 2Н), 8.13 (д, J=7.8 Гц, 2Н), 7.98-8.04 (м, 1Н), 7.62-7.67 (м, 1Н), 7.55 (д, J=7.82 Гц, 2Н), 7.22-7.34 (м, 4Н). MS (ESI) вычислено для C17H13N3O2S 323.1, найдено 324.0 [М+Н]+.
Синтез 1-(фенилсульфонил)-3-(1-(фенилсульфонил)-1Н-имидазол-2-ил)-1Н-индола (10ya): К раствору 3-(1Н-имидазол-2-ил)-1-(фенилсульфонил)-1Н-индола (9ya) (20 ммоль) в безводном ТГФ (300 мл) при 0°С добавляли гидрид натрия (60%-ная дисперсия в минеральном масле, 1.2 экв, 24 ммоль) и перемешивали в течение 20 мин. Добавляли бензолсульфонил хлорид (1.2 экв, 24 ммоль), и реакционную смесь перемешивали в течение ночи. После добавления 200 мл насыщенного водного раствора NaHCO3, реакционную смесь экстрагировали этилацетатом (600 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 5:1), получая белое твердое вещество. Выход: 40%. 1Н-ЯМР (CDCl3, 300 МГц) [δ] 8.02-8.08 (м, 4Н), 7.72 (д, J=1.5 Гц, 1Н), 7.35-7.60 (м, 8Н), 7.23 (д, J=1.5 Гц, 1Н), 7.10-7.16 (м, 3Н). MS (ESI) вычислено для C23H17N3O4S2 463.1, найдено 486.0 [M+Na]+.
Синтез (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17yaa): К раствору 1-(фенилсульфонил)-3-(1-(фенилсульфонил)-1Н-имидазол-2-ил)-1Н-индола (10ya) (5.0 ммоль) в безводном ТГФ (100 мл) при -78°С добавляли 1.7 М раствор трет-бутиллития в пентане (1.2 экв, 6.0 ммоль) и перемешивали в течение 10 мин. Добавляли раствор 3,4,5-триметоксибензоил хлорида (1.2 экв, 6.0 ммоль) в ТГФ при -78°С и перемешивали в течение ночи. Реакционную смесь гасили добавлением 100 мл насыщенного водного раствора NaHCO3 и экстрагировали этилацетатом (300 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 3:1), получая белое твердое вещество. Выход: 30%. 1Н-ЯМР (500 МГц, CDCl3) [δ] 8.09 (д, 7=10 Гц, 1Н), 8.04 (д, 7=10 Гц, 2Н), 7.91 (c, 1H), 7.76 (д, J=5 Гц, 2H), 7.65 (т, J=10 Гц, 1H), 7.55-7.58 (м, 5H), 7.40 (c, 2H), 7.33-7.36 (м, 3H), 7.25 (т, 7=10 Гц, 1Н), 4.05 (с, 3H), 4.03 (c, 6H). MS (ESI) вычислено для C33H27N3O8 657.0, найдено 680.1 [M+Na]+.
Синтез (2-(1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17ya): К раствору (1-(фенилсульфонил)-2-(1-(фенилсульфонил)-1Н-индол-3-ил)-1Н-имидазол-4-ил)(3,4,5-триметоксифенил)метанона (17yaa) (1 ммоль) в этаноле (40 мл) и воде (4 мл) добавляли гидроксид натрия (10 экв, 10 ммоль) и перемешивали в течение ночи при кипячении в темноте. Реакционную смесь разбавляли добавлением 50 мл воды и экстрагировали этилацетатом (200 мл). Органический слой сушили над сульфатом магния и упаривали. Остаток очищали методом колоночной флэш-хроматографии (гексан:этилацетат 1:1), получая желтое твердое вещество. Выход: 60%. 1Н-ЯМР (500 МГц, CD3OD) [δ] 8.31 (д, J=6.5 Гц, 1Н), 7.99 (с, 1Н), 7.90 (с, 1Н), 7.48-7.52 (м, 3Н), 7.24-7.28 (м, 2Н), 4.00 (с, 6Н), 3.93 (с, 3Н). MS (ESI) вычислено для C21H19N3O4 377.1, найдено 400.1 [M+Na]+. Т.пл. 208-210°С.
Пример 8
Синтез (2-(1h-индол-5-иламино)тиазол-4-ил(3,4,5-триметоксифенил)метанона (соединение 55) (фиг.15)
Смесь 5-нитро-1H-индола (11 г, 67.9 ммоль) и Pd/C (5%; 1 г), растворенную в этаноле (50 мл), гидрировали 3 часа под давлением 40 фунт/кв. дюйм. Реакционную смесь фильтровали, и избыток этанола упаривали при пониженном давлении. Твердый продукт перекристаллизовывали из гексана, получая чистое соединение - 5-аминоиндол (55-1). Выход: 92.5%. 1Н-ЯМР (500 МГц, CDCl3): [δ] 7.96 (ушир, 1Н), 7.20 (д, 1Н), 7.13 (с, 1Н), 6.95 (с, 1Н), 6.67 (дд, 1Н), 6.37 (с, 1Н), 3.50 (с, 2Н). MS (ESI) m/z 133.0 (M+H)+.
Раствор 5-аминоиндола (8 г, 60.6 ммоль) в ацетоне (150 мл) вводили в реакцию с бензоилизотиоцианатом (9.88 г, 60. ммоль) при комнатной температуре в течение примерно 4 часов. Полученное твердое вещество отфильтровывали и обрабатывали 2 н. раствором NaOH в ТГФ (120 мл). Смесь кипятили примерно 6 часов и доводили до комнатной температуры. Растворитель упаривали в вакууме. Остаток разбавляли добавлением воды (20 мл) и доводили до нейтрального рН 7 добавлением 1 н. раствора HCl. Полученное твердое вещество отфильтровывали и сушили в вакууме, получая 5-индолилтиомочевину (55-2). 5-Индолилтиомочевину (0.01 моль) и этилбромпируват (0.011 моль) растворяли в 3 мл этанола и кипятили 2 часа. Реакционную смесь охлаждали, кристаллический этил 2-(1Н-индол-5-иламино)тиазол-4-карбоксилат (55-3) отфильтровывали и промывали этанолом. Кипячением смеси этиловых эфиров с раствором NaOH-этанол получали 2-(1Н-индол-5-иламино)тиазол-4-карбоновую кислоту (55-4), которую использовали напрямую в последующих стадиях. К смеси неочищенной кислоты (2.5 ммоль), EDCI (2.9 ммоль), HOBt (2.6 ммоль) и NMM (5.3 ммоль) в CH2Cl2 (30 мл) добавляли HNCH3OCH3 гидрохлорид (2.6 ммоль) и продолжали перемешивать при комнатной температуре в течение ночи. Реакционную смесь разбавляли добавлением CH2Cl2 (20 мл) и последовательно промывали водой, насыщенным раствором NaHCO3, насыщенным раствором соли и сушили над MgSO4. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение - 2-(1Н-индол-5-иламино)-N-метокси-N-метилтиазол-4-карбоксамид (55-5) (45.6% общий выход за 5 стадий). При -78°С к раствору 5-бром-1,2,3-триметоксибензола (1.235 г, 5.0 ммоль) в 30 мл ТГФ добавляли н-BuLi в гексане (2.5 н. раствор, 2.4 мл, 6 ммоль) в атмосфере аргона, и перемешивали в течение 10 мин. К полученному литиевому реагенту добавляли амид Вайнреба (1 ммоль) в 10 мл ТГФ и перемешивали при комнатной температуре 2 часа. Реакционную смесь гасили добавлением насыщенного раствора NH4Cl, экстрагировали диэтиловым эфиром, сушили сульфатом магния. Растворитель удаляли при пониженном давлении, получая неочищенный продукт, который очищали методом колоночной хроматографии, получая чистое соединение 55 (51.7% выход). 1Н-ЯМР (300 МГц, CDCl3) [δ] 8.29 (ушир, 1Н), 7.68 (д, 1Н), 7.46 (с, 2Н), 7.39 (с, 1Н), 7.36 (с, 1Н), 7.28-7.26 (м, 1Н), 7.15-7.12 (м, 1Н), 6.55 (м, 1Н), 3.93 (с, 3Н), 3.89 (с, 6Н). MS (ESI) m/z 432.1 (M+Na)+, 408.0 (M-H)-.
Пример 9
Синтез хинолин- и изохинолин-арильных соединений (фиг.16)
Ряд соединений был получен реакцией Сузуки 7-бром-1-хлоризохинолина с различными арилбороновыми кислотами.
Синтез 1-хлор-7-(1Н-индол-5-ил)-изохинолина (6d) (фиг.16С):
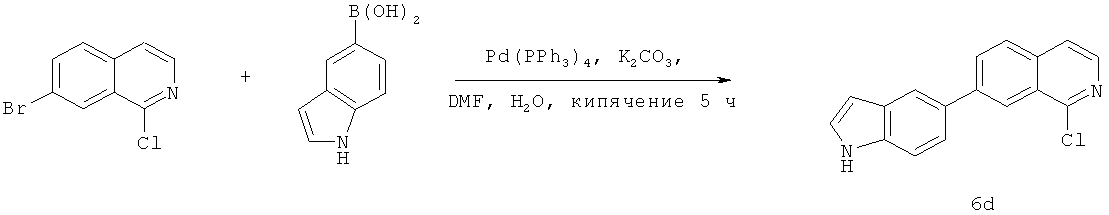
Смесь 7-бром-1-хлоризохинолина (0.50 г, 2.1 ммоль), 5-индолбороновой кислоты (0.40 г, 2.5 ммоль), тетракис(трифенилфосфин)палладия (0.035 г, .08 ммоль), карбоната калия (2.1 мл, 2 М, 4.1 ммоль), N,N-диметилформамида (11 мл) перемешивали при пропускании через реакционный сосуд аргона в течение 30 мин. Затем смесь кипятили 16 часов и охлаждали до комнатной температуры. Смесь фильтровали через слой силикагеля, разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделяли и промывали раствором NaOH (2×20 мл, 10% водн.), водой (5×30 мл, до исчезновения видимых рефракционных изменений на границе раздела водной и органической фаз) и раствором хлорида аммония (20 мл, насыщ.). Органический слой затем адсорбировали на силикагеле и проводили колоночную флэш-хроматографию (этилацетат/гексан), получая 0.14 г (25%) желтого твердого вещества. MS (ESI): вычислено для C17H11ClN2, 278.1, найдено 301.0 [M+Na]+. 1Н-ЯМР (300 МГц, ДМСО-d6) [δ] 6.56-6.58 (м, 1Н), 7.44 (т, J=2.77 Гц, 1Н), 7.57-7.59 (м, 2Н), 7.93 (м, 1Н), 8.04 (с, 1Н), 8.13-8.20 (м, 1Н), 8.27-8.31 (м, 2Н), 8.43 (м, 1Н), 11.25 (ушир. с, 1Н).
1,7-Бис-(1Н-индол-5-ил)-изохинолин (6b) (фиг.16Е):
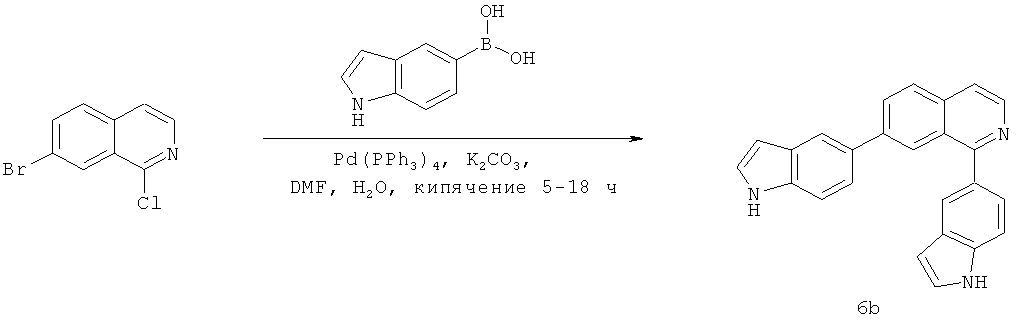
Смесь 7-бром-1-хлоризохинолина (0.20 г, 2.1 ммоль), 5-индолбороновой кислоты (0.80 г, 5.0 ммоль), тетракис(трифенилфосфин)палладия (0.19 г, 0.16 ммоль), карбоната калия (2.1 мл, 2 М, 4.1 ммоль), N,N-диметилформамида (11 мл) перемешивали при пропускании через реакционный сосуд аргона в течение 30 мин. Затем смесь кипятили 16 часов и охлаждали до комнатной температуры. Смесь фильтровали через слой силикагеля, разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделяли и промывали раствором NaOH (2×20 мл, 10% водн.), водой (5×30 мл, до исчезновения видимых рефракционных изменений на границе раздела водной и органической фаз) и раствором хлорида аммония (20 мл, насыщ.). Органический слой затем адсорбировали на силикагеле и проводили колоночную флэш-хроматографию (этилацетат/гексан), получая 0.29 г (39%) желтого твердого вещества. MS (ESI): вычислено для C25H17N3, 359.1, найдено 360.2 [M+H]+ 382.1 [M+Na]+, и 358.0 [М-Н]-. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 6.46-6.50 (м, 1Н), 6.52-6.59 (м, 1Н), 7.34-7.36 (м, 1Н), 7.36-7.41 (м, 2Н), 7.42-7.52 (м, 3Н), 7.58 (д, J=8.30 Гц, 1Н), 7.81 (дд, J=5.49, 5.00 Гц, 2Н), 7.92 (с, 1Н), 8.08-8.17 (м, 2Н), 8.33 (с, 1Н), 8.54 (д, J=5.61 Гц, 1Н), 11.18 (ушир. с, 1Н), 11.30 (ушир. с, 1Н) м.д.
1-(4-фтор-фенил)-7-(1Н-индол-5-ил)-изохинолин (6с) (фиг.16D):
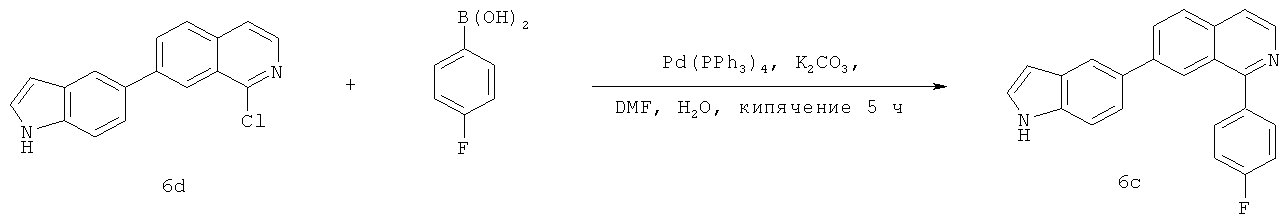
Смесь 6d (0.20 г, 0.72 ммоль), 4-фторфенилбороновой кислоты (0.12 г, 0.86 ммоль), тетракис(трифенилфосфин)палладия (0.033 г, 0.03 ммоль), карбоната калия (0.72 мл, 2-М, 1.4 ммоль), N,N-диметилформамида (22 мл) перемешивали при пропускании через реакционный сосуд аргона в течение 30 мин. Затем смесь кипятили 16 часов и охлаждали до комнатной температуры. Смесь фильтровали через слой силикагеля, разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделяли и промывали раствором NaOH (2×20 мл, 10% водн.), водой (5×30 мл, до исчезновения видимых рефракционных изменений на границе раздела водной и органической фаз) и раствором хлорида аммония (20 мл, насыщ.). Органический слой затем адсорбировали на силикагеле и проводили колоночную флэш-хроматографию (этилацетат/гексан), получая 0.038 г (16%) желтого твердого вещества. MS (ESI): вычислено для C23H15FN2, 338.12, найдено 339.2 [М+Н]+ и 361.2 [M+Na]+. 1Н-ЯМР (300 МГц, ДМСО-d6) [δ] 6.47-6.55 (м, 1H), 6.80 (д, J=9.16 Гц, 2H), 7.38-7.45 (м, 2H), 7.47-7.62 (м, 3H), 7.72 (д, J=8.85 Гц, 2H), 7.79-7.96 (м, 3Н), 11.18 (ушир. с, 1Н).
1,7-Бис-(4-фтор-фенил)-изохинолин (40) (фиг.16А).
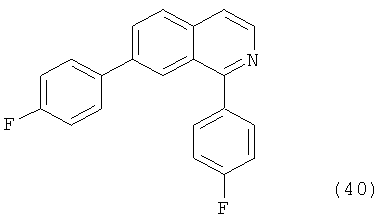
MS (ESI): вычислено для C21H13F2N, 317.10, найдено 318.1 [М+Н]+, 340.1 [M+Na]+, и 315.9 [М-Н]-. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 7.31 (ушир. с, 1H), 7.31-7.37 (м, 2H) 7.39 (ушир. с, 1H), 7.41 (т, J=8.54 Гц, 2H), 7.72-7.77 (м, 2H), 7.78-7.84 (м, 2H), 7.89 (ушир. с, 1H), 7.90-7.99 (м, 1H), 8.09-8.19 (м, 3H), 8.59 (ушир. с, 1H), 8.60-8.65 (м, 1H) м.д.
Синтез 7-бром-1-(4-фтор-бензолсульфонил)-1,2,3,4-тетрагидрохинолина (43) и 1-(4-фтор-бензолсульфонил)-7-(1Н-индол-5-ил)-1,2,3,4-тетрагидрохинолина (41) (фиг.16В).
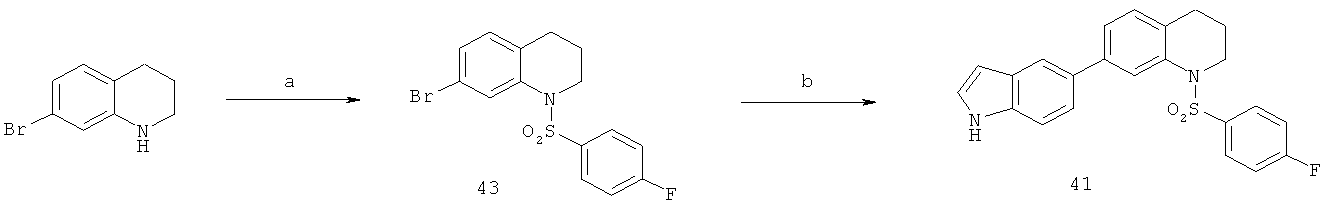
7-Бром-1,2,3,4-тетрагидрохинолин (0.60 г, 2.8 ммоль) перемешивали с 4-фторфенилсульфонил хлоридом (1.65 г, 8.49 ммоль) в пиридине (5 мл) при 80°С 3 часа. Смесь охлаждали, упаривали, и остаток хроматографировали (EtOAc/гексан на SiO2), получая 845 мг (81%) соединения 43 в виде коричневого твердого вещества. C15H13BrFNO2S 368.98, найдено 394.0 [M+Na]+ и 367.8 [М-Н]-. 1Н-ЯМР (500 МГц, CDCl3-d) [δ] 1.58-1.67 (м, 2H) 2.41 (т, J=6.71 Гц, 2H) 3.72-3.82 (м, 2H) 6.89 (д, J=8.30 Гц, 1H) 7.08-7.17 (м, 2H) 7.18-7.24 (м, 1H) 7.59-7.68 (м, 2H) 7.92-8.01 (м, 1H) м.д.
43 (0.46 г, 1.3 ммоль), 5-индолбороновую кислоту (0.26 г, 1.6 ммоль), тетракис(трифенилфосфин)палладия (0.031 г, 0.03 ммоль), карбонат калия (1.35 мл, 2-М, 2.7 ммоль), и N,N-диметилформамид (135 мл) перемешивали при пропускании через реакционный сосуд аргона в течение 30 мин. Затем смесь кипятили 16 часов и охлаждали до комнатной температуры. Смесь фильтровали через слой силикагеля, разбавляли водой (50 мл) и экстрагировали этилацетатом (50 мл). Органический слой отделяли и промывали раствором NaOH (2×20 мл, 10% водн.), водой (5×30 мл, до исчезновения видимых рефракционных изменений на границе раздела водной и органической фаз) и раствором хлорида аммония (20 мл, насыщ.). Органический слой затем адсорбировали на силикагеле и проводили колоночную флэш-хроматографию (этилацетат/гексан), получая 0.38 г (77%) соединения 41 в виде белого кристаллического вещества. MS (ESI): вычислено для C23H19FN2O2S, 406.12, найдено 404.9 [М-Н]- и 429.1 [M+Na]+. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 1.56-1.66 (м, 2H) 2.48 (т, J=6.59 Гц, 2H) 3.76-3.86 (м, 2H) 6.46-6.56 (м, 1H) 7.14 (м, J=7.81 Гц, 1H) 7.33-7.37 (м, 1H) 7.38-7.45 (м, 4H) 7.49 (м, J=8.54 Гц, 1H) 7.66-7.74 (м, 2H) 7.74-7.81 (м, 1H) 7.85-7.94 (м, 1H) 11.17 (ушир. с, 1H) м.д.
7-бром-2-(4-фтор-бензолсульфонил)-1,2,3,4-тетрагидроизохинолин (фиг.16В).
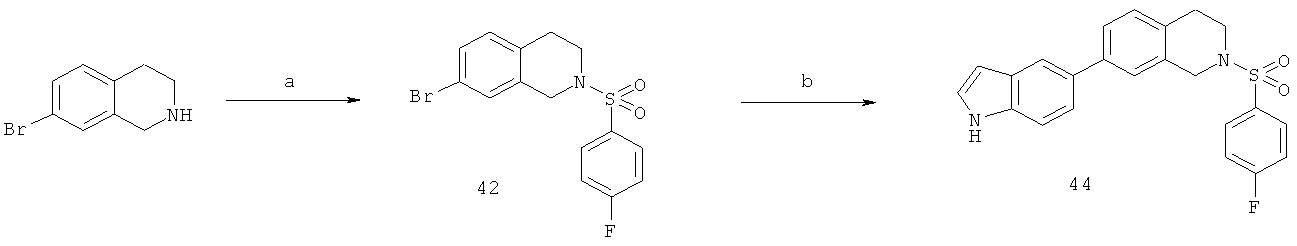
Выход 23%. C15H13BrFNO2S, 369.0, найдено 392.0 [M+Na]+ и 367.7 [М-Н]-. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 2.75-2.82 (м, 2H) 3.32 (т, J=6.10 Гц, 2H) 4.24 (с, 2H) 7.07 (д, J=8.30 Гц, 1Н) 7.29-7.37 (м, 1Н) 7.37-7.43 (м, 1Н) 7.47 (т, J=8.79 Гц, 2Н) 7.87-7.93 (м, 2Н) м.д.
2-(4-фтор-бензолсульфонил)-7-(1Н-индол-5-ил)-1,2,3,4-тетрагидроизохинолин (44).
Выход 77%. 1Н-ЯМР (500 МГц, ДМСО-d6) [δ] 2.84-2.91 (м, 2Н) 3.35 (т, J=5.98 Гц, 2Н) 4.30 (с, 2Н) 6.44-6.48 (м, 1Н) 7.17 (д, J=7.81 Гц, 1Н) 7.32-7.40 (м, 2Н) 7.41-7.51 (м, 3Н) 7.75-7.79 (м, 1Н) 7.89-7.96 (м, 1Н) 11.13 (ушир. с, 1Н) м.д.
Пример 10
Растворимость в воде арил-бензоил-имидазольных (abi) соединений (фиг.17)
Определение растворимости в воде. Для определения растворимости в воде, 1 мг каждого соединения суспендировали в 1 мл воды и встряхивали 48 часов при комнатной температуре. Суспензию центрифугировали при 10000 об/мин и фильтровали на фильтре с диаметром пор 0.22 мкм. Концентрации каждого соединения измеряли на приборе LC-MS, состоящем из ВЭЖХ прибора HP S1100 (Agilent, Foster ceity, CA) и Bruker ESQUIRE масс-детектора с электроспрей/ионная ловушка прибором в режиме регистрации положительно заряженных ионов (Bruker, Fremont, CA). Для ВЭЖХ применяли обращенно-фазную Nova-pak С18 колонку (150 мм × 3.9 мм, Waters, Milford, MA). Подвижная фаза имела состав 20:80 (об/об) вода/ацетонитрил. При масс-спектрометрии выделяли пик 382 m/z (для имидазольных соединений) и 399 m/z (для тиазольных соединений), соответственно. Концентрацию каждого соединения вычисляли по площади MS-пика согласно следующему калибровочному уравнению: y=1.3295х+114.24 (R2=1.00). Для построения калибровочной кривой (фиг.17), на основе которой было выведено указанное уравнение, 50, 100 мкл растворов ABI соединения 12ga концентрации 100 мкг/мл и 10 мкг/мл, и их соответствующих тиазольных аналогов, а также СА-4 (структуру см. на фиг.19), в ацетонитриле вводили в установку ВЭЖХ и проводили масс-спектральный анализ. Количество (нг) в каждом введении откладывали на графике относительно площади соответствующего ему масс-пика, получая калибровочную кривую на фиг.17.
Времена удерживания ВЭЖХ для ABI соединения 12ga (1.5 мин) сравнивали с соответствующим ему тиазольным аналогом (2.2 мин) при использовании подвижной фазы состава 80/20 метанол/вода, скорости потока 1 мл/мин и обращенно-фазной колонки, что свидетельствует о более гидрофильном характере имидазольного производного, чем соответствующего ему тиазольного аналога. Вычисленные значения logP для ABI соединения 12ga и соответствующего ему тиазольного аналога составили приблизительно 2.9 и 4.4, соответственно. Растворимость в воде соединения 12ga составила 13 мкг/мл или примерно в 200 раз больше, чем его тиазольного аналога (72 нг/мл).
Пример 11
Биологические испытания соединений по настоящему изобретению
Пример 11А: Подавление роста клеток in vitro
Анализ культуры клеток и цитотоксичности в отношении рака предстательной железы и меланомы. Все клеточные линии получали от АТСС (American Type Culture Collection, Manassas, VA, USA), а культуры клеток покупали в Cellgro Mediatech (Herndon, VA, USA). Авторы настоящего изобретения изучали антипролиферативную активность новых анти-тубулиновых соединений на четырех клеточных линиях рака предстательной железы человека (LNCaP, DU 145, PC-3 и РРС-1) и двух клеточных линиях меланомы человека (А375 и WM-164). Линия клеток яичника OVCAR-8 и соответствующая ей резистентная клеточная линия, сверхэкспрессирующая P-gp (NCI/ADR-RES) использовались в качестве моделей MDR. Обе линии клеток яичника получали от Национального Института Рака (NCI). Все клеточные линии тестировали и проверяли на аутентичность в АТСС или NCI. Все клеточные линии рака предстательной железы и рака яичника выращивали в RPMI 1640, с добавлением 10% эмбриональной бычьей сыворотки (FBS).
Клетки меланомы выращивали в DMEM с добавлением 5% FBS, 1% смеси антибиотика/противогрибкового средства (Sigma-Aldrich, Inc., St. Louis, МО, USA) и бычьего инсулина (5 мкг/мл, Sigma-Aldrich). Цитотоксический потенциал анти-тубулиновых соединений определяли анализом с сульфородамином В (SRB) через 96 часов после введения.
Все описываемые соединения сначала проверяли на цитотоксичность в отношении клеточной линии мышиной меланомы B16-F1, клеточной линии человеческой меланомы (А375 и WM-164) и клеточных линий рака предстательной железы (DU145, РС-3, LNCaP, РРС-1). Соединение 1h и соединение АВТ-751 (Е7010, Abbott Laboratories/Eisai Co Ltd), которое проходит фазу II клинических испытаний в лечении пациентов с различными видами рака, включали в исследование как примеры средств, связывающихся с колхициновым сайтом. Значения IC50 для ингибирования роста клеток представлены в таблицах 1, 2 и 3.
Результаты:
|
|
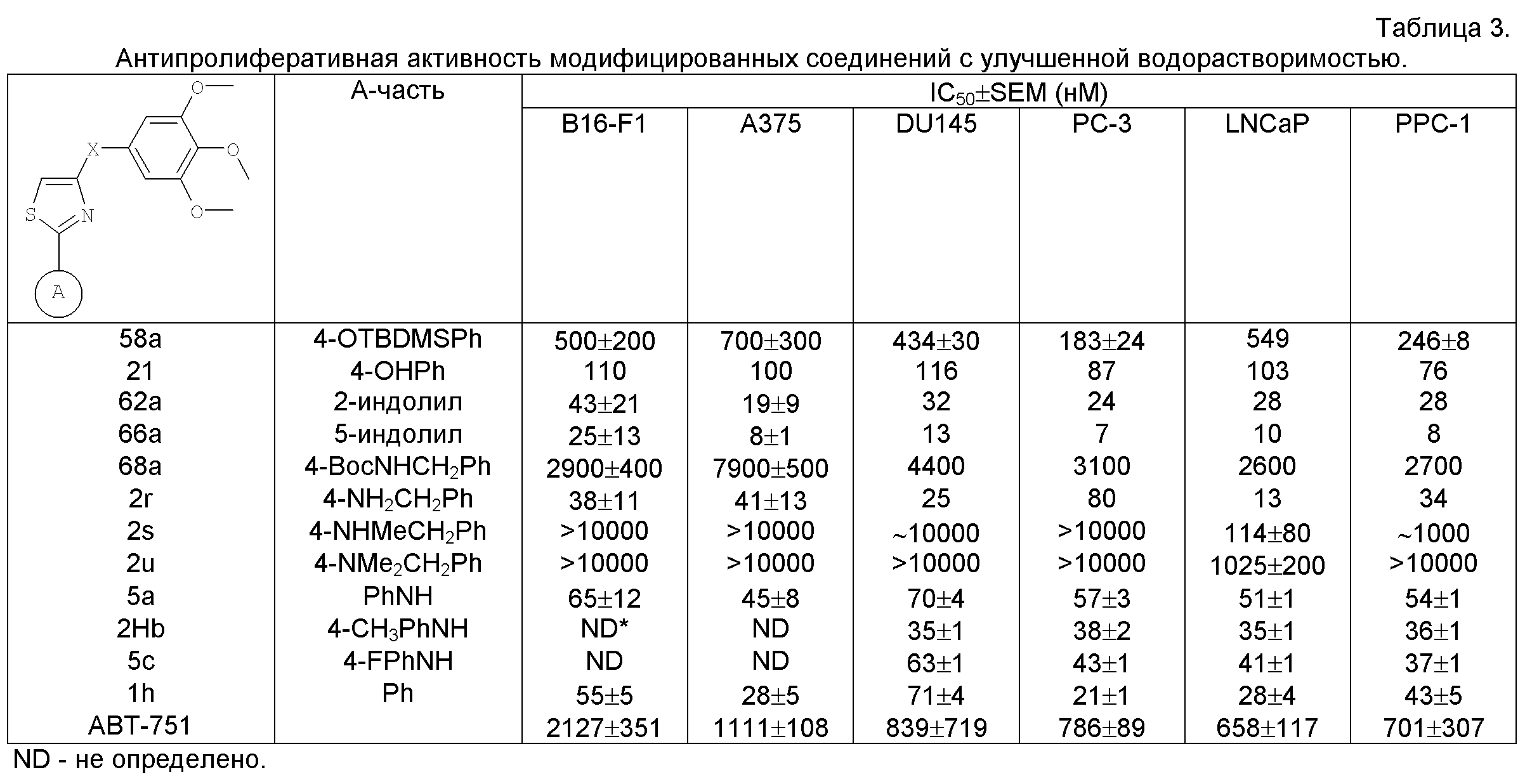
Взаимосвязь строения и свойств (SAR) для молекул с альтернативными циклами В. Первый ряд исследований имел целью подбор альтернативы тиазольному циклу «В». Соответственно, была изучена серия гетероциклических циклов «В». Как показано в таблице 1, успешной заменой тиазолу являются пиридин 1с, фуран 1d и тиофен 1f. Значения IC50 (12 нМ - 35 нМ в отношении клеток рака предстательной железы) близки к соответствующему значению для тиазольного соединения 1h. Введение фенила (1а), оксазолина (35а) и оксазола (36а) сохраняло активность в диапазоне сотен наномолей. Но введение пиримидина (1b, IC50 3.7-8.3 мкМ), обращенного 2,5-тиазола и 3,5-изоксазола (1е и 1i, IC50 >10 мкМ) вызвало заметное падение активности. Модификация цикла «В» в насыщенный пиперидиновый цикл (1g) также привела к полной потере активности (IC50 >20 мкМ).
SAR для молекул с альтернативными линкерами. In vitro исследования метаболической устойчивости в печени показали, что карбонильные линкеры между циклами «В» и «С» в SMART соединениях являются причинами короткого времени их полужизни (5-17 мин), главным образом из-за восстановления карбонила. Для блокирования восстановления кетона в неактивное соединение 2b с гидроксильным линкером, во второй серии исследований модифицировали карбонильный линкер (таблица 2). Карбонильный линкер заменяли на двойные связи (2а, 3а и 3b), амиды (2g, 2h), оксимы (2е-цис,транс и 2f-цис,транс), гидразид (2d-цис, 2d-транс), акрилонитрилы (2с-цис, 2с-транс), цианоимин (2j), сульфониламид (4d), эфир серной кислоты (4а), сульфонильное и сульфинильное соединение (4b, 4с). Также было получено соединение 2i с прямой связью без какого-либо линкера между циклами «В» и «С». Из этих модификаций линкера, только цианоиминовый линкер (2j) показал обнадеживающий потенциал (20-60 нМ) по сравнению с карбонильным соединением 1h, но исследование метаболизма in vitro показало, что время полужизни 2j в микросомах печени человека составило менее 5 минут. Предполагается, что, несмотря на блокировку восстановления кетона, такое изменение структуры могло привести к новому механизму метаболической нестабильности в соединении 2j. Пары изомеров соединений, содержащих двойные связи, оксимы и гидразиды, были разделены. Соединение 3а создавалось как имитация структуры СА-4 (фиг.19), которая содержит цис С=С связь между двумя арильными циклами, но, к сожалению, 3а и другие пары изомеров потеряли активность после замены С=О линкера. Наблюдался интересный феномен, состоящий в том, что син-изомер 2е-цис (0.1-0.3 мкМ) показал активность в 10 раз выше, чем анти-изомер 2е-транс (>10 мкМ). Время полужизни 2е-цис в микросомах печени человека увеличилось до 35 минут, в то время как времена полужизни соединений 2d увеличились до 55 минут. Однако активность 2d (~1 мкМ) ухудшилась.
Пример 11B: Растворимость соединений по настоящему изобретению в воде.
Растворимость лекарственных средств определяли на Multiscreen Solubility Filter Plate (Millipore Corporate, Billerica, MA), соединенном с LC-MS/MS. Вкратце, 198 мкл фосфатно-солевого буферного раствора (PBS) (рН 7.4) помещали в 96-луночный планшет и диспергировали и смешивали с 2 мкл 10 мМ раствора испытуемых соединений (в ДМСО) при аккуратном встряхивании (200-300 об/мин) в течение 1.5 часов при комнатной температуре (N=3). Планшет центрифугировали при 800 g в течение 5 минут, и фильтрат использовали для определения его концентрации и растворимости испытуемого соединения методом LC-MS/MS, как описано ниже.
Введение в анти-тубулиновые средства полярных и ионизирующихся групп. Главным ограничением для SMART средств является их низкая растворимость в воде. Для изучения поведения SMART in vivo применяли поверхностно-активные вещества, такие как Tween 80, и были получены положительные результаты. Но подобные поверхностно-активные вещества обладают биологической активностью и вызывают множество побочных эффектов. Кроме того, полагают, что низкая растворимость соединения 1h в воде приводит к низкой оральной биодоступности (3.3%, таблица 4). В третьей серии исследований растворимость соединений в воде успешно увеличивали без влияния на активность путем введения полярных групп, таких как гидроксил и индолилы. Кроме того, в параположение цикла «А» также вводили ионизирующиеся группы, такие как амино- и алкиламино-группы. Как показано на фиг.5 и в таблице 3, введение индолильных групп в цикл «А», в особенности 5-индолильной группы (66а, 7-25 нМ) повышало активность по сравнению с 4-ОН соединением 2l (76-116 нМ). Аминометил -CH2NH2 в параположении цикла «А» также сохранял активность (2r, 13-80 нМ), но наличие пара-NHMe (2s) или пара-NMe2 (2u) сводило активность на нет. Как показано на фиг.18, аналитическое определение растворимости в воде показало, что у индолильного соединения 66а растворимость в PBS повышалась с 1.1 мкг/мл (соединение 1h) до 3.8 мкг/мл. Аминометильное соединение 2r превращали в соль с HCl, что повышало растворимость в 35 раз (>35 мкг/мл). Хотя соединение 2r продемонстрировало удовлетворительную водорастворимость, фармакокинетические исследования показали, что это соединение все равно обладает очень плохой биодоступностью (F%=0.2%). Авторы полагают, что соединение 2r ионизируется в желудке и поэтому не всасывается в кровеносную систему.
Пример 11С: Фармакокинетические исследования
Исследование фармакокинетики. Самок крыс Sprague-Dawley (n=3 или 4; 254±4 г) покупали в Harlan Inc. (Indianapolis, IN). Торакальные катетеры для яремной вены крыс покупали в Braintree Scientific Inc. (Braintree, MA). После прибытия, животные акклиматизировались в течение 3 дней в комнате с контролируемой температурой (20-22°С) с 12-часовым циклом темнота/свет перед какими либо экспериментами. Соединение 1h вводили внутривенно (i.v.) через катетеры в яремной вене в дозировке 2.5 мг/кг (в ДМСО/PEG300, 2/8), а 5На и 5Нс - в дозировке 5 мг/кг (в ДМСО/PEG300, 1/9). Равные объемы гепаринизированного физраствора инъецировали для замены забираемой крови, и отбирали образцы крови (250 мкл) через катетеры в яремной вене через 10, 20, 30 минут, и 1, 2, 4, 8, 12, 24 часа. Соединения 1h, 5На и 5Нс принудительно вводили перорально (р.о.) в дозировке 10 мг/кг (в Tween80/ДМСО/H2O, 2/1/7). Все образцы крови (250 мкл) после перорального введения отбирали через катетеры в яремной вене через 30, 60, 90 минут, 120 минут, 150 минут, 180 минут, 210 минут, 240 минут, и через 8, 12, 24 часа. Гепаринизированные шприцы и виалы готовили до забора крови. Образцы плазмы готовили центрифугированием образцов крови при 8000 g в течение 5 минут. Все образцы плазмы сразу замораживали и хранили при -80°С до момента анализа.
Аналиты экстрагировали из 100 мкл плазмы 200 микролитрами ацетонитрила, содержащего 200 нМ внутреннего стандарта ((3,5-диметоксифенил)(2-фенил-1Н-имидазол-4-ил)метанон). Образцы тщательно перемешивали, центрифугировали, и органический экстракт переносили в автосэмплер для LC-MS/MS анализа. Для наблюдения наиболее чувствительных сигналов применяли режим мониторинга нескольких реакций (multiple reaction monitoring (MRM)), сканируя m/z 356 → 188 (соединение 1h), m/z 371 → 203 (соединение 5На), m/z 389 → 221 (соединение 5Нс) и m/z 309 → 171 (внутренний стандарт). Фармакокинетические параметры определяли с применением анализа без учета компартментов (WinNonlin, Pharsight Corporation, Mountain View, CA).
Результаты:
|
Модификация замещенных метоксибензоил-арил-тиазольных молекул (Substituted Methoxybenzoyl Aryl Thiazole (SMART)) для улучшения оральной биодоступности. Многие известные противораковые средства, воздействующие на тубулин, такие как таксаны и винбластин, требуют внутривенного введения вследствие своей низкой оральной биодоступности. Оральная биодоступность представляет собой комплексный параметр, охватывающий многие химические и физиологические процессы, такие как растворимость, проницаемость и метаболическую устойчивость. Растворимость рассматриваемых ингибиторов тубулина повышали введением аминового линкера между циклами «А» и «В», как в соединениях 5а-с (фиг.6). В таблице 3 показано, что NH-мостиковые соединения (5а-с) имеют сходную активность с 1h при повышенной растворимости (15 и 19 мкг/мл для 5а и 5с, соответственно (фиг.18)), но они более чем в 20 раз активнее, чем АВТ-751 (структуру АВТ-571 смотри в таблице 3 и на фиг.19).
Проводили исследования фармакокинетики на крысах, чтобы выяснить - обладают ли описываемые новые соединения лучшей биодоступностью по сравнению с соединением 1h (таблица 4). Полученные данные ясно показывают, что 5Нс (соль с HCl 5с) показывает в 4.3 раза более высокую концентрацию (площадь под кривой, AUC) при пероральном введении по сравнению с 1h, подтверждая, что повышенная вследствие наличия аминового линкера водорастворимость успешно улучшает оральную биодоступность. Кроме того, максимальная концентрация (Смакс) 5На и 5Нс при пероральном введении составила 814 и 1262 нг/мл, соответственно. В то же время Смакс соединения 1h составила всего 212 нг/мл. В целом, биодоступность 5На и 5Нс возросла с 3.3% для 1h до 11% и 21%, соответственно (таблица 4). Соединение 5Нс продемонстрировало умеренный клиренс, средний объем распределения и приемлемую оральную биодоступность. Полученные данные подтверждают, что новые синтезированные амино-линкерные соединения имеют активность и фармакокинетический профиль, позволяющий разрабатывать их в качестве нового класса орально биодоступных антитубулиновых средств.
Пример 11D: ингибирование полимеризации тубулина in vitro соединениями по настоящему изобретению.
In Vitro анализ полимеризации тубулина. Тубулин бычьего мозга (0.4 мг, >97% чистота) (Cytoskeleton, Denver, CO) смешивали с 10 мкМ анализируемого соединения и инкубировали в 100 мкл общего тубулинового буфера (80 мМ PIPES, 2.0 мМ MgCl2, 0.5 мМ EGTA и 1 мМ GTP) при рН 6.9. Поглощение при длине волны 340 нм измеряли каждую минуту в течение 20 минут на микропланшетном ридере SYNERGY 4 (Bio-Tek Instruments, Winooski, VT). Температура в спектрофотометре поддерживалась равной 37°С для полимеризации тубулина.
Результаты
Ингибирование полимеризации тубулина выбранными активными соединениями 1с, 2j, 66а и 5а исследовали для всех трех стратегий дизайна (альтернативные В-циклы, новые линкеры и солюбилизирующие фрагменты) и сравнивали с 1h. Тубулин бычьего мозга (>97% чистота) инкубировали с индивидуальными соединениями (10 мкМ) для анализа их действия на полимеризацию тубулина (фиг.20). После 20 минут инкубирования, полимеризация тубулина ингибировалась на 47% в случае 1h, по сравнению с растворителем. 1с и 2j ингибировали 64% полимеризации через 20 минут, с различными моделями ингибирования. Соединения 5а и 66а обеспечивали более высокие значения ингибирования - 78% и 81%, соответственно. Полученные данные подтверждают, что данные соединения проявляют высокую активность в плане ингибирования полимеризации тубулина, которая хорошо согласуется с их цитотоксичностью.
Пример 11Е: Замещенные метоксибензоил-арил-тиазольные (SMART) соединения преодолевают обуславливаемую Р-гликопротеином множественную лекарственную устойчивость.
Р-гликопротеиновая (P-gp) система считается основным физиологическим механизмом возникновения множественной лекарственной устойчивости (MDR), эта система работает как АТФ-зависимый насос, выкачивающий из клетки лекарственные средства, активно удаляющий разнообразные структурно различающиеся цитотоксичные соединения. Усиленное выведение данных соединений снижает их внутриклеточную концентрацию и уменьшает таким образом их цитотоксичность. Поэтому новые соединения, не подверженные лекарственной устойчивости, могут иметь большую терапевтическую и экономическую ценность. Помимо P-gp, используемые в клинике антитубулиновые средства характеризуются другими механизмами возникновения устойчивости, такими как изменения в динамике микротрубочек и мутации β-тубулина, которые являются известным ограничивающим фактором в применении таксанов. Антитубулиновые соединения по настоящему изобретению анализировали в отношении клеточной линии рака яичника OVCAR-8 (родительская) и P-gp сверхвырабатывающей клеточной линии NCI/ADR-RES (таблица 5).
Результаты:
|
Следует отметить, что анти-тубулиновые соединения по настоящему изобретению демонстрируют эквипотентное антипролиферативное действие в отношении клеточных линий OVCAR-8 и NCI/ADR-RES, подтверждая, что они не являются P-gp субстратами и что они работают независимым от P-gp образом. Это свойство отличает данные соединения от паклитаксела, винбластина и колхицина на клетках NCI/ADR-RES.
Была открыта новая серия ингибиторов полимеризации тубулина с приемлемыми характеристиками оральной биодоступности и эквипотентной активностью в отношении клеточных линий опухолей с множественной лекарственной устойчивостью. Работа медицинских химиков началась с оптимизации SMART соединения 1h. Исследовали химические модификации различных замещенных арилов в цикле «В» и в линкерах между циклами «В» и «С» по результатам биологических испытаний на раковых клетках in vitro. Изучение SAR показало, что оптимальные циклы «В» включают пиридин (1с), тиофен (1f) и фуран (1d), которые сохраняют прекрасную активность in vitro. Замена карбонильного линкера на цианоимин (2j) между циклами «В» и «С» увеличивает активность. Проводили модификации структуры для повышения растворимости в воде и биодоступности. Введение амино-группы между циклами «А» и «В» дало соединения 5а-с, которые продемонстрировали сходную антипролиферативную активность в отношении протестированных раковых клеток, а также MDR(+) и MDR(-) клеточных линий; кроме того, растворимость и in vivo биодоступность сильно увеличилась по сравнению с 1h. Поэтому указанные новые анти-тубулиновые соединения представляют собой новое семейство соединений, которые могут оказаться весьма полезными в лечении рака.
Пример 12
Антипролиферативная активность соединений по настоящему изобретению
Данные по антипролиферативной активности аналогов, полученных способами по настоящему изобретению, представлены в таблицах 6 и 6А.
|
|
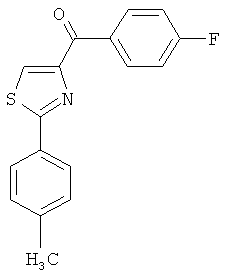
|
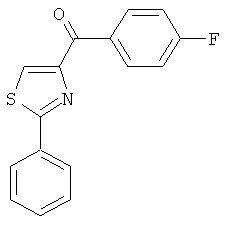
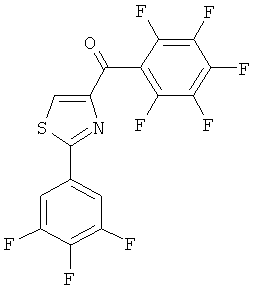
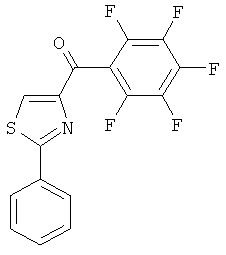
|
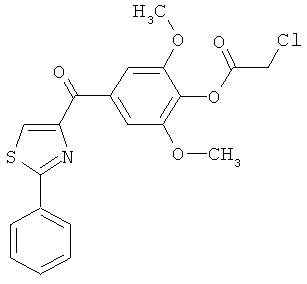
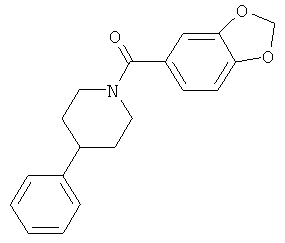
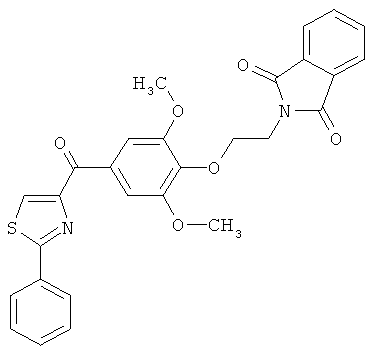
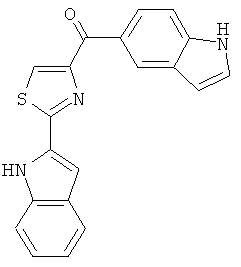
|
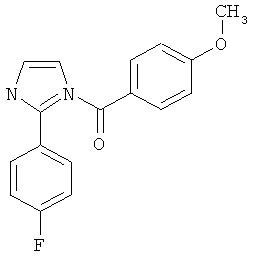
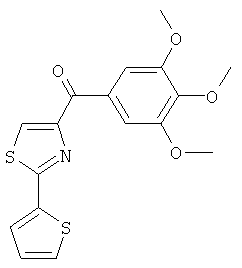
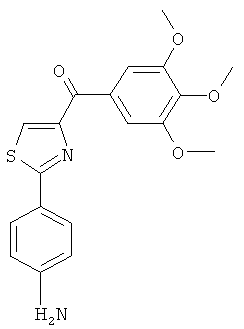
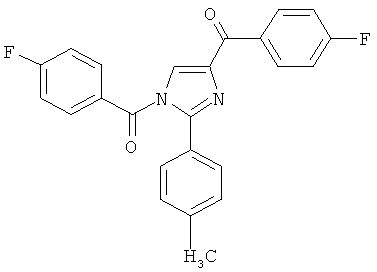
|
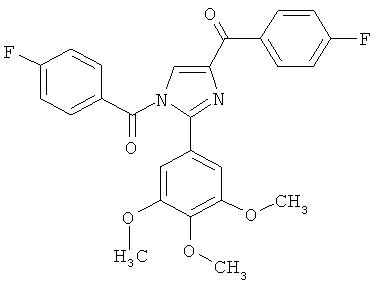
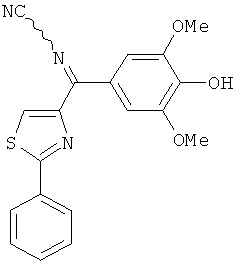
|
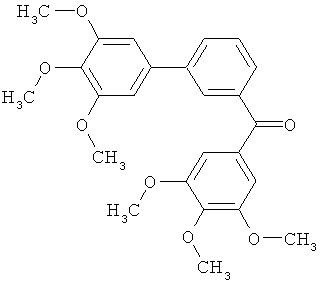
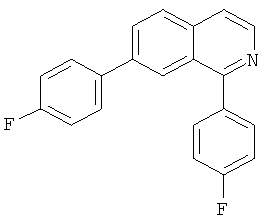
|
Пример 13
Биологическое исследование изохинолиновых производных по настоящему изобретению
Культура клеток
LNCaP, PC-3, DU-145, PPC-1, MES-SA и MES-SA/DX5 были получены от АТСС (Rockville, MD). Все клетки, полученные от АТСС, немедленно выращивали и замораживали, так чтобы все клеточные линии можно было вырастить заново каждые 2-3 месяца из замороженной виалы той же партии клеток. Для ксенографических in vivo экспериментов проводили аутентификацию РС-3 в Research Animal Diagnostic Laboratory (Columbia, МО) в период до четырех месяцев перед исследованиями. Межвидовое загрязнение проверяли методом ПЦР, и идентичность клеточных линий подтверждали генерацией генетического профиля. MES-SA и MES-SA/DX5 выращивали в среде Маккоя 5А, содержащей 2 мМ L-глутамина с добавлением 10% эмбриональной бычьей сыворотки (FBS). Все другие клетки выращивали в среде RPMI-1640 с 2 мМ L-глутамина и 10% FBS.
Исследование ингибирования роста
Цитотоксическую или анти-пролиферативную активность испытуемых соединений изучали на нескольких клеточных линиях, применяя анализ по сульфородамину В (SRB). Культуру клеток помещали в 96-луночный планшет и инкубировали со средой, содержащей различные концентрации испытуемых соединений, в течение 96 часов. Определяли оптическую плотность при 540 нм на микропланшетном ридере (Dynex Technologies, Chantilly, VA). Строили диаграммы зависимости процента ингибирования роста клеток от концентрации лекарственного средств, и определяли концентрацию, ингибирующую рост клеток на 50% относительно контрольного образца без добавления веществ (IC50), нелинейной регрессией по методу наименьших квадратов с применением программного обеспечения WinNonlin (Pharsight Corporation, Cary, NC).
Анализ клеточного цикла
Распределение по фазам клеточного цикла определяли окрашиванием пропидий иодидом (PI). Обработанные клетки промывали PBS и фиксировали 70%-ным ледяным этанолом в течение ночи. Затем зафиксированные клетки окрашивали PI с концентрацией 20 мкг/мл в присутствии RNase A (300 мкг/мл) при 37°С в течение 30 минут. Распределение по фазам клеточного цикла анализировали методом флуоресцентной сортировки клеток в University of Tennessee Health Science Center, TN.
Исследование метаболизма In Vitro
Для обеих фаз I инкубационная смесь в 65 мМ фосфатно-солевом буферном растворе (рН 7.4) состояла из 1 мг/мл микросомных белков печени, 3 мМ НАДФН и 0.5 мкМ испытуемого соединения. Концентрация метанола (использовался для растворения субстрата) составляла 1% (об/об). Общий объем инкубируемой смеси составлял 200 мкл, и реакционные смеси инкубировали при 37°С. Для построения кривых устойчивости для испытуемых соединений, инкубирование различных смесей останавливали через 10, 20, 30, 60 и 90 минут для анализа оставшихся соединений. Все реакции останавливали добавлением 200 мкл ледяного ацетонитрила. Затем образцы центрифугировали 5 минут при 3000 g и анализировали надосадочный раствор методом LC-MS/MS.
Исследование фармакокинетики на мышах.
Использовали самцов ICR мышей (5-6 недель, 20-25 г). Для 6а, 6b и 6с дозу 5 мг/кг вводили внутривенно (i.v.), внутрибрюшинно (i.p.) и перорально (p.o.). I.v. дозы вводили в хвостовую вену. Пероральные дозировки вводили принудительно. В каждую регистрируемую точку времени, 3-4 мыши подвергали эвтаназии изофлураном (Baxter Healthcare, Deerfield, IL) и отбирали образцы крови (до 600 мкл каждый) из задней полой вены. Образцы плазмы хранили при -20°С до момента анализа. Белки крови осаждали добавлением ацетонитрила (150 мкл, содержал внутренний стандарт) к 100 мкл плазмы крови мышей. Образцы встряхивали на вихревой мешалке и затем центрифугировали 10 минут при 8000 g. Надосадочный раствор переносили в чистую виалу для введения в масс-спектрометр для проведения анализа.
Исследование противоопухолевой эффективности in vivo.
Клетки PC-3 (2.5×106 клеток/сайт) плюс Matrigel (BD biosciences, San Jose, CA) вводили подкожной инъекцией в бок самцов nu/nu мышей. Размер опухоли измеряли штангенциркулем каждые 2-4 дня и вычисляли как V=π/6×(длина)×(ширина)2 [13]. Когда опухоли достигали размера примерно 100-150 мм3, начинали введение лекарств. Контрольной группе вводили носитель (20% Captex200 в Tween80). Во время лечения размер опухоли и вес тела измеряли каждые 2-4 дня.
Подсчет числа белых кровяных телец
Цельную кровь забирали у безтимусных мышей в конце исследования эффективности. Для подсчета числа белых кровяных телец (WBC) с помощью гемацитометра, 10 мкл образца цельной крови разбавляли добавлением 190 мкл 2%-ной уксусной кислоты. При правильном подборе света, лейкоциты в гематоцитометре видны как темные точки. Число WBC в каждом образце подсчитывали дважды в течение одного часа после разбавления и вычисляли среднее значение.
Результаты
|
|
|
|
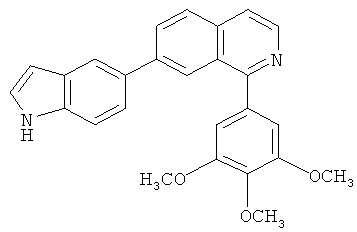
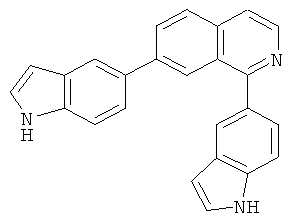
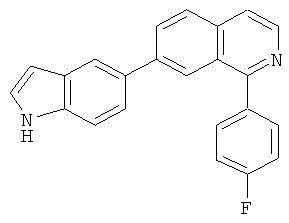
Эффективность и переносимость 6b и 6с определяли на ксенографических моделях после i.p. инъекции (фиг.34). PC-3 ксенотрансплантатам вводили носитель (один раз в день), 6b (40 мг/кг, один раз в день) или 6с (40 мг/кг, один раз в день) в течение 3 недель. Носитель состоял из 20% Captex200 в Tween80. Строили диаграммы зависимости объемов опухолей (мм3, средние значения для 8 животных ±SD) от времени. Объемы опухолей и уровни выживания или веса тела представлены на фиг.34А. Размер печени (г) каждой безтимусной мыши определяли после 3-недельного введения соединений, полученные данные приведены на фиг.34В. Подсчитывали число белых кровяных телец в цельной крови, взятой у животного после 3-недельного введения соединений, полученные данные приведены на фиг.34С.
Пример 14
Анти-пролифератвная активность избранных abi соединений по настоящему изобретению
Анализ цитотоксичности на культуре клеток
Материалы и методы
Исследовали анти-пролиферативную активность ABI соединений на трех клеточных линиях меланомы (A375 и WM-164, клеточная линия меланомы человека; В16-F1, клеточная линия меланомы мыши) и четырех клеточных линиях рака предстательной железы человека (LNCaP, DU145, PC-3 и РРС-1). Все линии клеток покупали в АТСС (American Type Culture Collection, Manassas, VA) за исключением клеточной линии РРС-1. MDA-MB-435 и MDA-MB-435/LCCMDR1 клетки любезно были предоставлены Робертом Кларком (Dr. Robert Clarke) из Georgetown University School of Medicine, Washington, DC. Клетки меланомы выращивали в DMEM (Cellgro Mediatech, Inc., Herndon, VA), а клетки рака предстательной железы выращивали в RPMI 1640 (Cellgro Mediatech, Inc., Herndon, VA) с добавлением 10% FBS (Cellgro Mediatech). Культуры выращивали при 37°С в увлажненной атмосфере, содержащей 5% CO2. 1000-5000 клеток помещали в каждую лунку 96-луночных планшетов в зависимости от скорости роста и добавляли различные концентрации испытуемого соединения на 48 часов (быстрорастущие клетки меланомы) или 96 часов (медленнорастущие клетки рака предстательной железы) в 3-5 повторах. Число клеток по окончании обработки лекарственным средством определяли анализом по сульфородамину В (SRB). Вкратце, клетки фиксировали 10%-ной трихлоруксусной кислотой и окрашивали 0.4%-ным SRB, и определяли поглощение при 540 нм на планшет-ридере (DYNEX Technologies, Chantilly, VA). Строили диаграмму зависимости процента выживаемости клеток от концентрации лекарства и вычисляли значения IC50 (концентрация, при которой рост клеток ингибируется на 50% относительно контрольного образца без добавления соединений) нелинейным регрессионным анализом с применением GraphPad Prism (GraphPad Software, San Diego, CA).
Результаты
Результаты исследования in vitro анти-пролиферативной активности соединений по настоящему изобретению с применением трех клеточных линий меланомы (одна клеточная линия мышиной меланомы B16-F1, и две клеточные линии метастатической меланомы человека, А375 и WM-164) и четырех клеточных линий рака предстательной железы человека (LNCaP, PC-3, Du145 и РРС-1) суммированы в таблицах 11-13.
|
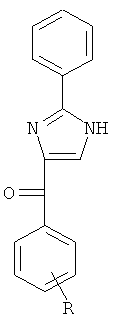
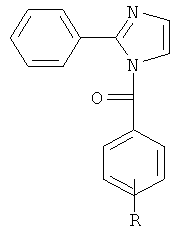
По данным таблицы 11 соединения 12aa-12ai продемонстрировали умеренную активность со значениями IC50 в микромолярном диапазоне (среднее значение для всех семи клеточных линий). Наиболее активным соединением из данной серии являлось 12аа, имеющее среднее значение IC50 160 нМ. Удаление одной из метокси-групп из 3,4,5-триметокси фрагмента в цикле С (12ad, 12ae) привело к значительному падению активности (IC50 >10 мкМ для 12ae и среднее IC50 3.1 мкм для 12ad). Соединение с 4-фтор заместителем в цикле С (12af) также показало относительно хорошую активность (IC50=0.91 мкМ), и это наблюдение имеет важное значение, поскольку замена триметокси-фрагмента на 4-фторную группу может обеспечить высокую активность и улучшенную метаболическую устойчивость. Положение фтора в цикле С критично для активности, поскольку сдвиг от 4-фтор к 3-фтор замещению привел к полной потере активности (IC50 >10 мкМ для 12ag, по сравнению с 0.91 мкМ для 12af). Этот результат позволяет предположить, что потенциальный донор водородной связи располагается недалеко от 4-положения данного цикла.
Как ясно следует из таблицы 11, положение циклов А и С является критичным. Простой сдвиг фрагмента С-цикла из положения 4 в положение 1 имидазольного цикла (цикл В) привел к полной потере активности (IC50 >10 мкМ для 12aba, 12ааа, 10а, 10х, 10j)
|
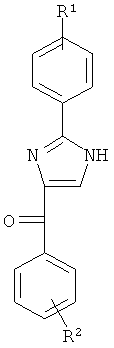
По данным таблицы 12 соединения с 3,4,5-триметокси и 4-фтор заместителями в цикле С показали хорошую активность с различными заместителями в цикле А. Данные соединения продемонстрировали прекрасную анти-пролиферативную активность со значениями IC50 до 8.0 нМ на клеточной линии WM164 (12da). В целом, соединения, имеющие единственный заместитель в параположении цикла А, были более активны, что можно увидеть по данным активности 12са, 12cb, 12da, 12db, 12fa, 12fb, 12ga и 12gb (IC50=7.9-110 нМ). Соль 12db-HCl (IC50=172 нМ) показала несколько пониженную активность по сравнению с соответствующим свободным основанием 12db (IC50=109 нМ). Соединение 12fb (IC50=63.7 нМ), имеющее единственный галогеновый заместитель в пара-положении циклов А и С, оказалось активным и не имело метокси-фрагмента. Соединения с 3,4,5-триметокси заместителями в цикле А полностью теряли активность (IC50 >10 мкМ для 12еа, 12eb), подтверждая очень разное связывающее окружение вокруг циклов А и С. Удаление 5-метокси заместителя из А-цикла значительно повышало активность (IC50=330 нМ и >10 мкМ для 12ha, 12еа соответственно). Деметилирование в 3,4,5-триметокси фрагменте резко снижало активность с 43 нМ (12fa) до 3.89 мкМ (13fa). Аналогичные результаты были получены для 13еа, 12ka, 12kb и 13ha при деметилировании заместителей на цикле А или С. Электроно-донорные группы (4-метокси, 4-диметиламино, 4-метил) и электрон-акцепторные группы (4-хлор, 2-трифторметил) в цикле А не продемонстрировали существенных различий в активности. Введение трифторметильной группы в орто-положение цикла А привело к полной потере активности (IC50 >10 мкМ для 12ia, 12ib). Наличие бензилокси-группы в параположении цикла А (IC50=75 нМ для 12jb) привело к 440-кратному росту активности по сравнению с пара-гидрокси соединением 12kb (IC50=33 мкМ). Стоит отметить, что соединение 12jb, имеющее 4-фторный заместитель в цикле С, имеет более высокую активность, чем его аналог 12ja, имеющий 3,4,5-триметокси группу в цикле С (IC50=75 нМ для 12jb, и 7.3 мкМ для 12ja).
|
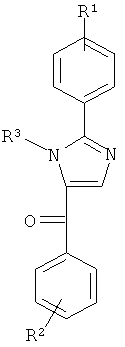
По данным таблицы 13 соединения с фенилсульфонильной защитной группой, присоединенной к атому азота имидазольного кольца (11cb, 11db, 11fb, 11ga, 11gb, 11ha, 11jb) также были активны и имели значения IC50 в наномолярном диапазоне (таблица 13). В целом, активность данных соединений сравнима с их соответствующими незащищенными аналогами, что можно подтвердить на примере сравнения активности 11cb (43 нМ), 11db (111 нМ), 11fb (72 нМ), 11ga (285 нМ), 11gb (87 нМ), 11ha (268 нМ) и 11jb (61 нМ) с соответствующими им незащищенным аналогами 12cb (36 нМ), 12db (109 нМ), 12fb (64 нМ), 12ga (131 нМ), 12gb (72 нМ), 12ha (330 нМ) и 12jb (75 нМ). Другие соединения (11ab-11ag, 11ea, 11eb, 11hb, 11ia и 11ib, 1-50 мкМ) были, в целом, намного менее активны, тоже проявляя сходство с их аналогами (12ab-12ag, 12ea, 12eb, 12hb, 12ia, и 12ib, 1-50 мкМ).
Пример 15
Активность арил-бензоил-имидазольных (abi) соединений в отношении клеток лекарственно-устойчивой меланомы
Обуславливаемый Р-гликопротеином (Pgp) вывод лекарства представляет собой основной механизм противодействия раковых клеток накоплению в клетке противораковых лекарственных средств в эффективных концентрациях. Активность ABI соединений сравнивали в отношении клеток меланомы с множественной лекарственной устойчивостью (MDR) (MDA-MB-435/LCCMDR1) и их родительских нерезистентных раковых клеток (MDA-MB-435). Хотя MDA-MB-435 изначально считалась клеточной линией рака груди, было однозначно показано, что она происходит от клеточной линии меланомы М14. Соединения 12da, 12fb, 12cb, 11cb и 11fb вместе с другими противо-тубулиновыми средствами, включая колхицин, паклитаксел и винбластин, тестировали на клеточной линии MDR меланомы и ее родительской клеточной линии меланомы (таблица 14). Паклитаксел и винбластин клинически применяются в качестве противораковых лекарственных средств, воздействующих на клеточный тубулин. Хотя колхицин не одобрен FDA в качестве лекарства для лечения рака, его пролекарство ZD6126 проходит клинические испытания против солидных опухолей. Бортезомиб является первым терапевтическим протеазомным ингибитором и был одобрен FDA в 2003 году для применения при множественной миеломе. Известно, что АВТ-751 действует через сайт связывания тубулина колхицина. Он является перспективным лекарственным кандидатом в клинических испытаниях для детей с рецидивной или не поддающейся лечению нейробластомой. Соединения 12da, 12fb, 12cb, 11cb, 11fb имеют значительно лучшие индексы резистентности (3.0 для 12da, 0.9 для 12fb, 1.3 для 12cb, 0.8 для 11cb, 0.7 для 11fb), чем колхицин (65.8), паклитаксел (69.3) и винбластин (27.5). Хотя колхицин, паклитаксел и винбластин показали прекрасную активность на нерезистентных клеточных линиях меланомы (0.5-10 нМ), данные соединения были значительно менее активны на клеточной линии MDR меланомы (277-658 нМ). Напротив, 12cb, 11cb, 11fb имели практически одинаковую активность как на MDR (15 нМ, 38 нМ, 30 нМ, 30 нМ, 35 нМ для 12da, 12fb, 12cb, 11cb и 11fb, соответственно), так и на нерезистентных клеточных линиях меланомы (5 нМ, 41 нМ, 24 нМ, 38 нМ, 50 нМ для 12da, 12fb, 12cb, 11cb и 11fb, соответственно). Соединение 12da было более активно, чем паклитаксел и колхицин, на клетках А375 и WM-164.
|
Представленные в таблице 14 результаты показали, что клеточная линия MDA-MB-435/LCCMDR1 очень устойчива к колхицину, паклитакселу и винбластину. Но ABI по настоящему изобретению показали одинаковую активность в отношении лекарственно-устойчивой клеточной линии и чувствительной клеточной линии. Этот результат подтверждает, что ABI не являются субстратом для P-gp. Таким образом, они преодолевают множественную лекарственную устойчивость, обнаруженную в клетках MDA-MB-435/LCCMDR1. Кривые зависимости доза/ответ показаны на фиг.21 для 12fb, 12da и 12cb.
Пример 16
Анализ in vitro полимеризации микротрубочек
Материалы и методы
Тубулин бычьего мозга (0.4 мг) (Cytoskeleton, Denver, CO) смешивали с 10 мкМ анализируемого соединения и инкубировали в 110 мкл общего тубулинового буфера (80 мМ PIPES, 2.0 мМ MgCl2, 0.5 мМ EGTA и 1 мМ GTP) при рН 6.9. Поглощение при длине волны 340 нм измеряли каждую минуту в течение 15 минут на микропланшетном ридере SYNERGY 4 (Bio-Tek Instruments, Winooski, VT). Температура в спектрофотометре поддерживалась равной 37°С для полимеризации тубулина.
Результаты
Исследовали ингибирование полимеризации Арил-Бензоил-Имидазольными (ABI) соединениями. Тубулин бычьего мозга (>97% чистота) инкубировали с тремя активными ABI соединениями, 12cb, 12da и 12db (10 мкМ) для анализа их действия на полимеризацию тубулина (фиг.22). Полимеризация тубулина полностью ингибировалась соединением 12da, в то время как при инкубировании с соединениями 12cb и 12db наблюдалось ингибирование около 80%.
Такое дестабилизирующее действие на микротрубочки было аналогично таковому для колхицина и винбластина, но было противоположно таковому для паклитаксела. Результаты не только подтвердили, что ABI могут напрямую взаимодействовать с тубулином, но также подтверждают, что они могут связываться с тем же сайтом связывания, что и колхицин (или винбластин).
Пример 17
Ингибирование меланомы in vitro
Материалы и методы
Клетки меланомы B16-F1 с колониеобразующей плотностью (2000 клеток на лунку в 6-луночных планшетах) помещали на поверхность 0.8% агара. Клетки выращивали в 0.4% агаре вместе со средой DMEM с добавлением эмбриональной бычьей сыворотки и раствора антибиотика-противогрибкового средства при 37°С в атмосфере, на 95% состоящей из воздуха и на 5% - из CO2. Клетки обрабатывали соединениями 12da, 12cb и 12fb в различных концентрациях (20, 100 и 500 нМ). Соединения добавляли в среду из 1-мМ стокового раствора в ДМСО, и соответствующее разбавление ДМСО использовали в качестве контроля. Клетки культивировали 14 дней. Планшеты фотографировали, и определяли количество колоний автоматическим счетчиком колоний Artek 880 (Artek Systems Corporation, Farmingdale, NY).
Результаты
Четыре репрезентативных фотографии изображены на фиг.23. После 14 дней инкубирования, в контрольном образце (без добавления веществ) сформировалось около 130 детектируемых колоний (диаметр больше 100 мкм).
Соединения 12cb и 12da эффективно подавляли формирование колоний меланомы B16-F1 даже при самой низкой из протестированных концентраций, 20 нМ (р<0.05 по сравнению с контролем), 12fb показало эффективное подавление при 100 нМ. Все три протестированных соединения показали полное подавление формирования колоний при 0.5 мкМ, дополнительно подтверждая противомеланомную эффективность ABI соединений.
Пример 18
In vivo противоопухолевая активность
Материалы и методы
Животные: самок мышей C57/BL, возраст 4-6 недель, покупали в Harlan Laboratories (Harlan Laboratories Inc., Indianapolis, IN). Условия содержания животных соответствовали требованиям Association for Assessment and Accreditation and Laboratory Animal Care. Все эксперименты проводились в соответствии с рекомендациями Комитета нашего института по содержанию и использованию животных.
Определение эффективности in vivo. Клетки меланомы B16-F1 готовили в среде DMEM без FBS (Cellgro Mediatech) в концентрации 5×106 живых клеток/мл. Суспензию клеток (100 мкл) вводили подкожной инъекцией в правую боковую часть спины каждой мыши. Когда размер опухоли достигал 100-150 мм, примерно через 7 дней после введения клеток, всех мышей с опухолями делили на контрольные и тестовые группы, в зависимости от размеров опухоли (n=5 в группе). Каждая группа имела сходный средний размер опухоли. Мышам в контрольных группах (отрицательный контроль) вводили внутрибрюшинной инъекцией 50 мкл только носителя или DTIC в концентрации 60 мг/кг (положительный контроль) один раз в сутки. Объем опухоли измеряли каждые 2 дня штангенциркулем с электронным цифровым отсчетом (Fisher Scientific, Inc., Pittsburgh, PA) и вычисляли по формуле а×b2×0.5, где а и b представляют собой наибольший и наименьший диаметр, соответственно. Объем опухоли измеряли в кубических миллиметрах. Данные представляли как средние значения ±SE для каждой группы и откладывали на графике как функцию от времени. Процент уменьшения опухоли по окончании эксперимента (14 дней после начала лечения) вычисляли по формуле 100-100×[(Т-Т0)/(С-С0)], где Т это средний объем опухоли тестовой группы в конкретный день, Т0 это средний объем опухоли той же группы в первый день лечения, С это средний объем опухоли контрольной группы в конкретный день, и С0 это средний объем опухоли той же группы в первый день лечения. Активность животных и средний вес тела в каждой группе отслеживали на протяжении всего периода эксперимента для оценки токсичности соединения. По окончании лечения, всех мышей подвергали эвтаназии CO2, с последующим смещением шейных позвонков и извлечением опухолей для дальнейшего изучения.
Результаты
Для оценки эффективности ABI аналогов in vivo авторы протестировали противоопухолевую активность соединения 12cb на мышиной ксенографической модели меланомы B16-F1 в сравнении с DTIC, золотым стандартом в лечении злокачественной меланомы, в качестве положительного контроля (фиг.24а). Двадцать самок мышей C57/BL разбивали на четыре группы: контрольная группа, которой вводили носитель, группа на DTIC (60 мг/кг), группа на 12cb (10 мг/кг), и группа на 12cb (30 мг/кг). Каждой мыши вводили подкожной инъекцией 0.5 миллиона клеток меланомы B16-F1. Через семь дней после инокулирования опухоли начинали лечение, ежедневно вводя внутрибрюшинно каждое тестируемое соединение (фиг.24). Через 14 дней лечения объем опухоли значительно (р<0.05) уменьшался на 47%, 51% и 73% в случае 12cb (10 мг/кг), DTIC (60 мг/кг) и 12cb (30 мг/кг), соответственно. Во время эксперимента ни в одной из групп не наблюдалось заметного снижения веса.
Были выбраны две дозировки - 15 и 45 мг/кг. DTIC в дозировке 60 мг/кг использовали в качестве положительного контроля. Сначала для исследования выбрали аллографическую модель B16-F1 меланомы на мышах C57BL/6. Через 13 дней лечения (фиг.24b), соединение 12fb ингибировало рост меланомной опухоли (значение TGI) на 32% при 15 мг/кг и на 82% при 45 мг/кг. Р-значение t-теста Стьюдента для 12fb при 45 мг/кг по сравнению с контрольной группой было меньше 0.001, указывая на значительное различие. Р-значение t-теста Стьюдента для 12fb при 15 мг/кг по сравнению с контрольной группой составило 0.08, указывая на неэффективность такой дозировки. В сравнении 12fb при 45 мг/кг с DTIC при 60 мг/кг, который показал TGI 51%, р-значение t-теста Стьюдента составило 0.001, указывая на значительно более высокую активность 12fb по сравнению с DTIC. В контрольной группе и группе на 12fb (15 мг/кг), средний вес тела немного увеличивался в ходе эксперимента.
Для дальнейшего подтверждения активности ABI in vivo, использовали ксенографическую модель меланомы человека А375 на SHO мышах, и тестировали 12fb в дозировке 25 мг/кг. В качестве положительного контроля снова применяли DTIC в дозировке 60 мг/кг. После 31 дня лечения (фиг.24с), 12fb ингибировал рост меланомной опухоли (значение TGI) на 69%, в то время как DTIC ингибировал рост на 52%. Р-значение t-теста Стьюдента для группы на 12fb в сравнении с контрольной группой составил менее 0.001, подтверждая, что 12fb заметно ингибировал рост меланомной опухоли в дозировке 25 мг/кг. Р-значение t-теста Стьюдента для группы на 12fb в сравнении с группой на DTIC составило менее 0.05, снова подтверждая, что 12fb имеет более высокую активность, чем DTIC. Средний вес тела во всех группах немного увеличивался в ходе эксперимента. Физическая активность мышей также выглядела нормально, подтверждая, что дозировка 25 мг/кг является хорошо переносимой для SHO мышей.
Пример 19
Связывание с колхицином
Материалы и методы
Каждое испытуемое соединение готовили в 20х концентрации в G-PEM буфере (Cytoskeleton Inc., Denver, CO), затем пипеткой распределяли 10 мкл испытуемого соединения в 96-луночные планшеты. Добавляли в каждую лунку десять микролитров тритий-меченого колхицина (Perkin-Elmer, Waltham, MA). Затем в каждую лунку добавляли 180 мкл суспензии бусины/тубулин (GE Healthcare Bio-Sciences Corp., Piscataway, NJ). Планшет инкубировали 45 мин при 37°С перед считыванием на планшет-ридере Topcount NXT (Perkin-Elmer, Waltham, MA). Немеченый радиоактивным изотопом "холодный" колхицин включали в качестве положительного контроля, и паклитаксел в качестве отрицательного контроля, поскольку паклитаксел связывается с другим сайтом в тубулине и не конкурирует за колхицин-связывающий сайт. Данные обрабатывали в программе GraphPad Prism.
Анализ клеточного цикла
Для изучения распределения по фазам клеточного цикла применяли поточную цитометрию. Клетки А375 выращивали в 10-см чашках для выращивания культур клеток до конфлуэнтности около 80%, и затем клетки обрабатывали 0, 10, 50, 200 и 1000 нМ колхицина, 12da, 12fb и 12cb в течение 24 часов в среде, используемой для роста. ДНК клеток красили PBS, содержащим 50 мкг/мл пропидий иодида и 100 мкг/мл RNase A. Распределение по фазам клеточного цикла анализировали на цитометре BD LSR-II (BD Biosciences, San Jose, CA) при подсчете 10000 клеток. Анализ данных и построение диаграмм осуществляли в программе Modfit 2.0 (Verity Software House, Topsham, ME).
Результаты
Известны три сайта связывания лигандов в α/β-гетеродимере тубулина: сайт связывания паклитаксела, сайт связывания винбластина и сайт связывания колхицина. Измеряли связывание соединения 12cb с применением 3H-меченого колхицина и сцинтилляционного анализа сближения при конкурентном связывании (SPA). Полученные результаты подтвердили сильное связывание 12cb, со связывающей способностью 3.4±1.5 мкМ (фиг.25а). Колхицин в этих условиях связывался с тубулином со значением IC50 1.8±0.5 мкМ. Полученные результаты четко указывают на то, что ABI соединения эффективно ингибируют полимеризацию тубулина.
Диаграмма связывания (фиг.25а) ясно показывает, что ABI могут конкурентно связываться с колхицин-связывающим сайтом тубулина. По мере повышения концентрации трех тестируемых соединений с 0.03 М до 100 мкМ, все большие количества тритий-меченого колхицина конкурентно вытеснялись с тубулина и давали меньшие значения в SPA. Отрицательный контроль, паклитаксел, дал только прямую линию, поскольку теоретически он не должен связываться с колхицин-связывающим сайтом в тубулине. Кроме того, ABI соединения имеют относительно высокое сродство к колхицин-связывающему сайту тубулина. Расчет на GraphPad Prism значений IC50 для связывания показал, что 12da имеет самое высокое связывание. Связывающая способность положительно коррелировала с противо-меланомной активностью in vitro; чем выше сродство к связыванию, тем выше противо-меланомная активность.
ABI соединения показали, что в анализе клеточного цикла они останавливают клетки в фазе G2/M, как указание на то, что они взаимодействуют с тубулином. Соединения 12da, 12fb и 12cb тестировали совместно с колхицином в качестве положительного контроля на клетках А375 (фиг.25b). Четыре различных концентрации - 10, 50, 200 и 1000 нМ - каждого соединения выбрали для демонстрации эффекта дозы (фиг.25С и 25D). Для контрольных образцов (не добавлялось вещество) без перекрывания примерно 16% клеток А375 было распределено в G2/M фазе. Для тестовой группы на колхицине, по мере увеличения концентрации с 10 нМ до 50 нМ, процент клеток, распределенных в G2/M фазе, вырос с 14% до 85%. ABI показали аналогичные результаты на клетках А375, останавливая их в фазе в G2/M дозо-зависимым образом. Активность различных концентраций в остановке клеточного цикла на в G2/M фазе положительным образом коррелировала с их активностью in vitro.
Пример 20
In vitro и in vivo фармакология соединений 17ya, 12fa и 55
Материалы и методы
Культура клеток и анализ цитотоксичности на клетках рака предстательной железы. Все клеточные линии рака предстательной железы (LNCaP, PC-3 и DU145, РРС-1) получали от АТСС (American Type Culture Collection, Manassas, VA, USA). Человеческие PC-3_TxR были устойчивы к паклитакселу и имели модель MDR, сравнимую с РС-3. Культуры клеток покупали в Cellgro Mediatech (Herndon, VA, USA). Все линии клеток применяли для анализа антипролиферативной активности соединений 17ya, 12fa и 55 по методу сульфородамина В (SRB). Все клеточные линии выращивали в среде RPMI 1640 с добавлением 2 мМ глутамина и 10% эмбриональной бычьей сыворотки (FBS).
Анализ полимеризации микротрубочек in vitro. Тубулин свиного мозга (0.4 мг) (Cytoskeleton, Denver, CO) смешивали с 1 и 5 мкМ испытуемых соединений или носителя (ДМСО) и инкубировали в 100 мкл буфера (80 мМ PIPES, 2.0 мМ MgCl2, 0.5 мМ EGTA, рН 6.9 и 1 мМ GTP). Поглощение при длине волны 340 нм отслеживали каждую минуту в течение 15 минут (SYNERGY 4 Microplate Reader, Bio-Tek Instruments, Winooski, VT). Температуру в спектрофотометре поддерживали равной 37°С для полимеризации тубулина.
Метаболическое инкубирование. Исследование метаболической устойчивости проводили инкубированием 0.5 мкМ испытуемых соединений в общем объеме реакционной смеси 1 мл, содержащем 1 мг/мл белков микросом в реакционном буфере [0.2М раствор фосфатного буфера (рН 7.4), 1.3 мМ НАДФ+, 3.3 мМ глюкоза-6-фосфата и 0.4 ед/мл глюкоза-6-фосфат дегидрогеназы] при 37°С при встряхивании в водяной бане. НАДФН-регенерирующую систему (раствор А и В) получали от BD Biosciences (Bedford, МА). Для исследования глюкуронирования, 2 мМ кофактора UDP-глюкуроновой кислоты (Sigma, St. Louis, МО) в деионизованой воде инкубировали с 8 мМ MgCl2, 25 мкг аламетицина (Sigma, St. Louis, МО) в деионизованной воде и НАДФН-регенерирующими растворами (BD Biosciences, Bedford, МА), как описано выше. Общая концентрация ДМСО в реакционном растворе составляла примерно 0.5% (об/об). Аликвоты (100 мкл) из реакционных смесей, используемых для определения метаболической устойчивости, отбирали через 5, 10, 20, 30, 60 и 90 минут. Добавляли ацетонитрил (150 мкл), содержащий 200 нМ внутреннего стандарта, для остановки реакции и осаждения белков. Затем образцы центрифугировали при 4000 g в течение 30 минут при комнатной температуре, и анализировали непосредственно надосадочные растворы методом LC-MS/MS.
Метод анализа. Раствор образца (10 мкл) вводили в ВЭЖХ систему Agilent series (Agilent 1100 Series Agilent 1100 Chemstation, Agilent Technology Co, Ltd). Все аналиты разделяли на narrow-bore С18 колонке (Alltech Alltima HP, 2.1×100 мм, 3 мкм, Fisher, Fair Lawn, NJ). Применяли два градиентных режима. Градиентный режим применяли для разделения аналитов в смесях подвижной фазы A [ACN/H2O (5/95, об/об), содержащей 0.1% муравьиной кислоты] и подвижной фазы В [ACN/H2O (95%/5%, об/об) содержащей 0.1% муравьиной кислоты] при скорости потока 300 мкл/мин. Подвижная фаза А использовалась в количестве 10% с 0 до 1 мин, затем линейный градиент до 100% подвижной фазы В в течение 4 мин, 100% подвижной фазы В выдерживали в течение 0.5 мин, затем быстрый переход на 10% подвижной фазы А. Подвижная фаза А подавалась еще 10 минут до конца анализа.
Использовали трехквадрупольный масс-спектрометр API Qtrap 4000™ (Applied Biosystems/MDS SCIEX, Concord, Ontario, Canada), работающий с источником TurboIonSpray. Напряжение на игле-распылителе составляло 5 кВ в режиме регистрации положительных ионов. Газовую завесу выставляли на 10, Газ 1 и газ 2 выставляли на 50. Газ активированной столкновениями диссоциации (CAD) на значение "medium", a температуру нагревателя образцов - на 500°С. Для наблюдения наиболее чувствительных сигналов применяли режим мониторинга нескольких реакций (multiple reaction monitoring (MRM)), сканируя m/z 378 → 210 (17ya), m/z 373 → 205 (12fa), m/z 410 → 242 (55) и m/z 309 → 171 (внутренний стандарт). Фармакокинетические параметры определяли с применением анализа без учета компартментов (WinNonlin, Pharsight Corporation, Mountain View, CA). Регистрацию данных и количественный обсчет значений проводили с применением программного обеспечения Analyst(TM) software, Ver. 1.4.1 (Applied Biosystems).
Растворимость в воде. Растворимость лекарственных средств определяли на Multiscreen Solubility Filter Plate (Millipore Corporate, Billerica, MA), соединенном с LC-MS/MS. Вкратце, 198 мкл фосфатно-солевого буферного раствора (PBS) (рН 7.4) помещали в 96-луночный планшет, и диспергировали и смешивали с 2 мкл 10 мМ раствора испытуемых соединений (в ДМСО) при аккуратном встряхивании (200-300 об/мин) в течение 1.5 часов при комнатной температуре (N=3). Планшет центрифугировали при 800 g в течение 5 минут, и фильтрат использовали для определения его концентрации и растворимости испытуемого соединения методом LC-MS/MS, как описано ранее.
Исследование фармакокинетики. Самцов ICR мышей (N=3 в группе) возраста 6-8 недель покупали в Harlan Inc. и использовали для исследования фармакокинетики 17ya, 12fa и 55. Все соединения (10 мг/кг) растворяли в ДМСО/PEG300 (1/9) и вводили однократной внутривенной (i.v.) инъекцией (50 мкл) в хвостовую цену. Отбирали образцы крови через 5, 15 и 30 мин, 1, 1.5, 2, 3, 4, 8, 12 и 24 часа после i.v. введения. Мышам принудительно вводили перорально (р.о.) 20 мг/кг (в Tween80/ДМСО/H2O, 2/2/6) каждого испытуемого соединения для оценки их оральной биодоступности. Образцы крови отбирали через 0.5, 1, 1.5, 2, 3, 4, 8, 12 и 24 часа после перорального введения.
Самок крыс Sprague-Dawley (n=3; 254±4 г) покупали в Harlan Inc. (Indianapolis, IN). Торакальные катетеры для яремной вены крыс покупали в Braintree Scientific Inc. (Braintree, MA). После прибытия, животные акклиматизировались в течение 3 дней в комнате с контролируемой температурой (20-22°С) с 12-часовым циклом темнота/свет перед какими либо экспериментами. Соединения 17ya, 12fa и 55 вводили внутривенно (i.v.) через катетеры в яремной вене в дозировке 5 мг/кг (в ДМСО/PEG300, 1/9). Равные объемы гепаринизированного физраствора инъецировали для замены забираемой крови, и отбирали образцы крови (250 мкл) через катетеры в яремной вене через 10, 20, 30 минут, и 1, 2, 4, 8, 12, 24 часа. Все испытуемые соединения принудительно вводили перорально (р.о.) в дозировке 10 мг/кг (в Tween80/ДМСО/H2O, 2/2/6) для оценки их оральной биодоступности. Все образцы крови (250 мкл) после перорального введения отбирали через катетеры в яремной вене через 30, 60, 90 минут, 120 минут, 150 минут, 180 минут, 210 минут, 240 минут, и через 8, 12, 24 часа. Гепаринизированные шприцы и виалы готовили до забора крови. Образцы плазмы готовили центрифугированием образцов крови при 8000 g в течение 5 минут. Все образцы плазмы сразу замораживали и хранили при -80°С до момента анализа.
Аналиты экстрагировали из 100 мкл плазмы 200 микролитрами ацетонитрила, содержащего 200 нМ внутреннего стандарта. Образцы тщательно перемешивали, центрифугировали, и органический экстракт переносили в автосэмплер для LC-MS/MS анализа.
Исследования на PC-3_TxR ксенографтах. PC-3_TxR клетки (10×107 в мл) готовили в среде для роста RPMI 1640, содержащей 10% FBS, и смешивали с Matrigel (BD Biosciences, San Jose, CA) в соотношении 1:1. Вводили опухоли инъекцией 100 мкл полученной смеси (5×106 клеток на животное) подкожно (s.c.) в бок самцов безтимусных мышей возраста 6-8 недель. Измеряли длину и ширину опухолей и вычисляли объем опухоли (мм3) по формуле π/6×L×W2, где длину (L) и ширину (W) измеряли в миллиметрах. Когда объем опухолей достигал размера 300 мм3, животным с опухолями PC-3_TxR начинали введение носителя [Tween80/ДМСО/H2O (2/2/6)] или 17ya (10 мг/кг) перорально. Режим дозирования: 3 раза в неделю в течение 4 недель.
Результаты
Таблица 15. In vitro эффективность 17ya, 12fa и 55 в отношении клеточных линий рака простаты (РС-3) и лекарственно-устойчивой линии (PC-3_TxR) (n=3, среднее ±SE). Паклитаксел использовался в качестве положительного контроля.
|
Соединения 17ya и 55 ингибируют рост линий раковых клеток со множественной лекарственной устойчивостью. Способность 17ya и 55 подавлять рост линий раковых клеток определяли анализом по SRB. Как показано в таблице 15, 17ya и 55 подавляли рост четырех клеточных линий рака предстательной железы, демонстрируя значения IC50 в нижнем наномолярном диапазоне. Полученные данные подтверждают, что оба соединения имеют цитотоксичность, сравнимую с паклитакселом. Кроме того, оценивали также действие 17ya и 55 на линии клеток PC-3 и PC-3_TxR (таблица 15). Оба соединения 17ya и 55 обладали одинаковой активностью в отношении MDR клеток (РС-3_TxR) и родительской клеточной линии (PC-3). Паклитаксел показал 20-кратное значение относительного коэффициент сопротивления. Полученные данные подтверждают, что 17ya и 55 преодолевают лекарственную устойчивость, обуславливаемую P-gp.
17ya и 55 ингибируют полимеризацию микротрубочек. Тубулин свиного мозга (>97% чистота) инкубировали с индивидуальными соединениями 17ya и 55 (1 и 5 мкМ) для анализа их воздействия на полимеризацию тубулина (фиг.26). Соединение 17ya ингибировало полимеризацию тубулина на 13% и 47% при 1 и 5 мкМ, соответственно. Соединение 55 ингибировало полимеризацию тубулина на 11% и 40% при 1 и 5 мкМ, соответственно. 5 мкМ колхицин применялся в качестве положительного контроля и продемонстрировал 32% ингибирования полимеризации тубулина. Полученные данные подтверждают, что 17ya и 55 оба обеспечивают несколько большее ингибирование полимеризации тубулина, чем колхицин, и ингибирование осуществляется дозо-зависимым образом.
Соединение 17ya устойчиво в микросомах печени человека. Соединение 12fa и 55 показывают приемлемую метаболическую стабильность.
|
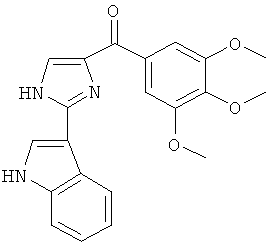
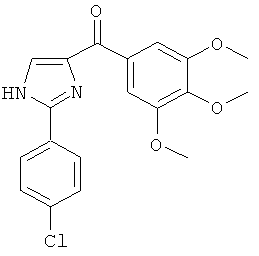
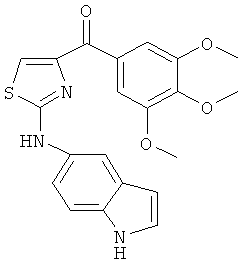
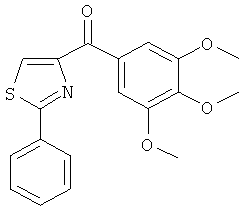
Как показано в таблице 16, 17ya имеет время полужизни 80 минут в реакции фазы I, подтверждая, что соединение 17ya устойчиво в метаболических процессах фазы I. Время полужизни (90 мин) в присутствии UDP-глюкуроновой кислоты было близко ко времени, наблюдаемому в ее отсутствие. Полученные данные подтверждают, что 17ya устойчиво в микросомах печени человека, и можно надеяться, что для человека будут получены низкое значение выведения и большое время полужизни. С другой стороны, время полужизни соединения 55 составило 30 и 43 минуты в присутствии и отсутствии UDP-глюкуроновой кислоты, соответственно. Время полужизни соединения 12fa в фазе I составило 44. Полученные данные подтверждают, что все три соединения показали приемлемую устойчивость в микросомах печени человека, и 17ya более устойчиво, чем 12fa и 55. При исследовании их метаболизма было обнаружено, что 12fa и 55 имеют более высокие уровни восстановления кетона (данные не представлены), подтверждая, что 12fa и 55 более неустойчивы, чем 17ya.
Соединение 17ya продемонстрировало прекрасную растворимость в воде. 12fa и 55 показали приемлемую растворимость.
Соединение 17ya содержит имидазольный цикл, и этот цикл улучшает растворимость в воде, обеспечивая водорастворимость >75 мкг/мл (таблица 16). Соединения 12fa и 55 показали более низкую растворимость в воде, равную 12 и 19 мкг/мл, соответственно. В целом, 17ya продемонстрировало прекрасную растворимость в воде, а 12fa и 55 показали приемлемую растворимость в воде, намного превышающую данное значение для 1h.
Все соединения 17ya, 12fa и 55 продемонстрировали прекрасные фармакокинетические характеристики и биодоступность на мышах и крысах.
Однократную дозу 17ya, 12fa и 55 вводили ICR мышам и Sprague-Dawley крысам. Их фармакокинетические параметры суммированы в таблице 16. In vivo общее значение выведения составило 19, 61 и 40 мл/мин/кг для 17ya, 12fa и 55 на мышах, соответственно. На крысах, их in vivo общее значение выведения составило 9.5, 16 и 10 мл/мин/кг для 17ya, 12fa и 55, соответственно. Соединение 17ya показало низкие значения выведения на мышах и на крысах. Соединения 12fa и 55 также имели низкие значения выведения на крысах, но среднее выведение на мышах. Полученные параметры выведения подтверждают, что все соединения могут преодолевать пресистемный метаболизм и имеют отличные шансы быть орально биодоступными средствами. Средние объемы распределения 2.9 и 1.8 л/кг были получены для 17ya; 4 и 1.9 л/кг были получены для 12fa, 1.3 и 1.0 л/кг были получены для 55 на мышах и крысах, соответственно. Оральная биодоступность составила 36% и 21% для 17ya на мышах и крысах. С другой стороны, 12fa имело оральную биодоступность 62% и 35%, соединение 55 показало оральную биодоступность 34% и 33% на мышах и крысах, соответственно. Полученные данные подтверждают, что все соединения 17ya, 12fa и 55 потенциально могут применяться перорально.
Соединение 17ya ингибирует рост паклитаксел-устойчивых ксенографов простаты. Опухоли паклитаксел-устойчивого рака предстательной железы PC-3_TxR на мышах доводили до объема 300 мм3, после чего мышам с опухолями перорально давали 10 мг/кг 17ya. Как показано на фиг.27, объемы опухолей в контрольной группе увеличивались до 1521±335 мм3 (среднее ±SE) за 13 дней. Мыши в группе на носителе теряли >20% веса тела и были умерщвлены на 13-й день. Объемы опухолей в группе на 17ya немного увеличивались до дня 6. Однако, размеры их опухолей уменьшались до объемов меньше изначального размера опухоли, подтверждая, что в группе на лекарстве достигалась частичная регрессия. Кроме того, вес тела в этой группе возрастал с течением времени, подтверждая, что лекарственное средство не проявляет очевидной токсичности.
Пример 21
Фармакокинетика соединений по настоящему изобретению
|
Пример 22
Биологическая активность 4-замещенных метоксибензоил-арил-тиазолов (SMART): активный ингибитор микротрубочек
Материалы и методы
In vitro анализ полимеризации микротрубочек. Тубулин бычьего мозга (0.4 мг) (Cytoskeleton, Denver, CO) смешивали с 10 мкМ испытуемого соединения или носителя (ДМСО) и инкубировали в 100 мкл буфера (80 мМ PIPES, 2.0 мМ MgCl2, 0.5 мМ EGTA, рН 6.9 и 1 мМ GTP). Поглощение при длине волны 340 нм измеряли каждую минуту в течение 15 минут на микропланшетном ридере (SYNERGY 4, Bio-Tek Instruments, Winooski, VT). Температура в спектрофотометре поддерживалась равной 37°С для полимеризации тубулина.
MS анализ конкурентного связывания. Колхицин, винбластин и паклитаксел (1.2 мкМ для каждого) инкубировали с тубулином (1.2 мг/мл) в инкубационном буфере (80 мМ PIPES, 2.0 мМ MgCl2, 0.5 мМ EGTA, рН 6.9) при 37°С в течение 1 часа. Тестировали 1h (0.5-125 мкМ) на предмет индивидуальной конкуренции с колхицин-, винбластин- и паклитаксел-тубулиновым связыванием. Свободные формы лигандов отделяли от тубулина или микротрубочек методом ультрафильтрования (микроконцентратор) (Microcon, Bedford, МА), отсечка молекул по размеру - 30 кДа. Колхицин, винбластин и паклитаксел определяли методом LCMS/MS. Способность 1h ингибировать связывание лигандов выражали в процентах от контрольного связывания в отсутствие какого-либо конкурента. Каждую реакцию проводили трижды.
Анализ культуры клеток и цитотоксичности в отношении рака предстательной железы и меланомы. Все клеточные линии рака предстательной железы и меланомы получали от АТСС (American Type Culture Collection, Manassas, VA, USA), a культуры клеток покупали в Cellgro Mediatech (Herndon, VA, USA). Изучали антипролиферативную активность соединений на четырех клеточных линиях рака предстательной железы человека (LNCaP, DU145, PC-3 и РРС-1) и двух клеточных линиях меланомы человека (A375 и WM-164). Линия клеток яичника OVCAR-8 и соответствующая ей резистентная клеточная линия, сверхэкспрессирующая P-gp (NCI/ADR-RES), использовались в качестве моделей MDR. Обе линии клеток яичника получали от Национального Института Рака (NCI). Все клеточные линии рака предстательной железы выращивали в 10%-ной эмбриональной бычьей сыворотке (FBS).
Анализ клеточного цикла. Для изучения влияния соединений на распределение по фазам клеточного цикла применяли поточную цитометрию. Клетки РС-3 и А375 обрабатывали в среде для роста указанными концентрациями соединений 1h, 2k, 2l в течение 24 часов. ДНК клеток красили 10 мкг/мл пропидия иодида и 100 мкг/мл RNase A в PBS, и проводили поточную цитометрию клеток для определения распределения по фазам клеточного цикла.
Детектирование апоптоза методом иммуно-ферментного анализа (ELISA), Количественную оценку обогащения цитоплазмы моно- и олиго-нуклеосомами применяли для определения способности соединений вызывать апоптоз (детектирование гибели клеток ELISA PLUS, Roche, Германия) по инструкциям производителя.
Исследование фармакокинетики. Самцов ICR мышей (N=3 или 4 в группе) возраста 6-8 недель покупали в Harlan Inc. и использовали для исследования фармакокинетики полученных соединений. 1h, 2k, 2l (15 мг/кг) растворяли в PEG300/ДМСО (1/4) и вводили однократной внутривенной (i.v.) инъекцией в хвостовую цену. Отбирали образцы крови через 2, 5, 15 и 30 мин, 1, 2, 4, 8, 16 и 24 часа после введения. Самцов крыс Sprague-Dawley (n=4; 254±4 г) покупали в Harlan Inc. (Indianapolis, IN). Соединения 1h, 2k вводили внутривенно через катетеры в яремной вене в дозировке 2.5 мг/кг (в ДМСО/PEG300, 1/4). Отбирали образцы крови (250 мкл) через 10, 20, 30 минут, и 1, 2, 4, 8, 12, 24, 48 часов. Для приготовления образцов применяли метод осаждения белков. Аликвоту (200 мкл) ацетонитрила добавляли к 100 мкл плазмы и интенсивно перемешивали 15 сек на вихревой мешалке. После центрифугирования, надосадочный раствор анализировали методом тандемной жидкостной хроматографии/масс-спектрометрии (LC-MS/MS). Фармакокинетические параметры определяли с применением анализа без учета компартментов (WinNonlin, Pharsight Corporation, Mountain View, CA).
Исследования ксенографтов опухолей PC-3 и A375. PC-3 и A375 клетки (5×107 на мл) готовили в несодержащей фенолов среде для роста, содержащей 10% FBS, и смешивали с Matrigel (BD Biosciences, San Jose, CA) в соотношении 1:1. Опухоли вводили инъекцией 100 мкл смеси (2.5×106 клеток на животное) подкожно (s.c.) в бок 6-8-недельных самцов безтимусных мышей. Измеряли длину и ширину опухолей, и вычисляли объем опухоли (мм3) по формуле π/6×L×W2, где длину (L) и ширину (W) измеряли в миллиметрах. Когда объем опухолей достигал размера 150 мм3, животным с опухолями РС-3 начинали введение носителя [Captex200/Tween80 (1/4)], 1h (5 и 15 мг/кг), 2k (5 и 15 мг/кг) и 2l (50 мг/кг) внутрибрюшинно в течение 21 дня. Винбластин (0.5 мг/кг) применяли в качестве положительного контроля и дозировали дважды в день носителем [ДМСО/PEG300 (1/9)]. С другой стороны, мышам с A375 опухолью 34 дня вводили носитель [Captex200/Tween80 (1/4)], 1h (20 мг/кг) или 2k (15 мг/кг). Дозировки подбирали на основе исследований острой токсичности 1h и 2k на ICR мышах (n=2/группа), показавших, что дозировки до 30 мг/кг и 15 мг/кг, соответственно, не вызывают потери веса тела более чем на 10% после 4 дней внутрибрюшинного введения подряд.
Противоопухолевая активность in vivo [ингибирование роста опухоли (% TIC), задержка роста опухоли (Т-С значение) и частичная гибель опухолевых клеток]. Доказательство лекарственного эффекта описывается следующими параметрами: % Т/С=[[Δ] объем опухоли в группе на лекарстве]/[[Δ] объем опухоли в контрольной группе]×100%. Значения Т-С (задержка роста опухоли) вычисляются по среднему времени (в днях) необходимому для опухолей в группе на лекарстве (Т) и в контрольной группе (С) для достижения определенного размера (600 мм в данном эксперименте). Полученные значения использовали для количественной оценки гибели опухолевых клеток по уравнению: частичная гибель клеток = (Т-С)/(3.32×Td). Td - это время, необходимое для двукратного увеличения объема опухоли (в днях). В настоящем исследовании, авторы определяли время, необходимое для двукратного увеличения объема опухоли с 300 до 600 мм3.
Испытание на вращающемся стержне. ICR мышей два дня тренировали три раза в день удерживаться на вращающемся стержне >120 секунд при скорости вращения 12 об/мин. Затем мышей ранжировали по времени, которое они могли удерживаться на стержне, и распределяли по 7-8 мышей в группе. Внутрибрюшинной инъекцией вводили 1h в дозировке 5 или 15 мг/кг в Captex200/Tween80 (1/4). Винбластин в дозировке 0.5 мг/кг/день использовали в качестве положительного контроля в тех же условиях. Испытание на вращающемся стержне проводили дважды в неделю. Введение лекарства прекращали на 31 день, и последующее наблюдение осуществляли в недели 1, 2 и 4 после окончания введения. Скорость вращения стержня увеличивали с 59 об/мин до 40 об/мин в течение 5 минут. Результат отсчитывали как длину периода времени, в течение которого мышь могла удерживаться на вращающемся стержне.
Исследование лекарственной устойчивости in vivo. По окончании исследований ксенографов РС-3, солидные опухоли из контрольной группы и группы на 1h (15 мг/кг) отделяли и подвергали обработке 0.1% коллагеназой (Тип I) и 50 мг/мл ДНК-азой (Worthington Biochemical Corp., Freehold, NJ). Диспергированные клетки распределяли в планшете в среде RPMI + 10% FBS и инкубировали при 37°С и 5% СО2 в течение 24 часов. Сравнивали анти-пролиферативное действие 1h для определения того, сохраняют ли опухолевые клетки, оставшиеся в PC-3 ксенографах, сензитивность к лекарству. PC-3 клетки, полученные от АТСС, использовали для контроля in vitro. Статистический анализ проводили с использованием простого t-теста.
Результаты
Основываясь на результатах исследования зависимости активности от структуры, выбирали три соединения (фиг.28А) для биологического исследования. В то время как 1h и 2k представляют собой высокоактивные молекулы с цитотоксичностью в низкой наномолярной области, соединение 2l, которое было рационально создано как потенциальный метаболит с улучшенной растворимостью, имело меньшую анти-пролиферативную активность (таблица 18).
|
SMART-соединения ингибируют полимеризацию микротрубочек путем связывания с колхицин-связывающим сайтом в тубулине.
Тубулин бычьего мозга (>97% чистота) инкубировали с индивидуальными соединениями (10 мкМ) для анализа их действия на полимеризацию тубулина (фиг.28в). В то время как 1h и 2k ингибировали полимеризацию тубулина на 90%, 2l ингибировало полимеризацию только на 55%. Более ранние исследования продемонстрировали концентрационно-зависимое ингибирование полимеризации тубулина соединением 1h. Кроме того, в тех же условиях значение IC50 для 1h (4.23 мкМ) близко к таковому для колхицина (4.91 мкМ). Полученные данные подтверждают, что указанные соединения оказывают сильное отрицательное действие на полимеризацию тубулина, что хорошо соответствует их цитотоксичности (таблица 18). Способность данных соединений конкурировать за известные сайты связывания в тубулине определяли по новой методике MS конкурентного связывания, разработанной в лаборатории авторов. Три тубулиновых лиганда, соответствующих трем сайтам связывания в тубулине - колхицин, винбластин и паклитаксел - применяли для исследования конкурентного связывания. Было обнаружено, что в диапазоне концентраций 0.1-125 мкМ, соединение 1h специфически конкурировало с колхицином за связывание с тубулином, но не конкурировало за связывание с тубулином с винбластином и паклитакселом (фиг.28С).
SMART-соединения ингибируют рост клеточных линий с множественной лекарственной устойчивостью.
Способность соединений ингибировать рост линий раковых клеток оценивали с помощью SRB анализа. Как показано в таблице 18, соединения ингибировали рост нескольких линий раковых клеток человека, включая четыре клеточных линии рака предстательной железы и две клеточные линии меланомы, показывая значения IC50 в нижнем наномолярном диапазоне. Из указанных трех соединений 2l было наименее активным (IC50 76-116 нМ). 2k продемонстрировало наилучшее анти-пролиферативное действие, показав значение IC50 от 6 до 43 нМ для клеточных линий рака предстательной железы и меланомы. Кроме того, оценивали также влияние указанных соединений на клеточные линии OVCAR-8 и NCI/ADR-RES (таблица 18). Соединения показали равную активность в отношении MDR клеток (NCI-ADR-RES) и родительской клеточной линии (OVCAR-8). Паклитаксел, винбластин и колхицин показали 1333-, 149- и 65-кратные значения относительной устойчивости, соответственно (таблица 18). Полученные данные указывают на то, что для описанных соединений не развивается лекарственная устойчивость, обусловленная P-gp.
SMART-соединения останавливают клетки РС-3 (предстательная железа) и А375 (меланома) на G2/M фазе клеточного цикла и индуцируют апоптоз клеток. Клетки РС-3 и А375 подвергали воздействию 10, 50, 200 и 1000 нМ соединений в течение 24 часов. Обработка SMART соединениями приводила к концентрационно-зависимому аккумулированию клеток PC-3 и А375 в фазе G2/M, сопровождающемуся уменьшением процента клеток в фазе G0/G1 (фиг.29А и 29В). Доля клеток в фазе G2/M заметно возрастала при обработке соединениями 1h, 2k, 2l в концентрациях от 50 до 200 нМ. Затем исследовали апоптоз, измеряя уровень ДНК-гистоновых комплексов в цитоплазме в клетках PC-3 и А375 после 24-часовой обработки. Повышение концентрации SMART соединений увеличивало концентрацию ДНК-гистоновых комплексов в цитоплазме в клетках PC-3 и А375 (фиг.29С). Данный эффект был более выражен на клетках А375, чем на PC-3, но апоптоз был очевиден для обоих типов клеток, 1h и 2k вызывали умеренный апоптоз в концентрации 50 нМ, а 2l вызывало апоптоз только при концентрациях 200 нМ или выше.
In vivo фармакокинетический профиль SMART-соединений. Однократную дозу каждого соединения (15 мг/кг) вводили инъекцией в хвостовую вену ICR мышам для исследования их фармакокинетики (фиг.30А). 1h и 2k показали сходные фармакокинетические характеристики, но для 2l была зафиксирована большая площадь под кривой (AUC), чем для 1h и 2k, что указывает на более низкое выведение 2l (таблица 19). 2l имел также значение Vss в 2-3 раза большее, чем 1h и 2k. Значения выведения для всех трех соединений были равны или превосходили 90 мл/мин/кг, скорость потока крови через печень у мышей, подтверждая, что помимо выведения через печень в выведении данных соединений могут быть задействованы другие пути деградации. Также исследовали фармакокинетику 1h и 2k (2.5 мг/кг) на крысах (фиг.30В). Интересно, что для обоих соединений были получены низкие значения выведения и скорости экстракции в печени, подтверждая, что данные соединения обладают видовой специфичностью в показателях выведения. На крысах соединение 1h показывало благоприятные фармакокинетические характеристики, которыми являются низкое выведение (6 мл/мин/кг), умеренный объем распределения (7.6 л/кг), большое время полужизни (24 ч) и высокая концентрация (AUC, 5.8 ч*мкг/мл) (таблица 19) при внутривенном введении.
|
SMART-соединения ингибируют рост ксенографов предстательной железы и меланомы без проявления нейротоксичности. Опухоли рака предстательной железы РС-3 и меланомы А375 у мышей доводили до объема 150 мм3, и затем мышам с развившимися опухолями вводили SMART-соединения. Как показано на фиг.31А, объемы опухолей в контрольной группе увеличились до 680 мм3 за 21 день эксперимента. Объемы опухолей у группы 1h выросли до 370 мм3 (5 мг/кг) и 176 мм3 (15 мг/кг) к 21-му дню, что указывает на сильную противоопухолевую активность данного соединения. Опухоли у животных группы 2k выросли до 269 мм3 (5 мг/кг) и 292 мм3 (15 мг/кг), а у животных группы 2l (50 мг/кг) объем опухолей составлял 331 мм3 к 21-му дню. Такое уменьшение объема опухолей менялось в противоположном направлении после отмены SMART-соединений (данные не представлены). В таблице 20 суммированы данные по in vivo эффективности (% Т/С, Т-С значения и частичная гибель клеток) для SMART-соединений.
|
Соединение 1h обеспечило % Т/С=29% и 4% при дозировке 5 и 15 мг/кг (все дозировки вводили внутрибрюшинно (i.p.)), соответственно, в то время как 2k обеспечило % Т/С равный 21% и 24% при дозировке 5 и 15 мг/кг, соответственно. Для высокой дозировки 2l (50 мг/кг) значение % Т/С составило 34%. Винбластин, положительный контроль, показал % Т/С 29% на 22-й день в РС-3 ксенографтах (фиг.31В). Измерения веса тела, проводимые для мониторинга токсичности, показали, что только 1 из 8 мышей на 1h (15 мг/кг), и 2 из 7 мышей на 2k (15 мг/кг) потеряли более 15% веса. Помимо противоопухолевого действия соединений на PC-3 опухоли предстательной железы, 1h (20 мг/кг) и 2k (15 мг/кг) обеспечили значительное уменьшение А375 опухолей. Как показано фиг.31С, объемы опухолей контрольной группы увеличились до 2183 мм3, в то время как 14 объемов в группах 1h и 2k выросли до 775 мм3 и 722 мм3, соответственно. Лечение соединениями 1h и 2k дало значения % Т/С 28% и 29%, соответственно. Проводили испытания на вращающемся стержне для оценки in vivo нейротоксичного действия соединения 1h. На основе результатов испытаний эффективности in vivo, были выбраны дозировки 5 или 15 мг/кг [i.p. введение, Captex200/Tween80 (1/4)] соединения 1h для изучения влияния на координацию движений. Введение 0.5 мг/кг винбластина использовали как положительный контроль в тех же условиях. Как показано на фиг.31D, винбластин плавно уменьшал время (в секундах), которое мышь могла удерживаться на вращающемся стержне, и данные приобретали статистическую значимость к 27-му или 31-му дню (р<0.05) в сравнении с группой на плацебо. Однако не было обнаружено значительной разницы с группой 1h, подтверждая, что 1h не оказывает нейротоксичного действия на ICR мышей в дозировках, обеспечивающих противоопухолевое действие.
В случае 1h не развивается лекарственной устойчивости у мышей с опухолью РС-3. Авторы вырезали РС-3 опухоли у безтимусных мышей после 21 дня введения носителя (n=3) или 15 мг/кг 1h (n=3). Солидные опухоли измельчали и диспергировали на клетки, как описано в разделе, посвященном методам. В качестве контроля использовали РС-3 клетки от АТСС (American Type Culture Collection, Manassas, VA, USA). Значения IC50 составили 29.1±1.1, 29.1±0.8 и 30.4±0.5 рМ для клеток PC-3 от АТСС, и диссоциированных клеток опухоли на носителе и на 1h, соответственно. Полученные данные показывают, что соединение 1h не вызывало развития лекарственной устойчивости в РС-3 опухолях после 21 дня непрерывного введения 1h.
Пример 23
Молекулярное моделирование
Методы
Все эксперименты по молекулярному моделированию проводили в программе Schrodinger Molecular Modeling Suite 2008 (Schrodinger LLC, New York, NY) на рабочей станции Dell Linux. Поскольку размер ABI соединений намного ближе к размеру АВТ-751, чем к DAMA-колхицину, авторы выбрали комплекс тубулина с АВТ-751 (PDB код: 3КНС) в качестве модельной системы. Структуры ABI строили и подготавливали с использование модуля Ligprep, и затем проводили их локирование в АВТ-751 сайт с использованием модуля Glide в Schrodinger Suite. Для наилучших локированных комплексов проводили оптимизацию методом молекулярной динамики, с ограничениями с помощью модуля Macromodel с силовым полем OPLS-2005, чтобы освободить их от всех напряжений. Лигандам и окружающим его фрагментам в пределах 15Ǻ позволяли двигаться свободно, а остатки за пределами радиуса в 15Ǻ делали жесткими.
Результаты
Изучали молекулярное моделирование связывания ABI соединений в тубулине. Несколько структур кристаллов комплекса лиганд-тубулин доступно в базе данных PDB, самая недавняя из которых описана Dorleans и др. В целом, колхицин-связывающий карман подходит для разнообразных молекулярных структур, что может указывать на значительные изменения конформации при связывании с лигандом. На самом деле, Dorleans и др. расшифровали кристаллические структуры и для пустого димера тубулина, и для комплекса лиганд-тубулин. Они обнаружили, что в отсутствие лиганда петля 7 (Т7, остатки 244-251, фиг.32) в бета-мономере складывается, занимая карман связывания, в то время как при связывании лиганда она разворачивается. Связанные с ней спираль 7 (Н7, остатки 224-243) и спираль 8 (Н8, остатки 252-260) смещались при связывании лиганда. Предположительно, степень смещения Т7 зависит от размера индивидуального лиганда. Такая гибкость обуславливает значительные трудности в понимании точной модели связывания для индивидуальных лигандов без расшифровки их истинных кристаллических структур. Тем не менее, аккуратный анализ возможных моделей связывания может пролить свет на связывание различных лигандов.
Модели связывания соединений 12cb и 11cb (стержневая модель) изображены на фиг.32А и 32В. Для сравнения, на фиг.32А изображены кристаллические структуры комплексов с АВТ-751 и DAMA-колхицином (каркасные модели) вместе с комплексом ABI-12cb/тубулин. Для ясности на фиг.32А представлены только вторичные структуры, принимающие участие в формировании кармана связывания в β-тубулине. Суммарные структуры 12cb, АВТ-751 и DAMA-колхицина хорошо вписываются в карман связывания. Были выявлены несколько потенциальных взаимодействий по типу водородных связей между соединением 12cb и тубулином. Карбонильная группа в 12cb находилась в достаточной близости для того, чтобы образовать две водородных связи с NH-фрагментом Leu-252 в Н8 и боковой цепью Asp-251 в Т7 β-мономера тубулина. Пара-фторный заместитель в С-цикле расположен близко к боковой цепи Cys241 в Т7 и Tyr202 в S6, возможно образуя одну или две водородных связи. Имидазольный протон расположен очень близко и, скорее всего, образует водородную связь с Thr179 в петле Т5 (остатки 173-182) α-мономера тубулина (фиг.32А). Вместе с гидрофобными взаимодействиями, обуславливаемыми ароматическими циклами, предположительное формирование указанных водородных связей вносит вклад в высокую способность к связыванию с тубулиновым димером, обеспечивая в результате высокую анти-пролиферативную активность.
Модель связывания соединения 11cb менее определенная, поскольку два из трех ароматических колец могут занимать карман связывания в β-мономере, а третье кольцо может быть направлено к границе соприкосновения α/β-мономеров, аналогично связыванию боковой цепи DAMA-колхицина. Построенная модель показывает, что защитная группа, скорее всего, направлена к границе соприкосновения димера тубулина, в то время как циклы А и С соединения 11cb занимают аналогичный карман связывания и имеют ту же ориентацию, как в 12cb (фиг.32В). Это может объяснять близкую активность двух данных соединений, несмотря даже на наличие в 11cb дополнительной циклической системы. Судя по исследованию молекулярных моделей, представленных на фиг.32А и 32В, донором водородной связи скорее всего является тиольная группа в Cys-241 в петле 7 β-субъединицы α/β-тубулинового димера.
Строили модель связывания ABI 12fb (не показано) и сравнивали с DAMA-колхицином (структуру колхицина см. на фиг.19) в α/β-тубулиновом гетеродимере. Общие структуры 12fb и DAMA-колхицина перекрываются очень хорошо. Пара-фторфенильный фрагмент перекрывается с триметоксифенильным фрагментом, который взаимодействует с Т7 петлей в β-субъединице. Аналогичным образом, пара-хлорфенильный фрагмент занимает другую сторону кармана, где находится 7-членный цикл DAMA-колхицина, при этом атом хлора занимает карман, в котором наблюдается взаимодействие метокси-фрагмента.
Пример 24
Визуализация микротрубочек
Материалы и методы
Для визуального подтверждения взаимодействия ABI с тубулином в клетках применяли набор Cellomics Cytoskeleton rearrangement kit (Thermo Scientific, Rockford, IL). Клетки WM-164 меланомы обрабатывали каждым соединением в течение 18 часов в двух повторах, используя 96-луночный планшет, покрытый коллагеном (Becton Dickinson Labware, Bedford, MA). Затем клетки фиксировали 4%-ным параформальдегидом (Thermo Scientific, Rockford, IL), и делали мембраны проницаемыми с помощью пермеабилизационного буфера из упомянутого выше набора. К клеткам последовательно добавляли первичное антитело и вторичное антитело с флуоресцентной меткой. Ядра клеток окрашивали флуоресцентным красителем DAPI. Все клетки также окрашивали красителем Whole Cell Stain Green. Все изображения получали с помощью инвертированного флуоресцентного микроскопа Olympus IX71 (Olympus Corp., Tokyo, Japan) с наложением отдельных изображений тубулина (красный), ядер (голубой) и целых клеток (зеленый). Для сравнения, наряду с ABI соединениями, включены паклитаксел, колхицин и АВТ-751.
Исследовали визуальные доказательства взаимодействия ABI с тубулином внутри клеток. Расположение микротрубочек в клетках WM-164 меланомы человека при обработке различными соединениями изображено на фиг.33. Изображения микротрубочек ясно показывают, что все пять протестированных соединений приводят к перераспределению микротрубочек. Наблюдалась значительная разница между паклитакселом и остальными четырьмя соединениями (колхицин, АВТ-751, 12cb и 12da). Обработка паклитакселом приводила к конденсации микротрубочек, упорядочение лежащих вокруг ядра, по сравнению с контролем, в соответствии с механизмом его действия, стабилизирующим микротрубочки. Напротив, обработка колхицином, АВТ-751, 12cb и 12da оказывала на микротрубочки схожее действие и приводила к некоторой фрагментации микротрубочек, в соответствии с их общим механизмом действия, дестабилизирующим микротрубочки. Полученные результаты подтверждали также, что ABI соединения имеют общую клеточную мишень с колхицином и оказывают аналогичное действие на клетки.
Все отличительные признаки, описанные в настоящем тексте (включая все пункты прилагаемой формулы изобретения, реферат и чертежи) и/или все стадии любого способа или процесса, описанного в настоящем тексте, могут комбинироваться с любым из описанных выше аспектов в любой комбинации, за исключением комбинаций, в которых по меньшей мере некоторые из таких отличительных признаков и/или стадий являются взаимоисключающими. Хотя в настоящем тексте были подробно описаны или изображены предпочтительные варианты осуществления, квалифицированному специалисту в данной области будет понятно, что могут применяться и их различные модификации, добавления, замены и т.п. без выхода за рамки объема настоящего изобретения, и поэтому они должны рассматриваться как входящие в объем настоящего изобретения, определенный в следующей далее формуле изобретения.
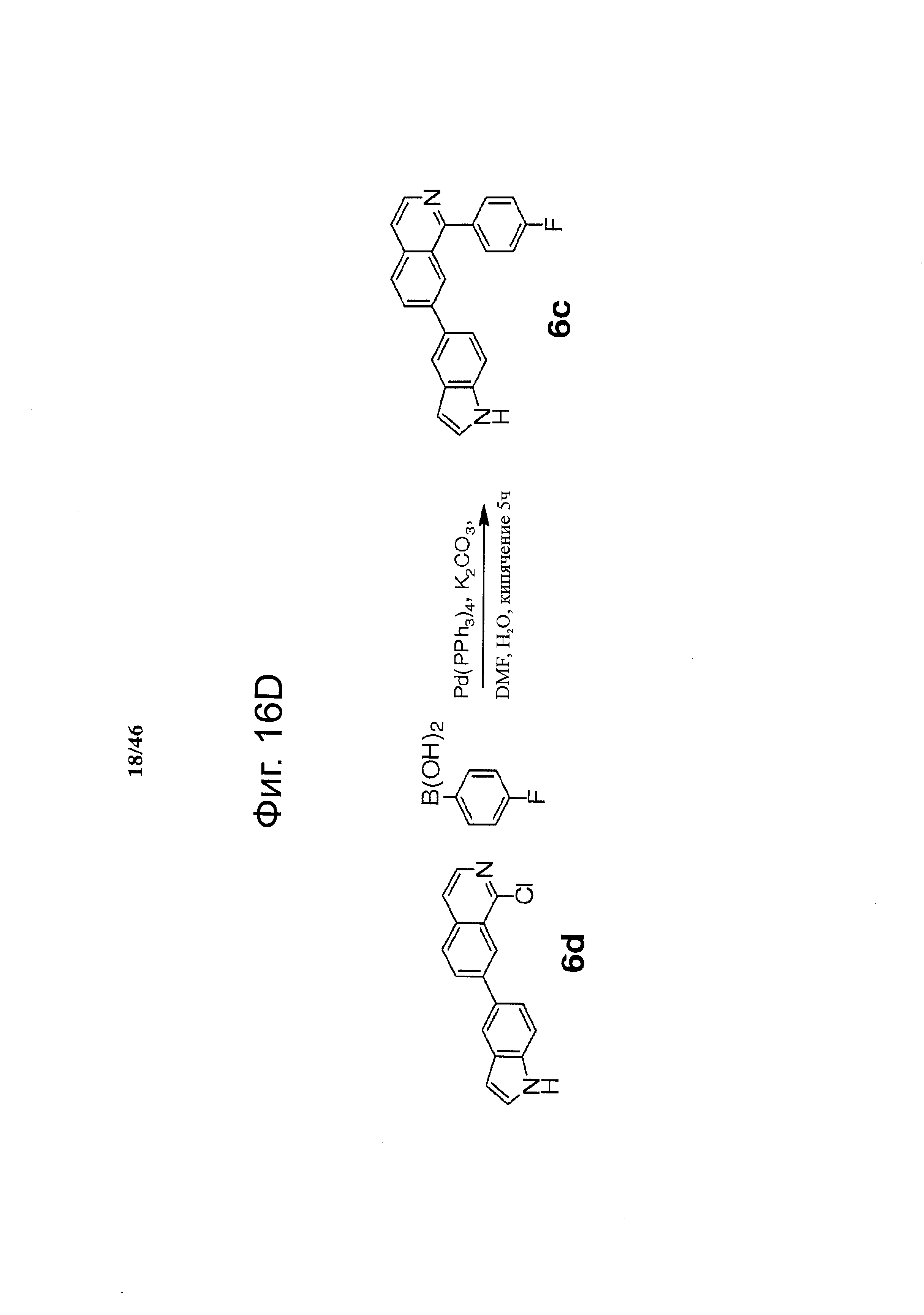
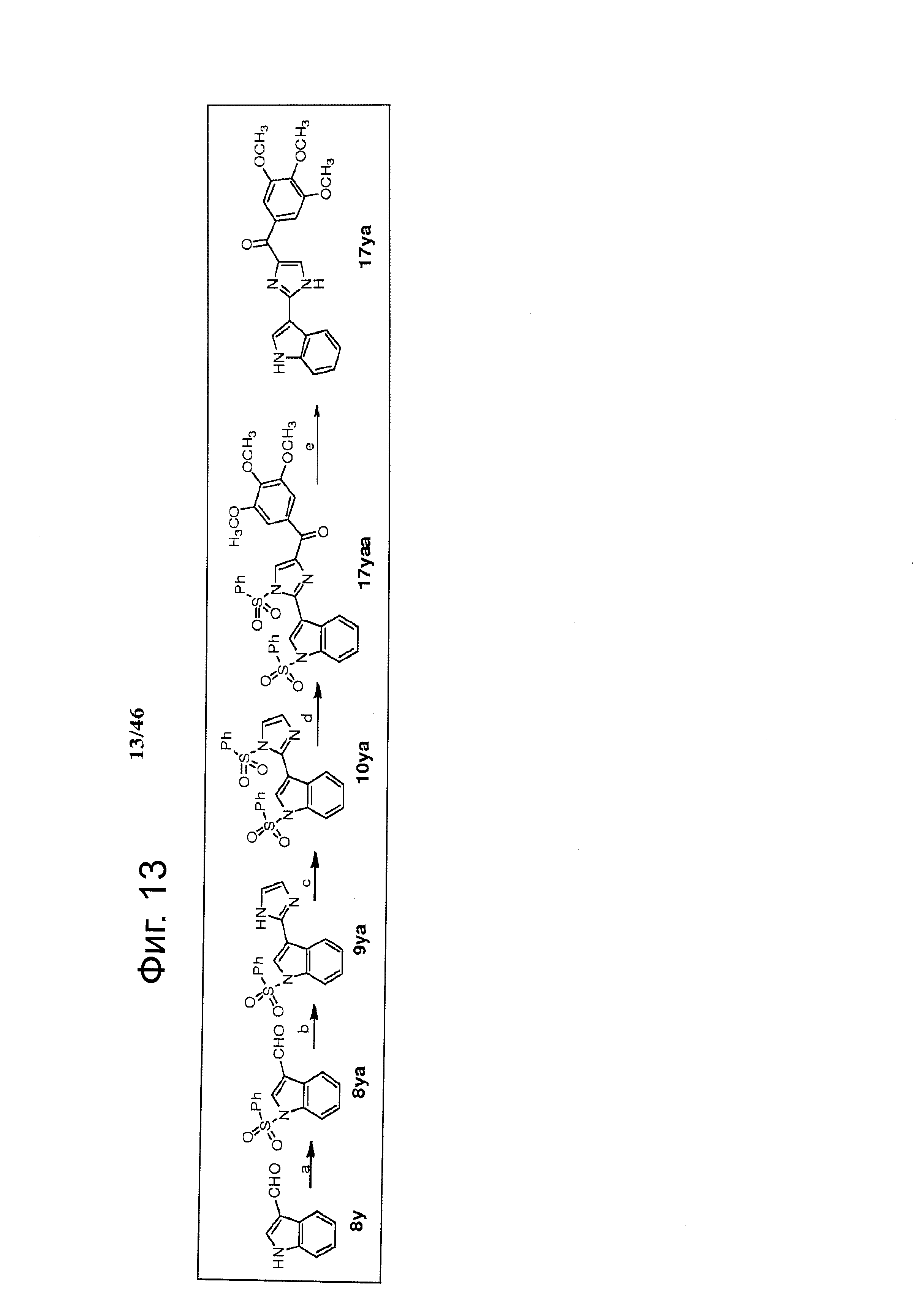
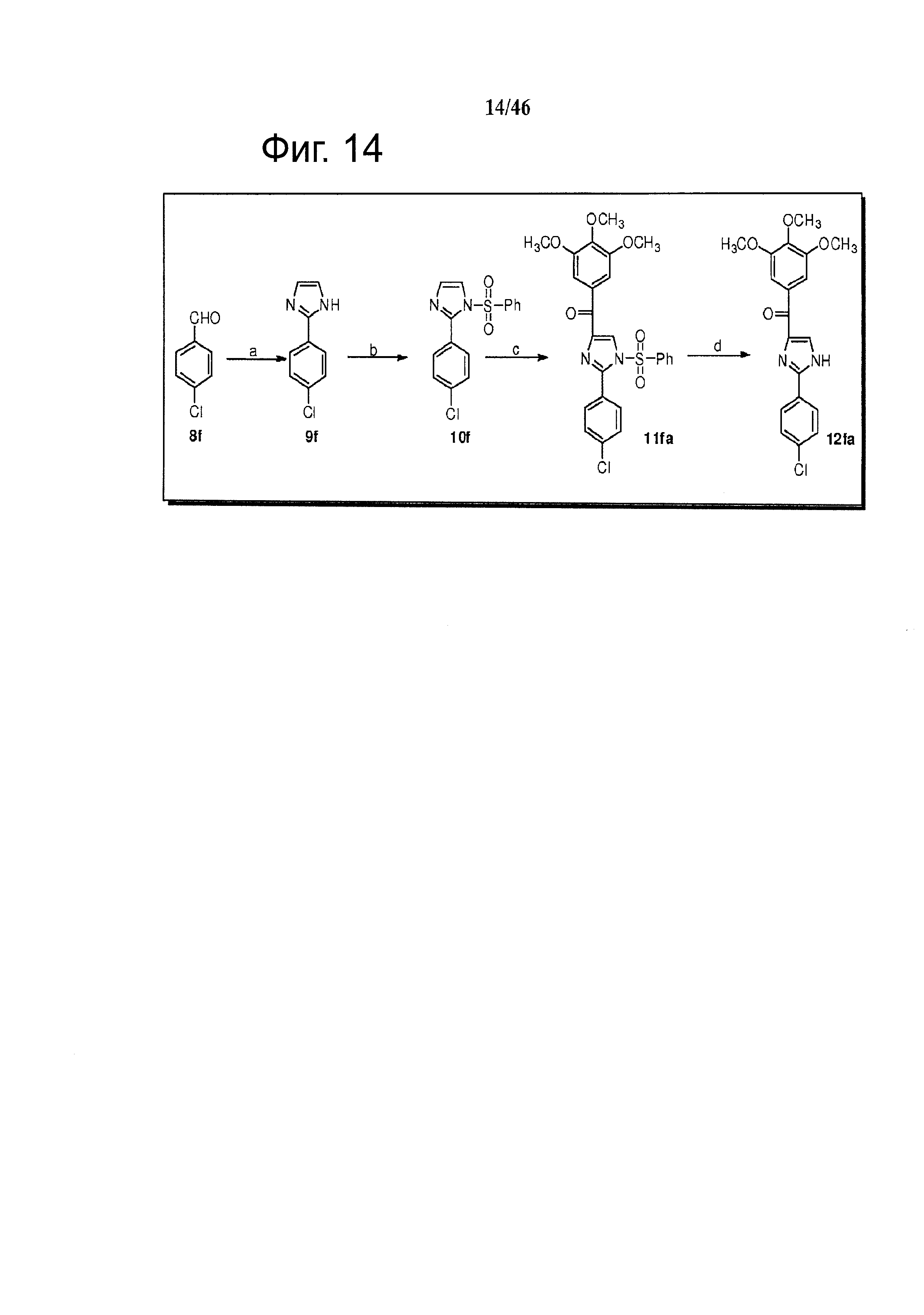
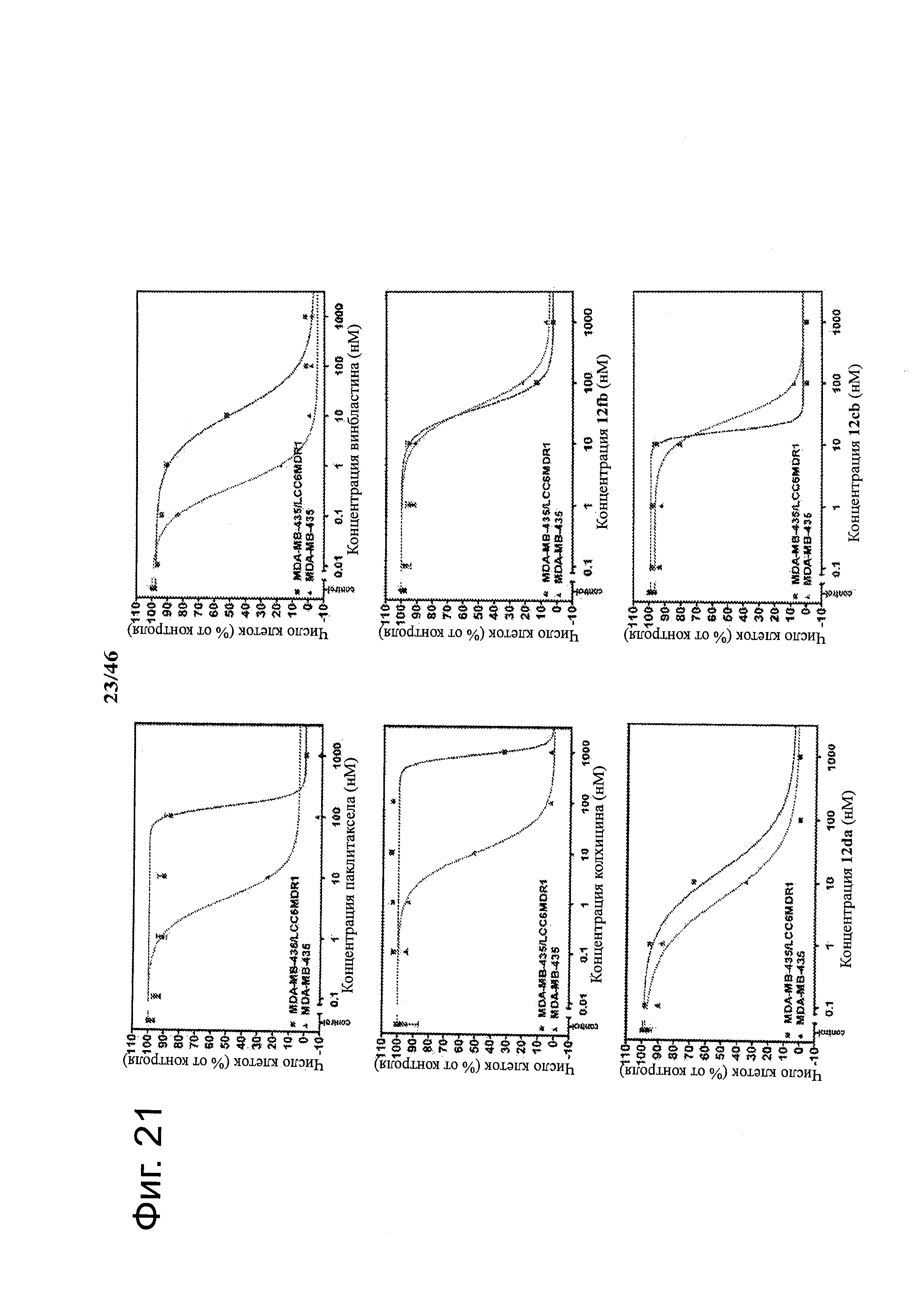
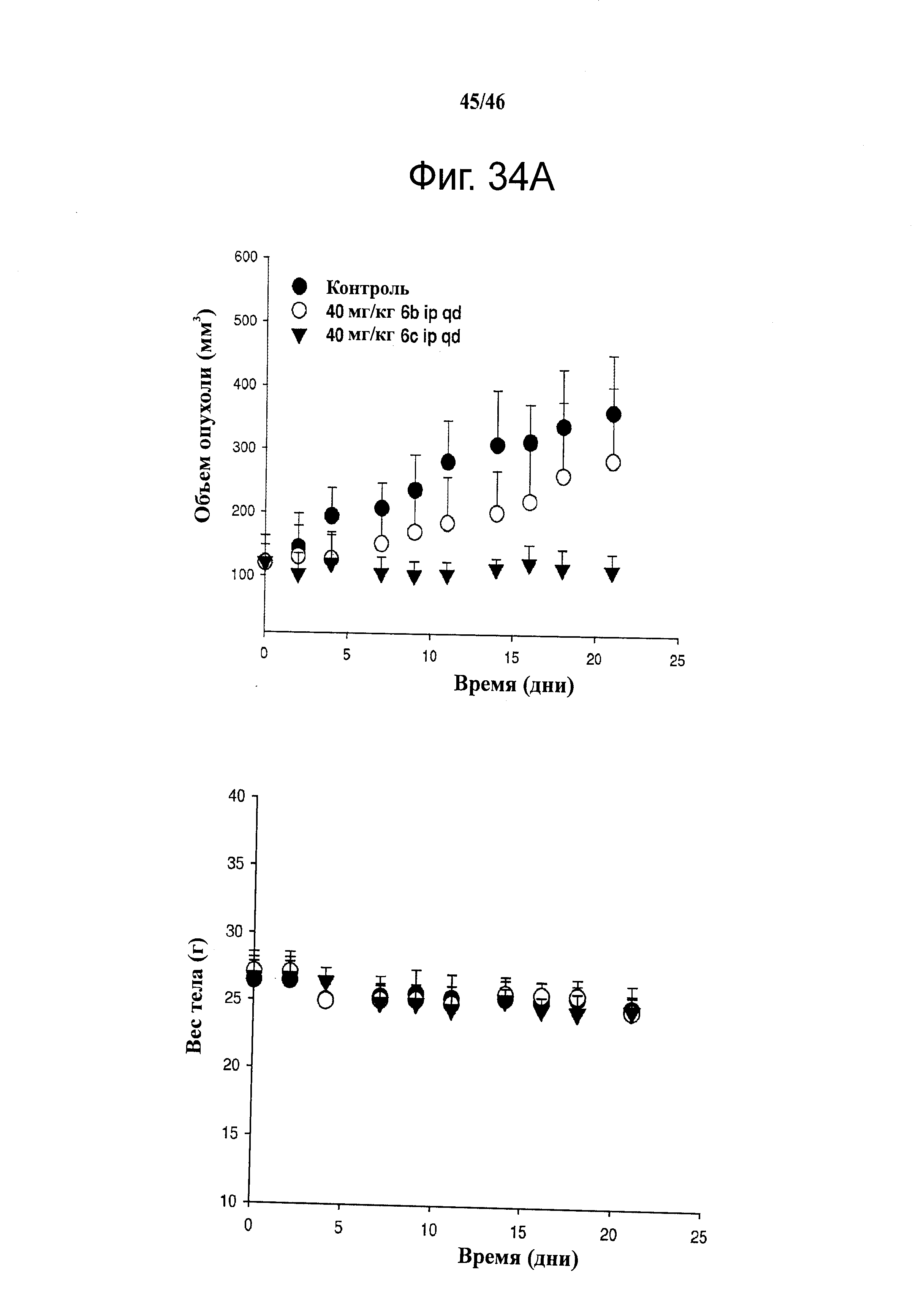
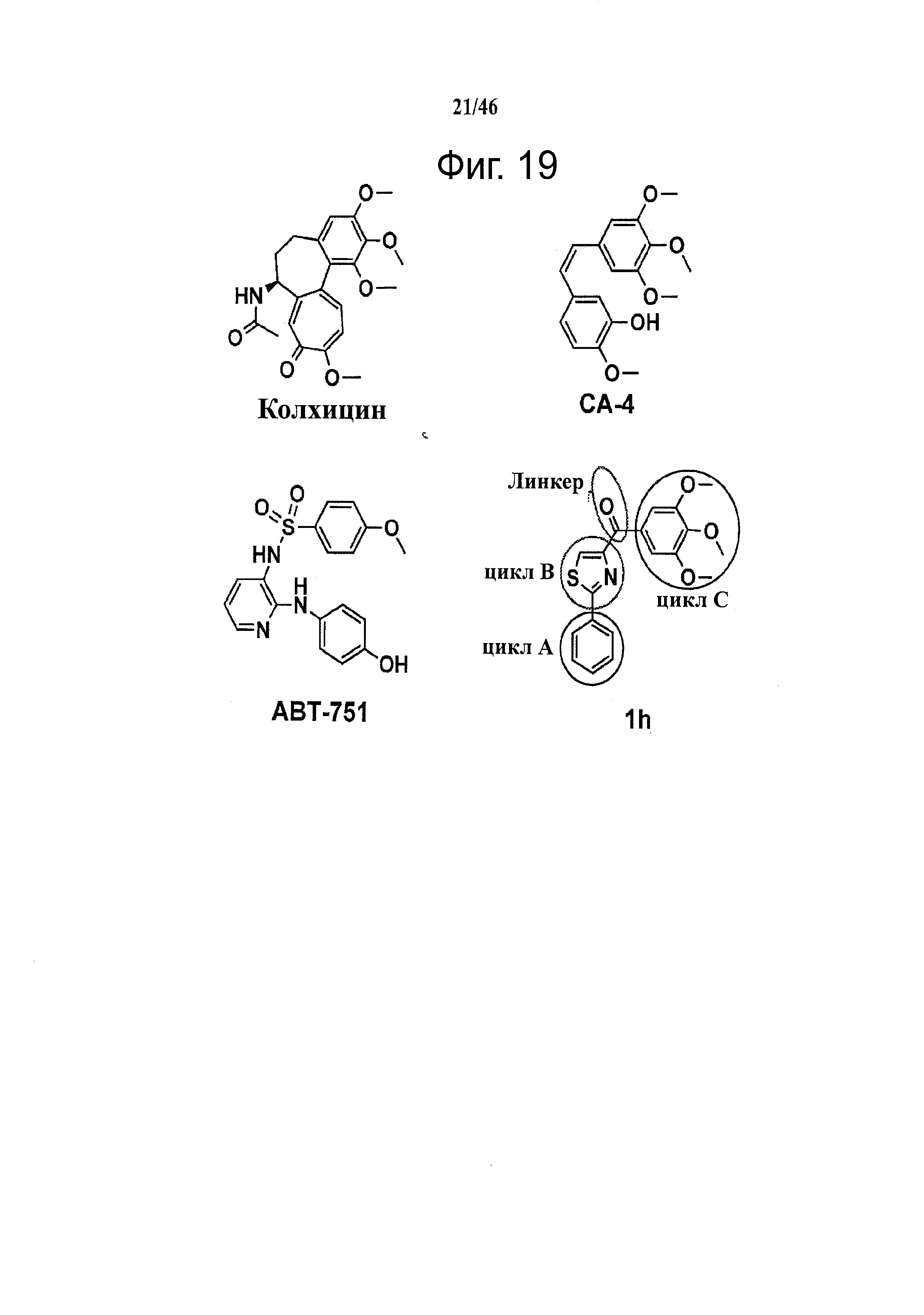
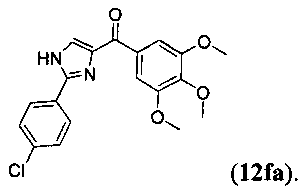

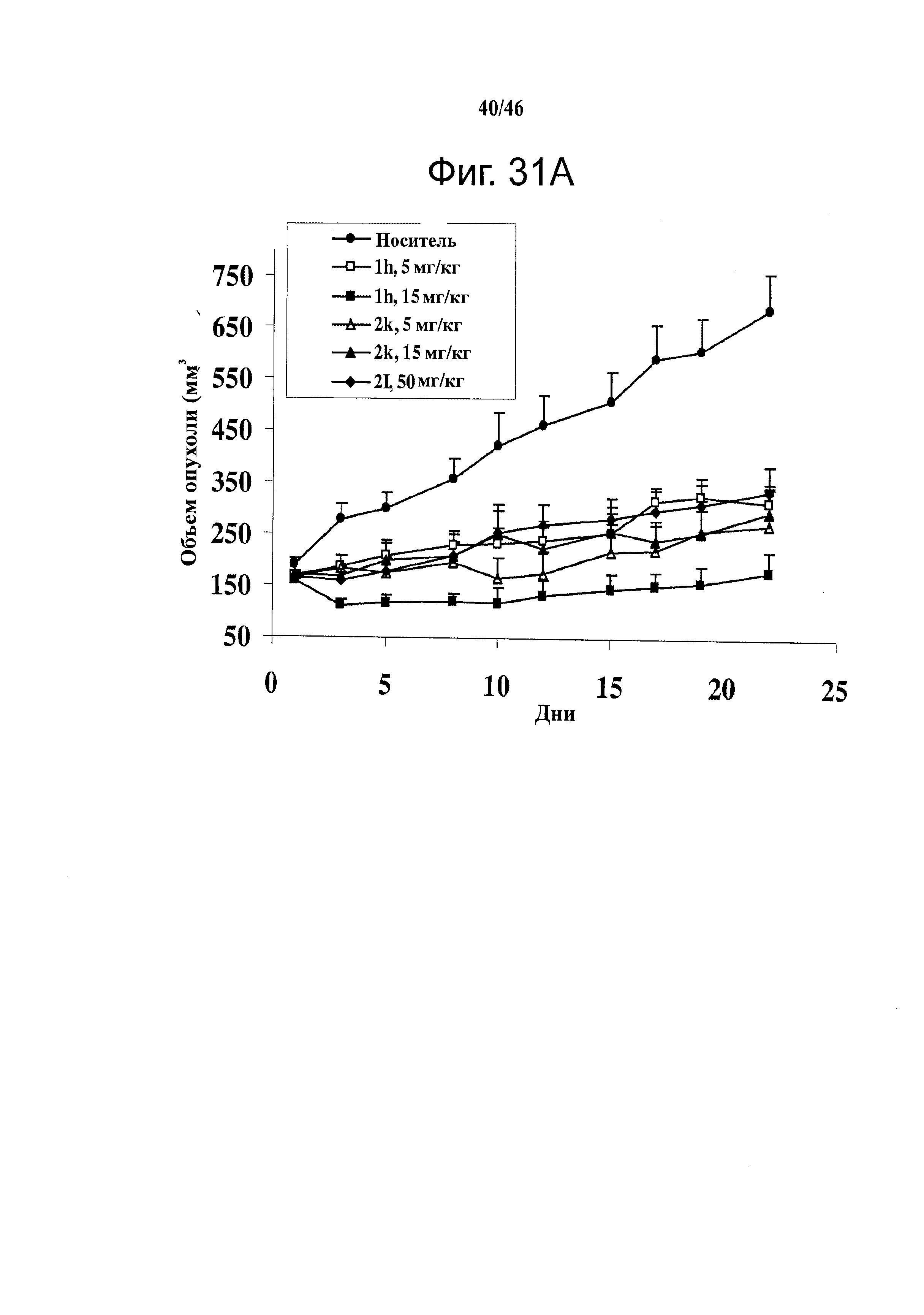
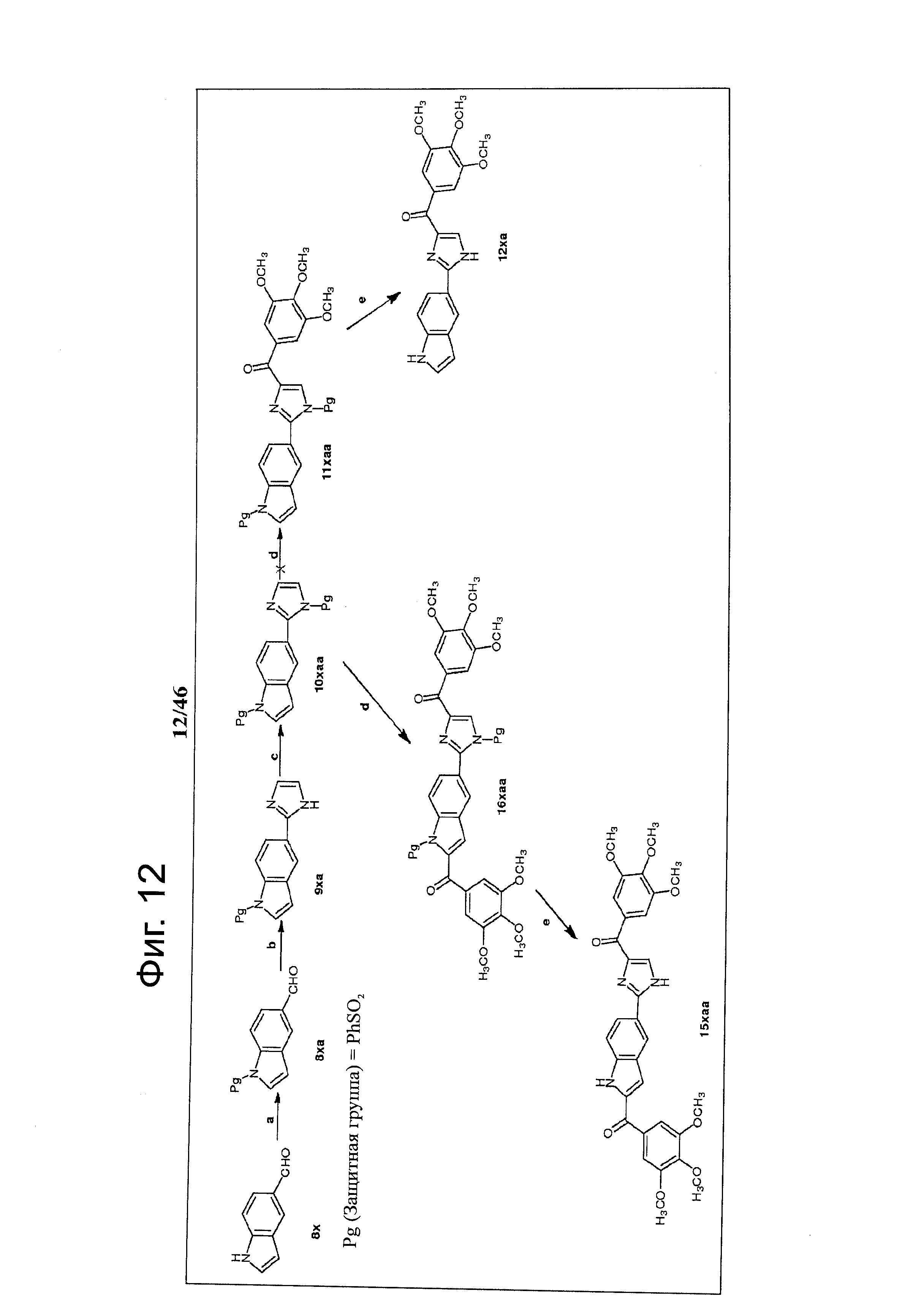
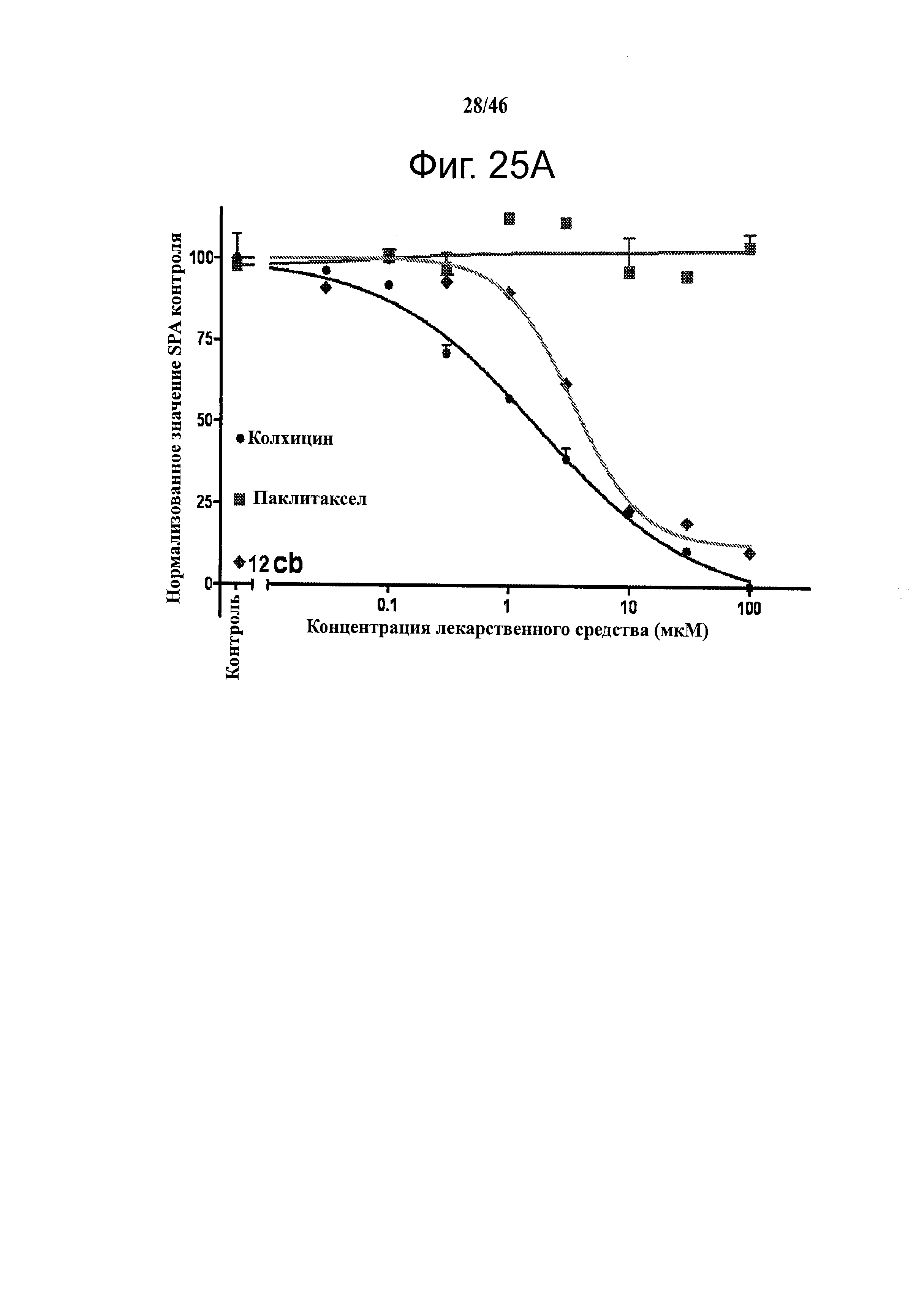
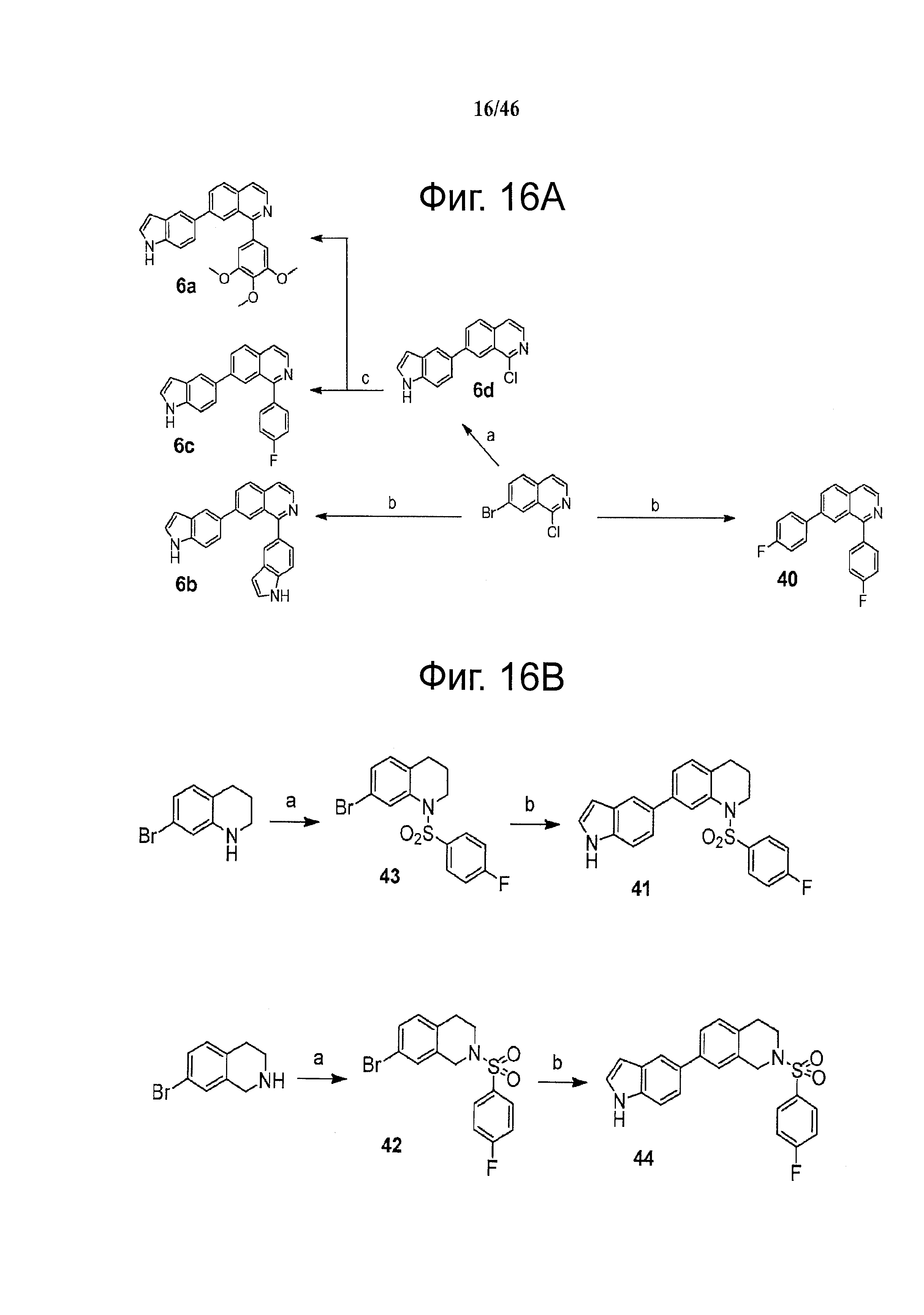
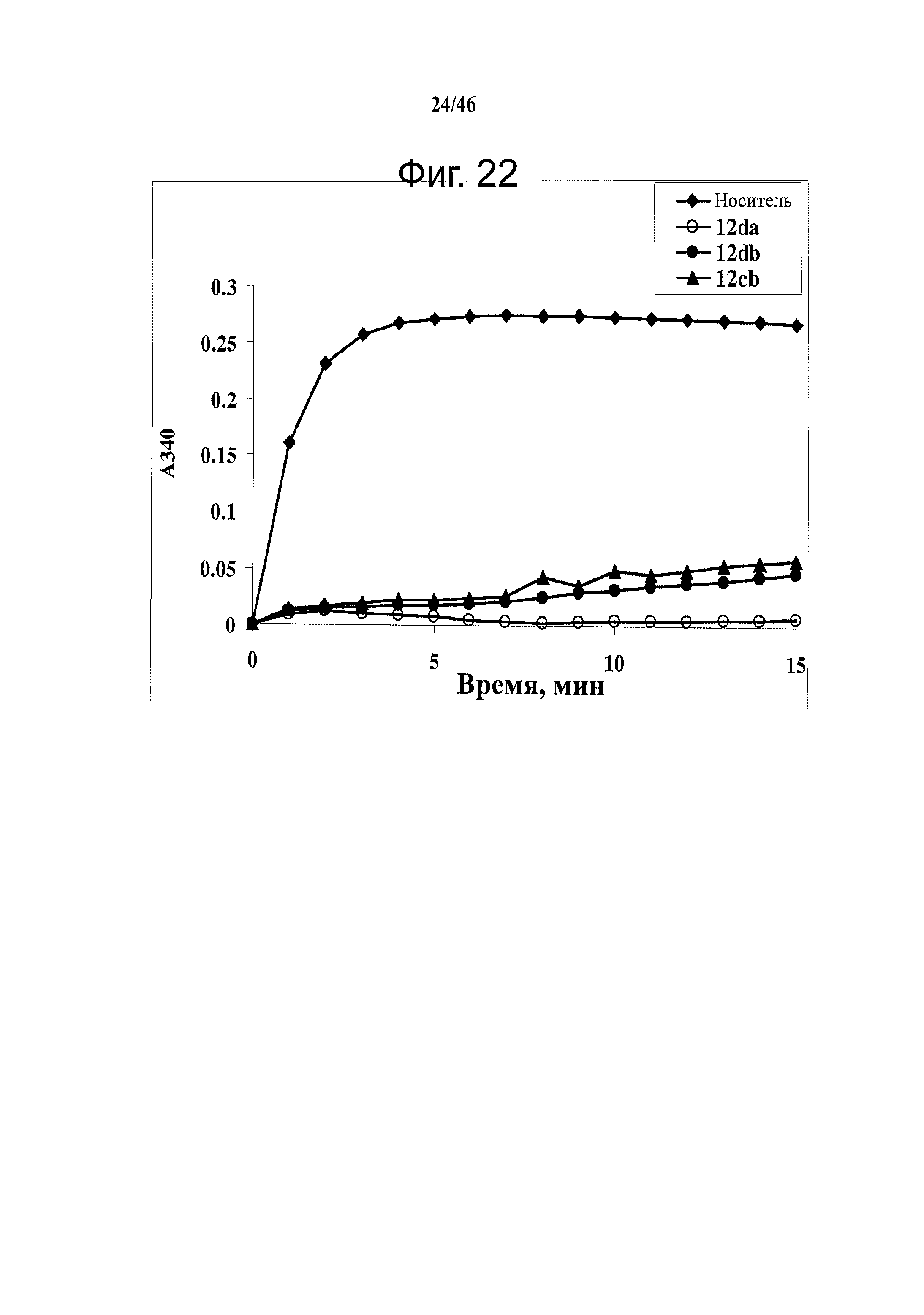
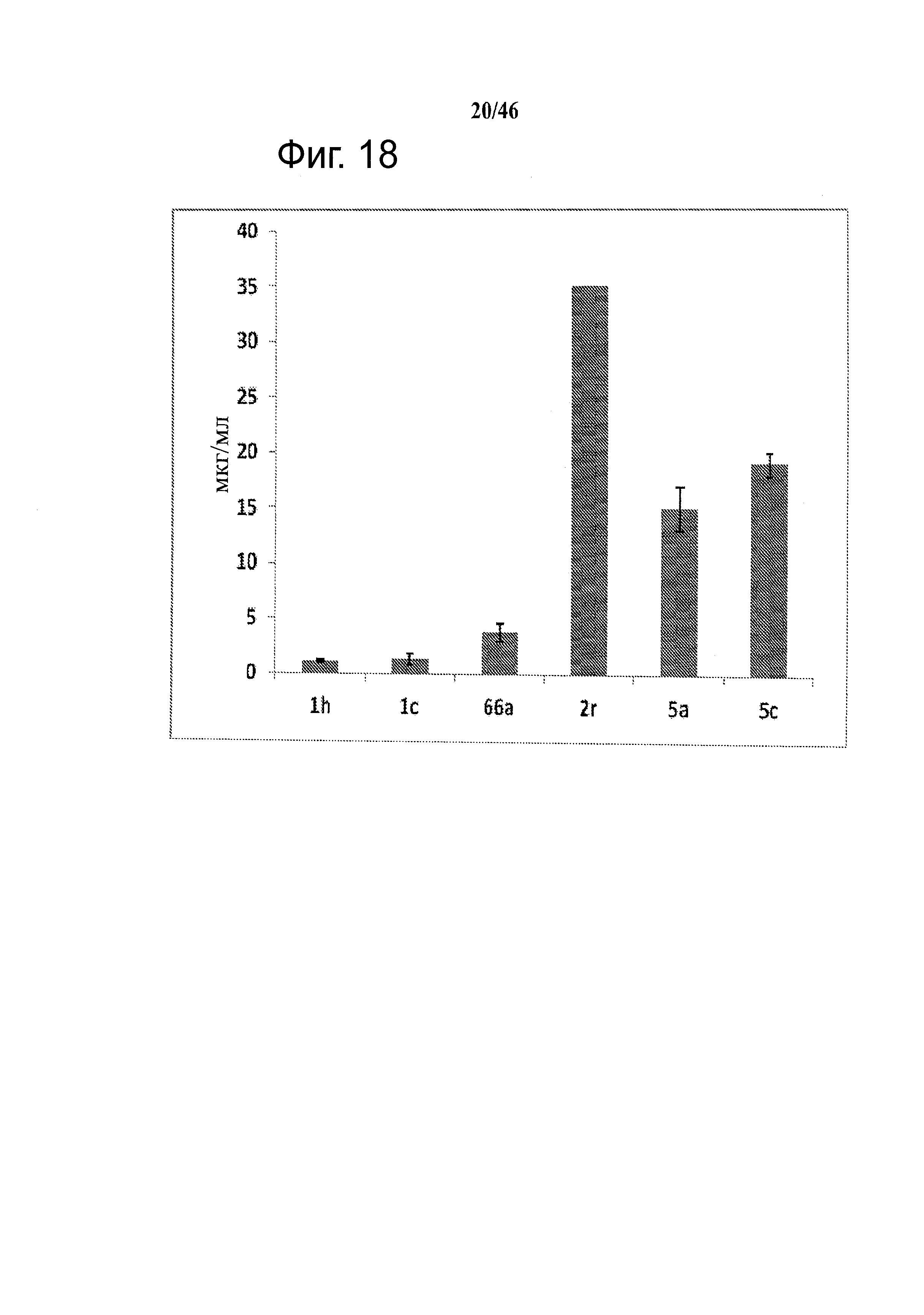
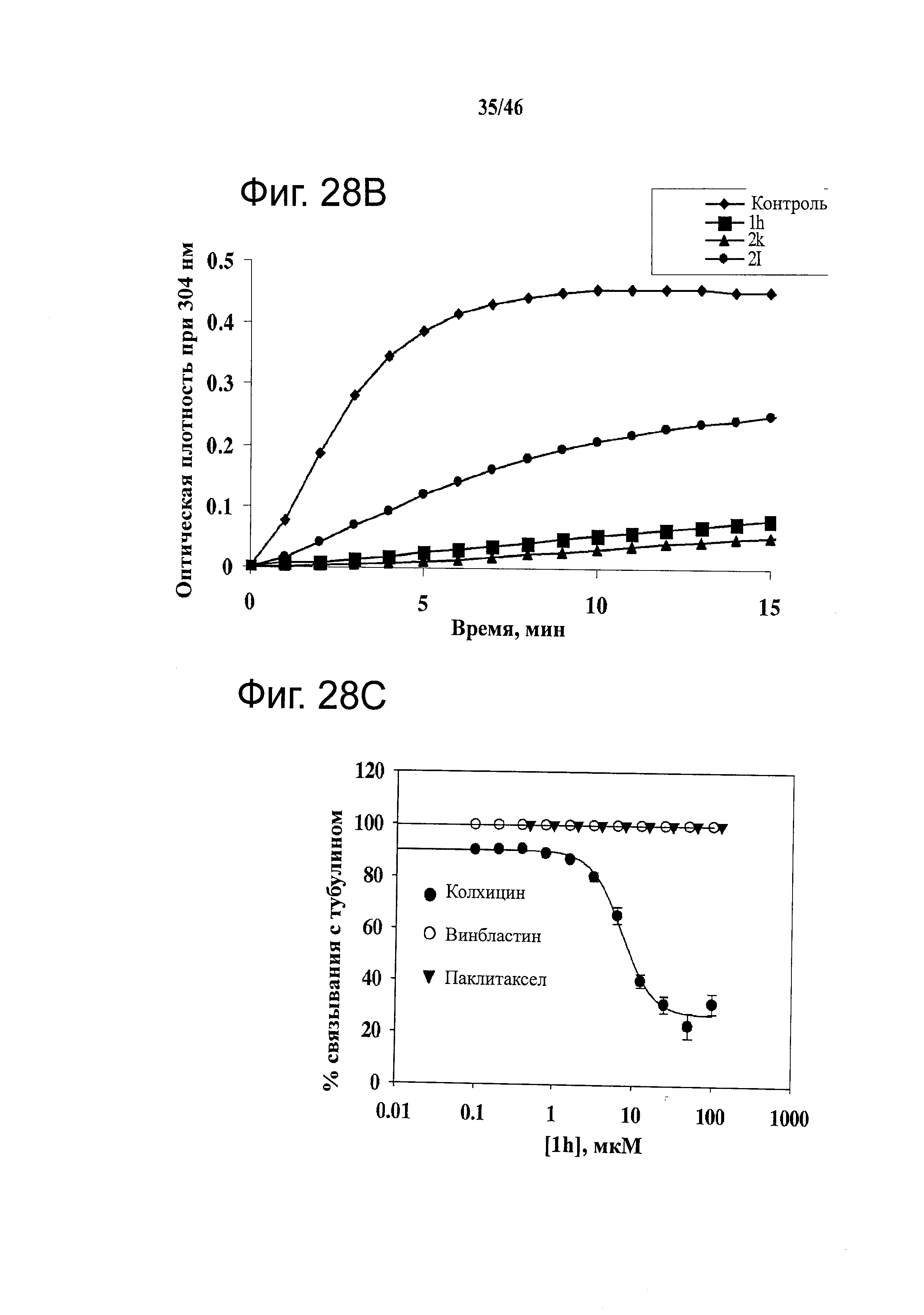
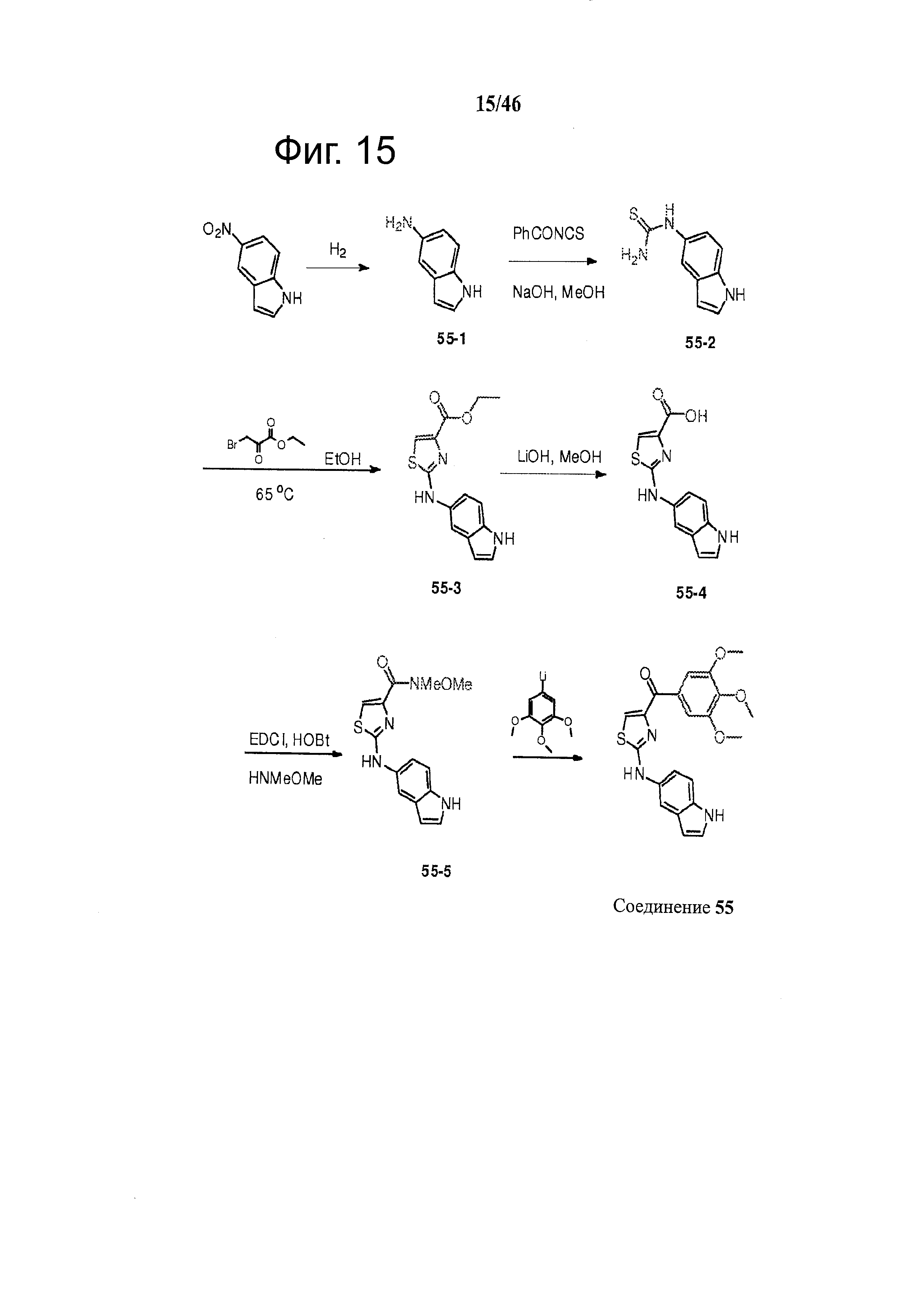
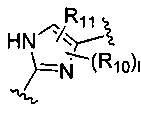
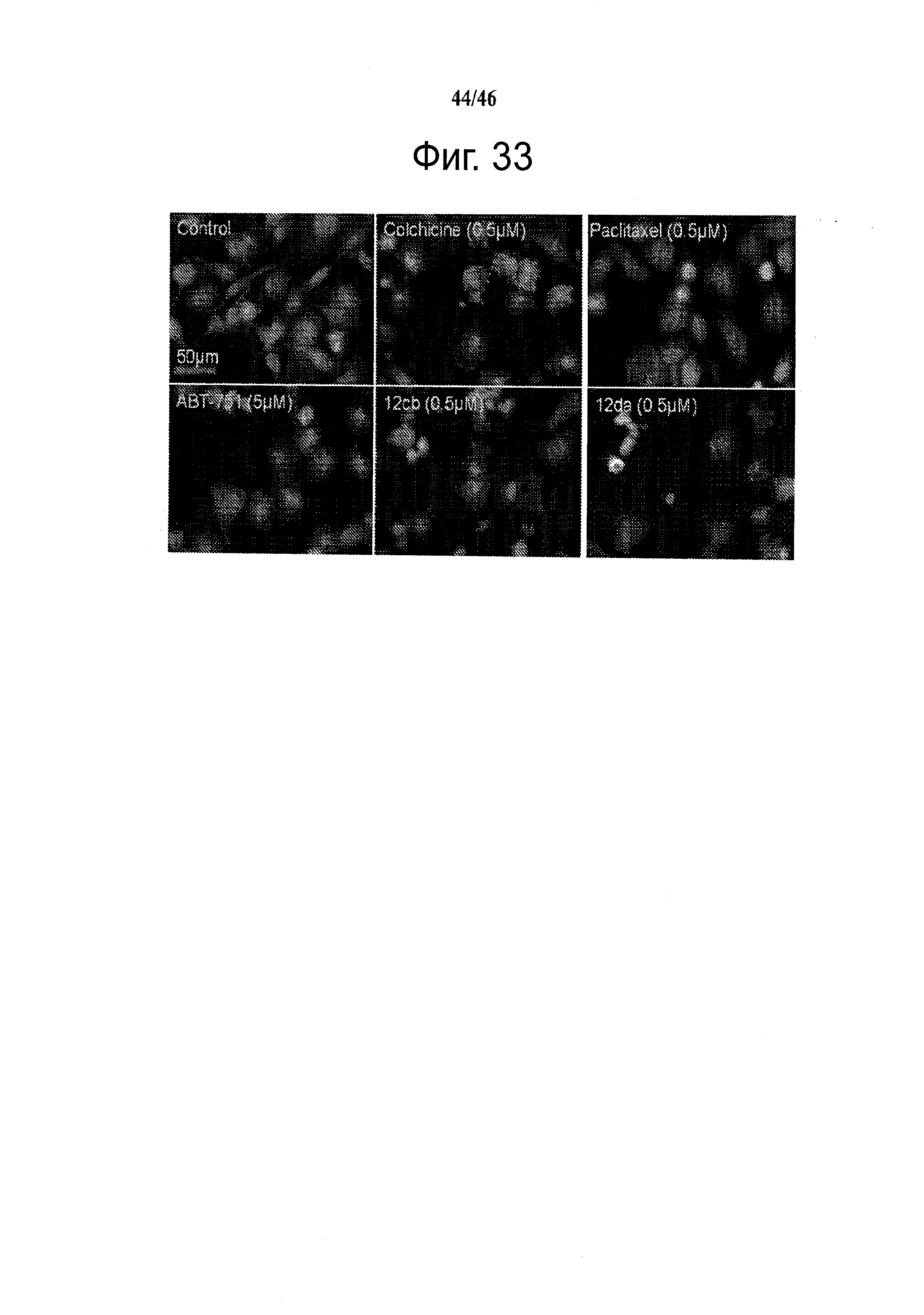
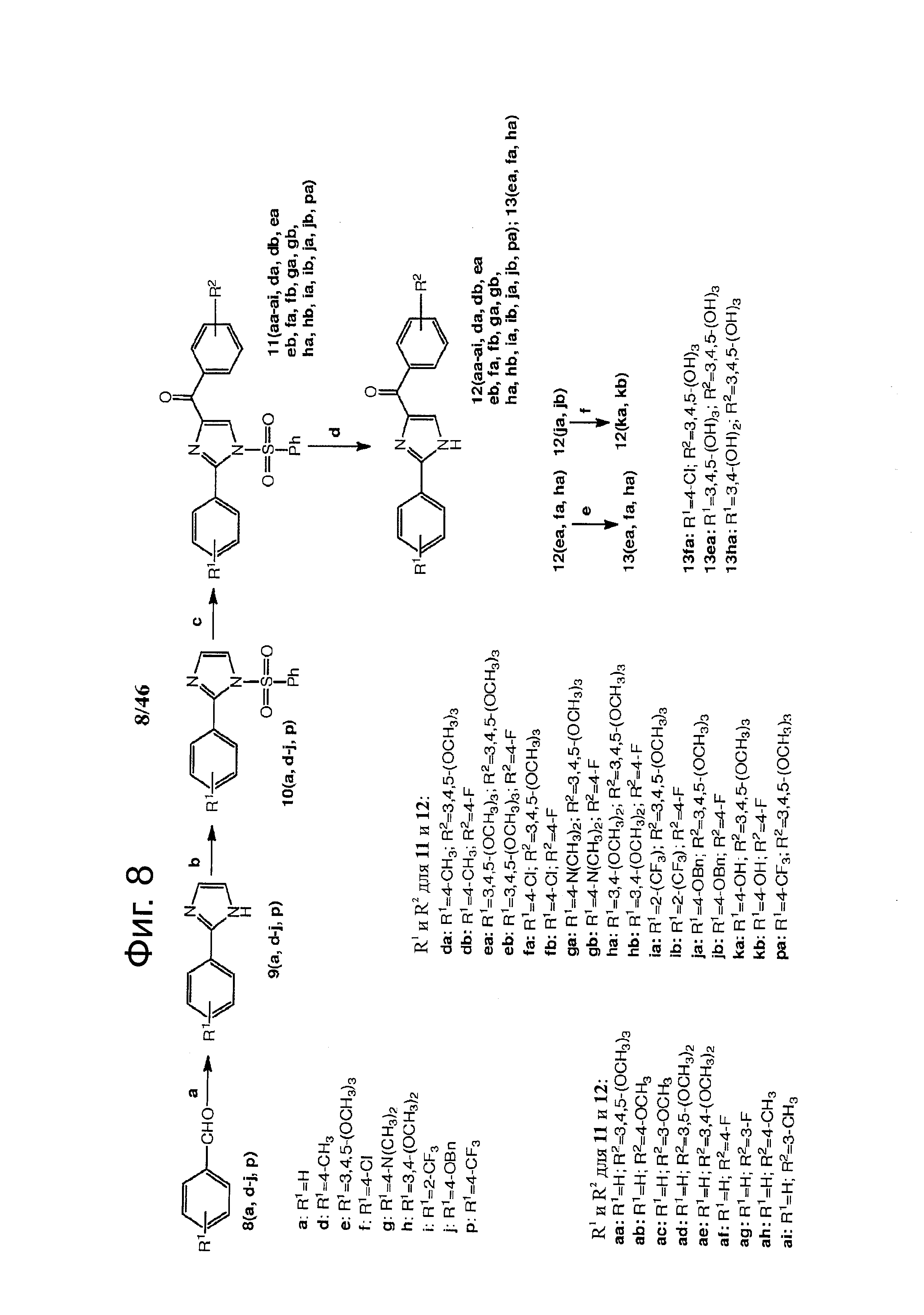
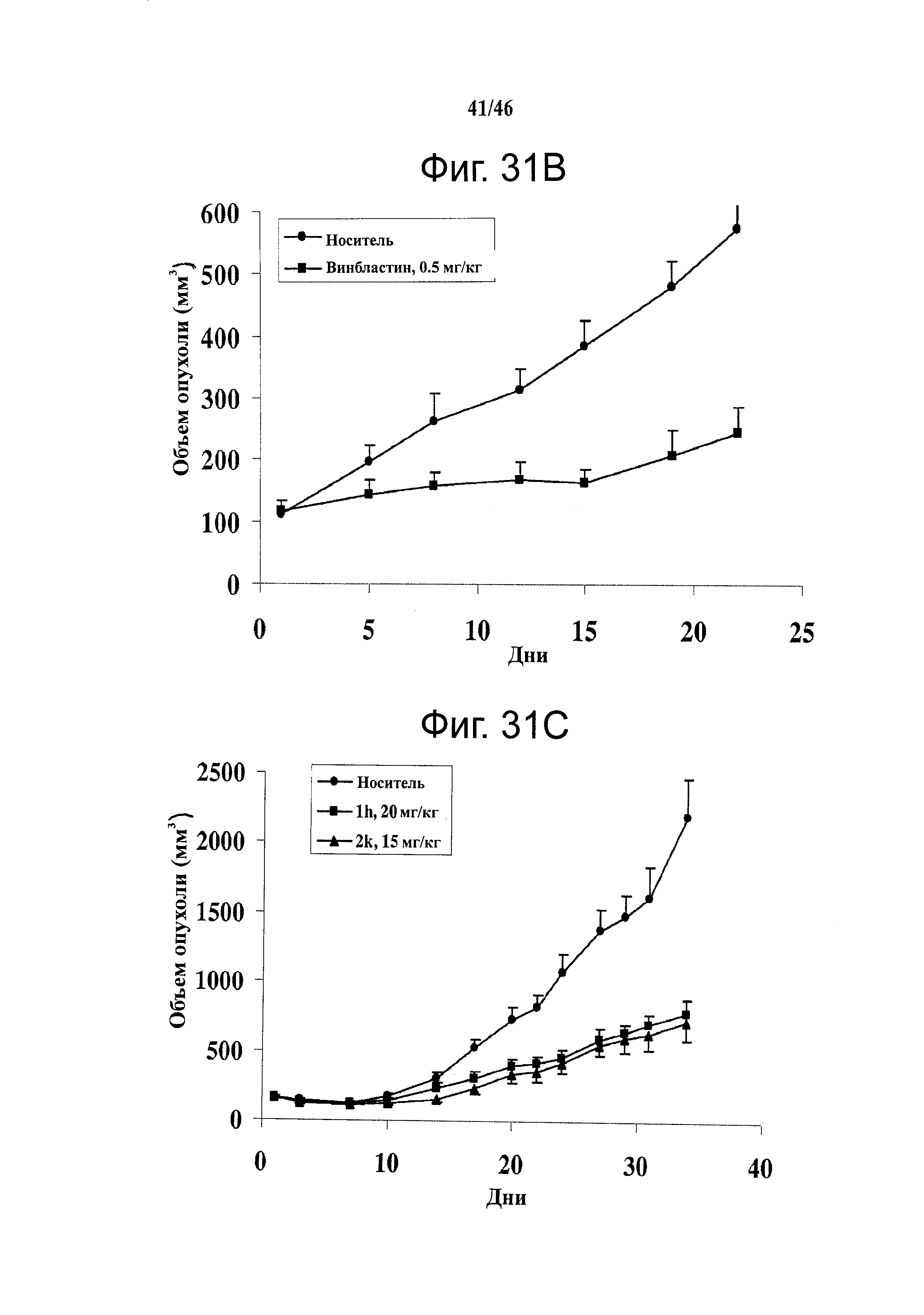
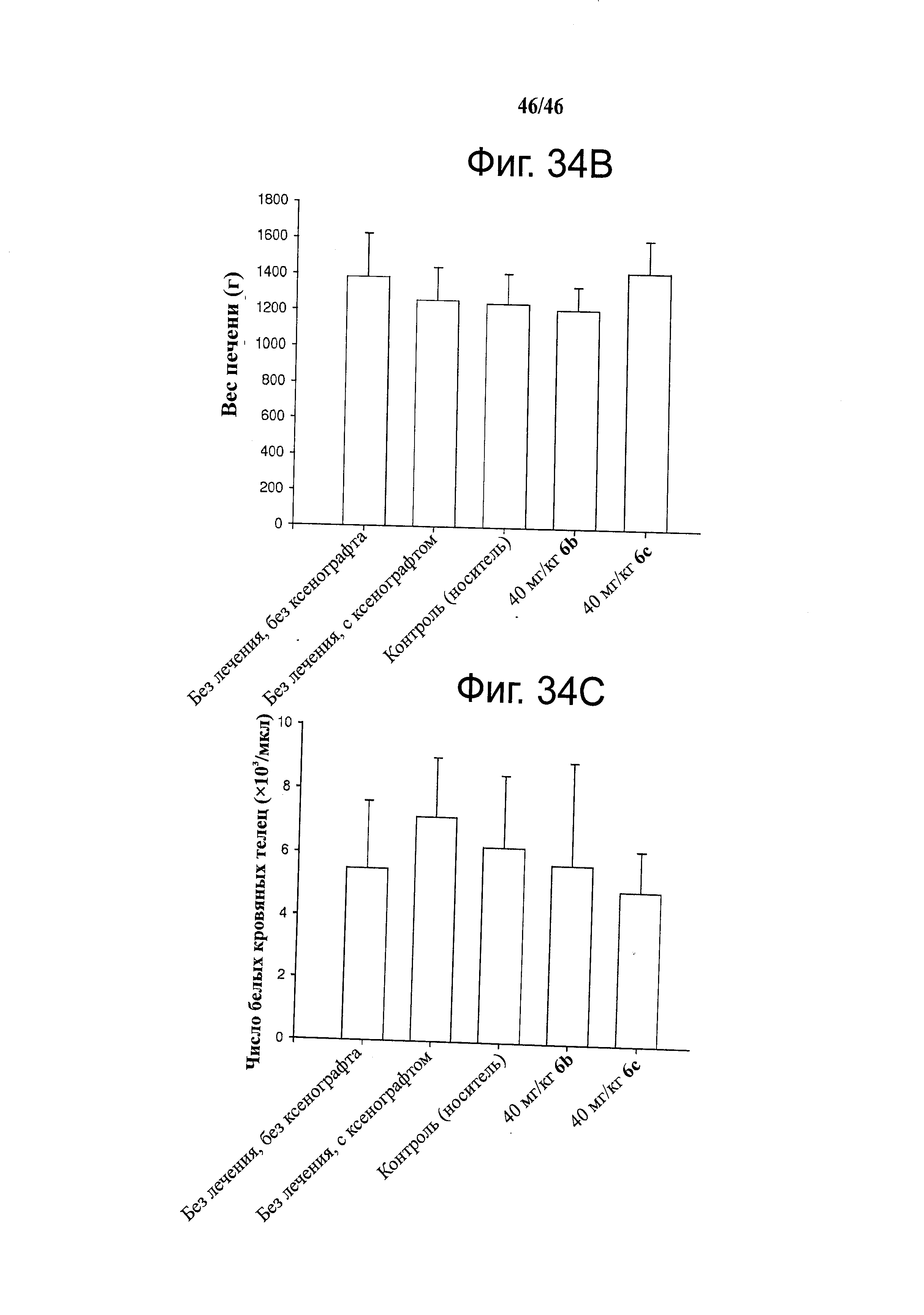
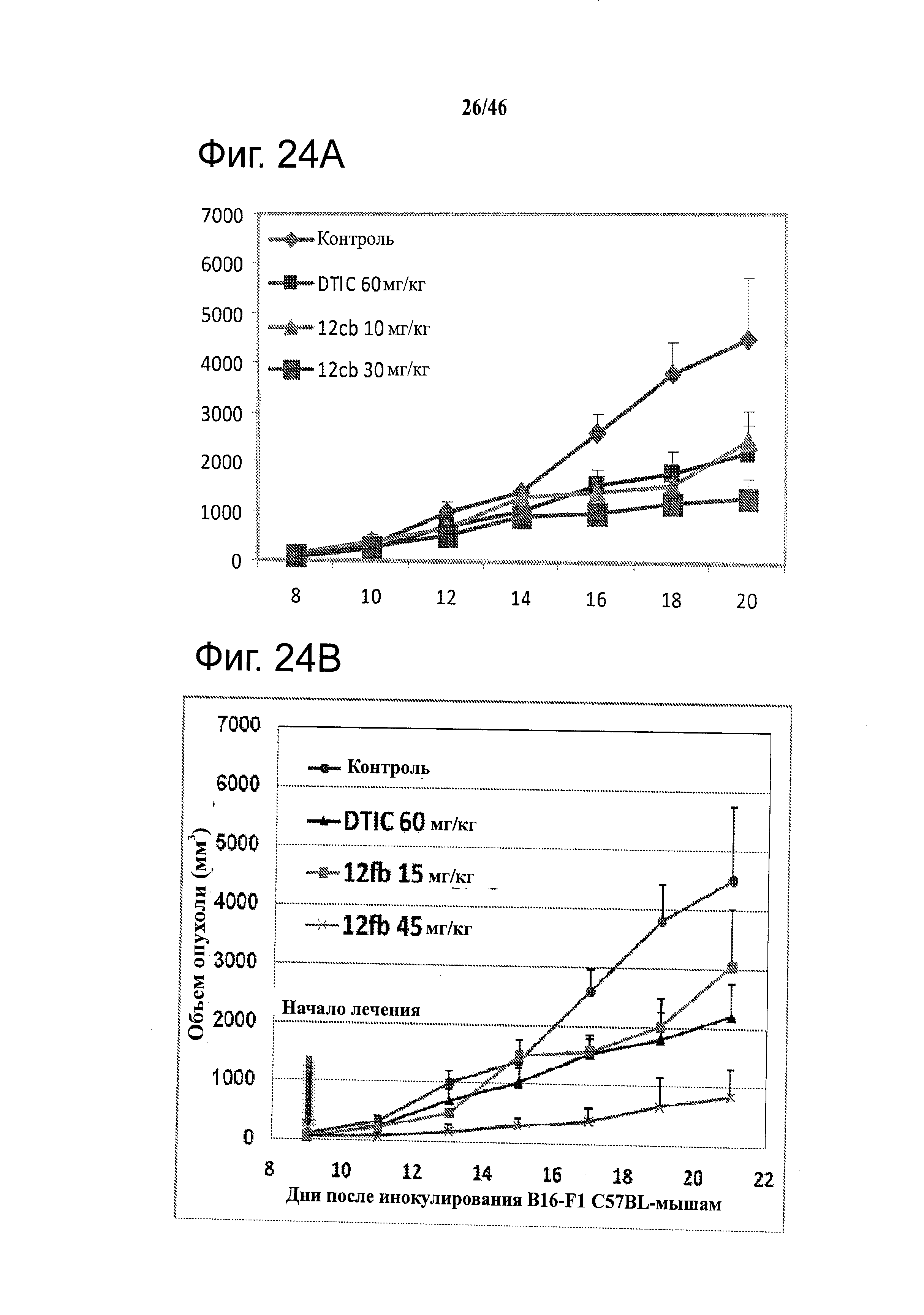
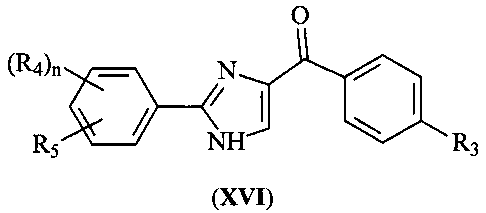
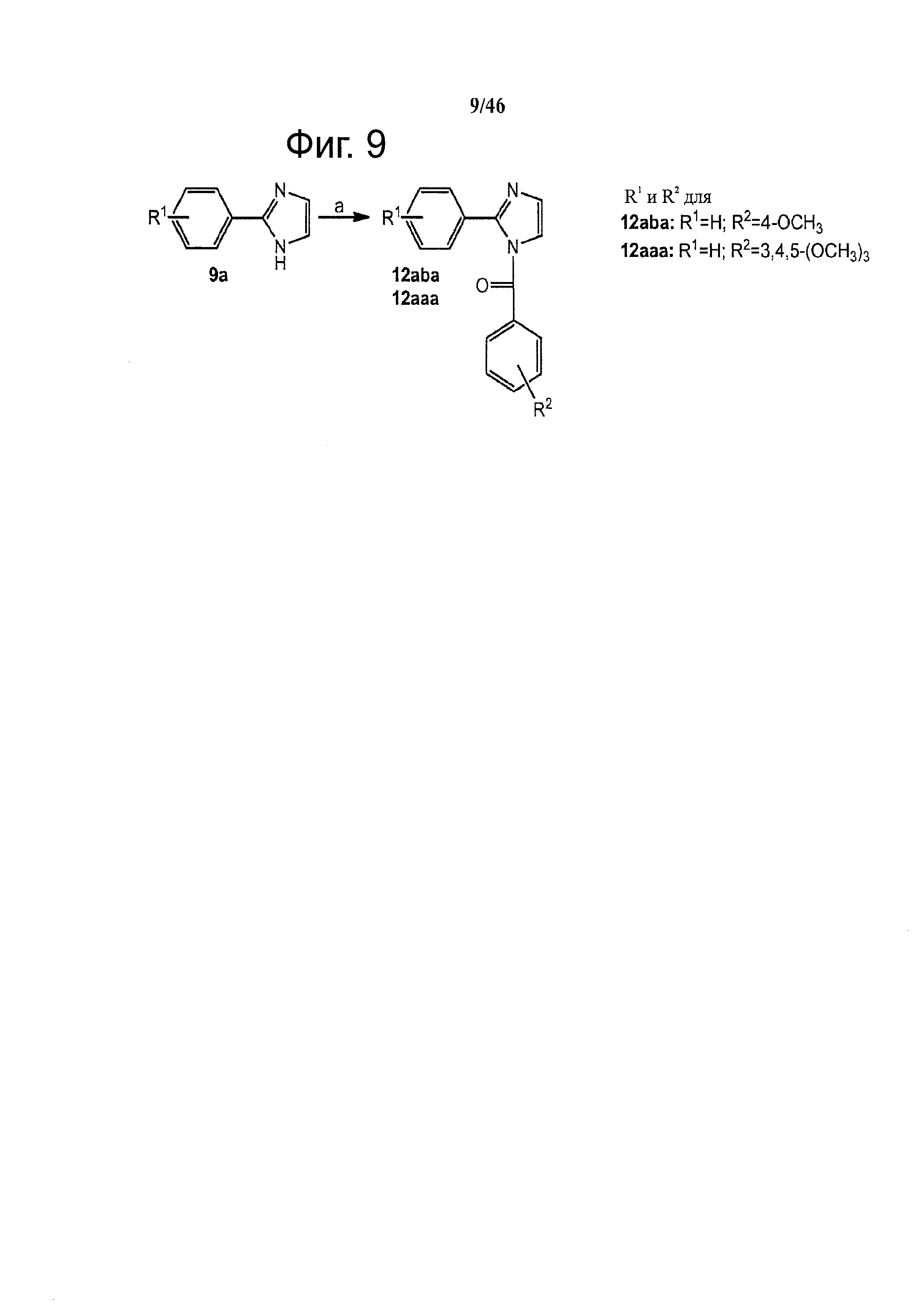
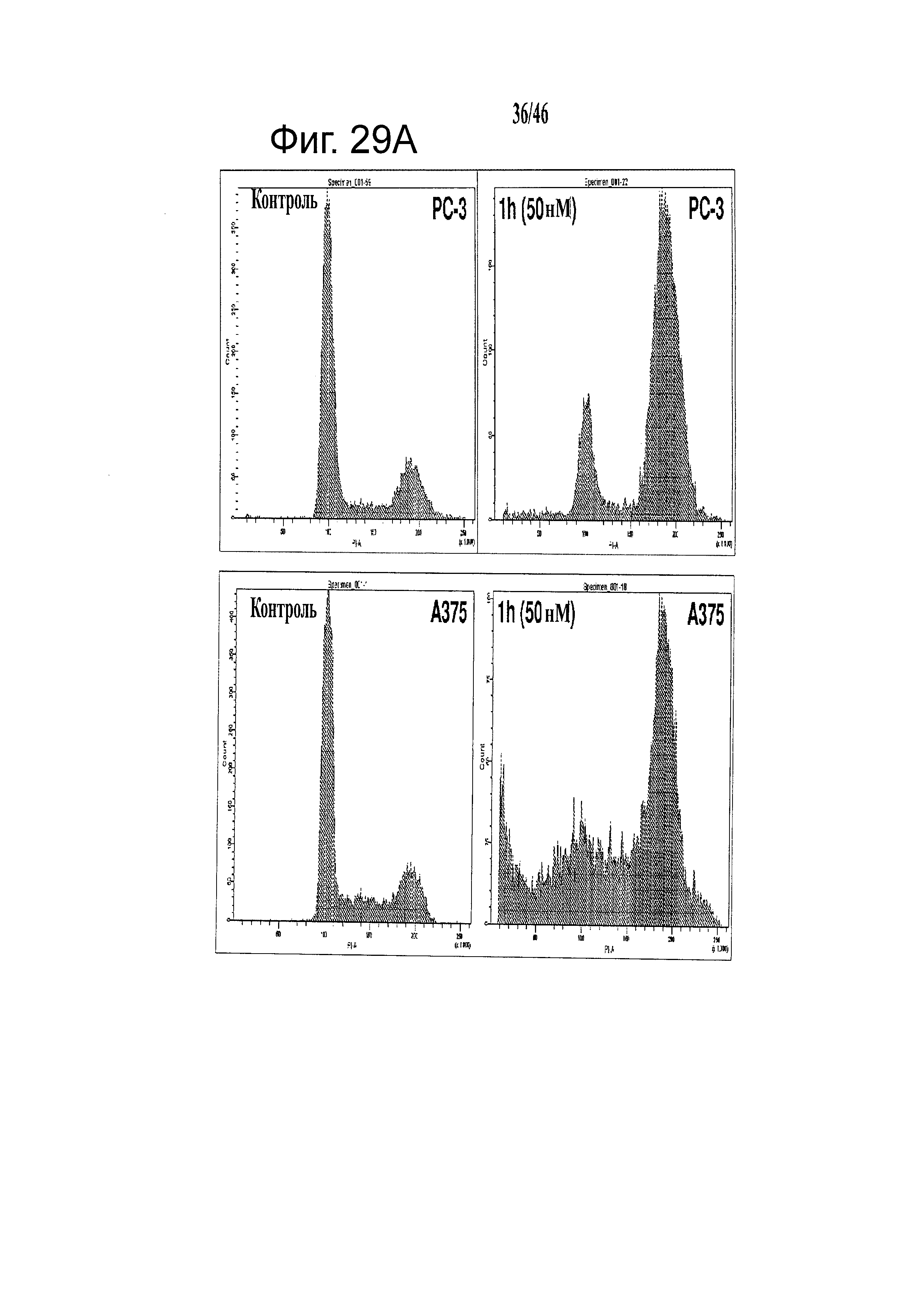
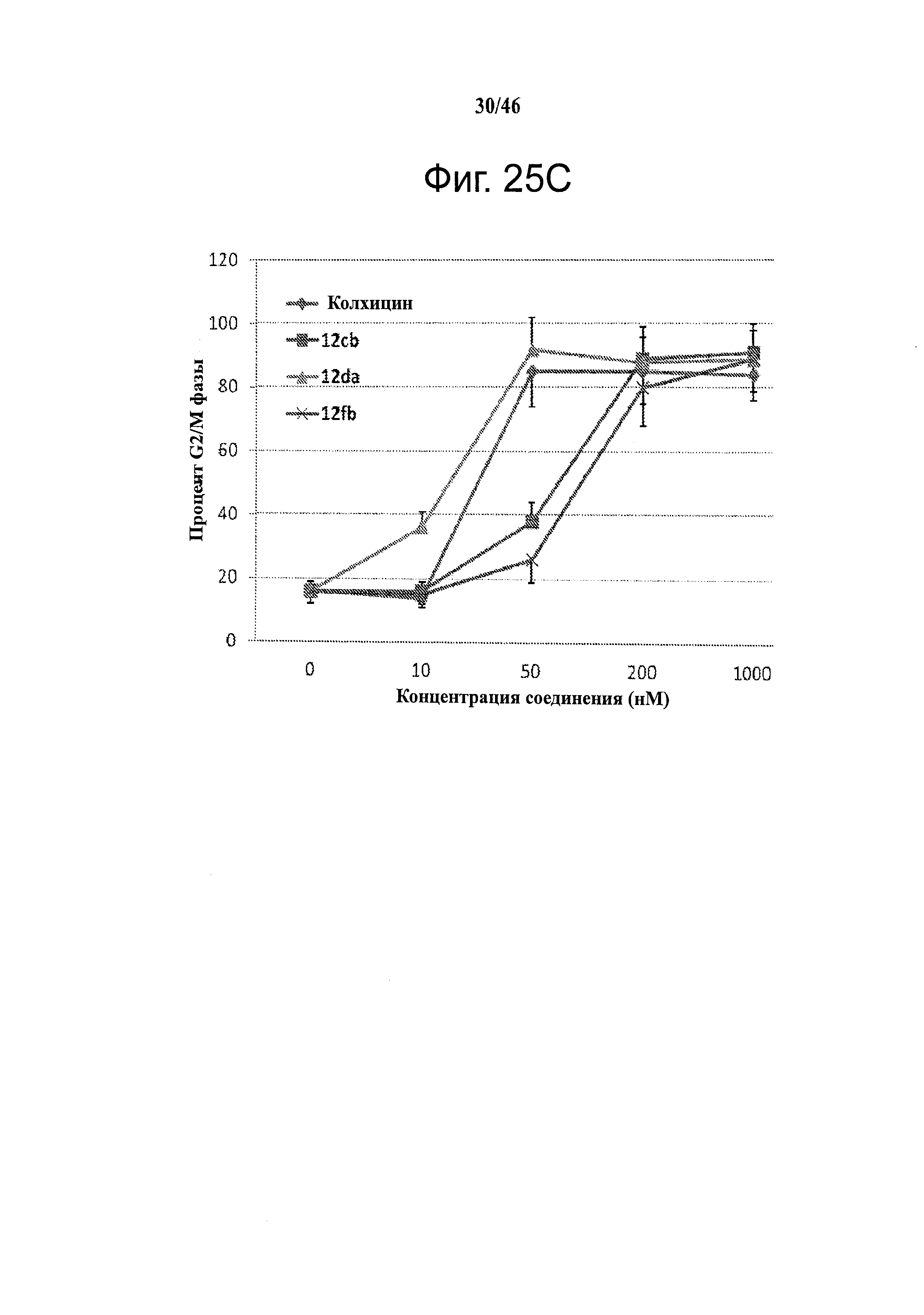
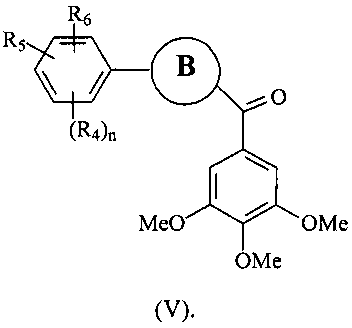
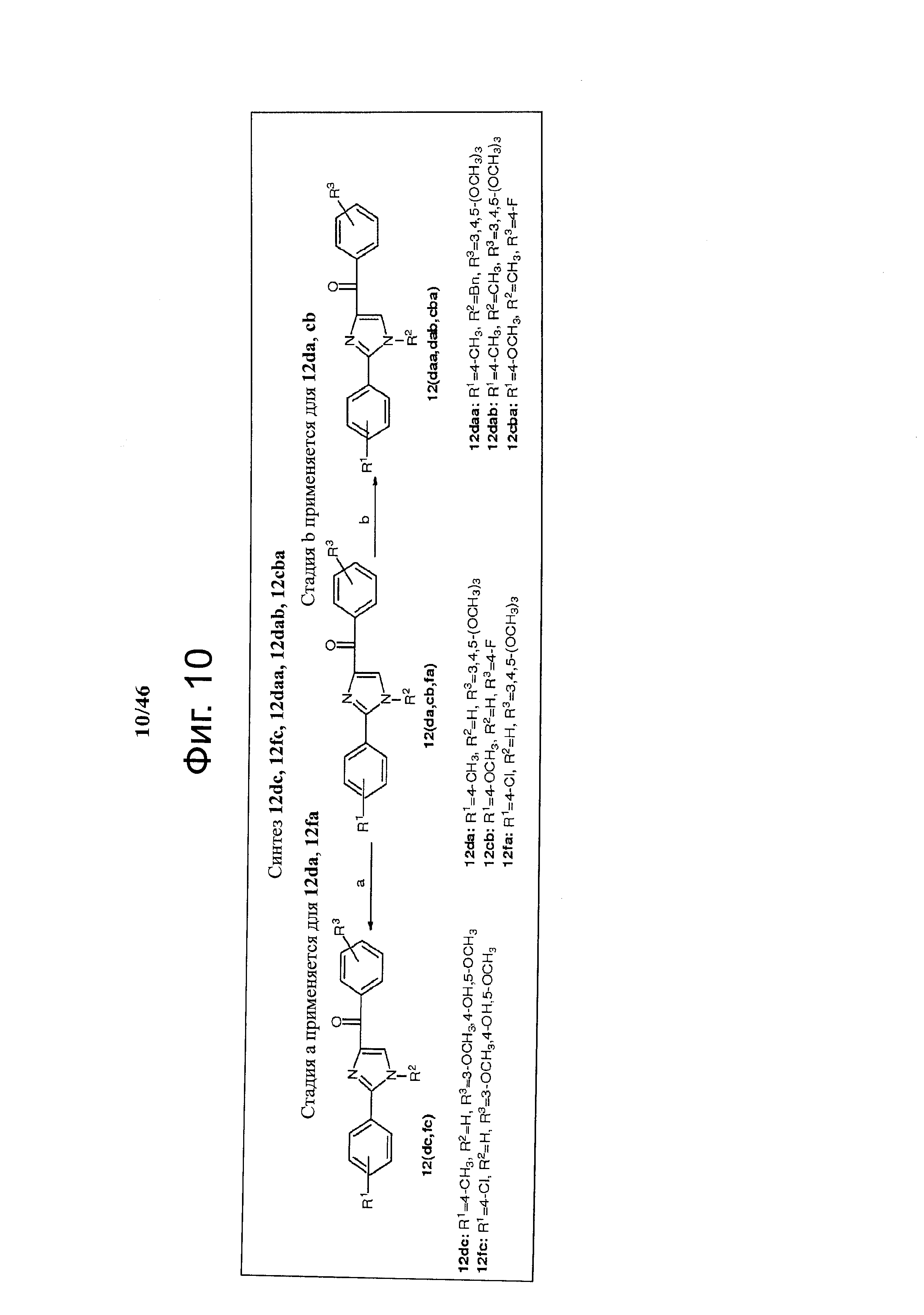

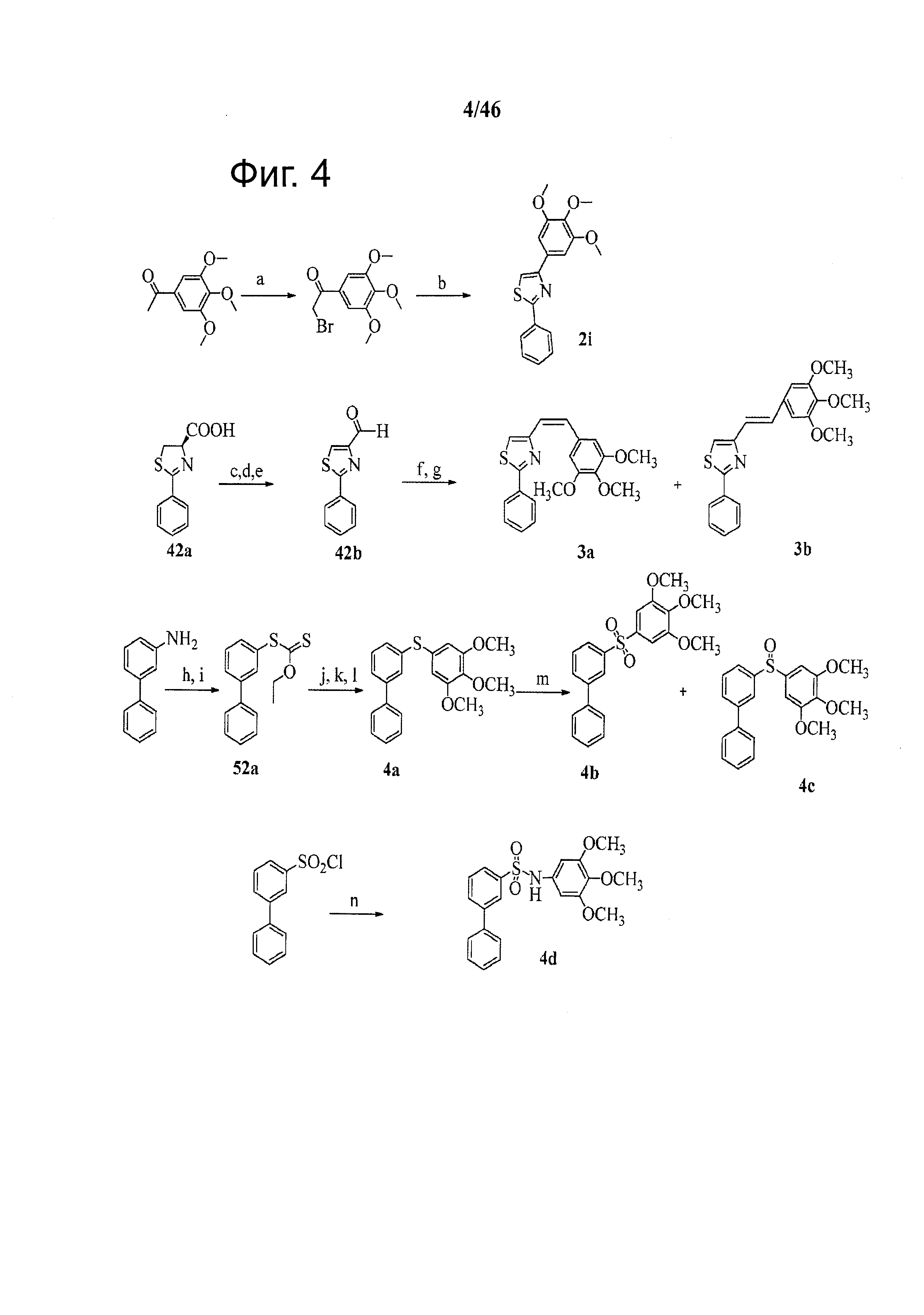
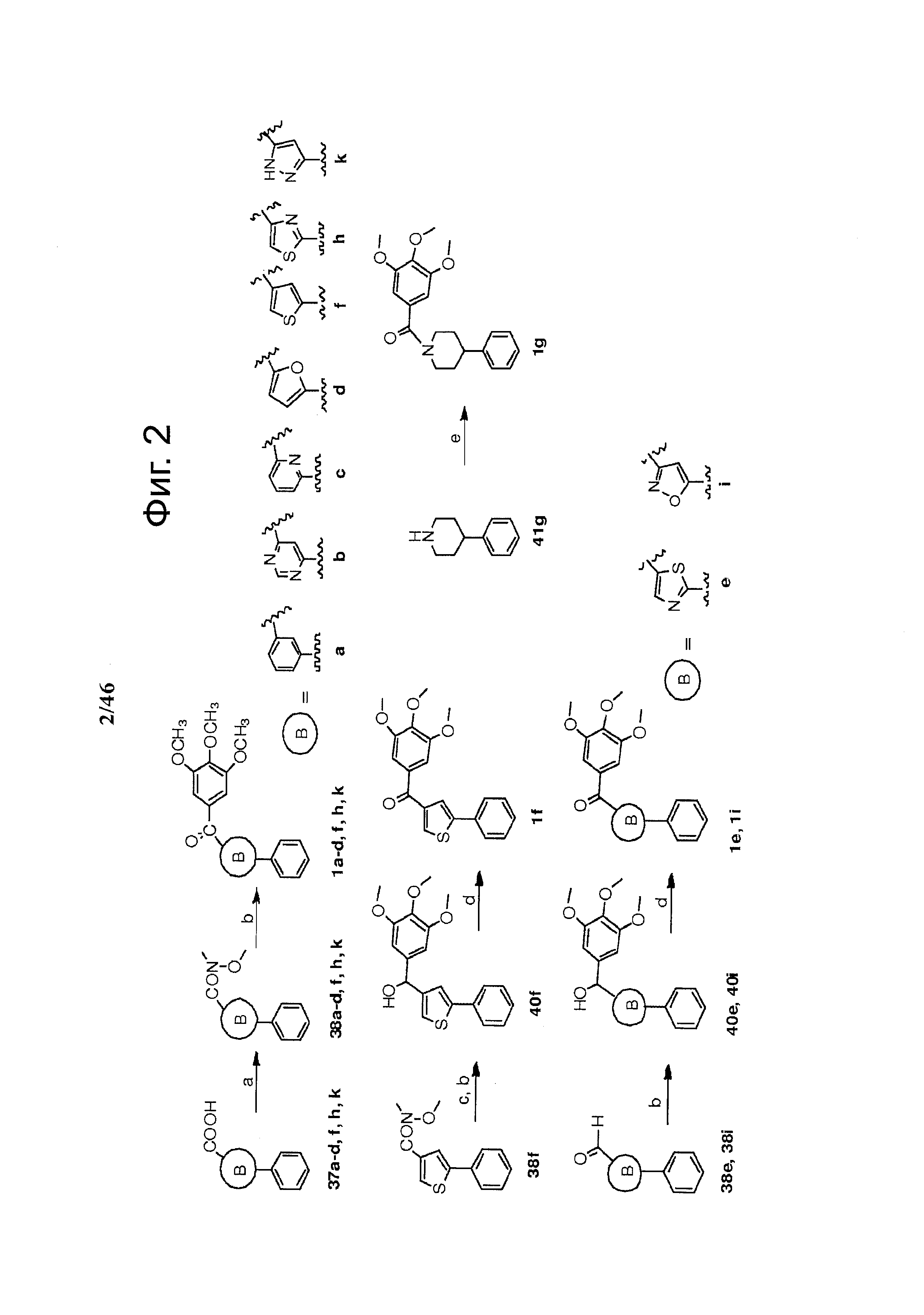

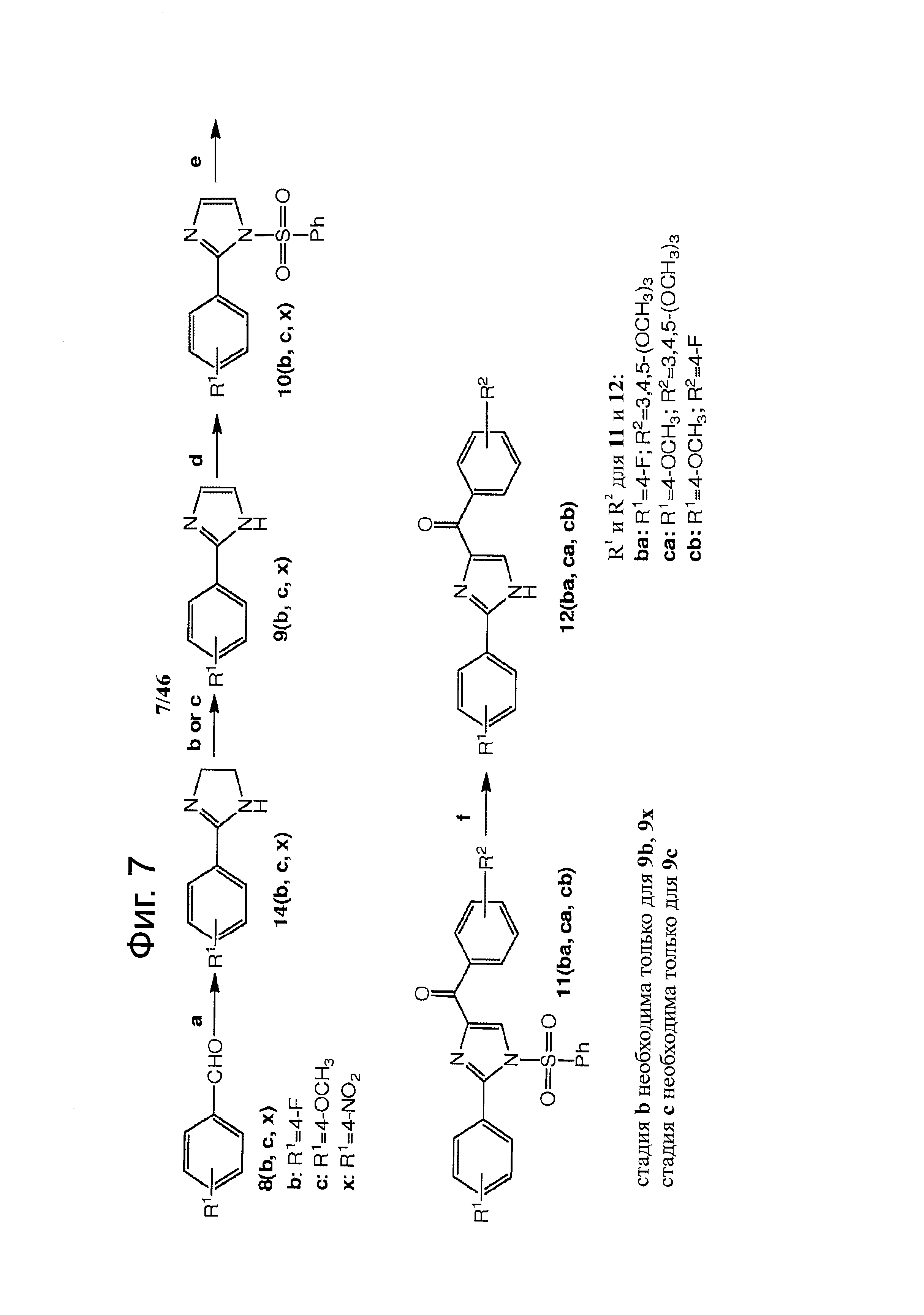
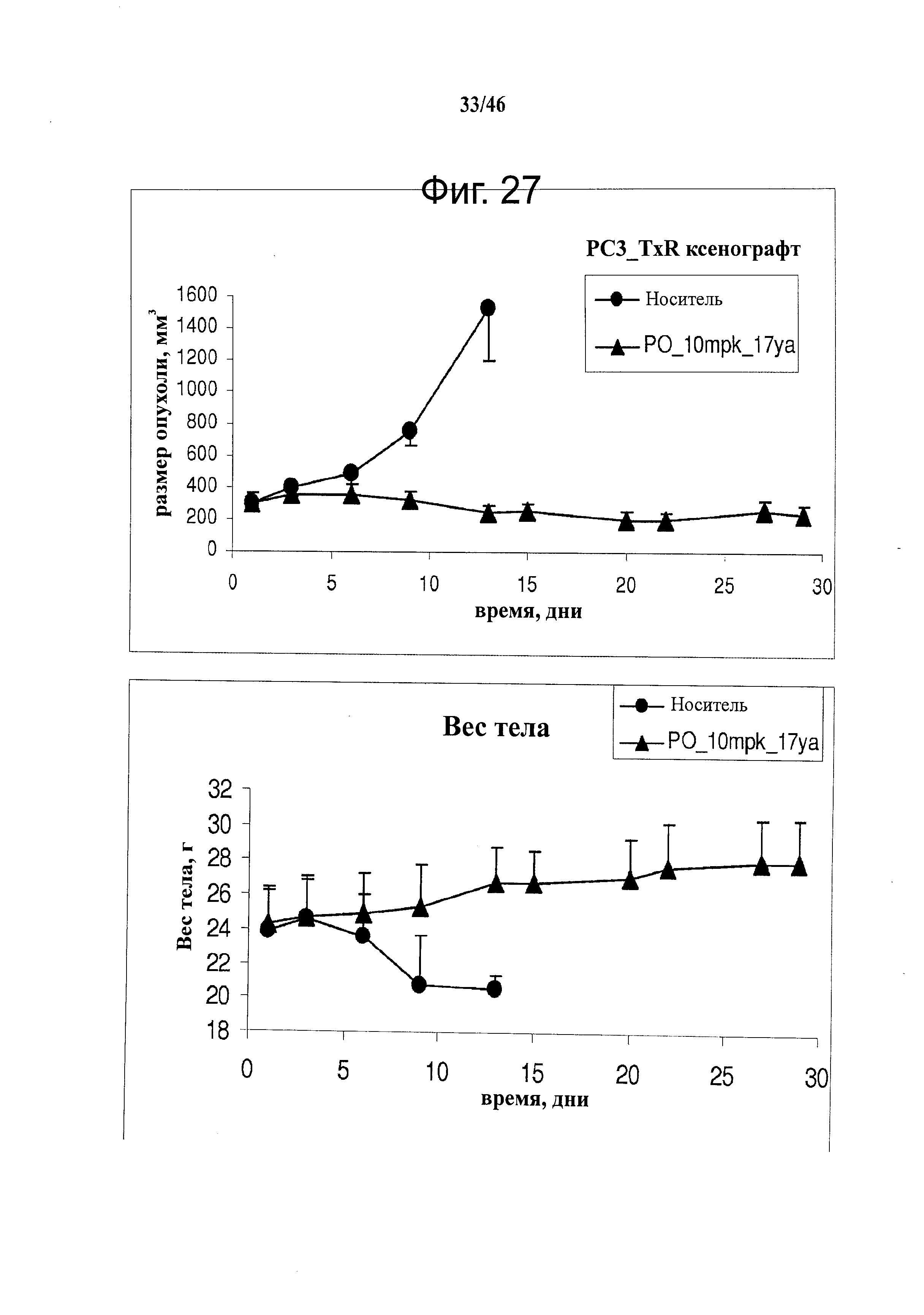
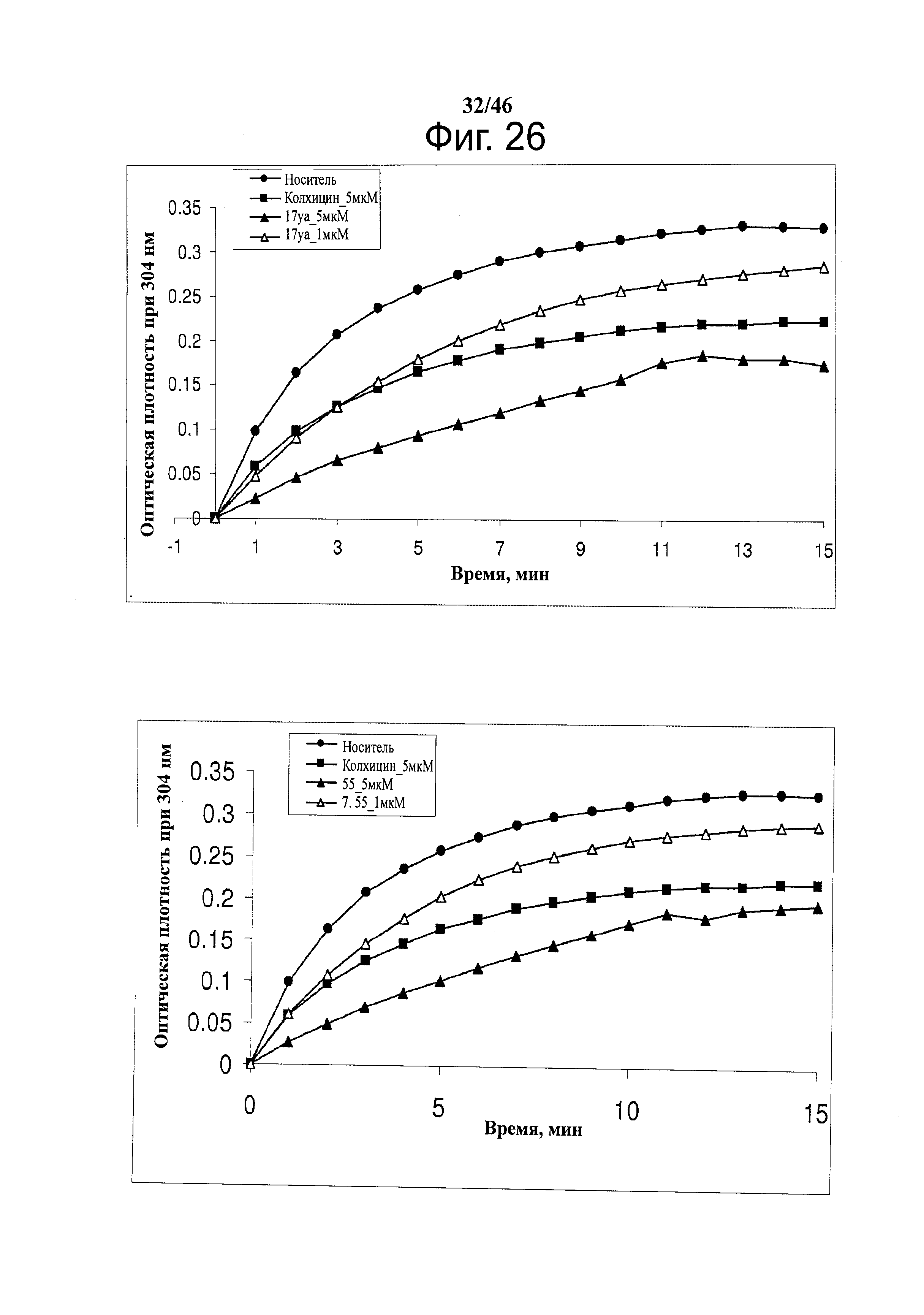
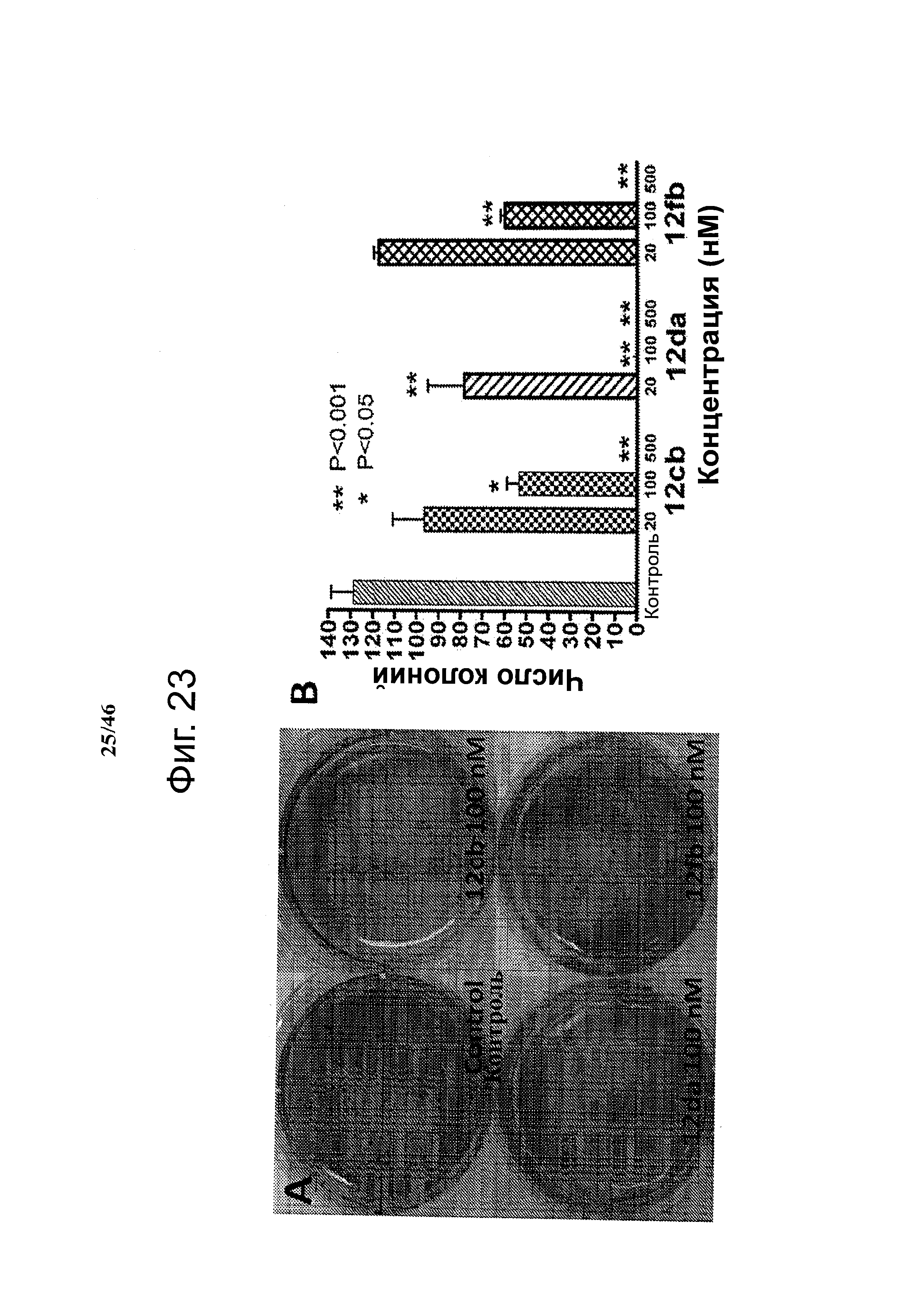
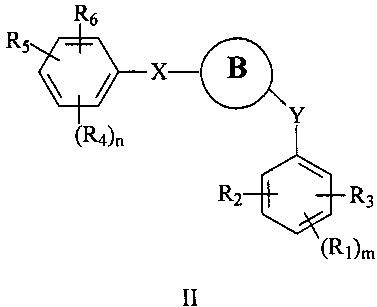

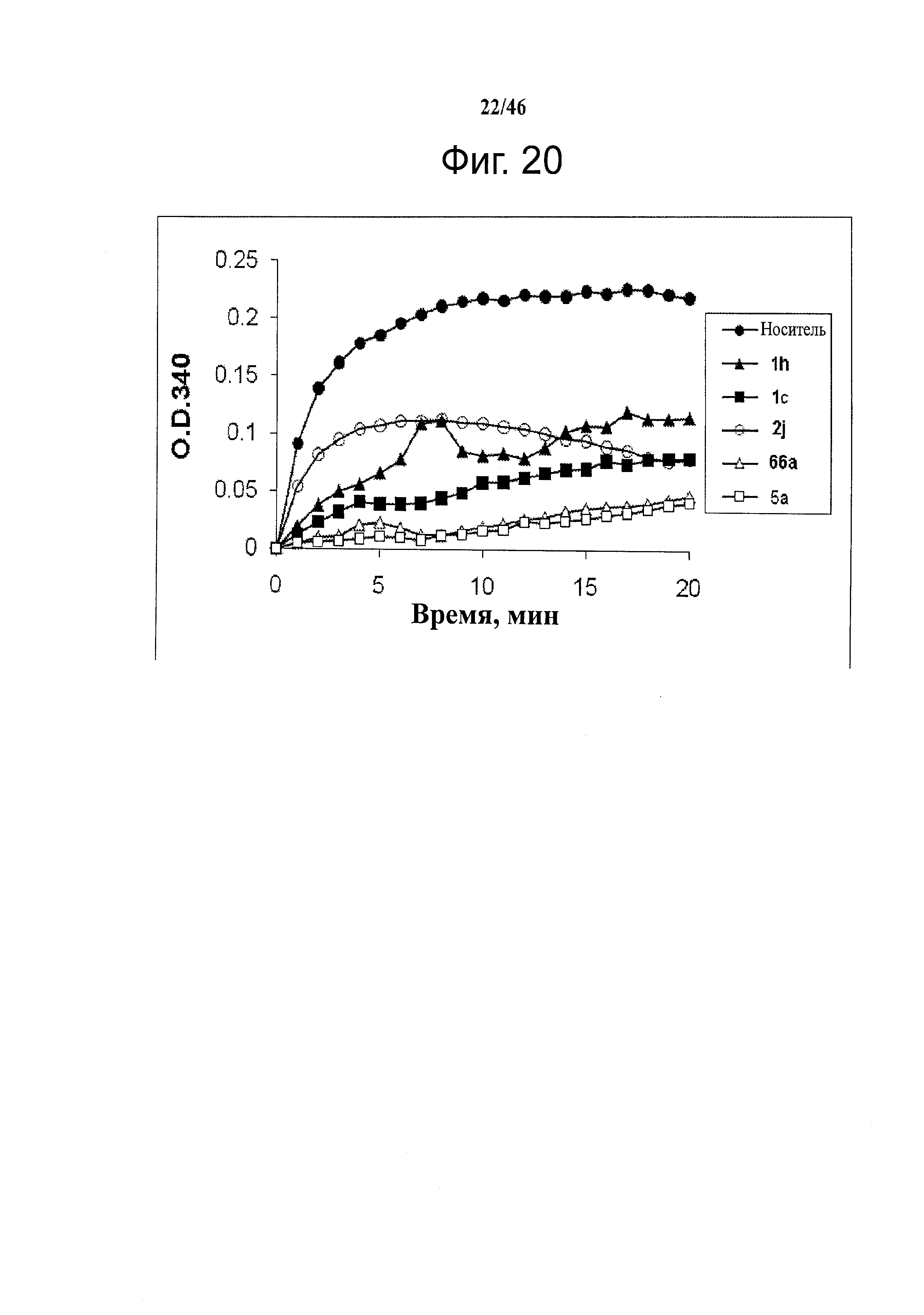
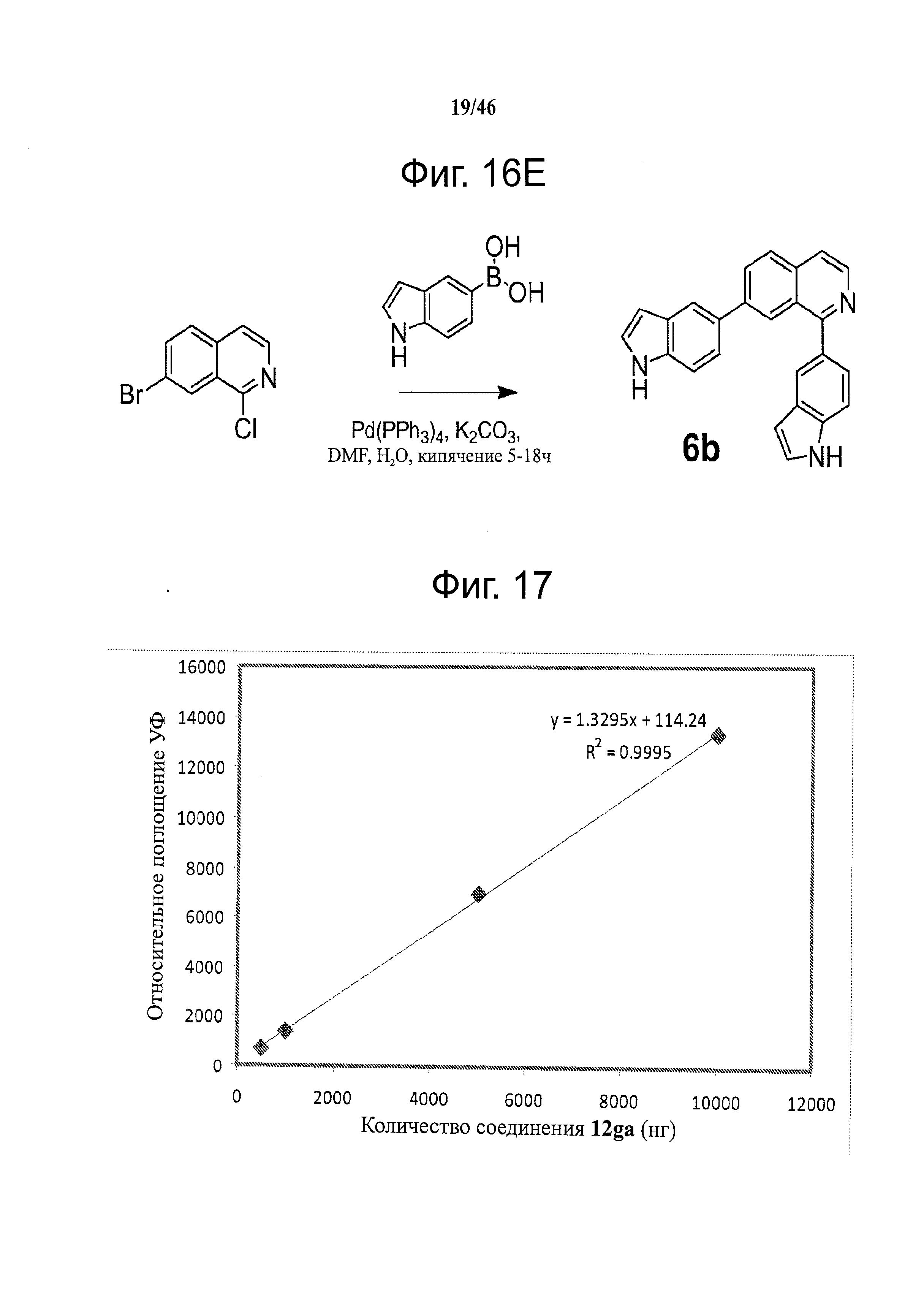
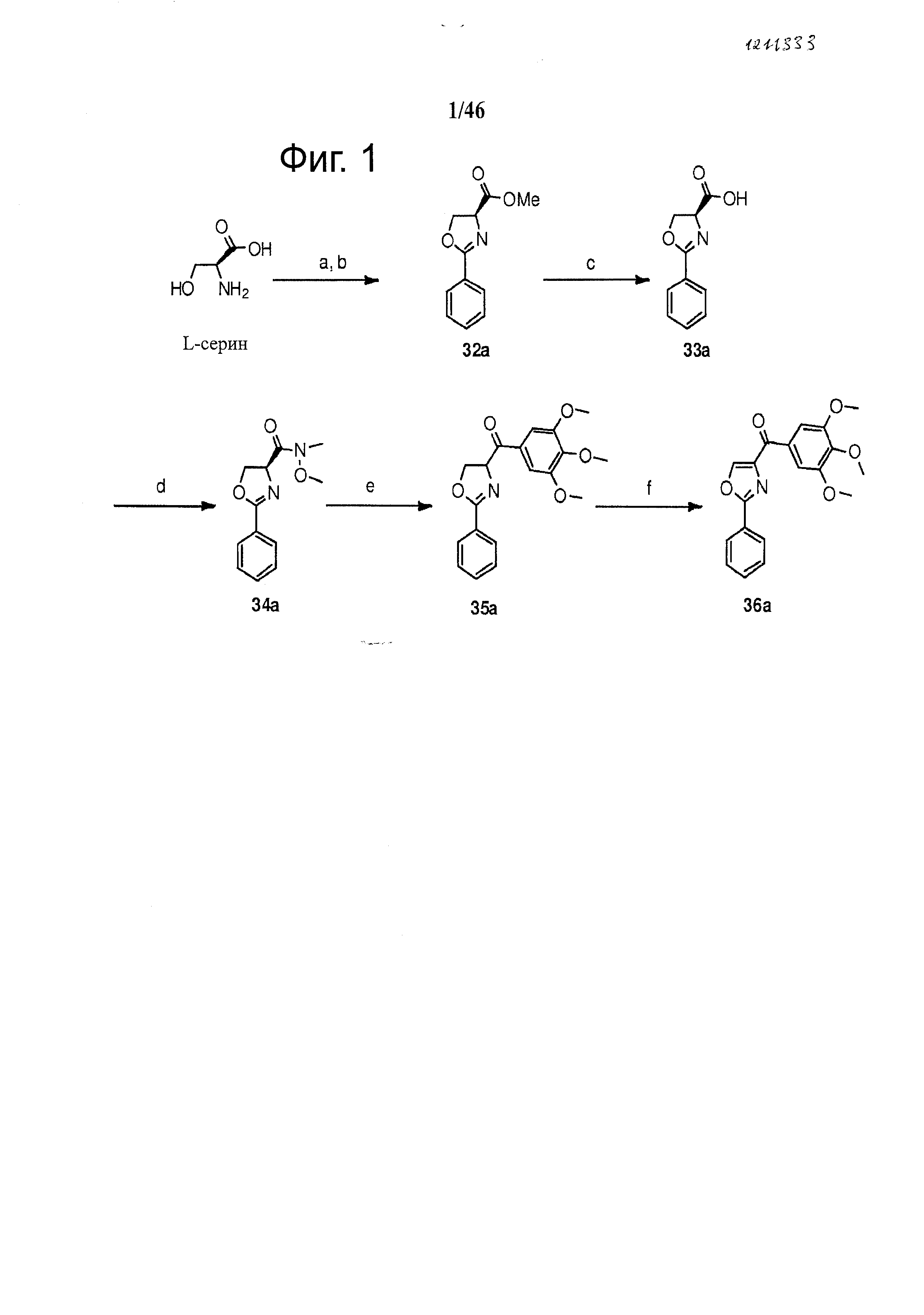
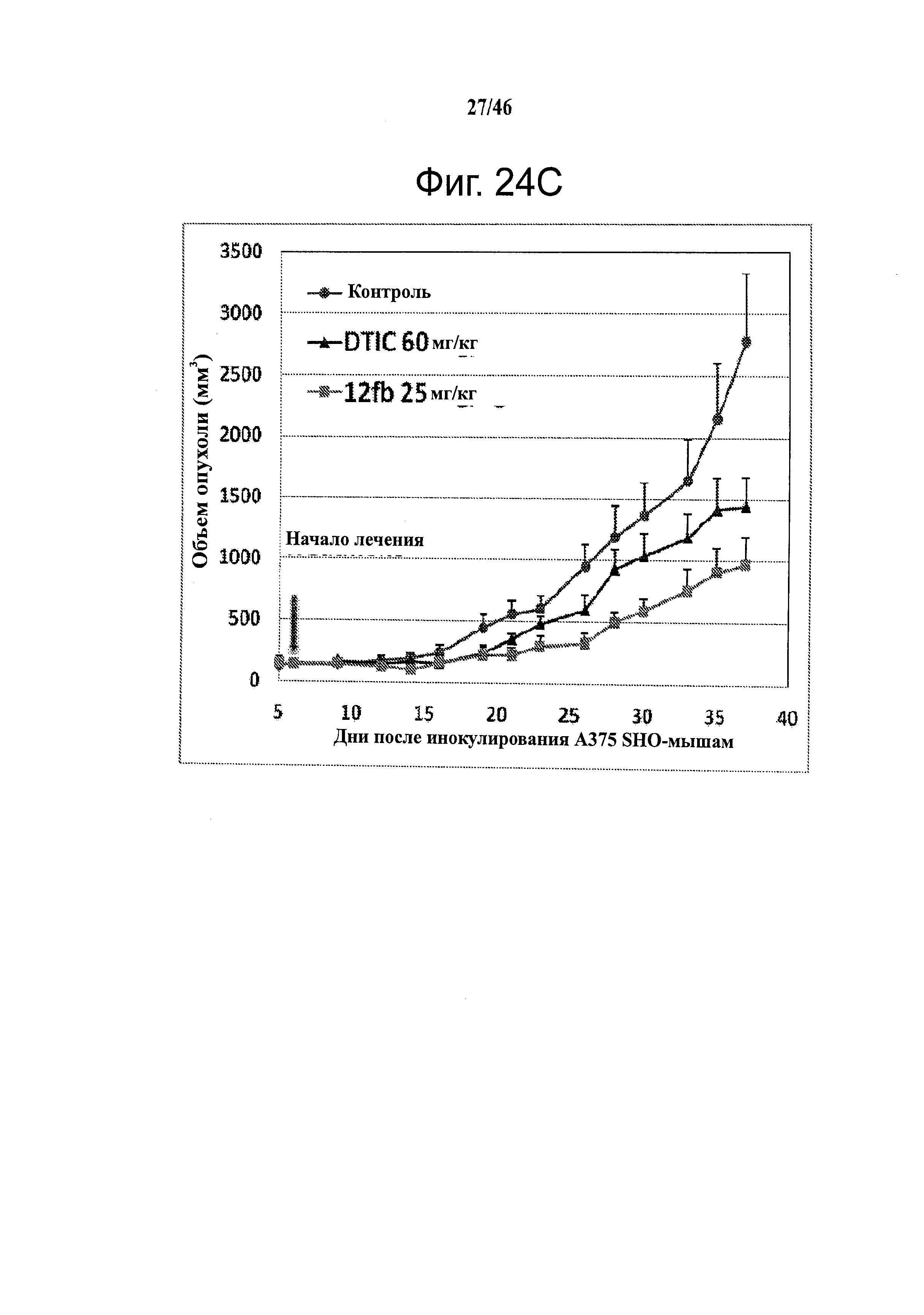
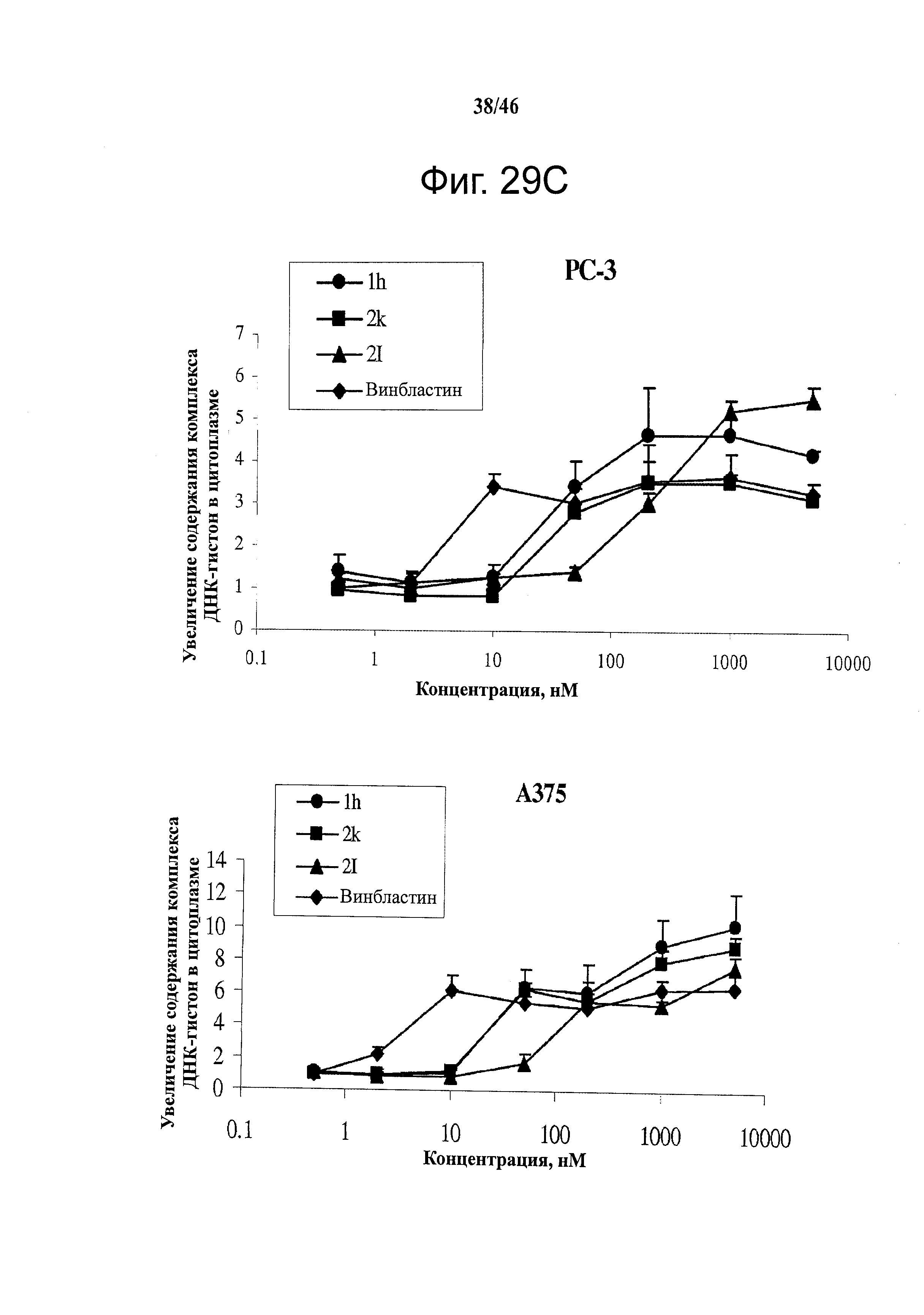
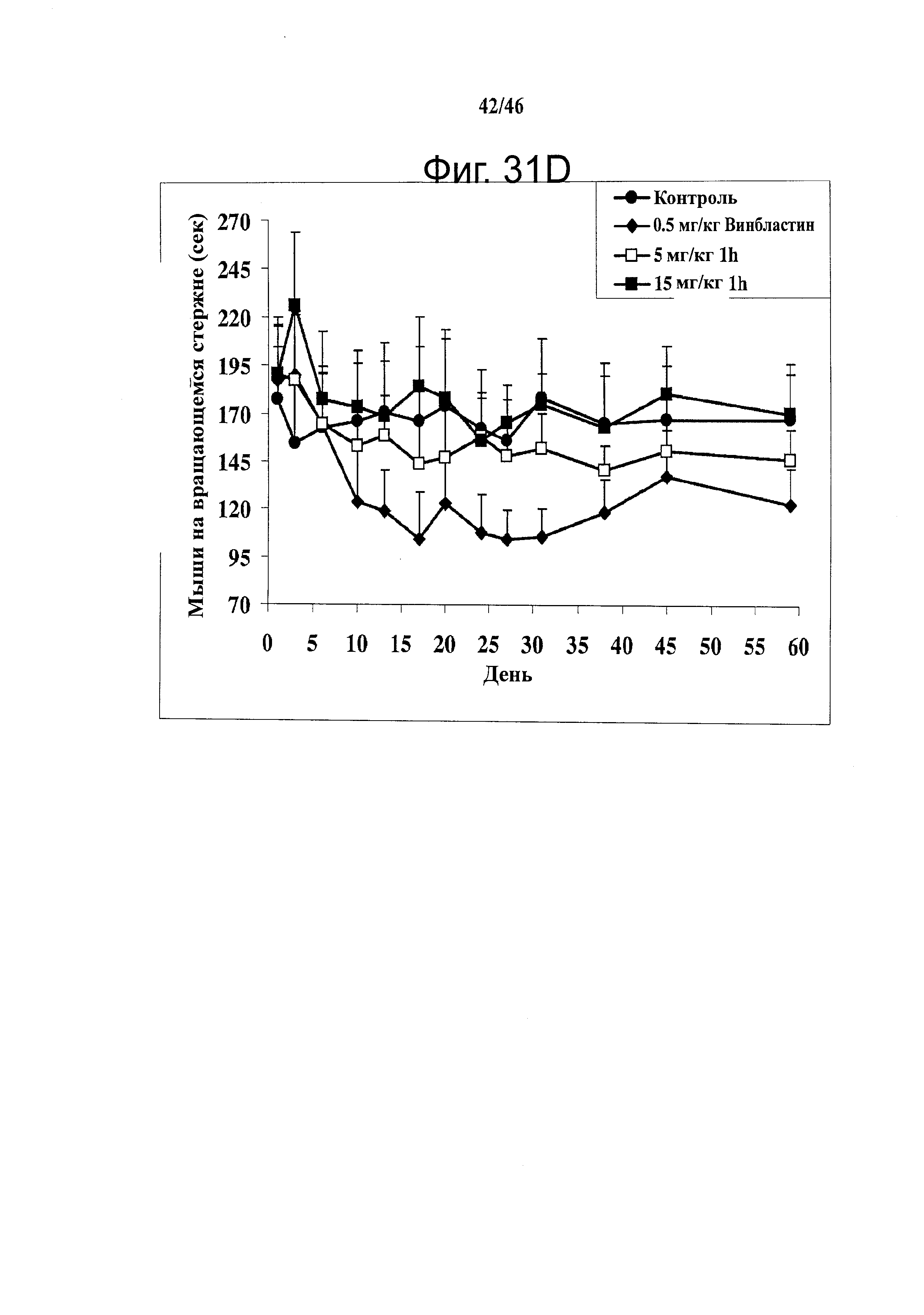
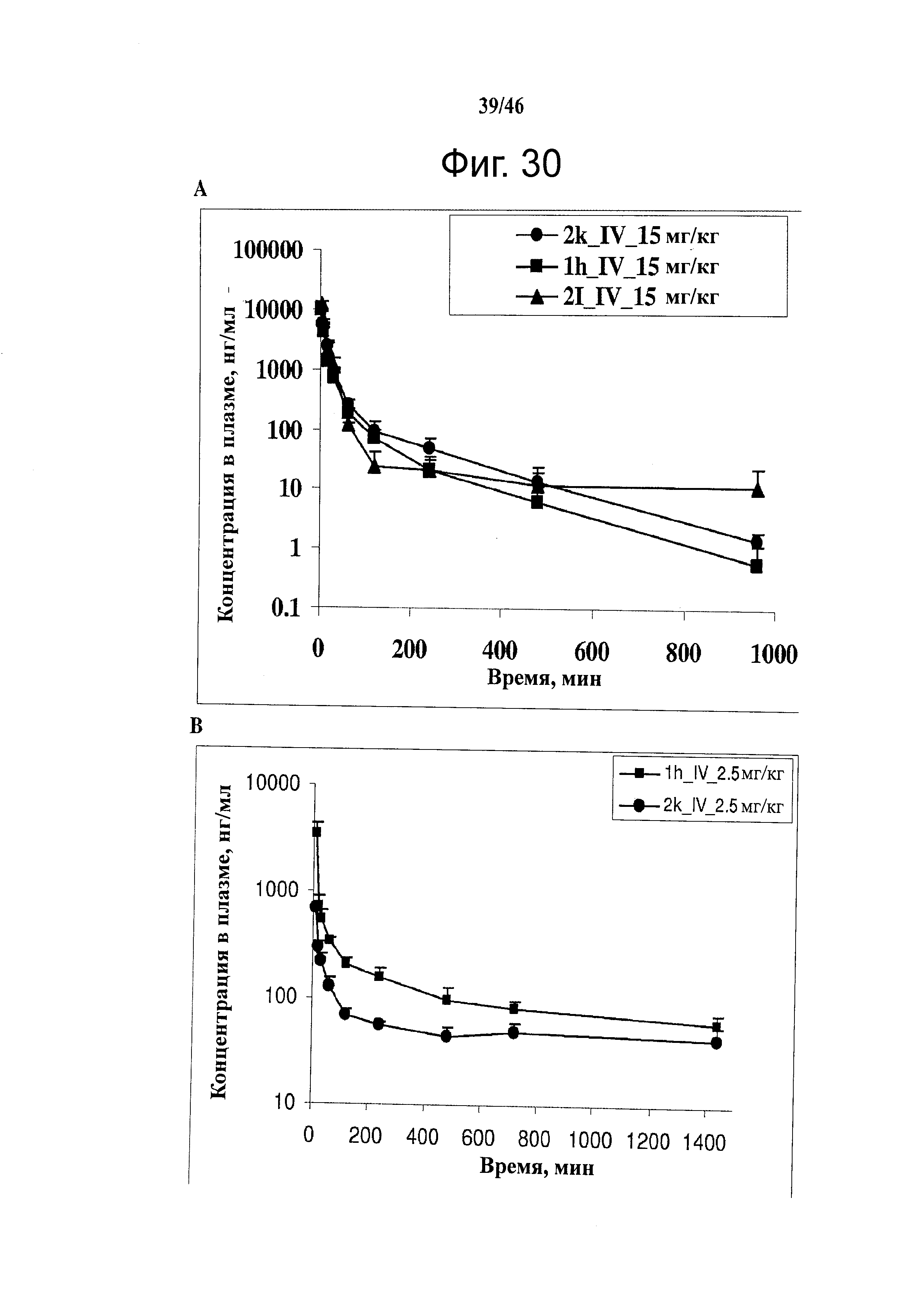
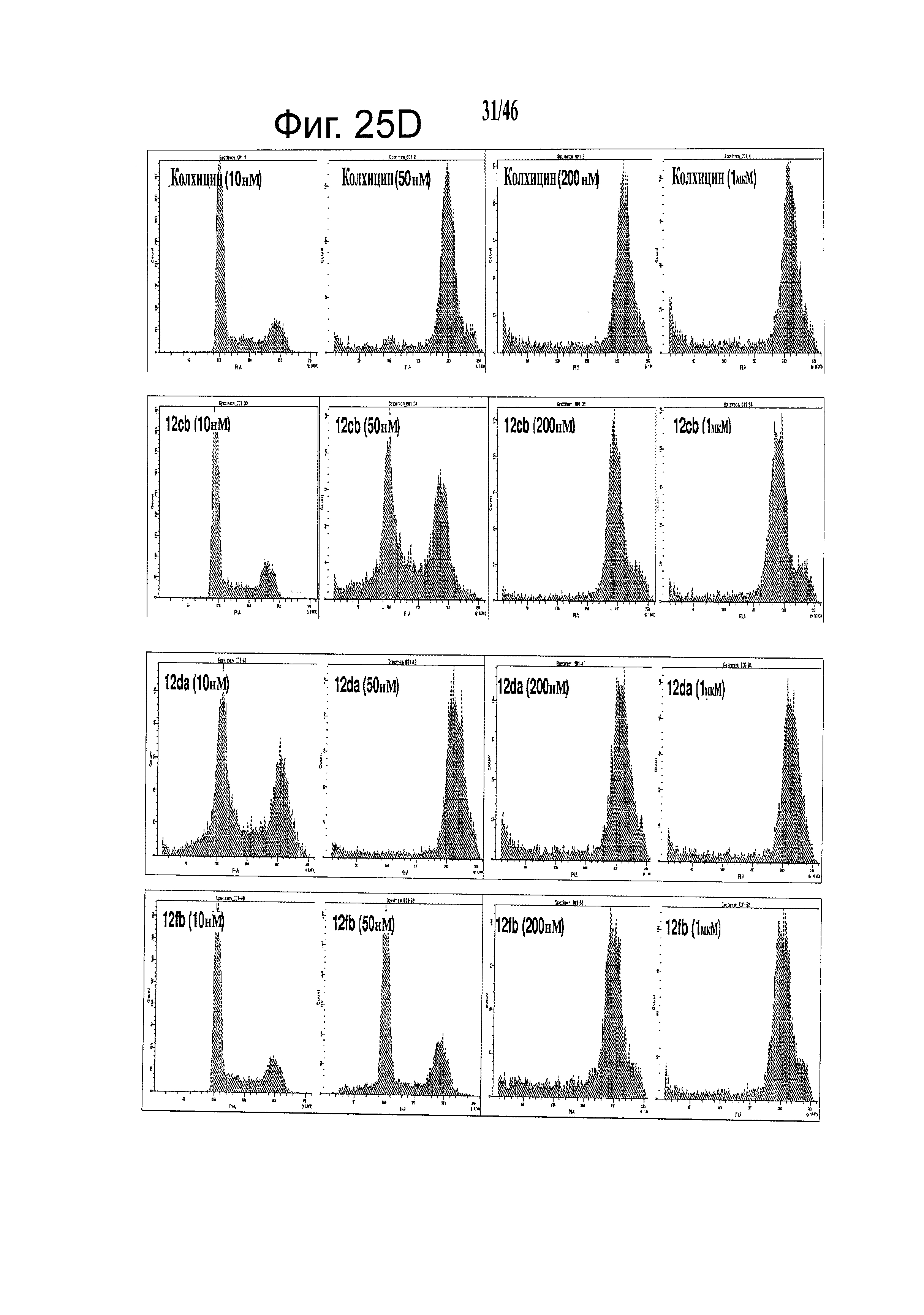
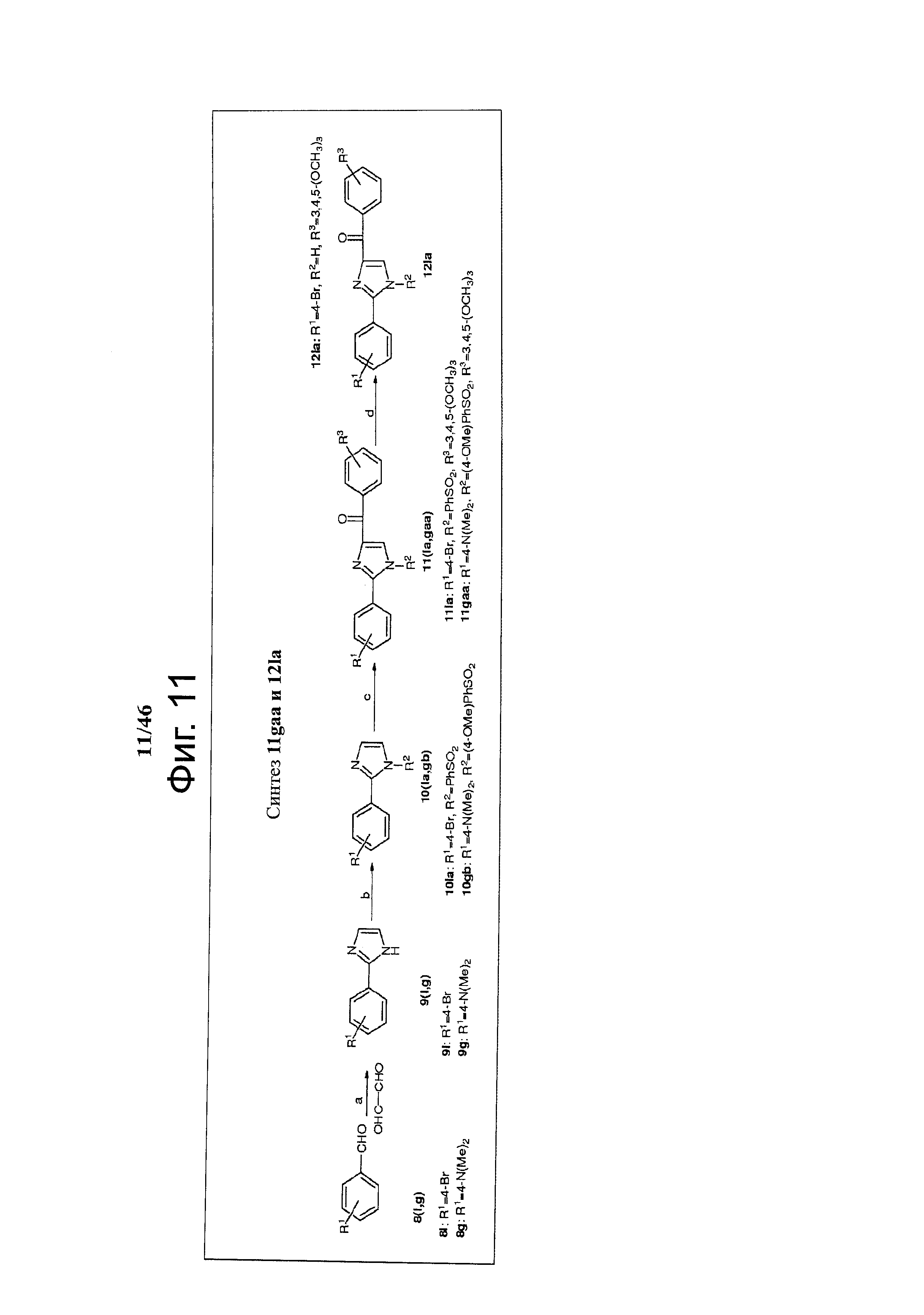
Пирролопиразиновые ингибиторы киназы
Новые пиридиноны и пиридазиноны
Соединения для лечения рака
Соединения sarm и способы их применения
Новые соединения и композиции для нацеливания на злокачественные стволовые клетки
Новые способы направленного воздействия на раковые стволовые клетки
Композиции вакцин и способы их применения
Соединения для лечения рака
Селективные лиганды-разрушители андрогенных рецепторов (sard) и способы их применения
Соединение для лечения рака
Пирролопиразиновые ингибиторы киназы
Новые пиридиноны и пиридазиноны
Соединения для лечения рака
Соединения sarm и способы их применения
Новые соединения и композиции для нацеливания на злокачественные стволовые клетки
Новые способы направленного воздействия на раковые стволовые клетки
Композиции вакцин и способы их применения
Соединения для лечения рака
Способ лечения андроген-рецептора(ar)-положительных форм рака молочной железы с использованием селективных модуляторов андрогенных рецепторов(sarm)
Новые способы направленного воздействия на раковые стволовые клетки

