Результат интеллектуальной деятельности: ПОЛУЧЕНИЕ ПРОМЕЖУТОЧНЫХ ПРОДУКТОВ ДЛЯ СИНТЕЗА ПОЗАКОНАЗОЛА
Вид РИД
Изобретение
Настоящее изобретение относится к получению хиральных соединений, в частности к получению хиральных соединений, которые можно использовать в качестве промежуточных продуктов для получения фунгицидных средств, предпочтительно позаконазола.
Уровень техники
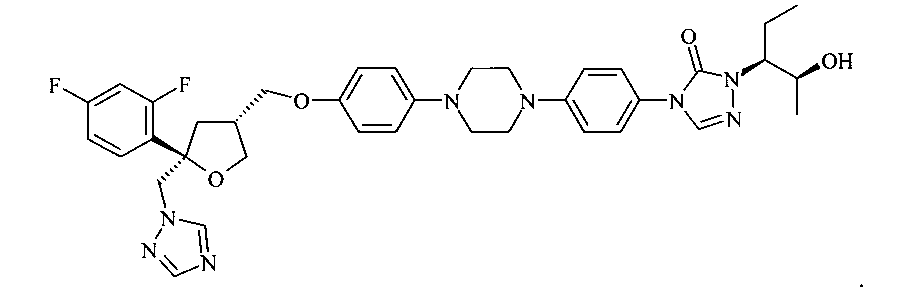
Позаконазол (регистрационный № CAS 171228-49-2; название CAS: 2,5-ангидро-1,3,4-тридезокси-2-C-(2,4-дифторфенил)-4-[[4-[4-[4-[1-[(1S,2S)-1-этил-2-гидроксипропил]-1,5-дигидро-5-оксо-4Н-1,2,4-триазол-4-ил]фенил]-1-пиперазинил]фенокси]метил]-1-(1Н-1,2,4-триазол-1-ил)-D-трео-пентит) является триазольным фунгицидным лекарственным средством, обладающим структурой:
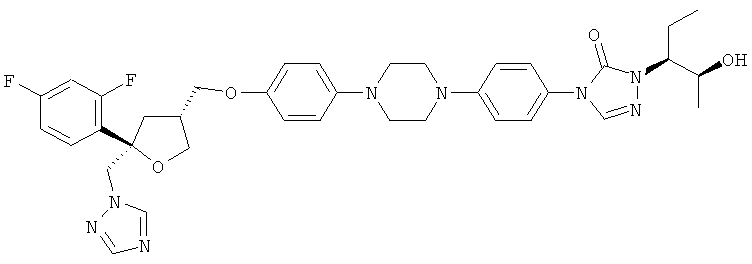 .
.
Позаконазол используется, например, для предупреждения и/или лечения инвазивных грибковых инфекций, вызванных штаммами Candida, штаммами Mucor, штаммами Aspergillus, штаммами Fusarium или штаммами Coccidioides, у пациентов с ослабленным иммунитетом и/или у пациентов, у которых заболевание устойчиво к воздействию других фунгицидных средств, таких как амфотерицин B, флуконазол или итраконазол, и/или у пациентов, которые не переносят эти фунгицидные средства.
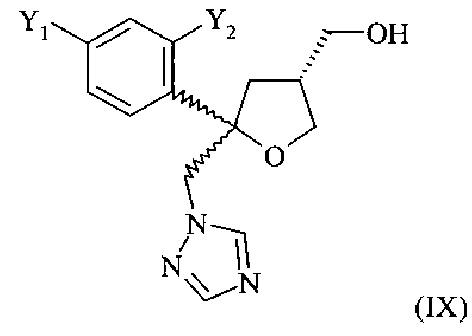
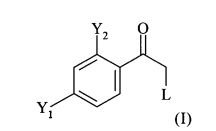
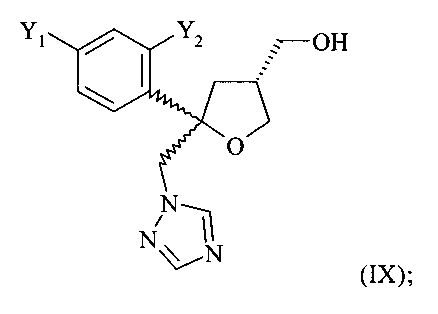
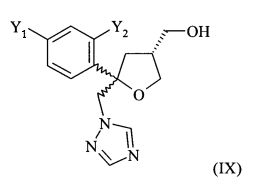
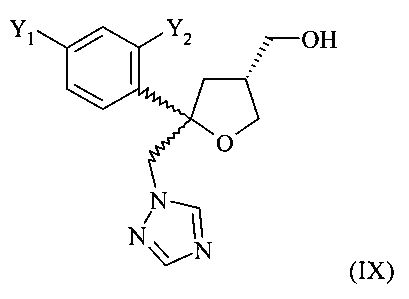
Одним из важных промежуточных продуктов для получения позаконазола является соединение формулы (IX)
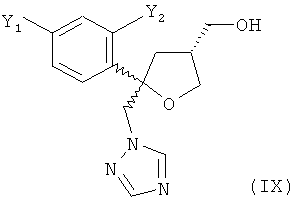
где оба остатка Y1 и Y2 обозначают F. Поэтому постоянно требуются эффективные способы получения этого промежуточного продукта.
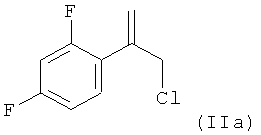
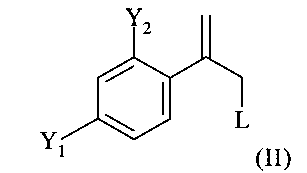
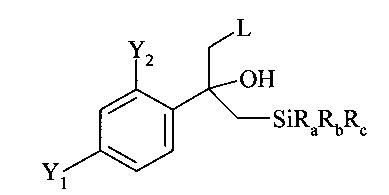
В соответствии с известными способами предшествующего уровня техники общим обычным исходным веществом для получения хирального соединения формулы (IX) является замещенный олефин следующей формулы (II)
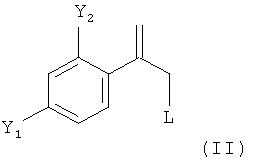
где L обозначает подходящую отщепляющуюся группу, такую как Cl, Br и сульфонаты.
Однако указанные способы предшествующего уровня техники обеспечивают только сравнительно сложные способы получения этих соединений формулы (II), ниже обозначаемые как Ar-C(=CH2)-CH2-L, в частности соединений в которых Y1 и Y2 оба обозначают F.
В WO 94/25452 раскрыт способ, в котором Ar-C(=CH2)-CH2-L получают по реакции соответствующего аллилового спирта Ar-C(=СН2)-СН2-OH с бромирующим реагентом или сульфирующим реагентом. В свою очередь, для получения аллилового спирта в литературе предложены несколько методик.
В одной методике, описанной в WO 94/25452, в качестве исходного вещества используют Ar-C(=O)-CH2-L, из которого по трехстадийной методике получают аллиловый спирт. На первой стадии Ar-C(=O)-CH2-Cl вводят в реакцию с KOAc (ацетат калия) и получают Ar-С(=O)-СН2-OAc, который затем вводят в реакцию с CH3Ph3PBr (метилтрифенилфосфоний бромид) и NaHMDS (гексаметилдисилазан натрия) в присутствии THF (тетрагидрофуран) и получают Ar-С(=CH2)-СН2-OAc. На третьей стадии Ar-С(=CH2)-СН2-ОАс дополнительно вводят в реакцию с KOH (гидроксид калия) и в заключение получают Ar-C(=СН2)-СН2-OH. Кроме того, что в этой методике используют 3 последовательные стадии, каждую из которых необходимо проводить в отдельном сосуде, следует отметить, что на второй стадии продукт реакции нелегко отделить от побочного продукта, трифенилфосфиноксида.
В другой методике, описанной в публикации P. Blundell et al., Synlett 1994, pp.263-265, в качестве исходного вещества используют Ar-Br, который на первой стадии превращают в реагент Гриньяра, который в свою очередь, вводят в реакцию с (Cl-СН2)2C=O (1,2-дихлорацетон), из которого получают Ar-С(OH)(CH2Cl)2, который на второй стадии обрабатывают карбонатом калия и получают эпоксид. Этот эпоксид затем, в свою очередь, на третьей стадии, превращают в Ar-С(=СН2)-CH2-OH. Кроме того, что в этой методике получения аллилового спирта используют 3 последовательные стадии, каждую из которых необходимо проводить в отдельном сосуде, известно, что реагент Гриньяра, полученный из Ar-Br, т.е. 2,4-дифторбромбензол, является потенциально опасным соединением.
В WO 95/16658 А1 предложен другой способ получения Ar-C(=CH2)-CH2-L, в котором в качестве исходного вещества используют Ar-C(=O)-CH3. В реакции Гриньяра с последующим элиминированием получают олефин Ar-С(=CH2)-CH3, который затем вводят в радикальное галогенирование и получают Ar-С(=СН2)-CH2-L (где L=Cl, Br). ПО сравнению с двумя методиками, описанными выше, эту методику, которая хотя и включает 3 стадии, можно провести только в 2 реакционных сосудах, поскольку реакцию Гриньяра и последующее элиминирование можно провести в одном сосуде. Однако основным недостатком этой методики является то, что галогенирование дает нежелательную смесь аллилгалогенидов и винилгалогенидов.
Поэтому объектом настоящего изобретения является разработка улучшенного способа получения хирального соединения формулы (IX), в котором исходное вещество - соединение формулы (II), получают новым способом, который лучше указанных известных способов предшествующего уровня техники.
Согласно изобретению было установлено, что указанное исходное вещество можно получить способом, который можно провести только в одном реакционном сосуде с хорошими выходами, причем способ включает только 2 стадии реакции. Согласно изобретению неожиданно было установлено, что для получения указанного выше аллилхлорида можно использовать методологию олефинирования, известную, как олефинирование по Петерсону. По этой методологии олефинирования, впервые описанной в публикации "D.J. Peterson, Carbonyl olefination reaction using silyl-substituted organometallic compounds; J. Org. Chem. (1968) 33 (2) pp.780-784", альфа-силильный карбанион вводят в реакцию с кетонами или альдегидами с получением бета-гидроксисиланов, которые можно подвергнуть элиминированию с получением алкенов. Большинство известных примеров олефинирования по Петерсону проводят в диэтиловом эфире, который нельзя использовать в промышленных способах по соображениям безопасности. В редких случаях в качестве альтернативного растворителя описано использование тетрагидрофурана.
Как отмечено выше, соединения формулы (IX) и их соли являются важными промежуточными продуктами для получения фунгицидных средств. По ряду причин является желательным присутствие таких промежуточных продуктов в виде кристаллических соединений. Однако по известным из литературы способам соединения формулы (IX) и их соли, в частности соединения формулы (IX), где оба Y1 и Y2 обозначают F, не образуются в виде по меньшей мере частично кристаллических соединений.
Поэтому другим объектом настоящего изобретения является разработка способа, с помощью которого соединение формулы (IX) и их соли, в частности соединения формулы (IX), где оба Y1 и Y2 обозначают F, получают в виде по меньшей мере частично кристаллического соединения.
Другим объектом настоящего изобретения является получение соединения формулы (IX) и их соли, в частности соединения формулы (IX), где оба Y1 и Y2 обозначают F, в виде по меньшей мере частично кристаллического соединения.
Краткое изложение сущности изобретения
Поэтому настоящее изобретение относится к способу получения хирального соединения формулы (IX)
или его соли, где Y1 и Y2 независимо обозначают F или Cl, предпочтительно F, который включает:
(1.1) реакцию соединения формулы (I)
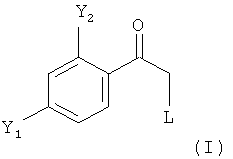
в которой L обозначает отщепляющуюся группу, предпочтительно галоген, более предпочтительно Cl, в растворителе с нуклеофильным соединением, содержащим нуклеофильный остаток RaRbRcSi-CH2, где Ra, Rb, и Rc являются одинаковыми или разными и выбраны из группы, включающей необязательно подходящим образом замещенные алкильные и арильные остатки, с получением реакционной смеси, содержащей в качестве промежуточного продукта бета-гидроксисилан формулы
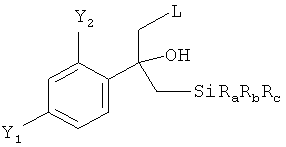
(1.2) обработку полученной реакционной смеси, предпочтительно без замены растворителя, реагентом промотирующим реакцию элиминирования, с получением реакционной смеси, содержащей соединение формулы (II)
.
В особенно предпочтительном варианте осуществления настоящее изобретение относится к способу, описанному выше, в котором получают соединение формулы (IX) или его соль
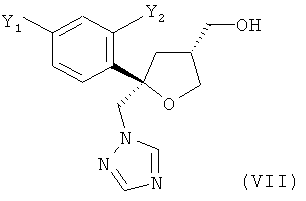
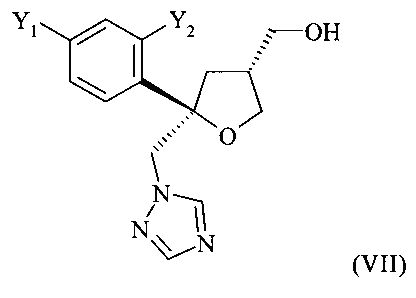
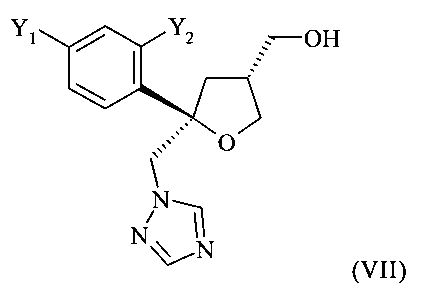
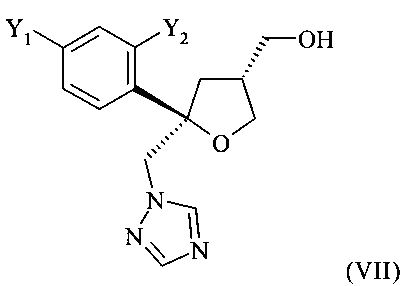
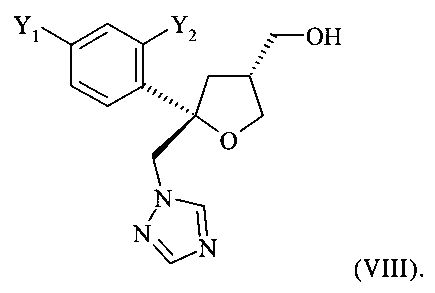
которое содержит цис-изомер формулы (VII) или его соль
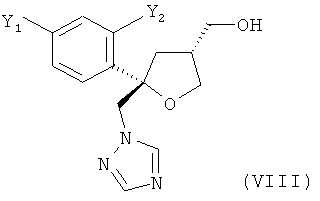
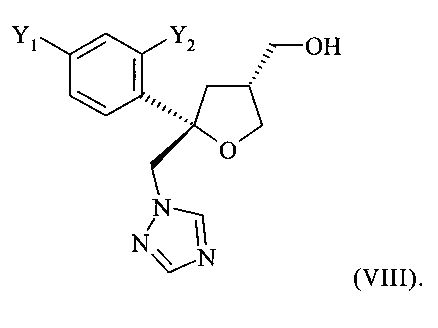
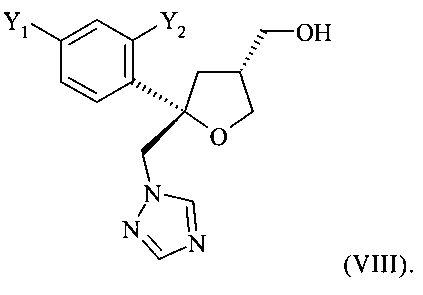
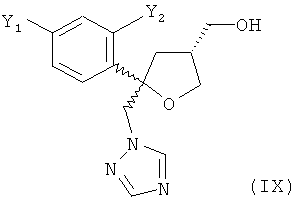
в виде смеси с его транс-изомеров формулы (VIII) или его солью
 ,
,
где предпочтительно от 80 до 95%, более предпочтительно от 85 до 95% молекул соединения формулы (IX) или его соли содержатся в виде цис-изомера формулы (VII) или его соли и предпочтительно от 20 до 5%, более предпочтительно от 15 до 5% молекул соединения формулы (IX) или его соли содержатся в виде транс-изомера формулы (VIII) или его соли.
Кроме того, настоящее изобретение относится к способу, в котором соединение (IX) и тем самым также соединение (VII), кристаллизуют из растворителя необязательно путем добавления подходящего антирастворителя, где растворителем предпочтительно является полярный не смешивающийся с водой растворитель, предпочтительно сложный эфир, такой как этилацетат или изопропилацетат, простой эфир, такой как тетрагидрофуран или метилтетрагидрофуран, кетон, такой как метилизобутилкетон, галогенированный растворитель, такой как дихлорметан, толуол или смесь двух или более этих растворителей, более предпочтительно сложный или простой эфир, более предпочтительно простой эфир, и еще более предпочтительно метилтетрагидрофуран, и где антирастворителем предпочтительно является насыщенный или ненасыщенный углеводород, такой как циклогексан, гексан или гептан или смесь двух или более из них.
Кроме того, настоящее изобретение относится к кристаллическому хиральному соединению формулы (IX) или его соли
где Y1 и Y2 независимо обозначают F или Cl, предпочтительно F, и где от 80 до 95%, предпочтительно от 85 до 95% молекул указанного кристаллического соединения или его соли содержатся в виде цис-изомера формулы (VII) или его соли
и от 20 до 5%, предпочтительно от 15 до 5% молекул указанного кристаллического соединения или его соли содержатся в виде транс-изомера формулы (VIII) или его соли
.
Кроме того, настоящее изобретение относится к применению соединения формулы (IX) или его соли, в частности предпочтительно по меньшей мере частично кристаллического, предпочтительно кристаллического соединения формулы (IX) или его соли, для получения фунгицидного средства, предпочтительно позаконазола:
Перечень чертежей
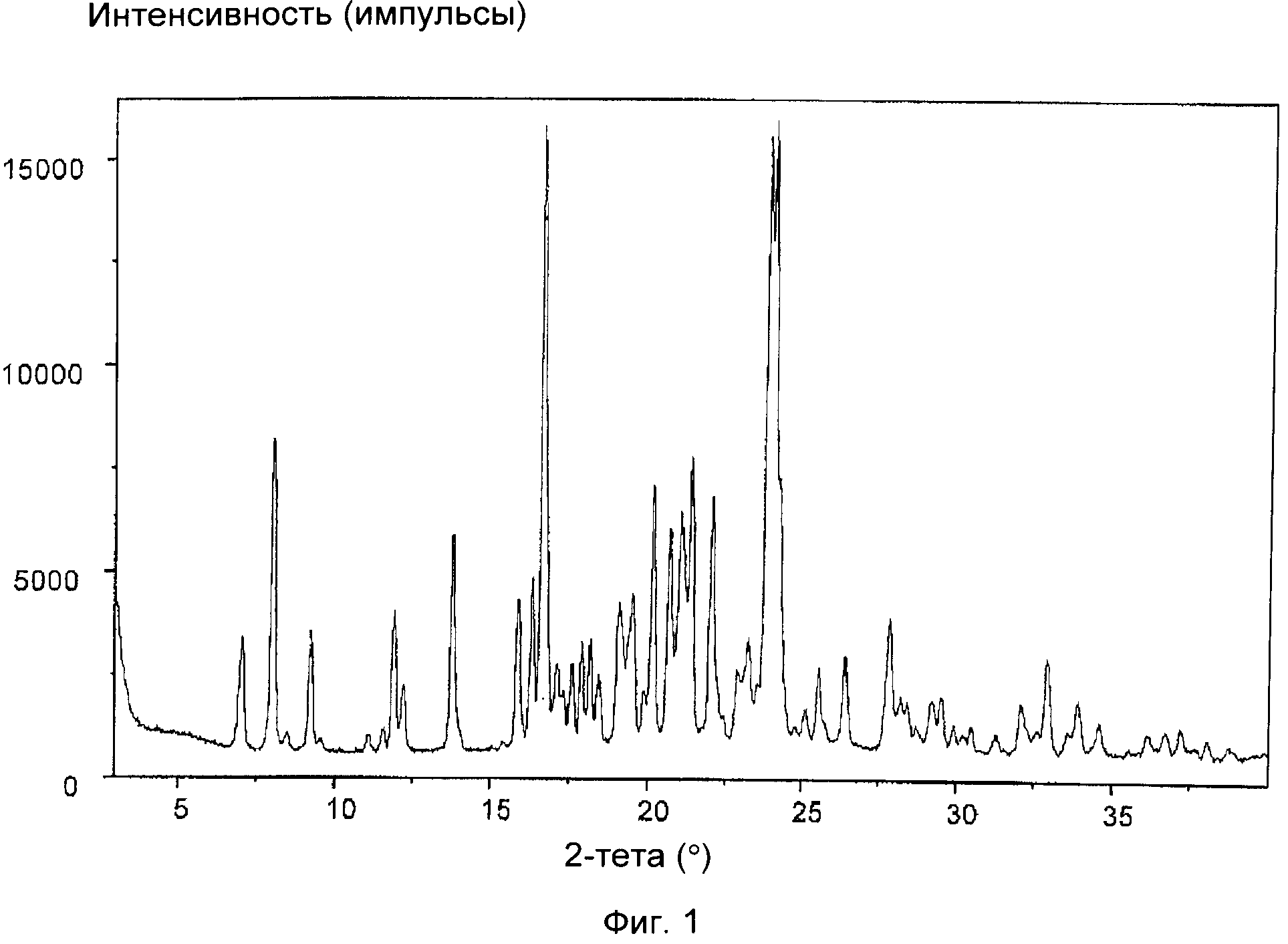
На фиг.1 приведена порошковая рентгенограмма (XRD) соединения формулы (IXa), полученного в примере 1. Отношение цис:транс, т.е. отношение соединение формулы (VIIa): соединение формулы (VIIIa) составляет 9:1.
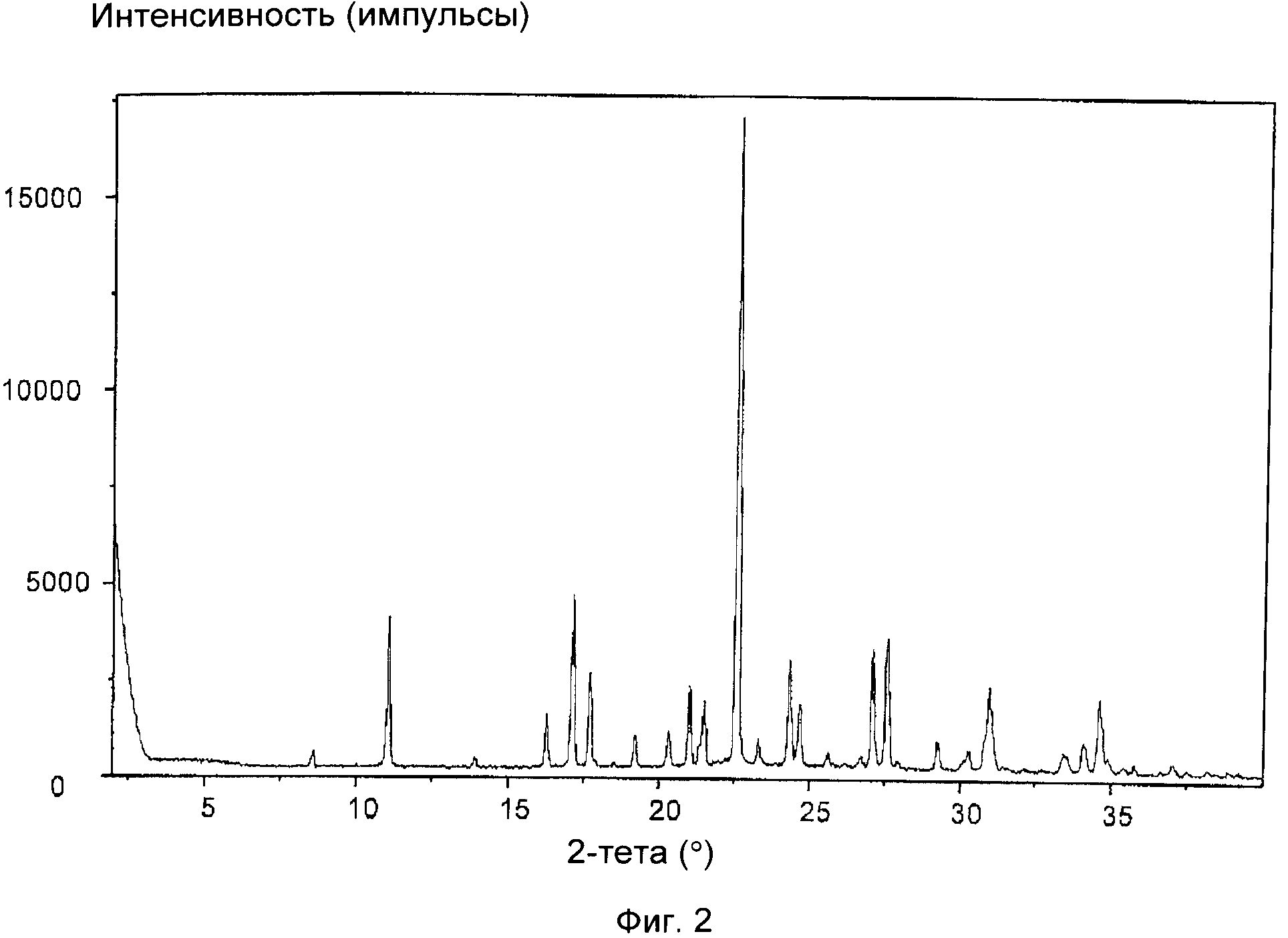
На фиг.2 приведена XRD соли с HCl соединения формулы (IXa), полученной в примере 2(a). Отношение цис:транс, т.е. отношение соль с HCl соединения формулы (VIIa): соль с HCl соединения формулы (VIIIa) составляет 9:1.
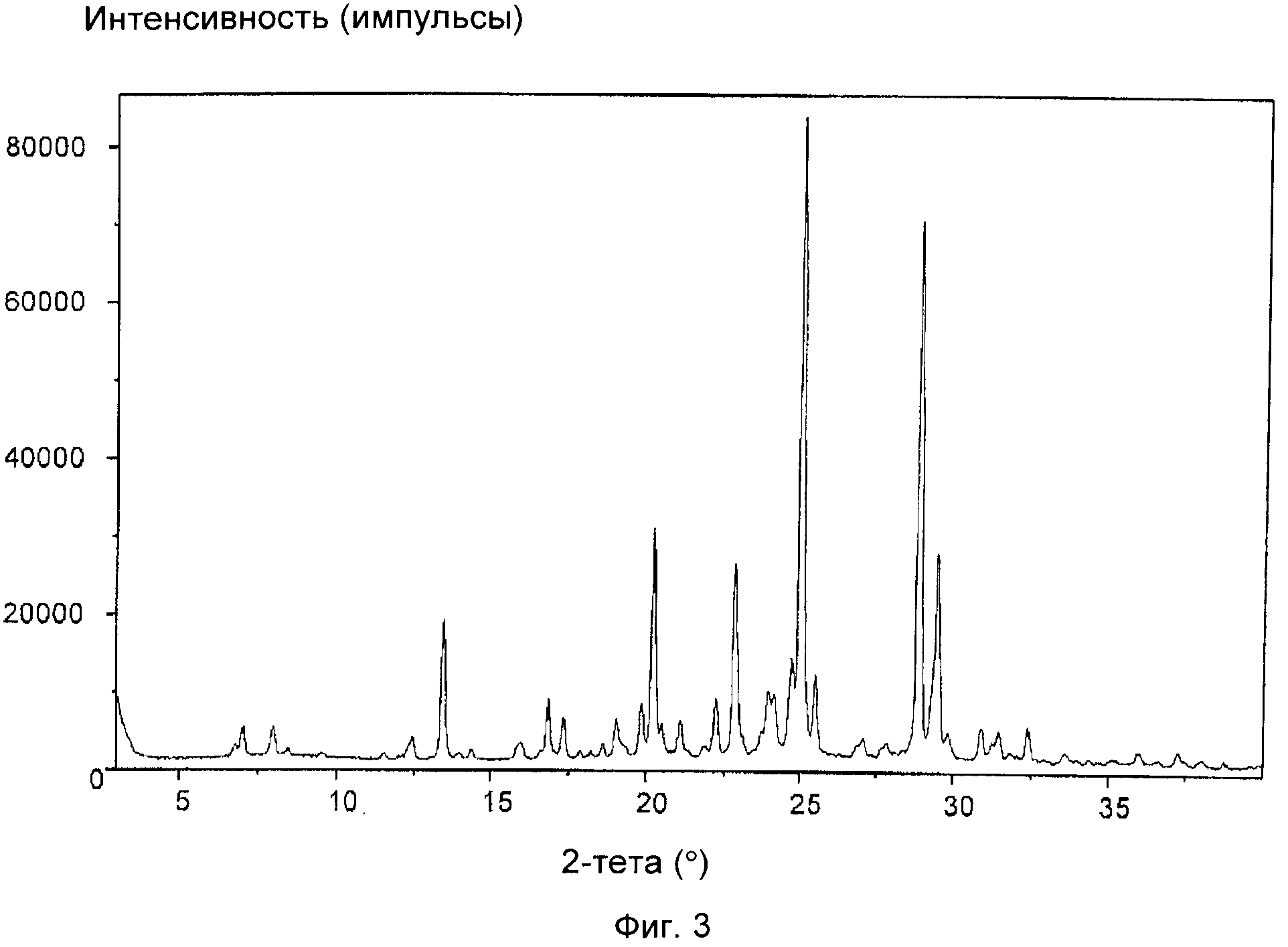
На фиг.3 приведена XRD соли фумаровой кислоты соединения формулы (IXa), полученной в примере 2(b). Отношение цис:транс приведено в таблице, представленной в примере 2(b).
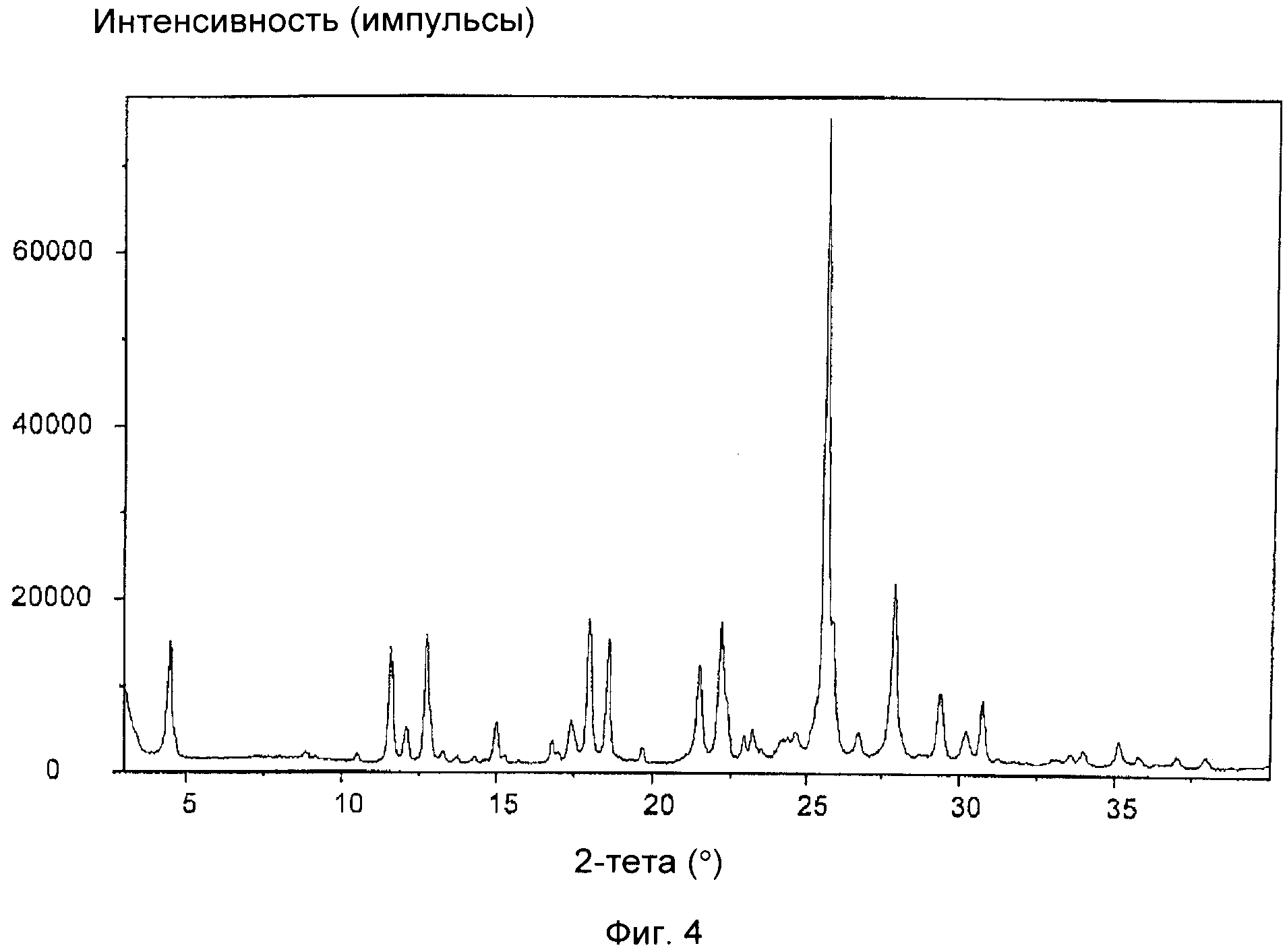
На фиг.4 приведена XRD соли щавелевой кислоты соединения формулы (IXa), полученной в примере 2(b). Отношение цис:транс приведено в таблице, представленной в примере 2(b).
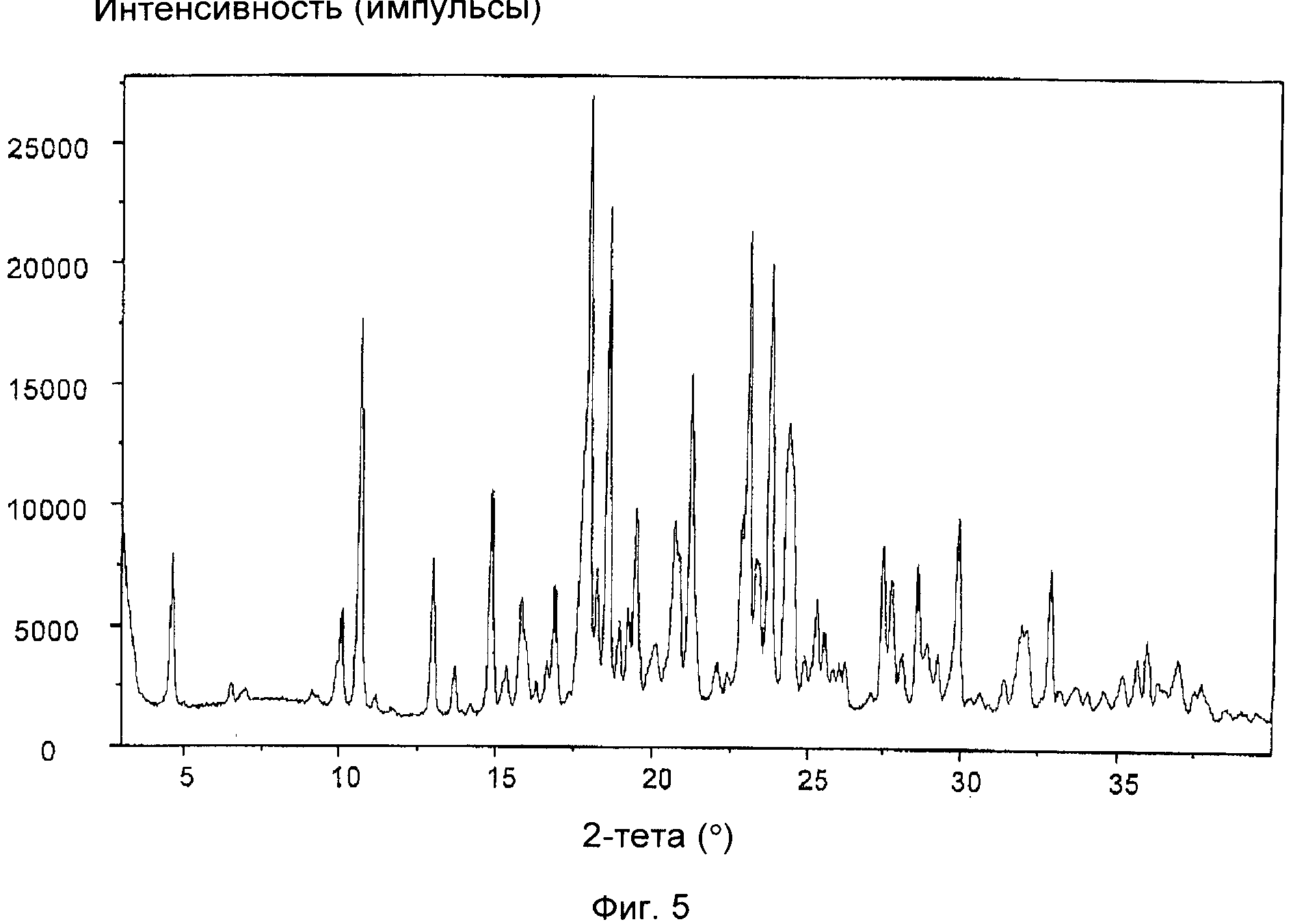
На фиг.5 приведена XRD соли винной кислоты соединения формулы (IXa), полученной в примере 2(b). Отношение цис:транс приведено в таблице, представленной в примере 2(b).
На фиг.1-5 приведены интенсивности, измеренные в импульсах в секунду (линейная шкала), указанные по оси y, а положения, выраженные в значениях 2-тета в градусах, указаны по оси x.
Подробное описание изобретения
Согласно настоящему изобретению, соединение формулы (II), в частности реакционную смесь, содержащую соединение формулы (II), получают способом, который включает:
(1.1) реакцию соединения формулы (I)
в которой L обозначает отщепляющуюся группу, предпочтительно галоген, более предпочтительно Cl, в растворителе с нуклеофильным соединением, содержащим нуклеофильный остаток RaRbRcSi-CH2, где Ra, Rb, и Rc являются одинаковыми или разными и выбраны из группы, включающей необязательно подходящим образом замещенные алкильные и арильные остатки, с получением реакционной смеси, содержащей в качестве промежуточного продукта бета-гидроксисилан формулы
(1.2) обработку полученной реакционной смеси, предпочтительно без замены растворителя, реагентом промотирующим реакцию элиминирования, с получением реакционной смеси, содержащей соединение формулы (II)
Стадии (1.1) и (1.2)
На стадии (1.1) способа, предлагаемого в настоящем изобретении, соединение формулы (I) содержит остатки Y1 и Y2. Согласно настоящему изобретению, Y1 и Y2 независимо обозначают F или Cl. Таким образом, Y1 может обозначать F или С1 и независимо от химической природы Y1, Y2 может обозначать F или Cl. Предпочтительно, если оба Y1 и Y2 обозначают F или Cl. Более предпочтительно, если оба Y1 и Y2 обозначают F.
Термин "отщепляющаяся группа L" при использовании на стадии (1.1) настоящего изобретения, означает любые химические фрагменты L, которые при подходящих условиях проведения реакции, отщепляются от соединения (I) с парой электронов путем гетеролитического расщепления связи. Для этой цели соединение (I), использующееся в настоящем изобретении, может содержать любую отщепляющуюся группу L. Предпочтительно, если отщепляющаяся группа L после отщепления является нейтральным или анионным фрагментом, более предпочтительно анионным фрагментом. Еще более предпочтительно, если L обозначает галоген, такой как, например Cl, Br, I. В еще более предпочтительном варианте осуществления настоящего изобретения L обозначает Cl.
Нуклеофильное соединение, с которым соединение (I) взаимодействует на стадии (1.1), содержит нуклеофильный остаток RaRbRcSi-CH2. На химическую природу этого остатка не налагают специальных ограничений при условии, что получают промежуточный бета-гидроксисилан формулы
.
Термин "промежуточный продукт" при использовании в настоящем изобретении обычно означает бета-гидроксисилан, который содержится в реакционной смеси, полученной на стадии (1.1) и который образуется из реагентов стадии (1.1) и дополнительно реагирует на стадии (1.2). Термин "промежуточный продукт" при использовании здесь не исключает такие бета-гидроксисиланы, которые можно выделить из реакционной смеси, полученной на стадии (1.1)
Нуклеофильным соединением, использующимся на стадии (1.1), может быть любое подходящее соединение, содержащее нуклеофильный остаток RaRbRcSi-CH2, который при реакции с соединением (I) прямо или косвенно приводит к образованию промежуточного бета-гидроксисилана, обсужденного выше. Ra, Rb и Rc, содержащиеся в нуклеофильном соединении, являются одинаковыми или разными и выбраны из группы, включающей необязательно подходящим образом замещенные алкильные и арильные остатки. Термин "необязательно подходящим образом замещенный арильный остаток" при использовании в настоящем изобретении означает арильные остатки, которые содержат, например, до 6 или до 12 атомов углерода. Если такой арильный остаток представляет собой замещенный арильный остаток, количество атомов углерода означает количество атомов углерода в соответствующем незамещенном арильном остатке. Термин "необязательно подходящим образом замещенный алкильный остаток" при использовании в настоящем изобретении означает алкильные остатки, которые содержат, например, от 1 до 20, предпочтительно от 1 до 10 атомов углерода. Если такой алкильный остаток представляет собой замещенный алкильный остаток, то количество атомов углерода означает количество атомов углерода в соответствующем незамещенном алкильном остатке.
В предпочтительном варианте осуществления настоящего изобретения Ra, Rb и Rc, содержащиеся в нуклеофильном соединении, являются одинаковыми или разными и выбраны из группы, включающей алкильные остатки, более предпочтительно незамещенные алкильные остатки, содержащие от 1 до 6 атомов углерода, предпочтительно от 1 до 4 атомов углерода, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил и трет-бутил, более предпочтительно 1 или 2 атома углерода, метил или этил, где предпочтительно Ra, Rb и Rc обозначают метил.
Предпочтительно, если нуклеофильным соединением, использующимся на стадии (1.1) является реагент Гриньяра. Термин "реагент Гриньяра" при использовании здесь означает любой подходящий нуклеофильный металлоорганический реагент, содержащий нуклеофильный остаток RaRbRcSi-СН2. Предпочтительно, если нуклеофильное соединение представляет собой соединение Гриньяра RaRbRcSi-CH2MgX, где X обозначает подходящий анион, который предпочтительно выбран из группы, включающей Cl, Br и I. Более предпочтительно, если соединением Гриньяра является соединение RaRbRcSi-CH2MgCl.
В качестве растворителя, который используется на стадии (1.1), можно использовать любой растворитель или смесь растворителей, предпочтительно растворитель или смесь растворителей, в котором можно провести реакцию Гриньяра. Приемлемыми растворителями являются, например, простые эфиры, такие как общеизвестные диэтиловый эфир и/или тетрагидрофуран (THF). Однако согласно изобретению неожиданно было установлено, что растворители, рассмотренные в предшествующем уровне техники для олефинирования по Петерсону, а именно диэтиловый эфир и THF, можно заменить на метил-трет-бутиловый эфир (МТВЕ). Этот растворитель обеспечивает значительное преимущество, поскольку по сравнению с соединениями, такими как диэтиловый эфир и THF, не образует пероксиды. Таким образом, применение МТВЕ является особенно подходящим в промышленных способах, для которых вопросы безопасности являются первостепенными по значению. Поэтому в особенно предпочтительном варианте осуществления растворитель, использующийся на стадии (1.1), является МТВЕ.
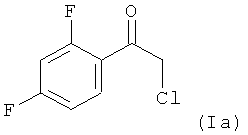
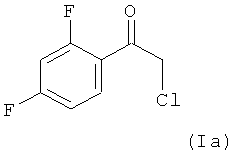
Поэтому в предпочтительном варианте осуществления настоящее изобретение относится к способу, определенному выше, в котором на стадии (1.1), соединением формулы (I) является соединение (Ia)
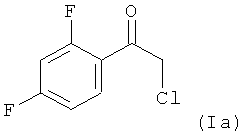
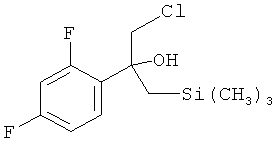
которое взаимодействует в MTBE в качестве растворителя с нуклеофильным соединением (H3C)3Si-CH2MgCl с получением реакционной смеси, содержащей в качестве промежуточного продукта бета-гидроксисилан формулы:
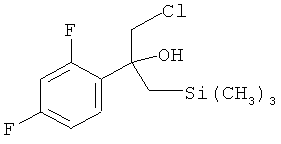
На температуру, при которой проводят реакцию на стадии (1.1), не налагают специальных ограничений при условии, что получают реакционную смесь, которая обеспечивает протекание реакции на стадии (1.2). Предпочтительно, если реакцию на стадии (1.1) проводят при температуре в диапазоне от -50 до +20°C, более предпочтительно от -40 до +15°C, более предпочтительно от -30 до +10°C, более предпочтительно от -20 до +10°C, более предпочтительно от -15 до +5°C, например при температуре в диапазоне от -15 до -10°C или от -10 до -5°C или от -5 до 0°C или от 0 до +5°C.
В качестве общей методики проведения олефинирования по Петерсону в литературе описан двухстадийный способ, в котором после проведения реакции Гриньяра проводят замену растворителя. Дается ссылка на публикацию Tetrahedron Letters 32 (1991), pp.7545-7548. Согласно изобретению неожиданно было установлено, что после стадии (1.1) настоящего изобретения не требуется замена растворителя и что промежуточный продукт, полученный на стадии (1.1), можно обработать подходящим реагентом, который промотирует реакцию элиминирования с помощью значительного упрощенного способа.
Поэтому согласно настоящему изобретению, реакционную смесь, полученную на стадии (1.1), обрабатывают на стадии (1.2), предпочтительно без замены растворителя, реагентом промотирующим реакцию элиминирования, с получением реакционной смеси, содержащей соединение формулы (II)
.
Поскольку по литературным данным вторая стадия олефинирования по Петерсону включает использование BF3∗Et2O (эфират трифторида бора), другим главным преимуществом настоящего изобретения является то, что на этой стадии реакции полностью исключено использование потенциально опасных химикатов, таких как эфират BF3. Как отмечено выше, осуществление способа, предлагаемого в настоящем изобретении, без замены растворителя после стадии (1.1) особенно предпочтительно, если в качестве растворителя на стадии (1.1) используют MTBE.
На температуру, при которой проводят реакцию на стадии (1.2), не налагают специальных ограничений при условии, что получают реакционную смесь, содержащую соединение формулы (II). Предпочтительно, если обработку на стадии (1.2) проводят при температуре в диапазоне от -20 до +70°C. Предпочтительные диапазоны температуры составляют, например, от -20 до -10°C или от -10 до 0°C, или от 0 до +10°C, или от +10 до +20°C, или от +20 до +30°C, или от +30 до +40°C, или от +40 до +50°C, или от +50 до +60°C, или от +60 до +70°C.
На реагент, промотирующий реакцию элиминирования, использующийся на стадии (12), не налагают специальных ограничений при условии, что получают соединение формулы (II), предпочтительно без замены растворителя после стадии (1.1). Предпочтительным реагентом является кислота или смесь двух или более кислот. Более предпочтительным реагентом является неорганическая кислота или смесь двух или более неорганических кислот. Особенно предпочтительным является использование серной кислоты. Предпочтительно, если в качестве реагента используют серную кислоту, температура, при которой проводят стадию (1.2), находится в диапазоне от +40 до +50°C.
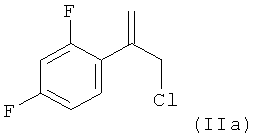
Поэтому в предпочтительном варианте осуществления настоящее изобретение относится к способу, определенному выше, в котором на стадии (1.2), реакционную смесь, полученную на стадии (1.1), без замены растворителя обрабатывают серной кислотой, промотирующей реакцию элиминирования, с получением реакционной смеси, содержащей, в качестве соединения формулы (II) соединение (IIa):
Таким образом, в еще более предпочтительном варианте осуществления настоящее изобретение относится к способу, определенному выше, который включает:
(1.1) реакцию соединения формулы (Ia)
с (H3C)3Si-CH2MgCl в MTBE в качестве растворителя с получением реакционной смеси, содержащей в качестве промежуточного продукта бета-гидроксисилан формулы
(1.2) обработку полученной реакционной смеси без замены растворителя МТВЕ серной кислотой, промотирующей реакцию элиминирования, с получением реакционной смеси, содержащей соединение формулы (IIa)
.
В еще более предпочтительном варианте осуществления настоящее изобретение относится к способу, определенному выше, который включает
(1.1) реакцию соединения формулы (Ia)
с (H3C)3Si-CH2MgCl в MTBE в качестве растворителя при температуре в диапазоне от -15 до +5°C с получением реакционной смеси, содержащей в качестве промежуточного продукта бета-гидроксисилан формулы
(1.2) обработку полученной реакционной смеси без замены растворителя MTBE при температуре в диапазоне от +40 до +50°C серной кислотой, промотирующей реакцию элиминирования, с получением реакционной смеси, содержащей соединение формулы (IIa)
.
Из соединения, содержащегося в реакционной смеси, полученной на стадии (1.2), обсужденного выше, получают соединение формулы (IX). На конкретные последовательности стадий реакции, приводящей от соединения формулы (II) к соединению формулы (IX), не налагают специальных ограничений. При использовании предпочтительной последовательности стадий реакции способ, предлагаемый в настоящем изобретении, определенный выше, дополнительно включает
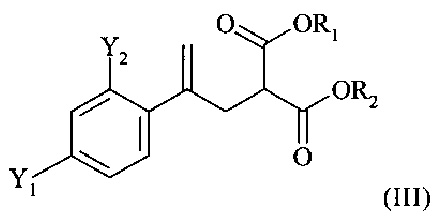
(2) реакцию соединения формулы (II) со сложным эфиром малоновой кислоты R1OOC-CH2-COOR2 с получением соединения формулы (III)
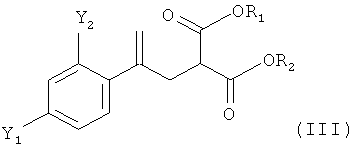 ,
,
в которой R1 и R2 независимо обозначают необязательно подходящим образом замещенную алкильную группу, содержащую от 1 до 5 атомов углерода;
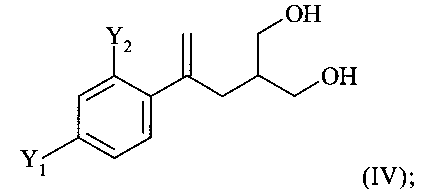
(3) восстановление соединения формулы (III) с получением соединения формулы (IV)
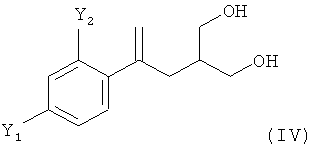 ;
;
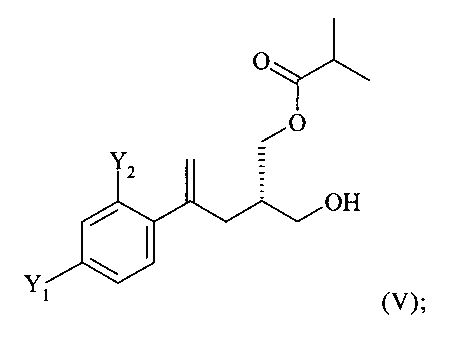
(4) ацилирование соединения формулы (IV) изомасляным ангидридом с получением соединения формулы (V)
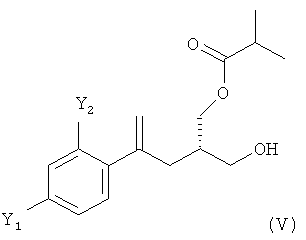 ;
;
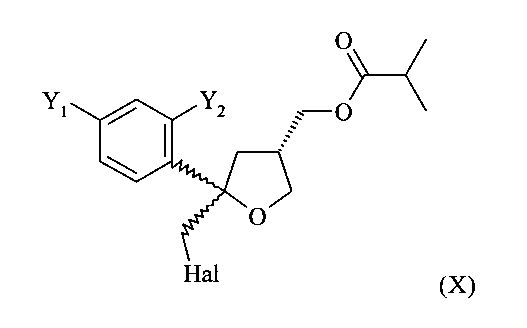
(5) реакцию соединения формулы (V) с галогеном Hal2, выбранным из группы, включающей Cl2, Br2 и I2, предпочтительно I2, в присутствии основания в растворителе с получением соединения формулы (X)
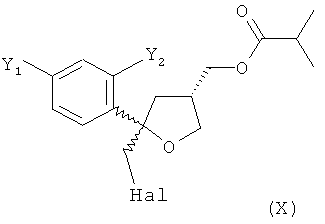 ;
;
(6.1) нагревание соединения формулы (X), предпочтительно при отсутствии DMPU (1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон) в растворителе предпочтительно DMSO (диметилсульфоксид), с солью щелочного металла 1,2,4-триазола, предпочтительно натриевой солью, и обработку полученной реакционной смеси основанием с получением соединения формулы (IX)
;
(6.2) выделение соединения формулы (IX) из реакционной смеси, полученной на стадии (6.1), путем экстракции подходящим растворителем.
Стадия (2)
На стадии (2) настоящего изобретения соединение формулы (II) предпочтительно вводят в реакцию со сложным эфиром малоновой кислоты R1OOC-CH2-COOR2, в котором R1 и R2 независимо обозначают необязательно подходящим образом замещенную алкильную группу, содержащую от 1 до 5 атомов углерода. Количество атомов углерода означает количество атомов углерода в незамещенном алкильном остатке. Предпочтительные алкильные группы R1 и R2 содержат от 1 до 4 атомов углерода, такие как метил, этил, н-пропил, изопропил, н-бутил, изобутил и трет-бутил. Еще более предпочтительные алкильные группы R1 и R2 содержат 1 или 2 атома углерода, такие как метил или этил, и этил является особенно предпочтительным. Еще более предпочтительные алкильные группы R1 и R2 представляют собой незамещенные алкильные группы.
На стадии (2) еще более предпочтительно вводить сложный эфир малоновой кислоты R1OOC-CH2-COOR2 в реакцию с соединением (II) в присутствии подходящего сильного основания, предпочтительно сильного основания щелочного металла, обеспечивающего реакцию соответствующего аниона CH(COOR1)(COOR2), полученного из сложного эфира малоновой кислоты R1OOC-CH2-COOR2. В качестве щелочного металла предпочтительным является натрий. Подходящими основаниями являются, например, NaH или NaOH и NaOH является предпочтительным. NaOH можно использовать в любом подходящем виде. В предпочтительном варианте осуществления NaOH используют в виде твердого вещества, такого как, например, чешуйки NaOH. Растворитель, в котором проводят стадию (2), можно выбрать, например, в соответствии с конкретной химической природой сильного основания, обсужденного выше. Приемлемыми растворителями являются, например THF, DMSO и т.п. Согласно настоящему изобретению, DMSO является предпочтительным. Температуры, при которых проводят реакцию на стадии (2), можно выбрать в соответствии с растворителем и основанием. Предпочтительные температуры находятся в диапазоне от 0 до 35°C, более предпочтительно от 25 до 30°C.
Продукт реакции на стадии (2), соединение формулы (III)
,
предпочтительно подходящим образом выделять из реакционной смеси, полученной на стадии (2). В предпочтительном варианте осуществления это выделение включает стадию, на которой соединение (III) выделяют путем экстракции в подходящем растворителе. Согласно настоящему изобретению, из подходящих растворителей предпочтительным является циклогексан.
Органический слой, полученный в результате экстракции, можно промыть в одну или большее количество стадий. В качестве реагентов для промывки следует отметить воду и водные растворы щелочей, такие как, например, водные растворы оснований щелочных металлов, таких как гидроксид щелочного металла, предпочтительно гидроксид натрия.
Стадия (3)
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (III), полученное на стадии (2), предпочтительно восстанавливают и из него получают соединение формулы (IV):
Восстановление на стадии (3) можно провести по любой подходящей методике с использованием любого подходящего восстановительного реагента. Согласно настоящему изобретению, использование гидридного восстановительного реагента является предпочтительным. Такими гидридными восстановительными реагентами является, например, борогидрид натрия (NaBH4), борогидрид лития (LiBH4), алюмогидрид лития (LiAlH4), диизобутилалюминийгидрид (ДИБАЛ) или триэтилборогидрид лития (LiEt3BH). В предпочтительном варианте осуществления настоящего изобретения на стадии (3) в качестве восстановительного реагента используют LiBH4.
В соответствии с предшествующим уровнем техники необходимо использовать не менее 3 мол. экв. LiBH4 в пересчете на соединение формулы (III) (см. WO 94/25452, стр.31, раздел "Получение 5"). Однако в отличие от данных предшествующего уровня техники, согласно изобретению неожиданно было установлено, что восстановительный реагент LiBH4 можно использовать в гораздо меньшем избытке в пересчете на сложный эфир малоновой кислоты (III). В улучшенном способе, предлагаемом в настоящем изобретении, используют не более 2 мол. экв. LiBH4 в пересчете на соединение формулы (III), что означает, что в отличие от предшествующего уровня техники можно сэкономить не менее 33% восстановительного реагента. Таким образом, в особенности для проводимого в промышленном масштабе способа, настоящее изобретение обеспечивает экономические и экологические преимущества. Таким образом, настоящее изобретение относится к способу, определенному выше, в котором LiBH4 используют в качестве восстановительного реагента и его предпочтительно используют в количестве, составляющем не более 2 мол. экв. в пересчете на соединение (III).
На растворитель, в котором проводят реакцию на стадии (3), не налагают специальных ограничений при условии, что получают соединение формулы (IV). Предпочтительные растворители выбраны из группы, включающей воду, спирт и смесь воды и по меньшей мере одного спирта. Предпочтительными спиртами являются метанол, этанол и изопропанол. Поэтому растворитель предпочтительно выбран из группы, включающей воду, метанол, этанол, изопропанол и смесь воды и по меньшей мере одного из этих спиртов, более предпочтительно из группы, включающей воду, этанол, изопропанол и смесь воды и по меньшей мере одного из этих спиртов, более предпочтительно из группы, включающей воду, изопропанол и смесь воды и изопропанола.
Согласно изобретению неожиданно было установлено, что в частности для наиболее предпочтительного восстановительного реагента, использующегося на стадии (3), LiBH4, смесь воды и изопропанола является наиболее подходящим растворителем. В отличие от известного факта, заключающегося в том, что вода разлагает гидридный восстановительный реагент, установлено, что присутствие воды благоприятно для стадии (3) способа, предлагаемого в настоящем изобретении. Если не ограничиваться какой-либо теорией, то можно полагать, что это может быть обусловлено тем, что некоторое количество воды повышает растворимость реагента LiBH4 и/или его предшественников NaBH4 и LiCl, и тем самым повышает скорость реакции и, таким образом, в свою очередь, с избытком компенсирует разложение восстановительного реагента. Поэтому в других предпочтительных вариантах осуществления растворитель, использующийся на стадии (3), содержит воду, причем растворитель предпочтительно содержит от 1 до 20 об.%, более предпочтительно от 5 до 15 об.% воды.
Температуры, при которых проводят реакцию на стадии (3), можно выбрать в соответствии с растворителем и восстановительным реагентом. Предпочтительные температуры находятся в диапазоне от 0 до 40°C, более предпочтительно от 20 до 35°C, более предпочтительно от 25 до 30°C.
Продукт восстановления на стадии (3), соединение формулы (IV), предпочтительно надлежащим образом выделяют из реакционной смеси, полученной на стадии (3). В предпочтительном варианте осуществления это выделение включает стадию, на которой соединение (IV) выделяют путем экстракции подходящим растворителем. Согласно настоящему изобретению, из подходящих растворителей толуол является предпочтительным.
Стадия (4)
На стадии (4) настоящего изобретения соединение формулы (IV) предпочтительно ацилируют изомасляным ангидридом с получением соединения формулы (V)
.
Более предпочтительно, если ацилирование на стадии (4) проводят в присутствии подходящего фермента, предпочтительно фермента Novo SP 435 в подходящем растворителе, предпочтительно ацетонитриле или толуоле, более предпочтительно толуоле, например по методике, аналогичной описанной в WO 97/22710. Выбор толуола в качестве растворителя также благоприятен для экстракционной обработки, поскольку не требуется дополнительный растворитель. При использовании ацетонитрила в качестве растворителя для экстракционной обработки требуется использование дополнительного несмешивающегося растворителя.
Температуры, при которых проводят ацилирование на стадии (4), можно выбрать в соответствии с растворителем, ацилирующим реагентом и ферментом. Предпочтительные температуры находятся в диапазоне от -20 до -5°C, более предпочтительно от -15 до -10°C, более предпочтительно от 25 до 30°C.
Полученную реакционную смесь предпочтительно дополнительно обрабатывают подходящим основанием, таким как, например, гидрокарбонат натрия.
В особенно предпочтительном варианте осуществления настоящего изобретения соединение формулы (V) предпочтительно кристаллизуют из реакционной смеси. Поэтому настоящее изобретение также относится к способу, описанному выше, в котором после стадии (4) и до стадии (5), соединение формулы (V) по меньшей мере частично кристаллизуют. Кристаллизацию можно провести по любой возможной методике. В предпочтительном варианте осуществления соединение формулы (V) кристаллизуют из н-гептана.
Стадия (5)
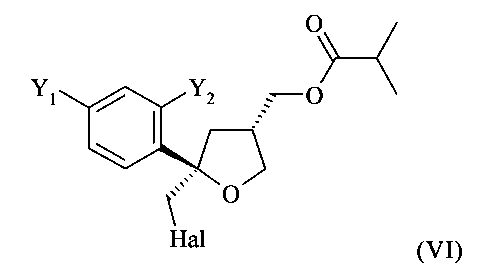
На стадии (5) настоящего изобретения соединение формулы (V) предпочтительно вводят в реакцию с галогеном Hal2, выбранным из группы, включающей Cl2, Br2 и I2, предпочтительно I2, в присутствии основания в растворителе с получением соединения формулы (VI)
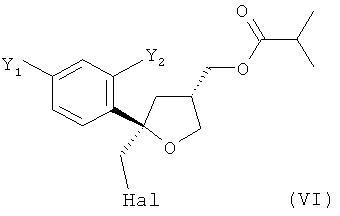 .
.
Обычно можно провести реакцию на стадии (5) в присутствии основания, такого как пиридин, и в подходящем растворителе, таком как ацетонитрил, THF, EtOAc (этилацетат) или CH2Cl2 (дихлорметан, ДХМ) при температуре в диапазоне от -20 до +30°C (см. WO 94/25452 A1, стр.16 и 35). Однако согласно изобретению было установлено, что реакцию предпочтительно проводить в этилацетате в качестве растворителя, причем в качестве основания используют гидрокарбонат натрия. Таким образом, настоящее изобретение относится к способу, в котором можно заменить небезопасное основание, пиридин. Кроме того, согласно изобретению было установлено, что температура, при которой проводят реакцию, предпочтительно равна ниже 0°C, более предпочтительно не выше -5°C и еще более предпочтительно не выше -10°C.
После завершения реакции органический слой, необязательно после соответствующей остановки реакции, необязательно можно промыть по меньшей мере один раз. Остановку реакции можно провести, например, с помощью 10% (мас./об.) водного раствора сульфита натрия.
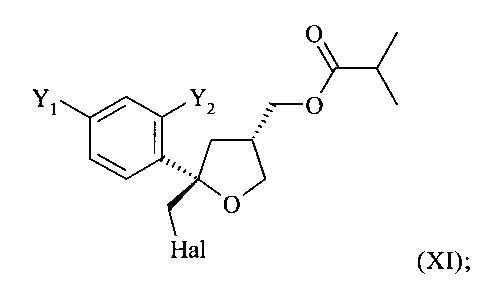
В особенно предпочтительном варианте осуществления настоящее изобретение относится к способу, описанному выше, в котором соединение формулы (VI), цис-изомер, получают на стадии (5) вместе с соединением формулы (XI), соответствующим транс-изомером
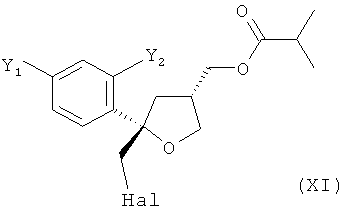 .
.
Эту смесь соединений формулы (VI) и (XI) ниже называют соединением формулы (X)
.
Согласно настоящему изобретению, в указанном соединении (X) предпочтительно от 80 до 95%, более предпочтительно от 85 до 95% молекул содержатся в виде цис-изомера формулы (VI) и предпочтительно от 20 до 5%, более предпочтительно от 15 до 5% молекул соединения (X) содержатся в виде транс-изомера формулы (XI).
Поэтому настоящее изобретение также относится к способу, определенному выше, дополнительно включающему:
(5) реакцию соединения формулы (V) с галогеном Hal2, выбранным из группы, включающей Cl2, Br2 и I2, предпочтительно I2, в присутствии основания в растворителе с получением соединения формулы (X)
где предпочтительно от 80 до 95%, более предпочтительно от 85 до 95% молекул соединения (X) содержатся в виде цис-изомера формулы (VI) и предпочтительно от 20 до 5%, более предпочтительно от 15 до 5% молекул соединения (X) содержатся в виде транс-изомера формулы (XI).
Соединение формулы (X) применимо для получения соединения формулы (IX), как это описано в настоящем изобретении.
Стадия (6.1)
На стадии (6.1) настоящего изобретения соединение формулы (X), т.е. предпочтительно соединение формулы (VI) и соединение формулы (XI) предпочтительно соответствующим образом нагревают в подходящем растворителе с подходящей солью 1,2,4-триазола. Предпочтительными солями 1,2,4-триазола являются соли щелочных металлов и натриевая соль является особенно предпочтительной. Предпочтительными растворителями являются полярные апротонные растворители, например DMF (N,N-диметилформамид) и DMSO, и DMSO является предпочтительным.
Температура, до которой нагревают реакционную смесь на стадии (6.1), предпочтительно находится в диапазоне от +70 до +100°C, предпочтительно от +80 до +95°C и более предпочтительно от +85 до +90°C.
По поводу таких реакций с солью 1,2,4-триазола в WO 94/25452 указано, что такое нагревание следует проводить в присутствии 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидинона (DMPU) (см. стр.17 стадия (1) и стр.39 стадия (b) в W094/25452). В отличие от данных, приведенных в WO 94/25452, согласно изобретению неожиданно было установлено, что нагревание соединения формулы (X) на стадии (6.1) можно провести при отсутствии DMPU. Таким образом, в значительно улучшенном способе, предлагаемом в настоящем изобретении, предложена упрощенная система растворителей, которая в предпочтительном варианте осуществления включает только DMSO, т.е. состоит только из одного растворителя в отличие от обязательной содержащей 2 соединения системы, указанной в WO 94/25452.
Затем смесь, полученную нагреванием, предпочтительно обрабатывают подходящим основанием для промотирования омыления сложноэфирного фрагмента. Такими основаниями являются, например, гидроксиды щелочных металлов, бикарбонаты щелочных металлов, карбонаты щелочных металлов, гидроксиды щелочноземельных металлов, бикарбонаты щелочноземельных металлов и карбонаты щелочноземельных металлов. Основания щелочных металлов являются предпочтительными. Предпочтительно, если основание добавляют в водных и/или спиртовых средах. Подходящими спиртами являются спирты, содержащие от 1 до 6, предпочтительно от 1 до 4, более предпочтительно от 1 до 3, наиболее предпочтительно от 1 до 2 атомов углерода. Согласно изобретению было установлено, что предпочтительным основанием является гидроксид натрия, предпочтительно использующийся в виде водного раствора в присутствии метанола.
Согласно настоящему изобретению, на стадии (6.1) получают соединение формулы (IX)
,
где предпочтительно от 80 до 95%, более предпочтительно от 85 до 95% молекул содержатся в виде цис-изомера формулы (VII)
и предпочтительно от 20 до 5%, более предпочтительно от 15 до 5% молекул содержатся в виде транс-изомера формулы (VIII)
.
В соответствии с предшествующим уровнем техники необходимо выделить соединения формулы (IX) и таким образом формулы (VII) после проведения стадий реакции, соответствующих стадии (6.1) настоящего изобретения, с помощью хроматографии (см. WO 94/25452, стр.39 стадия (b)). Таким образом, в предшествующем уровнем техники явно указано, что необходимо провести дорогостоящую и длительную очистку, которая делает известный способ убыточным при использовании в промышленном масштабе.
В отличие от предшествующего уровня техники, согласно изобретению было установлено, что не требуется проводить такое разделение с помощью хроматографии, если проводят определенную последовательность стадий (6.1) и экстракции на стадии (6.2), предпочтительно с последующей кристаллизацией на стадии (7) и/или образованием соли на стадии (8), как это описано ниже. Таким образом, эта модификация обеспечивает значительное преимущество по сравнению со способами предшествующего уровня техники.
Стадия (6.2)
На стадии (6.2) настоящего изобретения соединение формулы (IX)
,
предпочтительно соединение формулы (VII)
и соединение формулы (VIII),
содержащиеся в смеси, полученной на стадии (6.1), надлежащим образом выделяют, предпочтительно путем экстракции подходящим растворителем.
Согласно настоящему изобретению, предпочтительными растворителями являются полярные не смешивающиеся с водой растворители. Более предпочтительным растворителем является сложный эфир, такой как этилацетат или изопропилацетат, простой эфир, такой как тетрагидрофуран или метилтетрагидрофуран, кетон, такой как метилизобутилкетон, галогенированный растворитель, такой как дихлорметан, толуол или смесь двух или более этих растворителей, более предпочтительно сложный или простой эфир, более предпочтительно простой эфир и еще более предпочтительно метилтетрагидрофуран.
После разделения путем экстракции можно провести по меньшей мере одну стадию промывки. В частности, можно отметить промывку водным раствором хлорида натрия.
Поэтому в предпочтительном варианте осуществления настоящее изобретение относится к способу, определенному выше, включающему
(1.1) реакцию соединения формулы (Ia)
с (H3C)3Si-CH2MgCl в MTBE в качестве растворителя с получением реакционной смеси, содержащей в качестве промежуточного продукта бета-гидроксисилан формулы
(1.2) обработку полученной реакционной смеси без замены растворителя МТВЕ серной кислотой, промотирующей реакцию элиминирования, с получением реакционной смеси, содержащей соединение формулы (IIa)
 ;
;
(2) реакцию соединения формулы (IIa) со сложным эфиром малоновой кислоты Н3СН2СООС-СН2-СООСН2СН3 с получением соединения формулы (IIIa)
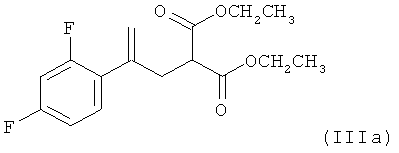 ,
,
(3) восстановление соединения формулы (IIIa) с использованием LiBH4 в качестве восстановительного реагента, который используют в количестве, составляющим не более 2 мол. экв. в пересчете на соединение (IIIa) в смеси воды и изопропанола в качестве растворителя с получением соединения формулы (IVa)
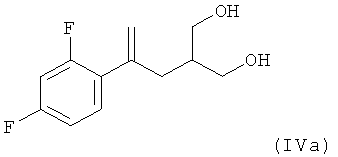 ;
;
(4) ацилирование соединения формулы (IVa) изомасляным ангидридом в присутствии фермента Novo SP 435 в толуоле в качестве растворителя с получением соединения формулы (Va)
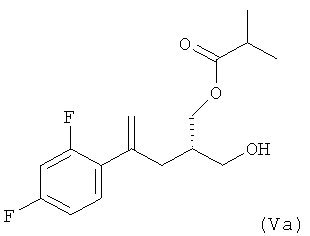 ;
;
(5) реакцию соединения формулы (Va) с I2 в присутствии гидрокарбонат натрия в качестве основания в этилацетате в качестве растворителя с получением соединения формулы (Xa)
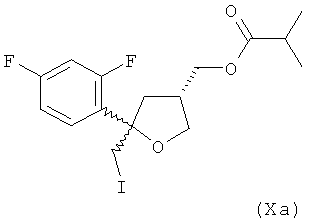
где предпочтительно от 80 до 95%, более предпочтительно от 85 до 95% молекул соединения (Ха) содержатся в виде цис-изомера формулы (VIa)

и предпочтительно от 20 до 5%, более предпочтительно от 15 до 5% молекул соединения (Xa) содержатся в виде транс-изомера формулы (XIA)
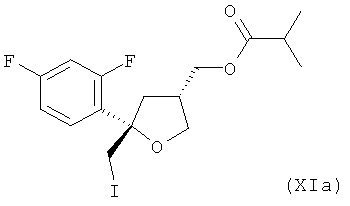 ;
;
(6.1) нагревание соединения формулы (Ха), предпочтительно при отсутствии DMPU (1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон), в DMSO в качестве растворителя с натриевой солью 1,2,4-триазола и обработку полученной реакционной смеси гидроксидом натрия с получением соединения формулы (IXa)
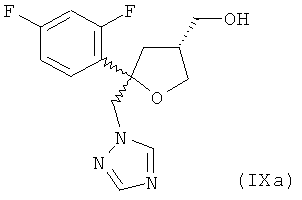 ;
;
где в соединении формулы (IXa), от 80 до 95%, предпочтительно от 85 до 95% молекул содержатся в виде цис-изомера формулы (VIIa)
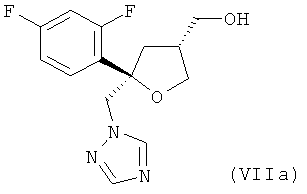
и от 20 до 5%, предпочтительно от 15 до 5% молекул содержатся в виде транс-изомера формулы (VIIIa)
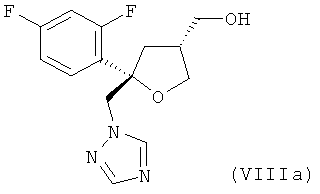 ;
;
(6.2) выделение соединения формулы (IXa)
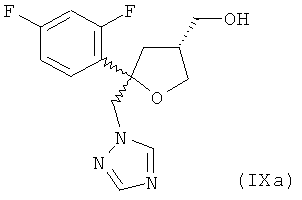
из реакционной смеси, полученной на стадии (6.1), путем экстракции метилтетрагидрофураном.
В одном предпочтительном варианте осуществления настоящего изобретения соединение формулы (IX) и тем самым также формулы (VII), полученное на стадии (6.2), предпочтительно и по меньшей мере частично кристаллизуют на стадии (7). Поэтому настоящее изобретение также относится к способу, определенному выше, который дополнительно включает:
(7) по меньшей мере частичную кристаллизацию соединения формулы (IX), предпочтительно соединения формулы (VII) и соединения формулы (VIII), после стадии (6.2).
Стадия (7)
На эту кристаллизацию не налагают специальных ограничений. В предпочтительном варианте осуществления настоящего изобретения растворителем, из которого соединение формулы (IX), предпочтительно соединение формулы (VII) и соединение формулы (VIII) кристаллизуют, является растворитель, который использовали на стадии (6.2), обсужденной выше, для экстракции. Поэтому предпочтительными растворителями на стадии (7) являются полярные не смешивающиеся с водой растворители. Более предпочтительным растворителем является сложный эфир, такой как этилацетат или изопропилацетат, простой эфир, такой как тетрагидрофуран или метилтетрагидрофуран, кетон, такой как метилизобутилкетон, галогенированный растворитель, такой как дихлорметан, толуол или смесь двух или более этих растворителей, более предпочтительно сложный или простой эфир, более предпочтительно простой эфир и еще более предпочтительно метилтетрагидрофуран.
Еще более предпочтительно, если после стадии (6.2) не проводят замену растворителя. Таким образом, в этом предпочтительном варианте осуществления соединение формулы (IX), предпочтительно соединение формулы (VII) и соединение формулы (VIII) кристаллизуют из смеси, полученной на стадии (6.2), без добавления другого подходящего растворителя, отличающегося от растворителя, использующегося на стадии (6.2).
Обычно такую кристаллизацию можно провести по любой подходящей методике. В частности, можно отметить охлаждение смеси, полученной на стадии (6.2), добавление антирастворителя к смеси, полученной на стадии (6.2), проведение химической реакции, изменение pH смеси, полученной на стадии (6.2), отгонку растворителя или комбинацию двух или большего количества этих методик.
В еще одном предпочтительном варианте осуществления эту кристаллизацию на стадии (7) проводят путем добавления подходящего антирастворителя. Обычно в зависимости от растворителя, использующегося на стадии (6.2), можно использовать каждый возможный антирастворитель при условии, что этот антирастворитель позволяет уменьшить растворимость растворенного соединения формулы (IX), предпочтительно соединения формулы (VII) и соединения (VIII), до такой степени, что соединение формулы (IX) по меньшей мере частично кристаллизуют. Предпочтительным антирастворителем является насыщенный или ненасыщенный углеводород, такой как циклогексан, гексан или гептан или смесь двух или более из них. Еще более предпочтительно, если антирастворитель, использующийся на стадии (7), выбран из группы, включающей циклогексан, гексан, гептан и смесь двух или более из них, предпочтительно гептан.
Обычно температуры, при которых проводят кристаллизацию на стадии (7), подбирают в соответствии с использующимся растворителем и предпочтительным антирастворителем. В предпочтительном варианте осуществления настоящего изобретения добавление антирастворителя проводят при температуре в диапазоне от 40 до 70°C, предпочтительно от 45 до 65°C, и более предпочтительно от 50 до 60°C. Затем полученную смесь предпочтительно непрерывно охлаждают до заданной температуры и охлаждение можно провести непрерывно или ступенчатым образом в две или большее количество стадий. В одном варианте осуществления настоящего изобретения заданная температура, до которой в конечном счете охлаждают смесь, находится в диапазоне от -15 до 0°C, предпочтительно от -10 до 0°C, более предпочтительно от -5 до 0°C. Охлаждение смеси до этой температуры может, как отмечено выше, включать по меньшей мере один диапазон температуры, до которой смесь охлаждают на первой стадии, выдерживание при этой температуре и последующее дополнительное охлаждение до конечной температуры, рассмотренной выше. Например, такой диапазон температуры, до которой смесь можно охладить на первой стадии, составляет от 20 до 35°C, предпочтительно от 25 до 30°C.
После кристаллизации закристаллизованное соединение (IX), в частности, закристаллизованное соединение формулы (VII) и закристаллизованное соединение формулы (VIII), предпочтительно выделяют из маточного раствора, например, с помощью подходящего фильтрования, и предпочтительно по меньшей мере один раз промывают подходящим промывочным реагентом. Предпочтительными промывочными реагентами являются смесь растворителей, использующаяся для кристаллизации, и антирастворителя, рассмотренная выше. После такого предпочтительного выделения закристаллизованное соединение формулы (IX), в частности, закристаллизованное соединение формулы (VII) и закристаллизованное соединение формулы (VIII), предпочтительно сушат при подходящих условиях сушки. Сушка в вакууме является предпочтительной и при этом температуры, предпочтительно находятся в диапазоне от 20 до 50°C, более предпочтительно от 30 до 45°C.
В способе, предлагаемом в настоящем изобретении и описанном выше, кристаллическое хиральное соединение формулы (VII), цис-изомер
получают в виде смеси с его диастереоизомерной формой, кристаллическим соединением формулы (VIII), а именно, с транс-изомером
.
Отношение количества молей цис-изомера к количеству молей трансизомера обычно зависит от общих условий проведения способа. При предпочтительных условиях проведения способа, предлагаемого в настоящем изобретении, закристаллизованное соединение формулы (IX), полученное после стадии (7),
содержит от 80 до 95%, предпочтительно от 85 до 95% цис-изомера (VII) и от 20 до 5%, предпочтительно от 15 до 5% транс-изомера (VIII). Поэтому настоящее изобретение также относится к способу, определенному выше, в котором получают по меньшей мере частично кристаллическое хиральное соединение формулы (IX)
,
в котором от 80 до 95%, предпочтительно от 85 до 95% молекул указанного кристаллического соединения содержатся в виде цис-изомера формулы (VII)
и от 20 до 5%, предпочтительно от 15 до 5% молекул указанного кристаллического соединения содержатся в виде транс-изомера формулы (VIII)
.
В предшествующем уровне техники не описано получение кристаллического соединения формулы (IX), т.е. кристаллической смеси, предпочтительно содержащей от 80 до 95%, более предпочтительно от 85 до 95% цис-изомера (VII) и предпочтительно от 20 до 5%, более предпочтительно от 15 до 5% транс-изомера (VIII) (см. WO 94/25452, стр.39 стадия (b)), где раскрыто, что необходимо проводить дорогостоящую и длительную очистку с помощью колоночной хроматографии, которая не дает по меньшей мере частично кристаллическое соединение (IX). Однако полагают, что смесь соединений (VII) и (VIII), полученную в настоящем изобретении, аналогичным образом можно использовать в качестве ключевого соединения для реакций, предпочтительно в качестве ключевого соединения для получения фунгицидного средства, такого как, предпочтительно позаконазол.
Таким образом, настоящее изобретение также относится к кристаллическому хиральному соединению формулы (IX) или его соли
в которой Y1 и Y2 независимо обозначают F или Cl, предпочтительно F, и где от 80 до 95%, предпочтительно от 85 до 95% молекул указанного кристаллического соединения или его соли содержатся в виде цис-изомера формулы (VII) или его соли
и от 20 до 5%, предпочтительно от 15 до 5% молекул указанного кристаллического соединения или его соли содержатся в виде транс-изомера формулы (VIII) или его соли
.
Кристаллическое хиральное соединение формулы (IX), как это описано в настоящем изобретении, где Y1 и Y2 обозначают F, предпочтительно обладает следующей рентгенограммой, содержащей по меньшей мере следующие отражения:
|
|
где 100% относится к интенсивности наиболее интенсивного пика на рентгенограмме.
На основе кристаллического соединения формулы (IX) предпочтительно содержащего от 80 до 95%, предпочтительно от 85 до 95% цис-изомера (VII) и от 20 до 5%, предпочтительно от 15 до 5% транс-изомера (VIII), можно получить подходящие соли кристаллического свободного основания (IX). Возможны все соли. Предпочтительно, если эти соли можно получить путем обработки соединения формулы (IX) и таким образом соединения формулы (VII) по меньшей мере одной подходящей неорганической кислотой и/или по меньшей мере одной подходящей органической кислотой, предпочтительно по меньшей мере одной подходящей неорганической кислотой Бренстеда и/или по меньшей мере одной подходящей органической кислотой Бренстеда, необязательно по меньшей мере в одном подходящем растворителе. Такие подходящие органические кислоты включают, но не ограничиваются только ими, фумаровую кислоту, щавелевую кислоту и винную кислоту. Подходящие неорганические кислоты включают, но не ограничиваются только ими, хлористоводородную кислоту.
Поэтому настоящее изобретение также относится к способу, определенному выше, который дополнительно включает:
(8) превращение соединения формулы (IX), предпочтительно соединения формулы (VII) и соединения формулы (VIII), в соответствующую соль путем обработки соединения неорганической или органической кислотой Бренстеда в подходящем растворителе и предпочтительно по меньшей мере частичную кристаллизацию соответствующей соли.
В частности, соль фумаровой кислоты или щавелевой кислоты или соль винной кислоты, или соль хлористоводородной кислоты соединения (IX) является предпочтительной. Соль хлористоводородной кислоты является еще более предпочтительной.
Согласно настоящему изобретению, подходящими растворителями являются полярные растворители. Более предпочтительным растворителем является сложный эфир, такой как этилацетат или изопропилацетат, простой эфир, такой как диоксан, тетрагидрофуран или метилтетрагидрофуран, кетон, такой как ацетон или метилизобутилкетон, галогенированный растворитель, такой как дихлорметан, толуол или смесь двух или более этих растворителей, более предпочтительно в качестве растворителя использовать кетон. Наиболее предпочтительным растворителем является ацетон.
Обычно такую кристаллизацию можно провести по любой подходящей методике. В частности, можно отметить охлаждение, добавление антирастворителя, отгонку растворителя или комбинацию двух или большего количества этих методик.
В предпочтительном варианте осуществления эту кристаллизацию проводят путем добавления подходящего антирастворителя вместе с кислотой или после добавления кислоты. Обычно можно использовать каждый возможный антирастворитель при условии, что этот антирастворитель позволяет уменьшить растворимость растворенной соли присоединения с кислотой соединения формулы (IX) и тем самым также формулы (VII) до такой степени, что соль присоединения с кислотой соединения (IX) и тем самым также соединение (VII) по меньшей мере частично кристаллизуют. Предпочтительным антирастворителем является простой эфир, такой как МТВЕ или насыщенный или ненасыщенный углеводород, такой как циклогексан, гексан или гептан. Более предпочтительным антирастворителем является МТВЕ.
В другом предпочтительном варианте осуществления настоящего изобретения соединение формулы (IX) и тем самым также формулы (VII), полученное на стадии (6.2), непосредственно превращают в подходящую соль, как это описано в настоящем изобретении, а именно, путем образования соли и предпочтительно также по меньшей мере частичной кристаллизации, описанной на стадии (8), без проведения кристаллизации на стадии (7). Замену растворителя необязательно проводят после стадии (6.2) до образования соли.
Кроме того, в этом варианте осуществления настоящего изобретения, в котором не проводят стадию (7), является предпочтительной соль фумаровой кислоты или соль щавелевой кислоты или соль винной кислоты, или соль хлористоводородной кислоты соединения (IX). Соль хлористоводородной кислоты является еще более предпочтительной. Согласно настоящему изобретению, подходящими растворителями являются полярные растворители. Более предпочтительным растворителем является сложный эфир, такой как этилацетат или изопропилацетат, простой эфир, такой как диоксан, тетрагидрофуран или метилтетрагидрофуран, кетон, такой как ацетон или метилизобутилкетон, галогенированный растворитель, такой как дихлорметан, толуол или смесь двух или более этих растворителей, более предпочтительно в качестве растворителя использовать кетон. Наиболее предпочтительным растворителем является ацетон. Обычно такую кристаллизацию можно провести по любой подходящей методике. В частности, можно отметить охлаждение, добавление антирастворителя, отгонку растворителя или комбинацию двух или большего количества этих методик. В предпочтительном варианте осуществления эту кристаллизацию проводят путем добавления подходящего антирастворителя вместе с кислотой или после добавления кислоты. Обычно можно использовать каждый возможный антирастворитель при условии, что этот антирастворитель позволяет уменьшить растворимость растворенной соли присоединения с кислотой соединения формулы (IX) до такой степени, что соль присоединения с кислотой соединения (IX) по меньшей мере частично кристаллизуют. Предпочтительным антирастворителем является простой эфир, такой как МТВЕ или насыщенный или ненасыщенный углеводород, такой как циклогексан, гексан или гептан. Более предпочтительным антирастворителем является МТВЕ.
Таким образом, настоящее изобретение также относится к кристаллической соли с хлористоводородной кислотой хирального соединения формулы (IX)
в которой Y1 и Y2 независимо обозначают F или Cl, предпочтительно F, и где от 80 до 95%, предпочтительно от 85 до 95% молекул указанного кристаллического соединения или его соли с хлористоводородной кислотой содержатся в виде цис-изомера формулы (VII)
и от 20 до 5%, предпочтительно от 15 до 5% молекул указанного кристаллического соединения или его соли с хлористоводородной кислотой содержатся в виде транс-изомера формулы (VIII)
.
Кристаллическая соль с хлористоводородной кислотой хирального соединения формулы (IX), как это описано в настоящем изобретении, где Y1 и Y2 обозначают F, предпочтительно обладает следующей рентгенограммой, содержащей по меньшей мере следующие отражения:
|
|
где 100% относится к интенсивности наиболее интенсивного пика на рентгенограмме.
Кроме того, настоящее изобретение относится к по меньшей мере частично кристаллическому хиральному соединению или его соли, которое можно получить или получают способом, определенным выше, способ предпочтительно включает стадии (1)-(8), еще более предпочтительно включает стадии (1)-(6.2), затем стадию (8).
Как уже отмечено выше, соединение формулы (IX) или его соль предпочтительно используют в качестве ключевого соединения для получения фунгицидного средства. Предпочтительным фунгицидным средством для получения которого можно использовать соединение формулы (IX) или его соль и тем самым соединение (VII) или его соль, является позаконазол, т.е. соединение следующей формулы:
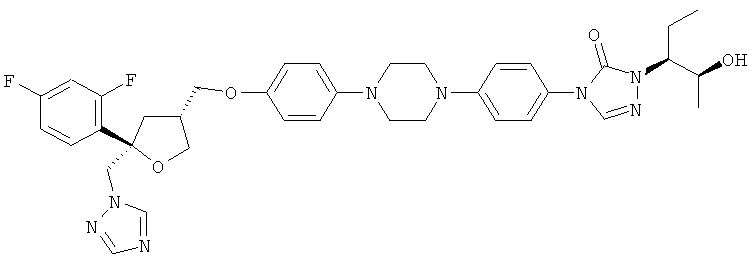
Таким образом, настоящее изобретение также относится к способу получения фунгицидного средства, предпочтительно позаконазола, в котором предпочтительно по меньшей мере частично кристаллическое, предпочтительно кристаллическое соединение формулы (IX) или его соль и тем самым (VII) или его соль используют в качестве исходного вещества.
В необязательном варианте осуществления настоящего изобретения соединение формулы (IX) или его соли можно надлежащим образом очистить и выделить цис-изомер (VII) или транс-изомер (VIII), предпочтительно цис-изомер (VII), до его использования для получения фунгицидного средства, предпочтительно позаконазола.
Настоящее изобретение иллюстрируется приведенными ниже примерами.
Примеры
Пример 1: Получение соединения формулы (IX)
(a) Получение соединения формулы (IIa)
В 20 мл МТВЕ суспендировали 3,8 г Mg. Температура суспензии равнялась 55°C. Затем добавляли 0,5 г реагента Гриньяра (CH3)3Si-CH2MgCl в МТВЕ из предыдущей партии для сушки системы (если не имелся такой реагент Гриньяра из первой партии, то можно использовать (CH3)3Si-CH2MgCl в диэтиловом эфире (регистрационный № CAS: 13170-43-9), продающийся в виде 1,0 М раствора фирмой Sigma-Aldrich), затем 1,0 мл хлорметилтриметилсилана (СМ-TMS; регистрационный №CAS: 2344-80-1; продающийся фирмой Sigma-Aldrich). В течение 2 ч при температуре, равной 55°C, медленно добавляли раствор 14 мл CM-TMS в 43 мл МТВЕ. Смесь перемешивали в течение 2 ч при температуре, равной 55°C, и затем охлаждали до температуры, равной -10°C. Затем добавляли 10,0 г имеющегося в продаже соединения формулы (Ia) (регистрационный № CAS: 51336-94-8; продающийся фирмой Sigma-Aldrich) в 30 мл МТВЕ и температуру поддерживали в диапазоне от 0 до -10°C. Реакцию останавливали с помощью 20% (мас./об.) водного раствора хлорида аммония. Полученный органический слой промывали с помощью 20% (мас./об.) водного раствора хлорида аммония. Затем этот промытый органический слой промывали водой.
К органическому слою добавляли 11,0 мл концентрированной серной кислоты и температуру поддерживали равной от 25 до 30°C. Затем реакционную смесь перемешивали при температуре, равной от 45 до 50°C, в течение 3 ч. Затем реакционную смесь охлаждали до 20°C и добавляли 25 мл воды и органический слой отделяли. Полученный органический слой экстрагировали 9% (мас./об.) водным раствором бикарбоната натрия, затем промывали водой. Растворители из промытого органического слоя удаляли путем отгонки при пониженном давлении и получали соединение формулы (IIa) в виде масла. Выход составлял 9,4 г, что соответствовало 95% от теоретического значения.
(b) Получение соединения формулы (IIIa)
10,0 г Соединения формулы (IIa) (в виде масла, полученного на стадии (a)) при перемешивании растворяли в 20 мл DMSO. Затем добавляли 3,2 г чешуек NaOH и 24,0 мл диэтилмалоната. Полученную суспензию перемешивали в течение 5 ч при температуре, равной от 25 до 30°C. Затем, добавляли 100 мл воды и полученную смесь перемешивали в течение 30 мин. Полученный таким образом раствор экстрагировали с помощью 80 мл циклогексана при температуре, равной от 25 до 30°C. После разделения слоев водный слой экстрагировали с помощью 40 мл циклогексана при температуре, равной от 25 до 30°C. Объединенные органические слои промывали 5% (мас./об.) водным раствором NaOH, затем промывали водой. После промывки растворители из органического слоя удаляли путем отгонки при пониженном давлении и получали соединение формулы (IIIa) в виде масла. Выход составлял 15,0 г, что соответствовало 90,0% от теоретического значения.
(c) Получение соединения формулы (IVa)
10,0 г Соединения формулы (IIIa) (в виде масла, полученного на стадии (b)) растворяли в 120 мл изопропилового спирта и 13,0 мл воды при перемешивании при температуре, равной от 25 до 30°C. Полученную смесь охлаждали до температуры, равной от 0 до -5°C. Затем добавляли 2,3 г хлорида лития и 2,1 г борогидрида натрия при температуре, равной от 0 до -5°C. Полученную суспензию перемешивали при температуре, равной от 25 до 30°C в течение 20 ч. Значение pH перемешанной смеси устанавливали равным 1 (измеряли калиброванным pH-метром) путем добавления 4 н. водного раствора HCl. Затем добавляли 20% (мас./об.) водный раствор NaOH для установления значения pH равным 10 (измеряли калиброванным pH-метром). Полученную смесь перемешивали в течение 1 ч. Затем нижний водный слой сливали. Из отделенного органического слоя отгоняли изопропиловый спирт и получали масло. К этому маслу добавляли 100 мл толуола и 100 мл воды и продукт экстрагировали толуольным слоем. Растворители из полученного толуольного слоя удаляли путем отгонки при пониженном давлении получали и соединение формулы (IVa) в виде масла. Выход составлял 6,0 г, что соответствовало 82,0% от теоретического значения.
(d) Получение соединения формулы (Va)
10,0 г Соединения формулы (IVa) (в виде масла, полученного на стадии (с)) растворяли в 80 мл толуола и охлаждали до -15°C. Затем добавляли 7,4 г бикарбоната натрия, 0,5 г фермента (Novo SP 435; Candida antarctica, Novozym 435, выпускающийся фирмой Novo Nordisk) и 7,9 мл ангидрида изомасляной кислоты. Полученную смесь перемешивали при -15°C в течение 24 ч. Затем твердые вещества отфильтровывали и фильтрат промывали 5% (мас./об.) водным раствором бикарбоната натрия, затем промывали водой. Растворители из полученного органического слоя удаляли путем отгонки при пониженном давлении и получали искомый продукт в виде масла. Это масло растворяли в 40 мл н-гептана при температуре, равной от 50 до 60°C. Прозрачный раствор постепенно охлаждали до температуры, равной 10°C. Соединение формулы (Va) кристаллизовали в виде бесцветных кристаллов. Полученное твердое вещество отфильтровывали и влажный осадок на фильтре промывали с помощью 20 мл н-гептана. Затем осадок на фильтре сушили при 40°C в вакууме и получали соединение формулы (Va) в виде бесцветных кристаллов. Выход составлял 9,2 г, что соответствовало 70,0% от теоретического значения.
(e) Получение соединения формулы (Xa)
10,0 г Кристаллов, полученных на стадии (d), при перемешивании растворяли в 80 мл этилацетата. Полученный раствор охлаждали до -15°C и добавляли 21,5 г йода и 7,0 г бикарбоната натрия. Полученную суспензию перемешивали при -15°C в течение 5 ч. Реакцию останавливали с помощью 200 мл 10% (мас./об.) водного раствора сульфита натрия. Органический слой промывали с помощью 100 мл 10% (мас./об.) водного раствора сульфита натрия, затем промывали водой. Растворители из полученного таким образом промытого органического слоя удаляли путем отгонки при пониженном давлении и получали соединение формулы (Xa) в виде масла. Выход составлял 13,5 г, что соответствовало 95,0% от теоретического значения.
(F) Получение соединения формулы (IXa)
10,0 г Соединения формулы (Xa) (в виде масла, полученного на стадии (e)) при перемешивании растворяли в 80 мл DMSO. Затем добавляли 10 г натриевой соли 1,2,4-триазола при температуре, равной от 25 до 30°C, и полученную реакционную смесь перемешивали в течение 24 ч при температуре, равной от 85 до 90°C. Затем смесь охлаждали до температуры, равной от 25 до 30°C, и добавляли 25 мл 5% (мас./об.) водного раствора гидроксида натрия. Затем смесь перемешивали в течение 3 ч при температуре, равной от 25 до 30°C. Добавляли 100 мл воды и продукт экстрагировали с помощью 150 мл метилтетрагидрофурана. Полученный таким образом органический слой промывали 10% (мас./об.) водным раствором хлорида натрия и затем растворители из полученного органического слоя удаляли путем отгонки при пониженном давлении и получали соединение формулы (IXa) в виде неочищенного масла. Выход составлял 6,0 г, что соответствовало 86,0% от теоретического значения.
10,0 г Неочищенного масла при перемешивании при температуре, равной от 50 до 60°C, растворяли в 100 мл метилтетрагидрофурана. Затем в течение 30 мин добавляли 300 мл н-гептана при температуре, равной от 50 до 60°C. Мутный раствор охлаждали до температуры, равной от 25 до 30°C, и перемешивали в течение еще 30 мин. Полученную суспензию охлаждали до температуры, равной от 0 до -5°C, и перемешивали в течение 2 ч. Продукт отфильтровывали и влажный осадок на фильтре промывали с помощью 20 мл н-гептана. Промытый продукт сушили при 40°C в вакууме и получали кристаллическое соединение формулы (IXa) в виде бесцветного твердого вещества. Выход составлял 7,0 г, что соответствовало 70,0% от теоретического значения.
Соединение формулы (IXa) получали в виде смеси цис-изомера с соответствующим транс-изомером при отношении цис:транс, составляющем 9:1.
Пример 2: Получение солей соединения формулы (IX)
(а) Получение соли с хлористоводородной кислотой
10,0 г Соединения формулы (IXa) в виде неочищенного масла, полученного в примере 1(f), до кристаллизации при перемешивании при температуре, равной от 30 до 40°C, растворяли в 200 мл ацетона. Полученный раствор охлаждали до температуры, равной от 25 до 30°C. Затем в течение 15 мин при температуре, равной от 25 до 30°C, добавляли HCl в МТВЕ (10 мас.%). При перемешивании смеси в течение 15 мин кристаллизовалось твердое вещество. Затем в течение 30 мин медленно добавляли 200 мл МТВЕ. Суспензию охлаждали до температуры, равной от 0 до -5°C, и перемешивали в течение 2 ч. Продукт отфильтровывали и влажный осадок на фильтре промывали с помощью 20 мл МТВЕ. После сушки при температуре, равной 70°C, в вакууме соль получали с HC1 соединения (IXa) в виде бесцветного твердого вещества. Выход составлял 9,5 г, что соответствовало 85,0% от теоретического значения.
Соль с HC1 соединения формулы (IXa) получали в виде смеси цис-изомера с соответствующим транс-изомером при отношении цис:транс, составляющем 9:1.
(b) Получение солей органических кислот
10,0 г Кристаллического соединения формулы (IXa), полученного выше в примере 1(f) (отношение цис:транс=9:1), растворяли в 20 мл ацетона. К полученному раствору при температуре окружающей среды, равной от 25 до 30°C, добавляли 1,1 мол. экв. органической кислоты (щавелевой кислоты, DL-винной кислоты, фумаровой кислоты). Полученные смеси перемешивали в течение 1 ч при температуре, равной от 25 до 30°C. Затем с помощью капельной воронки в течение 15 мин добавляли 150 мл МТВЕ. Твердые вещества кристаллизовались. Перемешивание продолжали в течение 2 ч при температуре, равной от 25 до 30°C. Твердые вещества выделяли фильтрованием, промывали с помощью 20 мл МТВЕ и сушили в вакууме при 45°C. Соли получали в виде бесцветного твердого вещества.
Получали следующие отношения цис:транс:
|
Определяли с помощью ВЭЖХ (высокоэффективная жидкостная хроматография).
Методика ВЭЖХ для определения чистоты и отношения цис/транс для соединения формулы (IXa):
|
Методика регистрации рентгенограмм
Образцы анализировали в держателе с нулевым фоном в режиме вращения при условиях окружающей среды. Типичная точность определения значений 2-тета находится в диапазоне примерно ±0,2 °2-тета. Таким образом, при использовании большинства дифрактометров при стандартных условиях дифракционный пик, который появляется при 8,6 °2-тета может появиться: диапазоне от 8,4 до 8,8 °2-тета.
Параметры прибора:
|
Оптика падающего пучка
|
Оптика дифрагирующего пучка
|
Перечень цитированных документов
- WO 94/25452 A1
- WO 95/16658 A1
- D.J. Peterson, Carbonyl olefination reaction using silyl-substituted organometallic compounds; J. Org. Chem. (1968) 33 (2) pp.780-784
- P. Blundell et al., Synlett 1994, pp.263-265
- Tetrahedron Letters 32 (1991), pp.7545-7548
- WO 97/22710 A1.

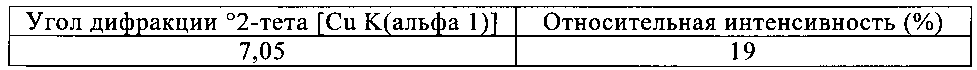

Устройство для перорального применения вещества
Кристаллические формы саксаглиптина
Способ и устройство для перорального введения композиции
Кристаллическая форма органического соединения
Очистка позаконазола и промежуточных продуктов для синтеза позаконазола
Способ получения хиральных триазолонов
Способ получения хиральных гидразидов
Региоселективное ацилирование рапамицина в положении с-42
Стабильные жидкие фармацевтические препараты слитого белка tnfr:fc
Порошковые смеси для составов сухого сиропа антибиотика
Устройство для перорального применения вещества
Кристаллические формы саксаглиптина
Способ и устройство для перорального введения композиции
Кристаллическая форма органического соединения
Очистка позаконазола и промежуточных продуктов для синтеза позаконазола
Способ получения хиральных триазолонов
Способ получения хиральных гидразидов
Региоселективное ацилирование рапамицина в положении с-42
Стабильные жидкие фармацевтические препараты слитого белка tnfr:fc

