Результат интеллектуальной деятельности: СПОСОБ ЭЛЕКТРОХИМИЧЕСКОГО СТЕРЕОСЕЛЕКТИВНОГО α-ГИДРОКСИАЛКИЛИРОВАНИЯ ГЛИЦИНА
Вид РИД
Изобретение
Область техники
Изобретение относится к области органической химии и электрохимии, конкретно к химии аминокислот, а именно к способу электрохимического стереоселективного α - гидроксиалкилирования глицина (Gly) - наиболее доступной аминокислоты - после введения его в виде основания Шиффа в координационную сферу комплекса никеля с хиральным лигандом ((S)-2N-(N'-бензилпролил)аминобензофеноном) {далее - [(<S)-BPB-Gly] Ni (II) комплекс} с целью получения ценных (S)- и (R)-β-гидрокси-α-аминокислот в оптически чистом виде, которые являются важными компонентами физиологически активных пептидов, циклических пептидов (Vancomycin, Cyclosporine) и ингибиторов ферментов, a (S)-серин и (5)-треонин также используются в составе аминокислотных смесей для парентерального питания.
Уровень техники
Стереоселективная функционализация природных α-аминокислот является важной синтетической задачей, поскольку такие соединения представляют большой практический интерес, в первую очередь как медицинские и биологически активные препараты. Известен способ решения этой задачи путем введения аминокислоты в координационную сферу металлокомплекса с хиральным лигандным окружением. Введение аминокислоты в координационную сферу комплекса Ni(II) позволяет осуществить ее функционализацию, поскольку повышает кислотность α-протонов, отщепление которых требуется в ходе реакции [Yu. N. Belokon', A.G. Bulychev, S.V. Vitt. General Method of Diastereo- and Enantioselective Synthesis of β-Hydroxy-α-amino Acids by Condensation of Aldehydes and Ketones with Glycine II J. Am. Chem. Soc, 1985, V. 107, P. 4252-4259]. Наличие хирального центра в непосредственной близости от места функционализации позволяет обеспечить ее строго определенную стереонаправленность.
До настоящего времени не известны примеры прямого стереоселективного электросинтеза с участием вышеуказанных хиральных комплексов. Способы создания новых хиральных центров в условиях электросинтеза до сих пор разработаны недостаточно, большинство из них приводит к невысоким оптическим выходам [Т. Nonaka, Т. Fuchigami, Stereochemistry of Organic Electrode Processes.// Organic Electrochemistry 4th ed. (ed. by H. Lund and O. Hammerich), New York: Marcel Dekker, Inc., 2001, p. 1051-1102].
Известен способ получения оптически чистых β-гидрокси-α-аминокислот, который включает взаимодействие хирального комплекса [(S)-BPB-Gly] Ni (II) с алифатическими альдегидами под действием метилата натрия, реакцию проводят при нагревании (40-50°С) в течение 2-3 часов [V.A. Soloshonok, D.V. Avilov, V.P. Kukhar', V.I. Tararov, T.F. Savereva, T.D. Churkina, N.S. Ikonnikov, K.A. Kochetkov, S.A. Orlova, A.P. Pysarevsky, Yu. T.Strachkov, N. I. Raevsky, Yu. N. Belokon1, Asymmetric Aldol Reactions of Chirai Ni(II)-Complex of Glycine with Aliphatic Aldehydes. Stereodivergent Synthesis of syn-(2S)- and syn-(2R)-β-Alkylserines//Tetrahedron: Asymmetry, 1995, V. 6, P. 1741-1756].
Известен способ получения оптически активной β-гидрокси-α-аминокислоты - (S)-серина (Ser) [А.С. Сагиян, С.М. Джамгарян, Б.С. Арутюнян, Ю.Н. Белоконь, М.Г. Рыжов, Оганесян А.Х. Способ получения серина, Авт. свид. СССР №1555324 (1990), С07С 229/00, опубл. 07.04.90. Бюл. №13], который включает образование хелатного комплекса глицина с (S)-2-N-(N'-бензилпролил)аминобензофеноном и нитратом никеля [(S)-ВРВ-Gly] Ni(II), его выделение и α-гидроксиметилирование глицинового фрагмента этого комплекса формальдегидом в присутствии метилата натрия, в результате образуется диастереомерная смесь новых хелатных комплексов [(S)-BPB-Ser] Ni (II), ее выделяют из реакционной смеси и затем разделяют комплексы серина. Выделение хелатного комплекса глицина и диастереомерной смеси комплексов серина осуществляют осаждением из разбавленных водой растворов при соотношении реакционная смесь: вода, равном 1:(20-25), на стадии выделения комплекса глицина и 1:(26-31) - на стадии выделения смеси комплексов серина, а разделение смеси комплексов S-серина и R-серина осуществляют дробной кристаллизацией из ацетона, взятого в массовом отношении к смеси комплексов (0.8-1):1. Полученную смесь комплексов серина [в соотношении (S):(R)=95:5] кристаллизуют из ацетона, выделяя комплекс S -энантиомера серина с выходом 86.8%. Выход целевого продукта S-серина на стадии разложения комплекса 96.2%.
Известен способ получения β-гидрокси-α-аминокислот [Yu. N. Belokon, К.А. Kochetkov, N.S. Ikonnikov, T.V. Strelkova, S.R. Harutyunyan A.S. Saghiyan, A new synthesis of enantiomerically pure syn-(S)-b-hydroxy-a-amino acids via asymmetric aldol reactions of aldehydes with a homochiral Ni(II)-glycine: (S)-BPB Schiff base complex, Tetrahedron Asymmetry, 2001, V. 12, P. 481-485], в котором глицин в виде основания Шиффа вводят в координационную сферу комплекса никеля с хиральным лигандом ((S)-2N-(N'-бензилпролил)аминобензофеноном), получают комплекс [(S)-BPB-Gly] Ni (II), затем осуществляют стереоселективное α-гидроксиалкилирование глицинового фрагмента этого комплекса путем его взаимодействия с альдегидом, причем в качестве основания используют гидрид натрия в тетрагидрофуране и реакцию проводят при комнатной температуре, в результате после осаждения получают комплексы целевых β-гидрокси-α-аминокислот с высокими выходами 70-96%. Выход аминокислоты на последующей стадии разложения комплекса 81-99%. Этот способ по совокупности существенных признаков наиболее близок к заявляемому способу и был выбран в качестве прототипа.
Все известные способы-аналоги и способ-прототип имеют ряд существенных недостатков: необходимость использования в качестве гидроксилирующих агентов достаточно дорогих и малоустойчивых альдегидов, а в качестве основания - дорогих и опасных метилата натрия или гидрида натрия. Альдегиды - активные реагенты, которые легко подвергаются окислению или вступают в другие побочные реакции. Кроме того, применение пожароопасного растворителя тетрагидрофурана является нежелательным. Все эти недостатки увеличивают стоимость целевых продуктов - β-гидрокси-α-аминокислот, а также тормозят внедрение способа-прототипа в промышленность, где приоритетными являются безотходные производства медицинских и биологически активных препаратов. В связи с этим разработка нового способа α-гидроксиалкилирования аминокислот, где в качестве исходных соединений используют не карбонильные соединения, а их предшественники - спирты, которые являются более доступными и дешевыми веществами, и в качестве основания используют дешевые и доступные щелочи, имеет большое практическое значение.
Задачей настоящего изобретения является разработка электрохимического способа α-гидроксиалкилирования глицина в составе комплекса [(S)-BPB-Gly] Ni(II) для получения β-гидрокси-α-аминокислот из доступного дешевого сырья.
Сущность изобретения
Поставленная задача решена заявляемым способом электрохимического стереоселективного α-гидроксиалкилирования глицина, включающим введение глицина в виде основания Шиффа в координационную сферу комплекса никеля с хиральным лигандом ((S)-2N-(N'-бензилпролил)аминобензофеноном) [(S)-BPB-Gly] Ni (II), α-гидроксиалкилирование глицинового фрагмента полученного комплекса путем взаимодействия с алифатическим спиртом в присутствии КОН с образованием диастереомерной смеси комплексов целевых β-гидрокси-α-аминокислот, выделение и разделение указанных комплексов известными приемами, при этом гидроксиалкилирование осуществляют как one-pot электрохимический процесс путем гальваностатического электролиза смеси комплекса [(S)-BPB-Gly] Ni (II), алифатического спирта и КОН. В качестве алифатического спирта можно использовать также этиленгликоль.
Подробное описание изобретения
Разработан новый удобный электросинтетический способ стереоселективного α-гидроксиалкилирования глицина путем введения оксиалкильных групп в глициновый фрагмент комплекса [(S)-BPB-Gly] Ni (II) с получением новых комплексов и выделением в качестве целевых продуктов β-гидрокси-α-аминокислот, при этом в качестве исходных реагентов используют соответствующие спирты.
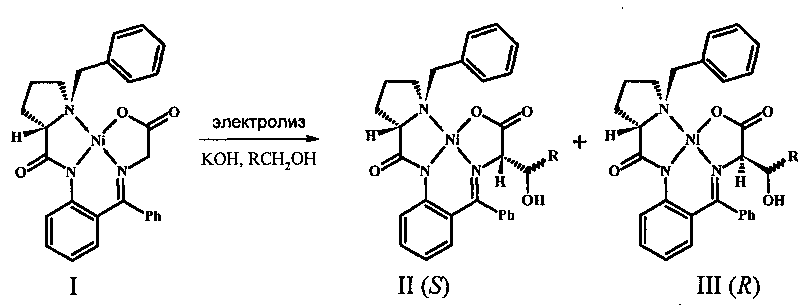
Способы получения комплекса [(S)-BPB-Gly] Ni (II), основного исходного соединения, хорошо отработаны и известны. Так, Ni (II) комплекс основания Шиффа глицина с хиральными производными (S)-2-N-(N'-бензилпропил)аминобензофенона [(S)-ВРВ- Gly] Ni (II) получают в щелочной среде в инертной атмосфере при 40-50°С в смеси метанол - диметилформамид (в объемном соотношении 85-60:15-40) и мольном соотношении компонентов [(S)-ВРВ]: Ni (II): Gly=(3-4):(1-2):1 с выходами более 95% [Рыжов М.Г., Лысова Л.А., Казика А.И., Носова Н.А., Мишин В.И., Белоконь Ю.Н., Способ получения никель (II) - комплексов основания Шиффа глицина с хиральными производными (S) или (R)-2-N-(N'-бензилпропил) аминобензофенона, Авторское свидетельство СССР №2027720 (1995), C07F 15/04. Опубл. 27.01.1995. Бюлл №3].
В комплексе [(S)-BPB- Gly] Ni (II) в мягких условиях происходит образование карбаниона [Y.N. Belokon', A.G. Bulychev, S.V. Vitt, General Method of Diastereo- and Enantioselective Synthesis of β-Hydroxy-α-amino Acids by Condensation of Aldehydes and Ketones with Glycine, J. Am. Chem. Soc, 1985, V. 107, 4252-4259], вступающего во взаимодействие с карбонильными соединениями (которые в нашем случае образуются путем электрохимического окисления спиртов и вступают в реакцию in situ). Термодинамическая энантиоселективность процесса обусловливается несвязывающим взаимодействием боковой цепи аминокислотного фрагмента (R)-конфигурации с бензильным фрагментом реагента, что приводит к диастереомерному избытку комплекса с (S)-аминокислотой. После кислотного гидролиза комплексов выделяют исходный хиральный реагент и энантиомерно обогащенную (S)-аминокислоту. При использовании (R)-хирального реагента получают соответственно (R)-аминокислоту. Способ универсален, т.к. варьируя природу электрофила, можно получать разнообразные β-гидрокси-α-аминокислоты.
Заявляемый способ отличается аппаратурной простотой и высокой эффективностью. Электролиз проводят при комнатной температуре в электрохимической ячейке с неразделенным электродным пространством в гальваностатическом режиме, в спиртовом растворе, в присутствии гидроксида калия, одновременно выполняющего роль основания и фонового электролита. Суммарный выход целевых комплексов практически количественный. После хроматографического разделения и очистки реакционной смеси на силикагеле выделяют два новых диастереомерных комплекса никеля с β-гидрокси-α-аминокислотами с общим выходом более 90%. Соотношение диастереомеров зависит от условий электросинтеза и может быть проконтролировано, что позволяет получить преимущественно тот или иной диастереомерный комплекс с (S)- или (R)-аминокислотой (см. ниже). Полученные диастереомерно чистые комплексы в результате сольволиза 1 М НСl в метаноле по стандартной методике дают соответствующие (S) или (R)-β-гидрокси-α-аминокислоты и регенерированный хиральный реагент (S)-ВРВ с практически количественным выходом.
Механизм электрохимического превращения можно представить следующим образом. В щелочной среде происходит депротонирование не только комплекса [(S)-ВРВ-Gly] Ni (II), но и спирта, с образованием соответствующего алкоголята в равновесной концентрации. Алкоголят легко окисляется на аноде с образованием соответствующего альдегида. На примере метанола образование альдегида на аноде выглядит следующим образом:
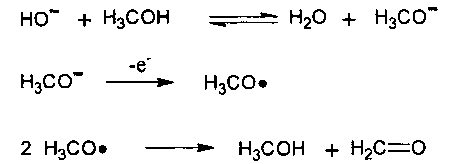
Генерируемый в растворе альдегид in situ реагирует по механизму нуклеофильного присоединения с депротонированным исходным комплексом [(S)-BPB-Gly] Ni (II), что приводит к его функционализации с образованием новых изомерных комплексов II и III, содержащих остаток β-гидрокси-α-аминокислоты. Таким образом, реакцию функционализации комплекса удается провести как one-pot процесс [термином «one-pot» обозначают одностадийный процесс, проводимый в одном сосуде, см. В.А. Смит, А.Д. Дильман. Основы современного органического синтеза, Бином. Лаборатория знаний, Москва, 2009, стр. 6].
В реакцию могут быть введены любые первичные алифатические спирты, за исключением тех, которые после окисления способны давать в щелочной среде устойчивые гидраты (например, полифторированные спирты; так, CF3CH2OH, для которого происходит дезактивация генерируемого in situ трифторацетальдегида вследствие взаимодействия с гидроксид-ионом и образования устойчивого гидрата, не вступает в реакцию), а также этиленгликоль. Строение продуктов реакции доказано спектральными методами (ЯМР 1Н, 13С, включая двумерные корреляционные спектры COSY и HMQC с полным отнесением сигналов, а также масс-спектрометрия высокого разрешения).
Для проведения электросинтеза подходит простейшая электрохимическая ячейка любой формы с неразделенным анодным и катодным пространством. В качестве рабочего электрода может быть использована платина или графит. Реакция может быть проведена как в потенциостатическом, так и в гальваностатическом режиме, однако в последнем случае процесс более прост и технологичен.
Преимущества по сравнению с прототипом и аналогами
Предлагаемый способ электрохимического стереоселективного α-гидроксиалкилирования глицина имеет ряд существенных преимуществ по сравнению с прототипом и аналогами.
Во-первых, вместо дорогостоящих альдегидов в реакцию вводят их прекурсоры - спирты, которые являются гораздо более дешевыми реагентами и более технологичны в использовании. Это позволяет избежать применения альдегидов, расширить ассортимент целевых продуктов за счет получения аминокислотных комплексов, которые ранее не были синтезированы.
Во-вторых, реакцию α-гидроксиалкилирования глицина проводят как технологичный one-pot процесс, поскольку альдегиды генерируются путем анодного гальваностатического окисления спиртов, используемых одновременно и в качестве растворителя (при этом также исключается использование пожароопасного тетрагидрофурана), и тем самым вводятся в реакцию in situ, что уменьшает вероятность характерных для них побочных процессов и позволяет снизить отходы при сохранении высокого выхода целевых комплексов II и III.
В третьих, в данном способе используется дешевая щелочь, в отличие от дорогого и огнеопасного гидрида натрия. Варьируя концентрацию КОН (который одновременно выполняет роль основания и фонового электролита) от 0.35 M до 0.1 M и время выдерживания реакционной смеси, полученной после пропускания заданного количества электричества, перед нейтрализацией щелочи (от 5 мин до 20 часов), можно широко варьировать соотношение образующихся изомеров с (S) или (R) конфигурацией α-аминокислотного центра (от 5:95 до 98:2). Меньшее количество щелочи и создание нейтральной рН сразу после окончания электролиза позволяет стереоселективно получать комплексы (S)-β-гидрокси-α-аминокислот. Увеличение рН и времени выдерживания реакционной смеси после электролиза приводит преимущественно к комплексам (R)-β-гидрокси-α-аминокислот. Дальнейшее увеличение количества щелочи ограничено ее растворимостью в спиртах, уменьшение количества щелочи <0.1 M уменьшает конверсию. Разложение комплексов по стандартной методике (пример 8) позволяет получать оптически чистые (S) и (R)- β-гидрокси-α-аминокислоты с выходами более 90%.
В ходе исследования были установлены оптимальные условия получения гидроксиалкилированных глициновых комплексов Ni (II) (примеры 1-7), выходы которых достигают 90%.
Заявляемое изобретение иллюстрируется следующими ниже примерами, которые не ограничивают объем настоящего изобретения.
Пример 1. Получение сериновых комплексов и их выделение
В двухэлектродную ячейку с неразделенным электродным пространством объемом 50 мл (рабочий электрод - пластина из стеклоуглерода площадью 300 мм2, вспомогательный электрод - платиновая пластина, размером 100 мм2) помещают раствор 900 мг (16 ммоль) гидроксида калия и 200 мг (0.4 ммоль) комплекса [(S)-ВРВ- Gly] Ni (II) в 30 мл метанола. Раствор продувают аргоном. Электролиз проводят в гальваностатическом режиме (при плотности тока J=5 мА/см2) в течение 2,5 ч в токе аргона при перемешивании магнитной мешалкой (пропускают 135 Кл электричества, 3.5 F/моль комплекса). К полученному после электролиза раствору добавляют 700 мкл (11.2 ммоль) ледяной уксусной кислоты и оставляют смесь на 20 часов. На следующий день добавляют еще 300 мкл ледяной уксусной кислоты (4.8 ммоль), раствор выливают в 80 мл воды и экстрагируют бензолом (3×30 мл). Объединенные бензольные фракции промывают водой и упаривают на роторном испарителе. Сухой остаток сушат в течение ночи в вакуум-эксикаторе над оксидом фосфора (V). Затем полученную смесь диастереомеров хроматографически разделяют на колонке с силикагелем (элюент - ацетон), выделяя две фракции. После упаривания растворителя и высушивания комплексов в вакууме над оксидом фосфора (V) из первой фракции получают 186 мг (88%) комплекса, содержащего фрагмент (R)-серина и 20 мг (9%) комплекса (S)-серина.
HRMS: m/z 528.1426 (М+Н+, вычислено для C28H28N3NiO4: 528.1432); 550.1252 (M+Na+, вычислено для C28H27N3NiO4Na: 550.1252).
Диастереомерный комплекс Ni(II)c (R)-серином
ЯМР 1H (CDCl3 δ, м.д.): 8.49 (дц, J=8.7, 1.1 Гц, 1Н (Н-8)), 8.10-8.05 (м, 2Н (Н-17,21)), 7.60-7.43 (м, 6Н (18,19,20,24,25,26)), 7.26 (ддд, J=8.7, 6.8, 1.8 Гц, 1Н (Н-7)), 7.22-7.18 (м, 1Н (Н-27)), 7.09-7.04 (м, 1H (Н-23)), 6.77 (дд, J=8.3, 1.8 Гц, 1H (Н-5)), 6.70 (ддд, J=8.3, 6.8, 1.2 Гц, 1H (Н-6)), 4.45 (д, J=13.0 Гц, 1H (Bn Н-15)), 4.18-4.09 (м, 1H (Pro Н-14)), 3.94 (дд, J=7.3, 4.6 Гц, 1H (Ser Н-2)), 3.76 (ддд, J=10.1, 7.3, 3.6 Гц, 1H (Ser β-СН2)), 3.63 (дд, J=9.6, 4.3 Гц, 1H (Pro Н-11)), 3.62-3.57 (м, 1H (Ser β-СН2)), 3.55 (д, J=13.0 Гц, 1Н (Вn Н-15)), 2.86 (дц, J=8.8, 3.6 Гц, 1H (Ser ОН)), 2.66-2.54 (м, 2Н (Pro Н-13,14)), 2.31-2.15 (м, 2Н (Pro Н-12)), 1.99-1.89 (м, 1Н (Pro Н-13)). (лит ЯМР 1Н [5]).
ЯМР 13С-{1Н} (CDCl3 δ, м.д.): 182.26, 179.52, 172.90, 143.10, 134.05, 133.94, 133.80, 132.86,131.74, 130.03, 129.40, 129.27, 128.96, 128.63, 128.33, 126.67, 125.84, 123.94, 120.91, 71.27, 69.42, 64.50, 62.28, 59.01, 30.68, 23.50.
Диастереомерный комплекс Ni(II) с (S)-серином
ЯМР 1H (CDCl3 δ, м.д.): 8.12 (дд, J=8.7, 1.1 Гц, 1H (Н-8)), 8.09-8.05 (м, 2Н (Н-17,21)), 7.53-7.42 (м, 3Н (Н-24,25,26)), 7.39-7.34 (м, 2Н (Н-18,20)), 7.26-7.23 (м, 1Н (Н-27)), 7.23-7.18 (м, 1Η (Η-19)), 7.14 (ддд, J=8.7, 6.6, 2.1 Гц, 1H (Н-7)), 7.02-6.98 (м, 1Н (Н-23)), 6.66 (ддд, J=8.3, 6.6,1.1 Гц, 1Н (Н-6)), 6.62 (дд, J=8.3,2.0 Гц, 1H (Н-5)), 4.35 (д, J=12.6 Гц, 1H (Вn Н-15)), 3.99 (т, J=4.5 Гц, 1H (Ser Н-2)), 3.84-3.72 (м, 3Н (Ser β-СН2, Pro Н-13)), 3.55 (д, J=12.6 Гц, 1H (Вn Н-15)), 3.49-3.45 (м, 1Н (Pro Н-14)), 3.45 (дц, J=11.1, 5.6 Гц, 1Н (Pro Н-11)), 2.98 (дд, J=7.0, 5.0 Гц, 1Н (Ser -ОН)), 2.80-2.71 (м, 1Н (Pro Н-12)), 2.54-2.42 (м, 1Н (Pro Н-12)), 2.15-2.06 (м, 1Н (Pro Н-13)), 2.05-1.97 (м, 1H (Pro Н-14)). (лит. ЯМР 1Н [5]).
ЯМР 13С-{1Н} (CDCl3 δ, м.д.): 180.55, 178.59, 171.64, 142.60, 133.84, 133.45, 133.40, 132.41, 131.66, 130.00, 129.22, 129.18, 129.07, 129.03, 127.88, 127.06, 126.71, 123.99, 120.84, 72.10,70.51,64.70,63.22, 57.38, 30.85,23.63.
Пример 2. Получение сериновых комплексов и их выделение
В двухэлектродную ячейку с неразделенным электродным пространством объемом 10 мл (рабочий электрод - платиновая пластина площадью 100 мм2, вспомогательный электрод - графитовая ткань марки «Урал», размером 10 мм×30 мм) помещают раствор 180 мг (3.2 ммоль) гидроксида калия и 40 мг (0.08 ммоль) комплекса [(S)-ВРВ- Gly] Ni (II) в 10 мл метанола. Раствор продувают аргоном. Электролиз проводят в гальваностатическом режиме (при плотности тока 5 мА/см2) в течение 1,5 ч в токе аргона при перемешивании магнитной мешалкой (пропущено 27 Кл электричества, 3.5 F/моль комплекса). К полученному в результате электролиза раствору сразу добавляют 200 мкл (3,2 ммоль) ледяной уксусной кислоты. Раствор упаривают, добавляют ацетон (10 мл), осадок ацетата калия отделяют фильтрованием. Суммарный выход целевых комплексов практически количественный. Реакционную смесь хроматографически разделяют на колонке с силикагелем (элюент - ацетон), выделяя две фракции. После упаривания растворителя из первой фракции получают 14 мг (33%) комплекса, содержащего фрагмент (R)-серина и 15 мг (35%) комплекса (S)-серина.
HRMS: m/z 528.1426 (М+Н+, вычислено для C28H28N3NiO4: 528.1432); 550.1252 (M+Na+, вычислено для C28H27N3NiO4Na: 550.1252).
Пример 3. Получение сериновых комплексов
В условиях, аналогичных условиям, описанным в примере 2, за исключением того, что вместо платиновой пластины в качестве рабочего электрода использовали графитовую ткань марки «Урал», получают 15 мг (35%) комплекса, содержащего фрагмент (R)-серина и 15 мг (35%) комплекса (S)-серина.
Пример 4. Получение комплекса с (S)серином.
В условиях, аналогичных условиям, описанным в примере 2, за исключением того, что при проведении электролиза 20 мг (0.04 ммоль) [(S)-BPB-Gly] Ni (II) в тех же условиях, но с меньшим количеством щелочи (56 мг (1.0 ммоль) КОН), получают практически только один продукт - (S)-диастереомер комплекса с (S)-серином (11 мг, выход 55%, выход в пересчете на прореагировавший [(S)-ВРВ- Gly] Ni (II) составляет 98%), соотношение комплексов с (S) и (R)-серином, определенное из спектра ЯМР 1Н, составляет 98:2.
Пример 5. Получение треониновых β-метилсериновых) комплексов и их выделение.
В ячейку с неразделенным электродным пространством помещают раствор 300 мг (5.4 ммоль) гидроксида калия и 20 мг (0.04 ммоль) комплекса [(S)-ВРВ- Gly] Ni (II) в 10 мл этанола. Раствор продувают аргоном. Электролиз проводили в гальваностатическом режиме (J=5 мА/см2) в течение 45 мин при перемешивании магнитной мешалкой (пропускают 13.5 Кл электричества, 3.5 F/моль комплекса). Затем к раствору добавляют 400 мкл (6.4 ммоль) ледяной уксусной кислоты. Раствор упаривают, добавляют четыреххлористый углерод (10 мл), осадок ацетата калия отделяют фильтрованием. Суммарный выход неразделенных диастереомерных комплексов составляет 95%, соотношение комплексов с (S) и (R)-треонином, определенное из спектра ЯМР 1Н, составляет 3:10. Реакционную смесь после фильтрования хроматографически разделяют на колонке с силикагелем (элюент - ацетон). Получают две фракции. После упаривания растворителя из первой фракции получают 13,2 мг (61%) комплекса, содержащего фрагмент (2R, 3S)-треонина и 6,9 мг (32%) комплекса (2S,3R)-треонина.
HRMS: m/z 542.1570 (М+Н+, вычислено для C29H30N3NiO4: 542.1589); 564.1401 (M+Na+, вычислено для C29H29N3NiO4Na: 564.1408).
Диастереомерный комплекс Ni(II) с (2R, 3S)-треонином:
ЯМР 1H (CDCl3 δ, м.д.): 8.47 (дд, J=8.8, 1.1 Гц, 1H), 7.86-7.83 (м, 2Н), 7.55-7.40 (м, 6Н), 7.28 (ддд, J=8.8, 6.9, 1.7 Гц, 1H), 7.24 (с, 1H), 7.10 (с, 1H), 6.85 (дд, J=8.3, 1.7 Гц, 1H), 6.74 (ддд, J=8.3, 6.9,1.1 Гц, 1H), 4.72 (д, J=13.2 Гц, 1Н), 4.07 (д, J=5.2 Гц, 1Н), 3.99 (ддд, J=11.1, 6.8, 4.0 Гц, 1Н), 3.90 (д, J=13.1 Гц, 1H), 3.81-3.72 (м, 1Н), 3.59 (дд, J=10.0,4.5 Гц, 1Н), 3.62-3.56 (м, 1Н), 2.76-2.61 (м, 1Н), 2.53 (ддд, J=11.5, 9.6,6.2 Гц, 1H), 2.29-2.20 (м, 1H), 2.13-2.01 (м, 1H), 1.91-1.80 (м, 1Н), 1.70 (д, J=6.3 Hz, 3Н).
ЯМР 13С-{1Н} (CDCl3 δ, м.д.): 182.33, 179.44, 173.83, 142.96, 134.19, 134.00, 132.88, 132.66, 131.89, 130.13, 129.34, 129.17, 128.84, 126.56, 125.91, 124.16, 121.07, 74.07, 68.96, 68.81,61.99, 57.60, 30.90, 23.64, 19.39.
Диастереомерный комплекс Ni(II) с (2S, 3R)-треонином:
HRMS: m/z 542.1583 (М+Н+, вычислено для C29H30N3NiO4 542.1589); 564.1409 (M+Na+, вычислено для С29Н29N3NiO4Na 564.1408).
ЯМР 1H (CDCl3 δ, м.д.): 8.26 (ддд, J=8.7, 1.2, 0.5 Гц, 1Н), 8.06-8.03 (м, 2Н), 7.58-7.51 (м, 3Н), 7.38-7.33 (м, 2Н), 7.22-7.14 (м, 3Н), 6.95-6.91 (м, 1Н), 6.67 (ддд, J=8.3, 6.7, 1.2 Гц, 1Н), 6.63 (ддд, J=8.3, 2.0, 0.5 Гц, 1Н), 4.37 (д, J=12.7 Гц, 1H), 4.12 (д, J=5.1 Гц, 1Н), 3.71-3.63 (м, 1Н), 3.62 (д, J=12.7 Гц, 1Н), 3.55-3.34 (м, 3Н), 2.85-2.75 (м, 1H), 2.60-2.51 (м, 1H), 2.16-2.02 (м, 2Н), 1.98 (д, J=6.3 Гц, 3Н).
Пример 6. Получение β-этилсериновых комплексов и их выделение.
В условиях, аналогичных примеру 5, за исключением того, что вместо этанола использовали н-пропанол, получают β-этилсериновые комплексы. Суммарный выход неразделенных диастереомерных комплексов составляет 95%. После хроматографического разделения получено 6.5 мг (30%) (S, 2R, 3S)-диастереомера (первая фракция) и 11 мг (50%) (S, 2S, 3S)-диастереомера (вторая фракция).
Диастереомерный комплекс Ni(II) с (2S, 3R)-β-этилсерином:
1H NMR (400 MHz, CDCl3) δ, м.д. 8.50 (дд, J=8.7, 1.2 Гц, 1H), 7.87-7.83 (м, 2Н), 7.55-7.41 (м, 6Н), 7.29 (ддд, J=8.7, 6.9, 1.7 Гц, 1H), 6.84 (дд, J=8.3, 1.7 Гц, 1H), 6.74 (ддд, J=8.3, 6.9, 1.2 Гц, 1Н), 4.66 (д, J=13.3 Гц, 1H), 4.09 (d, J=5.3 Гц, 1Н), 4.07-4.01 (м, 1H), 3.85 (d, J=13.3 Гц, 1Н), 3.62 (дд, J=9.9, 4.3 Гц, 1Н), 3.48-3.40 (м, 1H), 3.38-3.31 (м, 1Н), 2.71-2.53 (м, 3Н), 2.29-2.20 (м, 1Н), 2.13-2.01 (м, 1H), 1.91-1.81 (м, 1H), 1.65-1.58 (м, 1H), 1.08 (т, J=7.3 Гц, 3Н).
Диастереомерный комплекс Ni(II) с (2R, 3S)-β-этилсерином:
1Н NMR (400 MHz, CDCl3) δ, м.д. 8.24 (дд, J=8.7, 0.8 Гц, 1H), 8.07-8.02 (м, 2Н), 7.58-7.41 (м, 4Н), 7.38-7.32 (м, 2Н), 7.22-7.12 (м, 2Н), 6.94-6.88 (м, 1H), 6.70 (ддд, J=8.20, 6.70, 1.20 Гц, 1H), 6.62 (дд, J=8.2, 1.9 Гц, 1Н), 4.37 (д, J=12.7 Гц, 1H), 4.14 (д, J=4.8 Гц, 1H), 3.61 (d, J=12.7 Гц, 1Н), 3.53-3.43 (м, 2Н), 3.43-3.27 (м, 3Н), 3.21-3.09 (м, 1H), 2.84-2.74 (м, 1Н), 2.60-2.48 (м, 1H), 2.16-2.01 (м, 2Н), 1.14 (т, J=7.3 Гц, 3Н).
13С NMR (101 MHz, CDCl3) δ, м.д. 180.38, 179.08, 172.58, 142.70, 133.88, 133.83, 133.34, 132.77, 131.82, 131.64, 130.21, 129.24, 129.23, 129.11, 129.06, 128.19, 126.87, 126.44, 123.60, 120.91, 73.45, 70.63, 63.56, 57.07, 30.82, 26.65, 23.36, 11.00.
Пример 7. Получение сериновых комплексов с использованием этиленгликоля и их выделение.
В ячейку с неразделенным электродным пространством помещают раствор 300 мг (5.4 ммоль) гидроксида калия и 20 мг (0.04 ммоль) комплекса [(S)-ВРВ- Gly] Ni (II) в 10 мл этиленгликоля. Раствор продувают аргоном. Электролиз проводят в гальваностатическом режиме (J=5 мА/см2) в течение 45 мин при перемешивании магнитной мешалкой (пропускают 13.5 Кл электричества, 3.5 F/моль комплекса). Затем к раствору добавляют 400 мкл (6,4 ммоль) ледяной уксусной кислоты и разбавляют 70 мл воды. Экстрагируют хлористым метиленом (3×15 мл), объединенную органическую фракцию промывают два раза насыщенным раствором хлорида натрия, затем сушат над хлоридом кальция. Суммарный выход неразделенных диастереомерных комплексов составил 95%, соотношение комплексов с (S)- и (R)-серином, определенное из спектра ЯМР 1H, составляет 3:5. Полученную смесь разделяют хроматографически, элюент - ацетон. Получают две фракции диастереомерных комплексов, содержащих остаток серина: 10,5 мг (5,7 г)-диастереомера (50%) и 6,2 мг (S,S)-диастереомера (29%).
Пример 8. Стандартная методика разложения комплексов Ni(II) и выделения β-гидрокси-α-аминокислот
К 10 мл кипящего 2Ν раствора НСl добавляют по каплям раствор 108 мг (0,2 ммоль) Ni (II) комплекса (S)-серина в 5 мл МеОН. Через 30 мин после исчезновения красной окраски комплекса раствор нейтрализуют 20% NH4OH до рН 8-9 и экстрагируют хлороформом хиральный реагент (S)-ВРВ, пригодный для повторного использования. Выход реагента, определенный спектрофотометрически по поглощению при 330 нм, составляет ~98%, оптический выход (S)-ВРВ >99% (согласно поляриметрическим данным). Аминокислоту из водного слоя выделяют на катионообменной смоле Dowex-50 в Н+-форме, сорбированную аминокислоту элюируют 5% NH4OH, элюат упаривают под вакуумом, аминокислоту кристаллизуют. Выход аминокислоты (S)-серина - 20,5 мг (98%). Энантиомерная чистота >99% (по данным энантиомерного ГЖХ-анализа). Удельное вращение полученного (S)-серина соответствует литературным данным [α]20 D+15.2 (С=5.1, 1N HCl).
Количества реагентов, используемые в примерах, могут быть масштабированы в десятки раз. Для этого достаточно просто увеличить объем электролизера и площадь электродов.
Таким образом, предлагается новый эффективный способ электрохимического стереоселективного α-гидроксиалкилирования глицина, который включает введение глицина в виде основания Шиффа в координационную сферу комплекса никеля с хиральным лигандом ((S)-2N-(N'-бензилпролил)аминобензофеноном) и последующего гальваностатического электролиза в спиртовом растворе в присутствии КОН, причем реакцию проводят как one-pot процесс, а в качестве реагентов используются алифатические спирты, вместо более дорогих и менее доступных альдегидов. Способ является удобной альтернативой известной реакции конденсации с данным комплексом. Способ отличается аппаратурной простотой и высокой эффективностью.
Технический результат
Техническим результатом данного способа являются
- использование дешевых и доступных реагентов - спиртов (вместо соответствующих альдегидов) и щелочи (вместо гидрида или метилата натрия),
- проведение реакции как технологичный one-pot процесс и
- расширение ассортимента целевых продуктов.
Возможность введения в реакцию вместо дорогостоящих альдегидов их прекурсоров - спиртов, которые являются гораздо более дешевыми реагентами и более технологичны в использовании, позволяет избежать применения альдегидов, расширить ассортимент целевых продуктов за счет получения аминокислотных комплексов, которые ранее не были синтезированы.
α-Гидроксиалкилирование глицина проводят электрохимически в одном реакторе (one-pot процесс), поскольку альдегиды генерируются путем анодного гальваностатического окисления спиртов, используемых одновременно в качестве растворителя (при этом исключается использование пожароопасного тетрагидрофурана), и тем самым альдегиды вводятся в реакцию in situ, что уменьшает вероятность характерных для них побочных реакций и позволяет снизить отходы при сохранении высокого выхода целевых комплексов II и III.
В заявляемом способе используют дешевую щелочь, а недорогие и огнеопасные гидрид натрия или метилат натрия. Изменяя концентрацию КОН (который одновременно выполняет роль основания и фонового электролита), можно широко варьировать соотношение образующихся Ni(II) комплексов с (S) или (R) конфигурацией α-аминокислотного центра, при сохранении высоких выходов.
Способ получения n-(4-бромфенил)-n-(2-адамантил)амина (бромантана)
Способ получения мономера для протонпроводящих полимерных мембран
Соолигофенолформальдегидные фталидсодержащие новолаки для получения сшитых фталидсодержащих сополимеров, способы их получения (варианты) и сшитые фталидсодержащие сополимеры в качестве конструкционных полимеров
Способ получения олигоорганосилоксанов
Способ получения привитых силоксановых покрытий с сорбционными n-аминоди(метиленфосфоновыми) группами на волокнах и модифицированные волокнистые материалы
Способ формования криогелей поливинилового спирта
Способ получения 1-(8-метокси-4,8-диметилнонил)-4-(1-метилэтил)бензола из изопрена (варианты)
Магнитный нанокомпозит и способ его получения
Способ получения триметилфторсилана
Способ получения циклосилоксановых полиолов
Способ получения n-(4-бромфенил)-n-(2-адамантил)амина (бромантана)
Способ получения мономера для протонпроводящих полимерных мембран
Соолигофенолформальдегидные фталидсодержащие новолаки для получения сшитых фталидсодержащих сополимеров, способы их получения (варианты) и сшитые фталидсодержащие сополимеры в качестве конструкционных полимеров
Способ определения следовых компонентов методом лазерно-искровой эмиссионной спектроскопии
Способ синтеза сополимеров акрилонитрила с акриловой кислотой
Наночастицы антиоксидантного фермента супероксиддисмутазы в виде полиэлектролитного комплекса состава фермент-поликатион-полианион и способ их получения
Катализатор паровой конверсии углеводородов и способ его получения
Катодные материалы для твердооксидных топливных элементов на основе никельсодержащих слоистых перовскитоподобных оксидов
Способ нагрева электродов и создания самостоятельного дугового разряда с поджигом от тонкой металлической проволочки в свободном пространстве в магнитном поле
Способ определения катехоламинов и их метаболитов с использованием твердофазного флуоресцентного биосенсора

