Результат интеллектуальной деятельности: АНАЛОГИ ГЛЮКАГОНОПОДОБНОГО ПЕПТИДА-1 И ИХ ПРИМЕНЕНИЕ
Вид РИД
Изобретение
Уровень техники
Диабет стал третьим по распространенности неинфекционным заболеванием после сердечнососудистых и цереброваскулярных заболеваний и опухолей. ВОЗ прогнозирует, что к 2030 году количество больных диабетом во всем мире будет превышать 360 миллионов человек, более 90% из которых будут иметь диабет II типа. Сахарный диабет второго типа (СДIIТ) относится к инсулиннезависимым сахарным диабетам или приобретенным диабетам и становится все более распространенным. Распространение диабета второго типа увеличивается с угрожающей скоростью. Несмотря на прогресс в лечении сахарного диабета, случаи гипогликемии часто являются ограничивающим фактором для достижения оптимального контроля уровня сахара в крови. В борьбе с существующими ограничениями лечения диабета значительный прогресс был достигнут при исследовании глюкагоноподобного пептида-1 (глюкагоноподобный пептид-1, ГПП-1). ГПП-1 представляет собой кишечный гормон, вырабатываемый L-клетками кишечника. Это сильный антигипергликемический гормон, вызывающий глюкозозависимую стимуляцию секреции инсулина, подавляя в то же время секрецию глюкагона. По-видимому ГПП-1 вызывает восстановление чувствительности β-клеток поджелудочной железы к глюкозе, благодаря, возможно, повышенной экспрессии GLUT2 и глюкокиназы. Также известно, что ГПП-1 ингибирует апоптоз панкреатических β-клеток и стимулирует пролиферацию и дифференцировку β-клеток, секретирующих инсулин. Секреция ГПП-1 играет важную роль в патогенезе сахарного диабета II типа. Было показано, что секреция ГПП-1 L-клетками снижается у пациентов с сахарным диабетом II типа, оставаясь инсулинотропной. Существует несколько гипотез о том, как это влияет на предполагаемую клиническую эффективность таких новых препаратов. Секреция ГПП-1 L-клетками кишечника в кровоток зависит от присутствия питательных веществ в просвете тонкой кишки. Стимуляторами секреции этого гормона являются основные питательные вещества, такие как углеводы, белки и липиды. Это сильный антигипергликемический гормон, вызывающий глюкозозависимую стимуляцию секреции инсулина, подавляя в то же время секрецию глюкагона. Такие глюкозозависимые эффекты являются особенно привлекательными для новых способов лечения диабета, потому что, когда концентрация глюкозы в плазме крови находится на нормальном уровне, ГПП-1 не стимулирует выработку инсулина и не вызывает гипогликемию.
Терапевтический потенциал ГПП-1 и его аналогов еще больше возрастает, если учесть его использование у пациентов с сахарным диабетом типа I. Ряд исследований показали эффективность нативного ГПП-1 в лечении инсулинозависимого сахарного диабета (ИЗСД). Как и у больных инсулиннезависимым сахарным диабетом (ИНСД), ГПП-1 является эффективным в снижении гипергликемии натощак за счет подавления секреции глюкагона. Дополнительные исследования показали, что ГПП-1 также снижает изменение уровня глюкозы при постпрандиальной гликемии при ИЗСД, скорее всего, путем замедления опорожнения желудка. Эти наблюдения позволяют предположить, что ГПП-1 может быть полезен в качестве средства для лечения ИЗСД, а также для ИНСД.
К сожалению, период полужизни нативного ГПП-1, который подвержен действию дипептидил-пептидазы IV (ДПП IV), является довольно коротким. Например, период полужизни ГПП-1 (7-37) ОН составляет от 3 до 5 минут. Устойчивое снижение концентрации глюкозы в крови наблюдается только при непрерывной инфузии, как было показано в исследованиях, в которых ГПП-1 вводили внутривенно капельно в течение 24-часового периода. Поэтому более длительное действие пептидов, полученных на основе ГПП-1, которые являются устойчивыми к ДПП IV, может иметь большой терапевтический потенциал для лечения сахарного диабета.
Краткое описание изобретения
Настоящее изобретение обеспечивает аналоги глюкагоноподобного пептида-1 и их применение.
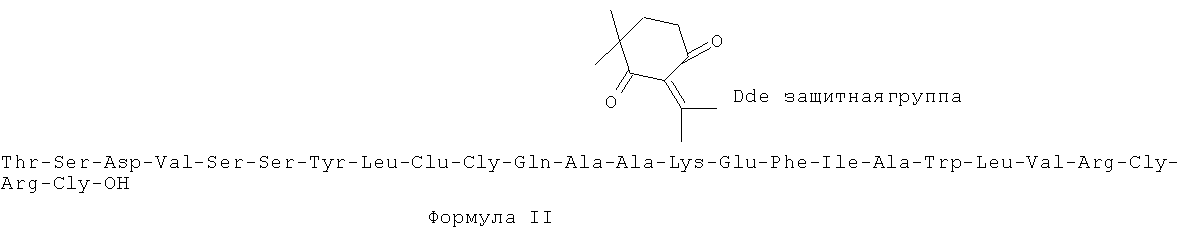
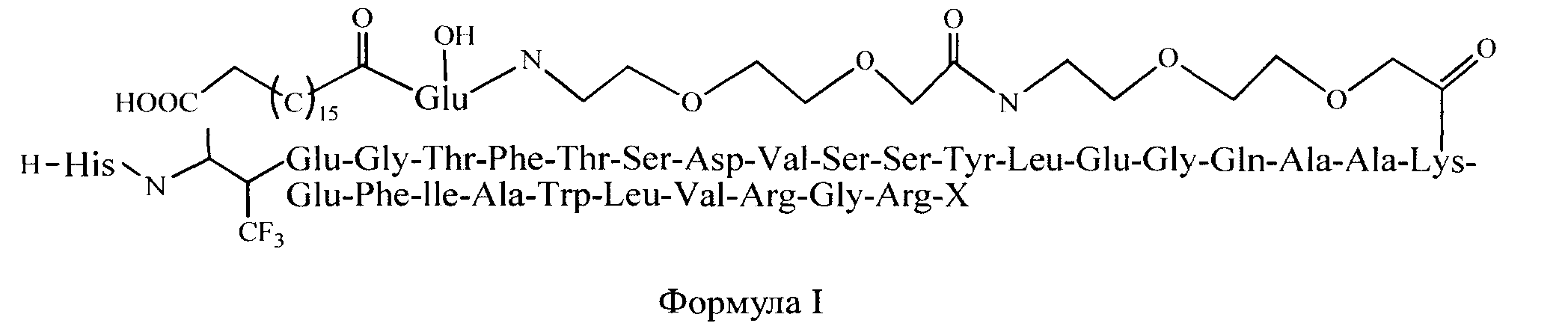
В первую очередь, настоящее изобретение относится к соединениям Формулы I, или ее фармацевтически приемлемой соли, сольвату, хелату, нековалентному комплексу, пролекарству, или смеси любых вышеуказанных соединений, где X - это глицин или глицинамид.
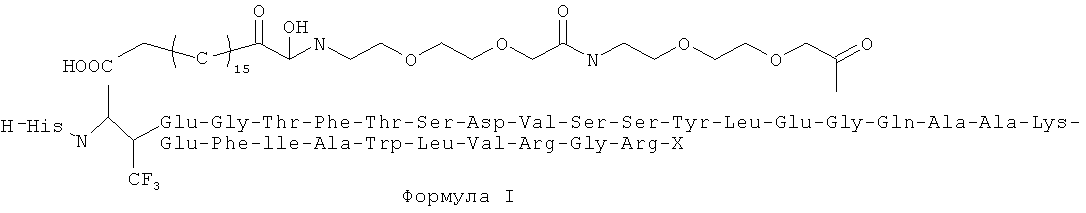
Настоящее изобретение также относится к фармацевтическим композициям, каждая из которых содержит терапевтически эффективное количество по меньшей мере одного соединения Формулы I и по меньшей мере один фармацевтически приемлемый наполнитель.
Настоящее изобретение также относится к применению фармацевтической композиции для получения лекарственного средства
В предпочтительном случае фармацевтические композиции применяют для приготовления лекарства для лечения или профилактики диабета II типа, нарушения толерантности к глюкозе, диабета I типа, ожирения, гипертонии, синдрома X, дислипидемии, когнитивных расстройств, атеросклероза, инфаркта миокарда, ишемической болезни сердца, сердечнососудистых заболеваний, инсульта, воспалительного заболевания кишечника и/или диспепсии или язвы желудка.
В предпочтительном случае фармацевтические композиции применяют для приготовления лекарства для замедления или предотвращения развития диабета II типа.
В предпочтительном случае фармацевтические композиции применяют для приготовления лекарства для снижения потребления пищи, уменьшения β-клеточного апоптоза, повышения функциональной активности β-клеток, увеличения массы β-клеток и/или для восстановления чувствительности β-клеток к глюкозе.
Настоящее изобретение также относится к применению соединений Формулы I для приготовления лекарственных средств для лечения или профилактики диабета II типа, нарушения толерантности к глюкозе, диабета I типа, ожирения, гипертонии, синдрома X, дислипидемии, когнитивных расстройств, атеросклероза, инфаркта миокарда, ишемической болезни сердца, сердечно-сосудистых заболеваний, инсульта, воспалительного заболевания кишечника и/или диспепсии, или язвы желудка.
В предпочтительном случае соединение Формулы I применяют для приготовления лекарства для замедления или предотвращения развития диабета II типа.
В предпочтительном случае соединение Формулы I применяют для приготовления лекарства для снижения потребления пищи, уменьшения β-клеточного апоптоза, повышения функциональной активности β-клеток, увеличения массы β-клеток и/или для восстановления чувствительности β-клеток к глюкозе.
Настоящее изобретение также обеспечивает способ изменения уровня глюкозы в крови у субъекта, который включает введение субъекту соединения Формулы I.
Настоящее изобретение подробно описано ниже и проиллюстрировано примерами вариантов реализации, представленными ниже.
Если не указано обратное, все числа, выражающие количества различных ингредиентов, условия реакции и т.д., используемые в описании и Формуле изобретения, следует понимать как характеризуемые термином "примерный" или "приблизительный". Соответственно, если прямо не указано обратное, числовые параметры, приведенные в следующем описании и прилагаемой Формуле изобретения, являются приблизительными и могут варьировать в зависимости от обычного отклонения, которое можно найти в соответствующих опытных измерениях.
Соединения, описанные в настоящем изобретении, могут содержать один или несколько хиральных центров и/или двойных связей и, следовательно, могут существовать в виде стереоизомеров, таких как изомеры по двойной связи (например, геометрические изомеры), энантиомеры или диастереомеры. Соответственно, любые химические структуры в рамках настоящего описания, полностью или их части, с относительной конфигурацией охватывают все возможные энантиомеры и стереоизомеры описанных соединений, включая стереоизомерически чистую форму (например, геометрически чистую, энантиомерно чистую или диастереомерно чистую) и смеси энантиомеров и стереоизомеров. Смеси энантиомеров и стереоизомеров могут быть разделены на отдельные энантиомеры или стереоизомеры с использованием методов разделения или методов синтеза хиральных соединений, хорошо известных специалистам в данной области.
Соединения Формулы I включают, но не ограничиваются перечисленными: оптические изомеры этих соединений, рацематы и другие смеси. В таких ситуациях, отдельные энантиомеры или диастереомеры, т.е. оптически активные формы, могут быть получены путем асимметрического синтеза или разделения рацематов. Разделение рацематов может быть достигнуто, например, обычными способами, такими как кристаллизация в присутствии разделяющего агента, или хроматография, с использованием хиральной высокоэффективной жидкостной хроматографической (ВЭЖХ) колонки. Кроме того, соединения Формулы I включают Z- и Е-формы (или цис- и транс-формы) соединений с двойными связями. Если соединения Формулы I существуют в различных таутомерных формах, химические объекты настоящего изобретения включают все таутомерные формы соединения.
Соединения согласно настоящему изобретению, включают, но не ограничиваются перечисленными: соединения Формулы I и все фармацевтически приемлемые формы. Фармацевтически приемлемые формы соединений, перечисленные здесь, включают фармацевтически приемлемые соли указанных соединений, сольваты указанных соединений, кристаллические формы (в том числе полиморфные кристаллы и клатраты) указанных соединений, хелаты указанных соединений, нековалентные комплексы указанных соединений, пролекарства указанных соединений и их смеси. В некоторых вариантах реализации соединения, описанные здесь, имеют форму фармацевтически приемлемой соли. В настоящем описании термин "соединение" включает не только само соединение, но также фармацевтически приемлемые соли указанного соединения, сольваты указанного соединения, хелаты указанного соединения, нековалентные комплексы указанного соединения, пролекарства указанного соединения, а также смеси любых вышеперечисленных веществ.
Как отмечалось выше, пролекарства также включены в объем химических соединений, например, сложноэфирные или амидные производные соединений Формулы I. Термин "пролекарства" включает любые соединения, которые превращаются в соединения Формулы I при введении пациенту, например, в результате метаболических превращений пролекарства. Примеры пролекарств включают, но не ограничиваются перечисленным: ацетаты, формиаты, бензоаты и другие производные функциональных групп (такие как спиртовые или аминогруппы) в соединениях Формулы I.
В различных разделах настоящего описания заместители соединений согласно изобретению перечислены в форме групп или классов. Это сделано специально, т.к. изобретение включает каждую отдельную комбинацию представителей таких групп и классов. Например, термин "C1-3алкил" специально предназначен для описания отдельно метил-, этил- и С3 алкила (включая н-пропил и изопропил).
Как показано в настоящем изобретении, структура Формулы I представляет химически стабильные соединения, менее восприимчивые к деградации под действием дипептидил-пептидазы IV (ДПП-IV), с периодом полужизни в плазме крови более 30 часов, тем самым превосходящие внутривенное введение ГПП-1 или непрерывное подкожное введение в лечение нарушений. Кроме того, настоящее изобретение относится к структуре, показанной в соединениях, или соединениям в качестве активного ингредиента для приготовления лекарств, используемых для снижения концентрации глюкозы в крови, с очень длительным периодом полужизни в плазме крови (30 часов), но также со значительным гипогликемическим эффектом.
Краткое описание чертежей
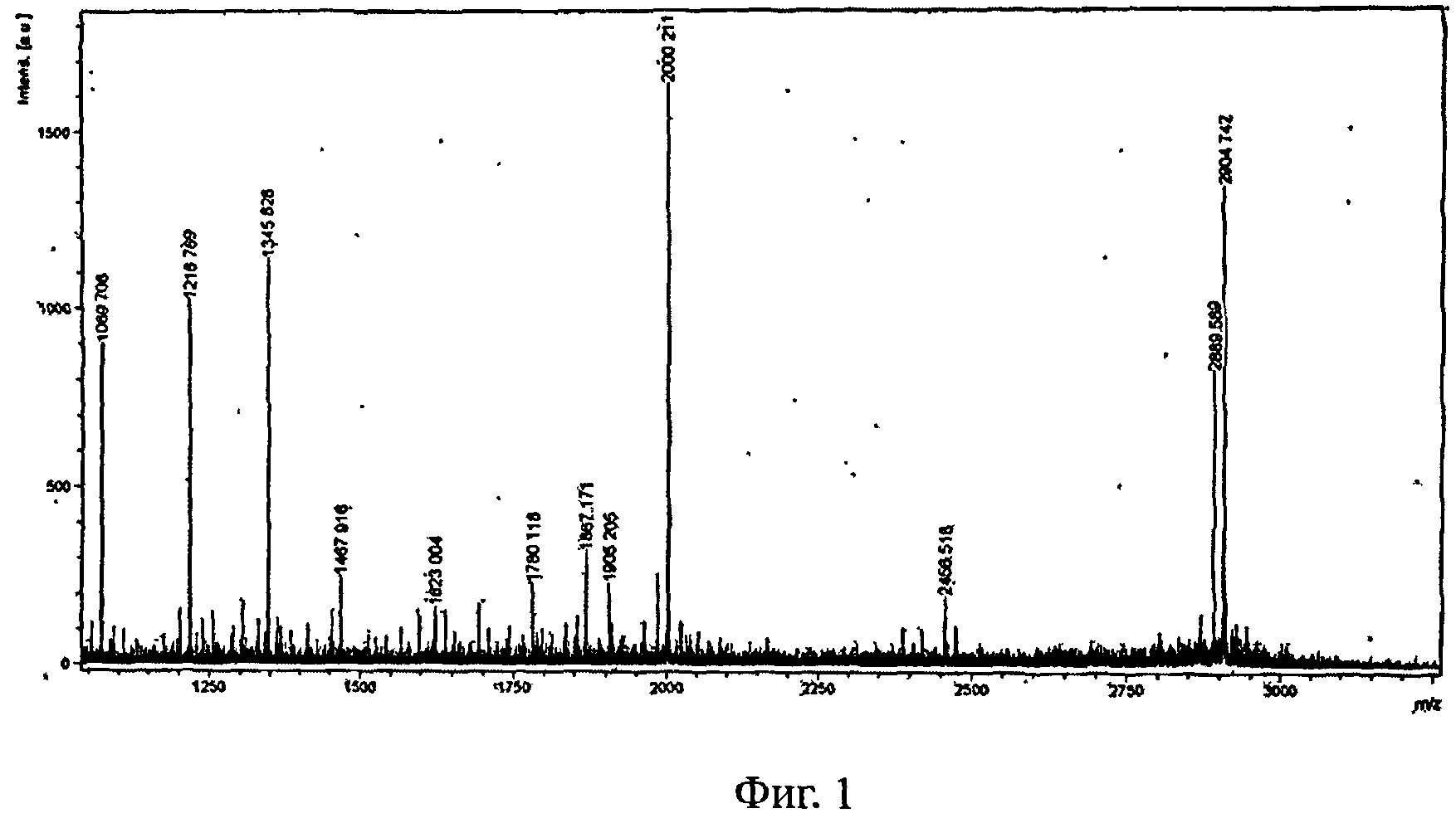
На Фигуре 1 представлена масс-спектрограмму соединения Формулы II
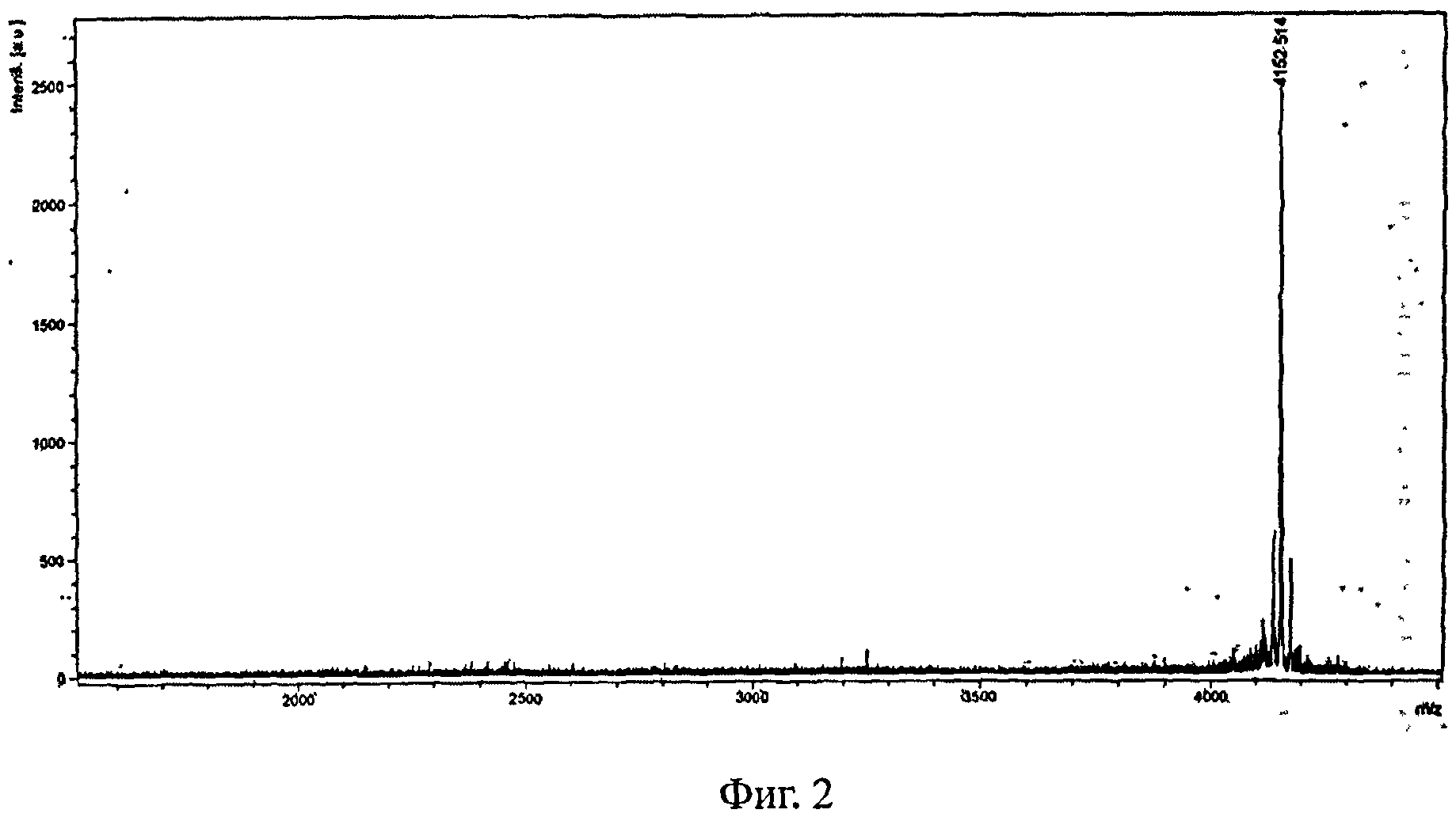
На Фигуре 2 представлена масс-спектрограмму соединения из Примера 1.
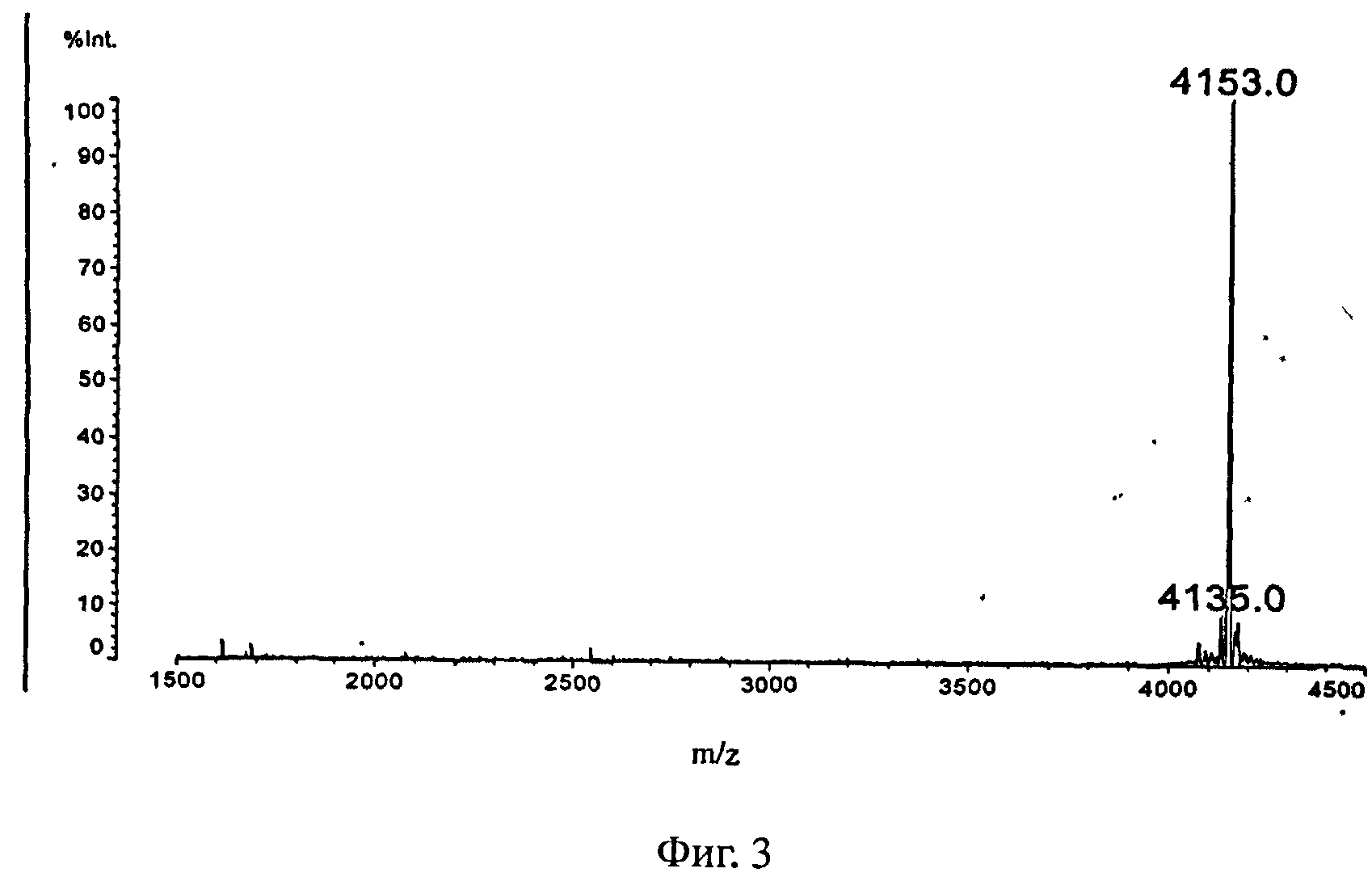
На Фигуре 3 представлена масс-спектрограмму соединения из Примера 2.
Варианты реализации изобретения
Настоящее изобретение проиллюстрировано, но не ограничивается, следующими примерами, которые описывают получение соединений Формулы I согласно настоящему изобретению.
В следующих примерах описаны только варианты реализации настоящего изобретения, для того, чтобы специалист в соответствующей области мог реализовать его, но настоящее изобретение не ограничивается этими вариантами. В приведенных ниже примерах, некоторые способы или методы не описаны в деталях, но понятно, что они представляют собой известные или используемые способы или методы, которые входят в число навыков специалиста в области, к которой относится настоящее изобретение.
Пример 1
I. Получение промежуточных соединений (Димер)
Структура Димера показана ниже:
Он может быть синтезирован следующим способом:
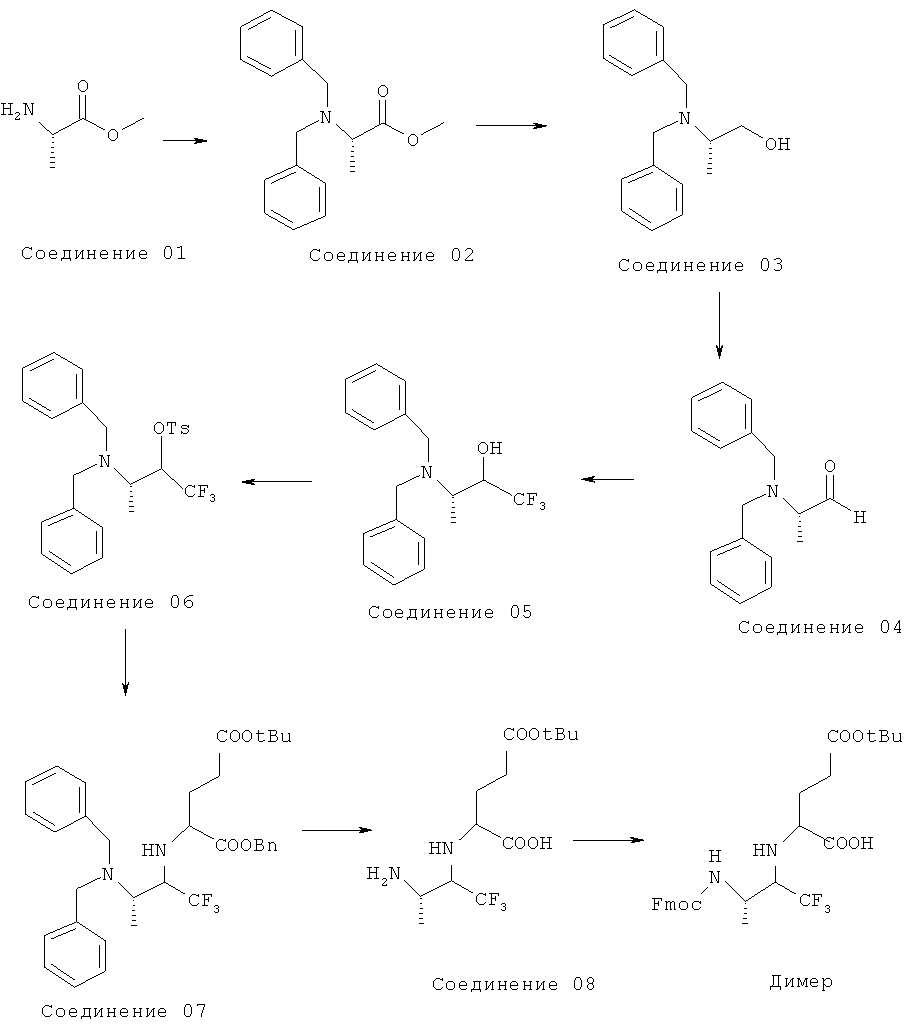
1. Приготовление Соединения 01·HCl
80 мл тионилхлорида добавляли по каплям к 250 мл метанола при -10°C в течение 2 часов. Затем смесь нагревали до комнатной температуры и перемешивали в течение 1 часа, добавляли 40 г L-аланина и перемешивали в течение ночи. Реакционную смесь нагревали с обратным холодильником в течение 4 часов, охлаждали до комнатной температуры и концентрировали с получением 80 г неочищенного Соединения 01·HCl.
2. Приготовление Соединения 02
113 г бензилбромида добавляли к 40 г смеси Соединения 01·HCl с 350 мл ДМФ. 150 г безводного карбоната калия добавляли к вышеуказанной смеси и перемешивали в течение 2 часов. Реакционную смесь нагревали до 50°C и перемешивали в течение 2 часов, затем охлаждали до комнатной температуры и экстрагировали 400 мл этилацетата и 1200 мл воды. Органический слой промывали 150 мл 6н HCl, водную фазу нейтрализовали бикарбонатом натрия до рН8 и экстрагировали этилацетатом. Органический слой сушили безводным сульфатом магния и затем концентрировали с получением остатка, который очищали колоночной хроматографией с получением 57,6 г Соединения 02.
3. Приготовление Соединения 03.
57,6 г раствора Соединения 02 в 100 мл эфира добавляли по каплям к смеси 11,7 г литийалюминийгидрида и 200 мл диэтилового эфира в течение 1 часа при температуре 0°C. Затем смесь нагревали до 30°C, и инкубировали в течение 0,5 часа. К реакционной смеси добавляли 18 г воды по каплям в течение 1 часа при температуре 0°C до прекращения реакции, затем смесь перемешивали в течение 1 часа. Удаляли фильтрацией преципитат и промывали этилацетатом. Весь этилацетат объединяли и концентрировали в вакууме с получением остатка, который очищали петролейным эфиром, и получали 36 г кристаллического Соединения 03.
4. Приготовление Соединения 05
Раствор 9,6 г диметилсульфоксида в 50 мл хлористого метилена добавляли по каплям к раствору 7,6 г оксалилхлорида в 150 мл дихлорметана в течение 0,5 часа при -65°C, затем смесь перемешивали в течение 0,5 часа. К этой смеси добавляли по каплям в течение часа раствор 11 г Соединения 03 в 100 мл хлористого метилена, и перемешивали раствор в течение 1 часа. 14 г триэтиламина по каплям добавляли к полученному раствору в течение 1 часа, затем перемешивали в течение еще 2 часов. Реакционную смесь нагревали до комнатной температуры, после чего добавляли к ней 50 мл воды. Органический слой отделяли и водную фазу экстрагировали 50 мл дихлорметана. Объединенные органические слои промывали 100 мл насыщенного раствора бикарбоната натрия и концентрированным раствором хлорида натрия, затем сушили безводным сульфатом магния. Растворитель удаляли и получали 11,7 г Соединения 04. 2,5 мл раствора тетрабутиламмония фторида в ТГФ (1 моль/л) добавляли в раствор Соединения 04 в 150 мл ТГФ при 0°C, затем раствор триметил(трифторметил)силана в ТГФ (9 г/50 мл) добавляли по каплям к смеси и перемешивали в течение 15 минут.35 мл концентрированной соляной кислоты добавляли в реакционную смесь и перемешивали в течение 30 минут. Затем добавляли воду и этилацетат к реакционной смеси и нейтрализовали раствором бикарбоната натрия до pH 8. Органический слой отделяли и сушили безводным сульфатом магния. После концентрирования полученный остаток очищали с помощью флэш-хроматографии на силикагеле с получением 9,0 г Соединения 05.
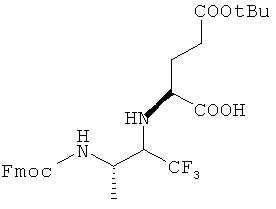
5. Приготовление Соединения 06А
17,6 г Соединения 06АО (коммерчески доступного) растворяли в 200 мл ДМФА и к раствору добавляли 24 г бензилбромида и 36 г бикарбоната натрия. Полученную смесь перемешивали в течение ночи. Затем добавляли в смесь 1200 мл воды и 400 мл этилацетата. Органический слой отделяли, сушили безводным сульфатом магния и затем концентрировали с получением остатка, который очищали с помощью колоночной хроматографии с получением Соединения 06А1. Смесь из 75 мл пиперидина и 350 мл этилацетата добавляли к Соединению 06А1 при перемешивании при 25°C в течение 2 часов. Затем смесь концентрировали с получением остатка, который очищали колоночной хроматографией с получением 10 г Соединения 06А.
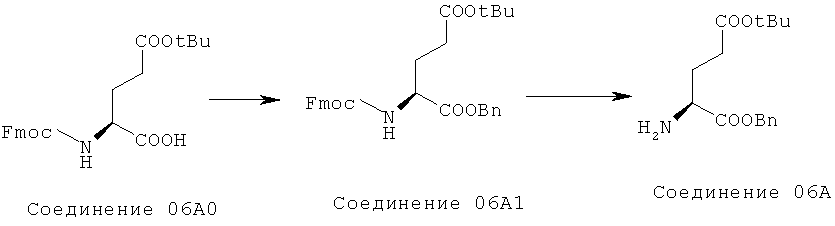
6. Приготовление Соединения 07
Раствор гидроксида натрия (5 г/30 мл) при 0°C добавляли к 30 мл раствора ТГФ, содержащего 2 г Соединения 05 и 0,2 г ТБАБ. Затем в раствор добавляли 2,5 г пара-толуолсульфонилхлорида и смесь перемешивали в течение 30 минут. Затем добавляли к смеси воду и этилацетат. Органический слой отделяли, сушили и концентрировали с получением 2,7 г Соединения 06. 20 мл раствора ацетонитрила, содержащего 2,7 г Соединения 06 и 3,3 г Соединения 06А кипятили в течение ночи. Затем ацетонитрил удаляли выпариванием, полученный остаток промывали петролейным эфиром и отфильтровывали. Фильтрат концентрировали, остаток очищали колоночной хроматографией с получением 1,8 г Соединения 07.
7. Приготовление Соединения 8
0,3 г катализатора Pd(OH)2/C добавляли к 400 мл раствора 1,8 г Соединения 07 в метаноле, и смесь гидрировали в течение ночи. Катализатор отфильтровывали, метанол концентрировали с получением 0,9 г Соединения 08.
8. Приготовление Димера.
Раствор Соединения 08 (0,9 г) и FmocCl (0,9 г) в диоксане (40 мл) перемешивали в течение ночи. Затем реакционную смесь отфильтровывали, осадок собирали и промывали диоксаном и петролейным эфиром с получением 0,59 г Димера.
1Н ЯМР (ДМСО-d6): 1,04 (d, 3Н), 1,38 (s, 9Н), 1,50-1,91 (m, 2Н), 2,21-2,30 (m, 2Н), 3,06-3,08 (m, 1Н), 3,30-3,37 (m, 1H), 4,26-4,38 (m, 4Н), 7,32-7,46 (m, 4Н), 7,72-7,76 (m, 2Н), 7,90 (d, 2Н), 8,04 (d, 1H).
13С ЯМР (ДМСО-d6): 15,20, 20,46, 27,16, 30,55, 46,08, 48,88, 54,75, 55,12, 55,90, 65,66, 78,98,119,54, 124,68, 124,79, 126,50, 126,75, 127,11, 140,21, 143,07, 143,24, 156,02, 171,37, 174,69.
19F ЯМР (ДМCO-d6): 70,023.
ЖХ/MC m/z: 551 (М+Н).
Путь синтеза Димера показан ниже:
II. Синтез главной цепи
1. Набухание.
Смолу Fmoc-Gly-Wang (0,27 ммоль/г, 0,2 моль) выдерживали в дихлорметане 20 минут, затем смолу собирали фильтрованием.
2. Снятие защиты Fmoc.
Описанную выше смолу добавляли к 10 мл смеси пиперидина с диметилформамидом (пиперидин/диметилформамид=1/4) и выдерживали в растворе в течение 8-10 минут, затем смесь фильтровали, чтобы собрать смолу.
Для выявления свободных аминогрупп был проведен нингидриновый тест Кайзера (КТ тест): 30-50 гранул смолы промывали дихлорметаном 3 раза. Затем добавляли около двух капель каждого из трех реагентов (А, В и С), смесь выдерживали при температуре 115°C в течение 5 минут. Снятие защиты считали полным, когда и раствор, и смола синели.
3. Промывка.
Смолу промывали дихлорметаном и диметилформамидом, каждым по 6 раз, а затем фильтровали.
4. Сочетание.
Диизопропилкарбодиимид (ДИК) является предпочтительным для реакции сочетания. Ее проводили следующим образом: 5 мл раствора Fmoc-Arg (PbF)-OH (0,8 ммоль, 0,52 г), HOBt (0,8 ммоль, 0,11 г) и ДИК (0,8 ммоль, 125 мкл) в диметилформамиде перемешивали в течение 1,5 часов. Затем повторяли процедуру обнаружения свободных аминогрупп с помощью КТ теста: Брали 30-50 гранул смолы, промывали дихлорметаном 3 раза. После добавления примерно двух капель каждого из трех реагентов (А, В и С), смесь выдерживали при температуре 115°C в течение 5 минут. Реакцию считали завершившейся, когда и раствор, и смола становились светло-желтыми или бесцветными. Реакция была незавершенной, если раствор или смола имели светло-синий цвет, в этом случае и процедуру обнаружения свободных аминогрупп повторяли в течение дополнительных 3 часов. Связующий реагент типа урония (аминокислоты/HATU/НОAt/DIEA=1/0,95-0,98/1/2) использовали, когда реакция оставалась незавершенной после трех часов.
5. Кэппирование
Если в пептиде общей длиной до 20 аминокислот КТ тесты показывали присутствие непрореагировавших аминогрупп после повторного сочетаия, было необходимо кэппировать их, чтобы избежать образования укороченных последовательностей. Это осуществляли путем короткой обработки (15 минут при комнатной температуре) смолы раствором ангидрида уксусной кислоты и DIEA (1:3 в молярном соотношении), при соотношении смола: реагент 1:5 с последующей фильтрацией для сухости.
6. Промывка.
Смолу промывали дихлорметаном, затем диметилформамидом. Стадию промывки повторяли 5 раз с последующей фильтрацией до сухости.
7. Этапы 2-6 повторяли до завершения синтеза главной цепи.
8. Для боковой цепи на Lys(12) пептида было необходимо избирательное снятие защиты (т.е., защита группы на одной молекуле может быть удалена селективно, без влияния на другие защитные группы). Fmoc-Lys (DDE)-OH может быть выбран, поскольку необходимо использовать защитные группы для аминогруппы, которые не могут быть удалены под действием пиперидина. Был применен способ сочетания с использованием диизопропилкарбодиимида (ДИК метод), описанный выше в Примере I,4.
9. Пептид Формулы II получали путем отщепления С-конца основной цепи до Thr 25. Его молекулярная масса была подтверждена масс-спектрометрией (расчетная масса 2905,18, экспериментальная масса 2904,74), как показано на рисунке 1.
10. Соединение согласно настоящему изобретению содержит аминокислоту в положении 29 от С-конца в основной цепи (Димер и его структура изображены выше). Для защиты его аминогруппы использовали Fmoc, а его карбоксильной группы - OtBu. Соединение получили путем сочетания с использованием диизопропилкарбодиимида как связующего реагента. Более конкретно, Димер (0,55 г, 1,0 ммоль, 5 экв смолы), HOBt (0,14 г, 1 ммоль) и диизопропилкарбодиимид (ДИК) (160 мкл, 1 ммоль) в 5 мл диметилформамида добавляли к смоле, реакцию проводили при комнатной температуре в течение 3 часов до присоединения Димера к основной цепи.
11. Присоединение последней С-концевой аминокислоты (His) осуществляли путем присоединения Boc-His (Trt)-OH и использовали диизопропилкарбодиимид (ДИК) в качестве связующего агента или реагенты урониевого типа.
III. Синтез боковой цепи и присоединение к главной цепи.
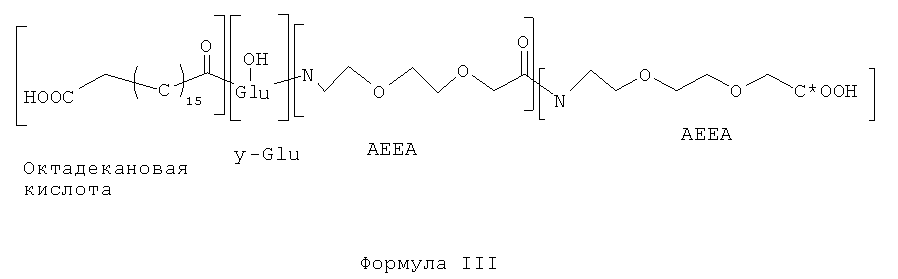
1. Боковая цепь, показанная в Формуле III, представляет собой соединение согласно настоящему изобретению. Она имеет структуру октадекановая кислота-γ-Glu-AEEA-AEEA, где С*ООН связана с основной пептидной цепью, показанной в Формуле II. Защитную группу Dde (например, 1-(4, 4-диметил-2,6-диоксициклогексилиден) этил) на Lys(12) основной пептидной цепи соединения формулы II удалили 2% гидразином. Боковую цепь присоединяли к Lys так же, как описано выше в разделе II, 2-6. Синтез боковой цепи, показанной в Формуле III, завершили путем присоединения Fmoc-AEEA-OH и Fmoc-Glu-OtBu и реагентов урониевого типа, используемых для связи γ-карбоксильной группы Glu с аминогруппой АЕЕА, с последующим присоединением 1,18-октадекановой кислоты с одной карбоксильной группой, защищенной tBu.
IV. Отщепление и очистка
1. Отщепление
Смолу с пептидом промывали и сушили путем фильтрации. Отщепление пептида осуществляли в соотношении 1 г пептид-смола на 10 мл лизата. Более конкретно, смолу с пептидом добавляли к раствору лизата (трифторуксусная кислота:тиоанизол:этандитиол:вода:фенол=82,5:5:2,5:5:5) и перемешивали в течение 3 часов. Смесь разбавляли 500 мл охлажденного льдом эфира и осадок собирали центрифугированием. Затем осадок промывали 4 раза и сушили на воздухе с получением неочищенного соединения Формулы I.
2. Очистка
Неочищенное соединение Формулы I растворяли в 90% растворе метанола в воде. Раствор обрабатывали ультразвуком, а затем фильтровали через ультрафильтрационную мембрану с размером пор 0,45 мкм. Фильтрат очищали с помощью препаративной хроматографии с получением чистого соединения Формулы I. Масс-спектрометрический анализ показал правильную молекулярную массу (расчетная масса 4152,1, экспериментальная масса 4152,5), как показано на рисунке 2.
Пример 2
Синтез основной пептидной цепи соединения формулы II осуществляли с использованием смолы Ринка вместо смолы Fmoc-Gly-Wang. Остальные условия были такими же, как описано в примере 1.
Если не указано обратное, температура, упомянутая в примере 1 и примере 2, была комнатной. Реагентами А, В и С были, соответственно, 80% раствор фенола в этаноле, дважды перегнанный пиридин и раствор нингидрина в этаноле (5 г на 100 мл).
Пример 3 - Оральная проба на толерантность к глюкозе (ОГТТ)
1. Группы
90 мышей ICR (Institute of Cancer Research), все самцы, разделили на три партии по массе тела, по 30 мышей в каждой партии. После голодания в течение ночи, каждая партия была разделена на две группы в зависимости от уровня сахара в крови: Контрольная группа и группа, получающая соединение из Примера 1 ("группа Соединения"). Контрольной группе вводили только физиологический раствор, а группе Соединения вводили Соединение из Примера 1, растворенное в физиологическом растворе.
2. Эксперименты
Партия 1: Мыши были разделены на две группы в зависимости от уровня сахара в крови после голодания в течение ночи. В 1-й день им вводили физиологический раствор или раствор Соединения в физиологическом растворе (0,3 мг/кг) подкожно. Через 2 часа после введения мышей подвергали принудительному кормлению с дозой глюкозы 2 г/кг, у мышей отбирали образцы крови из кончика хвоста через 30 минут и через 60 минут после введения глюкозы. Измеряли уровень глюкозы в крови. На 4-й день (72 часа после пробного введения Соединения) после голодания в течение ночи, мышей обеих групп подвергали принудительному кормлению с дозой глюкозы 2 г/кг. Образцы крови отбирали через 30 минут после введения глюкозы, измеряли уровень глюкозы в крови.
Партия 2: Мышей разделили на группы в зависимости от уровня сахара в крови после голодания в течение ночи. На 1-й день мышам вводили исследуемое Соединение подкожно в дозе 0,3 мг/кг. На 2-й день после голодания в течение 8 часов, мышей подвергали принудительному кормлению с дозой глюкозы 2 г/кг, через 30 минут после введения глюкозы отбирали образцы крови и измеряли уровень глюкозы. Следующее принудительное кормление проводили на 5-й день (90 часов после пробного введения соединения) после ночного голодания, и измеряли уровень глюкозы в крови через 30 минут после введения глюкозы.
Партия 3: Мышей разделили на группы в зависимости от уровня сахара в крови после голодания в течение ночи. В 1-й день мышам вводили исследуемое Соединение подкожно в дозе 0,3 мг/кг. На 3-й день (42 часа после введения исследуемого Соединения) после голодания в течение ночи, мышей подвергали принудительному кормлению с дозой глюкозы 2 г/кг. Через 30 минут после введения глюкозы отбирали образцы крови и измеряли уровень глюкозы в крови.
Измерение уровня глюкозы в крови проводили с использованием интегрированной системы обнаружения глюкозы в крови Roche's ACCU-CHEK ®.
3. Результаты
(1) 2 часа после введения соединения
|
(2) 25 часов после введения соединения
|
(3) 42 часа после введения соединения
|
После еды: постпрандиальный уровень глюкозы в крови, измеренный перед голоданием.
(4)72 часа после введения соединения
|
(5) 90 часов после введения соединения
|
Приведенные выше данные показывают, что исследуемое соединение обладает способностью значительно снижать уровень глюкозы в крови и способно оказывать этот эффект в течение 90 часов после введения.
Кроме того, соединения согласно настоящему изобретению имеют период полужизни в плазме крови более 30 часов, а период полувыведения ГПП-1 составляет 1~2 минуты.
Разумеется, описанные выше варианты реализации являются лишь иллюстративными примерами настоящего изобретения и не ограничивают объем настоящего изобретения, который определяется прилагаемой формулой изобретения. Различные модификации или изменения в изобретении, которые может сделать специалист в данной области, не изменяющие сути этого изобретения, также включены в объем данного изобретения.
Способы получения икотиниба и гидрохлорида икотиниба, а также их промежуточных соединений
Новые конденсированные пиридиновые производные, применимые в качестве ингибиторов тирозинкиназы с-met
Способы получения икотиниба и гидрохлорида икотиниба, а также их промежуточных соединений
Новые конденсированные пиридиновые производные, применимые в качестве ингибиторов тирозинкиназы с-met
Модуляторы протеин-тирозинкиназы и способы их применения
Икотиниб-содержащие местнодействующие накожные фармацевтические композиции и их применения
Полиморфные формы икотиниба малеата и их применения
Полиморфные формы икотиниба и их применения
Новые полиморфные формы икотиниба фосфата и их применения
Кристаллическая форма малеата конденсированного пиридинового производного и способы ее применения

