Результат интеллектуальной деятельности: ФТОРСОДЕРЖАЩИЕ ПРОИЗВОДНЫЕ ТЕВИНОЛА И ОРВИНОЛА И СПОСОБЫ ИХ ПОЛУЧЕНИЯ (ВАРИАНТЫ)
Вид РИД
Изобретение
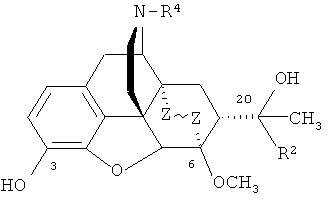
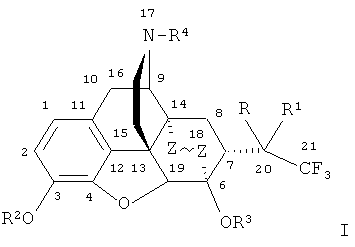
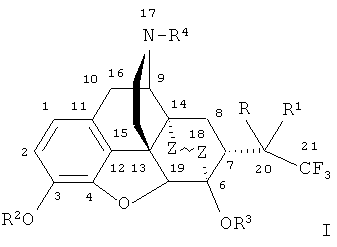
Изобретение относится к новым соединениям, содержащим циклическую систему 4аН-8,9с-иминоэтанфенантро[4,5-b,c,d] фурана, конденсированную с карбоциклическим кольцом, более точно, с мостиковой группой в положениях 6 и 14, состоящей только из двух атомов углерода, а именно к фторсодержащим производным тевинона, тервинола и орвинола и их фармацевтически приемлемым солям, конкретно к производным, содержащим атомы фтора в положении С(21), общей формулы
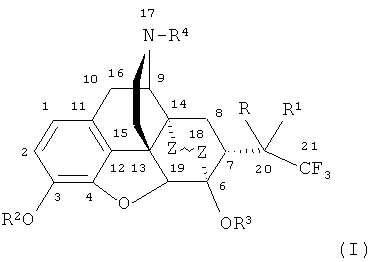 ,
,
где R=ОН; R1=Н, CF3, C1-C4-алкил или арил; R2=Н, СН3; R3=H, СН3; R4=СН3, циклопропилметил, аллил; Z~Z=CH2CH2, CH=CH,
и к способам их получения.
Заявляемые соединения, их свойства и способы получения в литературе не описаны. Ранее не были известны фторсодержащие соединения, описываемые общей формулой I.
Заявляемые соединения представляют собой фторсодержащие производные этеноизоморфинана и наиболее эффективно могут быть использованы (в виде фармацевтически приемлемых солей) в качестве лекарственных средств, механизм действия которых основан на агонистическом и/или антагонистическом взаимодействии с опиоидными рецепторами. Опиоидная система человека и животных модулирует ряд физиологических и поведенческих процессов, таких как восприятие боли, ответ на стрессовые состояния, иммунный ответ, нейроэндокринную функцию [Aldrich, J.V.; Vigil-Cruz, S.С. Narcotic Analgesics. In Burger's Medicinal Chemistry and Drug Discovery; Abraham, D.J, Ed.; John Wiley & Sons: New York, 2003; Vol.6, Chapter 7, pp 329-481]. По этой причине соединения, проявляющие способность связываться с опиоидными рецепторами (лиганды опиоидных рецепторов), используются в качестве действующего начала лекарственных средств, применяемых, в частности, для лечения боли, наркомании, алкоголизма, некоторых видов шока, диареи.
Наиболее близкими по структуре к заявляемым соединениям являются их известные аналоги, не содержащие фтора, формулы
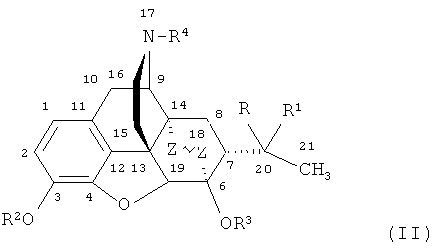 ,
,
где R=ОН; R1=Н, алкильная, алкенильная, алкинильная, циклоалкильная, арильная или другая органическая группа; R2=Н, CH3; R3=Н, СН3; R4=Н, алкильная, аллильная, пропаргильная, циклоалкилметильная, либо другая органическая группа.
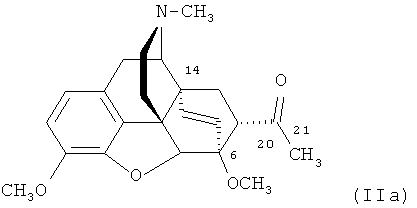
Известен «тевинон» (17-метил-3,6-диметокси-7α-ацетил-4,5α-эпокси-6α,14α-этеноизоморфинан) формулы
 ,
,
который широко используется как предшественник разнообразных не содержащих фтора производных этеноизоморфинанов формулы (II). Тевинон IIa представляет собой ранее описанный аддукт тебаина и винилметилкетона [В.Е. Allen, Е.Т. Jarvi, D.J. Kalota, J.R. Meyer, K.G. Tomazi, A. Mannmo, B. Orr. //WO 2010/039220 A1 (2010)], [В. Zhong, Z. Gong, Y. Wang, Y. Liu. // Заявка на патент США US 2005/70564 A1 (2005)]. Тривиальное название «тевинон» является основой краткой номенклатуры данного класса соединений [K.W. Bentley, D.G. Hardy, В. Meek. Novel Analgesics and Molecular Rearrangements in the Morphine-Thebaine Group. II. Alcohols Derived from 6,14-endo-Etheno- and 6,14-endo-Ethanotetrahydrothebaine. // J. Amer. Chem. Soc., 89 (13), 3273-3280 (1967)], согласно которой его аналог, содержащий три атома фтора в положении 21 формулы (Ia), следует называть 21,21,21-трифтортевинон.
Известно, что тевинон (IIa) является ключевым исходным соединением для получения веществ формулы (II), относящихся к классу тевинолов формулы
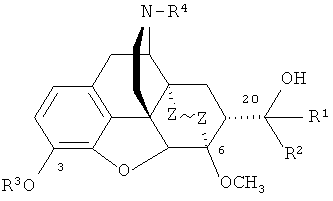 ,
,
где R1=СН3; R2=Н, алкильная, алкенильная, алкинильная, циклоалкильная, арильная или другая органическая группа; R3=СН3; R4=СН3; Z~Z=CH=CH, СН2СН2, и орвинолов (получаемых из тевинолов) формулы
 ,
,
где R2=Н, алкильная, алкенильная, алкинильная, циклоалкильная, арильная или другая органическая группа; R4=Н, алкильная, аллильная, пропаргильная, циклоалкилметильная, либо другая органическая группа; Z~Z=СН=СН, СН2СН2 [A.F. Casy, R.Т. Parfitt. Opioid Analgesics: Chemistry and Receptors. Plenum Press: New York, London, 1986], [K.W. Bentley. Brit. Pat. 1136214 (1968). Chem. Abstr., 70: 78218s (1969)], [Н.S. Park, Н.Y. Lee, Y.Н. Kim, J.K. Park, E.E. Zvartau, Н. Lee. A highly selective κ-opioid receptor agonist with low addictive potential and dependence liability. // Bioorg. Med. Chem. Lett., 16, (13), 3609-3613 (2006)].
Известно, что тевинолы являются предшественниками орвинолов, которые представляют собой деметилированные производные тевинолов и содержат гидроксильную группу в положении 3 гетероциклической системы.
Орвинолы представляют собой один из важнейших типов высокоэффективных лигандов опиоидных рецепторов [А.F. Casy, R.T. Parfitt. Opioid Analgesics: Chemistry and Receptors. Plenum Press: New York, London, 1986], [G.R. Lenz. S.M. Evans, D.E. Walters, A.J. Hopfinger, in: Opiates, Academic Press, Orlando - London. 1986] и являются субстанциями лекарственных средств, широко применяемых в медицине (бупренорфин, дигидроэторфин) и ветеринарии (эторфин, дипренорфин) в качестве обезболивающих средств для лечения острых и хронических болей средней и высокой интенсивности и для лечения наркомании, а также для обездвиживания крупных животных, выведения их из этого состояния и лечения острых передозировок наркотических анальгетиков.
Известный тевинон На является общим ключевым предшественником соединений формулы II (где R=ОН). Высокая степень взаимного структурного сходства соединений формул I и II (где R=ОН), наличие в них одной и той же циклической системы 4аН-8,9с-иминоэтанфенантро[4,5-b,c,d] фурана с мостиковой группой в положениях 6 и 14, состоящей только из двух атомов углерода, делает способность связываться с опиоидными рецепторами, известную для соединений II, общим свойством данного класса соединений,
Размеры атома фтора не очень сильно отличаются от размеров атома водорода. В частности, ван-дер-ваальсовы радиусы групп СН3 и CF3 равны 1.29 А и 1.35 А, соответственно, что позволяет в значительной степени сохранять геометрические параметры молекулы при частичной замене в ней атомов водорода на фтор. При этом известно, что введение в молекулы фармакологически активных соединений атомов фтора влияет на их фармакологическую активность, в частности, за счет повышения липофильности, улучшения их транспорта in vivo и большей устойчивости связей C-F к метаболизму [S.V. Druzhinin, E.S. Balenkova, V.G. Nenajdenko. Recent advances in the chemistry of α,β-unsaturated trifluoromethylketones. // Tetrahedron, 63 (33), 7753-7808 (2007)].
Опиоидные рецепторы широко распространены как в центральной, так и в периферической нервной системе.
Замена в соединениях общей формулы II (где R=ОН) некоторых атомов водорода на атомы фтора, привела бы к опиоидным лигандам общей формулы I (где R=ОН) с пониженной гидрофильностью и, как следствие, с повышенной способностью к преодолению гемато-энцефалического барьера и облегчению доступа к опиоидным рецепторам центральной нервной системы.
Стимулирование опиоидных рецепторов центральной нервной системы приводит к аналгезии, которая, однако, часто сопровождается рядом негативных побочных эффектов (угнетение дыхания, седация, толерантность, физическая зависимость, влияние на желудочно-кишечный тракт). Между тем, клинически значимая аналгезия, а также ряд других полезных терапевтических эффектов могут быть достигнуты и путем активации опиоидных рецепторов периферической нервной системы [E.K. Ryu, Zh. Wu, K. Chen, L.H. Lazarus, E.D. Marczak, Y. Sasaki, A. Ambo, S. Salvadori, C. Ren, H. Zhao, G. Balboni, X. Chen. Synthesis of a Potent and Selective 18F-Labeled δ-Opioid Receptor Antagonist Derived from the Dmt-Tic Pharmacophore for Positron Emission Tomography Imaging. // J. Med. Chem., 51 (6), 1817-1823 (2008)], [С. Stein, M. Schäfer, H. Machelska, Attaching pain at its source: new perspectives on opioids. Nat. Med., 9, (8), 1003-1008 (2003)], [S. Furst, P. Riba, T. Friedmann, J. Timar, M. Al-Khrasani, I. Obara, W. Makuch, M. Spetea, J. Schutz, R. Przewlocki, B. Przewlocka, H. Schmidhammer. Peripheral versus central antinociceptive actions of 6-amino acid-substituted derivatives of 14-O-methyloxymorphone in acute and inflammatory pain in the rat. J. Pharmacol. Exp. Ther., 312, (2), 609-618 (2005)]. Поэтому для использования в медицинских целях необходимы не только опиоидные лиганды рецепторов центральной нервной системы, обладающие пониженным уровнем нежелательных побочных эффектов, но и опиоидные лиганды, ориентированные на селективное взаимодействие с рецепторами периферической нервной системы, для того, чтобы минимизировать активацию центральных опиоидных рецепторов, связанные с ней нежелательные побочные эффекты и тем самым повысить безопасность опиоидных лекарственных средств. [I. Obara, W. Makuch, M. Spetea, J. Schutz, H. Schmidhammer, R. Przewlocki, B. Przewlocka. Local peripheral antinociceptive effects of 14-O-methyloxymorphone derivatives in inflammatory and neuropathic pain in the rat. // Eur. J. Pharm., 558 (1), 60-67 (2007)], [D.L. DeHaven-Hudkins, R.E. Dolle, Peripherally restricted opioid agonists as novel analgesic agents, Curr. Pharm. Des., 10, (7), 743-757 (2004)], [С. Stein, M. Schafer, H. Machelska, Attaching pain at its source: new perspectives on opioids. Nat. Med., 9, (8), 1003-1008 (2003)].
Одним из способов уменьшения доступа опиоидных лигандов в центральную нервную систему является их химическая модификация с целью повышения гидрофильности, препятствующая преодолению гемато-энцефалического барьера [J. Schutz, W. Brandt, M. Spetea, K. Wurst, G. Wunder, H. Schmidhammer. Synthesis of 6-amino acid substituted derivatives of the highly potent analgesic 14-O-methyloxymorphone. Helv. Chim. Acta, 86, (6), 2142-2148 (2003)], [M. Spetea, T. Friedmann, P. Riba, J. Schutz, G. Wunder, T. Langer, H. Schmidhammer, S. Furst. In vitro opioid activity profiles of 6-amino acid substituted derivatives of 14-O-methyloxymorphone. Eur. J. Pharmacol., 483, (2-3), 301-308 (2004)]. [M. Al-Khrasani, M. Spetea, T. Friedmann, P. Riba, K. Kiraly, H. Schmidhammer, S. Furst. DAMGO and 6β-glycine substituted 14-O-methyloxymorphone but not morphine show peripheral, preemptive antinociception after systemic administration in a mouse visceral pain model and high intrinsic efficacy in the isolated rat vas deferens. // Brain Res. Bull., 74, 369-375 (2007)].
Переход от соединений общей формулы I (где R=ОН; R3=СН3) к 6-O-деметилированным производным общей формулы I (где R=ОН; R3=H) повышает гидрофильность молекул, при сохранении в значительной степени их геометрических параметров, что должно способствовать взаимодействию фторсодержащих лигандов формулы I (где R=ОН; R3=H) с опиоидными рецепторами периферической нервной системы.
Известен способ получения не содержащих фтора аналогов заявляемых соединений общей формулы II (где R=ОН; R1=Н, замещенная или незамещенная алкильная, алкенильная или алкинильная группа, содержащая до 8 атомов углерода, либо циклоалкильная группа с числом атомов углерода от 4 до 8; R2=Н, СН3, CnH2n+1CO (n=1-3), С6Н5СО, никотиноил; R3=СН3; R4=Н, алкильная, алкенильная или алкинильная группа, содержащая до 8 атомов углерода, либо циклоалкилзамещенная метильная группа, общее число атомов углерода в которой составляет от 4 до 8; Z~Z=СН2СН2) [K.W. Bentley. Патент Великобритании GB 1136214, C07D 99/04 (1968). Chem. Abstr., 70: 78218s (1969)], из тевинона IIa, который подвергают действию реактива Гриньяра R1gX с последующим гидролизом образующегося магниевого алкоголята.
Однако получить аналогичным способом фторсодержащие тевинолы I (где R=ОН; R1=Н, алкильная, арильная или другая органическая группа; R2=Me; R3=Me; R4=Me; Z~Z=CH=CH, СН2СН2) не представляется возможным из-за чрезвычайной нестабильности металлоорганических соединений CF3MgX (где Х=галоген) [R.N. Haszeldine. Neuere Chemie des Fluors. Organometall- und Organometalloid-Verbindungen des Fluors. Angew. Chem., 66, (22), 693-701 (1954)], [R.N. Hasseldine. Perfluoroalkyl Grignard and Grignard-type Reagents. Part IV. Trifluoromethylmagnesium Iodide. J. Chem. Soc., 1273-1279 (1954)], [E.Т. McBee, R.D. Battershell, Н.P. Braendlin. A New Synthesis of Perfluoroalkylmagnesium Halides. J. Org. Chem., 28, (4), 1131-1133 (1963)], [K.J. Klabunde, Н.F. Efner, L. Satek and W. Donley. Preparation of an extremely active magnesium slurry for Grignard reagent preparations by metal atom-solvent condensation. J. Organometal. Chem., 71, 309-313 (1974)] и CF3Li [R.N. Haszeldine. Neuere Chemie des Fluors. Organometall- und Organometalloid-Verbindungen des Fluors. Angew. Chem., 66, (22), 693-701 (1954)], [O.R. Pierce, E.T. McBee, G.F. Judd. Preparation and reactions of perfluoroalkyllithiums. J. Amer. Chem. Soc., 76, (2), 474-478 (1954)].
Задачей настоящего изобретения является получение ранее неизвестных фторсодержащих производных тевинола и орвинола, которые составили бы новый тип лигандов опиоидных рецепторов с широкими возможностями варьирования их гидрофильно-гидрофобного баланса а также разработка способов получения таких соединений, исходя из небольшого числа предшественников.
Поставленная задача решается новыми соединениями - фторсодержащими производными тевинола и орвинола формулы I, в которых R представляет собой группу ОН; R1 выбран из группы Н, CF3, C1-C4 алкил, арил, или (R+R1) обозначает O=; R2 и R3 представляют собой атомы водорода или метальные группы, Z~Z выбран из группы: СН=СН или СН2СН2, а R4 выбран из группы: СН3, циклопропилметил, аллил, а также их фармацевтически приемлемыми солями, и способами получения соединений формулы (I).
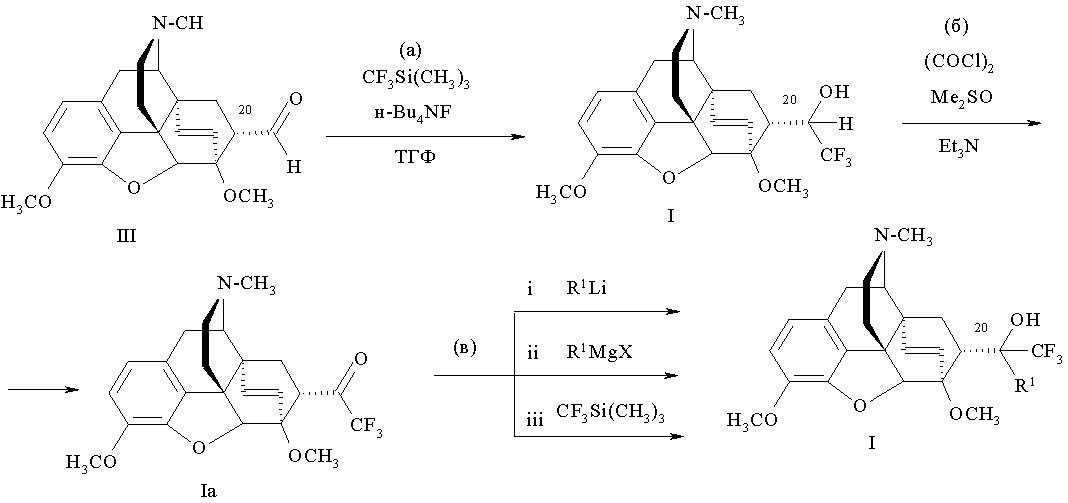
Способ получения соединений формулы (I), где R=ОН; R1=Н, CF3, C1-C4 алкил, арил, или (R+R1)=О; R2=СН3; R3=СН3; R4=СН3, Z~Z=СН=СН, представленный на схеме 1 включает следующие стадии:
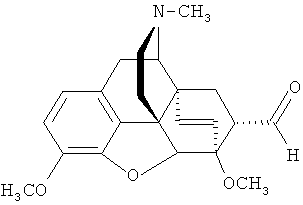
(а) трифторметилирование альдегида формулы (III)
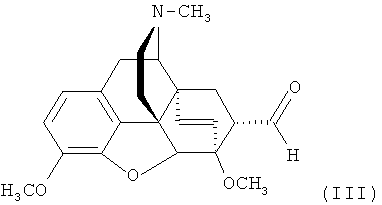
с образованием 21,21,21-трифтортевинола (7α-(1-гидрокси-2,2,2-трифторэтил)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этеноизоморфинана) Ib;
(б) окисление образовавшегося трифтортевинола Iб продуктом взаимодействия оксалилхлорида и ДМСО, с последующей обработкой основанием и образованием соответствующего кетона Ia;
(в) взаимодействие полученного кетона или (i) с соответствующим литийорганическим соединением формулы R1Li, или (ii) с реактивом Гриньяра формулы R1MgX, или с (iii) CF3Si(СН3)3, и последующее выделение фторсодержащих продуктов известными приемами, представлен на следующей ниже схеме 1.
Схема 1
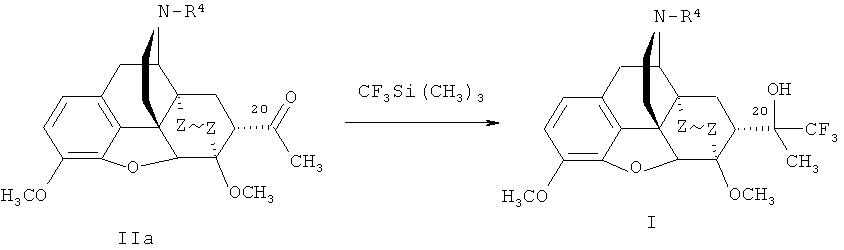
Второй способ получения соединений формулы I, где R=ОН; R1=Н, CF3, C1-C4 алкил, арил, или (R+R1)=О; R2=R3=СН3; R4=СН3, циклопропилметил, аллил; Z~Z=CH2CH2, СН=СН, представленный на схеме 2, заключается во взаимодействии трифторметилирующего агента такого, как CF3Si(СН3)3, с производными тевинона формулы (II), в которых R+R1=О, R2 R2=СН3; R3=СН3; R4=СН3, циклопропилметил, аллил; Z-Z=СН2СН2, СН=СН, последующей обработке реакционной смеси кислотой и выделении соответствующих тевинолов:
Схема 2
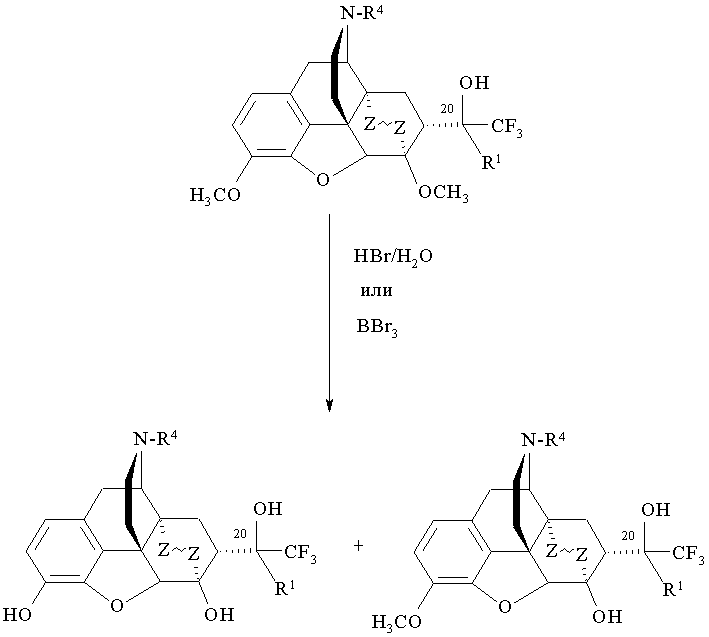
Еще один вариант способа получения соединений формулы I представлен на схеме 3, способ предназначен для получения фторсодержащих производных орвинола формулы I, где R=ОН; R1=СН3, CF3, C1-С4 алкил, арил, R2=Н; R3=Н, СН3; R4=СН3, циклопропилметил, аллил; Z~Z=СН2СН2, СН=СН, и состоит в деметилировании тевинолов, полученных двумя указанными выше способами (схемы 1 и 2), где R=ОН; R1=Н, CF3, С1-C4 алкил, арил, R2=СН3; R3=СН3; R4=СН3, циклопропилметил, аллил; Z~Z=СН2СН2, СН=СН, и выделении целевых продуктов известными приемами.
Деметилирование указанных тевинолов формулы I осуществляют действием BBr3 или HBr (Схема 3).
Схема 3
Первой стадией (а) способа по схеме 1 является взаимодействие альдегида формулы (III) с реактивом Руперта-Пракаша [CF3Si(CH3)3] в присутствии источника F-, такого, как тетрабутиламмонийфторид, в растворителе, таком, как тетрагидрофуран, затем реакционную смесь обрабатывают кислотой и выделяют вторичный фторсодержащий спирт 1b (21,21,21-трифтортевинол) (пример 1).
В качестве исходных веществ для получения вторичного фторсодержащего спирта 1b используют коммерчески доступный Me3SiCF3 [I.Ruppert, К.Schlich, W.Volbach. Tetrahedron Lett, 25, (21), 2195-2198 (1984)], [G.K.S.Prakash, R.Krishnamurti, G.A.Olah. J.Amer. Chem. Soc, 111, (7), 393-395 (1989)]). и альдегид (III), который является доступным соединением: его легко и с высоким выходом получают в одну стадию из природного сырья (тебаина) [J.J.Kopcho, J.C.Schaeffer. Selective O-Demethylation of 7-α-(Aminomethyl)-6,14-endo-ethenotetrahydrothebaine. // J: Org. Chem., 51(9), 1620-1622 (1986)].
На следующей стадии (б) указанного способа (схема 1) вторичный фторсодержащий спирт 1b, полученный на стадии (а), окисляют продуктом взаимодействия оксалилхлорида и диметилсульфоксида при температуре от -70°C до -78°C, затем обрабатывают реакционную смесь основанием, таким, как триэтиламин. В результате получают соответствующий фторсодержащий кетон (17-метил-3,6-диметокси-7α-(трифторацетил)-4,5α-эпокси-6α,14α-этеноизоморфинан (21,21,21-трифтортевинон) формулы I, где R+R1=О, R2=R3=R4=СН3; Z~Z=СН=СН, обозначаемый далее Ia (примеры 2-1, 2-2).
Следует отметить, что не содержащий фтор аналогичный вторичный спирт IIb можно окислить действием перманганата калия и получить соответствующий кетон -тевинон, однако ожидаемая аналогичная реакция с фторсодержащим спиртом 1b при действии перманганата калия не происходит.
На стадии (в) способа, представленного на схеме 1, происходит взаимодействие полученного на стадии (б) фторсодержащего кетона Ia с реагентом, выбранным из группы, включающей R1Li (i, пример 10), R1MgX (ii, пример 11), CF3Si(CH3)3 (iii, пример 5), что позволяет варьировать заместители R1 в положении 20 гетероциклической системы; это делает возможным расширение круга получаемых таким способом тевинолов и далее продуктов их деметилирования.
Второй вариант способа получения новых соединений формулы I, представленный на схеме 2, позволяет получать тевинолы непосредственно из тевинона IIa (пример 3), который проще всего получать не через альдегид (III), а минуя его, в одну стадию из того же природного сырья - тебаина [К.W.Bentley. Патент Великобритании GB 902659А) (1962)], или из продукта гидрирования тевинона - дигидротевинона (пример 4), или из N-норпроизводных тевинона (пример 12) и дигидротевинона (примеры 13 и 14).
Получение фторпроизводных орвинола формулы I, из тевинолов, полученных по схемам 1 и 2, способом, представленным на схеме 3, путем деметилирования тевинолов под действием HBr (пример 7) или BBr3 (примеры 6, 8, 9 и 15) делает возможным осуществить деметилирование тевинолов в 6 положении и получить производные орвинолов, содержащие не только трифторметильную группу, но и дополнительную гидроксильную группу в положении 6, что может определить появление неожиданных свойств у таких лигандов.
Изобретение иллюстрируется конкретными примерами его осуществления, приведенными ниже.
Пример 1. Получение 21,21,21-трифтортевинола [7α-(1-гидрокси-2,2,2-трифторэтил)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этеноизоморфинана] (1b).
К раствору 0.45 г (1.22 ммоль) альдегида формулы (III) в 10 мл безводного ТГФ добавляют 0.36 мл (2.45 ммоль) (СН3)3SiCF3. Реакционную смесь охлаждают до 0-10°C и при перемешивании прибавляют раствор 0.01 г тригидрата (h-Bu)4NF в 2 мл ТГФ. Охлаждающую баню удаляют, реакционной смеси позволяют нагреться до комнатной температуры, затем смесь выдерживают еще 1 час, прибавляют 5 мл 20% водного раствора HCl и интенсивно перемешивают в течение 20 минут. К смеси добавляют 15 мл хлороформа и доводят pH водного слоя до 10, прибавляя 25% водный раствор аммиака. Органический слой отделяют, водный слой экстрагируют хлороформом (3×10 мл). Органический слой и экстракты объединяют, сушат над безводным Na2S04 и растворитель отгоняют в вакууме. Получают 0.36 г (67%) соединения формулы 1b в виде смеси (20R)-и (20S)-изомеров в соотношении 17:1, представляющей собой прозрачное масло желтоватого цвета. Индивидуальные (20R)- и (20S)-изомеры из полученной смеси выделяют хроматографически на колонке с силикагелем [элюент -25%-ный водный раствор аммиака: CH3OH:CHCl=1:15:1600 (по объему)]. Оба изомера представляют собой масла желтого цвета.
(20R)-изомер: Масс-спектр (электроспрей) (m/z): 438 [М+1]+. Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 1.51 (дд, 2J=13.2 Гц, 3J=6.7 Гц, 1H, Н-8α), 1.86 (дд, 1H, Н-15эк), 2.00-2.05 (м, 1Н, Н-15ак), 2.21 (м, 1Н, Н-7β), 2.39 (с, 3H, NCH3), 2.35-2.44 (м, 2Н, Н-10α+Н-16ак), 2.56 (дд, 1Н, Н-16эк), 2.85 (дд, 2J=13.2 Гц, 3J=9.6 Гц, 1Н, H-8β), 3.21-3.24 (м, 2Н, Н-9, H-10β), 3.59 (с, 3H, 6-OCH3), 3.81 (с, 3H, 3-ОСН3), 4.45 (кв, 1H, H-20), 4.57 (д, 1H, H-5), 5.50 (д, J18,19)=8.7 Гц, 1Н, H-19), 5.83 (уш. д, 1Н, Н-18), 6.62+6.53 (AB система, JAB=8.3 Гц, 2Н, Н-1+Н-2). Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): - 76.44 (д, J=8.0 Гц, 3F, CF3).
(20S)-изомер: Масс-спектр (электроспрей) (m/z): 438 [М+1]+.
Спектр 1H ЯМР (CDC13, δ, м.д., относительно (CH3)4Si): 1.00-1.07 (м, 1H, Н-8а), 1.83 (дд, 1H, Н-15эк), 1.95-2.06 (м, 1H, Н-15ак), 2.11-2.19 (м, 1H, Н-7β), 2.35 (с, 3H, NCH3), 2.35-2.43 (м, 2Н, Н-10α+Н-16ак), 2.51 (дд, 1H, Н-16эк), 2.89 (дд, 2J=13.5 Гц, 3J=9.1 Гц, 1H, Н-8β), 3.14 (д, 1Н, Н-9), 3.21 (д, 1H, 2J=18.5 Гц, H-10β), 3.76 (с, 3H, 6-ОСН3), 3.81 (с, 3H, 3-ОСН3), 3.74-3.83 (м, 1Н, Н-20), 4.57 (д, 1H, Н-5), 5.59 (д, J18,19=8.9 Гц, 1H, Н-19), 5.95 (уш. д, 1H, Н-18), 5.93 (уш. с, 1H, OH), 6.62+6.53 (АВ система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): - 76.71 (д, J=8.0 Гц, 3F, CF3).
Пример 2-1. Получение 21,21,21-трифтортевинона [3,6-диметокси-17-метил-7α-(трифторацетил)-4,5α-эпокси-6α,14α-этеноизоморфинана] (Ia).
К перемешиваемому раствору 0.42 мл (0.005 моль) оксалилхлорида в CH2Cl2 (6 мл) при температуре от -70°C до -78°C добавляют по каплям в течение 20 минут раствор 0.76 мл (0.01 моль) диметилсульфоксида в CH2Cl2 (1 мл). Смесь перемешивают 30 минут при той же температуре, добавляют к ней по каплям в течение 20 минут раствор 1.8 г (0.004 моль) смеси (20R)- и (20S)-изомеров спирта 1b, полученного в Примере 1, в CH2CI2 (3 мл), перемешивают 30 минут при той же температуре, добавляют 2.86 мл (0.02 моль) Et3N, и нагревают до температуры 20-25°C. К реакционной смеси добавляют воду (10 мл) и перемешивают в течение 10 минут.Органический слой отделяют, водный слой экстрагируют хлороформом (2×20 мл). Органический слой и экстракты объединяют и сушат над безводным Na2SO4. Растворитель отгоняют и остаток перекристаллизовывают из смеси бензол: гексан (1:3). Получают 1.48 г (85%) соединения (Ia). Температура плавления: 118-120°C.
Масс-спектр (электроспрей) (m/z): 436 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 1.38 (дд, 2J=12.7 Гц, 3J=6.7 Гц, 1H, Н-8а), 1.87-1.92 (м, 1Н, Н-15эк), 1.99-2.04 (м, 1Н, Н-15ак), 2.38 (с, 3Н, NCH3), 2.42-2.48 (м, 2Н, H-10ct+Н-16ак), 2.55 (дд, 1H, 2J=12.1 Гц, 3J=5.2 Гц, Н-16эк), 3.08 (дд, 2J=12.6 Гц, 3J -9.5 Гц, 1H, Н-8у9), 3.22 (д, 3J=6.4 Гц, 1H, Н-9), 3.27 (д, 2J=18.5 Гц, 1Н, H-10β), 3.38 (дд, J7β,8α=6.7 Гц, J7β8β=9.5 Гц, 1Н, Н-7β), 3.63 (с, 3Н, 6-ОСН3), 3.84 (с, 3Н, 3-ОСН3), 4.60 (д, 4J=1.2 Гц, 1H, Н-5), 5.62 (д, J18,19=8.9 Гц, 1H, Н-19), 6.01 (уш. д, 1Н, Н-18), 6.66+6.57 (АВ-система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19F-ЯМР (CDC13, 8, м.д., относительно CFCl3): -78.83 (с, 3F, CF3).
Спектр 13С ЯМР (CDCl3, S, м.д., относительно (CH3)4Si): 22.60 (С-10), 31.64 (С-8), 33.35 (С-15), 43.48 (NCH3), 44.79 (С-7), 45.41 (С-16), 47.48 (С-13), 54.45 (6-ОСН3), 56.72 (3-OCH3), 59.90 (С-9), 82.22 (С-6), 95.85 (С-5), 113.83 (С-2), 115.24 (кв, CF3), 119.63 (С-1), 125.22 (С-18), 128.11 (С-11), 133.72 (С-12), 136.34 (С-19), 141.92 (С-3), 147.87 (С-4), 192.47 (кв, СО).
ИК-спектр: νC=o 1760 см-1 (в таблетке с КВг).
Пример 2-2. Получение 21,21,21-трифтортевинона [3,6-диметокси-17-метил-7α-(трифторацетил)-4,5α-эпокси-6,14α-этеноизоморфинана] (Ia) из (20R)-изомера спирта (Ib)
По методике, описанной в примере 2-1, из 0.23 мл (0.003 моль) оксалилхлорида, 0.42 мл (0.006 моль) диметилсульфоксида, 1.00 г (0.002 моль) (20R)-изомера спирта (Ib) и 1.59 мл (0.012 моль) Et3N получают 0.82 г (82%) соединения (Ia), свойства которого идентичны свойствам продукта, получение которого описано в примере 2-1.
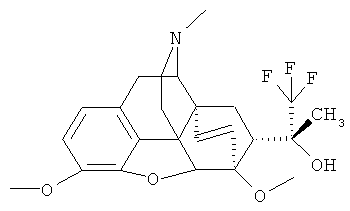
Пример 3. Получение (20R)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этено-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана и (20S)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этено-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R1=ОН; R1=CH3; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН=CH.
 .
.
К раствору 5.00 г (13.12 ммоль) тевинона На (R+R1=О; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН=СН) в 60 мл безводного ТГФ добавляют 4.00 мл (26.24 ммоль) (CH3)3SiCF3. Реакционную смесь охлаждают до 5-6°C и при перемешивании добавляют раствор 0.08 г тригидрата (h-Bu)4NF в 2 мл ТГФ. Охлаждающую баню убирают, реакционной смеси позволяют нагреться до комнатной температуры, после чего выдерживают смесь еще 1 час, добавляют к ней 40 мл 20%-ного раствора HCl и интенсивно перемешивают в течение 20 минут. К смеси добавляют 50 мл хлороформа и доводят pH водного слоя до 10 25%-ным раствором аммиака или 2N раствором NaOH. Органический слой отделяют, водный слой экстрагируют хлороформом (3×40 мл). Органический слой и экстракты объединяют, высушивают над безводным Na2S04, растворитель отгоняют в вакууме и остаток перекристаллизовывают из метанола. Получают 3.70 г (63%) (20R)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этено-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)-изоморфинана (I, R=ОН; R1=СН3; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН=СН).
Температура плавления: 162-163°C.
Масс-спектр (электроспрей) (m/z): 452 [М+1]+
Спектр 1H ЯМР (CDC13, 8, м.д., относительно (CH3)4Si): 1.24-1.32 (м, 1H, Н-8α), 1.33 (с, 3Н, СН3), 1.82-1.88 (м, 1H, Н-15эк), 1.88-1.99 (м, 1H, Н-15ак), 2.10 (м, 1H, H-7β), 2.36 (с, 3Н, NCH3), 2.34-2.42 (м, 2Н, Н-10а+Н-16ак), 2.51 (дд, 1H, Н-16эк), 2.86 (дд, 2J=12.5 Гц, 3J=9.8 Гц, 1Н, Н-8β), 3.14 (д, 3J=6.4 Гц, 1H, Н-9), 3.20 (д, 2J=18.7 Гц, 1Н, Н-10β), 3.77 (с, 3Н, 6-ОСН3), 3.81 (с, 3Н, 3-ОСНз), 4.47 (д, 1Н, Н-5), 5.49 (д, J18,19=9.0 Гц, 1H, Н-19), 5.94 (с, 1H, ОН), 6.04 (уш. д., 1H, Н-18), 6.62+6.52 (АВ-система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19F-ЯМР (CDCIl3, δ, м.д., относительно CFC13): -74.49 (с, 3F, CF3).
Растворитель из маточного раствора отгоняют в вакууме, остаток хроматографируют на колонке с силикагелем (элюент: хлороформ/метанол/аммиак=1600:15:1). После перекристаллизации из метанола выделенного хроматографически вещества получают 0.074 г (1.2%) (20S)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этено-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=СН3; R2=СН3; R3=СН3; R4
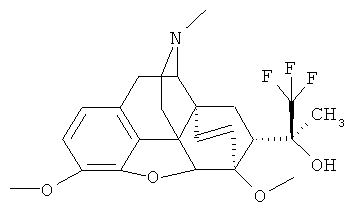
Температура плавления: 196-198°C.
Масс-спектр (электроспрей) (m/z): 452 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 1.03-1.09 (м, 1H, Н-8α), 1.18 (с, 3Н, СН3), 1.81-1.86 (м, 1H, Н-15эк), 1.96 (м, 1Н, Н-15ак), 2.27 (дд, 1H, Н-7 В), 2.35 (с, 3Н, NCH3), 2.33-2.41 (м, 2Н, Н-10а+Н-16ак), 2.50 (дд, 1H, Н-16эк), 2.90 (дд, 2J=13.2 Гц, 3J=9.0 Гц, 1Н, Н-8β, 3.13 (д, 3J=6.5 Гц, 1Н, Н-9), 3.21 (д, 2J=18.6 Гц, 1H, Н-10β), 3.80 (с, 3Н, 6-OCH3), 3.81 (с, 3Н, 3-OCH3), 4.54 (д, 4J=1.2 Гц, 1H, Н-5), 5.52 (д, J18;19=8.9 Гц, 1H, Н-19), 5.63 (с, 1Н, ОН), 5.95 (уш. д., 1Н, Н-18), 6.62+6.52 (АВ-система, JAB=8.2 Гц, 2Н, Н-1+Н-2).
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): -79.54 (с, 3F, CF3).
Пример 4. Получение (20R)-17-метил-3,6-диметокси-4,5а-эпокси-6α,14α-этано-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=СН3; R2=СН3; R3=CH3; R4=СН=CH3; Z~Z=СН2СН2
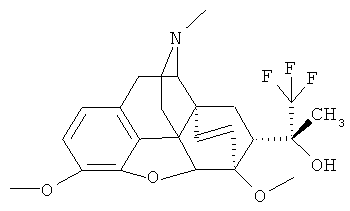
По методике, описанной в примере 1, из 4.00 г (10.44 ммоль) II (где R+R1=О; R=СН3; R3=СН3; R4=СН3; Z~Z=СН2СН2) и 3.00 мл (19.68 ммоль) (CH3)3SiCF3 получают 2.72 г (60%) соединения I (R=ОН; R1=СН3; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН2СН2) в результате перекристаллизации из метанола.
Температура плавления: 151-152°C
Масс-спектр (электроспрей) (m/z): 454 [М+1]+
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 0.67-0.78 (м, 1H, Н-19л), 1.03-1.13 (м, 1Н, Н-19п), 1.41 (с, 3Н, СН3), 1.52 (дд, 1H, H-8α), 1.61-2.01 (м, 4Н, 2Н-18+2Н-15), 2.07 (дд, 1H, Н-7β), 2.17-2.33 (м, 2Н, Н-16ак+Н-10α), 2.30 (с, 3Н, NCH3), 2.43 (дд, 1Н, 3J=5.4 Гц, 2J=11.7 Гц, Н-16эк), 2.65 (д, 1H, 3J=6.4 Гц, Н-9), 2.75-2.85 (м, 1Н, H-8β, 3.10 (д, 2J=18.2 Гц, 1H, Н-10β), 3.51 (с, 3Н, 6-ОСН3), 3.87 (с, 3Н, 3-ОСН3), 4.31 (д, 1H, 4J=1.9 Гц, Н-5), 5.88 (с, 1H, ОН), 6.71+6.57 (АВ система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): - 73.89 (с, 3F, CF3)
Пример 5. Получение 17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этено-7α-[1-гидрокси-1-(трифторметил)-2,2,2-трифторэтил]изоморфинана (I, где R=ОН; R1=CF3; R2=CH3; R3=CH3; R4=СН3; Z~Z=СН=СН).
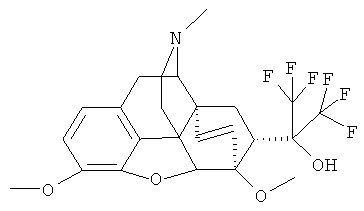
По методике, описанной в примере 1 из 1.50 г (3.44 ммоль) Ia (где R+R1=О; R=СН3; R3=СН3; R4=СН3; Z~Z=СН=СН) и 1.53 мл (10.30 ммоль) (CH3)3SiCF3 получают 1.00 г (57%) соединения I (где R=ОН; R1=CF3; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН=СН; Y=F) в результате перекристаллизации из этанола.
Температура плавления: 122-124°C
Масс-спектр (электроспрей) (m/z): 506 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 1.47-1.53 (м, 1H, Н-8α), 1.83-1.97 (м, 2Н, Н-15эк+Н-15ак), 2.35 (с, 3Н, NCH3), 2.34-2.45 (м, 3Н, Н-10α+H-16ак+Н-7β), 2.51 (дд, 1H, 2J=11.7 Гц, Н-16эк), 2.92 (дд, 2J=13.1 Гц, 3J=9.8 Гц, 1H, H-8β, 3.16 (д, 3J=6.5 Гц, 1Н, Н-9), 3.22 (д, 2J=18.6 Гц, 1H, Н-10β), 3.80 (с, 3Н, 6-ОСН3), 3.81 (с, 3Н, 3-OCH3), 4.49 (д, J=1.3 Гц, 1H, Н-5), 5.54 (д, J18,19=9.1 Гц, 1Н, Н-19), 6.03 (уш. д, 1H, Н-18), 6.53+6.63 (АВ система, JAB=8.1 Гц, 2Н, Н-1+Н-2), 6.82 (с, 1H, OH)
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): -70.34 (кв, J=9.9 Гц, 3F, CF3), -74.11 (кв, J=9.9 Гц, 3F, CF3)
Пример 6. Получение (20R)-3,6-дигидрокси-17-метил-4,5α-эпокси-6α,14α-этено-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=СН3; R=Н; R II: R4=СН3: Z~Z=СН=СН)
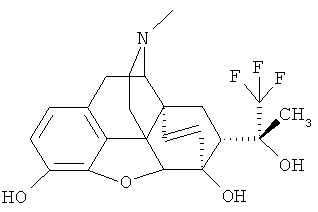
К раствору 1.00 г (2.22 ммоль) I (R=ОН; R1=СН3; R2=СН3; R3=СН3; R4=СН3; Z-Z=СН=СН) в дихлорметане (20 мл) при температуре от -70°C до -78°C в течение 10 минут добавляют 22.2 мл 1 М раствора BBr3 (22.2 ммоль) в дихлорметане. Реакционную смесь оставляют при постоянном перемешивании нагреваться до комнатной температуры, после чего добавляют метанол, доводят pH смеси до нейтральной реакции 25%-ным раствором аммиака и экстрагируют продукты хлороформом (3×50 мл). Органические экстракты объединяют, высушивают над безводным Na2SO4, растворитель отгоняют и остаток хроматографируют на колонке с силикагелем (элюент: NH4OH:СН3ОН: CHCl3=1:15:1600. Масс-спектр и ЯМР-спектры зарегистрированы для продукта I (R=ОН; R1=СН3, R2=Н, R3=Н, R4=СН3, Z~Z=СН=СН) в виде основания, полученного непосредственно в результате хроматографии в виде масла желтого цвета. Масс-спектр (электроспрей) (m/z): 424 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 1.31-1.35 (м, 1Н, Н-8о), 1.37 (с, 3Н, СН3), 1.88 (дд, 1H, Н-15эк), 1.92-2.00 (м, 1Н, Н-15ак), 2.13 (м, 1H, Н-70), 2.36 (с, 3Н, NCH3), 2.34-2.42 (м, 2Н, Н-10α+Н-16ак), 2.53 (дд, 1Н, 2J=11.7 Гц, 3J=4.8 Гц, Н-16эк), 2.86 (дд, 2J=12.8 Гц, 3J=9.8 Гц, 1H, Н-8β), 3.16 (д, 3J=7.4 Гц, 1H, Н-9), 3.20 (д, 2J=19.3 Гц, 1H, Н-10β), 4.33 (с, 1H, Н-5), 4.59 (с, 1H, OH), 5.38 (д, J18,19=8.7 Гц, 1H, Н-19), 5.51 (с, 1H, ОН), 5.63 (уш. д, 1H, Н-18), 6.59+6.49 (АВ-система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19Р-ЯМР (CDCl3, δ, м.д., относительно CFCl3): -74.36 (с, 3F, CF3).
В результате подкисления и осаждения из смеси этанол/эфир вещества I (R=ОН; R1=СН3, R2=Н, R3=Н, R4 - СН3, Z~Z=СН=СН) получают 0.43 г (46%) продукта I (R=ОН; R1=СН3, R2=Н, R3=Н, R4=СН3, Z-Z=СН=СН) в виде гидрохлорида с т.пл. 260°C (разл.).
Пример 7. Получение (20R)-3,6-дигидрокси-17-метил-4,5α-эпокси-6а,14α-этано-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R=СН3; R=Н; R=Н; R4=СН3; Z-Z=СН2СН2) и (20R)-6-гидрокси-17-метил-3-метокси-4,5α-эпокси-6α,14α-этано-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=CH3; R2=H, R3=H, R4=CH3, Z~Z=CH2CH2)
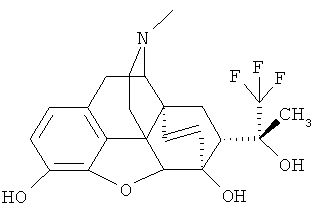
1.00 г (2.21 моль) I (R=ОН; R1=СН3; R2=СН3; R3=СН3; R4=СН3; Z-Z=СН2СН2) растворяют в 20 мл 48%-ного водного раствора HBr при нагревании и кипятят полученную реакционную смесь 15 минут. После охлаждения до комнатной температуры смесь разбавляют водой (10 мл), доводят значение pН до 9-10 с помощью 25%-ного водного раствора аммиака и экстрагируют хлороформом (3×50 мл). Органический слой высушивают над безводным Na2S04 и растворитель отгоняют в вакууме. Остаток хроматографируют на колонке с силикагелем (элюент: NH4OH: CH3OH:CHCl3=1:15:1600) и получают два основных продукта: a) I (R=ОН; R1=СН3; R2=СН3; R3=Н; R4=СН3; Z~Z=СН2СН2) Спектр 1Н ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 0.60-0.67 (м, 1Н, Н-19 л), 1.01 - 1.09 (м, 1H, Н-19п), 1.27-1.35 (м, 1H, Н-18л), 1.44 (с, ЗН, СН3), 1.56 (м, 1Н, Н-8а), 1.66-1.72 (м, 1Н, Н-15эк), 1.91-2.03 (м, 2Н, Н-15ак+Н-18п), 2.11 (м, 1Н, И-7/J), 2.22 (дд, 1Н, Н-10а), 2.30 (с, 3Н, NCH3), 2.29 - 2,35 (м, 1H, Н-16ак), 2.46 (д, 1Н, Н-16эк), 2.68 (д, 1H, Н-9), 2.81 (м, 1H, H-8β, 3.12 (д, 2J=18.3 Гц, 1H, Н-10β), 3.86 (с, 3Н, 3-OCH3), 4.19 (с, 1H, Н-5), 5.53 (с, 1H, ОН), 6.72+6.60 (АВ система, JAB=8.0 Гц, 2Н, Н-1+Н-2).
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): -73.72 (с, 3F, CF3). После подкисления полученного вещества концентрированной соляной кислотой и осаждения из смеси этанол/эфир получают 0.12 г (13%) гидрохлорида соединения I (R=ОН; R1=СН3; R2=СН3; R3=Н; R4=СН3; Z~Z=СН2СН2) в виде бесцветных кристаллов с т.пл. 240°C (разложение).
Масс-спектр (электроспрей) (m/z): 440[М+1]+.
б) I (R=ОН; R1=СН3; R2=Н; R3-Н; R4=СН3; Z-Z=СН2СН2)
Спектр 1H ЯМР (CDCl3, 3, м.д., относительно (CH3)4Si): 0.59-0.66 (м, 1Н, Н-19 л), 1.01-1.09 (м, 1Н, Н-19п), 1.24-1.34 (м, 1Н, Н-18 л), 1.44 (с, 3Н, СН3), 1.57-1.73 (м, 2Н, Н-8а+Н-15эк), 1.91-2.03 (м, 2Н, Н-15ак+Н-18п), 2.11 (дд, 1Н, Н-7Д), 2.21 (дд, 1Н, 3J=5.8 Гц, 2J=18.5 Гц, Н-10β), 2.30 (с, 3Н, NCH3), 2.31-2,35 (м, 1Н, Н-16ак), 2.46 (дд, 1H, 3J=5.2 Гц, 2J=12.3 Гц, Н-16эк), 2.56 (уш.с, 1Н, ОН), 2.67 (д, 1Н, 3J=5.8 Гц, Н-9), 2.78-2.85 (м, 1H, И-Щ, 3.10 (д, 2J=18.5 Гц, 1H, Н-10β), 4.21 (с, 1H, Н-5), 5.44 (с, 1H, ОН), 6.69+6.55 (АВ система, JAB=8.1 Гц, 2Н, Н-1+Н-2);
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): -75.78 (с, 3F, CF3).
После подкисления полученного вещества концентрированной соляной кислотой и осаждения из смеси этанол/эфир получают 0.26 г (28%) гидрохлорида продукта I (R=ОН; R1=СН3; R2=Н; R3=Н; R4=СН3; Z~Z=СН2СН2) в виде белого рыхлого порошка с т.пл. 239°C (разложение). Масс-спектр (электроспрей) (m/z): 426 [М+1]+.
Пример 8. Получение ^26Ш-3.6-дигидрокси-17-(циклопропилметил)-4,5α-эпокси-6α,14α-этено-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=CH3: R2=Н; R3 - Н; R4=циклопропилметил: Z~Z=СН=СН)
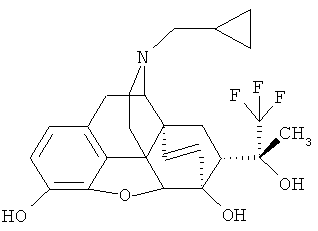
По методике, описанной в примере 6, из 0.83 г (1.70 ммоль) I (R=ОН; R1=СН3; R2=СН3; R3=СН3; R4=циклопропилметил; Z-Z=СН=СН) получают 0.30 г (38%) I (R=ОН; R1=СН3; R2=Н; R3=Н; R4=циклопропилметил; Z-Z=СН=СН) в результате осаждения соединения в виде соли гидрохлорида из смеси этанол/эфир. Температура плавления: 255°C (гидрохлорид плавится с разложением). Масс-спектр и ЯМР-спектры зарегистрированы с использованием I (R=ОН; R1=СН3; R2=Н; R3=Н; R4=циклопропилметил; Z~Z=СН=СН) в виде основания, полученного непосредственно в результате хроматографии в виде масла желтого цвета.
Масс-спектр (электроспрей) (m/z): 464 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 0.11-0.13 и 0.50-0.55 (м+м, 2Н+2Н, 2CH2 (из цикло-C3H5), 0.82-0.87 (м, 1Н, СН (из цикло-C3H5), 1.27-1.33 (м, 1H, Н-8α), 1.38 (с, 3Н, СН3), 1.74-1.77 (м, 1H, Н-15эк), 1.88-1.95 (м, 1Н, Н-15ак), 2.13 (м, 1H, H-7β), 2.27-2.44 (м, 4Н, Н-10α+Н-16ак+2Н (цикло-C3H3-CH2)), 2.69 (дд, 1Н, 2J=11.7 Гц, 3J=4.7 Гц, Н-16эк), 2.91 (дд, 2J=12.9 Гц, 3J=10.0 Гц, 1Н, Н-8β, 3.06 (д, 2J=18.4 Гц, 1Н, H-10β), 3.51 (д, 3J=6.4 Гц, 1Н, Н-9), 4.31 (д, 1H, Н-5), 5.37 (д, J18,19=8.8 Гц, 1Н, Н-19), 5.62 (уш. д, 1Н, Н-18), 5.73 (с, 1Н, ОН), 6.60+6.46 (АВ-система, JAB=8.0 Гц, 2Н, Н-1+Н-2). Спектр
19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): -74.26 (с, 3F, CF3).
Пример 9. Получение 3,6-дигидрокси-17-метил-4,5α-эпокси-6α, 14α-этено-7α-[1-гидрокси-1-(трифторметил)-2,2,2-трифторэтил1изоморфинана (I, R=ОН; R1=CF3; R2=Н: R3=Н; R4=CH3; Z~Z=СН=CH).
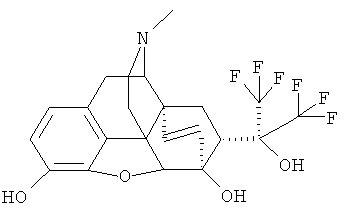
По методике, описанной в примере 6, из 0.54 г (1.07 ммоль) I (R=ОН; R1=CF3, R2=СН3, R3=СН3, R4=СН3, Z-Z=СН=СН) получают 0.39 г (76%) I (R=ОН; R1=CF3; R2=Н; R3=Н; R4=СН3; Z-Z=СН=СН) в результате осаждения соединения в виде соли гидрохлорида из смеси этанол/эфир.
Температура плавления: 280°C (гидрохлорид плавится с разложением) Масс-спектр и ЯМР-спектры зарегистрированы с использованием I (R=ОН; R1=CF3; R2=Н; R3=Н; R4=СН3; Z~Z=СН=СН) в виде основания, полученного непосредственно в результате хроматографии в виде масла желтого цвета.
Масс-спектр (электроспрей) (m/z): 478 [М+1]+.
Спектр 1Н ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 1.53 (дд, 2J=13.0 Гц, 3J=7.9 Гц, 1Н, Н-8α), 1.80 (дд, 1Н, Н-15эк), 1.89-1.96 (м, 1H, Н-15ак), 2.35 (с, 3Н, NCH3), 2.34-2.40 (м, 2Н, Н-Юсс+Н-16ак), 2.44 (м, 1H, Н-7Д, 2.51 (дд, 1H, Н-16эк), 2.91 (дд, 1Н, H-8β), 3.17-3.21 (м, 2Н, Н-9+Н-10β), 4.34 (с, 1H, Н-5), 5.40 (д, J18,i9=8.8 Гц, 1H, Н-19), 5.61 (уш. д, 1Н, Н-18), 6.50+6.60 (АВ система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19Р-ЯМР (CDCl3, м.д., относительно CFC13): -70.09 (кв, J=9.9 Гц, 3F, CF3), -74.30 (кв, J=9.9 Гц, 3F, CF3).
Пример 10. Получение (20S)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этено-7α-[1-гидрокси-1-(трифторметил)бутил1изоморфинана (I, R=ОН; R=; R=CN; R3=СН2; R4=СН3: Z~Z=СН=CH.
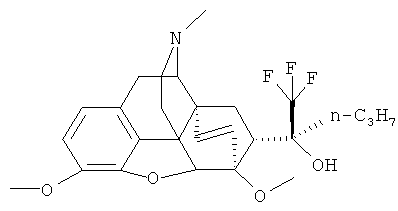
К охлажденному от -70°C до -78°C раствору 1.00 г (2.3 ммоль) la (R+R1=О; R2=CH3; R3=CH3; R4=CH3; Z~Z=CH=CH) в ТГФ (15 мл) прибавляют по каплям 9 мл 0,34 М раствора н-PrLi в гексане. Охлаждающую баню убирают, позволяют реакционной смеси нагреться до комнатной температуры и выливают ее в насыщенный водный раствор NH4CI (25 мл). Полученную смесь экстрагируют хлороформом (3×20 мл). Органические экстракты объединяют, высушивают над безводным Na2S04 и растворитель отгоняют.После перекристаллизации остатка из метанола получают 0.60 г (54%) продукта I (R=ОН; R1=СН2СН2СН3; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН=СН).
Температура плавления: 193-196°С. Масс-спектр (электроспрей) (m/z): 480 [М+1]+.
Спектр 1Н ЯМР (CDCД3, δ, м.д., относительно (CH3)4Si): 0.86 (т, ЗН, СН3СН2СН2), 1.08-1.14 (м, 1Н, Н-8а), 1.27-1.34 и 1.52-1.66 (м+м, 2Н+2Н, CH3CH2CH2), 1-83 (м, 1Н, Н-15эк), 1.95 (м, 1Н, Н-15ак), 1.28 (м, 1Н, K-7β), 2.35 (с, 3Н, NCH3), 2.32-2.40 (м, 2Н, Н-10α+Н-16ак), 2.50 (дд, 1H, 2J=11.6 Гц, 3J=5.2 Гц, Н-16эк), 2.89 (дд, 2J=13.1 Гц, 3J=9.3 Гц, 1H, Н-8β, 3.12 (д, 3J=6.4 Гц, 1Н, Н-9), 3.21 (д, 2J=18.5 Гц, 1Н, Н-10β), 3.79 (с, 3Н, 6-OCH3), 3.81 (с, 3Н, 3-ОСН3), 4.54 (д, 4J=1.0 Гц, 1H, Н-5), 5.30 (с, 1H, ОН), 5.49 (д, J,8,i9=8.9 Гц, 1Н, Н-19), 5.94 (уш. д, 1H, Н-18), 6.62+6.52 (АВ-система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): -74.09 (с, 3F, CF3).
Пример 11. Получение 17-метил-3,6-диметокси-4,5α-эпокси-6α,14α-этено-7α-(1-гидрокси-1-фенил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=C6H5; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН=CH.
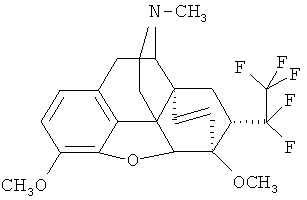
К раствору 0.20 г (0.46 ммоль) Ia (R+R1=О; R2=СН3; R3=СН3; R4=СН3; Z-Z=СН=СН) в ТГФ (15 мл) прибавляют по каплям 1.63 мл 0,56 М раствора (0,92 ммоль) PhMgBr в ТГФ. Смесь перемешивают 16 ч, выливают ее в насыщенный водный раствор NH4Cl (25 мл), добавляют воду (25 мл) и полученную смесь экстрагируют эфиром (3×20 мл). Органические экстракты объединяют, высушивают над безводным Na2SO4 и растворитель отгоняют. Продукт отделяют хроматографией на колонке с силикагелем [элюент - петролейный эфир/этилацетат (1:1)]. После перекристаллизации остатка из метанола получают 0.09 г (37%) продукта I (R=ОН; R1=С6Н5; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН=СН).
Температура плавления: 213-219°C (разложение).
Масс-спектр (электроспрей) (m/z): 514 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 1.36 (дд, 2J=13.0 Гц, 3J=7.8 Гц, 1Н, Н-8а), 1.65-1.71 (м, 1H, Н-15эк), 1.73-1.76 (м, 1H, Н-15ак), 2.13-2.39 (м, 6Н), 2.23 (с, 3Н, NCH3), 3.08 (д, 3J=6.4 Гц, 1H, Н-9), 3.16 (д, 2J=18.6 Гц, 1H, Н-10β), 3.82 (с, 3Н, ОСН3), 3.83 (с, 3H, ОСН3), 4.48 (д, 1H, Н-5), 5.55 (д, J18jl9=9.1 Гц, 1Н, Н-19), 6.13 (уш. д., 1H, Н-18), 6.52+6.63 (АВ-система, JAB=8.2 Гц, 2Н, Н-1+Н-2), 6.66 (с, 1Н, ОН), 7.30-7.40 (м, 3Н, 1Hn-+2Нм-, С6Н5), 7.61 (д, 3J=7.7, 2Н, 2Н o-, С6Н5).
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFCl3): - 68.46 (с, 3F, CF3).
Пример 12. Получение (20R)-3,6-диметокси-17-(пропен-2-ил-1)-4,5α-эпокси-6α,14α-этено-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=СН3; R2=CH3; R3=СН3; R4=СН2-СН=СН2; Z~Z=СН=CH.
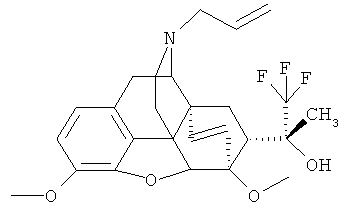
По методике, описанной в примере 1, из 1.6 г (3.93 ммоль) II (R+R=О; R=СН3; R=СН3; R4=СН2-СН=СН2; Z~Z=СН=СН) и 2.4 мл (16.23 моль) (CH3)3SiCF3 после разделения продуктов реакции хроматографией на колонке с силикагелем (элюент -25%-ный водный раствор NH3: СН3ОН: СНС13: гексан=1:15:800:800) получают 0.50 г (26.7%) I (R=ОН; R1=СН3; R2=СН3; R3=СН3; R4=СН2-СН=СН2; Z~Z=СН=СН) в виде желтого масла.
Масс-спектр (электроспрей) (m/z): 478 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 1.23-1.29 (м, 1H, Н-8а), 1.33 (с, 3H, СН3), 1.83-1.87 (м, 1H, Н-15эк), 1.88-1.95 (м, 1H, Н-15ак), 2.11 (м, 1Н, Н-7β), 2.35-2.43 (м, 2Н, Н-16ак+Н-10α), 2.60 (дд, 2J=12.4 Гц, 3J=4.4 Гц, 1H, Н-16ак), 2.90 (дд, 1H, 2J=12.6 Гц, 3J=9.9 Гц, Н-8β), 3.11-3.15 (м, 3H, Н-10β+CH2(allyl)), 3.27 (д, 3J=6.4 Гц, 1H, Н-9), 3.77 (с, 3Н, 6-OCH3), 3.81 (с, 3H, 3-ОСН3), 4.47 (уш.с, 1Н, Н-5), 5.14 (д, J=10.1 Гц, 1H, Hallyl, (-СН2-СН=CH2(cis)), 5.21 (дд, J=17.1 Гц, 1Н, Hallyl(-CH2-CH=CH2(trans)), 5.47 (д, J18,19=9.1 Гц, 1H, Н-19), 5.77-5.87 (м, 1Н, Hallyl(-СН2-СН=СН2), 5.94 (с, 1H, ОН), 6.03 (уш. д, 1H, Н-18), 6.62+6.51 (АВ-система, JAB=8.2 Гц, 2Н, Н-1+Н-2)
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFC13): -74.49 (с, 3F, CF3).
Пример 13. Получение (20R)-3,6-диметокси-17-(пропен-2-ил-1)-4,5α-эпокси-6α,14α-этано-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=СН3; R2=CH3: R3=CH3; R4=СН2-СН=CH2; Z~Z=СН2СН2).
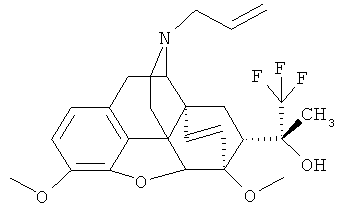
По методике, описанной в примере 1, из 0.80 г (1.96 ммоль) II (R+R=О; R=СН3; R=СН3; R4=СН2-СН=СН2; Z~Z=СН2СН2) и 1.30 мл (8.79 ммоль) (CH3)3SiCF3 получают 0.50 г (53%) I (R=ОН; R1=СН3; R2=СН3; R3=СН3; R4=СН2-СН=СН2; Z-Z=СН2СН2) в виде бесцветных кристаллов после перекристаллизации из метанола.
Температура плавления: 119-120°c.
Масс-спектр (электроспрей) (m/z): 480 [М+1]+.
Спектр 1H ЯМР (CDCl3, 8, м.д., относительно (CH3)4Si): 0.67-0.73 (м, 1Н, Н-19 л), 1.04-1.09 (м, 1H, Н-19п), 1.42 (с, 3Н, СН3), 1.49 (дд, 2J=13.0 Гц, 3J=9.8 Гц, 1Н, Н-8β), 1.68 (дд, 2J=13.0 Гц, 3J=2.52 Гц, 1H, Н-15эк), 1.70-1.75 (м, 1Н, Н-18 л), 1.80-1.85 (м, 1H, Н-18п), 1.92-1.97 (м, 1Н, Н-15ак), 2.08 (м, 1Н, Н-7β), 2.22 (дд, 2J=18.3 Гц, 3J=6.4 Гц, 1H, Н-10α), 2.28-2.32 (м, 1Н, Н-16ак), 2.52 (дд, 2J=12.2 Гц, 3J=7.7 Гц, 1Н, Н-16ак), 2.78 (д, 3J=6.4 Гц, 1Н, Н-9), 2.82-2.87 (м, 1Н, H-8β), 3.01-3.06 (м, 3Н, Н-10β+2Hallyl(-СН2-СН=СН2)), 3.51 (с, 3Н, 6-ОСНз), 3.87 (с, 3Н, 3-ОСН3), 4.32 (д, 4J=2.3 Гц, 1Н, Н-5), 5.11 (дд, 2J=10.0 Гц, 3J=1.3 Гц, 1Н, Hallyl(-СН2-СН=СН2(cis)), 5.17 (дд, 2J=17.2 Гц, 3J=1.9 Гц, 1Н, Hallyl(-CH2-CH=CH2(trans)), 5.74-5.81 (м, 1H, Hallyl(-CH2-CH=CH2), 5.87 (с, 1H, OH), 6.71+6.56 (АВ-система, JAB=8.0 Гц, 2Н, Н-1+Н-2). Спектр 19Р-ЯМР (CDCI3, 8, м.д., относительно CFCl3): -73.89 (с, 3F, CF3).
Пример 14. Получение (20R)-3,6-диметокси-17-(циклопропилметил)-4,5α-эпокси-6α,14α-этано-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=СН3: R2=CH3; R3=СН3; R4=циклопропилметил: Z~Z=СН2СН2).
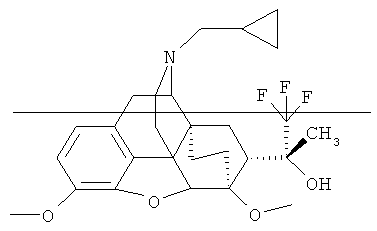
По методике, описанной в примере 1, из 3.20 г (7.30 ммоль) II (R+R1=О; R2=СН3; R3=СН3; R4=цикло-C3H5CO; Z~Z=СН2СН2) и 3.23 мл (22.0 моль) (CH3)3SiCF3 получают 3.54 г продукта, содержащего в основном I (R=ОН; R1=СН3; R2=СН3; R3=СН3, R4=цикло-С3Н5СО; Z~Z=СН2СН2), который растворяют в ТГФ (25 мл). Полученный раствор при интенсивном перемешивании добавляют по каплям в суспензию 1.10 г (0.03 моль) LiAlH4 в ТГФ (20 мл) в течение 30 минут, а затем смесь перемешивают еще 10 мин. В нее осторожно по каплям при постоянном перемешивании добавляют насыщенный раствор хлорида аммония (20 мл). Органический слой отделяют, водный экстрагируют эфиром (3×50 мл). Органический раствор и экстракты объединяют и сушат над безводным Na2S04. Растворитель отгоняют досуха, остаток хроматографируют на колонке с силикагелем (элюент - 25%-ный водный раствор NH3:СН3ОН:CHCl3:гексан=1:15:1600:1600). Получают 3.00 г (83%) продукта I (R=ОН; R1=СН3; R2=СН3; R3=СН3; R4=циклопропилметил; Z~Z=СН2СН2) в виде желтоватого масла.
Масс-спектр (электроспрей) (m/z): 494 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 0.07-0.11 и 0.43-0.52 (м+м, 2Н+2Н, 2СН2(из цикло-C3H5)), 0.61-0.68 (м, 1Н, Н-19 л), 0.78-0.87 (м, 1H, СН (из цикло-C3H5)), 1.07-1.15 (м, 1H, Н-19п), 1.30-1.35 (м, 1H, Н-18 л), 1.61-1.66 (м, 1Н, Н-8а), 1.70 (с, 3Н, CH3), 1.71-1.78 (м, 1H, Н-18п), 1.92 (дд, 1H, Н-15эк), 2.01-2.09 (м, 2Н, Н-16ак+Н-15ак), 2.22-2.31 (м, 4Н, 2Н (цикло-C3H5-CH2)+Н-10α+Н-7β), 2.62-2.71 (м, 2Н, Н-16эк+Н-8β, 3.01 (д, 3J=6.4 Гц, 1Н, Н-9), 2.97 (д, 2J=18.4 Гц, 1H, Н10β), 3.38 (с, 3Н, 6-ОСН3), 3.86 (с, 3Н, 3-OCH3), 4.43 (д, 4J=1.9 Гц, 1Н, Н-5), 6.67+6.54 (АВ-система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19F-ЯМР (CDCl3, δ, м.д., относительно CFC13): -76.78 (с, 3F, CF3)
Пример 15. Получение (20R)-3,6-дигидрокси-17-(пропен-2-ил-1)-4,5α-эпокси-6α,14α-этано-7α-(1-гидрокси-1-метил-2,2,2-трифторэтил)изоморфинана (I, R=ОН; R1=СН3; R2=Н; R3=Н; R4=СН?-СН=СН2; Z~Z=СН-СН2-CH2)
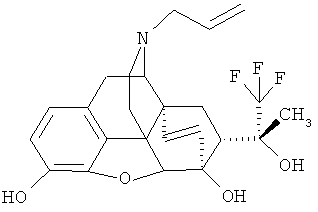
По методике, описанной в примере 6, из 0.40 г (0.84 ммоль) I (R=ОН; R1=СНз; R2=СН3; R3=СН3; R4=СН2-СН=СН2; Z-Z=СН2-СН2) получают 0.33 г (88%) I (R=ОН; R1=СН3; R2=Н; R3=Н; R4=СН2-СН=СН2; Z~Z=СН2-СН2) в результате осаждения соединения в виде соли гидрохлорида из смеси этанол/эфир.
Температура плавления: 246°C (гидрохлорид плавится с разложением).
Масс-спектр и ЯМР-спектры зарегистрированы с использованием I (R=ОН; R1=СН3; R2=Н; R3=Н; R4=СН2-СН=СН2; Z~Z=СН2-СН2) в виде основания, полученного непосредственно в результате хроматографии в виде масла желтого цвета.
Масс-спектр (электроспрей) (m/z): 452 [М+1]+.
Спектр 1H ЯМР (CDCl3, δ, м.д., относительно (CH3)4Si): 0.56-0.62 (м, 1Н, Н-19л), 0.98-1.06 (м, 1H, Н-19п), 1.27-1.35 (м, 1H, 2Н-18), 1.45 (с, 3Н, СН3), 1.47-1.54 (м, 1Н, Н-8а), 1.86-1.96 (м, 2Н, Н-15эк+Н-15ак), 2.07-2.12 (м, 1Н, Н-7β), 2.20 (дд, 2J=18.6 Гц, 3J=6.4 Гц, 1Н, Н-10а), 2.21-2.29 (м, 1H, Н-16ак), 2.50 (дд, 2 J=11.5 Гц, 3J=4.6 Гц, 1Н, Н-1 бак), 2.78 (д, J=6.5 Гц, 1Н, Н-9), 2.78-2.85 (м, 1H, H-8β), 2.98-3.05 (м, 3Н, H-10β+2Hallyl(-СН2-СН=СН2)), 4.16 (д, 4J=2.3 Гц, 1H, Н-5), 5.11 (уш.д, 2J=10.0 Гц, 1H, Ha11yl(-СН2-СН=CH2(cis)), 5.17 (уш.д, 2J=17.2 Гц, 1Н, Hallyl(-СН2-СН=СН2(trans)), 5.69-5.81 (м, 1H, Hallyl(-CH2-CH=CH2), 5.69 (с, 1Н, ОН), 6.71+6.53 (АВ-система, JAB=8.1 Гц, 2Н, Н-1+Н-2).
Спектр 19Р-ЯМР (CDCl3, δ, м.д., относительно CFCl3): -73.63 (с, 3F, CF3).
Техническим результатом изобретения является получение первых фторсодержащих производных этеноморфинанов, которые относятся к классу лигандов опиодных рецепторов или являются предшественниками таких соединений, структура которых позволяет осуществлять дальнейшие химические модификации для получения продуктов с заданными свойствами, содержащих трифторметильную группу. Предложенные способы получения орвинолов и их предшественников обеспечивают введение трифторметильной группы в доступные исходные соединения, а наличие других реакционных центров делает возможным направленное введение конкретных заместителей для получения фторпроизводных тевинола и орвинола, проявляющих свойства лигандов опиоидных рецепторов.
Преимуществом заявляемых соединений формулы I по сравнению с не содержащими фтора аналогами является более широкая возможность варьирования гидрофильно-гидрофобного баланса молекулы опиоидного лиганда в соответствии с потребностью достижения желаемой биомишени (опиодного рецептора, локализованного в центральной либо в периферической нервной системе) без существенного изменения геометрических параметров молекулы.
Все заявляемые фторсодержащие соединения могут быть получены, исходя из нескольких доступных предшественников: тевинона (I, R+R1=О; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН=СН; Y=Н), синтезируемого из природного алкалоида тебаина в одну стадию [К.W.Bentley. Brit. Pat. GB 902659(A) (1962)] или более длинным путем [Н. Саксена. Авт.свид. СССР 1362734 (1987)]; либо исходя из 18,19-дигидротевинона (I, R+R1=О; R2=СН3; R3=СН3; R4=СН3; Z~Z=СН2СН2; Y=Н), получаемого гидрированием тевинона [K.W.Bentley. Brit. Pat. 1136214 (1968). Chem. Abstr., 70: 78218s (1969)]; либо исходя из 21,21,21-трифтортевинона (I, R+R1=О; R2=CH3; R3=СН3; R4=СНз; Z~Z=CH=CH; Y=F), который также получают из природного сырья (тебаина).
Способ получения 1,3-дикарбонильных производных адамантанов
Реабилитационная противопролежневая кровать для ожоговых больных
1-алкокси-2,7-диметилокта-2,7-диены, обладающие акарицидной активностью, и способ их получения
Способ ингибирования активности фермента рнк-полимеразы
Твердый полимерный электролит для литиевых источников тока
3,6-диметокси-17-метил-7альфа-(трифторацетил)-4,5альфа-эпокси-6альфа,14альфа-этеноизоморфинан и способ его получения
7α-(1-гидрокси-2,2,2-трифторэтил)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14αэтеноизоморфинан и способ его получения
Способ получения полифторарил(триметил)силанов
4-замещенные n-арил-1,8-нафталимиды, проявляющие свойства флуоресцентных сенсоров на катионы металлов, и способы их получения
Цинковые димерные комплексы краунсодержащих стирилфенантролинов в качестве оптических сенсоров на катионы щелочноземельных и тяжелых металлов и способ их получения
Способ получения 1,3-дикарбонильных производных адамантанов
Реабилитационная противопролежневая кровать для ожоговых больных
1-алкокси-2,7-диметилокта-2,7-диены, обладающие акарицидной активностью, и способ их получения
Способ ингибирования активности фермента рнк-полимеразы
Твердый полимерный электролит для литиевых источников тока
3,6-диметокси-17-метил-7альфа-(трифторацетил)-4,5альфа-эпокси-6альфа,14альфа-этеноизоморфинан и способ его получения
7α-(1-гидрокси-2,2,2-трифторэтил)-17-метил-3,6-диметокси-4,5α-эпокси-6α,14αэтеноизоморфинан и способ его получения
Способ получения полифторарил(триметил)силанов
4-замещенные n-арил-1,8-нафталимиды, проявляющие свойства флуоресцентных сенсоров на катионы металлов, и способы их получения
Цинковые димерные комплексы краунсодержащих стирилфенантролинов в качестве оптических сенсоров на катионы щелочноземельных и тяжелых металлов и способ их получения

