Результат интеллектуальной деятельности: ЦИКЛИЧЕСКИЕ АЗАИНДОЛ-3-КАРБОКСАМИДЫ, ИХ ПОЛУЧЕНИЕ И ПРИМЕНЕНИЕ В КАЧЕСТВЕ ЛЕКАРСТВЕННЫХ ПРЕПАРАТОВ
Вид РИД
Изобретение
Настоящее изобретение относится к циклическим азаиндол-3-карбоксамидам формулы I:
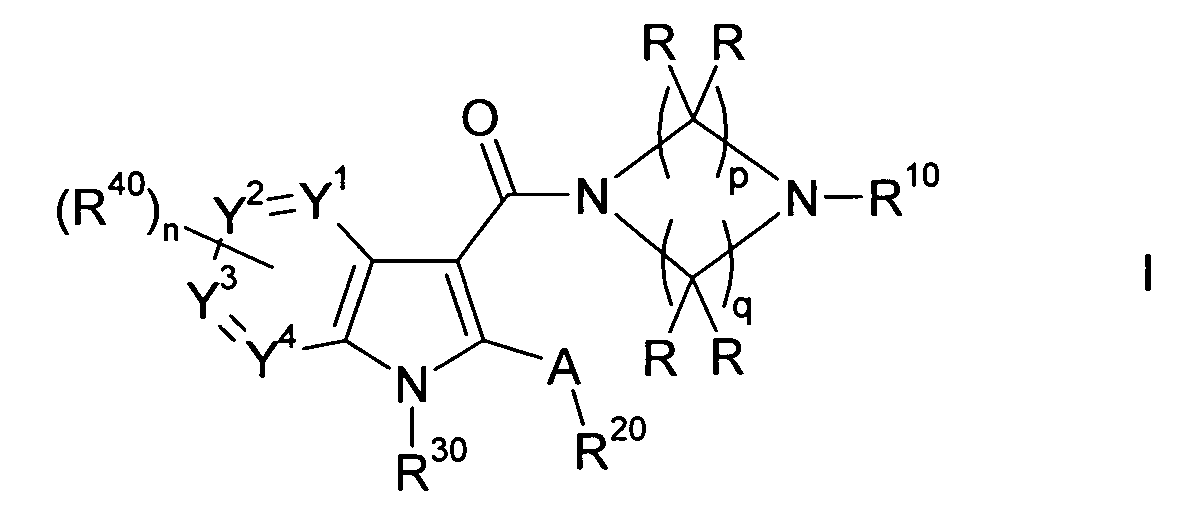 ,
,
в которой A, R, R10, R20, R30, R40, Y1, Y2, Y3, Y4, n, p и q имеют значения, показанные ниже, которые представляют собой ценные фармацевтически активные соединения. Конкретно, они ингибируют фермент ренин и модулируют активность ренин-ангиотензивной системы, и являются пригодными для лечения заболеваний, таких как, например, гипертензия. Кроме того, настоящее изобретение относится к способу получения соединений формулы I, их применению и содержащим их фармацевтическим композициям.
Ренин-ангиотензивная система (RAS; также обозначаемая, как система ренин-ангиотензин-альдостерон, RAAS) является ключевым регулятором сердечно-сосудистых функций, а также баланса электролитов и поддержания объема жидкостей тела, и является определяющим фактором кровяного давления (см., например, E. Lonn, Can. J. Cardiol. 20 (Suppl. B) (2004), 83B; I. A. Reid, Am. J. Physiol.: Advances in Physiology Education 20 (1998), S236). Она действует через ангиотензин II, октапептидный гормон, который связывается с ангиотензиновыми рецепторами. Образование ангиотензина II включает две основных стадии. На первой стадии ренин (EC 3.4.23.15; раньше EC 3.4.99.19 и EC 3.4.4.15), аспартилпротеиназа, состоящая из 340 аминокислот, расщепляет ангиотензиноген, образуя биологически неактивный декапептид ангиотензин I. На второй стадии ангиотензин I превращается в ангиотензин II с помощью цинк-зависимого протеазного ангиотензин-конвертирующего фермента (ACE). Ренин синтезируется в юкстагломерулярных клетках почек, первоначально в форме биологически неактивного проренина. Он высвобождается из почек и активируется, и последующая RAS активация у людей с нормальных артериальным давлением стимулируется уменьшением объема или снижением количества ионов натрия, или снижением кровяного давления.
RAS активность является главным определяющим фактором нескольких патологических состояний, т.к. ангиотензин II, главная эффекторная молекула данной системы, увеличивает кровяное давление или непосредственно сужением артериальных сосудов, или косвенно высвобождением альдостерона, являющегося задерживающим натрий гормоном из надпочечников, что сопровождается увеличением объема внеклеточной жидкости, а также обладает ускоряющим рост эффектом на ткани сосудов, сердца и почек, что способствует повреждению органов-мишеней.
Фармакологическая блокада RAS представляет собой хорошо известный способ лечения различных заболеваний, например, гипертензии (см., например, Handbook of Hypertension, W. H. Birkenhager et al. (ed.), Elsevier Science Publishers, Amsterdam (1986), vol.8, 489). Однако терапевтический ответ, достигаемый с помощью применяемых в настоящее время типов RAS блокаторов, ACE ингибиторов и блокаторов рецептора ангиотензина, хотя и эффективен, но является ограниченным. Это может быть результатом увеличения концентрации ренина, которое вызывается данными агентами и приводит в результате к увеличению концентрации ангиотензина I, который превращается в ангиотензин II через другие пути, чем посредством ACE. Ингибирование ренина, который контролирует первоначальную и ограничивающую скорость стадию в RAS катализированием расщепления Leu 10-Val 11 пептидной связи ангиотензиногена, что приводит в результате к образованию ангиотензиновых пептидов, будет ингибировать полную RAS и, таким образом, будет более эффективным. Кроме того, тогда как ингибирование ACE также воздействует на концентрацию других пептидов, которые расщепляются ACE, таких как, например, брадикинин, что связано с побочными эффектами ACE ингибиторов, подобно кашлю или ангиоэдеме, ренин является специфическим в том, что ангиотензиноген является его единственным природным субстратом. Таким образом, ингибирование ренина предлагает специфический и мощный способ снижения кровяного давления (см., M. Moser et al., J. Clin. Hypertension, 9 (2007), 701), а также обеспечивает защиту органов, таких как сердце, почки и мозг, и кроме лечения гипертензии, таким образом, является пригодным для лечения расстройств сердечно-сосудистой системы, таких как паралич сердца, сердечная недостаточность, порок сердца, инфаркт миокарда, гипертрофия сердца, гипертрофия сосудов, дисфункция левого желудочка, в частности дисфункции левого желудочка после инфаркта миокарда, рестеноз и стенокардия; заболевания почек, такие как фиброз почечной ткани, почечная недостаточность и слабость почек; диабетические осложнения, такие как нефропатия и ретинопатия; глаукома и церебральные заболевания, такие как внутримозговое кровоизлияние, например (в отношении эффекта RAS на заболевания почек и сердечные нарушения, см., например, U. C. Brewster, Am. J. Med. 116 (2004), 263; J. Gaedeke et al., Expert Opin. Pharmacother. 7 (2006), 377; B. Pilz et al., Hypertension 46 (2005), 569).
Большое количество пептидных и пептидо-миметиковых ингибиторов человеческого ренина с различными стабильными аналогами в переходном состоянии с расщепляющейся пептидной связью разработано приблизительно с 1980, и они способствовали признанию ренина в качестве терапевтической мишени (см., например, B. B. Scott et al., Curr. Protein Pept. Sci. 7 (2006), 241; J. Maibaum et al., Expert Opin. Ther. Patents 13 (2003), 589). Однако данные соединения обычно обладают недостатками, такими как недостаточная биодоступность (см., H. D. Kleinert, Cardiovasc. Drugs Therapy 9 (1985), 645) или продолжительность действия, или высокая стоимость получения. Недавно появился в продаже пероральный активный ингибитор ренина, алискирен (см., Drugs Fut. 26 (2001), 1139; J. Wood et al., J. Hypertens. 23 (2005), 417; M. Azizi et al., J. Hypertens. 24 (2006), 243). Но профиль свойств алискирена еще не является идеальным, например, что касается пероральной биодоступности, и конкретным недостатком алискирена является его сложная молекулярная структура с четырьмя хиральными центрами и его многостадийный синтез. Таким образом, еще имеется необходимость в новых, непептидных, небольших по размеру ингибиторах ренина, которые обладают подходящими свойствами, например, что касается пероральной биодоступности, или имеют несложную структуру и которые легко синтезировать. Настоящее изобретение удовлетворяет данную необходимость предоставлением ренин-ингибирующих циклических азаиндол-3-карбоксамидов формулы I.
Различные азаиндольные производные уже описаны. Например, в WO 01/62255 описывают антивирусные азаиндольные производные, пригодные для лечения человеческого вируса иммунодефицита 1, которые содержат в 3-положении азаиндольного кольца карбоксамидную или глиоксиламидную группу, в которой амидный атом азота является кольцевым членом пиперазиновой молекулы, которая несет при втором кольцевом атоме азота бензоильную группу, пиридин-2-карбонильную группу, фуран-2-карбонильную группу или тиофен-2-карбонильную группу, и которую необязательно можно заместить во 2-положении азаиндольного кольца заместителем, например, таким как насыщенный или ненасыщенный алкил или циклоалкил. В EP 1452525 описывают азаиндольные производные, которые, наряду с прочим, могут содержать в 3-положении азаиндольного кольца карбоксамидную группу, в которой амидный атом азота является кольцевым членом диазациклоалкана, который несет при втором кольцевом атоме азота пиридиновую, пиразиновую, пиридазиновую или пиримидиновую группу, и которые являются ингибиторами трансформирующего фактора роста β (TGF-β), пригодными для лечения, например, фибропролиферативных заболеваний. WO 2005/121175 относится к CD4 миметическим соединениям, которые образуют комплекс с оболочечными белками вируса иммунодефицита человека и являются пригодными для вызывания иммунного ответа, в общем они включают азаиндольные производные, которые могут содержать карбоксамидную группу, амидный атом азота которой является частью кольца. В US 2005/0054631 описывают определенные азаиндольные производные, которые содержат аминогруппу во 2-положении азаиндольного кольца и которые являются ингибиторами полимеразы поли(аденозин 5'-дифосфатрибозы) (PARP), пригодные для лечения различных заболеваний, включая заболевания, связанные с центральной нервной системой, и сердечно-сосудистые заболевания. WO 93/20078, которая относится к бициклическим гетероциклам, пригодным для лечения различных заболеваний, таких как повреждения головы, субарахноидальное кровоизлияние или астма, обычно включают, наряду с прочим, азаиндолы, которые замещают двумя аминозаместителями. Азаиндол-3-карбоксамиды по настоящему изобретению, в которых амидный атом азота является кольцевым членом 1,4- или 1,5-диазациклоалкеновой кольцевой системы, атом азота в положении 1 азаиндольной кольцевой системы несет циклическую группу, и атом углерода в положении 2 азаиндольной кольцевой системы несет (гетеро)ароматическую группу, еще не описаны.
Таким образом, объектом настоящего изобретения являются соединения формулы I в любой из их стереоизомерных форм или в виде смеси стереоизомерных форм в любом соотношении, и их физиологически приемлемые соли и физиологически приемлемые сольваты любого из них:
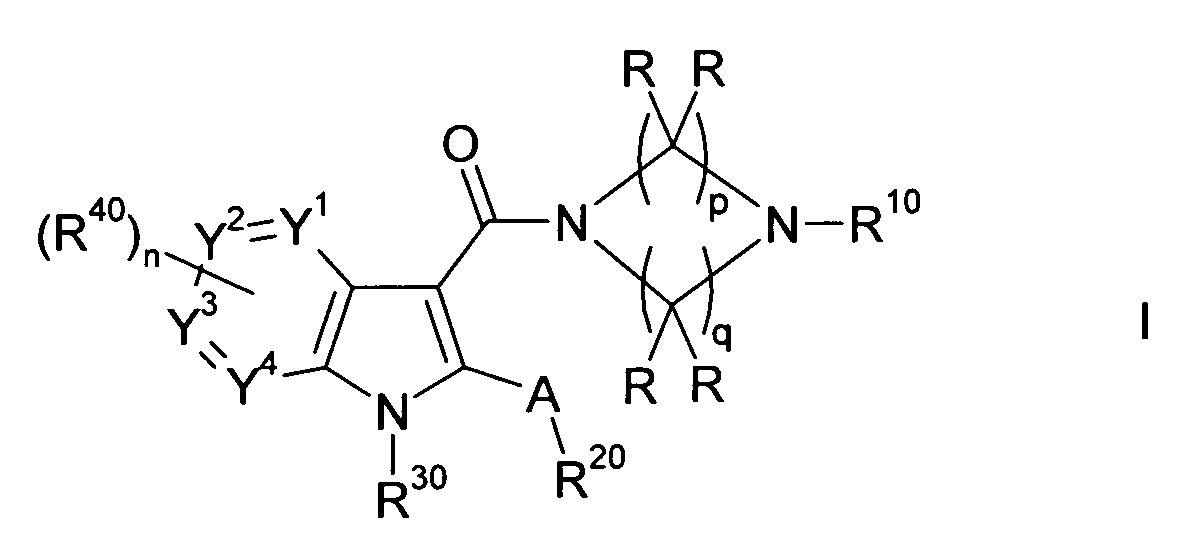 ,
,
в которой
A выбирают из O, S, N((C1-C4)-алкила) и C(Ra)2;
Ra выбирают из водорода, фтора и (C1-C4)-алкила, где две группы Ra являются независимыми друг от друга и могут быть одинаковыми или различными, или две группы Ra вместе представляют собой дивалентную (C2-C8)-алкильную группу;
R выбирают из водорода, фтора, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, фенил-(C1-C4)-алкила-, гетероарил-(C1-C4)-алкила-, (C1-C4)-алкил-O-CO-CuH2u-, R1-NH-CO-CuH2u- и (C1-C4)-алкил-O-, где все группы R являются независимыми друг от друга и могут быть одинаковыми или различными;
R1 выбирают из водорода, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила- и H2N-CO-(C1-C4)-алкила-;
R10 выбирают из водорода, (C1-C6)-алкил-O-CO- и (C3-C7)-циклоалкил-CvH2v-O-CO-;
R20 выбирают из фенила и гетероарила, которые необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, гидрокси и циано;
R30 выбирают из (C3-C7)-циклоалкила, (C5-C7)-циклоалкенила, тетрагидропиранила, фенила и гетероарила, где циклоалкил и циклоалкенил необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из фтора, (C1-C4)-алкила и гидрокси, и фенил и гетероарил необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C6)-алкила, (C3-C7)-циклоалкил-CvH2v-, гидрокси-(C1-C6)-алкила-, (C1-C4)-алкил-O-(C1-C6)-алкила-, (C3-C7)-циклоалкил-CvH2v-O-(C1-C6)-алкила-, (C1-C4)-алкил-CO-NH-(C1-C6)-алкила-, гидрокси, (C1-C6)-алкил-O-, (C3-C7)-циклоалкил-CvH2v-O-, гидрокси-(C1-C6)-алкил-O-, (C1-C4)-алкил-O-(C1-C6)-алкил-O-, (C3-C7)-циклоалкил-CvH2V-O-(C1-C6)-алкил-O-, (C1-C4)-алкил-CO-NH-(C1-C6)-алкил-O-, (C1-C6)-алкил-S(O)m- и циано;
R40 выбирают из галогена, (C1-C4)-алкила, (C3-C7)-циклоалкил-CvH2v-, фенил-(C1-C4)-алкила-, гетероарил-(C1-C4)-алкила-, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, (C3-C7)-циклоалкил-CvH2V-O-(C1-C4)-алкила-, фенил-O-(C1-C4)-алкила-, гетероарил-O-(C1-C4)-алкила-, ди((C1-C4)-алкил)N-(C1-C4)-алкила-, HO-CO-(C1-C4)-алкила-, (C1-C4)-алкил-O-CO-(C1-C4)-алкила-, H2N-CO-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O-, (C3-C7)-циклоалкил-CvH2v-O-, фенил-(C1-C4)-алкил-O-, гетероарил-(C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, (C3-C7)-циклоалкил-CvH2v-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, гетероарил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O-, H2N-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C3-C7)-циклоалкил-CvH2v-CO-O-, (C1-C4)-алкил-NH-CO-O-, (C3-C7)-циклоалкил-CvH2v-NH-CO-O-, (C1-C4)-алкил-S(O)m-, нитро, амино, (C1-C4)-алкиламино, ди((C1-C4)-алкил)амино, (C1-C4)-алкил-CO-NH-, (C3-C7)-циклоалкил-CvH2v-CO-NH-, (C1-C4)-алкил-S(O)2-NH-, HO-CO-, (C1-C4)-алкил-O-CO-, H2N-CO-, ((C1-C4)-алкил)-NH-CO-, ди((C1-C4)-алкил)N-CO-, циано, HO-S(O)2-, H2N-S(O)2-, ((C1-C4)-алкил)-NH-S(O)2- и ди((C1-C4)-алкил)N-S(O)2-, где все заместители R40 являются независимыми друг от друга и могут быть одинаковыми или различными;
одна из групп Y1, Y2, Y3 и Y4 представляет собой N, и другие являются одинаковыми или различными группами CH или CR40;
гетероарил представляет собой ароматическую моноциклическую, 5-членную или 6-членную гетероциклическую группу, которая содержит 1, 2 или 3 одинаковых или различных кольцевых гетероатома, выбранных из N, O и S, где один из кольцевых атомов азота может нести атом водорода или (C1-C4)-алкильную группу, и где гетероарильная группа присоединена через кольцевой атом углерода;
m выбирают из 0, 1 и 2, где все значения m являются независимыми друг от друга и могут быть одинаковыми или различными;
n выбирают из 0, 1, 2 и 3;
p и q, которые являются независимыми друг от друга и могут быть одинаковыми или различными, выбирают из 2 и 3;
u выбирают из 0, 1 и 2, где все значения u являются независимыми друг от друга и могут быть одинаковыми или различными;
v выбирают из 0, 1 и 2, где все значения v являются независимыми друг от друга и могут быть одинаковыми или различными;
где все алкильные группы, независимо друг от друга, необязательно замещают одним или более атомами фтора;
где все циклоалкильные группы, независимо друг от друга, необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из фтора и (C1-C4)-алкила, если не указано особо;
где все фенильные и гетероарильные группы, присутствующие в R и R40, независимо друг от друга, необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)2- и циано.
Если структурные элементы, такие как группы, заместители или значения, могут встречаться несколько раз в соединениях формулы I, они все являются независимыми друг от друга и могут в каждом случае иметь любое из указанных значений, и могут в каждом случае быть одинаковыми или отличными от любого другого данного элемента.
Алкильные группы, т.е. насыщенные углеводородные остатки, могут быть с нормальной цепью (линейными) или разветвленными. Это также применяют, если данные группы замещают, или если они являются частью другой группы, например, алкил-O-группы (алкилокси группы, алкокси группы) или алкил-S(O)m-группы. В зависимости от соответствующего определения, число атомов углерода в алкильной группе может составлять 1, 2, 3, 4, 5, 6, 7 или 8. Примерами алкила являются метил, этил, пропил, включая н-пропил и изопропил, бутил, включая н-бутил, втор-бутил, изобутил и трет-бутил, пентил, включая н-пентил, 1-метилбутил, изопентил, неопентил и трет-пентил, гексил, включая н-гексил, 3,3-диметилбутил и изогексил, гептил, включая н-гептил, и октил, включая н-октил. Примерами алкил-O- являются метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси, трет-бутокси и н-пентокси. Примерами алкил-S(O)m- являются метансульфанил- (CH3-S-, метилсульфанил-), метансульфинил- (CH3-S(O)-), метансульфонил- (CH3-S(O)2-), этансульфанил- (CH3-CH2-S-, этилсульфанил-), этансульфинил- (CH3-CH2-S(O)-), этансульфонил- (CH3-CH2-S(O)2-), 1-метилэтансульфанил- ((CH3)2CH-S-, 1-метилэтилсульфанил-), 1-метилэтансульфинил- ((CH3)2CH-S(O)-) и 1-метилэтансульфонил-((CH3)2CH-S(O)2-). В одном варианте осуществления настоящего изобретения значение m выбирают из 0 и 2, где все значения m являются независимыми друг от друга и могут быть одинаковыми или различными.
Замещенную алкильную группу можно заместить в любых положениях, при условии, что полученное в результате соединение является достаточно стабильным и является пригодным в качестве фармацевтически активного соединения. Обязательное условие, чтобы конкретная группа и соединение формулы I было достаточно стабильным и являлось пригодным в качестве фармацевтически активного соединения, применяют обычно относительно всех групп в соединениях формулы I. Если алкильную группу можно монозаместить или полизаместить фтором, ее можно не замещать, т.е. она не несет атомы фтора, или замещать, например 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11 атомами фтора, предпочтительно 1, 2, 3, 4 или 5 атомами фтора, которые могут присутствовать в любых положениях. Например, в фтор-замещенной алкильной группе одна или более метильных групп могут нести три атома фтора каждая и могут присутствовать в виде трифторметильных групп, и/или одна или более метиленовых групп (CH2) могут нести два атома фтора каждая и присутствовать в виде дифторметиленовых групп. Пояснения относительно замещения группы фтором является также применимыми, если группа дополнительно несет другие заместители и/или является частью другой группы, например, алкил-O-группы. Примерами фтор-замещенных алкильных групп являются трифторметил, 2-фторэтил, 1,1-дифторэтил, 2,2,2-трифторэтил, пентафторэтил, 3,3,3-трифторпропил, 2,2,3,3,3-пентафторпропил, 4,4,4-трифторбутил и гептафторизопропил. Примерами фтор-замещенных алкил-O-групп являются трифторметокси, 2,2,2-трифторэтокси, пентафторэтокси и 3,3,3-трифторпропокси. Примерами фтор-замещенных алкил-S(O)m-групп являются трифторметансульфанил- (CF3-S-, трифторметилсульфанил-), трифторметансульфинил- (CF3-S(O)-) и трифторметансульфонил- (CF3-S(O)2-).
Если возможно, вышеуказанные пояснения относительно алкильных групп применяют соответственно к дивалентным алкильным группам (алкандиильным группам), включая дивалентные алкильные группы CuH2u и CvH2v, где группы также могут считаться алкильной частью замещенной алкильной группы. Таким образом, дивалентные алкильные группы, включая дивалентные алкильные группы CuH2u и CvH2v, могут также быть с нормальной цепью или разветвленными, связи с соседними группами могут находиться в любых положениях и могут начинаться с одного атома углерода или с различных атомов углерода, и их можно заместить фтором. Примерами дивалентных алкильных групп являются -CH2-, -CH2-CH2-, -CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-CH2-, -CH2-CH2-CH2-CH2-CH2-CH2-, -CH(CH3)-, -C(CH3)2-, -CH(CH3)-CH2-, -CH2-CH(CH3)-, -C(CH3)2-CH2- и -CH2-C(CH3)2-. Примерами фтор-замещенных дивалентных алкильных групп, которые могут содержать 1 2, 3, 4, 5 или 6 атомов фтора, например, являются -CHF-, -CF2-, -CF2-CH2-, -CH2-CF2-, -CF2-CF2-, -CF(CH3)-, -C(CF3)2-, -C(CH3)2-CF2- и -CF2-C(CH3)2-. Если значение u в дивалентной алкильной группе CuH2u или значение v в дивалентной алкильной группе CvH2v равно 0 (нулю), две соседние группы, которые присоединены к данной группе, являются непосредственно связанными друг с другом одинарной связью. Например, если группа R40 представляет собой группу (C3-C7)-циклоалкил-CvH2v-, где группа соединена с остатком молекулы через CvH2v группу, как символически показано линией на конце (дефис) после CvH2v группы, представляющей свободную связь, и значение v в ней равно 0, (C3-C7)-циклоалкильная группа связана непосредственно через одинарную связь с атомом углерода, который несет группу R40. В одном варианте осуществления настоящего изобретения значение v выбирают из 0 и 1, где все значения v являются независимыми друг от друга и могут быть одинаковыми или различными.
Число кольцевых атомов углерода в циклоалкильной группе может составлять 3, 4, 5, 6 или 7. Число кольцевых атомов углерода в циклоалкенильной группе может составлять 5, 6 или 7. Примерами циклоалкила являются циклопропил, циклобутил, циклопентил, циклогексил и циклогептил, примерами циклоалкенила являются циклопентенил, циклогексенил и циклогептенил. Двойная связь в циклоалкенильной группе может присутствовать в любом положении относительно атома углерода в положении 1, через который данная группа соединена с азаиндольной группой, и, таким образом, циклоалкенил может, например, представлять собой циклопент-1-енил, циклопент-2-енил, циклопент-3-енил, циклогекс-1-енил, циклогекс-2-енил, циклогекс-3-енил, циклогепт-1-енил, циклогепт-2-енил, циклогепт-3-енил, циклогепт-4-енил. В предпочтительных вариантах осуществления настоящего изобретения циклоалкильную группу, такую как (C3-C7)-циклоалкил, в определении любой группы, выбирают из подгруппы любых двух или более из указанных конкретных циклоалкильных групп, например, из циклопропила и циклобутила, или из циклопропила, циклобутила и циклопентила, или из циклопропила, циклопентила и циклогексила, или из циклопентила и циклогексила, или из циклопентила, циклогексила и циклогептила. Аналогично, в предпочтительных вариантах осуществления циклоалкенильную группу выбирают из подгруппы любых двух или более из указанных конкретных циклоалкенильных групп, например, из циклопентенила и циклогексенила, или из циклогексенила и циклогептенила, или из циклопент-1-енила, циклопент-2-енила, циклогекс-1-енила, циклогекс-2-енила, циклогепт-1-енила и циклогепт-2-енила, или из циклопент-2-енила, циклопент-3-енила, циклогекс-2-енила, циклогекс-3-енила, циклогепт-2-енила, циклогепт-3-енила и циклогепт-4-енила, или из циклопент-2-енила и циклогекс-2-енила, или из циклопент-2-енила, циклогекс-2-енила и циклогепт-2-енила. В одном варианте осуществления настоящего изобретения, атом углерода, через который циклоалкенильная группа, представляющая R30, соединена с азаиндольным кольцом, не является частью двойной связи, т.е. циклоалкенильная группа не является циклоалк-1-енильной группой. Циклоалкильные группы и циклоалкенильные группы обычно необязательно замещают одним или более (C1-C4)-алкильными заместителями. Т.е. их не замещают, т.е. они не несут алкильные заместители, или замещают, например, 1, 2, 3 или 4 одинаковыми или различными (C1-C4)-алкильными заместителями, например, метильными группами и/или этильными группами и/или изопропильными группами и/или трет-бутильными группами, в особенности метильными группами, где заместители могут присутствовать в любых положениях. Примерами алкил-замещенных циклоалкильных групп являются 1-метилциклопропил, 2,2-диметилциклопропил, 1-метилциклопентил, 2,3-диметилциклопентил, 1-метилциклогексил, 4-метилциклогексил, 4-изопропилциклогексил, 4-трет-бутилциклогексил и 3,3,5,5-тетраметилциклогексил. Примерами алкил-замещенных циклоалкенильных групп являются 1-метилциклопент-2-енил, 2-метилциклопент-2-енил, 3-метилциклопент-2-енил, 3,4-диметилциклопент-3-енил, 1-метилциклогекс-2-енил, 2-метилциклогекс-2-енил, 3-метилциклогекс-2-енил, 4-метилциклогекс-2-енил, 2-метилциклогекс-3-енил, 3-метилциклогекс-3-енил, 4-метилциклогекс-3-енил, 2,3-диметилциклогекс-2-енил, 4,4-диметилциклогекс-2-енил, 3,4-диметилциклогекс-3-енил. Циклоалкильные группы и циклоалкенильные группы обычно также необязательно замещают одним или более атомами фтора. Т.е. их не замещают, т.е. они не несут атомы фтора, или замещают, например, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10 или 11 атомами фтора, предпочтительно 1, 2, 3, 4, 5 или 6 атомами фтора. Циклоалкильные группы и циклоалкенильные группы также замещают одновременно фтором и алкилом. Атомы фтора могут присутствовать в любых положениях и могут также присутствовать в алкильном заместителе. Примерами фтор-замещенных циклоалкильных групп являются 1-фторциклопропил, 2,2-дифторциклопропил, 3,3-дифторциклобутил, 1-фторциклогексил, 4,4-дифторциклогексил и 3,3,4,4,5,5-гексафторциклогексил. Примерами фтор-замещенных циклоалкенильных групп являются 1-фторциклопент-2-енил, 1-фторциклогекс-2-енил, 4-фторциклогекс-2-енил, 4,4-дифторциклогекс-2-енил. В одном варианте осуществления настоящего изобретения циклоалкильные группы необязательно не замещают заместителями, выбранными из фтора и (C1-C4)-алкила. Если циклоалкильную группу или циклоалкенильную группу можно заместить дополнительными заместителями, подобными гидрокси, как в случае циклоалкильной группы или циклоалкенильной группы, представляющих R30, их можно заместить одним или более данными дополнительными заместителями, подобными только гидрокси, и нельзя заместителями, выбранными из фтора и (C1-C4)-алкила, или одним или более данными дополнительными заместителями и одновременно одним или более заместителями, выбранными из фтора и (C1-C4)-алкила. Число данных дополнительных заместителей, подобных гидрокси, которые могут присутствовать в циклоалкильной или циклоалкенильной группе, предпочтительно равно 1, 2 или 3, более предпочтительно 1 или 2, например, 1. Суммарное количество всех заместителей в циклоалкильной группе или циклоалкенильной группе предпочтительно равно 1, 2, 3, 4, 5, 6, 7 или 8, более предпочтительно 1, 2, 3, 4 или 5, например, 1, 2 или 3. Данные дополнительные заместители, подобные гидрокси, могут присутствовать в любых положениях, при условии, что полученное в результате соединение является достаточно стабильным и является подходящим в качестве подгруппы в фармацевтически активном соединении. Предпочтительно гидрокси заместитель не присутствует в положении 1 циклоалкенильной группы или циклоалкильной группы, представляющих R30, и в циклоалкенильной группе гидрокси заместитель не присутствует при атоме углерода, который является частью двойной связи. Примерами гидрокси-замещенных циклоалкильных групп являются 3-гидроксициклобутил, 2-гидроксициклопентил, 3-гидроксициклопентил, 3,4-дигидроксициклопентил, 2-гидроксициклогексил, 3-гидроксициклогексил, 4-гидроксициклогексил, 2,3-дигидроксициклогексил, 2,4-дигидроксициклогексил, 3,4-дигидроксициклогексил, 3,5-дигидроксициклогексил, 3,4,5- тригидроксициклогексил, 2-гидроксициклогептил, 3-гидроксициклогептил, 4-гидроксициклогептил. Примерами гидрокси-замещенных циклоалкенильных групп являются 5-гидроксициклопент-2-енил, 4-гидроксициклогекс-2-енил, 5-гидроксициклогекс-2-енил, 6-гидроксициклогекс-2-енил, 6-гидроксициклогекс-3-енил. Примерами группы циклоалкилалкил-, которая может присутствовать в группе (C3-C7)-циклоалкил-CvH2v-, являются циклопропилметил-, циклобутилметил-, циклопентилметил-, циклогексилметил-, циклогептилметил-, циклопропилдифторметил-, циклобутилдифторметил-, циклопентилдифторметил-, циклогексилдифторметил-, циклогептилдифторметил-, 1-циклопропилэтил-, 2-циклопропилэтил-, 1-циклобутилэтил-, 2-циклобутилэтил-, 1-циклопентилэтил-, 2-циклопентилэтил-, 1-циклогексилэтил-, 2-циклогексилэтил-, 1-циклогептилэтил-, 2-циклогептилэтил-.
Тетрагидропиранильная группа, представляющая R30, где группу можно также обозначить, как оксанильную группу или тетрагидро-2H-пиранильную группу, может быть присоединена через любой атом углерода и может представлять тетрагидропиран-2-ил, тетрагидропиран-3-ил или тетрагидропиран-4-ил. Предпочтительно тетрагидропиранил представляет собой тетрагидропиран-3-ил или тетрагидропиран-4-ил. В одном варианте осуществления настоящего изобретения тетрагидропиранил представляет собой тетрагидропиран-4-ил.
В замещенных фенильных группах заместители могут присутствовать в любых положениях. В монозамещенных фенильных группах заместитель может присутствовать во 2-положении, в 3-положении или в 4-положении. В дизамещенных фенильных группах заместители могут присутствовать в 2,3-положении, 2,4-положении, 2,5-положении, 2,6-положении, 3,4-положении или 3,5-положении. В тризамещенных фенильных группах заместители могут присутствовать в 2,3,4-положении, 2,3,5-положении, 2,3,6-положении, 2,4,5-положении, 2,4,6-положении или 3,4,5-положении. Если фенильная группа несет четыре заместителя, из которых один, два, три или четыре заместителя, например, могут быть атомами фтора, незамещенный кольцевой атом углерода может присутствовать во 2-положении, в 3-положении или в 4-положении. Если полизамещенная фенильная группа или гетероарильная группа несет различные заместители, каждый заместитель может присутствовать в любом подходящем положении, и настоящее изобретение включает все изомеры положения. Число заместителей в замещенной фенильной группе может составлять 1, 2, 3, 4 или 5. Предпочтительно замещенная фенильная группа, и аналогично замещенная гетероарильная группа, несет 1, 2 или 3, в особенности 1 или 2, одинаковых или различных заместителя. В предпочтительных вариантах осуществления настоящего изобретения заместители в замещенных фенильных и гетероарильных группах выбирают из любой подгруппы заместителей, перечисленных в соответствующем определении, например, из заместителей, выбранных из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O- и (C1-C4)-алкил-S(O)m-, или из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O- и циано, или из галогена, (C1-C4)-алкила и (C1-C4)-алкил-O-, в случае фенильной группы или гетероарильной группы, представляющих R20, где все алкильные группы можно не замещать или замещать одним или более атомами фтора и, в качестве примера заместителей, содержащих фтор-замещенный алкил, заместители, содержащие группу CF3 (трифторметил), такие как сама CF3, CF3-O- или CF3-S-, могут быть включены в каждый список заместителей в добавление к заместителям, содержащим незамещенный алкил.
В гетероарильной группе, которая является остатком ароматической моноциклической, 5-членной или 6-членной гетероциклической кольцевой системы, кольцевые гетероатомы, приведенные в определении данной группы, могут присутствовать в любой комбинации и могут присутствовать в любом подходящем положении, при условии, что данная группа соответствует ее определению и полученное в результате соединение формулы I является стабильным и подходящим в качестве фармацевтически активного соединения. Один из кольцевых атомов азота, специально упоминаемый в определении гетероарильной группы, который может нести атом водорода или заместитель, такой как алкил, представляет собой кольцевой атом азота в 5-членной кольцевой системе, такой как пиррол, пиразол, имидазол или триазол, к которому присоединен экзоциклический атом или группа. Примерами кольцевых систем, из которых получают гетероарильную группу, являются пиррол, фуран, тиофен, имидазол, пиразол, триазолы, такие как [1,2,3]триазол и [1,2,4]триазол, оксазол ([1,3]оксазол), изоксазол ([1,2]оксазол), тиазол ([1,3]тиазол), изотиазол ([1,2]тиазол), оксадиазолы, такие как [1,2,4]оксадиазол, [1,3,4]оксадиазол и [1,2,5]оксадиазол, тиадиазолы, такие как [1,3,4]тиадиазол, пиридин, пиридазин, пиримидин, пиразин, триазины, такие как [1,2,3]триазин, [1,2,4]триазин и [1,3,5]триазин. В одном варианте осуществления настоящего изобретения гетероарильная группа содержит один или два одинаковых или различных кольцевых гетероатома, в другом варианте осуществления настоящего изобретения гетероарил содержит один кольцевой гетероатом, который определяют, как показано. В другом варианте осуществления гетероарил выбирают из тиофенила, тиазолила и пиридинила. В другом варианте осуществления гетероарил выбирают из тиофенила и пиридинила. В другом варианте осуществления гетероарил представляет собой тиофенил. Гетероарильные группы можно присоединить через любой кольцевой атом углерода. Например, тиофенильная группа (тиенильная группа) может представлять собой тиофен-2-ил (2-тиенил) или тиофен-3-ил (3-тиенил), фуранил может представлять собой фуран-2-ил или фуран-3-ил, пиридинил (пиридил) может представлять собой пиридин-2-ил, пиридин-3-ил или пиридин-4-ил, пиразолил может представлять собой 1H-пиразол-3-ил, 1H-пиразол-4-ил или 2H-пиразол-3-ил, имидазолил может представлять собой 1H-имидазол-2-ил, 1H-имидазол-4-ил или 3H-имидазолил-4-ил, тиазолил может представлять собой тиазол-2-ил, тиазол-4-ил или тиазол-5-ил, [1,2,4]триазолил может представлять собой 1H-[1,2,4]триазол-3-ил, 2H-[1,2,4]триазол-3-ил или 4H-[1,2,4]триазол-3-ил.
В замещенных гетероарильных группах заместители могут присутствовать в любых положениях, например, в тиофен-2-ильной группе или фуран-2-ильной группе в 3-положении и/или в 4-положении и/или в 5-положении, в тиофен-3-ильной группе или фуран-3-ильной группе во 2-положении и/или в 4-положении и/или в 5-положении, в пиридин-2-ильной группе в 3-положении и/или в 4-положении и/или в 5-положении и/или в 6-положении, в пиридин-3-ильной группе во 2-положении и/или в 4-положении и/или в 5-положении и/или в 6-положении, в пиридин-4-ильной группе во 2-положении и/или в 3-положении и/или в 5-положении и/или в 6-положении. Предпочтительно замещенную гетероарильную группу замещают одним, двумя или тремя, в особенности одним или двумя, например, одним, одинаковыми или различными заместителями. Если присутствует кольцевой атом азота, который может нести атом водорода или заместитель, заместитель при данном атоме азота может представлять собой, например, метильную группу, этильную группу, пропильную группу или трет-бутильную группу, причем группы можно также монозаместить или полизаместить фтором. Обычно подходящие кольцевые атомы азота в ароматическом кольце гетероарильной группы, например, атом азота в пиридинильной группе или атом азота в [1,2,5]оксадиазолильной группе, и кольцевой атом азота в 6-членном кольце азаиндольной группы может также нести оксидо заместитель -O-, и соединения формулы I, таким образом, могут присутствовать в форме N-оксида.
Галоген представляет собой фтор, хлор, бром или йод, предпочтительно фтор, хлор или бром, в особенности фтор или хлор.
Настоящее изобретение включает все стереоизомерные формы соединений формулы I, например, все возможные энантиомеры и диастереомеры, включая цис/транс изомеры. Настоящее изобретение также включает смеси двух или более стереоизомерных форм, например, смеси энантиомеров и/или диастереомеров, включая цис/транс изомеры, в любых соотношениях. Асимметрические центры, содержащиеся в соединениях формулы I, например, в незамещенных или замещенных алкильных группах или в диазациклоалкановом кольце, изображенном в формуле I, могут все независимо друг от друга иметь S-конфигурацию или R-конфигурацию. Настоящее изобретение относится к энантиомерам, и к левовращающему и правовращающему антиподу, в энантиомерно чистой форме и практически энантиомерно чистой форме и в форме рацематов и в форме смесей двух энантиомеров во всех соотношениях. Настоящее изобретение также относится к диастереомерам в форме чистых и практически чистых диастереомеров и в форме смесей двух или более диастереомеров во всех соотношениях. Настоящее изобретение также включает все цис/транс изомеры соединений формулы I в чистой форме и практически чистой форме и в форме смесей цис изомера и транс изомера во всех соотношениях. Цис/транс изомеризм может возникать, например, в замещенных циклоалкановых кольцах и в диазациклоалкановом кольце, изображенном в формуле I. Получение отдельных стереоизомеров, при желании, может осуществляться разделением смеси согласно общепринятым способам, например, хроматографией или кристаллизацией, или применением стереохимически однородных исходных соединений в синтезе или стереоселективными реакциями. Необязательно, перед разделением стереоизомеров можно осуществлять дериватизацию. Разделение смеси стереоизомеров можно осуществлять на стадии соединения формулы I или на стадии промежуточного соединения в процессе синтеза. Настоящее изобретение также включает все таутомерные формы соединений формулы I.
Физиологически приемлемые соли соединений формулы I представляют собой в особенности соли с нетоксичными солевыми компонентами, и предпочтительно они являются фармацевтически пригодными к применению солями. Они могут содержать неорганические или органические солевые компоненты. Данные соли можно получить, например, из соединений формулы I, которые содержат кислотную группу, например, карбоксильную группу (HO-CO-) или сульфоновую группу (HO-S(O)2-), и нетоксичного неорганического или органического основания. Подходящие основания представляют собой, например, соединения щелочных металлов или соединения щелочноземельных металлов, такие как гидроксид натрия, гидроксид калия, карбонат натрия или гидрокарбонат натрия, или аммиак, органические аминосоединения и гидроксиды четвертичного аммония. Реакции соединений формулы I с основаниями для получения солей обычно осуществляют согласно общепринятым методикам в растворителе или разбавителе. Исходя из физиологической и химической стабильности предпочтительными солями кислых групп являются, во многих случаях, натриевые, калиевые, магниевые или кальциевые соли или аммониевые соли, которые могут также нести одну или более органических групп при атоме азота. Соединения формулы I, которые содержат основную группу, т.е. группу, способную протонироваться, например, аминогруппу, диазациклоалкановую группу, изображенную в формуле I, в случае когда R10 представляет собой водород или другой основный гетероцикл, такой как 6-членное кольцо в азаиндольной группе, могут присутствовать в форме их солей присоединения кислоты с физиологически приемлемыми кислотами, например, в виде соли с хлороводородной кислотой, бромоводородной кислотой, фосфорной кислотой, серной кислотой, уксусной кислотой, бензойной кислотой, метансульфоновой кислотой, п-толуолсульфоновой кислотой, которые обычно получают из соединений формулы I реакцией с кислотой в растворителе или разбавителе согласно общепринятым методиками. Как принято, в особенности в случае солей присоединения кислот соединения, содержащего две или более основных групп, в полученной соли соотношение солевых компонентов может отклоняться в большую или меньшую сторону от стехиометрического соотношения, такого как молярное отношение 1:1 или 1:2, в случае соли присоединения кислоты соединения формулы I, содержащего одну или две основных группы с моновалентной кислотой, и изменяется в зависимости от применяемых условий. Настоящее изобретение включает также соли, содержащие компоненты при нестехиометрическом соотношении, и указание, что соль присоединения кислоты соединения формулы I содержит кислоту в двукратном молярном отношении, например, также применимо к меньшему или большему количеству кислоты в полученной соли, например, приблизительно 1,8 или приблизительно 2,1 моль кислоты на моль соединения формулы I. Если соединения формулы I одновременно содержат кислотную и основную группу в молекуле, настоящее изобретение также включает внутренние соли (бетаины, цвиттерионы) в добавление к упомянутым солевым формам. Настоящее изобретение также включает все соли соединений формулы I, которые, из-за низкой физиологической переносимости, не подходят непосредственно для применения в качестве фармацевтического препарата, но являются пригодными в качестве промежуточных соединений для химических реакций или для получения физиологически приемлемых солей, например, посредством анионного обмена или катионного обмена. Предметом настоящего изобретения также являются сольваты соединений формулы I и их соли, такие как гидраты и аддукты со спиртами, подобными (C1-C4)-алканолам, в особенности к физиологически приемлемым сольватам, а также активным метаболитам соединений формулы I и пролекарствам соединений формулы I, т.е. соединениям, которые in vitro могут необязательно обладать фармакологической активностью, но которые in vivo превращаются в фармакологически активные соединения формулы I, например, соединениям, которые превращаются при метаболическом гидролизе в соединения формулы I. Примерами данных пролекарств являются соединения, в которых атом азота, который можно ацилировать, например, атом азота, несущий группу R10 в диазациклоалкановой группе, изображенной в формуле I, в случае когда R10 представляет собой водород, несет алкил-O-CO-группу или ацильную группу, такую как алкил-CO-группа, например, и, таким образом, превращен в карбаматную группу или амидную группу, или соединения, в которых карбоксильная группа этерифицирована.
Группу A предпочтительно выбирают из O, S, NCH3 и C(Ra)2, более предпочтительно из O, S и C(Ra)2, особенно предпочтительно из O и C(Ra)2. В одном варианте осуществления настоящего изобретения группу A выбирают из O и S. В другом варианте осуществления настоящего изобретения группа A представляет собой O, в другом варианте осуществления группа A представляет собой C(Ra)2.
Если две группы Ra вместе представляют собой дивалентную (C2-C8)-алкильную группу, упомянутую алкильную группу предпочтительно соединяют с атомом углерода, несущим группы Ra через два различных атома углерода, и она образует вместе с атомом углерода, несущим группы Ra, циклоалкановое кольцо, с которым связано азаиндольное кольцо, изображенное в формуле I, и группа R20 связана в том же положении в кольце. Упомянутое циклоалкановое кольцо, подобное циклоалкановому кольцу в соединениях формулы I в целом, может нести одну или более (C1-C4)-алкильных групп, например одну, две, три или четыре метильных группы, и/или один или более, например, один, два, три или четыре атома фтора. Предпочтительно упомянутое циклоалкановое кольцо представляет собой циклопропановое, циклобутановое, циклопентановое или циклогексановое кольцо, которые все можно не замещать или замещать алкилом и/или фтором, как показано. В одном варианте осуществления настоящего изобретения упомянутое циклоалкановое кольцо представляет собой циклопропановое кольцо, которое можно не замещать или замещать алкилом и/или фтором, как показано, т.е. в данном варианте осуществления дивалентная (C2-C8)-алкильная группа представляет собой этан-1,2-диильную группу (1,2-этиленовую группу), которую не замещают или замещают алкилом и/или фтором, как показано. Предпочтительно дивалентная (C2-C8)-алкильная группа представляет собой (C2-C5)-алкильную группу, более предпочтительно (C2-C4)-алкильную группу, например, C2-алкильную группу. В одном варианте осуществления настоящего изобретения группы Ra выбирают из водорода и фтора, в другом варианте осуществления из водорода и (C1-C4)-алкила, где две группы Ra являются независимыми друг от друга и могут быть одинаковыми или различными, или во всех данных вариантах осуществления две группы Ra вместе представляют собой дивалентную (C2-C8)-алкильную группу. В одном варианте осуществления настоящего изобретения группы Ra являются одинаковыми или различными группами, выбранными из водорода и фтора, в другом варианте осуществления они являются одинаковыми или различными группами, выбранными из водорода и (C1-C4)-алкила. В другом варианте осуществления настоящего изобретения группы Ra являются одинаковыми и их выбирают из водорода, фтора и (C1-C4)-алкила, или две группы Ra вместе представляют собой дивалентную (C2-C8)-алкильную группу. В другом варианте осуществления настоящего изобретения обе группы Ra представляют собой водород, или две группы Ra вместе представляют собой дивалентную (C2-C8)-алкильную группу. В следующем варианте осуществления настоящего изобретения, обе группы Ra представляют собой водород, т.е. группа C(Ra)2, представляющая группу A, представляет собой группу CH2. (C1-C4)-алкильная группа, представляющая Ra, предпочтительно представляет собой метил.
В диазациклоалкановой группе, изображенной в формуле I, предпочтительно одну, две, три или четыре, более предпочтительно одну, две или три, особенно предпочтительно одну или две, например, одну, группы R, которые являются независимыми друг от друга и могут быть одинаковыми или различными, определяют, как показано выше или ниже, и выбирают из всех значений, включенных определением, включая водород, и все другие группы R представляют собой водород. В одном варианте осуществления настоящего изобретения все группы R представляют собой водород, и диазациклоалкановая группа, изображенная в формуле I, представляет собой пиперазиновое кольцо, гомопиперазиновое кольцо или 1,5-диазокановое кольцо, в особенности пиперазиновое кольцо, которое несет группу R10, но которое не замещают заместителями по кольцевым атомам углерода. Группы R, которые являются отличными от водорода, могут присутствовать в любых положениях диазациклоалкановой группы при условии, что полученное в результате соединение формулы I является стабильным и пригодным в качестве подгруппы в фармацевтически активном соединении. В одном варианте осуществления настоящего изобретения (C1-C4)-алкил-O-группы, представляющие R, не связаны с атомами углерода в диазациклоалкановом кольце, изображенном в формуле I, которые являются соседними кольцевому атому азота. Предпочтительно только одна или две, например, только одна, из групп R представляет собой (C1-C4)-алкил-O-.
В одном варианте осуществления настоящего изобретения группы R выбирают из водорода, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, фенил-(C1-C4)-алкила-, (C1-C4)-алкил-O-CO-CuH2u- и R1-NH-CO-CuH2u-, в другом варианте осуществления из водорода, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила-, фенил-(C1-C4)-алкила- и R1-NH-CO-CuH2u-, в другом варианте осуществления из водорода, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила- и R1-NH-CO-CuH2u-, в другом варианте осуществления из водорода, (C1-C4)-алкила и гидрокси-(C1-C4)-алкила-, в другом варианте осуществления из водорода, (C1-C4)-алкила и R1-NH-CO-CuH2U-, в другом варианте осуществления из водорода и (C1-C4)-алкила, в другом варианте осуществления из водорода и R1-NH-CO-CuH2u-, где все группы R являются независимыми друг от друга и могут быть одинаковыми или различными, и фенил необязательно замещают, как показано. В одном варианте осуществления настоящего изобретения одну из групп R выбирают из (C1-C4)-алкил-O-CO-CuH2u- и R1-NH-CO-CuH2u-, и в особенности она представляет собой R1-NH-CO-CuH2U-, и все другие группы R представляют собой водород. Группы R, которые являются отличными от водорода, могут быть соединены с любыми кольцевыми атомами углерода в диазациклоалкановом кольце, изображенном в формуле I. В случае когда присутствуют две или более группы R, которые являются отличными от водорода, кольцевой атом углерода может нести или одну или две данных группы R, которые являются отличными от водорода. В случае когда диазациклоалкановое кольцо, изображенное в формуле I, представляет собой пиперазиновое кольцо, несущее одну группу R, которая является отличной от водорода, данная группа R может присутствовать во 2-положении или в 3-положении относительно кольцевого атома азота, который связан с CO группой, изображенной в формуле I. В случае когда диазациклоалкановое кольцо, изображенном в формуле I, представляет собой пиперазиновое кольцо, несущее две группы R, которые являются отличными от водорода, обе данные группы R могут присутствовать во 2-положении, или они обе могут присутствовать в 3-положении, или они обе могут присутствовать в положениях 2 и 3, или в положениях 2 и 5, или в положениях 2 и 6, или в положениях 3 и 5, относительно кольцевого атома азота, который связан с CO группой, изображенной в формуле I, где в случае двух различных групп R каждая из них может присутствовать в каждом положении. В одном варианте осуществления настоящего изобретения значение u выбирают из 0 и 1, в другом варианте осуществления u выбирают из 1 и 2, в другом варианте осуществления u равно 0, в другом варианте осуществления u равно 1, в другом варианте осуществления u равно 2, где все значения u являются независимыми друг от друга и могут быть одинаковыми или различными.
В одном варианте осуществления настоящего изобретения R1 выбирают из (C1-C4)-алкила, гидрокси-(C1-C4)-алкила- и H2N-CO-(C1-C4)-алкила-, в другом варианте осуществления из (C1-C4)-алкила и гидрокси-(C1-C4)-алкила-, в другом варианте осуществления из (C1-C4)-алкила и H2N-CO-(C1-C4)-алкила. В одном варианте осуществления настоящего изобретения R1 представляет собой водород, в другом варианте осуществления R1 представляет собой (C1-C4)-алкил, в другом варианте осуществления R1 представляет собой гидрокси-(C1-C4)-алкил-, в другом варианте осуществления R1 представляет собой H2N-CO-(C1-C4)-алкил-.
R10 предпочтительно выбирают из водорода и (C1-C6)-алкил-O-CO-, более предпочтительно из водорода и (C1-C4)-алкил-O-CO-. В одном варианте осуществления настоящего изобретения R10 представляет собой водород.
В одном варианте осуществления настоящего изобретения R20 выбирают из фенила и гетероарила, где гетероарил выбирают из тиофенила, тиазолила и пиридинила, в другом варианте осуществления из фенила и гетероарила, где гетероарил представляет собой тиофенил, все из которых необязательно замещают, как показано. В другом варианте осуществления настоящего изобретения R20 представляет собой фенил, который необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, гидрокси и циано. Предпочтительно число заместителей в замещенной группе R20 равно одному, двум, трем или четырем, более предпочтительно одному, двум или трем, например, одному или двум. Заместители в замещенной группе R20 могут присутствовать при атомах углерода в любых положениях, как показано выше, относительно замещенных фенильных и гетероарильных групп в общем. Таким образом, например, в случае монозамещенной фенильной группы, представляющей R20, заместитель может присутствовать во 2-положении, в 3-положении или в 4-положении, и в случае дизамещенной фенильной группы заместители могут присутствовать в положениях 2 и 3, или положениях 2 и 4, или положениях 2 и 5, или положениях 2 и 6, или положениях 3 и 4, или положениях 3 и 5. Также тризамещенная фенильная группа, представляющая R20, может нести заместители в любых положениях и может быть группой, такой как 3-хлор-2,6-диметилфенил, 3-фтор-2,6-диметилфенил, 6-хлор-3-фтор-2-метилфенил или 2-хлор-3-фтор-6-метилфенил, например, в случае фенильной группы, тризамещенной фтором и/или хлором и метилом. Заместители, которые могут присутствовать в группе R20, предпочтительно выбирают из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m- и циано, более предпочтительно из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O- и (C1-C4)-алкил-S(O)m-, особенно предпочтительно из галогена, (C1-C4)-алкила и (C1-C4)-алкил-O-, более предпочтительно из галогена и (C1-C4)-алкила, например, из хлора, фтора и метила, где в одном варианте осуществления настоящего изобретения алкильную группу в заместителях в группе R20 можно не замещать или замещать одним или более атомами фтора и, в качестве примера заместителей, содержащих фтор-замещенный алкил, заместители, содержащие трифторметильную группу, такую как сама CF3, CF3-O- или CF3-S-, можно включить в каждый список заместителей в добавление к заместителям, содержащим незамещенный алкил, и в другом варианте осуществления настоящего изобретения алкильные группы в заместителях в группе R20 не замещают фтором и в данном последнем варианте осуществления упомянутый алкил, таким образом, обозначает незамещенный алкил. Конкретные группы в добавление к вышеупомянутым конкретным группам, которые могут представлять группу R20 и из которых, или из любой подгруппы которых, можно выбрать R20 в соединениях формулы I, включают, например, фенил, т.е. незамещенный фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 2-метилфенил (о-толил), 3-метилфенил (м-толил), 4-метилфенил (п-толил), 2-этилфенил, 3-этилфенил, 4-этилфенил, 2-метоксифенил, 3-метоксифенил, 4-метоксифенил, 2,3-дифторфенил, 2,4-дифторфенил, 2,5-дифторфенил, 2,6-дифторфенил, 3,4-дифторфенил, 3,5-дифторфенил, 2,3-дихлорфенил, 2,4-дихлорфенил, 2,5-дихлорфенил, 2,6-дихлорфенил, 3,4-дихлорфенил, 3,5-дихлорфенил, 2-хлор-3-фторфенил, 2-хлор-4-фторфенил, 2-хлор-5-фторфенил, 2-хлор-6-фторфенил, 3-хлор-2-фторфенил, 3-хлор-4-фторфенил, 3-хлор-5-фторфенил, 4-хлор-2-фторфенил, 4-хлор-3-фторфенил, 5-хлор-2-фторфенил, 2,3-диметилфенил, 2,4-диметилфенил, 2,5-диметилфенил, 2,6-диметилфенил, 3,4-диметилфенил, 3,5-диметилфенил, 2-фтор-3-метилфенил, 2-фтор-4-метилфенил, 2-фтор-5-метилфенил, 2-фтор-6-метилфенил, 3-фтор-2-метилфенил, 3-фтор-4-метилфенил, 3-фтор-5-метилфенил, 4-фтор-2-метилфенил, 4-фтор-3-метилфенил, 5-фтор-2-метилфенил, 2-хлор-3-метилфенил, 2-хлор-4-метилфенил, 2-хлор-5-метилфенил, 2-хлор-6-метилфенил, 3-хлор-2-метилфенил, 3-хлор-4-метилфенил, 3-хлор-5-метилфенил, 4-хлор-2-метилфенил, 4-хлор-3-метилфенил, 5-хлор-2-метилфенил, 2-метокси-3-метилфенил, 2-метокси-4-метилфенил, 2-метокси-5-метилфенил, 2-метокси-6-метилфенил, 3-метокси-2-метилфенил, 3-метокси-4-метилфенил, 3-метокси-5-метилфенил, 4-метокси-2-метилфенил, 4-метокси-3-метилфенил, 5-метокси-2-метилфенил.
В одном варианте осуществления настоящего изобретения R30 выбирают из (C3-C7)-циклоалкила, (C5-C7)-циклоалкенила, тетрагидропиранила и фенила, в другом варианте осуществления из (C3-C7)-циклоалкила, (C5-C7)-циклоалкенила и фенила, в другом варианте осуществления из (C3-C7)-циклоалкила, (C3-C7)-циклоалкенила и тетрагидропиранила, в другом варианте осуществления из (C3-C7)-циклоалкила, (C5-C7)-циклоалкенила, фенила и гетероарила, в другом варианте осуществления из (C3-C7)-циклоалкила, фенила и гетероарила, в другом варианте осуществления из (C3-C7)-циклоалкила и (C5-C7)-циклоалкенила, в другом варианте осуществления из (C3-C7)-циклоалкила и фенила, где все циклоалкильные, циклоалкенильные, фенильные и гетероарильные группы необязательно замещают, как показано, и циклоалкил предпочтительно представляет собой (C5-C7)-циклоалкил, более предпочтительно (C5-C6)-циклоалкил, например, циклогексил, циклоалкенил предпочтительно представляет собой (C5-C6)-циклоалкенил, например, циклогексенил, и гетероарил предпочтительно выбирают из тиофенила и пиридинила и более предпочтительно представляет собой тиофенил. В другом варианте осуществления настоящего изобретения R30 представляет собой фенил, который необязательно замещают, как показано. Предпочтительно число заместителей в замещенной группе R30 равно одному, двум, трем или четырем, более предпочтительно одному, двум или трем, особенно предпочтительно одному или двум, например, одному. Заместители в замещенной группе R30 могут присутствовать при атомах углерода в любых положениях, как показано выше, относительно замещенных циклоалкильных, циклоалкенильных, фенильных и гетероарильных групп в общем. Например, в случае монозамещенной фенильной группы, представляющей R30, заместитель может присутствовать во 2-положении, в 3-положении или в 4-положении, и в случае дизамещенной фенильной группы заместители могут присутствовать в положениях 2 и 3, или положениях 2 и 4, или положениях 2 и 5, или положениях 2 и 6, или положениях 3 и 4, или положениях 3 и 5. Заместители, которые могут присутствовать в циклоалкильной или циклоалкенильной группе, представляющей R30, предпочтительно выбирают из фтора, метила и гидрокси, например, из фтора и метила. В одном варианте осуществления настоящего изобретения заместители в циклоалкильной или циклоалкенильной группе, представляющей R30, представляют собой гидрокси. В другом варианте осуществления настоящего изобретения циклоалкильную или циклоалкенильную группу, представляющую R30, не замещают. Заместители, которые могут присутствовать в фенильной или гетероарильной группе, представляющей R30, предпочтительно выбирают из галогена, (C1-C6)-алкила, гидрокси-(C1-C6)-алкила-, (C1-C4)-алкил-O-(C1-C6)-алкила-, (C1-C4)-алкил-CO-NH-(C1-C4)-алкила-, гидрокси, (C1-C6)-алкил-O-, гидрокси-(C1-C6)-алкил-O-, (C1-C4)-алкил-O-(C1-C6)-алкил-O-, (C1-C4)-алкил-CO-NH-(C1-C4)-алкил-O-, (C1-C6)-алкил-S(O)m- и циано, более предпочтительно из галогена, (C1-C6)-алкила, (C1-C4)-алкил-O-(C1-C6)-алкила-, гидрокси, (C1-C6)-алкил-O-, (C1-C4)-алкил-O-(C1-C6)-алкил-O-, (C1-C6)-алкил-S(O)m- и циано, особенно предпочтительно из галогена, (C1-C6)-алкила, (C1-C4)-алкил-O-(C1-C6)-алкила-, гидрокси, (C1-C6)-алкил-O- и (C1-C4)-алкил-O-(C1-C6)-алкил-O-, более предпочтительно из галогена, (C1-C6)-алкила, гидрокси, (C1-C6)-алкил-O- и (C1-C4)-алкил-O-(C1-C6)-алкил-O-, особенно предпочтительно из галогена, (C1-C6)-алкила, (C1-C6)-алкил-O- и (C1-C4)-алкил-O-(C1-C6)-алкил-O-, например, из галогена, (C1-C6)-алкил-O- и (C1-C4)-алкил-O-(C1-C6)-алкил-O- или из галогена, (C1-C6)-алкила и (C1-C6)-алкил-O- или из галогена и (C1-C4)-алкила, где в одном варианте осуществления настоящего изобретения алкильные группы в заместителях в фенильных и гетероарильных группах, представляющих R30 , можно не замещать или замещать одним или более атомами фтора, и, в качестве примера заместителей, содержащих фтор-замещенный алкил, заместители, содержащие трифторметильную группу, такую как сама CF3, CF3-O- или CF3-S-, можно включать в каждый список заместителей в добавление к заместителям, содержащим незамещенный алкил, и в другом варианте осуществления настоящего изобретения алкильные группы в заместителях в группе R30 не замещают фтором, и в последнем варианте осуществления упомянутый алкил, таким образом, обозначает незамещенный алкил. В одном варианте осуществления настоящего изобретения (C1-C6)-алкильная группа в заместителе в R30 представляет собой (C1-C4)-алкильную группу. В одном варианте осуществления настоящего изобретения заместители, которые присутствуют в фенильной или гетероарильной группе, представляющей R30, выбирают из галогена, предпочтительно из фтора, хлора и брома, более предпочтительно из фтора и хлора. Конкретные группы, которые могут присутствовать в качестве группы R30 и из которых, или из любой подгруппы которых, можно выбрать R30 в соединениях формулы I, включают, например, циклопентил, циклогексил, циклогептил, циклопент-2-енил, циклогекс-2-енил, циклогепт-2-енил, 4-фтор-циклогексил, 4-метилциклогексил, 2-гидроксициклопентил, 3-гидроксициклопентил, 2-гидроксициклогексил, 3-гидроксициклогексил, 4-гидроксициклогексил, 2-гидроксициклогептил, 3-гидроксициклогептил, 4-гидроксициклогептил, 4,4-дифторциклогексил, 3,3-диметилциклогексил, 4,4-диметилциклогексил, тетрагидропиран-3-ил, тетрагидропиран-4-ил, фенил, т.е. незамещенный фенил, 2-фторфенил, 3-фторфенил, 4-фторфенил, 2-хлорфенил, 3-хлорфенил, 4-хлорфенил, 3-бромфенил, 4-бромфенил, 2-метилфенил, 3-метилфенил, 4-метилфенил, 2-гидроксифенил, 3-гидроксифенил, 4-гидроксифенил, 2-метоксифенил, 3-метоксифенил, 4-метоксифенил, 2-(2-метоксиэтокси)фенил, 3-(2-метоксиэтокси)фенил, 4-(2-метоксиэтокси)фенил, 2-(3-метоксипропокси)фенил, 3-(3-метоксипропокси)фенил, 4-(3-метоксипропокси)фенил, тиофен-2-ил, тиофен-3-ил, пиридин-2-ил, пиридин-3-ил, пиридин-4-ил, 2-гидроксипиридин-3-ил, 4-гидроксипиридин-3-ил, 5-гидроксипиридин-3-ил, 6-гидроксипиридин-3-ил, 2-метоксипиридин-3-ил, 4-метоксипиридин-3-ил, 5-метоксипиридин-3-ил, 6-метоксипиридин-3-ил, 2-гидроксипиридин-4-ил, 3-гидроксипиридин-4-ил, 2-метоксипиридин-4-ил, 3-метоксипиридин-4-ил.
Заместители R40 могут присутствовать при кольцевых атомах углерода в любом из положений 4 и/или 5 и/или 6 и/или 7 в 6-членном кольце азаиндольной группы, изображенной в формуле I, при условии, что кольцевой атом в соответствующем положении представляет собой атом углерода. В случае когда значение n заместителей R40 является меньшим, чем 3, все атомы углерода в положениях 4, 5, 6 и 7 азаиндольного кольца, которые не несут заместитель R40, несут атом водорода, т.е. соответствующие группы Y1, Y2, Y3 и Y4 представляют собой CH группы. В случае когда значение n равно 0, все кольцевые атомы углерода в положениях 4, 5, 6 и 7 азаиндольного кольца несут атомы водорода. Предпочтительно значение n заместителей R40 равно 0, 1 или 2, более предпочтительно 0 или 1. В одном варианте осуществления настоящего изобретения значение n равно 1. В другом варианте осуществления значение n равно 0, т.е. заместитель R40 не присутствует в соединении формулы I. R40 предпочтительно выбирают из галогена, (C1-C4)-алкила, фенил-(C1-C4)-алкила-, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C1-C4)-алкил-NH-CO-O-, (C1-C4)-алкил-S(O)m-, HO-CO-, (C1-C4)-алкил-O-CO-, H2N-CO- и циано, более предпочтительно из галогена, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C1-C4)-алкил-NH-CO-O-, (C1-C4)-алкил-S(O)m-, HO-CO-, (C1-C4)-алкил-O-CO-, H2N-CO- и циано, особенно предпочтительно из галогена, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C1-C4)-алкил-NH-CO-O-, HO-CO-, (C1-C4)-алкил-O-CO- и H2N-CO-, более предпочтительно из галогена, (C1-C4)-алкила, гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C1-C4)-алкил-NH-CO-O-, (C1-C4)-алкил-O-CO- и H2N-CO-, особенно предпочтительно из галогена, (C1-C4)-алкила, гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO- и H2N-CO-, где все заместители R40 являются независимыми друг от друга и могут быть одинаковыми или различными, и где все фенильные группы независимо друг от друга необязательно замещают, как показано. В одном варианте осуществления настоящего изобретения R40 выбирают из галогена, (C1-C4)-алкила, гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C1-C4)-алкил-NH-CO-O-, (C1-C4)-алкил-O-CO-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O- и H2N-CO-, предпочтительно из галогена, (C1-C4)-алкила, гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO- и H2N-CO-, где все заместители R40 являются независимыми друг от друга и могут быть одинаковыми или различными. В другом варианте осуществления настоящего изобретения R40 выбирают из галогена, (C1-C4)-алкила, фенил-(C1-C4)-алкила-, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C1-C4)-алкил-NH-CO-O- и (C1-C4)-алкил-S(O)m-, предпочтительно из галогена, (C1-C4)-алкила, фенил-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O- и (C1-C4)-алкил-S(O)m-, более предпочтительно из галогена, (C1-C4)-алкила, фенил-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O- и HO-CO-(C1-C4)-алкил-O-, особенно предпочтительно из галогена, (C1-C4)-алкила, гидрокси, (C1-C4)-алкил-O- и HO-CO-(C1-C4)-алкил-O-, более предпочтительно из галогена, (C1-C4)-алкила, гидрокси и (C1-C4)-алкил-O-, где все заместители R40 являются независимыми друг от друга и могут быть одинаковыми или различными, и где все фенильные группы независимо друг от друга необязательно замещают, как показано. Предпочтительно не более чем два заместителя R40 представляют собой NO2. В одном варианте осуществления настоящего изобретения значение n выбирают из 1, 2 и 3, предпочтительно из 1 и 2, и оно может быть равно, например, 1. Т.е., в данном последнем варианте осуществления, по меньшей мере один заместитель R40 присутствует в соединениях формулы I, предпочтительно один или два заместителя R40, например, один заместитель R40.
В одном варианте осуществления настоящего изобретения, по меньшей мере один заместитель R40, который может присутствовать в соединениях формулы I, предпочтительно один или два заместителя R40, например, один заместитель R40, представляет собой заместитель, в котором атом в заместителе, через который он связан с атомом углерода в 6-членном кольце азаиндольной группы, представляет собой атом кислорода, т.е. его выбирают из гидрокси, (C1-C4)-алкил-O-, (C3-C7)-циклоалкил-CvH2v-O-, фенил-(C1-C4)-алкил-O-, гетероарил-(C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, (C3-C7)-циклоалкил-CvH2v-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, гетероарил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O-, H2N-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C3-C7)-циклоалкил-CvH2v-CO-O-, (C1-C4)-алкил-NH-CO-O- и (C3-C7)-циклоалкил-CvH2v-NH-CO-O-, где данные заместители являются независимыми друг от друга и могут быть одинаковыми или различными, и где все фенильные и гетероарильные группы можно независимо друг от друга замещать, как показано. Предпочтительно данные заместители выбирают из гидрокси, (C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O- и (C1-C4)-алкил-NH-CO-O-, и более предпочтительно из гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O- и (C1-C4)-алкил-CO-O-, особенно предпочтительно из гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O- и ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, более предпочтительно выбирают из гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O- и фенил-O-(C1-C4)-алкил-O-, особенно предпочтительно из гидрокси, (C1-C4)-алкил-O- и (C1-C4)-алкил-O-(C1-C4)-алкил-O-, более предпочтительно из гидрокси и (C1-C4)-алкокси. В одном варианте осуществления данные заместители выбирают из гидрокси, (C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-O-алкил-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O-, H2N-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O- и (C1-C4)-алкил-NH-CO-O-, предпочтительно из гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O- и H2N-CO-(C1-C4)-алкил-O-, более предпочтительно из гидрокси, (C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O- и H2N-CO-(C1-C4)-алкил-O-, особенно предпочтительно из гидрокси, (C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O- и (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O-, более предпочтительно из гидрокси, (C1-C4)-алкил-O и HO-CO-(C1-C4)-алкил-O-, где данные заместители являются независимыми друг от друга и могут быть одинаковыми или различными. Если помимо данных заместителей, присоединенных через атом кислорода, дополнительные заместители R40 присутствуют в соединении формулы I, их выбирают из всех других значений R40, перечисленных выше, и предпочтительно выбирают из галогена и (C1-C4)-алкила, где все данные дополнительные заместители являются независимыми друг от друга и могут быть одинаковыми или различными. В одном варианте осуществления один данный заместитель R40, присоединенный через атом кислорода, присутствует при кольцевом атоме углерода в положении 5 или при кольцевом атоме углерода в положении 6.
В соединении формулы I, которое содержит один заместитель R40, заместитель может присутствовать при кольцевом атоме углерода в положении 4 или в положении 5 или в положении 6 или в положении 7 азаиндольного кольца. В соединении формулы I, которое содержит два заместителя R40, заместители могут присутствовать при кольцевых атомах углерода в положениях 4 и 5 или положениях 4 и 6, или положениях 4 и 7, или в положениях 5 и 6, или положениях 5 и 7, или положениях 6 и 7 азаиндольного кольца. В одном варианте осуществления настоящего изобретения соединения формулы I содержат ноль, один или два заместителя R40, где заместители R40 присутствуют при кольцевых атомах углерода в положении 4 или положении 5 или в положениях 4 и 5, и другие кольцевые атомы углерода в положениях 4, 5, 6 и 7 несут атомы водорода. В другом варианте осуществления настоящего изобретения соединения формулы I содержат ноль, один или два заместителя R40, где заместители R40 присутствуют при кольцевых атомах углерода в положении 4 или положении 6 или в положениях 4 и 6, и другие кольцевые атомы углерода в положениях 4, 5, 6 и 7 несут атомы водорода. В другом варианте осуществления настоящего изобретения соединения формулы I содержат ноль, один или два заместителя R40, где заместители R40 присутствуют при кольцевых атомах углерода в положении 4 или положении 7 или в положениях 4 и 7, и другие кольцевые атомы углерода в положениях 4, 5, 6 и 7 несут атомы водорода. В другом варианте осуществления настоящего изобретения соединения формулы I содержат ноль, один или два заместителя R40, где заместители R40 присутствуют при кольцевых атомах углерода в положении 5 или положении 6 или в положениях 5 и 6, и другие кольцевые атомы углерода в положениях 4, 5, 6 и 7 несут атомы водорода. В другом варианте осуществления настоящего изобретения соединения формулы I содержат ноль, один или два заместителя R40, где заместители R40 присутствуют при кольцевых атомах углерода в положении 5 или положении 7 или в положениях 5 и 7, и другие кольцевые атомы углерода в положениях 4, 5, 6 и 7 несут атомы водорода. В другом варианте осуществления настоящего изобретения соединения формулы I содержат ноль, один или два заместителя R40, где заместители R40 присутствуют при кольцевых атомах углерода в положении 6 или 7 или в положениях 6 и 7, и другие кольцевые атомы углерода в положениях 4, 5, 6 и 7 несут атомы водорода.
В одном варианте осуществления настоящего изобретения группа Y1 представляет собой атом азота, и группы Y2, Y3 и Y4 являются одинаковыми или различными группами CH или CR40, т.е. соединение формулы I представляет собой 4-азаиндол(1H-пирроло[3,2-b]пиридиновое) производное формулы Ia. В другом варианте осуществления настоящего изобретения группа Y2 представляет собой атом азота, и группы Y1, Y3 и Y4 являются одинаковыми или различными группами CH или CR40, т.е. соединение формулы I представляет собой 5-азаиндол(1H-пирроло[3,2-c]пиридиновое) производное формулы Ib. В другом варианте осуществления настоящего изобретения группа Y3 представляет собой атом азота, и группы Y1, Y2 и Y4 являются одинаковыми или различными группами CH или CR40, т.е. соединение формулы I представляет собой 6-азаиндол(1H-пирроло[2,3-c]пиридиновое) производное формулы Ic. В другом варианте осуществления настоящего изобретения группа Y4 представляет собой атом азота, и группы Y1, Y2 и Y3 являются одинаковыми или различными группами CH или CR40, т.е. соединение формулы I представляет собой 7-азаиндол(1H-пирроло[2,3-b]пиридиновое) производное формулы Id. A, R, R10, R20, R30, R40, n, p и q в формулах Ia, Ib, Ic и Id определяют, как в формуле I.
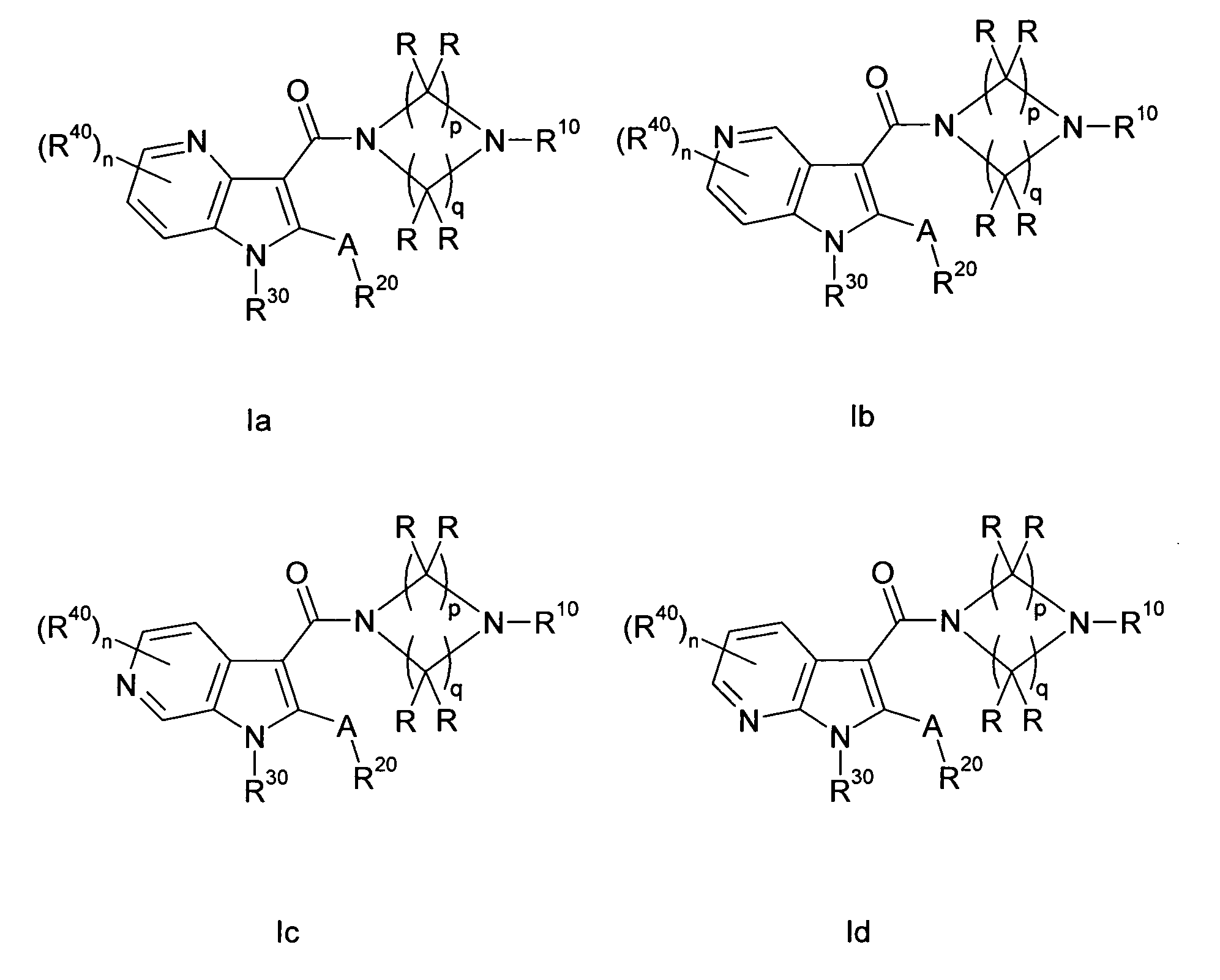
В другом варианте осуществления настоящего изобретения соединение формулы I представляет собой соединение из любых двух или трех формул Ia, Ib, Ic и Id, например, соединение формулы Ia или формулы Id, или соединение формулы Ib или формулы Ic, или соединение формулы Ia или формулы Ic или формулы Id. Иначе, в данных типичных последних трех вариантах осуществления одна из групп Y1 и Y4 в формуле I представляет собой N, и другие из Y1 и Y4, а также Y2 и Y3, являются одинаковыми или различными группами CH или CR40, или одна из групп Y2 и Y3 в формуле I представляет собой N, и другие из Y2 и Y3, а также Y1 и Y4, являются одинаковыми или различными группами CH или CR40, или одна из групп Y1, Y3 и Y4 в формуле I представляет собой N, и другие из Y1, Y3 и Y4, а также Y2, являются одинаковыми или различными группами CH или CR40.
В одном варианте осуществления настоящего изобретения значение p равно 2, и значение q выбирают из 2 и 3. В другом варианте осуществления настоящего изобретения и p и q равно 2, т.е. диазациклоалкановое кольцо, изображенное в формуле I, представляет собой пиперазиновое кольцо, и соединение формулы I представляет собой соединение формулы Ie. В другом варианте осуществления настоящего изобретения p равно 2, и q равно 3, т.е. диазациклоалкановое кольцо, изображенное в формуле I, представляет собой гомопиперазиновое кольцо, и соединение формулы I представляет собой соединение формулы If. В другом варианте осуществления настоящего изобретения и p и q равно 3, т.е. диазациклоалкановое кольцо, изображенное в формуле I, представляет собой 1,5-диазокановое кольцо, и соединение формулы I представляет собой соединение формулы Ig. A, R, R10, R20, R30, R40, Y1, Y2, Y3, Y4 и n в формулах Ie, If и Ig определяют, как в формуле I.
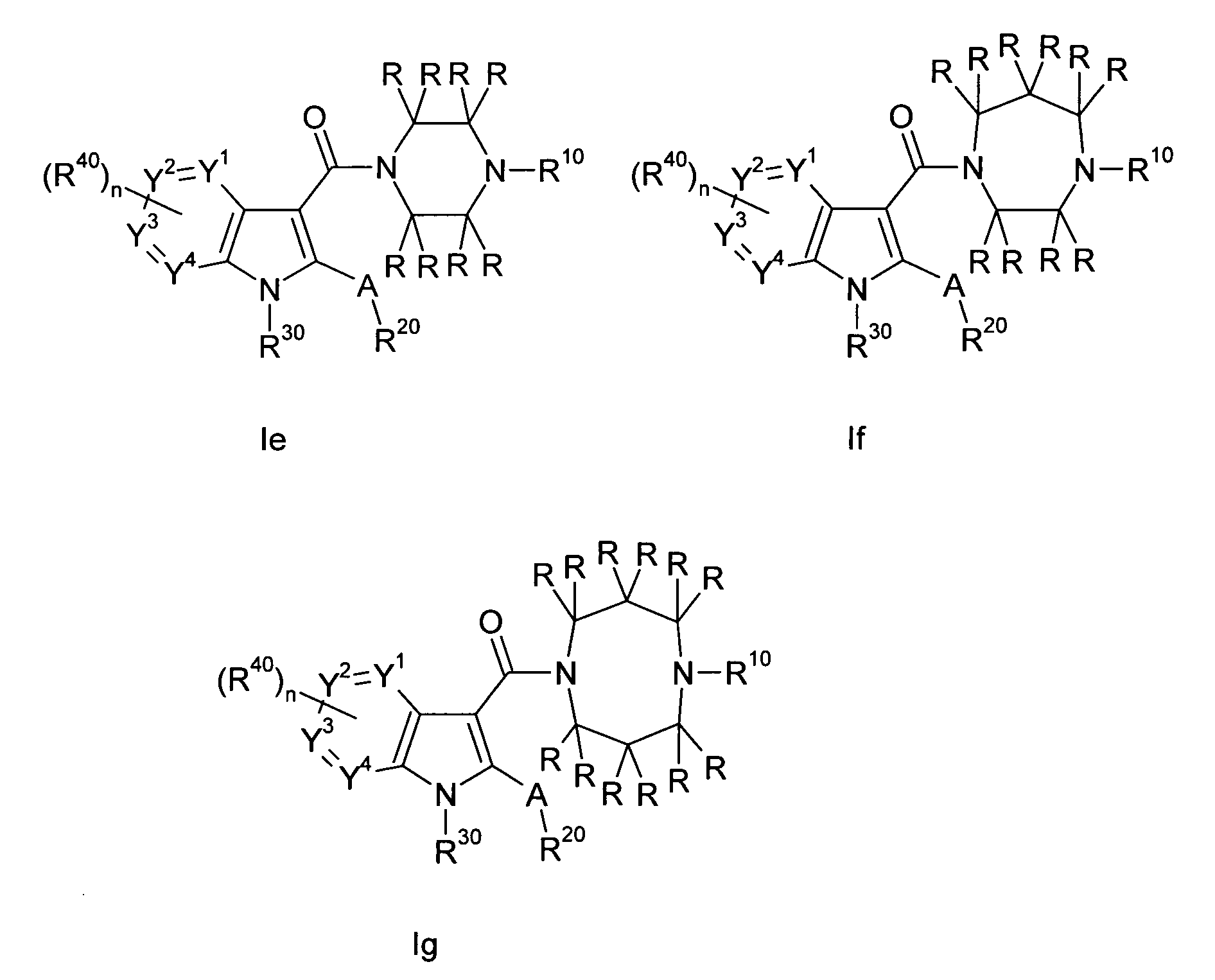
В предпочтительных соединениях настоящего изобретения любой один или более структурных элементов, таких как группы, заместители и значения, определяют, как в любом из предпочтительных определений элементов или в любом упомянутом варианте осуществления, и/или они могут иметь одно или более конкретных значений, которые упоминаются в качестве примеров элементов, где все комбинации одного или более предпочтительных определений и вариантов осуществления и/или конкретных значений являются объектом настоящего изобретения. Также относительно всех предпочтительных соединений формулы I, все их стереоизомерные формы и смеси стереоизомерных форм во всех соотношениях, и их физиологически приемлемые соли, и физиологически приемлемые сольваты любого из них являются объектом настоящего изобретения. Аналогично, также относительно всех конкретных соединений, описанных в настоящем изобретении, таких как примерные соединения, которые представляют варианты осуществления настоящего изобретения, где различные группы и значения в общем определении соединений формулы I имеют конкретные значения, представленные в соответствующем конкретном соединении, все их стереоизомерные формы и смеси стереоизомерных форм во всех соотношениях, и их физиологически приемлемые соли, и физиологически приемлемые сольваты любого их них являются объектом настоящего изобретения. В частности, объектом настоящего изобретения являются все конкретные соединения, описанные в настоящем изобретении, независимо от того, описаны ли они в виде свободного соединения и/или в виде конкретной соли, или в форме свободного соединения, или в форме всех его физиологически приемлемых солей, и если описывают конкретную соль, дополнительно в форме его конкретной соли и его физиологически приемлемых сольватов.
В качестве примера соединений по настоящему изобретению, в которых любой один или более структурных элементов определяют, как в предпочтительных определениях, можно упомянуть соединения формулы I, в которых и p и q равно 2, R10 представляет собой водород и A выбирают из O и C(Ra)2, т.е. соединения формулы Ie, в которых R10 представляет собой водород, и A выбирают из O и C(Ra)2, и все другие группы и значения определяют, как в общем определении соединений формулы I или в любом из предпочтительных определений или вариантов осуществления настоящего изобретения, в любой из их стереоизомерных форм или в виде смеси стереоизомерных форм в любом соотношении, и их физиологически приемлемые соли, и физиологически приемлемые сольваты любого из них.
Другим данным примером являются соединения формулы I, в любой из их стереоизомерных форм или в виде смеси стереоизомерных форм в любом соотношении, и их физиологически приемлемые соли, и физиологически приемлемые сольваты любого из них, в которых
A выбирают из O, S, NCH3 и C(Ra)2;
Ra выбирают из водорода, фтора и метила, где две группы Ra являются независимыми друг от друга и могут быть одинаковыми или различными, или две группы Ra вместе представляют собой дивалентную (C2-C5)-алкильную группу;
R выбирают из водорода, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, фенил-(C1-C4)-алкила-, (C1-C4)-алкил-O-CO-CuH2u- и R1-NH-CO-CuH2u-, где все группы R являются независимыми друг от друга и могут быть одинаковыми или различными;
R1 выбирают из (C1-C4)-алкила, гидрокси-(C1-C4)-алкила- и H2N-CO-(C1-C4)-алкила-;
R10 выбирают из водорода, (C1-C6)-алкил-O-CO- и (C3-C7)-циклоалкил-CvH2v-O-CO-;
R20 выбирают из фенила и гетероарила, которые необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, гидрокси и циано;
R30 выбирают из (C3-C7)-циклоалкила, (C5-C7)-циклоалкенила, тетрагидропиранила, фенила и гетероарила, где циклоалкил и циклоалкенил необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из фтора, (C1-C4)-алкила и гидрокси, и фенил и гетероарил необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C6)-алкила, (C1-C4)-алкил-O-(C1-C6)-алкила-, гидрокси, (C1-C6)-алкил-O-, (C1-C4)-алкил-O-(C1-C6)-алкил-O-, (C1-C6)-алкил-S(O)m- и циано;
R40 выбирают из галогена, (C1-C4)-алкила, фенил-(C1-C4)-алкила-, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, фенил-O-(C1-C4)-алкил-O-, ди((C1-C4)-алкил)N-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C1-C4)-алкил-NH-CO-O-, (C1-C4)-алкил-S(O)m-, HO-CO-, (C1-C4)-алкил-О-CO-, H2N-CO- и циано, где все заместители R40 являются независимыми друг от друга и могут быть одинаковыми или различными;
одна из групп Y1, Y2, Y3 и Y4 представляет собой N, и другие являются одинаковыми или различными группами CH или CR40;
гетероарил выбирают из тиофенила и пиридинила;
m выбирают из 0, 1 и 2, где все значения m являются независимыми друг от друга и могут быть одинаковыми или различными;
n выбирают из 0, 1 и 2;
p равно 2, и q выбирают из 2 и 3;
u выбирают из 0, 1 и 2, где все значения u являются независимыми друг от друга и могут быть одинаковыми или различными;
v выбирают из 0, 1 и 2;
где все алкильные группы, независимо друг от друга, необязательно замещают одним или более атомами фтора;
где циклоалкильную группу необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из фтора и (C1-C4)-алкила, если не указано особо;
где все фенильные группы, присутствующие в R и R40, независимо друг от друга, необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)2- и циано.
Другим данным примером являются соединения формулы I, в любой из их стереоизомерных форм или в виде смеси стереоизомерных форм в любом соотношении, и их физиологически приемлемые соли, и физиологически приемлемые сольваты любого из них, где
A выбирают из O, S и C(Ra)2;
Ra выбирают из водорода, фтора и метила, где две группы Ra являются независимыми друг от друга и могут быть одинаковыми или различными, или две группы Ra вместе представляют собой дивалентную (C2-C5)-алкильную группу;
R выбирают из водорода, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, фенил-(C1-C4)-алкила-, (C1-C4)-алкил-O-CO-CuH2u- и R1-NH-CO-CuH2u-, где все группы R являются независимыми друг от друга и могут быть одинаковыми или различными;
R1 выбирают из (C1-C4)-алкила, гидрокси-(C1-C4)-алкила- и H2N-CO-(C1-C4)-алкила-;
R10 выбирают из водорода, (C1-C6)-алкил-O-CO- и (C3-C7)-циклоалкил-CvH2v-O-CO-;
R20 представляет собой фенил, который необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)m-, гидрокси и циано;
R30 выбирают из (C3-C7)-циклоалкила, (C5-C7)-циклоалкенила и фенила, где циклоалкил и циклоалкенил необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из фтора, (C1-C4)-алкила и гидрокси, и фенил необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C6)-алкила, (C1-C4)-алкил-O-(C1-C6)-алкила-, гидрокси, (C1-C6)-алкил-O-, (C1-C4)-алкил-O-(C1-C6)-алкил-O-, (C1-C6)-алкил-S(O)m- и циано;
R40 выбирают из галогена, (C1-C4)-алкила, фенил-(C1-C4)-алкила-, гидрокси-(C1-C4)-алкила-, (C1-C4)-алкил-O-(C1-C4)-алкила-, гидрокси, (C1-C4)-алкил-O-, гидрокси-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, HO-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO-(C1-C4)-алкил-O-, (C1-C4)-алкил-CO-O-, (C1-C4)-алкил-NH-CO-O- и (C1-C4)-алкил-S(O)m-, где все заместители R40 являются независимыми друг от друга и могут быть одинаковыми или различными;
одна из групп Y1, Y2, Y3 и Y4 представляет собой N, и другие являются одинаковыми или различными группами CH или CR40;
m выбирают из 0, 1 и 2, где все значения m являются независимыми друг от друга и могут быть одинаковыми или различными;
n выбирают из 0, 1 и 2;
p и q равны 2;
u выбирают из 0, 1 и 2, где все значения u являются независимыми друг от друга и могут быть одинаковыми или различными;
v выбирают из 0, 1 и 2;
где все алкильные группы, независимо друг от друга, необязательно замещают одним или более атомами фтора;
где циклоалкильную группу необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из фтора и (C1-C4)-алкила, если не указано особо;
где все фенильные группы, присутствующие в R и R40, независимо друг от друга, необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C4)-алкила, (C1-C4)-алкил-O-, (C1-C4)-алкил-S(O)2- и циано.
Другим данным примером являются соединения формулы I, в любой из их стереоизомерных форм или в виде смеси стереоизомерных форм в любом соотношении, и их физиологически приемлемые соли, и физиологически приемлемые сольваты любого из них, где
A выбирают из O и C(Ra)2;
Ra представляет собой водород;
R выбирают из водорода, (C1-C4)-алкила, гидрокси-(C1-C4)-алкила- и R1-NH-CO-CuH2u-, где все группы R являются независимыми друг от друга и могут быть одинаковыми или различными;
R1 выбирают из (C1-C4)-алкила, гидрокси-(C1-C4)-алкила- и H2N-CO-(C1-C4)-алкила-;
R10 представляет собой водород;
R20 представляет собой фенил, который необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена и (C1-C4)-алкила;
R30 выбирают из (C5-C7)-циклоалкила, (C5-C7)-циклоалкенила и фенила, где фенил необязательно замещают одним или более одинаковыми или различными заместителями, выбранными из галогена, (C1-C6)-алкила, гидрокси, (C1-C6)-алкил-O- и (C1-C4)-алкил-O-(C1-C6)-алкил-O-;
R40 выбирают из галогена, (C1-C4)-алкила, гидрокси, (C1-C4)-алкил-O-, (C1-C4)-алкил-O-(C1-C4)-алкил-O-, (C1-C4)-алкил-O-CO- и H2N-CO-, где все заместители R40 являются независимыми друг от друга и могут быть одинаковыми или различными;
одна из групп Y1, Y2, Y3 и Y4 представляет собой N, и другие являются одинаковыми или различными группами CH или CR40;
n выбирают из 0, 1 и 2;
p и q равны 2;
u выбирают из 0, 1 и 2, где все значения u являются независимыми друг от друга и могут быть одинаковыми или различными;
где все алкильные группы, независимо друг от друга, необязательно замещают одним или более атомами фтора.
Другим объектом настоящего изобретения являются способы получения соединений формулы I, включая их соли и сольваты, которые изложены ниже и с помощью которых можно получить соединения. Например, получение соединений формулы I можно осуществлять первой реакцией азаиндола формулы II по кольцевому атому азота в 5-членном кольце алкилирующим или арилирующим соединением формулы III для того, чтобы получить соединение формулы IV, которое затем превращают в 1,3-дигидроазаиндол-2-он (азаоксиндол) формулы V.
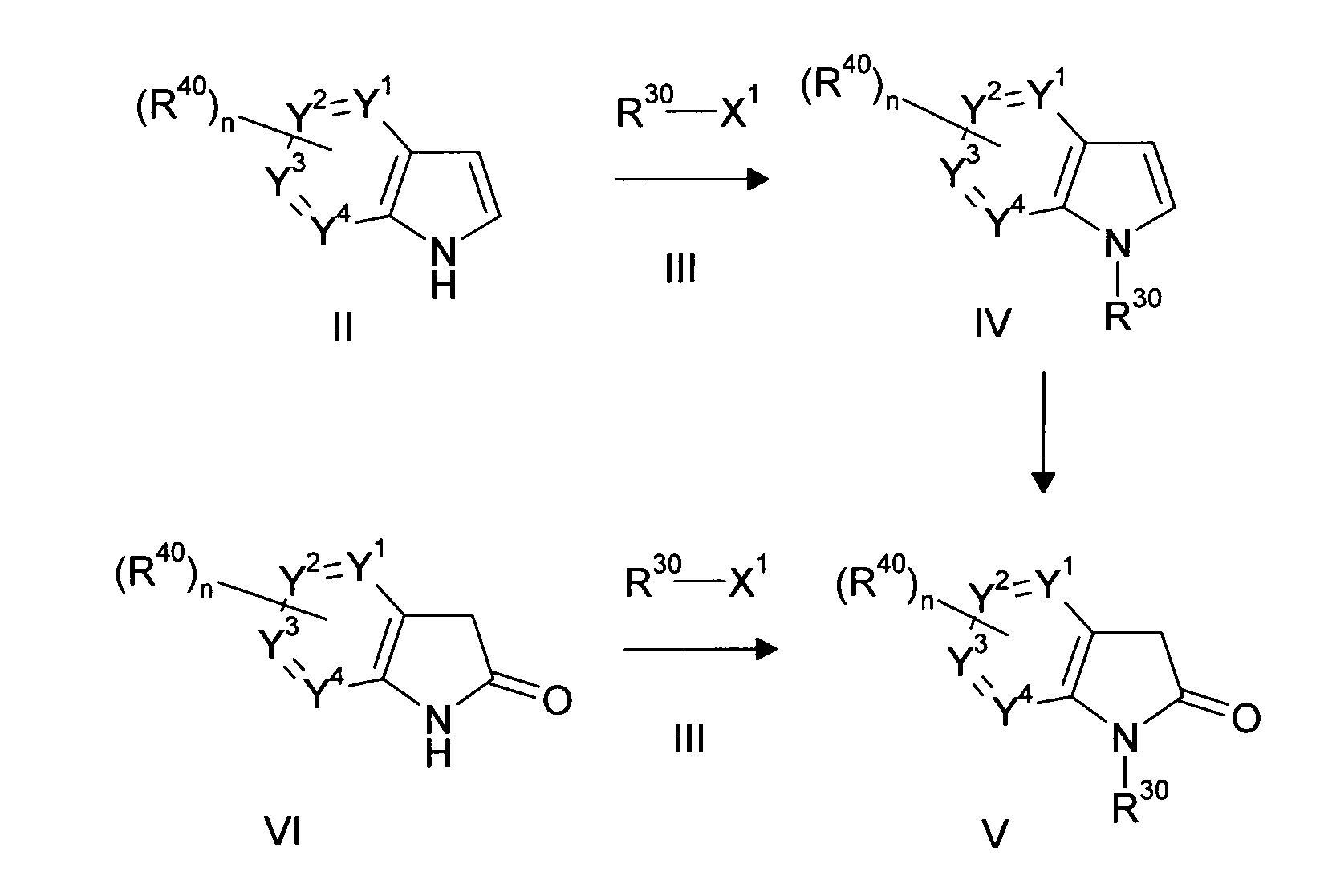
Группы R30, R40, Y1, Y2, Y3 и Y4 и значение n в соединениях формул II, III, IV и V определяют, как в соединениях формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которая впоследствии превращается в целевую группу. Группа X1 в соединениях формулы III представляет собой замещаемую группу, позволяющую проводить реакцию нуклеофильного замещения или реакцию другого типа механизма, включая радикальные реакции и реакции, катализируемые переходными металлами, которая приводит в результате к замене данной замещаемой группы кольцевым атомом азота в 5-членном кольце в соединении формулы II, например, галоген или арилсульфонилокси или алкилсульфонилокси группу или группу, содержащую бор.
В случае когда R30 необязательно замещают фенилом или гетероарилом, который замещают подходящей электрон-оттягивающей группой, или она содержит обедненное электронной плотностью гетероциклическое кольцо, или R30 необязательно замещают циклоалкилом или циклоалкенилом, или она представляет собой тетрагидропиранил, X1 может представлять собой галоген, в особенности хлор, бром или йод, или арилсульфонилокси или алкилсульфонилокси группу, такую как бензолсульфонилокси, толуолсульфонилокси, нитробензолсульфонилокси, метансульфонилокси или трифторметансульфонилокси, и реакцию можно проводить в условиях реакции нуклеофильного замещения, обычно в растворителе, например, инертном апротонном растворителе, таком как эфир, подобном тетрагидрофурану (THF), диоксану (1,4-диоксан) или диметиловому эфиру этиленгликоля (DME), амид, подобном диметилформамиду (DMF) или N-метилпирролидин-2-ону (NMP), или диметилсульфоксиду (DMSO), или их смеси, и в присутствии основания, такого как алкоголят, подобный этоксиду натрия или трет-бутоксиду калия, гидрид, подобный гидриду натрия, амид, подобный амиду натрия или диизопропиламиду лития, карбонат, подобный карбонату калия или карбонату цезия, или амин, подобный этилдиизопропиламину.
В случае когда R30 необязательно замещают фенилом или гетероарилом, X1 может представлять собой хлор, бром или йод, т.е. соединение формулы III может представлять собой необязательно замещенный хлорбензол, бромбензол, йодбензол, хлоргетероарен, бромгетероарен или йодгетероарен, и реакцию соединений формул II и III можно проводить в условиях реакции арилирования Ульмана в присутствии каталитического соединения, содержащего медь, например, бромид меди(I), йодид меди(I) или ацетилацетон меди(II), при повышенных температурах, например, при температурах от приблизительно 100°C до приблизительно 150°C, обычно в инертном апротонном растворителе, таком как DMSO, DMF, NMP, ацетонитрил, диоксан или толуол, в присутствии основания, такого как карбонат, подобный карбонату калия или карбонату цезия, или фосфат, подобный фосфату калия, и предпочтительно амин, подобный N,N'-диметилэтилендиамину, 1,2-диаминоциклогексану, пролину или 8-гидроксихинолину. Реакцию арилирования, подобно другим реакциям, проводимым при синтезе соединений формулы I, можно также проводить в микроволновом реакторе.
В другом способе получения соединений формулы IV соединение формулы II может реагировать с соединением формулы III, в котором R30 необязательно замещают фенилом или гетероарилом, и X1 представляет собой галоген, в особенности хлор, бром или йод, или алкилсульфонилокси группу, такую как трифторметансульфонилокси, в присутствии палладиевого катализатора, который может образовываться из трис(дибензилиденацетон)дипалладия(0), и, например, фосфинового лиганда, и основания, такого как трет-бутоксид натрия или трехосновный фосфат калия в инертном растворителе, таком как углеводород, подобный толуолу, или эфир, подобный диоксану, при температурах от приблизительно 60°C до приблизительно 120°C, как описано, например, в D. W. Old et al., Org. Lett. 2 (2000), 1403.
В следующем способе получения соединений формулы IV, соединение формулы II может реагировать с бороновой кислотой, т.е. соединение формулы III, в котором X1 представляет собой остаток бороновой кислоты B(OH)2, в реакции, катализируемой переходными металлами, например, согласно модификации Чена-Эванса-Лэма реакции конденсации Судзуки-Мияура в присутствии медного соединения, такого как ацетат меди(II), в растворителе, таком как хлорированный углеводород, подобный дихлорметану или хлороформу, при температурах от приблизительно 20°C до приблизительно 40°C, например, при комнатной температуре, и в присутствии третичного амина, такого как триэтиламин, этилдиизопропиламин или пиридин, как описано, например, в D. M. T. Chan et al., Tetrahedron Lett. 39 (1998), 2933. Вместо применения бороновой кислоты, соединение формулы IV можно также получить из соединения формулы II с помощью органотрифторборатной соли, т.е. соединения формулы III, в котором X1 представляет собой отрицательно заряженную трифторборатную группу BF3 -, содержащую катион, такой как катион щелочного металла, подобный катиону цезия, калия, натрия или лития или катиону четвертичного аммония или фосфония, в особенности катиона калия, в качестве противоиона (см., R. A. Batey et al., Tetrahedron Lett. 42 (2001), 9099), в присутствии каталитического соединения, содержащего переходный металл, такого как медное соединение, подобное ацетату меди(II), в растворителе, таком как хлорированный углеводород, подобный дихлорметану или хлороформу, при температурах от приблизительно 20°C до приблизительно 50°C в присутствии кислорода и молекулярных сит, как описано, например, в T. D. Quach et al., Org. Lett. 5 (2003), 4397.
Последующее превращение соединения формулы IV в азаоксиндол формулы V можно осуществлять первой обработкой соединения формулы IV N-хлорсукцинимидом в растворителе, таком как хлорированный углеводород, подобный дихлорметану, при температурах от приблизительно 10°C до приблизительно 30°C, например, при комнатной температуре, и затем обработкой неочищенного промежуточного продукта 85% фосфорной кислотой в уксусной кислоте при повышенных температурах от приблизительно 110°C до приблизительно 140°C, как описано в R. Sarges et al., J. Med. Chem. 32 (1989), 437. Превращение соединения формулы IV в азаоксиндол формулы V можно также осуществлять первой обработкой соединения формулы IV бромом или источником брома, таким как N-бромсукцинимид или бромид пербромид пиридиния (трибромид пиридиния), в растворителе, таком как хлорированный углеводород, подобный дихлорметану, или спирт, подобный трет-бутанолу или амиловому спирту, или смесь спирта и воды или водный буферный раствор, подобный фосфатному буферу, имеющему pH, приблизительно равный 5, например, при температурах от приблизительно 0°C до приблизительно 50°C. Затем восстановление промежуточных бром-содержащих продуктов или гидролиз до азаоксиндола формулы V можно осуществлять обработкой металлом, таким как цинк или железо в уксусной кислоте или смеси уксусной кислоты и растворителя, такого как спирт, подобный метанолу, этанолу или трет-бутанолу, или эфир, подобный диэтиловому эфиру или THF, или гидрированием в присутствии гидрогенизирующего катализатора, такого как гидроксид палладия или палладий на угле или никель Ренея, например, в растворителе, таком как спирт, подобный метанолу или этанолу, или эфир, подобный этилацетату, при температурах от приблизительно 0°C до приблизительно 60°C и давлении водорода от приблизительно 1 бар до приблизительно 100 бар, как описано, например, в J. Parrick et al., Tetrahedron Lett. 25 (1984), 3099; A. Marfat et al., Tetrahedron Lett. 28 (1987), 4027; или R. P. Robinson et al., J. Org. Chem. 56 (1991), 4805.
Соединения формулы V можно также получить реакцией азаоксиндола формулы VI, в котором группы R40, Y1, Y2, Y3 и Y4 и значение n определяют, как в соединении формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которая впоследствии превращается в целевую группу, с соединением формулы III, как определено выше, в котором X1 представляет собой галоген или арилсульфонилокси или алкилсульфонилокси группу или группу, содержащую бор, такую как остаток бороновой кислоты, или группу BF3 -, содержащую катион, подобный катиону калия, в качестве противоиона, в реакции нуклеофильного замещения или реакции Ульмана или другой реакции, катализируемой переходными металлами, как изложено выше. Пояснения, данные выше относительно реакции соединений формул II и III, например, касающиеся палладий-каталируемой и медь-катализируемой реакций, являются применимыми, соответственно, относительно реакции соединений формул VI и III.
Затем, в процессе синтеза соединений формулы I, азаоксиндолы формулы V можно подвергать формилированию по Вильсмейеру с сопутствующим хлорированием для того, чтобы получить 1-R30-2-хлоразаиндол-3-карбоксальдегиды формулы VII, в которых группы R30, R40, Y1, Y2, Y3 и Y4 и значение n определяют, как в соединении формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которая впоследствии превращается в целевую группу.
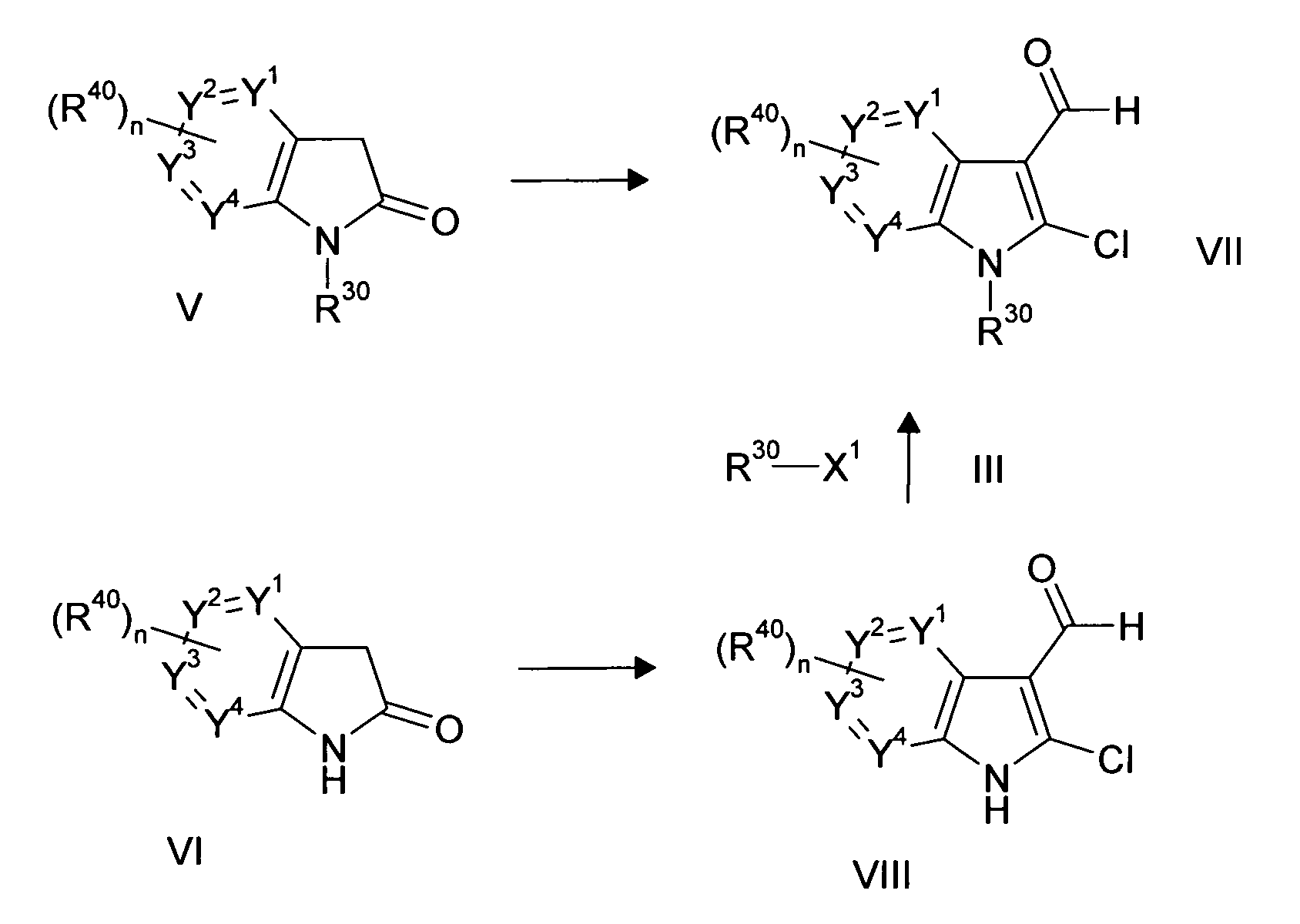
Реагент для формилирования по Вильсмейеру можно удобно получить in situ из диметилформамида и подходящего неорганического или органического хлорида, такого как фосген, оксалилхлорид или хлорокись фосфора, в инертном апротонном растворителе, таком как углеводород или хлорированный углеводород, подобный бензолу, дихлорметану или хлороформу, эфир, подобный DME или избыток DMF, или их смесь, при температурах от приблизительно 0°C до приблизительно 10°C. Предпочтительно применяют хлорокись фосфора. Реакцию реагента Вильсмейера с соединением формулы V обычно проводят при температурах от приблизительно 0°C до приблизительно 30°C, предпочтительно в присутствии основания, такого как пиридин. Гидролитическая обработка реакционной смеси, которую подобно обработке всех реакций получения соединений формулы I можно обычно проводить в стандартных условиях, затем дает альдегид формулы VII.
Соединения формулы VII можно также получить, вначале подвергая азаоксиндол формулы VI формилированию по Вильсмейеру с сопутствующим хлорированием во 2-положение аналогично, как указано выше, для того чтобы получить 2-хлоразаиндол-3-карбоксальдегид формулы VIII, в котором группы R40, Y1, Y2, Y3 и Y4 и значение n определяют, как в соединении формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которая впоследствии превращается в целевую группу, и затем введением группы R30 в 1-положение азаиндольного кольца в соединении формулы VIII реакцией с соединением формулы III, как определено выше, в котором X1 представляет собой галоген или арилсульфонилокси или алкилсульфонилокси группу или группу, содержащую бор, такую как остаток бороновой кислоты или группу BF3 -, содержащую катион, подобный катиону калия, в качестве противоиона, в реакции нуклеофильного замещения или реакции Ульмана или другой реакции, катализируемой переходными металлами, как указано выше. Пояснения, данные выше относительно реакции соединений формул II и III, например, касающиеся палладий-катализируемых и медь-катализируемых реакций, можно применять, соответственно, относительно реакции соединений формул VIII и III.
Затем азаиндол-3-карбоксальдегиды формулы VII можно окислить в стандартных условиях для окисления альдегидов до карбоновых кислот, для того чтобы получить азаиндол-3-карбоновые кислоты формулы IX, в которой группы R30, R40, Y1, Y2, Y3 и Y4 и значение n определяют, как в соединении формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которая впоследствии превращается в целевую группу. Например, окисление можно проводить перманганатом, таким как перманганат калия, в смеси воды и инертного органического растворителя, такого как кетон, подобный ацетону, или эфир, подобный THF, при температурах от приблизительно 10°C до приблизительно 30°C, например, при комнатной температуре, при приблизительно нейтральных значениях pH. Удобно также проводить окисление хлоритом, таким как хлорит натрия, в присутствии 2-метилбут-2-ен в смеси воды и инертного органического растворителя, такого как спирт, подобный трет-бутанолу, или эфир, подобный THF, при температурах от приблизительно 10°C до приблизительно 30°C, например, при комнатной температуре, при слабо кислых значениях pH, например, в присутствии дигидрофосфата.
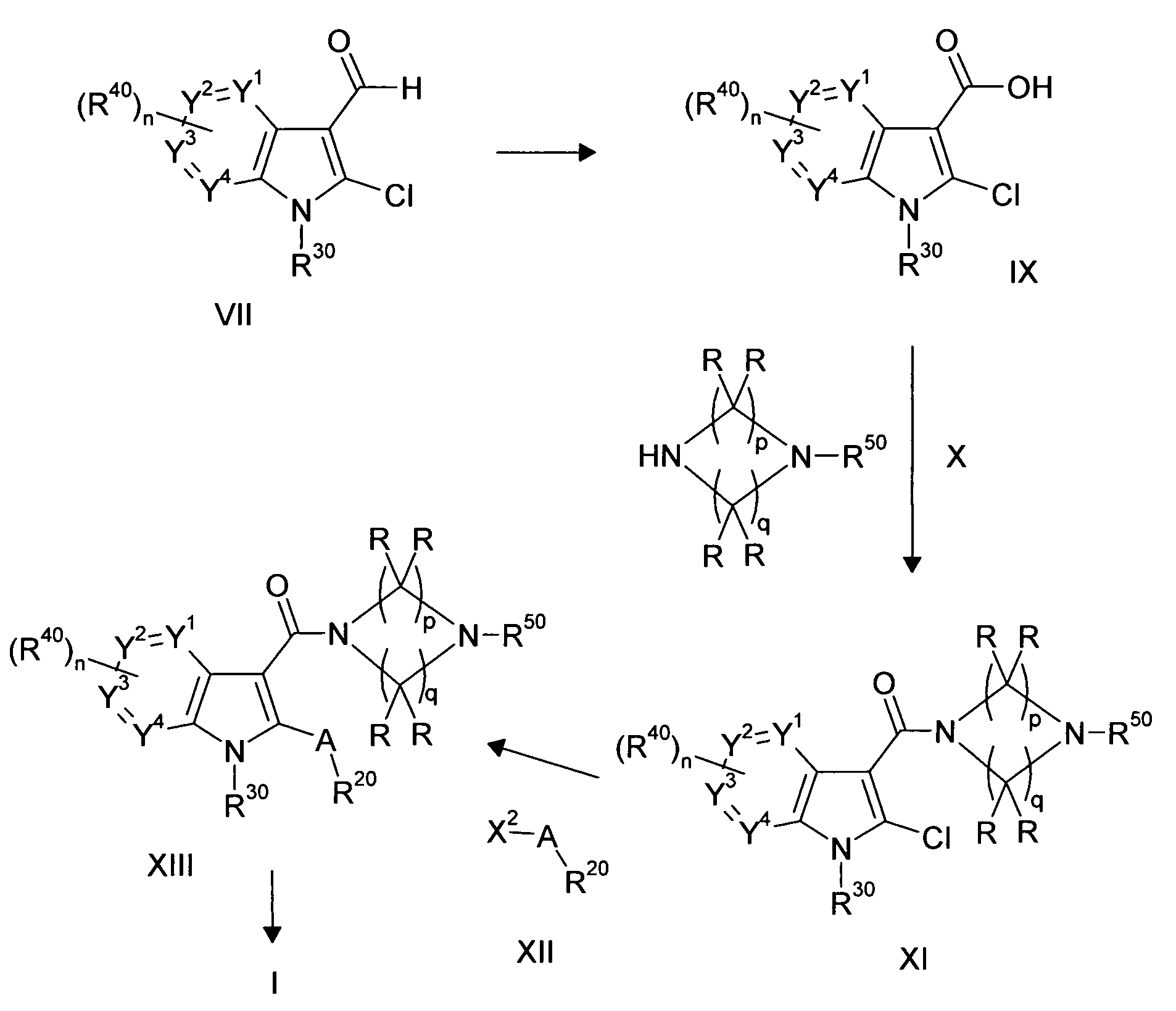
Затем карбоновую кислоту формулы IX можно конденсировать в стандартных условиях для образования амидной связи с диазациклоалканом формулы X, для того чтобы получить соединение формулы XI. Группы R, R30, R40, Y1, Y2, Y3 и Y4 и значения n, p и q в соединениях формул X и XI определяют, как в соединениях формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которая впоследствии превращается в целевую группу. Соединения формулы VII определяют, как выше. Группа R50 в соединениях формул X и XI может иметь значения группы R10 в соединениях формулы I, за исключением водорода, т.е. она может представлять собой (C1-C6)-алкил-O-CO- или (C3-C7)-циклоалкил-CvH2v-O-CO-, данные группы защищают атом азота, несущий R50 от реакции с соединением формулы IX, или R50 может представлять собой другую защитную группу, которая препятствует реакции по упомянутому атому азота и может впоследствии удаляться для того, чтобы получить целевое соединение формулы I, в котором R10 представляет собой водород. Примерами групп, которые препятствуют реакции по упомянутому атому азота, являются бензилоксикарбонильная группа, которую можно впоследствии отщеплять гидрированием в присутствии катализатора, такого как палладиевый катализатор, трет-бутилоксикарбонильная группа, которую можно впоследствии отщеплять обработкой кислотой, такой как трифторуксусная кислота или хлороводородная кислота, или флуорен-9-илоксикарбонильная группа, которую можно впоследствии отщеплять обработкой пиперидином. Для образования амидной связи карбоновую кислоту формулы IX обычно превращают в реакционноспособное производное, которое можно выделить или получить in situ, или которое активируют in situ общепринятым реагентом для амидной конденсации. Например, соединение формулы IX можно превратить в хлорангидрид кислоты обработкой тионилхлоридом, оксалилхлоридом или (1-хлор-2-метилпропенил)диметиламином, в реакционноспособный эфир, или в смешанный ангидрид обработкой алкилхлороформиатом, подобным этилхлороформиату или изобутилхлороформиату, или его можно активировать реагентом, таким как пропанфосфоновый ангидрид, N,N'-карбонилдиазол, подобный N,N'-карбонилдиимидазолу (CDI), карбодиимид, подобный N,N'-диизопропилкарбодиимиду (DIC), N,N'-дициклогексилкарбодиимиду (DCC) или гидрохлорид N-этил-N'-(3-диметиламинопропил)карбодиимиду (EDC), карбодиимиду вместе с добавкой, подобной 1-гидроксибензотриазолу (HOBT) или 1-гидрокси-7-азабензотриазолу (HOAT), конденсирующий реагент на основе урония, подобный гексафторфосфату O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HATU), гексафторфосфату O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония (HBTU) или тетрафторборату O-(циано(этоксикарбонил)метиленамино)-N,N,N',N'-тетраметилурония (TOTU), конденсирующий реагент на основе фосфония, подобный гексафторфосфату (бензотриазол-1-илокси)трис(диметиламино)фосфония (BOP), гексафторфосфату (бензотриазол-1-илокси)трипирролидинофосфония (PyBOP) или гексафторфосфату бромтрипирролидинофосфония (PyBroP). Активацию соединения формулы IX и реакцию активированного соединения формулы IX или реакционноспособного производного соединения формулы IX с соединением формулы X обычно осуществляют в инертном растворителе, таком как эфир, подобный THF, диоксану или DME, углеводород, такой как толуол, хлорированный углеводород, подобный дихлорметану или хлороформу, или амид, такой как DMF или NMP, например, или в смеси растворителей, при температурах от приблизительно 0°C до приблизительно 60°C в присутствии подходящего основания, такого как третичный амин, подобный триэтиламину, этилдиизопропиламину, N-метилморфолину или пиридину, или в присутствии основного соединения щелочного металла, такого карбонат щелочного металла, подобный, например, карбонату натрия, карбонату калия или карбонату цезия.
Затем полученное соединение формулы XI может реагировать с соединением формулы XII, для того чтобы получить соединение формулы XIII. Группы A, R, R20, R30, R40, Y1, Y2, Y3 и Y4 и значения n, p и q в соединениях формул XII и XIII определяют, как в соединениях формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которая впоследствии превращается в целевую группу. Группу R50 в соединении формулы XIII определяют, как в соединениях формул X и XI. В случае когда группа A в соединении формулы XII представляет собой O, S или N((C1-C4)-алкил), группа X2 представляет собой водород, и реакция соединений формул XI и XII представляет собой реакцию нуклеофильного замещения. В данном случае, как применяют в общем ко всем исходным соединениям и промежуточным соединениям в синтезе соединений формулы I, включая соединения формул IX, X и XII, например, упомянутое соединение формулы XII, в котором X2 представляет собой водород, можно также применять в форме соли. Также все продукты, полученные в процессе синтеза соединений формулы I, включая конечные соединения формулы I, можно получить в форме соли. Примерами подходящих солей соединений формулы XII, которые можно также получить in situ, являются соли щелочных металлов, такие как натриевые соли и калиевые соли и соли, содержащие инертный аммониевый катион в виде четвертичной аммониевой соли. Реакцию соединения формулы XII, в котором A представляет собой O, S или N((C1-C4)-алкил) и X2 представляет собой водород, с соединением формулы XI обычно проводят в растворителе, например, инертном апротонном растворителе, таком как амид, подобный DMF или NMP, или DMSO, или в смеси растворителей, в присутствии основания, такого как алкоголят, подобный этоксиду натрия или трет-бутоксиду калия, гидрид, подобный гидриду натрия или гидриду калия, или амид, подобный амиду натрия или диизопропиламиду лития, при повышенных температурах от приблизительно 80°C до приблизительно 180°C. Предпочтительно реакцию можно осуществлять в микроволновом реакторе. В случае когда группа A в соединении формулы XII представляет собой C(Ra)2, реакцию соединений формул XI и XII, для того чтобы получить соединение формулы XIII, предпочтительно проводят через органометаллическое соединение. Например, в данном случае соединение формулы XII может представлять собой органометаллическое соединение, такое как цинкорганическое соединение, подобное хлориду органоцинка или бромиду органоцинка, причем, затем, группа X2 в соединении формулы XII представляет собой группу Zn-Cl или Zn-Br, или борорганическое соединение, подобное 9-органо-9-борбицикло[3,3,1]нонану, причем, затем, группа X2 в соединении формулы XII представляет собой 9-борбицикло[3,3,1]нонан-9-ильную группу. Относительно соединения формулы XII, которое действительно применяют в реакции, в случае когда A представляет собой C(Ra)2, группа X2 в соединении формулы XII может также рассматриваться как галоген, такой как хлор или бром, и затем данное соединение формулы XII превращают in situ обработкой цинком в соответствующее цинкорганическое соединение или в борорганическое соединение. Реакцию цинкорганического соединения формулы XII с соединением формулы XI обычно проводят в инертном апротонном растворителе, таком как углеводород, подобный гексану, бензолу или толуолу, эфир, подобный THF или диоксану, или амид, подобный DMF или NMP, или в смеси растворителей, при температурах от приблизительно 0°C до приблизительно 120°C, предпочтительно в присутствии катализатора на основе переходного металла, такого как в присутствии палладиевого соединения, подобного ацетату палладия(II), трис(дибензилиденацетон)дипалладия(0) или бис(дибензилиденацетон)палладия(0) вместе с фосфиновым лигандом, подобным 2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенилу, например, и дополнительно алкоксиборанового производного, подобного B-метокси-9-борбицикло[3.3.1]нонану, или в присутствии никелевого соединения, подобного ацетилацетону никеля. В реакцию борорганического соединения формулы XII с соединением формулы XI обычно добавляют основание, такое как, например, фосфат калия.
В другом способе получения соединения формулы XIII из соединения формулы XI и соединения формулы XII, в которых A представляет собой C(Ra)2, соединение формулы XI вначале превращают в соответствующее органолитиевое соединение, которое содержит атом лития вместо атома хлора во 2-положении, например, реакцией с алкиллитиевым соединением, таким как н-бутиллитий, и данное промежуточное органолитиевое соединение затем вступает в реакцию замещения с соединением формулы XII, в котором группа X2 представляет собой замещаемую нуклеофилом уходящую группу, такую как галоген, в особенности хлор, бром или йод, или арилсульфонилокси или алкилсульфонилокси группу, такую как бензолсульфонилокси, толуолсульфонилокси, метансульфонилокси или трифторметансульфонилокси. Литиирование соединения формулы XI и последующее алкилирование обычно проводят в инертном апротонном растворителе, таком как углеводород, подобный гексану или бензолу, или эфир, подобный THF или диоксану, или в смеси растворителей, при температурах от приблизительно -80°C до приблизительно 30°C.
В случае когда группа R50 в соединении формулы XIII имеет любое из значений группы R10 в соединениях формулы I, и все другие группы имеют требуемые значения, включенные в определение соединений формулы I, таким образом полученное соединение формулы XIII уже представляет собой конечное соединение формулы I. В случае когда R50 представляет собой защитную группу, и нужно получить соединение формулы I, в котором R10 представляет собой водород, и/или любые другие группы присутствуют в защищенной форме или в форме группы, являющейся предшественником, таким образом полученное соединение формулы XIII можно в конце превратить в требуемое соединение формулы I удалением защитных групп и/или превращением любых других групп. Как показано выше, для того чтобы избежать нежелательного направления реакции или побочных реакций, в любой одной или более стадий синтеза соединений формулы I функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником. Кроме того, на конечной стадии синтеза соединения формулы I можно удалять защитные группы и превращать группы, являющиеся предшественниками, также на любой стадии синтеза. Соответствующие стратегии синтеза и информация о подходящих защитных группах и их введении и удалении являются хорошо известными специалистам в данной области техники и их можно найти, например, в P. G. M. Wuts и T. W. Greene, Greene's Protective Groups in Organic Synthesis, 4. ed. (2007), John Wiley & Sons. Примерами защитных групп, которые можно упомянуть, являются бензильные защитные группы, такие как в бензиловых эфирах гидроксигрупп и бензиловых эфирах карбоксильных групп, из которых бензильную группу можно удалить каталитическим гидрированием в присутствии палладиевого катализатора, трет-бутиловые защитные группы, такие как в трет-бутиловых эфирах карбоксильных групп, из которых трет-бутильную группу можно удалить обработкой трифторуксусной кислотой, ацильные защитные группы, которые применяют для защиты гидроксигрупп и аминогрупп в форме эфиров и амидов и которые можно отщепить кислотным или основным гидролизом, и алкилоксикарбонильные защитные группы, такие как в трет-бутоксикарбонильных производных аминогрупп, включая циклическую аминогруппу, являющуюся частью диазациклоалкановой группы, изображенной в формуле I, в случае когда R10 представляет собой водород, которую можно удалить обработкой трифторуксусной кислотой. Примерами групп, являющихся предшественниками, которые можно упомянуть, являются нитрогруппы, которые можно превратить в аминогруппы каталитическим гидрированием или восстановлением дитионитом натрия, например, и цианогруппы, которые можно превратить в карбоксамидные группы и карбоксильные группы гидролизом.
Кроме того, для того чтобы получить дополнительные соединения формулы I, различные другие трансформации функциональной группы можно осуществлять в соединениях формулы I или соединениях формулы XIII или других соединениях, встречающихся в синтезе соединений формулы I. Например, гидроксигруппу в соединении формулы I или XIII можно превратить в простой эфир или сложный эфир, или она может реагировать с изоцианатом для того, чтобы получить карбамат в стандартных условиях. Превращение в простые эфиры гидроксигрупп можно предпочтительно проводить алкилированием соответствующим галогеновым соединением, в особенности бромидом или йодидом, в присутствии основания, такого как карбонат щелочного металла, подобного карбонату калия или карбонату цезия, в инертном растворителе, таком как амид, подобный DMF или NMP, или кетон, подобном ацетону или бутан-2-ону, или реакцией с соответствующим спиртом в условиях реакции Мицунобу в присутствии азодикарбоксилата, подобного диэтилазодикарбоксилату или диизопропилазодикарбоксилату, и фосфина, подобного трифенилфосфину или трибутилфосфину, в инертном апротонном растворителе, таком как эфир, подобный THF или диоксану (см., O. Mitsunobu, Synthesis (1981), 1). Аминогруппу в соединении формулы I или XIII можно модифицировать в стандартных условиях для алкилирования, например, реакцией с галогеновым соединением или восстановительным аминированием карбонильного соединения, или в стандартных условиях для ацилирования или сульфонилирования, например, реакцией с активированной карбоновой кислотой или производным карбоновой кислоты, подобным хлорангидриду карбоновой кислоты или ангидриду или хлориду сульфоновой кислоты. Карбоксильную группу в соединении формулы I или XIII можно активировать или превратить в реакционноспособное производное, как указано выше относительно соединений формулы IX, и подвергнуть реакции со спиртом или амином для того, чтобы получить эфир или амид. Алкил-S-группу в соединении формулы I или XIII можно окислить пероксидом, подобным перекиси водорода, или надкислотой для того, чтобы получить алкил-S(O)- или алкил-S(O)2-группу, и из защищенной меркаптогруппы в соединении формулы XIII можно удалить защитную группу и окислить ее для того, чтобы получить сульфоновую кислоту, которую затем можно активировать и подвергнуть реакции с амином в стандартных условиях для того, чтобы получить сульфонамид.
Порядок, в котором вводят группы в процессе синтеза соединения формулы I, может также отличаться от порядка, указанного выше. Например, вместо первоначального введения диазациклоалкановой группы и затем группы -A-R20 реакцией соединения формулы IX с соединением формулы X и реакцией полученного соединения формулы XI с соединением формулы XII, также возможно ввести вначале группу -A-R20 и затем диазациклоалкановую группу реакцией соединения формулы IX или его защищенной формы, такой как эфир, с соединением формулы XII и, необязательно, после удаления защитной группы, реакцией полученного соединения формулы XIV с соединением формулы X для того, чтобы получить соединение формулы XIII, которое в конце превращают в требуемое соединение формулы I, например удалением защитной группы R50 в случае получения соединения формулы I, в котором R10 в соединении формулы I представляет собой водород.
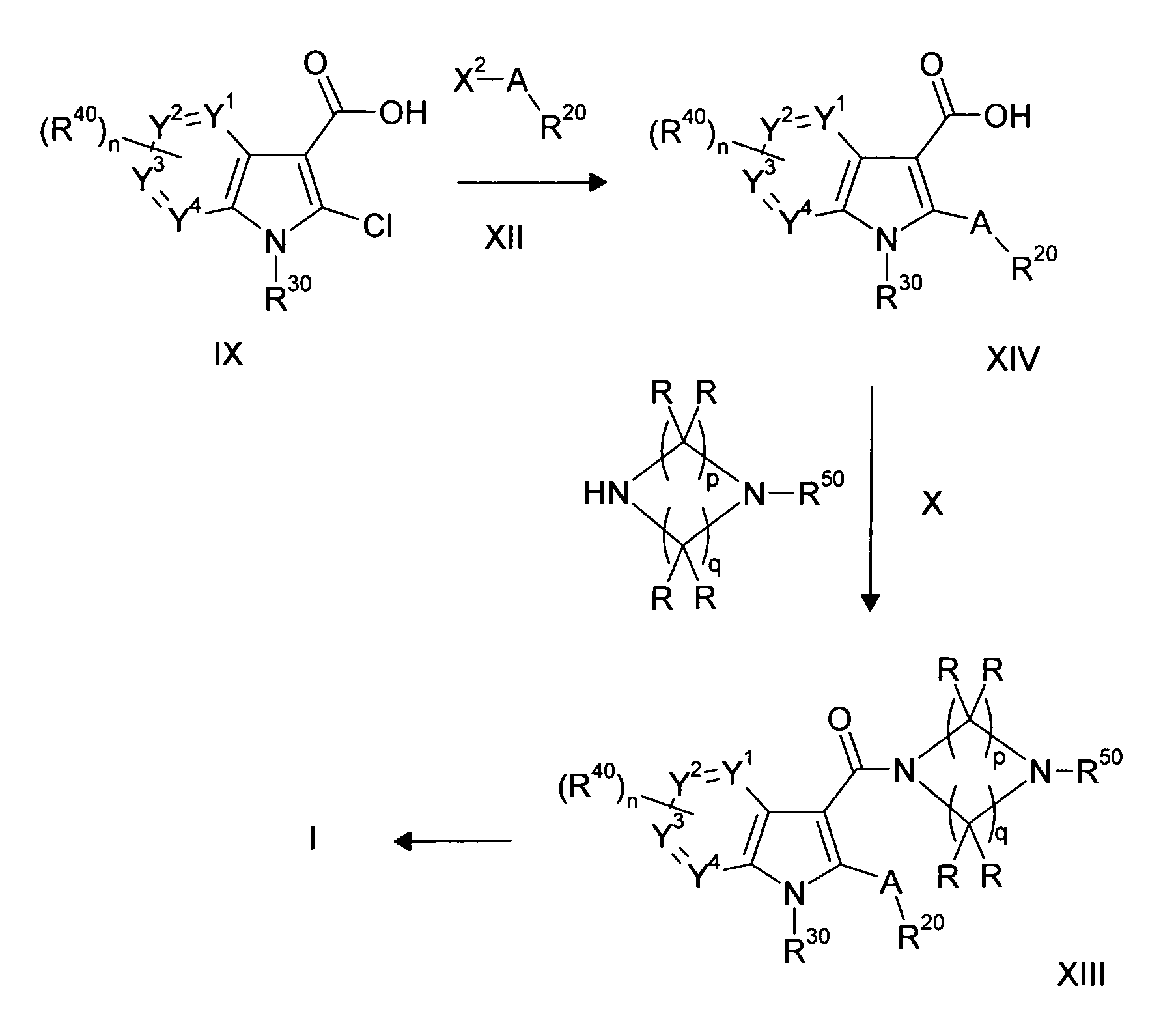
Группы A, R20, R30, R40, Y1, Y2, Y3 и Y4 и значение n в соединениях формулы XIV определяют, как в соединениях формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которые впоследствии превращают в целевую группу. Мало того, что, как упоминалось, в примененных соединениях формулы IX карбоновая кислота, изображенная формулой, может присутствовать в защищенной форме, например, в форме эфира, подобного трет-бутиловому эфиру или бензиловому эфиру, при реакции соединения формул IX и XII, и карбоксильная группа в соединении формулы XIV может, таким образом, также присутствовать в защищенной форме, и из нее можно удалять защитную группу перед реакцией соединения формул X и XIV. Соединения формул IX, X, XII и XIII определяют, как выше. Все пояснения, данные выше относительно реакции соединений формулы XI с соединениями формулы XII и реакции соединений формулы IX с соединениями формулы X, являются применимыми к реакции соединений формулы IX с соединениями формулы XII и реакции соединений формулы X с соединениями формулы XIV, соответственно. Таким образом, например, для образования амидной связи в реакции соединений формул X и XIV карбоксильную группу обычно превращают в реакционноспособное производное или активируют посредством общепринятого конденсирующего реагента для образования амидной связи и подвергают реакции с соединением формулы X в присутствии основания, как указано выше.
В следующей стратегии для синтеза соединений формулы I, группу -A-R20 можно также ввести в альдегид формулы VII реакцией его с соединением формулы XII для того, чтобы получить соединение формулы XV, альдегидную группу в соединении формулы XV затем окисляли для того, чтобы получить соединение формулы XIV, и последнее соединение затем подвергали реакции с соединением формулы X, чтобы в конце получить соединение формулы I, как уже упомянуто выше.
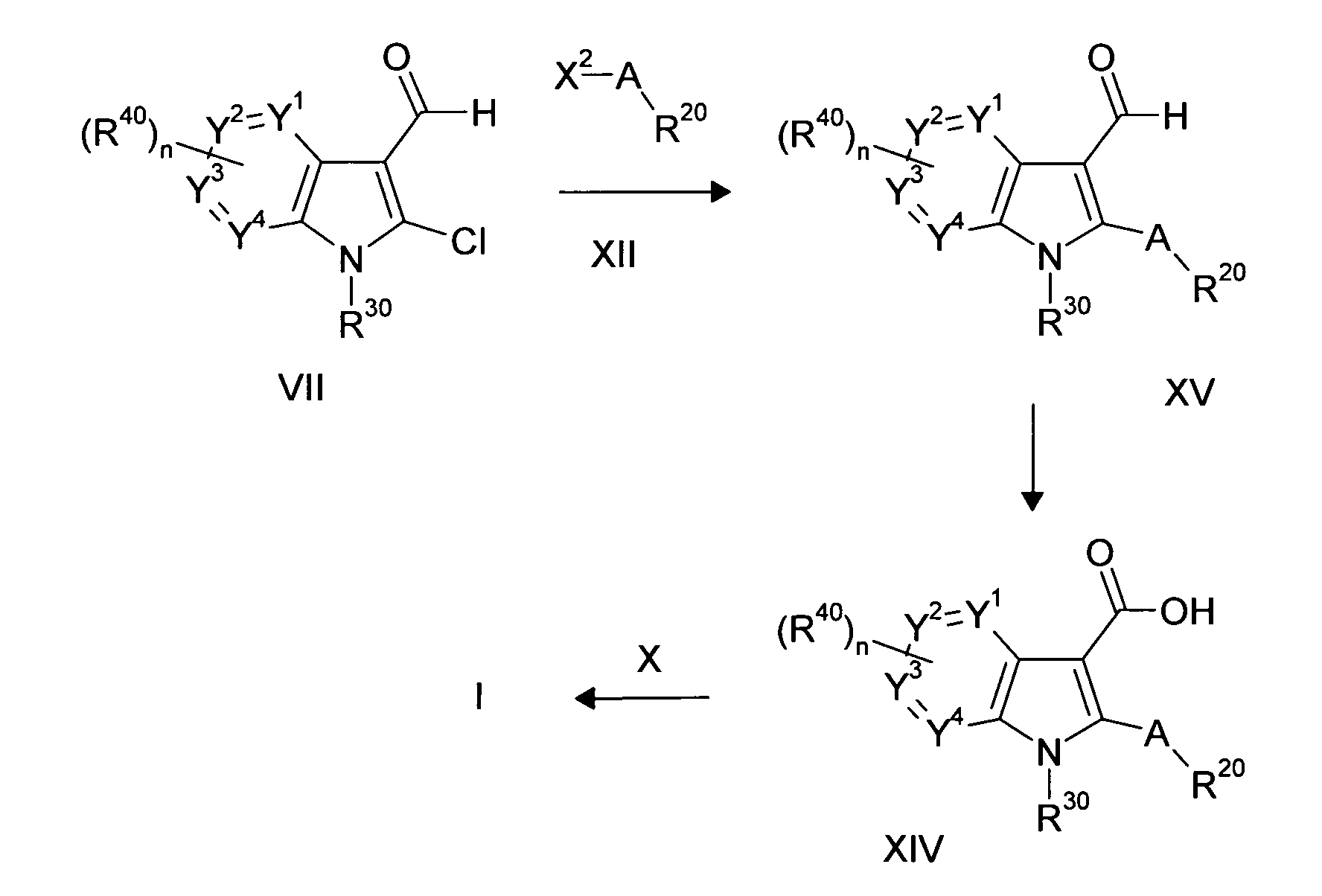
Группы A, R20, R30, R40, Y1, Y2, Y3 и Y4 и значение n в соединениях формулы XV определяют, как в соединениях формулы I, и дополнительно функциональные группы могут присутствовать в защищенной форме или в форме группы, являющейся предшественником, которые впоследствии превращают в целевую группу. Соединения формул VII, X, XII и XIV определяют, как выше. Все пояснения, данные выше относительно реакции соединений формулы XI или формулы IX с соединениями формулы XII и относительно окисления соединений формулы VII до соединений формулы IX, являются применимыми к реакции соединений формулы VII с соединениями формулы XII и окислению соединений формулы XV до соединений формулы XIV, соответственно. Таким образом, например, окисление альдегидной группы в соединениях формулы XIV можно удобно осуществлять хлоритом натрия в присутствии 2-метилбут-2-ена или перманганатом калия в смеси воды и органического растворителя, как указано выше.
Все реакции, проводимые при получении соединений формулы I, являются известными per se, и их можно осуществлять в порядке, известном специалистам в данной области техники, или аналогично методикам, которые описывают в стандартной литературе, например, в Houben-Weyl, Methods of Organic Chemistry, Thieme; или Organic Reactions, John Wiley & Sons; или R. C. Larock, Comprehensive Organic Transformations: A Guide to Functional Group Preparations, 2. ed. (1999), John Wiley & Sons, и ссылках, указанных в настоящем изобретении.
Исходные соединения и строительные блоки для синтеза соединений формулы I являются имеющимися в продаже, или их можно получить согласно методикам, описанным в литературе, или аналогично данным методикам. В качестве примеров статей, в которых описывают синтез и реакции 4-азаиндолов, 5-азаиндолов, 6-азаиндолов и 7-азаиндолов, можно упомянуть L. N. Yakhontov, Russ. Chem. Rev. 37 (1968), 551; L. N. Yakhontov et al., Russ. Chem. Rev. 49 (1980), 428; F. Popowycz et al., Tetrahedron 63 (2007), 8689; и F. Popowycz et al., Tetrahedron 63 (2007), 1031. Например, азаиндолы формулы II можно удобно получить из подходящим образом замещенных пиридинов в качестве исходных соединений, таких как нитро-замещенные пиридины или амино-замещенные пиридины. В нитро-замещенных пиридинах, которые несут метильную группу в соседнем положении, 5-членное кольцо азаиндольного кольца можно получить реакцией метильной группы с производным ортомуравьиной кислоты, таким как диметиламинодиметоксиметан или трет-бутокси-бис(диметиламино)метан, или с диэфиром щавелевой кислоты, восстановлением нитрогруппы, например, водородом, в присутствии катализатора, такого как никель Ренея или палладий на угле, и омылением и декарбоксилированием в случае реакции с диэфиром щавелевой кислоты, как описано в I. Mahadevan et al., J. Heterocycl. Chem 29 (1992), 359; K-H. Buchheit et al., Bioorg. Med. Chem. Lett. 5 (1995), 2495; и B. Frydman et al., J. Org. Chem 33 (1968), 3762. Нитро-замещенные пиридины можно непосредственно превратить в 4-азаиндолы и 6-азаиндолы формулы II реакцией с винилмагнийбромидом, как описано в Zhang et al., J. Org. Chem. 67 (2002), 2345. Амино-замещенные пиридины, которые несут атом галогена, такой как хлор, бром или йод, в соседнем положении, могут реагировать с триметилсилилацетиленом в присутствии палладиевого катализатора, такого как хлорид бис(трифенилфосфин)палладия(II), и медного соединения, такого как йодид меди(I), для того чтобы получить 1-(амино-замещенные пиридил)-3-триметилсилилацетилены, которые затем циклизуют до азаиндолов, как описано, например, в Mazéas et al., Heterocycles 50 (1999), 1065; и Song et al., Chem Soc. Rev. 36 (2007), 1120. В качестве другого примера методик получения исходного соединения и строительных блоков, можно упомянуть методики получение замещенных фенолов, описанных в US 2006/0160786 и в Organikum, 12. ed., VEB Deutscher Verlag der Wissenschaften, Berlin (1973), 588, согласно которым можно получить соединения формулы XII, в которых X2 представляет собой водород, A представляет собой O, и R20 замещают фенилом, такие как, например, 3-фтор-2-метилфенол, 2-фтор-6-метилфенол или 3,5-дифтор-2-метилфенол.
Другим объектом настоящего изобретения являются новые исходные соединения и промежуточные соединения, фигурирующие в синтезе соединений формулы I, включая соединения формул II, III, IV, V, VI, VII, VIII, IX, X, XI, XII, XIII, XIV и XV, в которых A, R, R20, R30, R40, R50, X1, X2, Y1, Y2, Y3, Y4, n, p и q определяют, как выше, в любой из их стереоизомерных форм или в виде смеси стереоизомерных форм в любом соотношении, и их соли, и сольваты любого из них, и их применение в качестве промежуточных соединений. Общие пояснения, предпочтительные определения групп и значений и варианты осуществления настоящего изобретения, данные выше относительно соединений формулы I, являются применимыми, соответственно, к упомянутым промежуточным соединениям и исходным соединениям. Объектом настоящего изобретения являются в особенности новые определенные исходные соединения и промежуточные соединения, описанные в настоящем изобретении. Независимо в данном изобретении, описывают ли их в виде свободного соединения и/или в виде конкретной соли, они представляют собой объект настоящего изобретения и в форме свободных соединений и в форме их солей, и если описывают конкретную соль, дополнительно в форме данной конкретной соли, и в форме сольватов любого из них.
Соединения формулы I ингибируют ренин, являющийся ферментом, как можно продемонстрировать в фармакологических тестах, описанных ниже, и в других фармакологических тестах, которые являются известными специалистам в данной области техники, например, в in vitro тестах, в которых определяют ингибирование человеческого ренина, или на моделях животных, в которых антигипертензивную активность и другие эффекты определяют in vivo. Соединения формулы I являются подходящими для лечения гипертензии, включая, например, легочную гипертензию, и других заболеваний сердечно-сосудистой системы и заболеваний сердца, таких как порок сердца, инфаркт миокарда, стенокардия, сердечная недостаточность, сердечная слабость, гипертрофия сердца, фиброз сердца, сосудистая гипертрофия, дисфункция левого желудочка, в особенности дисфункция левого желудочка после инфаркта миокарда, эндотелиальная дисфункция, ишемическое расстройство и расстройство с обструктивным периферическим кровообращением и рестеноз, включая рестеноз после пластической операции на сосудах, для лечения заболеваний почек, таких как почечный фиброз, почечная ишемия, слабость почек, почечная недостаточность, и для лечения других заболеваний, например, диабетических осложнений, таких как нефропатия и ретинопатия, заболеваний головного мозга, таких как внутримозговое кровоизлияние, глаукома, и повреждение органа-мишени. Лечение заболеваний следует понимать в смысле и терапии существующих патологических изменений или дисфункций организма или существующих симптомов с целью ослабления, облегчения или лечения, и профилактики или предотвращения патологических изменений или дисфункций организма или симптомов у людей или животных, которые подвержены им и нуждаются в данной профилактике или предотвращении, с целью предотвращения или подавления их появления или ослабления в случае их появления. Например, у пациентов, которые, исходя из их истории болезни, являются подверженными дисфункции желудочка после инфаркта миокарда, посредством профилактического или превентивного лекарственного лечения возникновение дисфункции желудочка может быть предотвращено, или снижена его степень и ослаблены последствия. Лечение заболеваний может осуществляться и в острых, и в хронических случаях.
Соединения формулы I и их физиологически приемлемые соли и их физиологически приемлемые сольваты можно, следовательно, применять на животных, в особенности на млекопитающих и конкретно на людях, в качестве фармацевтического препарата или лекарственного средства как таковых, в смеси с другим фармацевтическим препаратом или лекарственным средством или в форме фармацевтической композиции. Объектом настоящего изобретения также являются соединения формулы I и их физиологически приемлемые соли и физиологически приемлемые сольваты для применения в качестве фармацевтического препарата, а также фармацевтические композиции и лекарственные средства, которые содержат эффективную дозу, по меньшей мере, одного соединения формулы I и/или его физиологически приемлемой соли и/или физиологически приемлемого сольвата любого из них в качестве активного ингредиента и фармацевтически приемлемый носитель, т.е. одну или более фармацевтически безопасную среду и/или вспомогательное вещество. Объектом настоящего изобретения, кроме того, являются соединения формулы I и их физиологически приемлемые соли и их физиологически приемлемые сольваты для применения в лечении заболеваний, упомянутых выше или ниже, например, гипертензии, или для ингибирования ренина, а также применение соединений формулы I и их физиологически приемлемых солей и их физиологически приемлемых сольватов для получения лекарственного средства для лечения заболеваний, упомянутых выше или ниже, например, гипертензии, или для получения лекарственного средства для ингибирования ренина, где лечение заболеваний включает их терапию и профилактику. Объектом настоящего изобретения также являются способы лечения заболеваний, упомянутых выше или ниже, которые включают введение эффективного количества, по меньшей мере, одного соединения формулы I или его физиологически приемлемой соли или физиологически приемлемого сольвата любого из них человеку или животному, который нуждается в нем. Соединения формулы I и фармацевтическая композиция и лекарственные средства, содержащие их, можно вводить энтерально, например, пероральным, трансбуккальным, сублингвальным или ректальным введением, парентерально, например внутривенной, внутримышечной или подкожной инъекцией или инфузией, или другими способами введения, такими как местное, чрескожное, трансдермальное, интратекальное, интраназальное или внутриглазное введение.
Фармацевтические композиции и лекарственные средства согласно настоящему изобретению обычно содержат от приблизительно 0,5 до приблизительно 90% вес. соединений формулы I и/или их физиологически приемлемых солей и/или их физиологически приемлемых сольватов. Количество активного ингредиента формулы I и/или его физиологически приемлемой соли и/или физиологически приемлемого сольвата любого из них в фармацевтической композиции и лекарственном средстве обычно составляет от приблизительно 0,2 мг до приблизительно 1000 мг, предпочтительно от приблизительно 0,2 мг до приблизительно 500 мг, особенно предпочтительно от приблизительно 1 мг до приблизительно 300 мг в однократной дозе. Получение фармацевтических композиций и лекарственных средств можно осуществлять способом, известным per se. Для этого соединения формулы I и/или его физиологически приемлемые соли и/или их физиологически приемлемые сольваты смешивают вместе с одной или более твердой или жидкой средой и/или вспомогательным веществом, при желании также в комбинации с одним или более другими активными ингредиентами, такими как, например, ингибитор ангиотензин-конвертирующего фермента, антагонист рецептора ангиотензина, диуретик, антагонист рецептора эндотелина, ингибитор эндотелин-конвертирующего фермента, ингибитор нейтральной эндопептидазы, блокатор кальциевых каналов, нитрат, подобный изосорбиддинитрату, блокатор β-рецептора, антагонист α1 адренорецептора, антагонист канабиноидного рецептора, модулятор калиевых каналов, ингибитор тромбоксансинтетазы, антисеротонинергический агент или другой агент, пригодный, например, для лечения гипертензии, порока сердца, сосудистых заболеваний, связанных с диабетом, или заболеваний почек, таких как острая или хроническая почечная недостаточность, и переводят в подходящую форму для дозирования и введения, которую затем можно применять в лекарственных препаратах для человека или животных. Объектом настоящего изобретения также является в особенности фармацевтическая композиция, которая содержит эффективную дозу, по меньшей мере, одного соединения формулы I и/или его физиологически приемлемой соли и/или физиологически приемлемого сольвата любого из них и один или более других активных ингредиентов и фармацевтически приемлемый носитель, где другие активные ингредиенты являются пригодными для лечения гипертензии, инфаркта миокарда, порока сердца, сосудистых заболеваний, связанных с диабетом, повреждений органов-мишеней, таких как сердечная недостаточность, заболеваний почек, таких как острая или хроническая сердечная недостаточность, рестеноз или глаукома, и где в качестве примеров данных других активных ингредиентов можно упомянуть ингибиторы ангиотензин-конвертирующего фермента, антагонисты рецептора ангиотензина, диуретики, антагонисты рецептора эндотелина, ингибиторы эндотелин-конвертирующего фермента, ингибиторы нейтральной эндопептидазы, блокаторы кальциевых каналов, нитраты, подобные изосорбиддинитрату, блокаторы β-рецептора, антагонисты α1 адренорецептора, антагонисты канабиноидного рецептора, модуляторы калиевых каналов, ингибиторы тромбоксансинтетазы, антисеротонинергические агенты.
В качестве сред и вспомогательных веществ можно применять подходящие органические и неорганические вещества, которые не реагируют нежелательным способом с соединениями формулы I. Примерами, которые можно упомянуть, являются вода, растительные масла, воски, спирты, такие как этанол, изопропанол, 1,2-пропандиол, бензиловые спирты или глицерин, полиолы, полиэтиленгликоли, полипропиленгликоли, триацетат глицерина, желатин, углеводы, такие как лактоза или крахмал, стеариновая кислота и ее соли, такие как стеарат магния, тальк, ланолин, вазелиновое масло, или их смеси, например, смеси воды с одним или более органическими растворителями, такими как смеси воды со спиртами. Для перорального и ректального применения можно применять, в частности, фармацевтические формы, такие как, например, таблетки, покрытые пленкой таблетки, таблетки с сахарным покрытием, гранулы, твердые и мягкие желатиновые капсулы, суппозитории, растворы, предпочтительно масляные, спиртовые или водные растворы, сиропы, соки или капли, кроме того, суспензии или эмульсии. Для парентерального применения, например посредством инъекции или инфузии, можно применять в частности фармацевтические формы, такие как растворы, предпочтительно водные растворы. Для местного применения можно применять, в частности, фармацевтические формы, такие как мази, крема, пасты, лосьоны, гели, спреи, пены, аэрозоли, растворы или порошки. Дополнительными подходящими фармацевтическими формами являются, например, имплантаты и пластыри и формы, приспособленные для ингаляции. Соединения формулы I и их физиологически приемлемые соли и физиологически приемлемые сольваты любого из них можно также лиофилизовать и применять полученные лиофилизаты, например, для получения инъецируемых композиций. Липосомальные композиции также являются пригодными в особенности для местного применения. В качестве примеров типов вспомогательных веществ или добавок, которые могут содержаться в фармацевтических композициях и лекарственных средствах, можно упомянуть смазывающие вещества, консерванты, загустители, стабилизаторы, разрыхлители, смачивающие вещества, агенты для достижения депо-эффекта, эмульгаторы, соли, например, для влияния на осмотическое давление, буферные вещества, красители и ароматизирующие вещества. Фармацевтические композиции и лекарственные средства могут также содержать один или более других активных ингредиентов и/или, например, один или более витаминов.
Как обычно, доза соединений формулы I зависит от обстоятельств конкретного случая и устанавливается лечащим врачом согласно общепринятым правилам и процедурам. Она зависит, например, от вводимого соединения формулы I и его активности и продолжительности действия, от природы и тяжести отдельного синдрома, от пола, возраста, веса и индивидуальной чувствительности человека или животного, которого будут лечить, от того, является ли лечение неотложным или затяжным или профилактическим, или от того, вводят ли дополнительные фармацевтические активные соединения в добавление к соединению формулы I. Обычно в случае введения взрослому, весящему приблизительно 75 кг, доза от приблизительно 0,1 мг до приблизительно 100 мг на кг в день, предпочтительно от приблизительно 1 мг до приблизительно 10 мг на кг в день (в каждом случае в мг на кг веса тела) является достаточной. Дневную дозу можно вводить в форме однократной дозы или разделять на ряд отдельных доз, например, две, три или четыре отдельные дозы. Введение можно также осуществлять непрерывно, например, непрерывной инъекцией или инфузией. В зависимости от обстоятельств конкретного случая, может быть необходимо увеличение или уменьшение дозы по сравнению с указанными дозами.
Помимо применения фармацевтически активного соединения в лекарственных препаратах для человека и животных, соединения формулы I можно также применять в качестве добавок в биохимических исследованиях или в качестве научного инструмента или для диагностических целей, например, в in vitro диагностике биологических образцов, если предполагается ингибирование ренина. Соединения формулы I и их соли можно также применять в качестве промежуточных соединений, например, для получения дополнительных фармацевтически активных веществ.
Следующие примеры иллюстрируют настоящее изобретение.
Сокращения:
ACN ацетонитрил
B-OM-9-BBN B-метокси-9-борбицикло[3.3.1]нонан
DCM дихлорметан
DMF диметилформамид
DMSO диметилсульфоксид
EA этилацетат
HEP н-гептан
MOH метанол
NMM N-метилморфолин
NMP N-метилпирролидин-2-он
S-PHOS 2-дициклогексилфосфино-2',6'-диметокси-1,1'-бифенил
TFA трифторуксусная кислота
THF тетрагидрофуран
Когда соединения, содержащие основную группу, очищают препаративной жидкостной хроматографией высокого давления (ВЭЖХ) на обращенно-фазовом (RP) колоночном материале и, как общепринято, элюент представляет собой смесь воды и ацетонитрила, содержащего трифторуксусную кислоту, их обычно получают в форме соли присоединения кислоты с трифторуксусной кислотой, в зависимости от условий обработки, таких как условия лиофилизации. Содержание трифторуксусной кислоты, чье количество может изменяться и может составлять вплоть до приблизительно двух эквивалентов кислоты в случае соединения, содержащего две основные группы, например, не указывают в названиях заголовков примеров и не изображают в структурных формулах, но указывают в описании примеров. Это применимо соответственно к соединениям, которые получают в форме другой соли присоединения кислоты, такой как соль присоединения кислоты с хлороводородной кислотой, чье количество может также изменяться и составлять вплоть до приблизительно двух эквивалентов кислоты в случае соединения, содержащего, например, две основные группы, и которую не указывают в названиях заголовков примеров и не изображают в структурных формулах, но указывают в описании примеров. Данные способа препаративной ВЭЖХ были следующими. Колонка: Waters Atlantis dC18 OBD, 30x100 мм, 5 мкм. Скорость потока: 60 мл/мин. Элюент A: ACN. Элюент B: вода + 0,1% TFA. Градиент: от 10% A + 90% B до 90% A + 10% B в течение 10 мин.
Получение характеристик соединений
Полученные соединения в общем охарактеризовывали спектроскопическими данными и хроматографическими данными, в особенности масс-спектрами (MS) и ВЭЖХ временем удержания (Rt; в минутах), которые получали комбинированным аналитическим ВЭЖХ/MS получением характеристик (LC/MS), и/или спектрами ядерного магнитного резонанса (ЯМР). Если не указано особо, 1H-ЯМР спектры регистрировали при 500 МГц и в DMSO-d6 в качестве растворителя. При получении ЯМР характеристик дают химический сдвиг δ (в ppm, м.д.), число атомов водорода и мультиплетность (с: синглет, д: дуплет, дд: дуплет дуплетов, т: триплет, дт: дуплет триплетов, кв: квартет, м: мультиплет; уш.: уширенный) пиков. При получении MS характеристик дают в общем массовое число (m/z) пика молекулярного иона (М, например, M+) или связанный с ним ион, такой как ион M+1 (например, M+1+; протонированный молекулярный ион M+H+) или ион M-17 (например, M-17+; протонированный молекулярный ион минус H2O), которые образовывались в зависимости от применяемого способа ионизации. Обычно способом ионизации был способ электроспрей ионизации (ESI). Данные применяемых LC/MS способов были следующими.
Способ LC1
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; скорость потока: 1,3 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: от 5% A + 95% B до 95% A + 5% B в течение 2,5 мин, затем 95% A + 5% B в течение 0,5 мин; MS способ ионизации: ESI+
Способ LC2
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; скорость потока: 1,0 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: от 5% A + 95% B до 95% A + 5% B в течение 3,4 мин, затем 95% A + 5% B в течение 1,0 мин; MS способ ионизации: ESI+
Способ LC3
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; скорость потока: 1,3 мл/мин; элюент A: ACN + 0,08% муравьиная кислота; элюент B: вода + 0,1% муравьиная кислота; градиент: от 5% A + 95% B до 95% A + 5% B в течение 2,5 мин, затем 95% A + 5% B в течение 0,5 мин; MS способ ионизации: ESI+
Способ LC4
Колонка: YMC J'sphere ODS H80, 20×2,1 мм, 4 мкм; скорость потока: 1,0 мл/мин; элюент A: ACN; элюент B: вода + 0,05% TFA; градиент: от 4% A + 96% B до 95% A + 5% B в течение 2,0 мин, затем 95% A + 5% B в течение 0,4 мин, затем до 96% A + 4% B в течение 0,05 мин; MS способ ионизации: ESI+
Способ LC5
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; скорость потока: 1,3 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 5% A + 95% B в течение 0,5 мин, затем до 95% A + 5% B в течение 3,0 мин, затем 95% A + 5% B в течение 0,5 мин; MS способ ионизации: ESI+
Способ LC6
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; скорость потока: 1,0 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 2% A + 98% B в течение 1 мин, затем до 95% A + 5% B в течение 4 мин, затем 95% A + 5% B в течение 1,25 мин; MS способ ионизации: ESI+
Способ LC7
Колонка: YMC Pack Pro C18 RS, 33×2,1 мм, 4 мкм; скорость потока: 1,0 мл/мин; элюент A: ACN + 0,1% TFA; элюент B: вода + 0,1% TFA; градиент: от 5% A + 95% B до 95% A + 5% B в течение 2,5 мин, затем 95% A + 5% B в течение 0,5 мин; MS способ ионизации: ESI+
Способ LC8
Колонка: Waters XBridge C18, 33×2,1 мм, 4 мкм; скорость потока: 1,0 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 5% A + 95% B в течение 0,3 мин, затем до 95% A + 5% B в течение 3,2 мин, затем 95% A + 5% B в течение 0,5 мин; MS способ ионизации: ESI+
Способ LC9
Колонка: YMC J'sphere H80, 33×2,1 мм, 4 мкм; скорость потока: 1,0 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: 5% A + 95% B в течение 0,5 мин, затем до 95% A + 5% B в течение 3,0 мин, затем 95% A + 5% B в течение 0,5 мин; MS способ ионизации: ESI+
Способ LC10
Колонка: Luna C18, 10×2 мм, 3 мкм; скорость потока: 1,1 мл/мин; элюент A: ACN + 0,05% TFA; элюент B: вода + 0,05% TFA; градиент: от 7% A + 93% B до 95% A + 5% B в течение 1,2 мин, затем 95% A + 5% B в течение 0,2 мин; MS способ ионизации: ESI+
Способ LC11
Колонка: Waters XBridge C18, 33×2,1 мм, 4 мкм; скорость потока: 1,0 мл/мин; элюент A: ACN + 0,1% TFA; элюент B: вода + 0,08% TFA; градиент: от 3% A + 97% B до 60% A + 40% B в течение 3,5 мин, затем до 98% A + 2% B в течение 1,5 мин; MS способ ионизации: ESI+
Пример 1
[1-Фенил-2-(2-метилфенокси)-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
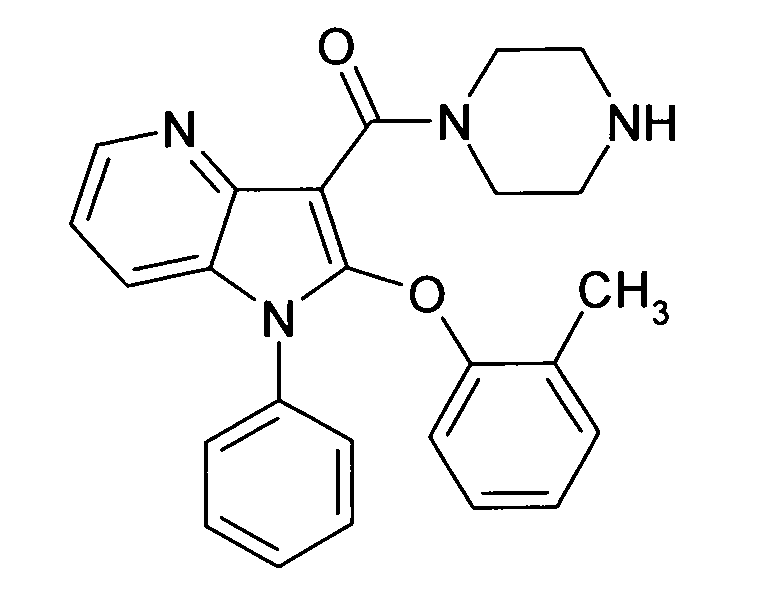
Стадия 1: 1-Фенил-1H-пирроло[3,2-b]пиридин
К смеси 4-азаиндола (1,20 г, 10,2 ммоль), йодида меди(I) (290 мг, 1,53 ммоль), 8-гидроксихинолина (221 мг, 1,53 ммоль) и карбоната калия (1,55 г, 11,2 ммоль) в DMSO (24 мл) добавляли йодбензол (1,25 мл, 11,2 ммоль). Реакционную смесь перемешивали при 130°C в течение 3 часов. Затем смесь охлаждали до комнатной температуры, и добавляли раствор гидроксида аммония (10% в воде) и EA. Органический слой отделяли, промывали дважды насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Колоночная хроматография на силикагеле (EA/HEP) давала 560 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=195
Стадия 2: 1-Фенил-1,3-дигидропирроло[3,2-b]пиридин-2-он
К перемешиваемому раствору 610 мг (3,14 ммоль) соединения стадии 1 в трет-бутаноле (23 мл) и воде (23 мл) добавляли по каплям в течение 20 мин бром (676 мкл, 13,2 ммоль). Затем реакционную смесь обрабатывали насыщенным раствором гидрокарбоната натрия до значения pH, равного приблизительно 6,5-7, и затем добавляли EA. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученный в результате твердый остаток растворяли в этаноле (45 мл), добавляли палладий на активированном угле (668 мг, 628 ммоль, 10%), и реакционную смесь гидрировали (6 бар H2) при комнатной температуре в течение ночи. Смесь фильтровали через целит, и растворитель удаляли при пониженном давлении для того, чтобы получить 660 мг неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=211
Стадия 3: 2-Хлор-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбальдегид
Раствор DMF (1,36 мл) в DCM (3,5 мл) охлаждали до 0°C и перемешивали под аргоном. В течение 15 мин добавляли хлорокись фосфора (1,32 мл, 14,1 ммоль), и смесь перемешивали в течение 30 мин при 0°C. Соединение стадии 2 (660 мг, 3,14 ммоль) растворяли в DCM (10 мл) и затем добавляли к охлажденному раствору пиридин (864 мкл, 10,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь медленно выливали на 300 мл льда, и через несколько минут добавляли DCM. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученный в результате твердый остаток растворяли в DCM (10 мл) и хлорокиси фосфора (1,32 мл, 14,1 ммоль) и нагревали при 100°C в течение 2 часов. После охлаждения смесь медленно выливали на 300 мл льда, и через несколько минут добавляли DCM. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Колоночная хроматография на силикагеле (EA/HEP) давала 508 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=257
Стадия 4: 2-Хлор-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбоновая кислота
Соединение стадии 3 (508 мг, 1,98 ммоль) растворяли в трет-бутаноле (25 мл) и 2-метил-2-бутене (5 мл), и добавляли раствор хлорита натрия (1,07 г, 11,9 ммоль) и дигидрофосфата натрия (950 мг, 7,92 ммоль) в воде (10 мл). Реакционную смесь перемешивали при комнатной температуре в течение 48 часов. Смесь разбавляли водой и экстрагировали EA. Органический слой сушили над сульфатом натрия, фильтровали, и растворитель удаляли при пониженном давлении для того, чтобы получить 474 мг неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=273
Стадия 5: трет-Бутиловый эфир 4-(2-хлор-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
К раствору 460 мг (1,69 ммоль) соединения стадии 4 в DMF (24 мл) и NMM (478 мкл, 4,35 ммоль) добавляли тетрафторборат O-(циано(этоксикарбонил)метиленамино)-N,N,N',N'-тетраметилурония (627 мг, 1,91 ммоль), и смесь перемешивали при комнатной температуре в течение 30 мин. Затем добавляли трет-бутил 1-пиперазинкарбоксилат (356 мг, 1,91 ммоль), и реакционную смесь перемешивали в течение ночи. Смесь гасили водой и экстрагировали EA. Органический слой отделяли, промывали насыщенным раствором гидрокарбоната натрия, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (EA (70-95%)/HEP). Получали 390 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=441
Стадия 6: трет-Бутиловый эфир 4-[1-фенил-2-(2-метилфенокси)-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
К раствору 2-метилфенола (36,8 мг, 133 мкмоль) в NMP (2 мл) добавляли гидрид натрия (15,0 мг, 375 мкмоль, 60% дисперсия в минеральном масле), и суспензию перемешивали при комнатной температуре под аргоном в течение 20 мин. После добавления 50,0 мг (113 мкмоль) соединения стадии 5 реакционную смесь перемешивали в течение 2 часов при 140°C в микроволновом реакторе. Смесь гасили водой и экстрагировали EA. Заявленное в заголовке соединение очищали хроматографией на силикагеле (EA (70-95%)/HEP) и непосредственно применяли в следующей стадии.
LC/MS (способ LC4): m/z=513
Стадия 7: [1-Фенил-2-(2-метилфенокси)-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Раствор соединения стадии 6 в DCM (12 мл) и TFA (3 мл) перемешивали при комнатной температуре в течение 2 часов. Растворители упаривали, и полученный в результате твердый остаток очищали препаративной ВЭЖХ. Фракции, содержащие заявленное в заголовке соединение, объединяли и лиофилизовали в течение ночи. Заявленное в заголовке соединение получали в форме биссоли трифторуксусной кислоты с [1-фенил-2-(2-метилфенокси)-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметаноном в виде твердого остатка белого цвета. Выход: 26,5 мг.
LC/MS (способ LC1): m/z=412,18; Rt=1,06 мин
1H-ЯМР: δ (ppm)=2,18 (с, 3H), 2,98 (уш. д, 4H), 3,60 (уш. д, 4H), 7,01-7,04 (м, 1H), 7,10-7,11 (м, 2H), 7,19 (д, 1H), 7,28 (кв, 1H), 7,50-7,54 (м, 1H), 7,56-7,61 (м, 4H), 7,67 (д, 1H), 8,46 (дд, 1H), 8,75 (уш. с, 2H).
Пример 2
(2-Бензил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил)пиперазин-1-илметанон
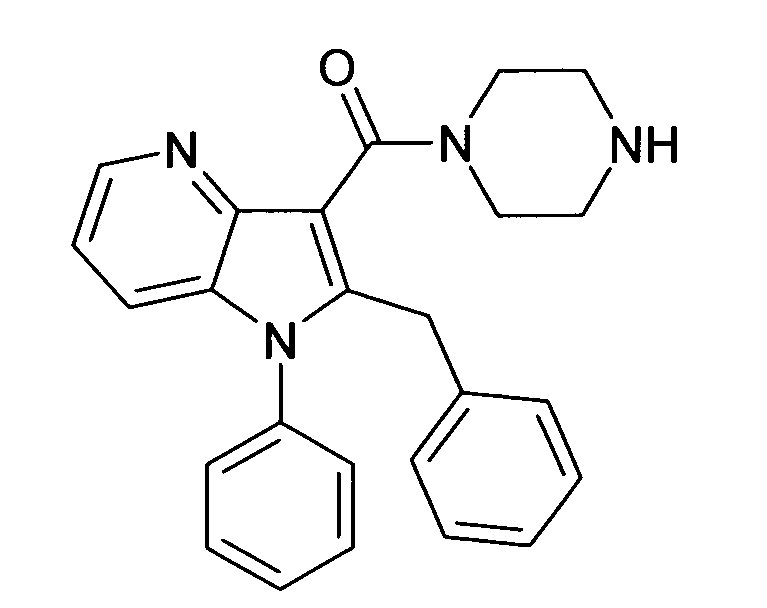
Стадия 1: трет-Бутиловый эфир 4-(2-бензил-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
К раствору ацетата палладия(II) (1,27 мг, 5,65 мкмоль) и S-PHOS (4,64 мг, 11,3 мкмоль) в DMF (1,5 мл) добавляли фосфат калия (71,9 мг, 339 мкмоль), соединение примера 1 стадии 5 (49,8 мг, 113 мкмоль) и B-бензил-9-борбицикло[3,3,1]нонан (452 мкл, 226 мкмоль, 0,5 M в THF). Реакционную смесь нагревали при 100°C в течение 1 часа и затем добавляли 2 н. раствор гидроксида натрия. Смесь экстрагировали EA, органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (EA (75-99%)/HEP). Получали 56,0 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=497
Стадия 2: (2-Бензил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил)пиперазин-1-илметанон
Из соединения стадии 1 (56,0 мг, 113 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с (2-бензил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил)пиперазин-1-илметаноном. Выход: 35,7 мг.
LC/MS (способ LC1): m/z=396,20; Rt=1,05 мин
1H-ЯМР: δ (ppm)=3,11 (уш. д, 4H), 3,60 (уш. с, 2H), 3,83 (уш. с, 2H), 4,22 (с, 2H), 6,86-6,88 (м, 2H), 7,10-7,14 (м, 3H), 7,24 (кв, 1H), 7,31-7,33 (м, 2H), 7,47 (д, 1H), 7,54-7,56 (м, 3H), 8,49 (дд, 1H), 8,82 (уш. с, 2H).
Пример 3
[2-(2-Метилбензил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
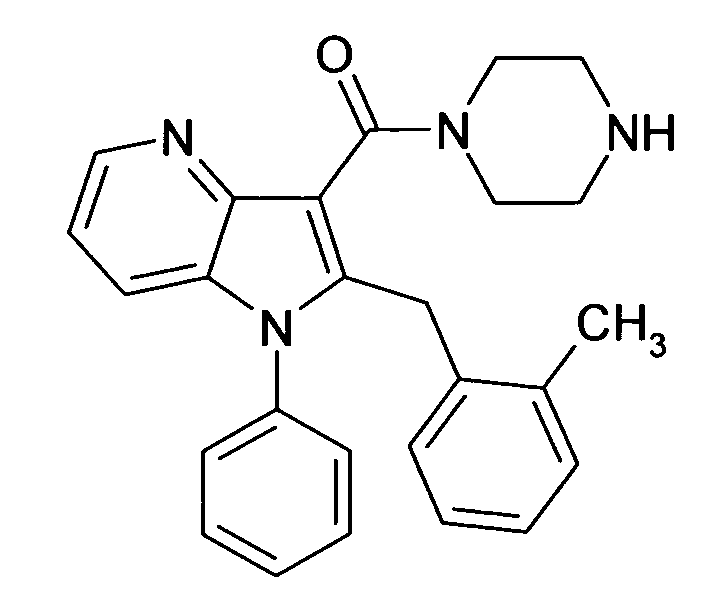
Стадия 1: трет-Бутиловый эфир 4-[2-(2-метилбензил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Раствор 2-метилбензилцинкхлорида (454 мкл, 227 мкмоль, 0,5 M в THF) добавляли по каплям при -78°C к раствору B-OM-9-BBN (1,13 мл, 1,13 ммоль, 1 M в гексане). Охлаждающую баню удаляли, и смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли DMF (2 мл), с последующим добавлением соединения примера 1, стадия 5, (50,0 мг, 113 мкмоль), ацетата палладия(II) (2,55 мг, 11,3 мкмоль) и S-PHOS (9,31 мг, 22,7 мкмоль). Реакционную смесь нагревали при 100°C с перемешиванием в течение 3,5 часов. После охлаждения смесь разбавляли водой и экстрагировали EA. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (EA/HEP). Фракции, содержащие заявленное в заголовке соединение, объединяли и упаривали для того, чтобы получить 38 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=511
Стадия 2: [2-(2-Метилбензил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Из соединения стадии 1 (38,0 мг, 74,4 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(2-метилбензил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 26,2 мг.
LC/MS (способ LC1): m/z=410,21 ; Rt=1,09 мин
1H-ЯМР: δ (ppm)=1,94 (с, 3H), 3,05 (уш. с, 4H), 3,56 (уш. с, 2H), 3,74 (уш. с, 2H), 4,15 (уш. с, 2H), 6,83 (д, 1H), 6,96-7,05 (м, 3H), 7,24 (кв, 1H), 7,32-7,34 (м, 2H), 7,47 (д, 1H), 7,51-7,54 (м, 3H), 8,49 (дд, 1H), 8,77 (уш. с, 2H).
Пример 4
[2-(3-Фтор-2-метилбензил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
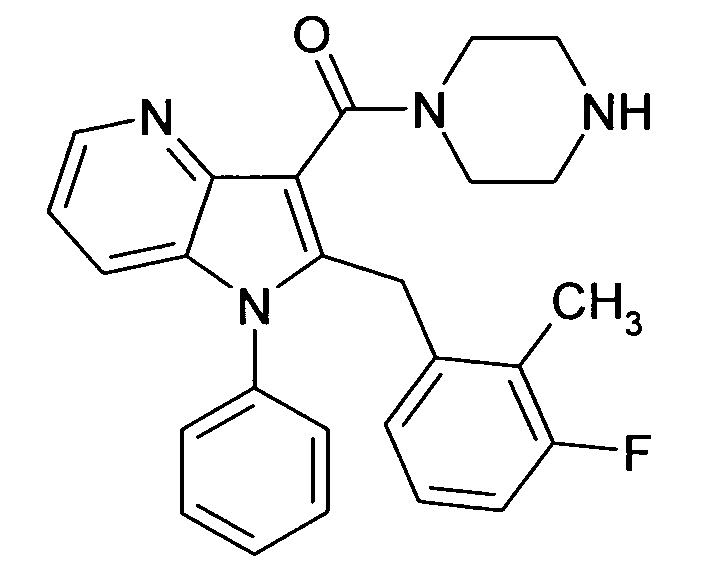
Стадия 1: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
К цинку (29,7 мг, 454 мкмоль) в сухом THF (300 мкл) в сухой колбе в атмосфере аргона добавляли 1,2-дибромэтан (0,49 мкл, 5,67 мкмоль). Смесь нагревали три раза до кипения термофеном. Через 5 мин колбу помещали в ледяную баню и медленно добавляли раствор 3-фтор-2-метилбензилбромида (23,0 мг, 227 мкмоль) в сухом THF (700 мкл) так, чтобы температура поддерживалась равной 0°C. Смесь перемешивали при 0°C в течение 3 часов. Затем охлажденную суспензию добавляли по каплям к предварительно охлажденному раствору (-78°C) B-OM-9-BBN (1,13 мл, 1,13 ммоль, 1 M в гексане). Смесь перемешивали при комнатной температуре в течение 30 мин. Затем добавляли DMF (4 мл), с последующим добавлением соединения примера 1, стадия 5, (50,0 мг, 113 мкмоль), ацетата палладия(II) (2,55 мг, 11,3 мкмоль) и S-PHOS (9,31 мг, 22,7 мкмоль). Реакционную смесь перемешивали при 100°C в течение 3,5 часов. После охлаждения смесь гасили водой и экстрагировали EA. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток очищали хроматографией для того, чтобы получить 44 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=529
Стадия 2: [2-(3-Фтор-2-метилбензил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Из соединения стадии 1 (43,0 мг, 81,3 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 30 мг.
LC/MS (способ LC1): m/z=428,20; Rt=1,13 мин
1H-ЯМР: δ (ppm)=1,85 (с, 3H), 3,10 (уш. с, 4H), 3,59 (уш. с, 2H), 3,76 (уш. с, 2H), 4,21 (с, 2H), 6,69 (д, 1H), 6,92 (т, 1H), 7,00 (кв, 1H), 7,25 (кв, 1H), 7,31-7,33 (м, 2H), 7,48 (д, 1H), 7,51-7,53 (м, 3H), 8,50 (дд, 1H), 8,82 (уш. с, 2H).
Пример 5
[2-(5-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
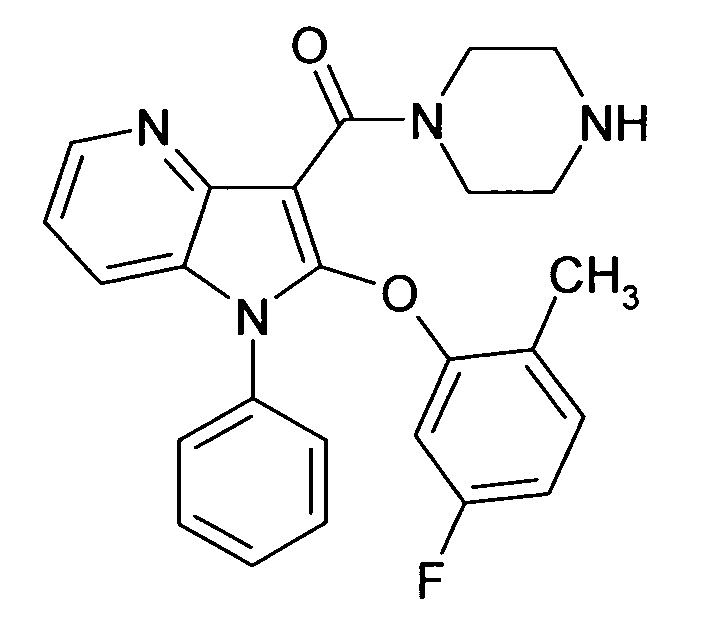
Стадия 1: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Из соединения примера 1, стадия 5, (50,0 мг, 113 мкмоль) и 5-фтор-2-метилфенола неочищенное заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 6.
LC/MS (способ LC4): m/z=531
Стадия 2: [2-(5-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из неочищенного соединения стадии 1 аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 30,6 мг.
LC/MS (способ LC1): m/z=430,18; Rt=1,10 мин
1H-ЯМР: δ (ppm)=2,13 (с, 3H), 3,04 (уш. д, 4H), 3,66 (уш. д, 4H), 6,88 (дт, 1H), 7,14 (дд, 1H), 7,22 (т, 1H), 7,28 (кв, 1H), 7,50-7,54 (м, 1H), 7,57-7,62 (м, 4H), 7,65 (д, 1H), 8,47 (дд, 1H), 8,81 (уш. с, 2H).
Пример 6
[2-(3-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
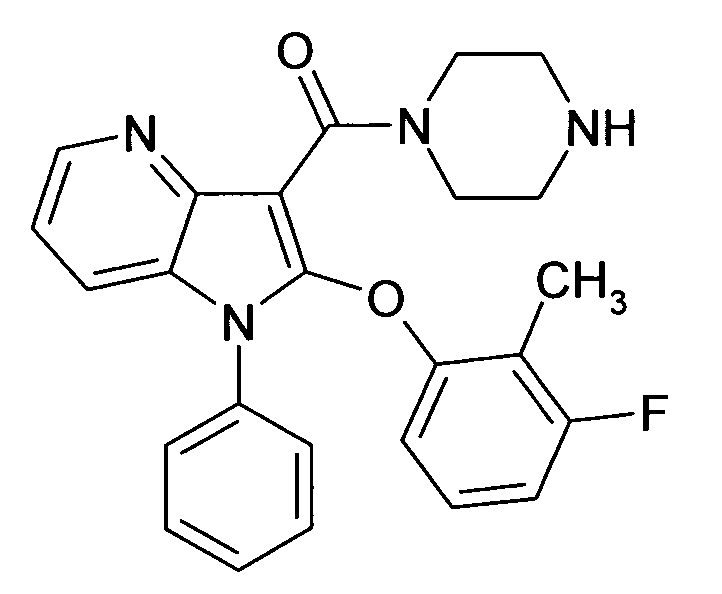
Стадия 1: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Из соединения примера 1, стадия 5, (50,0 мг, 113 мкмоль) и 3-фтор-2-метилфенола неочищенное заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 6.
LC/MS (способ LC4): m/z=531
Стадия 2: [2-(3-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из неочищенного соединения стадии 1 аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(3-фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 29,9 мг.
LC/MS (способ LC1): m/z=430,18; Rt=1,11 мин
1H-ЯМР: δ (ppm)=2,09 (с, 3H), 3,02 (уш. д, 4H), 3,63 (уш. д, 4H), 6,95 (т, 1H), 7,00 (д, 1H), 7,13 (кв, 1H), 7,28 (кв, 1H), 7,50-7,54 (м, 1H), 7,56-7,61 (м, 4H), 7,65 (д, 1H), 8,47 (дд, 1H), 8,73 (уш. с, 2H).
Пример 7
[2-(2-Фтор-6-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
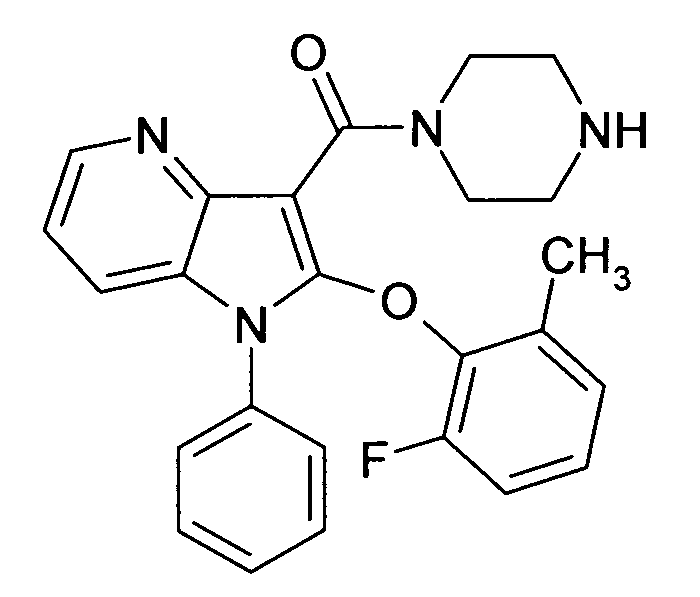
Стадия 1: трет-Бутиловый эфир 4-[2-(2-фтор-6-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Из соединения примера 1, стадия 5, (30,0 мг, 68 мкмоль) и 2-фтор-6-метилфенола неочищенное заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 6.
LC/MS (способ LC4): m/z=531
Стадия 2: [2-(2-Фтор-6-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из неочищенного соединения стадии 1 аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(2-фтор-6-метилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 6,1 мг.
LC/MS (способ LC4): m/z=431,1; Rt=0,934 мин
1H-ЯМР (500 МГц, CDCl3): δ (ppm)=2,16 (с, 3H), 3,19 (уш. с, 4H), 3,70 (уш. с, 4H), 6,79 (д, 1H), 6,90 (т, 1H), 7,13 (кв, 1H), 7,40 (кв, 1H), 7,46 (дд, 2H), 7,56-7,61 (м, 3H), 7,83 (д, 1H), 8,58 (д, 1H), 9,90 (уш. с, 2H).
Пример 8
[2-(3-Фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
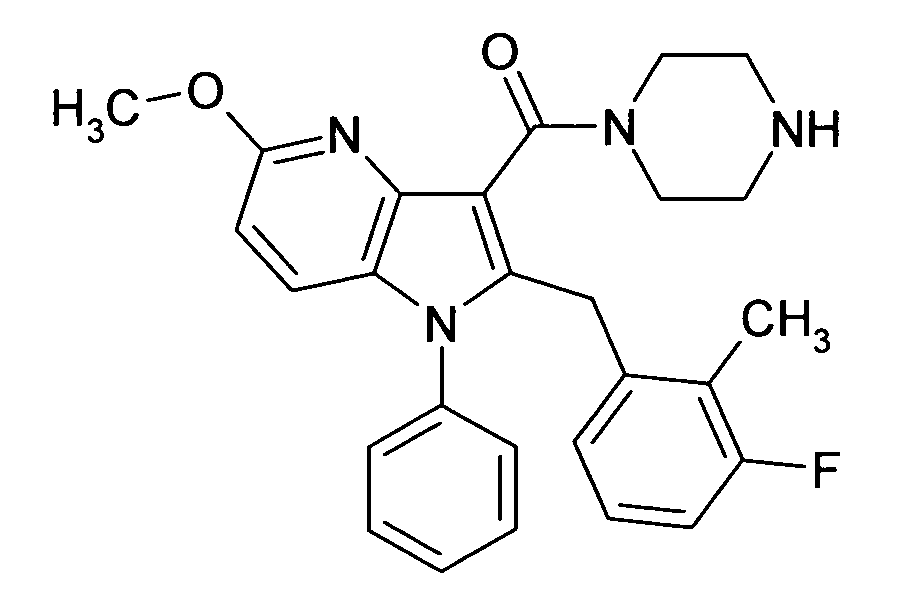
Стадия 1: 5-Метокси-1-фенил-1H-пирроло[3,2-b]пиридин
Заявленное в заголовке соединение получали из 5-метокси-1H-пирроло[3,2-b]пиридина (1,59 г, 10,7 ммоль; см., D. Mazeas et al., Heterocycles 50 (1999), 1065) аналогично, как описано в примере 1, стадия 1. Выход: 1,83 г.
LC/MS (способ LC4): m/z=225
Стадия 2: 5-Метокси-1-фенил-1,3-дигидропирроло[3,2-b]пиридин-2-он
Заявленное в заголовке соединение получали из соединения стадии 1 (1,70 г, 7,58 ммоль) аналогично, как описано в примере 1, стадия 2. Выход: 2,30 г.
LC/MS (способ LC4): m/z=241
Стадия 3: 2-Хлор-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 2 (790 мг, 3,29 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 820 мг.
LC/MS (способ LC4): m/z=287
Стадия 4: 2-Хлор-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбоновая кислота
Заявленное в заголовке соединение получали из соединения стадии 3 (920 мг, 3,21 ммоль) аналогично, как описано в примере 1, стадия 4, за исключением того, что реакционную смесь перемешивали при 100°C в течение 2 часов. Выход: 1,09 г.
LC/MS (способ LC4): m/z=303
Стадия 5: трет-Бутиловый эфир 4-(2-хлор-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 4 (658 мг, 3,53 ммоль) аналогично, как описано в примере 1, стадия 5. Выход: 1,12 г.
LC/MS (способ LC4): m/z=471
Стадия 6: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 5 (100 мг, 212 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 83 мг.
LC/MS (способ LC4): m/z=559
Стадия 7: [2-(3-Фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 6 (23 мг, 41,2 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 15,9 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC3): m/z=458,21; Rt=1,55 мин
1H-ЯМР: δ (ppm)=1,84 (с, 3H), 3,11 (уш. с, 4H), 3,70 (уш. д, 4H), 3,93 (с, 3H), 4,16 (с, 2H), 6,63 (д, 1H), 6,66 (д, 1H), 6,91 (т, 1H), 7,00 (кв, 1H), 7,28-7,30 (м, 2H), 7,35 (д, 1H), 7,49-7,52 (м, 3H), 9,04 (уш. с, 2H).
Пример 9
[2-(3-Фтор-2-метилбензил)-5-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
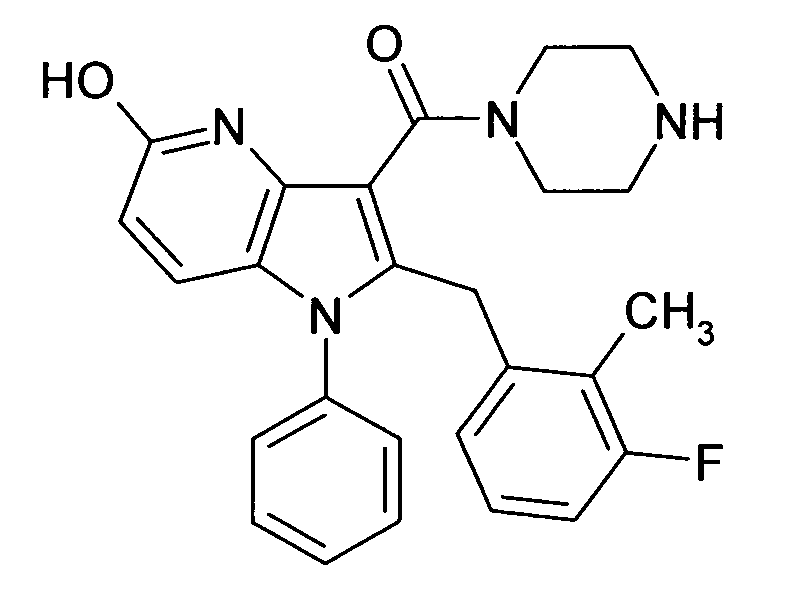
Раствор трибромида бора (537 мкл, 537 мкмоль, 1 M в DCM) добавляли по каплям при -78°C к раствору соединения примера 8, стадия 6, (50,0 мг, 89,5 мкмоль) в DCM (2 мл). Охлаждающую баню удаляли, и смесь перемешивали при комнатной температуре в течение ночи. Смесь снова охлаждали до -78°C и медленно добавляли трибромид бора (3,58 мл, 3,58 ммоль, 1 M в DCM). Охлаждающую баню удаляли, и смесь перемешивали при 65°C в течение 8 дней. Смесь медленно выливали в лед, и через несколько минут добавляли DCM. Водный слой отделяли и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (7 M аммиак в MOH (1,5-15%)/DCM). Полученный твердый остаток растворяли в небольшом количестве MOH, добавляли хлороводородную кислоту (0,1 M), и смесь лиофилизовали в течение ночи для того, чтобы получить 21,3 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-5-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC1): m/z=444,20; Rt=1,17 мин
1H-ЯМР (400 МГц, DMSO-d6): δ (ppm)=1,84 (с, 3H), 2,98 (уш. с, 4H), 4,04 (с, 2H), 6,29 (д, 1H), 6,67 (д, 1H), 6,91 (т, 1H), 7,01 (кв, 1H), 7,31-7,33 (м, 3H), 7,50-7,53 (м, 3H), 9,29 (уш. с, 2H).
Пример 10
[2-(5-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
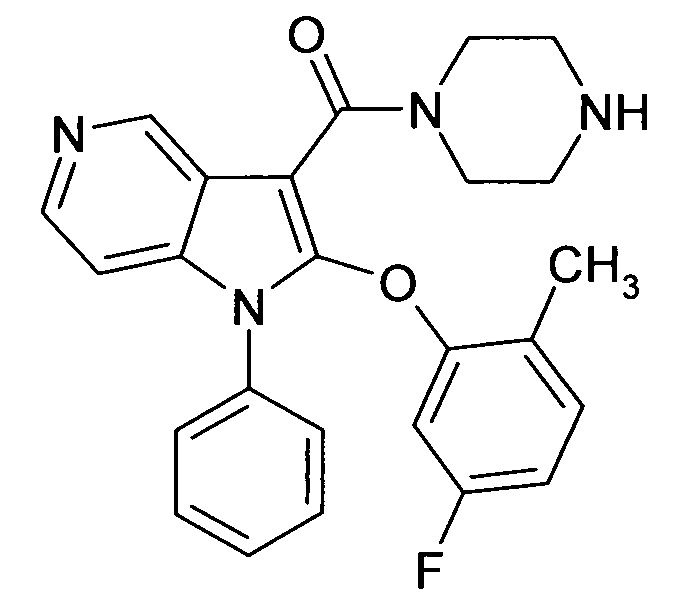
Стадия 1: 1-Фенил-1H-пирроло[3,2-c]пиридин
К смеси 5-азаиндола (780 мг, 6,60 ммоль), йодида меди(I) (25,1 мг, 132 мкмоль), (1S,2S)-(+)-1,2-диаминоциклогексана (162 мкл, 1,35 ммоль) и фосфата калия (2,52 г, 11,9 ммоль) в диоксане (24 мл) добавляли йодбензол (739 мкл, 6,60 ммоль). Реакционную смесь перемешивали в течение ночи при 110°C. Затем смесь охлаждали до комнатной температуры, фильтровали через силикагель, и силикагель промывали EA. Объединенные фильтраты упаривали при пониженном давлении, и полученный в результате твердый остаток очищали препаративной ВЭЖХ. Фракции, содержащие заявленное в заголовке соединение, объединяли и лиофилизовали в течение ночи. Выход: 1,28 г.
LC/MS (способ LC4): m/z=195
Стадия 2: 1-Фенил-1,3-дигидропирроло[3,2-c]пиридин-2-он
Неочищенное заявленное в заголовке соединение получали из соединения стадии 1 (1,28 г, 6,60 ммоль) аналогично, как описано в примере 1, стадия 2.
LC/MS (способ LC4): m/z=211
Стадия 3: 2-Хлор-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из неочищенного соединения стадии 2 аналогично, как описано в примере 1, стадия 3. Выход: 480 мг.
LC/MS (способ LC4): m/z=257
Стадия 4: 2-Хлор-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбоновая кислота
Заявленное в заголовке соединение получали из соединения стадии 3 (480 мг, 1,87 ммоль) аналогично, как описано в примере 1, стадия 4, за исключением того, что реакционную смесь перемешивали при 100°C в течение 90 мин. Выход: 1,14 г.
LC/MS (способ LC4): m/z=273
Стадия 5: трет-Бутиловый эфир 4-(2-хлор-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 4 (460 мг, 1,69 ммоль) аналогично, как описано в примере 1, стадия 5. Выход: 325 мг.
LC/MS (способ LC4): m/z=441
Стадия 6: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 5 (187 мг, 424 мкмоль) и 5-фтор-2-метилфенола аналогично, как описано в примере 1, стадия 6. Выход: 173 мг.
LC/MS (способ LC4): m/z=531
Стадия 7: [2-(5-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 6 (173 мг, 327 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 66 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC3): m/z=430,18; Rt=0,93 мин
1H-ЯМР (400 МГц, DMSO-d6): δ (ppm)=2,14 (с, 3H), 3,01 (уш. с, 4H), 3,70 (уш. с, 4H), 6,92 (дт, 1H), 7,12 (дд, 1H), 7,24 (т, 1H), 7,57-7,72 (м, 6H), 8,54 (д, 1H), 9,32 (с, 1H), 9,45 (уш. с, 2H).
Пример 11
(2-Бензил-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил)пиперазин-1-илметанон
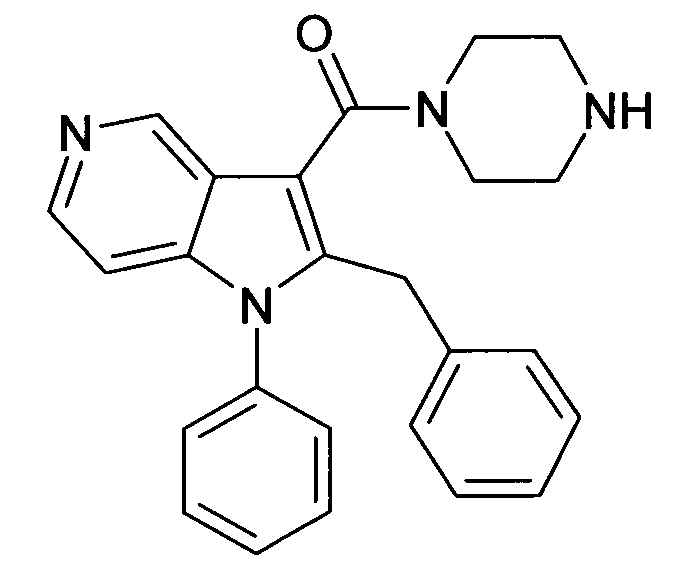
Стадия 1: трет-Бутиловый эфир 4-(2-бензил-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Из соединения примера 10, стадия 5, (49,8 мг, 113 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 2, стадия 1. Выход: 37 мг.
LC/MS (способ LC4): m/z=497
Стадия 2: (2-Бензил-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил)пиперазин-1-илметанон
Из соединения стадии 1 (37,0 мг, 74,5 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с (2-бензил-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил)-пиперазин-1-илметаноном. Выход: 10,9 мг.
LC/MS (способ LC1): m/z=396,20; Rt=1,07 мин
1H-ЯМР: δ (ppm)=3,72 (уш. с, 4H), 4,19 (с, 2H), 6,82-6,84 (м, 2H), 7,11-7,13 (м, 3H), 7,39 (уш. с, 2H), 7,49 (д, 1H), 7,57-7,63 (м, 3H), 8,47 (д, 1H), 8,91 (уш. с, 2H), 9,33 (с, 1H).
Пример 12
[2-(2-Метилбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
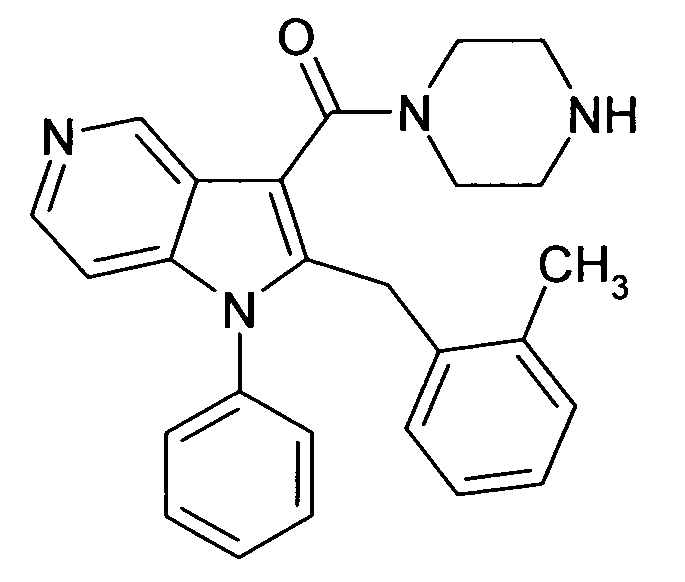
Стадия 1: трет-Бутиловый эфир 4-[2-(2-метилбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Из соединения примера 10, стадия 5, (49,8 мг, 113 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 3, стадия 1. Выход: 44 мг.LC/MS (способ LC4): m/z=511
Стадия 2: [2-(2-Метилбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Из соединения стадии 1 (44,0 мг, 86,2 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(2-метилбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 17,7 мг.
LC/MS (способ LC1): m/z=410,21; Rt=1,08 мин
1H-ЯМР: δ (ppm)=1,92 (с, 3H), 2,95 (уш. с, 2H), 3,16 (уш. с, 2H), 4,13 (с, 2H), 6,81 (д, 1H), 6,96-7,06 (м, 3H), 7,40 (уш. с, 2H), 7,48 (уш. д, 1H), 7,55-7,60 (м, 3H), 8,47 (д, 1H), 8,89 (уш. с, 2H), 9,31 (с, 1H).
Пример 13
[2-(2-Хлор-6-фторбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
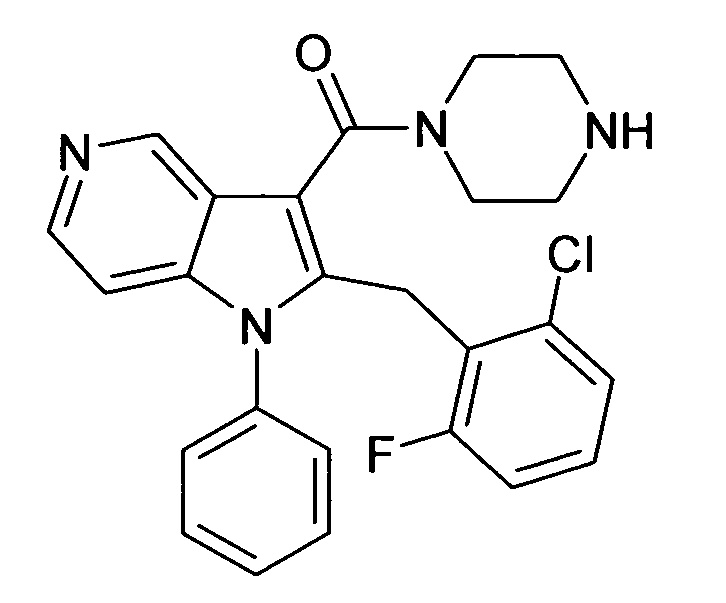
Стадия 1: трет-Бутиловый эфир 4-[2-(2-хлор-6-фторбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Из соединения примера 10, стадия 5, (49,8 мг, 113 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 4, стадия 1. Выход: 26 мг.
LC/MS (способ LC4): m/z=550
Стадия 2: [2-(2-Хлор-6-фторбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Из соединения стадии 1 (26,0 мг, 47,4 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(2-хлор-6-фторбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 2,7 мг.
LC/MS (способ LC4): m/z=449,10; Rt=0,865 мин
1H-ЯМР (400 МГц, MOH-D4): δ (ppm)=3,12 (уш. с, 2H), 3,65 (уш. с, 2H), 3,92 (уш. с, 2H), 4,39 (с, 2H), 7,00 (дт, 1H), 7,15 (д, 1H), 7,24-7,30 (м, 1H), 7,45-7,48 (м, 2H), 7,56 (уш. с, 1H), 7,61-7,66 (м, 3H), 8,41 (уш. с, 1H), 9,21 (уш. с, 1H).
Пример 14
[2-(3-Фтор-2-метилбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
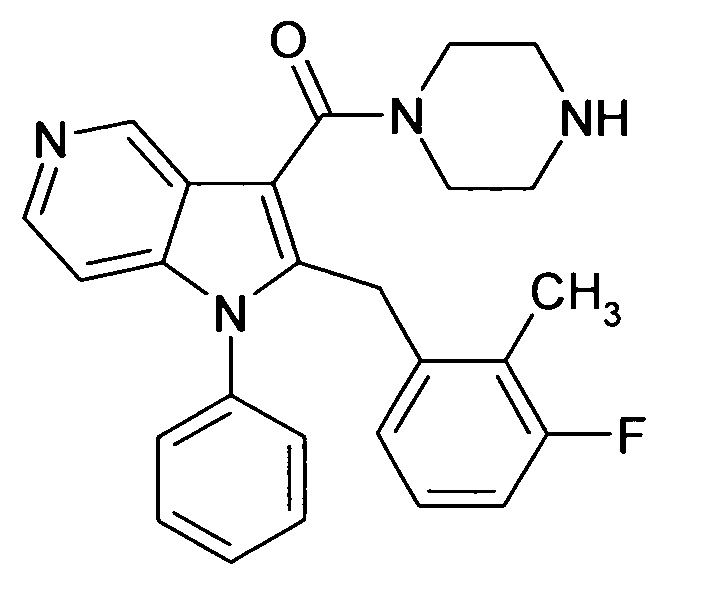
Стадия 1: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Из соединения примера 10, стадия 5, (97 мг, 220 мкмоль) заявленное в заголовке соединение получали аналогично, как описано в примере 4, стадия 1. Выход: 30 мг.
LC/MS (способ LC4): m/z=529
Стадия 2: [2-(3-Фтор-2-метилбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (28,3 мг, 53,5 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 20,1 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC1): m/z=428,20; Rt=1,04 мин
1H-ЯМР: δ (ppm)=1,82 (с, 3H), 3,08 (уш. д, 4H), 3,71 (уш. д, 4H), 4,20 (с, 2H), 6,67 (д, 1H), 6,93 (т, 1H), 7,00 (кв, 1H), 7,41 (уш. с, 2H), 7,52 (д, 1H), 7,55-7,60 (м, 3H), 8,47 (д, 1H), 9,43 (с, 1H).
Пример 15
[2-(5-Фтор-2-метилфенокси)-1-(4-фторфенил)-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
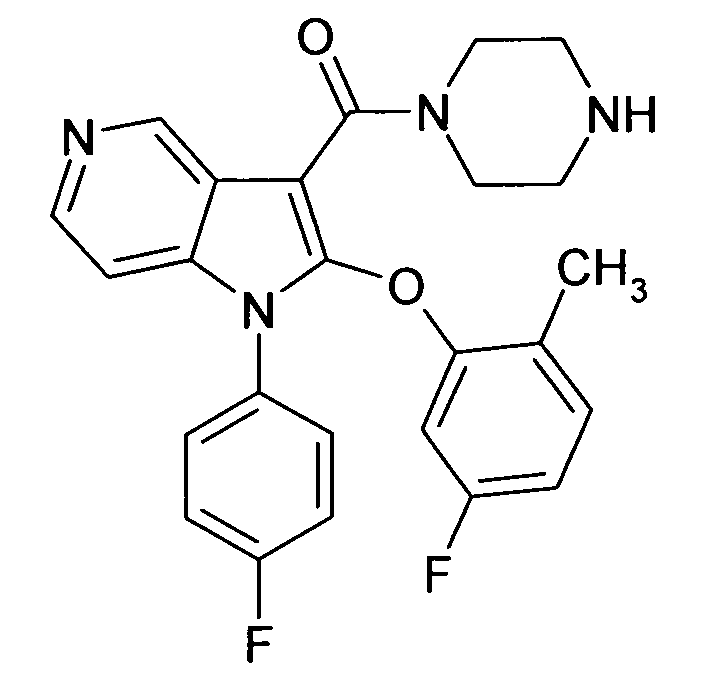
Стадия 1: 1-(4-Фторфенил)-1H-пирроло[3,2-c]пиридин
Заявленное в заголовке соединение получали из 5-азаиндола (1,00 г, 8,47 ммоль) и 1-фтор-4-йодбензола аналогично, как описано в примере 1, стадия 1. Выход: 1,23 г.
LC/MS (способ LC4): m/z=213
Стадия 2: 1-(4-Фторфенил)-1,3-дигидропирроло[3,2-c]пиридин-2-он
Заявленное в заголовке соединение получали из соединения стадии 1 (1,23 г, 5,79 ммоль) аналогично, как описано в примере 1, стадия 2. Выход: 1,27 г.
LC/MS (способ LC4): m/z=229
Стадия 3: 2-Хлор-1-(4-фторфенил)-1H-пирроло[3,2-c]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 2 (1,27 г, 5,56 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 278 мг.
LC/MS (способ LC4): m/z=275
Стадия 4: 2-Хлор-1-(4-фторфенил)-1H-пирроло[3,2-c]пиридин-3-карбоновая кислота
Неочищенное заявленное в заголовке соединение получали из соединения стадии 3 (278 мг, 1,01 ммоль) аналогично, как описано в примере 1, стадия 4, за исключением того, что реакционную смесь перемешивали при 100°C в течение 2 часов.
LC/MS (способ LC4): m/z=291
Стадия 5: трет-Бутиловый эфир 4-[2-хлор-1-(4-фторфенил)-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из неочищенного соединения стадии 4 аналогично, как описано в примере 1, стадия 5. Выход: 386 мг.
LC/MS (способ LC4): m/z=459
Стадия 6: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-1-(4-фторфенил)-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Неочищенное заявленное в заголовке соединение получали из соединения стадии 5 (50,0 мг, 109 мкмоль) и 5-фтор-2-метилфенола аналогично, как описано в примере 1, стадия 6.
LC/MS (способ LC4): m/z=549
Стадия 7: [2-(5-Фтор-2-метилфенокси)-1-(4-фторфенил)-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из неочищенного соединения стадии 6 аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(5-фтор-2-метилфенокси)-1-(4-фторфенил)-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 25,3 мг.
LC/MS (способ LC1): m/z=448,17; Rt=1,10 мин
1H-ЯМР: δ (ppm)=2,14 (с, 3H), 3,04 (уш. с, 4H), 3,66 (уш. с, 4H), 6,93 (дт, 1H), 7,11 (дд, 1H), 7,26 (т, 1H), 7,48-7,52 (м, 2H), 7,74-7,77 (м, 3H), 8,59 (д, 1H), 9,04 (уш. с, 2H), 9,29 (с, 1H).
Пример 16
[1-Фенил-2-(2-метилфенокси)-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
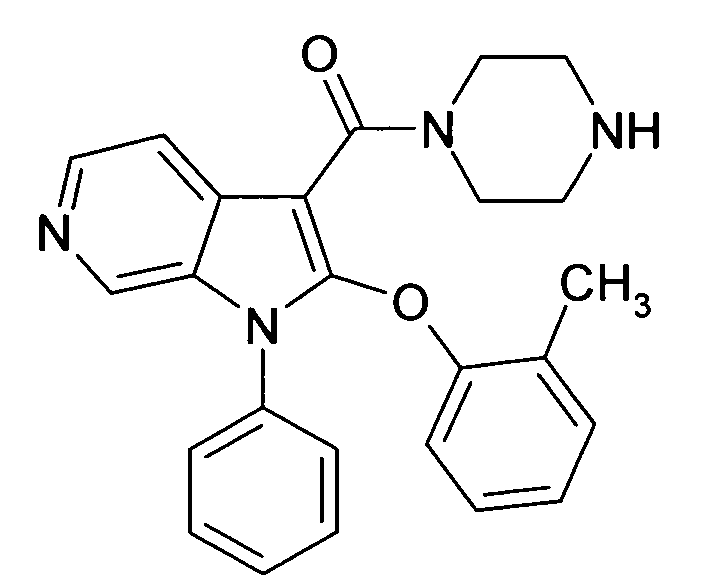
Стадия 1: 1-Фенил-1H-пирроло[2,3-c]пиридин
Заявленное в заголовке соединение получали из 6-азаиндола (1,00 г, 8,47 ммоль) аналогично, как описано в примере 10, стадия 1. Выход: 1,20 г.
LC/MS (способ LC4): m/z=195
Стадия 2: 1-Фенил-1,3-дигидропирроло[2,3-c]пиридин-2-он
Неочищенное заявленное в заголовке соединение получали из соединения стадии 1 (400 мг, 2,06 ммоль) аналогично, как описано в примере 1, стадия 2.
LC/MS (способ LC4): m/z=211
Стадия 3: 2-Хлор-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из неочищенного соединения стадии 2 аналогично, как описано в примере 1, стадия 3. Выход: 111 мг.
LC/MS (способ LC4): m/z=257
Стадия 4: 2-Хлор-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбоновая кислота
Заявленное в заголовке соединение получали из соединения стадии 3 (218 мг, 0,85 ммоль) аналогично, как описано в примере 1, стадия 4, за исключением того, что реакционную смесь перемешивали при 100°C в течение 2 часов. Выход: 217 мг.
LC/MS (способ LC4): m/z=272
Стадия 5: трет-Бутиловый эфир 4-(2-хлор-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 4 (217 мг, 796 мкмоль) аналогично, как описано в примере 1, стадия 5. Выход: 191 мг.
LC/MS (способ LC4): m/z=441
Стадия 6: трет-Бутиловый эфир 4-[1-фенил-2-(2-метилфенокси)-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 5 (43,0 мг, 97,5 мкмоль) аналогично, как описано в примере 1, стадия 6, и очисткой препаративной ВЭЖХ. Выход: 27 мг.
LC/MS (способ LC4): m/z=513
Стадия 7: [1-Фенил-2-(2-метилфенокси)-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-ил-метанон
Заявленное в заголовке соединение получали из соединения стадии 6 (27,0 мг, 52,7 мкмоль) аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [1-фенил-2-(2-метилфенокси)-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 24 мг.
LC/MS (способ LC1): m/z=412,19; Rt=0,98 мин
1H-ЯМР: δ (ppm)=2,21 (с, 3H), 2,96 (уш. с, 4H), 7,11-7,19 (м, 2H), 7,23 (д, 1H), 7,27 (д, 1H), 7,61-7,64 (м, 1H), 7,66-7,70 (м, 2H), 7,73-7,76 (м, 2H), 8,05 (д, 1H), 8,46 (д, 1H), 8,88 (уш. с, 3H).
Пример 17
(2-Бензил-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил)пиперазин-1-илметанон
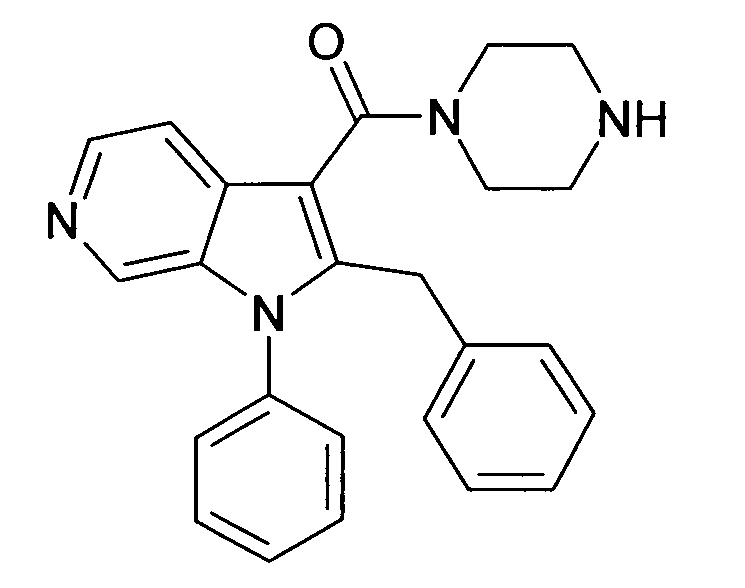
Стадия 1: трет-Бутиловый эфир 4-(2-бензил-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Неочищенное заявленное в заголовке соединение получали из соединения примера 16, стадия 5, (50,0 мг, 113 мкмоль) аналогично, как описано в примере 2, стадия 1. Выход: 105 мг.
LC/MS (способ LC4): m/z=497
Стадия 2: (2-Бензил-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил)пиперазин-1-илметанон
Заявленное в заголовке соединение получали из соединения стадии 1 (56,3 мг, 113 мкмоль) аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с (2-бензил-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил)пиперазин-1-илметаноном. Выход: 25 мг.
LC/MS (способ LC1): m/z=396,20; Rt=0,93 мин
1H-ЯМР: δ (ppm)=2,90 (уш. с, 2H), 3,21 (уш. с, 2H), 4,25 (с, 2H), 6,87-6,89 (м, 2H), 7,13-7,16 (м, 3H), 7,51 (уш. с, 2H), 7,61-7,64 (м, 3H), 8,13 (д, 1H), 8,45 (д, 1H), 8,77 (с, 1H), 8,99 (уш. д, 2H).
Пример 18
[2-(3-Фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
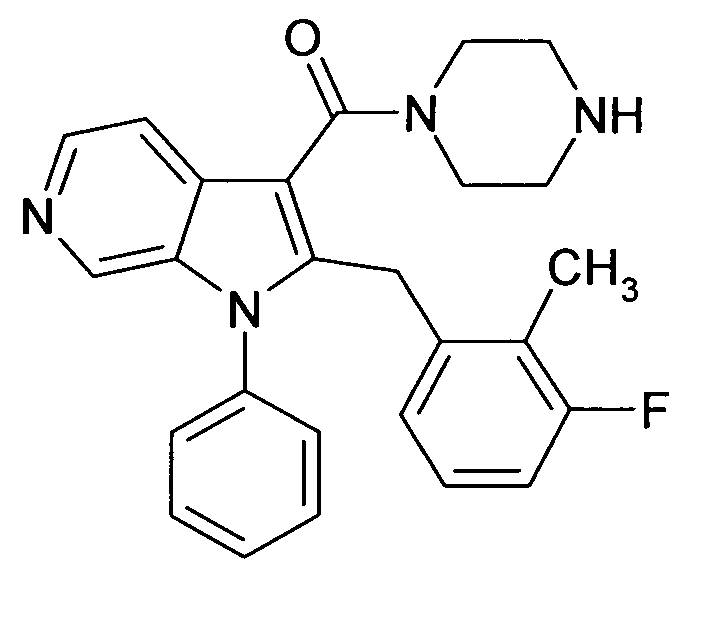
Стадия 1: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 16, стадия 5, (100 мг, 227 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 180 мг.
LC/MS (способ LC4): m/z=529
Стадия 2: [2-(3-Фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (120 мг, 226 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 36,1 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC3): m/z=428,20; Rt=1,06 мин
1H-ЯМР: δ (ppm)=1,85 (с, 3H), 2,90 (уш. с, 2H), 3,18 (уш. с, 2H), 3,74 (уш. с, 2H), 4,28 (уш. с, 2H), 6,70 (д, 1H), 6,95 (т, 1H), 7,01 (кв, 1H), 7,51 (уш. с, 2H), 7,58-7,61 (м, 3H), 8,22 (д, 1H), 8,47 (д, 1H), 8,77 (с, 1H), 9,41 (уш. с, 1H), 9,60 (уш. с, 1H).
Пример 19
[2-(5-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
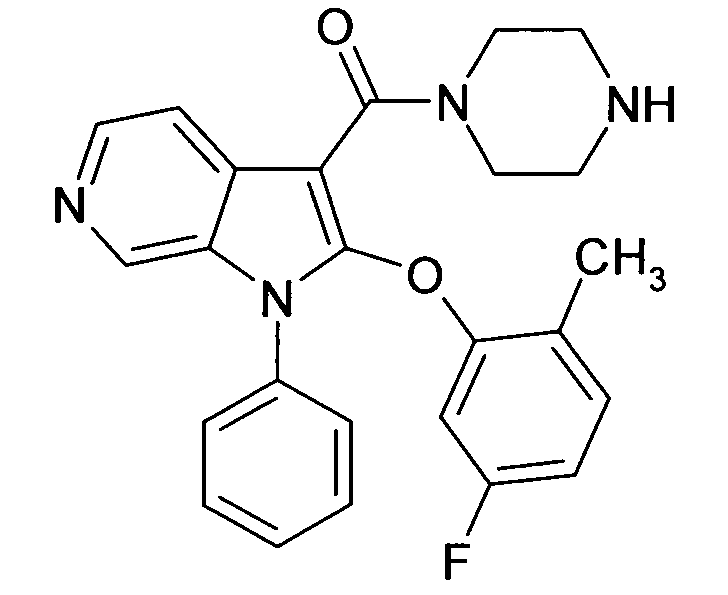
Стадия 1: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Неочищенное заявленное в заголовке соединение получали из соединения примера 16, стадия 5, (100 мг, 227 мкмоль) и 5-фтор-2-метилфенола аналогично, как описано в примере 1, стадия 6.
LC/MS (способ LC4): m/z=531
Стадия 2: [2-(5-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Неочищенное соединение стадии 1 реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 25,9 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC3): m/z=430,18; Rt=1,04 мин
1H-ЯМР: δ (ppm)=2,17 (с, 3H), 2,98 (уш. с, 4H), 3,62 (уш. с, 4H), 6,99 (дт, 1H), 7,30 (т, 2H), 7,62 (т, 1H), 7,68 (т, 2H), 7,75 (д, 2H), 8,10 (д, 1H), 8,47 (д, 1H), 8,87 (с, 1H), 9,40 (уш. с, 2H).
Пример 20
[2-(5-Фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
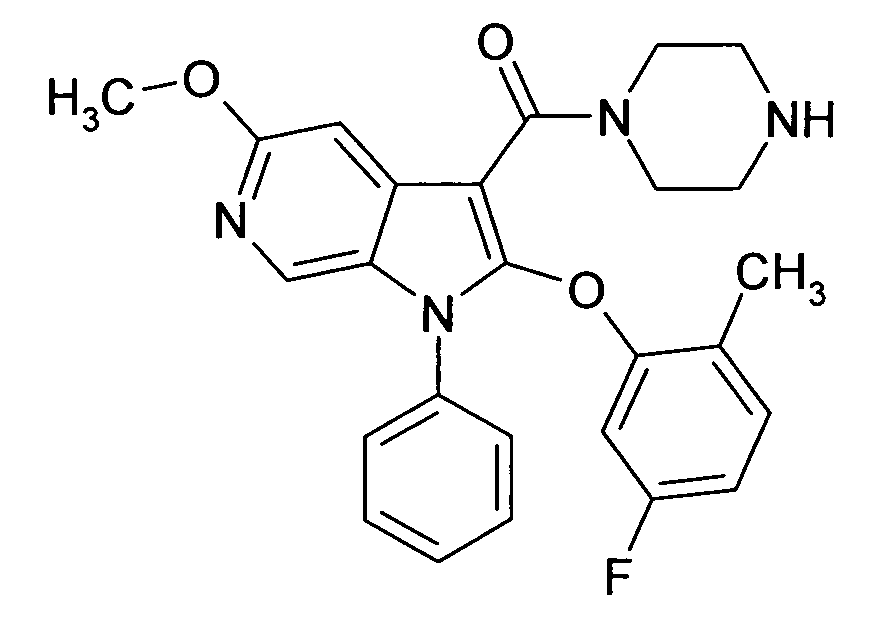
Стадия 1: 5-Метокси-1-фенил-1H-пирроло[2,3-c]пиридин
К смеси 5-метокси-1H-пирроло[2,3-c]пиридина (6,00 г, 40,5 ммоль; см., D. Mazeas et al., Heterocycles 50 (1999), 1065), ацетилацетоната меди(II) (1,06 г, 4,05 ммоль) и карбоната калия (11,2 г, 81,0 ммоль) в DMSO (63 мл) добавляли йодбензол (4,99 мл, 44,6 ммоль). Реакционную смесь перемешивали при 130°C в течение 10 часов. Затем смесь охлаждали до комнатной температуры, и добавляли раствор хлорида аммония (20% в воде). Смесь фильтровали через целит, и фильтрат экстрагировали три раза EA. Органические слои объединяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Колоночная хроматография остатка на силикагеле (EA/HEP) давала 8,43 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=225
Стадия 2: 5-Метокси-1-фенил-1,3-дигидропирроло[2,3-c]пиридин-2-он
Заявленное в заголовке соединение получали из соединения стадии 1 (7,68 г, 34,3 ммоль) аналогично, как описано в примере 1, стадия 2. Выход: 2,28 г.
LC/MS (способ LC4): m/z=241
Стадия 3: 2-Хлор-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 2 (2,23 г, 9,29 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 914 мг.
LC/MS (способ LC4): m/z=287
Стадия 4: 2-Хлор-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбоновая кислота
Неочищенное заявленное в заголовке соединение получали из соединения стадии 3 (790 мг, 2,76 ммоль) аналогично, как описано в примере 1, стадия 4, за исключением того, что реакционную смесь перемешивали при 60°C в течение 2 часов.
LC/MS (способ LC4): m/z=303
Стадия 5: трет-Бутиловый эфир 4-(2-хлор-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
К раствору неочищенного соединения стадии 4, трет-бутилу 1-пиперазинкарбоксилата (565 мг, 3,03 ммоль), гидрохлориду N-этил-N'-(3-диметиламинопропил)карбодиимида (581 мг, 3,03 ммоль) и гидрата 1-гидроксибензотриазола (312 мг, 2,29 ммоль) в DMF (20 мл) добавляли NMM (911 мкл, 8,27 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение ночи. Смесь гасили водой и экстрагировали EA. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (EA (5-35%)/HEP) для того, чтобы получить 653 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=471
Стадия 6: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 5 (100 мг, 212 мкмоль) и 5-фтор-2-метилфенола аналогично, как описано в примере 1, стадия 6. Выход: 98,0 мг.
LC/MS (способ LC4): m/z=561
Стадия 7: [2-(5-Фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 6 (27,0 мг, 48,2 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 21,9 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC1): m/z=460,19; Rt=1,06 мин
1H-ЯМР: δ (ppm)=2,13 (с, 3H), 2,98 (уш. с, 4H), 3,89 (с, 3H), 6,89 (дт, 1H), 6,96 (с, 1H), 7,04 (дд, 1H), 7,23 (т, 1H), 7,50 (т, 1H), 7,57-7,63 (м, 4H), 8,14 (с, 1H), 9,06 (уш. с, 2H).
Пример 21
[2-(5-Фтор-2-метилфенокси)-5-гидрокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
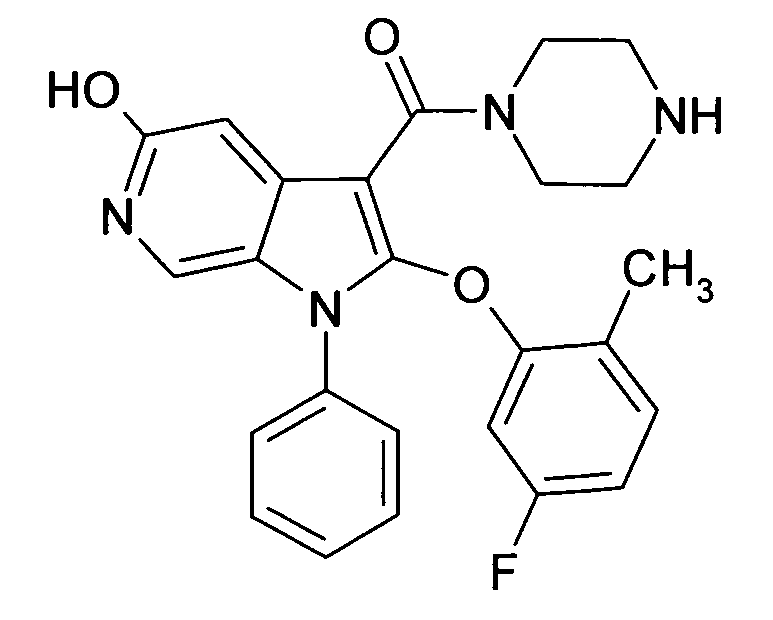
Заявленное в заголовке соединение получали из соединения примера 20, стадия 6, (67,0 мг, 119 мкмоль) аналогично, как описано в примере 9, и получали в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-5-гидрокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона. Выход: 35,5 мг.
LC/MS (способ LC1): m/z=446,18; Rt=1,07 мин
1H-ЯМР: δ (ppm)=2,15 (с, 3H), 2,99 (уш. с, 4H), 3,61 (уш. с, 4H), 6,95 (дт, 1H), 7,03 (с, 1H), 7,21 (д, 1H), 7,27 (т, 1H), 7,54 (т, 1H), 7,60-7,67 (м, 4H), 8,10 (с, 1H), 9,27 (уш. с, 2H).
Пример 22
[2-(3-Фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
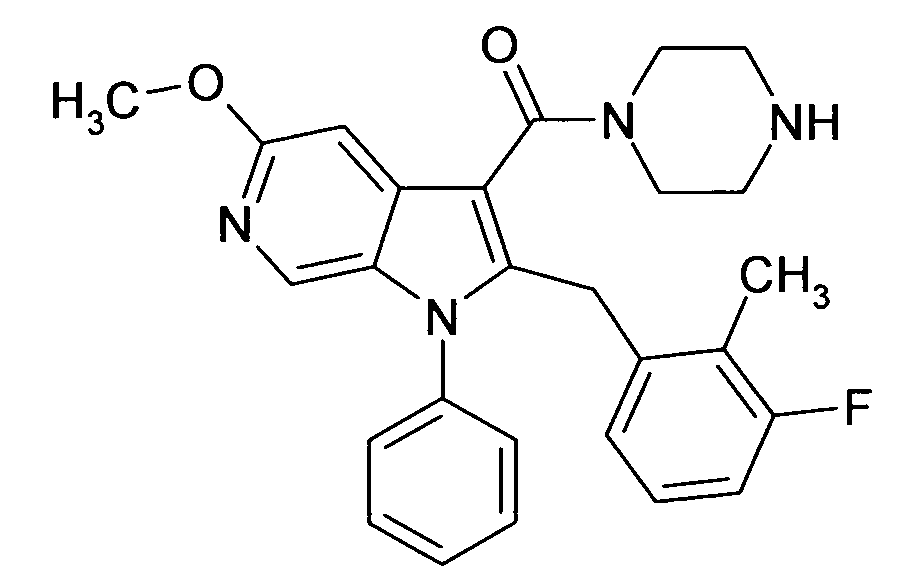
Стадия 1: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 20, стадия 5, (104 мг, 220 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 89 мг.
LC/MS (способ LC4): m/z=559
Стадия 2: [2-(3-Фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 ((25,0 мг, 44,7 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 18,4 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC1): m/z=458,21 ; Rt=1,08 мин
1H-ЯМР: δ (ppm)=1,85 (с, 3H), 2,86 (уш. с, 2H), 3,18 (уш. с, 2H), 3,90 (с, 3H), 4,15 (с, 2H), 6,67 (д, 1H), 6,92 (т, 1H), 6,98-7,02 (м, 2H), 7,37-7,39 (м, 2H), 7,51-7,54 (м, 3H), 7,99 (с, 1H), 9,19 (уш. с, 2H).
Пример 23
[2-(3-Фтор-2-метилбензил)-5-гидрокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
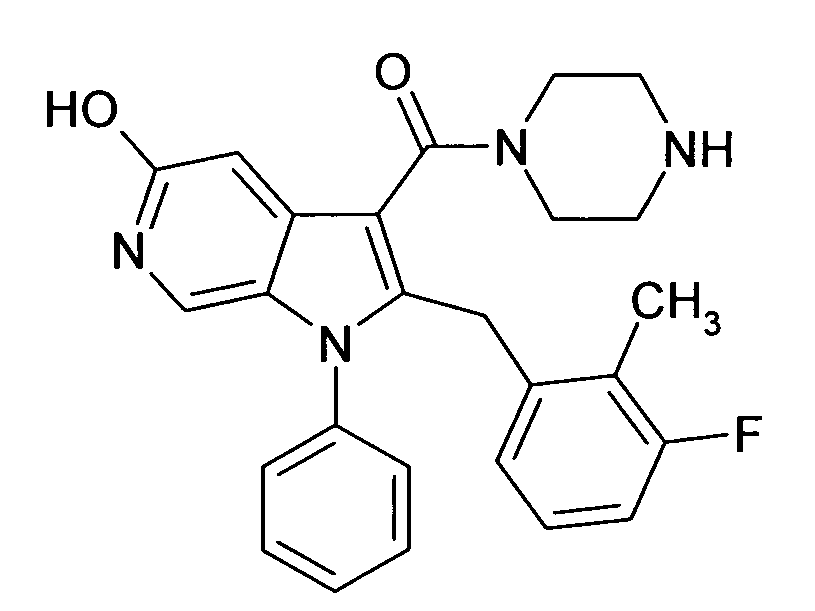
Заявленное в заголовке соединение получали из соединения примера 22, стадия 1, (60,0 мг, 107 мкмоль) аналогично, как описано в примере 9. и получали в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-5-гидрокси-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона. Выход: 29,3 мг.
LC/MS (способ LC1): m/z=444,20; Rt=1,09 мин
1H-ЯМР: δ (ppm)=1,84 (с, 3H), 2,93 (уш. с, 2H), 3,17 (уш. с, 2H), 3,56 (уш. с, 2H), 3,37 (уш. с, 2H), 4,20 (с, 2H), 6,68 (д, 1H), 6,94 (т, 1H), 7,01 (кв, 1H), 7,17 (с, 1H), 7,41-7,43 (м, 2H), 7,53-7,55 (м, 3H), 8,01 (с, 1H), 9,28 (уш. с, 1H), 9,56 (уш. с, 1H).
Пример 24
[2-(5-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
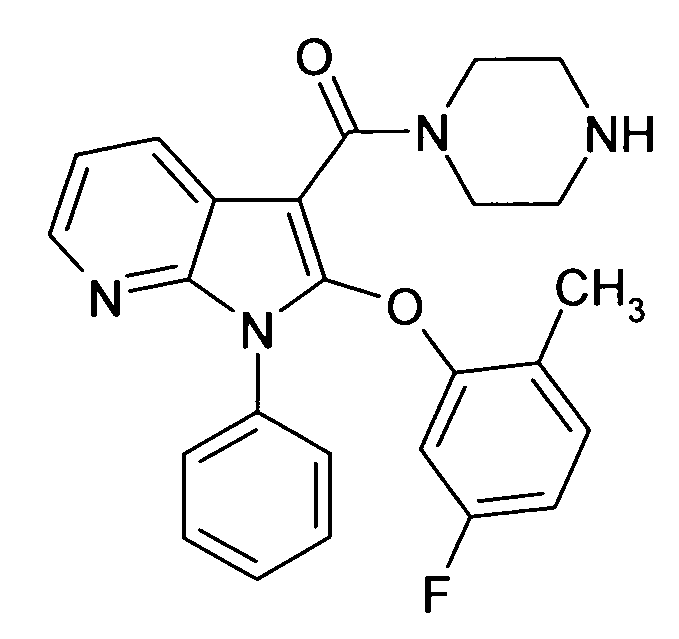
Стадия 1: 1-Фенил-1H-пирроло[2,3-b]пиридин
Заявленное в заголовке соединение получали из 7-азаиндола (1,18 г, 10,0 ммоль) аналогично, как описано в примере 10, стадия 1. Выход: 960 мг.
LC/MS (способ LC4): m/z=195
Стадия 2: 3,3-Дибром-1-фенил-1,3-дигидропирроло[2,3-b]пиридин-2-он
К перемешиваемому раствору соединения стадии 1 (960 мг, 4,94 ммоль) в трет-бутаноле (36 мл) под аргоном добавляли бромидпербромид пиридиния (6,32 г, 19,8 ммоль) в течение 2 часов небольшими порциями при температуре между 30°C и 35°C. Суспензию перемешивали при комнатной температуре в течение ночи. Растворитель упаривали, и полученный в результате твердый остаток растворяли в EA и воде. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (EA (15-40%)/HEP). Фракции, содержащие заявленное в заголовке соединение, объединяли и упаривали. Выход: 1,46 г.
LC/MS (способ LC4): m/z=369
Стадия 3: 1-Фенил-1,3-дигидропирроло[2,3-b]пиридин-2-он
Соединение стадии 2 (1,40 г, 3,80 ммоль) растворяли в этаноле (160 мл), и добавляли палладий на активированном угле (700 мг, 658 мкмоль, 10%). Реакционную смесь гидрировали (1 бар H2) при комнатной температуре в течение ночи. Смесь фильтровали через целит, и фильтрат упаривали при пониженном давлении. Колоночная хроматография остатка на силикагеле (EA (50%)/HEP) давала 1,10 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=211
Стадия 4: 2-Хлор-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 3 (799 мг, 3,80 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 290 мг.
LC/MS (способ LC4): m/z=257
Стадия 5: 2-Хлор-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбоновая кислота
Заявленное в заголовке соединение получали из соединения стадии 4 (280 мг, 1,09 ммоль) аналогично, как описано в примере 1, стадия 4, за исключением того, что реакционную смесь перемешивали при 100°C в течение 2 часов. Выход: 220 мг.
LC/MS (способ LC4): m/z=273
Стадия 6: трет-Бутиловый эфир 4-(2-хлор-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 5 (220 мг, 807 мкмоль) аналогично, как описано в примере 1, стадия 5. Выход: 292 мг.
LC/MS (способ LC4): m/z=441
Стадия 7: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 6 (50,0 мг, 113 мкмоль) и 5-фтор-2-метилфенола аналогично, как описано в примере 1, стадия 6, и очисткой препаративной ВЭЖХ. Выход: 28 мг.
LC/MS (способ LC4): m/z=531
Стадия 8: [2-(5-Фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из соединения стадии 6 (28,0 мг, 52,7 мкмоль) аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 30 мг.
LC/MS (способ LC1): m/z=430,18; Rt=1,23 мин
1H-ЯМР: δ (ppm)=2,13 (с, 3H), 3,01 (уш. с, 4H), 3,67 (уш. с, 4H), 6,85 (дт, 1H), 6,93 (дд, 1H), 7,21 (т, 1H), 7,32 (кв, 1H), 7,42-7,46 (м, 1H), 7,50-7,57 (м, 4H), 8,06 (д, 1H), 8,27 (д, 1H), 8,82 (уш. с, 2H).
Пример 25
(2-Бензил-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил)пиперазин-1-илметанон
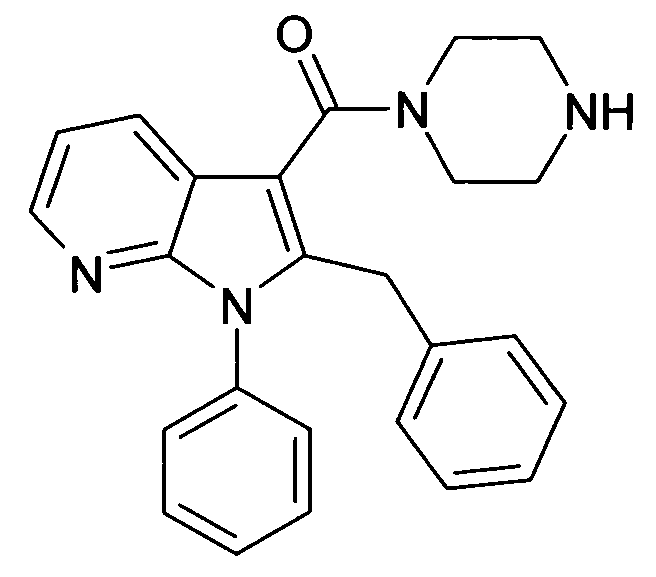
Стадия 1: трет-Бутиловый эфир 4-(2-бензил-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 24, стадия 6, (49,8 мг, 113 мкмоль) аналогично, как описано в примере 2, стадия 1. Выход: 45 мг.
LC/MS (способ LC4): m/z=497
Стадия 2: (2-Бензил-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил)пиперазин-1-илметанон
Заявленное в заголовке соединение получали из соединения стадии 1 (40,0 мг, 80,5 мкмоль) аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с (2-бензил-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил)пиперазин-1-илметаноном. Выход: 19 мг.
LC/MS (способ LC1): m/z=396,20; Rt=1,17 мин
1H-ЯМР: δ (ppm)=2,93 (уш. с, 2H), 3,17 (уш. с, 2H), 4,18 (с, 2H), 6,83-6,85 (м, 2H), 7,10-7,12 (м, 3H), 7,26-7,29 (м, 3H), 7,48-7,50 (м, 3H), 8,05 (дд, 1H), 8,21 (дд, 1H), 8,93 (уш. с, 2H).
Пример 26
[2-(2-Метилбензил)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
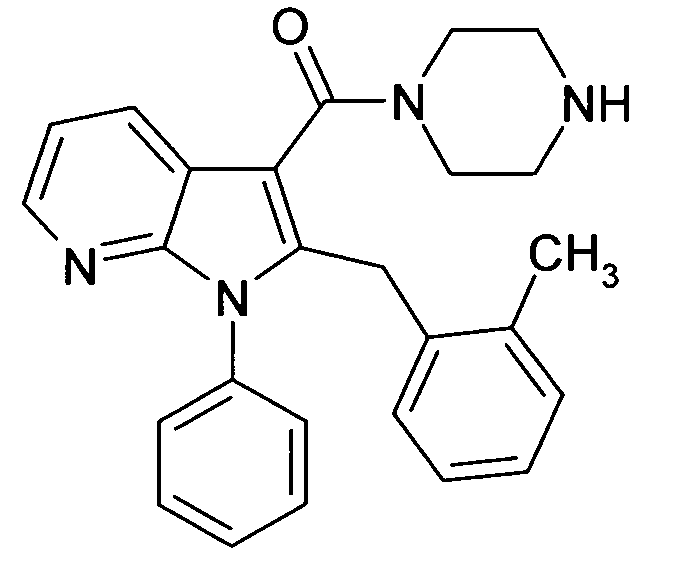
Стадия 1: трет-Бутиловый эфир 4-[2-(2-метилбензил)-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 24, стадия 6, (66,0 мг, 150 мкмоль) аналогично, как описано в примере 3, стадия 1, и очисткой препаративной ВЭЖХ в виде порошка белого цвета. Выход: 32 мг.
LC/MS (способ LC4): m/z=511
Стадия 2: [2-(2-Метилбензил)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из соединения стадии 1 (30,0 мг, 58,8 мкмоль) аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(2-метилбензил)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 29,4 мг.
LC/MS (способ LC1): m/z=410,21 ; Rt=1,17 мин
1H-ЯМР: δ (ppm)=1,93 (с, 3H), 2,86 (уш. с, 2H), 3,15 (уш. с, 2H), 4,12 (с, 2H), 6,82 (д, 1H), 6,95-7,04 (м, 3H), 7,26-7,29 (м, 3H), 7,45-7,48 (м, 3H), 8,04 (дд, 1H), 8,22 (дд, 1H), 8,84 (уш. с, 2H).
Пример 27
[2-(3-Фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
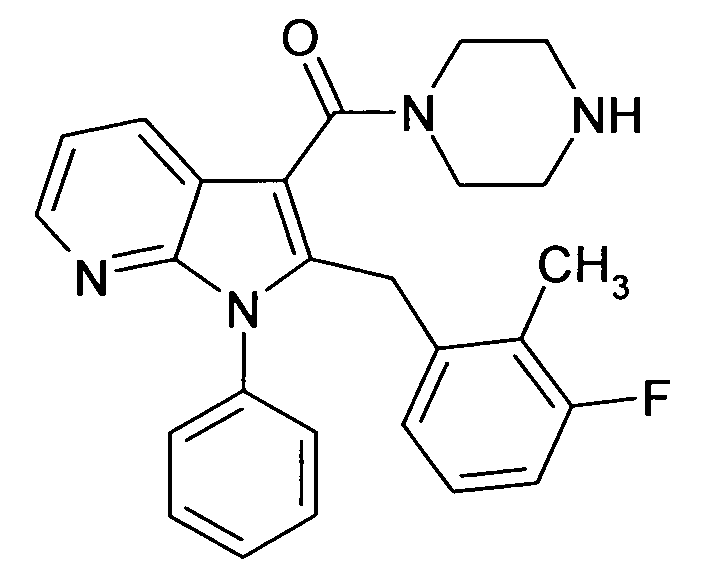
Стадия 1: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 24, стадия 6, (66,0 мг, 150 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 20 мг.
LC/MS (способ LC4): m/z=529
Стадия 2: [2-(3-Фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из соединения стадии 1 (20,0 мг, 37,8 мкмоль) аналогично, как описано в примере 1, стадия 7, и получали в форме биссоли трифторуксусной кислоты с [2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметаноном. Выход: 15 мг.
LC/MS (способ LC1): m/z=428,20; Rt=1,23 мин
1H-ЯМР: δ (ppm)=1,84 (с, 3H), 2,95 (уш. с, 2H), 3,16 (уш. с, 2H), 3,62 (уш. с, 4H), 4,17 (с, 2H), 6,67 (д, 1H), 6,91 (т, 1H), 6,99 (кв, 1H), 7,26-7,29 (м, 3H), 7,45-7,47 (м, 3H), 8,06 (дд, 1H), 8,22 (дд, 1H), 8,80 (уш. с, 2H).
Пример 28
[2-(5-Фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
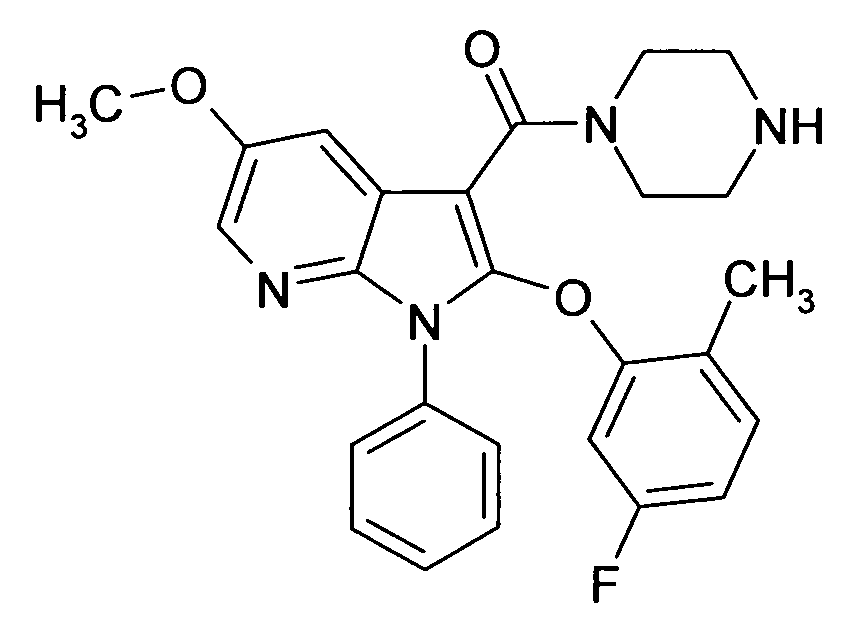
Стадия 1: 5-Метокси-1-фенил-1H-пирроло[2,3-b]пиридин
Заявленное в заголовке соединение получали из 5-метокси-1H-пирроло[2,3-b]пиридина (6,00 г, 40,5 ммоль) аналогично, как описано в примере 20, стадия 1. Выход: 5,69 г.
LC/MS (способ LC4): m/z=225
Стадия 2: 5-Метокси-1-фенил-1,3-дигидропирроло[2,3-b]пиридин-2-он
Заявленное в заголовке соединение получали из соединения стадии 1 (5,68 г, 25,3 ммоль) аналогично, как описано в примере 1, стадия 2. Выход: 1,71 г.
LC/MS (способ LC4): m/z=241
Стадия 3: 2-Хлор-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 2 (1,56 г, 6,49 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 475 мг.
LC/MS (способ LC4): m/z=287
Стадия 4: 2-Хлор-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбоновая кислота
Заявленное в заголовке соединение получали из соединения стадии 3 (500 мг, 1,74 ммоль) аналогично, как описано в примере 1, стадия 4, за исключением того, что реакционную смесь перемешивали при 45°C в течение 5 часов. Выход: 490 мг.
LC/MS (способ LC4): m/z=303
Стадия 5: трет-Бутиловый эфир 4-(2-хлор-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 4 (490 мг, 1,62 ммоль) аналогично, как описано в примере 20, стадия 5. Выход: 580 мг.
LC/MS (способ LC4): m/z=471
Стадия 6: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 5 (100 мг, 212 мкмоль) и 5-фтор-2-метилфенола аналогично, как описано в примере 1, стадия 6. Выход: 99,0 мг.
LC/MS (способ LC4): m/z=561
Стадия 7: [2-(5-Фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 6 (30,0 мг, 53,5 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 20,4 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC1): m/z=460,19; Rt=1,31 мин
1H-ЯМР: δ (ppm)=2,13 (с, 3H), 2,99 (уш. с, 4H), 3,70 (уш. с, 4H), 3,88 (с, 3H), 6,81-6,87 (м, 2H), 7,20 (т, 1H), 7,42 (м, 1H), 7,48-7,55 (м, 4H), 7,60 (д, 1H), 8,02 (д, 1H), 9,13 (уш. с, 2H).
Пример 29
[2-(5-Фтор-2-метилфенокси)-5-гидрокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
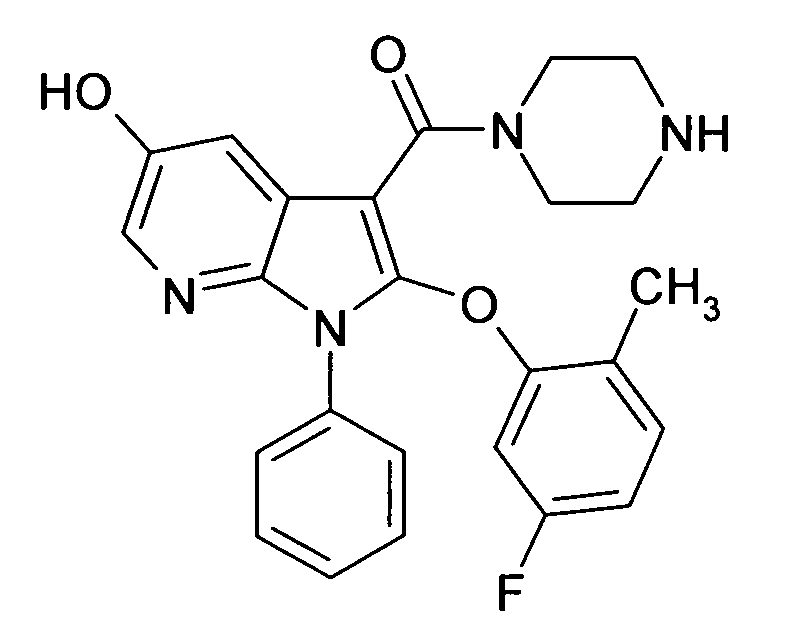
Заявленное в заголовке соединение получали из соединения примера 28, стадия 6, (70,0 мг, 125 мкмоль) аналогично, как описано в примере 9, и получали в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-5-гидрокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]-пиперазин-1-илметанона. Выход: 32,0 мг.
LC/MS (способ LC 1): m/z=446,18; Rt=1,26 мин
1H-ЯМР: δ (ppm)=2,12 (с, 3H), 2,98 (уш. с, 4H), 3,69 (уш. с, 4H), 6,80-6,84 (м, 2H)1 7,19 (кв, 1H), 7,38-7,42 (м, 2H), 7,47-7,54 (м, 4H), 7,88 (д, 1H), 9,20 (уш. с, 2H), 9,65 (уш. с, 1H).
Пример 30
[2-(3-Фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
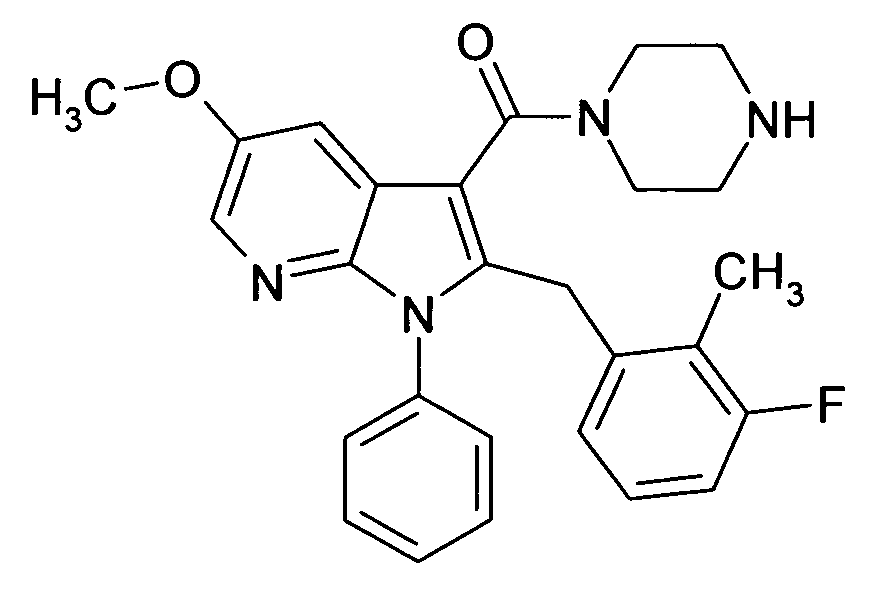
Стадия 1: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 28, стадия 5, (100 мг, 212 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 110 мг.
LC/MS (способ LC4): m/z=559
Стадия 2: [2-(3-Фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (35,0 мг, 62,6 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 17,6 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-5-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC2): m/z=458,21 ; Rt=2,68 мин
1H-ЯМР: δ (ppm)=1,84 (с, 3H), 2,90 (уш. с, 2H), 3,19 (уш. с, 2H), 3,72 (уш. с, 2H), 3,87 (с, 3H), 4,15 (с, 2H), 6,65 (д, 1H), 6,90 (т, 1H), 6,99 (кв, 1H), 7,25-7,27 (м, 2H), 7,44-7,46 (м, 3H), 7,56 (д, 1H), 7,97 (д, 1H), 9,05 (уш. с, 2H).
Пример 31
[2-(3-Фтор-2-метилбензил)-5-гидрокси-1-фенил-1H-пирроло[2, 3-b]пиридин-3-ил]пиперазин-1-илметанон
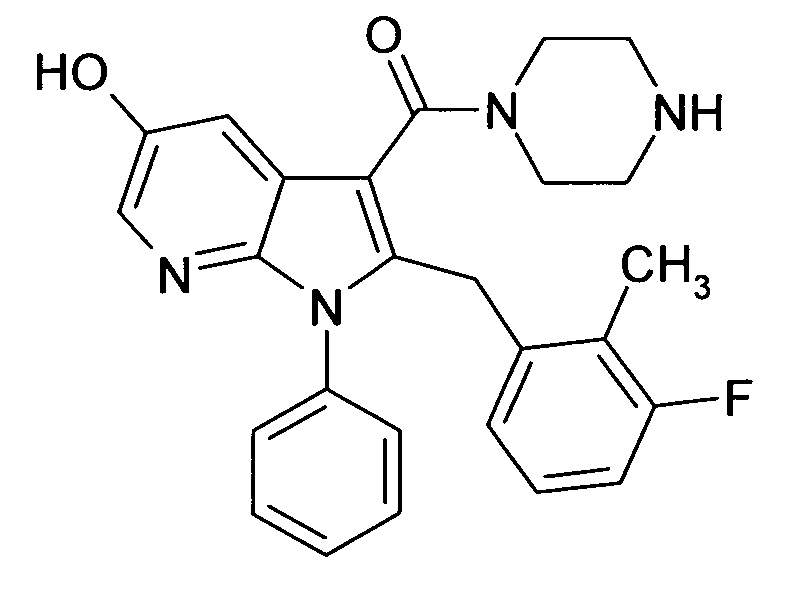
Заявленное в заголовке соединение получали из соединения примера 22, стадия 1, (72,0 мг, 129 мкмоль) аналогично, как описано в примере 9, и получали в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-5-гидрокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанона. Выход: 11,2 мг.
LC/MS (способ LC1): m/z=444,20; Rt=1,21 мин
1H-ЯМР: δ (ppm)=1,83 (с, 3H), 2,92 (уш. с, 2H), 3,14 (уш. с, 2H), 4,15 (с, 2H), 6,64 (д, 1H), 6,90 (т, 1H), 6,99 (кв, 1H), 7,23-7,25 (м, 2H), 7,35 (т, 1H), 7,42-7,45 (м, 3H), 6,84 (д, 1H), 9,05 (уш. д, 2H), 9,57 (уш. с, 1H).
Пример 32
[2-(3-Фтор-2-метилбензил)-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
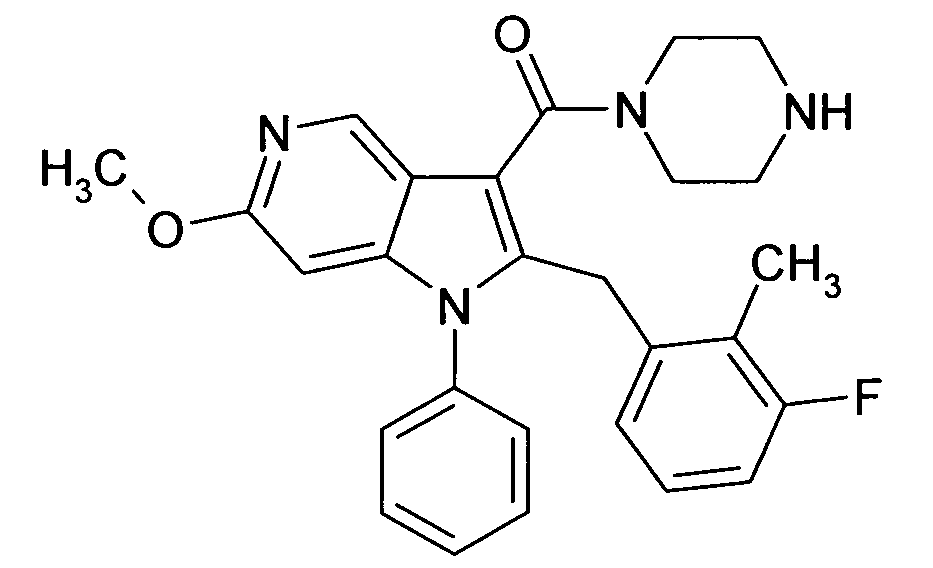
Стадия 1: 6-Метокси-1-фенил-1H-пирроло[3,2-c]пиридин
6-Метокси-1H-пирроло[3,2-c]пиридин (4,25 г, 28,6 ммоль) реагировал аналогично, как описано в примере 1, стадия 1, для того, чтобы получить 4,92 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=225
Стадия 2: 3,3-Дибром-6-метокси-1-фенил-1,3-дигидропирроло[3,2-c]пиридин-2-он
К перемешиваемому раствору 4,92 г (21,9 ммоль) соединения стадии 1 в трет-бутаноле (177 мл) и воде (177 мл) добавляли по каплям в течение 10 мин бром (5,06 мл, 98,7 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 30 мин. Смесь обрабатывали насыщенным раствором гидрокарбоната натрия до того момента, как значение pH становилось приблизительно равным 6,5-7, и затем добавляли EA. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении для того, чтобы получить 9,40 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=398
Стадия 3: 6-Метокси-1-фенил-1,3-дигидропирроло[3,2-c]пиридин-2-он
К раствору 8,73 г соединения стадии 2 в уксусной кислоте (180 мл) добавляли цинк, и суспензию перемешивали при комнатной температуре в течение ночи. Смесь экстрагировали EA, органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Полученный в результате твердый остаток растворяли в этаноле (245 мл), и добавляли палладий на активированном угле (1,75 г, 1,64 ммоль, 10%). Реакционную смесь гидрировали (5,2 бар H2) при комнатной температуре в течение ночи. Смесь фильтровали через целит, растворитель удаляли при пониженном давлении, и твердый остаток очищали хроматографией на силикагеле (EA (10-70%)/HEP). Получали 870 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=241
Стадия 4: 2-Хлор-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбальдегид
Из соединения стадии 3 (870 мг, 3,62 ммоль) заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 3. Выход: 300 мг.
LC/MS (способ LC4): m/z=287
Стадия 5: 2-Хлор-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбоновая кислота
Соединение стадии 4 (300 мг, 1,05 ммоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали в течение 3 часов при 45°C. Получали 304 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=303
Стадия 6: трет-Бутиловый эфир 4-(2-хлор-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Из соединения стадии 5 (304 мг, 1,00 ммоль) заявленное в заголовке соединение получали аналогично, как описано в примере 20, стадия 5. Выход: 300 мг.
LC/MS (способ LC4): m/z=471
Стадия 7: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
К цинку (167 мг, 2,55 ммоль) в сухом THF (500 мкл) в сухой колбе в атмосфере аргона добавляли 1,2-дибромэтан (5,49 мкл, 63,7 мкмоль). Смесь нагревали три раза до кипения термофеном и охлаждали до комнатной температуры. Затем добавляли хлортриметилсилан (0,27 мкл, 2,12 мкмоль), и смесь перемешивали при комнатной температуре в течение 20 мин. Затем колбу помещали в ледяную баню, и добавляли медленно раствор 3-фтор-2-метилбензилбромида (259 мг, 1,27 ммоль) в сухом THF (1 мл), так что температуру поддерживали равной 0°C. Смесь перемешивали при 0°C в течение 4,5 часов и выдерживали в холодильнике в течение ночи. Затем охлажденную смесь добавляли по каплям к предварительно охлажденному раствору (-78°C) B-OM-9-BBN (2,12 мл, 2,12 ммоль, 1 M) в гексане. Смесь перемешивали при комнатной температуре в течение 30 мин. Добавляли DMF (5 мл), с последующим добавлением соединения стадии 6 (100 мг, 212 мкмоль), ацетата палладия(II) (4,77 мг, 21,2 мкмоль) и S-PHOS (17,4 мг, 42,5 мкмоль). Реакционную смесь перемешивали при 100°C в течение 3 часов. После охлаждения смесь гасили водой и экстрагировали EA. Органический слой сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (EA (10-60%)HEP) для того, чтобы получить 90 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=559
Стадия 8: [2-(3-Фтор-2-метилбензил)-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 7 (33 мг, 59,1 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 22,3 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC6): m/z=458,21; Rt=2,45 мин
1H-ЯМР (400 МГц, DMSO-D6): δ (ppm)=1,81 (д, 3H), 3,08 (уш. с, 4H), 3,88 (с, 3H), 4,09 (с, 2H), 6,37 (с, 1H), 6,66 (д, 1H), 6,91 (т, 1H), 6,99 (кв, 1H), 7,28-7,31 (м, 2H), 7,49-7,51 (м, 3H), 8,64 (с, 1H), 9,28 (уш. с, 2H).
Пример 33
[2-(3-Фтор-2-метилбензил)-6-гидрокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
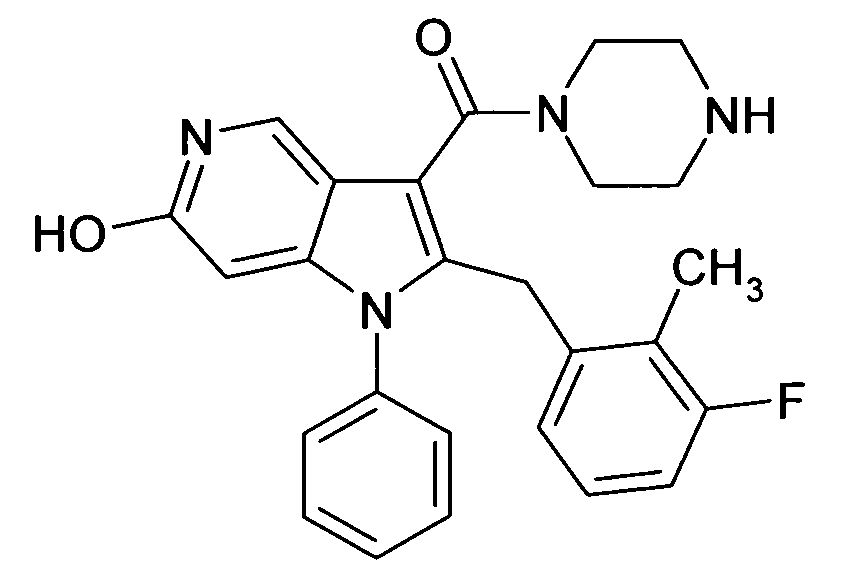
Заявленное в заголовке соединение получали из соединения примера 32, стадия 7, (60,8 мг, 109 мкмоль) аналогично, как описано в примере 9, и получали в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-6-гидрокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона. Выход: 18,7 мг.
LC/MS (способ LC5): m/z=444,20; Rt=1,67 мин
1H-ЯМР: δ (ppm)=1,80 (д, 1H), 4,05 (с, 2H), 6,27 (с, 1H), 6,68 (д, 1H), 6,91 (т, 1H), 7,00 (кв, 1H), 7,31 (с, 2H), 7,50-7,53 (м, 3H), 8,53 (с, 1H), 9,28 (уш. с, 2H).
Пример 34 [2-(5-Фтор-2-метилфенокси)-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
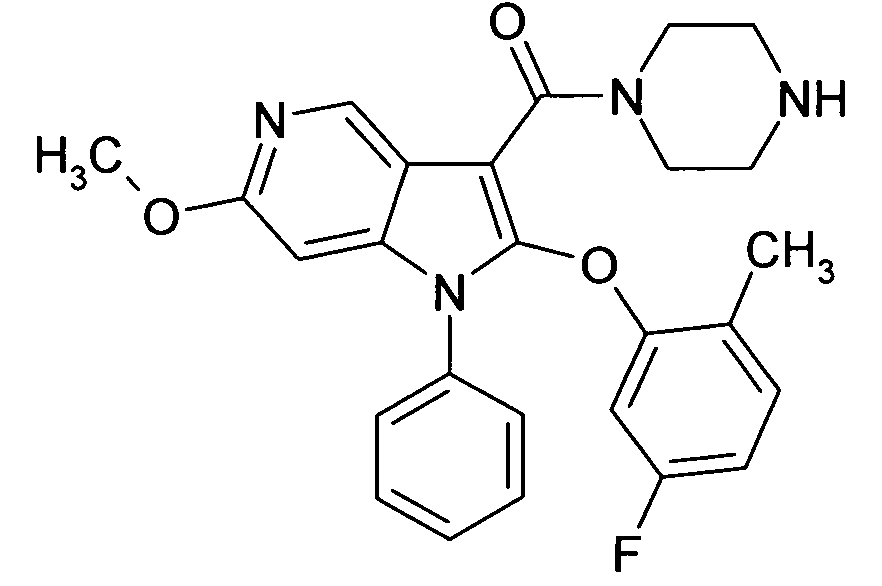
Стадия 1: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 6, из соединения примера 32, стадия 6, (100 мг, 212 мкмоль) и 5-фтор-2-метилфенола. Выход: 88 мг.
LC/MS (способ LC4): m/z=561
Стадия 2: [2-(5-Фтор-2-метилфенокси)-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (27,0 мг, 48,1 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 15,5 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-6-метокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC4): m/z=461,20; Rt=0,95 мин
1H-ЯМР: δ (ppm)=2,11 (с, 3H), 3,14 (м, 4H), 3,19 (м, 4H), 3,90 (м, 3H), 6,51 (м, 1H), 6,86 (м, 2H), 7,28 (м, 1H), 7,48 (м, 1H), 7,53 (м, 4H), 8,56 (м, 1H), 9,17 (м, 2H).
Пример 35
[2-(5-Фтор-2-метилфенокси)-6-гидрокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
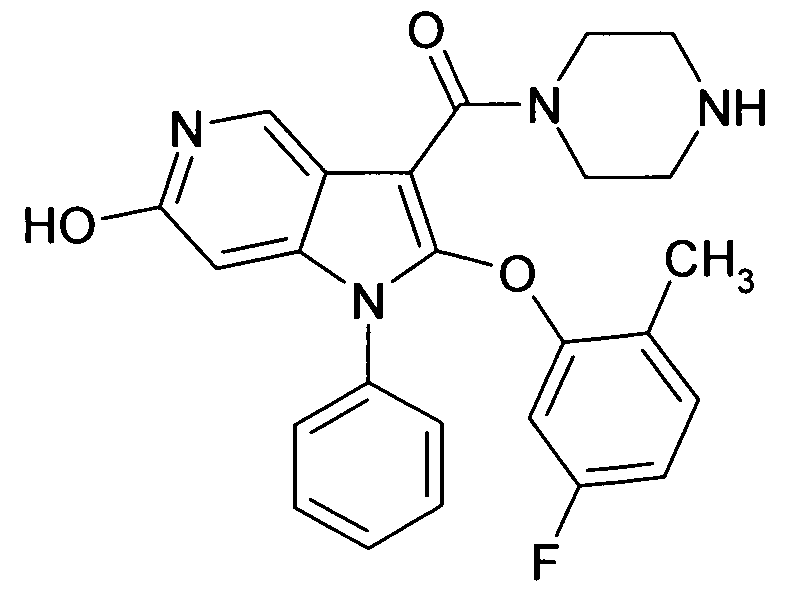
Заявленное в заголовке соединение получали из соединения примера 34, стадия 1, (61 мг, 108 мкмоль) аналогично, как описано в примере 9, и получали в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-6-гидрокси-1-фенил-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона. Выход: 25,9 мг.
LC/MS (способ LC6): m/z=446,18; Rt=2,37 мин
1H-ЯМР: δ (ppm)=2,10 (с, 3H), 3,00 (м, 4H), 3,68 (м, 4H), 6,48 (м, 1H), 6,88 (м, 1H), 6,97 (м, 1H), 7,22 (м, 1H), 7,51 (м, 1H), 7,54 (м, 4H), 8,40 (м, 1H), 9,28 (м, 2H).
Пример 36
[2-(2,6-Диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
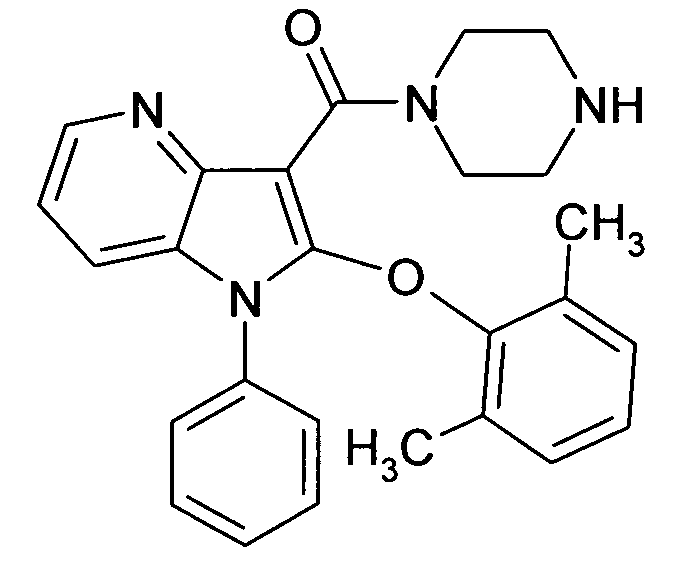
Стадия 1: трет-Бутиловый эфир 4-[2-(2,6-диметилфенокси)-1 -фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 6, из соединения примера 1, стадия 5, (100,0 мг, 227 мкмоль) и 2,6-диметилфенола. Выход: 96 мг.
LC/MS (способ LC4): m/z=500
Стадия 2: [2-(2,6-Диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из соединения стадии 1 аналогично, как описано в примере 1, стадия 7, и получали в форме дигидрохлорида [2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона. Выход: 51 мг.
LC/MS (способ LC5): m/z=426,21; Rt=1,71 мин
1H-ЯМР: δ (ppm) =2,16 (с, 6H), 2,94 (м, 4H), 3,38 (м, 2H), 3,50 (м, 2H), 7,10 (м, 3H), 7,44 (м, 1H), 7,64 (м, 1H), 7,72 (м, 4H), 7,92 (м, 1H), 8,48 (м, 1H), 9,24 (м, 2H).
Пример 37
[2-(3-Фтор-2,6-диметилфенокси]-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
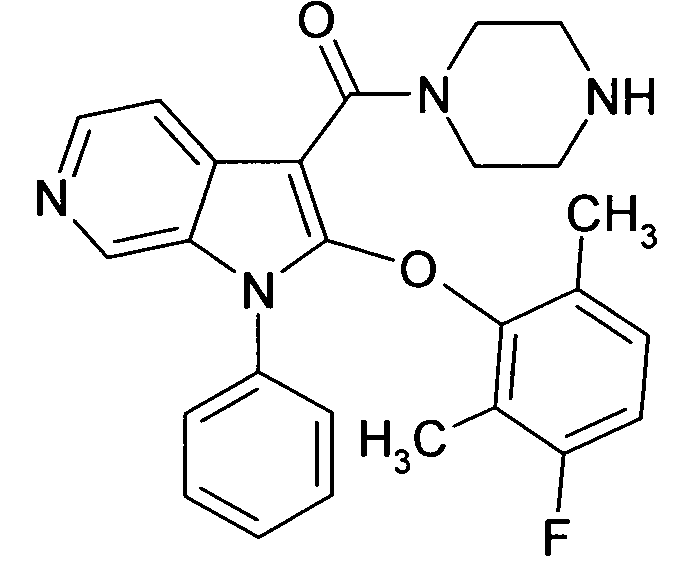
Стадия 1: трет-Бутиловый эфир 4-[2-(3-Фтор-2,6-диметилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 6, из соединения примера 16, стадия 5, (100 мг, 227 мкмоль) и 3-фтор-2,6-диметилфенола.
LC/MS (способ LC4): m/z=545
Стадия 2: [2-(3-Фтор-2,6-диметилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Неочищенное соединение стадии 1 реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 42 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2,6-диметилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC8): m/z=445,30; Rt=2,43 мин
1H-ЯМР: δ (ppm)=2,10 (м, 6H), 2,94 (м, 4H), 3,45 (м, 4H), 7,09 (м, 1H), 7,16 (м, 1H), 7,59 (м, 1H), 7,63 (м, 2H), 7,31 (м, 2H), 8,05 (м, 1H), 8,43 (м, 1H), 8,81 (м, 1H), 9,41 (м, 2H).
Пример 38 [7-Хлор-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
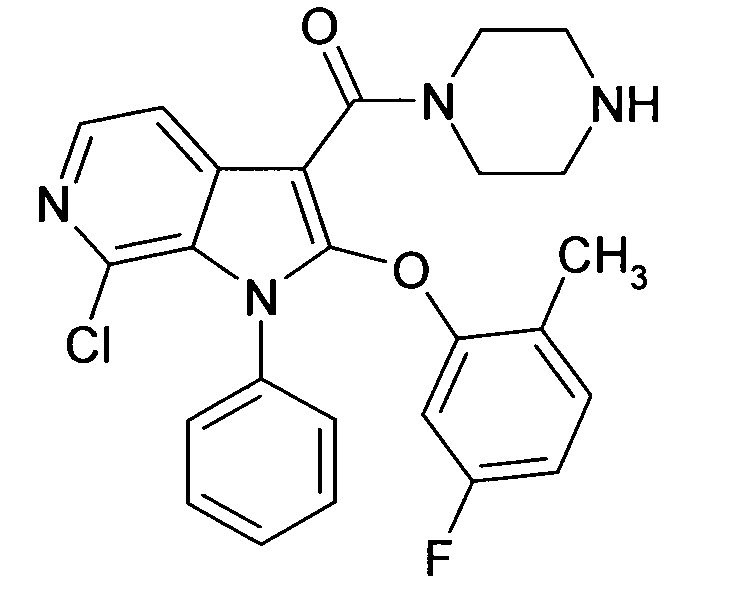
Стадия 1: 7-Метокси-1-фенил-1H-пирроло[2,3-c]пиридин
Заявленное в заголовке соединение получали из 7-метокси-1H-пирроло[2,3-c]пиридина (1 г, 6,75 ммоль) аналогично, как описано в примере 1, стадия 1. Выход: 1,27 г.
LC/MS (способ LC4): m/z=225
Стадия 2: 7-Метокси-1-фенил-1,3-дигидропирроло[2,3-c]пиридин-2-он
Заявленное в заголовке соединение получали из соединения стадии 1 (1,19 г, 5,31 ммоль) аналогично, как описано в примере 1, стадия 2. Выход: 0,74 г.
LC/MS (способ LC4): m/z=241
Стадия 3: 2,7-Дихлор-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 2 (639 мг, 2,66 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 490 мг.
LC/MS (способ LC4): m/z=291
Стадия 4: 2,7-Дихлор-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбоновая кислота
Соединение стадии 3 (691 мг, 2,37 ммоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали при 40°C в течение 2 часов. Получали 1,11 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=273
Стадия 5: трет-Бутиловый эфир 4-(2,7-дихлор-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Неочищенное соединение стадии 4 (1,11 г) реагировало аналогично, как описано в примере 1, стадия 5. Получали 587 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=475
Стадия 6: трет-Бутиловый эфир 4-[7-хлор-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 5 (160 мг, 336 мкмоль) аналогично, как описано в примере 5, стадия 1. Выход: 102 мг.
LC/MS (способ LC4): m/z=566
Стадия 7: [7-Хлор-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 6 (35 мг, 62 мкмоль) реагировало аналогично, как описано в примере 5, стадия 2. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 32 мг заявленного в заголовке соединения в форме дигидрохлорида [7-хлор-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC5): m/z=464,14; Rt=1,92 мин
1H-ЯМР: δ (ppm)=2,02 (с, 3H), 2,98 (м, 4H), 3,66 (м, 4H), 6,88 (м, 1H), 7,05 (м, 1H), 7,20 (м, 1H), 7,52 (м, 3H), 7,58 (м, 2H), 7,68 (м, 1H), 8,13 (м, 1H), 9,20 (м, 2H).
Пример 39
[7-Хлор-2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
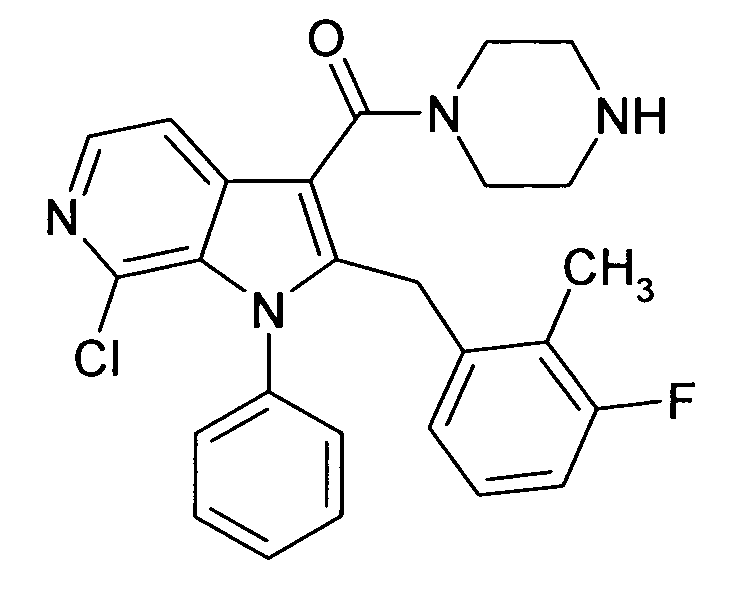
Стадия 1: трет-Бутиловый эфир 4-[7-хлор-2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Соединение примера 38, стадия 5, (100 мг, 210 мкмоль) реагировало аналогично, как описано в примере 4, стадия 1, для того, чтобы получить смесь заявленного в заголовке соединения и трет-бутилового эфира 4-[2,7-бис-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты, которые разделяли препаративной ВЭЖХ. Получали 30 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=564
Стадия 2: [7-Хлор-2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (30 мг, 53 мкмоль) реагировало аналогично, как описано в примере 4, стадия 2. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 8,6 мг заявленного в заголовке соединения в форме дигидрохлорида [7-хлор-2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC8): m/z=462,16; Rt=1,99 мин
1H-ЯМР: δ (ppm)=1,68 (с, 3H), 3,19 (м, 4H), 3,84 (м, 4H), 3,98 (м, 2H), 6,17 (м, 1H), 7,00 (м, 2H), 7,36 (м, 2H), 7,40 (м, 2H), 7,56 (м, 1H), 8,10 (м, 1H), 8,48 (м, 1H).
Пример 40
[2,7-Бис-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
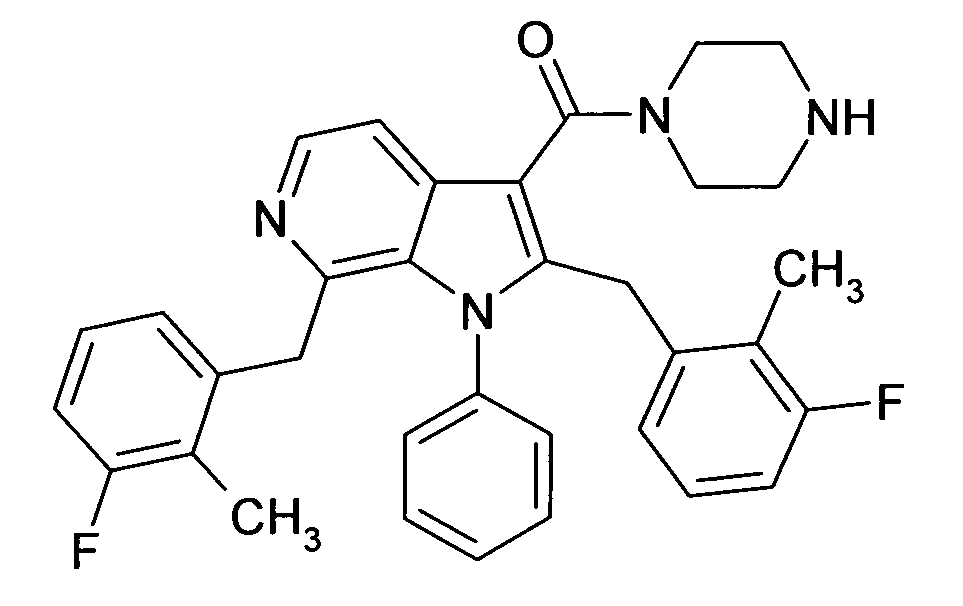
Стадия 1: трет-Бутиловый эфир 4-[2,7-бис(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Соединение примера 38, стадия 5, (100 мг, 210 мкмоль) реагировало аналогично, как описано в примере 4, стадия 1, для того, чтобы получить смесь заявленного в заголовке соединения и трет-бутилового эфира 4-[7-хлор-2-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты, которые разделяли препаративной ВЭЖХ. Получали 40 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=651
Стадия 2: [2,7-Бис-(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (40 мг, 61 мкмоль) реагировало аналогично, как описано в примере 4, стадия 2. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 30 мг заявленного в заголовке соединения в форме дигидрохлорида [2,7-бис(3-фтор-2-метилбензил)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC7): m/z=550,25; Rt=1,35 мин
1H-ЯМР: δ (ppm)=1,16 (с, 3H), 1,25 (с, 3H), 2,98 (м, 2H), 3,23 (м, 2H), 3,62 (м, 2H), 3,79 (м, 2H), 3,98 (м, 2H), 4,01 (м, 2H), 6,12 (м, 1H), 6,70 (м, 1H), 6,95 (м, 1H), 7,03 (м, 4H), 7,18 (м, 2H), 7,48 (м, 1H), 8,21 (м, 1H), 8,50 (м, 1H), 9,37 (м, 1H).
Пример 41 [7-Бензил-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
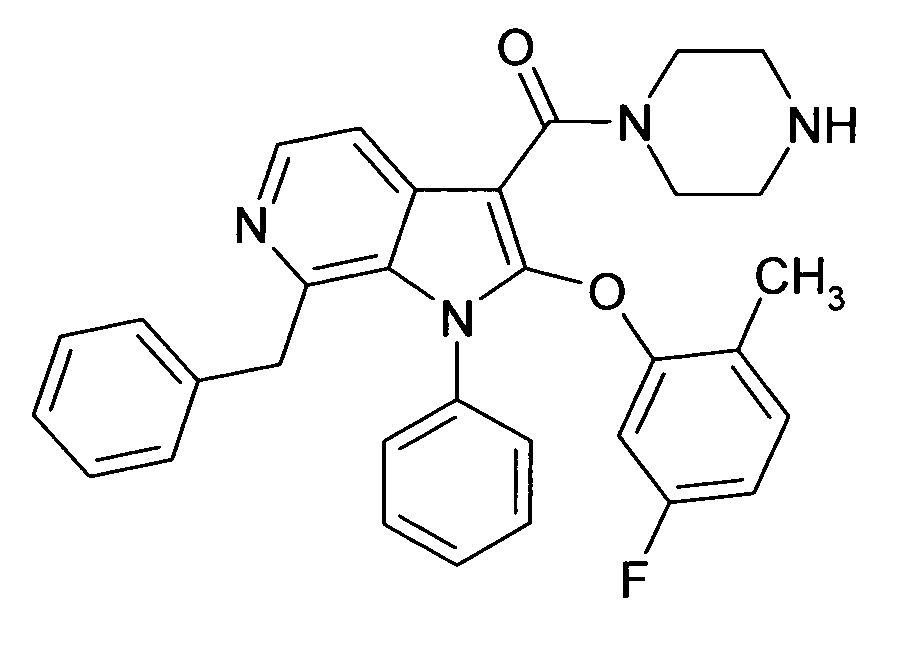
Стадия 1: трет-Бутиловый эфир 4-[7-бензил-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 38, стадия 6, (45 мг, 80 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 37 мг.
LC/MS (способ LC4): m/z=621
Стадия 2: [7-Бензил-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (37 мг, 60 мкмоль) реагировало аналогично, как описано в примере 4, стадия 2. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 35 мг заявленного в заголовке соединения в форме дигидрохлорида [7-бензил-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC6): m/z=520,23; Rt=2,59 мин
1H-ЯМР: δ (ppm)=2,06 (с, 3H), 2,97 (м, 4H), 3,61 (м, 4H), 4,12 (с, 2H), 6,68 (м, 2H), 6,98 (м, 1H), 7,18 (м, 3H), 7,25 (м, 2H), 7,48 (м, 2H), 7,58 (м, 3H), 8,08 (м, 1H), 8,46 (м, 1H), 9,33 (м, 2H).
Пример 42
[7-Этил-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
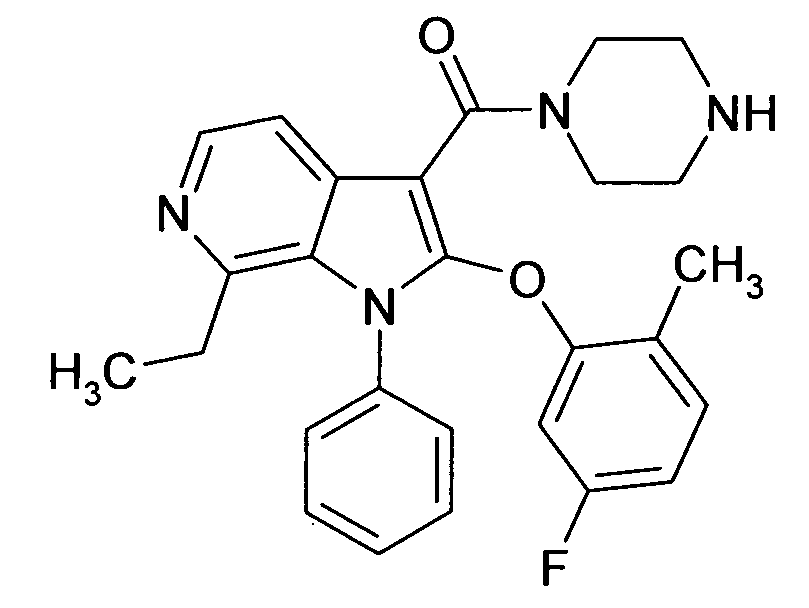
Стадия 1: трет-Бутиловый эфир 4-[7-этил-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Смесь соединения примера 38, стадия 6, (65 мг, 115 мкмоль), ацетилацетоната железа(III) (2,03 мг, 5,7 мкмоль) и NMP (102,6 мг, 1,04 ммоль) в THF (5 мл) охлаждали до 0°C. Добавляли этилмагнийхлорид (115 мкл, 230 мкмоль, 2 M в THF), и полученный в результате раствор перемешивали в течение 5 мин. Смесь коричневого цвета гасили водой и экстрагировали EA. Органические слои сушили над сульфатом натрия и концентрировали. Остаток очищали хроматографией на силикагеле (EA/HEP) для того, чтобы получить 29 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=558,66
Стадия 2: [7-Этил-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (29 мг, 52 мкмоль) реагировало аналогично, как описано в примере 4, стадия 2. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 12 мг заявленного в заголовке соединения в форме дигидрохлорида [7-этил-2-(5-фтор-2-метилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC6): m/z =458,21; Rt=2,38 мин
1H-ЯМР: δ (ppm)=1,06 (т, 3H), 2,08 (с, 3H), 2,58 (кв, 2H), 2,98 (м, 4H), 3,61 (м, 4H), 6,98 (м, 1H), 7,25 (м, 2H), 7,68 (м, 3H), 7,80 (м, 2H), 8,00 (м, 1H), 8,38 (м, 1H), 9,42 (м, 1H).
Пример 43
[2-(5-Фтор-2-метилфенокси)-6-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
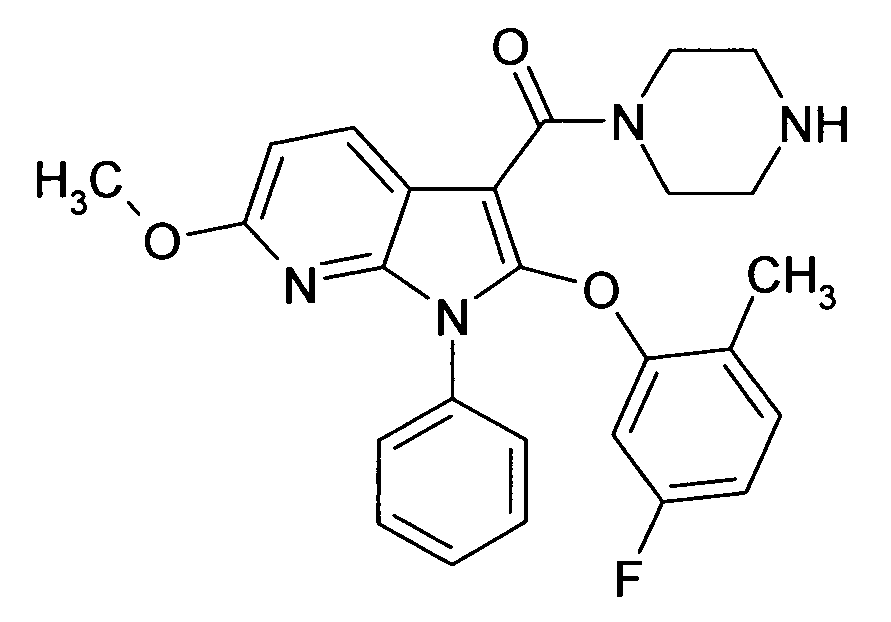
Стадия 1: 6-Метокси-1-фенил-1H-пирроло[2,3-b]пиридин
6-Метокси-1H-пирроло[2,3-b]пиридин (1 г, 6,75 ммоль) реагировал аналогично, как описано в примере 20, стадия 1, для того, чтобы получить 9,64 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=225
Стадия 2: 6-Метокси-1-фенил-1,3-дигидропирроло[2,3-b]пиридин-2-он
К перемешиваемому раствору 9,64 г (43 ммоль) соединения стадии 1 в DCM (250 мл) добавляли N-хлорсукцинимид (6,02 г, 45 ммоль) одной порцией при комнатной температуре. Реакционную смесь перемешивали в течение 12 часов, и растворитель упаривали. Оставшийся остаток растворяли в смеси уксусной кислоты (180 мл) и фосфорной кислоты (31 мл) и нагревали при 125°C в течение 1 часа. Раствор охлаждали до комнатной температуры и концентрировали в вакууме. Оставшийся остаток выливали на лед, и водную фазу экстрагировали EA. Объединенные органические фазы промывали насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали, и растворитель удаляли при пониженном давлении. Оставшееся масло очищали колоночной хроматографией на силикагеле (EA/HEP) для того, чтобы получить 4,80 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=241
Стадия 3: 2-Хлор-6-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 2 (4,32 г, 18,0 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 1,77 г.
LC/MS (способ LC4): m/z=287
Стадия 4: 2-Хлор-6-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбоновая кислота
Соединение стадии 3 (1,77 г, 6,17 ммоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали при 40°C в течение 2 часов. Получали 2,62 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=303
Стадия 5: трет-Бутиловый эфир 4-(6-хлор-2-метокси-7-фенил-7H-пирроло[2,3-c]пиридазин-5-карбонил)пиперазин-1-карбоновой кислоты
Неочищенное соединение стадии 4 (2,62 г) реагировало аналогично, как описано в примере 20, стадия 5. Получали 2,71 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=471
Стадия 6: трет-Бутиловый эфир 4-[2-(5-фтор-2-метилфенокси)-6-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 5 (153 мг, 325 мкмоль) аналогично, как описано в примере 5, стадия 1. Выход: 80 мг.
LC/MS (способ LC4): m/z=562
Стадия 7: [2-(5-Фтор-2-метилфенокси)-6-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 6 (30 мг, 54 мкмоль) реагировало аналогично, как описано в примере 5, стадия 2. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 16 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-6-метокси-1-фенил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC6): m/z=460,19; Rt=2,92 мин
1H-ЯМР: δ (ppm)=2,12 (с, 3H), 3,00 (м, 4H), 3,72 (м, 4H), 3,78 (с, 3H), 6,69 (м, 1H), 6,80 (м, 2H), 7,19 (м, 1H), 7,40 (м, 1H), 7,51 (м, 2H), 7,59 (м, 2H), 8,00 (м, 1H), 9,15 (м, 1H).
Пример 44
[2-(2,6-Диметилфенокси)-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
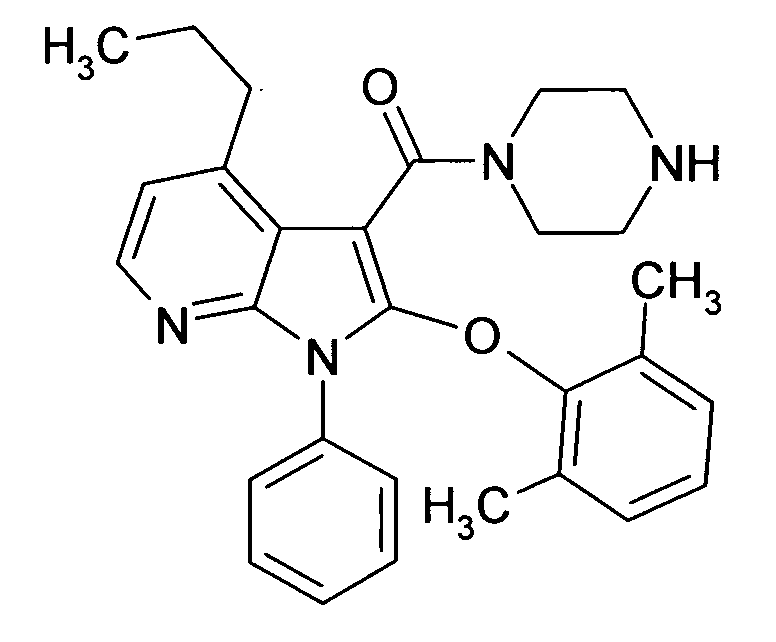
Стадия 1: 4-Хлор-1-фенил-1H-пирроло[2,3-b]пиридин
4-Хлор-1H-пирроло[2,3-b]пиридин (10 г, 65,5 ммоль) реагировал аналогично, как описано в примере 20, стадия 1, для того, чтобы получить 9,81 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=229
Стадия 2: 1-Фенил-4-пропил-1H-пирроло[2,3-b]пиридин
Заявленное в заголовке соединение получали из соединения стадии 1 (4,69 г, 20,5 ммоль) и хлорида пропилмагния аналогично, как описано в примере 42, стадия 1. Выход: 3,57 г.
LC/MS (способ LC4): m/z=237
Стадия 3: 3,3-Дибром-1-фенил-4-пропил-1,3-дигидропирроло[2,3-b]пиридин-2-он
Заявленное в заголовке соединение получали из соединения стадии 2 (3,57 г, 15,1 ммоль) аналогично, как описано в примере 32, стадия 2. Выход: 8,5 г.
LC/MS (способ LC4): m/z=410
Стадия 4: 1-Фенил-4-пропил-1,3-дигидропирроло[2,3-b]пиридин-2-он
Заявленное в заголовке соединение получали из соединения стадии 3 (8,5 г) аналогично, как описано в примере 32, стадия 3. Выход: 3,53 г.
LC/MS (способ LC4): m/z=253
Стадия 5: 2-Хлор-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 4 (3,53 г, 14,0 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 2,59 г.
LC/MS (способ LC4): m/z=299
Стадия 6: 2-Хлор-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-карбоновая кислота
Соединение стадии 5 (1,59 г, 5,32 ммоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали при 40°C в течение 2 часов. Получали 1,73 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=315
Стадия 7: трет-Бутиловый эфир 4-(2-Хлор-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Неочищенное соединение стадии 6 (1,73 г) реагировало аналогично, как описано в примере 1, стадия 5. Получали 1,28 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=483
Стадия 8: [2-(2,6-Диметилфенокси)-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
К раствору 2,6-диметилфенола (152 мг, 1,24 ммоль) в NMP (3 мл) добавляли гидрид натрия (50 мг, 1,24 ммоль, 60% дисперсия в минеральном масле), и суспензию перемешивали при комнатной температуре в атмосфере аргона в течение 20 мин. После добавления 100 мг (207 мкмоль) соединения стадии 7 реакционную смесь перемешивали в течение 2 часов при 140°C. После охлаждения реакционную смесь гасили водой и экстрагировали EA. Органические фазы концентрировали, и оставшийся остаток растворяли в DCM (12 мл) и TFA (3 мл) и перемешивали при комнатной температуре в течение 2 часов. Растворители упаривали, и полученный в результате твердый остаток очищали препаративной ВЭЖХ. Фракции, содержащие заявленное в заголовке соединение, объединяли и лиофилизовали в течение ночи. Полученный твердый остаток растворяли в небольшом количестве MOH, смешивали с хлороводородной кислотой (0,1 M) и лиофилизовали в течение ночи для того, чтобы получить 34 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(2,6-диметилфенокси)-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC8): m/z=468,25; Rt=2,60 мин
1H-ЯМР: δ (ppm)=0,91 (т, 3H), 1,58 (м, 2H), 2,05 (м, 3H), 2,19 (м, 3H), 2,55 (м, 1H), 2,62 (м, 2H), 2,75 (м, 1H), 2,88 (м, 2H), 3,22 (м, 1H), 3,35 (м, 1H), 3,50 (м, 2H), 7,04 (м, 4H), 7,50 (м, 1H), 7,59 (м, 4H), 8,06 (м, 1H), 9,13 (м, 2H).
Пример 45
[2-(3-Фтор-2-метилбензил)-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
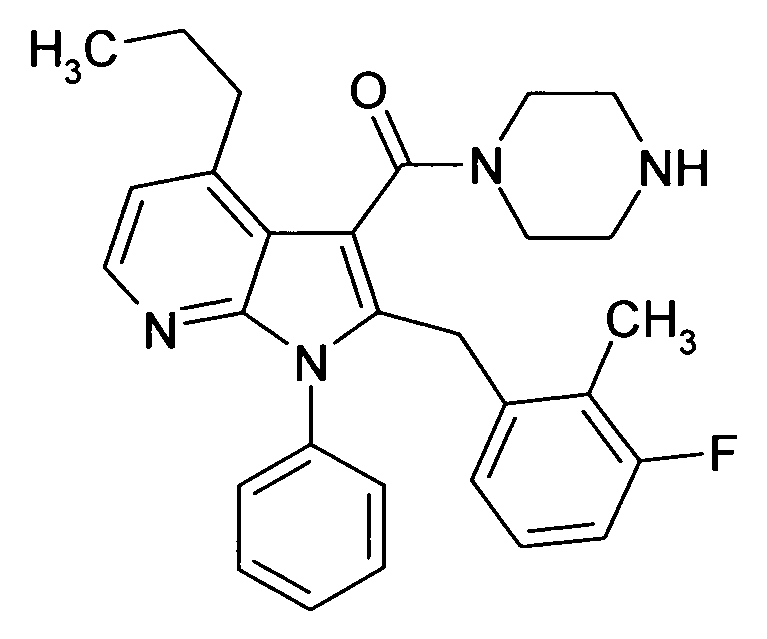
Стадия 1: трет-Бутиловый эфир 4-[2-(3-фтор-2-метилбензил)-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 44, стадия 7, (150 мг, 310 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 175 мг.
LC/MS (способ LC4): m/z=572
Стадия 2: [2-(3-Фтор-2-метилбензил)-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (175 мг, 307 мкмоль) реагировало аналогично, как описано в примере 4, стадия 2. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 90 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-1-фенил-4-пропил-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC8): m/z=470,25; Rt=2,61 мин
1H-ЯМР: δ (ppm)=0,95 (т, 3H), 1,62 (м, 2H), 1,71 (с, 3H), 2,70 (м, 1H), 2,83 (м, 2H), 2,96 (м, 1H), 3,14 (м, 2H), 4,04 (м, 3H), 6,65 (м, 1H), 6,88 (м, 1H), 7,07 (м, 1H), 7,25 (м, 2H), 7,42 (м, 3H), 8,10 (м, 1H), 9,23 (м, 2H).
Пример 46
[1-Циклогексил-2-(3-фтор-2-метилбензил)-6-метокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
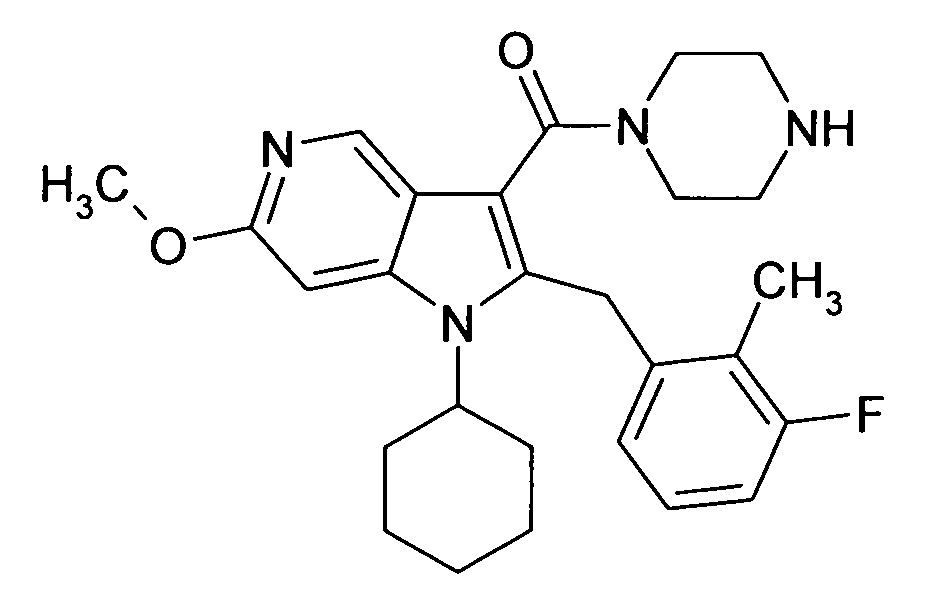
Стадия 1: 1-Циклогекс-2-енил-6-метокси-1H-пирроло[3,2-c]пиридин
Димер хлорида аллилпалладия(II) (558 мг, 1,52 ммоль) и трифенилфосфина (1,75 г, 6,68 ммоль) растворяли в сухом DMF (210 мл) и перемешивали при комнатной температуре в течение 30 мин. Добавляли метиловый эфир циклогекс-2-енилового эфира угольной кислоты (9,47 г, 60,74 ммоль), и смесь перемешивали в течение дополнительных 30 мин. Добавляли 6-метокси-1H-пирроло[3,2-c]пиридин (4,5 г, 30,37 ммоль) и карбонат цезия (19,79 г, 60,74 ммоль), и реакционную смесь перемешивали при комнатной температуре в течение 16 часов. Затем смесь распределяли между водой и EA, водную фазу экстрагировали EA, и объединенные органические фазы сушили над сульфатом натрия и концентрировали. Остаток очищали колоночной хроматографией на силикагеле (EA/HEP) для того, чтобы получить 5,6 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=229
Стадия 2: 1-Циклогексил-6-метокси-1H-пирроло[3,2-c]пиридин
Соединение стадии 1 (5,6 г, 24,5 ммоль) и палладий на угле (1,12 г, 10%) перемешивали в этаноле (160 мл) в атмосфере водорода в течение 3 часов. Катализатор отфильтровывали, и растворитель удаляли в вакууме для того, чтобы получить 5,34 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=231
Стадия 3: 3,3-Дибром-1-циклогексил-6-метокси-1,3-дигидропирроло[3,2-c]пиридин-2-он
Соединение стадии 2 (5,34 г, 23,2 ммоль) реагировало аналогично, как описано в примере 32, стадия 2, для того, чтобы получить 34,5 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=405
Стадия 4: 1-Циклогексил-6-метокси-1,3-дигидропирроло[3,2-c]пиридин-2-он
Неочищенное соединение стадии 3 (34,4 г) реагировало аналогично, как описано в примере 32, стадия 3, для того, чтобы получить 8,68 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=247
Стадия 5: 2-Хлор-1-циклогексил-6-метокси-1H-пирроло[3,2-c]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из неочищенного соединения стадии 4 (8,68 г) аналогично, как описано в примере 1, стадия 3. Выход: 3,60 г.
LC/MS (способ LC4): m/z=294
Стадия 6: 2-Хлор-1-циклогексил-6-метокси-1H-пирроло[3,2-c]пиридин-3-карбоновая кислота
Соединение стадии 5 (1,60 г, 5,47 ммоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали при 40°C в течение 2 часов. Получали 1,60 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=310
Стадия 7: трет-Бутиловый эфир 4-(2-хлор-1-циклогексил-6-метокси-1H-пирроло[3,2-c]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Неочищенное соединение стадии 6 (1,60 г) реагировало аналогично, как описано в примере 1, стадия 5. Получали 540 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=478
Стадия 8: трет-Бутиловый эфир 4-[1-циклогексил-2-(3-фтор-2-метилбензил)-6-метокси-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения стадии 7 (180 мг, 377 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 86 мг.
LC/MS (способ LC4): m/z=566
Стадия 9: [1-Циклогексил-2-(3-фтор-2-метилбензил)-6-метокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 8 (38 мг, 67 мкмоль) реагировало аналогично, как описано в примере 4, стадия 2. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 21 мг заявленного в заголовке соединения в форме дигидрохлорида [1-циклогексил-2-(3-фтор-2-метилбензил)-6-метокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC8): m/z=464,26; Rt=2,55 мин
1H-ЯМР: δ (ppm)=1,05 (м, 2H), 1,32 (м, 1H), 1,42 (м, 2H), 1,53 (м, 1H), 1,70 (м, 2H), 2,08 (м, 2H), 2,30 (с, 3H), 2,97 (м, 2H), 3,18 (м, 2H), 3,57 (м, 2H), 3,71 (м, 2H), 3,97 (м, 3H), 4,28 (м, 2H), 6,63 (м, 1H), 7,19 (м, 3H), 8,58 (м, 1H), 9,20 (м, 1H), 9,30 (м, 1H).
Пример 47
[1-Циклогексил-2-(3-фтор-2-метилбензил)-6-гидрокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
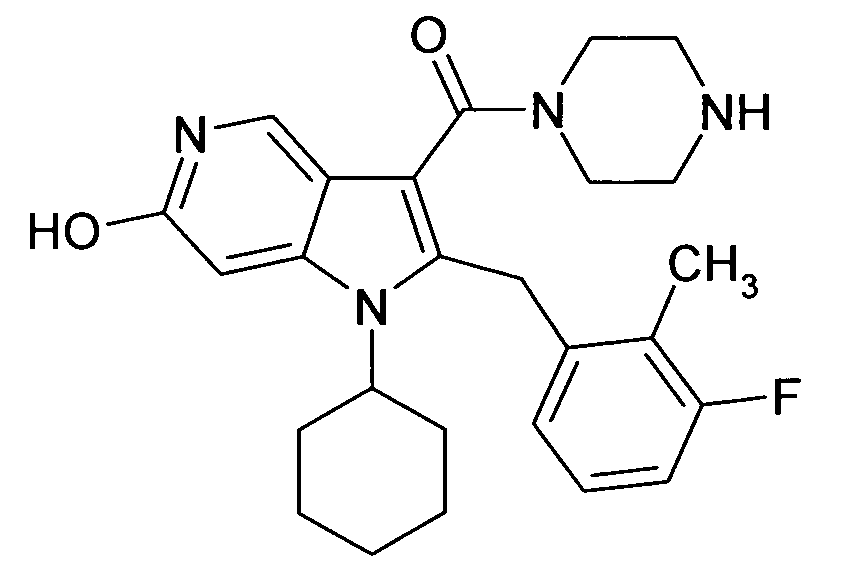
Заявленное в заголовке соединение получали из соединения примера 46, стадия 8, (42 мг, 74 мкмоль) аналогично, как описано в примере 9, и получали в форме дигидрохлорида [1-циклогексил-2-(3-фтор-2-метилбензил)-6-гидрокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона. Выход: 8 мг.
LC/MS (способ LC4): m/z=451,20; Rt=1,05 мин
1H-ЯМР (400 МГц, MOH-D4): δ (ppm)=1,21 (м, 4H), 1,68 (м, 3H), 1,84 (м, 2H), 2,08 (м, 2H), 2,34 (м, 3H), 3,10 (м, 2H), 3,32 (м, 2H), 3,88 (м, 4H), 3,96 (м, 1H), 6,72 (м, 1H), 6,98 (м, 1H), 7,10 (м, 2H), 8,51 (м, 1H).
Пример 48
[1-Циклогексил-2-(2,6-диметилфенокси)-6-метокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
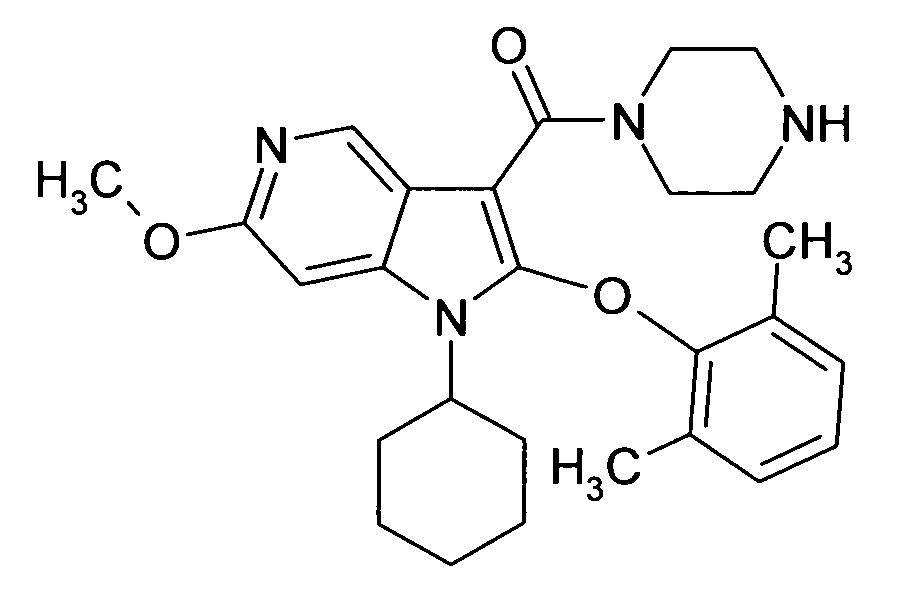
Стадия 1: трет-Бутиловый эфир 4-[1-циклогексил-2-(2,6-диметилфенокси)-6-метокси-1H-пирроло[3,2-c]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 46, стадия 7, (180 мг, 377 мкмоль) и 2,6-диметилфенола аналогично, как описано в примере 1, стадия 6. Выход: 117 мг.
LC/MS (способ LC4): m/z=564
Стадия 2: [1-Циклогексил-2-(2,6-диметилфенокси)-6-метокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 1 (52 мг, 92 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 33 мг заявленного в заголовке соединения в форме дигидрохлорида [1-циклогексил-2-(2,6-диметилфенокси)-6-метокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC8): m/z=464,26; Rt=2,55 мин
1H-ЯМР: δ (ppm)=1,33 (м, 1H), 1,54 (м, 2H), 1,69 (м, 1H), 1,93 (м, 4H), 2,17 (с, 6H), 2,28 (м, 2H), 2,85 (м, 4H), 3,29 (м, 4H), 4,02 (м, 3H), 4,68 (м, 1H), 7,11 (м, 3H), 7,39 (м, 1H), 8,37 (м, 1H), 9,18 (м, 1H).
Пример 49
[1-Циклогексил-2-(2,6-диметилфенокси)-6-гидрокси-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон

Заявленное в заголовке соединение получали из соединения примера 48, стадия 1, (63 мг, 112 мкмоль) аналогично, как описано в примере 9, и получали в форме дигидрохлорида [1-циклогексил-2-(2,6-диметилфенокси)-6-гидрокси-1H-пирроло[3,2-c]пиридин-3-ил]-пиперазин-1-илметанона. Выход: 10 мг.
LC/MS (способ LC4): m/z=449,20; Rt=1,08 мин
1H-ЯМР (400 МГц, MOH-D4): δ (ppm)=1,35 (м, 2H), 1,61 (м, 2H), 1,82 (м, 1H), 2,06 (м, 4H), 2,29 (с, 6H), 2,36 (м, 2H), 2,98 (м, 4H), 3,48 (м, 4H), 4,61 (м, 1H), 7,12 (м, 1H), 7,18 (м, 3H), 8,22 (м, 1H).
Пример 50
[1-Фенил-2-(1-фенилэтил)-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
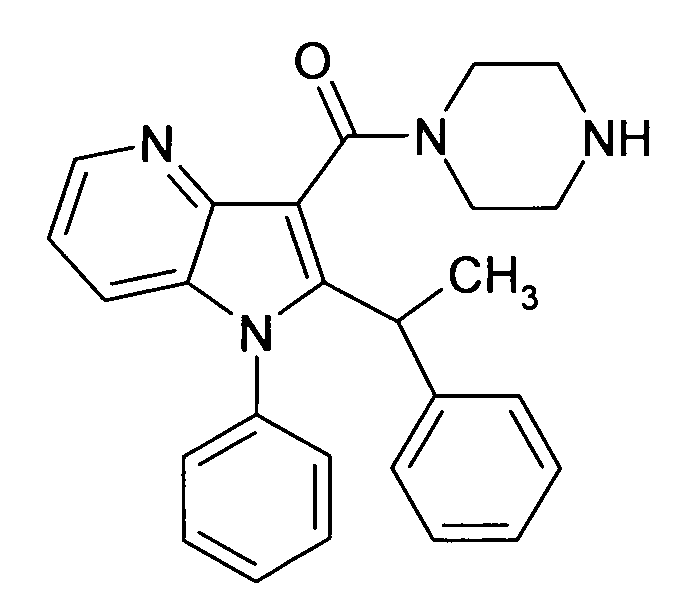
Раствор соединения примера 1, стадия 5, (120 мг, 27 мкмоль), бромида α-метилбензилцинка (820 мкл, 408 мкмоль, 0,5 M в THF), тетрафторбората три(трет-бутил)фосфония (15,6 мг, 54 мкмоль) и бис(дибензилиденацетон)палладия (15,6 мг, 27 мкмоль) в THF (5 мл) перемешивали в течение 12 часов при 80°C. Реакционную смесь разбавляли водой и экстрагировали EA. Органический слой сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Промежуточное соединение очищали препаративной ВЭЖХ, и оно реагировало аналогично, как описано в примере 1, стадия 7. Полученный твердый остаток растворяли в небольшом количестве MOH, смешивали с хлороводородной кислотой (0,1 M) и лиофилизовали в течение ночи для того, чтобы получить 20 мг заявленного в заголовке соединения в форме дигидрохлорида [1-фенил-2-(1-фенилэтил)-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC5): m/z=410,21; Rt=1,65 мин
1H-ЯМР: δ (ppm)=1,65 (м, 3H), 2,99 (м, 2H), 3,20 (м, 2H), 3,82 (м, 4H), 4,28 (м, 1H), 7,13 (м, 2H), 7,22 (м, 3H), 7,41 (м, 1H), 7,52 (м, 1H), 7,66 (м, 4H), 8,54 (м, 1H), 9,23 (м, 2H).
Пример 51
[2-(3-Фтор-2-метилбензил)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
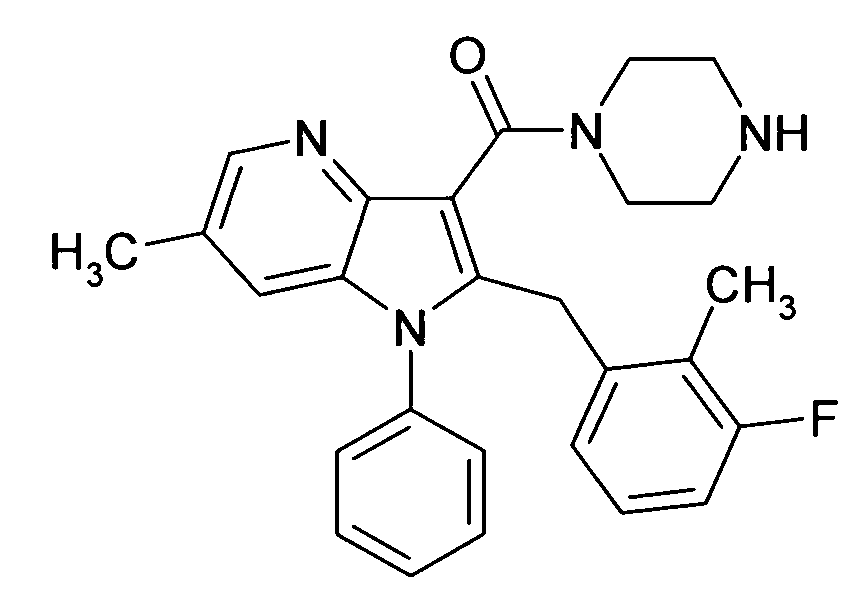
Стадия 1: 5-Метил-2-триметилсиланилэтинилпиридин-3-иламин
2-Хлор-5-метилпиридин-3-иламин (10 г, 70,13 ммоль), триметилсилилацетилен (13,8 г, 140,3 ммоль), йодид меди(I) (534 мг, 2,81 ммоль) и хлорид бис(трифенилфосфин)палладия(II) (1,97 г, 2,81 ммоль) растворяли в триэтиламине (140 мл) и перемешивали при 80°C в течение 5 часов. После охлаждения до комнатной температуры реакционную смесь фильтровали через слой целита, и растворитель удаляли в вакууме. Остаток очищали колоночной хроматографией (силикагель, EA/HEP) для того, чтобы получить 5,12 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=205
Стадия 2: 6-Метил-1H-пирроло[3,2-b]пиридин
Раствор соединения стадии 1 (5,12 г, 25,1 ммоль) в NMP (125 мл) добавляли по каплям при комнатной температуре к трет-бутилату калия (5,91 г, 52,6 ммоль) в NMP (125 мл). Реакционную смесь перемешивали в течение 4 часов при комнатной температуре, затем добавляли воду, и водную фазу экстрагировали диэтиловым эфиром. Объединенные органические фазы сушили над сульфатом натрия, фильтровали и концентрировали для того, чтобы получить 2,5 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=133
Стадия 3: 6-Метил-1-фенил-1H-пирроло[3,2-b]пиридин
Соединение стадии 2 (2,50 г, 18,9 ммоль) реагировало аналогично, как описано в примере 20, стадия 1, для того, чтобы получить 966 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=209
Стадия 4: 3,3-Дибром-6-метил-1-фенил-1,3-дигидропирроло[3,2-b]пиридин-2-он
Соединение стадии 3 (800 мг, 3,84 ммоль) реагировало аналогично, как описано в примере 24, стадия 2. Получали 1,97 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=383
Стадия 5: 6-Метил-1-фенил-1,3-дигидропирроло[3,2-b]пиридин-2-он
Заявленное в заголовке соединение получали из неочищенного соединения стадии 4 (1,97 г) аналогично, как описано в примере 32, стадия 3. Выход: 770 мг.
LC/MS (способ LC4): m/z=224
Стадия 6: 2-Хлор-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 5 (760 мг, 3,39 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 910 мг.
LC/MS (способ LC4): m/z=271
Стадия 7: 2-(3-Фтор-2-метилбензил)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 6 (150 мг, 554 мкмоль) аналогично, как описано в примере 4, стадия 1. Выход: 22 мг.
LC/MS (способ LC4): m/z=359
Стадия 8: 2-(3-Фтор-2-метилбензил)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбоновая кислота
Соединение стадии 7 (20 мг, 56 мкмоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали при 40°C в течение 2 часов. Получали 60 мг неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=375
Стадия 9: [2-(3-Фтор-2-метилбензил)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Неочищенное соединение стадии 8 (60 мг) реагировало аналогично, как описано в примере 1, стадия 5, и затем, как описано в примере 4, стадия 2. Полученный твердый остаток растворяли в небольшом количестве MOH, смешивали с хлороводородной кислотой (0,1 M) и лиофилизовали в течение ночи для того, чтобы получить 6,4 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(3-фтор-2-метилбензил)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC4): m/z=443,20; Rt=0,98 мин
1H-ЯМР (400 МГц, MOH-D4): δ (ppm)=1,96 (с, 3H), 2,61 (с, 3H), 3,22 (м, 4H), 4,02 (м, 4H), 4,35 (м, 2H), 6,83 (м, 1H), 6,98 (м, 1H), 7,11 (м, 1H), 7,48 (м, 2H), 7,66 (м, 3H), 8,09 (м, 1H), 8,53 (м, 1H).
Пример 52
[2-(5-Фтор-2-метилфенокси)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
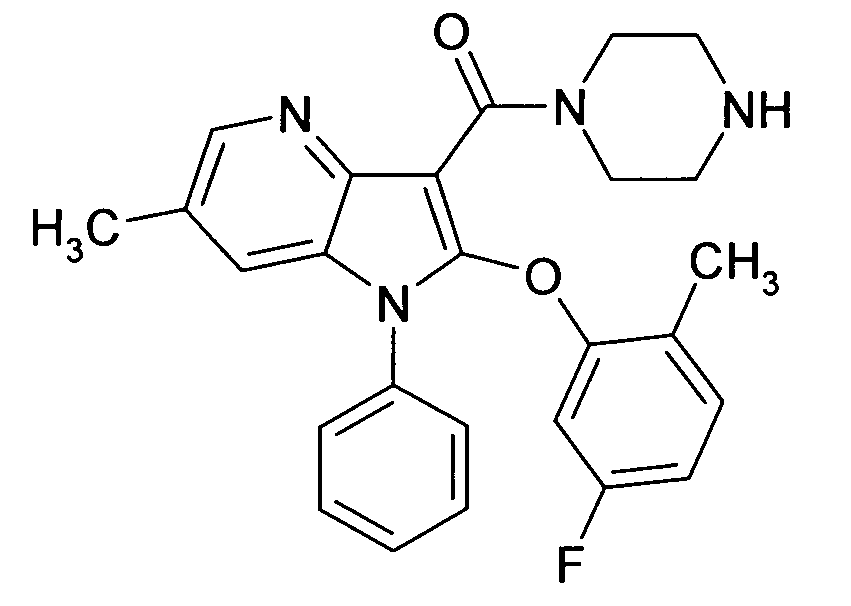
Стадия 1: 2-(5-Фтор-2-метилфенокси)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали аналогично, как описано в примере 1, стадия 6, из соединения примера 51, стадия 6, (150 мг, 554 мкмоль) и 5-фтор-2-метилфенола. Выход: 25 мг.
LC/MS (способ LC4): m/z=361
Стадия 2: 2-(5-Фтор-2-метилфенокси)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбоновая кислота
Соединение стадии 1 (25 мг, 69 мкмоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали при 40°C в течение 2 часов. Получали 17 мг неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=377
Стадия 3: трет-Бутиловый эфир 4-[2-(5-Фтор-2-метилфенокси)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Неочищенное соединение стадии 2 (17 мг) реагировало аналогично, как описано в примере 1, стадия 5. Получали 19 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=545
Стадия 4: [2-(5-Фтор-2-метилфенокси)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 3 (19 мг, 35 мкмоль) реагировало аналогично, как описано в примере 1, стадия 7. Растворение полученного твердого остатка в небольшом количестве MOH, добавление хлороводородной кислоты (0,1 M) и лиофилизация в течение ночи давали 2,4 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-6-метил-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC4): m/z=445,20; Rt=1,01 мин
1H-ЯМР (400 МГц, MOH-D4): δ (ppm)=2,20 (с, 3H), 2,58 (с, 3H), 3,21 (м, 4H), 3,84 (м, 4H), 6,89 (м, 2H), 7,23 (м, 1H), 7,60 (м, 5H), 8,09 (м, 1H), 8,48 (м, 1H).
Пример 53
[1-Циклогексил-2-(2,6-диметилфенокси)-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
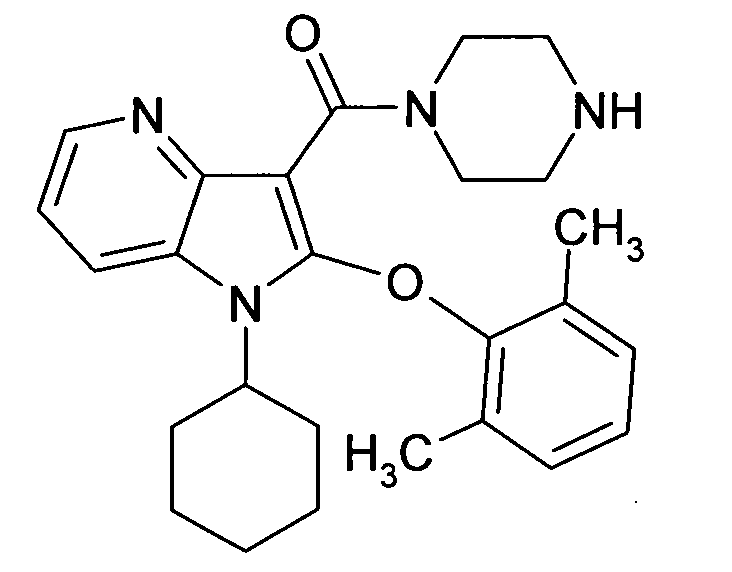
Стадия 1: 1-Циклогекс-2-енил-1H-пирроло[3,2-b]пиридин
Заявленное в заголовке соединение получали из 1H-пирроло[3,2-b]пиридина (5,7 г, 48,2 ммоль) аналогично, как описано в примере 46, стадия 1. Выход: 7,3 г.
LC/MS (способ LC4): m/z=199
Стадия 2: 1-Циклогексил-1H-пирроло[3,2-b]пиридин
Заявленное в заголовке соединение получали из соединения стадии 1 (7,30 г, 36,8 ммоль) аналогично, как описано в примере 46, стадия 2. Выход: 7,36 г.
LC/MS (способ LC4): m/z=201
Стадия 3: 3,3-Дибром-1-циклогексил-1,3-дигидропирроло[3,2-b]пиридин-2-он
Соединение стадии 2 (7,63 г, 38,1 ммоль) реагировало аналогично, как описано в примере 24, стадия 2. Получали 20 г неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=375
Стадия 4: 1-Циклогексил-1,3-дигидропирроло[3,2-b]пиридин-2-он
Заявленное в заголовке соединение получали из неочищенного соединения стадии 3 (20 г) аналогично, как описано в примере 32, стадия 3. Выход: 2,1 г.
LC/MS (способ LC4): m/z=217
Стадия 5: 2-Хлор-1-циклогексил-1H-пирроло[3,2-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 4 (1,60 г, 7,40 ммоль) аналогично, как описано в примере 1, стадия 3. Выход: 1,20 г.
LC/MS (способ LC4): m/z=263
Стадия 6: 2-Хлор-1-циклогексил-1H-пирроло[3,2-b]пиридин-3-карбоновая кислота
Соединение стадии 5 (700 мг, 2,66 ммоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали при 40°C в течение 2 часов. Получали 740 мг неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=279
Стадия 7: трет-Бутиловый эфир 4-(2-хлор-1-циклогексил-1H-пирроло[3,2-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Неочищенное соединение стадии 6 (740 мг) реагировало аналогично, как описано в примере 1, стадия 5. Получали 660 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=448
Стадия 8: [1-Циклогексил-2-(2,6-диметилфенокси)-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 7 (200 мг, 447 мкмоль) и 2,6-диметилфенол реагировали аналогично, как описано в примере 1, стадия 6, и продукт затем реагировал, как описано в примере 1, стадия 7. Полученный твердый остаток растворяли в небольшом количестве MOH, смешивали с хлороводородной кислотой (0,1 M) и лиофилизовали в течение ночи для того, чтобы получить 32 мг заявленного в заголовке соединения в форме дигидрохлорида [1-циклогексил-2-(2,6-диметилфенокси)-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC8): m/z=432,25; Rt=2,43 мин
1H-ЯМР: δ (ppm)=1,47 (м, 1H), 1,58 (м, 2H), 1,72 (м, 1H), 1,92 (м, 2H), 2,04 (м, 2H), 2,20 (с, 6H), 2,41 (м, 2H), 2,91 (м, 4H), 3,49 (м, 4H), 4,80 (м, 1H), 7,16 (м, 3H), 7,55 (м, 1H), 8,41 (м, 1H), 8,76 (м, 1H), 9,26 (м, 2H).
Пример 54
[2-(2,6-Диметилфенокси)-6-этокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
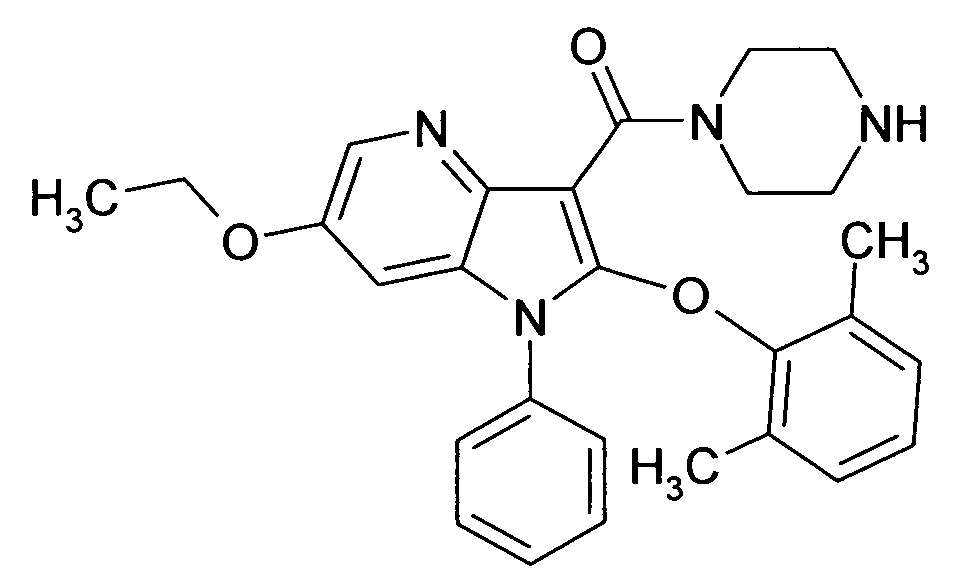
Стадия 1: 5-Этокси-2-триметилсиланилэтинилпиридин-3-иламин
Заявленное в заголовке соединение получали из 2-бром-5-этоксипиридин-3-иламина (20 г, 92,1 ммоль) аналогично, как описано в примере 51, стадия 1. Выход: 15,9 г.
LC/MS (способ LC4): m/z=235
Стадия 2: 6-Этокси-1H-пирроло[3,2-b]пиридин
Заявленное в заголовке соединение получали из соединения стадии 1 (12,5 г, 53,3 ммоль) аналогично, как описано в примере 52, стадия 2. Выход: 6,0 г.
LC/MS (способ LC4): m/z=163
Стадия 3: 6-Этокси-1-фенил-1H-пирроло[3,2-b]пиридин
Йодбензол (5,85 мл, 28,7 ммоль) добавляли к смеси соединения стадии 2 (3 г, 18,5 ммоль), йодида меди(I) (387,6 мг, 2,04 ммоль), хлорида лития (941,1 мг, 22,2 ммоль), N,N'-диметилэтилендиамина (505,5 мг, 5,74 ммоль) и карбоната калия (9,10 г, 65,9 ммоль) в DMF (50 мл). Реакционную смесь перемешивали при 120°C в течение 6 часов. После охлаждения до комнатной температуры добавляли раствор гидроксида аммония (10% в воде) и EA. Органический слой отделяли, промывали дважды насыщенным солевым раствором, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток очищали колоночной хроматографией на силикагеле (EA/HEP) для того, чтобы получить 3,40 г заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=239
Стадия 4: 6-Этокси-1-фенил-1,3-дигидропирроло[3,2-b]пиридин-2-он
Соединение стадии 3 (2,05 г, 8,60 ммоль) растворяли в DCM (30 мл) и добавляли N-хлорсукцинимид (1,26 г, 9,46 ммоль). Реакционную смесь перемешивали при комнатной температуре в течение 3 дней. Растворитель удаляли, и полученный твердый остаток растворяли в уксусной кислоте (10 мл) и нагревали при 70°C. После добавления фосфорной кислоты (7,31 мл, 107 ммоль, 85%), реакционную смесь нагревали при 120°C в течение 3 дней. После охлаждения смесь разбавляли водой и экстрагировали EA. Экстракты сушили над сульфатом натрия, фильтровали и упаривали. Остаток очищали хроматографией на силикагеле (EA/HEP 1:6). Получали 650 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=255
Стадия 5: 2-Хлор-6-этокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбальдегид
Заявленное в заголовке соединение получали из соединения стадии 4 (80 мг, 317 мкмоль) аналогично, как описано в примере 1, стадия 3. Выход: 50 мг.
LC/MS (способ LC4): m/z=201
Стадия 6: 2-Хлор-6-этокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбоновая кислота
Соединение стадии 5 (50 мг, 166,3 мкмоль) реагировало аналогично, как описано в примере 1, стадия 4. Реакционную смесь перемешивали при 40°C в течение 2 часов. Получали 47 мг неочищенного заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=317
Стадия 7: трет-Бутиловый эфир 4-(2-хлор-6-этокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил)пиперазин-1-карбоновой кислоты
Неочищенное соединение стадии 6 (47 мг) реагировало аналогично, как описано в примере 1, стадия 5. Получали 31 мг заявленного в заголовке соединения.
LC/MS (способ LC4): m/z=486
Стадия 8: [2-(2,6-Диметилфенокси)-6-этокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Соединение стадии 7 (28 мг, 57,7 мкмоль) и 2,6-диметилфенол реагировали аналогично, как описано в примере 1, стадия 6, и продукт затем реагировал, как описано в примере 1, стадия 7. Полученный твердый остаток растворяли в небольшом количестве MOH, смешивали с хлороводородной кислотой (0,1 M) и лиофилизовали в течение ночи для того, чтобы получить 15 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(2,6-диметилфенокси)-6-этокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC4): m/z=471,20; Rt=0,75 мин
1H-ЯМР: δ (ppm)=1,32 (т, 3H), 2,13 (с, 3H), 2,87 (м, 4H), 3,43 (м, 4H), 4,10 (кв, 2H), 7,08 (м, 3H), 7,29 (м, 1H), 7,59 (м, 1H), 7,68 (м, 4H), 8,18 (м, 1H), 9,21 (м, 2H).
Пример 55
[2-(2,6-Диметилфенокси)-6-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
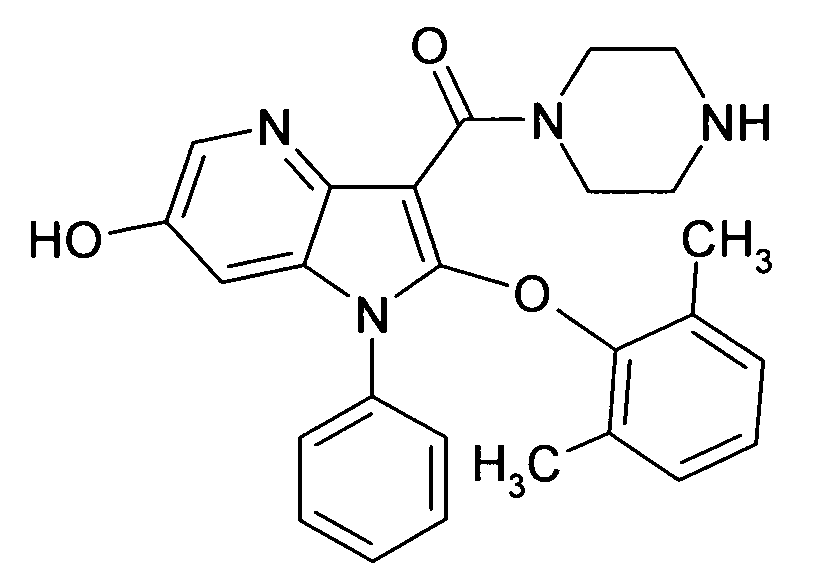
Стадия 1: трет-Бутиловый эфир 4-[2-(2,6-диметилфенокси)-6-этокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 54, стадия 7, (290 мг, 598 мкмоль) и 2,6-диметилфенола аналогично, как описано в примере 1, стадия 6. Выход: 170 мг.
LC/MS (способ LC10): m/z=571
Стадия 2: [2-(2,6-Диметилфенокси)-6-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали из соединения стадии 1 (170 мг, 298 мкмоль) аналогично, как описано в примере 9, и получали в форме дигидрохлорида [2-(2,6-диметилфенокси)-6-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона. Выход: 25,9 мг.
LC/MS (способ LC9): m/z=442,20; Rt=2,47 мин
1H-ЯМР: δ (ppm)=2,13 (с, 6H), 2,86 (м, 2H), 2,94 (м, 2H), 3,38 (м, 2H), 3,48 (м, 2H), 6,08 (с, 3H), 7,30 (м, 1H), 7,62 (м, 1H), 7,70 (м, 4H), 8,11 (м, 1H), 9,25 (м, 2H).
Пример 56
трет-Бутиловый эфир 4-[2-(2,6-диметилфенокси)-6-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-1-карбоновой кислоты
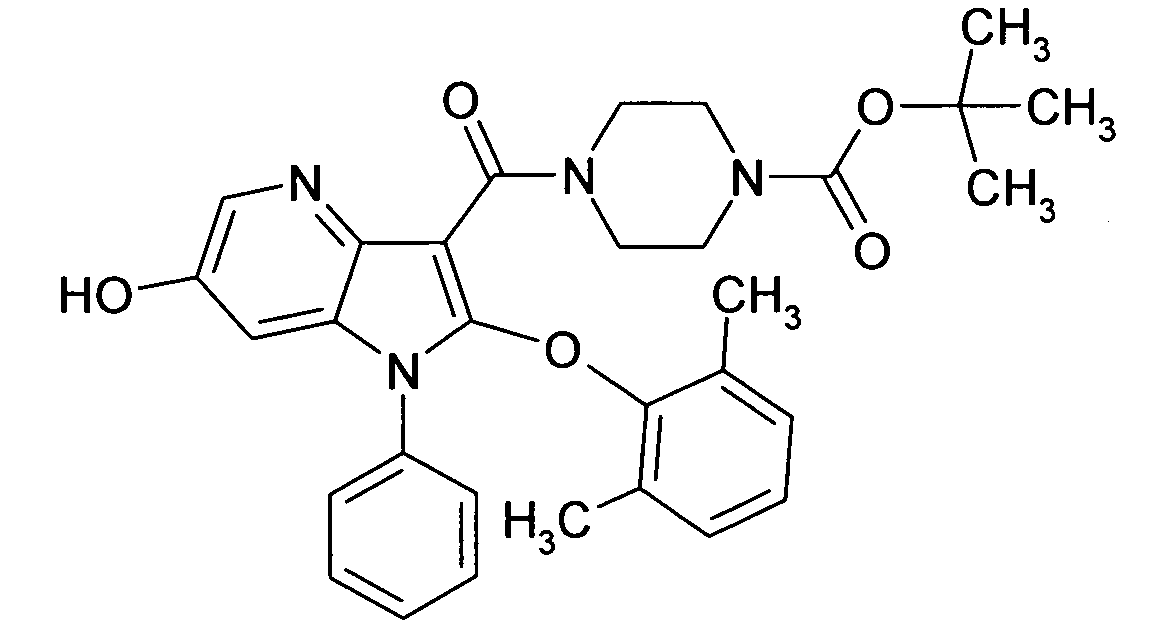
К раствору 80 мг (181 мкмоль) соединения примера 55, стадия 2, в MOH (1,0 мл) и THF (2,0 мл) добавляли гидрокарбонат натрия (45,6 мг, 542 мкмоль) и раствор ди-трет-бутилдикарбоната (43,4 мг, 199 мкмоль) в THF (2 мл). Реакционную смесь перемешивали при комнатной температуре в течение ночи. Растворители упаривали, и полученный твердый остаток растворяли в воде и EA. Органический слой отделяли, сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Получали 90 мг заявленного в заголовке соединения.
LC/MS (способ LC10): m/z=543
Пример 57
[2-(2,6-Диметилфенокси)-1-фенил-3-(пиперазин-1-карбонил)-1H-пирроло[3,2-b]пиридин-6-илокси]уксусная кислота
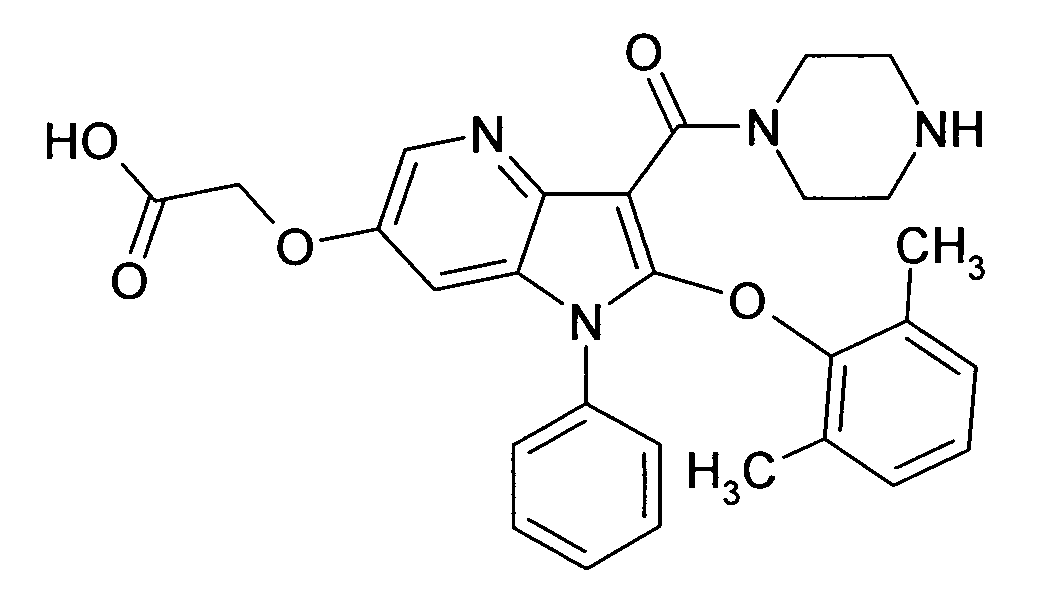
Стадия 1: трет-Бутиловый эфир 4-[6-трет-бутоксикарбонилметокси-2-(2,6-диметилфенокси)-1-фенил-1H-индол-3-карбонил]пиперазин-1-карбоновой кислоты
Соединение примера 56 (60 мг, 111 мкмоль), карбонат цезия (108 мг, 332 мкмоль) и трет-бутилбромацетат (17,9 мкл, 23,7 мкмоль) перемешивали в DMF при комнатной температуре в течение 2 часов. Смесь разбавляли водой и экстрагировали EA. Органический слой сушили над сульфатом натрия, фильтровали и упаривали при пониженном давлении. Остаток очищали хроматографией на силикагеле (EA/HEP). Выход: 67 мг.
LC/MS (способ LC10): m/z=657
Стадия 2: [2-(2,6-Диметилфенокси)-1-фенил-3-(пиперазин-1-карбонил)-1H-пирроло[3,2-b]пиридин-6-илокси]уксусная кислота
Соединение стадии 1 (67 мг, 102 мкмоль) реагировало аналогично, как описано в примере 1, стадия 3, полученный твердый остаток очищали хроматографией на силикагеле (EA/HEP), растворяли в небольшом количестве MOH, смешивали с хлороводородной кислотой (0,1 M) и лиофилизовали в течение ночи для того, чтобы получить 44 мг заявленного в заголовке соединения в форме дигидрохлорида [2-(2,6-диметилфенокси)-1-фенил-3-(пиперазин-1-карбонил)-1H-пирроло[3,2-b]пиридин-6-илокси]уксусной кислоты.
LC/MS (способ LC8): m/z=500,21; Rt=2,55 мин
1H-ЯМР (400 МГц, DMSO-D6): δ (ppm)=2,12 (с, 6H), 2,81 (м, 2H), 2,91 (м, 2H), 3,38 (м, 2H), 3,47 (м, 2H), 4,70 (с, 2H), 7,05 (с, 3H), 7,28 (м, 1H), 7,59 (м, 1H), 7,68 (м, 4H), 8,20 (м, 1H), 9,15 (м, 2H).
Пример 58
Метиловый эфир {4-[2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-2-ил}уксусной кислоты
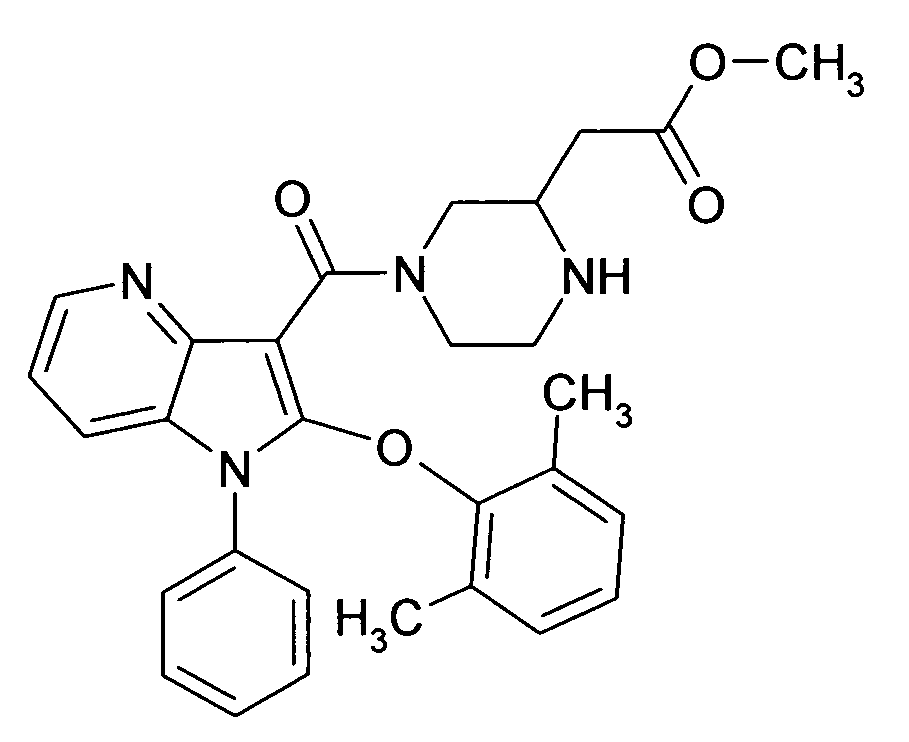
Стадия 1: трет-Бутиловый эфир 4-(2-хлор-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил)-2-метоксикарбонилметилпиперазин-1-карбоновой кислоты
Заявленное в заголовке соединение получали из соединения примера 1, стадия 4, (199,9 мг, 737 мкмоль) и трет-бутилового эфира 2-метоксикарбонилметилпиперазин-1-карбоновой кислоты (189,3 мг, 737 мкмоль) аналогично, как описано в примере 1, стадия 5. Выход: 254 мг.
LC/MS (способ LC10): m/z=512,9
Стадия 2: трет-Бутиловый эфир 4-[2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]-2-метоксикарбонилметилпиперазин-1-карбоновой кислоты
Соединение стадии 1 (254 мг, 507 мкмоль) и 2,6-диметилфенол реагировали аналогично, как описано в примере 1, стадия 6. Получали 340 мг неочищенного заявленного в заголовке соединения.
LC/MS (способ LC10): m/z=598,9
Стадия 3: Метиловый эфир {4-[2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-2-ил}уксусной кислоты
Неочищенное соединение стадии 2 (40 мг) реагировало аналогично, как описано в примере 1, стадия 7, полученный продукт растворяли в небольшом количестве MOH, смешивали с хлороводородной кислотой (0,1 M) и лиофилизовали в течение ночи для того, чтобы получить 13 мг заявленного в заголовке соединения в форме дигидрохлорида метилового эфира {4-[2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-2-ил}уксусной кислоты.
LC/MS (способ LC10): m/z=498,9; Rt=0,66 мин
1H-ЯМР: δ (ppm)=2,16 (м, 6H), 2,64-2,97 (м, 4H), 3,11-3,35 (м, 3H), 3,70 (с, 3H), 3,84 (м, 1H), 4,04 (м, 1H), 7,09 (м, 3H), 7,38 (м, 1H), 7,63 (м, 1H), 7,71 (м, 4H), 7,87 (м, 1H), 8,45 (м, 1H).
Пример 59
2-{4-[2-(2,6-Диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-2-ил}-N-метилацетамид
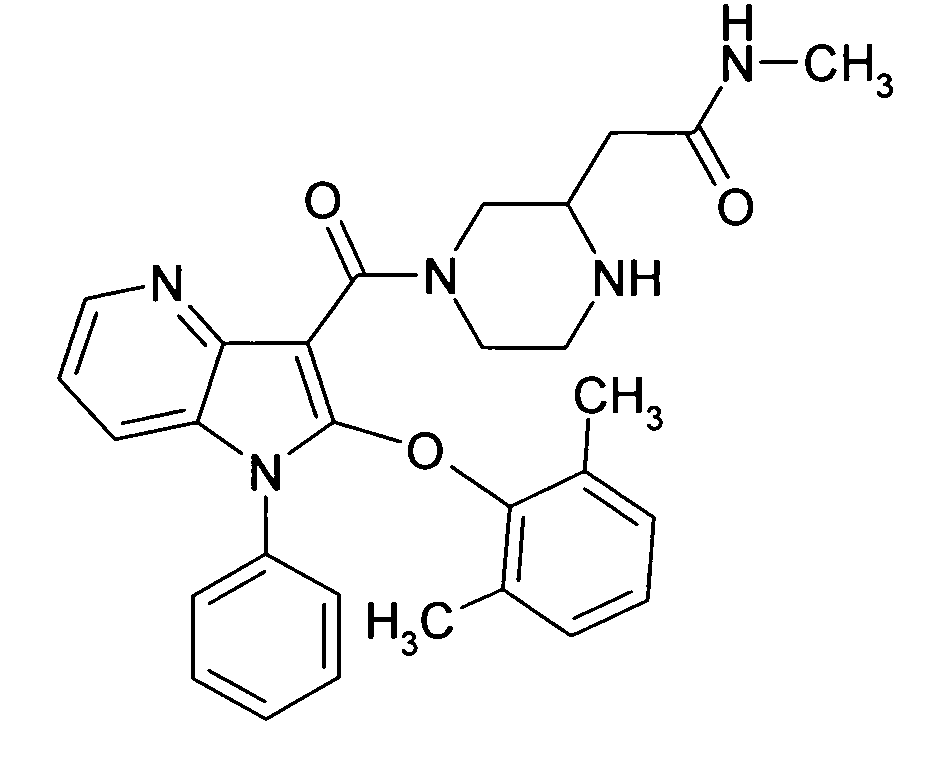
Стадия 1: трет-Бутиловый эфир 4-[2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]-2-метилкарбамоилметилпиперазин-1-карбоновой кислоты
Раствор неочищенного соединения примера 58, стадия 2, (140 мг, 234 мкмоль) в MOH (1 мл) смешивали с 2 M метанольным раствором метиламина (2,60 мл, 5,19 ммоль). Реакционную смесь перемешивали при 40°C в течение 7 дней. После охлаждения до комнатной температуры смесь нейтрализовали водным раствором лимонной кислоты и экстрагировали EA. Объединенные органические фазы сушили над сульфатом натрия и упаривали. Получали 45 мг неочищенного заявленного в заголовке соединения.
LC/MS (способ LC10): m/z=598,0
Стадия 2: 2-{4-[2-(2,6-Диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-2-ил}-N-метилацетамид
Неочищенное соединение стадии 1 реагировало аналогично, как описано в примере 1, стадия 7, полученный продукт растворяли в небольшом количестве MOH, смешивали с хлороводородной кислотой (0,1 M) и лиофилизовали в течение ночи для того, чтобы получить 27,6 мг заявленного в заголовке соединения в форме дигидрохлорида 2-{4-[2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[3,2-b]пиридин-3-карбонил]пиперазин-2-ил}-N-метилацетамида.
LC/MS (способ LC10): m/z=498,0; Rt=0,67 мин
1H-ЯМР: δ (ppm)=2,09-2,22 (м, 6H), 2,64 (м, 2H), 2,90 (м, 1H), 3,11-3,24 (м, 3H), 3,48 (м, 3H), 3,75 (м, 1H), 3,34 (м, 1H), 4,08 (м, 1H), 7,09 (м, 3H), 7,42 (м, 1H), 7,63 (м, 1H), 7,72 (м, 4H), 7,91 (м, 1H), 8,13 (м, 1H), 8,49 (м, 1H).
Аналогично, как описано в примерах выше, получали соединения формулы Ip, перечисленные в таблице 1, и их получали в форме биссоли трифторуксусной кислоты или в форме дигидрохлорида, соответственно. Соединения можно называть как [2-(R20-окси)-1-R30-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон, в случае когда группа A представляет собой O, или [2-(R20-сульфанил)-1-R30-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон, в случае когда группа A представляет собой S, или [2-(R20-метил)-1-R30-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон, в случае когда группа A представляет собой CH2, допуская модификации в связи с правилами номенклатуры, такие как обозначение группы R20-метил в виде бензильной группы.
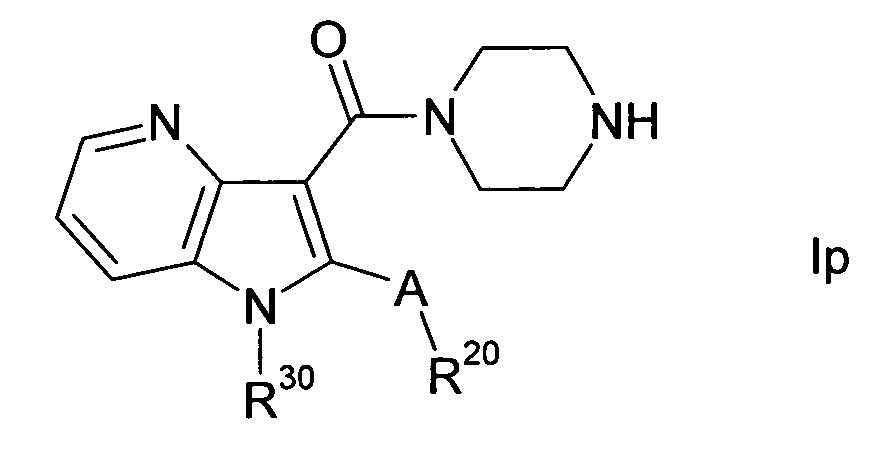
|
Аналогично. как описано в примерах выше, получали соединения формулы Iq, перечисленные в таблице 2, и их получали в форме дигидрохлорида. Соединения можно называть как [2-(R20-окси)-1-R30-1H-пирроло[3,2-b]пиридин-3-ил]-R100-метанон, допуская модификации в связи с правилами номенклатуры.
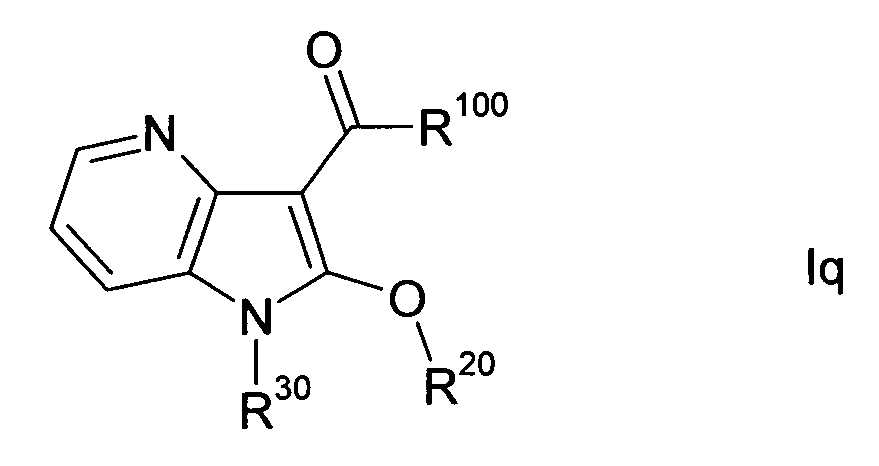
|
Аналогично. как описано в примерах выше, получали соединения формулы Ir, перечисленные в таблице 3, и их получали в форме биссоли трифторуксусной кислоты. Соединения можно называть как [1-фенил-2-(R20-окси)-1H-пирроло[3,2-c]пиридин-3-ил]пиперазин-1-илметанон.
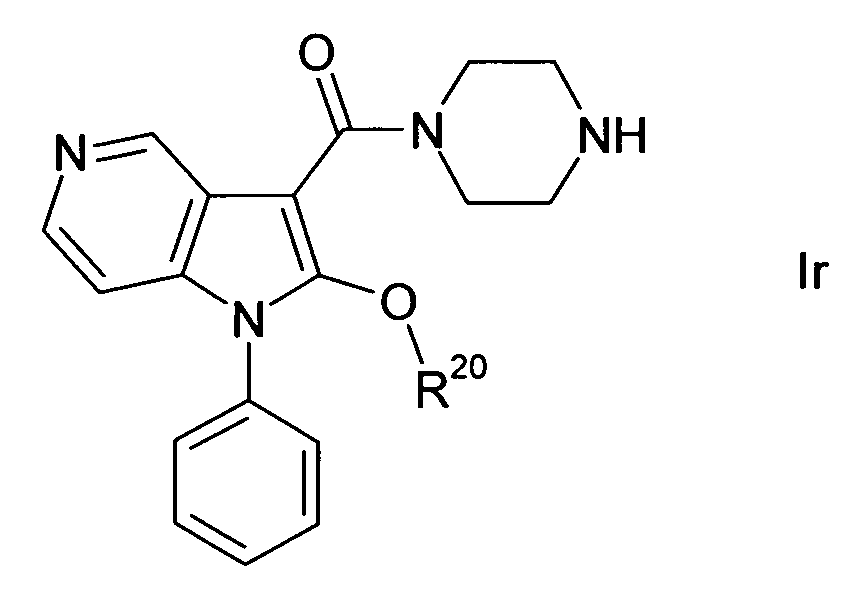
|
Аналогично, как описано в примерах выше, получали соединения формулы Is, перечисленные в таблице 4, и их получали в форме дигидрохлорида. Соединения можно называть как [1-циклогексил-2-(R20-окси)-6-R40-1H-пирроло[3,2-c]пиридин-3-ил]-R100-метанон, в случае когда группа A представляет собой O, или [1-циклогексил-2-(R20-метил)-6-R40-1H-пирроло[3,2-c]пиридин-3-ил]-R100-метанон, в случае когда группа A представляет собой CH2, допуская модификации в связи с правилами номенклатуры, такие как обозначение группы R20-метил в виде бензильной группы.
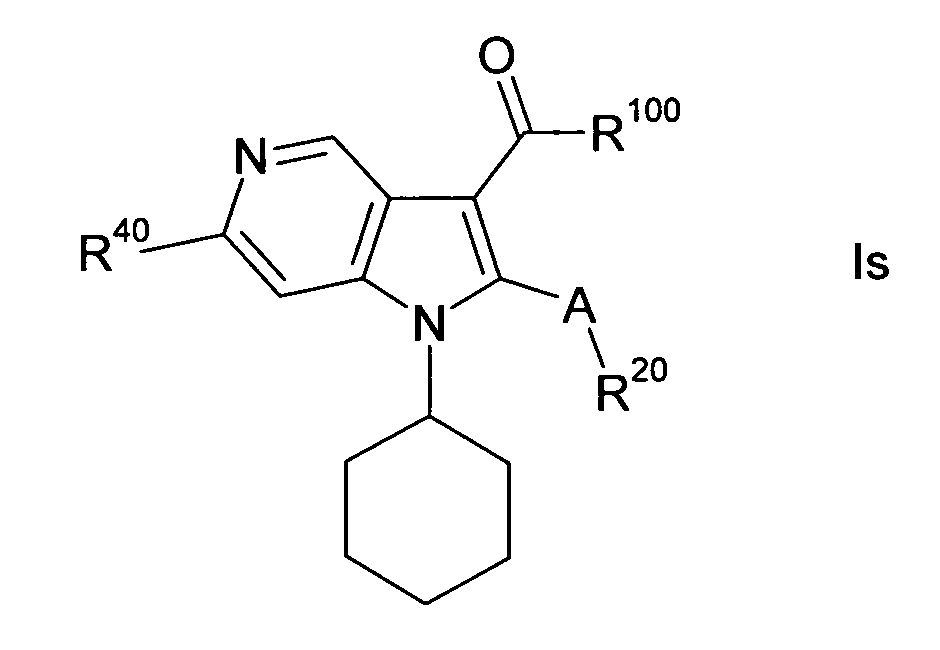
|
Аналогично, как описано в примерах выше, получали соединения формулы It, перечисленные в таблице 5, и их получали в форме биссоли трифторуксусной кислоты или дигидрохлорида, соответственно. Соединения можно называть как [2-(R20-окси)-1-R30-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон.
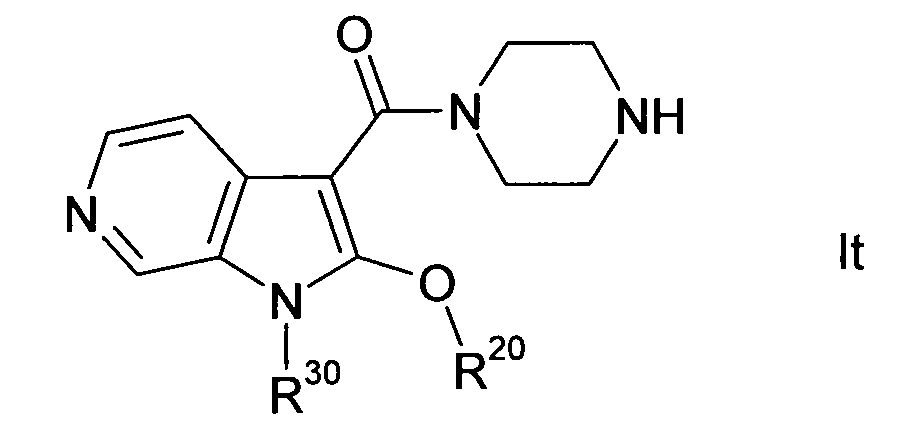
|
 Rt (мин)
Rt (мин)Аналогично, как описано в примерах выше, получали соединения формулы Iu, перечисленные в таблице 6, и их получали в форме биссоли трифторуксусной кислоты. Соединения можно называть как [1-фенил-2-(R20-окси)-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон.
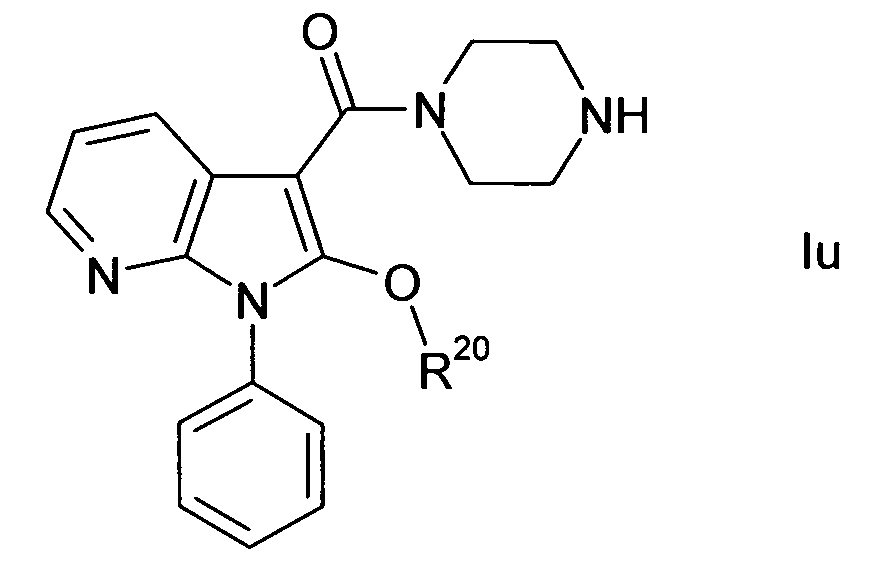
|
Аналогично, как описано в примерах выше, получали соединения формулы Iw, перечисленные в таблице 7, и их получали в форме дигидрохлорида. Соединения можно называть как [1-фенил-2-(R20-окси)-((4- или 5- или 6)-R40)-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон, в случае когда группа A представляет собой O, или [1-фенил-2-(R20-метил)-((4- или 5- или 6)-R40)-1H-пирроло[2,3-b]пиридин-3-ил]пиперазин-1-илметанон, в случае когда группа A представляет собой CH2, допуская модификации в связи с правилами номенклатуры, такие как обозначение группы R20-метил в виде бензильной группы.
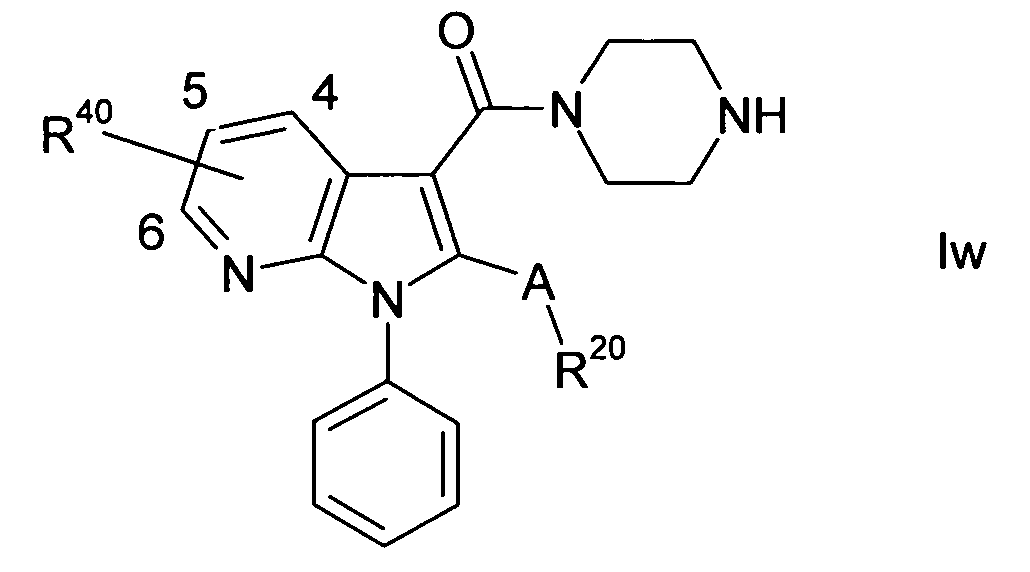
|
Пример 136
[2-(5-Фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали аналогично, как описано в примерах выше, и получали в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-5-метокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC3): m/z=460,19; Rt=1,52 мин
Пример 137
[2-(5-Фтор-2-метилфенокси)-5-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанон
Заявленное в заголовке соединение получали аналогично, как описано в примерах выше, и получали в форме дигидрохлорида [2-(5-фтор-2-метилфенокси)-5-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC1): m/z=446,18; Rt=1,18 мин
Пример 138
[2-(2,6-Диметилфенокси)-6-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]-((S)-3-метилпиперазин-1-ил)метанон
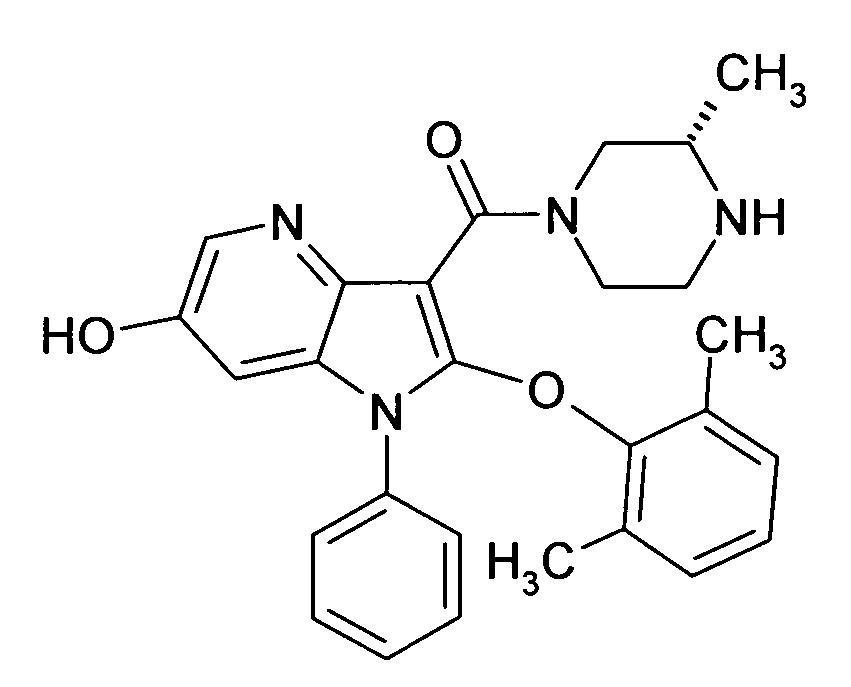
Заявленное в заголовке соединение получали аналогично, как описано в примерах выше и получали в форме дигидрохлорида [2-(2,6-диметилфенокси)-6-гидрокси-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]-((S)-3-метилпиперазин-1-ил)метанона.
LC/MS (способ LC10): m/z=456,22; Rt=2,45 мин
Пример 139
[2-(5-Фтор-2-метилфенилсульфанил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]-((S)-3-метилпиперазин-1-ил)метанон
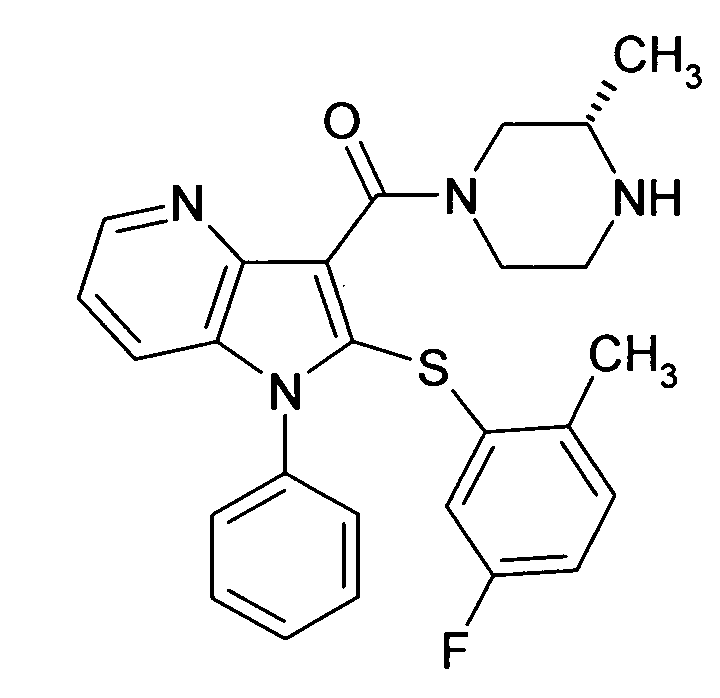
Заявленное в заголовке соединение получали аналогично, как описано в примерах выше, и получали в форме дигидрохлорида [2-(5-фтор-2-метилфенилсульфанил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]-((S)-3-метилпиперазин-1-ил)метанона.
LC/MS (способ LC4): m/z=461,2; Rt=1,05 мин
Пример 140
[2-(2,6-Диметилфенилсульфанил)-1-фенил-1H-пирроло[3,2-b]пиридин-3-ил]-((S)-3-метилпиперазин-1-ил)метанон
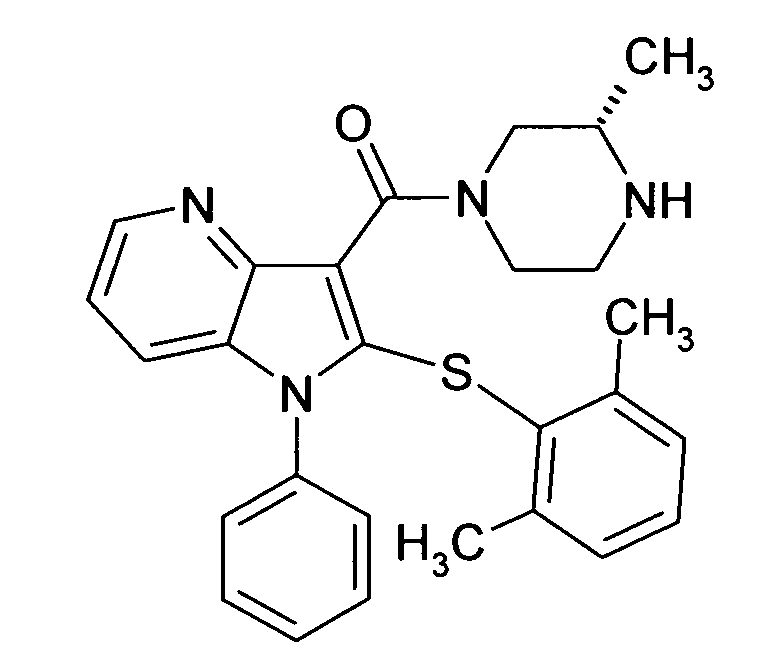
Заявленное в заголовке соединение получали аналогично, как описано в примерах выше, и получали в форме дигидрохлорида [2-(2,6-диметилфенилсульфанил)-1-фенил-1H- пирроло[2,3-b]пиридин-3-ил]-((S)-3-метилпиперазин-1-ил)метанона.
LC/MS (способ LC4): m/z=457,2; Rt=1,13 мин
Пример 141
[7-Хлор-2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанон
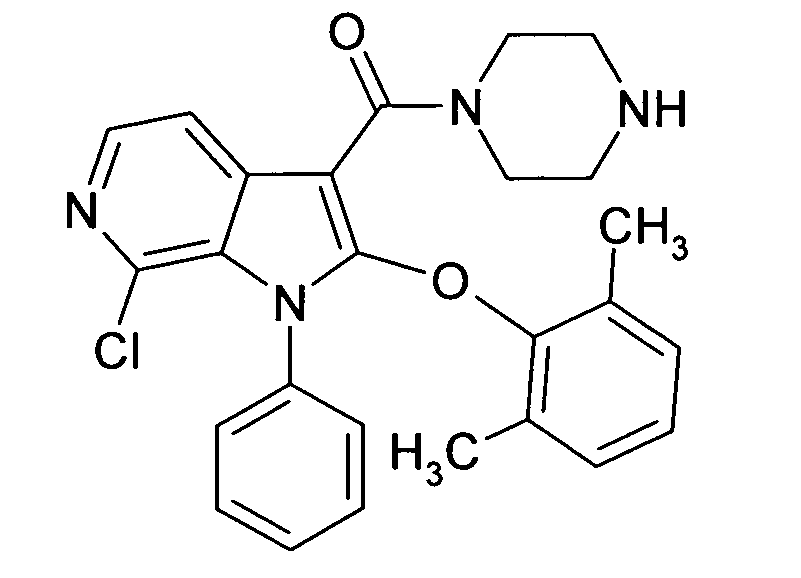
Заявленное в заголовке соединение получали аналогично, как описано в примерах выше, и получали в форме дигидрохлорида [7-хлор-2-(2,6-диметилфенокси)-1-фенил-1H-пирроло[2,3-c]пиридин-3-ил]пиперазин-1-илметанона.
LC/MS (способ LC5): m/z=460,17; Rt=1,94 мин
Фармакологические испытания
A) Ингибирование ренина
Ингибирующую ренин активность соединений по настоящему изобретению демонстрировали в in vitro испытании, в котором неэндогенный флуорогенный пептидный субстрат расщепляли ренином специфично по Leu-Val связи, которая соответствует сайту расщепления ангиотензиногена.
Рекомбинантный человеческий ренин (Cayman, № 10006217) с концентрацией 5 нМ инкубировали с испытуемыми соединениями при различных концентрациях и синтетическим субстратом Дабцил-γ-Abu-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-Thr-EDANS (Bachem, № M-2050; Дабцил относится к 4-(4-диметиламинофенилазо)бензоильной группе и EDANS относится к амиду 5-[(2-аминоэтил)амино]нафталин-1-сульфоновой кислоты) при концентрации 10 мкМ в течение 2 часов при комнатной температуре в 0,05 M Tris буфере (pH 8), содержащем 0,1 M NaCl, 2,5 мМ EDTA и 1,25 мг/мл бычьего сывороточного альбумина. Усиление флуоресценции, которое является результатом резонансного переноса энергии флуоресценции, фиксировали при длине волны возбуждения, равной 330 нм, и длине волны излучения, равной 485 нм, в микропланшетном спектрофотометре. Ингибирующие концентрации IC50 рассчитывали из процента ингибирования активности ренина, как функции концентрации испытуемого соединения. В данном испытании примерные соединения обычно ингибировали ренин с IC50 величиной, меньше чем приблизительно 10 микромоль/л (10 мкМ). Характерные IC50 величины, которые определяли с соединениями в форме полученной соли, показанными в примерах выше, перечислены в таблице 8.
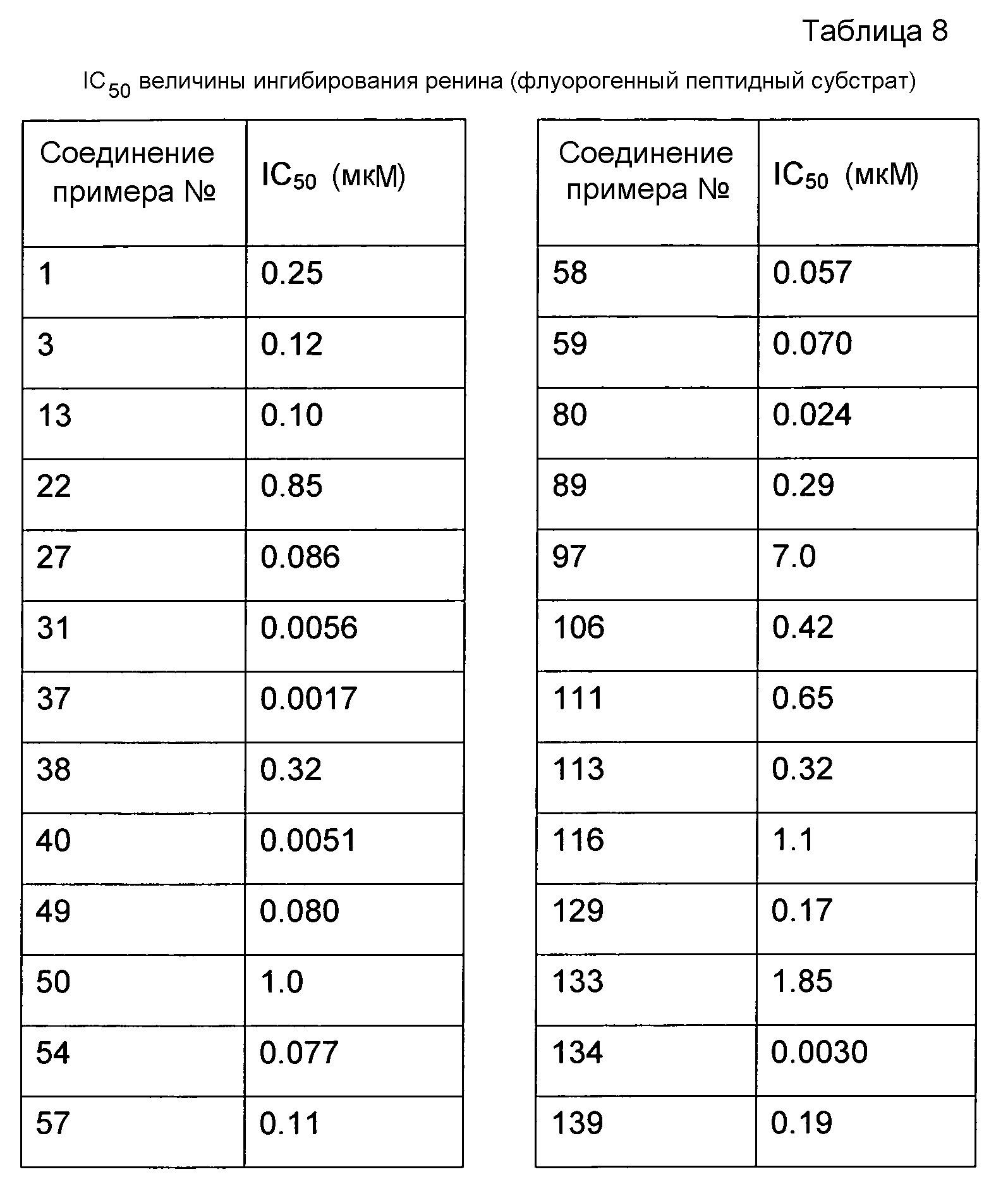
B) Ингибирование ренина в плазме человека
Ингибирующую ренин активность соединений по настоящему изобретению также демонстрировали в in vitro испытании в присутствии плазмы человека. Методика является аналогичной методике, описанной в фармакологическом испытании A, за исключением того, что человеческий рекомбинантный ренин при концентрации 30 нМ инкубировали с испытуемыми соединениями при различных концентрациях и флуорогенным субстратом Дабцил-γ-Abu-Ile-His-Pro-Phe-His-Leu-Val-Ile-His-Thr-EDANS при концентрации 25 мкМ в течение 30 мин при 37°C и 30 мин при комнатной температуре в плазме человека (Innovative Research, объединенная нормальная плазма человека, собранная на EDTA K3 в качестве антикоагулянта, № IPLA-5).
C) Антигипертензивная активность
Антигипертензивную активность in vivo соединений по настоящему изобретению можно продемонстрировать на дважды трансгенных мышах, сверхэкспрессирующих гены и человеческого ренина и ангиотензиногена (dTghRenhAgt мыши; см., D. C. Merrill et al., J. Clin. Invest. 97 (1996), 1047; R. L. Davisson et al., J. Clin. Invest. 99 (1997), 1258; J. L. Lavoie et al., Acta Physiol. Scand. 81 (2004), 571; полученные скрещиванием штаммов, несущих трансген ренина человека и трансген ангиотензиногена человека, соответственно). Вкратце, в данном испытании артериальное давление у свободно перемещающихся dTghRenhAgT мышей определяли телеметрическим наблюдением. Для этой цели катетер радиопередатчика (модель TA11 PA-10, DSI) имплантировали в левую сонную артерию dTghRenhAgT мышей при анестезии. Животных держали при 12-часовом цикле день/ночь и обеспечивали им свободный доступ к пище и воде. После одной недели восстановительного периода отслеживали артериальное давление и частоту сердечных сокращений в течение 24 часов для определения базовых величин. Затем животным вводили перорально через зонд или дневную дозу испытуемого соединения в среде (вода, содержащая 0,6% метилцеллюлозы и 0,5% Tween® 80), или в качестве контроля только среду. Гемодинамические параметры записывали непрерывно в течение дополнительных 24 часов, и определяли максимальный снижающий среднее артериальное давление эффект и продолжительность антигипертензивной активности (среднее артериальное давление=диастолическое давление + 1/3·(систолическое давление - диастолическое давление)). Соединения скринировали при различных дозах, таких как 3 мг/кг веса тела и 10 мг/кг веса тела в день.
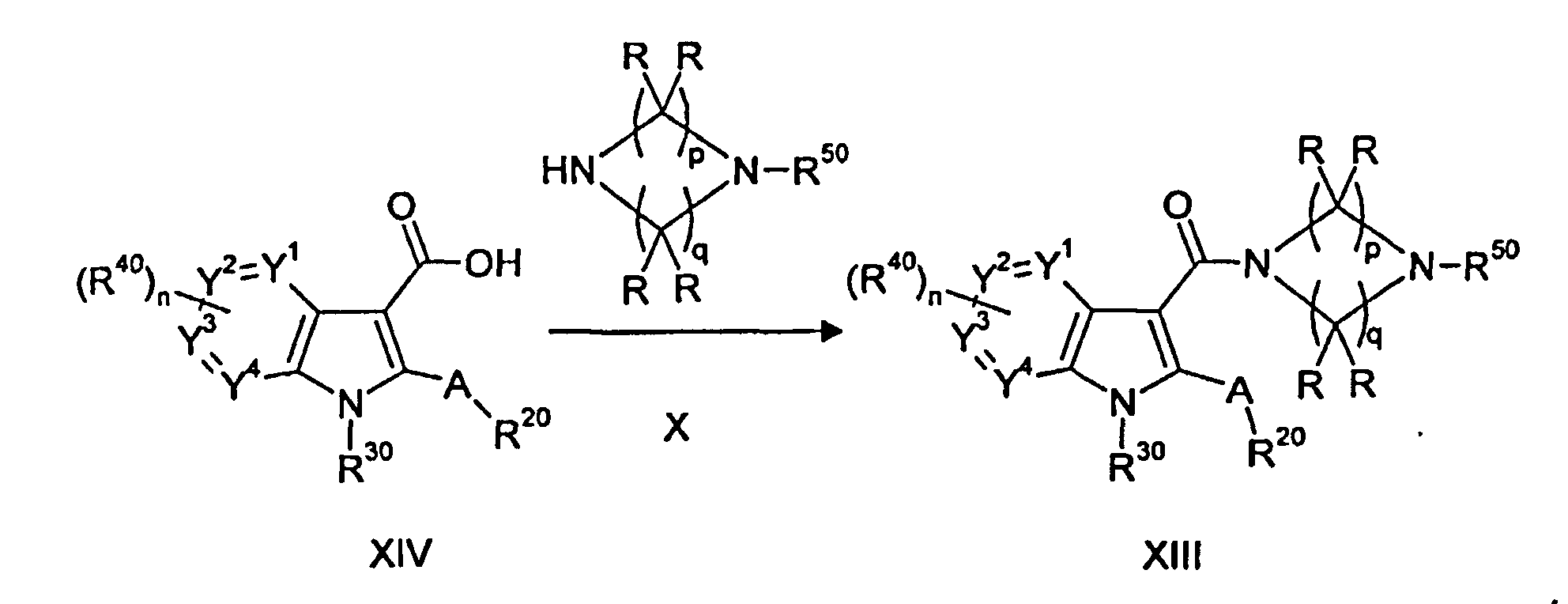

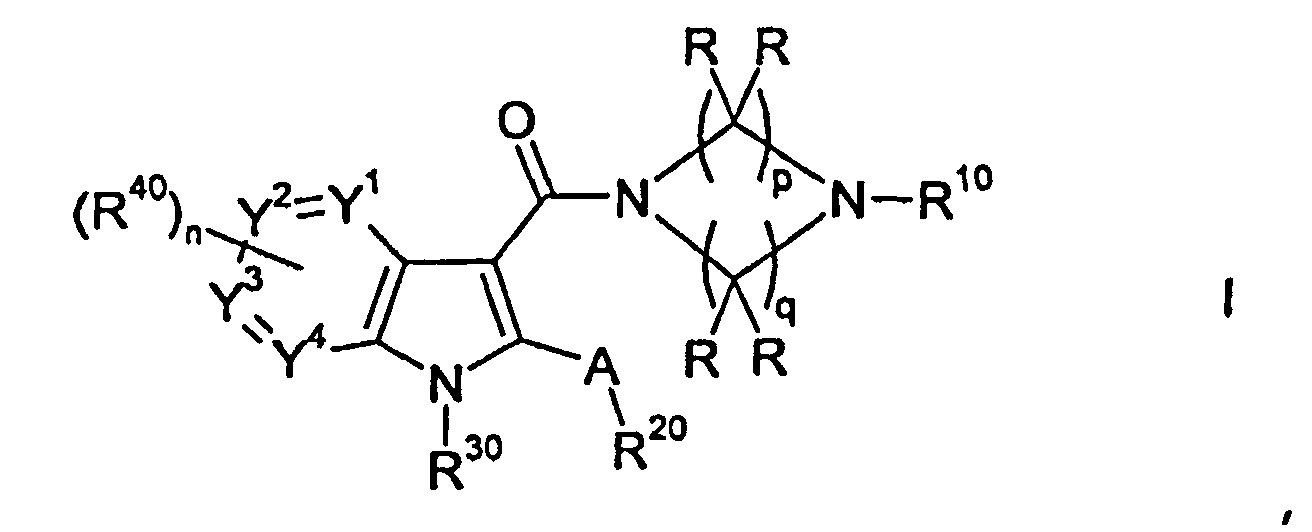
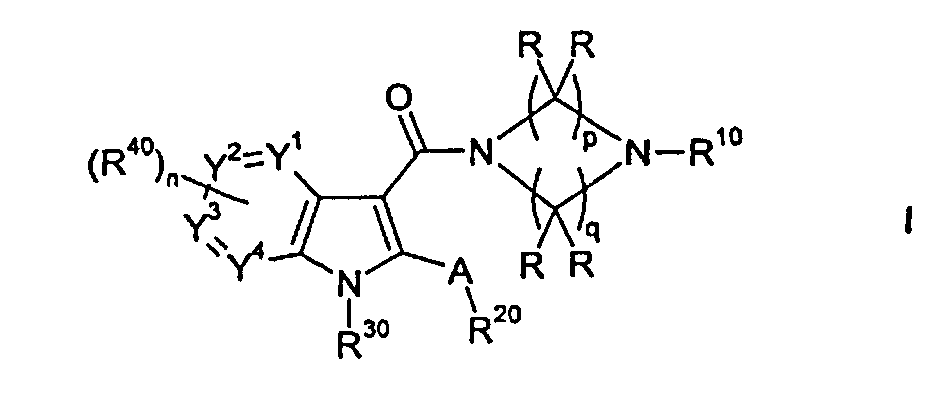
Замещенные циклоалкиламином производные изохинолина и изохинолинона
Производные циклогексиламинизохинолона в качестве ингибиторов rho-киназы
Производные алкиламинотиазола, их получение и их применение в терапии
Пиперидинил-замещенные производные изохинолона как ингибиторы rho-киназы
Антитела, связывающие il-4 и/или il-13, и их применение
Антитела, связывающие il-4 и/или il-13, и их применение
Замещенные изихинолины и изохинолиноны в качестве ингибиторов rho-киназы
Циклические (аза) индолизинкарбоксамиды, их получение и их примнение в качестве фармацевтических средств
Кислородзамещенные производные 3-гетероароиламинопропионовых кислот и их применение в качестве фармацевтических средств
Гуманизированные антитела к cxcr5, их производные и их применение
Антитела, связывающие il-4 и/или il-13, и их применение
Спироциклические нитрилы в качестве ингибиторов протеазы

