Результат интеллектуальной деятельности: ПРОИЗВОДНОЕ БЕНЗОПИПЕРИДИНА, СПОСОБ ЕГО ПОЛУЧЕНИЯ И ЕГО МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ
Вид РИД
Изобретение
Область изобретения
Настоящее изобретение принадлежит к области медицины и относится к производному бензопиперидина, способу его получения и его медицинскому применению. В настоящем изобретении раскрыто его применение в качестве модулятора рецептора эстрогена в профилактике и/или лечении опосредованных рецептором эстрогена или зависимых от него заболеваний или состояний, особенно предпочтительно рака молочной железы.
Предшествующий уровень техники
После длительного периода фундаментального исследования и клинического мониторинга обнаружено, что заболевания, такие как рак молочной железы, рак яичника, остеопороз, шизофрения и болезнь Альцгеймера, тесно связаны с нарушениями эстрогенового сигнального пути. Эстроген представляет собой стероидный гормон, секретируемый эндокринной системой, и играет важную роль в репродуктивной системе, костной ткани, сердечно-сосудистой системе, иммунной системе и центральной нервной системе. Система передачи сигнала эстрогена играет важную роль в регуляции роста, дифференцировки и апоптоза клеток. Возникновение и развитие эстроген-зависимых опухолей, таких как рак молочной железы, рак яичника и рак эндометрия, тесно связано с эстрогеном. В настоящее время основная химиотерапия рака молочной железы представляет собой применение антиэстрогенных агентов, таких как тамоксифен. Тем не менее тамоксифен проявляет свойства агониста эстрогена в матке, стимулируя, таким образом, раковые клетки в матке. В связи с данными серьезными побочными эффектами существует необходимость в поиске нового безопасного и эффективного лечения.
Одним из важных белков эстрогенового сигнального пути является рецептор эстрогена (ER - от англ. estrogen receptor). ER представляет собой рецептор стероидного гормона и принадлежит к активируемому лигандом транскрипционному фактору суперсемейства ядерных рецепторов, который содержит два подтипа: ERα (открытый в 1950 г.) и ERβ (открытый в 1996 г.), соответственно кодируемых разными генами. ERα и ERβ демонстрируют высокую степень сходства на аминокислотном уровне, и их сходство в ДНК-связывающем домене составляет вплоть до 97%, а сходство в лиганд-связывающем домене составляет вплоть до 56%, и только 24% низкую гомологию в N-конце. ER содержит 6 доменов от А до F, которые содержат четыре основные функциональные области. Функциональная область N-концевого домена А/В обладает лиганд-независимой функцией активации транскрипции AF-1, и AF-1 обладает конститутивной активностью. Транскрипция генов-мишеней активируется в результате взаимодействия с основными факторами транскрипции, факторами реактивации и другими факторами транскрипции. В этом функциональном участке имеются множественные сайты фосфорилирования, и сообщают, что роль AF-1 зависит от фосфорилирования белка. ДНК-связывающий домен (DBD - от англ. DNA binding domain), состоящий из домена С, высоко консервативен и содержит 2 домена типа «цинковых пальцев», которые могут специфично связываться с ДНК-мишенью одновременно, и данный домен играет важную роль в димеризации рецепторов. Домен D представляет собой шарнирную область, которая соединяет DBD и лиганд-связывающий домен (LBD - от англ. ligand binding domain), обладающую низкой консервативностью (только 30% гомология между двумя подтипами). Лиганд-связывающий домен (LBD), состоящий из С-концевого домена Е, определяет специфичное связывание ER с лигандами, такими как эстроген, селективный модулятор рецепторов эстрогена (SERM - от англ. selective estrogen receptor modulator) и селективный супрессор эстрогеновых рецепторов (SERD - от англ. selective estrogen receptor downregulator). LBD обладает лиганд-зависимой функцией активации транскрипции AF-2, которая имеет синергетическую реакцию с AF-1 с выполнением роли рецептора ER в активации транскрипции генов-мишеней. В то же время LBD имеет выраженную область контакта при димеризации и также может функционировать в отсутствие лигандов. Таким образом, LBD является ключевым сайтом димеризации рецептора.
ERα главным образом распространен в матке, яичнике, семеннике, гипофизе, почке, придатке яичка и надпочечной железе, тогда как ERβ главным образом распространен в предстательной железе, яичнике, легком, мочевом пузыре, головном мозге и кровеносных сосудах. Исследование SERM начинается в связи с серьезными побочными эффектами полных агонистов или полных антагонистов. «Селективность» означает, что SERM действует как агонист в некоторых тканях, таких как кость, печень и сердечно-сосудистая система, которые обогащены ERβ, тогда как в некоторых других тканях, таких как молочные железы, он действует как антагонист. В матке, являющейся значимой областью локализации ERα, он может быть либо агонистом, либо антагонистом. К настоящему времени имеющиеся в продаже SERM включают тамоксифен, ралоксифен, базедоксифен, торемифен и т.п. Тем не менее в исследованиях обнаружено, что имеющиеся в продаже SERM все еще обладают серьезными побочными эффектами, например, длительное применение тамоксифена и торемифена может вызывать гиперплазию эндометрия, полипы и рак эндометрия, и обычные побочные эффекты ралоксифена включают приливы, боль в ногах, болезненность в молочной железе и тромбоз вен и т.п. Поэтому исследование и разработка новых соединений все еще являются проблемами, требующими немедленного решения.
Тамоксифен принадлежит к классу соединений, известных как селективные модуляторы рецепторов эстрогена (SERM), и обладает способностью к стабилизации ERα и осуществлению легкой повышающей регуляции уровня рецепторов ERα. Напротив, фулвестрант индуцирует быструю деградацию ERα и усиливает блокирование сигнального пути рецептора ER, и такие соединения называют селективными супрессорами эстрогеновых рецепторов (SERD). Различия механизмов действия данных SERM и SERD, по-видимому, являются механизмами, ответственными за резистентность к этим соединениям. Большое число опухолей, которые являются резистентными к тамоксифену и ER-позитивными, все же чувствительны к фулвестранту. На основании клинических данных обнаружено, что SERD, такими как фулвестрант, можно эффективно лечить некоторые виды рака молочной железы, которые являются ERα-позитивными и резистентными к тамоксифену. Таким образом, соединения, ответственные за деградацию ERα, можно применять для продления периода эффективности у пациентов с раком молочной железы, подвергавшихся успешному лечению антиэстрогенной терапией, тогда как можно последовательно применять разные SERM, ингибиторы ароматазы и SERD.
Заявки на патенты, в которых раскрыты селективные модуляторы рецепторов эстрогена, включают WO 2014165723, WO 2014151899, WO 2014141292, WO 2014191726, WO 2015092634, WO 2014135834 и WO 2014106848, и в ЕР 1113007 А раскрыты сходные по структуре агонисты/антагонисты эстрогена.
Для достижения лучших терапевтических эффектов и для лучшего удовлетворения потребностей рынка авторы изобретения надеются разработать новое поколение высокоэффективных и низкотоксичных SERD, нацеленных на эстрогеновый сигнальный путь. Таким образом, в свете текущего исследования в данной области техники, особенно соединения AZD-9496 компании AstraZeneca в I фазе клинических исследований, согласно настоящему изобретению предложена новая структура SERD, которая показывает хорошую активность в ингибировании связывания Е (эстроген) с ER, деградации ER и пролиферации клеток MCF7 и т.д., особенно более выраженное преимущество в значении Emax деградации ER по сравнению с AZD-9496.
Краткое изложение сущности изобретения
Настоящее изобретение направлено на соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь или их фармацевтически приемлемую соль, где структура соединения формулы (I) является такой, как показано ниже:
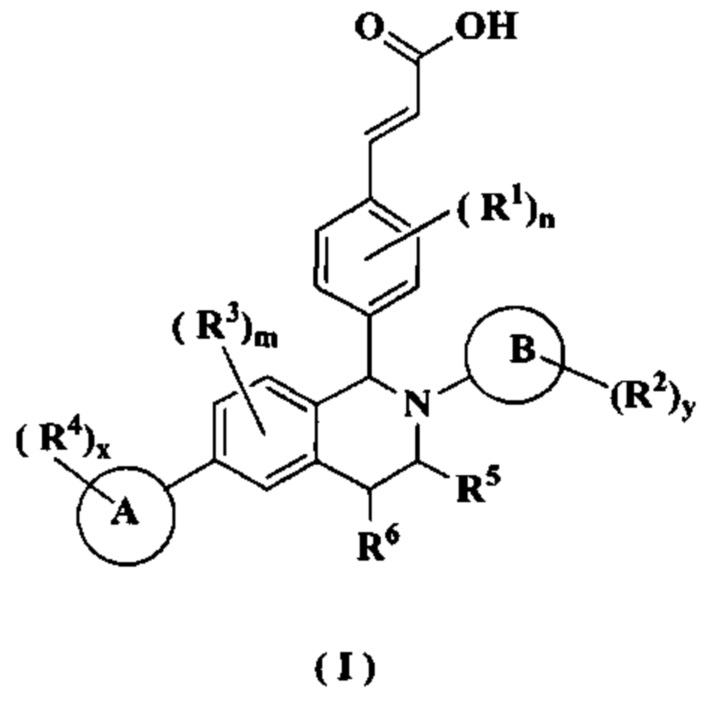
где:
кольцо А выбрано из группы, состоящей из циклоалкила, гетероциклила, арила и гетероарила;
кольцо В представляет собой арил или гетероарил;
каждый R1 является идентичным или отличным, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, алкокси, амино, циклоалкила, атома галогена, циано, карбокси, альдегида, гидрокси, нитро, арила и гетероарила, где каждый алкил, циклоалкил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, амино, нитро, циано, гидрокси, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила;
каждый R2 является идентичным или отличным, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, алкокси, амино, циклоалкила, атома галогена, циано, карбокси, альдегида, гидрокси, нитро, арила и гетероарила, где каждый алкил, циклоалкил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, амино, нитро, циано, гидрокси, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила;
каждый R3 является идентичным или отличнымо, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, галогеналкила, гидроксиалкила, алкокси, амино, циклоалкила, атома галогена, циано, карбокси, альдегида, гидрокси, нитро, арила и гетероарила, где каждый алкил, циклоалкил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, амино, нитро, циано, гидрокси, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила;
каждый R4 является идентичным или отличным, и каждый независимо выбран из группы, состоящей из атома водорода, алкила, дейтериоалкила, галогеналкила, гидроксиалкила, алкокси, амино, циклоалкила, атома галогена, циано, карбокси, альдегида, гидрокси, нитро, арила и гетероарила, где каждый алкил, циклоалкил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, амино, нитро, циано, гидрокси, гидроксиалкила, алкокси, циклоалкила, гетероциклила, арила и гетероарила;
R5 выбран из группы, состоящей из атома водорода, алкила, циклоалкила, гетероциклила, арила и гетероарила, где каждый алкил, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
R6 выбран из группы, состоящей из атома водорода, алкила, гидрокси, атома галогена, циано, амино, нитро, алкокси, циклоалкила, гетероциклила, арила и гетероарила, где каждый алкил, алкокси, циклоалкил, гетероциклил, арил и гетероарил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, гидрокси, амино, нитро, циано, алкокси, гидроксиалкила, циклоалкила, гетероциклила, арила и гетероарила;
m равен 0, 1, 2 или 3;
n равен 0, 1, 2, 3 или 4;
х равен 0, 1, 2 или 3; и
y равен 0, 1, 2, 3, 4 или 5;
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли кольцо А представляет собой гетероарил, предпочтительно пиразолил или тиазолил.
В предпочтительном воплощении настоящего изобретения соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль необязательно представляет собой соединение формулы (II):
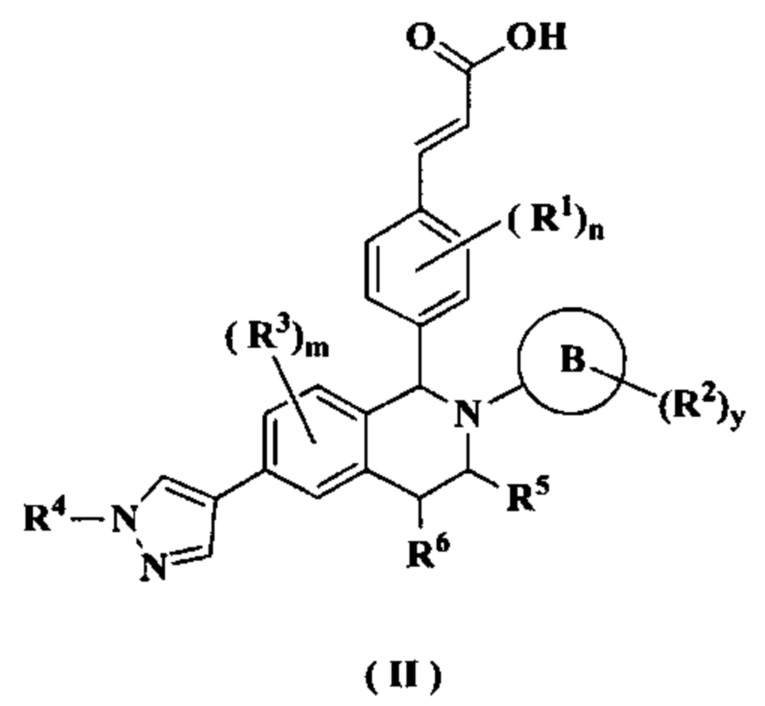
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемую соль,
где:
кольцо В, от R1 до R6, m, n, x и y являются такими, как определено в формуле (I)
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли кольцо В представляет собой арил.
В предпочтительном воплощении настоящего изобретения соединение формулы (II) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль необязательно представляет собой соединение формулы (III):
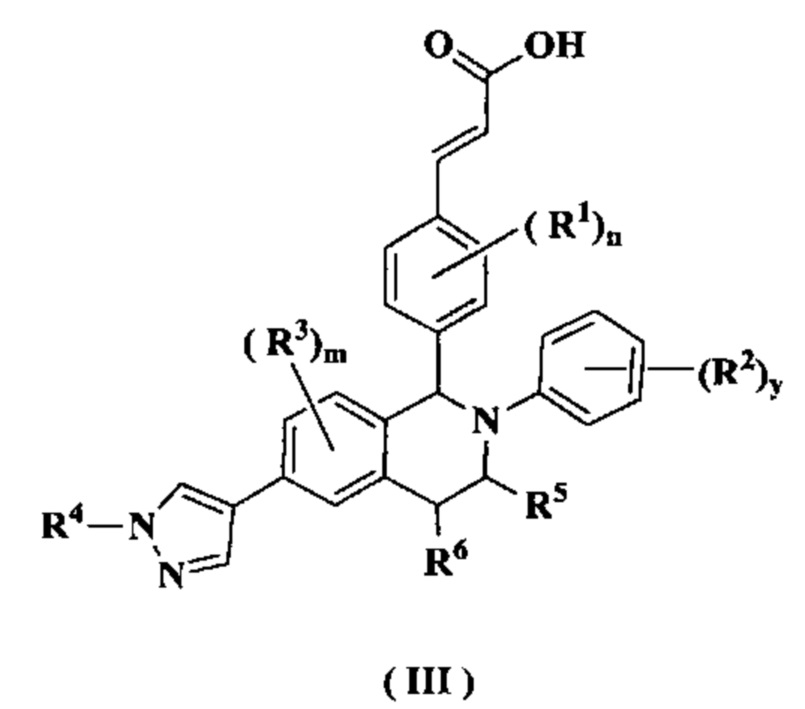
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемую соль,
где:
от R1 до R6, m, n, и у являются такими, как определено в формуле (I).
В предпочтительном воплощении настоящего изобретения соединение формулы (III) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль необязательно представляет собой соединение формулы (III-1):
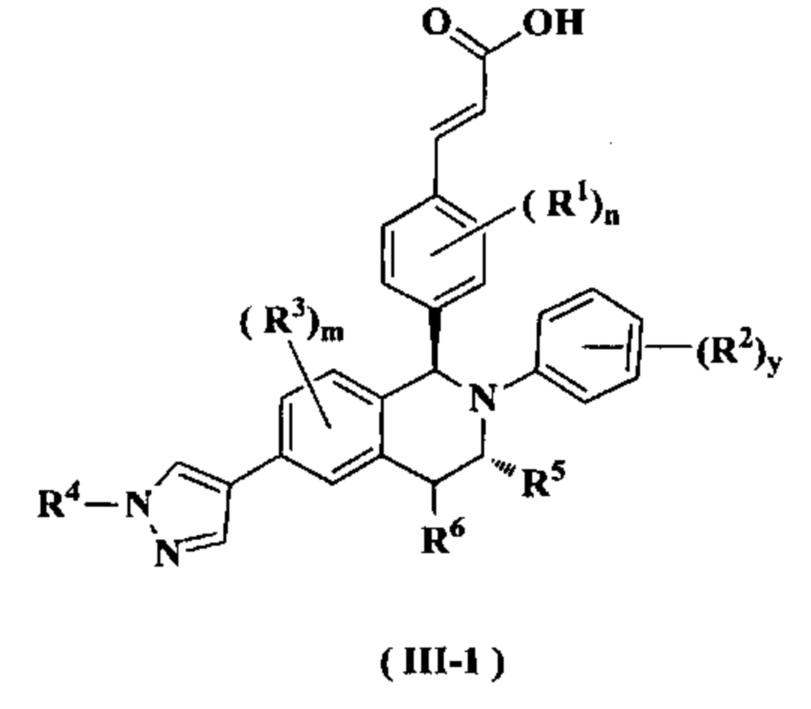
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемую соль,
где:
от R1 до R6, m, n, и y являются такими, как определено в формуле (I). В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R1 представляет собой атом водорода или атом галогена.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R2 выбран из группы, состоящей из атома водорода, атома галогена, алкила, галогеналкила, алкокси и циклоалкила, где алкил необязательно замещен одной или более группами, выбранными из группы, состоящей из атома галогена, алкокси и циклоалкила.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R3 представляет собой атом водорода.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R4 выбран из группы, состоящей из атома водорода, дейтероалкила, галогеналкила и алкила.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R5 представляет собой атом водорода или алкил.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли R6 представляет собой атом водорода.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли n равен 0 или 2.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли y равен 0, 1 или 2.
В предпочтительном воплощении настоящего изобретения в соединении формулы (I) или его таутомере, мезомере, рацемате, энантиомере, диастереомере или их смеси, либо их фармацевтически приемлемой соли m равен 0.
Типичные соединения формулы (I) включают без ограничений:

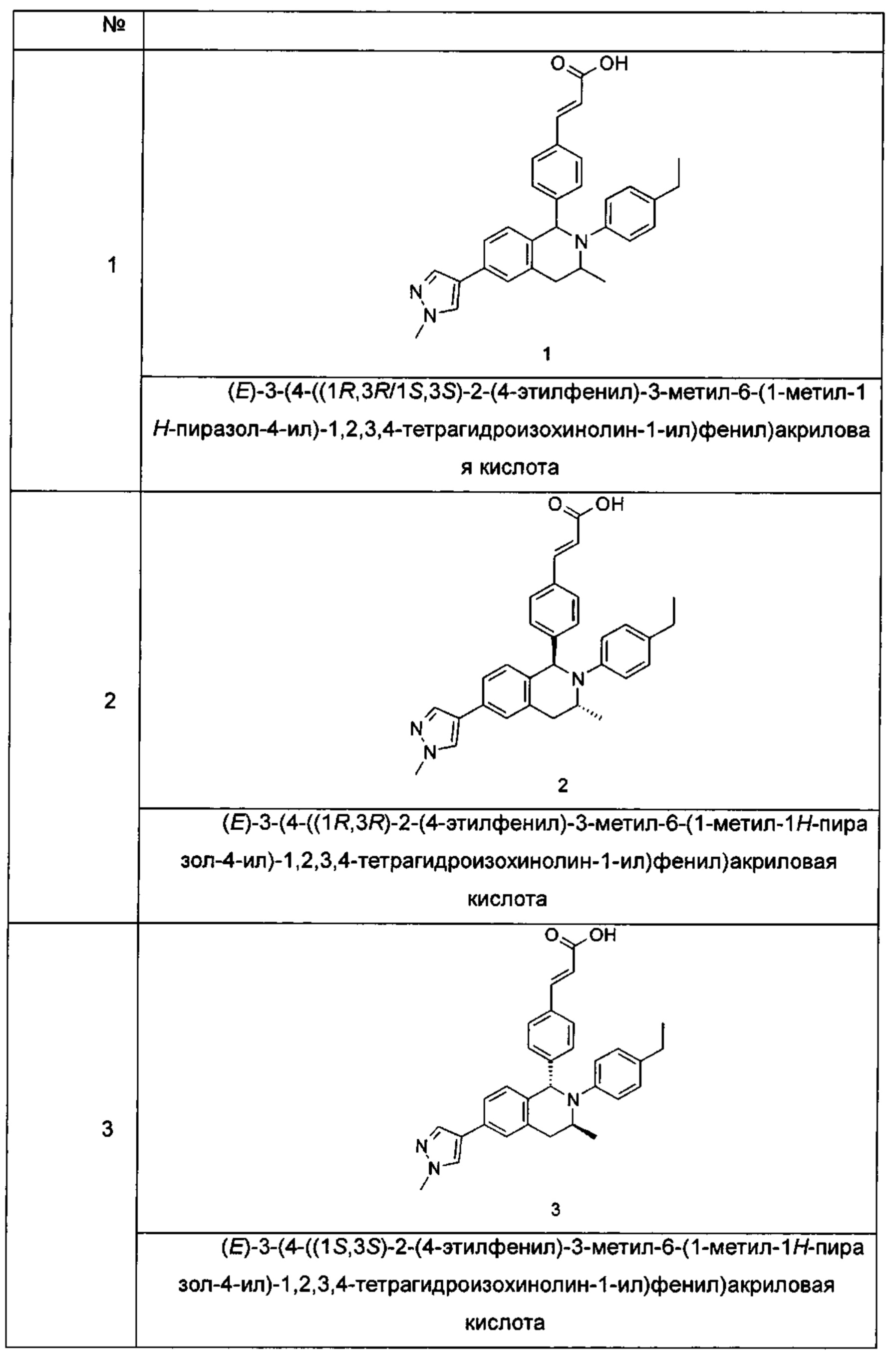
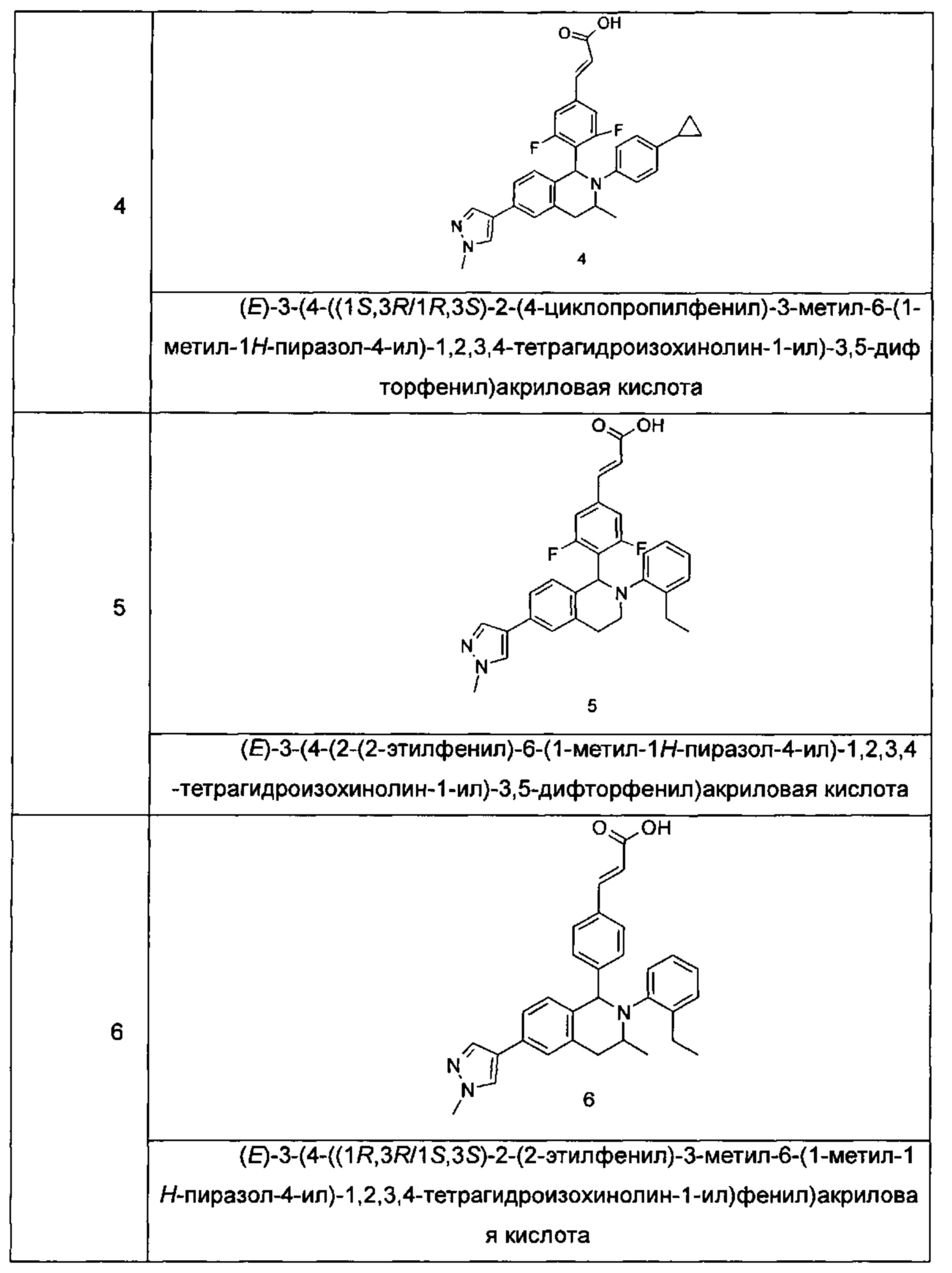
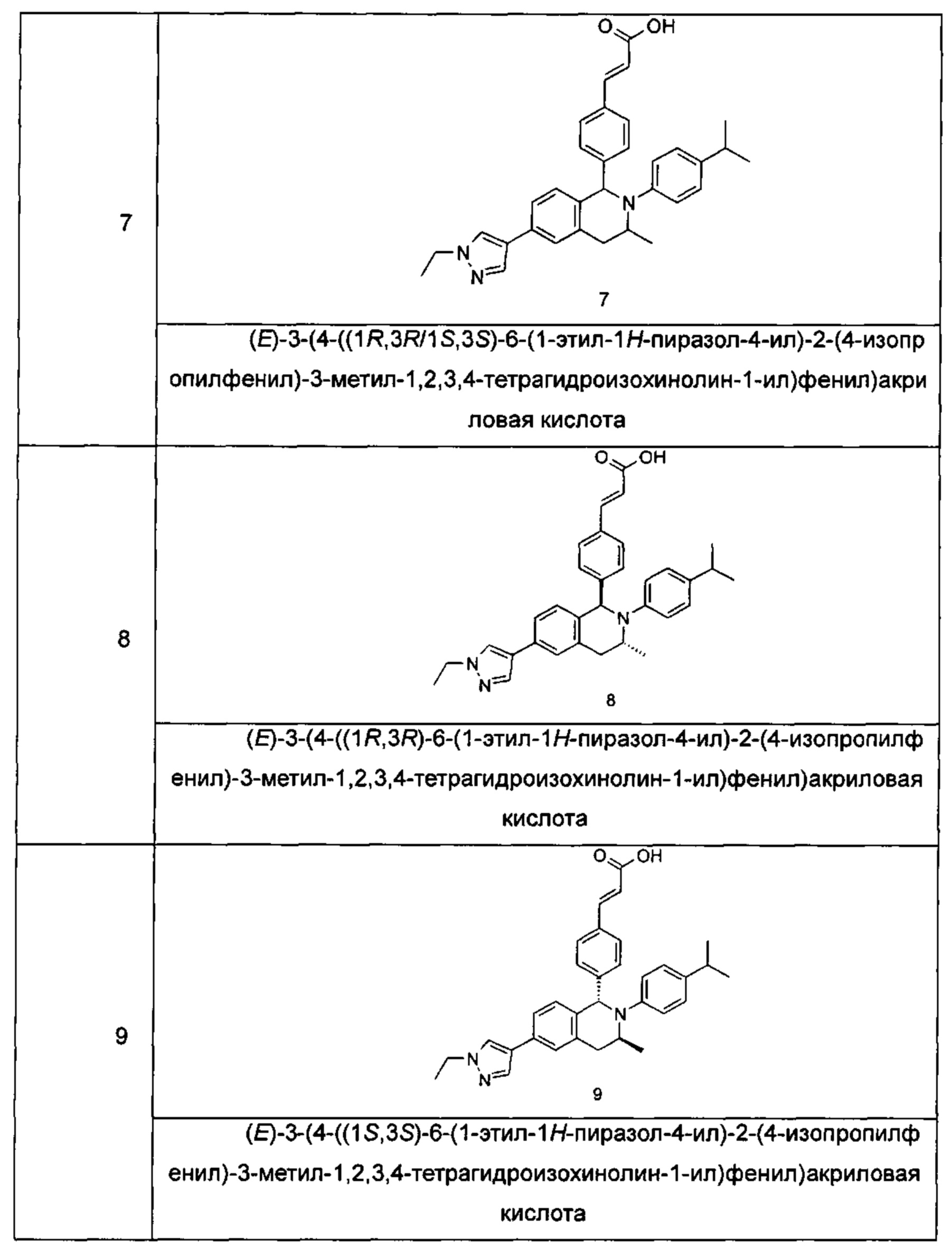
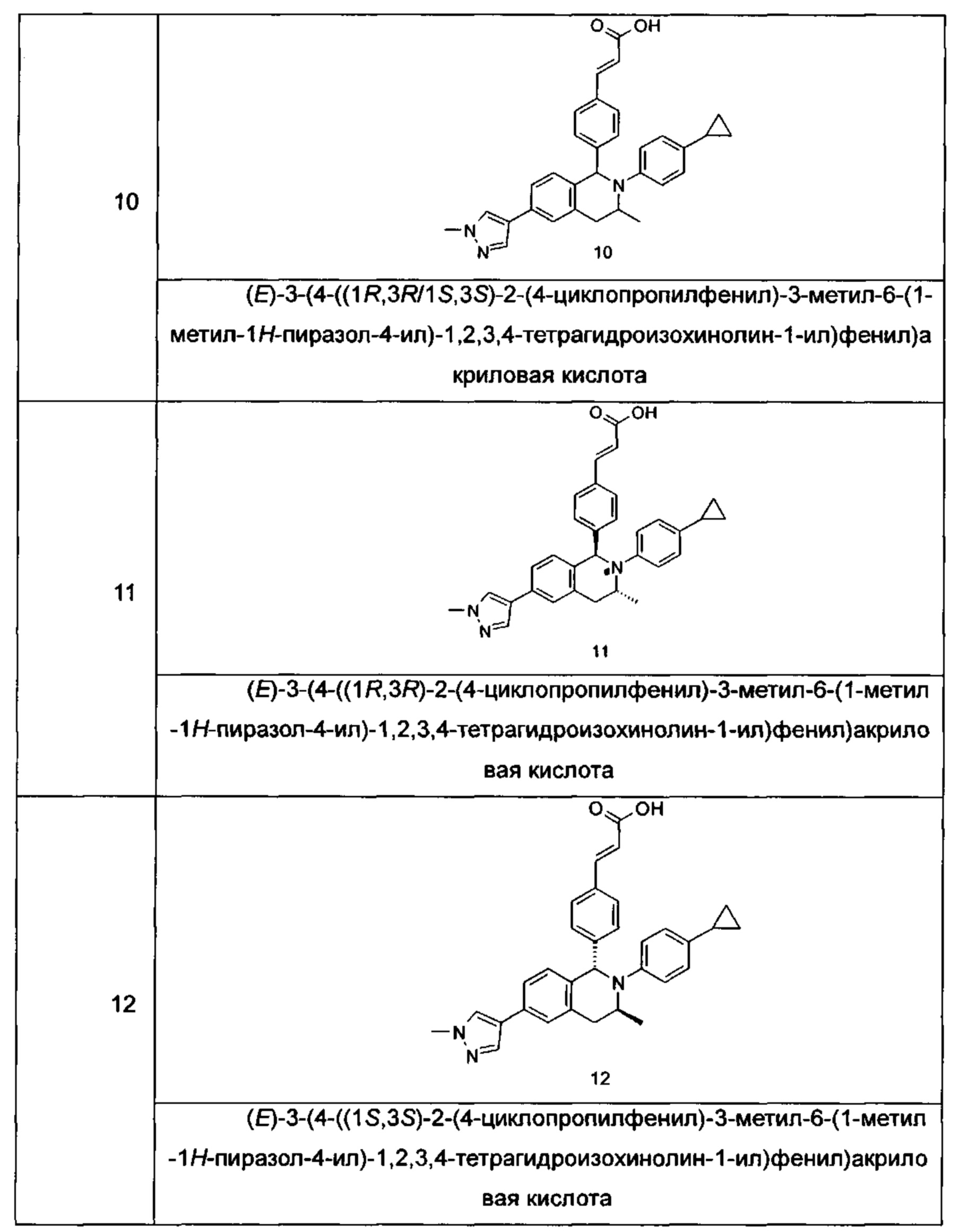
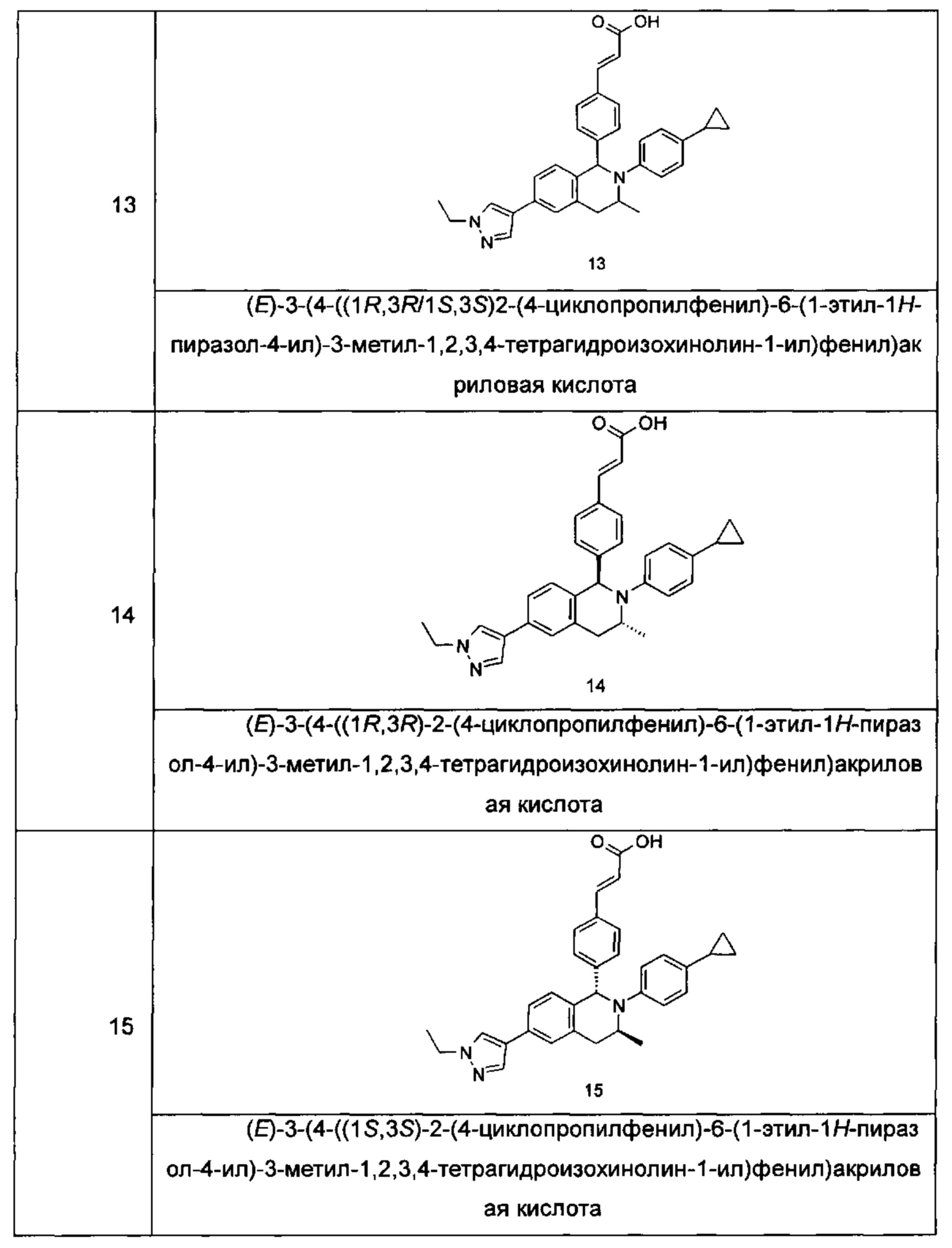
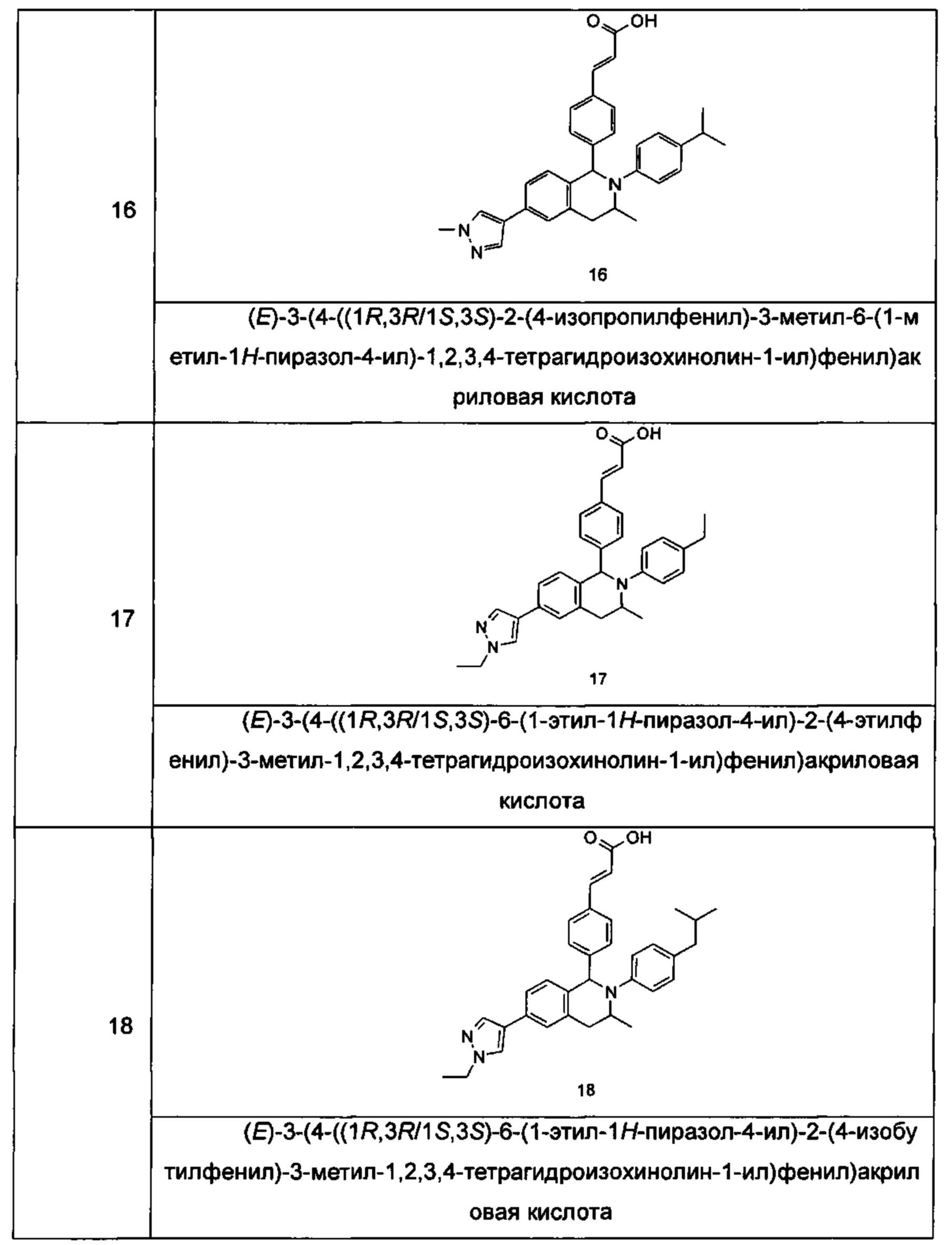
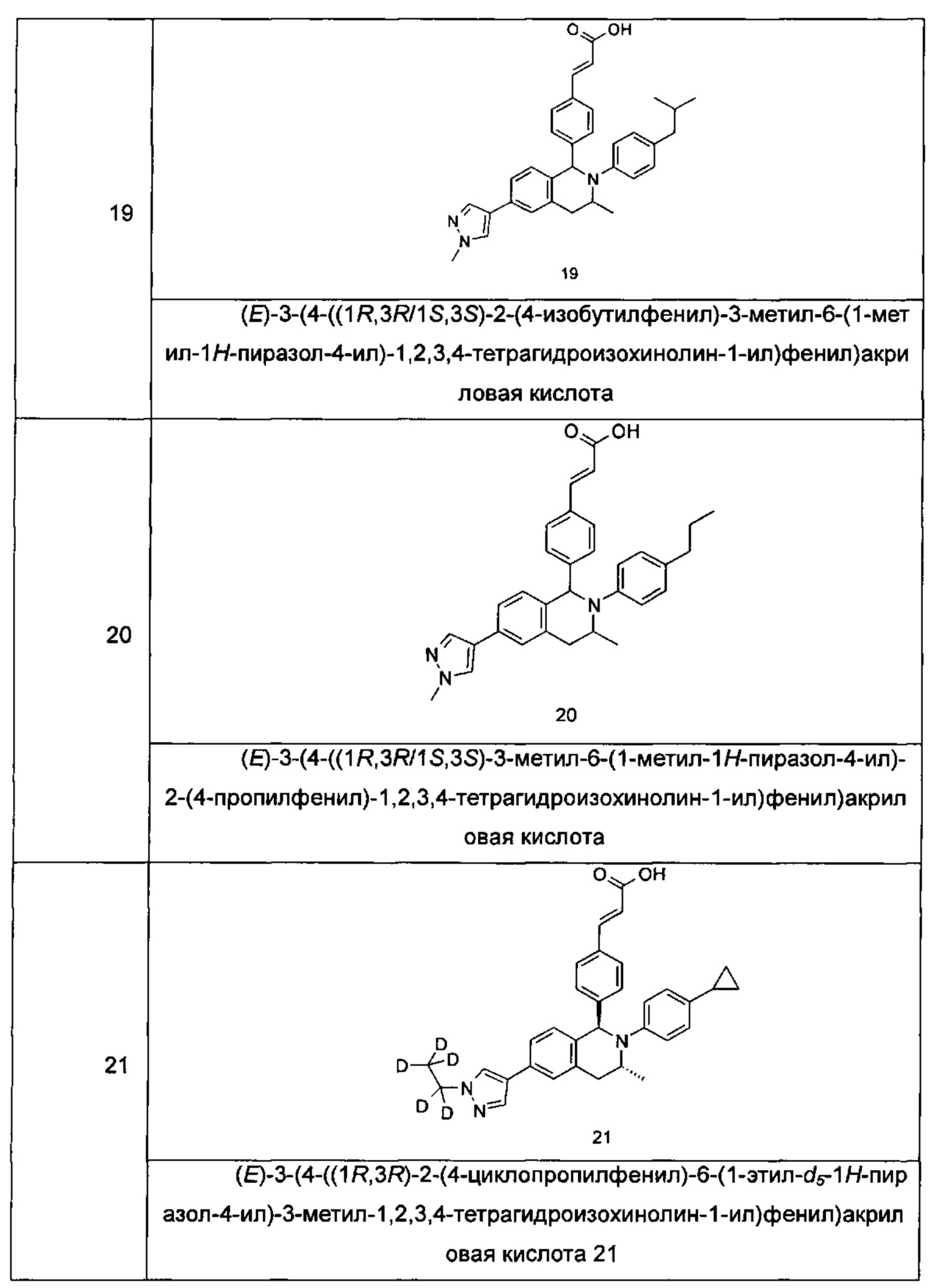
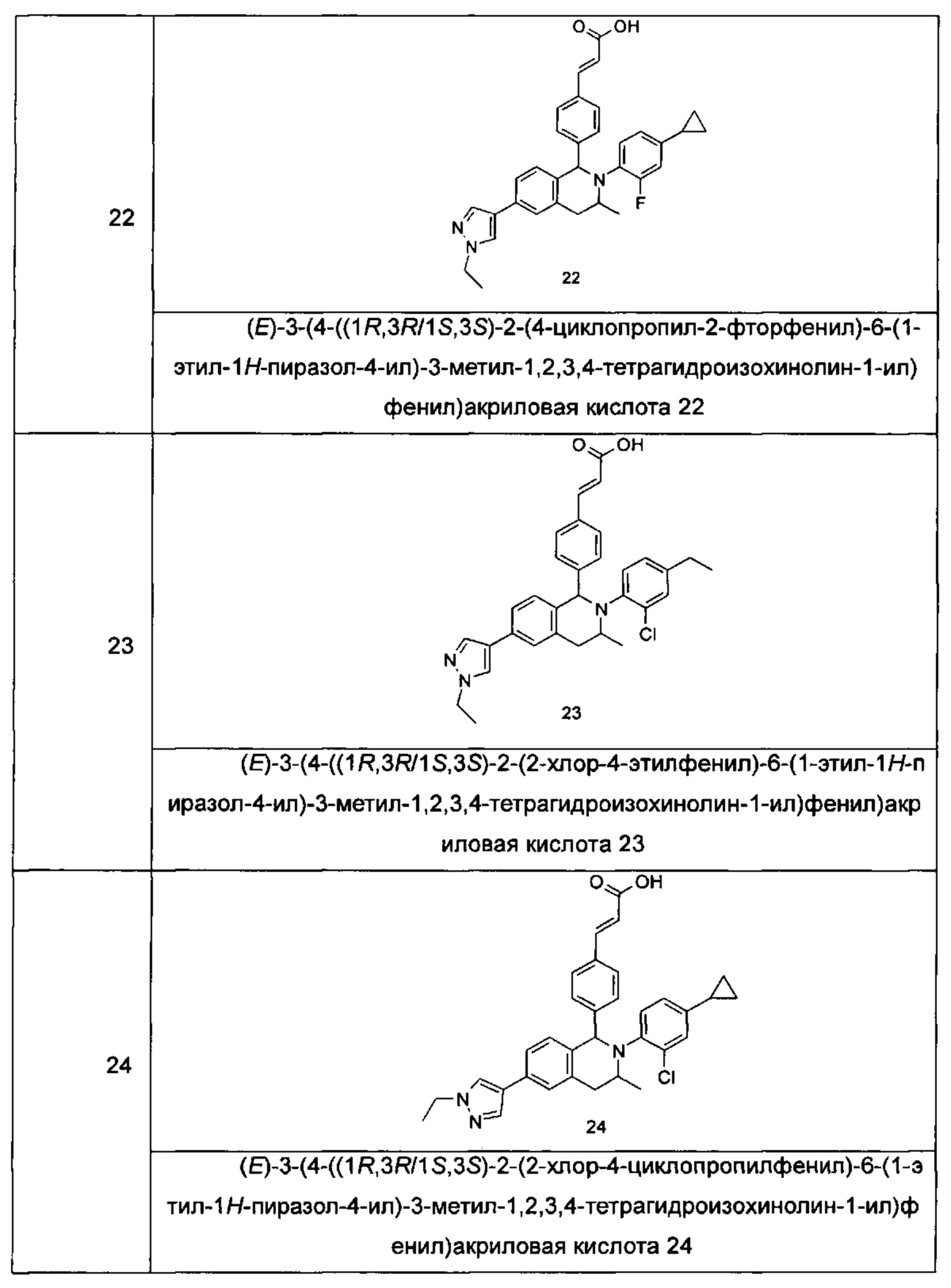
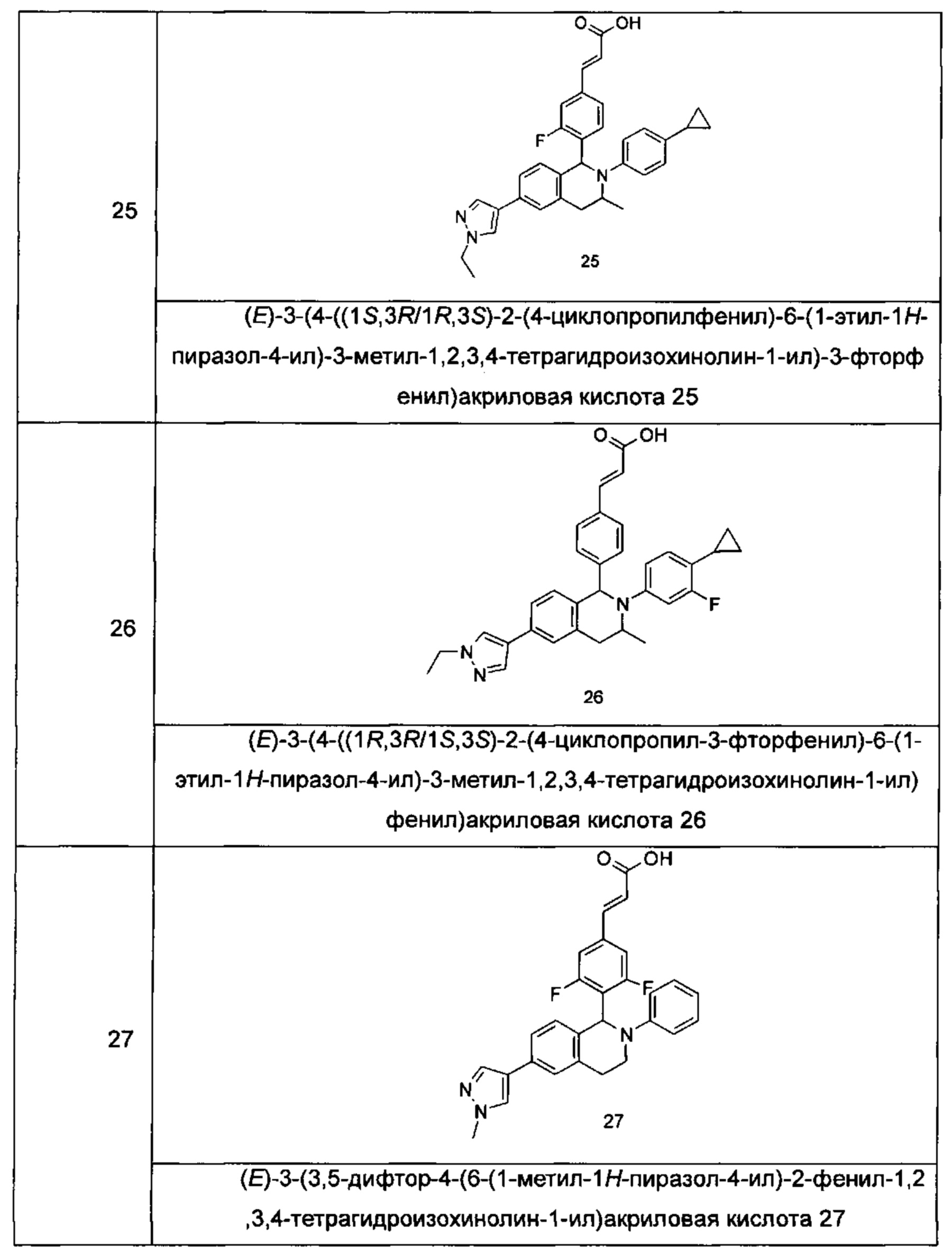
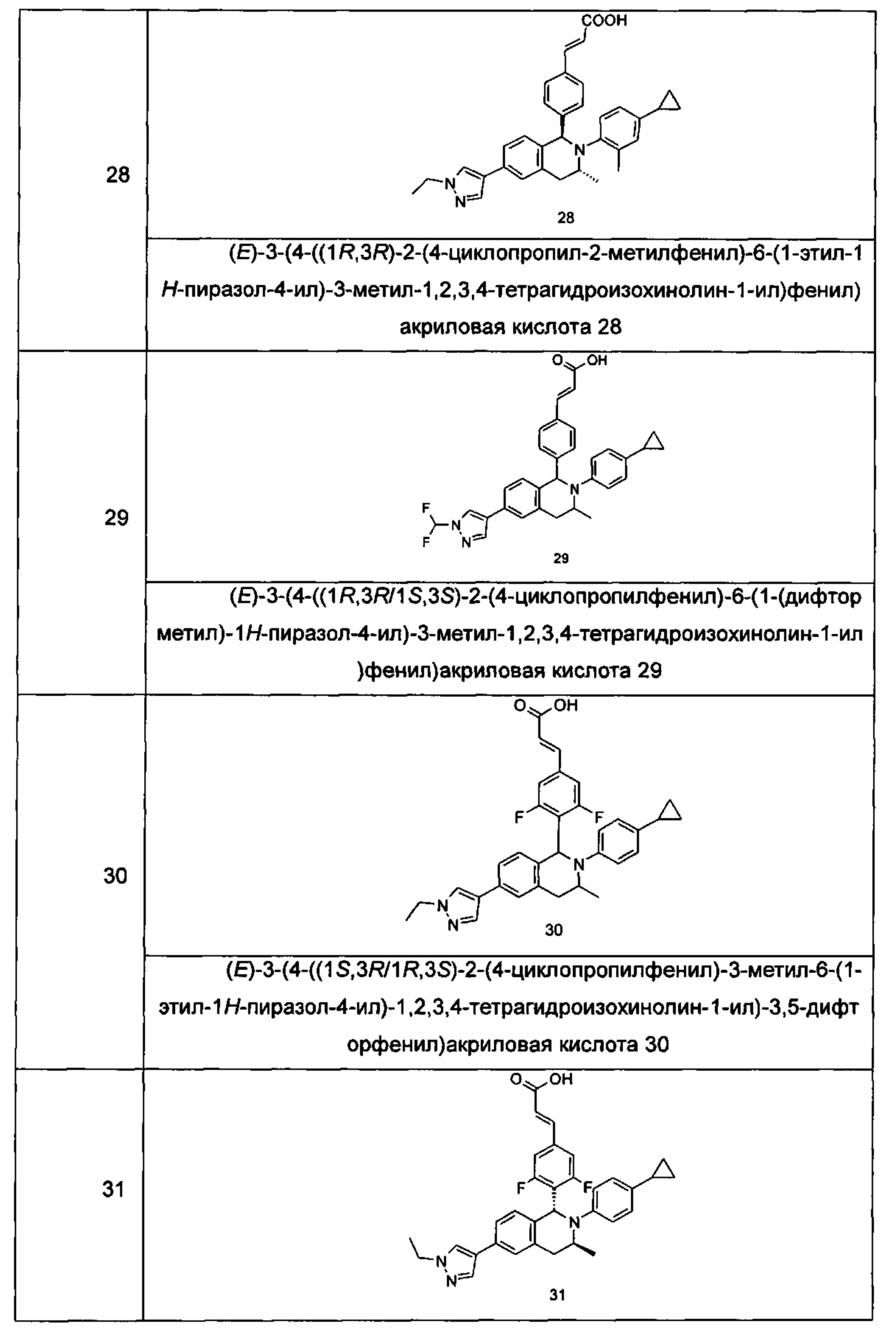
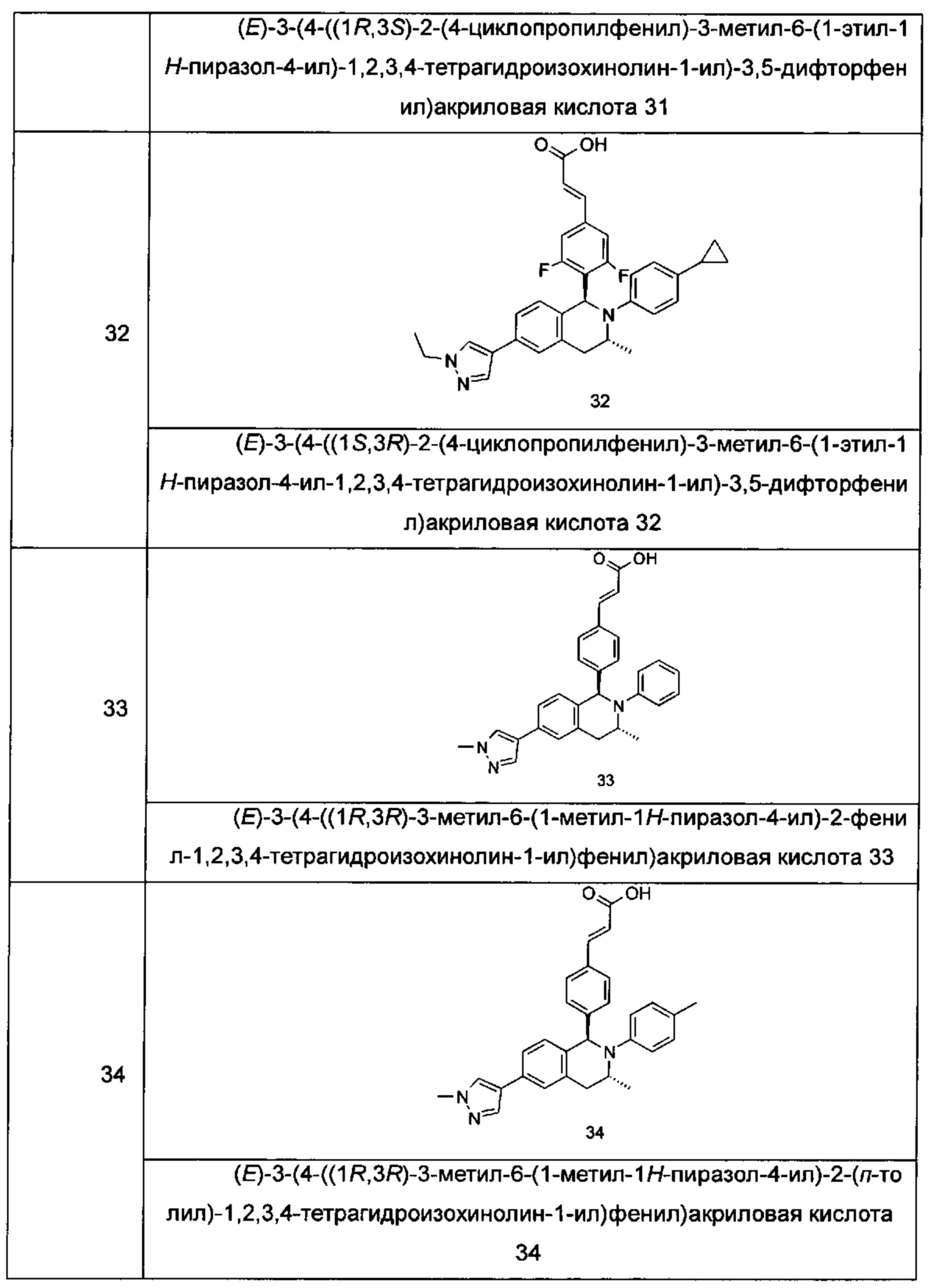
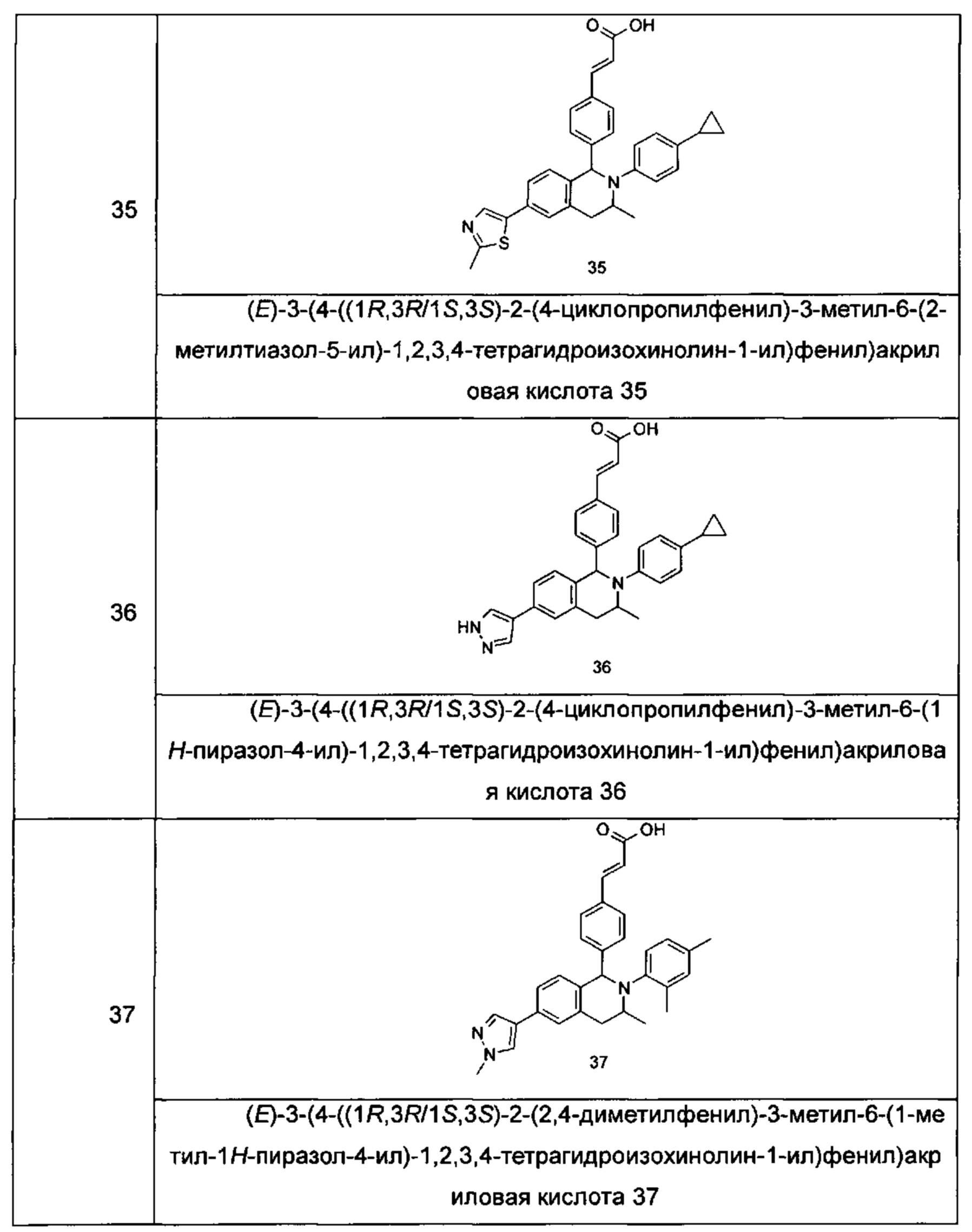
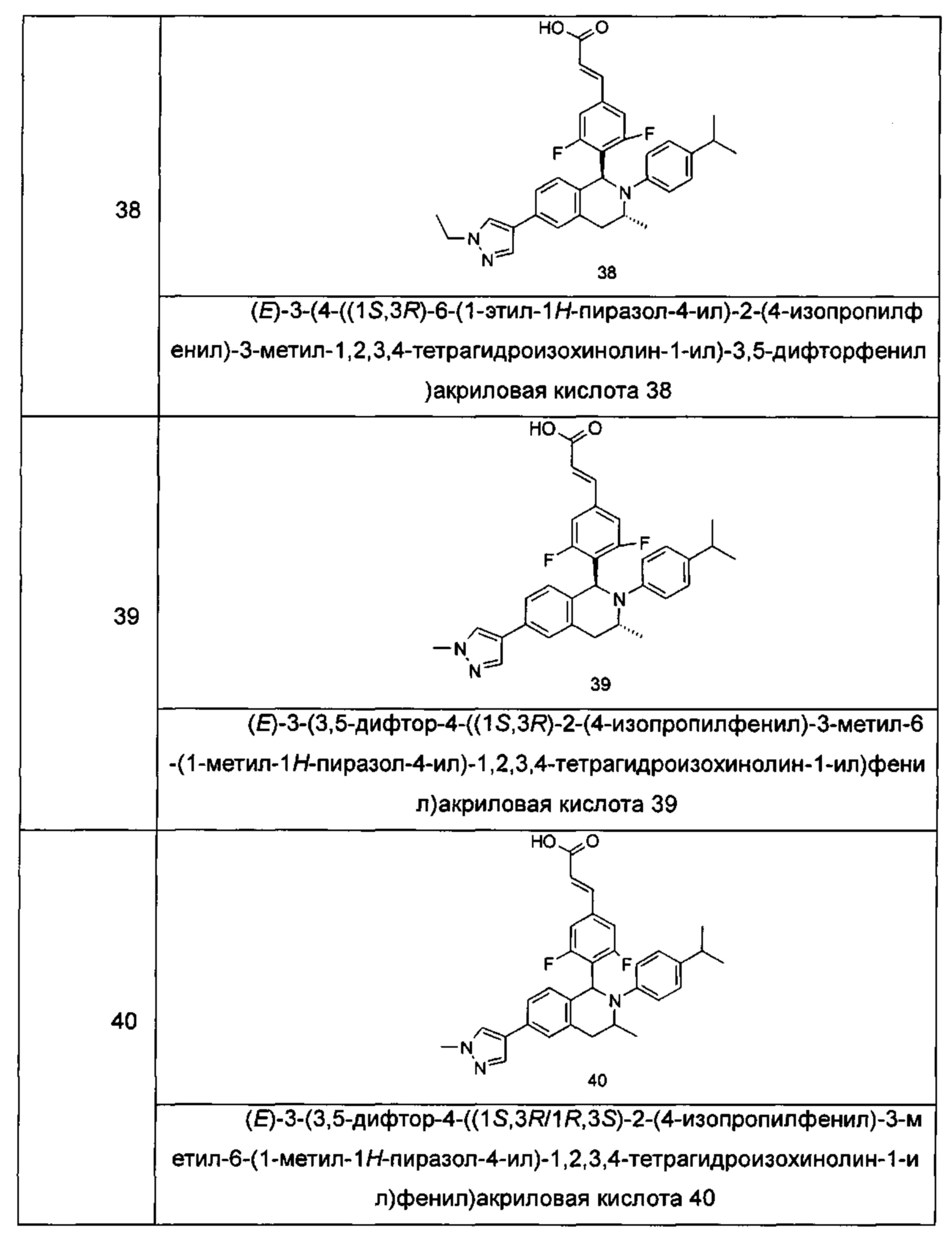
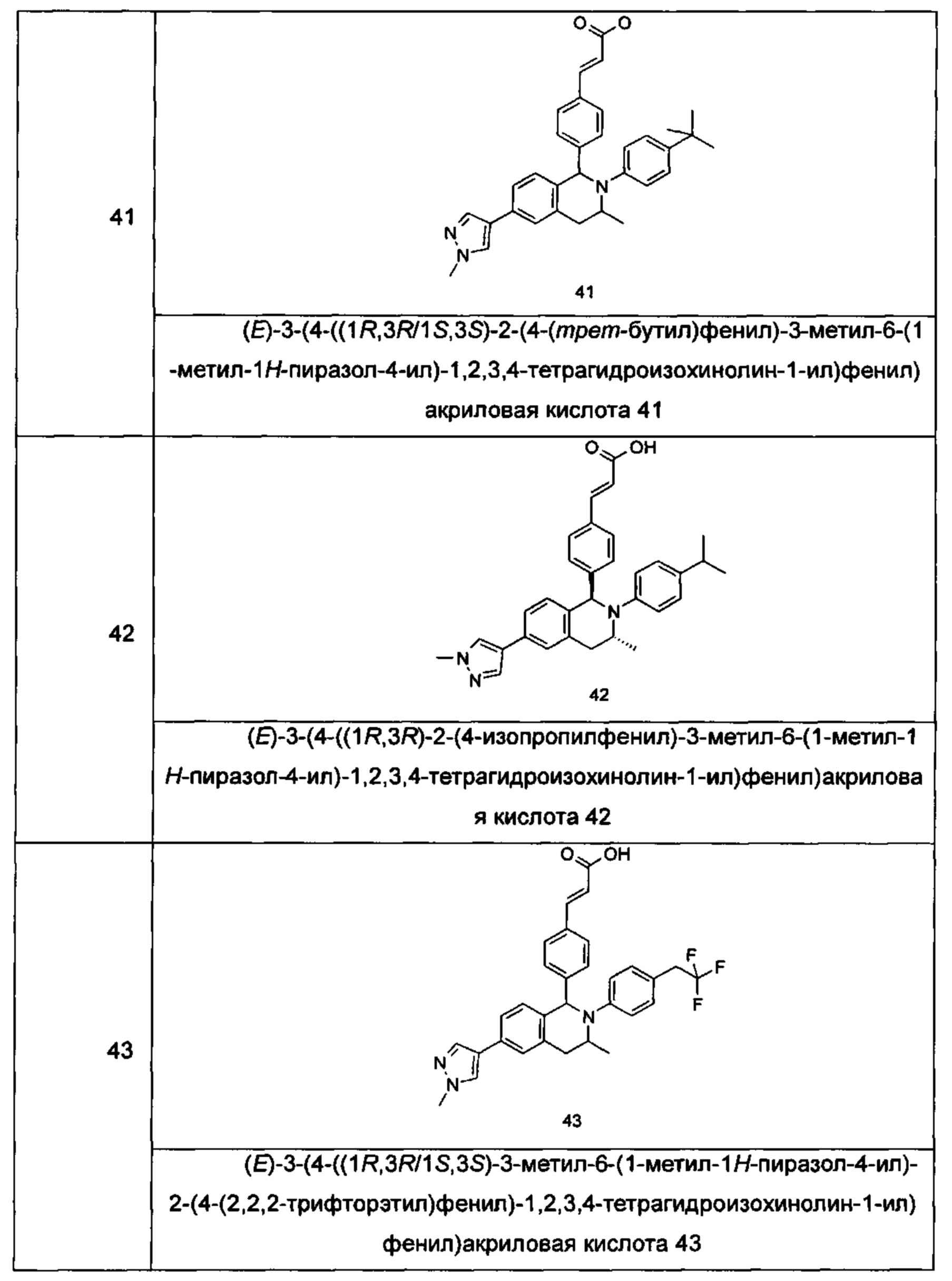
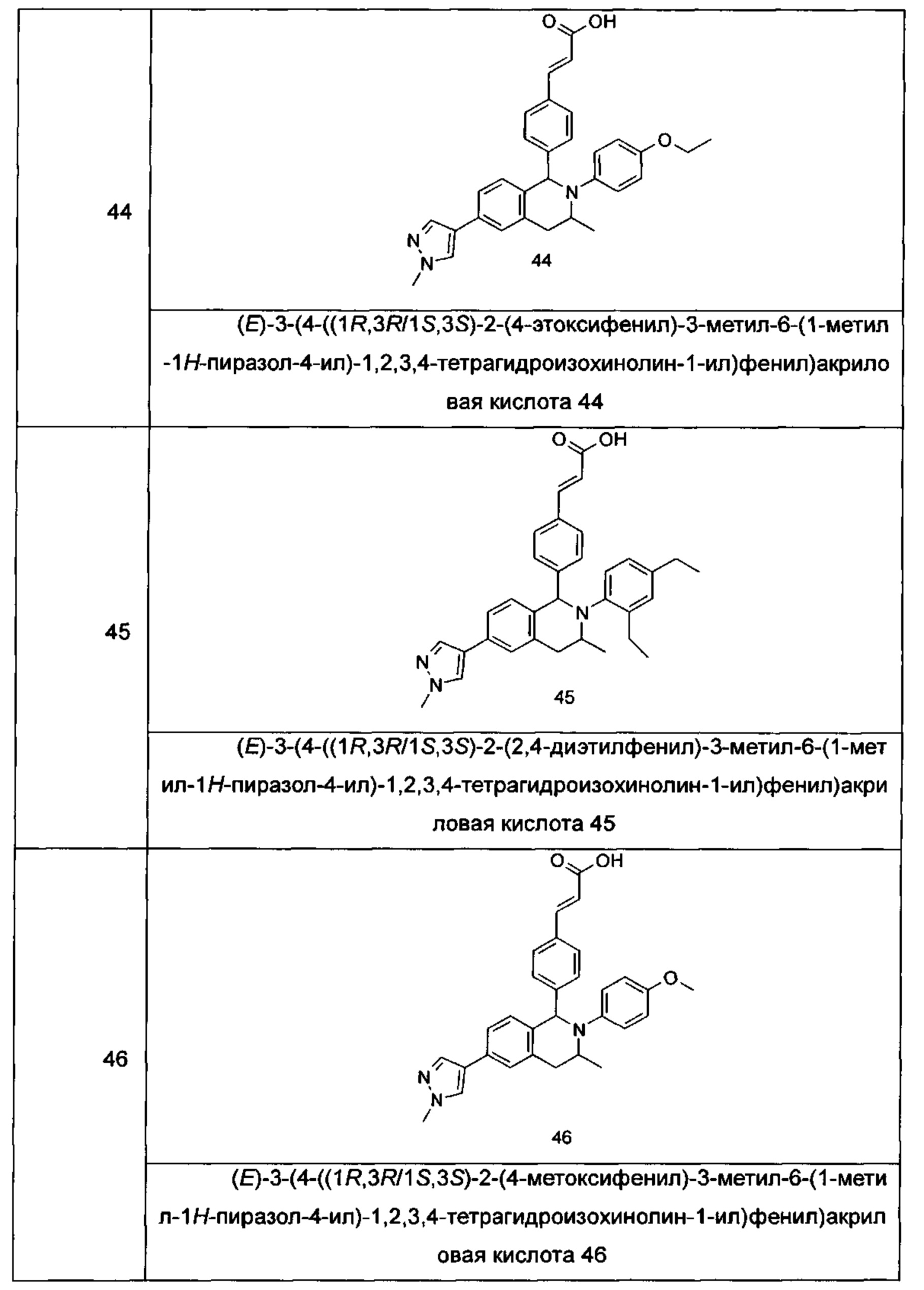
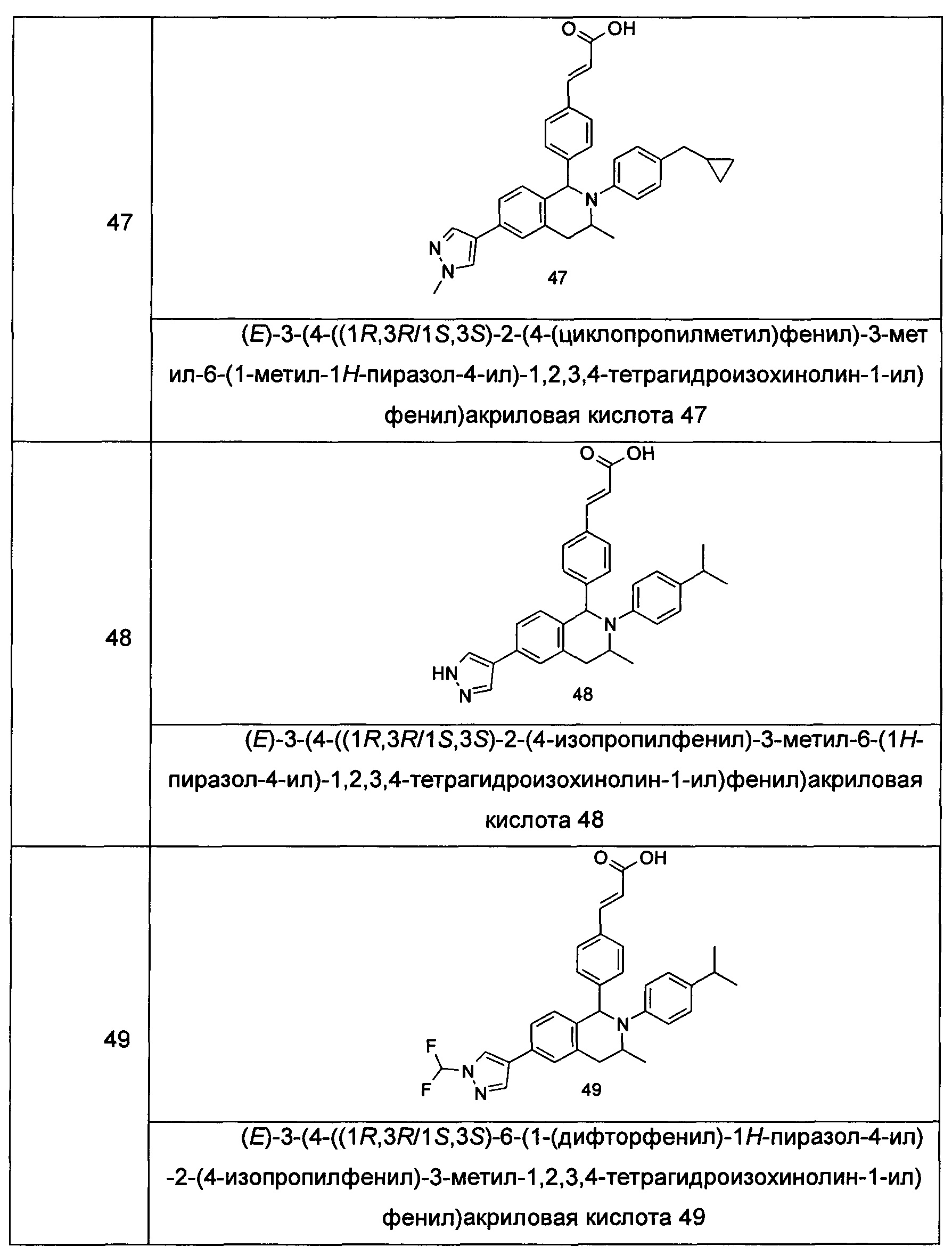
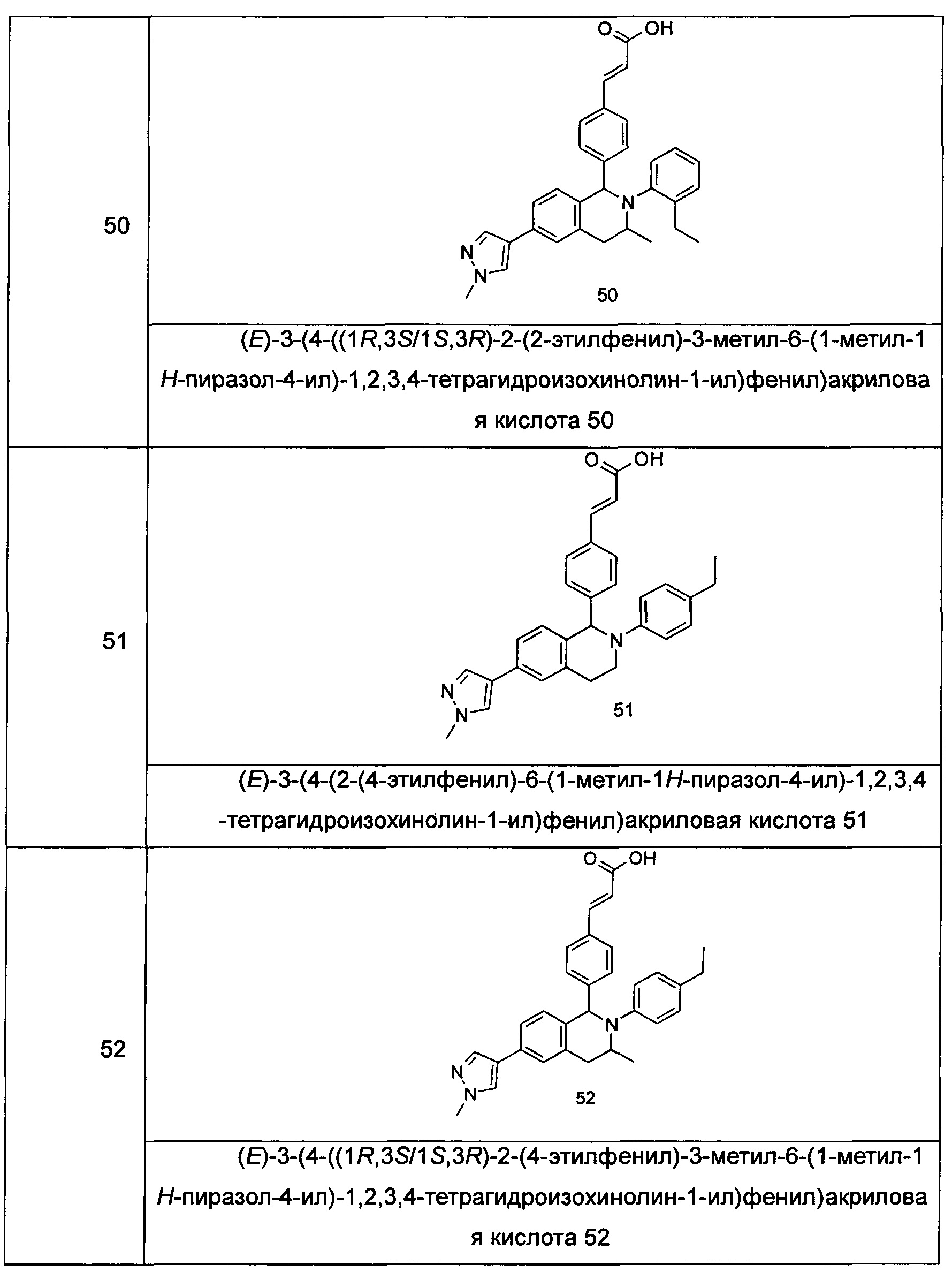
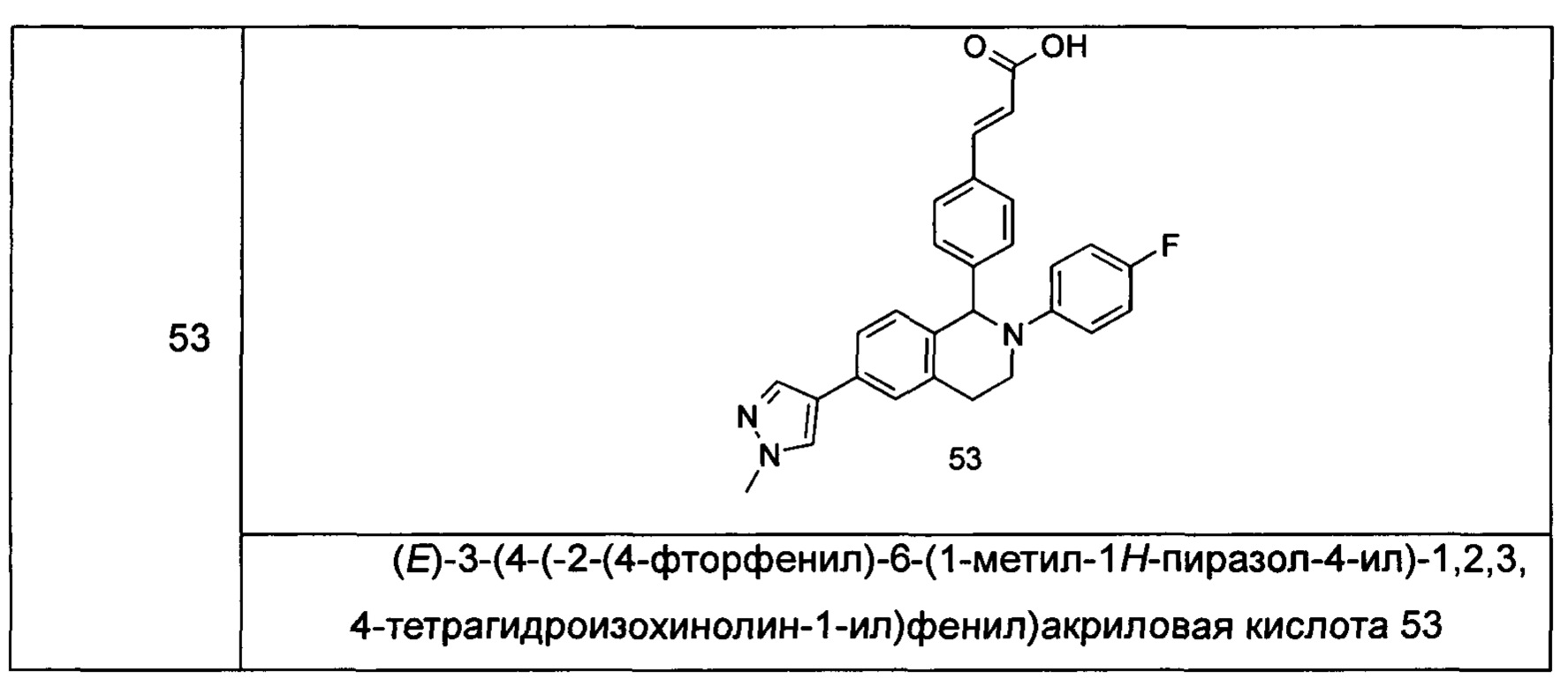
или их таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемую соль.
Согласно настоящему изобретению дополнительно предложено соединение формулы (IV):
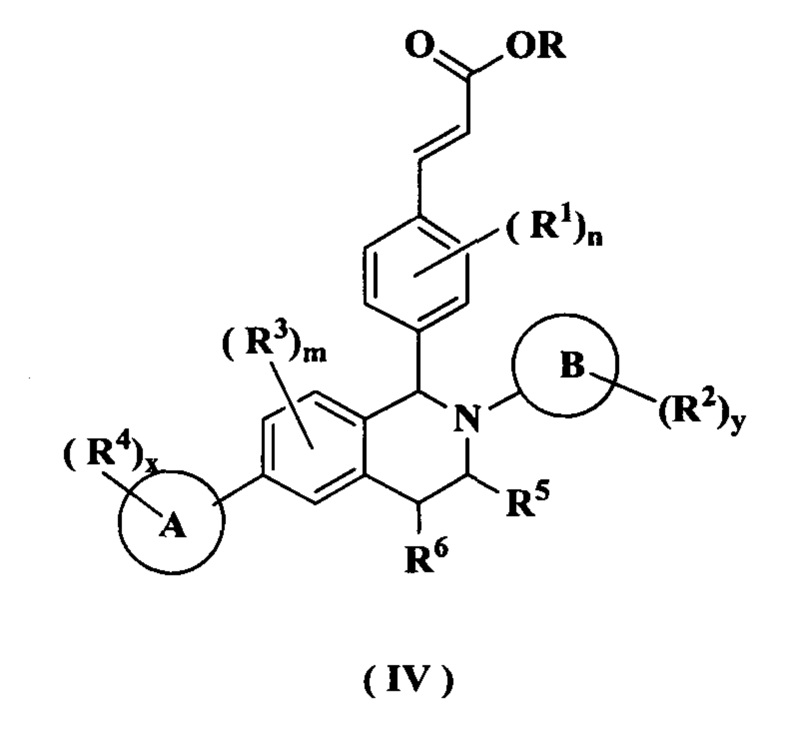
или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемая соль,
где:
R представляет собой алкил или циклоалкил, где каждый алкил и циклоалкил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, амино, циано, гидрокси, алкокси, карбокси и циклоалкила; и
кольцо А, кольцо В, от R1 до R6, m, n, x и у являются такими, как определено в формуле (I).
Другой аспект данного изобретения направлен на способ получения соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли, включающий стадию:
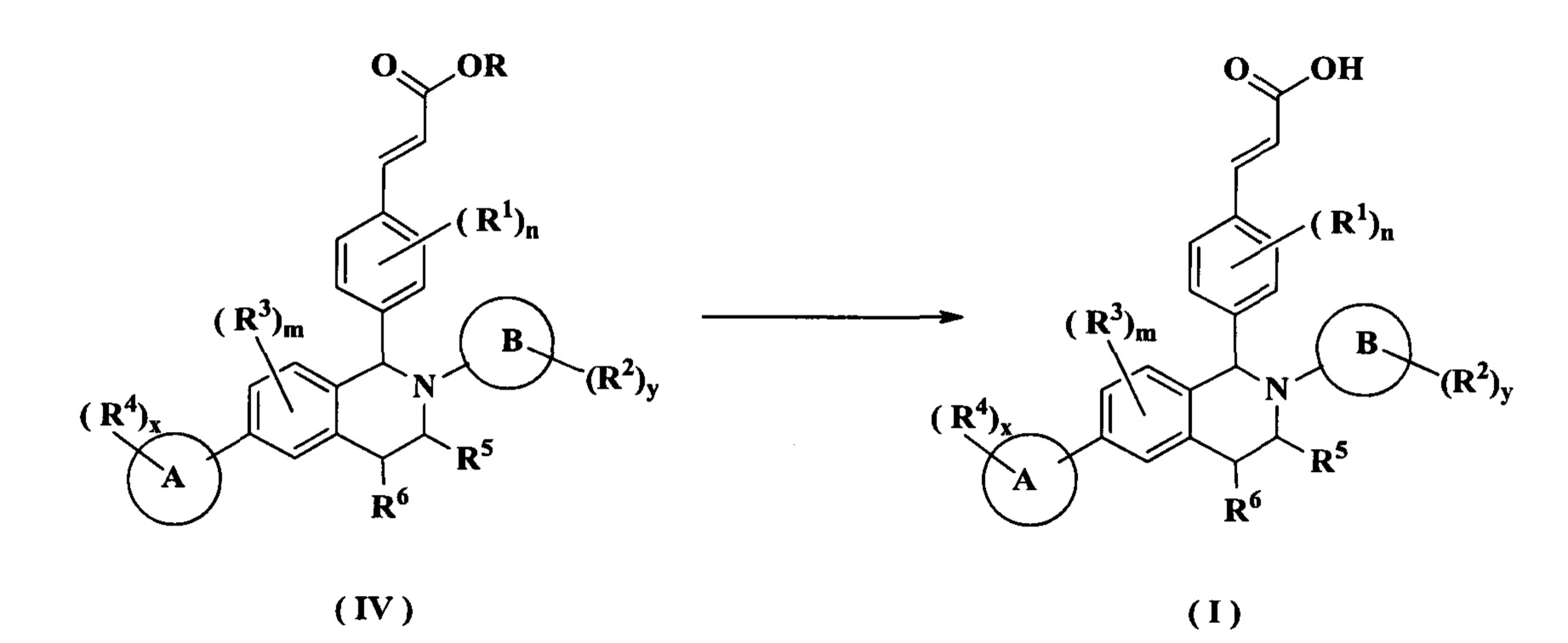
осуществления гидролиза соединения формулы (IV) в щелочных условиях с получением соединения формулы (I);
где:
R представляет собой алкил или циклоалкил, где каждый алкил и циклоалкил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, амино, циано, гидрокси, алкокси, карбокси и циклоалкила; и
кольцо А, кольцо В, от R1 до R6, m, n, x и у являются такими, как определено в формуле (I).
Другой аспект данного изобретения направлен на фармацевтическую композицию, содержащую терапевтически эффективное количество соединения каждой из указанных выше формул или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли и один или более фармацевтически приемлемых носителей, разбавителей или вспомогательных веществ. Настоящее изобретение дополнительно направлено на способ получения указанной выше фармацевтической композиции, включающий стадию смешивания соединения каждой из указанных выше формул или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли с одним или более фармацевтически приемлемыми носителями, разбавителями или вспомогательными веществами.
Настоящее изобретение дополнительно направлено на применение соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или их фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции в получении модулятора рецептора эстрогена.
Настоящее изобретение дополнительно направлено на применение соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или их фармацевтически приемлемой соли, либо содержащей их фармацевтической композиции в получении лекарственного средства для профилактики и/или лечения опосредованного или зависимого от рецептора эстрогена заболевания или состояния, где опосредованное или зависимое от рецептора эстрогена заболевание или состояние выбрано из группы, состоящей из рака, нарушений со стороны центральной нервной системы (ЦНС), нарушений со стороны сердечно-сосудистой системы, нарушений со стороны гематологической системы, иммунных и воспалительных заболеваний, восприимчивости к инфекции, нарушений метаболизма, неврологических нарушений, нарушений психики и нарушений со стороны репродуктивной системы;
где рак может представлять собой рак молочной железы, рак эндометрия, рак шейки матки, рак кожи, рак предстательной железы, рак яичника, опухоль фаллопиевой трубы, оофорому, гемофилию, лейкоз или лейомиоматоз (например, лейомиомы матки); предпочтительно рак молочной железы, рак яичника, рак эндометрия, рак предстательной железы или рак матки; более предпочтительно рак молочной железы; где нарушения со стороны центральной нервной системы (ЦНС) могут представлять собой алкоголизм или мигрень; где нарушения со стороны сердечно-сосудистой системы могут представлять собой аневризму аорты, подверженность инфаркту миокарда, склероз аортального клапана, сердечно-сосудистое заболевание, болезнь коронарных артерий или гипертензию;
где иммунные и воспалительные заболевания могут представлять собой болезнь Грейвса, артрит, рассеянный склероз или цирроз; где восприимчивость к инфекции может представлять собой гепатит В или хроническое заболевание печени; где нарушения метаболизма могут представлять собой холестаз, гипоспадии, ожирение, остеоартрит, остеопению или остеопороз; где неврологические нарушения могут представлять собой болезнь Альцгеймера, болезнь Паркинсона, мигрень или головокружение; где нарушения психики могут представлять собой нервную анорексию, синдром дефицита внимания и гиперактивности (СДВГ), деменцию, тяжелое депрессивное расстройство или психоз; и где нарушения со стороны репродуктивной системы могут представлять собой возраст менархе, эндометриоз и бесплодие и т.п.
Настоящее изобретение дополнительно направлено на соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или их фармацевтически приемлемую соль для применения в качестве лекарственного средства.
Настоящее изобретение дополнительно направлено на соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или их фармацевтически приемлемую соль для применения в качестве лекарственного средства для лечения опосредованного или зависимого от рецептора эстрогена заболевания или состояния, где опосредованное или зависимое от рецептора эстрогена заболевание или состояние является таким, как определено выше.
Настоящее изобретение дополнительно направлено на способ лечения заболевания или состояния, опосредованного рецептором эстрогена или зависимого от рецептора эстрогена, включающий стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или их фармацевтически приемлемой соли. Этот способ показывает исключительную эффективность и меньшие побочные эффекты. Опосредованное или зависимое от рецептора эстрогена заболевание или состояние является таким, как определено выше.
В другом аспекте настоящее изобретение направлено на применение соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, или их фармацевтически приемлемой соли в получении лекарственного средства для профилактики и/или лечения рака. Рак является таким, как определено выше.
В другом аспекте настоящее изобретение направлено на соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, или их фармацевтически приемлемую соль для применения в качестве лекарственного средства для лечения рака. Этот способ показывает исключительную эффективность и меньшие побочные эффекты в лечении рака. Рак является таким, как определено выше.
В другом аспекте настоящее изобретение направлено на способ лечения рака, включающий стадию введения пациенту, нуждающемуся в этом, терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси или их фармацевтически приемлемой соли. Этот способ показывает исключительную эффективность и меньшие побочные эффекты. Рак является таким, как определено выше.
В другом аспекте настоящее изобретение направлено на соединение формулы (I) или его таутомер, мезомер, рацемат, энантиомер, диастереомер или их смесь, либо их фармацевтически приемлемую соль для применения в качестве лекарственного средства для лечения рака кости, рака молочной железы, колоректального рака, рака эндометрия, рака предстательной железы, рака яичника, рака матки, рака шейки матки, рака легкого, лейомиоматоза, лейомиом матки, алкоголизма, мигрени, аневризмы аорты, подверженности инфаркту миокарда, склероза клапанов аорты, сердечно-сосудистого заболевания, ишемической болезни сердца, гипертензии, тромбоза глубоких вен, болезни Грейвса, артрита, рассеянного склероза, цирроза, гепатита В, хронического заболевания печени, холестаза, гипоспадий, ожирения, остеоартрита, остеопороза, болезни Альцгеймера, болезни Паркинсона, мигрени, головокружения, нервной анорексии, синдрома дефицита внимания и гиперактивности (СДВГ), деменции, тяжелого депрессивного расстройства, психоза, возраста менархе, эндометриоза или бесплодия у млекопитающих.
В другом аспекте настоящее изобретение направлено на способ лечения рака кости, рака молочной железы, колоректального рака, рака эндометрия, рака предстательной железы, рака яичника, рака матки, рака шейки матки, рака легкого, лейомиоматоза, лейомиом матки, алкоголизма, мигрени, аневризмы аорты, подверженности инфаркту миокарда, склероза клапанов аорты, сердечно-сосудистого заболевания, ишемической болезни сердца, гипертензии, тромбоза глубоких вен, болезни Грейвса, артрита, рассеянного склероза, цирроза, гепатита В, хронического заболевания печени, холестаза, гипоспадий, ожирения, остеоартрита, остеопороза, болезни Альцгеймера, болезни Паркинсона, мигрени, головокружения, нервной анорексии, синдрома дефицита внимания и гиперактивности (СДВГ), деменции, тяжелого депрессивного расстройства, психоза, возраста менархе, эндометриоза или бесплодия у млекопитающих, включающий стадию введения нуждающемуся в этом пациенту терапевтически эффективного количества соединения формулы (I) или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли.
Содержащие активный ингредиент фармацевтические композиции могут быть представлены в форме, приемлемой для перорального введения, например, таблетки, пастилки, леденца, водной или масляной суспензии, диспергируемого порошка или гранулы, эмульсии, твердой или мягкой капсулы, либо сиропа или эликсира. Пероральные композиции можно получать в соответствии с любым известным в данной области техники способом получения фармацевтических композиций. Такие композиции могут содержать одну или более добавок, выбранных из группы, состоящей из подсластителей, корригентов, красителей и консервантов, с целью получения привлекательного и приятного на вкус фармацевтического препарата. Таблетки содержат активный ингредиент и нетоксичные фармацевтически приемлемые вспомогательные вещества, пригодные для производства таблеток. Эти вспомогательные вещества могут представлять собой инертные вспомогательные вещества, гранулирующие агенты, разрыхлители и смазывающие вещества. Таблетка может не иметь покрытия или быть покрытой оболочкой посредством известной методики, чтобы замаскировать вкус лекарственного средства или отсрочить распад и всасывание лекарственного средства в желудочно-кишечном тракте, обеспечивая, таким образом, замедленное высвобождение в течение продолжительного периода времени. Например, можно использовать водорастворимые вещества, маскирующие вкус.
Пероральные композиции могут быть также представлены в виде мягких желатиновых капсул, в которых активный ингредиент смешан с инертным твердым разбавителем, либо активный ингредиент смешан с водорастворимым носителем.
Водная суспензия содержит активный ингредиент в смеси с вспомогательными веществами, пригодными для производства водной суспензии. Такие вспомогательные вещества представляют собой суспендирующие агенты, диспергирующие агенты или увлажнители и могут представлять собой природные фосфолипиды. Водная суспензия может также содержать один или более консервантов, один или более красителей, один или более корригентов и один или более подсластителей.
Масляная суспензия может быть приготовлена путем суспендирования активного ингредиента в растительном масле или в минеральном масле. Масляная суспензия может содержать загуститель. Для обеспечения препарата приятного вкуса можно добавлять указанные выше подсластители и корригенты. Эти композиции можно сохранять путем добавления антиоксиданта.
Активный ингредиент и диспергирующие агенты или смачивающие вещества, суспендирующий агент или один или более консервантов можно получать в виде диспергируемого порошка или гранулы, пригодных для получения водной суспензии путем добавления воды. Примерами пригодных диспергирующих агентов или смачивающих веществ и суспендирующих агентов являются диспергирущие агенты и смачивающие вещества, уже приведенные выше. Могут быть также добавлены дополнительные вспомогательные вещества, такие как подсластители, корригенты и красители. Эти композиции можно сохранять путем добавления антиоксиданта, такого как аскорбиновая кислота.
Настоящая фармацевтическая композиция может быть также представлена в форме эмульсии типа масло-в-воде. Масляная фаза может представлять собой растительное масло, либо минеральное масло, либо их смесь. Пригодные эмульгирующие агенты могут представлять собой природные фосфолипиды. Можно использовать подсластители. Такие композиции могут также содержать замедлители, консерванты, красители и антиоксиданты.
Фармацевтическая композиция может быть представлена в форме стерильного инъекционного водного раствора. Приемлемыми носителями и растворителями, которые можно применять, являются вода, раствор Рингера и изотонический раствор хлорида натрия. Стерильный инъекционный препарат может также представлять собой стерильную, инъекционную микроэмульсию типа масло-в-воде, в которой активный ингредиент растворен в масляной фазе. Инъекционный раствор или микроэмульсию можно вводить в кровоток индивида путем локальной болюсной инъекции. В качестве альтернативы преимуществом может обладать введение раствора или микроэмульсии таким путем, чтобы поддерживать постоянную концентрацию циркулирующего настоящего соединения. Для поддержания такой постоянной концентрации можно использовать устройство для непрерывной внутривенной доставки. Примером такого устройства является насос для внутривенных инъекций Deltec CADD-PLUS. ТМ. 5400.
Фармацевтическая композиция может быть представлена в форме стерильной инъекционной водной или масляной суспензии для внутримышечного и подкожного введения. Такая суспензия может быть приготовлена с пригодными диспергирующими агентами или смачивающими веществами и суспендирующими агентами, как описано выше, в соответствии с известными методиками. Стерильный инъекционный препарат может также представлять собой стерильный инъекционный раствор или суспензию, приготовленную в нетоксичном разбавителе или растворителе, приемлемом для парентерального введения. Кроме того, в качестве растворителя или суспензионной среды можно легко использовать стерильные нелетучие масла, а для получения инъекций можно также использовать жирные кислоты.
Настоящее соединение можно вводить в форме суппозитория для ректального введения. Эти фармацевтические композиции можно получать путем смешивания лекарственного средства с пригодным нераздражающим вспомогательным веществом, который является твердым при обычных температурах, но жидким в прямой кишке, таким образом, плавясь в прямой кишке с высвобождением лекарственного средства.
Специалистам в данной области техники хорошо известно, что дозировка лекарственного средства зависит от целого ряда факторов, включая следующие факторы: активность конкретного соединения, возраст, массу, общее состояние здоровья, поведение, рацион пациента, время введения, путь введения, скорость экскреции, комбинация лекарственных средств и т.п., но, не ограничиваясь ими. Кроме того, лучший вариант лечения, как например метод лечения, суточная доза соединения формулы (I) или тип его фармацевтически приемлемой соли, может быть подтвержден на основании традиционных схем лечения.
Подробное описание изобретения
Если не указано иное, термины, используемые в описании и формуле изобретения, имеют описанные ниже значения.
«Алкил» относится к насыщенной алифатической углеводородной группе, включающей C1-C20 линейные и разветвленные группы, предпочтительно алкил, имеющий от 1 до 12 атомов углерода. Неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил, н-гептил, 2-метилгексил, 3-метилгексил, 4-метилгексил, 5-метилгексил, 2,3-диметилпентил, 2,4-диметилпентил, 2,2-диметилпентил, 3,3-диметилпентил, 2-этилпентил, 3-этилпентил, н-октил, 2,3-диметилгексил, 2,4-диметилгексил, 2,5-диметилгексил, 2,2-диметилгексил, 3,3-диметилгексил, 4,4-диметилгексил, 2-этилгексил, 3-этилгексил, 4-этилгексил, 2-метил-2-этилпентил, 2-метил-3-этилпентил, н-нонил, 2-метил-2-этилгексил, 2-метил-3-этилгексил, 2,2-диэтилпентил, н-децил, 3,3-диэтилгексил, 2,2-диэтилгексили и их разветвленные изомеры. Более предпочтительно алкильная группа представляет собой низший алкил, имеющий от 1 до 6 атомов углерода, и неограничивающие примеры включают метил, этил, н-пропил, изопропил, н-бутил, изобутил, трет-бутил, втор-бутил, н-пентил, 1,1-диметилпропил, 1,2-диметилпропил, 2,2-диметилпропил, 1-этилпропил, 2-метилбутил, 3-метилбутил, н-гексил, 1-этил-2-метилпропил, 1,1,2-триметилпропил, 1,1-диметилбутил, 1,2-диметилбутил, 2,2-диметилбутил, 1,3-диметилбутил, 2-этилбутил, 2-метилпентил, 3-метилпентил, 4-метилпентил, 2,3-диметилбутил и т.п. Алкильная группа может быть замещенной или незамещенной. При замещении замещающая(ие) группа(-ы) может(-гут) быть замещена(-ы) в любой доступной точке соединения. Замещающая(ие) группа(-ы) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Циклоалкил» относится к насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей от 3 до 20 атомов углерода, предпочтительно от 3 до 12 атомов углерода и более предпочтительно от 3 до 6 атомов углерода. Неограничивающие примеры моноциклического циклоалкила включают циклопропил, циклобутил, циклопентил, циклопентенил, циклогексил, цикпогексенил, циклогексадиенил, циклогептил, циклогептатриенил, циклооктил и т.п. Полициклический циклоалкил включает циклоалкил, имеющий спирокольцо, конденсированное кольцо или мостиковое кольцо.
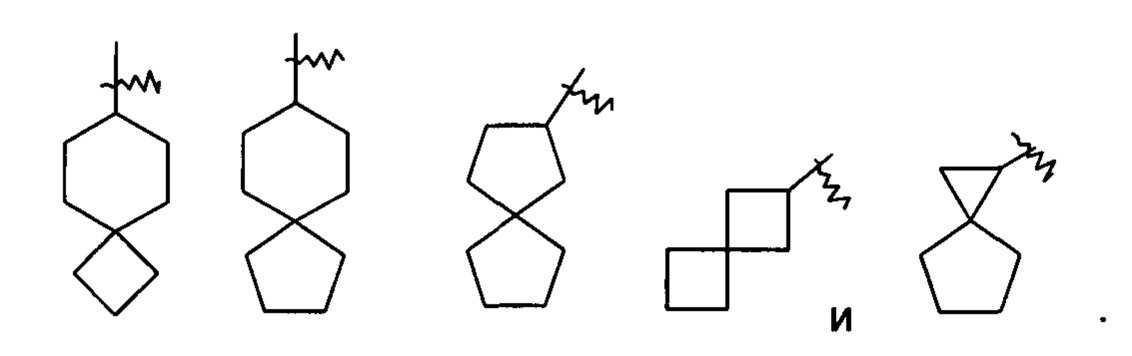
«Спироциклоалкил» относится к 5-20-членной полициклической группе с кольцами, соединенными посредством одного общего атома углерода (называемого спироатомом), где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно 6-14-членному спироциклоалкилу и более предпочтительно 7-10-членному спироциклоалкилу. В соответствии с числом спироатомов, общих у колец, спироциклоалкил можно подразделить на моноспироциклоалкил, диспироциклоалкил или полиспироциклоалкил, предпочтительно моноспироциклоалкил или диспироциклоалкил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспироциклоалкил. Неограничивающие примеры спироциклоалкилов включают:
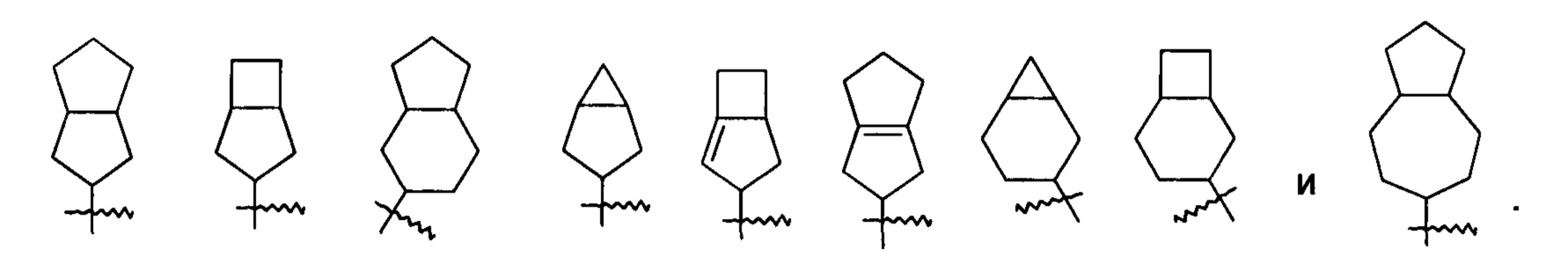
«Конденсированный циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, в которой каждое кольцо в системе имеет общую пару примыкающих атомов углерода с другим кольцом, где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно 6-14-членному конденсированному циклоалкилу, более предпочтительно 7-10-членному конденсированному циклоалкилу. В соответствии с числом колец, содержащих определенное число членов конденсированный циклоалкил можно подразделить на бициклический, трициклический, тетрациклический или полициклический конденсированный циклоалкил, предпочтительно бициклический или трициклический конденсированный циклоалкил и более предпочтительно 5-членный/5-членный или 5-членный/б-членный бициклический конденсированный циклоалкил. Неограничивающие примеры конденсированного циклоалкила включают:
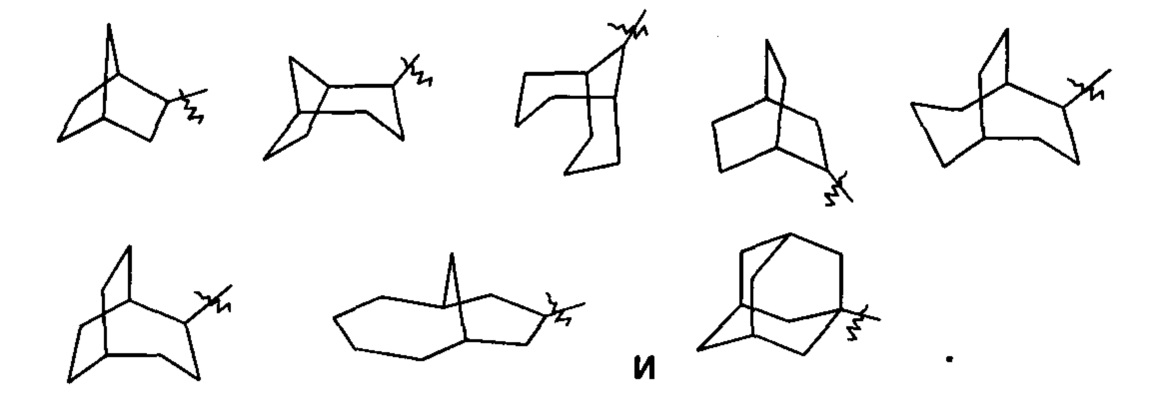
«Мостиковый циклоалкил» относится к 5-20-членной полностью углеродной полициклической группе, в которой каждые два кольца в системе имеют два общих разъединенных атома углерода, где кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно 6-14-членному мостиковому циклоалкилу и более предпочтительно 7-10-членному мостиковому циклоалкилу. В соответствии с числом колец, содержащих определенное число членов, мостиковый циклоалкил можно подразделить на бициклический, трициклический, тетрациклический или полициклический мостиковый циклоалкил, предпочтительно бициклический, трициклический или тетрациклический мостиковый циклоалкил и более предпочтительно бициклический или трициклический мостиковый циклоалкил. Неограничивающие примеры мостиковых циклоалкилов включают:
Циклоалкильное кольцо может быть конденсировано с кольцом арила, гетероарила или гетероциклила, где кольцо, связанное с исходной структурой, представляет собой циклоалкил. Неограничивающие примеры включают инданил, тетрагидронафтил, бензоциклогептил и т.п. Цикпоалкил может быть необязательно замещенным или незамещенным. При замещении замещающая(ие) группа(-ы) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Гетероциклил» относится к 3-20-членной насыщенной и/или частично ненасыщенной моноциклической или полициклической углеводородной группе, имеющей один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве кольцевых атомов, но за исключением -O-O-, -O-S- или -S-S- в кольце, причем остальные кольцевые атомы представляют собой атомы углерода. Предпочтительно гетероциклил имеет от 3 до 12 атомов, где от 1 до 4 атомов представляют собой гетероатомы, и более предпочтительно от 3 до 6 атомов. Неограничивающие примеры моноциклического гетероциклила включают пирролидинил, имидазолидинил, тетрагидрофуранил, тетрагидротиенил, дигидроимидазолил, дигидрофурил, дигидропиразолил, дигидропирролил, пиперидинил, пиперазинил, морфолинил, тиоморфолинил, гомопиперазинил и т.п., и предпочтительно пиперидинил или пирролидинил. Полициклический гетероциклил включает гетероциклил, имеющий спирокольцо, конденсированное кольцо или мостиковое кольцо.
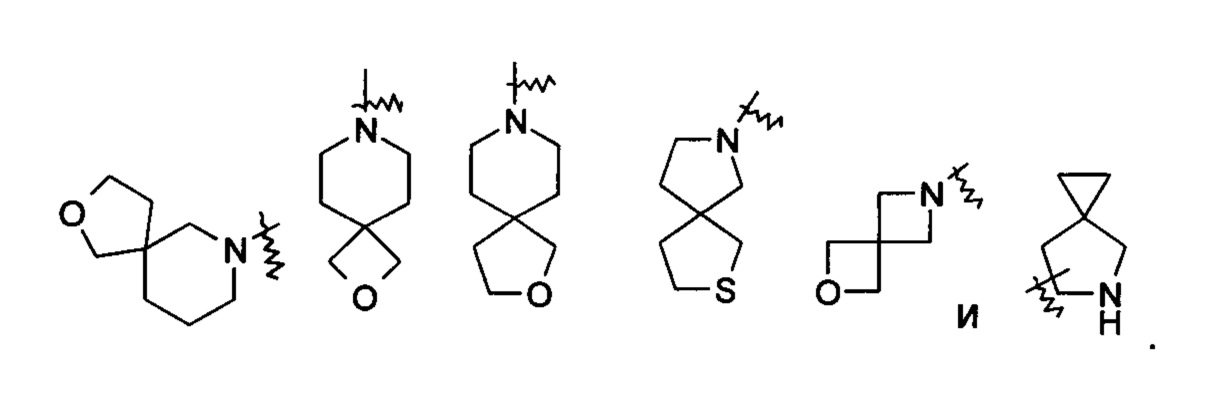
«Спирогетероциклил» относится к 5-20-членному полициклическому гетероциклилу, кольца которого соединены через один общий атом (называемый спироатомом), где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве кольцевых атомов, причем остальные кольцевые атомы представляют собой атомы углерода, где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, предпочтительно 6-14-членному спирогетероциклилу и более предпочтительно 7-10-членному спирогетероциклилу. В соответствии с числом общих у колец спироатомов спирогетероциклил можно подразделить на моноспирогетероциклил, диспирогетероциклил или полиспирогетероциклил, предпочтительно моноспирогетероциклил или диспирогетероциклил и более предпочтительно 4-членный/4-членный, 4-членный/5-членный, 4-членный/6-членный, 5-членный/5-членный или 5-членный/6-членный моноспирогетероциклил. Неограничивающие примеры спирогетероциклилов включают:
«Конденсированный гетероциклил» относится к 5-20-членной полициклической гетероциклильной группе, где каждое кольцо в системе имеет общую пару смежных атомов с другим кольцом, где одно или более колец могут содержать одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и где кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве кольцевых атомов, причем остальные кольцевые атомы представляют собой атомы углерода, предпочтительно 6-14-членному конденсированному гетероциклилу и более предпочтительно 7-10-членному конденсированному гетероциклилу. В соответствии с числом колец, содержащих определенное число членов, конденсированный гетероциклил можно подразделить на бициклический, трициклический, тетрациклический или полициклический конденсированный гетероциклил, предпочтительно бициклический или трициклический конденсированный гетероциклил и более предпочтительно 5-членный/5-членный или 5-членный/6-членный бициклический конденсированный гетероциклил. Неограничивающие примеры конденсированного гетероциклила включают:
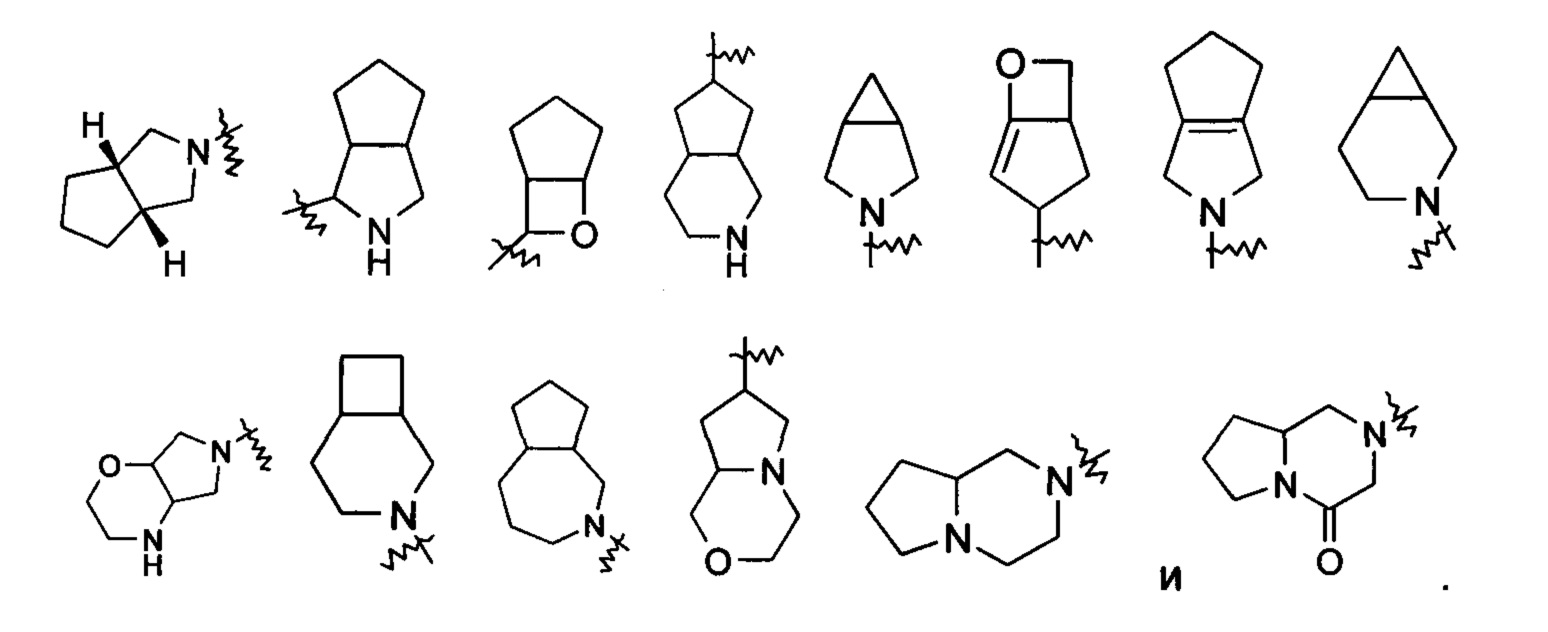
«Мостиковый гетероциклил» относится к 5-14-членной полициклической гетероциклильной группе, где каждые два кольца в системе имеют два общих разъединенных атома, где данные кольца могут иметь одну или более двойных связей, но ни одно из колец не имеет полностью конъюгированной пи-электронной системы, и кольца имеют один или более гетероатомов, выбранных из группы, состоящей из N, О и S(O)m (где m представляет собой целое число от 0 до 2), в качестве кольцевых атомов, причем остальные кольцевые атомы представляют собой атомы углерода, предпочтительно 6-14-членному мостиковому гетероциклилу и более предпочтительно 7-10-членному мостиковому гетероциклилу. В соответствии с числом колец, содержащих определенное число членов, мостиковый гетероциклил можно подразделить на бициклический, трициклический, тетрациклический или полициклический мостиковый гетероциклил, и предпочтительно бициклический, трициклический или тетрациклический мостиковый гетероциклил и более предпочтительно бициклический или трициклический мостиковый гетероциклил. Неограничивающие примеры мостиковых гетероциклилов включают:
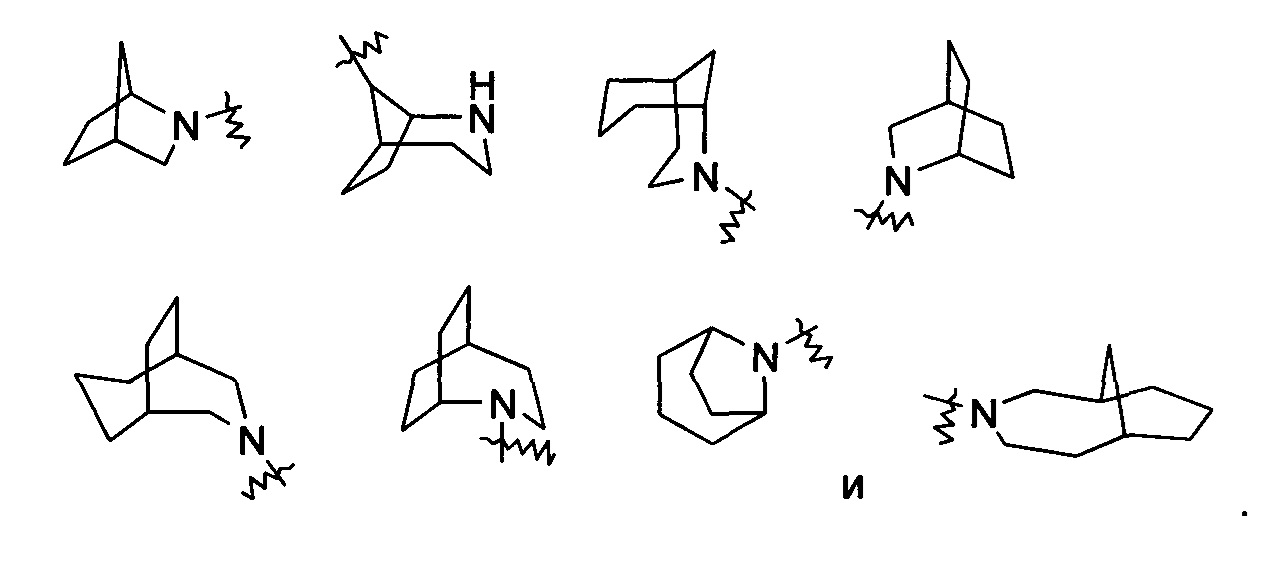
Гетероциклильное кольцо может быть конденсировано с кольцом арила, гетероарила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероциклил. Неограничивающие примеры включают:
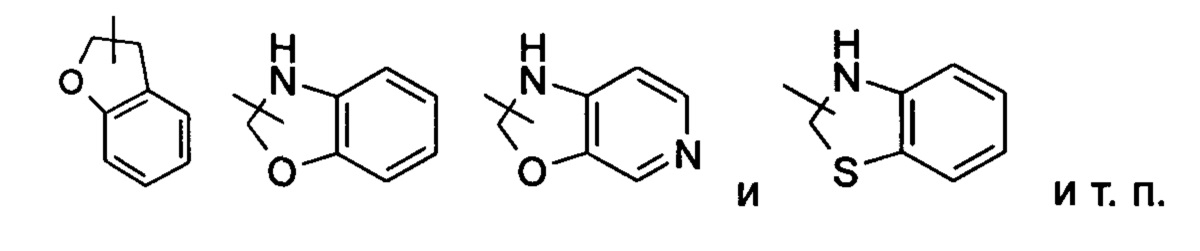
Гетероциклил может быть необязательно замещенным или незамещенным. При замещении замещающая(ие) группа(-ы) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, оксо, карбокси и алкоксикарбонила.
«Арил» относится к 6-14-членному полностью углеродному моноциклическому кольцу или полициклическому конденсированному кольцу (т.е. каждое кольцо в системе имеет общую пару смежных атомов углерода с другим кольцом в системе), имеющему полностью конъюгированную пи-электронную систему, предпочтительно 6-10-членному арилу, например фенилу и нафтилу. Арильное кольцо может быть конденсировано с кольцом гетероарила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой арильное кольцо. Неограничивающие примеры включают:
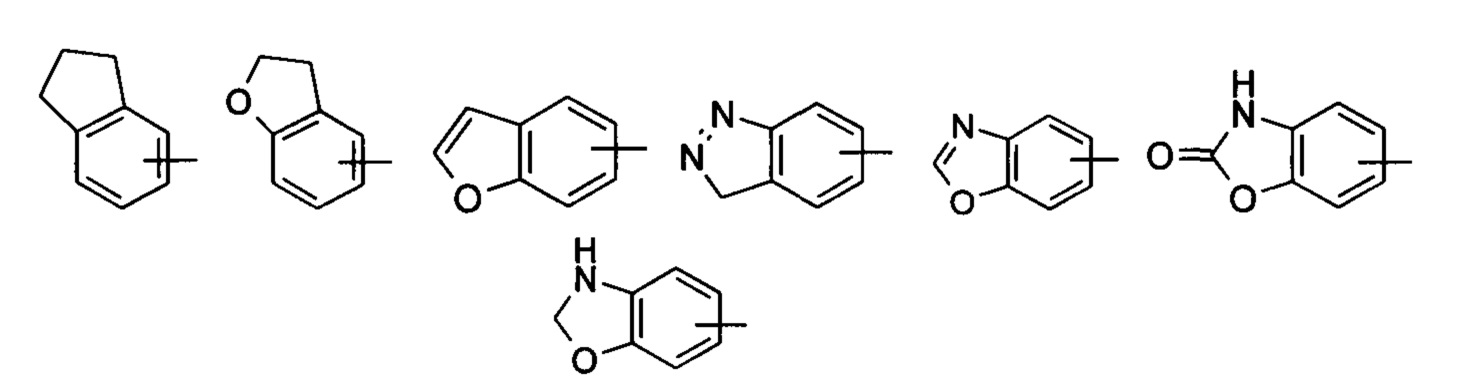
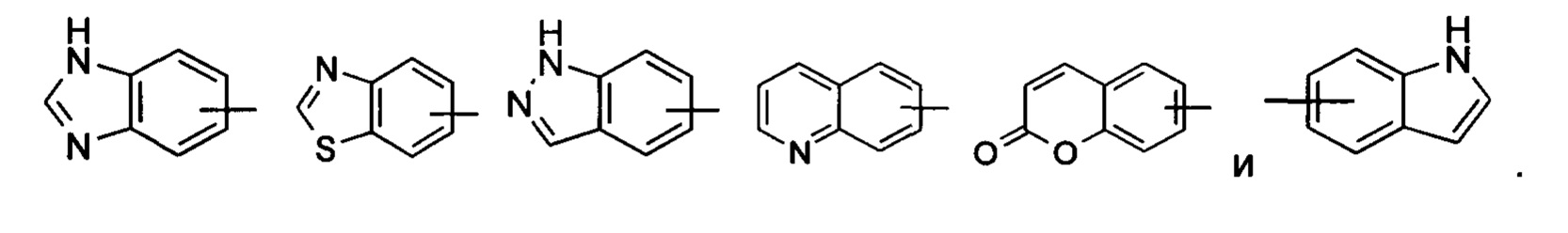
Арил может быть необязательно замещенным или незамещенным. При замещении замещающая(ие) группа(-ы) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила, и предпочтительно фенила.
«Гетероарил» относится к 5-14-членной гетероароматической системе, имеющей от 1 до 4 гетероатомов, выбранных из группы, состоящей из О, S и N, в качестве кольцевых атомов, предпочтительно 5-10-членному гетероарилу и более предпочтительно 5- или 6-членному гетероарилу, например, к имидазолилу, фурилу, тиенилу, тиазолилу, пиразолилу, оксазолилу, пирролилу, тетразолилу, пиридинилу, пиримидинилу, тиадиазолу, пиразинилу и т.п., предпочтительно к имидазолилу, пиразолилу, пиримидинилу или тиазолилу, и более предпочтительно пиразолилу или тиазолилу. Гетероарильное кольцо может быть конденсировано с кольцом арила, гетероциклила или циклоалкила, где кольцо, связанное с исходной структурой, представляет собой гетероарильное кольцо. Неограничивающие примеры включают:
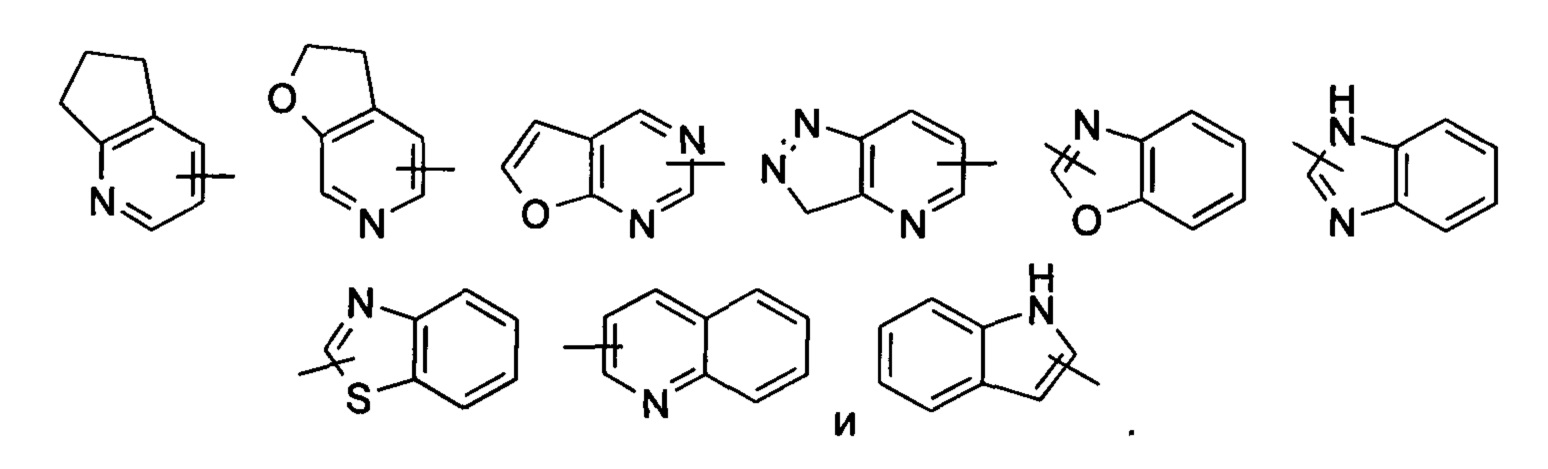
Гетероарил может быть необязательно замещенным или незамещенным. При замещении замещающая(ие) группа(-ы) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила.
«Алкокси» относится к группе -О-(алкил) или -O-(незамещенный циклоалкил), где алкил является таким, как определено выше. Неограничивающие примеры включают метокси, этокси, пропокси, бутокси, циклопропилокси, циклобутилокси, циклопентилокси, циклогексилокси. Алкоксигруппа может быть необязательно замещенной или незамещенной. При замещении заместитель(-и) предпочтительно представляет(-ют) собой одну или более групп, независимо выбранных из группы, состоящей из алкила, алкенила, алкинила, алкокси, алкилтио, алкиламино, атома галогена, тиола, гидрокси, нитро, циано, циклоалкила, гетероциклила, арила, гетероарила, циклоалкокси, гетероциклического алкокси, циклоалкилтио, гетероциклического алкилтио, карбокси и алкоксикарбонила.
«Гидроксиалкил» относится к алкилу, замещенному гидрокси, где алкил является таким, как определено выше.
«Галогеналкил» относится к алкилу, замещенному атомом галогена, где алкил является таким, как определено выше.
«Дейтероалкил» относится к алкилу, замещенному атомом дейтерия, где алкил является таким, как определено выше.
«Гидрокси» относится к группе -ОН.
«Атом галогена» относится к атому фтора, хлора, брома или йода.
«Амино» относится к группе -NH2.
«Циано» относится к группе -CN.
«Нитро» относится к группе -NO2.
«Карбокси» относится к группе -С(O)ОН.
«Альдегид» относится к группе -СНО.
«Алкоксикарбонил» относится к группе -С(O)O(алкил) или -С(O)O(циклоалкил), где алкил и циклоалкил являются такими, как определено выше.
«Ацилгалогенид» относится к соединению, содержащему группу -С(O)-атом галогена.
«Необязательный» или «необязательно» означает, что описанное впоследствии событие или обстоятельство может произойти, но необязательно произойдет, и такое описание включает ситуацию, в которой данное событие или обстоятельство происходит или не происходит. Например, фраза «гетероциклильная группа, необязательно замещенная алкилом» означает, что алкильная группа может присутствовать, но необязательно присутствует, и такое описание включает ситуацию, в которой гетероциклильная группа замещена алкилом и в которой гетероциклильная группа не замещена алкилом.
«Замещенный» относится к одному или более атомам водорода в группе, предпочтительно вплоть до 5 и более предпочтительно 1-3 атомам водорода, независимо замещенным соответствующим количеством заместителей. Совершенно очевидно, что заместители существуют только в их возможном химическом положении. Специалист в данной области техники способен определить, является ли замещение возможным или невозможным, с помощью экспериментов или теории, не прилагая слишком больших усилий. Например, комбинация амино- или гидроксигруппы, имеющей свободный атом водорода, и атомов углерода, имеющих ненасыщенные связи (такие как олефиновые), может быть нестабильной.
«Фармацевтическая композиция» относится к смеси одного или более соединений по настоящему изобретению или их физиологически/фармацевтически приемлемых солей или пролекарств и других химических компонентов, и других компонентов, таких как физиологически/фармацевтически приемлемые носители и вспомогательные вещества. Цель фармацевтической композиции состоит в том, чтобы облегчать введение соединения в организм, которое способствует поглощению активного ингредиента, демонстрируя, таким образом, биологическую активность.
«Фармацевтически приемлемая соль» относится к соли соединения по настоящему изобретению, которая является безопасной и эффективной у млекопитающих и обладает желаемой биологической активностью.
В настоящем изобретении различные термины, такие как «X выбран из группы, состоящей из А, В или С», «X выбран из группы, состоящей из А, В и С», «X представляет собой А, В или С» и «X представляет собой А, В и С», имеют одно и то же значение. Это означает, что Х может представлять собой любой один или более из А, В и С.
Способ синтеза соединения по настоящему изобретению
Для достижения цели настоящего изобретения в настоящем изобретении применяют следующие технические решения синтеза.
Способ получения соединения формулы (I) по настоящему изобретению или его таутомера, мезомера, рацемата, энантиомера, диастереомера или их смеси, либо их фармацевтически приемлемой соли включает следующие стадии:
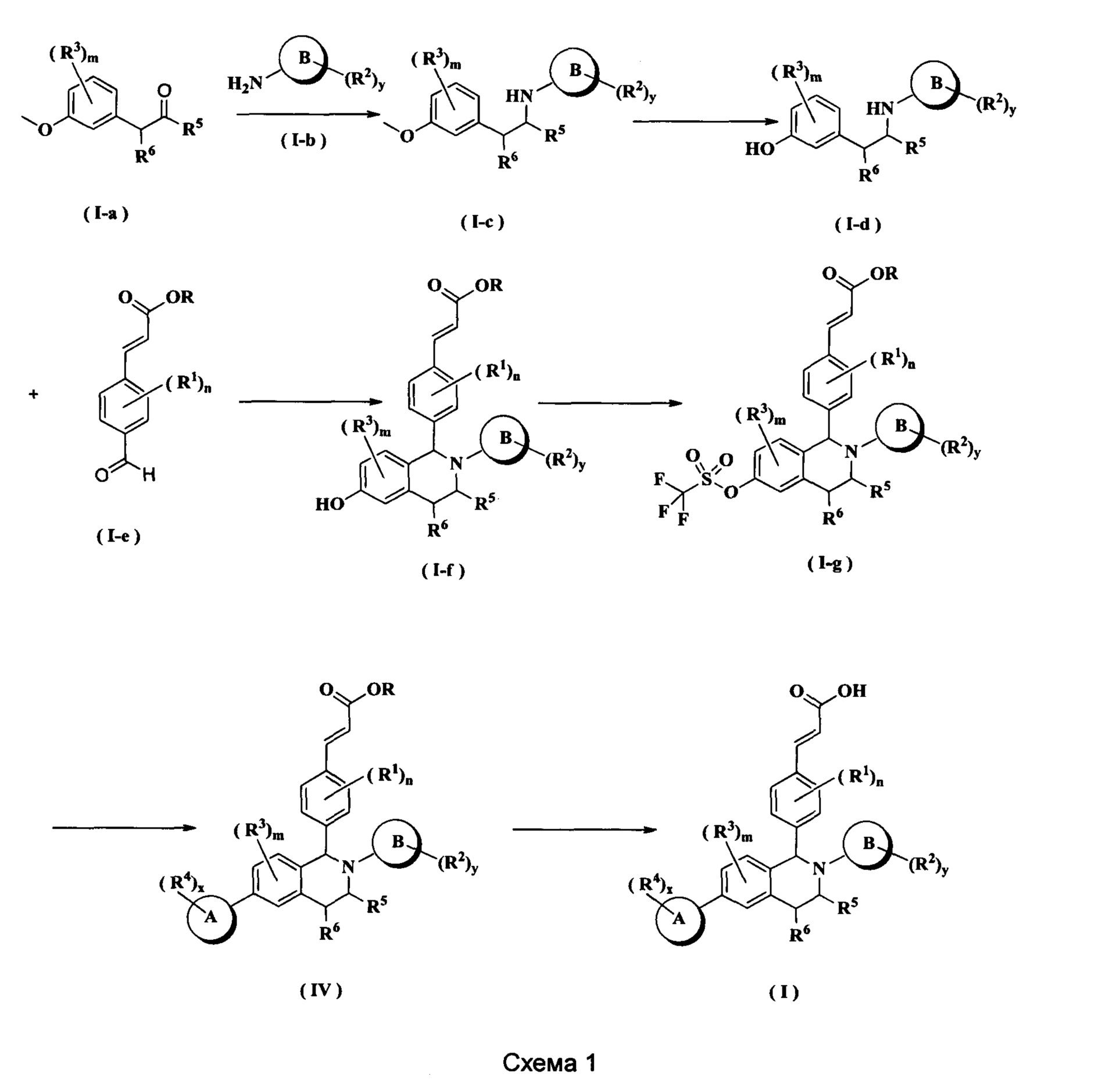
Соединение формулы (I-а) подвергают взаимодействию с соединением формулы (I-b) в щелочных условиях с получением соединения формулы (I-е), где щелочной реагент для этой реакции предпочтительно представляет собой триацетоксиборгидрид натрия. Образующееся соединение формулы (I-е) подвергают взаимодействию с трибромидом бора при комнатной температуре с получением соединения формулы (I-d). Полученное в результате соединение формулы (I-d) подвергают взаимодействию с соединением формулы (I-е) и триизопропилсилилхлоридом при нагревании с получением соединения формулы (I-f). Полученное в результате соединение формулы (I-f) дополнительно подвергают взаимодействию с трифторметансульфоновым ангидридом при низкой температуре с получением соединения формулы (I-g). Полученное в результате соединение формулы (I-g) подвергают взаимодействию с соединением борана в присутствии катализатора с получением соединения формулы (IV), где катализатор для данной реакции предпочтительно представляет собой [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий. Полученное в результате соединение формулы (IV) подвергают гидролизу в щелочных условиях с получением соединения формулы (I), где щелочной реагент для этой реакции предпочтительно представляет собой гидроксид лития или гидроксид натрия.
Реагент, который обеспечивает щелочные условия, включает органические основания и неорганические основания, где органические основания включают триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия, но не ограничиваются ими, и где неорганические основания включают гидрид натрия, фосфат калия, карбонат натрия, карбонат калия или карбонат цезия, гидроксид натрия и гидроксид лития, но не ограничиваются ими.
Задействованный катализатор включает 2-дициклогексилфосфино-2',4',6'-триизопропилбифенил, (±)-2,2'-бис(дифенилфосфино)-1,1'-динафталин, трис(дибензилиденацетон)дипалладий, ацетат палладия, [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий, трифенилфосфин, тетракистрифенилфосфин палладий, но не ограничивается ими.
Используемый в данном документе растворитель включает уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и N,N-диметилформамид, но не ограничивается ими.
Где:
R представляет собой алкил или циклоалкил, где каждый алкил и циклоалкил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, амино, циано, гидрокси, алкокси, карбокси и циклоалкила; и
кольцо А, кольцо В, от R1 до R6, m, n, x и у являются такими, как определено в формуле (I).
Соединения формулы (I) по настоящему изобретению также могут быть получены следующим образом:
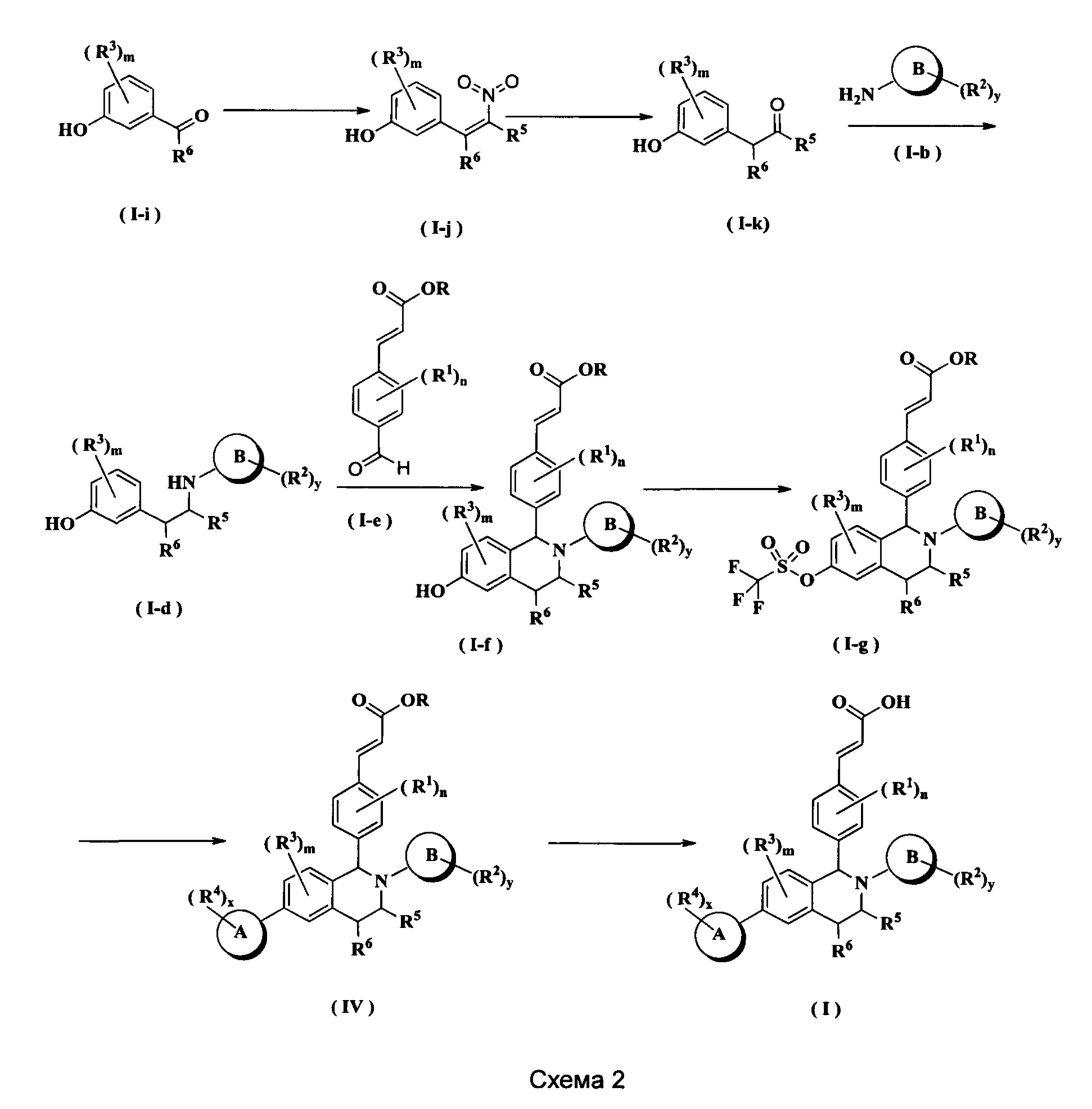
Соединение формулы (I-i) подвергают взаимодействию с соединением нитроалкана, метиламином и ацетатом аммония при нагревании с получением соединения формулы (I-j). Полученное в результате соединение формулы (I-j) подвергают взаимодействию с никелем Ренея в кислотных условиях с получением соединения формулы (I-k), где кислотный реагент для этой реакции предпочтительно представляет собой уксусную кислоту. Полученное в результате соединение формулы (I-k) подвергают взаимодействию с соединением формулы (I-b) в щелочных условиях с получением соединения формулы (I-d), где щелочной реагент для этой реакции предпочтительно представляет собой триацетоксиборгидрид натрия и триэтиламин. Полученное в результате соединение формулы (I-d) подвергают взаимодействию с соединением формулы (I-е) и триизопропилсилилхлоридом при нагревании с получением соединения формулы (I-f). Полученное в результате соединение формулы (I-f) дополнительно подвергают взаимодействию с трифторметансульфоновым ангидридом при низкой температуре с получением соединения формулы (I-g). Полученное в результате соединение формулы (I-g) подвергают взаимодействию с соединением борана в присутствии катализатора с получением соединения формулы (IV), где катализатор для данной реакции предпочтительно представляет собой [1,1'-бис(дифенилфосфино)ферроцен]дихпорпалладий. Полученное в результате соединение формулы (IV) подвергают гидролизу в щелочных условиях с получением соединения формулы (I), где щелочной реагент для этой реакции предпочтительно представляет собой гидроксид лития или гидроксид натрия.
Реагент, который обеспечивает щелочные условия, включает органические основания и неорганические основания, где органические основания включают триэтиламин, N,N-диизопропилэтиламин, н-бутиллитий, диизопропиламид лития, ацетат калия, трет-бутоксид натрия и трет-бутоксид калия, но не ограничиваются ими, и где неорганические основания включают гидрид натрия, фосфат калия, карбонат натрия, карбонат калия или карбонат цезия, гидроксид натрия и гидроксид лития, но не ограничиваются ими.
Используемый в данном документе растворитель включает уксусную кислоту, метанол, этанол, толуол, тетрагидрофуран, дихлорметан, диметилсульфоксид, 1,4-диоксан, воду и N,N-диметилформамид, но не ограничивается ими.
Где:
R представляет собой алкил или циклоалкил, где каждый алкил и циклоалкил необязательно замещен одной или более группами, выбранными из группы, состоящей из алкила, атома галогена, амино, циано, гидрокси, алкокси, карбокси и циклоалкила; и
кольцо А, кольцо В, от R1 до R6, m, n, x и у являются такими, как определено в формуле (I).
Предпочтительные воплощения
Далее настоящее изобретение будет описано со ссылкой на следующие примеры, но данные примеры не следует рассматривать как ограничивающие объем данного изобретения.
Примеры
Структуры соединений идентифицировали посредством ядерного магнитного резонанса (ЯМР) и/или масс-спектрометрии (МС). Химические сдвиги ЯМР (δ) приведены в 10-6 (млн-1). ЯМР определяли с помощью прибора Bruker AVANCE-400. Растворители для определения представляли собой дейтерированный диметилсульфоксид (ДМСО-d6), дейтерированный хлороформ (CDCl3) и дейтерированный метанол (CD3OD), и внутренний стандарт представлял собой тетраметилсилан (ТМС).
МС определяли с помощью масс-спектрометра FINNIGAN LCQAd (ионизация электрораспылением (ИЭР)) (производитель: Thermo, тип: Finnigan LCQ advantage MAX).
Высокоэффективную жидкостную хроматографию (ВЭЖХ) осуществляли на спектрометре для жидкостной хроматографии высокого давления Agilent 1200DAD (хроматографическая колонка Sunfire С18 150×4,6 мм) и на спектрометре для жидкостной хроматографии высокого давления Waters 2695-2996 (хроматографическая колонка Gimini С18 150×4,6 мм).
Хиральную ВЭЖХ осуществляли на приборе LC-10A vp (Shimadzu) или SFC-analytical (Berger Instruments Inc.).
Средние скорости ингибирования киназы и значения IC50 (концентрация полумаксимального ингибирования) определяли посредством набора для твердофазного иммуноферментного анализа (ELISA - от англ. Enzyme-linked immunosorbent assay) NovoStar (BMG Co., Германия).
Для тонкослойной хроматографии на силикагеле (ТСХ) использовали пластину с силикагелем Yantai Huanghai HSGF254 или Qingdao GF254. Размеры пластины с силикагелем, используемой для ТСХ, составляли от 0,15 мм до 0,2 мм, а размеры пластины с силикагелем, используемой для очистки продукта, составляли от 0,4 мм до 0,5 мм.
В качестве носителя для колоночной хроматографии использовали силикагель Yantai Huanghai от 200 до 300 меш.
Для хиральной препаративной колоночной хроматографии использовали Prep Star SD-1 (Varian Instruments Inc.) или SFC-multigram (Berger Instruments Inc.).
Известные исходные вещества по настоящему изобретению могут быть получены традиционными способами синтеза в данной области техники или могут быть приобретены у ABCR GmbH & Co. KG, Acros Organnics, Aldrich Chemical Company, Accela ChemBio Inc. или Dari Chemical Company и т.д.
Если не указано иное, реакции проводили в атмосфере азота или в атмосфере аргона.
Термин «атмосфера азота» или «атмосфера аргона» означает, что реакционная колба оборудована 1 л баллоном аргона или азота.
Термин «атмосфера водорода» означает, что реакционная колба оборудована 1 л баллоном водорода.
Реакции гидрогенизации под давлением проводили с помощью аппарата для гидрогенизации Parr 3916EKX и генератора водорода QL-500 или аппарата для гидрогенизации HC2-SS.
В реакциях гидрогенизации в реакционной системе обычно создают вакуум и заполняют ее водородом, причем описанную выше операцию повторяют три раза.
В микроволновых реакциях использовали микроволновый реактор типа СЕМ Discover-S 908860.
Если не указано иное, раствор, используемый в реакциях, относится к водному раствору.
Если не указано иное, температура реакции в реакциях относится к комнатной температуре от 20°С до 30°С.
Реакционный процесс отслеживали посредством тонкослойной хроматографии (ТСХ), и система растворителей для проявления включала: А: дихлорметан и метанол, В: н-гексан и этилацетат, С: петролейный эфир и этилацетат, D: ацетон. Объемную долю данного растворителя регулировали в соответствии с полярностью соединений. Система элюции для очистки соединений колоночной хроматографией и тонкослойной хроматографией включала: А: дихлорметан и метанол, В: н-гексан и этилацетат, С: дихлорметан и ацетон. Объемную долю данного растворителя регулировали в соответствии с полярностью соединений и иногда добавляли слабощелочной реагент, такой как триэтиламин, или кислотный реагент, такой как уксусная кислота.
Пример 1
(E)-3-(4-((1R,3R,/1S,3S)-2-(4-Этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
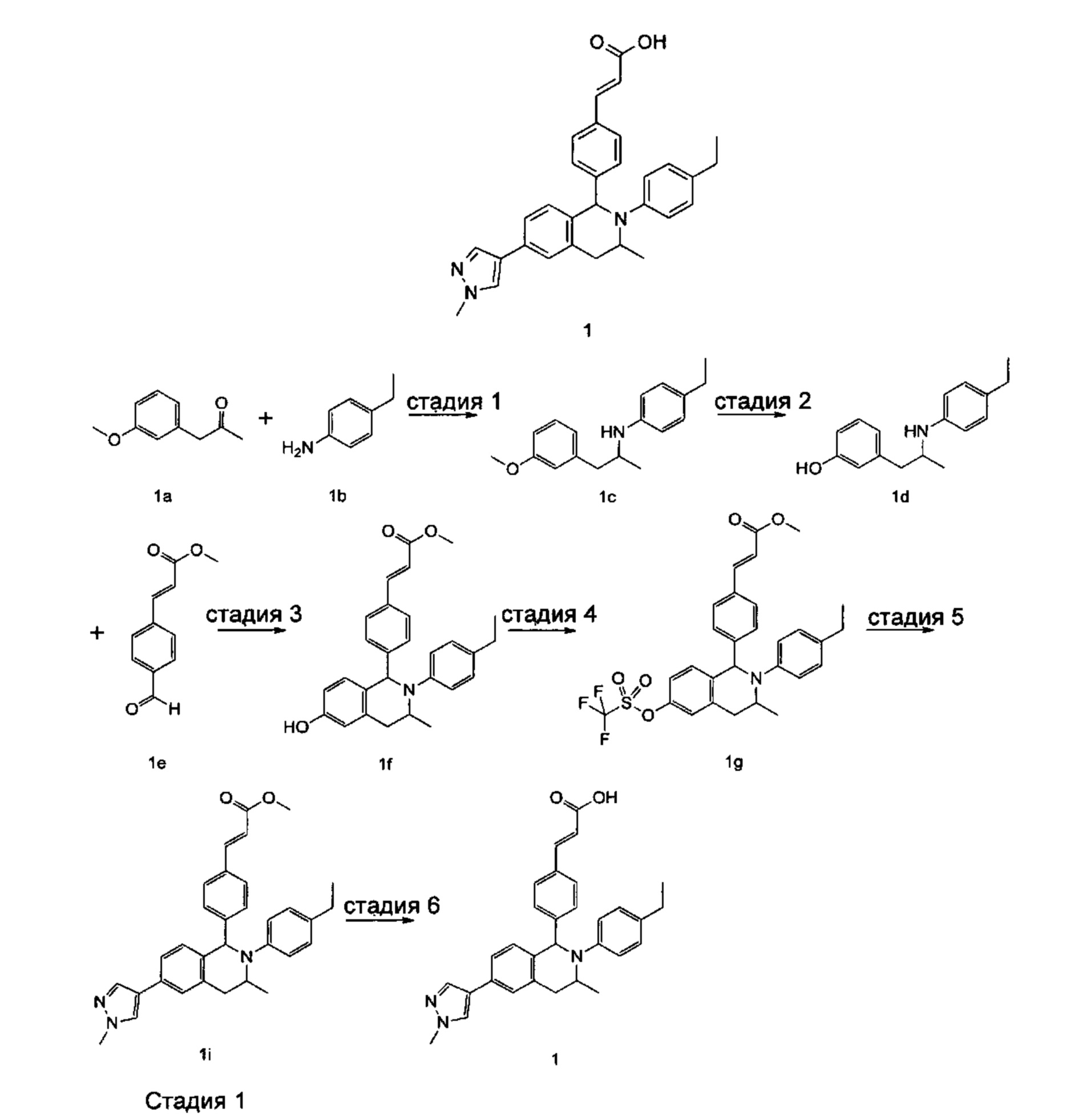
4-Этил-N-(1-(3-метоксифенил)пропан-2-ил))анилин 1с
1-(3-Метоксифенил)пропан-2-он 1а (820 мг, 5 ммоль), 4-этиланилин 1b (0,75 мл, 6 ммоль) и триацетоксиборгидрид натрия (1,58 г, 7,5 ммоль) растворяли в 30 мл 1,2-дихлорэтана. Смесь перемешивали в течение 16 часов. Для гашения реакции добавляли 30 мл воды. Реакционный раствор экстрагировали дихлорметаном (30 мл × 2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 1с (700 мг, выход 51,9%) в виде желтого масла.
МС m/z (ИЭР): 270,2 [М+1]
Стадия 2
3-(2-((4-Этил фенил)амино)пропил)фенол 1d
Соединение 1с (640 мг, 2,37 ммоль) растворяли в 18 мл дихлорметана, впоследствии по каплям добавляли раствор 1 М трибромида бора в дихлорметане (4,7 мл, 4,7 ммоль) в ледяной бане. После завершения добавления реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 30 мл воды. Реакционный раствор концентрировали при пониженном давлении для удаления дихлорметана. Добавляли еще 30 мл воды. Смесь перемешивали до однородного состояния и экстрагировали этилацетатом (25 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 1d (540 мг, выход 89,4%) в виде желтого масла.
Стадия 3
(E)-Метил-3-(4-(2-(4-этилфенил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 1f
1d (270 мг, 1,06 ммоль), (E)-метил-3-(4-формилфенил)акрилат 1е (402 мг, 2,11 ммоль) и тризопропилсилилхлорид (1,02 г, 5,29 ммоль) добавляли к 10 мл N,N-диметилформамид. После завершения добавления смесь нагревали до 120°С и перемешивали в течение 3 часов. После прекращения нагревания реакционный раствор охлаждали до комнатной температуры и добавляли 30 мл воды. Смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 1f (292 мг, выход 64,6%) в виде желтого твердого вещества.
Стадия 4
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-этилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 1g
1f (292 мг, 0,68 ммоль) растворяли в 30 мл дихлорметана, затем добавляли 2,6-лутидин (110 мг, 1,02 ммоль). После завершения добавления реакционную смесь охлаждали до 0°С в ледяной бане и по каплям добавляли трифторметансульфоновый ангидрид (289 мг, 1,02 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 30 мл воды и разделяли две фазы. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 1д (163 мг, выход 42,7%) в виде светло-желтого твердого вещества.
Стадия 5
(Е)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 1i
Соединение 1g (163 мг, 0,29 ммоль),
1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (91 мг, 0,44 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (21 мг, 0,03 ммоль) растворяли в 12 мл смеси 1,4-диоксана и воды (V:V составляет 7:1). Добавляли 2М раствор карбоната натрия (0,29 мл, 0,58 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 40 часов. После охлаждения до комнатной температуры добавляли 30 мл воды, и смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл × 2), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 1i (143 мг) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии.
Стадия 6
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 1
Неочищенное соединение 1i (143 мг, 0,29 ммоль) растворяли в 5 мл метанола, впоследствии добавляли 2 М раствор гидроксида натрия (0,73 мл, 1,45 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. По каплям добавляли 1 н. соляную кислоту для доведения рН реакционного раствора до 2. Смесь экстрагировали этилацетатом (20 мл × 2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 1 (85 мг, выход 61,2%) в виде почти белого твердого вещества.
МС m/z (ИЭР): 478,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 4.47-4.53 (m, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.70-2.78 (m, 1Н), 2.51-2.59 (m, 2Н), 1.26 (d, 3Н), 1.19 (t, 3Н).
Примеры 2, 3
(Е)-3-(4-((1R,3R)-2-(4-Этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 2
(E)-3-(4-((1S,3S)-2-(4-Этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 3
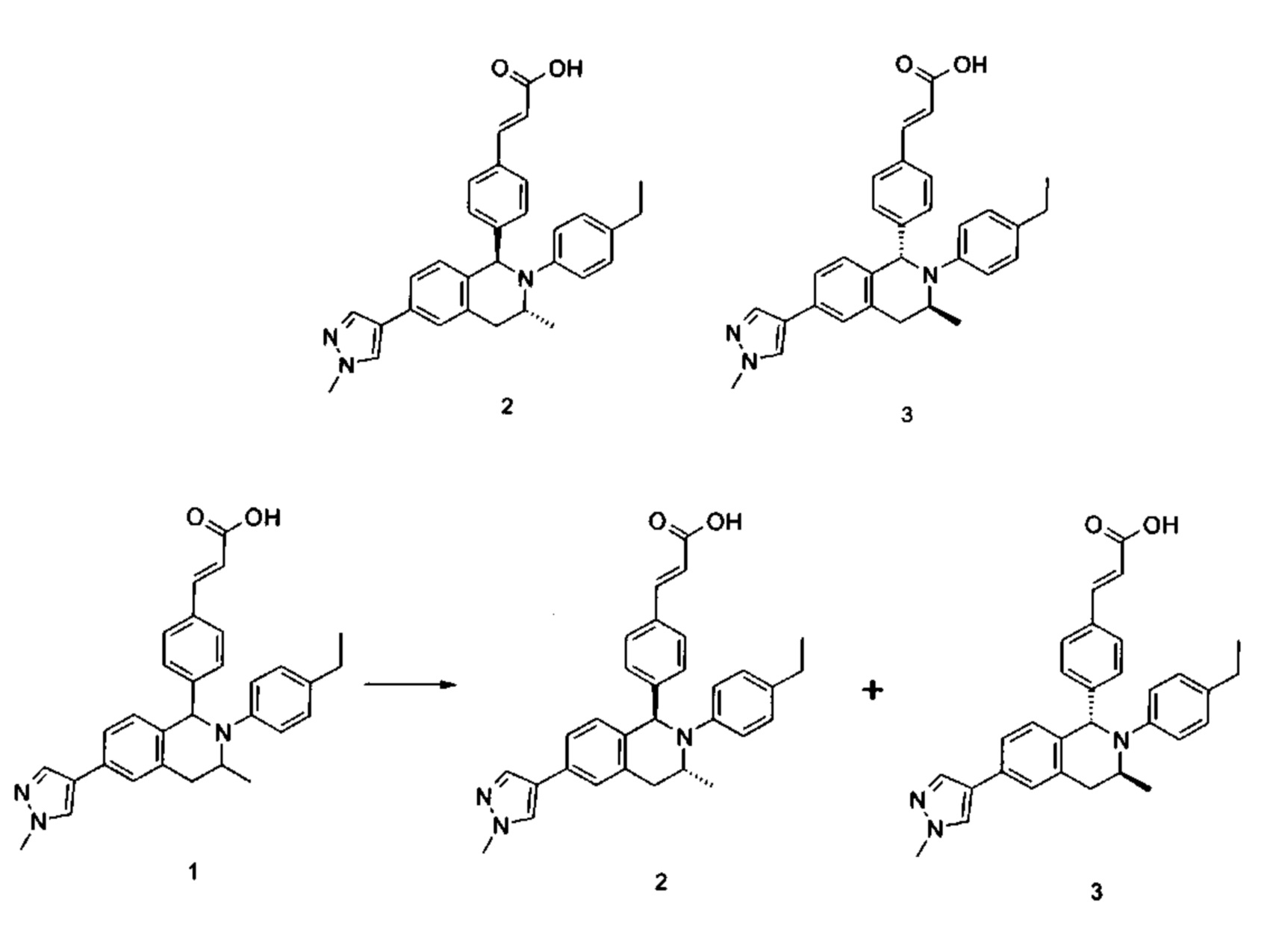
Соединение 1 (310 мг, 0,65 ммоль) подвергали хиральному разделению (условия разделения: хиральная препаративная колонка Superchiral S-AD (Chiralway), внутренний диаметр 2 см * 25 см, 5 мкм; подвижная фаза: н-гексан: этанол: трифторуксусная кислота составляет 50:50:0,01, скорость потока: 10 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанных в заголовке соединений 2 (108 мг, желтое твердое вещество) и 3 (95 мг, желтое твердое вещество).
Соединение 2:
МС m/z (ИЭР): 478,5 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 17,940 минут, хиральная чистота: 99,1% (хроматографическая колонка: Superchiral S-AD (Chiralway), внутренний диаметр 0,46 см * 25 см, 5 мкм; подвижная фаза: н-гексан: этанол: трифторуксусная кислота составляет 50:50:0,01 (v/v/v)).
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 4.47-4.53 (m, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.70-2.78 (m, 1Н), 2.51-2.59 (m, 2Н), 1.16-1.20 (m, 3Н), 0.95 (d, 3Н).
Соединение 3:
МС m/z (ИЭР): 478,5 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 23,198 минут, хиральная чистота: 99,3% (хроматографическая колонка: Superchiral S-AD (Chiralway), внутренний диаметр 0,46 см * 25 см, 5 мкм; подвижная фаза: н-гексан: этанол: трифторуксусная кислота составляет 50:50:0,01 (v/v/v)).
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 4.47-4.53 (m, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.70-2.78 (m, 1Н), 2.51-2.59 (m, 2Н), 1.16-1.20 (m, 3Н), 1.06 (d, 3Н).
Пример 4
(E)-3-(4-((1S,3R/1R,3S)-2-(4-Циклопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
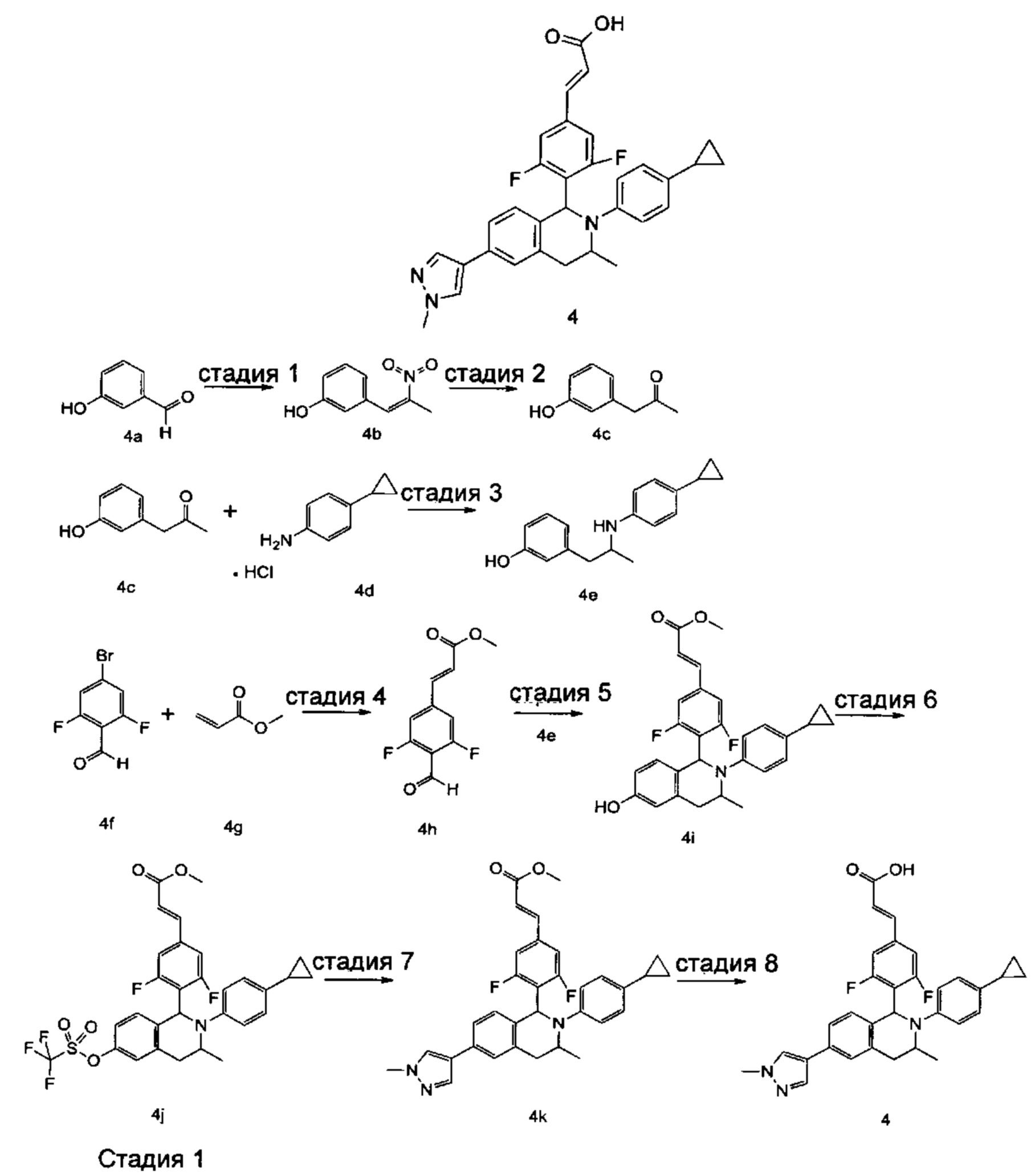
3-(2-Нитропроп-1-ен-1-ил)фенол 4b
В реакционную колбу добавляли 3-гидроксибензальдегид 4а (10 г, 81,9 ммоль), нитроэтан (60 г, 819 ммоль) и ацетат аммония (1,54 г, 20 ммоль). Смесь нагревали до 80°С и добавляли метиламин (1 г, 32,2 ммоль). После завершения добавления реакционную смесь перемешивали в течение 2 часов. Добавляли 50 мл воды, и смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 4b (9,5 г, выход 64,6%) в виде желтого твердого вещества.
Стадия 2
1-(3-Гидроксифенил)пропан-2-он 4с
Соединение 4b (9,5 г, 53 ммоль) добавляли к 110 мл смеси метанола и воды (V:V составляет 10:1), впоследствии добавляли никель Ренея (10%, 9,5 г) и уксусную кислоту (3,2 г, 53 ммоль). После завершения добавления реакционную систему три раза продували водородом, и реакционную смесь перемешивали в течение 16 часов. После фильтрования фильтрат упаривали с удалением большей части растворителя, и смесь экстрагировали этилацетатом (50 мл*3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 4с (3,7 г, выход 46,8%) в виде желтого масла.
Стадия 3
3-(2-((4-Циклопропилфенил)амино)пропил)фенол 4е
4-Циклопропиланилина гидрохлорид (390 мг, 2,30 ммоль, Bidepharmatech) растворяли в 10 мл дихлорметана, впоследствии добавляли триэтиламин (233 мг, 2,30 ммоль). После перемешивания смеси в течение 5 минут добавляли соединение 4 с (345 мг, 2,30 ммоль) и триацетоксиборгидрид натрия (730 мг, 3,45 ммоль), и реакционную смесь перемешивали в течение 12 часов. Добавляли 10 мл воды, и смесь экстрагировали дихлорметаном (10 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 4е (540 мг, выход 87,8%) в виде коричневого вязкого вещества.
Стадия 4
(E)-Метил-3-(3,5-дифтор-4-формилфенил)акрилат
В атмосфере аргона добавляли 4-бром-2,6-дифторбензальдегид 4f (10 г, 45,2 ммоль), метилакрилат 4д (6,1 мл, 67,9 ммоль), три(о-метилфенил)фосфин (1,4 г, 4,52 ммоль), ацетат палладия (507 мг, 2,26 ммоль) и триэтиламин (12,5 мл, 90,4 ммоль) к 100 мл N',N'-диметиланилина. После завершения добавления смесь нагревали до 80°С и перемешивали в течение 16 часов. После прекращения нагревания реакционный раствор естественным путем охлаждали до комнатной температуры и добавляли 300 мл воды. Реакционный раствор фильтровали, и фильтрационный кек последовательно промывали водой (50 мл × 3) и н-гексаном (50 мл × 3) и высушивали с получением указанного в заголовке соединения 4h (10 г, выход 98,0%) в виде желтого твердого вещества.
Стадия 5
(E)-Метил-3-(4-(2-(4-циклопропилфенил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 4i
Соединение 4е (4,8 г, 18,7 ммоль), 4h (8,5 г, 37,4 ммоль) и триизопропилсилилхлорид (7,2 г, 37,4 ммоль) добавляли к 120 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 120°С и перемешивали в течение 3 часов. После прекращения нагревания реакционный раствор охлаждали до комнатной температуры и добавляли 50 мл воды. Смесь экстрагировали этилацетатом (100 мл × 4). Органические фазы объединяли, высушивали над безводным сульфатом магния и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 4i (7,0 г, выход 85,4%) в виде желтого масла.
Стадия 6
(E)-Метил-3-(4-((1S,3R/1R,3S)-2-(4-циклопропилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 4j
Соединение 4i (3 г, 6,8 ммоль) растворяли в 50 мл дихлорметана, затем добавляли 2,6-лутидин (1,1 г, 10,2 ммоль). После завершения добавления смесь охлаждали до 0°С в ледяной бане и по каплям добавляли трифторметансульфоновый ангидрид (2,5 г, 8,87 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Добавляли 30 мл воды, и реакционный раствор экстрагировали дихлорметаном (15 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 4j (1,6 г, выход 39,0%) в виде желтого масла.
Стадия 7
(E)-Метил-3-(4-((1S,3R/1R,3S)-2-(4-циклопропилфенил)-3-метил-6-(1-метил-1H-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 4k
Соединение 4j (1,0 г, 1,65 ммоль),
1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)1H-пиразол (514 мг, 2,47 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (120 мг, 0,165 ммоль) растворяли в 14 мл смеси 1,4-диоксана и воды (V:V составляет 5:2). Добавляли 2М раствор карбоната натрия (1,65 мл, 3,3 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 1 часа. После охлаждения до комнатной температуры добавляли 50 мл воды, и смесь экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом магния и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 4k (420 мг, выход 47,3%) в виде желтого масла.
Стадия 8
(E)-3-(4-((1S,3R/1R,3S)-2-(4-Циклопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 4
Соединение 4k (420 мг, 0,78 ммоль) растворяли в 20 мл смеси метанола и тетрагидрофурана (V:V составляет. 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (2 мл, 4,0 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. По каплям добавляли 1 н. соляную кислоту для доведения рН реакционного раствора до 3. Смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом магния и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 4 (180 мг, выход 61,5%) в виде желтого твердого вещества.
МС m/z (ИЭР): 525,6 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 12.05 (3, 1Н), 7.75 (s, 1Н), 7.60 (s, 1Н), 7.52 (d, 1Н), 7.29 (s, 1Н), 7.21 (d, 1Н), 7.01-6.90 (m, 6Н), 6.32 (d, 1Н), 6.12 (s, 1Н), 4.35-4.28 (m, 1Н), 3.94 (s, 3Н), 3.69-3.60 (m, 1Н), 2.81-2.68 (m, 2Н), 1.83-1.74 (m, 1Н), 1.07-0.96 (m, 2Н), 0.91-0.84 (m, 2Н), 0.60 (d, 3Н).
Пример 5
(E)-3-(4-(2-(2-Этилфенил)-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота
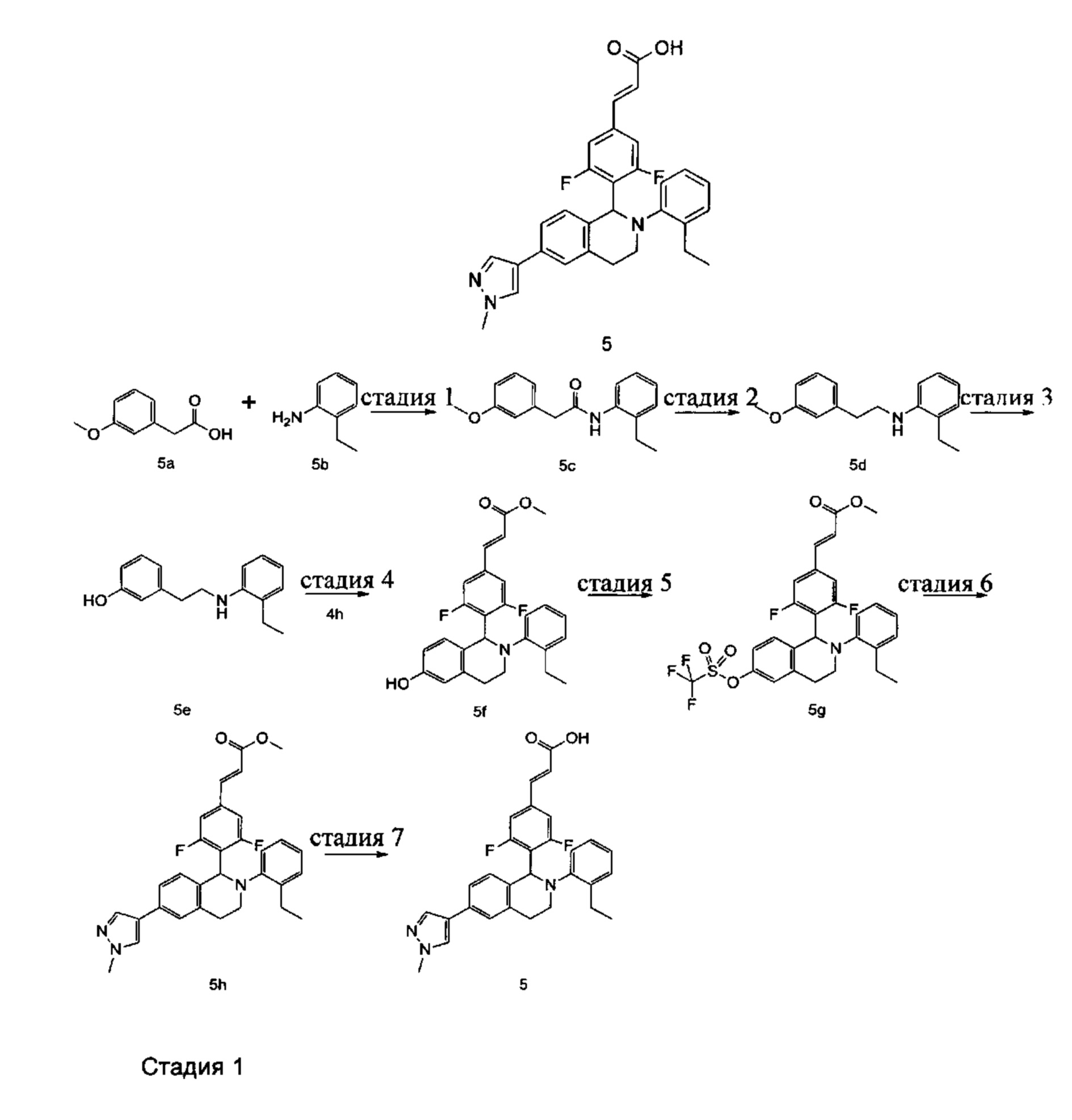
N-(2-Этилфенил)-2-(3-метоксифенил)ацетамид 5с
3-Метоксифенилуксусная кислота 5а (1,66 г, 10 ммоль), 2-этиланилин 5b (1,21 г, 10 ммоль), 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорид (2,3 г, 12 ммоль) и 4-диметиламинопиридин (122 мг, 1 ммоль) добавляли к 30 мл N,N-диметилформамида. После завершения добавления реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 5с (2,45 г, выход 91,1%) в виде белого твердого вещества.
Стадия 2
2-Этил-N-(3-метоксифенэтил)анилин 5d
Соединение 5с (2,45 г, 9,1 ммоль) добавляли к 60 мл тетрагидрофурана, затем порциями добавляли тетрагидроалюминат лития (1,73 г, 45,5 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. Для гашения реакции добавляли 2 мл воды и по каплям добавляли раствор гидроксида натрия (5%, 5,2 мл). Смесь перемешивали до однородного состояния, фильтровали через целит, и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 5d (1,05 г, выход 45,0%) в виде коричневого масла.
Стадия 3
3-(2-((2-Этилфенил)амино)этил)фенол 5е
Соединение 5d (1,05 г, 4,11 ммоль) растворяли в 30 мл дихлорметана. После охлаждения реакционной смеси до -78°С в бане с сухим льдом и ацетоном по каплям добавляли раствор бромида бора в дихлорметане (1M, 8,2 мл). После завершения добавления баню с сухим льдом и ацетоном удаляли. Реакционный раствор подогревали до комнатной температуры и перемешивали в течение 16 часов. Для гашения реакции добавляли 30 мл воды. Реакционный раствор выпаривали при пониженном давлении для удаления дихлорметана. Добавляли еще 30 мл воды. Смесь перемешивали до однородного состояния и экстрагировали этилацетатом (25 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 5е (620 мг, выход 62,6%) в виде желтого твердого вещества.
Стадия 4
(Е)-Метил-3-(4-(2-(2-этилфенил)-6-гидрокси-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 5f
Соединение 5е (520 мг, 2,15 ммоль), 4h (975 мг, 4,31 ммоль) и триизопропилсилилхлорид (2,07 г, 10,75 ммоль) добавляли к 15 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 120°С и перемешивали в течение 2 часов. После прекращения перемешивания реакционный раствор охлаждали до комнатной температуры и добавляли 50 мл воды. Смесь экстрагировали этилацетатом (80 мл × 2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 5f (720 мг, выход 74,6%) в виде бесцветного масла. Стадия 5
(E)-Метил 3-(4-(2-(2-этилфенил)-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 5g
Соединение 5f (720 мг, 1,6 ммоль) растворяли в 50 мл дихлорметана, впоследствии добавляли 2,6-лутидин (0,28 мл, 2,4 ммоль) и трифторметансульфоновый ангидрид (0,4 мл, 2,4 ммоль). После завершения добавления реакционную смесь перемешивали в течение 3 часов. Добавляли 0,5 мл воды, и реакционный раствор экстрагировали дихлорметаном (15 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 5g (660 мг, выход 71,0%) в виде белого твердого вещества.
Стадия 6
(E)-Метил-3-(4-(2-(2-этилфенил)-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 5h
Соединение 5g (104,6 мг, 0,18 ммоль),
1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (56,1 мг, 0,27 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (13 мг, 0,018 ммоль) растворяли в 2,4 мл смеси 1,4-диоксана и воды (V:V составляет 7:1), впоследствии добавляли 2М раствор карбоната натрия (0,18 мл, 0,36 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 1 часа. После охлаждения до комнатной температуры добавляли 20 мл воды, и смесь экстрагировали этилацетатом (15 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом магния и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 5h (60 мг, выход 65,0%) в виде бесцветного масла.
Стадия 7
(Е)-3-(4-(2-(2-Этилфенил)-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 5
Соединение 5h (60 мг, 0,12 ммоль) растворяли в 6 мл смеси метанола и тетрагидрофурана (V:V составляет. = 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (1 мл, 2,0 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. По каплям добавляли 1 н. соляную кислоту для доведения рН реакционного раствора до 3. Смесь фильтровали, и фильтрационный кек высушивали с получением указанного в заголовке соединения 5 (40 мг, выход 68,5%) в виде желтого твердого вещества.
МС m/z(M3P): 500,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.08 (s, 1Н), 7.81 (s, 1Н), 7.42 (s, 1Н), 7.20-7.29 (m, 4Н), 7.10-7.13 (m, 2Н), 7.00-7.02 (d, 1Н), 6.69-6.71 (d, 1Н), 6.51-6.55 (d, 1Н), 5.90 (s, 1Н), 3.85 (s, 3Н), 3.12-3.21 (m, 2Н), 2.84-2.89 (m, 1Н), 2.60-2.64 (m, 2Н), 1.22-1.25 (m, 2Н), 1.01-1.06 (t, 3Н).
Пример 6
(E)-3-(4-((1R,3R/1S,3S)-2-(2-Этилфенил)-3-метил-6-(1-метил-1H-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
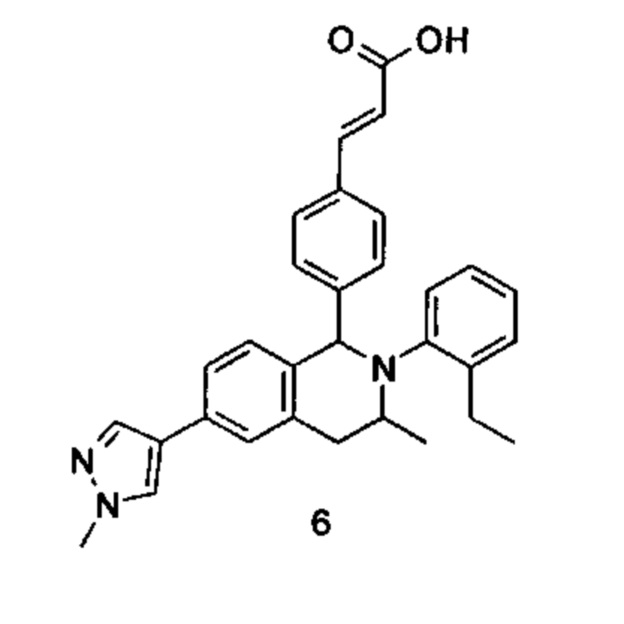
Стадия 1
2-Этил-N-(1-(3-метоксифенил)пропан-2-ил)анилин 6а
Соединение 1а (820 мг, 5 ммоль), 5b (0,75 мл, 6 ммоль) и триацетоксиборгидрид натрия (1,58 г, 7,5 ммоль) растворяли в 30 мл трихлорэтана. Смесь перемешивали в течение 16 часов. Для гашения реакции добавляли 30 мл воды. Реакционный раствор экстрагировали дихлорметаном (25 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 6а (800 мг, выход 59,3%) в виде желтого масла.
Стадия 2
3-(2-((2-Этилфенил)амино)пропил)фенол 6b
Соединение 6а (800 мг, 2,97 ммоль) растворяли в 20 мл дихлорметана, впоследствии по каплям добавляли раствор 1 Мтрибромида бора в дихлорметане (6 мл, 6,0 ммоль) в ледяной бане. После завершения добавления реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 15 мл воды. Реакционный раствор выпаривали при пониженном давлении, чтобы удалить дихлорметан, и впоследствии экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении с получением указанного в заголовке соединения 6b (540 мг, выход 71,2%) в виде желтого масла.
Стадия 3
(E)-Метил-3-(4-(2-(2-этилфенил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 6с
Соединение 6b (400 мг, 1,56 ммоль), 1е (596 мг, 3,13 ммоль) и триизопропилсилилхлорид (1,5 г, 7,8 ммоль) добавляли к 10 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 120°С и перемешивали в течение 3 часов. После прекращения нагревания реакционный раствор охлаждали до комнатной температуры и добавляли 30 мл воды. Смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 6 с (440 мг, выход 65,7%) в виде желтого твердого вещества.
Стадия 4
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(2-этилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 6d Соединение 6с (440 мг, 1,03 ммоль) растворяли в 50 мл дихлорметана в ледяной бане, впоследствии добавляли 2,6-лутидин (165 мг, 1,54 ммоль) с последующим добавлением трифторметансульфонового ангидрида (436 мг, 1,54 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. К реакционному раствору добавляли 50 мл воды и разделяли две фазы. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 6d (61 мг, выход 10,6%) в виде светло-желтого твердого вещества.
Стадия 5
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(2-этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 6е
Соединение 6d (47 мг, 0,084 ммоль),
1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (26 мг, 0,13 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (6 мг, 0,0084 ммоль) растворяли в 2,4 мл смеси 1,4-диоксана и воды (V:V составляет 7:1), впоследствии добавляли 2М раствор карбоната натрия (0,08 мл, 0,16 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 40 часов. После охлаждения до комнатной температуры добавляли 20 мл воды, и смесь экстрагировали этилацетатом (20 мл*3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл* 2), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 6е (40 мг, выход 97,6%) в виде желтого твердого вещества.
Стадия 6
(Е)-3-(4-((1R,3R/1S,3S)-2-(2-Этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 6
Неочищенное соединение 6е (40 мг, 0,08 ммоль) растворяли в 5 мл метанола, впоследствии добавляли 2 М раствор гидроксида натрия (0,2 мл, 0,4 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. По каплям добавляли 1 н. соляную кислоту для доведения рН реакционного раствора до 2. Смесь экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 6 (4 мг, выход 10,3%) в виде почти белого твердого вещества.
МС m/z (ИЭР): 478,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.92 (s, 1Н), 7.78 (s, 1Н), 7.66-7.62 (m, 1Н), 7.50-7.44 (m, 2Н), 7.37 (s, 1Н), 7.30 (d, 2Н), 7.20-7.17 (m, 2Н), 7.04-7.00 (m, 2Н), 6.58 (d, 1Н), 6.36 (d, 1Н), 5.33 (s, 1Н), 3.91 (s, 3Н), 3.05 (d, 1Н), 2.95 (d, 1Н), 2.62-2.56 (m, 2Н), 2.38-2.35 (m, 1Н), 0.84 (d, 3Н), 0.81 (t, 3Н).
Пример 7
(Е)-3-(4-((1R,3R/1S,3S)-6-(1-Этил-1Н-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
4-Изопропил-N-(1-(3-метоксифенил)пропан-2-ил)анилин 7b Соединение 1а (2,0 г, 12 ммоль), 4-изопропиланилин 7а (1,95 г, 18 ммоль) и триацетоксиборгидрид натрия (3,81 г, 18 ммоль) растворяли в 50 мл дихлорметана. Смесь перемешивали в течение 5 часов. Для гашения реакции добавляли 20 мл воды. Реакционный раствор экстрагировали дихлорметаном (20 мл × 2). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 7b (2,5 г, выход 75,3%) в виде коричневого масла.
Стадия 2
3-(2-((4-Изопропилфенил)амино)пропил)фенол 7с
Соединение 7b (2,5 г, 8,82 ммоль) растворяли в 70 мл дихлорметана, впоследствии по каплям добавляли раствор 1 М трибромида бора в дихлорметане (17,6 мл, 17,6 ммоль) в ледяной бане. После завершения добавления реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 50 мл воды. Реакционный раствор экстрагировали дихлорметаном (20 мл × 2). Органические фазы объединяли, промывали насыщенным раствором бикарбоната натрия (20 мл) и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 7 с (2,3 г, выход 96,6%) в виде коричневого масла.
Стадия 3
(Е)-Метил-3-(4-(6-гидрокси-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7d
Соединение 1е (2,44 г, 12,8 ммоль), 7 с (2,3 г, 8,6 ммоль) и триизопропилсилилхлорид (8,23 г, 42,69 ммоль) добавляли к 50 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 120°С и перемешивали в течение 3 часов. После прекращения нагревания реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 20 мл воды, впоследствии смесь перемешивали до однородного состояния и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 7d (2,3 г, выход 61,0%) в виде желтого твердого вещества.
Стадия 4
(Е)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-изопропилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 7е
Соединение 7d (2,3 г, 5,21 ммоль) растворяли в 50 мл дихлорметана, затем добавляли 2,6-лутидин (840 мг, 7,82 ммоль). После завершения добавления реакционную смесь охлаждали до 0°С в ледяной бане. Впоследствии по каплям добавляли трифторметансульфоновый ангидрид (1,91 г, 6,77 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 20 мл воды, и реакционный раствор экстрагировали дихлорметаном (20 мл × 2). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 7е (1,36 г, выход 45,5%) в виде желтого твердого вещества.
Стадия 5
(E)-Метил-3-(4-((1R,3R/1S,3S)-6-(1-этил-1Н-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 7f
Соединение 7е (300 мг, 0,52 ммоль),
1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (173 мг, 0,78 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (38 мг, 0,052 ммоль) растворяли в 4,8 мл смеси 1,4-диоксана и воды (V:V составляет 7:1), впоследствии добавляли 2М раствор карбоната натрия (0,52 мл, 1,04 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 40 минут. После охлаждения до комнатной температуры добавляли 10 мл воды, и смесь экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением неочищенного указанного в заголовке соединения 7f (200 мг, выход 74,1%) в виде светло-желтого твердого вещества.
Стадия 6
(E)-3-(4-((1R,3R/1S,3S)-6-(1-Этил-1Н-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 7
Соединение 7f (200 мг, 0,38 ммоль) растворяли в 12,8 мл смеси метанола и тетрагидрофурана (V:V составляет 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (1 мл, 2 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 10 мл воды, впоследствии смесь перемешивали до однородного состояния. Впоследствии по каплям добавляли 2 н. соляную кислоту для доведения рН реакционного раствора до 3. Смесь экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл × 1), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 7 (180 мг, выход 92,3%) в виде желтого твердого вещества.
MC m/z (ИЭР): 504,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.76 (s, 1Н), 7.65 (d, 1Н), 7.62 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.06 (d, 2Н), 6.73-6.77 (m, 2Н), 6.34 (d, 1Н), 5.68 (s, 1Н), 4.47-4.53 (m, 1Н), 4.20 (q, 2Н), 3.32-3.43 (m, 1Н), 2.70-2.81 (m, 2Н), 1.51 (t, 3Н), 1.19 (d, 6Н), 1.06 (S, 3Н).
Примеры 8, 9
(E)-3-(4-((1R,3R)-6-(1-Этил-1Н-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 8
(E)-3-(4-((1S,3S)-6-(1-Этил-1Н-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 9
Соединение 7 (500 мг, 0,96 ммоль) подвергали хиральному разделению (условия разделения: Superchiral S-AD, внутренний диаметр 2 см × 25 см, 5 мкм; подвижная фаза: диоксид углерода: этанол: диэтиламин составляет 60:40:0,05, скорость потока: 50 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанных в заголовке соединений 8 (160 мг, желтое твердое вещество) и 9 (160 мг, желтое твердое вещество).
Соединение 8:
MC m/z (ИЭР): 504,5 [М+1]
Анализ хиральной ВЭЖХ: время удерживания 9,84 минут, хиральная чистота: 100% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 2 см × 25 см, 5 мкм; подвижная фаза: диоксид углерода: этанол: диэтиламин составляет 60:40:0,05).
1Н ЯМР (400 МГц, CDCl3) δ 7.76 (s, 1Н), 7.65 (d, 1Н), 7.62 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.06 (d, 2Н), 6.73-6.77 (m, 2Н), 6.34 (d, 1Н), 5.68 (s, 1Н), 4.47-4.53 (m, 1Н), 4.20 (q, 2Н), 3.32-3.43 (m, 1Н), 2.70-2.81 (m, 2Н), 1.51 (t, 3Н), 1.19 (d, 6Н), 1.06 (s, 3Н).
Соединение 9:
МС m/z(ИЭР): 508,1 [М+1]
Анализ хиральной ВЭЖХ: время удерживания 14,26 минут, хиральная чистота: 99,0% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см × 25 см, 5 мкм; подвижная фаза: диоксид углерода: этанол: диэтиламин составляет 60:40:0,05).
1Н ЯМР (400 МГц, CDCl3) δ 7.76 (s, 1Н), 7.65 (d, 1Н), 7.62 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.06 (d, 2Н), 6.73-6.77 (m, 2Н), 6.34 (d, 1Н), 5.68 (s, 1Н), 4.47-4.53 (m, 1Н), 4.20 (q, 2Н), 3.32-3.43 (m, 1Н), 2.70-2.81 (m, 2Н), 1.51 (t, 3Н), 1.19 (d, 6Н), 1.06 (S, 3Н).
Пример 10
(Е)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропилфенил)-3-метил-6-(1-метил-1H-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
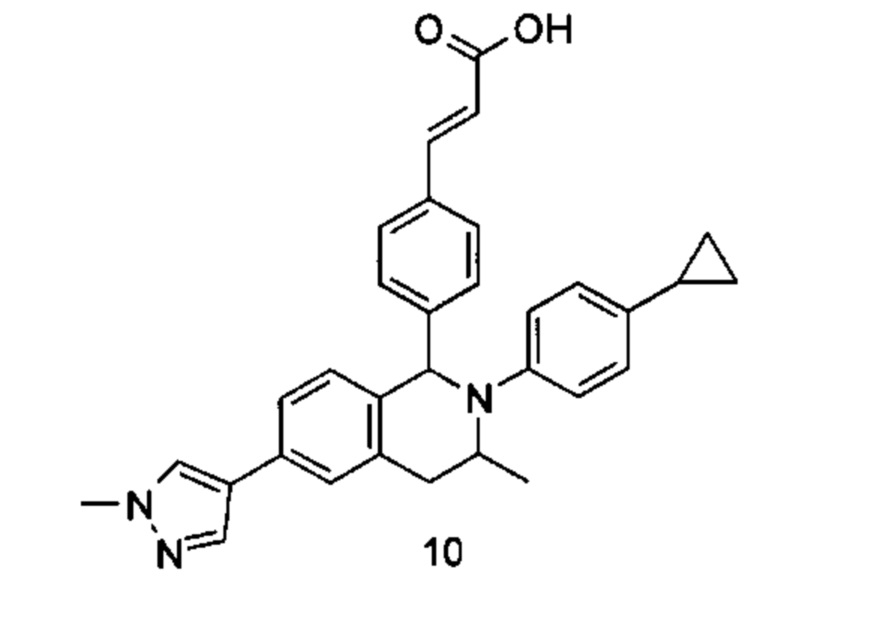
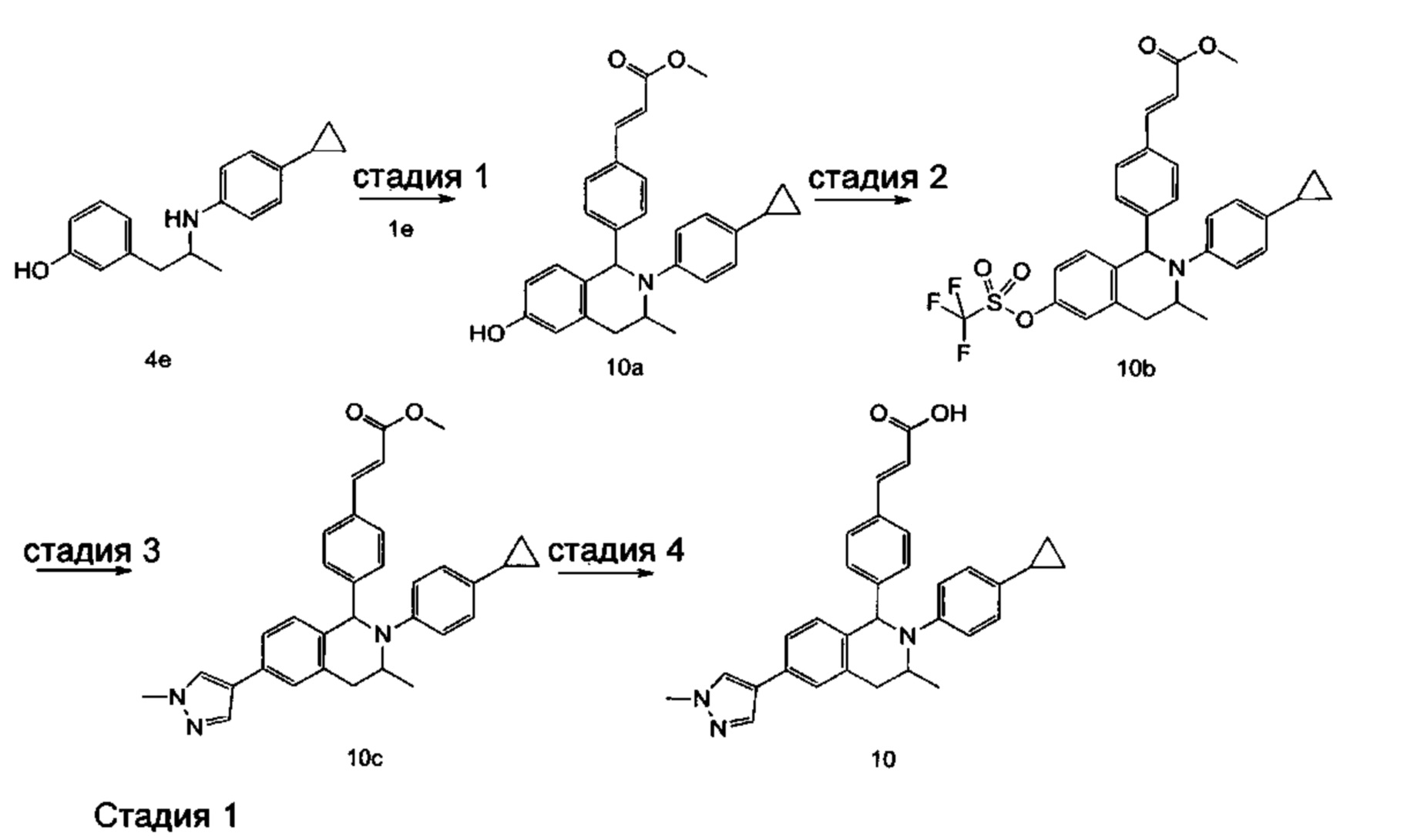
(E)-Метил-3-(4-(2-(4-циклопропилфенил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 10а
Соединение 4е (540 мг, 2,02 ммоль), 1е (576 мг, 3,03 ммоль) и триизопропилсилилхлорид (1,95 г, 10,10 ммоль) добавляли к 10 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 120°C и перемешивали в течение 3 часов. Реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. К полученному в результате остатку добавляли 20 мл воды, впоследствии смесь перемешивали до однородного состояния и экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 10а (490 мг, выход 55,2%) в виде коричневого твердого вещества.
Стадия 2
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-циклопропилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 10b
Соединение 10а (490 мг, 1,11 ммоль) растворяли в 10 мл дихлорметана, затем последовательно добавляли 2,6-лутидин (180 мг, 1,67 ммоль) и трифторметансульфоновый ангидрид (409 мг, 1,45 ммоль) в ледяной бане. После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов. Впоследствии для гашения реакции добавляли 10 мл воды, и реакционный раствор экстрагировали дихлорметаном (10 мл×2). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 10b (230 мг, выход 36,3%) в виде желтого твердого вещества.
Стадия 3
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-циклопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-12,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 10с
Соединение 10b (100 мг, 0,17 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (55 мг, 0,26 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (12 мг, 0,017 ммоль) растворяли в 1,6 мл смеси 1,4-диоксана и воды (V:V составляет 7:1), впоследствии добавляли 2М раствор карбоната натрия (0,17 мл, 0,34 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°C в течение 30 часов. После охлаждения до комнатной температуры добавляли 10 мл воды, и смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 10 с (55 мг, выход 64%) в виде желтого твердого вещества.
Стадия 4
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 10
Соединение 10с (55 мг, 0,1 ммоль) растворяли в 4 мл смеси метанола и тетрагидрофурана (V:V составляет 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (0,25 мл, 0,5 ммоль). После завершения добавления реакционную смесь перемешивали в течение 60 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 10 мл воды, впоследствии смесь перемешивали до однородного состояния. Впоследствии по каплям добавляли 2 н. соляную кислоту для доведения рН реакционного раствора до 2~3. Смесь экстрагировали этилацетатом (10 мл×3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 10 (50 мг, выход 100%) в виде желтого твердого вещества.
МС m/z (ИЭР): 490,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.65 (d, 1Н), 7.58 (s, 1Н), 7.38 (s, 3Н), 7.30 (s, 2Н), 7.24 (s, 2Н), 6.92 (d, 2Н), 6.71-6.73 (m, 2Н), 6.33 (d, 1Н), 5.68 (s, 1Н), 4.46 (m, 1Н), 3.93 (s, 3Н), 3.32-3.43 (m, 1Н), 2.72 (d, 1Н), 1.76-1.79 (m, 1Н), 1.05 (d, 3Н), 0.83 (m, 2Н), 0.57 (m, 2Н).
Примеры 11, 12
(Е)-3-(4-((1R,3R)-2-(4-Циклопропилфенил)-3-метил-6-(1-метил-1H-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 11
(E)-3-(4-((1S,3S)-2-(4-Циклопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 12
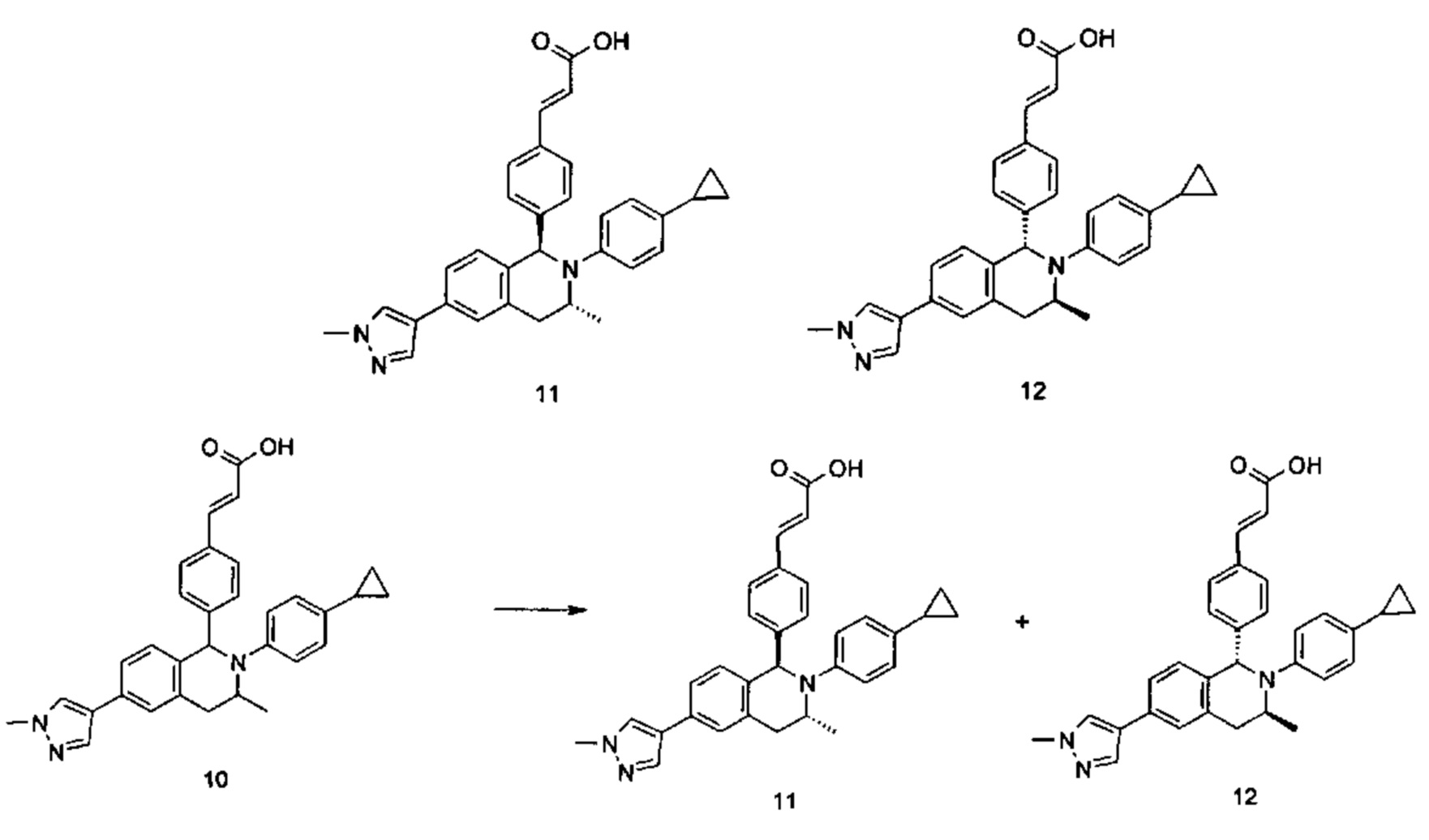
Соединение 10 (390 мг, 0,797 ммоль) подвергали хиральному разделению (условия разделения: хиральная колонка: Superchiral S-AD, внутренний диаметр 2 см * 25 см, 5 мкм; подвижная фаза: диоксид углерода: этанол составляет 60:40, скорость потока: 50 мл/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанных в заголовке соединений 11 (165 мг, желтое твердое вещество) и 12 (165 мг, желтое твердое вещество).
Пример 11:
МС m/z (ИЭР): 490,5 [М+1]
Анализ хиральной ВЭЖХ: время удерживания 8,822 минут, хиральная чистота: 100% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см * 15 см, 5 мкм; подвижная фаза: диоксид углерода: этанол составляет 60:40).
1Н ЯМР (400 МГц, CD3OD) δ 7.92 (s, 1Н), 7.80 (d, 1Н), 7.58 (d, 1Н), 7.36-7.42 (m, 7Н), 6.90 (d, 2Н), 6.77 (d, 2Н), 6.36 (d, 1Н), 5.74 (s, 1Н), 4.48-4.52 (m, 1Н), 3.92 (s, 3Н), 3.38-3.43 (m, 1Н), 2.82-2.77 (d, 1Н), 1.73-1.80 (m, 1Н), 1.04 (d, 3Н), 0.81-0.84 (m, 2Н), 0.55-0.53 (m, 2Н).
Пример 12:
МС m/z (ИЭР): 490,5 [М+1]
Анализ хиральной ВЭЖХ: время удерживания 12,539 минут, хиральная чистота: 99,4% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см * 15 см, 5 мкм; подвижная фаза: диоксид углерода: этанол составляет 60:40).
1Н ЯМР (400 МГц, CD3OD) δ 7.92 (s, 1Н), 7.80 (d, 1Н), 7.58 (d, 1Н), 7.36-7.42 (m, 7Н), 6.90 (d, 2Н), 6.77 (d, 2Н), 6.36 (d, 1Н), 5.74 (s, 1Н), 4.48-4.52 (m, 1Н), 3.92 (s, 3Н), 3.38-3.43 (m, 1Н), 2.82-2.77 (d, 1Н), 1.73-1.80 (m, 1Н), 1.04 (d, 3Н), 0.81-0.84 (m, 2Н), 0.55-0.53 (m, 2Н).
Пример 13
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота
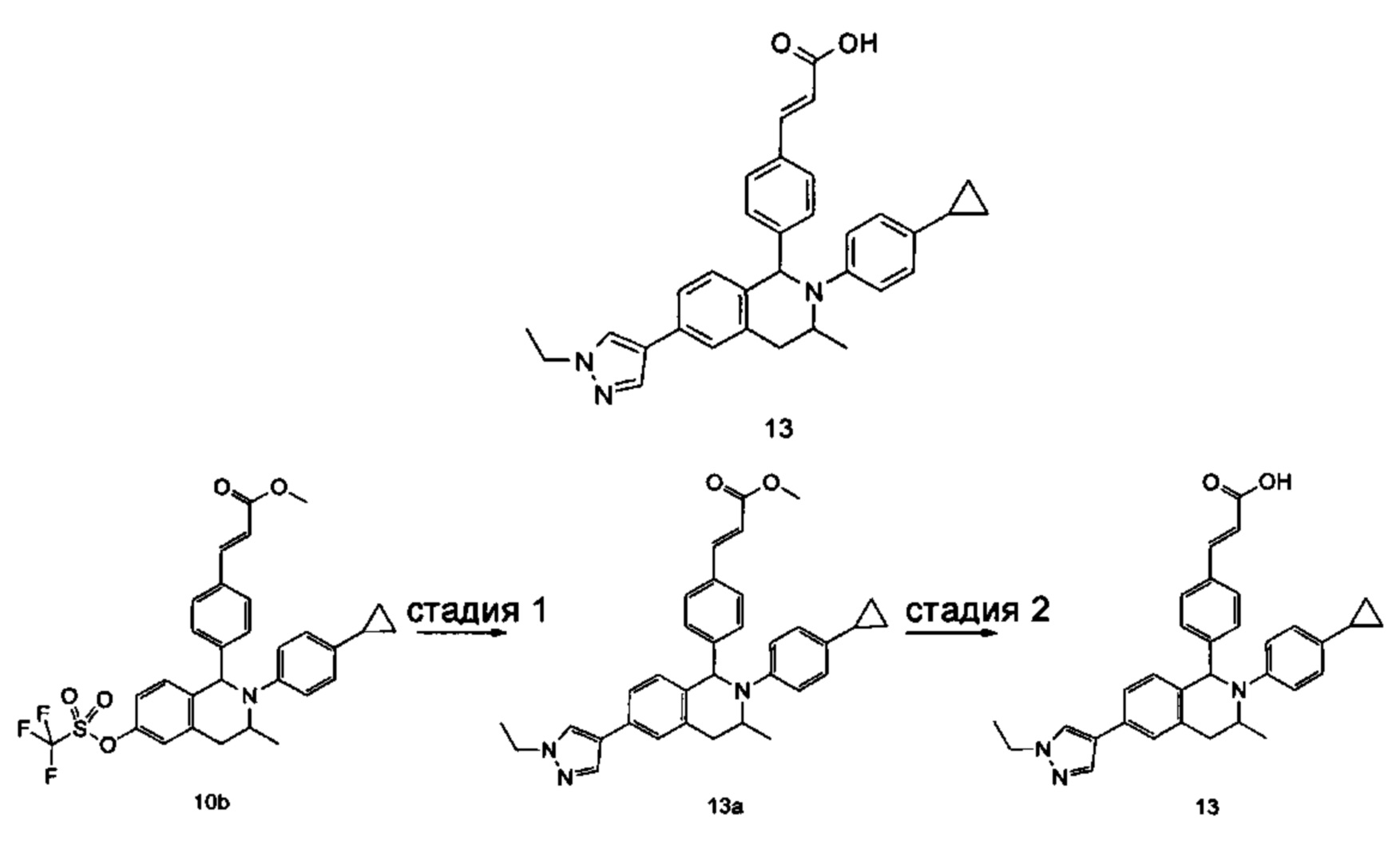
Стадия 1
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 13а
Соединение 10b (485 мг, 0,85 ммоль),
1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (283 мг, 1,275 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (63 мг, 0,085 ммоль) растворяли в 8 мл смеси 1,4-диоксана и воды (V:V составляет 7:1), впоследствии добавляли 2М раствор карбоната натрия (0,85 мл, 1,7 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 1 часа. После охлаждения до комнатной температуры добавляли 20 мл воды, и смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением неочищенного указанного в заголовке соединения 13а (352 мг, выход 80%) в виде желтого твердого вещества.
МС m/z (ИЭР): 518,5 [М+1]
Стадия 2
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 13
Соединение 13а (350 мг, 0,676 ммоль) растворяли в 28 мл смеси метанола и тетрагидрофурана (V:V составляет 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (1,7 мл, 3,38 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 10 мл воды, впоследствии смесь перемешивали до однородного состояния. Впоследствии по каплям добавляли 2 н. соляную кислоту для доведения рН реакционного раствора до 2~3. Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали под пониженным давлением. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 13 (260 мг, выход 76%) в виде желтого твердого вещества.
МС m/z (ИЭР): 504,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.95 (s, 1Н), 7.79 (s, 1Н), 7.54-7.58 (d, 1Н), 7.32-7.42 (m, 7Н), 6.86-6.88 (d, 2Н), 6.75-6.77 (d, 2Н), 6.34-6.38 (d, 1Н), 5.72 (s, 1Н), 4.72 (m, 1H), 4.16-4.22 (m, 2H), 3.36-3.41 (m, 1H), 2.75-2.79 (d, 1H), 1.73-1.77 (m, 1H), 1.45-1.49 (m, 3Н), 1.00-1.02 (d, 3Н), 0.78-0.80 (m, 2H), 0.50-0.51 (m, 2H).
Примеры 14, 15
(Е)-3-(4-((1R,3R)-2-(4-Циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 14
(E)-3-(4-((1S,3S)-2-(4-Циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 15
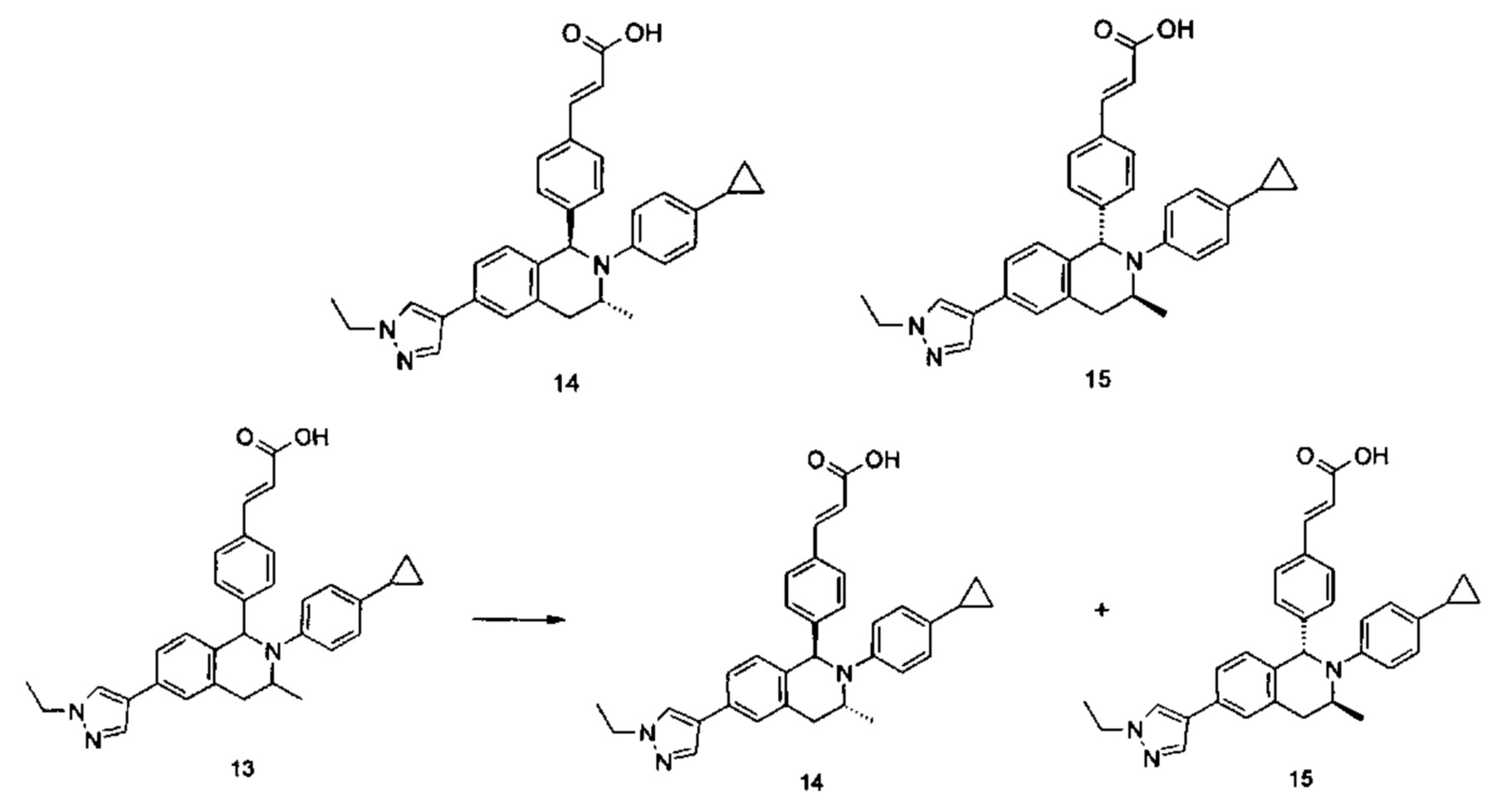
Соединение 13 (250 мг, 0,497 ммоль) подвергали хиральному разделению (условия разделения: хиральная колонка: Superchiral S-AD, внутренний диаметр 2 см * 25 см, 5 мкм; подвижная фаза: диоксид углерода : этанол составляет 60:40, скорость потока: 50 г/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанных в заголовке соединений 14 (105 мг, желтое твердое вещество) и 15 (110 мг, желтое твердое вещество).
Соединение 14:
МС m/z (ИЭР): 504,5 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 9,317 минут, хиральная чистота: 100% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см * 15 см, 5 мкм; подвижная фаза: диоксид углерода: этанол составляет 60:40).
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.95 (s, 1Н), 7.79 (s, 1Н), 7.54-7.58 (d, 1Н), 7.32-7.42 (m, 7Н), 6.86-6.88 (d, 2Н), 6.75-6.77 (d, 2Н), 6.34-6.38 (d, 1Н), 5.72 (s, 1Н), 4.72 (m, 1Н), 4.16-4.22 (m, 2Н), 3.36-3.41 (m, 1Н), 2.75-2.79 (d, 1Н), 1.73-1.77 (m, 1Н), 1.45-1.49 (m, 3Н), 1.00-1.02 (d, 3Н), 0.78-0.80 (m, 2Н), 0.50-0.51 (m, 2Н).
Соединение:
МС m/z (ИЭР): 504,5 [М+1];
Анализ хиральной ВЭЖХ: время удерживания 14,061 минут, хиральная чистота: 100% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см * 15 см, 5 мкм; подвижная фаза: диоксид углерода: этанол составляет 60:40).
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.95 (s, 1Н), 7.79 (s, 1Н), 7.54-7.58 (d, 1Н), 7.32-7.42 (m, 7Н), 6.86-6.88 (d, 2Н), 6.75-6.77 (d, 2Н), 6.34-6.38 (d, 1Н), 5.72 (s, 1Н), 4.72 (m, 1Н), 4.16-4.22 (m, 2Н), 3.36-3.41 (m, 1Н), 2.75-2.79 (d, 1Н), 1.73-1.77 (m, 1Н), 1.45-1.49 (m, 3Н), 1.00-1.02 (d, 3Н), 0.78-0.80 (m, 2Н), 0.50-0.51 (m, 2Н).
Пример 16
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Изопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 16
В соответствии с путем синтеза примера 7 исходные вещества, используемые на стадии 5, заменяли соединением 7е и
1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, соответственно, получали указанное в заголовке соединение 16.
МС m/z (ИЭР): 492,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.67 (d, 1Н), 7.58 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.25 (s, 2Н), 7.06 (d, 2Н), 6.73-6.77 (m, 2Н), 6.33 (d, 1Н), 5.68 (s, 1Н), 4.47-4.53 (m, 1Н), 3.93 (s, 3Н), 3.32-3.43 (m, 1Н), 2.71-2.83 (m, 2Н), 1.19 (d, 6H), 1.06 (s, 3Н).
Пример 17
(Е)-3-(4-((1R,3R/1S,3S)-6-(1-Этил-1Н-пиразол-4-ил)-2-(4-этилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 17
В соответствии с путем синтеза примера 1 исходные вещества, используемые на стадии 5, заменяли соединением 1g и 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, соответственно, получали указанное в заголовке соединение 17.
МС m/z (ИЭР): 492,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.17 (d, 1Н), 7.83 (d, 1Н), 7.33-7.55 (m, 8Н), 6.98 (d, 2Н), 6.73 (d, 2Н), 6.43 (d, 1Н), 5.81 (s, 1Н), 4.54-4.63 (m, 1Н), 4.10-4.21 (m, 2Н), 3.32-3.35 (m, 1Н), 2.78 (d, 1Н), 2.39-2.51 (m, 2Н), 1.37-1.41 (m, 3Н), 1.08-1.12 (m, 3Н), 0.95 (d, 3Н).
Пример 18
(Е)-3-(4-((1R,3R/1S,3S)-6-(1-Этил-1Н-пиразол-4-ил)-2-(4-изобутилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 18
В соответствии с путем синтеза примера 7 исходные вещества, используемые на стадии 1, заменяли соединением 1а и 4-изопропиланилином, соответственно, получали указанное в заголовке соединение 18.
MC m/z (ИЭР): 520,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.96 (s, 1Н), 7.79 (s, 1Н), 7.57 (d, 1Н), 7.37-7.44 (m, 6Н), 6.92 (d, 2Н), 6.77 (d, 2Н), 6.37 (d, 1Н), 5.74 (s, 1Н), 4.51 (m, 1Н), 4.19 (q, 2Н), 3.38 (dd, 1Н), 2.78 (dd, 1Н), 2.31 (d, 2Н), 1.75 (m, 1Н), 1.47 (t, 3Н), 1.03 (d, 3Н), 0.90 (m, 1Н), 0.85 (d, 6Н).
Пример 19
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Изобутилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 19
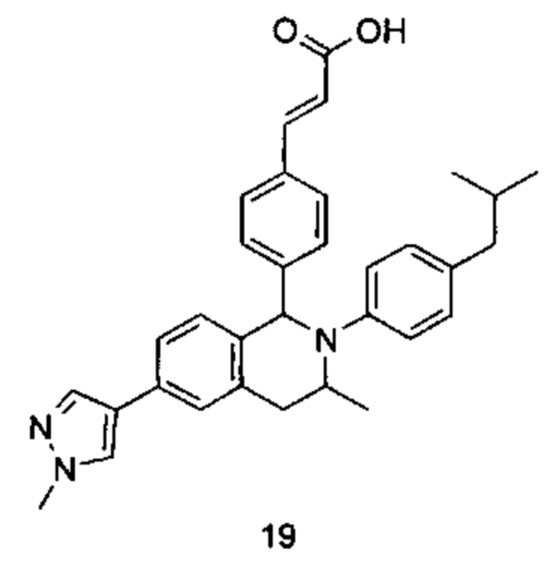
В соответствии с путем синтеза примера 1 исходные вещества, используемые на стадии 1, заменяли соединением 1а и 4-изопропиланилином, соответственно, получали указанное в заголовке соединение 19.
MC m/z (ИЭР): 506,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.90 (s, 1Н), 7.78 (s, 1Н), 7.55 (d, 1Н), 7.389-7.34 (m, 6Н), 6.92 (d, 2Н), 6.76 (d, 2Н), 6.36 (d, 1Н), 5.73 (s, 1Н), 4.50 (m, 1Н), 3.90 (s, 3Н), 3.38 (dd, 1Н), 2.77 (dd, 1Н), 2.31 (d, 2Н), 1.74 (m, 1Н), 1.02 (d, 3Н), 0.90 (m, 1Н), 0.85 (d, 6Н).
Пример 20
(E)-3-(4-((1R,3R/1S,3S)-3-Метил-6-(1-метил-1Н-пиразол-4-ил)-2-(4-пропилфенил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 20
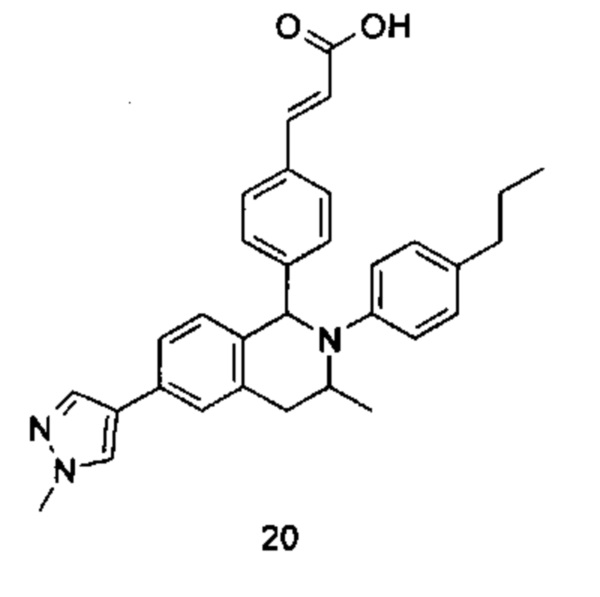
В соответствии с путем синтеза примера 1 исходные вещества, используемые на стадии 1, заменяли соединением 1а и 4-изопропиланилином, соответственно, получали указанное в заголовке соединение 20.
МС m/z(M3P): 492,5 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.08 (s, 1Н), 7.81 (s, 1Н), 7.43-7.53 (m, 6Н), 7.37-7.39 (m, 2Н), 6.93-6.95 (d, 2Н), 6.70-6.72 (d, 2Н), 6.36-6.42 (d, 1Н), 5.80 (s, 1Н), 4.51-4.52 (m, 1Н), 3.81 (s, 3Н), 3.32-3.34 (m, 2Н), 2.74-2.78 (d, 1Н), 2.35-2.39 (m, 2Н), 1.45-1.51 (m, 2Н), 0.92-0.94 (d, 2Н), 0.82-0.86 (m, 3Н).
Пример 21
(E)-3-(4-((1R,3R)-2-(4-Циклопропилфенил)-6-(1-этил-d6-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 21
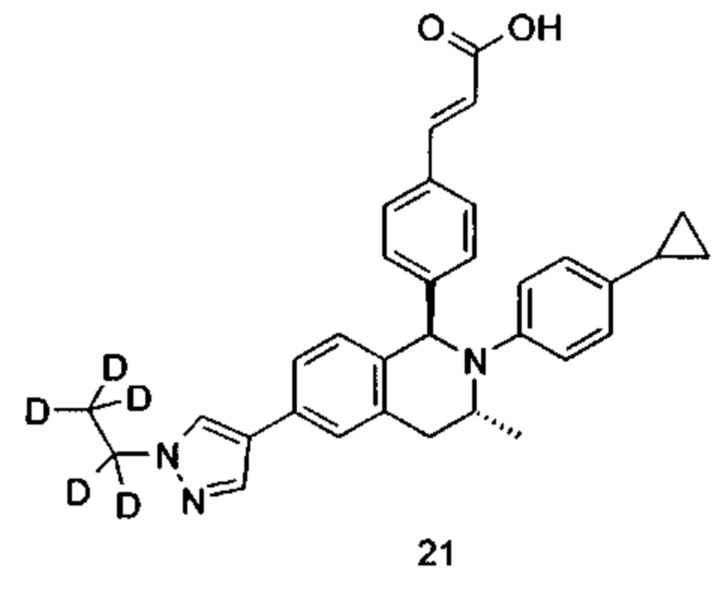
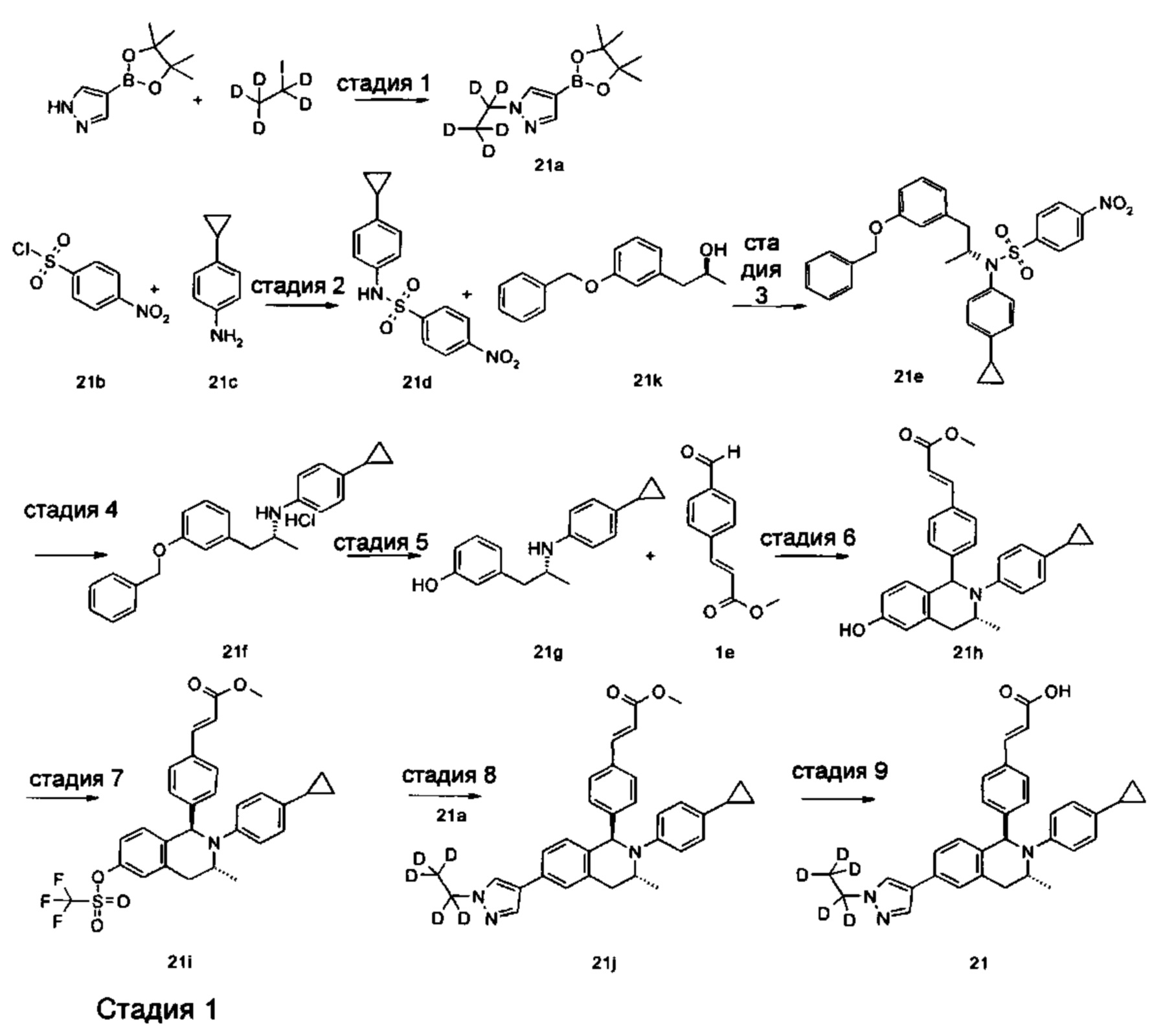
1-Этил-d6-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол 21a
4-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (2 г, 10,3 ммоль) и карбонат калия (2,14 г, 15.5 ммоль) добавляли к 20 мл N,N-диметилформамида, впоследствии добавляли йодэтан-d5 (2 г, 12,4 ммоль). Реакционную смесь перемешивали в течение 16 часов. Поскольку исходные вещества не прореагировали полностью, реакционную смесь нагревали до 50°C и перемешивали в течение 3 часов. Реакционный раствор фильтровали, и фильтрат концентрировали при пониженном давлении. Остаток промывали 50 мл этилацетата и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 21а (2,13 г, смесь твердого и жидкого вещества), которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 228,5 [М+1]
Стадия 2
N-(4-Циклопропилфенил)-4-нитробензолсульфонамид 21d
4-Циклопропиланилин 21 с (80,8 г, 0,61 моль, получен хорошо известным способом, описанным в Journal of the American Chemical Society, 2016, 138(27), 8533-8537) и 2,6-лутидин (97,5 г, 0,91 моль) растворяли в 400 мл дихлорметана. Реакционный раствор охлаждали до 0-5°C в атмосфере аргона. Впоследствии к указанному выше реакционному раствору по каплям добавляли раствор 4-нитробензол-1-сульфонилхлорида 21b (134,5 г, 0,61 моль) в 40 мл дихлорметана. После завершения добавления реакционную смесь перемешивали в течение 20 минут. Реакционный раствор нагревали до комнатной температуры и добавляли 500 мл воды. Смесь перемешивали до выпадения в осадок твердого вещества. После фильтрования фильтрационный кек и фильтрат собирали с получением неочищенного указанного в заголовке соединения 21d (340,3 г, черная вязкая жидкость), которое использовали на следующей стадии без дополнительной очистки.
Стадия 3
(R)-N-(1-(3-(Бензилокси)фенил)пропан-2-ил)-N-(4-циклопропилфенил)-4-нитробензолсульфонамид 21е
Неочищенное соединение 21d (305,5 г, 0,78 моль), (S)-1-(3-(бензилокси)фенил)пропан-2-ол 21k (188,8 г, 0,78 моль, получен способом, раскрытым в заявке на патент WO 2014133361 A1) и трифенилфосфин (306,5 г, 1,17 моль) растворяли в 2 л тетрагидрофурана. Реакционный раствор охлаждали до 0°C в атмосфере аргона, впоследствии к указанному выше реакционному раствору по каплям добавляли диэтилазодикарбоксилат (203,5 г, 1,17 моль). Температуру реакционной смеси поддерживали при 0-10°C. После завершения добавления реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Реакционный раствор концентрировали при пониженном давлении и последовательно растирали с 2 л трет-бутилметилового эфира и 2 л петролейного эфира с получением неочищенного указанного в заголовке соединения 21е (701,7 г) в виде светло-желтого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 4
(R)-N-(1-(3-(Бензилокси)фенил)пропан-2-ил)-4-циклопропиланилина гидрохлорид 21f
Неочищенное соединение 21е (550 г, 0,66 моль), гидрат гидроксида лития (273,1 г, 6,65 моль) и 2-амино-3-меркаптопропионовую кислоту (96,6 г, 0,80 моль) растворяли в 3,5 л N,N-диметилформамида. После перемешивания в течение 18 часов добавляли 6 мл воды, и реакционный раствор экстрагировали этилацетатом (1,5 л×3). Органические фазы объединяли, промывали водой (500 мл) и концентрировали при пониженном давлении. К полученному в результате остатку добавляли 170 мл 6 н. соляной кислоты и 2 л смеси этилацетата и петролейного эфира (V:V составляет 1:1). Смесь фильтровали, и фильтрационный кек собирали с получением неочищенного указанного в заголовке соединения 21h (120,1 г) в виде светло-желтого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 5
(R)-3-(2-((4-Циклопропилфенил)амино)пропил)фенол 21g
Неочищенное соединение 21f (120,1 г, 0,30 моль) и йодид натрия (182,8 г, 1,22 моль) растворяли в 840 мл ацетонитрила, впоследствии по каплям добавляли триметилсилилхлорид (132,5 г, 1,22 моль) в атмосфере аргона. Смесь перемешивали в течение 12 часов и добавляли 360 мл 1 н. соляной кислоты. Реакционную смесь перемешивали в течение 5 минут. Добавляли насыщенный раствор дитионита натрия, пока не исчезнет красный цвет йода. Смесь экстрагировали этилацетатом (300 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (200 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 21g (140,6 г) в виде бесцветного вязкого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 6
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-циклопропилфенил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 21h
Неочищенное соединение 21g (127,2 г, 0,48 ммоль), соединение 1е (144,8 г, 0,76 моль) и триизопропилсилилхлорид (183,4 г, 0,95 моль) добавляли к 1200 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 140°C в атмосфере аргона и перемешивали в течение 2 часов. После прекращения нагревания реакционный раствор концентрировали при пониженном давлении. Затем последовательно добавляли 500 мл воды и 500 мл этилацетата и фильтровали. Две фазы разделяли, и водную фазу промывали этилацетатом (300 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (300 мл), высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции C с получением указанного в заголовке соединения 21h (78,7 мг, выход 37,6%) в виде оранжевого твердого вещества.
Стадия 7
(E)-Метил-3-(4-((1R,3R)-2-(4-Циклопропилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 21i
Соединение 21h (20 г, 45,5 ммоль) и 2,6-лутидин (7,3 г, 68,3 ммоль) растворяли в 400 мл дихлорметана. После завершения добавления реакционную смесь охлаждали до 0°C в ледяной бане в атмосфере аргона. Впоследствии по каплям добавляли трифторметансульфоновый ангидрид (16,7 г, 59,2 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Для гашения реакции добавляли 100 мл воды. Две фазы разделяли, и водную фазу экстрагировали дихлорметаном (100 мл). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (100 мл), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции C с получением указанного в заголовке соединения 21i (4,5 г, выход 17,3%) в виде оранжевого вязкого твердого вещества.
МС m/z (ИЭР): 572,2 [М+1]
Стадия 8
(Е)-Метил-3-(4-((1R,3R)-2-(4-циклопропилфенил)-6-(1-этил-d5-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 21j
Неочищенное соединение 21а (2,04 г, 9 ммоль), 21i (4,1 г, 7,2 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (1,31 г, 1,8 ммоль) растворяли в 44 мл смеси 1,4-диоксана и воды (V:V составляет 10:1), впоследствии добавляли карбонат калия (3,72 мл, 26,9 ммоль). Смесь перемешивали в течение 12 часов при 80°С. После охлаждения до комнатной температуры добавляли 100 мл воды, и смесь экстрагировали этилацетатом (100 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции C с получением неочищенного указанного в заголовке соединения 21j (1,8 г, выход 38,3%) в виде желтого твердого вещества.
МС m/z (ИЭР): 523,6 [М+1]
Стадия 9
(E)-3-(4-((1R,3R)-2-(4-Циклопропилфенил)-6-(1-этил-d5-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 21
Соединение 21j (1,71 г, 3,3 ммоль) растворяли в 24 мл смеси метанола и тетрагидрофурана (V:V составляет 1:5), впоследствии добавляли гидроксид натрия (982 мг, 24,5 ммоль) и 20 мл воды. Реакционную смесь перемешивали в течение 16 часов в атмосфере аргона в темноте. Реакционный раствор концентрировали при пониженном давлении. К данному остатку добавляли 20 мл насыщенного раствора лимонной кислоты, впоследствии смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали высокоэффективной жидкостной хроматографией с получением указанного в заголовке соединения 21 (1,05 г, выход 63,2%) в виде желтого твердого вещества.
МС m/z (ИЭР): 509,6 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 7.98 (s, 1Н), 7.82 (s, 1Н), 7.59 (d, 1Н), 7.41-7.33 (m, 7Н), 6.90 (d, 2Н), 6.79 (d, 2Н), 6.39 (d, 1Н), 5.76 (s, 1Н), 4.48-4.51 (m, 1Н), 3.42 (dd, 1Н), 2.82 (d, 1Н), 1.74-1.81 (m, 1Н), 1.04 (d, 3Н), 0.82-0.84 (d, 2Н), 0.53-0.54 (m, 2Н).
Пример 22
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропил-2-фторфенил)-6-(1-этил-1H-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 22
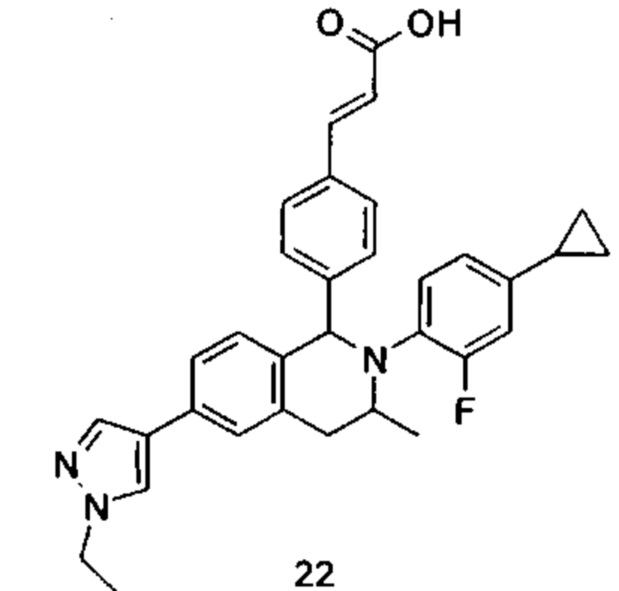
В соответствии с путем синтеза примера 7 используемое на стадии 1 исходное вещество 7а заменяли 4-циклопропил-2-фторанилином (получен хорошо известным способом, раскрытым в Tetrahedron Lett, 2002, 43, 6987), соответственно, получали указанное в заголовке соединение 22 (270 мг, желтое твердое вещество).
МС m/z (ИЭР): 522,6 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.96 (s, 1Н), 7.80 (s, 1Н), 7.57 (d, 1Н), 7.41-7.35 (m, 5Н), 7.24 (dd, 1Н), 7.07 (t, 1Н), 6.87 (d, 1Н), 6.69 (s, 1Н), 6.69-6.62 (m, 1Н), 6.38 (d, 1Н), 5.69 (s, 1H), 4.21 (q, 2H), 3.91 (dd, 1H), 3.59 (dd, 1H), 2.78 (dd, 1H), 1.78 (ddd, 1H), 1.49 (t, 3Н), 1.07 (d, 3Н), 0.93-0.84 (m, 2H), 0.61-0.51 (m, 2H).
Пример 23
(E)-3-(4-((1R,3R/1S,3S)-2-(2-Хлор-4-этилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 23
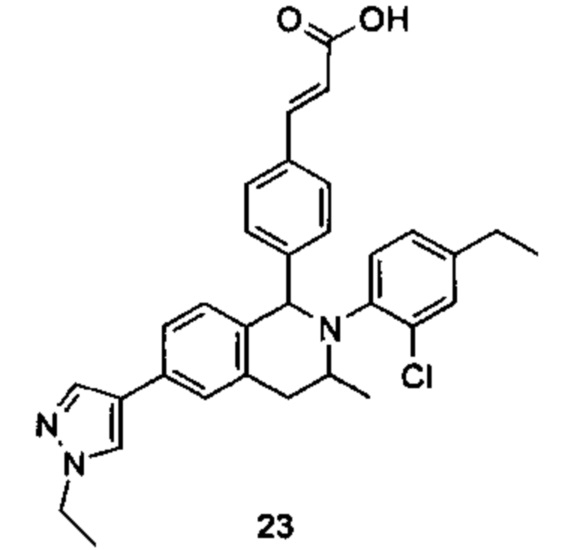
В соответствии с путем синтеза примера 7 используемое на стадии 1 исходное вещество 7а заменяли 2-хлор-4-этиланилином (получен хорошо известным способом, раскрытым в Journal of Chemical and Engineering Data, 1963, 8, 122-130), соответственно, получали указанное в заголовке соединение 23 (390 мг, желтое твердое вещество).
МС m/z (ИЭР): 527,1 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.96 (s, 1Н), 7.80 (s, 1Н), 7.54 (d, 1Н), 7.47-7.42 (m, 2Н), 7.41-7.35 (m, 3Н), 7.33-7.26 (m, 1Н), 7.21 (d, 1Н), 7.15 (s, 1Н), 6.95 (d, 1Н), 6.83 (d, 1Н), 6.36 (dd, 1Н), 5.75 (s, 1Н), 4.20 (q, 2Н), 3.90-3.85 (m, 1Н), 3.80-3.72 (m, 1Н), 2.77 (d, 1Н), 2.50 (q, 2Н), 1.48 (t, 3Н), 1.14 (t, 3Н), 1.08 (d, 3Н).
Пример 24
(Е)-3-(4-((1R,3R/1S,3S)-2-(2-Хлор-4-циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 24
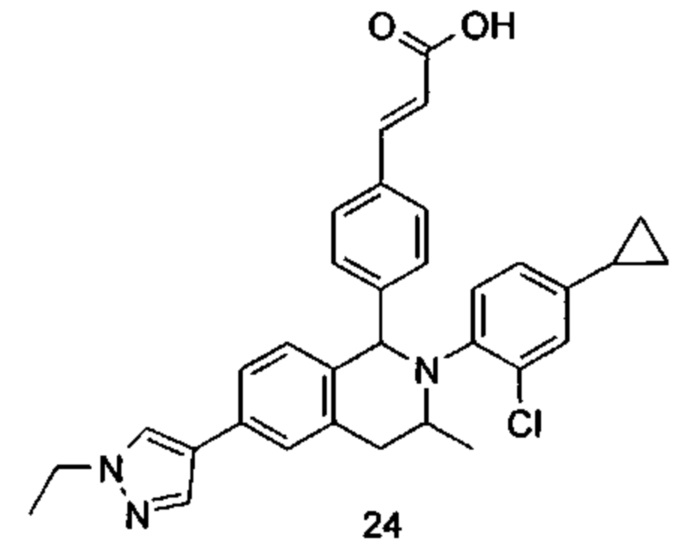
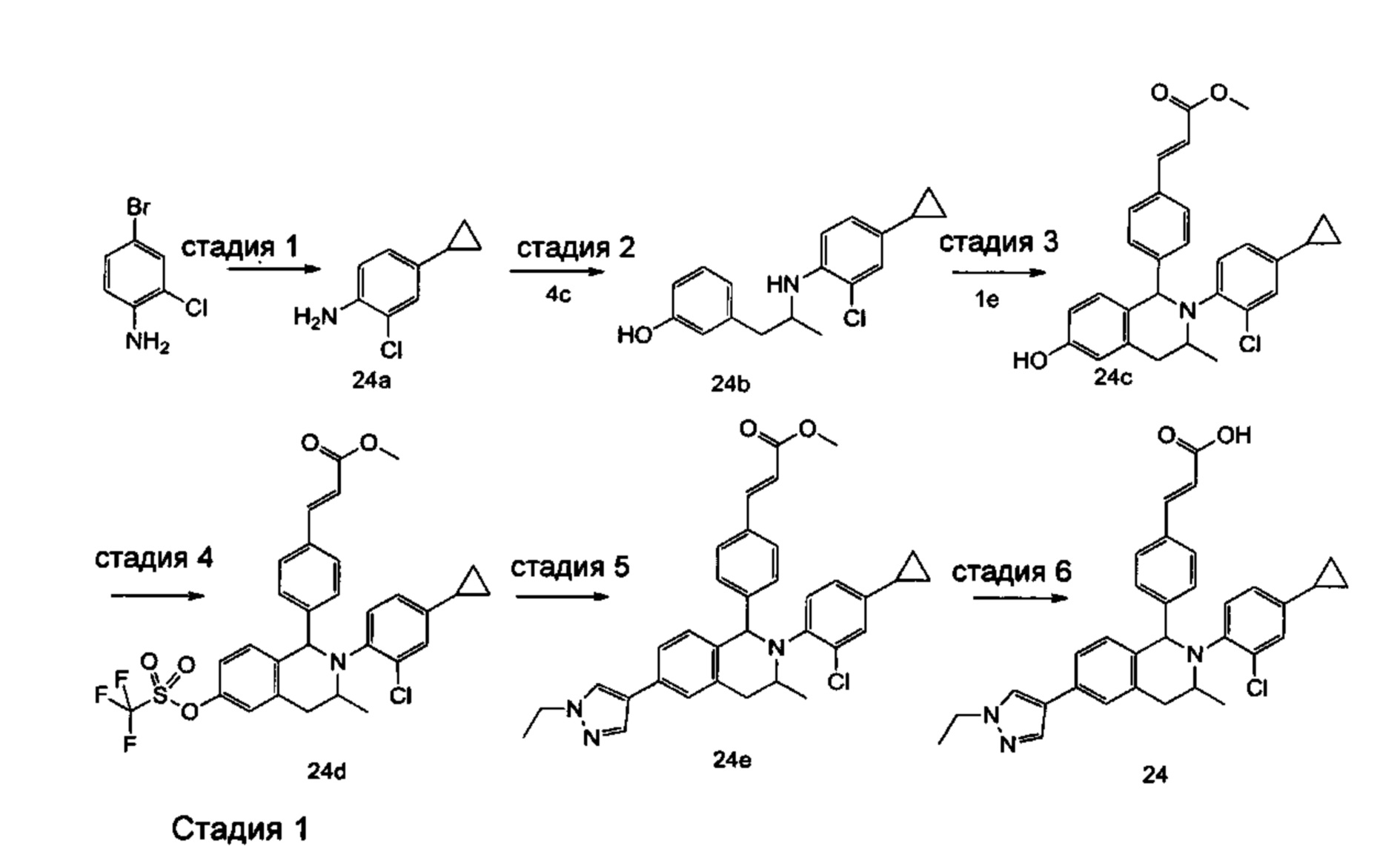
2-Хлор-4-циклопропиланилин 24а
4-Бром-2-хлоранилин (5 г, 24,2 ммоль), циклопропилбороновую кислоту (4 г, 48,4 ммоль), трициклогексилфосфин (680 мг, 2,42 ммоль), ацетат палладия (271 мг, 1,21 ммоль) и карбонат калия (15,4 г, 72,6 ммоль) добавляли к 130 мл смеси толуола и воды (V:V составляет 25:1). Смесь нагревали до 100°C и перемешивали в течение 12 часов. Реакционный раствор фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 24а (2,5 г, выход 61,7%) в виде желтой жидкости.
MC m/z (ИЭР): 168,1 [М+1]
Стадия 2
3-(2-((2-Хлор-4-изопропилфениламино)пропил)фенол 24b Соединение 4 с (2,46 г, 16,4 ммоль), соединение 24а (2,5 г, 14,9 ммоль) и триацетоксиборгидрид натрия (5,2 г, 24,6 ммоль) растворяли в 60 мл дихлорэтана. Смесь перемешивали в течение 12 часов. Для гашения реакции добавляли 20 мл воды. Реакционный раствор экстрагировали дихлорметаном (20 мл×2). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 24а (1,5 г, выход 33,5%) в виде желтого масла.
MC m/z (ИЭР): 302,1 [М+1]
Стадия 3
(Е)-Метил-3-(4-((1R,3R/1S,3S)-2-(2-хлор-4-циклопропилфенил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 24с
Соединение 24b (1,5 г, 5 ммоль), 1е (1,9 г, 10 ммоль) и триизопропилсилилхлорид (1,9 г, 10 ммоль) добавляли к 30 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 140°C и перемешивали в течение 3 часов. После прекращения нагревания реакционный раствор концентрировали при пониженном давлении. Впоследствии к полученному в результате остатку добавляли 20 мл воды, затем смесь перемешивали до однородного состояния и экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли и высушивали над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 24 с (1,5 г, выход 65,2%) в виде желтого твердого вещества.
МС m/z (ИЭР): 474,2 [М+1]
Стадия 4
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(2-хлор-4-циклопропилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 24d
Соединение 24с (1,5 г, 3,16 ммоль) and 2,6-лутидин (508 мг, 4,74 ммоль) растворяли в 30 мл дихлорметана. После охлаждения реакционной смеси до 0°C по каплям добавляли трифторметансульфоновый ангидрид (1,16 г, 4,11 ммоль). Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Для гашения реакционной смеси добавляли 20 мл воды, и реакционный раствор экстрагировали дихлорметаном (50 мл×2). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 24d (0,95 г, выход 49,7%) в виде желтого твердого вещества.
МС m/z (ИЭР): 606,2 [М+1]
Стадия 5
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(2-хлор-4-циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 24е
Соединение 24d (750 мг, 1,24 ммоль), 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол (412 мг, 1,86 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (91 мг, 0,124 ммоль) растворяли в 13,5 мл смеси 1,4-диоксана и воды (V:V составляет 8:1), впоследствии добавляли 2М раствор карбоната натрия (1,24 мл). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 45 минут. После охлаждения до комнатной температуры добавляли 20 мл воды, и смесь экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли и высушивали над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 24е (480 мг, выход 70,2%) в виде желтого твердого вещества.
МС m/z (ИЭР): 552,3 [М+1]
Стадия 6
(Е)-3-(4-((1R,3R/1S,3S)-2-(2-Хлор-4-циклопропилфенил)-6-(1-этил-1H-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 24
Соединение 24е (480 мг, 0,87 ммоль) растворяли в 10 мл смеси метанола и тетрагидрофурана (V:V составляет 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (2,17 мл). После завершения добавления реакционную смесь перемешивали в течение 12 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 10 мл воды, впоследствии смесь перемешивали до однородного состояния. Впоследствии по каплям добавляли 1 н. соляную кислоту для доведения рН реакционного раствора до 5. Смесь экстрагировали этилацетатом (30 мл×2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 24 (200 мг, выход 64,1%) в виде желтого твердого вещества.
МС m/z (ИЭР): 538,4 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.95 (s, 1Н), 7.79 (s, 1Н), 7.56 (d, 1Н), 7.47-7.40 (m, 2Н), 7.38-7.35 (m, 3Н), 7.27-7.20 (m, 2Н), 7.00 (s, 1Н), 6.83 (d, 2Н), 6.63 (d, 1Н), 5.73 (s, 1Н), 4.19 (q, 2Н), 3.87-3.83 (m, 1Н), 3.80-3.72 (m, 1Н), 2.81-2.71 (m, 1Н), 1.79-1.72 (m, 1Н), 1.48 (t, 3Н), 1.08 (d, 3Н), 0.88-0.86 (m, 2Н), 0.59-0.54 (m, 2Н).
Пример 25
(E)-3-(4-((1S,3R/1R,3S)-2-(4-Циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3-фторфенил)акриловая кислота 25
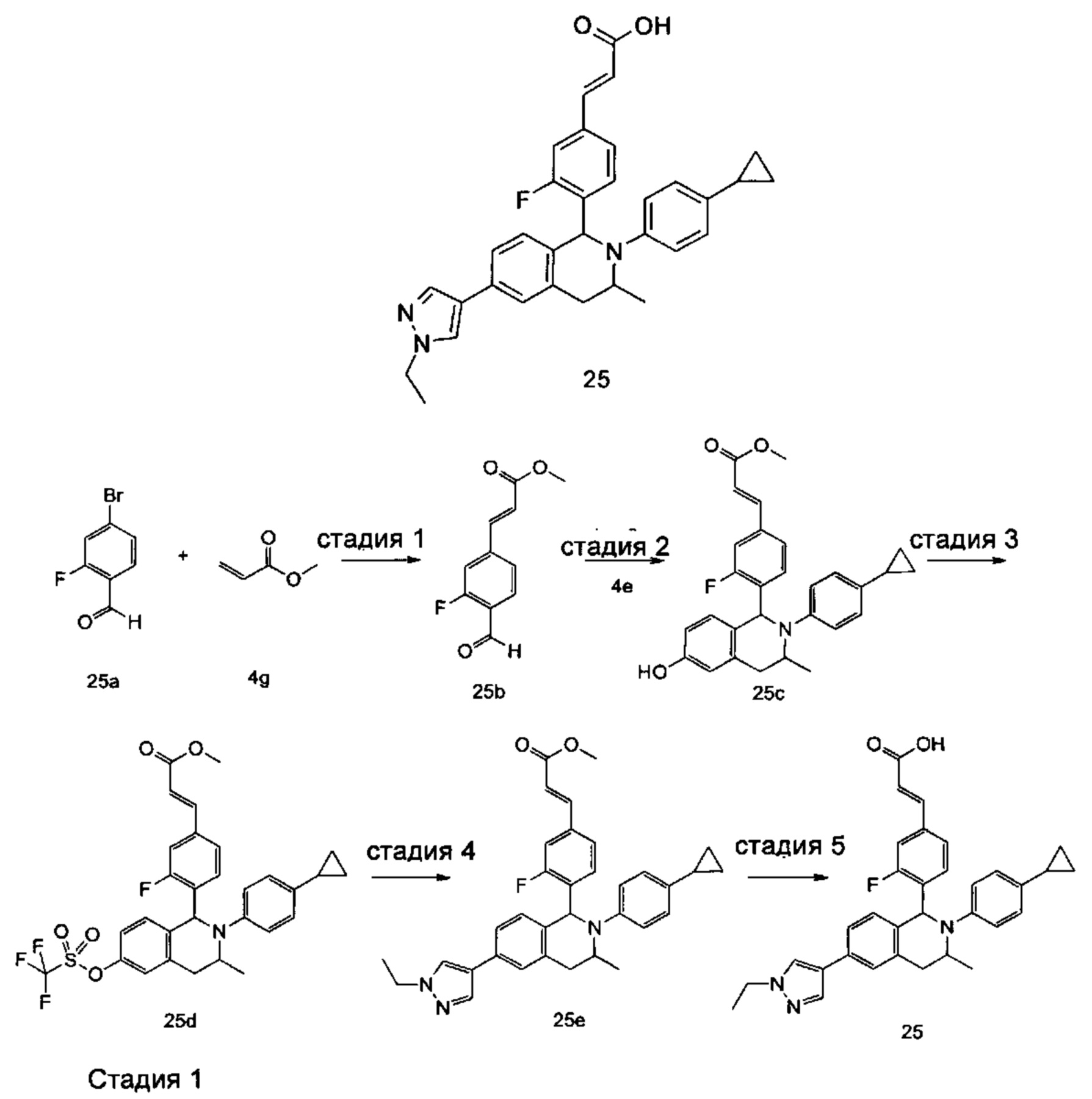
(Е)-Метил-3-(3-фтор-4-формилфенил)акрилат 25b
В атмосфере аргона добавляли 4-бром-2-фторбензальдегид 25а (10 г, 50 ммоль), соединение 4g (6,8 мл, 75 ммоль), трис(2-метилфенил)фосфин (1,52 г, 5 ммоль), ацетат палладия (561 мг, 2,5 ммоль) и триэтиламин (14 мл, 100 ммоль) к 100 мл N,N-диметилацетамида. После завершения добавления смесь нагревали до 80°C и перемешивали в течение 16 часов. После прекращения нагревания реакционный раствор естественным путем охлаждали до комнатной температуры, добавляли 300 мл воды и фильтровали. Фильтрационный кек последовательно промывали водой (50 мл×3) и н-гексаном (50 мл×3) и высушивали с получением указанного в заголовке соединения 25b (8 г, выход 77,0%) в виде желтого твердого вещества.
Стадия 2
(E)-Метил-3-(4-((1S,3R/1R,3S)-2-(4-циклопропилфенил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3-фторфенил)акрилат 25с
Соединение 4е (2,02 г, 7,6 ммоль), 25b (3,2 г, 15,2 ммоль) и триизопропилсилилхлорид (2,93 г, 15,2 ммоль) добавляли к 50 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 140°C и перемешивали в течение 3 часов. После прекращения нагревания реакционный раствор охлаждали до комнатной температуры и концентрировали при пониженном давлении. Впоследствии к остатку добавляли 50 мл воды и экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом магния и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 25с (968 мг, выход 31,0%) в виде желтого масла.
МС m/z (ИЭР): 458,3 [М+1]
Стадия 3
(E)-Метил-3-(4-((1S,3R/1R,3S)-2-(4-циклопропилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)-3-фторфенил) акрилат 25d
Соединение 25с (968 мг, 2,1 ммоль) растворяли в 20 мл дихлорметана, затем добавляли 2,6-лутидин (0,37 мл, 3,15 ммоль). После завершения добавления реакционную смесь охлаждали до 0°C в ледяной бане и по каплям добавляли трифторметансульфоновый ангидрид (0,45 мл, 2,73 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 3 часов при комнатной температуре. Добавляли 30 мл воды, и реакционный раствор экстрагировали дихлорметаном (15 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 25d (530 мг, выход 42,4%) в виде желтого масла.
МС m/z (ИЭР): 590,2 [М+1]
Стадия 4
(E)-Метил-3-(4-((1S,3R/1R,3S)-2-(4-циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3-фторфенил)акрилат 25е
Соединение 25d (530 мг, 0,9 ммоль), 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (300 мг, 1,35 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (66 мг, 0,09 ммоль) растворяли в 8 мл смеси 1,4-диоксана и воды (V:V составляет 7:1), впоследствии добавляли 2М раствор карбоната натрия (0,9 мл, 1,8 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°C в течение 45 минут. После охлаждения до комнатной температуры добавляли 50 мл воды, и смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом магния и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 25е (306 мг, выход 64,0%) в виде желтого масла.
MC m/z (ИЭР): 536,2 [М+1]
Стадия 5
(E)-3-(4-((1S,3R/1R,3S)-2-(4-Циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3-фторфенил)акриловая кислота 25
Соединение 25е (306 мг, 0,57 ммоль) растворяли в 10 мл смеси метанола и тетрагидрофурана (V:V составляет 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (1,4 мл, 2,8 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. По каплям добавляли 1 М соляную кислоту для доведения рН реакционного раствора до 3. Смесь экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом магния и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 25 (135 мг, выход 47,0%) в виде желтого твердого вещества.
MC m/z (ИЭР): 522,6 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.78 (s, 1Н), 7.65 (s, 1Н), 7.63 (d, 1Н), 7.30 (s, 1Н), 7.23 (d, 1Н), 7.13 (d, 1Н), 6.96 (d, 2Н), 6.90-6.74 (m, 4Н), 6.36 (d, 1Н), 6.06 (s, 1Н), 4.56-4.48 (m, 1Н), 4.24 (q, 2Н), 3.64-3.52 (m, 1Н), 2.82 (d, 2Н), 1.85-1.75 (m, 1Н), 1.56 (t, 3Н), 1.09 (d, 3Н), 0.88 (q, 2Н), 0.61 (q, 2Н).
Пример 26
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропил-3-фторфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 26
В соответствии с путем синтеза примера 24 исходное вещество 4-бром-2-хлоранилин, используемое на стадии 1, заменяли 4-бром-3-фторанилином, соответственно, получали указанное в заголовке соединение 26 (140 мг, желтое твердое вещество.
МС m/z (ИЭР): 522,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.75 (s, 1Н), 7.66 (d, 1Н), 7.62 (s, 1Н), 7.41-7.34 (m, 6Н), 6.73 (t, 1Н), 6.52-6.38 (m, 2Н), 6.34 (d, 1Н), 5.66 (s, 1Н), 4.52-4.43 (m, 1Н), 4.20 (q, 2Н), 3.34 (dd, 1Н), 2.73 (d, 1Н), 2.07-1.90 (m, 1Н), 1.51 (t, 3Н), 1.06 (d, 3Н), 0.88-0.82 (m, 3Н), 0.58 (q, 2Н).
Пример 27
(E)-3-(3,5-Дифтор-4-(6-(1-метил-1Н-пиразол-4-ил)-2-фенил-1,2,3,4-тетрагидро изохинолин-1-ил)акриловая кислота 27
В соответствии с путем синтеза примера 5 исходное вещество 5b, используемое на стадии 1, заменяли анилином, соответственно, получали указанное в заголовке соединение 27 (36 мг, желтое твердое вещество).
MC m/z (ИЭР): 472,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.91 (s, 1Н), 7.78 (s, 1Н), 7.49 (d, 1Н), 7.40 (s, 1Н), 7.28 (d, 1Н), 7.17-7.11 (m, 4Н), 7.02 (d, 2Н), 6.90 (d, 1Н), 6.78 (t, 1Н), 6.46 (d, 1Н), 6.23 (s, 1Н), 3.91 (s, 3Н), 3.67-3.61 (m, 2Н), 3.18-3.05 (m, 2Н).
Пример 28
(E)-3-(4-((1R,3R)-2-(4-Циклопропил-2-метилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 28
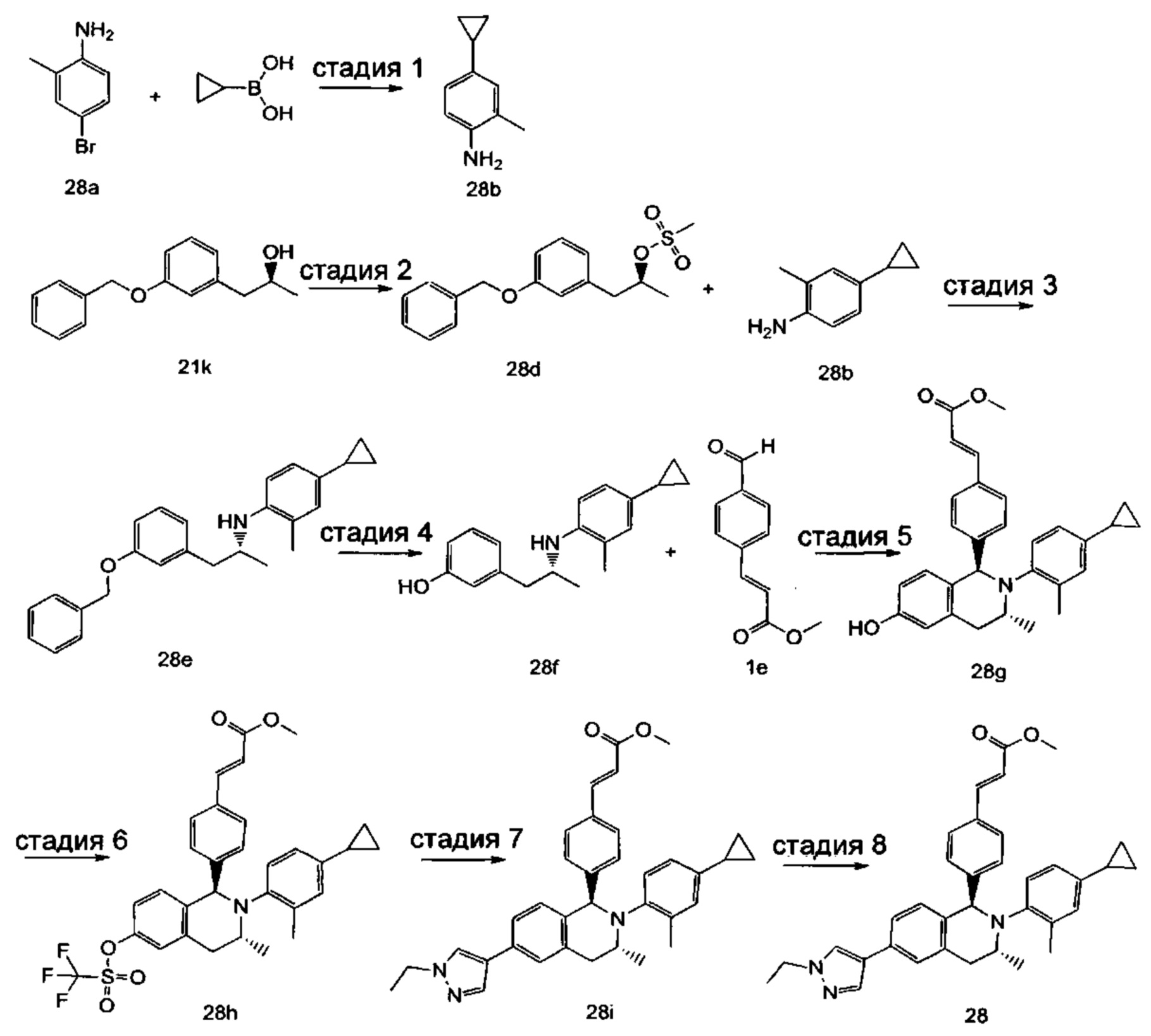
Стадия 1
4-Циклопропил-2-метиланилин 28b
4-Бром-2-метиланилин 28а (5 г, 26,9 ммоль), циклопропилбороновую кислоту (4,6 г, 53,8 ммоль), трициклогексилфосфин (754 мг, 2,69 ммоль), ацетат палладия (302 мг, 1,35 ммоль) и фосфат калия (17,1 г, 80,7 ммоль) добавляли к 105 мл смеси толуола и воды (V:V составляет 20:1). Смесь нагревали до 100°С и перемешивали в течение 12 часов. Реакционный раствор охлаждали до комнатной температуры, фильтровали, и фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 28b (3,5 г, выход 43,8%) в виде желтой жидкости.
МС m/z (ИЭР): 148,4 [М+1]
Стадия 2
(S)-1-(3-(Бензилокси)фенил)пропан-2-илметансульфонат 28d
Соединение 21k (25 г, 103 ммоль) и триэтиламин (29 мл, 206 ммоль) добавляли к 500 мл дихлорметана. После охлаждения до 0°С медленно по каплям добавляли метансульфонилхлорид (13,5 мл, 154 ммоль). Реакционную смесь перемешивали в течение 1 часа при 0°С. Добавляли 100 мл воды для гашения реакционной смеси, и реакционный раствор экстрагировали дихлорметаном (500 мл × 2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 28d (35 г) в виде оранжевого масла, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 3
(R)-N-(1-(3-(Бензилокси)фенил)пропан-2-ил)-4-циклопропил-2-метиланилин 28е
Неочищенное соединение 28d (2,4 г, 7,5 моль), 28b (3,3 г, 22,4 ммоль) и N,N-диизопропилэтиламин (1,9 г, 15 ммоль) добавляли в реакционную колбу и перемешивали в течение 12 часов при 80°С. После охлаждения до комнатной температуры добавляли 20 мл воды, и реакционный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 28е (1,3 г, выход 46,4%) в виде желтого масла.
МС m/z (ИЭР): 372,4 [М+1]
Стадия 4
(R)-3-(2-((4-Циклопропил-2-метилфенил)амино)пропил)фенол 28f
Соединение 28е (2 г, 5,4 ммоль) добавляли к 20 мл смеси толуола и трифторуксусной кислоты (V:V составляет 1:1). Смесь нагревали до 130°С и перемешивали в течение 5 часов. К реакционному раствору добавляли 20 мл воды и добавляли 2 М раствор гидроксида натрия для доведения рН до 7-8. Смесь экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 28f (1 г, выход 44,4%) в виде желтого масла.
МС m/z (ИЭР): 282,2 [М+1]
Стадия 5
(Е)-Метил-3-(4-((1R,3R)-2-(4-циклопропил-2-метилфенил)-6-гидрокси-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 28g
Соединение 28g (1 г, 3,6 ммоль), 1е (1,37 г, 7,2 ммоль) и триизопропилсилилхлорид (1,4 г, 7,2 ммоль) добавляли к 20 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 130°С и перемешивали в течение 4 часов. После прекращения нагревания реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 20 мл воды, впоследствии смесь перемешивали до однородного состояния и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 28g (800 мг, выход 50,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 454,4 [М+1]
Стадия 6
(E)-Метил-3-(4-((1R,3R)-2-(4-Циклопропил-2-метилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 28h
Соединение 28g (700 мг, 1,54 ммоль) растворяли в 30 мл дихлорметана, затем добавляли 2,6-лутидин (7,3 г, 68,3 ммоль). После завершения добавления реакционную смесь охлаждали до 0°С в ледяной бане. Впоследствии по каплям добавляли трифторметансульфоновый ангидрид (654 мг, 2,31 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 20 мл воды и разделяли две фазы. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 28h (420 мг, выход 40,8%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 586,3 [М+1]
Стадия 7
(E)-Метил-3-(4-((1R,3R)-2-(4-циклопропил-2-метилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 28i
Соединение 28h (420 мг, 0,72 ммоль),
1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (240 мг, 1,08 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (53 мг, 0,072 ммоль) растворяли в 13 мл смеси 1,4-диоксана и воды (V:V составляет 10:3), впоследствии добавляли карбонат натрия (152 мг, 1,44 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 45 часов. После охлаждения до комнатной температуры добавляли 10 мл воды, и смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 28i (200 мг, выход 52,5%) в виде желтого масла.
МС m/z (ИЭР): 532,4 [М+1]
Стадия 8
(Е)-3-(4-((1R,3R)-2-(4-Циклопропил-2-метилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 28
Соединение 28i (200 мг, 0,38 ммоль) растворяли в 7 мл смеси метанола и тетрагидрофурана (V:V составляет 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (0,9 мл, 1,9 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 10 мл воды, впоследствии смесь перемешивали до однородного состояния. По каплям добавляли 1 М соляную кислоту для доведения рН реакционного раствора до 5. Смесь экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл × 1), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 28 (130 мг, выход 66,3%) в виде желтого твердого вещества.
МС m/z (ИЭР): 518,3 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 8.11 (s, 1Н), 7.79 (s, 1Н), 7.43-7.34 (m, 5Н), 7.24-7.20 (m, 2Н), 6.76-6.65 (m, 3Н), 6.37 (d, 1Н), 5.74 (d, 1Н), 4.12 (q, 2Н), 3.65-3.56 (m, 1Н), 3.50-3.41 (m, 1Н), 3.37-3.28 (m, 1Н), 2.73-2.67 (m, 1Н), 2.25 (s, 3Н), 1.73-1.67 (m, 1Н), 1.38 (t, 3Н), 0.94 (d, 3Н), 0.82-0.79 (m, 2Н), 0.57-0.49 (m, 2Н).
Пример 29
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропилфенил)-6-(1-(дифторметил)-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 29
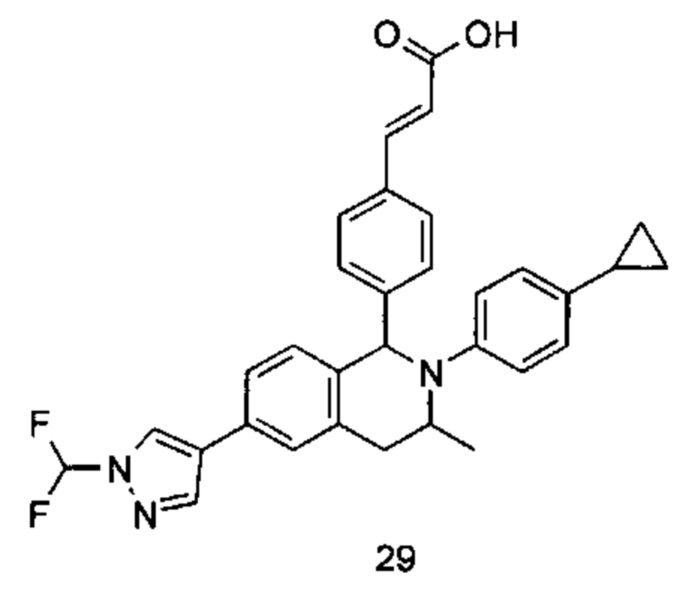
В соответствии с путем синтеза примера 10 исходное вещество 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол, используемое на стадии 3, заменяли 1-(дифторметил)-4-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом (получен способом, раскрытым в заявке на патент US 20100197651 A1), соответственно, получали указанное в заголовке соединение 29 (60 мг, желтое твердое вещество).
МС m/z (ИЭР): 526,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.36 (s, 1Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.32-3.43 (m, 1Н), 2.67-2.57 (m, 1Н), 2.42-2.32 (m, 1Н), 1.63-1.52 (m, 1Н), 1.26 (d, 3Н), 1.28-1.21 (m, 2Н), 0.13-0.94 (m, 2Н).
Пример 30
(E)-3-(4-((1S,3R/1R,3S)-2-(4-Циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-фторфенил)акриловая кислота 30
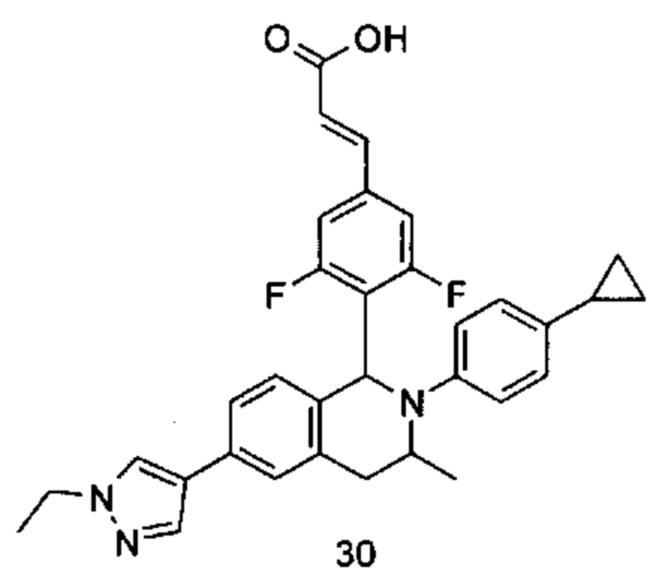
В соответствии с путем синтеза примера 4 исходное вещество 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол, используемое на стадии 7, заменяли 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, соответственно, получали указанное в заголовке соединение 30 (200 мг, желтое твердое вещество).
МС m/z (ИЭР): 540,6 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.75 (s, 1Н), 7.60 (s, 1Н), 7.52 (d, 1Н), 7.29 (s, 1Н), 7.21 (d, 1Н), 7.01-6.90 (m, 6Н), 6.32 (d, 1Н), 6.12 (s, 1Н), 4.35-4.28 (m, 1Н), 3.97 (q, 2Н), 3.69-3.60 (m, 1Н), 2.81-2.68 (m, 2Н), 1.83-1.74 (m, 1Н), 1.29 (t, 3Н), 1.07-0.96 (m, 2Н), 0.91-0.84 (m, 2Н), 0.60 (d, 3Н).
Примеры 31, 32
(Е)-3-(4-((1R,3S)-2-(4-Циклопропилфенил)-6-(1-этил-1H-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 31
(E)-3-(4-((1S,3R)-2-(4-Циклопропилфенил)-6-(1-этил-1Н-пиразол-4-ил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 32
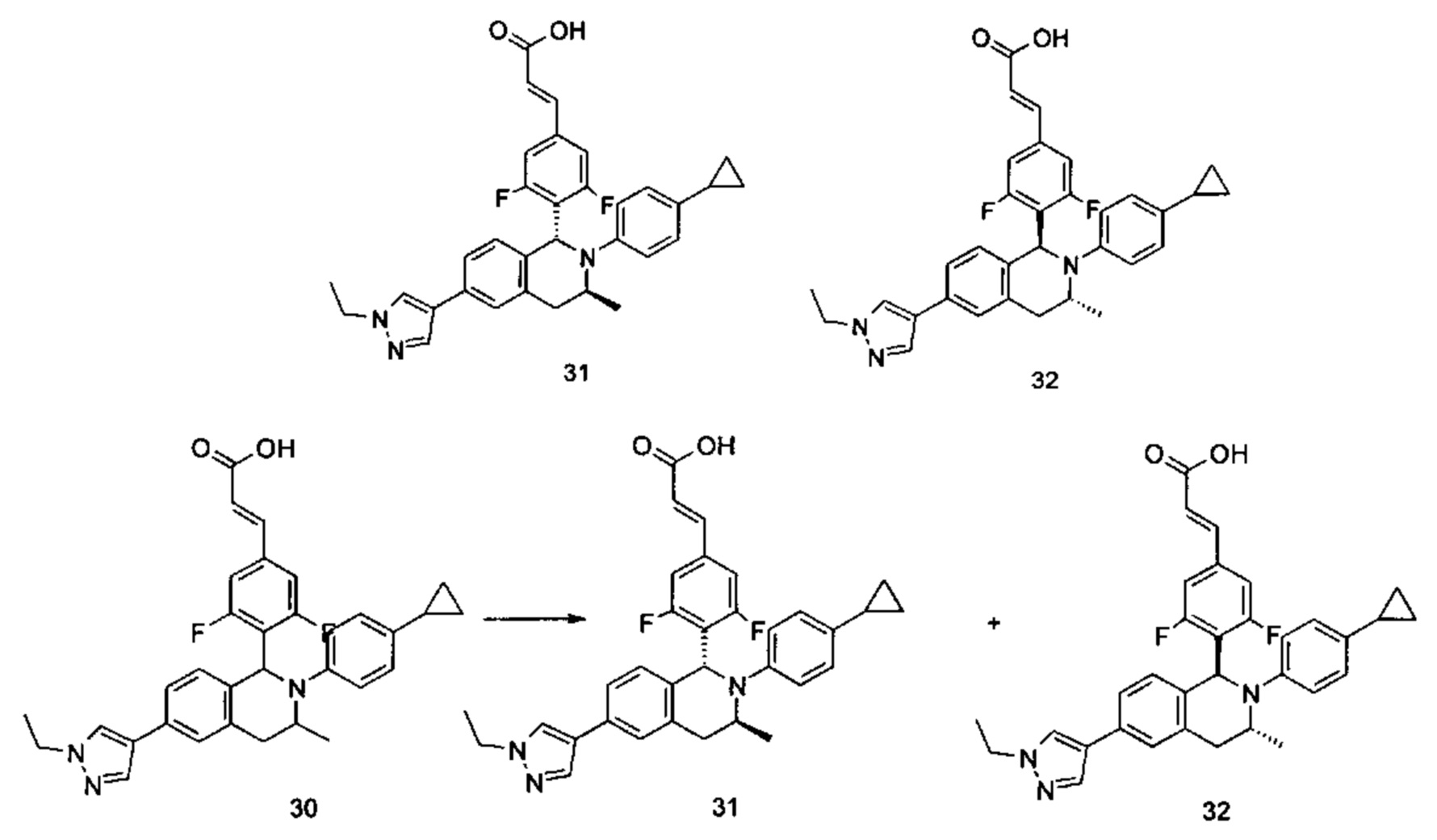
Соединение 30 (200 мг, 0,44 ммоль) подвергали хиральному разделению (условия разделения: хиральная колонка: Superchiral S-AD (Chiralway), внутренний диаметр 2 см * 25 см, 5 мкм; подвижная фаза: диоксид углерода : этанол : диэтиламин составляет 60:40:0,05, скорость потока: 50 г/мин). Соответствующие фракции собирали и концентрировали при пониженном давлении с получением указанных в заголовке соединений 31 (105 мг, желтое твердое вещество) и 32 (65 мг, желтое твердое вещество).
Соединение 31:
Анализ хиральной ВЭЖХ: время удерживания 2,15 минут, хиральная чистота: 99,0% (хроматографическая колонка: Superchiral S-AD, внутренний диаметр 0,46 см * 25 см, 5 мкм; подвижная фаза: диоксид углерода : этанол : диэтиламин = 60:40:0,05).
МС m/z (ИЭР): 540,6 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.75 (s, 1Н), 7.60 (s, 1Н), 7.52 (d, 1Н), 7.29 (s, 1Н), 7.21 (d, 1Н), 7.01-6.90 (m, 6Н), 6.32 (d, 1Н), 6.12 (s, 1Н), 4.35-4.28 (m, 1Н), 3.97 (q, 2Н), 3.69-3.60 (m, 1Н), 2.81-2.68 (m, 2Н), 1.83-1.74 (m, 1Н), 1.29 (t, 3Н), 1.07-0.96 (m, 2Н), 0.91-0.84 (m, 2Н), 0.60 (d, 3Н).
Соединение 32:
Анализ хиральной ВЭЖХ: время удерживания 4,06 минут, хиральная чистота: 100% (хроматографическая колонка: Superchiral S-AD (Chiralway), внутренний диаметр 0,46 см * 25 см, 5 мкм; подвижная фаза: диоксид углерода : этанол : диэтиламин составляет 60:40:0,05).
МС m/z (ИЭР): 540,6 [М+1]
1Н ЯМР (400 МГц, CDCl3): δ 7.75 (s, 1Н), 7.60 (s, 1Н), 7.52 (d, 1Н), 7.29 (s, 1Н), 7.21 (d, 1Н), 7.01-6.90 (m, 6Н), 6.32 (d, 1Н), 6.12 (s, 1Н), 4.35-4.28 (m, 1Н), 3.97 (q, 2Н), 3.69-3.60 (m, 1Н), 2.81-2.68 (m, 2Н), 1.83-1.74 (m, 1Н), 1.29 (t, 3Н), 1.07-0.96 (m, 2Н), 0.91-0.84 (m, 2Н), 0.60 (d, 3Н).
Пример 33
(E)-3-(4-((1R,3R)-3-Метил-6-(1-метил-1H-пиразол-4-ил)-2-фенил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 33
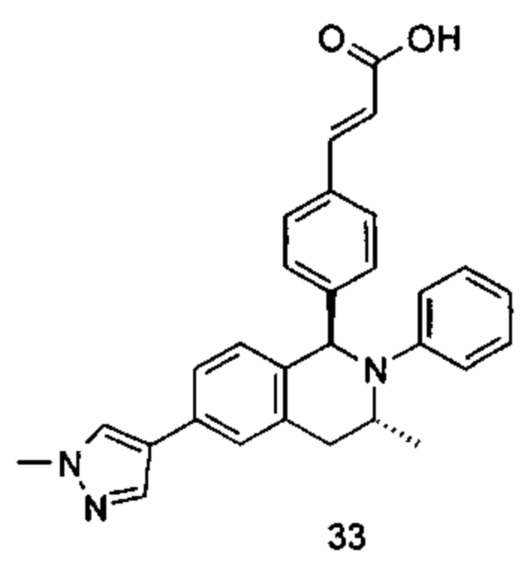
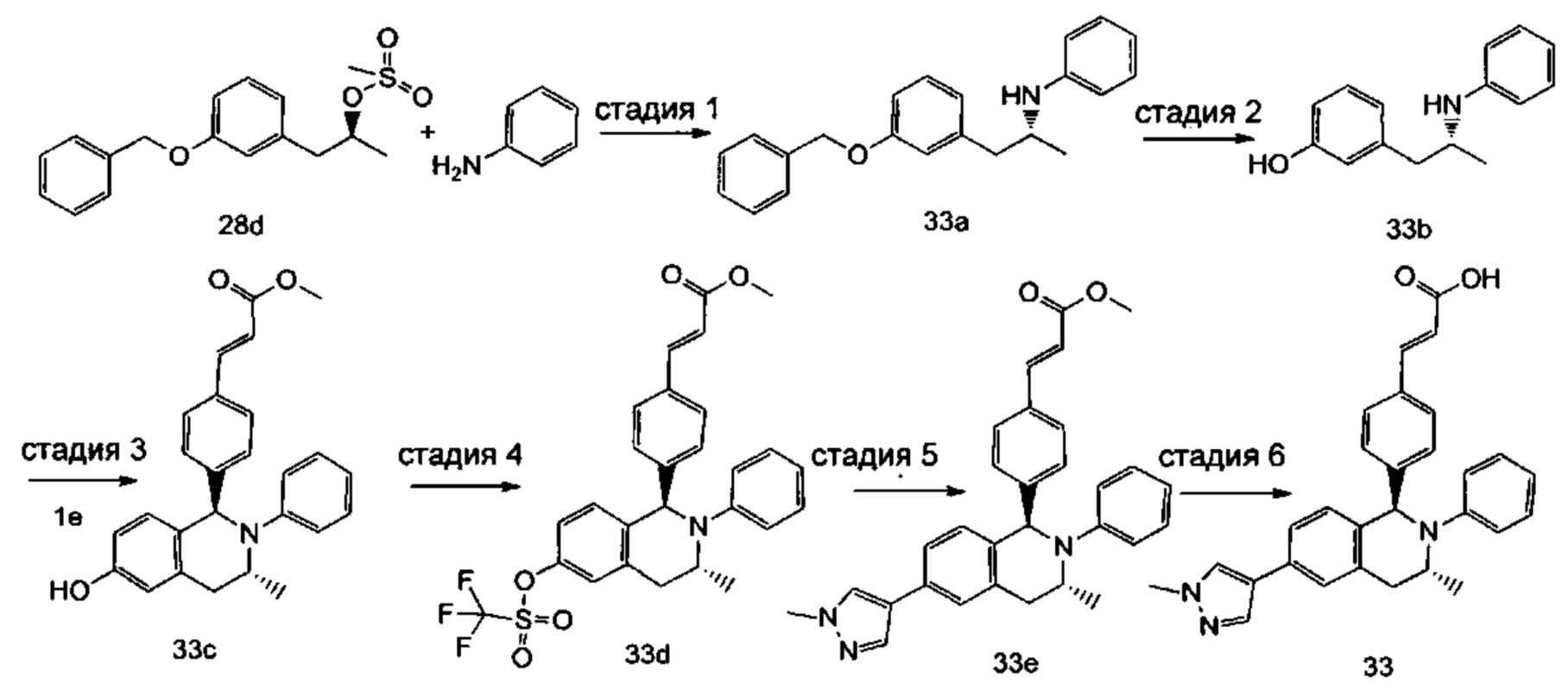
Стадия 1
(R)-N-(1-(3-(Бензилокси)фенил)пропан-2-ил)анилин 33а
В реакционную колбу добавляли неочищенное соединение 28d (1 г, 1,89 ммоль) и анилин (0,52 г, 6,2 ммоль). Смесь перемешивали в течение 12 часов при 70°С. После охлаждения до комнатной температуры добавляли 20 мл воды, и реакционный раствор экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 33а (630 мг, выход: 64,1%) в виде желтого масла.
МС m/z (ИЭР): 318,2 [М+1]
Стадия 2
(R)-3-(2-(фениламино)пропил)фенол 33b
Соединение 33а (600 мг, 1,89 ммоль) добавляли к 15 мл метанола, впоследствии добавляли палладий на углероде (120 мг). В реакционной системе создавали вакуум и заполняли ее водородом, причем описанную выше операцию повторяли три раза. Реакционную смесь перемешивали в течение 12 часов. После фильтрования фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 33b (300 мг, выход 70,0%) в виде желтого масла.
Стадия 3
(E)-Метил-3-(4-((1R,3R)-6-гидрокси-3-метил-2-фенил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 33с
Соединение 33b (300 мг, 1,32 ммоль), 1е (501 мг, 2,64 ммоль) и триизопропилсилилхлорид (509 мг, 2,64 ммоль) добавляли к 10 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 140°С и перемешивали в течение 2 часов. После прекращения нагревания реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 20 мл воды, и смесь перемешивали до однородного состояния и экстрагировали этилацетатом (20 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 33с (300 мг, выход 56,9%) в виде желтого твердого вещества.
МС m/z (ИЭР): 400,2 [М+1]
Стадия 4
(E)-Метил-3-(4-((1R,3R)-3-метил-2-фенил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 33d
Соединение 33с (100 мг, 0,25 ммоль) растворяли в 5 мл дихлорметана, затем добавляли 2,6-лутидин (54 мг, 0,5 ммоль). После завершения добавления реакционную смесь охлаждали до 0°С в ледяной бане и по каплям добавляли трифторметансульфоновый ангидрид (106 мг, 0,38 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 20 мл воды и разделяли две фазы. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 33d (80 мг, выход 60,6%) в виде желтого твердого вещества.
Стадия 5
(E)-Метил-3-(4-((1R,3R)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-2-фенил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 33е
Соединение 33d (50 мг, 0,056 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (18 мг, 0,085 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (4 мг, 0,0056 ммоль) растворяли в 3 мл смеси 1,4-диоксана и воды (V:V составляет 2:1), впоследствии добавляли карбонат натрия (12 мг, 0,112 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 45 минут.
После охлаждения до комнатной температуры добавляли 10 мл воды, и смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 33е (40 мг, выход 93,0%) в виде желтого твердого вещества.
Стадия 6
(E)-3-(4-((1R,3R)-3-Метил-6-(1-метил-1Н-пиразол-4-ил)-2-фенил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 33
Соединение 33е (40 мг, 0,086 ммоль) растворяли в 5 мл смеси метанола и тетрагидрофурана (V:V составляет 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (0,2 мл, 0,4 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 10 мл воды, и смесь перемешивали до однородного состояния. Впоследствии по каплям добавляли 1M соляную кислоту для доведения рН реакционного раствора до 5. Смесь экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 33 (30 мг, выход 77,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 450,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.69 (t, 1Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.67-2.57 (m, 1Н), 2.42-2.32 (m, 1Н), 1.26 (d, 3Н).
Пример 34
(E)-3-(4-((1R,3R)-3-Метил-6-(1-метил-1Н-пиразол-4-ил)-2-п-толил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 34
В соответствии с путем синтеза примера 33 исходное вещество анилин, используемое на стадии 1, заменяли метиланилином, соответственно, получали указанное в заголовке соединение 34 (9 мг, желтое твердое вещество.
МС m/z (ИЭР): 464,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.67-2.57 (m, 1Н), 2.42-2.32 (m, 1Н), 2.34 (s, 3Н), 1.26 (d, 3Н).
Пример 35
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропилфенил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 35
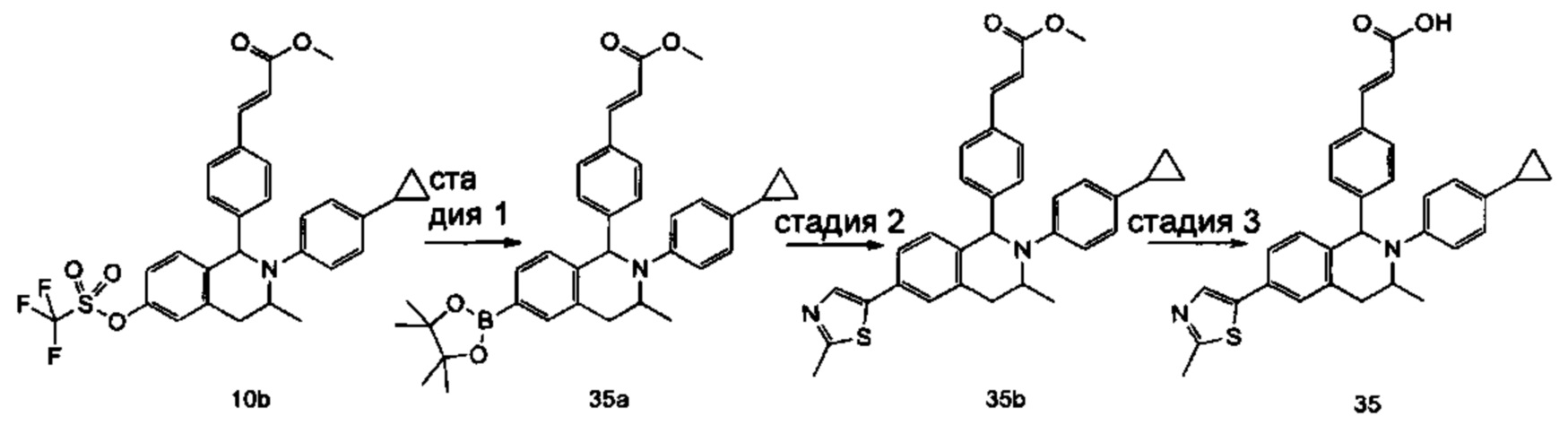
Стадия 1
(E)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-циклопропилфенил)-3-метил-6-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 35а
Соединение 10b (300 мг, 0,52 ммоль), бис(пинаколато)дибор (160 мг, 0,63 ммоль), ацетат калия (102 мг, 1,05 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (4 мг, 0,0056 ммоль) добавляли к 15 мл N,N-диметилформамида. Смесь нагревали до 90°С и перемешивали в течение 2 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 10 мл воды, и смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл × 1), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 35а (55 мг, выход: 77,0%) в виде желтого масла.
МС m/z (ИЭР): 550,3 [М+1]
Стадия 2
(Е)-Метил-3-(4-((1R,3R/1S,3S)-2-(4-циклопропилфенил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат
Соединение 35а (30 мг, 0,054 ммоль), 5-бром-2-метилтиазол (15 мг, 0,082 ммоль), 2-дициклогексилфосфино-2,4,6-триизопропилбифенил (3 мг, 0,007 ммоль) и трис(дибензилиденацетон)дипалладий (2 мг, 0,002 ммоль) растворяли в 2 мл смеси 1,4-диоксана и воды (V:V составляет 4:1), впоследствии добавляли фосфат калия (23 мг, 0,11 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 100°С в течение 45 минут. После охлаждения до комнатной температуры добавляли 10 мл воды, и смесь экстрагировали этилацетатом (30 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 35b (10 мг, выход 35,7%) в виде желтого масла.
МС m/z (ИЭР): 521,2 [М+1]
Стадия 3
(Е)-3-(4-((1R,3R/1S,3S)-2-(4-Циклопропилфенил)-3-метил-6-(2-метилтиазол-5-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 35
Соединение 35b (10 мг, 0,02 ммоль) растворяли в 5 мл смеси метанола и тетрагидрофурана (V:V составляет 1:1), впоследствии добавляли 2 М раствор гидроксида натрия (0,05 мл, 0,096 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 10 мл воды, и смесь перемешивали до однородного состояния. Впоследствии по каплям добавляли 1 M соляную кислоту для доведения рН реакционного раствора до 5. Смесь экстрагировали этилацетатом (10 мл × 3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (10 мл × 1), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 35 (6 мг, выход 60,0%) в виде желтого твердого вещества.
МС m/z (ИЭР): 507,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 7.00 (s, 1Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.32-3.43 (m, 1Н), 2.67-2.57 (m, 1Н), 2.42-2.32 (m, 1Н), 2.34 (s, 3Н), 1.63-1.52 (m, 1Н), 1.26 (d, 3Н), 1.28-1.21 (m, 2Н), 0.13-0.94 (m, 2Н).
Пример 36
(Е)-3-(4-((1R,3R/1S,3S)-2-(4-циклопропилфенил)-3-метил-6-(1H-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 36
В соответствии с путем синтеза примера 10 исходное вещество 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол, используемое на стадии 3, заменяли трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол-1-карбоксилатом, соответственно, получали указанное в заголовке соединение 36 (30 мг, желтое твердое вещество) после удаления трет-бутилоксикарбонила.
МС m/z (ИЭР): 476,58 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.32-3.43 (m, 1Н), 2.67-2.57 (m, 1Н), 2.42-2.32 (m, 1Н), 1.63-1.52 (m, 1Н), 1.26 (d, 3Н), 1.28-1.21 (m, 2Н), 0.13-0.94 (m, 2Н).
Пример 37
(Е)-3-(4-((1R,3R/1S,3S)-2-(2,4-Диметилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 37
В соответствии с путем синтеза примера 6 исходное вещество 2-этиланилин, используемое на стадии 1, заменяли 2,4-диметиланилином, соответственно, получали указанное в заголовке соединение 37 (60 мг, желтое твердое вещество).
МС m/z (ИЭР): 478,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.73 (s, 1Н), 7.66 (d, 1Н), 7.58 (s, 1Н), 7.38 (s, 3Н), 7.30 (s, 2Н), 7.24 (s, 1Н), 6.92 (d, 2Н), 6.71 (s, 1Н), 6.32 (d, 1Н), 6.66 (d, 1Н), 4.46 (d, 1Н), 3.93 (s, 3Н), 3.35 (d, 1Н), 2.28 (s, 3Н), 2.72 (d, 1Н), 2.17 (s, 3Н), 1.25 (s, 1Н), 1.04 (d, 3Н).
Пример 38
(E)-3-(4-((1S,3R)-6-(1-Этил-1Н-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 38
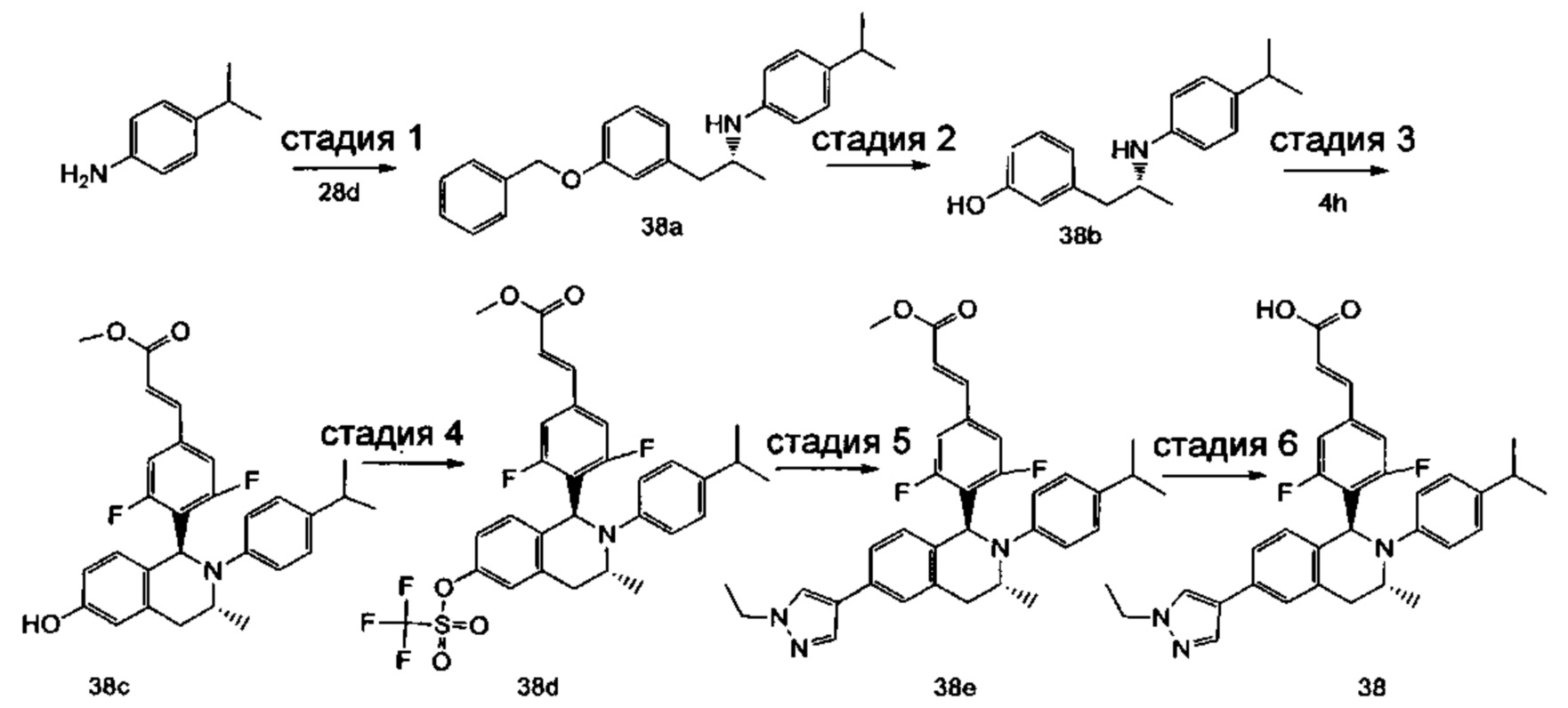
Стадия 1
(R)-N-(1-(3-(Бензилокси)фенил)пропан-2-ил)-4-изопропиланилин 38а
В реакционную колбу добавляли неочищенное соединение 28d (29 г, 85 ммоль) и 4-изопропиланилин (20 г, 153 ммоль). Смесь перемешивали в течение 2 часов при 100°С. После охлаждения до комнатной температуры добавляли 150 мл воды, и смесь экстрагировали этилацетатом (300 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 38а (21 г, выход: 70,0%) в виде желтого масла.
Стадия 2
(R)-3-(2-((4-Изопропилфенил)амино)пропил)фенол 38b
Соединение 38а (10,5 г, 29,2 ммоль) добавляли к 200 мл метанола, впоследствии добавляли палладий на углероде (10%, 2 г). В реакционной системе создавали вакуум и заполняли ее водородом, причем описанную выше операцию повторяли три раза. Реакционную смесь перемешивали в течение 12 часов. После фильтрования фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 38b (7 г, выход 88,6%) в виде красного твердого вещества.
Стадия 3
(E)-Метил
3-(3,5-дифтор-4-((1S,3R)-6-гидрокси-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 38с
Соединение 38b (5 г, 18,6 ммоль), 4h (8,4 г, 37,2 ммоль) и триизопропилсилилхлорид (7,2 г, 37,2 ммоль) добавляли к 100 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 140°С и перемешивали в течение 2 часов. После прекращения нагревания реакционный раствор концентрировали при пониженном давлении. Впоследствии к полученному в результате остатку добавляли 50 мл воды, и смесь перемешивали до однородного состояния. Смесь экстрагировали этилацетатом (50 мл × 4). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 38с (8,8 г, выход 100%) в виде желтого твердого вещества.
Стадия 4
(E)-Метил-3-(3,5-дифтор-4-((1S,3R)-2-(4-изопропилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 38d
Соединение 38с (8,8 г, 18,6 ммоль) растворяли в 200 мл дихлорметана, затем добавляли 2,6-лутидин (3 г, 27,9 ммоль). После завершения добавления реакционную смесь охлаждали до 0°С в ледяной бане и по каплям добавляли трифторметансульфоновый ангидрид (6,82 г, 24,2 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 100 мл воды и разделяли две фазы. Водную фазу экстрагировали дихлорметаном (150 мл × 3). Органическую фазу высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 38d (3,5 г, выход 30,9%) в виде желтого твердого вещества.
Стадия 5
(Е)-Метил-3-(4-((1S,3R)-6-(1-этил-1Н-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акрилат 38с
Соединение 38d (1,6 г, 2,62 ммоль), 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (0,87 г, 3,94 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (190 мг, 0,26 ммоль) растворяли в 25 мл смеси 1,4-диоксана и воды (V:V составляет 4:1), впоследствии добавляли карбонат натрия (555 мг, 5,24 ммоль). Смесь перемешивали в микроволновом реакторе при 120°С в течение 1 часа. После охлаждения до комнатной температуры добавляли 50 мл воды, и смесь экстрагировали этилацетатом (100 мл × 3). Органические фазы объединяли и концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 38е (1,0 г, выход 69,0%) в виде желтого масла.
МС m/z (ИЭР): 556,5 [М+1]
Стадия 6
(E)-3-(4-((1S,3R)-6-(1-Этил-1Н-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)-3,5-дифторфенил)акриловая кислота 38
Соединение 38е (1 г, 1,8 ммоль) растворяли в 30 мл смеси метанола и тетрагидрофурана (V:V составляет 1:2), впоследствии добавляли 2 М раствор гидроксида натрия (5,4 мл, 10,8 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. Реакционный раствор концентрировали при пониженном давлении. К полученному в результате остатку добавляли 50 мл воды, и смесь перемешивали до однородного состояния. По каплям добавляли 2 М соляну кислоту для доведения рН реакционного раствора до 5. Смесь экстрагировали этилацетатом (50 мл × 3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 38 (700 мг, выход 71,8%) в виде желтого твердого вещества.
МС m/z (ИЭР): 542,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.93 (s, 1Н), 7.78 (s, 1Н), 7.36-7.43 (m, 3Н), 7.27-7.25 (dd, 1Н), 7.05-6.98 (m, 5Н), 6.94 (d, 1Н), 6.38 (d, 1Н), 6.11 (s, 1Н), 4.25-4.35 (m, 1Н), 4.18 (q, 2Н), 3.64 (dd, 1Н), 2.68-2.81 (m, 2Н), 1.47 (t, 3Н), 1.11-1.18 (m, 6Н), 0.96 (d, 3Н).
Пример 39
(Е)-3-(3,5-Дифтор-4-((1S,3R)-2-(4-изопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 39
В соответствии с путем синтеза примера 38 исходное вещество 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол, используемое на стадии 5, заменяли 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразолом, соответственно, получали указанное в заголовке соединение 39 (330 мг, желтое твердое вещество).
МС m/z (ИЭР): 528,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 12.53 (br, 1Н), 8.09 (s, 1Н), 7.82 (s, 1Н), 7.45-7.31 (m, 5Н), 7.06 (d, 2Н), 6.98-6.94 (m, 3Н), 6.58 (d, 1Н), 6.03 (d, 1Н), 4.37 (s, 1Н), 3.85 (s, 3Н), 3.58-3.51 (m, 1Н), 2.79-2.73 (m, 2Н), 1.13 (d, 6Н), 0.87 (d, 3Н).
Пример 40
(E)-3-(3,5-Дифтор-4-((1S,3R/1R,3S)-2-(4-изопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 40
В соответствии с путем синтеза примера 4 исходное вещество 4d, используемое на стадии 3, заменяли 4-изопропиланилином, соответственно, получали указанное в заголовке соединение 40 (60 мг, светло-желтое твердое вещество).
МС m/z (ИЭР): 528,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.94 (s, 1Н), 7.78 (s, 1Н), 7.38-7.42 (m, 3Н), 7.27-7.25 (dd, 1Н), 7.05-6.98 (m, 5Н), 6.96 (d, 1Н), 6.38 (d, 1Н), 6.12 (s, 1Н), 4.25-4.35 (m, 1Н), 4.2 (s, 3Н), 3.64 (dd, 1Н), 2.68-2.81 (m, 2Н), 1.47 (t, 3Н), 1.11-1.18 (m, 6Н).
Пример 41
(E)-3-(4-((1R,3R/1S,3S)-2-(4-(трет-Бутил)фенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 41
В соответствии с путем синтеза примера 1 исходное вещество 1b, используемое на стадии 1, заменяли 4-трет-бутиланилином, соответственно, получали указанное в заголовке соединение 41 (15 мг, желтое твердое вещество).
МС m/z (ИЭР): 506,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.92 (s, 1Н), 7.79 (s, 1Н), 7.57 (d, 1H), 7.43 (s, 4Н), 7.35-7.38 (m, 3Н), 7.17 (d, 2Н), 6.79 (d, 2Н), 6.38 (d, 1Н), 5.76 (s, 1Н), 4.54 (m, 1Н), 3.91 (s, 3Н), 3.40 (dd, 1Н), 2.80 (dd, 1Н), 1.24 (s, 9Н), 1.02 (d, 3Н).
Пример 42
(E)-3-(4-((1R,3R)-2-(4-Изопропилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 42
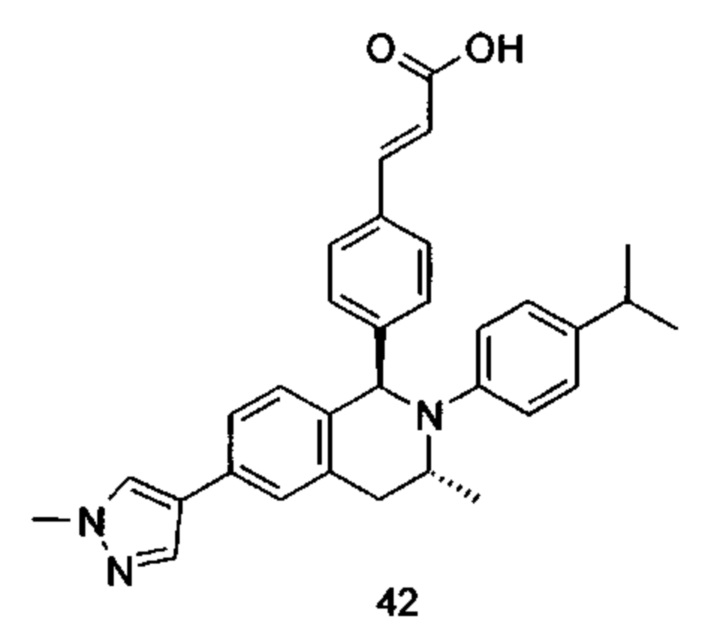
В соответствии с путем синтеза примера 33 исходное вещество анилин, используемое на стадии 1, заменяли 4-изопропиланилином, соответственно, получали указанное в заголовке соединение 42 (457 мг, желтое твердое вещество).
МС m/z (ИЭР): 492,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.89-2.78 (m, 1Н), 2.67-2.57 (m, 1Н), 2.42-2.32 (m, 1Н), 1.26-1.20 (m, 9Н).
Пример 43
(E)-3-(4-((1R,3R/1S,3S)-3-Метил-6-(1-метил-1Н-пиразол-4-ил)-2-(4-(2,2,2-трифторэтил)фенил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 43
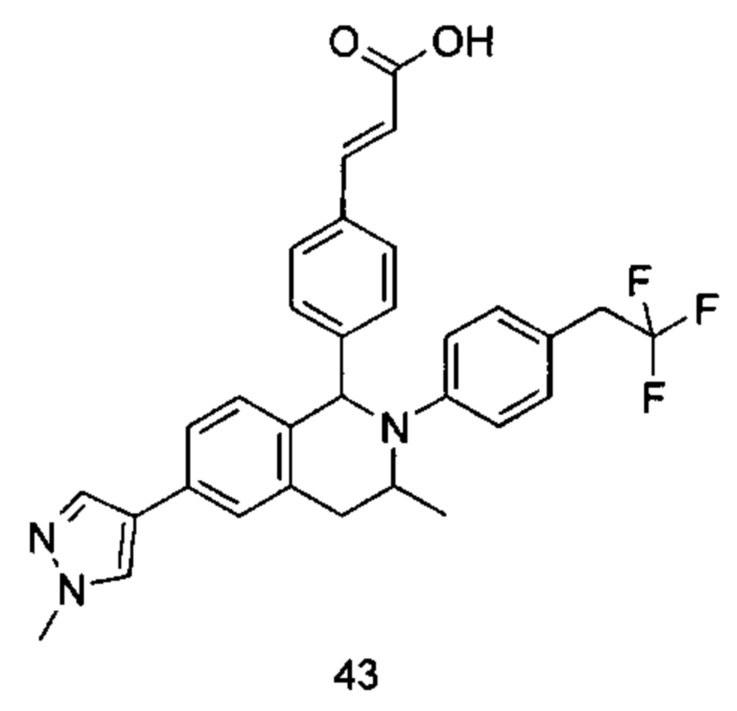
В соответствии с путем синтеза примера 1 исходное вещество 1b, используемое на стадии 1, заменяли 4-(2,2,2-трифторэтил)анилином, соответственно, получали указанное в заголовке соединение 43 (40 мг, желтое твердое вещество).
МС m/z (ИЭР): 532,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 3.12-3.03 (m, 2Н), 2.67-2.57 (m, 1Н), 2.42-2.32 (m, 1Н), 1.26 (d, 3Н).
Пример 44
(Е)-3-(4-((1R,3R/1S,3S)-2-(4-Этоксифенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 44
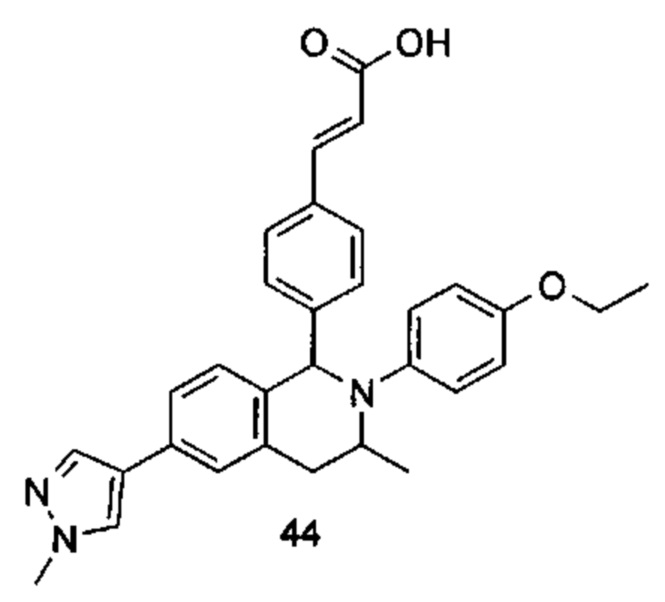
В соответствии с путем синтеза примера 1 исходное вещество 1b, используемое на стадии 1, заменяли 4-этоксианилином, соответственно, получали указанное в заголовке соединение 44 (20 мг, желтое твердое вещество).
МС m/z (ИЭР): 494,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 4.09 (q, 2Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.67-2.57 (m, 1Н), 2.42-2.32 (m, 1Н), 1.32 (t, 3Н), 1.26 (d, 3Н).
Пример 45
(E)-3-(4-((1R,3R/1S,3S)-2-(2,4-Диэтилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 45
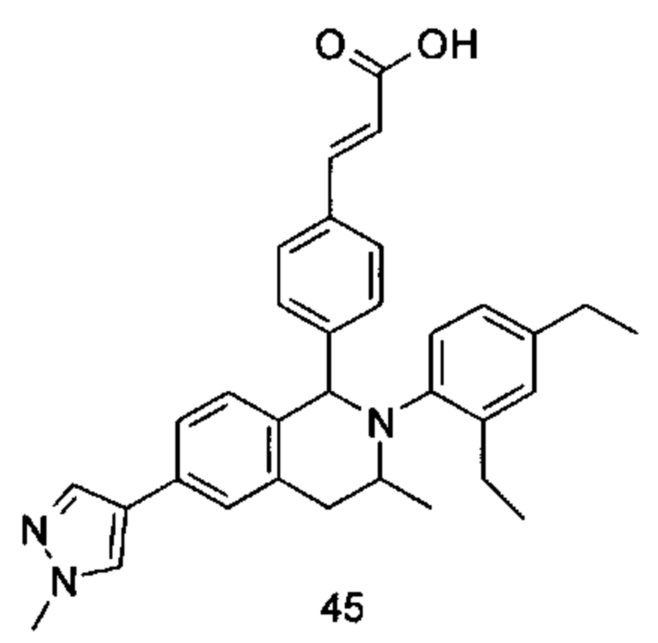
В соответствии с путем синтеза примера 1 исходное вещество 1b, используемое на стадии 1, заменяли 2,4-диэтиланилином, соответственно, получали указанное в заголовке соединение 45 (53 мг, желтое твердое вещество).
МС m/z (ИЭР): 506,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 1Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.67-2.56 (m, 5Н), 2.42-2.32 (m, 1Н), 1.26 (d, 3Н), 1.22-1.16 (m, 6Н).
Пример 46
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Метоксифенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 46
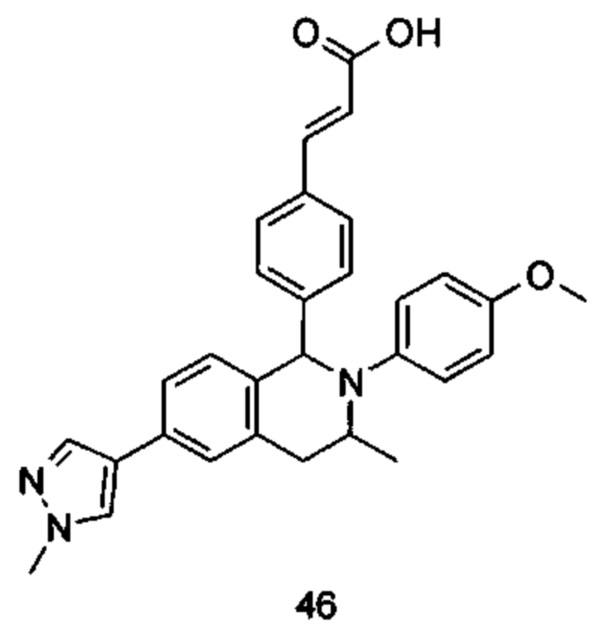
В соответствии с путем синтеза примера 1 исходное вещество 1b, используемое на стадии 1, заменяли 4-метоксианилином, соответственно, получали указанное в заголовке соединение 46 (50 мг, желтое твердое вещество).
МС m/z (ИЭР): 480,4 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.73 (s, 1Н), 7.66 (d, 1Н), 7.58 (s, 1Н), 7.38 (s, 3Н), 7.30 (s, 2Н), 7.24 (s, 1Н), 6.92 (d, 2Н), 6.71 (d, 2Н), 6.32 (d, 1Н), 6.66 (d, 1Н), 4.46 (d, 1Н), 4.36 (s, 3Н), 3.93 (s, 3Н), 3.35 (d, 1Н), 2.72 (d, 1Н), 1.25 (s, 1Н), 1.04 (d, 3Н).
Пример 47
(E)-3-(4-((1R,3R/1S,3S)-2-(4-(Циклопропилметил)фенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 47
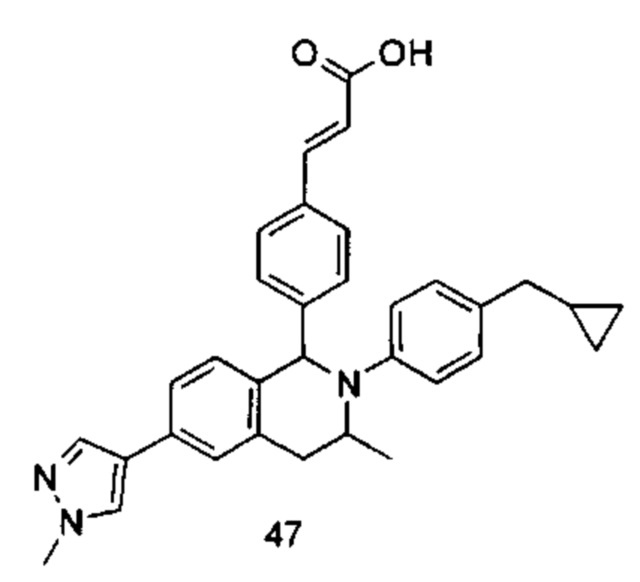
В соответствии с путем синтеза примера 1 исходное вещество 1b, используемое на стадии 1, заменяли 4-(циклопропилметил)анилином (получен способом, раскрытым в заявке на патент US 5455252 A1), соответственно, получали указанное в заголовке соединение 47 (120 мг, желтое твердое вещество).
МС m/z (ИЭР): 504,6 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.74 (s, 1Н), 7.66 (d, 1Н), 7.59 (s, 1Н), 7.39 (s, 3Н), 7.31 (s, 2Н), 7.26 (s, 2Н), 7.04 (d, 2Н), 6.73-6.77 (m, 2Н), 6.36 (d, 1Н), 5.70 (s, 1Н), 3.95 (s, 3Н), 3.32-3.43 (m, 1Н), 2.67-2.57 (m, 1Н), 2.54 (d, 2Н), 2.42-2.32 (m, 1Н), 1.26 (d, 3Н), 0.67-0.62 (m, 1Н), 0.33-0.27 (m, 2Н), 0.07-0.03 (m, 2Н).
Пример 48
(E)-3-(4-((1R,3R/1S,3S)-2-(4-Изопропилфенил)-3-метил-6-(1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 48
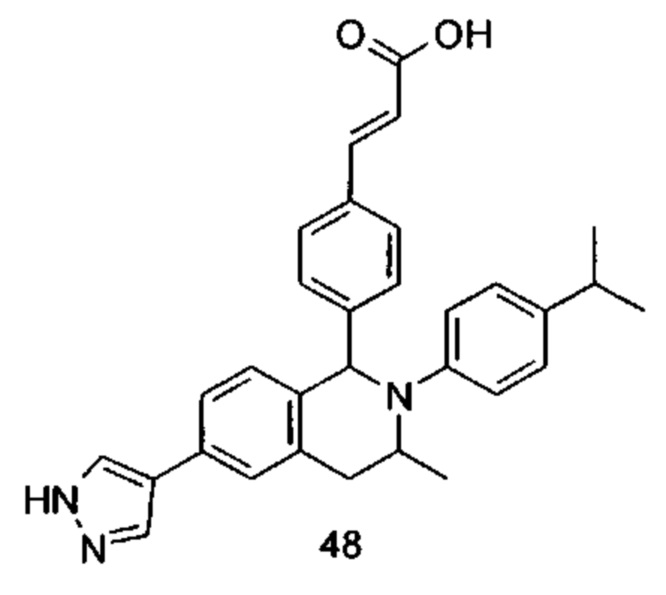
В соответствии с путем синтеза примера 7 исходное вещество 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол, используемое на стадии 5, заменяли трет-бутил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом, соответственно, получали указанное в заголовке соединение 48 (130 мг, желтое твердое вещество) после удаления трет-бутилоксикарбонила.
МС m/z(ИЭР): 478,4 [М+1]
1Н ЯМР (400 МГц, ДМСО-d6) δ 12.58 (br, 1Н), 8.02 (s, 2Н), 7.55-7.43 (m, 7Н), 6.99 (d, 2Н), 6.74 (d, 2Н), 6.43 (d, 1Н), 5.81 (s, 1Н), 4.59 (br, 1Н), 2.80-2.70 (m, 2Н), 1.24 (s, 2Н), 1.11 (d, 6Н), 0.94 (d, 3Н).
Пример 49
(Е)-3-(4-((1R,3R/1S,3S)-6-(1-(Дифторметил)-1H-пиразол-4-ил)-2-(4-изопропилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 49
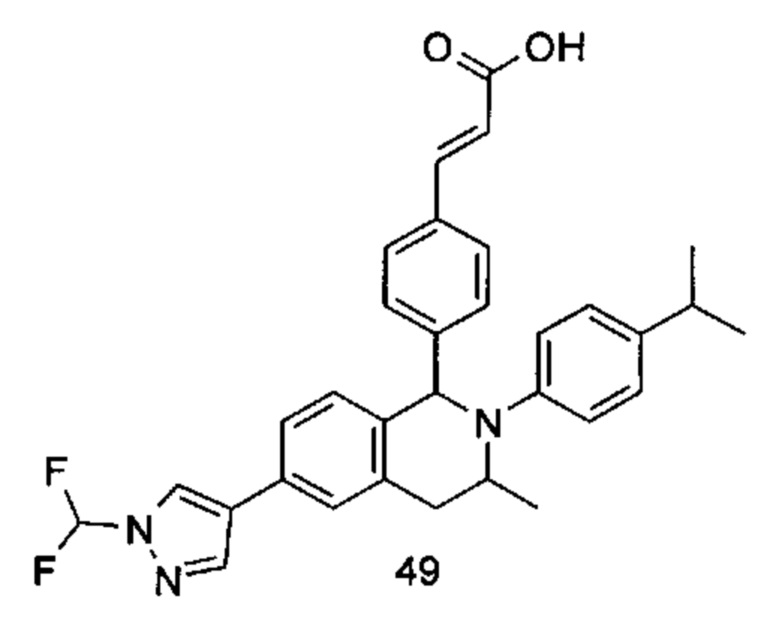
В соответствии с путем синтеза примера 7 исходное вещество 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол, используемое на стадии 5, заменяли 1-(дифторметил)-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразолом, соответственно, получали указанное в заголовке соединение 49 (35 мг, желтое твердое вещество).
МС m/z (ИЭР): [М+1] 528,6
1Н ЯМР (400 МГц, ДМСО-d6) δ 10.9 (s, 1Н), 8.80 (s, 1Н), 8.20 (s, 1Н), 7.74 (s, 1Н), 7.57-7.63 (m, 3Н), 7.11-7.25 (m, 7Н), 6.67-6.71 (m, 2Н), 6.31-6.33 (d, 1Н), 5.18-5.22 (m, 1Н), 3.15-3.18 (m, 1Н), 2.85-2.88 (m, 1Н), 2.63-2.65 (m, 1Н), 2.35-2.37 (m, 1Н), 1.20-1.25 (m, 9Н).
Пример 50
(E)-3-(4-((1R,3S/1S,3R)-2-(2-Этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 50
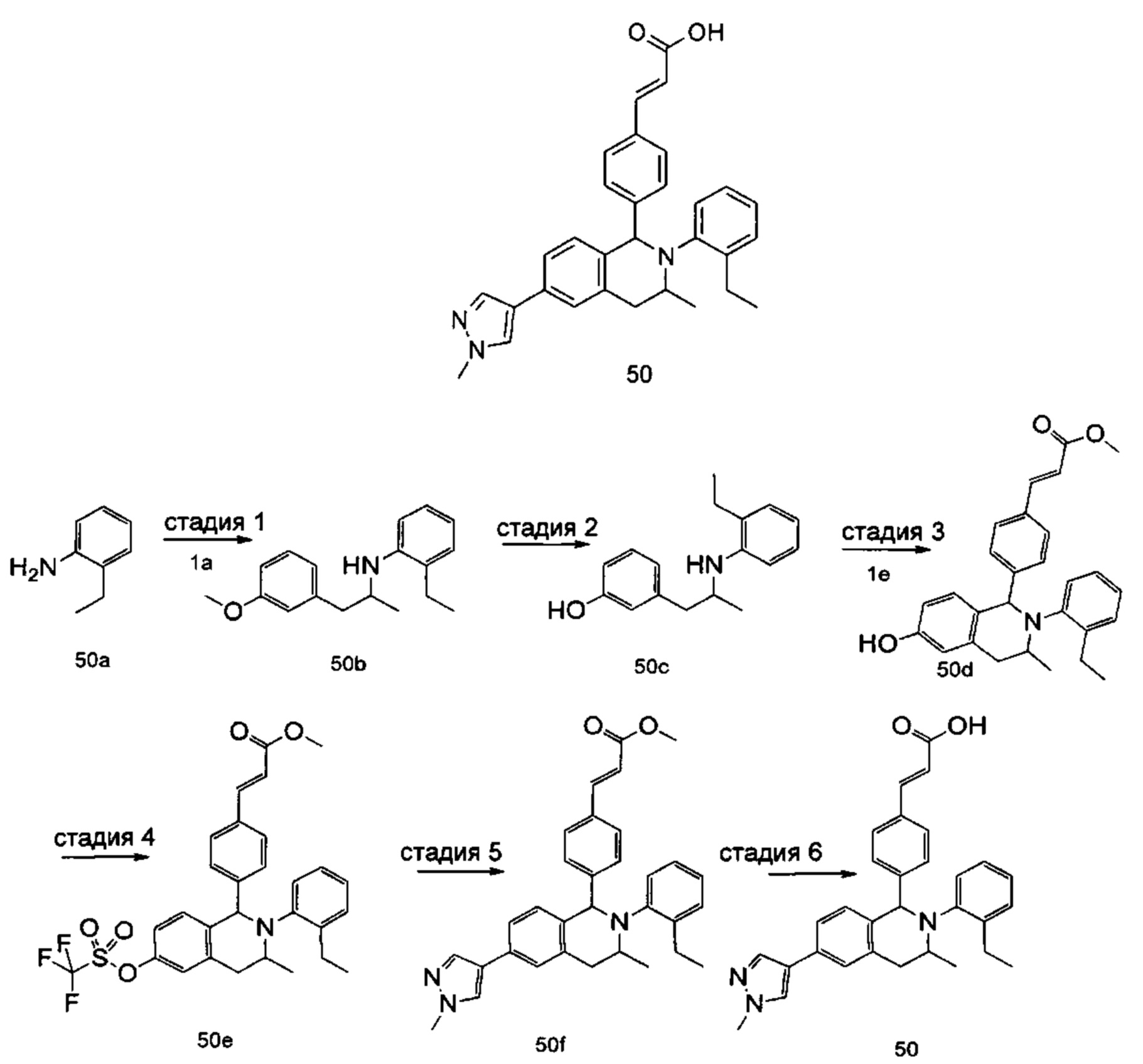
Стадия 1
2-Этил-N-(1-(3-метоксифенил)пропан-2-ил)анилин 50b
Соединение 1а (820 мг, 5 ммоль), 2-этиланилин 50а (0,75 мл, 6 ммоль) и триацетоксиборгидрид натрия (1,58 г, 7,5 ммоль) растворяли в 30 мл 1,2-дихлорэтана. Смесь перемешивали в течение 16 часов. Для гашения реакции добавляли 30 мл воды. Реакционный раствор экстрагировали дихлорметаном (25 мл×3). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 50b (0,8 г, выход 59,3%) в виде желтого масла.
МС m/z(ИЭР): 270,1 [М+1]
Стадия 2
3-(2-((2-Этилфенил)амино)пропил)фенол 50с
Соединение 50b (800 мг, 2,97 ммоль) растворяли в 20 мл дихлорметана, впоследствии по каплям добавляли 6 мл раствора 1 М трибромида бора в дихлорметане в ледяной бане. После завершения добавления реакционную смесь перемешивали в течение 16 часов при комнатной температуре. Для гашения реакции добавляли 15 мл воды. Реакционный раствор концентрировали при пониженном давлении. Остаток экстрагировали этилацетатом (30 мл×3), высушивали над безводным сульфатом магния и фильтровали. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 50с (500 мг) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 3
(Е)-Метил-3-(4-((1R,3S/1S,3R)-6-гидрокси-2-(2-этилфенил)-3-метил-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 50d
Неочищенное соединение 50с (400 мг, 1,57 ммоль), 1е (596 мг, 3,13 ммоль) и триизопропилсилилхлорид (1,51 г, 7,83 ммоль) добавляли к 10 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 120°С и перемешивали в течение 3 часов. После прекращения нагревания добавляли 20 мл воды, и реакционный раствор экстрагировали этилацетатом (20 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 50d (440 мг, выход 65,7%) в виде желтого твердого вещества.
МС m/z (ИЭР): 428,3 [М+1]
Стадия 4
(E)-Метил-3-(4-((1R,3S/1S,3R)-2-(2-этилфенил)-3-метил-6-(((трифторметил)сульфонил)окси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 50е
Соединение 50d (440 мг, 1,03 ммоль) и 2,6-лутидин (165 мг, 1,54 ммоль) растворяли в 50 мл дихлорметана. После охлаждения реакционной смеси до 0°С по каплям добавляли трифторметансульфоновый ангидрид (436 г, 1,54 ммоль). Реакционную смесь перемешивали в течение 12 часов при комнатной температуре. Для гашения реакции добавляли 20 мл воды, и реакционный раствор экстрагировали дихлорметаном (50 мл×2). Органические фазы объединяли и высушивали над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 50е (176 мг, выход 30,6%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 560,2 [М+1]
Стадия 5
(Е)-Метил-3-(4-((1R,3R/1S,3S)-2-(2-этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 50f
Соединение 50е (101 мг, 0,18 ммоль), 1-этил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (56 мг, 0,27 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (13 мг, 0,018 ммоль) растворяли в 3,6 мл смеси 1,4-диоксана и воды (V:V составляет 5:1), впоследствии добавляли 0,18 мл 2 н. раствора карбоната натрия. После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 40 минут. После охлаждения до комнатной температуры добавляли 20 мл воды, и смесь экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия и высушивали над безводным сульфатом натрия. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 50f (80 мг) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 492,3 [М+1]
Стадия 6
(E)-3-(4-((1R,3S/1S,3R)-2-(2-Этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 50
Соединение 50f (80 мг, 0,16 ммоль) растворяли в 5 мл метанола, впоследствии добавляли 0,4 мл 2 М раствора гидроксида натрия. После завершения добавления реакционную смесь перемешивали в течение 16 часов. По каплям добавляли 1 М соляную кислоту для доведения рН реакционного раствора до 3. Смесь экстрагировали этилацетатом (10 мл×2). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции А с получением указанного в заголовке соединения 50 (44 мг, выход 56,4%) в виде желтого твердого вещества.
МС m/z(ИЭР): 478,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.76 (s, 1Н), 7.62 (d, 1Н), 7.59 (s, 1Н), 7.31-7.27 (m, 5Н), 7.26-7.24 (m, 1Н), 7.17-7.13 (m, 2Н), 7.05-7.03 (m, 1Н), 7.01-6.98 (m, 1Н), 6.76 (d, 1Н), 6.31 (d, 1Н), 5.67 (s, 1Н), 3.95 (s, 3Н), 3.72-3.67 (m, 1Н), 3.58-3.56 (m, 1Н), 2.93-2.87 (m, 1Н), 2.75-2.63 (m, 2Н), 1.17 (t, 3Н), 1.09 (d, 3Н).
Пример 51
(E)-3-(4-(2-(4-Этилфенил)-6-(1-метил-1H-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 51
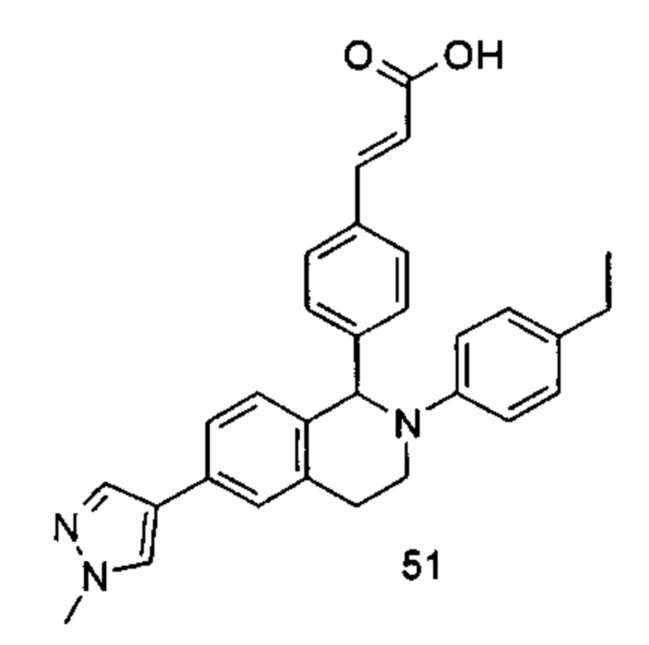
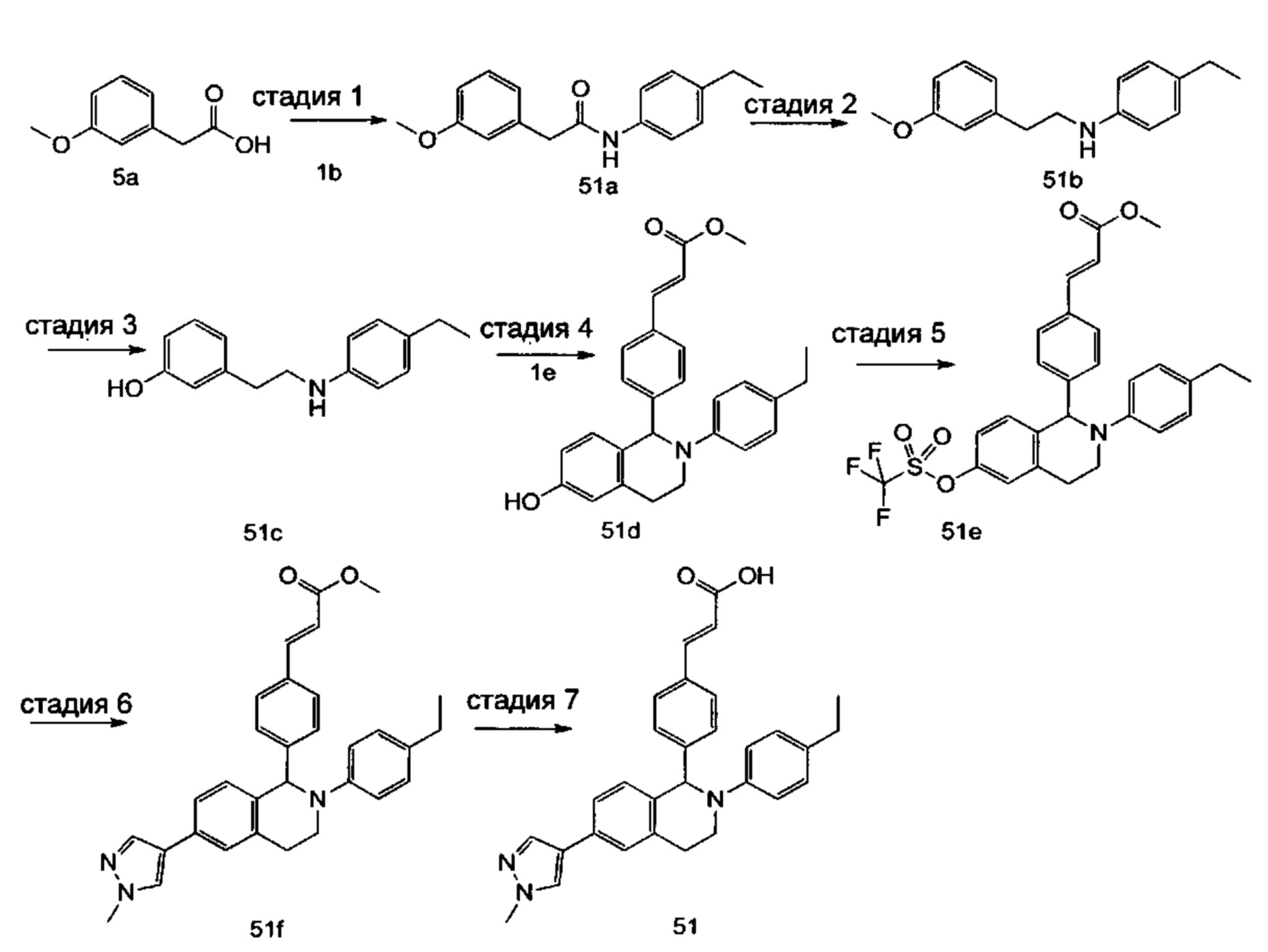
Стадия 1
N-(4-Этилфенил)-2-(3-метоксифенил)ацетамид 51а
Соединение 5а (1,77 г, 10,65 ммоль), 1b (1,29 г, 10,65 ммоль), 1-этил-(3-диметиламинопропил)карбодиимида гидрохлорид (2,45 г, 12,78 ммоль) и 4-диметиламинопиридин (130 мг, 1,07 ммоль) добавляли к 20 мл N,N-диметилформамида. Реакционную смесь перемешивали в течение 18 часов. Добавляли 30 мл воды, и смесь экстрагировали этилацетатом (100 мл×2). Органические фазы объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 51а (2,35 г, выход 81,9%) в виде почти белого твердого вещества.
Стадия 2
4-Этил-N-(3-метоксифенэтил)анилин 51b
Тетрагидроалюминат лития (1,33 г, 34,90 ммоль) суспендировали в 50 мл тетрагидрофурана, впоследствии добавляли соединение 51а (2,35 г, 8,73 ммоль). Реакционную смесь перемешивали в течение 18 часов, гасили водой и фильтровали. Фильтрационный кек промывали дихлорметаном. Фильтрат объединяли, высушивали над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 51b (1,68 г, выход: 75,3%) в виде желтого масла.
МС m/z (ИЭР): 256,2 [М+1]
Стадия 3
3-(2-((4-Этилфенил)амино)этил)фенол 51с
Соединение 51b (1,68 г, 6,58 ммоль) растворяли в 50 мл дихлорметана. После охлаждения до -78°С по каплям добавляли раствор 1M трибромида бора в дихлорметане (13,2 мл, 13,20 ммоль). После завершения добавления реакционную смесь нагревали до комнатной температуры и перемешивали в течение 16 часов. Для гашения реакции добавляли 2 мл 2М раствора соляной кислоты, впоследствии добавляли 5 мл воды для осаждения твердого вещества. Смесь фильтровали, и фильтрационный кек промывали дихлорметаном. Твердое вещество высушивали с получением указанного в заголовке соединения 51с (1,19 г, выход 74,8%) в виде желтого твердого вещества.
МС m/z (ИЭР): 242,1 [М+1]
Стадия 4
(E)-Метил-3-(4-(2-(4-этилфенил)-6-гидрокси-1,2,3,4-тетрагидроизохинолин-1-ил)фенил) акрилат 51d
Соединение 51с (367 мг, 1,52 ммоль), 1е (578 мг, 3,04 ммоль) и триизопропилсилилхлорид (1,47 г, 7,60 ммоль) добавляли к 10 мл N,N-диметилформамида. После завершения добавления смесь нагревали до 120°С и перемешивали в течение 3 часов. После прекращения нагревания реакционный раствор охлаждали до комнатной температуры и добавляли 30 мл воды. Смесь экстрагировали этилацетатом (50 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (30 мл), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 51d (437 мг, выход 69,5%) в виде желтого твердого вещества.
МС m/z (ИЭР): 414,2 [М+1]
Стадия 5
(E)-Метил-3-(4-(2-(4-этилфенил)-6-(трифторметилсульфонилокси)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 51е
Соединение 51d (437 мг, 1,06 ммоль) растворяли в 50 мл дихлорметана, затем добавляли 2,6-лутидин (170 мл, 1,59 ммоль). После завершения добавления смесь охлаждали до 0°С в ледяной бане и по каплям добавляли трифторметансульфоновый ангидрид (447 мг, 1,59 ммоль). После завершения добавления ледяную баню удаляли, и реакционную смесь перемешивали в течение 16 часов. Для гашения реакции добавляли 30 мл воды и разделяли две фазы. Органическую фазу высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 51е (359 мг, выход 62,2%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 546,2 [М+1]
Стадия 6
(E)-Метил-3-(4-(2-(4-этилфенил)-6-(1-метил-1H-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акрилат 51f
Соединение 51е (359 мг, 0,66 ммоль), 1-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1Н-пиразол (205 мг, 0,99 ммоль) и [1,1'-бис(дифенилфосфино)ферроцен]дихлорпалладий (48 мг, 0,07 ммоль) растворяли в 12 мл смеси 1,4-диоксана и воды (V:V составляет 5:1), впоследствии добавляли 2М раствор карбоната натрия (0,66 мл, 1,32 ммоль). После завершения добавления смесь перемешивали в микроволновом реакторе при 120°С в течение 40 минут. После охлаждения до комнатной температуры добавляли 30 мл воды, и смесь экстрагировали этилацетатом (30 мл×3). Органические фазы объединяли, промывали насыщенным раствором хлорида натрия (20 мл×2), высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении с получением неочищенного указанного в заголовке соединения 51f (190 мг) в виде желтого твердого вещества, которое использовали непосредственно на следующей стадии без дополнительной очистки.
МС m/z (ИЭР): 478,3 [М+1]
Стадия 7
(Е)-3-(4-(2-(4-Этилфенил)-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизох инолин-1-ил)фенил)акриловая кислота 51
Неочищенное соединение 51f (190 мг, 0,40 ммоль) растворяли в 5 мл метанола, впоследствии добавляли 2 М раствор гидроксида натрия (1 мл, 2,0 ммоль). После завершения добавления реакционную смесь перемешивали в течение 16 часов. По каплям добавляли 1М соляную кислоту для доведения рН реакционного раствора до 4, и смесь экстрагировали этилацетатом (30 мл×2). Органические фазы объединяли, промывали 10 мл насыщенного раствора хлорида натрия, высушивали над безводным сульфатом натрия и фильтровали для удаления высушивающего агента. Фильтрат концентрировали при пониженном давлении, и полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой элюции В с получением указанного в заголовке соединения 51(141 мг, выход 76,6%) в виде желтого твердого вещества.
МС m/z (ИЭР): 464,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.91 (s, 1Н), 7.79 (s, 1Н), 7.59 (d, 1Н), 7.43 (d, 2Н), 7.36 (d, 2Н), 7.25-7.20 (m, 3Н), 7.02 (d, 2Н), 6.81 (d, 2Н), 6.40 (d, 1Н), 5.79 (s, 1Н), 3.91 (s, 3Н), 3.65-3.61 (m, 1Н), 3.46-3.40 (m, 1Н), 2.99-2.93 (m, 2Н), 2.55-2.49 (dd, 2Н), 1.16 (t, 3Н).
Пример 52
(E)-3-(4-((1R,3S/1S,3R)-2-(4-Этилфенил)-3-метил-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 52
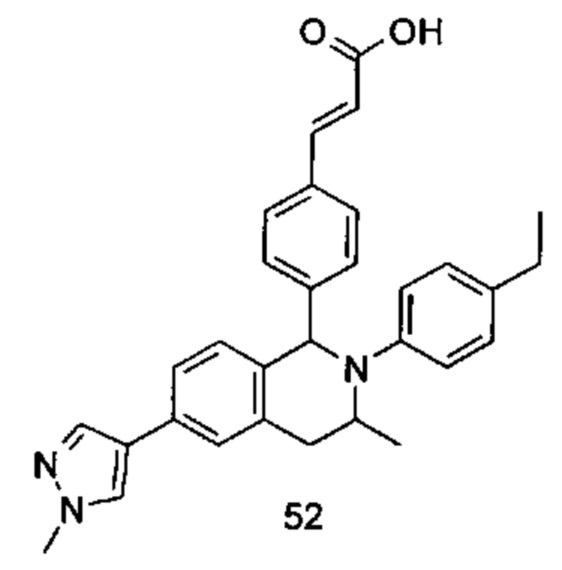
В соответствии с путем синтеза примера 50, исходное вещество 50а, используемое на стадии 1, заменяли 4-этиланилином, соответственно, получали указанное в заголовке соединение 52 (58 мг, желтое твердое вещество).
МС m/z (ИЭР): 478,5 [М+1]
1Н ЯМР (400 МГц, CDCl3) δ 7.77 (s, 1Н), 7.60 (s, 1Н), 7.45-7.36 (m, 4Н), 7.33-7.28 (m, 2Н), 7.18 (s, 1Н), 7.10-7.04 (m, 3Н), 6.83 (d, 2Н), 6.40 (d, 1Н), 5.62 (s, 1Н), 3.96 (s, 3Н), 3.83-3.80 (m, 1Н), 2.88-2.85 (m, 1Н), 2.67-2.54 (m, 3Н), 1.32 (d, 3Н), 1.20 (t, 3Н).
Пример 53
(E)-3-(4-(2-(4-Фторфенил)-6-(1-метил-1Н-пиразол-4-ил)-1,2,3,4-тетрагидроизохинолин-1-ил)фенил)акриловая кислота 53
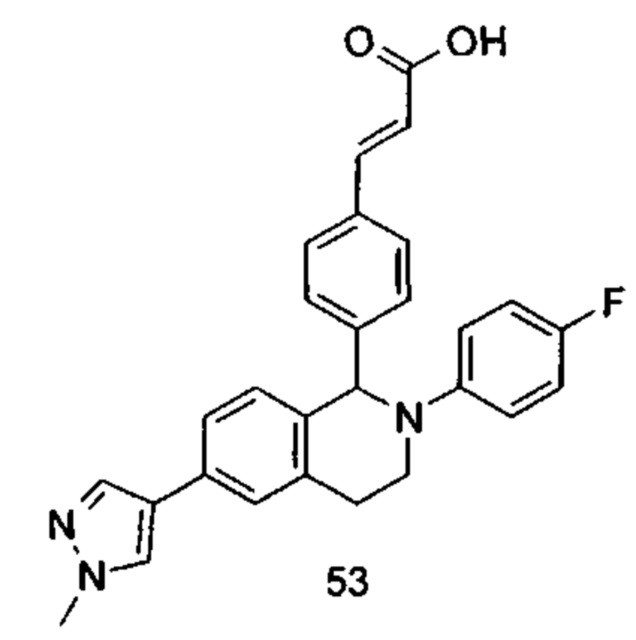
В соответствии с путем синтеза примера 51 исходное вещество 1b, используемое на стадии 1, заменяли 4-фторанилином, соответственно, получали указанное в заголовке соединение 53 (108 мг, желтое твердое вещество).
МС m/z (ИЭР): 453,5 [М+1]
1Н ЯМР (400 МГц, CD3OD) δ 7.92 (s, 1Н), 7.80 (s, 1Н), 7.60(d, 1Н), 7.46 (d, 2Н), 7.39-7.36 (m, 2Н), 7.25-7.18 (m, 3Н), 6.94-6.84 (m, 4Н), 6.41 (d, 1Н), 5.76 (s, 1Н), 3.91 (s, 3Н), 3.66-3.60 (m, 1Н), 3.45-3.39 (m, 1Н), 3.03-2.97 (m, 2Н).
Биологический анализ
Далее настоящее изобретение будет описано со ссылкой на следующие тестовые примеры, но данные примеры не следует рассматривать как ограничивающие объем изобретения.
Тестовый пример 1. Ингибирующее действие соединения по настоящему изобретению на связывание эстрогена (Е) с рецептором эстрогена (ER)
1. Цель эксперимента
Соединения по настоящему изобретению оказывают ингибирующее действие на связывание Е (эстрогена) с ER (рецептором эстрогена), блокируя, таким образом, связывание комплекса Е и ER с ERE (эстроген-чувствительным элементом) и впоследствии блокируя экспрессию последующего белка люциферазы.
Ингибиторующее действие соединений на связывание Е с ER in vitro тестировали следующим способом.
Цель данного эксперимента состояла в определении ингибирующего действия соединений на связывание Е с ER, и оценивали активность соединений in vitro в соответствии со значениями IC50.
2. Экспериментальные материалы и приборы
2.1 Экспериментальные приборы
2.2 Экспериментальные материалы
3. Экспериментальный способ
ERE клонировали выше гена люциферазы, и моноклональные клетки MCF-7I ERE-люцифераза отбирали путем трансфекции MCF-7 (Банк клеток Комитета по сохранению типовых клеточных культур Китайской Академии Наук, TCHu74). Клетки МСР-7/ERE-люцифераза инокулировали в минимальную питательную среду (MEM - от англ. Minimal essential medium) (Hyclone, SH30024.01B), содержащую 10% фетальную телячью сыворотку (ФТС), очищенную на активированном угле (Moregate, FBSF), 1% пируват натрия (Sigma, S8636), 1% заменимые аминокислоты (Sigma, М7145) и 500 мкг/мл G418, в 96-луночных планшетах при плотности 30000 клеток/лунка, и клетки инкубировали в условиях 37°С и 5% CO2. Лекарственное средство получали в виде 20 мМ исходного раствора, который позже разводили 100% ДМСО с получением 10-кратного градиента концентраций и впоследствии разводили 20-кратной питательной средой DMEM (от англ. Dulbecco's Modified Eagle's medium - среда Игла, модифицированная по Дульбекко). После инкубации в течение 24 часов питательную среду удаляли, затем в каждую лунку добавляли 0,1 нМ эстрадиол (Sigma, Е2758) и 10 мкл лекарственного средства, разведенного питательной средой, а в контрольную группу добавляли ДМСО. Планшет аккуратно встряхивали и инкубировали в инкубаторе при 37°С, 5% CO2. Через 24 часа среду для культивирования клеток удаляли. Впоследствии в каждую лунку добавляли 50 мкл подготовленного субстрата люциферазы (Promega, Е6110), и планшет помещали в темное место при комнатной температуре на 10-15 минут, затем определяли значение хемилюминесцентного сигнала.
4. Результат теста
В описанном выше эксперименте тестировали ингибирующее действие соединений по настоящему изобретению на связывание Е с ER. Значения хемилюминесцентного сигнала относительно логарифмических значений концентрации данных соединений наносили на график с использованием программы Graghpad Prism, и измеренные значения IC50 показаны в таблице 1.
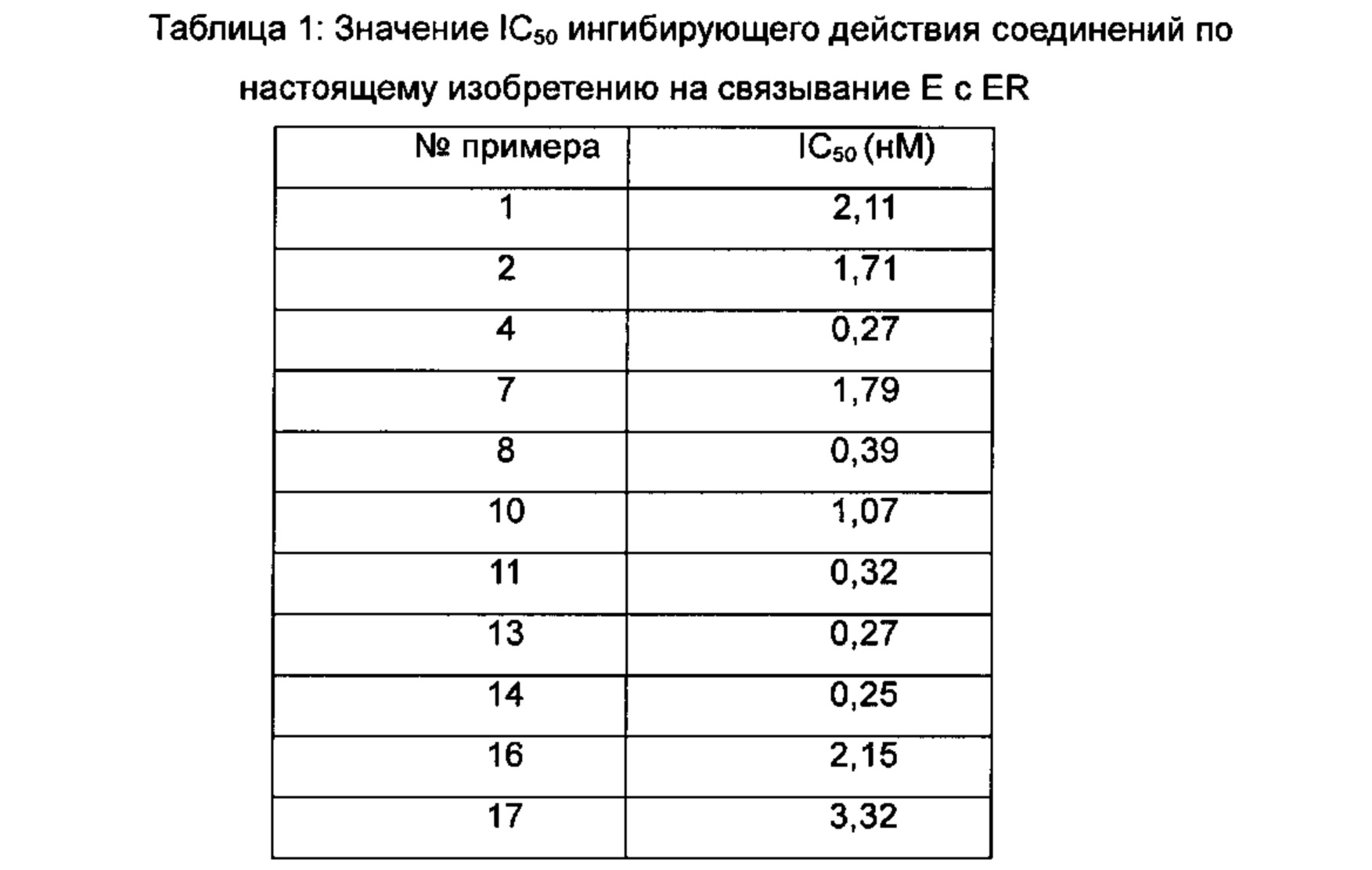
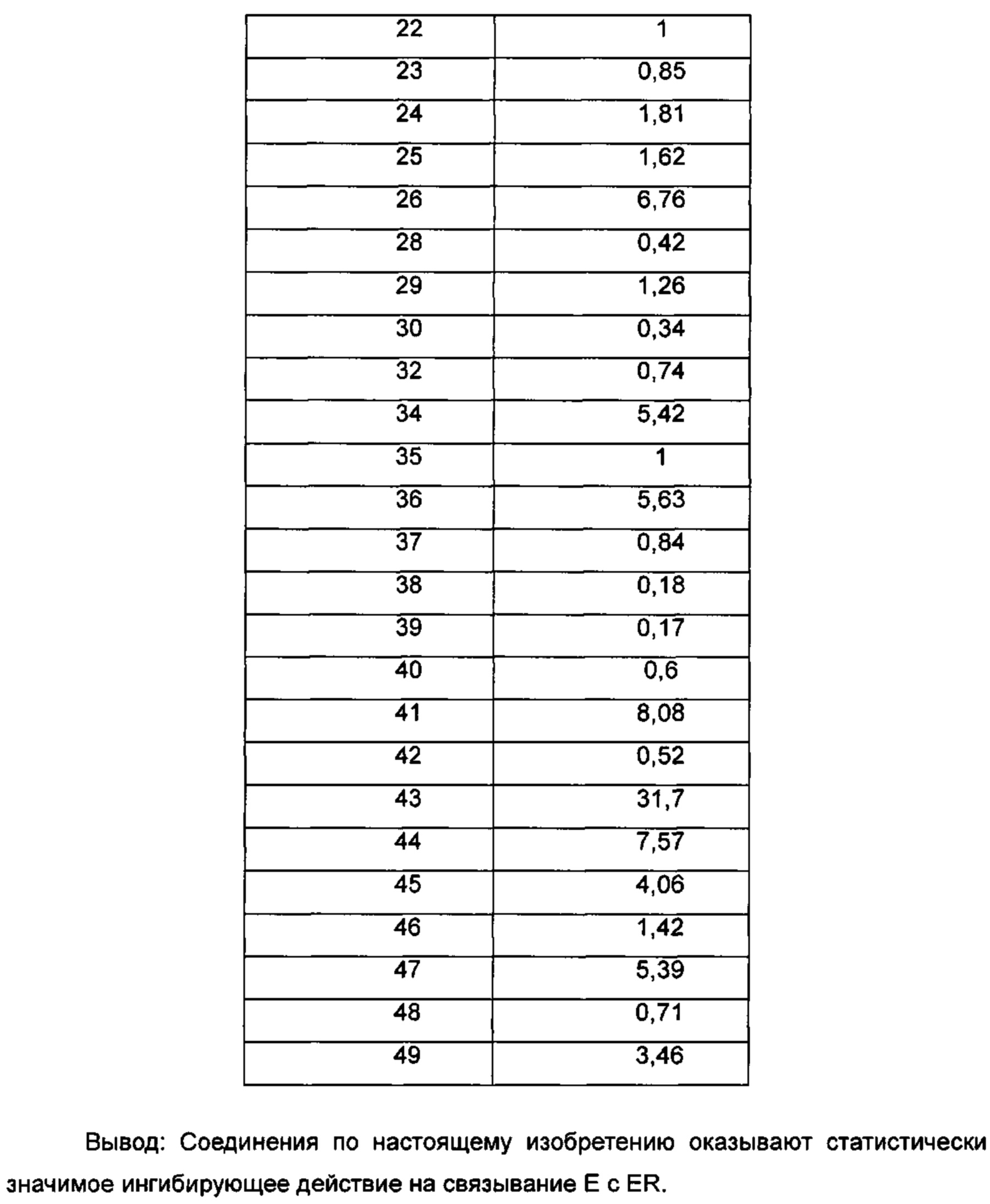
Тестовый пример 2. Ингибирующее действие соединений по настоящему изобретению на пролиферацию клеток MCF7
1. Цель эксперимента
Цель данного эксперимента состояла в определении ингибирующего действия соединений на активность пролиферации клеток MCF7, и активность данных соединений in vitro оценивали в соответствии со значениями IC50.
2. Экспериментальные реактивы и материалы
3. Экспериментальный способ
Клетки MCF-7 (Банк клеток Комитета по сохранению типовых клеточных культур Китайской Академии Наук, TChu 74) инокулировали в питательную среду MEM (Hyclone, SH30024.01B), содержащую 10% ФТС (Gibco, 10099-141), 1% пируват натрия (Sigma, S8636) и 1% заменимые аминокислоты (Sigma, М7145), в 96-луночном планшете при плотности 40000 клеток/лунка, и клетки инкубировали в условиях 37°С и 5% CO2. Соединение получали в виде 20 мМ исходного раствора, который позже разводили до 1000-кратной конечной концентрации 100% ДМСО и впоследствии разводили 20-кратной питательной средой, содержащей 2% ФТС. После инкубации в течение 24 часов питательную среду удаляли, и в каждую лунку добавляли 90 мкл питательной среды, содержащей 2% ФТС и 10 мкл лекарственного средства, в контрольную группу добавляли 10 мкл ДМСО, а холостая проба содержала только 100 мкл питательной среды, содержащей 2% ФТС. Планшет аккуратно встряхивали и инкубировали в инкубаторе при 37°С, 5% CO2. Через 72 часа в каждую лунку добавляли 50 мкл смешанного реагента Cell Titer-Glo (Promega, G7571). Планшет встряхивали до однородного перемешивания ингредиентов и помещали при комнатной температуре на 10 минут, затем определяли величину хемилюминесцентного сигнала. Ингибирущее действие соединения AZD9496 при концентрации 100 нМ на пролиферацию клеток MCF-7 принимали за 100%, и максимальная степень ингибирования других соединений = (величина хемилюминесцентного сигналаотрицательный контроль - величина хемилюминесцентного сигналасоединение 100 нм)/(величина хемилюминесцентного сигналаотрицательный контроль - величина хемилюминесцентного сигналаАZD9496 100 нм)×100%, и отрицательный контроль представлял собой лунку с 0,5% ДМСО.
4. Анализ результатов
Значения хемилюминесцентных сигналов относительно логарифмических значений концентрации соединений наносили на график с использованием программы Graghpad Prism с получением значений IC50. Результаты показаны в таблице 2.
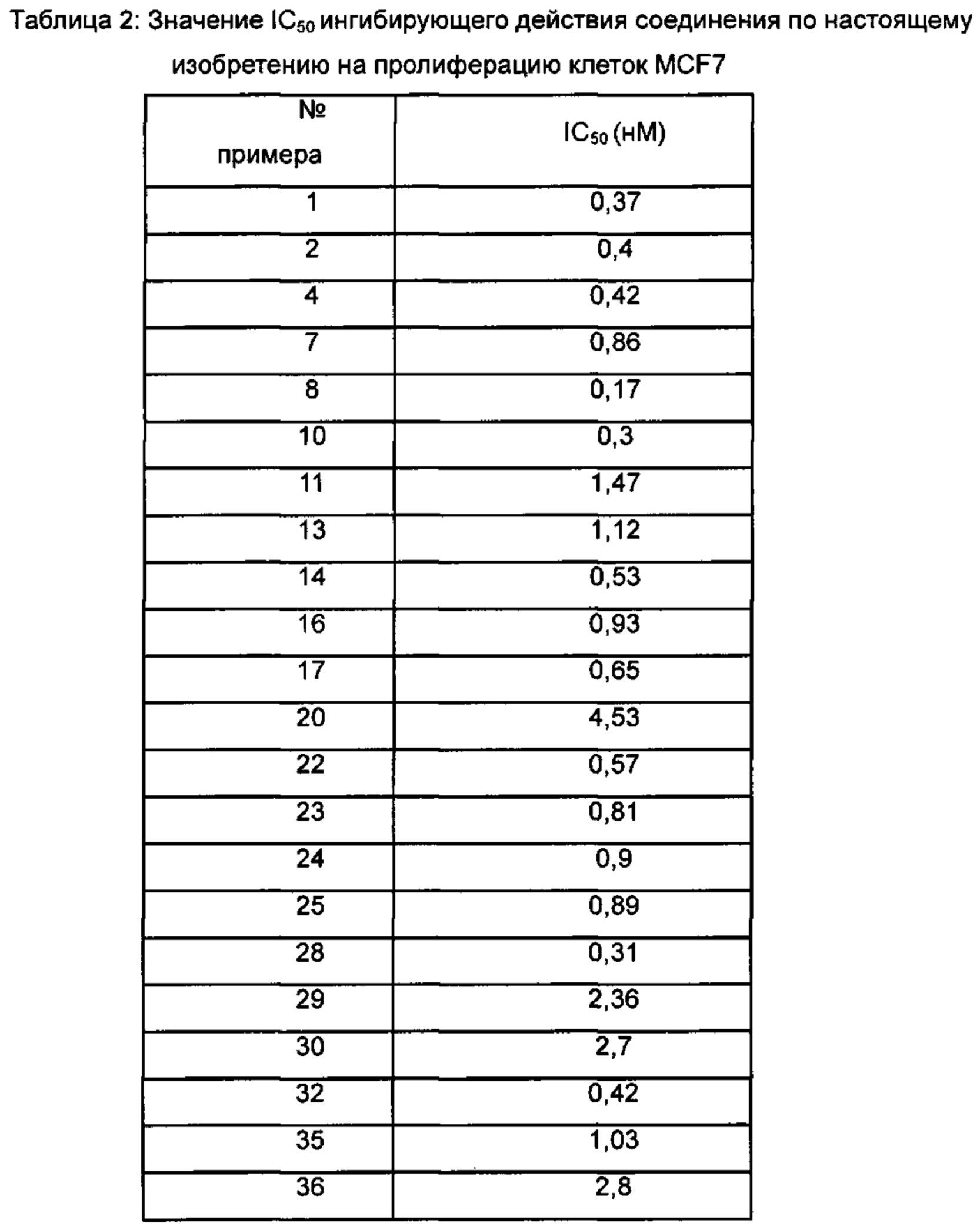
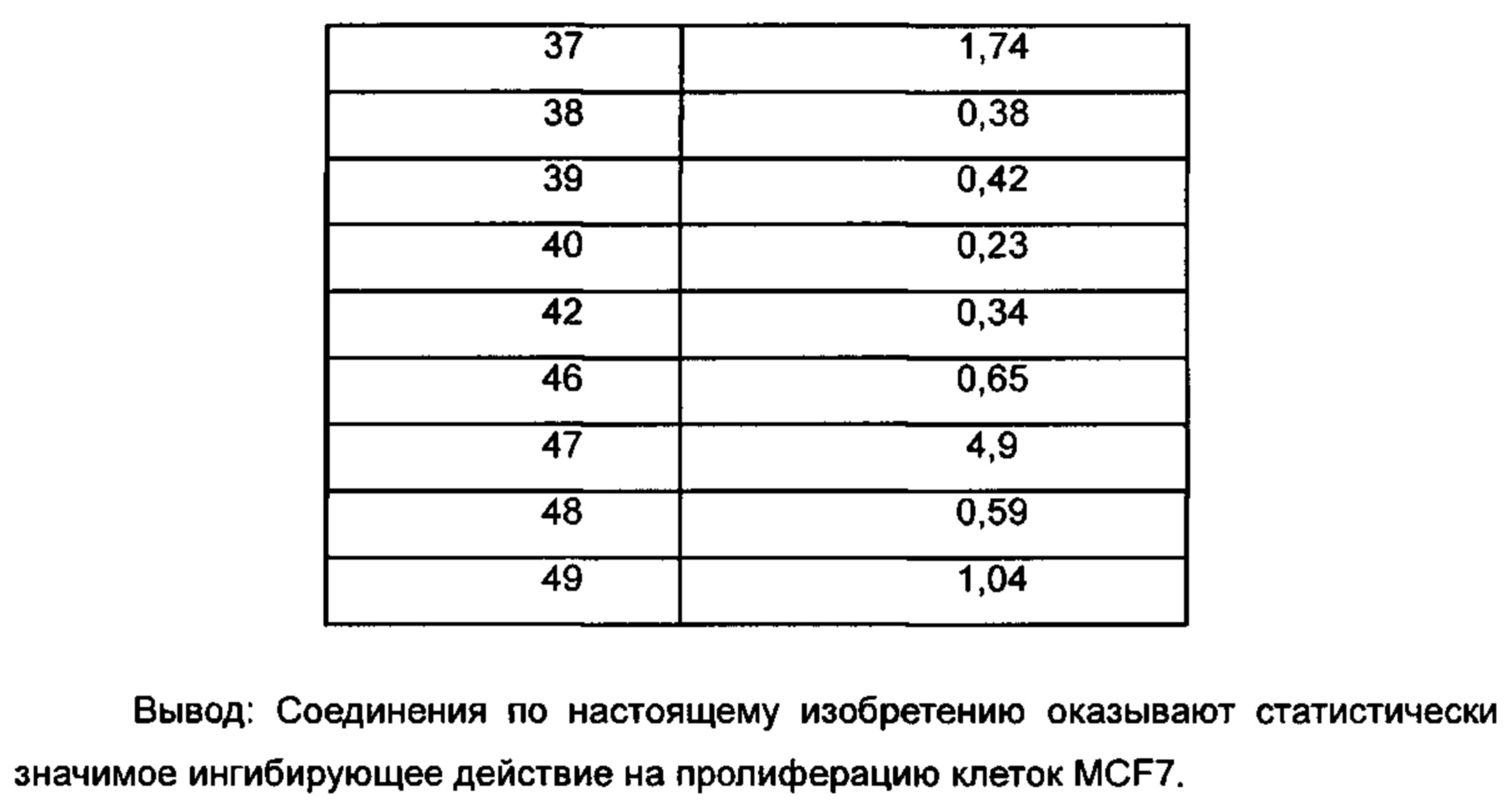
Тестовый пример 3. Действие соединений по настоящему изобретению, вызывающее деградацию ERα
1. Цель эксперимента
Для определения действия соединения по настоящему изобретению, вызывающего деградацию ER, использовали следующий способ определения действия соединений по настоящему изобретению, вызывающего деградацию ER.
2. Материалы и оборудование
Планшетный ридер BioTek Synergy НТ
Линия клеток MCF-7 (TChu 74, Банк клеток Комитета по сохранению типовых клеточных культур Китайской Академии Наук)
Набор реагентов ERα Duoset (#DYC5715E, R&D System)
3. Экспериментальный метод
Клетки MCF-7 инкубировали в питательной среде DMEM/F-12, содержащей 10% ФТС.
В первые сутки эксперимента клетки MCF-7 ресуспендировали в питательной среде DMEM/F-12, содержащей 10% ФТС, очищенную активированным углем, затем инокулировали в 48-луночный планшет при плотности 50000 клеток/лунка и инкубировали в течение 22-24 часов.
На вторые сутки эксперимента тестируемое соединение разводили питательной средой и добавляли в 48-луночный планшет.Антитело, захватывающее ERα, разводили до 1 мкг/мл фосфатно-солевым буферным раствором (ФСБ) и добавляли в 96-луночный планшет в количестве 100 мкл/лунка. Планшет блокировали и покрывали в течение ночи при комнатной температуре.
На третьи сутки эксперимента покрытый 96-луночный планшет дважды промывали ФСБ, добавляли блокирующий раствор (1% бычий сывороточный альбумин (БСА) в ФСБ) в количестве 110 мкл/лунка и блокировали в течение 1 часа при комнатной температуре. 48-луночный планшет промывали один раз и удаляли остаточную жидкость. Впоследствии в каждую лунку добавляли 60 мкл лизирующего буфера (6 М мочевина, 1 мМ ЭДТА (этилендиаминтетрауксусная кислота), 0,5% Тритон Х-100, 1 мМ фенилметилсульфонилфторид (ФМСФ), смесь ингибиторов протеаз). После лизиса на льду в течение 15 минут добавляли разбавитель (1 мМ этилендиаминтетрауксусная кислота (ЭДТА), 0,5% Тритон Х-100, растворенный в ФСБ). Разведенный лизат клеток переносили в 96-луночный планшет в количестве 100 мкл/лунка, затем планшет инкубировали при комнатной температуре в течение 2 часов. Разведенное первичное антитело добавляли после того, как планшет промывали 4 раза промывочной жидкостью (фосфатно-солевой буферный раствор с Твином (ФСБТ)). После инкубации в течение 1 часа 96-луночный планшет промывали 4 раза и добавляли второе антитело, затем планшет инкубировали в течение 30 минут. После промывания планшета промывочной жидкостью добавляли хромогенный раствор тетраметилбензидина (ТМБ) и инкубировали в течение 15 минут. Реакцию останавливали добавлением 1 М H2SO4, затем считывали поглощение света при длине волны 450 нм. Вызывающее деградацию действие соединения AZD9496 в концентрации 3 мкМ принимали за 100%, Emax других соединений = (ОDотрицательный контроль - ОDсоединение 3 мкМ)/(ODотрицательный контроль - ODAZD9496 3 мкМ)×100%, и в качестве отрицательного контроля использовали лунку с ДМСО. 4.
Результат теста
Значения IC50, измеренные для действия соединений по настоящему изобретению, вызывающего деградацию ERα, показаны в таблице 3.
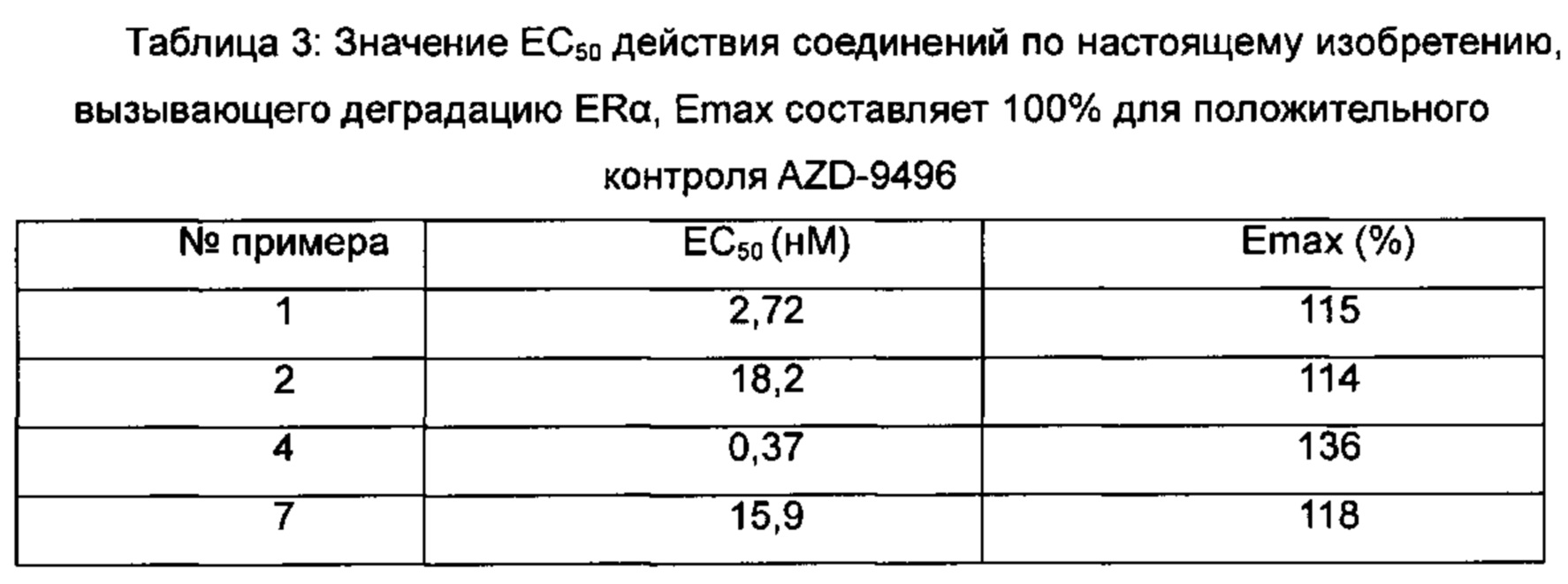
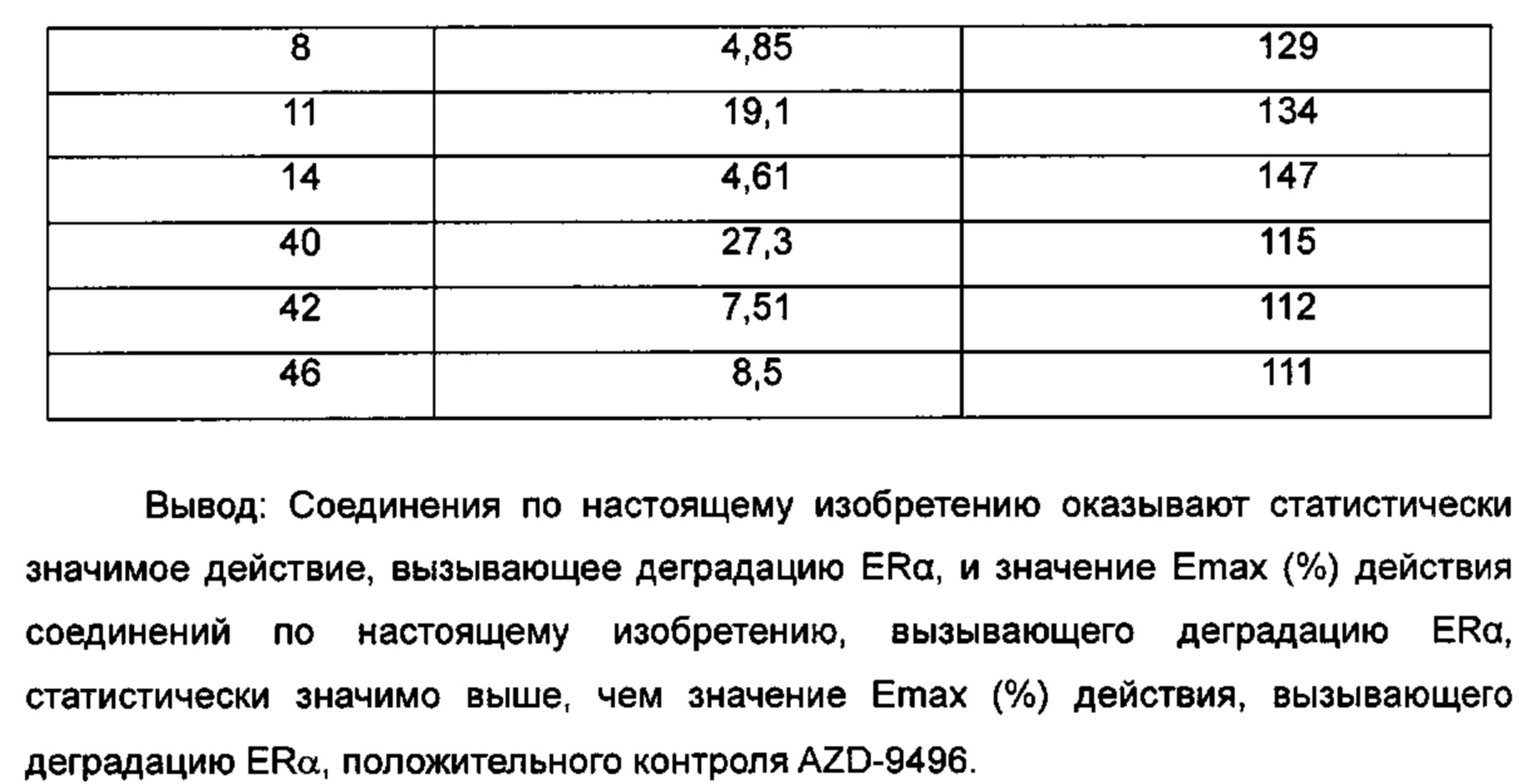
Фармакокинетический анализ
Тестовый пример 4. Фармакокинетический анализ соединений примеров 1, 7, 8, 10, 11, 14, 16, 39 и 42 по настоящему изобретению
1. Краткий обзор
В качестве экспериментальных животных использовали мышей линии BALB/C. Концентрацию лекарственного средства в плазме в различные моменты времени определяли методом жидкостной хроматографии с тандемной масс-спектрометрией (ЖХ/МС/МС) после внутрижелудочного введения соединений примеров 1, 7, 8, 10, 11, 14, 16, 39 и 42 голым мышам BALB/C. Фармакокинетические свойства соединений по настоящему изобретению изучали и оценивали у голых мышей BALB/C.
2. Протокол
2.1 Образцы
Соединения примеров 1, 7, 8, 10, 11, 14, 16, 39 и 42
2.2 Экспериментальные животные
Восемьдесят одну (81) самку голых мышей BALB/C, которые были приобретены у компании SINO-BRITSH SIPPR/BK LAB. ANIMAL LTD., СО, с №сертификата: SCXK (г. Шанхай) 2008-0016, делили поровну на 9 групп.
2.3 Получение тестируемых соединений
Взвешивали соответствующее количество каждого тестируемого соединения и к нему последовательно добавляли смесь 9% полиэтиленгликоля (ПЭГ) 400, 0,5% Твина 80, 0,5% поливинилпирролидона (ПВП) и 90% водного раствора 0,5% карбоксиметилцеллюлозы (КМЦ).
2.4 Введение препарата
После голодания в течение ночи 81 самку голых мышей BALB/C поровну делили на 9 групп и внутрижелудочно вводили тестируемые соединения в объеме введения 0,2 мл/10 г.
3. Способ
81 самке голых мышей Balb/C внутрижелудочно вводили тестируемые соединения после голодания в течение ночи. Забор крови (0,1 мл) (3 животных на каждый момент времени) осуществляли через 0,5, 1,0, 2,0, 4,0, 6,0, 8,0, 11,0, 24,0 часа после введения. Образцы хранили в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин, чтобы отделить плазму крови. Образцы плазмы хранили при -20°С.
Концентрацию прототипического лекарственного средства в плазме и в растворе, подлежащем введению, определяли посредством жидкостной хроматографии с тандемной масс-спектрометрией (ЖХ/МС МС).
4. Результаты фармакокинетических параметров у голых мышей BALB/C
Фармакокинетические параметры соединений примеров 1, 7, 8, 10, 11, 14, 16, 39 и 42 по настоящему изобретению показаны ниже.
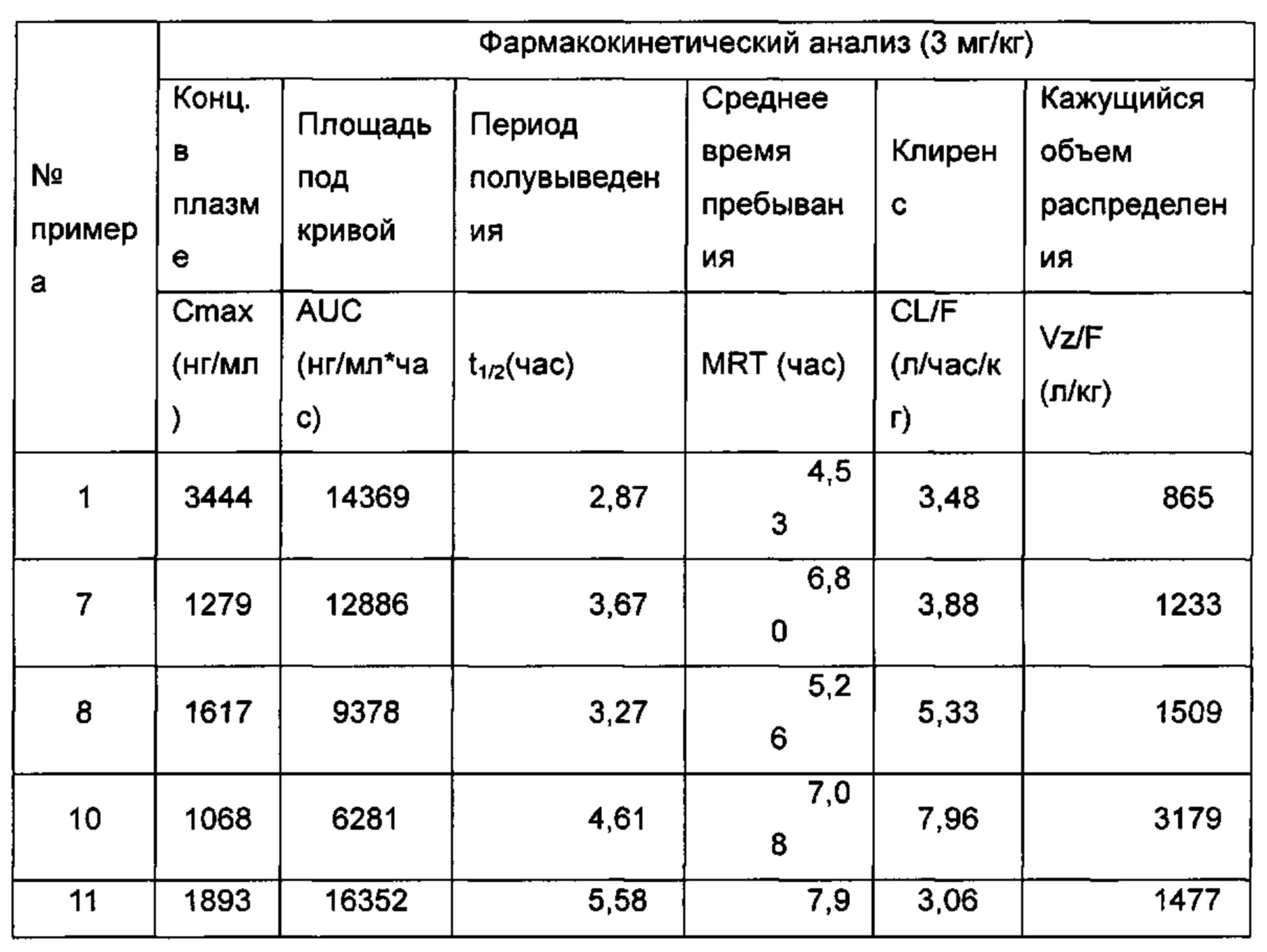
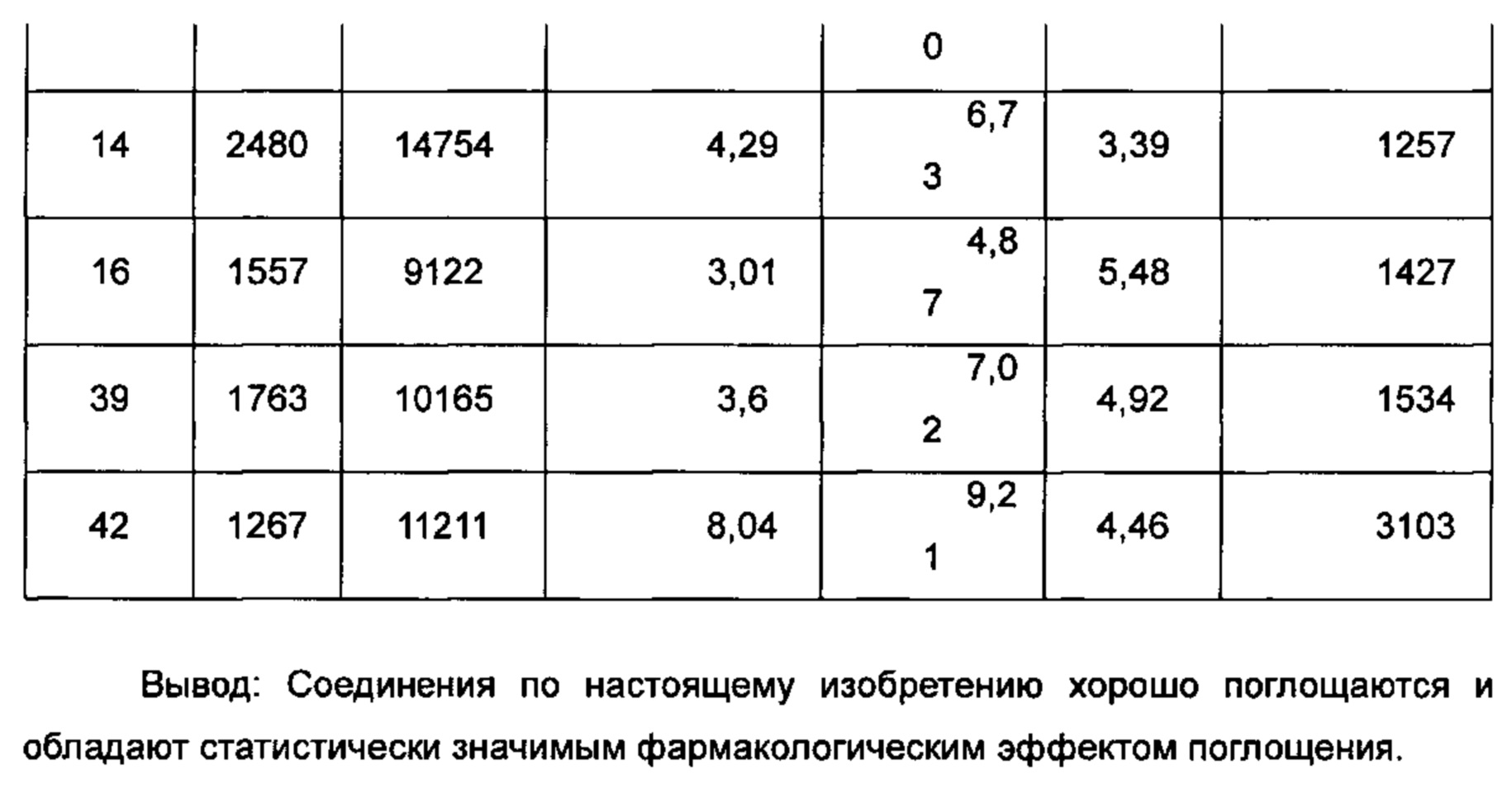
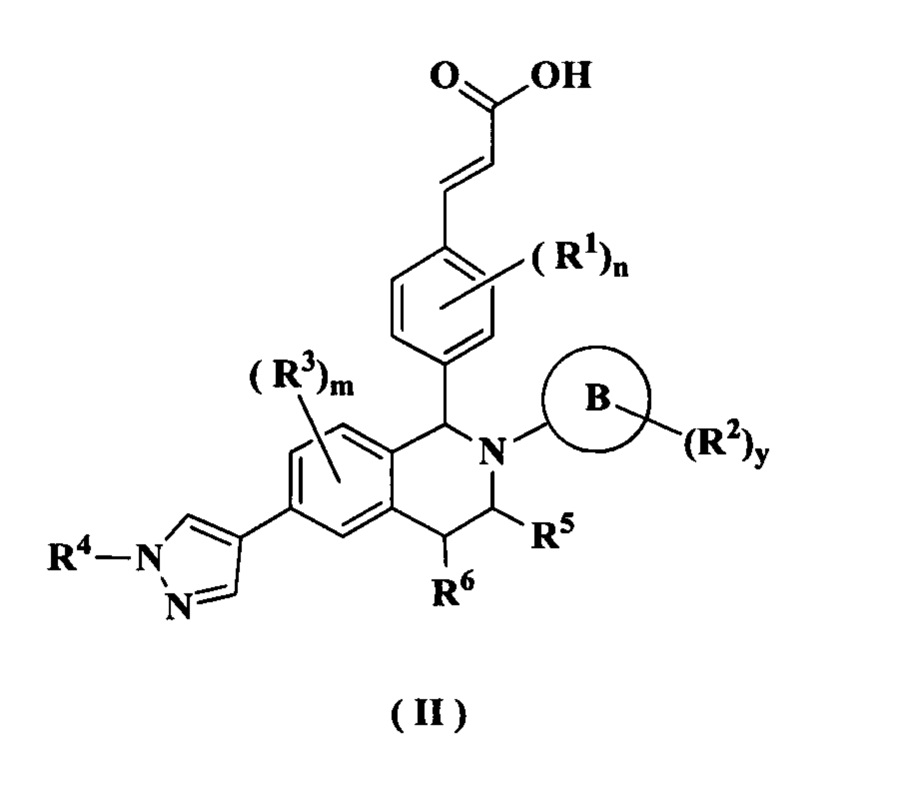
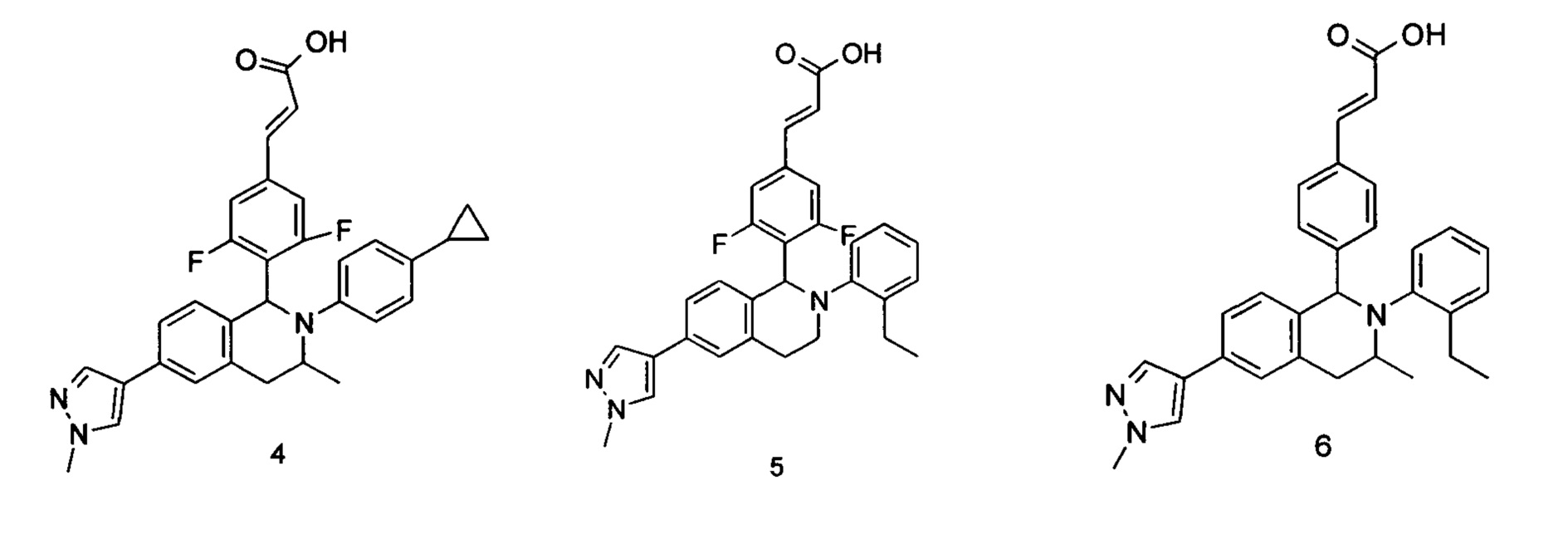
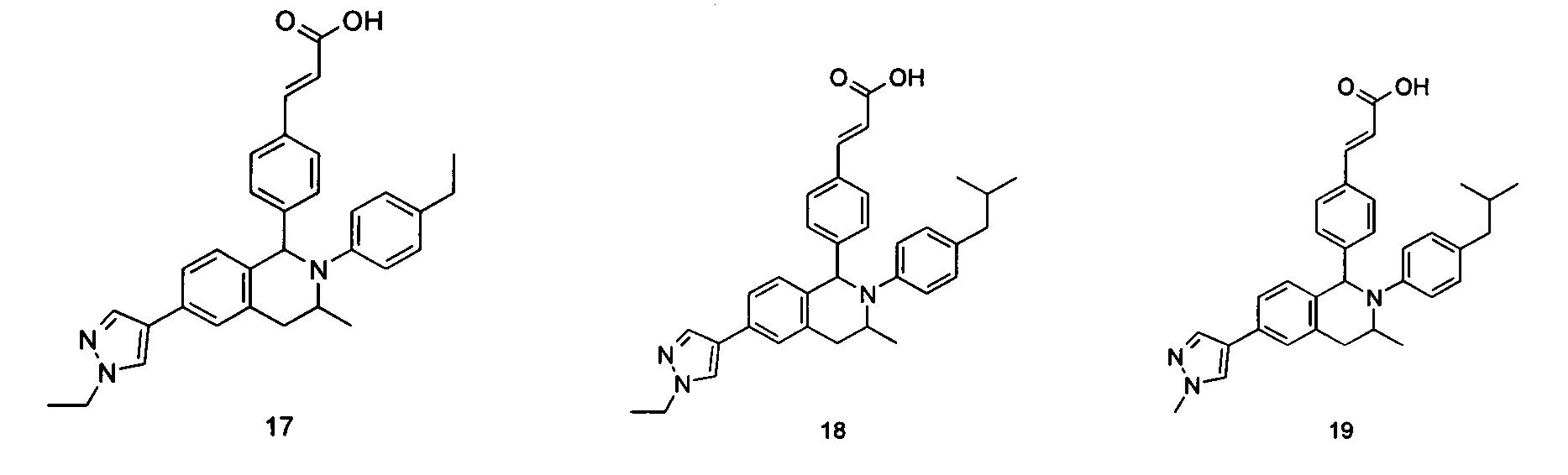
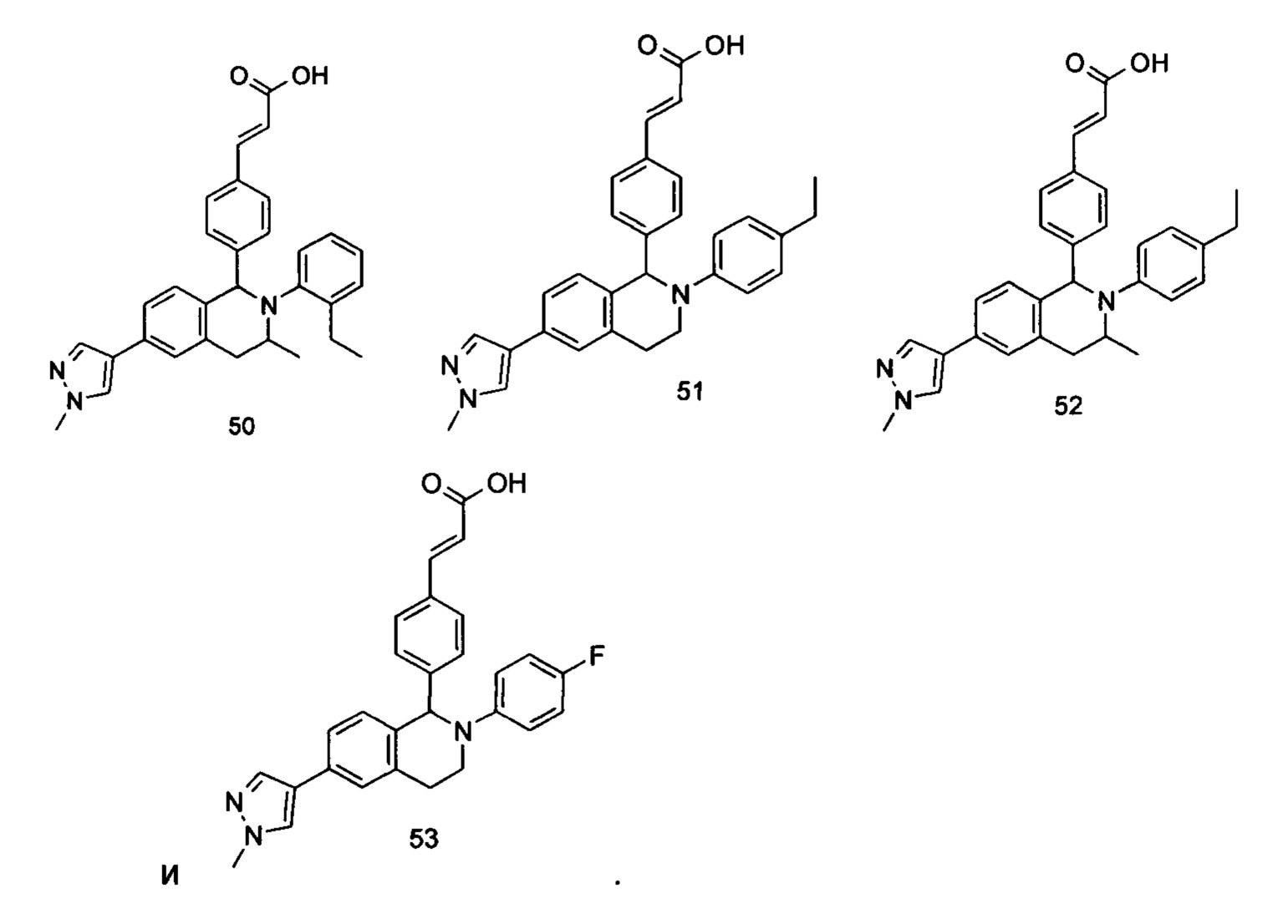
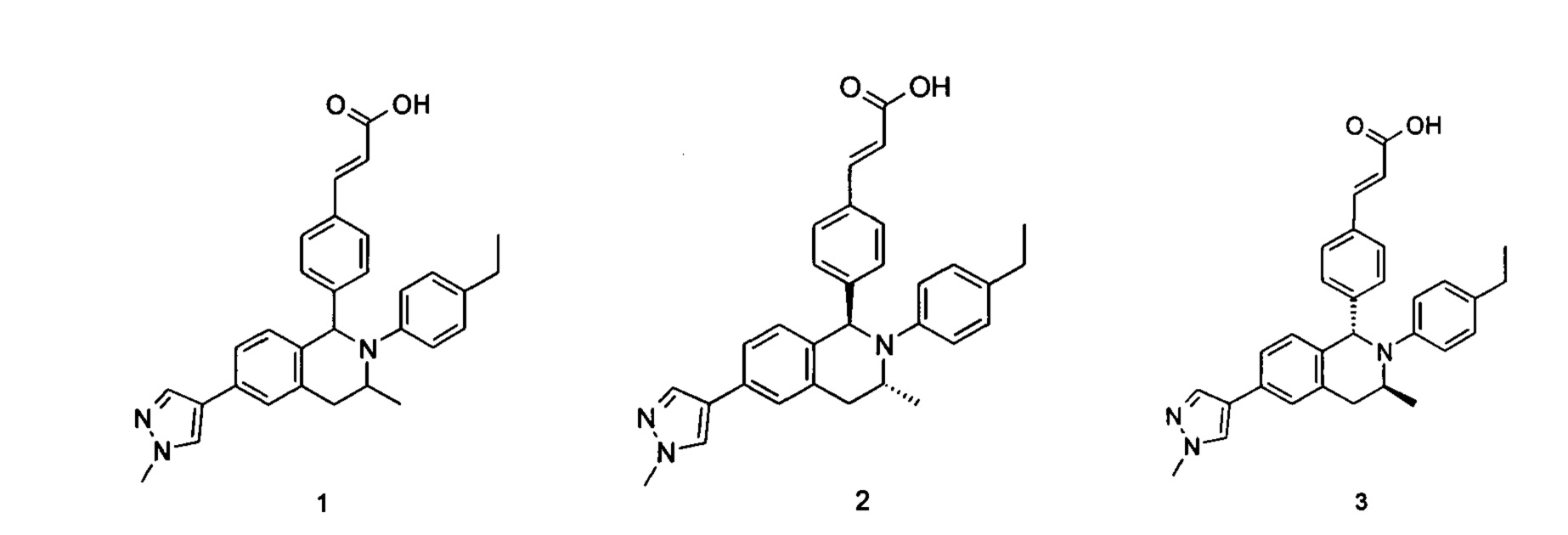
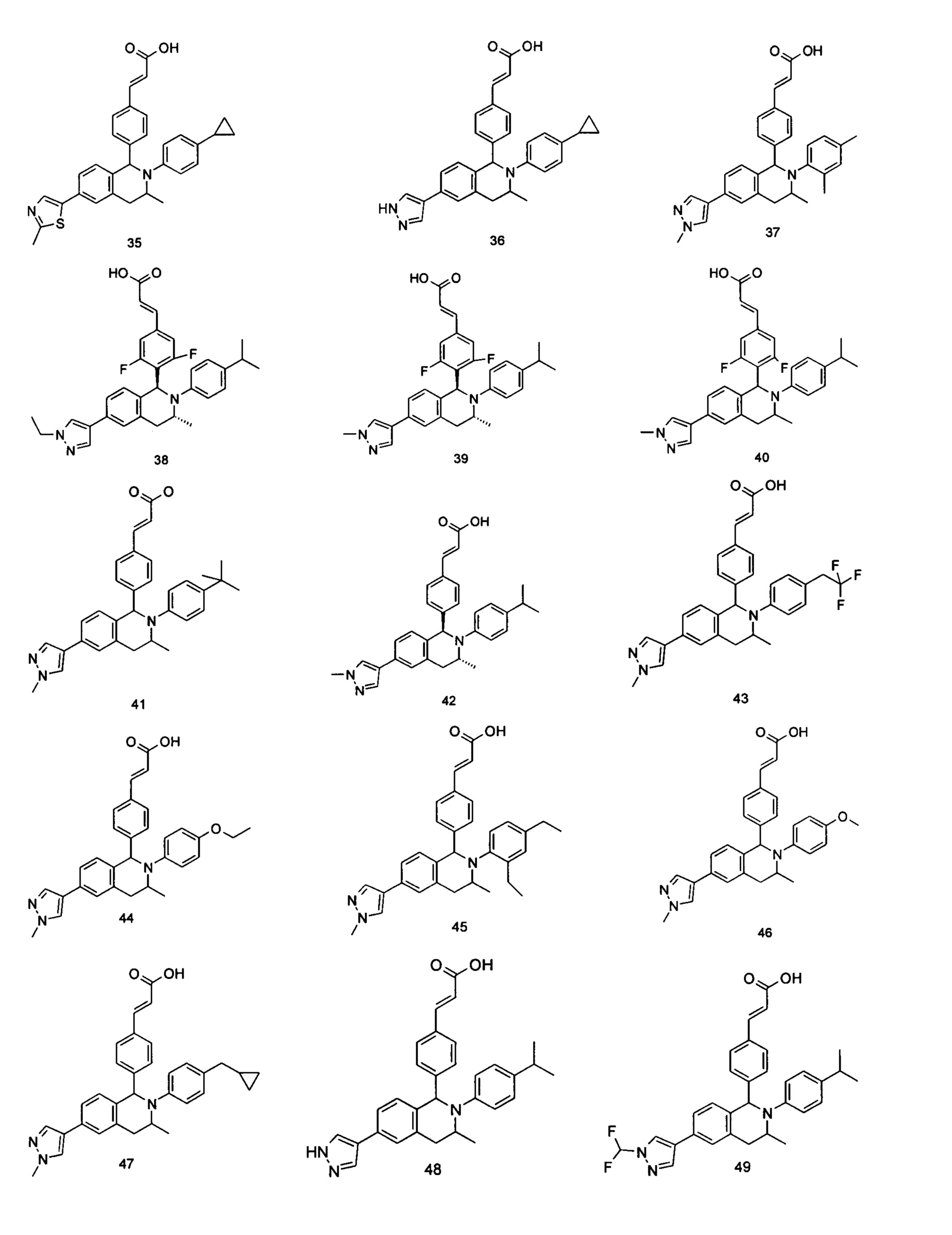
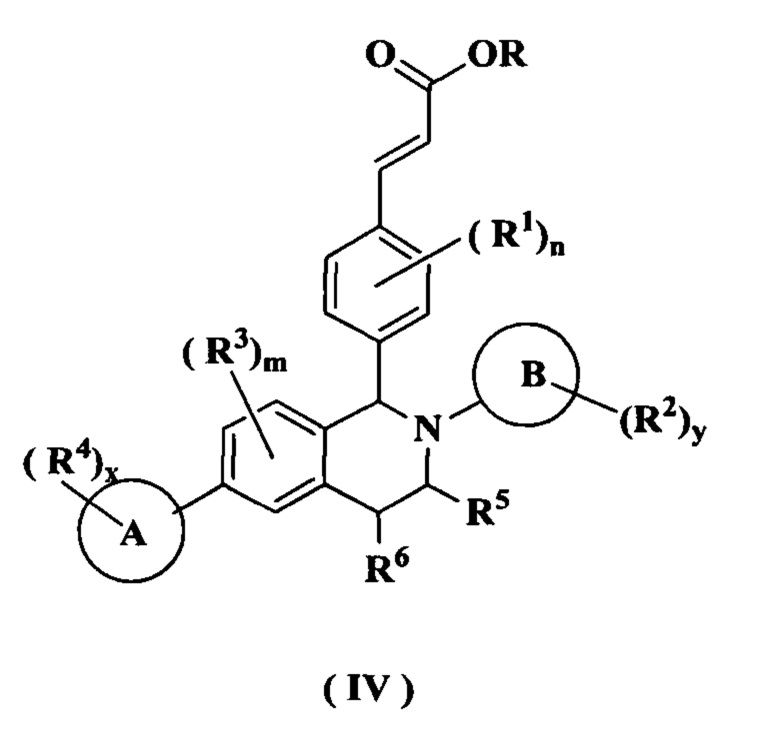
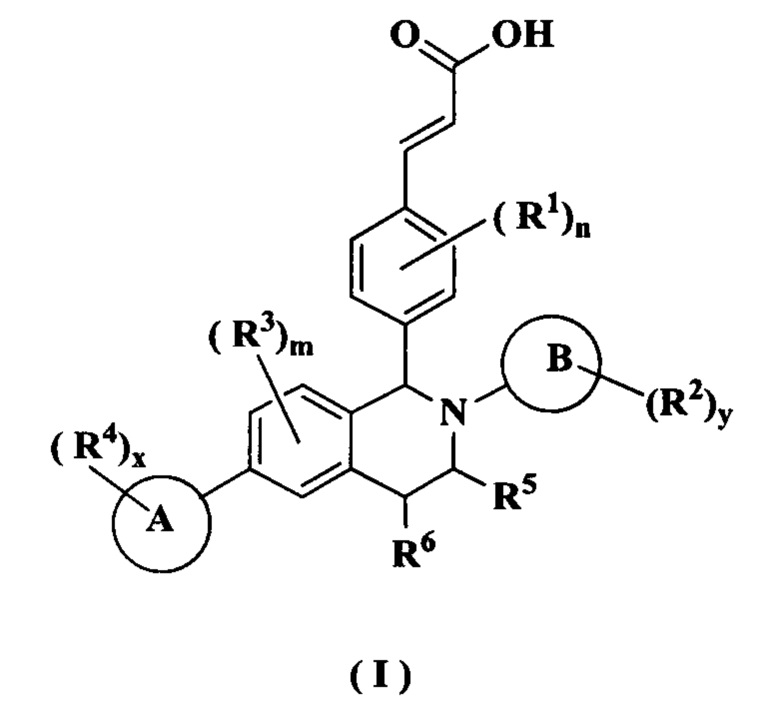
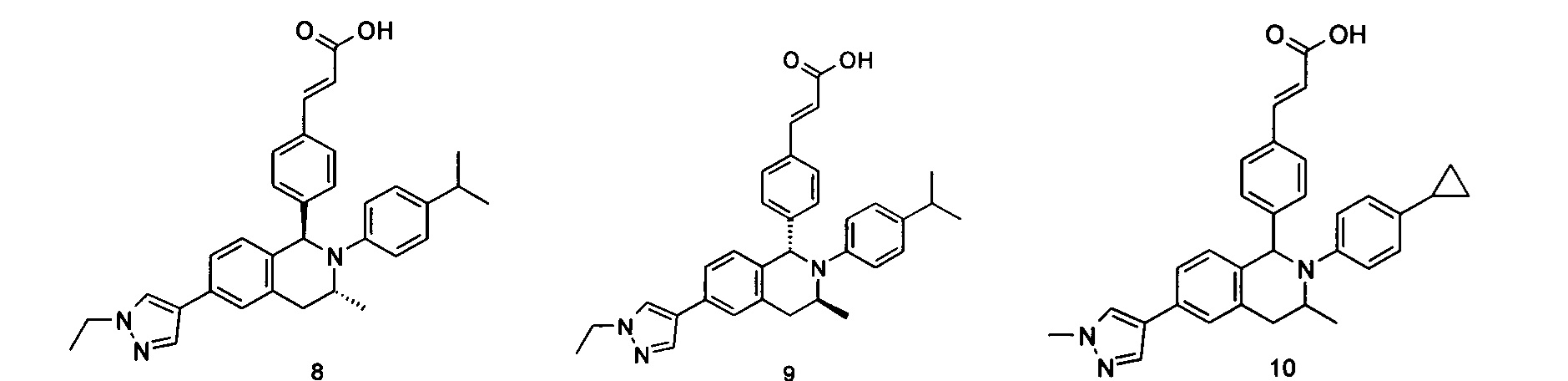
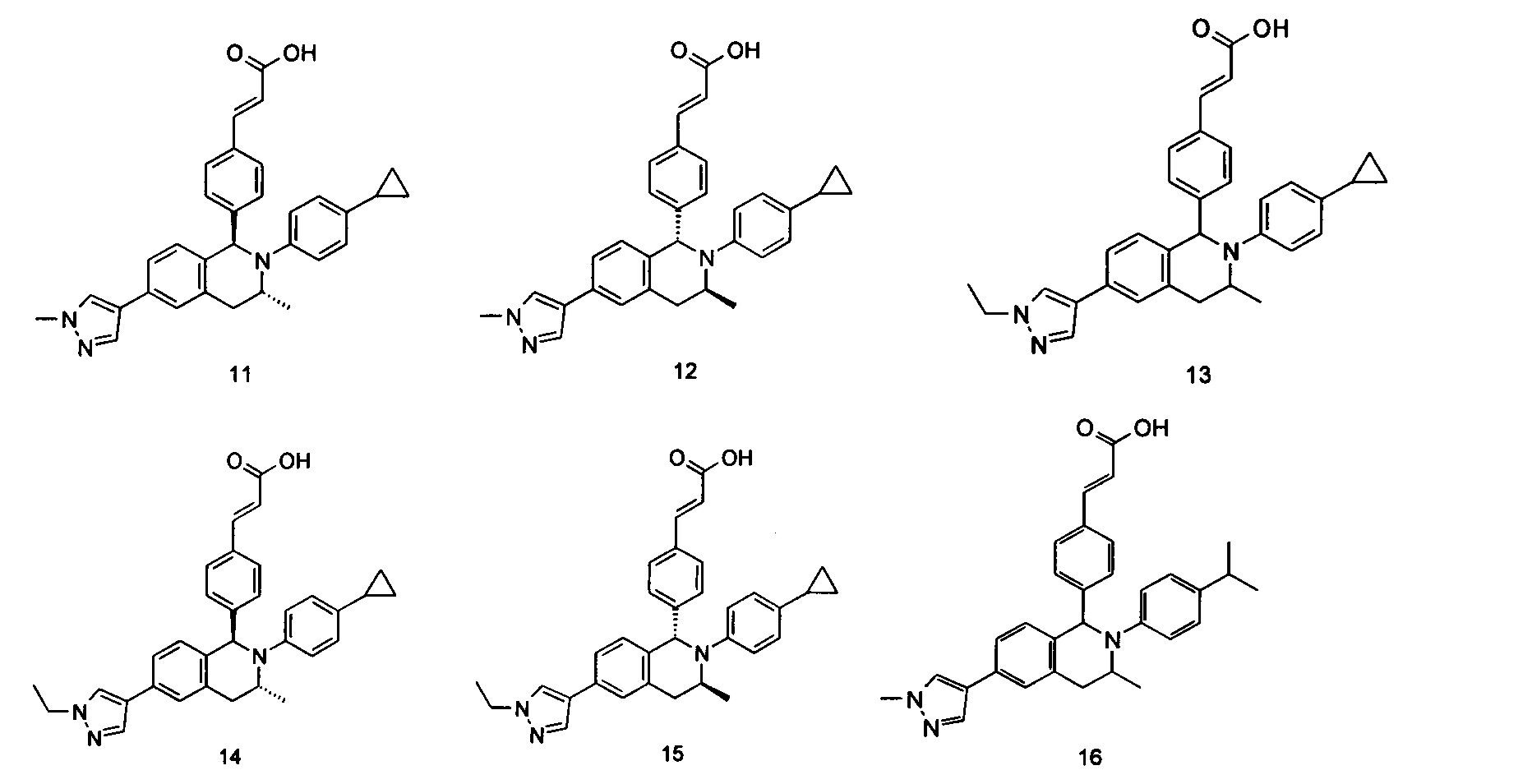
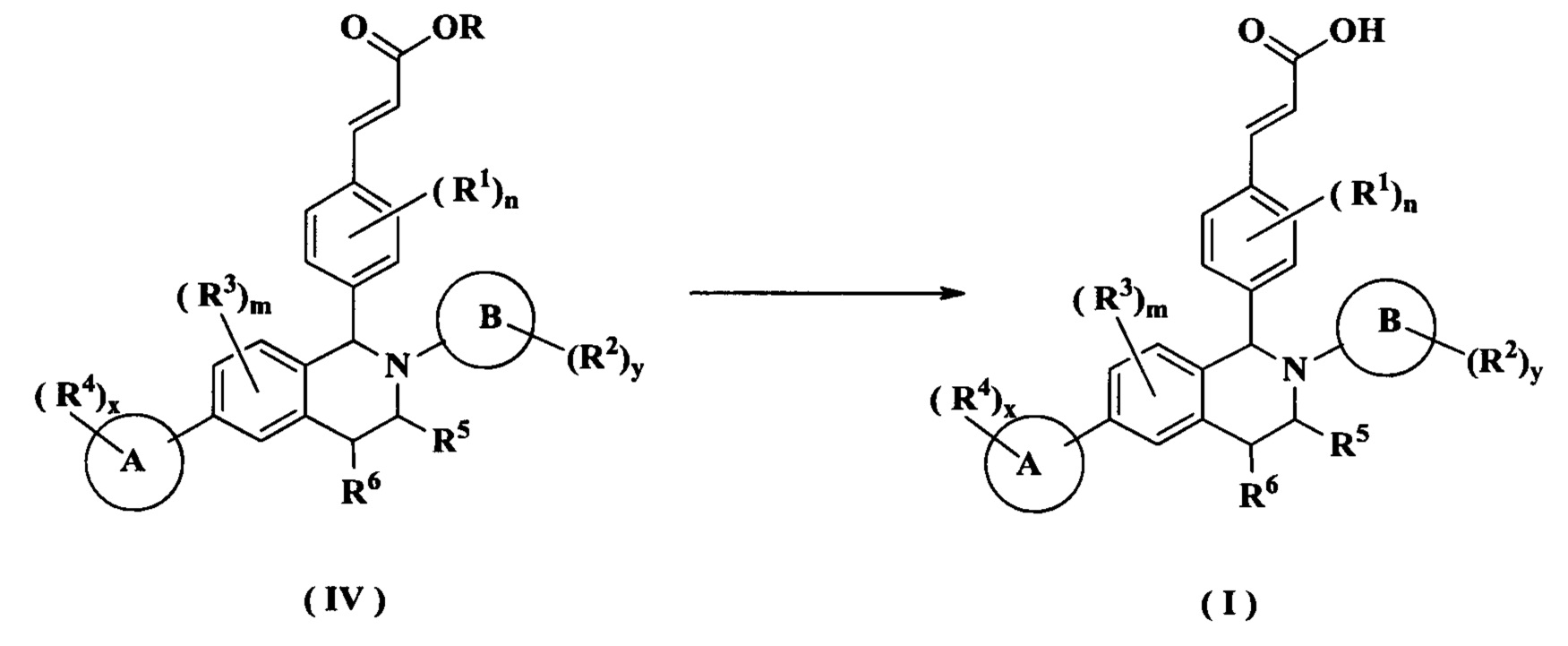
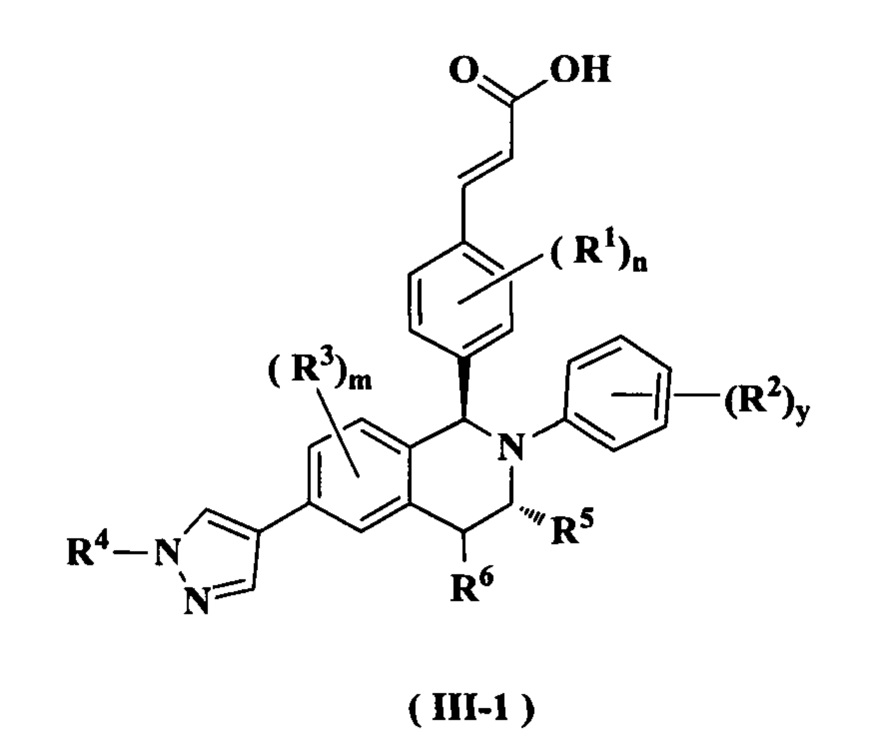
Соединения 2-(2-оксоиндолин-3-илиден)метил-5-(2-гидрокси-3-морфолин-4-илпропил)-6,7 дигидро-1-н-пиррол[3,2-с]пиридин-4(5н)-она и их применение в качестве ингибиторов протеинкиназы
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Производные дициклоазаалкана, способы их получения и их применение в медицине
Бициклозамещенные азопроизводные пиразолона, способ их получения и фармацевтическое применение
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Липосомы иринотекана или его солей, способ их получения
Соли метил(r)-7-[3-амино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-a]пиразин-1-карбоксилата
Фармацевтическая композиция для лечения диабета 2 типа
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Производные дициклоазаалкана, способы их получения и их применение в медицине
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Соли производных тетерагидро-имидазо[1,5-a]пиразина, способы их получения и их медицинское применение
Способ и система идентификации и обеспечения доступа в сеть домашнего шлюза
Фармацевтическая композиция для лечения диабета 2 типа
Промежуточное соединение для синтеза каспофунгина и способ его получения
Производные с-арилглюкозидов, способ их получения и фармацевтическое применение
Полициклические производные, способ их получения и их фармацевтическое применение

