Результат интеллектуальной деятельности: СОЛИ ПРОИЗВОДНЫХ ТЕТЕРАГИДРО-ИМИДАЗО[1,5-A]ПИРАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
Настоящее изобретение относится к фармацевтически приемлемым солям (R)-7-[3-амино-4-(2,4,5-трифторфенил)-бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты, к способам их получения, к фармацевтическим композициям, содержащим их, и к их применению в качестве терапевтического агента, в частности к их применению в качестве ингибитора дипептидилпептидазы IV (DPP-IV).
ПРЕДШЕСТВУЮЩИЙ УРОВЕНЬ ТЕХНИКИ
Диабет был зарегистрирован давно. Он является метаболическим заболеванием, характеризующимся хронической гипергликемией в связи с абсолютным или относительным недостатком инсулина в организме человека, приводящим в результате к повышенным концентрациям глюкозы в крови, и резкими выбросами глюкозы в моче параллельно с расстройством метаболизма сахаров, липидов и белков, и физиологически выражается в повышенном питье, повышенном мочеиспускании, повышенном приеме пищи, потере массы, головокружении, слабости и других симптомах.
Персистентная или неконтролируемая гипергликемия связана с повышенной заболеваемостью и смертностью. Часто аномальный гомеостаз глюкозы прямо или косвенно связан с изменениями метаболизма липидов, липопротеинов и аполипопротеинов и с другими метаболическими и гемодинамическими заболеваниями. Пациенты с сахарным диабетом 2 типа обладают особенно повышенным риском макрососудистых и микрососудистых осложнений, таких как коронарная болезнь сердца, удар, периферическое сосудистое заболевание, гипертензия, нефропатия, невропатия и ретинопатия и т.д. Поэтому терапевтический контроль гомеостаза глюкозы, липидного метаболизма, гипертензии и тому подобного очень важен для клинического лечения сахарного диабета.
Существует две общепризнанных формы диабета. При диабете 1 типа, то есть инсулинозависимом сахарном диабете (ИЗСД), пациенты продуцируют мало или не продуцируют инсулин, который является гормоном, регулирующим утилизацию глюкозы. При диабете 2 типа, то есть инсулиннезависимом сахарном диабете (ИНЗСД), пациенты часто имеют уровни инсулина в плазме, которые являются такими же или даже повышенными по сравнению с пациентами, не являющимися диабетиками. Однако эти пациенты обладают развитой устойчивостью к инсулину, который обладает стимулирующим эффектом на метаболизм глюкозы и липидов в основных тканях, чувствительных к инсулину, таких как мышцы, печень и 25 жировых тканей, и уровни инсулина в плазме, даже если они повышены, недостаточны для преодоления выраженной устойчивости к инсулину.
Устойчивость к инсулину, в основном, не связана с пониженным числом рецепторов инсулина, а связана также с нарушением связывания пост-инсулиновых рецепторов, которое еще недостаточно понято. Эта устойчивость к реактивности на инсулин приводит в результате к недостаточной инсулинозависимой активации захвата глюкозы, ее окисления и запасания в мышцах, а также к неадекватному подавлению липолиза в жировой ткани и регуляции продуцирования и секреции глюкозы в печени.
Дипептидилпептидаза-IV (DPP-IV) является сериновой протеазой, которая отщепляет N-концевые дипептиды от полипептида, содержащего остаток пролина в предпоследнем положении. Хотя биологическая роль DPP-IV в системах млекопитающих не полностью установлена, считают, что она играет важную роль в метаболизме нейропептидов, T-клеточной активации, адгезии и инвазии раковых клеток в эндотелии и в поступлении БИЧ в лимфоидные клетки (WO 98/19998).
Недавно открыто, что DPP-IV ответственна за предотвращение секреции глюкагоноподобного пептида-1 (GLP-1). Более конкретно DPP-IV отщепляет N-концевой дипептид His-Ala GLP-1, приводя, таким образом, к распаду активного GLP-1(7-36)NH2 до неактивного GLP-1(9-36)NH2 (Endocrinology, 1999, 140: 5356-5363). В физиологических условиях период полувыведения полноразмерного GLP-1 в кровообращении является коротким. Неактивный метаболит GLP-1 после расщепления DPP-IV может связываться с рецепторами GLP-1, следовательно, антагонизирует GLP-1 и сокращает физиологические ответы на GLP-1. Однако ингибиторы DPP-IV могут защитить эндогенный или даже экзогенный GLP-1 от инактивации, и, следовательно, значительно повысить биологическую активность GLP-1 (в 5-10 раз). Поскольку GLP-1 является главным стимулятором секреции инсулина поджелудочной железой и обладает прямыми полезными эффектами на удаление глюкозы, ингибирование DPP-IV, по-видимому, представляет собой привлекательный подход к лечению инсулиннезависимого сахарного диабета (ИНЗСД) (US 6110949).
Хотя некоторые ингибиторы DPP-IV раскрыты, к настоящему времени все еще не существует эффективного лекарства. Усовершенствованные ингибиторы DPP-IV все еще необходимы.
Целью настоящего изобретения является разработка серии соединений, которые обладают ингибиторной активностью в отношении DPP-IV и могут применяться для лечения диабета или подобного заболевания либо применяться в качестве паллиативных лекарств.
В заявке PCT/CN2008/001936, поданной заявителем настоящего изобретения 4 января 2009, раскрыты новые производные тетрагидро-имидазо[1,5-а]пиразина и их применения в качестве ингибитора DPP-IV. Пример 10 из этой заявки относится к (R)-7-[3-амино-4-(2,4,5-трифторфенил)-бутаноил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты гидрохлориду, в отношении которого было подтверждено, что он обладает отличной ингибиторной активностью против DPP-IV в соответствии с тестом. Поэтому эта заявка полностью включена в данную заявку посредством ссылки.
Другой целью настоящего изобретения является разработка фармацевтически приемлемых солей соединений, представленных формулой (I), и их композиций для улучшения их растворимости, биодоступности, гипогликемической активности и фармакокинетики.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Настоящее изобретение направлено на разработку новых фармацевтически приемлемых солей (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты, способов их получения, фармацевтических композиций, содержащих их, и их применения в качестве терапевтического агента, в частности их применения в качестве ингибитора дипептидилпептидазы IV. Соли соединения, представленного формулой (I), обладают великолепной активностью при лечении диабета, улучшенной растворимостью, хорошей активностью и биодоступностью in vivo, а также более низкой токсичностью и являются хорошими кандидатами для получения лекарственного средства для лечения диабета.
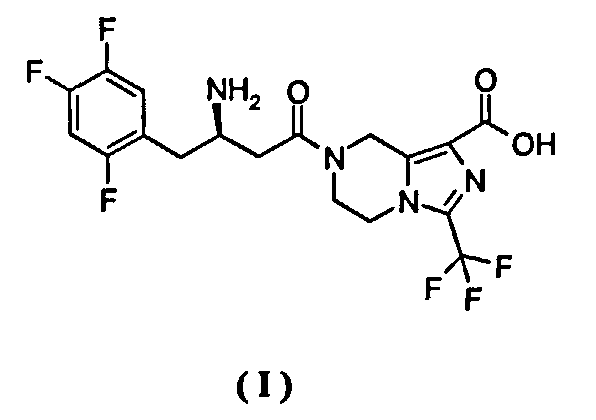
В настоящем изобретении предложены фармацевтически приемлемые соли соединений формулы (I). Термин "фармацевтически приемлемые соли" относится к фармацевтическим солям присоединения нетоксичной кислоты или к солям присоединения основания. Соли присоединения кислоты представляют собой соли, образованные соединениями формулы (I) и органическими или неорганическими кислотами, включающие гидрохлорид, фосфат, гидрофосфат, сульфат, бисульфат, сульфит, ацетат, оксалат, малонат, пентаноат, глутамат, олеат, пальмитат, стеарат, лаурат, борат, пара-толуолсульфонат, метансульфонат, малат, тартрат, бензоат, памоат, салицилат, ванилат, манделат, сукцинат, глюконат, лактобионат и лаурилсульфонат, предпочтительно фосфат. Соли присоединения основания представляют собой соли, образованные из соединений формулы (I) и органического или неорганического основания, включающие соли, образованные из щелочных металлов, амина или четвертичного аммония, такие как соль натрия, соль лития, соль калия, соль кальция, соль магния, соль амина, соль четвертичного тетраметиламмония, соль четвертичного тетраэтиламмония, соль холина, предпочтительно соль холина. Соли аминов представляют собой соли, образованные из соединений формулы (I) и амина, включающего аммиак, первичный амин, вторичный амин и третичный амин, такие как соль метанамина, соль диметиламина, соль триметиламина, соль триэтиламина, соль этиламина, соль этаноламина, соль лизина и соль аргинина, предпочтительно соль этаноламина.
Репрезентативные фармацевтически приемлемые соли соединений формулы (I) по настоящему изобретению включают, но не ограничены ими:
|
|
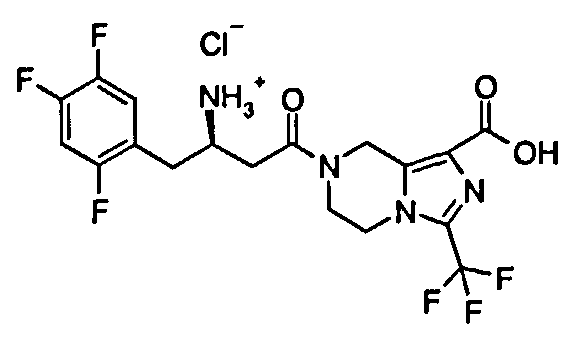
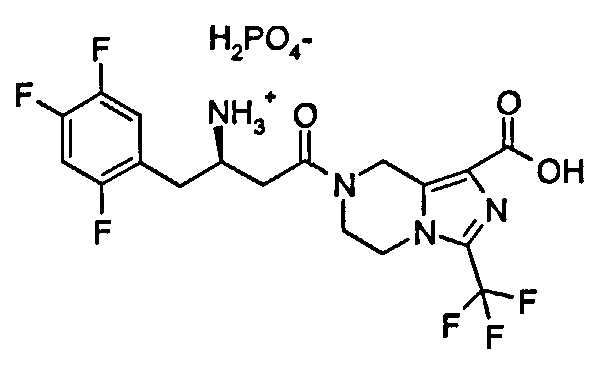
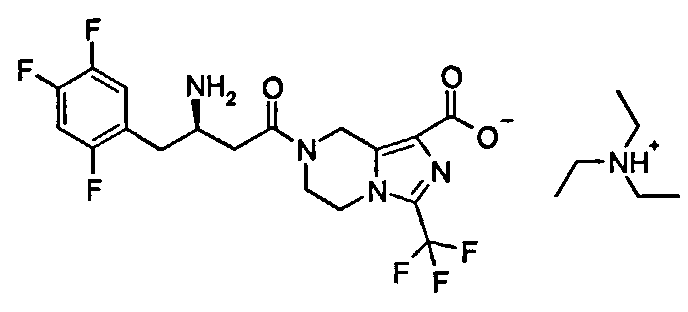
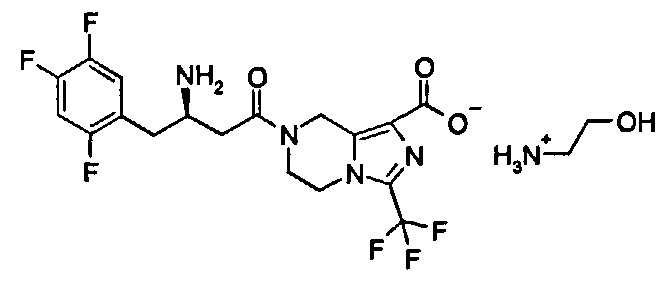
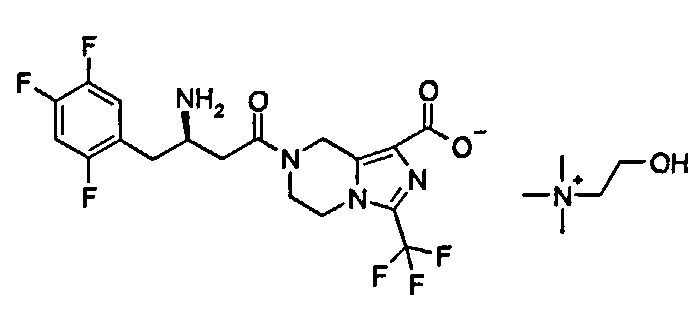
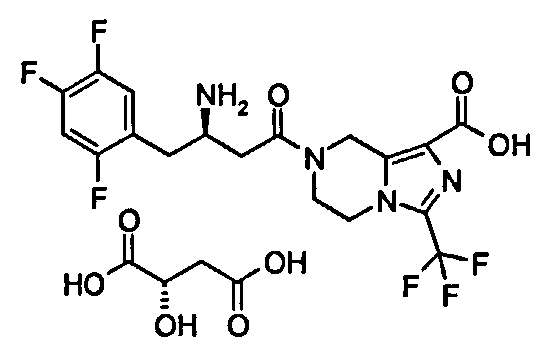
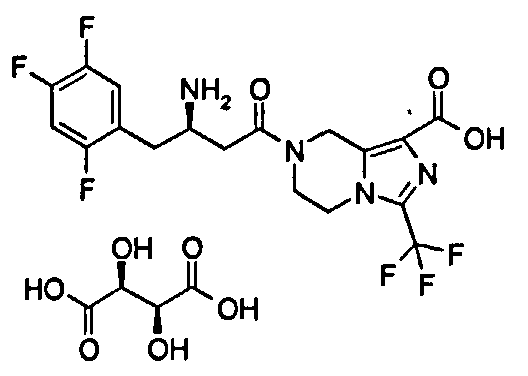
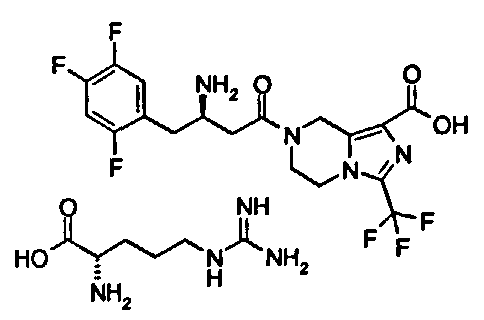
Вышеописанные реакции образования соли обычно готовят в условиях охлаждения, комнатной температуры или нагревания. Однако примечательно, что температура реакции имеет некоторое влияние на различные реакции образования соли, которые хорошо известны специалисту в данной области техники. Температуры реакций образования соли по настоящему изобретению находятся в интервале от комнатной температуры до точки кипения растворителя реакционной смеси, предпочтительно 0~40°С. Специалист в данной области техники может легко определить наиболее предпочтительную температуру реакции образования соли общепринятыми методами.
Настоящее изобретение относится к способу получения фармацевтически приемлемых солей соединения формулы (I), где этот способ включает присоединение кислоты и присоединение основания с образованием солей. Способ получения солей присоединения кислоты включает приведенные ниже стадии взаимодействия (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты гидрохлорида со щелочным раствором и взаимодействия полученной в результате (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты с органической или неорганической кислотой, где органическая или неорганическая кислота выбрана из группы, состоящей из соляной кислоты, фосфорной кислоты, серной кислоты, сернистой кислоты, уксусной кислоты, щавелевой кислоты, малоновой кислоты, пентановой кислоты, глутаминовой кислоты, олеиновой кислоты, пальмитиновой кислоты, стеариновой кислоты, лауриновой кислоты, ортоборной кислоты, пара-толуолсульфоновой кислоты, метансульфоновой кислоты, яблочной кислоты, винной кислоты, бензойной кислоты, памовой кислоты, салициловой кислоты, ванилиновой кислоты, миндальной кислоты, янтарной кислоты, глюконовой кислоты, лактобионовой кислоты и лаурилсульфоновой кислоты. Способ получения солей присоединения основания включает взаимодействие (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты с гидроксидом щелочного металла, замещенным амином или четвертичным аммонием, где гидроксид щелочного металла выбран из гидроксида натрия, гидроксида лития, гидроксида калия, гидроксида кальция, гидроксида магния, а амин или четвертичный аммоний выбран из группы, состоящей из четвертичного тетраметиламмония, четвертичного тетраэтиламмония, этаноламина, холина, лизина, аргинина, метанамина, диметиламина, триметиламина, триэтиламина и этиламина.
Настоящее изобретение относится к применению фармацевтически приемлемых солей соединений формулы (I) для получения лекарственного средства для лечения диабета 2 типа, гипергликемии, ожирения или устойчивости к инсулину.
Настоящее изобретение относится к применению фармацевтически приемлемых солей соединений формулы (I) при получении ингибитора дипептидилпептидазы (DPP-IV).
Настоящее изобретение относится к способу лечения диабета 2 типа, гипергликемии, ожирения или устойчивости к инсулину, включающему введение субъекту терапевтически эффективного количества фармацевтически приемлемых солей соединений формулы (I).
Настоящее изобретение относится к способу ингибирования каталитической активности дипептидилпептидазы IV, включающему приведение в контакт дипептидилпептидазы IV с фармацевтически приемлемыми солями соединений формулы (I).
Настоящее изобретение относится к применению фармацевтически приемлемых солей соединений формулы (I) в качестве лекарства для лечения диабета 2 типа, гипергликемии, ожирения или устойчивости к инсулину.
Настоящее изобретение относится к применению фармацевтически приемлемых солей соединений формулы (I) в качестве лекарства, ингибирующего дипептидилпептидазу IV (DPP-IV).
Настоящее изобретение относится к фармацевтической композиции, которая содержит терапевтически эффективное количество фармацевтически приемлемой соли соединений формулы (I) и фармацевтически приемлемый носитель. Настоящее изобретение также относится к применению этой композиции для получения лекарственного средства для лечения диабета 2 типа, гипергликемии, ожирения или устойчивости к инсулину.
Настоящее изобретение относится к способу лечения диабета 2 типа, гипергликемии, ожирения или устойчивости к инсулину, включающему введение субъекту терапевтически эффективного количества фармацевтической композиции, содержащей фармацевтически приемлемые соли соединений формулы (I).
Настоящее изобретение относится к применению фармацевтической композиции, содержащей фармацевтически приемлемые соли соединений формулы (I), в качестве лекарства при лечении диабета 2 типа, гипергликемии, ожирения или устойчивости к инсулину.
Если не указано иное, приведенные ниже термины, используемые в описании и в формуле изобретения, имеют значения, описанные ниже.
Термин "фармацевтическая композиция" относится к смеси одной или более чем одной из фармацевтически приемлемых солей соединения, описанного в данной заявке, или его пролекарств с другими химическими ингредиентами, такими как физиологически/фармацевтически приемлемые носители. Цель фармацевтической композиции состоит в облегчении введения соединения в организм.
Чтобы достичь целей изобретения, изобретение использует приведенные ниже технические решения:
Способ синтеза соединения формулы (I) относится к способу получения, описанному в примере 10 заявки PCT/CN2008/001936, поданной заявителем 27 ноября 2008. Таким образом, ее описание было полностью включено в данную заявку посредством ссылки.
Способ реакции присоединения кислоты или основания соединения (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты включает:
Способ реакции присоединения кислоты, включающий взаимодействие (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты гидрохлорида со щелочным раствором и взаимодействие полученной в результате (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты с органической или неорганической кислотой;
Способ реакции присоединения основания, включающий взаимодействие (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты с органическим или неорганическим основанием, таким как гидроксид щелочного металла, замещенный амин или четвертичный аммоний, в органическом растворителе, растворимом в воде.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Приведенные ниже примеры предложены для дополнительной иллюстрации изобретения, но их не следует считать ограничивающими изобретение.
Структура соединений была подтверждена ядерной магнитно-резонансной спектроскопией (1Н-ЯМР) или масс-спектрометрией (МС). Сдвиги 1H ЯМР (δ) приведены в ppm (млн-1). 1H ЯМР определяли с помощью оборудования Bruker AVANCE- 400. Растворители представляли собой дейтерированный метанол (CD3OD). Химические сдвиги приведены в 10-6 (млн-1).
МС определяли с помощью масс-спектрометра FINNIGAN LCQAd (ESI) (изготовитель: Thermo, тип: Finnigan LCQ advantage MAX).
Тонкослойная пластина силикагеля представляла собой пластину силикагеля Yantai Huanghai HSGF254 или Qingdao GF254. Пластина, используемая в ТСХ, имела размер 0,15 мм~0,2 мм, а пластина, используемая для очистки продуктов, имела размер 0,4 мм~0,5 мм.
При колоночной хроматографии, как правило, использовали силикагель Yantai Huanghai 200-300 меш в качестве носителя.
Исходные вещества по настоящему изобретению известны, и их покупали у фирм ABCR GmbH & Со. KG, Acros Organics, Aldrich Chemical Company, Accela ChemBio Inc, Darui Finechemical Co., Ltd и т.д., либо они могут быть получены способами синтеза, общепринятыми в данной области техники.
Если не указано иное, приведенные ниже реакции проводили в атмосфере азота.
"Атмосфера азота" относится к тому, что реакционная колба оборудована баллоном азота примерно 1 л.
"Атмосфера водорода" относится к тому, что реакционная колба оборудована баллоном водорода примерно 1 л.
Если не указано иное, раствор, используемый в приведенных ниже реакциях, относится к водному раствору.
Если не указано иное, температура реакции представляла собой комнатную температуру.
Подходящая комнатная температура составляла 20°С~30°С.
Мониторинг хода реакций примеров осуществляли с помощью тонкослойной хроматографии (ТСХ). Хроматографическая система растворителей включала систему дихлорметана и метанола, систему гексана и этилацетата, систему петролейного эфира и этилацетата и ацетон. Соотношение объемов растворителей регулировали в соответствии с полярностью соединений.
Система элюирования колоночной хроматографии включала: А) систему дихлорметана и метанола, В) систему гексана и этилацетата. Соотношение объемов растворителей регулировали в соответствии с полярностью соединений и иногда также добавляли аммиак или уксусную кислоту.
ВЭЖХ относится к высокоэффективной жидкостной хроматографии.
ВЭЖХ осуществляли с помощью прибора для высокоэффективной жидкостной хроматографии Agilent 2695-2996 (колонка Gimini C18 150×4,6 мм).
Условия теста ВЭЖХ: время пробега 30 минут; температура колонки 30°С; ФДМ 230 нм; подвижная фаза метанол:вода (0,1% водный аммиак) = 25:75; скорость тока 1,0 мл/мин.
Пример 1
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)-бутаноил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоновой кислоты гидрохлорид
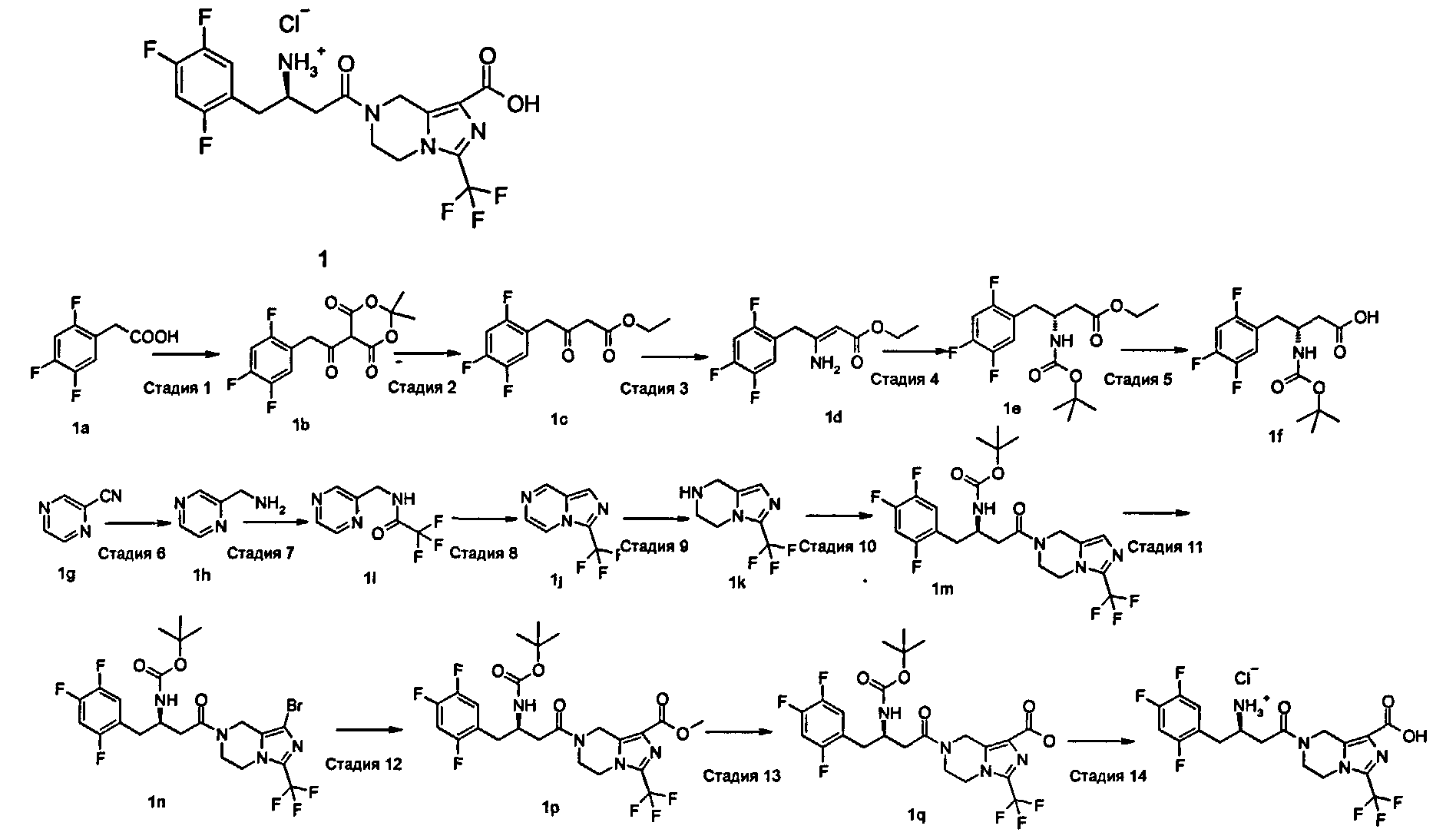
Стадия 1
2,2-Диметил-5-[2-(2,4,5-трифторфенил)-ацетил]-[1,3]диоксан-4,6-дион
2,2-Диметил-[1,3]диоксан-4,6-дион (5,69 г, 39,5 ммоль) растворяли в 400 мл дихлорметана при перемешивании с последующим добавлением 2,4,5-трифторфенилуксусной кислоты 1а (7,15 г, 37,6 ммоль) и 4-диметиламинопиридина (7,35 г, 60,2 ммоль) в бане лед-вода. Затем медленно добавляли по каплям суспензию 1-(3-диметиламинопропил)-3-этилкарбодиимида гидрохлорида (8,28 г, 43,2 ммоль) в 250 мл дихлорметана. После перемешивания при комнатной температуре в течение 36 часов реакционную смесь промывали раствором 5% бисульфата калия (250 мл×7) и насыщенным рассолом (250 мл×2), высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 2,2-диметил-5-[2-(2,4,5-трифторфенил)-ацетил]-[1,3]диоксан-4,6-диона 1b (11,4 г, выход 96%) в виде белого твердого вещества. МС m/z (ИЭР): 315,5 [М-1].
Стадия 2
Этил-3-оксо-4-(2,4,5-трифторфенил)-бутират
2,2-Диметил-5-[2-(2,4,5-трифторфенил)-ацетил]-[1,3]диоксан-4,6-дион 1b (15,72 г, 49,6 ммоль) растворяли в 280 мл этанола при перемешивании, затем реакционную смесь нагревали до 70°С в масляной бане в течение 12 часов. После охлаждения до комнатной температуры смесь концентрировали при пониженном давлении. Полученный в результате остаток очищали колоночной хроматографией на силикагеле с системой В в качестве элюента с получением соединения, указанного в заголовке, этил-3-оксо-4-(2,4,5-трифторфенил)-бутирата 1c (12 г, выход 88%) в виде желтого масла. МС m/z (ИЭР): 259 [М-1].
Стадия 3
Этил 3-амино-4-(2,4,5-трифторфенил)-бут-2-еноат
Этил-3-оксо-4-(2,4,5-трифторфенил)-бутират 1с (24,6 г, 94,5 ммоль) растворяли в 240 мл метанола и к раствору добавляли ацетат аммония (36,4 г, 473 ммоль). Реакционную смесь нагревали до образования флегмы в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении, а затем к остаткам добавляли 100 мл воды. Смесь экстрагировали этилацетатом (200 мл×3) и объединенную органическую фазу промывали 200 мл насыщенного рассола, высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением светло-желтого твердого вещества. Полученное в результате твердое вещество растворяли в 50 мл этилацетата при 80°С, а затем к раствору добавляли 50 мл н-гексана и затравочные кристаллы. Смесь охлаждали до комнатной температуры и через полчаса добавляли 100 мл н-гексана. Смесь хранили в холодильнике в течение 12 часов, а затем фильтровали с получением соединения, указанного в заголовке, этил-3-амино-4-(2,4,5-трифторфенил)-бут-2-еноата 1d (19,5 г, выход 80%) в виде белого твердого вещества. МС m/z (ИЭР): 260,1 [М+1].
Стадия 4 этил-3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-бутират
Этил-3-амино-4-(2,4,5-трифторфенил)-бут-2-еноат 1d (4,1 г, 15,8 ммоль) добавляли в автоклав с последующим добавлением 70 мл метанола, ди-трет-бутилдикарбоната (3,8 г, 17,4 ммоль), хлор-(1,5-циклооктадиен)родия (I) димера (32 мг, 0,0632 ммоль) и (R)-1-[(S)-2-(дифенилфосфино)ферроценил]-этил-трет-бутилфосфина (68 мг, 0,13 ммоль). Реакционную смесь подвергали взаимодействию в атмосфере водорода в течение 24 часов при 6,67 атмосфер при 30°С. Смесь фильтровали и фильтрат концентрировали при пониженном давлении. Затем к остаткам добавляли 34 мл метанола при 50°С с последующим добавлением 12 мл воды после полного растворения. После охлаждения до комнатной температуры смесь хранили в холодильнике в течение 12 часов, а затем фильтровали. Твердый продукт промывали смесью метанола/воды (об:об = 1:1), высушивали в вакууме с получением соединения, указанного в заголовке, этил-3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-бутирата 1е (4 г, выход 70%) в виде светло-желтого твердого вещества.
МС m/z (ИЭР): 362,4 [М+1].
Стадия 5
(R)-3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-масляная кислота
Ссылка на общеизвестный способ Tetrahedron Asymmetry, 2006, 17(2), 205-209. 3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-масляной кислоты этиловый эфир 1е (10 г, 27,7 ммоль) и гидроксид натрия (3,32 г, 83,1 ммоль) растворяли в 150 мл смеси метанола и воды (об:об = 1:1). Реакционную смесь перемешивали при 40-45°С в течение 1-1,5 часов, затем часть раствора выпаривали при пониженном давлении. К остаткам добавляли небольшое количество воды, затем рН доводили до 2-3 1 М соляной кислотой в бане лед-вода. Смесь экстрагировали этилацетатом (200 мл×3) и объединенные органические фазы промывали 200 мл насыщенного рассола, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении, а затем остатки перекристаллизовали из этилацетата/н-гексана с получением соединения, указанного в заголовке, (R)-3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-масляной кислоты 1f (9,2 г) в виде белого твердого вещества, которое непосредственно использовали в следующей стадии. МС m/z (ИЭР): 332,3 [М-1].
Стадия 6
С-Пиразин-2-илметиламин
Пиразин-2-карбонитрил 1g (10,5 г, 100 ммоль) растворяли в 150 мл 1,4-диоксана, затем добавляли никель Ренея (1,0 г) в 250 мл автоклав. Реакционную смесь подвергали взаимодействию в атмосфере водорода в течение 8 часов при 40 атмосферах при 60°С и фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, С-пиразин-2-илметиламина 1h (10,7 г, выход 98%) в виде коричневого масла. МС m/z (ИЭР): 110 [М+1].
Стадия 7
2,2,2-Трифтор-N-пиразин-2-илметилацетамид
С-Пиразин-2-илметиламин 1h (10,9 г, 100 ммоль) добавляли в реакционную колбу, затем медленно добавляли по каплям 20 мл трифторуксусного ангидрида в течение часа при 0°С в бане лед-вода. Реакционную смесь подвергали взаимодействию при комнатной температуре в течение 2 часов. Затем смесь концентрировали при пониженном давлении. Полученные в результате остатки очищали колоночной хроматографией на силикагеле с системой А в качестве элюента с получением соединения, указанного в заголовке, 2,2,2-трифтор-N-пиразин-2-илметилацетамида 1i (21,0 г) в виде коричневого масла. МС m/z (ИЭР): 206,1 [М+1].
Стадия 8
3-Трифторметилимидазо[1,5-а]пиразин
2,2,2-Трифтор-N-пиразин-2-илметилацетамид 1i (21,0 г, 100 ммоль) добавляли в реакционную колбу с последующим добавлением 100 мл оксихлорида фосфора. После перемешивания в течение 30 минут к раствору добавляли пентоксид фосфора (17,8 г, 125 ммоль). Реакционную смесь нагревали до образования флегмы в течение 5 часов. Смесь концентрировали при пониженном давлении и реакционную систему гасили деионизованной водой. Смесь доводили до рН 5-6 20% раствором гидроксида натрия в бане лед-вода, а затем экстрагировали этилацетатом (250 мл×4). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении. Полученные в результате остатки очищали колоночной хроматографией на силикагеле с системой А в качестве элюента с получением соединения, указанного в заголовке, 3-трифторметилимидазо[1,5-а]пиразина 1j (12,0 г, выход 65%) в виде желтого твердого вещества. MC m/z (ИЭР): 188,0 [М+1].
1H ЯМР (400 МГц, CDCl3, млн-1): δ 9.15 (s, 1Н), 8.06 (d, 1H), 7.92 (s, 1H), 7.81 (d, 1H).
Стадия 9
3-Трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин
3-Трифторметилимидазо[1,5-а]пиразин 1j (12,0 г, 64,2 ммоль) растворяли в 150 мл безводного этанола, затем к раствору добавляли 10% Pd/C (500 мг). Реакционную смесь перемешивали при комнатной температуре в атмосфере водорода в течение 12 часов. Реакционный раствор фильтровали через слой крупного силикагеля и фильтрат концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразина 1k (12,2 г, выход 99%) в виде коричневого твердого вещества.
1H ЯМР (400 МГц, CDCl3, млн-1): δ 6.84 (s, 1H), 4.10 (m, 4H), 3.26 (m, 2H), 1.81 (s, 1H).
Стадия 10
трет-бутил-(R)-[3-оксо-1-(2,4,5-трифторбензил)-3-(3-трифторметил-5,6-дигидро-8Н-имидазо[1,5-а]пиразин-7-ил)-пропил]-карбамат
(R)-3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-масляную кислоту 1f (8,6 г, 45 ммоль) и 9,4 мл триэтиламина растворяли в 300 мл дихлорметана. После перемешивания в течение 5 минут к раствору последовательно добавляли 3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин 1k (15,0 г, 45 ммоль) и бис(2-оксо-3-оксазолидинил)фосфинхлорид (17,1 г, 67,3 ммоль). Реакционную смесь подвергали взаимодействию в течение 2 часов, а затем концентрировали при пониженном давлении. Полученные в результате остатки очищали колоночной хроматографией на силикагеле с системой В в качестве элюента с получением соединения, указанного в заголовке, трет-бутил-(R)-[3-оксо-1-(2,4,5-трифторбензил)-3-(3-трифторметил-5,6-дигидро-8Н-имидазо[1,5-а]пиразин-7-ил)-пропил]-карбамата 1m (20,0 г, выход 88%) в виде белого твердого вещества. 1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.25 (m, 1H), 7.11 (m, 1H), 7.032 (s, 1H), 4.93 (m, 2H), 4.35 (m, 3Н), 4.05 (m, 2H), 2.99 (m, 2H), 2.73 (m, 2H), 1.34 (s, 9H).
Стадия 11
трет-бутил-(R)-[3-оксо-1-(2,4,5-трифторбензил)-3-(1-бром-3-трифторметил-5,6-дигидро-8Н-имидазо[1,5-а]пиразин-7-ил)-пропил]-карбамат
трет-бутил-(R)-[3-оксо-1-(2,4,5-трифторбензил)-3-(3-трифторметил-5,6-дигидро-8М-имидазо[1,5-а]пиразин-7-ил)-пропил]-карбамат 1m (20,0 г, 39,6 ммоль) растворяли в 300 мл безводного этанола, а затем к раствору добавляли N-бромсукцинимид (14,1 г, 79,2 ммоль). После перемешивания в течение часа к смеси добавляли карбонат калия (10,9 г, 79,2 ммоль) и ди-трет-бутилдикарбонат (8,6 г, 39,6 ммоль) и смесь перемешивали еще в течение одного часа. Реакционную смесь фильтровали через слой крупного силикагеля, а затем фильтрат концентрировали при пониженном давлении. Полученные в результате остатки очищали колоночной хроматографией на силикагеле с системой В в качестве элюента с получением соединения, указанного в заголовке, трет-бутил-(R)-[3-оксо-1-(2,4,5-трифторбензил)-3-(1-бром-3-трифторметил-5,6-дигидро-8Н-имидазо[1,5-а]пиразин-7-ил)-пропил]-карбамата 1n (20,0 г, выход 86%) в виде белого твердого вещества.
1H ЯМР (400 МГц, CDCl3, млн-1): δ 7.063 (m, 1H), 6.88 (m, 1H), 4.72 (s, 1H), 4.56 (s, 1H), 4.13 (m, 3Н), 3.88 (m, 2H), 2.94 (m, 2H), 2.62 (m, 2H), 1.36 (s, 9H).
Стадия 12
Метил-(R)-7-[3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-формиат
Ссылка на Journal of Organometallic Chemistry, 1985, 285(1-3), 293-303. Октакарбонилдикобальт (4,02 г, 11,76 ммоль), этилхлорацетат (0,71 г, 5,88 ммоль), карбонат калия (1,62 г, 11,76 ммоль) и 50 мл метанола добавляли в реакционную колбу. После перемешивания в течение 5 минут добавляли трет-бутил-(R)-[3-оксо-1-(2,4,5-трифторбензил)-3-(1-бром-3-трифторметил-5,6-дигидро-8Н-имидазо[1,5-а]пиразин-7-ил)-пропил]-карбамат 1n (2,3 г, 3,92 ммоль). Реакционную смесь перемешивали при 60°С в течение 2 часов, а затем концентрировали при пониженном давлении. Полученные в результате остатки очищали колоночной хроматографией на силикагеле с системой В в качестве элюента с получением соединения, указанного в заголовке, метил-(R)-7-[3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-формиата 1p (1,1 г, выход 50%) в виде белого твердого вещества. МС m/z (ИЭР): 565,0 [М+1].
Стадия 13
(R)-7-[3-трет-бутоксикарбонил-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновая кислота
Метил-(R)-7-[3-трет-бутоксикарбониламино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-формиат 1p (1,8 г, 3,2 ммоль) растворяли в 50 мл метанола с последующим добавлением 10 мл водного гидроксида натрия (4 М). Реакционную смесь перемешивали в течение 1 часа. Реакционную смесь доводили до рН=3-5 2 М соляной кислотой в ледяной бане. Смесь экстрагировали этилацетатом (100 мл×3). Объединенную органическую фазу высушивали над безводным сульфатом магния, фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (R)-7-[3-трет-бутоксикарбонил-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты 1q (1,76 г, выход 100%) в виде светло-желтого твердого вещества. МС m/z (ИЭР): 550,9 [М+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.29-7.23 (m, 1Н), 7.121-7.08 (m, 1Н), 5.15-5.03 (m, 2Н), 4.41-4.06 (m, 5Н), 2.98-2.77 (m, 4Н), 1.42-1.26 (m, 9Н).
Стадия 14
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты гидрохлорид
(R)-7-[3-трет-Бутоксикарбонил-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 1q (1,76 г, 3,2 ммоль) добавляли, в реакционную колбу с последующим добавлением 10 мл хлорида водорода в этилацетате. Реакционную смесь перемешивали в течение 1 часа, а затем концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (R)-7-[3-амино-4-(2,4,5-трифторфенил)-бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты гидрохлорида 1 (1,56 г, выход 100%) в виде белого твердого вещества. МС m/z (ИЭР): 451,2 [М+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.42-7.37 (m, 1Н), 7.28-7.23 (m, 1Н), 5.19-5.05 (m, 2Н), 4.36-4.29 (m, 1Н), 4.15-4.00 (m, 2Н), 3.94-3.93 (m, 2Н), 3.21-2.88 (m, 2Н), 2.86-2.81 (m, 2Н).
Пример 2
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты фосфат
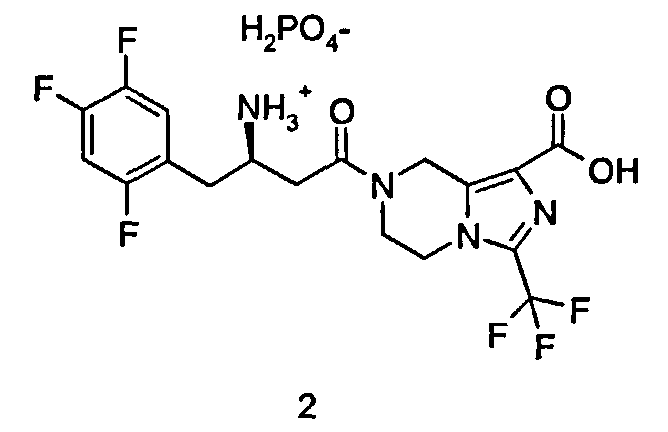
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)-бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты гидрохлорид 1 (1,45 г, 2,97 ммоль) растворяли в 14 мл дихлорметана. Затем смесь промывали 6 мл водного бикарбоната натрия, и водный слой экстрагировали 5,6 мл дихлорметана. Объединенную органическую фазу промывали 6 мл насыщенного рассола, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением маслянистых остатков (1,38 г). Эти остатки растворяли в 40 мл изопропилового спирта с последующим добавлением 85% фосфорной кислоты (342,8 мг, 2,97 ммоль) в 2 мл изопропилового спирта при перемешивании до осаждения твердого вещества. После перемешивания в течение 2 часов смесь фильтровали, и фильтрационный кек промывали изопропиловым спиртом и высушивали при пониженном давлении при 40°С с получением сырого соединения (1,44 г, 88,6%). Это сырое соединение (1,44 г, 2,63 ммоль) растворяли в 26 мл изопропилового спирта и перемешивали в течение 1 часа. Смесь фильтровали и фильтрационный кек промывали изопропиловым спиртом. Твердое вещество растворяли в деионизованной воде. Смесь концентрировали при пониженном давлении при 40°С и высушивали в вакууме при 40°С с получением соединения, указанного в заголовке, (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты фосфата 2 (1,33 г, 92,6%) в виде белого порошка. МС m/z (ИЭР): 451,2 [М+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.36-7.42 (m, 1Н), 7.19-7.25 (m, 1Н), 5.01-5.15 (m, 2Н), 4.24-4.34 (m, 2Н), 4.06-4.11 (m, 1Н), 3.91-3.98 (m, 1Н), 3.07-3.12 (m, 2Н), 2.8-3.09 (m, 2Н).
Пример 3
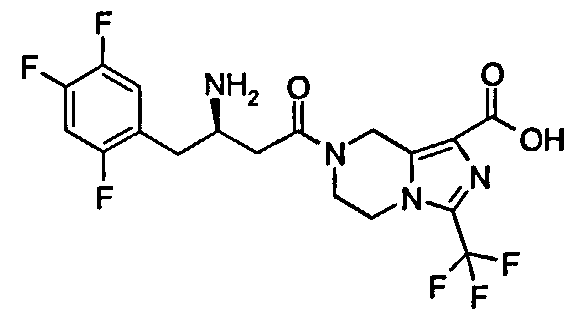
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновая кислота
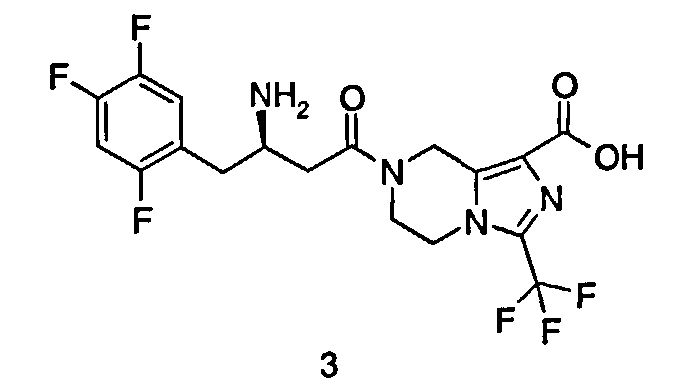
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)-бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты гидрохлорид 1 (1,02 г, 2,1 ммоль) растворяли в 30 мл метанола с последующим добавлением водного раствора гидроксида натрия (2,1 мл, 2,1 ммоль). Реакционную смесь перемешивали в течение 15 минут. Реакционную смесь концентрировали при пониженном давлении и полученное в результате твердое вещество растворяли в 15 мл смеси растворителей дихлорметан/метанол (об:об = 1:3). Смесь фильтровали и концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты 3 (943 мг, 100%) в виде белого твердого вещества. ВЭЖХ: 99,89%. МС m/z (ИЭР): 451,2 [М+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.32-7.41 (m, 1Н), 7.11-7.21 (m, 1Н), 5.00-5.07 (m, 2Н), 4.16-4.24 (m, 2Н), 4.05-4.08 (m, 1Н), 3.85-3.97 (m, 2Н), 3.05-3.17 (m, 2Н), 2.91-2.93 (m, 2Н).
Пример 4
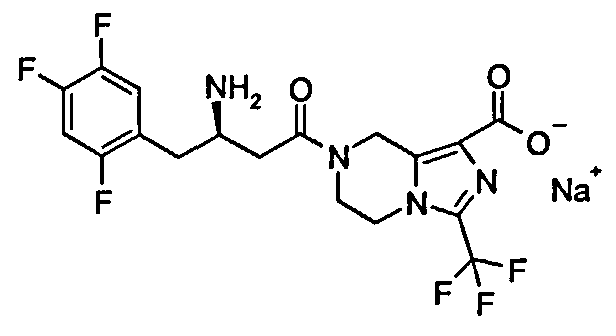
Натриевая соль (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-а]пиразин-1-карбоксилата
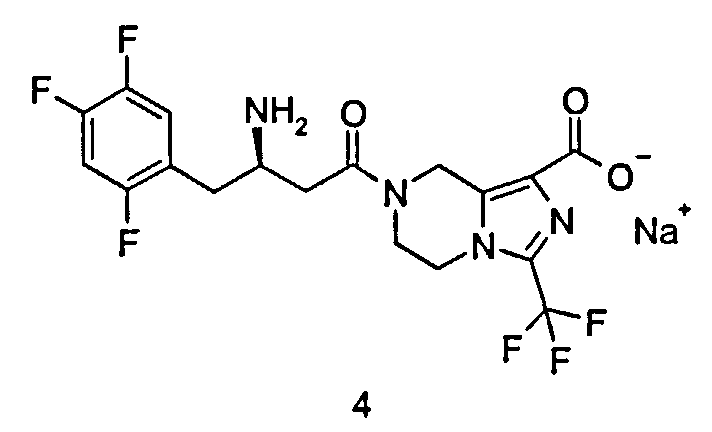
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 5 мл метанола с последующим добавлением водного гидроксида натрия (0,44 мл, 0,22 ммоль). Реакционную смесь перемешивали в течение 15 минут. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, натриевой соли (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата 4 (104 мг, 99,7%) в виде белого твердого вещества. ВЭЖХ: 99,65%. MC m/z (ИЭР): 451,2 [M+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.26-7.30 (m, 1Н), 7.08-7.13 (m, 1H), 5.00-5.20 (m, 2H), 4.26-4.27 (m, 2H), 4.00-4.11 (m, 2H), 3.44-3.48 (m, 1H), 2.72-2.83 (m, 2H), 2.59-2.60 (m, 2H).
Пример 5
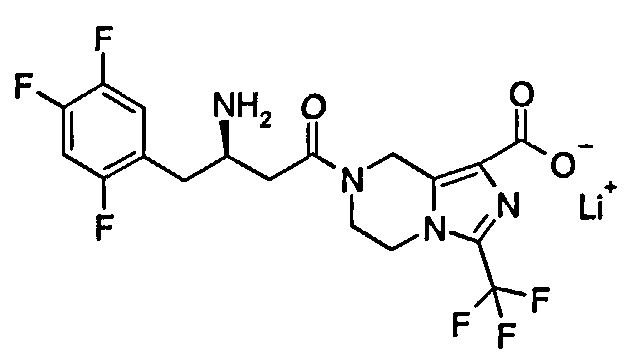
Литиевая соль (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата
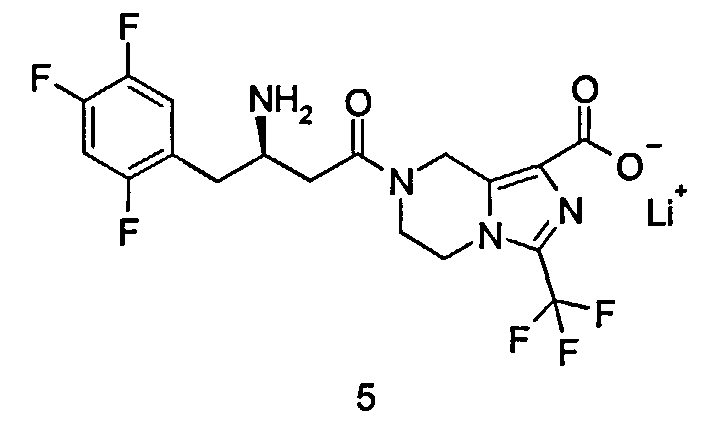
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 5 мл метанола с последующим добавлением водного раствора гидроксида лития (0,44 мл, 0,22 ммоль). Реакционную смесь перемешивали в течение 15 минут. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, литиевой соли (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата 5 (48 мг, 98,6%) в виде белого твердого вещества.
ВЭЖХ: 99,66%.
MC m/z (ИЭР): 451,2 [M+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.31-7.38 (m, 1Н), 7.13-7.22 (m, 1Н), 5.07-5.27 (m, 2Н), 4.26-4.36 (m, 2Н), 4.01-4.15 (m, 2Н), 3.53-3.60 (m, 1Н), 2.80-2.91 (m, 2Н), 2.59-2.72 (m, 2Н).
Пример 6
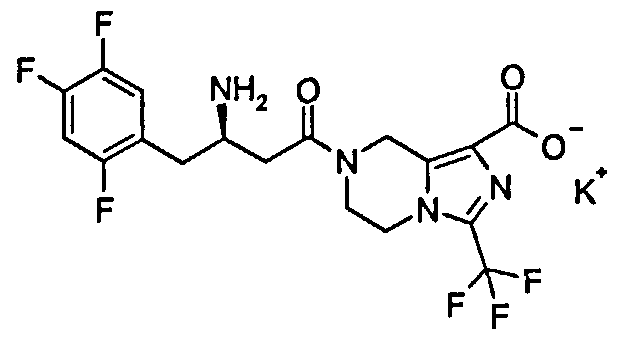
Калийная соль (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата
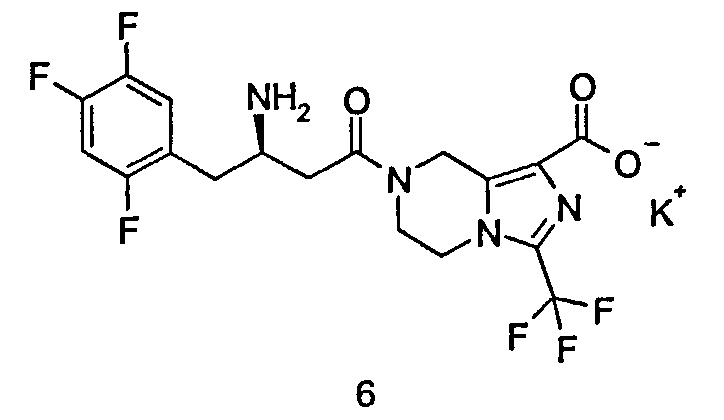
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 5 мл метанола с последующим добавлением водного гидроксида калия (0,44 мл, 0,22 ммоль). Реакционную смесь перемешивали в течение 15 минут. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, калийной соли (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата 6 (108 мг, 100%) в виде белого твердого вещества.
ВЭЖХ: 92,78%.
МС m/z (ИЭР): 451,2 [М+1].
1Н ЯМР (400 МГц, CD3OD, млн-1): δ 7.31-7.38 (m, 1Н), 7.14-7.23 (m, 1Н), 5.07-5.27 (m, 2Н), 4.26-4.36 (m, 2Н), 4.01-4.15 (m, 2Н), 3.52-3.60 (m, 1Н), 2.80-2.92 (m, 2Н), 2.59-2.74 (m, 2Н).
Пример 7
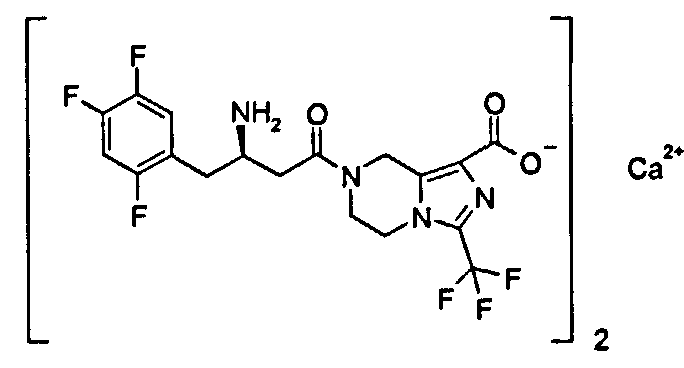
Кальциевая соль (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата
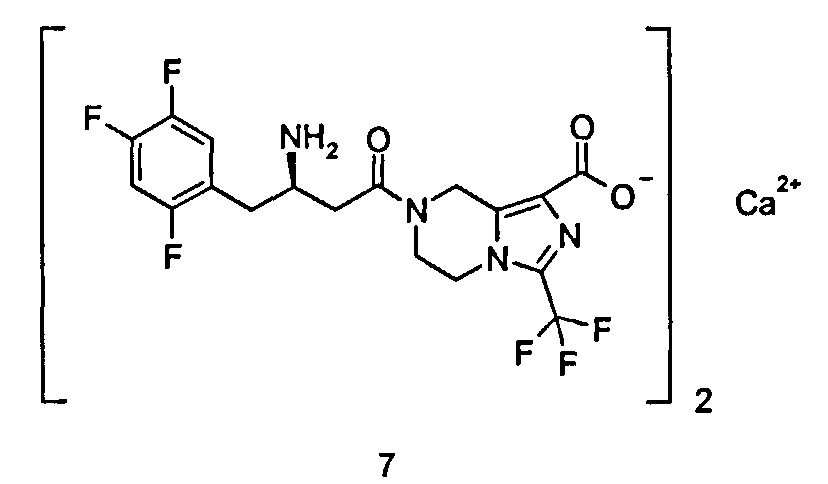
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 10 мл метанола с последующим добавлением водного раствора гидроксида кальция (8,1 мг, 0,11 ммоль). Смесь перемешивали в течение 20 часов. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, кальциевой соли (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата 7 (103 мг, 100%) в виде белого твердого вещества.
ВЭЖХ: 99,60%.
MC m/z (ИЭР): 451,2 [M+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.28-7.34 (m, 1H), 7.11-7.21 (m, 1H), 5.10-5.21 (m, 2H), 4.22-4.36 (m, 2H), 4.03-4.09 (m, 2H), 3.55-3.59 (m, 1H), 2.76-2.85 (m, 2H), 2.60-2.71 (m, 2H).
Пример 8
Триэтиламмониевая соль (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата
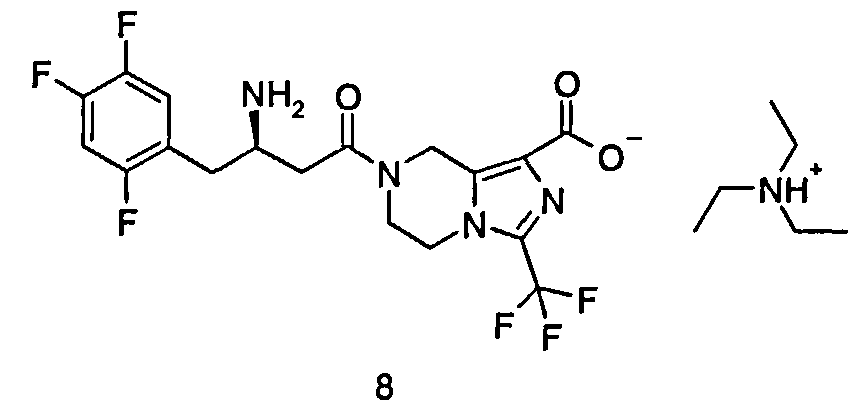
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 5 мл метанола с последующим добавлением раствора триэтиламина в метаноле (0,767 мл, 0,22 ммоль, раствор готовили путем добавления 1 мл триэтиламина к метанолу с получением 25 мл раствора триэтиламина в метаноле). Смесь перемешивали в течение 40 часов. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, триэтиламмониевой соли (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а] пиразин-1-карбоксилата 8 (112 мг, 99,8%) в виде белого твердого вещества.
ВЭЖХ: 99,8%.
MC m/2 (ИЭР): 451,2 [М+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.28-7.35 (m, 1Н), 7.10-7.19 (m, 1Н), 5.03-5.13 (m, 2Н), 4.17-4.25 (m, 2Н), 3.88-4.08 (m, 2Н), 3.70-3.73 (m, 1Н), 3.12-3.17 (m, 6Н), 2.93-2.95 (m, 2Н), 2.71-2.80 (m, 2Н), 1.27-1.30 (m, 9Н).
Пример 9
2-Гидроксиэтиламмониевая соль (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата
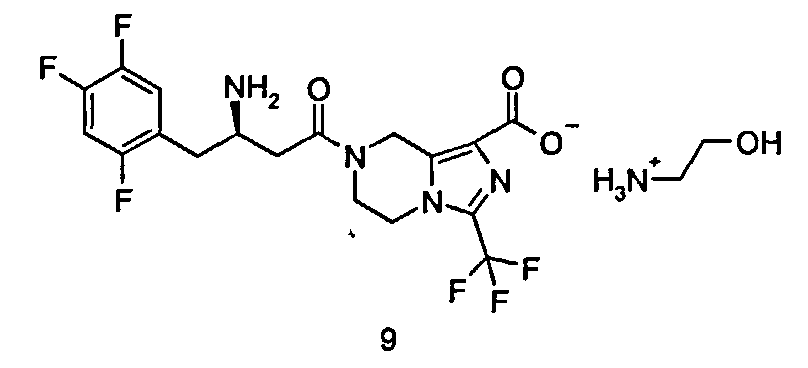
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 5 мл метанола с последующим добавлением раствора этаноламина в метаноле (0,33 мл, 0,22 ммоль, раствор готовили путем добавления 1 мл этаноламина к метанолу с получением 25 мл раствора этаноламина в метаноле). Смесь перемешивали в течение 40 часов. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 2-гидроксиэтиламмониевой соли (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата 9 (114 мг, 99,62%) в виде белого твердого вещества.
ВЭЖХ: 99,62%.
MC m/z (ИЭР): 451,2 [M+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.27-7.32 (m, 1H), 7.07-7.16 (m, 1H), 4.96-5.17 (m, 2H), 4.12-4.26 (m, 2H), 3.91-4.09 (m, 2H), 3.70-3.72 (t, 2H), 3.56-3.57 (m,1H), 2.95-2.98 (t, 2H), 2.80-2.89 (m, 2H), 2.58-2.70 (m, 2H).
Пример 10
2-Гидроксиэтил(триметил)аммониевая соль (R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоксилата
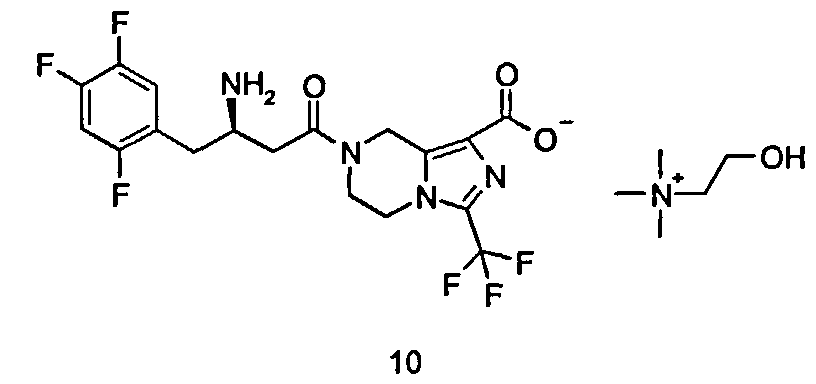
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 5 мл метанола с последующим добавлением раствора холина в метаноле (1,55 мл, 0,22 ммоль, раствор готовили путем добавления 1 мл холина к метанолу с получением 25 мл холина в метаноле). Смесь перемешивали в течение 3 часов. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, 2-гидроксиэтил(триметил)аммониевой соли (R)-7-[3-амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а] пиразин-1-карбоксилата 10 (120 мг, 98,5%) в виде белого твердого вещества.
ВЭЖХ: 99,41%.
MC m/z (ИЭР):451,2 [M+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.23-7.30 (m, 1H), 7.06-7.15 (m, 1H), 4.99-5.19 (m, 2H), 4.19-4.26 (m, 2H), 3.89-4.07 (m, 4H), 3.60-3.71 (m, 1H), 3.50-3.55 (m, 2H), 3.21 (s, 9H), 2.72-2.84 (m, 2H), 2.55-2.66 (m, 2H).
Пример 11
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты (2S)-2-гидроксибутандикислота
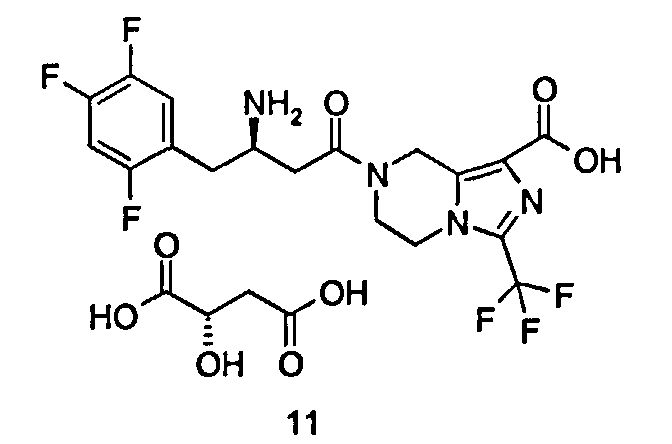
L-Яблочную кислоту (368 мг, 2,74 ммоль) растворяли в 25 мл смеси растворителей метанола/воды (об:об = 4:1) с получением 0,11 М раствора, используемого в последующих стадиях. (R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 10 мл метанола с последующим добавлением 2 мл вышеупомянутого раствора. Смесь перемешивали в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты (2S)-2-гидроксибутандикислоты 11 (129 мг, 98,92%) в виде белого твердого вещества.
ВЭЖХ: 98,92%.
MC m/z (ИЭР):451,1 [M+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.32-7.41 (m, 1Н), 7.16-7.23 (m, 1H), 4.96-5.13 (m, 2H), 4.35-4.39 (m, 1H), 4.20-4.30 (m, 2H), 4.04-4.13 (m, 1H), 3.90-4.00 (m, 2H), 3.07-3.13 (m, 2H), 2.77-2.97 (m, 3H), 2.56-2.62 (m, 1H).
Пример 12
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты (2S,3S)-2,3-дигидроксибутандикислота
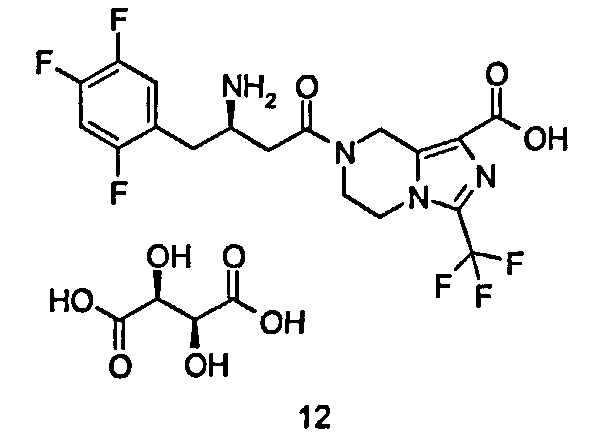
D-Винную кислоту (413 мг, 2,75 ммоль) растворяли в 25 мл смеси растворителей метанола/воды (об:об = 4:1) с получением 0,11 М раствора, используемого на последующих стадиях. (R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 10 мл метанола с последующим добавлением 2 мл вышеупомянутого раствора. Смесь перемешивали в течение 30 минут. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты (2S,3S)-2,3-дигидроксибутандикислоты 12 (131 мг, 99%) в виде белого твердого вещества. ВЭЖХ: 99,35%.
MC m/z (ИЭР):451,1 [M+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.32-7.41 (m, 1Н), 7.17-7.26 (m, 1Н), 5.01-5.14 (m, 2Н), 4.51 (s, 1Н), 4.20-4.35 (m, 2Н), 4.00-4.13 (m, 1Н), 3.89-3.96 (m, 2Н), 3.04-3.13 (m, 2Н), 2.90-3.00 (m, 1Н), 2.77-2.87 (m, 1Н).
Пример 13
(R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты (2S)-2-амино-5-гуанидинопентановая кислота
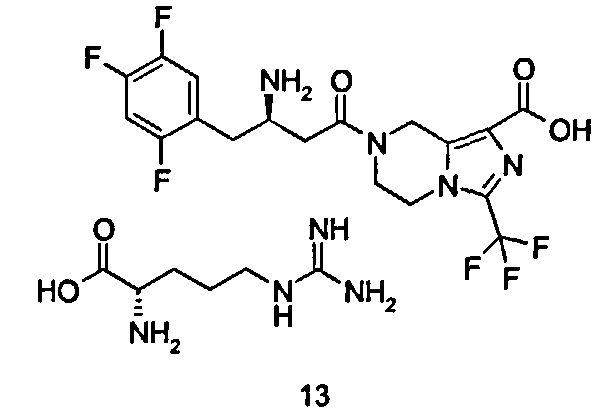
L-Аргинин (239 мг, 1,37 ммоль) растворяли в 25 мл смеси растворителей метанола/воды (об:об = 4:1) с получением 0,055 М раствора, используемого на последующих стадиях. (R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-(трифторметил)-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновую кислоту 3 (100 мг, 0,22 ммоль) растворяли в 15 мл метанола с последующим добавлением 4 мл вышеупомянутого раствора. Смесь перемешивали в течение 4 часов. Реакционную смесь концентрировали при пониженном давлении с получением соединения, указанного в заголовке, (R)-7-[3-Амино-4-(2,4,5-трифторфенил)бутаноил]-3-трифторметил-5,6,7,8-тетрагидроимидазо[1,5-а]пиразин-1-карбоновой кислоты (2S)-2-амино-5-гуанидинопентановой кислоты 13 (139 мг, 100%) в виде белого твердого вещества.
ВЭЖХ: 98,89%.
MC m/z (ИЭР)□451,1 [М+1].
1H ЯМР (400 МГц, CD3OD, млн-1): δ 7.24-7.32 (m, 1Н), 7.09-7.14 (m, 1Н), 4.98-5.18 (m, 2Н), 4.25-4.28 (m, 1Н), 4.18-4.19 (m, 1Н), 3.93-4.04 (m, 2Н), 3.50-3.54 (m, 2Н), 3.18-3.23 (m, 2Н), 2.76-2.87 (m, 2Н), 2.58-2.69 (m, 2Н), 1.77-1.90 (m, 2Н), 1.67-1.75 (m, 2Н).
Примеры тестов:
Эксперимент по растворимости
В соответствии с общепринятым измерением растворимости растворимость тестируемых образцов определяли в четырех различных системах: фосфатном буферном растворе (ФСБ, рН=7,4), метаноле, 0,1% соляной кислоте и воде. Результаты представлены в таблице 1:
|
Вывод: растворимость примера 2, примера 4 и примера 5 была улучшена очевидно.
Фармакологические анализы
Соединения по настоящему изобретению тестировали на определение их ингибиторной активности в отношении DPP-IV, DPP-VIII, DPP-IX в соответствии с приведенными ниже способами. Половинные ингибиторные концентрации IC50 (концентрация тестируемого соединения, показывающая 50% ингибирование активности фермента) каждого соединения определяли с помощью контрольной реакции и вычисления фиксированных количеств фермента и субстрата с несколькими различными концентрациями тестируемых соединений.
Соединения по настоящему изобретению тестировали на определение их ингибиторной активности в отношении DPP-IV, DPP-VIII, DPP-IX в приведенных ниже тестах, используя набор для анализа протеазы DPPIV-Glo™ фирмы Promega (№ по каталогу G8350/G8351), где:
Фермент DPP-IV покупали в фирме Calbiochem, № по каталогу 317630;
Фермент DPP-VIII покупали в фирме Bioscience, № по каталогу 80080;
Фермент DPP-IX покупали в фирме Bioscience, № по каталогу 80090.
Ссылку на методику приготовления общепринятых реагентов, таких как буфер DPPIV-GIo, реагенты люциферина, необходимые в тесте, и подробное описание проведения теста можно найти в описаниях к набору. Анализ осуществляли в виде приведенных ниже стадий.
Тестируемые соединения растворяли в ДМСО с получением серии различных концентраций раствора тестируемого соединения. Буфер DPPIV-Glo и замороженный реагент для обнаружения люциферина уравновешивали до комнатной температуры. Реагент для обнаружения люциферина растворяли в умеренном буфере в коричневой колбе с получением раствора. Затем DPP-IV-Glo растворяли в воде высшей степени очистки до получения соответствующей концентрации. Раствор субстрата и раствор реагента для обнаружения люциферина перемешивали достаточно в соответствующем соотношении (соотношение по изобретению составляет 1:49) и смесь оставляли при комнатной температуре на 30-60 минут. Трис буфер (2 мМ, рН 8,0), тестируемые соединения и DPP-IV (DPP-VIII или DPP-IX) смешивали, а затем переносили в 96-луночный планшет. Каждый тест содержит контроль двойной лунки или тройной лунки. Такой же объем ДМСО добавляли в отрицательный контроль и в чистый контроль. Затем в 96-луночный планшет добавляли перемешанный раствор реагента для обнаружения люциферина и субстрата для инициации реакции. 96-луночный планшет инкубировали при комнатной температуре на встряхивателе для планшетов в течение 40 минут после герметичного закрытия. Интенсивность флуоресцентного сигнала в каждой лунке определяли с помощью считывающего устройства для микропланшетов и процент ингибирования в отношении фермента соединения при данной концентрации вычисляли по формуле, приведенной ниже:
Процент ингибирования: IR=[1-(S-B)/(N-B)]·100%
S: значение образца
В: значение чистого контроля
N: значение отрицательного контроля
IC50 тестируемых соединений можно вычислить на основании процента ингибирования при различных концентрациях.
|
Вывод: свободная форма солей каждого соединения проявляла отличную ингибиторную активность в отношении DPP-IV. Соединение примера 2 показало более предпочтительную селективность.
ФАРМАКОКИНЕТИЧЕСКИЙ АНАЛИЗ
Пример тестирования 1: фармакокинетический анализ соединений по настоящему изобретению
1. Цель теста
Соединение Примера 3 вводили крысам внутрижелудочно или инъекцией в хвостовую вену и соединения Примеров 1-4, Примера 9 и Примеров 11-12 вводили внутрижелудочно, чтобы определить концентрацию лекарства в плазме в различные моменты времени на основании измерений ЖХ/МС/МС. Фармакокинетику соединений по настоящему изобретению исследовали и оценивали на крысах. Также исследовали пероральную абсолютную биодоступность.
2. Протокол
2.1. Образцы
Соединения Примеров 1-4, Примера 9 и Примеров 11-12.
2.2. Подопытные животные
28 здоровых взрослых крыс SD, половину самцов и половину самок, покупали в фирме SINO-BRITSH SIPPR/BK LAB.ANIMAL LTD., CO, номер лицензии: SCXK (Shanghai) 2008-0016.
2.3. Приборы
Линейный ионный масс-спектрометр API 4000 Q-trap, Applied Biosystems Corp., США;
Высокоэффективный жидкостный хроматограф Agilent 1200, Agilent Corp., США.
2.4. Приготовление тестируемых соединений
Группа инъекции в вену: точную навеску тестируемого соединения растворяли в 0,5 мл ДМСО с помощью обработки ультразвуком, а затем разводили нормальным физиологическим раствором до 15 мл с получением 0,3 мг/мл раствора.
Группа внутрижелудочного введения: точную навеску тестируемого соединения растворяли в 0,5% КМЦ-Na с помощью обработки ультразвуком с получением 0,3 мг/мл суспензии.
2.5. Введение
32 здоровых взрослых крысы SD, половину самцов и половину самок, делили на 8 групп, так что каждая группа состояла из 4 крыс. После ночного голодания крысам инъецировали в хвостовую вену соединение Примера 3 и вводили внутрижелудочно соединение Примера 3 и его соль в дозе 3,0 мг/кг (вычисленной по форме свободного основания) и объеме 10 мл/кг.
2.5. Отбор образцов
Образцы крови (0,2 мл) крыс в группе инъекции в вену брали из глазницы до введения и через 2, 15, 30 минут и 1,0, 2,0, 4,0, 6,0, 8,0, 12,0, 24,0 часа после введения, и образцы хранили в гепаринизированных пробирках и центрифугировали в течение 10 минут при 3500 об/мин. Образцы плазмы хранили при -20°С до анализа. Крыс кормили через 2 часа после введения.
Образцы крови крыс в группе внутрижелудочного введения брали до введения и через 0,5, 1,0, 2,0, 3,0, 4,0, 6,0, 8,0, 12,0, 24,0 часа после введения. Образцы обрабатывали таким же способом, как упомянуто выше.
2.7. Аналитические методы
50 мкл раствора внутреннего стандарта и 150 мкл метанола добавляли к 50 мкл плазмы крыс, полученной в различные моменты времени после введения. Затем смесь перемешивали в течение 3 минут, используя вортекс, и центрифугировали в течение 10 минут при 13500 об/мин. 10 мкл супернатанта анализировали с помощью ЖХ/МС/МС.
2.8. Вычисление фармакокинетических параметров
Камерную модель фармакокинетики приспосабливали к тестируемым соединениям, и основные фармакокинетические параметры вычисляли с помощью программного обеспечения DAS 2.0, в котором Cmax и tmax являлись действительно измеренными значениями. Абсолютную биодоступность вычисляли с помощью AUC0-t, снятой после введения и инъекции в вену.
1. Результаты фармакокинетических параметров
Фармакокинетические параметры соединений по настоящему изобретению представлены в таблице 3.
Вывод: по сравнению с другими соединениями фармакокинетика и биодоступность соединения Примера 2 были очевидно улучшены и это соединение было явно лучшим в отношении фармакокинетики.
|
Предварительная оценка гипогликемических эффектов соединений по настоящему изобретению
1. Цель теста
Для наблюдения эффектов на пероральную толерантность к глюкозе соединений Примеров 1-4 и Примеров 8-12 у нормальных мышей ICR (SINO-BRITSH SIPPR/BK LAB.ANIMAL LTD., CO) оценивали гипогликемические эффекты in vivo с использованием измерителя глюкозы в крови для измерения и анализа содержания сахара в образцах в различные моменты времени в течение 2 часов. Образцы брали из хвоста мыши.
2. Способ
2.1. дозировка
Доза введения составляла 10 мг/кг, и чистый контроль вводили с водой. Обе группы содержали 5% ДМСО.
2.2. Способ введения
Введение мышам осуществляли с помощью зонда. 4 г/кг 10%-ного раствора глюкозы вводили (0,8 мл каждой мыши) через 15 минут после введения.
2.3. Уровень сахара в крови
Введение мышам осуществляли в соответствии с вышеуказанной дозой (группе чистого контроля вводили 5%-ный водный раствор ДМСО) и измеряли значение сахара в крови (-15 минут).
Через 15 минут после введения мышам вводили 4 г/кг 20%-ного раствора глюкозы и значения сахара в крови тестировали с помощью прибора Roche ACCU-CHEK через 0,15, 30, 45, 60, 120 минут.
2.4. Результаты представлены в таблице 4:
|
Вывод: по сравнению с гипогликемическим эффектом других соединений соединение Примера 2 обладало значительным гипогликемическим эффектом.
![СОЛИ ПРОИЗВОДНЫХ ТЕТЕРАГИДРО-ИМИДАЗО[1,5-A]ПИРАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ](https://fips.edrid.ru/images/rid/ac/f4/61/400faab28e48592a5667c2a83810cce3.png)
![СОЛИ ПРОИЗВОДНЫХ ТЕТЕРАГИДРО-ИМИДАЗО[1,5-A]ПИРАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ](https://fips.edrid.ru/images/rid/ac/f4/61/e6212fc9c352669b3e71931706b7bad3.png)
![СОЛИ ПРОИЗВОДНЫХ ТЕТЕРАГИДРО-ИМИДАЗО[1,5-A]ПИРАЗИНА, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ИХ МЕДИЦИНСКОЕ ПРИМЕНЕНИЕ](https://fips.edrid.ru/images/rid/ac/f4/61/bbafe9da9d06d8b163b5e379ef5a1b4a.png)
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Производные дициклоазаалкана, способы их получения и их применение в медицине
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Липосомы иринотекана или его солей, способ их получения
Соли метил(r)-7-[3-амино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-a]пиразин-1-карбоксилата
Фармацевтическая композиция для лечения диабета 2 типа
Производные 6-аминохиназолина или 3-цианохинолина, способы их получения и их применение в качестве ингибитора рецепторных тирозинкиназ egfr или her-2
Соли бициклозамещенных производных азопиразолона, способ их получения и применение
Производные дигидроптеридинона, способ их получения и фармацевтическое применение
Производные тетрагидроимидазо[1,5-a]пиразина, способ их получения и применение их в медицине
Производные дициклоазаалкана, способы их получения и их применение в медицине
Способ получения производных r-бета-аминофенилмасляной кислоты
Соли n-[4-(1-цианоциклопентил)фенил]-2-(4-пиридилметил)амино-3-пиридинкарбоксамида
Липосомы иринотекана или его солей, способ их получения
Соли метил(r)-7-[3-амино-4-(2,4,5-трифторфенил)-бутирил]-3-трифторметил-5,6,7,8-тетрагидро-имидазо[1,5-a]пиразин-1-карбоксилата
Фармацевтическая композиция для лечения диабета 2 типа
Производные 6-аминохиназолина или 3-цианохинолина, способы их получения и их применение в качестве ингибитора рецепторных тирозинкиназ egfr или her-2
Соли бициклозамещенных производных азопиразолона, способ их получения и применение
Производные дигидроптеридинона, способ их получения и фармацевтическое применение

