Результат интеллектуальной деятельности: НОВЫЙ ИНГИБИТОР ГЛУТАМИНИЛЦИКЛАЗ И ЕГО ПРИМЕНЕНИЕ ДЛЯ ЛЕЧЕНИЯ ЗАБОЛЕВАНИЙ ЛЕГКИХ И ДЫХАТЕЛЬНЫХ ПУТЕЙ
Вид РИД
Изобретение
Область техники
Данное изобретение относится к химии органических соединений, фармакологии и медицине и касается терапии заболеваний, связанных с аберрантной активностью клеток иммунной системы, предпочтительно терапии заболеваний легких и дыхательных путей, в частности терапии бронхиальной астмы, острого и хронического бронхита, фарингита, эмфиземы легкого, ринита, риносинусита и хронической обструктивной болезни легких посредством применения соединения, обладающего эффективностью в ингибировании фермента глутаминилциклазы, вовлеченной, в частности, в процессы пост-трансляционной модификации хемокинов и хемотаксиса клеток иммунной системы.
Уровень техники
Хемотаксис или направленное движение клеток иммунной системы по градиенту концентрации некоторых эндогенных и экзогенных веществ (хемоаттрактантов) является одной из важнейших составных частей функционирования клеток иммунной системы. Избыточный приток клеток иммунной системы как правило вызывает избыточную активность клеток иммунной системы и повреждению окружающих органов и тканей. Понимание участников и процессов, связанных с процессом хемотаксиса, на молекулярном уровне может привести к новым эффективным подходам в лечении и профилактике целого ряда заболеваний, связанных с аберрантной активностью клеток иммунной системы.
Хемокины семейства CCL (CCL2, CCL7, CCL8, CCL13), являющиеся лигандами рецептора ССR2, являются наиболее мощными факторами хемотаксиса моноцитов и макрофагов в организме млекопитающих (Biochem. J. 2012 Mar 1; 442(2):403-12). Хемокины семейства CCL составляют важный класс цитокинов, необходимых для активации нейтрофилов и моноцитов и привлечения этих клеток в очаг воспаления. Однако высокие концентрации хемокинов как привило вызывают избыточный приток клеток иммунной системы. Аберрантная активность клеток иммунной системы может привести к серьезным повреждением окружающих органов и тканей. Например, в ряде случаев продукты окисления липидов могут активировать клетки эндотелия сосудов (интиму), что приводит к выделению CCL2, привлечению макрофагов которые, в свою очередь выделяют маркеры воспаления, провоцирующие повреждение артериальной стенки и развитие атеросклероза (Mol Cell. 1998 Aug;2(2):275-81; Nature. 1998 Aug 27;394(6696):894-7). Известна роль хемокинов семейства CCL в патофизиологии целого ряда аутоиммунных и аллергических состояний, опосредованных аберрантной активностью моноцитов CCR2+, CD14+ и CD16lo (ревматоидный артрит, рассеянный склероз). Кроме того, хемокины семейства CCL (в частности CCL2) вовлечены в патогенез метаболического синдрома, хронической боли, фиброза и некоторых форм рака.
Подавление аберрантной активности клеток иммунной системы, за счет ингибирования CCL-опосредованного хемотаксиса, может быть крайне востребовано для терапии целого круга заболеваний дыхательной системы, таких как бронхиальная астма, бронхит, хроническая обструктивная болезнь легких и т.д. Например воздействие бактериальных липополисахаридов, липотейхоевой кислоты или других раздражителей на слизистую оболочку органов дыхательной системы приводит к увеличению секреции CCL2 клетками гладкой мускулатуры бронхов (Am. J. Physiol. Lung Cell Mol. Physiol. 2012 Apr 15;302(8):L785-92) и росту концентрации CCL2 в бронхоальвеолярном лаваже (Mol. Immunol. 2011 Jul;48(12-13):1468-76). Увеличение концентрации CCL2 в свою очередь вызывает миграцию эозинофилов, моноцитов и базофилов и развитие аберрантного ответа ассоциированного с выделением большего количества хемокинов (TNFα, IL-1, IL-6, IL-4) и активных форм кислорода, повреждающих окружающие клетки бронхов и органов дыхания (Immunobiology. 2016 Feb;221(2):182-7; Int. J. Biol Sci. 2012; 8(9):1281-90; Mol. Immunol. 2013 Nov; 56(1-2):57-63). Повреждение бронхов приводит к развитию и сохранению аберрантной активности клеток иммунной системы и дальнейшему разрушению тканей органов дыхания. В in vivo моделях аллергической астмы блокада взаимодействия CCL2/CCR2 низкомолекулярными антагонистами показала значительную эффективность (Int Arch Allergy Immunol. 2015;166(1):52-62).
Члены семейства CCL (CCL2, CCL7, CCL8, CCL13), а также ряда других гормонов и секретируемых белков, содержат остаток пироглутаминовой кислоты (pE), роль которого заключается в защите от деградации аминопептидазами (Chem. Immunol. 1999;72:42-56; Biochemistry, 1999 Oct 5; 38(40):13013-25). Пироглутаминирование N-концевого остатка глутамина является пост-трансляционной модификацией белка, и катализируется ферментами глутаминилциклазой (QPCT или QC) (J. Biol. Chem. 2003 Dec 12; 278(50):49773-9) и изо-глутаминилциклазой (QPCTL или isoQC) (J. Mol. Biol. 2008 Jun 20;379(5):966-80). Обе изоформы фермента представляют собой близкие по структуре и субстратной специфичности белки с различной локализацией: глутаминилциклаза является секретируемым белком, в то время как изоглутаминилциклаза находится внутри клетки и закреплена в аппарате Гольджи (J. Mol. Biol. 2008 Jun 20;379(5):966-80).
В ходе экспериментальных исследований было показано, что ингибирование глутаминилциклаз приводит к резкому снижению хемоаттракторной активности непироглутаминированных форм хемокинов CCL2, CCL7, CCL8 и CCL13 (Biochem. J. (2012) 442, 403-412). Таким образом, пироглутаминирование хемокинов семейства CCL, является необходимым этапом CCL-опосредованного хемотаксиса моноцитов, а стратегия направленная на ингибирование глутаминилциклазы является возможной стратегией модулирования аберрантной активности клеток иммунной системы.
К настоящему времени известны ингибиторы глутаминилциклазы, включающие сульфолипиды (WO 2017/046256), производные флаваноидов (Bioorg. Med Chem. 2016 May 15;24(10):2280-6), производные пиридина (US 2015/0291632) и некоторые небольшие молекулы, описанные в работах последнего времени (J Med Chem. 2017 Mar 23;60(6):2573-2590; WO 2014/193974, US 2015/0291557). Наиболее близкие аналоги, соединения, являющегося предметом настоящего изобретения приведены в публикациях компании Probiodrug Aktiengesellschaft (J Biol Chem. 2003 Dec 12;278(50):49773-9). В данной работе описаны ингибиторы глутаминилциклазы на основе производных имидазола. Однако, в структурах соединений опубликованных компанией Probiodrug Aktiengesellschaft имидозол содержит алифатический заместитель по одному из атомов азота. Введение алифатического заместителя снижает метаболическую стабильность соединений. Кроме того, введение алифатического заместителя увеличивает гидрофобность соединений и облегчает проникновение соединения через гемато-энцефалический барьер, что явно излишне для подавления аберрантной активности клеток иммунной системы и потенциально может привести к возникновению побочных эффектов.
На сегодняшний день нет ни одного препарата, действующего как ингибитор глутаминилциклазы, который бы применяли в терапии заболеваний, связанных с аберрантной активностью клеток иммунной системы, поэтому сохраняется потребность в создании и внедрении в клинику новых эффективных лекарственных средств на основе ингибиторов глутаминилциклазы.
Данное изобретение касается применения соединения, обладающего эффективностью в ингибировании глутаминилциклазы, в терапии заболеваний, связанных с аберрантной активностью клеток иммунной системы, предпочтительно терапии заболеваний легких и дыхательных путей, в частности терапии бронхиальной астмы, острого и хронического бронхита, фарингита, эмфиземы легкого, ринита, риносинусита и хронической обструктивной болезни легких, а так же других заболеваний органов дыхания, связанных с аберрантной активностью клеток иммунной системы.
Краткое описание чертежей.
Фиг.1 - Приток клеток воспаления в бронхоальвеолярное пространство при изучении специфической фармакологической активности Соединения 1 на модели острой астмы у морских свинок (M±m, n=10).
Раскрытие изобретения
Задачей настоящего изобретения является разработка нового средства - ингибитора внутриклеточной и секретируемой изоформ глутаминилциклазы, эффективного для лечения заболеваний, связанных с аберрантной активностью клеток иммунной системы, в особенности заболеваний легких и дыхательных путей, таких как бронхиальная астма, острый и хронический бронхит, фарингит, эмфизема легкого, ринит, риносинусит и хроническая обструктивная болезнь легких, а так же прочих заболеваний, связанных с аберрантной активности клеток иммунной системы.
Техническим результатом данного изобретения является разработка и получение эффективного модулятора глутаминилциклазы, и в частности, эффективного ингибитора глутаминилциклазы характеризующегося высокой ингибирующей активностью и фармакокинетическими характеристиками (в частности, существенным превышением экспозиции ингибитора в органах мишенях (легкие, носогубный треугольник) по сравнению с плазмой крови), позволяющих использовать данный ингибитор для лечения заболеваний, связанных с аберрантной активностью клеток иммунной системы, в особенности заболеваний легких и дыхательных путей, таких как бронхиальная астма, острый и хронический бронхит, фарингит, ринит (в частности aллергический ринит), риносинусит, хроническая обструктивная болезнь легких и ее проявления (в частности эмфизема легкого, бронхиальная обструкция), а так же прочих заболеваний, связанных с аберрантной активностью клеток иммунной системы, в частности с аберрантным хемотаксисом клеток иммунной системы.
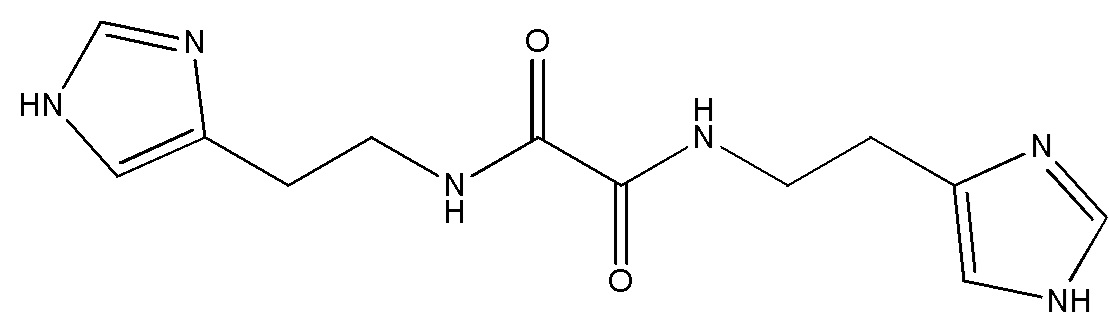
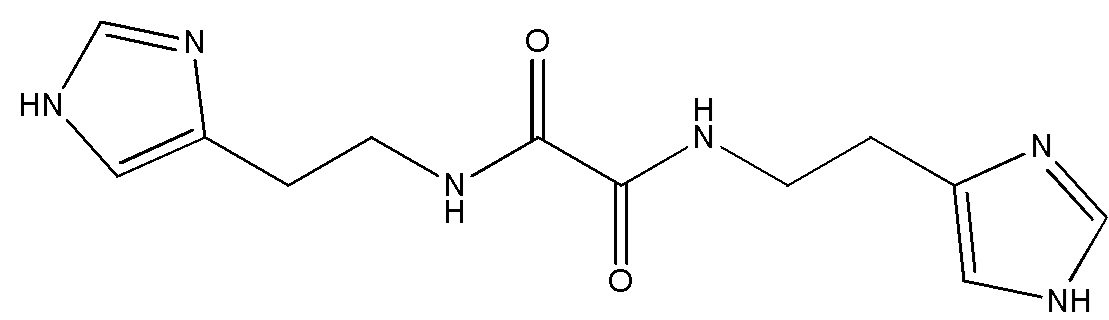
Указанный технический результат достигается путем применения соединения N,N'-бис[2-(1H-имидазол-4-ил)этил]оксаламида
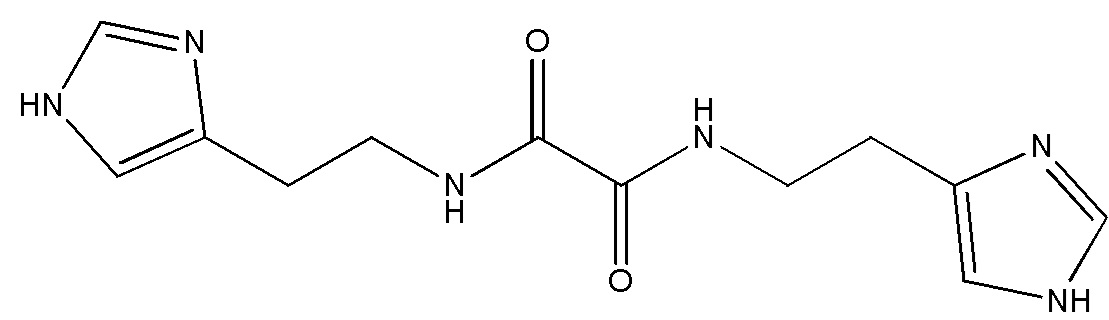
или его соли, гидрата, сольвата в качестве ингибитора внутриклеточной и секретируемой изоформ глутаминилциклазы.
Указанный технический результат достигается также посредством применения соединения N,N'-бис[2-(1H-имидазол-4-ил)этил]оксаламида или его соли, гидрата, сольвата для получения фармацевтической композиции для предупреждения и/или лечения расстройства, связанного с активностью глутаминилциклазы.
Указанный технический результат достигается также посредством применения соединения N,N'-бис[2-(1H-имидазол-4-ил)этил]оксаламида или его соли, гидрата, сольвата для получения фармацевтической композиции для предупреждения и/или лечения расстройства, связанного с аберрантной активностью клеток иммунной системы, в частности с аберрантным хемотаксисом клеток иммунной системы.
Кроме того, изобретение предусматривает фармацевтические композиции и лекарственные средства для предупреждения и/или лечения расстройства, связанного с активностью глутаминилциклазы и/или с аберрантной активностью клеток иммунной системы, в частности с аберрантным хемотаксисом клеток иммунной системы, и характеризующиеся тем, что они содержит эффективное количество соединения по изобретению и, по меньшей мере, одно фармацевтически приемлемое вспомогательное вещество. В некоторых вариантах воплощениях изобретения вспомогательное вещество представляет собой фармацевтически приемлемый носитель и/или эксципиент.
Изобретение также включает способ предупреждения и/или лечения расстройства, связанного с активностью глутаминилциклазы в организме, включающий введение в указанный организм фармацевтической композиции по изобретению. Такое расстройство, связанное с активностью глутаминилциклазы, представляет собой заболевание, связанное с аберрантной активностью клеток иммунной системы, в частности с аберрантным хемотаксисом клеток иммунной системы, в особенности заболевание легких и дыхательных путей. В некоторых неограничивающих вариантах воплощения изобретения заболевание легких и дыхательных путей представляет собой бронхиальную астму, острый и хронический бронхит, фарингит, эмфизему легкого, ринит, риносинусит или хроническую обструктивную болезнь легких. В частных случаях воплощения изобретения организм представляет собой человека или животного.
Соединение N,N'-бис[2-(1H-имидазол-4-ил)этил] оксаламида описано в заявке на изобретение RU 2013/116822.
Подробное раскрытие изобретения
Получение Соединения 1, медицинское применение которого является предметом настоящего изобретения, описано в заявке на изобретение RU 2013/116822. В указанной патентной заявки описаны производные бисамидов дикарбоновых кислот, обладающие способностью к комплексообразованию или хелатированию ионов металлов, а также их применение в качестве средства для профилактики и/или лечения вирусного гепатита, ВИЧ-инфекции, онкологических, нейродегенеративных, сердечнососудистых, воспалительных заболеваний, диабета, геронтологических заболеваний, заболеваний, вызываемых токсинами микроорганизмов, а также алкоголизма, алкогольного цирроза печени, анемии, поздней порфирии, отравлений солями переходных металлов.
В ходе исследований специфической фармакологической активности Соединения 1 в моделях различных заболеваний было установлено, что применение Соединения 1 достоверно снижает приток клеток иммунной системы. Таким образом, показано, что Соединение 1 влияет на хемотаксис клеток иммунной системы. Снижение притока клеток иммунной системы может применяться в терапии целого ряда заболеваний связанных с аберрантной активностью клеток иммунной системы, в частности заболеваний легких и дыхательных путей, таких как бронхиальная астма, острый и хронический бронхит, фарингит, эмфизема легкого, ринит, риносинусит и хроническая обструктивная болезнь легких. Поскольку влияние на хемотаксис не может быть предсказано или объяснено способностью соединения к комплексообразованию или хелатированию ионов металлов, была предпринята попытка поиска возможных терапевтических мишени Соединения 1.
Поиск возможных терапевтических мишеней, с использованием методов вычислительной химии, молекулярного моделирования и in vitro испытаний на ферментном препарате позволил обнаружить, что наблюдаемый терапевтический эффект Соединения 1 связан со способностью данного соединения ингибировать активность глутаминилциклазы.
В ходе дальнейших исследований фармакокинетики соединения 1 неожиданно оказалось, что после перорального применения вещество распределяется по органам и тканям, причем высокие концентрации достигаются в органах дыхания, в частности, в легких и носовом треугольнике.
Таким образом, Соединение 1 является новым ингибитором глутаминилциклазы, который влияет на хемотаксис клеток иммунной системы и может применяться для терапии заболеваний, связанных с аберрантной активностью клеток иммунной системы, предпочтительно заболеваний легких и дыхательных путей, в частности бронхиальной астмы, острого и хронического бронхита, фарингита, эмфиземы легкого, ринита, риносинусита и хронической обструктивной болезни легких, а также других заболеваний легких и дыхательных путей, связанных с аберрантной активностью клеток иммунной системы.
Термины и определения
Термин «Соединение 1» относится к N,N'-бис[2-(1H-имидазол-4-ил)этил]оксаламиду, также представленному структурной формулой:
.
Термин «С», когда он используется со ссылкой на температуру, означает температурную шкалу Цельсия.
Термин «IC50» означает концентрацию тестируемого соединения, при которой достигается полумаксимальное ингибирование фермента.
Термин «фармацевтически приемлемые соли» или «соли» включает соли активных соединений, которые получены с помощью относительно нетоксичных кислот. Примерами фармацевтически приемлемых нетоксичных солей могут служить соли, образованные неорганическими кислотами, такими как соляная, бромоводородная, фосфорная, серная и хлорная кислоты, или органическими кислотами, такими как уксусная, щавелевая, малеиновая, винная, янтарная, лимонная или малоновая кислоты, или полученные другими методами, используемыми в данной области, например, с помощью ионного обмена. К другим фармацевтически приемлемым солям относятся адипинат, альгинат, аскорбат, аспартат, бензолсульфонат, бензоат, бисульфат, борат, бутират, камфорат, камфорсульфонат, цитрат, циклопентанпропионат, диглюконат, додецилсульфат, этансульфонат, формиат, фумарат, глюкогептонат, глицерофосфат, глюконат, гемисульфат, гептанат, гексанат, гидройодид, 2-гидрокси-этансульфонат, лактобионат, лактат, лаурат, лаурил сульфат, малат, малеат, малонат, метансульфонат (мезилат), 2-нафталинсульфонат, никотинат, нитрат, олеат, оксалат, пальмитат, памоат, пектинат, персульфат, 3-фенилпропионат, фосфат, пикрат, пивалат, пропионат, полуфумарат, стеарат, сукцинат, сульфат, тартрат, тиоцианат, п-толуолсульфонат (тозилат), ундеканат, валериат и подобные.
Термин «сольват» используется для описания молекулярного комплекса, содержащего соединение по изобретению и одну или более молекул фармацевтически приемлемого растворителя, например этанола. Термин «гидрат» используется, когда указанным растворителем является вода.
Термин «аберрантная активность» клеток иммунной системы в настоящем документе означает активность, существенно отличающуюся от базового уровня активности клеток иммунной системы в организме при отсутствии патологии. Аберрантная активность может быть вызвана избыточным притоком клеток иммунной системы к органу или ткани, нарушением процессов, приводящих к активации клеток иммунной системы, дерегулированием процессов связанных с гибелью клеток иммунной системы, а также другими факторами.
Термин «вспомогательное вещество» означает любое фармацевтически приемлемое вещество неорганического или органического происхождения, входящее в состав лекарственного препарата или используемое в процессе производства, изготовления лекарственного препарата для придания ему необходимых физико-химических свойств.
Термин «AUC» (area under the curve) означает фармакокинетический параметр, характеризующий суммарную концентрацию лекарственного препарата в плазме крови в течение всего времени наблюдения. Математически определяется как интеграл от 0 до ∞ функции концентрации препарата (фармакокинетической кривой) в плазме крови от времени и равен площади фигуры, ограниченной фармакокинетической кривой и осями координат.
Термин «среда RPMI» (англ. Roswell Park Memorial Institute medium) означает среду для культур клеток и тканей. RPMI традиционно используется для выращивания лимфоидных клеток человека. Среда содержит значительное количество фосфата и имеет состав для выращивания в атмосфере с содержанием углекислого газа 5%.
Термин «глутаминилциклаза» означает фермент аминоацилтрансферазу участвующую в преобразовании N-концевой глутамина в пироглутамин в различных пептидных субстратах. Образование N-концевого пироглутамата защищает биологически активные пептиды, гормоны и хемокины (например, тиреотропин-высвобождающий гормон, β-хемокиновый лиганд-2) от деградации экзопептидазами и в некоторых случаях может увеличивать аффинность лигандов к их рецепторам.
Термин «секретируемая глутаминилциклаза» означает одну из изоформ фермента глутаминилциклазы, содержащая секреторный N-концевой домен и преимущественно секретируемая клетками человека. Секретируемая глутаминилциклаза преимущественно экспрессируются в клетках нервной и иммунной систем.
Термин «внутриклеточная глутаминилциклаза» означает одну из изоформ фермента глутаминилциклазы, содержащая якорный N-концевой домен и преимущественно связанная с аппаратом Гольджи. Внутриклеточная глутаминилциклаза распространена повсеместно в клетках млекопитающих и человека
Термин «хемотаксис» означает направленное движение клеток в ответ на химический раздражитель. В основе хемотаксиса лежит способность клетки отвечать на градиент концентрации хемотаксического медиатора. Хемотаксис является тем процессом, благодаря которому клетки иммунной системы покидают сосудистое русло и мигрируют в поврежденную ткань. Ведущую роль в хемотаксисе играют хемотаксические вещества (хемоатрактанты). Одним из наиболее мощных хемоатрактантов для моноцитов и макрофагов является хемокин CCL2.
Термины «лечение», «терапия» охватывают лечение патологических состояний у млекопитающих, предпочтительно у человека, и включают: а) снижение, б) блокирование (приостановку) течения заболевания, в) облегчение тяжести заболевания, т.е. индукцию регрессии заболевания, г) реверсирование заболевания или состояния, к которому данный термин применяется, или одного или более симптомов данного заболевания или состояния.
Термин «профилактика», «предотвращение» охватывает устранение факторов риска, а также профилактическое лечение субклинических стадий заболевания у млекопитающих, предпочтительно у человека, направленное на уменьшение вероятности возникновения клинических стадий заболевания. Пациенты для профилактической терапии отбираются на основе факторов, которые, на основании известных данных, влекут увеличение риска возникновения клинических стадий заболевания по сравнению с общим населением. К профилактической терапии относится а) первичная профилактика и б) вторичная профилактика. Первичная профилактика определяется как профилактическое лечение у пациентов, клиническая стадия заболевания у которых еще не наступила. Вторичная профилактика - это предотвращение повторного наступления того же или близкого клинического состояния заболевания.
Соединение 1, перспективно для лечения заболеваний, связанных с аберрантной активностью клеток иммунной системы, в особенности заболеваний связанных с аберрантным хемотаксисом клеток иммунной системы, предпочтительно терапии заболеваний легких и дыхательных путей, таких как бронхиальная астма, острый и хронический бронхит, фарингит, эмфизема легкого, ринит, риносинусит и хроническая обструктивная болезнь легких, имеющих как системный, так и локальный характер, в том числе, обусловленных первичными патологическими изменениями в тканях легких и дыхательных путей, или связанных с различными заболеваниями или длительным приемом некоторых лекарственных препаратов. В некоторых частных вариантах соединения по изобретению могут быть использованы для лечения других заболеваний легких и дыхательных путей, связанных с аберрантной активностью клеток иммунной системы.
Способ терапевтического применения соединений
Предмет данного изобретения также включает введение субъекту, нуждающемуся в соответствующем лечении, терапевтически эффективного количества соединения по изобретению. Под терапевтически эффективным количеством подразумевается такое количество соединения, вводимого или доставляемого пациенту, при котором у пациента с наибольшей вероятностью проявится желаемая реакция на лечение (профилактику). Точное требуемое количество может меняться от субъекта к субъекту в зависимости от возраста, массы тела и общего состояния пациента, тяжести заболевания, методики введения препарата, комбинированного лечения с другими препаратами и т.п.
Соединение по изобретению или фармацевтическая композиция, содержащая соединение, может быть введено в организм пациента в любом количестве (предпочтительно, суточная доза действующего вещества составляет до 0,5 г на пациента в сутки, наиболее предпочтительно, суточная доза составляет 5-50 мг/сутки) и любым путем введения (предпочтительно, пероральный путь введения), эффективным для лечения или профилактики заболевания.
После смешения лекарственного препарата с конкретным подходящим фармацевтически допустимым носителем в желаемой дозировке, композиции, составляющие суть изобретения, могут быть введены в организм человека или других животных перорально, парентерально, местно и т.п.
Введение может осуществляться как разово, так и несколько раз в день, неделю (или любой другой временной интервал), или время от времени. Кроме того, соединение может вводиться в организм пациента ежедневно в течение определенного периода дней (например, 2-10 дней), а затем следует период без приема вещества (например, 1-30 дней).
В том случае, когда соединение по изобретению используется как часть режима комбинированной терапии, доза каждого из компонентов комбинированной терапии вводится в течение требуемого периода лечения. Соединения, составляющие комбинированную терапию, могут вводиться в организм пациента как единовременно, в виде дозировки, содержащей все компоненты, так и в виде индивидуальных дозировок компонентов.
Фармацевтические композиции
Изобретение также относится к фармацевтическим композициям, которые содержат соединение 1 по изобретению (или пролекарственную форму или другое фармацевтически приемлемое производное) и один или несколько фармацевтически приемлемых носителей, адъювантов, растворителей и/или наполнителей, таких, которые могут быть введены в организм пациента совместно с соединением, составляющем суть данного изобретения, и которые не влияют на фармакологическую активность этого соединения, и являются нетоксичными при введении в дозах, достаточных для доставки терапевтического количества соединения.
Фармацевтические композиции, заявляемые в данном изобретении, содержат соединение 1 совместно с фармацевтически приемлемыми носителями, которые могут включать в себя любые растворители, разбавители, дисперсии или суспензии, поверхностно-активные вещества, изотонические агенты, загустители и эмульгаторы, консерванты, вяжущие вещества, скользящие материалы и т.д., подходящие для конкретной формы дозирования. Материалы, которые могут служить фармацевтически приемлемыми носителями, включают, но не ограничиваются, моно- и олигосахаридами, а также их производными; желатин; тальк; эксципиенты, такие как какао-масло и воск для суппозиториев; масла, такие как арахисовое, хлопковое, сафроловое, кунжутное, оливковое, кукурузное и соевое масло; гликоли, такие как пропиленгликоль; сложные эфиры, такие как этилолеат и этиллаурат; агар; буферные вещества, такие как гидроксид магния и гидроксид алюминия; альгиновая кислота; апирогенная вода; изотонический раствор, раствор Рингера; этиловый спирт и фосфатные буферные растворы. Также в составе композиции могут быть другие нетоксичные совместимые скользящие вещества, такие как лаурилсульфат натрия и стеарат магния, а также красители, пленкообразователи, подсластители, вкусовые добавки и ароматизаторы, консерванты и антиоксиданты.
Предметом данного изобретения являются также лекарственные -средства, под которыми подразумевают фармацевтические композиции, состав которых оптимизирован для определенного пути введения в организм в терапевтически эффективной дозе, например, для введения в организм орально, местно, ингаляционно, например, в виде ингаляционного спрея, или внутрисосудистым способом, интраназально, подкожно, внутримышечно, а также инфузионным способом, в рекомендованных дозировках.
Лекарственные средства данного изобретения могут содержать составы, полученные методами использования липосом, методами микрокапсулирования, методами приготовления наноформ препарата, или другими методами, известными в фармацевтике.
При получении композиции, например в форме таблетки, активное начало смешивают с одним или несколькими фармацевтическими эксципиентами, такими как желатин, крахмал, лактоза, стеарат магния, тальк, кремнезем, аравийская камедь, маннит, микрокристаллическая целлюлоза, гипромеллоза или аналогичные соединения.
Таблетки можно покрыть сахарозой, целлюлозным производным или другими веществами, подходящими для нанесения оболочки. Таблетки могут быть получены различными способами, такими как непосредственное сжатие, сухое или влажное гранулирование или горячее сплавление в горячем состоянии.
Фармацевтическую композицию в форме желатиновой капсулы можно получить, смешивая активное начало с другими веществами и заполняя полученной смесью мягкие или твердые капсулы.
Для введения парентеральным путем используются водные суспензии, изотонические солевые растворы или стерильные растворы для инъекций, которые содержат фармакологически совместимые агенты, например пропиленгликоль или бутиленгликоль.
Примеры фармацевтических композиций
Вещество, описанные в данном изобретении, может быть использовано для профилактики и/или лечения болезней человека, или животных в виде следующих составов (под «Веществом» понимается активный ингредиент):
Таблетка I мг/таблетка
Вещество 2.0
Микрокристаллическая целлюлоза 73,2
Карбоксиметилкрахмал натрия 4.0
Магния стеарат 0,8
Таблетка II мг/таблетка
Вещество 10.0
Микрокристаллическая целлюлоза 366
Карбоксиметилкрахмал натрия 20
Магния стеарат 4.0
Таблетка III мг/таблетка
Вещество 20
Микрокристаллическая целлюлоза 732
Карбоксиметилкрахмал натрия 40
Магния стеарат 8.0
Таблетка IV мг/таблетка
Вещество 50
Лактоза Ph. Eur 223.75
Кроскармеллоза натрия 6.0
Кукурузный крахмал 15
Поливинилпироллидон (5% w/v паста) 2.25
Стеарат магния 3.0
Таблетка V мг/таблетка
Вещество 200
Лактоза Ph. Eur 182.75
Кроскармеллоза натрия 12.0
Кукурузный крахмал (5% w/v паста) 2.25
Стеарат магния 3.0
Капсула мг/капсула
Вещество 10
Лактоза Ph. Eur 488.5
Магнезия 1.5
Данные составы могут быть приготовлены в соответствии со стандартными фармацевтическими методиками. Таблетки (I)-(II) могут быть покрыты кишечнорастворимой оболочкой с использованием, например, фталата ацетата целлюлозы.
Применение Соединения 1 в комбинированной терапии
Несмотря на то, что Соединение 1 по данному изобретению может вводиться в качестве индивидуального активного фармацевтического средства, его также можно использовать в сочетании с одним или несколькими другими агентами, в частности, другой агент может представлять собой антибиотик, НПВС или другое противовоспалительное средство, антигипертензивные средство, α-блокатор, цитостатический препарат и т.д. При совместном приеме внутрь терапевтические агенты могут представлять собой разные лекарственные формы, которые вводятся одновременно или последовательно в разное время, либо терапевтические агенты могут быть объединены в одну лекарственную форму.
Фраза «комбинированная терапия» в отношении соединения данного изобретения в сочетании с другими фармацевтическими агентами, означает одновременный или последовательный прием всех агентов, который так или иначе обеспечит благоприятное воздействие сочетания лекарств. Совместное введение подразумевает, в частности, совместную доставку, например, в одной таблетке, капсуле, инъекции или в другой форме, имеющий фиксированное соотношение активных веществ, также как и одновременную доставку в нескольких, отдельных лекарственных формах для каждого соединения соответственно.
Таким образом, введение соединений данного изобретения может быть осуществлено в сочетании с дополнительными методами лечения, известными специалистам в области профилактики и лечения соответствующих заболеваний, включающими применение антибактериальных, цитостатических и цитотоксических препаратов, препаратов для подавления симптомов или побочных эффектов одного из лекарств.
Если лекарственная форма представляет собой фиксированную дозу, такая комбинация использует соединения данного изобретения в приемлемом дозовом диапазоне. Соединение 1 по данному изобретению также может быть введено в организм пациента последовательно с другими агентами, в том случае, когда комбинация этих препаратов невозможна. Изобретение не ограничено последовательностью введения; соединение данного изобретения может быть введено в организм пациента совместно, до или после введения другого препарата.
Примеры
1) Получение соединения 1
Получение Соединения 1 описано в заявке на изобретение RU 2013/116822. В той же заявке описана способность Соединения 1 к комплексообразованию или хелатированию ионов металлов.
2) Характеристика биологической активности соединения по изобретению
Биологическая активность Соединения 1, являющегося предметом настоящего изобретения, была изучена в различных in vitro и in vivo экспериментах. В частности, при изучении активности Соединения 1 в различных in vitro и in vivo моделях было показано ингибирующее действие Соединения 1 на хемотаксис моноцитов, макрофагов и других клеток иммунной системы. Данное биологическое действие Соединения 1 не может быть предсказано или объяснено на основе предшествующих знаний о способности Соединения 1 к хелатированию ионов металлов.
Исследования биологической активности Соединения 1 in vitro, позволили установить, что Соединение 1 является ингибитором фермента глутаминилциклазы и, таким образом, действие Соединения 1 на хемотаксис клеток иммунной системы может быть опосредованно ингибированием активности глутаминилциклазы.
2.1) Исследование влияния Соединения 1 на ферментативную активность внутриклеточной глутаминилциклазы человека in vitro.
В ходе исследований влияния Соединения 1, являющегося предметом настоящего изобретения, на ферментативную активность внутриклеточной глутаминилциклазы in vitro впервые было обнаружено прямое ингибирующее действие Соединения 1 на рекомбинантную внутриклеточную глутаминилциклазу человека.
Активность глутаминилциклазы при различных концентрациях Соединения 1 изучалась при 25°C с использованием флуоресцентного субстрата L-глутаминил 2-нафтиламида (Gln-bNA) (Anal. Biochem. 2002 Apr 1;303(1):49-56). Реакционная смесь объемом 100 мкл содержала 50микромоль флуорогенного субстрата; ~0,2 единицы пироглутаминиламинопептидазы человека (1 единица определяется как количество, гидролизующее 1 микромоль pGlu-bNA в минуту), и аликвоту рекомбинантной внутриклеточной глутаминилциклазы человека (gQC) в 50 милимоль трисаминометан-HCl и 5% глицерине, pH 8,0. Реакцию инициировали добавлением к реакционной смеси аликвоты глутаминилциклазы, инкубированной с Соединением 1 в течение 5 минут. Дальнейшее протекание реакции отслеживали спектрофотометрически (длина волны возбуждения и эмиссии составляли 320 и 410 нм). Ферментативную активность определяли по количеству выделившегося 2-нафтиламида (bNA), рассчитанному по калибровочной кривой. Значения IC50 рассчитывали с помощью нелинейной регрессии кривой ʺконцентрация ингибитораʺ-ʺферментативная активностьʺ. В качестве вещества сравнения использовали известный ингибитор глутаминилциклаз -- соединение PBD150 (J. Med. Chem. 2006 Jan 26;49(2):664-77).
В результате эксперимента было установлено, что Соединение 1 ингибирует активность внутриклеточной глутаминилциклазы с IC50=2,7 мкМ.
2.2) Исследование влияния Соединения 1 на ферментативную активность секретируемой глутаминилциклазы человека in vitro.
В лунки 96-луночного ПЦР планшета (Bio-Rad) добавляли по 25 мкл раствора Соединения 1. В каждую лунку, содержащую Соединение 1, помещали 25 мкл 320 мкМ раствора (2S)-2-амино-N-(4-метил-2-оксо-хромен-7-ил)пентандиамида (Q-AMC) в 25 мМ HEPES, pH 7.0 и перемешивали. В каждую лунку (кроме контролей холостого гидролиза) помещали 50 мкл раствора секретируемой глутаминилциклазы (QPCT) с концентрацией 0,2 мкг/мл в буфере (25 мМ HEPES, pH 7.0), либо аналогичный буферный раствор (контроль холостого гидролиза). Планшет инкубировали при комнатной температуре в течение 20 минут, затем помещали в термостат, нагревали при 100°C в течение 5 минут и затем охлаждали во льду в течение 3 минут. В каждую лунку добавляли 100 мкл раствора пироглутаминилпептилазы (PGPEP) с концентрацией 1 мкг/мл в буфере (0.1 мМ трисаминометан, 5 мМ дитиотреитол, pH 9.0) и инкубировали при комнатной температуре в течение 10 минут. Из каждой лунки отбирали 2 раза по 90 мкл раствора, помещали в 3 лунки планшета для флуориметрии Greiner Bio и определяли флуоресценцию (возбуждение - 355 нм, испускание - 460 нм). Полученную кривую ингибирования (зависимости I от концентрации вещества) аппроксимировали дозозависимой кривой (Dose-response curve) c нижней асимптотой=0 и верхней асимптотой =1 с помощью программного обеспечения Origin Pro 8.0, что позволяло получить значение IC50.
В результате эксперимента было установлено, что Соединение 1 ингибирует активность секретируемой глутаминилциклазы с IC50=1,4 мкМ.
2.3) Исследование влияния Соединения 1 на миграцию моноцитов in vitro
Влияние Соединения 1 на миграцию моноцитов in vitro было изучено с использованием клеток линии U937, подрощенных до концентрации (2-3)×106 клеток/мл на среде RPMI 1640 с добавлением 10% термоинактивированной фетальной коровьей сыворотки (Biochem. J. 2012 Mar 1;442(2):403-12). Примерно 1×107 клеток U937 инкубировали с различными концентрациями Соединения 1 (всего 7 концентраций) при 37°C в течение 2 часов и затем обрабатывали липополисахаридом E.coli (O111:B4). Супернатант (кондиционированная среда) использовали для изучения миграции моноцитов.
Свежую порцию клеток U937 окрашивали флуоресцентным красителем Calcein AM в течение 1 часа при 37°C. После этого аликвоту окрашенных клеток помещали верхнее отделение лунок планшета BD FalconTM HTS FluoroBlok, ячейки которого разделены полупроницаемыми оптически непрозрачными мембранами. В нижнее отделение лунок планшета помещали кондиционированную среду, полученную после инкубирования клеток с ингибитором и липополисахаридом. Планшеты инкубировали 2 часа при 37°C, количество (% относительно эксперимента без ингибитора) клеток, мигрировавших в нижний отсек лунок определяли флуориметрически. В качестве вещества сравнения использовали известный ингибитор глутаминилциклаз -- соединение PBD150 (J. Med. Chem. 2006 Jan 26;49(2):664-77).
В результате эксперимента было установлено, что Соединение 1 оказывает ингибирующий эффект на миграцию моноцитов in vitro, при этом эффективность подавления миграции, близкая к 60%, сохраняется в широком диапазоне концентраций от 1 мкМ до 300 мкМ.
2.4) Исследование влияния Соединения 1 на хемотаксис лейкоцитов in vivo на модели бронхиальной астмы у морских свинках
Индукцию бронхиальной астмы у морских свинок осуществляли по стандартной методике (Current Drug Targets. 2008 Jun; 9(6):452-65). Животных иммунизировали однократным внутрибрюшинным введением 0,5 мл раствора, содержащего 100 мкг/мл овальбумина (Sigma) и 100 мг/мл гидрокиси алюминия. Интактным животным внутрибрюшинно вводили физ. раствор в объеме 0,5 мл.
На 29, 30 и 31 день эксперимента проводили провокацию гиперреактивности дыхательных путей путем ингаляционного введения овальбумина в возрастающих концентрациях - 0,1, 0,3 и 0,5 мг/мл (на 29, 30 и 31 день соответственно). Ингаляцию осуществляли в течение 5 минут или до появления выраженных признаков асфиксии (падение на бок). На 32-ой день животным вводили разрешающую дозу овальбумина - 1 мг/мл в течение 5 минут с оценкой бронхоспастической реакции.
Исследуемое соединение вводили животным внутрижелудочно ежедневно один раз в день в течение 10 суток, заканчивая за 24 ч до введения разрешающей дозы антигена.
Через 24 часа после введения разрешающей дозы овальбумина у животных отбирали бронхоальвеолярный лаваж (БАЛ). Взятие БАЛ проводили под наркозом путем промывания легких 5-ю мл подогретого до 37°С физиологического раствора через трахею при помощи шприцевого дозатора.
В жидкости бронхоальвеолярного смыва с помощью камеры Горяева подсчитывали абсолютное количество клеточных элементов в 1 мкл смыва. Затем бронхоальвеолярный лаваж центрифугировали при 200 g в течение 10 минут. Из осадка клеток готовили мазки, которые в дальнейшем фиксировали в метаноле и окрашивали по Романовскому-Гимзе для подсчета эндопульмональной цитограммы.
Цитологический анализ бронхоальвеолярного лаважа (БАЛ) выявил многократное увеличение клеточных элементов в БАЛ сенсибилизированных морских свинок (рисунок 1). Таким образом, данная модель характеризуется воспалительной реакцией респираторного тракта экспериментальных животных. Анализ отдельных типов клеток показал, что наиболее выраженный приток клеток приходился на эозинофилы. Полученные результаты подтверждают литературные данные и позволяют сделать заключение о том, что смоделированное воспаление носит аллергический характер.
Ежедневное внутрижелудочное введение Соединения 1 в течение 10 дней снизило приток клеток воспаления в бронхоальвеолярное пространство. Соединение 1 оказывало терапевтический эффект во всем тестируемом диапазоне доз (0.14-14 мг/кг), снижало как общее количество лейкоцитов, так и количество отдельных клеточных типов: эозинофилов, нейтрофилов, макрофагов (Фиг.1). При этом на фигуре 1, знаком * обозначены различия, статистически значимые по сравнению с интактной группой (р<0,05); и знаком - & - различия, статистически значимые по сравнению с группой контроля (р<0,05).
Полученные результаты свидетельствуют о том, что Соединение 1 оказывает выраженный терапевтический эффект на модели бронхиальной астмы.
2.5) Исследование влияния Соединения 1 на хемотаксис макрофагов, нейтрофилов и эозинофилов in vivo на модели неинфекционного воспаления легких крыс
Модель сефадекс-индуцированного неинфекционного воспаления легких у крыс реализовали по стандартной методике (Int. Arch. Allergy Immunol. 2011; 154(4):286-94). Крысам-самцам линии Вистар однократно ингаляционно вводили Cефадекс G-200 (Pharmacia, Sweden) в дозе 5 мг/кг. Исследуемые соединения вводили животным внутрижелудочно четырех-кратно: за 24 и 1 ч до, а также 24 и 45 ч после введения сефадекса. Препарат сравнения будесонид вводили по той же схеме ингаляционно в дозе 0,5 мг/кг. Через 48 ч после ингаляции сефадекса производили забор бронхоальвеолярного лаважа. В лаваже оценивали суммарное количество лейкоцитов и определяли лейкоцитарную формулу.
Анализ бронхоальвеолярного лаважа показал, что однократное ингаляционное введение сефадекса G-200 крысам вызывает выраженный приток лейкоцитов в легкое. Количество всех клеточных типов было увеличено в группе контроля по сравнению с интактными (таблица 1).
|
Внутрижелудочное введение Соединения 1 крысам снизило содержание клеток воспаления в БАЛ до уровня интактных животных. Полученные результаты свидетельствуют о том, что Соединение 1 оказывает терапевтический эффект при воспалении нижних дыхательных путей, в частности при неинфекционном воспалении легких.
2.6) Исследование активности Соединения 1 in vivo на модели аллергического ринита у морских свинок
Модель аллергического ринита реализовали по стандартной методике (Int. Immunopharmacol. 2013 Sep;17(1):18-25). Морских свинок (250-300 гр) иммунизировали 4х кратным (на 0, 7, 14 и 21 сутки) внутрибрюшинным введением смеси овальбумина (100 мкг/свинка) и гидроксида алюминия (5 мг/свинка), разведенных и суспендированных в физиологическом растворе. На 28-е сутки исследования раствор овальбумина (60 мг/мл) животным вводили интраназально по 20 мкл в каждую ноздрю. На 35-е сутки животным вводили раствор овальбумина (200 мкг/мл, 25 мкл) подкожно, предварительно выбрив участок кожи на спине. Подтверждением наличия сенсибилизации было формирование отека и покраснения в месте инъекции. На 42-е сутки исследования проводили интраназальное введение раствора овальбумина (60 мг/мл, 20 мкл/ноздря). С целью контроля формирования именно аллергического воспаления была сформирована группа ложноиммунизированных животных: на 0, 7, 14 и 21 сутки свинки получали раствор гидроксида аллюминия (5 мг/свинка), на 28-е и 35-е сутки - физ. раствор, на 42-е овальбумина (60 мг/мл, 20 мкл/ноздря).
Соединение 1 (0.014, 0.14, 1.4 мг/кг) вводили животным внутрижелудочно однократно в двух режимах: за 3 и 1 ч до последнего интраназального введения овальбумина. Препарат сравнения (дексаметазон) вводили внутрижелудочно однократно за 3 ч до последнего интраназального введения овальбумина.
В течение 2 ч после последнего введения овальбумина проводили оценку клинических проявлений ринита: подсчитывали количество чихов, почесываний носа. Через 24 часа после последнего введения овальбумина производили забор назального смыва. В назальном смыве оценивали суммарное количество лейкоцитов и определяли лейкоцитарную формулу.
Анализ назального смыва показал, что аллергический ринит сопровождается выраженным притоком лейкоцитов в полость носа. Максимальное увеличение было отмечено в отношении количества эозинофилов (таблица 2).
Внутрижелудочное введение Соединения 1 морским свинкам снизило содержание эозинофилов в назальном смыве. При введении Соединения 1 в дозе 0.014 мг/кг за 1 час до последнего интраназального введения овальбумина уровень эозинофилов снижался до уровня ложноиммунизированных животных. По выраженности действия Соединение 1 не уступало дексаметазону.
|
Учет клинических проявлений аллергического ринита в течение 2х часов после последнего интраназального введения овальбумина животным показал выраженное увеличение у экспериментальных животных количества чихов и почесываний носа, что свидетельствует о корректности реализованной модели аллергического ринита. При терапии Соединением 1 наблюдалась тенденция к снижению клинических проявлений ринита. При терапии Соединением 1 в дозах 0.14, 1.4 мг/кг уровень почесываний носа снижался статистически значимо (таблица 3). Препарат сравнения (дексаметазон) оказал сходное действие.
|
2.7) Исследование активности Соединения 1 in vivo на модели формалин-индуцированного острого риносинусита у крыс
Индукцию острого риносинусита проводили у крыс-самцов Вистар путем интраназального введения 20 мкл 7,5% формалина в каждый носовой ход. Соединение 1 вводили в дозах 1.8 мг/кг и 18 мг/кг ежедневно один раз в сутки, начиная через 24 ч после введения формалина, последнее введение происходило на 7-е сутки. Дексаметазон (0.6 мг/кг) вводили в том же режиме. На 8-е сутки производили забор назального смыва. В назальном смыве оценивали суммарное количество лейкоцитов и определяли лейкоцитарную формулу.
Анализ назального смыва показал, что острый риносинусит сопровождается выраженным притоком лейкоцитов в полость носа. Максимальное увеличение было отмечено в отношении количества макрофагов (таблица 4).
Внутрижелудочное введение исследуемого соединения крысам снизило содержание макрофагов и нейтрофилов в назальном смыве до уровня интактных животных. По выраженности действия Соединение 1 не уступало дексаметазону (Таблица 4).
|
Таким образом, полученные результаты показали, что Соединение 1 оказывает выраженное терапевтическое действие при риносинусите.
2.8) Исследование активности Соединения 1 in vivo на модели неинфекционного фарингита у крыс
Модель неинфекционного фарингита реализовали на крысах-самцах линии Вистар. Крысам производили анестезию натрий-тиопентоном (50 мг/кг, внутрибрюшинно) и в наружную яремную вену вставляли канюлю с трубкой из силиконового каучука RenaSil® (SIL 037, Braintree Scientific, Inc., Braintree, MA) с помощью гепаринизированного физиологического раствора (40 ЕД/мл). Краситель Эванс голубой (Evans Blue, ЭГ) (30 мг/кг) вводили всем животным внутривенно через катетер; через 10 мин после введения красителя ЭГ на поверхность слизистой глотки наносили 30%-ый раствор формалина следующим образом: язык слегка вытаскивали, область глотки открывали глубоко в полости рта с помощью тупых щипцов и осторожно стерильным ватным тампоном 3 раза наносили раствор формалина (50 мкл) в течение 5 сек при каждом нанесении. В интактной группе применяли физиологический раствор.
Через 60 минут после нанесения 30% раствора формалина животных эвтаназировали путем обескровливания. Головную часть каждой крысы перфузировали гепаринизированным физиологическим раствором (40 ЕД/мл), чтобы удалить внутрисосудистый краситель ЭГ.
Степень воспаления оценивали по тесту экссудации красителя Эванса голубого (ЭГ). Краситель ЭГ из ткани экстрагировали в формамид при 55°С в течение 24 часов, и поглощение определяли спектрофотометрически при 620 нм. Количество красителя в ткани рассчитывали, используя стандартную кривую для красителя Эванса голубого, и выражали в микрограммах красителя на грамм сырого веса ткани (мкг/г).
|
Соединение 1 вводили внутрижелудочно за 24 и 1 ч до нанесения формальдегида в дозах 6 и 18 мг/кг. Контрольной группе вводили раствор формальдегида с концентрацией 30%. В качестве препаратов сравнения использовали дексаметазон и диклофенак. Дексаметазон (0,6 мг/кг) и диклофенак (8 мг/кг) вводили внутрижелудочно за 24 и 1 ч до нанесения формальдегида. Результаты исследования представлены в таблице 5.
Из таблицы видно, что введение формальдегида (контроль) приводит значительному увеличению концентрации красителя в ткани, что говорит о воспалении глотки у крыс - фарингите. Исследуемое соединение проявило значительную защиту от вызываемого формальдегидом фарингита. Соединение 1 оказало действие во всех тестируемых дозах (6 и 18 мг/кг) -концентрация красителя в ткани снизилась в 2.8 и 6.4 раза, соответственно, по сравнению с контролем. Введение дексаметазона (0,6 мг/кг) также приводило к уменьшению воспаления: концентрация красителя снизилась в 2,1 раза. Диклофенак эффекта не оказал.
Таким образом, полученные результаты показали, что Соединение 1 оказывает выраженное противовоспалительное действие при фарингите
2.8) Исследование активности Соединения 1 in vivo на модели эмфиземы легкого у мышей
Воспаление и эмфизему легких вызывали однократной эндотрахеальной инъекцией свиной панкреатической эластазы мышам- самкам линии C57Bl/6 [Lüthje L., Raupach T., Michels H. et al. Exercise intolerance and systemic manifestations of pulmonary emphysema in a mouse model// Respiratory Research 2009, 10(1), 7]. Эластазу вводили однократно эндотрахеально в дозе 0,75 Ед/мышь в 30 мкл NaCl 0,9%. Введение эластазы принимали за 0 день эксперимента.
Соединение 1 вводили в дозах 0.3, 1 и 3 мг/кг ежедневно внутрижелудочно один раз в сутки на 8-21-е сутки эксперимента. На 21е сутки исследования животных эвтаназировали в CO2-камере и выделяли легкие. Для оценки динамики и выраженности развития эмфиземы в ткани легких из левого легкого изготавливались гистологические препараты. Для этого легкое фиксировали в 10% растворе нейтрального формалина и затем заливали в парафин по стандартной методике. Из депарафинизированных срезов получали препараты верхушки легкого, среднего легочного поля и нижнего легочного поля толщиной 5 мкм и окрашивали гематоксилином и эозином по стандартной методике. Далее делали фотографии верхушки легкого, среднего легочного поля и нижнего легочного поля. С помощью средств компьютерной обработки графических данных исследовали локализацию и площадь эмфизематозно-расширенной ткани легких (% от нормальной ткани), из расчетов исключались сосуды и бронхи (Int. J. Biomed. Imaging. 2012;2012:734734; Front Physiol. 2015 May 12;6:14).
При гистологическом исследовании на 21 сутки после введения эластазы во всех зонах легкого мышей обнаруживалось умеренно выраженное полнокровие сосудов микроциркулярного русла и капилляров межальвеолярных перегородок. Кроме этого, эластаза приводила к утолщению стенок альвеол за счет лимфо-макрофагальной инфильтрации, а также воспалительной инфильтрации интерстициальной ткани. Просвет отдельных альвеол также заполнен макрофагами и лимфоцитами. В результате действия повреждающего агента на эластические волокна бронхиол, альвеолярных ходов и альвеол в паренхиме легких отмечались растяжение альвеол и разрывы альвеолярных перегородок, развивалась диффузная эмфизема. Анализ препаратов показал, что на 21-е сутки опыта значительная часть ткани левого легкого была занята эмфизематозно-расширенными альвеолами с разрушением межальвеолярных перегородок. Эмфизема локализовалась во всех легочных полях. Наиболее выраженные поражения наблюдались в нижнем легочном поле (таблица 6).
Таблица 6
Площадь эмфизематозно расширенной ткани легких (% от нормальной) у мышей в условиях интратрахеального введения эластазы на 21-е сутки эксперимента (M±m, n=15)
|
Примечание: - различия статистически значимы по сравнению с интактной группой (р<0,05), & - различия статистически значимы по сравнению с контрольной группой (р<0,05).
Введение Соединения 1 уменьшало относительную площадь эмфизематозно-расширенных альвеол по сравнению с группой патологического контроля (таблица 6). Полученные данные дают основание заключить, что Соединение 1 оказывает терапевтический эффект при эмфиземе легкого.
2.9) Исследование терапевтической активности Соединения 1 на модели ХОБЛ у морских свинок
Исследование проводили на морских свинках самцах. Хроническую обструктивную болезнь легких вызывали курсовым эндотрахеальным введением липополисахарида клеточной стенки E. Coli (ЛПС) и экстрактом табачного дыма (ЭТД) (Biol. Pharm. Bull. 2009 Sep;32(9):1559-64). ЭТД получали из сигарет марки Hi-Lite (Япония) (состав 1 сигареты: смола 17 мг/сиг, никотин 1,4 мг/сиг). Перед получением экстракта удаляли сигаретный фильтр, длина сигареты с фильтром 80 мм, при удалении фильтра 55 мм. Экстракцию производили путем протягивания дыма зажженной сигареты через PBS с постоянной скоростью, при помощи вакуумного насоса (40 мл/40 сигарет). Время сжигания одной сигареты составляло 180 секунд. Для удаления частиц полученный экстракт фильтровали через бактериальный фильтр с величиной поры 45 нм. ЭСД ингалировали морским свинкам (0.3 мл/мин, 40 мин) на ежедневно один раз в сутки на 1-4, 6-9, 11-14, 16-19 сутки. ЛПС ингалировали морским свинкам (25 мкг/мл, 0.3 мл/мин, 1ч) один раз в сутки на 5, 10 и 15е сутки. Через 2 ч после последней ингаляции ЭСД проводили забор бронхоальвеолярного лаважа (БАЛ) и ткани легкого. Взятие БАЛ проводили под наркозом путем промывания легких 5-ю мл подогретого до 37°С физиологического раствора через трахею при помощи шприцевого дозатора.
В жидкости бронхоальвеолярного смыва с помощью камеры Горяева подсчитывали абсолютное количество клеточных элементов в 1 мкл смыва (цитоз). Затем БАЛ центрифугировали при 200 g в течение 10 минут. Из осадка клеток готовили мазки, которые в дальнейшем фиксировали в метаноле и окрашивали по Романовскому-Гимзе для подсчета эндопульмональной цитограммы.
Для оценки динамики и выраженности развития эмфиземы в ткани легких из левого легкого изготавливались гистологические препараты. Для этого легкое фиксировали в 10% растворе нейтрального формалина и затем заливали в парафин по стандартной методике. Из депарафинизированных срезов получали препараты верхушки легкого, среднего легочного поля и нижнего легочного поля толщиной 5 мкм и окрашивали гематоксилином и эозином по стандартной методике. Оценивали площадь эмфизематозно-расширенной ткани легких (% от нормальной ткани), из расчетов исключались сосуды и бронхи (Int J Biomed Imaging. 2012;2012:734734; Front Physiol. 2015 May 12;6:14).
Соединение 1 вводили животным внутрижелудочно ежедневно 1 раз в сутки на 10-19е сутки исследования (последнее введение - за 1 ч до последней ингаляции ЭСД). Группе ложной патологии вместо ЭТД и ЛПС ингалировали физ. раствор. Контрольные животные вместо исследуемого вещества получали растворитель в эквивалентном объеме.
Проведенное исследование показало, что на модели ХОБЛ Соединение 1 снижает приток клеток воспаления в бронхоальвеолярное пространство морских свинок. Наиболее выражено Соединение 1 снижает приток нейтрофилов, макрофагов и эозинофилов (Таблица 7). Снижение притока клеток воспаления в легкое ведет к снижению площади эмфизематозно расширенной ткани соответствующего органа. Соединение 1 снизило площадь эмфизематозно расширенной ткани легкого в 1.5 раза (с 45,7±3,84% до 31,0±4,4%).
|
Таблица 8
Площадь эмфизематозно расширенной ткани легких (% от нормальной) у морских свинок на модели ХОБЛ (M±m, n=10)
|
Примечания:
# - отличие от группы ложной патологии по t-критерию Стьюдента при p<0,05;
& - отличие от группы контроля по t-критерию Стьюдента при p<0,05.
Обобщая полученные результаты, можно заключить, что Соединение 1 оказывает выраженное терапевтическое действие при ХОБЛ.
Таким образом, проведенные исследования показали, что Соединение 1 обладает эффективностью в ингибировании фермента глутаминилциклазы (IC50=2,7 мкМ для внутриклеточной глутаминилциклазы и IC50=1,4 мкМ для секретируемой глутаминилциклазы) и в микромолярном диапазоне концентраций ингибирует хемотаксис макрофагов in vitro. На моделях бронхиальной астмы у морских свинок и сефадекс-индуцированного неинфекционного воспаления легких у крыс Соединение 1 ингибирует приток клеток воспаления (эозинофилов, нейтрофилов, макрофагов) в бронхоальвеолярное пространство. На моделях аллергического ринита, риносинусита, фарингита, эмфиземы легкого и ХОБЛ введение Соединения 1 приводит к снижению выраженности заболевания.
2.10) Исследование фармакокинетики Соединения 1 при однократном пероральном введении
Для анализа применимости Соединения 1 в качестве лекарственного препарата были исследованы фармакокинетические параметры этого соединения. Поскольку соединение оказывает выраженное действие на хемотаксис клеток иммунной системы, для достижения максимального терапевтического эффекта представляется разумным достичь высоких концентраций Соединения 1 в органе-мишени. В то же время с точки зрения минимизации возможных негативных эффектов, связанных с системным подавлением хемотаксиса клеток иммунной системы представляется обоснованным стараться создать по возможности низкие концентрации Соединения 1 в системном кровотоке. Таким образом, органы обеспечивающие «накопление» Соединения 1 будут наиболее благоприятными органами мишенями. В качестве меры «накопления» Соединения 1 была выбрана характеристика «тканевая доступность» определяемая отношением значения площади под фармакокинетической кривой, которая рассчитывается от момента введения до конкретной временной точки (AUC0-t) в ткани к соответствующей величине AUC0-t в плазме крови.
Фармакокинетика Соединения 1 была изучена после однократного внутрижелудочного введения Соединения 1 крысам в дозе 15.4 мг/кг. Определение концентрации Соединения 1 при фармакокинетических исследованиях было проведено методом высокоэффективной жидкостной хроматографии с масс-спектрометрическим детектированием (ВЭЖХ-МС/МС).
При изучении фармакокинетики Соединения 1 неожиданно оказалось, что концентрации Соединения 1 в органах дыхания (в легком и носовом треугольнике) в 3-27 раз выше, чем в плазме (см таблицу 9).
|
Тканевая доступность Соединения 1 в легком составляет 5, биодоступность Соединения 1 в носовом треугольнике составляет 29 (Таблица 10). Таким образом применение Соединения 1 перспективно для лечения заболеваний легких и дыхательных путей.
|
Несмотря на то, что изобретение описано со ссылкой на раскрываемые варианты воплощения, для специалистов в данной области должно быть очевидно, что конкретные подробно описанные эксперименты приведены лишь в целях иллюстрирования настоящего изобретения, и их не следует рассматривать как каким-либо образом ограничивающие объем изобретения. Должно быть понятно, что возможно осуществление различных модификаций без отступления от сути настоящего изобретения

Новый способ получения n,n'-бис[2-(1н-имидазол-4-ил)этил]малонамида
Новый способ получения (s)-2-(2-(4-гидроксифенил)ацетамидо)-3-фенилпропановой кислоты
Производные глутаримидов, их применение, фармацевтическая композиция на их основе, способы их получения
Циклическое производное гемина с антимикробными свойствами и способ его синтеза
Новый ингибитор глутаминилциклаз и его применение
Амидное соединение и его применение в качестве средства для лечения и профилактики заболеваний, вызываемых рнк-содержащими вирусами
Производные бисамидов дикарбоновых кислот, их применение, фармацевтическая композиция на их основе, способы их получения
Способ восстановления плотности интерфероновых рецепторов для профилактики или лечения заболеваний, связанных с пониженной плотностью интерфероновых рецепторов
Новый способ получения n,n'-бис[2-(1н-имидазол-4-ил)этил]малонамида
Применение бисамидного производного малоновой кислоты для лечения аллергических и других заболеваний человека и животных
Новый антагонист тахикининовых рецепторов и его применение
Новый цинковый комплекс, его получение и применение для терапии заболеваний человека и животных

