Результат интеллектуальной деятельности: Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака
Вид РИД
Изобретение
Настоящее изобретение относится к новым замещенным N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)фенил]-5-хлор-пиримидин-2,4-диаминам, ингибиторам киназы анапластической лимфомы ALK и рецептора эпидермального фактора роста EGFR, предназначенным для лечения рака.
Протеинкиназы представляют собой большое семейство белков, которые играют центральную роль в регуляции широкого круга клеточных процессов и осуществляют контроль над клеточными функциями. Аномальная активность протеинкиназы связана с несколькими заболеваниями, в том числе псориазом и раковыми заболеваниями.
Известны ингибиторы киназы анапластической лимфомы (ALK), представляющие собой производные фосфора, запатентованные ARIAD Pharmaceuticals [США 2014/0066406 A1, WO 2009/143389]. В этой серии соединений самым известным является АР26113, также известный как Бригатиниб (Brigatinib), оральный ингибитор рецепторов тирозинкиназ, таких как киназы анапластической лимфомы (ALK) и рецептора эпидермального фактора роста (EGFR), предназначенный для лечения рака. Двойной ALK/EGFR ингибитор АР26113 связывает и ингибирует ALK и гибридные ALK белки, а также EGFR и его мутантные формы.
Следует отметить, что в публикациях имеет место путаница в химической структуре АР26113. Под этим номером опубликованы две структуры: CAS # 197958-12-5 и CAS #197953-54-0) [http://www.medkoo.com/Anticancer-trials/AP26113.htm]. Поэтому ниже эти ингибиторы будут обозначаться как АР26113(1) и АР26113(2).
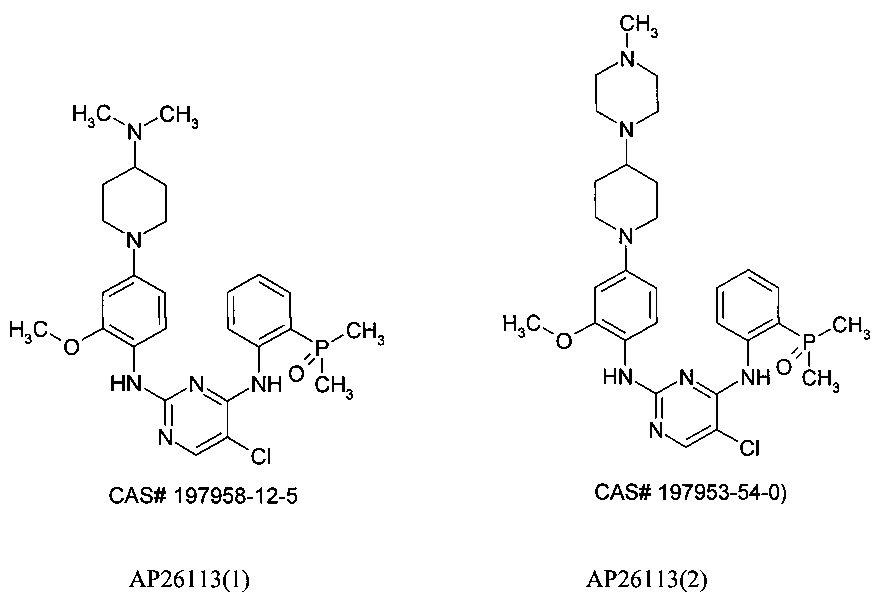
В настоящее время Бригатиниб (АР26113) демонстрирует в фазе 2 клинических испытаний эффективную внутричерепную противоопухолевую активность по отношению к ALK-положительному немелкоклеточному раку легких (NSCLC) у больных с метастазами в головном мозге, последовавшими после лечения Кризатинибом [http://www.esmo.org/Conferences/Past-Conferences/ELCC-2015-Lung-Cancer/News-Press-Releases/Brigatinib-AP26113-Shows-Intracranial-Anti-tumour-Activity-in-ALK-positive-NSCLC-patients-with-Brain-Metastasis-Following-Crizotinib].
В связи с большим количеством протеинкиназ и множеством связанных с ними заболеваний, создание новых классов селективных ингибиторов протеинкиназ для лечения заболеваний, обусловленных повышенной активностью протеинкиназ, остается актуальной задачей.
Ниже приведены определения терминов, которые использованы в описании данного изобретения.
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» (С1-С6)алкильных заместителей. Предпочтительными алкильными группами являются (С1-С6)алкил, еще более предпочтительными (С1-С3)алкил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклопропилметил, циклобутилметил, циклопентилметил, н-пентил, 2-пентил, 3-пентил, нео-пентил, н-гексил, циклогексил. Алкил может иметь заместители.
«Замещенный алкил» - замещенный алкил может иметь один или несколько одинаковых или различных заместителей, включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, гетероароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонил, гетероаралкилокси или RkaRk+1aN-, где Rka и Rk+1a независимо друг от друга представляют собой «заместители аминогруппы», значение которых определено в данном разделе, например, атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rka и Rk+1a вместе с атомом N, с которым они связаны, образуют через Rka и Rk+1a 4-7 членный гетероциклил или гетероцикленил. Предпочтительными «алкильными заместителями» являются арил, гетероарил, гетероциклил, гидрокси, C1-C5 алкокси, С1-С5 алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или RkaRk+1aN-, RkaRk+1aNC(=O)-, аннелированный арилгетероцикленил, аннелированный арил гетероциклил.
«Аминогруппа» означает R1R2N- группу, замещенную или незамещенную необязательно одинаковыми заместителями R1 и R2. Аминогруппа может иметь заместители.
«Активный компонент» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Галоген» означает фтор, хлор, бром и йод. Предпочтительными являются фтор, хлор и бром.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и других готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» обозначает композицию, включающую в себя активный компонент и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких, как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, такие как мази и кремы, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., "Pharmaceutical Salts" J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Цель настоящего изобретения заключается в создании новых ингибиторов киназы анапластической лимфомы ALK и рецептора эпидермального фактора роста EGFR для лечения рака.
Поставленная цель достигается новыми замещенными N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)фенил]-5-хлор-пиримидин-2,4-диаминами, их таутомерами, стереоизомерами, фармацевтически приемлемыми солями и сольватами.
Предметом настоящего изобретения являются замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)фенил]-5-хлор-пиримидин-2,4-диамины общей формулы 1, их таутомеры, стереоизомеры, фармацевтически приемлемые соли и сольваты:
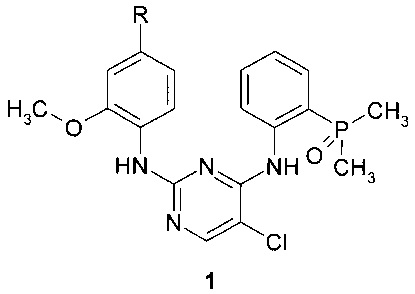
где R представляет собой заместитель, выбранный из ряда (а)-(о):
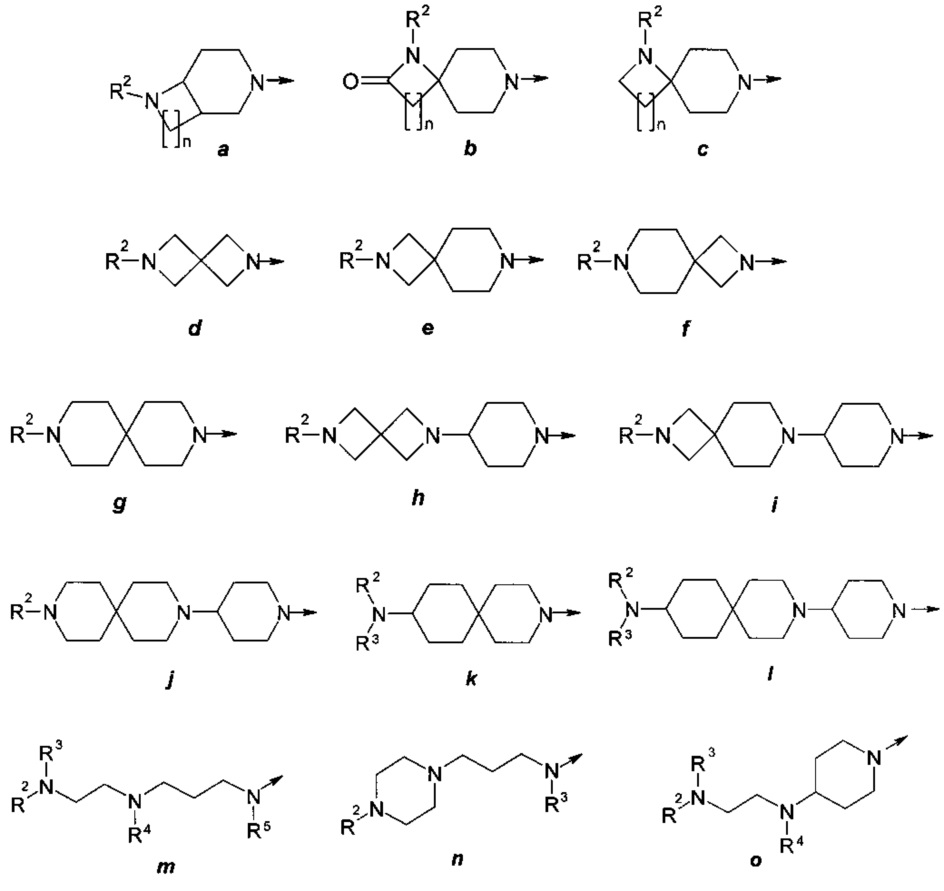
где: R2, R3, R4 и R5 представляют собой необязательно одинаковые и необязательно замещенные С1-С4алкилы;
n представляют собой необязательно одинаковое число 1 или 2; стрелка указывает точку присоединения заместителя R.
Более предпочтительным соединением является ингибитор ALK и EGFR, выбранный из ряда:
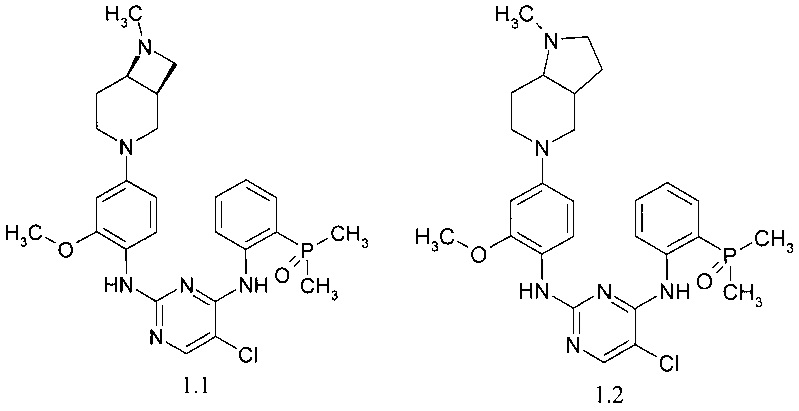
N4-[2-(диметилфосфорил)фенил]-N2-[4-((1S,6R)-7-метил-3,7-диазабицикло[4.2.0]окт-3-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.1);
N4-[2-(диметилфосфорил)фенил]-N2-[4-(1-метилоктагидро-пирроло[3,2-с]пиридин-5-ил)-фенил]-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.2);
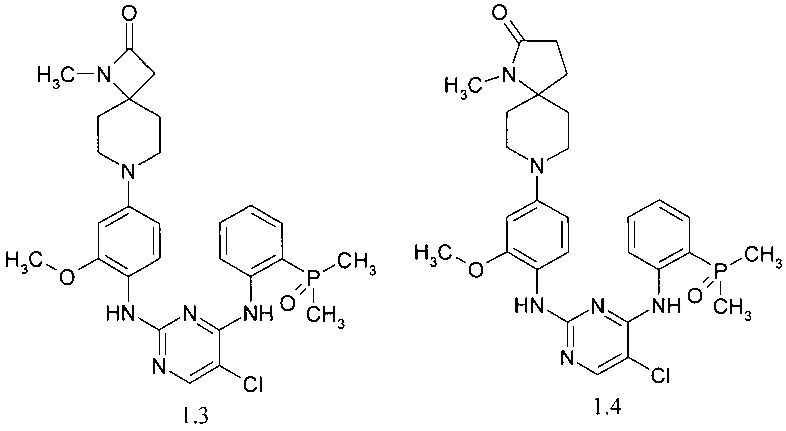
7-(4-{4-[2-(диметилфосфорил)фениламино]-5-хлор-пиримидин-2-иламино}-3-метоксифенил)-1-метил-1,7-диаза-спиро[3.5]нонан-2-он (1.3);
8-(4-{4-[2-(диметилфосфорил)-фениламино]-5-хлор-пиримидин-2-иламино}-3-метоксифенил)-1-метил-1,8-диаза-спиро[4.5]декан-2-он (1.4);
N4-[2-(диметилфосфорил)фенил]-N2-[4-(1-метил-1,7-диаза-спиро[3.5]нон-7-ил)-2-метокси-фенил]-5-хлор-пиримидин-2,4-диамин (1.5);
N4-[2-(диметилфосфорил)фенил]-N2-[4-(1-метил-1,7-диаза-спиро[4.5]дек-8-ил)-2-метокси-фенил]-5-хлор-пиримидин-2,4-диамин (1.6);
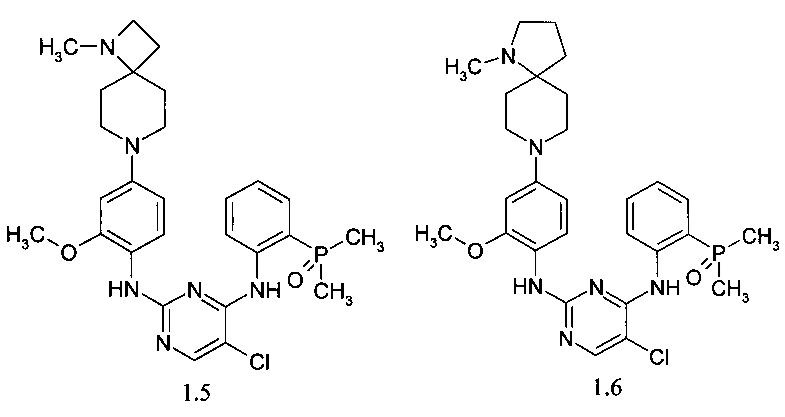
N2-[4-(9-диметиламино-3-аза-спиро[5.5]ундец-3-ил)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин (1.7);
N2-{4-[4-(9-диметиламино-3-аза-спиро[5.5]ундец-3-ил)-пиперидин-1-ил]-2-метоксифенил}-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин (1.8);
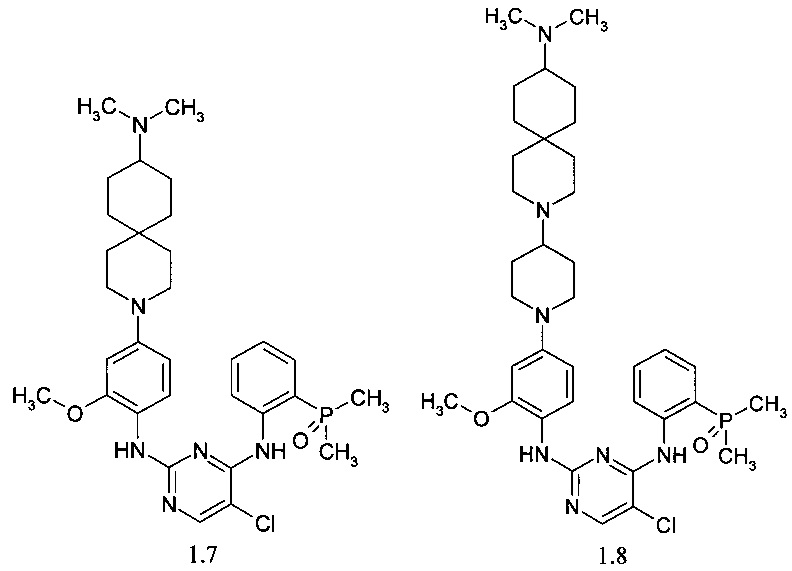
N4-[2-(диметилфосфорил)-фенил]-N2-{4-[4-(1-метил-1,8-диаза-спиро[4.5]дец-8-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамин (1.9);
N4-[2-(диметилфосфорил)-фенил]-N2-{2-метокси-4-[4-(9-метил-3,9-диаза-спиро[5.5]ундец-3-ил)-пиперидин-1-ил]-фенил}-5-хлор-пиримидин-2,4-диамин (1.10);
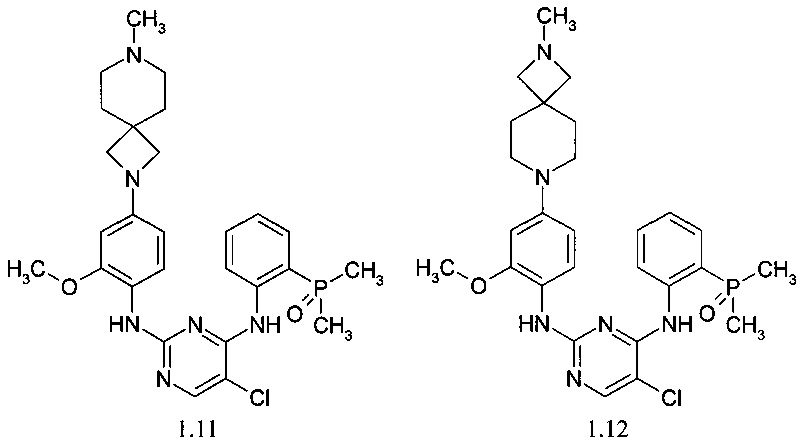
N4-[2-(диметилфосфорил)-фенил]-N2-[4-(7-метил-2,7-диаза-спиро[3.5]нон-2-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.11);
N4-[2-(диметилфосфорил)-фенил]-N2-[4-(2-метил-2,7-диаза-спиро[3.5]нон-7-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.12);
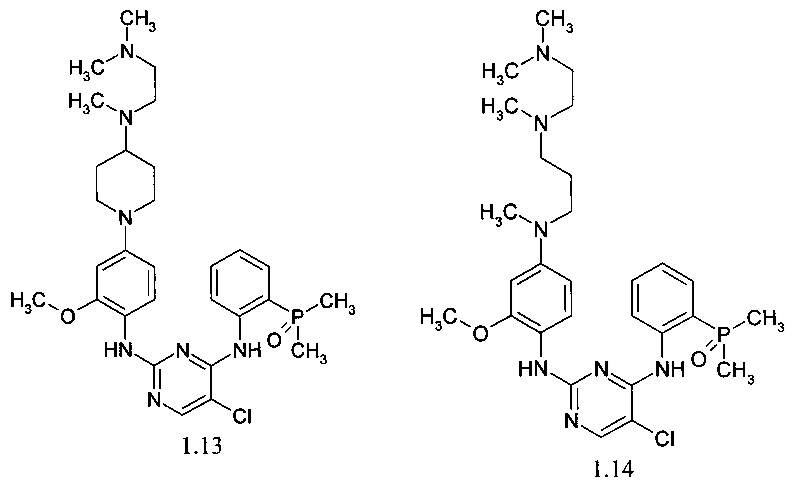
N2-(4-{4-[(2-диметиламиноэтил)-метиламино]-пиперидин-1-ил}-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин (1.13);
N2-[4-({3-[(2-диметиламиноэтил)-метиламино]-пропил}-метиламино)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин (1.14);
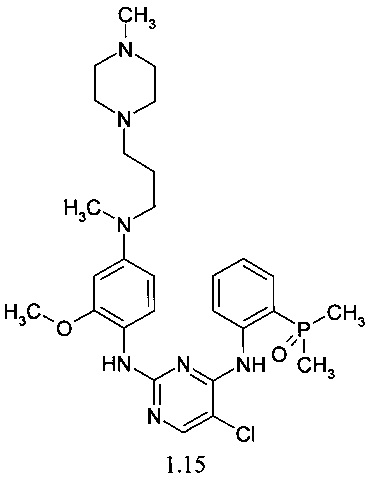
N4-[2-(диметилфосфорил)-фенил]-N2-(4-{метил-[3-(4-метилпиперазин-1-ил)-пропил]-амино}-2-метоксифенил)-5-хлор-пиримидин-2,4-диамин (1.15)
или их таутомеры, стереоизомеры, фармацевтически приемлемые соли и сольваты.
Настоящее изобретение также включает фармацевтически приемлемые соли соединений общей формулы 1. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, соли минеральных или органических кислот соединений формулы 1, таких как карбоновых кислот, например, дихлоруксусной кислоты, а также соли хлористого водорода, фосфорной кислоты или сульфокислоты. Фармацевтически приемлемые соли по настоящему изобретению включают обычные нетоксичные соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основную или кислотную группу, обычными химическими способами. Как правило, такие соли могут быть получены взаимодействием свободных кислотных и основных форм соединений общей формулы 1 со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе или в смеси двух растворителей. Как правило, предпочтительны неводные среды, такие как эфир, этилацетат, этанол, изопропанол, ацетон или ацетонитрил (ACN). Списки подходящих солей можно найти в справочнике фармацевтических солей [Р.Н. Stahl, C.G. Wermuth (Eds.). Handbook of Pharmaceutical Salts, Properties, Selection, and Use. VHCA, Verlag Helvetica Chimica Acta, Zurich, Switzerland, and Wiley-VCH, Weinheim, Germany. 2002.].
Предметом настоящего изобретения являются модуляторы киназ, в том числе и ALK и EGFR, которые представляют собой соединения общей формулы 1, или их таутомеры, стереоизомеры, фармацевтически приемлемые соли и сольваты.
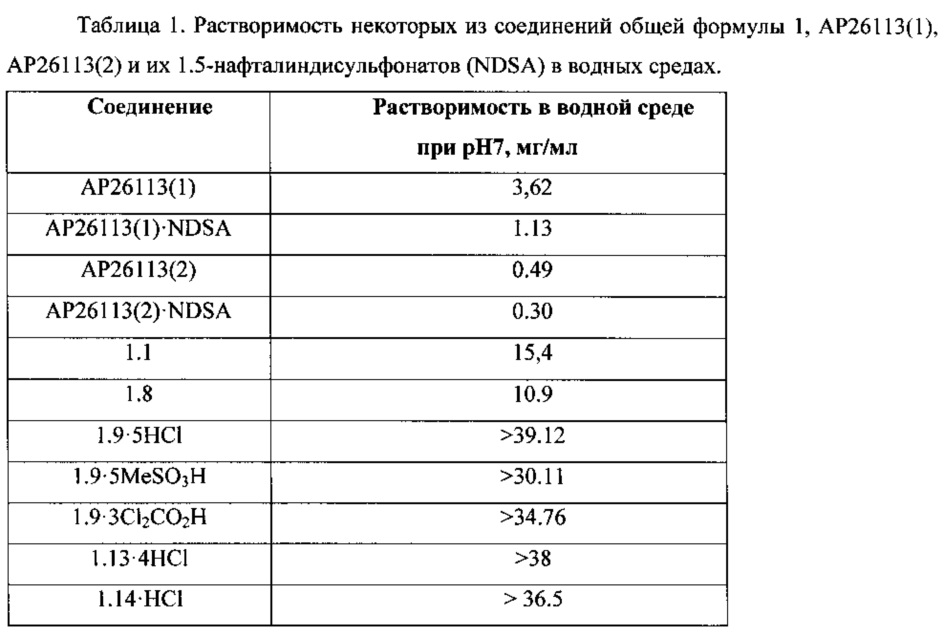
Новые соединения общей формулы 1, их таутомеры, стереоизомеры и фармацевтически приемлемые соли имеют в водных средах прекрасную и более высокую растворимость, чем растворимость известных ингибиторов АР26113(1) и АР26113(2) и их 1.5-нафталиндисульфонатов (NDSA) (Таблица 1). Так, например, в сопоставимых условиях растворимость ингибитора 1.8 в водной среде при рН7 имеет значение 10.9 мг/мл и превышает растворимость известных ингибиторов АР26113(1) и АР26113(2) в 3 и в 22 раза, соответственно, а их солей АР26113(1)⋅NDSA и АР26113(2)⋅NDSA в 8,4 и в 36,3 раза, соответственно. Растворимость ингибиторов 1.1, 1.9⋅HCl, 1.9⋅3MeSO3H, 1.9⋅3Cl2CO2H, 1.13⋅HCl и 1.14⋅HCl еще выше, чем растворимости ингибитора 1.8.
В сопоставимых условиях активность новых соединений общей формулы 1, их таутомеров, стереоизомеров, фармацевтически приемлемых солей и сольватов по отношению к ALK и EGFR, как правило, несколько выше или сопоставима с активностью известных ингибиторов АР26113 (Таблица 2).
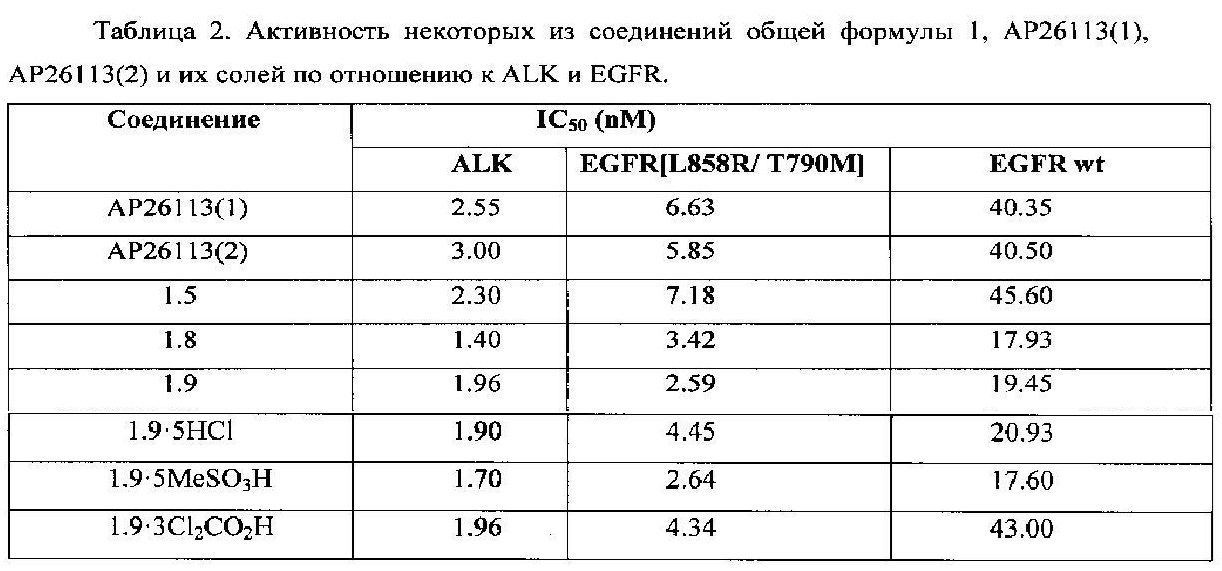
Предметом настоящего изобретения являются соединения общей формулы 1, предназначенные для лечения у пациента рака и других заболеваний, в том числе для лечения немелкоклеточного рака легких (NSCLC), в том числе с метастазами в головном мозге.
Предметом изобретения является фармацевтическая композиция, содержащая, по меньшей мере, одно соединение общей формулы 1 или его таутомер, стереоизомер, фармацевтически приемлемую соль или его сольват, и, по меньшей мере, один фармацевтически приемлемый наполнитель или добавку. Такая композиция может быть введена субъекту в случае необходимости подавлять рост, развитие рака и/или метастазов рака, в том числе твердых опухолей (например, рака предстательной железы, рака толстой кишки, поджелудочной железы и яичников, рака молочной железы, немелкоклеточного рака легкого (NSCLS), нейронных опухолей, таких как глиобластомы и нейробластомы; карциномы, рака мягких тканей); различных форм лимфомы, различных форм лейкемии и в том числе раковых заболеваний, устойчивых к другой обработке, в том числе тех, которые устойчивы к обработке другими ингибиторами киназ, и, как правило, для лечения и профилактики заболеваний или нежелательных состояний, обусловленных одной или более киназой, которые ингибируются соединениями по настоящему изобретению.
В соответствии с данным изобретением фармацевтическая композиции может быть в форме, подходящей для перорального применения (например, в виде таблеток, пастилок, твердых или мягких капсул, водных или масляных суспензий, эмульсий, диспергируемых порошков или гранул, сиропов или эликсиров), для местного применения (например, в виде кремов, мазей, гелей или водных или масляных растворов или суспензий), для введения путем ингаляции (например, в виде тонкоизмельченного порошка или жидкого аэрозоля), для введения путем инсуффляции (например, в виде тонкоизмельченного порошка) или для парентерального введения (например, в виде стерильного водного или масляного раствора для внутривенного, подкожного, внутримышечного введения дозы или в виде суппозиториев для ректального введения дозы).
Фармацевтическая композиция по изобретению может быть получена обычными способами с использованием обычных фармацевтических наполнителей, хорошо известных в данной области. Таким образом, фармацевтические композиции, предназначенные для перорального применения, могут содержать, например, один или несколько окрашивающих, подслащивающих, вкусовых компонентов и/или консервант.
Соединения общей формулы 1 обычно вводят теплокровному животному в единичной дозе в диапазоне 5-5000 мг/м2 площади тела животного, то есть примерно 0,1-100 мг/кг, и это обычно обеспечивает терапевтически эффективную дозу. Форма единичной дозы, такой как таблетка или капсула, обычно содержит, например, 1-250 мг активного ингредиента. Суточная доза обязательно будет варьироваться в зависимости от пациента, конкретного пути введения и тяжести заболевания, которое лечат. Соответственно, врач, который лечит конкретного пациента, может определить оптимальную дозу, учитывая ингибирующую активность против ALK и EGFR соединений общей формулы 1 и их таутомеров, стереоизомеров, фармацевтически приемлемых солей и сольватов, предназначенных для лечения заболеваний или медицинских состояний, обусловленных активностью ALK и EGFR, например, рака. Типы рака, которые могут быть восприимчивы к лечению с использованием соединений общей формулы 1 и их таутомеров, стереоизомеров, фармацевтически приемлемых солей и сольватов включают, но не ограничиваются лечением пациентов с немелкоклеточным раком легких (NSCLC), в том числе с метастазами в головном мозге.
Согласно другому аспекту настоящего изобретения соединения общей формулы 1, как определено выше, или их таутомеры, стереоизомеры, фармацевтически приемлемые соли и сольваты предназначены для использования в качестве лекарственного средства.
В соответствии с настоящим изобретением предлагаются соединения общей формулы 1 и их таутомеры, стереоизомеры, фармацевтически приемлемые соли и сольваты для лечения заболевания, обусловленного активностью ALK и EGFR, в том числе для лечения ракового заболевания, обусловленного активностью ALK и EGFR.
Предметом настоящего изобретения является также способ производства лекарственного средства для лечения заболевания, обусловленного ALK и EGFR, предусматривающий использование соединения общей формулы 1 и его таутомера, стереоизомера, фармацевтически приемлемой соли и сольвата.
Предметом настоящего изобретения является также способ производства лекарственного средства для лечения рака, обусловленного ALK и EGFR, предусматривающий использование соединения общей формулы 1 и его таутомера, стереоизомера, фармацевтически приемлемой соли и сольвата.
Предметом настоящего изобретения является также способ лечения рака, обусловленного ALK и EGFR, предусматривающий использование соединения общей формулы 1 и его таутомера, стереоизомера, фармацевтически приемлемой соли и сольвата.
Предметом настоящего изобретения является также способ получения противоракового эффекта у пациента, нуждающегося в таком лечении, который включает введение указанному пациенту эффективного количества соединения общей формулы 1 или его таутомера, стереоизомера, фармацевтически приемлемой соли и сольвата.
В соответствии с настоящим изобретением предложен способ лечения человека, страдающего от заболевания, в том числе рака, при котором предпочтительно ингибирование ALK и EGFR. Лечение осуществляется путем введения нуждающемуся в этом человеку терапевтически эффективного количества соединения общей формулы 1 или его таутомера, стереоизомера, фармацевтически приемлемой соли и сольвата.
В любом из аспектов или вариантов упомянутый в данном описании рак, указанный в общем смысле, может быть выбран из твердых опухолей (например, рака предстательной железы, рака толстой кишки, поджелудочной железы и рака яичников, рака молочной железы, немелкоклеточного рака легкого (NSCLS), нейронных опухолей, таких как глиобластомы и нейробластомы; карциномы, рака мягких тканей); различных форм лимфомы, таких как неходжкинская лимфома (NHL), известная как анапластическая крупноклеточная (ALCL), различные формы лейкемии; и в том числе рака, который устойчив к другим лекарствам, в том числе тех, которые устойчивы к ингибированию других киназ, и, как правило, для лечения и профилактики заболеваний или нежелательных состояний, обусловленных одной или более киназами, которые ингибируются соединениями по настоящему изобретению.
Настоящее изобретение относится также к способу лечения рака. Способ включает введение (в виде монотерапии или в комбинации с одним или несколькими другими противораковыми агентами, одним или несколькими агентами для облегчения побочных эффектов, излучение и т.д.) терапевтически эффективного количества соединения по настоящему изобретению нуждающемуся в этом человеку или животному, чтобы ингибировать, замедлить или уменьшить рост, развитие или распространение рака у реципиента, в том числе твердых опухолей или других форм рака, таких как лейкозы. Такое введение представляет собой способ лечения или профилактики заболеваний, обусловленных одной или несколькими киназами, путем их ингибирования одним из соединений общей формулы 1 или или его таутомером, стереоизомером, фармацевтически приемлемой солью или его сольватом или его фармацевтически приемлемым производным. Термин "Введение" включает доставку соединения общей формулы 1 или его пролекарства или другого его фармацевтически приемлемого производного с использованием любого подходящего состава или способа введения. Обычно соединение вводят один или несколько раз в месяц, часто один или более раз в неделю, например, ежедневно, через день, 5 дней/неделя, и т.д. Наиболее предпочтительными являются оральный и внутривенный способы введения.
Способ лечения рака, описанный выше, может быть применен в качестве единственной терапии или может включать в дополнение использование соединения по настоящему изобретению к обычной хирургии или лучевой терапии или химиотерапии или иммунотерапии. Такая химиотерапия может вводиться одновременно, одновременно-последовательно или раздельно с лечением соединением по изобретению и может включать один или более из следующих категорий противоопухолевых агентов: антипролиферативные/противоопухолевые препараты и их комбинации, используемые в медицинской онкологии; цитостатические агенты; агенты антивторжение; ингибиторы функции фактора роста; антиангиогенные агенты; сосудистые средства; антагонисты эндотелиновых рецепторов; антисмысловые терапии; подходы генной терапии и подходы иммунотерапии.
Предметом настоящего изобретения является также фармацевтическая композиция для комбинированного лечения рака, включающая соединение общей формулы 1 или его таутомер, стереоизомер, фармацевтически приемлемую соль и сольват и дополнительно противоопухолевое вещество, как определено выше.
Здесь, где термин "совместное лечение" используют по отношению к комбинированной терапии, следует понимать, что это может относиться к одновременному, раздельному или последовательному введению. В одном аспекте изобретения "комбинированное лечение" относится к одновременному введению. В другом аспекте настоящего изобретения "комбинированное лечение" относится к раздельному введению. В третьем аспекте настоящего изобретения "комбинированное лечение" относится к последовательному введению. Если введение последовательное или раздельное, задержка во введении второго компонента не должна быть такой, чтобы сохранить эффективность эффекта, возникающего в результате использования комбинацию.
Таким образом, одним из вариантов изобретения является применение соединения формулы 1 или его таутомера, стереоизомера, фармацевтически приемлемой соли и сольвата и дополнительно противоопухолевого вещества для совместной терапии рака.
Предметом настоящего изобретения является также способ получения противоракового эффекта у нуждающегося в таком лечении теплокровного животного и человека, который включает введение указанным млекопитающим соединения общей формулы 1 или его таутомера, стереоизомера, фармацевтически приемлемой соли и сольвата и одновременное, раздельное или последовательное введение дополнительного противоопухолевого вещества указанному млекопитающему в количествах, совместно обеспечивающих получение противоракового эффекта.
Предметом данного изобретения являются замещенные анилины общей формулы 2 и их таутомеры, стереоизомеры, соли и сольваты:
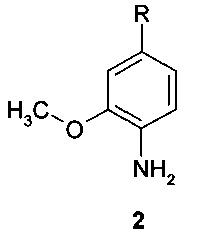
где R представляет собой заместитель, выбранный из ряда (а) - (о):
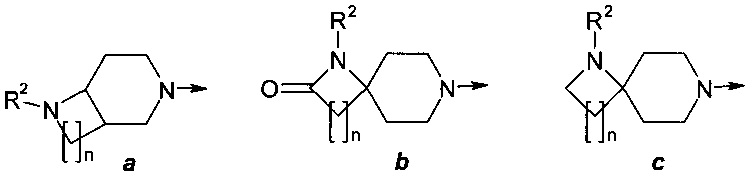
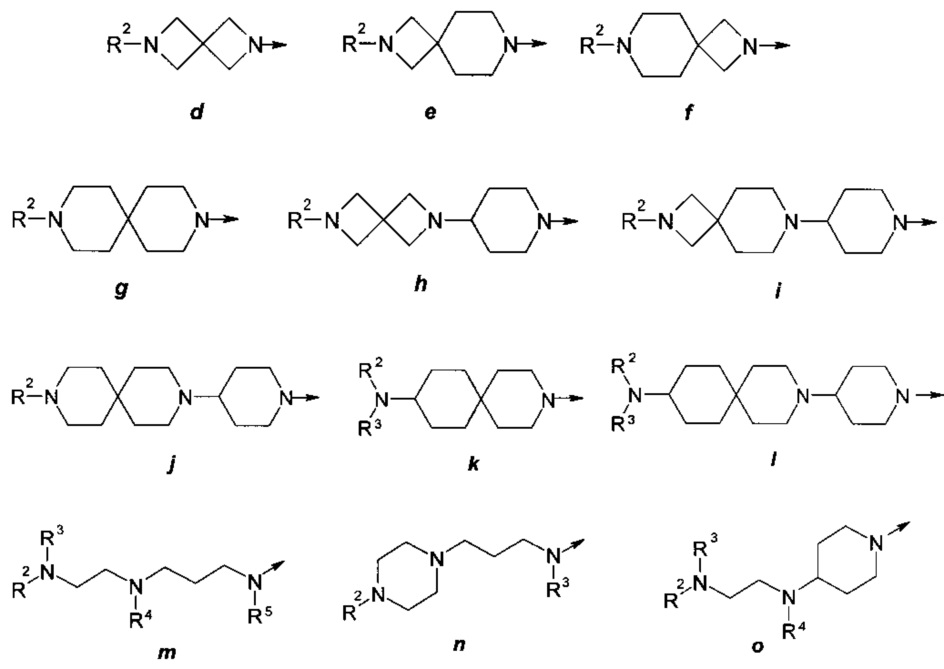
где: R2, R3, R4 и R5 представляют собой необязательно одинаковые и необязательно замещенные С1-С4алкилы;
n представляют собой необязательно одинаковое число 1 или 2; стрелка указывает точку присоединения заместителя.
Предметом данного изобретения является способ получения соединения общей формулы 1 путем взаимодействия соединения общей формулы 2 с 2,5-дихлор-N-[2-(диметилфосфорил)фенил]пиримидин-4-амином формулы 1.1(17).
Ниже изобретение описано более подробно с помощью конкретных примеров. Следующие примеры представлены с целью иллюстрации и не предназначены для ограничения изобретения каким-либо образом. Специалисты в данной области техники легко поймут различие некритических параметров, которые могут быть изменены или модифицированы, чтобы получить те же результаты.
Пример 1. N4-[2-(Диметилфосфорил)фенил]-N2-[4-((1S,6R)-7-метил-3,7-диазабицикло[4.2.0]окт-3-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.1) был получен в соответствии со Схемой 1.
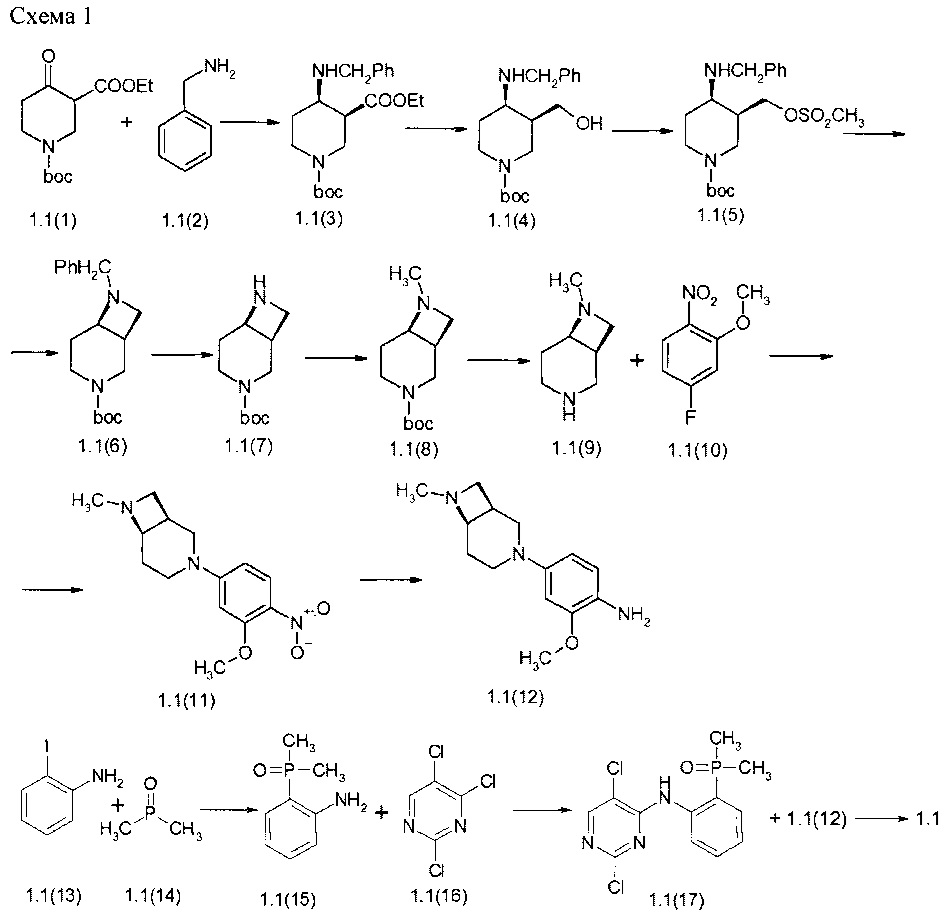
Смесь соединения 1.1(1) (15,0 г 55,3 ммоль), бензиламина (1,1(2): 6,3 г, 58,0 ммоль) и TsOH (0,6 г) в толуоле (500 мл) перемешивали и нагревали с обратным холодильником в колбе, снабженной ловушкой Дина-Старка до прекращения отделения воды. Реакционную смесь охлаждали (в ледяной бане) и добавляли NaBH(OAc)3 (51,5 г, 221 ммоль) в виде одной порции. Затем медленно по каплям добавляли уксусную кислоту (4 мл, 66,3 ммоль) к перемешиваемой реакционной смеси. Реакционную смесь перемешивали в течение ночи и нейтрализовали насыщенным водным раствором. NaHCO3 до рН8-9. Органический слой отделяли и водный слой экстрагировали этилацетатом (3×150 мл). Органический слой промывали водным, солевым раствором Na2CO3, сушили над Na2SO4 и концентрировали с получением 1-трет-6утил 3-этил (3S,4R)-4-[(фенилметил)амино]-пиперидин-1,3-дикарбоксилата (1.1(3): 20,0 г, 99%), который использовали на следующей стадии без дополнительной очистки. LC-MS (ESI) (m/z): 363 (М+Н)+.
К раствору соединения 1.1(3) (20,0 г, 55,2 ммоль) в 400 мл ТГФ добавляли, LiBH4 (6,0 г, 276 ммоль) при 0-5°С. Смесь перемешивали с обратным холодильником в течение ночи. Смесь осторожно гасили водой при температуре 0-5°С и разбавляли насыщенным раствором соли. Органический слой отделяли и упаривали в вакууме. Остаточное масло разбавляли дихлорметаном, сушили над Na2SO4, фильтровали и упаривали. Остаток очищали колоночной хроматографией на силикагеле (гексан:этилацетат=1:1). Выход трет-бутил (3S 4R)-3-(гидроксиметил)-4-[(фенилметил)амино]пиперидин-1-карбоксилата 1.1(4) 11.0 г, (62%), LC-MS (ESI) (m/z): 321 (М+Н)+
К раствору соединения 1.1(4) (10,4 г, 32,4 ммоль) и N-метилморфолина (10,7 мл, 97,2 ммоль) в 400 мл ТГФ добавляли по каплям при 0-5°С метансульфонилхлорид (5,0 мл, 64,8 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Неорганический твердый осадок отфильтровывали фильтрат концентрировали при пониженном давлении. Остаток растворяли в CH2Cl2 (150 мл) и промывали 10% водным раствор лимонной кислоты и водой. После сушки над Na2SO4, фильтровали и выпаривали в вакууме, остаток очищали с помощью колоночной хроматографии на силикагеле (гексан:этилацетат=1:1). Выход трет-бутил (3S,4R)-3-{[(метилсульфонил)окси]метил}-4-[(фенилметил)амино]пиперидин-1-карбоксилата (1,1(5): 10,2 г, 78%), LC-MS (ESI) (m/z): 399 (М+Н)+.
К раствору соединения 1.1(5) (9,2 г, 23,0 ммоль) в ДМФ (150 мл), добавляли DIPEA (40 мл, 230 ммоль). Смесь перемешивали при 110°С в течение 14 ч. После охлаждения смесь упаривали в вакууме, остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2: МеОН=60:1). Выход трет-бутил (1S,6R)-7-(фенилметил)-3,7-диазабицикло[4.2.0]октан-3-карбоксилат 1,1(6): 2,8 г, 40%); 1H-NMR, (DMSO-d6, 400 МГц): 7.45-7.20 (m, 5H), 4.20-3.98 (m, 1H), 3.80-3.50 (m, 3H), 3.40-3.20 (m, 3H), 3.12-2.97 (m, 1H), 2.97-2.85 (m, 1H), 2.52-2.35 (m, 1H), 1.55-1.40 (m, 10H), 1.37-1.25 (m, 1H).
Раствор соединения 1.1(6) (2,7 г, 8,9 ммоль) в 100 мл этанола гидрировали с 0,6 г 10% Pd на угле в течение 14 ч при 30 атм водорода при 60°С. Формалин (3,6 мл 37% водного раствора, 44,5 ммоль) и 0.6 г 10% Pd на угле добавляли в реакционную смесь и перемешивали в течение 14 ч при 20 атм водорода при комнатной температуре. Смесь фильтровали через целит, фильтрат выпаривали, и получили трет-бутил (1S,6R)-7-метил-3,7-диазабицикло[4.2.0]октан-3-карбоксилата (1.1(8): 1,8 г, 88%), который использовали на следующей стадии без дополнительной очистки. LC-MS (ESI) (m/z): 227 (М+Н)+.
К раствору соединения 1.1(8) (1,75 г, 7,7 ммоль) в диоксане (50 мл), добавляли 30 мл 3М HCl в диоксане. Смесь перемешивали при 50°С в течение 14 ч. После охлаждения смесь упаривали в вакууме, остаток использовали на следующей стадии без дополнительной очистки. Выход (1S,6R)-7-метил-3,7-диазабицикло[4.2.0]октана дигидрохлорида (1.1(9)⋅2HCl) 1,5 г (99%).
К перемешиваемой смеси соединения 1.1(9)⋅2HCl (1,54 г, 7,7 ммоль) и DIPEA (5,4 мл, 30,8 ммоль) в ДМФ (30 мл) добавляли соединение 1.1(10) (1,46 г, 8,5 ммоль) и смесь перемешивали в течение 14 ч при 70°С. Реакционную смесь концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2: МеОН 40:1). Выход (1S,6R)-7-метил-3-[3-(метилокси)-4-нитрофенил]-3,7-диазабицикло[4.2.0]октана (1.1(11)) 1,7 г (79%). LC-MS (ESI) (m/z): 278 (М+Н)+.
К раствору соединения 1.1(11) (1,43, 5,2 ммоль) в смеси этанола (30 мл), воды (4 0 мл) и уксусной кислоты (5 мл 1), добавляли карбонильное железо (1,4 г, 26,0 ммоль). Смесь перемешивали при 45°С в течение 1 ч. Смесь фильтровали через целит и упаривали. Остаток растворяли в дихлорметане, промывают 10% Na2CO3 и сушили над Na2SO4. К фильтрату добавляли избыток 3М HCl в диоксане и полученный раствор упаривали в вакууме. Получали 1,7 г (92%) [4-[(1S,6R)-7-метил-3,7-диазабицикло[4.2.0]окт-3-ил]-2-(метилокси)фенил]амина тригидрохлорид (1.1(12)⋅3HCl, (LC-MS (ESI) (m/z): 248 (М+Н)+.
К раствору 2-иоданилина (5,0 г, 22,8 ммоль) и диметилфосфин оксида (2,0 г, 25,0 ммоль) в 100 мл ДМФ добавляли фосфат калия (5,4 г, 22,8 ммоль), Хантфос (1,3 г, 2,2 ммоль) и ацетат палладия (0,52 г, 2,2 ммоль). Реакционную смесь перемешивали при 140°С в течение 3 ч и охлаждали до комнатной температуры Растворитель выпаривали в вакууме, а остаток обрабатывали смесью дихлорметан/вода. Органический слой оделяли, сушили над Na2SO4, фильтровали и концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, EtOAc/МеОН=20:1). Выход 2-(диметилфосфорил)анилина (1.1(15)) 3,0 г (77%). 1H-NMR, (DMSO-d6, 400 МГц): 7.25-7.10 (m, 2Н), 6.70-6.60 (m, 1Н), 6.60-6.50 (m, 1H), 1.66 (s, 3H), 1.62 (s, 3H).
К раствору соединения 1.1(15) (1,5 г, 8,9 ммоль) в 30 мл ДМФА добавляли карбонат калия (4,0 г, 28,5 ммоль) и соединение 1.1(16) (2,6 г, 14,2 ммоль). Реакционную смесь перемешивали при 60°С в течение 14 ч и охлаждали до комнатной температуры. Реакционную смесь разбавляли водой и экстрагировали CH2Cl2 (50×5 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, EtOAc/MeOH=20:1). Выход N-[2-(диметилфосфорил)фенил]-2,5-дихлор-пиримидин-4-амин (1.1(17)) 1,5 г (53%). 1H-NMR, (DMSO-d6, 400 МГц): 11.8 (s, 1Н), 8.44 (s, 1H), 8.43-8.37 (m, 1H), 7.70-7.55 (m, 2H), 7.28-7.18 (m, 1H), 1.82 (s, 3H), 1.79 (s, 3H).
Раствор соединения 1.1(17) (0,25 г, 0,8 ммоль) и соединения 1.1(12) (0,28 г, 0,8 ммоль) в 8 мл 2-метоксиэтанол и 2,0 мл воды перемешивали при 120°С в течение 14 ч и охлаждали до комнатной температуры. Реакционную смесь упаривали в вакууме. Остаток разбавляли 10% водным K2CO3 и экстрагировали CH2Cl2 (50×5 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью обращенно-фазовой ВЭЖХ (градиента MeCN-H2O с 0,1% TFA). Целевые фракции подщелачивали NaHCO3, ацетонитрил частично выпаривали и экстрагировали продукт дихлорметаном. Объединенные органические экстракты сушили над Na2SO4 и упаривали. Получали N4-[2-(диметилфосфорил)фенил]-N2-[4-((1S,6R)-7-метил-3,7-диазабицикло[4.2.0]окт-3-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.1). 1Н-NMR, (DMSO-d6, 400 МГц): 11.16 (s, 1Н), 8.49 (br.s, 1H), 8.05 (s, 1H), 8.02 (s, 1H), 7.58-7.46 (m, 1H), 7.38-7.25 (m, 2H), 7.12-7.05 (m, 1H), 6.58-6.53 (m, 2H), 6.43 (dd, J1=8.7, J2=9.0Гц, 1H), 3.97-3.86 (m, 1H), 3.74 (s, 3H), 3.64-3.54 (m, 1H), 3.30-3.15 (m, 3H), 3.15-2.95 (m, 2H), 2.90-2.70 (m, 1H), 2.24 (s, 3H), 1.77 (s, 3H), 1.74 (s, 3H), 1.73-1.60 (m, 1H), 1.57-1.44 (m, 1H).
Пример 2. N4-[2-(Диметилфосфорил)фенил]-N2-[4-(1-метилоктагидро-пирроло[3,2-с]пиридин-5-ил)-фенил]-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.2) был получен в соответствии со Схемой 2.
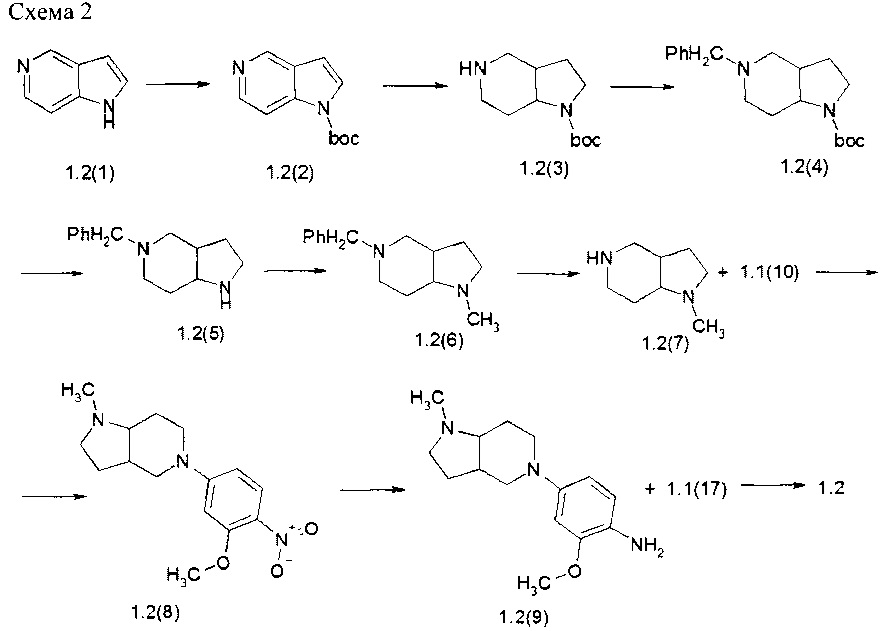
К перемешиваемой смеси соединения 1.2(1) (7 г, 59,2 ммоль) и N, N-диметилпиридин-4-амина (7,2 г, 59,2 ммоль) в ацетонитриле (250 мл) добавляли Вос-ангидрид (12,9 г, 59,2 ммоль). Смесь перемешивали в течение 24 ч при комнатной температуре. Реакционную смесь концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, гексан/EtOAc=1:1). Получали 11,8 г (91%) трет-бутил пирроло[3,2-с]пиридин-1-карбоксилата (1,2(2). 1H-NMR, (DMSO-d6, 400 МГц): 8.9 (s, 1Н), 8.43 (d, J=5.7Гц, 1H), 7.91 (d, J=5.7ГЦ, 1H), 7.73 (d, J=3.7Гц, 1H), 6.84 (d, J=3.7Гц, 1H), 1.63 (s, 9H).
Раствор соединения 1.2(2) (11,8 г, 54,1 ммоль) в 150 мл этанола и 100 мл уксусной кислоты гидрировали с 3,8 г 20% Pd(OH)2 на угле в течение 14 ч при 70°С при 50 атм водорода, Смесь фильтровали через целит, фильтрат выпаривали, получали 11,6 г (94%)%) трет-бутил октагидропирроло[3,2-с]пиридин-1-карбоксилата (1,2(3)), который использовали на следующей стадии без дополнительной очистки. LC-MS (ESI) (m/z): 227 (М+Н)+.
К раствору соединения 1.2(3) (11,6 г, 51,2 ммоль) и Et3N (28,5 мл, 206,0 ммоль) в 400 мл CH2Cl2 добавляли по каплям при 0-5°С бензилхлорформиат (11,0 мл, 76,8 ммоль). Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Неорганический твердый осадок отфильтровывали, а фильтрат концентрировали при пониженном давлении. Полученный концентрат промывают 10% водным раствором лимонной кислоты и водой. После сушки над Na2SO4, фильтровали и выпаривали в вакууме, остаток использовали на следующей стадии без дополнительной очистки. Выход трет-бутил 5-бензил-октагидро-пирроло[3,2-с]пиридин-1-карбоксилата (1.2(4) 18,5 г (100%). LC-MS (ESI) (m/z): 361 (М+Н)+.
К раствору соединения 1.3(4) (18,5 г, 51,3 ммоль) в диоксане (400 мл) добавляли 170 мл 3М HCl в диоксане. Смесь перемешивали при 40°С в течение 14 ч. После охлаждения смесь упаривали в вакууме, остаток использовали на следующей стадии без дополнительной очистки. Выход 5-бензил-октагидро-пирроло[3,2-c]пиридин гидрохлорида (1,2(5) HCl) 15,2 г, (100%). LC-MS (ESI) (m/z): 261 (М+Н)+.
К раствору соединения 1,2(5)/⋅HCl (15,2 г, 51,2 ммоль) в МеОН (300 мл) добавляли формальдегид (20,8 мл 37% водного раствора, 256 ммоль). Смесь перемешивали при комнатной температуре в течение около 1 ч и добавляли NaBH3CN (7,1 г, 112,6 ммоль) в виде одной порции. Смесь перемешивали при комнатной температуре в течение примерно 24 ч, а затем упаривали в вакууме. Остаток растворяли в воде, доводили рН до 11 с помощью 2N водного раствора NaOH. Смесь экстрагировали CH2Cl2 (4×100 мл). Объединенные органические слои сушили над Na2SO4, фильтровали и концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, СН2Сl2: МеОН 40:1). Выход 5-бензил-1-метилоктагидро-пирроло[3,2-с]пиридина (1.2(6) 8,2 г (58%). 1H-NMR, (DMSO-d6, 400 МГц): 7.45-7.27 (m, 5Н), 5.1 (s, 2Н), 3.85-3.65 (m, 1Н), 3.65-3.45 (m, 1H), 3.45-3.25 (m, 1H), 3.15-2.95 (m, 2H), 2.29 (s, 3H), 1.93-1.75 (m, 2H), 1.75-1.65 (m, 1H), 1.47-1.30 (m, 1H).
Раствор соединения 1.2(6) (7,7 г, 28,1 ммоль) в 150 мл этанола гидрировали с 2,1 г 10% Pd на угле в течение 14 ч при комнатной температуре и 1 атм водорода. Смесь фильтровали через целит, фильтрат выпаривали и получали 3,7 г (94%) 1-метилоктагидро- 5Н-пирроло[3,2-с]пиридина (1.2(7), который использовали на следующей стадии без дополнительной очистки. 1H-NMR, (DMSO-d6, 400 МГц): 4.05-3.55 (m, 3Н), 3.20-3.10 (m, 1Н), 3.00-2.85 (m, 2H), 2.76-2.64 (m, 1H), 2.36-2.16 (m, 5H), 1.95-1.70 (m, 2H), 1.40-1.20 (m, 1H).
К перемешиваемой смеси соединения 1.2(7) (3,7 г, 26,4 ммоль) и DIPEA (9,2 г, 52,8 ммоль) в ДМФА (100 мл) добавляли соединение 1.1(10) (5,0 г, 29,0 ммоль). Смесь перемешивали в течение 14 ч при 70°С. Реакционную смесь концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2:МеОН=40:1). Выход 1-метил-5-[3-(метилокси)-4-нитрофенил]октагидро-1Н-пирроло[3,2-с]пиридина (1.2(8)) 5,5 г (72%). 1H-NMR, (DMSO-d6, 400 МГц): 8.03 (d, J=9.4Гц, 1H), 6.29 (d, J=9.4Гц, 1H), 6.14 (s, 1H), 3.95 (s, 3H), 3.60-3.45 (m, 2H), 3.40-3.23 (m, 2H), 3.15-3.00 (m, 1H), 2.55-2.44 (m, 2H), 2.38 (s, 3H), 2.32-2.20 (m, 1H), 2.10-2.00 (m, 1H), 2.00-1.90 (m, 1H), 1.90-1.80 (m, 1H), 1.56-1.44 (m, 1H).
Раствор соединения 1.2(8) (5,5 г, 18,9 ммоль) в 150 мл этанола гидрировали с 1,0 г 10%) Pd на угле в течение 14 ч при 50°С и 30 атм водорода. Смесь фильтровали через целит. К фильтрату добавляли избыток 3М HCl в диоксане и раствор упаривали в вакууме. Получали 6,9 г (98%) 4-(1-метилоктагидро-5Н-пирроло[3,2-с]пиридин-5-ил)-2-(метилокси)анилин тригидрохлорида (1.2(9)⋅3HCl), LC-MS (ESI) (m/z): 262 (М+Н)+.
Раствор соединения 1.1(17) (0,124 г, 0,4 ммоль) и соединения 1,2(9) (0,16 г, 0,44 ммоль) в 3 мл 2-метоксиэтанола и 1,5 мл воды перемешивали при 120°С в течение 14 ч и охлаждали до комнатной температуры. Реакционную смесь упаривали в вакууме. Остаток разбавляли водой и экстрагировали CH2Cl2 (5×50 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью обращенно-фазовой ВЭЖХ (градиента MeCN-Н2О с 0,1% TFA). Целевые фракции подщелачивали NaHCO3, ацетонитрил частично выпаривали и продукт экстрагировали дихлорметаном. Объединенные органические экстракты сушили над Na2SO4 и упаривали. Получали N4-[2-(диметилфосфорил)фенил]-N2-[4-(1-метилоктагидро-пирроло[3,2-с]пиридин-5-ил)-фенил]-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.2), LC-MS (ESI) (m/z): 542 (М+Н).
Пример 3. 7-(4-{4-[-(Диметилфосфорил)фенил-фениламино]-1-метил-5-хлор-пиримидин-2-иламино}-3-метоксифенил)-1,7-диаза-спиро[3.5]нонан-2-он (1.3) был получен в соответствии со схемой 3.
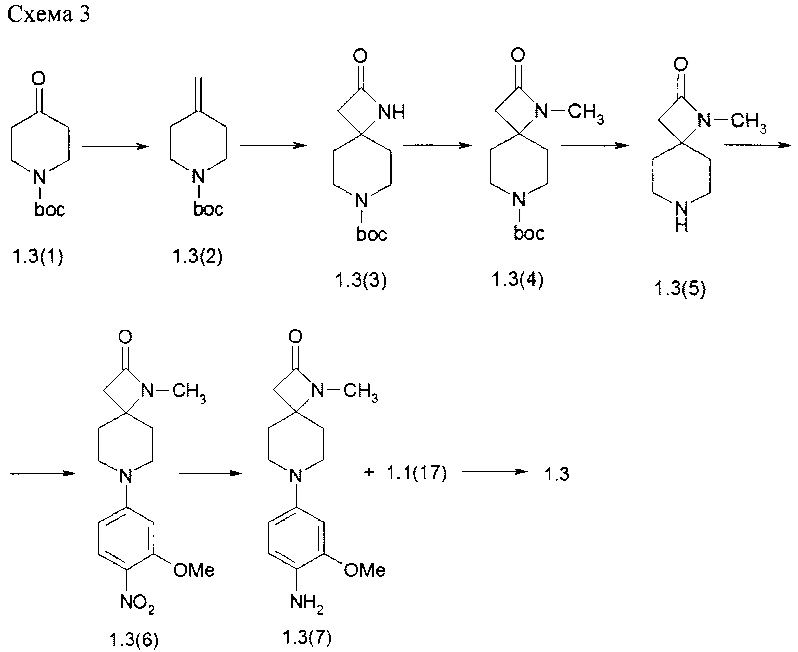
Суспензию (метил)трифенилфосфоний йодида (45,7 г, 0,113 моль) в безводном тетрагидрофуране (150 мл) охлаждали в бане со льдом и добавляли трет-бутоксид калия (12.67 г, 0,113 моль). Смесь перемешивали в течение 30 мин и затем давали нагреться до комнатной температуры в течение 30 мин. Реакционную смесь снова охлаждали на бане со льдом и обрабатывали раствором трет-бутил 4-оксопиперидина-1-карбоксилатом (1.3(1): 15 г, 0,075 моль) в ТГФ (45 мл). Смесь оставляли нагреваться до комнатной температуры и перемешивали в течение ночи. Затем растворитель удаляли в вакууме, а остаток экстрагировали гексаном (3×200 мл). Объединенные органические слои концентрировали и хроматографировали на силикагеле (10% EtOAc в гексане). Получали 14,1 г, (95%) трет-бутил 4-метиленпиперидин-1-карбоксилат (1.3(2)).
К раствору 1.3(2) (14,1 г, 0,0715 моль) в сухом диэтиловом эфире (140 мл) при комнатной температуре в добавляли хлорсульфонил изоцианат (7,38 мл, 12 г, 0,0858 моль) в атмосфере аргона в течение 20 мин. Смесь перемешивали при комнатной температуре в течение ночи в атмосфере аргона. Затем реакционную смесь по каплям добавляли в течение 10 мин к энергично перемешиваемой смеси воды, бикарбоната натрия (28 г), сульфита натрия (19 г) и льда (общий объем около 200 мл). Смесь перемешивали в течение 1 ч, а затем экстрагировали диэтиловым эфиром (3×200 мл). Объединенные экстракты промывали водой (2×30 мл) и сушили над MgSO4. Растворитель удаляли в вакууме. Остаток в виде белого твердого вещества растирали с гексаном и получали 12 г (69%) трет-бутил 2-оксо-1,7-диазаспиро[3,5]нонан-7-карбоксилата (1.3(3)).
Раствор соединения 1.3(3) (3,5 г; 14,5 ммоль) в 10,5 мл ДМФА, охлаждали до 0°С на ледяной бане и постепенно добавляли гидрид натрия (0,45 г, 18,9 ммоль). Смесь перемешивали в течение 30 мин, затем доводили реакционную массу до комнатной температуры и перемешивали в течение 30 мин. После этого добавляли метилиодид (2,88 г, 20 ммоль) и перемешивание продолжают в течение ночи. После того как реакция была завершена, смесь выливали в 30 мл воды, и продукт экстрагировали этилацетатом (3×50 мл). Объединенный органический слой сушили над сульфатом магния и затем концентрировали при пониженном давлении. Полученный сырой продукт очищали на колонке с силикагелем и получали 2,1 г (56%) трет-бутил 1-метил-2-оксо-1,7-диазаспиро[3,5]нонан-7-карбоксилата (1.3(4). LC-MS (ESI) 255 (М+Н)+.
Раствор 1.3(4) (2 г) в 10 мл 10% CF3COOH в CH2Cl2 перемешивали при комнатной температуре в течение 3 ч. Затем растворители выпаривали в вакууме и получали с количественным выходом 1-метил-1,7-диазаспиро[3,5]нонан-2-он трифторацетат (1,3(5)⋅CF3CO2H), который далее использовали без очистки.
К раствору 1.3(5)⋅CF3CO2H (800 мг, 5,2 ммоль) и DIPEA (2,98 мл, 17.12 ммоль) в 3 мл ДМФ добавляли 4-фтор-2-метокси-1-нитробензол (1.1(17)): 976 мг, 5,71 ммоль) и полученную смесь перемешивали при 70°С в течение 15 ч. Когда реакция была завершена (контроль с помощью LS-MS) Растворитель выпаривали в вакууме, остаток фракционировали на колонке с силикагелем (EtOAc/MeOH) и получали 1-метил-7-(3-метокси-4-нитрофенил)-1,7-диазаспиро[3,5]нонан-2-он (1.3(6)). LC-MS (ESI) 306 (М+Н)+.
К раствору 1.3(6) (150 мг, 0,49 ммоль) в 3 мл метанола добавляли 15 мг 10% Pd/C. Полученную смесь энергично перемешивали в атмосфере водорода в течение 5 ч. Затем добавляли CF3COOH (150 мг), катализатор отфильтровывали, растворитель удаляли при пониженном давлении. Полученный 1-метил-7-(4-амино-3-метоксифенил)-1,7-диазаспиро[3,5]нонан-2-он трифторацетат (1.3(7)⋅CF3CO2H) сушили в вакууме и использовали в следующей реакции без очистки. Выход 1.3(7)⋅CF3CO2H близок к количественному. LC-MS (ESI) 276 (М+Н)+.
К раствору 1.3(7)°CF3CO2H (0,49 ммоль) в 3 мл 2-метоксиэтанола добавляли 155,4 мг (0,492 ммоль) 2,5-дихлор-N-(2-(диметилфосфорил)фенил)пиримидин-4-амина (1.1(17)). Полученную смесь перемешивали с обратным холодильником в течение 15 ч. Когда реакция была завершена (контроль с помощью LC-MS) растворители удаляли при пониженном давлении, а остаток очищали с помощью обращенно-фазовой ВЭЖХ (градиента MeCN-H2O с 0,1% TFA). Целевые фракции подщелачивали NaHCO3, ацетонитрил частично выпаривали, а продукт экстрагировали ДХМ. Объединенные органические экстракты сушили над Na2SO4 и упаривали в вакууме. Получали 7-(4-{4-[-(диметилфосфорил)фенил-фениламино]-1-метил-5-хлор-пиримидин-2-иламино}-3-метоксифенил)-1,7-диаза-спиро[3.5]нонан-2-он (1.3). LC-MS (ESI) 555 (М+Н)+. 1Н NMR (DMSO-d6, 400 МГц) δ 11.17 (s, 1Н), 8.49 (br, 1H), (s, 1H) 8.07 (s, 1H), 8.05 (s, 1H), 7.53 (m, 1H), 7.42 (d, J=8Гц, 1H), 7.36 (m, 1H), 7.11 (m, 1H), 6.66 (s, 1H), 6.51 (m, 1H), 3.75 (br, 5H), 2.77 (br, 4H), 2.64 (s, 3H), 2.03 (m, 2H), 1.78 (s, 3H), 1.75 (s, 3H) 1.60 (br, d, J=12.4 Гц, 2H).
Пример 4. 8-(4-{4-[2-(Диметилфосфорил)-фениламино]-5-хлор-пиримидин-2-иламино}-3-метоксифенил)-1-метил-1,8-диаза-спиро[4.5]декан-2-он (1.4) был получен в соответствии со схемой 4.
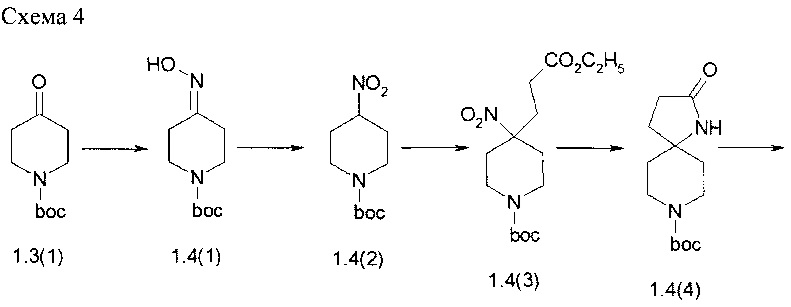
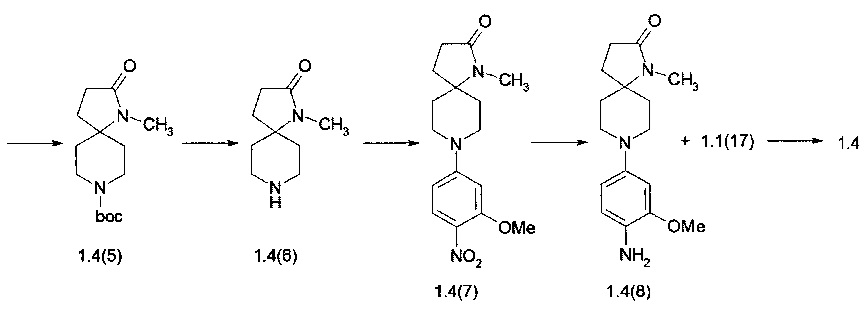
К раствору 1.3(1) (50 г, 0,25 моль) в 1,5 л этанола добавляли гидрохлорид гидроксиламина (22,7 г, 0,32 моль) и ацетата натрия (28,8 г, 0,35 моль), Реакционную смесь перемешивали при комнатной температуре в течение 1 ч. После завершения реакции (контроль ТСХ), реакционный раствор фильтровали и концентрировали при пониженном давлении. Остаток растворяли в этилацетате (300 мл), фильтровали, фильтрат промывали рассолом, сушили над сульфатом магния, концентрировали, а затем разбавляли гексаном. Выпавшие белые кристаллы отфильтровывали, промывали гексаном и сушили. Получали 48,5 г трет-бутил 4-(гидроксиимино)пиперидин-1-карбоксилата (1,4 (1)). 1Н NMR (CDCl3, 400 МГц) δ 3.55 (m, 4Н) 2.64 (t, J=6.4 Гц, 2H), 2.35 (t, J=6.4 Гц, 2H), 1.49 (s, 9H).
Смесь 1.4 (1) (45 г, 0,21 моль), мочевины (113,5 г, 1,89 моль) и Na2HPO4 (179 г, 1,26 моль) в ацетонитриле (750 мл) нагревали с обратным холодильником в течение 0,5 ч, а затем добавляли по каплям в течение 1 ч раствор м-хлорпербензойной кислоты (108 г, 0,631 моль) в ацетонитриле (750 мл). После того как добавление было закончено, реакционную смесь продолжали кипятить с обратным холодильником в течение 2 ч. За ходом реакции следили с помощью ТСХ. После завершения реакции реакционную смесь охлаждали до комнатной температуры, промывали насыщенным водным раствором сульфита натрия, а затем концентрировали при пониженном давлении. Остаток экстрагировали этилацетатом, экстракт промывали водой, сушили над сульфитом магния и концентрировали в вакууме. Сырой продукт фракционировали с помощью колоночной хроматографии и получали 23,7 г (49%) трет-бутил 4-нитропиперидин-1-карбоксилат (1.4(2)) в виде прозрачного бледно-желтого масла. LC-MS (ESI) 231 (М+Н)+. 1Н NMR (CDCl3, 400 МГц) δ 4.51 (m, 1Н), 4.05 (d, J=14 Гц, 2H), 3.01 (m, 2H), 2.22 (m, 3H) 2.06 (m, 2H) 1.48 (s, 9H).
К раствору 10 г (0,0435 моль) 1.4(2) в 100 мл этанола добавляли этилакрилат (5,32 мл, 0,05 моль) и сухой K2CO3 (9 г, 0,0652 моль). Полученную смесь перемешивали при комнатной температуре в течение ночи. После завершения реакции (контроль ТСХ) осадок отфильтровывали, раствор выпаривали в вакууме, а остаток сушат в вакууме. Полученный трет-бутил 4-(3-этокси-3-оксопропил)-4-нитропиперидин-1-карбоксилат (1.4(3)) использовали в последующей реакции без дополнительной очистки. LC-MS (ESI) 331 (М+Н)+. 1Н NMR (CDCl3, 400 МГц) δ 4.14 (q, J=7.2 Гц, 2H), 3.91 (br, 2H), 2.96 (br. t., J=12 Гц, 2H), 2.47 (br. d.,J=13.2 Гц, 2H), 2.31 (m, 2H), 2.22 (m, 2H), 1.74 (m, 2H), 1.46 (s, 9H), 1.27 (t, J=6.8 Гц, 3H).
К раствору соединения 1.4(3) в 150 мл метанола добавляли 1 г Ni-Re и реакционную смесь перемешивали в атмосфере водорода при 50°С в течение 24 ч. Затем катализатор отфильтровывали, раствор концентрировали при пониженном давлении, а остаток сушили в вакууме. Получали 11 г трет-бутил 2-оксо-1,8-диазаспиро[4,5]декан-8-карбоксилата (1.4(4)), который не требует какой-либо очистки. LC-MS (ESI) 255 (М+Н)+.
Раствор 1.4(4) (11 г, 0,0433 моль) в 33 мл DMF охлаждали до 0°С на ледяной бане и постепенно прибавляли в течение 30 мин гидрид натрия (1,35 г, 0,0563 моль). Смесь перемешивали в течение 30 мин, затем нагревали до комнатной температуры и дополнительно перемешивали в течение 1 ч. После этого добавляли метилиодид (9,16 г, 0,065 моль) и перемешивание продолжали в течение ночи. После того как реакция была завершена, смесь выливали в 100 мл воды и продукт экстрагировали этилацетатом (3×50 мл). Объединенный органический слой сушили над сульфатом магния и затем концентрировали при пониженном давлении. Полученный сырой продукт очищали на колонке с силикагелем и получали 7,1 г (61,2%) трет-бутил 1-метил-2-оксо-1,8-диазаспиро[4,5]декан-8-карбоксилата (1. (5)). LC-MS (ESI) 269 (М+Н)+.
Раствор соединения 1.4(6) (2 г) в 10 мл 1М HCl в диоксане перемешивали при комнатной температуре 1 ч. Затем образовавшийся осадок отфильтровывали, промывали диоксаном и сушили в вакууме. Полученный 1-метил-1,8-диазаспиро[4,5]декан-2-она гидрохлорид (1.4(6)⋅HCl) использовали в дальнейших реакций без очистки. Выход 1,3 г (90%).
К раствору соединения 1.4(6)⋅HCl (1,3 г, 6,7 ммоль) и DIPEA (3,8 мл, 22,11 ммоль) в 5 мл ДМФ добавляли 4-фтор-2-метокси-1-нитробензола (1.1(10): 1.317 мг, 7,71 ммоль) и полученную смесь перемешивали при 70°С в течение 15 ч. Когда реакция была завершена (контроль LC-MS), растворитель выпаривали в вакууме, а остаток фракционировали на колонке с силикагелем (EtOAc/МеОН). Получали 970 мг (45%) 1-метил-8-(3-метокси-4-нитрофенил)-1,8-диазаспиро[4,5]декан-2-она (1,4 (7)). LC-MS (ESI) 319 (М+Н)+.
К раствору соединения 1.4(7) (150 мг, 0,47 ммоль) в 3 мл метанола прибавляли 15 мг 10% Pd/C. Полученную смесь энергично перемешивали в атмосфере водорода в течение 5 часов. Затем добавляли CF3CO2H (150 мг), катализатор отфильтровывали, растворитель удаляли при пониженном давлении. Получали 8-(4-амино-3-метоксифенил)-1-метил-1,8-диазаспиро[4,5]декан-2-она трифторацетат (1.4(8)⋅CF3CO2H), который сушили в вакууме и использовали в следующей реакции без очистки. Выход 1.4(8)⋅CF3CO2H близко к количественному. LC-MS (ESI) 290 (М+Н)+.
К раствору соединения 1.4(8)⋅CF3CO2H (0,47 ммоль) в 3 мл 2-метоксиэтанола добавляли 149 мг (0,47 ммоль) 2,5-дихлор-N-(2-(диметилфосфорил)фенил)пиримидин-4-амина (1.1(17)) и полученную смесь перемешивали при кипении с обратным холодильником в течение 15 ч. Когда реакция была завершена (контроль LC-MS) растворители удаляли при пониженном давлении, а остаток очищали с помощью обращенно-фазовой ВЭЖХ (градиента MeCN-H2O с 0,1% TFA). Целевые фракции подщелачивали NaHCO3, ацетонитрил частично выпаривали, а продукт экстрагировали дихлорметаном. Объединенные органические экстракты сушили над Na2SO4 и выпаривали в вакууме. Получали 8-(4-{4-[2-(диметилфосфорил)-фениламино]-5-хлор-пиримидин-2-иламино}-3-метоксифенил)-1-метил-1,8-диаза-спиро[4.5]декан-2-он (1.4). LC-MS (ESI) 569 (М+Н)+. 1Н NMR (DMSO-d6, 400 МГц) δ 11.17 (s, 1Н), 8.50 (br, 1H), (s, 1H) 8.07 (s, 1H), 8.05 (s, 1H), 7.54 (m, 1H), 7.43 (d, J=8.4 Гц, 1H), 7.37 (t, J=6 Гц, 1H), 7.11 (t,J=6.8Гц, 1H), 6.67 (s, 1H), 6.51 (d, J=8.4 Гц, 1H), 3.77 (s, 3H), 3.69 (br. d, J=11.2 Гц, 2H), 2.82 (t,J=12.4 Гц, 2H), 2.64 (s, 3H), 2.28 (m, 2H), 1.98 (m, 4H), 1.78 (s, 3H), 1.75 (s, 3H) 1.47 (br, d,J=12.8 Гц, 2H).
Пример 5. N4-[2-(Диметилфосфорил)фенил]-N2-[4-(1-метил-1,7-диаза-спиро[3.5]нон-7-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.5) был получен в соответствии со схемой 5.
Схема 5
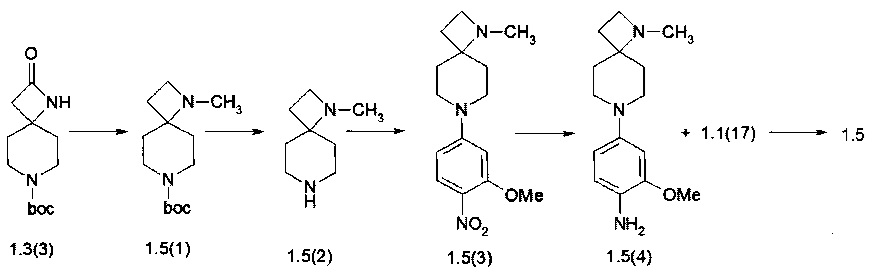
К раствору соединения 1.3(3) (850 мг, 3,75 ммоль) в МеОН добавляли 10% Pd/C (85 мг) и водный раствор формальдегида (4,875 ммоль). Реакционную смесь перемешивали в атмосфере водорода при температуре окружающей среды. После того как реакция была завершена, катализатор отфильтровывали, а растворитель удаляли при пониженном давлении. Остаток растворяли в EtOAc, фильтровали через тонкий слой силикагеля, сушили над Na2SO4, а затем упаривали в вакууме. Получали 690 мг (77%) трет-бутил 1-метил-1,7-диазаспиро[3,5]нонан-7-карбоксилата (1.5(1)). LC-MS (ESI) 241 (М+Н)+.
Раствор соединения 1.5(1) (690 г) в 10 мл 10% CF3COOH в CH2Cl2 перемешивали при комнатной температуре в течение 3 ч. Затем растворители выпаривали в вакууме и получали 1-метил-1,7-диаза-спиро[3,5]нонан трифторацетат (1.5(2)⋅CF3CO2H), который использовали в последующей реакции без какой-либо дополнительной очистки. Выход количественный.
К раствору 1.5(2)⋅CF3CO2H (1270 мг, 5 ммоль) и DIPEA (2,87 мл, 16,6 ммоль) в 3 мл ДМФ прибавляли 4-фтор-2-метокси-1-нитробензола (940 мг, 5,5 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 48 ч. После завершения реакции (контроль LC-MS) растворитель выпаривали в вакууме, а остаток фракционировали на колонке с силикагелем (EtOAc/MeOH). Получали 500 мг (34%) 1-метил-7-(3-метокси-4-нитрофенил)-1,7-диаза-спиро[3,5]нонан (1,5 (3)). LC-MS (ESI) 292 (М+Н)+.
К раствору соединения 1.5(3) (500 мг, 1,7 ммоль) в 5 мл метанола прибавляли 50 мг 10% Pd/C и смесь энергично перемешивали в атмосфере водорода в течение 3 ч. Затем добавляли CF2CO2H (300 мг), катализатор отфильтровывали, растворитель удаляли при пониженном давлении и получали 2-метокси-4-(1-метил-1,7-диаза-спиро[3.5]нонан-7-ил)анилин трифторацетат (1.5(4)⋅CF3CO2H), который сушили в вакууме и использовали в следующей реакции без очистки. Выход 1.5(4)⋅CF3CO2H близок к количественныму. LC-MS (ESI) 262 (М+Н)+.
К раствору соединения 1.5(4)⋅CF3CO2H (1,7 ммоль) в 3 мл 2-метоксиэтанол 590 мг (1,8 ммоль) прибавляли N-(2-(диметилфосфорил)фенил)-2,5-дихлор-пиримидин-4-амина (1.1(17)) и полученную смесь перемешивали при 70°С в течение 15 ч. После завершения реакции (контроль LC-MS) растворитель удаляли при пониженном давлении, а остаток очищали колоночной хроматографией (EtOAc:MeOH:+3% Net3). Выход N4-[2-(диметилфосфорил)фенил]-N2-[4-(1-метил-1,7-диаза-спиро[3.5]нон-7-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамина (1.5) 28 mg. LC-MS (ESI) 541 (М+Н)+. 1Н NMR (DMSO-d6, 400 МГц) δ 11.09 (br, 2H), 8.47 (br, 1H), 8.06 (s, 1H), 8.04 (s, 1H), 7.53 (m, 1H), 7.36 (m, 1H), 7.09 (m, 1H), 6.63 (m, 1H), 6.47 (m, 1H), 3.75 (s, 3H), 3.63 (m, 2H) 3.24 (m, 2H) 2.64 (m, 2H), 2.21 (s, 3H), 1.99 (m, 2H), 1.84 (br, 2H), 1.73 (s, 3H), 1.79 (s, 3H), 1.71 (br, 2H).
Пример 6. N4-[2-(Диметилфосфорил)фенил]-N2-[4-(1-метил-1,7-диаза-спиро[4.5]дек-8-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.6) был получен в соответствии со схемой 6.
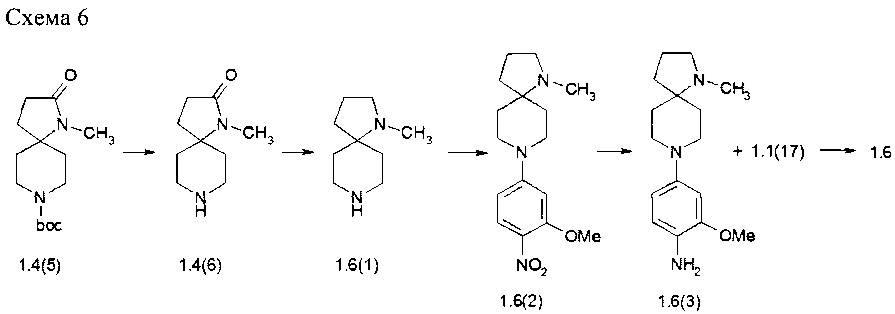
Перемешивали 500 мг соединения 1.4(5) в 2,5 мл 1М HCl в диоксане при комнатной температуре в течение 1 ч. Затем образовавшийся осадок отфильтровывали, промывали диоксаном и сушили в вакууме. Полученный продукт 1,4(6)⋅HCl растворяли в 5 мл сухого ТГФ и при перемешивании охлаждали на ледяной бане, добавляли по каплям суспензию 213 мг LiAlH4 в 5 мл сухого ТГФ. Затем реакционную смесь кипятили с обратным холодильником и перемешивали в течение 2 ч при кипении в атмосфере аргона. После завершения реакции смесь охлаждали до комнатной температуры и осторожно добавляли 0,5 мл воды, осадок отфильтровывают, маточный раствор концентрировали при пониженном давлении, сушили в вакууме и получали 303 мг 1-метил-1,8-диазаспиро[4,5]декана (1.6(1)), который был использован в последующей реакции без дополнительной очистки.
К раствору соединения 1.6(1) (300 мг, 1,95 ммоль) и DIPEA (1,12 мл, 6,43 ммоль) в 3 мл ДМФ добавляли 2-метокси-4-фтор-1-нитробензола (1.1(10): 383 мг, 2,24 ммоль). Полученную смесь перемешивали при 70°С в течение 15 ч. После завершения реакции (контроль LC-MS) растворитель выпаривали в вакууме и остаток фракционировали на колонке с силикагелем (EtOAc/МеОН). Получали 414 мг (69%) 1-метил-8-(3-метокси-4-нитрофенил)-1,8-диаза-спиро[4,5]декана (1.6(2). LC-MS (ESI) 306 (М+Н)+.
К раствору соединения 1,6(2) (200 мг, 0656 ммоль) в 2 мл метанола добавляли 20 мг 10%) Pd/C и смесь энергично перемешивали в атмосфере водорода в течение 5 ч. Затем добавляли CF3CO2H (150 мг), катализатор отфильтровывали, растворитель удаляли при пониженном давлении. Получали 4-(1-метил-1,8-диазаспиро[4.5]декан-8-ил)-2-метоксианилина трифторацетат (1.6(3)⋅CF3CO2H), который сушили в вакууме и использовали в последующей реакции без очистки. Выход 1.6(3)⋅CF3CO2H близок к количественному. LC-MS (ESI) 276 (М+Н)+.
К раствору соединения 1.6(3)⋅CF3CO2H (0,656 ммоль) в 2 мл 2-метоксиэтанола прибавляли 218 мг (0,689 ммоль) N-(2-(диметилфосфорил)фенил)-2,5-дихлор-пиримидин-4-амина (1.1(17)). Полученную смесь кипятили с обратным холодильником в течение 15 ч. После завершения реакции (контроль LC-MS) растворителя удаляли при пониженном давлении и остаток очищали с помощью обращенно-фазовой ВЭЖХ (градиента MeCN-Н2О с 0,1%) TFA). Целевые фракции подщелачивали NaHCO3, ацетонитрил частично выпаривали и продукт экстрагировали дихлорметаном. Объединенные органические экстракты сушили над Na2SO4 и выпаривали. Получали N4-[2-(диметилфосфорил)фенил]-N2-[4-(1-метил-1,7-диаза-спиро[4.5]дек-8-ил)-2-метоксифенил]-5-хлор-пиримидин-2,4-диамин (1.6). LC-MS (ESI) 555 (М+Н)+. 1Н NMR (DMSO-d6, 400 МГц) δ 11.16 (s, 1Н), 8.49 (br, 1H), 8.07 (s, 1H) 8.04 (s, 1H), 7.53 (m, 1H), 7.40 (d,J=8.8Гц, 1H), 7.35 (t, J=8 Гц, 1H), 7.10 (t, J=7.6 Гц, 1H), 6.63 (m, 1H), 6.48 (m, 1H), 3.76 (s, 3H), 3.68 (br. d, J=12.8 Гц, 2H), 2.70 (m, 4H), 2.21 (s, 3H), 1.78 (s, 4H), 1.75 (s, 4H), 1.71 (s, 4H), 1.29 (br, d, J=12.8 Гц, 2H).
Пример 7. N2-[4-(9-диметиламино-3-аза-спиро[5.5]ундец-3-ил)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин (1.7) был получен в соответствии со схемой 7.
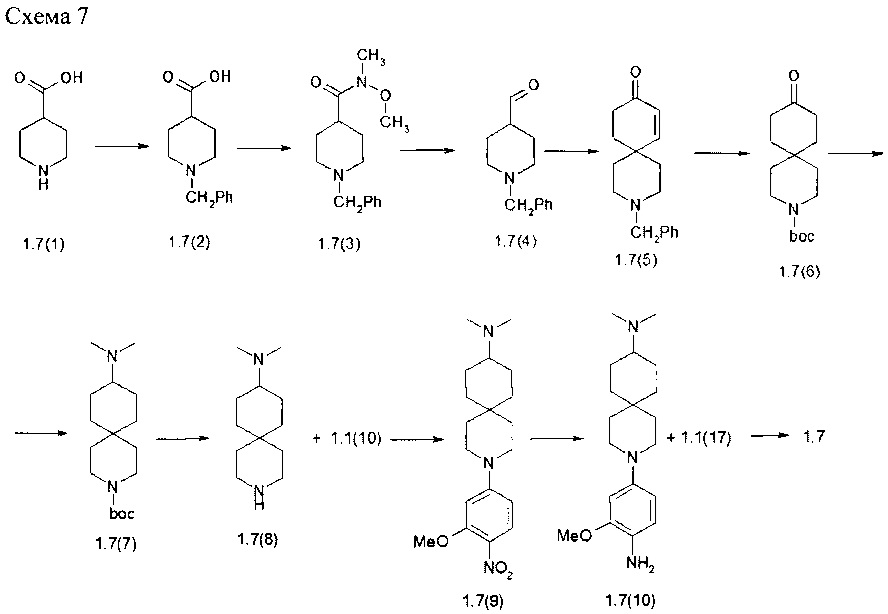
К раствору 1.7(1) (10,0 г, 77,4 ммоль) и NaOH (7,7 г, 193,5 ммоль) в 200 мл воды, добавляли бензилхлорида (11,6 мл, 81,3 ммоль) при 0-5°С. Смесь перемешивали при комнатной температуре в течение ночи, затем промывали диэтиловым эфиром. Водный слой подкисляли соляной кислотой до рН 3-4. Белое твердое вещество отфильтровывали. Выход 1-{[(фенилметил)окси]карбонил}пиперидин-4-карбоновой кислоты (1,7(2)) составил 20.1 г (98%). 1H-NMR, (DMSO-d6, 400 МГц): 12.25 (br.s, 1Н), 7.45-7.25 (m, 5H), 5.06 (s, 2H), 4.10-3.90 (m, 2H), 3.10-2.80 (m, 2H), 2.47-2.37 (m, 1H), 1.90-1.70 (m, 2H), 1.50-1.33 (m, 2H).
К раствору соединения 1.7(2) (20,1 г, 76,3 ммоль) в 400 мл CH2Cl2 добавляли N,N-карбодиимидазол (13,6 г, 83,9 ммоль), смесь перемешивали при комнатной температуре в течение 3 ч, после чего был добавлен N,O-диметилгидроксиламина (9,7 г, 99,2 ммоль) и триэтиламин (13,8 мл, 99,2 ммоль). Смесь перемешивали при комнатной температуре в течение 14 ч, затем промывали 10% водным раствором Na2CO3 и 3% HCl. Органический слой сушили над Na2SO4, фильтровали и выпаривали в вакууме. Получали 20,6 г (88%) бензил 4-[метил(метилокси)амино]карбонил}пиперидин-1-карбоксилата (1.7(3)), который использовали на следующей стадии без дополнительной очистки. 1H-NMR, (CDCl3, 400 МГц): 7.40-7.28 (m, 5Н), 5.13 (s, 2Н), 4.38-4.10 (m, 2Н), 3.71 (s, 3Н), 3.19 (s, 3Н), 3.00-2.70 (m, 3Н), 1.85-1.60 (m, 4Н).
К смеси LiAlH4 (3,9 г, 100,8 ммоль) в 400 мл ТГФ, добавляли соединение 1.7(3) (20,6 г, 67,2 ммоль) при -60°С. Реакционную смесь оставляли самопроизвольно нагреваться до 0-5°С и затем снова охлаждали до -60°С, затем добавляли целит и смесь KHSO4 (10 г) в воде (50 мл) и фильтровали через целит. Фильтрат промывали холодным 1N HCl, насыщенным раствором NaHCO3, рассолом, сушили над MgSO4 и концентрировали в вакууме. Остаток очищали колоночной хроматографией на силикагеле (гексан: этилацетат =3:1). Выход 4-фенилметил формилпиперидин-1-карбоксилата (1.7(4)) составлял 12 г (72%).. 1H-NMR, (DMSO-d6, 400 МГц): 9.58 (s, 1Н), 7.40-7.28 (m, 5H), 5.06 (s, 2H), 3.94-3.80 (m, 2H), 3.15-2.85 (m, 2H), 2.60-2.51 (m, 1H), 1.93-1.76 (m, 2H), 1.48-1.30 (m, 2H).
К бензольному (250 мл) раствору соединения 1.7(4) (16,6 г, 67,1 ммоль) при перемешивании прибавляли п-толуолсульфокислоту (1,9 г, 10 ммоль). Реакционную смесь нагревали до 70°С с насадкой Дина-Старка и добавляли 3-бутен-2-он (9,9 г, 140,9 ммоль). Смесь кипятили с обратным холодильником в течение 24 часов собирая выделяющуюся воду в насадке Дина-Старка. Реакционную смесь охлаждали до комнатной температуры и промывали 500 мл насыщенного водного раствора бикарбоната натрия. Органическую фазу сушили над сульфатом натрия, фильтровали и концентрировали. Остаток очищают колоночной хроматографией на силикагеле (гексан: этилацетат = 2:1). Выход бензил 9-оксо-аза-спиро[5,5]ундец-7-ен-3-карбоксилата (1.7. (5)) составил 15,6 г (78%). 1H-NMR, (DMSO-d6, 400 МГц): 7.50-7.25 (m, 5Н), 6.97 (d, J=10.5Гц, 1H), 5.84 (d, J=10.5Гц, 1H), 5.08 (s, 2H), 3.67-3.50 (m, 2H), 3.47-3.33 (m, 2H), 2.45-2.30 (m, 2H), 1.95-1.85 (m, 2H), 1.70-1.40 (m, 4H).
Раствор соединения 1.7(5) (15,6 г, 52,1 ммоль) и ди-трет-бутилдикарбоната (14,8 г, 67,7 ммоль) в 400 мл EtOAc гидрировали с 1,1 г 10% Pd/C в течение 24 ч при 1 атм водорода. Смесь фильтровали через целит, фильтрат концентрировали. Остаток очищали колоночной хроматографией на силикагеле (гексан: этилацетат =3:1). Выход трет-бутил 9-оксо-аза-спиро[5,5]ундекан-3-карбоксилата (1.7(6)) составил 8,6 г (62%). 1H-NMR, (CDCl3, 400 МГц): 3.54-3.38 (m, 4Н), 2.44-2.28 (m, 4Н), 1.83-1.73 (m, 4H), 1.58-1.52 (m, 4H), 1.47 (s, 9H).
Раствор соединения 1.7(6) (2,0 г, 7,5 ммоль) и 33% раствора диметиламина в воде перемешивали при комнатной температуре в течение 3 ч.. Затем добавляли триацетоксиборгидрид натрия (4,8 г 22.5 ммоль) и смесь перемешивали при комнатной температуре в течение 14 ч. Смесь промывали 10%-ным раствором Na2CO3, сушили над Na2SO4, фильтровали и выпаривали в вакууме. Получали 2,2 г (100%) трет-бутил 9-(диметиламино)-3-аза-спиро[5,5]ундекан-3-карбоксилата (1.7(7)), который использовали в следующей стадии без дополнительной очистки. LC-MS (ESI) (m/z): 297 (М+Н)+.
К смеси соединения 1.7(7) в 30 мл диоксана добавили 10 мл 3М HCl в диоксане (2,2 г, 7,4 ммоль) и полученную смесь выдерживали при температуре окружающей среды в течение 14 ч. Диоксан выпаривали в вакууме досуха, твердый остаток сушили и получали 2,0 г (100%) N,N-диметил-3-аза-спиро[5,5]ундекан-9-амин дигидрохлорида (1.7(8)⋅2HCl). 1H-NMR, (D2O, 400 МГц): 3.20-3.10 (m, 5Н), 2.78 (s, 6Н), 1.95-1.83 (m, 4Н), 1.77-1.70 (m, 2Н), 1.61-1.52 (m, 4Н), 1.35-1.23 (m, 2Н).
К перемешиваемой смеси 1.7(8)⋅2HCl (0,5 г, 1,8 ммоль) и DIPEA (1,3 мл, 2,5 ммоль) в ДМФ (15 мл) добавляли соединение 1.1(10) (0,35 г, 2,0 ммоль). Смесь перемешивали в течение 14 ч при 70°С. Реакционную смесь концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2: МеОН 40:1). Выход N,N-диметил-3-[3-(метилокси)-4-нитрофенил]-3-аза-спиро[5,5]ундекан-9-амина (1.7(9)) составил 0,4 г (62%). 1H-NMR, (DMSO-d6, 400 МГц): 7.87 (d, J=9.4Гц, 1H), 6.56 (d, J=9.7Гц, 1H), 6.46 (s, 1H), 3.89 (s, 3H), 3.47-3.40 (m, 2H), 2.31 (s, 6H), 1.80-1.60 (m, 4H), 1.60-1.50 (m, 2H), 1.45-1.33 (m, 4H), 1.20-0.95 (m, 5H).
Раствор соединения 1.7(9) (0,4 г, 1,15 ммоль) в 15 мл этанола гидрировали с 0,06 г 10% Pd/C в течение 14 ч при 60°С и 40 атм водорода. Смесь фильтровали через целит. К фильтрату добавляли избыток 3М HCl в диоксане и полученный раствор упаривали. Получали 0,33 г (67%) 3-[4-амино-3-(метилокси)фенил]-N,N-диметил-3-аза-спиро[5,5] ундекан-9-амина тригидрохлорида (1.7(10)⋅3HCl).
Раствор соединения 1.7(10)⋅3HCl (0,33 г, 0,77 ммоль) и соединения 1.1(17) (0,3 г, 0,92 ммоль) в 8 мл 2-метоксиэтанола и 2,0 мл воды перемешивали при 120°С в течение 14 ч и охлаждали до комнатной температуры. Реакционную смесь упаривали в вакууме. Остаток разбавляли 10% водным K2CO3 и экстрагировали CH2Cl2 (5×50 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью обращенно-фазовой ВЭЖХ (градиент MeCN-H2O с 0,1% TFA). Целевые фракции подщелачивали водным NaHCO3, ацетонитрил частично выпаривали и продукт экстрагировали дихлорметаном. Объединенные органические экстракты сушили над Na2SO4 и упаривали в вакууме. Получали 0.22 г N2-[4-(9-диметиламино-3-аза-спиро[5.5]ундец-3-ил)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамина (1.7). 1H-NMR, (DMSO-d6, 400 МГц): 11.16 (s, 1Н), 8.50 (br.s, 1H), 8.10-8.00 (m, 2H), 7.60-7.47 (m, 1H), 7.45-7.28 (m, 2H), 7.14-7.04 (m, 1H), 6.61 (s, 1H), 6.46 (d, J=8.8, 1H), 3.75 (s, 3H), 3.18-3.06 (m, 4H), 2.17 (s, 6H), 2.12-2.02 (m, 1H), 1.77 (s, 3H) 1.74 (s, 3H), 1.73-1.65 (m, 2H), 1.65-1.55 (m, 4H), 1.47-1.40 (m, 2H), 1.40-1.30 (m, 2H), 1.17-1.05 (m, 2H).
Пример 8. N2-{4-[4-(9-Диметиламино-3-аза-спиро[5.5]ундец-3-ил)-пиперидин-1-ил]-2-метоксифенил}-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин (1.8) был получен в соответствии со схемой 8.
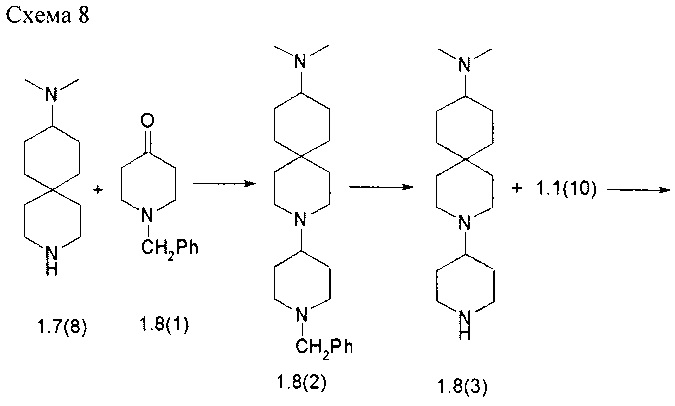
Смесь соединения 1.7(8)⋅2HCl (1,0 г, 3,7 ммоль) и соединение 1.8(1) (0,77 г, 4,07 ммоль) в CH2Cl2 (70 мл) перемешивали при комнатной температуре в течение 3 ч, добавляли триацетоксиборгидрид натрия (4,0 г, 18,5 ммоль) и TEA (1,0 мл, 7,4 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 14 ч. Полученную реакционную смесь последовательно промывали 10% водным раствором K2CO3 и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали и выпаривали в вакууме. Получали 8,9 г (61%) N,N-диметил-3-[1-(фенилметил)пиперидин-4-ил]-3-азапиро[5,5]ундекан-9-амин (1.8(2)).
Раствор соединения 1.8(2) (1,3 г, 3,5 ммоль) в 50 мл этанола гидрировали с 0,19 г 10% Pd/C в течение 14 ч при 60°С и 40 атм водорода. Полученную смесь фильтровали через целит и выпаривали в вакууме. Получали 0,98 г (100%) N,N-диметил-3-пиперидин-4-ил-3-аза-спиро[5,5]ундекан-9-амина (1.8(3)).
К перемешиваемой смеси соединения 1.8(3) (0,98 г, 3,5 ммоль) и DIPEA (1,6 мл, 8,75 ммоль) в ДМФ (20 мл) добавляли соединение 1.1(10) (0,66 г, 3,85 ммоль) и перемешивали в течение 14 ч при 70°С.Реакционную смесь концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2: МеОН 40:1). Получали 0,28 г N,N-диметил 3-{1-[3-(метилокси)-4-нитрофенил]пиперидин-4-ил}-3-аза-спиро[5,5]ундекан-9-амина (1.8(4)). LC-MS (ESI) (m/z): 431 (М+Н)+.
Раствор соединения 1.8(4) (0,28 г, 0,65 ммоль) в 15 мл этанола гидрировали с 0,04 г 10% Pd/C в течение 14 ч при 60°С и 40 атм водорода. Смесь фильтровали через целит. К фильтрату добавляли избыток 3М HCl в диоксане и полученный раствор упаривали в вакууме. Получали 0,31 г (87%) 3-{1-[4-амино-3-(метокси)фенил]пиперидин-4-ил}-N,N-диметил-3-аза-спиро[5,5]ундекан-9-амина тетрагидрохлорида (1,8(5)⋅4HCl). LC-MS (ESI) (m/z): 401 (М+Н)+.
Раствор соединения 1.8(5)⋅4HCl (0,31 г, 0,56 ммоль) и соединения 1.1(17) (0,21 г, 0,67 ммоль) в 8 мл 2-метоксиэтанол и 2,0 мл воды перемешивали при 120°С в течение 14 ч и охлаждали до комнатной температуры. Реакционную смесь упаривали в вакууме, остаток разбавляли 10% водным раствором K2CO3 и экстрагировали CH2Cl2 (5×50 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2:MeOH:NH4OH=10:1:0,1). Получали 0,1 г N2-{4-[4-(9-диметиламино-3-аза-спиро[5.5]ундец-3-ил)-пиперидин-1-ил]-2-метоксифенил}-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамина (1.8). 1H-NMR, (DMSO-d6, 400 МГц): 11.17 (s, 1H), 8.50 (br.s, 1H), 8.10-8.00 (m, 2H), 7.60-7.47 (m, 1H), 7.45-7.28 (m, 2H), 7.14-7.04 (m, 1H), 6.61 (s, 1H), 6.46 (d, J=8.8, 1H), 3.75 (s, 3H), 3.75-3.68 (m, 2H), 2.71-2.58 (t, J=11.1Гц, 2H), 2.48-2.40 (m, 4H), 2.37-2.28 (m, 1H), 2.14 (s, 6H), 2.07-1.96 (m, 1H), 1.88-1.70 (m, 8H), 1.67-1.60 (m, 2H), 1.60-1.47 (m, 4H) 1.47-1.40 (m, 2H), 1.32-1.24 (m, 4H), 1.06-0.95 (m, 2H).
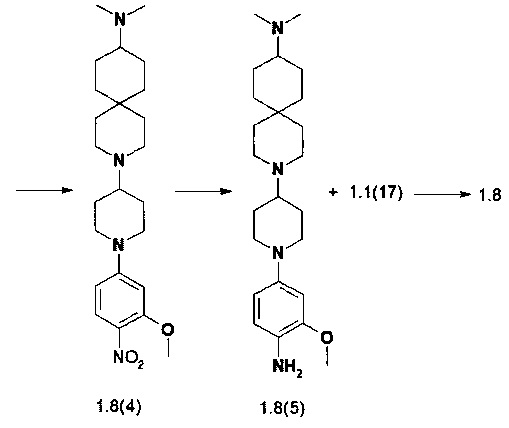
Пример 9. N4-[2-(Диметилфосфорил)-фенил]-N2-{4-[4-(1-метил-1,8-диаза-спиро[4.5]дец-8-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамин (1.9) и его соли 1.9⋅5HCl, 1.9⋅5MeSO3H и 1.9⋅3Cl2CO2H были получены в соответствии со схемой 9.
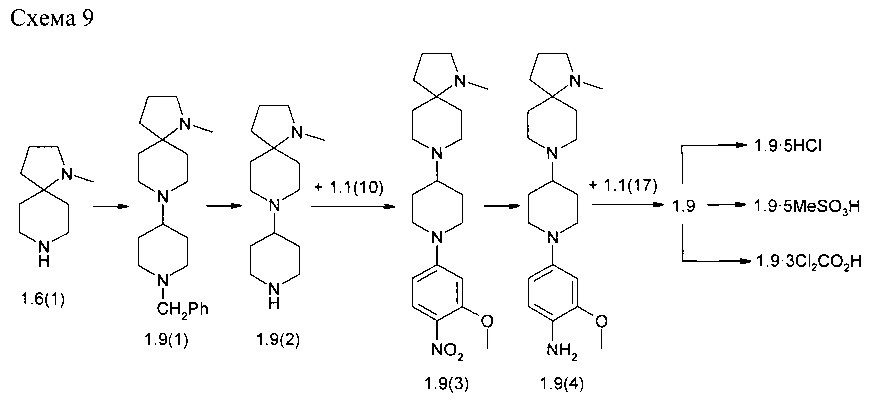
К раствору соединения 1.6(1) 900 мг (5,85 ммоль) в 27 мл CH2Cl2 и 1-бензилпиперидин-4-она (1160 мг, 6.136 ммоль) добавляли АсО3ВН (2765 мг, 14,625 ммоль) и перемешивали полученную реакционную смесь в течение ночи при комнатной температуре. После того как реакция была завершена (контроль LC-MS) осадок отфильтровывали, маточный раствор промывали рассолом, сушили над Na2SO4 и упаривали в вакууме. Полученное масло фракционировали на колонке с силикагелем и получали 552 мг 8-(1-бензилпиперидин-4-ил)-1-метил-1,8-диа-заспиро[4,5]декана (1.9(1)). LC-MS (ESI) 328 (М+Н)+.
К раствору соединения 1.9(1) (552 мг) в МеОН (5 мл) добавляли 50 мг 10% Pd/C и полученную смесь энергично перемешивали в атмосфере водорода в течение 5 ч. После того как реакция была завершена, катализатор отфильтровывали, растворитель удаляли при пониженном давлении, а остаток 1.9(2) был использован в дальнейшей реакции без очистки. Выход 1-метил-8-(пиперидин-4-ил)-1,8-диаза-спиро[4,5]декана (1.9(2)) количественный. LC-MS (ESI) 238 (М+Н)+.
К раствору соединения 1.9(2) (400 мг, 1,68 ммоль) и DIPEA (0,616 мл, 3,54 ммоль) в 4 мл диметилформамида добавляли 2-метокси-1-нитро-4-фтор-бензола (1.1(10): 316 мг, 1,848 ммоль) и полученную смесь перемешивали при комнатной температуре в течение 15 ч. После завершения реакции (контроль LC-MS) растворитель выпаривали в вакууме и остаток фракционировали на колонке с силикагелем (EtOAc/MeOH). Получали 315 мг (48%) 1-метил-8-(1-(3-метокси-4-нитрофенил)пиперидин-4-ил)-1,8-диаза-спиро[4,5]декана (1.9(3)). LC-MS (ESI) 389 (М+Н)+.
К раствору соединения 1.9(3) (315 мг, 0812 ммоль) в 3 мл метанола добавляли 30 мг 10% Pd/C и полученную смесь энергично перемешивали в атмосфере водорода в течение 5 ч. Затем добавляли CF3CO2H (150 мг), катализатор отфильтровывали, растворитель удаляли при пониженном давлении. Полученный с близким к количественному выходом 4-(4-(1-метил-1,8-диаза-спиро[4,5]декан-8-ил)пиперидин-1-ил)-2-метоксианилин трифторацетат (1.9(4)⋅CF3CO2H) сушили в вакууме и использовали в последующей реакции без дополнительной очистки. LC-MS (ESI) 359 (М+Н)+.
К раствору соединения 1.9(4)⋅CF3CO2H (0,812 ммоль) в 12 мл 2-метоксиэтанола добавляли 282 мг (0,893 ммоль) N-(2-(диметилфосфорил)фенил)-2,5-дихлор-пиримидин-4-амина (1.1(17)) и полученную смесь перемешивали при 75°С в течение 15 ч. После завершения реакции (контроль LC-MS) растворитель удаляли при пониженном давлении, а остаток очищали колоночной хроматографией. Целевые фракции объединяли, растворитель удаляли в вакууме и получали 247 мг, (47%) N4-2-(диметилфосфорил)-фенил]-N2-{4-[4-(1-метил-1,8-диаза-спиро[4.5]дек-8-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамин (1.9). LC-MS (ESI) 638 (М+Н)+. 1Н NMR (DMSO-d6, 400 МГц) δ 11.17 (s, 1Н), 8.48 (br, 1H), 8.06 (s, 1H) 8.05 (s, 1H), 7.53 (m, 1H), 7.36 (m, 2H), 7.09 (t, J=7.6 Гц, 1H), 6.61 (s, 1H), 6.46 (m, 1H), 3.76 (s, 3H), 3.72 (d, J=10.8 Гц, 2H), 2.85 (d, J=10.8 Гц, 2H), 2.63 (m, 4H), 2.35 (m, 1H) 2.17 (s, 3H), 2,13 (m, 2H), 1.80 (m, 2H), 1.78 (s, 3H), 1.74 (s, 3H), 1.59 (m, 8H) 1.14 (d,J=14 Гц, 2H).
К раствору ингибитора 1.9 (40 мг, 0.06 ммоль) в дихлорметане (4 мл) при 25°С прибавляют 2 мл 2 М HCl в эфире. Полученную смесь перемешивают в течение 20 ч. Образовавшийся осадок отильтровывают и сушат на воздухе в течение ночи. Получают 41 мг N4-2-(диметилфосфорил)-фенил]-N2-{4-[4-(1-метил-1,8-диаза-спиро[4.5]дек-8-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамин пента-гидрохлорида (1.9⋅5HCl) в виде белых кристаллов. 1H-NMR (D2O, 400 МГц): 8.01 (s, 1H), 7.70-7.58 (m, 2H), 7.58-7.51 (m, 1H), 7.46-7.39 (m, 1H), 7.23 (d, J=8.7Гц, 1H), 6.80 (d, J=2.2Гц, 1H), 6.64 (dd, J1=8.7ГЦ, J2=2.2Гц, 1H), 3.85-3.65 (m, 8H), 3.61-3.50 (m, 1H), 3.30-3.15 (m, 3H), 3.11-2.99 (m, 2H), 2.80 (s, 3H), 2.38-2.01 (m, 10H), 2.00-1.88 (m, 2H), 1.73 (s, 3H), 1.70 (s, 3H).
К раствору ингибитора 1.9 (40 мг, 0.06 ммоль) в ацетоне (4 мл) при 25°С прибавляют метансульфокислоту (42 мг, 0,42 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивают в течение 20 ч. Образовавшийся осадок отфильтровывают и сушат на воздухе в течение ночи. Получают 50 мг N4-2-(диметилфосфорил)-фенил]-N2-{4-[4-(1-метил-1,8-диаза-спиро[4.5]дец-8-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамин пента-мезилата (1.9⋅5MeSO3H) в виде белых кристаллов. 1H-NMR (D2O, 400 МГц): 8.06 (s, 1H), 7.69-7.54 (m, 3H), 7.50-7.43 (m, 1H), 7.33 (d, J=8.7Гц, 1H), 7.00 (d, J=2.2Гц, 1H), 6.82 (dd, J1=8.7Гц, J2=2.2Гц, 1H), 3.90-3.61 (m, 9H), 3.55-3.39 (m, 2H), 3.35-3.15 (m, 3H), 2.80 (s, 3H), 2.71 (s, 15H), 2.52-2.20 (m, 4H), 2.20-2.00 (m, 8H), 1.71 (s, 3H), 1.68 (s, 3H).
К раствору ингибитора 1.9 (40 мг, 0.06 ммоль) в ацетоне (4 мл) при 25°С прибавляют дихлоруксусную кислоту (60 мг, 0,42 ммоль) в виде раствора в ацетоне (1 мл). Полученную смесь перемешивают в течение 20 ч. Образовавшийся осадок отфильтровывают и сушат на воздухе в течение ночи. Получают 30 мг N4-2-(диметилфосфорил)-фенил]-N2-{4-[4-(1-метил-1,8-диаза-спиро[4.5]дец-8-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамин трис-дихлорацетата (1.9⋅3Cl2CO2H) в виде белых кристаллов. 1H-NMR (D2O, 400 МГц): 7.97 (s, 1H), 7.74-7.66 (m, 1H), 7.64-7.54 (m, 1H), 7.53-7.46 (m, 1H), 7.43-7.34 (m, 1H), 7.20 (d, J=8.8Гц, 1H), 6.74-6.68 (m, 1H), 6.60-6.51 (m, 1H), 5.96 (s, 3H), 3.85-3.65 (m, 8H), 3.55-3.42 (m, 1H), 3.30-3.14 (m, 3H), 2.98-2.84 (m, 2H), 2.79 (s, 3H), 2.34-2.00 (m, 10H), 1.95-1.80 (m, 2H), 1.73 (s, 3H), 1.70 (s, 3H).
Пример 10. N2-(4-{4-[(2-Диметиламиноэтил)-метиламино]-пиперидин-1-ил}-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин тетрагидрохлорид (1.13⋅4HCl) был получен в соответствии со схемой 10.
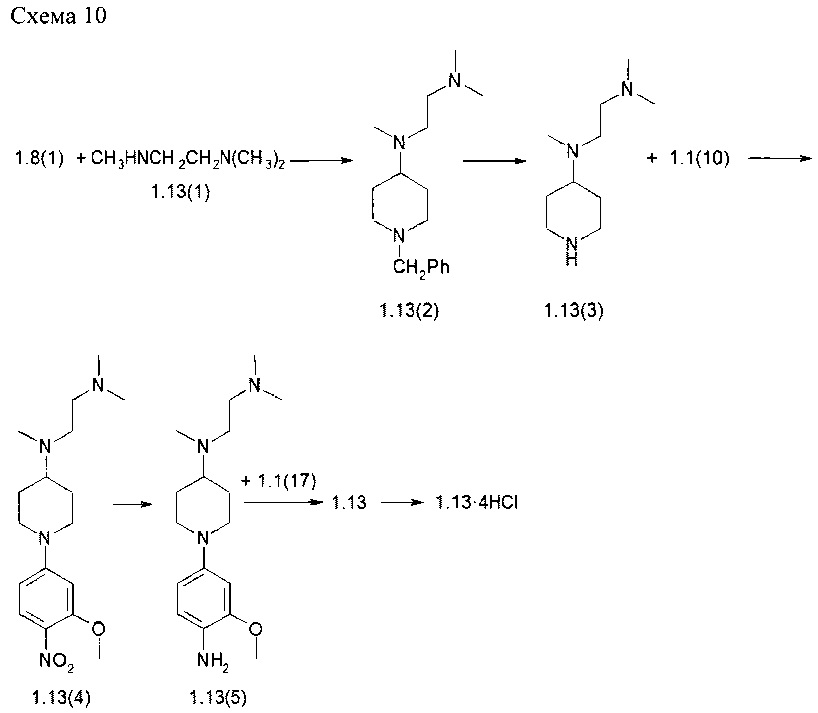
Смесь соединения 1.8(1) (2,0 г, 10,6 ммоль) и 1.13(1) (1,2 г, 11,7 ммоль) в CH2Cl2 (50 мл) перемешивали при комнатной температуре в течение 3 ч. Затем добавляли триацетоксиборгидрид натрия (6,7 г, 31,8 ммоль) и смесь перемешивали при комнатной температуре в течение 14 ч. Смесь последовательно промывали 10% водным раствором K2CO3 и насыщенным раствором соли. Органический слой сушили над Na2SO4, фильтровали и упаривали. Остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2: МеОН=40:1). Получали 1,8 г (62%) N,N,N'-триметил-N'-[l-(фенилметил)пиперидин-4-ил]этан-1,2-диамина (1.13(2)). 1H-NMR, (CDCl3, 400 МГц): 7.36-7.21 (m, 5Н), 3.49 (s, 2Н), 3.00-2.90 (m, 2Н), 2.59-2.51 (m, 2Н), 2.43-2.31 (m, 2Н), 2.28 (s, 3Н), 2.24 (s, 6Н), 2.00-1.90 (m, 2Н), 1.77-1.67 (m, 2Н), 1.65-1.52 (m, 2Н).
Раствор соединения 1.13(2) (1,8 г, 6,5 ммоль) в 40 мл ЕОН гидрировали 0,35 г 10% Pd на угле в течение 14 ч при 60°С и 40 атм водорода. Реакционную смесь фильтровали через целит, фильтрат выпаривали, и получали 1,2 г (100%) N,N,N'-триметил-N'-пиперидин-4-илэтан-1,2-диамина (1.13(3), который был использован на следующей стадии без дополнительной очистки. 1H-NMR, (CDCl3, 400 МГц): 3.21-3.09 (m, 2Н), 2.63-2.53 (m, 2Н), 2.52-2.43 (m, 1Н), 2.42-2.35 (m, 2H), 2.28 (s, 3H), 2.24 (s, 6H), 1.83-1.71 (m, 2H), 1.50-1.35 (m, 2H).
К перемешиваемой смеси соединения 1.13(3) (1,2 г, 6,4 ммоль) и DIPEA (2,3 мл, 12,8 ммоль) в ДМФ (15 мл) добавляли соединение 1.1(10) (1,4 г, 7,7 ммоль). Смесь перемешивали в течение 14 ч при 70°С.Реакционную смесь концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2: МеОН 30:1). Получали 2,2 г (100%) N,N,N'-триметил-N'-{1-[3-(метилокси)-4-нитрофенил]пиперидин-4-ил}этан-1,2-диамина (1.13(4)). 1H-NMR, (CDCl3, 400 МГц): 8.00 (d, J=9.4Гц, 1H), 6.42 (dd, J1=9.4Гц, J2=2.6Гц, 1H), 6.31 (d, J=2.6Гц, 1H), 4.10-3.90 (m, 5H), 3.02-2.90 (m, 2H), 2.71-2.55 (m, 3H), 2.46-2.39 (m, 2H), 2.30 (s, 3H), 2.27 (s, 6H), 1.96-1.86 (m, 2H), 1.69-1.54 (m, 2H).
Раствор полученного соединения 1.13(4) (2,2 г, 6,5 ммоль) в 50 мл этанола гидрировали 0,35 г 10% Pd на угле в течение 14 ч при 60°С при 40 атм водорода. Смесь фильтровали через целит. К фильтрату добавляли избыток 3М HCl в диоксане и раствор упаривали в вакууме. Получали 2,6 г (87%) N-{1-[4-амино-3-(метилокси)фенил]пиперидин-4-ил}-N,N',N'-триметилэтан-1,2-диамина тригидрохлорида (1.13 (5)⋅3HCl). 1H-NMR, (D2O, 400 МГц): 7.46 (d, J=8.7Гц, 1H), 7.24 (br.s, 1H), 7.14 (d, J=8.7Гц), 3.96-3.84 (m, 6H), 3.76-3.55 (m, 6H), 2.95 (s, 6H), 2.93 (s, 3H), 2.48-2.38 (m, 2H), 2.32-2.18 (m, 2H);
Раствор соединения 1.13(5)⋅3HCl (0,4 г, 0,88 ммоль) и соединения 1.1(17) (0,33 г, 1,06 ммоль) в 8 мл 2-метоксиэтанола и 2,0 мл воды перемешивали при 120°а С в течение 14 ч, а затем охлаждали до комнатной температуры. Реакционную смесь упаривали в вакууме. Остаток разбавляли 10% водным раствором K2CO3 и экстрагировали CH2Cl2 (5×50 мл).
Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2:МеОН:NH4OH=10:1:0,1). Полученный продукт 1.13 растворяли в CH2Cl2 и добавляли избыток 3М HCl в диоксане. Реакционную смесь выпаривали в вакууме и получили N2-(4-{4-[(2-диметиламиноэтил)-метиламино]-пиперидин-1-ил}-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамина тетрагидрохлорид ((1.13)⋅4HCl). 1H-NMR, (D2O, 400 МГц): 8.06 (s, 1Н), 7.71-7.62 (m, 2H), 7.62-7.54 (m, 1H), 7.51-7.44 (m, 1H), 7.35-7.27 (m, 1H), 6.97-6.93 (m, 1H), 6.81-6.74 (m, 1H), 3.87-3.75 (m, 6H), 3.68 (s, 4H), 3.40-3.27 (m, 2H), 2.97 (s, 6H), 2.93 (s, 3H), 2.41-2.30 (m, 2H), 2.20-2.05 (2H).
Пример 11. N2-[4-({3-[(2-Диметиламиноэтил)-метиламино]-пропил}-метиламино)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин тригидрохлорид (1.14⋅3HCl) был получен в соответствии со схемой 11.
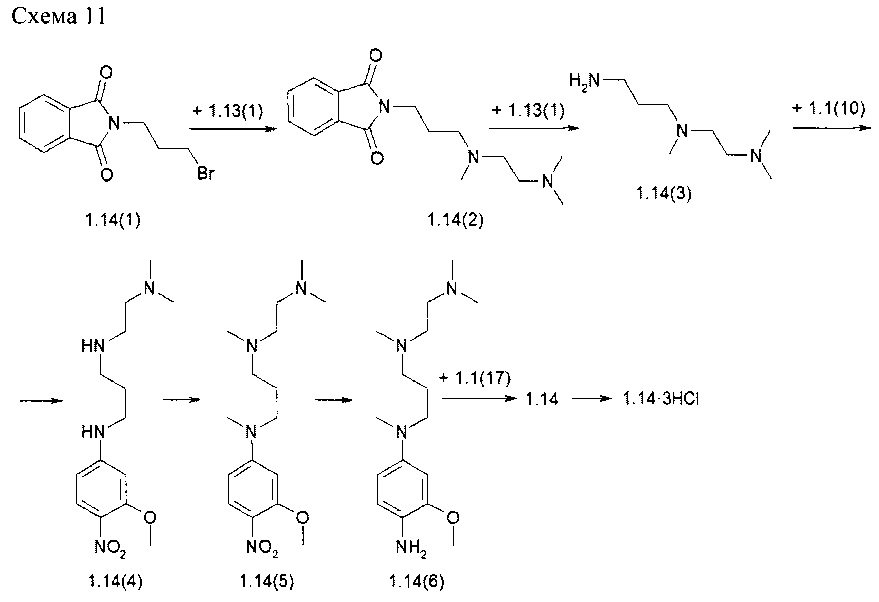
К раствору соединения 1.13(1) (3,0 г, 29,4 ммоль) в 100 мл CH3CN добавляли K2CO3 (12,2 г, 88,2 ммоль) и N-(3-бромпропил)фталимид (1.14(1): 9,1 г, 32,4 ммоль). Смесь перемешивали в течение 24 ч при 50°С.Затем к реакционной массе добавляли EtOAc и H2O, слои разделяли, органический слой сушили над Na2SO4, фильтровали и упаривали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2: МеОН=30:1). Получали 2,4 г 2-{3-[[2-(диметиламино)этил]-метиламино]пропил}-1Н-изоиндол-1,3(2Н)-дион (1.14(2)). 1H-NMR, (CdCl3, 400 МГц): 7.90-7.80 (m, 2Н), 7.75-7.65 (m, 2Н), 3.80-3.67 (m, 2Н), 2.50-2.33 (m, 6Н), 2.24 (s, 6Н), 2.22 (s,3H), 1.90-1.80 (m, 2H).
К раствору соединения 1.14(2) (2,4 г, 8,3 ммоль) в 200 мл этанола добавляли 51% водный гидразин гидрат (2,7 мл, 41,5 ммоль) и перемешивали смесь с обратным холодильником при кипячении в течение ночи. Твердое вещество отфильтровывали, фильтрат концентрировали при пониженном давлении, остаток растворяли в CH2Cl2 (150 мл), фильтровали и упаривали в вакууме. Получали 1,32 г (100%) N-[2-(диметиламино)этил]-N-метилпропан-1,3-диамина (1.14(3)), который использовали на следующей стадии без дополнительной очистки. 1H-NMR, (DMSO-d6, 400 МГц): 2.65-2.55 (m, 2Н), 2.40-2.20 (m, 6Н), 2.11 (s, 9Н), 1.56-1.44 (m, 2Н);
Соединение 1.1(10) (1,5 г, 8,7 ммоль) добавляли к перемешиваемой смеси соединения 1.14(3) (1,32 г, 8,3 ммоль) и DIPEA (3,6 мл, 20,8 ммоль) в ДМФ (20 мл). Смесь перемешивали в течение 14 ч при 70°С. Реакционную смесь концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2: МеОН 30:1). Получали 2,1 г (82%) N-[2-(диметиламино)этил]-N,N'-диметил-N'-[3-метилокси)-4-нитрофенил]пропан-1,3-диамина (1.14(4)). LC-MS (ESI) (m/z): 311 (М+Н)+.
Параформальдегид (2 г, 67 ммоль) добавляли к раствору соединения 1.14(4) (2,1 г 6,7 ммоль) в уксусной кислоте (15 мл), смесь перемешивали при комнатной температуре в течение 3 ч. К этой смеси добавляют порциями цианоборгидрид натрия (2,1 г 32,2 ммоль) и перемешивали при комнатной температуре в течение ночи. Полученную смесь выливали на лед, нейтрализовали 20% водным раствором K2СO3 до рН 8,0 и экстрагировали CH2Cl2. Органический слой промывали водой и солевым раствором. После сушки над Na2SO4, фильтровали и выпаривали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2: МеОН=30:1). Получали 0,8 г (37%) N-[2-(диметиламино)этил]-N-метил-N'-[3-метилокси)-4-нитрофенил]пропан-1,3-диамина (1.14(5)). LC-MS (ESI) (m/z): 325 (М+Н)+.
Раствор соединения 1.14(5) (0,8 г, 2,5 ммоль) в 50 мл этанола гидрировали с 0,13 г 10% Pd на угле в течение 14 ч при 60°С при 40 атм водорода. Смесь фильтровали через целит. К фильтрату добавляли избыток 3М HCl в диоксане и полученный раствор упаривали в вакууме. Получали 0,33 г (67%) N4-{3-[(2-диметиламиноэтил)-метиламино]-пропил}-N4-метил-2-метоксибензол-1,4-диамин виде гидрохлорида 1.14(6)⋅4HCl.
Раствор соединения 1.14(6) (0,51 г, 0,16 ммоль) и соединения 1.1(17) (0,44 г, 0,19 ммоль) в 8 мл 2-метоксиэтанол и 2,0 мл воды перемешивали при 120°С в течение 14 ч и охлаждали до комнатной температуры. Реакционную смесь упаривали в вакууме. Остаток разбавляли 10% водным раствором K2CO3 и экстрагировали CH2Cl2 (5×50 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью обращенно-фазовой ВЭЖХ (градиент MeCN-H2O с 0,1% TFA). Целевые фракции подщелачивали NaHCO3, реакционную массу частично выпаривали в вакууме и продукт экстрагировали дихлорметаном. Объединенные органические экстракты сушили над Na2SO4 и выпаривали в вакууме. Остаток 1.14 разбавляли CH2Cl2 и добавляли избыток 3М HCl в диоксане. Реакционную смесь выпаривали в вакуме и получали N2-[4-({3-[(2-диметиламиноэтил)-метиламино]-пропил}-метиламино)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин тригидрохлорид (1.14⋅3HCl). 1H-NMR, (D2O, 400 МГц): 8.08 (s, 1Н), 7.75-7.55 (m, 3H), 7.53-7.46 (m, 1H), 7.45-7.36 (m, 1H), 7.01 (br.s, 1H), 6.86-6.76 (m, 1H), 3.81 (s, 3H), 3.67-3.53 (m, 6H), 3.32-3.23 (m, 2H), 3.18 (s, 3H), 2.91 (s, 6H), 2.84 (s, 3H), 2.05-1.92 (m, 2H), 1.74 (s, 3H), 1.71 (s, 3H).
Пример 12. N4-[2-(Диметилфосфорил)-фенил]-N2-(4-{метил-[3-(4-метилпиперазин-1-ил)-пропил]-амино}-2-метоксифенил)-5-хлор-пиримидин-2,4-диамин (1.15) был получен в соответствии со схемой 12.
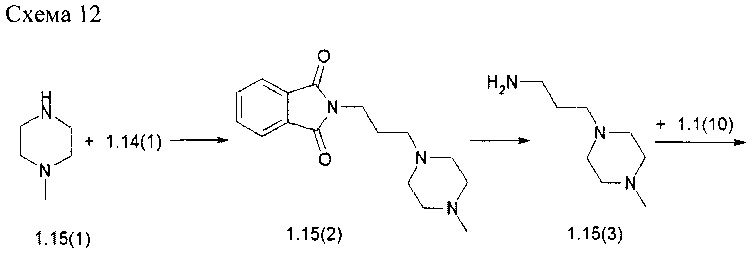
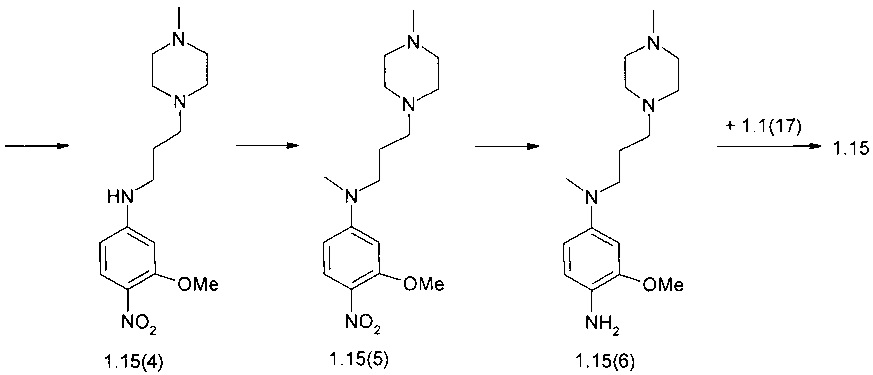
Раствор N-(3-бромпропил)фталимида (1.14(1): 6,5 г, 24.2 ммоль) в 60 мл ксилола добавляли по каплям к раствору 1-метилпиперазина (1.15(1): 5.9 мл, 53.3 ммоль) в 90 мл ксилола при 70°С. Затем смесь нагревали с обратным холодильником в течение 14 ч. Осадок удаляли фильтрованием, а фильтрат концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2:МеОН=30:1). Получали 5,3 г (76%) 2-[3-(4-метилпиперазин-1-ил]пропил]-1Н-изоиндол-1,3(2Н)-диона (1.15(2)). 1H-NMR, (CDCl3, 400 МГц): 7.88-7.80 (m, 2Н), 7.75-7.67 (m, 2Н), 3.80-3.72 (t, J=6.8Гц, 2H), 2.60-2.20 (m, 10Н), 2.19 (s, 3Н), 1.92-1.80 (m, 2H).
К раствору соединения 1.15(2) (5,3 г, 18.4 ммоль) в 400 мл этанола добавляли 51% водный раствор гидразин гидрата (6,0 мл, 92.0 ммоль). Смесь перемешивали с обратным холодильником в течение ночи. Белое твердое вещество отфильтровывали, а фильтрат концентрировали при пониженном давлении. Остаток растворяли в CH2Cl2 (150 мл), твердое вещество отфильтровывали, а фильтрат упаривали в вакууме. Получали 2,9 г (100%) [3-(4-метилпиперазин-1-ил]пропил]амина (1.15(3)), который использовали на следующей стадии без дополнительной очистки. 1H-NMR, (CDCl3, 400 МГц): 7.88-7.80 (m, 2Н), 7.75-7.67 (m, 2Н), 3.80-3.72 (t, J=6.8Гц, 2H), 2.60-2.20 (m, 10Н), 2.19 (s, 3Н), 1.92-1.80 (m, 2H).
К перемешиваемой смеси соединения 1.15(3) (2,9 г, 18.4 ммоль) и DIPEA (8.0 мл, 46.0 ммоль) в ДМФ (40 мл) добавляли соединение 1.1(10) (3,3 г, 19.32 ммоль). Смесь перемешивали в течение 14 ч при 70°С.Реакционную смесь концентрировали в вакууме, а затем очищали с помощью колоночной хроматографии (силикагель, CH2Cl2:МеОН=30:1). Получали 2,9 г, (51%) (3-метокси)-4-нитрофенил)-[3-(4-метилпиперазин-1-ил)пропил]амина (1.15(4)). 1H-NMR, (CDCl3, 400 МГц): 7.96 (d, J=9.2Гц, 1Н), 6.49 (br.s, 1H), 6.09 (dd, J1=9.2Гц, J2=2.2Гц, 1H), 5.99 (d, J=2.2Гц, 1H), 3.91 (s, 3H), 3.34-3.22 (m, 2H), 2.85-2.32 (m, 10H), 2.31 (s, 3H), 1.90-1.76 (m, 2H).
К раствору соединения 1.15(4) (2,8 г 9.1 ммоль) в уксусной кислоте (40 мл) добавляли параформальдегид (2,7 г, 91 ммоль). Смесь перемешивали при комнатной температуре в течение 3 ч. Затем добавляли порциями цианоборгидрид натрия (2,7 г 43.7 ммоль) и перемешивали при комнатной температуре в течение ночи. Полученную смесь выливали на лед, нейтрализовали 20% водным раствором K2CO3 до рН 8,0 и экстрагировали CH2Cl2. Органический слой промывали водой и солевым раствором. После сушки над Na2SO4, фильтровали и выпаривали в вакууме, остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2:МеОН=30:1). Получали 2,6 г (88%) метил-[3-(4-метилпиперазин-1-ил)-пропил]-(3-метокси-4-нитрофенил)-амина 1.15(5). 1H-NMR, (CDCl3, 400 МГц): 8.00 (d, J=9.4Гц, 1H), 6.31 (dd, Jl=9.4Гц, J2=2.6Гц, 1H), 6.08 (d, J=2.6Гц, 1H), 3.95 (s, 3H), 3.54-3.44 (m, 2H), 3.06 (s, 3H), 2.63 2.41 (m, 7H), 2.40-2.34 (m, 3H), 2.33 (s, 3H), 1.86-1.74 (m, 2H).
Раствор соединения 1.15(5) (2,6 г, 8.1 ммоль) в 50 мл этанола гидрировали с 0,43 г 10%) Pd на угле в течение 14 ч при 60°С при 40 атм водорода. Смесь фильтровали через целит. К фильтрату добавляли избыток 3М HCl в диоксане и полученный раствор упаривали в вакууме. Получали 3,3 г (93%) N4-метил-N4-[3-(4-метилпиперазин-1-ил)-пропил]-2-метокси-бензол-1,4-диамина (1.15(6)) в виде тетрагидрохлорида 1.15(6)⋅4HCl, LC MS (ECI): m/z 293 (М+Н)+.
Раствор соединения 1.15(6) (0,60 г, 1,4 ммоль) и соединения 1.1(17) (0,52 г, 1.68 ммоль) в 8 мл 2-метоксиэтанол и 2,0 мл воды перемешивали при 120°С в течение 14 ч, а затем охлаждали до комнатной температуры. Реакционную смесь упаривали в вакууме. Остаток разбавляли 10% водным раствором K2CO3 и экстрагировали CH2Cl2 (5×50 мл). Объединенные органические экстракты сушили над Na2SO4, фильтровали и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии на силикагеле (CH2Cl2:МеОН=20:1). Получали 0,39 г (49%) N4-[2-(диметилфосфорил)-фенил]-N2-(4-{метил-[3-(4-метилпиперазин-1-ил)-пропил]-амино}-2-метоксифенил)-5-хлор-пиримидин-2,4-диамина (1.15). 1H-NMR, (DMSO-d6, 400 МГц): 11.17 (s, 1Н), 8.50 (br.s, 1H), 8.03 (s, 2H), 7.60-7.43 (m, 1H), 7.35-7.17 (m, 2H), 7.10-7.00 (m, 1H), 6.40-6.21 (m, 2H), 3.74 (s, 3Н), 3.47-3.33 (m, 4Н), 2.89 (s, 3Н), 2.42-2.20 (m, 8Н), 2.13 (s, 3Н), 1.77 (s, 3Н), 1.74 (s, 3Н), 7.71-1.60 (m, 2Н).
Пример 13. Способ получения ингибиторов-прототипов АР26113(1), АР26113(2) и их солей: АР26113(1) NDSA, АР26113(1)-3PTSA и АР26113(2) NDSA.
А). Ингибиторы АР26113(1) и АР26113(2) получены по аналогии с протоколом, описанным в патентной заявке США [США 2014/0066406 А1]. N2-[4-(4-диметиламино-пиперидин-1-ил)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин (АР26113(1)): LC-MS (ESI) 530 (М+Н)+. 1Н NMR (DMSO-d6, 400 МГц) δ 11.17 (s, 1Н), 8.48 (br, 1H), 8.06 (s, 1H) 8.04 (s, 1H), 7.52 (m, 1H), 7.39 (d, J=10.4 Гц, 1H), 7.34 (t, J=8 Гц, 1H), 7.09 (t, J=6.8 Гц, 1H), 6.63 (m, 1H), 6.47 (m, 1H), 3.76 (s, 3H), 3.71 (br. d, J=12.4 Гц, 2H), 2.67 (t, 11.2 Гц, 2H) 2.22 (br, 7H) 1.85 (d, J=12.8 Гц, 2H), 1.78 (s, 3H), 1.74 (s, 3H), 1.50 (m, 2H). N4-[2-(диметилфосфорил)-фенил]-N2-{4-[4-(4-метилпиперазин-1-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамин (AP26113(2)): 1H-NMR, (CDCl3, 400 МГц): 10.81 (br.s, 1H), 8.68-8.60 (m, 1H), 8.13-8.06 (m, 2H), 7.55-7.45 (m, 1H), 7.35-7.23 (m, 2H), 7.16-7.08 (m, 1H), 6.56 (d, J=2.3Гц, 1H), 6.50 (dd, J1=8.9Гц, J2=8.9Гц, 1H), 3.87 (s, 3H), 3.67 (d, J=12.2Гц, 2H), 2.80-2.36 (m, 11H), 2.34 (s, 3H), 2.04-1.95 (m, 2H), 1.85 (s, 3H), 1.82 (s, 3H), 1.80-1.66 (m, 2H).
Б). Общий способ получения N2-[4-(4-диметиламино-пиперидин-1-ил)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин нафталин-1,5-дисульфоната (АР26113(1)⋅NDSA) и N2-[4-(4-диметиламино-пиперидин-1-ил)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамин трис-пара-толуолсульфоната (АР26113(1)⋅3PTSA). К раствору (,05 г (0,1 ммоль) N2-[4-(4-диметиламино-пиперидин-1-ил)-2-метоксифенил]-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамина (АР26113(1)) в 4 мл ацетона при 25°С добавляли раствор 0.034 г (0,1 ммоль) нафталин-1,5-дисульфокислоты в 1 мл ацетона или раствор 0.036 г (0,2 ммоль) 4-метилбензолсульфокислоты в мл ацетона. Полученную смесь перемешивали в течение 20 ч. Осадок отфильтровывали и сушили на воздухе до постоянного веса. Получали соответственно ингибитор АР26113(1)⋅NDSA: 1H-NMR (DMSO-d6, 400 МГц): 1.60-1.76 (m, 2H),1.78 (s,3H), 1.81 (s, 3H), 2.02-2.12 (m, 2H), 2.70-2.77 (m, 2H),2.77 (s, 3H), 2.78 (s, 3H), 3.26-3.39 (m, 1H), 3.79 (s, 3H), 3.89 (d, J=12.8Гц, 2H), 6.59 (d, J=8.2, 1H), 6.74 (s, 1H), 7.19-7.27 (m, 1H) 7.33 (d, J=8.1Гц, 1H), 7.41 (dd, J1=7.2, J2=8.6Гц, 3H), 7.58-7.68(m, 1H), 7.94 (d, J=7.2Гц, 2H), 8.21 (s, 1H), 8.43 (br.s, 1H), 8.87 (d, J=8.6Гц, 2H), 9.45 (br.s, 1H), 12.03 (br.s, 1H) или ингибитор AP26113(1)⋅3PTSA: 1H-NMR (DMSO-d6, 400 МГц): 1.66-1.75 (m, 2H), 1.78 (s, 3H), 1.82 (s, 3H), 2.05-2.16 (m, 2H), 2.28 (s, 9H), 2.79 (s, 3H), 2.81 (s, 3H), 2.81-2.90 (m, 2H), 3.31-3.42 (m, 1H), 3.79 (s, 3H), 3.92 (d, 12.5Гц, 2H), 6.62 (d, J=8.7Гц, 1H), 6.76 (br.s, 1H), 7.11 (d, J=8.1Гц, 6H), 7.22-7.30 (m, 1H), 7.33 (d, J=7.7Гц, 1H), 7.39-7.46 (m, 1H), 7.48 (d, J=8.1Гц, 6H), 7.60-7.70 (m, 1H), 8.24 (s, 1H), 8.42 (br.s, 1H), 9.45 (br.s, 1H), 12.15 (br.s, 1H).
В). Способ получения N4-[2-(диметилфосфорил)-фенил]-N2-{4-[4-(4-метилпиперазин-1-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамин нафталин-1,5-дисульфоната (АР26113(2)NDSA). К раствору 0,1 г (0,17 ммоль) N4-[2-(диметилфосфорил)-фенил]-N2-{4-[4-(4-метилпиперазин-1-ил)-пиперидин-1-ил]-2-метоксифенил}-5-хлор-пиримидин-2,4-диамина (АР26113(2)) в смеси 4 мл ацетона и 4 мл этанола при 25°С добавляли раствор 0.062 г (0,17 ммоль) 1,5-нафталин-дисульфокислоты в 1 мл ацетона. Полученную смесь перемешивают в течение 20 ч, осадок отфильтровывали и сушили на воздухе до постоянного веса. Получали ингибитор АР26113(2) NDSA: 'H-NMR (DMSO-d6, 400 МГц): 11.52 (br.s, 1H), 8.9 (d, J=8.4Гц, 2H), 8.80-8.30 (m, 2H), 8.14 (br.s, 1H), 7.98 (d, J=7.1Гц, 2H), 7.64-7.54 (m, 1H), 7.52-7.36 (m, 4H), 7.16 (t, J=7.2Гц, 1H), 6.90-6.50 (m, 2H), 4.00-2.70 (m, 19H), 2.05-1.90 (m, 2H), 1.79 (s, 3H), 1.76 (s, 3H), 1.70-1.55 (m, 2H).
Пример 14. Эссей 1. Вещества тестировали по влиянию на активность киназы ALK с помощью скрининговой платформы Z'-LYTE (Life Technologies). Концентрация ДМСО в реакционной смеси составляла 1%. 100 нл 100-кратных стоков исследуемых веществ в 100% ДМСО разбавляли в 2.4 мкл киназного буфера (50 мМ HEPES рН 7.5, 0.01% BRIJ-35, 10 мМ MgCl2, 1 мМ EGTA) и добавляли к 5 мкл 2-кратной смеси Субстрат/Киназа (ALK/Tyr01, финальные концентрации - 4.24-96 нг ALK и 2 мкМ Tyr01) в 384-луночном планшете (черные, малого объема, производство Corning, кат. #3676). Вещества преинкубировали с киназами в течение 10 мин при комнатной температуре. После этого для начала реакции добавляли 2.5 мкл 4-кратного раствора АТФ (конечная концентрация АТФ в реакционной смеси 25 мкМ). После 30 сек инкубации на шейкере реакцию инкубировали в течение 60 мин при комнатной температуре. После этого добавляли 5 мкл Реагента В (разбавленного 1:256) и инкубировали еще 60 мин при комнатной температуре. Измеряли флуоресценцию при возбуждении длиной волны 400 нм и эмиссии при 445 и 520 нм. Степень фосфорилирования пептидного субстрата рассчитывали по формуле ниже (если отношение эмиссии низкое - пептид фосфорилирован, т.е. нет ингибирования активности киназы, если отношение высокое - пептид не фосфорилирован, т.е. киназа ингибирована)
% фосфорилирования рассчитывается по формуле, указанной на странице 6 в стандартном протоколе скрининга Z'-LYTE (Life Technologies).
Полученные результаты представлены в Таблице 2.
Пример 15.
Эссей 2. Соединения общей формулы (1) тестировали по влиянию на активность киназ EGFR(L858R/T790M) b EGFR wt (все ферменты предоставлены Invitrogen Corp., каталожные номера PV3872, PV4128 and PV4803, соответственно) с помощью скрининговой платформы Z'-LYTE (производство Life Technologies). Концентрация ДМСО в реакционной смеси составляла 1%. 5 мкл 2-кратной смеси Субстрат/Киназа (Tyr4/EGFRwt или EGFR-L858R или EGFR-T790M, конечные концентрации - 0.5 мкМ для субстрата Туr 4 и 1000, 250, 1000 нг/мл для EGFRwt или EGFR-L858R или EGFR-T790M соответственно) добавляли в 384-луночные планшеты (черные, малого объема, производство Corning, кат.#3676). 1.5 мкл 100-кратных стоков исследуемых веществ в 100% ДМСО разбавляли в 10 раз в 13.5 мкл киназного буфера (50 мМ HEPES рН 6.5, 0.01% BRIJ-35, 10 мМ MgCl2, 1 мМ EGTA, 0.02% NaN3) и затем 1 мкл разбавленных веществ добавляли к смеси Субстрат/Киназа. Вещества преинкубировали с киназами в течение 10 мин при комнатной температуре. После этого для начала реакции добавляли 4 мкл 2.5-кратного раствора АТФ (конечная концентрация АТФ в реакционной смеси была 180 или 100 или 40 мкМ для EGFRwt, EGFR-L858R и EGFR-Т790М соответственно). После 30 сек инкубации на шейкере реакцию инкубировали в течение 60 мин при комнатной температуре. После этого добавляли 5 мкл Реагента В (разбавленного 1:500) и инкубировали еще 60 мин при комнатной температуре. Измеряли флуоресценцию при возбуждении длиной волны 400 нм и эмиссии при 445 и 520 нм. Степень фосфорилирования пептидного субстрата рассчитывали по формуле ниже (если отношение эмиссии низкое - пептид фосфорилирован, т.е. нет ингибирования активности киназы, если отношение высокое - пептид не фосфорилирован, т.е. киназа ингибирована)
% фосфорилирования рассчитывается по формуле, указанной на странице 6 в стандартном протоколе скрининга Z'-LYTE (Life Technologies).
Концентрационная кривая зависимости активности киназы от концентрации тестируемых веществ была построена с использованием сигмоидной модели.
Полученные результаты представлены в Таблице 2.
Пример 16.
Определение термодинамической растворимости соединения общей формулы (1) и прототипов АР26113 (CAS # 197958-12-5 и CAS #197953-54-0). Смешивали 5 мг исследуемого соединения с 1 мл универсального буфера (pION) с рН 2,0, 4,0 или 7,0 в течение 15 мин при 25°С. Дополнительные количества веществ добавляли до тех пор, пока раствор не становился мутным. Виалы с раствором инкубировали при перемешивании в течение 24 ч при 25°С для достижения равновесия между раствором и осадком при насыщении. После уравновешивания 200 мкл раствора (в 2-х повторах) фильтровали через 96-луночный фильтровальный планшет (Millipore) для отделения осадка. Концентрацию соединений в фильтрате определяли спектрофотометрически с помощью стандартной калибровочной кривой. Проводили измерение спектра оптического поглощения вещества и построение калибровочной кривой при выбранной длине волны (обычно, соответствующей максимуму поглощения вещества λmax). Концентрацию вещества в фильтрате (т.е. растворимость) рассчитывали по нижеприведенной формуле:
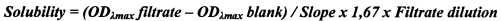 ,
,
где
ODλmax filtrate - оптическая плотность фильтрата,
ODλmax blank - оптическая плотность холостого раствора без вещества,
Slope - наклон калибровочной кривой,
1,67 - фактор разведения фильтрата ацетонитрилом,
Filtrate dilution - фактор разведения фильтрата буфером
Полученные результаты представлены в Таблице 1.
Пример 17. Приготовление лекарственного вещества в виде таблеток. Смешивали, а затем спрессовывали в брусок 1600 мг крахмала, 1600 мг измельченной лактозой, 400 мг талька и 1000 мг соединения 1.8. Полученный брусок измельчали в гранулы и просеивают через сита, собирая гранулы размером 14-16 меш. Полученные гранулы таблетировали в таблетки, пригодных форм весом 500 мг каждая.
Пример 18. Приготовление лекарственного вещества в виде капсул. Соединение 1.8 тщательно смешивали с порошком лактозы в соотношении 2:1. Полученную порошкообразную смесь упаковывали по 600 мг в желатиновые капсулы подходящего размера.
Пример 19. Приготовление лекарственного вещества в виде композиций для внутримышечных, внутрибрюшинных или подкожных инъекций. Смешивали 500 мг соединения 1.8, 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл воды для инъекций. Раствор фильтровали и помещали в 1 мл в ампулы, которые укупоривали.
Специалисты в данной области техники легко поймут, различие некритических параметров, которые могут быть изменены или модифицированы, чтобы достичь тех же результатов, представленных в изобретении. Различные модификации изобретения, в дополнение к описанным здесь, станут очевидными специалистам в данной области техники из приведенного выше описания.
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/3a532451f329c9c590ce871113f73a41.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/fb40de973d68bf0e2e65ab3988921cce.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/08242ef0fbff3ff1d6cf0e2d2acd0130.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/4d29ffa947f214ee04b66dab7c5fbcc0.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/c90d98148e2e075540ac35684a926b01.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/9e3448e50a1446ddc7355d5ebf18c609.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/1c028c086fd5efbfcc3af50936db9249.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/fb3199d99fecc31e9e3d3bb79528dea2.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/793a3562dce8ba208ac9e144f8455d90.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/0bd587326fcbb7405ac0e4610e669abc.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/66128ba54a10da393c04d28ec0ff475d.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/024097f6e943ce3d4747b1d9fed17c5f.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/95b6d97826d4938a4c44a613f000bb3f.jpg)
![Замещенные N2-(4-амино-2-метоксифенил)-N4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов ALK и EGFR, предназначенные для лечения рака](https://fips.edrid.ru/images/rid/77/dd/e6/0ffe0871cf7a58c324a1d0a9e7c92571.jpg)
Замещенные феноксиуксусные кислоты, их эфиры и амиды, включающие 2,6-диоксо-2,3,6,7-тетрагидро-1н-пурин-8-иловый фрагмент, - антагонисты аденозинового a рецептора и их применение
(3-арилсульфонилхинолин-8-ил)-диалкил-амины - селективные антагонисты серотониновых 5-ht рецепторов, способы их получения и применения
Алкил [2-(2-{5-[4-(4-{2-[1-(2-метоксикарбониламино-ацетил)-пирролидин-2-ил]-3н-имидазол-4-ил}-фенил)-бута-1,3-диинил]-1н-имидазол-2-ил}-пирролидин-1-ил)-2-оксо-этил]-карбамат, фармацевтическая композиция, лекарственное средство, способ лечения вирусных заболеваний
Замещенные (r)-3-(4-метилкарбамоил-3-фторфениламино)-тетрагидро-фуран-3-енкарбоновые кислоты и их эфиры, способ их получения и применения
Замещенные (2r,3r,5r)-3-гидрокси-(5-пиримидин-1-ил)тетрагидрофуран-2-илметил арил фосфорамидаты
Замещенные 4-{ [4-(3,3-диоксидо-1,3-бензоксатиол-6-ил)арилокси]метил} пиперидины как агонисты рецепторов gpr119, фармацевтическая композиция, способы их получения и применения
{3-[(7h-пирроло[2,3-d]пиримидин-4-ил)азолил]азетидин-3-ил}ацетонитрилы в качестве ингибиторов янус киназ
Замещенные пиразинопиримидиноны как блокаторы trpa1 каналов, фармацевтическая композиция, способы их получения и применения
Производные 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1h,6h-пиридо[4,3-d]пиримидин-2,4,7-триона в качестве ингибиторов мек1/2
Дихлорацетат n,n-дизамещенного n-[4-(1-метил-1н-индол-3-ил)-пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина в качестве модулятора egfr для лечения рака
Замещенные феноксиуксусные кислоты, их эфиры и амиды, включающие 2,6-диоксо-2,3,6,7-тетрагидро-1н-пурин-8-иловый фрагмент, - антагонисты аденозинового a рецептора и их применение
(3-арилсульфонилхинолин-8-ил)-диалкил-амины - селективные антагонисты серотониновых 5-ht рецепторов, способы их получения и применения
Алкил [2-(2-{5-[4-(4-{2-[1-(2-метоксикарбониламино-ацетил)-пирролидин-2-ил]-3н-имидазол-4-ил}-фенил)-бута-1,3-диинил]-1н-имидазол-2-ил}-пирролидин-1-ил)-2-оксо-этил]-карбамат, фармацевтическая композиция, лекарственное средство, способ лечения вирусных заболеваний
Замещенные (r)-3-(4-метилкарбамоил-3-фторфениламино)-тетрагидро-фуран-3-енкарбоновые кислоты и их эфиры, способ их получения и применения
Замещенные (2r,3r,5r)-3-гидрокси-(5-пиримидин-1-ил)тетрагидрофуран-2-илметил арил фосфорамидаты
Замещенные 4-{ [4-(3,3-диоксидо-1,3-бензоксатиол-6-ил)арилокси]метил} пиперидины как агонисты рецепторов gpr119, фармацевтическая композиция, способы их получения и применения
{3-[(7h-пирроло[2,3-d]пиримидин-4-ил)азолил]азетидин-3-ил}ацетонитрилы в качестве ингибиторов янус киназ
Замещенные пиразинопиримидиноны как блокаторы trpa1 каналов, фармацевтическая композиция, способы их получения и применения
Производные 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1h,6h-пиридо[4,3-d]пиримидин-2,4,7-триона в качестве ингибиторов мек1/2
Дихлорацетат n,n-дизамещенного n-[4-(1-метил-1н-индол-3-ил)-пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина в качестве модулятора egfr для лечения рака

