Результат интеллектуальной деятельности: {3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ
Вид РИД
Изобретение
Настоящее изобретение относится к {3-[(7H-пирроло[2,3-d]пиримидин-4-ил)-азолил]азетидин-3-ил}-ацетонитрилам, а также их композициям, способам применения и получения. Эти соединения и их композиции являются ингибиторами Янус киназ (JAK) и могут использоваться при лечении JAK ассоциированных заболеваний, включая, например, воспалительные и аутоиммунные расстройства, а также рак.
Онкогенные протеинкиназы представляют собой одну из самых крупных и наиболее привлекательных групп белковых мишеней для разработки лекарственных средств. JAK играют важную роль в цитокин-зависимой регуляции пролиферации и функции клеток, участвующих в иммунной реакции. В настоящее время известны четыре JAK: JAK1, JAK2, JAK3 (также известная как Янус-киназа лейкоцитов; JAKL; L-JAK) и TYK2 (также известна как тирозинкиназа T).
Блокировка передачи сигнала на уровне JAK перспективна для разработки методов лечения воспалительных заболеваний, аутоиммунных заболеваний, миелопролиферативных и раковых заболеваний человека. Предполагается, что ингибирование JAK имеет терапевтический эффект у пациентов, страдающих иммунными расстройствами кожи, такими как псориаз и сенсибилизация кожи. Эффективные ингибиторы JAK представляют безусловный интерес для разработки новых лекарственных средств. Некоторые JAK ингибиторы, в том числе пирролопиримидины, представлены в WO 2009/114512. В этой серии соединений Барицитиниб (Baricitinib) является самым передовым лекарственным кандидатом. Барицитиниб показал быструю (через 6 месяцев лечения) и устойчивую эффективность при лечении ревматоидного артрита в трех исследованиях III фазы [NCT02340104, NCT02263911, NCT01710358. http://www.medpagetoday.com/MeetingCoverage/EULAR/52084. http://www.fiercepharma.com/press-releases/lilly-and-incyte-announce-positive-top-line-results-phase-3-trial-baricitin].
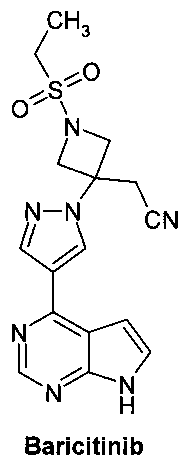
Таким образом, поиск новых улучшенных JAK ингибиторов является одним из основных направлений для разработки новых, более эффективных лекарственных препаратов для лечения ревматоидного артрита, рака и других заболеваний. Соединения и способы, описанные здесь, направлены на удовлетворение этих потребностей.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» (C1-C6)алкильных заместителей. Предпочтительными алкильными группами являются (C1-C6)алкил, еще более предпочтительными (C1-C3)алкил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклопропилметил, циклобутилметил, циклопентилметил, н-пентил, 2-пентил, 3-пентил, нео-пентил, н-гексил, циклогексил. Алкил может иметь заместители.
«Замещенный алкил» - замещенный алкил может иметь один или несколько одинаковых или различных заместителей, включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, гетероароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонил, гетероаралкилокси или Rk aRk+1 aN-, где Rk a и Rk+1 a независимо друг от друга представляют собой «заместители аминогруппы», значение которых определено в данном разделе, например, атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rk a и Rk+1 a вместе с атомом N, с которым они связаны, образуют через Rk a и Rk+1 a 4-7-членный гетероциклил или гетероцикленил. Предпочтительными «алкильными заместителями» являются арил, гетероарил, гетероциклил, гидрокси, C1-C5 алкокси, C1-C5 алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или Rk aRk+1 aN-, Rk aRk+1 aNC(=O)-, аннелированный арилгетероцикленил, аннелированный арилгетероциклил.
«Аминогруппа» означает R1R2N-группу, замещенную или незамещенную необязательно одинаковыми заместителями R1 и R2. Аминогруппа может иметь заместители.
«Активный компонент» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Галоген» означает фтор, хлор, бром и йод. Предпочтительными являются фтор, хлор и бром.
«Метин» представляет собой группу из одного атома углерода и одного атома водорода CH.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и других готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» обозначает композицию, включающую в себя активный компонент и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлимых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные такие как мази и кремы, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, дихлорацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей можно найти в справочнике фармацевтических солей [P.H. Stahl, C.G. Wermuth (Eds.). Handbook of Pharmaceutical Salts, Properties, Selection, and Use. VHCA, Verlag Helvetica Chimica Acta,  , Switzerland, and Wiley-VCH, Weinheim, Germany. 2002.]. Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
, Switzerland, and Wiley-VCH, Weinheim, Germany. 2002.]. Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Цель настоящего изобретения заключается в создании новых ингибиторов JAK для лечения ревматоидного артрита, рака и других заболеваний.
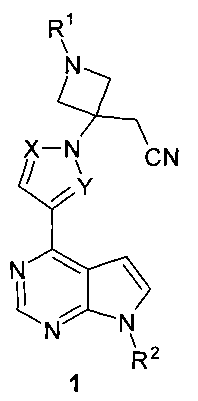
Поставленная цель достигается {3-[(7H-пирроло[2,3-d]пиримидин-4-ил)-азолил]азетидин-3-ил}-ацетонитрилами общей формулы 1:
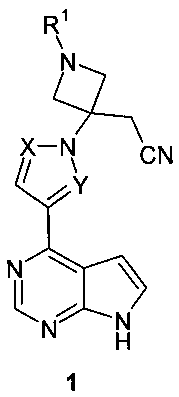
или их фармацевтически приемлемыми солями, где:
R1 представляет собой SO2(CH2)nCH2OH, SO2(CH2)nCH2F, SO2(CH2)nCHF2, SO2(CH2)nCF3, SO2(CH2)nCO2R1a, SO2(CH2)nCO2H, CH2CH2SO2CH3, CH2CH2SO3H, CH2CH2SO2NH2; n=1 или 2.
R1a представляет собой алкил или циклоалкил, предпочтительно C1-С3алкил;
X и Y представляют собой атом азота; или X представляет собой атом азота, a Y представляет собой метин; или X представляет собой метин, a Y представляет собой атом азота;
или R1 представляет собой SO2CH2CH3; X и Y представляют собой атом азота, или X представляет собой метин, a Y представляет собой атом азота.
Предметом настоящего изобретения является новый JAK ингибитор общей формулы 1.
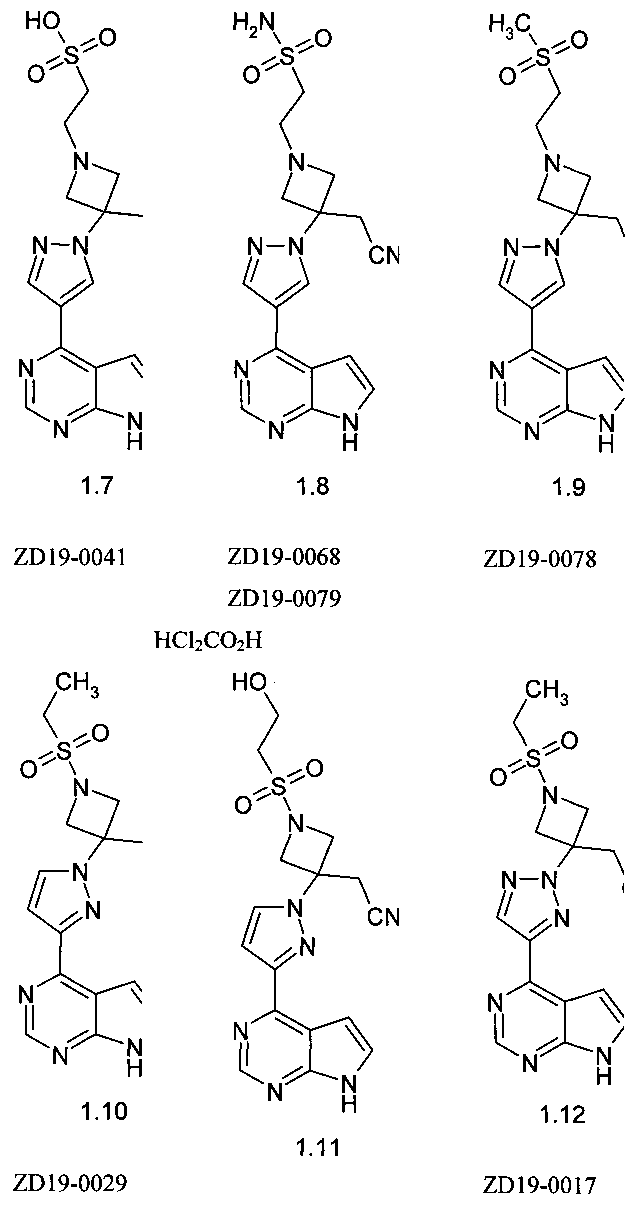
Настоящее изобретение относится, в частности, к JAK ингибитору выбранному из: {1-(2-гидрокси-этансульфонил)-3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-азетидин-3-ил}-ацетонитрила (1.1);
{3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-(2-фтор-этансульфонил)-азетидин-3-ил}-ацетонитрила (1.2);
{3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-(2,2-дифтор-этансульфонил)-азетидин-3-ил}-ацетонитрила (1.3);
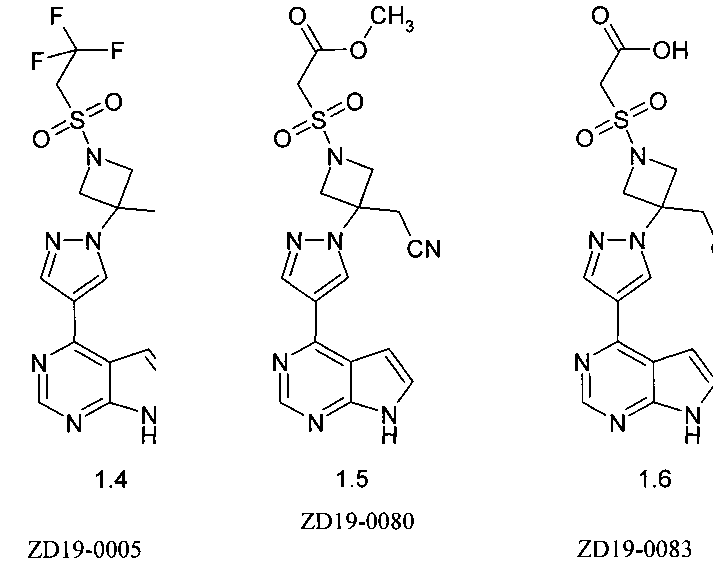
[3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-(2,2,2-трифтор-этансульфонил)-азетидин-3-ил]-дифторэтансульфонил (1.4);
метилового эфира {3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил}уксусной кислоты (1.5);
{3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил}уксусной кислоты (1.6);
2-{3-[4(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]-3-цианометил-азетидин-1-ил}-этансульфокислоты (1.7);
амида 2-{3[4(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]-3-цианометил-азетидин-1-ил}-этансульфокислоты (1.8);
{1-(2-метансульфонил-этил)-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]-азетидин-3-ил}-ацетонитрила (1.9);
{3-[3-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]-1-этансульфонил-азетидин-3-ил}-ацетонитрила (1.10);
{1-(2-гидрокси-этансульфонил)-3-[3-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]-азетидин-3-ил}-ацетонитрила (1.11);
{3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-[1,2,3]триазол-2-ил]-1-этансульфонил-азетидин-3-ил}-ацетонитрила (1.12);
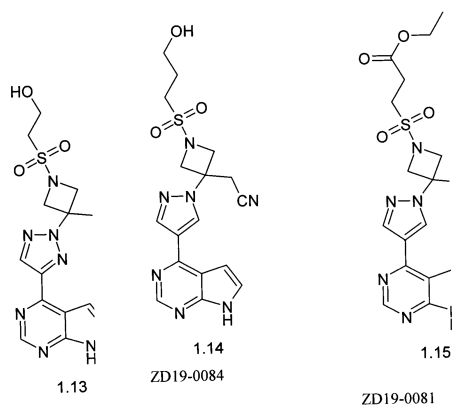
{1-(2-гидрокси-этансульфонил)-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил)-[1,2,3]триазол-2-ил]-азетидин-3-ил}-ацетонитрила (1.13);
{1-(3-гидрокси-пропан-1-сульфонил)-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил)]-азетидин-3-ил}-ацетонитрила (1.14);
метилового эфира {3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил}пропионовой кислоты (1.15);
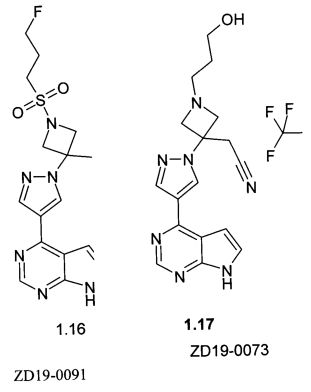
{3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-(3-фторпропансульфонил)-азетидин-3-ил}-ацетонитрила (1.16)
{1-(3-Гидроксипропил)-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]-азетидин-3-ил}ацетонитрила трифторацетата (1.17)
или их фармацевтически приемлемых солей.

Предметом настоящего изобретения являются также гидраты и сольваты соединений общей формулы 1 по настоящему изобретению, а также безводные и несольватированные формы.
Термин "соединение", как он использован здесь, означает все стереоизомеры, геометрические изомеры, таутомеры и изотопы соединений общей формулы 1. Все соединения и фармацевтические приемлемые соли могут быть выделены вместе с другими веществами, такими как вода и растворители (например гидратами и сольватами).
Соединения по настоящему изобретению могут также включать все изотопы атомов, присутствующих в промежуточных или конечных соединений. Изотопы включают атомы, имеющие одинаковый атомный номер, но разные массовые числа. Например, изотопы водорода включают тритий и дейтерий.
Фраза "фармацевтически приемлемый" используется здесь для обозначения соединений, материалов, композиций и/или лекарственных форм, которые в пределах погрешности медицинской оценки пригодны для использования в контакте с тканями человека и животных без чрезмерной токсичности, раздражения, аллергической реакции или другой проблемы или осложнения, соразмерных с разумным соотношением польза/риск.
Настоящее изобретение также включает фармацевтически приемлемые соли соединений, описанных здесь. Примеры фармацевтически приемлемых солей включают, но не ограничиваются ими, соли минеральных или органических кислот; щелочные или органические соли кислотных остатков соединений формулы 1, таких как карбоновые или сульфокислоты. Фармацевтически приемлемые соли по настоящему изобретению включают обычные нетоксичные соли исходного соединения, образованные, например, из нетоксичных неорганических или органических кислот. Фармацевтически приемлемые соли по настоящему изобретению могут быть синтезированы из исходного соединения, которое содержит основную или кислотную группу, обычными химическими способами. Как правило, такие соли могут быть получены взаимодействием свободных кислотных или основных форм указанных соединений со стехиометрическим количеством соответствующего основания или кислоты в воде или в органическом растворителе или в смеси двух. Как правило, предпочтительны неводные среды, такие как эфир, этилацетат, этанол, изопропанол, ацетон или ацетонитрил (ACN). Списки подходящих солей можно найти в справочнике фармацевтических солей [P.H. Stahl, C.G. Wermuth (Eds.). Handbook of Pharmaceutical Salts, Properties, Selection, and Use. VHCA, Verlag Helvetica Chimica Acta, , Switzerland, and Wiley-VCH, Weinheim, Germany. 2002.].
Соединения по настоящему изобретению, включая их соли, могут быть получены с использованием известных методов органического синтеза и могут быть синтезированы в соответствии с любым из многочисленных возможных путей синтеза. Реакция для получения соединений по изобретению может быть осуществлена в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Подходящие растворители могут по существу реагировать с исходными материалами (реагентами), промежуточными продуктами при температурах проведения реакции, например, температура которой может варьироваться от температуры замерзания растворителя до температуры кипения растворителя. Данная реакция может быть осуществлена в одном растворителе или в смеси растворителей. В зависимости от конкретной стадии реакции, подходящие растворители для конкретной стадии реакции могут быть выбраны специалистом в данной области.
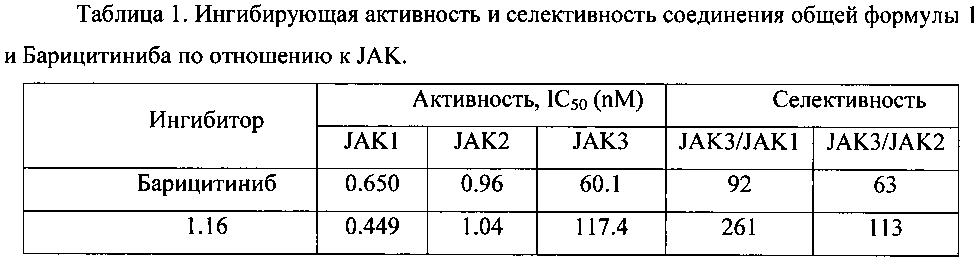
Ингибирующая активность и селективность соединений общей формулы 1 по отношению к JAK, как правило, сопоставимы с активностью и селективностью Барицитиниба, который в настоящее время проходит фазу III клинических испытаний [NCT02340104; NCT02263911; NCT01710358; J.D. Clark, M.E. Flanagan, J.-B. Telliez. Discovery and Development of Janus Kinase (JAK) Inhibitors for Inflammatory Diseases. J. Med. Chem. 2014, 57, 5023-5038]. Однако некоторые ингибиторы имеют более высокую селективность взаимодействия с JAK. Так, например, ингибитор 1.16 менее активен по отношению к JAK3 (IC50=117 nM), чем Барицитиниб (IC50=92 nM). В результате селективность JAK3/JAK1=261 и селективность JAK3/JAK2=113 ингибитора 1.16 в два раза выше селективности Барицитиниба (Таблица 1).
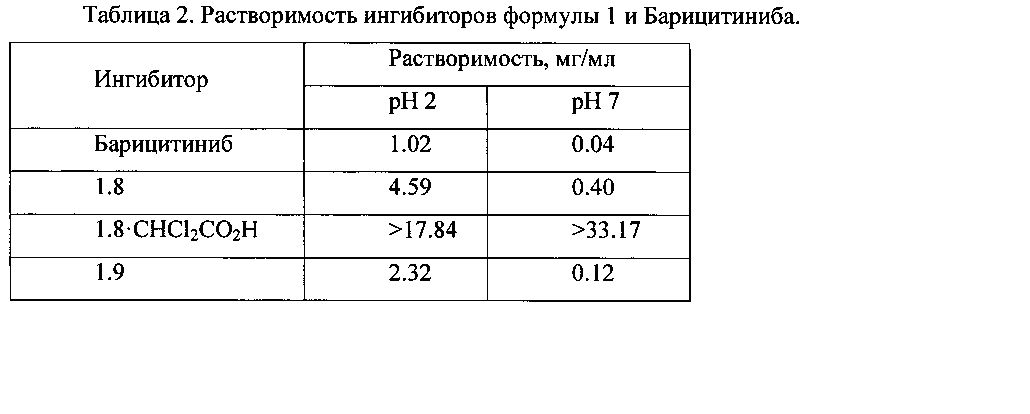
Преимуществом новых соединений общей формулы 1 является и значительно более высокая их растворимость в водных растворах, чем у Барицитиниба (Таблица 2). Так, например, растворимость ингибитора 1.8·CHCl2CO2H в воде при pH7 (>33.17 мг/мл) более чем в 829 раз, а при pH2 (>17.84 мг/мл) более чем в 17 раз выше растворимости Барицитиниба.
Способ получения соединений согласно изобретению может включать в себя защитную группу и ее снятие. Необходимость защитной группы и ее снятия, и выбор соответствующих защитных групп, могут быть легко определены специалистом в данной области. Химию защитных групп можно найти, например, в книге Т.W. Greene and P.G.M. Wuts, Protective Groups in Organic Synthesis, 3rd Ed., Wiley & Sons, Inc., New York (1999), которая включена в настоящее описание посредством ссылки во всем ее объеме.
Настоящее изобретение относится к способу получения соединения общей формулы 1, который заключается в обработке соединения формулы 1a, приводящей к удалению защитной группы R2 (схема 1).
где:
R1 представляет собой SO2(CH2)nCH2OH, SO2(CH2)nCH2F, SO2(CH2)nCHF2, SO2(CH2)nCF3, SO2(CH2)nCO2Rla, SO2(CH2)nCO2H, CH2CH2SO2CH3, CH2CH2SO3H, CH2CH2CH2OH, CH2CH2SO2NH2; n=1 или 2;
Rla представляет собой алкил или циклоалкил, предпочтительно С1-С3алкил;
X и Y представляют собой атом азота; или X представляет собой атом азота, a Y представляет собой метин; или X представляет собой метин, a Y представляет собой атом азота;
или R1 представляет собой SO2CH2CH3; X и Y представляют собой атом азота, или X представляет собой метин, a Y представляет собой атом азота;
R2 обозначает защитные группы, которые включают в себя, но не ограничиваются защитными группами для аминов, представленными в книге [Wuts and Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, 696-887, 2007], которая включена в настоящее описание посредством ссылки во всей своей полноте.
В некоторых вариантах изобретения защитная группа R2 является стабильной в условиях удаления защитных групп R3 и R4 (схема 1 и схема 2). В некоторых вариантах осуществления изобретения защитная группа R2 представляет собой группу, которая не удаляется от 1N до 5N соляной кислотой при комнатной температуре, при температуре от около 10°С до 40°С, при температуре от около 15°С до 40°С или при температуре от 15°С до 30°С. В некоторых вариантах осуществления изобретения R2 представляет собой бензилоксикарбонил (Cbz), 2,2,2-трихлорэтоксикарбонил (Troc), 2-(триметилсилил)этоксикарбонил (Teoc), 2-(4-трифторметилфенилсульфонил)этоксикарбонил (TSC), трет-бутоксикарбонил (BOC), 1-адамантилоксикарбонил (Adoc), 2-адамантилкарбонил (2-Adoc), 2,4-диметилпент-3-илоксикарбонил (Doc), циклогексилоксикарбонил (Специальный), L, L-диметил-2,2,2-трихлорэтоксикарбонил (TcBOC), винил, 2-хлорэтил, 2-фенилсульфонилэтил, аллил, бензил, 2-нитробензил, 4-нитробензил, 4-дифенил-пиридилметил, N′,N′-диметилгидразинил, метоксиметил, трет-бутоксиметил (Bum), бензилоксиметил (BOM), или 2-тетрагидропиранил (THP). гидроксиметил (HM). В некоторых вариантах осуществления R2 представляет собой 2-(триметилсилил)этоксиметил (SEM). В некоторых вариантах осуществления R2 представляет собой N-пивалоилоксиметил (полиоксиметилен) или гидроксиметил (HM).
Для удаления защитной группы R2 у соединения формулы 1а могут быть использованы способы, известные в данной области для удаления конкретных защитных групп аминов [Wuts and Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, 696-887, 2007]. Например, в некоторых вариантах изобретения защитная группа R2 может быть удалена обработкой фторид-ионом (например, обработкой фторидом тетрабутиламмония), соляной кислотой или кислотой Льюиса (например, литий тетрафторборатом). В некоторых вариантах изобретения обработка включает обработку тетрафторборатом лития с последующей обработкой гидроксидом аммония (например, когда R2 обозначает 2-(триметилсилил)-этоксиметил)). Полученное соответствующее гидроксиметил-промежуточное затем обрабатывают водным раствором гидроксида аммония при комнатной температуре с получением соединения общей формулы 1.
Схема 1
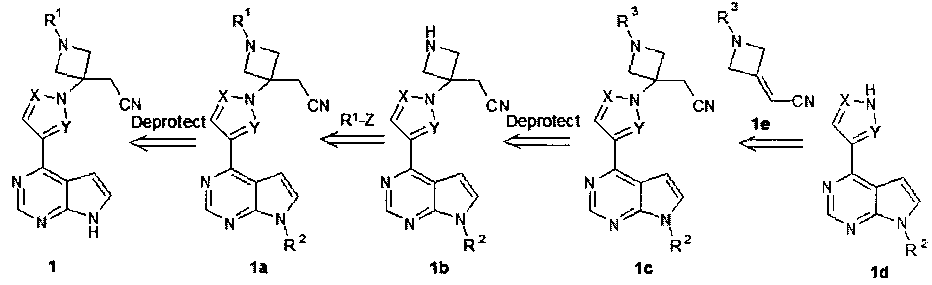
Схема 2
Соединение формулы 1a может быть получено взаимодействием соединения 1b и соединения формулы R1-Z (Схема 1). Z в соединении R1-Z представляет собой уходящую группу. В некоторых вариантах изобретения уходящая группа Z представляет собой общеизвестную в данной области группу, например галоген. Более предпочтительной уходящей группой Z является хлор или фтор.
Соединение формулы 1b может быть получено удалением защитной группы R3 у соединения 1c (Схема 1).
Защитные группы R3 включают, но не ограничиваются защитными группами для аминов, представленными в книге [Wuts and Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, 696-887, 2007], которая включена в настоящее описание посредством ссылки во всей своей полноте. R3 представляет собой защитную группу, которая может быть селективно удалена в условиях, которые не вытесняют защитную группу R2. В некоторых вариантах изобретения R3 представляет собой защитную группу, которую можно удалить в кислых средах при комнатной температуре, при температуре от 15°C до 40°C или при температуре от 15°C до приблизительно 30°C. В некоторых вариантах осуществления изобретения R3 представляет собой алкоксикарбонил, например, трет-бутоксикарбонил. Для удаления группы R3 обработкой соединения формулы 1c используют общеизвестные способы, например, представленные в книге [Wuts and Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, 696-887, 2007], которая включена в настоящее описание посредством ссылки во всей своей полноте. Подходящие условия обработки не вытесняют защитную группу R2. В некоторых вариантах осуществления изобретения обработка соединения формулы 1c осуществляется в кислых средах при комнатной температуре, при температуре от 15°C до 40°C или при температуре от 15°C до приблизительно 30°C. В некоторых вариантах осуществления изобретения обработка соединения формулы 1c включает обработку хлористоводородной кислотой в 1,4-диоксане.
На схеме 1 соединение формулы 1a получают по способу, включающему взаимодействие соединения формулы 1d и соединения формулы 1e.
Реакции присоединения Михаэля между соединением формулы 1d и соединением формулы 1e (схема 1) может проводиться в присутствии основания (катализатора Михаэля). В некоторых вариантах осуществления изобретения в качестве катализатора Михаэля используют галогениды тетраалкиламмония, гидроксид тетраалкиламмония, гуанидин, амидин, гидроксид или алкоксид щелочного металла, фосфат щелочного металла, третичный амин, карбонат щелочного металла, бикарбонат щелочного металла, гидрофосфат щелочного металла, фосфин или соль щелочного металла карбоновой кислоты и др. В некоторых вариантах осуществления изобретения в качестве катализатора Михаэля используют тетраметилгуанидин, 1,8-диазабицикло(5.4.0)ундец-7-ен, 1,5-диазабицикло(4.3.0)нон-5-ен, 1,4-диазабицикло-(2.2.2)октан, трет-бутил гидроксид аммония, гидроксид натрия, гидроксид калия, метоксид натрия, этоксид натрия, трикалийфосфат, силикат натрия, оксид кальция, триэтиламин, карбонат натрия, карбонат калия, бикарбонат натрия, бикарбонат калия, гидрофосфат калия, трифенилфосфин, триэтилфосфин, ацетат калия, или акрилат калия. В некоторых вариантах осуществления изобретения используют в качестве катализатора Михаэля 1,8-диазабицикло(5.4.0)ундец-7-ен (DBU). В некоторых вариантах осуществления изобретения используют стехиометрическое или каталитическое количество основания для облегчения реакции присоединения Михаэля. В некоторых вариантах осуществления изобретения реакцию проводят в органическом растворителе, таком как ацетонитрил или диметилацетамид, при комнатной температуре в течение от двух до шести часов.
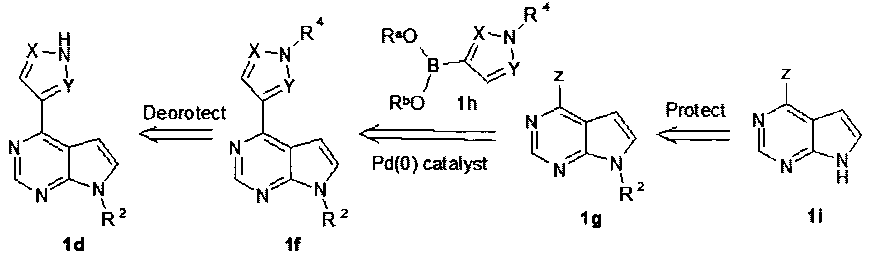
Соединение формулы 1f получают посредством способа, включающего обработку соединения формулы 1d (схема 2).
Если у соединения 1f удалить группу R4, образуется соединение формулы 1d. R4 представляет собой защитную группу. Соответствующие R4 группы включают, но не ограничиваются защитными группами для аминов, представленными в книге [Wuts and Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, 696-887, 2007], которая включена в настоящее описание посредством ссылки во всей своей полноте. R4 представляет собой защитную группу, которая может быть селективно удалена в условиях, которые не вытесняют защитную группу R2. В некоторых вариантах осуществления R4 представляет собой защитную группу, которую можно удалить кислотой при комнатной температуре, при температуре от 15°C до 40°C или при температуре от 15°C до 30°C. В некоторых вариантах осуществления изобретения R4 представляет собой 1-(этокси)этил, три(C1-С6алкил)силил (например, трет-бутилдиметилсилил или триизопропилсилил), п-метоксибензил (PMB), трифенилметил (Tr), дифенилметил, гидроксиметил, метоксиметил (MOM), диэтоксиметил, или трет-бутилдиметилсилил. В некоторых вариантах осуществления изобретения R4 представляет собой L-(этокси)этил.
Удаление у соединения формулы 1f защитной группы R4 может быть достигнуто способами, известными в данной области для удаления конкретных защитных групп аминов, представленными в книге [Wuts and Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, 696-887, 2007], которая включена в настоящее описание посредством ссылки во всей своей полноте. В некоторых вариантах осуществления изобретения обработку соединения формулы 1f проводят кислотой (например, соляной кислотой или трифторуксусной кислотой) при комнатной температуре, при температуре от 15°C до 40°C или при температуре от 15°C до 30°C. Соединение формулы 1f получено с использованием способа, включающего взаимодействие соединения формулы 1g с соединением формулы 1h (Схема 2).
Соединения формулы 1f получают реакцией Сузуки из соединений формулы 1g и 1h в присутствии палладиевого катализатора и основания (Схема 2), где Z представляет тозилат группу, трифлат группу, йод, хлор, бром, a Ra и Rb в соединении 1h независимо друг от друга представляют собой водород или алкил; или Ra и Rb вместе с атомами кислорода, к которым они присоединены, и атомом бора образуют 5- или 6-членное гетероциклическое кольцо, которое необязательно замещено одной, двумя, тремя или четырьмя C1-C4 алкильными группами.
Реакцию Сузуки можно проводить с использованием ряда катализаторов Pd(0) и Pd(II) в известных в данной области условиях, (смотреть, например, обзор Miyaura and Suzuki, Chem. Rev. 1995, 95, 2457-2483, который включен в описание во всей своей полноте). В некоторых вариантах используют в качестве палладиевого катализатора Pd(DPPF)2Cl2 или тетракис(трифенилфосфин)палладий(0) или тетракис(три(о-толил)фосфин)палладий(0), или тетракис(трифенилфосфин)палладий (0).
В некоторых вариантах осуществления изобретения используют примерно от 0,0010 до примерно 0,0015 эквивалентов палладиевого катализатора. В некоторых вариантах осуществления изобретения используют приблизительно стехиометрическое соотношение соединений формулы 1g и 1h, например от приблизительно 1 до приблизительно 1,05, или приблизительно от 1 до приблизительно 1,35. В некоторых вариантах для этой стадии синтеза используют органический растворитель, содержащий воду. В качестве органического растворителя используют 1,4-диоксан, 1-бутанол, 1,2-диметоксиэтан (DME), 2-пропанол, толуол или этанол, или их комбинации. В некоторых вариантах осуществления изобретения органический растворитель содержит комбинацию 1-бутанола и простого диметилового эфира.
В некоторых вариантах осуществления изобретения основание представляет собой неорганическое основание, а в некоторых - органическое основание. В некоторых вариантах основание представляет собой карбонат щелочного металла или гидрокарбонат щелочного металла. В некоторых вариантах осуществления изобретения основанием является карбонат калия (K2CO3). В некоторых вариантах осуществления изобретения используют от двух до пяти эквивалентов основания (например, K2CO3) или от двух до пяти эквивалентов основания (например, NaHCO3).
В некоторых вариантах осуществления изобретения реакцию Сузуки проводят при температуре примерно от 80°C до 100°C. В некоторых вариантах осуществления изобретения реакцию проводят в течение от двух до двенадцати часов.
В некоторых вариантах осуществления изобретения, Z в соединении формулы 1g представляет собой хлор, бром или йод.
Соединение формулы 1g (Схема 2) получают из соединения формулы 1i введением защитной группы R2, которая описана выше, предпочтительно (триметилсилил)этоксиметил или N-пивалоилоксиметил.
Введение защитной группы R2 проводят с помощью известных в области химии защитных групп для аминов [Wuts and Greene, Protective Groups in Organic Synthesis, 4th ed., John Wiley & Sons: New Jersey, 696-887, 2007]. Например, азот индола депротонируют основанием (например, гидридом натрия) в органическом растворителе (например, ТГФ, 1,4-диоксан, 1,2-диметоксиэтан (DME), или N,N-диметилацетамид (ДМАЦ)) при низкой температуре (например, от 0°C до 5°C), а затем обрабатывают хлоридом триметилсилилэтоксиметил (SEM-Cl) или пивалоилоксиметилхлоридом (ПОМ-Cl). SEM- или ПОМ-защищенные 4-хлор-7H-пирроло[2,3-d]пиримидин (1i:Z=Cl) затем выделяют и используют в последующей реакции Сузуки 1i с или без дальнейшей очистки.
Способ получения соединения формулы 1 включает стадию взаимодействия соединения формулы 1j с соединением формулы R1-Z; где Z представляет собой уходящую группу. В некоторых вариантах изобретения Z представляет собой галоген, в том числе хлор или фтор.
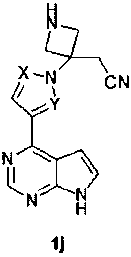
Реакцию соединения формулы 1j с соединением формулы R1-Z проводят в присутствии основания, например, третичного амина, такого как триэтиламин, диизопропилэтиламин, N-метилморфолин и тому подобного.
Соединение формулы 1j получают удалением защитной группы R2 у соединения формулы 1b (Схема 1).
Представленные выше методы синтеза могут быть использованы для получения любого из описанных здесь соединений или их комбинаций или любого из соединений, описанных в примерах. Настоящее изобретение относится к любой комбинации отдельных методов для получения соединения общей формулы 1, соединения формулы 1a и т.д.
Настоящее изобретение дополнительно относится к любому из промежуточных соединений, описанных выше, или их солей.
Способ получения солей соединений общей формулы 1 включает взаимодействие соединения формулы 1a с хлористоводородной кислотой, дихлоруксусной кислотой, метансульфоновой кислотой, фосфорной кислотой, бензолсульфоновой кислотой (PhSO3H), пара-толуолсульфоновой кислотой (n-CH3PhSO3H) или с 1,5-нафталин дисульфокислотой (1,5-NDSA) с образованием соответствующей соли. Соли соединения общей формулы 1 могут быть получены путем обработки раствора соответствующего свободного основания формулы 1a в органическом растворителе, таком как этанол (EtOH), раствором кислоты в органическом растворителе, таком как этанол, при комнатной температуре или при повышенной температуре (например, от 60°C до 70°C). Полученная соль в случае необходимости может быть дополнительно очищена перекристаллизацией или другими общеизвестными методами, например переосаждением или промывкой.
Соединения общей формулы 1 и их соли могут быть получены в соответствии с синтетическими протоколами, описанными ниже в разделе примеров.
Соединения по настоящему изобретению могут модулировать активность одной или более JAK. Термин "модулировать" означает способность увеличивать или уменьшать активность одного или нескольких членов семьи JAK. Соответственно, соединения по изобретению могут быть использованы в способах модуляции активности JAK любым одним или более из соединений общей формулы 1 или их композиций.
В соответствии с настоящим изобретением соединения общей формулы 1 могут действовать в качестве ингибиторов одной или нескольких JAK.
В соответствии с настоящим изобретением соединения общей формулы 1 могут быть использованы для модуляции активности JAK у пациента, который нуждается в такой модуляции рецептора, путем введения модулирующего количества соединения общей формулы 1.
Предметом настоящего изобретения является способ лечения заболеваний или расстройств у пациентов, связанных с JAK, путем введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения по настоящему изобретению или его фармацевтической композиции.
JAK-связанное заболевание может включать в себя любое заболевание, расстройство или состояние, непосредственно или косвенно связанные с экспрессией или активностью в JAK, в том числе гиперэкспрессией и/или аномальным уровнем активности. JAK-сопутствующее заболевание может также включать любое заболевание, расстройство или состояние, которое может быть предотвращено, облегчено или вылечено путем модуляции JAK активности.
Предметом настоящего изобретения является способ лечения JAK-ассоциированных аутоиммунных заболеваний, таких как рассеянный склероз, ревматоидный артрит, ювенильный артрит, псориатический артрит, диабет типа I, волчанка, псориаз, воспалительное заболевание кишечника, язвенный колит, болезнь Крона, миастения, нефропатия иммуноглобулинов, аутоиммунные заболевания щитовидной железы и тому подобное.
В соответствии с настоящим изобретением соединения общей формулы 1 могут быть введены пациенту в виде фармацевтических композиций. Эти композиции могут быть получены способом, хорошо известным в фармацевтической области, и могут быть введены различными способами в зависимости от того, требуется местное или системное лечение и на площади, подлежащей обработке. Композиции могут быть в форме таблеток, пилюль, порошков, лепешек, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как твердое вещество или в жидкой среде), мазей, содержащих, например, до 10% по весу активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных инъекционных растворов и стерильно упакованных порошков. Композиции могут быть приготовлены в виде единичной дозы, причем каждая доза содержит от примерно 5 мг до примерно 1000 мг, обычно примерно от 100 мг до примерно 500 мг, активного ингредиента. Термин "единичные дозы" относится к физически дискретным единицам, пригодным в качестве единичных доз для человека и других млекопитающих, причем каждая единица содержит заданное количество активного материала, рассчитанное на получение желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим наполнителем.
Активное соединение может быть эффективным в широком диапазоне доз и обычно вводится в фармацевтически эффективном количестве. Следует понимать, однако, что количество фактически принимаемого соединения обычно будет определяться лечащим врачом в соответствии с состоянием пациента, подлежащего лечению.
Ниже изобретение будет описано более подробно с помощью конкретных примеров, которые представлены с целью иллюстрации и не предназначены для ограничения изобретения каким-либо образом. Специалисты в данной области техники легко поймут различие некритических параметров, которые могут быть изменены или модифицированы, чтобы получить те же результаты.
Пример 1. {1-(2-Гидрокси-этансульфонил)-3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-азетидин-3-ил}-ацетонитрил (1.1) получали в соответствии со схемой 3.
Схема 3
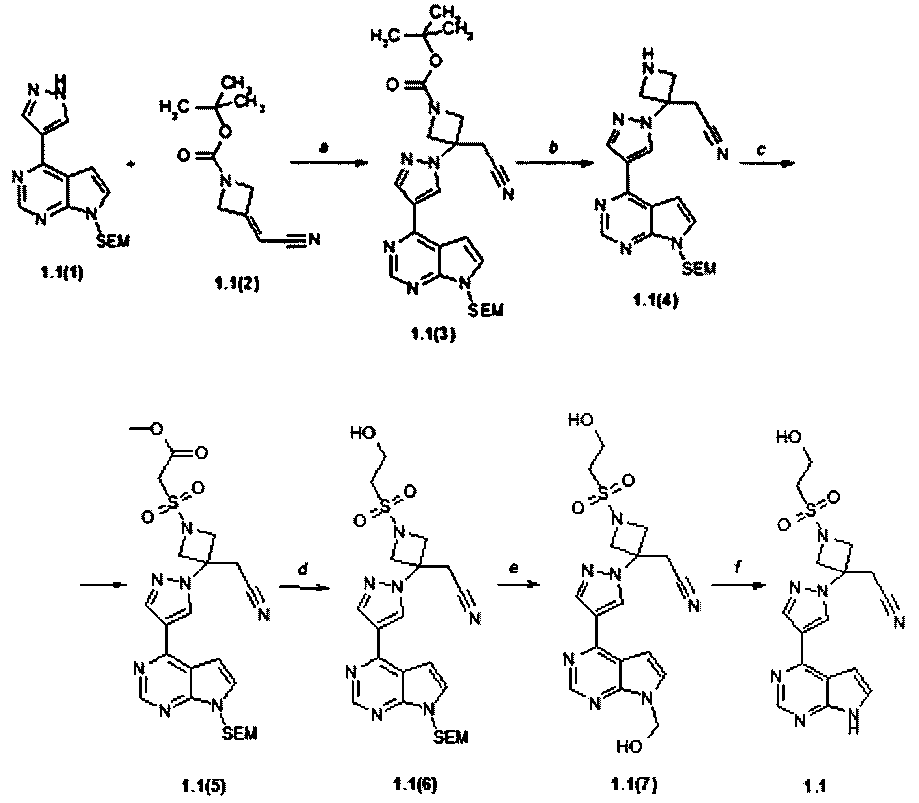
Стадия (a). Трет-бутиловый эфир 3-{4-[7-(2-триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-карбоновой кислоты (1.1(3)). В круглодонную колбу помещали 4-(1H-пиразол-3-ил)-7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин (1.1(1): 300 мг, 0,951 ммоль, 1 экв) и трет-бутиловый эфир 3-(цианометилен)азетидин-1-карбоновой кислоты (1.1(2): 208 мг, 1,071 ммоль, 1,126 экв) в ацетонитриле (V=18 мл). К полученной суспензии добавляли при комнатной температуре 1,8-диазабицикло[5.4.0]ундец-7-ен (ДБУ, 15 мг, 0,095 ммоль, 0,10 экв). Полученный раствор перемешивали при комнатной температуре в течение ночи. После завершения реакции (контроль с помощью ТСХ-элюент CHCl3:MeOH:25% NH4OH = 200:10:1) реакционную смесь упаривали на роторном испарителе досуха, к остатку добавляли воду (V=50 мл) и экстрагировали EtOAc (три раза 50 мл), объединенные экстракты промывали рассолом, сушили над Na2SO4 и выпаривали на роторном испарителе досуха. Остаток очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 4:1-3:1-2:1-1:1). Получали трет-бутиловый эфир 3-{4-[7-(2-триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-карбоновой кислоты (1.1(3)) в виде желтой пены (350 мг, 72%). 1H-NMR (DMSO-d6, 400 MHz): 0.0 (s, 9H), 0.84 (t, J=8 Hz, 2H), 1.42 (s, 9H), 3.53 (t, J=8 Hz, 2H), 3.7 (s, 2H), 4.27 (d, J=9.2 Hz, 2H), 4.48 (d, J=9.2 Hz, 2H), 5.66 (s, 2H), 7.17 (d, J=2.4 Hz, 1H), 7.23 (d, J=3.6 Hz, 1H), 7.76 (d, J=3.6 Hz, 1H), 8.29 (d, J=2.4 Hz, 1H), 8.85 (s, 1H).
Стадия (b). (3-{4-[7-(2-Триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-3-ил)-ацетонитрила дигидрохлорид (1,1(4)·HCl). К раствору трет-бутилового эфира 3-{4-[7-(2-триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-карбоновой кислоты (1.1(3): 350 мг, 0,687 ммоль) в 10 мл ТГФ добавляли 10 мл 3N HCl в диоксане. Раствор перемешивали при комнатной температуре в течение ночи и концентрировали в вакууме (контроль с помощью ТСХ-гексан:EtOAc = 1:1). Продукт 1.1(4) был использован в следующей реакции без очистки. Выход 300 мг, 91% в виде желтого порошка. 1H-NMR (DMSO-d6, 400 MHz): 0.0 (s, 9H), 0.85 (t, J=8 Hz, 2H), 3.55 (t, J=8 Hz, 2H), 3.99 (s, 2H), 4.33-4.44 (m, 2H), 4.64-4.78 (m, 2H), 5.69 (s, 2H), 7.39-7.48 (m, 2H), 7.95 (d, J=3.6 Hz, 1H), 8.47 (d, J=2.4 Hz, 1H), 8.97 (s, 1H), 9.89 (br.s, 1H), 10.14 (br.s, 1H).
Стадия (c). Метиловый эфир (3-цианометил-3-{4-[7-(2-триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил)-уксусной кислоты (1.1(5)). К раствору (3-{4-[7-(2-триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-3-ил)-ацетонитрила дигидрохлорида (1,1(4)·HCl (1,1(4)·HCl: (300 мг, 0,622 ммоль, 1 экв) в дихлорметане (V=20 мл), содержащему Et3N (220 мг, 2,18 ммоль, 3,5 экв) добавляли метил (хлорсульфонил)ацетат (161 мг, 0,933 ммоль, 1,5 экв). Смесь перемешивают в течение ночи при комнатной температуре и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 1:2). Соответствующие фракции объединяли и концентрировали в вакууме и получали соединения 1.1(5) (200 мг, 59%). 1H-NMR (DMSO-d6, 400 MHz): -0.05 (s, 9H), 0.92 (t, J=8.3 Hz, 2H), 3.43 (s, 2H), 3.56 (t, J=8.3 Hz, 2H), 3.83 (s, 3H), 4.16 (s, 2H), 4.40 (d, J=9.3 Hz, 2H), 4.74 (d, J=9.3 Hz, 2H), 5.68 (s, 2H), 6.81 (d, J=3.7 Hz, 1H), 7.42 (d, J=3.7 Hz, 1H), 8.36 (s, 1H), 8.53 (s, 1H), 8.85 (s, 1H).
Стадия (d). (1-(2-Гидрокси-этансульфонил)-3-{4-[7-(2-триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-3-ил)-ацетонитрил (1,1(6)). К раствору метилового эфира (3-цианометил-3-{4-[7-(2-триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил)-уксусной кислоты (1.1(5): 300 мг, 0,55 ммоль, 1 экв) в ТГФ (V=20 мл) добавляли LiBH4 (36 мг, 1,65 ммоль, 3 экв). Смесь перемешивали в течение ночи при комнатной температуре и концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель, метанол:этилацетат = 1:20). Соответствующие фракции объединяли и концентрировали в вакууме и получали соединения 1,1(6) (160 мг, 56%). 1H-NMR (DMSO-d6, 400 MHz): -0.07 (s, 9H), 0.91 (t, J=8.3 Hz, 2H), 3.36 (t, J=5.7 Hz, 2H), 3.43 (s, 2H), 3.54 (t, J=8.3 Hz, 2H), 4.06 (t, J=5.7 Hz, 2H), 4.31 (d, J=9.3 Hz, 2H), 4.67 (d, J=9.3 Hz, 2H), 5.64 (s, 2H), 6.77 (d, J=3.7 Hz, 1H), 7.39 (d, J=3.7 Hz, 1H), 8.30 (s, 1H), 8.49 (s, 1H), 8.78 (s, 1H).
Стадия (e). {1-(2-Гидрокси-этансульфонил)-3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-азетидин-3-ил}-ацетонитрил (1.1). Раствор (1-(2-гидрокси-этансульфонил)-3-{4-[7-(2-триметилсилил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-3-ил)-ацетонитрила (1.1(6): 160 мг, 0,309 ммоль) в метиленхлориде (V=10 мл) и трифторуксусной кислоте (V=5 мл) перемешивали в течение 2 часов. Растворители удаляли в вакууме. Остаток {1-(2-гидрокси-этансульфонил)-3-[4-(7-гидроксиметил-7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-азетидин-3-ил}-ацетонитрила (1.1(7)) растворяли в метаноле (V=30 мл), к полученному раствору добавляли раствор 25% NH4OH (V=1 мл) в воде (V=2 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель, метанол:этилацетат = 1:20). Соответствующие фракции объединяли и концентрировали в вакууме и получали целевой продукт 1.1 (72 мг 1,1, 60%) в виде белого порошка. 1H-NMR (DMSO-d6, 400 MHz): 3.41 (t, J=6.1 Hz, 2H), 3.67 (s, 2H), 3.78 (qw, J=6.1 Hz, 2H), 4.28 (d, J=9.3 Hz, 2H), 4.62 (d, J=9.3 Hz, 2H), 5.15 (t, J=6.1 Hz, 2H), 7.08 (d, J=3.7 Hz, 1H), 7.62 (d, J=3.7 Hz, 1H), 8.47 (s, 1H), 8.71 (s, 1H), 8.92 (s, 1H), 12.1 (s, 1H).
Пример 2. {1-(2,2-Дифторэтан-сульфонил)-3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-азетидин-3-ил}-ацетонитрил (1.3) получали в соответствии с схеме 4.
Схема 4
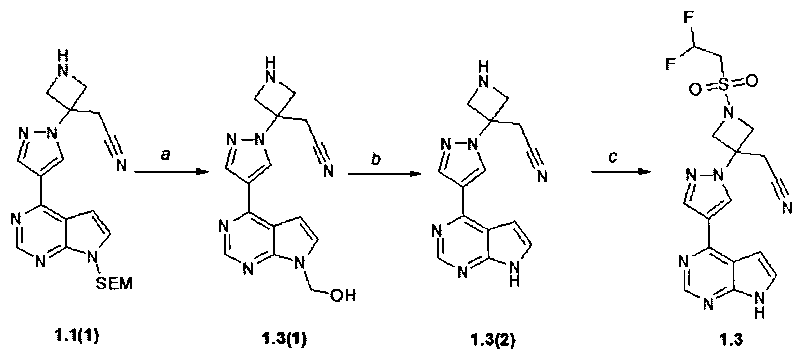
Стадия (a, b). {3-[4-(7H-Пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]азетидин-3-ил}ацетонитрил (1.3(2)). Раствор 4-(1H-пиразол-4-ил)-7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидина (1.1(1): 400 мг, 0,977 ммоль) в метиленхлориде (V=10 мл) и трифторуксусной кислоте (V=5 мл) перемешивали в течение 2 ч. Растворители удаляли в вакууме. Остаток (смесь 1.3(1) и 1.3(2)) растворяют в метаноле (V=30 мл), к полученному раствору добавляли раствор 25% NH4OH (V=1 мл) в воде (V=2 мл). Смесь перемешивали при комнатной температуре в течение ночи и затем упаривали на роторном испарителе досуха. Остаток очищали с помощью колоночной хроматографии (силикагель, CHCl3:MeOH:25% NH4OH = 200:10:1-150:10:1). Получали продукт 1.3(2) в виде белой пены (200 мг, 73%). 1H-NMR (DMSO-d6, 400 MHz): 3.59 (s, 2H), 3.66 (d, J=9.6 Hz, 2H), 4.1 (d, J=9.6 Hz, 2H), 7.06 (d, J=3.6 Hz, 1H), 7.6 (d, J=3.6 Hz, 1H), 8.41 (s, 1H), 8.69 (s, 1H), 8.8 (s, 1H), 12.11 (br.s, 1H).
Стадия (c). {1-(2,2-Дифторэтан-сульфонил)-3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-азетидин-3-ил}-ацетонитрил (1.3). К раствору {3-[4-(7H-Пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]азетидин-3-ил}ацетонитрила (1.3(2): 260 мг, 0.931 ммоль, 1 экв) в DCM (V=30 мл), содержащему Et3N (94 мг, 0.931 ммоль, 1 экв) добавляли 2,2-дифторэтансульфонилхлорид (153 мг, 1 экв 0.931 ммоль) при -10°C. Смесь перемешивали при комнатной температуре 12-15 ч. Осадок отфильтровывали и очищали колоночной хроматографией (силикагель-CHCl3:MeOH = 19:1-9:1). Получали продукт 1.3 в виде белого кристаллического твердого вещества. 1H-NMR (DMSO-d6, 400 MHz): 3.71 (s, 2H), 4.14-4.27 (m, 2H), 4.35 (d, J=9.2 Hz, 2H), 4.70 (d, J=9.2 Hz, 2H), 6.26-6.61 (m, 1H), 7.08 (s, 1H), 7.62 (s, 1H), 8.48 (s, 1H), 8.71 (s, 1H), 8.93 (s, 1H), 12.14 (br.s, 1H).
Пример 3. {3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-[(2,2,2-трифторэтан-сульфонил]-азетидин-3-ил}ацетонитрил (1.4) получали в соответствии со схемой 5.
Схема 5

3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]-1-[(2,2,2-трифторэтил)сульфонил]-азетидин-3-ил}ацетонитрил (1.4). К раствору {3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]азетидин-3-ил}ацетонитрила (1.3(2): 100 мг, 0,358 ммоль, 1 экв) в DCM (V=4 мл), содержащему Et3N (40 мг, 0,4 ммоль, 1,1 экв), добавляли 2,2,2-трифторэтансульфонилхлорида (65,4 мг, 0,358 ммоль, 1 экв). Полученную смесь перемешивали при комнатной температуре в течение ночи. Осадок отфильтровывали, промывают DCM (V=2 мл). Продукт 1.4 очищали с помощью колоночной хроматографии (гель кремнезема CHCl3:MeOH = 19:1-9:1). Получали продукт 1,4 в виде белого кристаллического вещества (48 мг, 31%). 1H-NMR (DMSO-d6, 400 MHz): 3.7 (s, 2H), 4.41 (d, J=9.2 Hz, 2H), 4.71-4.88 (m, 4H), 7.05-7.1 (m, 1H), 7.6-7.65 (m, 1H), 8.49 (s, 1H), 8.71 (s, 1H), 8.94 (s, 1H), 12.14 (br.s, 1H).
Пример 4. Метиловый эфир {3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил}-уксусной кислоты (1.5) получали в соответствии со схемой 6.
Схема 6.
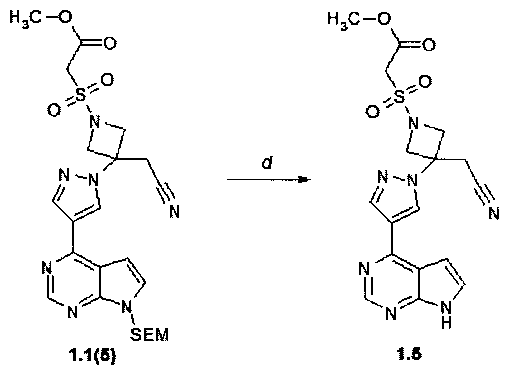
Метиловый эфир {3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил}-уксусной кислоты (1.5) был получен взаимодействием {3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]азетидин-3-ил}ацетонитрил (1.4(2)) с метил(хлорсульфонил)-ацетатом в условиях синтеза {3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]-1-[(2,2,2-трифторэтил)сульфонил]-азетидин-3-ил}ацетонитрил (1.4) (пример 3). 1H-NMR (DMSO-d6, 400 MHz) ингибитора 1.5: 3.69 (s, 2H), 3.70 (s, 3H), 4.38 (d, J=9.3 Hz, 2H), 4.64 (s, 2H), 4.72 (d, J=9.3 Hz, 2H), 7.07-7.10 (m, 1H), 7.61-7.64 (m, 1H), 8.48 (s, 1H), 8.71 (s, 1H), 8.94 (s, 1H), 12.14 (s, 1H)
Пример 5. {3-[4-(7H-Пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил}-уксусную кислоту (1.6) получали в соответствии со схемой 7.
Схема 7.
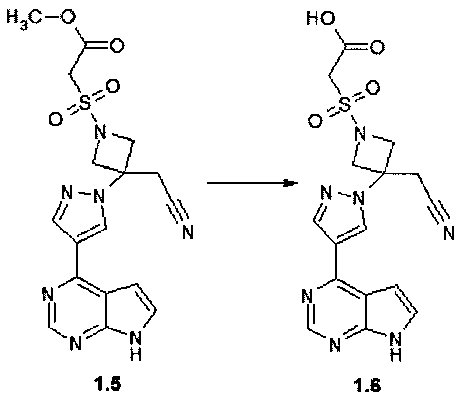
К суспензии 110 мг (0,26 ммоль) соединения 1.5 в 5 мл MeOH добавляли раствор 16 мг (0,39 ммоль) NaOH в 5 мл воды. Смесь перемешивали при комнатной температуре в течение ночи и затем MeOH удаляли в вакууме. Водный остаток подкислили 2ТЧ соляной кислотой до pH5 при охлаждении льдом. Осадок отфильтровывали и сушили на воздухе. Получали 92 мг (90%) соединения 1.6 в виде белого порошка. 1H-NMR (DMSO-d6, 400 MHz): 3.68 (s, 2H), 4.37 (d, J=9.3 Hz, 2H), 4.47 (s, 2H), 4.72 (d, J=9.3 Hz, 2H), 7.07-7.10 (m, 1H), 7.61-7.64 (m, 1H), 8.47 (s, 1H), 8.71 (s, 1H), 8.94 (s, 1H), 13.46 (br.s, 1H).
Пример 6. 2-{3-[4-(7H-Пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-нл}-этансульфоновую кислоту (1.7) получали в соответствии со схемой 8.
Схема 8.
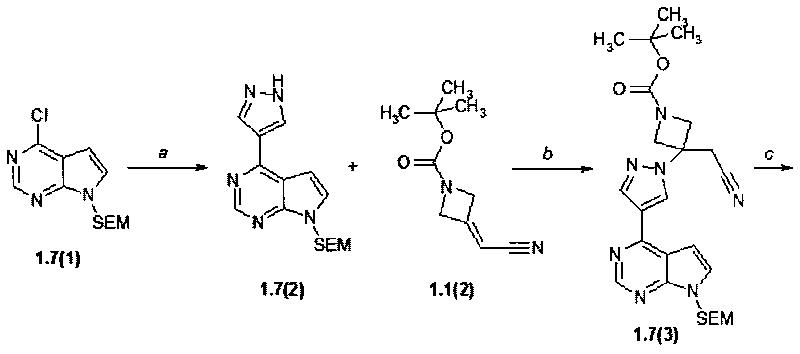
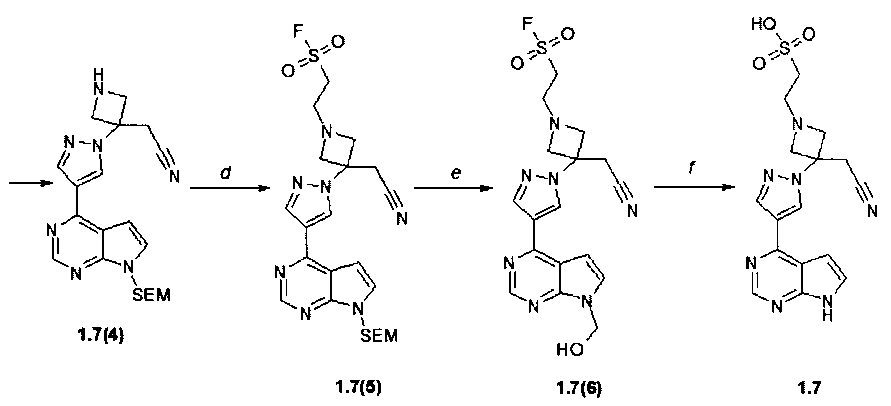
Стадия (a). 4-(1H-Пиразол-4-ил)-7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин (1.7(2)). К раствору 4-хлор-7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидина (1.7(1): 1,0 г, 3,5 ммоль, 1 экв) в диоксане (V=10 мл) были добавлены 1H-пиразол-5-илборная кислота (0,51 г, 4,6 ммоль, 1,30 экв) и Na2CO3 (1,11 г, 10,5 ммоль, 3,0 экв) и вода (V=5 мл). Раствор дегазируют пропусканием через раствор потока аргона в течение 3 минут, после чего добавляют тетракис(трифенилфосфин)палладий(0) (0,23 г, 1,4 ммоль, 0,05 экв) и полученную реакционную смесь нагревают в микроволновой системе (120°C, 2 ч). Когда по данным ТСХ (гексан:этилацетат = 1:1) реакция была завершена, реакционную смесь охладили до комнатной температуры, разбавили этилацетатом (V=30 мл) и водой (V=40 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (дважды по 30 мл). Органические слои объединяли, промывали насыщенным раствором NaHCO3 (V=20 мл), насыщенным раствором соли (V=20 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток растирали с метил-трет-бутиловым эфиром (МТБЭ, V=40 мл), твердое вещество собирали фильтрованием и промывали МТБЭ (дважды по 20 мл). Получали в виде белого кристаллического вещества продукт 1.7(2) (133 мг). 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.94 (t, J=8 Hz, 2H), 3.58 (t, J=8 Hz, 2H), 5.71 (s, 2H), 7.08 (d, J=3.6 Hz, 1H), 7.18 (d, J=1.6 Hz, 1H), 7.47 (d, J=3.6 Hz, 1H), 7.78 (d, J=1.6 Hz, 1H), 8.95 (s, 1H).
Стадия (b). Трет-бутиловый эфир 3-{4-[7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-3-цианометил-азетидин-1-карбоновой кислоты (1,7(3)). В круглодонную колбу при комнатной температуре помещали 4-(1H-пиразол-4-ил)-7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин (1.7(2): 300 мг, 0,951 ммоль, 1 экв) и трет-бутил-3-(цианометилен)азетидин-1-карбоксилат (1.1(2): 208 мг, 1,071 ммоль, 1,126 экв) в ацетонитриле (V=18 мл). К полученной розовой суспензии добавили 1,8-диазабицикло[5.4.0]ундец-7-ен (DBU: 15 мг, 0,095 ммоль, 0,10 экв). Полученный коричневый раствор перемешивали при комнатной температуре 12-15 ч. После того, как по данным ТСХ (CHCl3:MeOH:25% NH4OH = 200:10:1) реакция была завершена, реакционную смесь упаривали на роторном испарителе досуха, к остатку добавляли воду (V=50 мл) и экстрагировали EtOAc (три раза 50 мл). Объединенные экстракты промывали рассолом, сушили над Na2SO4 и выпаривали досуха. Остаток очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 4:1-3:1-2:1-1:1). Получали продукт 1,7(3) в виде желтой пены (350 мг, 72%). 1H-NMR (DMSO-d6, 400 MHz): 0.0 (s, 9H), 0.84 (t, J=8 Hz, 2H), 1.42 (s, 9H), 3.53 (t, J=8 Hz, 2H), 3.7 (s, 2H), 4.27 (d, J=9.2 Hz, 2H), 4.48 (d, J=9.2 Hz, 2H), 5.66 (s, 2H), 7.17 (d, J=2.4 Hz, 1H), 7.23 (d, J=3.6 Hz, 1H), 7.76 (d, J=3.6 Hz, 1H), 8.29 (d, J=2.4 Hz, 1H), 8.85 (s, 1H).
Стадия (c). (3-{4-[7-(2-Триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-азетидин-ацетонитрил (1.7(4)). К раствору трет-бутилового эфира 3-{4-[7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-3-цианометил-азетидин-1-карбоновой кислоты (1.7(3): 350 мг, 0,687 ммоль) в 10 мл ТГФ добавляли 10 мл 3N HCl в диоксане. Раствор перемешивали при комнатной температуре в течение ночи и концентрировали в вакууме. Получали продукт 1,7(4) (300 мг, 91%) в виде желтого порошка, который был использован в следующей стадии без очистки. 1H-NMR (DMSO-d6, 400 MHz): 0.0 (s, 9H), 0.85 (t, J=8 Hz, 2H), 3.55 (t, J=8 Hz, 2H), 3.99 (s, 2H), 4.33-4.44 (m, 2H), 4.64-4.78 (m, 2H), 5.69 (s, 2H), 7.39-7.48 (m, 2H), 7.95 (d, J=3.6 Hz, 1H), 8.47 (d, J=2.4 Hz, 1H), 8.97 (s, 1H), 9.89 (br.s, 1H), 10.14 (br.s, 1H).
Стадия (d). 2-(3-{4-[7-(2-Триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-3-цианометил-азетидин-1-ил)-этансульфофторид (1.7(5)). К раствору (3-{4-[7-(2-Триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-азетидин-ацетонитрила (1.7(4): 300 мг, 0,622 ммоль, 1 экв) в DCM (V=20 мл), содержащему Et3N (251 мг, 2,49 ммоль, 4 экв) добавляли 2-фторэтансульфонилфторидфторида (82 мг, 0,622 ммоль, 1 экв). Смесь перемешивали при комнатной температуре 12-15 ч. Остаток очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 1:2). Соответствующие фракции объединяли и концентрировали в вакууме. Получали продукт 1.7(5) (150 мг, 46%). 1H-NMR (CDCl3, 400 MHz): -0.07 (s, 9H), 0.91 (t, J=8 Hz, 2H), 3.14 (t, J=6.6 Hz, 2H), 3.39 (s, 2H), 3.49 (t, J=6.6 Hz, 2H), 3.54 (t, J=8 Hz, 2H), 3.74 (d, J=8.3 Hz, 2H), 3.83 (d, J=8.3 Hz, 2H), 5.66 (s, 2H), 6.80 (d, J=3.7 Hz, 1H), 7.39 (d, J=3.7 Hz, 1H), 8.31 (s, 1H), 8.45 (s, 1H), 8.84 (s, 1H).
Стадия (e, f). 2-{3-[4-(7H-Пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил]-3-цианометил-азетидин-1-ил}-этансульфокислота (1-7). Раствор 2-(3-{4-[7-(2-триметилсиланил-этоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-3-цианометил-азетидин-1-ил)-этансульфофторида (1.7(5): 150 мг, 0.289 mmol) в метиленхлориде (V=10 мл) и трифторуксусной кислоте (V=5 мл) перемешивали в течение 2 ч. Растворители удаляли в вакууме. Остаток 1.7(6) растворяли в MeOH (V=30 мл), к раствору добавляли раствор 25% NH4OH (V=1 мл) в воде (V=2 мл). Смесь перемешивали при комнатной температуре 12-15 ч и затем концентрировали в вакууме. Остаток очищали с помощью колоночной хроматографии (силикагель, метанол:этилацетат = 1:2). Соответствующие фракции объединяли, концентрировали в вакууме и получали желаемый продукт 1.7 (55 мг, 49%) в виде серого порошка. 1H-NMR (CH3OD, 400 MHz): 2.91-2.98 (m, 2H), 3.02-3.09 (m, 2H), 3.51 (s, 2H), 3.80 (d, J=8.9 Hz, 2H), 3.87 (d, J=8.9 Hz, 2H), 7.00 (d, J=3.7 Hz, 1H), 7.51 (d, J=3.7 Hz, 1H), 8.40 (s, 1H), 8.68 (s, 1H), 8.71 (s, 1H).
Пример 7. Амид 2-{3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-нл}-этансульфоновой кислоты (1.8) и его дихлорацетат 1.8·CHCl2CO2H получали в соответствии со схемой 9.
Схема 9.
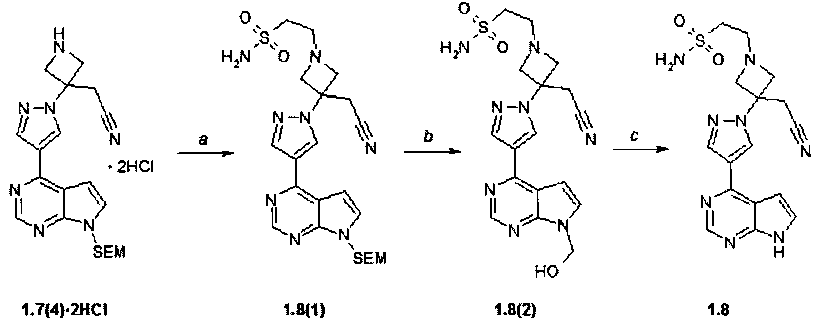
Стадия (a). Амид 2-{3-[4-(7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]-3-(цианометил)-азетидин-1-ил}этансульфоновой кислоты (1.8(1)). К суспензии {3-[4-(7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]-3-(цианометил)-азетидин-1-ил}ацетонитрила дигидрохлорида 1.7(4)·2HCl: 508 мг, 1.05 ммол, 1 экв) в ацетонитриле (15 мл=V), содержащей триэтиламин (213 mg, 2.11 mmol, 2 экв), добавили этиленсульфамид (140 мг, 1.16 ммол, 1.1 экв). Смесь перемешивали и нагревали с обратным холодильником 12-15 ч. Окончание реакции контролировали с помощью ТСХ (CHCl3:MeOH:25% NH4OH = 100:10:1). реакционную смесь упаривали на роторном испарителе досуха, к остатку добавляли воду (V=50 мл) и экстрагировали EtOAc (3 раза по 20 мл). Объединенные экстракты промывали солевым раствором, сушили над Na2SO4 и упаривали на роторном испарителе досуха. Продукт очищали с помощью колоночной хроматографии (силикагель, CHCl3:MeOH:25% NH4OH = 200:10:1). Получали продукт 1.8(1) в виде желтого масла (300 мг, 55%). 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.93 (t, J=8 Hz, 2H), 3.13-3.32 (m, 4H), 3.37 (s, 2H), 3.5 (s, 2H), 3.56 (t, J=8 Hz, 2H), 3.8-3.99 (m, 4H), 5.68 (s, 2H), 6.82 (d, J=3.6 Hz, 1H), 7.44 (d, J=3.6 Hz, 1H), 8.31 (s, 1H), 8.53 (s, 1H), 8.84 (s, 1H).
Стадия (b, c). Амид 2-{3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-нл}-этансульфоновой кислоты (1.8). Перемешивали 2 ч амид 2-{3-[4-(7-{[2-(триметилсилил)этокси]метил}-7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]-3-(цианометил)-азетидин-1-ил}этансульфоновой кислоты (1.8(1): 300 мг, 0.581 ммол) в метиленхлориде (V=10 мл) и трифторуксусной кислоте (V=5 мл). Растворители удаляли в вакууме. Остаток растворяли в метаноле (V=15 мл), к полученному раствору добавляли раствор 25% NH4OH (V=1 мл) в воде (V=2 мл). Смесь перемешивали 12-15 ч. Осадок отфильтровывали и сушили на воздухе. Получали 70 мг соединения 1.8. 1H-NMR (DMSO-d6, 400 MHz): 2.88-2.97 (m, 2H), 3.03-3.12 (m, 2H), 3.54-3.67 (m, 4H), 3.73-3.82 (m, 2H), 6.78 (s, 2H), 7.03-7.08 (m, 1H), 7.57-7.62 (m, 1H), 8.42 (s, 1H), 8.7 (s, 1H), 8.81 (s, 1H), 12.12 (br.s, 1H). К раствору 50 мг (0.129 ммоль). В 10 мл тетрагидрофурана добавляют 16.7 мг (0.129 ммоль) дихлоруксусной кислоты. Смесь упаривают на роторном испарителе досуха. Дихлорацетат 1.8·CHCl2CO2H получают в виде белого твердого вещества.
Пример 8. {1-(2-Метасульфонил-этил)-3-[4-(7H-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-азетидин-3-ил}ацетонитрил (1.9) получали в соответствии со схемой 10.
Схема 10
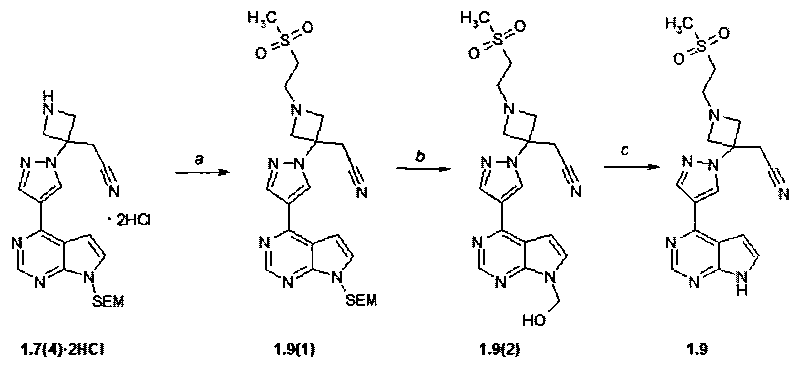
Стадия (a). К суспензии 588 мг (1.22 ммоль) соединения 1.7(4)·2HCl в 15 мл ацетонитрила добавляют 247 мг (2.44 ммоль) триэтиламина и 194 мг (1.83 ммоль) метилвинилсульфона. Смесь кипятили при перемешивании с обратным холодильником в течение ночи. Реакционную смесь упаривали в вакууме досуха, к остатку добавляли 50 мл воды и экстрагировали этилацетатом (трижды по 20 мл). Объединенные экстракты промывали солевым раствором, сушили над Na2SO4 и выпаривали в вакууме досуха. Полученный продукт очищали с помощью колоночной хроматографии (силикагель, CHCl3:MeOH:25% водный NH4OH = 800:10:1-500:10:1-300:10:1). Получали 400 мг (63%) продукта 1.9(1)_в виде желтого масла. 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.94 (t, J=8 Hz, 2H), 3.04 (s, 3H), 3.07-3.18 (m, 4H), 3.37 (s, 2H), 3.57 (t, J=8 Hz, 2H), 3.77 (d, J=8.4 Hz, 2H), 3.86 (d, J=8.4 Hz, 2H), 5.7 (s, 2H), 6.85 (d, J=4 Hz, 1H), 7.45 (d, J=4 Hz, 1H), 8.33 (s, 1H), 8.59 (br.s, 1H), 8.87 (s, 1H).
Стадия (b, c). Перемешивали в течение 2 ч 400 мг (0,78 ммоль) продукта 1.9(1) в смеси 20 мл метиленхлорида и 10 мл трифторуксусной кислоты. Растворители удаляли в вакууме. Остаток 1.9(2) растворяли в 20 мл метанола, добавляли раствор 1 мл 25% NH4OH в 2 мл воды. Смесь перемешивали при комнатной температуре в течение ночи. Смесь упаривали в вакууме досуха, к остатку добавляют 70 мл воды и экстрагировали этилацетатом (трижды по 30 мл). Объединенный органический экстракт промывали насыщенным раствором соли, сушили над Na2SO4 и выпаривали в вакууме досуха. Получали 184 мг (61%) ингибитора 1.9 в виде белого твердого вещества. 1H-NMR (DMSO-d6, 400 MHz): 2.94 (t, J=6.8 Hz, 2H), 3.04 (s, 3H), 3.20 (t, J=6.8 Hz, 2H), 3.55 (s, 2H), 3.64 (d, J=8.4 Hz, 2H), 3.79 (d, J=8.4 Hz, 2H), 7.03-7.08 (m, 1H), 7.58-7.62 (m, 1H), 8.42 (s, 1H), 8.69 (s, 1H), 8.81 (s, 1H), 12.11 (br.s, 1H).
Пример 9. {3-[4-(7H-Пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-этансульфонил-азетидин-3-ил}ацетонитрил (1.10) получали в соответствии со схемой 11.
Схема 11.

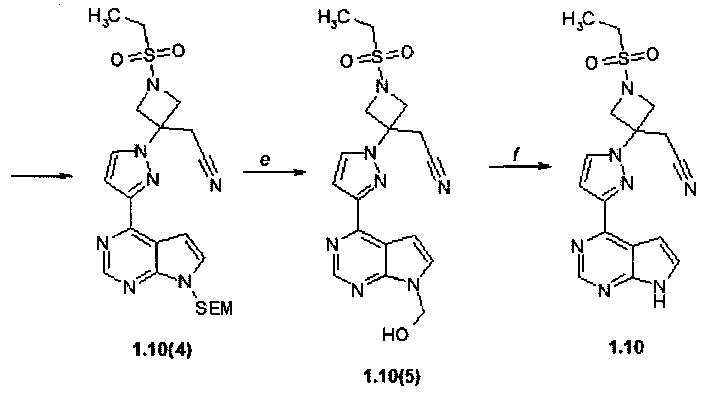
Стадия (a). 4-(1H-Пиразол-3-ил)-7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин (1.10(1)). К раствору 4-хлор-7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидина (1.7(1): 1,0 г, 3,5 ммоль, 1 экв) в диоксане (V=10 мл) добавляли 1H-пиразол-5-илборную кислоту (0,51 г, 4,6 ммоль, 1,30 экв), Na2CO3 (1,11 г, 10,5 ммоль, 3,0 экв) и воду (V=5 мл). Раствор дегазировали пропусканием потока аргона через раствор в течение 3 минут, после чего обрабатывали тетракис(трифенилфосфин)палладием(0) (0,23 г, 1,4 ммоль, 0,05 экв). Полученную реакционную смесь нагревали в микроволновой системе (MW 120°C, 2 ч). После окончания реакции (контролировали с помощью ТСХ-гексан:этилацетат = 1:1) реакционную смесь охлаждали до комнатной температуры, разбавляли этилацетатом (V=30 мл) и водой (V=40 мл). Два слоя разделяли и водный слой экстрагировали этилацетатом (дважды по 30 мл). Соединенные органические слои промывали насыщенным раствором NaHCO3 (V=20 мл), насыщенным раствором соли (V=20 мл), сушили над сульфатом натрия и концентрировали при пониженном давлении. Остаток растирали с метил-трет-бутиловым эфиром (МТБЭ, V=40 мл), твердое вещество собирали фильтрованием и промывали МТБЭ (дважды 20 мл). Получали продукт 1.10(1) (133 мг, выход 13%) в виде белого кристаллического вещества. 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.94 (t, J=8 Hz, 2H), 3.58 (t, J=8 Hz, 2H), 5.71 (s, 2H), 7.08 (d, J=3.6 Hz, 1H), 7.18 (d, J=1.6 Hz, 1H), 7.47 (d, J=3.6 Hz, 1H), 7.78 (d, J=1.6 Hz, 1H), 8.95 (s, 1H).
Стадия (b). Трет-бутиловый эфир 3-{3-[7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-3-цианометил-азетидин-1-карбоновой кислоты (1.10(2)).
В круглодонную колбу помещали 4-(1H-пиразол-3-ил)-7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин (1,10 (1): 300 мг, 0,951 ммоль, 1 экв) и трет-бутил-3-(цианометилен)-азетидин-1-карбоксилат (208 мг, 1,071 ммоль, 1,126 экв) в ацетонитриле (V=18 мл) при комнатной температуре. К полученной розовой суспензии добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (15 мг, 0,095 ммоль, 0,10 экв). Полученный коричневый раствор перемешивали при комнатной температуре 12-15 ч. После окончания реакции (контроль с помощью ТСХ-CHCl3:MeOH:25% NH4OH = 200:10:1) реакционную смесь упаривали на роторном испарителе досуха, к остатку добавляли воду (V=50 мл) и экстрагировали EtOAc (три раза по 50 мл). Объединенные экстракты промывали рассолом, сушили над Na2SO4 и выпаривают досуха. Остаток очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 4:1-3:1-2:1-1:1). Получали продукт 1,10(2) в виде желтой пены (350 мг, 72%). 1H-NMR (DMSO-d6, 400 MHz): 0.0 (s, 9H), 0.84 (t, J=8 Hz, 2H), 1.42 (s, 9H), 3.53 (t, J=8 Hz, 2H), 3.7 (s, 2H), 4.27 (d, J=9.2 Hz, 2H), 4.48 (d, J=9.2 Hz, 2H), 5.66 (s, 2H), 7.17 (d, J=2.4 Hz, 1H), 7.23 (d, J=3.6 Hz, 1H), 7.76 (d, J=3.6 Hz, 1H), 8.29 (d, J=2.4 Hz, 1H), 8.85 (s, 1H).
Стадия (c). (3-{3-[7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-азетидин-3-ил)-ацетонитрил (1.10(3)·2HCl). К раствору трет-бутилового эфира 3-{3-[7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-3-цианометил-азетидин-1-карбоновой кислоты (1.10(2): 350 мг, 0,687 ммоль) в 10 мл ТГФ добавляли 10 мл 3N HCl в диоксане. Раствор перемешивали при комнатной температуре 12-15 ч и концентрировали в вакууме. Окончание реакции контролировали с помощью ТСХ-гексан:EtOAc = 1:1. Полученный продукт 1.10(3)·2HCl (300 мг, 91%) в виде желтого порошка, который использовали в следующей стадии без очистки. 1H-NMR (DMSO-d6, 400 MHz): 0.0 (s, 9H), 0.85 (t, J=8 Hz, 2H), 3.55 (t, J=8 Hz, 2H), 3.99 (s, 2H), 4.33-4.44 (m, 2H), 4.64-4.78 (m, 2H), 5.69 (s, 2H), 7.39-7.48 (m, 2H), 7.95 (d, J=3.6 Hz, 1H), 8.47 (d, J=2.4 Hz, 1H), 8.97 (s, 1H), 9.89 (br.s, 1H), 10.14 (br.s, 1H).
Стадия (d). (3-{3-[7-(2-Триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-1-этансульфонил-азетидин-3-ил)-ацетонитрил (1.10(4)). К раствору (3-{3-[7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-азетидин-3-ил)-ацетонитрил (1.10(3)·2HCl: 300 мг, 0,622 ммоль, 1 экв) в DCM (V=30 мл), содержащем Et3N (251 мг, 2,49 ммоль, 4 экв) добавляли этансульфонилхлорида (80 мг, 0,622 ммоль, 1 экв). Смесь перемешивали при комнатной температуре 12-15 ч. Остаток очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 3:1-2:1-1:1-1:2). Получали продукт 1.10(4) (200 мг, 64%) в виде белого твердого вещества. 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.84 (t, J=8 Hz, 2H), 1.25 (t, J=7.2 Hz, 3H), 3.25 (q, J=7.2 Hz, 2H), 3.53 (t, J=8 Hz, 2H), 3.74 (s, 2H), 4.28 (d, J=9.6 Hz, 2H), 4.59 (d, J=9.6 Hz, 2H), 5.66 (s, 2H), 7.2 (d, 2.4 Hz, 2H), 7.27 (d, 3.6 Hz, 2H), 7.78 (d, 3.6 Hz, 2H), 8.31 (d, 2.4 Hz, 2H), 8.86 (s, 1H).
Стадия (e, f). (3-{3-[7H-Пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-1-этансульфонил-азетидин-3-ил)-ацетонитрил (1.10). Раствор (3-{3-[7-(2-триметилсиланилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-пиразол-1-ил}-1-этансульфонил-азетидин-3-ил)-ацетонитрила (1.10(4): 150 мг, 0,298 ммоль) в метиленхлориде (V=10 мл) и трифторуксусной кислоте (V=5 мл) перемешивали в течение 2 ч. Растворители удаляли в вакууме. Остаток 1.10(5) растворяли в метаноле (V=30 мл), добавляли раствор 25% NH4OH (V=1 мл) в воде (V=2 мл) и перемешивали при комнатной температуре 12-15 ч. Осадок отфильтровывали, промывали водой (V=5 мл) и сушили на воздухе. Получали продукт 1.10 (61 мг, 41%) в виде белого твердого вещества. 1H-NMR (DMSO-d6, 400 MHz): 1.25 (t, J=7.2 Hz, 3H), 3.25 (q, J=7.2 Hz, 2H), 3.73 (s, 2H), 4.27 (d, J=9.6 Hz, 2H), 4.58 (d, J=9.6 Hz, 2H), 7.18 (s, 1H), 7.6 (s, 1H), 8.28 (s, 1H), 8.78 (s, 1H), 12.11 (br.s, 1H).
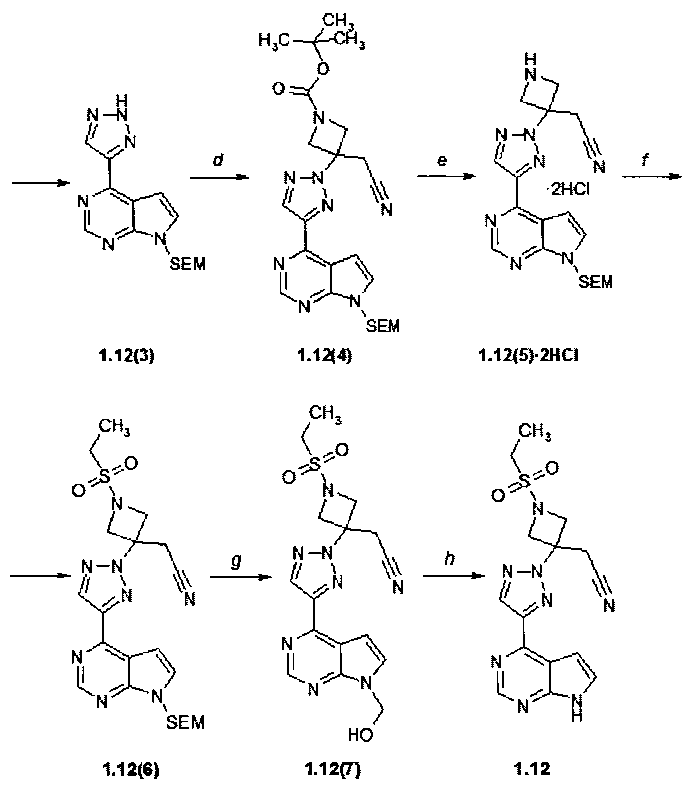
Пример 10. {3-[4-(7H-Пирроло[2,3-d]пиримидин-4-ил)-триазол-2-ил]-1-этансульфонил-азетидин-3-ил}ацетонитрил (1.12) получали в соответствии со схемой 12.
Схема 12
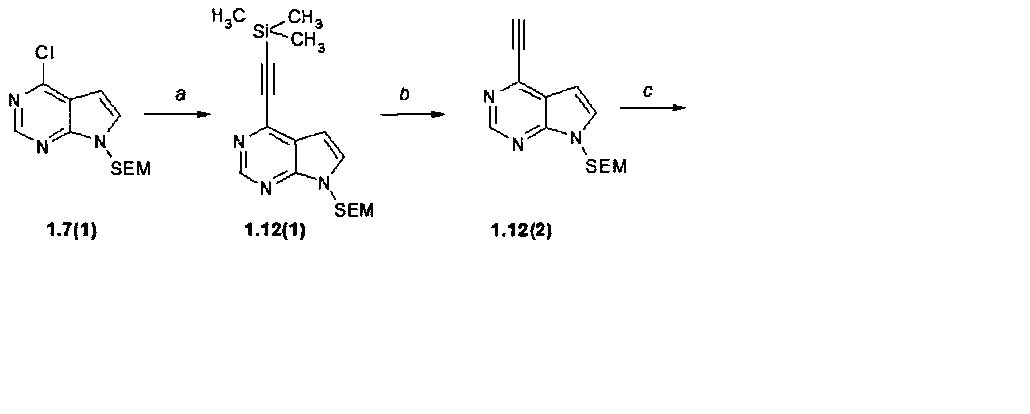
Стадия (a). 7-(2-Триметилсиланилэтоксиметил)-4-триметилсиланилэтинил-7H-пирроло[2,3-d]пиримидин (1.12(1)). К смеси 7-{[2-триметилсилил-этокси]метил}-4-хлор-7H-пирроло[2,3-d]пиримидина (1.7(1): 5,3 г, 18,7 ммоль, 1 экв), этинилтриметидсилана (3,67 г, 37,4 ммоль, 2 экв), PdCl2(PPh3)2 (1,33 г, 1,9 ммоль, 0,1 экв), CuI (0,36 г, 1,9 ммоль, 0,1 экв) в TEA (V=95 мл) добавляли безводный ДМФА (V=19 мл) и нагревали с обратным холодильником в течение 3 ч. Реакционную смесь фильтровали в горячем состоянии и концентрировали в вакууме. Полученный остаток растворяют в EtOAc (V=100 мл) и фильтровали через целит. Этилацетатный слой промывают водой и солевым раствором. Органическую фазу упаривают на роторном испарителе досуха. Продукт очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 30:1-20:1). Получали продукт 1.12(1) в виде коричневого масла (1,6 г, 25%). 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.34 (s, 9H), 0.91 (t, J=8 Hz, 2H), 3.52 (t, J=8 Hz, 2H), 5.66 (s, 2H), 6.71 (d, J=3.6 Hz, 1H), 7.4 (d, J=3.6 Hz, 1H), 8.86 (s, 1H).
Стадия (b). 7-{[2-триметилсилилэтокси]метил}-4-этинил-7H-пирроло[2,3-d]пиримидин (1.12(2)). 7-(2-Триметилсиланилэтоксиметил)-4-триметилсиланилэтинил-7H-пирроло[2,3-d]пиримидин (1.12(1): 1,6 г, 4,6 ммоль, 1 экв) растворяли в ТГФ (V=50 мл), полученный раствор охлаждали до 0°C и добавляли воду (V=3,4 мл), а затем добавляли по каплям 75% TBAF в воде (1,88 г, 1,41 г, 5,4 ммоль, 1,17 экв). Реакционную смесь оставляли нагреваться от 0°C до 15°C в течение 1 ч. Окончание реакции контролировали с помощью ТСХ (гексан:EtOAc = 4:1). К реакционной смеси прибавляли при перемешивании воду (V=150 мл) и EtOAc (V=50 мл), водную фазу отделяли, промывают дополнительным этилацетатом (дважды по 50 мл). Объединенные органические фазы промывали водой (три раза по 50 мл), затем насыщенным раствором соли, затем сушат над Na2SO4 и упаривали на роторном испарителе досуха. Получали продукт 1.12(2) в виде коричневого масла (1,26 г, 99%).
Стадия (c). 4-(1H-1,2,3-Триазол-4-ил)-7-{[2-триметилсилилэтокси]метил}-7H-пирроло[2,3-d]пиримидин (1.12(3)). К перемешиваемому раствору 7-{[2-триметилсилилэтокси]метил}-4-этинил-7H-пирроло[2,3-d]пиримидин (1.12(2): 1,26 г, 4,6 ммоль, 1 экв) и CuI (40 мг, 0,2 ммоль, 0,05 экв) в растворе ДМФ/MeOH (V=30 мл, 9:1) добавляли в атмосфере аргона триметилсилилазид (0,79 г, 6,9 ммоль, 1,5 экв). Полученный раствор перемешивали при 100°C в течение 24 ч. После исчезновения в реакционной смеси соединения 1.12(2) (ТСХ контроль - гексан:EtOAc = 1:1) смесь охлаждали до комнатной температуры и упаривали на роторном испарителе досуха. Остаток очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 2:1-1:1). Получали продукт 1.12(3) в виде коричневого масла (300 мг, 20%).
1H-NMR (DMSO-d6, 400 MHz): 0.0 (s, 9H), 0.84 (t, J=8 Hz, 2H), 3.54 (t, J=8 Hz, 2H), 5.67 (s, 2H), 7.21 (br.s, 1H), 7.81 (s, 1H), 8.87 (s, 1H), 15.52 (br.s, 1H).
Стадия (d). Трет-бутиловый эфир 3-{4-[7-(2-триметилсилилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-[1,2,3]триазол-2-ил}-3-цианометил-азетидин-1-карбоновой кислоты (1.12(4)). В круглодонную колбу при 20±5°C поместили 4-(1H-1,2,3-триазол-4-ил)-7-{[2-триметилсилилэтокси]метил}-7H-пирроло[2,3-d]пиримидин (1.12(3): 300 мг, 0,948 ммоль, 1 экв) и трет-бутил-3-(цианомнтилен)азетидин-1-карбоксилат (207 мг, 1,07 ммоль, 1,126 экв) в ацетонитриле (V=6 мл). К полученному раствору добавляли 1,8-диазабицикло[5.4.0]ундец-7-ен (15 мг, 0,095 ммоль, 0,10 экв). Коричневый раствор перемешивают при 20°C 12-15 ч. Окончание реакции контролировали с помощью ТСХ (CHCl3:MeOH:25% NH4OH = 200:10:1) После окончания реакции добавили воду (V=35 мл) при перемешивании в течение 80 минут при 20°C. Смесь перемешивали при комнатной температуре в течение 12 ч. Если осадок не образуется, в смесь добавляли воду (V=50 мл) и экстрагировали EtOAc (три раза по 20 мл). Объединенные экстракты промывали рассолом, сушили над Na2SO4 и выпаривают на роторном испарителе досуха. Остаток очищали с помощью колоночной хроматографии (силикагель, гексан:EtOAc = 4:1-3:1-2:1). Получали продукт 1.12(4) (200 мг, 41%) в виде темно-желтого кристаллического вещества. 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.94 (t, J=8 Hz, 2H), 1.51 (s, 9H), 3.51 (s, 2H), 3.57 (t, J=8 Hz, 2H), 4.35 (d, J=9.6 Hz, 2H), 4.79 (d, J=9.6, 2H), 5.72 (s, 2H), 7.23 (d, J=3.6 Hz, 1H), 7.48 (d, J=3.6 Hz, 1H), 8.57 (s, 1H), 8.97 (s, 1H).
Стадия (e). (3-{4-[7-(2-Триметилсилилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-[1,2,3]триазол-2-ил}-азетидин-3-ил)-ацетонитрила дигидрохлорид (1.12(5)·2HCl). К раствору трет-бутилового эфира 3-{4-[7-(2-триметилсилилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-[1,2,3]триазол-2-ил}-3-цианометил-азетидин-1-карбоновой кислоты (1.12(4): 200 мг, 0,39 ммоль) в 15 мл ТГФ добавляли 15 мл 3N HCl в диоксане. Раствор перемешивали 12-15 ч при комнатной температуре и концентрировали в вакууме. Окончание реакции контролировали с помощью ТСХ (гексан:EtOAc = 1:1). Полученный продукт 1.12(5)·2HCl (189 мг, 99%) в виде желтого порошка был использован в следующей стадии без очистки.
Стадия (f). {1-(Этилсульфонил)-3-[4-(7-{[2-триметилсилилэтоксиметил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2H-1,2,3-триазол-2-ил]азетидин-3-ил)-ацетонитрил (1.12(6)). К раствору (3-{4-[7-(2-триметилсилилэтоксиметил)-7H-пирроло[2,3-d]пиримидин-4-ил]-[1,2,3]триазол-2-ил}-азетидин-3-ил)-ацетонитрила дигидрохлорида (1.12(5)·2HCl): 189 мг, 0,391 ммоль, 1 экв) в DCM (V=30 мл), содержащему Et3N (0,22 мл, 159 мг, 1,56 ммоль, 4 экв) при -10°C добавляли этансульфонилхлорида (50,3 мг, 0,391 ммоль, 1 экв). Смесь перемешивали при комнатной температуре 12-15 ч. Реакционную смесь выпаривали на роторном испарителе досуха, к остатку добавляли насыщенный раствор NaHCO3 (V=30 мл) и экстрагировали этилацетатом (три раза по 20 мл). Объединенный экстракт промывали насыщенным раствором соли и упаривают на роторном испарителе досуха. Остаток промывали гексаном, фильтровали, сушили на воздухе и очищали с помощью колоночной хроматографии (силикагель, гексан : EtOAc = 3:1-2:1-1:1). Получали продукт 1.12(6) (150 мг, 76%) в виде белого твердого вещества. 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.94 (t, J=11.2 Hz, 2H), 1.46 (t, J=9.6 Hz, 3H), 3.07-3.2 (m, 2H), 3.51-3.53 (m, 4H), 4.31 (d, J=12.4 Hz, 2H), 4.93 (d, J=12.4 Hz, 2H), 5.73 (s, 2H), 5.51 (d, J=4 Hz, 1H), 8.66 (br.s, 1H), 8.99 (s, 1H).
Стадии (g, h). {3-[4-(7Н-Пирроло[2,3-d]пиримидин-4-ил)-триазол-2-ил]-1-этансульфонил-азетидин-3-ил}ацетонитрил (1.12). {1-(этилсульфонил)-3-[4-(7-{[2-триметилсилилэтоксиметил}-7H-пирроло[2,3-d]пиримидин-4-ил)-2H-1,2,3-триазол-2-ил]азетидин-3-ил)-ацетонитрил (1.12(6): 150 мг, 0,298 ммоль) в метиленхлориде (V=10 мл) и трифторуксусной кислоте (V=5 мл) перемешивали в течение 2 ч. Растворители удаляли в вакууме. Остаток 1.12(7) растворяли в метаноле (V=30 мл), к раствору добавляют раствор 25% NH4OH (V=1 мл) в воде (V=2 мл). Смесь перемешивали при комнатной температуре 12-15 ч, осадок отфильтровывали, промывали водой (V=5 мл) и сушили на воздухе. Получали продукт 1.12 (55 мг, 47%) в виде белого твердого вещества. 1H-NMR (DMSO-d6, 400 MHz): 1.28 (t, J=7.2 Hz, 3H), 3.22-3.33 (m, 2H), 3.89 (s, 2H), 4.31 (d, J=9.2 Hz, 2H), 4.69 (d, J=9.2 Hz, 2H), 7.17 (s, 1H), 7.71 (s, 1H), 8.7 (s, 1H), 8.85 (s, 1H), 12.3 (br, s. 1H).
Пример 11. {1-(3-Гидроксипропил)-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]-азетидин-3-ил}ацетонитрила трифторацетат (1.17·CF3CO2H) получали в соответствии со схемой 13.
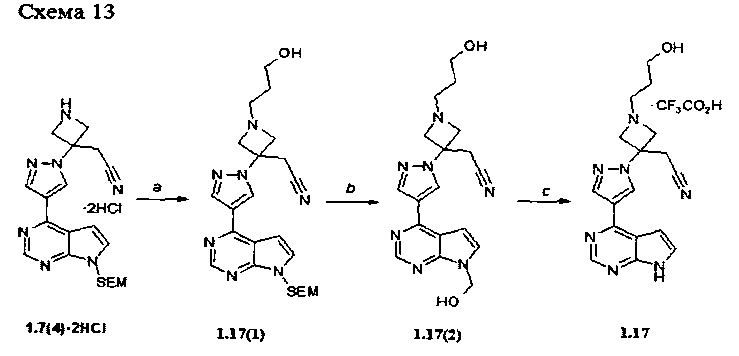
Стадия (а). {1-(3-Гидроксипропил)-3-[4-(7-[2-триметилсилилэтоксиметил]-7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]азетидин-3-ил}ацетонитрил (1.17(1)). К раствору {3-[4-(7-{[2-триметилсилилэтоксиметил}-7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]азетидин-3-ил}ацетонитрила дигидрохлорида (1.7(4)·2HCl: 500 мг, 1.036 ммоль, 1, 1 экв) в ДМФ (15 мл V=), содержащем K2CO3 (430 мг, 3.11 ммоль, 3 экв), добавляли 3-бромпропан-1-ол (173 мг, 1.24 ммоль, 1.2 экв). Смесь перемешивали при комнатной температуре 48 ч. К реакционной смеси добавляли воду и экстрагировали EtOAc (трижды по 20 мл). Объединенный экстракт промывали насыщенным раствором соли, сушили над Na2SO4 и выпаривали на роторном испарителе досуха. Продукт 1.17(1) используют на следующей стадии без дополнительной очистки.
Стадия (b, с). {1-(3-Гидроксипропил)-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил]-азетидин-3-ил}ацетонитрила трифторацетат (1.17·CF3CO2H).
{1-(3-Гидроксипропил)-3-[4-(7-[2-триметилсилилэтоксиметил]-7Н-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил]азетидин-3-ил}ацетонитрил (1.17(1): 410 мг, 0.876 ммоль) в метиленхлориде (V=10 мл) и трифторуксусной кислоте (V=5 мл) перемешивали в течение 2 ч. Растворители удаляли в вакууме. Остаток (смесь 1.17(1) и 1.17(2)) растворяли в метаноле (V=15 мл), к раствору добавляли раствор 25% NH4OH (V=1 мл) в воде (V=2 мл). Смесь перемешивали при комнатной температуре 12-15 ч. Реакционную смесь выпаривали на роторном испарителе досуха, к остатку добавляли 5% водный раствор K2CO3 (V=40 мл), экстрагировали EtOAc (трижды по 20 мл) и объединенный экстракт выпаривают на роторном испарителе досуха. Остаток очищали с помощью препаративной ВЭЖХ. Продукт 1.17·CF3CO2H получали в виде белого твердого вещества. 1H-NMR (DMSO-d6, 400 MHz): 0.33-0.44 (m, 2Н), 2.07 (t, J=7.2Hz, 2H), 2.18-2.30 (m, 4H), 3.22-3.38 (m, 4H), 3.54 (d, J=12Hz, 2H), 5.71 (d, J=3.6Hz, 1H), 6.27 (d, J=3.6Hz, 1H), 7.09 (s, 1H), 7.37 (s, 1H), 7.52 (s, 1H).
Аналогично было получено соединение{1-(3-гидрокси-пропан-1-сульфонил)-3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)пиразол-1-ил)]-азетидин-3-ил}-ацетонитрила (1.14).
Пример 12. Метиловый эфир {3-[4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-3-цианометил-азетидин-1-сульфонил}пропионовой кислоты (1.15) получали в соответствии со схемой 14.
Схема 14
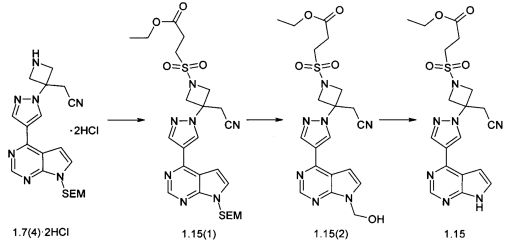
К раствору 2,6 г (5.4 ммоль) соединения 1.7(4)·2HCl в 100 мл дихлорметана добавляют 2,2 г (21.6 ммоль) триэтиламина, охлаждают до -20°С и добавляют по каплям 1,6 г (8.1 ммоль) этил 3-(хлорсульфонил)пропаноата. Полученную смесь перемешивают при комнатной температуре в течение ночи, упаривают в вакууме досуха, к остатку добавляют 70 мл воды, экстрагируют этилацетатом (трижды по 30 мл). Объединенный органический экстракт промывают насыщенным раствором соли, сушат над Na2SO4 и выпаривают в вакууме досуха. Остаток очищают с помощью колоночной хроматографии (силикагель, гексан: EtOAc = 2:1-1:1-1:20:1). Получают 2,3 г (74%) соединения 1.15(1) в виде бесцветного масла. 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9Н), 0.94 (t, J=8.4 Hz, 2H), 1.27 (t, J=7.2 Hz, 3H), 2.84 (t, J=7.2 Hz, 2H), 3.36-3.46 (m, 4H), 3.57 (t, J=8.4 Hz, 2H), 4.19 (q, J=7.2 Hz, 2H), 4.28 (d, J=9.2 Hz, 2H), 4.63 (d, J=9.2 Hz, 2H), 5.7 (s, 2H), 6.85 (d, J=4 Hz, 1H), 7.47 (d, J=4 Hz, 1H), 8.37 (s, 1H), 8.66 (br.s, 1H), 8.88 (s, 1H). Раствор 445 мг (0.776 ммоль) соединения 1.15(1) в 20 мл метиленхлорида и 10 мл трифторуксусной кислоты перемешивают в течение 3 ч. Растворители удаляли в вакууме. Остаток 1.15(2) растворяют в 15 мл этанола, к полученному раствору добавляют раствор 1 мл 25% NH4OH в 2 мл воды. Полученную смесь перемешивают при комнатной температуре в течение ночи. Осадок отфильтровывают и очищают с помощью колоночной хроматографии (силикагель, CHCl3 : МеОН = 19:1-10:1). Получают 150 мг соединения 1.15 в виде белого твердого вещества. 1H-NMR (DMSO-d6, 400 MHz): 1.16 (t, J=7.2 Hz, 3H), 2.75 (t, J=7.2 Hz, 2H), 3.5 (t, J=7.2 Hz, 2H), 3.69 (s, 2H), 4.05 (q, J=7.2 Hz, 2H), 4.26 (d, J=9.2 Hz, 2H), 4.6 (d, J=9.2 Hz, 2H), 7.06-7.10 (m, 1H), 7.60-7.64 (m, 1H), 8.47 (s, 1H), 8.71 (s, 1H), 8.93 (s, 1H), 12.14 (br.s, 1H).
Пример 13. {3-[4-(7Н-Пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-(3-фторпропансульфонил)-азетидин-3-ил}-ацетонитрил (1.16) и его дихлорацетат 1.16·CHCl2CO2H получали в соответствии со схемой 15.
Схема 15
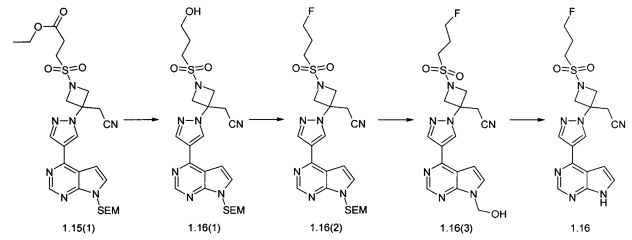
К раствору 1,7 г (3 ммоль) соединения 1.15(1) в 50 мл тетрагидрофурана добавляют 0,2 г (9.0 ммоль) LiBH4. Смесь перемешивают при комнатной температуре в течение ночи, затем добавляют 10 мл метанола и 250 мл воды. Полученную смесь экстрагируют этилацетатом (трижды по 30 мл), объединенные экстракты промывают водой, рассолом, сушат над Na2SO4, выпаривают в вакууме досуха, остаток очищают колоночной хроматографией (силикагель, гексан: EtOAc = 1:1-0:1, затем этилацетат : метанол = 9:1). Получают соединение 1.16(1). 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9Н), 0.94 (t, J=8.4 Hz, 2H), 2.06-2.16 (m, 2H), 2.71-2.80 (m, 1H), 3.28 (t, J=7.6 Hz, 2H), 3.45 (s, 2H), 3.57 (t, J=8 Hz, 2H), 3.79 (t, J=6 Hz, 2H), 4.32 (d, J=9.6 Hz, 2H), 4.6 (d, J=9.6 Hz, 2H), 5.71 (s, 2H), 6.88 (d, J=3.6 Hz, 1H), 7.5 (d, J=3.6 Hz, 1H), 8.36 (s, 1H), 8.81 (br.s, 1H), 8.87 (s, 1H). К раствору 800 мг (1.505 ммоль) соединения 1.16(1) в 20 мл дихлорметана добавляют при температуре -20°С 0,5 мл (606 мг, 3.76 ммоль) DAST. Смесь перемешивают при комнатной температуре в течение ночи, добавляют при 0°С 100 мл насыщенного водного раствора NaHCO3 и перемешивают в течение 20 мин. Полученную реакционную массу экстрагируют этилацетатом (трижды по 30 мл), объединенный экстракт промывают насыщенным раствором соли, сушат над Na2SO4 и выпаривают в вакууме досуха. Остаток очищают колоночной хроматографией (силикагель, гексан:EtOAc = 1:1-1:2). Получают соединение 1.16(2). 1H-NMR (CDCl3, 400 MHz): 0.0 (s, 9H), 0.94 (t, J=8 Hz, 2H), 2.17-2.34 (m, 2H), 3.18-3.26 (m, 2H), 3.42 (s, 2H), 3.57 (t, J=8 Hz, 2H), 4.28 (d, J=9.6 Hz, 2H), 4.54 (t, J=5.6 Hz, 1H), 4.62-4.7 (m, 3H), 5.7 (s, 2H), 6.82 (d, J=3.6 Hz, 1H), 7.45 (d, J=3.6 Hz, 1H), 8.37 (s, 1H), 8.51 (s, 1H), 8.87 (s, 1H).
Раствор 300 мг (0.562 ммоль) соединения 1.16(2) в 20 мл метиленхлориде и 10 мл трифторуксусной кислоты перемешивают при комнатной температуре в течение 3 ч. Растворители удаляют в вакууме, к остатку 1.16(3) добавляют 30 мл метанола и 3 мл раствора, полученного из 1 мл 25% NH4OH и 2 мл воды. Смесь перемешивают при комнатной температуре в течение ночи и упаривают в вакууме досуха. Остаток очищали с помощью колоночной хроматографии (силикагель, CHCl3:MeOH = 19:1-15:1). Получают 60 мг соединения 1.16 в виде белого твердого вещества. 1H-NMR (DMSO-d6, 400 MHz): 2.01-2.18 (m, 2H), 3.30-3.39 (m, 2H), 2.7 (s, 2H), 4.27 (d, J=9.2 Hz, 2H), 4.49 (t, J=6 Hz, 1H), 4.57-4.66 (m, 3H), 7.06-7.1 (m, 1H), 7.59-7.64 (m, 1H), 8.48 (s, 1H), 8.71 (s, 1H), 8.93 (s, 1H), 12.13 (br.s, 1H). К раствору 80,7 мг (0.2 ммоль) соединения 1.16 в 10 мл тетрагидрофурана добавляют 25.8 мг (0.2 ммоль) дихлоруксусной кислоты. Смесь упаривают на роторном испарителе досуха. Дихлорацетат 1.16·CHCl2CO2H получают в виде белого твердого вещества.
Пример 14. Определение ингибирующей активности соединений по отношению к JAK. Соединения тестировали в соответствии со стандартным протоколом Z`-Lyte скрининга компании Life Technologi. Тест соединения проверяются в 1% ДМСО (окончательный) в скважине. 100 нл 100x тестируемых соединений в 100% ДМСО добавляют 2,4 мкл киназного буфера (50 мМ HEPES, pH6.5, 0,01% BRIJ-35, 10 мМ MgCl2, 1 мМ EGTA, 0,02% NaN3) в условиях низкой объемной NBS, черный 384 -Ну пластины (Corning Кат. # 4514). После этого 5 мкл 2X пептида/киназы смесь (Tyr06/JAK1, окончательные концентрации 21.2-91.5 нг JAK1 и 2 мкм Тир 06) и 2,5 мкл 4xATP (конечная концентрация 75 мкМ) решения в киназы буфера добавляют. Через 30 сек на шейкере реакция киназы инкубировали в течение 60 мин при комнатной температуре. После этого 5 мкл 1:128 разбавлении развития реагента A и инкубируют в течение еще 60 мин при комнатной температуре. Флуоресценции была измерена при возбуждении при 400 нм и эмиссией при 445 нм и 520. Степень фосфорилирования FRET-пептида может быть рассчитана из соотношения выбросов. Коэффициент выбросов будет оставаться низким, если лада-пептид не фосфорилируется (т.е., не киназы ингибирование) и будет высоким, если лада-пептид нефосфорилированный (т.е. киназы ингибирование). % фосфорилирования рассчитывается по формуле, указанной на странице 6 в стандартном протоколе скрининга Z′-LYTE (Life Technologies).
Концентрационная кривая зависимости активности киназы от концентрации тестируемых веществ построена с использованием сигмоидной модели.
Данные активности соединений общей формулы 1 по отношению к JAK1 представлены в таблице 1.
Пример 15. Определение термодинамической растворимости соединения формулы (1) и прототипа Baricitinib. Смешивали 5 мг исследуемого соединения с 1 мл универсального буфера (pION) с рН 2,0, 4,0 или 7,0 в течение 15 мин при 25°С. Дополнительные количества веществ добавляли до тех пор, пока раствор не становился мутным. Виалы с раствором инкубировали при перемешивании в течение 24 ч при 25°С для достижения равновесия между раствором и осадком при насыщении. После уравновешивания 200 мкл раствора (в 2-х повторах) фильтровали через 96-луночный фильтровальный планшет (Millipore) для отделения осадка. Концентрацию соединений в фильтрате определяли спектрофотометрически с помощью стандартной калибровочной кривой. Проводили измерение спектра оптического поглощения вещества и построение калибровочной кривой при выбранной длине волны (обычно, соответствующей максимуму поглощения вещества λmax). Концентрацию вещества в фильтрате (т.е. растворимость) рассчитывали по нижеприведенной формуле:
Solubility = (ODλmaxfiltrate - ODλmax blank) / Slope × 1,67 × Filtrate dilution,
где: ODλmax filtrate - оптическая плотность фильтрата,
ODλmax blank - оптическая плотность холостого раствора без вещества,
Slope - наклон калибровочной кривой,
1,67 - фактор разведения фильтрата ацетонитрилом,
Filtrate dilution - фактор разведения фильтрата буфером.
Полученные результаты представлены в таблице 2.
Пример 16. Получение лекарственного средства в виде таблеток. Спрессовывали смесь 1600 мг крахмала, 1600 мг измельченной лактозы, 400 мг талька и 1000 мг ингибитора 1.16·CHCl2CO2H. Полученный брусок измельчали в гранулы и просеивали через сито, собирая гранулы размером 14-16 меш. Полученные гранулы таблетировали в таблетки, пригодных форм весом 500 мг каждая.
Пример 17. Получение лекарственного средства в форме капсул. Ингибитор 1.16·CHCl2CO2H тщательно смешивали с лактозой в соотношении 2: 1. Полученную порошкообразную смесь упаковывали по 600 мг в желатиновые капсулы подходящего размера.
Пример 18. Получение лекарственного средства в форме композиций для внутримышечных, внутрибрюшинных или подкожных инъекций. Смешали 500 мг ингибитора 1.16·CHCl2CO2H, 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл воды для инъекций. Раствор фильтровали и помещали в 1 мл в ампулы, которые укупоривали.
Из приведенного выше описания специалистам в данной области техники очевидны различные модификации изобретения, в дополнение к описанным здесь.
![{3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ](https://fips.edrid.ru/images/rid/ef/f8/79/4516e947601be9f057cfdcf65af43fd2.png)
![{3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ](https://fips.edrid.ru/images/rid/ef/f8/79/e5cd4d68c86d12d3be7fb631f29ae71c.png)
![{3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ](https://fips.edrid.ru/images/rid/ef/f8/79/d9c9ca033a1823ee02ded22f7696983e.png)
![{3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ](https://fips.edrid.ru/images/rid/ef/f8/79/ce69a66e3086d6cd29c271101f22f730.png)
![{3-[(7H-ПИРРОЛО[2,3-d]ПИРИМИДИН-4-ИЛ)АЗОЛИЛ]АЗЕТИДИН-3-ИЛ}АЦЕТОНИТРИЛЫ В КАЧЕСТВЕ ИНГИБИТОРОВ ЯНУС КИНАЗ](https://fips.edrid.ru/images/rid/ef/f8/79/94689ff1609ada077ff470b292727226.png)
Замещенные феноксиуксусные кислоты, их эфиры и амиды, включающие 2,6-диоксо-2,3,6,7-тетрагидро-1н-пурин-8-иловый фрагмент, - антагонисты аденозинового a рецептора и их применение
(3-арилсульфонилхинолин-8-ил)-диалкил-амины - селективные антагонисты серотониновых 5-ht рецепторов, способы их получения и применения
Алкил [2-(2-{5-[4-(4-{2-[1-(2-метоксикарбониламино-ацетил)-пирролидин-2-ил]-3н-имидазол-4-ил}-фенил)-бута-1,3-диинил]-1н-имидазол-2-ил}-пирролидин-1-ил)-2-оксо-этил]-карбамат, фармацевтическая композиция, лекарственное средство, способ лечения вирусных заболеваний
Замещенные (r)-3-(4-метилкарбамоил-3-фторфениламино)-тетрагидро-фуран-3-енкарбоновые кислоты и их эфиры, способ их получения и применения
Замещенные (2r,3r,5r)-3-гидрокси-(5-пиримидин-1-ил)тетрагидрофуран-2-илметил арил фосфорамидаты
Замещенные 4-{ [4-(3,3-диоксидо-1,3-бензоксатиол-6-ил)арилокси]метил} пиперидины как агонисты рецепторов gpr119, фармацевтическая композиция, способы их получения и применения
Замещенные пиразинопиримидиноны как блокаторы trpa1 каналов, фармацевтическая композиция, способы их получения и применения
Производные 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1h,6h-пиридо[4,3-d]пиримидин-2,4,7-триона в качестве ингибиторов мек1/2
Дихлорацетат n,n-дизамещенного n-[4-(1-метил-1н-индол-3-ил)-пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина в качестве модулятора egfr для лечения рака
Дихлорацетат {3-[4-(7h-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-этилсульфонил-азетидин-3-ил}-ацетонитрила в качестве ингибитора янус киназ
Замещенные феноксиуксусные кислоты, их эфиры и амиды, включающие 2,6-диоксо-2,3,6,7-тетрагидро-1н-пурин-8-иловый фрагмент, - антагонисты аденозинового a рецептора и их применение
(3-арилсульфонилхинолин-8-ил)-диалкил-амины - селективные антагонисты серотониновых 5-ht рецепторов, способы их получения и применения
Алкил [2-(2-{5-[4-(4-{2-[1-(2-метоксикарбониламино-ацетил)-пирролидин-2-ил]-3н-имидазол-4-ил}-фенил)-бута-1,3-диинил]-1н-имидазол-2-ил}-пирролидин-1-ил)-2-оксо-этил]-карбамат, фармацевтическая композиция, лекарственное средство, способ лечения вирусных заболеваний
Замещенные (r)-3-(4-метилкарбамоил-3-фторфениламино)-тетрагидро-фуран-3-енкарбоновые кислоты и их эфиры, способ их получения и применения
Замещенные (2r,3r,5r)-3-гидрокси-(5-пиримидин-1-ил)тетрагидрофуран-2-илметил арил фосфорамидаты
Замещенные 4-{ [4-(3,3-диоксидо-1,3-бензоксатиол-6-ил)арилокси]метил} пиперидины как агонисты рецепторов gpr119, фармацевтическая композиция, способы их получения и применения
Замещенные пиразинопиримидиноны как блокаторы trpa1 каналов, фармацевтическая композиция, способы их получения и применения
Производные 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1h,6h-пиридо[4,3-d]пиримидин-2,4,7-триона в качестве ингибиторов мек1/2
Дихлорацетат n,n-дизамещенного n-[4-(1-метил-1н-индол-3-ил)-пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина в качестве модулятора egfr для лечения рака
Дихлорацетат {3-[4-(7h-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-этилсульфонил-азетидин-3-ил}-ацетонитрила в качестве ингибитора янус киназ

