Результат интеллектуальной деятельности: МОДУЛЯТОРЫ ФОСФОДИЭСТЕРАЗЫ 10
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новым соединениям формулы (I)
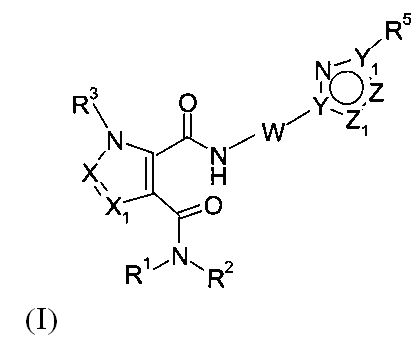 ,
,
в которых:
X и X1 независимо представляют собой CR4 или N;
Y и Y1 независимо представляют собой C или N;
Z и Z1 независимо представляют собой CR6, NR7, N, O или S;
R1 и R2 независимо выбраны из водорода, C1-C7-алкила, C3-C8-циклоалкила, C1-C7-галогеналкила, C1-C7-алкокси-C1-C7-алкила, гетероциклоалкила или C1-C7-алкила, возможно, замещенного арилом или гетероарилом, или
R1 и R2, совместно с атомом азота, к которому они присоединены, образуют бициклическую кольцевую систему или гетероциклоалкил, который может содержать от 1 до 3 заместителей, независимо выбранных из группы, включающей гидроксил, галоген, C1-C7-алкил, C1-C7-алкоксигруппу, C1-C7-галогеналкил и оксогруппу;
R3 представляет собой водород или C1-C7-алкил;
R4 представляет собой водород, C1-C7-алкил, C3-C8-циклоалкил, C1-C7-галогеналкил или галоген;
R5 представляет собой арил или гетероарил, при этом обе группы, возможно, содержат в качестве заместителя C1-C7-алкил, C3-C8-циклоалкил, галоген, C1-C7-галогеналкил, C1-C7-алкоксигруппу, гидроксил, C1-C7-гидроксиалкил, C1-C7-алкоксиалкил, ацетил, цианогруппу, аминогруппу, возможно, замещенную одной или двумя C1-C7-алкильными группами;
R6 представляет собой водород, галоген, C1-C7-алкил, C3-C8-циклоалкил, C1-C7-алкоксигруппу, C1-C7-галогеналкил, C1-C7-алкоксиалкил, гетероциклоалкил, арил, гетероарил, или C1-C7алкил, возможно, замещенный арилом, гетероарилом, гетероциклоалкилом, цикпоалкилом, или
R5 и R6, совместно с Υ1 и атомом Ζ, к которому они присоединены, образуют арил или гетероарил, при этом обе группы, возможно, замещены C1-C7-алкилом, галогеном, C1-C7-алкоксигруппой, C1-C7-галогеналкилом;
R7 представляет собой водород, галоген, C1-C7-алкил, C3-C8-циклоалкил, C1-C7-алкоксигруппу, C1-C7-галогеналкил, C1-C7-алкоксиалкил, гетероциклоалкил, арил, гетероарил, или C1-C7-алкил, возможно, замещенный арилом, гетероарилом, гетероциклоалкилом, цикпоалкилом, или
R5 и R7, совместно с Υ1 и атомом Ζ, к которому они присоединены, образуют арил или гетероарил, при этом обе группы, возможно, замещены C1-C7-алкилом, галогеном, C1-C7-алкоксигруппой и C1-C7-галогеналкилом;
W выбирают из этилена, этенилена, при этом обе группы, возможно, замещены C1-C7-алкилом или галогеном, или W представляет собой -N=CH-.
Кроме того, настоящее изобретение относится к способу получения раскрытых выше соединений, к фармацевтическим препаратам, содержащим такие соединения, а также к применению этих соединений для изготовления фармацевтических препаратов.
СУЩНОСТЬ ИЗОБРЕТЕНИЯ
Шизофрения представляет собой прогрессирующее и пагубное неврологическое заболевание, которое характеризуется эпизодическими позитивными симптомами, такими как бред, галлюцинации, нарушение процессов мышления и психоз, и постоянными негативными симптомами, такими как эмоциональная тупость, нарушение внимания и социальное очуждение, а также нарушениями когнитивной функции (Lewis DA and Lieberman JA, Neuron, 28:325-33, 2000). На протяжении десятилетий внимание исследователей было сфокусировано на гипотезе "дофаминергической гиперактивности", которая привела к терапевтическим вмешательствам, включающим блокаду дофаминергической системы (Vandenberg RJ and Aubrey KR., Exp. Opin. Ther. Targets, 5(4):507-518, 2001; Nakazato A and Okuyama S, et al., Exp. Opin. Ther. Patents, 10(1):75-98, 2000). Данный фармакологический подход, хотя и смягчает положительные симптомы у пациентов с шизофренией, оказывает слабый эффект на негативные и когнитивные симптомы, которые представляют собой наилучшие критерии, позволяющие оценить функциональный результат (Sharma Т., Br. J. Psychiatry, 174 (suppl. 28):44-51, 1999). Кроме того, современное антипсихотическое лечение связано с побочными действиями, такими как увеличение веса, экстрапирамидальные симптомы или воздействие на метаболизм глюкозы и липидов, которые связаны с его неспецифической фармакологией.
Таким образом, до сих пор сохраняется необходимость в разработке новых антипсихотиков с улучшенными эффективностью и профилем безопасности. В середине 1960-х годов была выдвинута комплементарная модель шизофрении, которая основана на психотомиметическом действии, вызванном блокадой глутаматной системы такими соединениями, как фенциклидин (PCP) и родственные агенты (кетамин), которые представляют собой неконкурентные антагонисты NMDA. Удивительно, что у здоровых добровольцев PCP-индуцированное психотомиметическое действие включает позитивные и негативные симптомы, а также когнитивную дисфункцию, и, таким образом, очень напоминает шизофрению у пациентов (Javitt DC et al., Biol. Psychiatry, 45:668-679, 1999).
Циклические нуклеотиды циклический аденозин монофосфат (cAMP) и циклический гуанозин монофосфат (cGMP) представляют собой распространенные вторичные мессенджеры, ответственные за передачу биологического ответа от различных внеклеточных сигналов, включая нейротрансмиттеры, свет и гормоны. cAMP и cGMP регулируют множество внутриклеточных процессов, в частности, в нейронах центральной нервной системы, путем активации cAMP- и cGMP-зависимых киназ, которые далее фосфорилируют белки, участвующие в регуляции синаптической передачи, дифференцировки нейронов и их выживаемости.
Ключевой механизм для контроля внутриклеточного уровня циклических нуклеотидов и, следовательно, сигнального пути циклических нуклеотидов, опосредован гидролизом 3′,5′-фософдиэфирной связи фосфодиэстеразами. Фосфодиэстеразы (PDEs) представляют собой семейство повсеместно экспрессируемых ферментов, которые кодируются 21 различными генами у человека, при этом каждый ген кодирует несколько сплайс-вариантов (Beavo, J., Physiol. Rev. 1995, 75, 725-748; Conti, M., Jin, S.L., Prog. Nucleic Acid Res. Mol. Biol. 1999, 63, 1-38; Soderling, S.H., Beavo, J.A., Curr. Opin. Cell Biol. 2000,12, 174-179, Manallack, D.T. et al. J. Med. Chem. 2005, 48(10), 3449-3462).
Семейства фосфодиэстераз (PDE) различаются по своей субстратной специфичности к циклическим нуклеотидам, их механизму регуляции и чувствительности к ингибиторам. Кроме того, они различным образом локализованы в организме, среди клеток органа, и даже внутри клеток. Эти различия приводят к дифференцированной роли семейств PDE в различных физиологических функциях.
PDE10A представляет собой двухсубстратную фосфодиэстеразу, кодируемую единственным геном, как сообщалось в 1999 году тремя независимыми исследовательскими группами (Fujishige K., et al., Eur J Biochem (1999) 266(3):1118-1127, Soderling S.H., et al., ProcNatl Acad Sci USA (1999) 96(12):7071-7076, Loughney K., et al., Gene (1999) 234(1):109-117). Фосфодиэстераза PDE10A обладает уникальной особенностью, по сравнению остальными членами мультигенного семейства, в отношении аминокислотной последовательности (779 а.к.), тканеспецифического профиля экспрессии, сродства к cAMP и cGMP и воздействия на фосфодиэстеразную активность специфических и общих ингибиторов.
Фосфодиэстераза PDE10A является одним из наиболее ограниченно распространенных членов семейства фосфодиэстераз, и этот фермент в первую очередь экспрессируется в головном мозге, в частности, в прилежащем ядре и дорсальном стриатуме. Кроме того, в таламусе, обонятельной луковице, гиппокампе и лобной доле присутствует умеренный уровень экспрессии фосфодиэстеразы PDE10A. Считается, что все эти участи мозга вовлечены в патофизиологию шизофрении и психоза, что свидетельствует о ключевой роли фосфодиэстеразы PDE10A в этом пагубном умственном заболевании. За пределами центральной нервной системы экспрессия транскрипта фосфодиэстеразы PDE10A наблюдается также и в периферических тканях, таких как щитовидная железа, гипофиз, инсулин-секретирующие клетки поджелудочной железы и семенники (Fujishige, K. et al., J. Biol. Chem. 1999, 274, 18438-18445, Sweet, L. (2005) WO 2005/012485). С другой стороны, экспрессия белка фосфодиэстеразы PDE10A, наблюдается только в кишечных ганглиях, в семенниках и сперме эпидидимиса (Coskran T.M; et al., J. Histochem. Cytochem. 2006, 54(11), 1205-1213).
В стриатуме как мРНК, так и белок экспрессируются только в содержащих GABA (γ-аминомасляную кислоту) средних проекционных шипиковых нейронах, что обуславливает их привлекательность в качестве мишени для лечения заболеваний центральной нервной системы (Fujishige, K. et al., Eur. J. Biochem. 1999, 266, 1118-1127; Seeger, T.F. et al., Brain Res. 2003, 985, 113-126). Стриарные средние шипиковые нейроны являются главным сайтом ввода и первым сайтом для интеграции информации в канале базальных ганглий головного мозга млекопитающих. Базальные ганглии представляют собой последовательность связанных между собой подкорковых ядер, которые интегрируют обширный кортикальный входной поток посредством дофаминергической сигнальной системы в плановые и исполняемые значимые двигательные образы и когнитивные профили, подавляя при этом нежелательные или несущественные образы (Graybiel, A.M. Curr. Biol. 2000, 10, R509-R511 (2000)).
Папаверин, относительно специфичный ингибитор фосфодиэстеразы PDE10A, и PDEIOA-нокаутированные мыши были использованы для исследования физиологии этого фермента и возможной терапевтической пользы ингибирования PDE10A. Ингибирование этого фермента, фармакологически или путем разрушения гена, вызывает уменьшение активности и сниженный отклик на психомоторные стимуляторы. Ингибирование также снижает условную реакцию избегания, поведенческую реакцию, которая является признаком клинической антипсихотической активности (Siuciak, J.A.; et al., Neuropharnacology 2006, 51(2), 386-396; Siuciak, J.A.; et al., Neuropharnacology 2006, 51(2), 374-385).
Кроме того, ингибирование фосфодиэстеразы PDE10A обладает потенциалом к улучшению негативных и когнитивных симптомов, связанных с шизофренией. Действительно, показали, что папаверин смягчает дефицит способности к обучению с экстрамерным сдвигом, индуцированный у крыс путем суб-хронического воздействия с помощью PCP, в животной модели гипофункции рецептора NMDA (Rodefer, J,S., et al., Eur. J. Neuroscience 2005, 2: 1070-1076). Кроме того, у PDE10A2-дефицитных мышей наблюдалось увеличение социального взаимодействия (Sano, H.J. Neurochem. 2008, 105, 546-556).
Заболевания, которые можно лечить с помощью ингибиторов PDE10A, включают, без ограничения, заболевания, которые предположительно опосредованы частично дисфункцией базальных ядер, других частей центральной нервной системы и других PDE10A-экспрессирующих тканей. В частности, можно лечить заболевания, при которых ингибирование фосфодиэстеразы PDE10A может оказывать терапевтическое воздействие.
Такие заболевания включают, без ограничения, конкретные психические расстройства, такие как шизофрения, позитивные, негативные и/или когнитивные симптомы, связанные с шизофренией, бредовое расстройство или интоксикационное психическое расстройство, тревожные расстройства, такие как паническое расстройство, синдром навязчивых состояний, острое стрессовое расстройство или общее тревожное расстройство, наркозависимость, двигательные расстройства, такие как болезнь Паркинсона или синдром беспокойных ног, расстройства с дефицитом познавательной способности, такие как болезнь Альцгеймера или мультиинфарктная деменция, расстройства настроения, такие как депрессия или биполярные расстройства, или психоневрологические состояния, такие как психоз, синдром гиперактивности с дефицитом внимания (СГДВ) или сопутствующие расстройства внимания.
Соединения по настоящему изобретению можно также применять для лечения диабета и связанных с ним расстройств, таких как тучность, посредством регулирования cAMP-опосредованной сигнальной системы.
Ингибиторы PDE10A могут быть также полезны для предотвращения апоптоза нейронов при повышении уровней cAMP и cGMP и, таким образом, могут обладать противовоспалительными свойствами. Нейродегенеративные расстройства, которые можно лечить с помощью ингибиторов PDE10A, включают, без ограничения, болезнь Альцгеймера, болезнь Гентингтона, болезнь Паркинсона, множественный склероз, инсульт или повреждение спинного мозга. Посредством cAMP и cGMP ингибируется рост раковых клеток. Поэтому, за счет повышения уровня cAMP и cGMP, ингибиторы PDE10A можно применять также для лечения различных твердых опухолей и гематологических новообразований, таких как почечно-клеточный рак или рак молочной железы.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Если не указано иное, ниже следующие понятия приведены, чтобы наглядно представить и определить значение и объем различных терминов, использованных в данном тексте для описания настоящего изобретения.
Следует заметить, что используемые в описании и формуле изобретения формы единственного числа включают ссылки на формы множественного числа, если из контекста однозначно не следует противоположное.
Если указано число заместителей, термин "один или более" обозначает от одного заместителя до максимально возможного числа заместителей, т.е. замещение от одного атома водорода и вплоть до замещения всех атомов водорода заместителями.
Термин "алкил" обозначает моновалентную линейную или разветвленную насыщенную углеводородную группу, включающую от 1 до 12 атомов углерода. В частных воплощениях алкил включает от 1 до 7 атомов углерода, и в более частных воплощениях - от 1 до 4 атомов углерода. Примеры алкила включают метил, этил, пропил, изопропил, н-бутил, изобутил, втор-бутил или трет-бутил.
Термин "алкилен" обозначает линейную насыщенную двухвалентную углеводородную группу, включающую от 1 до 7 атомов углерода, или двухвалентную разветвленную насыщенную двухвалентную углеводородную группу, включающую от 3 до 7 атомов углерода. Примеры алкиленовых групп включают метилен, этилен, пропилен, 2-метилпропилен, бутилен, 2-этилбутилен, пентилен, гексилен.
Термин "алкенилен" обозначает линейную двухвалентную углеводородную цепь, включающую от 2 до 7 атомов углерода, или разветвленную двухвалентную углеводородную цепь, включающую от 3 до 7 атомов углерода, включая по меньшей мере одну двойную связь. Примеры алкенилена включают этенилен, 2,2-диметилэтенилен, пропенилен, 2-метилпропенилен, бутенилен, и пентенилен. Термин "алкокси" обозначает группу формулы -O-R′, где R′ представляет собой алкильную группу. Примеры алкоксигрупп включают метокси-, этокси-, изопропокси- и трет-бутоксигруппу.
Термин "алкоксиалкил" обозначает алкильную группу, в которой по меньшей мере один из атомов водорода алкильной группы замещен алкоксигруппой. Примеры алкоксиалкильных групп включают 2-метоксиэтил, 3-метоксипропил, 1-метил-2-метоксиэтил, 1-(2-метоксиэтил)-3-метоксипропил и 1-(2-метоксиэтил)-3-метоксипропил.
Термин "амино" обозначает группу формулы -NR′R″, в которой R′ и R″ независимо представляют собой водород, алкил, алкоксигруппу, циклоалкил, гетероциклоалкил, арил или гетероарил. Как вариант, R′ и R″, совместно с атомом азота, к которому они присоединены, могут образовывать гетероциклоалкил. Термин "первичная аминогруппа" обозначает группу, в которой оба радикала R′ и R″ представляют собой водород. Термин "вторичная аминогруппа" обозначает группу, в которой R′ представляет собой атом водорода, a R″ не является атомом водорода. Термин "третичная аминогруппа" обозначает группу, в которой оба R′ и R″ не являются атомами водорода. Примерами вторичных и третичных аминов являются метиламин, этиламин, пропиламин, изопропиламин, фениламин, бензиламин, диметиламин, диэтиламин, дипропиламин и диизопропиламин.
Термин "циклоалкил" обозначает моновалентную насыщенную моноциклическую или бициклическую углеводородную группу, включающую от 3 до 10 кольцевых атомов углерода. В частных воплощениях циклоалкил обозначает моновалентную насыщенную моноциклическую углеводородную группу, включающую от 3 до 8 кольцевых атомов углерода. Термин "бициклическая" означает, что такая группа состоит из двух насыщенных карбоциклов, имеющих один или более общих атомов углерода. В частности, циклоалкильные группы являются моноциклическими. Примерами моноциклического циклоалкила являются циклопропил, циклобутил, циклопентил, циклогексил или циклогептил. Примерами бициклического циклоалкила являются бицикло[2.2.1]гептил или бицикло[2.2.2]октил.
Термин "галоген" обозначает фтор, хлор, бром и иод, в частности, фтор, хлор и бром.
Термин "галогеналкил" обозначает алкильную группу, в которой по меньшей мере один из атомов водорода алкильной группы замещен одинаковыми или разными атомами галогена, в частности, атомами фтора. Примеры галогеналкила включают монофтор-, дифтор- или трифтор-метил, -этил или -пропил, например 3,3,3-трифторпропил, 2-фторэтил, 2,2,2-трифторэтил, фторметил или трифторметил. Термин "пергалогеналкил" обозначает алкильную группу, в которой все атомы водорода алкильной группы замещены одинаковыми или разными атомами галогена.
Термин "галогеналкокси" обозначает алкоксигруппу, в которой по меньшей мере один из атомов водорода алкоксигруппы замещен одинаковыми или разными атомами галогена, в частности, атомами фтора. Примеры галогеналкоксила включают монофтор-, дифтор- или трифтор-метокси-, -этокси- или -пропоксигруппу, например 3,3,3-трифторпропокси-, 2-фторэтокси-, 2,2,2-трифторэтокси-, фторметокси- или трифторметоксигруппу. Термин "пергалогеналкокси" обозначает алкоксигруппу, в которой все атомы водорода алкоксигруппы замещены одинаковыми или разными атомами галогена.
Термин "гидроксиалкил" обозначает алкильную группу, в которой по меньшей мере один из атомов водорода алкильной группы замещен гидроксигруппой. Примеры гидроксиалкила включают гидроксиметил, 2-гидроксиэтил, 2-гидроксипропил, 3-гидроксипропил, 1-(гидроксиметил)-2-метилпропил, 2-гидроксибутил, 3-гидроксибутил, 4-гидроксибутил, 2,3-дигидроксипропил, 2-гидрокси-1-гидроксиметилэтил, 2,3-дигидроксибутил, 3,4-дигидроксибутил или 2-(гидроксиметил)-3-гидроксипропил.
Термин "гетероциклоалкил" обозначает моновалентную насыщенную или частично ненасыщенную моно- или бициклическую кольцевую систему, включающую от 3 до 9 кольцевых атомов, в том числе 1, 2, или 3 кольцевых гетероатома, выбранных из N, O и S, при этом оставшиеся кольцевые атомы представляют собой атомы углерода. В частных воплощениях гетероциклоалкил представляет собой моновалентную насыщенную моноциклическую кольцевую систему, включающую от 4 до 7 кольцевых атомов, в том числе 1, 2 или 3 кольцевых гетероатома, выбранных из N, O и S, при этом оставшиеся кольцевые атомы представляют собой атомы углерода. Примерами моноциклического насыщенного гетероциклоалкила являются азиридинил, оксиранил, азетидинил, оксетанил, пирролидинил, тетрагидрофуранил, тетрагидротиенил, пиразолидинил, имидазолидинил, оксазолидинил, изоксазолидинил, тиазолидинил, пиперидинил, тетрагидропиранил, тетрагидротиопиранил, пиперазинил, морфолинил, тиоморфолинил, 1,1-диоксотиоморфолин-4-ил, азепанил, диазепанил, гомопиперазинил или оксазепанил. Примерами бициклического насыщенного гетероциклоалкила являются 8-аза-бицикло[3.2.1]октил, хинуклидинил, 8-окса-3-аза-бицикло[3.2.1]октил, 9-аза-бицикло[3,3.1]нонил, 3-окса-9-аза-бицикло[3,3.1]нонил или 3-тиа-9-аза-бицикло[3,3.1]нонил. Примеры частично ненасыщенного гетероциклоалкила являются дигидрофурил, имидазолинил, дигидрооксазолил, тетрагидропиридинил или дигидропиранил.
Термин "оксо", в случае когда он относится к заместителям при гетероциклоалкиле, означает, что к гетероциклоалкильному кольцу присоединен атом кислорода. При этом "оксо"-группа может либо замещать два атома водорода при атоме углерода, либо просто быть присоедина к атому серы, при этом сера присутствует в окисленной форме, т.е. несет один или два атома кислорода.
Термин "арил" обозначает моновалентную ароматическую карбоциклическую моно- или бициклическую кольцевую систему, включающую от 6 до 10 кольцевых атомов углерода. Примеры арильных групп включают фенил и нафтил.
Термин "гетероарил" обозначает моновалентную ароматическую гетероциклическую моно- или бициклическую кольцевую систему, включающую от 5 до 12 кольцевых атомов, включая 1, 2, 3 или 4 гетероатома, выбранных из N, O и S, при этом оставшиеся кольцевые атомы представляют собой атомы углерода. Примеры гетероарильных групп включают пирролил, фуранил, тиенил, имидазолил, оксазолил, тиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиразинил, пиразолил, пиридазинил, пиримидинил, триазинил, азепинил, диазепинил, изоксазолил, бензофуранил, изотиазолил, бензотиенил, индолил, изоиндолил, изобензофуранил, бензимидазолил, бензоксазолил, бензоизоксазолил, бензотиазолил, бензоизотиазолил, бензооксадиазолил, бензотиадиазолил, бензотриазолил, пуринил, хинолинил, изохинолинил, хиназолинил или хиноксалинил.
Термин "бициклическая кольцевая система" обозначает два кольца, которые конденсированы друг с другом через общую одинарную или двойную связь (аннелированная бициклическая кольцевая система), через последовательность из трех или более общих кольцевых атомов (мостиковая бициклическая кольцевая система) или через общий единственный атом (спиро-бициклическая кольцевая система). Бициклические кольцевые системы могут быть насыщенными, частично ненасыщенными, ненасыщенными или ароматическими. Бициклические кольцевые системы могут включать гетероатомы, выбранные из N, O и S.
Термин "возможный" или "возможно" означает, что описанное далее событие или обстоятельство может, но необязательно, иметь место, и что данное описание включает случаи, в которых данное событие или обстоятельство имеют место, и случаи, в которых они не имеют место.
Соединения формулы (I) могут образовывать фармацевтически приемлемые соли. Примерами таких фармацевтически приемлемых солей являются соли соединений формулы (I) с физиологически совместимыми минеральными кислотами, такими как хлороводородная кислота, серная кислота, сернистая кислота или фосфорная кислота; или с органическими кислотами, такими как метансульфоновая кислота, п-толуолсульфоновая кислота, уксусная кислота, молочная кислота, трифторуксусная кислота, лимонная кислота, фумаровая кислота, малеиновая кислота, винная кислота, янтарная кислота или салициловая кислота.
Термин "фармацевтически приемлемые соли" относится к таким солям. Соединения формулы (I), включающие кислотную группу, такую как, например, группа COOH, могут дополнительно образовывать соли с основаниями. Примерами таких солей являются соли щелочных металлов, щелочно-земельных металлов и аммония, например, такие как соли Na-, Κ-, Ca- и триметиламмония. Термин "фармацевтически приемлемые соли" также относится к таким солям. В частности, соли представляют собой такие соли, которые получены за счет добавления кислоты.
Термин "фармацевтически приемлемые эфиры" обозначает производные соединений по настоящему изобретению, в котором карбоксигруппа была превращена в эфирную группу, при этом карбоксигруппа обозначает -C(O)O-. Примерами таких подходящих эфиров являются метиловые, этиловые, метоксиметиловые, метилтиометиловые и пивалоилоксиметиловые эфиры. Кроме того, термин "фармацевтически приемлемые эфиры" охватывает производные соединений по настоящему изобретению, в которых гидроксигруппы были превращены в соответствующие эфиры с неорганическими или органическими кислотами, такими как азотная кислота, серная кислота, фосфорная кислота, лимонная кислота, муравьиная кислота, малеиновая кислота, уксусная кислота, янтарная кислота, винная кислота, метансульфоновая кислота или п-толуолсульфоновая кислота, и которые не токсичны для живых организмов.
Настоящее изобретение касается новых соединений формулы (I):
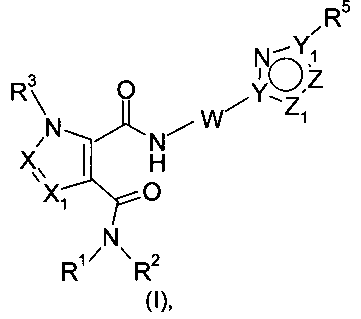
где:
X и X1 независимо представляют собой CR4 или N;
Y и Y1 независимо представляют собой C или N;
Z и Z1 независимо представляют собой CR6, NR7, N, O или S;
R1 и R2 независимо выбраны из водорода, C1-C7-алкила, C3-C8-циклоалкила, C1-C7-галогеналкила, C1-C7-алкокси-C1-C7-алкила, гетероциклоалкила или C1-C7-алкила, возможно, замещенного арилом или гетероарилом, или
R1 и R2, совместно с атомом азота, к которому они присоединены, образуют бициклическую кольцевую систему или гетероциклоалкил, который может содержать от 1 до 3 заместителей, независимо выбранных из группы, включающей гидроксил, галоген, C1-C7-алкил, C1-C7-алкоксигруппу, C1-C7-галогеналкил и оксогруппу;
R3 представляет собой водород или C1-C7-алкил;
R4 представляет собой водород, C1-C7-алкил, C3-C8-циклоалкил, C1-C7-галогеналкил или галоген;
R5 представляет собой арил или гетероарил, при этом обе группы, возможно, содержат в качестве заместителя C1-C7-алкил, C3-C8-циклоалкил, галоген, C1-C7-галогеналкил, C1-C7-алкоксигруппу, гидроксил, C1-C7-гидроксиалкил, C1-C7-алкоксиалкил, ацетил, цианогруппу, аминогруппу, возможно, замещенную одной или двумя C1-C7-алкильными группами;
R6 представляет собой водород, галоген, C1-C7-алкил, C3-C8-циклоалкил, C1-C7-алкоксигруппу, C1-C7-галогеналкил, C1-C7-алкоксиалкил, гетероциклоалкил, арил, гетероарил или C1-C7-алкил, возможно, замещенный арилом, гетероарилом, гетероциклоалкилом, циклоалкилом, или
R5 и R6, совместно с Y1 и атомом Z, к которому они присоединены, образуют арил или гетероарил, при этом обе группы, возможно, замещены C1-C7-алкилом, галогеном, C1-C7-алкоксигруппой и C1-C7-галогеналкилом;
R7 представляет собой водород, галоген, C1-C7-алкил, C3-C8-циклоалкил, C1-C7-алкоксигруппу, C1-C7-галогеналкил, C1-C7-алкоксиалкил, гетероциклоалкил, арил, гетероарил, или C1-C7-алкил, возможно, замещенный арилом, гетероарилом, гетероциклоалкилом, циклоалкилом, или
R5 и R7, совместно с Y1 и атомом Z, к которому они присоединены, образуют арил или гетероарил, при этом обе группы, возможно, замещены C1-C7-алкилом, галогеном, C1-C7-алкоксигруппой, C1-C7-галогеналкилом;
W выбирают из этилена и этенилена, при этом обе группы, возможно, замещены C1-C7-алкилом или галогеном, или W представляет собой -N=CH-.
В одном частном воплощении настоящее изобретение относится к соединениям формулы (I), описанным выше, в которых W представляет собой этилен, возможно замещенный C1-C7-алкилом.
В другом частном воплощении настоящее изобретение относится к соединениям формулы (I), описанным выше, в которых R1 и R2, совместно с атомом азота, к которому они присоединены, образуют 4-, 5- или 6-членный гетероциклоалкил, содержащий два гетероатома, выбранных из N и O, предпочтительно азетидинил или морфолинил,
R3 представляет собой водород или метил и
X представляет собой азот и X1 представляет собой CR4, при этом R4 представляет собой водород.
В другом частном воплощении настоящее изобретение относится к соединениям формулы (I), описанным выше, в которых R1 и R2, совместно с атомом азота, к которому они присоединены, образуют 4, 5 или 6-членный гетероциклоалкил, содержащий два гетероатома, выбранных из N и O, предпочтительно азетидинил или морфолинил,
R3 представляет собой водород или метил, и
X представляет собой CR4, при этом R4 представляет собой метил или галоген, и X1 представляет собой азот.
В одном воплощении настоящее изобретение относится к соединениям формулы (I), описанным выше, в которых Y представляет собой C, и Y1 представляет собой C или N.
В одном воплощении настоящее изобретение относится к соединениям формулы (I), описанным выше, в которых Y представляет собой N, и Y1 представляет собой C.
Еще в одном частном воплощении настоящее изобретение относится к соединениям формулы (I), описанным выше, в которых Z представляет собой C, а Z1 представляет собой N, или Z представляет собой N, а Z1 представляет собой C или O.
В другом частном воплощении настоящее изобретение относится к соединениям формулы (I), описанным выше, в которых Y1 представляет собой C, Z представляет собой CR6 или NR7, при этом R5 и R6 или R5 и R7 вместе с Y1 и атомом Z, к которому они присоединены, образуют гетероарил, выбранных из возможно замещенного имидазопиридинила и возможно замещенного бензоимидазолила.
В другом частном воплощении настоящее изобретение относится к соединениям формулы (I), описанным выше, в которых R5 выбирают из фенила, пиридинила, возможно, замещенного галогеном или C1-C7 алкоксигруппой.
Еще в одном частном воплощении настоящее изобретение относится к соединениям формулы (Ib):
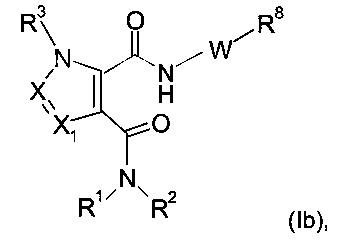
где R8 выбирают из группы, включающей:
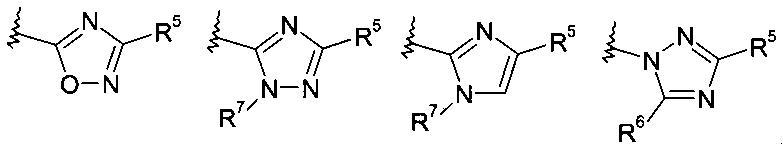
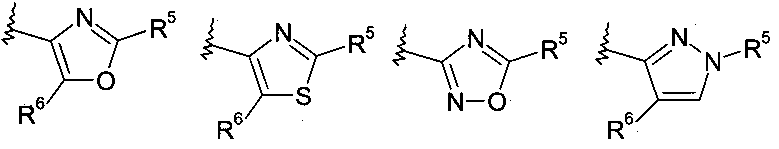
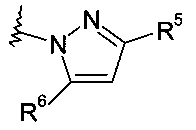 .
.
В другом частном воплощении настоящее изобретение относится к соединениям формулы (Ib), где R8 выбирают из группы, включающей:
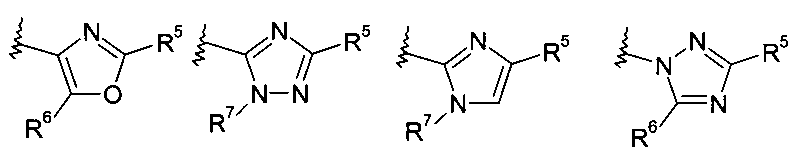
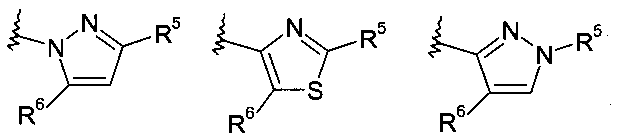 .
.
В другом частном воплощении настоящее изобретение относится к соединению формулы (I) и (Ib), в котором R6 выбирают из водорода, C1-C7-алкила и гетероарила.
Еще в одном частном воплощении настоящее изобретение относится к соединениям формулы (I) и (Ib), в которых R7 выбирают из водорода, C3-C8-циклоалкила, C1-C7-алкоксигруппы, C1-C7-галогеналкила, C1-C7-алкоксиалкила, гетероциклоалкила, арила, гетероарила или C1-C7-алкила, возможно, замещенного арилом, гетероарилом, гетероциклоалкилом, C3-C8-циклоалкилом. Еще в одном частном воплощении настоящее изобретение относится к соединению как описано выше, в котором R5 выбирают из фенила, пиридинила, возможно, замещенного галогеном или C1-C7алкоксигруппой.
В частности, соединения формулы (I) выбирают из группы, включающей следующие соединения:
(2-имидазо[1,2-a]пиридин-2-ил-этил)-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(3-фенил-[1,2,4]оксадиазол-5-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(8-метил-имидазо[1,2-a]пиридин-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
{2-[3-(4-хлорфенил)-[1,2,4]оксадиазол-5-ил]-этил}-амид 4-(азетидин-1-карбонил)-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 2,3-диметил-5-(морфолин-4-карбонил)-3H-имидазол-4-карбоновой кислоты
(2-бензотиазол-2-ил-этил)-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-3H-имидазол-4-карбоновой кислоты
[2-(1,5-диметил-1H-бензоимидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(3-фенилпиразол-1-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-2-хлор-метил-3H-имидазол-4-карбоновой кислоты
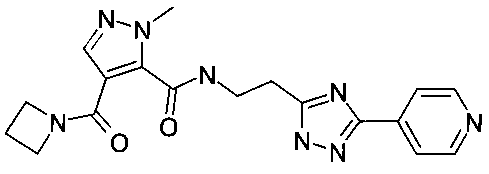
[2-(5-пиридин-4-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
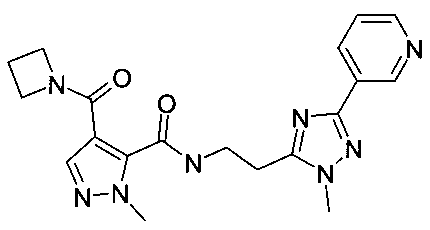
[2-(2-метил-5-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
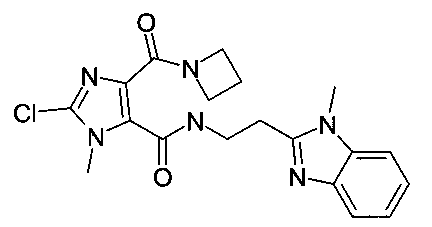
[2-(5-хлор-1-метил-1H-бензоимидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-метил-5-пиридин-4-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(3-фенил-[1,2,4]триазол-1-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-фенил-2H-[1,2,3]триазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-фенил-[1,2,4]оксадиазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-метил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[1,1-диметил-2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 2-метил-4-(морфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты
[1,1-диметил-2-(3-фенил-[1,2,4]оксадиазол-5-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-пиримидин-2-илтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-4-пиридин-3-ил-1H-имидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-фенил-1H-пиразол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-фенилтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
{2-[4-(3-метоксифенил)-1-метил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
{2-[4-(2-метоксифенил)-1-метил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
{2-[1-(2-метоксиэтил)-4-фенил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-2,3-диметил-3H-имидазол-4-карбоновой кислоты
{2-[2-(2-метоксиэтил)-5-фенил-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-циклопропилметил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
2-метил-4-(морфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты [2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид
[2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 2-метил-4-(пирролидин-1-карбонил)-2H-пиразол-3-карбоновой кислоты
[2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 4-(3-фторазетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
4-(циклопропилметиламид)3-{[2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид}2-метил-2H-пиразол-3,4-дикарбоновой кислоты
[2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 2-метил-4-(2-окса-6-аза-спиро[3,3]гептан-6-карбонил)-2H-пиразол-3-карбоновой кислоты
[2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 2-метил-4-(тиоморфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты
[2-(5-метил-2-фенилтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-метил-3-фенил-[1,2,4]триазол-1-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-бензил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-метил-2-п-толилтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(4-метил-1-фенил-1H-пиразол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
{2-[2-(2-этилпиридин-4-ил)-5-метилтиазол-4-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
{2-[5-фенил-2-(2,2,2-трифторэтил)-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 2-метил-4-(морфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 2-метил-4-(пирролидин-1-карбонил)-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-3H-[1,2,3]триазол-4-карбоновой кислоты
4-(циклопропилметиламид) 3-{[2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид}2-метил-2H-пиразол-3,4-дикарбоновой кислоты
[2-(5-метил-4-фенилтиазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2,5-дифенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-циклопропил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-фенил-2-пиридин-2-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2,5-дифенилоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-фенил-2-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
{2-[5-(3-фторфенил)-2-фенил-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
4-(циклопентилметиламид)-3-{[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид}2-метил-2H-пиразол-3,4-дикарбоновой кислоты
4-(циклобутилметиламид)-3-{[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид}2-метил-2H-пиразол-3,4-дикарбоновой кислоты
[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-2-трифторметил-3H-имидазол-4-карбоновой кислоты
(2-[1,2,4]триазоло[1,5-a]пиридин-2-ил-этил)-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-фенил-5-пиридин-3-илоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
В частности, соединения формулы (I) выбирают также из группы, включающей следующие соединения:
{2-[1-(2-метоксиэтил)-4-фенил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(1-метил-4-пиридин-3-ил-1H-имидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-метил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-метил-5-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-хлор-1-метил-1H-бензоимидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-метил-5-пиридин-4-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-фенил-2H-[1,2,3]триазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-фенил-[1,2,4]оксадиазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[1,1-диметил-2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[1,1-диметил-2-(3-фенил-[1,2,4]оксадиазол-5-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-циклопропилметил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2,5-дифенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(2-циклопропил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-фенил-2-пиридин-2-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
[2-(5-фенил-2-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
4-(циклобутилметиламид)-3-{[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид}2-метил-2H-пиразол-3,4-дикарбоновой кислоты.
Кроме того, настоящее изобретение касается способа получения соединения формулы (I), раскрытого выше, включающего введение в реакцию соединения формулы (II):
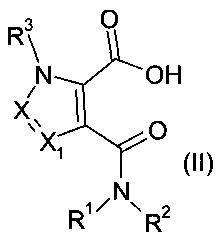
с соединением формулы (III)
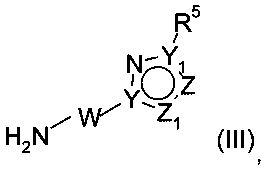
где R1, R2, R3, R5, W, X, X1, Y, Y1, Z и Z1 таковы, как определено выше.
Реакцию соединения формулы (II) с соединением формулы (III) можно проводить в условиях, описанных в примерах, или в условиях, хорошо известных специалисту в данной области техники. Например, эту реакцию можно проводить в растворителях, таких как диметилформамид (DMF), тетрагидрофуран (THF), диоксан, дихлорметан, этилацетат, 1-метил-2-пирролидон (NMP) и т.п., при температуре в интервале, например, от -10 до 120°C, но, как правило, от 0°C до комнатной температуры, при атмосферном давлении или повышенном давлении. Реакцию можно проводить в одну стадию или в несколько стадий. Если реакцию проводят в одну стадию, то конверсия обычно достигается с помощью сшивающего реагента, такого как N,N′-дициклогексилкарбодиимид (DCC), N-(3-диметиламинопропил)-N′-этилкарбодиимид гидрохлорид (EDC), O-(бензотриазол-1-ил)-N,N,N′,N′-тетраметилуроний тетрафторборат (TBTU), O-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилуроний гексафторфосфат (HATU), (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламиноморфолинокарбений гексафторфосфат (COMU), пропилфософорный ангидрид и т.п. (в литературе описано большое количество различных по химической структуре сшивающих реагентов). Если реакцию проводят в несколько стадий, то кислоту (II) обычно превращают в реакционноспособное вещество, например в хлорид кислоты или ангидрид кислоты, например реакцией с тионилхлоридом, сульфурилхлоридом, фосфороксихлоридом, оксалилхлоридом и т.п., в присутствии или отсутствии растворителя, такого как дихлорметан, в присутствии или отсутствии добавки, например DMF. Эти реакционноспособные вещества далее превращают на другой стадии, путем добавления амина (III), в продукт (I). Вторую стадию обычно проводят в растворителе, таком как диметилформамид (DMF), тетрагидрофуран (THF), диоксан, дихлорметан, этилацетат, 1-метил-2-пирролидон (NMP) и т.п., при температуре в интервале, например, от -10 до 120°C, но, как правило, при температуре от 0°C до комнатной, при атмосферном или повышенном давлении. Часто бывает удобно добавлять к реакционной смеси основание, такое как триэтиламин или диизопропилэтиламин.
Соединения формулы (II) и (III) можно получать способами, известными в данной области техники, или описанными ниже, или аналогичными им. Если не указано иное, R1, R2, R3, R5, W, X, X1, Y, Y1, Ζ и Ζ1 таковы, как описано выше.
ОБЩИЕ СПОСОБЫ СИНТЕЗА
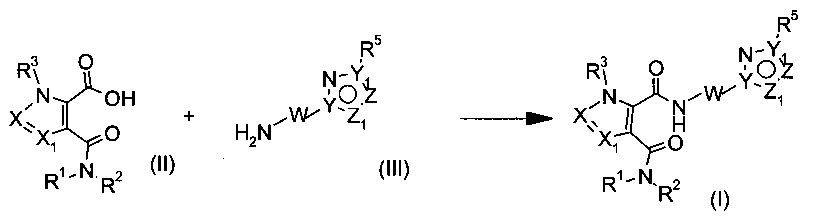
Соединения формулы (I) можно получить из строительных блоков (II) и (III) согласно Схеме 1. Данное превращение, которое хорошо известно как "амидная конденсация", можно проводить несколькими путями. По одному способу, кислоту (II) активируют с помощью сшивающего реагента, такого как 2-(1H-бензотриазол-1-ил)-1,1,3,3-тетраметилуроний тетрафторборат (TBTU), и превращают путем добавления амина (III) в желаемый продукт (I). По другому способу, кислоту (II) активируют путем превращения ее в хлорид кислоты, например, реакцией с тионилхлоридом. Полученный хлорид кислоты затем превращают путем добавления амина (III) в желаемый продукт (I). Как правило, для связывания выделяющегося HCl добавляют основание, например диизопропилэтиламин (DIPEA).
Схема 1
Соединения формулы (I), содержащие пиразолдикарбоксилатное ядро (X=N, X1=CR4), можно получить по Схеме 2 путем следующих превращений, по близкой аналогии с известными способами.
Схема 2
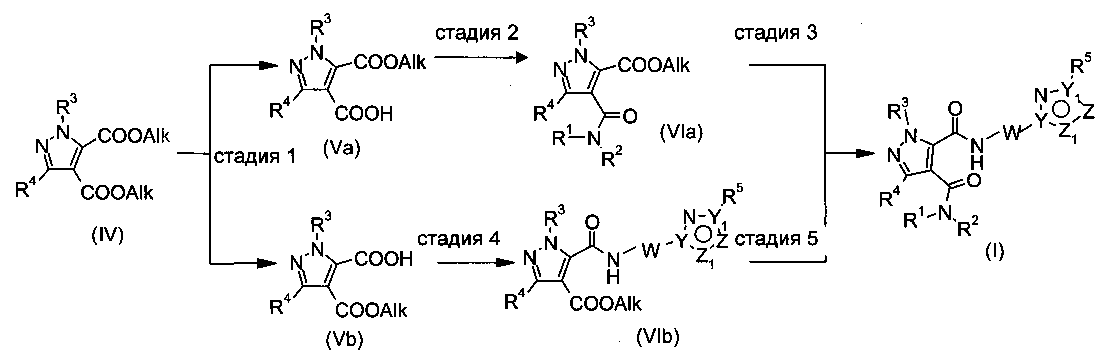
Стадия 1: соединения формулы (IV) имеется в продаже или их можно получить, например, в соответствии с US 2011/0071128. Соединения формулы (IV) можно селективно омылять по одной карбоксигруппе с получением моноэфира (Va) или (Vb) в подходящих биохимических (для Va) или химических (для Vb) условиях. Ферментативное расщепление соединения (IV) с помощью, например, подходящей эстеразы, и водного раствора неорганического основания, дает соединение (Va), тогда как классическое омыление соединения (IV) с помощью неорганического основание (например гидроксида лития, гидроксида натрия) в органическом растворителе (например в этаноле, диоксане, THF) дает соединение (Vb).
Стадия 2: промежуточные соединения формулы (Va) можно превращать в соединения формулы (VIa) путем образования амидной связи свободного карбоксилата с аминами R1-NH-R2, сшивающего реагента (например, ангидрида пропилфософорной кислоты, HATU, TBTU) и органического основания (например, N,N-диизопропилэтиламина, N-метилморфолина или триэтиламина) в органическом растворителе (например в DMF, этилацетате, THF).
Стадия 3: соединения формулы (I) можно получать либо прямым аминолизом эфирной группы с помощью аминов (III)
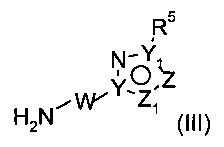
и кислоты Льюиса (например, хлорида триметилалюминия или диметилалюминия) в органическом растворителе (например, в толуоле или диоксане), или путем омыления соединения (VIa), как описано выше для соединения (Vb), и последующего образования амидной связи свободного карбоксилата с аминами (III)
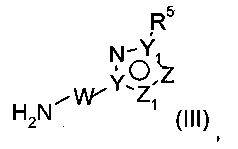
как описано выше. Соединения формулы (I) можно выделять и очищать стандартными способами.
Стадия 4: промежуточные соединения формулы (Vb) можно превращают в соединения формулы (VIb) путем образования амидной связи свободного карбоксилата с аминами (III)
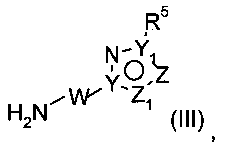
как описано выше.
Стадия 5: соединения формулы (I) можно получать либо прямым аминолизом эфирной группы соединения (VIb) с помощью аминов R1-NH-R2, как описано выше, либо путем омыления соединения (VIb), как описано выше для соединения (Vb), и последующего образования амидной связи свободного карбоксилата с аминами R1-NH-R2, как описано выше. Соединение формулы (I) можно выделять и очищать стандартными способами.
Соединения формулы (I), содержащие триазолдикарбоксилатное ядро (X и X1=N), можно получить по Схеме 3 путем следующих превращений, по близкой аналогии с известными способами.
Схема 3

Соединения формулы (VII) имеется в продаже или их можно получить, например, как описано в следующих источниках: Chem. Lett. 1983, 1131; или J. Heterocycl. Chem. 2002, 39, 889. Соединения формулы (VII) можно превращают в соединения формулы (I) таким же способом (стадия 1 - стадия 5), как описано выше для превращений, исходя из соединений формулы (IV).
Соединения формулы (I), содержащие имидазолдикарбоксилатное ядро (X=CR4, X1=Ν), можно получить по Схеме 4 путем следующих превращений, по близкой аналогии с известными способами.
Схема 4
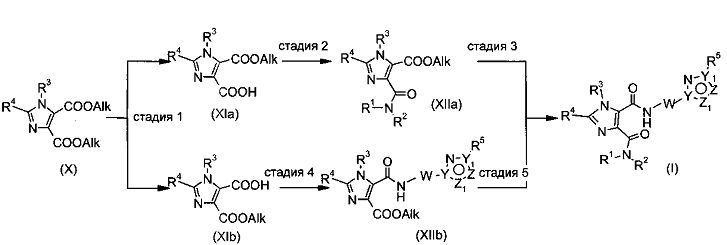
Соединения формулы (X) имеются в продаже или их можно получить из имеющихся в продаже соединений-предшественников (имидазол-4,5-дикарбоксилат) или, как описано, например, в источнике: J. Med. Chem. 1989, 32, 119 (2-метилимидазол-4,5-дикарбоксилат). Соединения формулы (X) можно превращать в соединения формулы (I) таким же способом (стадия 1 - стадия 5), как описано выше для превращений, исходя из соединений формулы (IV).
Все реакции обычно проводят в подходящем растворителе и в атмосфере аргона или азота.
Кроме того, настоящее изобретение относится к применению соединений формулы (I), описанных выше, в качестве терапевтически активного вещества.
Кроме того, настоящее изобретение относится к фармацевтической композиции, включающей соединения формулы (I), раскрытые выше, и терапевтически инертный носитель.
Как говорилось выше, обнаружили, что новые соединения по настоящему изобретению ингибируют активность PDE10A. Таким образом, соединения по настоящему изобретению можно применять, как в отдельности, так и в комбинации с другими лекарственными препаратами, для лечения или профилактики психических расстройств, шизофрении, позитивных, негативных и/или когнитивных симптомов, связанных с шизофренией, бредового расстройства, интоксикационного психического расстройства, тревожных расстройств, панического расстройства, синдрома навязчивых состояний, острого стрессового расстройства, общего тревожного расстройства, наркозависимости, двигательных расстройств, болезни Паркинсона, синдрома беспокойных ног, расстройства с дефицитом познавательной способности, болезни Альцгеймера, мультиинфарктной деменции, расстройств настроения, депрессии, биполярных расстройств, психоневрологических состояний, психоза, синдрома гиперактивности с дефицитом внимания, расстройств внимания, диабета и связанных расстройств, сахарного диабета 2 типа, нейродегенеративных расстройств, болезни Гентингтона, множественного склероза, инсульта, повреждения спинного мозга, твердых опухолей, гематологических новообразований, почечно-клеточной карциномы или рака молочной железы.
Еще в одном воплощении настоящее изобретение относится к применению соединения по настоящему изобретению для изготовления лекарственного средства для лечения или профилактики психических расстройств, шизофрении, позитивных, негативных и/или когнитивных симптомов, связанных с шизофренией, бредового расстройства, интоксикационного психического расстройства, тревожных расстройств, панического расстройства, синдрома навязчивых состояний, острого стрессового расстройства, общего тревожного расстройства, наркозависимости, двигательных расстройств, болезни Паркинсона, синдрома беспокойных ног, расстройства с дефицитом познавательной способности, болезни Альцгеймера, мультиинфарктной деменции, расстройств настроения, депрессии, биполярного расстройства, психоневрологических состояний, психоза, синдрома гиперактивности с дефицитом внимания, расстройств внимания, диабета и связанных расстройств, сахарного диабета 2 типа, нейродегенеративных расстройств, болезни Гентингтона, множественного склероза, инсульта, повреждения спинного мозга, твердых опухолей, гематологических новообразований, почечно-клеточной карциномы или рака молочной железы.
Настоящее изобретение также относится к соединению, описанному выше, для лечения или профилактики психических расстройств, шизофрении, позитивных, негативных и/или когнитивных симптомов, связанных с шизофренией, бредового расстройства, интоксикационного психического расстройства, тревожных расстройств, панического расстройства, синдрома навязчивых состояний, острого стрессового расстройства, общего тревожного расстройства, наркозависимости, двигательных расстройств, болезни Паркинсона, синдрома беспокойных ног, расстройства с дефицитом познавательной способности, болезни Альцгеймера, мультиинфарктной деменции, расстройств настроения, депрессии, биполярных расстройств, психоневрологических состояний, психоза, синдрома гиперактивности с дефицитом внимания, расстройств внимания, диабета и связанных расстройств, сахарного диабета 2 типа, нейродегенеративных расстройств, болезни Гентингтона, множественного склероза, инсульта, повреждения спинного мозга, твердых опухолей, гематологических новообразований, почечно-клеточной карциномы или рака молочной железы.
Соединения формулы (I) могут содержать один или несколько асимметрических атомов углерода, и поэтому могут существовать в виде энантиомерной смеси, смеси стереоизомеров или в виде оптически чистых соединений. Соединения формулы (I) включают все диастереомеры, таутомеры, рацематы и их смеси.
В частности, соединения формулы (I) описаны в примерах как индивидуальные соединения, а также их фармацевтически приемлемые соли, а также фармацевтически приемлемые эфиры. Кроме того, заместители, присутствующие в конкретных примерах, описанных далее, в отдельности составляют воплощения настоящего изобретения.
ФАРМАЦЕВТИЧЕСКИЕ КОМПОЗИЦИИ И ИХ ВВЕДЕНИЕ
В другом воплощении предложены фармацевтические композиции или лекарственные средства, содержащие соединения по настоящему изобретению и терапевтически инертный носитель, разбавитель или эксципиент, а также способы применения соединений по настоящему изобретению для изготовления таких композиций и лекарственных средств. Например, можно приготовить лекарственную форму с соединениями формулы (I) путем их смешивания при комнатной температуре при подходящем значении pH и при желаемой степени чистоты, с физиологически приемлемыми носителями, т.е., носителями, которые не токсичны для реципиента в дозировках и концентрациях, применяемых в галеновой форме для введения. Значение pH лекарственной формы главным образом зависит от конкретного применения и концентрации соединения, но предпочтительно в любом случае находится в интервале от 3 до примерно 8. В одном примере соединение формулы (I) присутствует в лекарственной форме в ацетатном буфере при pH 5. В другом осуществлении соединения формулы (I) стерильны. Соединение можно хранить, например, в виде твердой или аморфной композиции, в виде лиофилизованной лекарственной формы или в виде водного раствора.
Изготовление лекарственных форм с композициями, их дозирование и введение осуществляют в соответствии с надлежащей медицинской практикой. Факторы, которые следует учитывать в данном контексте, включают конкретное расстройство, на которое направлено лечение, конкретное млекопитающее, которому проводят лечение, клиническое состояние конкретного пациента, причину расстройства, сайт доставки действующего вещества, способ введения, схему введения и другие факторы, известные практикующим врачам. "Эффективное количество" соединения, которое нужно ввести, будет определяться из этих соображений, и оно представляет собой минимальное количество, которое необходимо для ингибирования PDE10 и контроля сигнального пути, опосредуемого cAMP. Например, такое количество может быть меньше количества, которое токсично для нормальных клеток или млекопитающего в целом.
В одном примере фармацевтически эффективное количество соединения по настоящему изобретению вводят парентерально, в дозе, которая находится в диапазоне примерено 0,01-100 мг/кг, как вариант, примерно 0,1-20 мг/кг массы тела пациента в сутки, при этом начальный диапазон дозировок соединения обычно составляет 0,3-15 мг/кг/сутки. В другом осуществлении пероральные дозированные лекарственные формы, такие как таблетки и капсулы, предпочтительно содержат примерно 25-100 мг соединения по настоящему изобретению.
Соединения по настоящему изобретению можно вводить любым удобным способом, включая оральное, местное (в т.ч. трансбуккальное и сублингвальное), ректальное, вагинальное, трансдермальное, парентеральное, подкожное, внутрибрюшинное, внутрилегочное, внутрикожное, интратекальное, эпидуральное и интраназальное, а также, в целях местного лечения, внутриочаговое введение. Парентеральные инфузии включают внутримышечное, внутривенное, внутриартериальное, внутрибрюшинное или подкожное введение.
Соединения по настоящему изобретению можно вводить в любой удобной форме для введения, например, в виде таблеток, порошков, капсул, растворов, дисперсий, суспензий, сиропов, спрэев, суппозиториев, гелей, эмульсий, пластырей и т.п. Такие композиции могут содержать компоненты, которые обычно применяются в фармацевтических препаратах, например, разбавители, носители, модификаторы pH, подсластители, объемообразующие агенты и дополнительные действующие вещества.
Типичную лекарственную форму изготавливают путем смешивания соединения по настоящему изобретению с носителем или эксципиентом. Подходящие носители и эксципиенты хорошо известны специалистам в данной области техники и подробно описаны, например, в следующих источниках: Ansel, Howard C, et al., Ansel′s Pharmaceutical Dosage Forms and Drug Delivery Systems. Philadelphia: Lippincott, Williams & Wilkins, 2004; Gennaro, Alfonso R., et al. Remington: The Science and Practice of Pharmacy. Philadelphia: Lippincott, Williams & Wilkins, 2000; Rowe, Raymond C. Handbook of Pharmaceurical Excipients. Chicago, Pharmaceutical Press, 2005. Такие лекарственные формы могут также включать один или более буферных агентов, стабилизаторов, поверхностно-активных веществ, увлажняющих агентов, лубрикантов, эмульгаторов, суспендирующих агентов, консервантов, антиоксидантов, кроющих агентов, скользящих веществ, вспомогательных веществ для производственного процесса, красителей, подсластителей, отдушек, ароматизаторов, разбавителей и других известных добавок, для обеспечения надлежащей формы выпуска лекарства (т.е. соединения по настоящему изобретению или его фармацевтической композиции) или вспомогательного средства при изготовлении фармацевтического продукта (т.е. лекарственного средства).
Примером подходящей пероральной лекарственной формы является таблетка, содержащая примерно 25 мг, 50 мг, 100 мг, 250 мг или 500 мг соединения по настоящему изобретению, объединенного примерно с 90-30 мг безводной лактозы, примерно 5-40 мг кроскармеллозы натрия, примерно 5-30 мг поливинилпирролидона (PVP) K30 и примерно 1-10 мг стеарата магния. Порошкообразные ингредиенты вначале смешивают вместе, а затем смешивают с раствором PVP. Полученную композицию можно высушить, гранулировать, смешать со стеаратом магния и спрессовать в форму таблетки с помощью стандартного оборудования. Образец аэрозольной лекарственной формы можно получить путем растворения, например, 5-400 мг соединения по изобретению в подходящем буферном растворе, например в монофосфатном буфере, и, по желанию, добавления агента для регуляции тоничности, например, соли, такой как хлорид натрия. Такой раствор можно фильтровать, например, через фильтр с размером пор 0,2 микрон, для удаления примесей и загрязнений.
Таким образом, воплощение настоящего изобретения включает фармацевтическую композицию, содержащую соединение формулы (I), или его стереоизомер, или фармацевтически приемлемую соль. Еще одно воплощение включает фармацевтическую композицию, содержащую соединение формулы (I), или его стереоизомер, или же его фармацевтически приемлемую соль, вместе с фармацевтически приемлемым носителем или эксципиентом.
Ниже следующий тест проводили с целью определения активности соединений по настоящему изобретению. Активность соединений по настоящему изобретению по отношению к PDE10 определяли с помощью метода, основанного на сцинтилляционном анализе сближения (SPA), по аналогии с описанным ранее (Fawcett, L. et al., ProcNatl Acad Sci USA (2000) 97(7):3702-3707).
Анализ полноразмерной фосфодиэстеразы PDE10A человека проводили на 96-луночных микропланшетах. Реакционная смесь объемом 50 мкл содержала 20 мМ HEPES pH=7.5/10 мМ MgCl2/0,05 мг/мл BSA (Sigma кат. № A-7906), 50 нМ cGMP (Sigma, кат. № G6129) и 50 нМ [3H]-cGMP (GE Healthcare, кат. № TRK392 S.A. 13,2 Ки/ммоль), 3,75 нг/лунку фермента PDE10A (Enzo Life Science, Лаузен, Швейцария, кат. № SE-534), включая или не включая специфическое тестируемое соединение. Использовали диапазон концентраций потенциального ингибитора для получения данных для расчета концентрации ингибитора, взывающей 50-процентный эффект (например IC50 - концентрация конкурента, которая ингибирует активность PDE10A на 50%). Тестировали неспецифическую активность без фермента. Реакцию инициировали добавлением раствора субстрата (cGMP и [3H]-cGMP) и оставляли для развития реакции на 20 минут при комнатой температуре. Реакцию останавливали добавлением 25 мкл сцинтилляционных шариков YSi-SPA (GE Healthcare, кат. № RPNQ0150) в 18 мМ растворе сульфата цинка (стоп-реагент). Встряхивали в течение 1 ч, после чего планшет центрифугировали в течение 1 мин при 170 g для осаждения шариков. После этого измеряли радиоактивность на сцинтилляционном считывателе для планшетов Perkin Elmer TopCount.
Для соединений формулы (I) характерна величина IC50 ниже 10 мкМ, в частности, ниже 5 мкМ, в частных случаях, ниже 1 мкМ. В ниже следующей таблице 1 представлены данные для некоторых примеров.
Ниже следующие примеры служат наглядным представлением настоящего изобретения в более детализированном виде. При этом подразумевается, что они ни коим образом не ограничивают объем изобретения.
МЕТОДИКА ЭКСПЕРИМЕНТА
В представленных выше схемах, а также в ниже следующих примерах получения используются следующие сокращения: ч - час(ы), мин - минута(ы), КТ - комнатная температура.
ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ
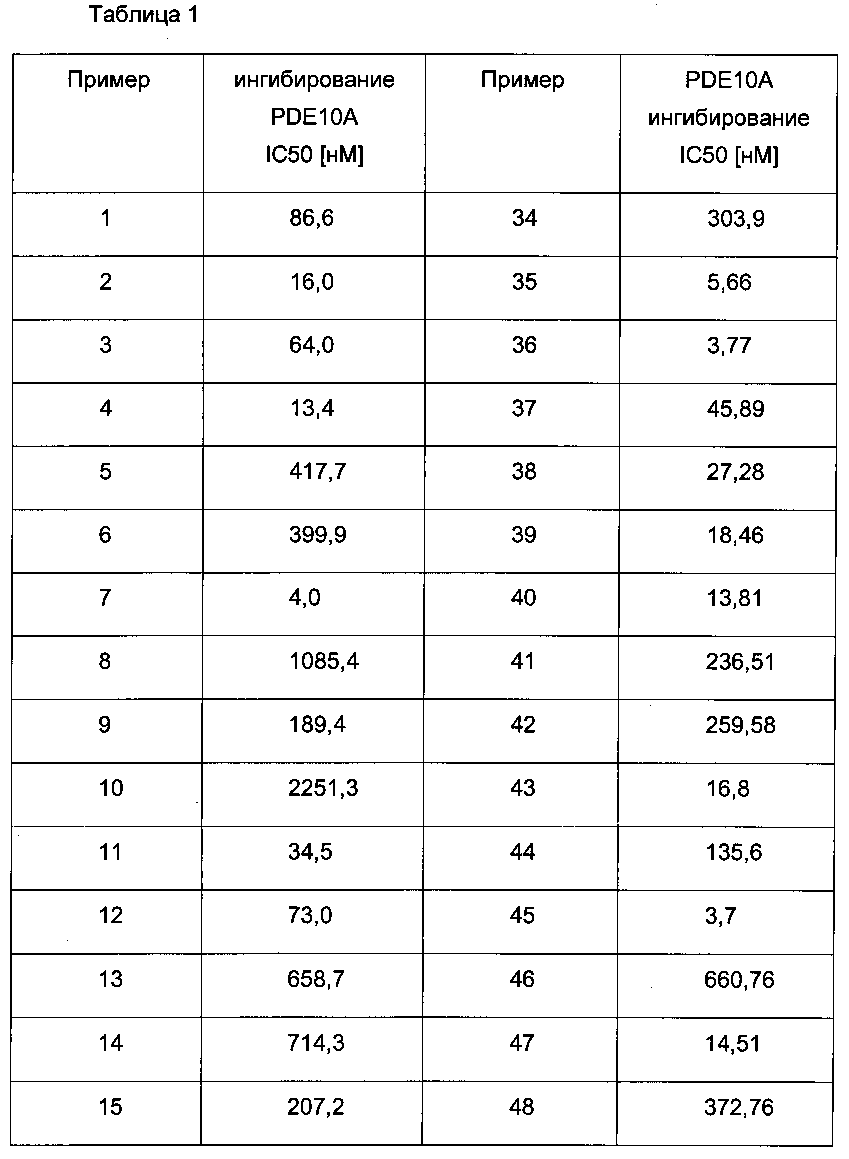
Промежуточное соединение A-1: 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновая кислота
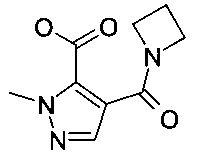
Промежуточное соединение A-1 получали, как описано в US 2011/0071128, пример 74, стадии 1-3.
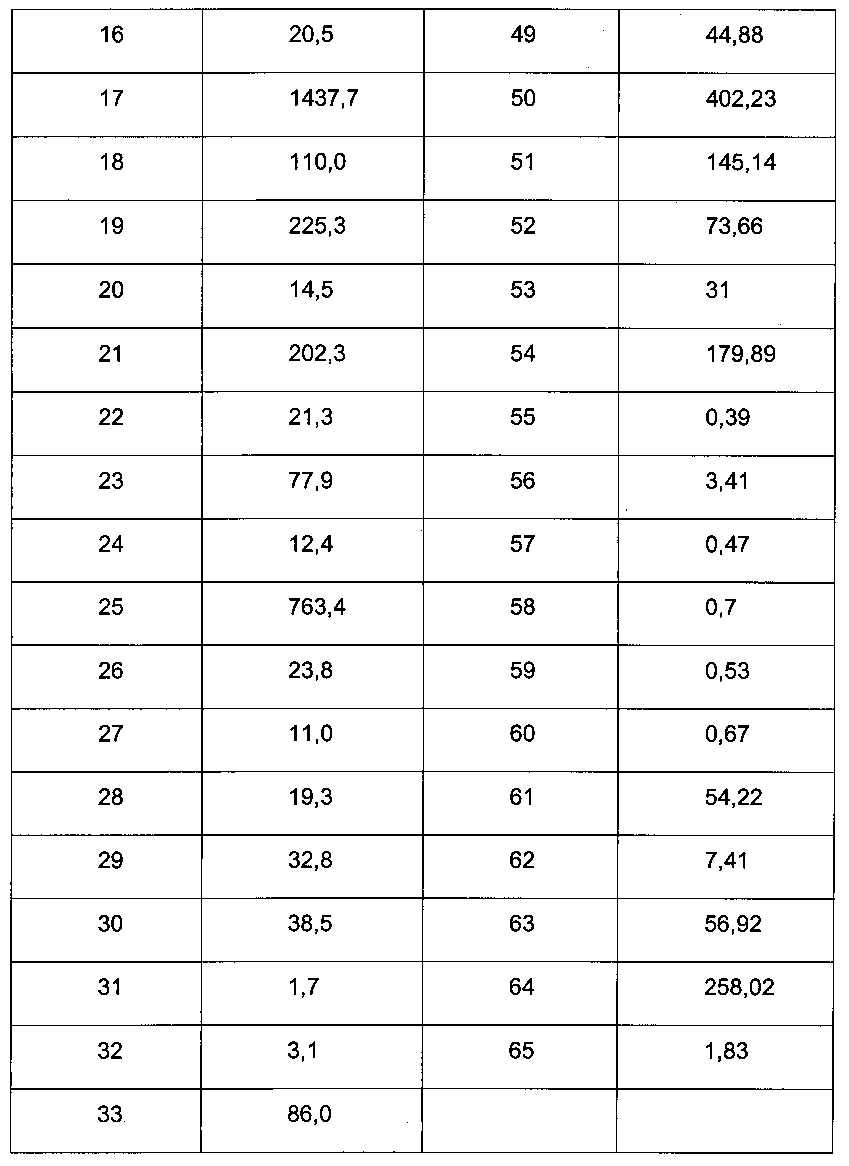
Промежуточное соединение А-2: 2-метил-4-(морфолин-4-карбонил)-2H-пиразол-3-карбоновая кислота
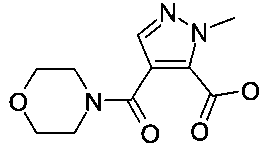
Промежуточное соединение A-2 получали в соответствии с US 2011/0071128, пример 74, стадии 1-3, с использованием морфолина вместо азетидина на стадии 2.
ПРИМЕРЫ
Пример 1: (2-имидазо[1,2-a]пиридин-2-ил-этил)-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
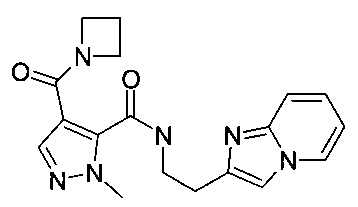
Смесь промежуточного соединения A-1 (32 мг; 153 мкмоль), 2-(имидазо[1,2-a]пиридин-2-ил)этанамина дигидрохлорида (46,7 мг; 199 мкмоль), O-(7-азабензотриазол-1-ил)-N,N,N′,N′-тетраметилурония гексафторфосфата (HATU; 64,1 мг; 169 мкмоль) и N-метилморфолина (84,3 мкл; 767 мкмоль) в THF (1,5 мл) нагревали в атмосфере азота при 70°C в течение 16 ч. Реакционную смесь охлаждали до КТ, вливали в этилацетат (50 мл) и экстрагировали водой (2×15 мл). Органическую фазу промывали солевым раствором (15 мл) и водные слои снова экстрагировали этилацетатом (1×50 мл). Органические слои высушивали над MgSO4 и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией (с помощью силикагеля и градиента MeOH/этилацетат) с получением указанного в заголовке соединения (18 мг; 51,1 мкмоль; 33,3%) в виде бесцветного твердого вещества. MS: M=353,2 (M+H)+
Пример 2: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
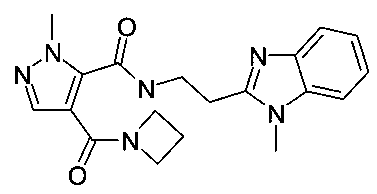
Данный продукт получали, исходя из промежуточного соединения A-1 (32 мг; 153 мкмоль) и 2-(1-метил-1H-бензо[d]имидазол-2-ил)этанамина (34,9 мг; 199 мкмоль), по способу, описанному в примере 1, в виде бесцветного твердого вещества (19,4 мг; 52,9 мкмоль; 34,5%). MS: M=367,2 (M+H)+
Пример 3: [2-(3-фенил-[1,2,4]оксадиазол-5-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
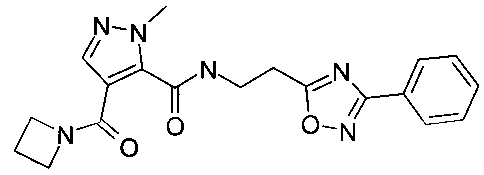
Данный продукт получали, исходя из промежуточного соединения A-1 (32 мг; 153 мкмоль) и 2-(3-фенил-1,2,4-оксадиазол-5-ил)этанамина (37,7 мг; 199 мкмоль), по способу, описанному в примере 1, после очистки с помощью препаративной ВЭЖХ, с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% муравьиную кислоту), в виде бесцветного твердого вещества (26 мг; 68,3 мкмоль; 44,6%). MS: M=381,3 (M+H)+
Пример 4: [2-(5-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
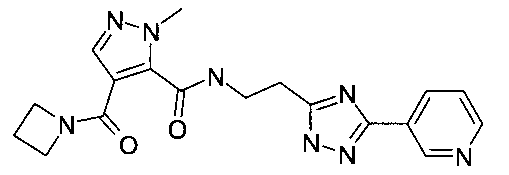
Данный продукт получали, исходя из промежуточного соединения A-1 (32 мг; 153 мкмоль) и 2-(3-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)этанамина дигидрохлорида (55,0 мг; 199 мкмоль), по способу, описанному в примере 1, после обработки неочищенного продукта этилацетатом, в виде светло-коричневого твердого вещества (17 мг; 44,7 мкмоль; 29,1%). MS: M=381,3 (M+H)+
Пример 5: [2-(8-метилимидазо[1,2-a]пиридин-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
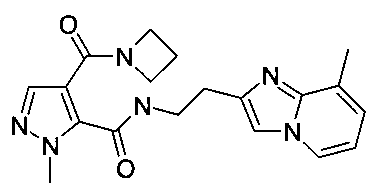
Данный продукт получали, исходя из промежуточного соединения A-1 (32 мг; 153 мкмоль) и 2-(8-метилимидазо[1,2-a]пиридин-2-ил)этанамина (34,9 мг; 199 мкмоль), по способу, описанному в примере 1, в виде светло-желтого твердого вещества (15 мг; 26,7%). MS: M=367,4 (M+H)+
Пример 6: {2-[3-(4-хлорфенил)-[1,2,4]оксадиазол-5-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
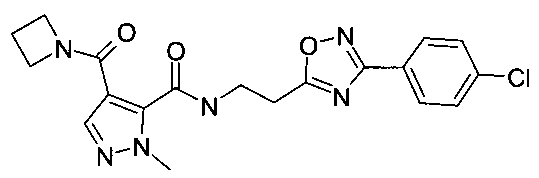
Данный продукт получали, исходя из промежуточного соединения A-1 (32 мг; 153 мкмоль) и 2-(3-(4-хлорфенил)-1,2,4-оксадиазол-5-ил)этанамина (44,6 мг; 199 мкмоль), по способу, описанному в примере 3, в виде бесцветного твердого вещества (9 мг; 21,7 мкмоль; 14,1%). MS: M=415,3 (M+H)+
Пример 7: [2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
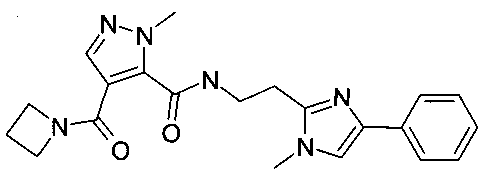
Стадия 1: 2-оксо-2-фенилэтил 3-(трет-бутоксикарбониламино)пропионат
К раствору 3-(трет-бутоксикарбониламино)пропионовой кислоты (1 г; 5,29 ммоль) в EtOH (20 мл) добавляли карбонат цезия (861 мг; 2,64 ммоль) и реакционную смесь перемешивали при 20°C в течение 1 ч. Растворитель удаляли в вакууме и полученный остаток растворяли в DMF (20,0 мл). Добавляли 2-бром-1-фенилэтанон (1,05 г; 5,29 ммоль) и реакционную смесь перемешивали при 20°C в течение 4 ч. Растворитель удаляли в вакууме и неочищенный продукт растворяли в этилацетате (20 мл). Бромид цезия отфильтровывали и промывали дважды этилацетатом (10 мл). Полученный фильтрат выпаривали и высушивали в вакууме с получением продукта в виде бесцветного полутвердого вещества (2,02 г; 5,26 ммоль; 99,4%). MS: M=208,1 (M-Boc+Н)+
Стадия 2: трет-бутил 2-(4-фенил-1H-имида-зол-2-ил)этилкарбамат
К раствору 2-оксо-2-фенилэтил-3-(трет-бутоксикарбониламино)-пропионата (1,954 г; 5,09 ммоль) в ксилоле (15 мл) добавляли ацетат аммония (7,84 г; 102 ммоль). Реакционную смесь нагревали до 140°C (происходило интенсивное кипение) и перемешивали в течение 1,5 ч. Реакционную смесь вливали в насыщенный раствор бикарбоната натрия (20 мл; выделение газа, pH=7) и экстрагировали этилацетатом (2×40 мл). Органические слои промывали насыщенным раствором бикарбоната натрия (20 мл; выделение газа, pH=8-9) и солевым раствором (10 мл), объединяли, высушивали над MgSO4 и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией (с использованием аминосиликагелевой фазы и градиента этилацетат/гептан) с получением продукта в виде желтой пены (1,233 г; 4,29 ммоль; 84,4%). MS: M=288,1 (M+H)+
Стадия 3: трет-бутил 2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамат
Суспензию трет-бутил-2-(4-фенил-1H-имидазол-2-ил)этилкарбамата (1,071 г; 3,73 ммоль) и карбоната калия (1,13 г; 8,2 ммоль) в DMF (10 мл) перемешивали при 20°C в течение 30 мин. Затем реакционную смесь охлаждали до 0-5°C, добавляли подметан (280 мкл; 4,47 ммоль) и полученную смесь перемешивали в течение 30 мин. Ледяную баню убирали, после чего реакционную смесь перемешивали при 20°C в течение 6 ч, вливали в воду (10 мл) и экстрагировали дихлорметаном (2×20 мл). Органические слои объединяли, высушивали над MgSO4 и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией (с использованием аминосиликагелевой фазы и градиента этилацетат/гептан) с получением продукта в виде светло-желтого твердого вещества (835 мг; 2,77 ммоль; 74,3%).
MS: M=302,2 (M+H)+
Стадия 4: 2-(1-метил-4-фенил-1H-имидазол-2-ил)этанамин дигидрохлорид
К суспензии трет-бутил-2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамата (831 мг; 2,76 ммоль) в диоксане (5 мл) добавляли по каплям HCl в диоксане (4 М; 6.89 мл; 27,6 ммоль). Реакционную смесь перемешивали при 20°C в течение 1,5 ч. Неочищенную реакционную смесь концентрировали в вакууме. Добавляли гептан, полученную суспензию перемешивали в течение 30 мин и выпаривали в вакууме с получением продукта в виде белого твердого вещества (760 мг; 2,72 ммоль; 98,5%). MS: M=202,3 (M-2HCl+H)+
Стадия 5: [2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
К суспензии промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(1-метил-4-фенил-1H-имидазол-2-ил)этанамина дигидрохлорида (57,7 мг; 210 мкмоль) в THF (1 мл) добавляли в атмосфере азота циклический ангидрид 1-пропанфософорной кислоты (50% в этилацетате, 287 мкл; 478 мкмоль) и Ν,Ν-диизопропилэтиламин (267 мкл; 1,53 ммоль). Реакционную смесь нагревали до 70°C и перемешивали в течение 2 ч. Неочищенный продукт очищали после удаления всех летучих веществ с помощью препаративной ВЭЖХ с использованием градиента ацетонитрил/вода, с получением белого твердого вещества (31 мг; 79,0 мкмоль; 41,3%). MS: M=393,2 (M+H)+
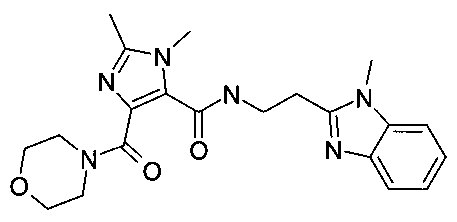
Пример 8: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 2,3-диметил-5-(морфолин-4-карбонил)-3H-имидазол-4-карбоновой кислоты
Стадия 1: диметиловый эфир 1,2-диметил-1H-имидазол-4,5-дикарбоновой кислоты
К суспензии диметил-2-метил-1H-имидазол-4,5-дикарбоксилата (2 г; 10,1 ммоль; полученного, как описано в: J. Med. Chem. 1989, 32, 119) в DMF (20 мл) добавляли в атмосфере азота трет-бутоксид калия (1,25 г; 11,1 ммоль) при комнатной температуре. Эту смесь перемешивали в течение 15 мин и добавляли подметан (694 мкл; 11,1 ммоль). Реакционную смесь перемешивали в течение 2 ч при этой же температуре. Реакционную смесь вливали в воду (75 мл) и экстрагировали этилацетатом (2×100 мл). Органическую фазу промывали водой (3×50 мл) и солевым раствором (20 мл), высушивали и концентрировали в вакууме с получением 720 мг неочищенного продукта. Объединенные водные фазы снова экстрагировали дихлорметаном (4×50 мл), высушивали над MgSO4 и концентрировали в вакууме с получением дополнительных 1,4 г неочищенного продукта. Объединенное неочищенное вещество очищали флэш-хроматографией (с использованием силикагеля и градиента MeOH/дихлорметан) с получением продукта в виде бесцветного масла (1.89 г; 8,91 ммоль; 88,3%). MS: M=213,1 (M+H)+
Стадия 2: 4-метиловый эфир 1,2-диметил-1H-имидазол-4,5-дикарбоновой кислоты
К раствору диметил-1,2-диметил-1H-имидазол-4,5-дикарбоксилата (1.89 г; 8,91 ммоль) в THF (35 мл) и MeOH (15 мл) добавляли при 0-5°C LiOH (1 М; 9,35 мл; 9,35 ммоль). Реакционную смесь перемешивали при КТ в течение 7 ч, и растворители выпаривали в вакууме. Полученный остаток вливали в воду (20 мл) и этилацетат (30 мл). Этилацетатную фазу промывали водой (10 мл). Объединенные водные фазы подкисляли хлороводородной кислотой (1 М; 10 мл) и экстрагировали дихлорметаном/MeOH (95:5, 8×60 мл). Фазы дихлорметана/MeOH высушивали над MgSO4, фильтровали и выпаривали с получением продукта в виде бесцветного твердого вещества (1,29 г; 6,51 ммоль; 73,1%). MS: M=199,1 (M+H)+
Стадия 3: метиловый эфир 1,2-диметил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты
Данный продукт получали, исходя из 4-метилового эфира 1,2-диметил-1H-имидазол-4,5-дикарбоновой кислоты (200 мг; 1,01 ммоль) и 2-(1-метил-1H-бензо[d]имидазол-2-ил)этанамина (195 мг; 1,11 ммоль), по способу, описанному в примере 7, стадия 5, после обработки водой и очистки флэш-хроматографией (с использованием аминосиликагелевой фазы и градиента MeOH/этилацетат) в виде светло-красной смолы (98 мг; 276 мкмоль; 27,3%). MS: M=356,1 (M+H)+
Стадия 4: 1,2-диметил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновая кислота
К раствору метил-1,2-диметил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоксилата (98 мг; 276 мкмоль) в THF (2 мл) и MeOH (1 мл) при комнатной температуре добавляли LiOH (1 M; 1,1 мл; 1,1 ммоль). Реакционную смесь перемешивали при КТ в течение 6 ч. Реакционную смеси подкисляли хлороводородной кислотой (1 М; 1,5 мл), все легколетучие вещества удаляли в вакууме и полученный остаток суспендировали в смеси дихлорметан/MeOH (95:5) и фильтровали. Эту жидкость выпаривали и высушивали в вакууме с получением продукта в виде бесцветного аморфного вещества (95 мг; 264 мкмоль; 95,9%), которое использовали без дополнительной очистки на следующей стадии. MS: M=340,2 (M-H)-
Стадия 5: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 2,3-диметил-5-(морфолин-4-карбонил)-3H-имидазол-4-карбоновой кислоты
Данный продукт получали, исходя из 1,2-диметил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты (51 мг; 149 мкмоль) и морфолина (39,0 мкл; 448 мкмоль), по способу, описанному в примере 8, стадия 3, в виде светло-коричневого воскообразного твердого вещества (17,3 мг; 42,1 мкмоль; 28,2%). MS: M=411,3 (M+H)+
Пример 9: (2-бензотиазол-2-ил-этил)-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
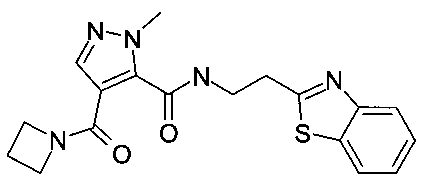
Данный продукт получали, исходя из промежуточного соединения A-1 (38 мг; 182 мкмоль; экв.: 1,00) и 2-(бензо[d]тиазол-2-ил)этанамина (35 мг; 196 мкмоль; экв.: 1,08), по способу, описанному в примере 7, стадия 5, в виде белого твердого вещества (26 мг; 70,4 мкмоль; 38,7%). MS: M=370,1 (M+H)+
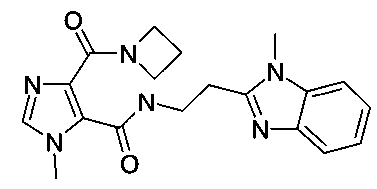
Пример 10: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-3H-имидазол-4-карбоновой кислоты
Стадия 1: диметил 1-метил-1H-имидазол-4,5-дикарбоксилат
Данный продукт получали, исходя из диметил-1H-имидазол-4,5-дикарбоксилата (2 г; 10,9 ммоль), по способу, описанному в примере 8, стадия 1, в виде бесцветного масла (1,76 г; 8.88 ммоль; 81.8%). MS: M=167,2 (M+H-CH3OH)+
Стадия 2: 4-метиловый эфир 1-метил-1H-имидазол-4,5-дикарбоновой кислоты
Данный продукт получали, исходя из диметил-1-метил-1H-имидазол-4,5-дикарбоксилата (1,76 г; 8,88 ммоль), по способу, описанному в примере 8, стадия 2, в виде белого твердого вещества (850 мг; 4,62 ммоль; 52,0%). MS: M=185,1 (M+H)+
Стадия 3: метиловый эфир 1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты
Данный продукт получали, исходя из 4-метилового эфира 1-метил-1H-имидазол-4,5-дикарбоновой кислоты (200 мг; 1,09 ммоль) и 2-(1-метил-1H-бензо[d]имидазол-2-ил)этанамина дигидрохлорида (296 мг; 1,19 ммоль), по способу, описанному в примере 8, стадия 3, в виде розовой пены (156 мг; 457 мкмоль; 42,1%). MS: M=342,1 (M+H)+
Стадия 4: 1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновая кислота
Данный продукт получали, исходя из метил-1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоксилата (151 мг; 442 мкмоль), по способу, описанному в примере 8, стадия 3, в виде фиолетового твердого вещества (231 мг; 423 мкмоль; 95,7%). MS: M=326,2 (M-H)-
Стадия 5: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-3H-имидазол-4-карбоновой кислоты
Данный продукт получали, исходя из 1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты (80 мг; 147 мкмоль) и азетидина (29,7 мкл; 440 мкмоль), по способу, описанному в примере 7, стадия 5, после очистки флэш-хроматографией (с использованием аминосиликагелевой фазы и градиента MeOH/этилацетат) в виде белого твердого вещества (22 мг; 60,0 мкмоль; 40,9%). MS: M=367,2 (M+H)+
Пример 11: [2-(1,5-диметил-1H-бензоимидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
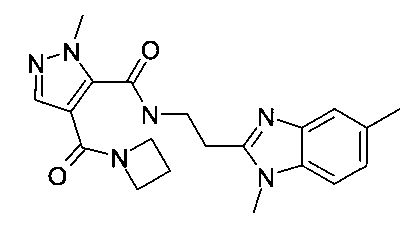
Данный продукт получали, исходя из промежуточного соединения A-1 (38 мг; 184 мкмоль; экв.: 1,00) и 2-(1,5-диметил-1H-бензо[d]имидазол-2-ил)этанамина дигидрохлорида (62,7 мг; 239 мкмоль; экв.: 1,3), по способу, описанному в примере 3, после перемешивания в течение 22 ч с 8 эквивалентами N-метилморфолина и после очистки с помощью препаративной ВЭЖХ, с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% триэтиламин), в виде бесцветного твердого вещества (30.8 мг; 81,0 мкмоль; 44,0%). MS: M=381,2 (M+H)+
Пример 12: [2-(3-фенилпиразол-1-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
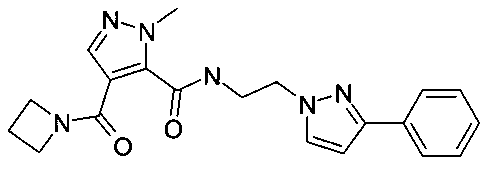
Данный продукт получали, исходя из промежуточного соединения A-1 (38 мг; 184 мкмоль) и 2-(3-фенил-1H-пиразол-1-ил)этанамина гидрохлорида (53,5 мг; 239 мкмоль), по способу, описанному в примере 3, в виде бесцветного твердого вещества (31 мг; 81,9 мкмоль; 44,5%). MS: M=379,3 (M+H)+
Пример 13: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-2-хлор-3-метил-3H-имидазол-4-карбоновой кислоты
Стадия 1: диметил-2-хлор-1-метил-1H-имидазол-4,5-дикарбоксилат
К бесцветному раствору диметил-1-метил-1H-имидазол-4,5-дикарбоксилата (500 мг; 2,42 ммоль; экв.: 1,00) в DMF (5,00 мл) добавляли в атмосфере азота 1,3-дихлор-5,5-диметилгидантоин (487 мг; 2,42 ммоль; экв.: 1,00). Реакционную смесь нагревали до 80°C и перемешивали в течение 2 ч, охлаждали до комнатной температуры, вливали в насыщенный раствор бикарбоната натрия (5 мл) и экстрагировали этилацетатом (2×10 мл). Органические слои промывали солевым раствором (5 мл), высушивали над MgSO4 и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) с получением продукта в виде бесцветного масла (415 мг; 1,78 ммоль; 73,7%). MS: M=233,0 (M+H)+
Стадия 2: 4-метиловый эфир 2-хлор-1-метил-1H-имидазол-4,5-дикарбоновой кислоты
Данный продукт получали, исходя из диметил-2-хлор-1-метил-1H-имидазол-4,5-дикарбоксилата (449 мг; 1,93 ммоль), по способу, описанному в примере 8, стадия 2, в виде белого твердого вещества (336 мг; 1,54 ммоль; 79,6%). MS: M=219,0 (M+H)+
Стадия 3: метиловый эфир 2-хлор-1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты
Данный продукт получали, исходя из 2-(1-метил-1H-бензо[d]имидазол-2-ил)этанамина дигидрохлорида (125 мг; 504 мкмоль) и 4-метилового эфира 2-хлор-1-метил-1H-имидазол-4,5-дикарбоновой кислоты (172 мг; 787 мкмоль), по способу, описанному в примере 8, стадия 3, в виде фиолетового твердого вещества (77 мг; 205 мкмоль; 40,7%). MS: M=376,3 (M+H)+
Стадия 4: 2-хлор-1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновая кислота
Данный продукт получали, исходя из метил-2-хлор-1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоксилата (73 мг; 194 мкмоль), по способу, описанному в примере 8, стадия 4, после осаждения из кислотной фазы, в виде белого твердого вещества (60 мг; 166 мкмоль; 85,4%). MS: M=360,2 (M-H)-
Стадия 5: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-2-хлор-3-метил-3H-имидазол-4-карбоновой кислоты
Данный продукт получали, исходя из 2-хлор-1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты (30 мг; 82,9 мкмоль) и азетидина (14,2 мг; 16,9 мкл; 249 мкмоль), по способу, описанному в примере 7, стадия 5 в виде белого воскообразного твердого вещества (17,5 мг; 41,5 мкмоль; 50,0%). MS: M=401,2 (M+H)+
Пример 14: [2-(5-пиридин-4-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (32 мг; 153 мкмоль) и 2-(3-(пиридин-4-ил)-1H-1,2,4-триазол-5-ил)этанамина дигидрохлорида (52,3 мг; 199 мкмоль), по способу, описанному в примере 1, после очистки с использованием аминосиликагелевой фазы и последующей обработки этилацетатом, в виде белого твердого вещества (14,7 мг; 38,6 мкмоль; 25,2%). MS: M=381,2 (M+H)+
Пример 15: [2-(2-метил-5-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Стадия 1: 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-3-пиридин-3-ил-[1,2,4] триазол
К суспензии 2-(3-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)этанамина дигидрохлорида (400 мг; 1,53 ммоль) в дихлорэтане (5 мл) добавляли N,N-диизопропилэтиламин (493 мг; 666 мкл; 3.81 ммоль) и DMAP (9,32 мг; 76,3 мкмоль) в атмосфере азота при КТ. Добавляли по каплям раствор ди-трет-бутил-дикарбоната (0,999 г; 1,06 мл; 4,58 ммоль) в дихлорэтане (5 мл) в течение 5 мин при КТ и продолжали перемешивать при этой же температуре в течение 2 ч. Реакционную смесь вливали в водный раствор гидросульфата калия (10%, 25 мл) и экстрагировали этилацетатом (2×75 мл). Объединенную органическую фазу промывали водой (2×25 мл) и солевым раствором (20 мл). Органический слой высушивали над MgSO4, концентрировали в вакууме, растворяли в дихлорметане и очищали флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан, 30 мл/мин, 254 нм) с получением продукта в виде светло-желтого масла (382 мг; 981 мкмоль; 64,3%). MS: M=390,3 (M+H)+
Стадия 2: 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-1-метил-3-пиридин-3-ил-[1,2,4] триазол
Данный продукт получали, исходя из 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-3-пиридин-3-ил-[1,2,4]триазола (382 мг; 981 мкмоль), по способу, описанному в примере 8, стадия 1, после очистки флэш-хроматографией (с использованием градиента этилацетат/гептан) в виде бесцветного масла (133 мг; 330 мкмоль; 33,7%). MS: M=404,2 (M+H)+
Стадия 3: 2-(1-метил-3-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)этанамин дигидрохлорид
5-[2-Бис(трет-бутоксикарбонил)аминоэтил]-1-метил-3-пиридин-3-ил-[1,2,4]триазол (133 мг; 330 мкмоль) суспендировали в хлороводородной кислоте (4 М раствор в диоксане, 2 мл; 8,00 ммоль) и эту смесь перемешивали при КТ в течение 1 ч. Реакционную смесь выпаривали и высушивали с получением продукта в виде бесцветного твердого вещества (118 мг; 342 мкмоль; 104%), которое использовали без дополнительной очистки на следующей стадии. MS: M=204,2 (M+H)+
Стадия 4: [2-(2-метил-5-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (38 мг; 184 мкмоль) и 2-(1-метил-3-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)этанамина дигидрохлорида (82,6 мг; 239 мкмоль), по способу, описанному в примере 3, осуществляя очистку с помощью ВЭЖХ (с использованием градиента смеси ацетонитрил/вода, содержащей 0,1% триэтиламин) и последующей флэш-хроматографией (с использованием градиента MeOH/этилацетат), в виде бесцветного аморфного вещества (6,2 мг; 14,9 мкмоль; 8,12%). MS: M=395,2 (M+H)+
Пример 16: [2-(5-хлор-1-метил-1H-бензоимидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (32 мг; 153 мкмоль) и 2-(5-хлор-1-метил-1H-бензо[d]имидазол-2-ил)этанамина гидрохлорида (49,1 мг; 199 мкмоль), по способу, описанному в примере 3, после очистки с помощью препаративной ВЭЖХ, с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% триэтиламин), в виде бесцветного твердого вещества (30 мг; 74.8 мкмоль; выход 48,8%). MS: M=401,2 (M+H)+
Пример 17: [2-(2-метил-5-пиридин-4-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
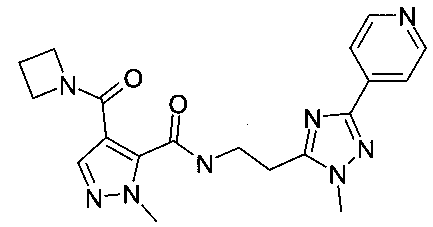
Стадия 1: 3-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-тиопропионамид
Суспензию 3-(1,3-диоксоизоиндолин-2-ил)-пропионитрила (2 г; 9,99 ммоль) и O,O′-диэтилового эфира дитиофософорной кислоты (2,05 г; 1,74 мл; 11,0 ммоль) в воде (4 мл) облучали при 80°C в течение 10 мин. Полученную суспензию разбавляли водой (5 мл), фильтровали, промывали водой и высушивали в вакууме с получением продукта в виде светло-желтого твердого вещества (2,13 г; 9,09 ммоль; 91,0%), которое использовали без дополнительной очистки на следующей стадии. MS: M=235,1 (M+H)+
Стадия 2: метиловый эфир 3-(1,3-диоксо-1,3-дигидро-изоиндол-2-ил)-тиопропионимидной кислоты гидроиодид
К суспензии 3-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-тиопропионамида (2,13 г; 9,09 ммоль) в ацетоне (30 мл) добавляли подметан (3,87 г; 1,71 мл; 27,3 ммоль) при КТ в атмосфере азота. Реакционную смесь перемешивали при КТ в течение 48 ч и полученную суспензию фильтровали, промывали диэтиловым эфиром и высушивали в вакууме с получением продукта в виде светло-желтого твердого вещества (2,3 г; 6,11 ммоль; 67,2%). MS: M=249,1 (M+H)+
Стадия 3: 2-(2-(5-(пиридин-4-ил)-4H-1,2,4-триазол-3-ил)этил)изоиндолин-1,3-дион
К суспензии метилового эфира 3-(1,3-диоксо-1,3-дигидроизоиндол-2-ил)-тиопропионимидной кислоты гидроиодида (880 мг; 2,34 ммоль) в 2-пропаноле (16 мл) добавляли изоникотиногидразид (337 мг; 2,46 ммоль) и карбонат натрия (248 мг; 2,34 ммоль). Реакционную смесь перемешивали при 85°C в течение 1 ч. Добавляли уксусную кислоту (32 мл) и реакционную смесь перемешивали при 130°C в течение 3 ч. После охлаждения до комнатной температуры все легколетучие вещества выпаривали. Остаток растворяли в уксусной кислоте (30 мл) и реакционную смесь перемешивали при 130°C в течение 16 ч. Реакционную смесь охлаждали до КТ и концентрировали в вакууме. Полученный остаток вливали в насыщенный раствор бикарбоната натрия (40 мл) и экстрагировали этилацетатом (2×80 мл). Объединенную органическую фазу промывали водой (40 мл) и солевым раствором (40 мл), высушивали над MgSO4 и концентрировали в вакууме. Полученный остаток суспендировали в дихлорметане, фильтровали, промывали дихлорметаном и высушивали в вакууме с получением продукта в виде коричневого твердого вещества (480 мг (чистота ~80%), 0,96 ммоль; 41%). Полученный фильтрат выпаривали и получали чистый продукт, после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан), в виде светло-коричневого твердого вещества (180 мг; 562 мкмоль; 24,0%). MS: M=321,1 (M+H)+
Стадия 4: 2-(2-(1-метил-3-(пиридин-4-ил)-1H-1,2,4-триазол-5-ил)этил)изоиндолин-1,3-дион
Данный продукт получали, исходя из 2-(2-(5-(пиридин-4-ил)-4H-1,2,4-триазол-3-ил)этил)изоиндолин-1,3-диона (300 мг; 752 мкмоль), по способу, описанному в примере 8, стадия 1, с использованием 1,3 эквивалента трет-бутоксида калия и подметана, со временем реакции 16 ч, и после очистки флэш-хроматографией (с использованием силикагеля и градиента MeOH/этилацетат), в виде белого твердого вещества (114,9 мг; 345 мкмоль; 45,9%). MS: M=334,1 (M+H)+
Стадия 5: 2-(1-метил-3-(пиридин-4-ил)-1H-1,2,4-триазол-5-ил)этанамин
2-(2-(1-Метил-3-(пиридин-4-ил)-1H-1,2,4-триазол-5-ил)этил)изоиндолин-1,3-дион (114,9 мг; 345 мкмоль) растворяли в растворе метиламина в EtOH (3,07 мл; 22,5 ммоль) и перемешивали при КТ в течение 1 ч. Реакционную смесь выпаривали с получением светло-коричневого твердого вещества. Полученное твердое вещество снова растворяли в растворе метиламина в EtOH (3,07 мл; 22,5 ммоль) и эту смесь нагревали до 50°C и перемешивали в течение 3 ч. Все легколетучие вещества удаляли в вакууме и полученный остаток суспендировали в дихлорметане (1,5 мл) и фильтровали. Полученный фильтрат выпаривали с получением продукта в виде светло-желтого твердого вещества (81,3 мг (80% чистота), 256 мкмоль; 74,2%), которое использовали без очистки на следующей стадии. MS: M=204,2 (M+H)+
Стадия 6: [2-(2-метил-5-пиридин-4-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (50 мг; 239 мкмоль) и 2-(1-метил-3-(пиридин-4-ил)-1H-1,2,4-триазол-5-ил)этанамина (77,9 мг; 307 мкмоль), по способу, описанному в примере 1, в виде светло-желтого твердого вещества (42,2 мг; 107 мкмоль; выход 44,8%). М5: M=395,2 (M+H)+
Пример 18: [2-(3-фенил-[1,2,4]триазол-1-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
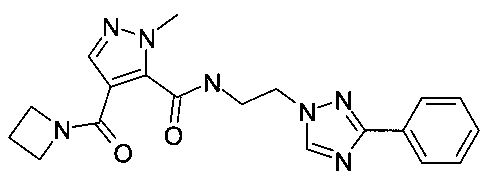
Стадия 1: 3-фенил-1H-1,2,4-триазол
К суспензии этилбензимидата гидрохлорида (2 г; 10,8 ммоль) в EtOH (10,0 мл) добавляли этоксид натрия (21% в EtOH, 4,02 мл; 10,8 ммоль). Выпавший в осадок хлорид натрия отфильтровывали, промывали с помощью EtOH (14,0 мл) и добавляли гидразид муравьиной кислоты (712 мг; 11,9 ммоль). Реакционную смесь перемешивали при 20°C в течение 92 ч и растворитель удаляли в вакууме. Добавляли гидроксид натрия (1 М; 50 мл) и полученную смесь экстрагировали трет-бутилметиловым эфиром (100 мл). Органический слой промывали водой и добавляли твердый диоксид углерода, чтобы довести pH до ~10. Полученную суспензию фильтровали и жидкую фазу выпаривали. Добавляли смесь дихлорметан/MeOH (9:1), реакционную смесь фильтровали и растворитель в фильтрате выпаривали с получением продукта в виде светло-коричневого полутвердого вещества (1,48 г; 9,18 ммоль; 85,2%). MS: M=146,1 (M+H)+
Стадия 2: 3-(3-фенил-1H-1,2,4-триазол-1-ил)пропионовая кислота
Смесь 3-фенил-1H-1,2,4-триазола (1,46 г; 9,05 ммоль), 3-хлорпропионовой кислоты (1,08 г; 9,96 ммоль) и гидроксида натрия (2 М; 9,05 мл; 18,1 ммоль) в воде (3 мл) нагревали до 115°C и перемешивали в течение 66 ч. Вносили еще по 0,5 эквивалентов каждого из реагентов, 3-хлорпропионовой кислоты и гидроксида натрия (значение pH возрастало до ~11) и продолжали перемешивать еще 24 ч. Добавляли хлороводородную кислоту (2 М; 10 мл), полученную суспензию фильтровали и экстрагировали дихлорметаном. Продукт получали после очистки в ходе суперкритической флюидной хроматографии в виде светло-коричневого твердого вещества (236 мг; 1,09 ммоль; 12,0%). MS: M=218,1 (M+H)+
Стадия 3: трет-бутил-2-(3-фенил-1H-1,2,4-триазол-1-ил)этилкарбамат
Смесь 3-(3-фенил-1H-1,2,4-триазол-1-ил)пропионовой кислоты (195 мг; 862 мкмоль), дифенилфосфоразидата (242 мкл; 1,12 ммоль) и триэтиламина (240 мкл; 1,72 ммоль) в трет-бутаноле (33 мл) нагревали при 50°C в течение 5 ч. Продолжали перемешивать при 90°C еще в течение 12 ч и все легколетучие вещества удаляли в вакууме. Неочищенный продукт очищали с помощью препаративной ВЭЖХ с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% муравьиную кислоту), с получением продукта в виде бесцветного масла (37 мг; 122 мкмоль; 14,1%). MS: M=289,1 (M+H)+
Стадия 4: 2-(3-фенил-1H-1,2,4-триазол-1-ил)этанамин дигидрохлорид
К раствору трет-бутил-2-(3-фенил-1H-1,2,4-триазол-1-ил)этилкарбамата (34 мг; 118 мкмоль) в диоксане (1 мл) добавляли хлороводородную кислоту (4 М в диоксане, 590 мкл; 2,36 ммоль). Реакционную смесь перемешивали при КТ в течение 18 ч. Все легколетучие вещества удаляли в вакууме, и продукт высушивали с получением белого твердого вещества (38 мг; 116 мкмоль; 98,7%), которое использовали без дополнительной очистки. MS:=189,3 M+H)+
Стадия 5: [2-(3-фенил-[1,2,4]триазол-1-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (20 мг; 95,6 мкмоль) и 2-(3-фенил-1H-1,2,4-триазол-1-ил)этанамина дигидрохлорида (34 мг; 104 мкмоль), по способу, описанному в примере 3, в виде белого твердого вещества (26 мг; 68,5 мкмоль; 71,7%). MS: M=380,2 (M+H)+
Пример 19: [2-(2-фенил-2H-[1,2,3]триазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (50 мг; 239 мкмоль) и 2-(2-фенил-2H-1,2,3-триазол-4-ил)этанамина (49,5 мг; 263 мкмоль), по способу, описанному в примере 3, в виде белого твердого вещества (69 мг; 182 мкмоль; 76,1%). MS: M=380,3 (M+H)+
Пример 20: [2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
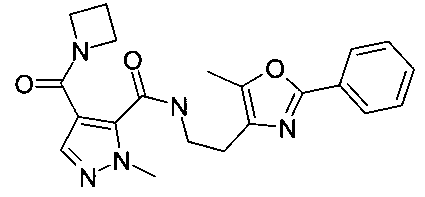
Данный продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(5-метил-2-фенилоксазол-4-ил)этанамина (47 мг; 232 мкмоль), по способу, описанному в примере 3, и в ходе последующей очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде бесцветного твердого вещества (7,2 мг; 18,3 мкмоль; 9,57%). MS: M=394,2 (M+H)+
Пример 21: [2-(5-фенил-[1,2,4]оксадиазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
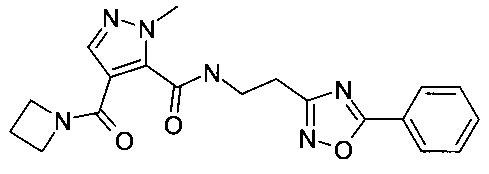
Данный продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(5-фенил-1,2,4-оксадиазол-3-ил)этанамина гидрохлорида (51,8 мг; 229 мкмоль), по способу, описанному в примере 20, в виде белого твердого вещества (46,6 мг; 123 мкмоль; 64,1%). MS: M=381,2 (M+H)+
Пример 22: [2-(2-метил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
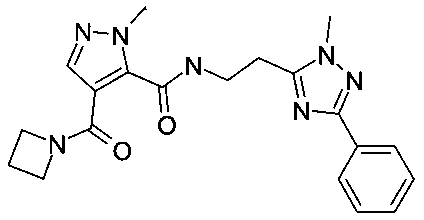
Стадия 1: 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-3-фенил-[1,2,4]триазол
Данный продукт получали, исходя из 2-(3-фенил-1H-1,2,4-триазол-5-ил)этанамина тригидрохлорида (500 мг; 1,68 ммоль), по способу, описанному в примере 15, стадия 1, в виде бесцветного масла (663 мг; 1,67 ммоль; 99,6%). MS: M=389,3 (M+H)+
Стадия 2: 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-1-метил-3-фенил-[1,2,4]триазол
Данный продукт получали, исходя из 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-3-фенил-[1,2,4]триазола (663 мг; 1,67 ммоль), по способу, описанному в примере 15, стадия 2, в виде бесцветного масла (251 мг; 0,62 ммоль; 37,3%). MS: M=403,2 (M+H)+
Стадия 3: 2-(1-метил-3-фенил-1H-1,2,4-триазол-5-ил)этанамин гидрохлорид
Данный продукт получали, исходя из 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-1-метил-3-фенил-[1,2,4]триазола (247,5 мг; 615 мкмоль), по способу, описанному в примере 15, стадия 3, в виде белого твердого вещества (143 мг; 599 мкмоль; 97,4%). MS: M=203,2 (M+H)+
Стадия 4: [2-(2-метил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(1-метил-3-фенил-1H-1,2,4-триазол-5-ил)этанамина гидрохлорида (59,3 мг; 249 мкмоль), по способу, описанному в примере 3, в виде бесцветного масла (51,1 мг; 130 мкмоль; 67,9%). MS: M=394,2 (M+H)+
Пример 23: [1,1-диметил-2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
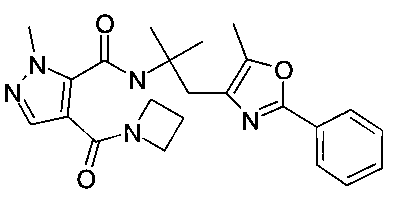
Данный продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-метил-1-(5-метил-2-фенилоксазол-4-ил)пропан-2-амина (36,3 мг; 158 мкмоль; полученного по WO 2003/037327, пример 60), по способу, описанному в примере 7, стадия 5, в виде бесцветного вязкого масла (46,1 мг; 109 мкмоль; 76,3%). MS: M=422,2 (M+H)+
Пример 24: [2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 2-метил-4-(морфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты
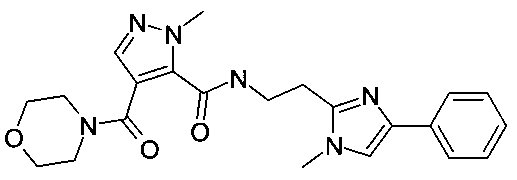
Данный продукт получали, исходя из промежуточного соединения A-2 (50 мг; 209 мкмоль) и 2-(1-метил-4-фенил-1H-имидазол-2-ил)этанамина дигидрохлорида (63,0 мг; 230 мкмоль; пример 7, стадии 1-4), по способу, описанному в примере 8, стадия 5, в виде белого твердого вещества (44 мг; 104 мкмоль; 49,8%). MS: M=423,2 (M+H)+
Пример 25: [1,1-диметил-2-(3-фенил-[1,2,4]оксадиазол-5-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
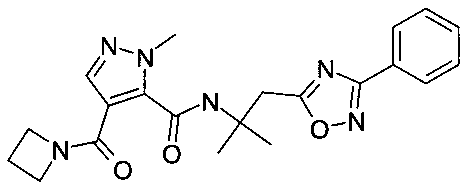
Стадия 1: трет-бутил-2-метил-1-(3-фенил-1,2,4-оксадиазол-5-ил)пропан-2-илкарбамат
Раствор 3-(трет-бутоксикарбониламино)-3-метилбутановой кислоты (2,75 г; 12,7 ммоль) и N,N′-карбонилдиимидазола (CDI; 2,05 г; 12,7 ммоль) в DMF (20,0 мл) перемешивали при КТ в атмосфере азота в течение 5 ч. Добавляли N-гидроксибензамидин (1,72 г; 12,7 ммоль) и реакционную смесь нагревали до 100°C и перемешивали в течение 15 ч. Неочищенную реакционную смесь концентрировали в вакууме, вливали в этилацетат (100 мл) и экстрагировали водой (3×100 мл). Объединенный водный слой снова экстрагировали этилацетатом (100 мл). Объединенный органический слой высушивали над MgSO4, концентрировали в вакууме и очищали флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) с получением продукта в виде белого твердого вещества (2,72 г; 8,57 ммоль; 67,5%). MS: M=318,1 (M+H)+
Стадия 2: 2-метил-1-(3-фенил-1,2,4-оксадиазол-5-ил)пропан-2-амин гидрохлорид
Раствор трет-бутил-2-метил-1-(3-фенил-1,2,4-оксадиазол-5-ил)пропан-2-илкарбамата (2,72 г; 8,57 ммоль) и HCl (4 М в диоксане; 21,4 мл; 85,7 ммоль) перемешивали при 25°C в течение 3 ч. Полученную белую суспензию разбавляли трет-бутилметиловым эфиром (100 мл) и перемешивали в течение 1 ч. Реакционную смесь фильтровали и полученный остаток высушивали в вакууме с получением продукта в виде белого твердого вещества (2,01 г; 7,92 ммоль; 92,4%). MS: M=218,1 (M+H-HCl)+
Стадия 3: [1,1-диметил-2-(3-фенил-[1,2,4]оксадиазол-5-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-метил-1-(3-фенил-1,2,4-оксадиазол-5-ил)пропан-2-амина гидрохлорида (40,0 мг; 158 мкмоль), по способу, описанному в примере 7, стадия 5, в виде белого твердого вещества (50 мг; 122 мкмоль; 85,4%). MS: M=409,3 (M+H)+
Пример 26: [2-(2-пиримидин-2-илтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
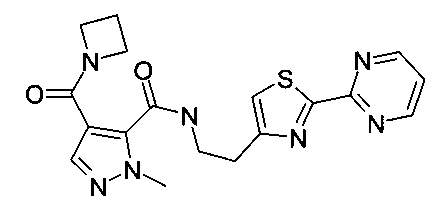
Данный продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(2-(пиримидин-2-ил)тиазол-4-ил)этанамина дигидрохлорида (40,0 мг; 143 мкмоль), по способу, описанному в примере 7, стадия 5, в виде коричневого воскообразного твердого вещества (32,5 мг; 81,8 мкмоль; 57,0%). MS: M=398,2 (M+H)+
Пример 27: [2-(1-метил-4-пиридин-3-ил-1H-имидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
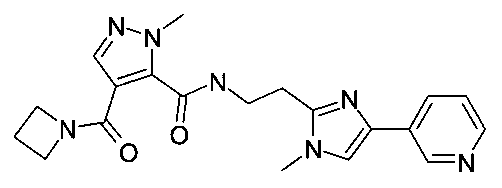
Стадия 1: 2-оксо-2-(пиридин-3-ил)этил 3-(трет-бутоксикарбониламино)пропионат
Данный продукт получали, исходя из 3-(трет-бутоксикарбониламино)пропионовой кислоты (1 г; 5,29 ммоль) и 2-бром-1-(пиридин-3-ил)этанона гидробромида (1,48 г; 5,29 ммоль), по способу, описанному в примере 7, стадия 1, после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан), в виде белого твердого вещества (1,255 г; 4,07 ммоль; 77,0%). MS: M=309,2 (M+H)+
Стадия 2: трет-бутил-2-(4-(пиридин-3-ил)-1H-имидазол-2-ил)этилкарбамат
Данный продукт получали, исходя из 2-оксо-2-(пиридин-3-ил)этил-3-(трет-бутоксикарбониламино)пропионата (1,252 г; 4,06 ммоль), по способу, описанному в примере 7, стадия 2, в виде светло-желтой пены (744 мг; 2,58 ммоль; 63,5%). MS: M=189,3(M-Boc+H)+
Стадия 3: 3-(2-(2-(трет-бутоксикарбониламино)этил)-1-метил-1H-имидазол-4-ил)-1-метилпиридиний иодид
Данный продукт получали, исходя из трет-бутил-2-(4-(пиридин-3-ил)-1H-имидазол-2-ил)этилкарбамата (732 мг; 2,54 ммоль), по способу, описанному в примере 7, стадия 3, после очистки флэш-хроматографией (с использованием аминосиликагелевой фазы и градиента MeOH/этилацетат), в виде коричневой пены (819 мг; 2,06 ммоль; 81,3%). MS: M=317,2 (M-I+H)+
Стадия 4: трет-бутил-2-(1-метил-4-(пиридин-3-ил)-1H-имидазол-2-ил)этилкарбамат
Раствор 3-(2-(2-(трет-бутоксикарбониламино)этил)-1-метил-1H-имидазол-4-ил)-1-метилпиридиния иодида (655 мг; 2,06 ммоль) в 1-метилимидазоле (2 мл; 25,1 ммоль) нагревали до 160°C и перемешивали в течение 9 ч. Реакционную смесь вливали в воду (5 мл) и экстрагировали дихлорметаном (2×5 мл). Органические слои объединяли, высушивали над MgSO4 и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией (с использованием аминосиликагелевой фазы и градиента этилацетат/гептан) с получением 1,04 г светло-желтой жидкости. Оставшийся 1-метилимидазол отгоняли на аппарате для вакуумной дистилляции "Kugelrohr" («шариковая трубка») при 100-120°C при ~0,2 мбар, с получением продукта в виде светло-желтого масла (102 мг; 337 мкмоль; 16,3%). MS: M=303,1 (M+H)+
Стадия 5: 2-(1-метил-4-(пиридин-3-ил)-1H-имидазол-2-ил)этанамин тригидрохлорид
Данный продукт получали, исходя из трет-бутил-2-(1-метил-4-(пиридин-3-ил)-1H-имидазол-2-ил)этилкарбамата (100 мг; 331 мкмоль), по способу, описанному в примере 7, стадия 4, в виде белого твердого вещества (110 мг; 318 мкмоль; 96,1%). MS: M=203,2 (M+H)+
Стадия 6: [2-(1-метил-4-пиридин-3-ил-1H-имидазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(1-метил-4-(пиридин-3-ил)-1H-имидазол-2-ил)этанамина тригидрохлорида (66,2 мг; 191 мкмоль), по способу, описанному в примере 10, стадия 5, в виде белого твердого вещества (35 мг; 89,0 мкмоль; 46,5%). MS: M=394,1 (M+H)+
Пример 28: [2-(1-фенил-1H-пиразол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
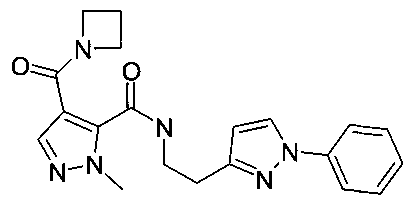
Данный продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(1-фенил-1H-пиразол-3-ил)этанамина (26,9 мг; 143 мкмоль), по способу, описанному в примере 7, стадия 5, в виде бесцветного твердого вещества (21 мг; 55,5 мкмоль; 38,7%). MS: M=379,3 (M+H)+
Пример 29: [2-(2-фенилтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
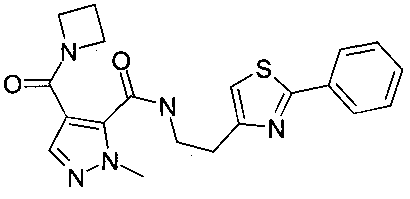
Данный продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(2-фенилтиазол-4-ил)этанамина (29,3 мг; 143 мкмоль), по способу, описанному в примере 7, стадия 5, в виде светло-желтого твердого вещества (23 мг; 58,2 мкмоль; 40,6%). MS: M=396,2 (M+H)+
Пример 30: {2-[4-(3-метоксифенил)-1-метил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
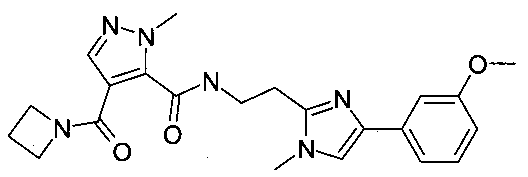
Стадия 1: 2-(3-метоксифенил)-2-оксоэтил-3-(трет-бутоксикарбониламино)пропионат
Данный продукт получали, исходя из 3-(трет-бутоксикарбониламино)пропионовой кислоты (400 мг; 2,11 ммоль) и 2-бром-1-(3-метоксифенил)этанона (484 мг; 2,11 ммоль), по способу, описанному в примере 7, стадия 1, в виде желтого масла (759 мг; 2,11 ммоль; 100%). MS: M=238,2 (M-Boc+H)+
Стадия 2: трет-бутил-2-(4-(3-метоксифенил)-1H-имидазол-2-ил)этилкарбамат
Данный продукт получали, исходя из 2-(3-метоксифенил)-2-оксоэтил-3-(трет-бутоксикарбониламино)пропионата (755 мг; 2,1 ммоль), по способу, описанному в примере 7, стадия 2, в виде желтой пены (533 мг; 1,68 ммоль; 79,8%). MS: M=318,3 (M+H)+
Стадия 3: трет-бутил-2-(4-(3-метоксифенил)-1-метил-1H-имидазол-2-ил)этилкарбамат
Данный продукт получали, исходя из трет-бутил-2-(4-(3-метоксифенил)-1H-имидазол-2-ил)этилкарбамата (528 мг; 1,66 ммоль), по способу, описанному в примере 7, стадия 3, в виде белого твердого вещества (350 мг; 1,06 ммоль; 63,5%). MS: M=332,2 (M+H)+
Стадия 4: 2-(4-(3-метоксифенил)-1-метил-1H-имидазол-2-ил)этанамин дигидрохлорид
Данный продукт получали, исходя из трет-бутил-2-(4-(3-метоксифенил)-1-метил-1H-имидазол-2-ил)этилкарбамата (344 мг; 1,04 ммоль), по способу, описанному в примере 7, стадия 4, в виде белого твердого вещества (321 мг; 1,03 ммоль; 99,6%). MS: M=232,1 (M-2HCl+H)+
Стадия 5: {2-[4-(3-метоксифенил)-1-метил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(4-(3-метоксифенил)-1-метил-1H-имидазол-2-ил)этанамина дигидрохлорида (64,0 мг; 210 мкмоль), по способу, описанному в примере 7, стадия 5, после очистки флэш-хроматографией с использованием силикагеля и градиента этилацетат/гептан, в виде белого пены (49 мг; 116 мкмоль; 60,7%). MS: M=423,3 (M+H)+
Пример 31: {2-[4-(2-метоксифенил)-1-метил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
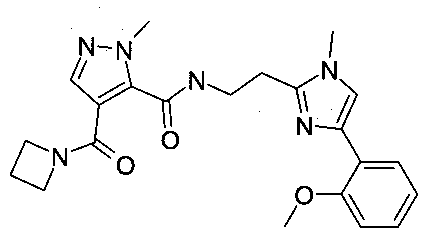
Стадия 1: 2-(2-метоксифенил)-2-оксоэтил 3-(трет-бутоксикарбониламино)пропионат
Данный продукт получали, исходя из 3-(трет-бутоксикарбониламино)пропионовой кислоты (400 мг; 2,11 ммоль) и 2-бром-1-(2-метоксифенил)этанона (484 мг; 2,11 ммоль), по способу, описанному в примере 7, стадия 1, в виде черного масла (786 мг; 2,1 ммоль; 99,2%). MS: M=238,2 (M-Boc+H)+
Стадия 2: трет-бутил-2-(4-(2-метоксифенил)-1H-имидазол-2-ил)этилкарбамат
Данный продукт получали, исходя из 2-(2-метоксифенил)-2-оксоэтил-3-(трет-бутоксикарбониламино)пропионата (784 мг; 2,09 ммоль), по способу, описанному в примере 7, стадия 2, в виде желтой пены (501 мг; 1,58 ммоль; 75,5%). MS: M=218,3 (M-Boc+H)+
Стадия 3: трет-бутил-2-(4-(2-метоксифенил)-1-метил-1H-имидазол-2-ил)этилкарбамат
Данный продукт получали, исходя из трет-бутил-2-(4-(2-метоксифенил)-1H-имидазол-2-ил)этилкарбамата (499 мг; 1,57 ммоль), по способу, описанному в примере 7, стадия 3, в виде коричневого твердого вещества (303 мг; 914 мкмоль; 58,2%). MS: M=332,2 (M+H)+
Стадия 4: 2-(4-(2-метоксифенил)-1-метил-1H-имидазол-2-ил)этанамин дигидрохлорид
Данный продукт получали, исходя из трет-бутил-2-(4-(2-метоксифенил)-1-метил-1H-имидазол-2-ил)этилкарбамата (293 мг; 884 мкмоль), по способу, описанному в примере 7, стадия 4, в виде светло-коричневого твердого вещества (271 мг; 873 мкмоль; 98,7%). MS: M=232,1 (M-2HCl+H)+
Стадия 5: {2-[4-(2-метоксифенил)-1-метил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(4-(2-метоксифенил)-1-метил-1H-имидазол-2-ил)этанамина дигидрохлорида (64,0 мг; 210 мкмоль), по способу, описанному в примере 30, стадия 5, в виде светло-коричневого твердого вещества (48 мг; 114 мкмоль; 59,4%). MS: M=423,2 (M+H)+
Пример 32: {2-[1-(2-метоксиэтил)-4-фенил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
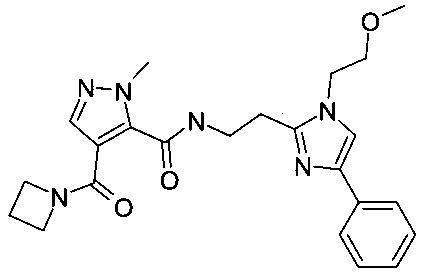
Стадия 1: трет-бутил-2-(1-(2-метоксиэтил)-4-фенил-1H-имидазол-2-ил)этилкарбамат
Суспензию трет-бутил-2-(4-фенил-1H-имидазол-2-ил)этилкарбамата (400 мг; 1,39 ммоль; полученного в соответствии с примером 7, стадии 1-2) и карбоната калия (423 мг; 3,06 ммоль) в DMF (4 мл) перемешивали в течение 30 мин при КТ. После охлаждения до 0-5°C добавляли 1-бром-2-метоксиэтан (157 мкл; 1,67 ммоль). Через 5 мин ледяную баню убирали и реакционную смесь перемешивали при КТ в течение 16 ч. Вносили еще по два эквивалента карбоната калия (618 мг; 4,45 ммоль) и 1-бром-2-метоксиэтана (262 мкл; 2,78 ммоль) при КТ, и реакционную смесь нагревали до 60°C и перемешивали в течение 20 ч. Реакционную смесь вливали в этилацетат (50 мл) и органическую фазу промывали водой (3×20 мл) и солевым раствором (25 мл). Водные слои снова экстрагировали этилацетатом (50 мл). Органические слои высушивали над MgSO4 и концентрировали в вакууме. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде бесцветного твердого вещества (359 мг; 1,04 ммоль; 74,7%). MS: M=346,2 (M+H)+
Стадия 2: 2-(1-(2-метоксиэтил)-4-фенил-1H-имидазол-2-ил)этанамин гидрохлорид
Данный продукт получали, исходя из трет-бутил-2-(1-(2-метоксиэтил)-4-фенил-1H-имидазол-2-ил)этилкарбамата (359 мг; 1,04 ммоль), по способу, описанному в примере 7, стадия 4, в виде светло-желтого твердого вещества (273 мг; 969 мкмоль; 93,2%). MS: M=246,2 (M-HCl+H)+
Стадия 3: {2-[1-(2-метоксиэтил)-4-фенил-1H-имидазол-2-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(1-(2-метоксиэтил)-4-фенил-1H-имидазол-2-ил)этанамина гидрохлорида (56,5 мг; 201 мкмоль), по способу, описанному в примере 7, стадия 5, с использованием 8 эквивалентов N,N-диизопропилэтиламина и проводя вторую очистку флэш-хроматографией (с использованием аминосиликагелевой фазы и градиента MeOH/этилацетат), в виде бесцветного полутвердого вещества (20,3 мг; 46,5 мкмоль; 32,4%). MS: M=437,3 (M+H)+
Пример 33: [2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-2,3-диметил-3H-имидазол-4-карбоновой кислоты
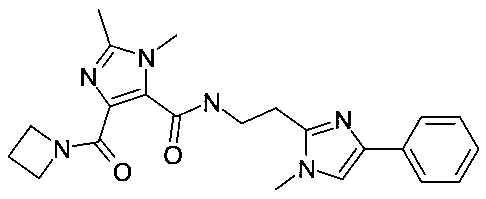
Стадия 1: метиловый эфир 1,2-диметил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты
Данный продукт получали, исходя из 4-метилового эфира 1,2-диметил-1H-имидазол-4,5-дикарбоновой кислоты (100 мг; 505 мкмоль; пример 8, стадии 1-2) и 2-(1-метил-4-фенил-1H-имидазол-2-ил)этанамина дигидрохлорида (152 мг; 555 мкмоль; пример 7, стадии 1-4), по способу, описанному в примере 10, стадия 5, после перемешивания при 70°C в течение 16 ч, обработки водой и очистки, в виде светло-желтого масла (111 мг; 291 мкмоль; 57,7%). MS: M=382,4 (M+H)+
Стадия 2: 1,2-диметил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновая кислота
Данный продукт получали, исходя из метилового эфира 1,2-диметил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты (110 мг; 288 мкмоль), по способу, описанному в примере 8, стадия 4, в виде бесцветной пены (60 мг; 163 мкмоль; 56,6%). MS: M=368,2 (M+H)+
Стадия 3: [2-(1 -метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-2,3-диметил-3H-имидазол-4-карбоновой кислоты
К раствору 1,2-диметил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-имидазол-4-карбоновой кислоты (30 мг; 81,7 мкмоль) в DMF (1 мл) добавляли в атмосфере азота при 0-5°C (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолинокарбений гексафторфосфат (COMU; 38,5 мг; 89,8 мкмоль) и N,N-диизопропилэтиламин (14,3 мкл; 81,7 мкмоль). Перемешивали в течение 5 мин, после чего добавляли азетидин (6,18 мкл; 89,8 мкмоль) и N,N-диизопропилэтиламин (14,3 мкл; 81,7 мкмоль), Еще через 10 мин ледяную баню убирали и реакционную смесь перемешивали при КТ в течение 2 ч. Продукт получали после очистки с помощью препаративной ВЭЖХ с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% муравьиную кислоту), в виде бесцветного воскообразного твердого вещества (1,9 мг; 4,67 мкмоль; 5,72%). MS: M=407,4 (M+H)+
Пример 34: {2-[2-(2-метоксиэтил)-5-фенил-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
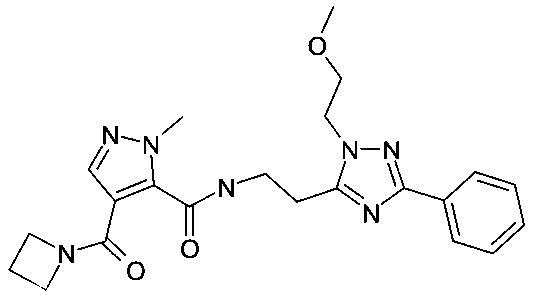
Стадия 1: трет-бутил-2-(1-(2-метоксиэтил)-3-фенил-1H-1,2,4-триазол-5-ил)этилкарбамат
5-[2-Бис(трет-бутоксикарбонил)аминоэтил]-3-фенил-[1,2,4]триазол (96 мг; 247 мкмоль; пример 22, стадия 1) и карбонат калия (85,4 мг; 618 мкмоль) объединяли с DMF (1 мл) и эту смесь перемешивали в течение 30 мин при КТ. Добавляли 1-бром-2-метоксиэтан (34,3 мг; 23,2 мкл; 247 мкмоль) и смесь перемешивали при 60°C в течение 16 ч. Реакционную смесь охлаждали до КТ, добавляли вторую порцию 1-бром-2-метоксиэтана (34,3 мг; 23,2 мкл; 247 мкмоль) и карбоната калия (34,2 мг; 247 мкмоль) и эту смесь перемешивали при 60°C еще в течение 4 ч. Реакционную смесь вливали в этилацетат (25 мл) и органическую фазу промывали водой (3×10 мл) и солевым раствором (1×15 мл). Водные слои снова экстрагировали этилацетатом (1×25 мл). Органические слои высушивали над MgSO4 и концентрировали в вакууме с получением 120 мг неочищенного продукта. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде бесцветного масла (29 мг; 83,7 мкмоль; 33,9%).
MS: M=347,2 (M+H)+
Стадия 2: 2-(1-(2-метоксиэтил)-3-фенил-1H-1,2,4-триазол-5-ил)этанамин гидрохлорид
Данный продукт получали, исходя из трет-бутил-2-(1-(2-метоксиэтил)-3-фенил-1H-1,2,4-триазол-5-ил)этилкарбамата (29 мг; 83,7 мкмоль), по способу, описанному в примере 15, стадия 3, в виде белого твердого вещества (27,2 мг; 81,8 мкмоль; 97,7%).
MS: M=247,2 (M-HCl+H)+
Стадия 3: {2-[2-(2-метоксиэтил)-5-фенил-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (12,6 мг; 60,2 мкмоль) и 2-(1-(2-метоксиэтил)-3-фенил-1H-1,2,4-триазол-5-ил)этанамина гидрохлорида (26,0 мг; 78,3 мкмоль), по способу, описанному в примере 3, в виде бесцветного масла (15,2 мг; 34,7 мкмоль; 57,7%).
MS: M=438,3 (M+H)+
Пример 35: [2-(2-циклопропилметил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
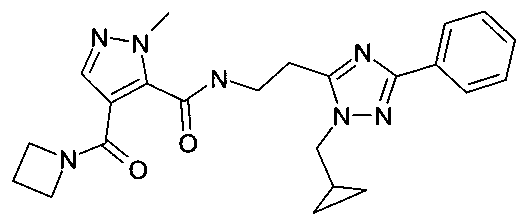
Стадия 1: трет-бутил-2-(1-(циклопропилметил)-3-фенил-1H-1,2,4-триазол-5-ил)этилкарбамат
Данный продукт получали, исходя из 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-3-фенил-[1,2,4]триазола (250 мг; 644 мкмоль; пример 22, стадия 1) и (бромметил)циклопропана (104 мг; 73,9 мкл; 772 мкмоль), по способу, описанному в примере 34, стадия 1, в виде бесцветного масла (69 мг; 201 мкмоль; 31,3%).
MS: M=343,3 (M+H)+
Стадия 2: 2-(1-(циклопропилметил)-3-фенил-1H-1,2,4-триазол-5-ил)этанамин гидрохлорид
Данный продукт получали, исходя из трет-бутил-2-(1-(циклопропилметил)-фенил-1H-1,2,4-триазол-5-ил)этилкарбамата (67 мг; 196 мкмоль), по способу, описанному в примере 15, стадия 3, в виде бесцветного твердого вещества (59 мг; 190 мкмоль; 97,3%).
MS: M=243,4 (M-HCl+H)+
Стадия 3: [2-(2-циклопропилметил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Данный продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(1-(циклопропилметил)-3-фенил-1H-1,2,4-триазол-5-ил)этанамина гидрохлорида (53,3 мг; 172 мкмоль), по способу, описанному в примере 3, в виде светло-коричневого твердого вещества (34 мг; 78,4 мкмоль; 54,7%).
MS: M=434,3 (M+H)+
Пример 36: [2-(5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
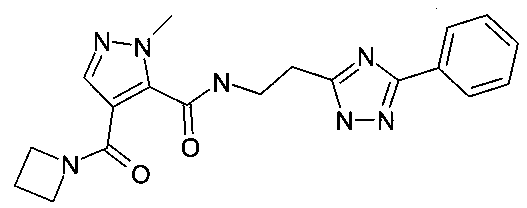
Данный продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(3-фенил-1H-1,2,4-триазол-5-ил)этанамина тригидрохлорида (53,9 мг; 172 мкмоль), по способу, описанному в примере 3, в виде светло-коричневого твердого вещества (29 мг; 76,4 мкмоль; 53,3%).
MS: M=380,3 (M+H)+
Пример 37: [2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 2-метил-4-(морфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты
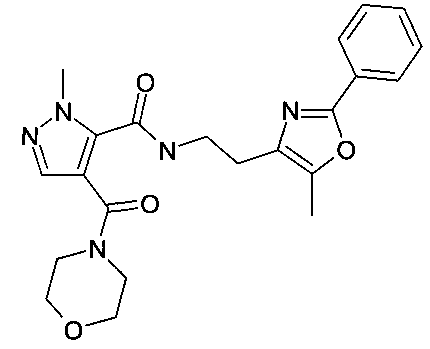
Стадия 1: этил-1 -метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоксилат
К суспензии 4-(этоксикарбонил)-1-метил-1H-пиразол-5-карбоновой кислоты (500 мг; 2,52 ммоль; полученной, как описано в US 2011/0071128), в THF (12,5 мл) добавляли 2-(5-метил-2-фенилоксазол-4-ил)этанамин гидрохлорид (602 мг; 2,52 ммоль), циклический ангидрид 1-пропанфософорной кислоты (4,01 г; 3,75 мл; 6,31 ммоль) и N,N-диизопропилэтиламин (2,61 г; 3,53 мл; 20,2 ммоль) при КТ. Эту смесь перемешивали при 70°C в течение 2 ч, вливали в KHSO4 (10% водный раствор, 50 мл) и экстрагировали этилацетатом (2×75 мл). Органические фазы промывали водой (50 мл) и солевым раствором (50 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 735 мг неочищенного продукта. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде белого твердого вещества (410 мг; 1,07 ммоль; 42,5%).
MS: M=383,2 (M+H)+
Стадия 2: 1-метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоновая кислота
К раствору этил-1-метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоксилата (407 мг; 1,06 ммоль) в THF (3,00 мл) и этанола (1,5 мл) добавляли LiOH (1 М водный раствор, 2,13 мл; 2,13 ммоль) при 0-5°C. Реакционную смесь перемешивали при КТ в течение 2 ч и вливали в HCl (1 М; 2,15 мл). Полученную густую суспензию фильтровали и этот фильтрат экстрагировали дихлорметаном (3×50 мл). Органические слои объединяли, высушивали над MgSO4 и концентрировали в вакууме с получением 341 мг (90,4%) грязно-белого твердого вещества в виде неочищенного продукта, которое использовали без дополнительной очистки.
MS: M=355,2 (M+H)+
Стадия 3: [2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 2-метил-4-(морфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты
К суспензии 1-метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (40 мг; 113 мкмоль) в DMF (1 мл) добавляли COMU (58,0 мг; 135 мкмоль) и N,N-диизопропилэтиламин (14,6 мг; 19,7 мкл; 113 мкмоль) при 0-5°C. Через 10 мин суспензия превращалась в прозрачный раствор, после чего добавляли морфолин (11,8 мг; 135 мкмоль) и N,N-диизопропилэтиламин (14,6 мг; 19,7 мкл; 113 мкмоль). Через 10 мин ледяную баню убирали и смесь перемешивали при КТ в течение 3 ч. Продукт получали после очистки с помощью препаративной ВЭЖХ с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% муравьиную кислоту) в виде грязно-белого воскообразного твердого вещества (31 мг; 73,2 мкмоль; 64,9%).
MS: M=424,2 (M+H)+
Пример 38: [2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 2-метил-4-(пирролидин-1-карбонил)-2H-пиразол-3-карбоновой кислоты
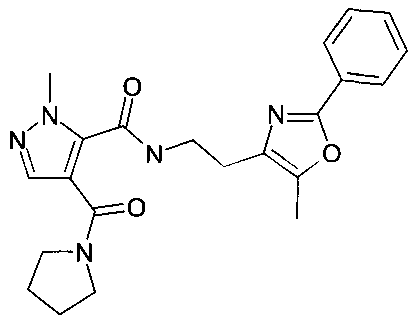
Продукт получали, исходя из 1-метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (40 мг; 113 мкмоль) и пирролидина (8,03 мг; 9,34 мкл; 113 мкмоль), по способу, описанному в примере 37, стадия 3, в виде грязно-белого твердого вещества (22,1 мг; 54,2 мкмоль; 48,0%).
MS: M=408,4 (M+H)+
Пример 39: [2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 4-(3-фторазетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
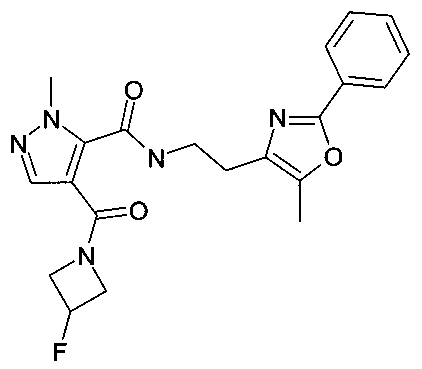
Продукт получали, исходя из 1-метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (40 мг; 113 мкмоль) и 3-фторазетидина гидрохлорида (15,1 мг; 135 мкмоль), по способу, описанному в примере 37, стадия 3, в виде белого твердого вещества (39 мг; 94,8 мкмоль; 84,0%).
MS: M=412,2 (M+H)+
Пример 40: 4-(циклопропилметиламид)-3-{[2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид} 2-метил-2H-пиразол-3,4-дикарбоновой кислоты
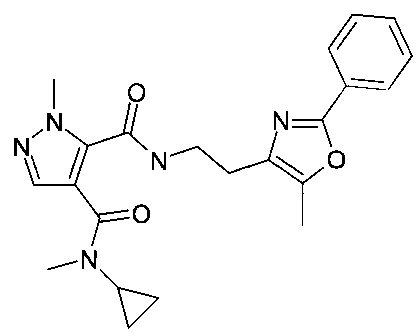
Продукт получали, исходя из 1-метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (40 мг; 113 мкмоль) и N-метилциклопропанамина (9,63 мг; 135 мкмоль), по способу, описанному в примере 37, стадия 3, в виде бесцветного масла (35,5 мг; 87,1 мкмоль; 77,2%).
MS: M=408,4 (M+H)+
Пример 41: [2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 2-метил-4-(2-окса-6-аза-спиро[3,3]гептан-6-карбонил)-2H-пиразол-3-карбоновой кислоты
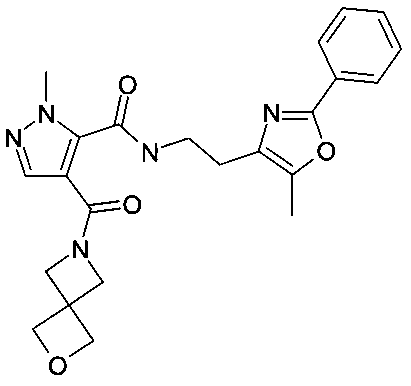
Продукт получали, исходя из 1-метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (40 мг; 113 мкмоль) и 2-окса-6-азаспиро[3,3]гептана гемиоксалата (32,5 мг; 112,9 мкмоль), по способу, описанному в примере 37, стадия 3, после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-желтого твердого вещества (24,6 мг; 50%).
MS: M=436,2 (M+H)+
Пример 42: [2-(5-метил-2-фенилоксазол-4-ил)-этил]-амид 2-метил-4-(тиоморфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты
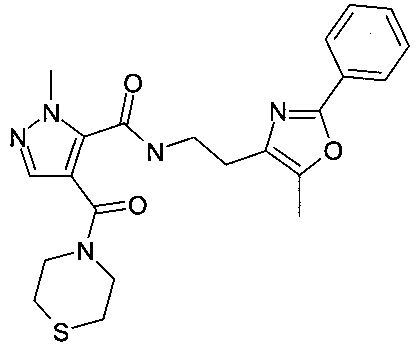
Продукт получали, исходя из 1-метил-5-(2-(5-метил-2-фенилоксазол-4-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (40 мг; 113 мкмоль) и тиоморфолина (14,4 мг; 14,3 мкл; 135 мкмоль), по способу, описанному в примере 37, стадия 3, в виде бесцветного масла (41,7 мг; 94,9 мкмоль; 84,0%).
MS: M=440,3 (M+H)+
Пример 43: [2-(5-метил-2-фенилтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
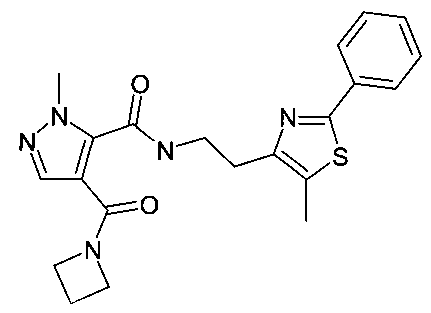
Стадия 1: 4-(2-азидоэтил)-5-метил-2-фенилтиазол
К коричневому раствору 2-(5-метил-2-фенилтиазол-4-ил)этилметансульфоната (200 мг; 673 мкмоль; полученному по WO 2001/021602) в DMF (2,5 мл) добавляли азид натрия (52,5 мг; 807 мкмоль) при КТ. Эту смесь нагревали до 60°C и перемешивали в течение 3 ч и затем медленно остужали до КТ. Реакционную смесь вливали в этилацетат (50 мл) и органическую фазу промывали водой (3×25 мл) и солевым раствором (1×25 мл). Водные слои снова экстрагировали этилацетатом (1×50 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 174 мг неочищенного продукта. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде бесцветного масла (107 мг; 438 мкмоль; 65,1%).
MS: M=245,2 (M+H)+
Стадия 2: 2-(5-метил-2-фенилтиазол-4-ил)этанамин
4-(2-Азидоэтил)-5-метил-2-фенилтиазол (104 мг; 426 мкмоль) объединяли с трифенилфосфином (123 мг; 468 мкмоль) и водой (1 капля, 4 мкл; 222 мкмоль) при КТ. Реакционную смесь перемешивали при КТ в течение ночи и растворитель удаляли в вакууме с получением светло-коричневого остатка. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и 1 М аммиака в градиенте метанол/дихлорметан) в виде бесцветного полутвердого вещества (72,3 мг; 331 мкмоль; 77,8%).
MS: M=219,2 (M+H)+
Стадия 3: [2-(5-метил-2-фенилтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(5-метил-2-фенилтиазол-4-ил)этанамина (31,3 мг; 143 мкмоль), по способу, описанному в примере 7, стадия 5, после очистки с помощью препаративной ВЭЖХ с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% муравьиную кислоту), в виде белого твердого вещества (36 мг; 87,9 мкмоль; 61,3%).
MS: M=410,2 (M+H)+
Пример 44: [2-(5-метил-3-фенил-[1,2,4]триазол-1-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
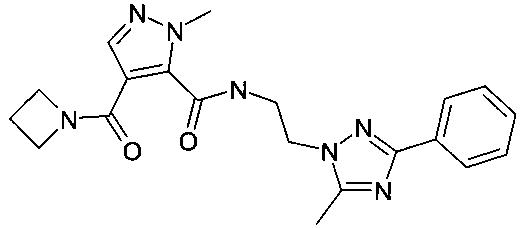
Стадия 1: 5-метил-3-фенил-1H-1,2,4-триазол
К суспензии этилбензимидата гидрохлорида (2,0 г; 10,8 ммоль) в EtOH (10 мл) добавляли этоксид натрия (3,49 г; 4 мл; 10,8 ммоль) при КТ. Выпавший в осадок NaCl отфильтровывали и промывали с помощью EtOH (14 мл). В полученный фильтрат вносили ацетогидразид (880 мг; 11,9 ммоль) и перемешивали при КТ в течение 6 суток. Неочищенную реакционную смесь концентрировали в вакууме. Неочищенный продукт суспендировали в NaOH (1 M водный раствор, 50 мл) и экстрагировали трет-бутил метиловым эфиром (100 мл). Органический слой промывали водой (50 мл) и водные слои снова экстрагировали трет-бутилметиловым эфиром (50 мл). Вносили сухой лед для доведения pH до ~7, при этом выяснилось, что органический слой практически не содержал продукт, и его отбросили. Водный слой доводили до pH=7 добавлением HCl (25% водный раствор) и экстрагировали дихлорметаном (6×50 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гексан) в виде белого твердого вещества (154 мг; 9%).
MS: M=160,1 (M+H)+
Стадия 2: трет-бутил-2-(5-метил-3-фенил-1H-1,2,4-триазол-1-ил)этилкарбамат
5-Метил-3-фенил-1H-1,2,4-триазол (120 мг; 754 мкмоль) растворяли в DMF (2,4 мл) и добавляли карбонат калия (208 мг; 1,51 ммоль). Через 20 мин добавляли трет-бутил-2-бромэтилкарбамат (186 мг; 829 мкмоль) при 0-5°C и реакционную смесь перемешивали при КТ в течение 25 ч. Реакционную смесь экстрагировали этилацетатом (30 мл) и промывали водой (3×50 мл). Водные слои снова экстрагировали этилацетатом (20 мл) и объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде белого твердого вещества (237 мг; 784 мкмоль; 83,2%).
MS: M=303,2 (M+H)+
Стадия 3: 2-(5-метил-3-фенил-1H-1,2,4-триазол-1-ил)этанамин гидрохлорид
трет-Бутил-2-(5-метил-3-фенил-1H-1,2,4-триазол-1-ил)этилкарбамат (235 мг; 777 мкмоль) суспендировали в HCl (4 M раствор в диоксане; 2,5 мл; 10,0 ммоль) и перемешивали при КТ в течение 2 ч. Полученную суспензию вливали в диэтиловый эфир (3 мл) и фильтровали. Полученные кристаллы промывали диэтиловым эфиром (2 мл) и высушивали. Эти белые кристаллы (193 мг; 777 мкмоль; 100%) использовали без дополнительной очистки на следующей стадии.
MS: M=203,2 (M-HCl+H)+
Стадия 4: [2-(5-метил-3-фенил-[1,2,4]триазол-1-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(5-метил-3-фенил-1H-1,2,4-триазол-1-ил)этанамина гидрохлорида (50,2 мг; 210 мкмоль), по способу, описанному в примере 43, стадия 3, и в ходе дополнительной очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/метанол) в виде светло-желтого полутвердого вещества (27,1 мг; 36%)
MS: M=394,2 (M+H)+
Пример 45: [2-(2-бензил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
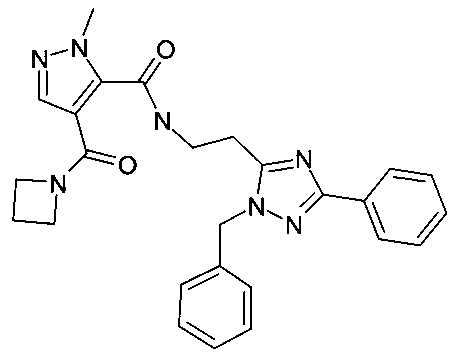
Стадия 1: 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-1-бензил-3-фенил-[1,2,4]триазол и трет-бутил-2-(1-бензил-3-фенил-1H-1,2,4-триазол-5-ил)этилкарбамат
К раствору 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-3-фенил-[1,2,4]триазола (232 мг; 597 мкмоль; пример 22, стадия 1) в DMF (2,83 мл) добавляли гидрид натрия (55%, 25 мг; 574 мкмоль) при 0-5°C. Полученную суспензию перемешивали в течение 30 мин. Затем добавляли бензилбромид (102 мг; 71 мкл; 597 мкмоль) при 0-5°C, через 45 мин ледяную баню убирали и продолжали перемешивать в течение 1 ч. Реакционную смеси экстрагировали этилацетатом (40 мл) и промывали водой (3×20 мл) и солевым раствором (1×20 мл). Водные слои снова экстрагировали этилацетатом (40 мл) и объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме. Неочищенное вещество очищали флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) с получением 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-1-бензил-3-фенил-[1,2,4]триазола (115 мг; 240 мкмоль; 73,2%, MS: M=479,3 (M+H)+) и трет-бутил-2-(1-бензил-3-фенил-1H-1,2,4-триазол-5-ил)этилкарбамата (43 мг; 114 мкмоль; 34,6%, MS: M=379,4 (M+H)+), оба продукта в виде бесцветных масел, которые объединяли на следующей стадии.
Стадия 2: 2-(1-бензил-3-фенил-1H-1,2,4-триазол-5-ил)этанамин гидрохлорид
Раствор 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-1-бензил-3-фенил-[1,2,4]триазола (103 мг; 215 мкмоль) и трет-бутил-2-(1-бензил-3-фенил-1H-1,2,4-триазол-5-ил)этилкарбамата (38,5 мг; 102 мкмоль) в HCl (4 М в диоксане, 3 мл; 12,0 ммоль) перемешивали при КТ в течение 1 ч. Реакционную смесь концентрировали в вакууме и полученное твердое вещество вливали в диэтиловый эфир (2,5 мл), отфильтровывали и промывали небольшим количеством диэтилового эфира. Полученные белые кристаллы высушивали (74 мг; 74%) и использовали без дополнительной очистки на следующей стадии.
MS: M=279,3 (M+H)+
Стадия 3: [2-(2-бензил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(1-бензил-3-фенил-1H-1,2,4-триазол-5-ил)этанамина гидрохлорида (66,2 мг; 210 мкмоль), по способу, описанному в примере 43, стадия 3, в виде бесцветного твердого вещества (40 мг; 85,2 мкмоль; 44,6%).
MS: M=470,4 (M+H)+
Пример 46: [2-(5-метил-2-п-толилтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
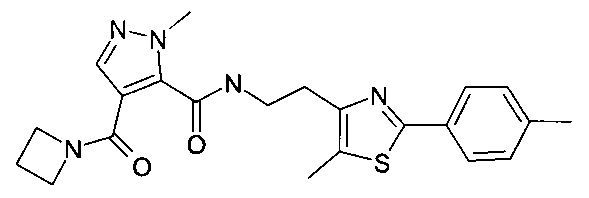
Стадия 1: 2-(5-метил-2-п-толилтиазол-4-ил)этил метансульфонат
К желтому раствору 2-(5-метил-2-п-толилтиазол-4-ил)этанола (200 мг; 857 мкмоль) в дихлорметане (3 мл) добавляли N,N-диизопропилэтиламин (166 мг; 225 мкл; 1,29 ммоль) при КТ. Добавляли метансульфонилхлорид (113 мг; 76,8 мкл; 986 мкмоль) при 0-5°C и продолжали перемешивать в течение 4 ч при 0°C. Реакционную смесь вливали в этилацетат (25 мл) и экстрагировали с помощью KHSO4 (10% водный раствор, 15 мл). Органическую фазу промывали водой (2×15 мл) и солевым раствором (1×20 мл). Водные слои снова экстрагировали этилацетатом (1×25 мл) и объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 280 мг (99%) желтого масла, которое использовали без дополнительной очистки на следующей стадии.
MS: M=312,2 (M+H)+
Стадия 2: 4-(2-азидоэтил)-5-метил-2-п-толилтиазол
Продукт получали, исходя из 2-(5-метил-2-п-толилтиазол-4-ил)этилметансульфоната (271 мг; 827 мкмоль), по способу, описанному в примере 43, стадия 1, в виде бесцветного масла (163 мг; 631 мкмоль; 76,3%).
MS: M=259,1 (M+H)+
Стадия 3: 2-(5-метил-2-п-толилтиазол-4-ил)этанамин
4-(2-Азидоэтил)-5-метил-2-п-толилтиазол (160 мг; 619 мкмоль), полимерсвязанный трифенилфосфин (270 мг; 803 мкмоль) и воду (2 капли, 8 мкл; 444 мкмоль) объединяли при КТ с получением коричневой суспензии. Эту смесь перемешивали при КТ в течение ночи. Суспензию фильтровали и полученную жидкость выпаривали с получением продукта в виде светло-желтого масла (151 мг; 617 мкмоль; 99,7%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=233,2 (M+H)+
Стадия 4: [2-(5-метил-2-п-толилтиазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(5-метил-2-п-толилтиазол-4-ил)этанамина (35,1 мг; 143 мкмоль), по способу, описанному в примере 43, стадия 3, в виде белого твердого вещества (41 мг; 96,8 мкмоль; 67,5%).
MS: M=424,3 (M+H)+
Пример 47: [2-(4-метил-1-фенил-1H-пиразол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
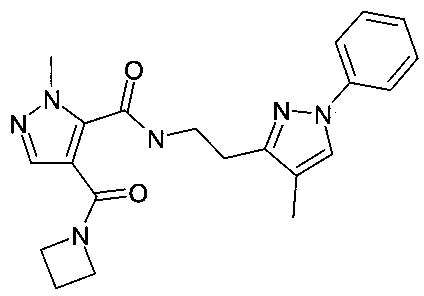
Стадия 1: (E)-1,1,1-трихлор-4-этокси-3-метилбут-3 ен-2-он
К раствору 2,2,2-трихлорацетилхлорида (10,6 г; 6,52 мл; 58,1 ммоль) в дихлорметане (15 мл), охлажденному до 10°C, добавляли смесь (E)-1-этоксипроп-1-ена (5 г; 6,43 мл; 58,1 ммоль) и пиридина (4,59 г; 4,7 мл; 58,1 ммоль) в течение 15 мин. По окончании внесения эту смесь перемешивали при КТ в течение 16 ч. Полученный осадок фильтровали и промывали дихлорметаном. Полученный фильтрат концентрировали в вакууме и высушивали при 40°C в высоком вакууме с получением продукта в виде коричневого полутвердого вещества (16,1 г с 80% чистотой, 95,8%). Неочищенное вещество использовали непосредственно на следующей стадии без дополнительной очистки.
Стадия 2: этил 4-метил-1-фенил-1H-пиразол-3-карбоксилат
Смесь (E)-1,1,1-трихлор-4-этокси-3-метилбут-3-ен-2-она (16,1 г с 80% чистотой, 55,6 ммоль) и фенилгидразина (8,09 г; 7,36 мл; 71,0 ммоль) в этаноле (70 мл) нагревали с обратным холодильником в течение 4 ч. Реакционную смесь вливали в дихлорметан (150 мл) и экстрагировали с помощью HCl (1 М; 1×75 мл). Органическую фазу промывали водой (1×75 мл), и водные слои снова экстрагировали дихлорметаном (1×150 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 11,94 г неочищенного продукта. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде коричневого масла (1,51 г; 6,56 ммоль; 11,1%)
MS: M=231,2 (M+H)+
Стадия 3: (4-метил-1-фенил-1H-пиразол-3-ил)метанол
К раствору этил-4-метил-1-фенил-1H-пиразол-3-карбоксилата (100 мг; 434 мкмоль) в толуоле (1 мл), охлажденному до -78°C, добавляли по каплям DIBAL-H (1 M раствор в толуоле, 868 мкл; 868 мкмоль), выдерживая температуру ниже -68°C. Реакционную смесь перемешивали при -78°C в течение 30 мин и при -15°C в течение 1 ч. При этой температуре добавляли по каплям KHSO4 (10% водный раствор, 10 мл) и поученную реакционную смесь экстрагировали этилацетатом (2×50 мл). Органическую фазы промывали водой (1×30 мл) и солевым раствором (1×20 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 112 мг неочищенного вещества. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде желтого вязкого масла (52 мг; 276 мкмоль; 63,6%).
MS: M=189,3 (M+H)+
Стадия 4: 3-(бромметил)-4-метил-1-фенил-1H-пиразол
К раствору (4-метил-1-фенил-1H-пиразол-3-ил)метанола (158 мг; 0,84 ммоль) и полимерсвязанного трифенилфосфина (422 мг; 1,26 ммоль) в дихлорметане (4,5 мл) добавляли тетрабромид углерода (416 мг; 1,26 ммоль) при 0-5°C. Через 15 мин ледяную баню убирали и эту смесь перемешивали при КТ в течение 2 ч. Смесь фильтровали, промывали дихлорметаном и растворитель выпаривали с получением 426 мг неочищенного вещества. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-желтого масла (115 мг; 458 мкмоль; 38,3%).
MS: M=251,2 (M+H)+
Стадия 5: 2-(4-метил-1-фенил-1H-пиразол-3-ил)ацетонитрил
Смесь 3-(бромметил)-4-метил-1-фенил-1H-пиразола (112 мг; 446 мкмоль) и цианида калия (33,3 мг; 491 мкмоль) в DMSO (2 мл) нагревали до 60°C в течение 2 ч. Реакционную смесь охлаждали до КТ, вливали в воду (25 мл) и экстрагировали этилацетатом (2×50 мл). Органические фазы промывали водой (3×25 мл) и солевым раствором 1×25 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 99,6 мг неочищенного вещества. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-желтого масла (58 мг; 294 мкмоль; 65,9%).
MS: M=198,3 (M+H)+
Стадия 6: 2-(4-метил-1-фенил-1H-пиразол-3-ил)этанамин
К борогидриду лития (2 М раствор в THF, 629 мкл; 1,26 ммоль) добавляли по каплям триметилхлорсилан (273 мг; 321 мкл; 2,51 ммоль) в течение 5 мин при 0-5°C. При этой температуре добавляли по каплям раствор 2-(4-метил-1-фенил-1H-пиразол-3-ил)ацетонитрила (62 мг; 314 мкмоль) в THF (0,5 мл). Эту смесь перемешивали при 70°C в течение 1,5 ч. После охлаждения до 0-5°C добавляли по каплям MeOH (0,5 мл). Через 10 мин ледяную баню убирали и смесь доводили до КТ. Реакционную смесь вливали в NaOH (1 М; 10 мл) и экстрагировали дихлорметаном (3×25 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением продукта в виде светло-желтого полутвердого вещества (46 мг; 206 мкмоль; 65,4%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=202,9 (M+H)+
Стадия 7: [2-(4-метил-1-фенил-1H-пиразол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (43,0 мг; 206 мкмоль) и 2-(4-метил-1-фенил-1H-пиразол-3-ил)этанамина (46 мг; 206 мкмоль), по способу, описанному в примере 43, стадия 3, в виде бесцветной пены (20,5 мг; 52,2 мкмоль; 25,4%).
MS: M=393,2 (M+H)+
Пример 48: {2-[2-(2-этилпиридин-4-ил)-5-метилтиазол-4-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
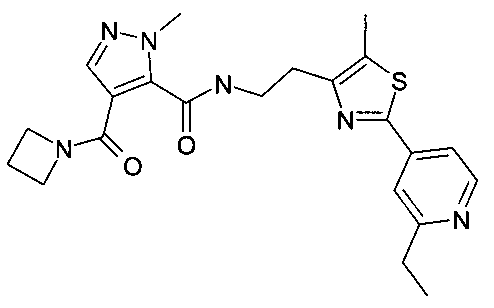
Стадия 1: 2-(2-(2-этилпиридин-4-ил)-5-метилтиазол-4-ил)этил метансульфонат
Продукт получали, исходя из 2-(2-(2-этилпиридин-4-ил)-5-метилтиазол-4-ил)этанола (200 мг; 805 мкмоль; получен, как описано в WO 2003/037327) и метансульфонилхлорида (111 мг; 75,3 мкл; 966 мкмоль), по способу, описанному в примере 46, стадия 1, в виде желтого масла (226 мг; 692 мкмоль; 86%), и его использовали без дополнительной очистки на следующей стадии.
MS: M=327,1 (M+H)+
Стадия 2: 4-(2-азидоэтил)-2-(2-этилпиридин-4-ил)-5-метилтиазол
Продукт получали, исходя из 2-(2-(2-этилпиридин-4-ил)-5-метилтиазол-4-ил)этилметансульфоната (226 мг; 692 мкмоль) и азида натрия (63,0 мг; 969 мкмоль), по способу, описанному в примере 46, стадия 2, в виде светло-желтого масла (138 мг; 505 мкмоль; 72,9%).
MS: M=274,1 (M+H)+
Стадия 3: 2-(2-(2-этилпиридин-4-ил)-5-метилтиазол-4-ил)этанамин
Продукт получали, исходя из 4-(2-азидоэтил)-2-(2-этилпиридин-4-ил)-5-метилтиазола (134 мг; 490 мкмоль) и полимерсвязанного трифенилфосфина (248 мг; 735 мкмоль), по способу, описанному в примере 46, стадия 3, в виде желтого масла (117 мг; 473 мкмоль; 96,5%).
MS: M=248,3 (M+H)+
Стадия 4: {2-[2-(2-этилпиридин-4-ил)-5-метилтиазол-4-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(2-(2-этилпиридин-4-ил)-5-метилтиазол-4-ил)этанамина (35,5 мг; 143 мкмоль), по способу, описанному в примере 43, стадия 3, в виде бесцветного полутвердого вещества (35 мг; 79,8 мкмоль; 55,7%).
MS: M=439,3 (M+H)+
Пример 49: {2-[5-фенил-2-(2,2,2-трифторэтил)-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
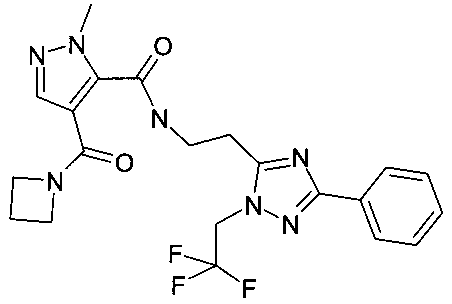
Стадия 1: трет-бутил-2-(3-фенил-1-(2,2,2-трифторэтил)-1H-1,2,4-триазол-5-ил)этилкарбамат
5-[2-Бис(трет-бутоксикарбонил)аминоэтил]-3-фенил-[1,2,4]триазол (200 мг; 515 мкмоль; пример 22, стадия 1) растворяли в DMF (3,57 мл) и добавляли карбонат калия (157 мг; 1,14 ммоль) при КТ. Эту смесь перемешивали в течение 20 мин, добавляли 2,2,2-трифторэтилтрифторметансульфонат (165 мг; 710 мкмоль) и продолжали перемешивать при КТ в течение ночи. Реакционную смесь экстрагировали этилацетатом (40 мл) и промывали водой (3×20 мл) и солевым раствором (1×20 мл). Водные слои снова экстрагировали этилацетатом (40 мл) и объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этил ацетат/гептан) в виде белого твердого вещества (140 мг; 378 мкмоль; 73,4%).
MS: M=371,2 (M+H)+
Стадия 2: 2-(3-фенил-1-(2,2,2-трифторэтил)-1H-1,2,4-триазол-5-ил)этанамин гидрохлорид
Продукт получали, исходя из трет-бутил-2-(3-фенил-1-(2,2,2-трифторэтил)-1H-1,2,4-триазол-5-ил)этилкарбамата (133 мг; 359 мкмоль), по способу, описанному в примере 45, стадия 2, в виде белого твердого вещества (108 мг; 352 мкмоль; 98%).
MS: M=271,3 (M-HCl+H)+
Стадия 3: {2-[5-фенил-2-(2,2,2-трифторэтил)-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (40 мг; 191 мкмоль) и 2-(3-фенил-1-(2,2,2-трифторэтил)-1H-1,2,4-триазол-5-ил)этанамина гидрохлорида (65 мг; 212 мкмоль), по способу, описанному в примере 43, стадия 3, в виде белого твердого вещества (43 мг; 93,2 мкмоль; 47,5%).
MS: M=462,2 (M+H)+
Пример 50: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 2-метил-4-(морфолин-4-карбонил)-2H-пиразол-3-карбоновой кислоты
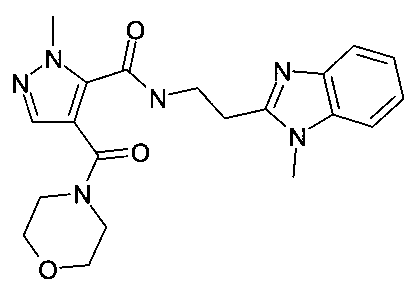
Продукт получали, исходя из промежуточного соединения A-2 (40 мг; 167 мкмоль) и 2-(1-метил-1H-бензо[d]имидазол-2-ил)этанамина дигидрохлорида (49,8 мг; 201 мкмоль), по способу, описанному в примере 8, стадия 3, после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-розового твердого вещества (22 мг; 55,5 мкмоль; 33,2%).
MS: M=397,4 (M+H)+
Пример 51: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 2-метил-4-(пирролидин-1-карбонил)-2H-пиразол-3-карбоновой кислоты
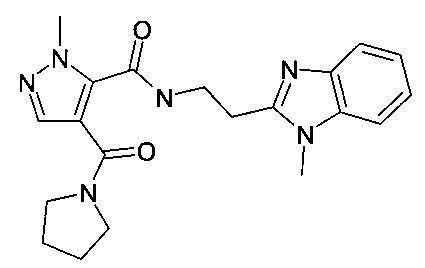
Стадия 1: этил-1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоксилат
Продукт получали, исходя из 4-(этоксикарбонил)-1-метил-1H-пиразол-5-карбоновой кислоты (150 мг; 757 мкмоль; получена, как описано в US 2011/0071128) и 2-(1-метил-1H-бензо[d]имидазол-2-ил)этанамина дигидрохлорида (225 мг; 908 мкмоль), по способу, описанному в примере 8, стадия 3, в виде красного вязкого масла (80 мг; 225 мкмоль; 29,7%).
MS: M=356,2 (M+H)+
Стадия 2: 1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоновая кислота
К раствору этил-1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоксилата (76 мг; 214 мкмоль) в THF (0,9 мл) и этанола (0,3 мл) добавляли LiOH (1 М водный раствор, 428 мкл; 428 мкмоль) при 0-5°C. Эту смесь перемешивали при КТ в течение 2 ч. Добавляли HCl (1 М водный раствор, 0,43 мл; 0,43 ммоль) и выпадающий в осадок продукт фильтровали, промывали смесью THF/вода 4:1 и высушивали в вакууме с получением продукта в виде бесцветного твердого вещества (52 мг; 159 мкмоль; 74,3%).
MS: M=328,3 (M+H)+
Стадия 3: [2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид 2-метил-4-(пирролидин-1-карбонил)-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из 1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (25 мг; 76,4 мкмоль) и пирролидина (6,52 мг; 7,58 мкл; 91,6 мкмоль), по способу, описанному в примере 37, стадия 3, в виде светло-розового твердого вещества (15 мг; 39,4 мкмоль; 51,6%).
MS: M=381,4 (M+H)+
Пример 52: [2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-3H-[1,2,3]триазол-4-карбоновой кислоты
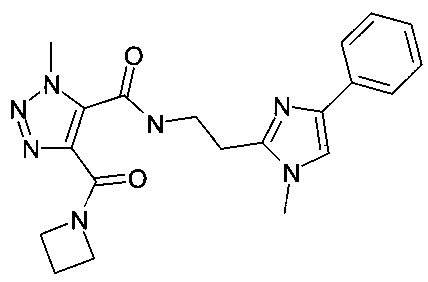
Стадия 1: диметиловый эфир 1-триметилсиланилметил-1H-[1,2,3]триазол-4,5-дикарбоновой кислоты
Раствор азидометилтриметилсилана (5,0 г; 38,69 ммоль) и диметилацетилендикарбоксилата (4,74 мл; 38,69 ммоль) в бензоле (200 мл) нагревали с обратным холодильником в течение 2 ч. Растворитель удаляли в вакууме и полученное неочищенное вещество разбавляли водой (100 мл) и экстрагировали этилацетатом (3×100 мл). Объединенный органический слой промывали солевым раствором (50 мл), высушивали над безводным Na2SO4, фильтровали, и выпаривали в вакууме. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и смеси 20% этилацетат/гексан) в виде бесцветной жидкости (10 г; 36,9 ммоль; 95,2%).
MS: M=272,0 (M+H)+
Стадия 2: диметиловый эфир 1-метил-1H-[1,2,3]триазол-4,5-дикарбоновой кислоты
К раствору диметилового эфира 1-триметилсиланилметил-1H-[1,2,3]триазол-4,5-дикарбоновой кислоты (10 г; 36,9 ммоль) в метаноле (100 мл) добавляли фторид калия (11,07 г; 184,5 ммоль) при КТ. Реакционную смесь перемешивали при КТ в течение 6 ч. После удаления растворителя в вакууме неочищенное вещество разбавляли водой (100 мл) и экстрагировали этилацетатом (2×150 мл). Объединенный органический слой промывали водой (2×50 мл) и солевым раствором (50 мл), высушивали над безводным Na2SO4, фильтровали и выпаривали в вакууме. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и 30% смеси этилацетат/гексан) в виде белого твердого вещества (4,3 г; 21,6 ммоль; 58,5%).
MS: M=200,2 (M+H)+
Стадия 3: 4-(метоксикарбонил)-1-метил-1H-1,2,3-триазол-5-карбоксилат лития
К раствору диметилового эфира 1-метил-1H-[1,2,3]триазол-4,5-дикарбоновой кислоты (1 г; 5,025 ммоль) в THF (20 мл) и этаноле (5 мл) добавляли при 0°C раствор гидроксида лития моногидрата (169 мг; 4,02 ммоль) в воде (5 мл) в течение 10 мин. Реакционную смесь оставляли перемешиваться при 0°C в течение 3 ч. Эту смесь разбавляли водой (10 мл) и промывали этилацетатом (2×50 мл). Водный слой выпаривали при пониженном давлении и полученное таким образом неочищенное вещество высушивали в ходе азеотропной перегонки с толуолом с получением продукта в виде белого твердого вещества (0,6 г; 3,24 ммоль; 64,5%).
Стадия 4: метил-1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-1,2,3-триазол-4-карбоксилат
Продукт получали, исходя из 4-(метоксикарбонил)-1-метил-1H-1,2,3-триазол-5-карбоксилата лития (150 мг; 785 мкмоль) и 2-(1-метил-4-фенил-1H-имидазол-2-ил)этанамина дигидрохлорида (226 мг; 824 мкмоль; пример 7, стадия 4), по способу, описанному в примере 7, стадия 5, после обработки водой и очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-желтого твердого вещества (79 мг; 214 мкмоль; 27,3%).
MS: M=369,2 (M+H)+
Стадия 5: 1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-1,2,3-триазол-4-карбоновая кислота
К раствору метил-1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-1,2,3-триазол-4-карбоксилата (77 мг; 209 мкмоль) в THF (0,9 мл) и MeOH (0,3 мл) добавляли LiOH (1 М водный раствор, 418 мкл; 418 мкмоль) при 0-5°C. Ледяную баню убирали и смесь перемешивали при КТ в течение 4 ч. Добавляли HCl (1 М водный раствор; 0,42 мл; 0,42 ммоль) и этот раствор вливали в дихлорметан (40 мл) с получением суспензии. Твердый осадок фильтровали, промывали смесью THF/вода 4:1 и высушивали в вакууме с получением продукта в виде бесцветного твердого вещества (52 мг; 147 мкмоль; 70,2%).
MS: M=355,2 (M+H)+
Стадия 6: [2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-3H-[1,2,3]триазол-4-карбоновой кислоты
Продукт получали, исходя из 1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-1,2,3-триазол-4-карбоновой кислоты (30 мг; 84,7 мкмоль) и азетидина (6,8 мг; 8 мкл; 119 мкмоль), по способу, описанному в примере 37, стадия 3, в виде бесцветного твердого вещества (11 мг; 28 мкмоль; 33%).
MS: M=394,2 (M+H)+
Пример 53: 4-(циклопропилметиламид)-3-{[2-(1-метил-1H-бензоимидазол-2-ил)-этил]-амид} 2-метил-2H-пиразол-3,4-дикарбоновой кислоты
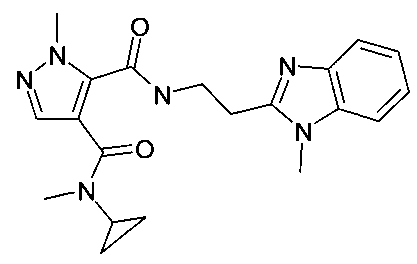
Продукт получали, исходя из 1-метил-5-(2-(1-метил-1H-бензо[d]имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (25 мг; 76,4 мкмоль; пример 51, стадия 2) и N-метилциклопропанамина гидрохлорида (10,4 мг; 91,6 мкмоль), по способу, описанному в примере 37, стадия 3, после очистки с помощью препаративной ВЭЖХ, с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% триэтиламин), в виде бесцветной пены (16,6 мг; 43,6 мкмоль; 57,1%).
MS: M=381,3 (M+H)+
Пример 54: [2-(5-метил-4-фенилтиазол-2-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
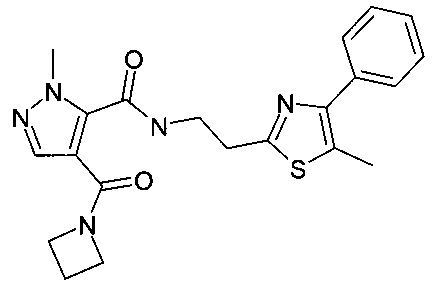
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(5-метил-4-фенилтиазол-2-ил)этанамина дигидрохлорида (45,9 мг; 158 мкмоль), по способу, описанному в примере 43, стадия 3, в виде светло-желтого воскообразного твердого вещества (31,6 мг; 77,2 мкмоль; 53,8%).
MS: M=410,3 (M+H)+
Пример 55: [2-(2,5-дифенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
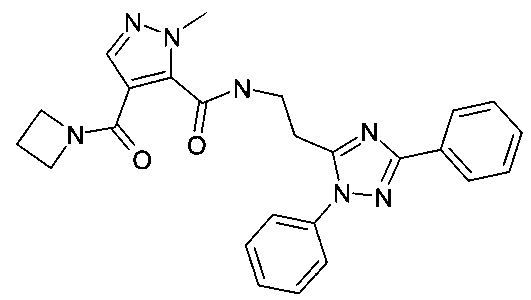
Стадия 1: 2-фенилоксазол-4(5H)-он
К раствору бензоилизоцианата (2 г; 12,2 ммоль) в ацетонитриле (40 мл) быстро добавляли (диазометил)триметилсилан (2 M раствор в гексане, 7,34 мл; 14,7 ммоль) при 0-5°C, при этом температура повышалась до 15°C (выделение пузырьков). Эту смесь перемешивали при 0-5°C в течение 1 ч. Выпаривали растворитель и получали продукт после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде желтого твердого вещества (1,84 г с 90% чистотой, 10,3 ммоль; 84,0%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=162,3 (M+H)+
Стадия 2: (1,3-дифенил-1H-1,2,4-триазол-5-ил)метанол
Смесь 2-фенилоксазол-4(5H)-она (600 мг; 3,35 ммоль) и фенилгидразина (420 мг; 382 мкл; 3,69 ммоль) в этаноле (10 мл) нагревали с обратным холодильником в течение 2 ч. Реакционную смесь концентрировали в вакууме. Полученную суспензию фильтровали, промывали с помощью EtOH и высушивали в вакууме с получением 513 мг светло-желтого твердого вещества, которое представляло собой продукт, что было подтверждено. Полученный фильтрат выпаривали с получением 430 мг желтого твердого вещества, которое также содержало значительное количество продукта. Таким образом, продукт получали после очистки выпаренного фильтрата флэш-хроматографией (с использованием силикагеля и градиента этилацетат/дихлорметан) и объединения с полученным выпавшем в осадок продуктом, в виде светло-желтого твердого вещества (711 мг; 2,83 ммоль; 84,4%).
MS: M=252,2 (M+H)+
Стадия 3: 5-(бромметил)-1,3-дифенил-1H-1,2,4-триазол
К суспензии (1,3-дифенил-1H-1,2,4-триазол-5-ил)метанола (350 мг; 1,39 ммоль) и полимерсвязанного трифенилфосфина (679 мг; 2,02 ммоль) в дихлорметане (6 мл) добавляли при 0-5°C тетрабромид углерода (670 мг; 2,02 ммоль). Ледяную баню убирали, и эту смесь перемешивали при КТ в течение 18 ч. Полученную суспензию фильтровали, промывали дихлорметаном и выпаривали с получением 844 мг желтого масла. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде бесцветного твердого вещества (267 мг; 850 мкмоль; 61,0%).
MS: M=316,0 (M+H)+
Стадия 4: 2-(1,3-дифенил-1H-1,2,4-триазол-5-ил)ацетонитрил
Смесь 5-(бромметил)-1,3-дифенил-1H-1,2,4-триазола (265 мг; 843 мкмоль) и цианида калия (62,3 мг; 928 мкмоль) в DMSO (5 мл) нагревали при 60°C в течение 1,5 ч. Реакционную смесь охлаждали до КТ, вливали в воду (25 мл) и экстрагировали этилацетатом (2×50 мл). Органическую фазы промывали водой (2×25 мл) и солевым раствором (1×25 мл), объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 251 мг неочищенного продукта. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-желтого воскообразного твердого вещества (120 мг; 461 мкмоль; 54,7%).
MS: M=261,2 (M+H)+
Стадия 5: 2-(1,3-дифенил-1H-1,2,4-триазол-5-ил)этанамин
К борогидриду лития (2 M раствор в THF, 899 мкл; 1,8 ммоль) добавляли по каплям триметилхлорсилан (391 мг; 460 мкл; 3,6 ммоль) в течение 5 мин при 0-5°C, а затем добавляли по каплям раствор 2-(1,3-дифенил-1H-1,2,4-триазол-5-ил)ацетонитрила (117 мг; 449 мкмоль) в THF (1,5 мл). Ледяную баню убирали через 10 мин и смесь перемешивали при 60°C в течение 1,5 ч. Реакционную смесь охлаждали до 0-5°C и медленно добавляли 1 мл MeOH (выделение пузырьков!). Через 10 мин ледяную баню убирали и смесь доводили до КТ. Эту смесь вливали в NaOH (1 M водный раствор, 15 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенную органическую фазу высушивали над MgSO4, фильтровали и выпаривали с получением продукта (118 мг с 90% чистотой, 402 мкмоль; 89,4%), в виде желтого вязкого масла, которое использовали без дополнительной очистки.
MS: M=265,2 (M+H)+
Стадия 6: [2-(2,5-дифенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(1,3-дифенил-1H-1,2,4-триазол-5-ил)этанамина (50,5 мг; 172 мкмоль) путем конденсации с HATU, по способу, описанному в примере 1, и в ходе прямой очистки с помощью препаративной ВЭЖХ с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% муравьиную кислоту), в виде бесцветного твердого вещества (36 мг; 79,0 мкмоль; 55,1%).
MS: M=456,5 (M+H)+
Пример 56: [2-(2-циклопропил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
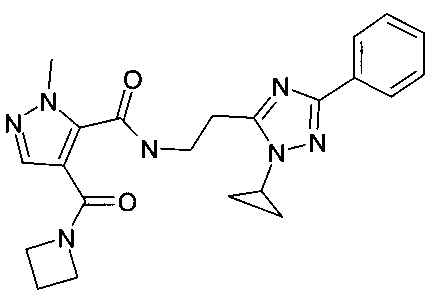
Стадия 1: трет-бутил-2-(1-циклопропил-3-фенил-1H-1,2,4-триазол-5-ил)этилкарбамат
Суспензию 5-[2-бис(трет-бутоксикарбонил)аминоэтил]-3-фенил-[1,2,4]триазола (218 мг; 561 мкмоль; пример 22, стадия 1), циклопропилбороновой кислоты (192,8 мг; 2,24 ммоль) и карбоната натрия (119 мг; 1,12 ммоль) в дихлорэтане (4 мл) вакуумировали и вновь наполняли аргоном. Добавляли горячую суспензию ацетата меди(II) (102 мг; 561 мкмоль) и 2,2′-бипиридина (87,6 мг; 561 мкмоль) в дихлорэтане (1,5 мл) и реакционную смесь перемешивали при 85°C в течение 40 ч. Реакционную смесь охлаждали до КТ, фильтровали и промывали дихлорметаном. Полученный фильтрат экстрагировали водой (2×20 мл), высушивали над MgSO4, фильтровали и выпаривали с получением 285 мг зеленого масла. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде бесцветного вязкого масла (105 мг; 320 мкмоль; 57,0%).
MS: M=329,3 (M+H)+
Стадия 2: 2-(1-циклопропил-3-фенил-1H-1,2,4-триазол-5-ил)этанамин гидрохлорид
трет-Бутил-2-(1-циклопропил-3-фенил-1H-1,2,4-триазол-5-ил)этилкарбамат (105 мг; 320 мкмоль) растворяли в HCl (4 М в диоксане, 2 мл; 8,00 ммоль). Эту смесь перемешивали при КТ в течение 2 ч. Полученную суспензию фильтровали, промывали диэтиловым эфиром и высушивали в вакууме с получением продукта в виде бесцветного твердого вещества (75,5 мг; 285 мкмоль; 89,2%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=229,4 (M+H)+
Стадия 3: [2-(2-циклопропил-5-фенил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(1-циклопропил-3-фенил-1H-1,2,4-триазол-5-ил)этанамина гидрохлорида (45,6 мг; 172 мкмоль) путем сочетания с HATU, по способу, описанному в примере 55, стадия 6, в виде бесцветного твердого вещества (34 мг; 81,1 мкмоль; 56,5%).
MS: M=420,3 (M+H)+
Пример 57: [2-(5-фенил-2-пиридин-2-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Стадия 1: (Z)-этил-N-2-хлорацетилбензимидат
К суспензии этилбензимидата гидрохлорида (500 мг; 2,69 ммоль) в дихлорэтане (25 мл) добавляли в атмосфере аргона триэтиламин (872 мг; 1,2 мл; 8,62 ммоль). Реакционную смесь охлаждали при перемешивании до 0°C и добавляли хлорацетилхлорид (608 мг; 431 мкл; 5,39 ммоль). Через 30 мин охлаждающую баню убирали и продолжали перемешивать при КТ в течение 4 ч. После фильтрации органическую фазу экстрагировали с помощью KHSO4 (10% водный раствор), высушивали и выпаривали с получением продукта в виде светло-коричневого полутвердого вещества (740 мг с 80% чистотой, 2,62 ммоль; 97%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=226,1 (M+H)+
Стадия 2: 2-(5-(хлорметил)-3-фенил-1H-1,2,4-триазол-1-ил)пиридин
К раствору (Z)-этил-N-2-хлорацетилбензимидата (160 мг; 709 мкмоль) в THF (5 мл) добавляли 2-гидразинилпиридин (139,2 мг; 1,28 ммоль) в THF (1,5 мл) при 0°C в атмосфере аргона. Полученный красно-коричневый раствор перемешивали при КТ в течение ночи. Легколетучие вещества удаляли и получали продукт, после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан), в виде белого твердого вещества (130 мг; 480 мкмоль; 67,7%).
MS: M=271,3 (M+H)+
Стадия 3: 2-(3-фенил-1-(пиридин-2-ил)-1H-1,2,4-триазол-5-ил)ацетонитрил
Смесь 2-(5-(хлорметил)-3-фенил-1H-1,2,4-триазол-1-ил)пиридина (130 мг; 480 мкмоль) и цианида калия (43,8 мг; 672 мкмоль) в DMSO (3,5 мл) нагревали до 50°C в течение 1,5 ч. Охлажденную реакционную смесь вливали в воду (25 мл) и экстрагировали этилацетатом (2×50 мл). Объединенную органическую фазу промывали водой (2×25 мл), высушивали над MgSO4 и концентрировали в вакууме с получением 110 мг неочищенного продукта. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде белого твердого вещества (30 мг; 114 мкмоль; 23,7%).
MS: M=262,1 (M+H)+
Стадия 4: 2-(3-фенил-1-(пиридин-2-ил)-1H-1,2,4-триазол-5-ил)этанамин
Раствор 2-(3-фенил-1-(пиридин-2-ил)-1H-1,2,4-триазол-5-ил)ацетонитрила (28 мг; 96,4 мкмоль) в THF (1,5 мл) в атмосфере аргона охлаждали до 0-5°C. Последовательно, очень медленно добавляли триметилхлорсилан (83,8 мг; 98,6 мкл; 772 мкмоль) и борогидрид лития (2 M раствор в THF, 193 мкл; 386 мкмоль). Через 10 мин ледяную баню убирали и смесь перемешивали при 60°C в течение 1,5 ч. Реакционную смесь охлаждали до 0-5°C и MeOH (1 мл) медленно добавляли (выделение пузырьков!). После доведения температуры до КТ, смесь вливали в NaOH (1 M водный раствор, 15 мл) и экстрагировали дихлорметаном (2×25 мл) и этилацетатом (1×25 мл). Объединенную органическую фазу высушивали над MgSO4, фильтровали и выпаривали с получением продукта в виде желтого воскообразного твердого вещества (18 мг; 67,8 мкмоль; 70%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=266,3 (M+H)+
Стадия 5: [2-(5-фенил-2-пиридин-2-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (25,5 мг; 121,4 мкмоль) и 2-(3-фенил-1-(пиридин-2-ил)-1H-1,2,4-триазол-5-ил)этанамина (18 мг; 67,8 мкмоль) путем сочетания с HATU (с использованием 2 экв. сшивающего реагента), по способу, описанному в примере 55, стадия 6, в виде белого полутвердого вещества (9,5 мг; 20,8 мкмоль; 30,7%).
MS: M=457,4 (M+H)+
Пример 58: [2-(2,5-дифенилоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Стадия 1: 4-метил-2,5-дифенилоксазол 3-оксид
К раствору бензальдегида (325 мг; 311 мкл; 3,06 ммоль) в HCl (4 М в диоксане, 10 мл) добавляли (E)-2-(гидроксиимино)-1-фенилпропан-1-он (500 мг; 3,06 ммоль) при 0-5°C. Ледяную баню убирали и смесь перемешивали при КТ в течение 24 ч. Все легколетучие вещества удаляли и полученный остаток растворяли в HCl (4 М в диоксане, 10 мл). Реакционную смесь перемешивали при КТ в течение 6 ч и концентрировали. Полученную суспензию фильтровали, промывали диэтиловым эфиром и высушивали в вакууме с получением продукта в виде бесцветного твердого вещества (398 мг; 1,58 ммоль; 51,7%).
MS: M=252,3 (M+H)+
Стадия 2: 4-(хлорметил)-2,5-дифенилоксазол
К раствору 4-метил-2,5-дифенилоксазола 3-оксида (395 мг; 1,57 ммоль) в хлороформе (2 мл) добавляли по каплям раствор оксихлорида фосфора (265 мг; 161 мкл; 1,73 ммоль) в хлороформе (2,00 мл) в течение 10 мин. Реакционную смесь перемешивали при 65°C в течение 4 ч и охлаждали до 0-5°C. При этой температуре добавляли по каплям NH4OH (25% водный раствор, 10 мл) и полученную смесь экстрагировали дихлорметаном (3×40 мл). Объединенную органическую фазу высушивали над MgSO4, фильтровали и концентрировали в вакууме. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента дихлорметан/гептан) в виде бесцветного твердого вещества (249 мг; 923 мкмоль; 58,7%).
MS: M=270,3 (M+H)+
Стадия 3: 2-(2,5-дифенилоксазол-4-ил)ацетонитрил
К суспензии цианида калия (67,1 мг; 0,999 ммоль) в DMSO (2 мл) добавляли по каплям при КТ раствор 4-(хлорметил)-2,5-дифенилоксазола (245 мг; 908 мкмоль) в DMSO (3 мл) в течение 5 мин. Реакционную смесь перемешивали при КТ в течение 2 ч и при 45°C еще в течение 3 ч. Реакционную смесь охлаждали до КТ, вливали в воду (25 мл) и экстрагировали этилацетатом (2×75 мл). Органическую фазы промывали водой (2×20 мл) и солевым раствором (1×20 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 245 мг неочищенного продукта. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-желтого твердого вещества (189 мг; 726 мкмоль; 79,9%).
MS: M=261,2 (M+H)+
Стадия 4: 2-(2,5-дифенилоксазол-4-ил)этанамин
Продукт получали, исходя из 2-(2,5-дифенилоксазол-4-ил)ацетонитрила (185 мг; 711 мкмоль), по способу, описанному в примере 55, стадия 5, в виде желтой пены (202 мг; 535 мкмоль; 75,3%).
MS: M=265,2 (M+H)+
Стадия 5: [2-(2,5-дифенилоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(2,5-дифенилоксазол-4-ил)этанамин (65,0 мг; 172 мкмоль) путем сочетания с HATU, по способу, описанному в примере 55, стадия 6, в виде бесцветного твердого вещества (31 мг; 68,1 мкмоль; 47,5%).
MS: M=456,2 (M+H)+
Пример 59: [2-(5-фенил-2-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Стадия 1: (3-фенил-1-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)метанол
К раствору 3-гидразинилпиридина дигидрохлорида (336 мг; 1,84 ммоль) и N,N-диизопропилэтиламина (476 мг; 644 мкл; 3,69 ммоль) в этаноле (7 мл) добавляли 2-фенилоксазол-4(5H)-он (300 мг; 1,68 ммоль; пример 55, стадия 1) и реакционную смесь нагревали с обратным холодильником в течение 2 ч. Все легколетучие вещества удаляли в вакууме и получали продукт после очистки флэш-хроматографией (с использованием силикагеля и градиента метанол/этилацетат) в виде светло-желтого твердого вещества (390 мг; 1,546 ммоль; 92,3%).
MS: M=253,1 (M+H)+
Стадия 2: 3-(5-(бромметил)-3-фенил-1H-1,2,4-триазол-1-ил)пиридин
Продукт получали, исходя из (3-фенил-1-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)метанола (390 мг; 1,55 ммоль), по способу, описанному в примере 55, стадия 3, в виде бесцветного твердого вещества (130 мг; 412 мкмоль; 26,7%).
MS: M=317,0 (M+H)+
Стадия 3: 2-(3-фенил-1-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)ацетонитрил
Смесь цианида калия (30,7 мг; 472 мкмоль) и 3-(5-(бромметил)-3-фенил-1H-1,2,4-триазол-1-ил)пиридина (124 мг; 393 мкмоль) в DMSO (3 мл) перемешивали при КТ в течение 2 ч. Реакционную смесь вливали в воду (20 мл) и экстрагировали этилацетатом (2×25 мл). Объединенную органическую фазу промывали водой и солевым раствором, высушивали, фильтровали и выпаривали. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-коричневого твердого вещества (47 мг с чистотой 80%, 180 мкмоль; 36,6%).
MS: M=262,5 (M+H)+
Стадия 4: 2-(3-фенил-1-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)этанамин
Продукт получали, исходя из 2-(3-фенил-1-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)ацетонитрила (47 мг; 144 мкмоль), по способу, описанному в примере 55, стадия 5, в виде желтого полутвердого вещества (28 мг с чистотой 80%; 84 мкмоль; 58,7%).
MS: M=266,5 (M+H)+
Стадия 5: [2-(5-фенил-2-пиридин-3-ил-2H-[1,2,4]триазол-3-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (15 мг; 71,7 мкмоль) и 2-(3-фенил-1-(пиридин-3-ил)-1H-1,2,4-триазол-5-ил)этанамина (28,5 мг; 86,0 мкмоль) путем сочетания с HATU, по способу, описанному в примере 55, стадия 6, в виде бесцветного воскообразного твердого вещества (7,7 мг; 16,9 мкмоль; 23,5%).
MS: M=457,5 (M+H)+
Пример 60: {2-[5-(3-фторфенил)-2-фенил-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Стадия 1: этил-3-фторбензимидат гидрохлорид
Раствор 3-фторбензонитрила (1 г; 8,26 ммоль) в этаноле (12 мл) охлаждали до 0-5°C. Через эту смесь барботировали HCl (газ) до насыщения и смесь перемешивали при КТ в течение 16 ч. Все легколетучие вещества удаляли и к полученному остатку добавляли диэтиловый эфир (20 мл). Полученную суспензию фильтровали, промывали диэтиловым эфиром и высушивали в вакууме с получением продукта в виде бесцветного твердого вещества (1,57 г; 7,71 ммоль; выход 93,4%).
MS: M=168,2 (M-HCl+H)+
Стадия 2: (Z)-этил-N-2-хлорацетил-3-фторбензимидат
К суспензии этил-3-фторбензимидата гидрохлорида (400 мг; 1,96 ммоль) в дихлорметане (15 мл) добавляли триэтиламин (497 мг; 684 мкл; 4,91 ммоль) при КТ. Реакционную смесь охлаждали до 0-5°C и добавляли по каплям 2-хлорацетилхлорид (333 мг; 234 мкл; 2,95 ммоль) в течение 5 мин. Через 20 мин ледяную баню убирали и смесь перемешивали при КТ в течение 2 ч. Реакционную смесь вливали в KHSO4 (10% водный раствор, 25 мл) и экстрагировали дихлорметаном (3×50 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением продукта в виде светло-коричневого масла (537 мг; 1,98 ммоль; 101%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=244,3 (M+H)+
Стадия 3: 5-(хлорметил)-3-(3-фторфенил)-1-фенил-1H-1,2,4-триазол
Продукт получали, исходя из (Z)-этил-N-2-хлорацетил-3-фторбензимидата (535 мг; 1,98 ммоль) и фенилгидразина (270 мг; 246 мкл; 2,37 ммоль), по способу, описанному в примере 57, стадия 2, в виде оранжевого твердого вещества (448 мг; 1,56 ммоль; 78,8%).
MS: M=288,1 (M+H)+
Стадия 4: 2-(3-(3-фторфенил)-1-фенил-1H-1,2,4-триазол-5-ил)ацетонитрил
Продукт получали, исходя из 5-(хлорметил)-3-(3-фторфенил)-1-фенил-1H-1,2,4-триазола (445 мг; 1,55 ммоль), по способу, описанному в примере 58, стадия 3, в виде коричневого вязкого масла (103 мг; 370 мкмоль; 23,9%).
MS: M=279,2 (M+H)+
Стадия 5: 2-(3-(3-фторфенил)-1-фенил-1H-1,2,4-триазол-5-ил)этанамин
Продукт получали, исходя из 2-(3-(3-фторфенил)-1-фенил-1H-1,2,4-триазол-5-ил)ацетонитрила (100 мг; 359 мкмоль), по способу, описанному в примере 55, стадия 5, в виде желтого масла (105 мг с чистотой 80%, 298 мкмоль; 82,8%).
MS: M=283,2 (M+H)+
Стадия 6: {2-[5-(3-фторфенил)-2-фенил-2H-[1,2,4]триазол-3-ил]-этил}-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (20 мг; 95,6 мкмоль) и 2-(3-(3-фторфенил)-1-фенил-1H-1,2,4-триазол-5-ил)этанамина (40,5 мг; 115 мкмоль) путем сочетания с HATU, по способу, описанному в примере 55, стадия 6, в виде бесцветного твердого вещества (21 мг; 44,4 мкмоль; 46,4%).
MS: M=474,3 (M+H)+
Пример 61: 4-(циклопентилметиламид)-3-{[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид}2-метил-2H-пиразол-3,4-дикарбоновой кислоты
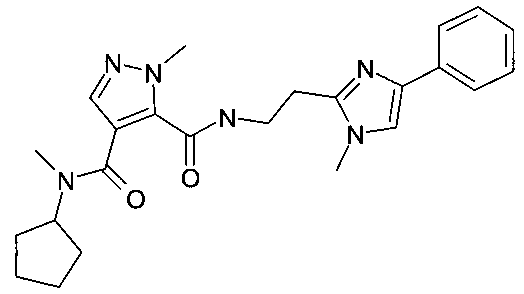
Стадия 1: этил-1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоксилат
Продукт получали, исходя из 4-(этоксикарбонил)-1-метил-1H-пиразол-5-карбоновой кислоты (200 мг; 1,01 ммоль; полученной, как описано в US 2011/0071128) и 2-(1-метил-4-фенил-1H-имидазол-2-ил)этанамина гидрохлорида (288 мг; 1,21 ммоль; пример 7, стадия 4), по способу, описанному в примере 37, стадия 1, в виде светло-желтого твердого вещества (252 мг; 661 мкмоль; 65,5%).
MS: M=382,3 (M+H)+
Стадия 2: 1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоновая кислота
Продукт получали, исходя из этил-1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоксилата (250 мг; 655 мкмоль), по способу, описанному в примере 37, стадия 2, в виде бесцветного твердого вещества (224 мг; 634 мкмоль; 96,7%).
MS: M=354,5 (M+H)+
Стадия 3: 4-(циклопентилметиламид)-3-{[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид}2-метил-2H-пиразол-3,4-дикарбоновой кислоты
Продукт получали, исходя из 1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (30 мг; 84,9 мкмоль) и N-метилциклопентанамина (10,1 мг; 102 мкмоль) путем конденсации с COMU в течение ночи, по способу, описанному в примере 37, стадия 3, и после очистки с помощью препаративной ВЭЖХ с использованием градиента смеси ацетонитрил/вода (содержащей 0,1% триэтиламин), в виде светло-розового твердого вещества (27 мг; 62,1 мкмоль; 73,2%).
MS: M=435,4 (M+H)+
Пример 62: 4-(циклобутилметиламид)-3-{[2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид} 2-метил-2H-пиразол-3,4-дикарбоновой кислоты
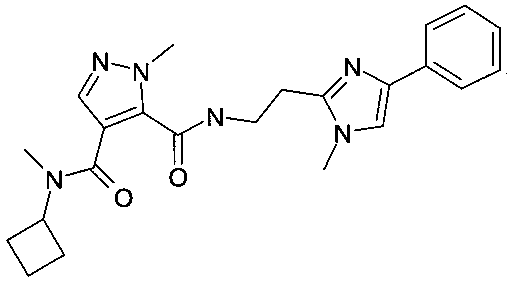
Продукт получали, исходя из 1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-1H-пиразол-4-карбоновой кислоты (30 мг; 84,9 мкмоль; пример 61, стадия 2) и N-метилциклобутанамина гидрохлорида (13,0 мг; 102 мкмоль), по способу, описанному в примере 61, стадия 3, в виде бесцветного твердого вещества (25 мг; 59,5 мкмоль; 70,0%).
MS: M=421,3 (M+H)+
Пример 63: [2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-2-трифторметил-3H-имидазол-4-карбоновой кислоты
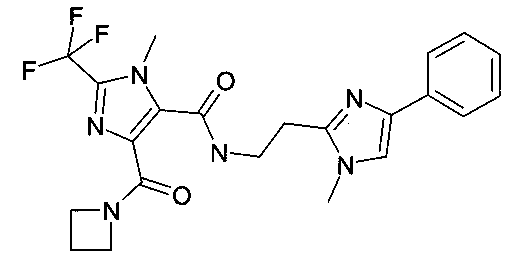
Стадия 1: 2-(трифторметил)-1H-имидазол-4,5-дикарбоновая кислота
К раствору 2-(трифторметил)-1H-бензо[d]имидазола (2 г; 10,7 ммоль) в H2SO4 (55,2 г; 30 мл; 535 ммоль) медленно добавляли пероксид водорода (35% раствор, 10,4 г; 9,41 мл; 107 ммоль) при 0-5°C. Реакционную смесь перемешивали при 120°C в течение 3 ч, охлаждали до КТ, вливали в воду (80 мл) при охлаждении на ледяной бане и экстрагировали диэтиловым эфиром (3×80 мл). Объединенную органическую фазу высушивали над MgSO4, фильтровали и выпаривали с получением продукта в виде бесцветного твердого вещества (1,98 г; 8,84 ммоль; 82,2%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=225,4 (M+H)+
Стадия 2: диметил-1-метил-2-(трифторметил)-1H-имидазол-4,5-дикарбоксилат
Смесь 2-(трифторметил)-1H-имидазол-4,5-дикарбоновой кислоты (1,98 г; 8,84 ммоль) и 1,1,1-триметоксиэтана (14,9 г; 15,5 мл; 124 ммоль) перемешивали при 120°C в течение 2 ч. Все легколетучие вещества удаляли с получением 2,58 г светло-коричневого масла, которое очищали флэш-хроматографией (с использованием силикагеля и градиента этил ацетат/гептан) с получением продукта в виде светло-желтого масла (1,94 г; 7,29 ммоль; 82,5%).
MS: M=267,4 (M+H)+
Стадия 3: 4-(метоксикарбонил)-1-метил-2-(трифторметил)-1H-имидазол-5-карбоновая кислота
Продукт получали, исходя из диметил-1-метил-2-(трифторметил)-1H-имидазол-4,5-дикарбоксилата (500 мг; 1,88 ммоль), по способу, описанному в примере 37, стадия 2, в виде бесцветного твердого вещества (460 мг; 1,82 ммоль; 97,1%).
MS: M=253,4 (M+H)+
Стадия 4: метил-1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-2-(трифторметил)-1H-имидазол-4-карбоксилат
Продукт получали, исходя из 4-(метоксикарбонил)-1-метил-2-(трифторметил)-1H-имидазол-5-карбоновой кислоты (200 мг; 793 мкмоль) и 2-(1-метил-4-фенил-1H-имидазол-2-ил)этанамина гидрохлорида (226 мг; 952 мкмоль; пример 7, стадия 4), по способу, описанному в примере 7, стадия 5, после обработки водой и очистки флэш-хроматографией (с использованием силикагеля и градиента этил ацетат/гептан), в виде белого пены (128 мг; 294 мкмоль; 37,1%).
MS: M=436,5 (M+H)+
Стадия 5: 1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-2-(трифторметил)-1H-имидазол-4-карбоновая кислота
Продукт получали, исходя из метил-1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-2-(трифторметил)-1H-имидазол-4-карбоксилата (125 мг; 287 мкмоль), по способу, описанному в примере 37, стадия 2, в виде белого твердого вещества (118 мг; 280 мкмоль; 97,5%).
MS: M=422,2 (M+H)+
Стадия 6: [2-(1-метил-4-фенил-1H-имидазол-2-ил)-этил]-амид 5-(азетидин-1-карбонил)-3-метил-2-трифторметил-3H-имидазол-4-карбоновой кислоты
Продукт получали, исходя из 1-метил-5-(2-(1-метил-4-фенил-1H-имидазол-2-ил)этилкарбамоил)-2-(трифторметил)-1H-имидазол-4-карбоновой кислоты (30 мг; 71,2 мкмоль) и азетидина (4,98 мг; 5,88 мкл; 85,4 мкмоль) путем конденсации с COMU), по способу, описанному в примере 61, стадия 3, в виде белого твердого вещества (20,5 мг; 44,5 мкмоль; 62,5%).
MS: M=461,5 (M+H)+
Пример 64: (2-[1,2,4]триазоло[1,5-a]пиридин-2-ил-этил)-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
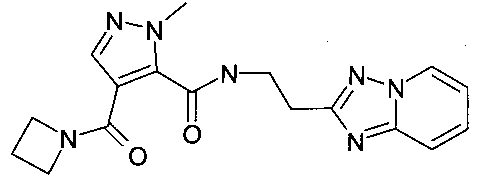
Стадия 1: O-(мезитилсульфонил)гидроксиламин
В атмосфере аргона растворяли (Z)-этил-N-мезитилсульфонилоксиацетимидат (15 г; 52,6 ммоль) в THF (13 мл) при 0°C. Затем добавляли по каплям перхлорную кислоту (70% водный раствор, 22,6 г; 13,6 мл; 158 ммоль) при 0-5°C в течение 50 мин. Через 10 мин начиналась преципитация, добавляли THF (3 мл). Перемешивали при 0°C в течение 30 мин, после чего эту смесь вливали в ледяную воду (200 мл) при интенсивном перемешивании, с получением белой суспензии. Перемешивали еще 15 мин, после чего суспензию фильтровали и полученный остаток промывали ледяной водой. Полученное белое твердое вещество растворяли в диэтиловом эфире (150 мл), высушивали над MgSO4 и выпаривали при КТ без нагревания, при этом колбу охлаждали (очень важно, так как это соединение может разлагаться при 25°C!) с получением O-(мезитилсульфонил)гидроксиламина в виде белого твердого вещества (9,2 г; 42,7 ммоль; 81,3%). Неочищенный продукт использовали далее без очистки, т.к. O-(мезитилсульфонил)гидроксиламин не стабилен при 25°C!
Стадия 2: 2,4,6-триметилбензолсульфонат 1,2-диаминопиридиния
К суспензии O-(мезитилсульфонил)гидроксиламина (9,2 г; 42,7 ммоль) в дихлорметане (110 мл) добавляли пиридин-2-амин (4,02 г; 42,7 ммоль) четырьмя порциями при 0-5°C с получением желтого раствора. Через 5 мин этот желтый раствор превращался в светло-желтую суспензию. Через 10 мин ледяную баню убирали и реакционную смесь перемешивали при КТ в течение 1 ч. Полученную суспензию разбавляли диэтиловым эфиром (100 мл), фильтровали, промывали диэтиловым эфиром и высушивали в вакууме с получением продукта (10,9 г; 35,2 ммоль; 82,4%) в виде светло-розового твердого вещества, которое использовали без дополнительной очистки на следующей стадии.
Стадия 3: этил [1,2,4]триазоло[1,5-a]пиридин-2-карбоксилат
К светло-розовому раствору 1,2-диаминопиридиния 2,4,6-триметилбензолсульфоната (10,9 г; 35,2 ммоль) в пиридине (50 мл) добавляли этил-2-хлор-2-оксоацетат (9,62 г; 7,84 мл; 70,5 ммоль) при КТ (экзотермическая реакция!). Светло-красный раствор превращался в темно-красный раствор. Эту смесь нагревали до 100°C и перемешивали в течение ночи. Реакционную смесь выпаривали и черный остаток обрабатывали в течение 30 мин с помощью Na2CO3 (насыщенный водный раствор, 300 мл). Реакционную смесь экстрагировали дихлорметаном (4×250 мл) и объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 6,64 г неочищенного продукта. Неочищенный продукт суспендировали в диэтиловом эфире (30 мл), фильтровали и промывали диэтиловым эфиром. Полученный осадок высушивали в вакууме с получением продукта в виде светло-коричневого твердого вещества (6,1 г; 31,9 ммоль; 90,6%).
MS: M=192,2(M+H)+
Стадия 4: [1,2,4]триазоло[1,5-a]пиридин-2-илметанол
В атмосфере аргона этил-[1,2,4]триазоло[1,5-a]пиридин-2-карбоксилат (1 г; 5,23 ммоль) объединяли с THF (10 мл) при КТ с получением коричневой суспензии. Добавляли борогидрид натрия (1,19 г; 31,4 ммоль) четырьмя порциями. Эту смесь нагревали до 65°C в течение 15 мин. После охлаждения до КТ добавляли по каплям этанол (10 мл) в течение 15 мин. Эту смесь перемешивали при 65°C в течение 4 ч. Смесь охлаждали до 0-5°C и добавляли по каплям NH4Cl (насыщенный водный раствор, 20 мл) в течение 10 мин (вспенивание!). Добавляли воду (20 мл) и полученную желтую суспензию вливали в дихлорметан (100 мл) и экстрагировали дихлорметаном (4×75 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением продукта в виде светло-желтого твердого вещества (720 мг; 4,76 ммоль; 91%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=150,1 (M+H)+
Стадия 5: 2-(бромметил)-[1,2,4]триазоло[1,5-a]пиридин
К коричневой суспензии [1,2,4]триазоло[1,5-a]пиридин-2-илметанола (870 мг; 5,83 ммоль) и полимерсвязанного трифенилфосфина (2,94 г; 8,75 ммоль) в дихлорметане (18 мл) медленно добавляли тетрабромид углерода (2,9 г; 8,75 ммоль) при 0-5°C. Через 5 мин ледяную баню убирали и реакционную смесь перемешивали при КТ в течение ночи. Растворитель выпаривали, коричневый остаток суспендировали в этилацетате (100 мл), органическую фазу промывали водой (3×50 мл) и солевым раствором (1×50 мл). Водные слои снова экстрагировали этилацетатом (1×100 мл), объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением продукта в виде бесцветного воскообразного твердого вещества (1,35 г с чистотой 90%; 5,7 ммоль; 98,2%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=214,0 (M+H)+
Стадия 6: 2-([1,2,4]триазоло[1,5-a]пиридин-2-ил)ацетонитрил
Продукт получали, исходя из 2-(бромметил)-[1,2,4]триазоло[1,5-a]пиридина (1,35 г; 5,73 ммоль), по способу, описанному в примере 47, стадия 5, в виде бесцветного твердого вещества (240 мг; 1,52 ммоль; 26,5%).
MS: M=159,1 (M+H)+
Стадия 7: 2-([1,2,4]триазоло[1,5-a]пиридин-2-ил)этанамин
Продукт получали, исходя из 2-([1,2,4]триазоло[1,5-a]пиридин-2-ил)ацетонитрила (235 мг; 1,49 ммоль), по способу, описанному в примере 55, стадия 5, в виде желтого масла (103 мг; 0,635 ммоль; 42,7%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=163,3 (M+H)+
Стадия 8: (2-[1,2,4]триазоло[1,5-a]пиридин-2-ил-этил)-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-([1,2,4]триазоло[1,5-a]пиридин-2-ил)этанамина (27,9 мг; 172 мкмоль) путем сочетания с HATU, по способу, описанному в примере 55, стадия 6, в виде белого твердого вещества (12,8 мг; 36,2 мкмоль; 25,3%).
MS: M=354,4 (M+H)+
Пример 65: [2-(2-фенил-5-пиридин-3-илоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
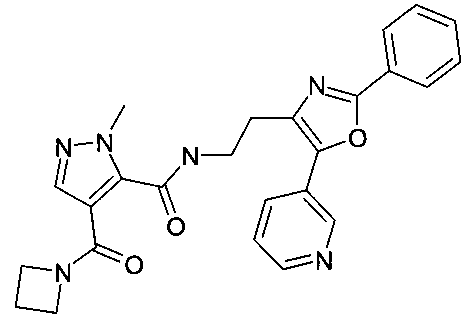
Стадия 1: метил-2-фенил-5-(пиридин-3-ил)оксазол-4-карбоксилат
К раствору метил-3-оксо-3-(пиридин-3-ил)пропионата (4 г; 22,3 ммоль) в этилацетате (200 мл) добавляли при КТ иодид тетрабутиламмония (1,65 г; 4,46 ммоль), бензиламин (4,78 г; 4,87 мл; 44,6 ммоль) и трет-бутилгидропероксид (5,5 М раствор в декане, 16,2 мл; 89,3 ммоль). Реакционную смесь перемешивали при 40°C в течение 19 ч. Добавляли тиосульфат натрия (10% водный раствор, 200 мл) при КТ и реакционную смесь перемешивали в течение 10 мин (до отрицательного теста на пероксиды). Водную фазу отделяли, органическую фазу промывали водой (2×100 мл) и солевым раствором (20 мл). Водные фазы снова экстрагировали этилацетатом (1×100 мл) и объединенную органическую фазу высушивали над MgSO4, фильтровали и выпаривали с получением 11,18 г желтого полутвердого вещества. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде желтого твердого вещества (1,33 г; 4,75 ммоль; выход 21,3%).
MS: M=281,2 (M+H)+
Стадия 2: (2-фенил-5-(пиридин-3-ил)оксазол-4-ил)метанол
К раствору метил-2-фенил-5-(пиридин-3-ил)оксазол-4-карбоксилата (1,33 г; 4,75 ммоль) в THF (25 мл) добавляли по каплям при 0-5°C алюмогидрид лития (1 M раствор в THF, 4,75 мл; 4,75 ммоль) в течение 15 мин. Реакционную смесь перемешивали при 0-5°C в течение 1 ч. Добавляли по каплям воду (20 мл) в течение 10 мин. Реакционную смесь вливали в KHSO4 (10% водный раствор, 75 мл) и экстрагировали этилацетатом (2×100 мл). Органическую фазу промывали водой (2×50 мл) и солевым раствором (1×50 мл), высушивали над MgSO4 и концентрировали в вакууме с получением 490 мг неочищенного продукта. Объединенные водные фазы доводили до pH 8 с помощью NaOH (1 M раствор) и экстрагировали дихлорметаном (3×100 мл). Дихлорметановую фазу высушивали над MgSO4, фильтровали и выпаривали с получением 480 мг коричневой пены. Продукт получали после очистки объединенных неочищенных продуктов флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан), в виде светло-коричневого твердого вещества (210 мг; 832 мкмоль; 17,5%).
MS: M=253,1 (M+H)+
Стадия 3: 4-(хлорметил)-2-фенил-5-(пиридин-3-ил)оксазол
К раствору (2-фенил-5-(пиридин-3-ил)оксазол-4-ил)метанола (210 мг; 832 мкмоль) и триэтиламина (253 мг; 348 мкл; 2,5 ммоль) в дихлорметане (4 мл) добавляли при 0-5°C метансульфонилхлорид (191 мг; 130 мкл; 1,66 ммоль). Через 10 мин ледяную баню убирали и реакционную смесь перемешивали при КТ в течение 2 ч. Реакционную смесь вливали в воду (25 мл) и экстрагировали этилацетатом (2×50 мл). Объединенный органический слой промывали водой (1×25 мл) и солевым раствором (1×25 мл), высушивали над MgSO4 и концентрировали в вакууме с получением продукта в виде светло-коричневого твердого вещества (197 мг; 728 мкмоль; 87,4%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=271,1 (M+H)+
Стадия 4: 2-(2-фенил-5-(пиридин-3-ил)оксазол-4-ил)ацетонитрил
К раствору 4-(хлорметил)-2-фенил-5-(пиридин-3-ил)оксазола (197 мг; 728 мкмоль) в DMSO (4 мл) добавляли при КТ цианид калия (56,9 мг; 873 мкмоль). Продолжали перемешивать в течение 1 ч и реакционную смесь вливали в воду (25 мл) и экстрагировали этилацетатом (2×50 мл). Органические фазы промывали водой (2×20 мл) и солевым раствором (1×20 мл). Объединенный органический слой высушивали над MgSO4 и концентрировали в вакууме с получением 168 мг неочищенного продукта. Продукт получали после очистки флэш-хроматографией (с использованием силикагеля и градиента этилацетат/гептан) в виде светло-желтого твердого вещества (80 мг; 306 мкмоль; 42,1%).
MS: M=262,2 (M+H)+
Стадия 5: 2-(2-фенил-5-(пиридин-3-ил)оксазол-4-ил)этанамин
Продукт получали, исходя из 2-(2-фенил-5-(пиридин-3-ил)оксазол-4-ил)ацетонитрила (95 мг; 364 мкмоль), по способу, описанному в примере 55, стадия 5, в виде светло-желтого вязкого масла (47 мг; 177 мкмоль; 48,7%), которое использовали без дополнительной очистки на следующей стадии.
MS: M=266,2 (M+H)+
Стадия 6: [2-(2-фенил-5-пиридин-3-илоксазол-4-ил)-этил]-амид 4-(азетидин-1-карбонил)-2-метил-2H-пиразол-3-карбоновой кислоты
Продукт получали, исходя из промежуточного соединения A-1 (30 мг; 143 мкмоль) и 2-(2-фенил-5-(пиридин-3-ил)оксазол-4-ил)этанамина (47 мг; 177 мкмоль), путем сочетания с HATU по способу, описанному в примере 55, стадия 6, и последовательной очистку флэш-хроматографией (с использованием силикагеля и градиента метанол/этилацетат) в виде светло-желтого вязкого масла (5,2 мг; 11,4 мкмоль; 8,0%).
MS: M=457,5 (M+H)+
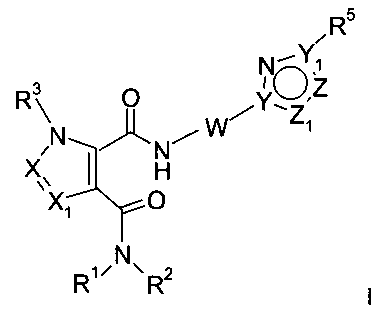
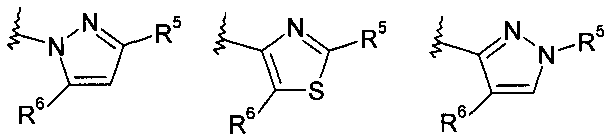
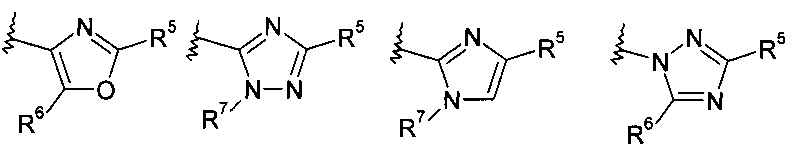
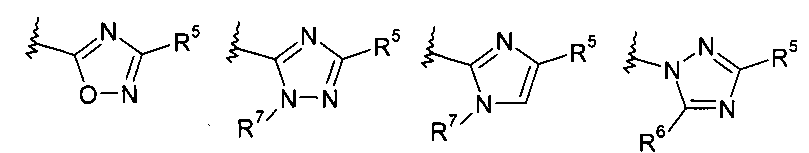
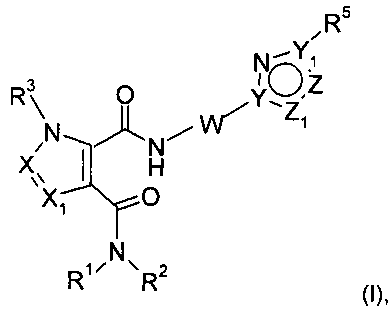
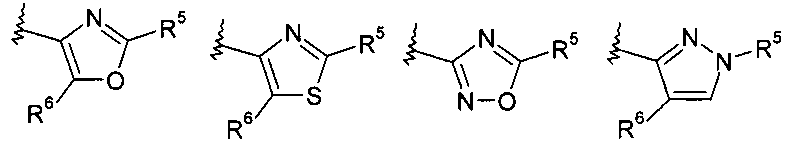
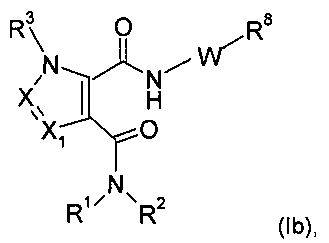
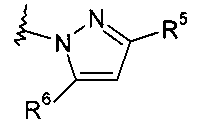
Новые 2-аминооксазолины в качестве лигандов taar1 для заболеваний цнс
Препарат антитела
Производные гетероарилпирролидинил- и пиперидинилкетона
Производные 1,1,1-трифтор-2-гидрокси-3-фенилпропана
Производные изоксазоло-пиридина
Карбоксил- или гидроксилзамещенные производные бензимидазола
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Ингибиторы jnk
Бензопирановые и бензоксепиновые ингибиторы рi3k и их применение
Арилциклогексилэфиры дигидротетраазабензоазуленов для применения в качестве антагонистов рецептора вазопрессина v1a
Новые 2-аминооксазолины в качестве лигандов taar1 для заболеваний цнс
Аналитический инструмент с тестовой лентой, содержащий двигатель постоянного тока и редуктор
Препарат антитела
Производные гетероарилпирролидинил- и пиперидинилкетона
Производные 1,1,1-трифтор-2-гидрокси-3-фенилпропана
Производные изоксазоло-пиридина
Карбоксил- или гидроксилзамещенные производные бензимидазола
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Ингибиторы jnk
Бензопирановые и бензоксепиновые ингибиторы рi3k и их применение

