Результат интеллектуальной деятельности: СПОСОБ ЭНАНТИОСЕЛЕКТИВНОГО СИНТЕЗА (S)-ПРЕГАБАЛИНА
Вид РИД
Изобретение
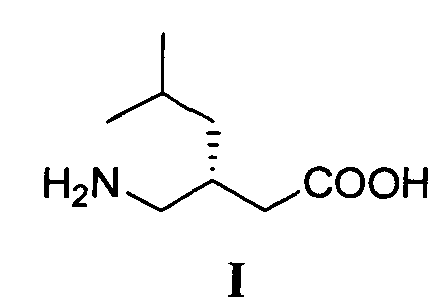
Изобретение относится к способу получения (S)-прегабалина формулы I
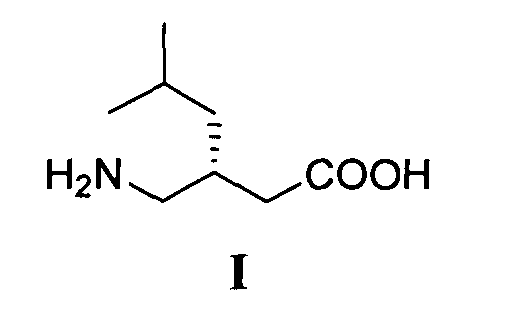
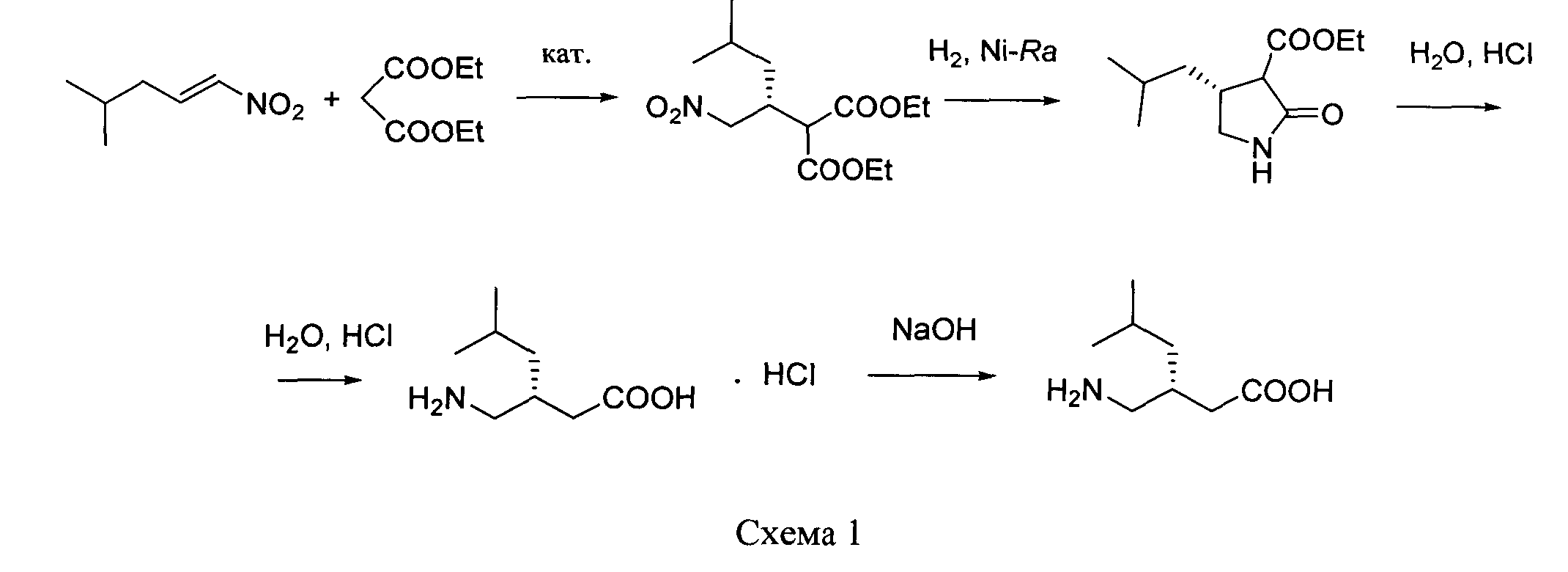
путем энантиоселективного присоединения диэтилмалоната к 4-метил-1-нитропентену-1 с последующим восстановлением и кислотным гидролизом продукта присоединения в соответствии со схемой 1,
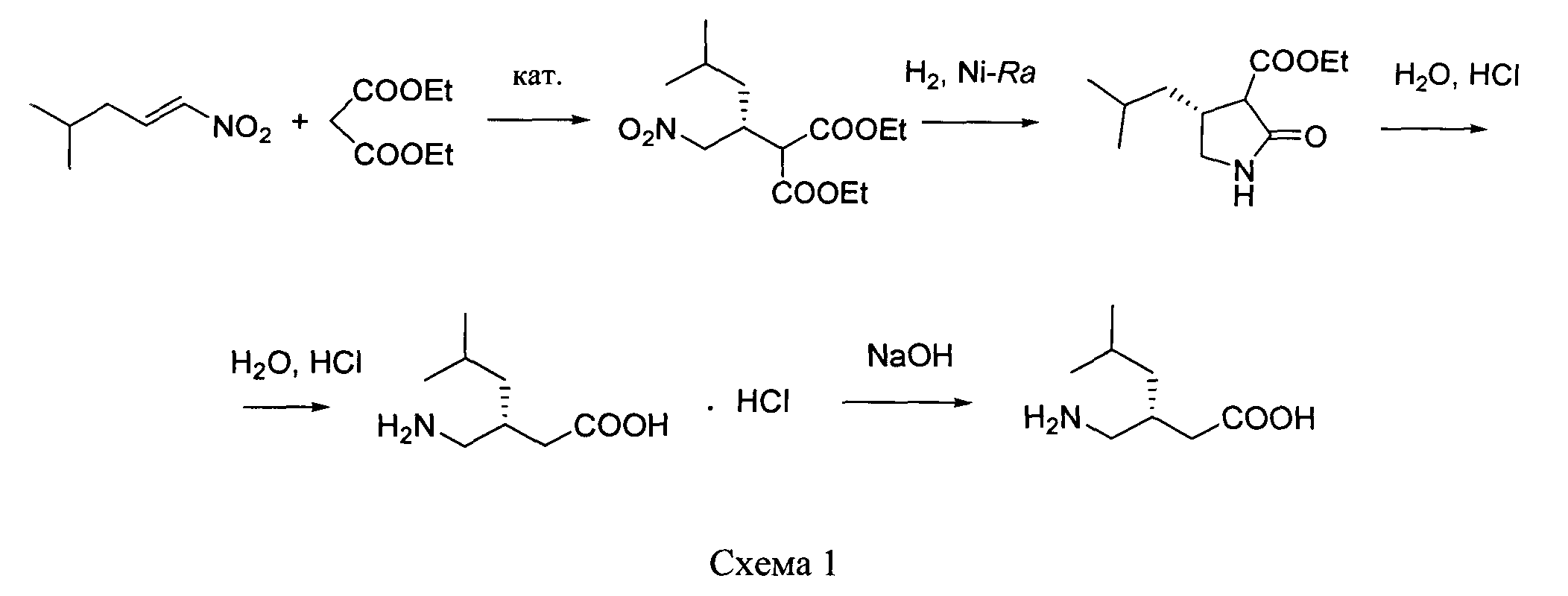
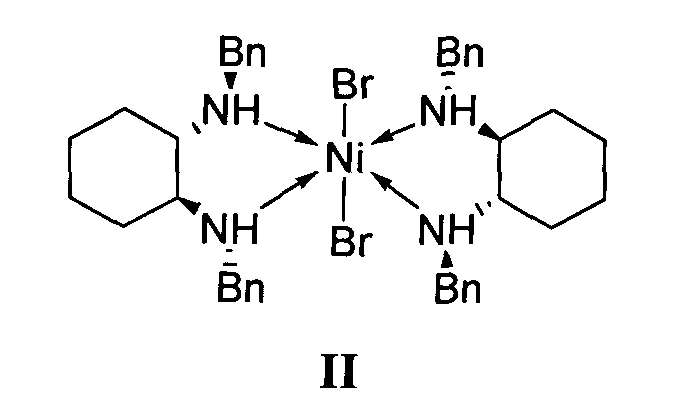
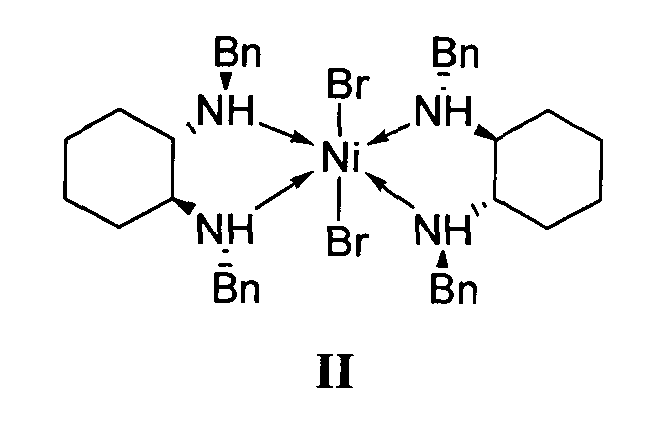
отличающийся тем, что используемый на стадии присоединения катализатор представляет собой комплекс никеля(II) формулы II с (S,S)-N,N′-дибензилциклогексан-1,2-диамином.
(S)-Прегабалин используется в терапии ряда нейропатических заболеваний и расстройств, в том числе невропатических болей, эпилепсии, фибромиалгии, мигрени, синдрома беспокойных ног, нарушений сна и др.
(Silverman R.B., Andruszkiewicz R. Pat. US 6359169 (B1) GABA and L-glutamic acid analogs for antiseizure treatment (дата публикации 19.03.2002); Forster E.R., Koppiker N.P. Pat. MY 138266 (A) Treatment of neuropathy (дата публикации 29.05.2009); Meergans D., Paetz J. Pat. WO 2012016683 (A2) Oral dosage form of pregabalin (дата публикации 09.02.2012); Kusuki N., Hiroshi O. Pat. KR 20120067783 (A) Pharmaceutical composition for treatment of fibromyalgia (дата публикации 26.06.2012)).
Данным изобретением решена задача энантиоселективного синтеза прегабалина с использованием легкодоступного и дешевого катализатора, что обеспечивает значительное удешевление и упрощение технологического процесса. Поскольку нейротропная активность (S)-изомера прегабалина значительно превосходит активность (R)-изомера (Silverman R.B., Andruszkiewicz R. Pat. US 6359169 (B1) GABA and L-glutamic acid analogs for antiseizure treatment, дата публикации 19.03.2002), формирование асимметрического центра требуемой конфигурации на стадии синтеза соединения формулы I исключает необходимость дополнительных стадий разделения рацемических смесей, обычно используемых в производстве данного фармацевтического препарата.
Известен ряд способов получения (S)-прегабалина.
Группа методов, основанных на разделении конечного продукта или синтетических интермедиатов на (R)- и (S)-энантиомеры. Такие методы были использованы при получении прегабалина с использованием промежуточной стадии разделения рацемата 3-(2-амино-2-оксоэтил)-5-метилкапроновой кислоты с помощью (R)-α-фенилэтиламина (Huckabee B.K., Sobieray D.M. Pat. US 5629447 Methods of making (S)-3-(aminomethyl)-5-methylhexanoic acid (дата публикации 13.05.1997).
Рацемат прегабалина может быть разделен на энантиомеры с помощью хиральных кислот: (R)-миндальной (Grote T.M., Huckabee B.K. Pat. US 6046353 Methods of making (S)-3-(aminomethyl)-5-methylhexanoic acid (дата публикации 07.06.1995)), L-винной и ди-пара-толуил-L-винной (Gore V., Datta D. Pat. WO 2009/122215 (A1) Novel process (дата публикации 02.04.2009)).
Преимуществом энантиоселективного синтеза прегабалина является то, что в этом случае не образуется нежелательный (R)-энантиомер, отсутствует необходимость в использовании стехиометрических количеств хиральных реагентов и, потенциально, общий выход может быть удвоен.
Асимметрическим гидрированием солей 3-циано-5-метилгекс-5-еновой кислоты получено хиральное цианопроизводное, из которого последующим восстановлением синтезирован прегабалин (Burk M.J., Goel О. Pat. US 6891059 Asymmetric synthesis of pregabalin (дата публикации 13.11.2003). Асимметрическое гидрирование проводилось в присутствии дорогостоящих металлокомплексных катализаторов с дифосфиновыми лигандами, такими как (R,R)-Me-DUPHOS.
Прегабалин также синтезирован с использованием хиральных вспомогательных реагентов, таких как (4R,5S)-4-метил-5-фенил-2-оксазолидинон (Pat. US 6359169, 6028214, 5847151, 5710304, 5684189, 5608090, 5599973).
Хотя эти методы обеспечивают получение (S)-прегабалина высокой хиральной чистоты, они трудно реализуемы в крупномасштабном промышленном синтезе, поскольку предполагают использование дорогостоящих реактивов (хиральных оксазолидинонов), а также специального криогенного оборудования для обеспечения требуемых рабочих температур (-78°С).
Хиральный оксазолидинон был использован в синтезе оптического антипода прегабалина (Armstrong A., Convine N.J., Popkin M.E. Diastereoselective conjugate addition of cyanide to α,β-unsaturated oxazolidinones: enantioselective synthesis of ent-pregabalin and backlofen Synlett, 2006, (10), 1589-1591). Присоединением цианида к α,β-непредельному оксазолидинону получено цианопроизводное - хиральный прекурсор прегабалина. Однако производное оксазолидинона, полученное из природного изомера валина, после цианирования приводит к продукту с противоположной абсолютной конфигурацией. Кроме того, для получения ненасыщенного субстрата в реакции цианирования требуется стехиометрическое количество хирального оксазолидинона. В качестве катализатора используется соединение самария. Эти факторы делают данную схему синтеза прегабалина экономически невыгодной и экологически опасной при реализации в промышленных масштабах.
Два других метода приводятся в патенте: Davies B.S., Guzman M.M. Pat. WO 2010/070593 Malonate esters (дата публикации 24.06.2010). В первом случае энантиоселективное присоединение цианида к алкилиденмалонату, приводящее к ключевому полупродукту - цианоалкилмалонату, осуществлено в условиях межфазного катализа с использованием хирального катализатора фазового переноса. Во втором случае используется ферментативный катализ для получения хирального циангидрина изовалерианового альдегида, из которого в две стадии получен цианоалкилмалонат.
Наиболее близким по технической сущности к заявляемому способу является способ получения (S)-прегабалина из диметил[(2S)-4-метил-1-нитропент-2-ил]малоната, описанный в патенте: Maymon A., Kansal V.K. Pat. WO 2006/110783 (A2) Process for making (S)-pregabalin (дата публикации 19.10.2006).
Диметил[(2S)-4-метил-1-нитропент-2-ил]малонат, в соответствии с Pat. WO 2006/110783 (A2), может быть получен реакцией 4-метил-1-нитропент-1-ена с диметилмалонатом в присутствии органокатализаторов на основе цинхонидина по методике, описанной в работе: Li H., Wang Y., Tang L., Deng L. Highly enantioselective conjugate addition of malonate and β-ketoester to nitroalkenes: asymmetric C-C bond formation with new bifunctional organic catalysts based on cinchona alkaloids // J. Am. Chem. Soc. 2004. Vol.126. N 32. P.9906-9907.
Описанный в Pat. WO 2006/110783 (A2) способ получения соединения формулы I заключается в том, что борогидрид натрия (3.3 г) добавляют к суспензии диметил[(2S)-4-метил-1-нитропент-2-ил]малоната (14 г) и гексагидрата хлорида никеля(II) (5 г) в 140 мл метанола при 0°С. Реакционную смесь перемешивают 6 ч, обрабатывают хлоридом аммония и экстрагируют 55 мл хлористого метилена. Органическую фазу отделяют и сушат над сульфатом магния, фильтруют и упаривают в вакууме. Полученный метиловый эфир (4S)-изобутилпирролидин-2-он-3-карбоновой кислоты растворяют в 350 мл этанола и к этому раствору добавляют 135 мл 1 N раствора гидроксида натрия при комнатной температуре, перемешивают 30 мин и упаривают в вакууме. К остатку добавляют 250 мл 6 N соляной кислоты, органическую фазу отделяют, а водную экстрагируют 120 мл хлористого метилена. Объединенную органическую часть сушат над сульфатом магния и упаривают в вакууме. Полученную (4S)-изобутилпирролидин-2-он-3-карбоновую кислоту декарбоксилируют кипячением в 120 мл толуола в течение 6 ч. Затем реакционную смесь упаривают и продукт - (4S)-изобутилпирролидин-2-он выделяют хроматографией на силикагеле. 10 г полученного пирролидинона растворяют в 440 мл 6 N соляной кислоты и кипятят 15 ч. После этого реакционную смесь разбавляют водой и трижды экстрагируют хлористым метиленом. Водную фазу упаривают и остаток сушат в вакууме, получая хлоргидрат (S)-прегабалина. Хлоргидрат растворяют в изобутаноле и добавляют триэтиламин. Образовавшийся осадок (S)-прегабалина фильтруют и промывают изобутанолом.
Указанный способ обладает целым рядом существенных недостатков:
1. Необходимость использования больших количеств дорогостоящих органокатализаторов (10-20 мольных % по отношению к реагентам).
2. Использование дорогостоящего и токсичного борогидрида натрия и больших объемов метанола на стадии восстановления диметил[(2S)-4-метил-1-нитропент-2-ил]малоната.
3. Многостадийность синтеза прегабалина, использование хроматографической очистки полупродукта, больших объемов токсичных растворителей (хлористый метилен, толуол) и многократных экстракций усложняет технологический процесс и делает его экологически опасным.
4. Применение изобутанола и триэтиламина на заключительной стадии синтеза прегабалина недопустимо с точки зрения обеспечения чистоты фармацевтической субстанции.
Технический результат - экономически выгодный и безопасный процесс получения (S)-прегабалина с энантиомерным избытком 96.2 %, который может быть реализован в промышленном масштабе с использованием легкодоступных и дешевых реагентов. Технический результат достигается тем, что (S)-прегабалин формулы I
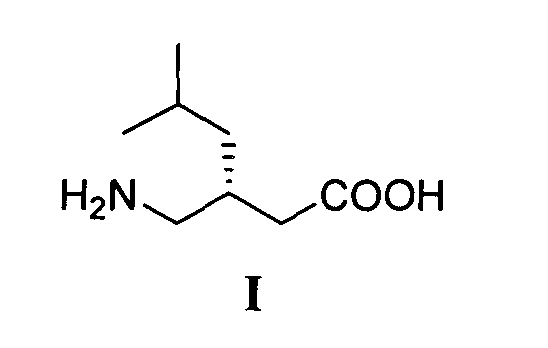
получают путем энантиоселективного присоединения диэтилмалоната к 4-метил-1-нитропентену-1 с последующим восстановлением и кислотным гидролизом продукта присоединения в соответствии со схемой 1:
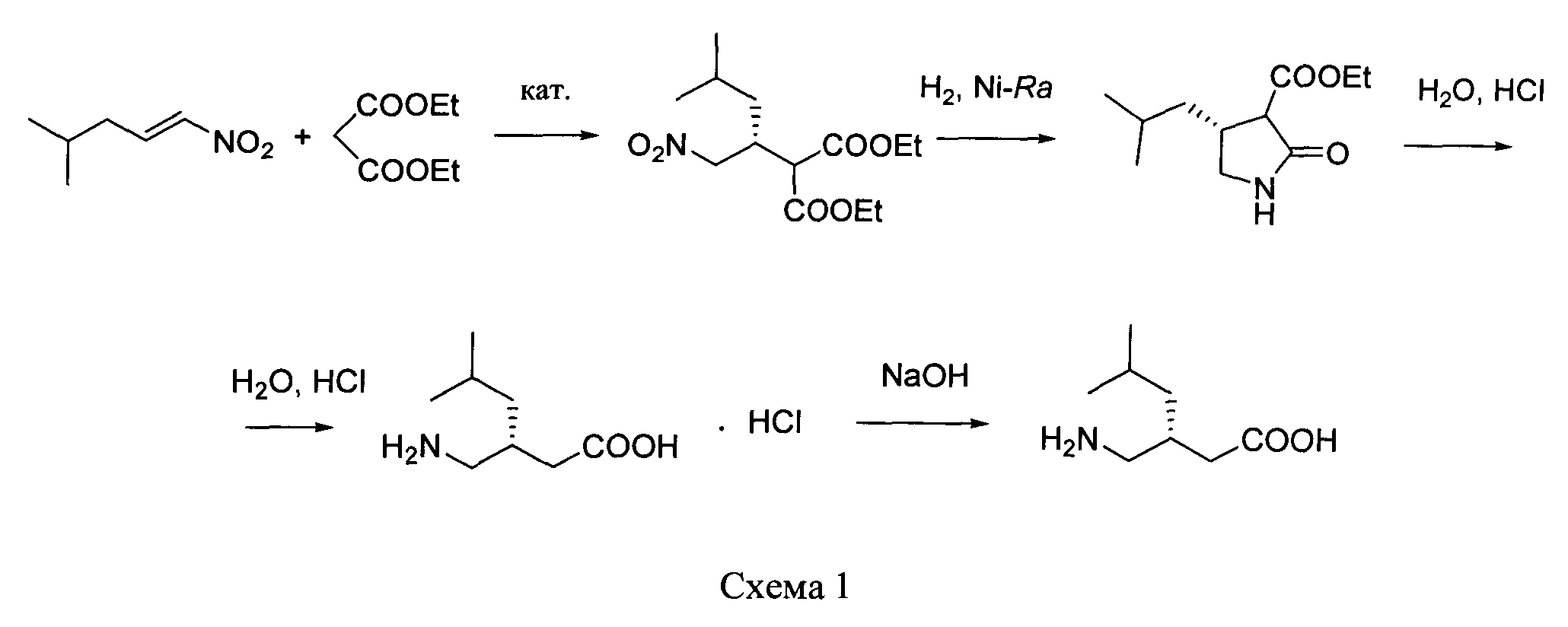
Отличительные признаки:
1. Используемый на стадии присоединения катализатор представляет собой комплекс никеля(II) формулы II с (S,S)-N,N′-дибензилциклогексан-1,2-диамином.
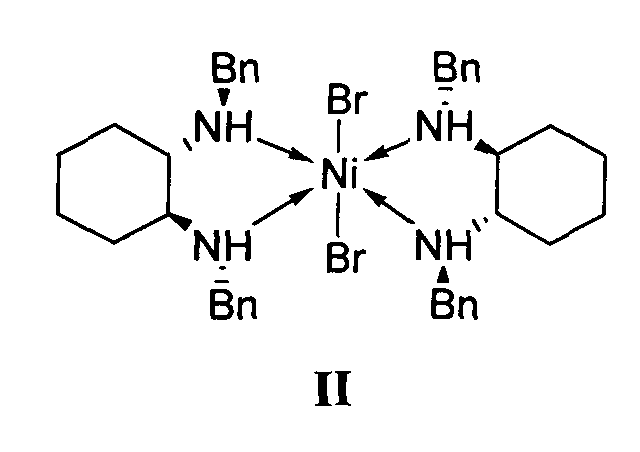
2. Реакция диэтилмалоната с 4-метил-1-нитропентеном-1 проводится без растворителя, а концентрация катализатора формулы II составляет 0.5-2.5 мол. %.
3. Обогащение (S)-изомером проводится путем перекристаллизации этилового эфир (4S)-изобутилпирролидин-2-он-3-карбоновой кислоты из петролейного эфира.
4. На стадии получения (S)-прегабалина из хлоргидрата используют раствор гидроксида натрия в метаноле.
Заявляемое изобретение имеет следующие преимущества.:
Удешевление производства за счет применения в качестве катализатора энантиоселективного присоединения диэтилмалоната к 4-метил-1-нитропент-1-ену комплекса никеля(II) формулы II и отказа от использования дорогостоящих производных цинхонидина в больших концентрациях.
Упрощение технологического процесса за счет сокращения общего числа стадий, отказа от использования многократных экстракций и хроматографического выделения полупродуктов.
Обеспечение экологической безопасности процесса за счет отказа от использования больших объемов токсичных растворителей.
Примеры выполнения способа
Спектры ЯМР регистрировали на приборе Jeol JNM-ECX400 с использованием растворителей CDCl3 и метанол-d4 [399.78 (1Н) и 100.53 МГц (13С)]. Измерения проводили без использования дополнительных эталонов с привязкой частоты к сигналу дейтерированного растворителя.
Масс-спектры синтезированных соединений получены на хромато-масс-спектрометре Finnigan Trace DCQ с использованием капиллярной колонки ВРХ-5 30×0.32 компании SGE при энергии ионизирующих электронов 70 эВ.
Определение удельных углов вращения проводилось на поляриметре Rudolph Research Analitical.
Энантиомерный состав продуктов реакции определен методом ВЭЖХ на жидкостном хроматографе Waters, оснащенном спектрофотометрическим детектором Waters 2487 и рефрактометрическим детектором Waters 2414. Условия анализа для диэтил[(2S)-4-метил-1-нитропент-2-ил]малоната: колонна Chiralcel AD; мобильная фаза: гексан: изопропанол (95:5), расход 1.0 мл / мин. Условия анализа для (S)-прегабалина: колонна YMC Pack Pro С18; мобильная фаза: формиатный раствор (рН 3.0) в метаноле в соотношении 4:6 и метанол. Расход: 0.6 мл/мин. Хиральный модификатор - реагент D-Марфея.
Диэтил [(2S)-4-метил-1-нитропент-2-ил] малонат
Пример 1.
К 12.1 г (93.5 ммоль) 4-метил-1-нитропент-1-ена и 30.0 г (187 ммоль) диэтилмалоната добавляли 1.89 г (2.34 ммоль, 2.5 мольн. %) катализатора - дибромобис[(S,S)-N,N′-дибензилциклогексан-1,2-диамин]никеля (II). Реакционную массу выдерживали при температуре 50°С в течение 15 ч. Избыток диэтилмалоната отгоняли в вакууме при остаточном давлении 20 мм рт.ст. Выход: 22.0 г (81.4 %). ЯМР 1Н (CDCl3), δ, м.д.: 0.85-0.87 м. (6Н), 1.20-1.29 м (8Н), 1.57-1.63 м (1Н), 2.87-2.92 м (1Н), 3.55 д (1Н, JHH 5.6 Гц), 4.13-4.19 м (4Н), 4.46 дд (1Н, JHH 13.2, 6.4 Гц), 4.65 дд (1Н, JHH 13.2, 4.8 Гц). δс, м.д.: 13.8, 13.9, 22.1, 22.2, 25.0, 34.7, 38.9, 52.6, 61.6, 61.7, 76.8, 167.7, 167.9. ГХ-МС, m/z (Iотн, %): 243 (6, M+ - NO2), 215 (10), 169 (25), 160 (100), 133 (30), 55 (35). ВЭЖХ: время выхода (R)-изомера 6.4 мин, (S)-изомера 8.8 мин; элюент гексан/изопропанол 98:2; расход мобильной фазы 1 мл/мин. По данным ВЭЖХ энантиомерный избыток (S)-изомера 81.8%.
Пример 2.
К 24.2 г (0.187 моль) 4-метил-1-нитропент-1-ена и 59.9 г (0.375 моль) диэтилмалоната добавляли 2.26 г (2.80 ммоль, 1.5 мольн. %) катализатора - дибромобис[(S,S)-N,N′-дибензилциклогексан-1,2-диамин]никеля (II). Реакционную массу выдерживали при температуре 50°С в течение 24 ч. Избыток диэтилмалоната отгоняли в вакууме при остаточном давлении 20 мм рт.ст. Выход: 44.0 г (81.0 %). По данным ВЭЖХ энантиомерный избыток (S)-изомера 81.8 %.
Пример 3.
К 50.0 г (0.387 моль) 4-метил-1-нитропент-1-ена и 124.0 г (0.774 моль) диэтилмалоната добавляли 1.56 г (1.94 ммоль, 0.5 мольн. %) катализатора - дибромобис[(S,S)-N,N′-дибензилциклогексан-1,2-диамин]никеля (II). Реакционную массу выдерживали при температуре 50°С в течение 72 ч. Избыток диэтилмалоната отгоняли в вакууме при остаточном давлении 20 мм рт.ст. Выход: 97.7 г (87.3 %). По данным ВЭЖХ энантиомерный избыток (S)-изомера 81.2 %.
Этиловый эфир (4S)-изобутилпирролидин-2-он-3-карбоновой кислоты
Пример 1.
Раствор 22.0 г (76.0 ммоль) диэтил[(2S)-4-метил-1-нитропент-2-ил]малоната в 280 мл изопропанола гидрировали в присутствии 5.0 г никеля Ренея при температуре 50°С и избыточном давлении водорода 10 атм в течение 13 ч. Раствор фильтровали, изопропанол упаривали. Остаток перекристаллизовывали из петролейного эфира. Выход: 8.14 г.
(50.2 %). Т.пл. 72-73°С. [α]D 20 - 51.7° (с 0.67, СНСl3). ЯМР 1Н (СDСl3), δ, м.д.: 0.90 т (6Н, JHH 6.4 Гц), 1.30 т (3Н, JHH 7.2 Гц), 1.34-1.46 м (2Н), 1.51-1.61 м (1Н), 2.06-2.89 м (3Н), 3.52-3.58 м (1Н), 4.21-4.28 м (2Н), 6.69 с (1Н). δС, м.д.: 14.1, 22.4, 22.5, 25.8, 37.4, 43.2, 46.8, 54.9, 61.4, 169.9, 173.7. ГХ-МС, m/z (Iотн, %): 213 (3, М+), 168 (12), 156 (100), 110 (18), 84 (20), 56 (25).
Пример 2.
Раствор 42.7 г (0.148 моль) диэтил[(2S)-4-метил-1-нитропент-2-ил]малоната в 280 мл изопропанола гидрировали в присутствии 5.0 г никеля Ренея при температуре 50°С и избыточном давлении водорода 10 атм в течение 13ч. Раствор фильтровали, изопропанол упаривали. Остаток перекристаллизовывали из петролейного эфира. Выход: 16.2 г
(51.2%).
Пример 3.
Раствор 97.7 г (0.338 моль) диэтил[(2S)-4-метил-1-нитропент-2-ил]малоната в 280 мл изопропанола гидрировали в присутствии 11.0 г никеля Ренея при температуре 50°С и избыточном давлении водорода 20 атм в течение 10ч. Раствор фильтровали, изопропанол упаривали. Остаток перекристаллизовывали из петролейного эфира. Выход: 40.2 г
(55.7 %).
Хлоргидрат (S)-прегабалина
Пример 1.
Смесь 16.2 г (75.9 ммоль) этилового эфира (4S)-изобутилпирролидин-2-он-3-карбоновой кислоты и 381 мл 6 N соляной кислоты кипятили с обратным холодильником в течение 18 часов. После охлаждения смесь экстрагировали этилацетатом (2×95 мл). Водный слой отделяли и упаривали в вакууме. Выход: 14,8 г (99.0 %). ЯМР 1Н (метанол-d4), δ, м.д.: 0.92-0.96 м (6Н), 1.25-1.29 м (1Н), 1.66-1.73 м (1Н), 2.18-2.24 м (1Н), 2.37-2.48 м (1Н), 2.96-2.98 м (1Н); δС, м.д.: 22.6, 23.1, 26.0, 32.4, 37.2, 41.9, 44.4, 175.7.
Пример 2.
Смесь 40.2 г (0.188 моль) этилового эфира (4S)-изобутилпирролидин-2-он-3-карбоновой кислоты и 950 мл 6 N соляной кислоты кипятили с обратным холодильником в течение 18 часов. После охлаждения смесь экстрагировали этилацетатом (2×200 мл). Водный слой отделяли и упаривали в вакууме. Выход: 35.4 г (96.3 %).
(S)-Прегабалин
Пример 1.
К раствору 7.0 г (35.8 ммоль) хлоргидрата (3S)-аминометил-5-метилкапроновой кислоты в 50 мл метанола при перемешивании и охлаждении добавляли насыщенный раствор гидроксида натрия в метаноле до рН 7. Реакционную смесь фильтровали через слой силикагеля и фильтрат упаривали. Выход: 5.3 г (93.0 %). Т.пл. 170-172°С. [α]D 20=+7.15° (с 1.03, Н2O). Спектр ЯМР 1Н (метанол-d4), δ, м.д.: 0.89-0.92 м 6Н), 1.15-1.28 м (2Н), 1.65-1.72 м (1Н), 2.02-2.12 м (1Н), 2.22-2.28 м (1Н), 2.39-2.44 м (1Н), 2.80-2.85 м (1Н), 2.94-2.97 м (1Н), 3.29 м (1Н). ВЭЖХ: время выхода (R)-изомера 6.0 мин, (S)-изомера 7.8 мин; По данным ВЭЖХ энантиомерный избыток (S)-изомера 96.2 %.
Пример 2.
К раствору 35.4 г (0.181 моль) хлоргидрата (3S)-аминометил-5-метилкапроновой кислоты в 250 мл метанола при перемешивании и охлаждении добавляли насыщенный раствор гидроксида натрия в метаноле до рН 7. Реакционную смесь фильтровали через слой силикагеля и фильтрат упаривали. Выход: 27.1 г (94.2 %). По данным ВЭЖХ энантиомерный избыток (S)-изомера 96.2 %.
Производные 2-r1-4-r2-6-полинитрометил-1,3,5-триазинов, обладающие антибактериальной активностью
Способ получения гидрохлоридов аминов адамантанового ряда
Способ энантиоселективного синтеза диэтил[3-метил-(1s)-(нитрометил)бутил]малоната формулы i
Способ получения 1-гидрокси-4-адамантанона
Антигололедная композиция
Способ получения трехосновных карбоновых кислот адамантанового ряда
Дезинфицирующая композиция
Способ получения 3-арил-1н-бензо[f]хроменов
Способ получения диэфиров 5,7-диметил-3-карбокси-1-адамантилуксусной кислоты
Способ получения нерацемического 1-(адамант-1-ил)-2-(2-нитро-1-фенилэтил)бутан-1,3-диона
Дезинфицирующая композиция
Способ получения 3-арил-1н-бензо[f]хроменов
Способ получения диэфиров 5,7-диметил-3-карбокси-1-адамантилуксусной кислоты
Способ получения нерацемического 1-(адамант-1-ил)-2-(2-нитро-1-фенилэтил)бутан-1,3-диона
Способ получения 1н-бензо[f]хромен-2-ил(арил)кетонов
Способ получения (s)-3-(аминометил)-5-метилгексановой кислоты
Способ получения производных хроман-2-аминов
Способ генерирования диоксида хлора
Применение гетероциклических гидразонов в качестве средств, обладающих антигликирующей активностью
Способ получения 1-(1н-бензохромен-2-ил)-2,2,2-трифторэтанонов

