Результат интеллектуальной деятельности: Композиция на основе пептида, подавляющего репликацию вируса гриппа А
Вид РИД
Изобретение
Изобретение относится к медицине и фармацевтике, а именно к разработке лекарственных средств, содержащих пептиды, гомологичные функционально значимым доменам вирус-специфических белков, пригодных для интраназального применения, и может быть использовано для лечения инфекции, вызванной вирусом гриппа А человека, в частности штамма субтипа A(H1N1)pdm09.
Несмотря на десятилетия наблюдений за распространением и борьбы с заболеваемостью гриппом с применением различных методов, в том числе с использованием фармацевтических терапевтических и профилактических средств, сезонный грипп поражает 5-15 % популяции людей и является причиной гибели 250000-500000 человек ежегодно [1]. Эволюционная изменчивость вирусов гриппа позволяет циркулирующим сезонным штаммам ускользать от специфического иммунного ответа, как естественного (индуцированного перенесенной инфекцией прошлых лет), так и искусственного (индуцированного вакциной, соответствующей штаммам прошлых сезонов) [2].
Вирусы гриппа относятся к семейству Orthomyxoviridae, род Influenzavirus, типы А, В, С и D. Вирионы гриппа А представляют собой сферические или слегка вытянутые частицы диаметром 80-100 нм, покрытые липидной оболочкой с интегрированными молекулами поверхностных гликопротеинов – гемагглютинина (далее по тексту – «HA») и нейраминидазы (далее по тексту – «NA»), а также поверхностного белка М2 (далее по тексту – «белок М2»), работающего как ионный канал и ассоциированного со слоем мембранного белка M1. Нуклеоид вириона представлен 8 сегментами однонитевой РНК отрицательной полярности (вРНК), каждый из которых образует комплекс с несколькими десятками молекул нуклеопротеина (NP) и тремя молекулами субъединиц полимеразного комплекса (PA, PB1, РВ2) (далее по тексту – «RNA-dependent RNA Polymerase», «RdRP»). При встрече с вирусом гриппа организма реципиента HA отвечает за связывание с молекулами сиаловой кислоты на поверхности клеток заражаемой ткани и последующее проникновение вируса внутрь клетки посредством слияния мембран. При формировании иммунного ответа HA является основной мишенью и поэтому наиболее сильно подвергается эволюционной изменчивости, направленной на ускользание от него. NA осуществляет гидролиз связи между HA и сиаловыми кислотами, способствуя выходу новых вирионов из инфицированных клеток, что позволяет вирусу осуществлять заражение новых и новых клеток заражаемой ткани. После проникновения в клетку и «раздевания» вириона сегменты генома попадают в цитоплазму, откуда транспортируются в ядро, где RdRP осуществляет транскрипцию и репликацию вРНК. В результате образуются молекулы вирусспецифической матричной РНК (мРНК) и вРНК, входящей затем в состав вирионов потомства. мРНК транслируется при помощи клеточных ферментов с образованием структурных и неструктурных белков, первые формируют структуру вириона, а последние выполняют важные регуляторные функции в ходе жизненного цикла вируса [3].
Не имеющая редакторской активности RdRP не способна исправлять ошибки в нуклеотидной последовательности сегментов в ходе репликации генома, что и приводит к возникновению изменчивости. Будучи подвергнутыми эволюционному отбору, в популяции вирусного потомства закрепляются варианты нуклеотидных последовательностей сегментов, кодирующие варианты поверхностных белков, избегающих воздействия иммунного ответа (антигенный дрифт). Кроме того, сегментированность генома вируса гриппа приводит к возможности обмена сегментами между различными штаммами (антигенный шифт) [4].
Вирусы гриппа А подразделяются на подтипы в зависимости от сочетания HA и NA. На сегодняшний день известно 18 подтипов по HA (у вирусов гриппа человека – Н1, Н2, Н3) и 11 подтипов по NA (у вирусов гриппа человека – N1, N2). Многие подтипы вируса гриппа А встречаются только у водоплавающих птиц, однако обладают потенциалом к межвидовой передаче. Вирус гриппа А также способен вызывать глобальные пандемии. К числу пандемических относят штаммы вируса, вызвавшие пандемии «испанского» гриппа («испанка») в 1918 г., «свиного» гриппа в 2009 г. A(H1N1)pdm 2009, «азиатского» гриппа в 1957 г. A(H2N2), «гонгконгского» гриппа в 1968 г. A(H3N2), с последующим распространением в популяции людей в виде сезонного гриппа. В настоящее время за сезонные эпидемии гриппа ответственны преимущественно вирусы гриппа A(H1N1) и A(H3N2). Вирус A(H5N1) считают самой большой пандемической опасностью нашего времени [5].
Вирус гриппа, в частности гриппа А, передается от человека к человеку воздушно-капельным путем, через прямой контакт, а также опосредованно через предметы, бывшие в контакте с патогенными микроорганизмами. При этом заражение воздушно-капельным путем происходит гораздо более эффективно, чем контактная передача. Входными воротами для вируса гриппа у человека являются клетки мерцательного эпителия верхних дыхательных путей – носа, трахеи, бронхов. В этих клетках вирус размножается и приводит к их разрушению и гибели. Этим, а также локализацией реакции организма вблизи пораженных вирусом тканей объясняется раздражение верхних дыхательных путей, кашель, чихание, заложенность носа. Общая реакция организма на проникновение и размножение вируса, а также на синтезируемые в процессе цикла размножения вируса регуляторные белки проявляется как повышение температуры, озноб, миалгия, головная боль. В тяжелых случаях вирус вызывает неадекватную реакцию иммунной системы организма («цитокиновый шторм»), респираторную недостаточность и смерть. В случае наличия у заболевшего сопутствующих сердечно-сосудистых и других хронических заболеваний велика вероятность их обострения, что также часто приводит к неблагоприятному исходу. Также при гриппе возможно возникновение осложнений, связанных с присоединением вторичной бактериальной инфекции, что связано с индуцированными гриппом изменениями в иммунной системе и разрушением эпителиальных клеток. У здоровых людей заражение гриппом приводит к развитию специфической иммунной реакции (цитотоксические Т-лимфоциты и антитела) в течение 1-2 недель [6].
Некоторые подтипы гриппа, заражающие людей, в том числе большинство штаммов пандемического гриппа A(H1N1)pdm09, сезонного гриппа A(H3N2) и «птичьего» гриппа A(H5N1), являются чувствительными к этиотропной терапии. Однако высокая скорость эволюционной изменчивости вируса гриппа, которая в настоящее время уже привела к появлению штаммов с лекарственной устойчивостью к существующим препаратам, в дальнейшем будет способствовать ускорению развития устойчивости к этим лекарственным препаратам при их широком применении [7]. В этой связи для лечения и профилактики гриппозной инфекции актуальной является разработка новых терапевтических средств, активных не только в отношении циркулирующих в человеческой популяции штаммов, устойчивых к действию уже существующих препаратов, но и в отношении вновь появляющихся штаммов. Кроме того, поскольку репликация вируса гриппа протекает в первую очередь в эпителиальных тканях верхних дыхательных путей, для адресной доставки противовирусного средства целесообразно использовать интраназальную форму препарата – капли или спрей [8].
Применяемые в настоящее время антивирусные средства можно условно подразделить на две группы – препараты, действующие на клеточные мишени и этиотропные препараты прямого противовирусного действия. К первой группе могут быть отнесены, в частности лекарственные средства, действующие на клеточные системы транспорта и на синтетический аппарат клетки. Так, известен рекомбинантный человеческий белок СС10 (называемый также утероглобин), снижающий титр вируса в легочной ткани экспериментальных животных при интраназальном применении. Указанный белок подавляет репликацию вируса, препятствуя осуществлению везикулярного транспорта в клетке [9].
Известно также лекарственное средство для профилактики и лечения вирусных болезней, содержащее метисазон, диметилсульфоксид (далее по тексту – «ДМСО») и полиэтиленгликоль-9 при соотношении компонентов, мас. %: метисазон – 2,0-2,5; ДМСО – 20-25; полиэтиленгликоль-9 – остальное [10]. Действие указанного средства направлено, в частности, на комплексы «полирибосомы-РНК», что выражается в ингибирующем влиянии препарата на репродукцию вирусов.
Общим недостатком препаратов первой группы является то, что они обладают относительно низкой специфичностью в отношении вирусов (в том числе вируса гриппа А человека), что значимо увеличивает вероятность побочных эффектов. Побочные эффекты, о6условленные, в частности, нарушением везикулярного транспорта вируса (для аналога [9]) или нарушением белкового синтеза клеточного генома (для аналога [10]), в дальнейшем могут приводить к нарушениям функционирования органов и тканей организма.
К антивирусным этиотропным средствам прямого противовирусного действия относятся лекарственные средства, направленные на конкретную молекулярную мишень, характерную для вируса и отсутствующую в клетке хозяина. На сегодняшний день известно использование в качестве мишеней для специфической химиотерапии трех структурных белков из множества вирусных белков, продуцирующихся в процессе жизненного цикла вирусов. Во-первых, это белок М2 вируса гриппа, играющий роль ионного канала в вирусной мембране, который блокируют препараты адамантанового ряда – ремантадин (а-метил-1-адамантил-метиламина гидрохлорид) и амантадин (1-аминоадамантан). Препараты адамантанового ряда эффективны в отношении вирусов гриппа А, однако их применение ограничено наличием серьезных побочных эффектов и быстрым возникновением и 100 % резистентности циркулирующих в настоящее время штаммов [11].
Другой мишенью для лекарственного воздействия является NA – фермент, необходимый для нормального почкования вирусных частиц и проявления инфекционных свойств вируса гриппа, против которой эффективны ингибиторы занамивир (5-(ацетиламино)-4-[(аминоиминометил)-амино]-2,6-ангидро-3,4,5-тридезокси-D-глицеро-D-галактонон-2-еноновая кислота), озельтамивир (Тамифлю™) [12] и перамивир ((1S,2S,3S,4R)-3-[(1S)-1-ацетамидо-2-этил-бутил]-4-(диаминометилиденамино)-2-гидрокси-циклопентан-1-карбоновая кислота) [13]. Первичная структура NA является низкоконсервативной, т.е. быстро изменяется в процессе эволюции вируса гриппа, что приводит к высокой вероятности появления штаммов, резистентных к действию ингибиторов NA [14]. В частности, имеются сведения о возникновении устойчивых штаммов пандемического вируса гриппа A(H1N1)pdm09 [15]. Серьезным недостатком указанных препаратов является их частичная или полная непригодность для интраназального введения. Так, занамивир выпускается в виде дискхалера – системы для вдыхания порошка [16], озельтамивир – в виде таблеток, а перамивир – в виде раствора для внутривенного применения [13].
И наконец, третьей мишенью является полимераза RdRP как фермент, катализирующий синтез вирусной РНК. RdRP является, с одной стороны, высококонсервативной среди вирусов гриппа А всех подтипов, с другой – жизненно необходимым компонентом для осуществления цикла вирусной репликации в клетке. В настоящее время в ряде работ продемонстрирована высокая противовирусная активность олигопептидов, гомологичных функционально значимым последовательностям субъединицы РВ1, конкурирующих за сайты связывания ее с другими субъединицами полимеразного комплекса. Так, известен олигопептид последовательности 531-540 белка полимеразного комплекса РВ1 вируса гриппа, имеющий аминокислотный состав H-Lys-Asn-Asn-Met-Ile-Asn-Asn-Asp-Leu-Gly-OH (KNNMINNDLG), ингибирующий репликацию вирусов. Установлено, что указанный полипептид обладает выраженной селективностью в отношении вирусов гриппа разных подтипов – H1 и Н5. Результаты исследований на модели экспериментальной гриппозной инфекции, вызванной вирусами гриппа человека A(H1N1) и гриппа птиц A(H5N2) (в культуре клеток MDCK), показали, что указанный пептид проявляет достаточно высокую антивирусную активность в отношении вируса гриппа человека A(H1N1) (50 % эффективная доза препарата /ED50/ составила 28,6 мкг/мл; индекс селективности /SI/ – 43,0) и несколько меньшую активность в отношении вируса гриппа птиц A(H5N2) (ED50 – 35,3 мкг/мл; SI – 34,8) при значительно меньшем (в 20 раз) уровне токсичности по сравнению препаратом ремантадином (50 % цитотоксическая концентрация /CTD50/ – 1229 мкг/мл) [17].
Наиболее близким к заявленной композиции является олигопептид последовательности 6-14 белка полимеразного комплекса РВ1 вируса гриппа, ацетилированный по N-концевой аминогруппе и амидированный по С-концевой карбоксильной группе формулы Ac-Thr-Leu-Leu-Phe-Leu-Lys-Val-Pro-Ala-NH2 (Ac-TLLFLKVPA-NH2) [18] (далее по тексту – «Пептид»). Испытания, проведенные на культуре клеток MDCK в отношении вируса гриппа A/California/07/2009 (H1N1)pdm09, показали, что Пептид проявляет относительно высокую противовирусную активность (CTD50 – 477 мкМ; ED50 – 4,3 мкМ; SI – 111). Кроме того, показано, что пептид обладает противовирусной активностью, в том числе и в отношении штаммов подтипов H5 и H3. Выявлено, что Пептид способен в присутствии ДМСО проникать через плазматическую мембрану клетки, а также что основной механизм его активности не связан с инактивацией внеклеточного вируса. Предполагается, что противовирусная активность Пептида реализуется путем нарушения сборки комплекса РНК-полимеразы, возможно, за счет связывания Пептида с белком РА, то есть ингибирование вируса гриппа происходит на ранних стадиях вирусной репродукции.
Общим недостатком олигопептидов полимеразного комплекса, в том числе пептида-аналога [17] и пептида-прототипа [18], является их низкая растворимость в воде и водных растворах солей, что продемонстрировано, в частности для олигопептида последовательности 6-13 белка полимеразного комплекса РВ1 [19]. Экспериментально установлено, что молекулы пептида склонны к взаимодействию с образованием амилоидоподобных фибрилл, присутствие которых значимо снижает долю растворенного пептида в растворе. Это существенно затрудняет приготовление пригодной для интраназального применения у человека и животных лекарственной формы препарата, в которой бы достигалась концентрация, необходимая и достаточная для эффективного терапевтического использования. На настоящий момент сведения о подобной лекарственной форме на основе пептидов полимеразного комплекса РВ-1 (в том числе, пептида-аналога [17] и пептида-прототипа [18]) отсутствуют.
Известно применение ДМСО в медицине как самостоятельно в виде раствора, в частности в качестве противовоспалительного средства местного применения (Димексид), регистрационное удостоверение ЛП-004005, ОАО «Кемеровская фармацевтическая фабрика», Россия), так и в качестве фармацевтически приемлемого носителя в составе лекарственных средств, в частности для лечения кожи или ткани слизистой оболочки млекопитающего [20]. Установлено, что в присутствии ДМСО, как правило, значительно повышается транспорт многих веществ, в том числе лекарственных, через биологические мембраны. При подобной ускоренной доставке нередко меняется не только фармакокинетика лекарств, но может иметь место потенцирование действия препарата. Благодаря этим качествам, сочетающимся с биологической безвредностью, димексид получил широкое распространение в технологии различных лекарственных форм [21]. Способность ДМСО повышать растворимость гидрофобных веществ в воде обусловливает его использование в качестве компонента, обеспечивающего растворимость труднорастворимых субстанций, а также стабильность растворенных форм этих субстанций [22]. Так, введение в состав лекарственной композиции ДМСО в концентрации 20-25 мас. % обеспечивает лучшее растворение метисазона и способствует проникновению лекарственного средства в инфицированную вирусом клетку [10]. В евразийском патенте № 011863 [23] описан стабилизирующий эффект ДМСО в отношении ряда температурно-чувствительных пептидов (гастрина, холецистокинина, секретина), который выражается в предотвращении разложения этих пептидов и сохранении их активности в образцах млекопитающих (в сыворотке и плазме) и проявляется в диапазоне концентраций от 1 до 10 об.%, преимущественно 4-6 об.%.
Известно использование этанола в медицине как в виде основного действующего вещества различных кожных антисептиков [24], так и в качестве одного из компонентов различных растворителей [25, 26]. Так, известно использование бинарного растворителя этанол–вода для растворения гидрофобных пептидов, а именно растворения DL-a-аланил-Р-аланина [25]. При этом любые изменения количественного состава данного растворителя влияют на его взаимодействия с пептидом. Кроме того, известно, что этанол повышает растворимость в составе трехкомпонентных растворителей, в частности растворимость мелоксикама в растворителе вода–N–метилпирролидон–этанол в сравнении с данными по растворимости мелоксикама в двухкомпонентной системе вода–N-метилпирролидон [26]. Также известно влияние этанола на повышения проницаемости мембран [27]. Однако в общедоступной литературе отсутствуют сведения о взаимодействиях в многокомпонентной системе Пептид–ДМСО–этанол–вода, которые в заявленных диапазонах концентраций обеспечивали бы повышение растворимости Пептида при стабилизации его активной формы в растворе в динамике.
Таким образом, при анализе патентной и научно-технической документации, определяющей уровень техники в области, к которой относится изобретение, не выявлено лекарственных средств, которым были бы присущи все признаки заявленного решения. Кроме того, при анализе указанных документов не обнаружено известности влияния признаков предложенного решения на технический результат, достигаемый при его реализации. Таким образом, заявленное изобретение соответствует, по мнению Заявителя, условиям патентоспособности «новизна» и «изобретательский уровень».
Технической проблемой является необходимость разработки лекарственного средства, обладающего высокой активностью в отношении устойчивых к действию существующих препаратов штаммов вируса гриппа А, а также вновь возникающих штаммов, пригодного для интраназального применения при лечении гриппа у людей и животных.
Техническим результатом является повышение растворимости Пептида и агрегативной стабильности раствора для достижения и сохранения в динамике концентрации активного компонента, необходимой и достаточной для эффективного интраназального применения.
Технический результат достигается тем, что композиция на основе пептида, подавляющего репликацию вируса гриппа А, последовательности 6-14 белка полимеразного комплекса РВ1 вируса гриппа, ацетилированного по N-концу и амидированного по С-концу, формулы Ac-Thr-Leu-Leu-Phe-Leu-Lys-Val-Pro-Ala-NH2 для лечения гриппа, согласно изобретению дополнительно содержит диметилсульфоксид, этанол и воду, при следующих соотношениях компонентов, мас. %:
|
Выбор оптимальных параметров заявленной композиции включал: подбор оптимального растворителя (состоящего из одного или несколько компонентов), выбор и обоснование оптимальных диапазонов концентраций растворителя (из одного или нескольких компонентов), а также выбор и обоснование терапевтически эффективной концентрации Пептида.
Используемый в экспериментах Пептид синтезировали, как это описано в работе [18], твердофазным методом на 2-Cl-тритилхлоридной смоле (емкостью 1,55 ммоль/г) по Fmoc-/t-Bu стратегии с использованием стандартных методик последовательного наращивания цепи. Очистку Пептида проводили методом препаративной обращеннофазной высокоэффективной жидкостной хроматографии (ВЭЖХ) в градиентном режиме в системе ацетонитрил (AN)–вода–трифторуксусная кислота (TFA). Фракции, содержащие целевой компонент, после объединения и концентрирования в вакууме подвергали лиофилизации. Чистота Пептида составила не менее 95 %. ВЭЖХ проводили в условиях: жидкостный хроматограф с УФ-детектором, колонка из нержавеющей стали с размерами 2,0х150 мм, заполненная сорбентом октадецилсиликагелем Triart C18 (YMC) с размером зерна 3 мк, в градиенте подвижной фазы: фаза А: 1,0 мл TFA в 1000 мл воды очищенной; фаза Б: 1,0 мл TFA в 1000 мл AN при скорости 0,2 мл/мин. Хроматографировали 5,0 мкл испытуемого раствора препарата. Структура Пептида подтверждена масс-спектрометрически (MALDI-TOF).
В результате предварительных исследований авторов заявленного решения в экспериментах на культуре клеток MDCK в отношении вируса гриппа A/California/07/2009 (H1N1)pdm09 было установлено, что эффективная концентрация Пептида при его применении in vitro составляла 50 мкг/мл (ED50 – 4,3 мкМ) [18].
Учитывая, что Пептид практически нерастворим в воде и в водных растворах солей, были проведены исследования в отношении двух потенциальных растворителей – диметилсульфоксида (Димексид, концентрат для приготовления растворов для наружного применения, регистрационное удостоверение ЛСР-001906/08-180308, 2017, ООО «Тульская фармацевтическая фабрика», Россия) и этанола (Спирт этиловый медицинский ректифицированный, ФС.2.1.0036.15; ООО «Росбио», Россия). Для разбавления использовали воду очищенную (по ФС.2.2.00.20.15).
Разведение веществ проводили в поддерживающей питательной среде для клеток MDCK. Состав поддерживающей среды: на 100 мл среды альфа-МЕМ (Питательная среда альфа-МЕМ с глутамином, ООО «Биолот», Санкт-Петербург) вносили 1,0 мл раствора антибиотика ципрофлоксацина («Синтез», Курган) и 0,1 мл раствора TPCK-трипсина (конечная концентрация в среде 2,0 мкг/мл, кат. номер Т1426, «Sigma», Германия).
Готовили серию разведений препаратов (10,0; 7,5; 5,0 и 2,5 об. %). Опыт проводили в 3 повторах для каждой концентрации.
Односуточную культуру клеток MDCK-FR, выращенную на 96-луночных планшетах («Orange Scientific», Китай), концентрация клеток 6Ч104/лунку планшета, проверяли визуально в инвертированном микроскопе на целостность монослоя. В работу отбирали планшеты, где сомкнутость клеток составляла 95 % и выше. Планшеты двукратно отмывали теплой средой альфа-МЕМ, не содержащей сыворотки, после чего на клетки монослоя в планшете вносили разведения биназы в соответствующей концентрации в объеме 100 мкл в каждую лунку в 3 повторах на каждую тестируемую концентрацию. Планшеты инкубировали 24 ч при температуре 37°С в присутствии 5 % СО2. На конечном сроке проводили оценку результата визуально в инвертированном микроскопе, оценивая состояние монослоя в присутствии разных концентраций препарата по сравнению с клетками в контрольных лунках.
На первом этапе для определения рабочего диапазона параметров раствора Пептида в ДМСО оценивали проникновение Пептида в клетки и цитотоксичность при различных концентрациях ДМСО. Для исследования проникновения Пептида использовали FITC–модифицированную производную этого пептида. Учитывая, что противовирусные свойства данного соединения не отличались от свойств Пептида, считали, что кинетика проникновения модельного флуоресцентно меченного соединения не отличается от кинетики проникновения Пептида в клетки на модели клеточной культуры А549. Растворы пептида добавляли к клеточной культуре A549 до конечной концентрации по Пептиду 50 мкг/мл. Проникновение Пептида в клетку детектировали при помощи конфокальной микроскопии, фиксируя флуоресценцию FITC-красителя внутри клетки. В экспериментах использовали растворы FITC-меченого Пептида в концентрации 50 мкг/мл в водном растворе, содержащем 2,5; 5,0; 7,5 и 10,0 об. % ДМСО.
О цитотоксичности растворов ДМСО судили по результатам микротетразолиевого теста (МТТ) [28]. МТТ-тест основан на восстановлении МТТ (желтый водорастворимый тетразолиевый краситель) под действием дегидрогеназ живых клеток с образованием голубых кристаллов формазана, количество которого измеряется спектрофотометрически. Раствор МТТ готовили в физиологическом растворе в концентрации 0,5 мг/мл. Перед внесением раствора МТТ клетки промывали 0,1 мл физиологического раствора. Далее вносили 0,1 мл раствора МТТ в каждую лунку. После 1,5 ч контакта МТТ при З7°С при концентрации СО2 5 % с клетками лунки промывали и заливали 0,1 мл этилового спирта 96 %, после чего оптическую плотность в лунках измеряли на ридере Victor 2 1440 (Perkin Elmer, США) при длине волны 535 нм. Основываясь на полученных данных рассчитывали ЦТД50 (дозу препарата в лунке, при которой погибает 50 % клеток).
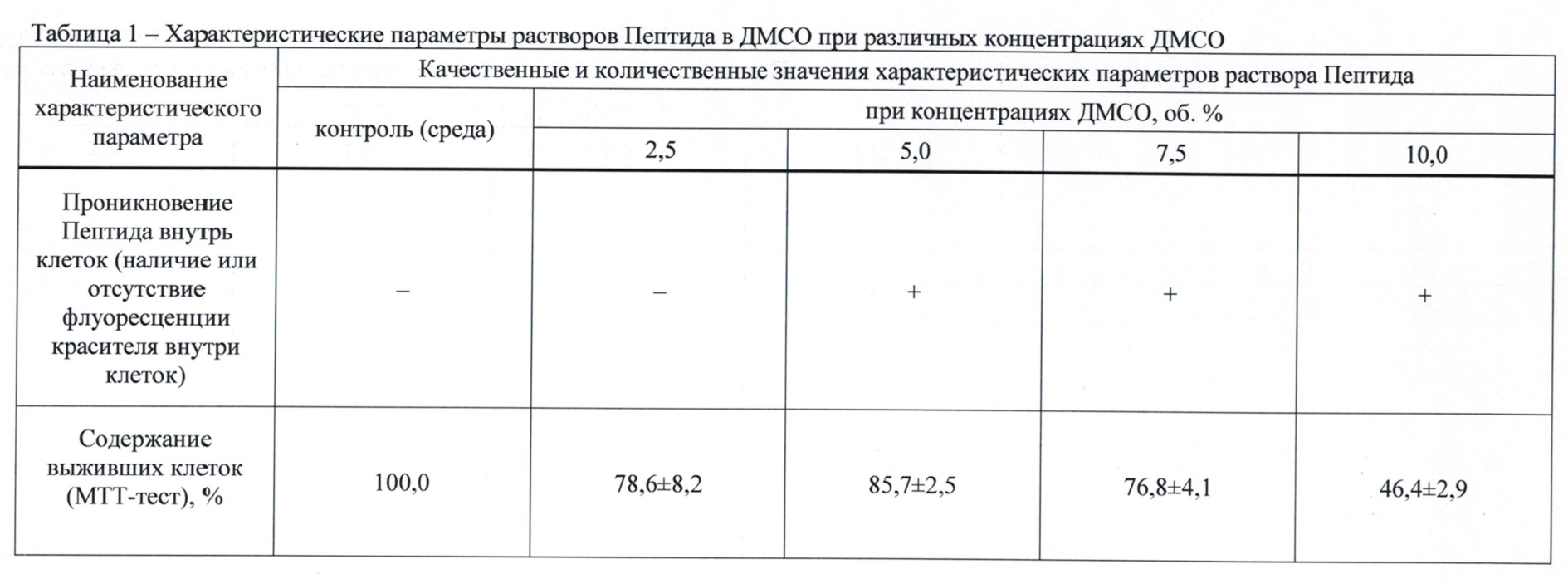
В табл. 1 представлены результаты качественной оценки проникновения Пептида в клетку и процентное содержание клеток, инкубировавшихся в присутствии различных концентраций ДМСО, определенное по оптической плотности раствора формазана, образовавшегося под действием дыхательных ферментов клеток, получавших вещества.
Данные табл. 1 показывают, что при концентрации ДМСО в исходном растворе менее 5,0 об. %, а также при его отсутствии при проведении конфокальной микроскопии наблюдали гранулы нерастворенного пептида, сорбировавшегося на поверхности клеток, при отсутствии характерной флуоресценции красителя внутри клеток. В диапазоне концентраций 5,0-7,5 об. % по ДМСО отмечали характерную флуоресценцию красителя внутри клеток, свидетельствовавшую о проникновении Пептида. При концентрации 10,0 об. % также выявлено проникновение красителя внутрь клеток. Однако значимое снижение (в 1,7 раза) содержания выживших клеток по сравнению с аналогичными показателями диапазона 2,5-7,5 об. % (достоверно не различавшихся в пределах диапазона), свидетельствовало о токсичности ДМСО в концентрации 10,0 об. %. Таким образом, был определен рабочий диапазон концентраций ДМСО в композиции (5-10 %), в котором имело место проникновение пептида внутрь клеток при отсутствии токсического действия ДМСО.
На втором этапе при переходе от исследований на культуре клеток к интраназальному применению у экспериментальных животных (мышей) принимали во внимание следующие факторы. Авторами изобретения было экспериментально установлено, что максимально допустимый суммарный объем раствора для разового введения в нос мыши составил 50 мкл (по 25 мкл в каждую ноздрю). На основании проведенных опытов оптимальный суммарный объем раствора для однократного введения в нос был определен как 30 мкл (по 15 мкл в каждую ноздрю). Первоначальную расчетную эффективную дозу для скрининга на мышах вычисляли с использованием однокамерной модели фармакокинетики лекарственного препарата при однократном введении [29]. Расчетная эффективная доза для мыши при оптимальном суммарном объеме введения составила 20 мг/мл, что значимо превышает эффективную дозу, установленную на культуре клеток.
В связи с тем, что гидрофобный Пептид склонен к образованию фибрилл молекулярных размеров, то оценить подлинную растворимость Пептида затруднительно. Количество растворенного Пептида при различных концентрациях (2,5-10,0 об. %) ДМСО оценивали, проводя серию последовательных разведений препаратов.
При этом приготовление образцов, не содержащих ДМСО, осуществляли следующим образом. Для контроля полной растворимости Пептида: навеску Пептида растворили в 40 % AN до концентрации 5,0 мг/мл; затем выполнили серию последовательных разбавлений до концентраций 2,0; 1,0; 0,5; 0,2; 0,1 мг/мл раствором 40 % AN. Для контроля растворимости Пептида без ДМСО: навеску пептида растворили в H2O до концентрации 5,0 мг/мл. Затем выполнили серию последовательных разбавлений полученной суспензии до концентраций 2,0; 1,0; 0,5; 0,2; 0,1 мг/мл в H2O.
Приготовление образцов, содержащих ДМСО, осуществляли следующим образом. Навеску Пептида растворили в 100 % ДМСО. Приготовили исходный раствор с концентрацией 10 мг/мл, также из него получили растворы 5,0; 2,0 и 1,0 мг/мл в 100 % ДМСО. В пробирки поместили необходимое количество мкл раствора Пептида, добавили 100 % ДМСО (для достижения необходимой итоговой концентрации), а затем H2O до конечного объема 100 мкл.
Все подготовленные образцы (содержащие и не содержащие ДМСО) центрифугировали при 20000g, 40 мин, +4°C для осаждения нерастворившегося Пептида. Надосадочную жидкость отобрали и провели обращенно-фазную хроматографию (колонка – YMC-Triart C18, 2,0х150 мм, размер гранул 3 мк; подвижная фаза А – 0,1 % TFA в деионизованной воде, подвижная фаза В – 0,1 % TFA в AN; скорость подвижной фазы – 0,2 мл/мин; элюция линейным градиентом 10-80 % «В» в течение 15 мин; детекция по длине волны 214 нм; объем вводимой пробы – 10 мкл). Пики, соответствующие времени выхода Пептида (время удерживания Rt составило /13,7±0,03/ мин), интегрировали. Сравнили площади пиков Пептида в растворителе и в AN, считая растворимость Пептида в AN равной 100 %.
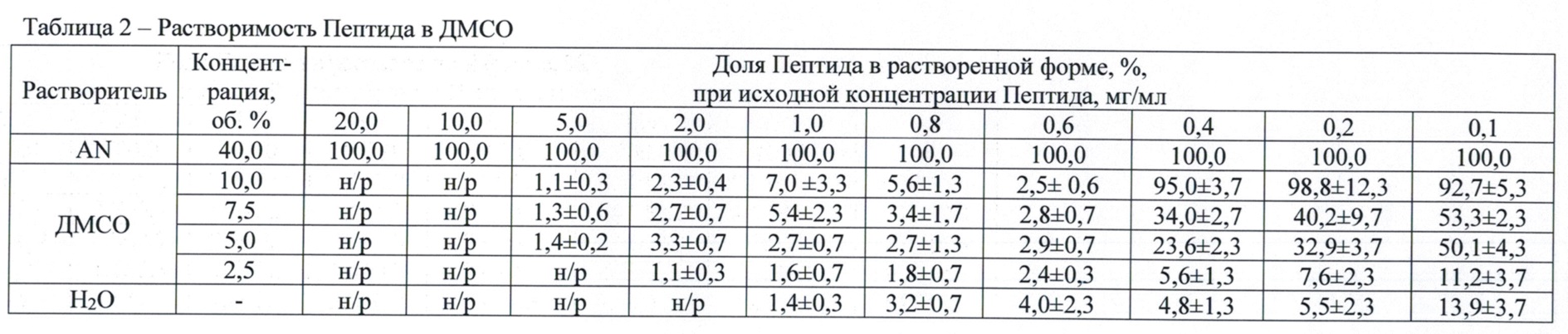
Доли Пептида в растворенной форме в зависимости от концентрации исходного раствора и концентрации ДМСО представлены в табл. 2.
Из данных табл. 2 видно, что в рабочем диапазоне концентраций ДМСО (5,0-7,5 об. %) наблюдаются низкие значения исследуемого параметра – доля растворенного Пептида превышает 50 % только при исходной концентрации Пептида 0,1 мг/мл. Увеличение исходной концентрации Пептида свыше 0,4 об. % приводит к резкому уменьшению исследуемого параметра (2,9-5,4 %), что свидетельствует о низкой растворимости Пептида в ДМСО в рабочем диапазоне концентраций ДМСО. Таким образом, при использовании в качестве растворителя только ДМСО достижение эффективной концентрации, которая оказалась бы необходимой и достаточной для эффективного интраназального применения не представляется возможным.
На основании полученных данных была испытана трехкомпонентная система растворителей – ДМСО+этанол+вода. На третьем этапе была исследована цитотоксичность растворителя, содержащего два активных компонента, при варьировании концентрации этанола. Цитотоксичность растворов ДМСО+этанол+вода оценивали с помощью МТТ-теста (по количеству выживших клеток) аналогично описанному на первом этапе.
Результаты представлены в табл. 3.
Данные табл. 3 свидетельствуют о том, что начиная с концентрации 7,5 об. %, проявляются токсические эффекты этанола – количество выживших клеток достоверно снижается с 98 до 84 %.
Количество растворенного Пептида при концентрациях ДМСО 5,0 об. % и 7,5 об. % и концентрации этанола 5,0 об. % оценивали аналогично описанному на втором этапе, проводя последовательно растворение в смеси растворителей при каждой определенной концентрации Пептида. При этом для контроля полной растворимости Пептида в AN образец приготовили, как это описано на втором этапе. Для приготовления растворов Пептида в воде навеску пептида растворили в 5,0 об. % этаноле, 5,0 об. % ДМСО до концентрации 5,0 мг/мл, затем выполнили серию последовательных разбавлений полученной суспензии до концентраций 2,0; 1,0; 0,5; 0,2; 0,1 мг/мл. Аналогичным образом готовили растворы Пептида в 5,0 об. % этаноле, 7,5 об. % ДМСО в воде.
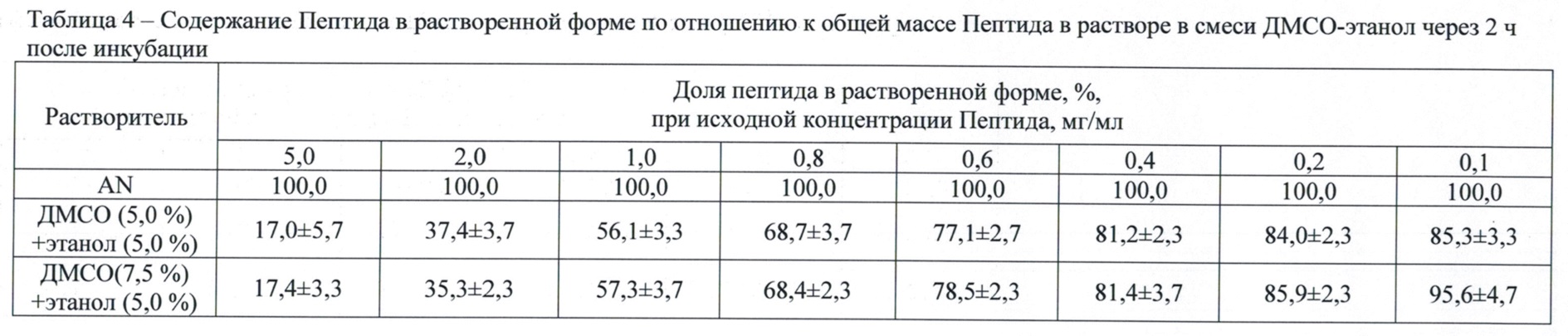
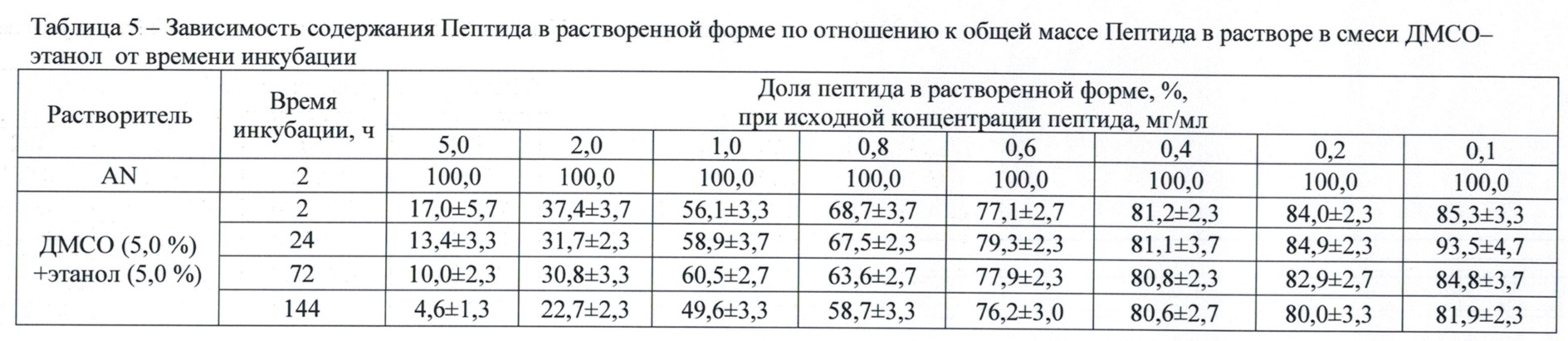
Через 2, 24, 72 и 144 ч после приготовления растворов, подвергавшихся инкубации при комнатной температуре, образцы перемешивали и отбирали из них по 100 мкл, центрифугировали и проводили обращенно-фазную хроматографию, как описано во втором этапе. Доли Пептида в растворенной форме в зависимости от концентрации исходного раствора и концентрации смеси ДМСО-этанол через 2 ч после инкубации представлены в табл. 4, а зависимость содержания Пептида в растворенной форме по отношению к его общей массе в растворе в смеси ДМСО–этанол от времени инкубации выборочно приведена в табл. 5.
Данные табл. 4 показывают, что применение трех-компонентной системы растворителей – ДМСО (5,0-7,5 об. %; 5,5-8,3 мас. %)–этанол (5,0 об. %; 3,9 мас. %)–вода повышает (в сравнении с двух-компонентной системой растворителей ДМСО-вода в тех же концентрациях) растворимость Пептида, увеличивая значение верхней границы диапазона рабочих исходных концентраций до 0,8 мг/мл (доля растворенного Пептида 68,4-68,7 мас. %). Из табл. 5 видно, что при исходных концентрациях Пептида свыше 1,0 мас. % имеет место достоверное снижение доли растворенного Пептида при инкубации свыше 72 ч, тогда как в диапазоне исходных концентраций Пептида 0,4-0,8 мас. % раствор сохраняет стабильность в течение периода наблюдения (144 ч). Это позволяет рассматривать вышеуказанные параметры как оптимальные для лекарственной композиции для интраназального применения.
Следует также отметить, что растворы Пептида в 5,0 об. % и 7,5 об. % ДМСО при содержании этанола менее 5,0 об. % были неустойчивы и количество растворенного пептида через 2 ч после растворения была ниже предела детектирования используемого метода (в таблицах не приведено).
Заявленная композиция поясняется примером.
Пример
Определяли противовирусную активность композиции при оптимальных концентрациях ДМСО и этанола в смеси, варьируя при этом концентрацию Пептида.
Мышей Balb/c линии (самки) массой 16-20 г (возраст 5-7 недель) получали из питомника «Столбовая» (Московская обл.). Животных содержали в стандартных условиях, соответствующих требованиям действующих нормативных и методических документов как РФ, так и международных. В период акклиматизации и эксперимента мышей размещали в поликарбонатных клетках (BENEX а.с., Чешская республика, тип Т3А, S=1200 см2), группами по 21 особь. Кормление животных проводили в соответствии с требованиями действующих в РФ нормативно-методических документов с использованием гранулированного корма для содержания мышей (ООО «Лабораторкорм», Москва) и воды, очищенной и нормированной в соответствии с требованиями действующих в РФ нормативных документов. Животных содержали в контролируемых условиях окружающей среды (температуры 18-22°C и относительной влажности воздуха 50-70 %), NH3=0,001 мг/м3, CO2=0,1 %. Световой режим составлял 12 ч света и 12 ч темноты. Никаких существенных отклонений этих параметров в период акклиматизации и в ходе эксперимента не зафиксировано. Лабораторных животных до начала исследования содержали 7 дней для адаптации при групповом содержании в клетках. Во время этого периода у животных каждый день контролировали клиническое состояние путем визуального осмотра. Животных с обнаруженными в ходе осмотра отклонениями в экспериментальные группы не включали.
В исследовании использовали штамм вируса гриппа A/California/07/09 (H1N1)pdm09, адаптированный к мышам, из коллекции ФГБУ «НИИ гриппа им. А.А. Смородинцева» Минздрава России.
Исследуемые образцы содержащей Пептид лекарственной формы композиции представляли собой растворы в смеси ДМСО (5,0 об. %) этанол (5,0 об. %) (при концентрации Пептида 1,0 мг/мл, 0,8 мг/мл, 0,6 мг/мл, 0,4 мг/мл и 0.2 мг/мл). В качестве базового препарата (препарата сравнения) был применен Осельтамивира фосфат («Тамифлю») («LaRoche», Швейцария), лекарственная форма: капсулы для приема внутрь по 75 мг.
Исследуемые образцы были приготовлены следующим образом. Навески Пептида (0,2; 0,4; 0,6; 0,8; 1,0 мг) помещали в пробирку типа Eppendorf объёмом 1,5 мл, затем добавляли водный раствор 5,0 об. % ДМСО 5,0 об. % этанола. После добавления раствор тщательно пипетировали, а затем подвергали перемешиванию при комнатной температуре (25°С) на перемешивателе ELMI Intellimixer RM1L в режиме С1 30 об/мин в течение 60 мин. Полученные растворы затем передавали в виварий для интраназального введения мышам. Хранение растворов между введениями в течение недели осуществляли при +4°С.
Исследуемые образцы композиции вводили животным интраназально раз в день в объеме 0,01 мл по лечебной (через 1, 2, 3 и 4 дня после инфицирования) схеме. Базовый препарат «Тамифлю» применяли для контроля специфичности патологического процесса. Навеску препарата растирали в фарфоровой ступке с 50 мкл Tween-20, добавляли необходимый объем физиологического раствора и вводили животным перорально при помощи желудочного зонда один раз в сутки в объеме 0,2 мл из расчета 20 мг/кг/сут по лечебной схеме (через 1, 2, 3 и 4 дня после инфицирования). Животным из группы отрицательного контроля вводили растворитель (смесь 5,0 % ДМСО и 5,0 % этанол) в том же объеме и по той же схеме, что и исследуемый пептид. Мышей заражали интраназально под легким эфирным наркозом вирусом в дозе 4,1Ч105 ТИД50 (50 % тканевая ингибирующая доза) на мышь в объеме 15 мкл, что составило 1,2 ЛД50 (21 мышь в группе). На 3-й день после заражения по 6 животных из каждой группы подвергали эвтаназии, вскрывали и изолировали их легкие и ткань носовой полости. Легкие и носовые полости гомогенизировали и использовали для титрования вируса.
Образцы ткани, изолированные на третий день после заражения у 5 животных из каждой группы, гомогенизировали в физиологическом растворе с помощью прибора TissueLyserII («Qiagen», США) и определяли в гомогенатах инфекционную активность вируса, как описано ниже.
Для оценки уровня репродукции вируса в образцах ткани животных проводили титрование его инфекционной активности в культуре клеток MDCK (4 лунки на каждое разведение). С этой целью клетки MDCK (105 кл./мл) рассевали в лунки культуральных планшетов с плоским дном «Corning», США, (Кат.№ 3585) и выдерживали в течение 24 ч в CO2-инкубаторе при 36°С в атмосфере 5 % CO2 до формирования монослоя. Клетки промывали 5 мин средой MEM (ООО «Биолот», Санкт-Петербург) и далее использовали для культивирования вируса.
Из гомогената образцов ткани готовили серию 10-кратных разведений (10-1 – 10-7) на среде AlphaМЕМ с добавлением трипсина (1 мкг/мл) и 20 мкг/мл ципрофлоксацина и вносили их в лунки планшета с клетками MDCK. Планшеты инкубировали в течение 72 ч при 36°С в атмосфере 5 % СО2. По окончании срока инкубации культуральную жидкость (100 мкл на лунку) переносили в лунки планшетов с круглым дном для иммунологических реакций («Медполимер», Санкт-Петербург) и добавляли по 100 мкл на лунку 1 % суспензии куриных эритроцитов в физиологическом растворе. Планшеты выдерживали 1 ч при 20°С, после чего визуально оценивали наличие или отсутствие гемагглютинации в лунках. Титр вируса рассчитывали по методу Рида и Менча и выражали в ТИД50 на 100 мкл объема.
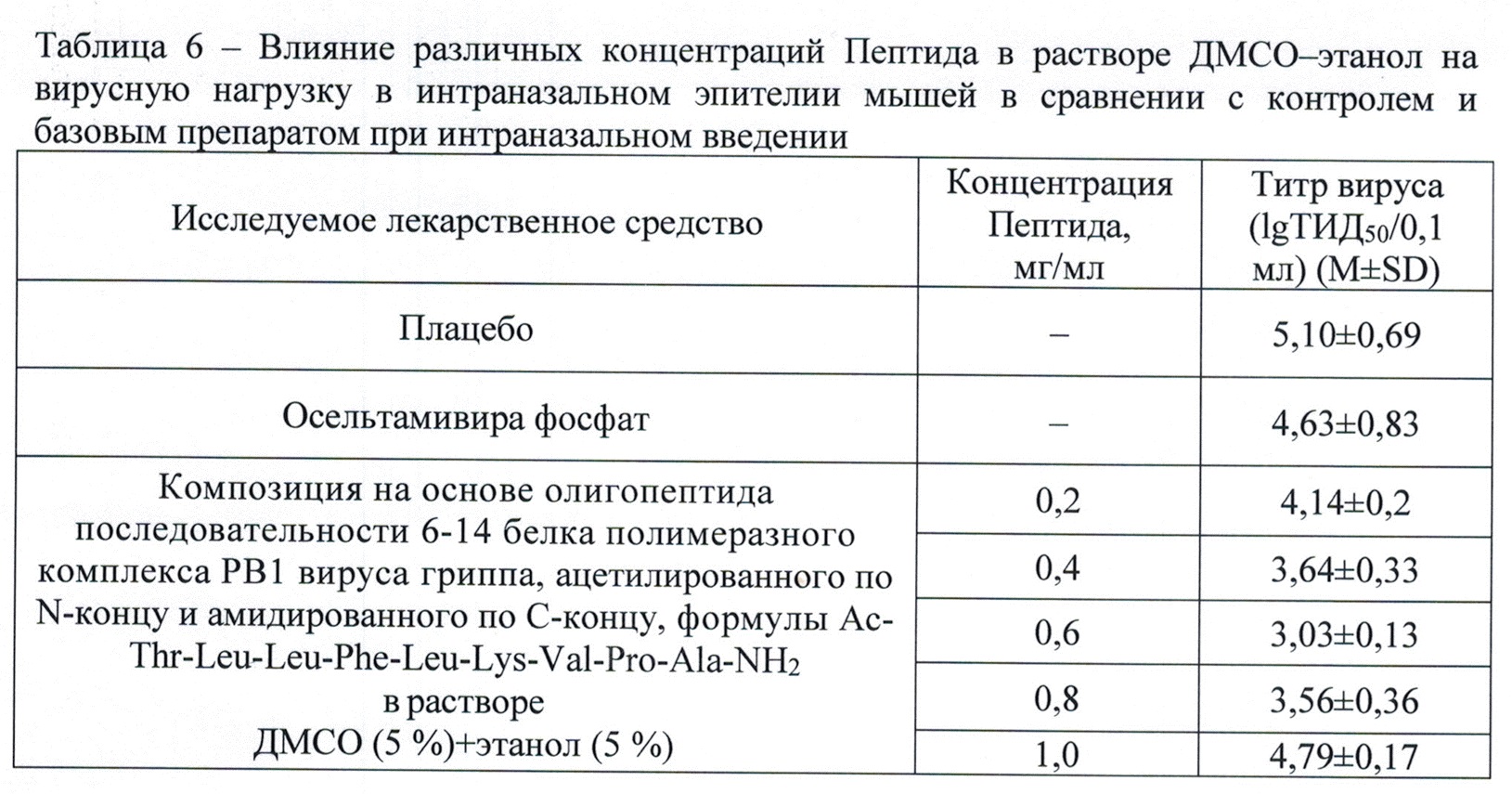
Результаты исследований противовирусной активности заявляемой композиции представлены в табл. 6.
Из данных табл. 6 следует, что при интраназальном применении Пептид (в концентрациях 0,4-0,8 мг/мл) в растворителе 5,0 об. % этанол 5,0 об. % ДМСО действует более эффективно, чем препарат сравнения озельтамивира фосфат.
Таким образом, проведенная оценка противовирусной активности заявленной лекарственной композиции показала её высокую эффективность при интраназальном применении у мышей.

Источники информации
1. V. N. Petrova and C. A. Russell, “The evolution of seasonal influenza viruses”, Nat. Rev. Microbiol., vol. 16, no. 1, pp. 47–60, 2018.
2. T. M. Sokolova, A. N. Shuvalov, V. V Poloskov, I. M. Shapoval, and M. P. Kostinov, “Grippol, Vaxigrip and influvac vaccines--inductors of innate and adaptive immunity factor genes in human blood cells.”, Zh. Mikrobiol. Epidemiol. Immunobiol., no. 5, pp. 37–43, Jan.
3. S. Chang et al., “Cryo-EM structure of influenza virus RNA polymerase complex at 4.3 Е resolution”, Mol. Cell, vol. 57, no. 5, pp. 925–35, Mar. 2015.
4. A. V Vasin, O. A. Temkina, V. V Egorov, S. A. Klotchenko, M. A. Plotnikova, and O. I. Kiselev, “Molecular mechanisms enhancing the proteome of influenza A viruses: an overview of recently discovered proteins,” Virus Res., vol. 185, pp. 53–63, Jun. 2014.
5. J. Romanova et al., “Preclinical evaluation of a replication-deficient intranasal DeltaNS1 H5N1 influenza vaccine,” PLoS One, vol. 4, no. 6, p. e5984, Jun. 2009.
6. О. И. Киселев, “Иммуносупрессия при беременности и риски при вирусных инфекциях”, vol. 85, no. 6, 2013.
7. A. Moscona, “Global Transmission of Oseltamivir-Resistant Influenza”, N. Engl. J. Med., vol. 360, no. 10, pp. 953–956, 2009.
8. J. P. Wong et al., “Aerosol and nasal delivery of vaccines and antiviral drugs against seasonal and pandemic influenza”, Expert Rev. Respir. Med., vol. 4, no. 2, pp. 171–177, 2010.
9. Рекомбинантный человеческий белок СС10 для лечения гриппа: патент 2554745, Рос. Федерация. № 2012120671; заявл. 13.10.10; опубл. 27.06.15, Бюл. № 18.
10. Лекарственное средство для профилактики и лечения вирусных болезней животных: патент 2168988, Рос. Федерация. № 99116456; заявл. 29.07.99; опубл. 10.06.01, Бюл. № 17.
11. C. Scholtissek, G. Quack, H. D. Klenk, and R. G. Webster, “How to overcome resistance of influenza A viruses against adamantane derivatives”, Antiviral Res., vol. 37, no. 2, pp. 83–95, 1998.
12. A. Moscona, “Neuraminidase Inhibitors for Influenz”, N. Engl. J. Med., vol. 353, no. 13, pp. 1363–1373, Sep. 2005.
13. C. E. Mancuso, M. P. Gabay, L. M. Steinke, and S. J. VanOsdol, “Peramivir: An intravenous neuraminidase inhibitor for the treatment of 2009 H1N1 influenza”, Ann. Pharmacother., vol. 44, no. 7–8, pp. 1240–1249, 2010.
14. T. C. M. Li, M. C. W. Chan, and N. Lee, “Clinical Implications of Antiviral Resistance in Influenza”, Viruses, vol. 7, no. 9, pp. 4929–4944, Jan. 2015.
15. Y. Abed and G. Boivin, “A Review of Clinical Influenza A and B Infections With Reduced Susceptibility to Both Oseltamivir and Zanamivir”, Open Forum Infect. Dis., vol. 4, no. 3, pp. 1–10, 2017.
16. M. Elliott, “Zanamivir: From drug design to the clinic”, Philos. Trans. R. Soc. B Biol. Sci., vol. 356, no. 1416, pp. 1885–1893, 2001.
17. Противовирусный пептид, подавляющий репликацию вируса гриппа: патент 2492178, Рос. Федерация. № 2012115794/10; заявл. 19.04.12; опубл.10.09.13, Бюл. № 25.
18. O. V Matusevich et al., “Synthesis and antiviral activity of PB1 component of the influenza A RNA polymerase peptide fragments”, Antiviral Res., vol. 113, pp. 4–10, Jan. 2015.
19. Y. A. Zabrodskaya et al., “The amyloidogenicity of the influenza virus PB1-derived peptide sheds light on its antiviral activity”, Biophys. Chem., vol. 234, pp. 16–23, 2018.
20. Короткие биоактивные пептиды для ускорения заживления ран: патент 2606753 Рос. Федерация. № 2015138925; заявл. 21.01.2014; опубл. 10.01.2017.
21. Григорович Н.А. и др. “Нанотехнологический лекарственный препарат – димексид. Химические, биологические свойства. Клиническое применение. Обзор литературы”, Н.А. Григорович, С.Ф. Дорофтиенко, Т.М. Григорович. URL: https://mductor.weebly.com/uploads/1/0/8/4/10843070/dmso.pdf (дата обращения 25.06.2018).
22. Гулякин И.Д. и др., “Основные методы повышения растворимости гидрофобных и труднорастворимых веществ”, Разработка и регистрация лекарственных средств, №2 (15). Фармконтракт, М, 52-59, 2016.
23. Применение диметилсульфоксида (ДМСО) для стабилизации гастрина, холецистокинина, секретина и способ стабилизации указанных пептидов: патент 011863, Евразийская патентная организация, № 200602088; заявл. 09.05.2005, опубл. 30.06.2009.
24. Волкова С.В. “Этиловый спирт как основное действующее вещество кожных антисептиков”, Поликлиника. –2008. –С.108.
25. Смирнов, В.И. “Энтальпии растворения DL–α–аланина в смесях вода–спирты при 298.15 К,” В.И. Смирнов, И.Н. Межевой, В.Г. Баделин, Журн. физ. хим. –2004. –Т.78, №2. – С. 280–283.
26. Ляпунов А.Н. “Исследование вязкости смешанных растворителей, состоящих из воды, этанола, n-метилпирролидона и растворимости в них мелоксикама”, А.Н. Ляпунов, Е.П. Безуглая, А.П. Краснопёрова, Научные ведомости Белгородского государственного университета. Серия Медицина. Фармация. –2015. –№ 22 (219). Выпуск 32 –С.138–145.
27. Михайлова Р.В. “Влияние этанола на образование внеклеточной и клеточно-связанной каталаз Penicillium Piceum БИМ F-371 Д”, Р.В. Михайлова, И.В. Мороз, А.Г. Лобанок, А. Радукан, Вестник национальной академии наук Беларуси. Серия биологических наук. –2013. –№1, – С. 43-47.
28. Киселев О.И. “Оценка метаболических показателей in vitro как модельная система тестирования цитотоксичности противовирусных препаратов”, О.И. Киселев, Е.М. Еропкина, Т.Д. Смирнова, М.Ю. Еропкин, Е.В. Ильинская, В.П. Сухинин, А.Р. Прочуханова, В.В. Зарубаев, Экспериментальная и клиническая фармакология. 2006. Т. 69. № 1. С. 65-70.29.
29. “Фармакология”, Под ред. проф. Р.Н. Аляутдина. - 4-е изд., перераб. и доп. - 2008. - 832 с. : ил.
Моноклональные антитела, специфичные к различным штаммам респираторно-синцитиального вируса
Моноклональные антитела, специфичные к различным штаммам вируса гриппа в
Способ диагностики возбудителей острых респираторных вирусных инфекций
Способ флуоресцентного мечения кднк вируса гриппа типа а
Способ лечения папилломавирусных инфекций, реализуемый через индукцию интерлейкина-18
Противовирусный пептид, подавляющий репликацию вируса гриппа
Штамм вируса гриппа а/гонконг/1/68/162/35 (h3n2)-универсальный донор внутренних генов для реассортантов и реассортантные штаммы а/спб/гк/09 (h1n1) и а/нк/astana/6:2/2010 (h5n1), полученные на его основе
Симметричные диимины на основе камфоры - ингибиторы репродукции вируса гриппа (штамм a/california/07/09 (h1n1)pdm09)
Способ получения 2-(1-адамантил)-5r-тетразолов, проявляющих активность против вируса гриппа а
Применение 1,7,7-триметилбицикло[2.2.1]гептан-2-илиден-аминоэтанола в качестве ингибитора репродукции вируса гриппа
N-адамантилбензотриазолы, проявляющие активность против вируса гриппа а, и способ их получения
Иминопроизводные камфоры - эффективные ингибиторы репродукции вируса гриппа (штамм a/california/07/09 (h1n1)pdm09)
Средство, представляющее собой амид глицирризиновой кислоты с 5-аминоурацилом, проявляющее противовирусную активность в отношении вируса гриппа a/h1n1

