Результат интеллектуальной деятельности: ФЕНИЛКАРБАМАТЫ И ИХ ПРИМЕНЕНИЯ В КАЧЕСТВЕ ИНГИБИТОРОВ ФЕРМЕНТА ГИДРОЛАЗЫ АМИДОВ ЖИРНЫХ КИСЛОТ (FAAH) И МОДУЛЯТОРОВ D3 ДОПАМИНОВОГО РЕЦЕПТОРА (D3DR)
Вид РИД
Изобретение
ПЕРЕКРЕСТНАЯ ССЫЛКА НА РОДСТВЕННЫЕ ЗАЯВКИ
По настоящей заявке испрашивается приоритет на основании временной заявки США с серийным номером 61/847807, поданной 18 июля 2013 года, раскрытие которой включено в настоящую заявку посредством ссылки.
ЗАЯВЛЕНИЕ ОТНОСИТЕЛЬНО ПРАВ НА ИЗОБРЕТЕНИЯ, ВЫПОЛНЕННЫЕ НА ОСНОВАНИИ ФИНАНСИРУЕМОГО ИЗ ФЕДЕРАЛЬНОГО БЮДЖЕТА ИССЛЕДОВАНИЯ ИЛИ РАЗРАБОТКИ
Настоящее исследование было осуществлено, частично, с правительственной поддержкой на основании NIH Гранта R01 DA12413, присужденного Национальным Институтом Здравоохранения; правительство Соединенных Штатов имеет некоторые права на настоящее изобретение.
Область, к которой относится изобретение
Настоящее изобретение раскрывает направленные на несколько мишеней лиганды (MTDLs), которые одновременно являются ингибиторами фермента гидролазы амидов жирных кислот (FAAH) и модуляторами D3 допаминового рецептора (D3DR), способы их получения, включающие их композиции в качестве лекарственных средств и их терапевтическое применение для лечения патологий, состояний и расстройств, для которых может быть клинически полезным сочетание ингибирования фермента гидролазы амидов жирных кислот и модуляции D3 допаминового рецептора.
Предпосылки создания изобретения
Будучи ответственным за более пяти миллионов смертей каждый год, табакокурение является хроническим и разрушительным синдромом, который представляет собой одну из наиболее серьезных угроз здоровью в западных странах. Недавно проведенный анализ (Syed and Chaudhari, Nature Rev. Drug Discovery, 2013; 12: 97-98) установил, что в мире сушествует более 1,3 миллиарда курильщиков, определив распространенность табакозависимости среди взрослого населения около 33%. При том, что это вызывает длительное воздействие многих вредных веществ, содержащихся в сигаретном дыме, что с течением времени приводит к сердечно-сосудистым состояниям, легочным заболеваниям, раку и другим расстройствам, табакозависимость вызывается никотином. Никотин представляет собой психоактивный алкалоид, который проявляет свое действие, повышая уровень допамина в мезолимбокортикальной системе, специфической нервной цепи головного мозга, сильно связанной с вознаграждением, мотивированным поведением и сигнал- и стресс-индуцированным пристрастием к психоактивным веществам (Caponnetto et al., Curr. Opin. Pharmacol., 2012; 12: 229-237). Кроме того, никотин является агонистом никотиновых ацетилхолиновых рецепторов в центральной нервной системе (nAChRs). При связывании, он повышает частоту пульсации допаминовых нейронов, которые из вентральной области покрышки выступают в направлении прилежащего ядра. Никотиновая зависимость представляет собой очень сильную форму зависимости, при этом большинство курильщиков, которые пытаются бросить курить, снова начинают курить в течение одного месяца.
Существует несколько лекарственных средств, одобренных регулирующими органами, для лечения никотиновой зависимости: i) продукты-заменители никотина, ii) Варениклин, антагонист α4β2 никотинового рецептора, и iii) Бупроприон, не-трициклический антидепрессант. Никотиновые вакцины, основанные на стратегиях активной и пассивной иммунизации, в настоящее время находятся в стадии клинических исследований. Существующие лечения показали многообещающие эффекты в облегчении симптомов никотиновой зависимости, но их успех в предотвращении рецидива и поддержании длительного отказа от употребления никотина был только маргинальным (Benowitz, Annu. Rev. Pharmacol. Toxicol., 2009; 49: 57-71). Важно отметить, что постоянно сообщалось о статистически значимой зависимости между никотиновой зависимостью и сопутствующиими заболеваниями, такими как пост-травматическое стрессовое расстройство (PTSD) и подверженность депресси. Например, среди ветеранов войны, страдающих PTSD, никотиновую зависимость положительно связывали с PTSD симптомами (Thorndike, Addict Behav., 2006; 31: 223-231). Курильщики с PTSD имеют намного большую вероятность стать заядлыми курильщиками, т.е. которые выкуривают более чем 25 сигарет в день. Было показано, что замедленное высвобождение бупропиона является эффективным для краткосрочного прекращения курения в исследованиях, в которых участвовали ветераны с диагностированным PTSD и другими сопутствующими психиатрическими состояниями (Hertzberg, J. Clin. Psychopharmacol., 2001; 21:91-98).
D3 допаминовый рецептор (D3DR) представляет собой молекулярную мишень, которая была всесторонне исследована для разработки новых и эффективных лекарственных средств для лечения никотиновой зависимости (Le Foil et al., Expert Op. Invest. Drugs, 2007; 16: 45-57). На самом деле, этот подтип рецептора преимущественно экспрессируется в мезолимбокортикальной системе. В предклинических животных моделях, модуляторы D3DR были способны снижать непреодолимое желание к самостоятельному введению никотина в режиме подкрепления и предотвращали установление поведенческих типов выработки рефлекса Павлова влечения к этому веществу. Это предполагает трансляционную стратегию, в которой модуляторы D3DR можно было бы использовать для ослабления эффектов ассоциированных с психоактивным средством стимулов, которые в результате приводят к восстановлению моделей поведения поиска психоактивного средства. Однако модуляторы D3DR не показали существенные эффекты на истинное действие никотина и только имели умеренные эффекты на никотиновую зависимость.
В последнее время на основании поведенческих и нейрохимических доказательств было установлено, что ингибирование фермента гидролазы амидов жирных кислот (FAAH) является эффективным для противодействия связанным с злоупотреблением эффектам никотина. FAAH фермент представляет собой мембраносвязанную серингидролазу - члена сигнатурного семейства амидаз, характеризующуюся необычной
Ser-Ser-Lys каталитической триадой. FAAH катализирует разложение N-ацилэтаноламидов некоторых жирных кислот (FAEs), эндогенных лигандов как для каннабиноидных (CB) рецепторов, так и для ядерных рецепторов, активируемых пролифератором пероксисом (PPAR) (Panlilio et al., Pharmacol. Ther., 2013; 138: 84-102). Селективные ингибиторы FAAH фермента были способны снижать никотин-индуцированное повышение допамина в мезолимбокортикальной системе, предотвращая установление поведения самостоятельному введения никотина и никотин-индуцированных преференциальных поведений. Эти ингибиторы действовали, повышая уровни олеоилэтаноламида (OEA) и пальмитоилэтаноламида (PEA), эндогенных агонистов ядерных рецепторов PPAR-альфа. Активация PPAR-альфа повышает активность некоторых тирозиновых киназ, что, в свою очередь, блокирует передачу сигнала, инициируемую связыванием никотина с nAChRs (Mascia et al., Biol. Psychiatry, 2011; 69: 633-641). Кроме того, блокирование расщепления анандамида, ингибирование FAAH фермента всегда связывали с анксиолитическим эффектом, который противодействует синдрому отмены и индуцируемому определенными веществами и сигналами рецидиву (Justinova et al., Biol. Psychiatry, 2008; 64: 930-937).
В этом сценарии можно предвидеть стратегии лечения, направленные как на симптомы пристрастия к никотину, так и на рецидив, путем сочетания ингибитора FAAH фермента с модулятором D3 рецептора. Кроме того, такая комбинация оказалась благоприятной для лечения других сопутствующих состояний, часто ассоциированных с никотиновой зависимостью, таких как указанное выше PTSD, а также состояние тревоги, патологические поведения и шизофрения (Wu et al., J. Clin. Psychopharmacol., 2013; 33: 319-28). В случае шизофрении, комбинация ингибитора FAAH фермента с модулятором D3 рецептора могла бы вызывать в специфических областях головного мозга повышение уровней анандамида, которые, как было показано, отрицательно коррелируют с психотическими симптомами, указывая на защитную роль анандамида (Leweke et al., Translational Psychiatry, 2012; 2: e94), и, в то же время, задействовать вызываемое допамином D3 улучшение когнитивной функции, эмоциональное регулирование, исполнительные функции, гибкость и социальное поведение, как продемонстрировано в in vivo экспериментах на животных моделях (Gross and Dresker, Handb. Exp. Pharmacol., 2012; 213: 167-210).
Однако эта комбинированная терапия имеет некоторые недостатки. Кроме того, что она предполагает затруднительное введение двух отдельных лекарственных средств, что обычно затрудняет соблюдение режима лечения, особенно пациентами, у которых диагностированы психотические симптомы, разные фармакокинетические свойства соответствующих лекарственных средств могут влиять на разную фармакодинакику. На практике клиницист сталкивается и имеет дело с комбинированной терапией с двумя разными ADME кривыми (Всасывание Распределение Метаболизм Выведение). Инновационной альтернативой комбинациям лекарственных средств являются лекарственные средства, которые могут прицельно действовать на несколько мишеней - так называемые направленные на несколько мишеней лиганды (MTDLs; Cavalli et al. J. Med. Chem., 2008; 51:347-72). Стратегия таргетирования двух или более белков одновременно одним соединением может обеспечить терапевтические эффекты, превосходящие эффекты селективного лекарственного средства (Zimmermann et al., Drug Discovery Today, 2007; 12: 34-42; Morphy R. and Rankovic Z. Drug Discovery Today, 2007, 12, 156-60; Hopkins A.L., Nat. Chem. Biol., 2008; 4: 682-90). Это можно объяснить рядом потенциальных преимуществ, предлагаемых применением MTDLs, по сравнению со смесями различных лекарственных препаратов или многокомпонентными лекарственными средствами. Преимущества MTDLs можно обобщенно указать следующим образом: 1) снижение неопределенности в клинической разработке, поскольку предсказать фармакокинетические свойства одного соединения намного проще, чем для смеси различных лекарственных препаратов, преодолевая при этом проблему разной биодоступности, фармакокинетических свойств и метаболизма; 2) определенность в отношении фармакодинамики; 3) улучшенный эффект одновременного ингибирования нескольких мишеней; 4) улучшенная безопасность путем снижения побочных эффектов, связанных с нагрузкой смеси различных лекарственных препаратов (уменьшение риска межлекарственного взаимодействия); это особенно относится к лекарственному метаболизму, где конкуренция различных лекарственных средств за один и тот же метаболический фермент влияет на их токсичность. Другим важным преимуществом является упрощенный терапевтический режим и улучшенное его соблюдение пациентом, что особенно важно для пациентов, у которых может возникнуть рецидив.
MTDLs стратегия представляет собой инновационный подход к разработке нового лекарственного средства центрального действия для лечения комплексных расстройств, особенно в свете того, что наиболее важные основные процессы, которые в конечном счете приводят к зависимости, являются многофакторными по своей природе (Gardner et al., Adv. Psychosom. Med., 2011; 30: 22-60). Такая стратегия основана на концепции, что одно многофункциональное соединение можно вводить, чтобы поразить несколько мишеней, которые кооперируют в установлении и поддержании никотиновой зависимости, и поэтому будет предотвращать нежелательную компенсацию между взаимодействующими путями. Действительно, MTDLs могли бы представлять собой практическую альтернативу использованию комбинаций лекарственных средств. Поскольку многие вызывающие зависимость вещества имеют общие для них основные механизмы, которые вызывают зависимость, такие MTDLs также можно использовать в качестве лекарственных средств для других состояний, для которых клинически полезным могло бы быть сочетание ингибирования фермента гидролазы амидов жирных кислот и модуляции D3 допаминового рецептора.
Одна проблема, связанная с MTDLs, состоит в том, что многие из них имеют низкую эффективность, что касается их энергии связи на единицу молекулярной массы. Это потому, что они содержат группы, являющиеся важными только для одной из мишеней, которые являются только лишь переносимыми для других. Это приводит к несбалансированному профилю (Morphy R. et al., Drug Discov. Today, 2007; 12: 156-160; Morphy R., J. Med. Chem., 2006; 4: 2969-2978). Путь слияния в одну молекулу двух фармакофорных элементов, одного отличного от FAAH молекулярной мишени, а другого, способного распознавать вторую молекулярную мишень (D3DR), не является очевидным, а также неочевидной является необходимая оптимизация этих двух активностей.
Авторы настоящего изобретения решили эту проблему и неожиданно обнаружилт класс соединений, способный одновременно ингибировать FAAH фермент и модулировать D3D рецептор, таким образом, предложив превосходное лекарственное средство для лечения синдромов, ассоциированных с зависимостью и пристрастием к никотину и другим веществам, вызывающим зависимость.
Известный уровень техники
Арилпиперазины и арилпиперидины составляют класс антагонистов допаминовых рецепторов, хорошо известных из уровня техники. Некоторые патенты, патентные заявки и научные публикации описывают структуры и терапевтические применения таких соединений в качестве модуляторов допаминовых рецепторов.
В US7056922 заявлены некоторые N-ациламиноциклопропаниларилпиперазиновые производные в качестве модуляторов допаминовых D3 рецепторов. В US8334289 заявлены некоторые пиридиноиларилпиперазиновые производные в качестве модуляторов допаминовых D3 рецепторов. WO2006/072608 и WO2008/043839 описывают арилпиперазиновые и арилпиперидиновые производные в качестве модуляторов допамин D2-подобных и серотониновых 5-HT2 подтипов рецепторов. WO2010/034648 описывает некоторые пиридин-2-ил-пиперазины, обладающие аффинностью и селективностью в отношении допаминовых D3 рецепторов. WO2009/112568, WO2009/095438 и WO2010/040808 раскрывают некоторые арилпиперазиновые производные в качестве модуляторов допаминовых D3 и серотониновых 5-HT2A рецепторов с потенциальной применимостью в медицине. WO2006/072608 описывает арилпиперазиновые производные, полезные в качестве модуляторов допаминовых и серотониновых рецепторов. В WO2003/028728 сообщается о некоторых замещенных пиперазинилбутилкарбоксамидах, полезных в качестве селективных лигандов допаминовых D3 рецепторов.
Leopoldo et al. (J. Med. Chem., 2002; 45: 5727-5735) описывают исследование взаимозависимости структура-аффинность на некоторых N-[4-(4-арилпиперазин-1-ил)бутил]арилкарбоксамидах, полезных в качестве сильных и селективных лигандов допаминовых D3 рецепторов. Campiani et al. (J. Med. Chem., 2003; 46: 3822-3839) описывают синтез и фармакологическую оценку некоторых сильных и высокоселективных лигандов D3 рецепторов. Hackling et al. (J. Med. Chem., 2003; 46:3883-3899) описывают ряд N-(омега-(4-(2-метоксифенил)пиперазин-1-ил)алкил)карбоксамидов в качестве лигандов допаминовых D2 и D3 рецепторов.
В WO2004/112729 и WO2004/033426 и EP409048 заявлены некоторые арилпиперазиновые и арилпиперидиновые производные, обладающие активностью на D2 подтипах допаминовых рецепторов.
WO96/02246, WO2006/058993 и WO2004/004729 описывают некоторые арилпиперазиновые и арилпиперидиновые производные, обладающие активностью на D3 подтипах допаминовых рецепторов. WO2004/024878 описывает селективные лиганды допаминовых D3 рецепторов.
В US4803203 заявлены некоторые гетероциклические пиперазинилалкокси-бензогетероциклические производные, полезные в качестве антипсихотических средств.
В US6100255 заявлены арилпиперазиновые производные, обладающие активностью на D4 подтипах допаминовых рецепторов. WO94/22839 описывает бензимидазольные производные, обладающие аффинностью в отношении D4 подтипа допаминовых рецепторов. WO2001/49677 описывает некоторые индолилпиперазиновые производные, обладающие активностью на D4 подтипах допаминовых рецепторов.
Leopoldo et al. (J. Med. Chem., 2006; 49: 358-365) описывает разработку, синтез и аффинность связывания потенциальных PET лигандов для визуализации допаминовых D3 рецепторов головного мозга.
В CN103073524A описаны некоторые замещенные фенилпиперазин-1-ил-бутилкарбаматные производные и заявлена их аффинность в отношении D3 рецептора. Соединения, описанные в этой заявке, демонстрируют от умеренной до низкой аффинности связывания с D3 рецептором (Ki, нМ), как показано в таблице на стр. 23 CN103073524A.
С годами был накоплен существенный объем знаний, поскольку открытие гидролазы амидов жирных кислот и некоторых классов соединений было заявлено в ряде патентов и патентных заявок. Класс O-арилкарбаматных ингибиторов FAAH хорошо известен из уровня техники; в US7176201 заявлен ряд бифениловых эфиров алкилкарбаминовой кислоты. В US8003693 заявлены нафтиловые эфиры алкилкарбаминовой кислоты. WO2008/020866 раскрывает сложные эфиры алкилкарбаминовой кислоты формулы (1) и (2)
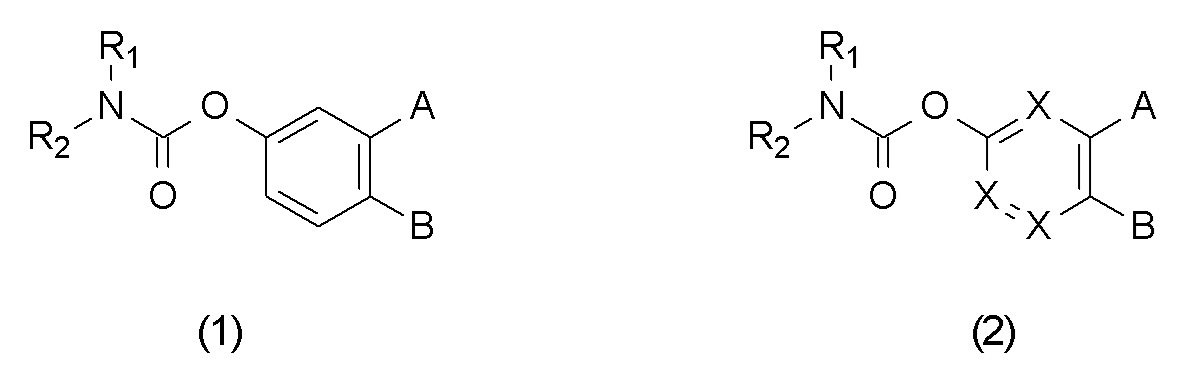
где R1 и R2 представляют собой необязательно замещенные алкильные или циклоалкильные группы, тогда как A и B представляют собой некоторые различные заместители.
В WO2008/063714 заявлены FAAH ингибиторы, имеющие структуры формул (3), (4) и (5)
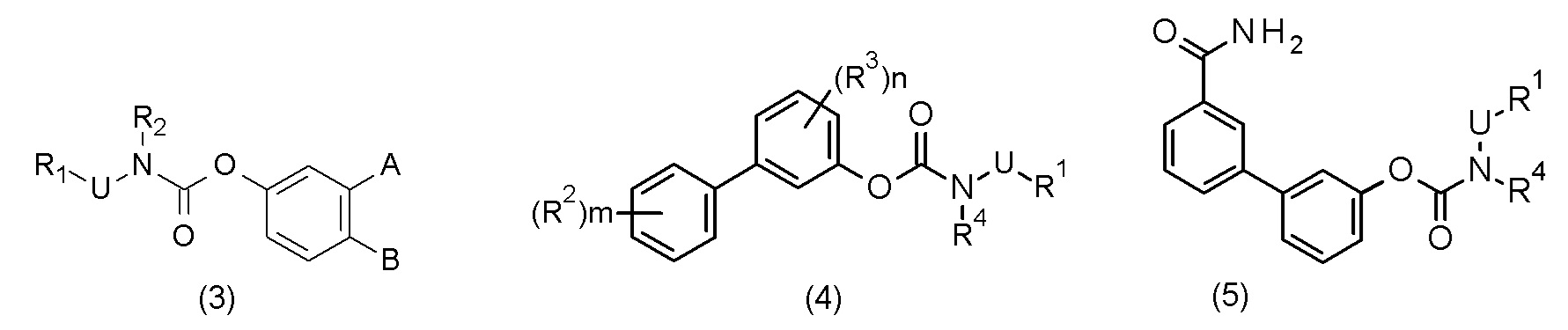
где R1 выбран из группы, состоящей из необязательно замещенных насыщенных циклоалкильных групп, U представляет собой связь или метиленовую группу, и R2 независимо представляет собой H или насыщенную алкильную группу. A и B, рассматриваемые независимо друг от друга, представляют собой ряд некоторых различных заместителей, или, взятые вместе с ароматическим кольцом, с которым они связаны, они представляют собой ароматический или неароматический карбоцикл или гетероцикл.
Другие ингибиторы FAAH на основе карбамата были раскрыты в WO2010/105930 как имеющие общую структурную формулу (6)
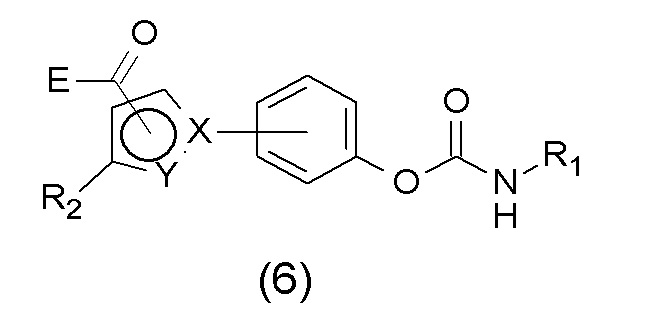
где R1 представляет собой H или (C1-C4)-алкил или (C3-C6)-циклоалкил или (C1-C6)-алкил-арил или (C1-C6)-алкил-(C2-C5)-алкинил; и ароматическое кольцо является моно-замещенным некоторыми гетероарильными группами, охватывающими различным образом замещенные 5-членные гетероциклические группы;
WO2008/129129 раскрывает некоторые сложные эфиры алкилкарбаминовых кислот общей структурной формулы (7)
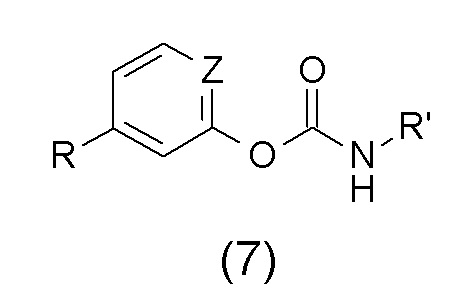
где R' выбран из группы, состоящей из H, замещенного или незамещенного алкила, включающего 1-24 атомов углерода, замещенного или незамещенного циклоалкила, замещенного или незамещенного гетероалкила, замещенного или незамещенного арила и замещенного или незамещенного гетероарила; Z представляет собой либо N либо CH, и R представляет собой некоторую гетероциклильную или гетероциклическую карбонильную группу, различным образом замещенную.
Авторам настоящего изобретения не известен ни один документ предшествующего уровня техники, описывающий каое-либо соединение в качестве модулятора нескольких мишеней, способное одновременно ингибировать FAAH фермент и модулировать D3 допаминовый рецептор.
Сущность изобретения
Авторами настоящего изобретения было обнаружено, что специфические соединения, содержащие O-фенилкарбамат, соответствующим образом связанный через подходящий спейсер с арилпиперазиновым или арилпиперидиновым кольцом, способны одновременно ингибировать FAAH фермент и модулировать D3 допаминовый рецептор и поэтому являются полезными для лечения синдромов, для которых могут быть клинически полезными активности ингибирования FAAH фермента и модуляции D3 допаминового рецептора.
В первом аспекте, настоящее изобретение обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль
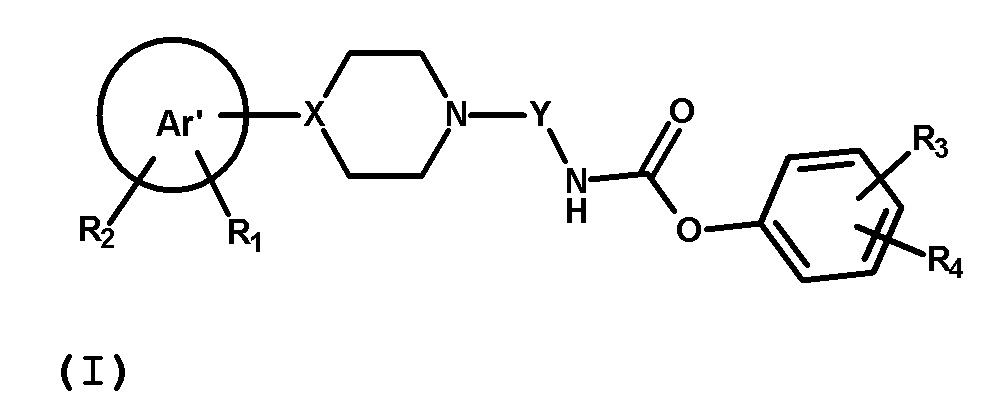
где Ar’, R1, R2, R3, R4, X, Y имеют значения, определенные ниже.
Во втором аспекте, настоящее изобретение обеспечивает фармацевтическую композицию, включающую одно или несколько соединений формулы I, определенной выше, или фармацевтически приемлемую соль такого соединения и фармацевтически приемлемые эксципиенты, носители или разбавители.
В третьем аспекте, настоящее изобретение обеспечивает способ модуляции уровней активности D3 допаминовых рецепторов и N-ацилэтаноламидов жирных кислот (FAEs) у субъекта путем введения композиции в соответствии с настоящим изобретением. В некоторых вариантах воплощения, настоящее изобретение обеспечивает способы лечения состояний, ассоциированных с несбалансированной активностью D3 допаминовых рецепторов, и для которых будет полезно повышение уровней AEA, OEA и PEA, включая никотин и другие психоактивные вещества, вызывающие сильную зависимость и привыкание, путем введения терапевтически эффективного количества соединения формулы I, определенной выше, или его фармацевтически приемлемой соли в соответствии с настоящим изобретением.
В четвертом аспекте, настоящее изобретение обеспечивает способы получения соединений формулы I, определенной выше, через процесс, состоящий из подходящих синтетических преобразований.
Подробное описание изобретения
В первом аспекте, настоящее изобретение обеспечивает соединение формулы (I) или его фармацевтически приемлемую соль
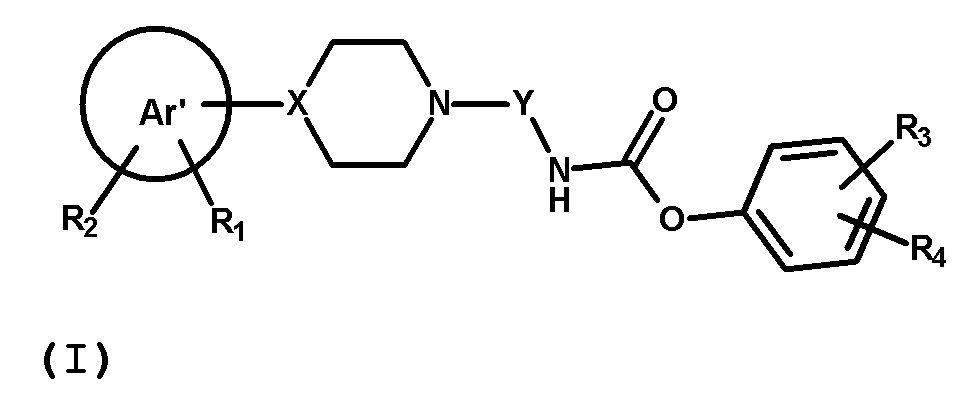
где:
Ar’ представляет собой 5-10-членное ароматическое или гетероароматическое одиночное или конденсированное кольцо, включающее до 3 гетероатомов, выбранных из N, O, S;
R1 и R2 независимо выбраны из группы, состоящей из водорода, галогена (предпочтительно F или Cl), линейного или разветвленного C1-6алкила (предпочтительно Me), C1-6алкокси (предпочтительно OMe), OH, CF3; R1 и R2 могут быть присоединены в любом положении Ar’ группы;
X представляет собой N или CH;
Y представляет собой алкиленовую или алкениленовую группу, выбранную из группы, состоящей из
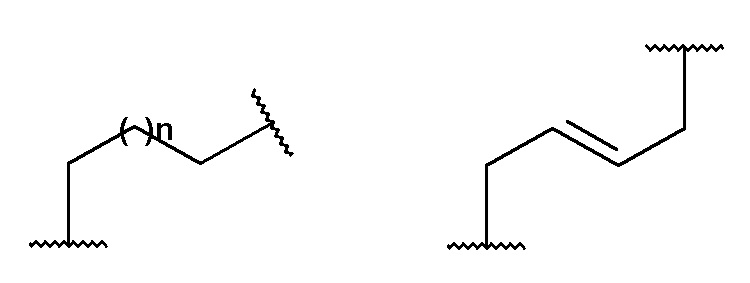
где n имеет значение 0 или представляет собой целое число, имеющее значение от 1 до 3, и где Y необязательно замещен в одинаковыми или отличными друг от друга заместителями B количестве до двух, присоединенными к любому положению Y группы и выбранными из группы, состоящей из F, OH, CH2OH, CH2F; предпочтительно n представляет собой целое число, имеющее значение от 1 до 2;
R3 представляет собой 5-6-членное ароматическое или гетероароматическое кольцо, бензильную или бензоильную группу, необязательно замещенную одинаковыми или отличными друг от друга заместителями R5 в количестве до двух, присоединенными к любому положению R3 кольца, где R5 выбран из OH, NH2, CN, галогена (предпочтительно F или Cl), линейного или разветвленного C1-6алкила (предпочтительно Me), C1-6алкокси (предпочтительно OMe), гидрокси C1-6алкила (предпочтительно CH2OH), SO2NH2, CONH2, CONR7R8, где R7 и R8 независимо представляют собой водород или C1-6алкил (предпочтительно Me);
или R3 представляет собой 5-7-членное алифатическое или гетероциклическое кольцо, включающее до 3 гетероатомов, выбранных из N, O, S, необязательно замещенное одинаковыми или отличными друг от друга заместителями R6 в количестве до двух, присоединенными к любому положению R3 кольца, где R6 выбран из F, гем-дифтора, Me, гем-диметила, =O, COMe, OH, CONH2;
и где группа R3 может быть присоединена к любому положению фенильного кольца;
R4 представляет собой водород, галоген (предпочтительно F или Cl), линейный или разветвленный C1-6алкил (предпочтительно Me), C1-6 алкокси (предпочтительно OMe), гидроксиC1-6алкил (предпочтительно CH2OH), CF3; группа R4 может быть присоединена к любому положению фенильного кольца;
или R3 и R4 вместе с фенильным кольцом, с которым они связаны, могут образовывать 9H-карбазольное кольцо.
Все технические и научные термины, используемые в настоящей заявке, имеют значения, широко известные среднему специалисту в данной области, если не определено иначе. Следующие термины, используемые в описании и в формуле изобретения настоящей заявки, имеют значения, определенные ниже, если не определено иначе.
Термин “алкил”, в контексте настоящей заявки, означает насыщенный алифатический углеводородный радикал, включая радикалы с прямой цепью и с разветвленной цепью, включающий от 1 до 6 атомов углерода. Неограничивающими примерами алкила вляются, например, метил, этил, пропил, изопропил, н-бутил, изобутил, трет-бутил, н-амил, изоамил, н-гексил и т.п.
Термин “алкенил”, в контексте настоящей заявки, означает алкильную группу, определенную выше, состоящую из по меньшей мере двух атомов углерода и содержащую по меньшей мере одну углерод-углеродную двойную связь. Репрезентативные примеры включают, но не ограничиваются этим, этенил, 1-пропенил, 2-пропенил, 1- или 2-бутенил и т.п.
Термин “алкинил”, в контексте настоящей заявки, означает алкильную группу, определенную выше, состоящую из по меньшей мере двух атомов углерода и содержащую по меньшей мере одну углерод-углеродную тройную связь. Репрезентативные примеры включают, но не ограничиваются этим, этинил, 1-пропинил, 2-пропинил, 1- или 2-бутинил и т.п.
Термин “циклоалкил”, в контексте настоящей заявки, означает 3-7-членное полностью углеродное моноциклическое кольцо, которое может содержать одну или несколько двойных связей, но не содержит полностью сопряженную пи-электронную систему. Примеры циклоалкильных групп включают, без ограничения, циклопропан, циклобутан, циклопентан, циклопентен, циклогексан, циклогексен, циклогексадиен и циклогептан.
Термин “арил”, в контексте настоящей заявки, означает углеводород, состоящий из моно-, би- или трициклической кольцевой системы, где кольца являются конденсированными вместе или связаны друг с другом ковалентно, и по меньшей мере одно из карбоциклических колец является ароматическим. Термин “арил” означает циклический ароматический, такой как 6-членный углеводород, два конденсированных шести-членных углеводорода и два шести-членных углеводорода, ковалентно связанных. Примеры арильных групп включают фенил, альфа- или бета-нафтил, 9,10-дигидроантраценил, инданил, флуоренил, бифенил и т.п.
Термин “гетероарил”, в контексте настоящей заявки, означает моно-, би- или трициклическую кольцевую систему, содержащую от одного до четарех гетероатомов, выбранных из азота, кислорода и серы, где кольца являются конденсированными вместе или связаны друг с другом ковалентно, и по меньшей мере одно из колец является ароматическим. Примеры гетероарильных групп включают пирролил, фурил, тиофенил, имидазолил, пиразолил, оксазолил, изоксазолил, тиазолил, изотиазолил, индолил, бензофуранил, бензотиофенил, бензимидазолил, бензопиразолил, бензоксазолил, бензоизоксазолил, бензотиазолил, бензоизотиазолил, триазолил, оксадиазолил, тетразолил, пиридил, пиразинил, пиримидинил, пиридазинил, хинолинил, изохинолинил, хиназолинил, хиноксалинил.
Термины “гетероциклил” или “гетероциклическое кольцо”, в контексте настоящей заявки, означают 3 - 7-членное насыщенное или частично ненасыщенное карбоциклическое кольцо, где один или несколько атомов углерода независимо замещены азотом, кислородом или серой. Гетероатомы азота и серы необязательно являются окисленными, и атом(атомы) азота необязательно являются кватернизированными. Примеры гетероциклильных групп включают, например, радикалы, образованные из оксирана, азиридина, оксетана, азетидина, тетрагидрофурана, дигидрофурана, тетрагидротиофена, дигидротиофена, пирролидина, дигидропиррола, пирана, дигидропирана, тетрагидропирана, тетрагидротиопирана, пиперидина, пиразолина, оксазолина, изоксазолидина, изоксазолина, тиазолидина, тиазолина, изотиазолина, диоксана, пиперазина, морфолина, тиоморфолина, гексаметиленимина, гомопиперазина и т.п.
Термин “ароматический” относится к группе, где составляющие ее атомы образуют ненасыщенную кольцевую систему, все атомы в кольцевой системе являются sp2 гибридизованными, и общее количество пи электронов равно 4n+2, где n представляет собой целое число.
Любая из указанных выше алкильных, алкенильных, алкинильных, циклоалкильных, арильных, гетероарильных, гетероциклильных или гетероциклических кольцевых групп может быть незамещенной или замещена одним или несколькими заместителями.
Если не указано иное, термин “замещенный”, в контексте настоящей заявки, означает, что один или несколько атомов водорода указанных выше групп замещены другим атомом или функциональной группой, включая, в качестве примера, алкил, замещенный алкил, арил, замещенный арил, арилалкил, алкокси, циклоалкилокси, арилокси, арилалкилокси, гидрокси, гетероарил, гетероарилокси, гетероциклилокси, трифторметил, трифторметокси, карбокси, ацил, ароил, гетероароил, галоген, нитро, циано, алкоксикарбонил, арилоксикарбонил, аралкилоксикарбонил, циклоалкилоксикарбонил, гетероарилоксикарбонил, ацилокси, алкилтио, арилтио, алкилсульфинил, арилсульфинил, алкилсульфонил, арилсульфонил, -O-ароил, -O-гетероароил, оксо (=O), -C(=O)-NRhRk и -NRpRq, где каждый из Rh, Rk, Rp и Rq независимо представляет собой водород, незамещенный или замещенный алкил, незамещенный или замещенный циклоалкил, незамещенный или замещенный арил, незамещенный или замещенный арилалкил, незамещенный или замещенный гетероарил, незамещенный или замещенный гетероциклил, ацил, ароил, гетероароил, и, когда Rh и Rk, или Rp и Rq взяты вместе с атомом азота, с которым они связаны, группа -NRhRk или группа NRpRq представляют собой гетероциклильный остаток, и где термины алкил, циклоалкил, арил, гетероарил, гетероциклил имеют значение, определенное выше.
В одном варианте воплощения, изобретение относится к соединениям формулы (I), где:
Ar’ представляет собой бензольное, индольное, нафтильное или пиридиновое кольцо;
R1 и R2 независимо представляют собой водород, галоген (предпочтительно Cl или F) или C1-6 алкокси (предпочтительно OMe), C1-6алкил (предпочтительно Me), CF3;
X представляет собой N или CH;
Y представляет собой CH2-(CH2)n-CH2, где n имеет значение 0 или представляет собой целое число, имеющее значение от 1 до 2, или группу CH2-CH=CH-CH2, и где Y необязательно замещен одинаковыми или отличными друг от друга заместителями B в количестве до двух, присоединенными к любому положению Y группы и выбранными из группы, состоящей из F, OH, CH2OH, CH2F;
R3 представляет собой фенильное, бензильное, бензоильное или пиридиновое кольцо, присоединенное к фенильному кольцу в мета или пара положении и необязательно замещенное одинаковыми или отличными друг от друга заместителями R5 в количестве до двух, где R5 представляет собой галоген (предпочтительно F), C1-6алкил (предпочтительно Me) или группу CONH2 или CONHCH3, присоединенную к R3 в мета или пара положении;
или R3 представляет собой 5-7-членное гетероциклическое кольцо, включающее до 2 гетероатомов, предпочтительно 1-пирролидинил или 1-пиперидинил или 1-пиперазинил или 4-морфолинил, присоединенный к фенильному кольцу в мета или пара положении и необязательно замещенный одинаковыми или отличными друг от друга заместителями R6 в количестве до двух, где R6 представляет собой гем-дифтор, гем-диметил, COMe, присоединенный к R3 в любом положении кольца;
R4 представляет собой водород, галоген (предпочтительно F), C1-6алкил (предпочтительно Me), C1-6алкокси (предпочтительно OMe), CF3; группа R4 может быть присоединена к фенильному кольцу в любом положении кольца;
или R3 и R4 вместе с фенильным кольцом, с которым они связаны, могут образовывать 9H-карбазольное кольцо.
Предпочтительные соединения в соответствии с этим вариантом воплощения представляют собой такие, в которых:
Ar’ представляет собой бензольное кольцо;
R1 и R2 независимо представляют собой H, Cl, OMe, Me, или CF3;
X представляет собой N или CH;
Y представляет собой CH2-(CH2)n-CH2, где n имеет значение 0 или представляет собой целое число, имеющее значение от 1 до 2, группу CH2-CH=CH-CH2, группу CH(CH2F)CH2 или группу CH2CH(F)CH2CH2;
R3 представляет собой фенильное, бензильное, бензоильное или пиридиновое кольцо, присоединенное к фенильному кольцу в мета или пара положении и необязательно замещенное группой CONH2 или CONHCH3, присоединенной к R3 в мета или пара положении;
или R3 представляет собой 1-пиперидинильное или 1-пиперазинильное или 4-морфолинильное кольцо, присоединенное к фенильному кольцу в мета или пара положении и необязательно замещенное гем-дифтором;
R4 представляет собой водород, F, Me или OMe; группа R4 может быть присоединена к фенильному кольцу в любом положении кольца;
или R3 и R4 вместе с фенильным кольцом, с которым они связаны, могут образовывать 9H-карбазольное кольцо.
Примеры соединений по настоящему изобретению включают следующие:
[3-(3-карбамоилфенил)фенил] N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил] карбамат;
[3-(3-карбамоилфенил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил] карбамат;
[3-(3-карбамоилфенил)фенил] N-[3-[4-(2,3-дихлорфенил)пиперазин-1-ил]пропил] карбамат;
[3-(3-карбамоилфенил)фенил] N-[3-(4-фенилпиперазин-1-ил)пропил] карбамат;
(3-фенилфенил) N-[3-(4-фенилпиперазин-1-ил)пропил] карбамат;
[4-(3-карбамоилфенил)фенил] N-[3-[4-(2,3-дихлорфенил)пиперазин-1-ил]пропил] карбамат;
[4-(3-карбамоилфенил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил] карбамат;
(3-морфолинoфенил) N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат гидрохлорид;
[3-(4,4-дифтор-1-пиперидинил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат гидрохлорид;
[4-(3-карбамоилфенил)фенил] N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат;
[4-(3-карбамоилфенил)фенил] N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат;
[4-(4,4-дифтор-1-пиперидинил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат гидрохлорид;
(3-морфолинoфенил) N-[3-[4-(o-толил)пиперазин-1-ил]пропил]карбамат гидрохлорид;
[3-(3-карбамоилфенил)фенил] N-[4-(4-фенил-1-пиперидинил)бутил]карбамат гидрохлорид;
(4-фенилфенил) N-[3-[4-(o-толил)пиперазин-1-ил]пропил] карбамат;
(4-фенилфенил) N-[3-[4-[2-(трифторметил)фенил]пиперазин-1-ил]пропил]карбамат;
9H-карбазол-2-ил N-[3-[4-(2,3-дихлорфенил)пиперазин-1-ил]пропил]карбамат;
[4-(4,4-дифтор-1-пиперидинил)фенил] N-[3-[4-[2-(трифторметил)фенил]пиперазин-1-ил]пропил]карбамат гидрохлорид;
[4-(3-карбамоилфенил)фенил] N-[(E)-4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бут-2-енил]карбамат;
[4-(3-карбамоилфенил)-3-фтор-фенил] N-[(E)-4-[4-(2,3-дихлорфенил) пиперазин-1-ил]бут-2-енил]карбамат;
[4-(3-карбамоилфенил)-3-фтор-фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат;
[4-(3-карбамоилфенил)фенил] N-[4-[4-[2-(трифторметил)фенил]пиперазин-1-ил]бутил]карбамат;
[3-(3-карбамоилфенил)фенил] N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат;
[4-(2-пиридил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат;
[4-(3-карбамоилфенил)фенил] N-[4-(4-фенилпиперазин-1-ил)бутил]карбамат;
(3-фенилфенил) N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат гидрохлорид;
(4-фенилфенил) N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат;
9H-карбазол-2-ил N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат;
(4-фенилфенил) N-[4-[4-(o-толил)пиперазин-1-ил]бутил]карбамат;
(4-фенилфенил) N-[4-[4-[2-(трифторметил)фенил]пиперазин-1-ил]бутил]карбамат;
(4-фенилфенил) N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат гидрохлорид;
(4-фенилфенил) N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат гидрохлорид;
9H-карбазол-2-ил N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат;
[4-(3-карбамоилфенил)-3-метокси-фенил] N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат;
(4-фенилфенил) N-[3-фтор-4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат гидрохлорид;
(4-фенилфенил) N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-фтор-бутил]карбамат;
(3-метокси-4-фенил-фенил) N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат гидрохлорид;
(3-метокси-4-фенил-фенил) N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат;
[4-(3-карбамоилфенил)фенил] N-[4-[4-(o-толил)пиперазин-1-ил]бутил]карбамат;
[4-(3-карбамоилфенил)-3-метокси-фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат;
(4-фенилфенил) N-[2-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-фтор-пропил]карбамат гидрохлорид;
(4-бензилфенил) N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат гидрохлорид;
(4-бензилфенил) N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат гидрохлорид;
(4-бензоилфенил) N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат гидрохлорид;
(4-бензоилфенил) N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат гидрохлорид.
Настоящее изобретение также обеспечивает способы получения соединений формулы (I), определенных выше, через процесс, состоящий из подходящих синтетических преобразований, описанных, например, в Michael Smith, Jerry March - March’s Advanced Organic Chemistry: reactions mechanisms and structure - 6th Edition, John Wiley & Sons Inc., 2007, который включен в настоящую заявку посредством ссылки. Среднему специалисту в данной области хорошо известно, что для преобразования химической функциональной группы в другую может потребоваться защита одного или нескольких реакционноспособных центров в соединении, содержащем эту функциональную группу, чтобы избежать нежелательных побочных реакций. Защиту таких реакционноспособных центров и последующее удаление защиты по окончании синтетических преобразований можно осуществить, следуя стандартным процедурам, описанным, например, в Theodora W. Green and Peter G.M. Wuts - Protective Groups in Organic Synthesis, Fourth Edition, John Wiley & Sons Inc., 2006, который включен в настоящую заявку посредством ссылки.
В одном варианте воплощения, соединение формулы (I) можно получить с использованием химических преобразований, показанных на схемах, описанных в настоящей заявке.
Синтез соединений формулы I
Конечные соединения формулы I можно получить в соответствии со Схемой Ia.
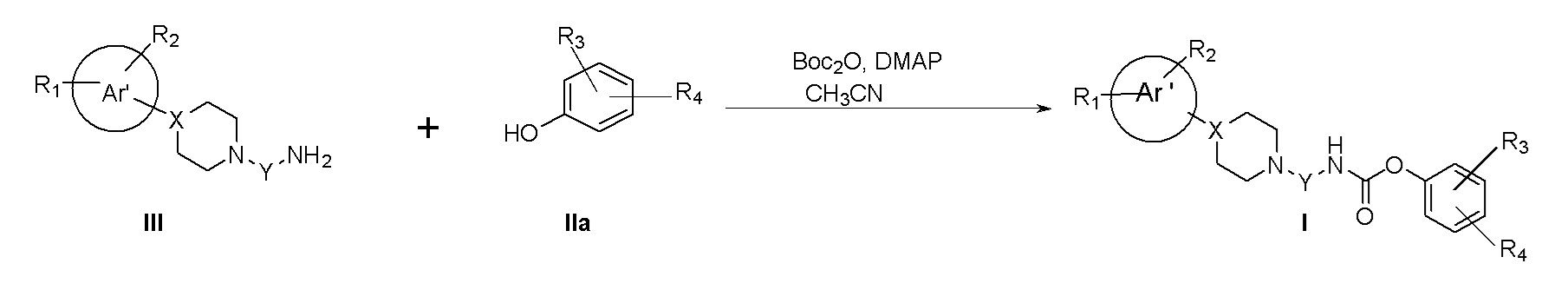
Схема Ia
Конечные соединения формулы (I) можно синтезировать при активации аминов формулы III в виде изоцианатов, путем взаимодействия с Boc ангидридом и 4-диметиламинопиридином в ацетонитриле, с последующим взаимодействием с соответствующим спиртом IIa (Tet. Lett., 1996; 4039(33): 5861-5864).
Альтернативно, амины формулы III могут быть активированы в виде карбаматов путем взаимодействия с пара-нитрофенилхлорформиатом и диизопропиламином в дихлорметане.
Другая процедура синтеза может включать преобразование спирта формулы IIa в хлорформиаты путем обработки трифосгеном и диизопропиламином в дихлорметане, с последующим взаимодействием с аминами III.
Синтез соединений формулы II
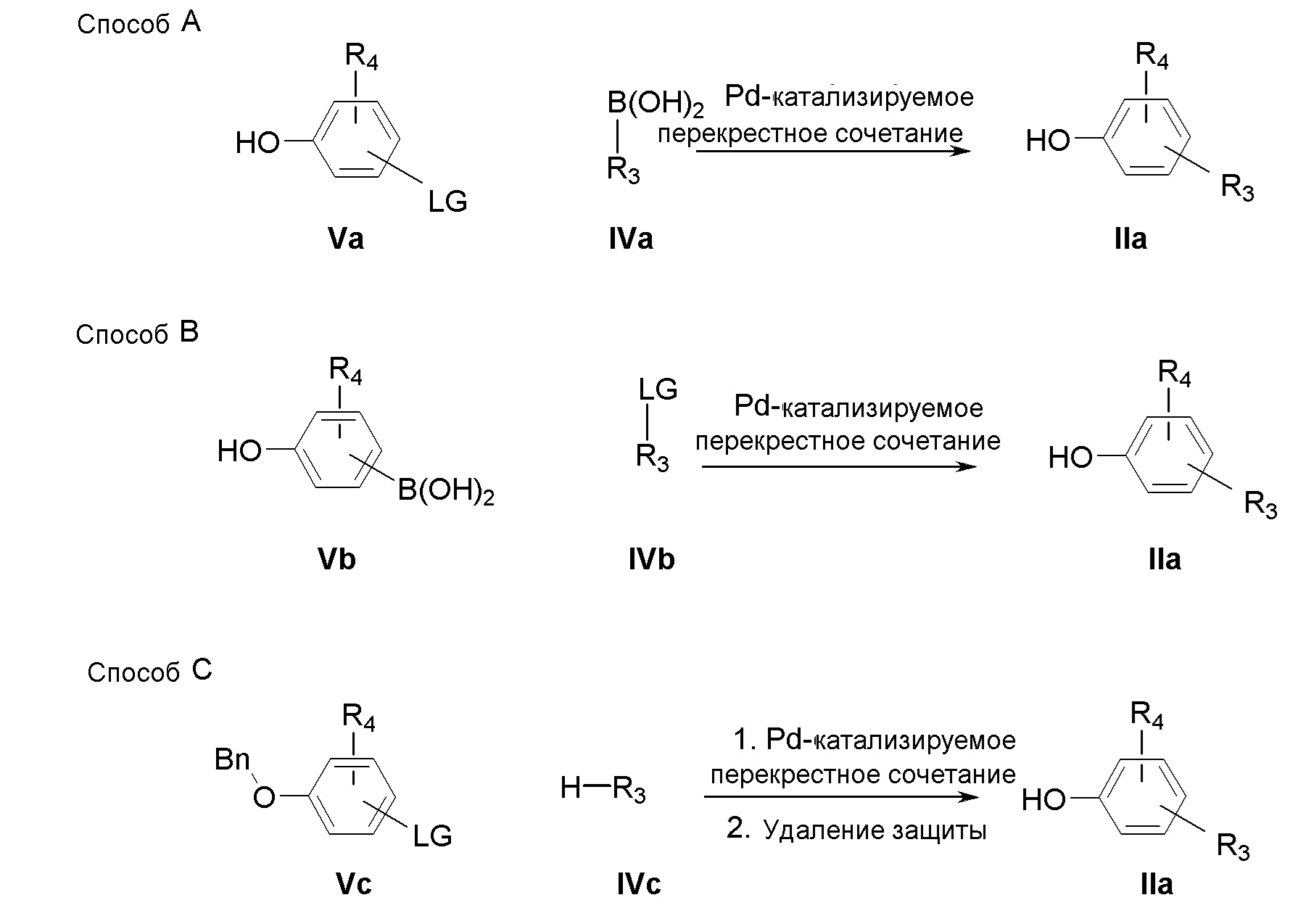
Схема II
В соответствии со Схемой II (Способ A и B), промежуточные соединения IIa можно получить через палладий-катализируемую реакцию перекрестного сочетания коммерчески доступных арил/гетероарилбороновых кислот с арил/гетероарил-производными, содержащими соответствующие удаляемые группы (LG). Например, галогенфенолы Va могут взаимодействовать с арилбороновыми кислотами IVa в присутствии каталитического ацетата палладия в среде, состоящей из воды и монометилового эфира этиленгликоля, следуя методу, разработанному Del Zotto et al. (Eur. J. Org. Chem., 2009; (1): 110-116). Соединения IIa могут быть выделены путем фильтрования катализатора из реакционной смеси и хроматографической очистки неочищенного реакционного продукта.
Когда R3 представляет собой 5-7-членное азот-содержащее гетероциклическое кольцо, включающее до 3 гетероатомов, промежуточные соединения IIa можно получить в соответствии со Схемой II (Способ C), путем взаимодействия бензилокси-замещенных арильных производных, содержащих соответствующие удаляемые группы (LG), с коммерчески доступными гетероциклическими соединениями IVc в присутствии палладиевого комплекса, как описано Buchwald and co-workers (Nature protocols, 2007; 2(11): 2881-7), с последующим удалением бензильной группы.
Синтез соединений формулы III
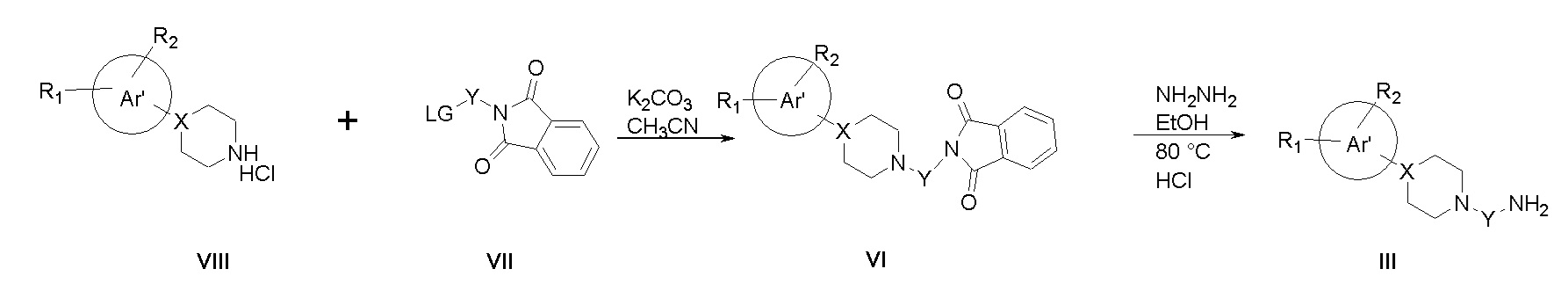
Схема IIIa
В соответствии со Схемой IIIa, амины общей формулы III можно синтезировать, исходя из коммерчески доступных арил-замещенных азот-содержащих гетероциклов общей формулы VIII путем обработки соответствующими коммерчески доступными фталимидами VII в присутствии карбоната калия и ацетонитрила при температуре кипения с обратным холодильником. Полученные таким образом фталимиды VI затем преобразовывают в первичные амины путем нагревания с обратным холодильником в спиртовом растворе гидразина. После подкисления и отделения нерастворимого 2,3-дигидрофталазин-1,4-диона желаемые амины могут быть выделены путем нейтрализации при помощи гидроксида натрия и экстракции органическими растворителями (например, этилацетатом или дихлорметаном).
Когда Y замещен группой B, амины общей формулы III можно синтезировать, следуя Схеме IIIb. Промежуточные соединения XI синтезировали, исходя из коммерчески доступных эпоксидных производных XII, при температуре кипения с обратным холодильником с фталимидом в щелочных условиях в DMF; промежуточные соединения XI выделяли путем экстракции органическими растворителями (DCM или этилацетат). Полученные таким образом промежуточные соединения XI подвергали взаимодействию с коммерчески доступными арил-замещенными азот-содержащими гетероциклами общей формулы VIII при нагревании в i-PrOH, с получением промежуточного соединения X. Также были разработаны процедуры с использованием микроволногого нагрева для синтеза этих соединений; чистые промежуточные соединения получали путем фильтрования реакционной смеси. Фторирование гидроксильной группы можно осуществить с использованием коммерчески доступных фторирующих агентов (например, DAST, DeOxo-Fluor®, Xtal Fluor-E®) в чистом виде или в аполярных растворителях (DCM или хлороформ). Не связывая это с какой-либо теорией, образование фторированных соединений формулы IX может происходить, как предполагается в Ji-Wang Chern et al, Tetrahedron Letters, 1998, 39: 8483-8486, т.е. через начальную нуклеофильную атаку гидрокси группы соединений формулы X по атому серы фторирующего агента с образованием -OSF2NEt2 группы в случае DAST или XTal Fluor-E®, или -OSF2N(CH2CH2OCH3)2 группы в случае DeOxo-Fluor®, с последующим межмолекулярным замещением указанной группы через анхимерное участие пиперазиновой группы, с образованием спиро азиридиниевого промежуточного соединения. Раскрытие кольца этого соединения при помощи фторидного аниона либо через менее пространственно затрудненный, либо более пространственно затрудненный углерод приведет к образованию соединения IX, где B представляет собой CH2F или F, соответственно. Удаление фталимидной защитной группы осуществляли, как описано для Схемы IIIa.
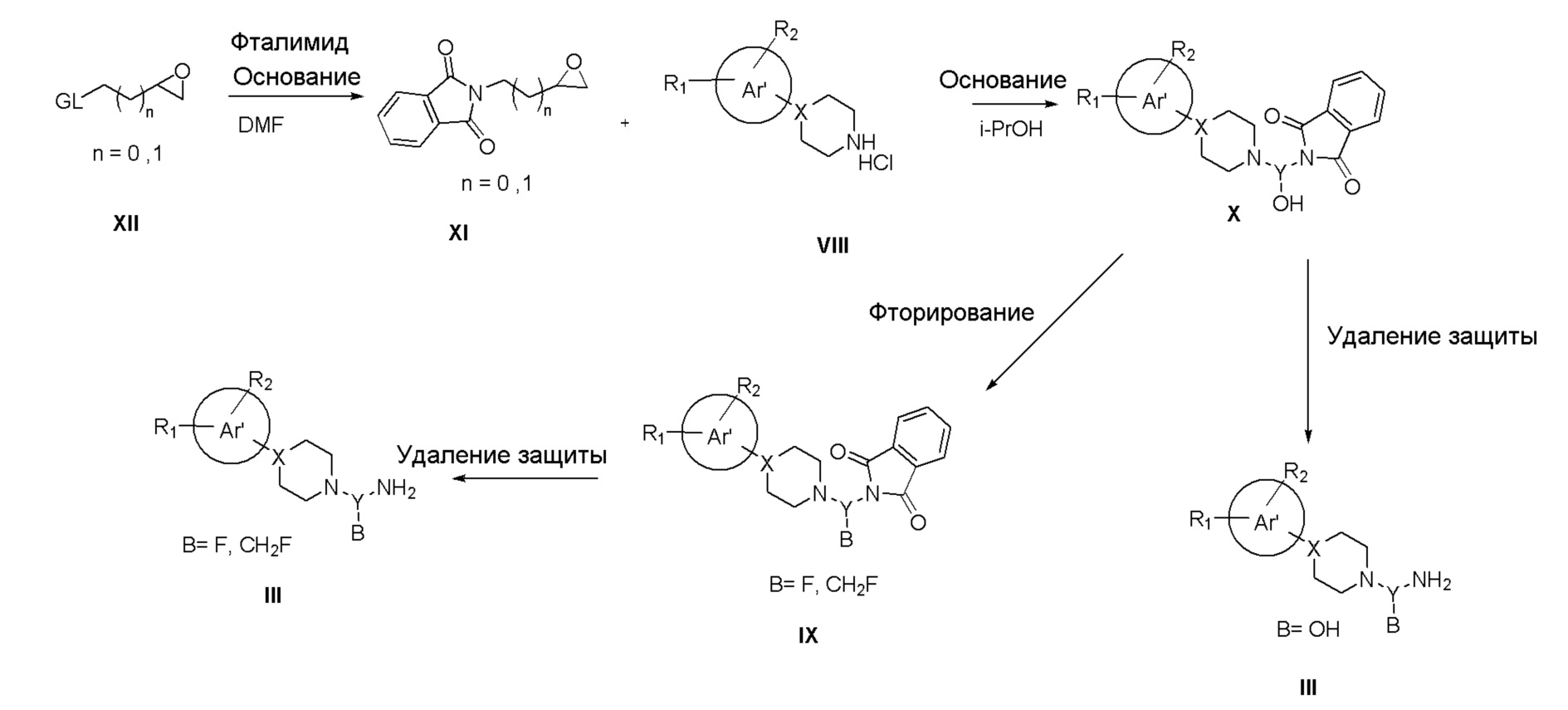 Схема IIIb
Схема IIIb
Коммерчески доступные исходные вещества IIb-c, IVa-c, Va-b, VII, VIII и XII могут быть закуплены у специальных поставщиков, таких как Sigma-Aldrich, Alfa Aesar и другие, или получены в соответствии со стандартными процедурами синтеза, как описано, например, в Michael Smith, Jerry March - March’s Advanced Organic Chemistry: reactions mechanisms and structure - 6th Edition, John Wiley & Sons Inc., 2007, который включен в настоящую заявку посредством ссылки.
Должно быть понятно, что используемые в настоящей заявке ссылки на соединения формулы (I) означают включение также их фармацевтически приемлемых солей или производных.
Кроме того, соединения формулы (I) могут образовывать кислотно-аддитивную соль или соль с основанием, в зависимости от типа заместителей, и эти соли включены в настоящее изобретение, при условии, что они являются фармацевтически приемлемыми солями.
Термины “соединение по настоящему изобретению” и “соединения по настоящему изобретению” и “соединения формулы (I)” относятся к каждому из соединений формулы (I) и предполагают включение их фармацевтически приемлемых солей, гидратов, сольватов и кристаллических форм, а также любых подходящих форм, как проиллюстрировано далее.
В контексте настоящей заявки, термин “соль” относится к любой соли соединения в соответствии с настоящим изобретением, полученной из неорганической или органической кислоты или основания, и к внутренне образованным солям. Типично, такие соли содержат физиологически приемлемый анион или катион.
Подходящие физиологически или фармацевтически приемлемые соли соединений по настоящему изобретению включают гидрохлорид, ацетат, цитрат, глюконат, лактат, тартрат, фосфат, борат, малеат, сульфат и нитрат, при этом гидрохлорид является предпочтительным.
Соли соединений формулы (I) можно получить путем взаимодействия щелочного соединения с желаемой кислотой в растворе.
Физиологически или фармацевтически приемлемые соли являются особенно подходящими для медицинских целей, благодаря их большей водорастворимости по сравнению с исходным соединением.
Фармацевтически приемлемые соли также можно получить из других солей, включая другие фармацевтически приемлемые соли соединений формулы (I), с использованием традиционных способов.
Специалистам в области органической химии должно быть понятно, что органические соединения могут образовывать комплексы с растворителями, в которых они взаимодействуют или из которых они осаждаются или кристаллизуются. Эти комплексы известны как “сольваты”. Например, комплекс с водой известен как “гидрат”. Сольваты соединений по настоящему изобретению охватываются объемом настоящего изобретения. Соединения формулы (I) легко могут быть выделены в ассоциации с молекулами растворителя путем кристаллизации или выпаривания соответствующего растворителя с получением соответствующих сольватов.
Соединения (I) могут быть в кристаллической форме. В некоторых вариантах воплощения, кристаллические формы соединений (I) представляют собой полиморфы.
Настоящее изобретение также включает изотопно-меченные соединения, которые идентичны соединениям, представленным в Формуле (I) и далее, но отличаются тем, что один или несколько атомов замещены атомом, имеющим атомную массу или массовое число, отличные от атомной массы или массового числа, обычно присутствующих в природе. Примеры изотопов, которые могут быть включены в соединения по настоящему изобретению и их фармацевтически приемлемые соли, включают изотопы водорода, углерода, азота, кислорода, фосфора, серы, фтора, иода и хлора, такие как 2H, 3H, 11C, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F, 36Cl, 123I, 125I.
Соединения по настоящему изобретению и фармацевтически приемлемые соли таких соединений, которые содержат указанные выше изотопы и/или другие изотопы других атомов, охватываются объемом настоящего изобретения. Изотопно-меченные соединения по настоящему изобретению, например такие, в которые включены радиоактивные изотопы, такие как 3H, 14C, являются полезными в анализах распределения лекарственного средства и/или субстрата в тканях. Изотопы трития, т.е. 3H, и углерода-14, т.е. 14C, являются особенно предпочтительными из-за простоты их получения и возможности детекции. 11C и 18F изотопы являются особенно полезными в PET (Позитрон-Эмиссионная Томография), и 125I изотопы являются особенно полезными в SPECT (Однофотонная Эмиссионная Компьютерная Томография), все они полезны для визуализации головного мозга. Кроме того, замещение более тяжелыми изотопами, такими как дейтерий, т.е. 2H, может дать некоторые терапевтические преимущества как результат лучшей метаболической стабильности, например, увеличение периода полужизни in vivo или снижение уровня необходимых доз, и, следовательно, может быть предпочтительных в некоторых обстоятельствах. Изотопно-меченные соединения формулы (I) и другие соединения по настоящему изобретению, как правило, можно получить путем осуществления процедур, раскрытых в Схемах и/или в примерах ниже, путем замещения не меченного изотопом реагента легко доступным изотопно-меченным реагентом.
Некоторые группы/заместители, включенные в настоящее изобретение, могут присутствовать в виде изомеров или в одной или нескольких таутомерных формах. Соответственно, в некоторых вариантах воплощения, соединения формулы (I) могут существовать в некоторых случаях в форме других таутомеров или геометрических изомеров, в зависимости от типа заместителей. В настоящем описании соединения могут быть описаны только в одной форме таких изомеров, но настоящее изобретение включает все такие изомеры, выделенные формы изомеров или их смесь. Кроме того, соединения формулы (I) могут в некоторых случаях содержать асимметричные атомы углерода или аксиальные асимметрии, соответственно, они могут существовать в форме оптических изомеров, например, в (R)-форме, (S)-форме и т.п. Настоящее изобретение охватывает своим объемом все такие изомеры, включая рацематы, энантиомеры и их смеси.
В частности, в объем настоящего изобретения включены все стереоизомерные формы, включая энантиомеры, диастереоизомеры и их смеси, включая рацематы, и общая ссылка на соединения формулы (I) включает все стереоизомерные формы, если не указано иное.
Как правило, соединения или соли по настоящему изобретению следует рассматривать как исключающие те соединения (если таковые имеются), которые являются настолько химически нестабильными, либо per se либо в воде, что они определенно являются неподходящими для фармацевтического применения через все пути введения, такими как пероральный, парентеральный или другие пути. Такие соединения известны специалистам в области химии. Однако включены пролекарства или соединения, которые стабильны ex vivo и которые могут преобразовываться в организме млекопитающего (например, человека) в соединения по настоящему изобретению.
Настоящее изобретение также охватывает активные метаболиты соединений формулы (I).
Еще один аспект настоящего изобретения относится к фармацевтическим композициям, содержащим соединение формулы (I).
Фармацевтические композиции по настоящему изобретению охватывают любые композиции, полученные путем смешивания соединения по настоящему изобретению и фармацевтически приемлемого носителя. Такие композиции являются подходящими для фармацевтического применения для животного или человека.
Фармацевтические композиции по настоящему изобретению включают терапевтически эффективное количество одного или нескольких соединений формулы (I) или фармацевтически приемлемой соли такого соединения и фармацевтически приемлемый носитель.
Фармацевтическая композиция необязательно содержит другие активные ингредиенты. Термин “носитель” относится к наполнителю, эксципиенту, разбавителям или адъюванту, с которыми вводят терапевтическое средство или активный ингредиент. Любой носитель и/или эксципиент, подходящий для получения желаемого для введения препарата, предусматривается для использования с соединениями, раскрываемыми в настоящей заявке.
Носитель может принимать самые различные формы, в зависимости от формы желаемого для введения препарата, например, пероральной или парентеральной (включая внутривенную). Для получения композиций для пероральных лекарственных форм можно использовать любую из обычных фармацевтических сред, такую как, например, вода, гликоли, масла, спирты, отдушки, консерванты, красители и т.п., в случае пероральных жидких препаратов, таких как, например, суспензии, эликсиры и растворы; или носители, такие как крахмалы, сахара, микрокристаллическая целлюлоза, разбавители, гранулирующие агенты, смазывающие вещества, связующие, разрыхлители и т.п., в случае пероральных твердых препаратов, таких как, например, порошки, твердые и мягкие капсулы и таблетки, при этом твердые пероральные препараты являются более предпочтительными, чем жидкие препараты.
В некоторых вариантах воплощения, соединения по настоящему изобретению можно объединять в качестве активного ингредиента в тесной смеси с подходящим фармацевтическим носителем и/или эксципиентом в соответствии с традиционными методами фармацевтического компаундирования.
Композиции включают композиции, подходящие для парентерального, включая подкожное, внутримышечное и внутривенное, пульмонального, назального, ректального, местного или перорального введения. Подходящий путь введения в каждом конкретном случае будет зависеть частично от природы и тяжести состояния, подлежащего лечению, и от природы активного ингредиента. Типичным путем введения является пероральный путь. Композиции могут быть для удобства представлены в стандартной лекарственной форме и могут быть получены любым из способов, хорошо известных в фармацевтике. Предпочтительные композиции включают композиции, подходящие для перорального, парентерального, местного, подкожного или пульмонального, в форме назальной или буккальной ингаляции, введения. Композиции могут быть получены любым из способов, хорошо известных в фармацевтике.
Фармацевтические композиции могут быть в форме таблеток, пилюль, капсул, растворов, суспензий, эмульсии, порошков, суппозитория и в виде композиций замедленного высвобождения.
Если желательно, на таблетки может быть нанесено покрытие стандартными водными или не-водными способами. В некоторых вариантах воплощения такие композиции и препараты могут содержать по меньшей мере 0,1 процента активного соединения. Процент активного соединения в этих композициях, конечно, может варьироваться и в целях удобства может составлять от около 1 процента до около 60 процентов в расчете на массу стандартной единицы. Количество активного соединения в таких терапевтически полезных композициях является таким, чтобы можно было получить терапевтически активную дозу. Активные соединения также можно вводить интраназально, например, в виде жидких капель или спрея.
Таблетки, пилюли, капсулы и т.п. также могут содержать связующее, такое как трагакант, аравийская камедь, кукурузный крахмал или желатин; эксципиенты, такие как дикальций фосфат; разрыхлитель, такой как кукурузный крахмал, картофельный крахмал, альгиновая кислота; смазывающее вещество, такое как стеарат магния; и подсластитель, такой как сахароза, лактоза или сахарин. Когда стандартная лекарственная форма представляет собой капсулу, она может содержать, помимо веществ указанного выше типа, жидкий носитель, такой как жирное масло. Различные другие вещества могут присутствовать в виде покрытий или для модификации физической формы единицы дозирования. Например, таблетки могут иметь покрытия из шеллака, сахара или и того и другого. Сироп или эликсир может содержать, помимо активного ингредиента, сахарозу в качестве подсластителя, метил и пропилпарабены в качестве консервантов, краситель и отдушку, такую как вишневая или апельсиновая отдушка. Для предотвращения разложения при прохождении через верхнюю часть желудочно-кишечного тракта, композиция может представлять собой лекарственную форму с энтеросолюбильным покрытием.
Композиции для местного введения включают, но не ограничиваются этим, мази, кремы, лосьоны, растворы, пасты, гели, карандаши, липосомы, наночастицы, пластыри, бандажи и повязки на раны. В некоторых вариантах воплощения, местная композиция включает вещество, способствующее проникновению.
Композиции для пульмонального введения включают, но не ограничиваются этим, композиции сухого порошка, состоящие из порошкообразного соединения формулы (I) или его соли и порошкообразного подходящего носителя и/или смазывающего вещества. Композиции для пульмонального введения можно вводить путем ингаляции из любого подходящего ингалятора сухого порошка, известного специалистам в данной области.
Введение композиций осуществляют в соответствии с протоколом и при дозе, достаточной для уменьшения воспаления и боли у субъекта. В некоторых вариантах воплощения, в фармацевтических композициях по настоящему изобретению активное вещество или активные вещества, как правило, сформулированы в виде единиц дозирования. Единица дозирования может содержать от 0,1 до 1000 мг соединения формулы (I) на единицу дозирования для ежедневного введения.
В некоторых вариантах воплощения, количества, эффективные для местной композиции, будут зависеть от тяжести заболевания, расстройства или состояния, ранее используемого лечения, состояния здоровья индивидуума и ответа на лекарственное средство. В некоторых вариантах воплощения, доза находится в пределах от 0,001% до около 60% в расчете на массу композиции.
При использовании в комбинации с одним или несколькими другими активными ингредиентами, соединение по настоящему изобретению и другой активный ингредиент можно использовать в более низких дозах по сравнению с дозами, когда каждое средство используется отдельно.
Что касается композиций, предназначенных для любых различных путей введения, способы и композиции для введения лекарственных средств раскрыты в Remington’s Pharmaceutical Sciences, 17th Edition, Gennaro et al. Eds., Mack Publishing Co., 1985, and Remington’s Pharmaceutical Sciences, Gennaro AR ed. 20th Edition, 2000, Williams & Wilkins PA, USA, and Remington: The Science and Practice of Pharmacy, 21st Edition, Lippincott Williams & Wilkins Eds., 2005; а также в Ansel’s Pharmaceutical Dosage Forms and Drug Delivery Systems, 8th Edition, Lippincott Williams & Wilkins Eds., 2005, которые включены в настоящую заявку посредством ссылки.
Следующий аспект настоящего изобретения относится к соединениям формулы (I) для применения в способе модуляции уровней активности D3 допаминовых рецепторов и N-ацилэтаноламидов жирных кислот (FAEs) у субъекта путем введения композиции в соответствии с настоящим изобретением. В некоторых вариантах воплощения, настоящее изобретение обеспечивает соединения (I) для применения в способе лечения состояний, ассоциированных с несбалансированной активностью D3 допаминовых рецепторов, и для которых будут полезны повышенные уровни арахидоноилэтаноламида (AEA) и/или олеоилэтаноламида (OEA) и/или пальмитоилэтаноламида (PEA), путем введения терапевтически эффективного количества соединения формулы (I), определенного выше, или его фармацевтически приемлемой соли в соответствии с настоящим изобретением.
В одном варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе, обеспечивающем прекращение курения млекопитающего, где указанный способ включает введение терапевтически эффективного количества соединения формулы (I) млекопитающему.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе уменьшения потребления табака млекопитающим, где указанный способ включает введение терапевтически эффективного количества соединения формулы (I) млекопитающему.
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе лечения или предупреждения синдромов, ассоциированных с расстройствами, связанными с употреблением каннабиса, включая пристрастие и привыкание к каннабиноидам, зависимость от каннабиноидов, синдром отмены каннабиноида, с расстройствами, связанными с употреблением кокаина, включая пристрастие и привыкание к кокаину, зависимость от кокаина, синдром отмены кокаина, с расстройствами, связанными с употреблением опиоидных средств, включая пристрастие и привыкание к опиоидным средствам, зависимость от опиоидных средств, синдром отмены опиоидного средства, с расстройствами, связанными с употреблением опиатов, включая пристрастие и привыкание к опиатам, зависимость от опиатов, синдром отмены опиата, с расстройствами, связанными с употреблением амфетамина, включая пристрастие и привыкание к амфетамину, зависимость от амфетамина, синдром отмены амфетамина, с расстройствами, связанными с употреблением метамфетамина, включая пристрастие и привыкание к метамфетамину, зависимость от метамфетамина, синдром отмены метамфетамина, с расстройствами, связанными с употреблением алкоголя, включая пристрастие и привыкание к алкоголю, алкогольную зависимость, абстинентный алкогольный синдром и вызванный алкоголем делириозный синдром, у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I).
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе модуляции аппетита или массы тела, или и того и другого, у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I).
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе предупреждения или лечения расстройств пищевого поведения, включая нейрогенную булимию, нервную анорексию, компульсивное переедание, расстройство пищевого поведения без дополнительных уточнений (EDNOS) у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I).
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе предупреждения или лечения состояния тревоги у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I).
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе предупреждения или лечения пост-травматического стрессового расстройства у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I).
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе предупреждения или лечения шизофрении у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I).
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе предупреждения или лечения расстройств настроения, включая биполярное расстройство типа I, биполярное расстройство типа II, циклотимию, вызванное химическими веществами расстройство настроения, депрессию, атипичную депрессию, психотическую глубокую депрессию, послеродовую депрессию, дистимию, кататоническую депрессию, меланхолическую депрессию, депрессивное расстройство неуточненного типа (DDNOS) у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I).
В другом варианте воплощения, настоящее изобретение обеспечивает соединения формулы (I) для применения в способе предупреждения или лечения патологических поведений, включая компульсивное влечение к азартным играм, непреодолимое влечение к покупкам, патологическое накопительство и клептоманию, у млекопитающего, где указанный способ включает введение млекопитающему терапевтически эффективного количества соединения формулы (I).
Примеры получения
Аббревиатуры, используемые в описании представленных далее примеров, имеют следующие значения:
Ацетонитрил (MeCN); хлорид аммония (NH4Cl); BnBr (бензилбромид); карбонилдиимидазол (CDI); карбонат цезия (Cs2CO3); циклогексан (Cy); хлороформ (CHCl3); дейтерированный хлороформ (CDCl3 или хлороформ-d); дейтерированный диметилсульфоксид (DMSO-d6); дихлорметан (DCM); диметилсульфоксид (DMSO); N,N-диизопропилэтиламин (DIPEA); 4-(диметиламино)-пиридин (DMAP); монометиловый эфир этиленгликоля (EGME); этанол (EtOH); электроспрей (ES); этилацетат (EtOAc); хлористоводородная кислота (HCl); масс-спектрометрия (MS); микроволновой (MW); серная кислота (H2SO4); иодметан (MeI); N,N-диметилформамид (DMF); гидроксид лития (LiOH); сульфат магния (MgSO4); метанол (MeOH); ядернный магнитный резонанс (ЯМР); комнатная температура (RT); бикарбонат натрия (NaHCO3); иодид тетрабутиламмония (TBAI); триэтилсилан (TES); тетрагидрофуран (ТГФ); тонкослойная хроматография (ТСХ); триэтиламин (Et3N); трифторуксусная кислота (TFA).
Очистки автоматизированным методом колоночной хроматографии осуществляли с использованием Teledyne ISCO устройства (CombiFlashTM Rf) с использованием колонок разных размеров с насадкой из силикагеля (от 4 г до 120 г). Смеси увеличивающейся полярности циклогексана и этилацетата или дихлорметана и метанола использовали в качестве элюентов. Препаративную ТСХ осуществляли с использованием предварительно покрытых Macherey-Nagel 0,05 мм ТСХ пластин (SIL G-50 УФ254). Реакции гидрирования осуществляли с использованием оборудования H-CubeTM для непрерывного гидрирования (SS-reaction line version), используя разовые картриджи с катализатором (CatCartTM), предварительно нагруженные необходимым гетерогенным катализатором. Микроволновой нагрев осуществляли с использованием ExplorerTM-48-позиционного устройства (CEM). ЯМР эксперименты осуществляли на Bruker Avance III 400 системе (400,13 МГц для 1H и 100,62 МГц для 13C), снабженной BBI зондом и Z-градиентами. Спектры получали при 300 K, с использованием дейтерированного диметилсульфоксида (DMSO-d6) или дейтерированного хлороформа (CDCl3) в качестве растворителей. Химические сдвиги для 1H и 13C спектров регистрировали в миллионных долях с использованием остаточного не-дейтерированного растворителя в качестве внутреннего стандарта (для CDCl3: 7,26 м.д., 1H и 77,16 м.д., 13C; для DMSO-d6: 2,50 м.д., 1H; 39,52 м.д., 13C).
ВЭЖХ/МС анализы осуществляли на Waters ACQUITY ВЭЖХ/МС системе, состоящей из SQD (одноквадрупольный детектор) масс-спектрометра, снабженного интерфейсом для ионизации электроспреем и детектором на фотодиодной матрице. PDA диапазон составлял 210-400 нм. Использовали ионизацию электроспреем в положительном и отрицательном режиме. ВЭЖХ подвижные фазы представляли собой следующие: (A) 10 мМ NH4OAc в H2O, pH 5; (B) 10 мМ NH4OAc в MeCN/H2O (95:5) pH 5. Анализы осуществляли с использованием способа A, B или C, описанного ниже.
Способ A (общий)
Градиент: 5 до 95% B в течение 3 минут. Скорость потока 0,5 мл/минута. Температура 40ºC
Предколонка: Vanguard BEH C18 (1,7 мкм 2,1×5 мм). Колонка: BEH C18 (1,7 мкм 2,1×50 мм)
Способ B (полярный)
Градиент: 0 до 50% B в течение 3 минут. Скорость потока 0,5 мл/минута. Температура 40°C
Предколонка: VanGuard HSS T3 C18 (1,7 мкм 2,1×5 мм). Колонка HSS T3 (1,8 мкм 2,1×50 мм)
Способ C (аполярный)
Градиент: 50 до 100% B в течение 3 минут. Скорость потока 0,5 мл/минута. Температура 40°C
Предколонка: Vanguard BEH C18 (1,7 мкм 2,1×5 мм). Колонка: BEH C18 (1,7 мкм 2,1×50мм)
Очистки методом препаративной ВЭЖХ/MS осуществляли на автоматизированной системе очистки Waters, состоящей из 3100 одноквадрупольного масс-спектрометра, снабженного интерфейсом для ионизации электроспреем и 2998 детектором на фотодиодной матрице. ВЭЖХ система включала 2747 устройство для отбора проб, 2545 бинарный градиентный модуль, органайзер для жидкостных систем и 515 ВЭЖХ насос. PDA диапазон составлял 210-400 нм. Очистки осуществляли на XBridgeTM Prep C18 OBD колонке (100×19 мм в.д., размер частиц 5 мкм) с XBridgeTM Prep C18 (10×19 мм в.д., размер частиц 5 мкм) Guard картриджем. Подвижные фазы представляли собой 10 мМ NH4OAc в H2O при pH 5, доведенном при помощи AcOH (A), и 10 мМ NH4OAc в MeCN-H2O (95:5) при pH 5 (B). Использовали ионизацию электроспреем в положительном и отрицательном режиме. Анализы методом хиральной ВЭЖХ осуществляли на Waters Alliance ВЭЖХ устройстве, состоящем из e2695 разделительного модуля и 2998 фотодиодного матричного детектора. PDA диапазон составлял 210-400 нм. Анализы осуществляли изократно на Daicel ChiralPak AD колонке (250×4,6 мм в.д., размер частиц 10мкм). Подвижная фаза представляла собой 0,1% TFA гептан-2-пропанол (75:25). Разделения с использованием препаративной хиральной ВЭЖХ осуществляли на Waters Alliance ВЭЖХ устройстве, состоящем из 1525 бинарного ВЭЖХ насоса, Waters отборника фракций III и 2998 фотодиодного матричного детектора. УФ детекцию осуществляли при 240 нм. Очистки осуществляли изократно на Daicel ChiralPak AD колонке (250×10 мм в.д., размер частиц 10мкм). Подвижная фаза представляла собой 0,1% TFA гептан-2-пропанол (75:25).
Общая процедура III для синтеза аминовых производных III (Схема IIIa).
Стадия A.
Смесь арилпиперазин.HCl (1 экв.), N-(бромалкил)фталимида (1,1 экв.) и K2CO3 (3 экв.) в ацетонитриле нагревали до температуры кипения с обратным холодильником в течение 6 часов. Горячую суспензию фильтровали и остаток промывали ацетоном несколько раз. Фильтраты концентрировали при пониженном давлении с получением фталимидных промежуточных соединений.
Стадия B.
Фталимидное производное (1 экв.) и гидрат гидразина (1,2 экв.) в метаноле нагревали до температуры кипения с обратным холодильником в течение 2 часов. К горячему раствору добавляли 2 н раствор HCl и нагревание при температуре кипения с обратным холодильником продолжали в течение еще одного часа. После охлаждения до температуры окружающей среды смесь фильтровали, остаток промывали метанолом и фильтрат упаривали досуха. Этот остаток суспендировали в воде и нейтрализовали при помощи 2 н раствора NaOH. Экстракция при помощи EtOAc давала маслянистый продукт, который был достаточно чистым для следующей стадии.
Общая процедура II для синтеза фенольных производных IIa (Схема II, A-B).
Способ A:
Производное бороновой кислоты IVa (1,2 экв.) добавляли к раствору арилгалогенида Va (1 экв.) в EGME/воде 3:1, с последующим добавлением Pd(OAc)2 (0,01 экв.) и K2CO3 (2,5 экв.). Через несколько минут желтоватая суспензия становилась темной, почти черной, и реакция достигла полной конверсии.
Смесь перемешивали еще в течение 5 минут, затем разбавляли водой, подкисляли при помощи 2 н раствора HCl и экстрагировали при помощи DCM. Органическую фазу сушили над Na2SO4 и концентрировали досуха. Неочищенный остаток адсорбировали на диоксиде кремния и очищали флэш-хроматографией (элюент: 5% MeOH в DCM).
Способ B:
Производное бороновой кислоты Vb (1,2 экв.) добавляли к раствору арил/гетероарилгалогенида IVb (1 экв.) в EGME/воде 3:1, с последующим добавлением Pd(OAc)2 (0,01 экв.) и K2CO3 (2,5 экв.); затем применяли такую же процедуру, как описано в способе A.
Общая процедура II для синтеза фенольных производных IIa (Схема II, C).
Способ C:
Стадия A.
Арилбензилоксигалогенид Vc (1 экв.) и амин IVc (1 экв.) суспендировали в безводном толуоле. Затем добавляли соединения Pd2(dba)3 (0,02 экв.), DavePhos (0,06 экв.) и t-BuO-K+ (2 экв.). Полученную смесь перемешивали в течение 2 часов при 100°C. Реакционную смесь промывали насыщенным раствором NH4Cl и органическую фазу собирали и сушили над сульфатом натрия, затем фильтровали. Упаривание при пониженном давлении органической фазы давало неочищенный продукт, который адсорбировали на силикагеле и очищали колоночной хроматографией (элюент: циклогексан/этилацетат 9:1). Фракции собирали и упаривали при пониженном давлении.
Стадия B.
К перемешиваемому раствору бензилоксипроизводного со стадии A (1 экв.) и 10% Pd-C (10% масс.) (0,1 экв.) в EtOH (4 мл) добавляли чистый TES (10 экв.) по каплям. Реакционную смесь перемешивали при комнатной температуре в течение 24-48 часов, затем смесь фильтровали через целит и растворитель удаляли в вакууме. Полученный неочищенный продукт подвергали хроматографической очистке на колонке с силикагелем (элюент: DCM/MeOH 95:5). Желаемое соединение выделяли путем упаривания органических фракций при пониженном давлении.
Общая процедура Ia для синтеза карбаматных производных I (Схема Ia).
Способ A.
К раствору ди-трет-бутилдикарбоната (1,4 экв.) в ацетонитриле добавляли последовательно: раствор DMAP (1 экв.) в ацетонитриле и раствор соответствующего амина III (1 экв.) в ацетонитриле. После перемешивания в течение 10 минут при комнатной температуре добавляли спиртовое производное IIa (1,2-1,4 экв.). Реакционную смесь перемешивали в течение 22 часов при комнатной температуре, после этого растворитель выпаривали при пониженном давлении. Неочищенный остаток растворяли в этилацетате и промывали насыщенным раствором NaHCO3. Органический слой сушили (Na2SO4), концентрировали и очищали флэш-хроматографией (элюент: 5% MeOH в DCM).
Способ B.
Аминовое производное III (1,0 экв.) обрабатывали п-нитрофенилхлорформиатом (1,1 экв.) и DIPEA (1,1 экв.) в 1:1 смеси DMA:DCM. Реакционную смесь перемешивали при температуре окружающей среды в течение 30 минут. К полученному раствору п-нитрофенилкарбамата добавляли спиртовое производное IIa (1,25 экв.) и DIPEA (1,1 экв., 2,2 всего) и полученную смесь перемешивали при комнатной температуре в течение 48-72 часов. Желаемый карбамат выделяли путем удаления нежелательного п-нитрофенольного побочного продукта и DMA путем промывки несколько раз насыщенным солевым раствором и водой, сбора и концентрации органической фазы и очистки флэш-хроматографией (элюент: 5% MeOH в DCM).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 1. 2-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]изоиндолин-1,3-дион
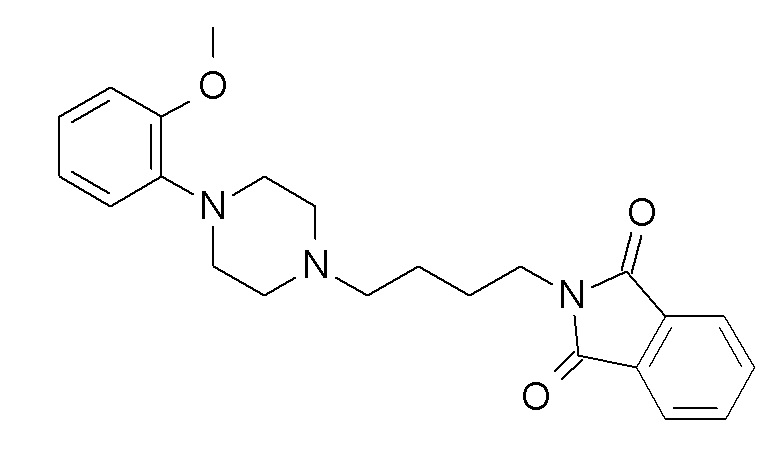
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-(2-метоксифенил)пиперазин гидрохлорида (2,62 ммоль, 600 мг), N-(4- бромбутил)фталимида (2,89 ммоль, 814 мг) и K2CO3 (7,87 ммоль, 1088 мг) в 7 мл ацетонитрила. Белое твердое вещество 953 мг (92%). ВЭЖХ-МС (способ A): Rt 1,27 мин; m/z 242 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,85 (дд, J=3,02, 5,46 Гц, 2H), 7,75-7,69 (м, 2H), 7,04-6,77 (м, 4H), 3,87 (с, 3H), 3,74 (т, J=7,06 Гц, 2H), 3,10 (с, 4H), 2,67 (с, 4H), 2,52-2,39 (м, 2H), 1,80-1,69 (м, 2H), 1,67-1,55 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 2. 2-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]изоиндолин-1,3-дион
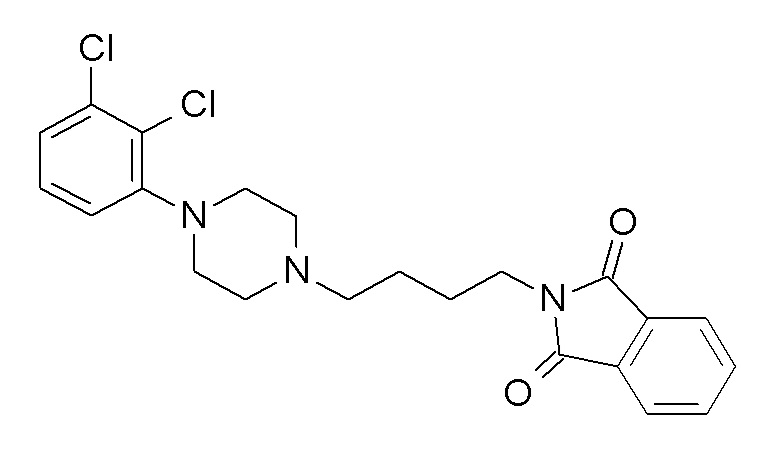
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-(2,3-дихлорфенил)пиперазин гидрохлорида (1,87 ммоль, 500 мг), N-(4-бромбутил)фталимида (2,06 ммоль, 579,9 мг) и K2CO3 (5,61 ммоль, 774,7 мг) в 7 мл ацетонитрила. Белое твердое вещество 771 мг (95%). ВЭЖХ-МС (способ A): Rt 2,66 мин; m/z 432 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,86 (дд, J=3,05, 5,42 Гц, 2H), 7,73 (дд, J=3,04, 5,45 Гц, 2H), 7,20-7,12 (м, 2H), 6,97 (дд, J=2,79, 6,76 Гц, 1H), 3,75 (т, J=7,07 Гц, 2H), 3,16-3,03 (м, 4H), 2,75-2,59 (м, 4H), 2,50 (т, J=7,60 Гц, 2H), 1,77 (тт, J=6,35, 7,59 Гц, 2H), 1,70-1,56 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 3. 2-[3-[4-(2,3-дихлорфенил)пиперазин-1-ил]пропил]изоиндолин-1,3-дион
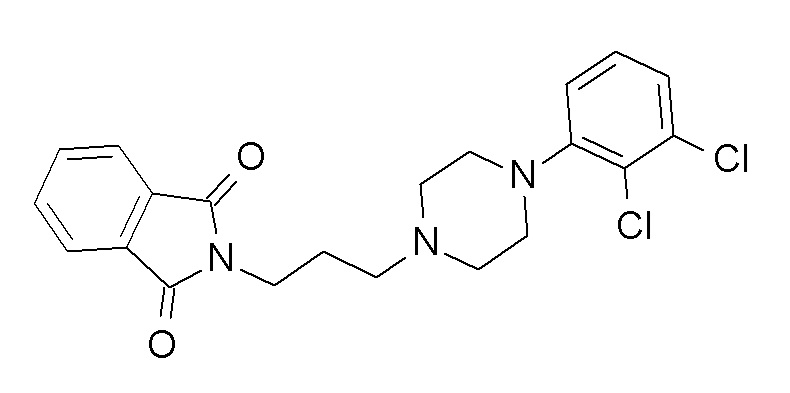
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-(2,3-дихлорфенил)пиперазин гидрохлорида (400,0 мг, 1,49 ммоль), N-(3-бромпропил)фталимида (440,8 мг, 1,64 ммоль) и K2CO3 (516,4 мг, 3,74 ммоль) в 7 мл ацетонитрила. Белое твердое вещество 534 мг (83%). ВЭЖХ-МС (способ A): Rt 2,92 мин; m/z 418 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,87 (дд, J=3,06, 5,44 Гц, 2H), 7,73 (дд, J=3,06, 5,44 Гц, 2H), 7,20-7,09 (м, 2H), 6,85 (дд, J=2,13, 7,37 Гц, 1H), 3,83 (т, J=6,86 Гц, 2H), 2,95 (м, 4H), 2,68-2,61 (м, 4H), 2,56 (т, J=7,05 Гц, 2H), 1,96 (п, J=6,79 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 4. 2-[3-(4-фенилпиперазин-1-ил)пропил]изоиндолин-1,3-дион
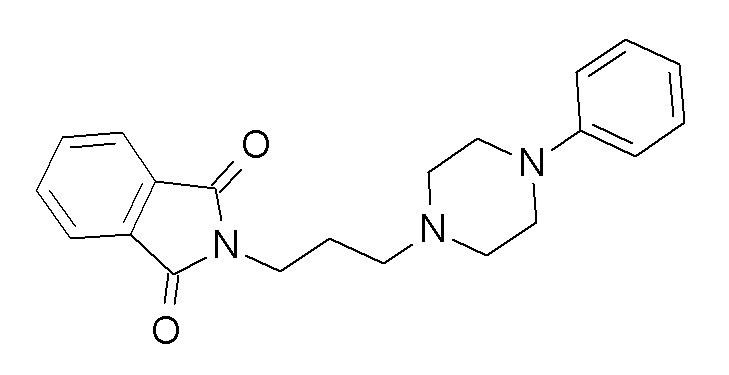
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-фенилпиперазин гидрохлорида (300,0 мг, 1,85 ммоль), N-(3-бромпропил)фталимида (545,4 мг, 2,03 ммоль) и K2CO3 (766,8 мг, 5,55 ммоль) в 7 мл ацетонитрила. Белое твердое вещество 600 мг (93%). ВЭЖХ-МС (способ A): Rt 2,14 мин; m/z 350 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,85 (дд, J=5,4, 3,0 Гц, 2H), 7,70 (дд, J=5,5, 3,0 Гц, 2H), 7,29-7,21 (м, 2H), 6,90-6,81 (м, 3H), 3,81 (т, J=6,9 Гц, 2H), 3,11-3,01 (м, 4H), 2,59-2,53 (м, 4H), 2,50 (т, J=6,9 Гц, 2H), 1,93 (п, J=6,9 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 5. 4-[4-(2-метоксифенил)пиперазин-1-ил]бутан-1-амин
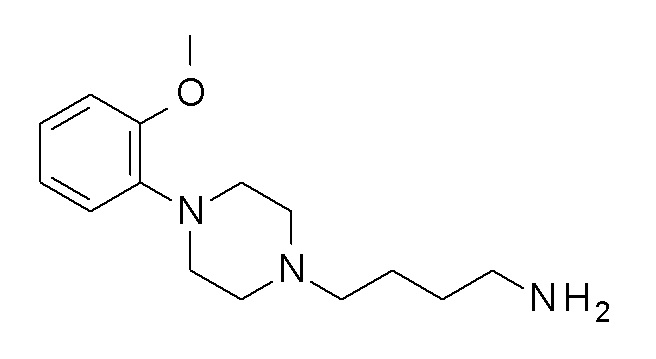
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 1 (953,0 мг, 2,42 ммоль) и гидрата гидразина (0,14 мл, 2,91 ммоль) в 3 мл метанола. Желтое масло 338 мг (53%). ВЭЖХ-МС (способ A): Rt 1,07 мин; m/z 263 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,04-6,90 (м, 3H), 6,87 (дт, J=8,0, 1,4 Гц, 1H), 3,88 (д, J=1,6 Гц, 3H), 3,12 (с, 4H), 2,74 (тд, J=6,8, 1,5 Гц, 2H), 2,68 (д, J=5,8 Гц, 4H), 2,49-2,39 (м, 2H), 1,64-1,54 (м, 2H), 1,50 (дтд, J=8,8, 6,9, 5,3 Гц, 2H), 1,36 (с, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 6. 4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутан-1-амин
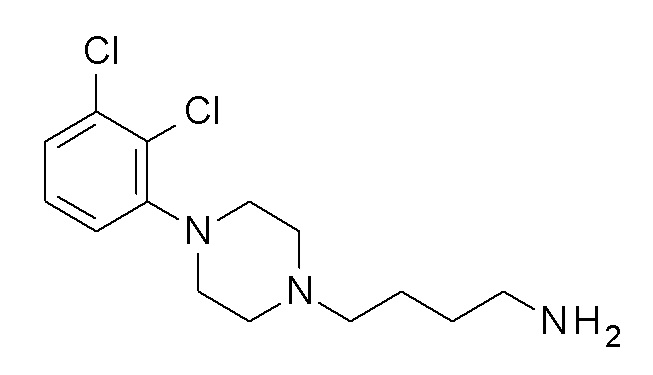
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 2 (771 мг, 1,78 ммоль) и гидрата гидразина (0,10 мл, 2,14 ммоль) в 3 мл метанола, затем 2 мл HCl. Желтое масло 351 мг (65%). ВЭЖХ-МС (способ A): Rt 1,64 мин; m/z 302 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,18-7,09 (м, 2H), 6,95 (дд, J=6,5, 3,1 Гц, 1H), 3,16-2,98 (м, 4H), 2,72 (т, J=6,8 Гц, 2H), 2,63 (д, J=7,0 Гц, 4H), 2,48-2,36 (м, 2H), 1,62-1,42 (м, 4H), 1,37 (с, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 7. 3-[4-(2,3-дихлорфенил)пиперазин-1-ил]пропан-1-амин
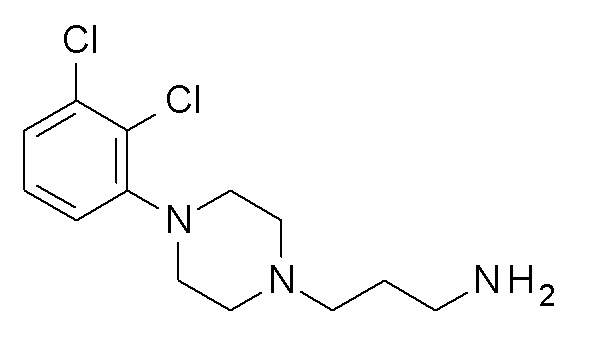
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 3 (534 мг, 1,28 ммоль) и гидрата гидразина (0,07 мл, 1,53 ммоль) в 4 мл метанола, затем 2 мл HCl. Желтое масло 233 мг (63%). ВЭЖХ-МС (способ A): Rt 1,81 мин; m/z 288 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,18-7,09 (м, 2H), 6,95 (дд, J=6,5, 3,1 Гц, 1H), 3,87 (с, 3H), 3,11 (с, 4H), 2,99 (т, J=6,2 Гц, 2H), 2,72 (с, 4H), 2,59 (т, J=6,5 Гц, 2H), 1,98 (с, 2H), 1,82 (п, J=6,4 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 8. 3-(4-фенилпиперазин-1-ил)пропан-1-амин
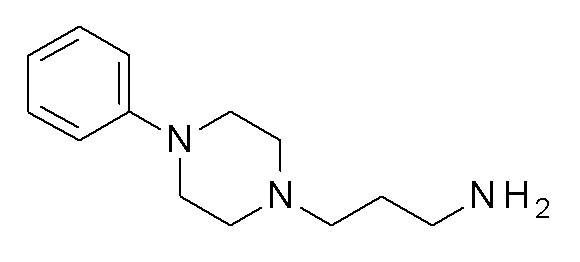
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 4 (600 мг, 1,72 ммоль) и гидрата гидразина (0,10 мл, 2,06 ммоль) в 4 мл метанола, затем 2 мл HCl. Желтое масло 278 мг (74%). ВЭЖХ-МС (способ A): Rt 1,09 мин; m/z 220 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,31-7,23 (м, 2H), 6,97-6,92 (м, 2H), 6,86 (тт, J=7,3, 1,1 Гц, 1H), 3,27-3,17 (м, 4H), 2,79 (т, J=6,8 Гц, 2H), 2,68-2,56 (м, 4H), 2,52-2,44 (м, 2H), 1,75-1,64 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 9. 3-(3-гидроксифенил)бензамид
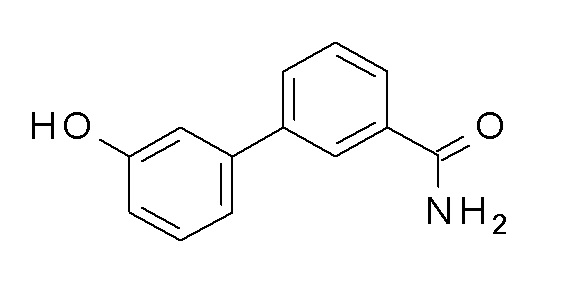
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой II, способ A, с использованием 3-бензамидбороновой кислоты (143,0 мг, 0,87 ммоль), 3-бромфенолы (100,0 мг, 0,58 ммоль) Pd(OAc)2 (1,3 мг, 0,01 ммоль) и K2CO3 (199,7 мг, 1,45 ммоль) в EGME/воде 3:1 (4 мл). Коричневое твердое вещество 119 мг (97%). ВЭЖХ-МС (способ A): Rt 1,59 мин; m/z 214 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 9,57 (с, 1H), 8,11 (т, J=1,86 Гц, 1H), 7,85 (дт, J=1,37, 7,80 Гц, 1H), 7,75 (дт, J=1,37, 7,75 Гц, 1H), 7,53 (т, J=7,72 Гц, 1H), 7,40 (с, 2H), 7,29 (т, J=7,84 Гц, 1H), 7,14 (дт, J=1,20, 7,74 Гц, 1H), 7,10 (т, J=2,10 Гц, 1H), 6,80 (дд, J=2,38, 7,96 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 10. 3-фенилфенол
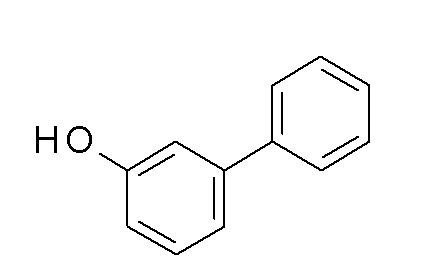
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой II, способ A, с использованием бензолбороновой кислоты (317,1 мг, 2,60 ммоль), 3-бромфенола (300,0 мг, 1,73 ммоль) Pd(OAc)2 (3,9 мг, 0,02 ммоль) и K2CO3 (599,2 мг, 4,34 ммоль) в EGME/воде 3:1 (8 мл). Коричневое твердое вещество 285 мг (97%). ВЭЖХ-МС (способ A): Rt 2,33 мин; m/z 169 [M]-. 1H ЯМР (400 МГц, DMSO-d6) δ 9,52 (с, 1H), 7,62-7,57 (м, 2H), 7,45 (ддд, J=7,8, 6,9, 1,2 Гц, 2H), 7,39-7,32 (м, 1H), 7,26 (т, J=7,8 Гц, 1H), 7,07 (ддд, J=7,7, 1,8, 1,0 Гц, 1H), 7,03 (т, J=2,0 Гц, 1H), 6,78 (ддт, J=8,3, 2,1, 1,0 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 11. 3-(4-гидроксифенил)бензамид
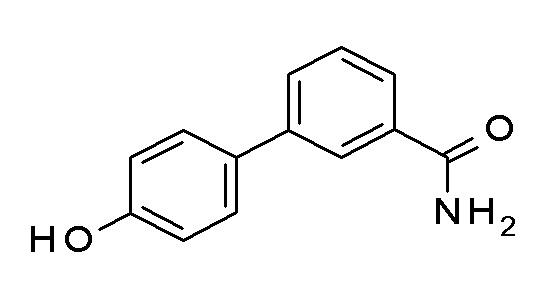
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой II, способ B, с использованием 4-гидроксибензолбороновой кислоты (450,0 мг, 3,26 ммоль), 3-бромбензамида (783,2 мг, 3,92 ммоль) Pd(OAc)2 (7,3 мг, 0,03 ммоль) и K2CO3 (1127,5 мг, 8,16 ммоль) в EGME/воде 3:1 (12 мл). Коричневое твердое вещество 625 мг (90%). ВЭЖХ-МС (способ A): Rt 1,53 мин; m/z 214 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 9,58 (с, 1H), 8,12-8,02 (м, 2H), 7,75 (дддд, J=20,2, 7,7, 1,8, 1,1 Гц, 2H), 7,61-7,52 (м, 2H), 7,48 (т, J=7,7 Гц, 1H), 7,37 (кв., J=3,6, 3,0 Гц, 1H), 6,92-6,83 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 12. 3-(4,4-дифтор-1-пиперидил)фенол
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой II, способ C, с использованием коммерчески доступного 1-бензилокси-3-бром-бензола (200,0 мг, 0,76 ммоль), 4,4-дифторпиперидин гидрохлорида (119,8 мг, 0,76 ммоль), Pd2(dba)3 (13,9 мг, 0,02 ммоль), DavePhos (17,9 мг, 0,06 ммоль) и t-BuO-K+ (170,6 мг, 1,52 ммоль) в безводном толуоле (5 мл). Вторую стадию осуществляли с использованием Pd-C (7,0 мг, 0,07 моль), триэтилсилана (1,05 мл, 6,59 ммоль) в EtOH (4 мл). Коричневое масло 63 мг (45%). ВЭЖХ-МС (способ A): Rt 2,10 мин; m/z 214 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,13 (т, J=8,1 Гц, 1H), 6,54 (ддд, J=8,2, 2,4, 0,8 Гц, 1H), 6,43 (т, J=2,3 Гц, 1H), 6,36 (ддд, J=8,0, 2,3, 0,8 Гц, 1H), 3,39-3,31 (м, 4H), 2,15-2,03 (м, 4H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 13. 2-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]изоиндолин-1,3-дион
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-(2-метоксифенил)пиперазин гидрохлорида (1000,0 мг, 4,37 ммоль), N-(3-бромпропил)фталимида (1289,4 мг, 4,81 ммоль) и K2CO3 (1510,7 мг, 10,93 ммоль) в 7 мл ацетонитрила. Белое твердое вещество 1549 мг (93%). ВЭЖХ-МС (способ A): Rt. 1,96 мин; m/z 380 [M-H]+, 1H ЯМР (400 МГц, DMSO-d6) δ 7,91-7,85 (м, 2H), 7,85-7,78 (м, 2H), 6,94-6,86 (м, 2H), 6,83 (ддд, J=7,8, 6,7, 2,1 Гц, 1H), 6,70 (дд, J=7,8, 1,5 Гц, 1H), 3,74 (с, 3H), 3,68 (т, J=6,7 Гц, 2H), 2,79-2,61 (м, 4H), 2,38 (т, J=6,5 Гц, 6H), 1,78 (п, J=6,6 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 14. 3-[4-(2-метоксифенил)пиперазин-1-ил]пропан-1-амин
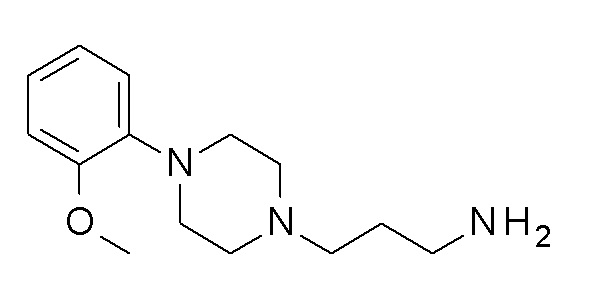
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 13 (1549 мг, 4,08 ммоль) и гидрата гидразина (0,24 мл, 4,90 ммоль) в 8 мл метанола. Желтое масло 631 мг (62%). ВЭЖХ-МС (способ A): Rt 1,13 мин; m/z 250 [M-H]+, 1H ЯМР (400 МГц, хлороформ-d) δ 7,01 (ддд, J=7,9, 6,4, 2,6 Гц, 1H), 6,98-6,90 (м, 2H), 6,87 (дд, J=8,0, 1,3 Гц, 1H), 3,87 (с, 3H), 3,11 (с, 4H), 2,99 (т, J=6,2 Гц, 2H), 2,72 (с, 4H), 2,59 (т, J=6,5 Гц, 2H), 1,98 (с, 2H), 1,82 (п, J=6,4 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 15. 4-(4,4-дифтор-1-пиперидил)фенол
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой II, способ C, с использованием коммерчески доступного 1-бензилокси-4-бром-бензола (200,0 мг, 0,76 ммоль), 4,4-дифторпиперидин гидрохлорида (119,8 мг, 0,76 ммоль), Pd2(dba)3 (13,9 мг, 0,02 ммоль), DavePhos (17,9 мг, 0,05 ммоль) и t-BuO-K+ (255,9 мг, 2,28 ммоль) в безводном толуоле (3 мл). Вторую стадию осуществляли с использованием Pd-C (7,0 мг, 0,07 моль), триэтилсилана (1,05 мл, 6,59 ммоль) в EtOH (4 мл). Коричневое масло 112 мг (69% за две стадии). ВЭЖХ-МС (способ A): Rt 2,00 мин; m/z 214 [M-H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 6,93-6,86 (м, 2H), 6,81-6,76 (м, 2H), 3,26-3,18 (м, 4H), 2,14 (тт, J=13,8, 5,7 Гц, 4H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 16. 2-[4-(4-фенил-1-пиперидил)бутил]изоиндолин-1,3-дион
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 4-фенилпиперидина (161 мг, 1,00 ммоль), N-(4-бромбутил)фталимида (296 мг, 1,05 ммоль), K2CO3 (152 мг, 1,50 ммоль) и MeCN (2 мл). Белое твердое вещество, 340 мг (94%). ВЭЖХ-МС (способ A): Rt 2,04 мин, m/z 363 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,91-7,80 (м, 2H), 7,76 (тд, J=5,2, 2,1 Гц, 2H), 7,38-7,20 (м, 5H), 3,76 (т, J=6,8 Гц, 2H), 3,46 (с, 2H), 2,92 (с, 2H), 2,78-2,53 (м, 3H), 2,41 (с, 2H), 1,91 (с, 4H), 1,81 (п, J=6,9 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 17. 2-[3-[4-[2-(трифторметил)фенил]пиперазин-1-ил]пропил]изоиндолин-1,3-дион
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-[2-(трифторметил)фенил]пиперазина (0,187 мл, 1,00 ммоль), N-(3-бромпропил)фталимида (281 мг, 1,00 ммоль), триэтиламина (0,348 мл, 2,50 ммоль) и MeCN (2 мл). Белое твердое вещество, 349 мг (84%). ВЭЖХ-МС (способ A): Rt 2,58 мин, m/z 418 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 7,95-7,84 (м, 4H), 7,61 (т, J=7,8 Гц, 2H), 7,30 (т, J=7,6 Гц, 1H), 7,21 (д, J=8,0 Гц, 1H), 3,69 (т, J=6,7 Гц, 2H), 2,59 (т, J=4,4 Гц, 4H), 2,39 (т, J=6,4 Гц, 6H), 1,78 (п, J=6,5 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 18. 2-[3-[4-(o-толил)пиперазин-1-ил]пропил]изоиндолин-1,3-дион
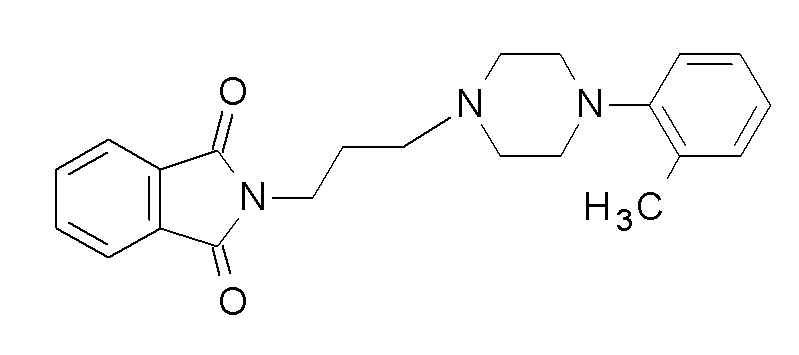
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-(o-толил)пиперазина (176 мг, 1,00 ммоль), N-(3-бромпропил)фталимида (281 мг, 1,00 ммоль), триэтиламина (0,348 мл, 2,50 ммоль) и MeCN (2 мл). Белое твердое вещество, 266 мг (73%). ВЭЖХ-МС (способ A): Rt 2,31 мин, m/z 364 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,09-7,57 (м, 4H), 7,18-7,01 (м, 2H), 6,91 (тд, J=7,4, 1,1 Гц, 1H), 6,77 (д, J=7,9 Гц, 1H), 3,69 (т, J=6,7 Гц, 2H), 2,56 (с, 3H), 2,40 (т, J=6,4 Гц, 5H), 2,18 (с, 3H), 1,79 (п, J=6,5 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 19. 4-(4-фенил-1-пиперидил)бутан-1-амин
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 16 (340 мг, 0,94 ммоль), гидрата гидразина (0,070 мл, 1,5 ммоль) в MeOH (5 мл), затем HCl (2 мл). Слегка желтоватое твердое вещество, 152 мг (70%). ВЭЖХ-МС (способ A): Rt 1,18 мин, m/z 233 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,35-7,14 (м, 5H), 3,08 (д, J=11,7 Гц, 2H), 2,75 (т, J=6,9 Гц, 2H), 2,51 (tq, J=12,5, 7,3, 6,1 Гц, 1H), 2,45-2,35 (м, 2H), 2,05 (тт, J=11,9, 5,8 Гц, 2H), 1,90-1,77 (м, 4H), 1,65-1,41 (м, 6H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 20. 3-[4-[2-(трифторметил)фенил]пиперазин-1-ил]пропан-1-амин
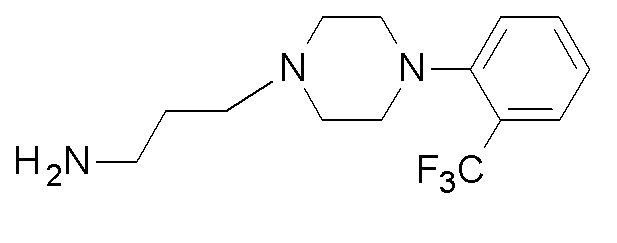
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 17 (349 мг, 0,84 ммоль), гидрата гидразина (0,059 мл, 1,5 ммоль) в MeOH (5 мл), затем HCl (2 мл). Бесцветный воск, 225 мг (94%). ВЭЖХ-МС (способ A): Rt 1,54 мин, m/z 288 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,63 (дд, J=7,9, 1,5 Гц, 1H), 7,55-7,46 (м, 1H), 7,39 (д, J=8,0 Гц, 1H), 7,22 (т, J=7,6 Гц, 1H), 2,98 (т, J=4,8 Гц, 4H), 2,80 (т, J=6,8 Гц, 2H), 2,62 (с, 4H), 2,54-2,43 (м, 2H), 1,70 (дт, J=14,1, 6,9 Гц, 2H), 1,58 (с, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 21. 3-[4-(o-толил)пиперазин-1-ил]пропан-1-амин
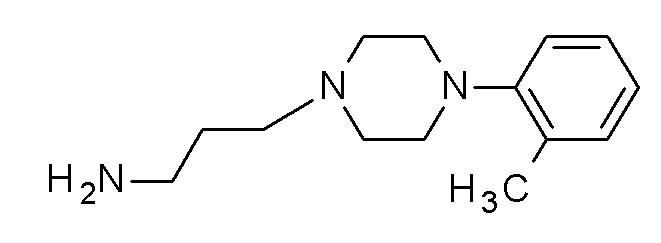
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 18 (266 мг, 0,73 ммоль), гидрата гидразина (0,059 мл, 1,5 ммоль) в MeOH (5 мл), затем HCl (2 мл). Бесцветный воск, 170 мг (99%). ВЭЖХ-МС (способ A): Rt 1,21 мин, m/z 234 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,18 (т, J=7,4 Гц, 2H), 7,05 (д, J=7,7 Гц, 1H), 6,99 (тд, J=7,4, 1,2 Гц, 1H), 2,96 (т, J=4,8 Гц, 4H), 2,80 (т, J=6,8 Гц, 2H), 2,63 (шир.с, 4H), 2,55-2,42 (м, 2H), 2,32 (с, 3H), 1,71 (дт, J=14,1, 6,9 Гц, 2H), 1,53 (шир.с, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 22. 3-(2-фтор-4-гидрокси-фенил)бензамид
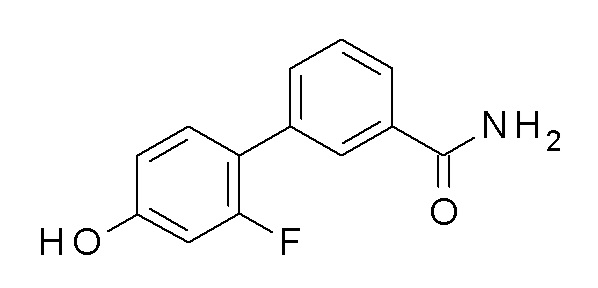
Указанное в заголовке соединение получали в соответствии с общей процедурой II, способ A. Исходя из 4-бром-3-фтор-фенола (250 мг, 1,32 ммоль), (3-карбамоилфенил)бороновой кислоты (260 мг, 1,58 ммоль), Pd(OAc)2 (3 мг, 0,01 ммоль) и 3:1 EGME/H2O (4 мл). Белое твердое вещество, 176 мг (58%). ВЭЖХ-МС (способ A): Rt 1,63 мин, m/z 232 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 10,07 (с, 1H), 8,03 (с, 1H), 8,00-7,93 (м, 1H), 7,83 (дт, J=7,7, 1,3 Гц, 1H), 7,66-7,58 (м, 1H), 7,51 (т, J=7,7 Гц, 1H), 7,44-7,35 (м, 2H), 6,78-6,65 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 23. 2-[(E)-4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бут-2-енил]изоиндолин-1,3 дион
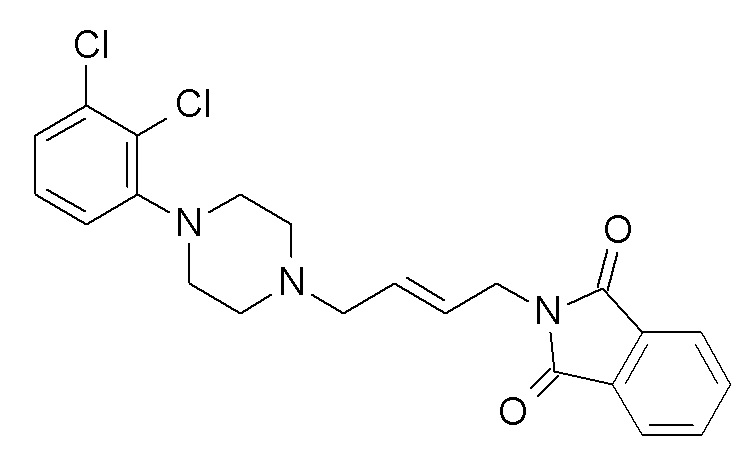
Указанное в заголовке соединение получали с использованием двухстадийного способа, исходя из калия изоиндолин-2-ид-1,3-диона и (E)-1,4-дихлорбут-2-ена, в присутствии DMF. Полученное таким образом производное использовали в общей процедуре III, стадия A, с использованием 1-(2,3-дихлорфенил)пиперазин гидрохлорида (250 мг, 0,93 ммоль), карбоната калия (323 мг, 2,34 ммоль) и MeCN (3 мл). Белое твердое вещество, 332 мг (83% за две стадии). ВЭЖХ-МС (способ A): Rt 2,73 мин, m/z 430 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,89-7,80 (м, 2H), 7,75-7,68 (м, 2H), 7,16-7,09 (м, 2H), 6,94 (тд, J=7,3, 6,7, 2,8 Гц, 1H), 5,74 (тт, J=4,4, 2,2 Гц, 2H), 4,33-4,28 (м, 2H), 3,12-2,99 (м, 6H), 2,71 (с, 1H), 2,60 (с, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 24. (E)-4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бут-2-ен-1-амин
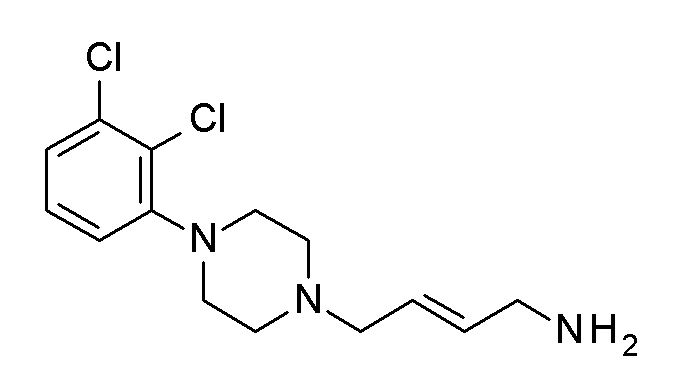
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 23 (332 мг, 0,77 ммоль), гидрата гидразина (0,040 мл, 0,93 ммоль) в MeOH (5 мл), затем HCl (2 мл). Слегка желтоватое твердое вещество, 203 мг (88%). ВЭЖХ-МС (способ A): Rt 1,57 мин, m/z 300 [M+H]+. 1H ЯМР (400 МГц, хлороформ-d) δ 7,10 (дд, J=5,5, 2,6 Гц, 2H), 6,95-6,89 (м, 1H), 5,74 (дт, J=15,3, 5,4 Гц, 1H), 5,63 (дддд, J=13,2, 8,2, 6,3, 3,0 Гц, 1H), 3,28 (д, J=5,1 Гц, 2H), 3,08-2,95 (м, 6H), 2,60 (с, 4H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 25. 2-[4-[4-[2-(трифторметил)фенил]пиперазин-1-ил]бутил]изоиндолин-1,3-дион
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-(2-трифторметилфенил)пиперазин гидрохлорида (1,52 ммоль, 350 мг), N-(4-бромбутил)фталимида (1,52 ммоль, 429 мг) и K2CO3 (3,8 ммоль, 525 мг) в 6 мл ацетонитрила. Белое твердое вещество 600 мг (91%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,86 (дд, J=5,4, 3,1 Гц, 2H), 7,77-7,69 (м, 2H), 7,62 (дд, J=7,9, 1,6 Гц, 1H), 7,54-7,48 (м, 1H), 7,38 (д, J=8,1 Гц, 1H), 7,25-7,17 (м, 1H), 3,75 (т, J=7,1 Гц, 2H), 2,95 (кв., J=4,7 Гц, 4H), 2,60 (с, 3H), 2,53-2,39 (м, 2H), 2,13 (с, 1H), 1,76 (тт, J=7,7, 6,5 Гц, 2H), 1,68-1,54 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 26. 4-[4-[2-(трифторметил)фенил]пиперазин-1-ил]бутан-1-амин
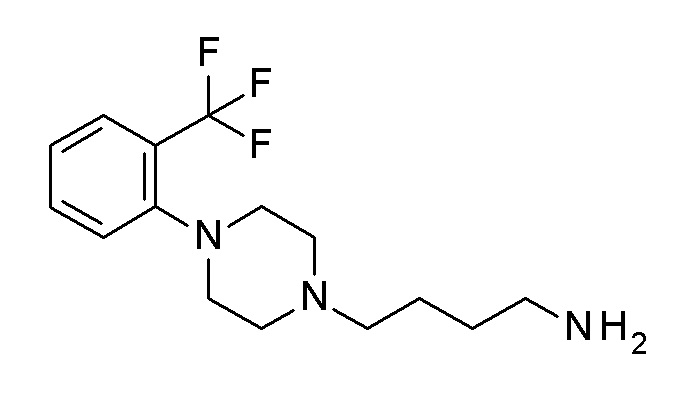
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 25 (600 мг, 1,39 ммоль) и гидрата гидразина (0,08 мл, 1,67 ммоль) в 4 мл метанола, затем 2 мл HCl. Желтое масло 312 мг (75%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,62 (дд, J=7,8, 1,6 Гц, 1H), 7,51 (тд, J=7,7, 1,6 Гц, 1H), 7,38 (д, J=7,8 Гц, 1H), 7,25-7,17 (м, 1H), 3,05-2,93 (м, 4H), 2,73 (т, J=6,8 Гц, 2H), 2,60 (м, 4H), 2,50-2,36 (м, 2H), 1,61-1,42 (м, 4H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 27. 4-(2-пиридил)фенол
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой II, способ A, с использованием 4-гидроксифенилбороновой кислоты (200,0 мг, 1,45 ммоль), 2-бромпиридина (0,14 мл, 1,45 ммоль) Pd(OAc)2 (3,3 мг, 0,01 ммоль) и K2CO3 (601 мг, 4,35 ммоль) в EGME/воде 3:1 (4 мл). Коричневое твердое вещество 137 мг (55%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,71 (с, 1H), 8,59 (ддд, J=4,8, 1,7, 1,1 Гц, 1H), 8,00-7,86 (м, 2H), 7,89-7,73 (м, 2H), 7,24 (ддд, J=6,7, 4,8, 1,8 Гц, 1H), 6,91-6,83 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 28. 2-[4-[4-(o-толил)пиперазин-1-ил]бутил]изоиндолин-1,3-дион
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия A, с использованием 1-(2-метилфенил)пиперазина (1,99 ммоль, 350 мг), N-(4- бромбутил)фталимида (1,99 ммоль, 560 мг) и K2CO3 (4,97 ммоль, 686 мг) в 6 мл ацетонитрила. Белое твердое вещество 750 мг (99%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,92-7,79 (м, 2H), 7,72 (дт, J=5,5, 3,1 Гц, 2H), 7,16 (т, J=7,6 Гц, 2H), 7,08-6,88 (м, 2H), 3,74 (т, J=7,1 Гц, 2H), 2,93 (т, J=4,8 Гц, 4H), 2,60 (с, 4H), 2,52-2,39 (м, 2H), 2,30 (с, 3H), 1,75 (п, J=7,3 Гц, 2H), 1,59 (тт, J=9,7, 5,9 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 29. 4-[4-(o-толил)пиперазин-1-ил]бутан-1-амин
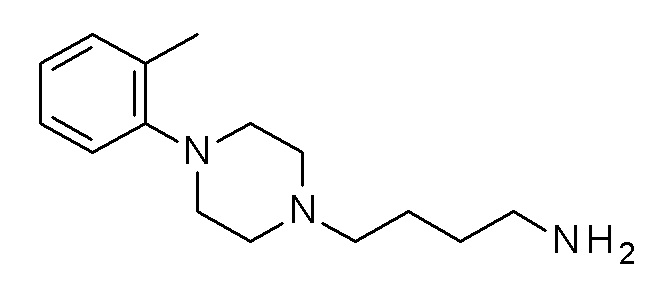
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 28 (750 мг, 1,99 ммоль) и гидрата гидразина (0,12 мл, 2,38 ммоль) в 4 мл метанола, затем 2 мл HCl. Желтое масло 384 мг (78%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,18 (т, J=7,4 Гц, 2H), 7,05 (дд, J=7,5, 1,5 Гц, 1H), 6,99 (тд, J=7,4, 1,3 Гц, 1H), 2,97 (т, J=4,8 Гц, 4H), 2,75 (т, J=6,8 Гц, 2H), 2,63 (с, 4H), 2,53-2,39 (м, 2H), 1,63-1,46 (м, 4H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 30. 3-метокси-4-фенил-фенол
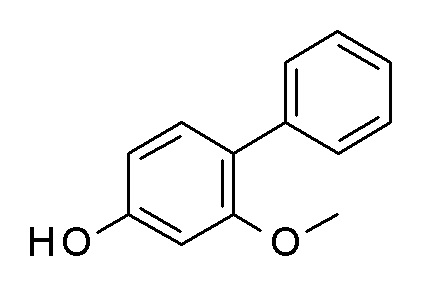
В 50-мл колбу, содержащую магнитную мешалку, загружали PdCl2(dppf) (21 мг, 0,03 ммоль), бензолбороновую кислоту (270 мг, 2,22 ммоль), K2CO3 (612 мг, 4,43 ммоль) и 4-бром-3-метокси-фенол (300 мг, 1,48 ммоль). Твердые вещества полностью растворяли в 3:1 смеси диоксана и воды (4 мл) и реакционную смесь интенсивно перемешивали при 90°C в течение 3 часов. Реакционную смесь разбавляли водой и подкисляли при помощи 2 н раствора HCl и экстрагировали при помощи DCM. Органическую фазу собирали и упаривали в вакууме. Неочищенное вещество очищали колоночной хроматографией на силикагеле (элюент: циклогексан/этилацетат). Органические фракции собирали и упаривали в вакууме. Желтоватое масло 250 мг (84%). 1H ЯМР (400 МГц, DMSO-d6) δ 9,59 (с, 1H), 7,43-7,39 (м, 2H), 7,35 (дд, J=8,4, 6,8 Гц, 2H), 7,27-7,21 (м, 1H), 7,08 (д, J=8,2 Гц, 1H), 6,51 (д, J=2,2 Гц, 1H), 6,44 (дд, J=8,2, 2,3 Гц, 1H), 3,71 (с, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 31. 2-(2-бромэтил)оксиран
4-Бромбут-1-ен (1000 мг, 7,41 ммоль) растворяли в DCM (30 мл) и затем в раствор добавляли по порциям m-CPBA (1917 мг, 11,11 ммоль). Смесь перемешивали в течение ночи при комнатной температуре, затем нейтрализовали при помощи 0,1 н раствора гидроксида натрия, экстрагировали при помощи DCM и сушили над безводным Na2SO4. Растворители удаляли с получением бесцветного масла, которое можно использовать непосредственно на следующей стадии. Бесцветное масло 1050 мг (94%). 1H ЯМР (400 МГц, хлороформ-d) δ 3,54 (дд, J=7,4, 5,9 Гц, 2H), 3,12 (дтд, J=6,7, 4,2, 2,7 Гц, 1H), 2,87 (дд, J=4,9, 4,0 Гц, 1H), 2,61 (дд, J=4,9, 2,6 Гц, 1H), 2,19 (дтд, J=14,8, 7,4, 4,5 Гц, 1H), 2,08 (дкв., J=14,7, 6,1 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 32. 2-[2-(оксиран-2-ил)этил]изоиндолин-1,3-дион
Суспензию соли фталимида калия (800 мг, 4,32 ммоль) в DMF (15 мл) обрабатывали ПРОМЕЖУТОЧНЫМ СОЕДИНЕНИЕМ 31 (978 мг, 6,48 ммоль), нагревали при 100°C в течение ночи. Охлажденную реакционную смесь фильтровали, разбавляли при помощи EtOAc и промывали при помощи H2O. Органическую фазу сушили над сульфатом натрия и летучие вещества удаляли в вакууме с получением елаемого соединения в виде пенистого вещества, которое использовали без дополнительной очистки. Белое пенистое вещество 890 мг (95%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,86 (дд, J=5,5, 3,0 Гц, 2H), 7,76-7,68 (м, 2H), 3,54 (дд, J=7,4, 5,9 Гц, 2H), 3,12 (дтд, J=6,8, 4,2, 2,7 Гц, 1H), 2,87 (дд, J=4,9, 4,0 Гц, 1H), 2,61 (дд, J=4,9, 2,6 Гц, 1H), 2,19 (дтд, J=14,8, 7,4, 4,5 Гц, 1H), 2,08 (дкв., J=14,7, 6,1 Гц, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 33. 2-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-гидрокси-бутил]изоиндолин-1,3-дион
1-(2,3-Дихлорфенил)пиперазин гидрохлорид (1,38 ммоль, 369 мг) в 2-PrOH (15 мл) перемешивали с ПРОМЕЖУТОЧНЫМ СОЕДИНЕНИЕМ 32 (300 мг, 1,38 ммоль) в присутствии Et3N (0,4 мл, 2,76 ммоль) при 60°C в течение 6 часов, затем при комнатной температуре в течение ночи. Неочищенное вещество упаривали и остаток адсорбировали на силикагеле и очищали колоночной хроматографией (элюент: циклогексан/этилацетат). Органические фракции собирали и упаривали в вакууме. Желтое масло 350 мг (57%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,86 (дд, J=3,05, 5,42 Гц, 2H), 7,73 (дд, J=3,04, 5,45 Гц, 2H), 7,20-7,12 (м, 2H), 6,97 (дд, J=2,79, 6,76 Гц, 1H), 3,10 (д, J=24,5 Гц, 8H), 2,61 (д, J=6,4 Гц, 1H), 2,50-2,34 (м, 3H), 1,86-1,74 (м, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 34. 2-[3-гидрокси-4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]изоиндолин-1,3-дион
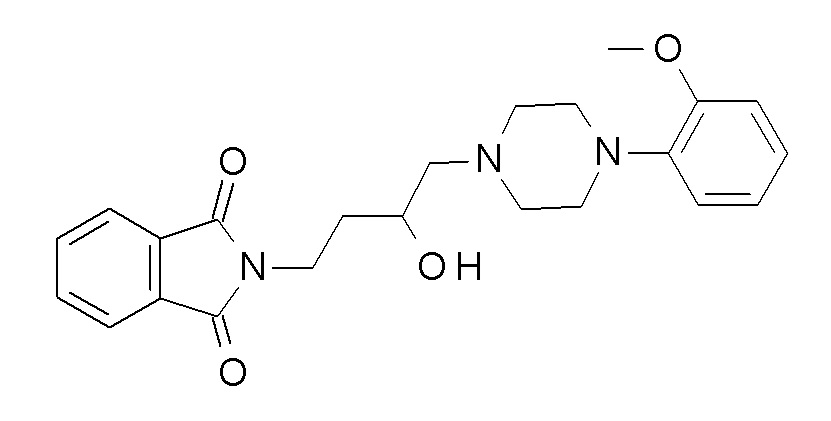
1-(2-Метоксифенил)пиперазин гидрохлорид (1,84 ммоль, 421 мг) в 2-PrOH (15 мл) подвергали взаимодействию в микроволновой печи (100°C, 90 мин) с ПРОМЕЖУТОЧНЫМ СОЕДИНЕНИЕМ 32 (400 мг, 1,84 ммоль) в присутствии Et3N (0,5 мл, 3,68 ммоль). Неочищенное вещество упаривали и остаток адсорбировали на силикагеле и очищали колоночной хроматографией (элюент: циклогексан/этилацетат). Органические фракции собирали и упаривали в вакууме. Желтое масло 409 мг (54%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,86 (дд, J=5,5, 3,0 Гц, 2H), 7,76-7,68 (м, 2H), 7,00 (ддд, J=7,9, 5,7, 3,4 Гц, 1H), 6,95-6,89 (м, 2H), 6,89-6,82 (м, 1H), 3,86 (с, 3H), 3,10 (д, J=24,5 Гц, 4H), 2,89 (д, J=0,6 Гц, 6H), 2,61 (д, J=6,4 Гц, 1H), 2,50-2,34 (м, 2H), 1,86-1,74 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 35. 2-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-фтор-бутил]изоиндолин-1,3-дион
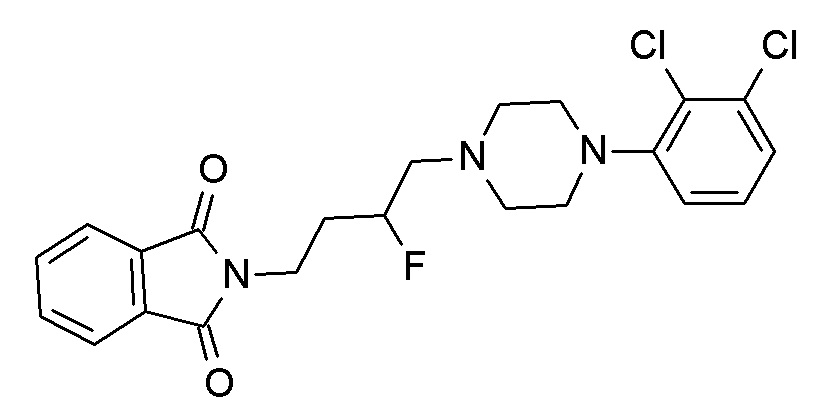
К раствору DeOxo-Fluor® (0,58 мл, 2,68 ммоль) в безводном DCM (10 мл) при -8°C добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 33 (300 мг, 0,67 ммоль). Эту реакционную смесь непрерывно перемешивали в течение 72 часов при комнатной температуре. Реакционную смесь разбавляли при помощи DCM и промывали водой и NaHCO3 и органический слой сушили над Na2SO4. Растворитель удаляли в вакууме с получением неочищенного продукта, который затем очищали колоночной хроматографией на силикагеле (элюент: циклогексан/этилацетат 1:1). Органические фракции собирали и растворитель удаляли при пониженном давлении. Желтое масло 150 мг (50%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,88 (ддд, J=5,4, 4,7, 3,0 Гц, 2H), 7,76 (ддд, J=9,7, 5,5, 3,0 Гц, 2H), 7,19-7,12 (м, 1H), 6,96 (дд, J=6,6, 3,0 Гц, 1H), 4,96-4,70 (м, 1H), 3,97-3,83 (м, 2H), 3,07 (м, 4H), 2,82-2,69 (м, 4H), 2,67-2,48 (м, 1H), 2,35-2,19 (м, 1H), 2,19-1,94 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 36. 2-[3-фтор-4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]изоиндолин-1,3-дион
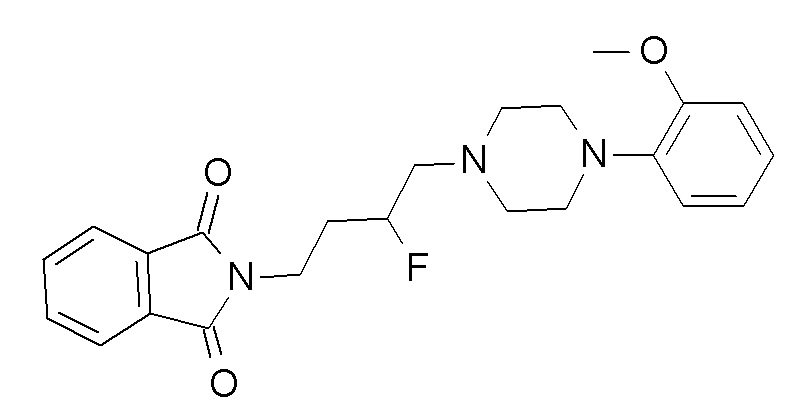
К раствору DeOxo-Fluor® (0,64 мл, 2,93 ммоль) в безводном DCM (10 мл) при -8°C добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 34 (300 мг, 0,73 ммоль). Эту реакционную смесь непрерывно перемешивали в течение 72 часов при комнатной температуре. Реакционную смесь разбавляли при помощи DCM и промывали водой и NaHCO3 и органический слой сушили над Na2SO4. Растворитель удаляли в вакууме с получением неочищенного продукта, который затем очищали колоночной хроматографией на силикагеле (элюент: циклогексан/этилацетат 1:1). Органические фракции собирали и растворитель удаляли при пониженном давлении. Желтое масло 200 мг (66%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,91-7,83 (м, 2H), 7,74 (дд, J=5,4, 3,0 Гц, 2H), 7,02 (ддд, J=7,8, 6,1, 2,9 Гц, 1H), 6,97-6,91 (м, 2H), 6,91-6,84 (м, 1H), 4,82 (дддд, J=49,9, 11,1, 7,1, 3,3 Гц, 1H), 3,91 (дд, J=7,1, 2,4 Гц, 2H), 3,88 (с, 3H), 3,10 (с, 4H), 2,82-2,69 (м, 5H), 2,67-2,49 (м, 1H), 2,21-2,07 (м, 1H), 1,65 (м, 1H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 37. 4-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-фтор-бутан-1-амин
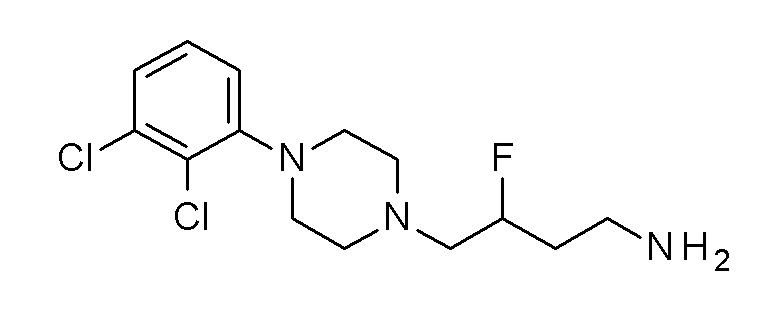
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 35 (150 мг, 0,33 ммоль) и гидрата гидразина (0,02 мл, 0,40 ммоль) в 4 мл метанола, затем 2 мл HCl. Желтое масло 80 мг (75%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,19-7,12 (м, 1H), 6,96 (дд, J=6,6, 3,0 Гц, 1H), 4,96-4,70 (м, 1H), 3,97-3,83 (м, 2H), 3,07 (м, 4H), 2,82-2,69 (м, 4H), 2,67-2,48 (м, 1H), 2,35-2,19 (м, 1H), 2,19-1,94 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 38. 3-фтор-4-[4-(2-метоксифенил)пиперазин-1-ил]бутан-1-амин
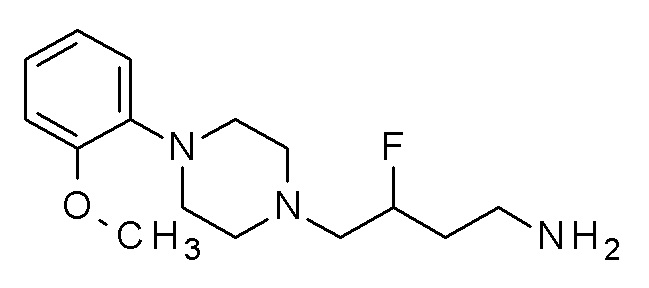
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 36 (200 мг, 0,49 ммоль) и гидрата гидразина (0,03 мл, 0,58 ммоль) в 4 мл метанола, затем 2 мл HCl. Желтое масло 130 мг (95%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,02 (ддд, J=7,7, 6,7, 2,3 Гц, 1H), 6,99-6,92 (м, 2H), 6,88 (дд, J=8,0, 1,3 Гц, 1H), 4,88 (дддд, J=50,4, 11,6, 7,2, 3,3 Гц, 1H), 3,88 (с, 3H), 3,13 (с, 4H), 2,98-2,84 (м, 2H), 2,84-2,69 (м, 5H), 2,56 (ддд, J=30,8, 13,9, 2,9 Гц, 1H), 1,96-1,64 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 39. 3-(4-гидрокси-2-метокси-фенил)бензамид
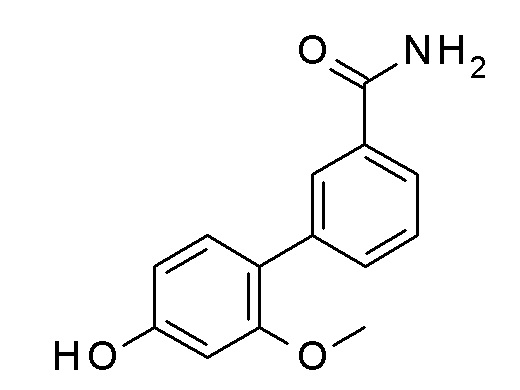
В 50-мл колбу, содержащую магнитную мешалку, загружали PdCl2(dppf) (21 мг, 0,03 ммоль), 3-бензамидбороновую кислоту (365 мг, 2,22 ммоль), K2CO3 (612 мг, 4,43 ммоль) и 4-бром-3-метокси-фенол (300 мг, 1,48 ммоль). Твердые вещества полностью растворяли в 3:1 смеси диоксана и воды (4 мл) и реакционную смесь интенсивно перемешивали при 90°C в течение 3 часов. Реакционную смесь разбавляли водой и подкисляли при помощи 2 н раствора HCl и экстрагировали при помощи DCM. Органическую фазу собирали и упаривали в вакууме. Неочищенное вещество очищали колоночной хроматографией на силикагеле (элюент: циклогексан/этилацетат). Органические фракции собирали и упаривали в вакууме. Желтоватое масло 100 мг (28%). 1H ЯМР (400 МГц, хлороформ-d) δ 8,19 (т, J=2,0 Гц, 1H), 7,73 (дт, J=7,5, 2,0 Гц, 1H), 7,66 (дт, J=7,5, 2,0 Гц, 1H), 7,50 (т, J=7,4 Гц, 1H), 7,41 (д, J=7,5 Гц, 1H), 7,00 (с, 1H), 6,68 (д, J=2,1 Гц, 1H), 6,64 (дд, J=7,5, 2,0 Гц, 1H), 6,21 (с, 2H), 3,90 (с, 3H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 40. 2-[3-[4-(2,3-дихлорфенил)пиперазин-1-ил]-2-гидрокси-пропил]изоиндолин-1,3-дион
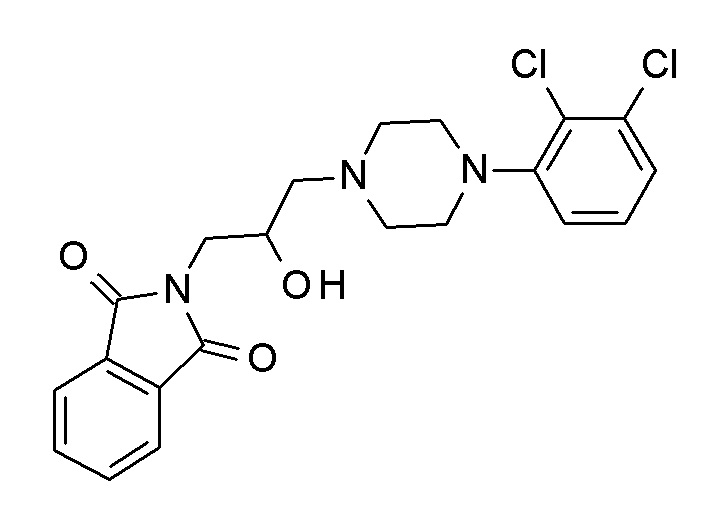
1-(2,3-Дихлорфенил)пиперазин моногидрохлорид (1185 мг, 4,43 ммоль) в 2-PrOH (10 мл) подвергали взаимодействию в микроволновой печи (сосуд высокого давления, Pmax, 200 Вт, охлаждение, 100°C, 90 мин) с N-(2,3-эпоксипропил)фталимидом (900 мг, 4,43 ммоль) в присутствии Et3N (1,2 мл, 8,86 ммоль). Неочищенное вещество фильтровали и остаток промывали водой. Белое твердое вещество 1900 мг (99%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,92-7,84 (м, 2H), 7,75 (дд, J=5,5, 3,0 Гц, 2H), 7,21-7,11 (м, 2H), 6,99-6,92 (м, 1H), 4,11 (ддт, J=9,1, 7,0, 4,5 Гц, 1H), 3,90-3,75 (м, 2H), 3,14-2,99 (м, 4H), 2,84 (дт, J=10,1, 4,7 Гц, 2H), 2,66 (д, J=8,4 Гц, 2H), 2,61-2,46 (м, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 41. 2-[2-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-фтор-пропил]изоиндолин-1,3-дион
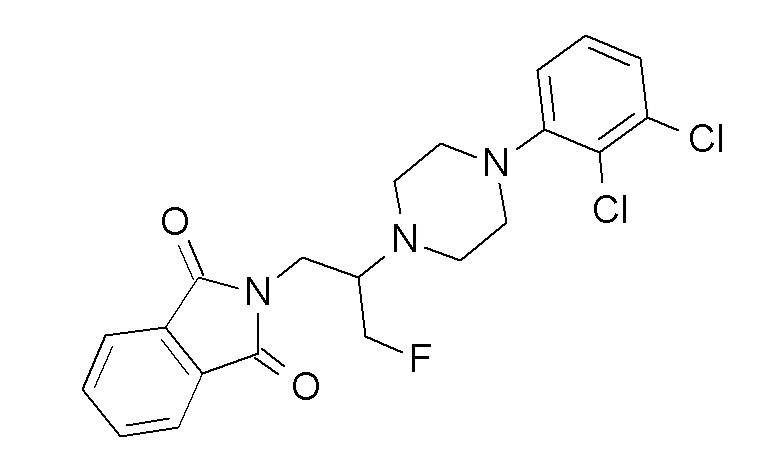
Это промежуточное соединение получали через фторирование ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 40 с использованием DeOxo-Fluor®. Это получение включает 1,2-перегруппировку 1,3-бифункционализированного амино-2-пропанола в 1,2-бифункционализированный амино-1-фторметилен, аналогично перегруппировкам, подобным тем, которые наблюдали с DAST, которые были описаны Ji-Wang Chern et al, Tetrahedron Letters, 1998, 39: 8483-8486.
К раствору DeOxo-Fluor® (1,40 мл, 6,45 ммоль) в безводном DCM (10 мл) при -8°C добавляли ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 40 (700 мг, 1,61 ммоль). Эту реакционную смесь непрерывно перемешивали в течение 24 часов при комнатной температуре. Реакционную смесь разбавляли при помощи DCM и промывали водой и NaHCO3 и органический слой сушили над Na2SO4. Растворитель удаляли в вакууме с получением неочищенного продукта, который затем очищали колоночной хроматографией на силикагеле (элюент: циклогексан/этилацетат 1:1). Органические фракции собирали и растворитель удаляли при пониженном давлении. Желтое масло 303 мг (43%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,94-7,85 (м, 2H), 7,79-7,73 (м, 2H), 7,18-7,08 (м, 2H), 6,88 (дд, J=7,5, 2,1 Гц, 1H), 4,83-4,56 (м, 2H), 4,18-4,04 (м, 1H), 3,72 (дд, J=14,1, 5,8 Гц, 1H), 3,41-3,26 (м, 1H), 3,09 (дт, J=10,1, 4,7 Гц, 2H), 2,93 (с, 4H), 2,74 (дт, J=9,9, 4,6 Гц, 2H).
ПРОМЕЖУТОЧНОЕ СОЕДИНЕНИЕ 42. 2-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-фтор-пропан-1-амин
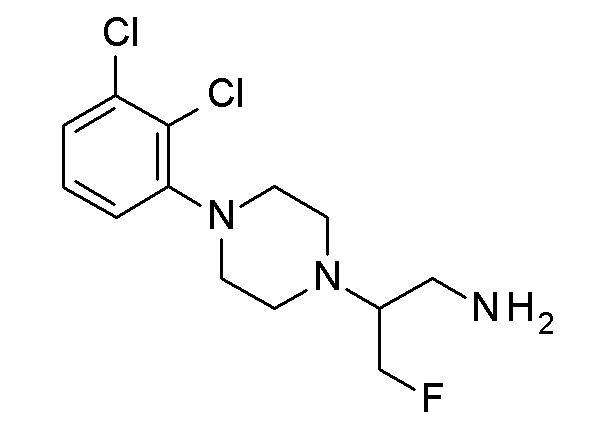
Указанное в заголовке соединение синтезировали, применяя общую процедуру III, стадия B, с использованием ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 41 (300 мг, 0,69 ммоль) и гидрата гидразина (0,04 мл, 0,83 ммоль) в 4 мл метанола, затем 2 мл HCl. Желтое масло 157 мг (75%). 1H ЯМР (400 МГц, хлороформ-d) δ 7,20-7,14 (м, 2H), 6,97 (дд, J=6,7, 2,9 Гц, 1H), 4,75-4,54 (м, 2H), 3,15-2,72 (м, 7H), 1,55 (с, 4H).
ПРИМЕР 1. [3-(3-карбамоилфенил)фенил] N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (153,1 мг, 0,70 ммоль), 4-диметиламинопиридина (61,2 мг, 0,50 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 5 (132,0 мг, 0,50 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 9 (149,6 мг, 0,70 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 51 мг (20%). ВЭЖХ-МС (способ A): Rt 1,90 мин; m/z 503 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,19-8,10 (м, 2H), 7,92-7,79 (м, 3H), 7,62-7,36 (м, 5H), 7,17-7,10 (м, 1H), 6,99-6,81 (м, 4H), 3,77 (с, 3H), 3,12 (кв., J=6,16, 6,56 Гц, 2H), 2,98 (м, 4H), 2,56 (м, 4H), 2,40 (м, 2H), 1,54 (м, 4H).
ПРИМЕР 2. [3-(3-карбамоилфенил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (50,5 мг, 0,23 ммоль), 4-диметиламинопиридина (20,2 мг, 0,17 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (50,0 мг, 0,17 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 9 (49,4 мг, 0,23 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 51 мг (57%). ВЭЖХ-МС (способ A): Rt 2,36 мин; m/z 541 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,19-8,11 (м, 2H), 7,91-7,80 (м, 3H), 7,62-7,42 (м, 5H), 7,35-7,26 (м, 2H), 7,19-7,07 (м, 2H), 3,12 (д, J=6,19 Гц, 2H), 2,99 (с, 4H), 2,55 (с, 4H), 2,38 (д, J=6,39 Гц, 2H), 1,56-1,49 (м, 4H).
ПРИМЕР 3. [3-(3-карбамоилфенил)фенил] N-[3-[4-(2,3-дихлорфенил)пиперазин-1-ил]пропил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (95,4 мг, 0,44 ммоль), 4-диметиламинопиридина (38,1 мг, 0,31 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 7 (90 мг, 0,31 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 9 (79,9 мг, 0,37 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 45 мг (27%). ВЭЖХ-МС (способ A): Rt 2,44 мин; m/z 527 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,17 (м, 1H), 8,12 (м, 1H), 7,92-7,84 (м, 1H), 7,84 (м, 2H), 7,62-7,45 (м, 4H), 7,41 (м, 1H), 7,30 (кв., J=3,71, 4,39 Гц, 2H), 7,20-7,08 (м, 2H) 3,16 (кв., J=6,51 Гц, 2H), 3,00 (т, J=4,84 Гц, 4H), 2,56 (м, 4H), 2,42 (т, J=7,04 Гц, 2H), 1,69 (п, J=6,95 Гц, 2H).
ПРИМЕР 4. [3-(3-карбамоилфенил)фенил] N-[3-(4-фенилпиперазин-1-ил)пропил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (167,2 мг, 0,77 ммоль), 4-диметиламинопиридина (66,8 мг, 0,55 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 8 (120,0 мг, 0,55 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 9 (140,0 мг, 0,66 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 40 мг (16%). ВЭЖХ-МС (способ A): Rt 1,94 мин; m/z 459 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,16 (т, J=1,7 Гц, 1H), 8,15-8,10 (м, 1H), 7,88 (дт, J=7,8, 1,4 Гц, 1H), 7,86 (м, 2H), 7,62-7,45 (м, 4H), 7,46-7,39 (м, 1H), 7,26-7,15 (м, 2H), 7,14 (ддд, J=8,0, 2,3, 1,0 Гц, 1H), 6,97-6,89 (м, 2H), 6,77 (тт, J=7,2, 1,0 Гц, 1H), 3,11-3,19 (м, 6H), 2,57-2,52 (м, 4H), 2,40 (т, J=7,1 Гц, 2H), 1,75-1,64 (м, 2H).
ПРИМЕР 5. (3-фенилфенил) N-[3-(4-фенилпиперазин-1-ил)пропил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (206,2 мг, 0,94 ммоль), 4-диметиламинопиридина (82,4 мг, 0,67 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 8 (148,0 мг, 0,67 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 10 (137,8 мг, 0,81 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 66 мг (24%). ВЭЖХ-МС (способ A): Rt 2,66 мин; m/z 416 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 7,81 (т, J=5,7 Гц, 1H), 7,71-7,64 (м, 2H), 7,56-7,44 (м, 4H), 7,42-7,31 (м, 2H), 7,25-7,17 (м, 2H), 7,13 (дддд, J=11,4, 7,7, 2,4, 1,2 Гц, 1H), 6,97-6,89 (м, 2H), 6,77 (тт, J=7,2, 1,0 Гц, 1H), 3,20-3,09 (м, 6H), 2,53-2,50 (м, 4H), 2,39 (кв., J=6,7 Гц, 2H), 1,70 (п, J=7,0 Гц, 2H).
ПРИМЕР 6. [4-(3-карбамоилфенил)фенил] N-[3-[4-(2,3-дихлорфенил)пиперазин-1-ил]пропил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (84,8 мг, 0,39 ммоль), 4-диметиламинопиридина (33,9 мг, 0,28 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 7 (80,0 мг, 0,28 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 11 (82,9 мг, 0,39 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 34 мг (23%). ВЭЖХ-МС (способ A): Rt 2,34 мин; m/z 527 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,15 (т, J=1,7 Гц, 1H), 8,09 (с, 1H), 7,86 (дт, J=7,7, 1,4 Гц, 1H), 7,84-7,78 (м, 2H), 7,78-7,70 (м, 2H), 7,55 (т, J=7,7 Гц, 1H), 7,41 (с, 1H), 7,35-7,27 (м, 2H), 7,27-7,20 (м, 2H), 7,16 (дд, J=6,6, 3,0 Гц, 1H), 3,15 (кв., J=6,6 Гц, 2H), 3,05-2,97 (м, 4H), 2,57 (с, 4H), 2,43 (т, J=7,0 Гц, 2H), 1,70 (кв., J=7,0 Гц, 2H).
ПРИМЕР 7. [4-(3-карбамоилфенил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (80,9 мг, 0,37 ммоль), 4-диметиламинопиридина (32,3 мг, 0,26 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (80,0 мг, 0,26 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 11 (79,0 мг, 0,37 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 59 мг (41%). ВЭЖХ-МС (способ A): Rt 2,32 мин; m/z 541 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,15 (т, J=1,8 Гц, 1H), 8,09 (с, 1H), 7,83 (дддд, J=17,5, 7,8, 2,3, 1,2 Гц, 3H), 7,78-7,70 (м, 2H), 7,55 (т, J=7,7 Гц, 1H), 7,41 (с, 1H), 7,34-7,27 (м, 2H), 7,26-7,19 (м, 2H), 7,15 (дд, J=6,5, 3,1 Гц, 1H), 3,12 (кв., J=6,3 Гц, 2H), 3,00 (д, J=5,0 Гц, 4H), 2,56 (с, 4H), 2,39 (д, J=6,8 Гц, 2H), 1,60-1,47 (м, 4H).
ПРИМЕР 8. (3-морфолинoфенил) N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (101,1 мг, 0,46 ммоль), 4-диметиламинопиридина (40,4 мг, 0,33 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (100,0 мг, 0,33 ммоль) и 3-морфолинoфенола (83,0 мг, 0,46 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире, и указанное в заголовке соединение выделяли в виде гидрохлоридной соли. Белое твердое вещество 30 мг (17%). ВЭЖХ-МС (способ A): Rt 2,44 мин; m/z 507 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 11,06-10,87 (м, 1H), 7,75 (т, J=5,6 Гц, 1H), 7,37 (д, J=7,0 Гц, 2H), 7,27-7,14 (м, 2H), 6,77 (дд, J=8,2, 2,6 Гц, 1H), 6,64 (с, 1H), 6,54 (д, J=8,1 Гц, 1H), 3,81-3,65 (м, 4H), 3,57 (д, J=10,5 Гц, 2H), 3,43 (д, J=11,2 Гц, 2H), 3,19 (дд, J=17,0, 10,1 Гц, 7H), 3,10 (т, J=5,0 Гц, 5H), 1,92-1,67 (м, 2H), 1,53 (п, J=7,4 Гц, 2H).
ПРИМЕР 9. [3-(4,4-дифтор-1-пиперидинил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (80,9 мг, 0,37 ммоль), 4-диметиламинопиридина (32,4 мг, 0,26 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (100,0 мг, 0,33 ммоль) и ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 12 (62,1 мг, 0,29 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире, и указанное в заголовке соединение выделяли в виде гидрохлоридной соли. Белое твердое вещество 57 мг (37%). ВЭЖХ-МС (способ A): Rt 2,87 мин; m/z 541 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 10,77 (с, 1H), 7,73 (т, J=5,5 Гц, 1H), 7,44-7,28 (м, 2H), 7,20 (т, J=8,2 Гц, 2H), 6,83 (дд, J=8,2, 2,4 Гц, 1H), 6,71 (т, J=2,1 Гц, 1H), 6,53 (дд, J=7,9, 2,0 Гц, 1H), 3,35 (с, 3H), 3,10 (кв., J=6,4 Гц, 7H), 2,03 (тт, J=14,1, 5,7 Гц, 5H), 1,56 (тт, J=18,5, 11,3 Гц, 5H), 1,92-1,67 (м, 2H), 1,53 (п, J=7,4 Гц, 2H).
ПРИМЕР 10. [4-(3-карбамоилфенил)фенил] N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ B, исходя из п-нитрофенилхлорформиата (84,2 мг, 0,42 ммоль), DIPEA (0,15 мл, 0,84 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 5 (100,0 мг, 0,38 ммоль) и ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 11 (101,2 мг, 0,47 ммоль) в 1:1 смеси DMA:DCM (4 мл). Белое твердое вещество 50 мг (26%). ВЭЖХ-МС (способ A): Rt 1,86 мин; m/z 503 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,15 (т, J=1,7 Гц, 1H), 8,09 (с, 1H), 7,88-7,78 (м, 3H), 7,77-7,69 (м, 2H), 7,54 (т, J=7,7 Гц, 1H), 7,41 (с, 1H), 7,25-7,18 (м, 2H), 6,97-6,82 (м, 4H), 3,77 (с, 3H), 3,11 (т, J=6,2 Гц, 2H), 2,96-3,02 (м, 4H), 2,56-2,51 (м, 4H), 2,36 (т, J=6,8 Гц, 2H), 1,60-1,44 (м, 4H).
ПРИМЕР 11. [4-(3-карбамоилфенил)фенил] N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ B, исходя из п-нитрофенилхлорформиата (103,8 мг, 0,52 ммоль), DIPEA (0,15 мл, 0,84 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 14 (70,0 мг, 0,47 ммоль) и ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 11 (99,9 мг, 0,47 ммоль) в 1:1 смеси DMA:DCM (4 мл). Белое твердое вещество 22 мг (10%). ВЭЖХ-МС (способ A): Rt 1,83 мин; m/z 489 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,15 (т, J=1,7 Гц, 1H), 8,10 (с, 1H), 7,86 (дт, J=7,8, 1,4 Гц, 1H), 7,84-7,78 (м, 2H), 7,78-7,70 (м, 2H), 7,55 (т, J=7,7 Гц, 1H), 7,42 (с, 1H), 7,27-7,19 (м, 2H), 6,99-6,91 (м, 2H), 6,91-6,84 (м, 2H), 3,78 (с, 3H), 3,15 (т, J=6,6 Гц, 2H), 2,97 (с, 4H), 2,53 (с, 4H), 2,40 (т, J=7,1 Гц, 2H), 1,69 (п, J=7,2 Гц, 2H).
ПРИМЕР 12. [4-(4,4-дифтор-1-пиперидинил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (63,5 мг, 0,29 ммоль), 4-диметиламинопиридина (35,6 мг, 0,29 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (80,0 мг, 0,26 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 15 (62,1 мг, 0,29 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества 19 мг (12%). ВЭЖХ-МС (способ A): Rt 2,83 мин; m/z 541 [M-H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 11,17 (д, J=11,2 Гц, 1H), 7,75 (т, J=5,4 Гц, 1H), 7,36 (д, J=6,0 Гц, 2H), 7,20 (д, J=7,1 Гц, 1H), 7,14 (д, J=8,1 Гц, 2H), 7,02 (д, J=7,4 Гц, 2H), 3,49-3,29 (м, 7H), 3,29-3,02 (м, 9H), 2,04-2,26 (м, 4H), 1,70-1,88 (м, 2H), 1,61-1,43 (м, 2H).
ПРИМЕР 13. (3-морфолинoфенил) N-[3-[4-(o-толил)пиперазин-1-ил]пропил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (51,4 мг, 0,24 ммоль), 4-диметиламинопиридина (26,2 мг, 0,21 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 21 (50,0 мг, 0,21 ммоль) и коммерчески доступного 3-морфолинoфенола (42,2 мг, 0,24 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества 25 мг (25%). ВЭЖХ-МС (способ A): Rt 2,15 мин; m/z 439 [M-H]+.
ПРИМЕР 14. [3-(3-карбамоилфенил)фенил] N-[4-(4-фенил-1-пиперидинил)бутил]карбамат гидрохлорид
Указанное в заголовке соединение получали в соответствии с общей процедурой Ia. Исходя из ди-трет-бутилкарбоната (135 мг, 0,62 ммоль), 4-диметиламинопиридина (63 мг, 0,52 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 19 (120 мг, 0,52 ммоль), ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 9 (154 мг, 0,72 ммоль) в MeCN (5 мл). Указанное в заголовке соединение (белое твердое вещество) выделяли в виде HCl соли основания после хроматографической очистки. 54 мг (22%), ВЭЖХ-МС (способ A): Rt 1,97 мин, m/z 472 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 9,30 (с, 1H), 8,16 (с, 1H), 7,92-7,85 (м, 2H), 7,82 (д, J=8,0 Гц, 1H), 7,61-7,44 (м, 4H), 7,38-7,30 (м, 2H), 7,23 (дт, J=7,0, 2,9 Гц, 3H), 7,13 (дд, J=7,6, 1,8 Гц, 1H), 3,59 (д, J=11,6 Гц, 2H), 3,22-2,91 (м, 6H), 2,83 (т, J=12,0 Гц, 1H), 2,01 (д, J=13,3 Гц, 2H), 1,89 (кв., J=12,8 Гц, 2H), 1,76 (с, 2H), 1,65-1,48 (м, 2H).
ПРИМЕР 15. (4-фенилфенил) N-[3-[4-(o-толил)пиперазин-1-ил]пропил]карбамат
Указанное в заголовке соединение получали в соответствии с общей процедурой Ia, исходя из ди-трет-бутилкарбоната (66 мг, 0,30 ммоль), 4-диметиламинопиридина (31 мг, 0,25 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 21 (59 мг, 0,25 ммоль), 4-фенил-фенола (52 мг, 0,30 ммоль) в MeCN (3 мл). Указанное в заголовке соединение (белое твердое вещество) выделяли в виде свободного основания после хроматографической очистки. 63 мг (58%). ВЭЖХ-МС (способ A): Rt 2,77 мин, m/z 430 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 7,81 (т, J=5,6 Гц, 1H), 7,70-7,62 (м, 4H), 7,47 (т, J=7,6 Гц, 2H), 7,41-7,32 (м, 1H), 7,24-7,10 (м, 4H), 7,05-6,90 (м, 2H), 3,15 (кв., J=6,7 Гц, 2H), 2,85 (т, J=4,3 Гц, 4H), 2,55 (с, 4H), 2,42 (т, J=6,9 Гц, 2H), 2,25 (с, 3H), 1,69 (п, J=7,0 Гц, 2H).
ПРИМЕР 16. (4-фенилфенил) N-[3-[4-[2-(трифторметил)фенил]пиперазин-1-ил]пропил]карбамат
Указанное в заголовке соединение получали в соответствии с общей процедурой Ia, исходя из ди-трет-бутилкарбоната (66 мг, 0,30 ммоль), 4-диметиламинопиридина (31 мг, 0,25 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 20 (70 мг, 0,25 ммоль), 4-фенил-фенола (52 мг, 0,30 ммоль) в MeCN (3 мл). Указанное в заголовке соединение(бесцветное масло) выделяли в виде свободного основания после хроматографической очистки. 78 мг (66%). ВЭЖХ-МС (способ A): Rt 2,93 мин, m/z 484 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 7,81 (т, J=5,6 Гц, 1H), 7,70-7,61 (м, 6H), 7,56 (д, J=7,8 Гц, 1H), 7,50-7,43 (м, 2H), 7,41-7,28 (м, 2H), 7,25-7,17 (м, 2H), 3,15 (кв., J=6,7 Гц, 2H), 2,89 (т, J=4,6 Гц, 4H), 2,55-2,48 (м, 4H), 2,41 (т, J=7,1 Гц, 2H), 1,69 (п, J=7,0 Гц, 2H).
ПРИМЕР 17. 9H-карбазол-2-ил N-[3-[4-(2,3-дихлорфенил)пиперазин-1-ил]пропил]карбамат
Указанное в заголовке соединение получали в соответствии с общей процедурой Ia, исходя из ди-трет-бутилкарбоната (73 мг, 0,33 ммоль), 4-диметиламинопиридина (34 мг, 0,28 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 7 (80 мг, 0,28 ммоль), 2-гидроксикарбазола (61 мг, 0,33 ммоль) в MeCN (3 мл). Указанное в заголовке соединение(не совсем белое твердое вещество) выделяли в виде свободного основания после хроматографической очистки. 14 мг (10%). ВЭЖХ-МС (способ A): Rt 2,88 мин, m/z 497 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 11,24 (с, 1H), 8,07 (т, J=8,3 Гц, 2H), 7,76 (т, J=5,6 Гц, 1H), 7,48 (д, J=8,1 Гц, 1H), 7,40-7,27 (м, 3H), 7,20 (д, J=2,0 Гц, 1H), 7,19-7,13 (м, 2H), 6,89 (дд, J=8,4, 2,1 Гц, 1H), 3,16 (кв., J=6,7, 6,0 Гц, 2H), 3,01 (с, 4H), 2,57 (с, 4H), 2,44 (т, J=7,0 Гц, 2H), 1,71 (п, J=6,9 Гц, 2H).
ПРИМЕР 18. [4-(4,4-дифтор-1-пиперидинил)фенил] N-[3-[4-[2-(трифторметил)фенил] пиперазин-1-ил]пропил]карбамат гидрохлорид
Указанное в заголовке соединение получали в соответствии с общей процедурой Ia, исходя из ди-трет-бутилкарбоната (64 мг, 0,29 ммоль), 4-диметиламинопиридина (36 мг, 0,29 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 20 (77 мг, 0,27 ммоль), ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 15 (63 мг, 0,29 ммоль) в MeCN (3 мл). Масло растворяли в небольшом количестве 1,4-диоксана, к которому добавляли 4M HCl в 1,4-диоксане. Выпаривание растворителя давало указанное в заголовке соединение в виде белого твердого вещества. 21 мг (14%). ВЭЖХ-МС (способ A): Rt 2,74 мин, m/z 527 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 10,97 (с, 1H), 7,81 (т, J=5,7 Гц, 1H), 7,72 (д, J=7,7 Гц, 2H), 7,56 (д, J=8,1 Гц, 1H), 7,42 (т, J=7,6 Гц, 1H), 7,10 (д, J=8,6 Гц, 2H), 7,02 (д, J=9,0 Гц, 2H), 3,33 (кв., J=7,2, 6,8 Гц, 6H), 3,27-3,02 (м, 10H), 2,13 (тт, J=13,9, 5,7 Гц, 4H), 1,96 (п, J=6,7 Гц, 2H).
ПРИМЕР 19. [4-(3-карбамоилфенил)фенил] N-[(E)-4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бут-2-енил]карбамат
Указанное в заголовке соединение получали в соответствии с общей процедурой Ia, исходя из ди-трет-бутилкарбоната (102 мг, 0,47 ммоль), 4-диметиламинопиридина (49 мг, 0,40 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 24 (100 мг, 0,33 ммоль), бифенильного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 11 (78 мг, 0,37 ммоль) в MeCN (3 мл). Белое твердое вещество 28 мг (16%). ВЭЖХ-МС (способ A): Rt 2,36 мин, m/z 539 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,15 (т, J=1,8 Гц, 1H), 8,13-8,07 (м, 1H), 8,00 (т, J=5,7 Гц, 1H), 7,86 (дт, J=7,8, 1,4 Гц, 1H), 7,83-7,79 (м, 1H), 7,78-7,70 (м, 2H), 7,55 (т, J=7,7 Гц, 1H), 7,42 (с, 1H), 7,30 (кв., J=5,2, 4,6 Гц, 2H), 7,26-7,20 (м, 2H), 7,15 (дд, J=6,7, 3,0 Гц, 1H), 5,68 (т, J=2,5 Гц, 2H), 3,77-3,69 (м, 2H), 3,01 (кв., J=4,7 Гц, 6H), 2,56 (с, 4H).
ПРИМЕР 20. [4-(3-карбамоилфенил)-3-фтор-фенил] N-[(E)-4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бут-2-енил]карбамат
Указанное в заголовке соединение получали в соответствии с общей процедурой Ia, исходя из ди-трет-бутилкарбоната (102 мг, 0,47 ммоль), 4-диметиламинопиридина (49 мг, 0,40 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 24 (100 мг, 0,33 ммоль), бифенильного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 22 (85 мг, 0,37 ммоль) в MeCN (3 мл). Белое твердое вещество 41 мг (22%). ВЭЖХ-МС (способ A): Rt 2,43 мин, m/z 557 [M+H]+. 1H ЯМР (400 МГц, DMSO-d6) δ 8,12-8,01 (м, 3H), 7,90 (дт, J=7,8, 1,4 Гц, 1H), 7,70 (д, J=7,9 Гц, 1H), 7,64-7,56 (м, 2H), 7,42 (д, J=2,3 Гц, 1H), 7,30 (кв., J=3,9 Гц, 2H), 7,22 (дд, J=11,7, 2,3 Гц, 1H), 7,19-7,09 (м, 2H), 5,68 (т, J=3,3 Гц, 2H), 3,74 (с, 2H), 3,05-2,94 (м, 6H), 2,56 (с, 4H).
ПРИМЕР 21. [4-(3-карбамоилфенил)-3-фтор-фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (73 мг, 0,33 ммоль), 4-диметиламинопиридина (41 мг, 0,33 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (92,0 мг, 0,30 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 22 (77 мг, 0,33 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 12 мг (7%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,06 (д, J=22,6 Гц, 1H), 8,01-7,87 (м, 2H), 7,70 (д, J=7,8 Гц, 1H), 7,56 (дп, J=24,1, 8,1, 7,7 Гц, 2H), 7,40 (дд, J=17,3, 8,1 Гц, 1H), 7,34-7,25 (м, 2H), 7,19 (дд, J=11,7, 2,3 Гц, 1H), 7,12 (ддд, J=14,7, 7,5, 2,5 Гц, 2H), 6,80-6,65 (м, 1H), 3,12 (кв., J=6,2 Гц, 2H), 2,54 (м, 4H), 2,35 (дт, J=20,4, 6,8 Гц, 4H), 1,60-1,48 (м, 4H), 1,47-1,34 (м, 2H).
ПРИМЕР 22. [4-(3-карбамоилфенил)фенил] N-[4-[4-[2-(трифторметил)фенил]пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (79 мг, 0,36 ммоль), 4-диметиламинопиридина (44 мг, 0,36 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 26 (100,0 мг, 0,33 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 11 (77 мг, 0,36 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 63 мг (35%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,22-8,04 (м, 2H), 7,94-7,83 (м, 1H), 7,80 (ддт, J=7,6, 5,9, 1,4 Гц, 1H), 7,77-7,69 (м, 1H), 7,64 (кв.дд, J=7,3, 5,8, 1,7 Гц, 2H), 7,59-7,36 (м, 4H), 7,36-7,28 (м, 2H), 7,27-7,20 (м, 1H), 6,93-6,86 (м, 1H), 2,84 (тт, J=16,8, 4,8 Гц, 6H), 2,48 (с, 4H), 2,33 (дт, J=21,6, 6,8 Гц, 2H), 1,54 (дп, J=8,4, 3,5, 2,9 Гц, 2H), 1,49-1,33 (м, 2H).
ПРИМЕР 23. [3-(3-карбамоилфенил)фенил] N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (115 мг, 0,53 ммоль), 4-диметиламинопиридина (64 мг, 0,53 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 14 (120,0 мг, 0,48 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 9 (112 мг, 0,53 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 70 мг (30%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,17 (т, J=1,8 Гц, 1H), 8,15-8,09 (м, 1H), 7,91-7,80 (м, 3H), 7,62-7,47 (м, 4H), 7,41 (д, J=12,9 Гц, 1H), 7,14 (ддд, J=7,9, 2,4, 1,0 Гц, 1H), 6,98-6,83 (м, 4H), 3,77 (д, J=2,8 Гц, 4H), 3,15 (кв., J=6,8 Гц, 2H), 2,97 (м, 4H), 2,52 (м, 4H), 2,40 (т, J=7,1 Гц, 2H), 1,69 (п, J=7,1 Гц, 2H).
ПРИМЕР 24. [4-(2-пиридил)фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (555 мг, 0,25 ммоль), 4-диметиламинопиридина (31 мг, 0,25 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (70,0 мг, 0,23 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 27 (43 мг, 0,25 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 34 мг (29%), 1H ЯМР (400 МГц, DMSO-d6) δ 8,66 (дт, J=4,8, 1,3 Гц, 1H), 8,13-8,06 (м, 2H), 7,98-7,82 (м, 3H), 7,37-7,32 (м, 1H), 7,30 (кв., J=3,1 Гц, 2H), 7,26-7,19 (м, 2H), 7,14 (тд, J=6,8, 3,2 Гц, 1H), 3,12 (кв., J=6,2 Гц, 2H), 2,99 (кв., J=9,0, 6,6 Гц, 4H), 2,55 (м, 4H), 2,37 (кв., J=6,5 Гц, 2H), 1,53 (п, J=3,5 Гц, 4H).
ПРИМЕР 25. [4-(3-карбамоилфенил)фенил] N-[4-(4-фенилпиперазин-1-ил)бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ B, исходя из п-нитрофенилхлорформиата (42 мг, 0,21 ммоль), DIPEA (0,07 мл, 0,42 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 19 (45 мг, 0,19 ммоль) и ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 11 (45 мг, 0,21 ммоль) в 1:1 смеси DMA:DCM (4 мл). Белое твердое вещество 14 мг (15%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,15 (д, J=2,0 Гц, 1H), 8,13-8,06 (м, 1H), 7,90-7,70 (м, 4H), 7,55 (т, J=7,8 Гц, 1H), 7,51-7,35 (м, 1H), 7,21 (ддд, J=9,1, 5,6, 3,4 Гц, 4H), 6,96-6,85 (м, 3H), 6,77 (т, J=7,2 Гц, 1H), 3,35 (с, 4H), 3,17-3,07 (м, 6H), 2,48 (д, J=5,9 Гц, 2H), 2,34 (кв., J=6,7 Гц, 2H), 1,53 (п, J=3,3 Гц, 2H).
ПРИМЕР 26. (3-фенилфенил) N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (111 мг, 0,51 ммоль), 4-диметиламинопиридина (53 мг, 0,44 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (110,0 мг, 0,36 ммоль) и 3-фенилфенола (68 мг, 0,40 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества, 52 мг (27%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,15 (с, 1H), 7,90 (т, J=5,7 Гц, 1H), 7,71-7,63 (м, 2H), 7,49 (дт, J=15,2, 7,7 Гц, 4H), 7,41-7,32 (м, 4H), 7,20 (дд, J=7,0, 2,6 Гц, 1H), 7,12 (дт, J=7,7, 1,7 Гц, 1H), 3,43 (с, 2H), 3,25 (т, J=12,0 Гц, 2H), 3,20-3,08 (м, 8H), 1,90-1,74 (м, 2H), 1,56 (п, J=7,5 Гц, 2H).
ПРИМЕР 27. (4-фенилфенил) N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (111 мг, 0,51 ммоль), 4-диметиламинопиридина (53 мг, 0,44 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (110,0 мг, 0,36 ммоль) и 4-фенилфенола (68 мг, 0,40 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 42 мг (23%). 1H ЯМР (400 МГц, DMSO-d6) δ 7,83 (т, J=5,7 Гц, 1H), 7,69-7,62 (м, 4H), 7,50-7,43 (м, 2H), 7,39-7,34 (м, 1H), 7,31-7,27 (м, 2H), 7,23-7,16 (м, 2H), 7,14 (дд, J=6,5, 3,2 Гц, 1H), 3,11 (кв., J=6,3 Гц, 2H), 2,99 (т, J=4,9 Гц, 4H), 2,54 (с, 4H), 2,38 (д, J=6,7 Гц, 2H), 1,53 (кв., J=3,7 Гц, 4H).
ПРИМЕР 28. 9H-карбазол-2-ил N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (121 мг, 0,56 ммоль), 4-диметиламинопиридина (58 мг, 0,48 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (120,0 мг, 0,40 ммоль) и 9H-карбазол-2-ола (80 мг, 0,44 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 43 мг (21%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,26 (с, 1H), 8,07 (т, J=8,2 Гц, 2H), 7,79 (т, J=5,7 Гц, 1H), 7,48 (д, J=8,0 Гц, 1H), 7,36 (т, J=7,6 Гц, 1H), 7,32-7,27 (м, 2H), 7,20 (д, J=2,1 Гц, 1H), 7,19-7,12 (м, 2H), 6,88 (дд, J=8,4, 2,1 Гц, 1H), 3,14 (т, J=6,0 Гц, 2H), 3,02 (д, J=4,9 Гц, 4H), 2,62 (с, 4H), 2,46 (д, J=6,2 Гц, 2H), 1,55 (г, J=3,2 Гц, 4H).
ПРИМЕР 29. (4-фенилфенил) N-[4-[4-(o-толил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (136 мг, 0,62 ммоль), 4-диметиламинопиридина (65 мг, 0,53 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 29 (110,0 мг, 0,44 ммоль) и 4-фенилфенола (83 мг, 0,49 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 27 мг (14%). 1H ЯМР (600 МГц, DMSO-d6) δ 7,89 (т, J=5,7 Гц, 1H), 7,69-7,63 (м, 4H), 7,47 (т, J=7,7 Гц, 2H), 7,39-7,35 (м, 1H), 7,23-7,18 (м, 2H), 7,18-7,11 (м, 2H), 7,02 (дд, J=8,0, 1,2 Гц, 1H), 6,95 (тд, J=7,3, 1,2 Гц, 1H), 3,12 (кв., J=6,3 Гц, 2H), 2,85 (т, J=4,7 Гц, 4H), 2,62-2,52 (м, 4H), 2,37 (д, J=6,5 Гц, 2H), 2,24 (д, J=4,8 Гц, 3H), 1,57-1,48 (м, 4H).
ПРИМЕР 30. (4-фенилфенил) N-[4-[4-[2-(трифторметил)фенил]пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (91 мг, 0,42 ммоль), 4-диметиламинопиридина (44 мг, 0,36 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 26 (90,0 мг, 0,30 ммоль) и 4-фенилфенола (56 мг, 0,33 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 47 мг (32%). 1H ЯМР (600 МГц, DMSO-d6) δ 7,88 (д, J=6,1 Гц, 1H), 7,66 (д, J=8,4 Гц, 6H), 7,55 (д, J=7,8 Гц, 1H), 7,47 (кв., J=7,0, 6,5 Гц, 2H), 7,35 (дт, J=24,8, 7,3 Гц, 2H), 7,20 (д, J=8,3 Гц, 2H), 3,12 (д, J=6,6 Гц, 2H), 2,88 (д, J=5,9 Гц, 4H), 2,51 (с, 4H), 2,36 (д, J=6,4 Гц, 2H), 1,52 (с, 4H).
ПРИМЕР 31. (4-фенилфенил) N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (122 мг, 0,56 ммоль), 4-диметиламинопиридина (59 мг, 0,48 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 14 (100,0 мг, 0,40 ммоль) и 4-фенилфенола (75 мг, 0,44 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества, 64 мг (33%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,23 (с, 1H), 8,00 (т, J=5,8 Гц, 1H), 7,70-7,63 (м, 4H), 7,47 (дд, J=8,4, 7,0 Гц, 2H), 7,39-7,33 (м, 1H), 7,26-7,20 (м, 2H), 7,07-6,89 (м, 4H), 3,80 (с, 3H), 3,53 (дд, J=22,4, 9,4 Гц, 4H), 3,27-3,09 (м, 8H), 2,07-1,91 (м, 2H).
ПРИМЕР 32. (4-фенилфенил) N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (116 мг, 0,53 ммоль), 4-диметиламинопиридина (56 мг, 0,46 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 5 (100,0 мг, 0,38 ммоль) и 4-фенилфенола (71 мг, 0,42 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества, 54 мг (29%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,16 (с, 1H), 7,93 (т, J=5,7 Гц, 1H), 7,72-7,62 (м, 4H), 7,47 (дд, J=8,4, 6,9 Гц, 2H), 7,41-7,33 (м, 1H), 7,25-7,19 (м, 2H), 7,09-6,88 (м, 4H), 3,80 (с, 3H), 3,52 (дд, J=18,8, 8,5 Гц, 4H), 3,15 (дкв., J=12,9, 6,9, 6,4 Гц, 8H), 1,82 (тд, J=11,0, 9,5, 6,2 Гц, 2H), 1,55 (п, J=7,2 Гц, 2H).
ПРИМЕР 33. 9H-карбазол-2-ил N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (116 мг, 0,53 ммоль), 4-диметиламинопиридина (56 мг, 0,46 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 5 (100,0 мг, 0,38 ммоль) и 9H-карбазол-2-ола (77 мг, 0,42 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 89 мг (50%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,27 (с, 1H), 8,08 (т, J=8,4 Гц, 2H), 7,82 (т, J=5,6 Гц, 1H), 7,49 (д, J=8,1 Гц, 1H), 7,44-7,31 (м, 1H), 7,20 (д, J=2,1 Гц, 1H), 7,16 (т, J=7,4 Гц, 1H), 6,91 (дтд, J=19,6, 5,4, 2,6 Гц, 5H), 3,77 (с, 3H), 3,13 (кв., J=6,3 Гц, 2H), 2,97 (с, 4H), 2,53 (д, J=5,0 Гц, 4H), 2,37 (д, J=6,8 Гц, 2H), 1,66-1,47 (м, 4H).
ПРИМЕР 34. [4-(3-карбамоилфенил)-3-метокси-фенил] N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (93 мг, 0,43 ммоль), 4-диметиламинопиридина (44 мг, 0,36 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 5 (80,0 мг, 0,30 ммоль) и ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 39 (81 мг, 0,33 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 35 мг (22%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,02 (с, 1H), 7,95 (т, J=1,8 Гц, 1H), 7,87 (т, J=5,6 Гц, 1H), 7,82 (дт, J=7,7, 1,5 Гц, 1H), 7,62 (дт, J=7,7, 1,4 Гц, 1H), 7,48 (т, J=7,7 Гц, 1H), 7,38 (с, 1H), 7,32 (д, J=8,3 Гц, 1H), 6,97-6,84 (м, 5H), 6,80 (дд, J=8,2, 2,2 Гц, 1H), 3,77 (с, 3H), 3,76 (с, 3H), 3,12 (кв., J=6,3 Гц, 2H), 2,97 (с, 4H), 2,57-2,51 (м, 4H), 2,36 (д, J=6,7 Гц, 2H), 1,53 (п, J=3,6 Гц, 4H).
ПРИМЕР 35. (4-фенилфенил) N-[3-фтор-4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (130 мг, 0,60 ммоль), 4-диметиламинопиридина (62 мг, 0,51 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 38 (120,0 мг, 0,43 ммоль) и 4-фенилфенола (80 мг, 0,47 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества, 46 мг (21%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,35 (с, 1H), 8,01 (т, J=5,7 Гц, 1H), 7,74-7,58 (м, 4H), 7,48 (дд, J=8,4, 6,9 Гц, 2H), 7,40-7,33 (м, 1H), 7,28-7,19 (м, 2H), 7,09-6,87 (м, 4H), 5,55-5,25 (м, 1H), 3,81 (с, 3H), 3,65-3,44 (м, 8H), 3,39-3,04 (м, 4H), 1,89 (дкв., J=24,7, 6,6, 6,2 Гц, 2H).
ПРИМЕР 36. (4-фенилфенил) N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-фтор-бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (76 мг, 0,35 ммоль), 4-диметиламинопиридина (37 мг, 0,30 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 37 (80,0 мг, 0,25 ммоль) и 4-фенилфенола (47 мг, 0,27 ммоль) в ацетонитриле (4 мл). Желтого твердое вещество 20 мг (16%). 1H ЯМР (400 МГц, DMSO-d6) δ 7,93 (т, J=5,7 Гц, 1H), 7,69-7,63 (м, 4H), 7,47 (дд, J=8,4, 6,9 Гц, 2H), 7,39-7,33 (м, 1H), 7,32-7,28 (м, 2H), 7,24-7,18 (м, 2H), 7,16 (дд, J=6,0, 3,6 Гц, 1H), 4,97-4,72 (м, 1H), 3,22 (дкв., J=16,2, 6,7 Гц, 2H), 3,00 (т, J=4,8 Гц, 4H), 2,67 (кв.д, J=13,3, 12,8, 6,4 Гц, 6H), 1,97-1,77 (м, 2H).
ПРИМЕР 37. (3-метокси-4-фенил-фенил) N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (122 мг, 0,56 ммоль), 4-диметиламинопиридина (59 мг, 0,48 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 14 (100,0 мг, 0,40 ммоль) и ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 30 (88 мг, 0,44 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества 12 мг (6%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,11 (с, 1H), 7,98 (т, J=5,9 Гц, 1H), 7,48-7,44 (м, 2H), 7,41 (дд, J=8,5, 6,8 Гц, 2H), 7,34-7,30 (м, 1H), 7,27 (д, J=8,2 Гц, 1H), 7,07-6,88 (м, 5H), 6,81 (дд, J=8,3, 2,2 Гц, 1H), 3,80 (с, 3H), 3,76 (с, 3H), 3,56-3,44 (м, 4H), 3,29-3,05 (м, 8H), 2,00 (дкв., J=11,6, 6,6 Гц, 2H).
ПРИМЕР 38. (3-метокси-4-фенил-фенил) N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (139 мг, 0,64 ммоль), 4-диметиламинопиридина (68 мг, 0,55 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 5 (120,0 мг, 0,46 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 30 (100 мг, 0,50 ммоль) в ацетонитриле (4 мл). Желтого твердое вещество 94 мг (42%). 1H ЯМР (400 МГц, DMSO-d6) δ 7,94 (т, J=5,8 Гц, 1H), 7,48-7,43 (м, 2H), 7,40 (дд, J=8,4, 6,6 Гц, 2H), 7,35-7,29 (м, 1H), 7,26 (д, J=8,3 Гц, 1H), 6,99 (дт, J=11,7, 4,6 Гц, 2H), 6,95-6,87 (м, 3H), 6,78 (дд, J=8,3, 2,2 Гц, 1H), 3,79 (с, 3H), 3,75 (с, 3H), 3,39-3,33 (м, 10H), 3,12 (кв., J=6,6 Гц, 2H), 1,81 (с, 2H), 1,53 (т, J=7,5 Гц, 2H).
ПРИМЕР 39. [4-(3-карбамоилфенил)фенил] N-[4-[4-(o-толил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (136 мг, 0,62 ммоль), 4-диметиламинопиридина (65 мг, 0,53 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 29 (110,0 мг, 0,44 ммоль) и фенольного ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 11 (83 мг, 0,38 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 30 мг (14%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,26 (т, J=2,0 Гц, 1H), 7,96 (дт, J=7,5, 2,1 Гц, 1H), 7,68 (дт, J=7,5, 2,0 Гц, 1H), 7,65-7,56 (м, 3H), 7,14-7,08 (м, 2H), 7,03 (ддт, J=5,4, 3,1, 1,0 Гц, 1H), 6,92-6,87 (м, 2H), 6,73-6,66 (м, 1H), 6,20 (с, 2H), 5,34 (с, 1H), 3,24 (т, J=7,5 Гц, 2H), 2,98 (т, J=5,1 Гц, 4H), 2,78 (т, J=5,1 Гц, 4H), 2,44 (т, J=5,4 Гц, 2H), 2,22 (д, J=1,1 Гц, 3H), 1,69 (тт, J=7,8, 5,4 Гц, 2H), 1,49 (п, J=7,7 Гц, 2H).
ПРИМЕР 40. [4-(3-карбамоилфенил)-3-метокси-фенил] N-[4-[4-(2,3-дихлорфенил)пиперазин-1-ил]бутил]карбамат
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (93 мг, 0,43 ммоль), 4-диметиламинопиридина (44 мг, 0,36 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 6 (80,0 мг, 0,26 ммоль) и ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 39 (81 мг, 0,33 ммоль) в ацетонитриле (4 мл). Белое твердое вещество 35 мг (23%). 1H ЯМР (400 МГц, DMSO-d6) δ 8,03 (т, J=2,0 Гц, 1H), 7,81 (дт, J=7,5, 2,0 Гц, 1H), 7,75 (д, J=7,5 Гц, 1H), 7,66 (дт, J=7,5, 1,9 Гц, 1H), 7,43 (т, J=7,5 Гц, 1H), 7,21-7,15 (м, 2H), 7,03 (д, J=2,1 Гц, 1H), 6,99 (дд, J=7,5, 2,0 Гц, 1H), 6,90 (дд, J=7,0, 2,6 Гц, 1H), 6,20 (с, 2H), 5,34 (с, 1H), 3,90 (с, 3H), 3,24 (т, J=5,1 Гц, 2H), 2,98 (т, J=5,1 Гц, 4H), 2,53 (т, J=5,1 Гц, 4H), 2,44 (т, J=7,5 Гц, 2H), 1,69 (тт, J=7,4, 5,1 Гц, 2H), 1,49 (п, J=5,1 Гц, 2H).
ПРИМЕР 41. (4-фенилфенил) N-[2-[4-(2,3-дихлорфенил)пиперазин-1-ил]-3-фтор-пропил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (55 мг, 0,25 ммоль), 4-диметиламинопиридина (31 мг, 0,25 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 42 (70,0 мг, 0,23 ммоль) и 4-фенилфенола (43 мг, 0,25 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества, 19 мг (15%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,50 (с, 1H), 8,29 (с, 1H), 7,67 (т, J=8,2 Гц, 4H), 7,47 (т, J=7,6 Гц, 2H), 7,43-7,34 (м, 3H), 7,27 (д, J=8,5 Гц, 2H), 7,22 (дд, J=6,9, 2,7 Гц, 1H), 5,28-4,85 (м, 2H), 3,88-3,45 (м, 1H), 3,41-3,24 (м, 10H).
ПРИМЕР 42. (4-бензилфенил) N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (116 мг, 0,53 ммоль), 4-диметиламинопиридина (56 мг, 0,46 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 5 (100,0 мг, 0,38 ммоль) и 4-бензилфенола (77 мг, 0,42 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества 97 мг (50%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,29 (с, 1H), 7,84 (т, J=5,7 Гц, 1H), 7,32-7,15 (м, 8H), 7,09-6,97 (м, 5H), 6,92 (тд, J=7,5, 1,6 Гц, 1H), 3,93 (с, 2H), 3,80 (с, 3H), 3,51 (дд, J=16,4, 8,5 Гц, 4H), 3,23-3,03 (м, 8H), 1,90-1,73 (м, 2H), 1,51 (п, J=7,2 Гц, 2H).
ПРИМЕР 43. (4-бензилфенил) N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (98 мг, 0,45 ммоль), 4-диметиламинопиридина (47 мг, 0,38 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 14 (80,0 мг, 0,32 ммоль) и 4-бензилфенола (65 мг, 0,35 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире,. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества, 66 мг (41%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,20 (с, 1H), 7,92 (т, J=5,8 Гц, 1H), 7,32-7,16 (м, 8H), 7,07-6,95 (м, 5H), 6,92 (ддд, J=8,0, 6,9, 1,6 Гц, 1H), 3,93 (с, 2H), 3,80 (с, 3H), 3,52 (дд, J=20,1, 9,6 Гц, 4H), 3,20-3,11 (м, 8H), 2,02-1,91 (м, 2H).
ПРИМЕР 44. (4-бензоилфенил) N-[3-[4-(2-метоксифенил)пиперазин-1-ил]пропил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (98 мг, 0,45 ммоль), 4-диметиламинопиридина (47 мг, 0,38 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 14 (80,0 мг, 0,32 ммоль) и (4-гидроксифенил)-фенил-метанона (70 мг, 0,35 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества, 37 мг (23%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,02 (с, 1H), 8,14 (т, J=5,8 Гц, 1H), 7,83-7,77 (м, 2H), 7,77-7,72 (м, 2H), 7,72-7,63 (м, 1H), 7,60-7,53 (м, 2H), 7,37-7,31 (м, 2H), 7,07-6,87 (м, 5H), 3,80 (д, J=2,1 Гц, 3H), 3,53 (дд, J=22,2, 11,7 Гц, 4H), 3,24-3,05 (м, 8H), 2,00 (дд, J=9,9, 6,4 Гц, 2H).
ПРИМЕР 45. (4-бензоилфенил) N-[4-[4-(2-метоксифенил)пиперазин-1-ил]бутил]карбамат гидрохлорид
Указанное в заголовке соединение синтезировали в соответствии с общей процедурой Ia, Способ A, исходя из ди-трет-бутилдикарбоната (128 мг, 0,58 ммоль), 4-диметиламинопиридина (61 мг, 0,50 ммоль), аминового ПРОМЕЖУТОЧНОГО СОЕДИНЕНИЯ 5 (110,0 мг, 0,42 ммоль) и (4-гидроксифенил)-фенил-метанона (91 мг, 0,46 ммоль) в ацетонитриле (4 мл). Полученное масло растворяли в небольшом количестве диэтилового эфира, к которому добавляли 2M HCl в диэтиловом эфире. Выпаривание растворителя давало указанное в заголовке соединение в виде желтого твердого вещества 83 мг (38%). 1H ЯМР (400 МГц, DMSO-d6) δ 11,20 (с, 1H), 8,08 (т, J=5,7 Гц, 1H), 7,81-7,76 (м, 2H), 7,76-7,72 (м, 2H), 7,71-7,66 (м, 1H), 7,60-7,55 (м, 2H), 7,35-7,31 (м, 2H), 7,06-6,88 (м, 5H), 3,80 (с, 3H), 3,51 (дд, J=18,5, 8,1 Гц, 4H), 3,19-3,09 (м, 8H), 1,92-1,75 (м, 2H), 1,55 (п, J=7,1 Гц, 2H).
|
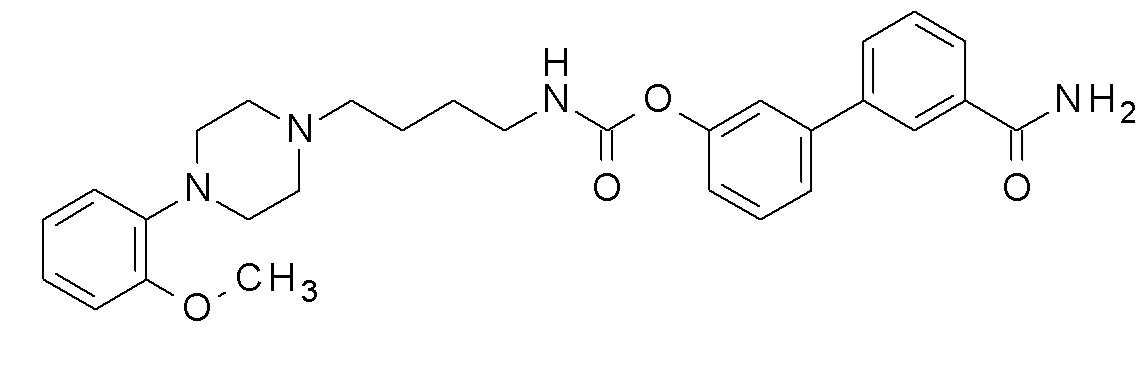
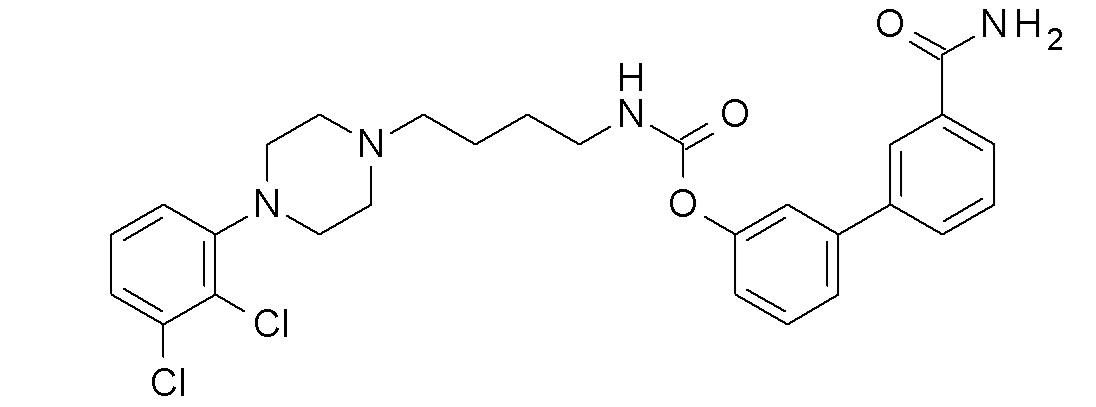
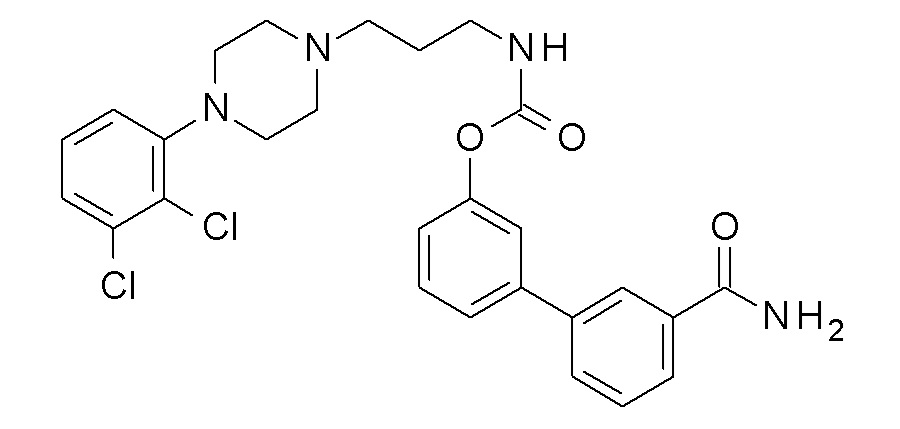
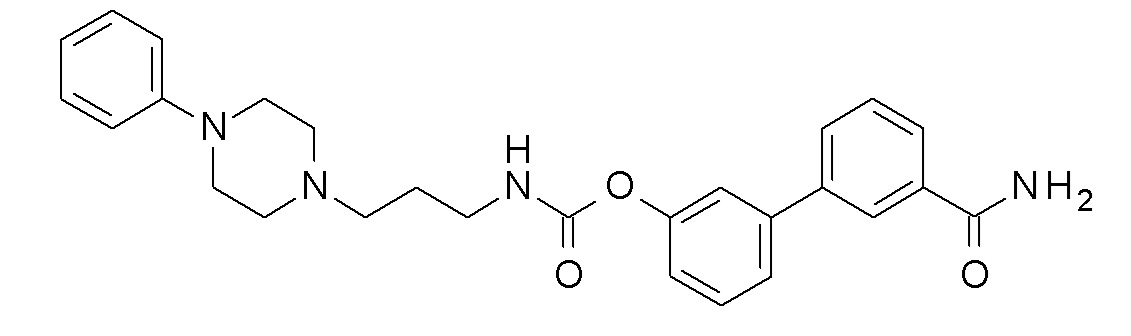
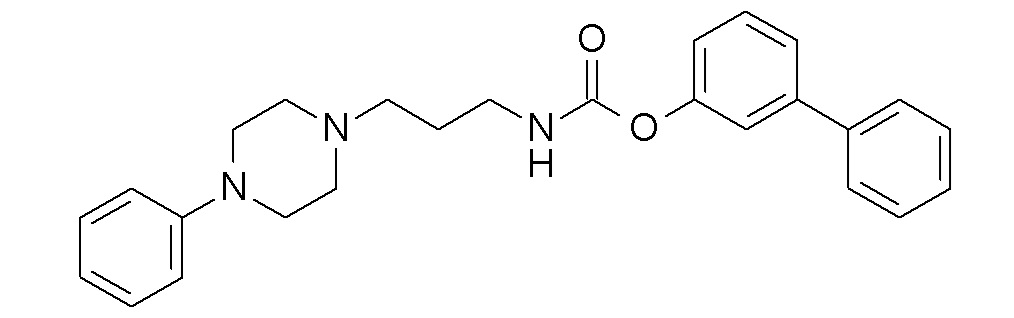
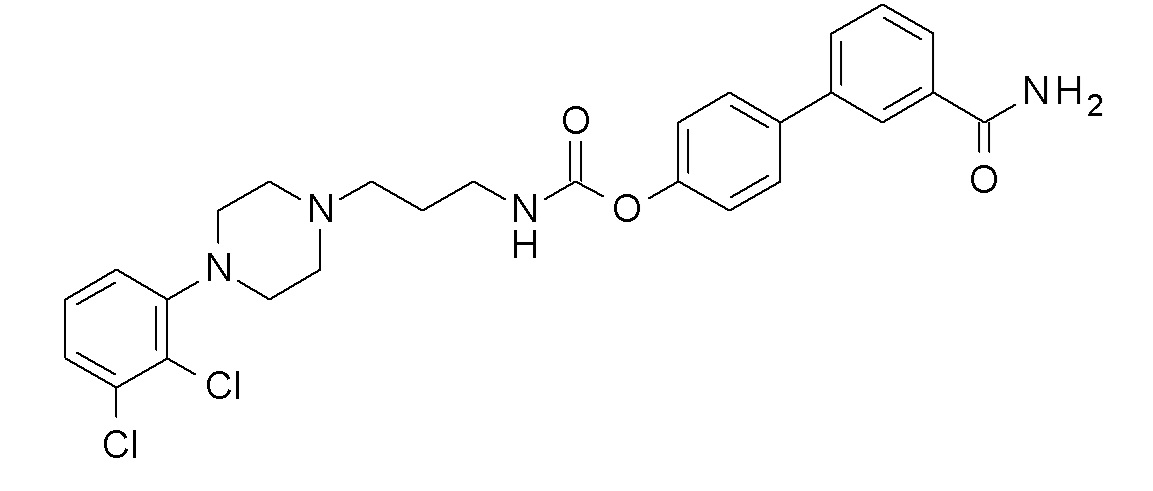
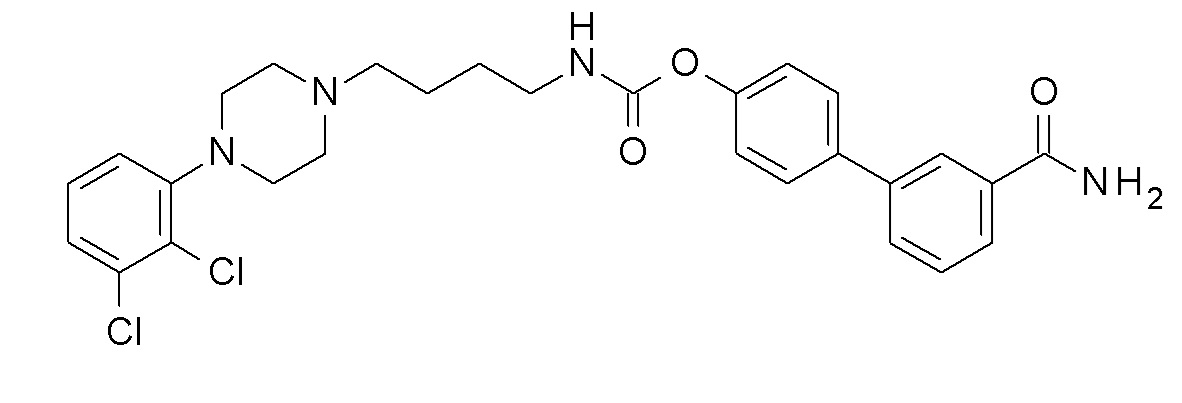
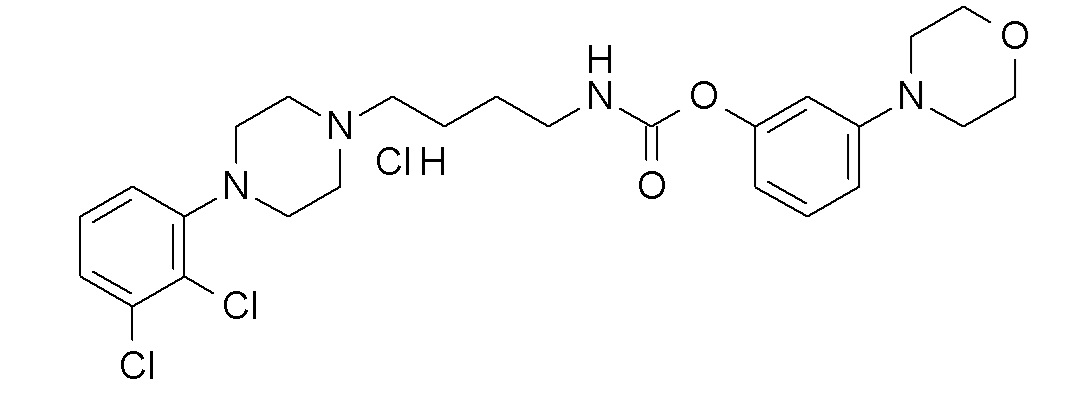
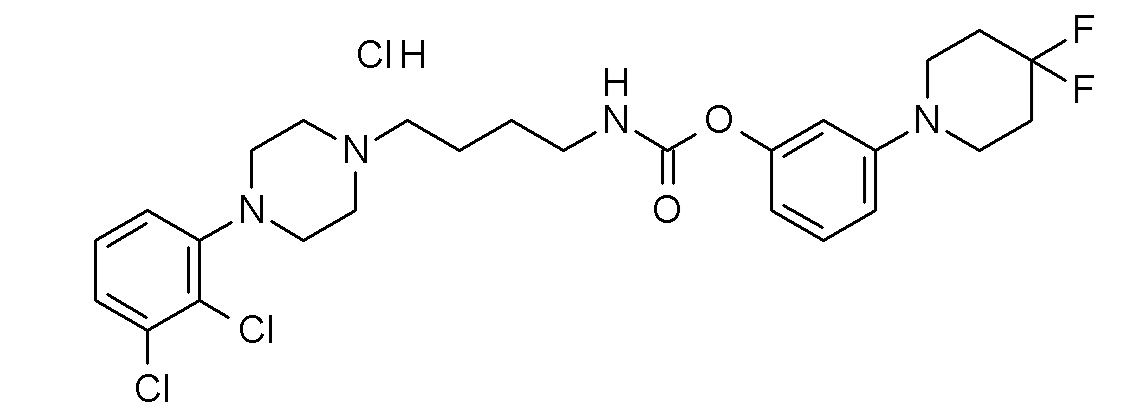
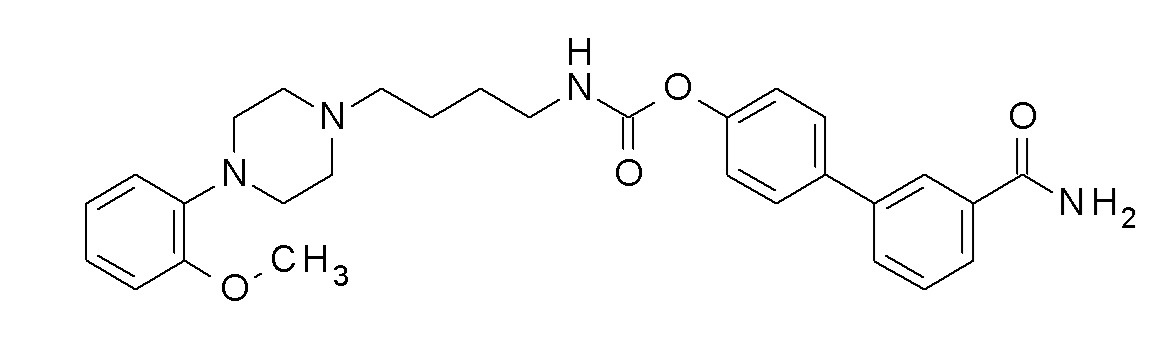
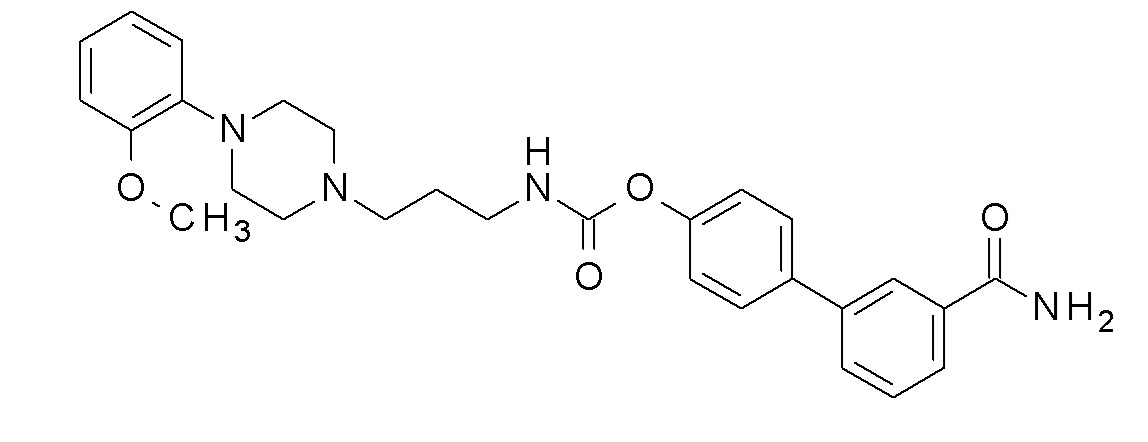
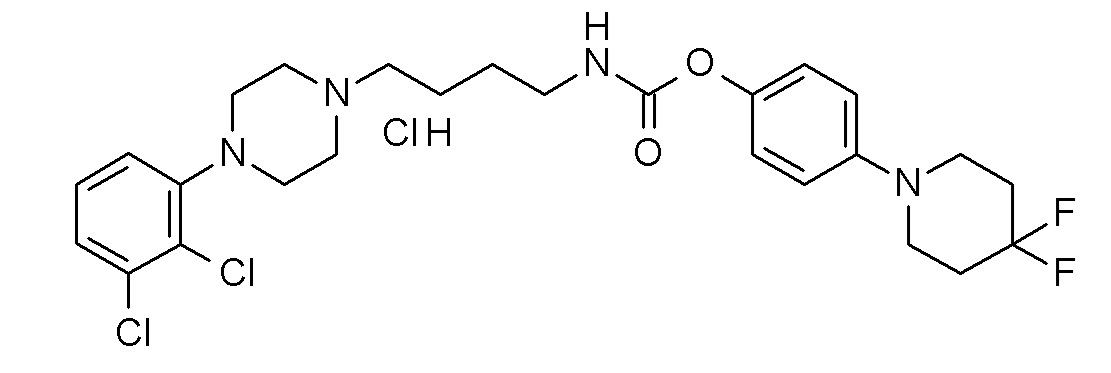
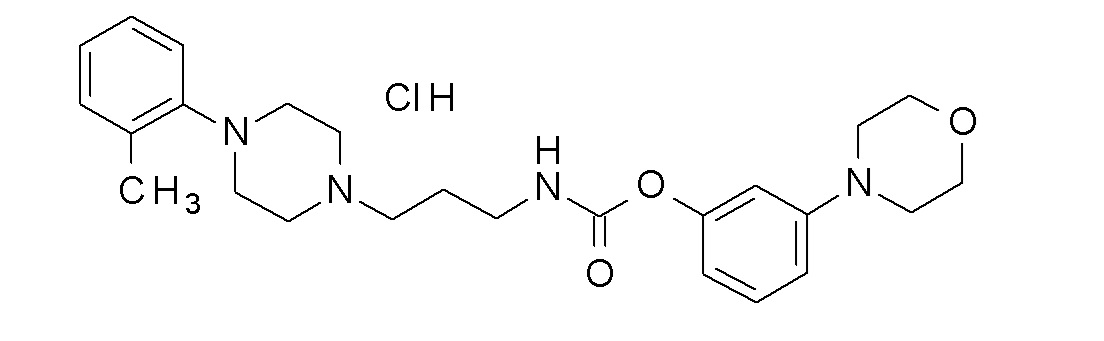
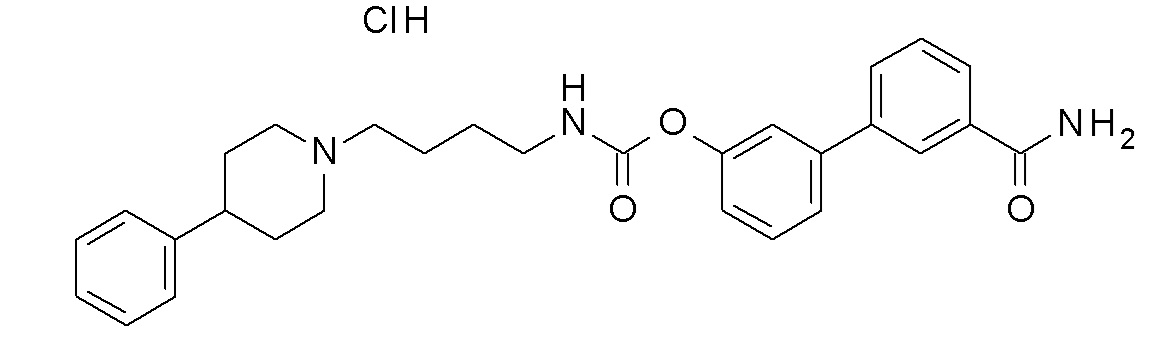
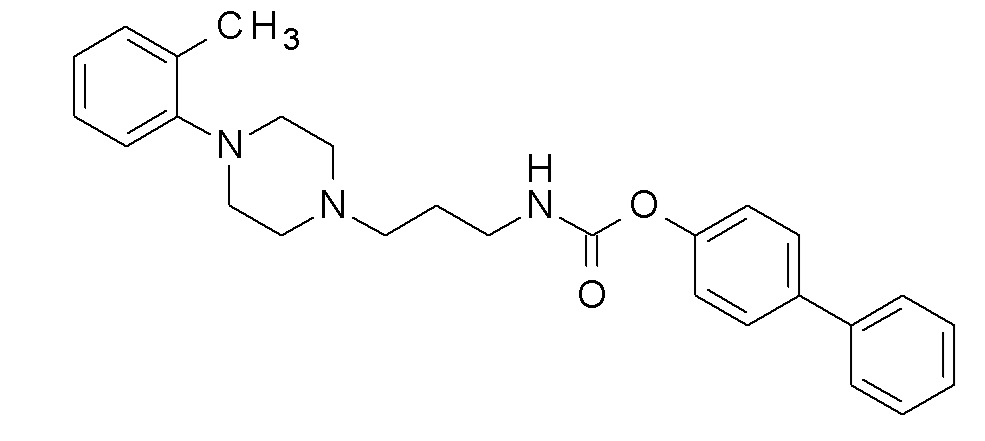
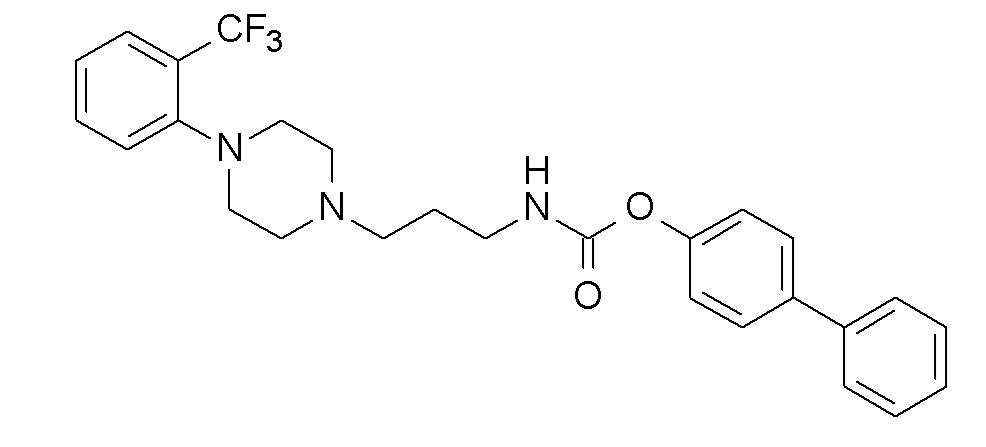
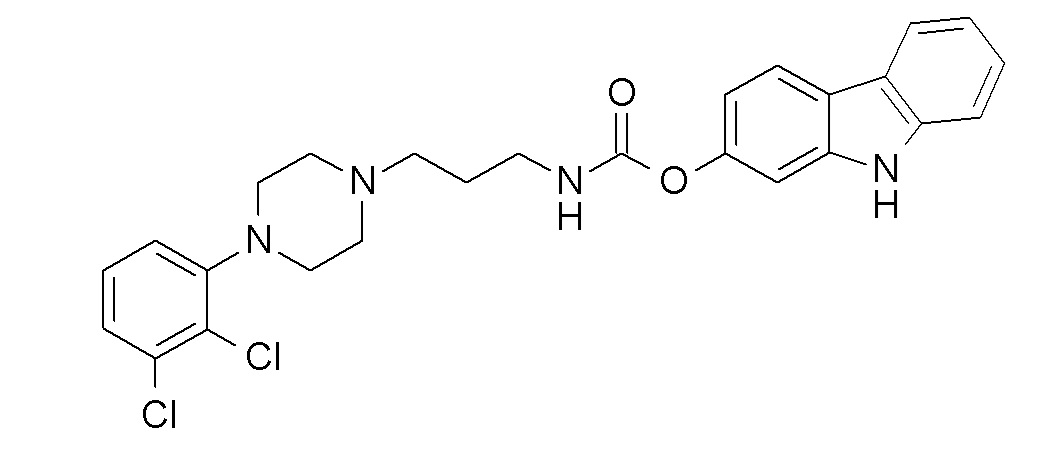
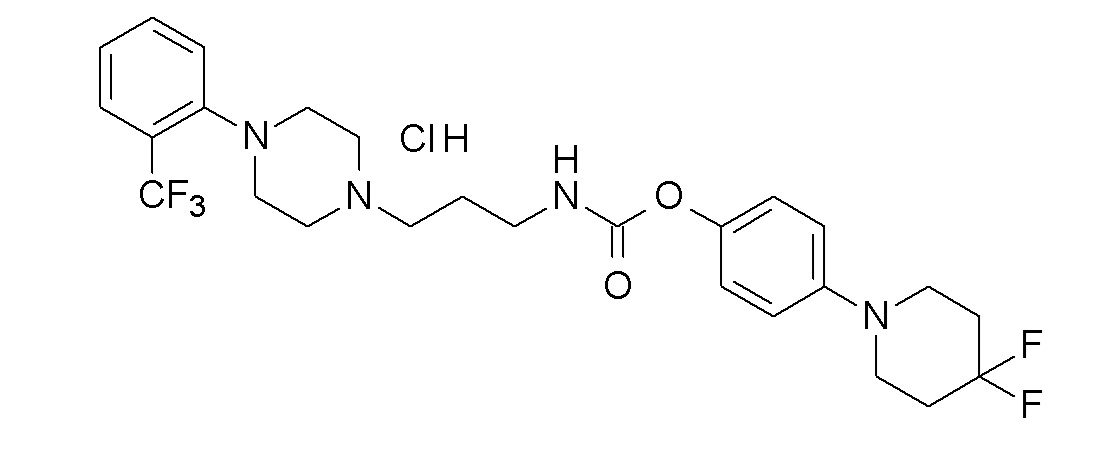
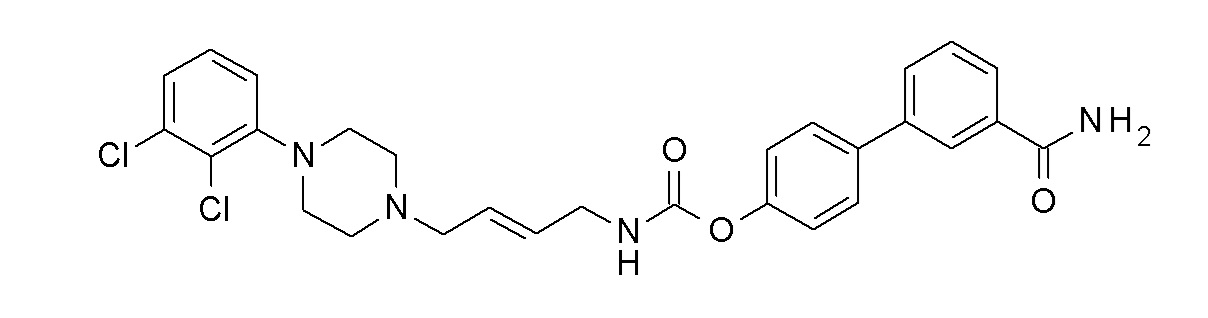
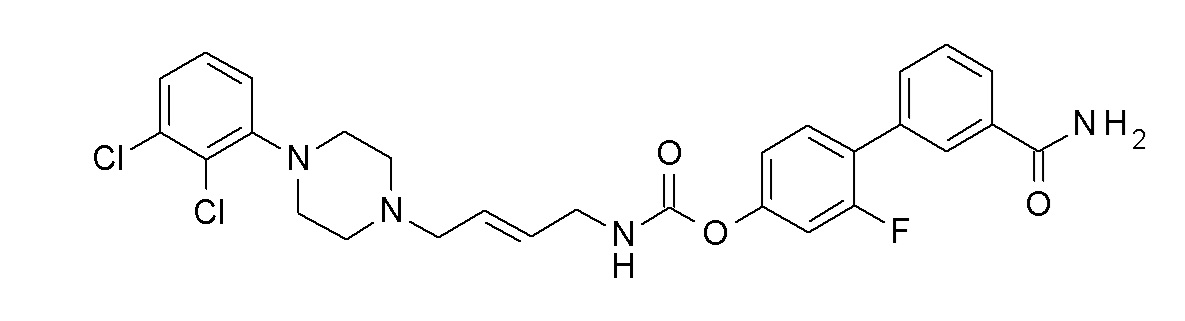
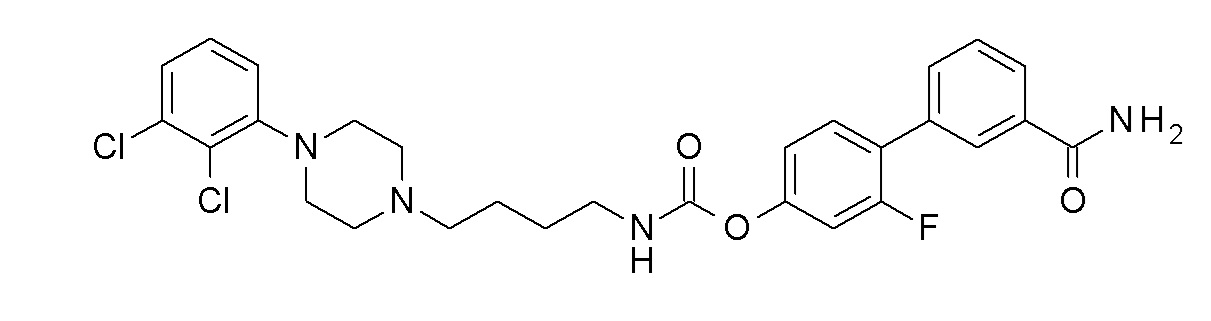
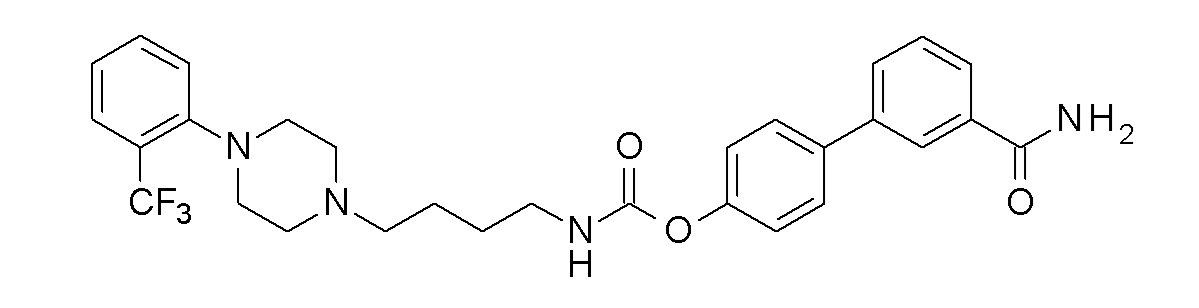
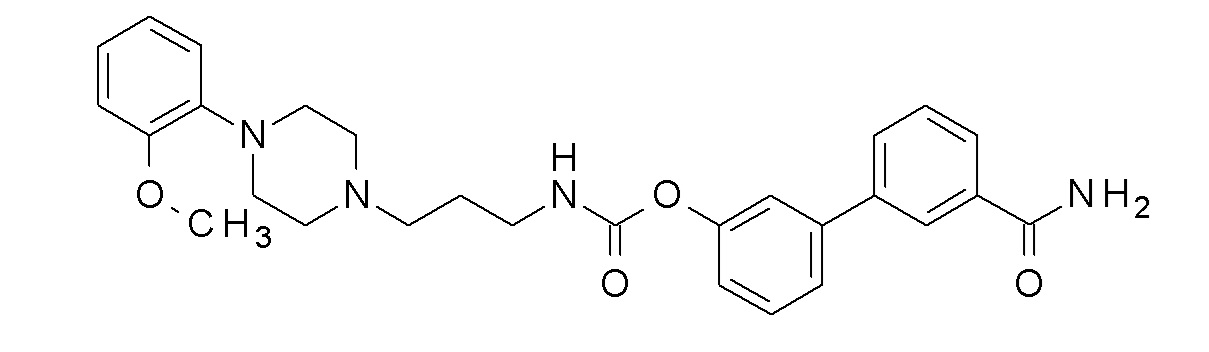
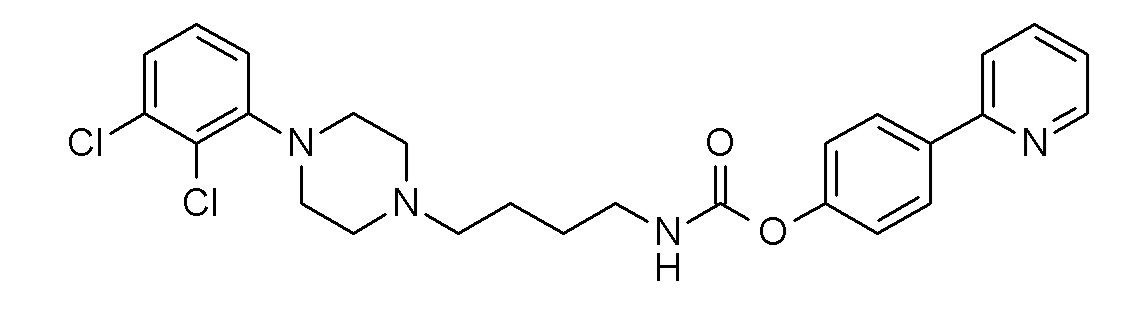
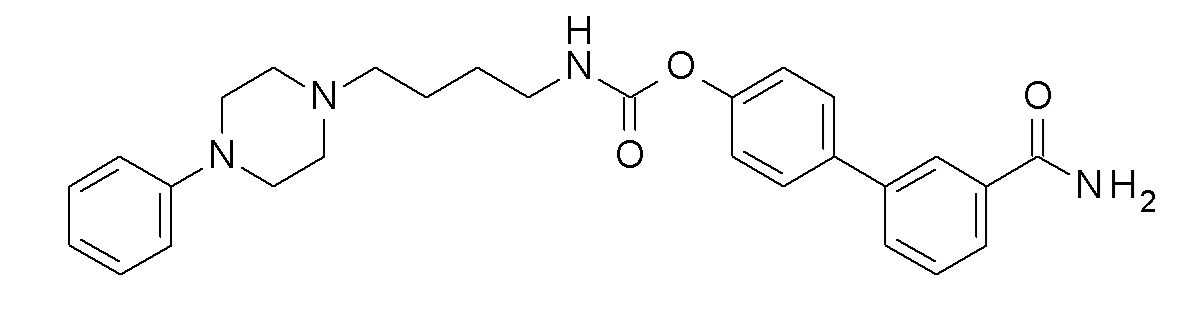
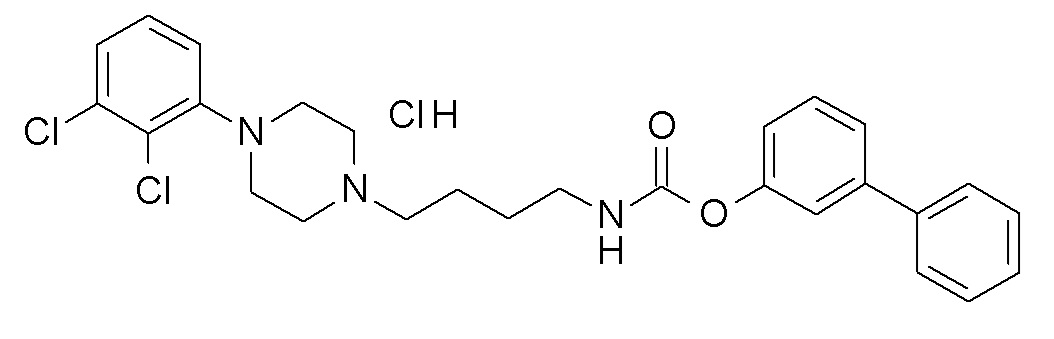
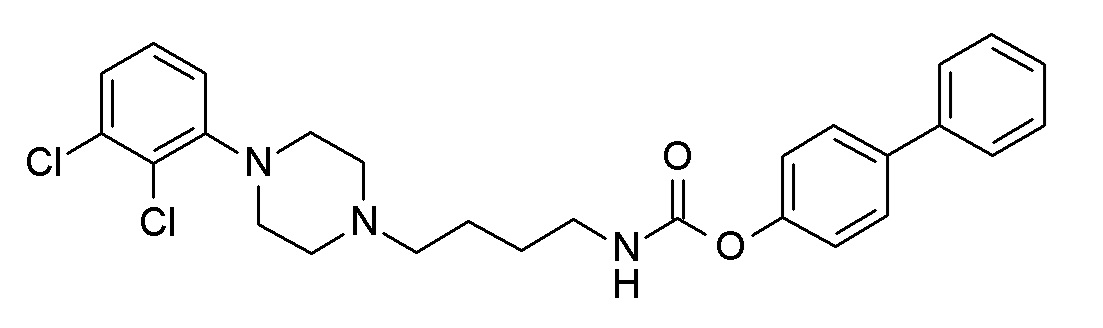
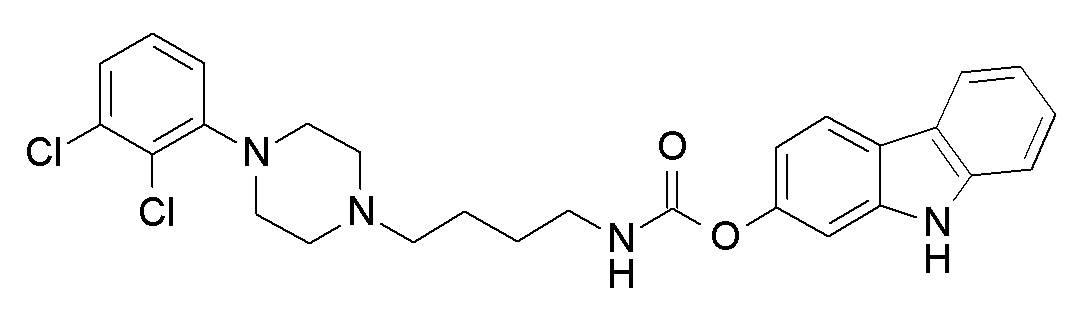
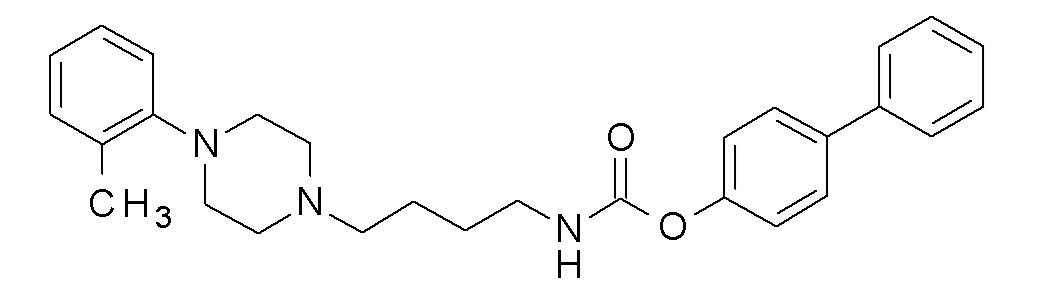
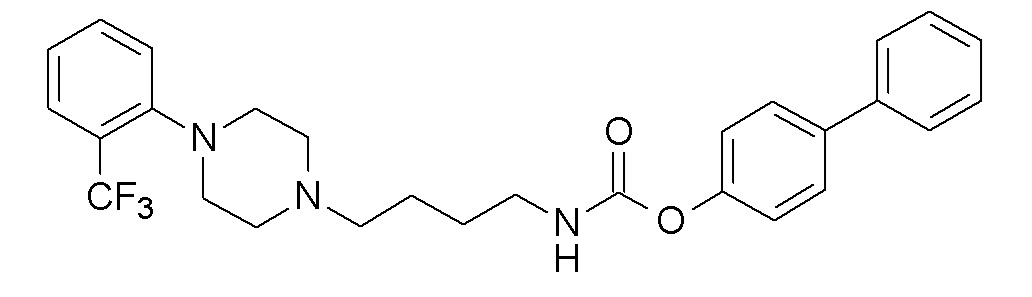
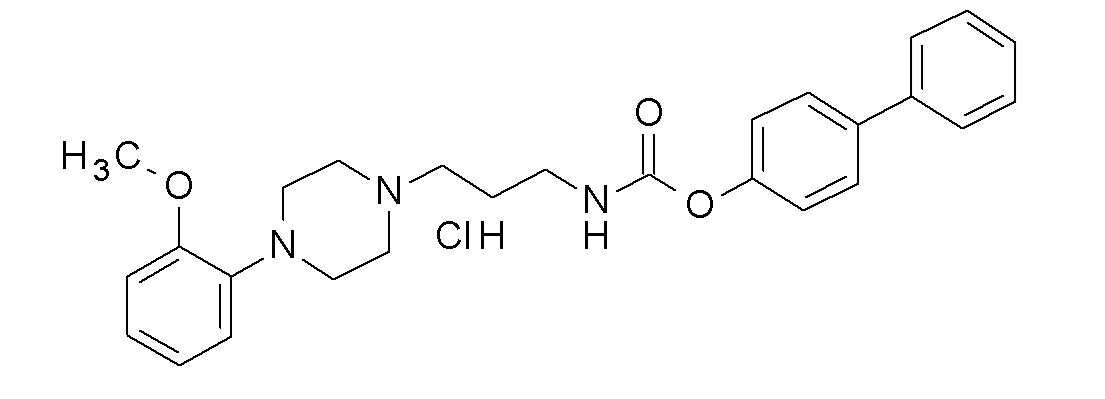
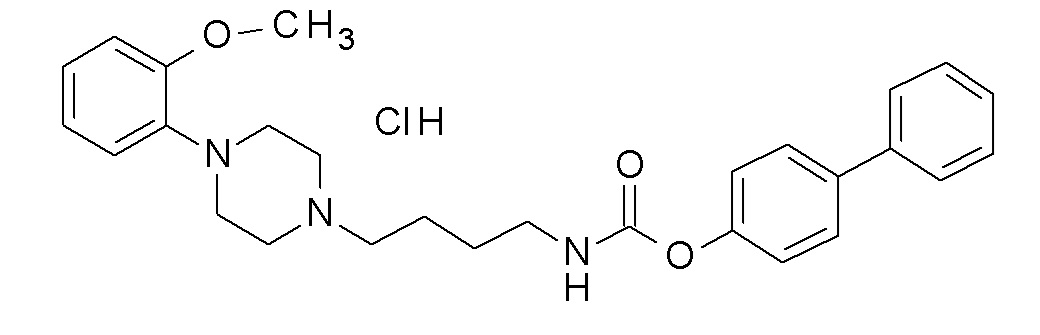
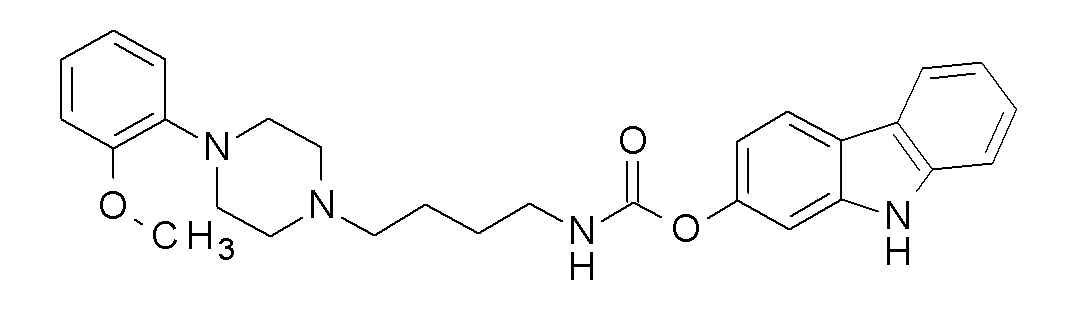
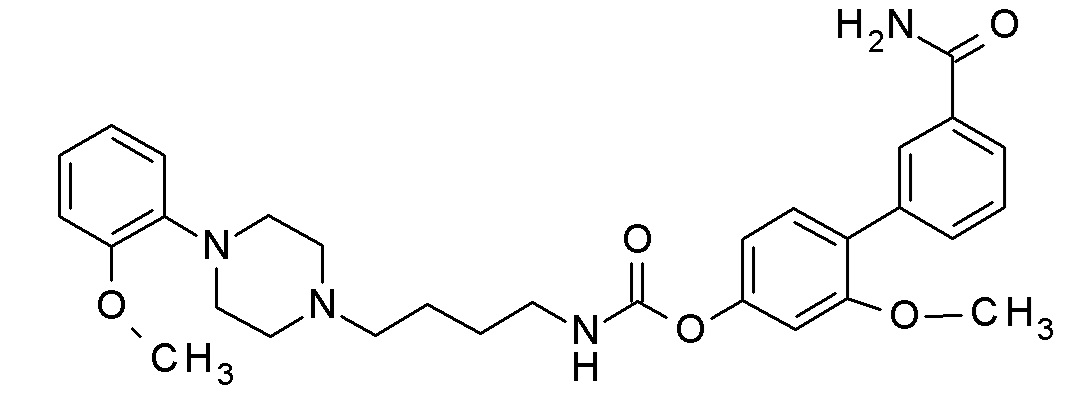
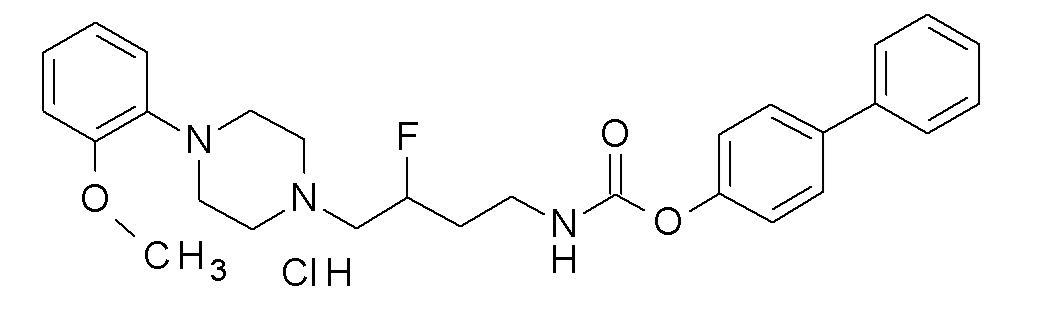
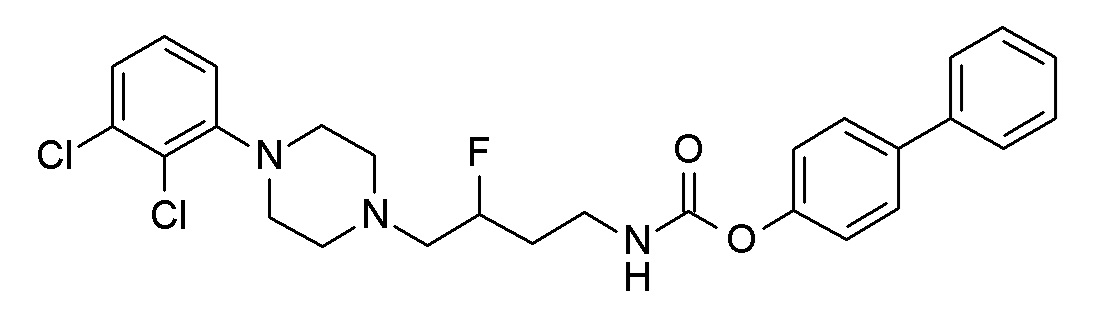
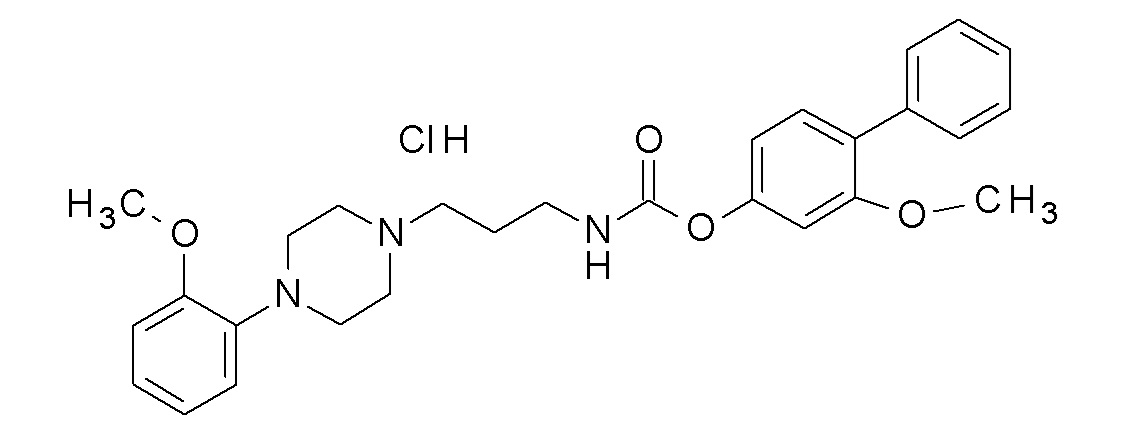
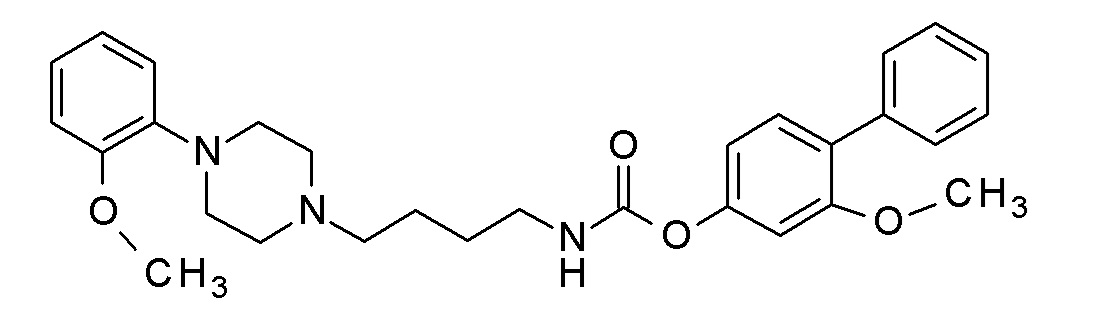
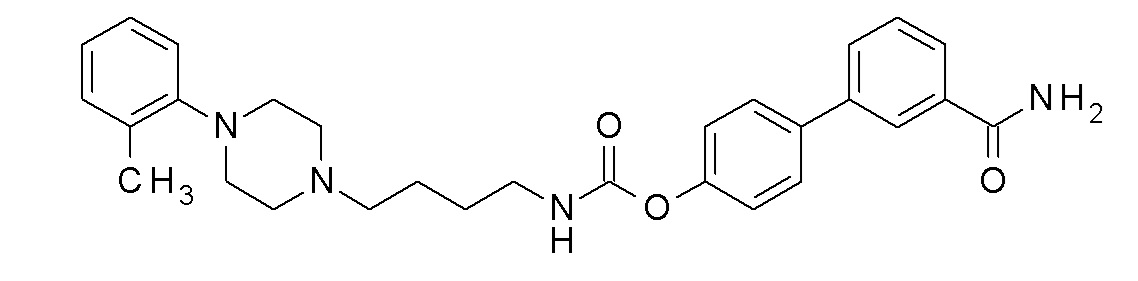
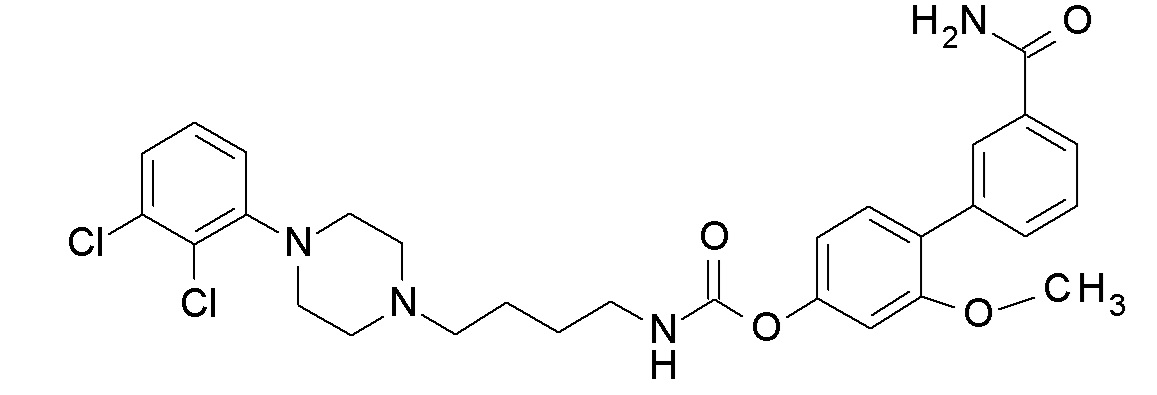
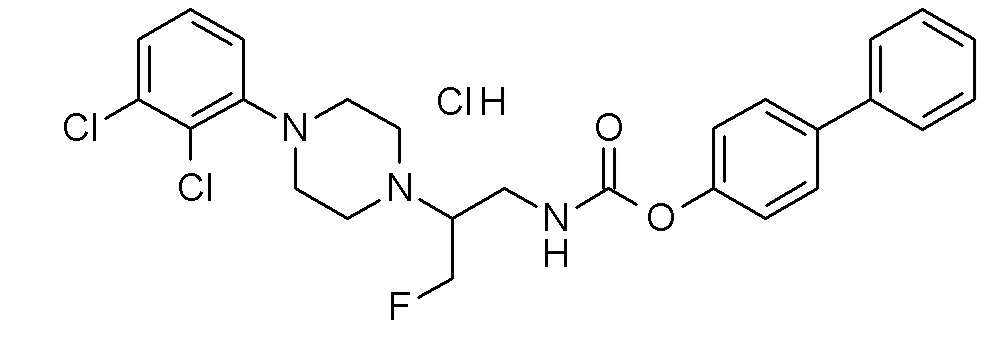
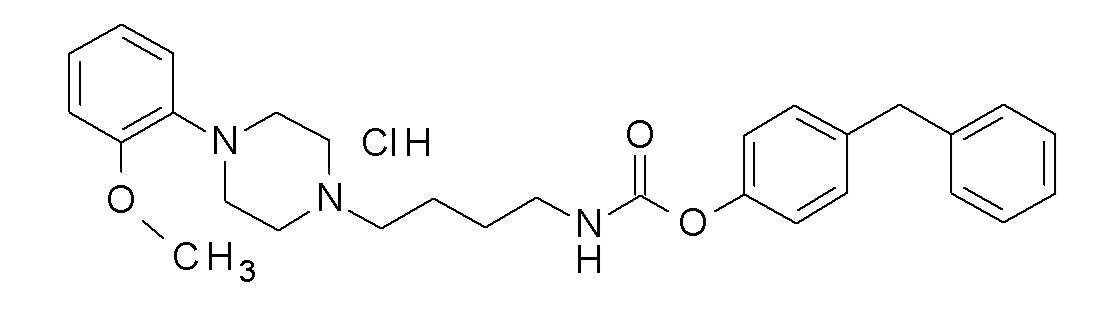
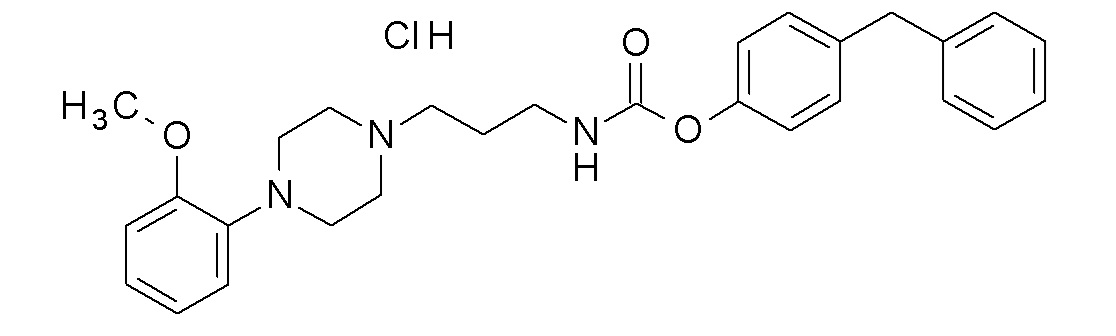
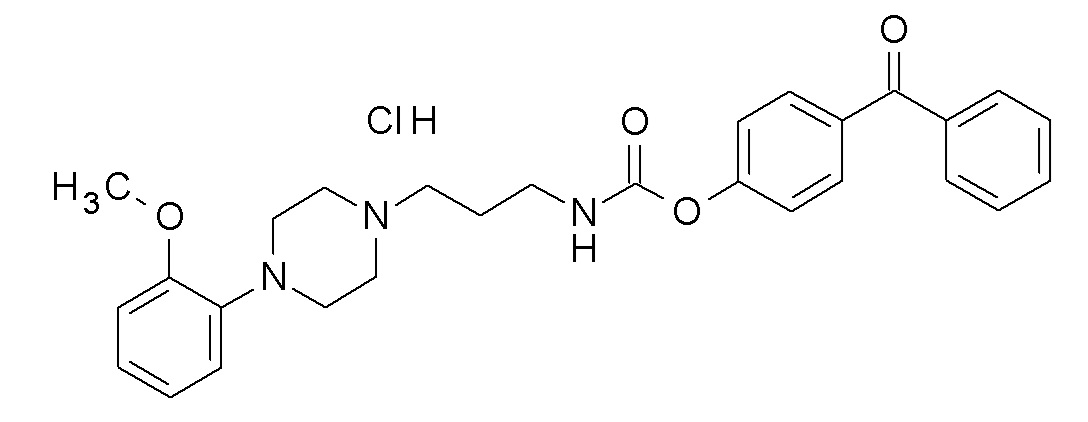
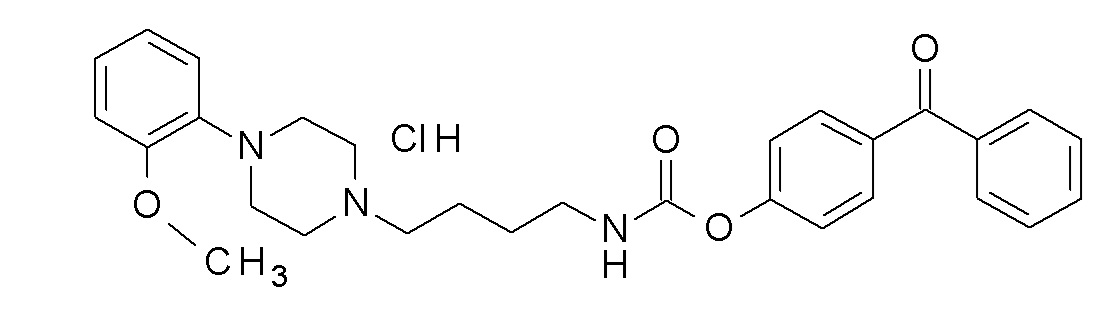
Биологические методы оценки активности соединений по настоящему изобретению
Анализ гидролазы амидов жирных кислот (FAAH) крысы
Головной мозг самцов крыс Sprague Dawley гомогенизировали в сосуде в 20 мМ Tris-HCl pH 7,4, 0,32M сахарозы, измеряли концентрацию белка и алкивоту проб хранили при -80°C до использования.
Активность FAAH измеряли с использованием 50 мкг общего гомогената головного мозга крыс, предварительно инкубированного в течение 10 минут при 37°C в 445,5 мкл буфера для анализа (50мМ Tris-HCl pH7,4, 0,05% не содержащего жирных кислот BSA) с 4,5 мкл ингибитора при соответствующей концентрации в DMSO, или только DMSO для измерения общей активности FAAH. Холостую пробу (без активности) получали с 445,5 мкл буфера для анализа и 4,5 мкл DMSO, без 50 мкг общего гомогената головного мозга крыс.
После 10 минут прединкубации начинали реакцию путем добавления 50 мкл субстрата в течение 30 мин при 37°C. Субстрат подготавливали в буфере для анализа для достижения конечной концентрации 1 мкМ арахидоноил-этаноламида (N,90050, Cayman Chemical) и 0,6 нМ анандамида [этаноламин-1-3H] (American Radiolabeled Chemicals Inc., ART,0626, 1мКи/мл, удельная активность 60 Ки/ммоль).
Реакцию останавливали путем добавления холодной смеси 1:1 CHCl3/метанол. После 10 минут центрифугирования (845×g при 4°C) 600 мкл водной фазы переносили в сцинтилляционные сосуды, предварительно заполненные 3 мл сцинтилляционной жидкости (ULTIMA GOLD, Cat,6013329, Perkin Elmer Inc.).
Радиоактивность измеряли методом жидкостного сцинтилляционного счета (Microbeta2 Lumijet, Perkin Elmer Inc.).
Флуоресцентный анализ человеческой рекомбинантной гидролазы амидов жирных кислот-1 (FAAH-1)
Hek293 клетки, стабильно трансфицированные человеческим FAAH-1, поддерживали в DMEM, содержащей 10% FBS, 1% пенициллина/стрептомицина и 500мкг/мл G418, с поддержанием селективного давления. Клетки соскребали с пластин с использованием холодного PBS1X pH 7,4 и собирали путем центрифугирования (500 g, 10 минут, 4°C); клеточный осадок после центрифугирования ресуспендировали в гомогенизирующем буфере (20 мМ Tris-HCl pH7,4, 0,32M сахарозы), разрушали путем воздействия ультразвуком (10 импульсов, 5 раз) и центрифугировали при 800×g в течение 15 минут при 4°C; полученные супернатанты затем центрифугировали при 105000×g в течение 1 часа при 4°C и осажденные центрифугированием мембраны ресуспендировали в PBS1X pH 7,4.
Измеряли концентрацию белка и алкивоту проб хранили при -80°C до использования.
Флуоресцентный анализ, используемый для измерения активности FAAH, осуществляли в 96-луночных планшетах (OptiPlate-96 Black, cat.6005279, Perkin Elmer Inc.) в объеме 180 мкл/лунка буфера для анализа (50мМ Tris-HCl pH7,4, 0,05% не содержащего жирных кислот BSA), содержащего 2,5 мкг/лунка Hek293-hFAAH-1 мембранного препарата с 10 мкл ингибитора при соответствующей концентрации в DMSO, или 10 мкл только DMSO, для измерения общей активности FAAH. Холостую пробу (без активности) получали с использованием 180 мкл буфера для анализа без Hek293-hFAAH-1 мембранного препарата и 10 мкл DMSO.
После 50 минут прединкубации соединений с ферментом при 37°C начинали реакцию путем добавления 10 мкл субстрата (AMC Арахидоил Амид, N.10005098, Cayman Chemical) в течение 30 минут при 37°C. Субстрат подготавливали в этаноле для достижения конечной концентрации 2 мкМ.
Флуоресценцию определяли при помощи Tecan Infinite M200 nanoquant (Tecan Group Ltd.) при длине волны возбуждения 350 нм и длине волны эмиссии 460 нм.
Клеточный анализ D3 допаминового рецептора (D3dR)
Активность на D3R испытывали с использованием cAMP функционального анализа на стабильно трансфицированной CHO клеточной линии, экспрессирующей человеческий D3R. В день осуществления анализа клетки промывали два раза при помощи PBS (без кальция и магния) и отделяли при помощи скребка для отделения клеток, переносили в PBS, центрифугировали и ресуспендировали в HBSS (без кальция и магния), дополненной 1 г/л глюкозы и 500нМ IBMX. Клеточную суспензию высевали в 384-луночный планшет (Greiner) и осуществляли анализ путем добавления соединений: либо только испытываемых соединений (для агонистической активности), либо испытываемого соединения после прединкубации с 10 нМ Допамина в качестве стимулятора (для антагонистической активности). После инкубации (30 минут при комнатной температуре) стимуляцию клеток останавливали путем добавления реагентов для детекции (cAMP-XL665 и анти-cAMP криптат), разбавленных в лизисном буфере. Количественное определение сигнала осуществляли методом гомогенной разрешенной по времени флуоресценции (HTRF) на Perkin Elmer Envision планшет-ридере после одного часа инкубации при комнатной температуре.
EC50 или IC50 значения (концентрации, вызывающие полумаксимальный ответ или ингибирование ответа контрольного агониста) определяли при помощи анализа нелинейной регрессии Log [концентрация]/ответ кривых, полученных с использованием средних репликативных значений четырех-параметрового уравнения Хилла, используя программу GraphPad Prism 5 (GraphPad Software Inc., CA - USA).
Биологическая активность отдельных соединений по настоящему изобретению
|
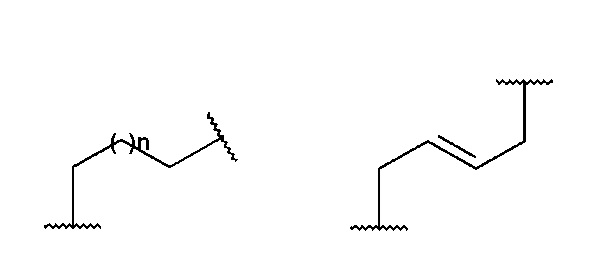
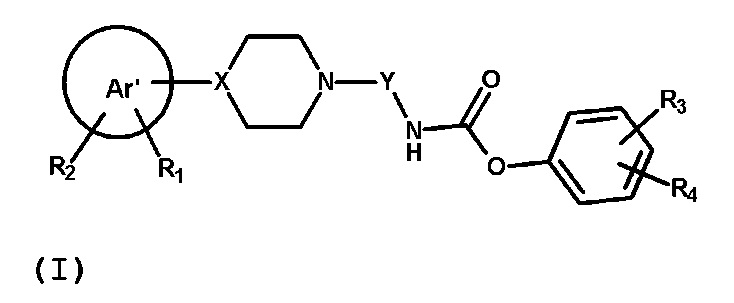
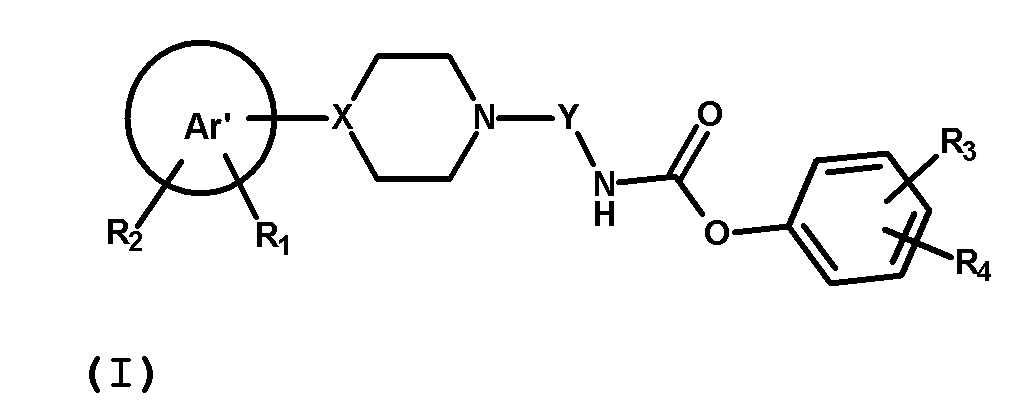
Профильное формование металлов посредством лазерной проковки
Композиции, содержащие антитела к cd38 и леналидомид
Устойчивые к хлору гидрофильные фильтрационные мембраны на основе полианилина
Модифицированные цитотоксины и их терапевтическое применение
Обнаружение изменений конформации полимеразы нуклеиновых кислот с помощью нанотрубки
Композиции, включающие антитела к cd38 и карфилзомиб
Соединения и композиции для ингибирования ire1
Соединения и композиции для ингибирования ire1
Замещенные пиримидинилпирролы, активные в качестве ингибиторов киназы

