Результат интеллектуальной деятельности: ТАКСАНОВЫЕ СОЕДИНЕНИЯ, А ТАКЖЕ СПОСОБ ИХ ПОЛУЧЕНИЯ И ИХ ПРИМЕНЕНИЯ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ, К КОТОРОЙ ОТНОСИТСЯ ИЗОБРЕТЕНИЕ
Настоящее изобретение относится к области фармацевтической химии, и конкретно относится к новому соединению, в частности, к таксановым соединениям. Настоящее изобретение также относится к способу получения таксановых соединений и их применениям в качестве активных ингредиентов в производстве противоопухолевых лекарственных препаратов для перорального введения.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Паклитаксел (РТХ) имеет структуру, представленную следующей формулой:
Паклитаксел был извлечен из коры Taxus brevifolia рода Taxus в 1971 г., он является активным противоопухолевым соединением с уникальным противораковым механизмом и обладает определенным терапевтическим эффектом в отношении ряда типов рака. В настоящее время в клинической практике паклитаксел обычно вводят путем внутривенной инъекции. Однако вследствие его низкой растворимости в воде паклитаксел обычно растворяют в смешанном растворителе из полиоксиэтилированного касторового масла (Chremophor EL) и этанола (1:1, об./об.) с целью получения инъекций паклитаксела, которые реализуют на рынке под торговым наименованием "Taxol" или "Paxene".
Несмотря на то, что в клиническом применении были достигнуты большие успехи, паклитаксел при этом также ограничен многими факторами: (1) во-первых, паклитаксел сам по себе обладает токсическими и побочными действиями, включая дозолимитирующую токсичность и подавление функции костного мозга (клинически необходимо, чтобы он применялся для лечения в комбинации с фактором роста), на нормальных тканях и клетках, и не может пересекать гематоэнцефалический барьер и т.д.; (2) с применением Кремофора EL вытекающей проблемой являются серьезные аллергические реакции, первичная гиперлипидемия, токсичность по отношению к центральной нервной системе (CNS) и изменение фармакокинетики паклитаксела [ten Tije AJ, et al, Clin Pharmacokinet 42, 655-685, 2003; H. Gelderblom, et al, Eur. J. Cancer 37 (13), 1590-1598, 2001; van Zuylen L, et al, Invest New Drugs 19, 125-141, 2001; R.B. Weiss, et al, J. Clin. Oncol.8 (7), 1263-1268, 1990]; (3) вследствие длительного приема возникает множественная устойчивость к лекарственным препаратам.
С целью решения вышеуказанных проблем многие ученые внутри страны и за рубежом провели фундаментальные исследования взаимосвязи структура-активность паклитаксела, включая изменение лекарственной формы, разработку пролекарства на основе паклитаксела, синтез таксановых производных, лечение в комбинации с ингибитором P-gp и т.п. Постоянно исследуются новые пути улучшения его растворимости в воде, усиления терапевтического эффекта, а также снижения токсических и побочных действий.
Огромное практическое значение имеет проведение исследований таксановых производных для перорального введения, поскольку изменение природы самих таксановых соединений может фундаментально решить проблемы, связанные с ними, такие как низкая растворимость в воде, высокая токсичность и т.п., таким образом, чтобы улучшить биодоступность при пероральном введении, снизить токсические и побочные действия и усилить их терапевтический эффект. Кроме того, это позволит избежать нежелательных реакций, вызванных сорастворителями и способствовать продлению терапевтического эффекта и улучшению переносимости у пациентов путем замены инъекционного введения пероральным введением.
Исследователи обнаружили, что при структурной модификации молекулы паклитаксела варьирование заместителей в положениях С7, С9 и С10 имеет незначительное воздействие на его активность, но эти положения являются активными центрами по отношению к белку P-gp. На сродство молекулы паклитаксела к белку P-gp влияет размер, электронное строение, способность к образованию водородной связи заместителей в этих положениях. Таким образом, модификация по этим группам могла бы преодолеть множественную устойчивость к лекарственным препаратам, вызванную повышенной экспрессией P-gp, и решить проблему низкой биодоступности при пероральном введении и т.п.
В связи с этим авторы настоящего изобретения занимались исследованиями производных паклитаксела и в итоге выявили ряд новых соединений с улучшенной биодоступностью при пероральном введении. Как показано в фармакологических экспериментах, по сравнению с предшествующим уровнем техники эти таксановые производные, синтезированные в настоящем изобретении, характеризуются сильной цитотоксичностью в отношении ряда линий раковых клеток человека и противоопухолевыми эффектами широкого спектра. Из данных активности in vitro в отношении линии клеток рака молочной железы MCF-7 можно видеть, что цитотоксичность сохраняется, при этом некоторые производные обладают еще лучшей цитотоксичностью, чем у паклитаксела. Поглощение и перенос таксановых производных in vivo предсказаны с применением модели клеточного монослоя выделенной из человека линии клеток колоректальной аденокарциномы Сасо-2. Из экспериментальных результатов можно видеть, что по сравнению с предыдущим уровнем техники большинство таких производных обладают улучшенной всасываемость пермеата, проходящего через мембрану, и пониженными эффектами активного выведения, поэтому предполагается, что они будут обладать улучшенной биодоступностью при пероральном введении. Поэтому цитотоксичность таких таксановых производных сохраняется (или даже увеличивается), кроме того, их биодоступность при пероральном введении также намного улучшается.
КРАТКОЕ ОПИСАНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
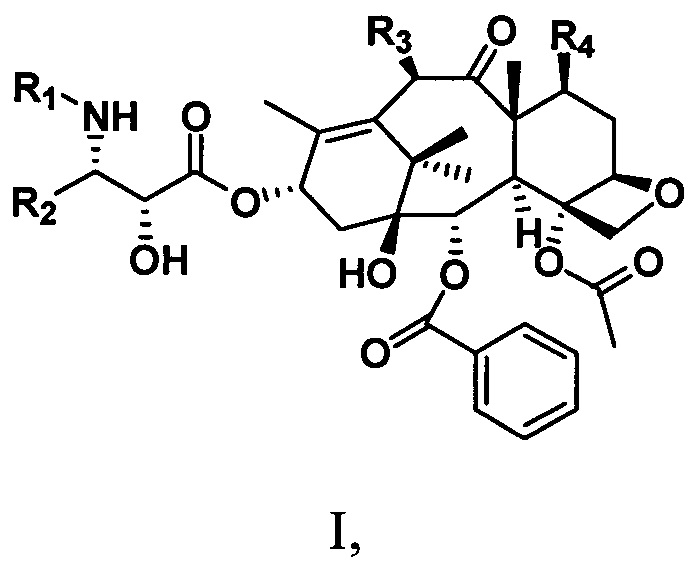
Настоящее изобретение предусматривает таксановые соединения, имеющие структуру, представленную следующей общей формулой I:
где
R1 представляет собой -COR6, -COOR6 или -CONR7aR7b;
R2 представляет собой алкильную С1-С6алкильную, С1-С6алкенильную группу, замещенную углеводородную группу, гетероциклическую группу, ароматическую группу или замещенную ароматическую группу;
R3 представляет собой -OR6, -OCOOR6, -OCOSR6 или -OCONR7aR7b;
R4 представляет собой -OR6, -OCOOR6, -OCOSR6, -OCONR7aR7b или H;
где R6 представляет собой С1-С6алкильную, С1-С6алкенильную, С1-С6алкинильную группу, замещенную углеводородную группу, ароматическую группу или гетероциклическую группу; R7a и R7b соответственно представляют собой водород, углеводородную группу, замещенную углеводородную группу или гетероциклическую группу.
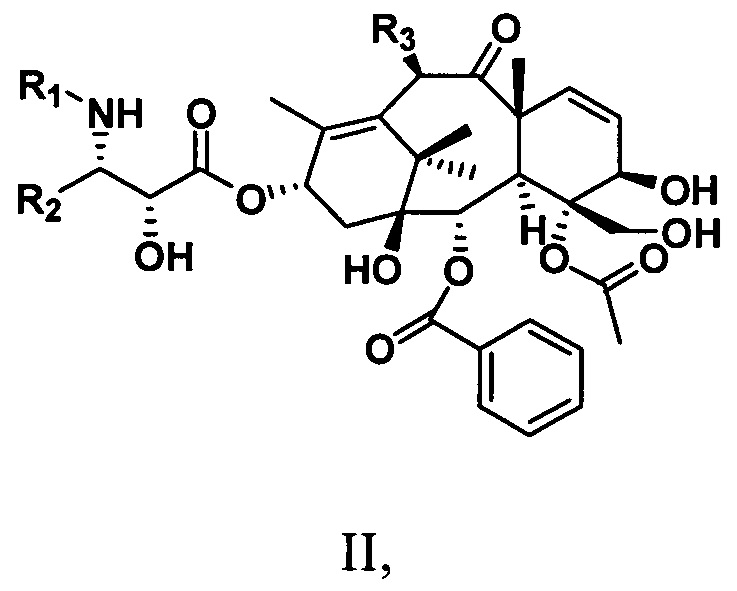
Кроме того, настоящее изобретение также предусматривает таксановые соединения, имеющие структуру, представленную следующей общей формулой II:
где
R1 представляет собой -COR6 или -COOR6;
R2 представляет собой ароматическую группу;
R3 представляет собой -OR6;
где R6 представляет собой С1-С6алкильную, С1-С6алкенильную, С1-С6алкинильную группу, замещенную углеводородную группу, ароматическую группу или гетероциклическую группу.
Кроме того, настоящее изобретение предусматривает способ получения таксановых соединений в соответствии с настоящим изобретением.
Указанный способ получения таксановых соединений в соответствии с настоящим изобретением включает следующие стадии:
стадия 1 синтеза предшественника боковой цепи из кислоты с пятичленным оксазолидиновым кольцом: предшественник боковой цепи из кислоты с пятичленным оксазолидиновым кольцом получают посредством ряда реакций, включающих введение защитных групп, конденсацию с присоединением, кислотный гидролиз, альдольную конденсацию, каталитическое гидрирование и т.п.;
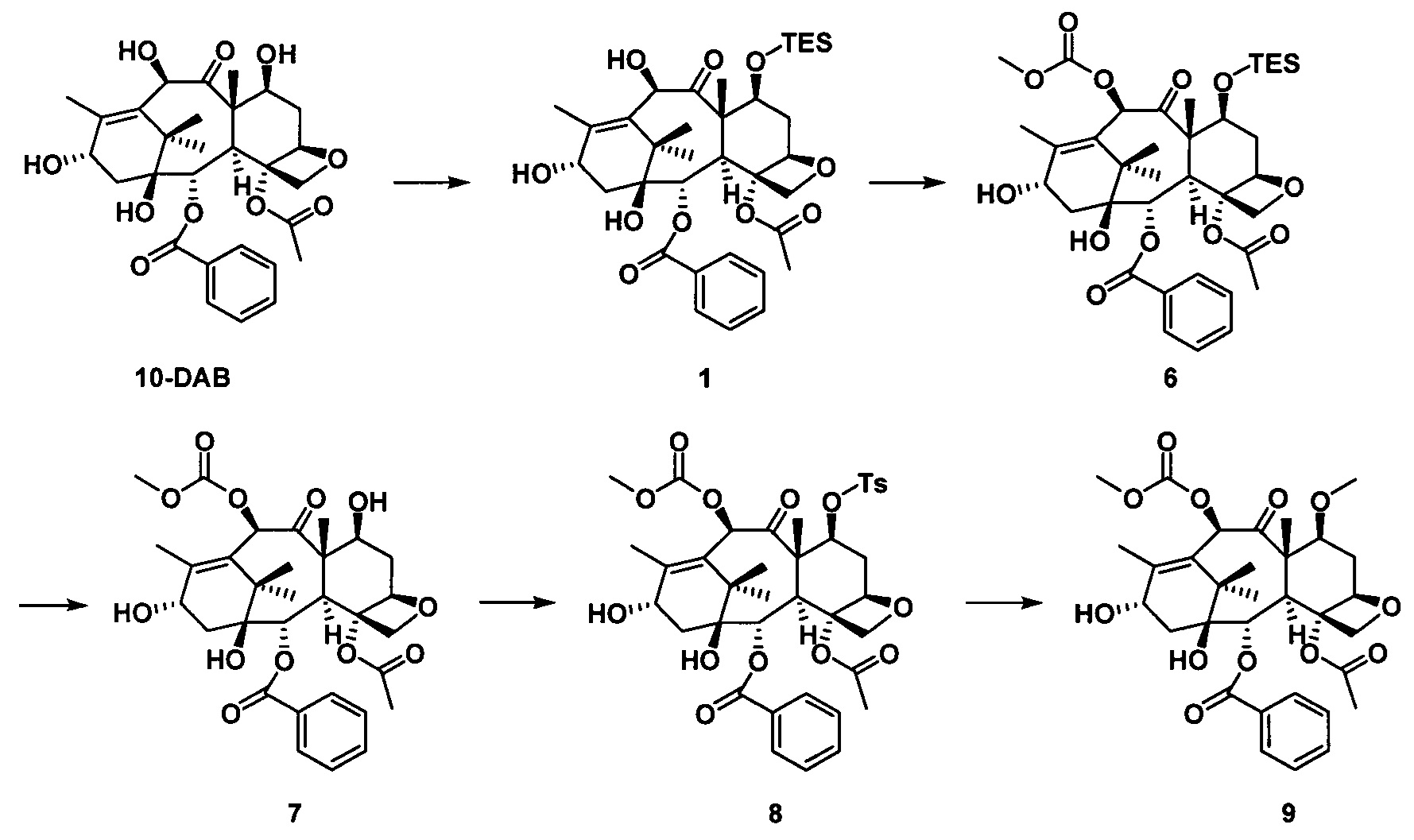
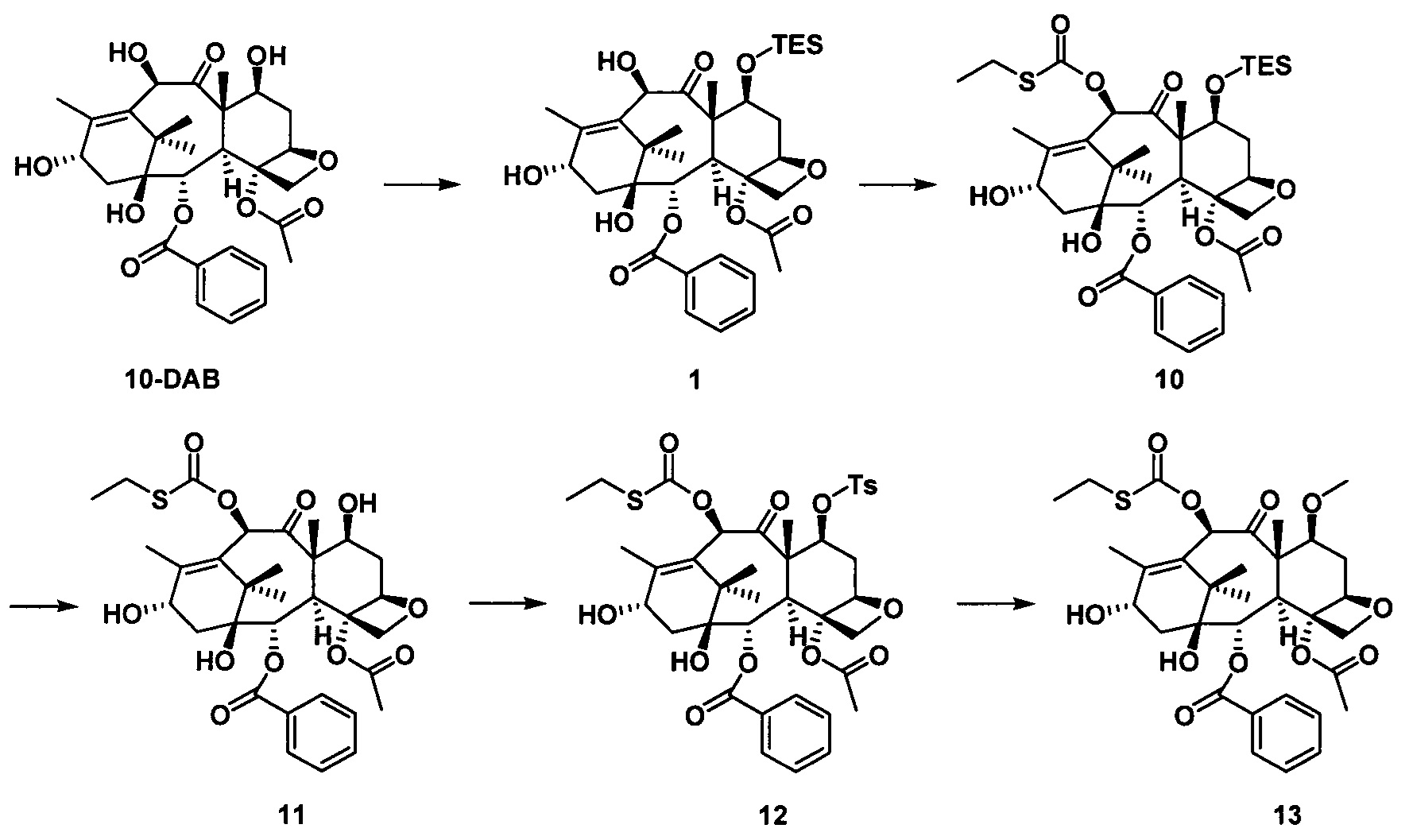
стадия 2 синтеза части таксанового исходного ядра: с применением 10-деацетилбаккатина III (10-DAB) в качестве исходного вещества гидроксильные группы в положениях С7 и С10 части исходного ядра селективно модифицируют исходя из их различных активностей с получением части таксанового исходного ядра;
стадия 3 синтеза таксановых производных: предшественник боковой цепи из кислоты с пятичленным оксазолидиновым кольцом присоединяют к части таксанового исходного ядра путем эстерификации и после удаления защитной группы кислотным гидролизом получают ряд таксановых производных.
Кроме того, настоящее изобретение предусматривает фармацевтическую композицию, содержащую соединения определенных выше общей формулы (I) и общей формулы (II), их фармацевтически приемлемые соли или сольваты в качестве активных ингредиентов, а также их применение в производстве противоопухолевых лекарственных препаратов для перорального введения.
Настоящее изобретение имеет следующие преимущества.
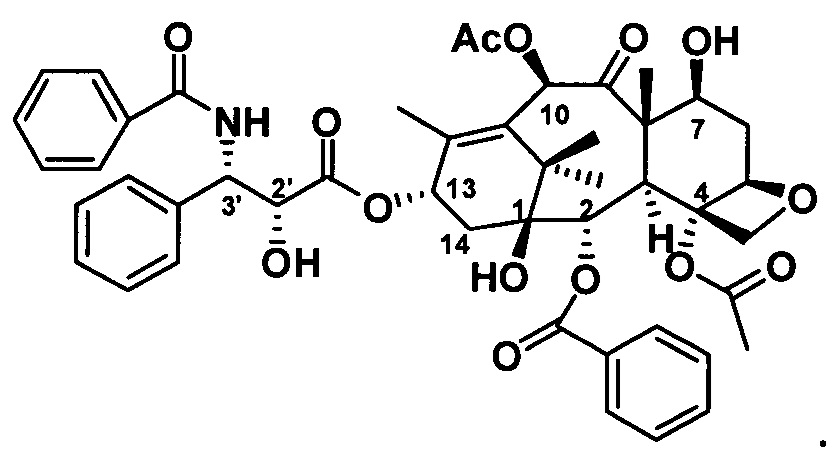
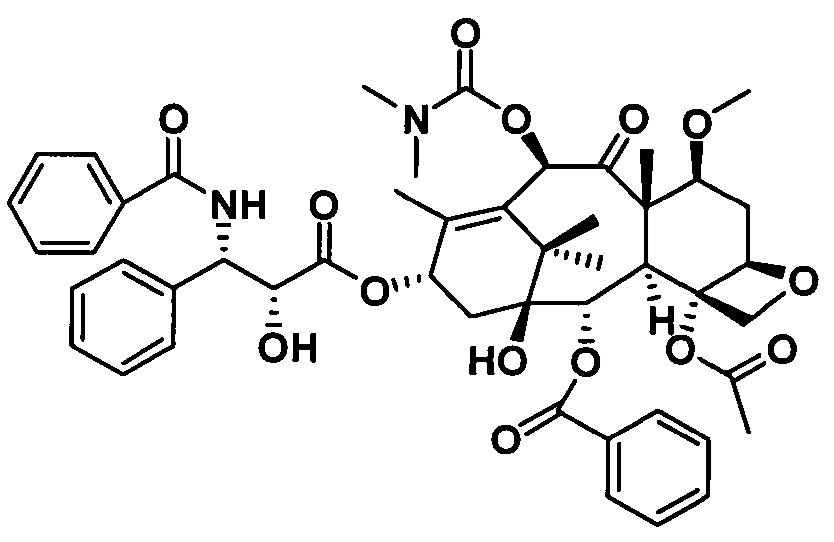
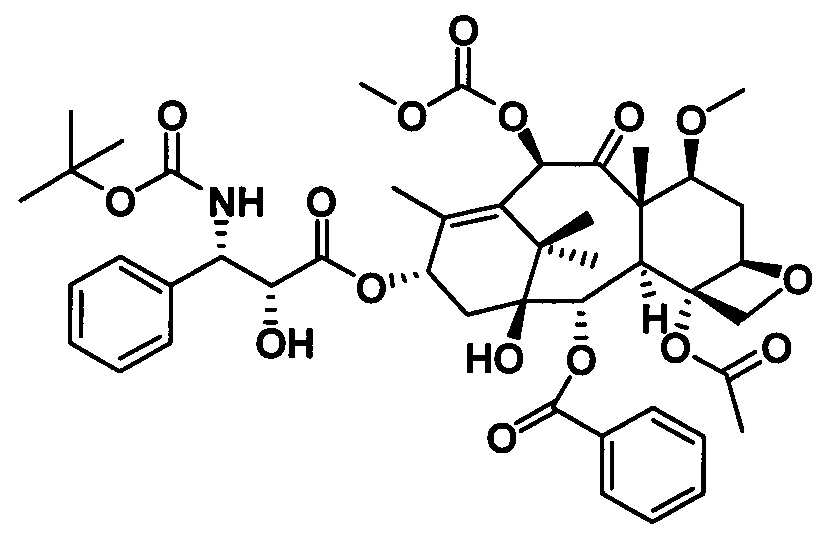
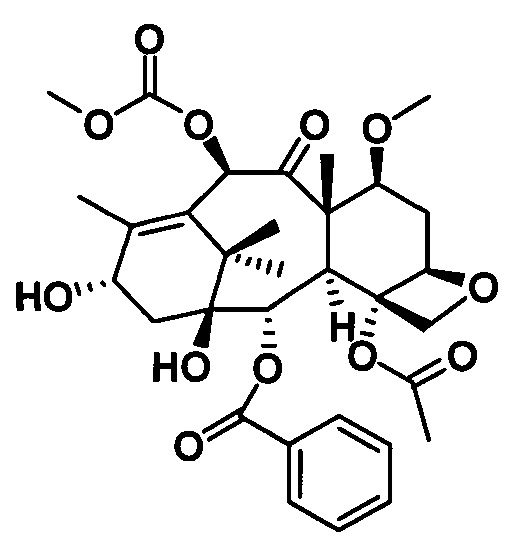
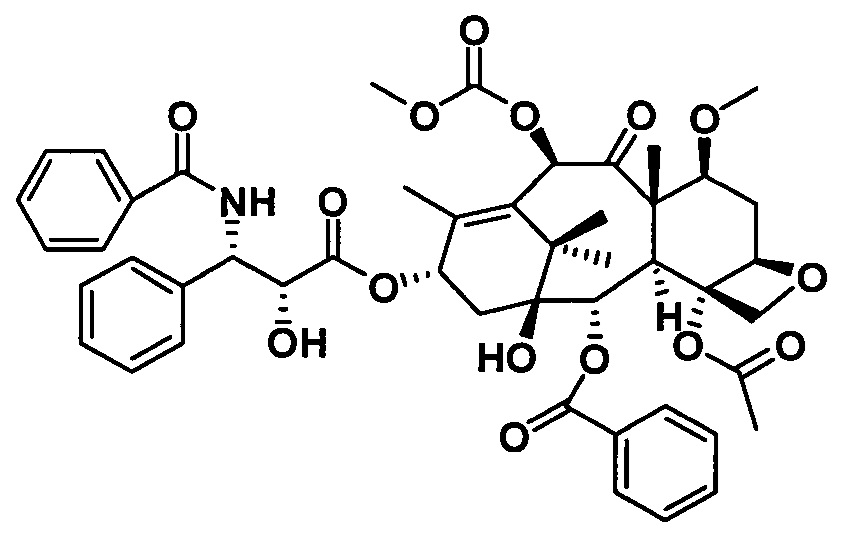
Ряд таксановых производных синтезировали путем одновременной замены заместителей в нескольких положениях С7, С10, C3'N и С3' паклитаксела. При анализе на цитотоксичность in vitro в ряде линий раковых клеток они проявили хорошие противоопухолевые активности. Биодоступность при пероральном введении таких таксановых производных in vitro была предсказана с применением испытания трансмембранного переноса в монослое клеток Сасо-2, демонстрирующего из экспериментальных результатов, что проницаемость через мембрану для всех таких производных была выше, чем у паклитаксела, при этом степень улучшения была значительной. Посредством анализа результата коэффициента активного выведения в испытании двунаправленного переноса было показано, что такие производные могут ингибировать эффект активного выведения от P-gp в различных степенях, и дополнительно подтверждено, что всасываемость при пероральном введении таких соединений была улучшена. Кроме того, соединение PCMI-31, проявившее наивысшую проницаемость через мембрану в анализе in vitro, было выбрано для испытания биодоступности при пероральном введении in vivo на крысах. Из экспериментальных результатов было показано, что его абсолютная биодоступность при пероральном введении увеличилась до 10,7%, показывая, что его всасываемость при пероральном введении in vivo улучшилась в некоторой степени по сравнению с таковой у паклитаксела. Соответственно, таксановые производные в соответствии с настоящим изобретением с такими структурами представляли собой потенциальные противоопухолевые препараты для перорального введения.
ОПИСАНИЕ ГРАФИЧЕСКИХ МАТЕРИАЛОВ
Фиг. 1 представляет собой кривую зависимости концентрации в плазме от времени для PCMI-31.
Фиг. 2 представляет собой спектр 1Н ЯМР для PCMI-22.
Фиг. 3 представляет собой масс-спектр для PCMI-22.
Фиг. 4 представляет собой спектр 1Н ЯМР для PCMI-23.
Фиг. 5 представляет собой спектр 13С ЯМР для PCMI-23.
Фиг. 6 представляет собой спектр 1Н ЯМР для PCMI-24.
Фиг. 7 представляет собой спектр 13С ЯМР для PCMI-24.
Фиг. 8 представляет собой масс-спектр для PCMI-24.
Фиг. 9 представляет собой спектр 1Н ЯМР для PCMI-25.
Фиг. 10 представляет собой спектр 13С ЯМР для PCMI-25.
Фиг. 11 представляет собой масс-спектр для PCMI-25.
Фиг. 12 представляет собой спектр 1Н ЯМР для PCMI-26.
Фиг. 13 представляет собой спектр 13С ЯМР для PCMI-26.
Фиг. 14 представляет собой ИК-спектр для PCMI-26.
Фиг. 15 представляет собой спектр 1Н ЯМР для PCMI-27.
Фиг. 16 представляет собой спектр 13С ЯМР для PCMI-27.
Фиг. 17 представляет собой масс-спектр для PCMI-27.
Фиг. 18 представляет собой спектр 1Н ЯМР для PCMI-28.
Фиг. 19 представляет собой спектр 13С ЯМР для PCMI-28.
Фиг. 20 представляет собой масс-спектр для PCMI-28.
Фиг. 21 представляет собой спектр 1Н ЯМР для PCMI-29.
Фиг. 22 представляет собой спектр 13С ЯМР для PCMI-29.
Фиг. 23 представляет собой масс-спектр для PCMI-29.
Фиг. 24 представляет собой спектр 1Н ЯМР для PCMI-30.
Фиг. 25 представляет собой спектр 13С ЯМР для PCMI-30.
Фиг. 26 представляет собой спектр 1Н ЯМР для PCMI-31.
Фиг. 27 представляет собой спектр 13С ЯМР для PCMI-31.
Фиг. 28 представляет собой масс-спектр для PCMI-31.
Фиг. 29 представляет собой ИК-спектр для PCMI-31.
Фиг. 30 представляет собой спектр 1Н ЯМР для PCMI-32.
Фиг. 31 представляет собой спектр 13С ЯМР для PCMI-32.
Фиг. 32 представляет собой масс-спектр для PCMI-32.
Фиг. 33 представляет собой спектр 1Н ЯМР для PCMI-33.
Фиг. 34 представляет собой спектр 13С ЯМР для PCMI-33.
Фиг. 35 представляет собой масс-спектр для PCMI-33.
Фиг. 36 представляет собой спектр 1H ЯМР для PCMI-34.
Фиг. 37 представляет собой спектр 13С ЯМР для PCMI-34.
Фиг. 38 представляет собой масс-спектр для PCMI-34.
Фиг. 39 представляет собой спектр 1Н ЯМР для PCMI-35.
Фиг. 40 представляет собой спектр 13С ЯМР для PCMI-35.
Фиг. 41 представляет собой масс-спектр для PCMI-35.
Фиг. 42 представляет собой спектр 1Н ЯМР для PCMI-36.
Фиг. 43 представляет собой спектр 13С ЯМР для PCMI-36.
Фиг. 44 представляет собой масс-спектр для PCMI-36.
Фиг. 45 представляет собой спектр 1Н ЯМР для PCMI-37.
Фиг. 46 представляет собой спектр 13С ЯМР для PCMI-37.
Фиг. 47 представляет собой масс-спектр для PCMI-37.
Фиг. 48 представляет собой спектр 1Н ЯМР для PCMI-38.
Фиг. 49 представляет собой спектр 13С ЯМР для PCMI-38.
Фиг. 50 представляет собой масс-спектр для PCMI-38.
Фиг. 51 представляет собой спектр 1Н ЯМР для PCMI-39.
Фиг. 52 представляет собой спектр 13С ЯМР для PCMI-39.
Фиг. 53 представляет собой масс-спектр для PCMI-39.
Фиг. 54 представляет собой спектр 1Н ЯМР для PCMI-40.
Фиг. 55 представляет собой спектр 13С ЯМР для PCMI-40.
Фиг. 56 представляет собой масс-спектр для PCMI-40.
Фиг. 57 представляет собой спектр 1Н ЯМР для PCMI-41.
Фиг. 58 представляет собой спектр 13С ЯМР для PCMI-41.
Фиг. 59 представляет собой масс-спектр для PCMI-41.
Фиг. 60 представляет собой ИК-спектр для PCMI-41.
Фиг. 61 представляет собой спектр 1Н ЯМР для PCMI-42.
Фиг. 62 представляет собой спектр 13С ЯМР для PCMI-42.
Фиг. 63 представляет собой масс-спектр для PCMI-42.
Фиг. 64 представляет собой спектр 1Н ЯМР для PCMI-43.
Фиг. 65 представляет собой масс-спектр для PCMI-43.
Фиг. 66 представляет собой спектр 1H ЯМР для PCMI-44.
Фиг. 67 представляет собой масс-спектр для PCMI-44.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Выражение "алкил", применяемое в настоящем документе, относится к группе, состоящей только из атомов углерода и водорода без какой-либо степени ненасыщенности (такой как двойные связи, тройные связи или кольца), при этом оно охватывает все типы ее возможных геометрических изомеров и стереоизомеров. Группы присоединены к остальной части молекулы посредством одинарной связи. Выражение "С1-С6алкил", применяемое в настоящем документе, относится к определенному выше алкилу с количеством атомов углерода 1-6. В качестве неограничивающих примеров С1-С6алкила можно перечислить следующие группы с прямой цепью или разветвленной цепью: метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, н-пентил и их изомеры, а также н-гексил и его изомеры.
Выражение "алкенил", применяемое в настоящем документе, относится к группе, образованной из вышеуказанной алкильной группы (за исключением метила) путем включения одной или более двойных связей. Выражение "С1-С6алкенил" относится к определенному выше алкенилу с количеством атомов углерода 1-6.
Выражение "алкинил", применяемое в настоящем документе, относится к группе, образованной из вышеуказанной алкильной группы (за исключением метила) путем включения одной или более тройных связей. Выражение "С1-С6алкинил" относится к определенному выше алкинилу с количеством атомов углерода 1-6.
Выражение "углеводородная группа", применяемое в настоящем документе, относится к группе, состоящей только из атомов углерода и водорода. Выражение "замещенная углеводородная группа" относится к определенной выше алкильной, алкенильной или алкинильной группе и т.п., содержащим заместители. Заместитель может представлять собой гидроксильную группу, аминогруппу и т.п.
Выражение "гетероциклическая группа", применяемое в настоящем документе, относится к ароматическому 5-14-членному кольцу или неароматическому 3-15-членному кольцу, включающему атомы углерода и гетероатомы, независимо выбранные из N, О или S. Ароматическое кольцо может быть моноциклическим, бициклическим или полициклическим, при этом бициклические и полициклические группы образованы из моноциклических групп посредством соединения друг с другом через одинарные связи или путем сочленения. В качестве неограничивающих примеров гетероарильных групп можно перечислить следующие группы: оксазолил, изоксазолил, имидазолил, фурил, индолил, изоиндолил, пирролил, триазолил, триазинил, тетразолил, тиенил, тиазолил, изотиазолил, пиридинил, пиримидинил, пиразинил, пиридазинил, бензофуранил, бензотиазолил, бензоксазолил, бензимидазолил, бензотиенил, бензопиранил, карбазолил, хинолинил, изохинолинил, хиназолинил, циннолинил, нафтиридинил, птеридинил, пуринил, хиноксалинил, тиадиазолил, индолизинил, акридинил, феназинил, фталазинил, кумаринил, пиразолопиридинил, пиридинопиридазинил, пирролопиридинил, имидазопиридинил, пиразолопиридазинил и группы, образованные из вышеуказанных гетероарильных групп посредством соединения друг с другом через одинарные связи или путем сочленения. Неароматическое кольцо может представлять собой моноциклическое, бициклическое или полициклическое и конденсированное кольцо, кольцо с внутренним мостиком или спирокольцо, которое может необязательно содержать одну или более двойных связей. В качестве неограничивающих примеров гетероциклических групп можно перечислить следующие группы: азепинил, акридинил, бензодиоксолил, бензодиоксанил, хроманил, диоксоланил, диоксафосфоланил, декагидроизохинолинил, инданил, индолинил, изоиндолинил, изохроманил, изотиазолидинил, изоксазолидинил, морфолинил, оксазолинил, оксазолидинил, оксадиазолил, 2-оксо-пиперазинил, 2-оксо-пиперидинил, 2-оксопирролидинил, 2-оксо-азепинил, октагидроиндолил, октагидроизоиндолил, пергидроазепинил, пиперазинил, 4-пиперидонил, пиперидинил, фенотиазинил, феноксазинил, хинуклидинил, тетрагидроизохинолинил, тетрагидрофуранил, тетрагидропиранил, тетрагидропирролидинил, тиазолинил, тиазолидинил, тиоморфолинил, тиоморфолинсульфоксид и тиоморфолинилсульфон.
Выражение "арил", применяемое в настоящем документе, относится к ароматическому кольцу, включающему по меньшей мере 6 атомов углерода, которое может быть моноциклическим, бициклическим или полициклическим, в котором бициклические и полициклические кольца могут быть образованы из моноциклических колец посредством соединения друг с другом через одинарные связи или путем сочленения. В качестве неограничивающих примеров арильных групп можно перечислить следующие группы: фенил, нафтил, антрил, фенантрил, инденил, пиренил, периленил, азуленил, аценафтенил, флуоренил, бензоаценафтенил, трифениленил, хризенил, бифенил, бинафтил и т.п.
Выражение "замещенная ароматическая группа", применяемое в настоящем документе, относится к определенной выше ароматической группе, содержащей заместители. Заместитель может представлять собой алкил, алкенил, алкинил, гидроксил, амин и т.п.
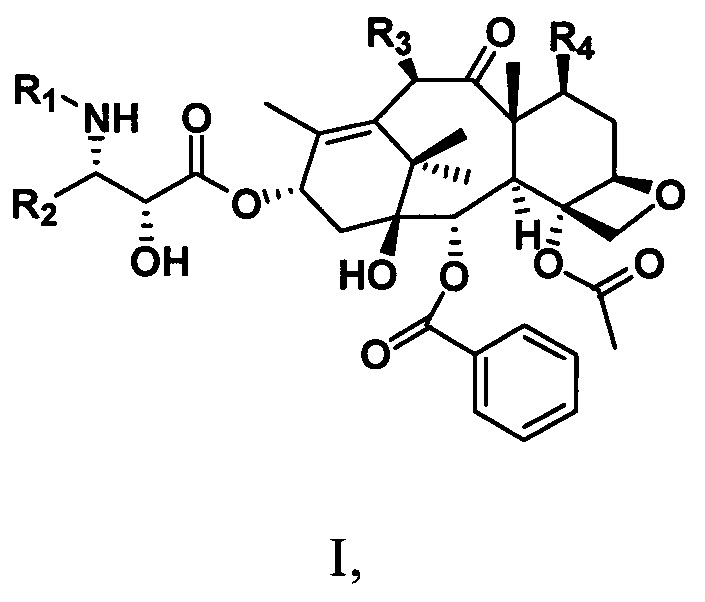
Настоящее изобретение предусматривает таксановые соединения, имеющие структуру, представленную следующей общей формулой I:
где
R1 представляет собой -COR6, -COOR6 или -CONR7aR7b;
R2 представляет собой алкильную С1-С6алкильную, С1-С6алкенильную группу, замещенную углеводородную группу, гетероциклическую группу, ароматическую группу или замещенную ароматическую группу;
R3 представляет собой -OR6, -OCOOR6, -OCOSR6 или -OCONR7aR7b;
R4 представляет собой -OR6, -OCOOR6, -OCOSR6, -OCONR7aR7b или H;
где R6 представляет собой С1-С6алкильную, С1-С6алкенильную, С1-С6алкинильную группу, замещенную углеводородную группу, ароматическую группу или гетероциклическую группу; R7a и R7b соответственно представляют собой водород, углеводородную группу, замещенную углеводородную группу или гетероциклическую группу.
Предпочтительно R1 представляет собой бензоил, трет-бутилоксикарбонил или N,N'-диметилкарбамоил;
R2 представляет собой фенил, или 
R3 представляет собой -ОМе, -ОСООСН3, -OCON(CH3)2 или -OCOSC2H5;
R4 представляет собой -ОМе, -ОСООСН3, -OCON(CH3)2, -OCOSC2H5 или Н.
Кроме того, настоящее изобретение предусматривает таксановые соединения, имеющие структуру, представленную следующей общей формулой II:
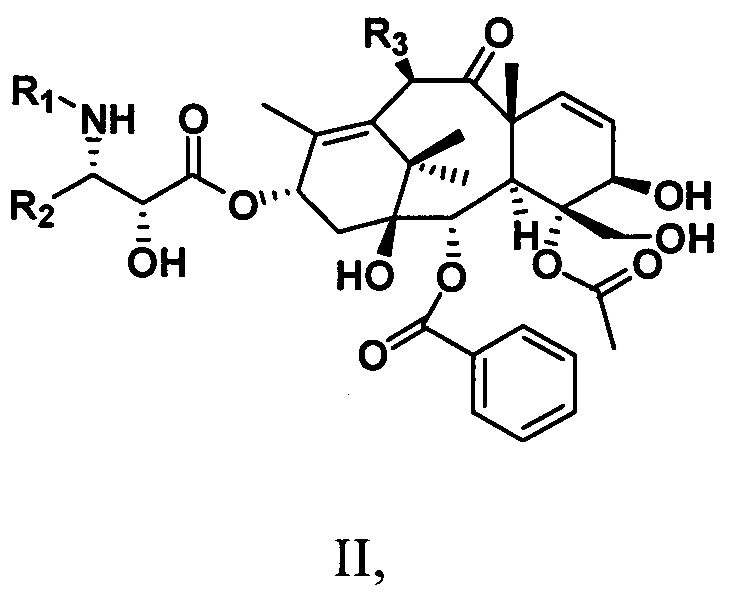
где
R1 представляет собой -COR6 или -COOR6;
R2 представляет собой ароматическую группу; R3 представляет собой -OR6;
где R6 представляет собой С1-С6алкил, С1-С6алкенил, С1-С6алкинил, замещенную углеводородную группу, ароматическую группу или гетероциклическую группу.
Предпочтительно R1 выбран из бензоила и трет-бутилоксикарбонила;
R2 выбран из фенила;
R3 выбран из -ОМе.
Наиболее предпочтительно таксановые соединения в соответствии с настоящим изобретением выбраны из соединений, имеющих следующие структуры:
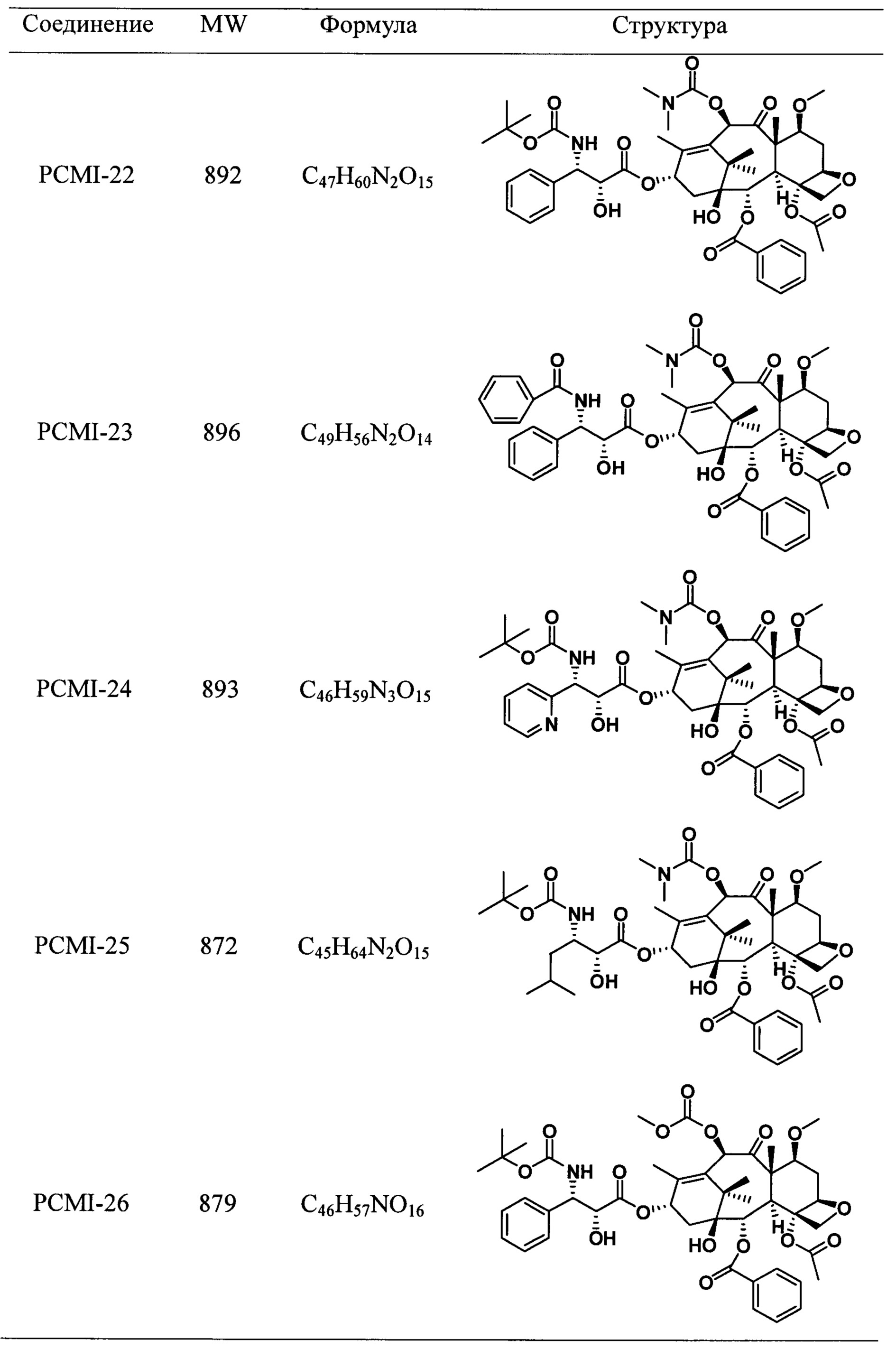
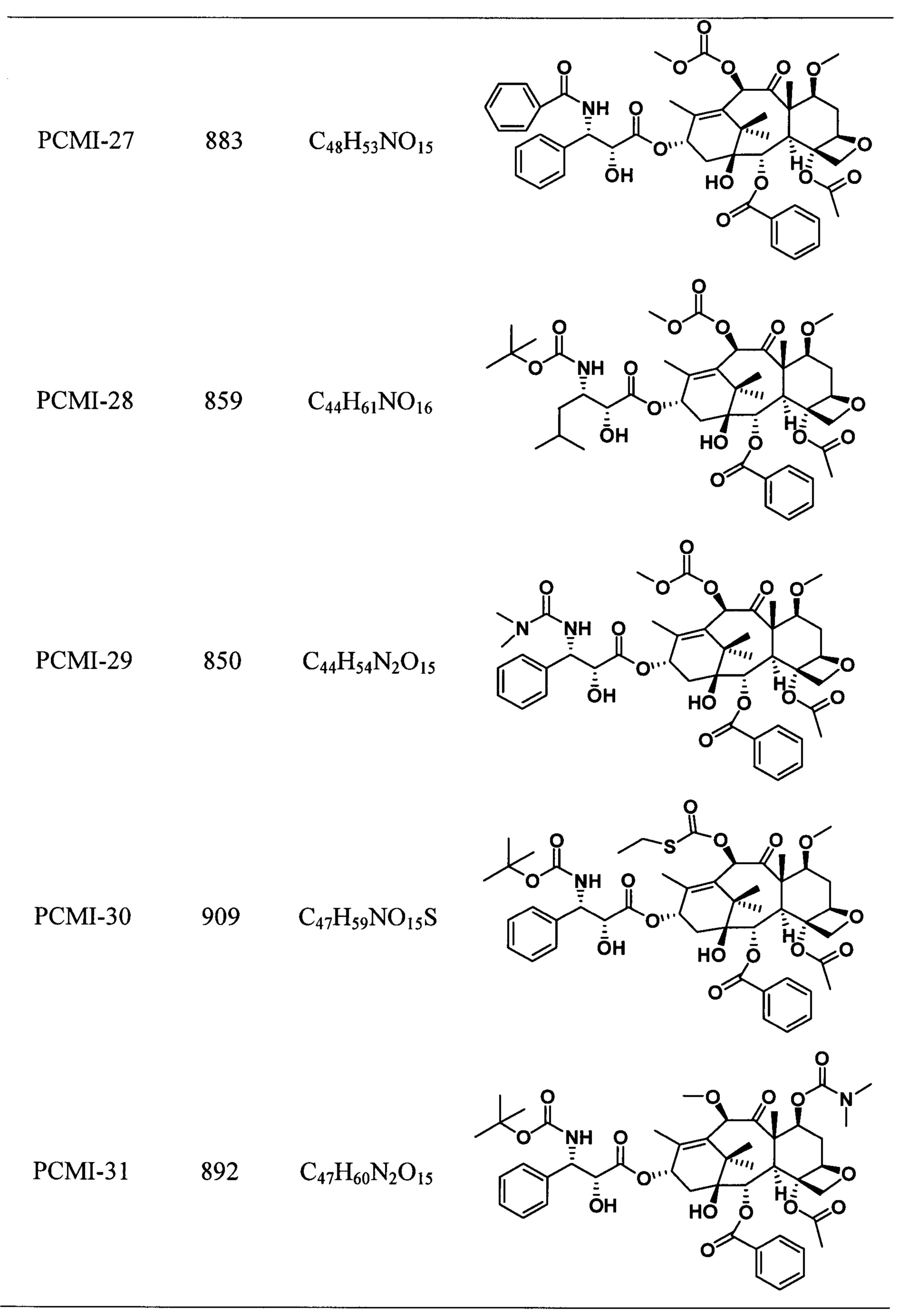
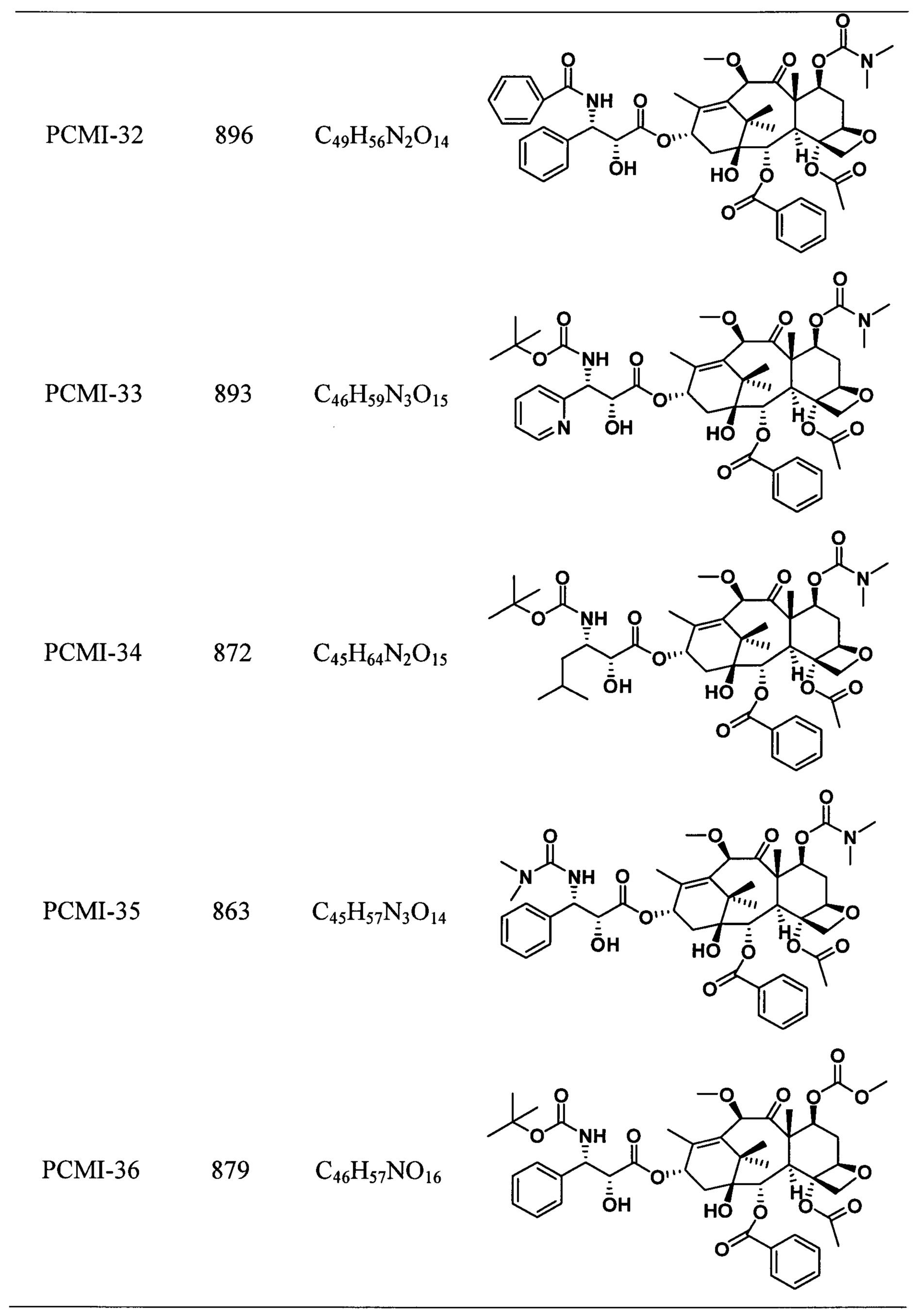
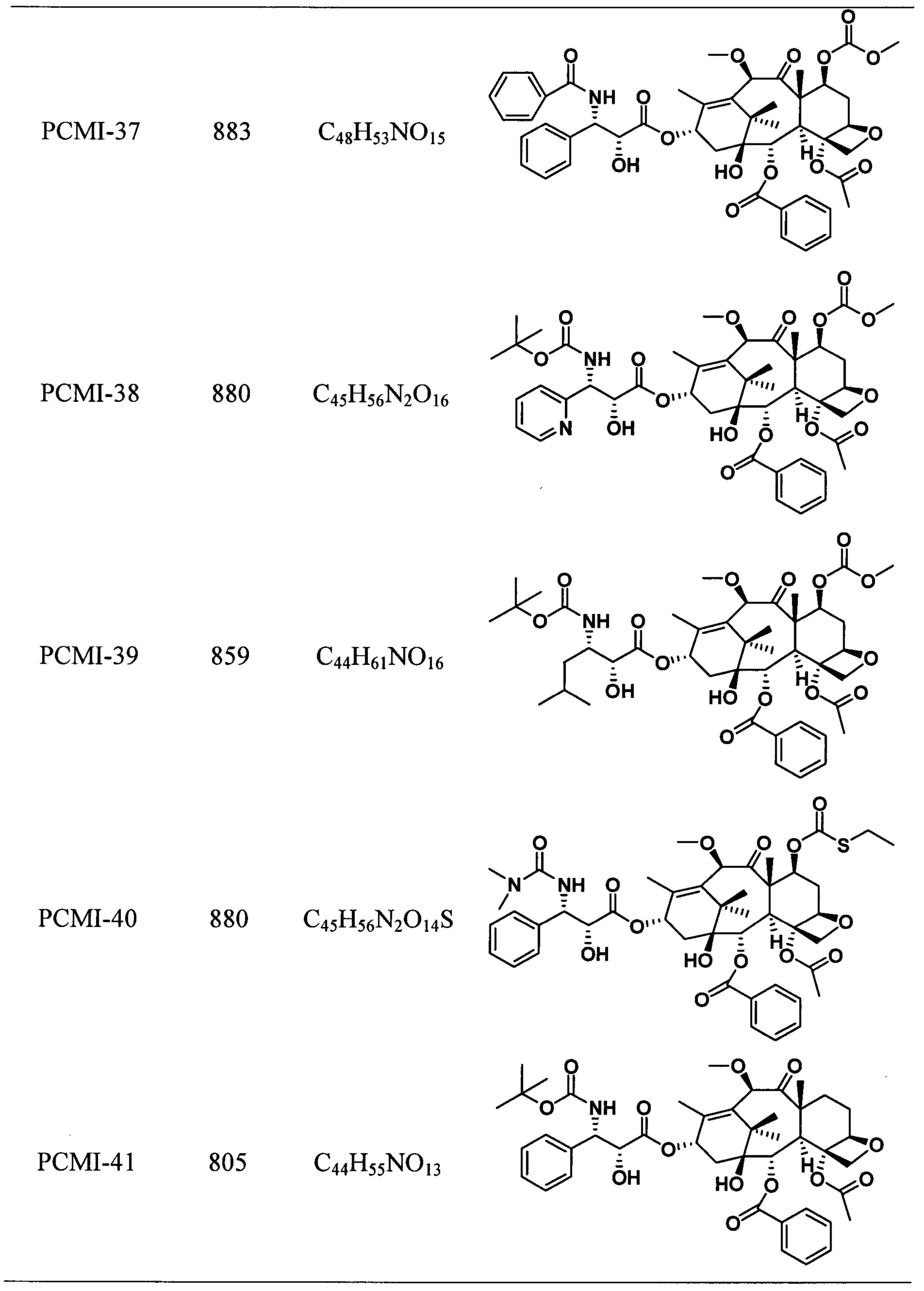
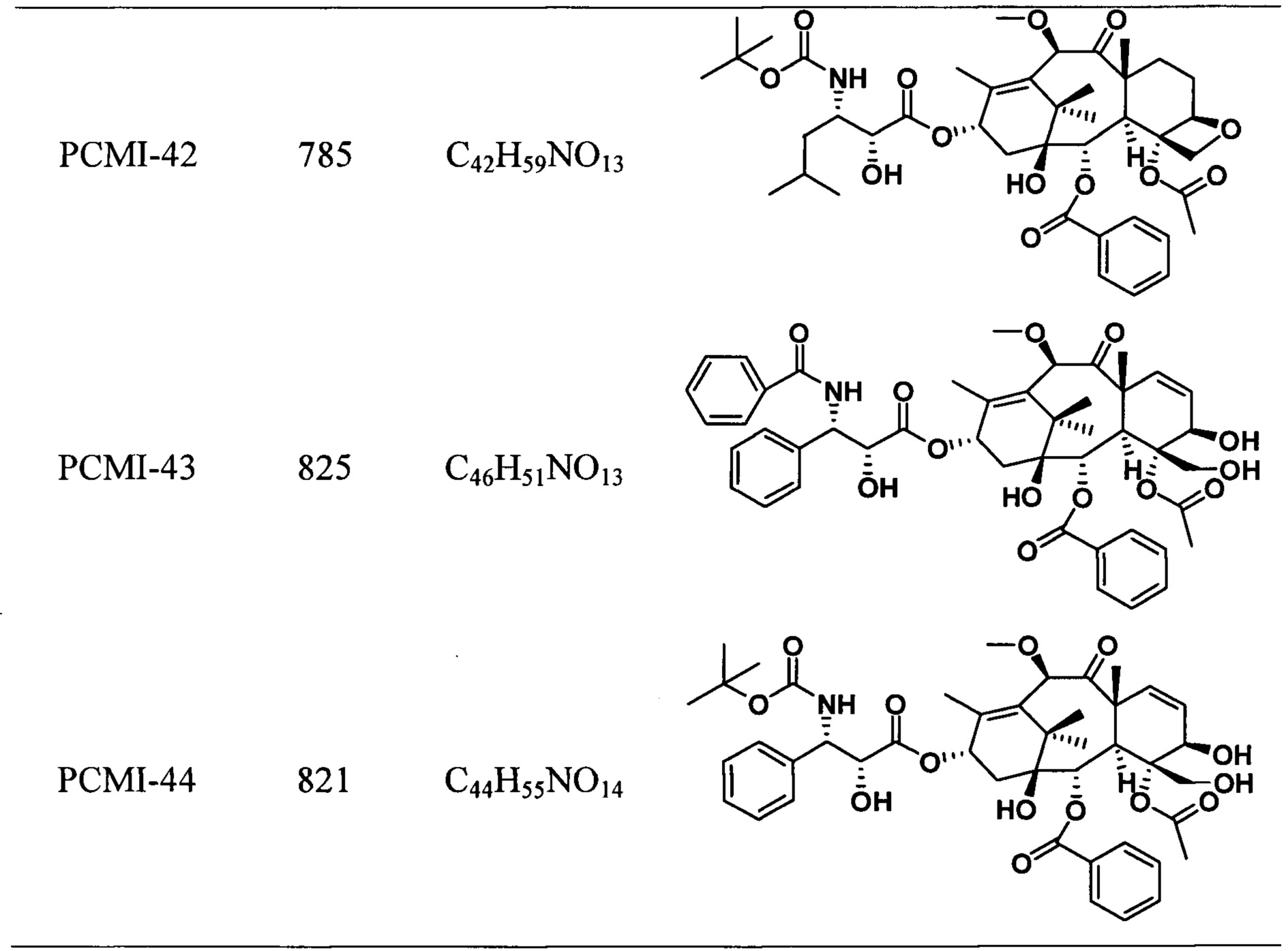
Согласно настоящему изобретению соединения, имеющие структуры, представленные общей формулой (I) и формулой (II), также включают все изомеры таких соединений и смеси изомеров.
При необходимости соединения, имеющие структуры, представленные общей формулой (I) и формулой (II), могут быть превращены в фармацевтически приемлемые нетоксичные соли.
Согласно настоящему изобретению соединения, имеющие структуры, представленные общей формулой (I) и формулой (II), могут необязательно существовать в виде сольватов (таких как гидраты). Поэтому такие сольваты (такие как гидраты) также включены в соединения в соответствии с настоящим изобретением.
Кроме того, настоящее изобретение предусматривает фармацевтическую композицию, содержащую соединения, имеющие структуры, представленные определенной выше общей формулой (I) и формулой (II), их фармацевтически приемлемые соли или сольваты в качестве активных ингредиентов, а также их применение в производстве противоопухолевых лекарственных препаратов для перорального введения.
В фармацевтической композиции в соответствии с настоящим изобретением весовая доля соединений в соответствии с настоящим изобретением составляет 0,01% - 99,99%, при этом остальное представляет собой фармацевтически приемлемые носители. Фармацевтическая композиция представлена в форме подходящих препаратов. Препараты включают: таблетки, капсулы, гранулы, драже, порошки, взвеси, суспензии, инъекционные растворы, порошки для инъекций, суппозитории, кремы, капли или трансдермальные терапевтические системы. При этом таблетки представляют собой таблетки, покрытые сахаром, таблетки, покрытые пленкой, таблетки, покрытые энтеросолюбильной оболочкой, или таблетки с замедленным высвобождением; капсулы представляют собой твердые капсулы, мягкие капсулы или капсулы с замедленным высвобождением; порошки для инъекций представляют собой лиофилизированные порошки для инъекций.
В лекарственной форме фармацевтической композиции в соответствии с настоящим изобретением каждая лекарственная форма содержит эффективное количество 0,1 мг - 1000 мг соединений в соответствии с настоящим изобретением. При этом каждая лекарственная форма относится к каждой ее единице, например, каждой таблетке в таблетках, каждой капсуле в капсулах. В качестве альтернативы, она может также относиться к дозе, вводимой каждый раз (например, доза 100 мг каждый раз).
Могут применяться твердые носители, если фармацевтическую композицию в соответствии с настоящим изобретением преобразуют в твердые или полужидкие препараты, такие как порошки, таблетки, диспергируемые порошки, капсулы, крахмальные облатки, суппозитории и мази. Пригодный твердый носитель предпочтительно выбирают из одного или более веществ из разбавителей, ароматизаторов, солюбилизаторов, смазывающих средств, суспендирующих средств, связующих, объемообразующих средств и т.п., или он может представлять собой инкапсулирующий материал. В порошковых препаратах он содержит 5-70 вес. % тонкодисперсного активного ингредиента в носителях. Подходящие твердые носители включают карбонат магния, стеарат магния, порошок талька, сахарозу, лактозу, пектин, декстрин, крахмал, желатин, метилцеллюлозу, натрий-карбоксиметилцеллюлозу, низкокипящий воск, кокосовое масло и т.п. Поскольку таблетки, порошки, крахмальные облатки и капсулы легко вводить, они представляют наиболее преимущественные твердые препараты для перорального введения.
Жидкие препараты в соответствии с настоящим изобретением включают растворы, суспензии и эмульсии. Например, инъекционные препараты для парентерального введения могут быть в виде водного раствора или водно-пропиленгликолевого раствора, который используется для регулировки изотоничности, рН и т.д., что делает его адаптированным к физиологическим условиям живого тела. В качестве альтернативы, жидкие препараты можно получать в виде полиэтиленгликолевого или водного раствора. Водный раствор для перорального введения можно получать путем растворения активных ингредиентов в воде и добавления к ним необходимых количеств красителей, ароматизаторов, стабилизаторов и загустителей. Кроме того, водные суспензии для перорального введения можно получать диспергированием тонкодисперсных активных ингредиентов в вязких материалах, таких как натуральные и синтетические камеди, метилцеллюлоза, натрий-карбоксиметил целлюлоза и другие известные суспендирующие средства.
Для удобства приема и равномерности доз особенно выгодно получать вышеуказанные фармацевтические препараты в виде единицы препарата. Единица препарата относится к физически отделяемой единице, содержащей одну дозу. Каждая единица содержит точно рассчитанное предварительно определенное количество активных ингредиентов, которое может обеспечивать необходимые терапевтические эффекты. Данная единица препарата может быть в упакованном виде, например, таблетки, капсулы, порошки в небольших тубах или пузырьках, или мази, гели или крема в тубах или пузырьках.
Несмотря на то, что количество активного ингредиента в единице препарата может варьироваться, в целом оно находится в диапазоне 1-1000 мг в зависимости от эффективности выбранного активного ингредиента.
Если соединения в соответствии с настоящим изобретением, представленные формулой (I) и формулой (II), применяются в качестве противоопухолевых средств, их доза может варьироваться в зависимости от потребностей пациентов, условий заболевания, выбранных соединений и т.п.
Согласно настоящему изобретению, таксановые соединения получают способом, включающим следующие стадии:
стадия 1 синтеза предшественника боковой цепи из кислоты с пятичленным оксазолидиновым кольцом: предшественник боковой цепи из кислоты с пятичленным оксазолидиновым кольцом получают посредством ряда реакций, включающих введение защитных групп, конденсацию с присоединением, кислотный гидролиз, альдольную конденсацию, каталитическое гидрирование и т.п.;
стадия 2 синтеза части таксанового исходного ядра: с применением 10-деацетилбаккатина III (10-DAB) в качестве исходного вещества гидроксильные группы в положениях С7 и С10 части исходного ядра селективно модифицируют исходя из их различных активностей с получением части таксанового исходного ядра;
стадия 3 синтеза таксановых производных: предшественник боковой цепи из кислоты с пятичленным оксазолидиновым кольцом присоединяют к части таксанового исходного ядра путем эстерификации и после удаления защитной группы кислотным гидролизом получают ряд таксановых производных.
Предпочтительно способ получения в соответствии с настоящим изобретением включает следующие стадии.
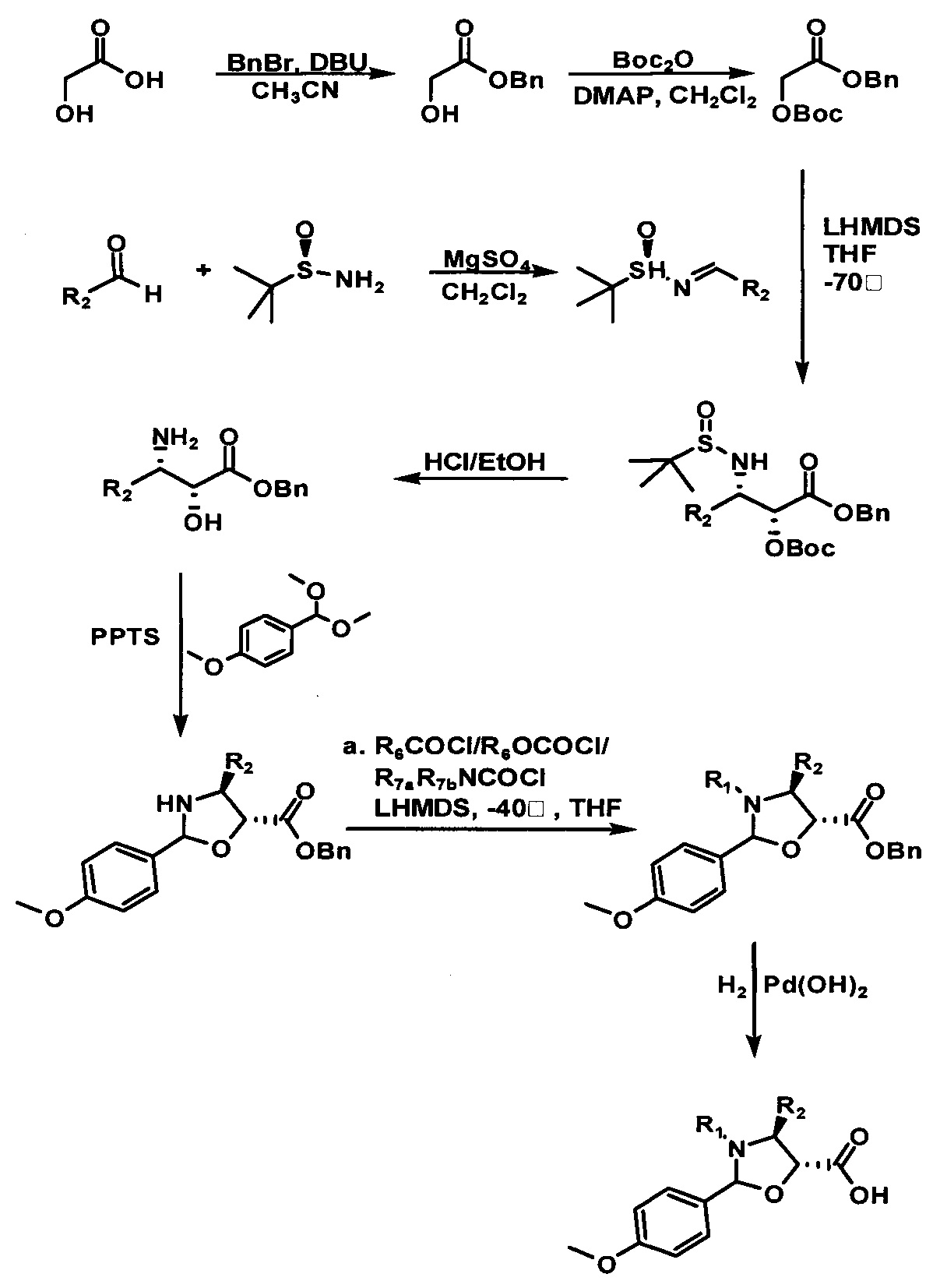
Стадия 1 синтеза предшественника боковой цепи из кислоты с пятичленным оксазолидиновым кольцом: гликолевую кислоту, используемую в качестве исходного вещества, защищают последовательно бензильной группой и трет-бутилоксикарбонильной группой (Boc группой) с образованием Вос-защищенного бензилгликолята; различные замещенные альдегиды конденсируют с (SR)-трет-бутилсульфинамидом с образованием соответствующих енаминных соединений. Вос-защищенный бензилгликолят и енаминное соединение вводя в реакцию присоединения в присутствии соли лития, а затем после кислотного гидролиза получают хиральное промежуточное соединение и полученное промежуточное соединение с 1,1'-(диметоксиметил)-n-метоксибензолом вводят в реакцию альдольной конденсации, катализируемую n-толуолсульфонатом пиридиния (PPTS) с получением соединения-продукта конденсации. Аминогруппу соединения-продукта конденсации замещают различными заместителями и после каталитического гидрирования в итоге получают предшественник боковой цепи из кислоты с пятичленным оксазолидиновым кольцом. Путь реакции следующий.
Стадия 2 синтеза части таксанового исходного ядра: в качестве исходного вещества используют 10-деацетилбаккатин III, С7-гидроксильную группу сначала защищают силильной группой, а С10-гидроксильную группу селективно защищают заместителями; После введения защитной группы в положение С10 защитную группу гидроксигруппы в положении С7 удаляют и затем гидроксигруппу в положении С7 замещают выбранными заместителями с получением части таксанового исходного ядра.
Стадия 3 синтеза таксановых производных: предшественник боковой цепи из кислоты с пятичленным оксазолидиновым кольцом присоединяют к части таксанового исходного ядра путем эстерификации и полученное соединение подвергают кислотному гидролизу для удаления защитной группы на боковой цепи с получением таксановых производных.
При этом на стадии 1 различные замещенные альдегиды включают: С1-С6гидрокарбильные альдегиды, замещенные С1-С6гидрокарбильные альдегиды, ароматические альдегиды, замещенные ароматические альдегиды и гетероароматические альдегиды;
на стадии 1 применяемую реакцию, при которой аминогруппу соединения-продукта конденсации замещают соответствующим ацилхлоридом, проводят при следующих условиях: щелочные условия; в качестве растворителя используют тетрагидрофуран, дихлорметан или диоксан, и температура составляет от комнатной температуры до -70°С;
на стадии 1 в реакции каталитического гидрирования в качестве катализатора используют палладированный уголь или гидроксид палладия; водород вводят при обычном давлении или в условиях повышенного давления в спиртах, тетрагидрофуране или дихлорметане и т.п. в качестве растворителя.
На стадии 2 С7- и С10-гидроксильные группы защищают заместителями:
(1) если R3 и R4 представляют собой -OR6, предусматривают следующую реакцию: сначала гидроксильную группу вводят в реакцию с n-толуолсульфонилхлоридом (TsCl) при температуре от комнатной до 0°С в тетрагидрофуране или дихлорметане в качестве растворителя, при этом пиридин применяют в качестве щелочи (Ру) с получением n-толуолсульфоната (C7/10-OTs), который далее вводят в реакцию с реактивом Гриньяра с получением соответствующего простого эфира -OR6;
(2) если R3 и R4 представляют собой -OCOOR6 или -OCONR7aR7b, предусматривают следующую реакцию: в щелочных условиях гидроксильную группу вводят в реакцию с соответствующим ацилхлоридом в тетрагидрофуране в качестве растворителя при температуре от комнатной до -70°С;
(3) если R3 и R4 представляют собой -OCOSR6, предусматривают следующую реакцию: гидроксильную группу вводят в реакцию с N,N'-карбонилдиимидазолом (CDI) в тетрагидрофуране в качестве растворителя при комнатной температуре и полученный продукт далее вводят с меркаптаном в реакцию замещения;
(4) если R4 представляет собой водород, предусматривают следующую реакцию: С7-гидроксильную группу вводят в реакцию с раствором N,N'-тиокарбонилдиимидазола (TCDI) в тетрагидрофуране при комнатной температуре с получением ксантата и полученный ксантат подвергают свободнорадикальной реакции деоксигенирования Бартона в смешанном растворе диоксана/тетрагидрофурана при 80-100°С, предпочтительно 85°С, катализируемой азобисизобутиронитрилом (AIBN) под действием гидрида н-бутилолова (Bu3SnH);
(5) если между положениями С6 и С7 образуется двойная связь (т.е. соединения общей формулы II), предусматривают следующую реакцию: С7-гидроксильную группу вводят в реакцию с трифторметансульфоновым ангидридом (Tf2O) в растворе дихлорметана с получением сульфоната С7-OTf с применением пиридина в качестве щелочи и сульфонат C7-OTf подвергают реакции элиминирования при 100°С в смешанном растворе диоксана/тетрагидрофурана под действием 1,8-диазабицикло[5.4.0]ундец-7-ена (DBU) с образованием двойной связи между положениями С6 и С7.
Предпочтительно,
на стадии 1 в реакции, применяемой при замещении аминогруппы соединения-продукта конденсации, в качестве щелочи предпочтительно применяют гексаметилдисилазид лития (LHMDS), а в качестве растворителя применяют тетрагидрофуран; температура составляет предпочтительно -40°С, ацилхлорид включает R6COCl, R6OCOCl и R7aR7bNCOCl; в реакции каталитического гидрирования в качестве катализатора предпочтительно применяют гидроксид палладия, водород вводят при 20 фунт/кв.дюйм и реакцию предпочтительно проводят в спиртовом растворе;
на стадии 2 С7- и С10-гидроксильные группы защищают заместителями:
(1) если R3 и R4 представляют собой -OR6, в качестве растворителя предпочтительно выбирают дихлорметан, при этом температура предпочтительно составляет 0°С, и реактив Гриньяра включает R6MgBr;
(2) если R3 и R4 представляет собой -OCOOR6 или -OCONR7aR7b, в качестве щелочи предпочтительно выбирают гексаметилдисилазид лития и температура составляет предпочтительно -40°С; ацилхлорид включает R6OCOCl и R7aR7bNCOCl;
(3) если R3 и R4 представляют собой -OCOSR6, меркаптан включает R6SH.
Таксановые соединения в соответствии с настоящим изобретением обладают противоопухолевой активностью при пероральном введении, при этом полезные эффекты настоящего изобретения проиллюстрированы ниже экспериментальными данными.
1. Проба на цитотоксичность с применением линий опухолевых клеток человека
Паклитаксел применяли в качестве лекарственного средства положительного контроля. Для изучения степени ингибирования пролиферации таксановых производных в соответствии с настоящим изобретением применяли МТТ-тест на 16 линиях раковых клеток (включая клетки рака молочной железы MCF-7, MDA-MB-436; немелкоклеточного рака легких А549, NCI-H460; рака яичников А2780; меланомы А375, В16; рака толстой кишки НСТ 116, НТ-29; рака шейки матки Hela; лейкемии HL-60, K562; рака предстательной железы LNCaP, Du145; рака желудка LN-18, BGC-823) при концентрации 1 мкмоль/л, и экспериментальные результаты приведены в таблице 1.
Таблица 1. Степень ингибирования пролиферации для таксановых соединений в соответствии с настоящим изобретением на 16 линиях раковых клеток
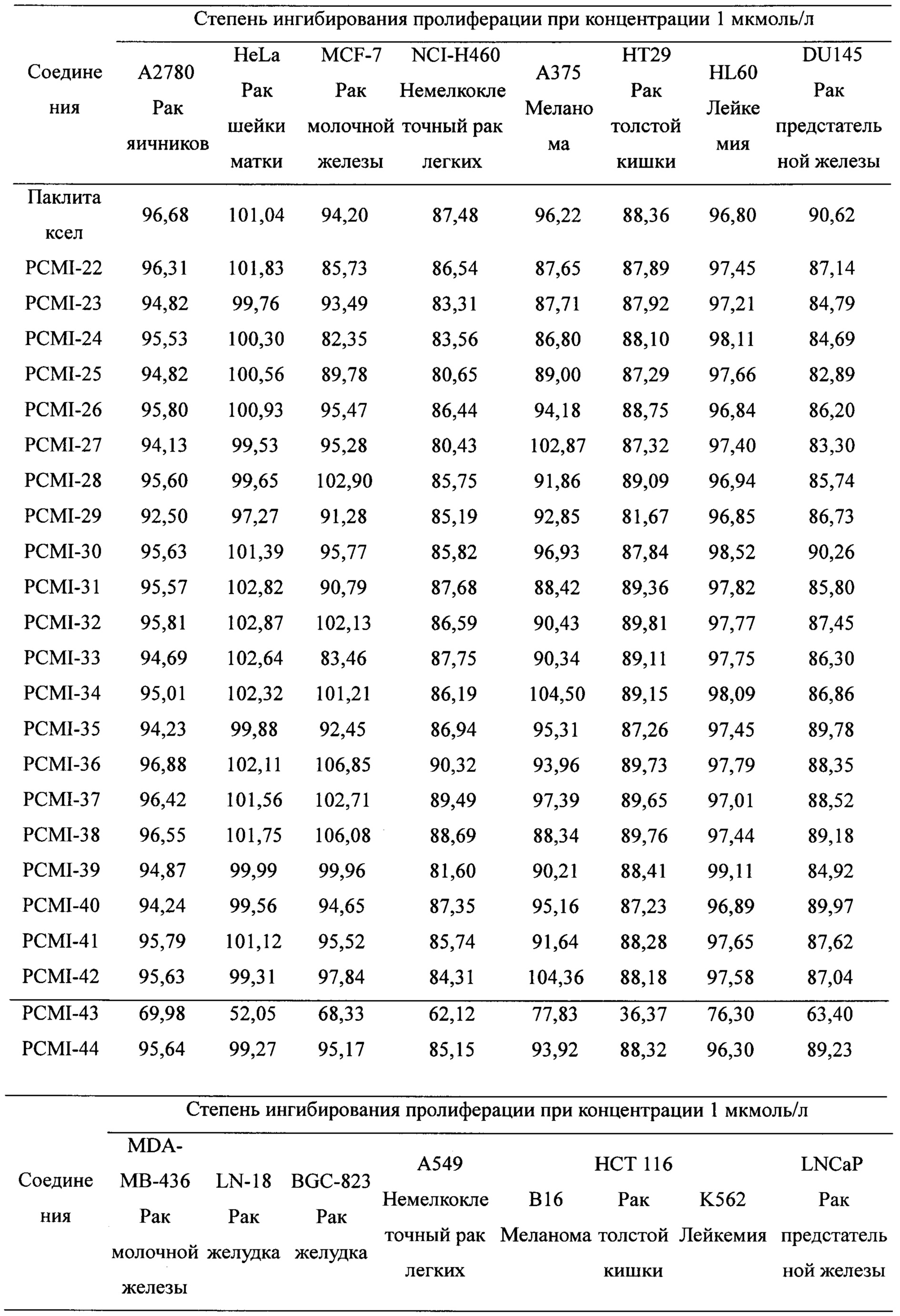
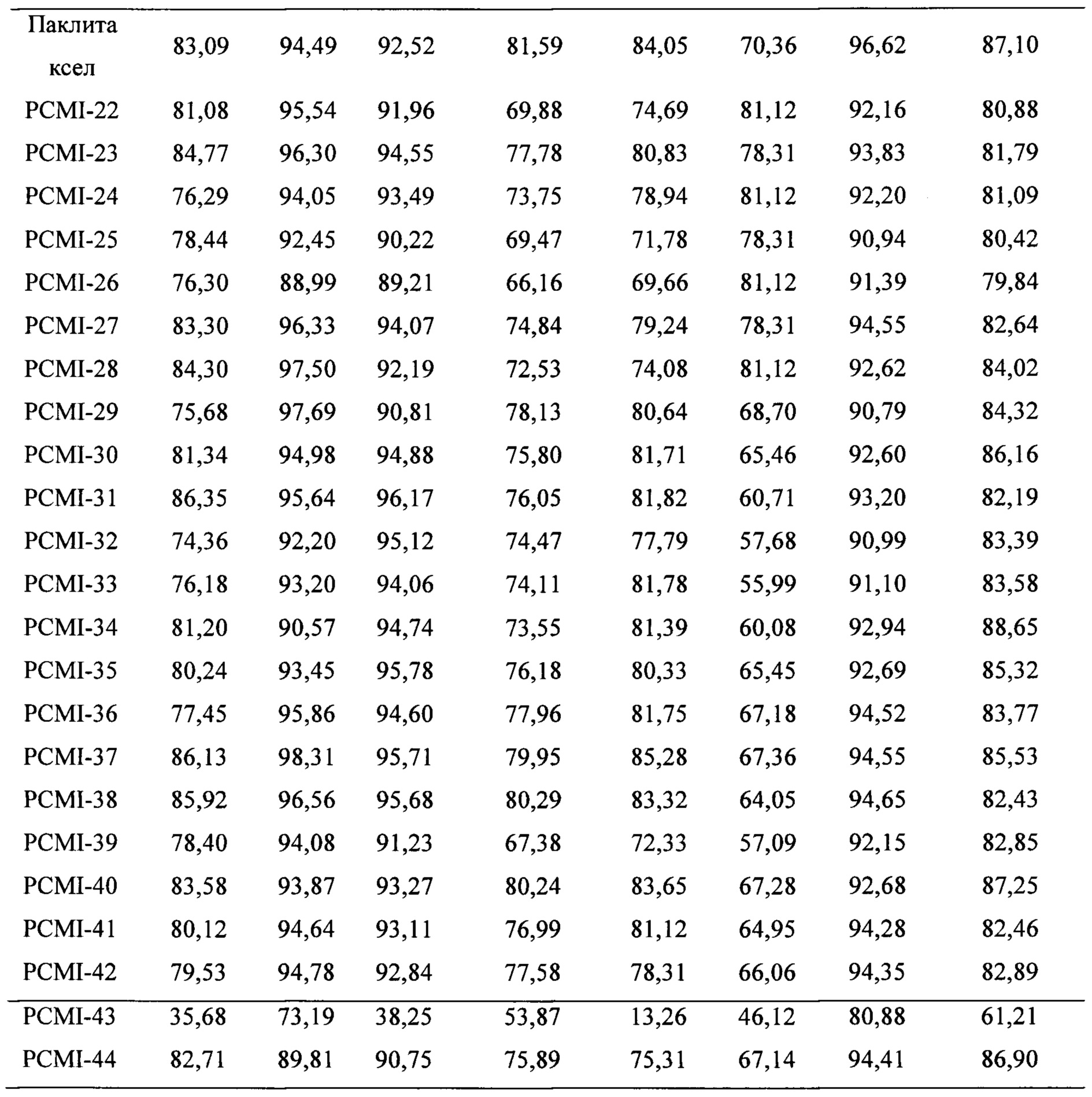
Предварительная оценка активности показывает, что подавляющее большинство таксановых производных проявляют близкую или более сильную цитотоксичность в отношении большинства линий раковых клеток по сравнению с лекарственным средством положительного контрольного. В линиях раковых клеток как А549, так и В16 цитотоксичность таксановых производных немного ниже по сравнению с лекарственным средством положительного контрольного. Цитотоксичность PCMI-43 ослаблена, при этом PCMI-44 сохраняет хорошую цитотоксичность, указывая на то, что с раскрытием D-кольца части исходного ядра паклитаксела его цитотоксичность не обязательно исчезает, и цитотоксичность также связана с функциональными группами на боковых цепях. Экспериментальные результаты показывают, что таксановые производные в соответствии с настоящим изобретением обладают исключительной активностью ингибирования опухолей.
Из данных оценки активности вышеуказанного предварительного скрининга можно видеть, что ряд таксановых производных, синтезированных в настоящем изобретении, обладают активностью (хотя активность PCMI-43 ослаблена). После этого большую часть соединений выбирали из таких производных для исследования их значений IC50 в отношении линии клеток рака молочной железы MCF-7. В качестве лекарственного средства положительного контроля применяли паклитаксел. Эксперименты для каждого соединения независимо повторяли три раза и в каждом эксперименте использовали несколько ячеек. Время воздействия лекарственных средств составляло 72 часа. Доза половинной смертности (IC50) выражена как среднее ±SD, и экспериментальные данные приведены в таблице 2.
Таблица 2. Значения IC50 таксановых соединений в соответствии с настоящим изобретением для линий клеток рака молочной железы MCF-7
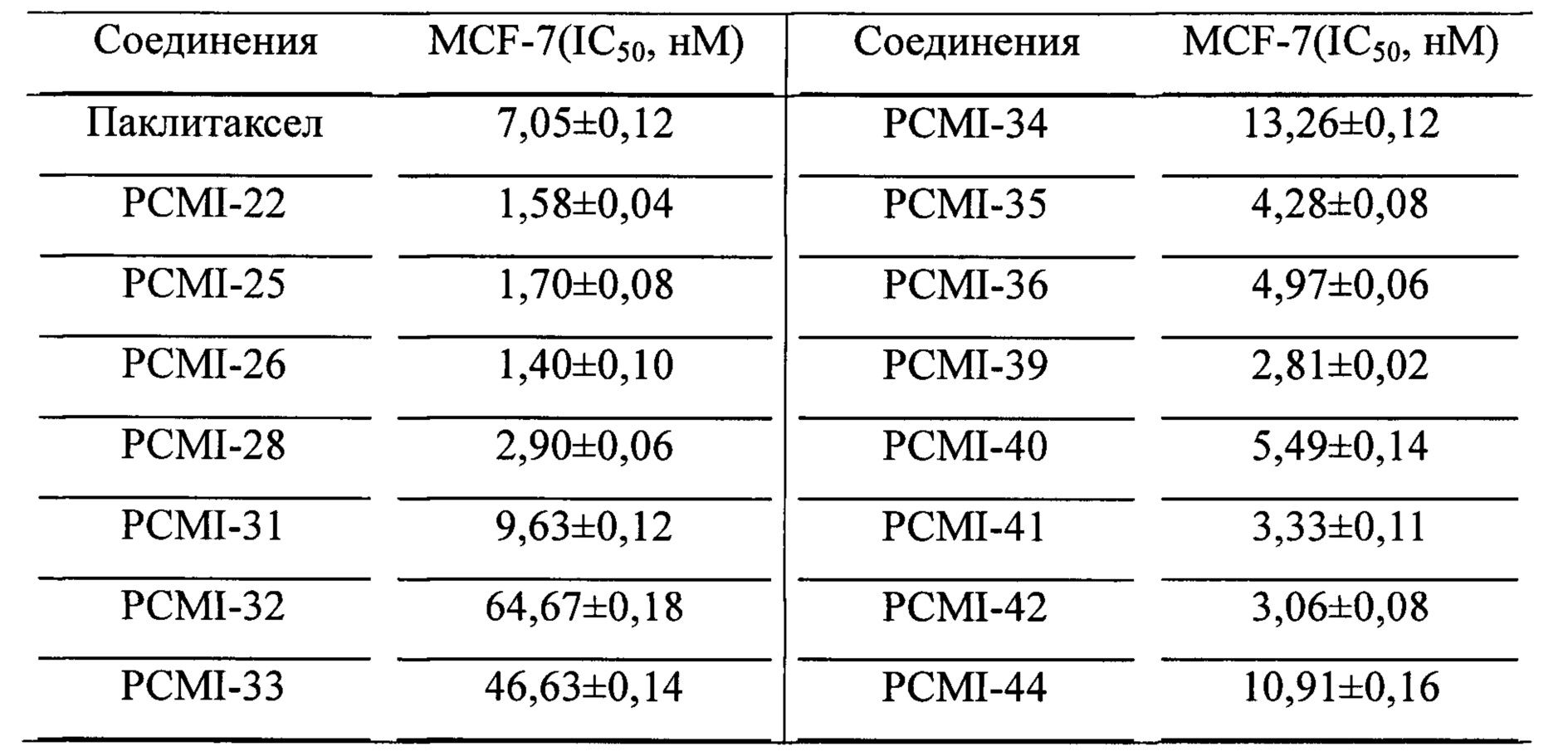
Как показано из данных таблицы 2, значение IC50 лекарственного средства положительного контроля паклитаксела составляет приблизительно 7,05 нМ. Если структуры боковых цепей ряда таксановых производных в соответствии с настоящим изобретением представляют собой R1, представляющий собой -COOR6 или -CONR7aR7b, R2, представляющий собой фенил или алкильную цепь, их значения IC50 почти равняются таковым для паклитаксела при сохранении на приблизительно таком же порядке величины. При этом значения IC50 для некоторых производных лучше, чем у паклитаксела. Так, можно видеть, что активность in vitro большей части производных в соответствии с настоящим изобретением остается неизменной или даже улучшается по сравнению с паклитакселом. Некоторые из соединений обладают немного более низкой активностью, чем у паклитаксела, представляющего собой лекарственное средство положительного контроля.
2. Испытания мембранного переноса в монослое клеток Сасо-2
Для изучения двунаправленного переноса целевых соединений с апикальной (АР) стороны на базолатеральную (BL) сторону и с BL стороны на АР сторону использовали модель клеточного монослоя выделенной из человека линии клеток колоректальной аденокарциномы Сасо-2. Для количественного анализа с целью расчета параметров переноса, предполагаемого коэффициента проницаемости (Рарр) и коэффициента активного выведения использовали ВЭЖХ. В качестве лекарственного средства положительного контроля использовали паклитаксел и субстраты P-gp, в качестве образца для сравнения с целью теоретической оценки биодоступности при пероральном введении in vivo таких таксановых производных и их сродства к P-gp использовали эритромицин.
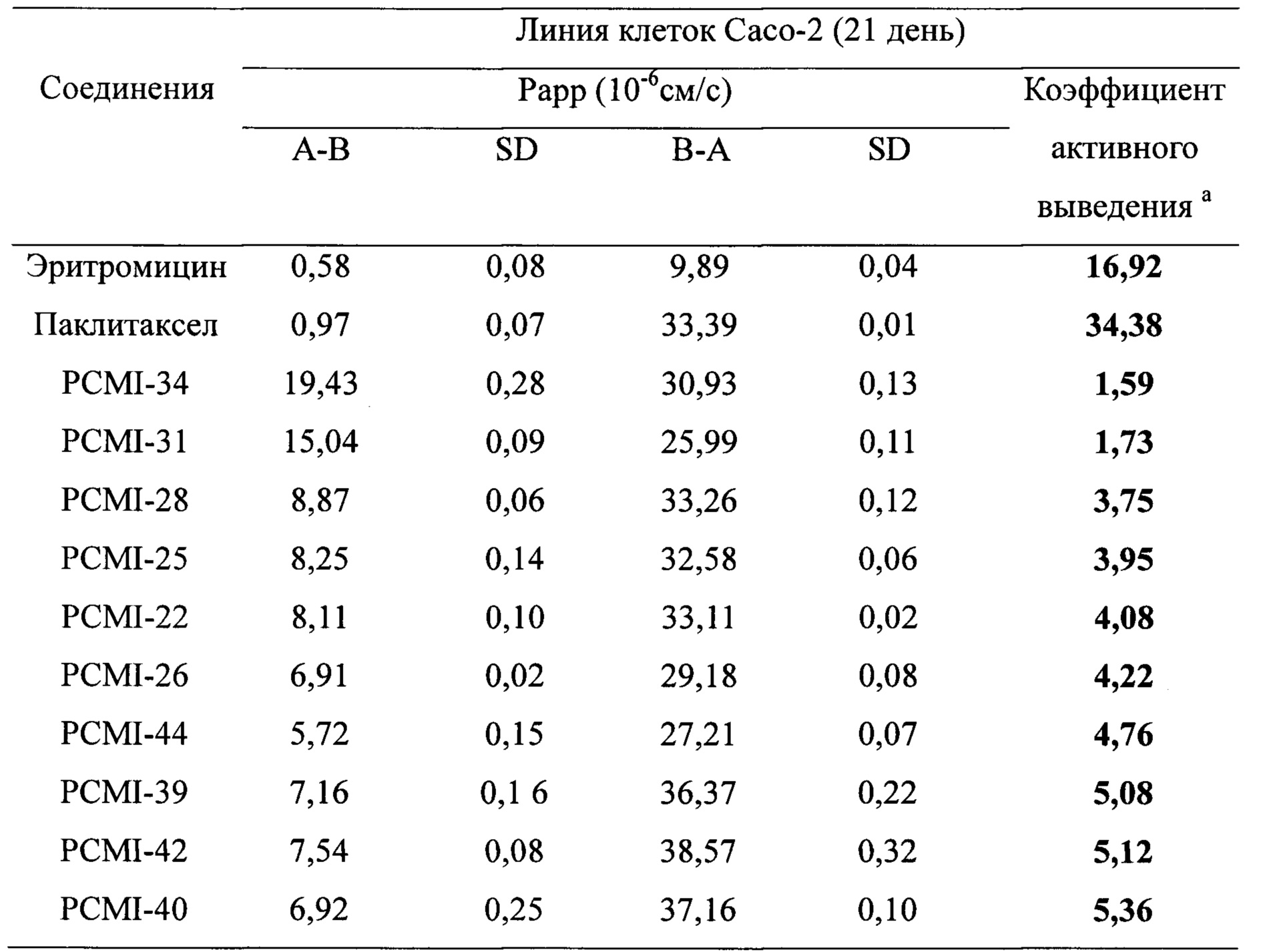
Таблица 3. Рарр А-к-В для таксановых производных в соответствии с настоящим изобретением в клеточной модели Сасо-2

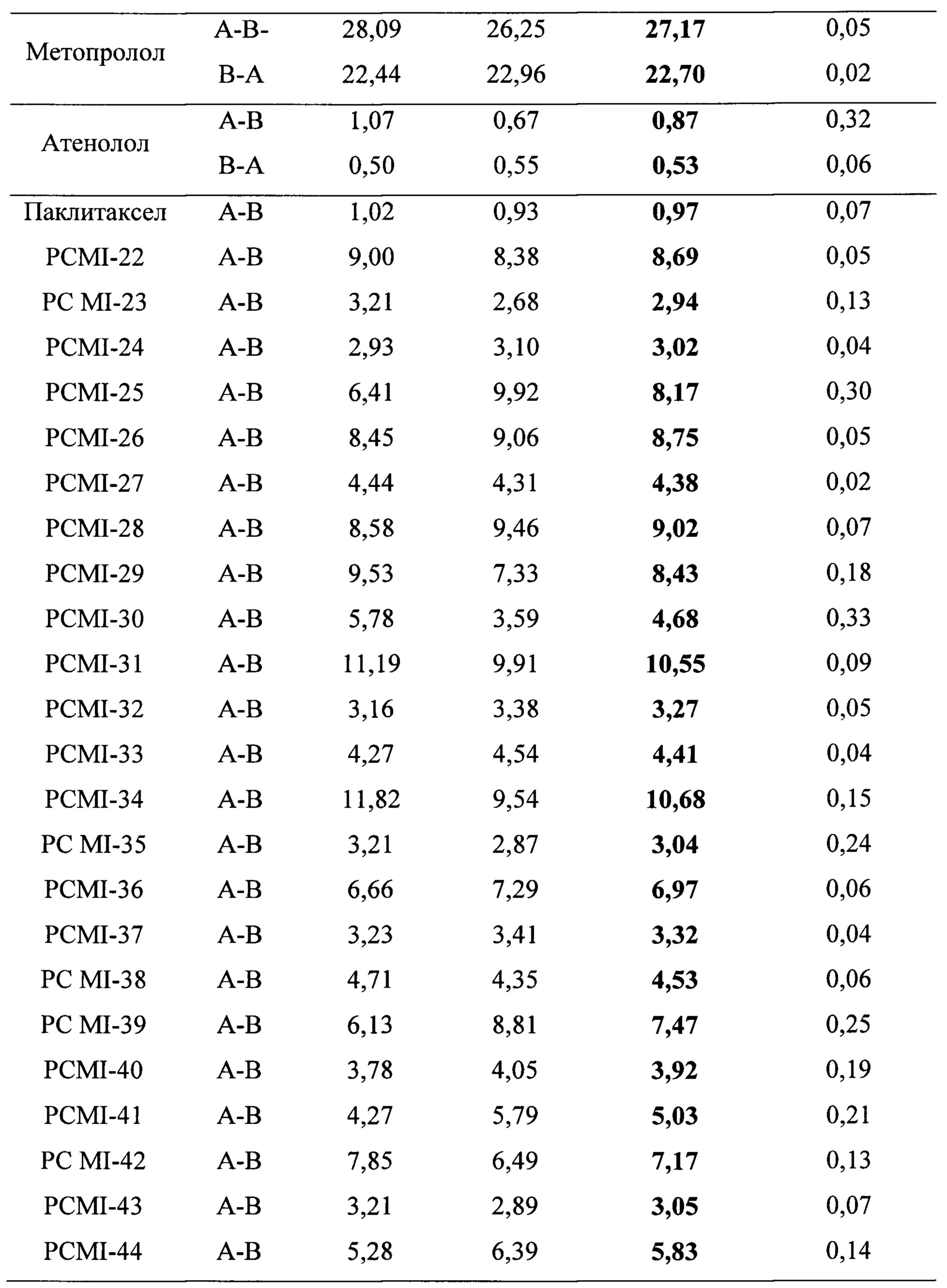
Таблица 4. Степень трансмембранного выделения в массе таксановых производных в соответствии с настоящим изобретением в клеточной модели Сасо-2
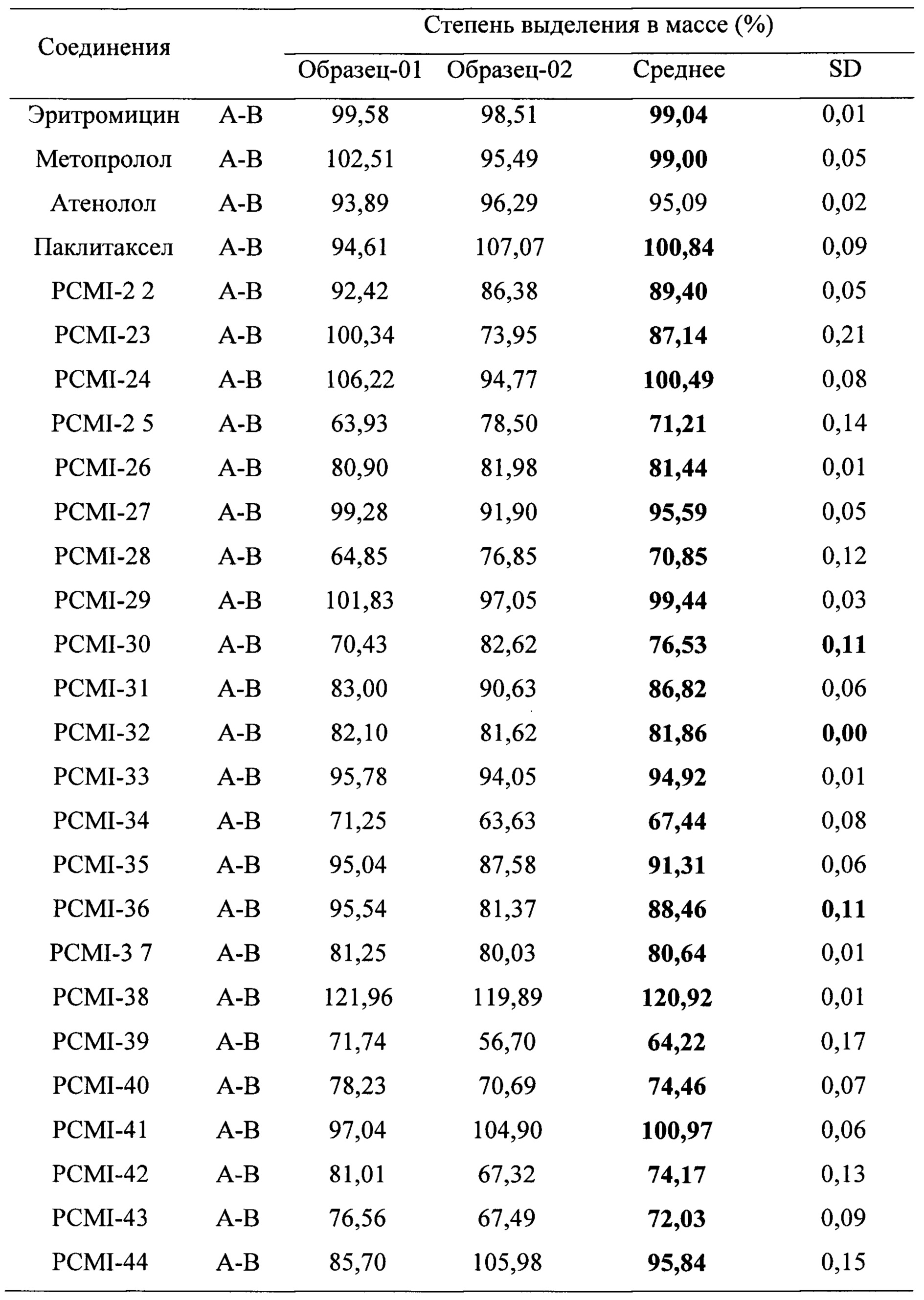
Таблица 5. Коэффициент активного выведения иллюстративных таксановых производных в соответствии с настоящим изобретением в клеточной модели Сасо-2
а. коэффициент активного выведения = Рарр В-А/Рарр А-В
Экспериментальные результаты приведены в таблице 3. Можно видеть, что значения А-к-В Рарр таких таксановых производных в соответствии с настоящим изобретением выше, чем у паклитаксела (Рарр А-к-В=0,97), в частности, для производных PCMI-34 и PCMI-31 их значения Рарр А-к-В>10×10-6 см/с, что относятся к субстратам с высокой проницаемостью. Эти данные показывают, что такие таксановые производные имеют хорошую трансмембранную способность, поэтому предполагалось, что они будут лучше поглощаться in vivo, чем паклитаксел.
Степени трансмембранного извлечения таких таксановых производных приведены в таблице 4. Оценены двунаправленные переносы 10 соединений, выбранных из 23 таксановых производных, при этом результаты приведены в таблице 5. Из коэффициентов активного выведения можно видеть, что по сравнению с паклитакселом коэффициенты активного выведения таких производных снижены в значительной степени, они значительно меньше, чем у паклитаксела (коэффициент активного выведения которого = 34,38). Соответственно, предполагается, что поглощение при пероральном введении in vivo подлежит улучшению.
3. Испытания биодоступности при пероральном введении in vivo
Материалы
Соединение PCMI-31 синтезировали и идентифицировали в соответствии со способами, предусмотренными в настоящем изобретении. Внутренний стандарт, паклитаксел, приобретали у China's National Institute for Control of Pharmaceutical and Biological Products (NICPBP). Ацетонитрил хроматографической чистоты приобретали у Sigma-Aldrich Inc. Tween 80 и этилацетат приобретали у Aladdin reagent Inc. Самцов крыс Спрег-Доули приобретали у Beijing Weitonglihua Inc. и разводили в виварии в течение двух недель.
Аппаратура
Установка ВЭЖХ серии Agilent 1100, автоматический дозатор Agilent G1313A, квадрупольный масс-спектрометр Thermo Finnigan TSQ (San Jose, Калифорния, США), программное обеспечение для анализа данных XcalIbur® (версия 1.3) (Thermo Finnigan).
Процедура эксперимента
Растворяли 200 мг PCMI-31 в 4 мл смешанного раствора Tween 80 и безводного этанола (1:1) с получением исходного раствора с концентрацией 50 мг/мл и добавляли изотонический раствор хлорида натрия для доведения до подходящей концентрации. Брали 12 самцов крыс Спрег-Доули (с массой тела приблизительно 300 г) и разделяли на две группы после голодной паузы в течение ночи. Одну группу обрабатывали внутривенной инъекцией (5 мг/кг), а другую группу обрабатывали посредством перорального введения (60 мг/кг). Отбирали пробы крови в группе внутривенного введения на 0й мин., 5й мин., 10й мин., 20й мин., 40й мин., 1й ч., 2й ч., 4й ч., 6й ч., 8й ч., 12й ч., 24й ч., при этом для пероральной группы отбирали пробы на 5й мин., 15й мин., 30й мин., 45й мин., 1й ч., 2й ч., 4й ч., 6й ч., 8й ч., 12й ч., 24й ч. После 10 мин. центрифугирования плазмы при 4500 об./мин. отбирали верхнюю сыворотку и переносили в соответствующую пробирку Eppendorf, помещали в морозильную камеру с температурой -40°С для испытаний.
Построение стандартной кривой для PCMI-31
Конфигурация устройства серии Agilent 1100: устройство автоматического дозатора ВЭЖХ Agilent G1313A, колонка для обращенно-фазовой хроматографии 150 мм × 2,1 мм С18 Thermo column (размер частиц 3 мкм), длина волны детектора 230 нм, температура колонки 30°С, подвижная фаза ацетонитрил/вода (7:3), расход 0,2 мл/мин., объем пробы 20 мкл. Комбинированную масс-спектрометрию (MS) проводили на тройном квадрупольном спектрометре Thermo Finnigan TSQ, выполненном с возможностью ионизации электрораспылением (ESI) в режиме определения положительных ионов. Параметры MS анализа были следующими: напряжение в камере распыления, 4,0 кВ; температура нагретого капилляра, 350°С; защитный газ (азот); 20 фунт/кв.дюйм; вспомогательный газ (азот): 5 фунт/кв.дюйм; газ для соударений (аргон); давление: 1,5 мм.рт.ст.; энергия соударений: СА 17 эВ; FA и IFA составляли 19 эВ; IS составлял 15 эВ.
В качестве внутреннего стандарта был выбран паклитаксел со временем удерживания 3,07 мин. Время удерживания PCMI-31 составляло 4,21 мин. Условия детектирования MS для PCMI-31 устанавливали следующими: масса/заряд 915→634; паклитаксел в качестве внутреннего стандарта. Условия детектирования: масса/заряд 876→308. Диапазон концентраций стандартной кривой PCMI-31 составлял 5-10000 нг/мл (γ2>0,99), и минимальный предел определения составлял 5 нг/мл.
Экстракция и анализ образцов плазмы
Отбирали 100-мкл образцы плазмы, в которые добавляли 100 мкл внутреннего стандарта (паклитаксел, раствор в ацетонитриле 500 нг/мл), с последующим добавлением 3 мл этилацетата после тщательной гомогенизации с помощью вихревой мешалки, через 5 мин. встряхивания центрифугировали в течение 8 мин. при скорости вращения 4500 об./мин. Надосадочную жидкость переносили в чистую пробирку Eppendorf с продувкой азотом для высушивания в условиях подогрева. После повторного разбавления 120 мкл подвижной фазы (CH3CN/H2O=7:3) раствор центрифугировали при 12000 об./мин. в течение 3 мин., отбирали 100 мкл надосадочной жидкости и переносили в ампулу автоматический дозатора. После анализа LC-MS/MS статистические данные и параметры фармакокинетики обрабатывали с помощью программного обеспечения XcalIbur® (версия 1.3) (Thermo Finnigan).
Результаты
Кривая зависимости концентрации лекарственного средства от времени для соединения PCMI-31 при пероральном или внутривенном введении показана на фиг. 1. Связанные с этим параметры фармакокинетики PCMI-31 приведены в таблице 6. Период полувыведения PCMI-31 является относительно коротким, в целом составляет приблизительно 3 ч., а абсолютная биодоступность при пероральном введении (F%) составляет 10,7%. По сравнению с приведенной абсолютной биодоступностью при пероральном введении паклитаксела, составляющей менее 5%, биодоступность при пероральном введении PCMI-31 у животных в некоторой степени улучшилась.
Таблица 6. Связанные параметры фармакокинетики PCMI-31 при внутривенном и пероральном введении
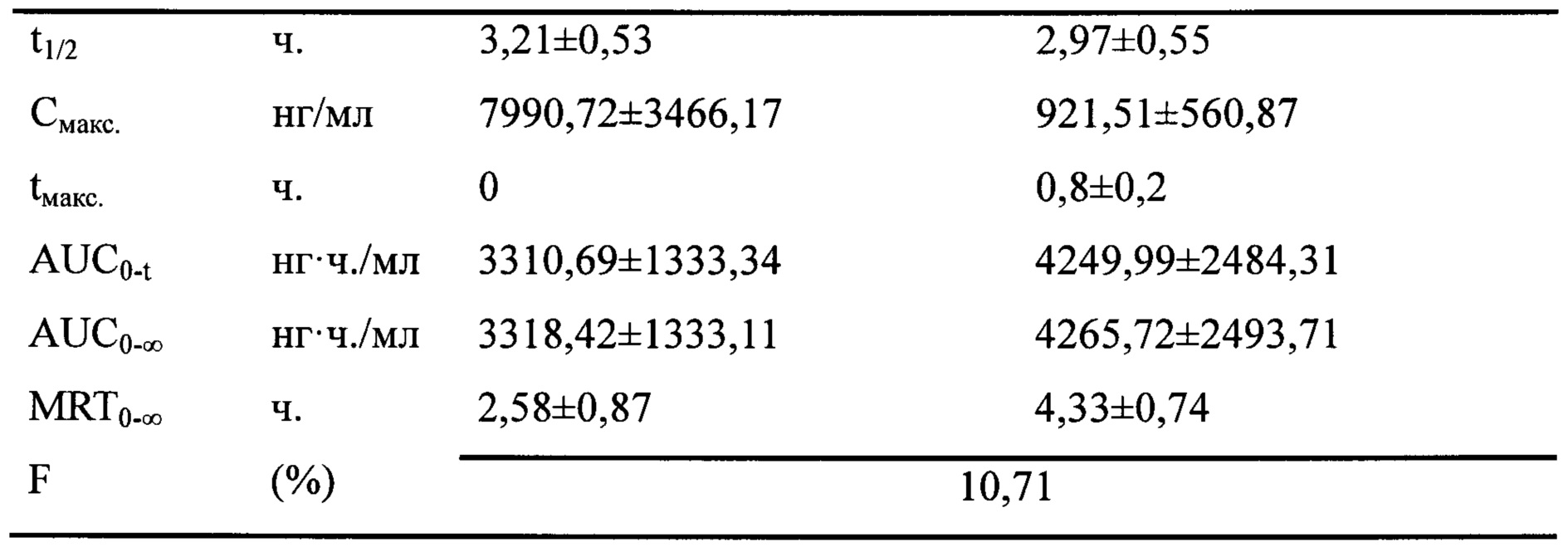
AUC0-t: площадь под кривой 0-24 часа; AUC0-∞: площадь под кривой; Смаск.: пиковая концентрация; tмакс.: время пика; MRT: среднее время удерживания; t1/2: период полувыведения; F: Абсолютная биодоступность при пероральном введении, F=(АUСп.o.×дозав.в.)/(АUСв.в.×дозап.о.)×100%
ПРИМЕРЫ
Следующие примеры предоставлены для дополнительной иллюстрации синтеза соединений в соответствии с настоящим изобретением и не предназначены для ограничения настоящего изобретения каким-либо образом.
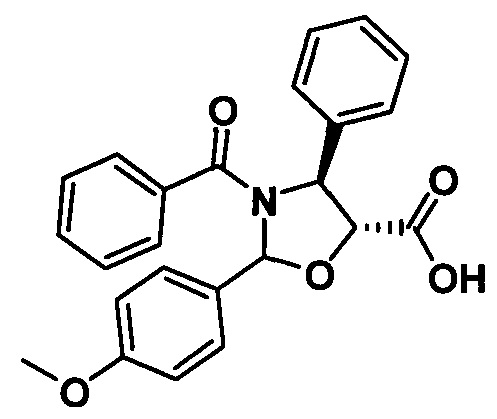
Пример 1. Получение PCMI-22
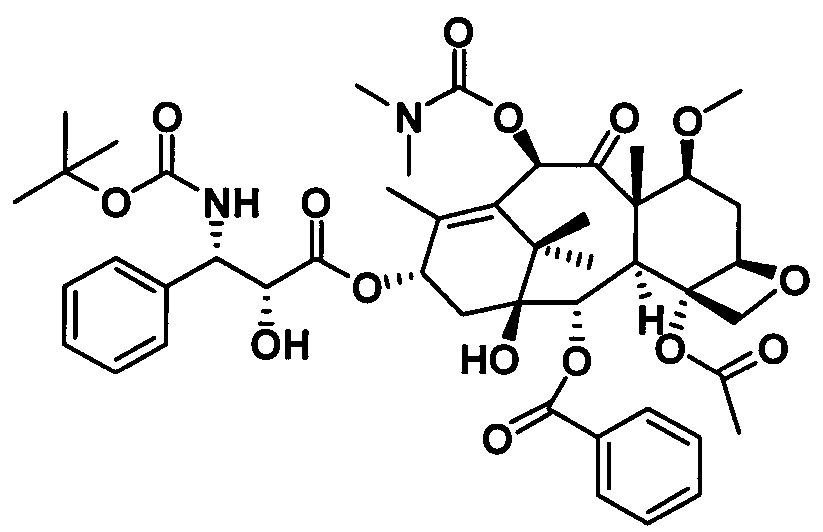
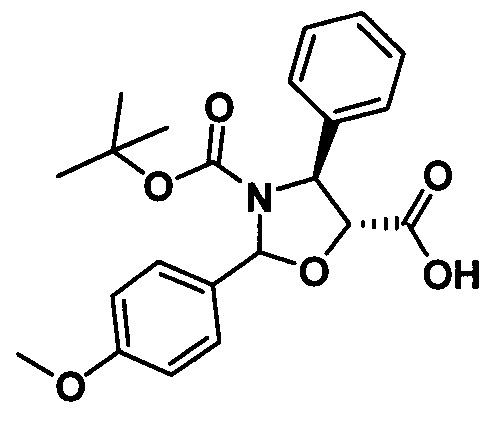
1) Получение (4S,5R)-3-трет-бутилоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
a. Получение бензилгликолята
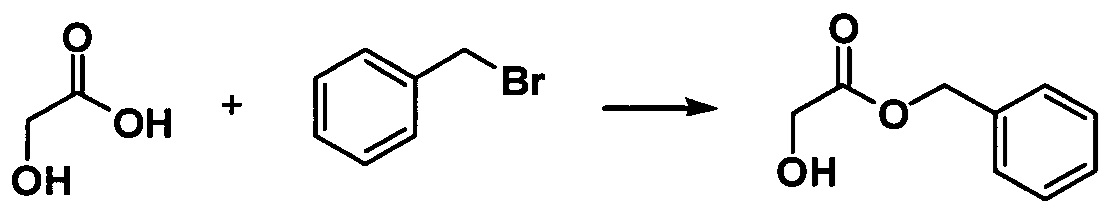
Гликолевую кислоту (7,60 г, 0,10 моль) растворяли в 10 мл ацетонитрила, в который добавляли бензилбромид (13,60 г, 0,08 моль), и равномерно перемешивали. В реакционную жидкость медленно добавляли по каплям DBU (12,16 г, 0,08 моль) при 0°С. После этого реакционную жидкость перемешивали в течение ночи при комнатной температуре. Реакционную жидкость выливали в ледяную воду, экстрагировали этилацетатом, полученную объединенную органическую фазу последовательно промывали 1 М раствором соляной кислоты и насыщенным водным раствором соли, высушивали с применением безводного сульфата натрия и концентрировали с помощью роторного испарителя с получением соединения в виде желтого масла (12,50 г, 94%).
b. Получение Вос-защищенного бензилгликолята
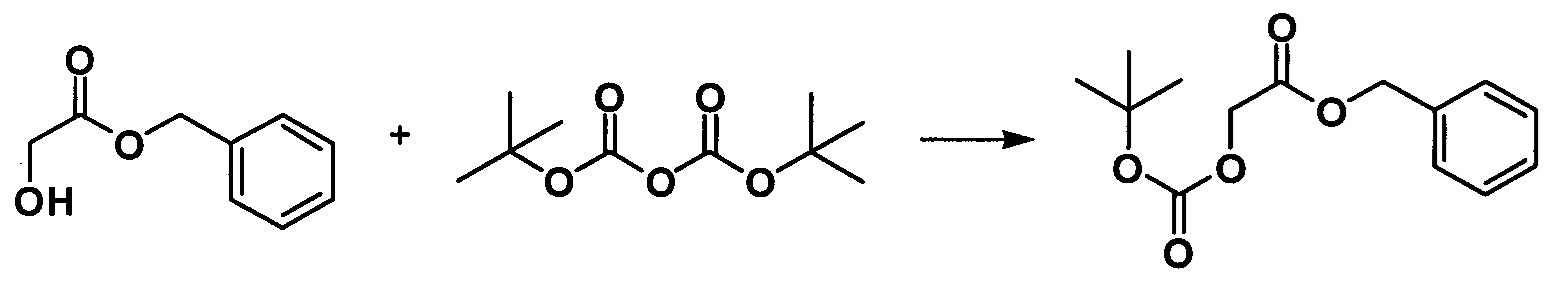
Бензилгликолят (30 г, 0,25 моль) и Вос-ангидрид (39,1 г, 0,19 моль) растворяли в 30 мл дихлорметана. По каплям добавляли 5 мл DMAP (4,62 г, 0,038 моль) в растворе дихлорметана в полученную реакционную жидкость при 80°С. После этого реакционную жидкость вводили в реакцию при 15°С в течение 0,5 ч. После завершения реакции реакционную жидкость выливали в ледяную воду, экстрагировали этилацетатом, полученную объединенную органическую фазу промывали последовательно водой и насыщенным водным раствором соли. Органическую фазу концентрировали и перекристаллизовывали с применением петролейного эфира/этилацетата при соотношении 10:1 с получением белого твердого вещества (32,5 г, 66%).
c. Получение N-трет-бутилсульфинилбензиленамина
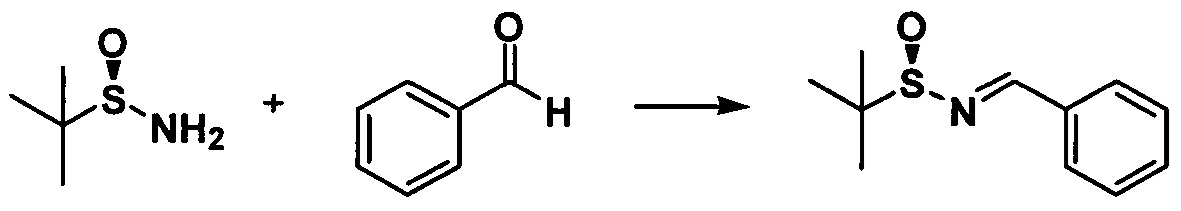
Растворяли (SR)-трет-бутилсульфинамид (5,22 г, 0,043 моль) и бензальдегид (5,51 г, 0,052 моль) в 20 мл дихлорметана и к раствору добавляли сульфат магния (25,90 г, 0,22 моль) и PPTS (0,54 г, 2,20 ммоль). Реакционную жидкость перемешивали при комнатной температуре в течение 24 ч., фильтровали и полученный осадок на фильтре промывали дихлорметаном 3 раза (20 мл × 3) с получением неочищенного продукта после концентрирования. Неочищенный продукт очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 15:1) с получением бесцветного масла (7,71 г, 85,8%).
d. Получение бензил-2R-трет-бутилоксикарбонил-3S-трет-бутилсульфинамид-фенилпропионата
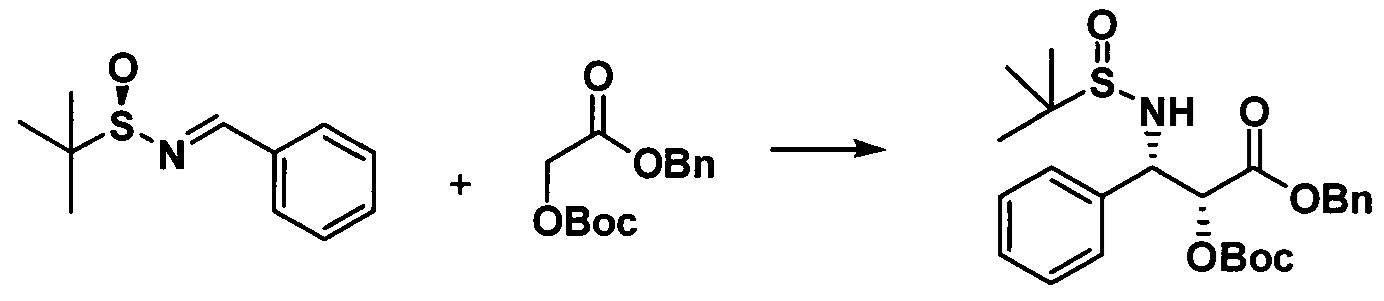
Растворяли Вос-защищенный бензилгликолят (32,5 г, 0,12 моль) в 15 мл тетрагидрофурана и в реакционную жидкость медленно по каплям добавляли LHMDS (120 мл, 0,12 моль) при -70°С. После завершения добавления реакционную жидкость перемешивали в течение 0,5 ч. с последующим медленным добавлением по каплям N-трет-бутилсульфинилбензиламина в растворе THF (5,02 г, 0,024 моль 8 мл раствора THF), и 4 через часа реакцию завершали. Реакционную жидкость выливали в 50 мл насыщенного раствора хлорида аммония и экстрагировали этилацетатом 3 раза (30 мл × 3). Объединенные органические фазы высушивали, концентрировали с помощью роторного испарителя и очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 10:1) с получением белого твердого вещества (5,25 г, 46%).
e. Получение бензил-2R-гидрокси-3S-аминофенилпропионата
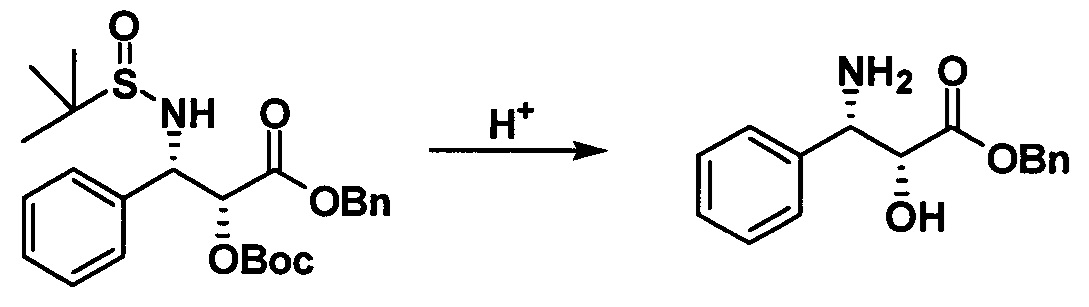
Продукт, полученный на предыдущей стадии (5,25 г, 0,011 моль), растворяли в 20 мл 2 н. раствора HCl/EtOAc и вводили в реакцию при комнатной температуре в течение 10 часов. После завершения реакции реакционную жидкость концентрировали и полученный концентрат экстрагировали смесью дихлорметан/вода (50 мл/100 мл). Водную фазу собирали, экстрагировали дихлорметаном и регулировали ее значение рН 28% водным раствором аммиака до 9-10. Наконец, водную фазу экстрагировали дихлорметаном 3 раза (20 мл × 3). Объединенную органическую фазу высушивали, фильтровали и концентрировали с получением белого твердого вещества (2,85 г, 95,7%).
f. Получение бензил (4S,5R)-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоксилата
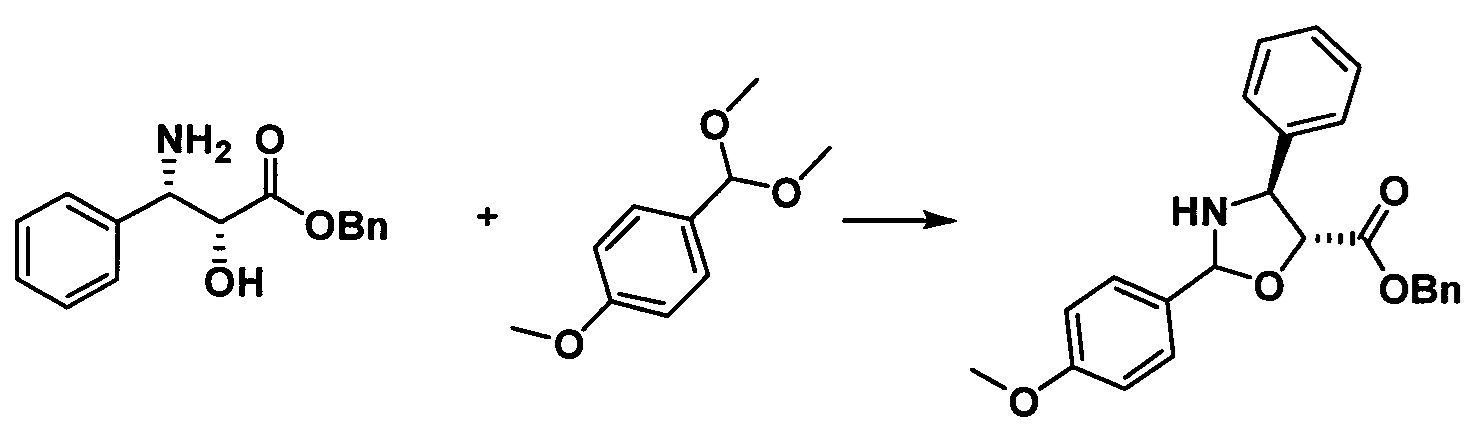
Растворяли бензил-2R-гидрокси-3S-амино-фенилпропионат (2,66 г, 9,84 ммоль) и катализатор PPTS (0,24 г, 0,93 ммоль) в 10 мл толуола и в реакционную жидкость медленно добавляли по каплям 1,1-диметоксиметил-4-метоксибензол (2,15 г, 11,79 ммоль) при 100°С. После этого реакцию поддерживали при температуре 90-100°С в течение 2 часов, что продолжили добавлением 2,4 г 1-диметоксиметил-4-метоксибензола и затем обеспечением протекания реакции в течение приблизительно 2 часов до завершения реакции. Полученную реакционную жидкость концентрировали, разделяли и очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 10:1) с получением желтого масла (3,52 г, 92%). Желтое масло содержало небольшое количество n-метоксибензальдегида.
g. Получение бензил-(4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоксилата
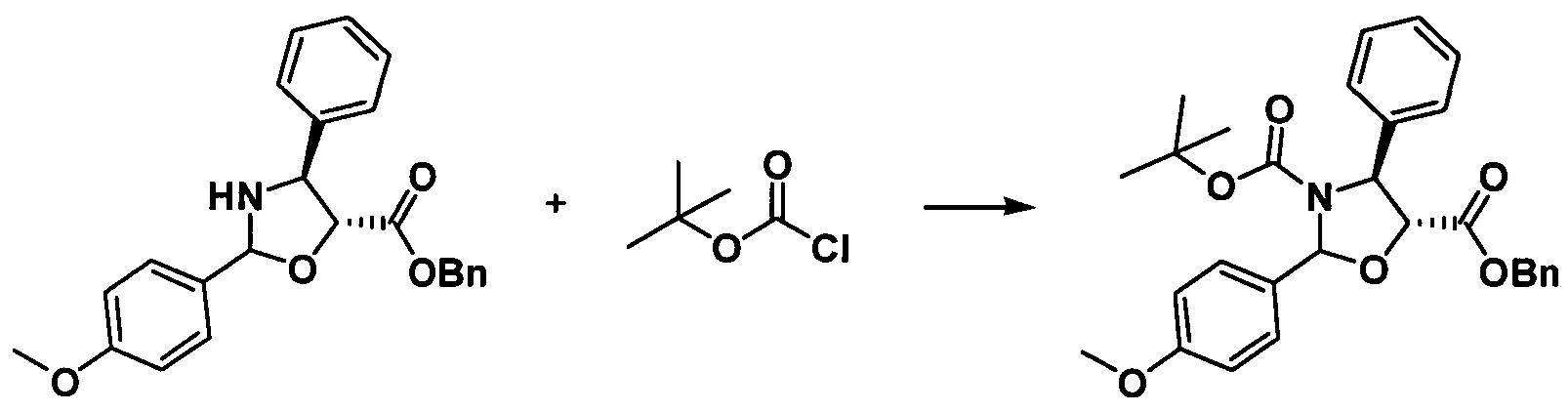
Растворяли масло, полученное на предыдущей стадии (4,07 г, 10,47 ммоль), трет-бутилоксиформилхлорид (1,56 г, 12,57 ммоль) и триэтиламин (2,64 г, 26,17 моль) в 10 мл дихлорметана и перемешивали в течение ночи при комнатной температуре. Реакционную жидкость концентрировали, разделяли и очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 10:1) с получением желтого масла (4,83 г, 94,4%).
h. Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
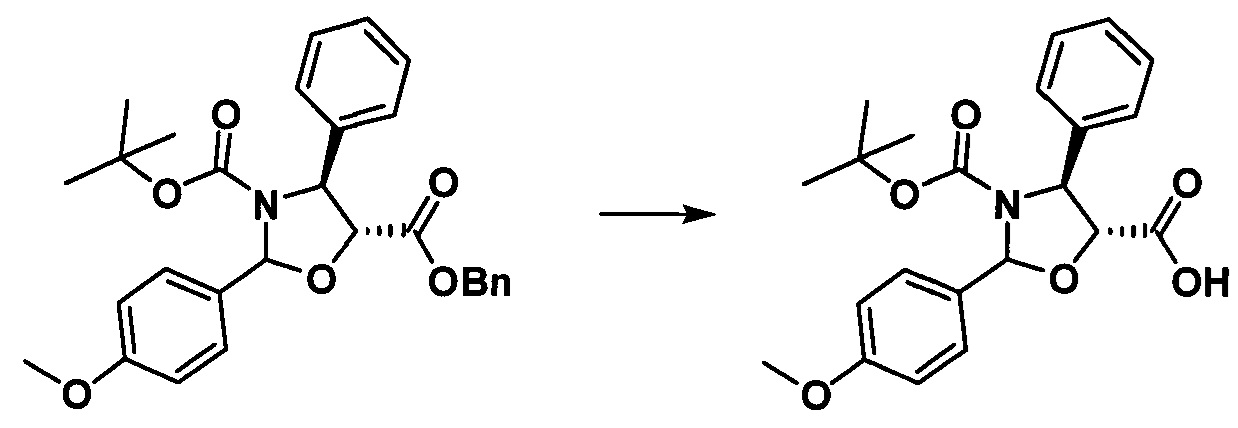
Продукт, полученный на предыдущей стадии (4,83 г, 9,88 ммоль), растворяли в 10 мл метанола, в который добавляли 1,0 г гидроксида палладия. Вводили водород (20 фунт/кв.дюйм) при комнатной температуре и проводили реакцию в течение приблизительно 1 ч., завершение реакции контролировали с помощью TLC. Реакционную жидкость фильтровали, концентрировали, разделяли и очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 5:1) с получением конечного продукта в виде белого твердого вещества (2,68 г, 67,9%).
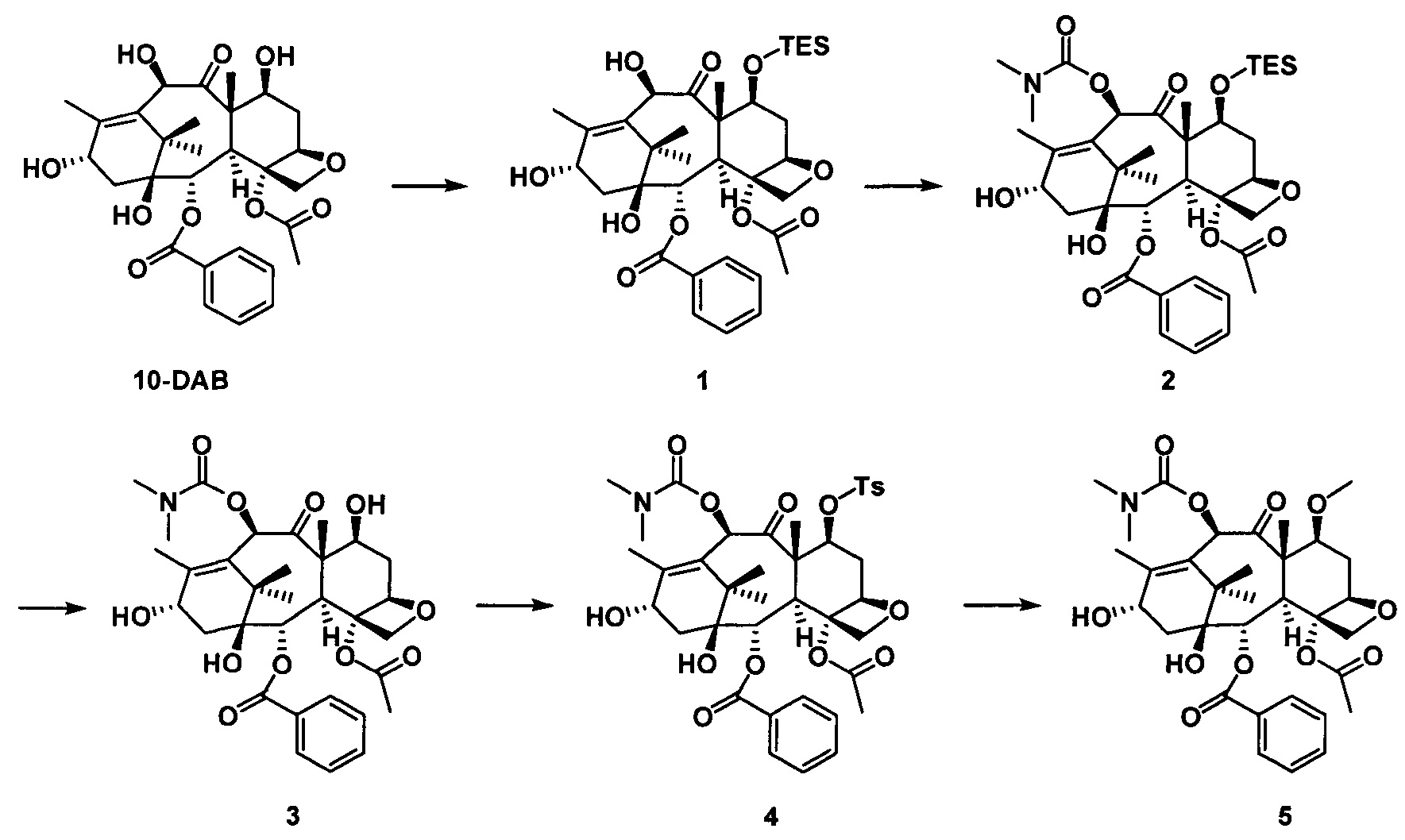
2) Получение 10-диметилкарбамоил-7-метоксибаккатина III
В качестве исходного вещества использовали 10-DAB (1 экв.), растворяли в DMF и добавляли последовательно 2,5 эквивалента имидазола и 2,5 эквивалента триэтилхлорсилана. После дополнительной обработки получали неочищенное соединение 1, приведенное на следующей схеме реакции.
Полученное соединение 1 (1 экв.) растворяли в сухом THF, который использовали в качестве растворителя, в который добавляли 1,5 эквивалента LHMDS при 0°С. Через 1 час протекания реакции к реакционной жидкости медленно добавляли по каплям 2 эквивалента диметилкарбамоилхлорида и проводили реакцию в течение 2 часов. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 2 с выходом 87%.
Полученное соединение 2 (1 экв.) растворяли в сухом THF, в который добавляли 1,5 эквивалента фторида тетрабутиламмония (в виде раствора в THF) при комнатной температуре. Спустя 1 час протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 3 с выходом 90%.
Полученное соединение 3 сначала вводили в реакцию с n-толуолсульфонилхлоридом в дихлорметане с применением пиридина в качестве щелочи с образованием соединения 4 при комнатной температуре.
В безводном тетрагидрофуране соединение 4 (1 экв.) вводили в реакцию с метилмагнийбромидом (2 экв.) при комнатной температуре в течение 3 часов в защитной атмосфере азота. После дополнительной обработки посредством очистки колоночной хроматографией получали соединение 5 с выходом 75%.
3) Получение PCMI-22
Растворяли 10-диметилкарбамоил-7-метоксибаккатин III (1 экв.) и (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновую кислоту (4 экв.) при комнатной температуре в дихлорметане, к которым последовательно добавляли 0,5 эквивалента 4-диметиламинопиридина (DMAP) и 2,0 эквивалента N,N-дициклогексилкарбодиимида (DCC) и проводили реакцию в течение ночи. Полученный продукт вводили в реакцию в 2 эквивалентах раствора ацетилхлорид/метанол с получением конечного таксанового производного PCMI-22. Общий выход двух стадий составлял 71%, а чистота продукта составляла 95% или выше.
PCMI-22: Т. пл.: 239-240°С;
MS (масса/заряд) ESI: 915,3 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,11 (d, J=7,4 Гц, 2Н), 7,62 (t, J=7,4 Гц, 1Н), 7,50 (t, J=7,7 Гц, 2Н), 7,44-7,30 (m, 5Н), 6,42 (s, 1Н), 6,21 (t, J=8,6 Гц, 1Н), 5,67 (d, J=6,9 Гц, 1Н), 5,50 (d, J=8,3 Гц, 1H), 5,28 (d, J=8,3 Гц, 1H), 4,98 (d, J=8,2 Гц, 1Н), 4,64 (s, 1Н), 4,31 (d, J=8,6 Гц, 1Н), 4,18 (d, J=8,4 Гц, 1H), 3,92-3,82 (m, 2Н), 3,55 (d, J=4,6 Гц, 1H), 3,37 (s, 3Н), 3,08 (s, 3Н), 2,98 (s, 3Н), 2,73 (m, 1H), 2,39 (s, 3Н), 2,31 (d, J=8,7 Гц, 2Н), 1,95 (s, 3Н), 1,82-1,76 (m, 1Н) 1,74 (s, 3Н), 1,37 (s, 9Н), 1,24 (s, 6Н).
Пример 2. Получение PCMI-23
1) Получение (4S,5R)-3-бензоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
(4S,5R)-3-бензоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновую кислоту получали, по сути, тем же способом, который показан в примере 1, за исключением стадии g. Другие стадии можно увидеть в реакции примера 1.
g. Получение бензил-(4S,5R)-3-бензоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоксилата
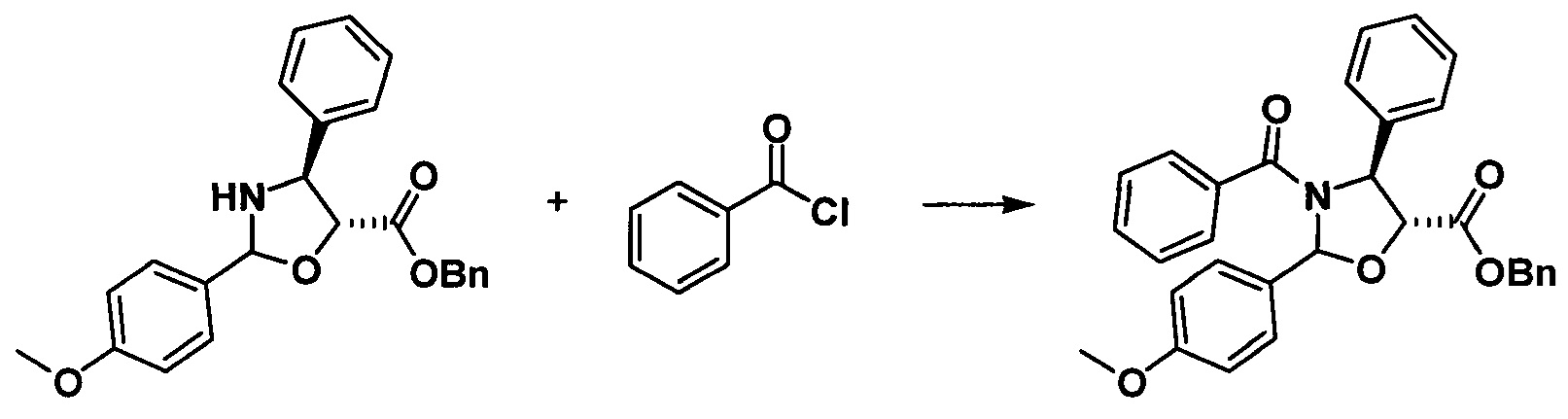
Бензил-(4S,5R)-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоксилат (1 экв.) растворяли в безводном тетрагидрофуране, в который добавляли 1,5 эквивалента LHMDS при -40°С. Через 1 час протекания реакции к реакционной жидкости добавляли по каплям 2 эквивалента бензоилхлорида, проводили реакцию в течение 3 часов перед завершением реакции. После дополнительной обработки посредством очистки колоночной хроматографией получали продукт с выходом 85%.
Получение 10-диметилкарбамоил-7-метоксибаккатина III на стадии 2) и PCMI-23 на стадии 3) проводили в соответствии с теми же процедурами, что и на стадии 2) и стадии 3) в примере 1. В частности, процедуры можно увидеть на стадии 2) и стадии 3) в примере 1, при этом чистота конечного продукта составляла 95% или выше.
PCMI-23: Т. пл.: 228-229°С;
MS (масса/заряд) ESI: 919,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,16-8,06 (m, 2Н), 7,81-7,72 (m, 2Н), 7,61 (t, J=7,4 Гц, 1H), 7,53-7,30 (m, 10Н), 7,16 (d, J=8,9 Гц, 1Н), 6,38 (s, 1Н), 6,18 (t, J=8,4 Гц, 1H), 5,80 (dd, J=8,8, 2,6 Гц, 1Н), 5,66 (d, J=6,9 Гц, 1Н), 4,97 (d, J=8,4 Гц, 1H), 4,79 (s, 1Н), 4,29 (d, J=8,1 Гц, 1Н), 4,18 (d, J=8,3 Гц, 1Н), 3,85 (dd, J=10,7, 6,8 Гц, 2Н), 3,81-3,70 (m, 1H), 3,35 (s, 3Н), 3,05 (s, 3Н), 2,96 (s, 3Н), 2,72 (ddd, J=16,0, 9,6, 6,7 Гц, 1H), 2,38 (s, 3Н), 2,31 (dd, J=8,8, 2,8 Гц, 2Н), 1,86 (s, 3Н), 1,79 (m, 1H), 1,74 (d, J=8,0 Гц, 3Н), 1,22 (s, 3Н), 1,19 (s, 3Н).
13С ЯМР (101 МГц, CDCl3) δ 203,72, 172,37, 171,22, 170,59, 166,98, 166,91, 155,15, 139,42, 138,00, 134,34, 133,72, 131,95, 130,17, 129,17, 128,95, 128,70, 128,32, 127,12, 127,07, 84,15, 81,75, 80,55, 78,57, 75,72, 74,44, 73,29, 72,24, 57,75, 57,23, 54,93, 47,30, 43,22, 36,74, 36,20, 35,53, 32,42, 29,70, 26,74, 22,69, 21,22, 14,62, 10,31.
Пример 3. Получение PCMI-24
1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-(2-пиридинил)-5-оксазолидинкарбоновой кислоты
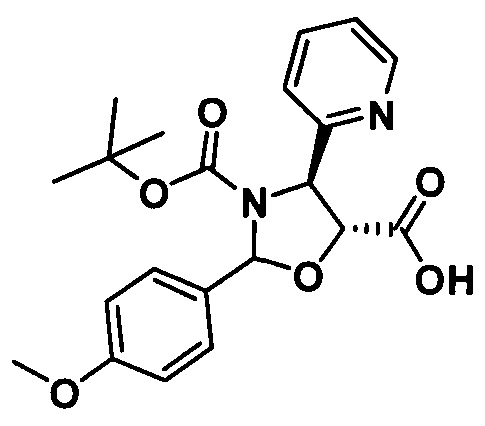
(4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-(2-пиридинил)-5-оксазолидинкарбоновую кислоту получали, по сути, тем же способом, который показан в примере 1, за исключением стадии с. Другие стадии можно увидеть в реакциях примера 1.
с. Получение N-трет-бутилсульфинил-2-пиридинилкарбоксаенамина
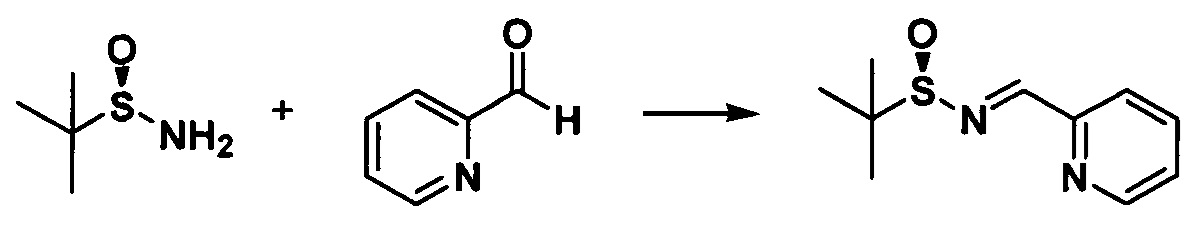
Растворяли (SR)-трет-бутилсульфинамид (5,22 г, 0,043 моль) и пиридин-2-карбальдегид (4,47 г, 0,052 моль) в 20 мл дихлорметана, в который добавляли сульфат магния (25,90 г, 0,22 моль) и PPTS (0,54 г, 2,20 ммоль). Реакционную жидкость перемешивали при комнатной температуре в течение 24 часов, фильтровали и осадок на фильтре промывали дихлорметаном 3 раза (20 мл × 3) и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 15:1) с получением бесцветного масла (7,13 г, 80,2%).
Получение 10-диметилкарбамоил-7-метоксибаккатина III на стадии 2) и PCMI-24 на стадии 3) проводили в соответствии с теми же процедурами, что и на стадии 2) и стадии 3) в примере 1. В частности, процедуры можно увидеть на стадии 2) и стадии 3) в примере 1, при этом чистота конечного продукта составляла 95% или выше.
PCMI-24: Т.пл.: 235-236°С;
MS (масса/заряд) ESI: 916,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,49 (dd, J=12,1, 4,7 Гц, 1H), 8,11 (dd, J=8,7, 7,5 Гц, 2Н), 7,77 (td, J=7,7, 1,7 Гц, 1H), 7,63 (t, J=7,4 Гц, 1H), 7,51 (t, J=7,7 Гц, 2Н), 7,44 (d, J=7,8 Гц, 1H), 7,28-7,23 (m, 1Н), 6,44 (s, 1H), 6,17 (t, J=8,7 Гц, 1Н), 5,90 (d, 1Н), 5,68 (d, J=7,1 Гц, 1Н), 5,24 (s, 1Н), 5,03 (d, J=9,5 Гц, 1Н), 4,80 (s, 1Н), 4,33 (d, J=8,5 Гц, 1Н), 4,18 (d, J=8,6 Гц, 1H), 3,99-3,94 (m, 1Н), 3,92 (d, J=7,2 Гц, 1Н), 3,87 (m, 1H), 3,39 (s, 3Н), 3,07 (s, 3Н), 2,98 (s, 3Н), 2,82-2,71 (m, 1Н), 2,49 (s, 3Н), 2,01 (s, 3Н), 1,86-1,77 (m, 4Н), 1,64 (s, 3Н), 1,28 (d, J=1,9 Гц, 12Н).
13С ЯМР (101 МГц, CDCl3) δ 203,86, 170,64, 167,06, 155,18, 148,40, 137,58, 133,69, 130,14, 129,21, 128,66, 122,96, 121,90, 84,25, 81,29, 80,54, 78,96, 75,72, 74,65, 60,41, 57,76, 57,15, 47,37, 43,23, 36,74, 36,18, 32,32, 31,94, 29,71, 28,31, 27,95, 26,50, 22,46, 21,06, 14,59, 14,17, 10,31.
Пример 4. Получение PCMI-25

1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-изобутил-5-оксазолидинкарбоновой кислоты
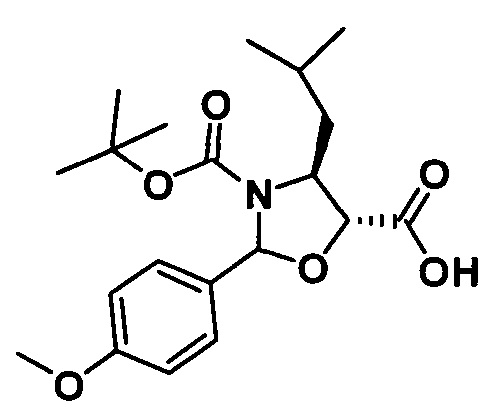
(4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-изобутил-5-оксазолидинкарбоновую кислоту получали, по сути, тем же способом, который показан в примере 1, за исключением стадии с. Другие стадии можно увидеть в реакции примера 1.
с. Получение N-трет-бутилсульфинилизобутилкарбоксаенамина
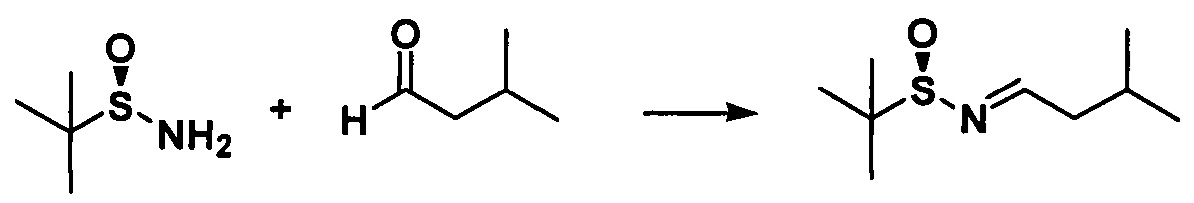
Растворяли (SR)-трет-бутилсульфинамид (5,22 г, 0,043 моль) и изовалериановый альдегид (5,51 г, 0,052 моль) в 20 мл дихлорметана, в который добавляли сульфат магния (25,90 г, 0,22 моль) и PPTS (0,54 г, 2,20 ммоль). Реакционную жидкость перемешивали в течение 24 часов при комнатной температуре, фильтровали и осадок на фильтре промывали дихлорметаном 3 раза (20 мл × 3) и концентрировали с получением неочищенного продукта. Неочищенный продукт очищали с помощью колоночной хроматографии (петролейный эфир/этилацетат = 15:1) с получением бесцветного масла (7,26 г, 89,3%).
Получение 10-диметилкарбамоил-7-метоксибаккатина III на стадии 2) и PCMI-25 на стадии 3) проводили в соответствии с теми же процедурами, что и на стадии 2) и стадии 3) в примере 1. В частности, процедуры можно увидеть на стадии 2) и стадии 3) в примере 1, при этом чистота конечного продукта составляла 95% или выше.
PCMI-25: Т. пл.: 221-222°С;
MS (масса/заряд) ESI: 873,0 (М+Н)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,11 (d, J=7,5 Гц, 2Н), 7,61 (t, J=7,4 Гц, 1Н), 7,48 (t, J=7,7 Гц, 2Н), 6,43 (s, 1Н), 6,16 (t, J=8,7 Гц, 1H), 5,67 (d, J=7,0 Гц, 1Н), 4,99 (d, J=8,2 Гц, 1Н), 4,63 (d, J=9,1 Гц, 1Н), 4,31 (d, J=8,4 Гц, 1H), 4,19 (d, J=8,0 Гц, 2Н), 3,93-3,85 (m, 2Н), 3,36 (s, 3Н), 3,32 (d, J=6,0 Гц, 1Н), 3,06 (s, 3Н), 2,97 (s, 3Н), 2,73 (ddd, J=15,9, 9,7, 6,6 Гц, 1H), 2,39 (s, 5Н), 2,02 (s, 3Н), 1,84-1,75 (m, 1Н), 1,73 (s, 3Н), 1,70-1,65 (m, 1Н), 1,33 (s, 10Н), 1,23 (s, 3Н), 1,22 (s, 3Н), 0,98 (dd, J=6,3, 2,8 Гц, 6Н).
13С ЯМР (101 МГц, CDCl3) δ 203,78, 173,78, 170,09, 166,91, 155,56, 155,16, 140,08, 133,99, 133,62, 130,19, 129,27, 128,63, 84,17, 81,60, 80,52, 79,7, 78,74, 76,42, 75,75, 74,66, 73,05, 72,62, 57,77, 57,14, 51,34, 47,29, 43,28, 41,11, 36,73, 36,18, 35,39, 32,34, 29,70, 28,20, 26,45, 24,72, 23,31, 22,60, 21,91, 21,46, 21,06, 14,65, 14,20, 10,32.
Пример 5. Получение PCMI-26
1) Получение (4S,5R)-3-трет-бутилоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен в примере 1.
2) Получение 10-метоксилкарбамоил-7-метоксибаккатина III
В качестве исходного вещества использовали 10-DAB (1 экв.), растворяли в DMF и добавляли последовательно 2,5 эквивалента имидазола и 2,5 эквивалента триэтилхлорсилана. После дополнительной обработки получали неочищенное соединение 1, приведенное на следующей схеме реакции.
Соединение 1 (1 экв.) растворяли в сухом THF, который использовали в качестве растворителя, в который добавляли 1,5 эквивалента LHMDS при 0°С. Через 1 час протекания реакции к реакционной жидкости медленно добавляли по каплям 2 эквивалента метоксилформилхлорида и проводили реакцию в течение 2 часов. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 6 с выходом 62%.
Соединение 6 (1 экв.) растворяли в сухом THF, в который добавляли 1,5 эквивалента фторида тетрабутиламмония (в виде раствора в THF) при комнатной температуре. Спустя 1 час протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 7 с выходом 90%.
Соединение 7 сначала вводили в реакцию с n-толуолсульфонилхлоридом в дихлорметане с применением пиридина в качестве щелочи с образованием соединения 8 при комнатной температуре.
В безводном тетрагидрофуране соединение 8 (1 экв.) вводили в реакцию с метилмагнийбромидом (2 экв.) в течение 3 часов при комнатной температуре в защитной атмосфере азота. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 9 с выходом 71%.
3) Получение PCMI-26
Конкретный способ был представлен на стадии 3) в примере 1, при этом чистота конечного продукта составляла 95% или выше.
PCMI-26: Т. пл.: 227-228°С;
MS (масса/заряд) ESI: 902,4 (M+Na)+;
1Н ЯМР (400 МГц, DMSO) δ 7,99 (d, 7,4 Гц, 2Н), 7,72 (t, J=7,4 Гц, 1Н), 7,63 (t, J=7,5 Гц, 2Н), 7,38 (m, 5Н), 7,22 (t, J=7,2 Гц, 1Н), 6,08 (s, 1Н), 5,95 (d, J=8,8 Гц, 1Н), 5,92-5,87 (m, 1Н), 5,43 (d, J=7,1 Гц, 1Н), 4,98 (d, J=9,9 Гц, 1Н), 4,92 (dd, J=8,8, 6,8 Гц, 1Н), 4,78 (s, 1H), 4,37 (t, J=7,1 Гц, 1Н), 4,04 (s, 2Н), 3,77 (s, 4Н), 3,62 (d, J=6,9 Гц, 1Н), 3,22 (s, 3Н), 2,73-2,68 (m, 1Н), 2,26 (s, 3Н), 1,85 (s, 3Н), 1,77-1,68 (m, 1Н), 1,56 (s, 3Н), 1,51 (d, J=9,8 Гц, 2Н), 1,36 (s, 9Н), 1,04 (s, 4Н), 1,00 (s, 3Н).
13С ЯМР (101 МГц, DMSO) δ 202,12, 173,31, 170,54, 165,64, 155,59, 154,53, 141,23, 139,98, 133,95, 133,04, 130,30, 130,05, 129,18, 128,68, 127,69, 83,55, 80,56, 78,63, 78,06, 77,14, 74,45, 70,21, 57,43, 56,84, 55,51, 47,97, 46,83, 43,35, 35,02, 33,81, 32,33, 28,61, 26,81, 25,79, 24,92, 22,88, 21,60, 14,60, 10,59.
Пример 6. Получение PCMI-27
1) Получение (4S,5R)-3-бензоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 2.
2) Получение 10-метоксилкарбамоил-7-метоксибаккатина III
Конкретный способ был представлен на стадии 2) в примере 5.
3) Получение PCMI-27
Конкретный способ был представлен на стадии 3) в примере 1, при этом чистота конечного продукта составляла 95% или выше.
PCMI-27: Т. пл.: 223-224°С;
MS (масса/заряд) ESI: 906,3 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,14-8,08 (m, 2Н), 7,81-7,74 (m, 2Н), 7,62 (t, J=7,4 Гц, 1Н), 7,50 (dd, J=12,7, 4,6 Гц, 5Н), 7,40 (ddd, J=17,6, 10,5, 5,1 Гц, 5Н), 7,23 (d, J=8,9 Гц, 1Н), 6,22-6,13 (m, 2Н, Н-13), 5,79 (dd, J=8,9, 2,7 Гц, 1Н), 5,69-5,62 (m, 1Н), 4,97 (d, J=8,4 Гц, 1Н), 4,80 (dd, J=4,7, 2,9 Гц, 1Н), 4,30 (d, J=8,4 Гц, 1Н), 4,18 (d, J=8,4 Гц, 1Н), 3,91-3,82 (m, 5Н), 3,80 (d, J=6,9 Гц, 1Н), 3,36 (s, 3Н), 2,73 (m, 1H), 2,39 (s, 3Н), 2,35-2,28 (m, 2H), 1,83 (s, 3Н), 1,78 (m, 1H), 1,76 (d, J=6,1 Гц, 3Н), 1,20 (s, 6H).
13C ЯМР (101 МГц, CDCl3) δ 202,07, 172,42, 171,28, 170,64, 167,08, 166,87, 154,77, 140,51, 137,94, 133,69, 133,39, 131,97, 130,16, 129,04, 128,69, 128,33, 127,11, 84,06, 81,61, 80,47, 78,56, 77,98, 76,47, 74,33, 73,35, 72,12, 57,60, 57,19, 55,17, 47,22, 43,14, 35,40, 32,30, 26,63, 22,66, 20,98, 14,70, 10,33.
Пример 7. Получение PCMI-28
1) Получение (4S,5R)-3-трет-бутилоксикарбонил-2-(4-метоксифенил)-4-изобутил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 4.
2) Получение 10-метоксилкарбамоил-7-метоксибаккатина III Конкретный способ был представлен на стадии 2) в примере 5.
3) Получение PCMI-28
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-28: Т. пл.: 235-236°С;
MS (масса/заряд) ESI: 882,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,12 (d, J=7,5 Гц, 2Н), 7,62 (t, J=7,4 Гц, 1Н), 7,49 (t, J=7,7 Гц, 2H), 6,23 (s, 1H), 6,17 (t, J=8,8 Гц, 1H), 5,67 (d, J=7,0 Гц, 1H), 5,01 (d, J=8,3 Гц, 1H), 4,65 (d, J=9,7 Гц, 1H), 4,33 (d, J=8,4 Гц, 1H), 4,20 (d, J=7,8 Гц, 2H), 3,95-3,81 (m, 5H), 3,42-3,32 (m, 4H), 2,76 (ddd, J=15.9, 9,6, 6,5 Гц, 1Н), 2,49-2,31 (m, 5H), 2,01 (s, 3Н), 1,88-1,77 (m, 1H), 1,78-1,64 (m, 5H), 1,35 (s, 10H), 1,23 (d, J=7,0 Гц, 6H), 1,00 (dd, J=6,2, 3,2 Гц, 6H).
13C ЯМР (101 МГц, CDCl3) δ 202,10, 173,81, 170,17, 166,88, 155,57, 154,80, 141,15, 133,64, 133,12, 130,19, 129,23, 128,64, 84,08, 81,54, 80,43, 79,77, 78,70, 78,04, 76,41, 74,54, 73,05, 72,57, 57,60, 57,13, 55,17, 51,36, 47,22, 43,21, 41,10, 35,24, 32,25, 29,70, 28,19, 26,36, 24,71, 23,29, 22,58, 21,91, 14,79, 10,34.
Пример 8. Получение PCMI-29
1) Получение (4S,5R)-3-диметилкарбамоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
(4S,5R)-3-диметилкарбамоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновую кислоту получали, по сути, тем же способом, который показан на стадии 1) примера 1, за исключением стадии g. Другие стадии можно увидеть в реакциях на стадии 1) примера 1.
g. Получение бензил (4S,5R)-3-диметилкарбамоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоксилата
Бензил-(4S,5R)-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоксилат (1 экв.) растворяли в сухом тетрагидрофуране, в который добавляли 1,5 эквивалента LHMDS при -40°С. Через 1 час протекания реакции к реакционной жидкости добавляли по каплям 2 эквивалента бензоилхлорида, проводили реакцию в течение 3 часов перед завершением реакции. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали продукт с выходом 80%.
2) Получение 10-метоксилформил-7-метоксибаккатина III
Конкретный способ был представлен на стадии 2) в примере 5.
3) Получение PCMI-29
Конкретный способ был представлен на стадии 3) в примере 1, при этом чистота конечного продукта составляла 95% или выше.
PCMI-29: Т. пл.: 227-228°С;
MS (масса/заряд) ESI: 873,0 (M+Na)+;
1Н ЯМР (400 МГц, DMSO) δ 7,99 (d, J=7,4 Гц, 2Н), 7,72 (t, J=7,4 Гц, 1Н), 7,63 (t, J=7,5 Гц, 2Н), 7,38 (m, 5Н), 7,22 (t, J=7,2 Гц, 1Н), 6,08 (s, 1Н), 5,95 (d, J=8,8 Гц, 1Н), 5,92-5,87 (m, 1Н), 5,43 (d, J=7,1 Гц, 1Н), 4,98 (d, J=9,9 Гц, 1Н), 4,92 (dd, J=8,8, 6,8 Гц, 1Н), 4,78 (s, 1Н), 4,37 (t, J=7,1 Гц, 1Н), 4,04 (s, 2Н), 3,77 (s, 4Н), 3,62 (d, J=6,9 Гц, 1Н), 3,22 (s, 3Н), 2,73-2,68 (m, 1Н), 2,26 (s, 3Н), 1,85 (s, 3Н), 1,77-1,68 (m, 1H), 1,56 (s, 3Н), 1,51 (d, J=9,8 Гц, 2Н), 1,36 (s, 9Н), 1,04 (s, 4Н), 1,00 (s, 3Н).
13С ЯМР (101 МГц, DMSO) δ 202,12, 173,31, 170,54, 165,64, 155,59, 154,53, 141,23, 139,98, 133,95, 133,04, 130,30, 130,05, 129,18, 128,68, 127,69, 83,55, 80,56, 78,63, 78,06, 77,14, 74,45, 70,21, 57,43, 56,84, 55,51, 47,97, 46,83, 43,35, 35,02, 33,81, 32,33, 28,61, 26,81, 25,79, 24,92, 22,88, 21,60, 14,60, 10,59.
Пример 9. Получение PCMI-30
1) Получение (4S,5R)-3-трет-бутилоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен в примере 1.
2) Получение 10-этилтиоформил-7-метоксибаккатина III
В качестве исходного вещества использовали 10-DAB (1 экв.), растворяли в DMF и добавляли последовательно 2,5 эквивалента имидазола и 2,5 эквивалента триэтилхлорсилана. После дополнительной обработки получали неочищенное соединение 1, приведенное на следующей схеме реакции.
Соединение 1 (1 экв.) растворяли в сухом THF, который использовали в качестве растворителя, и сначала вводили в реакцию с 2 эквивалентами N,N-карбонилдиимидазола при комнатной температуре в течение 2 часов. К реакционной жидкости затем добавляли 2 эквивалента этантиола и проводили реакцию в течение 4 часов. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 10 с выходом 72%.
Соединение 10 (1 экв.) растворяли в сухом THF, в который добавляли 1,5 эквивалента фторида тетрабутиламмония (в виде раствора в THF) при комнатной температуре. Спустя 1 час протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 11 с выходом 90%.
Соединение 11 сначала вводили в реакцию с n-толуолсульфонилхлоридом с образованием соединения 12.
В безводном тетрагидрофуране соединение 12 (1 экв.) вводили в реакцию с метилмагнийбромидом (2 экв.) в течение 3 часов при комнатной температуре в защитной атмосфере азота. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 13 с выходом 69%.
3) Получение PCMI-30
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-30: Т. пл.: 235-236°С;
MS (масса/заряд) ESI: 910,6 (М+Н)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,13-8,03 (m, 2Н), 7,62 (t, J=7,4 Гц, 1Н), 7,53-7,40 (m, 4Н), 6,96 (d, J=8,7 Гц, 2Н), 6,39 (s, 1Н), 6,32 (t, J=8,8 Гц, 1H), 5,72 (d, J=7,0 Гц, 1Н), 5,45 (dd, J=10,5, 7,3 Гц, 1Н), 5,21 (s, 1Н), 4,98 (d, J=9,1 Гц, 1H), 4,59 (s, 1Н), 4,54-4,44 (m, 1H), 4,33 (d, J=8,4 Гц, 1H), 4,21 (d, J=8,4 Гц, 1Н), 4,01 (d, J=6,9 Гц, 1Н), 3,86 (s, 3Н), 3,77 (s, 3Н), 3,41 (s, 3Н), 2,70-2,59 (m, 1Н), 2,39-2,21 (m, 5Н), 2,11 (s, 3Н), 2,08-1,99 (m, 1H), 1,83 (s, 3Н), 1,38 (s, 9Н), 1,26 (s, 3Н), 1,20 (s, 3Н), 1,05 (dd, J=13,9, 5,9 Гц, 6Н).
13С ЯМР (101 МГц, CDCl3) δ 202,12, 171,91, 171,24, 170,81, 164,68, 155,71, 154,81, 151,92, 138,16, 137,39, 134,64, 134,15, 129,93, 128,95, 128,12, 128,01, 126,63, 88,01, 84,02, 81,04, 80,60, 80,16, 79,68, 75,91, 75,00, 74,74, 74,34, 69,12, 57,81, 57,36, 46,68, 41,83, 36,81, 36,21, 32,12, 28,26, 25,87, 22,75, 22,62, 14,55, 10,39.
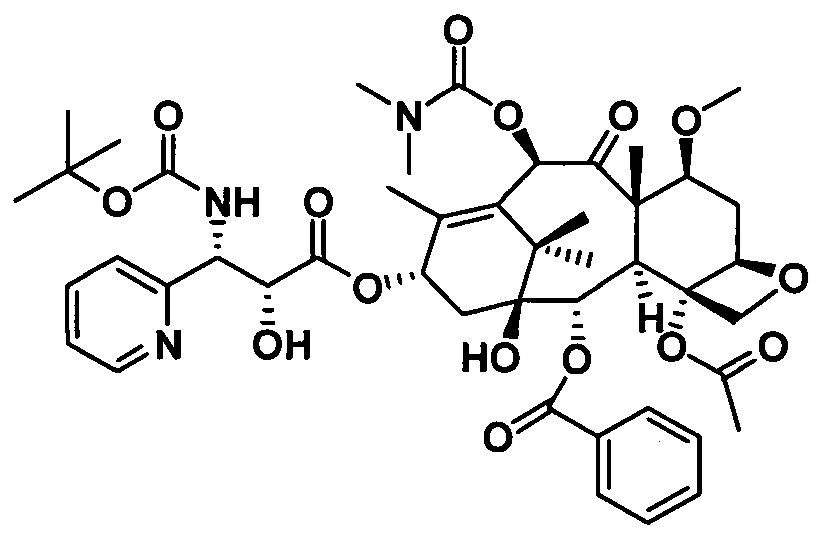
Пример 10. Получение PCMI-31
1) Получение (4S,5R)-3-трет-бутилоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен в примере 1.
2) Получение 7-диметилкарбамоил-10-метоксибаккатина III
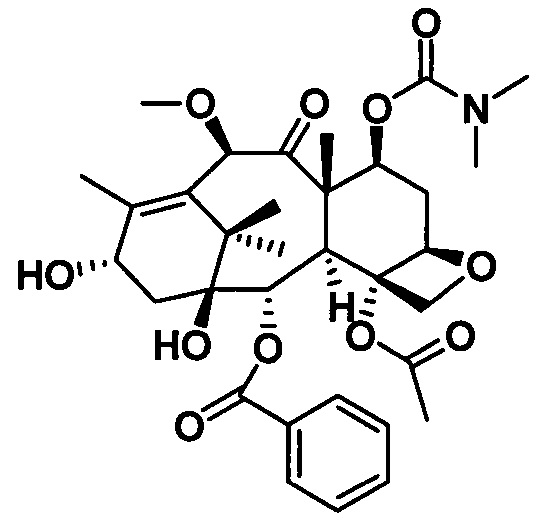
В качестве исходного вещества использовали 10-DAB (1 экв.), растворяли в DMF и добавляли последовательно 2,5 эквивалента имидазола и 2,5 эквивалента триэтилхлорсилана. После дополнительной обработки получали неочищенное соединение 1, приведенное на следующей схеме реакции.
Соединение 1 растворяли в дихлорметане, который использовали в качестве растворителя, в который добавляли 2 эквивалента пиридина при 0°С. Затем в реакционную жидкость добавляли по каплям 2 эквивалента р-толуолсульфонилхлорида. После 4 часов протекания реакции с помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 14 с выходом 90%.
Соединение 14 (1 экв.) растворяли в безводном тетрагидрофуране и вводили в реакцию с метилмагнийбромидом (2 экв.) в течение 3 часов при комнатной температуре в защитной атмосфере азота. С помощью дополнительной обработки получали неочищенное соединение 15 после высушивания.
Соединение 15 (1 экв.) растворяли в сухом THF, в который добавляли 1,5 эквивалента фторида тетрабутиламмония (в виде раствора в THF) при комнатной температуре. Спустя 1 час протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 16 с выходом 90%.
Соединение 16 (1 экв.) растворяли в сухом растворителе THF, в который добавляли 1,5 эквивалента LHMDS при 0°С. Через 1 час протекания реакции к реакционной жидкости медленно добавляли по каплям 2 эквивалента диметилкарбамоилхлорида и проводили реакцию в течение 2 часов. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 17 с выходом 87%.
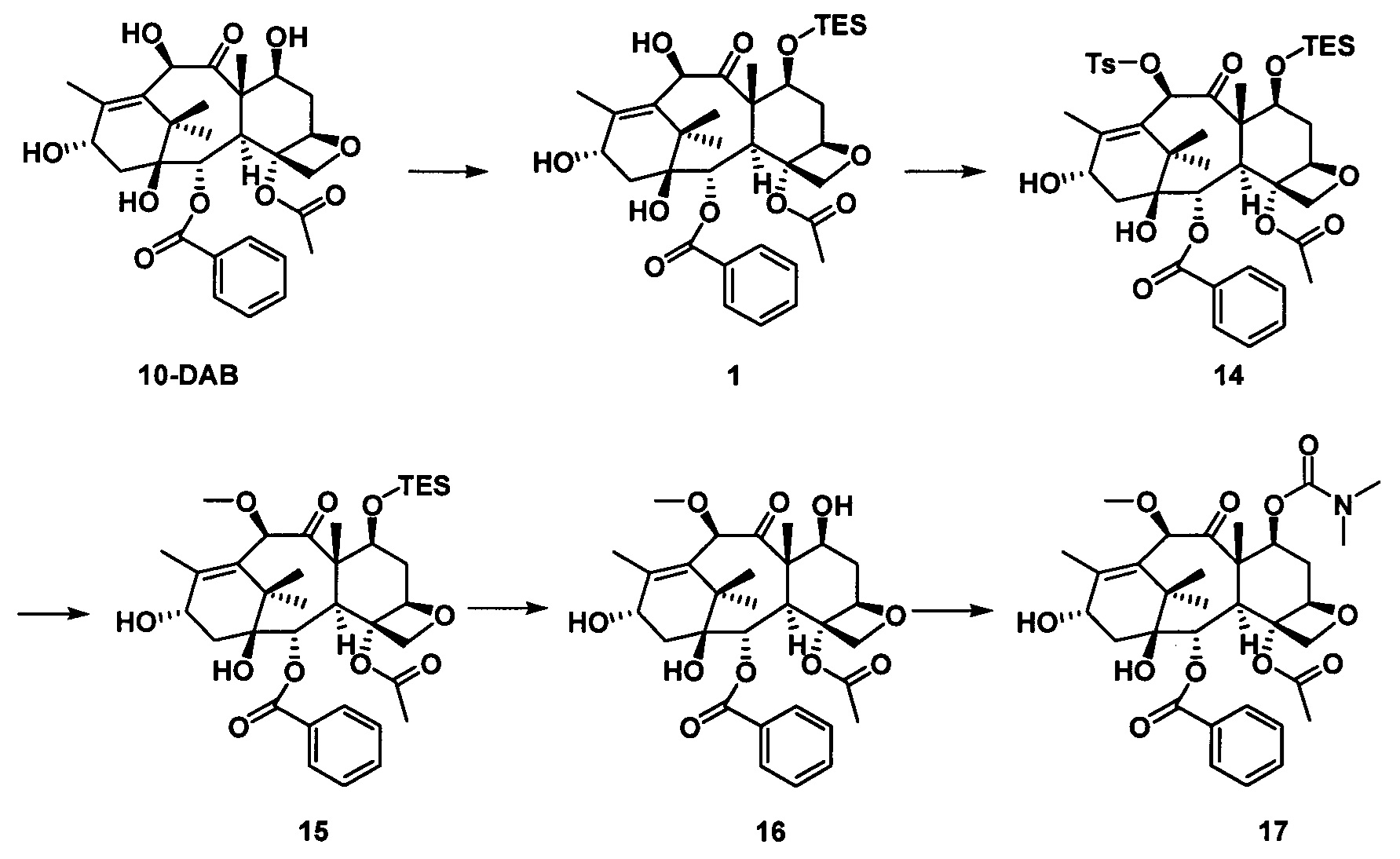
3) Получение PCMI-31
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-31: Т. пл.: 247-249°С;
MS (масса/заряд) ESI: 914,6 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,12 (d, J=7,4 Гц, 2Н), 7,62 (t, J=1,4 Гц, 1Н), 7,51 (t, J=7,7 Гц, 2Н), 7,41 (d, J=4,6 Гц, 4Н), 7,33 (dd, J=8,8, 4,4 Гц, 1Н), 6,24 (t, J=8,4 Гц, 1Н), 5,69 (d, J=6,9 Гц, 1Н), 5,51 (d, J=9,2 Гц, 1Н), 5,45 (dd, J=10,5, 7,4 Гц, 1Н), 5,30 (d, J=9,2 Гц, 1Н), 5,21 (s, 1Н), 4,95 (d, J=8,3 Гц, 1Н), 4,65 (s, 1Н), 4,32 (d, J=8,4 Гц, 1Н), 4,21 (d, J=8,3 Гц, 1Н), 3,96 (d, J=6,9 Гц, 1Н), 3,57 (d, J=4,0 Гц, 1Н), 3,38 (s, 3Н), 2,87 (d, J=4,5 Гц, 6Н), 2,57 (m, 1Н), 2,39 (s, 3Н), 2,31 (d, J=9,0 Гц, 2Н), 2,01-1,89 (m, 4Н), 1,81 (s, 3Н), 1,36 (s, 9Н), 1,22 (s, 3Н), 1,19 (s, 3Н).
13С ЯМР (101 МГц, CDCl3) δ 206,26, 170,00, 167,03, 156,84, 155,32, 155,03, 139,40, 134,91, 133,66, 130,18, 129,20, 128,74, 127,98, 126,79, 84,06, 82,76, 80,95, 80,11, 78,78, 76,57, 74,75, 73,63, 72,74, 72,47, 60,42, 56,77, 49,17, 46,48, 43,18, 36,53, 35,89, 35,37, 33,95, 29,70, 28,21, 26,38, 25,61, 24,94, 22,58, 20,62, 14,44, 10,79.
Пример 11. Получение PCMI-32
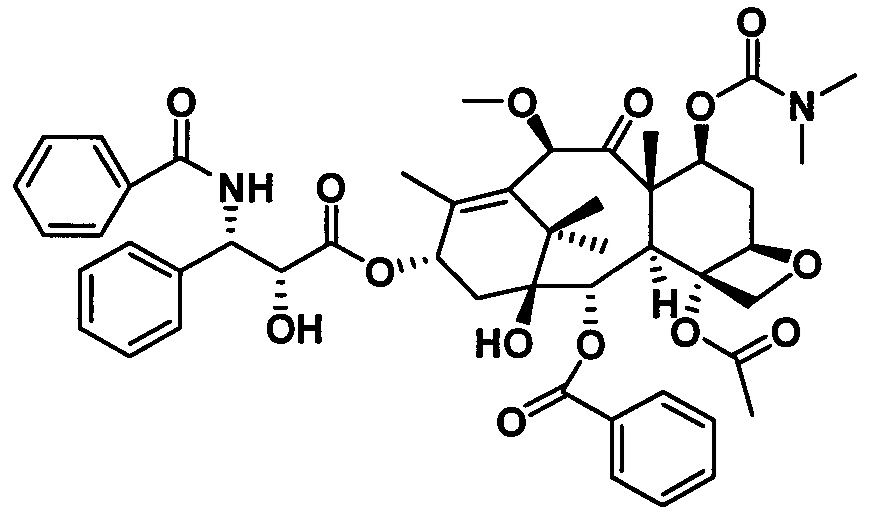
1) Получение (4S,5R)-3-бензоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 2.
2) Получение 7-диметилкарбамоил-10-метоксибаккатина III
Конкретный способ был представлен на стадии 2) в примере 10.
3) Получение PCMI-32
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-32: Т. пл.: 233-235°С;
MS (масса/заряд) ESI: 919,0 (M+Na)+;
1H ЯМР (400 МГц, CDCl3) δ 8,12 (d, J=7,2 Гц, 2Н), 7,77 (d, J=7,2 Гц, 2Н), 7,61 (t, J=7,4 Гц, 1Н), 7,54-7,46 (m, 5Н), 7,44-7,36 (m, 4Н), 7,34 (d, J=7,3 Гц, 1Н), 7,20 (d, J=8,8 Гц, 1Н), 6,21 (t, J=8,7 Гц, 1Н), 5,80 (dd, J=8,9, 2,4 Гц, 1Н), 5,67 (d, J=6,9 Гц, 1Н), 5,43 (dd, J=10,5, 7,4 Гц, 1Н), 5,16 (s, 1Н), 4,93 (d, J=8,7 Гц, 1Н), 4,79 (dd, J=5,1, 2,7 Гц, 1Н), 4,31 (d, J=8,4 Гц, 1Н), 4,21 (d, J=8,4 Гц, 1Н), 3,93 (d, J=6,8 Гц, 1Н), 3,84 (d, J=5,2 Гц, 1Н), 3,34 (s, 3Н), 2,85 (d, J=3,1 Гц, 6Н), 2,63-2,49 (m, 1Н), 2,38 (s, 3Н), 2,35-2,27 (m, 2Н), 1,88 (s, 4Н), 1,80 (s, 3Н), 1,17 (d, J=5,6 Гц, 6H).
13С ЯМР (101 МГц, CDCl3) δ 206,21, 172,54, 171,18, 170,19, 166,97, 166,93, 155,03, 139,05, 138,17, 135,13, 133,78, 133,66, 131,85, 130,17, 129,25, 128,92, 128,69, 128,65, 128,21, 127,09, 126,92, 84,04, 82,76, 81,01, 78,68, 76,59, 74,70, 73,29, 72,77, 72,34, 56,84, 56,73, 54,94, 46,48, 43,14, 36,51, 35,88, 35,55, 34,00, 33,87, 29,69, 26,47, 25,58, 24,90, 22,58, 20,55, 14,43, 10,79.
Пример 12. Получение PCMI-33
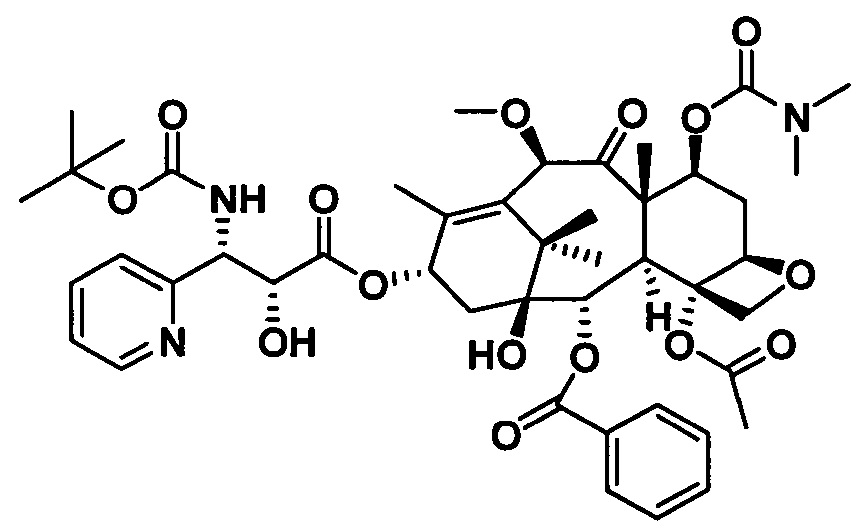
1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-(2-пиридинил)-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 3.
2) Получение 7-диметилкарбамоил-10-метоксибаккатина III
Конкретный способ был представлен на стадии 2) в примере 10.
3) Получение PCMI-33
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-33: Т. пл.: 236-239°С;
MS (масса/заряд) ESI: 916,4 (M+Na)+;
1H ЯМР (400 МГц, CDCl3) δ 8,54 (dd, J=14,0, 4,6 Гц, 1Н), 8,14-8,07 (m, 2Н), 7,79-7,70 (m, 1H), 7,61 (t, J=7,4 Гц, 1H), 7,49 (t, J=7,8 Гц, 2Н), 7,43-7,37 (m, 1Н), 7,25 (d, J=9,4 Гц, 1H), 6,18 (t, J=8,9 Гц, 1H), 5,88 (d, J=9,6 Гц, 1H), 5,67 (d, J=7,1 Гц, 1H), 5,48 (dd, J=10,5, 7,4 Гц, 1H), 5,37 (d, J=8,9 Гц, 1H), 5,21 (s, 1H), 4,97 (d, J=8,2 Гц, 1H), 4,80 (d, J=2,6 Гц, 1H), 4,32 (d, J=8,3 Гц, 1H), 4,18 (d, J=8,3 Гц, 1H), 3,98 (d, J=7,0 Гц, 1H), 3,85 (d, J=5,3 Гц, 1H), 3,35 (s, 3H), 2,88-2,83 (d, 6H), 2,66-2,53 (m, 1H), 2,46 (s, 3H), 2,38-2,31 (m, 2H), 1,97 (s, 4H, H-6), 1,80 (s, 3H), 1,25 (s, 9H), 1,15 (d, J=5,6 Гц, 6H).
13C ЯМР (101 МГц, CDCl3) δ 206,43, 171,16, 170,28, 167,06, 158,81, 155,04, 148,51, 139,81, 137,51, 134,61, 133,65, 130,14, 130,07, 129,27, 128,66, 122,94, 121,82, 84,13, 82,72, 80,60, 80,20, 79,02, 74,89, 73,66, 72,75, 71,40, 56,09, 46,48, 43,15, 43,06, 36,53, 35,91, 35,47, 33,99, 31,93, 31,44, 29,70, 29,36, 28,29, 27,94, 26,25, 22,70, 22,18, 20,67, 14,38, 14,13, 10,79.
Пример 13. Получение PCMI-34
1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-изобутил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 4.
2) Получение 7-диметилкарбамоил-10-метоксибаккатина III Конкретный способ был представлен на стадии 2) в примере 10.
3) Получение PCMI-34
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-34: Т. пл.: 229-231°С;
MS (масса/заряд) ESI: 895,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,12 (d, J=7,4 Гц, 2Н), 7,61 (t, J=7,4 Гц, 1Н), 7,48 (t,J=7,7 Гц, 2Н), 6,19 (t, J=8,7 Гц, 1Н), 5,68 (d, J=7,0 Гц, 1Н), 5,47 (dd, J=10,5, 7,3 Гц, 1Н), 5,22 (s, 1Н), 4,97 (d, J=8,1 Гц, 1Н), 4,67 (d, J=9,8 Гц, 1Н), 4,32 (d, J=8,4 Гц, 1Н), 4,26-4,14 (m, 3Н), 3,97 (d, J=6,9 Гц, 1Н), 3,38 (s, 3Н), 3,34 (d, J=5,8 Гц, 1Н), 2,86 (d, J=2,7 Гц, 6Н), 2,57 (ddd, J=14,5, 9,4, 7,4 Гц, 1Н), 2,40 (d, J=5,3 Гц, 5Н), 2,02 (d, J=0,6 Гц, 3Н), 1,92 (ddd, J=14,5, 10,9, 1,9 Гц, 1Н), 1,81 (s, 3Н), 1,69 (m, 1Н), 1,33 (s, 11Н), 1,19 (d, J=9,1 Гц, 6Н), 0,99 (d, J=6,0 Гц, 6Н).
13С ЯМР (101 МГц, CDCl3) δ 206,25, 173,76, 169,75, 166,93, 155,46, 155,05, 139,59, 134,86, 133,52, 130,18, 129,39, 128,60, 84,07, 82,81, 80,94, 79,57, 78,85, 76,53, 74,95, 73,02, 72,74, 72,65, 56,80, 56,68, 51,29, 46,49, 43,18, 41,26, 36,49, 35,86, 35,45, 33,97, 28,19, 26,23, 24,72, 23,27, 22,45, 21,91, 20,69, 14,43, 10,79.
Пример 14. Получение PCMI-35
1) Получение (4S,5R)-3-диметилкарбамоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 8.
2) Получение 7-диметилкарбамоил-10-метоксибаккатина III
Конкретный способ был представлен на стадии 2) в примере 10.
3) Получение PCMI-35
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-35: Т. пл.: 229-231°С;
MS (масса/заряд) ESI: 902,3 (М+K)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,07-7,97 (m, 2Н), 7,61 (t, J=7,4 Гц, 1H), 7,52-7,36 (m, 10Н), 6,93 (d, J=8,7 Гц, 2Н), 6,39 (s, 1Н), 6,14 (s, 1H), 5,58 (d, J=7,1 Гц, 1H), 5,42 (s, 1H), 4,89 (d, J=8,7 Гц, 1H), 4,71 (s, 1H), 4,58 (d, J=4,9 Гц, 1H), 4,22 (d, J=8,4 Гц, 1H), 4,10 (d, J=8,4 Гц, 1H), 3,85-3,77 (m, 4H), 3,71 (d, J=7,1 Гц, 1H), 3,40 (s, 3H), 3,26 (s, 3H), 2,64 (ddd, J=14,4, 9,7, 6,4 Гц, 1H), 2,19 (dd, J=15,3, 9,1 Гц, 1H), 2,11-2,04 (m, 1H), 1,97-1,89 (m, 1H), 1,80 (s, 3H), 1,66 (s, 3H), 1,58 (s, 3H), 1,19 (s, 3H), 1,15 (s, 3H), 1,05 (s, 9H).
13C ЯМР (101 МГц, CDCl3) δ 204,87, 171,16, 169,91, 169,55, 166,88, 160,38, 156,99, 151,48, 139,05, 135,07, 133,68, 130,04, 129,29, 128,97, 128,68, 128,57, 128,17, 126,57, 113,87, 92,60, 84,09, 82,35, 81,23, 80,85, 80,56, 79,06, 76,27, 74,68, 71,83, 63,68, 57,10, 57,05, 56,70, 55,30, 49,12, 47,30, 43,18, 35,42, 33,88, 31,86, 29,68, 27,81, 26,65, 25,61, 24,93, 21,60, 20,89, 13,86, 10,34.
Пример 15. Получение PCMI-36
1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен в примере 1.
2) Получение 7-метоксиформил-10-метоксибаккатина III
В качестве исходного вещества использовали 10-DAB (1 экв.), растворяли в DMF и добавляли последовательно 2,5 эквивалента имидазола и 2,5 эквивалента триэтилхлорсилана. После дополнительной обработки получали неочищенное соединение 1, приведенное на следующей схеме реакции.
Соединение 1 (1 экв.) растворяли в дихлорметане, который использовали в качестве растворителя, в который добавляли 2 эквивалента пиридина при 0°С. Затем к реакционной жидкости добавляли по каплям 2 эквивалента n-толуолсульфонилхлорида. После 4 часов протекания реакции с помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 14 с выходом 90%.
Соединение 14 (1 экв.) растворяли в безводном тетрагидрофуране и вводили в реакцию с метилмагнийбромидом (2 экв.) в течение 3 часов при комнатной температуре в защитной атмосфере азота. После дополнительной обработки получали неочищенное соединение 15 после высушивания.
Соединение 15 (1 экв.) растворяли в сухом THF, в который добавляли 1,5 эквивалента фторида тетрабутиламмония (в виде раствора в THF) при комнатной температуре. Спустя 1 час протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 16 с выходом 90%.
Соединение 16 (1 экв.) растворяли в растворителе, представляющем собой сухой THF, в который добавляли 1,5 эквивалента LHMDS при 0°С. Через 1 час протекания реакции к реакционной жидкости медленно добавляли по каплям 2 эквивалента метоксиформилхлорида и проводили реакцию в течение 2 часов. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 18 с выходом 71%.
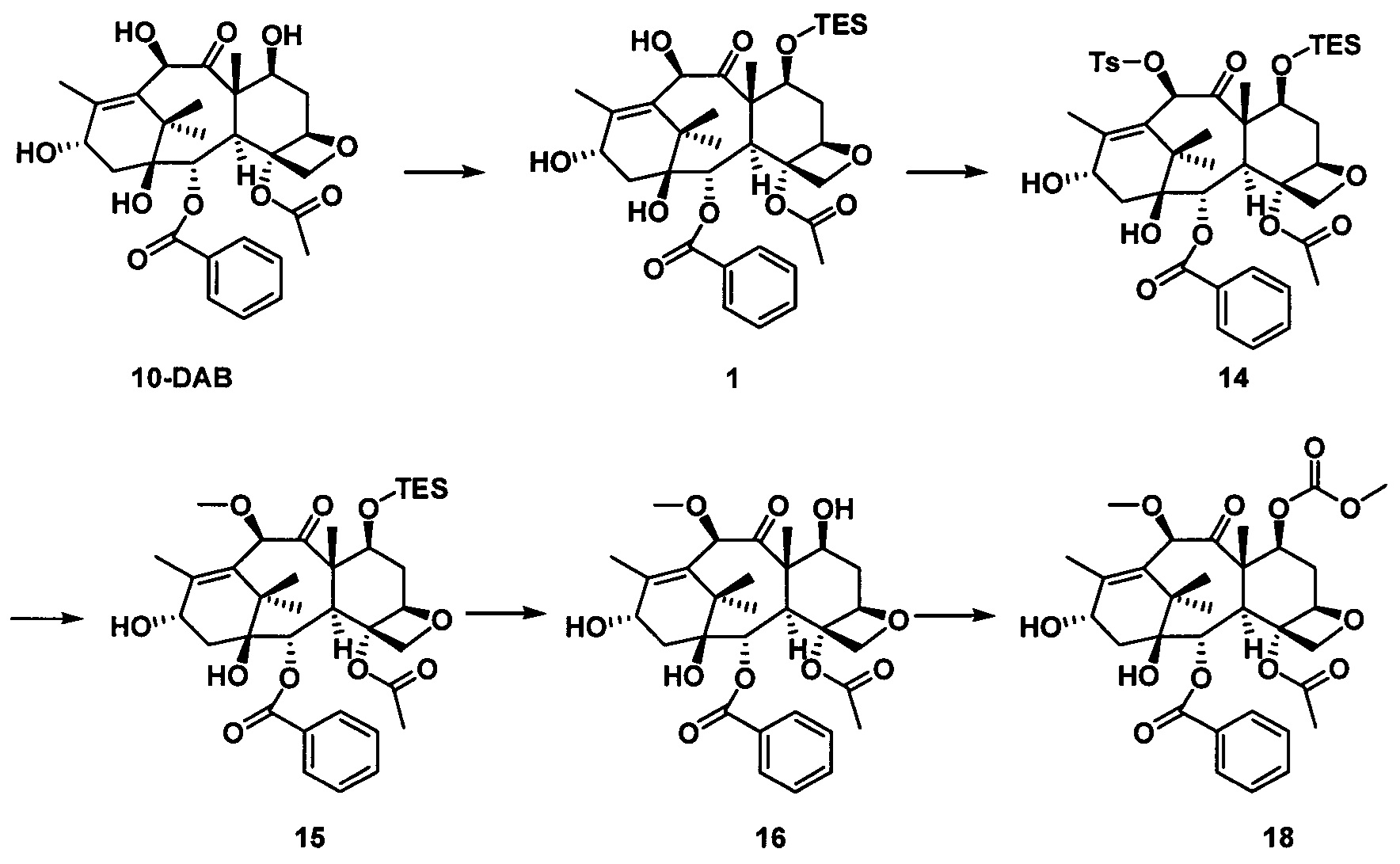
3) Получение PCMI-36
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-36: Т. пл.: 232-234°С;
MS (масса/заряд) ESI: 902,3 (M+Na)+;
1Н ЯМР (400 МГц, DMSO) δ 8,00 (d, J=7,4 Гц, 2Н), 7,71 (t, J=7,4 Гц, 1Н), 7,63 (q, J=7,4 Гц, 2H), 7,47-7,28 (m, 5H), 7,21 (t, J=7,1 Гц, 1H), 5,92 (t, J=9,6 Гц, 2H), 5,46 (d, J=7,1 Гц, 1H), 5,28 (dd, J=10,5, 7,4 Гц, 1H), 4,95 (t, J=11,3 Гц, 2H), 4,90 (s, 1H), 4,67 (s, 1H), 4,39 (t, J=7,1 Гц, 1H), 4,08 (s, 2H), 3,75 (d, J=7,0 Гц, 1H), 3,68 (s, 3H), 3,25 (s, 3H), 2,54-2,46 (m, 1H), 2,27 (s, 3Н), 1,83 (s, 3Н), 1,87-1,68 (m, 6Н), 1,63 (s, 3Н), 1,37 (s, 9Н), 1,03 (s, 3Н), 0,99 (s, 3Н).
13С ЯМР (101 МГц, DMSO) δ 205,78, 173,22, 170,65, 165,68, 155,56, 154,87, 140,02, 138,98, 135,26, 133,88, 130,17, 129,12, 128,61, 127,68, 83,21, 82,77, 80,15, 79,73, 79,40, 79,07, 78,58, 77,33, 76,22, 75,82, 74,78, 74,38, 70,24, 60,18, 57,76, 57,18, 56,04, 55,57, 46,27, 43,25, 35,30, 33,82, 33,36, 28,61, 26,67, 24,93, 22,79, 21,48, 21,17, 14,42, 10,85.
Пример 16. Получение PCMI-37
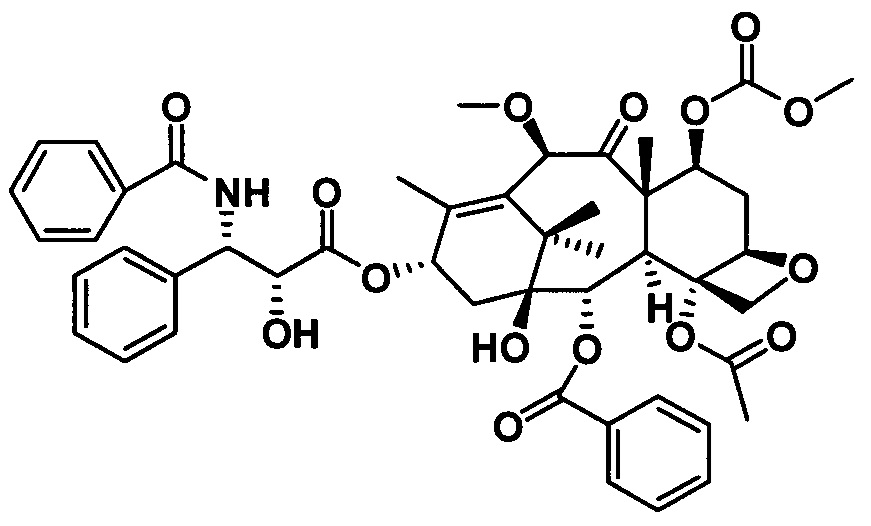
1) Получение (4S,5R)-3-бензоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 2.
2) Получение 7-метоксиформил-10-метоксибаккатина III
Конкретный способ был представлен на стадии 2) в примере 15.
3) Получение PCMI-37
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-37: Т. пл.: 222-224°С;
MS (масса/заряд) ESI: 906,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,16-8,09 (m, 2Н), 7,80-7,73 (m, 2Н), 7,63 (t, J=7,4 Гц, 1H), 7,56-7,46 (m, 5H), 7,46-7,32 (m, 5H), 7,16 (d, J=8,9 Гц, 1H), 6,22 (t, J=8,6 Гц, 1H), 5,81 (dd, J=8,9, 2,5 Гц, 1H), 5,69 (d, J=7,0 Гц, 1H), 5,36 (dd, J=10,5, 7,4 Гц, 1H), 5,11 (s, 1H), 4,94 (d, J=8,3 Гц, 1H), 4,80 (dd, J=5,3, 2,8 Гц, 1H), 4,33 (d, J=8,4 Гц, 1H), 4,21 (d, J=8,5 Гц, 1H), 3,93 (d, J=6,8 Гц, 1H), 3,80-3,73 (m, 4H), 3,38 (s, 3H), 2,59 (m, 1H), 2,40 (s, 3H), 2,32 (dd, J=8,9, 3,3 Гц, 2H), 2,05-1,96 (m, 1H), 1,87 (s, 3H), 1,81 (s, 3H), 1,18 (d, J=7,7 Гц, 6H).
13C ЯМР (101 МГц, CDCl3) δ 205,31, 172,62, 170,47, 166,99, 154,99, 139,01, 138,02, 135,17, 133,71, 131,95, 130,19, 129,07, 128,72, 128,31, 127,08, 83,68, 82,62, 80,85, 78,74, 76,56, 76,06, 74,56, 73,27, 72,37, 56,92, 56,61, 55,23, 55,03, 46,44, 43,08, 35,54, 33,44, 26,45, 22,58, 20,66, 14,28, 10,66.
Пример 17. Получение PCMI-38
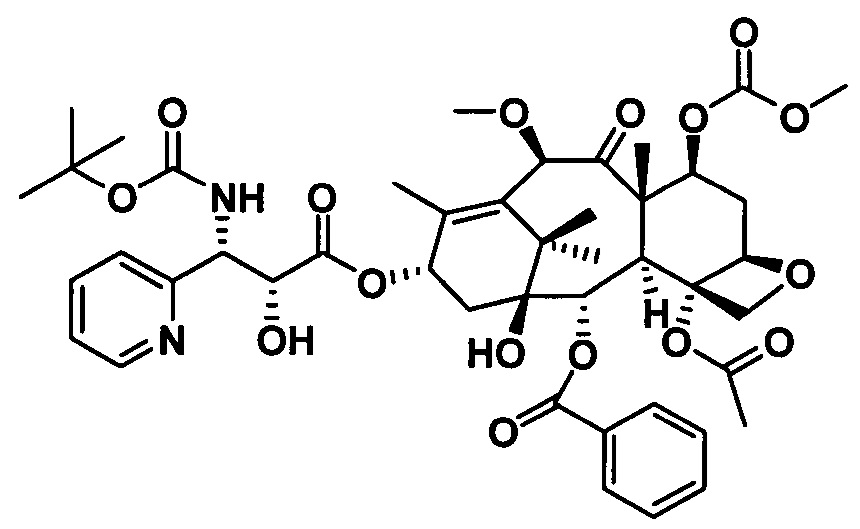
1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-(2-пиридинил)-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 3.
2) Получение 7-метоксиформил-10-метоксибаккатина III
Конкретный способ был представлен на стадии 2) в примере 15.
3) Получение PCMI-38
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-38: Т. пл.: 235-237°С;
MS (масса/заряд) ESI: 903,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,53 (d, J=4,4 Гц, 1Н), 8,12 (d, J=7,2 Гц, 2Н), 7,78 (td, J=7,7, 1,6 Гц, 1Н), 7,63 (t, J=7,4 Гц, 1Н), 7,51 (t, J=7,7 Гц, 2Н), 7,44 (d, J=7,9 Гц, 1Н), 7,31-7,26 (m, 1Н), 6,20 (t, J=8,8 Гц, 1Н), 5,88 (d, J=9,9 Гц, 1Н), 5,70 (d, J=7,0 Гц, 1Н), 5,47-5,36 (m, 2Н), 5,17 (s, 1Н), 4,98 (d, J=8,1 Гц, 1Н), 4,82 (s, 1Н), 4,35 (d, J=8,4 Гц, 1Н), 4,20 (d, J=8,4 Гц, 1Н), 3,99 (d, J=7,0 Гц, 1Н), 3,77 (s, 3Н), 3,39 (s, 3Н), 2,68-2,56 (m, 1Н), 2,50 (s, 3Н), 2,31 (dd, J=14,3, 8,9 Гц, 2Н), 2,06-1,93 (m, 4Н), 1,82 (s, 3Н), 1,46 (s, 9Н), 1,19 (d, J=6,1 Гц, 6Н).
13С ЯМР (101 МГц, CDCl3) δ 205,49, 170,61, 167,01, 154,97, 148,46, 139,76, 137,57, 134,71, 133,70, 130,14, 129,20, 128,69, 122,97, 121,89, 83,77, 82,59, 80,51, 79,03, 76,08, 74,73, 71,33, 56,87, 56,61, 55,18, 54,38, 54,32, 46,47, 43,09, 35,47, 33,45, 31,93, 31,44, 30,19, 29,70, 29,37, 28,30, 26,24, 22,70, 22,18, 20,74, 14,35, 14,13, 10,64.
Пример 18. Получение PCMI-39
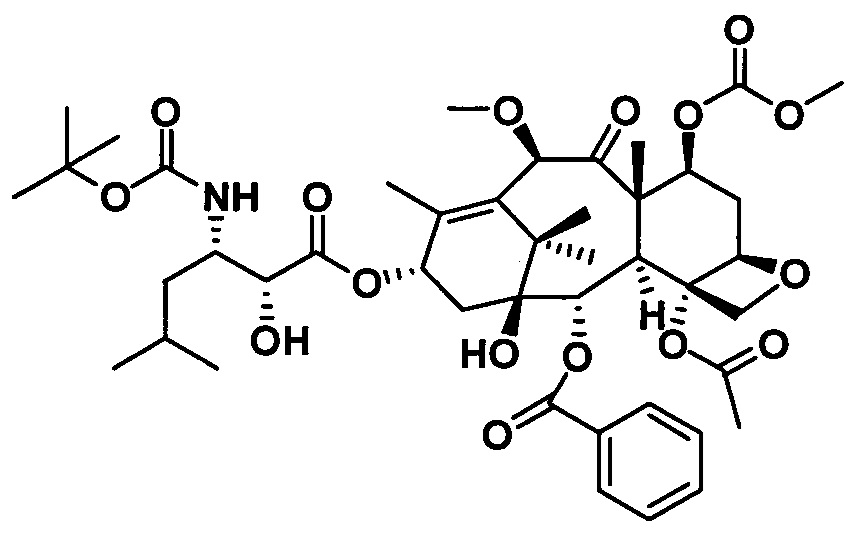
1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-изобутил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 4.
2) Получение 7-метоксиформил-10-метоксибаккатина III
Конкретный способ был представлен на стадии 2) в примере 15.
3) Получение PCMI-39
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-39: Т. пл.: 237-239°С;
MS (масса/заряд) ESI: 882,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,11 (d, J=7,5 Гц, 2Н), 7,61 (t, J=1,4 Гц, 1Н), 7,48 (t, J=1,1 Гц, 2Н), 6,19 (t, J=8,8 Гц, 1Н), 5,68 (d, J=7,0 Гц, 1Н), 5,38 (dd, J=10,6, 7,4 Гц, 1Н), 5,15 (s, 1Н), 4,95 (d, J=8,2 Гц, 1H), 4,60 (d, J=9,7 Гц, 1Н), 4,32 (d, J=8,5 Гц, 1Н), 4,26-4,13 (m, 3Н), 3,95 (d, J=6,9 Гц, 1Н), 3,75 (s, 3Н), 3,39 (s, 3Н), 3,25 (d, J=5,9 Гц, 1Н), 2,68-2,52 (m, 1Н), 2,39 (s, 5Н), 1,99 (s, 4Н), 1,80 (s, 3Н), 1,69 (dd, J=17,2, 7,1 Гц, 2Н), 1,33 (s, 10Н), 1,20 (s, 3Н), 1,17 (s, 3Н), 1,02-0,95 (m, 6Н).
13С ЯМР (101 МГц, CDCl3) δ 205,35, 173,90, 170,06, 166,91, 155,00, 130,21, 128,66, 83,70, 82,65, 80,80, 78,86, 76,02, 74,75, 73,02, 72,72, 56,85, 56,59, 55,20, 51,31, 46,43, 43,13, 41,23, 33,44, 29,71, 28,19, 26,18, 24,72, 23,29, 22,49, 21,92, 20,77, 14,40, 10,68.
Пример 19. Получение PCMI-40
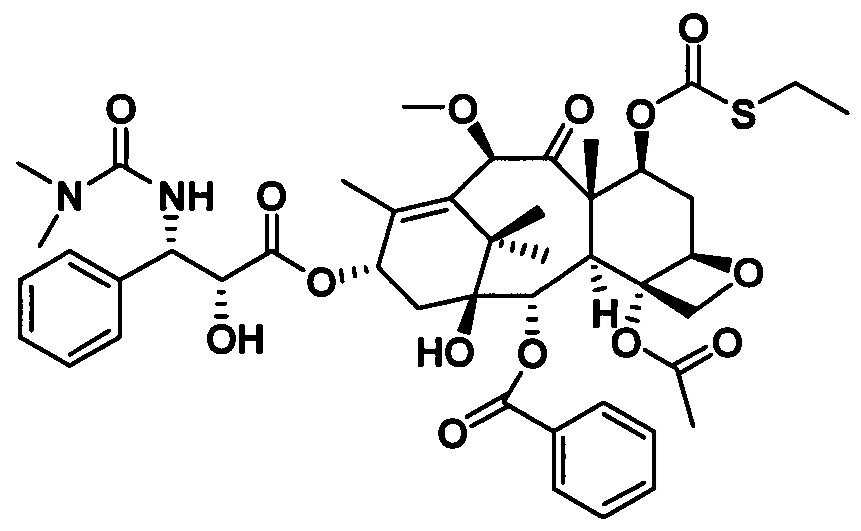
1) Получение (4S,5R)-3-диметилкарбамоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 8.
2) Получение 7-этилтиоформил-10-метоксибаккатина III
В качестве исходного вещества использовали 10-DAB (1 экв.), растворяли в DMF и добавляли последовательно 2,5 эквивалента имидазола и 2,5 эквивалента триэтилхлорсилана. После дополнительной обработки получали неочищенное соединение 1.
Соединение 1 растворяли в дихлорметане, который использовали в качестве растворителя, в который добавляли 2 эквивалента пиридина при 0°С. Затем к реакционной жидкости добавляли по каплям 2 эквивалента n-толуолсульфонилхлорида. После 4 часов протекания реакции с помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 14 с выходом 90%.
Соединение 14 (1 экв.) растворяли в безводном тетрагидрофуране и вводили в реакцию с метилмагнийбромидом (2 экв.) в течение 3 часов при комнатной температуре в защитной атмосфере азота. После дополнительной обработки получали неочищенное соединение 15 после высушивания.
Соединение 15 (1 экв.) растворяли в сухом THF, в который добавляли 1,5 эквивалента фторида тетрабутиламмония (в виде раствора в THF) при комнатной температуре. Спустя 1 час протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 16 с выходом 90%.
Соединение 16 (1 экв.) растворяли в сухом THF, который использовали в качестве растворителя, и сначала вводили в реакцию с 2 эквивалентами N,N-карбонилдиимидазола в течение 2 часов. К реакционной жидкости затем добавляли 2 эквивалента этантиола. После 4 часов протекания реакции с помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 19 с выходом 78%.
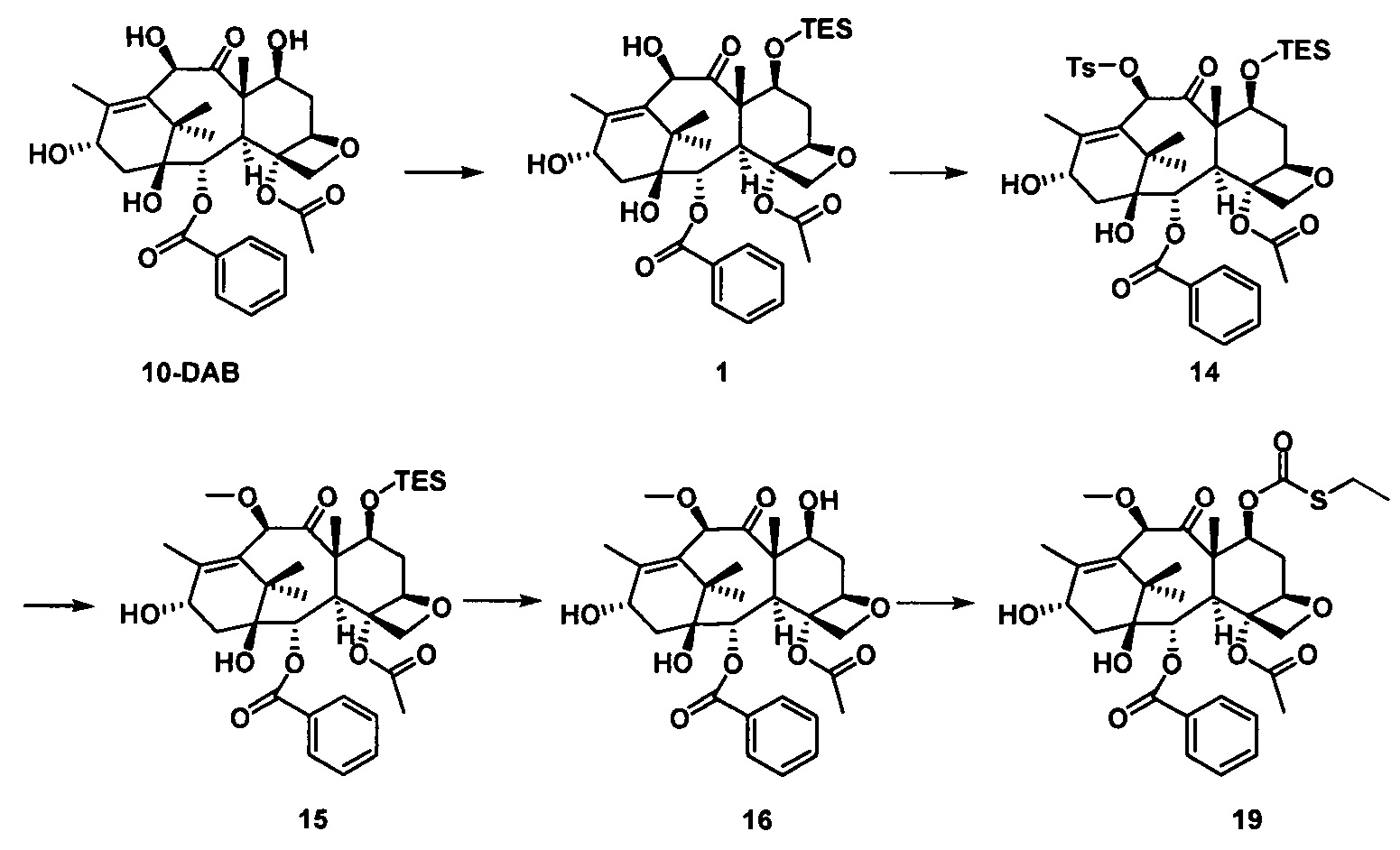
3) Получение PCMI-40
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-40: Т. пл.: 241-242°С;
MS (масса/заряд) ESI: 882,4 (М+Н)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,08-8,00 (m, 2Н), 7,64 (t, J=7,5 Гц, 1Н), 7,50 (t, J=7,8 Гц, 2Н), 6,11 (d, J=7,2 Гц, 1Н), 5,41 (dd, J=10,7, 7,2 Гц, 1Н), 5,19 (s, 1H), 5,06 (t, J=5,0 Гц, 1Н), 5,00-4,93 (m, 1Н), 4,82 (d, J=5,8 Гц, 1Н), 4,34 (d, J=8,4 Гц, 1Н), 4,23 (d, J=8,6 Гц, 1Н), 3,88 (d, J=7,1 Гц, 1Н), 3,77 (s, 3Н), 3,43 (s, 3Н), 3,36 (d, J=5,5 Гц, 1Н), 2,60 (ddd, J=14,5, 9,5, 7,2 Гц, 1Н), 2,34 (s, 3Н), 2,17 (d, J=1,1 Гц, 3Н), 2,06-1,98 (m, 1Н), 1,83 (s, 3Н), 1,26 (s, 3Н), 1,16 (s, 3Н).
13С ЯМР (101 МГц, CDCl3) δ 204,92, 170,54, 164,78, 155,00, 152,90, 141,17, 134,24, 129,85, 128,96, 128,07, 88,53, 84,18, 83,50, 82,74, 80,01, 76,01, 75,52, 71,84, 69,37, 57,32, 56,86, 55,29, 46,16, 41,28, 33,24, 25,61, 22,24, 21,41, 14,82, 10,74.
Пример 20. Получение PCMI-41
1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен в примере 1.
2) Получение 10-метокси-7-дигидробаккатина III
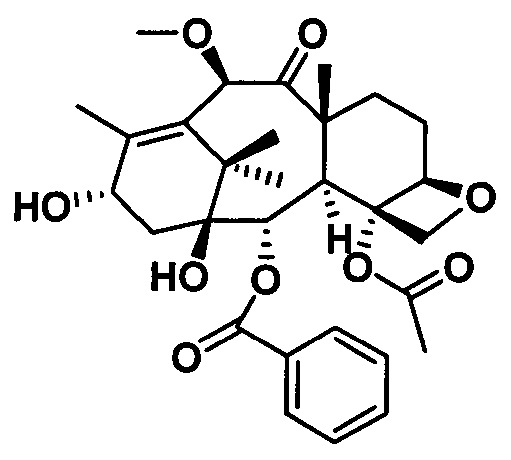
В качестве исходного вещества использовали 10-DAB (1 экв.), растворяли в DMF и добавляли последовательно 2,5 эквивалента имидазола и 2,5 эквивалента триэтилхлорсилана. После дополнительной обработки получали неочищенное соединение 1, приведенное на следующей схеме реакции.
Соединение 1 растворяли в дихлорметане, который использовали в качестве растворителя, в который добавляли 2 эквивалента пиридина при 0°С. Затем к реакционной жидкости добавляли по каплям 2 эквивалента n-толуолсульфонилхлорида. После 4 часов протекания реакции с помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 14 с выходом 90%.
Соединение 14 (1 экв.) растворяли в безводном тетрагидрофуране и вводили в реакцию с метилмагнийбромидом (2 экв.) в течение 3 часов при комнатной температуре в защитной атмосфере азота. После дополнительной обработки получали неочищенное соединение 15 после высушивания.
Соединение 15 (1 экв.) растворяли в сухом THF, в который добавляли 1,5 эквивалента фторида тетрабутиламмония (в виде раствора в THF) при комнатной температуре. Спустя 1 час протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 16 с выходом 92%.
Соединение 16 (1 экв.) растворяли в растворе сухого THF и к реакционной жидкости добавляли 6 эквивалентов N,N'-тиокарбонилдиимидазола. Спустя 2 часа протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 21 с выходом 86%.
Соединение 21 (1 экв.) растворяли в растворе диоксана/тетрагидрофурана (10:1). Для инициирования свободнорадикальной реакции добавляли 0,2 эквивалента азобисизобутиронитрила в качестве катализатора при 100°С. После этого к реакционной жидкости добавляли 4 эквивалента гидрида n-бутилолова и после 1 часа протекания реакции охлаждали в течение ночи при комнатной температуре. С помощью очистки колоночной хроматографией получали соединение 22 с выходом 52%.
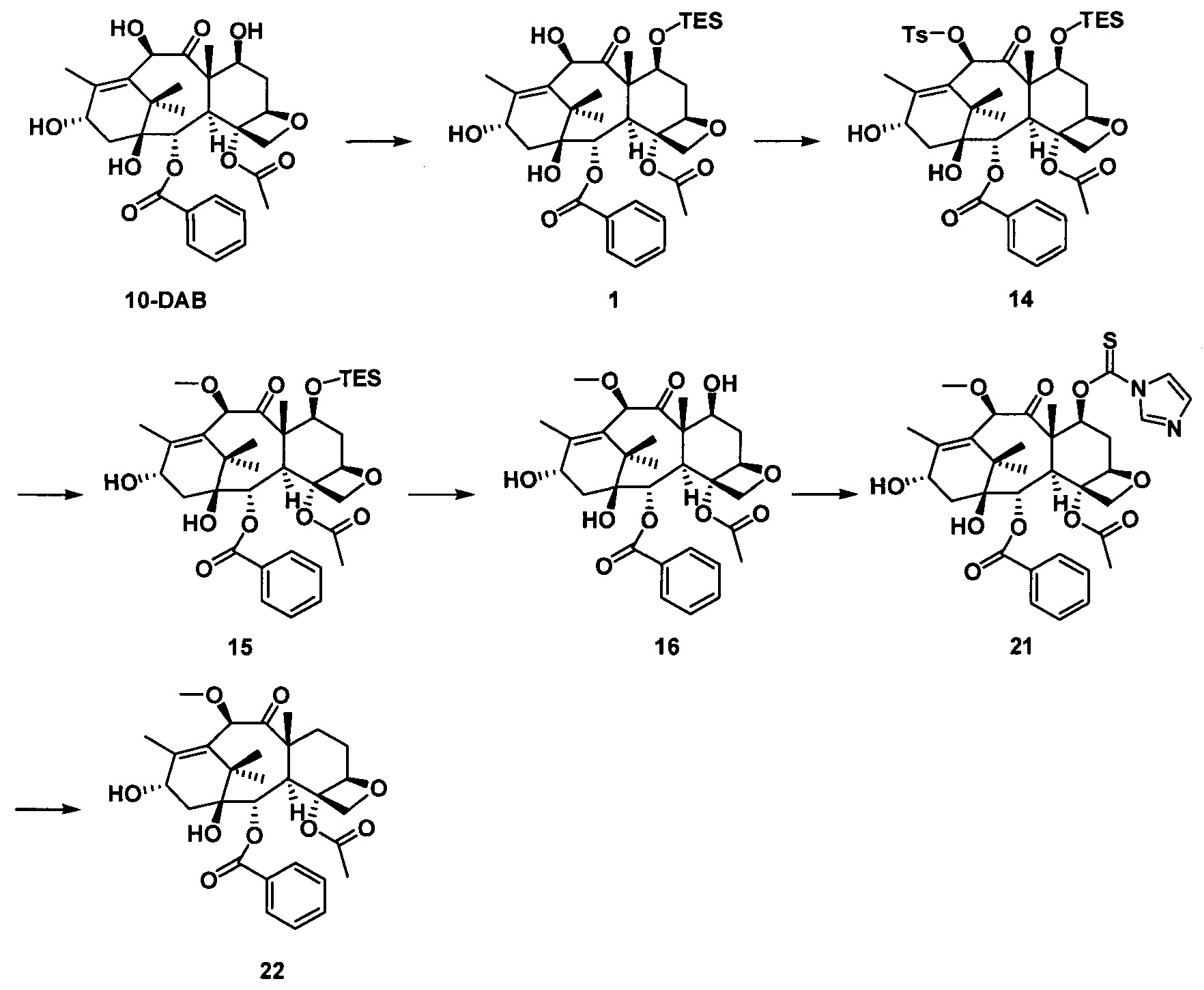
3) Получение PCMI-41
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-41: Т. пл.: 217-219°С;
MS (масса/заряд) ESI: 844,2 (М+K)+;
ИК: 3384, 2977, 2927, 2850, 1712, 1627, 1367, 1313, 1269, 1245, 1170, 1097, 987, 711.
1Н ЯМР (400 МГц, CDCl3) δ 8,13 (d, J=7,4 Гц, 2Н), 7,61 (t, J=7,4 Гц, 1H), 7,50 (t, J=7,6 Гц, 3Н), 7,39 (q, J=7,9 Гц, 4Н), 7,35-7,29 (m, 1H), 6,27 (t, J=8,7 Гц, 1Н), 5,66 (d, J=7,4 Гц, 1Н), 5,41 (d, J=9,4 Гц, 1H), 5,29 (s, 1Н), 4,98-4,88 (m, 2Н), 4,61 (s, 1Н), 4,31 (d, J=8,4 Гц, 1Н), 4,21 (d, J=8,4 Гц, 1Н), 3,77 (d, J=7,3 Гц, 1H), 3,42 (d, J=8,2 Гц, 4Н), 2,32 (m, 4Н), 2,23 (dd, J=13,3, 9,4 Гц, 1H), 2,04-1,93 (m, 1Н), 1,87 (s, 3Н), 1,75 (s, 3Н), 1,60-1,52 (m, 1H), 1,32 (s, 9H), 1,22 (s, 3Н), 1,16 (s, 3Н).
13C ЯМР (101 МГц, CDCl3) δ 209,18, 171,19, 170,14, 167,23, 155,31, 139,05, 138,44, 134,72, 133,59, 130,23, 129,36, 128,85, 128,67, 128,05, 126,72, 84,46, 82,27, 81,39, 80,19, 79,02, 75,85, 73,75, 72,43, 56,49, 53,08, 45,22, 43,02, 35,58, 29,70, 28,17, 27,20, 26,09, 22,74, 20,87, 14,82.
Пример 21. Получение PCMI-42

1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-изобутил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 4.
2) Получение 10-метокси-7-дигидробаккатина III
Конкретный способ был представлен на стадии 2) в примере 20.
3) Получение PCMI-42
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-42: Т. пл.: 213-215°С;
MS (масса/заряд) ESI: 808,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,14 (d, J=7,4 Гц, 2Н), 7,60 (t, J=7,4 Гц, 1Н), 7,47 (t, J=7,7 Гц, 2Н), 6,21 (dd, J=9,1, 7,9 Гц, 1Н), 5,66 (d, J=7,4 Гц, 1H), 5,00-4,90 (m, 2Н), 4,62 (d, J=9,8 Гц, 1Н), 4,31 (d, J=8,5 Гц, 1Н), 4,25 (d, J=8,4 Гц, 1H), 4,18 (d, J=5,6 Гц, 1H), 4,14 (m, 1H), 3,79 (d, J=7,7 Гц, 1H), 3,43 (s, 3H), 3,29 (d, J=6,1 Гц, 1H), 2,49-2,31 (m, 5H), 2,29-2,18 (m, 1H), 2,04-1,93 (m, 1H), 1,91 (d, J=0,9 Гц, 3Н), 1,76 (s, 3Н), 1,72-1,64 (m, 1H), 1,62-1,50 (m, 1H), 1,48-1,36 (m, 2H), 1,31 (s, 9H), 1,20 (s, 3H), 1,16 (s, 3H), 0,99 (d, J=6,4 Гц, 6H).
13C ЯМР (101 МГц, CDCl3) δ 209,29, 173,95, 169,98, 167,13, 155,44, 139,36, 134,55, 133,50, 130,24, 129,50, 128,62, 84,45, 82,23, 81,41, 79,65, 79,07, 75,96, 73,05, 72,67, 56,44, 53,03, 51,26, 45,21, 42,98, 41,28, 35,68, 29,70, 28,17, 27,20, 25,92, 24,71, 23,27, 22,63, 21,92, 20,89, 14,89, 14,28.
Пример 22. Получение PCMI-43
1) Получение (4S,5R)-3-бензоил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен на стадии 1) в примере 2.
2) Получение 10-метокси-6,7-двойная-связь-баккатина III
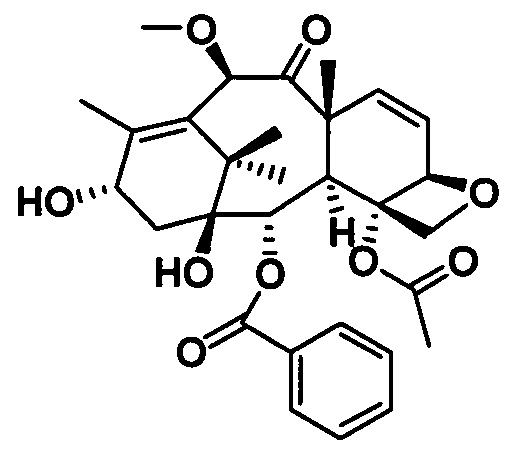
В качестве исходного вещества использовали 10-DAB (1 экв.), растворяли в DMF и добавляли последовательно 2,5 эквивалента имидазола и 2,5 эквивалента триэтилхлорсилана. После дополнительной обработки получали неочищенное соединение 1.
Соединение 1 растворяли в дихлорметане, который использовали в качестве растворителя, в который добавляли 2 эквивалента пиридина при 0°С. Затем к реакционной жидкости добавляли по каплям 2 эквивалента n-толуолсульфонилхлорида. После 4 часов протекания реакции с помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 14 с выходом 90%.
Соединение 14 (1 экв.) растворяли в безводном тетрагидрофуране и вводили в реакцию с метилмагнийбромидом (2 экв.) в течение 3 часов при комнатной температуре в защитной атмосфере азота. После дополнительной обработки получали неочищенное соединение 15 после высушивания.
Соединение 15 (1 экв.) растворяли в сухом THF, в который добавляли 1,5 эквивалента фторида тетрабутиламмония (в виде раствора в THF) при комнатной температуре. Спустя 1 час протекания реакции реакцию завершали. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 16 с выходом 92%.
Соединение 16 (1 экв.) растворяли в сухом дихлорметане. К реакционной жидкости сначала добавляли 2 эквивалента пиридина на ледяной бане, а затем добавляли по каплям 2 эквивалента трифторметансульфонового ангидрида. После 2 часов протекания реакции с помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 23 с выходом 78%.
Соединение 23 (1 экв.) растворяли в растворе диоксана/тетрагидрофурана (10:1). Добавляли 2 эквивалента DBU при 100°С и проводили реакцию элиминирования сульфоната в течение 0,5 часа. С помощью дополнительной обработки посредством очистки колоночной хроматографией получали соединение 24 с выходом 64%.
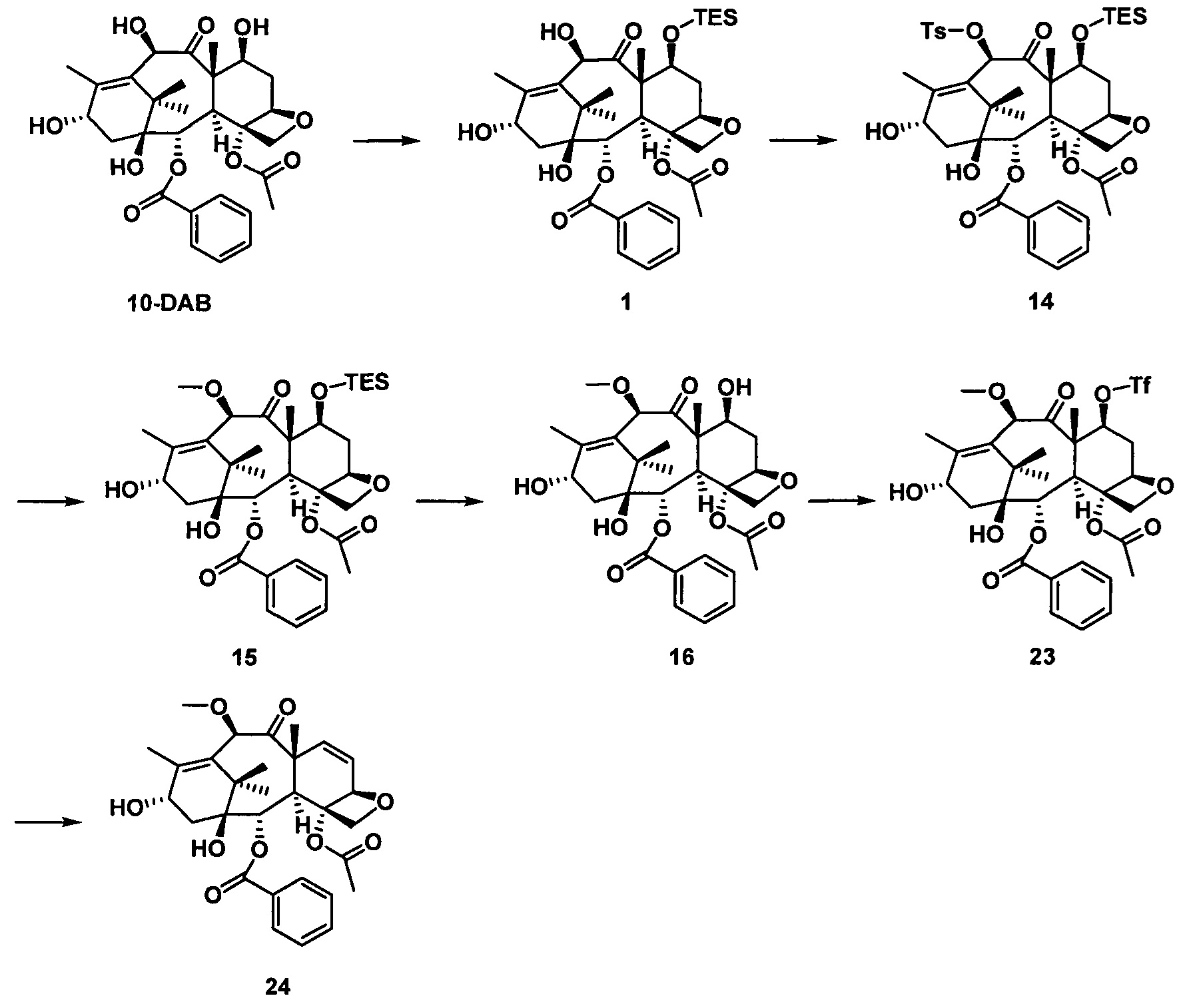
3) Получение PCMI-43
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-43: Т. пл.: 198-200°С;
MS (масса/заряд) ESI: 848,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,12 (d, J=1,3 Гц, 2Н), 7,81-7,75 (m, 2Н), 7,58-7,47 (m, 4Н), 7,47-7,32 (m, 5Н), 7,26 (t, J=1,9 Гц, 2Н), 7,11 (d, J=9,6 Гц, 1Н), 6,17 (t, J=7,6 Гц, 1Н), 6,08 (d, J=9,6 Гц, 1Н), 5,94-5,86 (m, 2Н), 5,52 (dd, J=10,6, 7,9 Гц, 2Н), 4,80 (dd, J=3,2, 1,6 Гц, 1Н), 4,72 (s, 1Н), 4,44 (s, 1H), 4,25 (d, J=5,5 Гц, 1Н), 3,71 (d, J=3,1 Гц, 1Н), 3,50 (s, 3Н), 3,42 (d, J=10,3 Гц, 1Н), 3,25 (t, J=10,7 Гц, 1Н), 3,05 (dd, J=15,4, 7,5 Гц, 1H), 2,47 (d, J=10,6 Гц, 1H), 2,15 (s, 3Н), 2,13 (s, 3Н), 1,52 (s, 3Н), 1,24 (s, 3Н), 1,18 (s, 3Н).
Пример 23. Получение PCMI-44
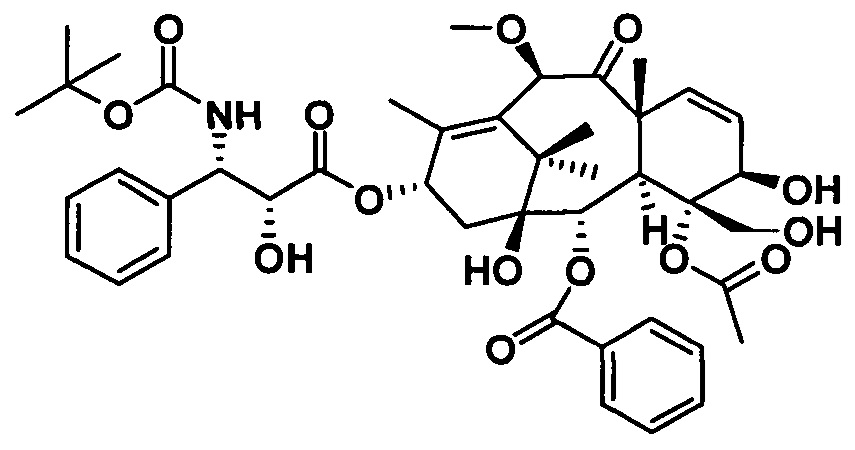
1) Получение (4S,5R)-3-трет-бутоксикарбонил-2-(4-метоксифенил)-4-фенил-5-оксазолидинкарбоновой кислоты
Конкретный способ был представлен в примере 1.
2) Получение 10-метокси-6,7-двойная-связь-баккатина III
Конкретный способ был представлен в примере 22.
3) Получение PCMI-44
Конкретный способ был представлен на стадии 3) в примере 1. Чистота конечного продукта составляла 95% или выше.
PCMI-44: Т. пл.: 186-187°С;
MS (масса/заряд) ESI: 844,4 (M+Na)+;
1Н ЯМР (400 МГц, CDCl3) δ 8,24 (t, 2Н), 7,61 (t, 1Н), 7,42 (m, 7Н), 6,25 (t, J=7,9 Гц, 1H), 5,88 (d, 2Н), 5,54 (d, 2Н), 5,40 (d, 1Н), 4,72 (s, 1Н), 4,58 (d, J=2,6 Гц, 1H), 4,31 (d, 1Н) 3,47 (d, 4Н), 3,30 (t, 1Н), 3,00 (d, 1Н), 2,41-2,34 (m, 1H), 2,07 (s, 3Н), 1,51 (s, 3Н), 1,35 (s, 9Н), 1,28 (s, 3Н), 1,23 (s, 3Н), 1,18 (s, 3Н).
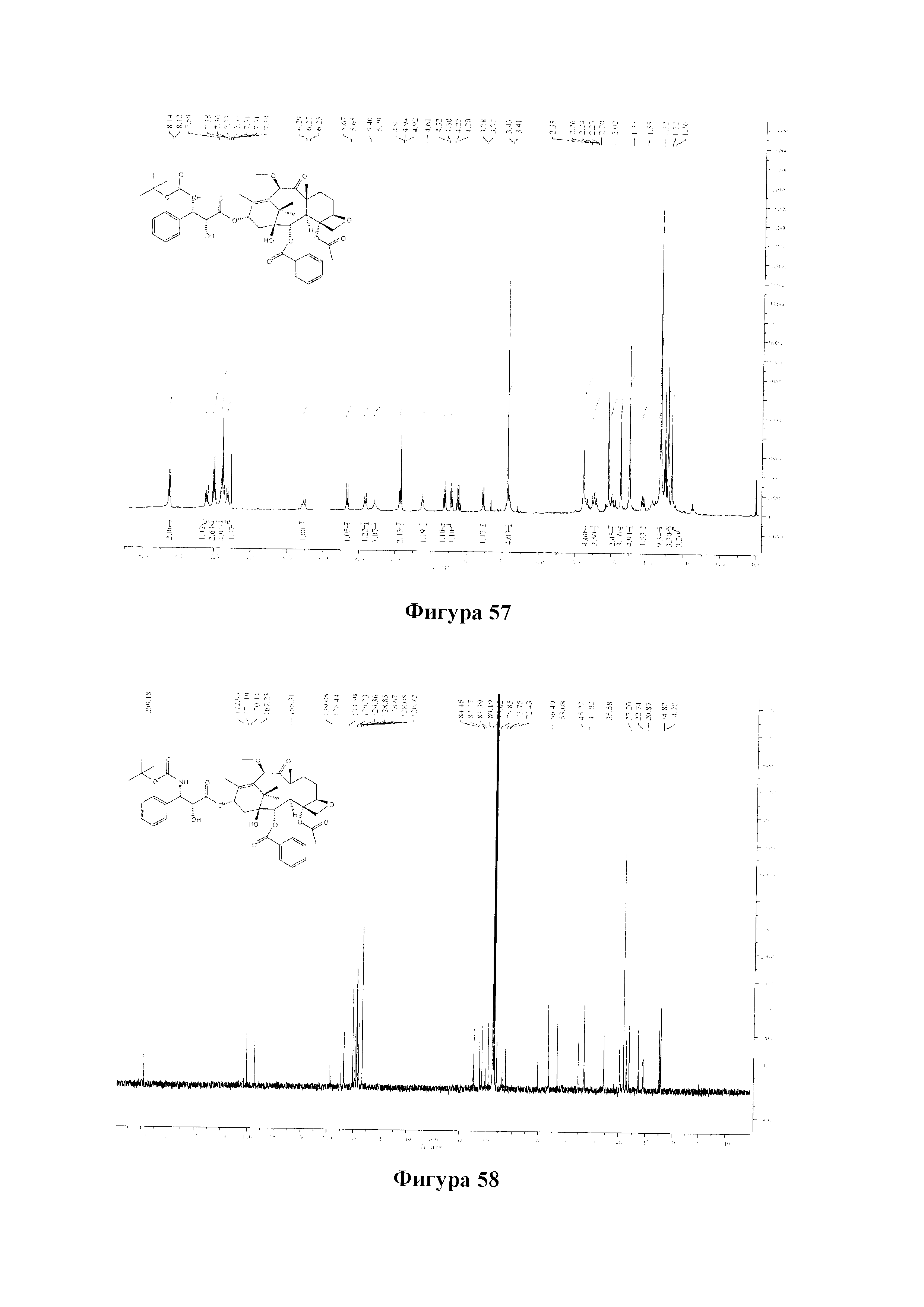
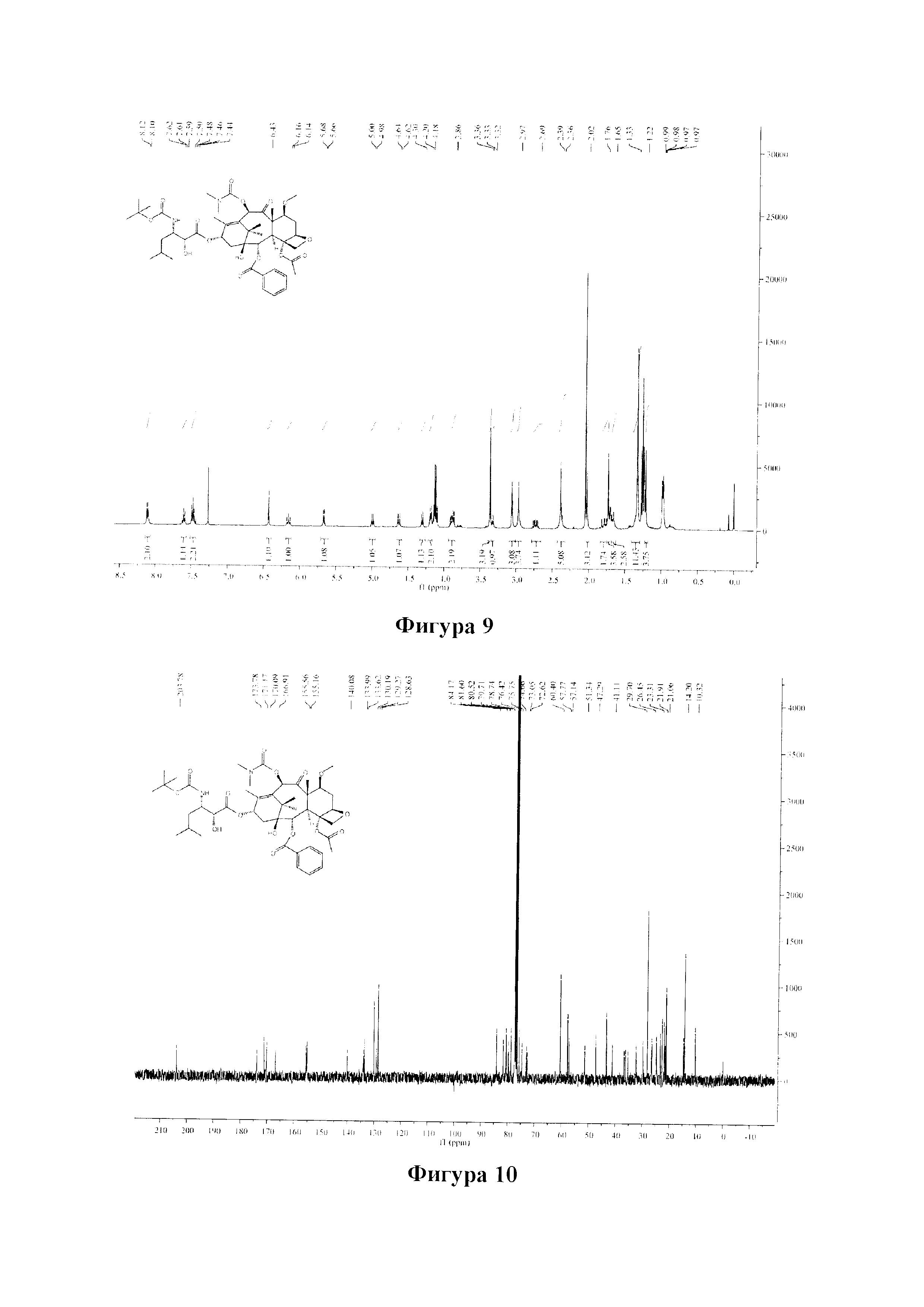
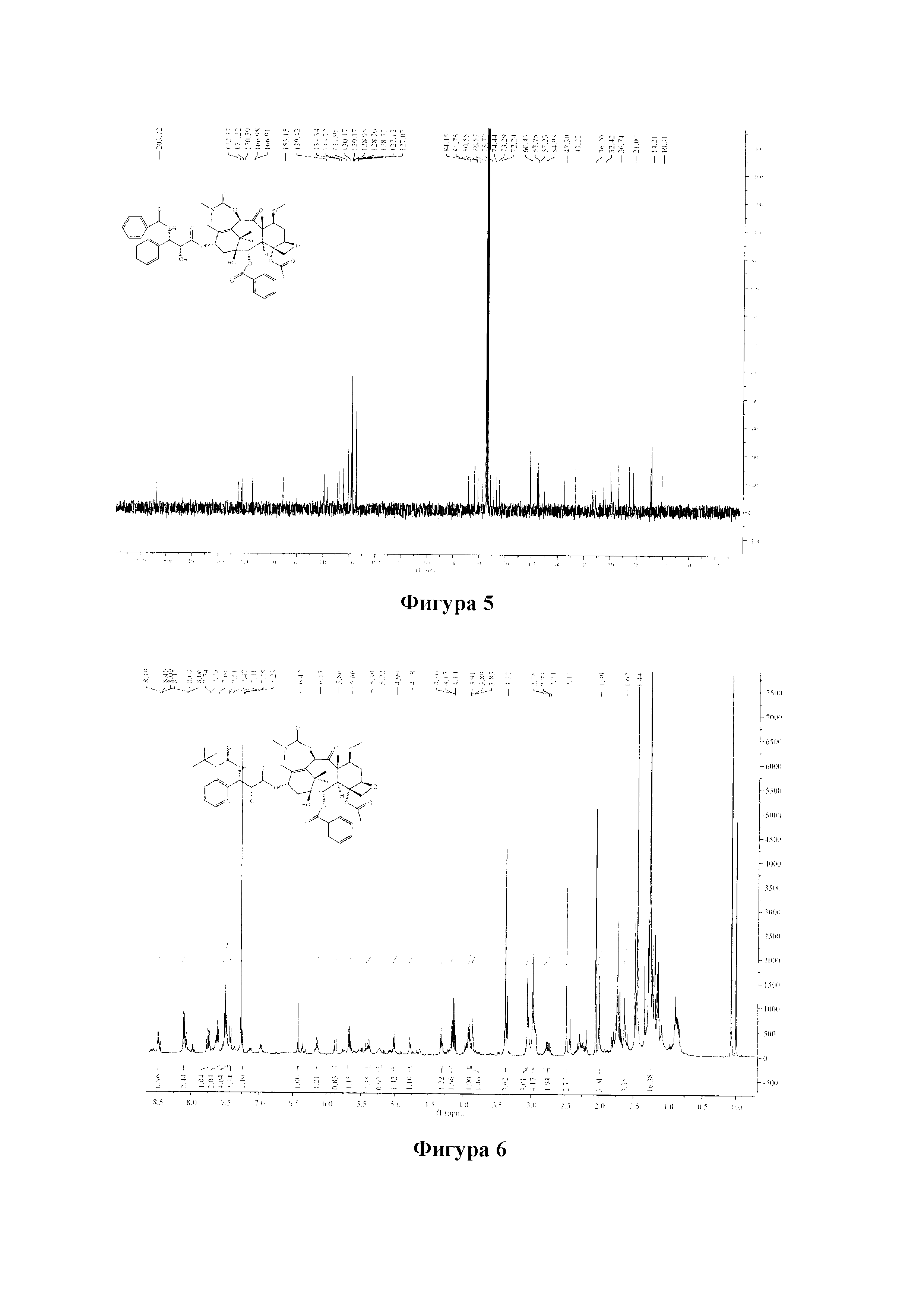
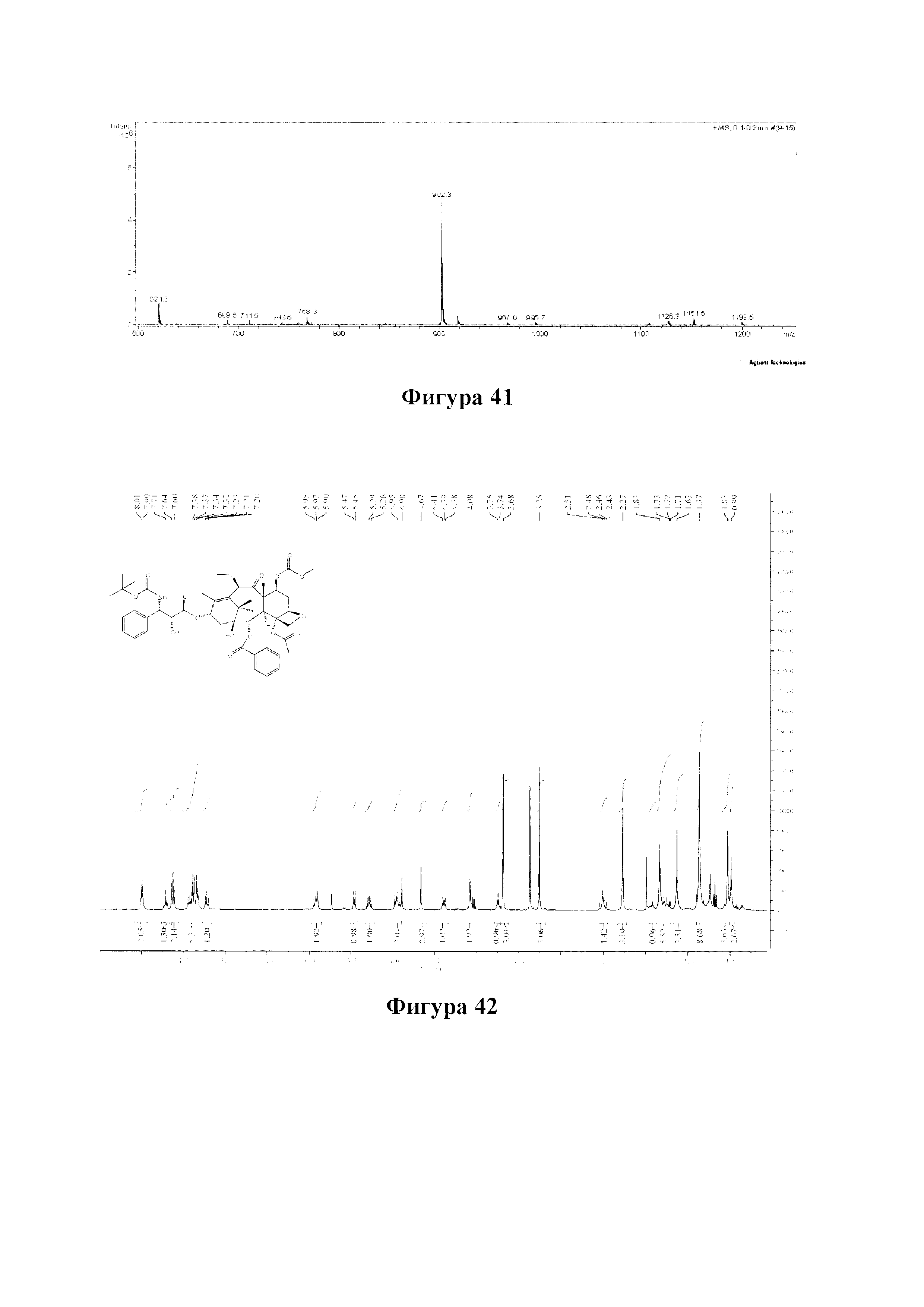
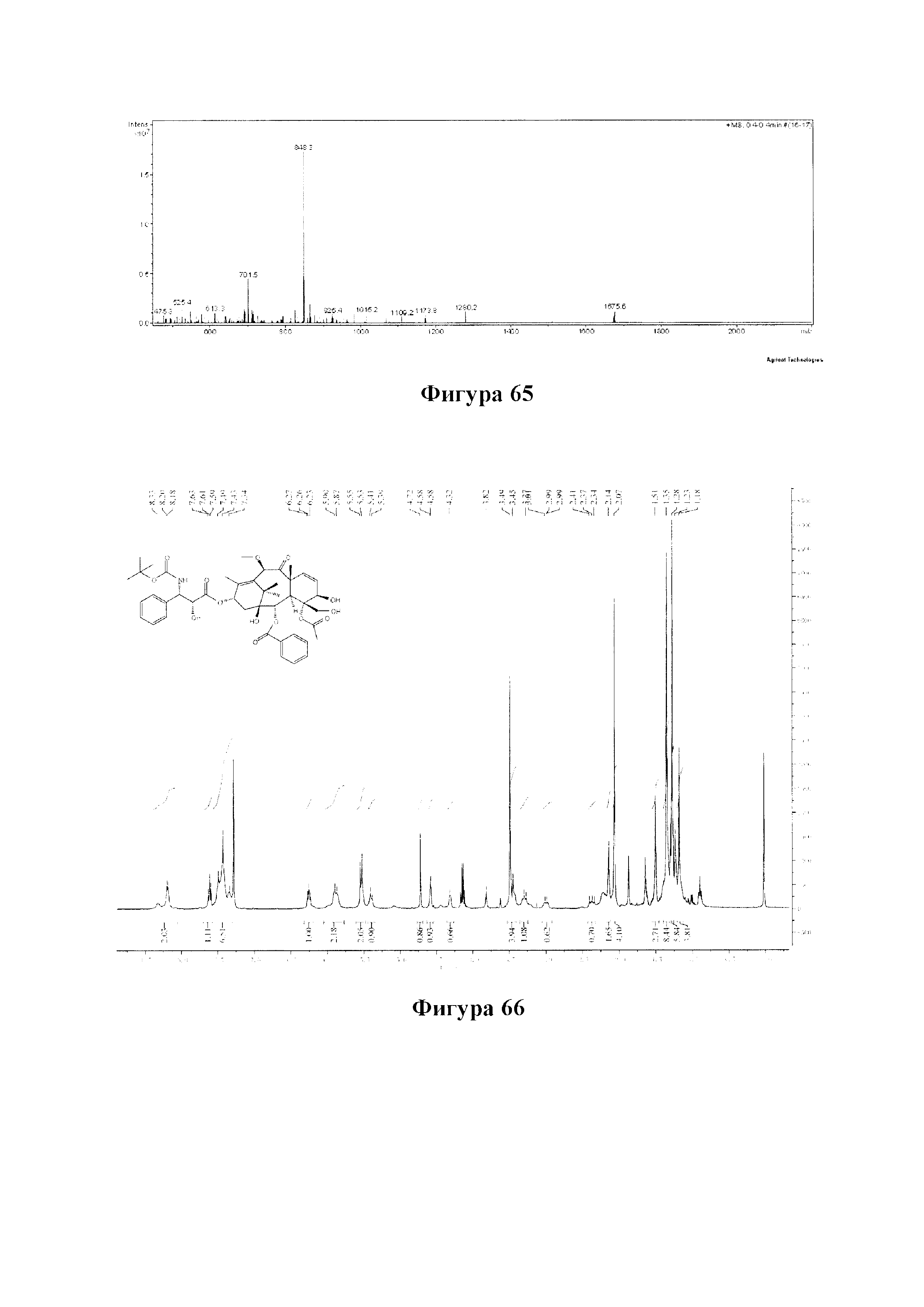
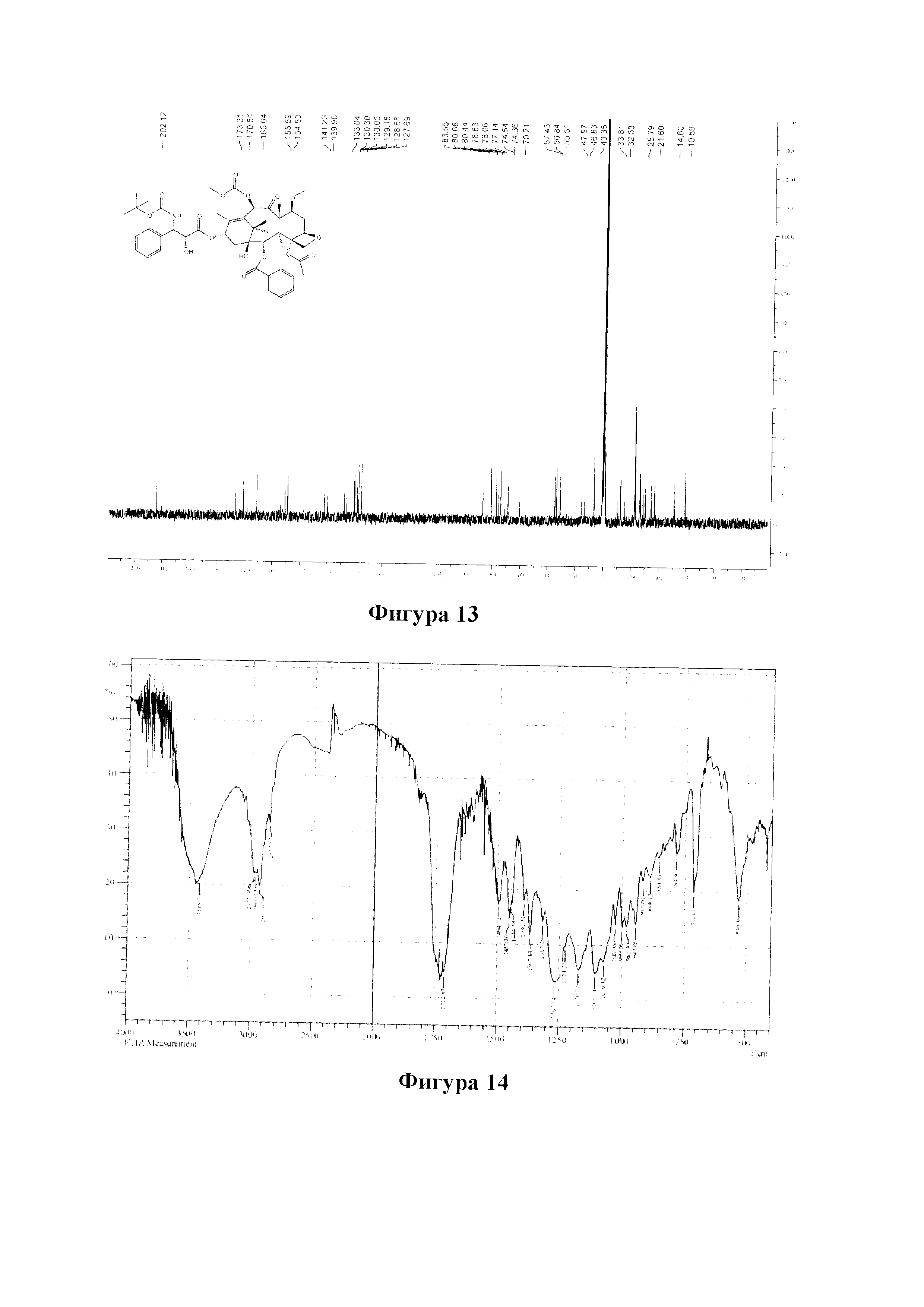

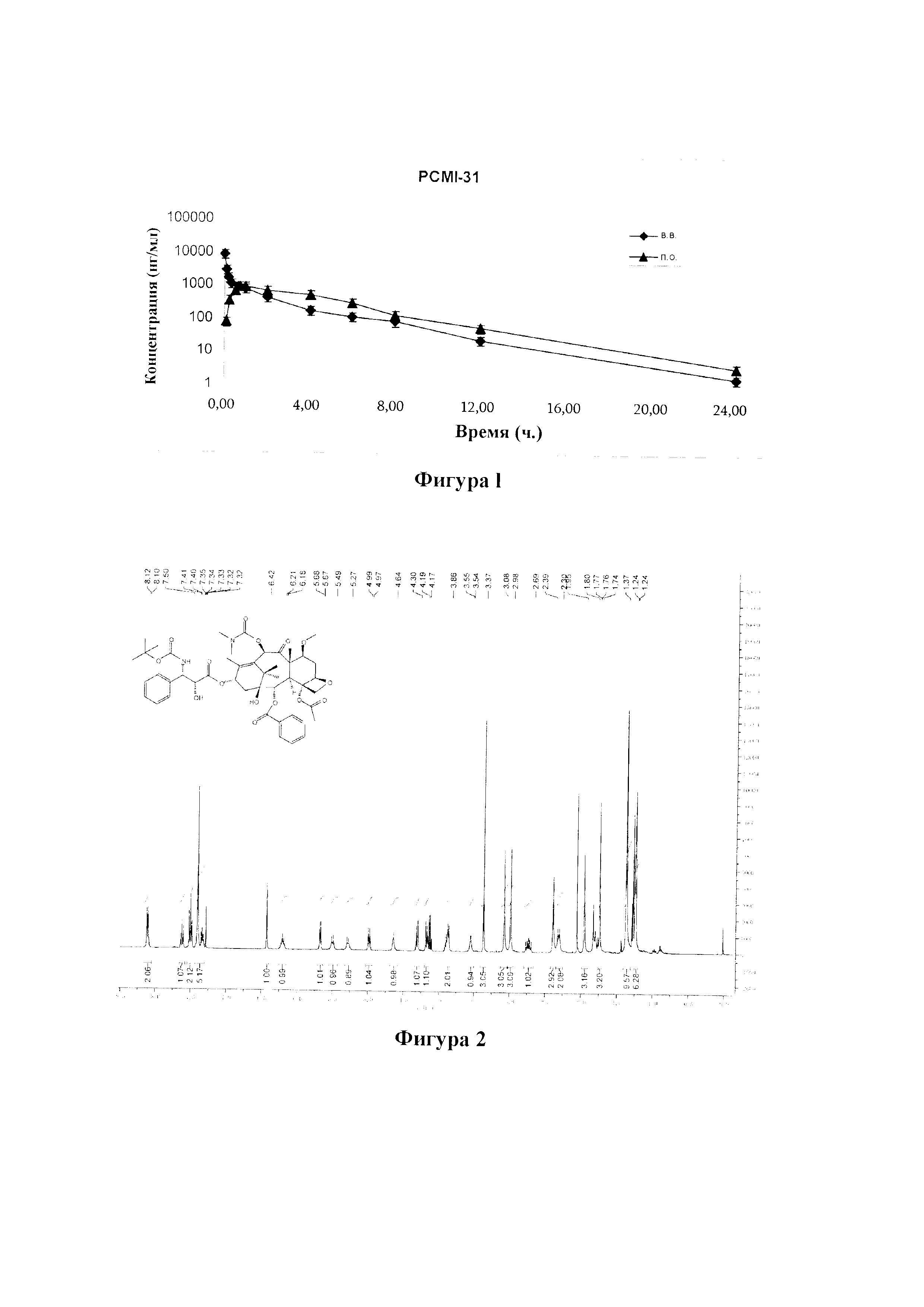
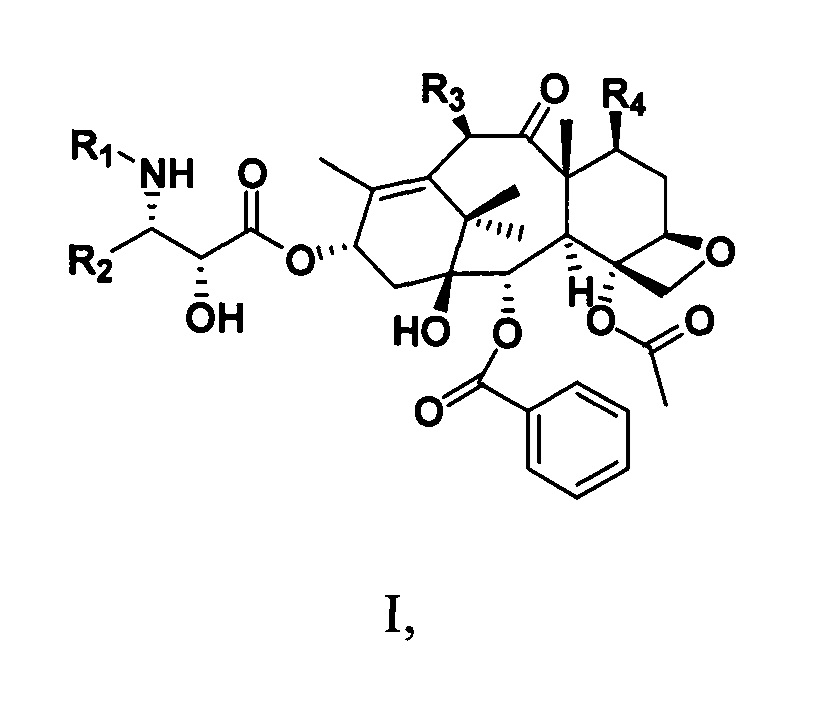
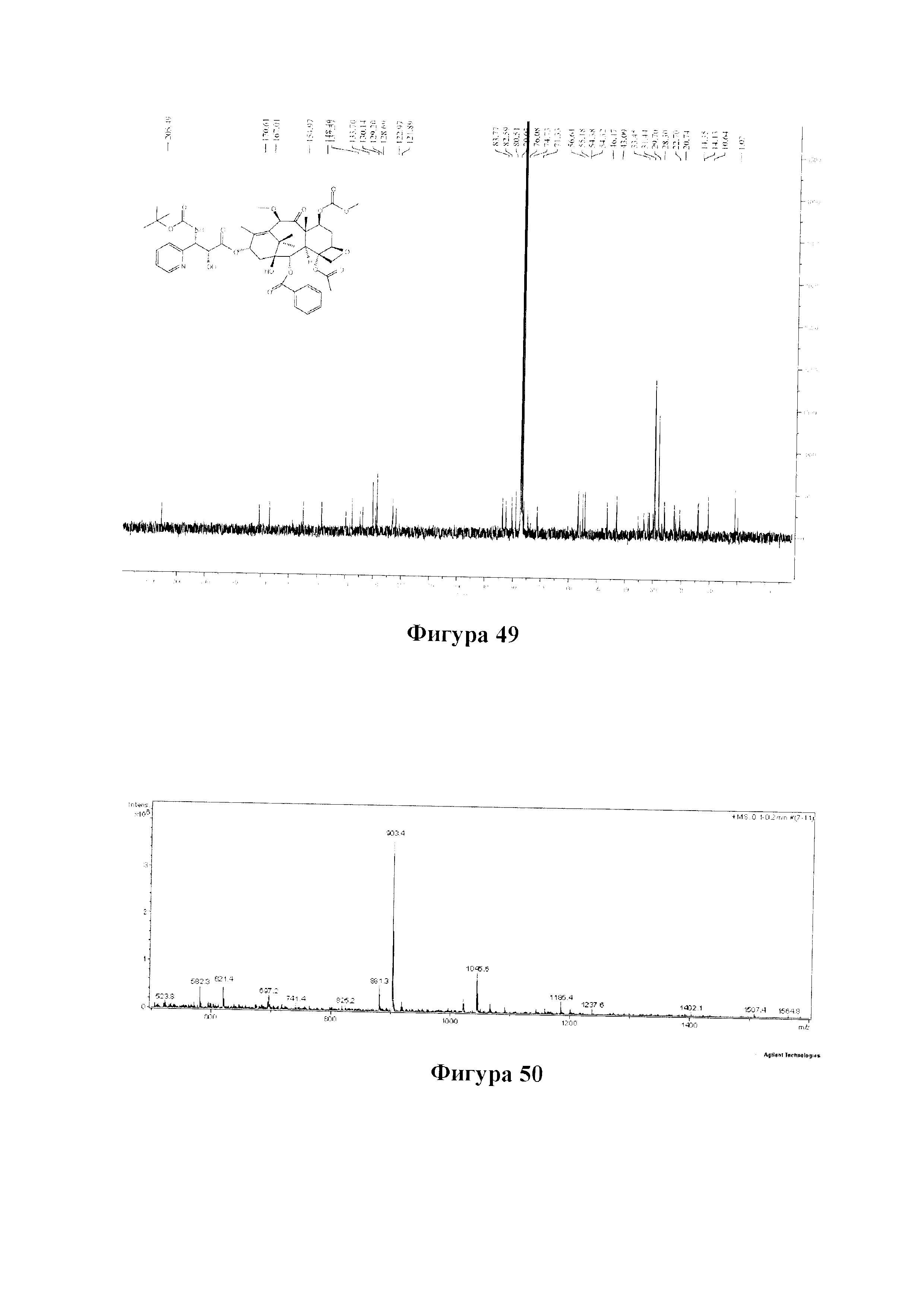
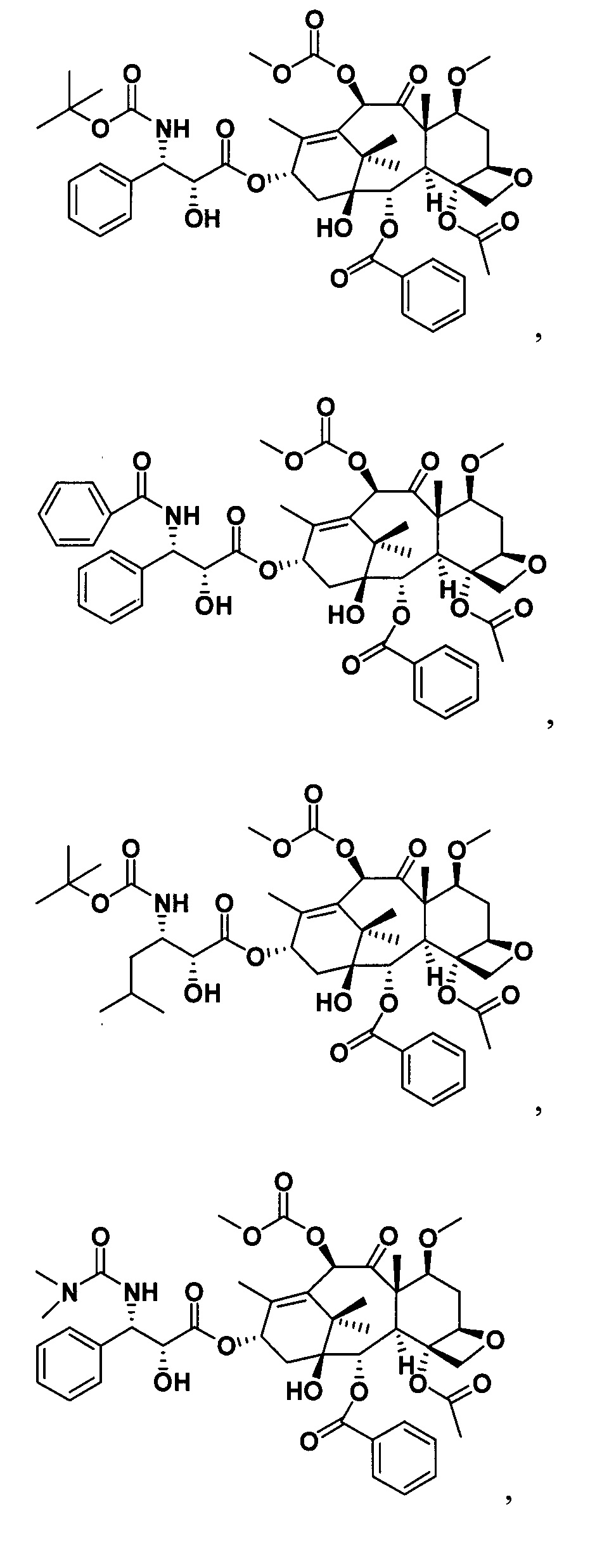
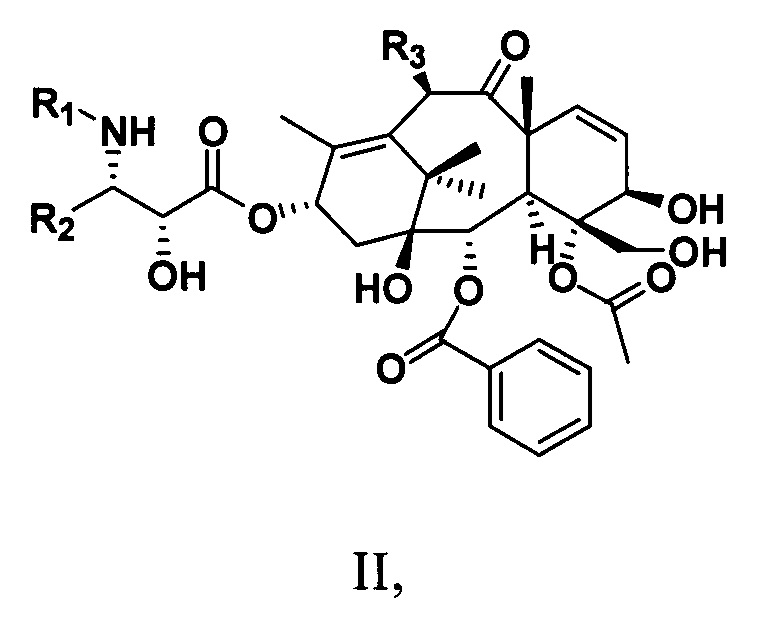

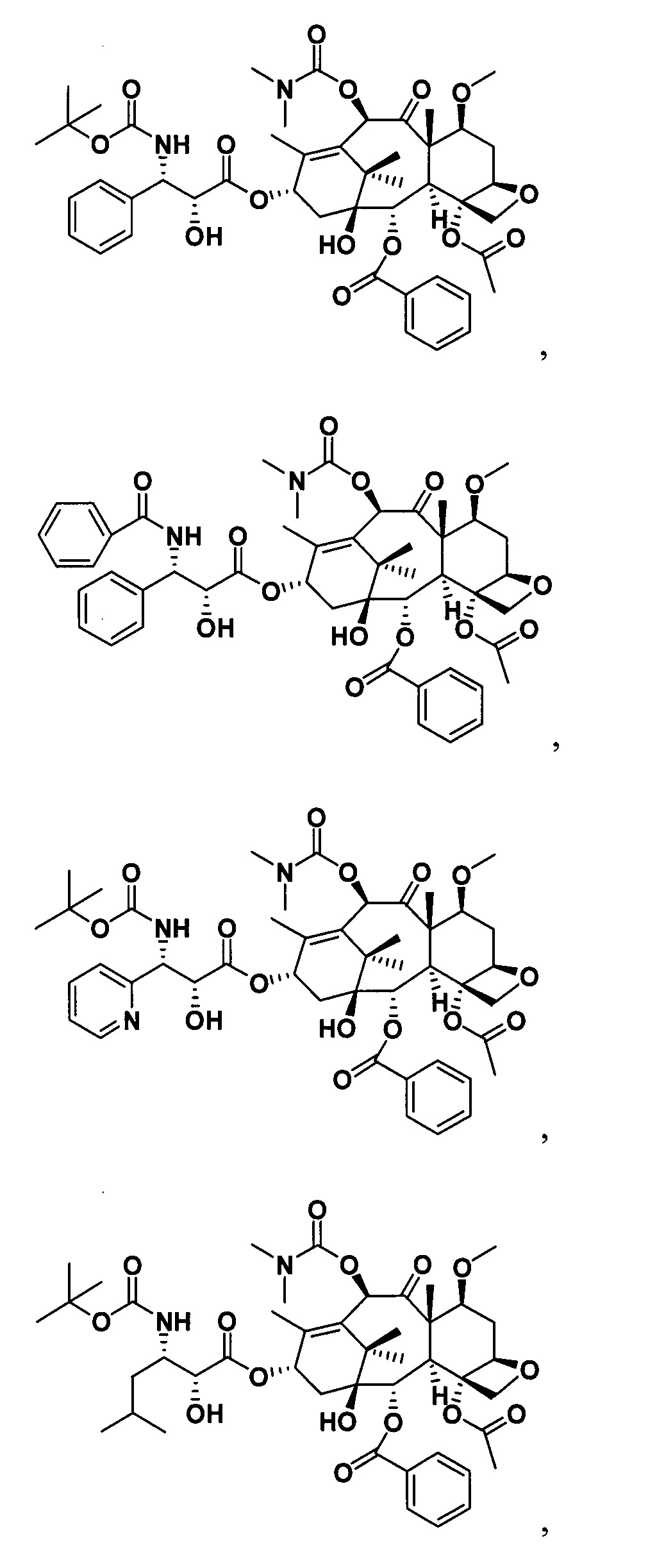

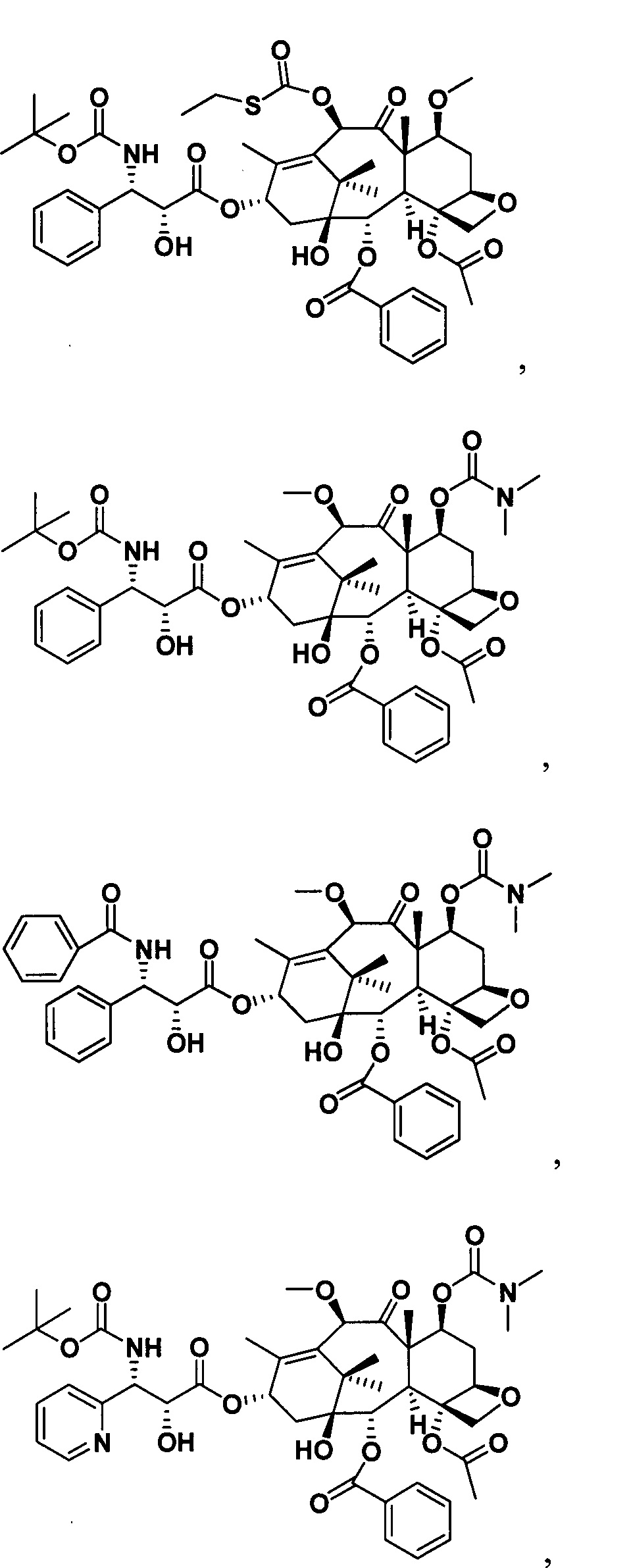
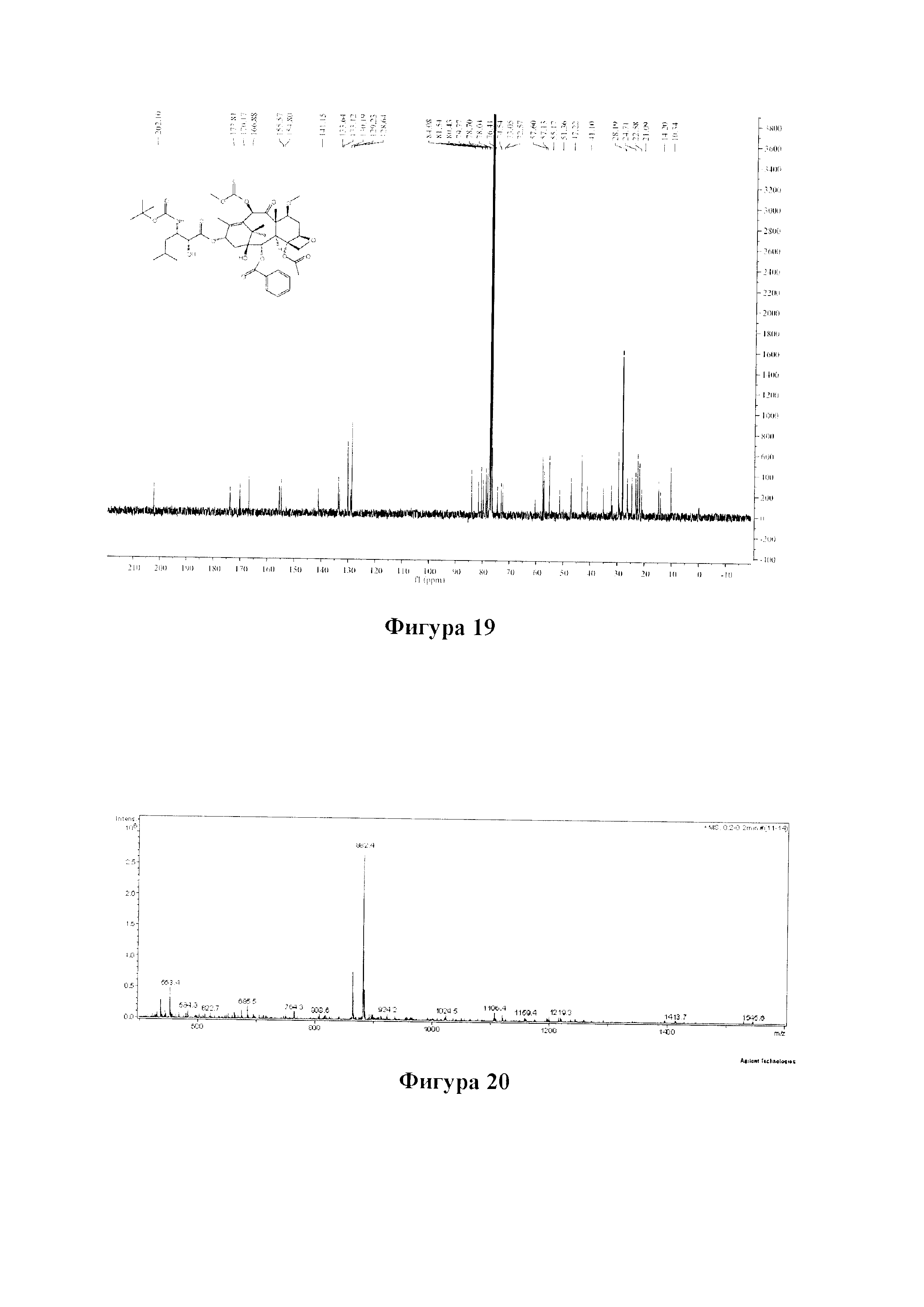
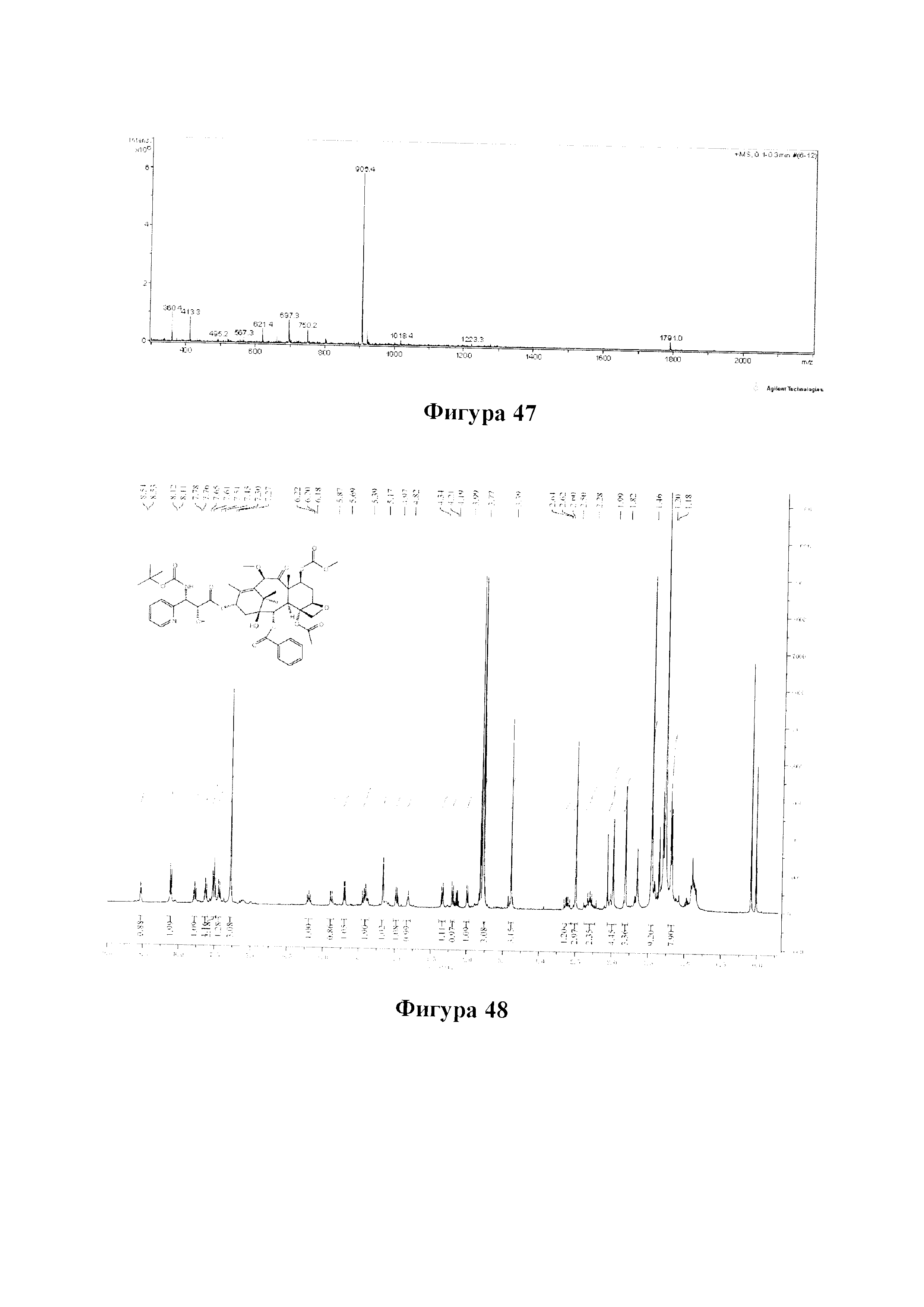
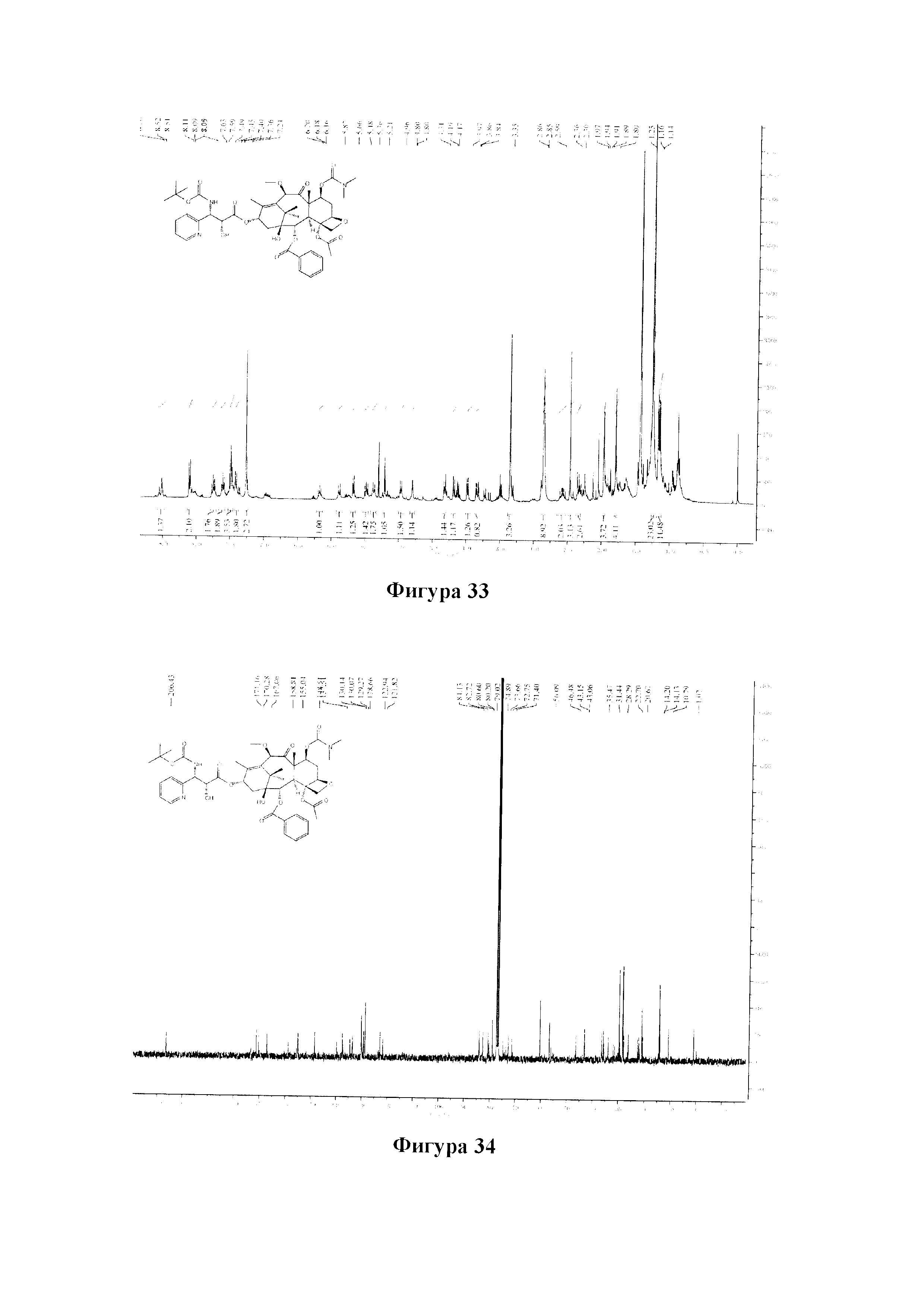
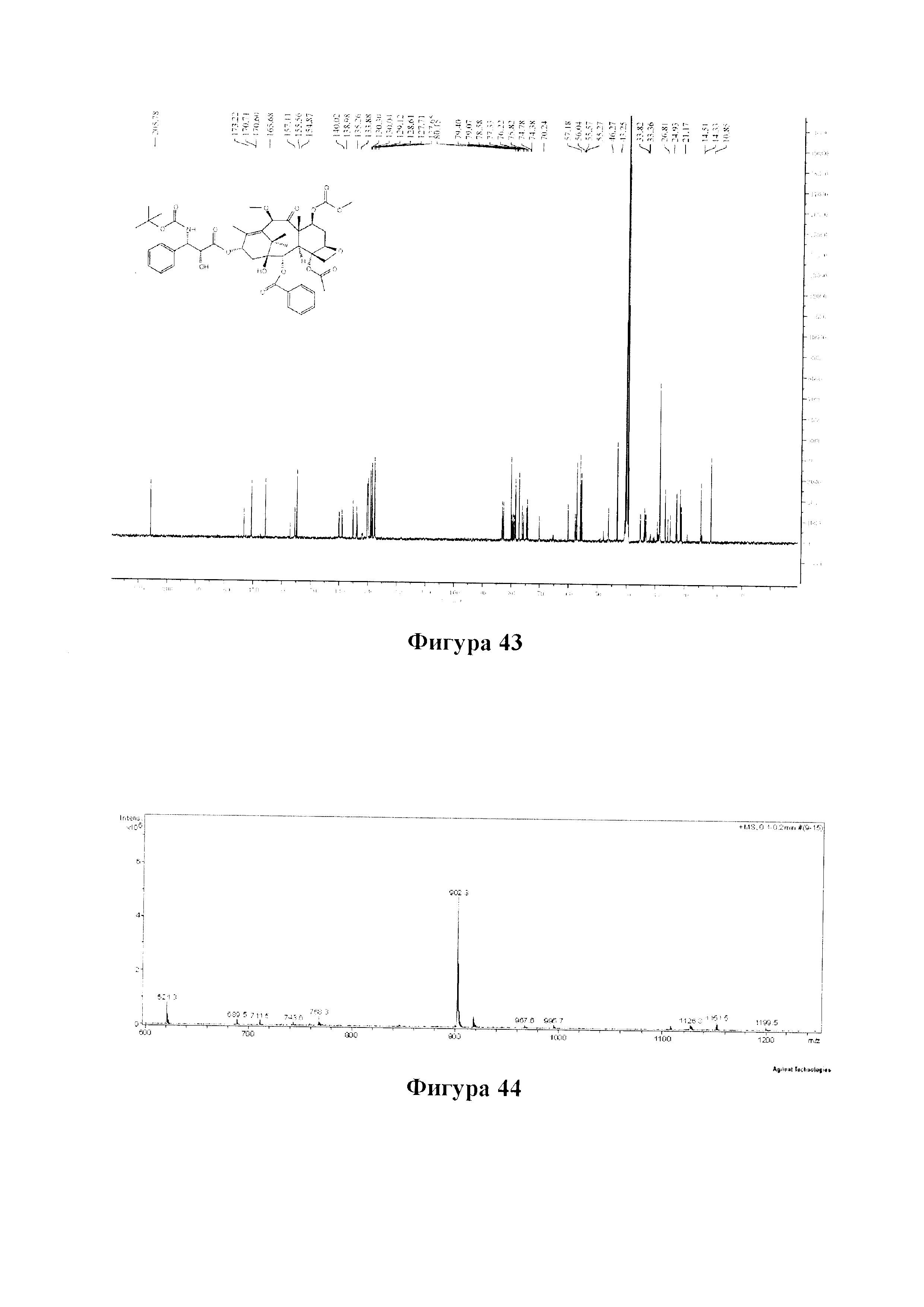
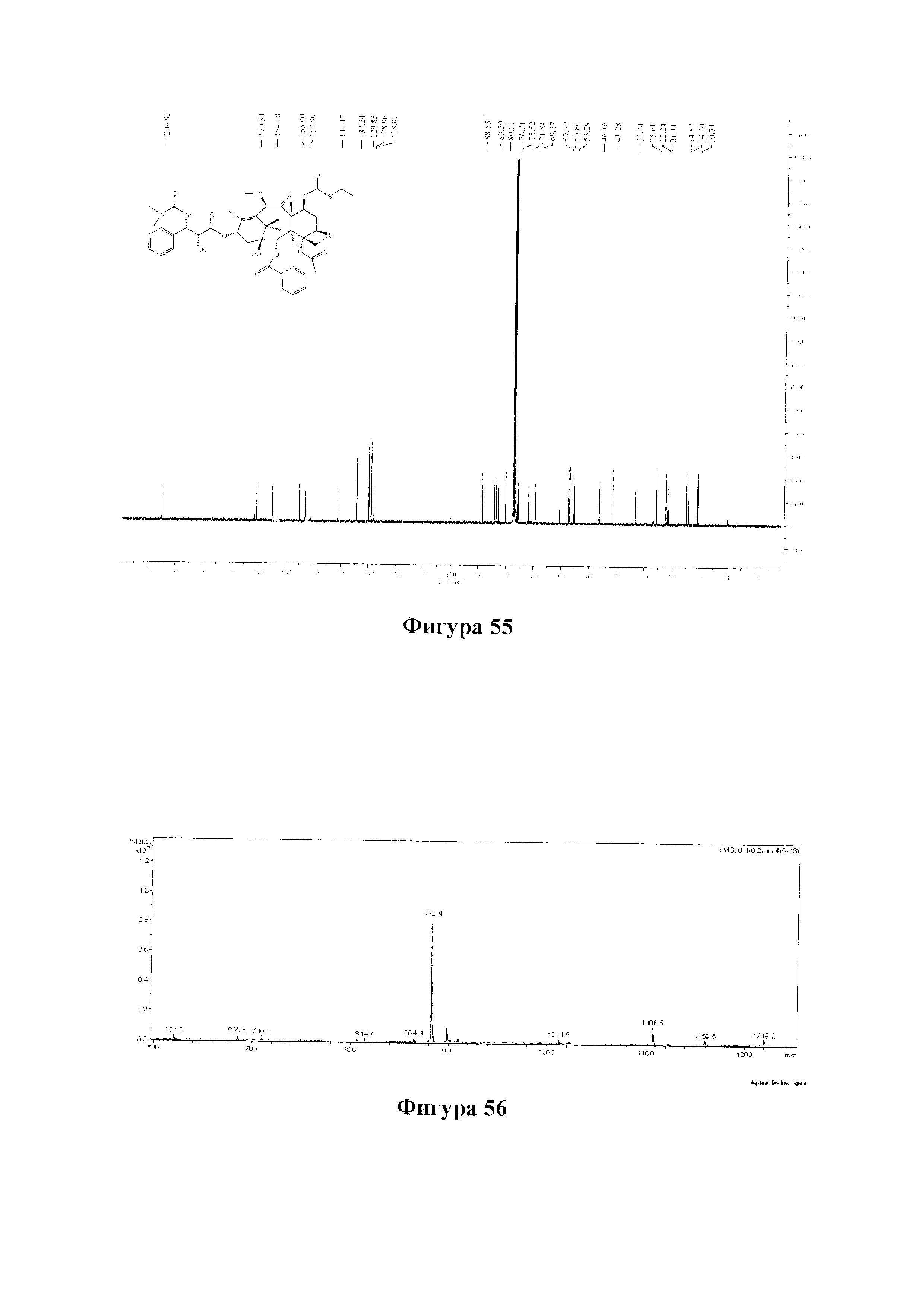

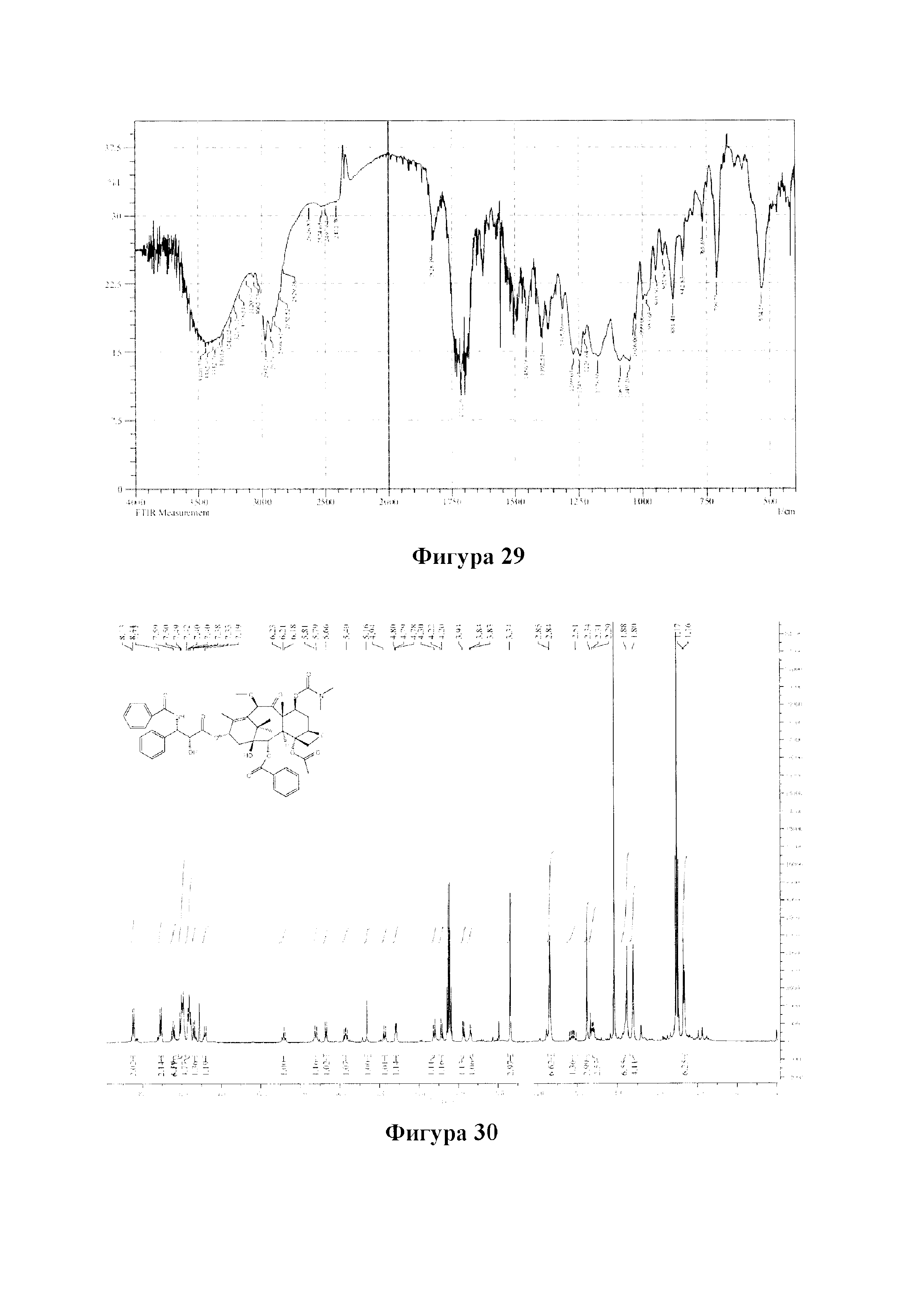

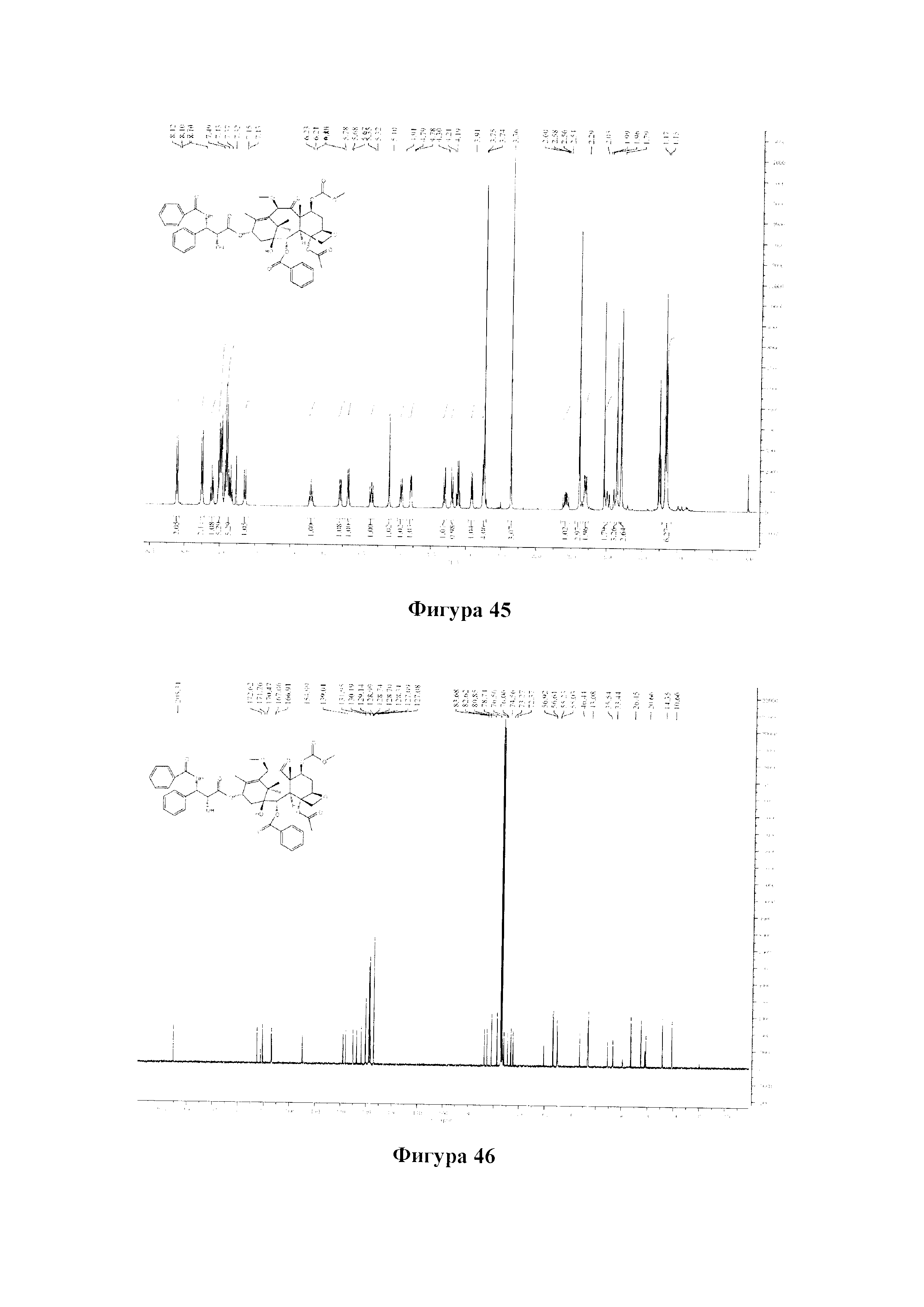
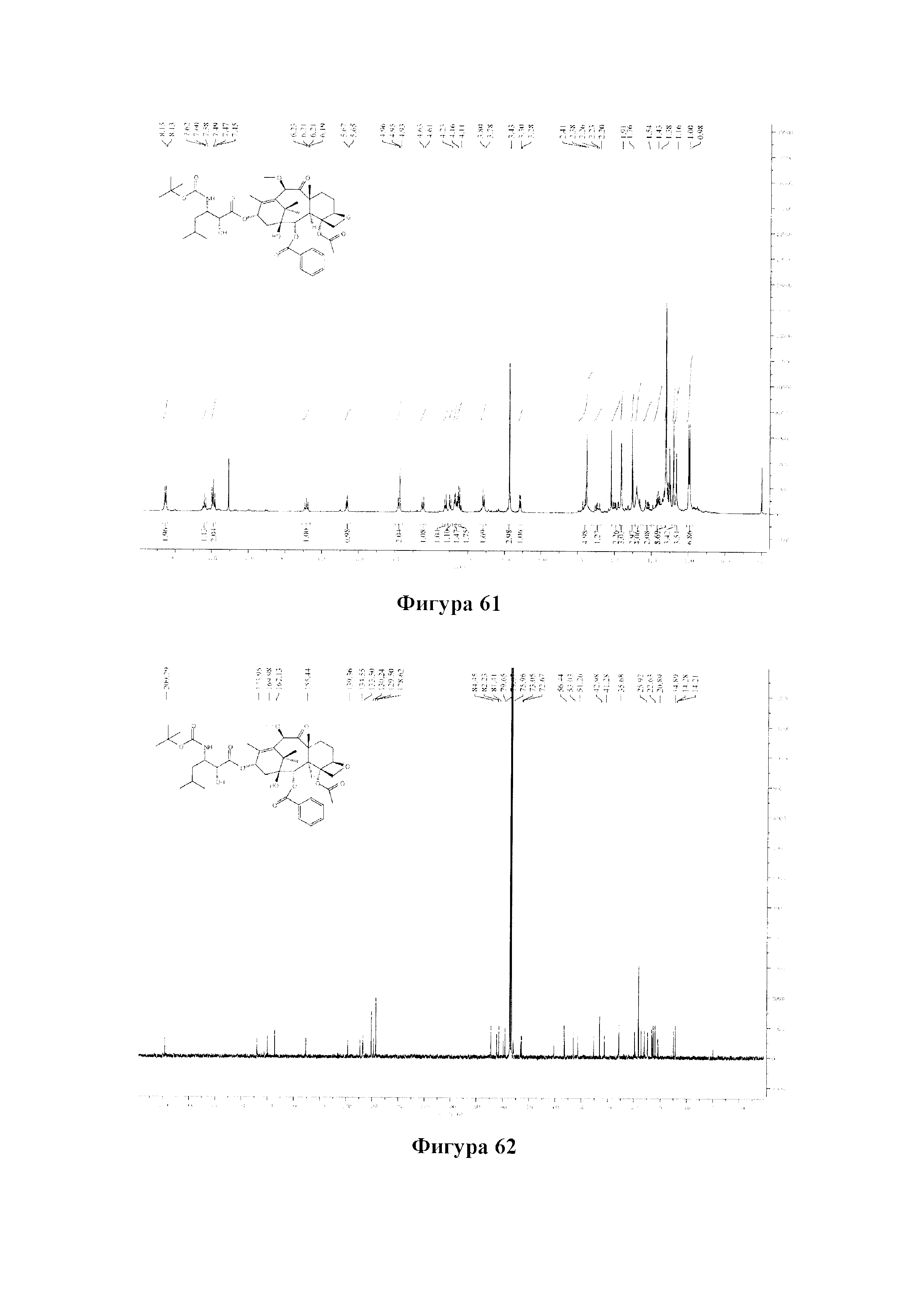
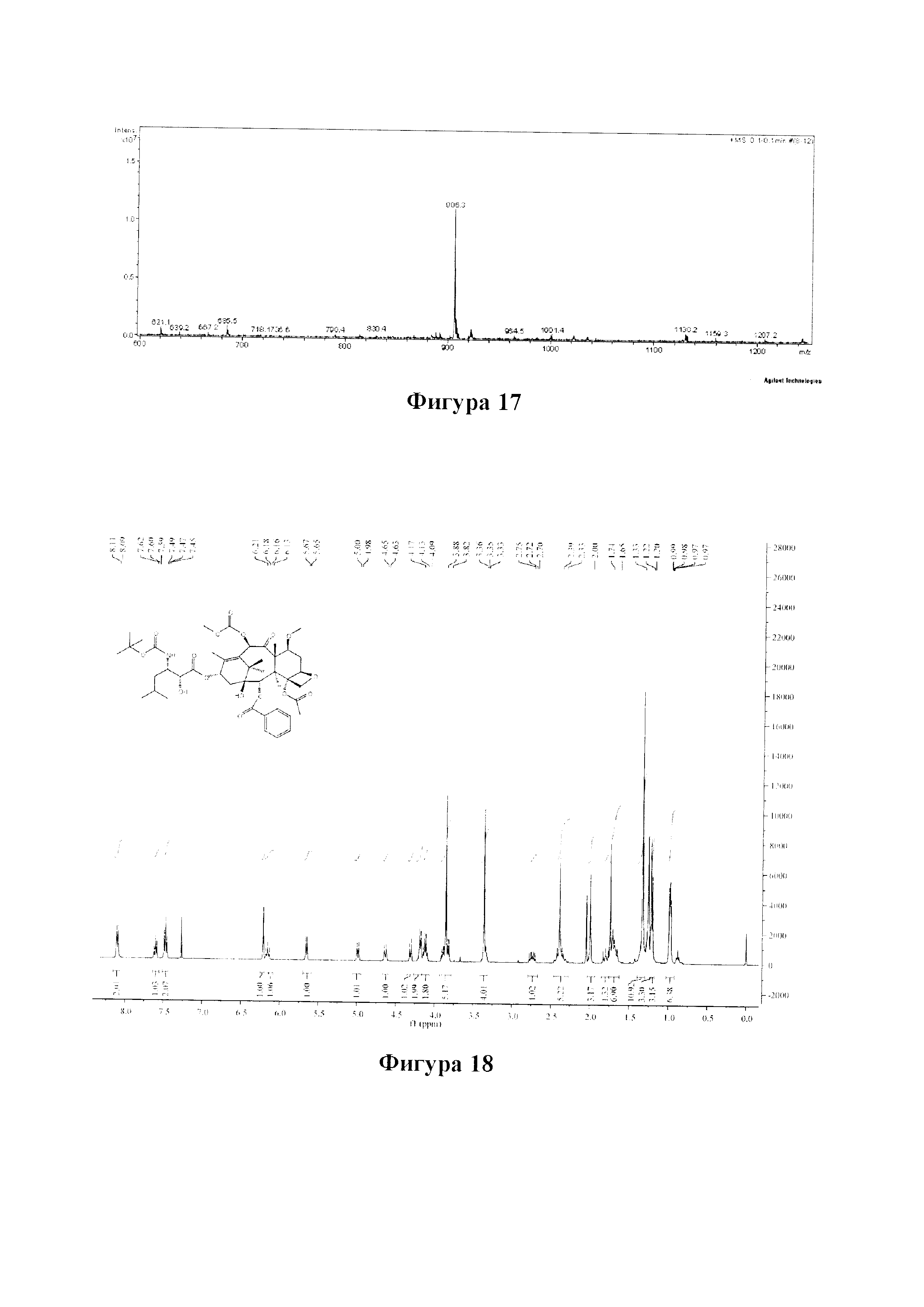
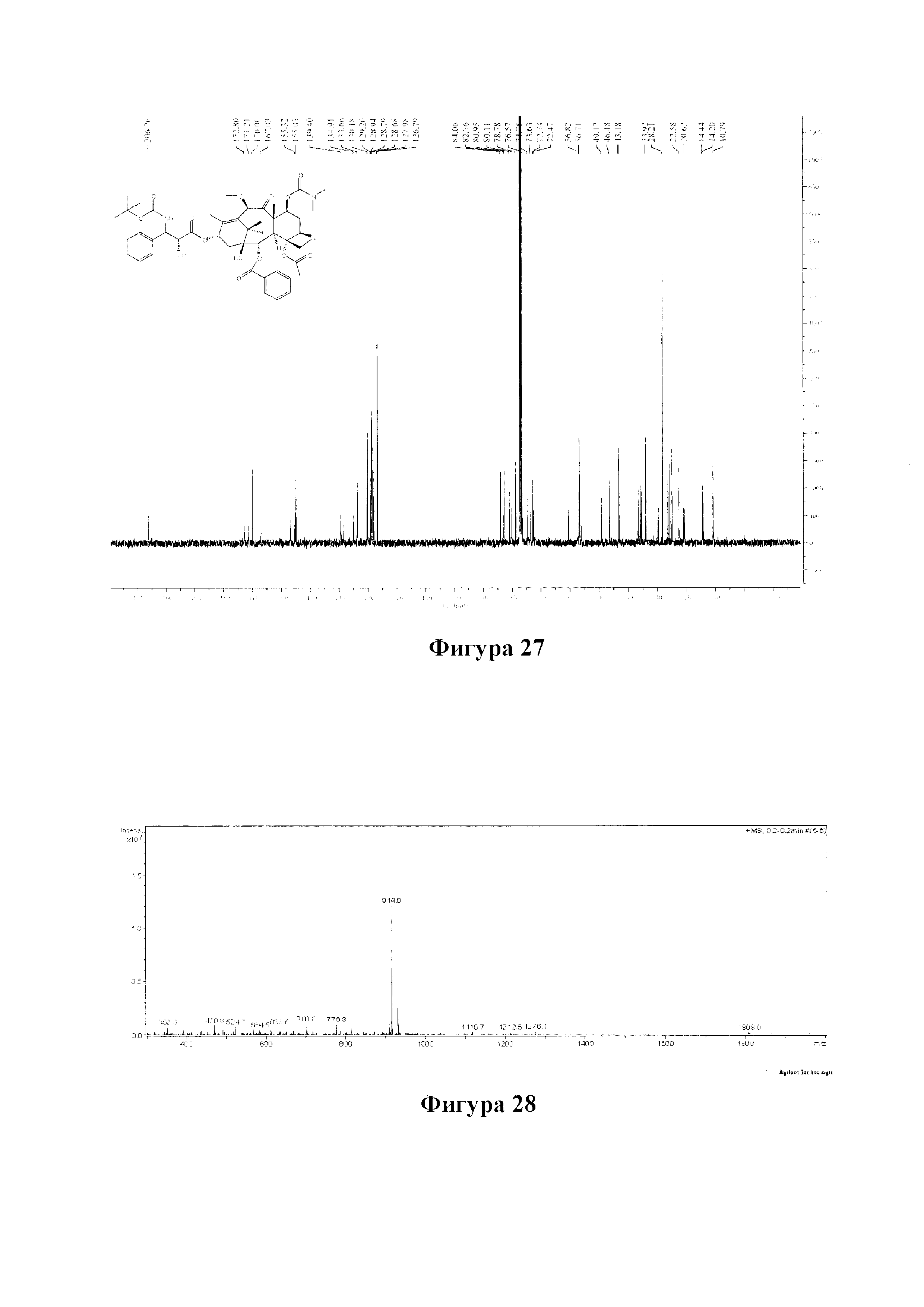
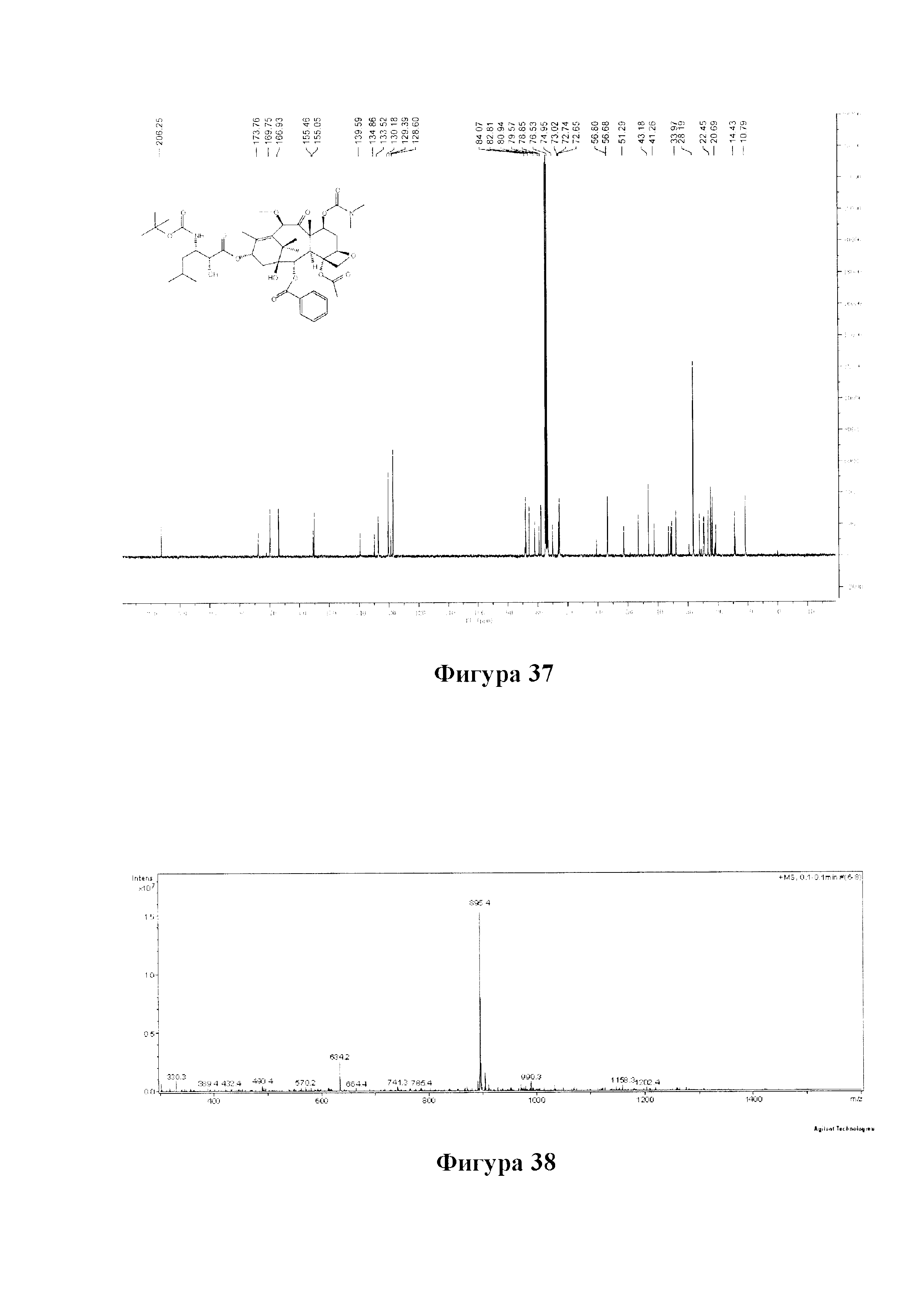
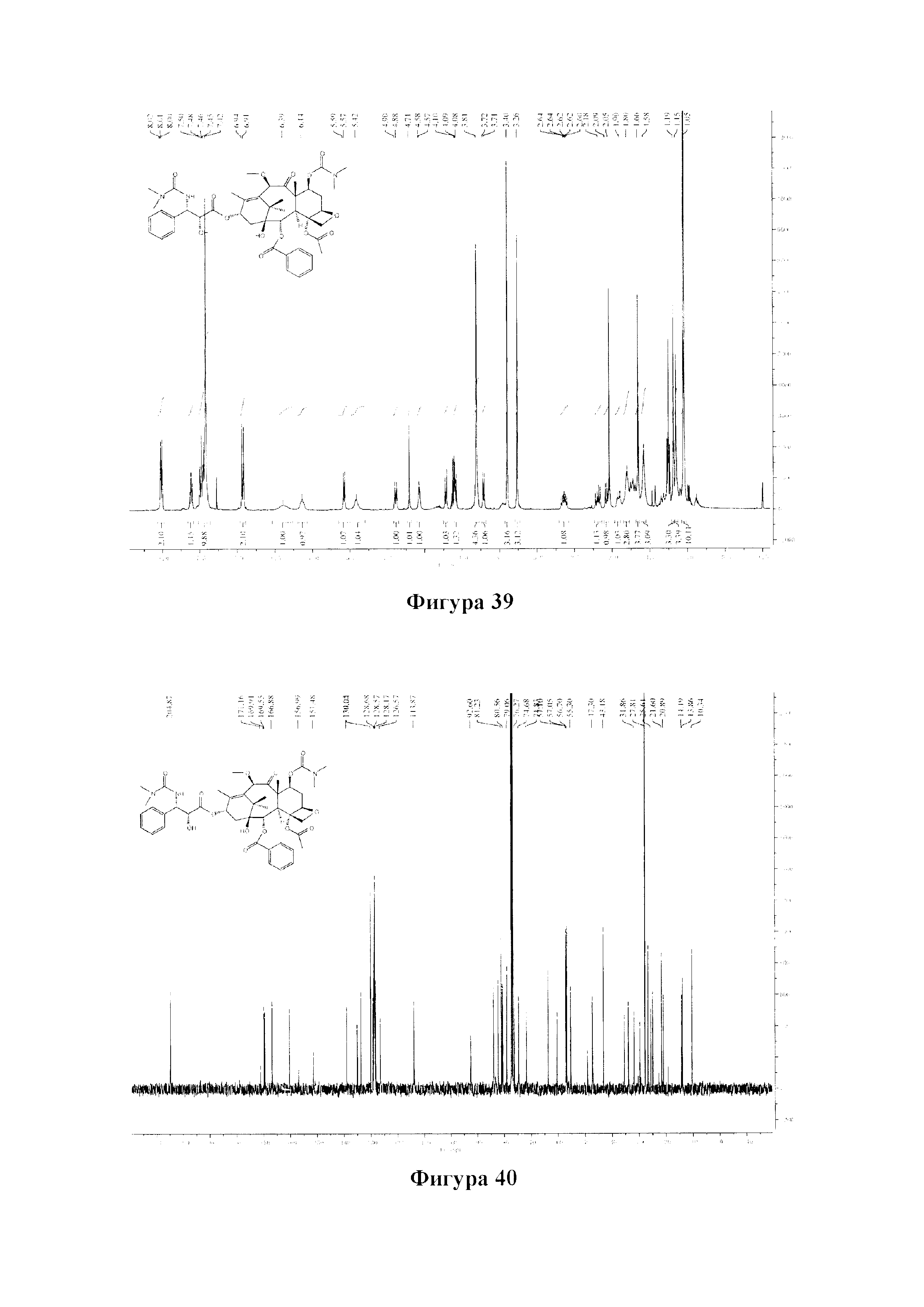
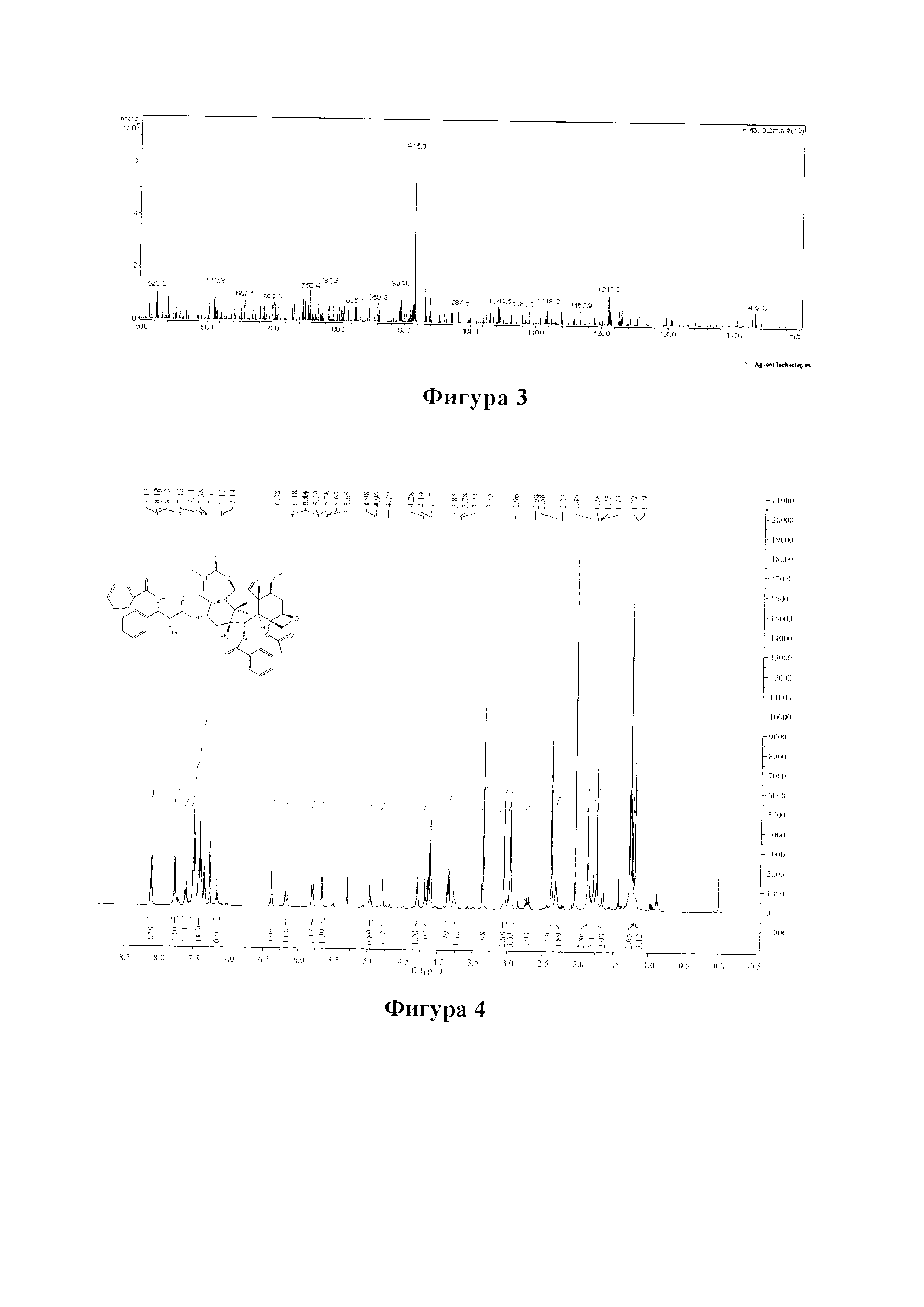
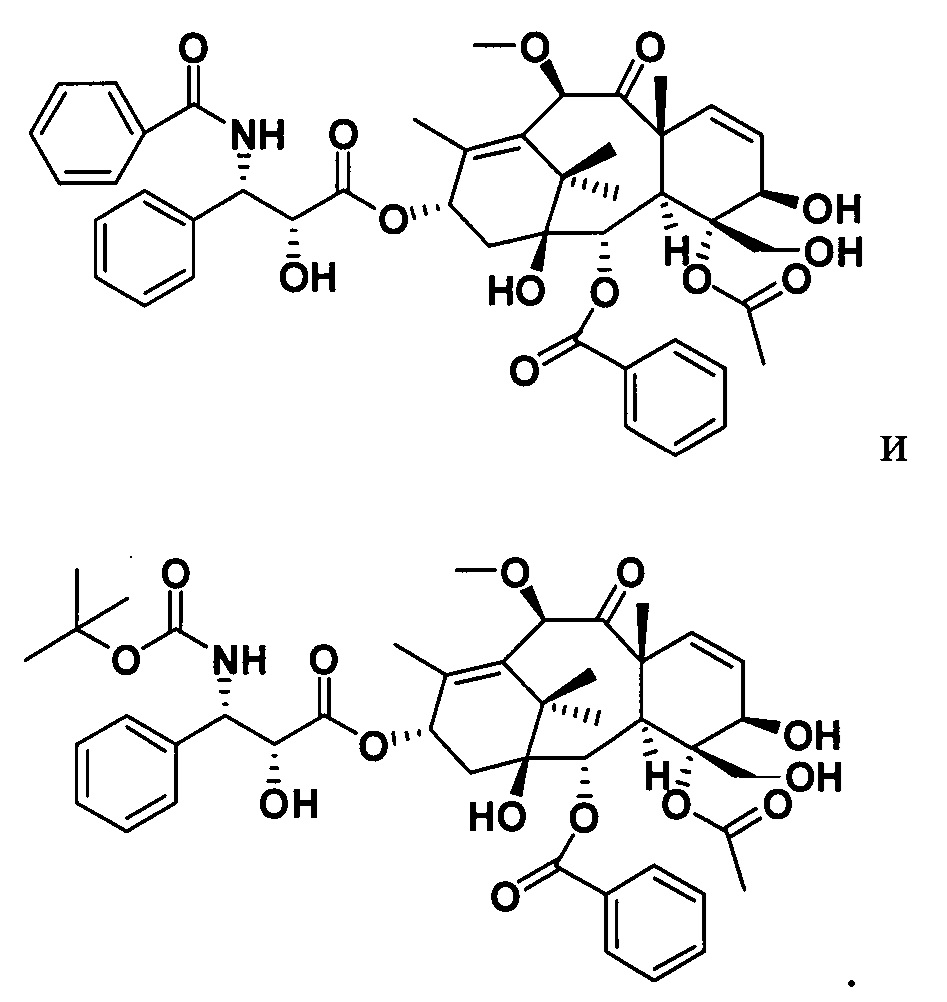
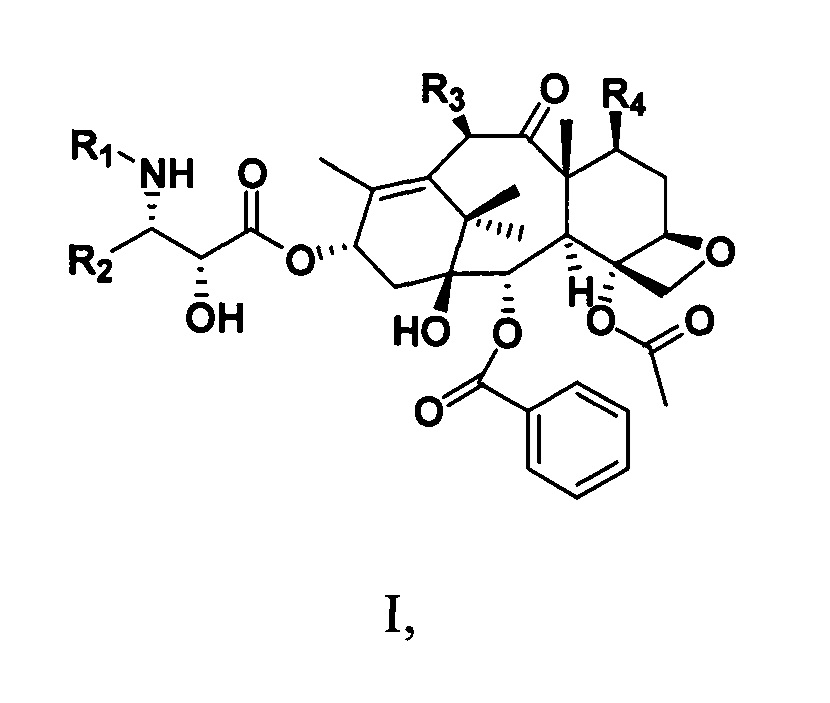
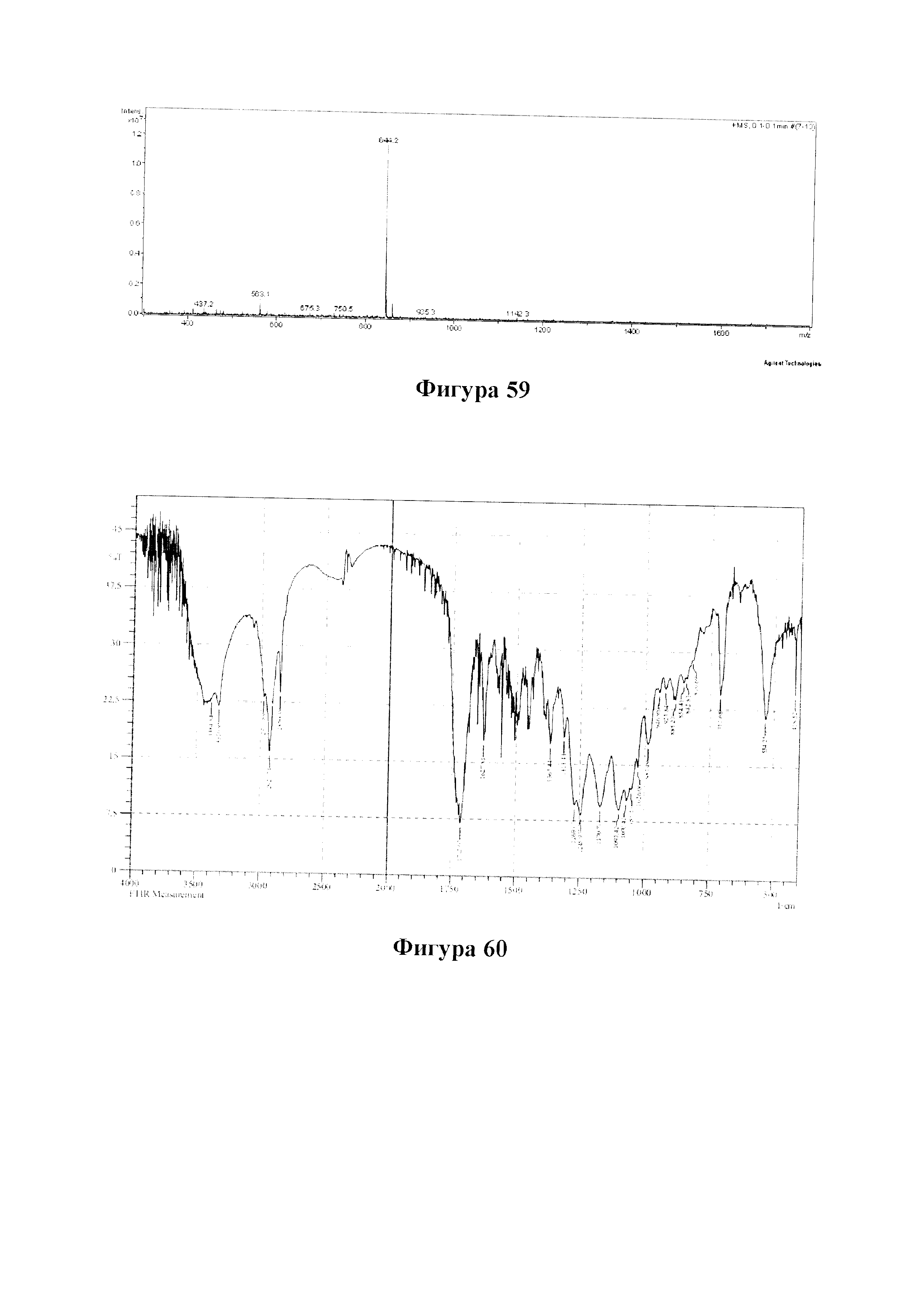
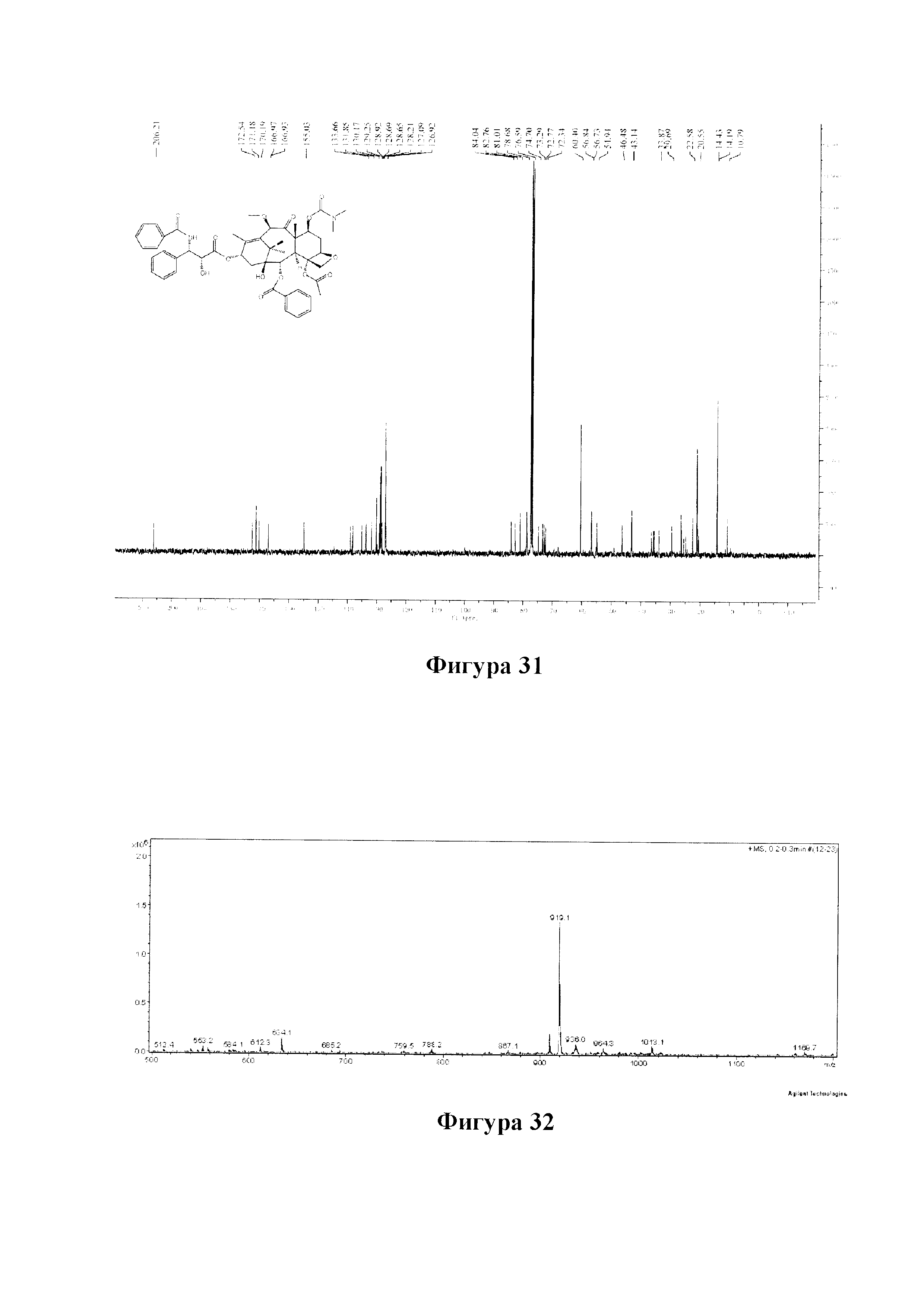
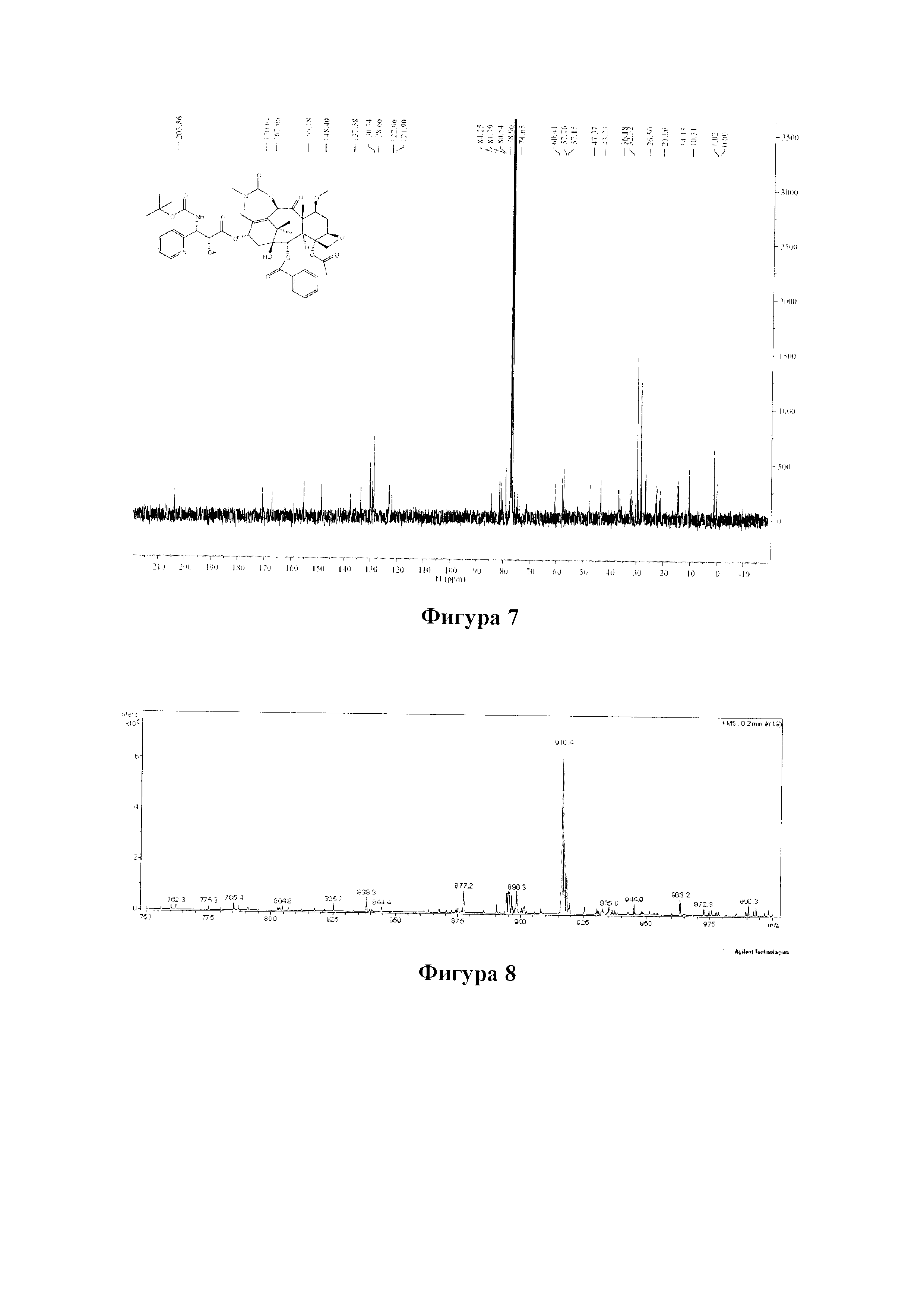
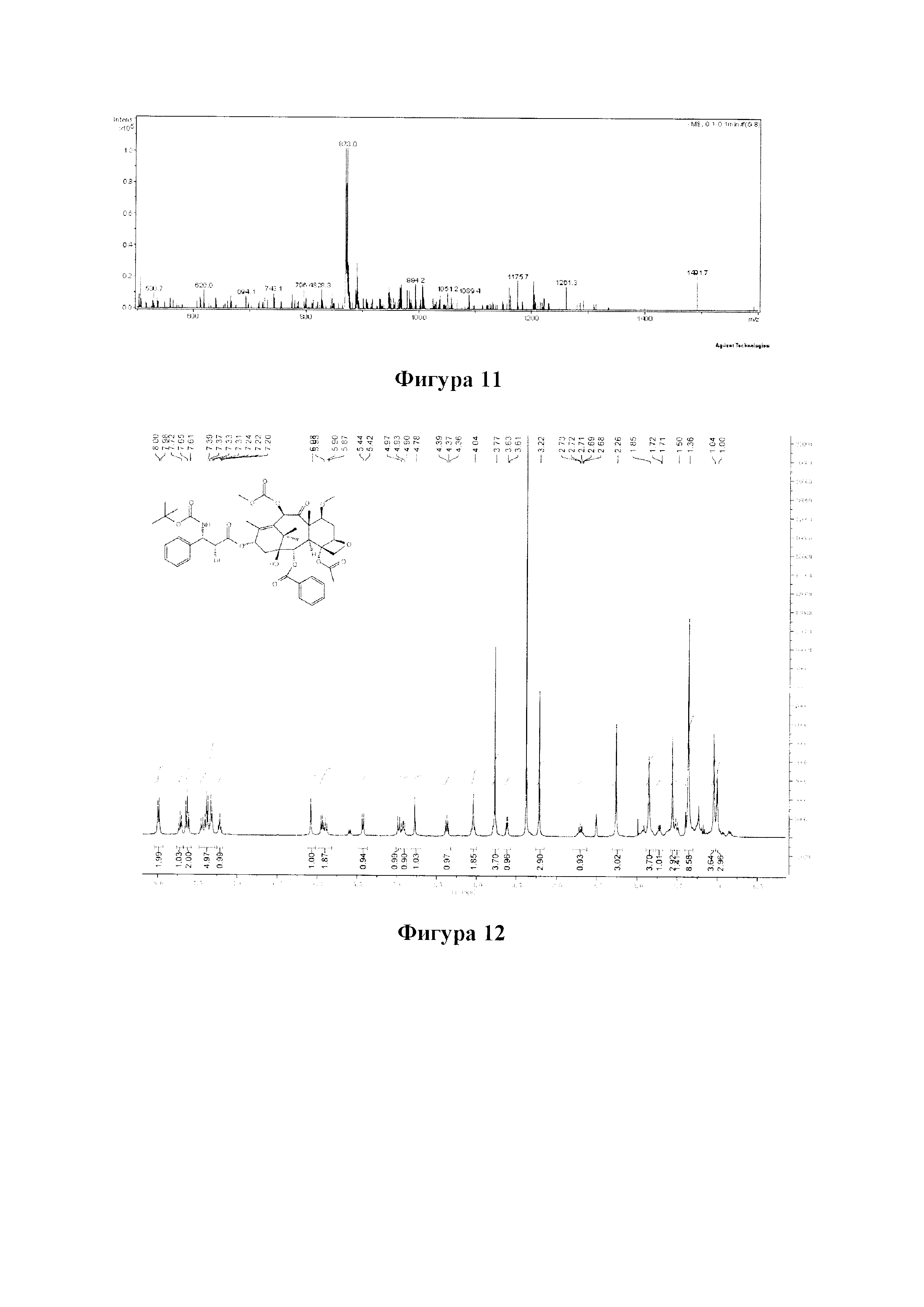
Кристаллические формы темозоломида и способ получения таковых
Таксановое соединение, а также способ его получения и его применение
Способ получения гидрохлорида ревапразана
Производное ксантина
Дейтерированные производные тиенопиперидина, способ их получения и применение
Состав для инъекции на основе жировой эмульсии кабазитаксела, способ его получения и применение
Пролекарство на основе фосфамида монобензилового сложного эфира тенофовира, способ его получения и применение
Способ, устройство и система оптимизации радиоресурсов
Производное замещенного циннамамида, способ его получения и применения
Способ, устройство и система контроля качества услуг интернет-доступа для мобильного терминала
Способ и устройство для распределения контента для просмотра с множеством экранов
Способ и устройство для распределения контента для просмотра с множеством экранов
Способ передачи пакетов, устройство и система связи
Способ и устройство балансировки сетевых ресурсов
Способ управления таблицей потока и соответствующие устройство и система
Новое соединение сальвианоловой кислоты т, способ его получения и его применение
Кристаллические формы темозоломида и способ получения таковых

