Результат интеллектуальной деятельности: БИЦИКЛИЧЕСКИЕ ИНГИБИТОРЫ
Вид РИД
Изобретение
Это изобретение относится к бициклическим производным, которые представляют собой ингибиторы калликреина плазмы крови, и к фармацевтическим композициям, содержащим такие производные, и к их применению.
Уровень техники изобретения
Бициклические производные настоящего изобретения представляют собой ингибиторы калликреина плазмы крови и имеют ряд терапевтических применений, прежде всего, при лечении нарушения проницаемости сосудов сетчатки глаза в связи с диабетической ретинопатией и диабетическим макулярным отеком.
Калликреин плазмы крови представляет собой трипсиноподобную сериновую протеазу, которая может высвобождать кинины из кининогенов (см. K. D. Bhoola et al., "Kallikrein-Kinin Cascade", Encyclopedia of Respiratory Medicine, p483-493; J. W. Bryant et al., "Human plasma kallikrein-kinin system: physiological and biochemical parameters" Cardiovascular and haematological agents in medicinal chemistry, 7, p.234-250, 2009; K. D. Bhoola et al., Pharmacological Rev., 1992, 44, 1; and D. J. Campbell, "Towards understanding the kallikrein-kinin system: insights from the measurement of kinin peptides", Brazilian Journal of Medical and Biological Research 2000, 33, 665-677). Он представляет собой принципиально значимый член внутреннего каскада свертывания крови, тем не менее, его роль в этом каскаде не включает высвобождение брадикинина или ферментативное расщепление. Прекалликреин плазмы крови закодирован одним единственным геном и синтезируется в печени. Его секретируют гепатоциты как неактивный прекалликреин плазмы крови, который циркулирует в плазме крови в виде гетеродимерного комплекса, будучи связанным с высокомолекулярным кининогеном, и который активируется, давая активный калликреин плазмы крови. Кинины представляют собой высокоактивные медиаторы воспалительного процесса, действующие при посредничестве рецепторов, связанных с G-белком, и антагонисты кининов (такие как антагонисты брадикинина) ранее были исследованы в качестве потенциальных терапевтических средств для лечения ряда нарушений (F. Marceau and D. Regoli, Nature Rev., Drug Discovery, 2004, 3, 845-852).
Калликреин плазмы крови, как считается, играет определенную роль в ряде воспалительных нарушений. Наиболее значимый ингибитор калликреина плазмы крови представляет собой серпин, ингибитор С1 эстеразы. Пациенты, у которых имеется генетически обусловленная недостаточность ингибитора C1 эстеразы, страдают от врожденного ангионевротического отека (HAE), что в результате выражается в виде периодического опухания лица, рук, горла, желудочно-кишечного тракта и гениталий. Волдыри, образующиеся во время приступа обострения, содержат высокие уровни калликреина плазмы крови, который отщепляет высоккомолекулярный кининоген, высвобождающий брадикинин, что приводит к повышенной сосудистой проницаемости. Лечение посредством увеличения количества белкового ингибитора калликреина плазмы крови, как было показано, эффективно излечивает HAE путем предотвращения высвобождения брадикинина, который вызывает повышение сосудистой проницаемости (A. Lehmann "Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery" Expert Opin. Biol. Ther. 8, p.1187-99).
Система калликреин-кинин в плазме крови присутствует в аномальном избытке у пациентов с прогрессирующим диабетическим макулярным отеком. Совсем недавно было опубликовано, что калликреин плазмы крови способствует сосудистой дисфункции сетчатки глаза у страдающих диабетом крыс (A. Clermont et al. "Plasma kallikrein mediates retinal vascular dysfunction and induces retinal thickening in diabetic rats" Diabetes, 2011, 60, p.1590-98). Кроме того, введение ингибитора ASP-440 калликреина плазмы крови улучшает как проницаемость сосудов сетчатки глаза, так и кровоток сетчатки глаза в случае патологических изменений у страдающих диабетом крыс. В этой связи, ингибитор калликреина плазмы крови следует практически применять при лечении для снижения проницаемости сосудов сетчатки глаза, в связи с диабетической ретинопатией и диабетическим макулярным отеком.
Другие осложнения диабета, такие как геморрагический инсульт, нефропатия, кардиомиопатия и нейропатия, которые связаны с калликреином плазмы крови, также можно рассматривать в качестве мишеней ингибитора калликреина плазмы крови.
Синтетические и небольшие молекулы ингибиторов калликреина плазмы крови ранее были описаны, например, Garrett et al. ("Peptide aldehyde…." J. Peptide Res. 52, p.62-71 (1998)), T. Griesbacher et al. ("Involvement of tissue kallikrein but not plasma kallikrein in the development of symptoms mediated by endogenous kinins in acute pancreatitis in rats" British Journal of Pharmacology 137, p.692-700 (2002)), Evans ("Selective dipeptide inhibitors of kallikrein " WO03/076458), Szelke et al. ("Kininogenase inhibitors" WO92/04371), D. M. Evans et al. (Immunolpharmacology, 32, p.115-116 (1996)), Szelke et al. ("Kininogen inhibitors" WO95/07921), Antonsson et al. ("New peptides derivatives" WO94/29335), J. Corte et al. (”Six membered heterocycles useful as serine protease inhibitors” WO2005/123680), J. Stürzbecher et al. (Brazilian J. Med. Biol. Res 27, p.1929-34 (1994)), Kettner et al. (US 5,187,157), N. Teno et al. (Chem. Pharm. Bull. 41, p.1079-1090 (1993)), W. B. Young et al. ("Small molecule inhibitors of plasma kallikrein" Bioorg. Med. Chem. Letts. 16, p.2034-2036 (2006)), Okada et al. ("Development of potent and selective plasmin and plasma kallikrein inhibitors and studies on the structure-activity relationship" Chem. Pharm. Bull. 48, p.1964-72 (2000)), Steinmetzer et al. ("Trypsin-like serine protease inhibitors and their preparation and use" WO08/049595), Zhang et al. ("Discovery of highly potent small molecule kallikrein inhibitors" Medicinal Chemistry 2, p.545-553 (2006)), Sinha et al. ("Inhibitors of plasma kallikrein " WO08/016883), Shigenaga et al. (“Plasma Kallikrein Inhibitors” WO2011/118672), и Kolte et al. (“Biochemical characterization of a novel high-affinity and specific kallikrein inhibitor”, British Journal of Pharmacology (2011), 162(7), 1639-1649). А также, в публикации Steinmetzer et al. (“Serine protease inhibitors” WO2012/004678) описываются циклизированные пептидные аналоги, которые представляют собой ингибиторы человеческого плазмина и калликреина плазмы крови.
К настоящему времени, ни один низкомолекулярный синтетический ингибитор калликреина плазмы крови не был зарегистрирован как разрешенный к применению в медицине. Молекулы, описанные в соответствии с известным уровнем техники, имеют недостатки из-за ограничений их применимости, такие как низкая селективность в отношении соответствующих ферментов, таких как KLK1, тромбин и другие сериновые протеазы, и недостаточная пригодность для перорального применения. Большие высокомолекулярные белковые ингибиторы калликреина плазмы крови привносят риски анафилактических реакций, как сообщалось в отношении лекарственного препарата Ecallantide. Таким образом, из изложенного следует, что существует потребность в соединениях, которые селективно ингибируют калликреин плазмы крови, так, что при этом не вызывают анафилаксию и при этом являются пригодными для перорального применения. Кроме того, подавляющее большинство молекул, известных из существующего уровня техники, характеризуются сильной способностью функциональных групп гуанидина или амидина к поляризации и ионизации. Общеизвестно, что такие реакционные способности функциональных групп могут ограничивать хорошую проникающую способность и, вследствие этого, пригодность для перорального применения. Например, сообщалось в публикации Tamie J. Chilcote and Sukanto Sinha (“ASP-634: An Oral Drug Candidate for Diabetic MacularEdema”, ARVO 2012 May 6th - May 9th, 2012, Fort Lauderdale, Florida, Presentation 2240), что ASP-440, бензамидин, имеет недостатки из-за низкой пригодности для перорального применения. Также сообщалось, что абсорбцию можно улучшить посредством создания пролекарства, такого как ASP-634. Однако, общеизвестно, что пролекарствам присущи некоторые недостатки, например, плохая химическая стабильность и потенциальная токсичность из-за инертного носителя или из-за непредвиденных метаболитов. В другом сообщении индоламиды заявлены в качестве соединений, способных преодолеть проблемы, связанные с плохим сохранением лекарств или с несоответствием требованиям всасывания, распределения, метаболизма и выведения токсических соединений (ADME-tox), и физико-химическими свойствами, хотя никакого ингибирования в отношении калликреина плазмы крови не выявлено или не заявлено (Griffioen et al, “Indole amide derivatives and related compounds for use in the treatment of neurodegenerative diseases”, WO2010, 142801).
BioCryst Pharmaceuticals Inc. сообщили об открытии пригодного для перорального применения ингибитора калликреина плазмы крови BCX4161 (“BCX4161, An Oral Кallikrein Inhibitor: Safety and Pharmacokinetic Results Of a Phase 1 Study In Healthy Volunteers”, Journal of Allergy and Clinical Immunology, Volume 133, Issue 2, Supplement, February 2014, page AB39 и “A Simple, Sensitive and Selective Fluorogenic Assay to Monitor Plasma Кallikrein Inhibitory Activity of BCX4161 in Activated Plasma”, Journal of Allergy and Clinical Immunology, Volume 133, Issue 2, Supplement February 2014, page AB40). Однако, дозы для человека довольно велики, в настоящее время проводится тестирование в рамках проведения ранней стадии клинического исследования при введении доз 400 мг три раза в сутки.
Существует всего несколько сообщений об ингибиторах калликреина плазмы крови, которые не имеют в качестве характерного признака функциональные группы гуанидина или амидина. Один из примеров представляет собой публикация Brandl et al. (“N-((6-amino-pyridin-3-yl)methyl)-heteroaryl-carboxamides as inhibitors of plasma kallikrein” WO2012/017020), в которой описаны соединения, имеющие аминопиридиновую функциональную группу. Эффективность при пероральном приеме на модели крыс продемонстрирована при достаточно высоких дозах, 30 мг/кг и 100 мг/кг, но не опубликованы данные о фармакокинетическом профиле. Таким образом, все еще не ясно, будут ли такие соединения обеспечивать достаточную возможность перорального применения или эффективность действия для продвижения в клинику. Другие примеры представляют собой публикации Brandl et al. (“Aminopyridine derivatives as plasma kallikrein inhibitors” WO2013/111107) и Flohr et al. (“5-membered heteroarylcarboxamide derivatives as plasma kallikrein inhibitors” WO2013/111108). Однако, никакой из этих документов не сообщает какие-либо результаты по исследованиям in vivo, и поэтому все еще не ясно, будут ли такие соединения обеспечивать достаточную возможность перорального применения или эффективность действия для продвижения в клинику.
В силу вышесказанного, сохраняется потребность в разработке новых ингибиторов калликреина плазмы крови, которые найдут применение для лечения широкого спектра патологических состояний, а именно, для снижения проницаемости сосудов сетчатки глаза, в связи с диабетической ретинопатией и диабетическим макулярным отеком. Предпочтительные соединения будут обладать хорошим фармакокинетическим профилем, а именно, будут приемлемыми в качестве лекарств для перорального введения.
Сущность изобретения
Настоящее изобретение относится к ряду бициклических производных, которые представляют собой ингибиторы калликреина плазмы крови. Эти соединения потенциально полезны для лечения снижения остроты зрения, диабетической ретинопатии, макулярного отека, врожденного ангионевротического отека, диабета, панкреатита, геморрагического инсульта, нефропатии, кардиомиопатии, нейропатии, воспалительного заболевания кишечника, артрита, воспаления, септического шока, гипотензии, рака, респираторного дистресс-синдрома взрослых, синдрома диссеминизированной внутрисосудистой коагуляции, при операции в условиях искусственного кровообращения и при кровотечении после хирургического вмешательства. Изобретение также относится к фармацевтическим композициям ингибиторов, к применению композиций в качестве лекарственных средств, и к способам лечения с использованием этих композиций.
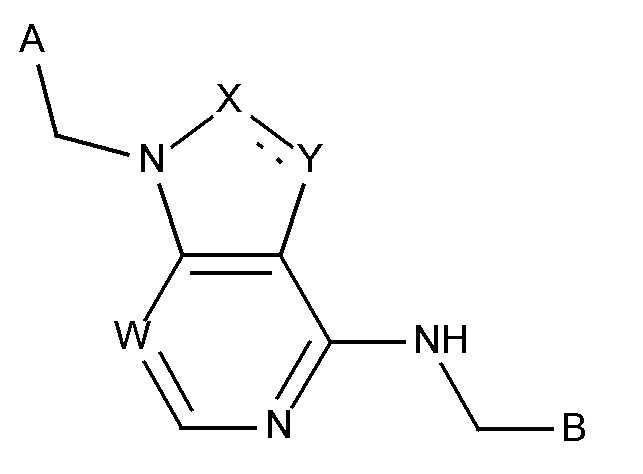
Соединение формулы (I),

Формула (I)
в которой
W выбирают из CH и N;
X выбирают из CH, CH2-CH2, CH=CH, N и NH;
Y выбирают из CH2, CH, N, NH и O;
в которой связь между X и Y  представляет собой либо насыщенную, либо ненасыщенную или ароматическую;
представляет собой либо насыщенную, либо ненасыщенную или ароматическую;
B выбирают из
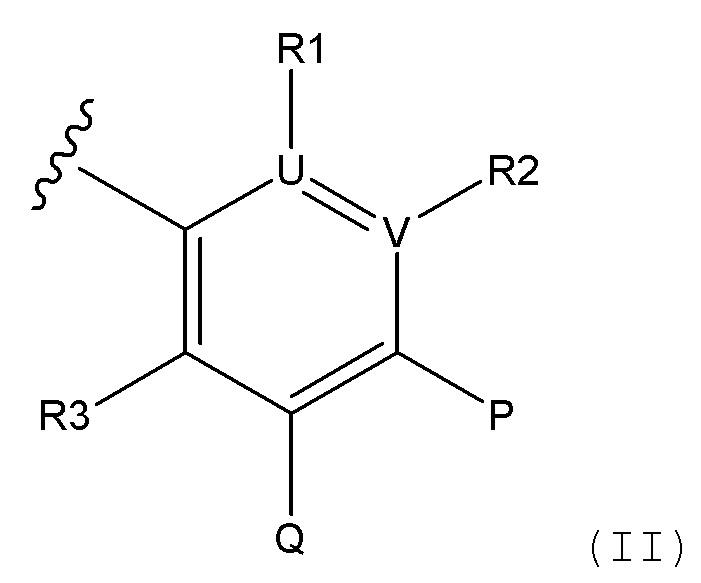
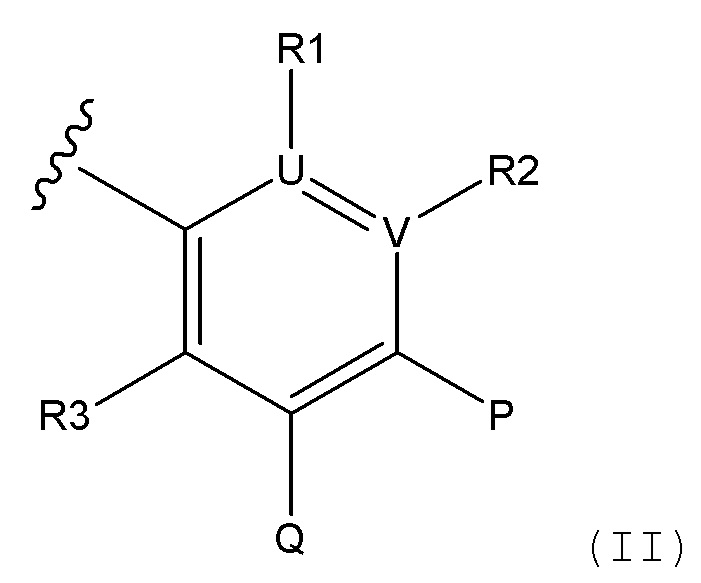
i) радикал формулы II

и
ii) конденсированная 6,5- или 6,6-гетероароматическая бициклическая кольцевая структура, содержащая N и, необязательно, один или два дополнительных гетероатома, которые независимо друг от друга выбирают из N, O и S, которая представляет собой, необязательно, моно-, ди- или три-замещенную заместителем, который выбирают из алкила, алкокси, OH, галогена, CN, COOR8, CONR8R9, CF3 и NR8R9;
P представляет собой H и Q представляет собой -C(R20)(R21)NH2, или P представляет собой -C(R20)(R21)NH2 и Q представляет собой H;
U и V, независимо друг от друга, выбирают из C и N, таким образом, ароматическое кольцо, содержащее U и V представляет собой фенил, пиридин или пиразин;
R1 отсутствует при условии, что U представляет собой N;
R2 отсутствует при условии, что V представляет собой N;
или, в случае присутствия, R1 и R2, независимо друг от друга, выбирают из H, алкила, алкокси, CN, галогена и CF3;
R3 выбирают из H, алкила, алкокси, CN, галогена и CF3;
A выбирают из -(CH2)0 9-гетероарила и -(CH2)0 9-арила;
R8 и R9, независимо друг от друга, выбирают из H и алкила;
R20 и R21, независимо друг от друга, выбирают из H и алкила, или они вместе могут образовать циклоалкильное кольцо или циклический простой эфир;
Алкил представляет собой насыщенный углеводород с прямой цепью, имеющий до 10 углеродных атомов (C1-C10), или насыщенный углеводород с разветвленной цепью, имеющий от 3 до 10 углеродных атомов (C3-C10); алкил, необязательно, может быть замещенным 1 или 2 заместителями, которые, независимо друг от друга выбирают из (C1-C6)алкокси, OH, CN, CF3, COOR10, CONR10R11, фтора и NR10R11;
Циклоалкил представляет собой моноциклический насыщенный углеводород, имеющий от 3 до 7 углеродных атомов;
Циклический простой эфир представляет собой моноциклический насыщенный углеводород, имеющий от 4 до 7 углеродных атомов, в котором один из кольцевых углеродов заменен кислородным атомом;
Алкокси представляет собой с О-связанный углеводород с прямой цепью, имеющий от 1 до 6 углеродных атомов (C1-C6), или с О-связанный углеводород с разветвленной цепью, имеющий от 3 до 6 углеродных атомов (C3-C6); алкокси, необязательно, может быть замещенным 1 или 2 заместителями, которые, независимо друг от друга, выбирают из OH, CN, CF3, COOR10, CONR10R11, фтора и NR10R11;
Арил представляет собой фенил, бифенил или нафтил; арил, необязательно, может быть замещенным 1, 2 или 3 заместителями, которые, независимо друг от друга, выбирают из алкила, алкокси, метилендиокси, этилендиокси, OH, галогена, CN, морфолинила, пиперидинила, гетероарила, -(CH2)0-3-O-гетероарила, арилb, -O-арилb, -(CH2)1-3-арилb, -(CH2)1-3-гетероарила, -COOR10, -CONR10R11, -(CH2)1-3-NR14R15, CF3 и -NR10R11;
арилb представляет собой фенил, бифенил или нафтил, которые, необязательно, могут быть замещенными 1, 2 или 3 заместителями, которые, независимо друг от друга, выбирают из алкила, алкокси, OH, галогена, CN, морфолинила, пиперидинила, -COOR10, -CONR10R11, CF3 и NR10R11;
гетероарил представляет собой 5, 6, 9 или 10 членную моно- или би-циклическую ароматическую кольцевую структуру, содержащую, там, где это возможно, 1, 2 или 3 кольцевых члена, которые выбирают, независимо друг от друга, из N, NR8, S и O; гетероарил, необязательно, может быть замещенным 1, 2 или 3 заместителями, независимо выбранными друг от друга из алкила, алкокси, OH, галогена, CN, арила, морфолинила, пиперидинила, -(CH2)1-3-арила, гетероарилb, -COOR10, -CONR10R11, CF3 и -NR10R11;
гетероарилb представляет собой 5, 6, 9 или 10 членную моно- или би-циклическую ароматическую кольцевую структуру, содержащую, там, где это возможно, 1, 2 или 3 кольцевых члена, которые выбирают, независимо друг от друга, из N, NR8, S и O; при этом гетероарилb, необязательно, может быть замещенным 1, 2 или 3 заместителями, которые, независимо друг от друга, выбирают из алкила, алкокси, OH, галогена, CN, морфолинила, пиперидинила, арила, -(CH2)1-3-арила, -COOR10, -CONR10R11, CF3 и NR10R11;
R10 и R11, независимо друг от друга, выбирают из H и алкила; или R10 и R11 вместе с азотом, к которому они присоединены, образуют 4-, 5-, 6- или 7-членную гетероциклическую кольцевую структуру, которая может быть насыщенной или ненасыщенной с 1 или 2 двойными связями;
R14 и R15, независимо друг от друга, выбирают из алкила, арилb и гетероарилb; или R14 и R15 вместе с азотом, к которому они присоединены, образуют 4-, 5-, 6- или 7- членную гетероциклическую кольцевую структуру, которая может быть насыщенной или ненасыщенной с 1 или 2 двойными связями, и, необязательно, может быть оксо-замещенной;
и таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереомеры и их рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В другом аспекте, в настоящем изобретении предлагается пролекарство соединения формулы (I), как определено в этом документе, или его фармацевтически приемлемая соль.
И еще в другом аспекте, в настоящем изобретении предлагается N-оксид соединения формулы (I,) как определено в этом документе, или пролекарство, или его фармацевтически приемлемая соль.
Следует понимать, что определенные соединения настоящего изобретения могут существовать в виде сольватированных, например, гидратированных, а также и в виде несольватированных форм. Нужно понимать, что настоящее изобретение охватывает все такие сольватированные формы.
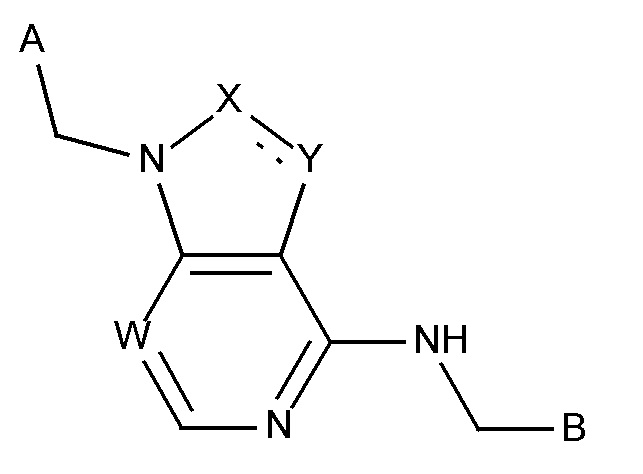
В некотором аспекте, изобретение включает в себя подгруппу соединений формулы I,

Формула (I)
в которой
W выбирают из CH и N;
X выбирают из CH, CH2-CH2 и N;
Y выбирают из CH, N и O;
в которой связь между X и Y  представляет собой либо насыщенную, либо ненасыщенную или ароматическую;
представляет собой либо насыщенную, либо ненасыщенную или ароматическую;
в которой A и B представляют собой такие, как предварительно определено выше;
и таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереомеры и их рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В некотором аспекте, изобретение включает в себя подгруппу соединений формулы I,

Формула (I)
в которой
W выбирают из CH и N;
X выбирают из CH и CH2-CH2;
Y выбирают из CH и O;
в которой связь между X и Y представляет собой либо насыщенную, либо ненасыщенную или ароматическую;
в которой A и B представляют собой такие, как предварительно определено выше;
и таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереомеры и их рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
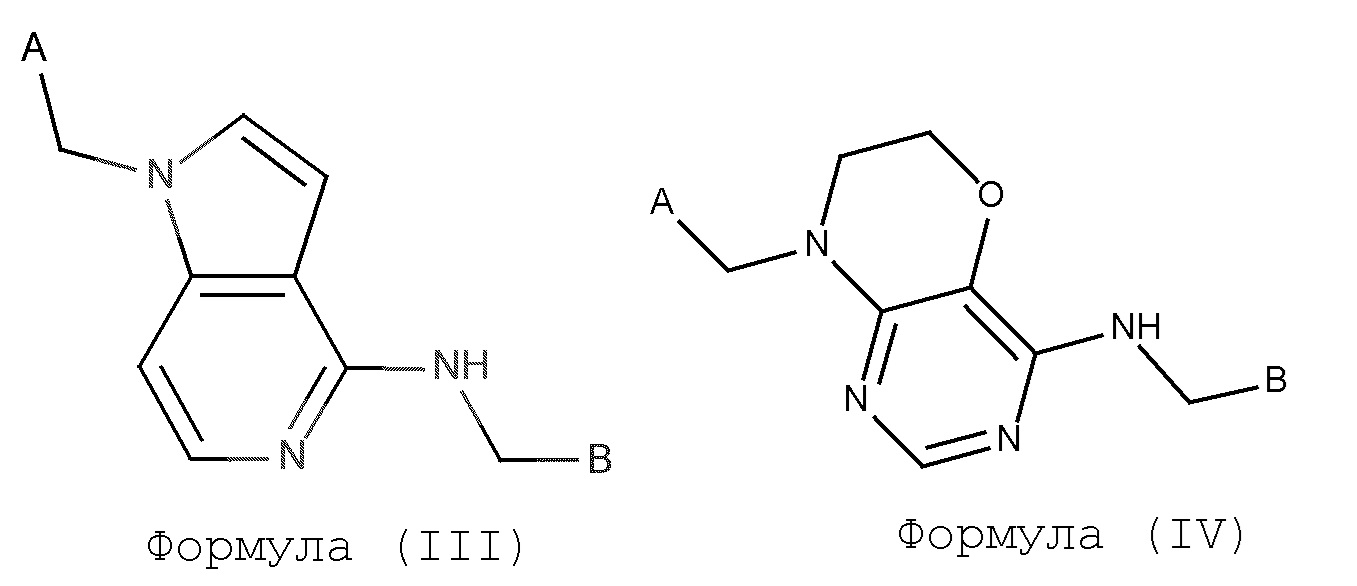
В некотором аспекте, изобретение включает в себя подгруппу соединений формулы I, как определено формулой (III),

Формула (III)
в которой A и B представляют собой такие, как предварительно определено выше;
и таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереомеры и их рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
В некотором аспекте, изобретение включает в себя подгруппу соединений формулы I, как определено формулой (IV),

Формула (IV)
в которой A и B представляют собой такие, как предварительно определено выше;
и таутомеры, изомеры, стереоизомеры (включая энантиомеры, диастереомеры и их рацемические и скалемические смеси), их фармацевтически приемлемые соли и сольваты.
Настоящее изобретение также включает в себя нижеследующие аспекты и их комбинации:
Соединения формулы (I), формулы (III) или формулы (IV), в которых, A выбирают из -(CH2)0 9-гетероарила и -(CH2)0 9-арила, в которых гетероарил и арил представляют собой такие, как определено в соответствии с формулой (I) выше.
Соединения формулы (I), формулы (III) или формулы (IV), в которых, A выбирают из гетероарила, замещенного фенилом; и -(CH2)0-3 фенила, замещенного гетероарилом, -(CH2)1-3-гетероарила и -(CH2)1-3-NR14R15; в которых гетероарил, R14 и R15 представляют собой такие, как определено в соответствии с формулой (I) выше.
Предпочтительными являются соединения формулы (I), формулы (III) или формулы (IV), в которых, A выбирают из -(CH2)0-3 фенила,

Наиболее предпочтительными являются соединения формулы (I), формулы (III) или формулы (IV), в которых A выбирают из:
 фенила, и
фенила, и 
Соединения формулы (I), формулы (III) или формулы (IV), в которых B выбирают из:
i) радикал формулы II

и
ii) конденсированная 6,5- или 6,6-гетероароматическая бициклическая кольцевая структура, содержащая N и, необязательно, один или два дополнительных гетероатома, которые, независимо друг от друга, выбирают из N, O и S, которая представляет собой, необязательно, моно-, ди- или три-замещенную заместителем, который выбирают из алкила, алкокси, OH, галогена, CN, COOR8, CONR8R9, CF3 и NR8R9;
в которых R1, R2, R3, R8, R9, P, Q, U, V, алкил и алкокси представляют собой такие, как определено в соответствии с формулой (I) выше.
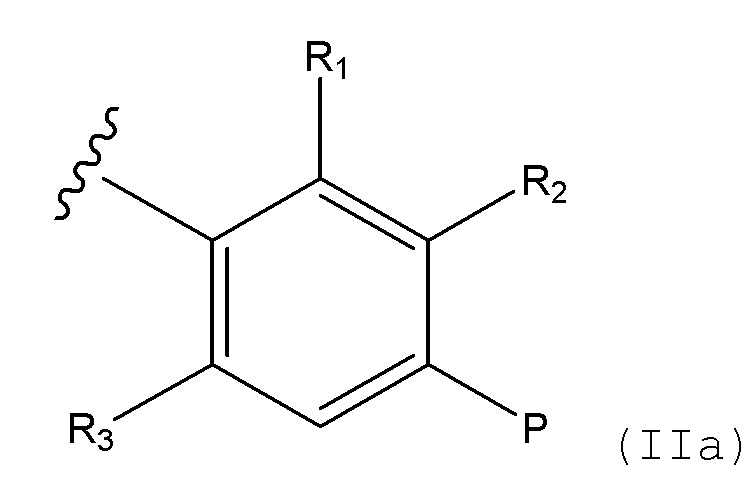
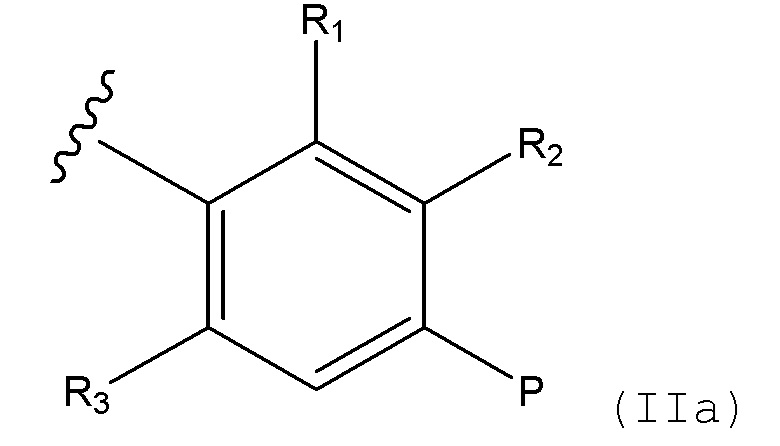
Соединения формулы (I), формулы (III) или формулы (IV), в которых, B выбирают из:
i) радикал формулы IIa
 (IIa)
(IIa)
в которой R1 выбирают из H и алкила, R2 представляет собой H, R3 выбирают из H и алкила, и P представляет собой -CH2NH2; и
ii) конденсированная 6,5- или 6,6-гетероароматическая бициклическая кольцевая структура, содержащая N и, необязательно, один или два дополнительных гетероатома, которые, независимо друг от друга, выбирают из N и O, которая представляет собой, необязательно, моно- или ди-замещенную заместителем, который выбирают из алкила, алкокси, OH, галогена, CN, CF3 и NR8R9;
в которых алкил, алкокси, R8 и R9 представляют собой такие, как определено в соответствии с формулой (I) выше.
Соединения формулы (I) или формулы (III), в которых B представляет собой радикал формулы IIa,
 (IIa)
(IIa)
в которых R1 выбирают из H и алкила, R2 представляет собой H, R3 выбирают из H и алкила, и P представляет собой -CH2NH2; в которых алкил представляет собой такой, как определено в соответствии с формулой (I) выше.
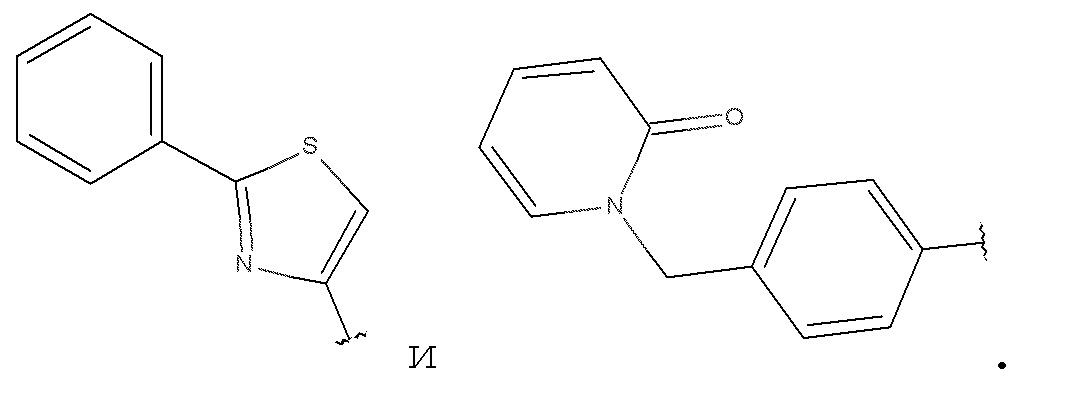
Соединения формулы (I), формулы (III) или формулы (IV), в которых, B выбирают из, необязательно, замещенного изохинолинила, в котором указанный необязательный заместитель выбирают из алкила, алкокси, OH, и NR8R9; и, необязательно, замещенного 1H-пирроло[2,3-b]пиридина, в котором указанный необязательный заместитель выбирают из алкила, алкокси, OH, F, Cl, CN, COOR8, CONR8R9, CF3; в которых R8 и R9, независимо друг от друга, выбирают из H и алкила, и алкил, и алкокси представляют собой такие, как определено в соответствии с формулой (I) выше.
Соединения формулы (I), формулы (III) или формулы (IV), в которых B выбирают из, необязательно, замещенного изохинолинила, в котором указанный необязательный заместитель представляет собой NH2; и 1H-пирроло[2,3-b]пиридина.
В некотором аспекте, изобретение включает в себя соединение, выбираемое из:
|
и их фармацевтически приемлемые соли и сольваты.
Терапевтическое применение
Как отмечалось выше, соединения настоящего изобретения представляют собой высокоактивные ингибиторы калликреина плазмы крови. Поэтому они подходят для применения при лечении болезненных патологических состояний, для которых повышенная активность калликреина плазмы крови представляет собой фактор, вызывающий болезнь.
В соответствии с этим, в настоящем изобретении предлагается соединение формулы (I) для применения в медицине.
В настоящем изобретении также предлагается использование соединения формулы (I) в промышленном производстве лекарства для лечения или профилактики болезни или патологического состояния, в которые вмешивается активность калликреина плазмы крови.
В настоящем изобретении также предлагается соединение формулы (I) для применения при лечении или профилактике болезни или патологического состояния, в которые вмешивается активность калликреина плазмы крови.
В настоящем изобретении также предлагается способ лечения болезни или патологического состояния, в которые вмешивается активность калликреина плазмы крови, включающий введение нуждающемуся в этом субъекту терапевтически эффективного количества соединения формулы (I).
В одном аспекте, болезнь или патологическое состояние, в которые вмешивается активность калликреина плазмы крови, включает снижение остроты зрения, диабетическую ретинопатию, диабетический макулярный отек, врожденный ангионевротический отек, диабет, панкреатит, геморрагический инсульт, нефропатию, кардиомиопатию, нейропатию, воспалительное заболевание кишечника, артрит, воспаление, септический шок, гипотензию, рак, респираторный дистресс-синдром взрослых, синдром диссеминизированной внутрисосудистой коагуляции, операцию в условиях искусственного кровообращения и кровотечение после хирургического вмешательства.
В другом аспекте, болезнь или патологическое состояние, в которые вмешивается активность калликреина плазмы крови, представляет собой связанное с проницаемостью сосудов сетчатки глаза при диабетической ретинопатии и диабетическом макулярном отеке.
Комбинированная терапия
Соединения настоящего изобретения можно вводить в комбинации с другими лекарственными препаратами. Подходящие комбинированные терапии включают соединение формулы (I) объединенное с одним или несколькими препаратами, выбранными из таких препаратов, которые ингибируют тромбоцитарный фактор роста (PDGF), эндотелиальный фактор роста (VEGF), интегрин альфа5бета1, стероиды, другими препаратами, ингибирующими калликреин плазмы крови, и другими ингибиторами воспаления. Конкретные примеры лекарственных препаратов, которые можно комбинировать с соединениями настоящего изобретения, включают такие, которые раскрыты в патенте EP2281885A и в публикации S. Patel in Retina, 2009 Jun; 29(6 Suppl):S45-8.
В случае использования комбинированной терапии соединения, настоящего изобретения и указанные комбинации препаратов могут быть в виде одинаковых или в виде различных фармацевтических композиций, and may be administered separately, sequentially or simultaneously.
В другом аспекте, соединения настоящего изобретения можно вводить в комбинации с лазерным лечением сетчатки. Комбинирование лазерного лечения с интравитреальной инъекцией ингибитора VEGF при лечении диабетического макулярного отека известно (Elman M, Aiello L, Beck R, et al. “Randomized trial evaluating ranibizumab plus prompt or deferred laser or triamcinolone plus prompt laser for diabetic macular edema”. Ophthalmology. 27 April 2010).
Определения терминов
Термин "алкил" включает насыщенные углеводородные остатки, включая:
- группы с прямой цепью, содержащие до 10 углеродных атомов (C1-C10), или до 6 углеродных атомов (C1-C6), или до 4 углеродных атомов (C1-C4). Примеры таких алкильных групп включают, но не ограничиваются только приведенными, C1 - метил, C2 - этил, C3 - пропил и C4 - н-бутил.
- группы с разветвленной цепью, содержащие от 3 до 10 углеродных атомов (C3-C10), или до 7 углеродных атомов (C3-C7), или до 4 углеродных атомов (C3-C4). Примеры таких алкильных групп включают, но не ограничиваются только приведенными,, C3 - изо-пропил, C4 - втор-бутил, C4 - изо-бутил, C4 - трет-бутил и C5 - нео-пентил.
каждый из которых, необязательно, замещенный, согласно вышеизложенному.
Циклоалкил представляет собой моноциклический насыщенный углеводород, содержащий от 3 до 7 углеродных атомов; при этом циклоалкил, необязательно, может быть замещенным, с заместителями, которые выбирают из алкила, алкокси и NR10R11; в при этом R10 и R11, независимо друг от друга, выбирают из H и алкила, или R10 и R11, вместе с азотом, к которому они присоединены, образуют 4-, 5-, 6- или 7-членную гетероциклическую кольцевую структуру, которая может быть насыщенной или ненасыщенной с 1 или 2 двойными связями. Циклоалкильные группы могут содержать от 3 до 7 углеродных атомов, или от 3 до 6 углеродных атомов, или от 3 до 5 углеродных атомов, или от 3 до 4 углеродных атомов. Примеры подходящих моноциклических циклоалкильных групп включают циклопропил, циклобутил, циклопентил, циклогексил и циклогептил.
Термин "алкокси" включает связанные с O углеводородные остатки, включая:
- группы с прямой цепью, содержащие от 1 до 6 углеродных атомов (C1-C6), или от 1 до 4 углеродных атомов (C1-C4). Примеры таких алкокси-групп включают, но не ограничиваются только приведенными, C1 - метокси, C2 - этокси, C3 - н-пропокси и C4 - н-бутокси.
- группы с разветвленной цепью, содержащие от 3 до 6 углеродных атомов (C3-C6), или от 3 до 4 углеродных атомов (C3-C4). Примеры таких алкокси-групп включают, но не ограничиваются только приведенными, C3 - изо-пропокси и C4 - втор-бутокси и трет-бутокси.
каждый из которых, необязательно, замещенный, согласно вышеизложенному.
Если не оговаривается иное, галоген выбирают из Cl, F, Br и I.
Арил представляет собой такой, как определено выше. Обычно, арил, необязательно, будет замещенным, с 1, 2 или 3 заместителями. Необязательные заместители выбирают из тех, которые указаны выше. Примеры подходящих арильных групп включают фенил и нафтил (каждый, необязательно, замещенный, как указано выше). Предпочтительно, арил выбирают из фенила, замещенного фенила (замещенного в соответствии с вышеуказанным) и нафтила.
Гетероарил представляет собой такой, как определено выше. Примеры подходящих гетероарильных групп включают тиенил, фуранил, пирролил, пиразолил, имизазолил, оксазолил, изоксазолил, тиазолил, изотиазолил, триазолил, оксадиазолил, тиадиазолил, тетразолил, пиридинил, пиридазинил, пиримидинил, пиразинил, индолил, бензимидазолил, бензотриазолил, хинолинил и изохинолинил (необязательно, замещенные, как указано выше). Предпочтительно, гетероарил выбирают из пиридила, бензотиазола, индола, N-метилиндола, тиазола, замещенного тиазола, тиофенила, фурила, пиразина, пиразола и замещенного пиразола; при этом заместители представляют собой такие, как указано выше.
Термин "связанный с N", такой как в "связанный с N гетероциклоалкил", обозначает, что гетероциклоалкильная группа присоединена к остальной части молекулы через кольцевой атом азота.
Термин "связанный с O", такой как в "связанный с O углеводородный остаток", обозначает, что углеводородный остаток присоединен к остальной части молекулы через атом кислорода.
В группах, таких как -COOR*, "-" указывает точку присоединения замещающей группы к остальной части молекулы.
"Фармацевтически приемлемая соль" обозначает физиологически или токсикологически допустимую соль и включает, в соответствующих случаях, фармацевтически приемлемые соли, образованные добавлением основания, и фармацевтически приемлемые соли, образованные добавлением кислоты. Например, (i) в случае, если соединение изобретения содержит одну или несколько кислотных групп, например, карбокси-групп, то могут быть образованы его фармацевтически приемлемые соли при добавлении основания, включая соли натрия, калия, кальция, магния и аммония, или соли с органическими аминами, такими как, диэтиламин, N метил-глюкамин, диэтаноламин или аминокислоты (например, лизин) и т.п.; (ii) в случае, если соединение изобретения содержит основную группу, такую как амино-группа, то могут быть образованы его фармацевтически приемлемые соли при добавлении кислоты, включая хлоргидраты, бромгидраты, сульфаты, фосфаты, ацетаты, цитраты, лактаты, тартраты, мезилаты, сукцинаты, оксалаты, фосфаты, эзилаты, тозилаты, бензолсульфонаты, нафталиндисульфонаты, малеаты, адипаты, фумараты, гиппураты, камфораты, ксинафоаты, п-ацетамидобензоаты, дигидроксибензоаты, гидроксинафтоаты, сукцинаты, аскорбаты, олеаты, бисульфаты и т.п.
Гемисоли кислот и оснований также могут быть образованы, например, гемисульфат и гемикальциевые соли.
Для обзора подходящих солей см. "Handbook of Pharmaceutical Salts: Properties, Selection and Use" by Stahl and Wermuth (Wiley-VCH, Weinheim, Germany, 2002).
“Пролекарство” относится к соединению, которое превращается in vivo посредством метаболических путей (например, посредством гидролиза, восстановления или окисления) в соединение изобретения. Подходящие группы для формирования пролекарств описаны в “The Practice of Medicinal Chemistry”, 2nd Ed. pp561-585 (2003) и в F. J. Leinweber, Drug Metab. Res., 1987, 18, 379.
Соединения изобретения могут существовать как в несольватированной, так и в сольватированной формах. Термин 'сольват' используется в этом документе для описания молекулярного комплекса, включающего соединение изобретения и стехиометрическое количество молекул одного или нескольких фармацевтически приемлемых растворителей, например, этанола. Термин 'гидрат' применяется в том случае, когда растворитель представляет собой воду.
Поскольку соединения изобретения существуют в одной или нескольких геометрических, оптических, энантиомерных, диастереомерных и таутомерных формах, включая, но не ограничиваясь только приведенными, цис- и транс-формы, E- и Z-формы, R-, S- и мезо-формы, кето- и енольные-формы, если не оговаривается иное, ссылка на конкретное соединение включает все такие изомерные формы, включая их рацемические и другие смеси. При необходимости, такие изомеры могут быть выделены в индивидуальном виде из их смесей посредством практического использования или применения общеизвестных способов (например, хроматографических методик и методов перекристаллизации). При необходимости, такие изомеры могут быть получены посредством практического использования или применения общеизвестных способов (например, ассиметрического синтеза).
В контексте настоящего изобретения, ссылки в этом документе на "лечение" включают ссылки на радикальное лечение, паллиативное лечение и профилактическое лечение.
Общие методы
Соединения формулы (I) должны оцениваться по их биофармацевтическим свойствам, таким как растворимость стабильность раствора (в широком диапазоне pH), всасываемость, и т.п., с целью выбрать наиболее подходящие готовую лекарственную форму и способ введения для лечения при предложенном показании к применению. Они могут вводиться в индивидуальном виде или в комбинации с одним или несколькими другими соединениями изобретения, или в комбинации с одним или несколькими другими лекарствами (или как любая их комбинация). Как правило, их будут вводить в виде лекарственного препарата в соединении с одним или несколькими фармацевтически приемлемыми эксципиентами. Термин ’эксципиент’ используется в этом документе для описания любого ингредиента, отличающегося от соединения(ий) изобретения, который может обеспечить придание как функциональных (т.e., регуляция скорости высвобождения лекарственного средства), так и/или нефункциональных (т.e., использование в производственном процессе вспомогательного вещества или разбавителя) характеристик лекарственным формам. Выбор эксципиента в большой степени будет зависеть от таких факторов, как определенный способ введения, воздействие эксципиента на растворимость и стабильность, и тип готовой лекарственной формы.
Соединения изобретения, предназначенные для фармацевтического применения, можно вводить в виде твердых веществ или жидкостей, в таких формах, как таблетки, капсулы или растворы. Фармацевтические композиции, подходящие для доставки соединений настоящего изобретения, и способы их получения будут вполне очевидны для специалистов в данной области техники. Такие композиции и способы их получения можно найти, например, в издании Remington’s Pharmaceutical Sciences, 19th Edition (Mack Publishing Company, 1995).
Таким образом, в настоящем изобретении предлагается фармацевтическая композиция, включающая соединение формулы (I) и фармацевтически приемлемые носитель, разбавитель или эксципиент.
Для лечения патологических состояний, таких как нарушение проницаемости сосоудов сетчатки глаза, связанное с диабетической ретинопатией и диабетическим макулярным отеком, соединения изобретения можно вводить в форме, подходящей для инъекций в область глазницы пациента, а именно, в форме, подходящей для интравитреальной инъекции. Предусматривается, что готовые лекарственные формы, подходящие для такого использования, будут в форме стерильных растворов соединения изобретения в подходящей водной среде для растворения. Композиции могут быть введены пациенту под надзором лечащего врача.
Соединения изобретения также могут быть введены прямо в кровоток, в подкожную клетчатку, в мышцу или во внутренний орган. Подходящие способы парентерального введения включают введение внутривенное, внутриартериальное, интраперитониальное, интратекальное, интравентрикулярное, интрауретральное, интрастернальное, интракраниальное, внутримышечное, внутрисуставное и подкожное. Подходящие медицинские устройства или изделия для парентерального введения включают устройства для инъекций с иглой (включая микроиглу), безыгольные устройства для инъекций и оборудование для инфузии.
Парентеральные лекарственные формы обычно представляют собой водные или масляные растворы. В том случае, когда раствор водный, тогда эксципиенты такие, как, например, сахара (включая, но не ограничиваясь приведенными, глюкозу, манитол, сорбитол и т.п.), соли, углеводы и буферные агенты (предпочтительно, для значений pH от 3 до 9), но, для некоторых применений, они могут быть получены в виде более удобных лекарственных форм, таких как стерильный неводный раствор или таких как высушенная форма, предназначенная для использования при соединении с подходящей средой для растворения, такой как стерильная, апирогенная вода.
Парентеральные лекарственные формы могут включать имплантанты, полученные из поддающихся разложению полимеров, таких как полиэфиры (т.e., полимер молочной кислоты, полилактид, сополимер полилактидгликолид, поликапролактон, полигидроксибутират), полиортоэфиры и полиангидриды. Эти лекарственные формы могут вводиться посредством хирургического разреза в подкожную клетчатку, мышечную ткань или прямо в конкретные органы.
Получение парентеральных лекарственных форм в стерильных условиях, например, путем лиофилизации, можно легко осуществить, используя стандартное фармацевтическое оборудование, хорошо известное специалисту в данной области техники.
Растворимость соединений формулы (I), используемых при получении парентеральных растворов, можно увеличить путем применения подходящих технологических приемов получения лекарственных форм, таких как включение в состав вспомогательных растворителей и/или усилителей растворимости, таких как поверхностно-активные вещества, мицеллярные структуры и циклодекстрины.
В одном варианте осуществления изобретения, соединения изобретения may be administered orally. Пероральное введение может включать проглатывание, таким образом соединение поступает в желудочно-кишечный тракт, и/или буккальное, лингвальное или сублингвальное введение, посредством которого соединение поступает в кровоток прямо непосредственно изо рта.
Лекарственные формы, подходящие для перорального введения, включают вкладыши из твердой прессованной массы, твердые микрочастицы, мягкие лекарственные формы и жидкие лекарственные формы (включая многофазные или дисперсные системы), такие как таблетки; мягкие или твердые капсулы, содержащие мульти или- нано-частицы, жидкие лекарственные формы, эмульсии или порошки; леденцы (включая леденцы с жидким наполнением); жвачки; гели; быстро диспергирующиеся лекарственные формы; пленки; суппозитории; спреи; и буккальные/мукоадгезивные пластыри.
Лекарственные формы, подходящие для перорального введения, также могут быть созданы для доставки соединений изобретения с быстрым высвобождением или с замедленной скоростью высвобождения, при этом графический профиль высвобождения может соответствовать высвобождению отсроченному, прерывистому, контролируемому, пролонгированному, или отсроченному и пролонгированному, или модифицированы таким образом, чтобы оптимизировать терапевтическую эффективность указанных соединений. Способы доставки соединений для обеспечения замедленной скорости высвобождения общеизвестны из уровня техники в данной области техники и включают использование медленно размыкающихся полимеров, которые могут включаться в состав лекарственной формы с указанными соединениями для контроля их высвобождения.
Примеры полимеров с замедленной скоростью высвобождения включают поддающиеся разложению и неподдающиеся разложению полимеры, которые могут использоваться для высвобождения указанных соединений путем диффузии или путем комбинирования диффузии и разрушения полимера. Примеры полимеров с замедленной скоростью высвобождения включают гидроксипропилметилцеллюлозу, гидроксипропилцеллюлозу, метилцеллюлозу, этилцеллюлозу, натриевая соль карбоксиметилцеллюлозы, поливиниловый спирт, поливинилпирролидон, ксантановую камедь, полиметакрилаты, полиэтиленоксид и полиэтиленгликоль.
Жидкие лекарственные формы (включая многофазные и дисперсные системы) включают эмульсии, растворы, сиропы и эликсиры. Такие лекарственные формы могут быть представлены в виде наполнителей в мягких или твердых капсулах (изготовленных, например, из желатина или гидроксипропилметилцеллюлозы) и, как правило, включают носитель, например, воду, этанол, полиэтиленгликоль, пропиленгликоль, метилцеллюлозу, или подходящее масло, и один или несколько эмульгаторов и/или суспендирующих веществ. Жидкие лекарственные формы также могут быть получены посредством разведения твердых, например, из сухого порошка.
Соединения изобретения также могут применяться в быстрорастворимых, рассасываемых готовых лекарственных формах, таких как описаны в публикации Liang and Chen, Expert Opinion in Therapeutic Patents, 2001, 11 (6), 981-986.
Лекарственная форма в виде таблеток рассматривается в издании Pharmaceutical Dosage Forms: Tablets, Vol. 1, by H. Lieberman and L. Lachman (Marcel Dekker, New York, 1980).
Для введения больным людям общая суточная доза соединений изобретения, как правило, находится в диапазоне 0,01 мг - 1000 мг, или 0,1 мг - 250 мг, или 1 мг - 50 мг, конечно же, в зависимости от способа введения лекарства.
Общая доза может вводиться вся одноразово или делиться на дозы и, по указанию лечащего врача, может выходить за пределы общепринятого диапазона, приведенного в этом документе. Эти дозы основаны на среднем значении массы человека от, приблизительно 60 кг до 70 кг. Лечащий врач легко определит дозы для пациентов, масса которых выходит за пределы этого диапазона, таких как дети и пожилые люди.
Методы синтеза
Соединения настоящего изобретения могут быть получены в соответствии с методиками по нижеприведенным схемам и примерам, при использовании соответствующих веществ, и дополнительно представленными конкретными примерами, приведенными в этом документе ниже. Кроме того, используя описанные в этом документе методики, средний специалист в данной области техники легко может получить дополнительные соединения, попадающие под действие приведенной в этом документе формулы изобретения настоящего изобретения. Соединения, проиллюстрированные в примерах, однако, не следует интерпретировать в качестве единственно возможного класса соединений, рассматриваемого как изобретение. Примеры дополнительно способствуют иллюстрации подробностей для получения соединений настоящего изобретения. Тот, кто является специалистом в данной области техники, легко поймет, что известные изменения условий и процессов осуществления нижеприведенных препаративных методик могут применяться для получения этих соединений.
Соединения изобретения могут быть выделены в виде их фармацевтически приемлемых солей, таких как предварительно описаны в этом документе выше.
Может возникнуть необходимость защиты реакционноспособных функциональных групп (например, гидрокси, амино, тио или карбокси групп) в промежуточных соединениях, используемых при получении соединений изобретения, чтобы избежать их нежелательного участия в реакции, приводящей к образованию целевых соединений. Обычно применяемые защитные группы, например, такие как описаны в издании T. W. Greene and P. G. M. Wuts in “Protective groups in organic chemistry” John Wiley and Sons, 4th Edition, 2006, могут применяться. Например, обычная защитная группа для амино-группы, подходящая для использования в данном изобретении, представляет собой трет-бутоксикарбонильную (Boc), которая легко удаляется при обработке кислотой, такой как трифторуксусная кислота или хлористый водород в органическом растворителе, таком как дихлорметан. Альтернативно защитной группой для амино-группы может быть бензилоксикарбонильная (Z) группа, которая может быть удалена посредством гидрирования с палладиевым катализатором в атмосфере водорода, или 9-флуоренилметоксикарбонильная (Fmoc) группа, которая может быть удалена с помощью растворов вторичных органических аминов, таких как диэтиламин или пиперидин, в органических растворителях. Карбоксильные группы обычно защищают в виде сложных эфиров, таких как метиловый, этиловый, бензиловый или трет-бутиловый, каждый из которых можно удалить с посредством гидролиза в присутствии оснований, таких как гидроксид лития или гидроксид натрия. Бензильные защитные группы также могут удаляться посредством гидрирования с палладиевым катализатором в атмосфере водорода, в то время как трет-бутильные группы также могут быть удалены с помощью трифторуксусной кислоты. Альтернативно, трихлорэтилового эфира защитная группа удаляется цинком в уксусной кислоте. Обычная защитная группа для гидрокси-группы для использования в этом изобретении представляет собой метиловый простой эфир, условия снятия защиты включают кипячение в 48% водной HBr в течение 1-24 часов, или путем перемешивания с трибромидом бора в дихлорметане в течение 1-24 часов. Альтернативно, если гидрокси-группа защищена в виде бензилового простого эфира, то условия снятия защиты включают гидрирование с палладиевым катализатором в атмосфере водорода.
Примеры
Изобретение иллюстрируется нижеприведенными, не имеющими ограничительного характера, примерами, в которых нижеследующие сокращения и определения используются:
|
Все реакции проводили в атмосфере азота, если нет иных конкретных указаний.
1H ЯМР спектры регистрировали на спектрометре Bruker (400 MHz) в дейтерированном растворителе при комн.т.
Молекулярные ионы получали, используя LCMS, которую проводили, используя Chromolith Speedrod RP-18e колонку, 50×4,6 мм, элюент с линейным градиентом 10%-90% 0,1% HCO2H/MeCN в 0,1% HCO2H/H2O в течение 13 мин, скорость потока элюента 1,5 мл/мин, или используя Agilent, X-Select, кислая среда, 5-95% MeCN/вода в течение 4 мин. Результаты получали, используя Thermofinnigan Surveyor MSQ масс-спектрометр с ионизацией электрораспылением в сочетании с Thermofinnigan Surveyor LC системой.
Химические названия составляли, используя программное обеспечение Autonom, предоставляемое как составная часть ISIS графического пакета программ MDL Information Systems, по форме требований IUPAC, используя программное обеспечение Chemaxon.
В случаях, когда продукты очищали посредством флэш-хроматографии, ‘силикагель’ имеет отношение к силикагелю для хроматографии, 0,035-0,070 мм (220-440 меш) (например, Merck silica gel 60), и применяемое давление азота до 10 фунт на кв. дюйм ускоряло элюирование колонки. Очистки с помощью обращеннофазовой препаративной ВЭЖХ проводили с использованием двойной насосной системы создания градиента Waters 2525 при скорости потока элюента, как правило, 20 мл/мин, используя детектор с фотодиодной матрицей Waters 2996.
Все растворители и коммерческие реагенты использовали в стандартном виде, в котором их приобретали.
Общие способы получения соединений нижеприведенных Таблиц описаны в этом документе: -
Общий способ алкилирования по бициклическому азоту
К гидриду натрия (2экв.) в DMF при 0°C добавляли бициклический амин в виде свободного основания (1экв.) и реакционную массу перемешивали в течение 20 мин, затем добавляли бензилбромид (1,1экв.) и реакционную массу перемешивали при комн.т. в течение 2-16 ч. При охлаждении реакционной смеси гасили протекание реакции водой и проводили экстракцию EtOAc (2x), объединенные органические фазы промывали водой и насыщенным солевым раствором, сушили (MgSO4) и концентрировали, и очищали по мере необходимости.
Общие методики замещения хлора первичными аминами
A: Арилхлорид (1экв.) и амин (1-5экв.) в этаноле выдерживали при нагревании до 130°C в течение 8-120 ч. Реакционную смесь концентрировали в вакууме и очищали по мере необходимости.
B: Арилхлорид (1экв.) и амин (1-5экв.) в н-бутаноле выдерживали при нагревании до 130°C в течение 8-120 ч. Реакционную смесь концентрировали в вакууме и очищали по мере необходимости.
C: К арилхлориду (1экв.) в пробирке, предназначенной для микроволновой печи, в сухом толуоле добавляли амин (1-1,4экв.), BINAP (0,8экв.) и трет-бутоксид натрия (1,4экв.). Струю N2 пропускали через реакционную смесь в течение 5 мин. Под конец добавляли Pd2dba3 (0,3экв.) и реакционную массу перемешивали в течение 1 мин перед тем, как сразу же поместить в микроволновую печь для выдерживания при 170°C в течение 30-90 мин. Реакционную смесь концентрировали и очищали либо с помощью флэш-хроматографии, либо с помощью обращеннофазовой препаративной ВЭЖХ.
Общий способ восстановления нитрила
К охлажденному нитрилу (1экв.) в метаноле добавляли гексагидрат хлорида никеля (II) (0,1экв.) и ди-трет-бутилдикарбонат (2экв.). Борогидрид натрия (7экв.) добавляли частями, контролируя газообразование. Реакционную смесь перемешивали при 0oC, постепенно доводя до комнатной температуры в течение 18 часов, по истечении этого времени MeOH удаляли посредством упаривания. Остаток растворяли в CHCl3, промывали насыщенным раствором NaHCO3, водой, насыщенным солевым раствором, сушили (Na2SO4) и фильтровали. Фильтрат упаривали и очищали по мере необходимости.
Общий метод удаления защитных Boc-групп
К Boc-защищенному бензиламину добавляли 4M HCl в диоксане и реакционную массу перемешивали при комнатной температуре в течение 1-16 ч. Растворитель удаляли в вакууме, что давало возможность получить целевое соединение в виде HCl соли.
Пример 7
1-{4-[4-(4-Аминометил-бензиламино)-пирроло[3,2-c]пиридин-1-илметил]-бензил}-1H-пиридин-2-он

A. 1-(4-Гидроксиметил-бензил)-1H-пиридин-2-он
4-(Хлорметил)бензиловый спирт (5 г, 31,93 ммоль) растворяли в ацетоне (150 мл) и добавляли 2-гидроксипиридин (3,6 г, 38,31 ммоль) и карбонат калия (13,2 г, 95,78 ммоль). Реакционную смесь перемешивали при 50°C в течение 3 часов. Растворитель удаляли в вакууме и остаток соединяли с хлороформом (100 мл). Органический слой промывали водой (30 мл), насыщенным солевым раствором (30 мл), сушили (Na2SO4), фильтровали и упаривали. Остаток очищали с помощью флэш-хроматографии, элюируя 5% MeOH-DCM, получали белое твердое вещество, идентифицированное как озаглавленное соединение, (5,4 г, 25,09 ммоль, выход 79%).
[M+Na]+=237,8
B. 1-(4-Бромметил-бензил)-1H-пиридин-2-он
1-(4-Гидроксиметил-бензил)-1H-пиридин-2-он (1 г, 4,65 ммоль) растворяли в DCM (75 мл) и добавляли трибромид фосфора (2,5 г, 9,29 ммоль). Реакционную массу перемешивали при комн.т. в течение 3 ч. По окончании реакции реакционную смесь разбавляли CHCl3 (75 мл) и промывали насыщенным раствором NaHCO3 (30 мл), водой (30 мл) и насыщенным солевым раствором (30 мл). Органический слой сушили (Na2SO4), фильтровали и упаривали, получая белое твердое вещество, идентифицированное как озаглавленное соединение, которое в дальнейшем использовали без дополнительной очистки (1,05 г, 3,78 ммоль, выход 81%).
[M+H]+=277,61 и 279,59
C. 4-[(1H-Пирроло[3,2-c]пиридин-4-иламино)-метил]-бензонитрил
4-(Аминометил)бензонитрил.HCl распределяли между хлороформом (50 мл) и насыщенным раствором NaHCO3 (10 мл), сушили над Na2SO4, фильтровали и упаривали, что давало возможность получить 4-(аминометил)бензонитрил в форме свободного основания в виде желтого маслообразного вещества. К 4-(аминометил)бензонитрилу (250 мг, 1,89 ммоль) добавляли 4-хлор-5-азаиндол (289 мг, 1,89 ммоль) в этаноле (1 мл) и смесь выдерживали при нагревании до 130°C в течение 35 часов, прибавляя минимальное количество этанола в случае его выпаривания. Неочищенный остаток подвергали очистке с помощью флэш-хроматографии, элюируя 4%-12% MeOH-DCM, получая в результате бледно-желтое смолистое вещество, идентифицированное как озаглавленное соединение (300 мг, 1,21 ммоль, выход 64%).
[M+H]+=248,7
D. {4-[(1H-Пирроло[3,2-c]пиридин-4-иламино)-метил]-бензил}-карбаминовой кислоты трет-бутиловый эфир
4-[(1H-Пирроло[3,2-c]пиридин-4-иламино)-метил]-бензонитрил (300 мг, 1,21 ммоль) растворяли в MeOH (30 мл) и охлаждали до 0oC. Гексагидрат хлорида никеля (II) (29 мг, 0,12 ммоль) и ди-трет-бутилдикарбонат (527 мг, 2,42 ммоль) добавляли, затем добавляли частями борогидрид натрия (320 мг, 8,46 ммоль) на протяжении 10 мин. Реакционную смесь перемешивали при 0oC, постепенно доводя температуру до комн.т. в течение 4 часов, после этого MeOH удаляли упариванием. Остаток растворяли в CHCl3 (100 мл), промывали насыщенным раствором NaHCO3 (30 мл), водой (30 мл), насыщенным солевым раствором (30 мл), сушили (Na2SO4), фильтровали и упаривали. Неочищенный остаток подвергали очистке с помощью флэш-хроматографии, элюируя 12% MeOH-DCM, получая в результате серовато-белое твердое вещество, идентифицированное как озаглавленное соединение (180 мг, 0,51 ммоль, выход 42%).
[M+H]+=352,8
E. [4-({1-[4-(2-Оксо-2H-пиридин-1-илметил)-бензил]-1H-пирроло[3,2-c]пиридин-4-иламино}-метил)-бензил]-карбаминовой кислоты трет-бутиловый эфир
{4-[(1H-Пирроло[3,2-c]пиридин-4-иламино)-метил]-бензил}-карбаминовой кислоты трет-бутиловый эфир (70 мг, 0,20 ммоль) растворяли в сухом DMF (7 мл), помещали в атмосферу азота и охлаждали до 0oC. Добавляли NaH (60% в вазелиновом масле, 16 мг, 0,40 ммоль), реакционную массу оставляли нагреваться до комн.т. и перемешивали в течение 15 мин при комн.т.. В течение этого времени происходило изменение цвета раствора от бледно-желтого до темно-красного/оранжевого. Реакционную смесь затем охлаждали до 0°C и 1-(4-бромметил-бензил)-1H-пиридин-2-он (66 мг, 0,24 ммоль) в DMF (3 мл) добавляли по каплям, затем реакционную массу оставляли нагреваться до комн.т. и перемешивали в течение 3 часов при комн.т. В реакционной смеси гасили протекание реакции водой и разбавляли этилацетатом (50 мл). Органический слой промывали насыщенным раствором NaHCO3 (20 мл), водой (3×20 мл), насыщенным солевым раствором (20 мл), сушили (Na2SO4) и упаривали под вакуумом. Неочищенное вещество подвергали очистке с помощью флэш-хроматографии, элюируя 8% MeOH-DCM, получая в результате бледно-желтое смолистое вещество, идентифицированное как озаглавленное соединение (40 мг, 0,073 ммоль, выход 37%).
[M+H]+=550,0
F. 1-{4-[4-(4-Аминометил-бензиламино)-пирроло[3,2-c]пиридин-1-илметил]-бензил}-1H-пиридин-2-он
[4-({1-[4-(2-Оксо-2H-пиридин-1-илметил)-бензил]-1H-пирроло[3,2-c]пиридин-4-иламино}-метил)-бензил]-карбаминовой кислоты трет-бутиловый эфир (40 мг, 0,07 ммоль) растворяли в MeOH (1мл) и обрабатывали 4Н HCl в диоксане (4 мл). Спустя три часа при комн.т. растворитель удаляли в вакууме и остаток подвергали азеотропной отгонке с толуолом (10 мл). Неочищенную реакционную смесь подвергали очистке с помощью препаративной ВЭЖХ, что давало возможность получить серовато-белое твердое вещество, идентифицированное как озаглавленное соединение в виде его соли с бис(трифторуксусной кислотой) (24 мг, 0,035 ммоль, выход 49%).
[M+H]+=449,8
ЯМР (d6-DMSO) δ 4,03 (2H, д, J=5,8 Гц), 4,76 (2H, д, J=6,2 Гц), 5,05 (2H, с), 5,48 (2H, с), 6,21-6,24 (1H, м), 6,38 (1H, д, J=8,9 Hz), 7,12 (1H, д,J=3,2 Гц), 7,21-7,28 (5H, м), 7,39-7,46 (5H,м), 7,53 (1H, т, J=6,2 Гц), 7,64 (1H, д, J=3,3 Гц), 7,77 (1H, дд, J=6,6, 1,7 Гц), 8,17 (3H, с), 9,39 (1H, д, J=5,8 Гц), 12,63 (1H, д, J=5,0 Гц) м.д.
Пример 10
(1-Амино-изохинолин-6-илметил)-{8-[4-(4-метил-пиразол-1-илметил)-бензил]-7,8-дигидро-6H-пиримидо[5,4-b][1,4]оксазин-4-ил}-амин

A. 2-((E)-2-Диметиламино-винил)-терефталонитриловый эфир
Метилтерефталонитрил (1,42 г, 9,99 ммоль) и трет-бутокси бис(диметиламино)метан (Bredereck's reagent) (3,48 г, 19,98 ммоль) растворяли в DMF (15 мл). Реакционную смесь выдерживали при нагревании до 75°C в атмосфере азота в течение 72 ч, после этого растворитель удаляли в вакууме. Растирание в петролейном эфире приводило к получению ярко-желтого твердого вещества, идентифицированного как 2-((E)-2-диметиламино-винил)-терефталонитриловый эфир (1,88 г, 0,95 ммоль, 95%).
1H ЯМР (CD3OD) δ: 3,20 (6H, с), 5,34 (1H, д, J=13,4 Гц), 7,21 (1H, дд, J=8,0 Гц, 1,4 Гц), 7,9 (1H, д, 13,4 Гц), 7,61 (1H, д, J=8,0 Гц), 7,94 (1H, д, J=1,2 Гц)
B. 1-Амино-2-(2,4-диметокси-бензил)-1,2-дигидро-изохинолин-6-карбонитрил
2-((E)-2-Диметиламино-винил)-терефталонитриловый эфир (1,85 г, 9,38 ммоль) растворяли в 1,3-диметил-3,4,5,6-тетрагидро-2(1H)-пиримидиноне (5 мл) и добавляли 2,4-диметоксибензиламин (2,35 г, 14,07 ммоль). Реакционную смесь выдерживали при нагревании до 75°C в атмосфере азота. По истечении 3 ч реакционную смесь охлаждали и добавляли диэтиловый эфир/петролейный эфир (15:85). Желтое твердое вещество отфильтровывали, сушили под вакуумом и идентифицировали как 1-амино-2-(2,4-диметокси-бензил)-1,2-дигидро-изохинолин-6-карбонитрил (2,65 г, 8,38 ммоль, 89%).
[M+H]+=320,0
1H ЯМР (CD3OD) δ: 3,85 (3H, с), 3,92 (3H, с), 5,02 (2H, с), 6,39 (1H, д, J=7,4 Гц), 6,57 (1H, дд, J=8,4 Гц, 2,4 Гц), 6,66 (1H, д, 2,4 Гц), 7,18 (1H, д, 8,4 Гц), 7,24(1H, д, 7,4 Гц), 7,72 (1H, дд, J=8,5 Гц, 1,4 Гц), 7,93 (1H,с), 8,45 (1H, д, J=8,5 Гц)
C. 1-Амино-изохинолин-6-карбонитрил
1-Амино-2-(2,4-диметокси-бензил)-1,2-дигидро-изохинолин-6-карбонитрил (1,6 г, 5,0 ммоль) растворяли в анизоле (17 мл) и трифторуксусной кислоте (20 мл). Реакционную смесь выдерживали при нагревании до 105°C в атмосфере азота в течение 12 ч, после этого реакционную смесь охлаждали, добавляли диэтиловый эфир/петролейный эфир (3:7), полученное в результате твердое вещество отфильтровывали, сушили под вакуумом и идентифицировали как 1-амино-изохинолин-6-карбонитрил (770 мг, 4,54 ммоль, 91%).
[M+H]+=170,0
1H ЯМР (CD3OD) δ: 7,23-7,25 (1H, д, J=6,9 Гц), 7,65 (1H, д, J=6,8 Гц), 8,11 (1H, дд, J=8,7 Гц, 1,6 Гц), 8,33 (1H, с), 8,45 (1H, д, J=8,7 Гц).
D. (1-Амино-изохинолин-6-илметил)-карбаминовой кислоты трет-бутиловый эфир
1-Амино-изохинолин-6-карбонитрил (200 мг, 1,18 ммоль) was растворяли в метаноле (20 мл). Этот раствор охлаждали до 0oC. Добавляли гексагидрат хлорида никеля (II) (28 мг, 0,12 ммоль) и ди-трет-бутилдикарбонат (516 г, 2,36 ммоль), затем частями добавляли борогидрид натрия (313 г, 8,22 ммоль). Реакционную смесь перемешивали при 0°C, постепенно нагревая до комн.т. в течение 3 дней. MeOH удаляли упариванием. Остаток растворяли в CHCl3 (70 мл), промывали насыщенным раствором NaHCO3 (1×30 мл), водой (1×30 мл), насыщенным солевым раствором (1×30 мл), сушили (Na2SO4) и упаривали под вакуумом, получая желтое маслообразное вещество, идентифицированное как (1-амино-изохинолин-6-илметил)-карбаминовой кислоты трет-бутиловый эфир (110 мг, 0,4 ммоль, 34%).
[M+H]+=274,1.
E. 6-Аминометил-изохинолин-1-иламина гидрохлорид
(1-Амино-изохинолин-6-илметил)-карбаминовой кислоты трет-бутиловый эфир (110 мг, 0,40 ммоль) растворяли в 4M HCl в диоксане (40 мл). По истечении 18 ч выдерживания при комн.т. растворитель удаляли под вакуумом, получая светло-коричневое твердое вещество, идентифицированное как 6-аминометил-изохинолин-1-иламина гидрохлорид (67 мг, 0,39 ммоль, 96%).
[M+H]+=174,3
F. (4-((4-Метил-1H-пиразол-1-итл)метил)фенил)метанол
В круглодонную колбу в атмосфере N2 добавляли: (4-(хлорметил)фенил)метанол (10,04 г, 60,9 ммоль), 4-метил-1H-пиразол (5,05 мл, 60,9 ммоль) и сухой MeCN (100 мл). Затем добавляли карбонат калия (9,26 г, 67,0 ммоль) и полученную белую суспензию выдерживали при нагревании до 60°C в течение 18 ч. Летучие компоненты удаляли под вакуумом. Остаток распределяли между EtOAc (100 мл) и водой (150 мл). Водный слой нейтрализовали до pH 7 с помощью 1Н HCl и экстрагировали EtOAc (2×100 мл). Объединенные органические слои промывали водой (100 мл), насыщенным солевым раствором (50 мл), затем сушили(MgSO4), фильтровали и концентрировали под вакуумом. Неочищенный продукт подвергали очистке с помощью хроматографии(10-80% EtOAc в изо-гексане), что давало возмохность получить (4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанол (2,9 г, 14,05 ммоль, выход 23,07%) в виде легкотекучего маслообразного, которое затвердевало при хранении
[M+H]+=203,2
G. 1-(4-(Бромметил)бензил)-4-метил-1H-пиразол
В колбу в атмосфере N2 добавляли: (4-((4-метил-1H-пиразол-1-ил)метил)фенил)метанол (250 мг, 1,236 ммоль), трифенилфосфин (373 мг, 1,421 ммоль) и сухой DCM (5,0 мл). Охлаждали смесь в бане со льдом перед тем, как добавить тетрабромметан (451 мг, 1,360 ммоль). Перемешивали смесь при комн.т. в течение 1 ч. Концентрировали под вакуумом и очищали с помощью колоночной хроматографии (0-20% EtOAc в изо-гексане), что давало возможность получить 1-(4-(бромметил)бензил)-4-метил-1H-пиразол (0,33 г, 1,182 ммоль, выход 96%) в виде маслообразного вещества, которое затвердевало при хранении, становилось белым твердым веществом.
[M+H]+=265,1/267,1
H. 2-[(6-Хлор-5-метокси-4-пиримидинил)амино]-этанол
К раствору 4,6-дихлор-5-метоксипиримидина (1,00 г, 5,59 ммоль) в диоксане (15 мл) добавляли 2-аминоэтанол (348 мг, 5,70 ммоль) и карбонат калия (926 мг, 6,70 ммоль). Реакционную смесь кипятили при 125°C. По окончании реакции охлаждали реакционную смесь до комнатной температуры, полученную в результате суспензию фильтровали, фильтрат концентрировали под вакуумом. И отфильтрованное твердое вещество, и твердое вещество, полученное в результате концентрирования фильтрата, были идентифицированы как озаглавленное соединение и объединены, что давало возможность получить 1,2 г (5,89 ммоль, количественный выход) озаглавленного соединения.
[M+H]+=203,9
I. 4-Хлор-7,8-дигидро-6H-пиримидо[5,4-b][1,4]оксазин
2-[(6-Хлор-5-метокси-4-пиримидинил)амино]-этпнол (1,2 г, 5,89 ммоль) растворяли в растворе трибромида бора (1,0 M в DCM, 40 мл), и полученную в результате реакционную массу нагревали до кипения и перемешивали в течение 3 ч. Реакционную смесь охлаждали до комн.т., обрабатывали ледяной водой (30 мл) и экстрагировали EtOAc (3×50мл). Органический слой сушили (MgSO4), фильтровали и концентрировали под вакуумом, что давало возможность получить озаглавленное соединение в виде HBr соли (0,96 г, 3,81 ммоль, выход 65%).
[M+H]+=172,0
J. 4-Хлор-8-[4-(4-метил-пиразол-1-илметил)-бензил]-7,8-дигидро-6H-пиримидо[5,4-b][1,4]оксазин
4-Хлор-7,8-дигидро-6H-пиримидо[5,4-b][1,4]оксазин (109 мг, 0,64 ммоль) растворяли в DMF (2 мл), добавляли к полученному раствору ди-изо-пропилэтиламин (332 мкл, 1,91 ммоль), затем добавляли 1-(4-(бромметил)бензил)-4-метил-1H-пиразол (168 мг, 0,64 ммоль). Реакционную массу перемешивали при комнатной температуре в течение ночи. Реакционную смесь разбавляли CHCl3 (40 мл) и последовательно промывали водой (5×40 мл) и насыщенным солевым раствором (40 мл). Органический слой сушили (MgSO4), фильтровали и концентрировали, что давало возможность получить желтое твердое вещество. Неочищенное вещество подвергали очистке с помощью флэш-хроматографии (8% MeOH/DCM - 10% MeOH/DCM/1% NH4OH). Фракции, содержащие озаглавленное соединение, концентрировапли, что давало возможность получить озаглавленное соединение в виде светло-желтого маслообразного вещества (119 мг, 0,33 ммоль, выход 52.6%).
LCMS: 355,9 @ 6,29 мин
1H ЯМР: (CDCl3) 2,10 (3H, с), 4,29 (2H, т, J=9,4 Гц), 4,64 (2H, ушир. с), 5,21 (2H, с), 5,25 (2H, с), 7,19 (2H, д, J=8,1 Гц), 7,26 (1H, ушир. с), 7,34-7,37 (2H, м), 7,69 (2H, д, J=7,9 Гц).
K. (1-Амино-изохинолин-6-илметил)-{8-[4-(4-метил-пиразол-1-илметил)-бензил]-7,8-дигидро-6H-пиримидо[5,4-b][1,4]оксазин-4-ил}-амин
4-Хлор-8-[4-(4-метил-пиразол-1-илметил)-бензил]-7,8-дигидро-6H-пиримидо[5,4-b][1,4]оксазин (100 мг, 0,28 ммоль) и 6-(аминометил)изохинолин-1-иламин (48,7 мг, 0,28 ммоль) суспендировали в этаноле (0,5 мл) и нагревали микроволновым излучением (CEM фокусирующая микроволновая печь, Power 300W, 120°C в течение 90 мин). Реакционную массу фильтровали и твердое вещество промывали этанолом. Фильтрат концентрировали при пониженном давлении. Неочищенное вещество подвергали очистке с помощью препаративной ВЭЖХ. Фракции, содержащие целевое соединение, объединяли и сушили посредством лиофилизации, что давало возможность получить серовато-белое твердое вещество, идентифицированное как озаглавленное соединение (19,5 мг, 0,027 ммоль, выход 10%).
[M+H]+=493,0
ВЭЖХ: 99% чистота
1H ЯМР d6-DMSO: 1,98 (3H, с), 3,84 (2H, т, J=8,9 Гц), 4,45 (2H, т, J=8,9 Гц), 4,76 (2H, д, J=5,9 Гц), 4,92 (2H, с), 5,24 (2H, с), 7,21-7,17 (3H, м), 7,25 (1H, с), 7,50 (1H, с), 7,52 (2H, д, J=2,3 Гц), 7,56 (1H, дд, J=8,7, 1,6 Гц), 7,66 (1H, д, J=6,8 Гц), 7,69 (1H, с), 8,36 (1H, с), 8,52-8,46 (2H, м), 8,98 (2H, с).
Соединения нижеприведенных таблиц были синтезированы, как описано в общих способах выше и как описано в вышеприведенных Примерах 7 и 10.
|



















|
|
Биологические методы
Способность соединений формулы (I) ингибировать калликреин плазмы крови можно определить, используя нижеуказанные биологические испытания:
Определение IC50 для калликреина плазмы крови
Ингибирующую активность в отношнении калликреина плазмы крови in vitro определяли, используя общепринятые опубликованные методы (см.,например, Johansen et al., Int. J. Tiss. Reac. 1986, 8, 185; Shori et al., Biochem. Pharmacol., 1992, 43, 1209; Stürzebecher et al., Biol. Chem. Hoppe-Seyler, 1992, 373, 1025). Человеческий калликреин плазмы крови (Protogen) инкубировали при 37°C с флуорогенным субстратом H-DPro-Phe-Arg-AFC и с тестируемым соединением в различных концентрациях. Остаточную ферментативную активность (начальную скорость реакции) определяли путем измерения изменения величины оптического поглощения при 410 нм и величину IC50 для тестируемого соединения определяли.
Результаты, полученные при этих испытаниях, представлены в нижеприведенной Таблице 6:
|
Отобранные соединения далее подвергались проверке на их ингибирующую активность в отношении родственного фермента KLK1. Способность соединений формулы (I) ингибировать KLK1 можно определить, используя нижеследующее биологическое испытание:
Определение IC50 для KLK1
Ингибирующую активность в отношении KLK1 in vitro определяли, используя общепринятые опубликованные методы (см., например, Johansen et al., Int. J. Tiss. Reac. 1986, 8, 185; Shori et al., Biochem. Pharmacol., 1992, 43, 1209; Stürzebecher et al., Biol. Chem. Hoppe-Seyler, 1992, 373, 1025). Человеческий KLK1 (Callbiochem) инкубировали при 37°C с флуорогенным субстратом H-DVal-Leu-Arg-AFC и с тестируемым соединением в различных концентрациях. Остаточную ферментативную активность (начальную скорость реакции) определяли путем измерения изменения величины оптического поглощения при 410 нм и величину IC50 для тестируемого соединения определяли.
Результаты, полученные при этих испытаниях, представлены в нижеприведенной Таблице 7:
|
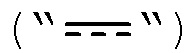
Бензиламиновые производные как ингибиторы калликреина плазмы
Бензиламиновые производные
Ингибиторы ферментов
N-((гет)арилметил)-гетероарил-карбоксамидные соединения в качестве ингибиторов плазменного калликреина
Азотсодержащие гетероциклические производные, полезные в качестве ингибитора калликреина плазмы
Полиморфные модификации n-[(3-фтор-4-метоксипиридин-2-ил)метил]-3-(метоксиметил)-1-({ 4-[(2-оксопиридин-1-ил)метил]фенил} метил)пиразол-4-карбоксамида и их соли
Производные пиразола в качестве ингибиторов калликреина
Бензиламиновые производные как ингибиторы калликреина плазмы
Бензиламиновые производные
Ингибиторы ферментов
N-((гет)арилметил)-гетероарил-карбоксамидные соединения в качестве ингибиторов плазменного калликреина
Азотсодержащие гетероциклические производные, полезные в качестве ингибитора калликреина плазмы
Полиморфные модификации n-[(3-фтор-4-метоксипиридин-2-ил)метил]-3-(метоксиметил)-1-({ 4-[(2-оксопиридин-1-ил)метил]фенил} метил)пиразол-4-карбоксамида и их соли
Производные пиразола в качестве ингибиторов калликреина

