Результат интеллектуальной деятельности: ЗАМЕЩЕННЫЙ 2-АМИНОПИРИДИН В КАЧЕСТВЕ ИНГИБИТОРА ПРОТЕИНКИНАЗЫ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
Настоящее изобретение относится к новым производным 2-аминопиридина, обладающим ингибирующей активностью в отношении протеинкиназы, к получению данных соединений и к фармацевтическим композициям, содержащим данные соединения, а также к применению указанных соединений и фармацевтических композиций для лечения заболеваний, ассоциированных с протеинкиназой.
УРОВЕНЬ ТЕХНИКИ
Пролиферация, апоптоз, метастаз опухолей и т.п. тесно связаны с анормальной активностью протеинкиназ в ряде внутриклеточных и внеклеточных путей сигнальной трансдукции. Анормальная активность протеинкиназ не только прямо ассоциирована с опухолью, но также приводит к возникновению ряда заболеваний человека, ассоциированных с воспалительными или пролиферативными ответами, таких как ревматоидный артрит, сердечно-сосудистые заболевания, заболевания нервной системы, астма, псориаз и тому подобное. В настоящее время известно более четырехсот заболеваний человека, которые прямо или косвенно ассоциированы с протеинкиназами, поэтому протеинкиназа является важной мишенью для лекарственных средств.
Киназа анапластической лимфомы (ALK), в качестве рецептора, обладающего тирозинкиназной активностью, является членом суперсемейства рецепторов инсулина и играет важную роль в росте и развитии опухолевых клеток. Ген ALK может образовывать гибридные гены с генами ряда других белков, экспрессироваться с образованием ALK-белка и также может генерировать изменения, такие как мутация, амплификация и тому подобное. В 1997 году впервые описана онкогенная перестройка гена ALK на коротком плече хромосомы 2 клеток анапластической (allobiosis) крупноклеточной лимфомы, затем данная перестройка также была найдена в клетках других злокачественных опухолей, включая диффузную крупноклеточную В-клеточную лимфому и злокачественный тканевой гистиоцитоз, а также ряд солидных опухолей, включая воспалительную миофибробластическую опухоль, плоскоклеточную карциному пищевода и, согласно недавно опубликованным данным, нейробластому и немелкоклеточную карциному легкого (NSCLC).
В 2007 впервые описано, что ген ALK может кодировать и продуцировать ALK после слияния с геном EML4, приводящего к образованию гибридного гена* и тем самым стимулировать рост клеток рака легкого. Образование гибридного гена EML4-ALK является следствием инсерции короткого плеча хромосомы 2, в настоящее время найдено много типов таких гибридных генов. Показано, что все найденные гибридные гены имеют биологические функции, и продукт, который они экспрессируют, представляет собой химерную тирозинкиназу; количество таких данных постоянно увеличивается, начиная с исследования NSCLC в 2007.
Обнаружение гибридного гена EML4-ALK и уникального действия ALK-ингибитора в подгруппах населения, несущих данный ген, позволило выделить несколько подтипов NSCLC, таких как NSCLC, обусловленная мутацией в гене EGFR (рецептора эпидермального фактора роста), мутацией в гене KRAS, гибридным геном EML4-ALK и т.п., в соответствии с разными молекулярными механизмами патогенеза. В основном только у приблизительно 3-7% пациентов с NSCLC присутствует гибридный ген EML4-ALK. Гибридный ген EML4-ALK присутствует главным образом у некурящих пациентов с аденокарциномой легкого, и он не несовместим ни с EGFR-мутацией, ни с KRAS-мутацией. Исследование, проведенное в Китае в 2010 году, показало, что гибридный ген EML4-ALK присутствует у 16,13% пациентов с аденокарциномой легкого, что значительно выше, чем у таких же пациентов в Европе и Америке; и у 19,23% некурящих пациентов с аденокарциномой легкого; причем гибридный ген EML4-ALK присутствует у 42,8% пациентов с аденокарциномой легкого, у которых отсутствуют мутации в генах EGFR и KRAS.
Уже изучено большое количество соединений, обладающих ингибирующей активностью в отношении протеинкиназ, и некоторые ингибиторы протеинкиназ уже используются в качестве противораковой терапии, однако может возникать устойчивость к данным лекарствам. Поэтому существует необходимость в разработке новых ингибиторов протеинкиназ, таких как ингибиторы ALK-киназы, для предупреждения, ослабления и/или лечения рака, обусловленного протеинкиназами (такими как ALK), такого как ALK-позитивная немелкоклеточная карцинома легкого (NSCLC) и тому подобное.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
В настоящем изобретении предложено соединение Формулы (I)
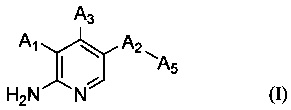 ,
,
где
А1 выбран из группы, состоящей из атома водорода, -O-(CHR1)-A4, -CH2OR2 и арила, имеющего в качестве заместителей один или более чем один R3;
R1 выбран из группы, состоящей из метила и метила, имеющего в качестве заместителей 1-3 атома галогена;
А4 выбран из группы, состоящей из арила, необязательно имеющего в качестве заместителей один или более чем один R4;
R2 выбран из группы, состоящей из арила, необязательно имеющего в качестве заместителей один или более чем один R3;
R3 выбран из группы, состоящей из атома галогена, -SO2(C1-6 алкил), -SO2NR6R7, -NR6R7, -NHSO2(C1-6 алкил) и -P(O)R6R7;
R4 выбран из группы, состоящей из атома галогена, C1-6 алкила, -NR6R7 и -P(O)R6R7;
каждый из R6 и R7 независимо выбран из группы, состоящей из атома водорода и С1-6 алкила, или R6 и R7 соединены и вместе с атомом, к которому они присоединены, образуют 3-12-членный гетероалициклил;
А2 выбран из группы, состоящей из фенила, пиридинила, пиримидинила и пиразолила, все из которых необязательно имеют один или более чем один заместитель, выбранный из группы, состоящей из атома галогена и -OC1-6 алкила, у которого каждый атом водорода C1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или 3-12-членным гетероалициклилом;
А5 представляет собой 3-12-членный гетероалициклил, который необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
=O,
незамещенного С1-6 алкила и
С1-6 алкила, имеющего один или более чем один заместитель, независимо выбранный из группы, состоящей из гидрокси, карбоксила и 3-12-членного гетероалициклила, и
3-12-членного гетероалициклила;
А3 выбран из группы, состоящей из атома водорода, -NH-арила, гетероарила, имеющего в качестве заместителя арил, гетероарила, имеющего в качестве заместителя гетероарил, гетероарила, имеющего в качестве заместителя арилалкил, гетероарила, имеющего в качестве заместителя гетероарилалкил, гетероарилэтинила, имеющего в качестве заместителя арилалкил, и гетероарилэтинила, имеющего в качестве заместителя, гетероарилалкил, где каждый арил и гетероарил необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
атома галогена,
С1-6 алкила, необязательно имеющего в качестве заместителей атом галогена, гидрокси или 3-12-членный гетероалициклил, и
-ОН, -OC1-6 алкила, -CN, -СООН, -C1-6-алкил-NH2, -С1-6-алкил-NH(C1-6 алкил), -С1-6-алкил-N(С1-6 алкил)2, -COO-C1-6 алкила, -SO2(C1-6 алкил), -SO2N(C1-6 алкил)2, -SO2NH(C1-6 алкил), -NR6R7, -NHSO2(C1-6 алкил) и -P(O)R6R7;
при условии, что
A1 и А3 одновременно не являются атомами водорода и один из А1 и А3 должен представлять собой атом водорода,
и его фармацевтически приемлемые соли, стереоизомеры и энантиомеры и их смеси.
Согласно некоторым вариантам осуществления изобретения, когда в соединении Формулы (I) A1 представляет собой -O-(CHR1)-A4 и R1 представляет собой метил, А2 имеет в качестве заместителей по меньшей мере один -ОС1-6 алкил; когда A1 представляет собой арил, имеющий в качестве заместителей один или более чем один R3, и R3 представляет собой -NR6R7, каждый из R6 и R7 независимо выбран из группы, состоящей из C1-6 алкила, или R6 и R7 соединены и вместе с атомом, к которому они присоединены, образуют 3-12-членный гетероалициклил.
Согласно некоторым вариантам осуществления изобретения А3 выбран из группы, состоящей из -NH-фенила, гетероарила, имеющего в качестве заместителя фенил, гетероарила, имеющего в качестве заместителя гетероарил, гетероарила, имеющего в качестве заместителя фенилметил, гетероарила, имеющего в качестве заместителя гетероарилметил, гетероарилэтинила, имеющего в качестве заместителя фенилметил, и гетероарилэтинила, имеющего в качестве заместителя гетероарилметил, где каждый фенил и гетероарил необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
атома галогена,
С1-6 алкила, необязательно имеющего в качестве заместителей атом галогена, гидрокси или 3-12-членный гетероалициклил, и
-ОН, -OC1-6 алкила, -CN, -СООН, -C1-6 алкил-NH2, -C1-6 алкил-NH(C1-6 алкил), -С1-6 алкил-N(С1-6 алкил)2, -COOC1-6 алкила, -SO2(C1-6 алкил), -SO2N(C1-6 алкил)2, -SO2NH(C1-6 алкил), -NR6R7, -NHSO2(C1-6 алкил) и -P(O)R6R7.
Согласно некоторым предпочтительным вариантам осуществления изобретения А3 выбран из группы, состоящей из -NH-фенила, гетероарила, имеющего в качестве заместителя фенил, гетероарила, имеющего в качестве заместителя гетероарил, гетероарила, имеющего в качестве заместителя фенилметил, гетероарила, имеющего в качестве заместителя гетероарилметил, гетероарилэтинила, имеющего в качестве заместителя фенилметил, и гетероарилэтинила, имеющего в качестве заместителя гетероарилметил, где каждый фенил и гетероарил необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
атома галогена,
C1-4 алкила, необязательно имеющего в качестве заместителей атом галогена, гидрокси или 5- или 6-членный гетероалициклил, и
-ОН, -ОС1-4 алкила, -CN, -СООН, -С1-4 алкил-Н2, -С1-4 алкил-Н(С1-4 алкил), -С1-4 алкил-N(С1-4 алкил)2, -COOC1-4 алкила, -SO2(C1-4 алкил), -SO2N(C1-4 алкил)2, -SO2NH(C1-4 алкил), -NH2, -NH(C1-4 алкил), -N(C1-4 алкил)2, -NHSO2(C1-4 алкил) и -Р(O)(С1-4 алкил)2.
Согласно некоторым более предпочтительным вариантам осуществления изобретения А3 выбран из группы, состоящей из -NH-фенила, пиразолила, имеющего в качестве заместителя фенил, пиразолила, имеющего в качестве заместителя фенилметил, и пиразолилэтинила, имеющего в качестве заместителя фенилметил, где фенил необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
атома галогена,
С1-4 алкила, имеющего в качестве заместителей атом галогена или гидрокси, и
-ОН, -ОС1-4 алкила, -CN, -СООН, -С1-4 алкил NH2, -С1-4 алкил NH(C1-4 алкил), -С1-4 алкил N(C1-4 алкил)2, -COOC1-4 алкила, -SO2(C1-4 алкил), -NH2, -NH(C1-4 алкил), -N(C1-4 алкил)2, -NHSO2(C1-4 алкил), -SO2N(C1-4 алкил)2, -SO2NH(C1-4 алкил) и -Р(O)(С1-4 алкил)2.
Согласно некоторым наиболее предпочтительным вариантам осуществления изобретения А3 выбран из группы, состоящей из -NH-фенила, пиразолила, имеющего в качестве заместителя фенил, пиразолила, имеющего в качестве заместителя фенилметил, и пиразолилэтинила, имеющего в качестве заместителя фенилметил, где фенил необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из F, Cl, трифторметила, -СООН, -СН2ОН, -ОСН3, -ОС2Н5, -CN, -SO2NHCH(CH3)2, -СООСН3, -SO2CH3, -NH2 и -Р(O)(СН3)2.
Согласно некоторым вариантам осуществления настоящего изобретения А3 представляет собой атом водорода.
Согласно некоторым вариантам осуществления настоящего изобретения, когда A3 представляет собой атом водорода, и А1 представляет собой арил, имеющий в качестве заместителей один или более чем один R3, и R3 представляет собой -NR6R7, каждый из R6 и R7 независимо выбран из группы, состоящей из С1-6 алкила, или R6 и R7 соединены и вместе с атомом, к которому они присоединены, образуют 3-12-членный гетероалициклил.
Согласно некоторым вариантам осуществления изобретения R2 выбран из группы, состоящей из фенила, необязательно имеющего в качестве заместителей один или более чем один R3. Согласно некоторым предпочтительным вариантам осуществления изобретения R2 выбран из группы, состоящей из фенила, необязательно имеющего в качестве заместителей один или более чем один R3, выбранный из группы, состоящей из атома галогена, -SO2(С1-6алкил), -SO2N(C1-6алкил)2, -SO2NH(C1-6алкил), -NH(С1-6алкил), -N(С1-6алкил)2, -NHSO2(С1-6алкил) и -Р(O)(С1-6алкил)2. Согласно более предпочтительным вариантам осуществления изобретения R2 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R3, выбранный из группы, состоящей из F, Cl, -SO2CH3, -SO2N(CH3)C2H5, -SO2NHCH(CH3)2, -NHCH3, -N(CH3)C2H5, -NHSO2CH3 и -P(O)(CH3)2.
Согласно некоторым вариантам осуществления изобретения R1 выбран из группы, состоящей из метила и трифторметила.
Согласно некоторым вариантам осуществления изобретения А4 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R4. Согласно некоторым предпочтительным вариантам осуществления изобретения А4 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R4, где R4 выбран из группы, состоящей из атома галогена, С1-6 алкила, имеющего в качестве заместителя атом галогена, -NR6R7 и -P(O)R6R7, где каждый из R6 и R7 независимо выбран из группы, состоящей из С1-6 алкила. Согласно некоторым более предпочтительным вариантам осуществления изобретения А4 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R4, где R4 выбран из группы, состоящей из F, Cl, метила, имеющего в качестве заместителя атом галогена, этила, имеющего в качестве заместителя атом галогена, -N(CH3)2 и -Р(O)(CH3)2. Согласно некоторым более предпочтительным вариантам осуществления изобретения А4 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R4, где R4 выбран из группы, состоящей из F, Cl, -CHF2, -CF3, -CF2CH3, -N(CH3)2 и -Р(O)(СН3)2, и А4 имеет в качестве заместителей по меньшей мере один атом F.
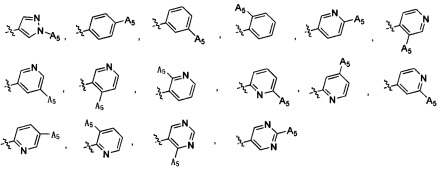
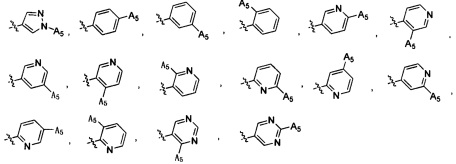
Согласно некоторым вариантам осуществления изобретения А2 выбран из группы, состоящей из фенила, пиридинила, пиримидинила и пиразолила. Согласно некоторым предпочтительным вариантам осуществления изобретения А2 выбран из группы, состоящей из  ,
,  ,
,  ,
, 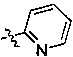 ,
, 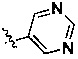 и
и 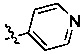 .
.
Согласно некоторым вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из атома галогена и -OC1-6 алкила, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом, морфолинилом, тетрагидрофурилом, пиперидинилом, пиперазинилом, тетрагидропиридилом, дигидропиридилом, тетрагидротиенилом, пирролидинилом, оксазолидинилом, тиазолидинилом, имидазолидинилом, изоксазолидинилом, изотиазолидинилом, пиразолидинилом, тиоморфолинилом, пиперазин-2-он-илом, пирролинилом, дигидрофуранилом или дигидротиенилом. Согласно некоторым предпочтительным вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из атома галогена и -ОС1-6 алкила, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или морфолинилом. Согласно некоторым более предпочтительным вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из F, Cl, метокси, этокси, -ОСН2СН2ОН и 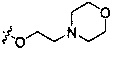 .
.
Согласно некоторым вариантам осуществления изобретения А5 представляет собой 5- или 6-членный гетероалициклил. Согласно более предпочтительным вариантам осуществления изобретения А5 представляет собой морфолинил, тетрагидрофурил, пиперидинил, пиперазинил, тетрагидропиридил, дигидропиридил, тетрагидротиенил, пирролидинил, оксазолидинил, тиазолидинил, имидазолидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, тиоморфолинил, пиперазин-2-он-ил, пирролинил, дигидрофурил или дигидротиенил. Согласно некоторым более предпочтительным вариантам осуществления изобретения А5 представляет собой морфолинил, 1,2,3,4-тетрагидропиридил, 1,2,3,6-тетрагидропиридил, 2,3,4,5-тетрагидропиридил, пиперазинил, пиперазин-2-он-ил или пиперидил. Согласно некоторым более предпочтительным вариантам осуществления изобретения А5 представляет собой пиперазин-1-ил, пиперазин-2-ил, пиперазин-3-ил, пиперидин-4-ил, пиперидин-1-ил, пиперидин-2-ил, пиперидин-3-ил, морфолин-4-ил, морфолин-2-ил, морфолин-3-ил, 1,2,3,4-тетрагидропиридин-4-ил, 1,2,3,6-тетрагидропиридин-4-ил, 2,3,4,5-тетрагидропиридин-4-ил или пиперазин-2-он-ил. Согласно некоторым наиболее предпочтительным вариантам осуществления изобретения А5 представляет собой 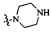 ,
, 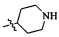 ,
, 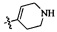 ,
, 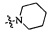 ,
, 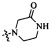 или
или 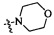 .
.
Согласно некоторым вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
=O,
незамещенного С1-6 алкила и
С1-6 алкила, имеющего один или более чем один заместитель, независимо выбранный из группы, состоящей из гидрокси, карбоксила и 3-12-членного гетероалициклила, и
3-12-членного гетероалициклила, где 3-12-членный гетероалициклил, в свою очередь, необязательно имеет заместители, выбранные из группы, состоящей из C1-6 алкила, =O, -ОН, -СООН, -CN, атома галогена, -NH(C1-6 алкил) и -N(C1-6 алкил)2.
Согласно некоторым предпочтительным вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из =O, метила, этила, н-пропила, изопропила и 5- или 6-членного гетероалициклила, где метил, этил, н-пропил и изопропил необязательно имеют один или более чем один заместитель, независимо выбранный из группы, состоящей из -ОН, -СООН и 5- или 6-членного гетероалициклила, где данный 5- или 6-членный гетероалициклил, в свою очередь, необязательно имеет заместитель(и), выбранный(ые) из группы, состоящей из метила, этила, н-пропила, изопропила, =O, -ОН, -СООН, -CN, атома галогена, -NH(C1-3 алкил) и -N(C1-3 алкил)2. Согласно некоторым более предпочтительным вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из метила, этила, н-пропила, изопропила, =O, пиперидила и пиперазинила, где пиперидил или пиперазинил необязательно имеет в качестве заместителя метил.
Согласно некоторым вариантам осуществления изобретения структура -А2-А5 представляет собой
 .
.
Согласно некоторым предпочтительным вариантам осуществления изобретения структура -А2-А5 представляет собой 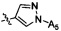 ,
, 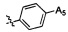 ,
, 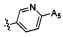 ,
, 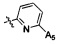 ,
, 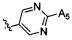 ,
, 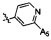 ,
, 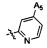 ,
, 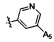 .
.
Согласно некоторым предпочтительным вариантам осуществления изобретения, когда A3 представляет собой атом водорода, A1 представляет собой -O-(CHR1)-A4 и R1 представляет собой метил, А2 выбран из группы, состоящей из фенила, пиридила, пиримидинила и пиразолила и А2 имеет в качестве заместителей по меньшей мере один -ОС1-6 алкил, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или 3-12-членным гетероалициклилом.
Согласно другому аспекту настоящего изобретения предложено соединение Формулы (II)
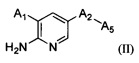 ,
,
где
A1 выбран из группы, состоящей из -O-(CHR1)-A4, -CH2OR2 и арила, имеющего в качестве заместителей один или более чем один R3;
R1 выбран из группы, состоящей из метила и метила, имеющего в качестве заместителей 1-3 атома галогена;
А4 выбран из группы, состоящей из арила, необязательно имеющего в качестве заместителей один или более чем один R4;
R2 выбран из группы, состоящей из арила, необязательно имеющего в качестве заместителей один или более чем один R3;
R3 выбран из группы, состоящей из атома галогена, -SO2(C1-6 алкил), -SO2NR6R7, -NR6R7, -NHSO2(C1-6 алкил) и -P(O)R6R7;
R4 выбран из группы, состоящей из атома галогена, С1-6 алкила, -NR6R7 и -P(O)R6R7;
R6 и R7 независимо выбраны из группы, состоящей из атома водорода и С1-6 алкила, или R6 и R7 соединены и вместе с атомом, к которому они присоединены, образуют 3-12-членный гетероалициклил;
А2 выбран из группы, состоящей из фенила, пиридинила, пиримидинила и пиразолила, все из которых необязательно имеют один или более чем один заместитель, выбранный из группы, состоящей из атома галогена и -ОС1-6 алкила, у которого каждый атом водорода C1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или 3-12-членным гетероалициклилом;
А5 представляет собой 3-12-членный гетероалициклил, который необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
=O,
незамещенного С1-6 алкила и
С1-6 алкила, имеющего один или более чем один заместитель, независимо выбранный из группы, состоящей из гидрокси, карбоксила и 3-12-членного гетероалициклила, и
3-12-членного гетероалициклила,
и его фармацевтически приемлемые соли, стереоизомеры и энантиомеры и их смеси.
Согласно некоторым вариантам осуществления изобретения, когда в соединении Формулы (II) A1 представляет собой арил, имеющий в качестве заместителей один или более чем один R3, и R3 представляет собой -NR6R7, каждый из R6 и R7 независимо выбран из группы, состоящей из С1-6 алкила, или R6 и R7 соединены и вместе с атомом, к которому они присоединены, образуют 3-12-членный гетероалициклил.
Согласно некоторым вариантам осуществления изобретения R2 выбран из группы, состоящей из фенила, необязательно имеющего в качестве заместителей один или более чем один R3. Согласно некоторым предпочтительным вариантам осуществления изобретения R2 выбран из группы, состоящей из фенила, необязательно имеющего в качестве заместителей один или более чем один R3, выбранный из группы, состоящей из атома галогена, -SO2(C1-6алкил), -SO2N(С1-6алкил)2, -SO2NH(С1-6алкил), -NH(С1-6алкил), -N(С1-6алкил)2, -NHSO2(С1-6алкил) и -Р(O)(С1-6алкил)2. Согласно некоторым более предпочтительным вариантам осуществления изобретения R2 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R3, выбранный из группы, состоящей из F, Cl, -SO2CH3, -SO2N(CH3)C2H5, -SO2NHCH(CH3)2, -NHCH3, -N(CH3)C2H5, -NHSO2CH3 и -Р(O)(СН3)2.
Согласно некоторым вариантам осуществления изобретения R1 выбран из группы, состоящей из метила и трифторметила.
Согласно некоторым вариантам осуществления изобретения А4 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R4. Согласно некоторым предпочтительным вариантам осуществления изобретения А4 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R4, выбранный из группы, состоящей из атома галогена, С1-6 алкила, имеющего в качестве заместителя атом галогена, -NR6R7 и -P(O)R6R7, где каждый из R6 и R7 независимо выбран из группы, состоящей из С1-6 алкила. Согласно некоторым предпочтительным вариантам осуществления изобретения А4 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R4, выбранный из группы, состоящей из F, Cl, метила, имеющего в качестве заместителя атом галогена, этила, имеющего в качестве заместителя атом галогена, -N(OH3)2 и -Р(O)(СН3)2. Согласно некоторым более предпочтительным вариантам осуществления изобретения А4 выбран из группы, состоящей из фенила, имеющего в качестве заместителей один или более чем один R4, выбранный из группы, состоящей из F, Cl, -CHF2, -CF3, -CF2CH3, -N(CH3)2 и -Р(O)(СН3)2, и А4 имеет в качестве заместителей по меньшей мере один атом F.
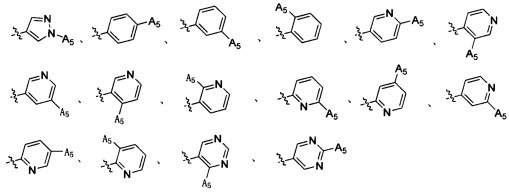
Согласно некоторым вариантам осуществления изобретения А2 выбран из группы, состоящей из фенила, пиридинила, пиримидинила и пиразолила. Согласно некоторым предпочтительным вариантам осуществления изобретения А2 выбран из группы, состоящей из 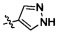 ,
, 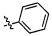 ,
, 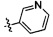 ,
, 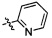 ,
, 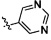 и
и 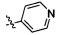 .
.
Согласно некоторым вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из атома галогена и -OC1-6 алкила, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом, морфолинилом, тетрагидрофурилом, пиперидинилом, пиперазинилом, тетрагидропиридилом, дигидропиридилом, тетрагидротиенилом, пирролидинилом, оксазолидинилом, тиазолидинилом, имидазолидинилом, изоксазолидинилом, изотиазолидинилом, пиразолидинилом, тиоморфолинилом, пиперазин-2-он-илом, пирролинилом, дигидрофурилом или дигидротиенилом. Согласно некоторым предпочтительным вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из атома галогена и -ОС1-6 алкила, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или морфолинилом. Согласно некоторым более предпочтительным вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из F, Cl, метокси, этокси, -ОСН2СН2ОН и 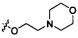 .
.
Согласно некоторым вариантам осуществления изобретения А5 представляет собой 5- или 6-членный гетероалициклил. Согласно более предпочтительным вариантам осуществления изобретения А5 представляет собой морфолинил, тетрагидрофурил, пиперидинил, пиперазинил, тетрагидропиридил, дигидропиридил, тетрагидротиенил, пирролидинил, оксазолидинил, тиазолидинил, имидазолидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, тиоморфолинил, пиперазин-2-он-ил, пирролинил, дигидрофурил или дигидротиенил. Согласно некоторым предпочтительным вариантам осуществления изобретения А5 представляет собой морфолинил, 1,2,3,4-тетрагидропиридил, 1,2,3,6-тетрагидропиридил, 2,3,4,5-тетрагидропиридил, пиперазинил, пиперазин-2-он-ил или пиперидил. Согласно некоторым более предпочтительным вариантам осуществления изобретения А5 представляет собой пиперазин-1-ил, пиперазин-2-ил, пиперазин-3-ил, пиперидин-4-ил, пиперидин-1-ил, пиперидин-2-ил, пиперидин-3-ил, морфолин-4-ил, морфолин-2-ил, морфолин-3-ил, 1,2,3,4-тетрагидропиридин-4-ил, 1,2,3,6-тетрагидропиридин-4-ил, 2,3,4,5-тетрагидропиридин-4-ил или пиперазин-2-он-ил. Согласно некоторым наиболее предпочтительным вариантам осуществления изобретения А5 представляет собой 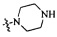 ,
, 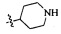 ,
, 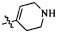 ,
, 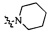 ,
, 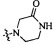 или
или 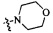 .
.
Согласно некоторым вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
=O,
незамещенного С1-6 алкила и
С1-6 алкила, имеющего один или более чем один заместитель, независимо выбранный из группы, состоящей из гидрокси, карбоксила и 3-12-членного гетероалициклила, и
3-12-членного гетероалициклила, где 3-12-членный гетероалициклил, в свою очередь, необязательно имеет заместитель(и), выбранный(ые) из группы, состоящей из С1-6 алкила, =O, -ОН, -СООН, -CN, атома галогена, -NH(C1-6 алкил) и -N(C1-6 алкил)2.
Согласно некоторым предпочтительным вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из =O, метила, этила, н-пропила, изопропила и 5- или 6-членного гетероалициклила, где метил, этил, н-пропил и изопропил необязательно имеют один или более чем один заместитель, независимо выбранный из группы, состоящей из -ОН, -СООН и 5- или 6-членного гетероалициклила, где данный 5-или 6-членный гетероалициклил, в свою очередь, необязательно имеет заместитель(и), выбранный(ые) из группы, состоящей из метила, этила, н-пропила, изопропила, =O, -ОН, -СООН, -CN, атома галогена, -NH(C1-3 алкил) и -N(C1-3 алкил)2. Согласно некоторым более предпочтительным вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из метила, этила, н-пропила, изопропила, =O, пиперидила и пиперазинила, где пиперидил и пиперазинил необязательно имеет в качестве заместителя метил.
Согласно некоторым вариантам осуществления структура -А2-А5 представляет собой
 .
.
 .
.
Согласно некоторым предпочтительным вариантам осуществления изобретения структура -А2-А5 представляет собой 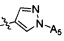 ,
, 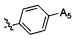 ,
, 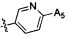 ,
, 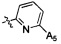 ,
, 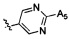 ,
, 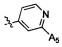 ,
, 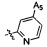 ,
, 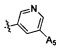 .
.
Согласно некоторым предпочтительным вариантам осуществления изобретения, когда A1 представляет собой -O-(CHR1)-A4 и R1 представляет собой метил, А2 выбран из группы, состоящей из фенила, пиридила, пиримидинила и пиразолила, и А2 имеет в качестве заместителей по меньшей мере один -ОС1-6 алкил, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или 3-12-членным гетероалициклилом.
Согласно другому аспекту настоящего изобретения предложено соединение Формулы (III) или Формулы (IV)
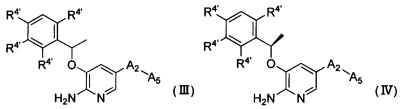 ,
,
где
R4' независимо выбран из группы, состоящей из атома водорода, атома галогена, C1-6 алкила, -NR6R7 и -P(O)R6R7;
R6 и R7 независимо выбраны из группы, состоящей из атома водорода и С1-6 алкила, или R6 и R7 соединены и вместе с атомом, к которому они присоединены, образуют 3-12-членный гетероалициклил;
А2 выбран из группы, состоящей из фенила, пиридинила и пиримидинила, все из которых необязательно имеют 1, 2, 3 или 4 заместителя, независимо выбранные из группы, состоящей из атома галогена и -ОС1-6 алкила, у которого каждый атом водорода C1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или 3-12-членным гетероалициклилом;
А5 представляет собой 3-12-членный гетероалициклил, который необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
=O,
незамещенного С1-6 алкила и
С1-6 алкила, имеющего один или более чем один заместитель,
независимо выбранный из группы, состоящей из гидрокси, карбоксила
и 3-12-членного гетероалициклила, и
3-12-членного гетероалициклила,
и его фармацевтически приемлемые соли, стереоизомеры и энантиомеры и их смеси.
Согласно некоторым вариантам осуществления изобретения предложено соединение Формулы (III) или Формула (IV), где R4' являются одинаковыми или разными, при условии, что по меньшей мере один R4' не является атомом водорода. Согласно некоторым предпочтительным вариантам осуществления изобретения заместитель R4' в 3-й позиции представляет собой атом галогена. Согласно некоторым предпочтительным вариантам осуществления изобретения заместитель R4' в 3-й позиции представляет собой F и каждый из остальных заместителей R4' независимо выбран из группы, состоящей из атома водорода, атома галогена, С1-6 алкила, имеющего в качестве заместителя атом галогена, -NR6R7 и -P(O)R6R7, где каждый из R6 и R7 независимо выбран из группы, состоящей из С1-6 алкила. Согласно некоторым предпочтительным вариантам осуществления изобретения заместитель R4' в 3-й позиции представляет собой F и каждый из остальных заместителей R4' независимо выбран из группы, состоящей из атома водорода, атома галогена, метила, имеющего в качестве заместителя атом галогена, этила, имеющего в качестве заместителя атом галогена, -N(CH3)2 и -Р(O)(CH3)2. Согласно некоторым предпочтительным вариантам осуществления изобретения заместитель R4' в 3-й позиции представляет собой F и каждый из остальных заместителей R4' независимо выбран из группы, состоящей из атома водорода, F, Cl, -CHF2, -CF2CH3, -N(CH3)2 и -Р(O)(СН3)2. Согласно более предпочтительным вариантам осуществления изобретения заместитель R4' в 3-й позиции представляет собой F и заместители R4' во 2-й позиции и 6-й позиции представляют собой Cl, заместитель R4' в 4-й позиции представляет собой атом водорода.
Согласно некоторым вариантам осуществления изобретения А2 выбран из группы, состоящей из 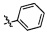 ,
, 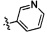 ,
, 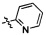 ,
, 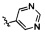 и
и 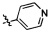 .
.
Согласно некоторым вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из атома галогена и -ОС1-6 алкила, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом, морфолинилом, тетрагидрофурилом, пиперидинилом, пиперазинилом, тетрагидропиридилом, дигидропиридилом, тетрагидротиенилом, пирролидинилом, оксазолидинилом, тиазолидинилом, имидазолидинилом, изоксазолидинилом, изотиазолидинилом, пиразолидинилом, тиоморфолинилом, пиперазин-2-он-илом, пирролинилом, дигидрофурилом или дигидротиенилом. Согласно некоторым предпочтительным вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из атома галогена и -ОС1-6 алкила, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или морфолинилом. Согласно более предпочтительным вариантам осуществления изобретения А2 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из F, Cl, метокси, этокси, -ОСН2СН2ОН и 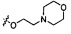 .
.
Согласно некоторым вариантам осуществления изобретения А5 представляет собой 5- или 6-членный гетероалициклил. Согласно более предпочтительным вариантам осуществления изобретения А5 представляет собой морфолинил, тетрагидрофурил, пиперидинил, пиперазинил, тетрагидропиридил, дигидропиридил, тетрагидротиенил, пирролидинил, оксазолидинил, тиазолидинил, имидазолидинил, изоксазолидинил, изотиазолидинил, пиразолидинил, тиоморфолинил, пиперазин-2-он-ил, пирролинил, дигидрофурил или дигидротиенил. Согласно некоторым более предпочтительным вариантам осуществления изобретения А5 представляет собой морфолинил, 1,2,3,4-тетрагидропиридил, 1,2,3,6-тетрагидропиридил, 2,3,4,5-тетрагидропиридил, пиперазинил, пиперазин-2-он-ил или пиперидинил. Согласно некоторым более предпочтительным вариантам осуществления изобретения А5 представляет собой пиперазин-1-ил, пиперазин-2-ил, пиперазин-3-ил, пиперидин-4-ил, пиперидин-1-ил, пиперидин-2-ил, пиперидин-3-ил, морфолин-4-ил, морфолин-2-ил, морфолин-3-ил, 1,2,3,4-тетрагидропиридин-4-ил, 1,2,3,6-тетрагидропиридин-4-ил, 2,3,4,5-тетрагидропиридин-4-ил или пиперазин-2-он-ил. Согласно некоторым наиболее предпочтительным вариантам осуществления изобретения А5 представляет собой 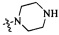 ,
, 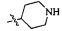 ,
, 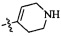 ,
, 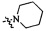 ,
, 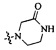 ,
, 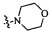 .
.
Согласно некоторым вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из
=O,
незамещенного С1-6 алкила и
С1-6 алкила, имеющего один или более чем один заместитель, независимо выбранный из группы, состоящей из гидрокси, карбоксила и 3-12-членного гетероалициклила, и
3-12-членного гетероалициклила, где 3-12-членный гетероалициклил, в свою очередь, необязательно имеет в качестве заместителей C1-6 алкил, =O, -ОН, -СООН, -CN, атом галогена, -NH(C1-6 алкил) или -N(C1-6 алкил)2.
Согласно некоторым предпочтительным вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из =O, метила, этила, н-пропила, изопропила и 5- или 6-членного гетероалициклила, где метил, этил, н-пропил и изопропил необязательно имеют один или более чем один заместитель, независимо выбранный из группы, состоящей из -ОН, -СООН и 5- или 6-членного гетероалициклила, и данный 5- или 6-членный гетероалициклил, в свою очередь, необязательно имеет заместитель(и), выбранный(ые) из группы, состоящей из метила, этила, н-пропила, изопропила, =O, -ОН, -СООН, -CN, атома галогена, -NH(C1-3 алкил) и -N(C1-3 алкил)2. Согласно некоторым более предпочтительным вариантам осуществления изобретения А5 необязательно имеет один или более чем один заместитель, выбранный из группы, состоящей из метила, этила, н-пропила, изопропила, =O, пиперидинила и пиперадинила, где пиперидинил и пиперазинил необязательно имеют в качестве заместителя метил.
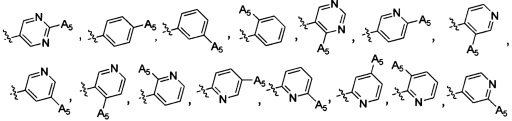
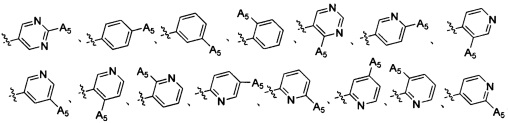
Согласно некоторым вариантам осуществления изобретения структура -А2-А5 представляет собой
 .
.
 .
.
Согласно некоторым предпочтительным вариантам осуществления изобретения структура -А2-А5 представляет собой  ,
,  ,
, 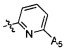 ,
, 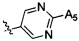 ,
, 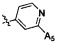 ,
, 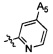 ,
,  .
.
Согласно некоторым вариантам осуществления изобретения А2 необязательно имеет 1 или 2 заместителя, выбранные из группы, состоящей из атома галогена и -OC1-6 алкила. Согласно некоторым предпочтительным вариантам осуществления изобретения А2 имеет в качестве заместителей по меньшей мере один -OC1-6 алкил, у которого каждый атом водорода С1-6 алкильной группировки необязательно замещен гидрокси, карбоксилом или 3-12-членным гетероалициклилом.
Конкретные примеры соединений, предложенных в настоящем изобретении, включают, без ограничения, следующие соединения, и их фармацевтически приемлемые соли, стереоизомеры и энантиомеры и их смеси:
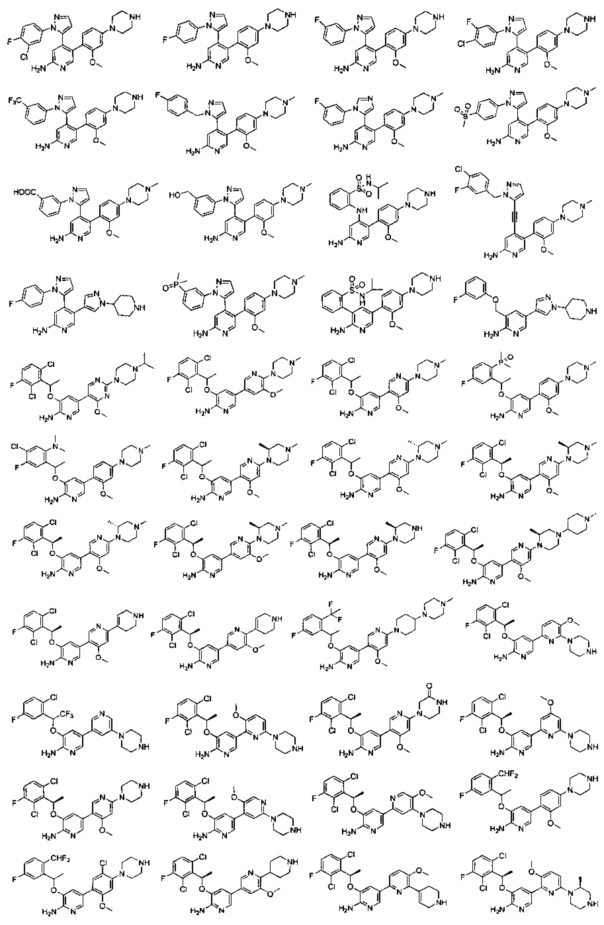
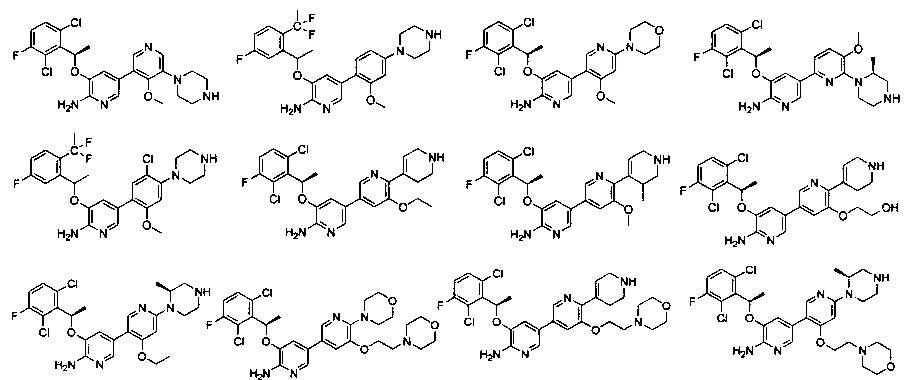 .
.
В настоящем изобретении также предложен способ получения соединений по настоящему изобретению, включающий следующие схемы синтеза.
Схема синтеза 1
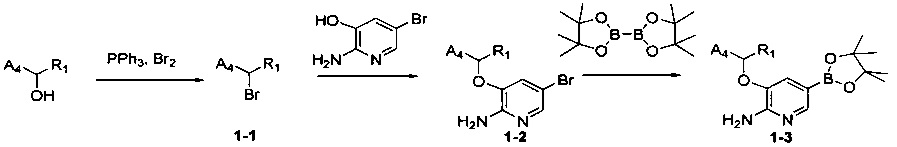
Соединения Формулы 1-3 могут быть синтезированы в соответствии со Схемой синтеза 1. К раствору трифенилфосфина в дихлорметане добавляют жидкий бром, и затем добавляют спирт с получением бром-замещенного Промежуточного соединения 1-1. Промежуточное соединение 1-1 подвергают взаимодействию с 2-амино-3-гидрокси-5-бромпиридином в растворителе (таком как N,N-диметилформамид или другой апротонный растворитель) с получением Промежуточного соединения 1-2. Промежуточное соединение 1-2, бис(пинаколато)дибор и палладиевый катализатор (такой как Pd(dppf)Cl2) подвергают взаимодействию в присутствии основания (такого как карбонат калия, ацетат калия) в растворителе (таком как диоксан, диметилсульфоксид) с получением соединения Формулы 1-3.
Схема синтеза 2
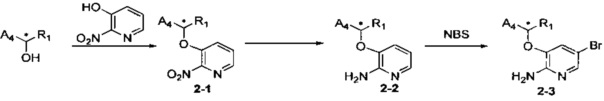
Соединения Формулы 2-3 могут быть синтезированы в соответствии со Схемой синтеза 2. Хиральный спирт и 3-гидрокси-2-нитро-пиридин вместе с DIAD (азодикарбоновой кислоты диизопропиловым эфиром) и трифенилфосфином подвергают взаимодействию в тетрагидрофуране с получением хирально инвертированного Промежуточного соединения 2-1. В стандартных условиях восстанавливают нитро-группу Промежуточного соединения 2-1 с получением Промежуточного соединения 2-2, и Промежуточное соединение 2-2 подвергают взаимодействию с бромсукцинимидом в органическом растворителе (таком как ацетонитрил, хлороформ, четыреххлористый углерод) с получением бром-замещенного Соединения 2-3.
Схема синтеза 3
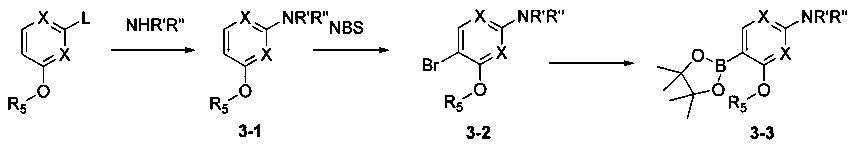
Соединения Формулы 3-3 могут быть синтезированы в соответствии со Схемой синтеза 3. L представляет собой уходящую группу (такую как Cl, Br); NR'R'' представляет собой 3-12-членный гетероалициклил, необязательно защищенный Boc; X независимо выбран из группы, состоящей из N и СН; и R5 выбран из группы, состоящей из атома водорода, замещенного C1-6 алкила и незамещенного C1-6 алкила. Промежуточное соединение 3-1 может быть получено в присутствии гидрида натрия и N,N-диметилформамида, Промежуточное соединение 3-2 может быть получено путем бромирования в присутствии органического растворителя (такого как ацетонитрил, хлороформ и четыреххлористый углерод) и бромсукцинимида, и затем Промежуточное соединение 3-2 может быть подвергнуто взаимодействию с бис(пинаколато)дибором с получением Промежуточного соединения 3-3.
Схема синтеза 3'
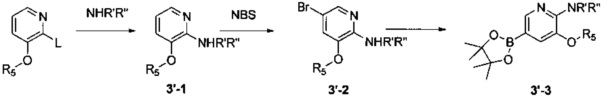
Соединения Формулы 3'-3 могут быть синтезированы в соответствии со Схемой синтеза 3'. L представляет собой уходящую группу (такую как Cl, Br); NR'R'' представляет собой 3-12-членный гетероалициклил, необязательно защищенный Boc; и R5 выбран из группы, состоящей из атома водорода, замещенного C1-6 алкила и незамещенного C1-6 алкила. Промежуточное соединение 3'-1 может быть получено в присутствии гидрида натрия и N,N-диметилформамида, Промежуточное соединение 3'-2 может быть получено путем бромирования в присутствии органического растворителя (такого как ацетонитрил, хлороформ и четыреххлористый углерод) и бромсукцинимида, и затем Промежуточное соединение 3-2 может быть подвергнуто взаимодействию с бис(пинаколато)дибором с получением Промежуточного соединения 3-3.
Схема синтеза 4
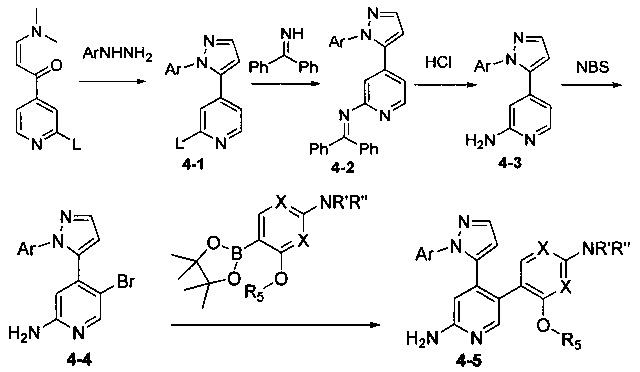
Соединения Формулы 4-5 могут быть синтезированы в соответствии со Схемой синтеза 4. Ar выбран из группы, состоящей из арила, гетероарила, арилалкила и гетероарилалкила; L представляет собой уходящую группу (такую как Cl, Br); NR'R'' представляет собой 3-12-членный гетероалициклил, необязательно защищенный Boc; X независимо выбран из группы, состоящей из N или СН; и R5 выбран из группы, состоящей из атома водорода, замещенного С1-6 алкила и незамещенного С1-6 алкила. В результате реакции замыкания цикла между 1-(2-галогенпиридин-4-ил)-3-(диметиламино)проп-2-ен-1-оном и гидразином в органическом растворителе, таком как этанол, получают Промежуточное соединение 4-1. Путем сочетания Промежуточного соединения 4-1 с бензофенонимином в присутствии палладиевого катализатора получают Промежуточное соединение 4-2, и затем бензофеноновую защитную группу удаляют с использованием кислоты (такой как разбавленная соляная кислота) с получением Промежуточного соединения 4-3. Промежуточное соединение 4-3 подвергают бромированию бромсукцинимидом с получением Промежуточного соединения 4-4, и затем в результате реакции сочетания Промежуточного соединения 4-4 с боратным эфиром в присутствии палладиевого катализатора получают Соединение 4-5. Если Соединение 4-5 содержит защитную группу (такую как Вое), может быть проведена дополнительная стадия снятия защитной группы с получением желаемого соединения.
Схема синтеза 5
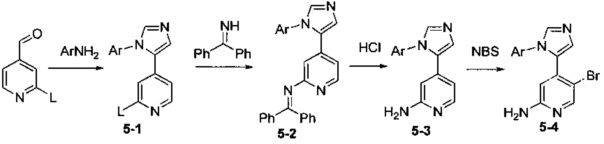
Соединения Формулы 5-4 могут быть синтезированы в соответствии со Схемой синтеза 5. Ar выбран из группы, состоящей из арила, гетероарила, арилалкила и гетероарилалкила, и L представляет собой уходящую группу (такую как Cl, Br). Сначала 2-галоген-4-пиридинкарбальдегид подвергают взаимодействию с амином в органическом растворителе, таком как толуол, и затем подвергают взаимодействию с тозилметилизоцианидом, используя реакцию замыкания цикла, в присутствии основания с получением Промежуточного соединения 5-1. Путем сочетания Промежуточного соединения 5-1 с бензофенонимином в присутствии палладиевого катализатора получают Промежуточное соединение 5-2, у которого затем удаляют бензофеноновую защитную группу с использованием кислоты (такой как разбавленная соляная кислота) с получением Промежуточного соединения 5-3. Промежуточное соединение 5-3 подвергают бромированию бромсукцинимидом с получением Промежуточного соединения 5-4.
Схема синтеза 6
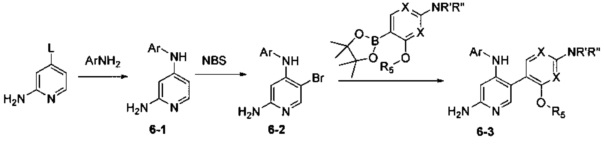
Соединения Формулы 6-3 могут быть синтезированы в соответствии со Схемой синтеза 6. Ar выбран из группы, состоящей из арила и гетероарила; L представляет собой уходящую группу (такую как Cl, Br); NR'R'' представляет собой 3-12-членный гетероалициклил, необязательно защищенный Boc; X независимо выбран из группы, состоящей из N и СН; и R5 выбран из группы, состоящей из атома водорода, замещенного С1-6 алкила и незамещенного C1-6 алкила. Путем сочетания 4-галоген-2-аминопиридина с амином в присутствии палладиевого катализатора получают Промежуточное соединение 6-1, затем Промежуточное соединение 6-1 бромируют бромсукцинимидом с получением Промежуточного соединения 6-2, из которого путем сочетания с боратным эфиром в присутствии палладиевого катализатора получают Соединение 6-3. Если Соединение 6-3 содержит защитную группу, может быть проведена дополнительная стадия снятия защитной группы в присутствии кислоты с получением желаемого соединения.
Схема синтеза 7
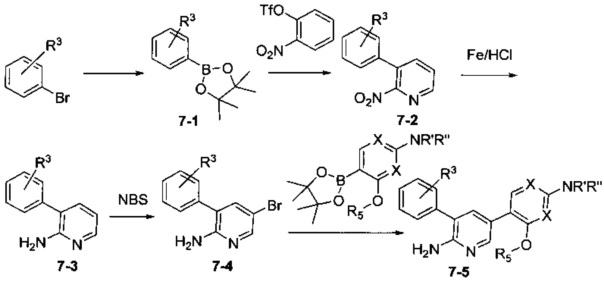
Соединения Формулы 7-5 могут быть синтезированы в соответствии со Схемой синтеза 7. NR'R'' представляет собой 3-12-членный гетероалициклил, необязательно защищенный Вос; X независимо выбран из группы, состоящей и: N и СН, и R5 выбран из группы, состоящей из атома водорода, замещенного С1-6 алкила и незамещенного C1-6 алкила. Боратный эфир 7-1 получают в присутствии бис(пинаколато)дибора и палладия. В результате реакции сочетания Промежуточного соединения 7-1 в присутствии палладиевого катализатора получают Промежуточное соединение 7-2, которое затем восстанавливают с использованием железа и разбавленной соляной кислоты с получением Промежуточного соединения 7-3. Промежуточное соединение 7-3 бромируют бромсукцинимидом с получением Промежуточного соединения 7-4, из которого путем сочетания с боратным эфиром в присутствии палладиевого катализатора получают Соединение 7-5. Если Соединение 7-5 содержит защитную группу, может быть проведена дополнительная стадия снятия защитной группы в присутствии кислоты с получением желаемого соединения.
Схема синтеза 8
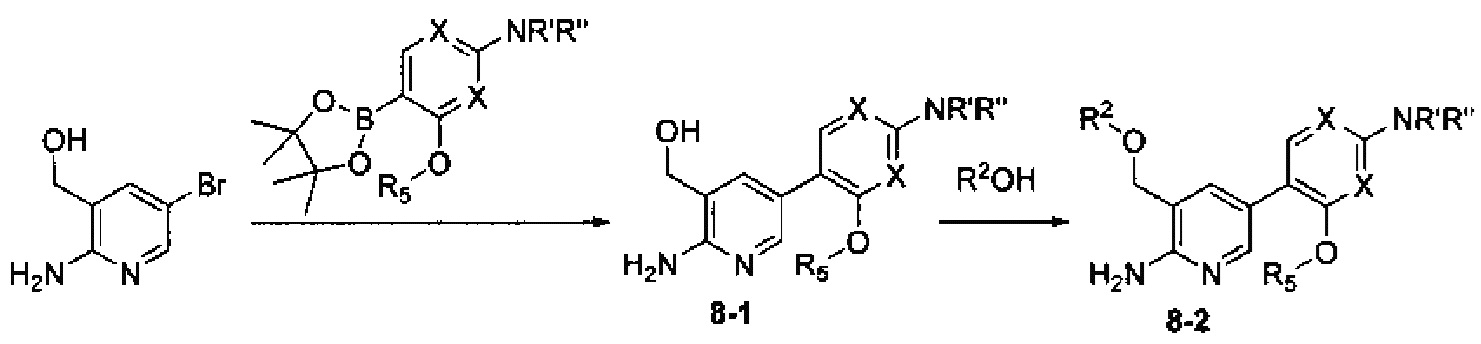
Соединения Формулы 8-2 могут быть синтезированы в соответствии со Схемой синтеза 8. NR'R'' представляет собой 3-12-членный гетероалициклил, необязательно защищенный Вос; X независимо выбран из группы, состоящей из N и СН; и R5 выбран из группы, состоящей из атома водорода, замещенного С1-6 алкила и незамещенного C1-6 алкила. Путем сочетания 3-гидроксиметил-5-бром-2-аминопиридина с боратным эфиром в присутствии палладиевого катализатора получают Промежуточное соединение 8-1, из которого затем получают соединение Формулы 8-2 с использованием реакции Мицунобу. Если Соединение 8-2 содержит защитную группу, может быть проведена дополнительная стадия снятия защитной группы в присутствии кислоты с получением желаемого соединения.
Схема синтеза 9
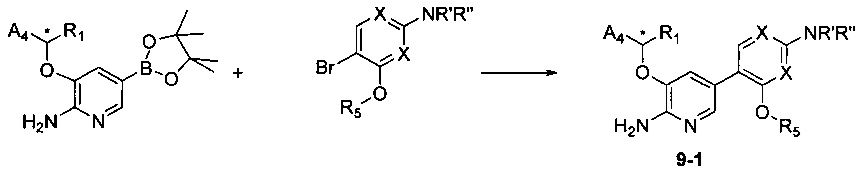
Соединения Формулы 9-1 могут быть синтезированы в соответствии со Схемой синтеза 9. NR'R'' представляет собой 3-12-членный гетероалициклил, необязательно защищенный Вос; X независимо выбран из группы, состоящей из N и СН; и R5 выбран из группы, состоящей из атома водорода, замещенного C1-6 алкила и незамещенного С1-6 алкила. Желаемое Соединение 9-1 получают путем сочетания пиридинборатного эфира в присутствии палладиевого катализатора. Если Соединение 9-1 содержит защитную группу, может быть проведена дополнительная стадия снятия защитной группы в присутствии кислоты с получением желаемого соединения. Когда в качестве реагента используется соединение Формулы 1-3, в результате реакции получают соответствующее соединение.
Схема синтеза 10
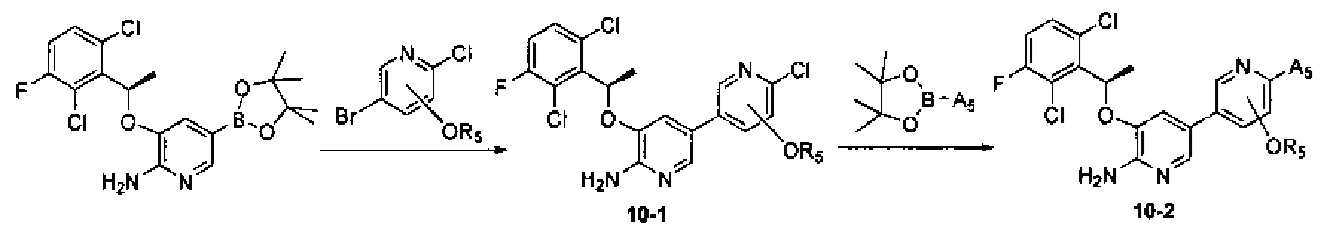
Соединения Формулы 10-2 могут быть синтезированы в соответствии со Схемой синтеза 10. A5 представляет собой 3-12-членный гетероалициклил, необязательно защищенный Вос; гетероатом гетероалициклила может быть соединен с атомом бора боратного эфира не напрямую; и R5 выбран из группы, состоящей из С1-6 алкила. В результате реакции сочетания боратного эфира в присутствии палладиевого катализатора получают Промежуточное соединение 10-1, из которого путем сочетания с другим боратным эфиром в присутствии палладиевого катализатора получают Соединение 10-2. Если Соединение 10-2 содержит защитную группу, может быть проведена дополнительная стадия снятия защитной группы в присутствии кислоты с получением желаемого соединения.
Если не указано иное, значения групп и терминов, использованных выше при описании схем синтеза, совпадают с соответствующими значениями, использованными при описании соединений Формулы (I), (II), (III) и (IV).
На описанных выше схемах синтеза приведены методики получения только части соединений по настоящему изобретению, однако специалист в данной области, опираясь на известный уровень техники и описанные схемы синтеза, сможет синтезировать также другие соединения по настоящему изобретению с использованием аналогичных методик.
Когда настоящее изобретение относится к соединениям Формулы (III) или Формулы (IV), атомы фенила пронумерованы следующим образом:
 ,
,  .
.
Термин "соединение" согласно настоящему изобретению включает все стереоизомеры, геометрические изомеры и таутомеры.
Соединения согласно настоящему изобретению могут быть асимметрическими, например, могут иметь один или более чем один стереоизомер. Если не указано иное, все стереоизомеры соединения, такие как энантиомеры и диастереоизомеры, включены в объем настоящего изобретения. Соединения по настоящему изобретению, содержащие асимметрический(ие) атом(ы) углерода, могут быть выделены в виде рацемической формы или оптически активной чистой формы. Оптически активная чистая форма может быть получена путем расщепления рацемических смесей или синтезирована из хиральных(ого) исходных(ого) веществ(а) или хиральных(ого) реагентов(а).
Соединения согласно настоящему изобретению также включают таутомерные формы. Таутомерные формы возникают в результате взаимного перемещения простой связи и соседней двойной связи, обусловленного миграцией протона.
При описании соединений Формул (I)-(IV) использованы термины, которые в контексте данной заявки имеют следующие значения.
Термин "галоген" относится к атому фтора, хлора, брома или иода, предпочтительно к атому фтора, хлора или брома.
Термин "гидрокси" относится к -ОН.
Термин "карбоксил" относится к -СООН.
Термин "амино" относится к -NH2, -NH(алкил) или -N(алкил)2. Конкретные примеры "амино" включают, без ограничения, -NH2, -NHCH3, -NHCH(CH3)2, -N(CH3)2, -NHC2H5, -N(CH3)C2H5 и тому подобное.
Термин "алкил" относится к нормальной или разветвленной насыщенной углеводородной группе, состоящей из атома(ов) углерода и атомов водорода, такой как С1-20 алкил, предпочтительно С1-6 алкил, такой как метил, этил, пропил (такой как н-пропил и изопропил), бутил (такой как н-бутил, изобутил, втор-бутил или трет-бугип), пентил (такой как н-пентил, изопентил, неопентил), н-гексил, 2-метил-гексил и тому подобное. Группа "алкил" может быть незамещенной или иметь заместитель(и), включая, без ограничения, алкокси, циано, карбоксил, арил, гетероарил, амино, атом галогена, сульфонил, сульфинил, фосфорил и гидрокси.
Термин "арил" относится к моноциклическому или конденсированному кольцу, содержащему в качестве кольцевых атомов только атомы углерода и имеющему полностью конъюгированную пи-электронную систему 6-14 атомов углерода, предпочтительно 6-12 атомов углерода, наиболее предпочтительно 6 атомов углерода. Арил может быть незамещенным или иметь один или более чем один заместитель. Примеры заместителей включают алкил, алкокси, арил, арилалкил, амино, атом галогена, гидрокси, сульфонил, сульфинил, фосфорил и гетероалициклил, но не ограничены ими. Неограничивающие примеры незамещенного арила включают фенил, нафтил и антраценил, но не ограничены ими.
Термин "арилалкил" относится к алкилу, имеющему в качестве заместителя арил, такой, как определено выше, предпочтительно к арил-замещенному С1-6 алкилу. Неограничивающие примеры арилалкила включают, без ограничения, -СН2-фенил, -(СН2)2-фенил, -(СН2)3-фенил, -СН(СН3)-фенил, -СН2-СН(СН3)-фенил, -(СН2)4-фенил, -СН2-СН(СН3)-СН2-фенил, -СН2-СН2-СН(СН3)-фенил и тому подобное.
Термин "гетероарил" относится к 5-12-членному моноциклическому или конденсированному кольцу, имеющему полностью конъюгированную пи-электронную систему 5, 6, 7, 8, 9, 10, И или 12 кольцевых атомов, 1, 2, 3 или 4 кольцевых атома которого выбраны из группы, состоящей из N, О и S, а другие кольцевые атомы представляет собой С. "Гетероарил" может быть незамещенным или иметь заместитель(и), включая, без ограничения, алкил, алкокси, арил, арилалкил, амино, атом галогена, гидрокси, циано, нитро, карбонил и гетероалициклил. Неограничивающие примеры незамещенных гетероарильных групп включают пирролил, фурил, тиенил, имидазолил, оксазолил, пиразолил, пиридинил, пиримидинил, пиразинил, хинолил, изо-хинолинил, тетразолил и триазинил, но не ограничены ими.
Термин "гетероарилалкил" относится к алкилу, имеющему в качестве заместителя гетероарил, такой, как определено выше, предпочтительно к гетероарил-замещенному С1-6 алкилу. Неограничивающие примеры гетероарилалкильных групп включают, без ограничения, -СН2-пиразолил, -(СН2)2-пиридинил, -(СН2)3-тиенил, -СН(СН3)-пиразинил, -СН2-СН(СН3)-фурил и тому подобное.
Термин "гетероалициклил" относится к 3-12-членному моноциклическому или конденсированному кольцу, имеющему 3, 4, 5, 6, 7, 8, 9, 10, 11 или 12 кольцевых атомов, 1 или 2 кольцевых атома которого представляют собой гетероатом(ы), выбранный(ые) из группы, состоящей из N, О и S(O)n (где n имеет значение 0, 1 или 2), а другой(ие) кольцевой(ые) атом(ы) представляет(ют) собой С. Такое кольцо может быть насыщенным или ненасыщенным (например, может иметь одну или более чем одну двойную связь), но у него отсутствует полностью конъюгированная пи-электронная система. 3-Членные насыщенные гетероалициклильные группы включают оксиранил, тииранил и азиранил, но не ограничены ими; 4-членные насыщенные гетероалициклильные группы включают азетидинил, диоксетанил и тиетанил, но не ограничены ими; 5-членные насыщенные гетероалициклильные группы включают тетрагидрофуранил, тетрагидротиенил, пирролидинил, изоксазолидинил, оксазолидинил, изотиазолидинил, тиазолидинил, имидазолидинил и тетрагидропиразолил, но не ограничены ими; 6-членные насыщенные гетероалициклильные группы включают пиперидинил, тетрагидропиранил, тетрагидротиопиранил, морфолинил, пиперазинил, 1,4-тиоксанил, 1,4-диоксанил, тиоморфолинил и 1,4-дитианил, но не ограничены ими; 7-членные насыщенные гетероалициклильные группы включают азепанил, оксепанил и тиепанил, но не ограничены ими; 5-членные ненасыщенные гетероалициклильные группы включают пирролинил, дигидрофурил и дигидротиенил, но не ограничены ими; и 6-членные ненасыщенные гетероалициклильные группы включают дигидропиридил, тетрагидропиридил, дигидропиранил, тетрагидропиранил и дигидротиопиранил, но не ограничены ими. Гетероалициклил может быть незамещенным, или каждый атом водорода гетероалициклила может быть замещен, при этом заместитель(и) включает(ют), без ограничения, алкил, алкокси, =O, арил, арилалкил, -COOH, -CN, амино, атом галогена и гидрокси.
Термин "терапевтически эффективное количество" относится к такому количеству соединения общей Формулы, которое является эффективным для лечения при введении млекопитающему, нуждающемуся в таком лечении. Терапевтически эффективное количество может изменяться в зависимости от специфической активности терапевтического агента и возраста, физиологического состояния, наличия других болезненных состояний и пищевого статуса пациента. Кроме того, одновременное использование другого лекарства при лечении будет оказывать влияние на определение терапевтически эффективного количества терапевтических агентов, которые должны быть назначены для введения.
"Лечение" означает любое воздействие на заболевание у млекопитающих, включая:
(i) предупреждение заболевания, что означает предотвращение появления клинических симптомов заболевания;
(ii) подавление заболевания, что означает предотвращение развития клинических симптомов; и/или
(iii) облегчение заболевания, что означает регрессию клинических симптомов.
Соединения согласно настоящему изобретению, или их соли, стереоизомеры или энантиомеры, или их смеси, могут быть введены как есть в качестве активного вещества, но предпочтительно вводятся в форме фармацевтических композиций.
Согласно другому аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая соединение Формулы (I), (II), (III) или (IV), или его фармацевтически приемлемую соль, стереоизомер или энантиомер. или их смесь, в качестве активного ингредиента, и один или более чем один фармацевтически приемлемый носитель.
"Фармацевтическая композиция" означает препарат, состоящий из одного или более чем одного соединения по настоящему изобретению, или его соли, стереоизомера или энантиомера, или их смеси, и носителя(ей), который(ые) обычно используют в данной области для доставки биологически активных соединений в организм (например в организм человека).
Термин "фармацевтически приемлемый носитель" относится к носителю, который не оказывает значительного действия на организм и не оказывает действия на биологическую активность и свойства активного соединения. "Фармацевтически приемлемый носитель" означает инертное вещество, вводимое вместе с активным ингредиентом и оказывающее на введение благоприятное действие, и включает, без ограничения, любые скользящие вещества, подслащивающие агенты, разбавители, консерванты, красящие вещества/пигменты, усилители вкуса и запаха, поверхностно-активные вещества, увлажняющие агенты, диспергирующие агенты, разрыхляющие агенты, суспендирующие агенты, стабилизаторы, изотонические агенты, растворители и эмульгаторы, которые лицензированы Государственным управлением по контролю за качеством медикаментов и продуктов питания и являются приемлемыми для человека или животного (например для сельскохозяйственных животных). Неограничивающие примеры носителей включают карбонат кальция, фосфат кальция, различные сахара и различные типы крахмала, производные целлюлозы, желатин, растительное масло и полиэтиленгликоль.
Фармацевтические композиции согласно настоящему изобретению могут быть изготовлены в виде твердых, полужидких, жидких или газообразных препаратов, таких как таблетки, пилюли, капсулы, порошки, гранулы, пасты, эмульсии, суспензии, растворы, суппозитории, инъекции, средства для ингаляции, гели, микросферы и аэрозоли и тому подобное.
Типичные пути введения соединения по настоящему изобретению, или его фармацевтически приемлемой соли, или фармацевтической композиции, содержащей данное соединение, включают, без ограничения, пероральное, ректальное, трансмукозальное, энтеральное введение и местное, трансдермальное; ингаляционное, парентеральное, сублингвальное, интравагинальное, интраназальное, внутриглазное, интраперитонеальное, внутримышечное, подкожное и внутривенное введение. Предпочтительным путем введения является пероральный.
Фармацевтические композиции по настоящему изобретению могут быть получены с использованием методик, хорошо известных в данной области техники, таких как стандартная методика смешивания, методика растворения, методика грануляции, методика изготовления драже, методика растирания, методика эмульгирования, методик лиофилизации и тому подобное.
Согласно предпочтительному варианту осуществления изобретения фармацевтические композиции представлены в пероральной форме. Фармацевтические композиции, предназначенные для перорального введения, могут быть получены в результате смешивания активных соединений с фармацевтически приемлемым(и) носителем(ями), хорошо известным(и) в данной области техники. Такой(ие) носитель(и) позволяет(ют) получать препараты соединений по настоящему изобретению, предназначенные для перорального введения пациенту, в виде таблеток, пилюль, лепешек, драже, капсул, жидкостей, гелей, сиропов, суспендирующих агентов и т.п.
Твердая пероральная фармацевтическая композиция может быть получена с использованием стандартной методики смешивания, наполнения или таблетирования. Например, фармацевтическая композиция может быть получена путем смешивания активного соединения с твердым эксципиентом, необязательного растирания полученной смеси, добавления при необходимости других подходящих вспомогательных веществ и превращения данной смеси в гранулы с последующим формированием таблеток или ядер драже. Подходящие вспомогательные вещества включают, без ограничения, связующие вещества, разбавители, разрыхляющие агенты, смазывающие вещества, скользящие вещества, подслащивающие агенты и корригенты и тому подобное. Примерами указанных веществ являются микрокристаллическая целлюлоза, раствор глюкозы, раствор гуммиарабика, раствор желатина, паста сахарозы и крахмала; тальк, крахмал, стеарат магния, стеарат кальция или стеариновая кислота; лактоза, сахароза, крахмал, маннит, сорбит или гидрофосфат кальция; диоксид кремния; сетчатая натрий-карбоксиметилцеллюлоза, желатинированный крахмал, натриевая соль гликолята крахмала, альгиновая кислота, кукурузный крахмал, картофельный крахмал, метилцеллюлоза, агар, карбоксиметилцеллюлоза, сетчатый поливинилпирролидон и тому подобное. На ядра драже необязательно может быть нанесено покрытие с использованием известных в фармацевтике методик, предпочтительно кишечно-растворимое покрытие.
Фармацевтические композиции могут быть адаптированы также для парентерального введения, примерами таких фармацевтических композиций являются стерильные растворы, суспензии или лиофилизированные продукты в подходящей стандартной лекарственной форме. Могут быть использованы подходящие эксципиенты, такие как наполнители, буферные агенты или поверхностно-активные вещества.
Согласно другому аспекту настоящего изобретения предложен способ регуляции активности протеинкиназы, включающий контактирование протеинкиназы с соединением Формулы (I), (II), (III) или (IV), или с его фармацевтически приемлемой солью, стереоизомером или энантиомером, или их смесью. Предпочтительно протеинкиназа выбрана из ALK. Кроме того, протеинкиназа включает мутированную киназу, при этом мутированная киназа выбрана из ALK-киназы.
Кроме того, в настоящем изобретении также предложено применение соединения Формулы (I), (II), (III) или (IV), или его фармацевтически приемлемой соли, стереоизомера или энантиомера, или их смеси, или фармацевтической композиции, содержащей данное соединение, или его фармацевтически приемлемую соль, стереоизомер или энантиомер, или их смесь, в изготовлении лекарства для терапевтического и/или профилактического лечения заболеваний, связанных с активностью протеинкиназы (такой как ALK), таких как анормальная клеточная пролиферация, с учетом того, что анормальная клеточная пролиферация включает рак. В настоящем изобретении также предложено применение соединения Формулы (I), (II), (III) или (IV), или его фармацевтически приемлемой соли, стереоизомера или энантиомера, их смеси или фармацевтической композиции, содержащей данное соединение, или его фармацевтически приемлемую соль, стереоизомер или энантиомер, или их смесь, в изготовлении лекарства для терапевтического и/или профилактического лечения заболеваний, опосредованных ALK.
Заболевания, опосредованные ALK, включают ALK-позитивную немелкоклеточную карциному легкого, анапластическую крупно клеточную лимфому, воспалительную миофибробластическую опухоль, назофарингеальную карциному, рак молочной железы, колоректальный рак, диффузную крупноклеточную В-клеточную лимфому, системный гистиоцитоз и нейробластому и тому подобное, предпочтительно ALK-позитивную немелкоклеточную карциному легкого.
Кроме того, в настоящем изобретении также предложен способ терапевтического и/или профилактического лечения заболеваний у млекопитающего (такого как человек), которые связаны с активностью протеинкиназы (такой как ALK), включающий введение млекопитающему (такому как человек) терапевтически эффективного количества соединения Формулы (I), (II), (III) или (IV), или его фармацевтически приемлемой соли, стереоизомера, энантиомера, или их смеси, или фармацевтической композиции, содержащей данное соединение, или его фармацевтически приемлемую соль, стереоизомер или энантиомер, или их смесь.
Кроме того, в настоящем изобретении также предложено соединение Формулы (I), (II), (III) или (IV), или его фармацевтически приемлемая соль, стереоизомер или энантиомер, их смесь или фармацевтическая композиция, содержащая данное соединение, или его фармацевтически приемлемую соль, стереоизомер или энантиомер, или их смесь, для модулирования активности протеинкиназы или для терапевтического и/или профилактического лечения заболеваний, ассоциированных с активностью протеинкиназы, у млекопитающего (такого как человек). Предпочтительной протеинкиназой является ALK. Протеинкиназа включает мутированную киназу, при этом мутированная киназа выбрана из ALK-киназы.
ПРИМЕРЫ
Цель приведенных ниже конкретных примеров - облегчить понимание и осуществление настоящего изобретения специалистам в данной области техники: Приведенные примеры не ограничивают объем настоящего изобретения каким-либо образом, являются исключительно иллюстративными и представляют собой типичные варианты осуществления изобретения. Специалистам в данной области техники должно быть понятно, что существуют другие пути синтеза соединений по настоящему изобретению, методики получения соединений, описанные ниже, приведены в качестве примеров и не ограничивают изобретение.
Все операции с использованием исходных веществ, которые легко окисляются или легко гидролизуются, выполняли в атмосфере азота. Если не указано иное, исходные вещества, использованные в изобретении, имеются в продаже и использованы без дополнительной очистки.
Для колоночной хроматографии использовали силикагель (200-300 меш) производства Qingdao Chemical Co., Ltd. Тонкослойную хроматографию выполняли с использованием готовых панелей (силикагель 60 PF254, 0,25 мм) производства Е. Merck. Разделение хиральных соединений и измерение энантиомерного избытка выполняли с использованием системы Agilent LC 1200 series (колонка: CHIRALPAK AD-H, ∅4,6×250 мм, 5 микрон, 30°С). ЯМР-спектр (ЯМР) получали с использованием ЯМР-спектрометра Varian VNMRS-400; ЖХ-МС (жидкостную хроматографию/масс-спектрометрию) выполняли с использованием FINNIGAN Thermo LCQ Advantage MAX, Agilent LC 1200 series (колонка: Waters Symmetry С18, ∅ 4.6×50 мм, 5 микрон, 35°C) и ESI (ионизации электрораспылением) в режиме положительных ионов.
ПРИМЕРЫ
Промежуточное соединение 1: трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилат
Стадия 1: трет-бутил 4-(3-метоксилфенил)пиперазин-1-карбоксилат
3-Броманизол (1,0 г, 5 ммоль), трет-бутил пиперазин-1-карбоксилат (1,2 г, 6 ммоль), Pd2(dba)3 (229 мг, 0,25 ммоль), BINAP (2,2'-бис(дифенилфосфино)-1,1'-бинафтил) (328 мг, 0,5 ммоль) и трет-бутилат натрия (0,72 г, 7,5 ммоль) добавляли в сухой толуол (20 мл). Данную смесь продували азотом и перемешивали при 80°С в течение ночи. Полученный раствор охлаждали, и затем концентрировали и очищали путем колоночной хроматографии на силикагеле с выделением трет-бутил 4-(3-метоксилфенил)пиперазин-1-карбоксилата (1,4 г, выход: 96%). МС m/z [ESI]: 293,2 [М+1].
Стадия 2: трет-бутил 4-(3-метоксил-4-бромфенил)пиперазин-1-карбоксилат
К перемешиваемому раствору трет-бутил 4-(3-метоксилфенил)пиперазин-1-карбоксилата (1,6 г, 5 ммоль) в CH2Cl2 (100 мл) добавляли по каплям раствор жидкого брома (0,87 г, 5 ммоль) в CH2Cl2 (10 мл) при 0°С. После завершения добавления полученную смесь перемешивали в течение 1 ч при 0°С. Затем смесь промывали насыщенным раствором бикарбоната натрия, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с выделением трет-бутил 4-(3-метоксил-4-бромфенил)пиперазин-1-карбоксилата (756 мг, выход: 40%). МС m/z [ESI]: 371,1 [М+1].
Стадия 3: трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилат
трет-Бутил 4-(3-метоксил-4-бромфенил)пиперазин-1-карбоксилат (740 мг, 2 ммоль), бис(пинаколато)дибор (1008 мг, 4 ммоль), Pd(dppf)Cl2 (73 мг, 0,1 ммоль) и безводный ацетат калия (588 мг, 6 ммоль) добавляли в сухой 1,4-диоксан (20 мл). Данную смесь продували азотом и затем перемешивали в течение 2 ч при 120°С. Полученный раствор охлаждали, и затем концентрировали и очищали путем колоночной хроматографии на силикагеле с выделением трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (640 мг, выход: 76%). МС m/z [ESI]: 419,3 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.212 (d, J=6.8 Гц, 1Н), 6.58-6.40 (m, 2Н), 3.861 (s, 3Н), 3.593-3.555 (m, 4Н), 3.125-3.110 (m, 4Н), 1.483 (s, 9Н), 1.240 (s, 12Н).
Промежуточное соединение 2: 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазин
Стадия 1: 1-(3-метоксил-4-нитрофенил)-4-метилпиперазин
5-Фтор-2-нитроанизол (14,0 г, 83 ммоль), N-метилпиперазин (9,1 г, 91 ммоль) и карбонат калия (34,5 г, 250 ммоль) добавляли в ДМСО (диметилсульфоксид) (200 мл). Полученную смесь перемешивали в течение ночи при 90°С. Затем смесь охлаждали, и добавляли воду (3 л), выпавшее в осадок твердое вещество собирали путем фильтрования и сушили с получением 1-(3-метоксил-4-нитрофенил)-4-метилпиперазина (20,9 г). МС m/z [ESI]: 252,1 [М+1].
Стадия 2: 1-(3-метоксил-4-аминофенил)-4-метилпиперазин
1-(3-Метоксил-4-нитрофенил)-4-метилпиперазин (20,8 г, 83 ммоль) и никель Ренея (4,0 г) добавляли в метанол (200 мл), и затем воздух вытесняли водородом. Полученную смесь перемешивали в течение ночи в атмосфере водорода. Затем смесь фильтровали и концентрировали с получением 1-(3-метоксил-4-аминофенил)-4-метилпиперазина (17,0 г, выход: 93%). МС m/z [ESI]: 222,2 [М+1].
Стадия 3: 1-(3-метоксил-4-бромфенил)-4-метилпиперазин
1-(3-Метоксил-4-аминофенил)-4-метилпиперазин (16,6 г, 75 ммоль) и CuBr (21,5 г, 0,15 моль) добавляли в тетрагидрофуран (200 мл), и затем при перемешивании добавляли по каплям амил нитрит (17,6 г, 0,15 моль). Полученную смесь перемешивали в течение 1 ч при комнатной температуре и подвергали дефлегмации в течение 3 ч. После охлаждения смесь фильтровали, и фильтрат концентрировали и очищали путем колоночной хроматографии на силикагеле (петролейный эфир: этилацетат = 1:1) с выделением 1-(3-метоксил-4-бромфенил)-4-метилпиперазина (5,98 г, выход: 28%). МС m/z [ESI]: 285,1 [М+1].
Стадия 4: 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазин
1-(3-Метоксил-4-бромфенил)-4-метилпиперазин (2,85 г, 10 ммоль), бис(пинаколато)дибор (3,78 г, 15 ммоль), Pd(dppf)Cl2 (366 мг, 0,5 ммоль) и безводный ацетат калия (1,96 г, 20 ммоль) добавляли в сухой 1,4-диоксан (100 мл). Полученную смесь продували азотом и перемешивали в течение 3 ч при 120°С. После охлаждения смесь концентрировали и очищали путем колоночной хроматографии на силикагеле с выделением 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина (1,86 г, выход: 56%). МС m/z [ESI]: 333,2 [М+1].
Промежуточное соединение 3: трет-бутил 4-(4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилат
Промежуточное соединение 4: 1-(2-хлорпиридин-4-ил)-3-(диметиламино)проп-2-ен-1-он
Стадия 1: 2-хлор-N-метоксил-N-метилизоникотинамид
К раствору 2-хлоризоникотиновой кислоты (18,0 г, 0,115 моль) в CH2Cl2 (250 мл) добавляли порциями при перемешивании N,N'-карбонилдиимидазол (17,0 г, 0,105 моль) при комнатной температуре. После завершения добавления полученную смесь перемешивали в течение 0,5 ч, затем добавляли N,O-диметилгидроксиламин (10,2 г, 0,167 моль), и данную смесь перемешивали в течение ночи при комнатной температуре. Добавляли диэтиловый эфир (200 мл), и данную смесь промывали водой, сушили и концентрировали с получением 2-хлор-N-метоксил-N-метилизоникотинамида (18,0 г, выход: 78%). МС m/z [ESI]: 201,0 [М+1].
Стадия 2: 1-(2-хлорпиридин-4-ил)этанон
К раствору 2-хлор-N-метоксил-N-метилизоникотинамида (10,0 г, 50 ммоль) в сухом тетрагидрофуране (50 мл), добавляли при перемешивании 3 М бромид метилмагния (50 мл, 150 ммоль) при 0°С. После завершения добавления полученную смесь перемешивали в течение ночи при комнатной температуре. Реакцию останавливали путем добавления насыщенного раствора хлорида аммония, реакционную смесь экстрагировали этилацетатом, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 1-(2-хлорпиридин-4-ил)этанона (7,5 г, выход: 96%). МС m/z [ESI]: 156,0 [М+1].
Стадия 3: 1-(2-хлорпиридин-4-ил)-3-(диметиламино)проп-2-ен-1-он
1-(2-Хлорпиридин-4-ил)этанон (7,5 г, 48 ммоль) добавляли в ДМФА/DMA (диметилформамид/диметилацеталь) (40 мл), и полученную смесь перемешивали в течение 2 ч при 100°С. После охлаждения смесь вливали в петролейный эфир (500 мл). Твердое вещество собирали путем фильтрования, промывали диэтиловым эфиром и сушили с получением 1-(2-хлорпиридин-4-ил)-3-(диметиламино)проп-2-ен-1-она (7,4 г, выход: 74%). МС m/z [ESI]: 211,1 [М+1].
Промежуточное соединение 5: 3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-аминопиридин
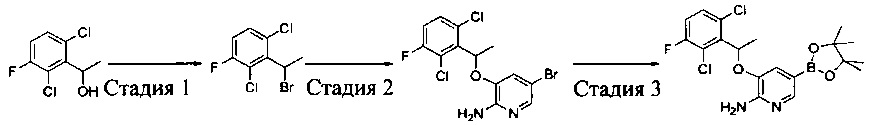
Стадия 1: 1,3-дихлор-2-(1-бромэтил)-4-фторбензол
К раствору трифенилфосфина (27,8 г, 0,106 моль) в CH2Cl2 (200 мл) при перемешивании медленно добавляли жидкий бром (16,8 г, 0,105 моль) при 0°С. После завершения добавления полученную смесь перемешивали в течение 10 мин, и затем к смеси добавляли 1-(2,6-дихлор-3-фторфенил)этанол (20,9 г, 0,10 моль). После завершения добавления смесь дополнительно перемешивали в течение 30 мин. Реакцию останавливали путем добавления этанола, и реакционную жидкость вливали в насыщенный раствор бикарбоната натрия и экстрагировали CH2Cl2. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 1,3-дихлор-2-(1-бромэтил)-4-фторбензола (25,8 г, выход: 95%). 1Н ЯМР (400 МГц, CDCl3): δ=7.28 (m, 1H), 7.05 (t, 1H), 5.97 (q, 1H), 2.16 (d, 3Н).
Стадия 2: 5-бром-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-аминопиридин
1,3-Дихлор-2-(1-бромэтил)-4-фторбензол (25,8 г, 95 ммоль), 2-амино-5-бром-пиридин-3-ол (28,7 г, 152 ммоль) и K2CO3 (26,2 г, 190 ммоль) добавляли в ДМФА (400 мл) при комнатной температуре. После завершения добавления реакционную смесь выдерживали в течение 6 ч в атмосфере азота. Полученный раствор концентрировали, остаток разбавляли CH2Cl2 и затем промывали водой, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с выходом 5-бром-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-аминопиридина (15,2 г, выход: 42%). МС m/z [ESI]: 380,9 [М+1].
Стадия 3: 3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-аминопиридин
5-Бром-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-аминопиридин (7,6 г, 20 моль), бис(пинаколато)дибор (7,56 г, 30 ммоль), Pd(dppf)Cl2 (732 мг, 1 ммоль) и безводный ацетат калия (4,90 г, 50 ммоль) добавляли в сухой 1,4-диоксан (200 мл), и полученную смесь продували азотом. Реакционную смесь выдерживали в течение 4 ч при 100°С. После охлаждения смесь концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-аминопиридина (5,12 г, выход: 60%). МС m/z [ESI]: 427,1 [М+1].
Промежуточное соединение 6: (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-аминопиридин
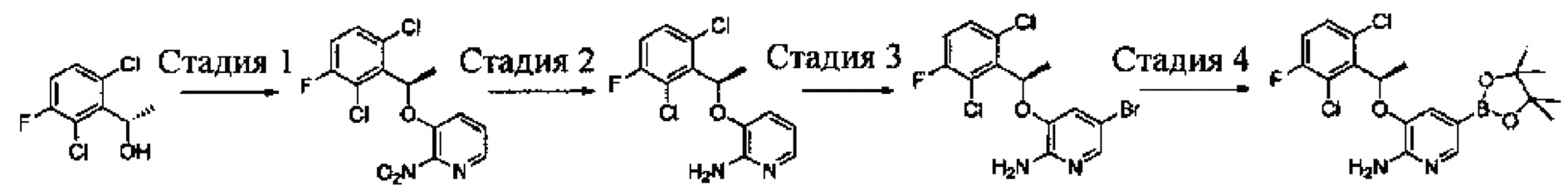
Стадия 1: (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-нитропиридин
(S)-3-(1-(2,6-Дихлор-3-фторфенил)этанол (20,9 г, 0,10 моль) растворяли в безводном тетрагидрофуране (200 мл), и затем в атмосфере азота последовательно добавляли 3-гидрокси-2-нитропиридин (16,0 г, 0,11 моль) и трифенилфосфин (40,0 г, 0,15 моль). Данную реакционную смесь перемешивали в течение 1 ч при комнатной температуре. Затем реакционную смесь охлаждали до 0°С, добавляли DIAD (40 мл, 0,15 моль), и данную смесь перемешивали в течение 12 ч. Растворитель выпаривали с получением неочищенного продукта в виде масла, который очищали путем колоночной хроматографии на силикагеле с получением (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-нитропиридина (20,2 г, выход: 61%).
Стадия 2: (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-аминопиридин
К раствору (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-нитропиридина (20,0 г, 60 ммоль) в этаноле (300 мл) добавляли при перемешивании 2 М HCl (15 мл) и порошок восстановленного железа (27 г, 480 ммоль) при 0°С. После завершения добавления реакционную смесь нагревали в течение 12 ч. После охлаждения до комнатной температуры реакционную смесь фильтровали, и фильтрат концентрировали с получением (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-аминопиридина (17,0 г, выход: 94%), который использовали на следующей стадии без дополнительной очистки. МС m/z [ESI]: 301,0 [М+1].
Стадия 3: (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-5-бром-2-аминопиридин
К раствору (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-аминопиридина (15,0 г, 50 ммоль) в ацетонитриле (200 мл) добавляли порциями при перемешивании N-бромбутанимид (10 г, 56 ммоль) при 0°С. После завершения добавления полученную смесь перемешивали в течение 1 ч. Растворитель выпаривали, и добавляли CH2Cl2. Данный раствор промывали насыщенным бикарбонатом натрия, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-5-бром-2-аминопиридина (9,88 г, выход: 52%). МС m/z [ESI]: 380,9 [М+1].
Стадия 4: (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-аминопиридин
(R)-3-(1-(2,6-Дихлор-3-фторфенил)ethyoxyl)-5-бром-2-аминопиридин (7,6 г, 20 моль), бис(пинаколато)дибор (7,56 г, 30 ммоль), Pd(dppf)Cl2 (732 мг, 1 ммоль) и безводный ацетат калия (4,90 г, 50 ммоль) добавляли в сухой 1,4-диоксан (200 мл), и данную смесь продували азотом. Затем смесь перемешивали в течение 4 ч при 100°С. После охлаждения смесь концентрировали и очищали путем колоночной хроматографии на силикагеле с получением (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-аминопиридина (5,46 г, выход: 64%). МС m/z [ESI]: 427,1 [М+1].
Промежуточное соединение 7: 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидин
Стадия 1: 2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидин
К раствору N-изопропилпиперазина (1,27 г, 10 ммоль) в безводном ДМФА (60 мл) добавляли NaH (600 мг, 60%, 15 ммоль). Полученную смесь перемешивали в течение 10 мин, и добавляли 2-хлор-4-метоксипиримидин (1,44 г, 10 ммоль) После завершения добавления реакционную смесь нагревали до 80°С в течение 3 ч. Растворитель выпаривали с получением неочищенного продукта в виде масла, который очищали путем колоночной хроматографии на силикагеле с получением 2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина (1,75 г, выход: 74%). МС m/z [ESI]: 237,2 [М+1].
Стадия 2: 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидин
К раствору 2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина (1,65 г, 7 ммоль) в ацетонитриле (50 мл) добавляли порциями при перемешивании N-бромбутанимид (1,37 г, 7,7 ммоль) при 0°С. Полученную смесь перемешивали в течение 1 ч при комнатной температуре. Растворитель выпаривали, остаток разбавляли CH2Cl2 и затем промывали насыщенным раствором бикарбоната натрия, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина (1,58 г, выход: 72%). МС m/z [ESI]: 315,1 [М+1].
Промежуточное соединение 8: 1-(5-бром-3-метоксилпиридин-2-ил)-4-метилпиперазин
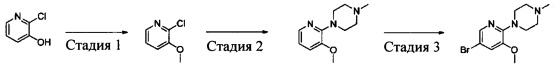
Стадия 1: 2-хлор-3-метоксилпиридин
2-Хлор-3-гидроксипиридин (2,59 г, 20 ммоль), иодметан (2,98 г, 21 ммоль) и K2CO3 (5,52 г, 40 ммоль) добавляли в ДМФА (50 мл), и полученную смесь перемешивали в течение 4 ч при 60°С. После охлаждения смесь вливали в воду и экстрагировали этилацетатом. Экстракт сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 2-хлор-3-метоксилпиридина (2,58 г, выход: 90%). МС m/z [ESI]: 144,0 [М+1].
Стадия 2: 1-(3-метоксилпиридин-2-ил)-4-метилпиперазин
2-Хлор-3-метоксилпиридин (2,58 г, 18 ммоль), N-метилпиперазин (2,7 г, 27 ммоль), Pd2(dba)3 (824 мг, 0,9 ммоль), BINAP (1,12 г, 1,8 ммоль) и Cs2CO3 (14,4 г, 45 ммоль) добавляли в сухой толуол (200 мл). Полученную смесь подвергали дефлегмации в течение 16 ч в атмосфере азота. Затем реакционную смесь фильтровали, и фильтрат концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 1-(3-метоксилпиридин-2-ил)-4-метилпиперазина (1,71 г, выход: 46%). МС m/z [ESI]: 208,1 [М+1].
Стадия 3: 1-(5-бром-3-метоксилпиридин-2-ил)-4-метилпиперазин
К раствору 1-(3-этоксилпиридин-2-ил)-4-метилпиперазина (1,66 г, 8 ммоль) в ацетонитрил е (50 мл) добавляли порциями при перемешивании N-бромбутанимид (1,57 г, 8,8 ммоль) при 0°С. После завершения добавления полученную смесь перемешивали в течение 2 ч при комнатной температуре. Растворитель выпаривали, остаток разбавляли CH2Cl2 и затем промывали насыщенным раствором бикарбоната натрия, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 1-(5-бром-3-метоксилпиридин-2-ил)-4-метилпиперазина (1,58 г, выход: 69%). МС m/z [ESI]: 286,1 [М+1].
Промежуточное соединение 9:1-(5-бром-4-метоксипиридин-2-ил)-4-метилпиперазин
Стадия 1: 1-(4-метоксипиридин-2-ил)-4-метилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2-хлор-4-метоксипиридина вместо 2-хлор-3-метоксипиридина (выход: 51%). МС m/z [ESI]: 208,1 [М+1].
Стадия 2: 1-(5-бром-4-метоксипиридин-2-ил)-4-метилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием 1-(4-метоксипиридин-2-ил)-4-метилпиперазина вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 83%). МС m/z [ESI]: 286,1 [М+1].
Промежуточное соединение 10: (S)-1-(5-бром-4-метоксипиридин-2-ил)-2,4-диметилпиперазин
Стадия 1: (S)-1-(4-метоксипиридин-2-ил)-2,4-диметилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2-хлор-4-метоксипиридина вместо 2-хлор-3-метоксипиридина и с использованием (S)-1,3-диметилпиперазина вместо N-метилпиперазина (выход: 43%). МС m/z [ESI]: 222,2 [М+1].
Стадия 2: (S)-1-(5-бром-4-метоксипиридин-2-ил)-2,4-диметилпиперазин
Указанное в заголовке соединения получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием (5)-1-(4-метоксипиридин-2-ил)-2,4-диметилпиперазина вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 80%). МС m/z [ESI]: 300,1 [М+1].
Промежуточное соединение 11: (R)-1-(5-бром-4-метоксипиридин-2-ил)-2,4-диметилпиперазин
Стадия 1: (R)-1-(4-метоксипиридин-2-ил)-2,4-диметилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2-хлор-4-метоксипиридина вместо 2-хлор-3-метоксипиридина и с использованием (R)-1,3-диметилпиперазина вместо N-метилпиперазина (выход: 42%). МС m/z [ESI]: 222,2 [М+1].
Стадия 2: (R)-1-(5-бром-4-метоксипиридин-2-ил)-2,4-диметилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием (R)-1-(4-метоксипиридин-2-ил)-2,4-диметилпиперазина вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 82%). МС m/z [ESI]: 300,1 [М+1].
Промежуточное соединение 12: (S)-1-(5-бром-3-метоксипиридин-2-ил)-2,4-диметилпиперазин
Стадия 1: (S)-1-(3-метоксипиридин-2-ил)-2,4-диметилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием (S)-1,3-диметилпиперазина вместо N-метилпиперазина (выход: 43%). МС m/z [ESI]: 222,2 [М+1].
Стадия 2: (S)-1-(5-бром-3-метоксипиридин-2-ил)-2,4-диметилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием (5)-1-(3-метоксипиридин-2-ил)-2,4-диметилпиперазина вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 82%). МС m/z [ESI]: 300,1 [М+1].
Промежуточное соединение 13: (S)-1-(5-бром-4-метоксипиридин-2-ил)-2-метил-4-(1-метилпиперидин-4-ил)пиперазин
Стадия 1: (S)-трет-бутил 4-бензил-3-метилпиперазин-1-карбоксилат
(S)-трет-Бутил 3-метилпиперазин-1 -карбоксилат (10,0 г, 50 ммоль), бензилбромид (8,89 г, 52 ммоль) и K2CO3 (13,8 г, 100 ммоль) добавляли в ацетонитрил (200 мл). Полученную смесь подвергали дефлегмации в течение 2 ч. Растворитель выпаривали, остаток разбавляли этилацетатом и затем промывали водой, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением (S)-трет-бутил 4-бензил-3-метилпиперазин-1-карбоксилата (12,2 г, выход: 84%). МС m/z [ESI]: 291,2 [М+1].
Стадия 2: (S)-1-бензил-2-метилпиперазин
(S)-трут-Бутил 4-бензил-3-метилпиперазин-1-карбоксилат (11,6 г, 40 ммоль) растворяли в CH2Cl2 (100 мл), добавляли по каплям трифторацетат (20 мл), и полученную смесь перемешивали в течение 30 мин. Смесь помещали в баню с ледяной водой, и добавляли концентрированный раствор NaOH, пока pH смеси не становился больше 13, затем смесь экстрагировали этилацетатом. Экстракт сушили и концентрировали с получением (S)-1-бензил-2-метилпиперазина (6,84 г, выход: 90%), который использовали на следующей стадии без дополнительной очистки. МС m/z [ESI]: 191,2 [М+1].
Стадия 3: (S)-1-бензил-2-метил-4-(1-метилпиперидин-4-ил)пиперазин
В безводный этанол (100 мл) последовательно добавляли (S)-1-Бензил-2-метилпиперазин (6,84 г, 36 ммоль), 1-метил-4-пиперидон (4,87 г, 43 ммоль) и ледяную уксусную кислоту (4,32 г, 72 ммоль). Полученную смесь перемешивали в течение 1 ч и затем охлаждали до 0°С. Добавляли порциями триацетоксиборгидрид натрия (31,6 г, 150 ммоль). Данную реакционную смесь выдерживали в течение 6 ч при комнатной температуре. Растворитель выпаривали, и остаток растворяли путем добавления этилацетата. Промывали водой, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением (S)-1-бензил-2-метил-4-(1-метилпиперидин-4-ил)пиперазина (7,86 г, выход: 76%). МС m/z [ESI]: 288,2 [М+1].
Стадия 4: (S)-3-метил-1-(1-метилпиперидин-4-ил)пиперазин
(S)-1-Бензил-2-метил-4-(1-метилпиперидин-4-ил)пиперазин (7,19 г, 25 ммоль) растворяли в метаноле (100 мл), добавляли Pd/C (1 г, 10%), и полученную смесь перемешивали в течение ночи в атмосфере водорода. Затем смесь фильтровали, и фильтрат концентрировали с получением (S)-3-метил-1-(1-метилпиперидин-4-ил)пиперазина (4,64 г, выход: 94%). МС m/z [ESI]: 198,2 [М+1].
Стадия 5: (S)-1-(4-метоксипиридин-2-ил)-2-метил-4-(1-метилпиперидин-4-ил)пиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2-хлор-4-метоксипиридина вместо 2-хлор-3-метоксипиридина и с использованием (S)-2-метил-4-(1-метилпиперидин-4-ил)пиперазина вместо N-метилпиперазина (выход: 37%). МС m/z [ESI]: 305,2 [М+1].
Стадия 6: (S)-1-(5-бром-4-метоксипиридин-2-ил)-2-метил-4-(1-метилпиперидин-4-ил)пиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием (S)-1-(4-метоксипиридин-2-ил)-2-метил-4-(1-метилпиперидин-4-ил)пиперазина вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 62%). МС m/z [ESI]: 383,1 [М+1].
Промежуточное соединение 14: (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Стадия 1: (S)-трет-бутил 4-(4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2-хлор-4-метоксипиридина вместо 2-хлор-3-метоксипиридина и с использованием (S)-трет-бутил 3-метилпиперазин-1-карбоксилата вместо N-метилпиперазина (выход: 50%). МС m/z [ESI]: 308,2 [М+1].
Стадия 2: (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием (S)-трет-бутил 4-(4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 75%). МС m/z [ESI]: 386,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.104 (s, 1H), 6.030 (s, 1H), 4.60-3.85 (br, 7H), 3.20-2.90 (br, 3H), 1.135 (d, J=6.8 Гц, 3H).
Промежуточное соединение 15: 4-(5-бром-4-метоксипиридин-2-ил)-1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин
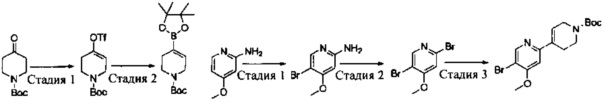
Стадия 1: 4-(трифторметансульфонилокси)-1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин
трет-Бутил 4-оксопиперидин-1-карбоксилат (22,8 г, 115 ммоль) растворяли в безводном тетрагидрофуране (150 мл), охлаждали до -78°С, и затем добавляли по каплям раствор лития диизопропиламида (126 ммоль) в тетрагидрофуране (100 мл). После завершения добавления полученный раствор перемешивали в течение 30 мин, и добавляли по каплям раствор бис(трифторметансульфонилокси)анилина (45,0 г) в тетрагидрофуране (126 ммоль). Данную реакционную смесь нагревали до комнатной температуры и перемешивали в течение ночи. Растворитель выпаривали, и остаток растворяли путем добавления диэтилового эфира. Данный раствор промывали 2 М раствором NaOH, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 4-(трифторметансульфонилокси)-1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридина (23,4 г, выход: 61%).
Стадия 2: трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат
трет-Бутил 4-(трифторметилсульфонилокси)-5,6-дигидропиридин-1(2Н)-карбоксилат (23,2 г, 70 ммоль), бис(пинаколато)дибор (25,4 г, 100 ммоль), Pd(dppf)Cl2 (2,93 г, 4 ммоль) и безводный ацетат калия (13,7 г, 140 ммоль) добавляли в сухой 1,4-диоксан (500 мл), и данную смесь продували азотом. Затем смесь перемешивали в течение 4 ч при 100°С. После охлаждения смесь концентрировали и очищали путем колоночной хроматографии на силикагеле с получением трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (13,0 г, выход: 60%).
Стадия 3: 5-бром-4-метоксил-2-аминопиридин
К раствору 4-метоксил-2-аминопиридина (12,4 г, 100 ммоль) в ацетонитриле (500 мл) добавляли порциями при перемешивании N-бромбутанимид (17,8 г, 100 ммоль) при 0°С. После завершения добавления данную смесь перемешивали в течение 1 ч при комнатной температуре. Растворитель выпаривали, и добавляли CH2Cl2. Данный раствор промывали насыщенным бикарбонатом натрия, сушили и концентрировали с получением 5-бром-4-метоксил-2-аминопиридина (20,3 г, выход: 100%), который использовали на следующей стадии без дополнительной очистки. МС m/z [ESI]: 203,0 [М+1].
Стадия 4: 2,5-дибром-4-метоксипиридин
5-Бром-4-метокси-2-аминопиридин (20,3 г, 100 ммоль) растворяли при перемешивании в 40% HBr (60 мл) и воде (40 мл) при -10°С, и затем добавляли раствор NaNO2 (17,3 г, 250 ммоль) в воде (25 мл). Данную смесь перемешивали при низкой температуре в течение 30 мин. Добавляли жидкий бром (48,0 и 300 ммоль), и перемешивание продолжали в течение 2 ч. Добавляли концентрированный раствор NaOH, пока pH смеси не становился больше 12, и затем смесь экстрагировали этилацетатом. Экстракт сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 2,5-дибром-4-метоксипиридина (13,8 г, выход: 52%). МС m/z [ESI]: 267,9 [М+1].
Стадия 5: трет-Бутил 4-(5-бром-4-метоксипиридин-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат
трет-Бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат (3,09 г, 10 моль), 2,5-дибром-4-метоксипиридин (2,67 г, 10 ммоль), Pd(PPh3)4 (578 мг, 0,5 ммоль) и K2CO3 (3,34 г, 24 ммоль) добавляли в смесь 1,4-диоксана (50 мл) и воды (10 мл), и полученную реакционную смесь продували азотом. Затем смесь перемешивали в течение ночи при 100°С. После охлаждения смесь очищали путем колоночной хроматографии на силикагеле с получением трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата (1,55 г, выход: 42%). МС m/z [ESI]: 369,1 [М+1]. 1 Н-ЯМР (400 МГц, CDCl3): δ=8.49 (s, 1Н), 7.26 (s, 1H), 6.58 (s, 1H), 4.13 (t, 2Н), 3.97 (s, 3Н), 2.62 (d, 2Н), 1.49 (s, 9Н).
Промежуточное соединение 16: 5-бром-2-хлор-3-метоксипиридин
Стадия 1: 5-бром-2-хлор-3-аминопиридин
5-Бром-2-хлор-3-нитропиридин (2,38 г, 10 ммоль) растворяли в метаноле (50 мл), и затем добавляли никель Ренея (0,5 г). Данную смесь перемешивали в течение ночи в атмосфере водорода. Никель Ренея удаляли путем фильтрования, и фильтрат концентрировали с получением 5-бром-2-хлор-3-аминопиридина (2,08 г, выход: 100%), который использовали на следующей стадии без дополнительной очистки. МС m/z [ESI]: 208,9 [М+1].
Стадия 2: 5-бром-2-хлор-3-гидроксипиридин
5-Бром-2-хлор-3-аминопиридин (2,08 г, 10 ммоль) растворяли при перемешивании в 4 М H2SO4 (50 мл) при 0°С, и затем добавляли раствор NaNO2 (760 мг, 11 ммоль) в воде (5 мл). После перемешивания в течение 30 мин при 0°С реакционную смесь нагревали до 80°С и перемешивали в течение 2 ч. Затем смесь охлаждали, и oH доводили до 7-8 путем добавления концентрированного раствора NaOH. Выпавшее в осадок твердое вещество отфильтровывали, промывали водой и сушили с получением 5-бром-2-хлор-3-гидроксипиридина (1,88 г, выход: 90%). МС m/z [ESI]: 209,9 [М+1].
Стадия 3: 5-бром-2-хлор-3-метоксипиридин
5-Бром-2-хлор-3-гидроксипиридин (1,88 г, 9 ммоль), иодметан (1,42 г, 10 ммоль) и K2CO3 (2,76 г, 20 ммоль) добавляли в ацетонитрил. Данный раствор перемешивали в течение 4 ч при 80°С. Растворитель выпаривали, и остаток растворяли путем добавления CH2Cl2. Данный раствор промывали водой, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 5-бром-2-хлор-3-метоксипиридина (1,62 г, выход: 81%). МС m/z [ESI]: 223,9 [М+1].
Промежуточное соединение 17: 1-(1-(4-метоксил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)пиперидин-4-ил)-4-метилпиперазин
Стадия 1: трет-бутил 4-(4-метилпиперазин-1-ил)пиперидин-1-карбоксилат
В безводный этанол (100 мл) последовательно добавляли трет-бутил 4-оксопиперидин-1-карбоксилат (4,0 г, 20 ммоль), N-метилпиперазин (2,4 г, 24 ммоль) и ледяную уксусную кислоту (2,4 г, 40 ммоль). Полученную смесь перемешивали в течение 1 ч, и затем охлаждали до 0°С. Добавляли порциями триацетоксиборгидрид натрия (17,0 г, 80 ммоль), и данную реакционную смесь выдерживали в течение 6 ч при комнатной температуре. Растворитель выпаривали. Остаток растворяли путем добавления этилацетата, промывали водой, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением mpem-бутил 4-(4-метилпиперазин-1-ил)пиперидин-1-карбоксилата (4,25 г, выход: 75%). МС m/z [ESI]: 284,2 [М+1].
Стадия 2: 1-метил-4-(пиперидин-4-ил)пиперазина гидрохлорид
трет-Бутил 4-(4-метилпиперазин-1-ил)пиперидин-1-карбоксилат (4,25 г, 15 ммоль) растворяли в метаноле (100 мл), и данный раствор насыщали газообразным хлористым водородом. Затем раствор перемешивали при температуре дефлегмации в течение 1 ч. Растворитель выпаривали на роторном испарителе с получением 1-метил-4-(пиперидин-4-ил)пиперазина гидрохлорида (4,39 г, выход: 100%), который использовали на следующей стадии без дополнительной очистки. МС m/z [ESI]: 184,2 [М+1].
Стадия 3: 1-(1-(4-метоксилпиридин-2-ил)пиперидин-4-ил)-4-метилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2-хлор-4-метоксипиридина вместо 2-хлор-3-метоксипиридина и с использованием 1-метил-4-(пиперидин-4-ил)пиперазина гидрохлорида вместо N-метилпиперазина (выход: 58%). МС m/z [ESI]: 291,2 [М+1].
Стадия 4: 1-(1-(5-бром-4-метоксилпиридин-2-ил)пиперидин-4-ил)-4-метилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием 1-(1-(4-метоксилпиридин-2-ил)пиперидин-4-ил)-4-метилпиперазина вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 62%). МС m/z [ESI]: 369,1 [М+1].
Стадия 5: 1-(1-(4-метоксил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)пиперидин-4-ил)-4-метилпиперазин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 1 (Стадия 3) с использованием 1-(1-(5-бром-4-метоксилпиридин-2-ил)пиперидин-4-ил)-4-метилпиперазина вместо трет-бутил 4-(3-метоксил-4-бромфенил)пиперазин-1-карбоксилата (выход: 81%). МС m/z [ESI]: 417,3 [М+1].
Промежуточное соединение 18: трет-бутил 4-(6-бром-3-метоксилпиридин-2-ил)пиперазин-1-карбоксилат
Промежуточное соединение 19: трет-бутил 4-(6-бром-5-метоксилпиридин-2-ил)пиперазин-1-карбоксилат
Указанные в заголовке соединения получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2,6-дибром-3-метоксилпиридина вместо 2-хлор-3-метоксилпиридина и с использованием N-метилпиперазина вместо N-трет-(бутоксикарбонил)пиперазина. Промежуточное соединение 18, трет-бутил 4-(6-бром-3-метоксилпиридин-2-ил)пиперазин-1-карбоксилат (выход: 21%), и Промежуточное соединение 19, трет-бутил 4-(6-бром-5-метоксилпиридин-2-ил)пиперазин-1-карбоксилат (выход: 43%), в заключение разделяли путем колоночной хроматографии на силикагеле. МС m/z [ESI]: 372,1 [М+1].
Промежуточное соединение 20: трет-бутил 4-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)пиперазин-1-карбоксилат
Промежуточное соединение 21: 4-(5-бром-4-метоксилпиридин-2-ил)пиперазин-2-он
Стадия 1: трет-Бутил 2-(4-метоксипиридин-2-иламино)этилкарбамат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2-хлор-4-метоксилпиридина вместо 2-хлор-3-метоксилпиридина и с использованием трет-бутил 2-аминоэтилкарбамата вместо N-метилпиперазина (выход: 53%). МС m/z [ESI]: 268,2 [М+1].
Стадия 2: N-(2-аминоэтил)-4-метоксилпиридин-2-амин
трет-Бутил 2-(4-метоксипиридин-2-иламино)этилкарбамат (2,66 г, 10 ммоль) растворяли в CH2Cl2 (50 мл), добавляли трифторацетат (10 мл), и данный раствор перемешивали в течение 1 ч. Реакционную смесь помещали в баню с ледяной водой, и добавляли концентрированный раствор NaOH, пока pH смеси не становился больше 12, и затем смесь экстрагировали этилацетатом. Экстракт сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением N-(2-аминоэтил)-4-метоксилпиридин-2-амина (1,27 г, выход: 76%). МС m/z [ESI]: 168,1 [М+1].
Стадия 3: 4-(4-метоксилпиридин-2-ил)пиперазин-2-он
N-(2-Аминоэтил)-4-метоксилпиридин-2-амин (1,17 г, 7 ммоль) и безводный K2CO3 (2,90 г, 21 ммоль) добавляли в безводный ацетонитрил, данную смесь охлаждали до 0°С, и затем добавляли по каплям хлорангидрид хлоруксусной кислоты (790 мг, 7 ммоль). После завершения добавления реакционную смесь перемешивали в течение 30 мин и затем подвергали дефлегмации в течение 6 ч. Затем смесь фильтровали, и фильтрат концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 4-(4-метоксилпиридин-2-ил)пиперазин-2-она (855 мг, выход: 59%). МС m/z [ESI]: 208,1 [М+1].
Стадия 4: 4-(5-бром-4-метоксилпиридин-2-ил)пиперазин-2-он
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием 4-(4-метоксилпиридин-2-ил)пиперазин-2-она вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 87%). МС m/z [ESI]: 286,0 [М+1].
Промежуточное соединение 22: трет-бутил 4-(6-хлор-4-метоксилпиридин-2-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2,6-дихлор-4-метоксилпиридина вместо 2-хлор-3-метоксилпиридина и с использованием трет-бутил пиперазин-1-карбоксилата вместо N-метилпиперазина (выход: 48%). МС m/z [ESI]: 328,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=6.266 (d, J=1.6 Гц, 1Н), 5.944 (d, J=1.6 Гц, 1Н), 3.804 (s, 3Н), 3.54-3.47 (m, 8Н), 1.481 (s, 9Н).
Промежуточное соединение 23: трет-бутил 4-(5-бром-4-метоксилпиридин-2-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием трет-бутил 4-(4-метоксилпиридин-2-ил)пиперазин-1-карбоксилата вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 87%). МС m/z [ESI]: 372,1 [М+1].
Промежуточное соединение 24: трет-бутил 4-(4-бром-5-метоксилпиридин-2-ил)пиперазин-1-карбоксилат
Промежуточное соединение 25: трет-бутил 4-(2-бром-5-метоксилпиридин-4-ил)пиперазин-1-карбоксилат
Указанные в заголовке соединения получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2,4-дибром-5-метоксилпиридина вместо 2-хлор-3-метоксилпиридина и с использованием трет-бутил пиперазин-1-карбоксилата вместо N-метилпиперазина. Промежуточное соединение 24, трет-бутил 4-(4-бром-5-метоксилпиридин-2-ил)пиперазин-1-карбоксилат (выход: 37%), и Промежуточное соединение 25, трет-бутил 4-(2-бром-5-метоксилпиридин-4-ил)пиперазин-1-карбоксилат (выход: 21%), в заключение разделяли путем хроматографии на силикагеле. МС m/z [ESI]: 372,1 [М+1].
Промежуточное соединение 26: 3-(1-(2-дифторметил-5-фторфенил)этокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-аминопиридин
Стадия 1: 1-бром-2-дифторметил-5-фторбензол
2-Бром-4-фторбензальдегид (8,04 г, 40 ммоль) добавляли в CH2Cl2 (80 мл), данную смесь охлаждали до 0°С, и затем добавляли по каплям диэтиламиносератрифторид (DAST) (7,96 мл, 60 ммоль). Полученную смесь перемешивали при низкой температуре в течение 30 мин и затем подвергали дефлегмации в течение ночи. Реакцию останавливали путем добавления этанола, реакционную смесь промывали водой, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 1-бром-2-(дифторметил)-5-фторбензола (8,36 г, выход: 93%). 1Н ЯМР (400 МГц, CDCl3): δ=7.579 (m, 1Н), 7.385 (m, 1H), 7.091 (m, 1H), 35 6.993-6.720 (t, J=54.6 Гц, 1H).
Стадия 2: 1-(2-(дифторметил)-5-фторфенил)этанон
1-Бром-2-(дифторметил)-5-фторбензол (8,36 г, 37,5 ммоль) растворяли в безводном диэтиловом эфире (100 мл), охлаждали до -78°С, и затем добавляли по каплям в атмосфере азота 2,4 М н-бутиллитий (18,7 мл, 45 ммоль). Данную смесь перемешивали в течение 1 ч. Добавляли N-метил-N-метоксилацетамид (7,73 г, 75 ммоль), поддерживая температуру -78°С, и полученную смесь перемешивали в течение 2 ч. Смесь нагревали до комнатной температуры и затем промывали насыщенным солевым раствором и экстрагировали этилацетатом. Экстракт сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 1-(2-(дифторметил)-5-фторфенил)этанона (4,2 г, выход: 60%). 1Н ЯМР (400 МГц, CDCl3): δ=7.45-7.40 (m, 1Н), 7.31-7.27 (m, 1H), 7.20-7.12 (m, 1H), 6.78-6.50 (t, J=56 Гц, 1H), 2.42 (s, 3H).
Стадия 3: 1-(2-(дифторметил)-5-фторфенил)этанол
1-(2-(Дифторметил)-5-фторфенил)этанон (3,0 г, 16 ммоль) добавляли в безводный этанол (100 мл), данную смесь охлаждали до 0°С, и затем добавляли порциями NaBH4 (1,22 г, 32 ммоль). Реакцию проводили при комнатной температуре в течение 4 ч, затем растворитель выпаривали, и остаток растворяли путем добавления этилацетата. Полученный раствор промывали водой, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 1-(2-(дифторметил)-5-фторфенил)этанола (1,82 г, выход: 60%). 1Н ЯМР (400 МГц, CDCl3): δ=7.598-7.562 (m, 1Н), 7.292-7.262 (m, 1H), 7.20-7.15 (m, 1H), 7.124-6.848 (t, J=45.2 Гц, 1H), 5.197 (t, J=6.4 Гц, 1Н), 1.97 (s, 1H), 1.515 (d, J=6.4 Гц, 3H).
Стадия 4: 2-(1-бромэтил)-1-(дифторметил)-4-фторбензол
PPh3 (1,57 г, 6 ммоль) растворяли в дихлорметаноле (30 мл), охлаждали до 0°С, добавляли Br2 (1,08 г, 6 ммоль), и полученную смесь перемешивали в течение 10 мин. Добавляли по каплям раствор 1-(2-(дифторметил)-5-фторфенил)этанола (950 мг, 5 ммоль) в CH2Cl2 (5 мл), и перемешивали в течение 30 мин. Затем реакционную смесь промывали водой, сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 2-(1-бромэтил)-1-(дифторметил)-4-фторбензола (1,25 г, выход: 100%).
Стадия 5: 5-бром-3-(1-(2-дифторметил-5-фторфенил)этокси)-2-амино-пиридин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 5 (Стадия 2) с использованием 2-(1-бромэтил)-1-(дифторметил)-4-фторбензола вместо 1,3-дихлор-2-(1-бромэтил)-4-фторбензола (выход: 62%). МС m/z [ESI]: 361,0 [М+1].
Стадия 6: 3-(1-(2-дифторметил-5-фторфенил)этокси-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-2-аминопиридин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 5 (Стадия 3) с использованием 5-бром-3-(1-(2-дифторметил-5-фторфенил)этокси)-2-аминопиридина вместо 5-бром-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-аминопиридина (выход: 54%). МС m/z [ESI]: 409,2 [М+1].
Промежуточное соединение 27: 4-(6-бром-3-метоксилпиридин-2-ил)-1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 15 (Стадия 5) с использованием 2,6-дибром-3-метоксилпиридина вместо 2,5-дибром-4-метоксилпиридина. Промежуточное соединение 27, 4-(6-бром-3-метоксилпиридин-2-ил)-1-(трет-бутоксикарбонил)-1,2,5,6-тетрагидропиридин, в заключение выделяли путем колоночной хроматографии на силикагеле (выход: 39%). МС m/z [ESI]: 369,1 [М+1].
Промежуточное соединение 28: (S)-трет-бутил 4-(6-бром-5-метоксилпиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2,6-дибром-3-метоксилпиридина вместо 2-хлор-3-метоксилпиридина и с использованием (S)-трет-бутил 3-метилпиперазин-1-карбоксилата вместо N-метилпиперазина, с последующим разделением изомеров путем колоночной хроматографии на силикагеле (выход: 38%). МС m/z [ESI]: 386,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.124 (d, J=2.0 Гц, 1Н), 6.470 (d, J=8.4 Гц, 1Н), 4.20-3.85 (br, 3H), 3.825 (s, 3H), 3.80-3.70 (br, 1H), 3.162 (d, J=10.8 Гц, 1Н), 3.085 (t, J=6.8 Гц, 1Н), 3.03-2.87 (br, 1H), 1.482 (s, 9H), 1.088 (d, J=6.8 Гц, 3Н).
Промежуточное соединение 29: трет-бутил 4-(5-бром-4-метоксилпиридин-3-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 3,5-дибром-4-метоксилпиридина вместо 2-хлор-3-метоксилпиридина и с использованием N-(трет-бутоксикарбонил)пиперазина вместо N-метилпиперазина (выход: 53%). МС m/z [ESI]: 372,1 [М+1].
Промежуточное соединение 30: 4-(5-бром-4-метоксилпиридин-2-ил)морфолин
Стадия 1: 4-(4-метоксилпиридин-2-ил)морфолин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием 2-хлор-4-метоксилпиридина вместо 2-хлор-3-метоксилпиридина и с использованием морфолина вместо N-метилпиперазина (выход: 76%). МС m/z [ESI]: 195,1 [М+1].
Стадия 2: 4-(5-бром-4-метоксилпиридин-2-ил)морфолин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием 4-(4-метоксилпиридин-2-ил)морфолина вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 91%). МС m/z [ESI]: 273,0 [М+1]. 1Н ЯМР (400 Мгц, CDCl3): δ=8.119 (s, 1H), 6.080 (s, 1H), 3.905 (s, 3H), 3.816 (t, J=4.8 Гц, 4H), 3.478 (t, J=5.0 Гц, 4Н).
Промежуточное соединение 31: (S)-трет-бутил 4-(6-бром-3-метоксилпиридин-2-ил)-3-метилпиперазин-1-карбоксилат
(S)-трет-Бутил 4-(6-бром-3-метоксилпиридин-2-ил)-3-метилпиперазин-1-карбоксилат выделяли путем колоночной хроматографии на силикагеле в виде изомера Промежуточного соединения 28 в процессе получения Промежуточного соединения 28. МС m/z [ESI]: 386,1 [М+1].
Промежуточное соединение 33: трет-бутил 5-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилат
Промежуточное соединение 34: 5-бром-2-хлор-3-(2-гидроксиэтокси)-пиридин
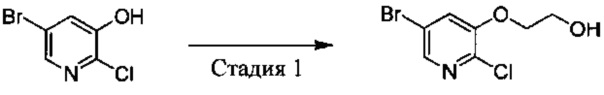
5-Бром-2-хлор-3-(2-гидроксиэтокси)-пиридин получали в соответствии с методикой синтеза Промежуточного соединения 16 (Стадия 3) с использованием 2-бромэтанола вместо подметана (выход: 50%). МС m/z [ESI]: 253,9 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.094 (d, J=2.0 Гц, 1Н), 7.378 (d, J=2.0 Гц, 1Н), 4.176-4.154 (t, J=4.4 Гц, 2Н), 4.056-4.018 (m, 2H), 2.055-2.023 (t, J=4.4 Гц, 1Н).
Промежуточное соединение 35: (S)-трет-бутил 4-(5-бром-4-этоксилпиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Стадия 1: (Ы)-трет-бутил 4-(4-этоксилпиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием (S)-трет-бутил 3-метилпиперазин-1-карбоксилата вместо N-метилпиперазина и с использованием 2-хлор-4-этоксилпиридина вместо 2-хлор-3-метоксилпиридина (выход: 76%). МС m/z [ESI]: 322,2 [М+1].
Стадия 2: (S)-трет-бутил 4-(5-бром-4-этоксилпиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием (S)-трет-бутил 4-(4-этоксилпиридин-2-ил)-3-метилпиперазин-1-карбоксилата вместо 1-(3-метоксипиридин-2-ил)-4-метилпиперазина (выход: 91%). МС m/z [ESI]: 400,1 [М+1].
Промежуточное соединение 36: 4-(2-((5-бром-2-морфолинопиридин-3-ил)окси)этил)морфолин
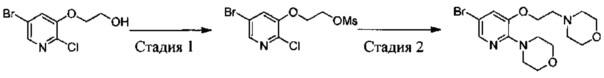
Стадия 1: 2-((5-бром-2-хлорпиридин-3-ил)окси)этилметансульфонат
Промежуточное соединение 34, 5-бром-2-хлор-3-(2-гидроксиэтокси)пиридин, и триэтиламин (487 мг, 4,82 ммоль) добавляли в CH2Cl2 (10 мл), данную смесь охлаждали до 0°С, затем добавляли метансульфохлорид (387 мг, 3,38 ммоль), и полученную смесь перемешивали в течение 2 ч. Затем реакционную смесь промывали водой, сушили и концентрировали с получением 2-((5-бром-2-хлорпиридин-3-ил)окси)этил метансульфоната (796 мг, выход: 100%), который использовали на следующей стадии без дополнительной очистки. MC m/z [ESI]: 331,9 [М+1].
Стадия 2: 4-(2-((5-бром-2-морфолинопиридин-3-ил)окси)этил)морфолин
2-((5-Бром-2-хлорпиридин-3-ил)окси)этилметансульфонат (796 мг, 2,41 ммоль) и Na2CO3 (511 мг, 4,82 ммоль) добавляли в морфолин (10 мл). Полученную смесь перемешивали в течение ночи при 100°С. Затем реакционную смесь концентрировали и очищали путем колоночной хроматографии на силикагеле (CH2Cl2/СН3ОН, 80:1) с получением 4-(2-((5-бром-2-морфолинопиридин-3-ил)окси)этил)морфолина (820 мг, выход: 92%). МС m/z [ESI]: 372,1 [М+1].
Промежуточное соединение 37: (S)-трет-бутил 4-(5-бром-4-(2-морфолиноэтокси)пиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Стадия 1: 2-хлор-4-(2-морфолиноэтокси)пиридин
N-(2-Гидроксиэтил)морфолин (3,15 г, 24 ммоль) растворяли в сухом ДМФА, добавляли порциями NaH (891 мг, 26 ммоль), и полученную смесь перемешивали в течение 30 мин. Затем добавляли 2-хлор-4-нитропиридин (3,17 г, 20 ммоль), и данную смесь перемешивали в течение 3 ч при комнатной температуре. Растворитель выпаривали на роторном испарителе, остаток растворяли путем добавления этилацетата и очищали путем колоночной хроматографии на силикагеле с получением 2-хлор-4-(2-морфолиноэтокси)пиридина (1,60 г, выход: 33%). МС m/z [ESI]: 243,1 [М+1].
Стадия 2: (S)-трет-бутил 3-метил-4-(4-(2-морфолиноэтокси)пиридин-2-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 2) с использованием (S)-трет-бутил 3-метилпиперазин-1-карбоксилата вместо N-метилпиперазина и с использованием 2-хлор-4-(2-морфолиноэтокси)пиридина вместо 2-хлор-3-метоксилпиридина (выход: 36%). МС m/z [ESI]: 407,3 [М+1].
Стадия 3: (S)-трет-бутил 4-(5-бром-4-(2-морфолиноэтокси)пиридин-2-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 8 (Стадия 3) с использованием (S)-трет-бутил 3-метил-4-(4-(2-морфолиноэтокси)пиридин-2-ил)пиперазин-1-карбоксилата вместо 1-(3-метоксилпиридин-2-ил)-4-метилпиперазина (выход: 71%). МС m/z [ESI]: 487,2 [М+1].
Промежуточное соединение 38: 2-хлор-4-((1-(4-хлор-3-фторбензил)-1H-имидазол-5-ил)этинил)пиридин
Стадия 1: 4-(бромметил)-1-хлор-2-фторбензол
К раствору трифенилфосфина (27,8 г, 0,103 моль) в CH2Cl2 (200 мл) добавляли по каплям при перемешивании бром (16,5 г, 0,103 моль) при 0°С. После завершения добавления полученную смесь перемешивали в течение 10 мин. Затем добавляли (4-хлор-3-фторфенил)метанол (15,8 г, 0,098 моль). Данную смесь перемешивали в течение 30 мин. Реакцию останавливали путем добавления этанола, реакционную смесь вливали в насыщенный раствор бикарбоната натрия и экстрагировали CH2Cl2. Органическую фазу сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 4-(бромметил)-1-хлор-2-фторбензола (18,6 г, выход: 85%). 1Н ЯМР (400 МГц, CDCl3): δ=7.27 (t, J=7.9 Гц, 1H), 7.10 (dd, J=7.9 Гц, 2.1 Гц, 1Н), 7.04-6.99 (m, 1Н), 4.32 (s, 2Н).
Стадия 2: 1-(4-хлор-3-фторбензил)-1H-пиразол
4-(Бромметил)-1-хлор-2-фторбензол (16,0 G, 72 ммоль), пиразол (5,35 г, 79 ммоль) и K2CO3 (20,0 г, 145 ммоль) добавляли в сухой ДМФА. Полученную смесь перемешивали в течение ночи при комнатной температуре. Растворитель выпаривали на роторном испарителе, остаток растворяли путем добавления этилацетата и очищали путем колоночной хроматографии на силикагеле с получением 1-(4-хлор-3-фторбензил)-1H-пиразола (13,7 г, выход: 91%). МС m/z [ESI]: 211,0 [М+1].
Стадия 3: 1-(4-хлор-3-фторбензил)-1H-пиразол-2-оксид
1-(4-Хлор-3-фторбензил)-1H-пиразол (9,7 г, 46 ммоль) и гидропероксид мочевины (9,1 г, 97 ммоль) добавляли в CH2Cl2 (200 мл), и данную смесь охлаждали до 0°С. Добавляли ангидрид трифторуксусной кислоты (19,4 г, 92 ммоль), и полученный раствор перемешивали в течение 5 ч при комнатной температуре. Реакционную смесь промывали раствором сульфита натрия и экстрагировали. Органический слой упаривали на роторном испарителе и очищали путем колоночной хроматографии на силикагеле с получением 1-(4-хлор-3-фторбензил)-1H-пиразол-2-оксида (7,30 г, выход: 70%). МС m/z [ESI]: 227,0 [М+1].
Стадия 4: 5-бром-1-(4-хлор-3-фторбензил)-1H-пиразол-2-оксид
1-(4-Хлор-3-фторбензил)-1H-пиразол-2-оксид (5,44 г, 24 ммоль) и K2CO3 (5,96 г, 43 ммоль) добавляли в CH2Cl2 (100 мл), данную смесь охлаждали до -80°С, и добавляли по каплям в течение 2 мин раствор жидкого брома (4,0 г, 25 ммоль) в CH2Cl2 (10 мл), предварительно охлажденный до -80°С. Перемешивание продолжали при этой же температуре в течение 15 мин. Смесь нагревали до 0°С, и перемешивание продолжали в течение 30 мин. Реакционную смесь промывали сульфитом натрия и экстрагировали. Органический слой упаривали на роторном испарителе и очищали путем колоночной хроматографии на силикагеле с получением 5-бром-1-(4-хлор-3-фторбензил)-1H-пиразол-2-оксида (6,67 г, выход: 91%). МС m/z [ESI]: 306,9 [М+1].
Стадия 5: 5-бром-1-(4-хлор-3-фторбензил)-1H-пиразол
5-Бром-1-(4-хлор-3-фторбензил)-1Я-пиразол-2-оксид (3,84 г, 12,6 ммоль) добавляли в CH2Cl2 (50 мл), данную смесь охлаждали до 0°С, и затем добавляли по каплям раствор PCl3 (3,97 г, 28,9 ммоль) в CH2Cl2 (10 мл). Полученную смесь перемешивали при этой же температуре в течение 1 ч, затем нагревали до 50°С и затем перемешивали в течение 3 ч. Добавляли раствор ацетата натрия в метаноле (1,21 М, 170 мл). Данную смесь упаривали на роторном испарителе и очищали путем колоночной хроматографии на силикагеле с получением 5-бром-1-(4-хлор-3-фторбензил)-1H-пиразола (3,10 г, выход: 85%). МС m/z [ESI]: 290,9 [М+1].
Стадия 6: 1-(4-хлор-3-фторбензил)-5-((триметилсилил)этинил)-1H-пиразол
5-Бром-1-(4-хлор-3-фторбензил)-1H-пиразол (2,0 г, 6,9 ммоль), этинил-триметилсилан (1,0 г, 10,4 ммоль), Pd(OAc)2 (155 мг, 0,69 ммоль), X-phos (657 мг, 1,38 ммоль), Cs2CO3 (3,4 г, 10 ммоль) и 1,4-диоксан (20 мл) вносили в реакционную пробирку для микроволновой печи. Реакцию проводили в течение 3 ч при 150°С в атмосфере азота. Растворитель выпаривали на роторном испарителе, и остаток очищали путем колоночной хроматографии на силикагеле с получением 1-(4-хлор-3-фторбензил)-5-((триметилсилил)этинил)-1H-пиразола (510 мг, выход: 24%). МС m/z [ESI]: 307,1 [М+1].
Стадия 7: 1-(4-хлор-3-фторбензил)-5-этинил-1H-пиразол
1-(4-Хлор-3-фторбензил)-5-((триметилсилил)этинил)-1H-пиразол (510 мг, 1,66 ммоль) и K2CO3 (460 мг, 3,33 ммоль) добавляли в метанол (30 мл), и данную смесь перемешивали в течение 2 ч при комнатной температуре. Твердое вещество удаляли путем фильтрования, и фильтрат упаривали на роторном испарителе и очищали путем колоночной хроматографии на силикагеле с получением 1-(4-хлор-3-фторбензил)-5-этинил-1H-пиразола (350 мг, выход: 90%). МС m/z [ESI]: 235,0 [М+1].
Стадия 8: 2-хлор-4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)пиридин
1-(4-Хлор-3-фторбензил)-5-этинил-1H-пиразол (348 мг, 1,48 ммоль), 2-хлорпиридин-4-ил-трифторметансульфонат (1,78 ммоль), [(C6H5)3P]2PdCl2 (0,148 ммоль), CuI (562 мг, 2,96 ммоль) и триэтиламин (300 мг, 2,96 ммоль) добавляли в ДМФА (20 мл). Данную реакционную смесь выдерживали при 70°С в течение ночи в атмосфере азота. Растворитель выпаривали на роторном испарителе, и остаток очищали путем колоночной хроматографии на силикагеле с получением 2-хлор-4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)пиридина (360 мг, выход: 70%). МС m/z [ESI]: 346,0 [М+1].
Промежуточное соединение 39: 5-бром-3-(1-(2-(диметилфосфорил)-5-фторфенил)этоксил)-2-аминопиридин
Стадия 1: 3-(1-(2-(диметилфосфорил)-5-фторфенил)этоксил)-2-нитропиридин
3-(1-(2-Бром-5-фторфенил)этоксил)-2-нитропиридин (371 мг, 1,1 ммоль), диметилфосфин-оксид  (94 мг, 1,2 ммоль), Pd(OAc)2 (22 мг, 0,1 ммоль), X-phos (95 мг, 0,2 ммоль) и K3PO4 (244 мг, 1,1 ммоль) растворяли в ДМФА (10 мл), и продували азотом. Реакционную смесь выдерживали при 130°С в течение 1 ч. Затем смесь охлаждали, растворитель выпаривали на роторном испарителе, и остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 35%). МС m/z [ESI]: 339,1 [М+1].
(94 мг, 1,2 ммоль), Pd(OAc)2 (22 мг, 0,1 ммоль), X-phos (95 мг, 0,2 ммоль) и K3PO4 (244 мг, 1,1 ммоль) растворяли в ДМФА (10 мл), и продували азотом. Реакционную смесь выдерживали при 130°С в течение 1 ч. Затем смесь охлаждали, растворитель выпаривали на роторном испарителе, и остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 35%). МС m/z [ESI]: 339,1 [М+1].
Стадия 2: 3-(1-(2-(диметилфосфорил)-5-фторфенил)этоксил)-2-аминопиридин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 6 (Стадия 2) с использованием 3-(1-(2-(диметилфосфорил)-5-фторфенил)этоксил)-2-нитропиридина вместо (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-нитропиридина (выход: 73%). МС m/z, [ESI]: 309,1 [М+1].
Стадия 3: 5-бром-3-(1-(2-(диметилфосфорил)-5-фторфенил)этоксил)-2-аминопиридин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 6 (Стадия 3) с использованием 3-(1-(2-(диметилфосфорил)-5-фторфенил)этоксил)-2-аминопиридина вместо (R)-3-(1-(2,6-дихлор-3-фторфенил)ethyoxyl)-2-аминопиридина (выход: 75%). МС m/z [ESI]: 387,0 [М+1].
Промежуточное соединение 40: 5-бром-3-(1-(4-хлор-2-(диметиламино)-5-фторфенил)этокси)-2-аминопиридин
Стадия 1: 2-бром-5-хлор-4-фторанилин
К раствору 3-хлор-4-фторанилина (5,82 г, 0,04 моль) в CH2Cl2 (150 мл) добавляли порциями при перемешивании N-бромсукцинимиде (7,12 г, 0,04 моль) при 0°С. После завершения добавления полученную смесь перемешивали в течение 30 мин и фильтровали. Фильтрат концентрировали и очищали путем колоночной хроматографии на силикагеле с получением 2-бром-5-хлор-4-фторанилина (6,16 г, выход: 69%). 1Н ЯМР (400 МГц, CDCl3): δ=7.234 (d, J=8.4 Гц, 1Н), 6.788 (d, J=6.4 Гц, 1H), 3.982 (br, 2H). MC m/z [ESI]: 225,9 [М+1].
Стадия 2: 2-бром-5-хлор-4-фтор-N,N-диметиланилин
Смесь 2-бром-5-хлор-4-фторанилина (2,25 г, 0,01 моль), 40% раствора формальдегида (10 мл, 0,133 моль) и муравьиной кислоты (4,6 г, 0,1 моль) выдерживали в течение 3 ч при 100°С. Добавляли NaOH, пока pH смеси не сдвигался в сильнощелочную область, и затем смесь экстрагировали CH2Cl2. Экстракт сушили, упаривали на роторном испарителе и очищали путем колоночной хроматографии на силикагеле с получением 2-бром-5-хлор-4-фтор-N,N-диметиланилина (2,46 г, выход: 97%). 1Н ЯМР (400 МГц, CDCl3): δ=7.368 (d, J=8.4 Гц, 1Н), 7.079 (d, J=6.8 Гц, 1H), 2.748 (s, 6Н). МС m/z [ESI]: 253,9 [М+1].
Стадия 3: 2-(диметиламино)-4-хлор-5-(фторфенил)этанон
К раствору 2-бром-5-хлор-4-фтор-N,N-диметиланилина (2,46 г, 9,7 ммоль) в сухом ТГФ (тетрагидрофуране) (50 мл), охлажденному до -78°С, добавляли в атмосфере азота 2,5 М н-бутиллитий (4,1 мл, 10,2 ммоль), и перемешивали в течение 2 ч при -78°С. Добавляли N-метил-N-метоксилацетамид (1,00 г, 9,7 ммоль), и раствор перемешивали в течение 2 ч при -78°С. Затем реакционную смесь нагревали до комнатной температуры и перемешивали в течение 2 ч. Растворитель выпаривали на роторном испарителе, и остаток очищали путем колоночной хроматографии на силикагеле с получением 2-(диметиламино)-4-хлор-5-(фторфенил)этанона (0,66 г, выход: 32%). 1Н ЯМР (400 МГц, CDCl3): δ=7.234 (d, J=9.2 Гц, 1Н), 7.032 (d, J=6.0 Гц, 1Н), 2.760 (s, 6Н), 2.597 (s, 3Н). МС m/z [ESI]: 216,0 [М+1].
Стадия 4: 1-(2-(диметиламино)-4-хлор-5-фторфенил)этанол
2-(Диметиламино)-4-хлор-5-(фторфенил)этанон (646 мг, 3 ммоль) растворяли в этаноле (10 мл), раствор помещали в ледяную баню, и добавляли порциями NaBH4 (342 мг, 9 ммоль). Полученную смесь перемешивали в течение 2 ч при комнатной температуре. Растворитель выпаривали, и остаток очищали путем колоночной хроматографии на силикагеле с получением 1-(2-(диметиламино)-4-хлор-5-фторфенил)этанола (458 мг, выход: 70%). 1Н ЯМР (400 МГц, CDCl3): δ=7.25 (d, J=6.6 Гц, 1H), 7.03 (d, J=10.0 Гц, 1Н), 5.77 (brs, 1Н), 5.06 (q, J=6.5 Гц, 1Н), 2.68 (s, 6Н), 1.51 (d, J=6.5 Гц, 3Н). МС m/z [ESI]: 218,1 [М+1].
Стадия 5: 2-(1-бромэтил)-5-хлор-4-фтор-N,N-диметиланилин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 5 (Стадия 1) с использованием 1-(2-(диметиламино)-4-хлор-5-фторфенил)этанола вместо 1-(2,6-дихлор-3-фторфенил)этанола (выход: 31%). МС m/z [ESI]: 282,0 [М+1].
Стадия 6: 5-бром-3-(1-(4-хлор-2-(диметиламино)-5-фторфенил)этокси)-2-аминопиридин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 5 (Стадия 2) с использованием 2-(1-бромэтил)-5-хлор-4-фтор-N,N-диметиланилина вместо 1,3-дихлор-2-(1-бромэтил)-4-фторбензола (выход: 22%). МС m/z [ESI]: 390,0 [М+1].
Промежуточное соединение 41: 4-(2-((5-бром-2-хлорпиридин-3-ил)окси)этил)морфолин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 16 (Стадия 3) с использованием 4-(2-хлорэтил)морфолина гидрохлорида вместо подметана (выход: 67%). МС m/z [ESI]: 323,0 [М+1].
Пример 1: 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
Общие методики синтеза
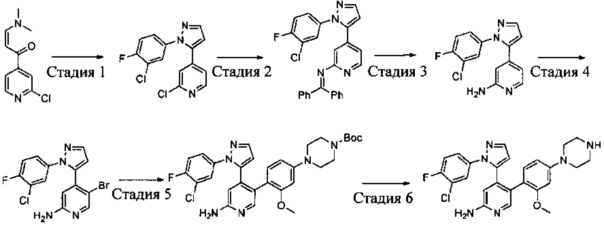
Стадия 1: 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин
1-(2-Хлорпиридин-4-ил)-3-(диметиламино)проп-2-ен-1-он (2,11 г, 10 ммоль), 3-хлор-4-фторфенилгидразин (1,61 г, 10 ммоль), несколько капель ледяной уксусной кислоты и воду (1 мл) добавляли в этанол (50 мл). Полученную смесь подвергали дефлегмации в течение 1,5 ч. Растворитель выпаривали на роторном испарителе. Остаток растворяли путем добавления этилацетата, и полученный раствор промывали водой. Органический слой сушили и очищали путем колоночной хроматографии на силикагеле с получением 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (2,50 г, выход: 81%). МС m/z [ESI]: 308.0 [М+1].
Стадия 2: 4-(1-(3-хлор-4-фторфенил)-1Н-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин
В герметически закрываемую пробирку вносили 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин (2,0 г, 6,5 ммоль), бензофенонимин (1,4 г, 7,7 ммоль), Pd2(dba)3 (297 мг, 0,325 ммоль), BINAP (0,42 г, 0,65 ммоль), трет-бутилат натрия (0,94 г, 9,75 ммоль) и толуол (30 мл), и данную смесь продували азотом. Затем смесь перемешивали в течение 3 ч при 120°С. Растворитель выпаривали на роторном испарителе, и добавляли этилацетат и воду. Органический слой сушили и очищали путем колоночной хроматографии на силикагеле с получением 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (1,4 г, выход: 48%). 1Н ЯМР (400 МГц, CDCl3): δ=8.235 (t, 1.8 Гц, 1Н), 7.803 (d, J=7.2 Гц, 2Н), 7.679 (d, J=1.6 Гц, 1Н), 7.502 (d, J=7.6 Гц 1Н), 7.44-7.26 (m, 6H), 7.155 (d, J=6.8 Гц, 2Н), 7.052 (t, J=8.3 Гц, 1Н), 6.92-6.88 (m, 1H), 6.551-6.536 (q, 2H), 6.389 (d, J=1.6 Гц, 1Н). МС m/z [ESI]: 453.1 [М+1].
Стадия 3: 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин
4-(1-(3-Хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин добавляли в 2 н. HCl (50 мл), и полученную смесь перемешивали в течение ночи. Реакционную смесь экстрагировали этилацетатом для удаления бензофенона, в водную фазу добавляли концентрированный раствор NaOH, пока pH не становился больше 12, и затем водную фазу экстрагировали этилацетатом. Органический слой сушили и упаривали на роторном испарителе с получением 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (0,77 г, выход: 87%), который использовали на следующей стадии без дополнительной очистки. МС m/z [ESI]: 289,1 [М+1].
Стадия 4: 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин
К раствору 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (0,72 г, 2,5 ммоль) в CH2Cl2 (25 мл), помещенному в ледяную баню, добавляли при перемешивании жидкий бром (400 мг, 2,5 ммоль). Полученную смесь перемешивали в течение 1 ч, промывали раствором Na2CO3 и экстрагировали CH2Cl2. Органический слой сушили над безводным сульфатом натрия, концентрировали и очищали путем колоночной хроматографии на силикагеле (CH2Cl2 : метанол = 50:1) с получением 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (588 мг, выход: 64%). 1Н ЯМР (400 МГц, CDCl3): δ=8.49 (s, 1Н), 7.746 (d, J=2.0 Гц, 1Н), 7.523 (dd, J=7.2 Гц, 2.0 Гц, 1Н), 7.066-7.048 (m, 2Н), 6.517 (d, J=2.0 Гц, 1Н), 6.424 (s, 1H), 4.622 (s, 2H). MC m/z [ESI]: 369,0 [М+1].
Стадия 5: трет-бутил 4-(4-(6-амино-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-3-ил)-3-метоксифенил)пиперазин-1-карбоксилат
5-Бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин (237 мг, 0,644 ммоль), трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилат (297 мг, 0,709 ммоль), Pd(PPh3)4 (75 мг, 0,064 ммоль) и Cs2CO3 (419 мг, 1,29 ммоль) добавляли в 1,4-диоксан (10 мл) и воду (1,5 мл), данную смесь продували азотом и перемешивали при 100°С в течение ночи. Полученный раствор охлаждали, и затем очищали путем колоночной хроматографии на силикагеле с выходом трет-бутил 4-(4-(6-амино-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-3-ил)-3-метоксифенил)пиперазин-1-карбоксилата (112 мг, выход: 30%). МС m/z [ESI]: 579,2 [М+1].
Стадия 6: 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
К раствору трет-бутил 4-(4-(6-амино-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-3-ил)-3-метоксифенил)пиперазин-1-карбоксилата (110 мг, 0,19 ммоль) в CH2Cl2 (10 мл) добавляли при перемешивании трифторацетат (1 мл), и данную смесь перемешивали в течение 1 ч. Добавляли раствор NaOH, пока pH водной фазы не становился больше 13, и затем смесь экстрагировали CH2Cl2. Органический слой сушили над безводным сульфатом натрия и фильтровали. Фильтрат концентрировали и очищали путем колоночной хроматографии на силикагеле (CH2Cl2 : метанол (об./об.) = 8:1) с получением 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амина (64 мг, выход: 70%). 1Н ЯМР (400 МГц, CDCl3): δ=7.650-7.631 (m, 2Н), 7.079-7.035 (m, 2Н), 6.872-6.850 (m, 1H), 6.812 (m, 1Н), 6.589 (d, J=1.6 Гц, 1Н), 6.355 (d, J=9.2 Гц, 1H), 6.295 (d, J=7.2 Гц, 2Н), 3.416-3.364 (m, 4Н), 3.341 (s, 3Н), 3.297-3.273 (m, 4Н). МС m/z [ESI]: 479,2 [М+1].
Пример 2: 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амина гидрохлорид
Стадия 1: 2-хлор-4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 1), с использованием 4-фторфенилгидразина вместо 3-хлор-4-фторфенилгидразина (выход: 86%). МС m/z [ESI]: 274,0 [М+1].
Стадия 2: 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин
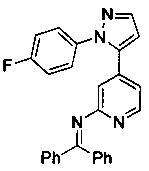
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-хлор-4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридина вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 56%). МС m/z [ESI]: 419,2 [М+1].
Стадия 3: 4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 77%). МС m/z [ESI]: 255,1 [М+1].
Стадия 4: 5-бром-4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 52%). МС m/z [ESI]: 333,0 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.176 (s, 1Н), 7.742 (d, J=2.0 Гц, 1Н), 7.291-7.256 (m, 2Н), 7.022 (t, J=8.6 Гц, 2Н), 6.521 (d, J=2.0 Гц, 1Н), 6.394 (s, 1Н), 4.502 (s, 2Н).
Стадия 5: 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 35%). МС m/z [ESI]: 545,3 [М+1].
Стадия 6: 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амина гидрохлорид
4-(1-(4-Фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амин (54 мг, 0,1 ммоль) растворяли в метаноле (20 мл), и затем данный раствор насыщали газообразным хлористым водородом. Перемешивание продолжали в течение 1 ч. Растворитель выпаривали на роторном испарителе, и остаток промывали диэтиловым эфиром и сушили с получением 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амина гидрохлорида (46 мг). МС m/z [ESI]: 445,2 [М+1].
Пример 3: 4-(1-(3-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амина гидрохлорид
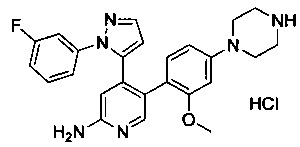
Стадия 1: 2-хлор-4-(1-(3-фторфенил)-1H-пиразол-5-ил)пиридин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 1), с использованием 3-фторфенилгидразина вместо 3-хлор-4-фторфенилгидразина (выход: 79%). МС m/z [ESI]: 274,0 [М+1].
Стадия 2: 4-(1-(3-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин
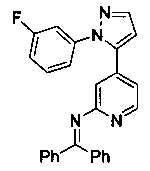
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-хлор-4-(1-(3-фторфенил)-1H-пиразол-5-ил)пиридина вместо 2-хлор-4-(1 -(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 69%). МС m/z [ESI]: 419,2 [М+1].
Стадия 3: 4-(1-(3-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-(1-(3-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 80%). МС m/z [ESI]: 255,1 [М+1].
Стадия 4: 5-бром-4-(1-(3-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-(1-(3-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 58%). МС m/z [ESI]: 333,0 [М+1].
Стадия 5: 4-(1-(3-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(3-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 30%). МС m/z [ESI]: 545,3 [М+1].
Стадия 6: 4-(1-(3-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амина гидрохлорид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 2 (Стадия 6), с использованием 4-(1-(3-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина вместо 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина (38 мг, выход: 77%). МС m/z [ESI]: 445,2 [М+1]. 1Н ЯМР (400 МГц, ДМСО-d6): δ=9.362 (br, 2Н), 8.233 (br, 2Н), 7.833 (s, 1Н), 7.769 (d, J=2.0 Гц, 1Н), 7.384-7.327 (m, 1H), 7.189-7.142 (m, 1H), 6.986 (s, 1H), 6.849-6.779 (m, 2Н), 6.508-6.467 (m, 2Н), 6.343 (d, J=6.8 Гц, 2Н), 3.451-3.356 (m, 4Н), 3.327 (s, 3Н), 3.185 (m, 4Н).
Пример 4: 4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
Стадия 1: 2-хлор-4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)пиридин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 1), с использованием 4-хлор-3-фторфенилгидразина вместо 3-хлор-4-фторфенилгидразина (выход: 79%). МС m/z [ESI]: 308,0 [М+1].
Стадия 2: 4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин
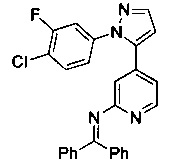
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-хлор-4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)пиридина вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 69%). МС m/z [ESI]: 453,1 [М+1].
Стадия 3: 4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 80%). МС m/z [ESI]: 289,1 [М+1].
Стадия 4: 5-бром-4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 58%). МС m/z [ESI]: 369,0 [М+1].
Стадия 5: 4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 30%). МС m/z [ESI]: 579,2 [М+1].
Стадия 6: 4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 2 (Стадия 6), с использованием 4-(1-(4-хлор-3-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина вместо 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина (56 мг, выход: 77%). МС m/z [ESI]: 479,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.049 (s, 1H), 7.598 (d, J=1.6 Гц, 1H), 7.157 (t, J=8.0 Гц, 1H), 6.873-6.842 (dd, J=10.0 Гц, 2.4 Гц, 1H), 6.756 (d, J=8.8 Гц, 1H), 6.502 (m, 2H), 6.315-6.281 (m, 2H), 6.168 (d, J=2.0 Гц, 1H), 4.491 (s, 2H), 3.359 (s, 3H), 3.138-3.114 (m, 4H), 3.052-3.028 (m, 4H).
Пример 5: 4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
Стадия 1: 2-хлор-4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)пиридин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 1), с использованием 3-трифторметилфенилгидразина вместо 3-хлор-4-фторфенилгидразина (выход: 40%). МС m/z [ESI]: 324,0 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.331 (d, J=8.8 Гц, 1H), 7.806 (s, 1H), 7.713 (s, 1H), 7.665 (d, J=7.6 Гц, 1H), 7.526 (t, J=7.6 Гц, 1H), 7.393 (d, J=8.0 Гц, 1H), 7.272-7.249 (m, 1H), 6.971-6.956 (m, 1H), 6.964 (s, 1H).
Стадия 2: 4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин
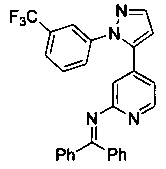
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-хлор-4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)пиридина вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 66%). МС m/z [ESI]: 469,2 [М+1].
Стадия 3: 4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 79%). МС m/z [ESI]: 305,1 [М+1].
Стадия 4: 5-бром-4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 57%). МС m/z [ESI]: 383,0 [М+1].
Стадия 5: 4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 42%). МС m/z [ESI]: 595,3 [М+1].
Стадия 6: 4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин получали в соответствии с методикой, описанной в Примере 2 (Стадия 6), с использованием 4-(1-(3-трифторметилфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина вместо 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина (65 мг, выход: 83%). МС m/z [ESI]: 495,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.82 (s, 1H), 7.76 (d, J=2.0 Гц, 1H), 7.67 (d, J=7.6 Гц, 1H), 7.57 (t, J=7.6 Гц, 1H), 7.33-7.30 (m, 2H), 6.48 (s, 1H), 6.43-6.40 (m, 2H), 6.28-6.27 (m, 2H), 6.12 (s, 2H), 3.31 (s, 3H), 3.16-3.14 (m, 4H), 2.98-2.96 (m, 4H).
Пример 6: 4-(1-(4-фторбензил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Стадия 1: 2-хлор-4-(1-(4-фторбензил)-1H-пиразол-5-ил)пиридин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 1), с использованием 4-фторбензилгидразина вместо 3-хлор-4-фторфенилгидразина (выход: 56%). МС m/z [ESI]: 288,1 [М+1].
Стадия 2: 4-(1-(4-фторбензил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин
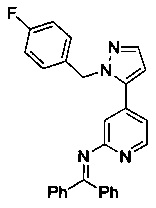
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-хлор-4-(1-(4-фторбензил)-1H-пиразол-5-ил)пиридина вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 65%). МС m/z [ESI]: 433,2 [М+1].
Стадия 3: 4-(1-(4-фторбензил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-(1-(4-фторбензил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 90%). МС m/z [ESI]: 269,1 [М+1].
Стадия 4: 5-бром-4-(1-(4-фторбензил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-(1-(4-фторбензил)-1H-пиразол-5-ил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 97%). МС m/z [ESI]: 347,0 [М+1].
Стадия 5: 4-(1-(4-фторбензил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(4-фторбензил)-1H-пиразол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина и с использованием 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина вместо трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (выход: 42%). МС m/z [ESI]: 473,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.065 (s, 1H), 7.798 (m, 1H), 7.696-7.552 (m, 3H), 7.428 (d, J=1.6 Гц, 1H), 6.82 (d, J=8.0 Гц, 1H), 6.396 (dd, J=8.0 Гц, 2.0 Гц, 1Н), 6.310 (d, J=3.6 Гц, 2Н), 6.065 (d, J=1.6 Гц, 1Н), 4.936 (s, 2Н), 4.762 (brs, 2Н), 3.969 (s, 3Н), 3.50 (m, 4Н), 3.14 (m, 4Н), 2.757 (s, 3Н).
Пример 7: 4-(1-(3-фторфенил-1H-имидазол-5-ил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Общие методики синтеза
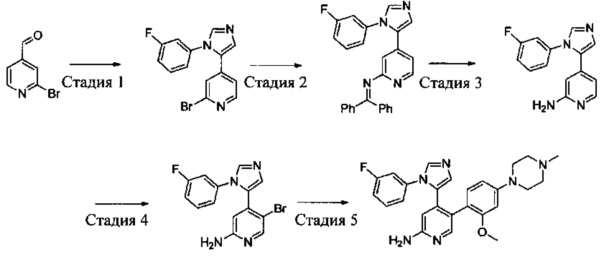
Стадия 1: 2-бром-4-(1-(3-фторфенил)-1H-имидазол-5-ил)пиридин
2-Бром-4-пиридинкарбоксальдегид (1,96 г, 10 ммоль), 3-фторанилин (1,11 г, 10 ммоль) и 4-метилбензолсульфоновую кислоту (50 мг) добавляли в толуол (50 мл). Полученную смесь подвергали дефлегмации в течение 12 ч с использованием водоотделителя для удаления воды, образующейся в ходе реакции. Растворитель выпаривали на роторном испарителе, и затем добавляли тозилметилизоцианид (1,95 г, 10 ммоль), Na2CO3 (9,66 г, 91 ммоль) и этанол (50 мл). Данную смесь подвергали дефлегмации в течение 6 ч. Растворитель выпаривали на роторном испарителе, и добавляли этилацетат и воду. Органический слой сушили и очищали путем колоночной хроматографии на силикагеле с получением 2-бром-4-(1-(3-фторфенил)-1H-имидазол-5-ил)пиридина (1,39 г, выход: 44%). МС m/z [ESI]: 318,0 [М+1].
Стадия 2: 4-(1-(3-фторфенил)-1H-имидазол-5-ил)-N-(дифенилметилен)пиридин-2-амин
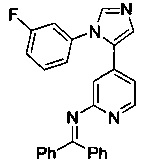
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-бром-4-(1-(3-фторфенил)-1H-имидазол-5-ил)пиридина вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 70%). 1Н ЯМР (400 МГц, CDCl3): δ=8.235 (t, 1.8 Гц, 1H), 7.803 (d, J=7.2 Гц, 2Н), 7.679 (d, J=1.6 Гц, 1H), 7.502 (d, J=7.6 Гц, 1Н), 7.44-7.26 (m, 6Н), 7.155 (d, J=6.8 Гц, 2Н), 7.052 (t, J=8.3 Гц, 1Н), 6.92-6.88 (m, 2H), 6.551-6.536 (q, 2H), 6.389 (d, J=1.6 Гц, 1H). МС m/z [ESI]: 419,2 [М+1].
Стадия 3: 4-(1-(3-фторфенил)-1H-имидазол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-(1-(3-фторфенил)-1H-имидазол-5-ил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 87%). МС m/z [ESI]: 255,1 [М+1].
Стадия 4: 5-бром-4-(1-(3-фторфенил)-1H-имидазол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-(1-(3-фторфенил)-1H-имидазол-5-ил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 64%). МС m/z [ESI]: 333,0 [М+1].
Стадия 5: 4-(1-(3-фторфенил)-1H-имидазол-5-ил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(3-фторфенил)-1H-имидазол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина и с использованием 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина вместо трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (выход: 33%). МС m/z [ESI]: 459,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.969 (s, 1H), 7.501 (d, J=1.2 Гц, 1H), 7.159-7.118 (m, 1H), 7.09 (s, 1H), 6.954-6.927 (m, 1H), 6.593 (d, J=8.0 Гц, 1H), 6.504-6.429 (m, 3H), 6.284-6.258 (dd, J=8.2 Гц, 2.2 Гц, 1H), 4.483 (s, 2H), 3.373 (s, 3H), 3.254 (m, 4H), 2.68 (m, 4H), 2.436 (s, 3H).
Пример 8: 4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Стадия 1: 2-хлор-4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)пиридин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 1), с использованием 4-метилсульфонилфенилгидразина вместо 3-хлор-4-фторфенилгидразина (выход: 40%). МС m/z [ESI]: 334,0 [М+1].
Стадия 2: 4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин
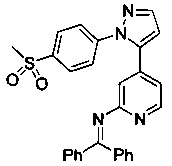
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-хлор-4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)пиридина вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 70%). МС m/z [ESI]: 315,1 [М+1].
Стадия 3: 4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 87%). МС m/z [ESI]: 315,1 [М+1].
Стадия 4: 5-бром-4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 64%). МС m/z [ESI]: 395,0 [М+1].
Стадия 5: 4-(1-(4-4-метилсульфонилфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(4-метилсульфонилфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина и с использованием 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина вместо трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (выход: 33%). МС m/z [ESI]: 519,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.99 (d, J=8.8 Гц, 2Н), 7.88 (s, 1H), 7.80 (d, J=5.6 Гц, 1H), 7.58 (d, J=8.8 Гц, 2H), 7.00 (d, J=8.4 Гц, 1H), 7.59 (d, J=2.0 Гц, 1H), 6.53 (dd, J=8.4 Гц, 2.0 Гц, 1H), 6.33 (s, 1H), 6.29 (d, J=5.6 Гц, 1H), 3.61 (s, 3H), 3.30-3.33 (m, 4H), 3.17 (s, 3H), 2.79-2.87 (m, 4H), 2.52 (s, 3H).
Пример 9: 3-(5-(2-амино-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-4-ил)-1H-пиразол-1-ил)бензойная кислота
Общие методики синтеза
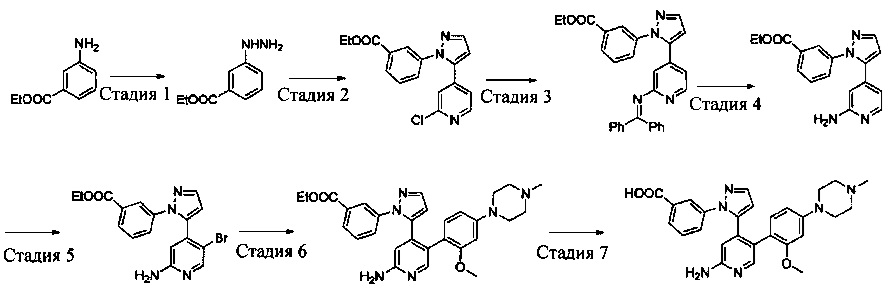
Стадия 1: этил 3-гидразинилбензоат
Этил 3-аминобензоат (5,0 г, 30 ммоль) растворяли в концентрированной соляной кислоте (25 мл), полученный раствор охлаждали до 0°С, и затем добавляли по каплям раствор NaNO2 (2,09 г, 30 ммоль) в воде (10 мл). Перемешивали в течение 30 мин при 0°С, и затем добавляли SnCl4 (12,86 г, 57 ммоль). Нагревали до комнатной температуры, и перемешивали в течение 1 ч. Добавляли концентрированный NaOH, пока pH раствора не сдвигался в сильнощелочную область, и затем раствор экстрагировали диэтиловым эфиром. Экстракт сушили, концентрировали и очищали путем колоночной хроматографии на силикагеле с получением этил 3-гидразинилбензоата (3,9 г, выход: 72%). МС m/z [ESI]: 181,1 [М+1].
Стадия 2: этил 3-(5-(2-хлорпиридин-4-ил)-1H-пиразол-1-ил)бензоат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 1), с использованием этил 3-гидразинилбензоата вместо 3-хлор-4-фторфенилгидразина (выход: 49%). МС m/z [ESI]: 328,1 [М+1].
Стадия 3: этил 3-(5-(2-((дифенилметилен)амино)пиридин-4-ил)-1H-пиразол-1-ил)бензоат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием этил 3-(5-(2-хлорпиридин-4-ил)-1H-пиразол-1-ил)бензоата вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 50%). МС m/z [ESI]: 473,2 [М+1].
Стадия 4: этил 3-(5-(3-аминофенил)-1H-пиразол-1-ил)бензоат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием этил 3-(5-(2-((дифенилметилен)амино)пиридин-4-ил)-1H-пиразол-1-ил)бензоата вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридан-2-амина (выход: 70%). МС m/z [ESI]: 309,1 [М+1].
Стадия 5: этил 3-(5-(5-амино-2-бромфенил)-1H-пиразол-1-ил)бензоат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием этил 3-(5-(3-аминофенил)-1H-пиразол-1-ил)бензоата вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 90%). 1Н ЯМР (400 МГц, CDCl3): δ=8.60-8.24 (brs, 2Н), 8.17 (s, 1H), 8.03 (t, J=1.7 Гц, 1H), 7.97 (dt, J=7.6, 1.4 Гц, 1H), 7.78 (d, J=1.8 Гц, 1H), 7.47 (ddd, J=8.0, 2.2, 1.3 Гц, 1H), 7.40 (t, J=7.8 Гц, 1H), 6.54 (s, 1H), 6.47-6.42 (m, 1H), 4.34 (q, J=7.1 Гц, 2H), 1.36 (t, J=7.1 Гц, 3Н). МС m/z [ESI]: 387,0 [М+1].
Стадия 6: этил 3-(5-(2-амино-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-4-ил)-1H-пиразол-1-ил)бензоат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием этил 3-(5-(5-амино-2-бромфенил)-1H-пиразол-1-ил)бензоата вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина и с использованием 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина вместо трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (выход: 30%). МС m/z [ESI]: 513,3 [М+1].
Стадия 7: 3-(5-(2-амино-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-4-ил)-1H-пиразол-1-ил)бензойная кислота
Этил 3-(5-(2-амино-5-(2-метокси-4-(4-метилпиперазйн-1-ил)фенил)пиридин-4-ил)-1H-пиразол-1-ил)бензоат (103 мг, 0,2 ммоль) растворяли в метаноле (10 мл), добавляли насыщенный раствор гидроксида натрия (5 мл), и затем полученный раствор перемешивали в течение 3 ч при комнатной температуре. Реакционную смесь нейтрализовали путем добавления соляной кислоты, и растворитель выпаривали на роторном испарителе. Остаток очищали путем колоночной хроматографии на силикагеле с получением 3-(5-(2-амино-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-4-ил)-1H-пиразол-1-ил)бензойной кислоты (63 мг, выход: 65%). МС m/z [ESI]: 485,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.54 (s, 1H), 7.89 (s, 1H), 7.78-7.83 (m, 2H), 7.57 (s, 1H), 7.27 (t, J=6.8 Гц, 1H), 6.14 (d, J=6.8 Гц, 1H), 6.77 (d, J=8.0 Гц,1Н), 6.38-6.42 (m, 2H), 6.14 (s, 1H), 6.03 (s, 1H), 5.91 (s, 2H), 3.41 (s, 3H), 3.12-3.24 (m, 4H), 2.44-2.51 (m, 4H), 2.26 (s, 3H).
Пример 10: 3-(5-(2-амино-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-4-ил)-1H-пиразол-1-ил)бензиловый спирт
Этил 3-(5-(2-амино-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-4-ил)-1H-пиразол-1-ил)бензоат (51 мг, 0,1 ммоль), полученный на Стадии 6 Примера 9, растворяли в сухом ТГФ (10 мл). Добавляли алюмогидрид лития (23 мг, 0,6 ммоль), и полученную смесь перемешивали в течение 4 ч при комнатной температуре. Реакцию останавливали путем добавления нескольких капель метанола, и растворитель выпаривали на роторном испарителе. Остаток очищали путем колоночной хроматографии на силикагеле с получением 3-(5-(2-амино-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-4-ил)-1H-пиразол-1-ил)бензилового спирта (23 мг, выход: 49%). МС m/z [ESI]: 471,2 [М+1]. 1Н ЯМР (400 МГц, ДМСО-d6): δ=7.793 (s, 1Н), 7.636 (s, 1Н), 7.251 (s, 2Н), 7.122 (s, 1Н), 6.923 (s, 1Н), 6.578 (d, J=8.8 Гц, 1Н), 6.354 (d, J=8.0 Гц, 3Н), 6.228 (s, 1H), 6.013 (s, 2Н), 5.29 (s, 1H), 4.496 (s, 2Н), 3.36 (br, 4Н), 3.23 (s, 3Н), 2.886 (br, 4Н), 2.56 (s, 3Н).
Пример 11: 2-((2-амино-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-4-ил)амино)-N-изопропилбензолсульфонамида гидрохлорид
Общие методики синтеза
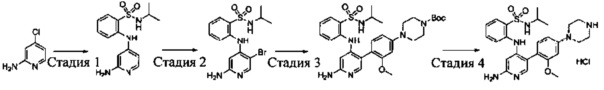
Стадия 1: 2-(2-аминопиридин-4-ил-амино)-N-изопропилбензолсульфонамид
В герметически закрываемую пробирку вносили 4-хлор-2-аминопиридин (1,28 г, 10 ммоль), 2-амино-N-изопропилбензолсульфонамид (2,35 г, 11 ммоль), Pd2(dba)3 (915 мг, 1 ммоль), BINAP (1,31 г, 2 ммоль), Cs2CO3 (6,50 г, 20 ммоль) и сухой толуол (80 мл), и данную смесь продували азотом. Затем смесь перемешивали в течение ночи при 130°С. После охлаждения смесь очищали путем колоночной хроматографии на силикагеле с получением 2-(2-аминопиридин-4-ил-амино)-N-изопропилбензолсульфонамида (740 мг, выход: 24%). МС m/z [ESI]: 307,1 [М+1].
Стадия 2: 2-(2-амино-5-бром-пиридин-4-ил-амино)-N-изопропилбензолсульфонамид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 2-(2-аминопиридин-4-ил-амино)-N-изопропилбензолсульфонамида вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 36%). МС m/z [ESI]: 387,0 [М+1].
Стадия 3: 2-(2-амино-5-(2-метокси-4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)фенил)пиридин-4-ил-амино)-N-изопропилбензолсульфонамид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 2-(2-амино-5-бром-пиридин-4-ил-амино)-N-изопропилбензолсульфонамида вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 21%). МС m/z [ESI]: 597,3 [М+1].
Стадия 4: 2-(2-амино-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-4-ил-амино)-N-изопропилбензолсульфонамида гидрохлорид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 2 (Стадия 6), с использованием 2-(2-амино-5-(2-метокси-4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)фенил)пиридин-4-ил-амино)-N-изопропилбензолсульфонамида вместо 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина (выход: 75%). МС m/z [ESI]: 497,2 [М+1]. 1Н ЯМР (400 МГц, ДМСО-d6): δ=13.02 (s, 1Н), 9.49 (s, 2Н), 7.81 (d, J=7.2 Гц, 1Н), 7.80 (s, 1Н), 7.63-7.67 (m, 2Н), 7.61 (d, J=7.2 Гц, 1Н), 7.53 (s, 1H), 7.50 (d, J=8.0 Гц, 1Н), 7.34 (td, J=8.0 Гц, 1.6 Гц, 1H), 7.17 (d, J=8.4 Гц, 1Н), 6.69 (s, 1H), 6.65 (dd, J=8.0 Гц, 1.6 Гц, 1Н), 6.48 (s, 1Н), 3.72 (s, 3Н), 3.46-3.49 (m, 4Н), 3.14-3.19 (m, 5Н), 0.89 (d, J=6.0 Гц, 6Н).
Пример 12: 4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Стадия 1: 4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)-N-(дифенилметилен)пиридин-2-амин
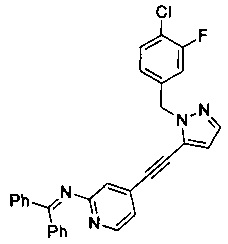
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-хлор-4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)пиридина вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 35%). МС m/z [ESI]: 491,1 [М+1].
Стадия 2: 4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 67%). МС m/z [ESI]: 327,1 [М+1].
Стадия 3: 5-бром-4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 76%). МС m/z [ESI]: 407,0 [М+1].
Стадия 4: 4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-((1-(4-хлор-3-фторбензил)-1H-пиразол-5-ил)этинил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина и с использованием 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина вместо трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (выход: 49%). МС m/z [ESI]: 531,2 [М+1].
Пример 13: 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)пиридин-2-амина гидрохлорид
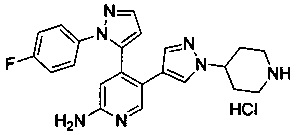
Стадия 1: трет-бутил 4-(4-(6-амино-4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридин-3-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина и с использованием трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-1-карбоксилата вместо трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (выход: 76%). МС m/z [ESI]: 504,2 [М+1].
Стадия 2: 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(1-(пиперидин-4-ил)-1Н-пиразол-4-ил)пиридин-2-амина гидрохлорид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 2 (Стадия 6), с использованием трет-бутил 4-(4-(6-амино-4-(1-(4-фторфенил)-1H-пиразол-5-ил)пиридин-3-ил)-1H-пиразол-1-ил)пиперидин-1-карбоксилата вместо 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина (выход: 49%). МС m/z [ESI]: 404,2 [М+1]. 1Н ЯМР (400 МГц, ДМСО-d6): δ=9.06-9.17 (1H, brs), 8.81-8.98 (1Н, brs), 8.13-8.24 (2Н, brs), 7.92 (1Н, s), 7.87 (1H, d, J=1.6 Гц), 7.17 (1Н, s), 7.05-7.10 (2Н, m), 7.00 (1Н, s), 6.90-6.94 (2Н, m), 6.83 (1Н, d, J=1.6 Гц), 6.64 (1Н, s), 4.23-4.34 (1Н, m), 3.33 (2Н, d, J=12.8 Гц), 2.95-3.05 (2Н, m), 1.92-2.08 (4Н, m).
Пример 14: 4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Стадия 1: 2-хлор-4-(1-(3-бромфенил)-1H-пиразол-5-ил)пиридин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 1), с использованием 3-бромфенилгидразина вместо 3-хлор-4-фторфенилгидразина (выход: 77%). МС m/z [ESI]: 336,0 [М+1].
Стадия 2: 2-хлор-4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)пиридин
2-Хлор-4-(1-(3-бромфенил)-1H-пиразол-5-ил)пиридин (335 мг, 1 ммоль), диметилфосфин-оксид (94 мг, 1,2 ммоль), Pd(OAc)2 (22 мг, 0,1 ммоль), X-phos (95 мг, 0,2 ммоль) и K3PO4 (244 Mg, 1,1 ммоль) добавляли в ДМФА (10 мл), и продували азотом. Реакционную смесь выдерживали в течение 2 ч при 150°С. Затем смесь охлаждали, растворитель выпаривали на роторном испарителе, и остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 36%). МС m/z [ESI]: 332,1 [М+1].
Стадия 3: 4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амин
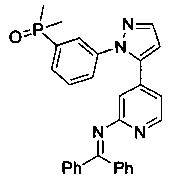
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 2), с использованием 2-хлор-4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)пиридина вместо 2-хлор-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридина (выход: 65%). МС m/z [ESI]: 477,2 [М+1].
Стадия 4: 4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 3), с использованием 4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-N-(дифенилметилен)пиридин-2-амина (выход: 90%). МС m/z [ESI]: 313,1 [М+1].
Стадия 5: 5-бром-4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 89%). МС m/z [ESI]: 391,0 [М+1].
Стадия 6: 4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 5-бром-4-(1-(3-(диметилфосфорил)фенил)-1H-пиразол-5-ил)пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина и с использованием 1-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина вместо трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (выход: 42%). МС m/z [ESI]: 517,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.24 (1Н, s), 8.18 (1H, d, J=12.4 Гц), 7.95 (1Н, d, J=8.0 Гц), 7.66-7.76 (3Н, m), 7.61 (1Н, s), 7.17 (1H, d, J=8.0 Гц), 6.70 (1H, d, J=8.0 Гц), 6.63 (1H, s), 6.98 (1H, d, J=1.6 Гц), 3.93-3.96 (2H, m), 3.59-3.62 (2H, m), 3.50 (3H, s), 3.10-3.26 (4H, m), 2.97 (3H, s), 1.84 (6H, d, J=13.2 Гц).
Пример 15: 2-(2-амино-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-3-ил)-N-изопропилбензолсульфонамид
Общие методики синтеза
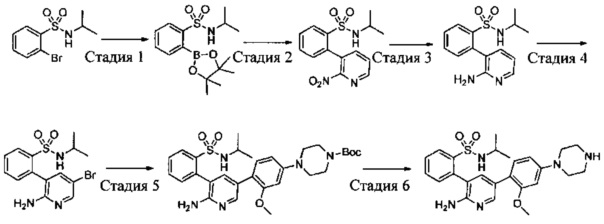
Стадия 1: 2-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-N-изопропилбензолсульфонамид
2-Бром-N-изопропилбензолсульфонамид (2,4 г, 8,6 ммоль), бис(пинаколато)дибор (3,3 г, 12,9 ммоль), Pd(dppf)Cl2 (630 мг, 0,86 ммоль) и безводный ацетат калия (1,7 г, 17,2 ммоль) добавляли в сухой 1,4-диоксан (100 мл), и затем продували азотом. Реакционную смесь перемешивали при 110°С в течение 2 суток. Затем смесь фильтровали. Полученный фильтрат упаривали на роторном испарителе и очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 37%).
Стадия 2: 2-(2-нитропиридин-3-ил)-N-изопропилбензолсульфонамид
2-(4,4,5,5-Тетраметил-1,3,2-диоксаборолан-2-ил)-N-изопропилбензолсульфонамид (390 мг, 1,2 ммоль), 2-нитро-3-(трифторметилсульфонилокси)пиридин (272 мг, 1 ммоль), Pd(PPh3)4 (58 мг, 0,05 ммоль) и Cs2CO3 (650 мг, 2 ммоль) добавляли в 1,4-диоксан (20 мл), полученную смесь продували азотом и затем перемешивали в течение 2 ч при 120°С в микроволновой печи. Полученный раствор фильтровали, и растворитель выпаривали на роторном испарителе. Остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 47%). МС m/z [ESI]: 322,1 [М+1].
Стадия 3: 2-(2-аминопиридин-3-ил)-N-изопропилбензолсульфонамид
2-(2-Нитропиридин-3-ил)-N-изопропилбензолсульфонамид (150 мг, 0,47 ммоль) растворяли в этаноле (15 мл), и затем добавляли 2 М HCl (0,5 мл) и порошок восстановленного железа (185 мг, 3,29 ммоль). Полученную смесь подвергали дефлегмации в течение 2 ч и затем фильтровали. Фильтрат упаривали на роторном испарителе и очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 80%). МС m/z [ESI]: 292,1 [М+1].
Стадия 4: 2-(2-амино-5-бромпиридин-3-ил)-N-изопропилбензолсульфонамид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 4), с использованием 2-(2-аминопиридин-3-ил)-N-изопропилбензолсульфонамида вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 58%). МС m/z [ESI]: 372,0 [М+1].
Стадия 5: 2-(2-амино-5-(2-метокси-4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)фенил)пиридин-3-ил-амино)-N-изопропилбензолсульфонамид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 2-(2-амино-5-бромпиридин-3-ил)-N-изопропилбензолсульфонамида вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина (выход: 30%). МС m/z [ESI]: 582,3 [М+1].
Стадия 6: 2-(2-амино-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-3-ил)-N-изопропилбензолсульфонамид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 6), с использованием 2-(2-амино-5-(2-метокси-4-(4-(трет-бутоксикарбонил)пиперазин-1-ил)фенил)пиридин-3-ил-амино)-N-изопропилбензолсульфонамида вместо 4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина (выход: 77%). МС m/z [ESI]: 482,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.15 (d, J=8.0 Гц, 1Н), 8.07 (d, J=1.6 Гц, 1H), 7.95 (d, J=2.0 Гц, 1H), 7.70-7.80 (m, 2H), 7.49 (d, J=7.2 Гц, 1H), 7.33 (d, J=8.4 Гц, 1H), 6.73 (d, J=1.6 Гц, 1H), 6.69 (dd, J=8.8 Гц, 2.4 Гц, 1H), 3.87 (s, 3H), 3.30-3.60 (m, 9H), 1.09 (d, J=6.4 Гц, 6H).
Пример 16: 3-((3-фторфенокси)метил)-5-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)пиридин-2-амина гидрохлорид
Общие методики синтеза
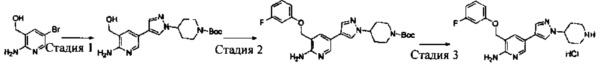
Стадия 1: 3-гидроксиметил-5-(1-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-пиразол-4-ил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 1 (Стадия 5), с использованием 3-гидроксиметил-5-бром-пиридин-2-амина вместо 5-бром-4-(1-(3-хлор-4-фторфенил)-1H-пиразол-5-ил)пиридин-2-амина и с использованием трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-1-карбоксилата вместо трет-бутил 4-(3-метоксил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата (выход: 37%). МС m/z [ESI]: 374,2 [М+1].
Стадия 2: 3-((3-фторфенокси)метил)-5-(1-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-пиразол-4-ил)пиридин-2-амин
3-Гидроксиметил-5-(1-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-пиразол-4-ил)пиридин-2-амин, 3-фторфенол (61,6 мг, 0,55 ммоль) и PPh3 (200 мг, 0,75 ммоль) добавляли в сухой ТГФ (20 мл), полученную смесь продували азотом и затем перемешивали в течение 1 ч. Затем смесь охлаждали до 0°С, добавляли по каплям DIAD (152 мг, 0,75 ммоль), и данную смесь перемешивали в течение ночи при комнатной температуре. Растворитель выпаривали на роторном испарителе, и остаток очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (50 мг, выход: 21%). МС m/z [ESI]: 468,2 [М+1].
Стадия 3: 3-((3-фторфенокси)метил)-5-(1-(пиперидин-4-ил)-1H-пиразол-4-ил)пиридин-2-амина гидрохлорид
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 2 (Стадия 6), с использованием 3-((3-фторфенокси)метил)-5-(1-(1-(трет-бутоксикарбонил)пиперидин-4-ил)-1H-пиразол-4-ил)пиридин-2-амина вместо 4-(1-(4-фторфенил)-1H-пиразол-5-ил)-5-(2-метокси-4-(4-трет-бутоксикарбонилпиперазин-1-ил)фенил)пиридин-2-амина (выход: 68%). МС m/z [ESI]: 368,2 [М+1]. 1Н ЯМР (400 МГц, ДМСО-d6): δ=8.93 (1H, brs), 8.75 (1Н, brs), 8.421 (1H, s), 8.343 (1Н, s), 8.301 (1H, s), 8.016 (1Н, s), 7.95 (2Н, brs), 7.417-7.358 (1H, m), 7.012 (1Н, dd, J=11.2 Гц, 2.4 Гц), 6.947 (1H, dd, J=8.4Н, 2.4 Гц), 6.852 (1Н, m), 5.085 (2Н, s), 4.508 (1Н, m), 3.384 (4Н, m), 2.17 (4Н, m).
Пример 17: 3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидин-5-ил)пиридин-2-амин Общие методики синтеза
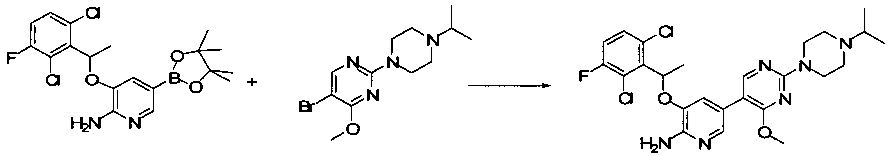
В соответствии с методикой, описанной в Примере 1 (Стадия 5), 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидин (158 мг, 0,5 ммоль), 3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин (235 мг, 0,55 ммоль), Pd(PPh3)4 (58 мг, 0,05 ммоль) и Cs2CO3 (325 мг, 1 ммоль) растворяли в смеси 1,4-диоксана (10 мл) и воды (1,5 мл), полученный раствор продували азотом и затем перемешивали в течение ночи при 100°С. После охлаждения реакционную смесь очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 58%). МС m/z [ESI]: 535,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.00 (s, 1Н), 7.69 (d, J=1.6 Гц, 1Н), 7.31-7.27 (m, 1H), 7.05 (t, J=8.2 Гц, 1Н), 6.94 (s, 1Н), 6.04 (q, J=6.5 Гц, 1Н), 4.79 (s, 2Н), 3.85 (s, 3Н), 3.84-3.79 (m, 4Н), 2.73 (dt, J=12.9, 6.4 Гц, 1Н), 2.58 (dd, J=9.9, 4.9 Гц, 4H), 1.83 (d, J=6.7 Гц, 3Н), 1.08 (d, J=6.5 Гц, 6Н).
Пример 18: 5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-6'-(4-метилпиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием 5-бром-2-(4-метилпиперазин-1-ил)-3-метоксипиридина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина (выход: 55%). МС m/z [ESI]: 506,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.71 (s, 1Н), 7.63 (s, 1H), 7.37 (dd, J=8.8 Гц, 4.8 Гц, 1Н), 7.15 (t, J=9.2 Гц, 1H), 7.06 (s, 1H), 6.84 (s, 1H), 6.11 (q, J=6.4 Гц, 1H), 3.81 (s, 3H), 3.37-3.52 (m, 4H), 2.87-2.97 (m, 4H), 2.56 (s, 3H), 1.79 (d, J=6.8 Гц, 3Н).
Пример 19: 5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-6'-(4-метилпиперазин-1-ил)-[3,3'-бипиридин1-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием 5-бром-2-(4-метилпиперазин-1-ил)-4-метоксипиридина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина (выход: 64%). МС m/z [ESI]: 506,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.91 (1Н, s), 7.69 (1Н, s), 7.28-7.31 (1H, m), 7.06 (1Н, t, J=8.0 Гц), 6.95 (1Н, s), 6.12 (1Н, s), 6.04 (1Н, q, J=6.8 Гц), 4.96 (2Н, s), 3.75 (3Н, s), 3.67-3.72 (4Н, m), 2.66-2.75 (4Н, m), 2.47 (3Н, s), 1.83 (3Н, d, J=6.8 Гц).
Пример 20: 3-(1-(2-(диметилфосфорил)-5-фторфенил)этокси)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием 5-бром-3-(1-(2-(диметилфосфорил)-5-фторфенил)этокси)пиридин-2-амина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина и с использованием 1-(3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина вместо 3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 64%). МС m/z [ESI]: 513,2 [М+1]. 1Н ЯМР (400 МГц, ДМСО-d6): δ=7.78-7.69 (m, 2Н), 7.65 (d, J=10.5 Гц, 1H), 7.58 (s, 1Н), 7.29 (dd, J=15.6, 6.8 Гц, 1Н), 7.20 (d, J=8.4 Гц, 1H), 7.09 (q, J=5.3 Гц, 1H), 6.64 (s, 1Н), 6.61 (q, J=6.5 Гц, 1Н), 3.67 (s, 3Н), 3.37-2.93 (m, 8H), 2.86 (s, 3H), 1.74 (dd, J=13.3, 5.6 Гц, 6H), 1.67 (d, 7=6.3 Гц, 3Н).
Пример 21: 3-(1-(4-хлор-2-(диметиламино)-5-фторфенил)этокси)-5-(2-метокси-4-(4-метилпиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием 5-бром-3-(1-(2-(диметиламино)-4-хлор-5-фторфенил)этокси)пиридин-2-амина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина и с использованием 1-(3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)-4-метилпиперазина вместо 3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 64%). МС m/z [ESI]: 513,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.035 (1Н, s), 7.534 (1Н, s), 7.105 (1Н, s), 6.987 (1Н, d, J=8.4 Гц), 6.546 (1Н, s), 6.460-6.422 (2Н, m), 6.385 (1Н, s), 5.564 (1Н, q), 3.612 (3Н, s), 3.453 (4Н, m), 2.983 (4Н, m), 2.815 (6Н, s), 2.620 (3Н, s), 1.64 (3Н, d, J=6.4 Гц).
Пример 22: 5-(1-(2,6-дихлор-3-фторфенил)этокси)-6'-((S)-2,4-диметилпиперазин-1-ил)-4'-метокси-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием (S)-1-(5-бром-4-метоксипиридин-2-ил)-2,4-диметилпиперазина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина (выход: 64%). МС m/z [ESI]: 520,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.90 (1Н, s), 7.63 (1H, s), 7.27-7.30 (1H, m), 7.05 (1Н, t, J=8.0 Гц), 6.95 (1H, s), 6.05 (1H, s), 6.02 (1Н, q, J=6.8 Гц), 5.29 (2Н, s), 4.40-4.50 (1Н, m), 3.87-4.15 (1Н, m), 3.74 (3Н, s), 3.12 (1Н, t, J=12.4 Гц), 2.93 (1Н, d, J=11.2 Гц), 2.77 (1Н, d, J=11.2 Гц), 2.32 (3Н, s), 2.30 (1Н, m), 2.07-2.14 (1H, m), 1.82 (3Н, d, J=6.8 Гц), 1.28 (3Н, d, J=6.8 Гц).
Пример 23: 5-(1-(2,6-дихлор-3-фторфенил)этокси)-6'-((R)-2,4-диметилпиперазин-1-ил)-4'-метокси-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием (R)-1-(5-бром-4-метоксипиридин-2-ил)-2,4-диметилпиперазина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина (выход: 64%). МС m/z [ESI]: 520,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.91 (s, 1Н), 7.69 (s, 1Н), 7.31-7.28 (m, 1H), 7.05 (t, J=8.4 Гц, 1H), 6.96 (s, 1Н), 6.06 (s, 1Н), 6.03 (q, J=6.5 Гц, 1H), 4.98 (s, 2Н), 4.54 (m, 1Н), 3.99 (d, J=12.4 Гц, 1Н), 3.74 (s, 3Н), 3.28 (t, J=13.8 Гц, 1Н), 2.99 (d, J=10.8 Гц, 1Н), 2.83 (d, J=11.2 Гц, 1H), 2.37 (s, 3Н), 2.32 (m, 1Н), 2.23-2.11 (m, 1Н), 1.83 (d, J=6.6 Гц, 3Н), 1.31 (d, J=6.4 Гц, 3Н).
Пример 24: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-6'-((S)-2,4-диметилпиперазин-1-ил)-4'-метокси-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием (S)-1-(5-бром-4-метоксипиридин-2-ил)-2,4-диметилпиперазина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина и с использованием (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина вместо 3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 64%). МС m/z [ESI]: 520,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.91 (1Н, s), 7.65 (1Н, s), 7.27-7.30 (1H, m), 7.05 (1H, t, J=8.4 Гц), 6.96 (1Н, s), 6.02-6.10 (2Н, m), 5.25 (2Н, s), 4.43-4.50 (1H, m), 3.97 (1H, d, J=12.8 Гц), 3.74 (3Н, s), 3.23 (1Н, td, J=12.8 Гц, 3.2 Гц), 2.94 (1Н, d, J=11.2 Гц), 2.78 (1H, d, J=11.2 Гц), 2.29-2.32 (4Н, m), 2.07-2.14 (1H, m), 1.82 (3Н, d, J=6.8 Гц), 1.28 (3Н, d, J=6.4 Гц).
Пример 25: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-6'-((R)-2,4-диметилпиперазин-1-ил)-4'-метокси-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием (R)-1-(5-бром-4-метоксипиридин-2-ил)-2,4-диметилпиперазина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина и с использованием (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина вместо 3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 64%). МС m/z [ESI]: 520,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.93 (1H, s), 7.70 (1Н, s), 7.28-7.31 (1Н, m), 7.07 (1Н, t, J=8.0 Гц), 6.97 (1Н, s), 6.04-6.13 (2Н, m), 4.98 (2Н, s), 4.43-4.52 (1Н, m), 3.98 (1Н, d, J=12.4 Гц), 3.76 (3Н, s), 3.24 (1H, td, J=12.4 Гц, 2.8 Гц), 2.93 (1H, d, J=10.8 Гц), 2.78 (1Н, d, J=10.4 Гц), 2.29-2.34 (4H, m), 2.09-2.14 (1H, m), 1.84 (3H, d, J=6.8 Гц), 1.29 (3Н, d, J=6.8 Гц).
Пример 26: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-6'-((S)-2,4-диметилпиперазин-1-ил)-5'-метокси-[3,3'-бипиридин1-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием (S)-1-(5-бром-3-метоксипиридин-2-ил)-2,4-диметилпиперазина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина и с использованием (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина вместо 3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 64%). МС m/z [ESI]: 520,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.89 (s, 1H), 7.78 (s, 1H), 7.33 (dd, 7=8.8, 4.8 Гц, 1H), 7.09 (t, J=8.4 Гц, 1H), 7.01 (s, 1H), 6.95 (s, 1H), 6.13 (q, J=6.6 Гц, 1H), 5.50 (s, 2H), 4.47 (m, 1H), 3.88 (s, 3H), 3.85 (t, J=4.2 Гц, 1H), 3.79 (d, J=11.8 Гц, 1H), 3.40 (m, 1H), 3.25 (m, 2H), 3.12 (m, 1H), 2.85 (s, 3H), 1.89 (d, J=6.6 Гц, 3Н), 1.26 (d, J=6.5 Гц, 3Н).
Пример 27: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-6'-((S)-2-метилпиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Общие методики синтеза
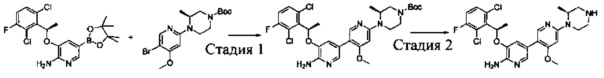
Стадия 1: (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилат
(S)-трет-Бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилат (106 мг, 0,275 ммоль), (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин (140 мг, 0,33 ммоль), Pd(PPh3)4 (32 мг, 0,0275 ммоль) и Cs2CO3 (179 мг, 0,55 ммоль) растворяли в смеси 1,4-диоксана (10 мл) и воды (1,5 мл), полученный раствор продували азотом и затем перемешивали в течение ночи при 100°С. После охлаждения реакционную смесь очищали путем колоночной хроматографии на силикагеле с получением (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (70 мг, выход: 42%). МС m/z [ESI]: 606,2 [М+1].
Стадия 2: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-6'-((S)-2-метилпиперазин-1-ил)-[3,3'-бипиридин]-6-амин
К перемешиваемому раствору (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (67 мг, 0,11 ммоль) в CH2Cl2 (10 мл) добавляли трифторацетат (1 мл), и затем данную смесь перемешивали в течение 1 ч. Добавляли концентрированный раствор NaOH, пока pH смеси не становился больше 13, и затем смесь экстрагировали CH2Cl2. Экстракт сушили над безводным сульфатом натрия, фильтровали, концентрировали и очищали путем колоночной хроматографии на силикагеле (CH2Cl2 : метанол = 8:1) с получением 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-6'-((S)-2-метилпиперазин-1-ил)-[3,3'-бипиридин]-6-амина (выход: 55%). МС m/z [ESI]: 506,1 [М+1]. 1H ЯМР (400 МГц, CDCl3): δ=7.94 (1H, s), 7.71 (1Н, s), 7.28-7.32 (1Н, m), 7.07 (1H, t, J=8.4 Гц), 6.97 (1H, s), 6.04-6.13 (2Н, m), 4.86 (2Н, s), 4.57-4.59 (1Н, m), 4.03 (1Н, d, J=14 Гц), 3.76 (3Н, s), 3.07-3.33 (4Н, m), 2.88-3.00 (1H, m), 1.84 (3Н, d, J=6.8 Гц), 1.34 (3Н, d, J=6.8 Гц).
Пример 28: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-6'-((S)-2-метил-4-(1-метилпиперидин-4-ил)пиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 17, с использованием (S)-1-(5-бром-4-метоксипиридин-2-ил)-2-метил-4-(1-метилпиперидин-4-ил)пиперазина вместо 5-бром-2-(4-изопропилпиперазин-1-ил)-4-метоксипиримидина и с использованием (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина вместо 3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 27%). МС m/z [ESI]: 603,2 [М+1]. 1H ЯМР (400 МГц, CDCl3): δ=7.88 (1Н, s), 7.66 (1H, s), 7.26-7.28 (1Н, m), 7.03 (1Н, t, J=8.0 Гц), 6.93 (1Н, s), 5.99-6.02 (2Н, m), 4.83 (2Н, s), 4.43-4.46 (1H, m), 3.98 (1H, d, J=12.4 Гц), 3.72 (3Н, s), 3.21-3.30 (2Н, m), 3.12 (1Н, t, J=11.6 Гц), 2.92 (1H, d, J=9.6 Гц), 2.79 (1H, d, J=10.8 Гц), 2.64-2.80 (4Н, m), 2.44-2.47 (3Н, m), 2.29-2.34 (1H, m), 2.06-2.14 (4H, m), 1.79 (3H, d, J=6.8 Гц), 1.21 (3Н, d, J=6.4 Гц).
Пример 29: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
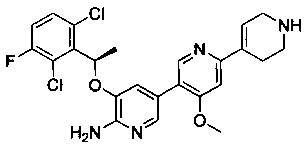
Стадия 1: (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 20%). МС m/z [ESI]: 589,2 [М+1].
Стадия 2: (К)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 41%). МС m/z [ESI]: 489,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.21 (1Н, s), 7.60 (1H, s), 7.26-7.29 (1Н, m), 7.04 (1Н, t, J=8.0 Гц), 6.96 (1H, s), 6.87 (1Н, s), 6.49 (1Н, s), 6.01 (1H, q, J=6.8 Гц), 5.33 (2Н, s), 3.94 (2Н, s), 3.74 (3Н, s), 3.49 (2Н, t, J=4.2 Гц), 3.00 (2Н, dt, J=4.2 Гц), 1.81 (3Н, d, J=6.8 Гц).
Пример 30: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
Общие методики синтеза
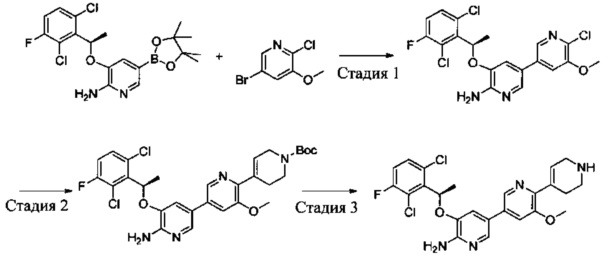
Стадия 1: (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-[3,3'-бипиридин]-6-амин
5-Бром-2-хлор-3-метоксипиридин (244 мг, 1,1 ммоль), (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амин (427 мг, 1,0 ммоль), Pd(PPh3)4 (116 мг, 0,1 ммоль) и Cs2CO3 (652 мг, 2,0 ммоль) растворяли в смеси 1,4-диоксана (10 мл) и воды (1,5 мл), полученный раствор продували азотом и затем перемешивали в течение ночи при 100°С. После охлаждения реакционную смесь очищали путем колоночной хроматографии на силикагеле с получением (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-[3,3'-бипиридин]-6-амина (252 мг, выход: 57%). МС m/z [ESI]: 442,0 [М+1].
Стадия 2: (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилат
(R)-6'-Хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-[3,3'-бипиридин]-6-амин (243 мг, 0,55 ммоль), трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-карбоксилат-1,2,5,6-тетрагидро-пиридин (204 мг, 0,66 ммоль), Pd(PPh3)4 (64 мг, 0,055 ммоль) и Cs2CO3 (359 мг, 1,1 ммоль) растворяли в смеси 1,4-диоксана (6 мл) и воды (1,5 мл), полученный раствор продували азотом, и затем реакционную смесь выдерживали при 100°С в течение ночи. После охлаждения смесь очищали путем колоночной хроматографии на силикагеле с получением (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата (65 мг, выход: 20%). МС m/z [ESI]: 589,2 [М+1].
Стадия 3: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 41%). МС m/z [ESI]: 489,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.21 (1Н, s), 7.84 (1Н, s), 7.28-7.32 (1Н, m), 7.04-7.08 (2Н, m), 6.93 (1H, s), 6.52 (1H, s), 6.10 (1Н, q, J=6.4 Гц), 5.21 (2Н, s), 3.86 (3Н, s), 3.74 (2Н, s), 3.28 (2Н, t, J=4.2 Гц), 2.77 (2Н, t, J=4.2 Гц), 1.87 (3Н, d, J=6.8 Гц).
Пример 31: 5-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)-4'-метокси-6'-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)-[3,3'-бипиридин]-6-амин
Общие методики синтеза
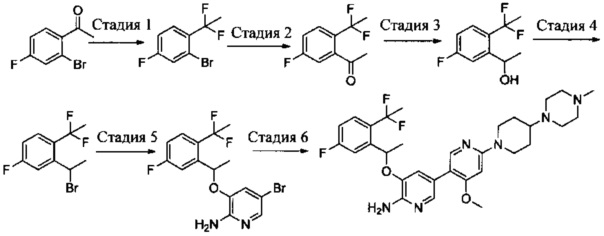
Стадия 1: 2-бром-1-(1,1-дифторэтил)-4-фторбензол
В герметически закрываемую пробирку вносили 1-(2-бром-4-фторфенил)этанон (3,5 г, 16 ммоль) и DAST (20 мл), и данную реакционную смесь выдерживали при 60°С в течение ночи. После охлаждения смесь осторожно выливали на дробленый лед и экстрагировали н-пентаном. Экстракт сушили и очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (2,3 г, выход: 60%).
Стадия 2: 1-(2-(1,1-дифторэтил)-5-фторфенил)этанон
В герметически закрываемую пробирку вносили 2-бром-1-(1,1-дифторэтил)-4-фторбензол (717 мг, 3 ммоль), н-бутилвиниловый эфир (3,0 г, 30 ммоль), Pd(OAc)2 (67 мг, 0,3 ммоль), 1,3-бис(дифенилфосфино)пропан (DPPP) (248 мг, 0,6 ммоль), триэтиламин (909 мг, 9 ммоль) и ДМФА (10 мл), данную реакционную смесь продували азотом и затем выдерживали при 120°С в течение ночи. После охлаждения смесь вливали в 10% соляную кислоту, перемешивали в течение 1 ч, нейтрализовали насыщенным NaHCO3 и экстрагировали CH2Cl2. Экстракт разделяли путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 33%).
Стадия 3:1-(2-(1,1-дифторэтил)-5-фторфенил)этанол
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 40 (Стадия 4) с использованием 1-(2-(1,1-дифторэтил)-5-фторфенил)этанона вместо 2-(диметиламино)-4-хлор-5-(фторфенил)этанона (выход: 30%).
Стадия 4: 2-(1-бромэтил)-1-(1,1-дифторэтил)-4-фторбензол
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 40 (Стадия 5) с использованием 1-(2-(1,1-дифторэтил)-5-фторфенил)этанола вместо 2-(диметиламино)-4-хлор-5-(фторфенил)этанола (выход: 50%).
Стадия 5: 5-бром-3-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 40 (Стадия 6) с использованием 2-(1-бромэтил)-1-(1,1-дифторэтил)-4-фторбензола вместо 2-(1-бромэтил)-5-хлор-4-фтор-N,N-диметиланилина (выход: 41%). МС m/z [ESI]: 375,0 [М+1].
Стадия 6: 5-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)-4'-метокси-6'-(4-(4-метилпиперазин-1-ил)пиперидин-1-ил)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием 5-бром-3-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)пиридин-2-амина вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата и с использованием 1-(1-(4-метоксил-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-ил)пиперидин-4-ил)-4-метилпиперазина вместо (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 32%). МС m/z [ESI]: 585,3 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.59-7.65 (2Н, m), 7.12-7.18 (2Н, m), 6.98 (1H, d, J=7.6 Гц), 6.47 (1H, d, J=6.4 Гц), 6.42 (1Н, s), 5.86 (2Н, s), 5.36 (1Н, q, J=6.8 Гц), 3.72-3.79 (2Н, m), 3.64 (3Н, s), 2.95-3.08 (4Н, m), 2.75 (2Н, t, J=12.4 Гц), 2.56-2.68 (4H, m), 2.22 (1Н, m), 2.07 (3Н, s), 2.027 (3Н, t, J=18.8 Гц), 1.93-1.83 (4H, m), 1.67 (3H, d, J=6.0 Гц).
Пример 32: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-6-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Стадия 1: (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-[2,3'-бипиридин]-6-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(6-бром-3-метоксипиридин-2-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 53%). МС m/z [ESI]: 592,2 [М+1].
Стадия 2: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-6-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-[2,3'-бипиридин]-6-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 65%). МС m/z [ESI]: 492,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.09 (1Н, s), 7.57-7.62 (1H, m), 7.43 (1H, t, J=8.0 Гц), 7.22-7.30 (2Н, m), 7.16 (1H, s), 6.01 (1H, q, J=6.4 Гц), 5.90 (2Н, s), 3.78 (3Н, s), 3.40-3.42 (4Н, m), 3.05-3.17 (4Н, m), 1.77 (3Н, d, J=6.4 Гц).
Пример 33: 5-(1-(2-хлор-5-фторфенил)-2,2,2-трифторэтокси)-5'-(пиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Общие методики синтеза
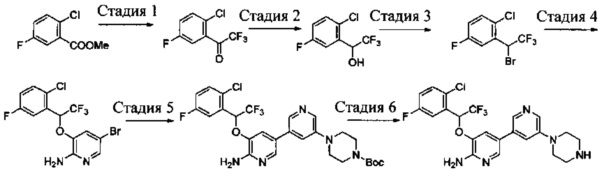
Стадия 1: 1-(2-хлор-5-фторфенил)-2,2,2-трифторэтанон
Смесь метил 2-хлор-5-фторбензоата (18,8 г, 0,1 моль), (трифторметил)триметилсилана (15,6 г, 0,11 моль) и CsF (2,0 г, 0,013 ммоль) перемешивали в течение 30 мин при комнатной температуре в атмосфере азота. Затем добавляли 5 М соляную кислоту (50 мл), и данную смесь перемешивали в течение ночи. Добавляли диметоксиэтан (50 мл), и смесь перемешивали в течение ночи при 120°С. После охлаждения смесь нейтрализовали путем добавления NaOH и экстрагировали CH2Cl2. Экстракт сушили и очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (3,5 г, выход: 15%).
Стадия 2: 1-(2-хлор-5-фторфенил)-2,2,2-трифторэтанол
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 40 (Стадия 4) с использованием 1-(2-хлор-5-фторфенил)-2,2,2-трифторэтанона вместо 2-(диметиламино)-4-хлор-5-(фторфенил)этанона (выход: 85%).
Стадия 3: 2-(1-бром-2,2,2-трифторэтил)-1-хлор-4-фторбензол
В герметически закрываемую пробирку вносили 1-(2-хлор-5-фторфенил)-2,2,2-трифторэтанол (2,0 г, 8,75 ммоль) и PBr5 (5,0 г, 11,6 ммоль), и данную реакционную смесь выдерживали при 140°С в течение ночи. Затем добавляли дробленый лед, смесь нейтрализовали путем добавления NaOH и экстрагировали CH2Cl2. Экстракт сушили и очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (выход: 33%).
Стадия 4: 5-бром-3-(1-(2-хлор-5-фторфенил)-2,2,2-трифторэтокси)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой синтеза Промежуточного соединения 40 (Стадия 6) с использованием 2-(1-бром-2,2,2-трифторэтил)-1-хлор-4-фторбензола вместо 2-(1 -бромэтил)-5-хлор-4-фтор-N,N-диметиланилина (выход: 27%). МС m/z [ESI]: 400,9 [М+1].
Стадия 5: трет-бутил 4-(6'-амино-5'-(1-(2-хлор-5-фторфенил)-2,2,2-трифторэтокси)-[3,3'-бипиридин]-5-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием 5-бром-3-(1-(2-хлор-5-фторфенил)-2,2,2-трифторэтокси)пиридин-2-амина вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата и с использованием трет-бутил 4-(5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-3-ил)пиперазин-1-карбоксилата вместо (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 41%). МС m/z [ESI]: 582,2 [М+1].
Стадия 6: 5-(1-(2-хлор-5-фторфенил)-2,2,2-трифторэтокси)-5'-(пиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием трет-бутил 4-(6'-амино-5'-(1-(2-хлор-5-фторфенил)-2,2,2-трифторэтокси)- [3,3'-бипиридин]-5-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 32%). МС m/z [ESI]: 482,1 [М+1]. 1H ЯМР (400 МГц, CDCl3): δ=8.07 (1Н, s), 7.97 (1Н, s), 7.80 (1Н, s), 7.40-7.50 (2Н, m), 7.27 (1H, s), 7.15 (1H, s) 7.07 (1Н, s), 6.43 (1H, m), 3.20 (4Н, m), 3.06 (4Н, m).
Пример 34: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-3-метокси-6-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Стадия 1: (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-3-метокси-[2,3'-бипиридин]-6-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(6-бром-5-метоксипиридин-2-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 592,2 [М+1].
Стадия 2: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-3-метокси-6-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-3-метокси-[2,3'-бипиридин]-6-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 492,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.20 (1Н, s), 7.54-7.58 (1Н, m), 7.45 (1H, t, J=8.8 Гц), 7.41 (1H, d, J=9.2 Гц), 7.22 (1H, s), 6.77 (1H, d, J=9.2 Гц), 6.04 (2Н, s), 5.98 (1H, q, J=6.6 Гц), 3.66 (3Н, s), 3.53-3.58 (4Н, m), 3.14-3.17 (4Н, m), 1.76 (3Н, d, J=6.4 Гц).
Пример 35: (R)-4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)пиперазин-2-он
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием 4-(5-бром-4-метоксипиридин-2-ил)пиперазин-2-она вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 506,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.16 (1Н, s), 7.59 (1H, s), 7.46 (1Н, dd, J=9.2 Гц, 4.8 Гц), 7.26 (1Н, t, J=8.4 Гц), 6.93 (1Н, s), 6.32 (1H, s), 6.10 (1H, q, J=6.8 Гц), 4.14 (2Н, s), 3.81 (2Н, t, J=5.2 Гц), 3.77 (3Н, s), 3.45 (2Н, t, J=5.6 Гц), 1.85 (3Н, d, J=6.4 Гц).
Пример 36: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-6-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Стадия 1: (R)-трет-6утил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[2,3'-бипиридин]-6-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(6-хлор-4-метоксипиридин-2-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 592,2 [М+1].
Стадия 2: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-6-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[2,3'-бипиридин]-6-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 492,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.15 (s, 1Н), 7.87 (s, 1H), 7.46 (dd, J=9.4, 4.6 Гц, 1H), 7.25 (t, J=8.5 Гц, 1H), 6.62 (d, J=1.6 Гц, 1H), 6.29 (d, J=1.6 Гц, 1H), 6.17 (q, J=6.3 Гц, 1H), 3.86 (s, 3H), 3.82 (m, 4H), 3.29 (m, 4H), 1.88 (d, J=6.6 Гц, 3Н).
Пример 37: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-6'-(пиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Стадия 1: (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 592,2 [М+1].
Стадия 2: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-6-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 492,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.91 (1H, s), 7.68 (1Н, s), 7.26-7.30 (1H, m), 7.05 (1Н, t, J=8.4 Гц), 6.94 (1Н, s), 6.12 (1Н, s), 6.04 (1Н, q, J=6.8 Гц), 4.9 (2Н, s), 3.66-3.75 (7Н, m), 3.11-3.18 (4Н, m), 1.83 (3Н, d, J=6.8 Гц).
Пример 38: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-2'-(пиперазин-1-ил)-[3,4'-бипиридин]-6-амин
Стадия 1: (R)-трет-бутил 4-(6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-[3,4'-бипиридин]-2'-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(4-бром-5-метоксипиридин-2-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 592,2 [М+1].
Стадия 2: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-2'-(пиперазин-1-ил)-[3,4'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 4-(6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-[3,4'-бипиридин]-2'-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 492,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.89 (1H, s), 7.85 (1Н, s), 7.30 (1Н, dd, J=8.8 Гц, 4.8 Гц), 7.08 (1Н, t, J=8.0 Гц), 7.01 (1H, s), 6.51 (1Н, s), 6.05 (1Н, q, J=6.8 Гц), 5.13 (2Н, s), 3.72-3.80 (4Н, m), 3.70 (3Н, s), 3.25-3.31 (4Н, m), 1.84 (3Н, d, J=6.4 Гц).
Пример 39: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-4-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Стадия 1: (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-[2,3'-бипиридин]-4-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(2-бром-5-метоксипиридин-4-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 592,2 [М+1].
Стадия 2: (R)-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-4-(пиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-[2,3'-бипиридин]-4-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 492,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.18 (1H, s), 8.11 (1Н, s), 7.35 (1H, s), 7.30-7.32 (1Н, m), 7.06 (1H, t, J=8.4 Гц), 6.88 (1Н, s), 6.15 (1H, q, J=6.8 Гц), 5.04-5.20 (2Н, brs), 3.93 (3Н, s), 3.41-3.48 (4Н, m), 3.28-3.32 (4Н, m), 1.85 (3Н, d,J=6.4 Гц).
Пример 40: 3-(1-(2-(дифторметил)-5-фторфенил)этокси)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
Стадия 1: трет-бутил 4-(4-(6-амино-5-(1-(2-(дифторметил)-5-фторфенил)этокси)пиридин-3-ил)-3-метоксифенил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата вместо (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина и с использованием 5-бром-3-(1-(2-(дифторметил)-5-фторфенил)этокси)пиридин-2-амина вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%) МС m/z [ESI]: 573,3 [М+1].
Стадия 2: 3-(1-(2-(дифторметил)-5-фторфенил)этокси)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием трет-бутил 4-(4-(6-амино-5-(1-(2-(дифторметил)-5-фторфенил)этокси)пиридин-3-ил)-3-метоксифенил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 473,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.68 (1H, s), 7.56 (1H, dd, J=8.8 Гц, 5.2 Гц), 7.21-7.23 (1H, m), 7.16 (1H, t, J=9.6 Гц), 7.03 (1H, d, J=8.0 Гц), 6.99 (1H, s), 6.81 (1H, t, J=54.8 Гц), 6.48 (1H, dd, J=8.4 Гц, 1.6 HZ), 6.42 (1H, d, J=2.0 Гц), 5.64 (1H, q, J=6.4 Гц), 5.08 (2H, s), 3.61 (3H, s), 3.43-3.46 (4H, m), 3.32-3.35 (4H, m), 1.67 (3H, d, J=6.4 Гц).
Пример 41: 5-(5-хлор-2-метокси-4-(пиперазин-1-ил)фенил)-3-(1-(2-(дифторметил)-5-фторфенил)этокси)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 40, в результате разделения путем колоночной хроматографии (выход: 7%). МС m/z [ESI]: 507,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.72 (1Н, s), 7.60 (1Н, dd, J=8.4 Гц, 5.6 Гц), 7.26 (1H, d, J=8.8 Гц), 7.20 (1Н, t, J=8.4 Гц), 7.17 (1H, s), 6.98 (1Н, s), 6.84 (1Н, t, J=52.0 Гц), 6.59 (1H, s), 5.68 (1H, q, J=6.4 Гц), 5.08 (2Н, s), 3.65 (3Н, s), 3.30-3.50 (8Н, m), 1.72 (3Н, d, J=6.4 Гц).
Пример 42: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-6'-(пиперидин-4-ил)-[3,3'-бипиридин]-6-амин
(R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин (98 мг, 0,2 ммоль), полученный в Примере 30, и Pd/C (10 мг) добавляли в метанол (20 мл), и данную реакционную смесь выдерживали в течение 6 ч в атмосфере водорода и затем фильтровали. Фильтрат концентрировали и очищали путем колоночной хроматографии на силикагеле с получением указанного в заголовке соединения (12 мг, выход: 12%). МС m/z [ESI]: 491,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.00 (d,J=1.8 Гц, 1Н), 7.70 (d, J=1.7 Гц, 1Н), 7.37 (dd, J=9.0, 4.9 Гц, 1Н), 7.18 (s, 1H), 7.14 (d, J=8.5 Гц, 1Н), 6.90 (d, J=1.7 Гц, 1Н), 6.14 (q, J=6.5 Гц, 1Н), 4.46 (m, 1Н), 3.83 (s, 3Н), 3.41 (m, 2Н), 3.11-2.99 (m, 2Н), 1.95 (dd, J=9.6, 3.6 Гц, 4Н), 1.81 (d, J=6.7 Гц, 3Н).
Пример 43: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-1'',2'',3'',6''-тетрагидро-[3,2':6',4''-терпиридин1-6-амин
Стадия 1: (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,2':6',4''-терпиридин]-1''(2''H)-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(6-бром-3-метоксипиридин-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 589,2 [М+1].
Стадия 2: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-1'',2'',3'',6''-тетрагидро-[3,2':6',4''-терпиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,2':6',4''-терпиридин]-1''(2''H)-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 489,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.09 (1H, s), 7.40-7.42 (2Н, m), 7.28-7.30 (1Н, s), 7.13 (1Н, d, J=8.8 Гц), 7.00 (1Н, t, J=8.8 Гц), 6.73 (1Н, s), 6.10 (1Н, q, J=6.8 Гц), 5.16 (2Н, s), 3.99 (2Н, t, J=5.6 Гц), 3.84 (2Н, m), 3.52 (2Н, X, J=5.6 Гц), 3.49 (3Н; s); 1.83 (3H, d, J=6.8 Гц)/
Пример 44: 5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-3-метокси-6-((S)-2-метилпиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Стадия 1: (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-3-метокси-[2,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием (S)-трет-бутил 4-(6-бром-5-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 606,2 [М+1].
Стадия 2: 5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-3-метокси-6-((S)-2-метилпиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-3-метокси-[2,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 506,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.38 (d, J=1.4 Гц, 1H), 7.45 (d, J=1.5 Гц, 1H), 7.30 (dd, J=8.9, 4.8 Гц, 1H), 7.21 (d, J=9.0 Гц, 1H), 7.11-7.01 (m, 1H), 6.53 (d, J=9.0 Гц, 1H), 6.08 (d, J=6.7 Гц, 1H), 5.23 (s, 2H), 4.62 (m, 1H), 4.03 (d, J=14.0 Гц, 1H), 3.73 (s, 3H), 3.59 (d, J=12.3 Гц, 1H), 3.45-3.29 (m, 3H), 3.11 (m, 1H), 1.82 (d, J=6.7 Гц, 3Н), 1.38 (d, J=6.9 Гц, 3Н).
Пример 45: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-5'-(пиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Стадия 1: (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-5-ил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием трет-бутил 4-(5-бром-4-метоксипиридин-3-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 592,2 [М+1].
Стадия 2: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-5'-(пиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (R)-трет-бутил 4-(6'-амино-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-5-ил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 492,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.13 (s, 1H), 8.10 (s, 1H), 7.75 (s, 1Н), 7.29 (d, J=5.0 Гц, 1H), 7.07 (t, J=8.4 Гц, 1Н), 6.96 (s, 1H), 6.04 (q, J=6.8 Гц, 1Н), 4.89 (s, 2Н), 3.43 (s, 3Н), 3.21 (d, J=7.3 Гц, 2Н), 3.07 (m, 6Н), 1.85 (d, J=6.7 Гц, 3Н).
Пример 46: 3-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
Стадия 1: трет-бутил 4-(4-(6-амино-5-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)пиридин-3-ил)-3-метоксифенил)пиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием 5-бром-3-(1-(2-(дифторэтил)-5-фторфенил)этокси)пиридин-2-амина вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата и с использованием трет-бутил 4-(3-метокси-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)фенил)пиперазин-1-карбоксилата вместо (R)-3-(1-(2,6-дихлор-3-фторфенил)этокси)-5-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)пиридин-2-амина (выход: 46%). МС m/z [ESI]: 587,3 [М+1].
Стадия 2: 3-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)-5-(2-метокси-4-(пиперазин-1-ил)фенил)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием трет-бутил 4-(4-(6-амино-5-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)пиридин-3-ил)-3-метоксифенил)пиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 487,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.63 (t, J=9.0 Гц, 2Н), 7.54 (s, 1H), 7.23 (t, J=8.2 Гц, 1Н), 6.99 (d, J=4.3 Гц, 2Н), 6.57 (s, 1Н), 6.52 (d, J=8.4 Гц, 1Н), 5.86 (s, 2Н), 5.81 (d, J=5.7 Гц, 1H), 3.60 (s, 3H), 3.17 (s, 4H), 3.12 (s, 4H), 2.08 (t, J=19.6 Гц, 3Н), 1.61 (d, J=6.1 Гц, 3Н).
Пример 47: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-4'-метокси-6'-морфолино-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием 4-(5-бром-4-метоксипиридин-2-ил)морфолина вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 493,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.93 (s, 1H), 7.70 (s, 1H), 7.31-7.26 (m, 1H), 7.05 (t, J=8.4 Гц, 1H), 6.95 (s, 1H), 6.10 (s, 1H), 6.04 (q, J=6.7 Гц, 1H), 4.94 (s, 2H), 3.86-3.81 (m, 4H), 3.75 (s, 3H), 3.56-3.50 (m, 4H), 1.83 (d, J=6.7 Гц, 3Н).
Пример 48: 5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-6-((S)-2-метилпиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Стадия 1: (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-[2,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием (S)-трет-бутил 4-(6-бром-3-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 606,2 [М+1].
Стадия 2: 5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-6-((S)-2-метилпиперазин-1-ил)-[2,3'-бипиридин]-6'-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-5-метокси-[2,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 506,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.13 (1H, s), 7.73 (1Н, m), 7.55 (1H, m), 7.32 (1Н, s), 7.20-7.09 (2Н, m), 6.12 (1Н, d, J=6.6 Гц), 5.21 (2Н, s), 4.54 (1H, m), 3.86 (3Н, s), 3.77 (1Н, m), 3.67 (1H, m), 3.51 (2H, m), 3.43 (1H, d, J=9.3 Гц), 3.24 (3Н, d, J=11.9 Гц), 1.87 (3Н, d, J=6.6 Гц), 1.38 (3Н, d, J=6.9 Гц).
Пример 49: 5-(5-хлор-2-метокси-4-(пиперазин-1-ил)фенил)-3-(1-(2-(1,1-дифторэтил)-5-фторфенил)этокси)пиридин-2-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 46, в результате очистки и разделения путем колоночной хроматографии на силикагеле (выход: 67%). МС m/z [ESI]: 521,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.65 (d, J=10.7 Гц, 2Н), 7.59 (d, J=13.4 Гц, 1H), 7.24 (s, 1H), 7.13 (s, 1H), 6.99 (s, 1H), 6.71 (s, 1H), 6.01 (s, 2H), 5.83 (d, J=5.8 Гц, 1H), 3.64 (s, 3H), 3.17 (s, 4H), 3.11 (s, 4H), 2.09 (t, J=19.6 Гц, 3Н), 1.62 (d, J=6.1 Гц, 3Н).
Пример 50: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-этокси-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
Стадия 1: (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-этокси-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 1), с использованием 5-бром-2-хлор-3-этоксипиридина вместо 5-бром-2-хлор-3-метоксипиридина (выход: 46%). МС m/z [ESI]: 456,0 [М+1].
Стадия 2: (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-этокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 2), с использованием (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-этокси-[3,3'-бипиридин]-6-амина вместо (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-[3,3'-бипиридин]-6-амина (выход: 46%). МС m/z [ESI]: 603,2 [М+1].
Стадия 3: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-этокси-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 3), с использованием (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-этокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата вместо (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата (выход: 67%). МС m/z [ESI]: 503,1 [М+1]. 1Н ЯМР (400 МГц, CD3OD): δ=8.06 (s, 1Н), 7.73 (s, 1Н), 7.36 (dd, J=9.0, 4.9 Гц, 1H), 7.23 (s, 1Н), 7.16 (t, J=8.6 Гц, 1Н), 6.90 (s, 1H), 6.48 (s, 1Н), 6.13 (d, J=6.7 Гц, 1H), 4.08 (q, J=6.8 Гц, 2Н), 3.79 (d, J=2.7 Гц, 2Н), 3.34 (t, J=6.1 Гц, 2Н), 2.82 (s, 2Н), 1.80 (d, J=6.6 Гц, 3Н), 1.38 (t, J=6.9 Гц, 3Н).
Пример 51: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-3''-метил-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
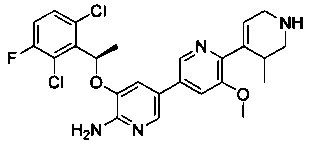
Стадия 1: трет-бутил 6-амино-5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5''-метил-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилат
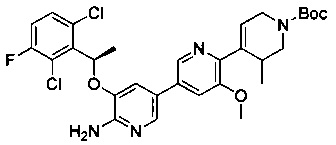
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 2), с использованием трет-бутил 5-метил-4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-5,6-дигидропиридин-1(2H)-карбоксилата вместо трет-бутил 4-(4,4,5,5-тетраметил-1,3,2-диоксаборолан-2-ил)-1-карбоксилат-1,2,5,6-тетрагидро-пиридина (выход: 46%). МС m/z [ESI]: 603,2 [М+1].
Стадия 2: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 3), с использованием трет-бутил 6-амино-5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5''-метил-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата вместо (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата (выход: 67%). МС m/z [ESI]: 503,1 [М+H]. 1Н ЯМР (400 МГц, CD3OD): δ=8.16 (dd, J=4.4, 1.7 Гц, 1H), 7.86 (s, 1Н), 7.46 (d, J=2.3 Гц, 1Н), 7.38 (s, 1Н), 7.25 (m, 1Н), 7.03 (s, 1H), 6.25 (q, J=6.5 Гц, 1Н), 6.12 (s, 1Н), 3.92 (s, 3Н), 3.84 (dd, J=5.9, 3.0 Гц, 2H), 3.54 (dd, J=12.3, 5.4 Гц, 1H), 3.48 (s, 1H), 3.10 (dd, J=12.3, 7.6 Гц, 1H), 1.91 (d, J=6.7 Гц, 3Н), 0.96 (d, J=7.0 Гц, 3Н).
Пример 52: (R)-2-((6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-5'-ил)окси)этанол
Стадия 1: (R)-2-((6'-амино-6-хлор-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-[3,3'-бипиридин]-5-ил)окси)этанол
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 1), с использованием 5-бром-2-хлор-3-(2-гидроксиэтокси)-пиридина вместо 5-бром-2-хлор-3-метоксипиридина (выход: 46%). МС m/z [ESI]: 472,0 [М+1].
Стадия 2: (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-(2-гидроксиэтокси)-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 2), с использованием (R)-2-((6'-амино-6-хлор-5'-(1-(2,6-дихлор-3-фторфенил)этокси)-[3,3'-бипиридин]-5-ил)окси)этанола вместо (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-[3,3'-бипиридин]-6-амина (выход: 46%). МС m/z [ESI]: 619,2 [М+1].
Стадия 3: (R)-2-((6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-5'-ил)окси)этанол
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 3), с использованием (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-(2-гидроксиэтокси)-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата вместо (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата (выход: 67%). МС m/z [ESI]: 519,1 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=8.23 (s, 1H), 7.87 (d, J=1.1 Гц, 1H), 7.31 (dd, J=8.9, 4.7 Гц, 1H), 7.11 (s, 1H), 7.07 (t, J=8.4 Гц, 1H), 6.94 (s, 1H), 6.41 (s, 1H), 6.11 (q, J=6.6 Гц, 1H), 4.96 (s, 2H), 4.18-4.04 (m, 2H), 4.01 (d, J=4.3 Гц, 2H), 3.64 (d, J=2.6 Гц, 2H), 3.16 (t, J=5.7 Гц, 2H), 2.68 (s, 2H), 1.88 (d, J=6.7 Гц, 3Н).
Пример 53: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4'-этокси-6'-((S)-2-метилпиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Стадия 1: (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-этокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием (S)-трет-бутил 4-(5-бром-4-этоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 620,2 [М+1].
Стадия 2: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4'-этокси-6'-((S)-2-метилпиперазин-1-ил)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-этокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 520,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.91 (s, 1Н), 7.78 (d, J=1.0 Гц, 1Н), 7.31-7.26 (m, 1Н), 7.03 (dd, J=17.1, 8.9 Гц, 2Н), 6.05 (m, 2Н), 4.77 (s, 2Н), 4.41 (m, 1Н), 4.05 (q, J=6.8 Гц, 2Н), 3.99-3.85 (m, 1Н), 3.20-3.02 (m, 3Н), 2.98-2.77 (m, 2Н), 1.82 (d, J=6.7 Гц, 3Н), 1.36 (t, J=7.0 Гц, 3Н), 1.23 (s, 3Н).
Пример 54: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-6'-морфолино-5'-(2-морфолиноэтокси)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием 4-(2-(5-бром-2-морфолинопиридин-3-ил-окси)этил)морфолина вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 592,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.93 (d, J=1.8 Гц, 1H), 7.81 (d, J=1.4 Гц, 1H), 7.31 (dd, J=8.9, 4.8 Гц, 1H), 7.11-7.03 (m, 1H), 6.99 (d, J=1.6 Гц, 1H), 6.91 (s, 1H), 6.10 (q, J=6.7 Гц, 1H), 4.91 (s, 2H), 4.19-4.06 (m, 2H), 3.88-3.83 (m, 4H), 3.74-3.69 (m, 4H), 3.48-3.38 (m, 4H), 2.85 (t, J=5.6 Гц, 2H), 2.60 (d, J=4.3 Гц, 4H), 1.87 (d, J=6.7 Гц, 3Н).
Пример 55: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-(2-морфолиноэтокси)-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
Стадия 1: (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-(2-морфолиноэтокси)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 1), с использованием 4-(2-(5-бром-2-хлорпиридин-3-ил-окси)этил)морфолина вместо 5-бром-2-хлор-3-метоксипиридина (выход: 46%). МС m/z [ESI]: 543,1 [М+1].
Стадия 2: (R)-трет-6утил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-(2-морфолиноэтокси)-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 2), с использованием (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-(2-морфолиноэтокси)-[3,3'-бипиридин]-6-амина вместо (R)-6'-хлор-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-[3,3'-бипиридин]-6-амина (выход: 46%). МС m/z [ESI]: 688,2 [М+1].
Стадия 3: (R)-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-(2-морфолиноэтокси)-1'',2'',3'',6''-тетрагидро-[3,3':6',4''-терпиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 30 (Стадия 3), с использованием (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-(2-морфолиноэтокси)-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата вместо (R)-трет-бутил 6-амино-5-(1-(2,6-дихлор-3-фторфенил)этокси)-5'-метокси-5'',6''-дигидро-[3,3':6',4''-терпиридин]-1''(2''H)-карбоксилата (выход: 67%). МС m/z [ESI]: 588,2 [М+1]. 1H ЯМР (400 МГц, CDCl3): δ=8.20 (s, 1H), 7.85 (s, 1H), 7.30 (dd, J=8.9, 4.8 Гц, 1H), 7.12-7.00 (m, 2H), 6.93 (s, 1H), 6.55 (s, 1H), 6.09 (d, J=6.7 Гц, 1H), 4.95 (s, 2H), 4.11 (dd, J=14.4, 5.8 Гц, 2H), 3.76-3.66 (m, 4H), 3.59 (d, J=2.6 Гц, 2H), 3.11 (t, J=5.6 Гц, 2H), 2.83 (t, J=5.7 Гц, 2H), 2.65-2.51 (m, 6H), 1.86 (d, J=6.7 Гц, 3Н).
Пример 56: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-6'-((S)-2-метилпиперазин-1-ил)-4'-(2-морфолиноэтокси)-[3,3'-бипиридин]-6-амин
Стадия 1: (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-(2-морфолиноэтокси)-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилат
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 1), с использованием (S)-трет-бутил 4-(5-бром-4-(2-морфолиноэтоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(5-бром-4-метоксипиридин-2-ил)-3-метилпиперазин-1-карбоксилата (выход: 46%). МС m/z [ESI]: 705,3 [М+1].
Стадия 2: 5-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-6'-((S)-2-метилпиперазин-1-ил)-4'-(2-морфолиноэтокси)-[3,3'-бипиридин]-6-амин
Указанное в заголовке соединение получали в соответствии с методикой, описанной в Примере 27 (Стадия 2), с использованием (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-(2-морфолиноэтокси)-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата вместо (S)-трет-бутил 4-(6'-амино-5'-((R)-1-(2,6-дихлор-3-фторфенил)этокси)-4-метокси-[3,3'-бипиридин]-6-ил)-3-метилпиперазин-1-карбоксилата (выход: 67%). МС m/z [ESI]: 605,2 [М+1]. 1Н ЯМР (400 МГц, CDCl3): δ=7.89 (s, 1H), 7.76 (d, J=1.5 Гц, 1H), 7.32-7.27 (m, 1H), 7.08-7.01 (m, 1H), 6.93 (d, J=1.5 Гц, 1H), 6.07 (s, 1H), 6.05 (d, J=6.7 Гц, 1H), 4.82 (s, 2H), 4.45-4.36 (m, 1H), 4.10 (dd, J=10.3, 6.1 Гц, 2H), 3.99-3.89 (m, 1H), 3.68-3.64 (m, 4H), 3.08 (dd, J=10.8, 7.5 Гц, 3Н), 2.94 (d, J=12.1 Гц, 1H), 2.85 (dd, J=13.5, 10.0 Гц, 1H), 2.72 (dd, J=10.1, 6.1 Гц, 2H), 2.49-2.44 (m, 4H), 1.82 (d, J=6.7 Гц, 3Н), 1.24 (d, J=6.7 Гц, 3Н).
Анализ уровня ингибирования ALK киназы
Для определения ингибирующей активности соединений по настоящему изобретению в отношении ALK-киназы использовали описанную ниже методику. Ингибирующая активность указана в виде ИК50 (концентрации соединения, требуемой для 50%-го ингибирования активности ALK-киназы). В настоящем патенте разработана и оптимизирована платформа для анализа активности ALK-киназы (полученной от Millipore) с использованием методики гомогенной флуоресценции с разрешением по времени (HTRF, Cisbio), позволяющая выполнять измерение активности соединений по настоящему изобретению.
Материалы и методики
Материалы
а. Белый планшет 384 Orifice (Perkin Elmer, №607290/99 по каталогу)
б. Буфер HEPES: 50 мл 0,05 М буфера HEPES получали из 1 М буфера HEPES (Invitrogen, №15630-080 по каталогу) путем добавления к 2,5 мл 1 М буфера HEPES подходящего количества дистиллированной воды (ddH2O), подведения pH до 7,0 с использованием NaOH и добавления ddH2O (бидистиллированной воды) до конечного объема 50 мл.
в. ALK-киназа (Millipore)
г. 0,1 M Na3VO4
д. 1 М MgCl2
е. 0,2 М DTT (дитиотрейтол)
ж. 10% BAS
з. ДМСО
и. ddH2O
к. Исследуемые соединения: соединения, описанные в Примерах
Исследование проводили с использованием следующей методики:
1. Готовили буфер для проведения ферментативной реакции с участием ALK: 50 мМ HEPES (pH=7,0), 0,1 мМ Na3VO4, 0,01% BAS, 5 мМ MgCl2, 1 мМ DTT (до использования буфер хранили на льду).
2. Делали ряд 3-кратных последовательных разбавлений соединения в 100% ДМСО, начиная с концентрации 1 мМ, добавляли 4 мкл из каждого разбавления в 96 мкл реакционного буфера, затем 2,5 мкл полученной смеси вносили в 384-луночный планшет (OptiPlate-384, PerkinElmer), и затем добавляли 5 мкл киназы, перемешивали до однородного состояния путем центрифугирования, затем для инициирования реакции добавляли 2,5 мкл смеси АТФ и TK-пептида (конечная концентрация АТФ имела значение Km (константы Михаэлиса)).
3. 384-Луночный планшет помещали в инкубатор (23°С) на 120 мин.
4. Добавляли 5 мкл антитела TK-антитело-Cryptate, 5 мкл стрептавидин-меченого XL-665 для остановки реакции.
5. Инкубировали в инкубаторе (22-23°С) в течение 1 ч.
6. Флуоресцентный сигнал реакционной смеси регистрировали с использованием считывающего устройства для микропланшетов Envision (PerkinElmer): для возбуждения использовали длину волны 320 нм, эмиссионный спектр регистрировали на длине волны 665 нм;
7. Получали ИК50 соединений для ALK: ИК50 соединений вычисляли с использованием GraFit6.
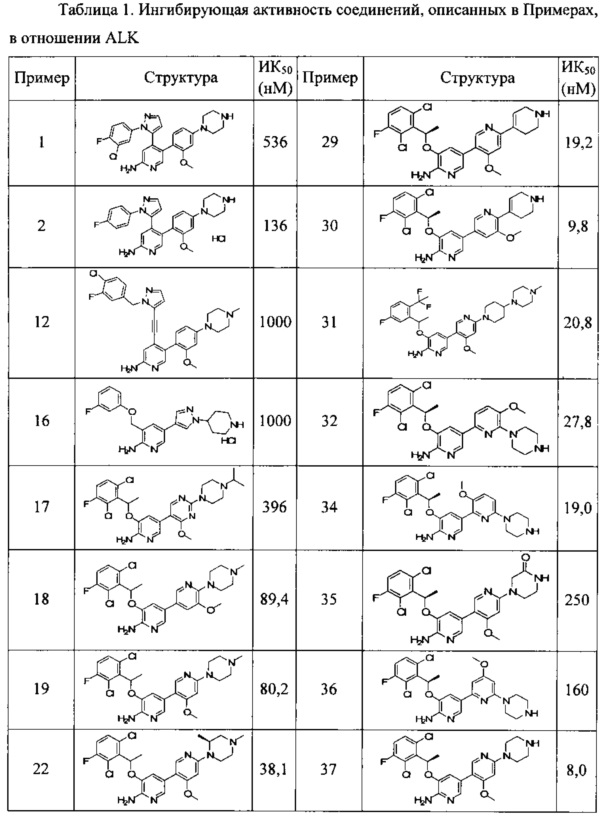
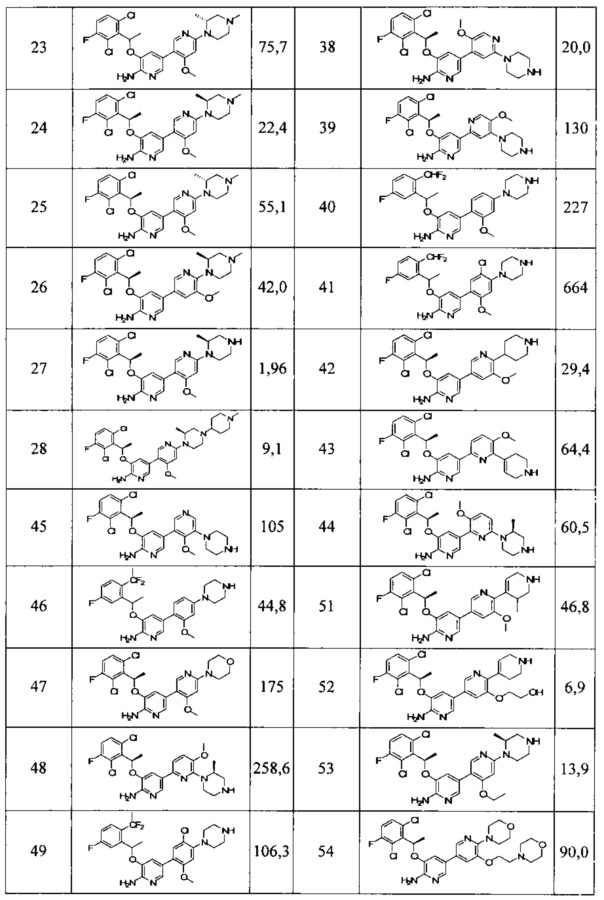
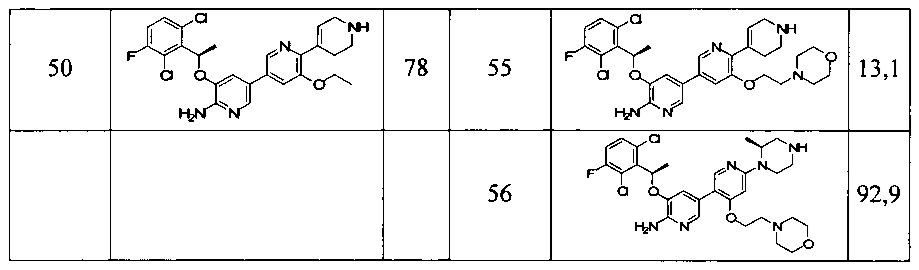
Результаты данного исследования, приведенные в Таблице 1, показывают, что соединения по настоящему изобретению обладают высокой ингибирующей активностью в отношении ALK.
В Таблице 2 приведены значения ингибирующей активности соединений, описанных в Примерах 27, 30 и 53, в отношении мутированной ALK-киназы. Мутированные ALK-киназы F1174L, L1196M, G1269S и R1275Q имеются в продаже.
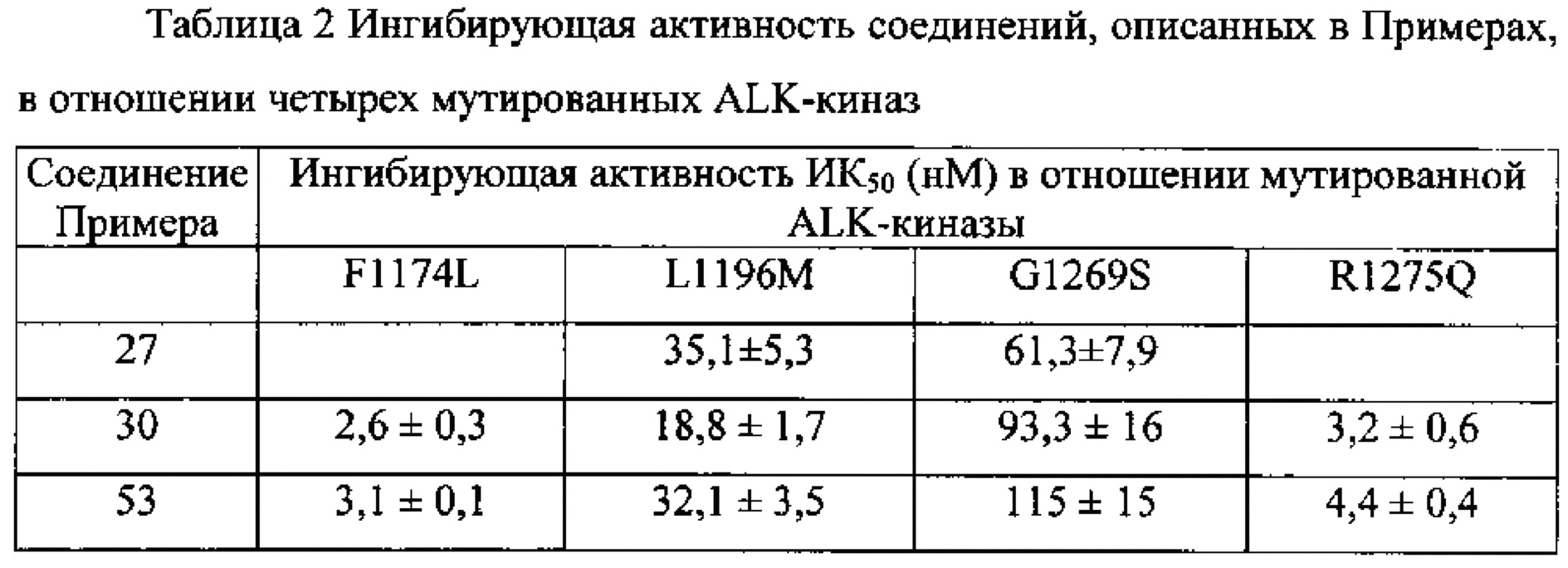
Соединения по настоящему изобретению имеют очень хорошие показатели in vivo метаболизма. В Таблице 3 приведены in vivo фармакокинетические данные для соединений, описанных в Примерах 27, 30 и 53 настоящего изобретения, полученные в исследовании, проводимом на SD-крысах.
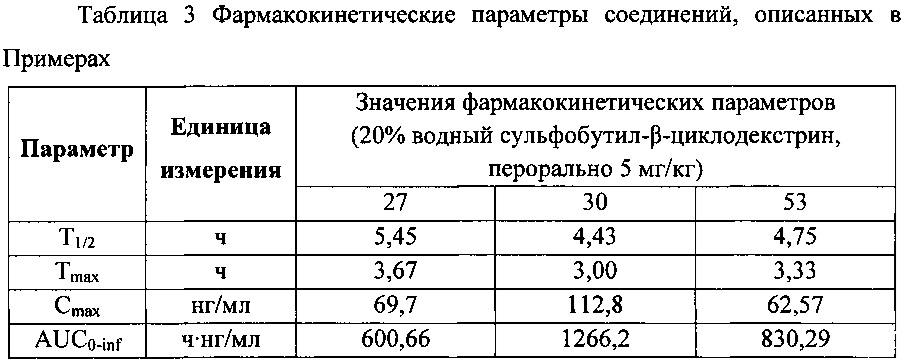
CYP-3A4 представляет собой важный метаболический фермент CYP-3A человека. Ингибирование данного фермента может приводить к неблагоприятному воздействию на метаболизм других лекарств, используемых в комбинированной терапии. Как показано в Таблице 4, соединения Примеров 27 и 30 не оказывают значительного ингибирующего действия на активность CYP-3А4, и поэтому влияние данных соединений на метаболизм других лекарств, используемых в комбинированной терапии, является незначительным или вообще отсутствует.

В Таблице 5 приведены результаты терапевтического действия соединения Примера 27 на немелкоклеточную карциному легкого человека NCI-H2228, полученные в исследовании, выполненном на "голых" мышах. Использовали следующую экспериментальную методику. "Голым" мышам подкожно инокулировали клетки NCI-H2228 немелкоклеточной карциномы легкого человека, после того, как опухоль вырастала до объема 80-200 мм3, мышей разделяли на группы методом случайной выборки (D0), и вводили соединение. Вводимая доза и режим введения приведены в Таблице 5. Дважды в неделю измеряли объем опухоли, мышей взвешивали и регистрировали полученные данные. Объем опухоли (V) рассчитывали следующим образом:
V=1/2xaxb2, где а и b представляют собой длину и ширину соответственно.
Т/С(%)=(Т-Т0)/(С-С0)x100, где Т, С, соответственно, представляют собой объем опухоли в конце эксперимента; Т0, С0, соответственно, представляют собой объем опухоли в начале эксперимента.
Когда наблюдается регресс опухоли, Т/С(%)=(Т-Т0)/Т0×100, где Т представляет собой объем опухоли в конце эксперимента; То представляет собой объем опухоли в начале эксперимента.
Процент ингибирования (%)=100-Т/С(%), частичный регресс указывает, что опухоль уменьшается, но не исчезает, полный регресс указывает, что опухоль исчезает.
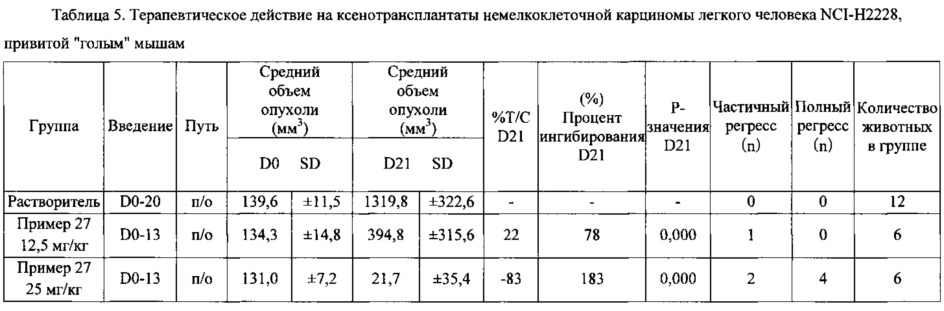
В таблице 5 группа "Растворитель" представляет собой контрольную группу; группам, получавшим лечение, вводили соединения, разбавленные дистиллированной водой, содержащей 0,1% Твин-80.
D0-20 означает, что делали одно введение в сутки, начиная с суток 0 (D0), и длительность введения составляла 21 сутки; D0-13 означает, что делали одно введение в сутки, начиная с суток 0 (D0), и длительность введения составляла 14 суток.
Р-значения получены с использованием t-критерия Стьюдента, для сравнения использованы данные контрольной группы.
n указывает количество мышей; количество тестируемых мышей в контрольной группе равно 12, и количество мышей во всех группах, получавших лечение, равно 6.
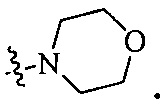
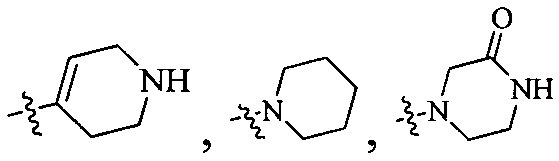
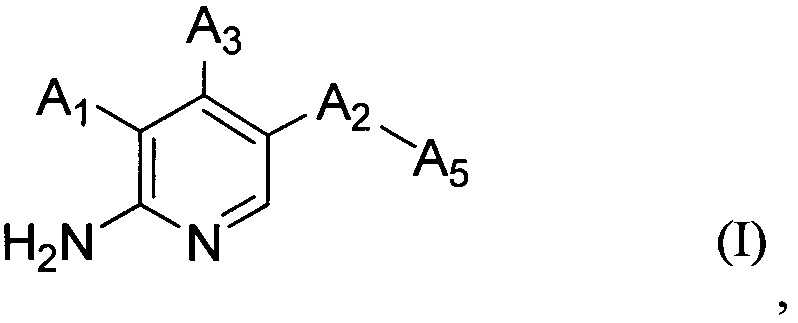
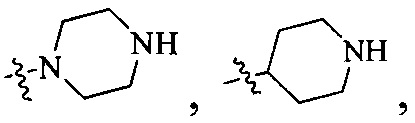
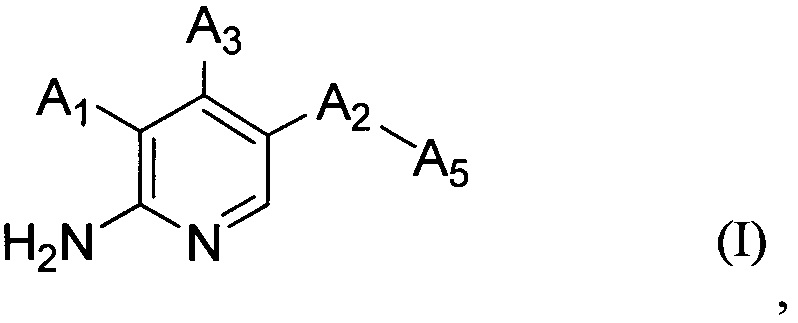
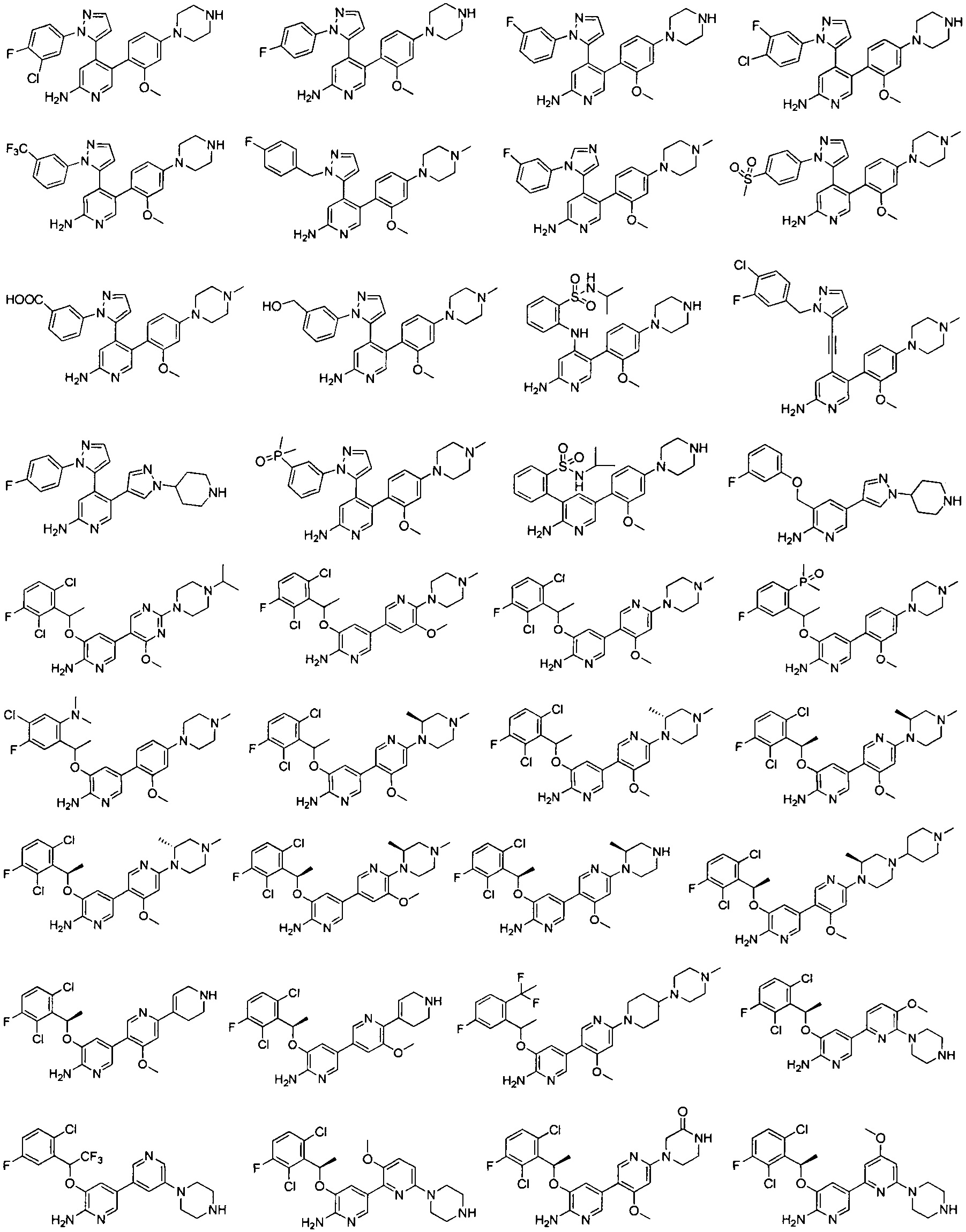

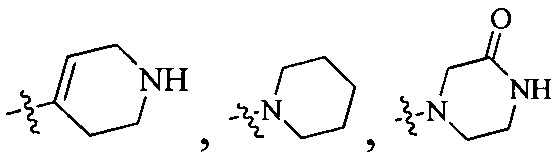
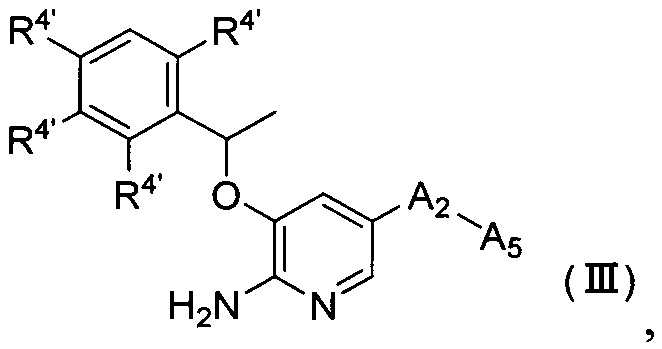
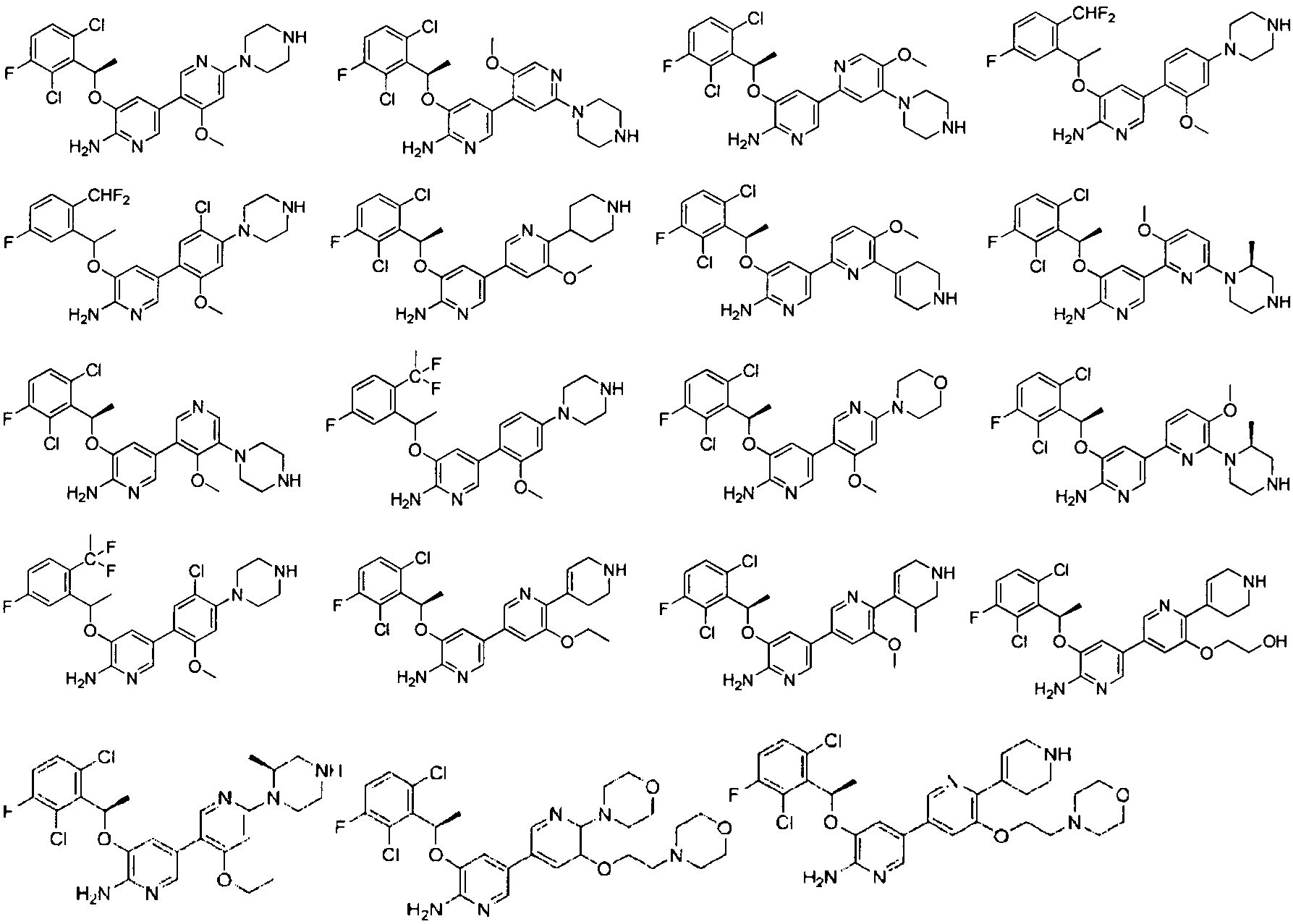
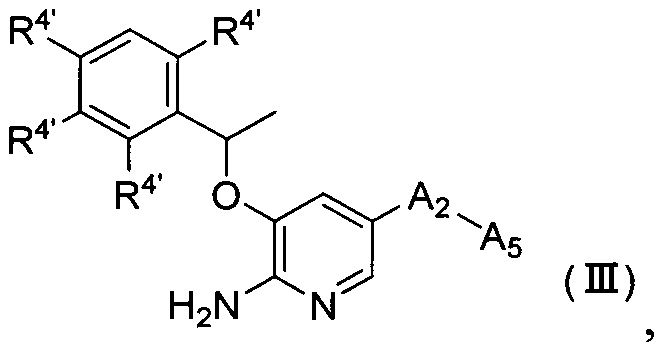
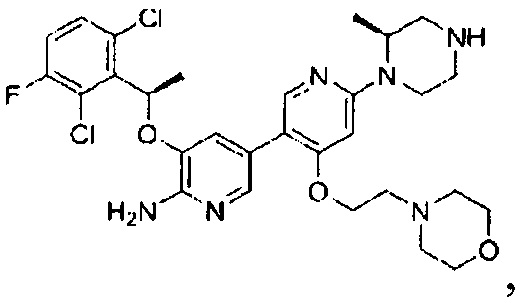
Фумарат пиридиламина и его кристаллы
Пирролопиримидиновое соединение
Способ получения ибрутиниба
Замещенные амино шестичленные насыщенные гетероалициклы в качестве ингибиторов dpp-iv длительного действия
Соли производного хиназолина и способ их получения
Производное 1,3,5-триазина и способ его применения
Кристалл ингибитора dpp-iv длительного действия и его соли
Кристалл пирролопиримидина для получения jak-ингибитора
Устройство для рекуперации отработанного тепла с комбинированной выработкой тепла и электроэнергии (снр) при пиковой электрической нагрузке и способ его работы
Пирролопиримидиновое соединение
Замещенные амино шестичленные насыщенные гетероалициклы в качестве ингибиторов dpp-iv длительного действия
Производное 1,3,5-триазина и способ его применения

