Результат интеллектуальной деятельности: ФУМАРАТ ПИРИДИЛАМИНА И ЕГО КРИСТАЛЛЫ
Вид РИД
Изобретение
ОБЛАСТЬ ТЕХНИКИ
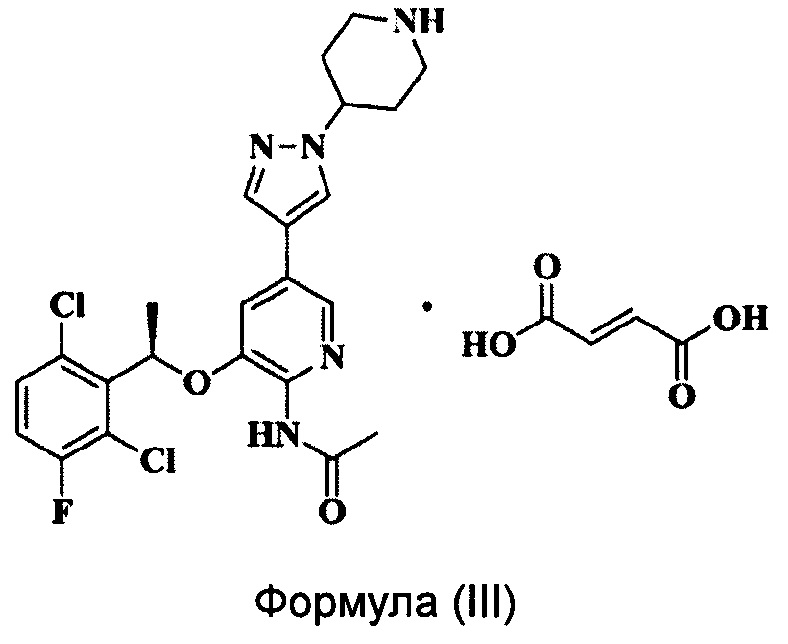
Настоящее изобретение относится к фумарату пиридиламина и его кристаллу и относится к области медицинской химии.
УРОВЕНЬ ТЕХНИКИ
Протеин-тирозин киназы (PTKs) играют чрезвычайно важную роль во внутриклеточных путях передачи сигнала. Они включены в регуляцию, передачу сигналов и развитие нормальных клеток, а также тесно связаны с пролиферацией, дифференцировкой, миграцией и апоптозом опухолевых клеток. Поэтому, ингибирующее воздействие на протеин-тирозин киназы оказывает положительное влияние на подавление и лечение опухолей. Семейство протеин-тирозин киназ имеет множество подтипов, включая подтип рецептора эпидермального фактора роста (EGFR), подтип рецептора сосудистого эндотелиального фактора роста (VEGFR), подтип рецептора тромбоцитарного фактора роста (PDGFR), киназу анапластической лимфомы, КАЛ (ALK) и т.д. Исследование показало, что аномальная активация и экспрессия КАЛ (ALK) киназ анапластической лимфомы существуют в различных опухолевых клетках, таких как немелкоклеточный рак легкого, рак молочной железы, глиобластомы и т.п.
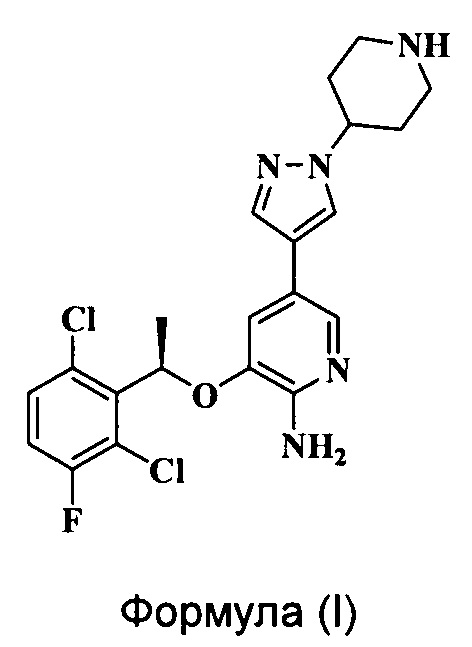
Кризотиниб (XALKORI™) является пероральным ингибитором киназы анапластической лимфомы (ALK), разработанным U.S. Pfizer Inc., который впервые был выпущен на рынок в США в августе 2011 года (Nat. Rev. Drug Disc, 10, 895-896, 2011). Клинически препарат в основном используют для лечения пациентов с киназа анапластической лимфомы (ALK)-позитивным местнораспространенным или метастатическим немелкоклеточным раком легкого (НМРЛ). Химическая структура Кризотиниба соответствует Формуле (I):
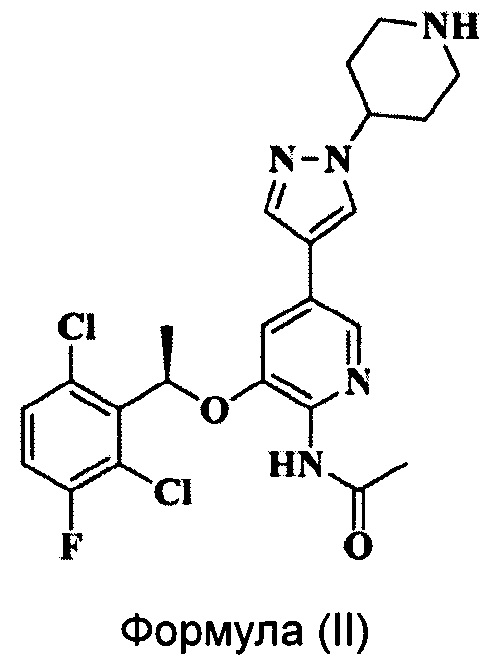
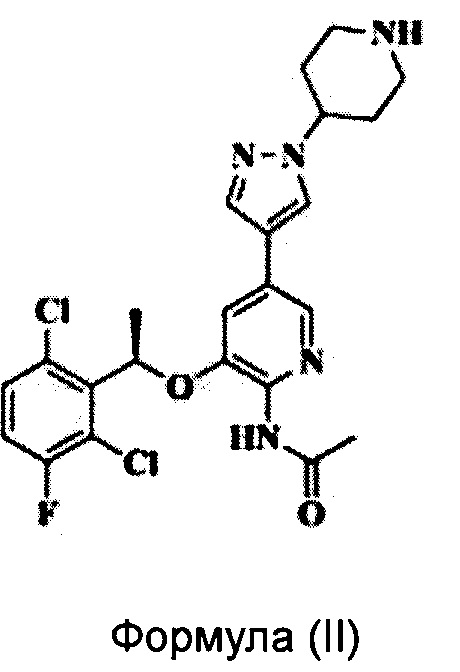
Заявка на патент CN 102850328 A раскрывает пиридиламин, имеющий химическую структуру, соответствующую Формуле (II), который является аналогом Кризотиниба и обладает хорошим ингибирующим действием на ALK. Однако существуют некоторые проблемы, связанные с соединением Формулы (II), такие как легкость поглощения влаги и появление деградации, жесткие условия хранения и т.д.
В дополнение к терапевтической эффективности стабильность, гигроскопичность, биодоступность и т.п. лекарственного средства в качестве лекарственного препарата при обработке, производстве и хранении имеют решающее значение для исследования и разработки лекарственных средств. Кроме того, химическая стабильность, стабильность в твердом состоянии и срок годности действующего вещества являются очень важными факторами с точки зрения получения коммерчески жизнеспособного способа производства или с точки зрения получения фармацевтической композиции, содержащей активное соединение. Поэтому для производства и хранения лекарственного средства очень важно обеспечить подходящую форму лекарственного средства, имеющую требуемые свойства.
РАСКРЫТИЕ ИЗОБРЕТЕНИЯ
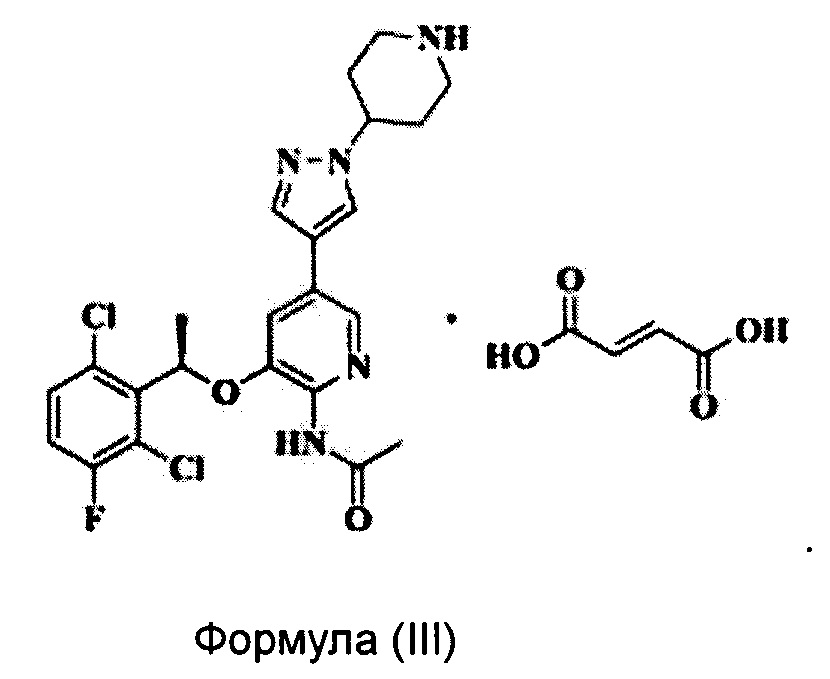
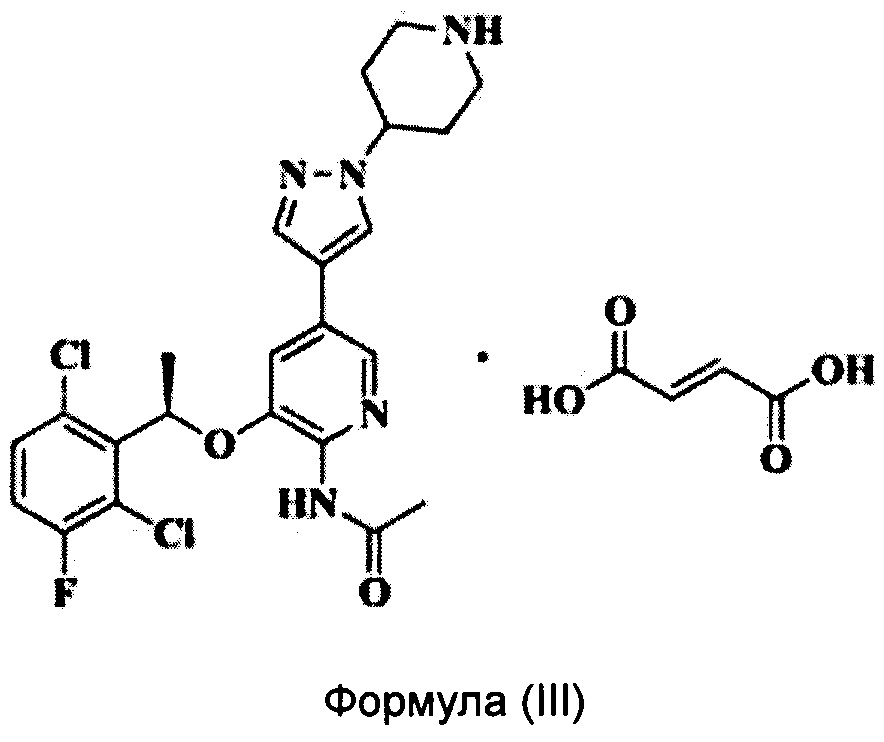
Согласно настоящему изобретению предложено соединение Формулы (III), имеющее следующую структуру, которое представляет собой фумарат соединения Формулы (II):
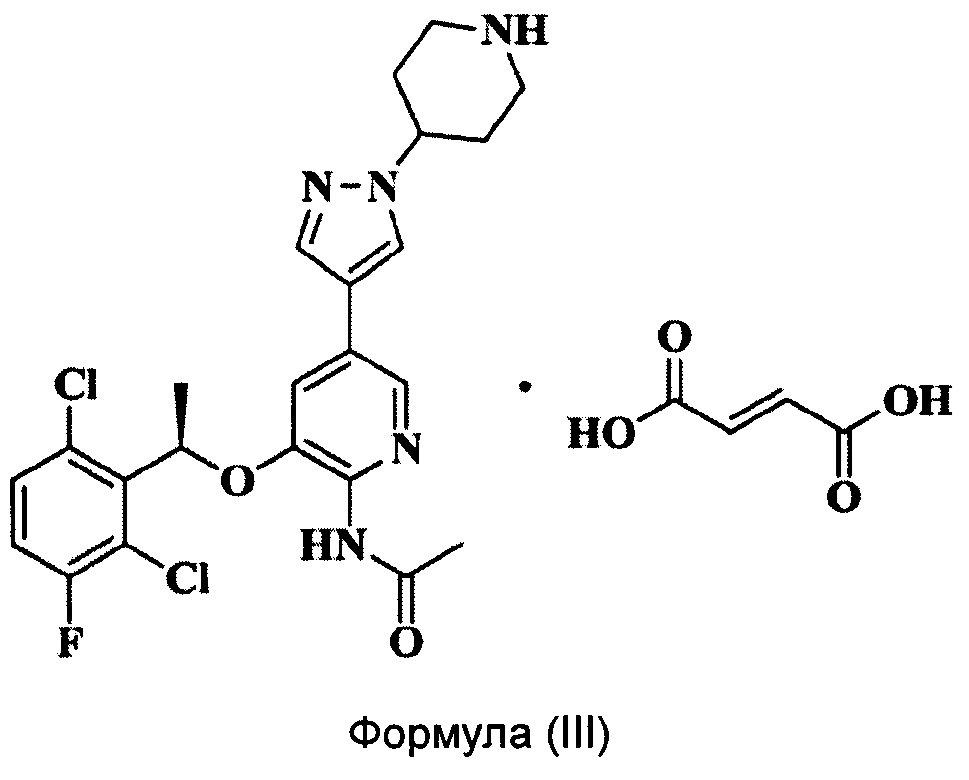
Согласно настоящему изобретению дополнительно предложен способ получения соединения Формулы (III), включающий следующие стадии: растворение соединения Формулы (II) в органическом растворителе и добавление раствора фумаровой кислоты в этаноле при температуре от 0°С до 80°С для проведения реакции получения соединения Формулы (III).
В некоторых вариантах реализации настоящего изобретения раствор фумаровой кислоты в этаноле добавляют при перемешивании, и реакцию проводят в течение от 0,5 часа до 2 часов.
В некоторых вариантах реализации настоящего изобретения после осаждения твердой фазы из реакционного раствора, твердую фазу фильтруют и сушат, предпочтительно сушат в вакууме.
В некоторых вариантах реализации настоящего изобретения органический растворитель представляет собой один или более растворителей, выбранных из группы, состоящей из метанола, этанола, дихлорметана и ацетона.
В некоторых вариантах реализации настоящего изобретения молярное соотношение добавленной фумаровой кислоты к соединению Формулы (II) составляет 1,2-1,5:1.
В некоторых вариантах реализации настоящего изобретения концентрация добавленного раствора фумаровой кислоты в этаноле составляет от 1,0 моль/л до 2,0 моль/л, предпочтительно от 1,0 моль/л до 1,5 моль/л.
В некоторых вариантах реализации настоящего изобретения реакцию предпочтительно проводят при температуре от 0°С до 50°С; в некоторых других вариантах реализации изобретения реакцию более предпочтительно проводят при температуре от 20°С до 50°С.
В некоторых частных вариантах реализации изобретения соединение Формулы (III) может быть получено в соответствии со следующими стадиями: растворение соединения Формулы (II) в этаноле, добавление раствора фумаровой кислоты в этаноле при температуре от 20°С до 50°С при перемешивании, проведение реакции в течение от 0,5 часа до 2 часов, осаждение твердой фазы из реакционного раствора, фильтрование твердой фазы и сушка твердой фазы в вакууме с получением соединения Формулы (III).
Фумаровая кислота может быть заменена разными неорганическими или органическими кислотами, и различные аддукты соединения Формулы (II) с кислотами могут быть получены за счет применения аналогичных способов, описанных выше.
Согласно настоящему изобретению дополнительно предложена фармацевтическая композиция, содержащая терапевтически эффективное количество соединения Формулы (III) и фармацевтически приемлемый носитель. Фармацевтически приемлемый носитель может быть твердым или жидким. Твердый носитель может содержать один или более веществ, выбранных из группы, состоящей из вкусовых добавок, скользящих веществ, солюбилизаторов, суспендирующих веществ, наполнителей, связывающих веществ, веществ для улучшения распадаемости таблеток или материалов для инкапсулирования. Подходящие твердые носители включают, например, стеарат магния, тальк, сахарозу, лактозу, декстрин, крахмал, желатин, целлюлозу, метилцеллюлозу, натрий-карбоксиметилцеллюлозу и поливинилпирролидон. Жидкие носители применяют для получения таких композиций, как растворы, суспензии, эмульсии, сиропы и т.п. Подходящие жидкие носители для перорального и парентерального введения включают воду, спирты, масло и т.п.
Согласно настоящему изобретению дополнительно предложено применение соединения Формулы (III) для получения лекарственного средства для профилактики или лечения опухолей. Соединение Формулы (III) по настоящему изобретению можно применять отдельно или в комбинации с другими лекарственными средствами для получения противоопухолевых лекарственных средств. Опухоли могут представлять собой раковые заболевания легкого, предпочтительно ALK-позитивные первичные или метастатические немелкоклеточные раковые заболевания легкого.
Согласно настоящему изобретению также предложена кристаллическая Форма А соединения Формулы (III),
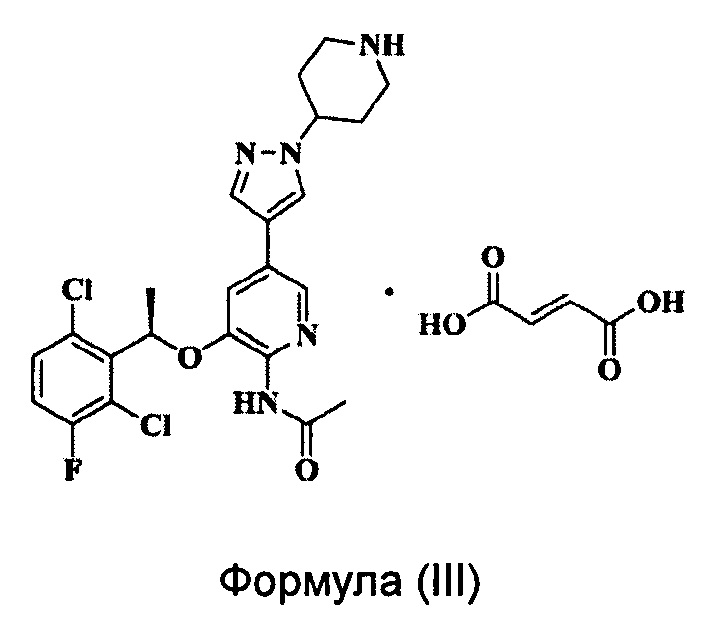
характеризующаяся тем, что ее рентгеновская порошковая дифрактрограмма имеет дифракционные пики при значениях 2θ около 6,3°, 11,7°, 12,5°, 14,1°, 22,6° и 23,3°; часто имеет дифракционные пики при значениях 2θ около 6,3°, 11,7°, 12,5°, 14,1°, 19,7°, 21,2°, 22,6°, 23,3°, 23,8° и 25,5°; наиболее часто имеет дифракционные пики при значениях 2θ около 6,3°, 11,7°, 12,5°, 14,1°, 15,0°, 15,9°, 17,0°, 19,7°, 20,6°, 21,2°, 21,6°, 22,6°, 23,3°, 23,8° и 25,5°; и наиболее часто имеет дифракционные пики при значениях 2θ около 6,3°, 9,9°, 11,7°, 12,5°, 14,1°, 15,0°, 15,9°, 17,0°, 19,7°, 20,6°, 21,2°, 21,6°, 22,6°, 23,3°, 23,8°, 24,6°, 25,1°, 25,5°, 27,1° и 28,7°.
Типичный, но неограничивающий пример кристаллической Формы А соединения Формулы (III) согласно настоящему изобретению имеет термограмму дифференциальной сканирующей калориметрии (ДСК) с пиком поглощения около 227,5°С.
Типичный, но неограничивающий пример кристаллической Формы А соединения Формулы (III) согласно настоящему изобретению имеет инфракрасный спектр (ИК), как показано на фиг. 5.
Согласно другому аспекту настоящего изобретения предложен кристаллический состав, в котором указанная выше кристаллическая Форма А соединения Формулы (III) составляет 50% масс. или более, предпочтительно 80% масс. или более, более предпочтительно 90% масс. или более и наиболее предпочтительно 95% масс. или более кристаллического состава.
Согласно другому аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество указанной выше кристаллической Формы А соединения Формулы (III) или указанного выше кристаллического состава.
Согласно другому аспекту настоящего изобретения предложено применение указанной выше кристаллической Формы А соединения Формулы (III), указанного выше кристаллической состава или указанной выше фармацевтической композиции для получения лекарственного средства для профилактики или лечения опухолей, предпочтительно для получении лекарственного средства для профилактики или лечения раковых заболеваний легкого и более предпочтительно для получения лекарственного средства для профилактики или лечения ALK-позитивных первичных или метастатических немелкоклеточных раковых заболеваний легкого.
Согласно другому аспекту настоящего изобретения предложен способ получения указанной выше кристаллической Формы А соединения Формулы (III) или указанного выше кристаллической состава, включающий:
(а) растворение соединения Формулы (III) в органическом растворителе и нагревание полученного раствора при перемешивании;
(b) добавление второго растворителя; и
(c) охлаждение полученной смеси для осаждения кристаллов.
На указанной выше стадии (а) органический растворитель представляет собой низший спирт, предпочтительно С1-С4-алкиловый спирт и более предпочтительно метанол; на стадии (а) полученный раствор может быть нагрет до температуры от 40°С до 65°С, предпочтительно от 50°С до 60°С; на стадии (а) отношение соединения Формулы (III) к органическому растворителю предпочтительно составляет от 1 г/50 мл до 1 г/10 мл, более предпочтительно от 1 г/15 мл до 1 г/10 мл; на стадии (а) скорость перемешивания предпочтительно составляет от 300 об/мин до 500 об/мин; на стадии (b), предпочтительно, вторым растворителем является ацетон, тетрагидрофуран, диоксан или вода; на стадии (b) объемное соотношение второго растворителя к органическому растворителю со стадии (а) может составлять 1-3:1, предпочтительно 2:1; на стадии (с) полученная смесь может быть охлаждена до температуры от -15°С или до 0°С, предпочтительно она может быть охлаждена до 0°С.
В некоторых вариантах реализации настоящего изобретения указанный выше способ получения кристаллической Формы А соединения Формулы (III) может дополнительно включать:
(d) фильтрование и сушку.
На стадии (d) сушка может быть проведена при 45°С в вакууме или продувкой воздухом при 45°С при атмосферном давлении.
Согласно настоящему изобретению также предложена кристаллическая Форма В соединения Формулы (III),
характеризующуюся тем, что рентгеновская порошковая дифрактрограмма кристаллической Формы В имеет дифракционные пики при значениях 2θ около 23,0°, 24,9°, 25,9°, 27,0°, 28,9°, 29,5°, 38,1° и 38,8°; часто имеет дифракционные пики при значениях 2θ около 18,7°, 23,0°, 24,9°, 25,9°, 27,0°, 28,0°, 28,9°, 29,5°, 36,0°, 38,1° и 38,7°.
Типичный, но неограничивающий пример кристаллической Формы В соединения Формулы (III) согласно настоящему изобретению имеет термограмму дифференциальной сканирующей калориметрии (ДСК) со значением пика около 230,6°С.
Типичный, но неограничивающий пример кристаллической Формы В соединения Формулы (III), обеспеченный в настоящем изобретении, имеет инфракрасный спектр (ИК), как показано на фиг. 6.
Согласно другому аспекту настоящего изобретения предложен кристаллический состав, в которой указанная выше кристаллическая Форма В соединения Формулы (III) составляет 50% масс. или более, предпочтительно 80% масс. или более, более предпочтительно 90% масс, или более и наиболее предпочтительно 95% масс. или более кристаллического состава.
Согласно другому аспекту настоящего изобретения предложена фармацевтическая композиция, содержащая терапевтически эффективное количество указанной выше кристаллической Формы В соединения Формулы (III) или указанного выше кристаллического состава.
Согласно другому аспекту настоящего изобретения предложено применение указанной выше кристаллической Формы В соединения Формулы (III), указанного выше кристаллического состава или указанной выше фармацевтической композиции для получения лекарственного средства для профилактики или лечения опухолей, предпочтительно для получения лекарственного средства для профилактики или лечения раковых заболеваний легкого и более предпочтительно для получения лекарственного средства для профилактики или лечения ALK-позитивных первичных или метастатических немелкоклеточных раковых заболеваний легкого.
Согласно другому аспекту настоящего изобретения предложен способ получения указанной выше кристаллической Формы В соединения Формулы (III) или указанного выше кристаллической состава, включающий:
(а) растворение соединения Формулы (III) в безводном метаноле при нагревании и перемешивании; и
(b) ступенчатое охлаждение полученного раствора для осаждения кристаллов. На указанной выше стадии (а) нагревание может быть проведено при температуре от 40°С до 70°С, и предпочтительно нагревание проводят с обратным холодильником; скорость перемешивания предпочтительно составляет от 300 об/мин до 500 об/мин; отношение соединения Формулы (III) к безводному метанолу предпочтительно составляет от 1 г/50 мл до 1 г/10 мл, более предпочтительно от 1 г/15 мл до 1 г/10 мл. На указанной выше стадии (b) ступенчатое охлаждение может быть таким, что полученный раствор охлаждают до температуры от 15°С до 25°С и далее охлаждают до температуры от -5°С до -20°С; предпочтительно охлаждают до комнатной температуры и далее охлаждают до -18°С.
В некоторых вариантах реализации настоящего изобретения указанный выше способ получения кристаллической Формы В соединения Формулы (III) может дополнительно включать:
(с) фильтрование и сушку.
На стадии (с) сушка может быть проведена при 45°С в вакууме или продувкой воздухом при 45°С при атмосферном давлении.
В настоящем изобретении рентгеновскую порошковую дифрактрограмму образца снимают при следующих условиях:
Прибор: рентгеновский дифрактометр Bruker D2; Условия анализа: 30 кВ 10 мА; Щель: 0,6 мм/3 мм/0,8 мм; Тип анода: Cu; Угловой диапазон: от 5°С до 40°С; Размер шага: 0,1 с/0,02°.
В настоящем изобретении ЯМР 1Н измеряют при следующих условиях:
Организация анализа: Аналитический и испытательный центр Китайского фармацевтического университета; Прибор: спектрометр ядерного магнитного резонанса типа BRUKER AV-500; Растворитель: ДМСО-d6; Внутреннее стандартное вещество: TMS; Температура: 303 K.
В настоящем изобретении термограмму ДСК измеряют при следующих условиях:
Прибор: дифференциальный термический анализатор Mettler типа 1; Диапазон температур: от 30°С до 270°С; Скорость нагрева: 10°С/мин.
В настоящем изобретении ИК-спектр регистрируют при следующих условиях:
Прибор: инфракрасный спектрометр Perkin Elmer spetrum типа 100; Калибровка прибора: волновое число прибора калибруют с помощью полосы поглощения инфракрасного спектра пленки полистирола; Метод: метод прессования таблеток с KBr.
В настоящем изобретении, если конкретно не указано иное, этанол, применяемый в настоящей заявке, представляет собой безводный этанол.
Следует отметить, что в рентгеновской порошковой дифрактрограмме (XRD) дифракционная картина кристаллического соединения обычно характерна для конкретной кристаллической формы. Относительные интенсивности полос (особенно при малых значениях углов) могут изменяться в зависимости от эффектов преимущественной ориентации, обусловленных различиями в состояниях кристаллов, размерах частиц и других условий измерения. Поэтому относительные интенсивности дифракционных пиков не характерны для конкретной кристаллической формы. При оценке того, является ли кристаллическая форма такой же, как известная кристаллическая форма, следует уделять больше внимания относительным положениям пиков, а не их относительным интенсивностям. В дополнение, как и для любой конкретной кристаллической формы, возможна небольшая ошибка в положении пика, которая также хорошо известна в области кристаллографии. Например, при анализе образца положение пика может быть изменено из-за изменения температуры, перемещения образца или калибровки прибора и т.д., и ошибка измерения значения 2θ иногда составляет около ± 0,2°. Соответственно, указанную ошибку следует учитывать при идентификации кристаллической структуры. Обычно положение пика выражают через угол 2θ или через межплоскостное расстояние d на XRD картине, при этом существует простая зависимость преобразования между ними: d=λ/2sinθ, где d представляет собой межплоскостное расстояние, λ представляет длину волны падающего рентгеновского луча, и θ представляет собой угол дифракции. Для той же кристаллической формы того же соединения положение пиков на его XRD дифрактрограмме имеет сходство в целом, а ошибка относительных интенсивностей может быть больше. Кроме того, необходимо отметить, что из-за некоторых факторов, таких как пониженное содержание, части дифракционных линий могут отсутствовать при идентификации смеси. В это время даже одна полоса может быть характерной для данной кристаллической формы, не завися от всех полос образца высокой чистоты.
ДСК применяют для измерения температуры теплового перехода при поглощении или высвобождении тепла вследствие изменения кристаллической структуры или плавления кристалла. При непрерывном анализе одной и той же кристаллической формы одного и того же соединения ошибка температуры теплового перехода и температуры плавления часто находится в диапазоне около ± 5°С, как правило в диапазоне около ± 3°С. Соединение с заданным положением ДСК пика или температурой плавления означает, что положение ДСК пика или температура плавления могут изменяться в диапазоне ± 5°С. ДСК обеспечивает дополнительный метод для распознавания разных кристаллических форм. Разные кристаллические формы могут быть идентифицированы по их отличающимся характерным температурам перехода. Необходимо отметить, что положение ДСК пика или температура плавления смеси могут изменяться в более широком диапазоне. Кроме того, из-за разложения вещества в процессе плавления температура плавления связана со скоростью нагрева.
В настоящем изобретении соединение Формулы (II) может быть получено со способом, раскрытым в CN 201210128875.0.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
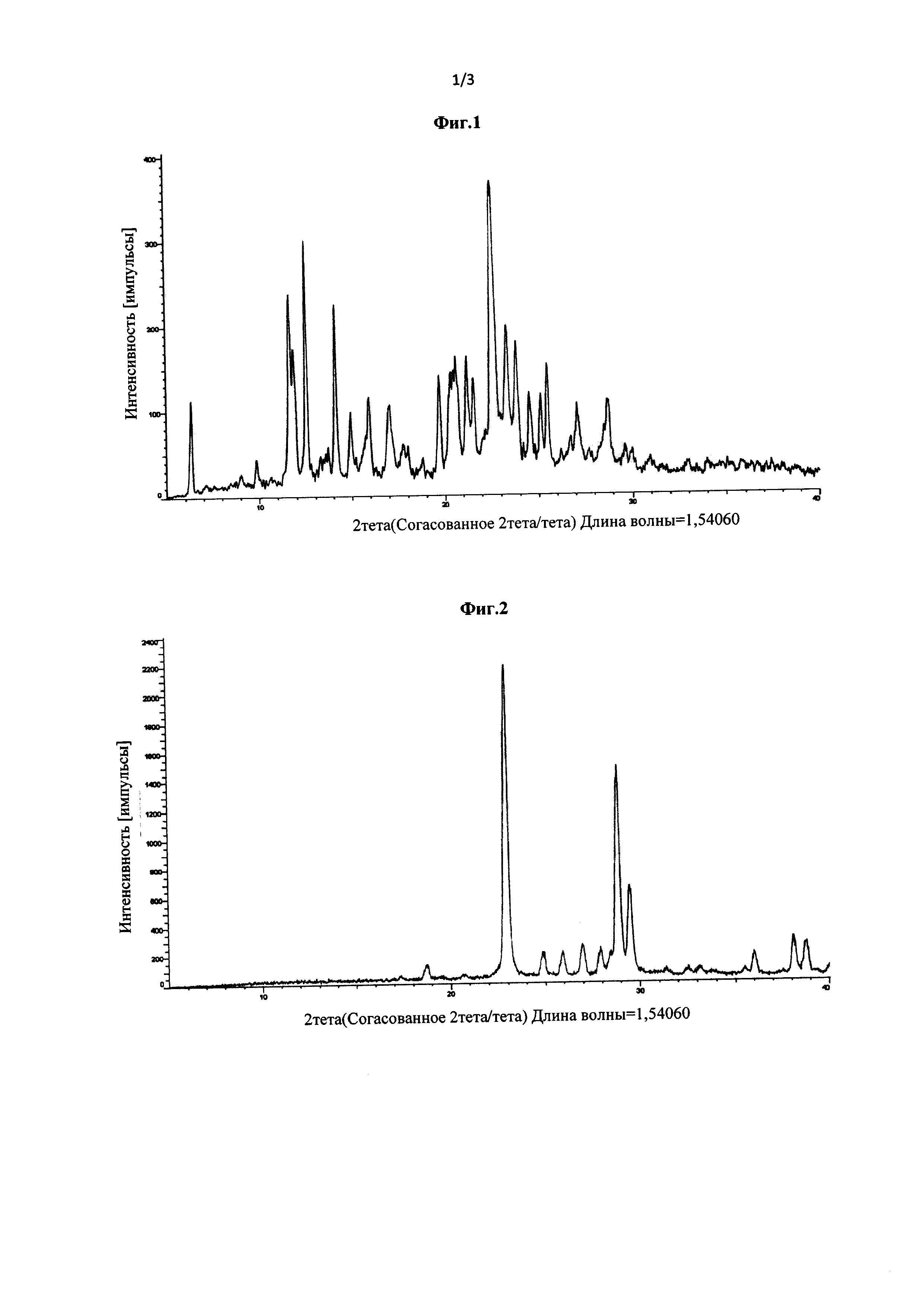
Фиг. 1 представляет собой рентгеновскую порошковую дифрактограмму кристаллической Формы А соединения Формулы (III), полученной в Примере 3.
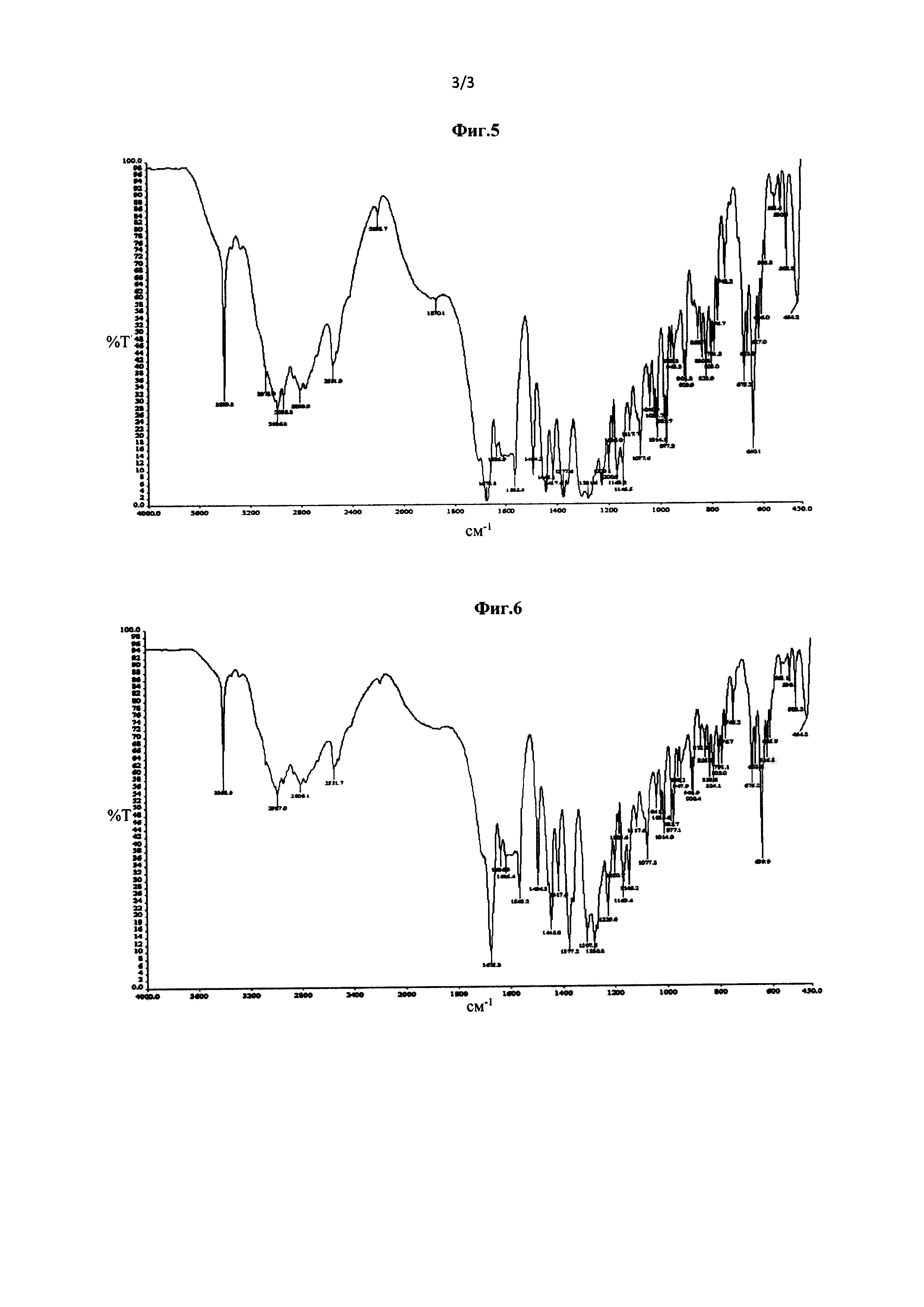
Фиг. 2 представляет собой рентгеновскую порошковую дифрактограмму кристаллической Формы В соединения Формулы (III), полученной в Примере 5.
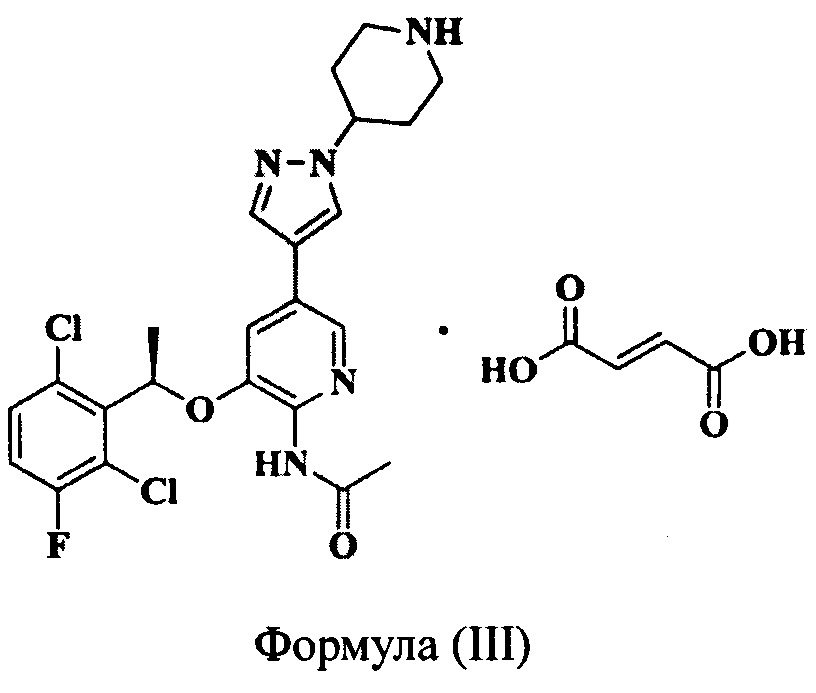
Фиг. 3 представляет собой термограмму дифференциальной сканирующей калориметрии (ДСК) кристаллической Формы А соединения Формулы (III), полученной в Примере 3.
Фиг. 4 представляет собой термограмму дифференциальной сканирующей калориметрии (ДСК) кристаллической Формы В соединения Формулы (III), полученной в Примере 5.
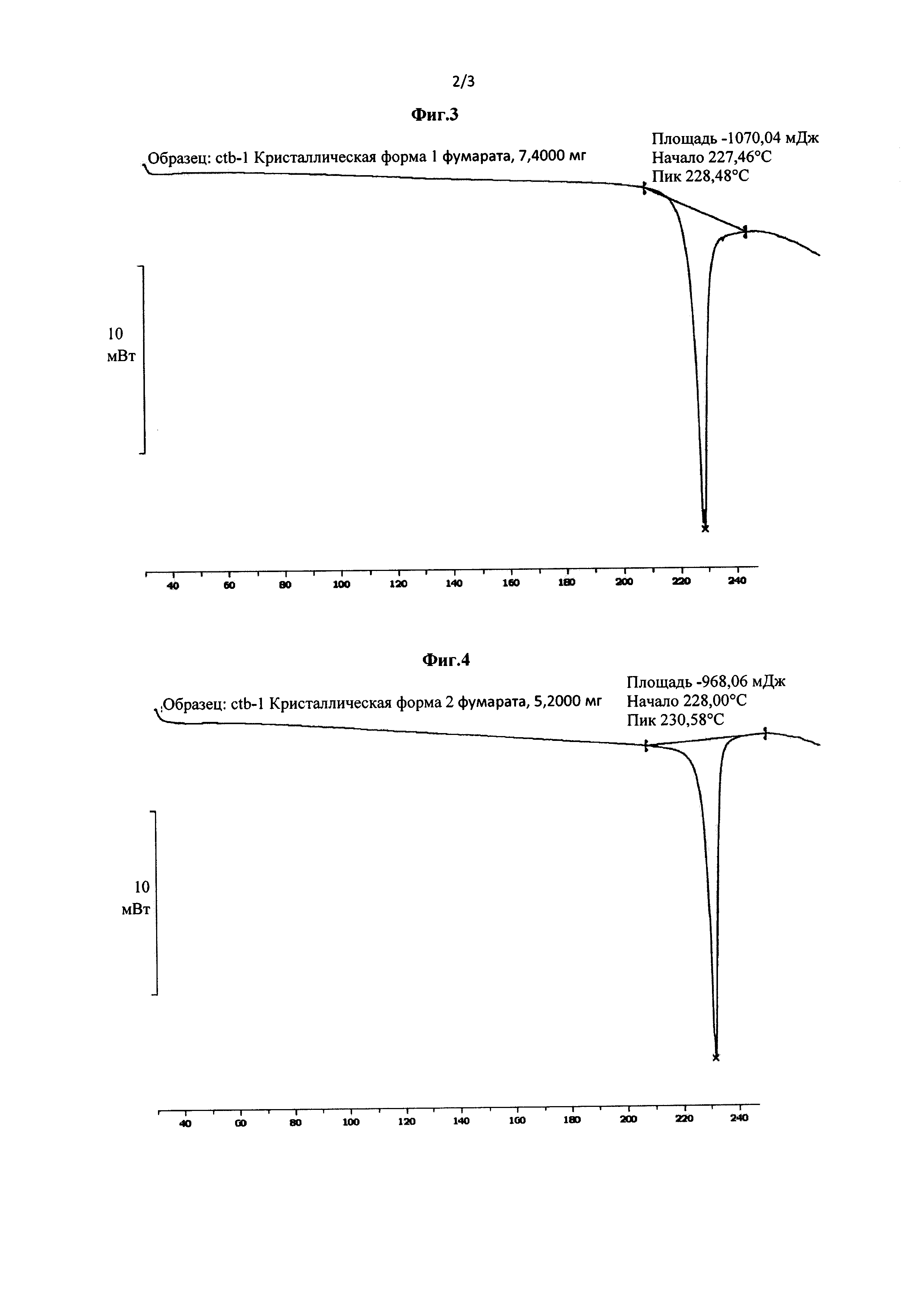
Фиг. 5 представляет собой инфракрасный (ИК) спектр кристаллической Формы А соединения Формулы (III), полученной в Примере 3.
Фиг. 6 представляет собой инфракрасный (ИК) спектр кристаллической Формы В соединения Формулы (III), полученной в Примере 5.
ВАРИАНТЫ РЕАЛИЗАЦИИ ИЗОБРЕТЕНИЯ
Настоящее изобретение будет описано более подробно со ссылкой на следующие примеры, но следующие примеры не ограничивают настоящее изобретение.
Пример 1. Получение соединения Формулы (II)
А. Получение N-ацетил-5-бром-3-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-2-пиридиламина
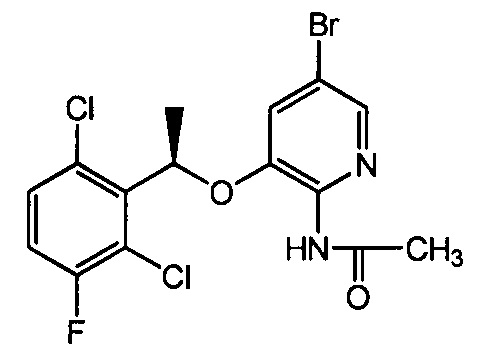
500 мл 5-бром-3-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-2-пиридиламина растворяли в 15 мл дихлорметана. Полученную смесь охлаждали до 0°С. Добавляли 1 мл триэтиламина, и полученную смесь непрерывно перемешивали в течение 5 минут. И затем по каплям добавляли 1,1 эквивалента ацетилхлорида. Полученную смесь нагревали до комнатной температуры, и смесь вступала в реакцию в течение 5 часов. Реакцию останавливали добавлением воды. Полученную смесь экстрагировали дихлорметаном, сушили над безводным сульфатом натрия, фильтровали и упаривали. Полученный сырой продукт очищали при помощи колоночной хроматографии (этилацетат: петролейный эфир = 1: 4) с получением 400 мг желто-белого твердого вещества (выход 72%).
5-Бром-3-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-2-пиридиламин может быть получен в соответствии со способами, описанными в литературе, например, в Organic Process Research & Development, 2011, 15 (5): 1018-1026 или в WO 2007066187 и др.
В. Получение N-ацетил-3-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-5-[1-(4-N-Вос-пиперидил)-1Н-пиразол-4-ил]-2-пиридиламина
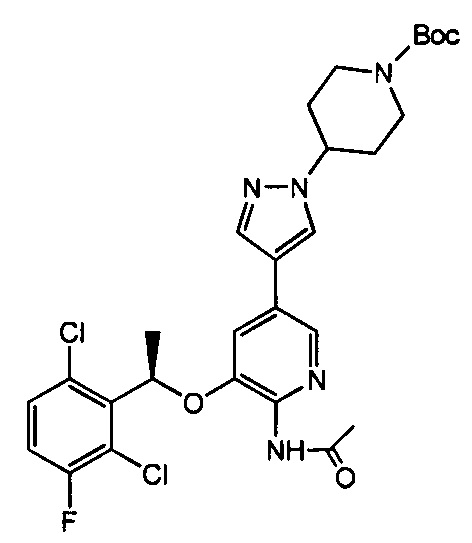
300 мг N-ацетил-5-бром-3-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-2-пиридиламина и 230 мг 1-(4-N-Вос-пиперидил)-4-(4,4,5,5-тетраметил-[1,3,2]диоксаборолан-2-ил)-1Н-пиразола растворяли в 5 мл ДМФА, и полученную смесь добавляли в 1 мл водного раствора, содержащего 300 мг карбоната цезия. Воздух заменяли азотом в течение трех раз и добавляли 20 мг Pd(PPh3)2Cl2, затем воздух снова заменяли азотом в течение трех раз. Полученную реакционную смесь нагревали до 75°С и перемешивали в течение 12 часов. После завершения реакции реакционную смесь охлаждали до комнатной температуры и затем разбавляли добавлением 20 мл этилацетата, фильтровали через целит и промывали этилацетатом. Объединенные фракции этилацетата сушили над безводным сульфатом натрия и затем упаривали. Полученный сырой продукт очищали при помощи колоночной хроматографии (этилацетат: петролейный эфир = 1: 1) с получением 330 мг твердой белой пены (выход 78%).
С. Получение N-ацетил-3-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-5-[1-(4-пиперидил)-1Н-пиразол-4-ил]-2-пиридиламина
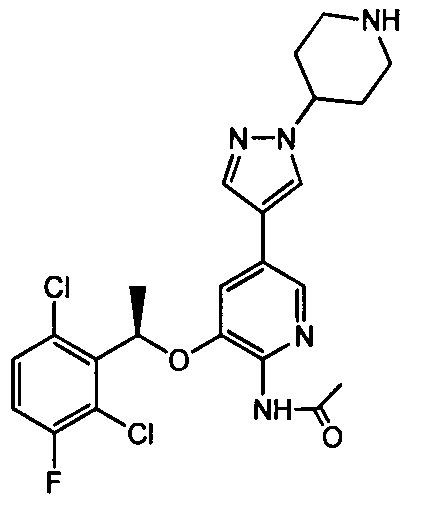
100 мг полученного N-ацетил-3-[(1R)-1-(2,6-дихлор-3-фторфенил)этокси]-5-[1-(4-N-Вос-пиперидил)-1Н-пиразола-4-ил]-2-пиридиламина растворяли в небольшом количестве дихлорметана. 2 мл раствора 4 н. HCl в диоксане добавляли при перемешивании при 0°С, полученную смесь перемешивали в течение 20 минут, и затем растворитель отгоняли при пониженном давлении. Добавляли 10 мл воды, и полученную смесь доводили с помощью бикарбоната натрия до рН=10, экстрагировали с применением дихлорметана и затем сушили, упаривали и очищали при помощи колоночной хроматографии с получением 71 мг белого твердого вещества (выход 85%). МС: m/е 492 (М+1).
Пример 2. Получение соединения Формулы (III)
0,01 моль соединения Формулы (II) растворяли в этаноле и добавляли 8,6 мл предварительно приготовленного 1,4 моль/л раствора фумаровой кислоты в этаноле при перемешивании при 20°С. После проведения реакции в течение 1 часа твердую фазу осаждали из реакционного раствора, фильтровали и сушили при 45°С в вакууме с получением почти белого твердого вещества (выход 85,5%).
ЯМР 1Н (ДМСО-D6) δ (м.д.): 1,79(3Н, -СН3), 2,10(3Н, -СН3), 2,19(4Н, -СН2-), 3,04(2Н, -СН2-), 3,38(2Н, -СН2-), 4,50(1Н, -СН-), 6,16(1Н, -СН-), 6,52(2Н, -СН), 7,38(1H, ArH), 7,44(1Н, ArH), 7,56(1Н, ArH), 7,83(1Н, =CH-N), 8,23(1Н, -CH=N), 8,24(1Н, -NHCO-), 9,39(1Н, ArH), 9-12(3Н, 2*-СООН, -NH-).
Пример 3. Получение кристаллической Формы А соединения Формулы (III)
2,0 г соединения Формулы (III) растворяли в 20 мл метанола. Полученный раствор нагревали до 50°С при перемешивании, и затем добавляли 40 мл ацетона. Полученную смесь охлаждали до 0°С и выдерживали в течение 48 ч, и кристаллы осаждали, фильтровали и сушили при 45°С в вакууме с получением кристаллической Формы А (выход 87,7%). Рентгеновская порошковая дифрактограмма полученного кристалла показана на фиг. 1. Термограмма дифференциальной сканирующей калориметрии (ДСК) полученного кристалла показана на фиг. 3. Инфракрасный (ИК) спектр полученного кристалла показан на фиг. 5.
Пример 4. Получение кристаллической Формы А соединения Формулы (III)
2,0 г соединения Формулы (III) растворяли в 20 мл метанола. Полученный раствор нагревали до 50°С при перемешивании и затем добавляли 40 мл тетрагидрофурана. Полученную смесь охлаждали до 0°С и выдерживали в течение 48 ч, и кристаллы осаждали, фильтровали и сушили продувкой воздухом при 45°С при атмосферном давлении. Анализ с использованием рентгеновской порошковой дифрактометрии показал, что полученный кристалл представляет собой кристаллическую Форму А (выход 86,0%).
Пример 5. Получение кристаллической Формы В соединения Формулы (III)
5,0 г соединения Формулы (III) добавляли в 50 мл безводного метанола. Полученную смесь нагревали с обратным холодильником при перемешивании, так что соединение Формулы (III) растворялось, и полученный раствор постепенно охлаждали до комнатной температуры и затем далее охлаждали до -18°С. Смесь выдерживали при низкой температуре в течение 48 часов, и кристаллы осаждали, фильтровали, сушили продувкой воздухом при 45°С при атмосферном давлении в течение 4 часов с получением кристаллической Формы В (выход 86,4%). Рентгеновская порошковая дифрактограмма полученного кристалла показана на фиг. 2. Термограмма дифференциальной сканирующей калориметрии (ДСК) полученного кристалла показана на фиг. 4. Инфракрасный (ИК) спектр полученного кристалла показан на фиг. 6.
Эталонный Пример 1 Получение других аддуктов соединения Формулы (II) с кислотами
Ряд аддуктов соединения Формулы (II) с кислотами получили с применением способа согласно Примеру 2 с заменой фумаровой кислоты разными органическими или неорганическими кислотами.
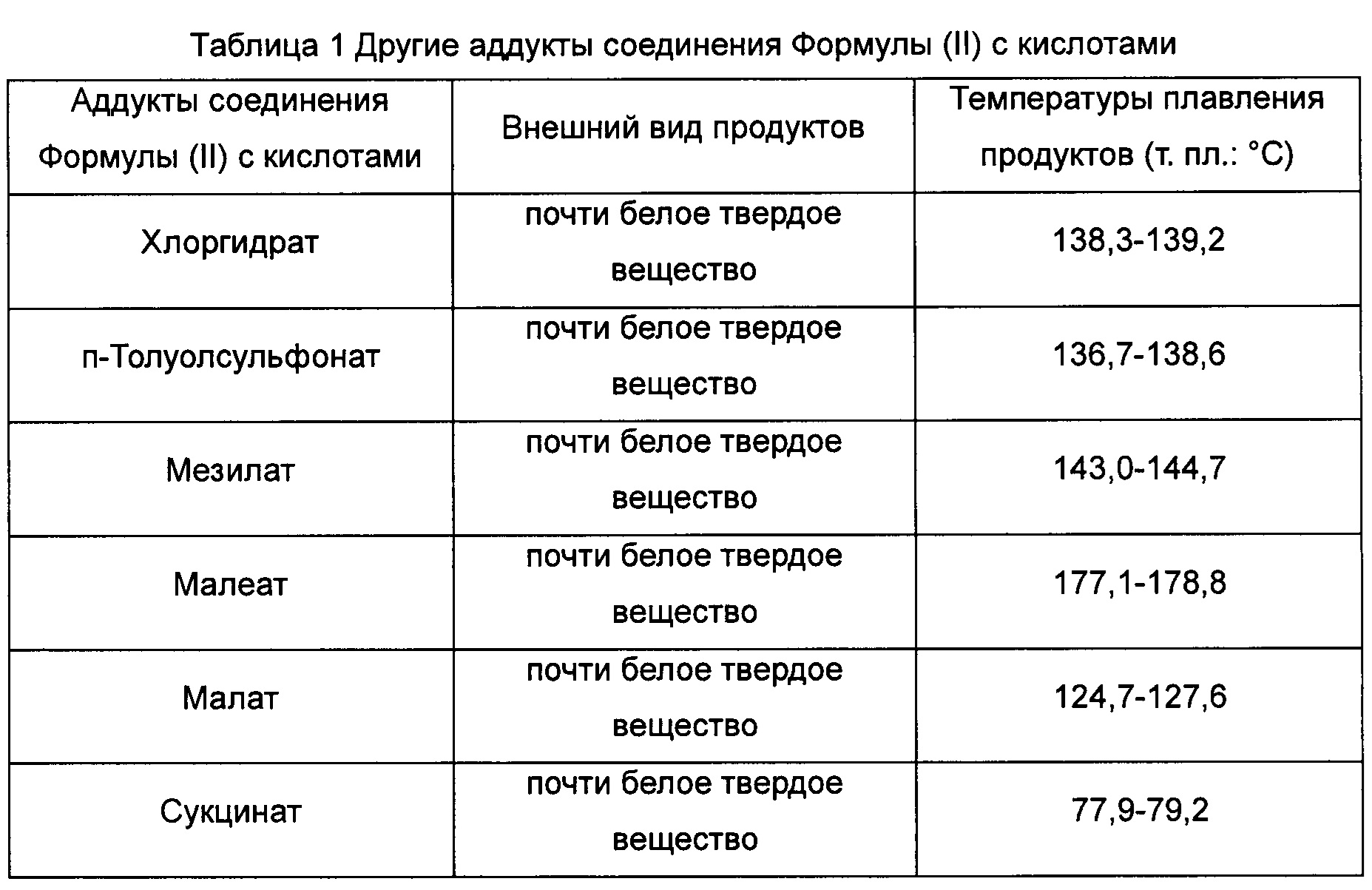
Пример 6. Испытание на гигроскопичность
Соединение Формулы (II) и различные соли соединения Формулы (II), полученные в Примере 2 и в эталонном Примере 1, анализировали в соответствии с «Основополагающими принципами испытания на гигроскопичность лекарственных средств», описанными в китайской фармакопее, издание 2010 года, Часть II, Приложение XIX J. Повышенные массы образцов вследствие их гигроскопичности были рассчитаны 5 соответственно, и результаты испытания показаны в Таблице 2.
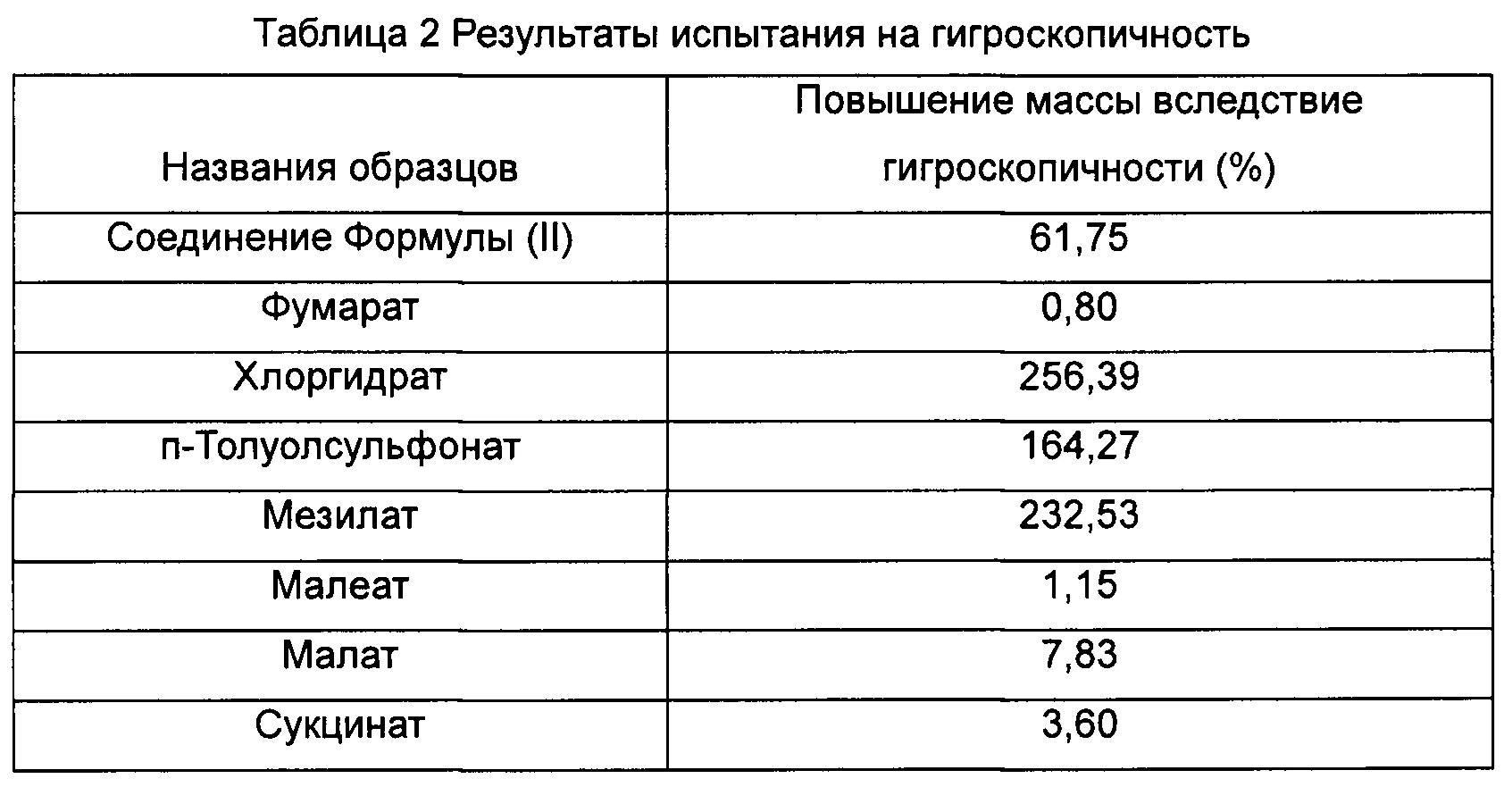
Пример 7. Фармакокинетическое исследование аддуктов соединения Формулы (II) с кислотами
Двадцать семь здоровых самцов крыс линии Спрег-Доули, весом 200 г-220 г, кормили стандартным рецептурным гранулированным кормом для крыс ежедневно в определенное время. Крыс не кормили в течение 12 ч перед экспериментом и снова кормили через 4 часа после введения лекарственного средства. Крысы могли свободно пить воду до и после эксперимента, а также в ходе эксперимента. Крыс произвольным образом разделили на 9 групп. Однократная доза Кризотиниба была интрагастрально введена первой группе; однократная доза соединения Формулы (II) была интрагастрально введена второй группе; и однократные дозы соединения Формулы (III), и гидрохлорида, п-толуолсульфоната, мезилата, малеата, малата и сукцината соединения Формулы (II) были интрагастрально введены с третьей группы по девятую группу, соответственно. Все дозы, введенные 9 группам крыс, составляли 0,11 ммоль/кг, и от 0,2 до 0,3 мл крови отбирали из венозного сплетения глазного дна перед введением лекарственного средства (0 ч) и через 0,5 ч, 1 ч, 2 ч, 4 ч, 6 ч, 8 ч, 10 ч и 24 ч после введения лекарственного средства. Гепарин применяли для антикоагуляции крови, и кровь центрифугировали для отделения плазмы крови. 0,1 мл плазмы крови точно измеряли и добавляли в пробирку с замком Eppendorf, и затем добавляли 1,2 мл этилацетата. Полученную смесь равномерно перемешивали с высокой скоростью в течение 5 мин при помощи Вортекс-миксера и центрифугировали в течение 5 мин (8000 обмин-1). Надосадочную жидкость собирали, и растворитель выпаривали при 30°С азотом на приборе продувки азотом. Осадок растворяли в 100 мкл подвижной фазы, равномерно перемешивали с высокой скоростью в течение 1 мин при помощи Вортекс-миксера и центрифугировали в течение 5 мин (14000 об мин-1). 80 мкл надосадочной жидкости переносили в виалу для образца, и 10 мкл надосадочной жидкости вводили для определения при помощи ВЭЖХ, и регистрировали хроматограмму ВЭЖХ. Результаты показаны в Таблице 3:
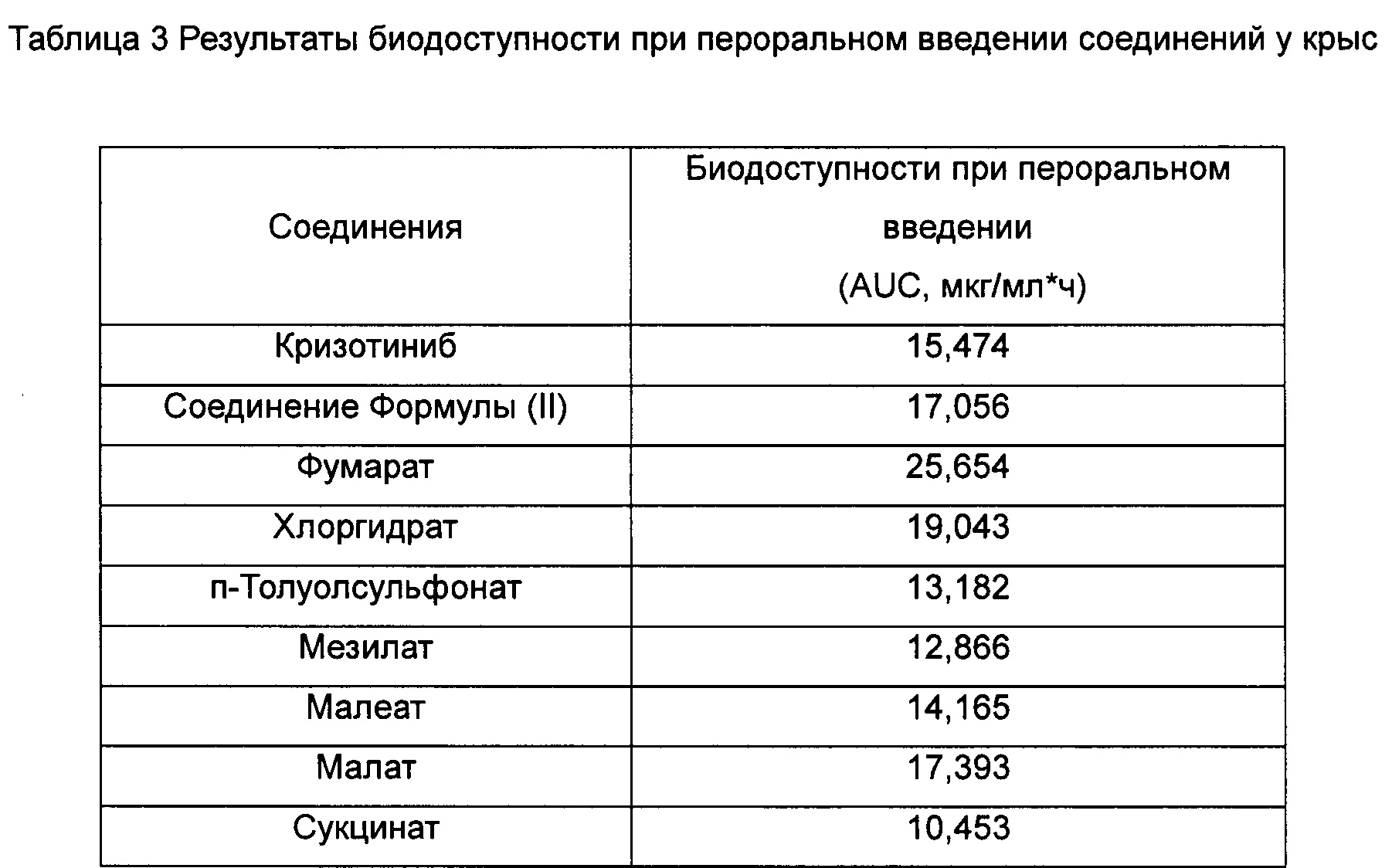
Условия ВЭЖХ определения были следующими:
Жидкостный хроматограф: Shimadzu сверхбыстрая высокоэффективная жидкостная хроматография на Prominence UFLC XR
Колонка анализа: Shim-pack XR-ODS II (2,0 *75 мм 2,2 мкм)
Подвижная фаза: 0,1% раствор муравьиной кислоты, содержащий 5 мМ формиата аммония/ацетонитрила = 80/20 (об./об.),
Скорость потока: 0,25 мл/мин, Температура колонки: 40°С
Объем пробы: 10 мкл, Время анализа: 10,5 мин
Спектральный диапазон PDA: от 260 нм до 275 нм, Температура детекторной ячейки: 40°С
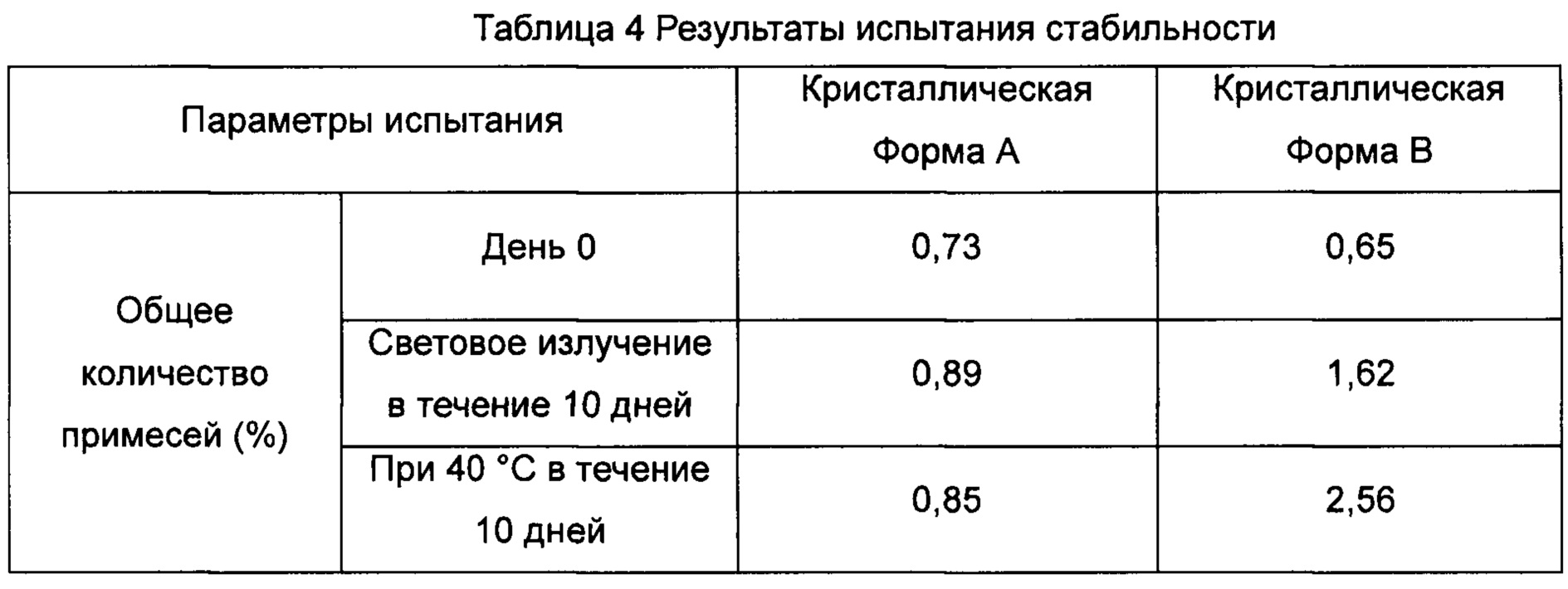
Пример 8 Испытание стабильности
В соответствии со способом испытания факторов воздействия на действующие фармацевтические вещества, описанного в китайской фармакопее, издание 2010 года, часть II, приложение XIX С, кристаллическую Форму А соединения Формулы (III), полученную в Примере 3, и кристаллическую Формы В соединения Формулы (III), полученную в Примере 5, подвергали высокотемпературному испытанию (40°С ± 2°С, относительная влажность составляла 75% ± 5%) и испытанию ярким световым излучением (4500 лк ± 500 лк) соответственно. Испытания проводили в течение 10 дней. Образцы были взяты на 0 и 10 дни для измерения общего количества примесей, чтобы определить их стабильность. Результаты испытания показаны в Таблице 4.
Замещенный 2-аминопиридин в качестве ингибитора протеинкиназы

