Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ДОЛАСТАТИНА 10 И АУРИСТАТИНОВ
Вид РИД
Изобретение
Объект настоящего изобретения относится к новым производным доластатина 10 и ауристатинов, к способам их получения, к содержащим их фармацевтическим композициям и к их применению в качестве лекарственного препарата, в частности, при лечении рака.
Доластатин 10 (D10) представляет собой производное цитотоксического пептида, выделенного из морского моллюска (Dolabella auricularia), абсолютная конфигурация которого была определена и впоследствии подтверждена после полного синтеза продукта (Pettit G.R.J. Am. Chem. Soc. 1987, 109, 6883; Pettit G.R.J. Am. Chem. Soc. 1987, 109, 7581; Pettit, G.R. Heterocycles 1989, 28, 553; Pettit, G.R.J. Am. Chem. Soc. 1989, 111, 5015; Pettit G.R.J. Am. Chem. Soc. 1991, 113, 6692). D10 образован из 5 звеньев, называемых долавалином (Dov), валином (Val), долаизолейцином (Dil), долапроином (Dap) и долафенином (Doe). Несколько аналогов этого соединения синтезировано путем модификации природы составляющих его аминокислот (Pettit G.R.J. Med. Chem. 1990, 33, 3133; Miyazaki K. Peptide Chemistry 1993, 31, 85; Miyazaki K. Chem. Pham. Bull. 1995, 43, 1706). Произведены также модификации его С-концевой части (правого конца), которые привели к различным производным, включающим ауристатин Е или F (Pettit G.R. Anticancer Drug Design, 1998, 13, 243; Pettit G.R. Antimicrobial Agents And Chemotherapy, 1998, 2961).
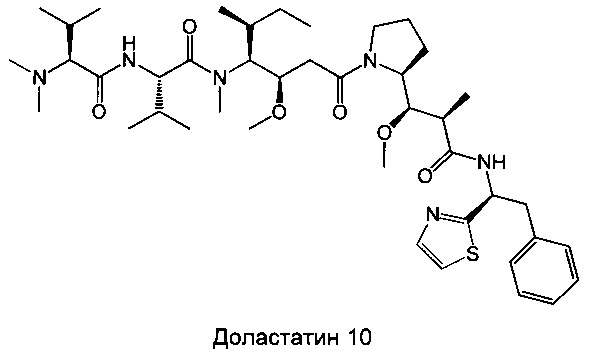
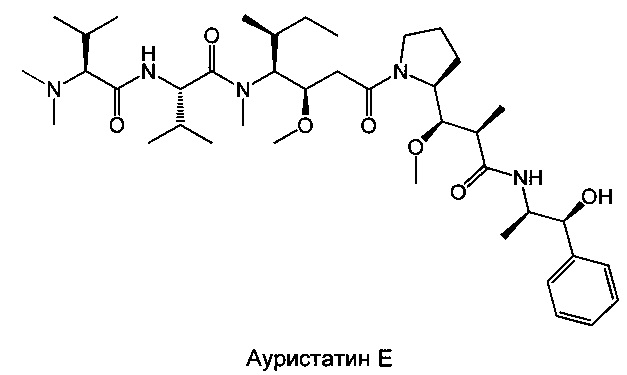
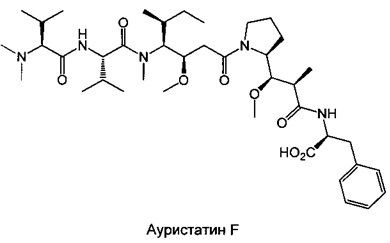
Настоящее изобретение сосредоточено на модификации N-концевой части (левого конца) производных доластатина 10 и ауристатинов Е и F. Несколько примеров, опубликованных в литературе, по модификациям, произведенным в этом положении, привело к потере активности (Miyazaki K. Chem. Pham. Bull. 1995, 43, 1706). Соединения, раскрытые в настоящем изобретении, отличаются от предшествующего уровня техники за счет их оригинальных химических структур, а также за счет их примечательного биологического свойства, которое оказалось абсолютно неожиданным с учетом элементов, опубликованных в литературе. Результат этих примечательных активностей делает эти соединения подходящими для применения при лечении рака.
Кроме того, эти соединения обладают преимуществом в том, что они являются и активными в качестве цитотоксических агентов, и более растворимыми, чем исходные соединения.
Таким образом, объектом настоящего изобретения является соединение приведенной ниже формулы (I):
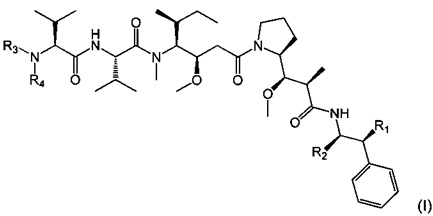
где:
- R1 представляет собой Н или ОН,
- R2 представляет собой группу: (С1-C6)алкил (например, метил), СООН, СОО-((С1-С6)алкил) (такой как СООМе) или тиазолил (такой как тиазол-2-ил),
- R3 представляет собой Н или (С1-С6)алкильную группу (такую как метил), в частности, (С1-С6)алкильную группу, и
- R4 представляет собой:
арил-(С1-С8)алкильную группу, замещенную одной или более групп (в частности, одной, предпочтительно на арильной части), выбранных из групп ОН и NR9R10, где R9 и R10, каждый независимо от другого, представляет собой Н или (C1-С6)алкильную группу (такую как метил),
или его фармацевтически приемлемая соль, гидрат или сольват.
Радикалы R2-R4 и, в частности, R4, могут представлять собой хиральные группы, и могут находиться в форме их различных стереоизомеров и необязательно в форме смеси стереоизомеров.
Под «стереоизомером» в значении настоящего изобретения подразумевают геометрический изомер или оптический изомер.
Геометрические изомеры являются результатом различного положения заместителей на двойной связи, которая может, таким образом, иметь Z или Е конфигурацию.
Оптические изомеры являются результатом, в частности, различного положения в пространстве заместителей на атоме углерода, содержащем 4 различных заместителя. В этом случае атом углерода образует хиральный или асимметрический центр. Оптические изомеры включают диастереоизомеры и энантиомеры. Оптические изомеры, являющиеся отображениями друг друга в зеркале, но которые не могут налагаться друг на друга, называют «энантиомерами». Оптические изомеры, не являющиеся налагаемыми отображениями друг друга в зеркале, называют «диастереоизомерами».
Смесь, содержащую равные количества двух индивидуальных энантиомерных форм противоположной хиральности, называют «рацемической смесью».
В настоящем изобретении под «фармацевтически приемлемым» подразумевают то, что можно применять при получении фармацевтической композиции, является по существу безопасным, нетоксичным и ни биологически, ни иначе нежелательным и приемлемым как для ветеринарного применения, так и для медицинского фармацевтического применения.
Под «фармацевтически приемлемой солью, гидратом или сольватом» соединения подразумевают соль, гидрат или сольват, которые являются фармацевтически приемлемыми, как определено в данном документе, и которые обладают желаемой фармакологической активностью исходного соединения.
Фармацевтически приемлемые соли, в частности, включают в себя:
(1) соли присоединения фармацевтически приемлемой кислоты, образованные с фармацевтически приемлемыми неорганическими кислотами, такими как соляная, бромисто-водородная, фосфорная, серная и подобные кислоты; или образованные с фармацевтически приемлемыми органическими кислотами, такими как уксусная, трифторуксусная, пропионовая, янтарная, фумаровая, яблочная, винная, лимонная, аскорбиновая, малеиновая, глутаминовая, бензойная, салициловая, толуолсульфоновая, метансульфоновая, стеариновая, молочная и подобные кислоты; и
(2) соли присоединения фармацевтически приемлемого основания, которые образуются, когда протон кислоты, присутствующий в исходном соединении, либо замещается ионом металла, например, ионом щелочного металла, ионом щелочноземельного металла или ионом алюминия; либо координируется с фармацевтически приемлемым органическим основанием, таким как лизин, аргинин и тому подобное; либо с фармацевтически приемлемым неорганическим основанием, таким как гидроксид натрия, гидроксид калия, гидроксид кальция и тому подобное.
Эти соли могут быть получены из соединений по изобретению, содержащих основную или кислотную функциональную группу, и соответствующих кислот или оснований с использованием традиционных химических способов.
Соединения формулы (I) по изобретению предпочтительно находятся в солевой форме и, в частности, в форме фармацевтически приемлемой соли присоединения кислоты.
Предпочтительно соединения формулы (I) согласно настоящему изобретению находятся в форме фармацевтически приемлемой соли присоединения кислоты, где кислота, возможно, представляет собой трифторуксусную кислоту, уксусную кислоту или соляную кислоту, например, и в частности, трифторуксусную кислоту.
Сольваты включают в себя традиционные сольваты, полученные на последней стадии получения соединений по изобретению благодаря присутствию растворителя, где растворитель, возможно, представляет собой, например, этанол.
Под «алкилом» в настоящем изобретении подразумевают прямоцепочечную или разветвленную насыщенную углеводородную цепь. Например, можно упомянуть метильную, этильную, пропильную, изопропильную, бутильную, изобутильную, втор-бутильную, трет-бутильную, пентильную или гексильную группы.
Под «(Сx-Сy)алкилом» в значении настоящего изобретения подразумевают алкильную цепь, такую как определено выше, содержащую от x до y атомов углерода. Таким образом, (С1-С6)лкильная группа представляет собой алкильную цепь, имеющую от 1 до 6 атомов углерода.
Под «арилом» в значении настоящего изобретения подразумевают ароматическую углеводородную группу, предпочтительно имеющую от 6 до 10 атомов углерода и способную содержать одно или два конденсированных кольца. Например, можно указать фенил или нафтил. Предпочтительно эта группа представляет собой фенил.
Под «арил-(С1-С8)алкилом» в контексте настоящего изобретения подразумевают арильную группу, такую как определено выше, связанную с остальной частью молекулы через алкильную группу, такую как определено выше, и содержащую от 1 до 8, в частности, от 1 до 6, преимущественно от 1 до 4, предпочтительно 1 или 2 атома углерода. Арильная группировка предпочтительно представляет собой фенильную группировку. (С1-С8)алкильная группировка преимущественно представляет собой (С1-С4)алкил, предпочтительно (С1-С2)алкил. В частности, арил-(С1-С8)алкильная группа представляет собой бензильную или фенетильную группу.
Среди соединений по изобретения один особенно приемлемый класс соединений соответствует соединениям формулы (I), в которых R1 представляет собой ОН, и R2 представляет собой (С1-С6)алкильную группу, такую как метил.
Другой особенно приемлемый класс соединений соответствует соединениям формулы (I), в которых R1 представляет собой атом водорода, и R2 представляет собой тиазол (в частности, тиазол-2-ильную группу).
Другой класс особенно приемлемых соединений соответствует соединениям формулы (I), в которых R1 представляет собой атом водорода, и R2 представляет собой СОО(С1-С6)алкильную группу, такую как СООМе.
Другой класс особенно приемлемых соединений соответствует соединениям формулы (I), в которых R1 представляет собой атом водорода, и R2 представляет собой группу СООН.
Таким образом, соединения по изобретению преимущественно представляют собой соединения формулы (I), в которых:
- R1=OH, и R2=Me (метил) или
- R1=H, и R2=COOH, СООМе или тиазол-2-ил.
Согласно одному конкретному воплощению настоящего изобретения R2 более конкретно представляет собой метильную группу, СООН, СООМе или тиазол-2-ильную группу.
Предпочтительно R1 представляет собой Н, и R2 представляет собой СООН или СОО(С1-С6)алкил, в частности, СООН или СООМе.
Согласно первому предпочтительному воплощению изобретения R1 представляет собой Н, и R2 представляет собой СООН.
Согласно второму предпочтительному воплощению изобретения R1 представляет собой Н, и R2 представляет собой СООМе.
R3 конкретно представляет собой Н или метильную группу, преимущественно метильную группу.
R4 преимущественно представляет собой арил-(С1-С8)алкильную группу, конкретно арил-(С1-С4)алкильную группу, такую как арил-(С1-С2)алкильная группа, замещенную одной группой, выбранной из ОН и NR9R10, и, в частности, представляющей собой NR9R10.
R4 преимущественно представляет собой арил-(С1-С8)алкильную группу, конкретно арил-(C1-С4)алкильную группу, такую как арил-(С1-С2)алкильная группа, замещенную одной группой на арильной группировке, выбранной из ОН и NR9R10, и, в частности, представляющей собой NR9R10.
Арильная группа преимущественно представляет собой фенильную группу.
Таким образом, R4 может, в частности, представлять собой фенил-(С1-С2)алкил, замещенный одной группой (предпочтительно на фенильной группировке), выбранной из ОН и NR9R10, и, в частности, представляющей собой NR9R10.
R4 может, таким образом, иметь следующую формулу:
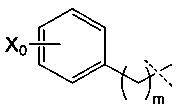
где Х0 представляет собой ОН или NR9R10, в частности, NR9R10, и m представляет собой целое число, составляющее от 1 до 8, конкретно от 1 до 4, и преимущественно представляет собой 1 или 2.
Согласно предпочтительному воплощению изобретения R4 имеет следующую формулу:
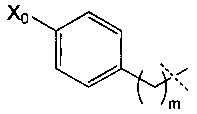
где Х0 и m являются такими, как определено выше, и, в частности, где X0=NR9R10 и m=1 или 2.
R4 может быть, в частности, выбран из:
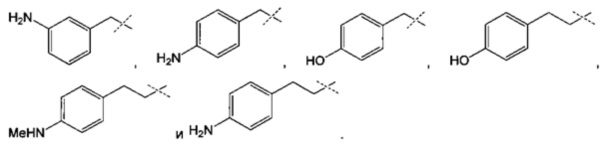
Преимущественно соединение формулы (I) выбрано из соединений 11-15, 19-20, 23-24, 27-29, 49-51 и 61-64, описанных ниже в примерах.
Следующим объектом настоящего изобретения является соединение формулы (I), такое как определено выше, применяемое в качестве лекарственного препарата, в частности, для лечения или профилактики рака или доброкачественных пролиферативных расстройств.
Настоящее изобретение также относится к применению соединение формулы (I), такого как определено выше, для получения лекарственного препарата, в частности, предназначенного для лечения или профилактики рака или доброкачественных пролиферативных расстройств.
Настоящее изобретение также относится к способу лечения или профилактики рака или доброкачественных пролиферативных расстройств, включающему введение человеку, нуждающемуся в этом, эффективного количества соединения формулы (I), такого как определено выше.
Рак, подлежащий лечению или профилактике, представляет собой более конкретно рак легкого, поджелудочной железы, кожи, головы, шеи, матки, яичников, ануса, желудка, ободочной кишки, молочной железы, пищевода, тонкого кишечника, щитовидной железы, лимфатической системы, предстательной железы, почки или мочевого пузыря, либо острый или хронический лейкоз, либо комбинацию двух или более из этих раков.
Под доброкачественными пролиферативными расстройствами подразумевают пролиферативные расстройства, не вызывающие метастазов, или еще не прогрессирующие в направлении рака (предраковые опухоли).
Следующим объектом настоящего изобретения является фармацевтическая композиция, содержащая соединение формулы (I), такое как определено выше, и по меньшей мере один фармацевтически приемлемый эксципиент.
Активный ингредиент можно вводить животным или людям в единичных дозированных формах введения в смеси с традиционными фармацевтическими носителями. Подходящие единичные дозированные формы введения включают формы введения посредством перорального пути, формы для сублингвального или трансбуккального введения, формы введения посредством парентерального пути (подкожного, внутрикожного, внутримышечного или внутривенного), формы для местного введения (на кожу и слизистую оболочку, включая интраназальное и внутриглазное введение) и формы для ректального введения.
Такие композиции могут иметь форму твердого вещества, жидкости, эмульсии, лосьона или крема.
В качестве твердых композиций для перорального введения можно применять таблетки, пилюли, порошки (твердые или мягкие желатиновые капсулы) или гранулы. В этих композициях активный ингредиент по изобретению смешивают с одним или более инертных разбавителей, таких как крахмал, целлюлоза, сахароза, лактоза или диоксид кремния, в потоке аргона. Эти композиции могут также содержать вещества, отличающиеся от разбавителей, например, одно или более смазывающих веществ, таких как стеарат магния или тальк, красящее вещество, покрытие (таблетки с покрытием) или глянцевое покрытие.
В качестве жидких композиций для перорального введения можно применять растворы, суспензии, эмульсии, сиропы и эликсиры, которые являются фармацевтически приемлемыми и содержат инертные разбавители, такие как вода, этанол, глицерин, растительные масла или вазелиновое масло. Эти композиции могут содержать вещества, отличающиеся от разбавителей; например, увлажняющие, подслащивающие, загущающие, корригирующие или стабилизирующие препараты.
Стерильные композиции для парентерального введения предпочтительно представляют собой водные или неводные растворы, суспензии или эмульсии. В качестве растворителя или основы можно применять воду, пропиленгликоль, полиэтиленгликоль, растительные масла, в частности, оливковое масло, инъекционные органические сложные эфиры, например, этилолеат или другие подходящие органические растворители. Эти композиции могут также содержать адъюванты, в частности, увлажняющие, изотонические, эмульгирующие, диспергирующие и стабилизирующие средства. Стерилизацию можно проводить несколькими способами, например, путем обеззараживающего фильтрования, путем включения стерилизующих средств в композицию, путем облучения или путем нагревания. Их можно также готовить в форме твердых стерильных композиций, которые можно растворять в момент применения в стерильной воде или любой другой инъекционной стерильной среде.
Композиции для ректального введения представляют собой суппозитории или ректальные капсулы, которые в дополнение к активному ингредиенту содержат эксципиенты, такие как масло какао, полусинтетические глицериды или полиэтиленгликоли.
Композиции для местного введения могут, например, представлять собой кремы, лосьоны, глазные капли, жидкости для полоскания рта, назальные капли или спреи.
Дозы зависят от желаемого эффекта, от продолжительности лечения и от применяемого пути введения. Как правило, врач определит подходящую дозу относительно возраста, массы и всех других факторов, конкретных для субъекта, подлежащего лечению.
В фармацевтических композициях согласно настоящему изобретению может содержаться другой активный ингредиент. В частности, он может представлять собой противораковое средство и, в частности, цитотоксическое противораковое средство, такое как навельбин, винфлунин, таксол, таксотер, 5-фторурацил, метотрексат, доксорубицин, камптотецин, гемцитабин, этопозид, цисплатин или кармустин (также называемый BCNU); или гормональное противораковое средство, такое как тамоксифен или медроксипрогестерон.
Радиационную терапию (рентгеновские лучи или гамма-лучи) можно также сочетать с введением соединения по настоящему изобретению. Такое облучение можно применять с использованием внешнего источника или путем имплантации крошечных внутренних источников радиоактивности.
Настоящее изобретение также относится к получению соединений формулы (I) согласно изобретению с использованием общих способов, описанных в приведенных ниже схемах синтеза, при необходимости необязательно с добавлением любой стандартной операции, описанной в литературе или хорошо известной специалистам в данной области техники, либо описанной в примерах в экспериментальном разделе.
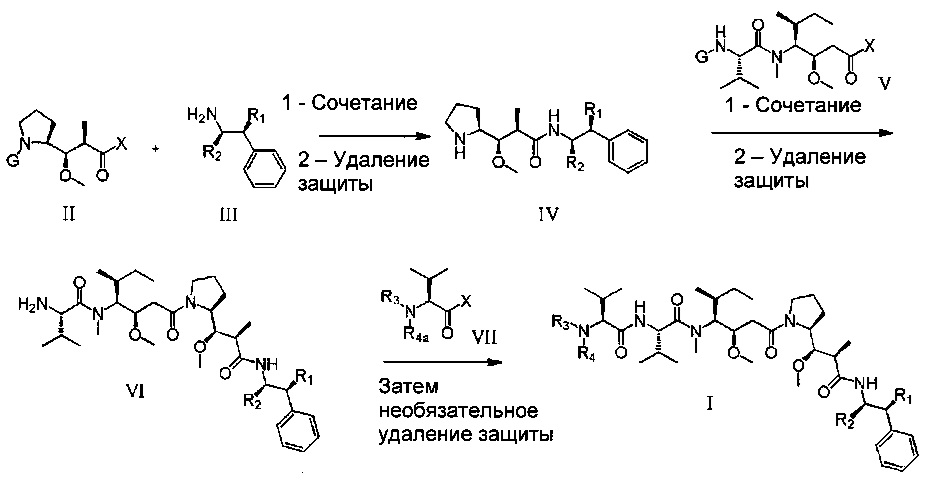
Схема 1
На схеме 1 проиллюстрирован первый общий способ, который можно применять для получения соединений формулы (I). В приведенных выше формулах R1, R2 и R3 являются такими, как определено выше, R4a представляет собой группу R4, такую как определено выше, необязательно в защищенной форме, и G представляет собой защитную группу.
Первая стадия состоит в конденсации соединения (II), защищенного на его аминной функциональной группе защитной группой G, с соединением (III). X может представлять собой уходящую группу, такую как атом хлора. В этом случае первая стадия состоит во взаимодействии между хлорангидридом и амином. Эту реакцию можно проводить с использованием способов и методов, хорошо известных специалистам в данной области техники. При одном особенно приемлемом способе вызывают взаимодействие этих двух химических соединений в присутствии органического или неорганического основания, например, Et3N, iPr2NEt, пиридина, NaH, Cs2CO3, K2СO3, в растворителе, таком как ТГФ, дихлорметан, ДМФ, ДМСО, при температуре, в частности, от -20°С до 100°С. X может также представлять собой гидроксил (ОН). В этом случае первая стадия представляет собой реакцию конденсации между карбоновой кислотой (II) и амином (III). Эту реакцию можно проводить путем следования способам и методам, хорошо известным специалистам в данной области техники. При одном особенно приемлемом способе вызывают взаимодействие этих двух химических соединений в присутствии агента сочетания, такого как 1-(3-диметиламинопропил)-3-этил-карбодиимид (EDC), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-он, третичный амин, такой как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре, в частности, от -15°С до 40°С. При другом, особенно приемлемом способе вызывают взаимодействие этих двух химических соединений в присутствии диэтилфосфорцианидата (DEPC), третичного амина, такого как триэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре от -15°С до 40°С. Другой особенно приемлемый способ состоит в том, чтобы вызвать взаимодействие этих двух химических соединений в присутствии О-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфата (HATU), третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре от -15°С до 100°С.
После удаления защиты промежуточного соединения с использованием методов, хорошо известных специалистам в данной области техники («Protective Groups in Organic Synthesis», T.W. Greene, John Wiley & Sons, 2006 и «Protecting Groups», P.J. Kocienski, Thieme Verlag, 1994), соединение (IV) можно конденсировать с соединением (V), следуя способам и методам, описанным выше, что приводит к соединению (VI) после стадии удаления защиты. Затем это соединение после конденсации с промежуточным соединением (VII) и необязательного удаления защиты может приводить к образованию соединений формулы (I). Соединение (VI) можно также подвергать сочетанию с соединением (VII'), в котором R'3 представляет собой предшественник R3, в частности, группу R3, защищенную защитной группой. Сочетание с последующим удалением защиты группы R'3 с получением R3 cмoжнo проводить, следуя таким же методикам, как описано выше.
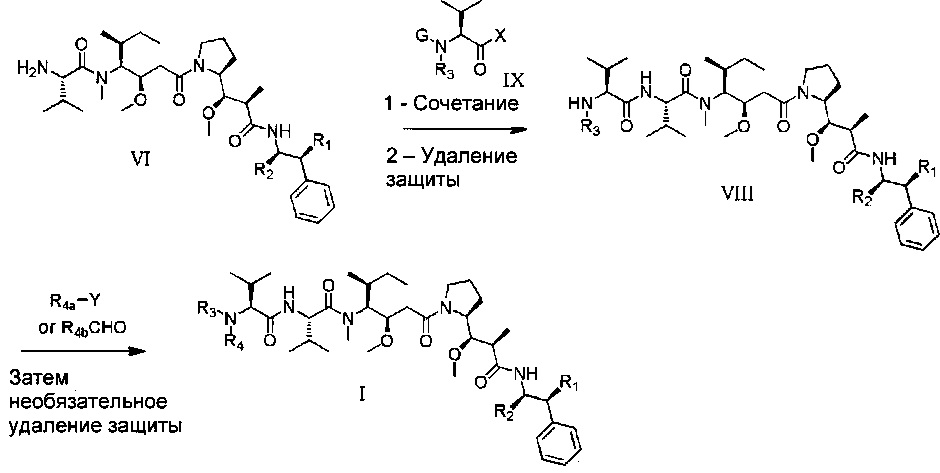
Схема 2
На схеме 2 проиллюстрирован второй общий способ, который можно использовать для получения соединений формулы (I). В приведенных выше общих формулах R1, R2 и R3 являются такими, как определено выше, R4a представляет собой группу R4, такую как определено выше, необязательно в защищенной форме, R4b представляет собой предшественник группы R4, и G представляет собой защитную группу.
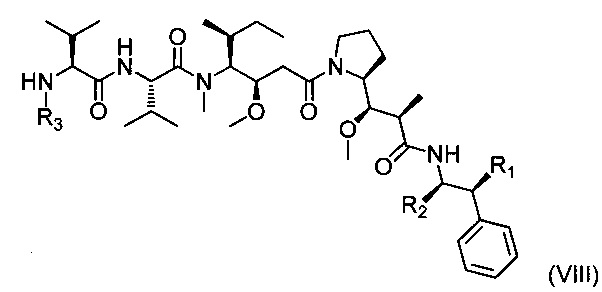
На первой стадии соединение (IX), защищенное на его аминной функциональной группе защитной группой G, конденсируют с соединением (VI). X может представлять собой уходящую группу, например, атом хлора. В данном случае первая стадия состоит во взаимодействии между хлорангидридом и амином. Эту реакцию можно проводить с использованием способов и методов, хорошо известных специалистам в данной области техники. При одном особенно приемлемом способе вызывают взаимодействие двух химических соединений в присутствии органического или неорганического основания, такого как Et3N, iPr2NEt, пиридин, NaH, Cs2CO3, K2СO3, в растворителе, таком как ТГФ, дихлорметан, ДМФ, ДМСО, при температуре конкретно от -20° до 100°С. X может также представлять собой гидроксил. В этом случае первая стадия представляет собой реакцию конденсации между карбоновой кислотой (IX) и амином (VI). Эту реакцию можно проводить, следуя способам и методам, хорошо известным специалистам в данной области техники. При одном особенно приемлемом способе вызывают взаимодействие этих двух химических соединений в присутствии 1-(3-диметиламинопропил)-3-этил-карбодиимида (EDC), 3-гидрокси-1,2,3-бензотриазин-4(3Н)-она, третичного амина, такого как диизопропилэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре конкретно от -15°С до 40°С. При другом, особенно приемлемом способе вызывают взаимодействие этих двух химических соединений в присутствии диэтилфосфорцианидата (DEPC), третичного амина, такого как триэтиламин, в полярном апротонном растворителе, таком как дихлорметан или ДМФ, при температуре конкретно от -15°С до 40°С.
После удаления защиты промежуточного соединения с использованием методов, хорошо известных специалистам в данной области техники, полученное соединение (VIII) может приводить к соединениям формулы (I) после взаимодействия с R4Y. В этом случае Y представляет собой уходящую группу, такую как Cl, Br, I, OSO2CH3, OSO2CF3 или О-тозил. Эту реакцию проводят в присутствии органического или неорганического основания, такого как Et3N, iPr2NEt, NaH, Cs2CO3, K2СO3, в полярном безводном растворителе, таком как дихлорметан, ТГФ, ДМФ, ДМСО, при температуре конкретно от -20° до 100°С. При другом особенно приемлемом способе вызывают взаимодействие соединения (VIII) с альдегидом формулы R4b-СНО, где R4b соответствует предшественнику R4. В этом случае реакция представляет собой восстановительное аминирование в присутствии восстанавливающего агента, такого как NaBH4, NaBH3CN, NaBH(OAc)3, в полярном растворителе, таком как 1,2-дихлорэтан, дихлорметан, ТГФ, ДМФ, МеОН, при необязательном присутствии изопропоксида титана (IV), при рН, который можно регулировать добавлением кислоты, такой как уксусная кислота, при температуре конкретно от -20°С до 100°С.
В приведенных выше схемах синтеза соединение формулы (I) может приводить к другому соединению формулы (I) после дополнительной стадии взаимодействия, такой как омыление, например, с использованием способов, хорошо известных специалистам в данной области техники, где группа R2, представляющая собой сложный эфир, предпочтительно метиловый эфир, изменяется на группу R2, представляющую собой карбоновую кислоту.
Если это желательно для выделения соединения формулы (I), содержащего по меньшей мере одну основную функциональную группу в состоянии соли присоединения кислоты, это возможно путем обработки свободного основания соединения формулы (I) (содержащего по меньшей мере одну основную функциональную группу) подходящей кислотой, предпочтительно в эквивалентном количестве. Подходящая кислота может представлять собой, в частности, трифторуксусную кислоту.
Таким образом, следующим объектом настоящего изобретения является первый способ получения соединения формулы (I), включающий реакцию конденсации между соединением приведенной ниже формулы (VI):

где R1 и R2 являются такими, как определено выше, и
соединением приведенной ниже формулы (VII):
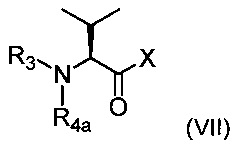
где R3 является таким, как определено выше, R4a соответствует группе R4, такой как определено выше, необязательно в защищенной форме, и X представляет собой ОН или Cl.
Когда X=ОН, реакцию сочетания можно проводить в условиях пептидного сочетания, хорошо известных специалистам в данной области техники.
Пептидное сочетание можно проводить в присутствии агента сочетания, такого как диизопропилкарбодиимид (DIC), дициклогексилкарбодиимид (DCC), гидрохлорид 1-(3-диметиламинопропил)-3-этилкарбодиимида (EDC), карбонилдиимидазол (CDI), 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HBTU), 2-(1Н-бензотриазол-1-ил)-1,1,3,3-тетраметилурония тетрафторборат (TBTU), O-(7-азабензотриазол-1-ил)-1,1,3,3-тетраметилурония гексафторфосфат (HATU), диэтилфосфорцианидат (DEPC) или (бензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат (РуВОР), необязательно в сочетании со вспомогательным агентом сочетания, таким как N-гидроксисукцинимид (NHS), N-гидроксибензотриазол (HOBt), 3,4-дигидро-3-гидрокси-4-оксо-1,2,3-бензотриазол (HOOBt), 1-гидрокси-7-азабензотриазол (HAt), N-гидроксисульфосукцинимид (сульфо-NHS) или диметиламинопиридин (DMAP). Предпочтительно агент сочетания представляет собой HATU или DEPC.
Реакцию можно также проводить в присутствии основания, такого как DIEA (диизопропилэтиламин).
В частности, пептидное сочетание проводят в присутствии HATU или DEPC и DIEA.
Данную реакцию можно проводить в полярном апротонном растворителе таком как дихлорметан (ДХМ) или диметилформамид (ДМФ), в частности, при температуре от -15°С до 40°С.
Когда X=Cl, реакцию конденсации следует проводить в присутствии основания, которое может быть органическим или неорганическим, такого как Et3N, iPr2NEt, пиридин, NaH, Cs2CO3 или K2СO3.
Реакцию можно проводить в растворителе, таком как тетрагидрофуран (ТГФ), дихлорметан (ДХМ), диметилформамид (ДМФ) или диметилсульфоксид (ДМСО), в частности, при температуре от -20° до 100°С.
Соединения формулы (VI) и (VII) могут быть получены путем следования протоколам синтеза, описанным в экспериментальном разделе ниже, или использования методов, известных специалистам в данной области техники.
Следующим объектом настоящего изобретения является второй способ получения соединения формулы (I), включающий реакцию замещения между соединением приведенной ниже формулы (VIII):
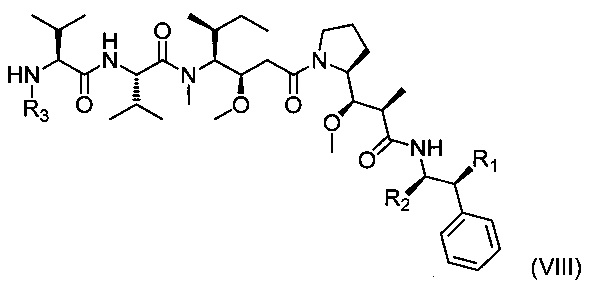
где R1, R2 и R3 являются такими, как определено выше, и
соединением приведенной ниже формулы (X):
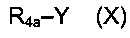
где R4a представляет собой группу R4, такую как определено выше, необязательно в защищенной форме, и Y представляет собой уходящую группу, такую как Cl, Br, I, OSO2CH3, OSO2CF3 или О-тозил.
Реакцию замещения конкретно следует проводить в присутствии основания, которое может быть органическим или неорганическим, таким как Et3N, iPr2NEt, NaH, Cs2CO3 или K2СO3.
Эту реакцию можно выполнять в полярном растворителе, предпочтительно безводном, таком как ДХМ, ТГФ, ДМФ или ДМСО, в частности, при температуре от -20° до 100°С.
Соединения формул (VIII) и (X) могут быть получены путем следования протоколам синтеза, описанным ниже в экспериментальном разделе, или использования методов, известных специалистам в данной области техники.
Следующим объектом настоящего изобретения является третий способ получения соединения формулы (I), в котором R4 представляет собой группу -СН2R4b, где R4b представляет собой арильную или арил-(С1-С7)алкильную группу, замещенную одной или более групп (в частности, одной, предпочтительно на арильной группировке), выбранной из групп ОН и NR9R10,
включающий реакцию восстановительного аминирования между соединением приведенной ниже формулы (VIII):
где R1, R2 и R3 являются такими, как определено выше, и
соединением приведенной ниже формулы (XI):
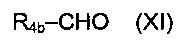
где R4b является таким, как определено выше.
Реакцию восстановительного аминирования можно проводить в присутствии восстанавливающего агента, такого как NaBH4, NaBH3CN или NaBH(OAc)3, и необязательно изопропоксида титана (IV).
рН можно регулировать добавлением кислоты, такой как уксусная кислота, в частности, до достижения рН от 4 до 5.
Эту реакцию можно выполнять в полярном растворителе, таком как ДХЭ (1,2-дихлорэтан), ДХМ, ТГФ, ДМФ или метанол, в частности, при температуре от -20° до 100°С.
Соединения формул (VIII) и (XI) могут быть получены путем следования протоколам синтеза, описанным ниже в экспериментальном разделе, или использования методов, известных специалистам в данной области техники.
Соединение, полученное после стадии конденсации/замещения/восстановительного аминирования одного из трех описанных выше способов, можно подвергать дополнительным стадиям удаления защиты, в частности, в отношении заместителей R2 и R4, и необязательно дополнительным стадиям функционализации с использованием способов, хорошо известных специалистам в данной области техники.
Когда R2 представляет собой группу СООН, упомянутую выше стадию конденсации/замещения/восстановительного аминирования можно проводить от соединения (VI), где группа R2 представляет собой сложноэфирную функциональную группу СОО-((С1-С6)алкил), где затем данную сложноэфирную функциональную группу, возможно, омыляют с получением соединения формулы (I), где R2=СООН.
Когда группа R4 содержит функциональную группу NH, ее можно защищать перед проведением реакции конденсации/замещения/восстановительного аминирования путем замещения атома азота N-защитной группой, такой как группа Boc или Fmoc.
Под «защитной группой» в настоящем изобретении подразумевают группу, которая селективно блокирует реакционный центр в многофункциональном соединении, так что химическую реакцию можно селективно проводить при другом незащищенном реакционном центре, в значении, традиционно связанном с ними в химическом синтезе.
Под «N-защитной группой» в настоящем изобретении подразумевают любой заместитель, который защищает группу NH от нежелательных реакций, такой как N-защитные группы, описанные в кн. Greene, «Protective Groups In Organic synthesis» (John Wiley & Sons, New York (1981)) и Harrison et al. «Compendium of Synthetic Organic Methods», Vols. 1 to 8 (J. Wiley & sons, 1971 to 1996). N-защитные группы включают карбаматы, амиды, N-алкилированные производные, аминоацеталевые производные, N-бензильные производные, иминные производные, енаминные производные и N-гетероатомные производные. N-защитные группы могут представлять собой формил; арил, такой как фенил, необязательно замещенный одной или несколькими метоксигруппами, такой как пара-метоксифенил (РМР); арил-(С1-С6)алкил, такой как бензил, арильную группировку, необязательно замещенную одной или несколькими метоксигруппами, такую как бензил (Bn), пара-метоксибензил (РМВ) или 3,4-диметоксибензил (DMPM); -CO-RGP1, такую как ацетил (Ас), пивалоил (Piv или Pv), бензоил (Bz) или пара-метоксибензилкарбонил (Moz); -CO2-RGP1, такую как трет-бутилоксикарбонил (Воc), трихлорэтоксикарбонил (TROC), аллилоксикарбонил (Alloc), бензилоксикарбонил (Cbz или Z) или 9-флуоренилметилоксикарбонил (Fmoc); -SO2-RGP1, такую как фенилсульфонил, тозил (Ts или Tos) или 2-нитробензолсульфонил (также называемый нозил - Nos или Ns); и тому подобное, где RGP1 представляет собой (С1-С6)алкил, необязательно замещенный одним или несколькими атомами галогена, такими как F или Cl; (С2-С6)алкенил, такой как аллил; арил, такой как фенил, необязательно замещенный одной или несколькими группами, выбранными из ОМе (метокси) и NO2 (нитро); арил-(С1-С6)алкил, такой как бензил, арильную группировку, необязательно замещенную одной или несколькими метоксигруппами; или 9-флуоренилметильную группу.
В частности, N-защитная группа включает формил, ацетил, бензоил, пивалоил, фенилсульфонил, бензил (Bn), трет-бутилоксикарбонил (Воc), бензилоксикарбонил (Cbz), пара-метоксибензилоксикарбонил, пара-нитробензил-оксикарбонил, трихлорэтоксикарбонил (TROC), аллилоксикарбонил (Alloc), 9-флуоренилметилоксикарбонил (Fmoc), трифторацетил, бензилкарбаматы (замещенные или незамещенные) и тому подобное. Она может представлять собой, в частности, группу Воc или Fmoc.
Защита функциональной группы амина NH группой Воc или Fmoc и последующее удаление ее защиты после реакции конденсации/замещения/восстановительного аминирования хорошо известна специалистам в данной области техники и описана, в частности, в экспериментальном разделе ниже.
Соединение формулы (I), полученное одним из трех упомянутых выше способов, можно также преобразовать в соль путем добавления фармацевтически приемлемого основания или кислоты, в частности, фармацевтически приемлемой кислоты, такой как трифторуксусная кислота. Данную стадию можно необязательно проводить одновременно с другой стадией реакции, в частности, одновременно со стадией удаления защиты, когда ее необходимо проводить, например, в кислой среде.
Соединение, полученное одним из этих трех способов, необязательно после дополнительной стадии (стадий) удаления защиты, функционализации и/или образования соли, можно выделить из реакционной среды с использованием способов, хорошо известных специалистам в данной области техники, например, путем экстракции, выпаривания растворителя или осаждения и фильтрования.
Соединение можно также при необходимости очищать с использованием методов, хорошо известных специалистам в данной области техники, например, путем перекристаллизации, если соединение является кристаллическим, путем перегонки, путем колоночной хроматографии на силикагеле или высокоэффективной жидкостной хроматографии (ВЭЖХ).
Приведенные ниже примеры иллюстрируют изобретение, не ограничивая, тем не менее, объем изобретения.
ПРИМЕРЫ
I - Синтез соединений по изобретению
В приведенных ниже примерах используют следующие сокращения:
водн. водный
э.и. энантиомерный избыток
экв. эквивалент
ИЭР Ионизация электрораспылением
ЖХ/МС Жидкостная хроматография, сопряженная с масс-спектрометрией
ВЭЖХ Высокоэффективная жидкостная хроматография
ЯМР Ядерный магнитный резонанс
нас. насыщенный
УФ ультрафиолет
Справочный пример 1
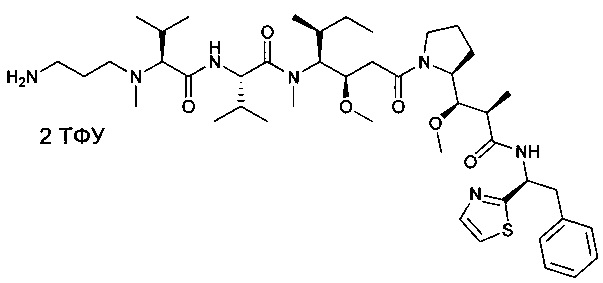
(S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, бис-трифторуксусная кислота
Пример 1А: (4R,5S)-4-метил-5-фенил-3-пропаноил-1,3-оксазолидин-2-он
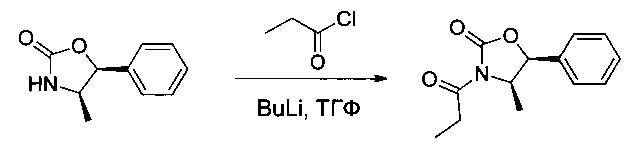
(4R,5S)-4-мeтил-5-фeнил-1,3-oкcaзoлидин-2-oн (5,8 г, 32,7 ммоль, 1,00 экв.) растворяли в тетрагидрофуране (ТГФ, 120 мл) в инертной атмосфере. Смесь охлаждали до -78°С и добавляли по каплям н-бутиллитий (14,4 мл). После перемешивания в течение 30 минут при -78°С добавляли пропаноилхлорид (5,7 мл). Перемешивание продолжали в течение 30 минут при -78°С, затем в течение ночи при температуре окружающей среды. Реакционную смесь концентрировали, затем снова растворяли в 200 мл воды. рН раствора доводили до 7 насыщенным водным раствором бикарбоната натрия. Эту водную фазу экстрагировали 3 раза 100 мл этилацетата (EtOAc). Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 6,8 г (89%) соединения 1А в форме желтого масла.
Пример 1В: трет-бутил-(2S)-2-[(1R,2R)-1-гидрокси-2-метил-3-[(4R,5S)-4-метил-2-оксо-5-фенил-1,3-оксазолидин-3-ил]-3-оксопропил]пирролидин-1-карбоксилат
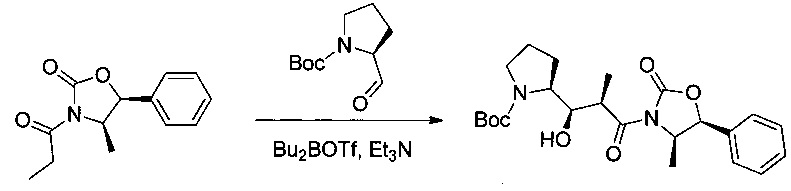
Соединение 1А (17,6 г, 75,45 ммоль, 1,00 экв.) растворяли в дихлорметане (ДХМ, 286 мл) в инертной атмосфере. Этот раствор охлаждали в ледяной бане. Добавляли по каплям триэтиламин (TEA, 12,1 мл, 1,15 экв.) и Bu2BOTf (78,3 мл, 1,04 экв.), при этом поддерживая температуру реакционной смеси ниже 2°С. Перемешивание продолжали при 0°С в течение 45 минут, после чего реакционную смесь охлаждали до -78°С. Добавляли по каплям раствор трет-бутил-(2S)-2-формилпирролидин-1-карбоксилата (8,5 г, 42,66 ммоль, 0,57 экв.) в ДХМ (42 мл). Перемешивание продолжали в течение 2 часов при -78°С, затем в течение 1 часа при 0°С и, наконец, 1 часа при температуре окружающей среды. Реакционную смесь нейтрализовали 72 мл фосфатного буфера (рН=7,2-7,4) и 214 мл метанола и охлаждали до 0°С. Добавляли по каплям 30% раствор перекиси водорода в метаноле (257 мл), поддерживая при этом температуру ниже 10°С. Перемешивание продолжали в течение 1 часа при 0°С. Реакционную смесь нейтрализовали 142 мл воды, затем концентрировали при пониженном давлении. Полученный в результате водный раствор экстрагировали 3 раза 200 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и петролейного эфира (EtOAc:РЕ = 1:8) с получением 13,16 г (40%) соединения 1В в форме бесцветного масла.
Пример 1С: (2R,3R)-3-[(2S)-1-[(трет-бутокси)карбонил]пирролидин-2-ил]-3-гидрокси-2-метилпропановая кислота
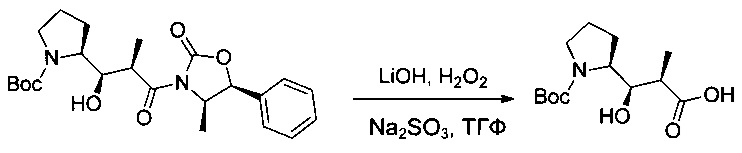
Соединение 1В (13,16 г, 30,43 ммоль, 1,00 экв.) растворяли в ТГФ (460 мл) в присутствии перекиси водорода (30% в воде, 15,7 мл), затем охлаждали в ледяной бане. Добавляли по каплям водный раствор гидроксида лития (0,4 моль/л, 152,1 мл), поддерживая при этом температуру реакционной смеси ниже 4°С. Реакционную смесь перемешивали в течение 2,5 часов при 0°С. Добавляли по каплям водный раствор Na2SO3 (1 моль/л, 167,3 мл), поддерживая при этом температуру реакционной смеси при 0°С. Реакционную смесь перемешивали в течение 14 часов при температуре окружающей среды, затем нейтрализовали 150 мл холодного насыщенного раствора бикарбоната натрия и промывали 3 раза 50 мл ДХМ. рН водного раствора доводили до 2-3 1 М водным раствором KHSO4. Этот водный раствор экстрагировали 3 раза 100 мл EtOAc. Органические фазы объединяли, промывали один раз насыщенным раствором NaCl, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 7,31 г (88%) соединения 1С в форме бесцветного масла.
Пример 1D: (2R,3R)-3-[(2S)-1-[(трет-бутокси)карбонил]пирролидин-2-ил]-3-метокси-2-метилпропановая кислота
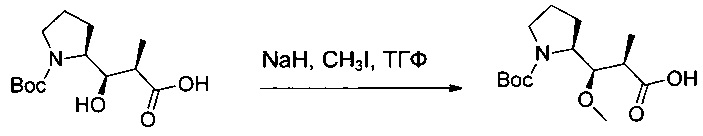
Соединение 1С (7,31 г, 26,74 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (135 мл) в присутствии йодметана (25,3 мл). Реакционную среду охлаждали в ледяной бане, после чего добавляли порциями NaH (60% в масле, 4,28 г). Реакционную смесь оставляли при перемешивании на 3 суток при 0°С, а затем нейтрализовали 100 мл насыщенного водного раствора бикарбоната натрия и промывали 3 раза 50 мл эфира. рН водного раствора доводили до 3 1 М водным раствором KHSO4. Этот водный раствор экстрагировали 3 раза 100 мл EtOAc. Органические фазы объединяли, промывали один раз 100 мл Na2S2O3 (5% в воде), один раз насыщенным раствором NaCl, затем высушивали над сульфатом натрия, фильтровали и концентрировали с получением 5,5 г (72%) соединения 1D в форме бесцветного масла.
Пример 1Е: N-метокси-N-метил-2-фенилацетамид
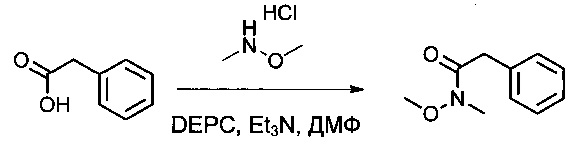
2-Фенилуксусную кислоту (16,2 г, 118,99 ммоль, 1,00 экв.) растворяли в диметилформамиде (ДМФ, 130 мл), затем охлаждали до -10°С. Добавляли диэтилфосфорцианидат (DEPC, 19,2 мл), гидрохлорид метокси(метил)амина (12,92 г, 133,20 ммоль, 1,12 экв.) и триэтиламин (33,6 мл). Реакционную смесь перемешивали в течение 30 минут при -10°С, затем 2,5 часов при температуре окружающей среды. Затем ее дважды экстрагировали 1 литром EtOAc. Органические фазы объединяли, дважды промывали 500 мл NaHCO3 (нас.), один раз 400 мл воды, затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и петролейного эфира (ПЭ) (от 1:100 до 1:3) с получением 20,2 г (95%) соединения 1Е в форме желтого масла.
Пример 1F: 2-фенил-1-(1,3-тиазол-2-ил)этан-1-он
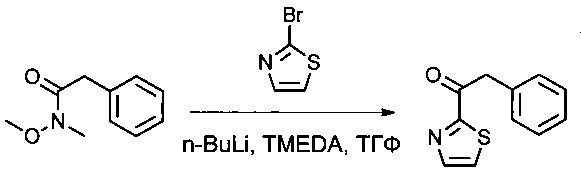
Тетраметилэтилендиамин (TMEDA, 27,2 мл) растворяли в ТГФ (300 мл) в инертной атмосфере, затем охлаждали до -78°С, после чего добавляли по каплям n-BuLi (67,6 мл, 2,5 М). Добавляли по каплям 2-бром-1,3-тиазол (15,2 мл), и перемешивание продолжали в течение 30 минут при -78°С. Добавляли по каплям соединение 1Е (25 г, 139,50 ммоль, 1,00 экв.), растворенное в ТГФ (100 мл). Перемешивание продолжали в течение 30 минут при -78°С, затем 2 часов при -10°С. Реакционную смесь нейтрализовали 500 мл KHSO4 (нас.), затем экстрагировали 3 раза 1 литром EtOAc. Органические фазы объединяли, дважды промывали 400 мл воды и дважды 700 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (от 1:100 до 1:10) с получением 25 г (88%) соединения 1F в форме желтого масла.
Пример 1G: (1R)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-ол
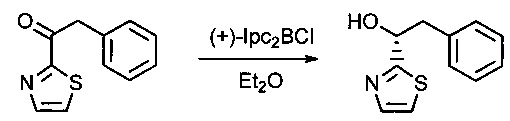
В инертной атмосфере раствор соединения 1F (15 г, 73,8 ммоль, 1,00 экв..) в эфире (300 мл) добавляли по каплям к (+)-В-хлордиизопинокамфеилборану ((+)-Ipc2BCl, 110,8 мл). Реакционную смесь перемешивали в течение 24 часов при 0°С, затем нейтрализовали 300 мл смеси (1:1) NaOH (10% в воде) и Н2O2 (30% в воде) и, наконец, экстрагировали три раза 500 мл EtOAc. Органические фазы объединяли, дважды промывали 300 мл K2СO3 (нас.) и один раз 500 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (от 1:20 до 1:2) с получением 6,3 г (42%) соединения 1G в форме белого твердого вещества.
Пример 1Н: 2-[(1S)-1-азидо-2-фенилэтил]-1,3-тиазол
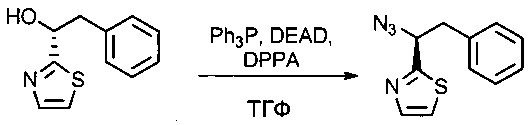
Соединение 1G (6 г, 29,23 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (150 мл) в присутствии трифенилфосфина (13 г, 49,56 ммоль, 1,70 экв.), затем охлаждали до 0°С. Добавляли по каплям диэтилазодикарбоксилат (DEAD, 7,6 мл), а затем дифенилфосфорилазид (DPPA, 11 мл), затем охлаждающую баню удаляли, и раствор оставляли при перемешивании на 48 часов при температуре окружающей среды. Среду концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (от 1:100 до 1:30) с получением 8 г частично очищенного соединения 1Н в форме желтого масла. Соединение 1Н использовали в таком виде на следующей стадии.
Пример 1I: трет-бутил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамат
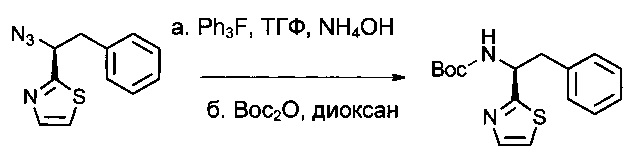
Соединение 1Н (6,5 г, 28,2 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (100 мл) в присутствии трифенилфосфина (6,5 г, 33,9 ммоль, 1,20 экв.) и нагревали до 50°С в течение 2 часов. Затем добавляли аммиак (70 мл), и нагревание продолжали в течение 3 часов. Реакционную смесь охлаждали, нейтрализовали 500 мл воды, затем экстрагировали 3 раза 500 мл EtOAc. Органические фазы объединяли и дважды экстрагировали 500 мл 1 н. HCl. Водные фазы объединяли, доводили до рН 8-9 добавлением раствора гидроксида натрия (10% в воде), затем экстрагировали 3 раза 500 мл ДХМ. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 4,8 г (83%) (1S)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-амина в форме желтого масла. Затем это соединение защищали группой Воc (трет-бутокси)карбонил) таким образом, чтобы ее можно было очистить. Его растворяли в инертной атмосфере в 1,4-диоксане (40 мл), затем охлаждали до 0°С. Добавляли по каплям (Вос)2O (10,26 г, 47,01 ммоль, 2,00 экв.), разведенный в 20 мл 1,4-диоксана. Охлаждающую баню удаляли, и раствор оставляли при перемешивании в течение ночи при температуре окружающей среды, после чего нейтрализовали 300 мл воды и дважды экстрагировали 500 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (от 1:100 до 1:20, э.и.=93%). Затем его перекристаллизовали в смеси гексан/ацетон (~ 5-10/1,1 г/10 мл) с получением 6 г (84%) соединения 1I в форме белого твердого вещества (э.и. более 99%).
Пример 1J: трет-бутил-(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-карбоксилат
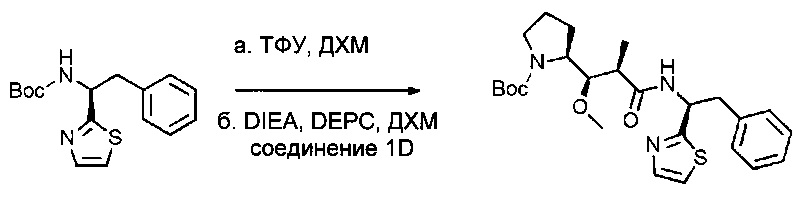
Соединение 1I (3 г, 9,86 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 10 мл ДХМ. Добавляли трифторуксусную кислоту (ТФУ, 10 мл), и раствор оставляли при перемешивании в течение ночи при температуре окружающей среды, затем концентрировали при пониженном давлении с получением 2,0 г (64%) (1S)-2-фенил-1-(1,3-тиазол-2-ил)этан-1-амина; трифторуксусная кислота в форме желтого масла. Это промежуточное соединение повторно растворяли в 20 мл ДХМ, после чего соединение 1D (1,8 г, 6,26 ммоль, 1,05 экв.), DEPC (1,1 г, 6,75 ммоль, 1,13 экв.) и добавляли диизопропилэтиламин (DIEA, 1,64 г, 12,71 ммоль, 2,13 экв.). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (от 1:100 до 1:3) с получением 2,3 г (81%) соединения 1J в форме бледно-желтого твердого вещества.
Пример 1K: (2R,3R)-3-метокси-2-метил-N-[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]-3-[(2S)-пирролидин-2-ил]пропанамид; трифторуксусная кислота
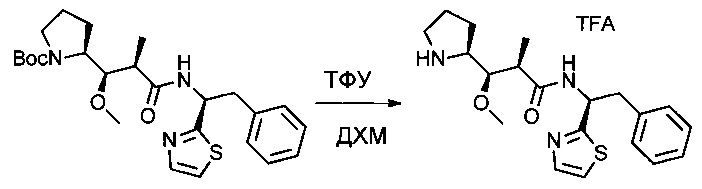
Соединение 1J (2,25 г, 4,75 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 10 мл ДХМ. Добавляли ТФУ (10 мл), и раствор оставляли при перемешивании в течение ночи при температуре окружающей среды, затем концентрировали при пониженном давлении с получением 2,18 г (94%) соединения 1K в форме желтого масла.
Пример 1L: (2S,3S)-2-(бензиламино)-3-метилпентановая кислота
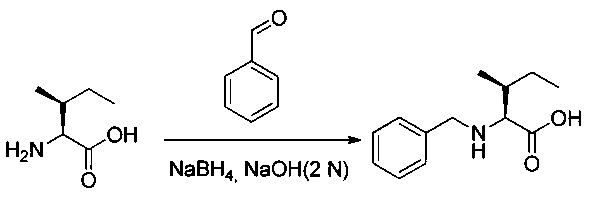
(2S,3S)-2-Амино-3-метилпентановую кислоту (98,4 г, 750 ммоль, 1,00 экв.) добавляли порциями при температуре окружающей среды к 2 н. раствору гидроксида натрия (375 мл). Быстро добавляли бензальдегид (79,7 г, 751,02 ммоль, 1,00 экв.), и полученный в результате раствор перемешивали в течение 30 минут. Добавляли боргидрид натрия (10,9 г, 288,17 ммоль, 0,38 экв.) небольшими порциями, при этом поддерживая температуру от 5 до 15°С. Перемешивание продолжали в течение 4 часов при температуре окружающей среды. Реакционную смесь разбавляли 200 мл воды, затем дважды промывали 200 мл EtOAc. рН водного раствора доводили до 7 2 н. раствором соляной кислоты. Образовавшийся осадок собирали фильтрованием и получили 149,2 г (90%) соединения 1L в форме белого твердого вещества.
Пример 1М: (2S,3S)-2-[бензил(метил)амино]-3-метилпентановая кислота
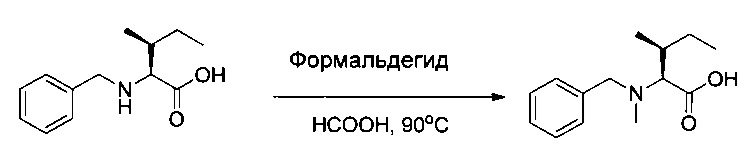
Соединение 1L (25 г, 112,97 ммоль, 1,00 экв.) растворяли в инертной атмосфере в муравьиной кислоте (31,2 г) в присутствии формальдегида (36,5% в воде, 22,3 г). Раствор перемешивали в течение 3 часов при 90°С, затем концентрировали при пониженном давлении. Остаток растирали в 250 мл ацетона, затем концентрировали. Эту операцию растирания/выпаривания повторяли дважды 500 мл ацетона с получением 21,6 г (81%) соединения 1 М в форме белого твердого вещества.
Пример 1N: (2S,3S)-2-[бензил(метил)амино]-3-метилпентан-1-ол
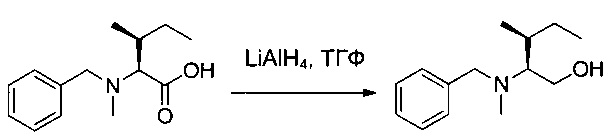
LiAlH4 (0,36 г) суспендировали в 10 мл ТГФ в инертной атмосфере при 0°С. Соединение 1М (1,5 г, 6,37 ммоль, 1,00 экв.) добавляли небольшими порциями, поддерживая при этом температуру от 0 до 10°С. Реакционную смесь перемешивали в течение 2 часов при 65°С, затем снова охлаждали до 0°С, после чего нейтрализовали последовательными добавлениями 360 мкл воды, 1 мл 15% гидроксида натрия и 360 мкл воды. Соли алюминия, выпавшие в осадок, удаляли фильтрованием. Фильтрат высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (1:50) с получением 820 мг (58%) соединения 1N в форме бледно-желтого масла.
Пример 1O: (2S,3S)-2-[бензил(метил)амино]-3-метилпентаналь
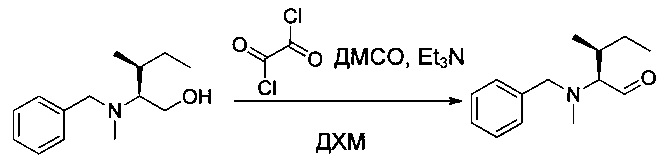
Оксалилхлорид (0,4 мл) растворяли в ДХМ (15 мл) в инертной атмосфере. Раствор охлаждали до -70°С и добавляли по каплям раствор диметилсульфоксида (ДМСО (0,5 мл) в ДХМ (10 мл)) в течение 15 минут. Реакционную смесь перемешивали в течение 30 минут, после чего добавляли по каплям раствор соединения 1N (820 мг, 3,70 ммоль, 1,00 экв.) в ДХМ (10 мл) в течение 15 минут. Реакционную смесь перемешивали еще 30 минут при низкой температуре, затем медленно добавляли триэтиламин (2,5 мл). Реакционную смесь перемешивали в течение 1 часа при -50°С, затем охлаждающую баню удаляли, и реакционную смесь нейтрализовали 25 мл воды, при этом давая возможность возврата температуры к нормальной. Раствор промывали один раз 30 мл насыщенного водного раствора NaCl, затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (1:200) с получением 0,42 г (52%) соединения 10 в форме желтого масла.
Пример 1Р: (2S,3S)-N-бензил-1,1-диметокси-N,3-диметилпентан-2-амин
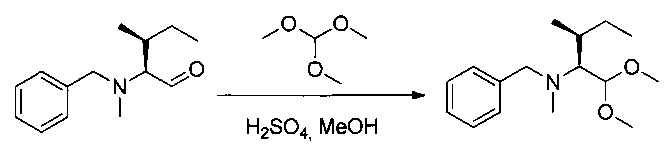
Соединение 1O (4,7 г, 21,43 ммоль, 1,00 экв.) растворяли в 20 мл метанола при 0°С. Добавляли по каплям концентрированную серную кислоту (4,3 мл), и перемешивание продолжали в течение 30 минут при 0°С. Добавляли триэтилортоформиат (21,4 мл), охлаждающую баню удаляли, и реакционную среду оставляли при перемешивании на 3 часа при температуре окружающей среды. Реакционную среду разбавляли 200 мл EtOAc, последовательно промывали 100 мл 10% Na2CO3 и 200 мл насыщенного NaCl, затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3,4 г (60%) соединения 1Р в форме бледно-желтого масла.
Пример 1Q: [[1-(трет-бутокси)этенил]окси](трет-бутил)диметилсилан
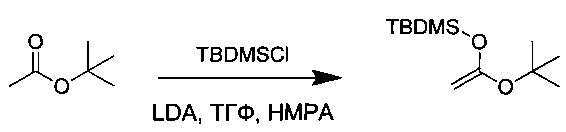
Диизопропиламин (20 г, 186,71 ммоль, 1,08 экв.) растворяли в 170 мл ТГФ в инертной атмосфере и охлаждали до -78°С. добавляли по каплям nBuLi (2,4 М, 78,8 мл), и раствор перемешивали в течение 30 минут при низкой температуре (с получением диизопропиламида лития (LDA)), после чего добавляли трет-бутилацетат (20 г, 172,18 ммоль, 1,00 экв.). Реакционную смесь перемешивали в течение 20 минут при -78°С, после чего добавляли гексаметилфосфорамид (НМРА, 25,8 мл) и раствор трет-бутилдиметилхлорсилана (TBDMSCl, 28 г, 185,80 ммоль, 1,08 экв.) в 35 мл ТГФ. Перемешивание продолжали дополнительно в течение 20 минут при низкой температуре, а затем охлаждающую баню удаляли. Раствор концентрировали при пониженном давлении. Остаток снова растворяли в 100 мл воды и экстрагировали 3 раза 100 мл ПЭ. Органические фазы объединяли, промывали один раз 500 мл насыщенного водного раствора NaCl, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали перегонкой с получением 16,6 г (83%) соединения 1Q в форме бесцветного масла.
Пример 1R: трет-бутил-(3R,4S,5S)-4-[бензил(метил)амино]-3-метокси-5-метилгептаноат
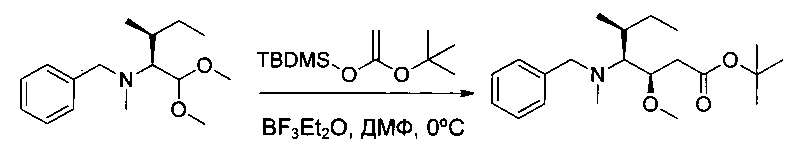
Соединение 1Р (2,0 г, 7,54 ммоль, 1,00 экв.) и соединение 1Q (2,6 г, 11,28 ммоль, 1,50 экв.) растворяли в 33 мл ДХМ в инертной атмосфере. Раствор охлаждали до 0°С. Добавляли по каплям ДМФ (1,2 г) вместе с раствором BF3⋅Et2O (2,1 г) в 7,5 мл ДХМ. Перемешивание продолжали в течение 24 часов при 0°С. Реакционную среду промывали один раз 30 мл карбоната натрия (10%) и дважды 50 мл насыщенного водного раствора NaCl, затем высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (1:100) с получением 1,82 г (91%) соединения 1R в форме желтого масла.
Пример 1S: гидрохлорид (3R,4S,5S)-3-метокси-5-метил-4-(метиламино)гептаноата
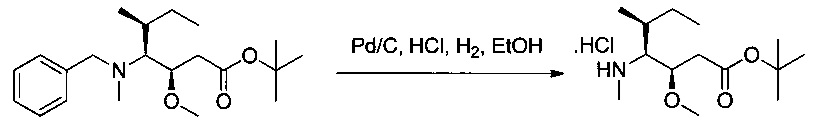
Соединение 1R (2,4 г, 6,87 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 35 мл этанола в присутствии Pd/C (0,12 г) и концентрированной соляной кислоты (0,63 мл). Атмосферу азота заменяли атмосферой водорода, и реакционную среду оставляли при перемешивании на 18 часов при температуре окружающей среды. Реакционную среду фильтровали и концентрировали при пониженном давлении. Остаток растирали в 50 мл гексана, и отделяли супернатант, из которого при пониженном давлении получили 1,66 г (82%) соединения 1S в форме белого твердого вещества.
Пример 1Т: трет-бутил-(3R,4S,5S)-4-[(2S)-2-[[(бензилокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
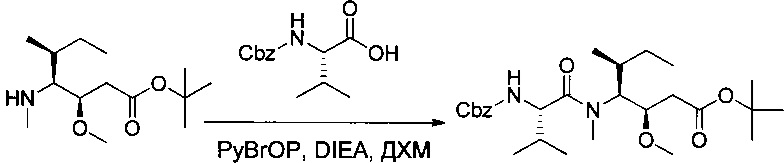
(2S)-2-[[(бензилокси)карбонил]амино]-3-метилбутановую кислоту (15 г, 0,40 ммоль, 1,00 экв.) растворяли в 300 мл ДХМ в присутствии DIEA (38,3 мл) и бромтрипирролидинофосфония гексафторфосфат (PyBrOP, 32,3 г). Раствор перемешивали в течение 30 минут при температуре окружающей среды, после чего добавляли соединение 1S (15,99 г, 0,42 ммоль, 1,07 экв.). Реакционную среду перемешивали в течение 2 часов, а затем концентрировали. Остаток очищали в обращенной фазе (С18) смесью ацетонитрила (ACN) и воды (от 30:70 до 100:0 за 40 минут) с получением 17 г (58%) соединения 1Т в форме бесцветного масла.
Пример 1U: трет-бутил-(3R,4S,5S)-4-[(2S)-2-амино-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
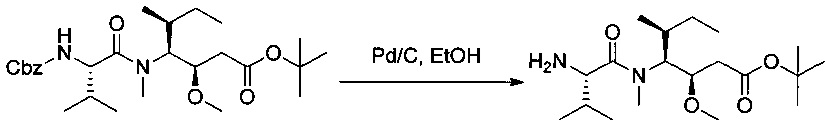
Соединение 1Т (76 мг, 0,15 ммоль, 1,00 экв.) растворяли в инертной атмосфере в 10 мл этанола в присутствии Pd/C (0,05 г). Атмосферу азота заменяли атмосферой водорода, и реакционную смесь перемешивали в течение 2 часов при температуре окружающей среды. Реакционную среду фильтровали и концентрировали при пониженном давлении с получением 64 мг соединения 1U в форме бесцветного масла.
Пример 1V: (3R,4S,5S)-4-[(2S)-2-[[(9Н-флуорен-9-илметокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноат
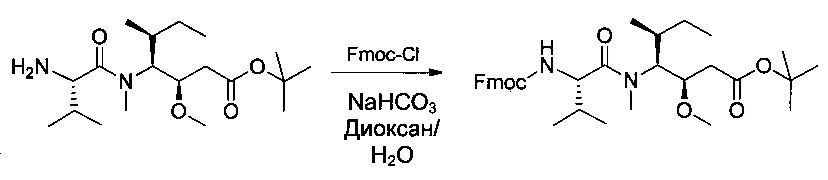
Соединение 1U (18,19 г, 50,74 ммоль, 1,00 экв.) растворяли в 400 мл смеси 1,4-диоксан/вода (1:1) в присутствии бикарбоната натрия (12,78 г, 152 ммоль, 3,00 экв.) и 9Н-флуорен-9-илметилхлорформиата (Fmoc-Cl, 19,69 г, 76 ммоль, 1,50 экв.), затем перемешивали в течение 2 часов при температуре окружающей среды. Затем реакционную среду разбавляли 500 мл воды и экстрагировали 3 раза 200 мл EtOAc. Органические фазы объединяли, промывали один раз 200 мл насыщенного водного раствора NaCl, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 40 г частично очищенного соединения 1V в форме бледно-желтого масла.
Пример 1W: (3R,4S,5S)-4-[(2S)-2-[[(9Н-флуорен-9-илметокси)карбонил]амино]-N,3-диметилбутанамидо]-3-метокси-5-метилгептановая кислота
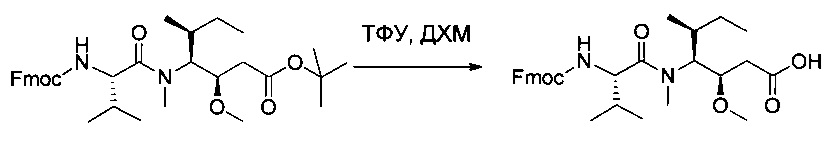
Соединение 1V (40 г, 68,88 ммоль, 1,00 экв.) растворяли в нейтральной атмосфере в 600 мл ДХМ. Добавляли ТФУ (300 мл). Раствор перемешивали в течение 2 часов при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью метанола и ДХМ (1:10) с получением 23,6 г (65%) соединения 1W в форме бесцветного масла.
Пример 1X: 9Н-флуорен-9-илметил-N-[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамат
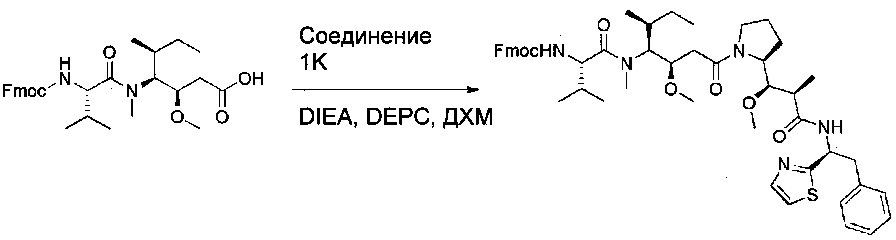
Соединение 1W (2,53 г, 4,82 ммоль, 1,08 экв.) растворяли в 20 мл ДХМ в присутствии соединения 1K (2,18 г, 4,47 ммоль, 1,00 экв.), DEPC (875 мг, 5,37 ммоль, 1,20 экв.) и DIEA (1,25 г, 9,67 ммоль, 2,16 экв.). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, затем последовательно промывали 50 мл насыщенного KHSO4 и 100 мл воды, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью метанола и ДХМ (от 1:200 до 1:40) с получением 2,8 г (71%) соединения 1Х в форме бледно-желтого твердого вещества.
Пример 1Y: (2S)-2-амино-N-[(3R,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил]-N,3-диметилбутанамид
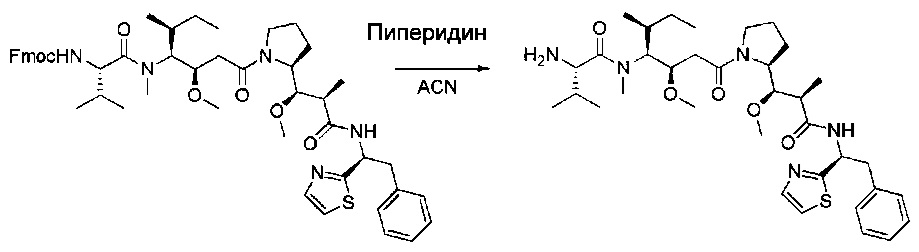
Соединение 1Х (2,8 г, 3,18 ммоль, 1,00 экв.) растворяли в ацетонитриле (ACN, 12 мл) в присутствии пиперидина (3 мл) и оставляли при перемешивании в течение 18 часов при температуре окружающей среды. Реакционную смесь нейтрализовали 50 мл воды, затем дважды экстрагировали 100 мл ДХМ. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью метанола и ДХМ (от 1:100 до 1:40) с получением 1,2 г (57%) соединения 1Y в форме желтого твердого вещества.
Пример 1ZA: (2S)-2-[[(трет-бутокси)карбонил](метил)амино]-3-метилбутановая кислота
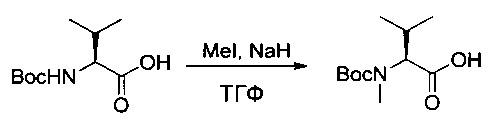
(2S)-2-[[(трет-бутокси)карбонил]амино]-3-метилбутановую кислоту (63 г, 289,97 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (1000 мл) в присутствии йодметана (181 мл). Раствор охлаждали до 0°С, после чего добавляли гидрид натрия (116 г, 4,83 моль, 16,67 экв.) небольшими порциями. Реакционную смесь перемешивали в течение 1,5 часов при 0°С, затем охлаждающую баню удаляли, и перемешивание продолжали в течение 18 часов. Реакционную смесь нейтрализовали 200 мл воды, а затем концентрировали при пониженном давлении. Остаточную водную фазу разбавляли 4 литрами воды, промывали один раз 200 мл EtOAc, и ее рН доводили до значения от 3 до 4 1 н. раствором соляной кислоты. Полученную смесь экстрагировали 3 раза 1,2 л EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 60 г (89%) соединения 1ZA в форме желтого масла.
Пример 1ZB: бензил-(2S)-2-[[(трет-бутокси)карбонил](метил)амино]-3-метилбутаноат
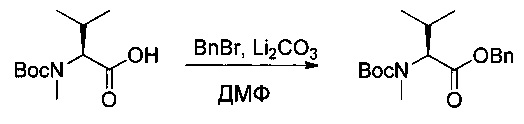
Соединение 1ZA (47 г, 203,21 ммоль, 1,00 экв.) растворяли в ДМФ (600 мл) в присутствии Li2CO3 (15,8 г, 213,83 ммоль, 1,05 экв.). Раствор охлаждали до 0°С, затем добавляли по каплям бензилбромид (BnBr 57,9 г, 338,53 ммоль, 1,67 экв.). Реакционную смесь оставляли при перемешивании в течение ночи, после чего нейтрализовали 400 мл воды и фильтровали. Полученный раствор дважды экстрагировали 500 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (от 1:100 до 1:20) с получением 22,5 г (34%) соединения 1ZB в форме желтого масла.
Пример 1ZC: гидрохлорид бензил-(2S)-3-метил-2-(метиламино)бутаноата
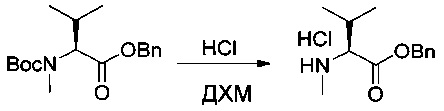
Соединение 1ZB (22,5 г, 70,00 ммоль, 1,00 экв.) растворяли в 150 мл ДХМ. Барботировали газообразную соляную кислоту. Реакционную смесь перемешивали в течение 1 часа при температуре окружающей среды, а затем концентрировали при пониженном давлении с получением 17 г (94%) соединения 1ZC в форме желтого твердого вещества.
Пример 1ZD: трет-бутил-N-(3,3-диэтоксипропил)карбамат
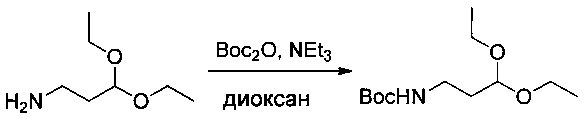
3,3-Диэтоксипропан-1-амин (6 г, 40,76 ммоль, 1,00 экв.) растворяли в 1,4-диоксане (30 мл) в присутствии TEA (4,45 г, 43,98 ммоль, 1,08 экв.), затем охлаждали до 0°С. Добавляли по каплям (Вос)2O (9,6 г, 43,99 ммоль, 1,08 экв.), разведенный в 20 мл 1,4-диоксана. Раствор перемешивали в течение 2 часов при 0°С, затем в течение ночи при температуре окружающей среды, после чего нейтрализовали 10 мл воды. рН доводили до 5 HCl (1%). Раствор экстрагировали 3 раза 50 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 8,21 г (81%) соединения 1ZD в форме бледно-желтого масла.
Пример 1Z: трет-бутил-N-(3-оксопропил)карбамат
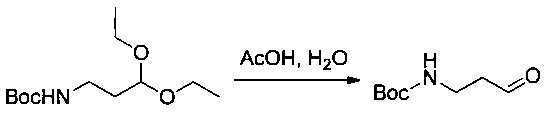
Соединение 1ZD (8,20 г, 33,15 ммоль, 1,00 экв.) растворяли в 18,75 мл уксусной кислоты и оставляли при перемешивании в течение ночи при температуре окружающей среды. Затем реакционную среду экстрагировали 3 раза 30 мл EtOAc. Органические фазы объединяли, промывали 3 раза 30 мл насыщенного раствора NaCl, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 5 г (87%) соединения 1ZE в форме темно-красного масла.
Пример 1ZF: (2S)-2-[(3-[[(трет-бутокси)карбонил]амино]пропил)(метил)амино]-3-метилбутановая кислота
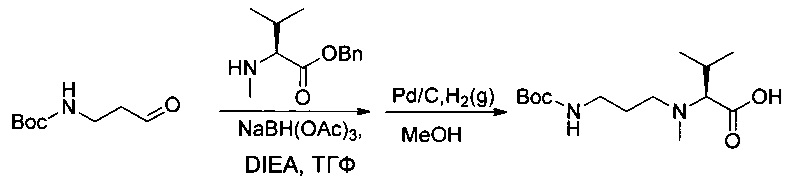
Соединение 1ZE (2,4 г, 13,86 ммоль, 1,00 экв.) растворяли в 50 мл ТГФ в присутствии соединения 1ZC (3,56 г, 13,81 ммоль, 1,00 экв.) и DIEA (9,16 мл, 4,00 экв.). Реакционную смесь перемешивали 30 минут при температуре окружающей среды, после чего добавляли триацетоксиборгидрид натрия (5,87 г, 27,70 ммоль, 2,00 экв.). Перемешивание продолжали в течение ночи, затем реакционную смесь нейтрализовали 100 мл воды и экстрагировали 3 раза 50 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток частично очищали на колонке силикагеля смесью EtOAc и ПЭ (1:4). Полученный неочищенный продукт повторно растворяли в 20 мл метанола в присутствии Pd/C (1,2 г) и гидрогенизировали в течение 20 минут при нормальных значениях температуры и давления. Реакционную среду фильтровали и концентрировали при пониженном давлении с получением 200 мг (5%) соединения 1ZF в форме белого твердого вещества.
Пример 1ZG: трет-бутил-N-(3-[[(1S)-1-[[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]тиил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамоил]-2-метилпропил](метил)амино]пропил)карбамат
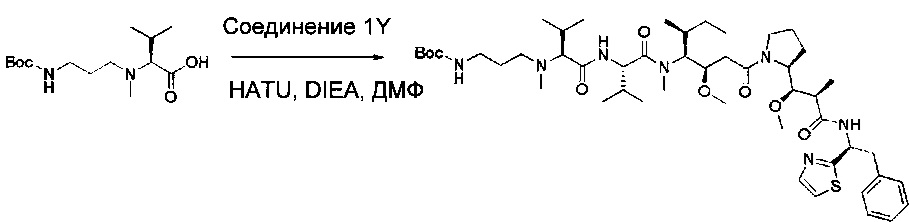
Соединение 1Y (50 мг, 0,08 ммоль, 1,00 экв.) растворяли в 2 мл ДМФ в присутствии соединения 1ZF (26,2 мг, 0,09 ммоль, 1,20 экв.), DIEA (37,7 мл) и O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HATU, 43,3 мг, 0,11 ммоль, 1,50 экв.). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, затем разбавляли 10 мл воды и экстрагировали 3 раза 5 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 100 мг соединения 1ZG в форме частично очищенного бесцветного масла.
Пример 1: Соединение 1ZG (90 мг, 0,10 ммоль, 1,00 экв.) растворяли в нейтральной атмосфере в 2 мл ДХМ, и раствор охлаждали в ледяной бане. Добавляли ТФУ (1 мл), и реакционную смесь перемешивали в течение 2 часов при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; градиент от 18% до 31% ACN за 7 минут, затем от 31% до 100% ACN за 2 минуты; УФ детектор Waters 2489 при 254 нм и 220 нм). Соединение 1 было получено при выходе 25% (23 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Atlantis Т3, 3 мкм, 4,6×100 мм; 35°С; 1 мл/мин, от 30% до 60% ACN в воде (20 мМ ацетат аммония за 6 минут); ИЭР (C44H73N7O6S, точная масса 827,53) m/z: 829 (МН+), 5,84 мин (93,7%, 254 нм).
1Н ЯМР (300 МГц, CD3OD, млн-1): δ (присутствие ротамеров) 7.85-7.80 (m, 1Н); 7.69-7.66 (m, 1Н), 7.40-7.10 (m, 5Н), 5.80-5.63 (m, 1Н), 4.80-4.65 (m, 2Н), 4.22-4.00 (m, 1Н), 3.89-0.74 (m, 58Н).
Справочный пример 2
(S)-2-((S)-2-(((2-аминопиридин-4-ил)метил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, трифторуксусная кислота
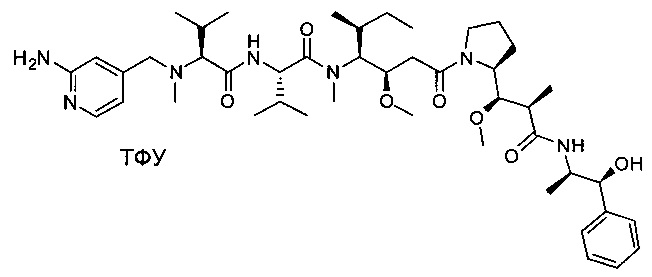
Пример 2А: трет-бутил-(S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-карбоксилат
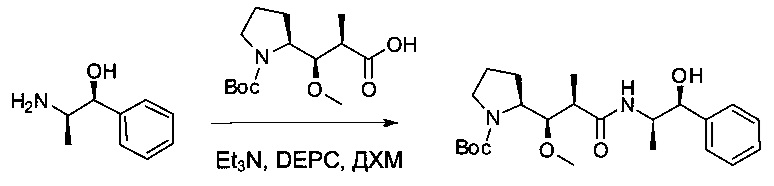
Соединение 1D (2,5 г, 8,70 ммоль, 1,00 экв.) и (1S,2R)-2-амино-1-фенилпропан-1-ол (1,315 г, 8,70 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ДМФ (35 мл). Раствор охлаждали до 0°С, затем добавляли по каплям DEPC (1,39 мл) и TEA (1,82 мл). Реакционную смесь перемешивали в течение 2 часов при 0°С, затем в течение 4 часов при температуре окружающей среды. Реакционную смесь разбавляли 200 мл воды и экстрагировали три раза 50 мл EtOAc. Органические фазы объединяли, промывали один раз 50 мл KHSO4 (1 моль/л), один раз 50 мл NaHCO3 (нас.), один раз 50 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3,6 г (98%) соединения 2А в форме желтого твердого вещества.
Пример 2В: (2R,3R)-N-((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)-3-метокси-2-метил-3-((S)-пирролидин-2-ил)пропанамида 2,2,2-трифторацетат
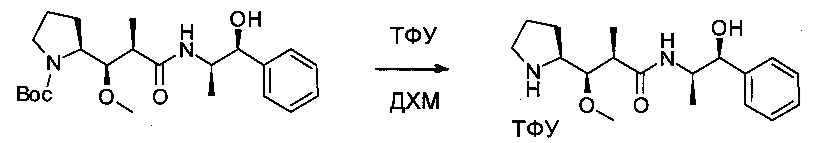
Соединение 2А (2,7 г, 6,42 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ДХМ (40 мл), затем охлаждали до 0°С. Добавляли ТФУ (25 мл), и раствор перемешивали в течение 2 часов при 0°С. Реакционную смесь концентрировали при пониженном давлении с получением 4,4 г соединения 2В в форме желтого масла.
Пример 2С: (9Н-флуорен-9-ил)метил-((S)-1-(((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)(метил)амино)-3-метил-1-оксобутан-2-ил)карбамат
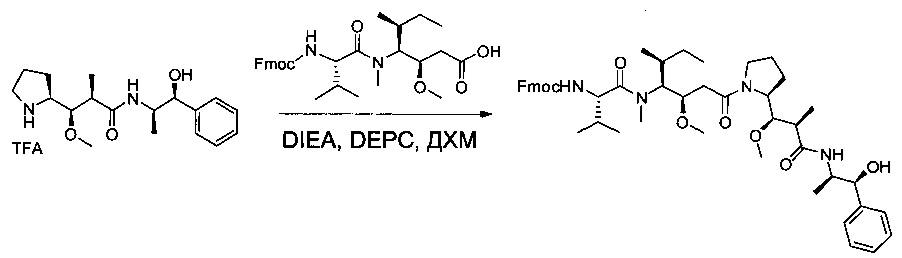
Соединения 2В (4,4 г, 10,13 ммоль, 1,00 экв.) и 1W (5,31 г, 10,12 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ДХМ (45 мл). Раствор охлаждали до 0°С, затем добавляли по каплям DEPC (1,62 мл) и DIEA (8,4 мл). Реакционную смесь перемешивали в течение 2 часов при 0°С, затем при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли 100 мл воды и экстрагировали три раза 50 мл ДХМ. Органические фазы объединяли, промывали один раз 50 мл KHSO4 (1 моль/л), один раз 50 мл NaHCO3 (нас.), один раз 50 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали под давлением с получением 3,3 г (39%) соединения 2С в форме желтого твердого вещества.
Пример 2D: (S)-2-амино-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид
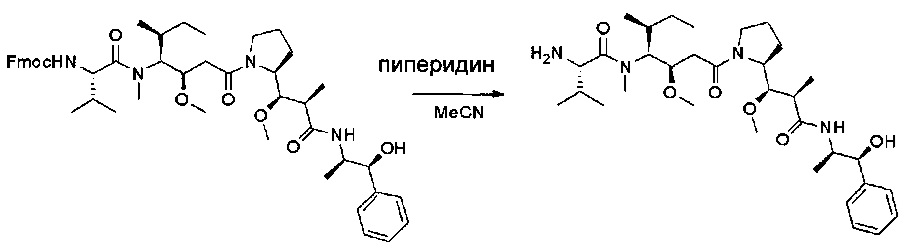
Соединение 2С (300 мг, 0,36 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ACN (2 мл) и пиперидине (0,5 мл). Раствор оставляли при перемешивании при температуре окружающей среды в течение ночи, затем выпаривали до сухого состояния при пониженном давлении. Остаток очищали на колонке силикагеля смесью ДХМ и МеОН (1:100) с получением 150 мг (68%) соединения 2D в форме белого твердого вещества.
Пример 2Е: метил-2-((трет-бутоксикарбонил)амино)изоникотинат
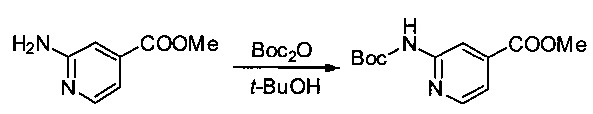
Метил-2-аминопиридин-4-карбоксилат (2 г, 13,14 ммоль, 1,00 экв.) растворяли в трет-бутаноле (20 мл), после чего добавляли ди-трет-бутилдикарбонат (4,02 г, 18,42 ммоль, 1,40 экв.). Реакционную смесь перемешивали при 60°С в течение ночи, затем реакцию останавливали путем добавления водного 1 М раствора NaHCO3 (50 мл). Твердое вещество выделяли фильтрованием, промывали 50 мл EtOH, затем высушивали в вакууме с получением 2,5 г (75%) соединения 2Е в форме белого твердого вещества.
Пример 2F: трет-бутил-(4-(гидроксиметил)пиридин-2-ил)карбамат
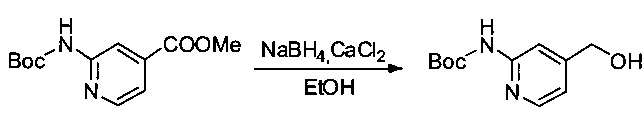
Соединение 2Е (2,5 г, 9,91 ммоль, 1,00 экв.) и CaCl2 (1,65 г) растворяли в EtOH (30 мл). Раствор охлаждали до 0°С, затем постепенно добавляли NaBH4 (1,13 г, 29,87 ммоль, 3,01 экв.). Раствор оставляли при перемешивании в течение ночи при температуре окружающей среды, затем реакцию останавливали добавлением воды (50 мл). Смесь экстрагировали три раза 20 мл EtOAc. Органические фазы объединяли, дважды промывали 20 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 2,0 г (90%) соединения 2F в форме бесцветного твердого вещества.
Пример 2G: трет-бутил-(4-формилпиридин-2-ил)карбамат
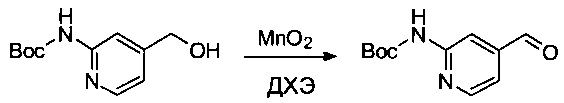
Соединение 2F (2,5 г, 11,15 ммоль, 1,00 экв.) растворяли в ДХЭ (25 мл), затем добавляли 19,4 г (223,14 ммоль, 20,02 экв.) MnO2. Смесь оставляли при перемешивании в течение ночи при 70°С, затем твердые вещества выделяли фильтрованием. Фильтрат выпаривали до сухого состояния с получением 1,4 г (57%) соединения 2G в форме белого твердого вещества.
Пример 2Н: бензил-(S)-2-(((2-((трет-бутоксикарбонил)амино)пиридин-4-ил)метил)(метил)амино)-3-метилбутаноат
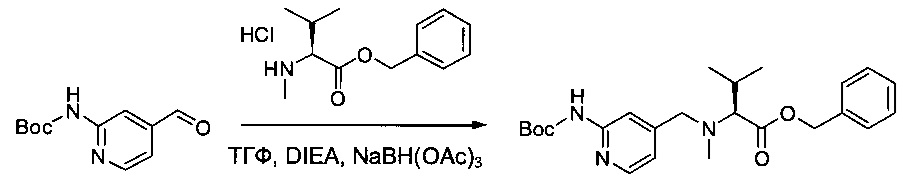
Соединение 2G (2,3 г, 10,35 ммоль, 1,00 экв.) растворяли в 25 мл ТГФ в присутствии соединения 1ZC (2,93 г, 11,37 ммоль, 1,10 экв.), DIEA (5,39 г, 41,71 ммоль, 4,03 экв.) и NaBH(OAc)3 (4,39 г, 20,71 ммоль, 2,00 экв.). Реакционную смесь перемешивали в течение 6 часов при температуре окружающей среды, затем нейтрализовали 60 мл NaHCO3 (нас.) и экстрагировали 3 раза 20 мл AcOEt. Органические фазы объединяли, дважды промывали 20 мл NaCl (нас.), высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (1:15) с получением 2,7 г (61%) соединения 2Н в форме белого твердого вещества.
Пример 2I: (S)-2-(((2-((трет-бутоксикарбонил)амино)пиридин-4-ил)метил)(метил)амино)-3-метилбутановая кислота
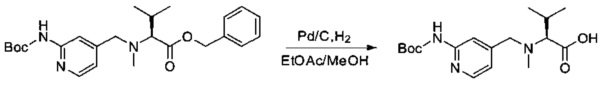
Соединение 2Н (500 мг, 1,17 ммоль, 1,00 экв.) растворяли в 10 мл AcOEt и 2 мл метанола в присутствии Pd/C (250 мг) и гидрогенизировали в течение 3 часов при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении с получением 254 мг (64%) соединения 2I в форме бесцветного твердого вещества.
Пример 2J: трет-бутил-(4-((3S,6S,9S,10R)-9-((S)-втор-бутил)-10-(2-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-2-оксоэтил)-3,6-диизопропил-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазадодецил)пиридин-2-ил)карбамат
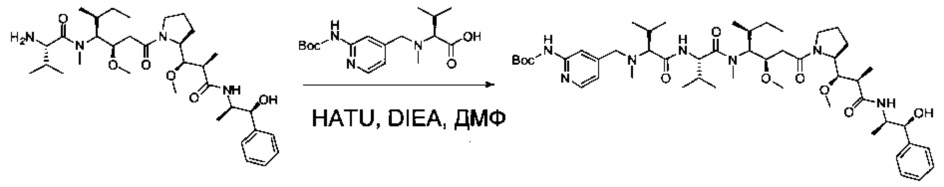
Соединение 2J было получено подобно соединению 1ZG из амина 2D (85,2 мг, 0,14 ммоль, 1,50 экв.), кислоты 2I (31,7 мг, 0,09 ммоль, 1,00 экв.), HATU (42,9 мг, 0,11 ммоль, 1,20 экв.) и DIEA (36,7 мг, 0,28 ммоль, 3,02 экв.) в ДМФ (3 мл). После выпаривания до сухого состояния было получено 100 мг неочищенного продукта в форме белого твердого вещества.
Пример 2: Соединение 2J (100 мг, 0,11 ммоль, 1,00 экв.) растворяли в 2 мл ДХМ и 1 мл ТФУ. Реакционную смесь перемешивали в течение 1 часа при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток (80 мг) очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2489 при 254 нм и 220 нм). Соединение 2 было получено при выходе 6% (6,3 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (Колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,8 мл/мин, от 10% до 95% ACN в воде (0,05% ТФУ) за 6 минут); ИЭР (С45Н73N7O7, точная масса 823,56) m/z: 824,5 (МН+) и 412,9 (М.2Н+/2, 100%), 3,21 мин (99,2%, 210 нм).
1Н ЯМР (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.81-7.79 (m, 1Н); 7.39-7.29 (m, 5Н); 6.61-6.59 (m, 2Н); 4.84-4.52 (m, 1Н); 4.32-4.02 (m, 1Н); 3.90-2.98 (m, 10Н); 2.90-2.78 (m, 1Н); 2.55-0.81 (m, 39Н).
Справочный пример 3
Метил-((S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трифторуксусная кислота

Пример 3А: трет-бутил-(S)-2-((1R,2R)-1-метокси-3-(((S)-1-метокси-1-оксо-3-фенилпропан-2-ил)амино)-2-метил-3-оксопропил)пирролидин-1-карбоксилат
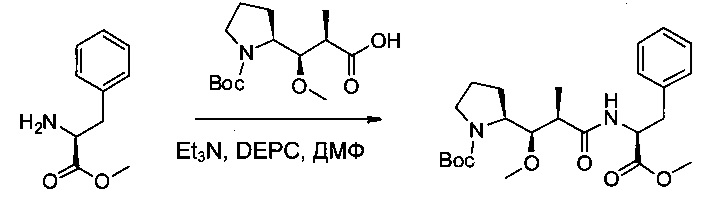
Соединение 1D (3 г, 10,44 ммоль, 1,00 экв.) и метил-(S)-2-амино-3-фенилпропаноат (2,25 г, 12,55 ммоль, 1,20 экв.) растворяли в инертной атмосфере в ДМФ (40 мл). Раствор охлаждали до 0°С, затем добавляли по каплям DEPC (1,67 мл, 1,05 экв.) и TEA (3,64 мл, 2,50 экв.). Реакционную смесь перемешивали в течение 2 часов при 0°С, затем при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли 100 мл воды и экстрагировали три раза 50 мл EtOAc. Органические фазы объединяли, промывали один раз 100 мл KHSO4 (1 моль/л), один раз 100 мл NaHCO3 (нас.), один раз 100 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали под давлением с получением 4 г (85%) соединения 3А в форме бесцветного масла.
Пример 3В: 2,2,2-трифторацетат метил-(S)-2-((2R,3R)-3-метокси-2-метил-3-((S)-пирролидин-2-ил)пропанамидо)-3-фенилпропаноата
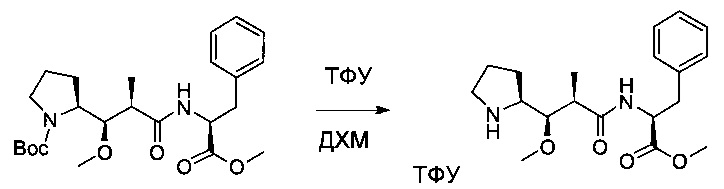
Соединение 3А (5 г, 11,15 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ДХМ (40 мл). Добавляли ТФУ (25 мл), и раствор перемешивали в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении с получением 8 г соединения 3В в форме желтого масла.
Пример 3С: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((((9Н-флуорен-9-ил)метокси)карбонил)амино)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
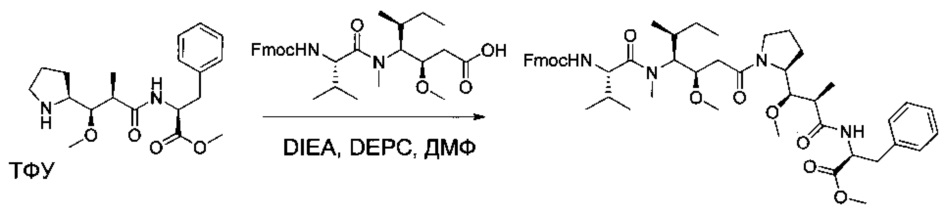
Соединения 3В (8,03 г, 17,36 ммоль, 1,00 экв.) и 1W (9,1 г, 17,34 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ДХМ (80 мл). Раствор охлаждали до 0°С, затем DEPC (2,8 мл) и добавляли по каплям DIEA (12 мл). Реакционную смесь перемешивали в течение 2 часов при 0°С, затем при температуре окружающей среды в течение ночи. Реакционную смесь разбавляли 200 мл воды и экстрагировали три раза 50 мл ДХМ. Органические фазы объединяли, промывали один раз 50 мл KHSO4 (1 моль/л), один раз 50 мл NaHСO3 (нас.), один раз 50 мл NaCl (нас.), затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 5 г (34%) соединения 3С в форме желтого твердого вещества.
Пример 3D: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-амино-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
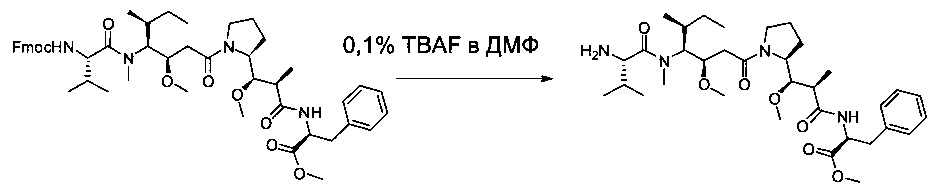
Соединение 3С (5,5 г, 6,43 ммоль, 1,00 экв.) растворяли в инертной атмосфере в растворе фторида тетрабутиламмония (TBAF, 2,61 г, 9,98 ммоль, 1,55 экв.) в ДМФ (100 мл). Раствор перемешивали при температуре окружающей среды в течение 2 часов, затем разбавляли 100 мл воды и экстрагировали три раза 50 мл EtOAc. Органические фазы объединяли, затем высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 3,3 г (81%) соединения 3D в форме желтого твердого вещества.
Пример 3Е: бензил-(S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутаноат
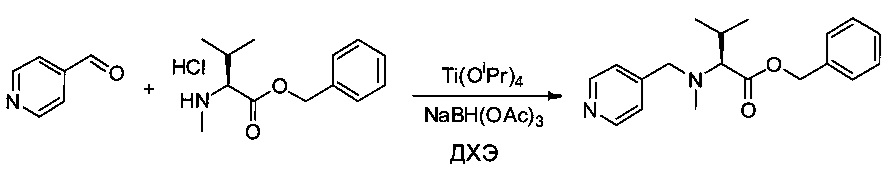
Пиридин-4-карбальдегид (1 г, 9,34 ммоль, 1,00 экв.) растворяли в 10 мл 1,2-дихлорэтана (ДХЭ) в присутствии соединения 1ZC (2,9 г, 11,25 ммоль, 1,21 экв.) и изопропоксида титана (IV) (4,19 мл, 1,40 экв.). Смесь перемешивали при температуре окружающей среды в течение 30 минут, затем добавляли 2,77 г NaBH(OAc)3 (13,07 ммоль, 1,40 экв.). Реакционную среду оставляли при перемешивании в течение ночи, затем нейтрализовали 100 мл воды, и смесь экстрагировали 3 раза 50 мл AcOEt. Органические фазы объединяли и выпаривали до сухого состояния. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (1:20) с получением 1,3 г (45%) соединения 3Е в форме бесцветного масла.
Пример 3F: (S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутановая кислота
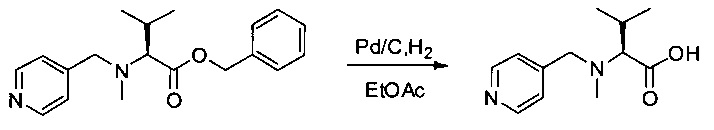
Соединение 3Е (800 мг, 2,56 ммоль, 1,00 экв.) растворяли в 30 мл AcOEt в присутствии Pd/C (300 мг) и гидрогенизировали в течение 3 часов при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью ДХМ и МеОН (от 100:1 до 5:1) с получением 100 мг (18%) соединения 3F в форме белого твердого вещества.
Пример 3: Соединения 3D (50 мг, 0,08 ммоль, 1,00 экв.) и 3F (26,34 мг, 0,12 ммоль, 1,50 экв.) растворяли в 3 мл ДХМ. Раствор охлаждали до 0°С, затем добавляли 0,018 мл DEPC и 0,0392 мл DIEA. Реакционную смесь перемешивали при 0°С в течение 2 часов, затем при температуре окружающей среды в течение ночи. Реакционную среду концентрировали при пониженном давлении, и остаток (70 мг) очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм). Соединение 3 было получено при выходе 27% (20 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (Колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,5 мл/мин, от 10% до 95% ACN в воде (0,05% ТФУ) за 8 минут); ИЭР (C46H72N6O8, точная масса 836,5) m/z: 837,5 (МН+) и 419,4 (М.2Н+/2 (100%)), 7,04 мин (90,0%, 210 нм)
1Н ЯМР (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.76-8.74 (m, 2Н); 8.53-8.48 (m, 0.4Н, NHCO неполный обмен); 8.29-8.15 (m, 0.8Н, NHCO неполный обмен); 8.01 (s, 2Н), 7.31-7.22 (m, 5Н), 4.88-4.68 (m, 3Н); 4.31-4.07 (m, 2H); 3.94-2.90 (m, 18H); 2.55-0.86 (m, 38H).
Справочный пример 4
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(пиридин-4-илметил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, трифторуксусная кислота
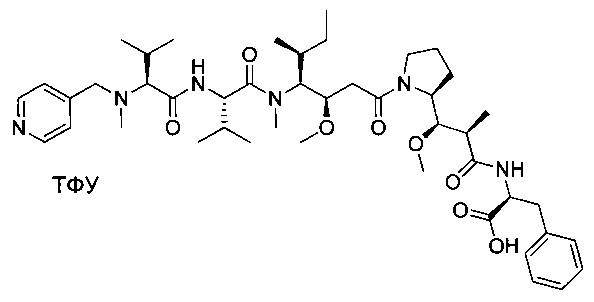
Пример 4: Соединение 3 (100 мг, 0,11 ммоль, 1,00 экв.) растворяли в смеси воды (5 мл), ACN (5 мл) и пиперидина (2,5 мл). Реакционную смесь оставляли при перемешивании в течение ночи, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм) с получением 20 мг (20%) соединения 4 в форме белого твердого вещества.
ЖХ/МС/УФ (Колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,5 мл/мин, от 10% до 95% ACN в воде (0,05% ТФУ) за 8 минут); ИЭР (C45H70N6O8, точная масса 822,5) m/z: 823,5 (МН+) и 412,4 (М.2Н+/2, 100%), 6,84 мин (89,1%, 210 нм).
1Н ЯМР (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.79-8.78 (m, 2Н); 8.09 (m, 2Н); 7.30-7.21 (m, 5Н); 4.80-4.80 (m, 1Н), 4.36-0.87 (m, 58Н).
Справочный пример 6
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, бис-трифторуксусная кислота
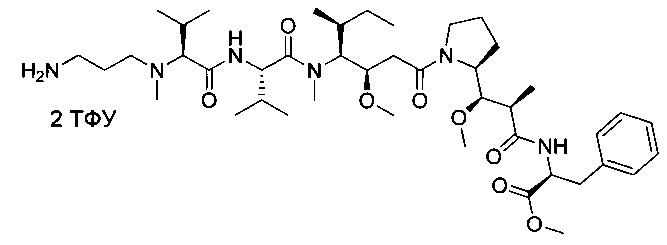
Пример 6А: метил-(2S)-2-[(2R)-2-[(R)-[(2S)-1-[(3R,4S,5S)-4-[(2S)-2-[(2S)-2-[(3-[[(трет-бутокси)карбонил]амино]пропил)(метил)амино]-3-метилбутанамидо]-N,3-диметилбутанамидо]-3-метокси-5-метилгептаноил]пирролидин-2-ил](метокси)метил]пропанамидо]-3-фенилпропаноат
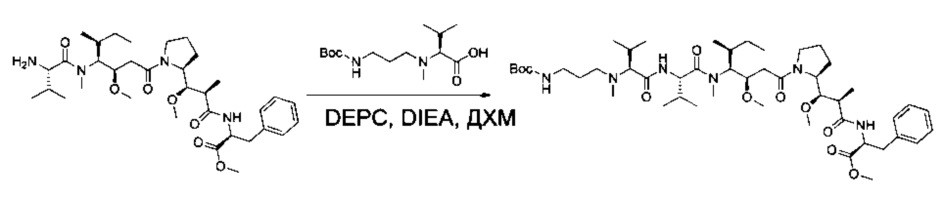
Соединение 3D (157,5 мг, 0,25 ммоль, 1,00 экв.) растворяли при 0°С в инертной атмосфере в 3 мл ДХМ в присутствии карбоновой кислоты 1ZF (78,7 мг, 0,27 ммоль, 1,10 экв.), DEPC (46 мкл) и DIEA (124 мкл). Реакционную смесь перемешивали в течение 2 часов при низкой температуре, а затем охлаждающую баню удаляли, и перемешивание продолжали в течение 4 часов. Затем ее концентрировали при пониженном давлении с получением 200 мг соединения 6А в форме неочищенного желтого масла. Его использовали в таком виде на следующей стадии.
Пример 6: Соединение 6А (200 мг, 0,22 ммоль, 1,00 экв.) растворяли в инертной атмосфере при 0°С в 2 мл ДХМ. Добавляли по каплям ТФУ (1 мл), и охлаждающую баню удаляли. Реакционную смесь перемешивали в течение 1 часа при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2489 при 254 нм и 220 нм) с получением 60 мг (26%, выход за 2 стадии) соединения 6 в форме белого твердого вещества.
ЖХ/МС/УФ (Zorbax Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл/мин, 40°С, от 30 до 80% метанола в воде (0,1% Н3РO4) за 18 минут); ИЭР (C43H74N6O8, точная масса 802,56) m/z: 804 (МН+); 11,50 мин (91,5%, 210 нм).
1Н ЯМР (300 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.52 (d, 0.3Н, NHCO неполный обмен); 8.25 (d, 0.5Н, NHCO неполный обмен); 7.30-7.22 (m, 5Н); 4.9-4.6 (m, 3Н); 4.2-4.0 (m, 1Н); 4.0-0.86 (m, 61Н).
Справочный пример 7
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминопропил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, бис-трифторуксусная кислота
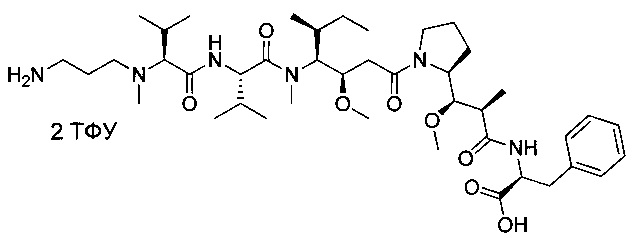
Пример 7: Соединение 6 (70 мг, 0,08 ммоль, 1,00 экв.) растворяли в смеси воды (5 мл), ACN (2,5 мл) и пиперидина (5 мл). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2489 при 254 нм и 220 нм) с получением 14,6 мг (21%) соединения 7 в форме белого твердого вещества.
ЖХ/МС/УФ (Ascentis Express C18, 2,7 мкм, 4,6×100 мм; 1,5 мл/мин, 40°С, от 0 до 80% метанола в воде (0,05% ТФУ) за 8 минут); ИЭР (C42H72N6O8, точная масса 788,54) m/z: 790 (МН+), 5,71 мин (96,83%, 210 нм).
1Н ЯМР (300 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.42 (d, 0.3Н, NHCO неполный обмен); 8.15 (d, 0.2Н, NHCO неполный обмен); 7.31-7.21 (m, 5Н); 4.9-4.6 (m, 3Н); 4.25-4.0 (m, 1Н); 4.0-0.86 (m, 59Н).
Пример 11
(S)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)фенетил)амино)бутанамидо)бутанамид, трифторуксусная кислота
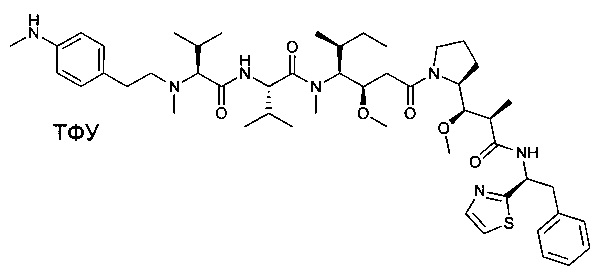
Пример 11А: трет-бутил N-[4-(2-гидроксиэтил)фенил]карбамат
Соединение 11А было получено при выходе 75% после взаимодействия при температуре окружающей среды 2-(4-аминофенил)этанола с ВОС2O в ТГФ.
Пример 11В: трет-бутил N-[4-(2-оксоэтил)фенил]карбамат
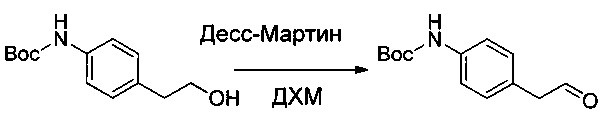
Соединение 11А (2,5 г, 10,5 ммоль, 1,00 экв.) растворяли в 25 мл ДХМ, затем охлаждали до -78°С. Добавляли по каплям раствор периодинана Десса-Мартина (DMP, 6,71 г, 15,8 ммоль, 1,5 экв.) в ДХМ (10 мл). Охлаждающую баню удаляли, и перемешивание продолжали в течение 1 часа при температуре окружающей среды. Реакционную смесь нейтрализовали 60 мл смеси 50/50 насыщенного водного раствора бикарбоната натрия и насыщенного водного раствора Na2S2O3. Полученный в результате раствор экстрагировали 3 раза 30 мл EtOAc. Органические фазы объединяли, дважды промывали насыщенным водным раствором NaCl, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (EtOAc/ПЭ 1/15) с получением 1,0 г (40%) соединения 11В в форме бледно-желтого твердого вещества.
Пример 11С: бензил-(2S)-2-[[2-(4-[[(трет-бутокси)карбонил]амино]фенил)этил](метил)амино]-3-метилбутаноат
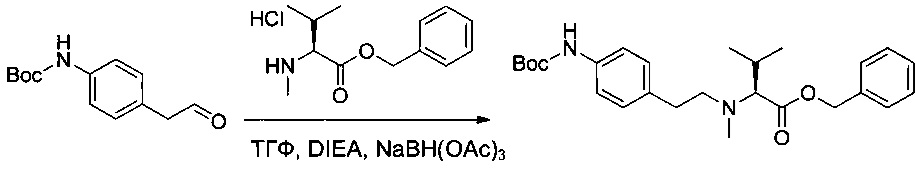
Соединение 1ZC (3,5 г, 13,6 ммоль, 1,1 экв.) растворяли в ТГФ (30 мл) в присутствии DIEA (6,4 г, 49,7 ммоль, 4,0 экв.), альдегид 11В (2,9 г, 12,3 ммоль, 1,0 экв.) и триацетоксиборгидрида натрия (5,23 г, 49,7 ммоль, 2,0 экв.). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, затем нейтрализовали 60 мл насыщенного раствора бикарбоната натрия. Полученный в результате раствор экстрагировали 3 раза 30 мл EtOAc. Органические фазы объединяли, дважды промывали Насыщенным водным раствором NaCl, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (EtOAc/ПЭ 1:20) с получением 3,7 г (68%) соединения 11С в форме желтого масла.
Пример 11D: (2S)-2-[[2-(4-[[(трет-бутокси)карбонил]амино]фенил)этил](метил)амино]-3-метилбутановая кислота
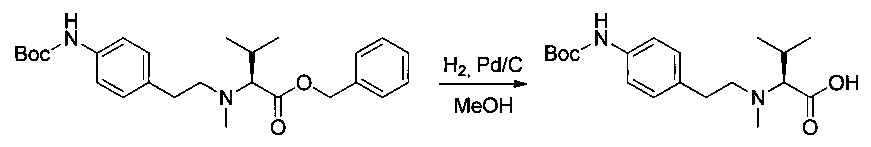
Соединение 11С (2 г, 4,5 ммоль, 1 экв.) растворяли в 10 мл метанола в присутствии Pd/C (2 г) и гидрогенизировали в течение 2 часов при нормальных значениях температуры и давления. Реакционную среду фильтровали и концентрировали при пониженном давлении с получением 1,2 г (75%) соединения 11D в форме желтого масла.
Пример 11Е: (2S)-2-[[2-(4-[[(трет-бутокси)карбонил](метил)амино]фенил)этил](метил)амино]-3-метилбутановая кислота
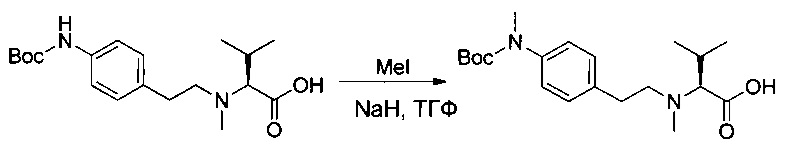
Соединение 11D (1,2 г, 3,4 ммоль, 1,00 экв.) растворяли в инертной атмосфере в ТГФ (20 мл). Реакционную среду охлаждали в ледяной бане, после чего добавляли порциями NaH (60% в масле, 549 мг, 13,7 ммоль, 4,0 экв.), а затем йодметан (4,9 г, 34 ммоль, 10 экв.). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, затем нейтрализовали водой и промывали 100 мл EtOAc. рН водного раствора доводили до 6-7 1 н. HCl. Этот водный раствор экстрагировали 3 раза 100 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали с получением 800 мг (64%) соединения 11Е в форме желтого твердого вещества.
Пример 11F: трет-бутил-N-[4-(2-[[(1S)-1-[[(1S)-1-[[(3R,4S,5S)-3-метокси-1-[(2S)-2-[(1R,2R)-1-метокси-2-метил-2-[[(1S)-2-фенил-1-(1,3-тиазол-2-ил)этил]карбамоил]этил]пирролидин-1-ил]-5-метил-1-оксогептан-4-ил](метил)карбамоил]-2-метилпропил]карбамоил]-2-метилпропил](метил)амино]этил)фенил]-N-метилкарбамат
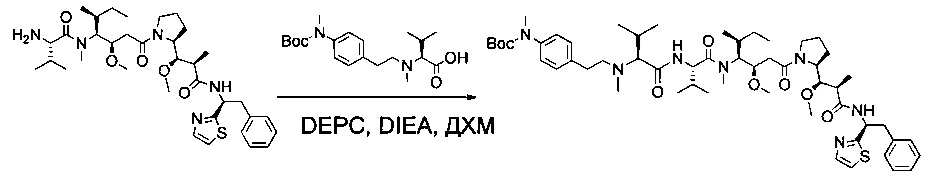
Соединение 11F было получено подобно соединению 6А из амина 1Y (150 мг, 0,22 ммоль, 1,2 экв.) и кислоты 11Е (70 мг, 0,19 ммоль, 1,0 экв.). После очистки на силикагеле (EtOAc/ПЭ 1:1) было получено 100 мг (52%) желаемого продукта в форме бледно-желтого твердого вещества.
Пример 11: Соединение 11 было получено таким же способом, как для соединения 1, из промежуточного соединения 11F (100 мг, 0,1 ммоль). Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2489 при 254 нм и 220 нм). Соединение 11 было получено при выходе 39% (39,7 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл/мин, 40°С, от 50 до 95% метанола в воде (0,05% ТФУ) за 18 минут); ИЭР (C50H77N7O6S, точная масса 903,57) m/z: 904,5 (МН+), 7,53 мин (93,68%, 254 нм).
1Н ЯМР (300 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.84 (d, 0.5Н, NHCO неполный обмен); 8.7-8.5 (m, 0.9Н, NHCO неполный обмен); 7.76-7.73 (m, 1Н); 7.55-7.4 (m, 1Н); 7.28-7.22 (m, 7Н); 7.08-7.05 (m, 2Н); 5.51-5.72 (m, 1Н); 4.9-4.80 (m, 2Н); 4.3-0.7 (m, 60Н).
Пример 12
Meтил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-димeтил-2-((S)-3-мeтил-2-(метил(4-(метиламино)фенетил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трифторуксусная кислота
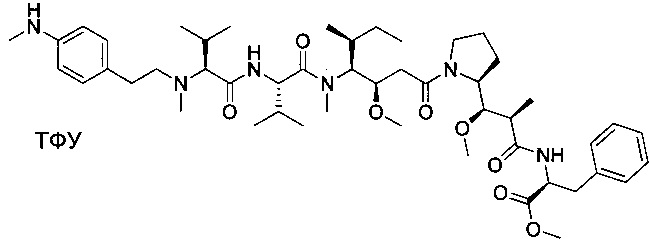
Пример 12: Таким же способом, как для конечных фаз в синтезе соединения 1, соединение 12 было получено в две стадии из амина 3D (118 мг, 0,19 ммоль) и кислоты 11Е (82 мг, 0,22 ммоль). Конечный остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2489 при 254 нм и 220 нм). Соединение 12 было получено при выходе 7% (13,7 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 1 мл/мин, 40°С, от 40 до 95% метанола в воде (0,05% ТФУ) за 18 минут); ИЭР (C49H78N6O8, точная масса 878,59) m/z: 879,7 (МН+), 10,07 мин (90,6%, 254 нм).
1Н: ЯМР (300 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.40 (se, 2Н); 7.38-7.22 (m, 7Н); 4.95-4.7 (m, 3Н); 4.2-4.0 (m, 1Н); 3.9-0.86 (m, 62Н).
Пример 13
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-N,3-диметил-2-((S)-3-метил-2-(метил(4-(метиламино)фенетил)амино)бутанамидо)бутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, трифторуксусная кислота
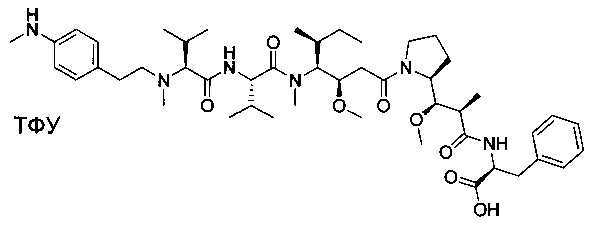
Пример 13: Соединение 13 было получено таким же способом, как для соединения 7, из соединения 12 (100 мг, 0,10 ммоль). Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2489 при 254 нм и 220 нм). Соединение 13 было получено при выходе 20% (20 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 1,5 мл/мин, 40°С, от 10 до 95% метанола в воде (0,05% ТФУ) за 8 минут); ИЭР (C48H76N6O8, точная масса 864,57) m/z: 865,6 (МН+), 6,05 мин (90,9%, 210 нм).
1Н ЯМР: (300 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.32-7.19 (m, 9Н); 4.9-4.65 (m, 3Н); 4.2-4.0 (m, 1Н); 3.9-0.86 (m, 59Н).
Пример 14
(S)-2-((S)-2-((3-aминoбeнзил)(мeтил)aминo)-3-мeтилбyтaнaмидo)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, трифторуксусная кислота

Пример 14А: трет-бутил-(3-(гидроксиметил)фенил)карбамат
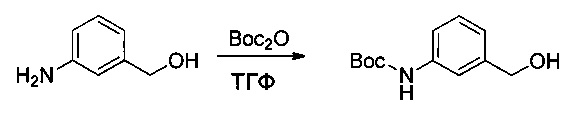
(3-Аминофенил)метанол (3 г, 24,36 ммоль, 1,00 экв.) растворяли в ТГФ (60 мл), после чего добавляли ди-трет-бутилдикарбонат (6,38 г, 29,23 ммоль, 1,20 экв.). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, а затем реакционную смесь разбавляли путем добавления 200 мл воды. Продукт экстрагировали 3 раза 100 мл AcOEt, а затем органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением неочищенного продукта (13,85 г соединения 14А) в форме желтого масла.
Пример 14В: трет-бутил-(3-формилфенил)карбамат
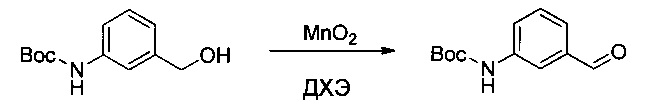
Соединение 14А (13,8 г, 61,81 ммоль, 1,00 экв.) растворяли в ДХЭ (400 мл), а затем добавляли МnO2 (54 г, 621,14 ммоль, 10,05 экв.). Смесь оставляли при перемешивании при температуре окружающей среды на 3 суток, после чего твердые вещества удаляли фильтрованием. Фильтрат выпаривали до сухого состояния, и остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (1:30) с получением 3 г (22%) соединения 14В в форме белого твердого вещества.
Пример 14С: бензил-(S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутаноат
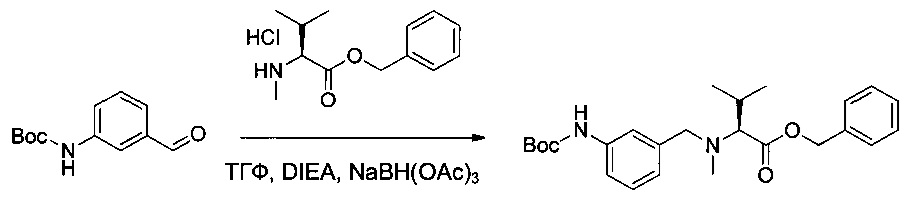
Соединение 14В (1 г, 4,52 ммоль, 1,00 экв.) растворяли в 20 мл ТГФ в присутствии соединения 1ZC (1,16 г, 4,50 ммоль, 1,00 экв.), DIEA (3 мл) и NaBH(OAc)3 (1,92 г, 9,06 ммоль, 2,01 экв.). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, а затем нейтрализовали 100 мл воды и экстрагировали 3 раза 50 мл AcOEt. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (1:50) с получением 1,9 г (99%) соединения 14С в форме белого твердого вещества.
Пример 14D: (S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутановая кислота
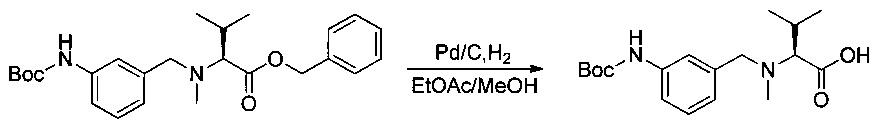
Соединение 14С (1 г, 2,34 ммоль, 1,00 экв.) растворяли в 30 мл AcOEt и 4 мл метанола в присутствии Pd/C (400 мг) и гидрогенизировали в течение 1 часа при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении с получением 680 мг (86%) соединения 14D в форме белого твердого вещества.
Пример 14Е: трет-бутил-(3-((3S,6S,9S,10R)-9-((S)-втор-бутил)-3,6-диизопропил-10-(2-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-2-оксоэтил)-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазадодецил)фенил)карбамат
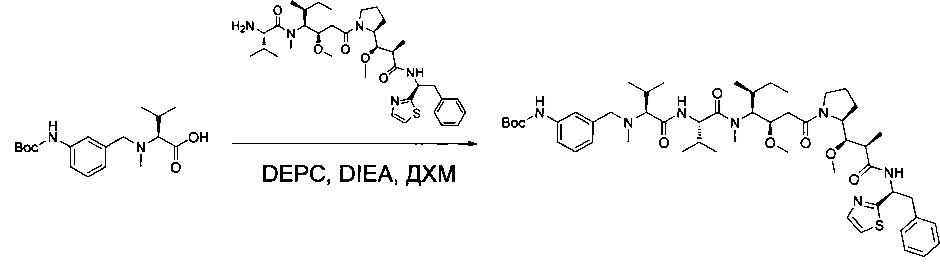
Соединение 14Е синтезировали таким же способом, как для соединения 3, из амина 1Y (100 мг, 0,15 ммоль, 1,00 экв.), кислоты 14D (102,27 мг, 0,30 ммоль, 2,00 экв.), DEPC (0,053 мл) и DIEA (0,046 мл) в ДХМ (3 мл). Неочищенный продукт (80 мг) очищали на колонке силикагеля смесью EtOAc и ПЭ (1:1) с получением 100 мг (67%) соединения 14Е в форме бледно-желтого твердого вещества.
Пример 14: Соединение 14 синтезировали таким же способом, как для соединения 2, из промежуточного соединения 14Е (100 мг, 0,10 ммоль, 1,00 экв.). Неочищенный продукт (80 мг) очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм). Соединение 14 было получено при выходе 10% (10 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Eclipse plus С8, 3,5 мкм, 4,6×150 мм; 40°С; 1,0 мл/мин, от 40% до 95% МеОН в воде (0,05% ТФУ) за 18 минут); ИЭР (C48H73N7O6S, точная масса 875,5) m/z: 876,5 (МН+) и 438,9 (М.2Н+/2, 100%), 11,35 мин (95,6%, 210 нм).
1Н ЯМР (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.92-8.86 (m, 0.4Н, NH неполный обмен); 8.70-8.54 (m, 0.6Н, NH неполный обмен); 7.88-7.78 (m, 1Н); 7.60-7.50 (m, 1Н); 7.45-6.97 (m, 9Н); 5.80-5.65 (m, 1Н); 4.85-4.70 (m, 1Н); 4.40-0.80 (m, 56Н).
Пример 15
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((S-аминобензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трифторуксусная кислота
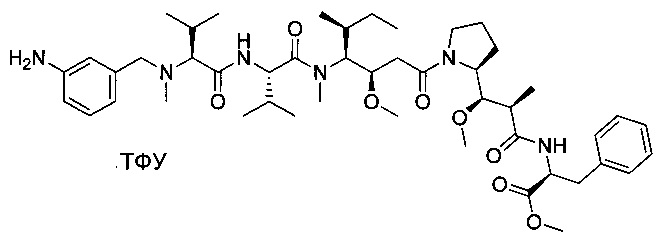
Пример 15А: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-((трет-бутоксикарбонил)амино)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
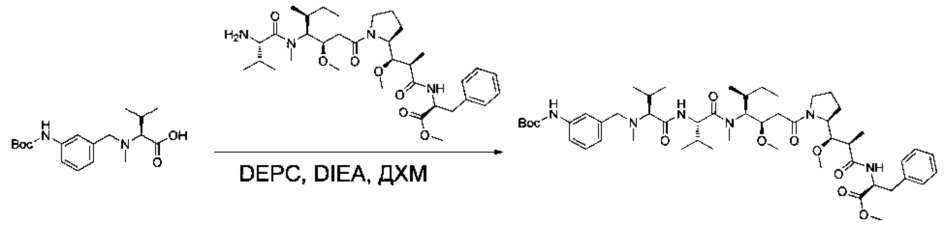
Соединение 15А синтезировали таким же способом, как для соединения 3, из амина 3D (200 мг, 0,32 ммоль, 1,00 экв.), кислоты 14D (212,6 мг, 0,63 ммоль, 2,00 экв.), DEPC (0,1103 мл) и DIEA (0,157 мл, 3,00 экв.) в ДХМ (5 мл). Неочищенный продукт очищали на колонке силикагеля смесью EtOAc и ПЭ (1:1) с получением 200 мг (67%) соединения 15А в форме желтого твердого вещества.
Пример 15: Соединение 15 синтезировали таким же способом, как для соединения 2, из промежуточного соединения 15А (200 мг, 0,21 ммоль, 1,00 экв.). Неочищенный продукт очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, Колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм). Соединение 15 было получено при выходе 19% (38,6 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (Колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% ТФУ) за 8 минут); ИЭР (C47H74N6O8, точная масса 850,5) m/z: 851,5 (МН+) и 426,4 (М.2Н+/2, 100%), 6,61 мин (91,1%, 210 нм).
1Н ЯМР (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.53-7.42 (m, 1Н); 7.35-7.18 (m, 8Н); 4.88-4.79 (m, 2Н); 4.42-4.00 (m, 3Н); 3.93-2.71 (m, 22Н); 2.61-0.81 (m, 33Н).
Примеры 19 и 20
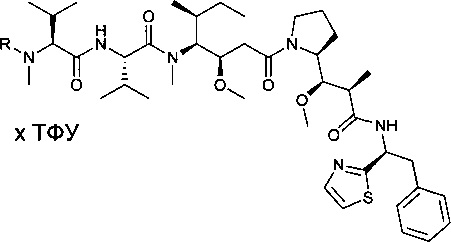
Соединения 19 и 20 были получены таким же способом, как для соединения 1, из аминов 1Y и 1ZC и соответствующих альдегидов.
трет-Бутил-4-формилфенилкарбонат, вовлеченный в получение соединения 19, был получен в одну стадию, как описано ниже: 4-гидроксибензальдегид (2,5 г, 20,5 ммоль, 1,0 экв.) растворяли в инертной атмосфере в ТГФ (20 мл) в присутствии 18-краун-6 (0,25 г) и карбоната калия (5 г). Реакционную смесь охлаждали до 0°С, а затем добавляли ди-трет-бутилдикарбонат (5,8 г, 26,58 ммоль, 1,30 экв.). Перемешивание продолжали в течение 1 часа при низкой температуре, после чего реакционную смесь нейтрализовали 30 мл воды. Полученный в результате раствор экстрагировали три раза 200 мл EtOAc. Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на силикагеле (EtOAc/ПЭ 1:10), и получили 4,2 г (92%) трет-бутил-4-формилфенилкарбоната в форме бледно-желтого твердого вещества.
4-Нитробензальдегид, вовлеченный в получение соединения 20, имелся в продаже.
Синтез соединения 20 выполняли путем восстановления нитрогруппы. Его проводили, как описано ниже: (2S)-N-[(3R,4S,5S)-1-[(2S)-2-[(1R,2R)-2-[[(1S,2R)-1-гидрокси-1-фенилпропан-2-ил]карбамоил]-1-метокси-2-метилэтил]пирролидин-1-ил]-3-метокси-5-метил-1-оксогептан-4-ил]-N,3-диметил-2-[(2S)-3-метил-2-[метил[(4-нитрофенил)метил]амино]бутанамидо]бутанамид (40 мг, 0,05 ммоль, 1,0 экв.) растворяли в 15 мл этанола. Добавляли дигидрат хлорида олова (II) (317 мг, 1,4 ммоль, 30 экв.), и раствор оставляли при перемешивании на 3 суток при температуре окружающей среды. Реакционную смесь нейтрализовали 50 мл воды, затем экстрагировали три раза 50 мл EtOAc. Органические фазы объединяли, высушивали над безводным сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением соединения 20 в неочищенном состоянии.
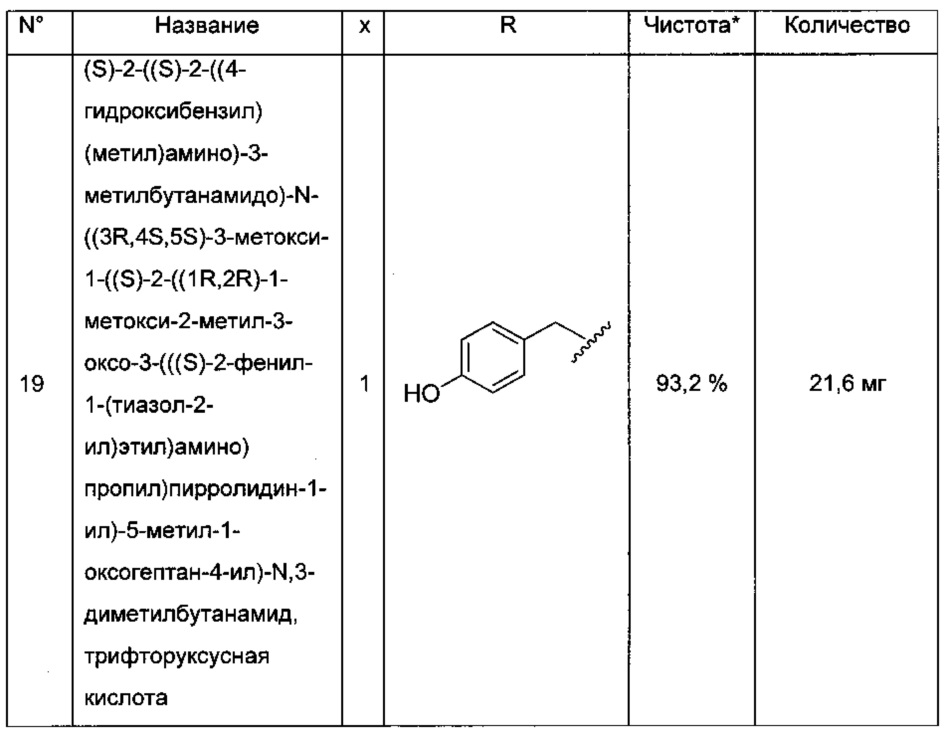
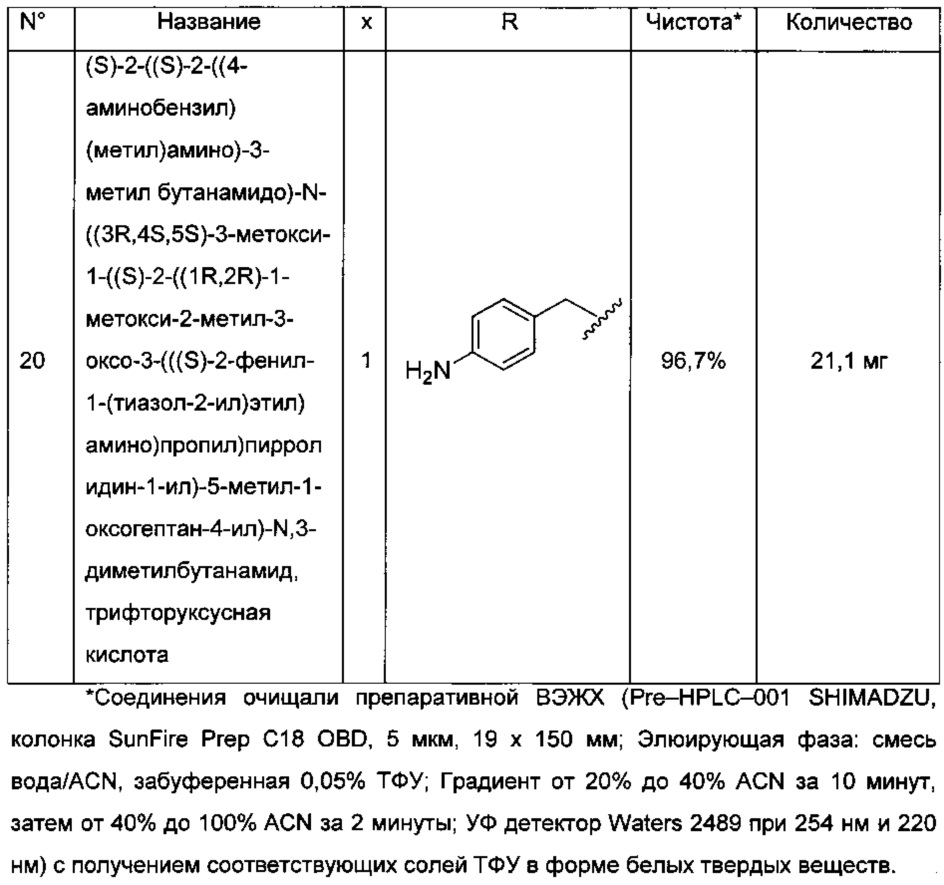
Характеризация конечных продуктов: Соединение 19 ЖХ/МС/УФ ИЭР: (C48H72N6O7S, точная масса 876,52) m/z 877 (МН+), 439 [100%, (М.2Н+)/2]; УФ: RT=1,76 мин (93,2%, 220 нм). Соединение 20 1Н ЯМР: (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.85-7.80 (m, 1Н); 7.6-7.5 (m, 1Н); 7.4-7.15 (m, 5Н); 7.1-7.05 (m, 2Н); 6.73-6.70 (m, 2Н); 5.8-5.55 (m, 1Н); 5.0-4.7 (m, 2Н); 4.25-4.05 (m, 1Н); 4.0-0.8 (m, 54Н). ЖХ/МС/УФ ИЭР: (C48H73N7O7S, точная масса 875,53) m/z 876 (МН+), 439 [75%, (М.2Н+)/2]; УФ: RT=4,83 мин (96,8%, 254 нм). 1Н ЯМР (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.85-7.80 (m, 1Н); 7.6-7.5 (m, 1Н); 7.4-7.1 (m, 7Н); 6.76-6.72 (m, 2Н); 5.8-5.55 (m, 1Н); 4.9-4.65 (m, 2Н); 4.25-4.05 (m, 1Н); 4.0-0.8 (m, 54Н).
Примеры 23 и 24
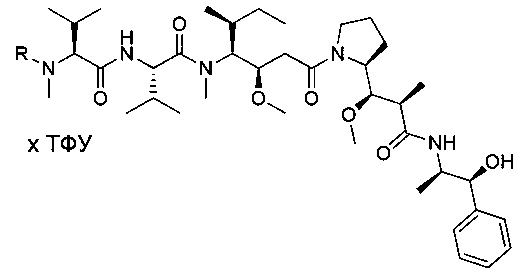
Соединения 23 и 24 были получены таким же способом, как для соединений 19 и 20, с заменой амина 1Y амином 2D.
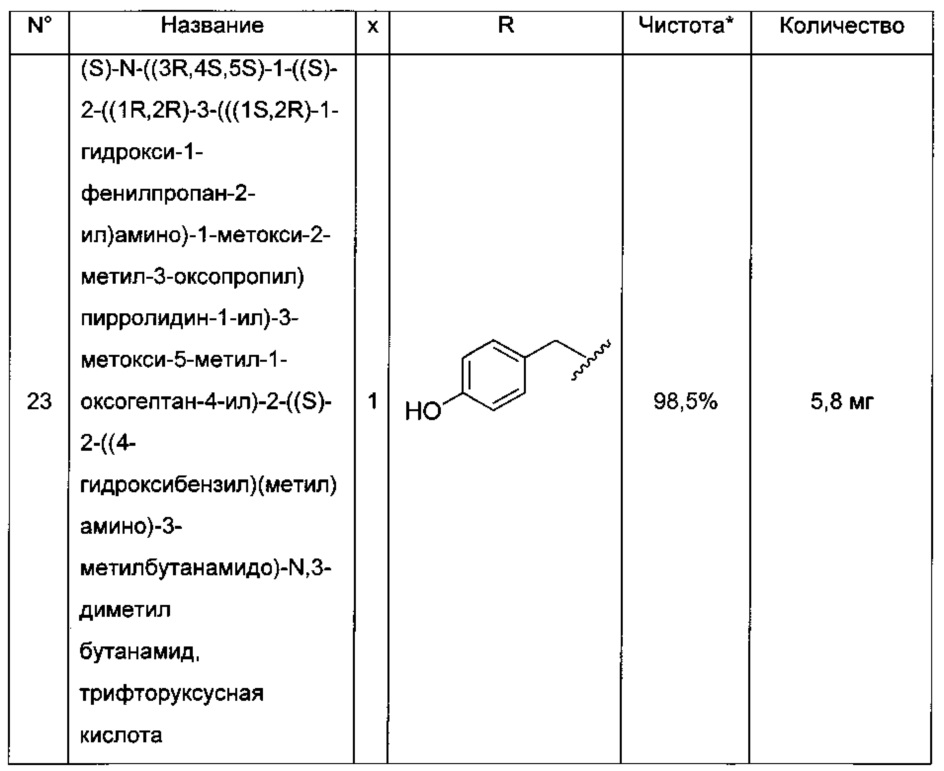
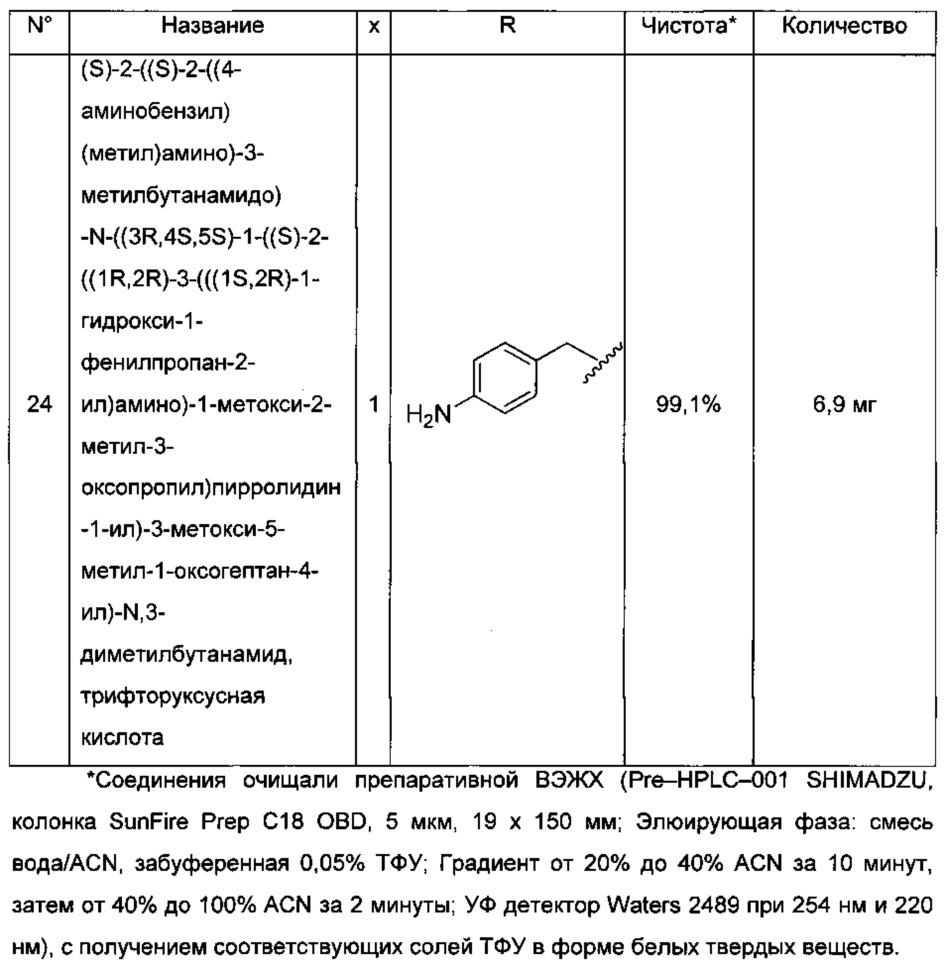
Характеризация конечных продуктов: Соединение 23 ЖХ/МС/УФ (ИЭР) (C46H73N5O8, точная масса 823,55) m/z 824 (МН+), 846 (MNa+), 413 (100%, (М.2Н+)/2); УФ: 4,76 мин (98,5%, 215 нм). 1Н ЯМР (400 МГц, CDCl3, млн-1): δ (Присутствие ротамеров) 7.5-7.2 (m, 5Н); 7.9-7.75 (m, 2Н); 5.5-5.3 (m, 1Н); 4.9-4.6 (m, 2Н); 4.55-4.15 (m, 4Н); 4.0-0.8 (m, 55Н). Соединение 24 ЖХ/МС/УФ (ИЭР) (C46H74N6O7, точная масса 822,56) m/z 823 (МН+), 845 (MNa+), 861 (МK+); УФ: 3,68 мин (99,15%, 254 нм). 1Н ЯМР (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.0-7.7 (m, 0.5Н, NHCO неполный обмен); 7.5-7.0 (m, 7Н); 6.75-6.65 (m, 2Н); 4.85-4.5 (m, 2Н); 4.4-4.05 (m, 2Н); 4.0-0.8 (m, 56Н).
Пример 27
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-гидроксифенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трифторуксусная кислота
Пример 27: Соединение 27 было получено таким же способом, как для соединения 3, из амина 3D (70 мг, 0,11 ммоль, 1,00 экв.), кислоты 49С (55,5 мг, 0,22 ммоль, 2,00 экв.), DEPC (0,034 мл, 2,00 экв.) и DIEA (0,055 мл, 3,00 экв.) в ДХМ (3 мл). Неочищенный продукт очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 45% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм). Соединение 27 было получено при выходе 3% (2,9 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 40°С; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% ТФУ) за 8 минут); ИЭР (C48H75N5O9, точная масса 866,56) m/z: 866,5 (МН+) и 433,9 (М.2Н+/2, 100%), 6,61 мин (89,1%, 210 нм).
1Н ЯМР (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.70-8.49 (m, 0.9 Н, NH/OH неполный обмен); 8.30-8.22 (m, 0.3Н, NH неполный обмен); 7.36-7.02 (m, 7Н); 6.86-6.62 (m, 2Н); 4.82-4.69 (m, 2Н); 4.20-4.03 (m, 1Н); 3.91-3.33 (m, 12Н); 3.30-2.90 (m, 17Н); 2.55-0.80 (m, 35Н).
Пример 28
(S)-2-((S)-2-((3-aминoбeнзил)(мeтил)aминo)-3-мeтилбyтaнaмидo)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, трифторуксусная кислота
Пример 28А: трет-бутил-(3-((3S,6S,9S,10R)-9-((S)-втор-бутил)-10-(2-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-2-оксоэтил)-3,6-диизопропил-2,8-диметил-4,7-диоксо-11-окса-2,5,8-триазадодецил)фенил)карбамат
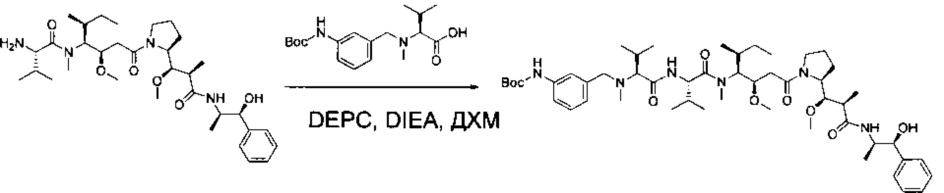
Соединение 28А было получено таким же способом, как для соединения 3, из амина 2D (100 мг, 0,17 ммоль, 1,00 экв.), кислоты 14D (111,25 мг, 0,33 ммоль, 2,00 экв.), DEPC (0,058 мл) и DIEA (0,05 мл) в ДХМ (3 мл). Остаток очищали на колонке силикагеля смесью EtOAc и гексана (1:1) с получением 100 мг (66%) соединения 28А в форме белого твердого вещества.
Пример 28: Соединение 28 синтезировали таким же способом, как для соединения 2, из промежуточного соединения 28А (100 мг, 0,11 ммоль, 1,00 экв.). Неочищенный продукт (80 мг) очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм). Соединение 28 было получено при выходе 20% (20 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Ascentis Express С18, 2,7 мкм, 4.6×100 мм; 40°С; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% ТФУ) за 8 минут); ИЭР (C46H74N6O7, точная масса 822,56) m/z: 823,5 (МН+) и 412,4 (М.2Н+/2, 100%), 12,45 мин (87,2%, 210 нм).
1Н ЯМР: (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.47-7.20 (m, 5Н); 7.10-7.01 (m, 1Н); 6.80-6.56 (m, 3Н); 4.82-4.52 (m, 3Н); 4.33-4.03 (m, 2Н); 3.91-3.82 (m, 0.5Н); 3.75-3.35 (m, 9.5Н); 3.28-3.10 (m, 2Н); 2.79-2.90 (m, 1Н); 2.60-2.40 (m, 2Н); 2.30-0.80 (m, 40Н).
Пример 29
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((3-аминобензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, трифторуксусная кислота
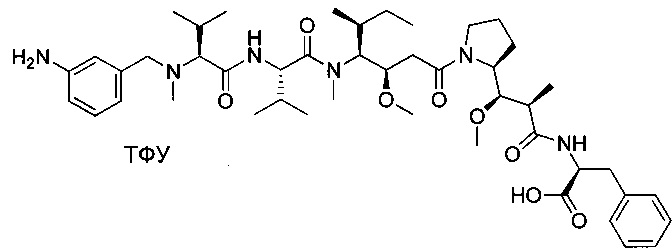
Пример 29: Соединение 15 (100 мг, 0,10 ммоль, 1,00 экв.) растворяли в смеси воды (5 мл), ACN (5 мл) и пиперидина (2,5 мл). Реакционную смесь оставляли при перемешивании в течение ночи при температуре окружающей среды, а затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм) с получением 20 мг (20%) соединения 29 в форме белого твердого вещества.
ЖХ/МС/УФ (колонка Eclipse Plus С8, 3,5 мкм, 4,6×150 мм; 40°С; 1,0 мл/мин, от 40% до 95% МеОН в воде (0,05% ТФУ) за 18 минут); ИЭР (C46H72N6O8, точная масса 836,54) m/z: 837,5 (МН+) и 419,4 (М.2Н+/2, 100%), 10,61 мин (92,5%, 210 нм).
1Н ЯМР: (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.38-7.15 (m, 6Н); 7.00-6.99 (m, 3Н); 4.85-4.68 (m, 2Н); 4.37-3.38 (m, 11Н); 3.31-2.70 (m, 8Н); 2.60-0.82 (m, 35Н).
Пример 49
(S)-2-((S)-2-((4-гидроксифенетил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)амино)пропил)пирролидин-1-ил)-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид, трифторуксусная кислота
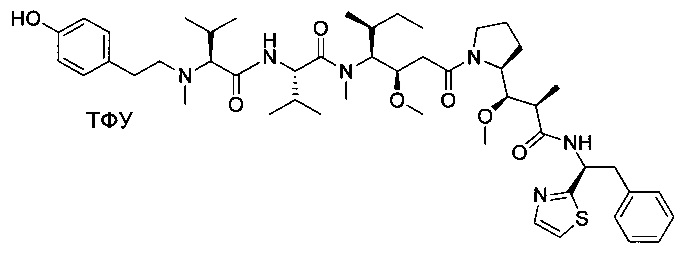
Пример 49А: 2-(4-гидроксифенил)ацетальдегид
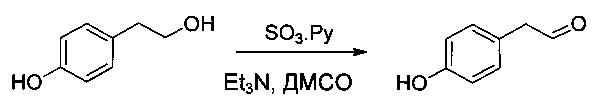
4-(2-Гидроксиэтил)фенол (4 г, 28,95 ммоль, 1,00 экв.) растворяли в ДМСО (32 мл), а затем добавляли по каплям TEA (8,8 мл, 2,20 экв.). Добавляли раствор SO3.Py (10 г, 2,20 экв.) в ДМСО (36 мл), и смесь оставляли при перемешивании в течение ночи при температуре окружающей среды. Реакционную смесь нейтрализовали 250 мл воды и экстрагировали 3 раза 100 мл AcOEt. Органические фазы объединяли, промывали 5 раз водой (100 мл), затем дважды 150 мл NaCl (нас.), высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на силикагеле (EtOAc/ПЭ (1:10)) с получением 1 г (25%) соединения 49А в форме бесцветного масла.
Пример 49В: бензил-(S)-2-((4-гидроксифенетил)(метил)амино)-3-метилбутаноат
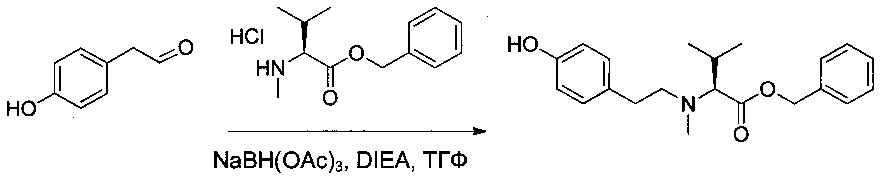
Соединение 49В синтезировали таким же способом, как для соединения 14С, из амина 1ZC (1,5 г, 5,82 ммоль, 0,99 экв.), альдегида 49А (800 мг, 5,88 ммоль, 1,00 экв.), NaBH(OAc)3 (2,7 г, 12,74 ммоль, 2,17 экв.) и DIEA (4,23 мл) в ТГФ (25 мл). Реакционную смесь нейтрализовали 50 мл воды и экстрагировали 3 раза 50 мл AcOEt. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали. Остаток очищали на силикагеле (EtOAc/ПЭ (1:10)) с получением 600 мг (37%) соединения 49В в форме белого твердого вещества.
Пример 49С: (S)-2-((4-гидроксифенетил)(метил)амино)-3-метилбутановая кислота
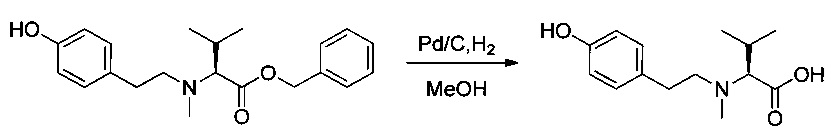
Соединение 49В (0,5 г, 1,46 ммоль, 1,00 экв.) растворяли в 40 мл МеОН в присутствии Pd/C (250 мг) и гидрогенизировали в течение 3 часов при температуре окружающей среды и атмосферном давлении. Реакционную среду фильтровали и концентрировали при пониженном давлении с получением 0,4 г соединения 49С в форме белого твердого вещества.
Пример 49: Соединение 49 синтезировали таким же способом, как для соединения 3, из амина 1Y (53,4 мг, 0,08 ммоль, 2,00 экв.), кислоты 49С (70 мг, 0,28 ммоль, 1,00 экв.), DEPC (0,032 мл, 2,00 экв.) и DIEA (0,053 мл, 3,00 экв.) в ДХМ (3 мл). Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка Atlantis Prep OBD Т3, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 45% ACN за 10 минут, затем от 45% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм) с получением 3 мг (1%) соединения 49 в форме белого твердого вещества.
ЖХ/МС/УФ (Колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% ТФУ) за 8 минут); ИЭР (C49H74N6O7S, точная масса 890,5) m/z: 891,5 (МН+) и 446,4 (М.2Н+/2, 100%), 6,69 мин (100%, 210 нм).
1Н ЯМР: (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.92-8.87 (m, 0.5 Н, NHCO, неполный обмен), 8.70-8.63 (m, 0.4 Н, NHCO, неполный обмен), 8.85-8.77 (m, 1Н), 7.59-7.51 (m, 1Н), 7.35-7.03 (m, 7Н), 6.82-6.71 (m, 2Н), 5.77-5.58 (m, 1Н), 5,81-5.70 (m, 1Н), 4.21-0.80 (m, 58Н).
Пример 50
(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-гидроксифенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропановая кислота, трифторуксусная кислота
Пример 50: Соединение 50 было получено таким же способом, как для соединения 4, из соединения 27 (100 мг, 0,10 ммоль, 1,00 экв.). Остаток очищали препаративной ВЭЖХ (Pre-HPLC-001 SHIMADZU, колонка Atlantis Prep OBD Т3, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 20% до 40% ACN за 10 минут, затем от 40% до 100% ACN за 2 минуты; УФ детектор Waters 2545 при 254 нм и 220 нм) с получением 10,7 мг (11%) соединения 50 в форме белого твердого вещества.
ЖХ/МС/УФ (Колонка Ascentis Express С18, 2,7 мкм, 4,6×100 мм; 40°С; 1,5 мл/мин, от 10% до 95% МеОН в воде (0,05% ТФУ) за 8 минут); ИЭР (C47H73N5O9, точная масса 851,5) m/z: 852,5 (МН+) и 426,8 (М.2Н+/2, 100%), 6,46 мин (91,7%, 210 нм).
1Н ЯМР: (400 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 7.34-7.15 (m, 5Н); 7.15-7.04 (se, 2Н), 6.82-6.83 (m, 2Н), 4.83-4.70 (m, 1Н), 4.21-4.00 (m, 1Н), 3.90-3.80 (m, 1Н), 3.74-3.62 (m, 1Н), 3.57-2.86 (m, 20Н), 2.56-0.80 (m, 36Н).
Пример 51
Метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-гидроксибензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат, трифторуксусная кислота
Пример 51 А: трет-бутил-(4-формилфенил)карбонат
4-Гидроксибензальдегид (3,0 г, 24 ммоль) растворяли в 30 мл ДХМ в присутствии 4-DMAP (300 мг, 2,46 ммоль, 0,1 экв.) и ди-трет-бутилдикарбоната (5,35 г, 24 ммоль, 1,0 экв.) и перемешивали в течение 1 часа при температуре окружающей среды. Затем раствор разбавляли 200 мл воды и экстрагировали 3 раза 100 мл ДХМ. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении с получением 5 г (92%) соединения 51А в форме белого твердого вещества.
Пример 51В: бензил-(S)-2-((4-((трет-бутоксикарбонил)окси)бензил)(метил)амино)-3-метилбутаноат
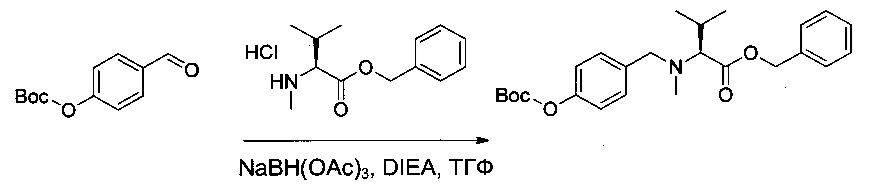
Соединение 51А (220 мг, 0,99 ммоль) растворяли в 5 мл ТГФ в присутствии соединения 1ZC (255 мг, 0,99 ммоль, 1,0 экв.), NaBH(OAc)3 (420 мг, 2 ммоль, 2,0 экв.) и DIEA (654 мкл) и перемешивали в течение ночи при температуре окружающей среды. Затем раствор разбавляли 100 мл воды и экстрагировали 3 раза 50 мл EtOAc. Органические фазы объединяли, высушивали над сульфатом натрия, фильтровали и концентрировали при пониженном давлении. Остаток очищали на колонке силикагеля смесью EtOAc и ПЭ (1:100) с получением 200 мг (47%) соединения 51В в форме белого твердого вещества.
Пример 51С: (S)-2-((4-((трет-бутоксикарбонил)окси)бензил)(метил)амино)-3-метилбутановая кислота
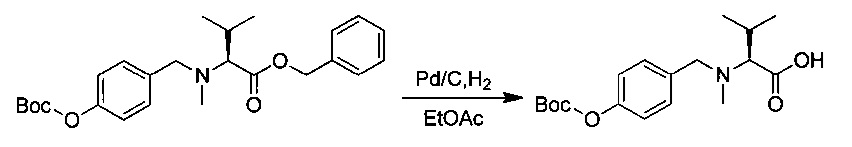
Соединение 51С было получено путем гидрогенизации соединения 51В (200 мг), следуя протоколу, используемому для получения соединения 3F.
Пример 51D: метил-(S)-2-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-((трет-бутоксикарбонил)окси)бензил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропанамидо)-3-фенилпропаноат
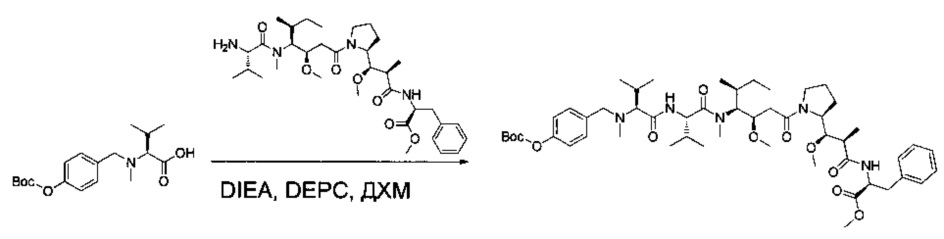
Соединение 51D было получено путем сочетания соединения 51С с амином 3D, следуя протоколу, используемому для получения соединения 3, с получением желаемого продукта в форме желтого масла при выходе 60%.
Пример 51: Соединение 51D (80 мг, 0,08 ммоль) растворяли в 1 мл ДХМ в присутствии 0,5 мл ТФУ, перемешивали в течение 2 часов при температуре окружающей среды, а затем концентрировали при пониженном давлении. Остаток очищали препаративной ВЭЖХ (Pre-HPLC-010, колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Элюирующая фаза: смесь вода/ACN, забуференная 0,05% ТФУ; Градиент от 23% до 40% ACN за 10 минут, затем от 40% до 95% ACN за 2 минуты; УФ детектор Waters 2489 при 254 нм и 220 нм). Соединение 51 было получено при выходе 24% (20 мг) в форме белого твердого вещества.
ЖХ/МС/УФ (Zorbax SB-Aq, 1,8 мкм, 4,6×100 мм; 2% МеОН в воде (0,05% ТФУ) в течение 1 минуты, затем от 2% до 95% МеОН за 13 минут); ИЭР (C47H73N5O9, точная масса 851,54) m/z: 874,5 (MNa+), 426,9 (М.2Н+/2); 12,48 мин (96%, 210 нм).
1Н ЯМР: (300 МГц, CD3OD, млн-1): δ (Присутствие ротамеров) 8.1-8.6 (m, 0.9Н, NHCO неполный обмен); 7.29-7.27 (m, 2Н), 7.25-6.86 (m, 5Н), 6.84-6.83 (m, 2Н), 4.83-4.72 (m, 3Н), 4.26-0.82 (m, 58Н).
Пример 61
(S)-2-((S)-2-((4-aминoфeнeтил)(мeтил)aминo)-3-мeтилбyтaнaмидo)-N-((3R,4S,5S)-3-метокси-1-((S)-2-((1R,2R)-1-метокси-2-метил-3-оксо-3-(((S)-2-фенил-1-(тиазол-2-ил)этил)aминo)пpoпил)пиppoлидин-1-ил)-5-мeтил-1-oкcoгeптaн-4-ил)-N,3-диметилбутанамид
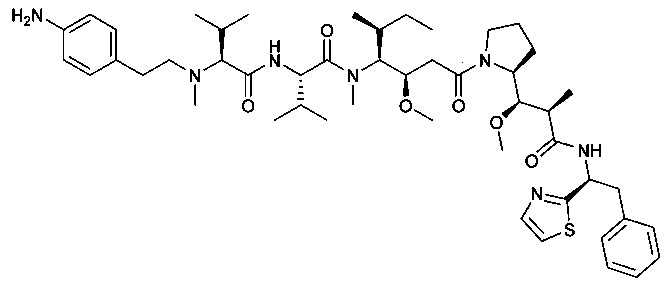
Пример 61А: дигидрохлорид N-(4-аминофенетил)-N-метил-L-валина
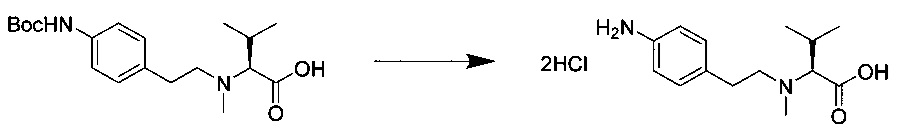
Соединение 11D (962 мг, 2,75 ммоль) растворяли в 10 мл имеющегося в продаже раствора HCl в пропан-2-оле (5-6 М) и перемешивали при комнатной температуре в течение 2 часов. Анализ ТСХ показал полное расходование исходного вещества. Растворитель выпаривали при пониженном давлении, и полученное в результате желтое твердое вещество растирали с Et2O (2×10 мл). Продукт высушивали в вакууме с получением соединения 61А в виде желтого твердого вещества (322 мг, 47%).
Пример 61: Карбоновую кислоту 61А (73 мг, 0,23 ммоль, 1 экв.) и амин 1Y (150 мг, 0,23 ммоль, 1 экв.) растворяли в безводном ДМФ (2 мл). Добавляли DIEA (158 мкл, 0,90 ммоль, 4 экв.) и DECP (51 мкл, 0,34 ммоль, 1,5 экв.), и реакционную смесь перемешивали в течение 4 часов при комнатной температуре. Анализ ЖХ-МС показал полное расходование исходного вещества. Растворитель выпаривали при пониженном давлении, и остаток очищали флэш-хроматографией на силикагеле (ДХМ/МеОН) с получением соединения 61 в виде желтого твердого вещества (83 мг, 40%).
1Н ЯМР: (500 МГц, ДМСО-d6, млн-1): δ (Присутствие ротамеров), 8.86 (d, 0.5Н, NHCO); 8.65 (d, 0.5Н, NHCO), 8.11-8.05 (m, 1H, NHCO), 7.80 (d, 0.5H, тиазол), 7.78 (d, 0.5H, тиазол), 7.65 (d, 0.5H, тиазол), 7.63 (d, 0.5H, тиазол), 7.32-7.12 (m, 5H), 6.83 (d, J=8.3 Гц, 2H), 6.45 (d, J=8.3 Гц, 2H), 5.56-5.49 (m, 0.5 H), 5.42-5.35 (m, 0.5H), 4.78 (s, 2H, NH2), 4.74-4.46 (m, 2H), 4.01-0.66 (m, 57H).
ВЭЖХ (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% ТФУ) в 2,25 минут, затем 95% MeCN в течение 0,5 минут, Tr=1,31 мин (96,5%, 220 нм). m/z (Q-TOF ИЭР+) 890,5558 (2%, МН+, для C49H76N7O6S требуется 890,5572), 445,7834 (100%, (МН2)2+, для C49H77N7O6S требуется 445,7823).
Пример 62
Метил-((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-аминофенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланинат
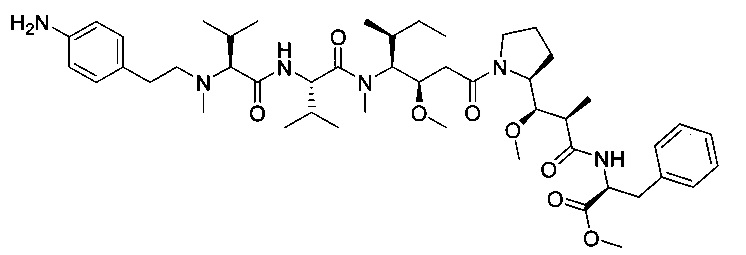
Пример 62: Соединение 62 было получено таким же способом, как для соединения 61, с использованием карбоновой кислоты 61А (69 мг, 0,21 ммоль, 1 экв.), амина 3D (135 мг, 0,21 ммоль, 1 экв.), DIEA (75 мкл, 0,43 ммоль, 2 экв.) и DECP (49 мкл, 0,32 ммоль, 1,5 экв.). Неочищенный продукт очищали флэш-хроматографией на силикагеле (ДХМ/МеОН) с получением соединения 62 в виде желтоватого твердого вещества (82 мг, 45%).
1Н ЯМР: (500 МГц, ДMCO-d6, млн-1): δ (Присутствие ротамеров), 8.50 (d, J=8.3, 0.5Н, NHCO); 8.27 (d, J=8.0, 0.5H, NHCO), 8.15-8.04 (m, 1H, NHCO), 7.27-7.13 (m, 5H), 6.86-6.79 (m, 2H), 6.48-6.42 (m, 2H), 4.78 (s, 2H, NH2), 4.74-4.44 (m, 3H), 4.01-3.72 (m, 1.5H), 3.66 (s, 1.5H, CO2Me), 3.63 (s, 1.5H, CO2Me), 3.57-0.65 (m, 55.5H).
ВЭЖХ (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% ТФУ) за 2,25 минуты, затем 95% MeCN в течение 0,5 минут, Tr=1,29 мин (95,3%, 220 нм).
m/z (Q-TOF ИЭР+) 865,5800 (2%, MH+, для C48H77N6O8 требуется 865,5797), 433,2937 (100%, (МН2)2+, для C48H78N6O8 требуется 433,2935).
Пример 63
((2R,3R)-3-((S)-1-((3R,4S,5S)-4-((S)-2-((S)-2-((4-аминофенетил)(метил)амино)-3-метилбутанамидо)-N,3-диметилбутанамидо)-3-метокси-5-метилгептаноил)пирролидин-2-ил)-3-метокси-2-метилпропаноил)-L-фенилаланин 2,2,2-трифторацетат
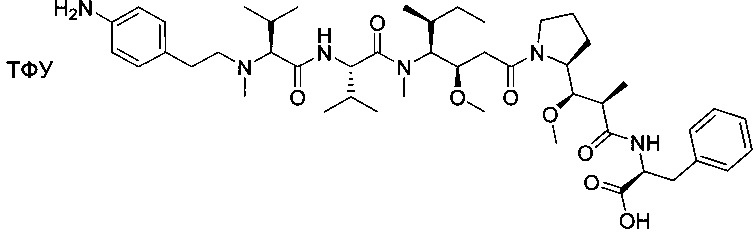
Пример 63: Соединение 62 (23 мг, 0,03 ммоль) растворяли в смеси воды (1 мл) и ацетонитрила (1 мл). Добавляли пиперидин (0,75 мл), и смесь перемешивали при комнатной температуре в течение 5 часов. Анализ ТСХ показал полное расходование исходного вещества. Растворитель выпаривали при пониженном давлении, и остаток очищали препаративной ВЭЖХ (колонка SunFire Prep С18 OBD, 5 мкм, 19×150 мм; Подвижная фаза: смесь вода/MeCN, забуференная 0,1% ТФУ; Градиент от 20% до 40% MeCN за 10 минут, затем от 40% до 100% MeCN за 2 минуты; Детектор УФ Waters 2545 при 254 нм и 220 нм). Соединение 63 было получено в виде белого твердого вещества (14 мг, 66%).
1Н ЯМР: (500 МГц, ДМСО-d6, млн-1): δ (Присутствие ротамеров), 12.7 (s(br), 1Н, СO2Н), 9.58 (m(br), 1Н); 9.04-8.89 (m, 1Н), 8.41 (d, 0.6Н, NHCO), 8.15 (d, 0.4Н, NHCO), 7.27-7.13 (m, 5H), 7.13-6.99 (m(br), 2H), 6.90-6.64 (s(br), 2H), 4.77-3.40 (m, 10H), 3.34-2.75 (m, 20H), 2.34-1.94 (m, 4H), 1.90-0.7 (m, 25H).
ВЭЖХ (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% ТФУ) за 2,25 минуты, затем 95% MeCN в течение 0,5 минут, Tr=1,24 мин (100%, 220 нм). m/z (Q-TOF ИЭР+) 851,5641 (6%, МН+, для C47H75N6O8 требуется 851,5641), 426,2854 (100%, (МН2)2+, для C47H76N6O8 требуется 426,2857).
Пример 64
(S)-2-((S)-2-((4-аминофенетил)(метил)амино)-3-метилбутанамидо)-N-((3R,4S,5S)-1-((S)-2-((1R,2R)-3-(((1S,2R)-1-гидрокси-1-фенилпропан-2-ил)амино)-1-метокси-2-метил-3-оксопропил)пирролидин-1-ил)-3-метокси-5-метил-1-оксогептан-4-ил)-N,3-диметилбутанамид
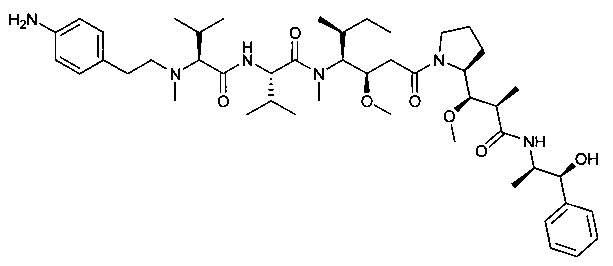
Соединение 64 было получено таким же способом, как для соединения 61, с использованием карбоновой кислоты 61А (93 мг, 0,29 ммоль, 1 экв.), амина 2D (174 мг, 0,29 ммоль, 1 экв.), DIEA (100 мкл, 0,58 ммоль, 2 экв.) и DECP (66 мкл, 0,43 ммоль, 1,5 экв.). Неочищенный продукт очищали флэш-хроматографией на силикагеле (ДХМ/МеОН) с получением соединения 64 в виде беловатого твердого вещества (51 мг, 21%).
1Н ЯМР: (500 МГц, ДMCO-d6, млн-1): δ (Присутствие ротамеров), 9.61 (m(br), 1Н); 9.05-8.89 (m, 1Н), 7.93 (d, 0.6Н, NHCO), 7.64 (d, 0.4Н, NHCO), 7.36-6.98 (m, 7H), 6.92-6.70 (m(br), 2H), 5.45 (s(br), 1H), 4.80-4.41 (m, 3H), 4.06-3.44 (m, 4H), 3.37-2.79 (m, 18H), 2.45-2.21 (m, 3H), 2.17-0.70 (m, 35H).
ВЭЖХ (Xbridge Shield C18, 3,5 мкм, 4,6×50 мм; 3,5 мл/мин, 40°C, от 0 до 95% MeCN в воде (0,1% ТФУ) за 2,25 минуты, затем 95% MeCN в течение 0,5 минут, Tr=1,20 мин (100%, 220 нм). m/z (Q-TOF ИЭР+) 837,5826 (33%, МН+, для C47H77N6O7 требуется 837,5848), 419,2956 (100%, (МН2)2+, для С47Н76N6O8 требуется 419,2961).
II - Биологическая активность соединений по изобретению
Способ:
Культура клеток. Клетки А549 (немелкоклеточный рак легкого - АТСС CCL-185) и MDA-MB-231 (аденокарцинома молочной железы - АТСС НТВ-26) культивировали в минимальной эссенциальной среде Игла (MEM) с 5% фетальной сывороткой теленка (ФСТ) и модифицированной Дульбекко среде Игла (DMEM) с 10% ФСТ соответственно. Клетки MCF7 (дуктальная карцинома молочной железы - АТСС НТВ-22) и SN-12C (карцинома почки - АТСС) поддерживали в среде RPMI1640 (без фенолового красного для клеток MCF7), содержащей 10% ФСТ. Во все среды добавляли фунгизон (1,25 мкг/мл) и пенициллин-стрептомицин (100 ед./100 мкг/мл). Клетки культивировали в стандартных условиях в термостате при 37°С, 5% СO2 и 95% атмосферной влажности.
Антипролиферативная активность на 4 опухолевых линиях клеток. Соединения согласно изобретению исследовали на их антипролиферативную активность с использованием анализа пролиферации ATPlite (Perkin Elmer, Villebon sur Yvette, France) на полной панели из 4 линий клеток. Клетки высевали в 96-луночные планшеты (103 клеток/лунка для А549, 2.103 для MCF7, MDA-MB-231 и SN12C) на сутки 0 при концентрации, гарантирующей, что клетки остаются в логарифмической фазе роста на протяжении 72 ч периода обработки лекарственным средством. После 24-часового периода инкубации все клетки обрабатывали серийными разведениями тестируемых соединений (11 мкл 10× раствора в 1% ДМСО - 6 лунок/условие). Во избежание прикрепления соединений к наконечникам наконечники меняли между двумя последовательными разведениями. Затем клетки помещали в термостат при 37°С, 5% СO2. На сутки 4 жизнеспособность клеток оценивали путем дозирования АТФ, высвобождаемой жизнеспособными клетками. Число жизнеспособных клеток анализировали в сравнении с числом клеток, обработанных растворителем. Значения ЕС50 определяли с помощью анализа соответствия кривой (модель нелинейной регрессии с сигмоидной кривой доза-ответ, вариабельных коэффициент наклона Хилла), проведенного с помощью алгоритма, предложенного программой GraphPad (GraphPad Software Inc., Калифорния, США).
Результаты:
Различные соединения:
Различные соединения согласно изобретению были протестированы для определения их антипролиферативной активности на линии клеток MDA-MB-231, следуя описанному выше способу. При измерении активностей получили значения ЕС50 менее 0,1 мкМ.
Несколько приведенных ниже примеров, выбранных из соединений согласно изобретению, иллюстрируют их полностью значимые антипролиферативные свойства:
Пример 12: ЕС50=5,80×10-10 М; Пример 13: ЕС50=7,95×10-8 М; Пример 15: ЕС50=1,70×10-10 М; Пример 27: ЕС50=1,20×10-10 М.
Различные линии клеток:
Соединение 15 было протестировано на различных линиях клеток (А549, MDA-MB-231, MCF-7, SN12C), следуя описанному выше способу. При измерении активностей получили значения ЕС50 менее 0,1 мкМ.

Сравнительные примеры:
Замещение на фенильном кольце (амино/гидроксил по сравнению с карбоксилом) было исследовано в приведенных ниже сравнительных примерах, показывающих улучшенную антипролиферативную активность лекарственных средств согласно изобретению, содержащих амино- или гидроксильный заместитель.
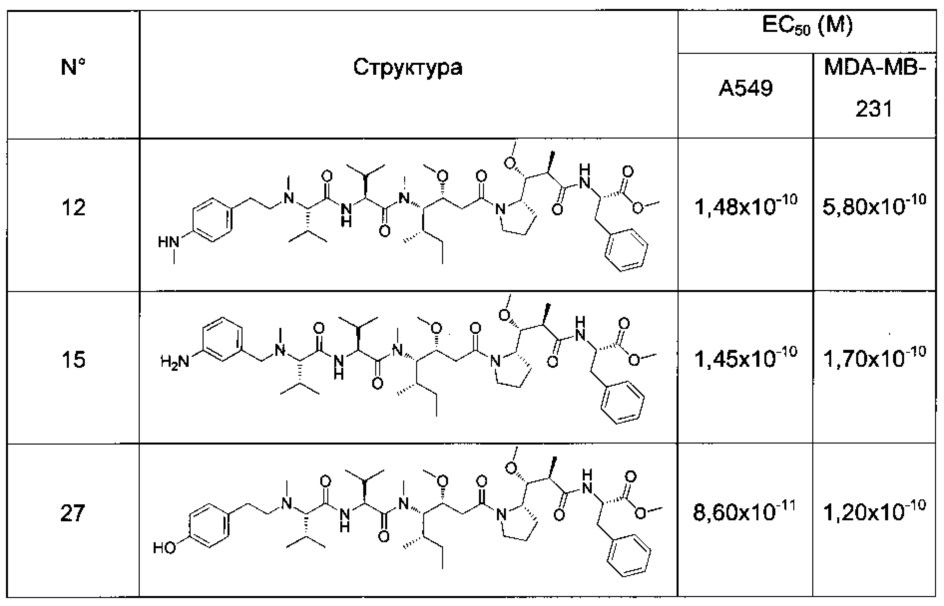
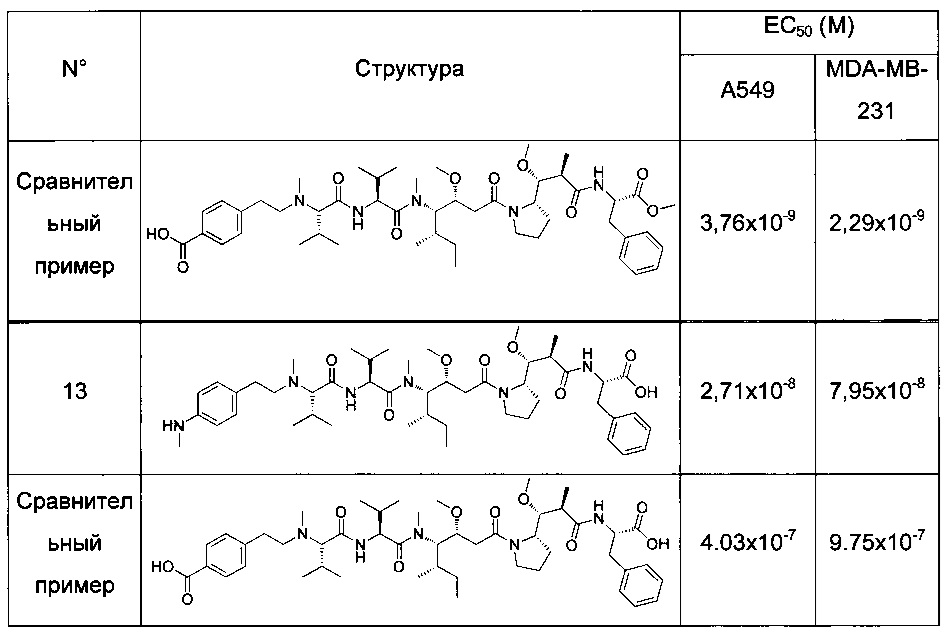
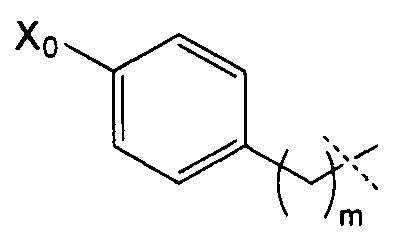
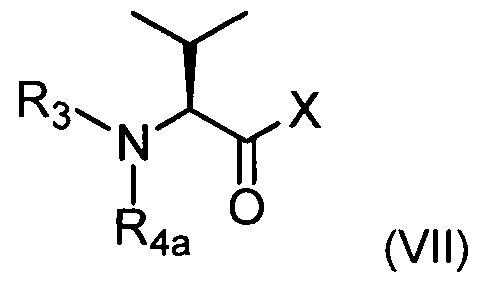
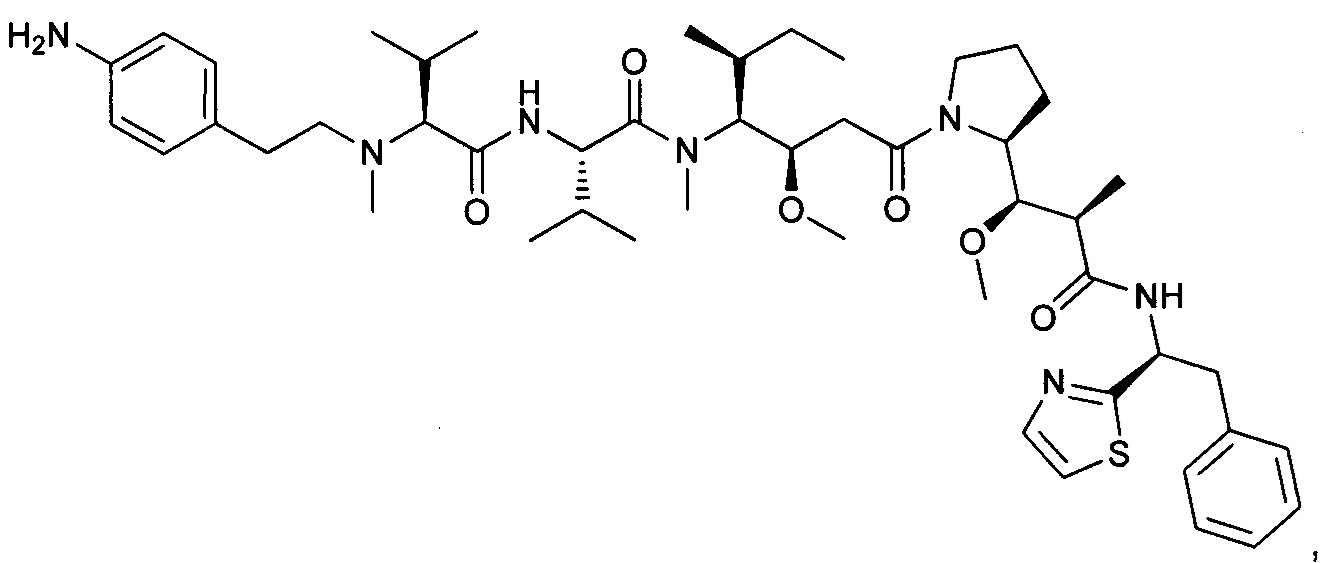
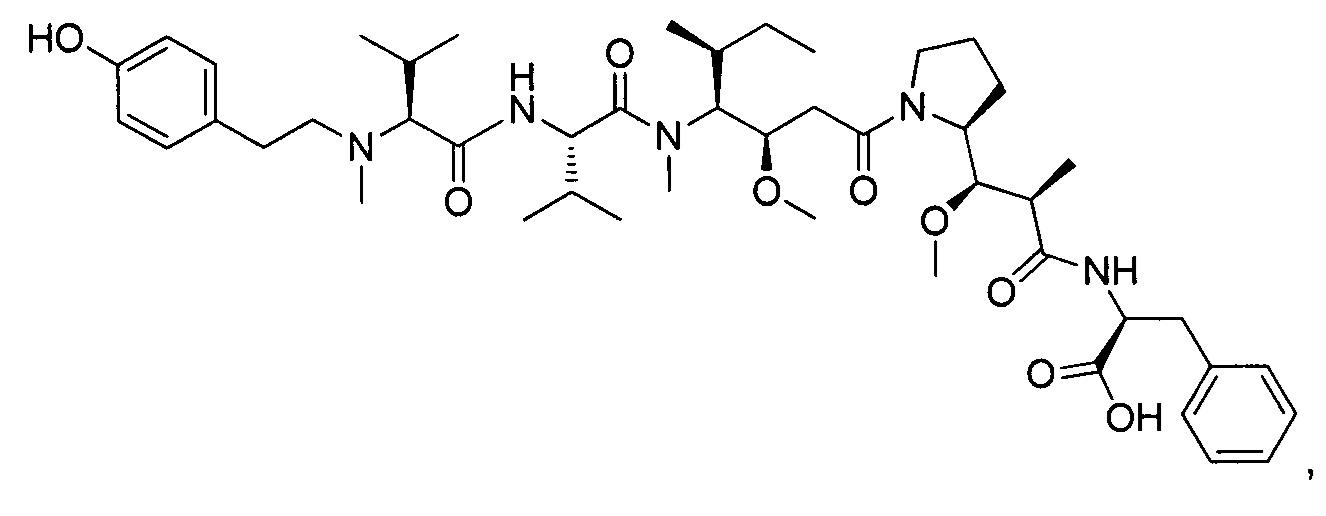
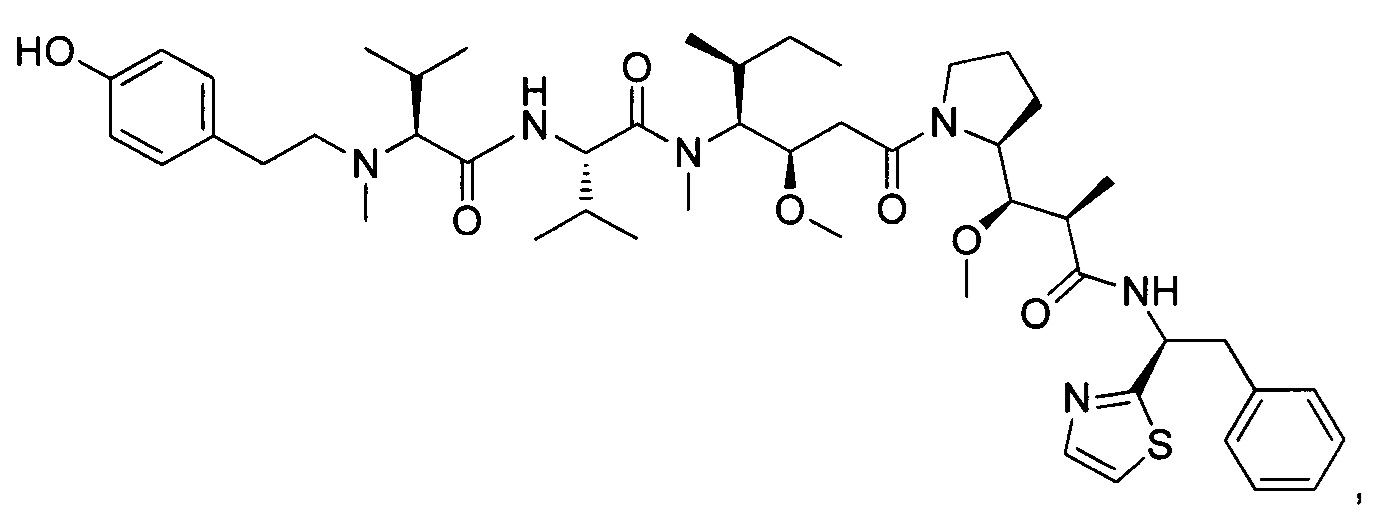
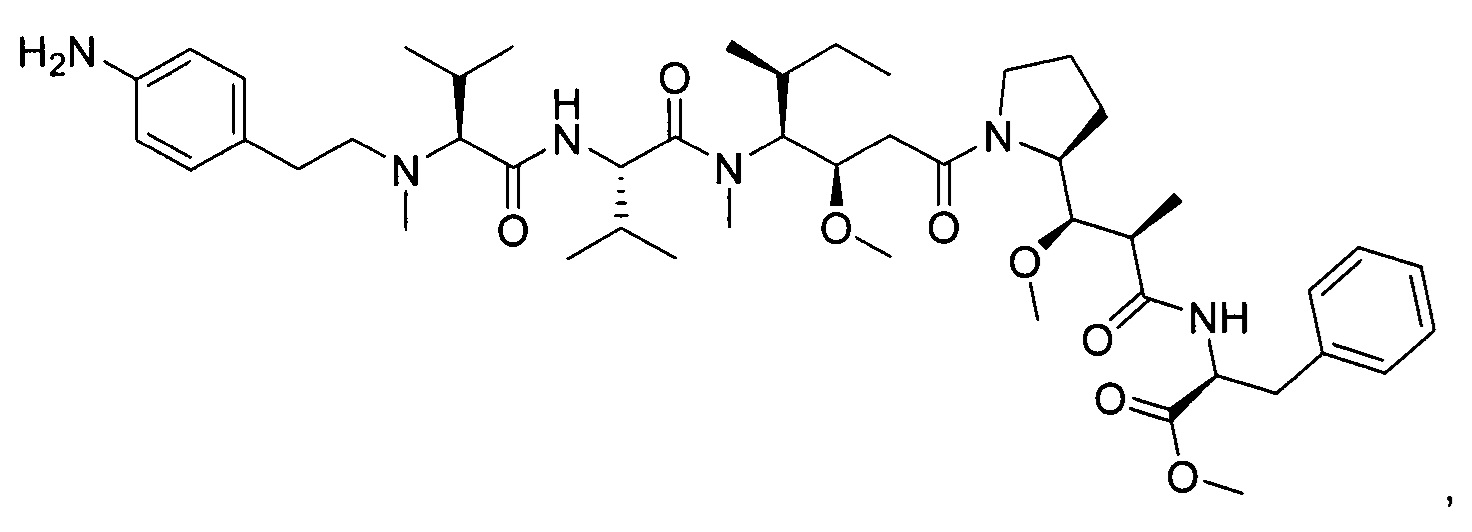
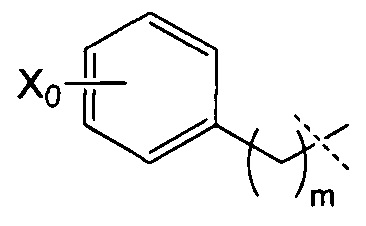
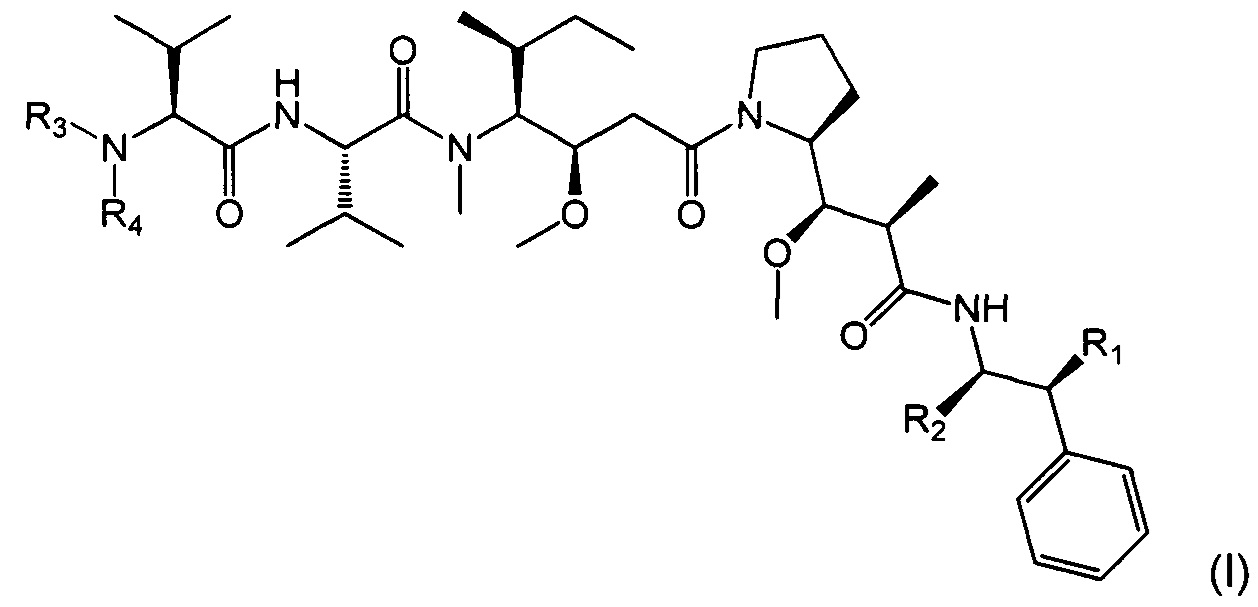
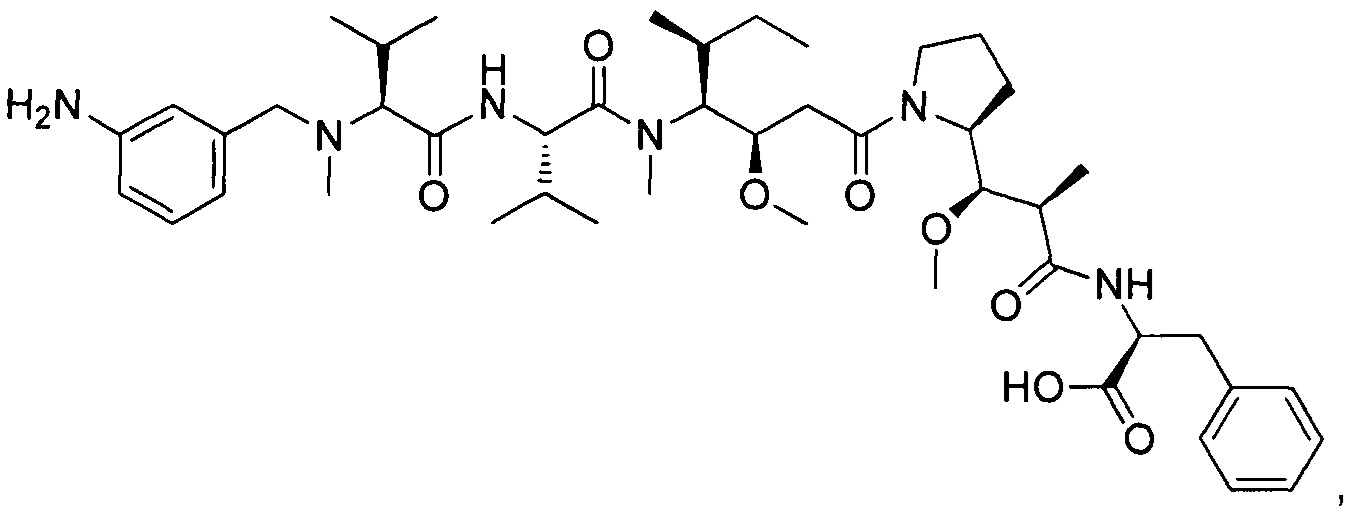
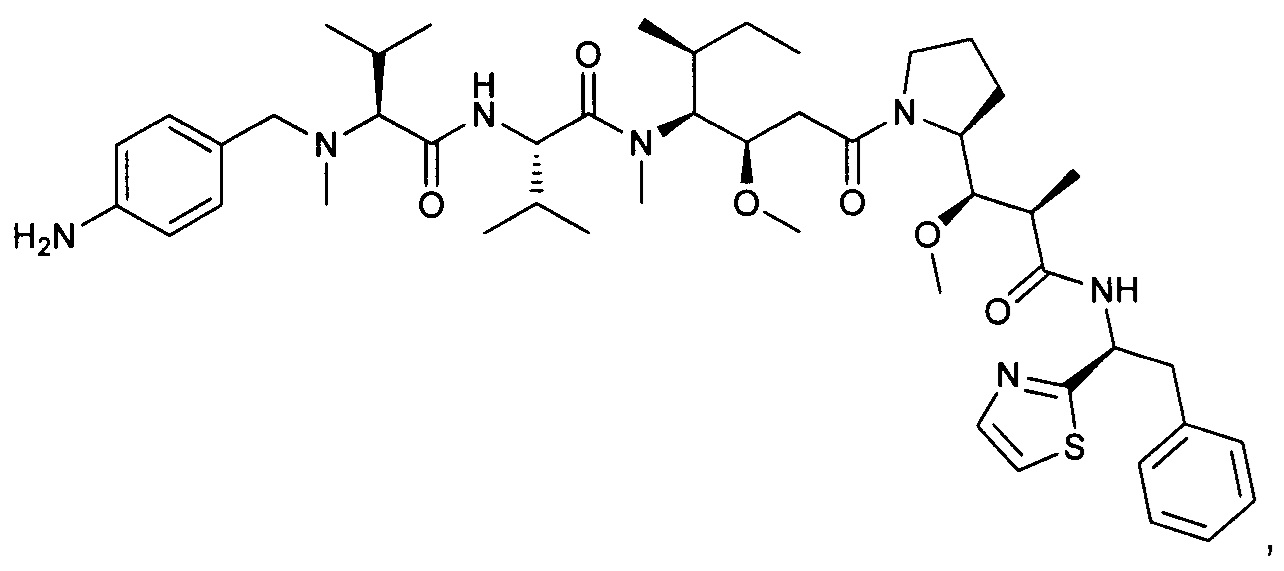
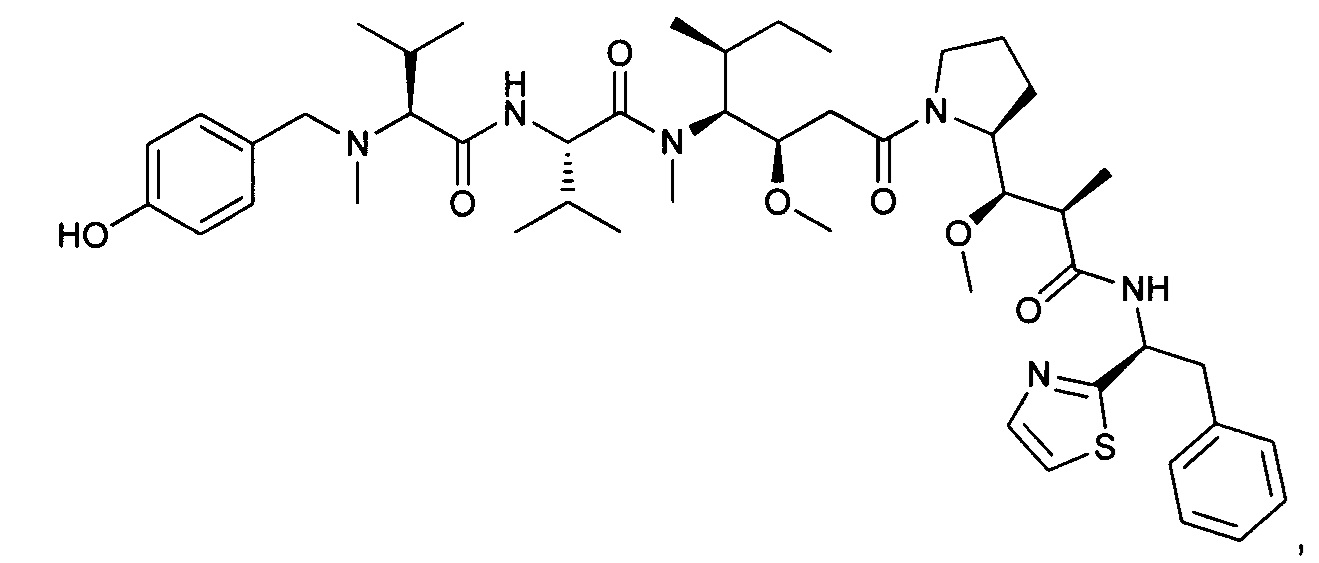
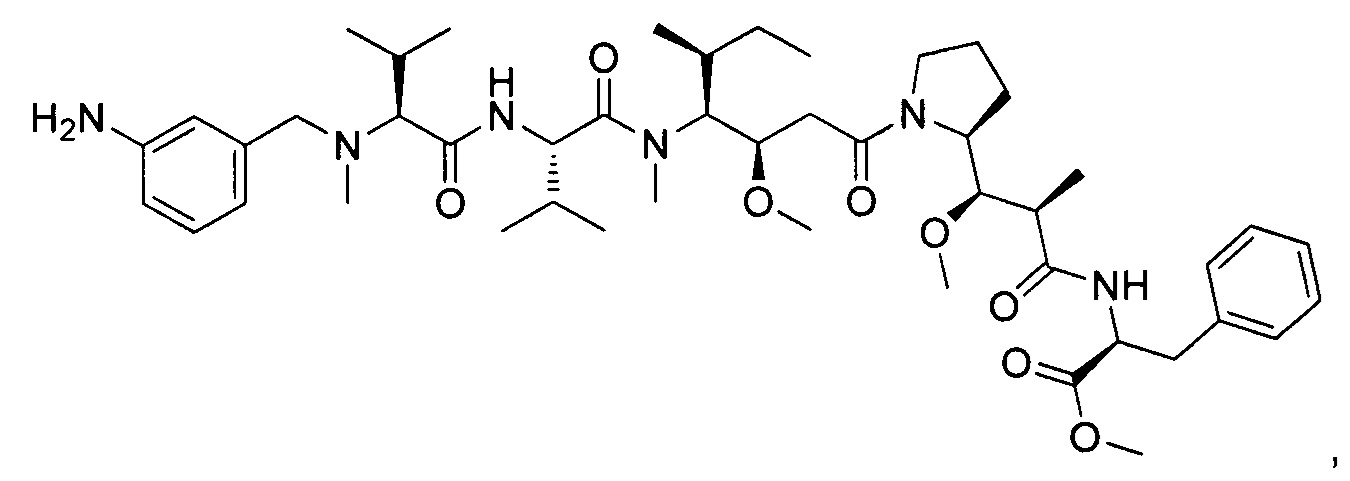
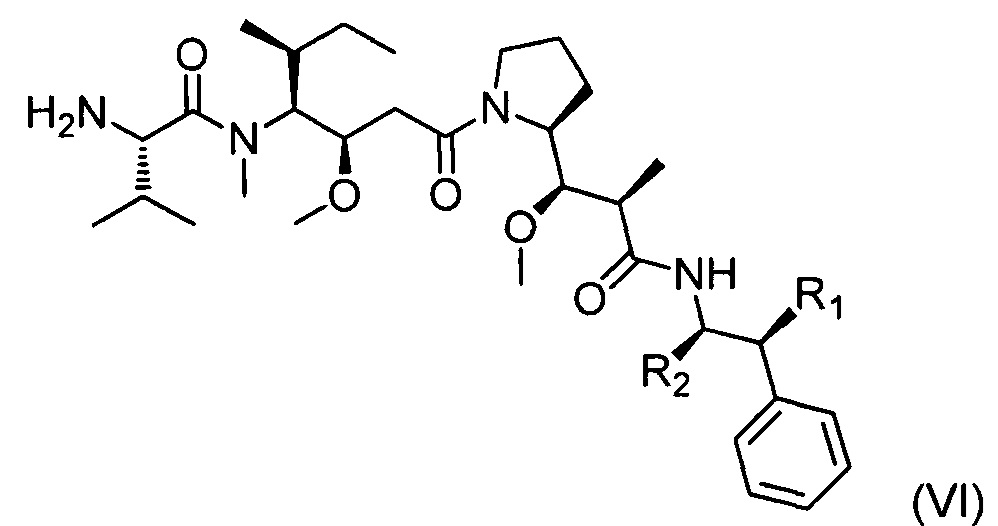
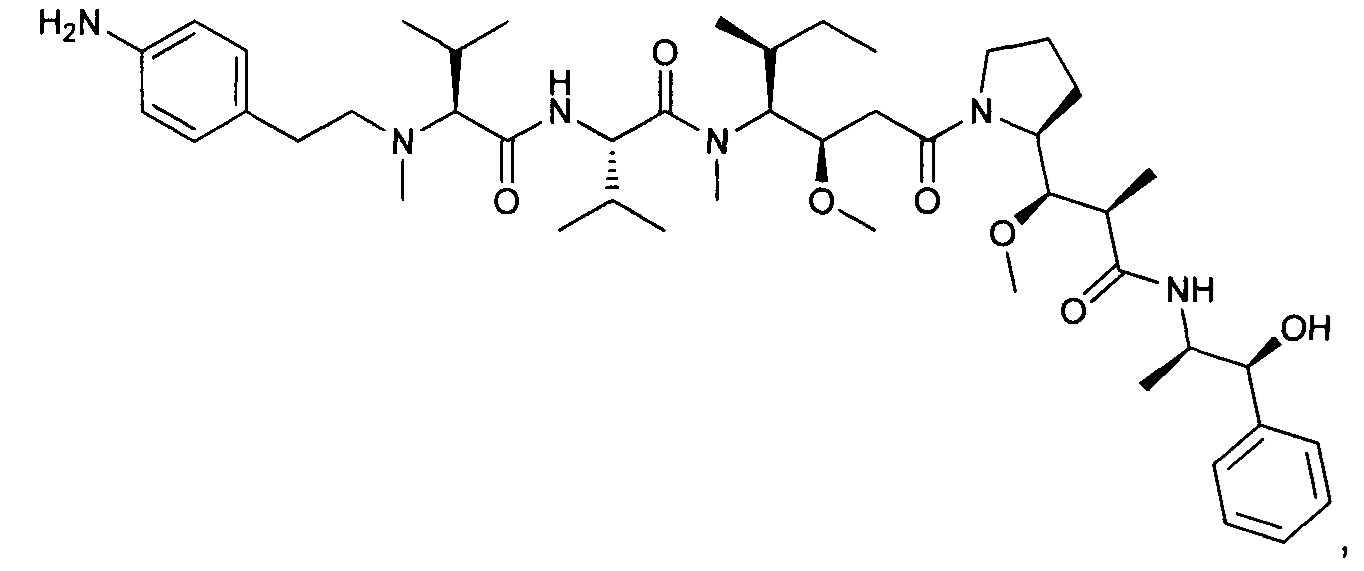
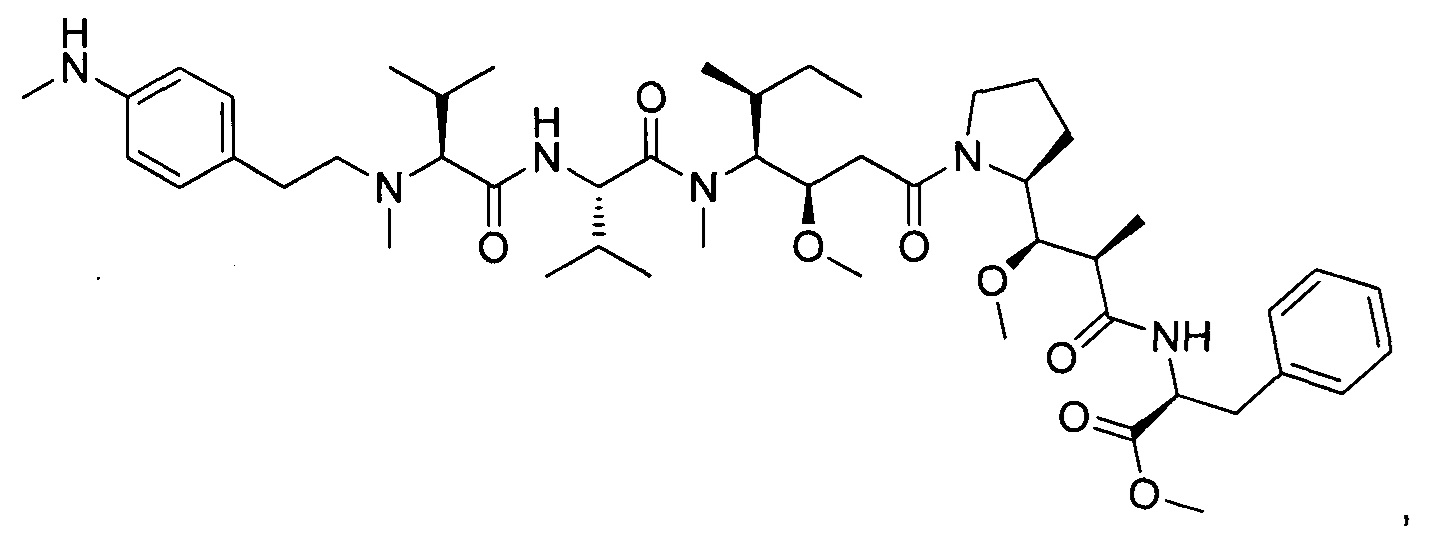
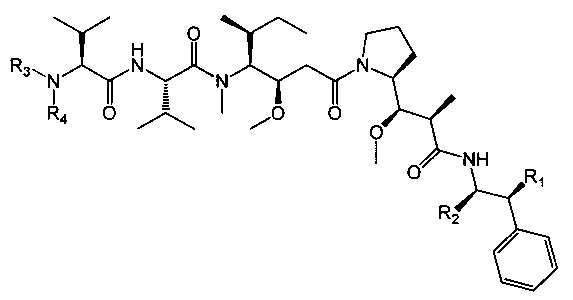
Меквитазин для лечения или предотвращения патологий, в которых задействованы н4 рецепторы гистамина
Способ генерирования активных антител к антигену устойчивости, антитела, полученные этим способом, и их применения
Способ синтеза (1s,2r)-милнаципрана
Производные бензотиазинов, их получение и применение в качестве лекарств
Комбинация антагониста с-мет и аминогетероарила для лечения рака
Новые (поли)аминоалкиламиноалкиламидные, алкил-мочевинные или алкил-сульфонамидные производные эпиподофиллотоксина, способ их получения и их применение в терапии в качестве противораковых средств
Фармацевтическая композиция, содержащая эфир дгк, для парентерального введения
Композиция, содержащая комбинацию экстракта бузины и штамма l. paracasel, l. casei, l. bulgaricus или s. thermophilics
Производные хромонов, способ их получения и их терапевтические применения
Новые антитела, ингибирующие димеризацию с-мет, и их применения
Производные бензотиазинов, их получение и применение в качестве лекарств
Производные азаиндазола или диазаиндазола в качестве медикамента
Конъюгат антитела и лекарственного средства и его применение для лечения рака
Производные циннамоил-пиперазина и их применение в качестве антагонистов par-1
Конъюгат антитела против igf-1r с лекарственным средством и его применение для лечения рака
Композиция для лечения рака, экспрессирующего igf-1r

