Результат интеллектуальной деятельности: Способ получения нитрата олова (IV)
Вид РИД
Изобретение
Изобретение относится к технологии получения солей олова и может быть использовано в различных областях химической и иных видов практики, в научных исследованиях и аналитическом контроле.
Известно получение нитрата олова (IV) путем растворения металла в 70%-ной азотной кислоте (atomistry.com. Periodic Table of Chemical Elements. http://tin.atomistry.com/stannic_nitrate.html).
Недостатками данного способа являются:
1. 70%-ная азотная кислота относится к категории специальных, поскольку не может быть получена в процессе перегонки разбавленной кислоты: препятствием является азеотропная смесь, содержащая 68,4%. HNO3. Следовательно для ее получения нужны соответствующие методы либо концентрированная (97-98%-ная) кислота для разбавления неконцентрированной.
2. Работа с такой кислотой требует специального оборудования и оснащения рабочего места. К тому же требуется соответствующие подготовка и опыт исполнителя.
3. Обозначенный метод не оформлен как конкретный способ. Нет временных характеристик в рекомендованных условиях проведения, которые также не приведены, нет сведений о выходе, степени превращения олова, селективности по целевому продукту, фазовому состоянию реакционной смеси, способу выделения, очистке и т.д.
Нитрат олова с химической формулой Sn(NO3)4 получают по реакции хлорида олова (IV) и пентаоксида азота (Ф. Коттон, ДЖ. Уилкинсон. Основы неоганической химии. - М: Мир, 1979, 678 с. (с. 323)).
Недостатками данного способа являются:
1. Обозначенный метод не оформлен как технологический процесс с конкретным указанием диапазонов варьирования условий проведения, в частности, температурных, наличия и природы растворителя, концентрационных, фазового состояния реакционной смеси, динамики изменения последнего по ходу процесса и т.д.
2. Нет определенных сведений, в каком виде накапливается целевой продукт и как его можно выделить из получаемых реакционных смесей.
3. Нет определенных сведений, насколько допустим контакт реакционных смесей с водой и влажным воздухом как по ходу получения продукта, так и при его выделении, сушке и т.д.
4. Реагентами в данном способе являются продукты глубокой переработки природного сырья, что существенно снижает технологическую привлекательность данного способа.
Наиболее близким к заявленному является способ получения сульфатов двух-, трех- и четырехвалентного марганца или их смесей (пат. РФ №2251529, опубл. 10.05.2005, Бюл. №13), в соответствии с которым взаимодействие оксидов марганца MnO, Mn2O3, Mn3O4 и MnO2 с концентрированной серной кислотой проводят в бисерной мельнице вертикального типа с высокооборотной механической мешалкой и стеклянным бисером в качестве перетирающего агента и места локализации получаемого(ых) продукта(ов) в присутствии жидкой фазы на основе кипящего менее 100°C низкомолекулярного парафинового углеводорода или смеси углеводородов при 5-25˚C в течение 5-10 мин при соотношении масс растворителя и реагентов (2,33:1)÷(10:1) с учетом того, что 79,8-90%-ную серную кислоту берут в мольном стехиометрическом избытке в отношении марганецсодержащего реагента 1,20-1,72. После завершения процесса бисер и продукт отделяют путем фильтрования, а локализованный в виде пленки на поверхности бисера и частично стенках реактора сульфат растворяют в серной кислоте соответствующей концентрации или вводят в новый процесс, проводимый в этой же бисерной мельнице с сульфатом марганца (III) или (IV) в качестве окислителя.
Недостатками данного способа являются:
1. Нет никаких оснований рассматривать во взаимодействии с кислотами диоксид марганца и диоксид олова как полностью аналогичные реагенты. Поэтому совсем не очевидно, что приведенные в цитируемом патенте условия проведения процесса будут приемлемыми при замене MnO2 на SnO2.
2. Отмеченная в п.1 неочевидность существенно возрастает при замене двухосновной сильной минеральной кислоты на одноосновную сильную минеральную кислоту, например азотную или хлороводородную, и, тем более, на значительно более слабую карбоновую кислоту.
3. В рассматриваемом способе в качестве растворителя жидкой фазы используется кипящий ниже 100°C парафиновый углеводород или смесь парафиновых углеводородов, что связано с их инертностью в отношении воздействия концентрированной серной кислоты, с одной стороны, и повышенной испаряемостью - с другой. При замене серной кислоты на иную первое требование полностью исчезает. А при увеличении длительности процесса в десятки раз в сравнении с приведенным в цитируемом процессе второе требование из-за повышенного уноса растворителя из полезного превращается во вредное. Следовательно нет никаких оснований ожидать, что обозначенный растворитель при замене MnO2 на SnO2 и концентрированную H2SO4 на иные кислоты останется не только целесообразным, но даже приемлемым.
4. В цитируемом способе в качестве основного места локализации продукта указана поверхность бисера и отчасти стенки корпуса реактора. Это довольно редко встречающийся и во многом уникальный благоприятный вариант, предопределяющий аномально быстрое протекание процесса образования соли при прямом взаимодействии диоксида с серной кислотой. При получении других, кроме сульфатов, солей марганца он не повторялся. Нет никаких оснований ожидать его повторение при замене MnO2 на SnO2 и H2SO4 на иную кислоту, тем более одноосновную.
Задачей предлагаемого решения является подобрать такие условия проведения процесса прямого взаимодействия оксида олова (IV) с азотной кислотой в бисерной мельнице с высокооборотной механической мешалкой и перетирающим агентом при комнатных и близких к ней температурах, которые бы обеспечили бы практически полное расходование оловосодержащего реагента и стремящуюся к 1 избирательность по целевому продукту, а также выделение последнего из реакционной смеси путем простого фильтрования с последующими промывками, сушкой и при необходимости очисткой.
Поставленная задача достигается тем, что оксид олова (IV) в количестве 0,05-0,5 моль/кг вводят в контакт с 0,63-2,05 моль/кг раствором азотной кислоты в начальной загрузке, который готовят растворением 54%-ной азотной кислоты в этилцеллозольве предварительно или непосредственно в реакторе перед вводом оксида олова (IV), при мольном соотношении кислота:оксид 4,0-12,50 в присутствии стеклянного бисера в качестве перетирающего агента и ведут процесс при интенсивном механическом перемешивании в отсутствие или в присутствии трибохимического катализатора при контроле за расходованием кислоты вплоть до достижения близкого к расчетному остаточного количества этого реагента, после чего перемешивание прекращают, проводят отделение и очистку перетирающего агента от остатков реакционной смеси, суспензию реакционной смеси фильтруют, осадок на фильтре промывают промывным растворителем, отжимают и сушат, а фильтрат направляют на уменьшение содержащейся в нем воды путем отгонки и далее возвращают на загрузку повторного процесса. При этом содержащие остаточную кислоту фильтраты могут служить жидкой фазой загрузок на последующие процессы, а в качестве трибохимических катализаторов используют п-аминоазобензол, п-толуидин, бензидин, 2-аминопиридин, 1-нафтиламин и п-аминофенол.
Характеристика используемого сырья:
Оксид олова (IV) ГОСТ 22516-77
Азотная кислота ГОСТ 4461-77
Этилцеллозольв ГОСТ 8313-88
п-Аминоазобензол ГОСТ 4681-70
п-Толуидин ТУ 6-09-66-75
Бензидин ТУ 6-09-4221-76
2-Аминопиридин ТУ 6-09-672-77
1- Нафтиламин ГОСТ 8827-74
п-Аминофенол ГОСТ 5209-77
Проведение процесса заявляемым способом следующее. В бисерную мельницу вертикального типа со стеклянным корпусом, высокооборотной механической мешалкой лопастного типа с лопастью из текстолита, стеклянным бисером в качестве перетирающего агента вводят стеклянный бисер, расчетные количества предварительно приготовленного раствора азотной кислоты или раздельно растворителя и концентрированной азотной кислоты включают механическое перемешивание, выводят последнее на стационарный режим, после чего дозируют оксид олова (IV) и, где предусмотрено, трибохимический катализатор. Последний ввод принимают за начало процесса и ведут его при стабильном интенсивном механическом перемешивании в течение требуемого времени, определяемого по результатам контроля расходования кислоты, осуществляемого методом отбора проб и их анализа. При приближении значения содержания этого реагента к расчетному в соответствии со стехиометрией при практически 100%-ном расходовании оксида олова (IV) в среднюю соль перемешивание прекращают, корпус реактора отсоединяют от крышки с сальниковой коробкой и мешалкой и опускают в гнездо каркасной рамы таким образом, чтобы лопасть мешалки оказалась выше уровня реакционной смеси в корпусе. Некоторое время выжидают, давая возможность остаткам реакционной смеси стечь с лопасти и вала механической мешалки. Затем корпус с реакционной смесью вынимают из гнезда каркасной рамы, перемещают к узлу отделения перетирающего агента и реакционную смесь и выливают содержимое в приемлемую воронку этого узла с сеткой с размерами ячеек 0,3×0,3 мм. Оставшийся на сетке перетирающий агент аккуратно возвращают в корпус бисерной мельницы, бисерную мельницу собирают вновь в своем же гнезде каркасной рамы, проверяют вручную работу перемешивающего устройства, вводят некоторое количество этилцеллозольва, включают перемешивание и проводят отмывку стенок реактора и его элементов, а также перетирающего агента от остатков суспензии реакционной смеси. По завершении этой операции проводят повторное отделение перетирающего агента, который далее сушат, взвешивают, внимательно осматривают и возвращают на загрузку повторного процесса. А получаемый в процессе отделения промывной растворитель используют при переработке суспензии реакционной смеси.
Отделенную от перетирающего агента реакционную смесь фильтруют, осадок на фильтре промывают фильтратом, затем промывным растворителем, тщательно отжимают, переносят с фильтра в емкость для сушки, измельчают и оставляют на воздухе до достижения постоянной массы. По ходу сушки измельчение повторяют несколько раз, достигая порошкообразного состояния продукта. Отбирают пробу полученного сухого продукта и определяют его эквивалент. По статистически подтвержденной величине его делают вывод о необходимости и целесообразности последующей очистки.
Объединенный фильтрат с промывным растворителем взвешивают и анализируют на содержание в нем продукта (обычно менее 0,005 моль/кг) и остаточной кислоты. По результатам анализов делают заключение, как использовать их: в загрузке повторного процесса непосредственно либо после снижения содержания воды путем отгонки.
Пример №1
В бисерную мельницу вертикального типа со стеклянным корпусом объемом 343 мл и высотой 126 мм, снабженную высокооборотной (3000 об/мин) мешалкой с текстолитовым валом диаметром 6,7 мм и лопастью из тестолита с размерами 42×38×1,9 мм, прижимной крышкой с сальниковой коробкой и отверстиями для датчика температуры, помещения пробоотборника и дозагрузок компонентов при работающем перемешивании, вводят 100 г стеклянного бисера, 95,9 г предварительно приготовленного раствора азотной кислоты с концентрацией 1,13 моль/кг, 3,90 г оксида олова (IV) и 0,197 г п-аминоазобензола как катализатора. Корпус реактора помещают в соответствующее гнездо каркасной рамы, соединяют с крышкой, должным образом крепят, прокручивают вал мешалки вручную, затем вводят механическое перемешивание и этот момент принимают за начало процесса. По ходу процесса отбирают пробы реакционной смеси, которые анализируют на остаточное количество кислоты. Кинетика расходования этого реагента представлена ниже
|
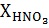 , моль/кг
, моль/кгНа 105 мин процесс прекращают, останавливают механическое перемешивание, корпус реактора отсоединяют от крышки и, не вынимая из гнезда, опускают вниз таким образом, чтобы лопасть мешалки оказалась выше уровня реакционной смеси в корпусе. В течение 5 мин дают возможность остаткам реакционной смеси стечь с вала и лопасти мешалки. Далее корпус с реакционной смесью и перетирающим агентом вынимают из гнезда каркасной рамы, содержимое выливают в приемлемую воронку узла отделения перетирающего агента с сеткой с размерами 0,3×0,3 мм и проводят отделение стеклянного бисера от суспензии реакционной смеси. Стеклянный бисер аккуратно снимают с сетки в качестве фильтровальной перегородки и возвращают в корпус реактора. Бисерную мельницу собирают на каркасной раме вновь, вводят 30 г растворителя – этилцеллозольва, включают механическое перемешивание и в течение 12 мин проводят отмывку перетирающего агента и внутренних элементов реактора от остатков реакционной смеси. По завершении этой операции проводят повторное отделение стеклянного бисера, который сушат и направляют в повторный процесс. Промывной растворитель оставляют в приемлемой емкости узла отделения перетирающего агента и далее используют в промывке продукта на фильтре.
Отделенную от перетирающего агента суспензию реакционной смеси фильтруют, осадок на фильтре промывают промывным растворителем, аккуратно отжимают, снимают с фильтра и оставляют на воздушную сушку до постоянного веса. Для ускорения сушки твердую массу периодически дробят, получая в конце порошкообразное состояние. Масса сухого продукта 9,14 г. На ее основе определяют выход выделенного продукта, который оказался равным 96,3%. Выполняют анализ на эквивалентную массу, после чего его помещают в емкость для хранения до использования либо направляют на очистку.
Смесь фильтрата и промывного растворителя возвращают на загрузку повторного процесса либо накапливают для проведения уменьшения содержания воды в нем или регенерации этилцеллозольва путем соответствующей перегонки.
Примеры №2-8
Реактор, исходные диоксид олова и предварительно приготовленный раствор азотной кислоты в этилцеллозольве с концентрацией 1,13 моль/кг, перетирающий агент, соотношение его массы с массой загрузки, мольное соотношение кислоты и диоксида олова, концентрация трибохимического катализатора где он есть, последовательность операций при загрузке, проведении процесса и контроле за ним, определение момента прекращения процесса, отделение перетирающего агента, выделение целевого продукта и использование фильтрата и промывного растворителя аналогичны описанным в примере №1. Отличаются природой используемого трибохимического катализатора, моментом ввода последнего по ходу процесса или отсутствием катализатора вообще. Указанные различия и другие характеристики процесса сведены в таблицу.
|
Примеры №9-18
Реактор, реагенты и трибохимический катализатор, предварительное приготовление раствора азотной кислоты в этилцеллозольве и концентрация этого раствора, перетирающий агент и его загрузка, последовательность операций при загрузке, проведении процесса и контроле за ним, определение момента прекращения процесса, отделение перетирающего агента, выделение целевого продукта и работа с ним аналогичны описанным в примере 1. Отличаются избытком кислоты в начальной загрузке, вводимой с предварительно приготовленным ее раствором в растворителе, дозагрузкой оксида олова (IV) после полного расходования ранее введенной порции, а также проведением процесса в фильтратах с непрореагировавшей кислотой как самостоятельных жидких фазах процесса. Указанные различия, а также характеристики проводимых процессов сведены в таблицу.
|
Примеры №19-23
Реактор, реагенты, трибохимический катализатор, растворитель жидкой фазы, перетирающий агент, а также последовательности операций при проведении процесса, контроле за его ходом, моменте определения целесообразности прекращения, отделении перетирающего агента и его подготовке к использованию в повторном процессе, выделении продукта и работе с ним аналогичны описанным в примере 1. Отличаются дозировками реагентов, приготовлением раствора кислоты непосредственно в реакторе для проведения взаимодействия, операциями и их последовательностью при приготовлении такого раствора, а также моментом ввода диоксида олова в зону реакции. Указанные различия и полученные характеристики процесса сведены в таблицу.
|
Положительный эффект предлагаемого решения состоит в том, что:
1. Предлагаемое решение предполагает использование доступных и дешевых исходных реагентов: диоксид олова вообще может быть природного происхождения (касситерит), а азотная кислота – крупнотоннажный продукт химической промышленности. Не является чем-то экзотическим и этилцеллозольв, производимый и используемый в промышленных масштабах.
2. Процесс протекает при комнатных температурах и довольно прост в аппаратурном оформлении. Никакого котлонадзорного оборудования для него не требуется.
3. Процесс характеризуется практически 100%-ным превращением исходного оловосодержащего реагента и практически 100%-ной избирательностью по целевому продукту. Следовательно выделенный продукт в большинстве случаев не требует каких-либо сложных дополнительных очисток от остаточных реагентов и побочных продуктов.
4. Растворимость целевого нитрата олова (IV) в используемой жидкой фазе реакционной смеси чрезвычайно мала, что предопределяет его накопление в суспендированной твердой фазе и довольно легкое выделение путем простого фильтрования.
5. Процесс не осложнен необходимостью утилизационных переработок фильтратов в смеси с промывным растворителем: они вполне могут служить жидкими фазами последующих процессов получения данного целевого продукта.
Устройство для поиска минимального значения интенсивности размещения в тороидальных системах при направленной передаче информации
Котел отопительный газовый
Способ регенерации скважин на воду
Автоматизированная система диагностики и контроля состояния изоляции силовых кабельных линий
Способ получения бензоата и замещенных бензоатов олова (iv)
Мостовой измеритель параметров двухполюсников
Ингибитор коррозии нефтяных труб и способ его получения
Способ измерения параметров фазового перехода жидкость-жидкость в водных растворах амфифилов
Трехслойная ресурсосберегающая железобетонная панель
Способ и устройство мобильного робота для прохождения замкнутых контуров и лабиринтов
Способ получения формиата цинка
Способ переработки прокорродировавших изделий из меди или ее сплава
Способ очистки поверхностей меди и ее сплавов от продуктов коррозии и окисления соединениями меди (ii)
Способ получения бензоата олова (ii)
Способ получения антибактериальной композиции, содержащей основной ацетат меди
Способ получения основного бензоата олова (ii)
Способ получения бензоата и замещенных бензоатов олова (iv)
Способ получения карбоксилатов олова (ii)
Способ получения карбоксилатов олова (ii)
Способ получения бензоата и замещенных бензоатов олова (iv) из вторичного сырья

