Результат интеллектуальной деятельности: 2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ
Вид РИД
Изобретение
Изобретение относится к 2-(3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонату, а также его композициям, способам применения и получения. Это соединение и его композиции являются ингибиторами Янус киназ (JAK) и могут использоваться при лечении JAK ассоциированных заболеваний, включая, например, воспалительные и аутоиммунные расстройства, а также рак.
Онкогенные протеинкиназы представляют собой одну из самых крупных и наиболее привлекательных групп белковых мишеней для разработки лекарственных средств. JAK играют важную роль в цитокин-зависимой регуляции пролиферации и функции клеток, участвующих в иммунной реакции. В настоящее время известны четыре JAK: JAK1, JAK2, JAK3 (также известная как Янус-киназа лейкоцитов; JAKL; L-JAK) и TYK2 (также известна как тирозинкиназа Т).
Блокировка передачи сигнала на уровне JAK перспективна для разработки методов лечения воспалительных заболеваний, аутоиммунных заболеваний, миелопролиферативных и раковых заболеваний человека. Предполагается, что ингибирование JAK имеет терапевтический эффект у пациентов, страдающих иммунными расстройствами кожи, такими как псориаз и сенсибилизация кожи. Эффективные ингибиторы JAK представляют безусловный интерес для разработки новых лекарственных средств. Некоторые JAK ингибиторы, в том числе пирролопиримидины, представлены в WO 2009/114512. В этой серии соединений Барицитиниб (Baricitinib) является самым передовым лекарственным кандидатом. Барицитиниб показал быструю (через 6 месяцев лечения) и устойчивую эффективность при лечении ревматоидного артрита в трех исследованиях III фазы [NCT02340104, NCT02263911, NCT01710358. http://www.medpagetoday.com/MeetingCoverage/EULAR/52084.http://www.fiercepharma.com/press-releases/lilly-and-incyte-announce-positive-top-line-results-phase-3-trial-baricitin].
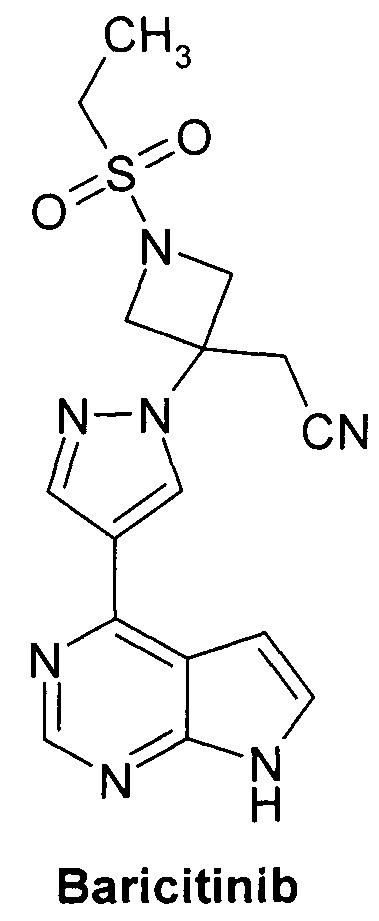
Таким образом, поиск новых улучшенных JAK ингибиторов является одним из основных направлений для разработки новых, более эффективных лекарственных препаратов для лечения ревматоидного артрита, рака и других заболеваний. Соединение и способы, описанные здесь, направлены на удовлетворение этих потребностей.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» (С1-C6)алкильных заместителей. Предпочтительными алкильными группами являются (С1-C6)алкил, еще более предпочтительными (С1-C3)алкил, метил, этил, н-пропил, изопропил, н-бутил, изобутил, втор-бутил, трет-бутил, циклопропилметил, циклобутилметил, циклопентилметил, н-пентил, 2-пентил, 3-пентил, нео-пентил, н-гексил, циклогексил. Алкил может иметь заместители.
«Замещенный алкил» - замещенный алкил может иметь один или несколько одинаковых или различных заместителей, включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, гетероароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонил, гетероаралкилокси или RkaRk+1aN-, где Rka и Rk+1a независимо друг от друга представляют собой «заместители аминогруппы», значение которых определено в данном разделе, например атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rka и Rk+1a вместе с атомом N, с которым они связаны, образуют через Rka и Rk+1a 4-7-членный гетероциклил или гетероцикленил. Предпочтительными «алкильными заместителями» являются арил, гетероарил, гетероциклил, гидрокси, C1-C5 алкокси, С1-С5 алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или RkaRk+1aN-, RkaRk+1aNC(=O)-, аннелированный арилгетероцикленил, аннелированный арилгетероциклил.
«Аминогруппа» означает R1R2N-группу, замещенную или незамещенную необязательно одинаковыми заместителями R1 и R2. Аминогруппа может иметь заместители.
«Активный компонент» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Галоген» означает фтор, хлор, бром и иод. Предпочтительными являются фтор, хлор и бром.
«Метин» представляет собой группу из одного атома углерода и одного атома водорода СН.
«Лекарственное средство (препарат)» - вещество (или смесь веществ в виде фармацевтической композиции) в виде таблеток, капсул, инъекций, мазей и других готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего.
«Фармацевтическая композиция» обозначает композицию, включающую в себя активный компонент и по крайней мере один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие как оливковое масло) и инъекционные органические сложные эфиры (такие как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, альгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, такие как мази и кремы, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, дихлорацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные. (Подробное описание свойств таких солей можно найти в справочнике фармацевтических солей [Р.Н. Stahl, C.G. Wermuth (Eds.). Handbook of Pharmaceutical Salts, Properties, Selection, and Use. VHCA, Verlag Helvetica Chimica Acta,  , Switzerland, and Wiley-VCH, Weinheim, Germany. 2002].) Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
, Switzerland, and Wiley-VCH, Weinheim, Germany. 2002].) Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
Цель настоящего изобретения заключается в создании новых ингибиторов JAK для лечения ревматоидного артрита, рака и других заболеваний.
Поставленная цель достигается 2-(3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонатом формулы 1:

Предметом настоящего изобретения является новый JAK ингибитор общей формулы 1.
Соединение 1 по настоящему изобретению может также включать все изотопы атомов, присутствующих в соединении 1. Изотопы включают атомы, имеющие одинаковый атомный номер, но разные массовые числа. Например, изотопы водорода включают тритий и дейтерий.
Соединение 1 по настоящему изобретению может быть получено с использованием известных методов органического синтеза и может быть синтезировано в соответствии с любым из многочисленных возможных путей синтеза. Реакция для получения соединения по изобретению может быть осуществлена в подходящих растворителях, которые могут быть легко выбраны специалистом в области органического синтеза. Данная реакция может быть осуществлена в одном растворителе или в смеси растворителей. В зависимости от конкретной стадии реакции подходящие растворители для конкретной стадии реакции могут быть выбраны специалистом в данной области.
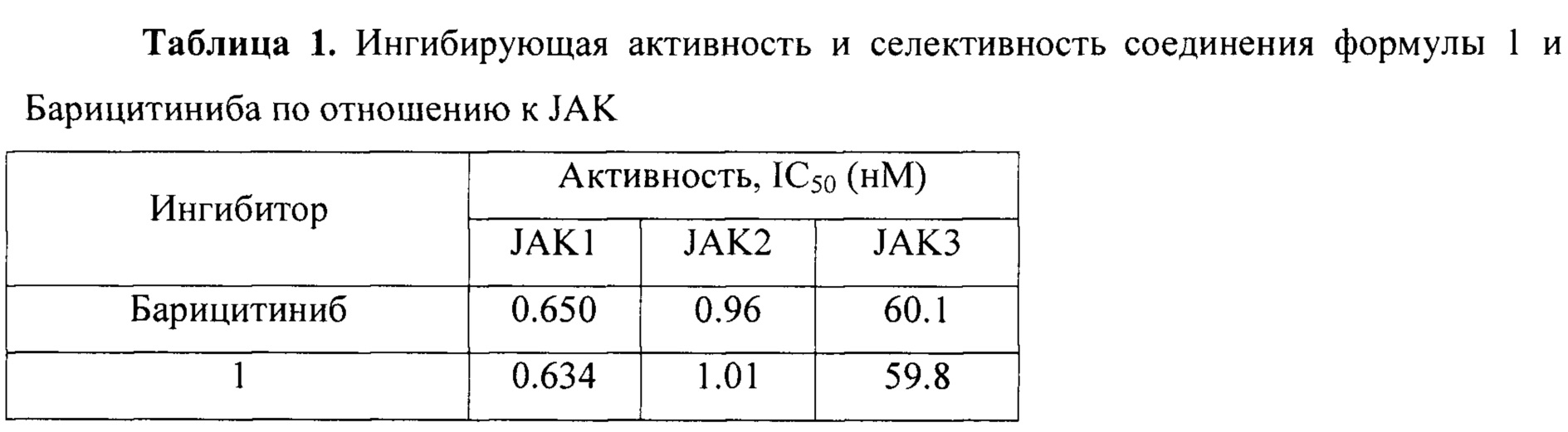
Ингибирующая активность и селективность соединения формулы 1 по отношению к JAK сопоставимы с активностью и селективностью Барицитиниба, который в настоящее время проходит фазу III клинических испытаний [NCT02340104; NCT02263911; NCT01710358; J.D. Clark, М.Е. Flanagan, J.-В. Telliez. Discovery and Development of Janus Kinase (JAK) Inhibitors for Inflammatory Diseases. J. Med. Chem. 2014, 57, 5023-5038].
Преимуществом нового соединения формулы 1 является и несколько более высокая растворимость в водных растворах, чем у Барицитиниба (Таблица 2). Так, например, растворимость ингибитора 1 в воде при рН=7 равна 1 мг/мл, в то время как у Барицитиниба в нейтральной среде растворимость полностью отсутствует.

Преимуществом соединения 1 являются двукратно лучшие по сравнению с Барицитинибом показатели АДМЕ теста Рарр А-В (СаСо-2), которые свидетельствуют о лучшем проникновении через клеточную мембрану в клетку, что, в свою очередь, является предпосылкой для лучшей биодоступности при пероральном применении.

Лучшие значения проникновения в клетку в тесте СаСо-2 могут свидетельствовать о потенциально лучшей биодоступности препарата при пероральном применении.
Основным преимуществом препарата формулы 1 является более высокая активность in vivo по сравнению с Барицитиниб-основанием, что может быть объяснено лучшей биодоступностью ввиду несколько большей растворимости и заметного ускорения проникновения к клетки СаСо-2. Лучшая in vivo активность препарата формулы 1 продемонстирована на модели ревматоидного артрита, вызванного коллагеном.
Динамика развития ревматоидного артрита по всем лапам животных приведена на Фиг. 1. На Фиг. 2 приведены данные показателей по группам в конце исследования. Видно, что наибольшую эффективность показала новая соль Барицитиниба - ZD19-0002 (препарат формулы 1) - снижение признаков ревматоидного артрита на 60%. На Фиг. 3 и 4 приведены данные по развитию ревматоидного артрита на терапевтических лапах (с признаками ревматоидного артрита на момент рандомизации по группам). Эффективность препаратов была ниже, чем при оценке по все лапам, но общая картина сохранилась - ZD19-0002 (препарат формулы 1) наиболее эффективен. На Фиг. 5 и 6 приведены данные по развитию ревматоидного артрита на профилактических лапах (без признаков ревматоидного артрита на момент рандомизации по группам). Сравнимый эффект наблюдается как в группе, получавшей ZD19-0002 (препарат формулы 1) в дозе 20 мг/кг 2 раза в день, так и в группе, получавшей ZD19-0001 (Барицитиниб-основание).
Настоящее изобретение относится к способу получения соединения формулы 1, который заключается в реакции двух молярных эквивалентов соединения формулы 1а (основания Барицитиниба) с одним эквивалентом нафталиндисульфокислоты в подходящем растворителе или смеси растворителей, при этом выход соединения формулы 1 может быть количественный, что выгодно отличает соединение формулы 1 от других солей Барицитиниба и существенно упрощает технологию синтеза.
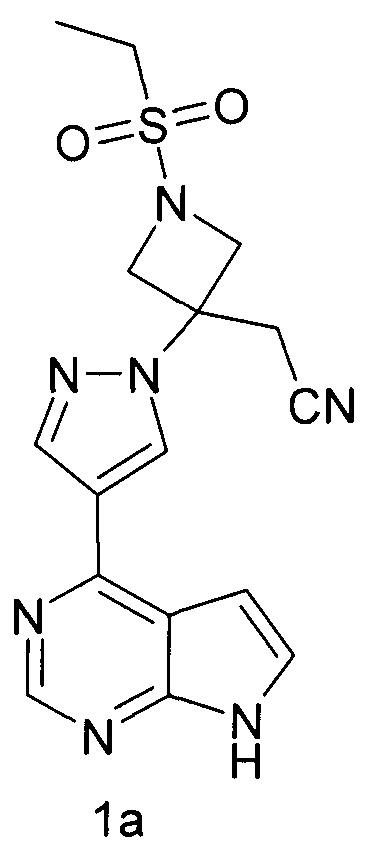
Способ получения соединения формулы 1 включает взаимодействие соединения формулы 1а с 1,5-нафталин дисульфокислотой путем обработки раствора свободного основания формулы 1а в органическом растворителе, таком как этанол (EtOH), раствором кислоты в органическом растворителе, таком как этанол, при комнатной температуре или при повышенной температуре (например, от 60°С до 70°С). Полученная соль в случае необходимости может быть дополнительно очищена перекристаллизацией или другими общеизвестными методами, например переосаждением или промывкой.
Соединение формулы 1 может быть получено в соответствии с синтетическими протоколами, описанными ниже в разделе примеров.
Соединение формулы 1 по настоящему изобретению может модулировать активность одной или более JAK. Термин «модулировать» означает способность увеличивать или уменьшать активность одного или нескольких членов семьи JAK. Соответственно, соединение формулы 1 по изобретению может быть использовано в способах модуляции активности JAK.
В соответствии с настоящим изобретением соединение формулы 1 может действовать в качестве ингибитора одной или нескольких JAK.
В соответствии с настоящим изобретением соединение формулы 1 может быть использовано для модуляции активности JAK у пациента, который нуждается в такой модуляции рецептора, путем введения модулирующего количества соединения формулы 1.
Предметом настоящего изобретения является способ лечения заболеваний или расстройств у пациентов, связанных с JAK, путем введения пациенту, нуждающемуся в таком лечении, терапевтически эффективного количества соединения формулы 1 по настоящему изобретению или его фармацевтической композиции.
JAK-связанное заболевание может включать в себя любое заболевание, расстройство или состояние, непосредственно или косвенно связанное с экспрессией или активностью в JAK, в том числе гиперэкспрессией и/или аномальным уровнем активности. JAK-сопутствующее заболевание может также включать любое заболевание, расстройство или состояние, которое может быть предотвращено, облегчено или вылечено путем модуляции JAK активности.
Предметом настоящего изобретения является способ лечения JAK-ассоциированных аутоиммунных заболеваний, таких как рассеянный склероз, ревматоидный артрит, ювенильный артрит, псориатический артрит, диабет типа I, волчанка, псориаз, воспалительное заболевание кишечника, язвенный колит, болезнь Крона, миастения, нефропатия иммуноглобулинов, аутоиммунные заболевания щитовидной железы и тому подобное.
В соответствии с настоящим изобретением соединение формулы 1 может быть введено пациенту в виде фармацевтических композиций. Эти композиции могут быть получены способом, хорошо известным в фармацевтической области, и могут быть введены различными способами в зависимости от того, требуется местное или системное лечение и на площади, подлежащей обработке. Композиции могут быть в форме таблеток, пилюль, порошков, лепешек, облаток, эликсиров, суспензий, эмульсий, растворов, сиропов, аэрозолей (как твердое вещество или в жидкой среде), мазей, содержащих, например, до 10% по весу активного соединения, мягких и твердых желатиновых капсул, суппозиториев, стерильных инъекционных растворов и стерильно упакованных порошков. Композиции могут быть приготовлены в виде единичной дозы, причем каждая доза содержит примерно от 5 мг до 1000 мг, обычно предпочтительно от 100 мг до 500 мг активного ингредиента. Термин «единичные дозы» относится к физически дискретным единицам, пригодным в качестве единичных доз для человека и других млекопитающих, причем каждая единица содержит заданное количество активного материала, рассчитанное на получение желаемого терапевтического эффекта, в сочетании с подходящим фармацевтическим наполнителем.
Активное соединение может быть эффективным в широком диапазоне доз и обычно вводится в фармацевтически эффективном количестве. Следует понимать, однако, что количество фактически принимаемого соединения обычно будет определяться лечащим врачом в соответствии с состоянием пациента, подлежащего лечению.
Данное изобретение поясняется чертежами.
Фиг. 1. Динамика развития ревматоидного артрита по всем лапам животных в модели ревматоидного артрита, вызванного коллагеном, у мышей при лечении ZD19-0002 (препарат формулы 1) и ZD19-0001(Барицитиниб-основание).
Фиг. 2. Усредненные значения клинических показателей по всем лапам животных в модели ревматоидного артрита, вызванного коллагеном, у мышей при лечении ZD19-0002 (препарат формулы 1) и ZD19-0001 (Барицитиниб-основание).
Фиг. 3. Динамика развития ревматоидного артрита по терапевтическим лапам животных в модели ревматоидного артрита, вызванного коллагеном, у мышей при лечении ZD19-0002 (препарат формулы 1) и ZD19-0001 (Барицитиниб-основание).
Фиг. 4. Усредненные значения клинических показателей по терапевтическим лапам животных в модели ревматоидного артрита, вызванного коллагеном, у мышей при лечении ZD19-0002 и ZD19-0001.
Фиг. 5. Динамика развития ревматоидного артрита по профилактическим лапам животных в модели ревматоидного артрита, вызванного коллагеном, у мышей при лечении ZD19-0002 (препарат формулы 1) и ZD19-0001 (Барицитиниб-основание).
Фиг. 6. Усредненные значения клинических показателей по профилактическим лапам животных в модели ревматоидного артрита, вызванного коллагеном, у мышей при лечении ZD19-0002 (препарат формулы 1) и ZD19-0001 (Барицитиниб-основание).
Ниже изобретение будет описано более подробно с помощью конкретных примеров, которые представлены с целью иллюстрации и не предназначены для ограничения изобретения каким-либо образом. Специалисты в данной области техники легко поймут различие некритических параметров, которые могут быть изменены или модифицированы, чтобы получить те же результаты.
Пример 1. Метод синтеза 2-(3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфоната (1)
2-(3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат (1) получали в соответствии со схемой 1:
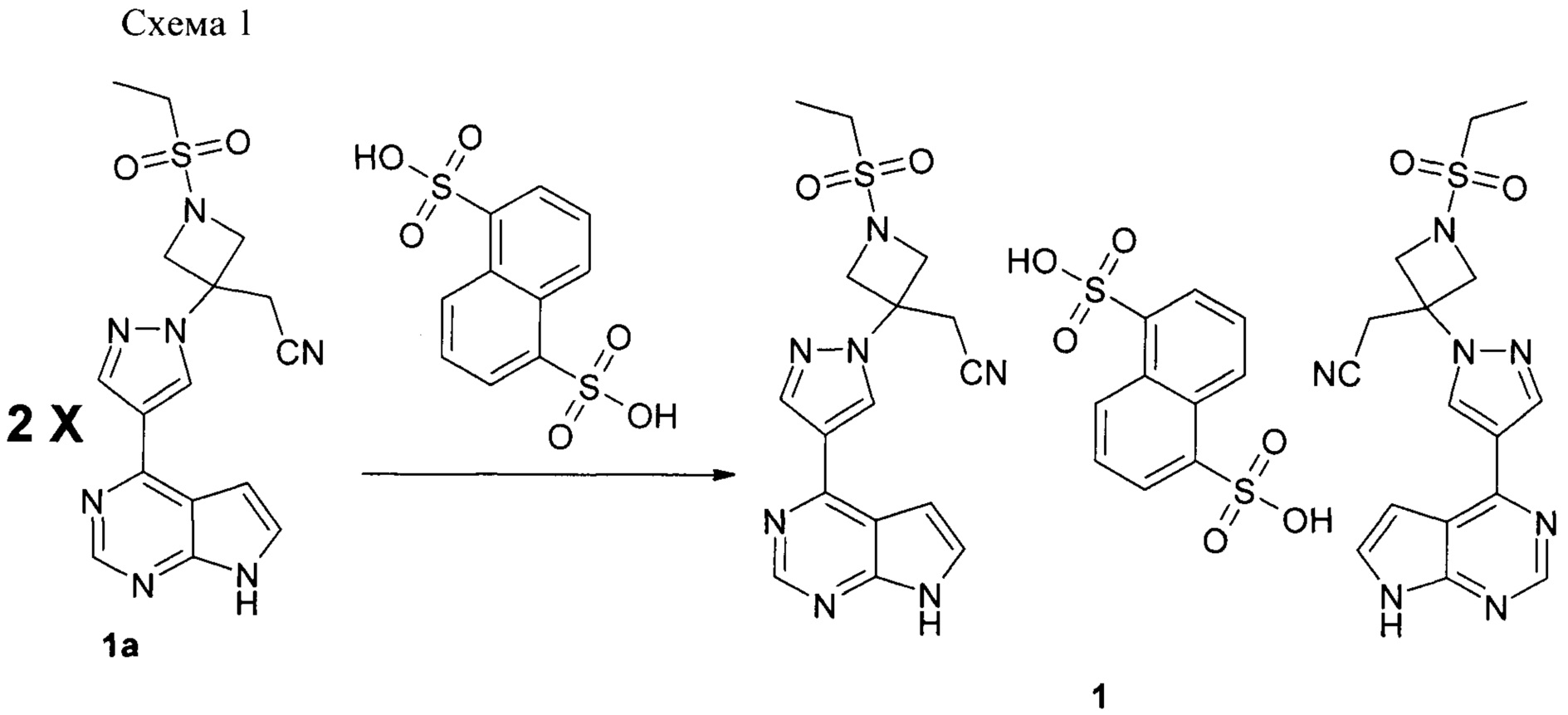
К нагретому до 60°С раствору {1-(этилсульфонил)-3-[4-(7Н-пирроло[2,3-d]пиpимидин-4-ил)-1H-пиpaзoл-1-ил]азетидин-3-ил}ацетонитрила (300 мг, 0.808 ммоль, 1 экв.) в ацетонитриле (40 мл) был прибавлен раствор тетрагидрата нафтил-1,5-дисульфокислоты (145.6 мг, 0.404 ммоль, 0.5 экв) в ацетонитриле (V=2 мл). Реакционная масса перемешивалась при 60°С 2 часа, затем выдержана при кипении 10 мин. Реакционная масса охлаждена до комнатной температуры, осадок отфильтрован и высушен. Выход 2-(3-(4-(7Н-пирроло[2,3-d]пиримидин-4-ил)-1Н-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфоната 379 мг, 91%. При охлаждении реакционной массы до -10°С в течение 16 часов выход продукта повышается до 408 мг, 98%.
Пример 2. Определение ингибирующей активности соединений по отношению к JAK
Соединения тестировали в соответствии со стандартным протоколом Z'-Lyte скрининга компании Life Technologies). Тест соединения проверяются в 1% ДМСО (окончательный) в скважине. 100 нл 100х тестируемых соединений в 100% ДМСО добавляют в 2,4 мкл киназного буфера (50 мМ HEPES, рН6.5, 0,01% BRIJ-35, 10 мМ MgCl2, 1 мМ EGTA, 0,02% NaN3) в условиях низкой объемной NBS, черный 384 -Ну пластины (Corning Кат. # 4514). После этого добавляют к 5 мкл 2Х пептида / киназы смесь (Туr06 / JAK1, окончательные концентрации 21.2 - 91.5 нг JAK1 и 2 мкМ Тир 06) и 2,5 мкл 4хАТР (конечная концентрация 75 мкМ) добавляют в киназный буфер. Через 30 сек инкубации на шейкере реакцию киназы инкубировали в течение 60 мин при комнатной температуре. После этого добавляют 5 мкл 1:128 разбавленного реагента А и инкубируют в течение еще 60 мин при комнатной температуре. Флуоресценция была измерена при возбуждении длиной волны 400 нм и эмиссии при 445 и 520 нм. Степень фосфорилирования FRET-пептида может быть рассчитана из соотношения выбросов. Коэффициент выбросов будет оставаться низким, если лада-пептид фосфорилируется (т.е. нет ингибирования киназы) и будет высоким, если лада-пептид не фосфорилирован (т.е. киназа ингибирована)
% фосфорилирования рассчитывается как
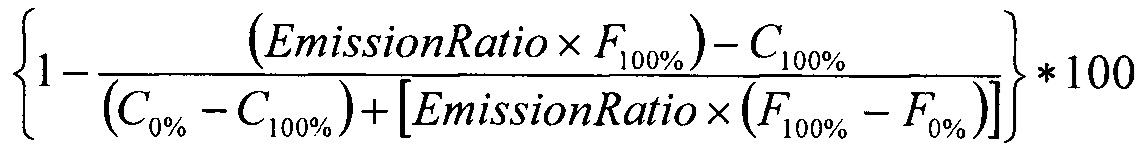 ,
,
% ингибирования рассчитывается как
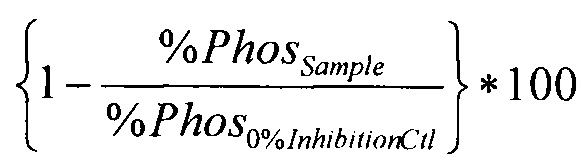 ,
,
где С100%=средний сигнал эмиссии кумарина контроль 100% фосфорилирования;
С0%=средний сигнал эмиссии кумарина контроль 0% фосфорилирования;
F100%=средний сигнал эмиссии флуоресцеина контроль 100% фосфорилирования;
F0%=средний сигнал эмиссии флуоресцеина контроль 0% фосфорилирования.
Концентрационная кривая зависимости активности киназы от концентрации тестируемых веществ построена с использованием сигмоидной модели.
Данные активности соединения формулы 1 и Барицитиниба по отношению к JAK1 представлены в таблице 1.
Пример 3. Определение термодинамической растворимости соединения формулы 1 и прототипа Барицитиниба
Смешивали 5 мг исследуемого соединения с 1 мл универсального буфера (pION) с рН=2,0; 4,0 или 7,0 в течение 15 мин при 25°С. Дополнительные количества веществ добавляли до тех пор, пока раствор не становился мутным. Виалы с раствором инкубировали при перемешивании в течение 24 ч при 25°С для достижения равновесия между раствором и осадком при насыщении. После уравновешивания 200 мкл раствора (в 2-х повторах) фильтровали через 96-луночный фильтровальный планшет (Millipore) для отделения осадка. Концентрацию соединений в фильтрате определяли спектрофотометрически с помощью стандартной калибровочной кривой. Проводили измерение спектра оптического поглощения вещества и построение калибровочной кривой при выбранной длине волны (обычно, соответствующей максимуму поглощения вещества λmax). Концентрацию вещества в фильтрате (т.е. растворимость) рассчитывали по нижеприведенной формуле:
Solubility=(ODλmax filtrate - ODλmax blank)/Slope × 1,67 × Filtrate dilution,
где ODλmax filtrate - оптическая плотность фильтрата;
ODλmax blank - оптическая плотность холостого раствора без вещества;
Slope - наклон калибровочной кривой;
1,67 - фактор разведения фильтрата ацетонитрилом;
Filtrate dilution - фактор разведения фильтрата буфером.
Полученные результаты представлены в Таблице 2.
Пример 4. Проницаемость в модели клеток кишечника линии Сасо-2
Культивирование Сасо-2 клеток
Линия клеток Сасо-2 была получена из АТСС (cat. # НТВ-37) и культивировалась согласно рекомендациям АТСС и Millipore [1] в 75 см2 флаконах при 37°С и 5% СO2. Клетки пересевали 2 раза в неделю в отношении 1:6.
Среда для Сасо-2 клеток: Dulbecco's Modified Eagle's Medium (DMEM), 0.584 г/л L-глутамина, 10% бычьей сыворотки, 100 U/мл пенициллина и 0.1 мг/мл стрептомицина, 2% незаменимые аминокислоты.
Посадка клеток в плашку
Для проведения эксперимента Сасо-2 клетки 10-50 пассажа в состоянии 80-90% покрытия флакона рассаживают в планшеты с двухкомпонентными ячейками Millicell 96 в количестве 10000 на 0.11 см2 лунку (90 909 клеток/см2). Дно внутренней части ячейки состоит из полупроницаемого фильтра, на котором образуется монослой клеток. Процедура посадки клеток в плашку:
- снять клетки с помощью трипсина-ЭДТА;
- посчитать количество живых клеток в суспензии с помощью автоматического счетчика клеток и приготовить 10 мл суспензии с концентрацией 1.33×105 клеток/мл;
- добавить по 250 мкл среды в нижние лунки планшета;
- добавить по 75 мкл суспензии клеток в верхние лунки планшета Millicell 96.
Сасо-2 клетки инкубируют в планшете Millicell 96 в течение 21 дня со сменой среды 2 раза в неделю в первые 2 недели и 3 раза в последнюю (по 75 мкл среды добавляют в верхние и по 250 мкл в нижние лунки).
Целостность монослоя проверяют путем измерения электрического сопротивления (TEER) с помощью прибора Millicell-ERS. Приемлемым является значение TEER не менее 3 кОм/лунку.
Протокол эксперимента
Рабочие растворы
1) 500 мл Буфера I (HBSS, 10 мМ HEPES, рН=7.4).
2) 50 мл Буфера II (HBSS, 10 мМ HEPES, рН=7.4, 1% ДМСО).
3) 50 мкл 100х стоков тестируемых веществ и контролей - 0.1 мМ в ДМСО.
4) 50 мкл 200х стока родамина 123 - 2 мМ в ДМСО.
5) 50 мкл 2 мМ стока циклоспорина А в ДМСО.
6) Приготовить 10 мл Буфера III (HBSS. 10 мМ HEPES, рН=7.4, 10 мкМ циклоспорин А из 2 мМ ДМСО стока).
7) Приготовить из 100х ДМСО стоков 1.6 мл исходных растворов тестируемых веществ в буфере I:
16 мкл (0.1 мМ ДМСО стока)+1584 мкл буфера I. Финальная концентрация веществ - 1 мкМ.
8) Приготовить из 100х ДМСО стоков 0.8 мл исходных растворов контролен в буфере I:
8 мкл (0.1 мМ ДМСО стока)+792 мкл буфера I.
9) Приготовить из 200х ДМСО стока 1600 мкл 10 мкМ исходных раствора родамина 123 с и без циклоспорина А в буфере I:
8 мкл (2 мМ ДМСО стока)+8 мкл (2 мМ ДМСО стока циклоспорина А или ДМСО)+1584 мкл буфера I.
10) Приготовить 500 мл раствора MeCN:H2O=1:1 с 50 нг/мл внутреннего стандарта толбутамида.
Прединкубация
1) Переместить Millicell 96 Сасо-2 систему из инкубатора и измерить TEER.
2) Удалить среду из всех 96 лунок с фильтром плашки Millicell 96 Сасо-2 и нижней части планшета.
3) Промыть клеточный монослой, добавляя по 100 мкл Буфера I в верхние и по 300 мкл в нижние лунки Millicell 96 Сасо-2 плашки. Удалить Буфер I сначала из верхних, затем из нижних лунок Millicell 96 Сасо-2 плашки. Повторить процесс промывки еще 2 раза. Оставить преинкубировать плашку с Буфером I в течение 30 минут при 37°С.
Изучение транспорта
Изучали транспорт соединений в двух направлениях с целью выяснить вклад мембранных транспортеров. Контроли с высокой (пропранолол) и низкой (ранитидин) проницаемостью тестировали в направлении А-В, контролем транспорта с участием Pgp служит родамин 123, его транспорт проводили в обоих направлениях с и без добавления ингибитора Pgp циклоспорина А.
4) Для определения скорости транспорта из апикальной (А) в базолатеральную (В) область [А-В] нужно: добавить по 90 мкл 1х тестируемого вещества в буфере с/без циклоспорина А в 3 лунки с фильтрами и по 250 мкл Буфера II или III в лунки нижней транспортной плашки.
5) Для определения скорости транспорта из базолатеральной (В) области в апикальную (А) [В-А] нужно: добавить по 90 мкл Буфера II или III в 3 лунки с фильтрами и по 250 мкл тестируемого вещества в буфере с/без циклоспорина А в нижние лунки плашки.
6) Инкубировать собранную Millicell 96 Сасо-2 систему в течение 2 ч при 37°С на шейкере с перемешиванием при 300 об/мин.
7) Разъединить верхнюю и нижнюю части плашки.
Работа с исходными растворами
8) Исходные растворы тестируемых веществ и контролей содержать в темном месте до конца инкубации (в течение 2 ч).
9) Перенести по 70 мкл исходных растворов тестируемых и контрольных веществ в подписанные пробирки на 1.1 мл, добавить по 210 мкл холодного 50% MeCN, содержащего 50 нг/мл толбутамида (внутренний стандарт). Перемешать и хранить при -80°С до анализа.
Измерение флуоресценции Родамина123
Для оценки проницаемости родамина 123 с и без циклоспорина А проводят измерения флуоресценции (Ех.485/Еm.535 нм) предварительно разведенных образцов:
1) 2 мкл (исходного раствора)+58 мкл (буфер II или III для проб с/без циклоспорина).
2) 2 мкл (донорный апикальный (DA) и базолатеральный (DB) образцы)+58 мкл (буфер II или III для проб с/без циклоспорина).
3) 60 мкл - акцепторный апикальный (А) и базолатеральный (В) образцы.
Подготовка образцов к анализу
По 70 мкл растворов отбираются из верхней и нижней плашек в отдельные пробирки в штативе, затем к ним добавляется по 210 мкл холодного 50% MeCN, содержащего 50 нг/мл толбутамида (внутренний стандарт). Манипуляции выполняются с помощью автоматизированной станции Biomek FX.
Образцы хранят при -80°С до ВЭЖХ-МС/МС анализа.
Обработка данных, расчеты
Для количественного определения использовали ВЭЖХ-МС/МС систему QTRAP 5500 (Applied Biosystems) с хроматографом Agilent Infinity 1290 (Agilent Technologies). Параметры хроматографии и детекции подбирались для достижения максимальной чувствительности при приемлемом времени анализа.
Площади под хроматографическими пиками аналита, нормированные на сигнал внутреннего стандарта (AUC/IS), считали в программе Analyst 1.5.2 и использовали для расчетов.
Проницаемость (Рарр, см/с) через монослой Сасо-2 клеток оценивается по формуле:
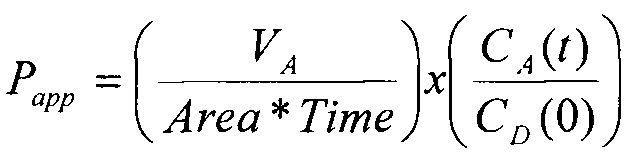 ,
,
где VA - объем (мл) в акцепторной лунке, который равен 0.25 для транспорта [А-В] и 0.09 для транспорта [В-А];
Area - площадь поверхности (см2), составляет 0,11 см2 для 96-луночной MultiScreen Сасо-2 плашки;
Time - время транспорта (7200 с);
СА(t) - AUC/IS вещества в акцепторной лунке после эксперимента;
CD(0) - AUC/IS исходного раствора вещества в донорной лунке.
Для родамина 123 отношение 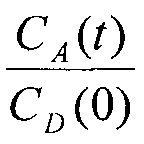 определяется по флуоресценции.
определяется по флуоресценции.
Считают проницаемость (Рарр) вещества.
Пример 5. Исследование эффективности в модели ревматоидного артрита, вызванного коллагеном
В дни 0 и 21 животных анестезировали Изофлураном и вводили подкожно в основание хвоста 100 мкл коллагена 2го типа в полном адъюванте Фрейнда. В период с 18 по 35 дни проявляются признаки ревматоидного артрита. Животных рандомизировали по группам после опухания хотя бы одной лапы (клинический показатель 1). Старались, чтобы средний показатель в группах на момент рандомизации был на уровне 0.25. Всего будет использовано как минимум 8 животных в группе.
После рандомизации (день 1 ревматоидного артрита) начинают лечение животных согласно таблице ниже. Ежедневно оценивали показатель лап животных. Эффективность исследуемых веществ оценивали на основе AUC, построенных на основе показателей лап, и по данным гистологии передних и задних лап. Оценивали как потенциальную терапевтическую эффективность (по индивидуальным лапам со значениями показателя >0 в момент рандомизации), так и потенциальную профилактическую эффективность (по индивидуальным лапам со значениями показателя =0 в момент рандомизации).
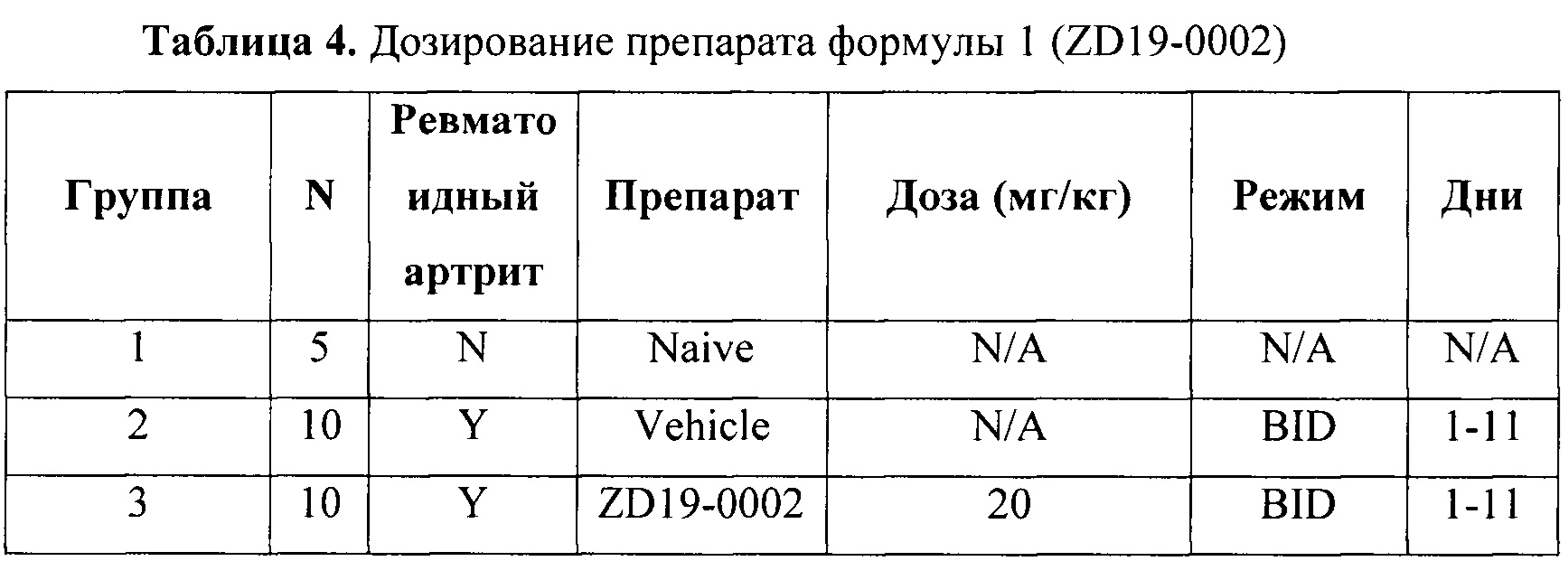
BID - введение 2 раза в сутки с интервалом 10-12 часов.
Также в ходе исследования определяли вес животных и оценивали признаки токсичности исследуемых веществ. В день 11 ревматоидного артрита животных анестезировали Изофлураном и подвергали некропсии.
Индукция ревматоидного артрита
Коллаген готовили в виде раствора с концентрацией 4 мг/мл в 0.01N уксусной кислоте. Равные объемы раствора коллагена (4 мг/мл) и полного адъюванта Фрейнда (5 мг/мл) с добавлением Micro-tuberculosis bacterium (МТВ) были смешаны для получения эмульсии в течение 5 мин.
Критерии клинических показателей
0 = норма;
1 = сустав одной передней или задней лапы нарушен или отмечена минимальная диффузная эритема или отек;
2 = сустав двух передних или задних лап нарушен или отмечена умеренная диффузная эритема или отек;
3 = сустав трех передних или задних лап нарушен или отмечена средняя диффузная эритема или отек;
4 = выраженная диффузная эритема или отек, или 4 сустава нарушены;
5 = резкая диффузная эритема или резкий отек полной лапы, невозможность двигать пальцами.
Пример 6. Получение лекарственного средства в виде таблеток
Спресовывали смесь 1600 мг крахмала, 1600 мг измельченной лактозы, 400 мг талька и 1000 мг ингибитора формулы 1. Полученный брусок измельчали в гранулы и просеивали через сито, собирая гранулы размером 14-16 меш. Полученные гранулы таблетировали в таблетки пригодных форм, весом 500 мг каждая.
Пример 7. Получение лекарственного средства в форме капсул
Ингибитор формулы 1 тщательно смешивали с лактозой в соотношении 2:1. Полученную порошкообразную смесь упаковывали по 600 мг в желатиновые капсулы подходящего размера.
Пример 8. Получение лекарственного средства в форме композиций для внутримышечных, внутрибрюшинных или подкожных инъекций
Смешали 500 мг ингибитора формулы 1, 300 мг хлорбутанола, 2 мл пропиленгликоля и 100 мл воды для инъекций. Раствор фильтровали и помещали в 1 мл ампулы, которые укупоривали.
Из приведенного выше описания специалистам в данной области техники очевидны различные модификации изобретения в дополнение к описанным здесь.
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/3bf8a1bcaf128500f51f19baf6454136.png)
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/64d3a7a3a6b082bb5f458b3fe8fc4375.png)
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/2d7a946ba6b3dbabdd8c8f9f08af474e.png)
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/7354d0b78a6b9c2ab299df4ae4436869.jpg)
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/2a3bcb9362161591d93cd9e40712f20e.png)
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/5bcebfebb9b4ac63eb5f5c7ebf91f5ba.png)
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/05d9ebe59b456292103ad3a804616a89.png)
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/51d01cc9b153a431d2682cf1ea30bde2.jpg)
![2-(3-(4-(7H-пирроло[2,3-d]пиримидин-4-ил)-1H-пиразол-1-ил)-1-(этилсульфонил)азетидин-3-ил)ацетонитрила геминафтилдисульфонат в качестве ингибитора Янус киназ](https://fips.edrid.ru/images/rid/ca/81/12/c40940b431cf4eb0bcafa28e40299f16.jpg)
Замещенные 4-{ [4-(3,3-диоксидо-1,3-бензоксатиол-6-ил)арилокси]метил} пиперидины как агонисты рецепторов gpr119, фармацевтическая композиция, способы их получения и применения
{3-[(7h-пирроло[2,3-d]пиримидин-4-ил)азолил]азетидин-3-ил}ацетонитрилы в качестве ингибиторов янус киназ
Замещенные пиразинопиримидиноны как блокаторы trpa1 каналов, фармацевтическая композиция, способы их получения и применения
Производные 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1h,6h-пиридо[4,3-d]пиримидин-2,4,7-триона в качестве ингибиторов мек1/2
Дихлорацетат n,n-дизамещенного n-[4-(1-метил-1н-индол-3-ил)-пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина в качестве модулятора egfr для лечения рака
Дихлорацетат {3-[4-(7h-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-этилсульфонил-азетидин-3-ил}-ацетонитрила в качестве ингибитора янус киназ
Замещенные n-{ 3-[4-(1-метил-1н-индол-3-ил)пиримидин-2-иламино]-4-метоксифенил} -амиды в качестве модуляторов egfr, предназначенные для лечения рака
Дихлорацетаты замещенных n4-[2-(диметилфосфорил)фенил]-n2-(2-метокси-4-пиперидин-1-илфенил)-5-хлорпиримидин-2,4-диаминов в качестве модуляторов alk и egfr, предназначенных для лечения рака
Замещенные n2-(4-амино-2-метоксифенил)-n4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов alk и egfr, предназначенные для лечения рака
Замещенные метил (2-{ 4-[3-(3-метансульфониламино-2-фтор-5-хлор-фенил)-1н-пиразол-4-ил]пиримидин-2-иламино} -этил)карбаматы, способ их получения и применения
Замещенные 4-{ [4-(3,3-диоксидо-1,3-бензоксатиол-6-ил)арилокси]метил} пиперидины как агонисты рецепторов gpr119, фармацевтическая композиция, способы их получения и применения
{3-[(7h-пирроло[2,3-d]пиримидин-4-ил)азолил]азетидин-3-ил}ацетонитрилы в качестве ингибиторов янус киназ
Замещенные пиразинопиримидиноны как блокаторы trpa1 каналов, фармацевтическая композиция, способы их получения и применения
Производные 1-(3-аминофенил)-6,8-диметил-5-(4-иод-2-фтор-фениламино)-3-циклопропил-1h,6h-пиридо[4,3-d]пиримидин-2,4,7-триона в качестве ингибиторов мек1/2
Дихлорацетат n,n-дизамещенного n-[4-(1-метил-1н-индол-3-ил)-пиримидин-2-ил]-5-метоксибензол-1,2,4-триамина в качестве модулятора egfr для лечения рака
Дихлорацетат {3-[4-(7h-пирроло[2,3-d]пиримидин-4-ил)-пиразол-1-ил]-1-этилсульфонил-азетидин-3-ил}-ацетонитрила в качестве ингибитора янус киназ
Замещенные n-{ 3-[4-(1-метил-1н-индол-3-ил)пиримидин-2-иламино]-4-метоксифенил} -амиды в качестве модуляторов egfr, предназначенные для лечения рака
Дихлорацетаты замещенных n4-[2-(диметилфосфорил)фенил]-n2-(2-метокси-4-пиперидин-1-илфенил)-5-хлорпиримидин-2,4-диаминов в качестве модуляторов alk и egfr, предназначенных для лечения рака
Замещенные n2-(4-амино-2-метоксифенил)-n4-[2-(диметилфосфорил)-фенил]-5-хлор-пиримидин-2,4-диамины в качестве модуляторов alk и egfr, предназначенные для лечения рака
Замещенные метил (2-{ 4-[3-(3-метансульфониламино-2-фтор-5-хлор-фенил)-1н-пиразол-4-ил]пиримидин-2-иламино} -этил)карбаматы, способ их получения и применения

