Результат интеллектуальной деятельности: ЗАМЕЩЕННЫЕ ПИРАЗИНОПИРИМИДИНОНЫ КАК БЛОКАТОРЫ TRPA1 КАНАЛОВ, ФАРМАЦЕВТИЧЕСКАЯ КОМПОЗИЦИЯ, СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ
Вид РИД
Изобретение
Изобретение относится к новым физиологически активным веществам, новым блокаторам каналов TRPA1, активным компонентам для фармацевтических композиций, к фармацевтическим композициям и лекарственным средствам, содержащим замещенные пиразинопиримидиноны в качестве блокаторов TRPA1 каналов, которые блокируют передачу болевого сигнала; способу лечения болезненных состояний, сопровождаемых острой и хронической болью различной этиологии.
Существуют различные протеины, именуемые ионными каналами, которые отвечают за проникновение через клеточные мембраны. Правильная экспрессия и функциональность ионных каналов важна для поддержания функционирования клеток, межклеточной коммуникации и передачи нервных импульсов. Многочисленные заболевания и болезненные состояния могут быть вызваны нарушением или сбоем регуляции мембранного потенциала. Ввиду исключительной важности ионных каналов для модулирования мембранного потенциала идентификация различных агентов, которые могут ускорять или ингибировать конкретные ионные каналы, очень важна с точки зрения создания новых терапевтических средств.
Одним из таких каналов является Transient Receptor Potential A1 (TRPA1) канал. TRPA1 канал - это кальциевый канал и конкретно неселективный кальциевый катионный канал. В дополнение к катионам кальция, TRPA1 канал может использоваться для проникновения через мембраны и других катионов, в частности натрия. Таким образом, TRPA1 каналы модулируют мембранный потенциал через модулирование проникновения через мембраны ионов кальция и натрия. Несмотря на то, что каналы TRPA1 модулируют проникновение ионов кальция в клетку, механистически они отличны от voltage-gated каналов.
В последние годы появилось ряд патентов, а также свидетельства о клинических испытаниях новых лекарственных средств для снятия боли различной этиологии на основе блокаторов TRPA1 канала. Так, американские компании Cubist Pharmaceuticals и Hydra Biosciences в 2012 году анонсировали первую фазу клинических испытаний препарата CB-189,625 как нового модулятора TRPA1 в качестве потенциального анальгизирующего средства. Glenmark Pharmaceuticals в августе 2014 года объявила о положительных результатах второй А фазы клинических испытаний соединения GRC 17536 - нового блокатора канала TRPA1 в качестве анальгизирующего средства при диабетической нейропатии. Исследования проводились на 138 пациентах в Европе и в Индии.
Таким образом, поиск эффективных блокаторов каналов TRPA1 представляется исключительно важной, актуальной и многообещающей задачей.
В качестве аналога данного изобретения можно указать вещество-блокатор каналов TRPA1 АР-18 [1]:

Вещество АР-18 обладает антагонитической активностью по отношению к TRPA1 каналам на уровне IC50 3.1 mM и проявляет некоторую анальгезирующую активность в экспериментах на крысах.
Недостатками аналога является ряд параметров молекулы АР-18, которые в совокупности делают его потенциальное применение в качестве лекарства невозможным: невысокая ин-витро активность предполагает введение в организм в высоких дозах, в то время как наличие двойной свзи в молекуле делает ее потенциально опасной с точки зрения токсикологии даже при минимальных дозировках.
[1] Petrus, M.; Peier, A.M.; Bandell, M.; Hwang, S.W.; Huynh, T.; Olney, N.; Jegla, T.; Patapoutian, A. A role of TRPA1 in mechanical hyperalgesia is revealed by pharmacological inhibition. Mol. Pain, 2007, 3, 40.
Общепризнанным подходом в разработке новых орально-биодоступных лекарств является следование так называемому «правилу пяти» Липински. Так, с целью получения “drug like” свойств молекулы рекомендуеся введение достаточного числи гетероатомов, в частности, в виде гетероциклических структур.
Следуя вышеизложенному, значительно большей перспективой стать потенциальным лекарственным препаратом обладает вещество НС-030031 (McNamara, C.R.; Mandel-Brehm, J.; Bautista, D.M.; Siemens, J.; Deranian, K.L.; Zhao, M.; Hayward, N.J.; Chong, J.A.; Julius, D.; Moran, M.M.; Fanger, C.M. TRPA1 mediates formalin-induced pain. Proc. Natl. Acad. Sci. USA, 2007, 104(33), 13525-13530), которое можно считать прототипом предлагаемого изобретения
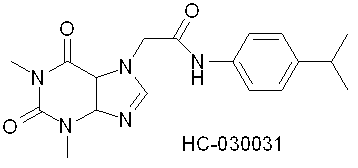
Вещество НС-030031 обладает антагонитической активностью по отношению к TRPA1 каналам на уровне IC50 1.0 mM и проявляет анальгезирующую активность в экспериментах на крысах.
К недостаткам прототипа можно отнести то, что в молекуле присутствует амидная связь, обычно быстро метаболизируемая в человеческом организме. Потенциальное лекарство с амидной связью может обладать очень коротким «временем жизни» и требовать многократного введения в течение дня. Также, активность ин-витро на уровне IC50 1.0 mM представляется недостаточной (повлечет введение в организм значительных количеств вещества для достижения терапевтического эффекта, что может оказать токсическое воздействие).
Таким образом, имеется задача создания новых анальгетических препаратов на основе блокаторов каналов TRPA1.
Для решения задачи разработки новых высокоэффективных блокаторов каналов TRPA1 авторами данного изобретения выполнены широкие скрининговые ин-витро исследования низкомолекулярных соединений различных классов, были обнаружены соединения-хиты и соединения-лидеры, относящиеся к нескольким семействам гетероциклических соединений, затем проводились направленные модификации многих позиций обнаруженных структур и их тестирование, установлена взаимосвязь «структура-активность» и выбраны наиболее перспективные ряды. В результате синтезирована серия новых производных, которые являются оригинальными и высокоэффективными блокаторов каналов TRPA1. Полученные физиологически активные гетероциклические соединения включают новые производные замещенных пиразинопиримидинонов, которые ранее не были описаны в литературе. До настоящего времени способность указанных соединений проявлять свойства блокаторов каналов TRPA1 также не была известной.
Ниже приведены определения терминов, которые использованы в описании этого изобретения.
«Алкил» означает алифатическую углеводородную линейную или разветвленную группу с 1-12 атомами углерода в цепи. Разветвленная означает, что алкильная цепь имеет один или несколько «низших алкильных» (С1-С6)алкильных заместителей. Предпочтительными алкильными группами являются низший (С1-С6) алкил или метил, этил, н-пропил, изо-пропил, н-бутил, изо-бутил, втор-бутил, трет-бутил, циклопропилметил, циклобутилметил, циклопентилметил, н-пентил, 2-пентил, 3-пентил, нео-пентил, н-гексил, циклогексил. Алкил может иметь заместители.
«Алифатический» радикал означает радикал, полученный удалением атома водорода из неароматической С-Н связи. Алифатический радикал может дополнительно содержать заместители - алифатические или ароматические радикалы, определенные в данном разделе. Представители алифатических радикалов включают алкил, алкенил, алкинил, циклоалкил, циклоалкенил, гетероциклил, гетероцикленил, аралкенил, аралкилоксиалкил, аралкилоксикарбонилалкил, аралкил, аралкинил, аралкилоксиалкенил, гетероаралкенил, гетероаралкил, гетероаралкилоксиалкенил, гетероаралкилоксиалкил, анелированный арилциклоалкил, анелированный гетероарилциклоалкил, анелированный арилциклоалкенил, анелированный гетероарилциклоалкенил, анелированный арилгетероциклил, анелированный гетероарилгетероциклил, анелированный арилгетероцикленил, анелированный гетероарилгетероцикленил.
«Алкенил» означает алифатическую линейную или разветвленную углеводородную группу, содержащую от 2 до 12 атомов углерода и включающую углерод-углеродную двойную связь. Разветвленная означает, что к линейной алкенильной цепи присоединены одна или несколько низших алкильных групп, таких как метил, этил или пропил. К алкенильной группе может быть присоединена одна или несколько ароматических, циклических или гетероциклических систем.
«Активный компонент» (лекарственное вещество, лекарственная субстанция, drug-substance) означает физиологически активное вещество синтетического или иного (биотехнологического, растительного, животного, микробного и прочего) происхождения, обладающее фармакологической активностью и являющееся активным началом фармацевтической композиции, используемой для производства и изготовления лекарственного препарата (средства).
«Антагонисты» означают лиганды, которые связываются с рецепторами определенного типа и не вызывают активного клеточного ответа. Антагонисты препятствуют связыванию агонистов с рецепторами и тем самым блокируют передачу специфического рецепторного сигнала.
«Алкилокси» или «Алкокси» означает АлкилО-группу, в которой алкил определен в данном разделе. Предпочтительными алкоксигруппами являются метокси, этокси, н-пропокси, изо-пропокси, н-бутокси и трет-бутокси.
«Аминогруппа» означает R′R″N-группу, замещенную или незамещенную необязательно одинаковыми заместителями R′ и R″. Аминогруппа может иметь заместители.
«Алкилтио» означает алкил-S группу, в которой алкильная группа определена в данном разделе.
«Аминокарбонил» означает С(=О)N Rk aRk+1 a-группу, замещенную или незамещенную необязательно одинаковыми заместителями карбамоильными Rk a и Rk+1 a, включая водород, метил, циклоалкил, который с атомом азота образует пирролидиновый цикл.
«Аннелированный цикл» (конденсированный цикл) означает би- или полициклическую систему, в которой аннелированный цикл и цикл или полицикл, с которым он «аннелирован», имеют как минимум два общих атома. Аннелированными могут быть ароматические, неароматические системы и гетероциклические системы.
«Аралкил» означает алкильную группу, замещенную одной или несколькими арильными группами, в которой значения арил и алкил определены в данном разделе. Примерами аралкильных групп являются необязательно замещенные бензил, фенетил, фенпропил.
«Арил» означает ароматическую моноциклическую или полициклическую систему, включающую от 6 до 14 атомов углерода, преимущественно от 6 до 10 атомов углерода. Арил может содержать один или более «заместителей циклической системы», которые могут быть одинаковыми или разными.
«Ароматический цикл» (ароматическая система) означает планарную циклическую систему, в которой все атомы цикла участвуют в образовании единой системы сопряжения, включающей, согласно правилу Хюккеля, (4n+2) п-электронов (n - целое неотрицательное число). Примерами ароматических циклов являются бензол, нафталин, антрацен и т.п. В случае «гетероароматических циклов» в системе сопряжения участвуют п-электроны и р-электроны гетероатомов, их суммарное число также равняется (4n+2). Примерами таких циклов являются пиридин, тиофен, пиррол, фуран, тиазол и т.п. Ароматический цикл может иметь один или более «заместителей циклической системы» и может быть анелирован с неароматическим циклом, гетероароматической или гетероциклической системой.
«Ацильная группа» (Ацил) означает H-C(=О)-, необязательно замещенные С1-С5алкил-С(=О)-, С1-С5алкенил-С(=О)-, С1-С5циклоалкил-С(=О)- (предпочтительно циклопропил-С(=О)-, циклобутил-С(=О)-); гетерциклил-С(=О)- (предпочтительно 2-метилфуран), арил-С(=О)- (ароил), аралкил-С(=О)- (предпочтительно 3-фенилпентан-С(=О)-), гетероарил-С(=О)- (гетероароил), гетероарилалкил-С(=О)- группы, в которых С1-С5алкил-, С1-С5алкенил-, С1-С5циклоалкил-, гетерциклил-, арил-, аралкил, гетероарил-, гетероарилалкил, метокси группа, указанные группы могут иметь заместители, см. «заместители циклической системы», «замещенный алкил», «замещенный алкенил», «заместители гетероциклической системы», определенные в данном разделе.
«Гетероарил» (гетарил) означает ароматическую моноциклическую или полициклическую систему, включающую от 5 до 14 атомов углерода, предпочтительно от 5 до 10, в которой один или больше атомов углерода замещены гетероатомом или гетероатомами, такими как азот, сера или кислород. Приставка «аза», «окса» или «тиа» перед «гетероарил» означает наличие в циклической системе атома азота, атома кислорода или атома серы, соответственно. Атом азота, находящийся в гетероариле, может быть окислен до N-оксида. Гетероарил может иметь один или несколько «заместителей циклической системы», которые могут быть одинаковыми или разными. Предпочтительно пирролил, фуранил, тиенил, пиридинил, пирролидин, имидазолил, оксазолил, бензотиадиазол, индолил, азаиндолил, бензимидазолил, бензотиазолил, хинолинил, имидазолил, тиенопиридил, хиназолинил, тиенопиримидинил, пирролопиридинил, имидазопиридил, изохинолинил, бензоазаиндолил, 1,2,4-триазинил, тиенопирролил, фуропирролил и др.
«Гетероциклил» означает ароматическую или неароматическую насыщенную или частично насыщенную моноциклическую или полициклическую систему, включающую от 3 до 10 атомов углерода, преимущественно от 4 до 6 атомов углерода, в которой один или несколько атомов углерода заменены на гетероатом, такой как азот, кислород, сера, фосфор. Приставка «аза», «окса» или «тиа» перед гетероциклилом означает наличие в циклической системе атома азота, атома кислорода или атома серы, соответственно. Гетероциклил может иметь один или несколько заместителей, которые могут быть одинаковыми или разными. Атомы азота и серы, находящиеся в гетероциклиле, могут быть окислены до N-оксида, S-оксида или S-диоксида. Представителями гетероциклилов являются пиперидинил, пирролидинил, пиперазинил, морфолинил, тиоморфолинил, тиазолидинил, 1,4-диоксан-2-ил, тетрагидрофурил, тетрагидротиенил и др.
«Замещенный алкенил» может также иметь один или несколько одинаковых или различных заместителей, включая галоген, алкенилокси, ароил, гетероароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонил, гетероаралкилокси и т.д. Предпочтительными алкенильными группами являются этенил, пропенил, н-бутенил, изо-бутенил, 3-метилбут-2-енил, н-пентенил и н-гексенил.
«Замещенный алкил» может иметь один или несколько одинаковых или различных заместителей, включая галоген, алкенилокси, циклоалкил, арил, гетероарил, гетероциклил, ароил, гетероароил, циано, гидрокси, алкокси, карбокси, алкинилокси, аралкокси, арилокси, арилоксикарбонил, алкилтио, гетероарилтио, аралкилтио, арилсульфонил, алкилсульфонил, гетероаралкилокси Rk aRk+1 aN-, где Rk a и Rk+1 a независимо друг от друга представляют собой «заместители аминогруппы», значение которых определено в данном разделе, например, атом водорода, алкил, арил, аралкил, гетероаралкил, гетероциклил или гетероарил, или Rk a и Rk+1 a вместе с атомом N, с которым они связаны, образуют через Rk a и Rk+1 a 4-7 членный гетероциклил или гетероцикленил. Предпочтительными «алкильными заместителями» являются арил, гетероарил, гетероциклил, гидрокси, С1-С5 алкокси, С1-С5 алкоксикарбонил, аралкокси, арилокси, алкилтио, гетероарилтио, аралкилтио, алкилсульфонил, арилсульфонил, алкоксикарбонил, аралкоксикарбонил, гетероаралкилоксикарбонил или Rk aRk+1 aN-, Rk aRk+1 aNC(=О)-, анелированный арилгетероцикленил, анелированный арилгетероциклил. А именно, гидроксигруппа, замещенная фторфенилом оксигруппа, моно- и диС1-С5алкиламиногруппа (диметиламиногруппа, метиламиногруппа), С1-С5алкилоксикарбонильная группа (этилоксикарбонил), фенил, хлорфенил, фторфенил, С1-С5алкил (метил) замещенный фенил, С1-С5циклоалкил (циклопентил) замещенный фенил, С1-С5алкокси замещенный фенил (метоксифенил), тиофенил, фуранил, пирролидин.
«Заместители циклической системы» могут быть представителями арильных групп предпочтительно фенил или нафтил, замещенный фенил или замещенный нафтил. Арил может быть аннелирован с неароматической циклической системой или гетероциклом. Предпочтительно земестителями циклической системы являются водород, галогены (хлор, фтор, бром), необязательно замещенный С1-С5алкил, необязательно замещенный циклоС1-С5алкил, С1-С5алкен, гидроскигруппа, С1-С5алкилоксигруппа (метокси, этокси, пропокси, диэфир этиленгликоль, диэфир метандиола), цианогруппа, С1-С5алкилоксикарбонил (метил, этил), алкилтиогруппа (метилтио), карбоксигруппа, аминокарбонил (см. «аминокарбонил»), фенил аннелированный с 5-7-членным насыщенным циклом, содержащим 1-3 гетероатома (атомы азота, кислорода и серы предпочтительно).
«Заместители гетероциклила» могут быть представителями арильных групп предпочтительно фенил или нафтил, замещенный фенил или замещенный нафтил. Арил может быть анелирован с неароматической циклической системой или гетероциклом. Предпочтительно земестителями циклической системы являются водород, галогены (хлор, фтор, бром), необязательно замещенный С1-С5алкил, необязательно замещенный циклоС1-С5алкил, С1-С5алкен, гидроскигруппа, С1-С5алкилоксигруппа (метокси, этокси, пропокси, диэфир этиленгликоля, диэфир метандиола), цианогруппа, С1-С5алкилоксикарбонил (метил, этил), алкилтиогруппа (метилтио), карбоксигруппа, аминокарбонил (см. «аминокарбонил»), фенил анелированный с 5-7 ти-членным насыщенным циклом, содержащим 1-3 гетероатома (атомы азота, кислорода и серы предпочтительно).
«Заместитель» означает химический радикал, который присоединяется к молекулярному остову (скэффолду, фрагменту), например, «заместитель алкильный», «заместитель аминогруппы», «заместитель карбамоильный», «заместитель циклической системы», значения которых определено в данном разделе.
«Замещенная амидиновая группа» означает R′R″N-(С=NR′″)- группу, в которой заместители R′, R″ и R′″ могут быть представлены необязательно замещенными алкилом, алкенилом, алкинилом, циклоалкилом, арилом, гетарилом и гетероциклилом, значение которых определено в данном разделе. R′R″N-фрагмент амидиновой группы может представлять неароматический азагетероцикл, предпочтительно азетидин, пирролидин, пиперидин, морфолин, тиоморфолин, пиперазин, гомопиперидин, гомопиперазин.
«Замещенная аминокарбонильная группа» (аминокарбонил) означает R′R″N-С(=О)- группу, в которой заместители R′ и R″ могут быть представлены необязательно замещенными алкилом, алкенилом, алкинилом, циклоалкилом, арилом, гетарилом и гетероциклилом, значение которых определено в данном разделе. Предпочтительными аминокарбонильными группами являются необязательно замещенный С1-С5алкил, С1-С5алкенил, С1-С5циклоалкил, необязательно замещенный арил (см. заместитель циклической системы), необязательно замещенный гетарил (см. заместитель циклической системы), необязательно замещенный гетероциклил (см. заместитель гетероциклила) или аминогруппой R'R”N.
«Замещенная аминотиокарбонильная группа» (аминотиокарбонил) означает R′R″N-С(=S)- группу, в которой заместители R′ и R″ могут быть представлены необязательно замещенными алкилом, алкенилом, алкинилом, циклоалкилом, арилом, гетарилом и гетероциклилом, значение которых определено в данном разделе. Предпочтительными аминокарбонильными группами являются алкиламинотиокарбонил, циклоалкиламинотиокарбонил, ариламинотиокарбонил, гетариламинотиокарбонил и бензиламинотиокарбонил.
«Замещенная оксигруппа» означает гидроксигруппу, в которой водород может быть замещен С1-С5алкилом, С1-С5алкенилом, С1-С5циклоалкилом, арилом, гетарилом и гетероциклилом, образуя простые эфиры, или ацилом, образуя сложные эфиры, или аминокарбонильной группой, образуя карбаматы, или оксикарбонильной группой, образуя карбонаты.
«Замещенная оксикарбонильная группа» (оксикарбонил) означает R-О-С(=О)- группу, в которой заместитель R может быть представлен необязательно замещенными алкилом, алкенилом, циклоалкилом, арилом, гетарилом и гетероциклилом, значение которых определено в данном разделе. Предпочтительными оксикарбонильными группами являются метоксикарбонил, этоксикарбонил, трет-бутилоксикарбонил и бензилоксикарбонил.
«Заместитель карбамоильный» означает заместитель, присоединенный к аминокарбонильной группе, значение которой определено в данном разделе. Заместитель карбамоильный представляет собой водород, алкил, циклоалкил, арил, гетероарил, гетероциклил, алкоксикарбонилалкил, аралкоксикарбонилалкил, гетероаралкилокси-карбонилалкил или Rk aRk+1 aN-, Rk aRk+1 aNC(=О)-алкил, аннелированный гетероарилциклоалкенил, аннелированный гетероарилциклоалкил, аннелированный гетероарилгетероцикленил, аннелированный гетероарилгетероциклил, аннелированный арилциклоалкенил, аннелированный арилциклоалкил, аннелированный арилгетероцикленил, аннелированный арилгетероциклил. Предпочтительными «заместителями карбамоильными» являются алкил, циклоалкил, арил, гетероарил, гетероциклил, алкоксикарбонилалкил, аралкоксикарбонилалкил, гетероаралкилокси-карбонилалкил или Rk aRk+1 aN-, Rk aRk+1 aNC(=О)-алкил, аннелированный арилгетероцикленил, аннелированный арилгетероциклил.
«Карбоксил» означает группу -CO2H.
«Лекарственное комбинированное средство (препарат)» - комбинация нескольких лекарственных веществ для одновременного использования в виде таблеток, капсул, инъекций, мазей, ректальных суспензий и гелей и др. готовых форм, предназначенное для восстановления, исправления или изменения физиологических функций у человека и животных, а также для лечения и профилактики болезней, диагностики, анестезии, контрацепции, косметологии и прочего. Лекарственные вещества в одном комплекте могут быть представлены в виде различных готовых форм, предназначенных для введения в организм животного или человека различными способами, например перорально и ректально.
«Метаболические заболевания» - группа заболеваний, связанных с нарушениями обмена веществ. Эти заболевания могут быть вызваны наследственной предрасположенностью, сбоями в эндокринной и нервной системах, а также нарушениями в работе метаболически активных органов. В настоящее время насчитывается большое количество метаболических нарушений, однако в данном изобретении рассматриваются главным образом болезни, связанные с сигнальным каскадом рецепторов GPR119, в частности такие как диабет, ожирение, диабетучность, метаболический синдром, гиперхолестеринемия, дислипидемия и т.д.
«Низший алкил» означает линейный или разветвленный алкил с 1-5 атомами углерода (С1-С5 алкил).
«Рецепторы» (от латинского recipere - получать, узнавать) представляют собой биологические макромолекулы, расположенные на цитоплазматической мембране клетки или внутриклеточно, способные специфически взаимодействовать с ограниченным набором физиологически активных веществ (лигандов) и трансформировать сигнал об этом взаимодействии в определенный клеточный ответ.
«Сульфонил» означает R-SO2- группу, в которой заместитель R может быть представлен необязательно замещенными С1-С5алкилом, С1-С5алкенилом, С1-С5циклоалкилом, необязательно замещенным арилом (см. заместитель циклической системы), необязательно замещенным гетарилом (см. заместитель циклической системы), необязательно замещенным гетероциклилом (см. заместитель циклической системы) или аминогруппой R′R″N, значение которых определено в данном разделе.
«Тиоацильная группа» (Тиоацил) означает необязательно замещенные алкил-С(=S)-, алкенил-С(=S)-, циклоалкил-С(=S)-, гетерциклил-С(=S)-, арил-С(=S)- аралкил-С(=S)-, гетероарил-С(=S)-, гетероарилалкил-С(=S)- группы.
«Циклоалкил» означает неароматическую моно- или полициклическую систему, включающую от 3 до 10 атомов углерода. Циклоалкил может иметь один или несколько «заместителей циклической системы», которые могут быть одинаковыми или разными. Представителями циклоалкильных групп являются циклопропил, циклобутил, циклопентил, циклогексил, декалин, норборнил, адамант-1-ил и т.п. Предпочтительными «заместителями циклической системы» являются алкил, алкокси, гидрокси или аминогруппа, значение которых определено в данном разделе.
«Циклоалкенил» означает неароматическую моно- или полициклическую систему, включающую от 3 до 10 атомов углерода и содержащую одну или более двойную углерод-углеродную связь. Представителями циклоалкенильных групп являются циклопентенил и циклогексенил.
«Фармацевтическая композиция» обозначает композицию, включающую в себя соединение общей формулы I и, по крайней мере, один из компонентов, выбранных из группы, состоящей из фармацевтически приемлемых и фармакологически совместимых наполнителей, растворителей, разбавителей, носителей, вспомогательных, распределяющих и воспринимающих средств, средств доставки, таких как консерванты, стабилизаторы, наполнители, измельчители, увлажнители, эмульгаторы, суспендирующие агенты, загустители, подсластители, отдушки, ароматизаторы, антибактериальные агенты, фунгициды, лубриканты, регуляторы пролонгированной доставки, выбор и соотношение которых зависит от природы и способа назначения и дозировки. Примерами суспендирующих агентов являются этоксилированный изостеариловый спирт, полиоксиэтилен, сорбитол и сорбитовый эфир, микрокристаллическая целлюлоза, метагидроксид алюминия, бентонит, агар-агар и трагакант, а также смеси этих веществ. Защита от действия микроорганизмов может быть обеспечена с помощью разнообразных антибактериальных и противогрибковых агентов, например, таких как, парабены, хлорбутанол, сорбиновая кислота и подобные им соединения. Композиция может включать также изотонические агенты, например, сахара, хлористый натрий и им подобные. Пролонгированное действие композиции может быть обеспечено с помощью агентов, замедляющих абсорбцию активного начала, например, моностеарат алюминия и желатин. Примерами подходящих носителей, растворителей, разбавителей и средств доставки являются вода, этанол, полиспирты, а также их смеси, растительные масла (такие, как оливковое масло) и инъекционные органические сложные эфиры (такие, как этилолеат). Примерами наполнителей являются лактоза, молочный сахар, цитрат натрия, карбонат кальция, фосфат кальция и им подобные. Примерами измельчителей и распределяющих средств являются крахмал, алгиновая кислота и ее соли, силикаты. Примерами лубрикантов являются стеарат магния, лаурилсульфат натрия, тальк, а также полиэтиленгликоль с высоким молекулярным весом. Фармацевтическая композиция для перорального, сублингвального, трансдермального, внутримышечного, внутривенного, подкожного, местного или ректального введения активного начала, одного или в комбинации с другим активным началом, может быть введена животным и людям в стандартной форме введения, в виде смеси с традиционными фармацевтическими носителями. Пригодные стандартные формы введения включают пероральные формы, такие как таблетки, желатиновые капсулы, пилюли, порошки, гранулы, жевательные резинки и пероральные растворы или суспензии, сублингвальные и трансбуккальные формы введения, аэрозоли, имплантаты, местные, трансдермальные, подкожные, внутримышечные, внутривенные, интраназальные или внутриглазные формы введения и ректальные формы введения. Фармацевтические композиции, как правило, получают с помощью стандартных процедур, предусматривающих смешение активного соединения с жидким или тонко измельченным твердым носителем. Для изготовления суппозиториев помимо активных компонентов используют также масло какао, сплавы его с парафином и гидрогенизированными жирами, растительные и животные гидрогенизированные жиры, твердый жир, ланоль, сплавы гидрогенизированных жиров с воском, твердым парафином и другие основы, разрешенные для медицинского применения.
«Фармацевтически приемлемая соль» означает относительно нетоксичные органические и неорганические соли кислот и оснований, заявленных в настоящем изобретении. Эти соли могут быть получены in situ в процессе синтеза, выделения или очистки соединений или приготовлены специально. В частности, соли оснований могут быть получены специально, исходя из очищенного свободного основания заявленного соединения и подходящей органической или неорганической кислоты. Примерами полученных таким образом солей являются гидрохлориды, гидробромиды, сульфаты, бисульфаты, фосфаты, нитраты, ацетаты, оксалаты, валериаты, олеаты, пальмитаты, стеараты, лаураты, бораты, бензоаты, лактаты, тозилаты, цитраты, малеаты, фумараты, сукцинаты, тартраты, мезилаты, малонаты, салицилаты, пропионаты, этансульфонаты, бензолсульфонаты, сульфаматы и им подобные (Подробное описание свойств таких солей дано в Berge S.M., et al., “Pharmaceutical Salts” J. Pharm. Sci. 1977, 66: 1-19). Соли заявленных кислот также могут быть специально получены реакцией очищенной кислоты с подходящим основанием, при этом могут быть синтезированы соли металлов и аминов. К металлическим относятся соли натрия, калия, кальция, бария, цинка, магния, лития и алюминия, наиболее желательными из которых являются соли натрия и калия. Подходящими неорганическими основаниями, из которых могут быть получены соли металлов, являются гидроксид, карбонат, бикарбонат и гидрид натрия, гидроксид и бикарбонат калия, поташ, гидроксид лития, гидроксид кальция, гидроксид магния, гидроксид цинка. В качестве органических оснований, из которых могут быть получены соли заявленных кислот, выбраны амины и аминокислоты, обладающие достаточной основностью, чтобы образовать устойчивую соль, и пригодные для использования в медицинских целях (в частности, они должны обладать низкой токсичностью). К таким аминам относятся аммиак, метиламин, диметиламин, триметиламин, этиламин, диэтиламин, триэтиламин, бензиламин, дибензиламин, дициклогексиламин, пиперазин, этилпиперидин, трис(гидроксиметил)аминометан и подобные им. Кроме того, для солеобразования могут быть использованы гидроокиси тетраалкиламмония, например, такие как, холин, тетраметиламмоний, тетраэтиламмоний и им подобные. В качестве аминокислот могут быть использованы основные аминокислоты - лизин, орнитин и аргинин.
«Фармацевтически приемлемые эксципиенты» под фармацевтически приемлимыми эксципиентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители.
Задача настоящего изобретения заключается в создании новых блокаторов TRPA1 канала как потенциальных обезболивающих препаратов.
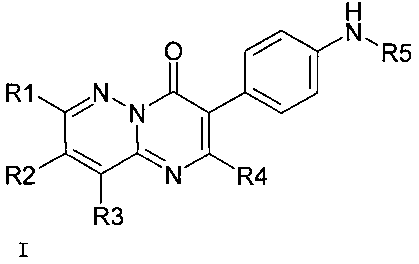
Поставленная задача достигается замещенными пиразинопиримидинонами общей формулы I в виде свободных оснований, а также в виде их фармацевтически приемлемых солей и/или гидратов и сольватов.
Предметом настоящего изобретения являются новые блокаторы TRPA1 канала, представляющие собой производные замещенных пиразинопиримидинонов общей формулы I в виде свободных оснований, а также в виде их фармацевтически приемлемых солей и/или гидратов, сольватов
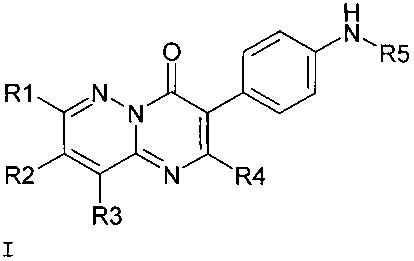
где
R1=H, (C1-C5) алкил,
R2=H, (C1-C5) алкил,
R3=H, (C1-C5) алкил,
R4=H, (C1-C7) алкил, замещенный (C1-C7) алкил, (C3-C7) циклоалкил,
R5=H, (C1-C5) алкил, гетероциклил, где гетероциклил означает насыщенную моноциклическую систему, включающую от 4 до 6 атомов углерода, в которой один атом углерода замещен на гетероатом, такой как азот.
Предметом данного изобретения являются фармацевтически приемлимые соли соединений общей формулы I. Соединения общей формулы I содержат ионогенные группы (первичные, вторичные амины) и могут образовывать соли, которые были получены способами, известными в данной области техники.
Предметом данного изобретения являются рацемические смеси и индивидуальные оптические изомеры соединений общей формулы I, поскольку ряд соединений общей формулы I имеет асимметрический центр, и вещества могут существовать в виде рацемических смесей и изомерных форм.
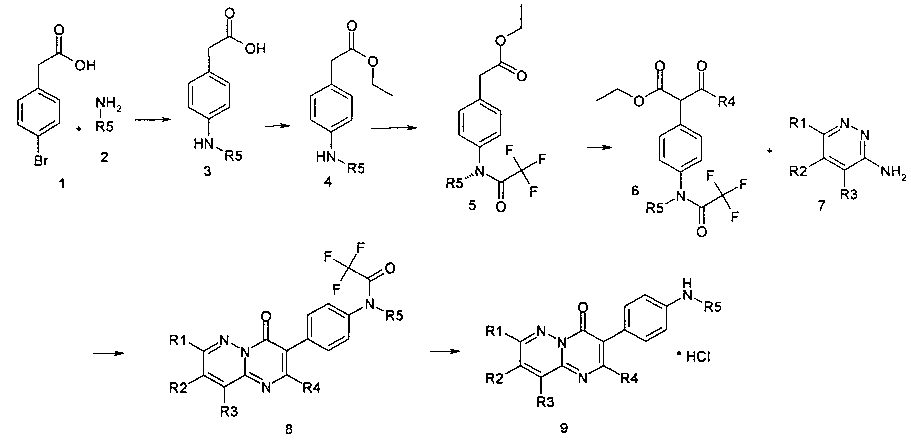
Предметом данного изобретения является способ получения производных замещенных пиразинопиримидинонов общей формулы I по следующей постадийной последовательности, отраженной на Схеме 1.
Схема 1
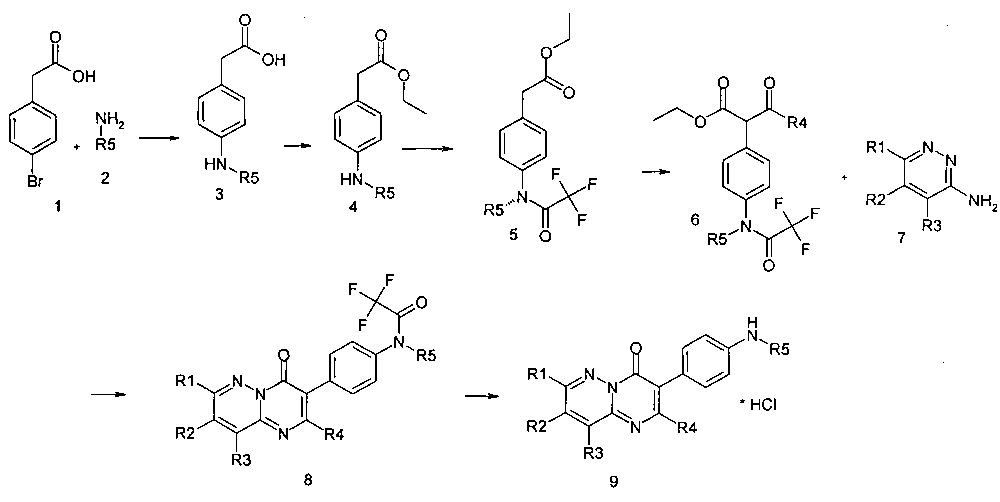
где R1, R2, R3, R4, R5
имеют вышеуказанные значения.
Предметом настоящего изобретения является активный компонент, обладающий свойством блокатора каналов TRPA1, представляющий собой производные замещенных пиразинопиримидинонов общей формулы I.
Предметом данного изобретения является фармацевтическая композиция, предназначенная для снятия болевого синдрома, содержащая в эффективном количестве активный компонент, обладающий свойством блокатора каналов TRPA1.
Предметом данного изобретения являются вышеуказанная фармацевтическая композиция в форме таблеток, капсул, мазей, гелей, инъекций и других готовых форм, помещенных в фармацевтически приемлемую упаковку.
Фармацевтическая композиция может включать фармацевтически приемлемые эксципиенты. Под фармацевтически приемлемыми эксципиентами подразумеваются применяемые в сфере фармацевтики разбавители, вспомогательные агенты и/или носители. Фармацевтическая композиция наряду с активным компонентом по настоящему изобретению может включать и другие активные ингредиенты, при условии, что они не вызывают нежелательных эффектов, например аллергических реакций.
При необходимости использования фармацевтических композиций по настоящему изобретению в клинической практике они могут смешиваться для изготовления различных форм, при этом они могут включать в свой состав традиционные фармацевтические носители; например, пероральные формы (такие как таблетки, желатиновые капсулы, пилюли, растворы или суспензии); формы для инъекций (такие как растворы или суспензии для инъекций, или сухой порошок для инъекций, который требует лишь добавления воды для инъекций перед использованием); местные формы (такие как мази или растворы).
Носители, используемые в фармацевтических композициях по настоящему изобретению, представляют собой носители, которые применяются в сфере фармацевтики для получения распространенных форм, в том числе: в пероральных формах используются связующие вещества, смазывающие агенты, дезинтеграторы, растворители, разбавители, стабилизаторы, суспендирующие агенты, бесцветные агенты, корригенты вкуса; в формах для инъекций используются антисептические агенты, солюбилизаторы, стабилизаторы; в местных формах используются основы, разбавители, смазывающие агенты, антисептические агенты.
Новая фармацевтическая композиции может быть получена смешением с инертным наполнителем и/или растворителем активного компонента, представляющего собой, по крайней мере, одно из соединений общей формулы I, а также их свободных оснований, рацемических смесей или индивидуальных оптических изомеров, а также их фармацевтически приемлемых солей и/или гидратов.
Предметом данного изобретения является лекарственное средство.
Лекарственные средства могут вводиться перорально или парентерально (например, внутривенно, подкожно, внутрибрюшинно, местно или ректально). Клиническая дозировка активного компонента (субстанции), фармацевтической композиции или лекарственного комбинированного средства, включающих фармацевтически эффективное количество активного компонента, у пациентов может корректироваться в зависимости от терапевтической эффективности и биодоступности активных ингредиентов в организме, скорости их обмена и выведения из организма, а также в зависимости от возраста, пола и стадии заболевания пациента, при этом суточная доза у взрослых обычно составляет 10-300 мг, предпочтительно 30-90 мг. Поэтому во время приготовления фармацевтических композиций по настоящему изобретению в виде единиц дозировки необходимо учитывать вышеназванную эффективную дозировку, при этом каждая единица дозировки препарата должна содержать 5-100 мг, предпочтительно 10-20 мг. В соответствии с указаниями врача или фармацевта данные препараты могут приниматься несколько раз в течение определенных промежутков времени (предпочтительно - от одного до шести раз).
Представленные ниже примеры иллюстрируют, но не ограничивают изобретение.
Структуры полученных соединений подтверждались данными химического, хроматографического и спектрального анализа.
Пример 1 Синтез соединения 18.1.
Общая методика синтеза соединения 18.1. приведена на Схеме 2
Схема 2
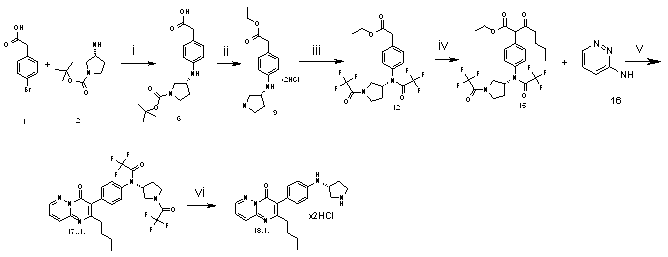
i - t-BuONa, Pd(OAc)2, BINAP, толуол, кипячение
ii - тионил хлористый, EtOH, кипячение
iii - трифторуксусный ангидрид, TEA, methylene chloride, 0°C
iv - LiHMDS, хлорангидрид валериановой кислоты, тетрагидрофуран, -78°C
v - Реагент Итона, 40°C
vi - NH3/MeOH, HCl/диоксан
Стадия 1:
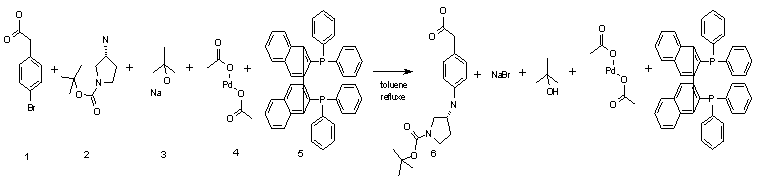
В 2 л коническую колбу помещают п-бромфенилуксусную кислоту 1 (25.0 г, 0.116 моль), N-бок-3-аминопирролидин 2 (26.0 г, 0.139 моль), третбутилат натрия 3 (27.9 г, 0.290 моль) и BINAP 5 (3.6 г, 0.006 моль) в 700 мл толуола. Реакционную массу при перемешивании продувают аргоном, затем вносят ацетат палладия (1.3 г, 0.006 моль) и нагревают до кипения. Смесь кипятят 12 ч, охлаждают до комнатной температуры и экстрагируют натриевую соль (4-{[(3R)-1-(трет-бутоксикарбонил)пирролидин-3-ил]амино}фенил)уксусной кислоты 6 водой 2×150 мл. Органическую фазу отбрасывают, а объединенную водную подкисляют до рН=7 2М соляной кислотой и экстрагируют 3×150 мл хлороформа. Хлороформный экстракт сушат над сульфатом натрия, удаляют растворитель на роторно-пленочном испарителе и получают (4-{[(3R)-1-(трет-бутоксикарбонил)пирролидин-3-ил]амино}фенил)уксусную кислоту 6 22.5 г с выходом 60%.
Стадия 2:
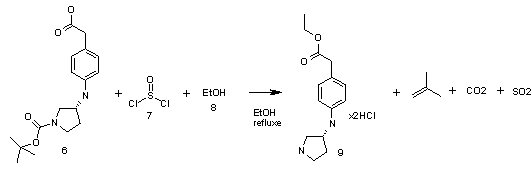
В 1 л конической колбе растворяют (4-{[(3R)-1-(трет-бутоксикарбонил)пирролидин-3-ил]амино}фенил)уксусную кислоту 6 (22.5 г, 0.070 моль) в 500 мл абс. этанола и при охлаждении на водно-ледяной бане прикапывают при 0-5°С хлористый тионил 7 (25.0 г, 0.210 моль). Охлаждение убирают и смесь кипятят с обратным холодильником в течение 3 ч. Затем реакционную массу охлаждают до комнатной температуры, этанол упаривают на РПИ, а остаток растирают с 300 мл диэтилового эфира. Полученный осадок фильтруют и сушат в эксикаторе при пониженном давлении. Получают дигидрохлорид этилового эфира (4-{[(3R)-1-(трет-бутоксикарбонил)пирролидин-3-ил]амино}фенил)уксусной кислоты 9 21.5 г с выходом 95%.
Стадия 3:
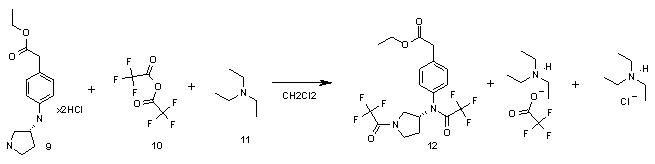
В 500 мл трехгорлой круглодонной колбе суспендируют дигидрохлорид этилового эфира (4-{[(3R)-1-(трет-бутоксикарбонил)пирролидин-3-ил]амино}фенил)уксусной кислоты 9 (21.5 г, 0.067 моль) в 300 мл хлористого метилена и при охлаждении на водно-ледяной бане прикапывают при 0-5°С триэтиламин (37.2 мл, 0.302 моль), затем трифторуксусный ангидрид (27.5 г, 0.147 моль). Охлаждение убирают, и после того как температура реакционной смеси поднимется до комнатной, промывают 3×100 мл воды. Органическую фазу сушат над сульфатом натрия, удаляют растворитель на роторно-пленочном испарителе и получают этил (4-{(трифторацетил)[(3R)-1-(трифторацетил)пирролидин-3-ил]амино}фенил)ацетат 12 24.0 г с выходом 81%.
Стадия 4:
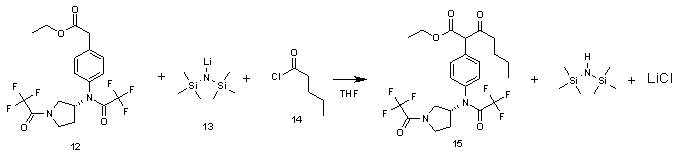
В 500 мл трехгорлую круглодонную колбу помещают 1М раствор LiHMDS 13 в ТГФ (93 мл, 0.065 моль), охлаждают до -78°С и порциями добавляют раствор этил (4-{(трифторацетил)[(3R)-1-(трифторацетил)пирролидин-3-ил]амино}фенил)ацетата 12 (24.0 г, 0.055 моль) в 100 мл ТГФ. Перемешивают 15 мин, затем при этой же температуре прикапывают раствор валероилхлорида 14 (7.2 г, 0.060 моль) в 50 мл ТГФ. Охлаждение убирают и реакционную массу перемешивают при комнатной температуре 12 ч, после чего добавляют 250 мл насыщенного раствора хлористого аммония и оставляют на 30 мин при интенсивном перемешивании. Перемешивание выключают, при этом реакционная масса разделяется на два слоя. Верхний слой отделяют и упаривают растворитель на РПИ. Остаток растворяют в 300 мл гексана, сушат над сульфатом натрия и растворитель упаривают на РПИ. Остаток чистят с помощью колоночной хроматографии на колонке (d=8 см, h=30 см) системой диэтиловый эфир - гексан 1:2. Получают этил 3-оксо-2-(4-{(трифторацетил)[(3R)-1-(трифторацетил)пирролидин-3-ил]амино}фенил)гептаноат 15 21.3 г с выходом 75%.
Стадия 5:
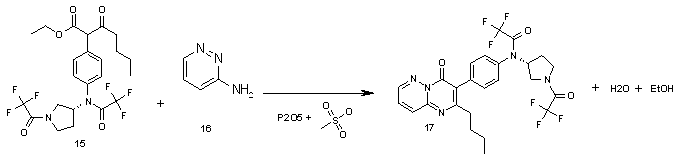
Получение 17: в коническую колбу на 10 мл помещают этил 3-оксо-2-(4-{(трифторацетил)[(3R)-1-(трифторацетил)пирролидин-3-ил]амино}фенил)гептаноат 15 (1.0 г, 1.9 ммоль), 3-аминопиридазин (272 мг, 2.9 ммоль) и 2.5 мл реактива Итона (раствор 10 масс. % P2O5 в метансульфокислоте). Смесь перемешивают при 50°С 24 ч, охлаждают до комнатной температуры, добавляют 20 мл хлороформа и выливают на смесь NaOH (2.4 г, 57 ммоль) и 30 г льда. Хлороформный слой отделают, сушат над сульфатом натрия и упаривают на РПИ. Остаток чистят колоночной хроматографией на колонке (d=1 см, h=20 см) системой хлороформ - метанол 100:1 и получают 850 мг N-[4-(2-бутил-4-оксо-4H-пиримидо[1,2-b]пиридазин-3-ил)фенил]-2,2,2-трифтор-N-[(3R)-1-(трифторацетил)пирроли дин-3-ил]ацетамид 17.1. с выходом 80%.
Стадия 6:
Получение 18.1.: В конической колбе на 20 мл растворяют 850 мг N-[4-(2-бутил-4-оксо-4H-пиримидо[1,2-b]пиридазин-3-ил)фенил]-2,2,2-трифтор-N-[(3R)-1-(трифторацетил)пирроли дин-3-ил]ацетамида 17.1. в 10 мл метанола и добавляют 3 мл водного концентрированного раствора аммиака. Смесь оставляют перемешиваться при комнатной температуре на 12 ч, затем упаривают растворители на РПИ. Остаток чистят колоночной хроматографией на колонке (d=1 см, h=20 см) системой хлороформ - метанол - водный аммиак 100:10:1. После упаривания элюента добавляют 10 мл эфира, затем 1 мл 5М солянокислого диоксана. Выпавший осадок фильтруют, сушат на воздухе. Получают 60 мг дигидрохлорид 2-бутил-3-{4-[(3R)-пирролидин-3-иламино]фенил}-4H-пиримидо[1,2-b]пиридазин-4-она. 18.1. с выходом 9%.
1H NMR (400 MHz, DMSO-D6) ppm 0.78 (t, 7.3Hz, 3H), 1.16-1.21 (m, 2H), 1.56-1.67 (m, 2H), 1.88-1.99 (m, 1H), 2.17-2.29 (m, 1H), 2.56 (t, 7.7Hz, 3H), 3.05-3.15 (m, 1H), 3.20-3.49 (m, 3H), 4.12-4.20 (m, 1H), 6.72 (d, 8.2Hz, 2H), 7.09 (d, 8.2Hz, 2H), 7.73-7.80 (m, 1H), 8.06-8.14 (m, 1H), 8.83 (br.s, 1H), 9.34 (br.s, 2H)
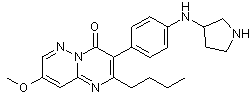
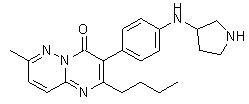
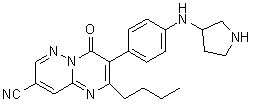
По аналогичной схеме, используя другие реагенты - аминопиразины 16, а также другие хлорангидриды кислот 14 и другие амины 2, были получены соединения 18.2-18.8, представленные в таблице 1
|
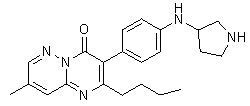
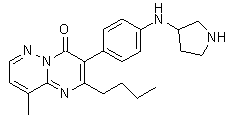
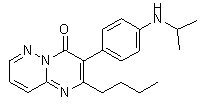
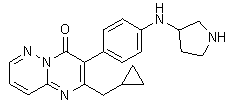
Пример 2. Общий метод синтеза солей соединений общей формулы I.
Часть соединений настоящего изобретения содержит ионогенные группы (первичные и вторичные амины) и может образовывать соли, которые получают способами, известными в данной области техники. Для этого, например, к раствору основания в инертном растворителе (спирт, ацетон, хлороформ, эфир, этилацетат) добавляли раствор эквивалентного количества (иногда - избытка) органической или неорганической кислоты в инертном растворителе и добивались осаждения искомой соли. Неорганическая кислота может представлять собой хлороводородную кислоту, фосфорную кислоту, серную кислоту, азотную кислоту, бромоводородную кислоту или йодоводородную кислоту. Органическая кислота может представлять собой метансульфокислоту, п-толуолсульфокислоту, уксусную кислоту, трифторуксусную кислоту, малеиновую кислоту, янтарную кислоту, щавелевую кислоту, бензойную кислоту, винную кислоту, фумаровую кислоту, миндальную кислоту, пропионовую кислоту, лимонную кислоту, молочную кислоту, гликолевую кислоту, глюконовую кислоту, галактуроновую кислоту, глютаминовую кислоту, глутаровую кислоту, глюкуроновую кислоту, аспарагиновую кислоту, аскорбиновую кислоту, карбоновую кислоту или ванилиновую кислоту, но не ограничиваясь ими.
Другим вариантом синтеза солей соединений I является снятие защитной группы в кислой среде (например, снятие третбутилоксикарбонильной группы) с одновременным образованием соли амина.
Определение антагонистической активности соединений по отношению к ионному каналу TRPA1.
Тестирование соединений осуществляли с использованием эмбриональных эпителиальных клеток почки человека, экзогенно экспресирующих ИПТГ-индуцируемый ионный канал TRPA-1. Антагонистическая активность соединений оценивалась по подавлению выброса внутриклеточного кальция. В качестве экспериментальной платформы была выбрана FLIPR (Molecular Devices). Выброс внутриклеточного кальция стимулировали с помощью Аллилизотоиционат (AITC), в качестве известного антагониста TRPA1 использовали Рутениевый красный (RR). Для всех соединений проводили анализ зависимости антагонистической активности вещества от концентрации вещества. Из полученных графиков зависимости определялись значения IC50 (концентрация вещества в мкмолях, которая вызывает 50% от максимального эффекта).
Клеточная линия: линия HEK293, стабильно экспрессирующая TRPA1 при индукции ИПТГ. Культуральная среда для клеток представляла собой смесь DMEM (Hyclone, USA), 10% FBS (Hyclone, USA), 1% NEAA, пируват Na, 2 мМ L-глутамина, 0.3 мкг/мл G418 (Sigma, USA), 20 мкг/мл гигромицина (Invitrogen, USA).
Подготовка клеток к эксперименту. Клетки суспендировали в культуральной среде с 1 мМ ИПТГ таким образом, чтобы получить концентрацию 8×105 клеток/мл, что даст 20000 клеток на лунку. Полученную клеточную суспензию помещали в 384-луночные экспериментальные культуральные плашки с оптическим дном при помощи автоматических роботизированных станций Biomek FX или Biomek NX, по 25 мкг суспензии в каждую лунку. 384-луночные плашки центрифурировали 200xg 5 минут. Далее экспериментальные плашки с клеточной суспензией перемещали в инкубатор 37°C, 5% CO2.
В день эксперимента клетки прогружали Ca2+- чувствительной краской, приготовленной по инструкции производителя. Вкратце, содержимое одной ампулы ФЛИПРR Кальций 4 тест-система (Molecular Devices, USA) растворяли в 20 мл буфера Хенкс/20mM ХЕПЕС и использовали в качестве 25-кратного стока. 25 мл рабочего раствора Ca2+- чувствительной краски добавляли в каждую лунку 384-луночной экспериментальной плашки. Экспериментальные плашки, загруженные Ca2+- чувствительной краской, инкубировали 1 час при комнатной температуре в темном месте. Далее приступали к считыванию на ФЛИПР.
Форматирование веществ.
- Вещества растворяли в ДМСО до концентрации 10 мМ.
- Готовили серийные разведения веществ в ДМСО с шагом 3.16 с помощью Biomek 2000. При разведении серии каждого вещества в 166.6 раз в буфере эссея Хенкс/20mM ХЕПЕС/0.6%Плюроник F-127/0.6%ДМСО получали 6-кратный стоковый растор серии вещества. Финальная максимальная тестируемая концентрация - 30 мкМ.
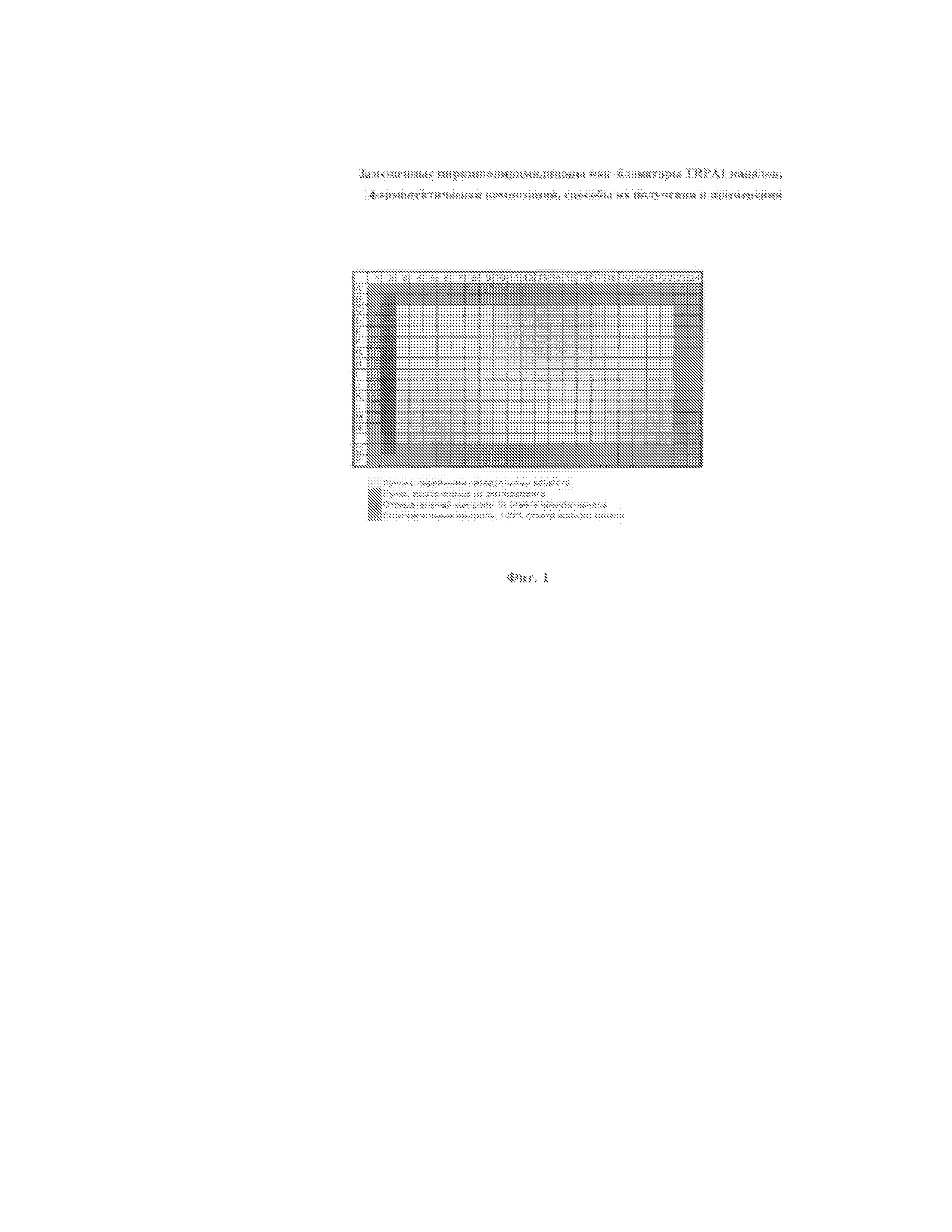
(Фиг. 1). Схема 384-луночной плашки.
Стимуляция кальциевого канала.
Для стимуляции ионного TRPA-1 канала мы использовали концентрацию EC80 агониста равную 30 мкМ. Готовили 6-кратный сток агониста 180 мкМ AITC на буфере эссея, Хенкс/20mM ХЕПЕС/0.6% Плюроник-127/0.6% ДМСО.
Контрольный антагонист.
Для ингибирования стимулированного кальциевого ответа использовали максимальную ингибирующую концентрацию контрольного антагониста IC100 равную 6 мкМ. Готовили 6-кратный сток контрольного антагониста 36 мкМ RR на буфере эссея, Хенкс/20mM ХЕПЕС/0.6% Плюроник-127/0.6% ДМСО.
Добавление тестируемых веществ и контрольного антагониста
Для детекции агонистической активности тестируемых веществ в каждую лунку 384-луночной плашки добавляли по 12.5 мкл вещества, согласно схеме плашки. Далее экспериментальную плашку считывали на ФЛИПР.
После инкубации в течение 667 секунд (11 минут) в каждую лунку 384-луночной экспериментальной плашки добавляли по 12.5 мкл агониста ионного канала TRPA-1, аллилизотоиционата AITC и немедленно начинали второе считывание на ФЛИПР для определения антагонистов.
Расчет величины IC50 для антагонистов
Для веществ была проанализирована зависимость антагонистической активности от концентрации. Величина IC50 была определена из полученной зависимости. Данные эксперимента анализировали в программе GraphPad Prizm (GraphPad Software, Inc., San Diego, CA). Для построения концентрационной зависимости было выбрано уравнение
Y = Нижнее плато кривой + (Верхнее плато - Нижнее плато) / (1+10∧((LogIC50-X)*Наклон кривой))
EC50 определялось исходя из построенного графика, как концентрация вещества, которая вызывает 50% от максимального эффекта. Диапазоны агонистической активности по отношению к GPR119 некоторых синтезированных соединений приведены в Таблице 2.
|
Где A - 5 nM<IC50<250 nM
B - 250 nM<IC50<1 uM
C - 1 uM<IC50<10 uM
Фармакологические свойства заявленного соединения
Пример 3. Исследование фармакологического (анальгетического) действия соединений 18.1, 18.3. и 18.6. с использованием теста температурной болевой чувствительности на хвосте.
Методика. Эксперименты по исследованию анальгетического действия соединений 18.1, 18.3. и 18.6. проведены с использованием теста температурной болевой чувствительности на хвосте с помощью аппаратно-программного комплекса компании Panlab, Harvard Apparatus, Analgesy-meter LE 7106, включающего пенал для фиксации животного с возможностью помещения хвоста над датчиком с фотоэлементов, фиксирующим отдергивание хвоста в результате болевого раздражения, вызванного нагреванием сфокусированной лампой (фокус в диапазоне 10-90, в эксперименте значение фокуса составляло 20 единиц во всех экспериментальных сериях), и программу SeDaCom.
Ингибиторы клеточного канала TRPA1 18.1, 18.3. и 18.6. вводили внутрижелудочно в дозе 3 мг/кг. В качестве контроля использовали данные, полученные при внутрижелудочном введении эквивалентного объема 1% крахмального раствора.
В данном тесте регистрировалось время от начала нагревания хвоста сфокусированной лампой до его отдергивания с поверхности над фотоэлементом.
Результаты.
При проведении серии экспериментов по исследованию анальгетической активности представленных ингибиторов клеточного канала TRPA1 обнаружено, что в исходе отличий в экспериментальных группах от показателей интактных животных нет (табл. 3).
При анализе абсолютных показателей активности (время до отдергивания хвоста) достоверно отличаются от времени отдергивания хвоста в группе интактных животных соединения 18.1, 18.3. и 18.6. (табл. 3).
|
Наиболее выраженной активностью в данном тесте обладало исследуемое соединение под лабораторным шифром 18.3.
Способ получения отбеленных износостойких отливок
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к эндотелиальной синтазе оксида азота человека
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к интерлейкину-6
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к фактору некроза опухоли-альфа
Способ коррекции ишемических нарушений, возникших в результате реперфузионной травмы печени
Способ коррекции эндотелиальной дисфункции смесью гелия с кислородом при l-name индуцированном дефиците оксида азота
Способ коррекции нарушения микроциркуляции в плаценте при adma-подобной модели гестоза в эксперименте однократным ишемическим эпизодом
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к с-концевому фрагменту рецептора ангиотензина ii
Дымообразующая композиция для электронных устройств, имитирующих табакокурение, способ ее получения и применения
Фармацевтическая композиция и набор для лечения бактериальных инфекций
Способ получения отбеленных износостойких отливок
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к эндотелиальной синтазе оксида азота человека
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к интерлейкину-6
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к фактору некроза опухоли-альфа
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к с-концевому фрагменту рецептора ангиотензина ii
Дымообразующая композиция для электронных устройств, имитирующих табакокурение, способ ее получения и применения
Фармацевтическая композиция и набор для лечения бактериальных инфекций
Способ фармакологической коррекции ишемии скелетной мышцы миноксидилом
Стартовая система предупреждения критических режимов одновинтового вертолета
Способ коррекции эндотелиальной дисфункции раствором гомеопатических разведений антител к эндотелиальной синтазе оксида азота человека в комплексе с раствором гомеопатических разведений антител к с-концевому фрагменту рецептора ангиотензина ii

