Результат интеллектуальной деятельности: ПРОИЗВОДНЫЕ ДИПЕПТИДА ЛИЗИН-ГЛУТАМИНОВАЯ КИСЛОТА
Вид РИД
Изобретение
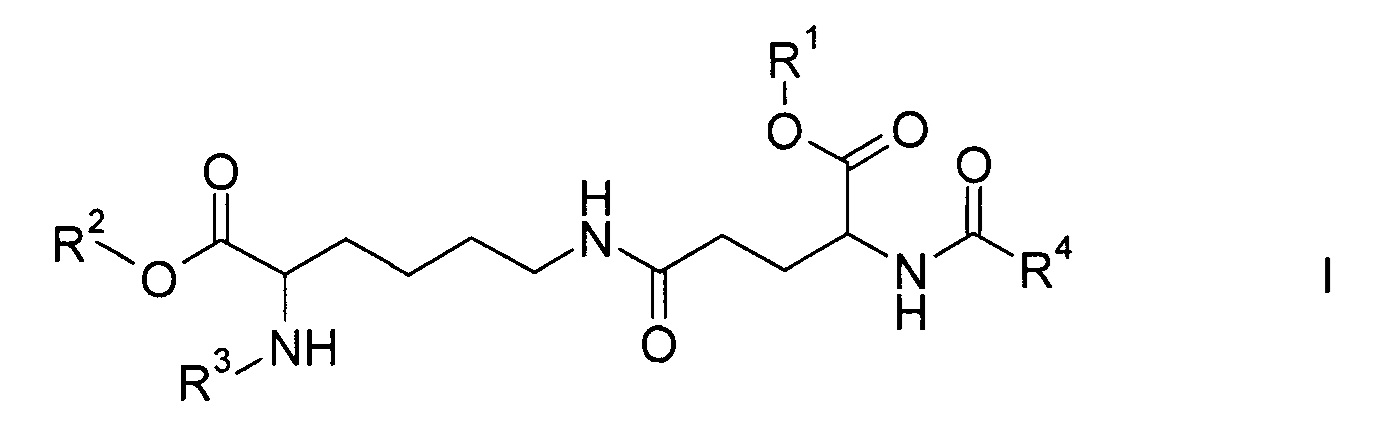
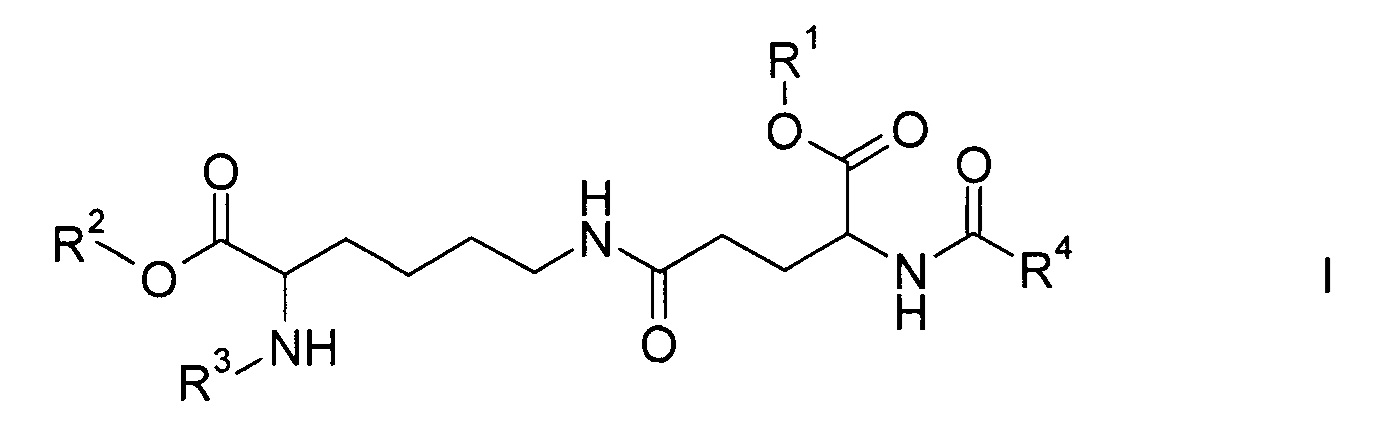
Настоящее изобретение относится к соединениям формулы
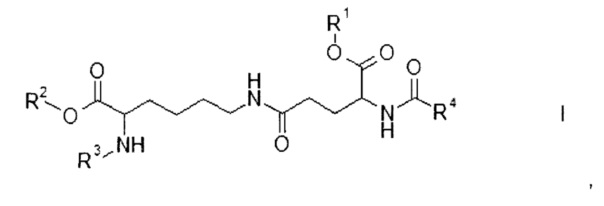
где
R1 и R2 являются одинаковыми или отличаются друг от друга и обозначают водород или сложноэфирную защитную группу,
R3 представляет собой водород или амино-защитную группу и
R4 представляет собой С12-20-алкил,
а также к их энантиомерам, диастереомерам и солям.
Согласно следующему варианту реализации настоящее изобретение относится к способу получения соединений формулы I и к применению соединений формулы I в твердофазном пептидном синтезе.
Обнаружили, что соединения согласно настоящему изобретению являются пептидными промежуточными соединениями (которые могут подвергаться различным превращениям) для твердофазного пептидного синтеза (ТФПС) пептидных лекарственных средств, содержащих боковую цепь со структурным блоком Glu-жирный алкил, присоединенным к остатку Lys пептидной цепи. К таким лекарственным средствам относится, например, Лираглутид, который представляет собой аналог ГПП-1 (глюкагоноподобного пептида-1) и является пептидным лекарственным средством для лечения диабета 2 типа. Лираглутид в положении Lys26 содержит боковую цепь со структурным блоком Glu - гексадеканоил (Википедия, свободная энциклопедия, 30.04.2012).
Целью настоящего изобретения является предложение новых пептидных промежуточных соединений, которые содержат боковую цепь Glu-жирный алкил и которые можно легко применять в процессе ТФПС.
Установили, что соединения согласно настоящему изобретению формулы
где
R1 и R2 являются одинаковыми или отличаются друг от друга и обозначают водород или сложноэфирную защитную группу,
R3 представляет собой водород или амино-защитную группу и
R4 представляет собой С12-20-алкил,
а также их энантиомеры, диастереомеры и соли можно эффективно использовать для указанной цели.
Термин «С12-20-алкил», который используют для обозначения заместителя R4, относится к разветвленному или неразветвленному моновалентному насыщенному алифатическому углеводородному радикалу, состоящему из от двенадцати до двадцати атомов углерода, в особенности к неразветвленному моновалентному насыщенному алифатическому углеводородному радикалу. Примерами данного термина могут служить радикалы додецил, тридецил, тетрадецил, пентадецил, гексадецил, гептадецил, октадецил, нонадецил и эйкозанил.
Согласно конкретным вариантам реализации настоящего изобретения R4 относится к С14-16-алкилу, еще более конкретно к С15-алкилу.
Более конкретно R4 представляет собой тетрадецил, пентадецил или гексадецил, в особенности пентадецил.
Термин «амино-защитная группа», который используют для обозначения заместителя R3, относится к общеизвестным заместителям, которые, как правило, применяют для препятствования реакционной способности аминогруппы. Подходящие амино-защитные группы описаны в руководстве "Fmoc Solid Phase Peptide Synthesis - A Practical Approach" W.C. Chan & P.D. White, Oxford University Press, 2000, переизданном в 2004 г, опубликованном в цифровом формате.
Более конкретно R3 представляет собой Fmoc (9Н-флуорен-9-илметоксикарбонил).
Термин «сложноэфирная защитная группа», который используют для обозначения заместителей R1 и R2, относится к любым заместителям, которые, как правило, применяют для препятствования реакционной способности гидроксильной группы. Подходящие гидроксильные защитные группы описаны в руководстве Green Т., "Protective Groups in Organic Synthesis", Chapter 1, John Wiley and Sons, Inc., 1991, 10-142 и могут быть выбраны из C1-4-алкила, в некоторых случаях замещенного фенилом, С2-4-алкенила, пиперидинила или диметиламиноборанила. Конкретные сложноэфирные защитные группы для R1 и R2 представляют собой С1-4-алкил или С2-4-алкенил.
Более конкретно R1 представляет собой трет-бутил и R2 представляет собой аллил.
Термин «соли» в связи с соединениями согласно настоящему изобретению охватывает общепринятые соли, которые может применять специалист в данной области техники, такие как гидрохлориды, ацетаты, трифторацетаты или формиаты.
Согласно конкретным вариантам реализации настоящего изобретения R1 представляет собой водород или С1-4-алкил и R2 представляет собой водород или С2-4-алкенил.
Согласно другому более конкретному варианту реализации настоящего изобретения R1 представляет собой трет-бутил и R2 представляет собой водород или аллил.
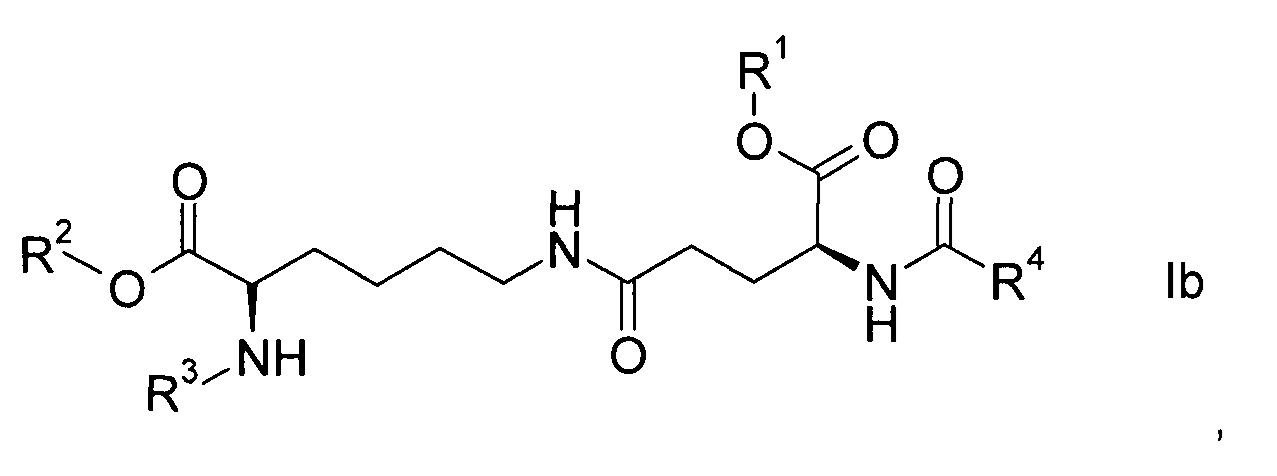
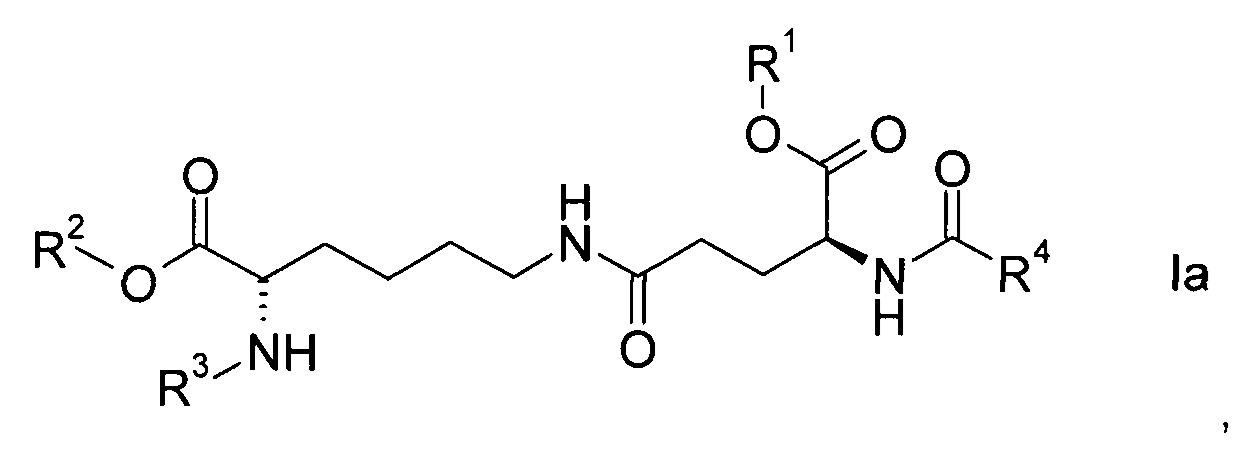
Согласно конкретным вариантам реализации настоящего изобретения соединения формулы I имеют формулу
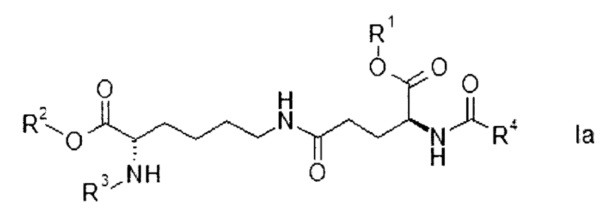 или
или
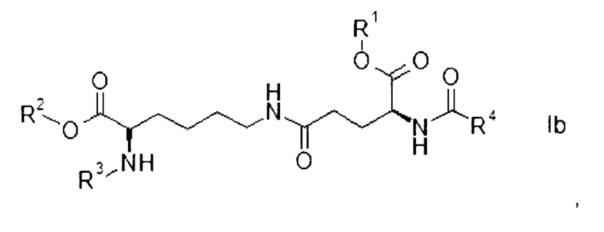
где
R1, R2, R3 и R4 представляют собой такие же, как указано выше, а также их энантиомеры, диастереомеры и соли.
Соединения формулы Ia или Ib с набором заместителей, указанным ниже, представляют собой еще более конкретные варианты реализации настоящего изобретения:
- R1 представляет собой трет-бутил, R2 представляет собой водород, R3 представляет собой Fmoc, R4 представляет собой С15-алкил, более конкретно пентадецил.
- R1 представляет собой трет-бутил, R2 представляет собой аллил, R3 представляет собой Fmoc, R4 представляет собой С15-алкил, более конкретно пентадецил.
Согласно более конкретному варианту реализации соединения формулы I имеют формулу Ia.
Соединения согласно настоящему изобретению можно получить в результате способов, которые по существу известны специалисту в области пептидного синтеза.
Способ получения соединений формулы I, в которых R2 представляет собой водород, включает:
а) объединение производного глутаминовой кислоты формулы
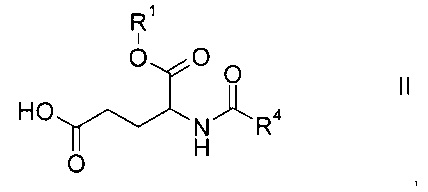
где R1 и R4 представляют собой такие же, как указано выше, или их соли, с производным лизина формулы
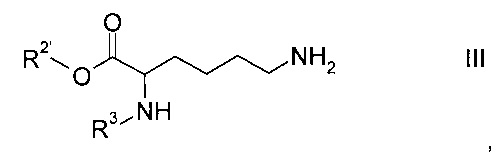
где R2' представляет собой сложноэфирную защитную группу и R3 представляет собой то же соединение, как указано выше, или его соль, с образованием соединения формулы
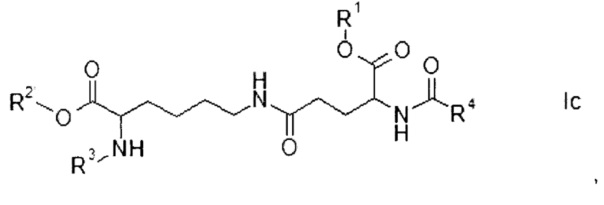
где R1, R2, R3 и R4 представляют собой такие же, как указано выше, и
b) удаление сложноэфирной защитной группы R2'.
Этап а)
Этап а) заключается в объединении производного глутаминовой кислоты формулы II с производным лизина формулы III.
Производное глутаминовой кислоты формулы II можно получить согласно схеме 1 ниже из коммерчески доступных исходных материалов.
Схема 1:
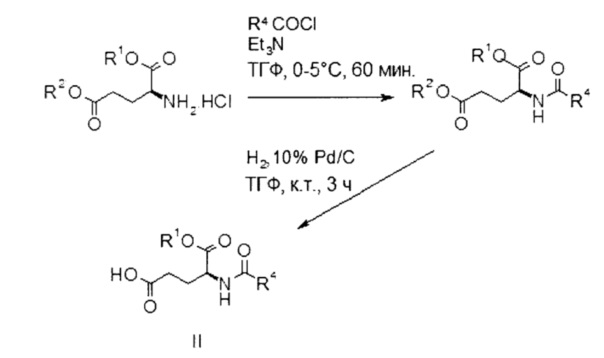
Подходящее коммерчески доступное производное глутаминовой кислоты формулы II представляет собой гидрохлорид (S)-5-бензил 1-трет-бутил 2-амино-пентандиоата.
Производные лизина формулы III являются коммерчески доступными. Подходящим производным является (S)-аллил 6-амино-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-аминогексаноат.
Объединение производного глутаминовой кислоты формулы II с производным лизина формулы III можно затем осуществить с применением классических методик пептидного синтеза.
Соответственно, производное глутаминовой кислоты формулы II сначала активируют с помощью активирующего агента, который представляет собой общепринятые в данной области техники агенты, такие как карбонилдиимидазол (КДИ), карбодиимиды, которые выбирают, например, из дициклогексилкарбодиимида (ДЦК) или диизопропилкарбодиимида (ДИК), или триазолы, которые выбирают, например, из 1-гидрокси-бензотриазола (ГОБт) или 1-гидрокси-7-аза-бензотриазола (ГОАт).
Положительные результаты получили с применением КДИ (1,1-карбонилдиимидазола) в подходящем органическом растворителе, таком как дихлорметан.
Затем можно провести объединение указанного производного глутаминовой кислоты формулы II с производным лизина формулы III в присутствии органического основания, такого как триэтиламин, как правило, при комнатной температуре.
Полученное в результате дипептидное соединение формулы Ib может быть выделено из органической фазы в результате выпаривания растворителя и последующей кристаллизации осадка в подходящем органическом растворителе, таком как диэтиловый эфир.
Соединения формулы Ib в качестве подкласса соединений формулы Ia, как обозначено выше, представляют собой конкретные варианты реализации настоящего изобретения.
Конкретным представителем соединений формулы Ib является (S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат, в котором R1 представляет собой трет-бутил, R2 представляет собой аллил, R3 представляет собой Fmoc и R4 представляет собой пентадецил.
Этап b)
Этап b) заключается в удалении сложноэфирной защитной группы R2' для образования соединения формулы Ia.
Вышеуказанная реакция хорошо известна специалисту в данной области техники.
Подходящие системы для удаления аллильной группы представляют собой, например, раствор источника Pd, такого как тетракис(трифенилфосфин)палладий (0), и фенилсилана в органическом растворителе, таком как дихлорметан, тетрагидрофуран или метилтетрагидрофуран.
Данная реакция проходит при комнатной температуре.
Полученный в результате дипептид формулы Ia может быть выделен из органической фазы в результате выпаривания растворителя и последующего расщепления неочищенного продукта подходящим органическим растворителем, таким как гептан и/или смесь гептана/дихлорметана.
Как отмечено выше, соединения формулы I можно применять в качестве промежуточных продуктов, которые могут подвергаться различным превращениям, в твердофазном пептидном синтезе, более конкретно - в синтезе пептидов, которые содержат боковую цепь со структурным блоком Glu-жирный алкил, присоединенную к остатку Lys пептидной цепи.
Еще более конкретно соединения формулы I можно применять в твердофазном пептидном синтезе на основе FMOC таких пептидов.
Примеры
Сокращения:
к.т. = комнатная температура, ДХМ = дихлорметан, ТГФ = тетрагидрофуран, ТБМЭ = трет-бутил метиловый эфир, EtOAc = этилацетат, ТСХ = тонкослойная хроматография.
Пример 1
(S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота
а) (S)-5-бензил 1-трет-бутил 2-пальмитамидопентандиоат
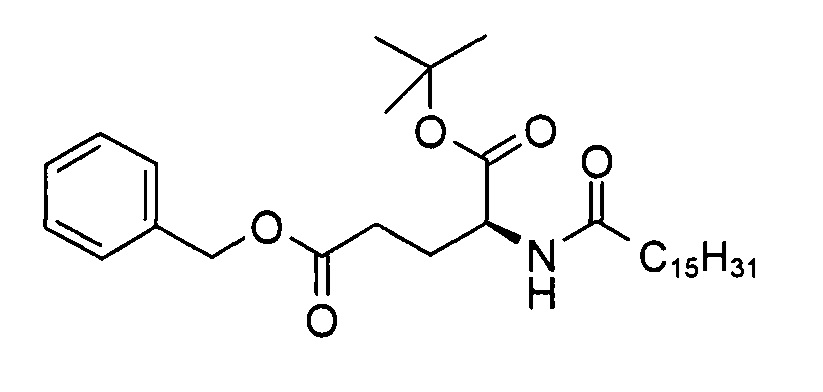
В 3-горлую колбу объемом 200 мл добавляли смесь гидрохлорида (S)-5-бензил 1-трет-бутил 2-амино-пентандиоата (5,00 г, 14,9 ммоль), триэтиламина (3,12 г, 30,7 ммоль) и тетрагидрофурана (100 мл) и перемешивали при температуре 0-5°С в течение 15 мин. Через 10 мин к суспензии с помощью шприца добавляли пальмитоил хлорид (4,35 г, 15,5 ммоль). Реакционную смесь перемешивали в течение 30 мин при температуре 0-5°С. Согласно результатам анализа методом ТСХ (ЭЭ/гептан 1:1, RF исходного вещества = 0,1, RF продукта = 0,6, продукт обнаруживали с помощью реактива Комаровского при длине волны 254 нм (сравн. P. Stevens, J. Chromatog. 1964, 14, 269)) превращение было полным. К реакционной смеси добавляли воду (60 мл) и трет-бутил метиловый эфир (70 мл), смесь перемешивали при к.т. в течение 5 мин. Органический слой отделяли, промывали солевым раствором (120 мл), высушивали над сульфатом натрия и выпаривали досуха с получением (S)-5-бензил 1-трет-бутил 2-пальмитамидопентандиоата (8,21 г, >99%) в виде белого твердого вещества с химической чистотой 98,9% (метод ЖХ см. ниже).
Т.п. 47°С; ЭИ-МС (масс-спектрометрия с ионизацией электронным ударом): m/z=531,39 (М+Н)+.
Метод ВЭЖХ: фенильная колонка X-Bridge №823, размер 50×4,6 мм, диаметр частиц 2,5 мкм; подвижная фаза А: вода/NCMe (95:5), В: NCMe, С: вода/глицин (рН 9); поток: 3 мл/мин; градиент от 50/4/55 (А/В/С) до 7/88/5 (А/В/С) в течение 2 мин, изократический режим 7/88/5 (А/В/С) в течение 0,8 мин. Времена удерживания: 0,54 мин ((S)- и (R)-5-бензил 1-трет-бутил 2-амино-пентандиоат), 2,17 мин ((S)- и (R)-5-бензил 1-трет-бутил 2-пальмитамидопентандиоат).
b) (S)-5-трет-бутокси-5-оксо-4-пальмитамидопентановая кислота
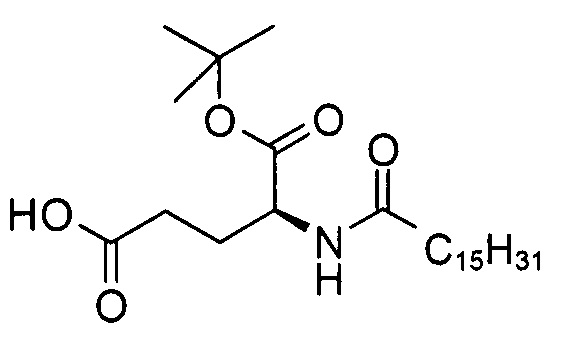
В 3-горлую колбу объемом 250 мл добавляли смесь неочищенного (S)-5-бензил 1-трет-бутил 2-пальмитамидопентандиоата (13,2 г, 24,8 ммоль), 10% палладия на углероде (1,31 г, 1,20 ммоль) и ТГФ (150 мл), перемешивали в атмосфере водорода при комнатной температуре. Согласно результатам анализа методом ТСХ (ЭЭ/гептан 1:1, RF исходного вещества = 0,5, RF продукта = 0,2, продукт обнаруживали с помощью реактива Комаровского (сравн. P. Stevens, J. Chromatog. 1964, 14, 269)) через 23 ч превращение было завершено. Черную суспензию пропускали через фильтр из стекловолокна, полученный в результате бесцветный фильтрат выпаривали досуха с получением неочищенного продукта (11,3 г), который очищали посредством кристаллизации из гептана с получением (S)-5-трет-бутокси-5-оксо-4-пальмитамидопентановой кислоты (8,78 г, выход 76%) в виде белого твердого вещества с химической чистотой 97,7% (метод ЖХ см. ниже).
Т.п. 63°С; ЭИ-МС: m/z=440,33 (М-Н)-.
Метод ВЭЖХ: фенильная колонка X-Bridge №823, размер 50×4,6 мм, диаметр частиц 2,5 мкм; подвижная фаза А: вода/NCMe (95:5), В: NCMe, С: вода/глицин (рН 9); поток: 3 мл/мин; градиент от 50/4/55 (А/В/С) до 7/88/5 (А/В/С) в течение 2 мин, изократический режим 7/88/5 (А/В/С) в течение 0,8 мин. Времена удерживания: 0,77 мин ((S)- и (R)-5-трет-бутокси-5-оксо-4- пальмитамидопентановая кислота), 2,17 мин ((S)- и (R)-5-бензил 1-трет-бутил 2-пальмитамидопентандиоат).
с) (S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат
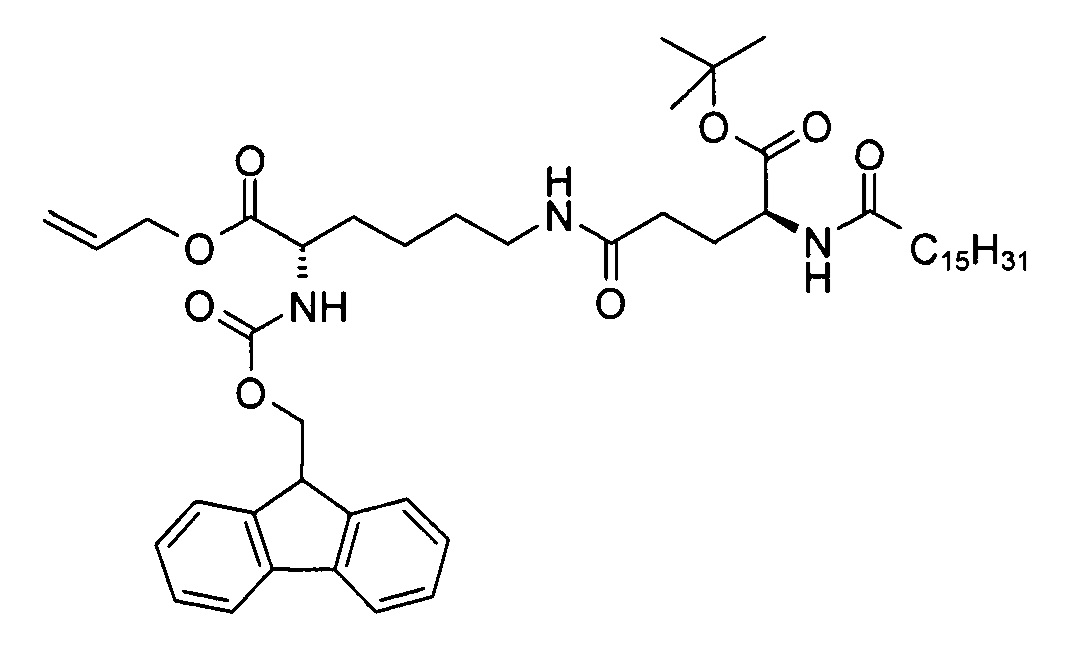
В 3-горлую колбу объемом 500 мл добавляли смесь (S)-5-трет-бутокси-5-оксо-4-пальмитамидопентановой кислоты (8,77 г, 19,4 ммоль), 1,1'-карбонилдиимидазола (3,30 г, 20,4 ммоль) и ДХМ (125 мл), перемешивали при комнатной температуре в течение 90 мин. Через 15 мин к полученной в результате белой суспензии добавляли раствор (S)-аллил 6-амино-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-аминогексаноата (8,68 г, 19,5 ммоль) и триэтиламина (1,96 г, 19,4 ммоль) в ДХМ (50 мл). Реакционную смесь перемешивали в течение 90 мин при к.т. для завершения превращения (которое определяли методом ТСХ (ЭЭ/гептан 1:1, RF исходного вещества = 0, RF продукта = 0,5, продукт обнаруживали с помощью реактива Комаровского (сравн. Р. Stevens, J. Chromatog. 1964, 14, 269)). Затем к смеси добавляли ДХМ (50 мл) и воду (40 мл) и проводили разделение слоев. Водный слой экстрагировали ДХМ (20 мл), объединенные органические слои высушивали над сульфатом натрия. После выпаривания растворителя неочищенный продукт (16,0 г), который находился в осадке, очищали путем кристаллизации из диэтилового эфира для получения
(S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноата (15,2 г, 91%) в виде белого твердого вещества с химической чистотой 96,5% (метод ЖХ см. ниже) и энантиомерной и диастереомерной чистотой >99,9% (метод хиральной ЖХ см. ниже).
Т.п. 118°С; ЭИ-МС: m/z=832,55 (М+Н)+.
Метод ВЭЖХ: фенильная колонка X-Bridge, размер 50×4,6 мм, диаметр частиц 2,5 мкм; подвижная фаза А: вода/NCMe (95:5), В: NCMe, С: 0,1% муравьиная кислота в воде; поток: 2 мл/мин; градиент от 65/25/10 (А/В/С) до 10/80/10 (А/В/С) в течение 10 мин, изократический режим 10/80/10 (А/В/С) в течение 2 мин. Время удерживания: 9,59 мин ((S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат)).
Метод хиральной ВЭЖХ: колонки Chiracel OD-RH №745 и №702, размер 150×4,6 мм, диаметр частиц 5 мкм; подвижная фаза A: NCMe, В: вода/HClO4 (рН 2); поток: 1 мл/мин, изократический режим 68:32 (А/В) в течение 32 мин, градиент от 68/32 (А/В) до 75/25 (А/В) в течение 0,5 мин, изократический режим 75/25 (А/В) в течение 29,5 мин. Времена удерживания: 45,39 мин ((R)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((R)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат), 47,75 мин ((R)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат), 51,98 мин ((S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((R)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат), 55,66 мин ((S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат).
Пример 2
(S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота
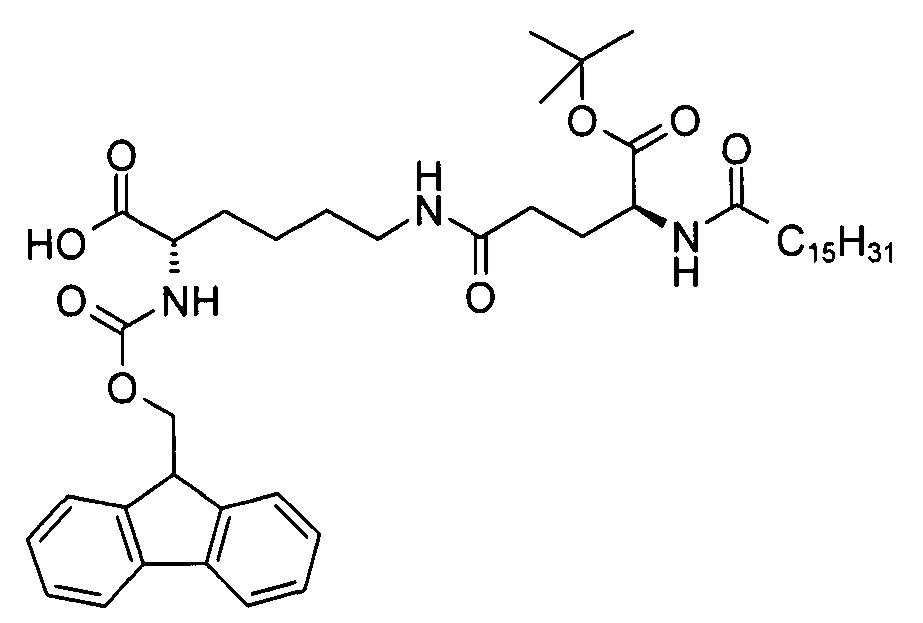
В 3-горлую колбу объемом 500 мл добавляли смесь (S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноата (10,0 г, 11,4 ммоль), фенилсилана (7,02 г, 62,9 ммоль), тетракис(трифенилфосфин) палладия (0) (1,00 г, 0,85 ммоль) и ДХМ (250 мл), перемешивали при комнатной температуре. Согласно результатам анализа методом ТСХ (ЭЭ/гептан 3:1, RF исходного вещества = 0,2, RF продукта = 0, продукт обнаруживали методом УФ-спектрофотометрии при длине волны 254 нм) через 11 мин превращение было завершено. Реакционную смесь разводили ДХМ (50 мл) и последовательно промывали водой (50 мл), водным раствором диэтилдитиокарбамата натрия (0,5%, 30 мл) и солевым раствором (30 мл), высушивали над сульфатом натрия и выпаривали на ротационном вакуумном испарителе досуха. Расщепление неочищенного продукта, который находился в осадке, сначала проводили гептаном (25 мл), а затем - гептаном/ДХМ (9:1) при к.т. и после фильтрации и высушивания получали неочищенную (S)-2-(((9H-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновую кислоту (8,92 г) с химической чистотой 77,2% (метод ЖХ см. ниже). Неочищенный продукт содержал 11% оксида трифенилфосфина в качестве основной примеси. В результате проведения препаративной сверхкритической флюидной хроматографии (СФХ, метод см. ниже) образца 1 г неочищенного продукта получали чистую (S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновую кислоту (0,75 г, 72%) в виде белого твердого вещества с химической чистотой 96,7% (метод ЖХ см. ниже), энантиомерной чистотой 98,0% и диастереомерной чистотой 99,8% (метод хиральной ЖХ см. ниже).
Т.п. 119°С; ЭИ-МС: m/z=792,52 (М+Н)+.
Метод ВЭЖХ: фенильная колонка X-Bridge №823, размер 50×4,6 мм, диаметр частиц 2,5 мкм; подвижная фаза А: вода/NCMe (95:5), В: NCMe, С: 0,1% муравьиная кислота в воде; поток: 2 мл/мин; градиент от 65/25/10 (А/В/С) до 10/80/10 (А/В/С) в течение 10 мин, изократический режим 10/80/10 (А/В/С) в течение 2 мин. Времена удерживания: 8,65 мин ((S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота), 9,59 мин ((S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат)).
Метод хиральной ВЭЖХ: колонки Chiracel OD-RH №745 и №702, размер 150×4,6 мм, диаметр частиц 5 мкм; подвижная фаза A: NCMe, В: вода/HClO4 (рН 2); поток: 1 мл/мин, изократический режим 68:32 (А/В) в течение 32 мин, градиент от 68/32 (А/В) до 75/25 (А/В) в течение 0,5 мин, изократический режим 75/25 (А/В) в течение 29,5 мин. Времена удерживания: 21,56 мин ((R)-2-(((9H- флуорен-9-ил)метокси)карбониламино)-6-((R)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота), 23,52 мин ((R)-2-(((9H-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота), 25,68 мин ((S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((R)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота), 28,32 мин ((S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота).
Метод препаративной СФХ: 2-этилпиридиновая колонка OBD Viridis, размер 150×30 мм, диаметр частиц 5 мкм; температура колонки 50°С; подвижная фаза А: CO2, В: МеОН; поток: 60 мл/мин, градиент от 80:20 (А/В) до 60/40 (А/В) в течение 10 мин.
Пример 3
(S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота
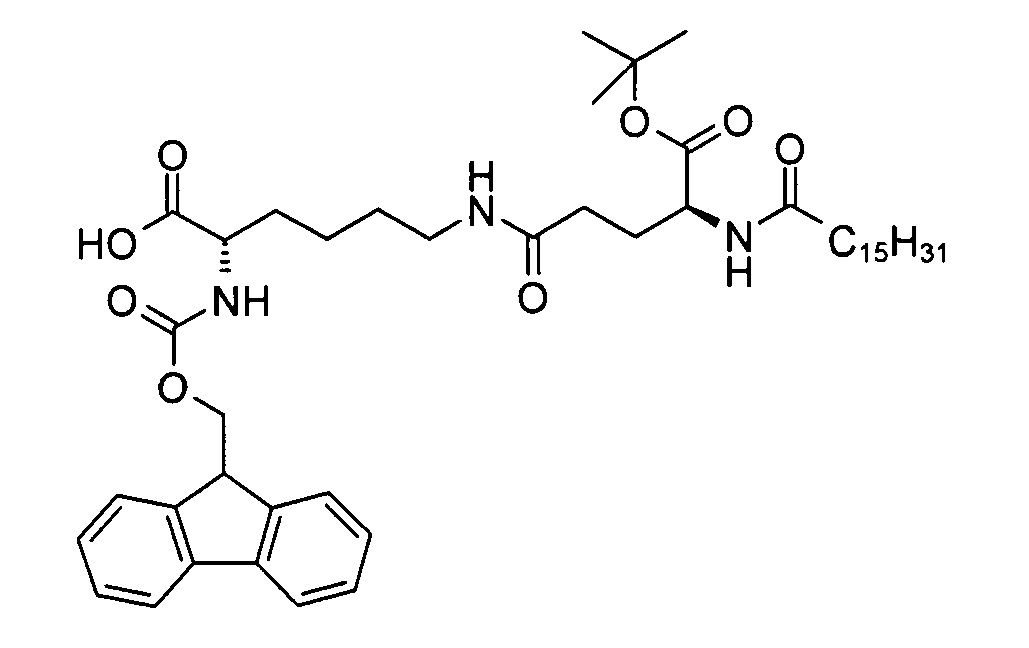
В 3-горлую колбу объемом 250 мл добавляли смесь (S)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноата (12,0 г, 13,7 ммоль), фенилсилана (2,28 г, 20,4 ммоль), тетракис(трифенилфосфин) палладия (0) (96,0 мг, 0,08 ммоль) и ДХМ (120 мл), перемешивали при к.т. Согласно результатам анализа методом ТСХ (ДХМ/МеОН 9:1, RF исходного вещества = 0,9, RF продукта = 0,3, продукт обнаруживали методом УФ-спектрофотометрии при длине волны 254 нм) через 3 ч превращение было завершено. Реакционную смесь затем последовательно промывали водным раствором диэтилдитиокарбамата натрия (0,5%, 20 мл) и солевым раствором (75 мл), высушивали над сульфатом натрия и выпаривали на ротационном вакуумном испарителе досуха с получением неочищенной (S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновой кислоты (11,6 г) с химической чистотой 93,5% (метод ЖХ см. в примере 2), энантиомерной чистотой >99,9% и диастереомерной чистотой 99,7% (метод хиральной ЖХ см. в примере 2), содержащей 1,2% остаточного оксида трифенилфосфина. Неочищенный продукт затем суспендировали в гептане (230 мл) в течение 1 ч при к.т., смесь фильтровали, отфильтрованный осадок промывали гептаном (50 мл) с получением (S)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновой кислоты (10,9 г, выход 97%) в виде желтоватого твердого вещества с химической чистотой 96,2% (метод ЖХ см. в примере 2), энантиомерной чистотой >99,9% и диастереомерной чистотой 99,8% (метод хиральной ЖХ см. в примере 2), содержащей 0,8% остаточного оксида трифенилфосфина.
Т.п. 119°С; ЭИ-МС: m/z=792,52 (М+Н)+.
Пример 4
((R)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат
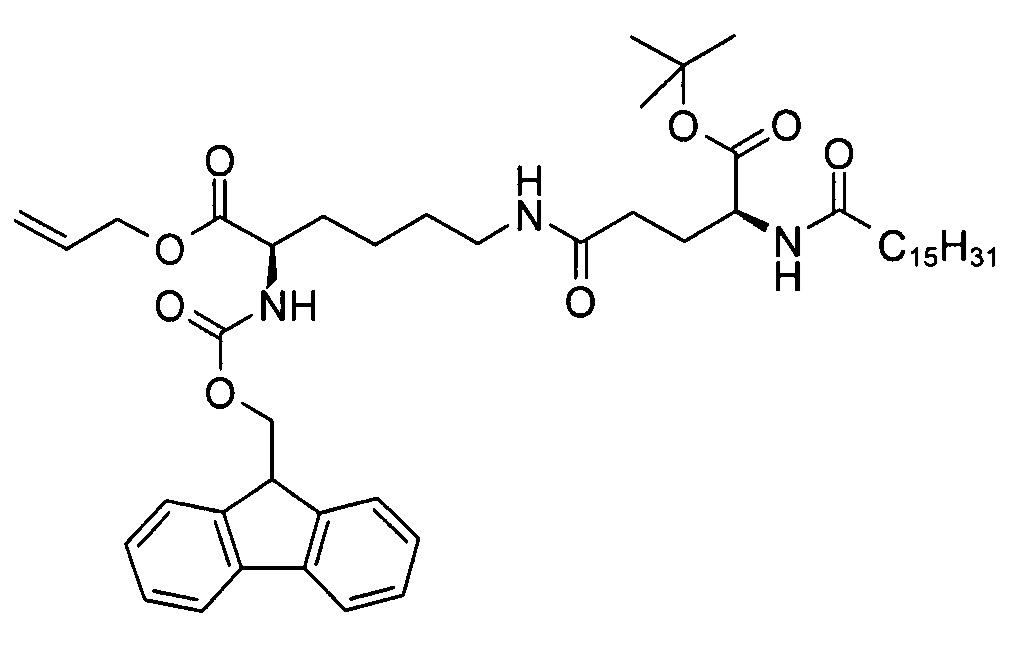
В 3-горлую колбу объемом 25 мл добавляли смесь (S)-5-трет-бутокси-5-оксо-4-пальмитамидопентановой кислоты (500 мг, 1,12 ммоль), 1-гидроксибензотриазола (175 мг, 1,14 ммоль), 1,1'-карбонилдиимидазола (200 мг, 1,23 ммоль) и ДХМ (10 мл), перемешивали при комнатной температуре в течение 90 мин. Через 5 мин к полученной в результате белой суспензии добавляли раствор (R)-аллил 6-амино-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-аминогексаноата (507 мг, 1,12 ммоль) и триэтиламина (113 мг, 1,12 ммоль) в ДХМ (5 мл). Реакционную смесь перемешивали в течение 60 мин при комнатной температуре для завершения превращения (что определяли методом ТСХ (ДХМ/МеОН 95:5, RF исходного вещества = 0, RF продукта = 0,2, продукт обнаруживали методом УФ-спектрофотометрии при длине волны 254 нм). Затем к смеси добавляли воду (10 мл) и проводили разделение слоев. Водный слой экстрагировали ДХМ (30 мл) и объединенные органические слои высушивали над сульфатом натрия. После выпаривания растворителя неочищенный продукт (983 мг), который находился в осадке, очищали путем кристаллизации из диэтилового эфира с получением (R)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноата (686 мг, 70%) в виде белого твердого вещества с химической чистотой 94,2% (метод ЖХ см. ниже) и энантиомерной и диастереомерной чистотой >99,9% (метод хиральной ЖХ см. в примере 1с).
Т.п. 114°С; ЭИ-МС: m/z=832,54 (М+Н)+.
Метод ВЭЖХ: фенильная колонка X-Bridge, размер 50×4,6 мм, диаметр частиц 2,5 мкм; подвижная фаза А: вода/NCMe (95:5), В: NCMe, С: 0,1% муравьиная кислота в воде; поток: 2 мл/мин; градиент от 65/25/10 (А/В/С) до 10/80/10 (А/В/С) в течение 10 мин, изократический режим 10/80/10 (А/В/С) в течение 2 мин. Время удерживания: 9,55 мин ((R)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат)).
Пример 5
(R)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота
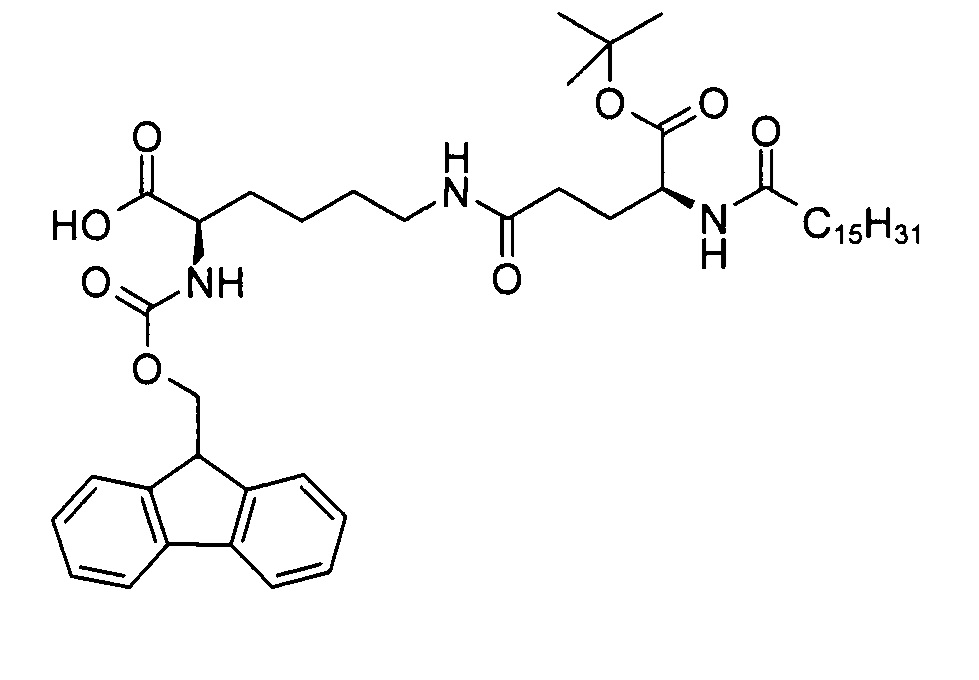
В 3-горлую колбу объемом 25 мл добавляли смесь (R)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноата (675 мг, 0,76 ммоль), фенилсилана (351 мг, 3,14 ммоль), тетракис(трифенилфосфина) палладия (0) (20,0 мг, 0,02 ммоль) и ДХМ (7 мл), перемешивали при температуре 10°С. Согласно результатам анализа методом ТСХ (ДХМ/МеОН 95:5, RF исходного вещества = 0,8, RF продукта = 0,2, продукт обнаруживали методом УФ-спектрофотометрии при длине волны 254 нм) через 25 мин превращение было завершено. Через 15 мин реакционную смесь разводили ДХМ (10 мл) и последовательно промывали водой (10 мл), водным раствором диэтилдитиокарбамата натрия (0,5%, 10 мл) и солевым раствором (10 мл). Органический раствор высушивали над сульфатом натрия и выпаривали на ротационном вакуумном испарителе досуха с получением неочищенной (R)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновой кислоты (631 мг) с химической чистотой 87,4 (метод ЖХ см. ниже)), энантиомерной чистотой >99,9% и диастереомерной чистотой 98,8% (метод хиральной ЖХ см. в примере 2). Неочищенный продукт содержал 6% оксида трифенилфосфина в качестве основной примеси. В результате проведения препаративной сверхкритической флюидной хроматографии (СФХ, метод см. в примере 2) образца 603 мг неочищенного продукта получали чистую (R)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновую кислоту (348 мг, 59%) в виде белого твердого вещества с химической чистотой 98,7% (метод ЖХ см. ниже), энантиомерной чистотой >99,9% и диастереомерной чистотой 99,4% (метод хиральной ЖХ см. в примере 2).
Т.п. 125°С; ЭИ-МС: m/z=792,52 (М+Н)+.
Метод ВЭЖХ: фенильная колонка X-Bridge, размер 50×4,6 мм, диаметр частиц 2,5 мкм; подвижная фаза А: вода/NCMe (95:5), В: NCMe, С: 0,1% муравьиная кислота в воде; поток: 2 мл/мин; градиент от 65/25/10 (А/В/С) до 10/80/10 (А/В/С) в течение 10 мин, изократический режим 10/80/10 (А/В/С) в течение 2 мин. Времена удерживания: 8,33 мин ((R)-2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)капроновая кислота), 9,35 мин ((R)-аллил 2-(((9Н-флуорен-9-ил)метокси)карбониламино)-6-((S)-5-трет-бутокси-5-оксо-4-пальмитамидопентанамидо)гексаноат)).
Разделение биспецифических антител и побочных продуктов процесса получения биспецифических антител с применением гидроксиапатитной хроматографии
Способ получения 2-трифторметилизоникотиновой кислоты и ее эфиров
Устройство, применяемое для детектирования аффинностей связывания
Ингибиторы тирозинкиназы брутона
Ингибиторы тирозинкиназы брутона
Устройство, применяемое для детектирования аффинностей связывания
Пирроло[2,3-в]пиразины в качестве ингибиторов syk
Полиморфы 2-(4-(2-(1-изопропил-3-метил-1h-1,2,4-триазол-5-ил)-5,6-дигидробензо[f]имидазо[1,2-d][1,4]оксазепин-9-ил)-1н-пиразол-1-ил)-2-метилпропанамида, способы их получения и фармацевтические применения
Способ получения промежуточных соединений бороновой кислоты
Крышка для емкости
Способ получения и отбора молекул, включающих по меньшей мере две различные группировки, и их применение
Катализируемое палладием сочетание пиразоламидов
Способы получения аналогов окситоцина

