Результат интеллектуальной деятельности: СПОСОБЫ ПОЛУЧЕНИЯ АНАЛОГОВ ОКСИТОЦИНА
Вид РИД
Изобретение
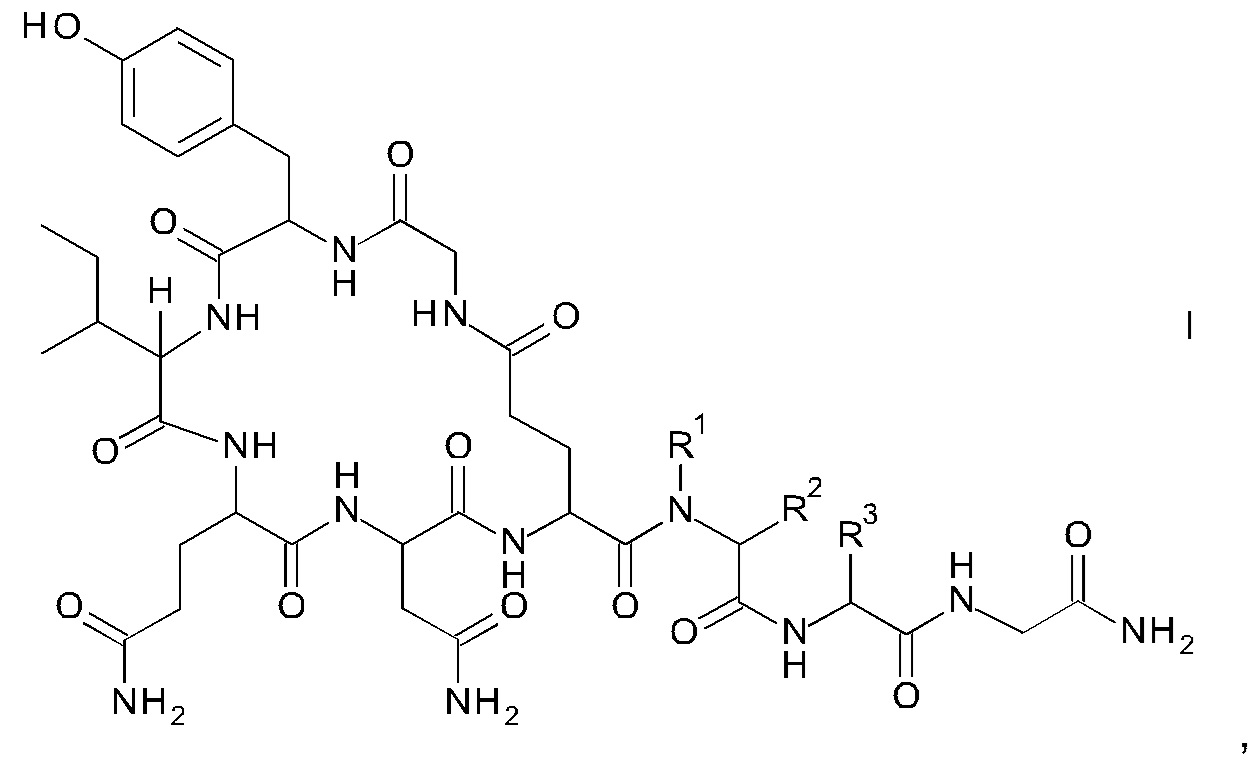
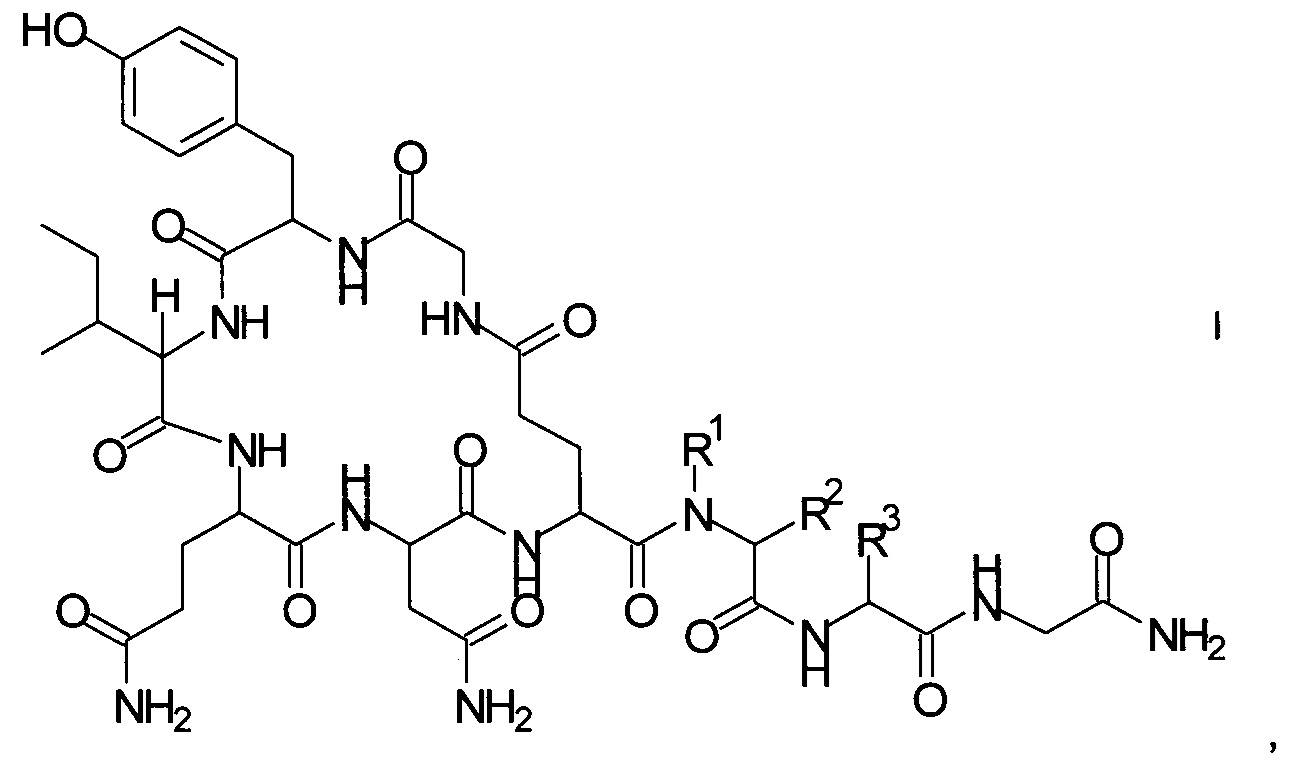
Данное изобретение относится к новому способу получения аналогов окситоцина формулы I
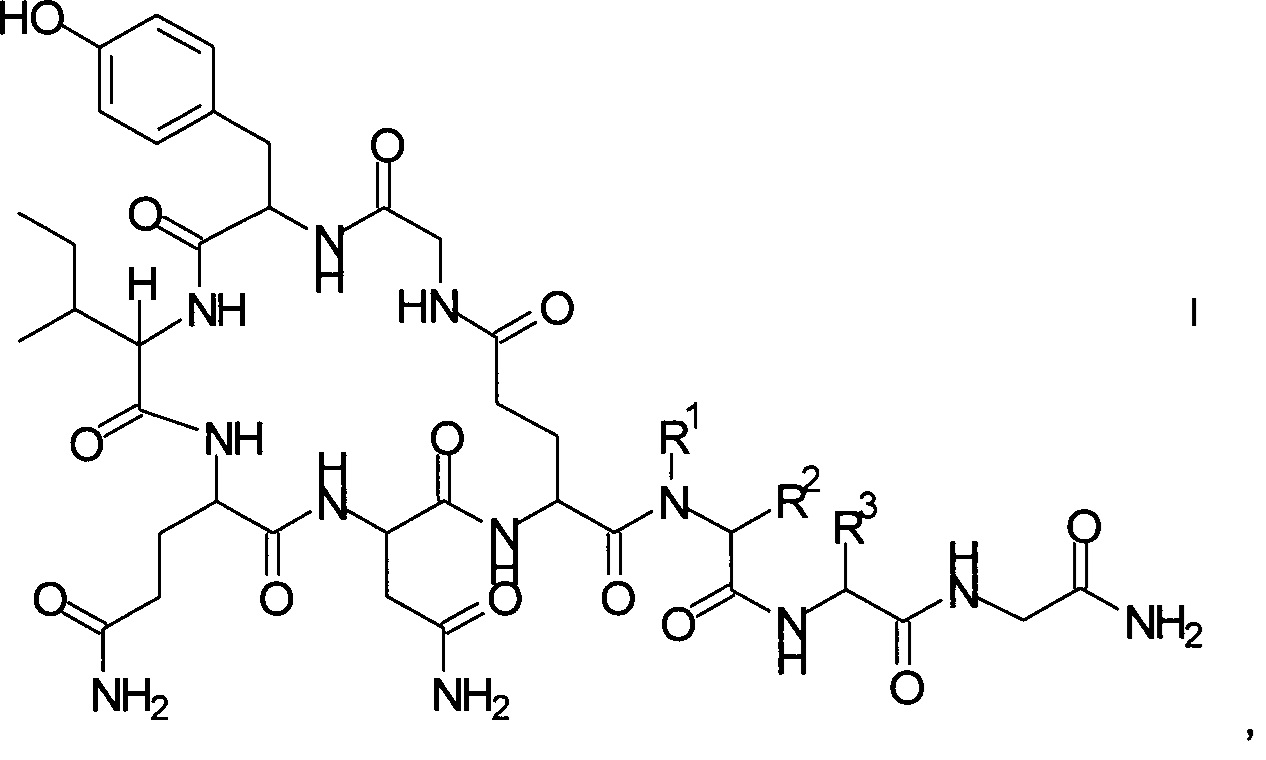
где
R1 представляет собой атом водорода или С1-7-алкил, и
R2 представляет собой атом водорода или С1-7-алкил; или
R1 и R2 вместе с атомом азота и атомом углерода, к которым они присоединены, образуют 5-членный гетероцикл, который возможно замещен группой гидрокси или атомом галогена;
R3 представляет собой C1-7-алкил;
и соответствующих им энантиомеров и/или их оптических изомеров.
Аналоги окситоцина формулы I действуют как агонисты рецепторов окситоцина и обладают потенциалом для применения в лечении неврологических расстройств, таких как аутизм, стресс, включая посттравматическое стрессовое расстройство, тревога, включая тревожные расстройства и депрессию, шизофрения, психические расстройства и потеря памяти, абстинентный алкогольный синдром, лекарственная зависимость, и для лечения синдрома Прадера-Вилли (публикация РСТ WO 2014/095773).
Получение аналогов окситоцина в соответствии со способом, описанным в публикации РСТ WO 2014/095773, характеризуется следующими стадиями:
х1) отщепление Fmoc (9-флуоренилметоксикарбонил) от связанного со смолой пептидного предшественника формулы X
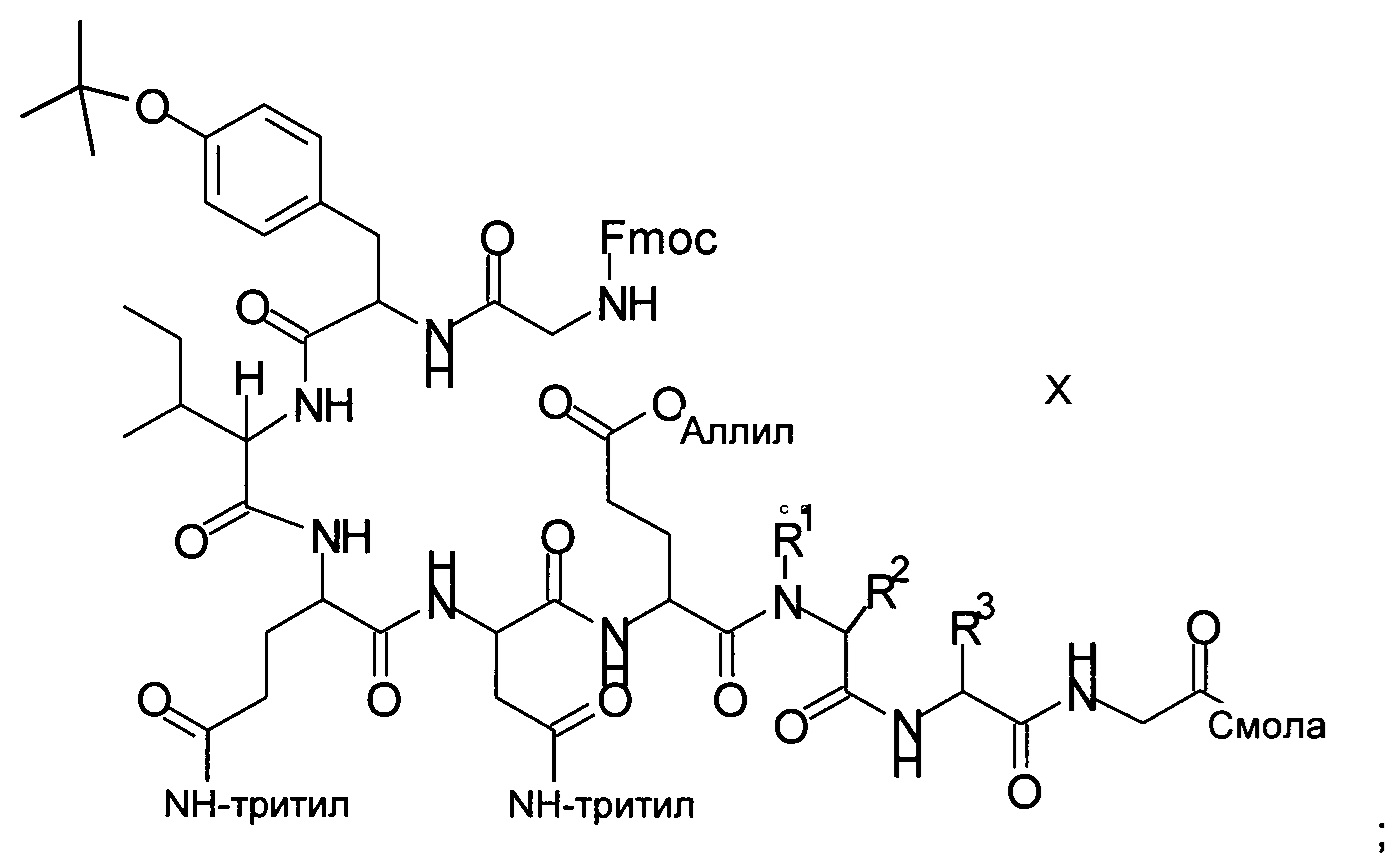
х2) отщепление аллильной группы на последующей стадии;
х3) циклизация на смоле с образованием кольца;
х4) удаление всех защитных групп и отщепление от смолы;
х5) очистка и выделение.
Было обнаружено, что этот известный в данной области техники способ имеет такие недостатки, как низкие общие выходы и селективность в отношении продукта.
Поэтому задача настоящего изобретения заключалась в улучшении способа синтеза касательно выхода и селективности в отношении желаемых аналогов окситоцина.
Данная задача может быть решена с использованием способа по настоящему изобретению, который указан ниже.
Способ получения аналогов окситоцина формулы I
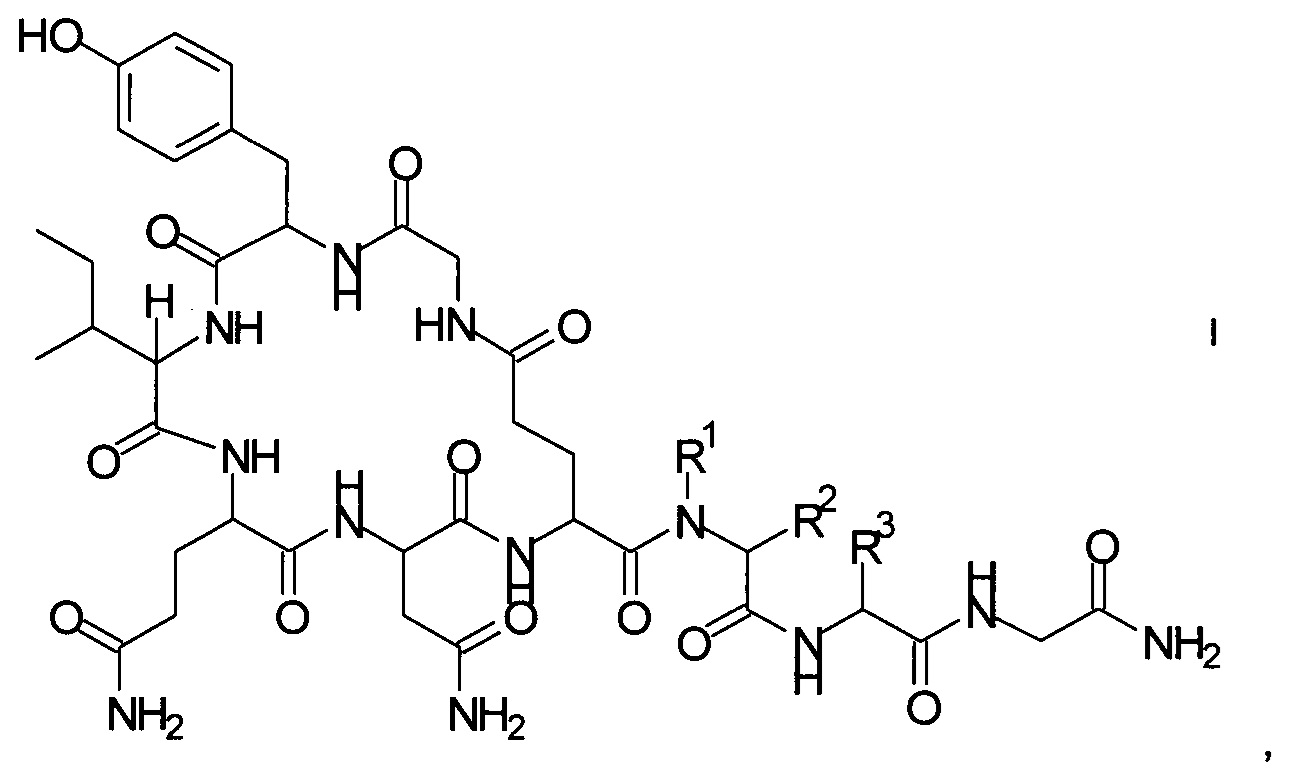
где
R1 представляет собой атом водорода или С1-7-алкил, и
R2 представляет собой атом водорода или C1-7-алкил; или
R1 и R2 вместе с атомом азота и атомом углерода, к которым они присоединены, образуют 5-членный гетероцикл, который возможно замещен группой гидрокси или атомом галогена;
R3 представляет собой С1-7-алкил;
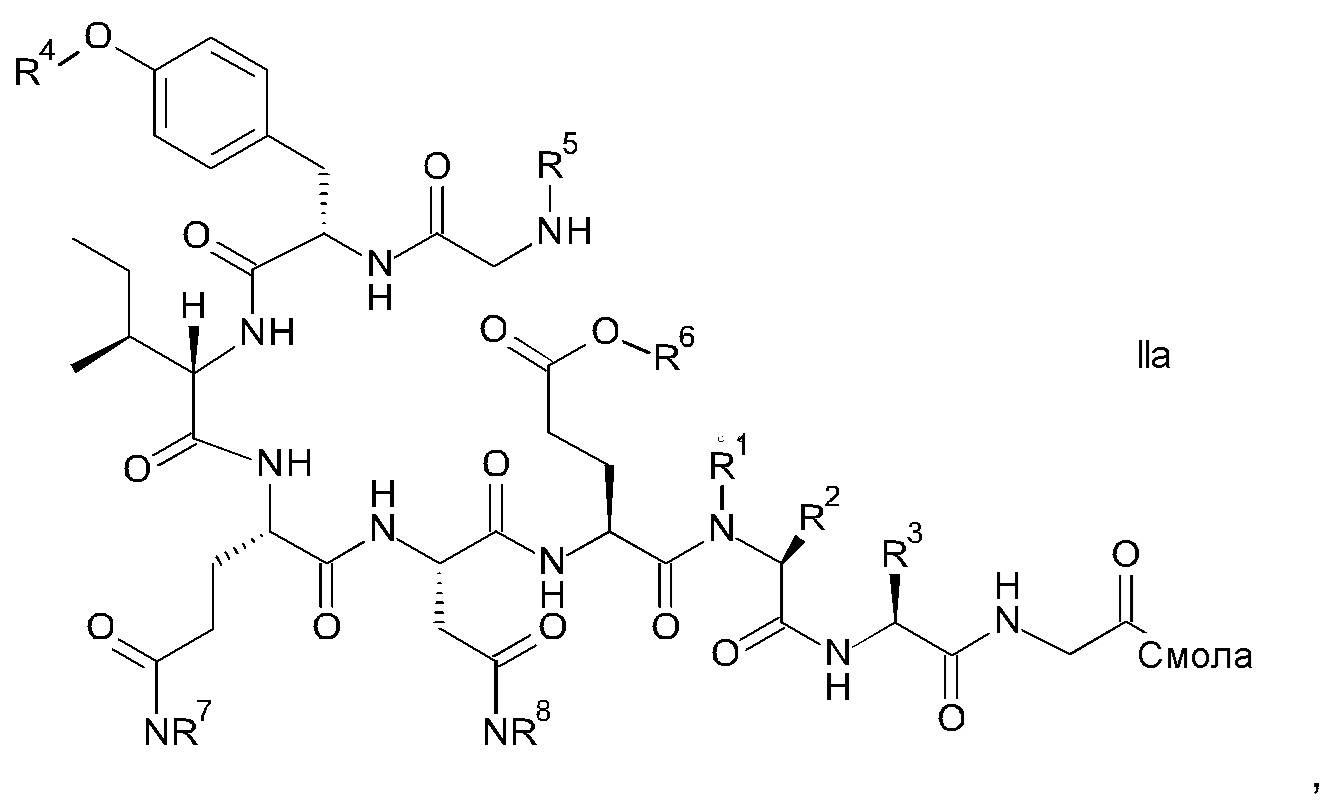
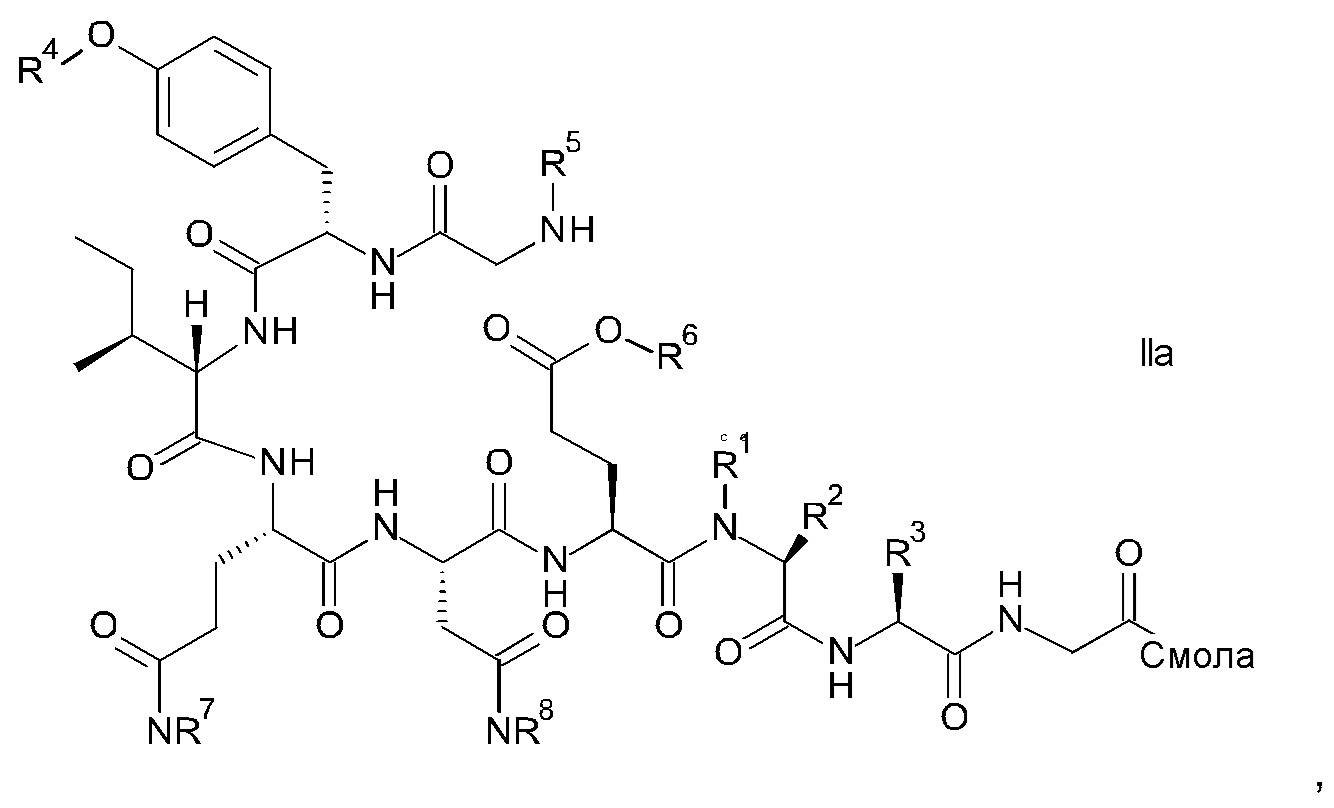
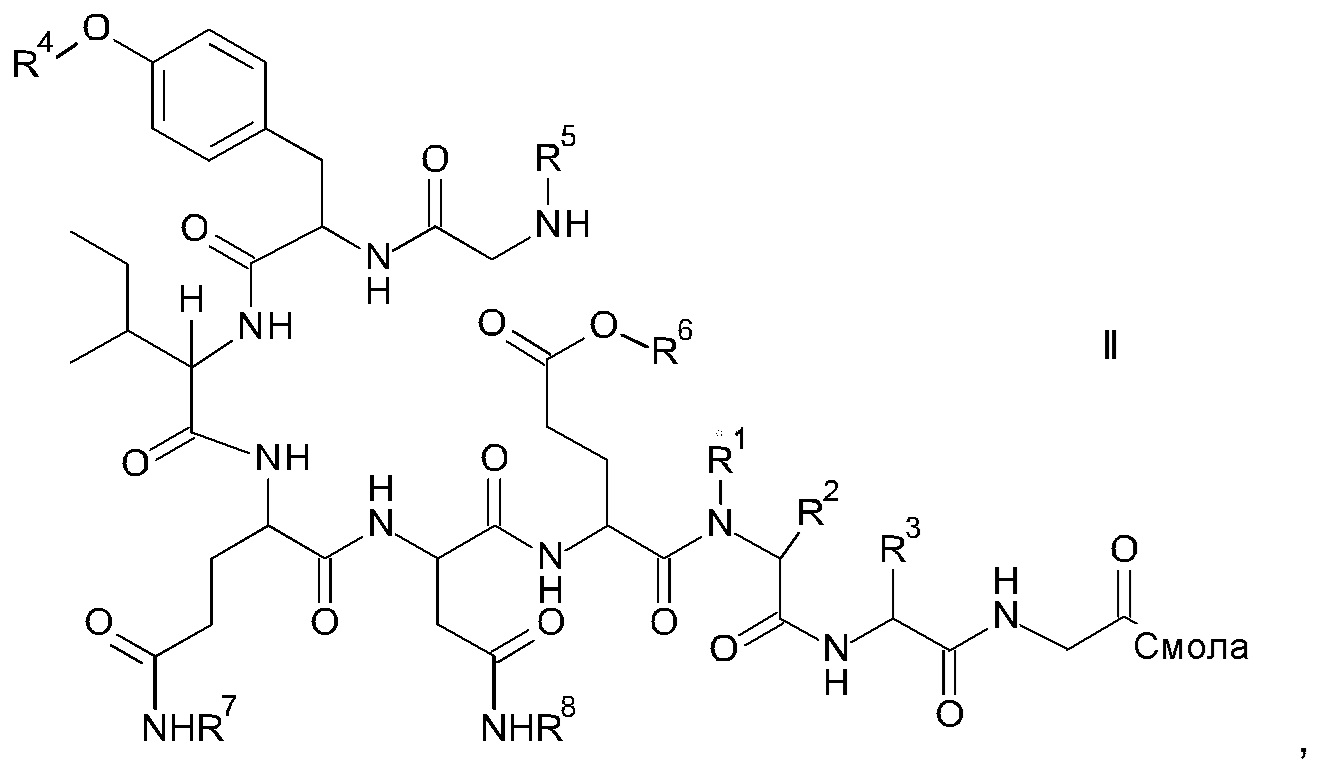
и соответствующих им энантиомеров и/или их оптических изомеров включает обработку связанного со смолой пептидного предшественника формулы II
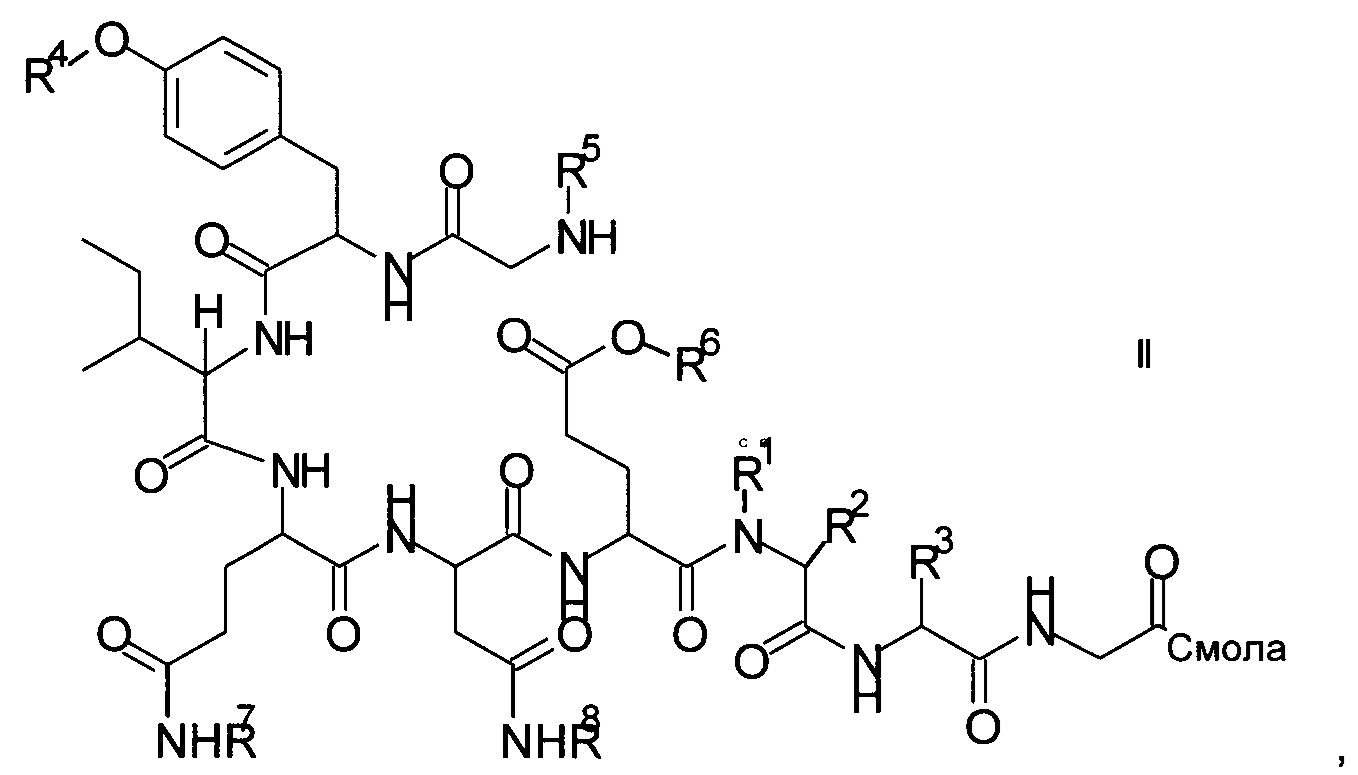
где
R1, R2 и R3 являются такими, как указано выше, и
R4 представляет собой гидрокси-защитную группу;
R5 представляет собой Fmoc;
R6 представляет собой аллил, трет-бутил, 1-адамантил, 4-{N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино}бензил или фенилизопропил;
R7 представляет собой амид-защитную группу; и
R8 представляет собой амид-защитную группу,
и соответствующих ему энантиомеров и/или их оптических изомеров,
либо в соответствии со способом:
а), где в случае, если R6 представляет собой аллил или 4-{N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино}бензил,
а1) отщепляют аллильную группу или 4-{N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино}бензильную группу R6, на последующей стадии
а2) отщепляют Fmoc группу R5, после чего
а3) на смоле осуществляют циклизацию с образованием кольца, на следующей стадии
а4) осуществляют удаление всех защитных групп и отщепление от смолы и, возможно,
а5) очищают и выделяют полученный таким образом аналог окситоцина формулы I;
либо в соответствии со способом:
b), где в случае, если R6 представляет собой трет-бутил, 1-адамантил или фенилизопропил,
b1) отщепляют Fmoc группу R5, после чего
b2) осуществляют удаление всех защитных групп и отщепление от смолы, на следующей стадии
b3) в растворе осуществляют циклизацию с образованием кольца, затем возможно
b4) выделяют и очищают полученный таким образом аналог окситоцина формулы I.
Следующие далее определения приведены для иллюстрации и установления значения и объема различных терминов, использованных для описания изобретения в данном документе.
Термин "C1-7-алкил" относится к одновалентному насыщенному алифатическому углеводородному радикалу с разветвленной или прямой цепью, содержащему от одного до семи атомов углерода, предпочтительно от одного до четырех, более предпочтительно один или два атома углерода. При этом в качестве примеров этого термина приводятся такие радикалы, как метил, этил, н-пропил, изопропил, н-бутил, втор-бутил, изобутил или трет-бутил, пентил и его изомеры, гексил и его изомеры и гептил и его изомеры.
Аналогичным образом термин "С1-4-алкил" относится к одновалентному насыщенному алифатическому углеводородному радикалу с разветвленной или прямой цепью, содержащему от одного до четырех атомов углерода, с предпочтениями и соответствующими примерами, упомянутыми выше.
Термин "C1-4-алкилокси" относится к С1-4-алкильной цепи, соединенной с атомом кислорода. При этом в качестве примеров этого термина приводятся такие радикалы, как метокси, этокси, н-пропокси, изопропокси, н-бутокси, изобутокси и трет-бутокси.
Термин "С1-4-алкилоксикарбонил" относится к цепи C1-4-алкокси, соединенной с карбонильной группой, и при этом в качестве примеров приводятся конкретные указанные выше алкокси-радикалы, соединенные с карбонильной группой.
Термин "С2-4-алкенил" относится к ненасыщенной прямой или разветвленной углеродной цепи, содержащий от 2 до 4 атомов углерода, содержащей по меньшей мере одну двойную связь. При этом в качестве примеров этого термина приводятся такие радикалы, как винил, аллил и бутенил и его изомеры.
Термин "галоген" относится к атому фтора, хлора, брома или йода.
Термин "5-членный гетероцикл", который образуется с участием R1 и R2 вместе с атомом азота и атомом углерода, к которым они присоединены, обозначает пирролидиновое кольцо, возможно замещенное группой гидрокси или атомом галогена, в частности, пирролидиновое кольцо пролина, которое замещено группой гидрокси или атомом фтора.
Термин "амид-защитная группа" относится к чувствительному к кислотам или кислотам Льюиса заместителю, традиционно используемому с целью воспрепятствования вступлению в реакцию амидной группы. Подходящие чувствительные к кислотам или кислотам Льюиса амид-защитные группы описаны в Isidro-Llobet A., Alvarez, М. and Albericio F., "Amino Acid-Protecting Groups", Chem. Rev., 2009, 109, 2455-2504; Chan W.C. and White P.D. "Fmoc Solid Phase Peptide Synthesis", Oxford University Press и Green Т., "Protective Groups in Organic Synthesis", 4oe изд., Wiley Interscience, 2007, глава 7, 696 и последующие страницы. Таким образом, подходящие амид-защитные группы могут быть выбраны из тритила, Tmob (2,4,6-триметоксибензил), Xan (9-ксантенил), Cpd (циклопропилдиметилкарбинил), Mbh (4,4'-диметоксибензгидрил) или Mtt (4-метилтритил).
Термин "гидрокси-защитная группа", использованный для заместителя R4, относится к любым заместителям, традиционно используемым с целью воспрепятствования вступлению в реакцию группы гидрокси. Подходящие гидрокси-защитные группы описаны в Isidro-Llobet A., Alvarez, М. and Albericio F., "Amino Acid-Protecting Groups", Chem. Rev., 2009, 109, 2455-2504; Chan W.C. and White P.D. "Fmoc Solid Phase Peptide Synthesis", Oxford University Press; Green Т., "Protective Groups in Organic Synthesis", глава 1, John Wiley и Sons, Inc., 1991, 10-142 и могут быть выбраны из С1-4-алкила, который возможно замещен фенилом или галогенированным фенилом; С2-4-алкенила; силила, который возможно замещен C1-4-алкилом или фенилом, или С1-4-алкилоксикарбонила.
Спиралевидная связь " " означает "
" означает " " или "
" или " ", указывая тем самым на хиральность молекулы.
", указывая тем самым на хиральность молекулы.
В случаях, когда в химической структуре присутствует хиральный атом углерода, подразумевается, что все стереоизомеры, ассоциированные с этим хиральным атомом углерода, охватываются данной структурой в виде чистых стереоизомеров, а также их смесей.
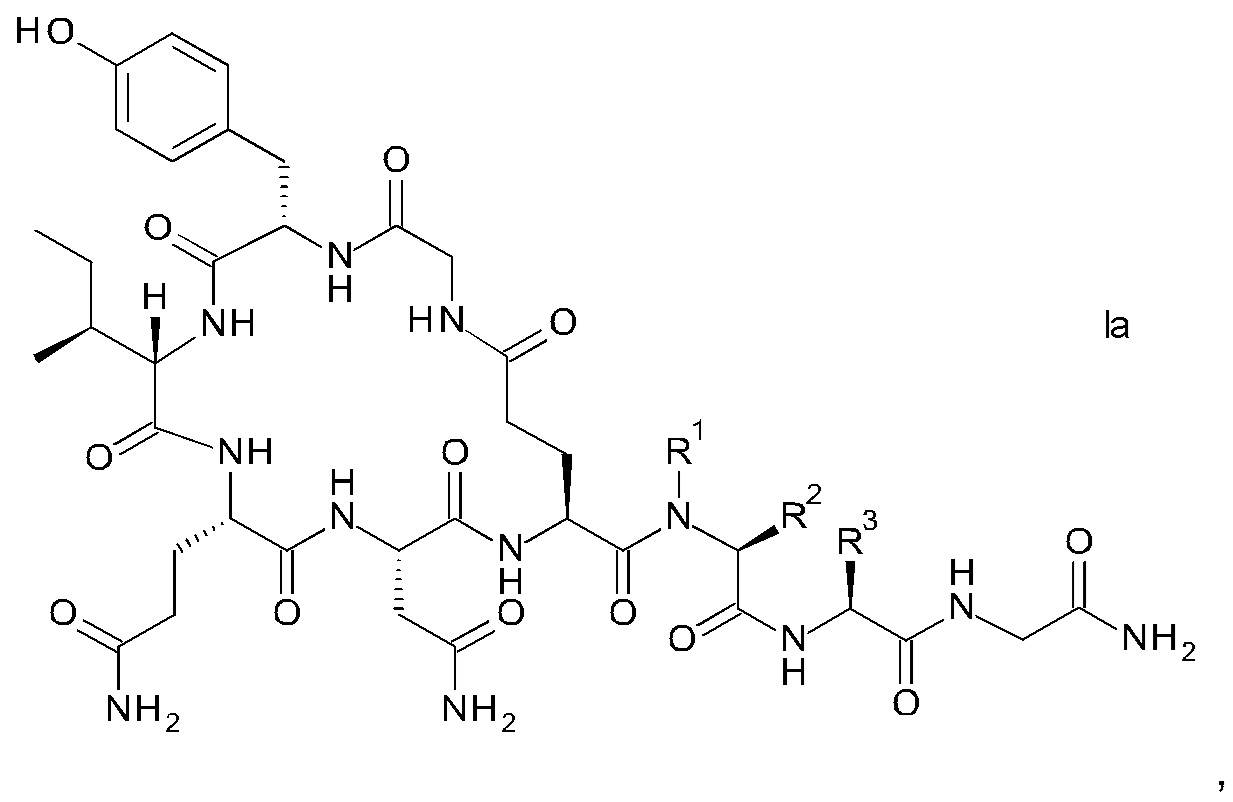
В конкретном воплощении настоящего изобретения аналоги окситоцина имеют формулу Iа
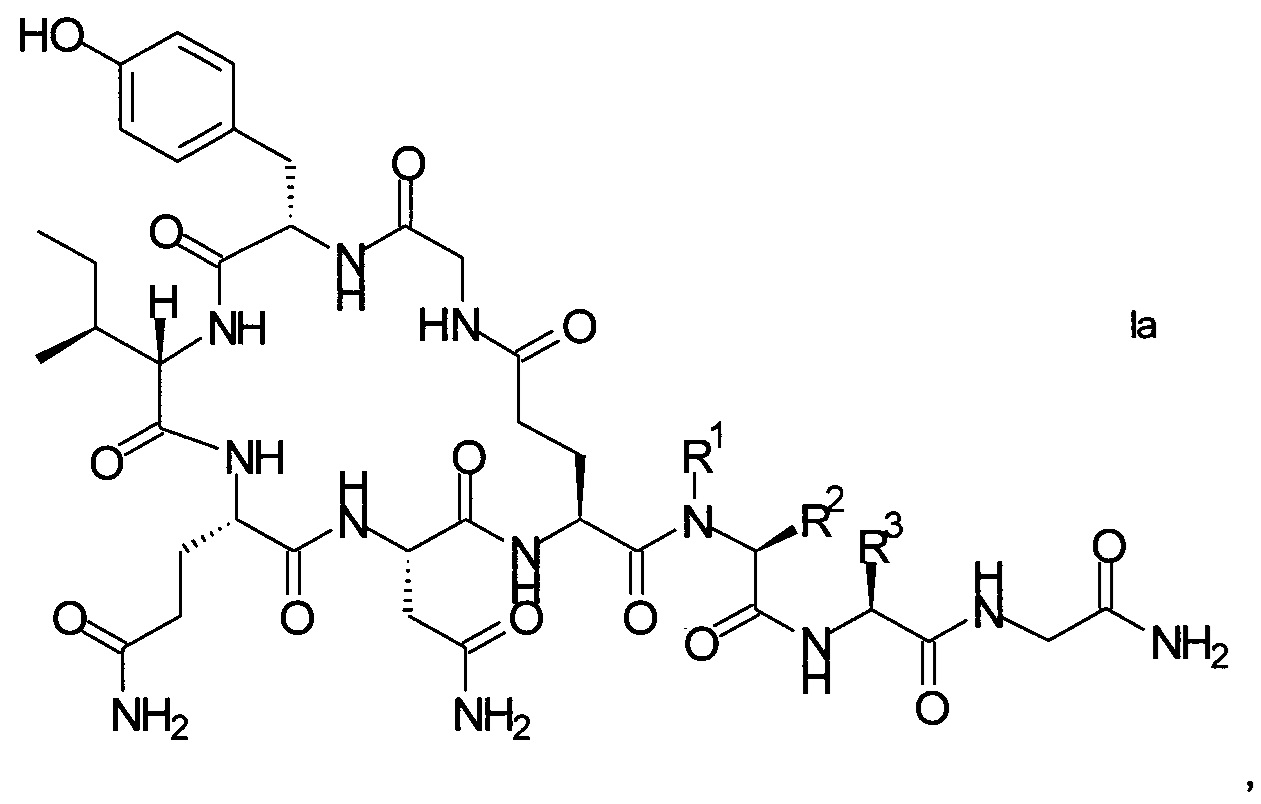
где R1, R2 и R3 являются такими, как указано выше.
R1 представляет собой, в частности, атом водорода или С1-4-алкил, более конкретно атом водорода или метил.
R2 представляет собой, в частности, атом водорода или С1-4-алкил, более конкретно атом водорода.
R1 и R2 вместе с атомом азота и атомом углерода, к которым они присоединены образуют, в частности, пирролидиновое кольцо пролина, которое возможно замещено группой гидрокси или атомом галогена, в частности, группой гидрокси или атомом фтора.
R3 означает, в частности, н-бутил или изобутил.
Еще более конкретные аналоги окситоцина приведены ниже:





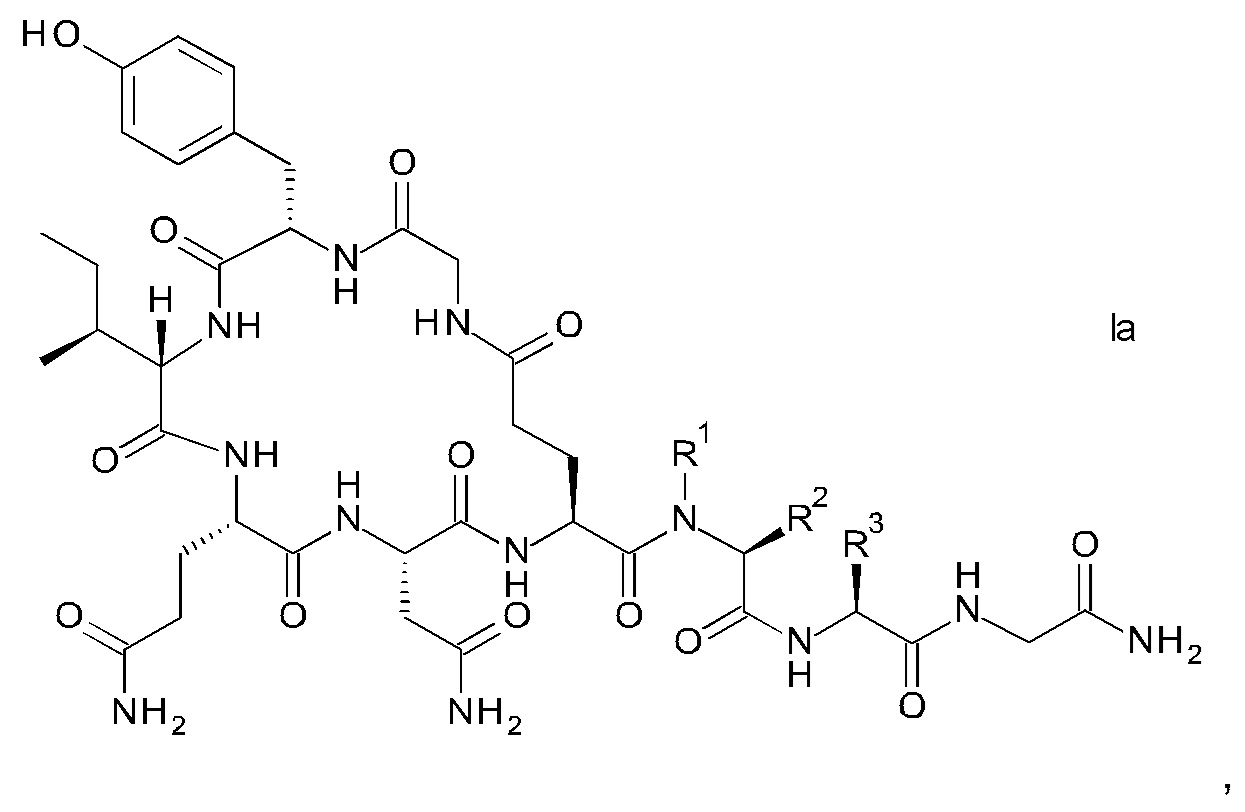
Связанный со смолой пептидный предшественник формулы II имеет формулу
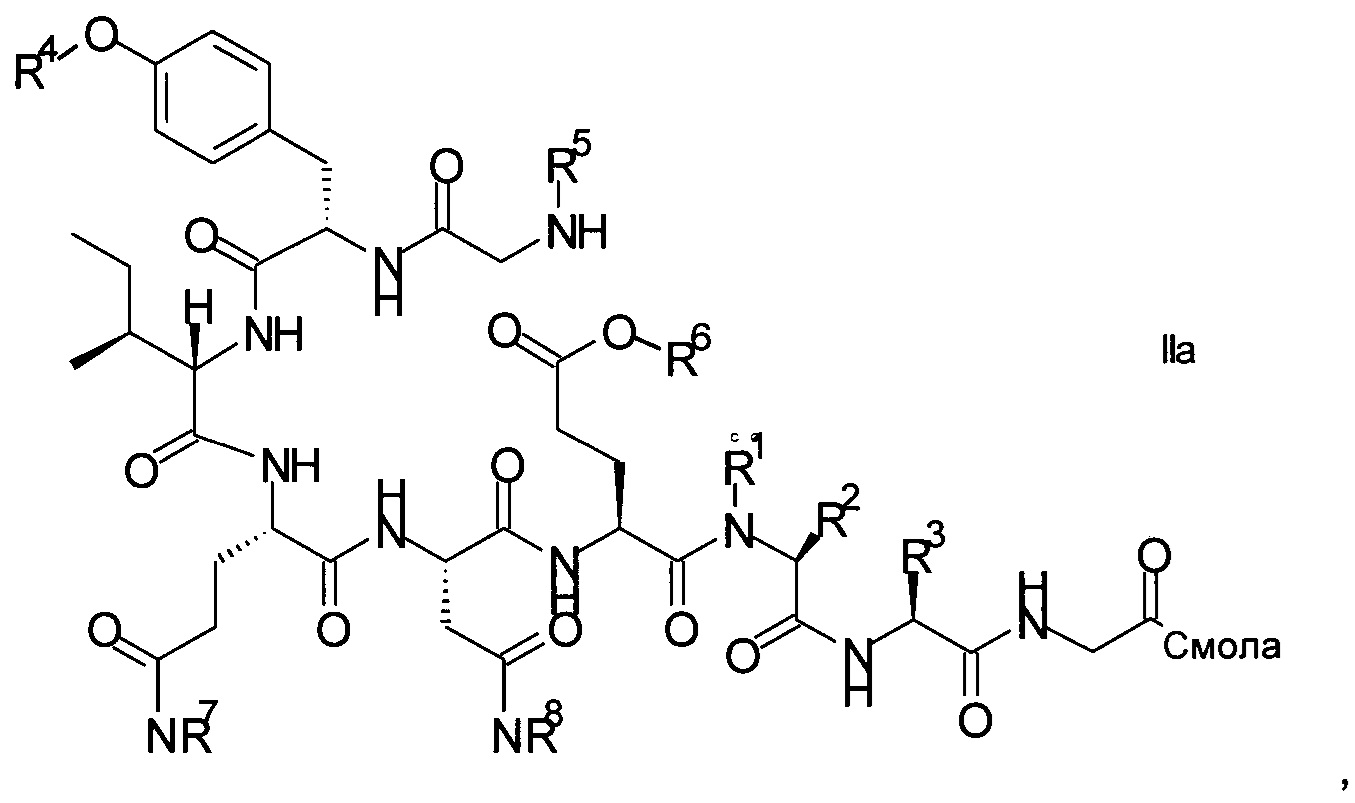
где R1, R2, R3, R4, R5, R6, R7 и R8 являются такими, как указано выше.
R1 представляет собой, в частности, атом водорода или С1-4-алкил, более конкретно атом водорода или метил.
R2 представляет собой, в частности, атом водорода или С1-4-алкил, более конкретно атом водорода.
R1 и R2 вместе с атомом азота и атомом углерода, к которым они присоединены образуют, в частности, пирролидиновое кольцо пролина, которое возможно замещено группой гидрокси или атомом галогена, в частности, группой гидрокси или атомом фтора;
R3 означает, в частности, н-бутил или изобутил;
R4 представляет собой, в частности, трет-бутил, аллил, тритил, 2-хлортритил, трет-бутилоксикарбонил, трет-бутилдифенилсилил или трет-бутилдиметилсилил, но более конкретно трет-бутил;
R5 представляет собой Fmoc;
R6 представляет собой, в частности, аллил, 1-адамантил, 4-{N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино}бензил, фенилизопропил или трет-бутил, но более конкретно аллил;
R7 представляет собой, в частности, тритил, 2-хлортритил, 4-метилтритил, но более конкретно тритил; и
R8 представляет собой, в частности, тритил, 2-хлортритил, 4-метилтритил, но более конкретно тритил.
Связанный со смолой пептидный предшественник формулы II может быть получен с использованием способов, известных специалистам в области твердофазного пептидного синтеза, обычно путем повторения отщепления Fmoc и связывания желаемых Fmoc-защищенных аминокислот.
Как правило, можно использовать имеющиеся в продаже амидные смолы, подходящие для твердофазного пептидного синтеза, в частности, для твердофазного пептидного синтеза с использованием Fmoc. Полезные смолы описаны, например, в Chan W.С. and White P.D. "Fmoc Solid Phase Peptide Synthesis", Oxford University Press. Например, обнаружено, что смола PL-Rink (4-[(2,4-диметоксифенил)Fmoc-аминометил]-феноксиацетамидометил-смола) от Agilent Technology особенно подходит для способа по настоящему изобретению.
Отщепление Fmoc может происходить с использованием раствора производных пиперидина в подходящем органическом растворителе. Предпочтительно, если существует возможность применения раствора пиперидина или 4-метилпиперидина в N,N-диметилформамиде или N-метилпирролидоне.
Реакция связывания на смоле Fmoc-защищенных аминокислот может протекать с участием связывающего агента, выбранного из бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфата (PyBOP), (7-азабензотриазол-1-илокси)трипирролидинофосфония гексафторфосфата (PyAOP), бромтрипирролидинофосфония гексафторфосфата (PyBrOP), гидроксибензотриазола (HOBt) и N,N'-диизопропилкарбодиимида (DIC), N,N,N',N'-тетраметил-O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HBTU), O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HATU), O-(6-хлорбензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HCTU), (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбения гексафторфосфата (COMU), тетраметилфторформамидиния гексафторфосфата (TFFH), 2-гидрокси-пиридина (HOPy) или 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния (DMTMM) хлорида в присутствии органического амина как основания и подходящего органического растворителя.
Обнаружено, что HOBt, HOPy и DIC в присутствии пиридина как органического амина в качестве основания и N,N'-диметилформамида как органического растворителя представляют собой предпочтительный связывающий агент.
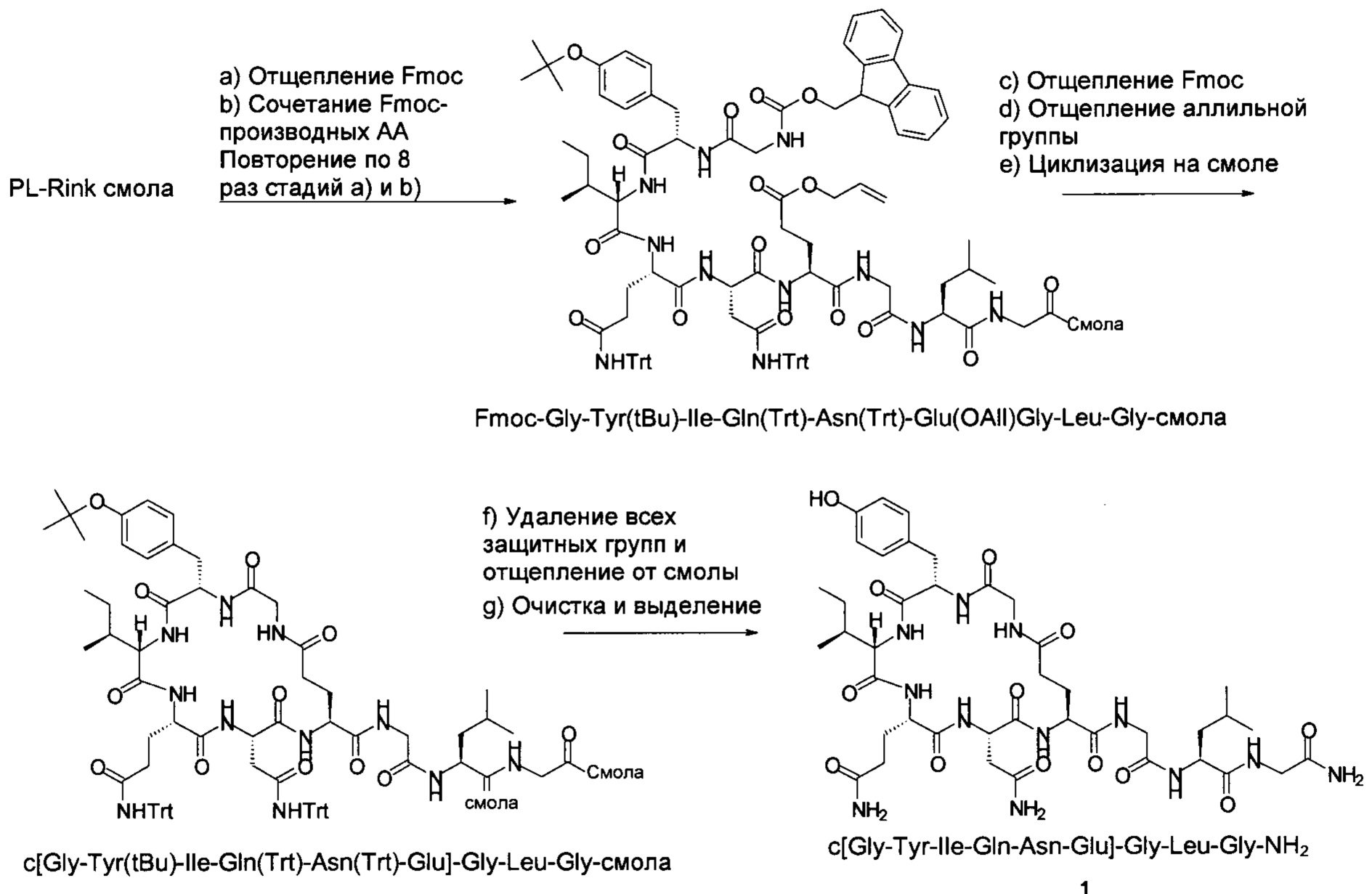
Fmoc-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(OAll)-Gly-Leu-Gly-смолу формулы X
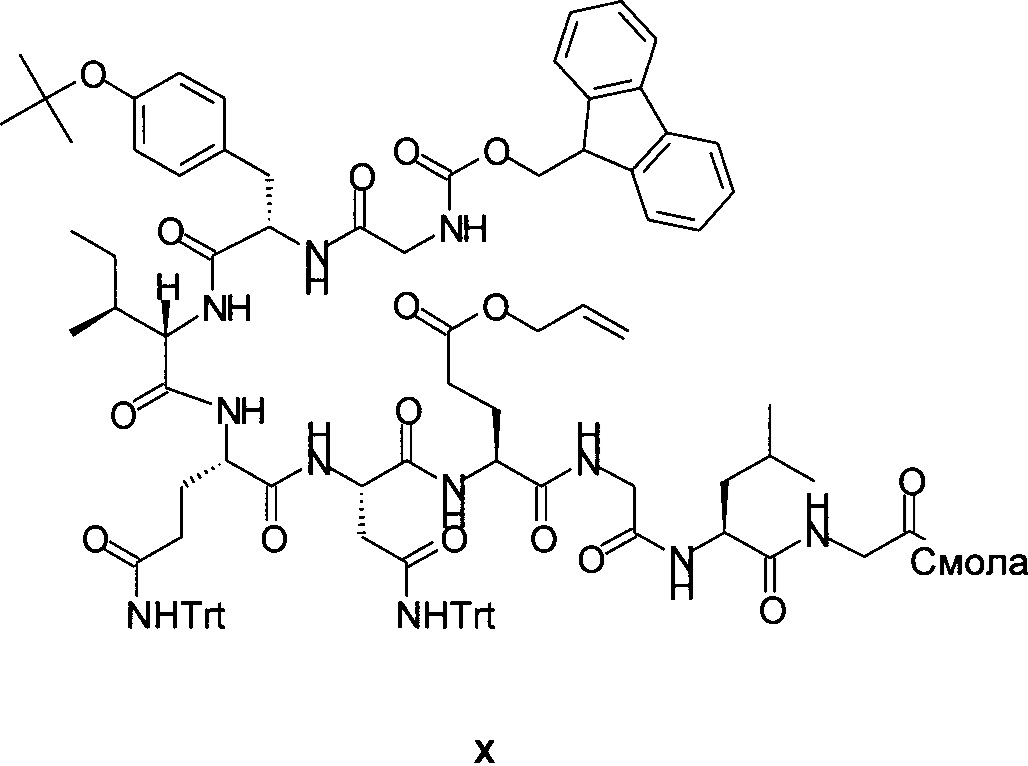
можно, например, создать на смоле PL-Rink посредством повторного отщепления Fmoc и повторного связывания следующих Fmoc-защищенных аминокислот в описанном далее порядке: Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-Gly-OH, Fmoc-Glu(OAll)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH и Fmoc-Gly-OH.
Как указано выше, способ по настоящему изобретению может быть осуществлен аналогично способу а), где R6 представляет собой аллил или 4-{N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино}бензил. В этом случае способ характеризуется следующими стадиями:
а1) отщепляют аллильную или 4-{N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино}бензильную группу R6, на последующей стадии
а2) отщепляют Fmoc группу R5, после чего
а3) на смоле осуществляют циклизацию с образованием кольца, на следующей стадии
а4) осуществляют удаление всех защитных групп и отщепление от смолы и, возможно,
а5) очищают и выделяют полученный таким образом аналог окситоцина формулы I.
Отщепление аллильной или 4-{N-[1-(4,4-диметил-2,6-диоксоциклогексилиден)-3-метилбутил]амино}бензильной группы на стадии а1) обычно выполняют в присутствии соединения палладия или родия либо в присутствии гидразина. Подходящие соединения палладия или родия могут быть выбраны из тетракис(трифенилфосфин)палладия, ацетата палладия/трифенилфосфина, ацетата палладия/триэтилфосфита, дихлорида бис(трифенилфосфин)палладия или хлорида трис(трифенилфосфин)родия. Предпочтительно используют соединения палладия, еще более предпочтительно тетракис(трифенилфосфин)палладий.
В дополнение к этому, обычно присутствует скавенджер, такой как фенилсилан, пирролидин, морфолин или N-метил-N-триметилсилил-трифторацетамид, в частности, фенилсилан.
Как правило, данная реакция может протекать при комнатной температуре в подходящем органическом растворителе, таком как метиленхлорид, ацетонитрил или тетрагидрофуран.
Отщепление Fmoc на стадии а2) может быть выполнено, как указано выше, в присутствии пиперидина или 4-метил-пиперидина в подходящем органическом растворителе.
Циклизацию с образованием кольца на стадии а3) осуществляют на смоле, целесообразно с использованием циклизующего агента, выбранного из бензотриазол-1-ил-окситрипирролидинофосфония гексафторфосфата (PyBOP), (7-азабензотриазол-1-илокси)трипирролидинофосфония гексафторфосфата (PyAOP), N,N,N',N'-тетраметил-O-(1Н-бензотриазол-1-ил)урония гексафторфосфата (HBTU), 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфата (HATU), O-(6-хлорбензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфата (HCTU), (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбения гексафторфосфата (COMU), 2-гидрокси-пиридина (HOPy) или 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорида (DMTMM), в присутствии органического амина как основания.
Подходящие в качестве оснований органические амины могут быть выбраны из пиридина, имидазола, N,N-диизопропилэтиламина, триэтиламина, N-метилморфолина, N,N-диметил-4-аминопиридина, 1,8-диазабицикло[5.4.0]ундец-7-ена или 1,4-диазабицикло[2.2.2]октана.
В предпочтительном воплощении стадия циклизации а3) может быть выполнена с использованием PyBOP или PyAOP в присутствии N,N-диизопропилэтиламина, имидазола или N-метилморфолина как органических аминов в качестве оснований при температурах от 0°С до 25°С.
Удаление всех защитных групп и отщепление от смолы на стадии а4) может быть осуществлено в присутствии трифторуксусной кислоты/воды и подходящего скавенджера, такого как тиоанизол, анизол, фенол, триизопропилсилан, триэтилсилан, этандитиол или дитиотреитол, обычно при температурах от 0°С до 25°С. Обнаружено, что предпочтительным скавенджером является триизопропилсилан.
На стадии а5) неочищенный аналог окситоцина может быть выделен посредством отфильтровывания смолы, удаления растворителя из фильтрата и далее внесения остатка в подходящий органический растворитель, например, в метил-трет-бутиловый эфир, 2-метилтетрагидрофуран или в их смеси, и в конце фильтрования и сушки.
Неочищенный аналог окситоцина далее может быть очищен препаративной высокоэффективной жидкостной хроматографией (ВЭЖХ) в растворе с использованием подходящего органического растворителя, например, с использованием водного ацетонитрила, и подходящих вспомогательных веществ, таких как трифторуксусная кислота, уксусная кислота или ацетат аммония.
Полученные фракции затем можно подвергнуть лиофилизации с получением чистого аналога окситоцина формулы I.
Альтернативно, способ по настоящему изобретению может быть осуществлен аналогично способу b), где R6 представляет собой трет-бутил, 1-адамантил или фенилизопропил. В этом случае способ характеризуется следующими стадиями:
b1) отщепляют Fmoc группу R5, после чего
b2) осуществляют удаление всех защитных групп и отщепление от смолы, на следующей стадии
b3) в растворе осуществляют циклизацию с образованием кольца, затем возможно
b4) выделяют и очищают полученный таким образом аналог окситоцина формулы I.
Отщепление Fmoc на стадии b1) может проходить так, как описано выше для стадии а2).
Удаление всех защитных групп и отщепление от смолы на стадии b2) может быть выполнено так, как описано выше на стадии а4). Предпочтительные воплощения, описанные для стадии а4, подобным же образом применимы для стадии b2).
Циклизацию с образованием кольца на стадии b3) осуществляют в растворе, но она может происходить в присутствии циклизующих агентов и органических аминов как оснований, приведенных выше для стадии а3). Предпочтительные воплощения, описанные для стадии а3, подобным же образом применимы для стадии b3).
Выделение и очистка на стадии b4) могут быть выполнены аналогично тому, как описано на стадии а5). Предпочтительные воплощения, описанные для стадии а5, подобным же образом применимы для стадии b4).
В конкретном воплощении настоящего изобретения альтернативный способ b) предпочтительнее альтернативного способа а).
ПРИМЕРЫ
Сокращения
SPPS означает твердофазный пептидный синтез, смола PL-Rink означает 4-[(2,4-диметоксифенил)Fmoc-аминометил]феноксиацетамидометил-смолу от Agilent Technology (PL1467-4749: 0,32 ммоль/г, 75-150⋅10-6 м; PL1467-4799: 0,55 ммоль/г, 75-150⋅10-6; PL1467-4689: 0,96 ммоль/г, 150-300⋅10-6 м), Fmoc означает 9-флуоренилметоксикарбонил, Gly означает глицин, Leu означает лейцин, Glu(OAll) означает аллил-защищенную глутаминовую кислоту, Glu(tBu) означает трет-бутил-защищенную глутаминовую кислоту, Asn(Trt) означает тритил-защищенный аспарагин, Gln(Trt) означает тритил-защищенный глутамин, Ilе означает изолейцин, Tyr(tBu) означает трет-бутил-защищенный тирозин, Sar означает N-метилглицин, Pro означает пролин, Nle означает норлейцин, ДМФА означает N,N-диметилформамид, HOBt означает 1-гидроксибензотриазол, HOPy означает 2-гидрокси-пиридин, DIC означает N,N'-диизопропилкарбодиимид, NEP означает N-этилпирролидон, PyBOP означает (бензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат, DIPEA означает диизопропилэтиламин, МеОН означает метанол, CH2Cl2 означает дихлорметан, МТВЕ означает метил-трет-бутиловый эфир, MeTHF означает 2-метилтетрагидрофуран, TFA означает трифторуксусную кислоту, MeCN означает ацетонитрил, РуАОР означает (7-азабензотриазол-1-илокси)трипирролидинофосфония гексафторфосфат, HBTU означает N,N,N',N'-тетраметил-O-(1Н-бензотриазол-1-ил)урония гексафторфосфат, HATU означает 1-[бис(диметиламино)метилен]-1Н-1,2,3-триазоло[4,5-b]пиридиний-3-оксида гексафторфосфат, HCTU означает O-(6-хлорбензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат, COMU означает (1-циано-2-этокси-2-оксоэтилиденаминоокси)диметиламино-морфолино-карбения гексафторфосфат, DMTMM означает 4-(4,6-диметокси-1,3,5-триазин-2-ил)-4-метилморфолиния хлорид, NMP означает 1-метил-2-пирролидинон, DMSO означает диметилсульфоксид, DMI означает 1,3-диметил-2-имидазолидинон, DMPU означает 1,3-диметил-3,4,5,6-тетрагидро-2(1Н)-пиримидинон, NMM означает N-метилморфолин, DMAP означает N,N-диметил-4-аминопиридин, DIPEA означает N,N-диизопропилзтиламин, DBU означает 1,8-диазабицикло[5.4.0]ундец-7-ен, DABCO означает 1,4-диазабицикло[2.2.2]октан.
Пример сравнения
Проводили сравнительный эксперимент для получения

по аналогии с синтезом, описание которого приведено в WO 2014/095773 (циклизация на твердой фазе), и как указано ниже на схеме 1.
Схема 1:
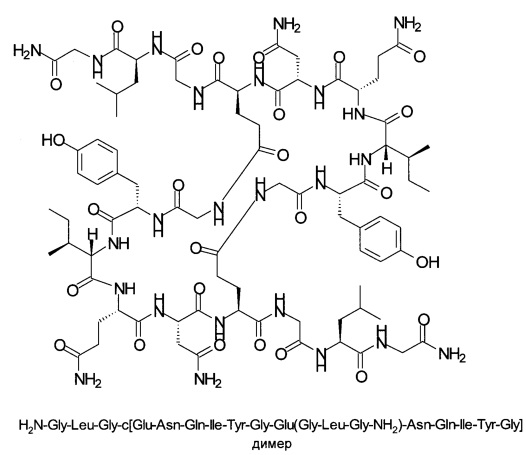
Эффективность синтеза была оценена по выходу продукта и соотношению продукта (1) и побочного продукта - димера формулы, показанной на схеме 2, приведенной ниже.
Схема 2:
a) Отщепление Fmoc
В реактор для SPPS (100 мл; пептидный синтезатор CS136XT от CSBio) загружали смолу PL-Rink (загрузка 0,55 ммоль/г; 5,00 г; 2,75 ммоль) и 20%-ный пиперидин в ДМФА (50,0 мл). Затем эту смесь перемешивали при 25°С в течение 10 мин. После слива растворителя добавляли другую порцию 20%-ного пиперидина в ДМФА (50,0 мл) и смесь перемешивали при 25°С в течение 30 мин. После слива растворителя полученную смолу промывали ДМФА (8×50,0 мл), получая PL-Rink смолу без Fmoc.
b) Связывание с Fmoc-производными аминокислот
К PL-Rink смоле без Fmoc группы добавляли раствор Fmoc-Gly-OH в смеси 0,35 М HOBt/ДМФА (32,0 мл; 11,2 ммоль), 0,92 М DIC в ДМФА (16,0 мл; 14,7 ммоль) и 10%-ный пиридин в ДМФА (16,0 мл; 19,8 ммоль) и перемешивали при 25°С в течение 3 ч. После слива растворителя полученную смолу промывали ДМФА (4×50,0 мл), получая Fmoc-Gly-смолу.
Стадии отщепления Fmoc и связывания с Fmoc-производными аминокислот повторяли 8 раз, используя вместо Fmoc-Gly-OH следующие Fmoc-производные аминокислот: Fmoc-Leu-OH, Fmoc-Gly-OH, Fmoc-Glu(OAll)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH, с получением Fmoc-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(OAll)-Gly-Leu-Gly-смолы. Образец отщепляли от смолы (см. ниже) для подтверждения правильности массы. MS (масс-спектр) (m/z): 1211,8 (М+Н)+.
c) Отщепление Fmoc
Отщепление Fmoc от концевого остатка Gly проводили так, как описано выше, получая H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(OAll)-Gly-Leu-Gly-смолу. Образец отщепляли от смолы (разбавитель см. ниже) для подтверждения правильности массы. MS (m/z): 989,8 (М+Н)+.
d) Отщепление аллильной группы
К H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(OAll)-Gly-Leu-Gly-смоле добавляли раствор тетракис-трифенилфосфин-палладия (159 мг; 0,138 ммоль) и фенилсилана (3,40 мл; 27,6 ммоль) в CH2Cl2 (50,0 мл) и перемешивали при 25°С в течение 30 мин. После слива растворителя эту стадию повторяли еще раз и промывали ДМФА (2×50,0 мл). Добавляли раствор дитиокарбамата натрия (250 мг) и DIPEA (0,250 мл) в ДМФА (50,0 мл) и смесь перемешивали при 25°С в течение 15 мин. После слива растворителя эту стадию повторяли еще раз. После слива растворителя полученную смолу промывали ДМФА (4×50,0 мл), получая H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu-Gly-Leu-Gly-смолу. Образец отщепляли от смолы (разбавитель см. ниже) для подтверждения правильности массы. MS (m/z): 949,7 (М+Н)+.
e) Циклизация на смоле
Раствор PyBOP (2,36 г; 4,54 ммоль) и DIPEA (2,40 мл; 13,8 ммоль) в NEP (60,0 мл) добавляли к H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu-Gly-Leu-Gly-смоле и смесь перемешивали при 25°С в течение 4 ч. После слива растворителя полученную смолу промывали ДМФА (4×50,0 мл), CH2Cl2 (3×50,0 мл) и МеОН (3×50,0 мл). Смолу сушили при давлении 10 мбар (1 кПа) при 25°С в течение 1 суток, получая с[Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu]-Gly-Leu-Gly-смолу (8,60 г).
f) Удаление всех защитных групп и отщепление от смолы
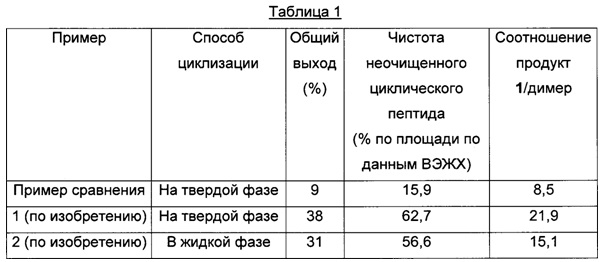
К предварительно охлажденному (10-15°С) раствору триизопропилсилана (2,80 мл) в TFA (40,0 мл) и воде (10,0 мл) добавляли c[Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu]-Gly-Leu-Gly-смолу (8,60 г) и перемешивали при 25°С в течение 3 ч. Смолу отфильтровывали и фильтрат концентрировали в вакууме. Остаток добавляли к МТВЕ (100 мл) и смесь перемешивали при 25°С в течение 15 ч. Смесь фильтровали и осадок на фильтре промывали МТВЕ (50,0 мл), затем сушили, получая неочищенный продукт c[Gly-Tyr-Ile-Gln-Asn-Glu]-Gly-Leu-Gly-NH2 1 (2,01 г; результаты анализа: 11,3 масс. %; общий выход 9%) в виде белого твердого вещества с чистотой 15,9% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1). Соотношение продукт 1/димер составляло 8,5.
Пример 1 (циклизация на твердой фазе)

Схема 3:
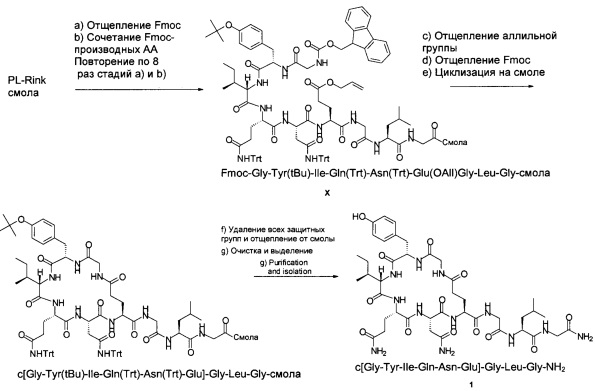
a) Отщепление Fmoc
В реактор для SPPS (100 мл; пептидный синтезатор CS136XT от CSBio) загружали смолу PL-Rink (загрузка 0,55 ммоль/г; 5,00 г; 2,75 ммоль) и 20%-ный пиперидин в ДМФА (50,0 мл). Затем эту смесь перемешивали при 25°С в течение 10 мин. После слива растворителя добавляли другую порцию 20%-ного пиперидина в ДМФА (50,0 мл) и смесь перемешивали при 25°С в течение 30 мин. После слива растворителя полученную смолу промывали ДМФА (8×50,0 мл), получая PL-Rink смолу без Fmoc группы.
b) Связывание с Fmoc-производными аминокислот
К PL-Rink смоле без Fmoc группы добавляли раствор Fmoc-Gly-OH в смеси 0,35 М HOBt/ДМФА (32,0 мл; 11,2 ммоль), 0,92 М DIC в ДМФА (16,0 мл; 14,7 ммоль) и 10%-ный пиридин в ДМФА (16,0 мл; 19,8 ммоль) и перемешивали при 25°С в течение 3 ч. После слива растворителя полученную смолу промывали ДМФА (4×50,0 мл), получая Fmoc-Gly-смолу.
Стадии отщепления Fmoc и связывания с Fmoc-производными аминокислот повторяли 8 раз, используя вместо Fmoc-Gly-OH следующие Fmoc-производные аминокислот: Fmoc-Leu-OH, Fmoc-Gly-OH, Fmoc-Glu(OAll)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH, с получением продукта X (Fmoc-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(OAll)-Gly-Leu-Gly-смолы). Образец отщепляли от смолы (см. ниже) для подтверждения правильности массы. MS (m/z): 1211,8 (М+Н)+.
c) Отщепление аллильной группы
К продукту X (Fmoc-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(OAll)-Gly-Leu-Gly-смоле) добавляли раствор тетракис-трифенилфосфин-палладия (159 мг; 0,138 ммоль) и фенилсилана (3,40 мл; 27,6 ммоль) в CH2Cl2 (50,0 мл) и перемешивали при 25°С в течение 30 мин. После слива растворителя эту стадию повторяли еще раз и промывали ДМФА (2×50,0 мл). Добавляли раствор дитиокарбамата натрия (250 мг) и DIPEA (0,250 мл) в ДМФА (50,0 мл) и смесь перемешивали при 25°С в течение 15 мин. После слива растворителя эту стадию повторяли еще раз. После слива растворителя полученную смолу промывали ДМФА (4×50,0 мл), получая Fmoc-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu-Gly-Leu-Gly-смолу. Образец отщепляли от смолы (разбавитель см. ниже) для подтверждения правильности массы. MS (m/z): 1171,8 (М+Н)+.
d) Отщепление Fmoc
Отщепление Fmoc от концевого остатка Gly проводили так, как описано выше, получая H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu-Gly-Leu-Gly-cмoлy. Образец отщепляли от смолы (разбавитель см. ниже) для подтверждения правильности массы. MS (m/z): 949,7 (М+Н)+.
e) Циклизация на смоле
Раствор PyBOP (2,36 г; 4,54 ммоль) и DIPEA (2,40 мл; 13,8 ммоль) в NEP (60,0 мл) добавляли к (H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu-Gly-Leu-Gly-cмoле и смесь перемешивали при 25°С в течение 4 ч. После слива растворителя полученную смолу промывали ДМФА (4×50,0 мл), CH2Cl2 (3×50,0 мл) и МеОН (3×50,0 мл). Смолу сушили при давлении 10 мбар (1 кПа) при 25°С в течение 1 суток, получая с[Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu]-Gly-Leu-Gly-cмoлy (9,17 г).
f) Удаление всех защитных групп и отщепление от смолы
К предварительно охлажденному (10-15°С) раствору триизопропилсилана (2,50 мл) в TFA (40,0 мл) и воде (10,0 мл) добавляли c[Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu]-Gly-Leu-Gly-cмoлy (9,17 г) и перемешивали при 25°С в течение 3 ч. Смолу отфильтровывали и фильтрат концентрировали в вакууме. Остаток добавляли к МТВЕ (100 мл) и смесь перемешивали при 25°С в течение 15 ч. Смесь фильтровали и осадок на фильтре промывали МТВЕ (50,0 мл), затем сушили, получая неочищенный продукт c[Gly-Tyr-Ile-Gln-Asn-Glu]-Gly-Leu-Gly-NH2 1 (2,39 г; результаты анализа: 40,9 масс. %, общий выход 38%) в виде белого твердого вещества с чистотой 62,7% (% по площади по данным ВЭЖХ; метод ВЭЖХ: С18-колонка для UPLC (сверхэффективная жидкостная хроматография) Aquity ВЕН130, 150×2,1 мм; подвижная фаза, А: 0,05% TFA в воде, В: 0,05% TFA в MeCN; скорость потока: 0,13 мл/мин в течение 40 мин; 0,25 мл/мин в течение 15 мин; изократический режим 90/10 (А/В) в течение 3 мин, градиент от 90/10 (А/В) до 62/38 (А/В) в пределах 37 мин, градиент от 62/38 (А/В) до 10/90 (А/В) в пределах 5 мин, изократический режим 10/90 (А/В) в течение 10 мин. Темп.: 60°С, УФ: 214 нм). Соотношение продукт 1/димер составляло 21,9.
Время удерживания: 23,2 мин (c[Gly-Tyr-Ile-Gln-Asn-Glu]-Gly-Leu-Gly-NH2), 18,8 мин (H-Gly-Tyr-Ile-Gln-Asn-Glu-Gly-Leu-Gly-NH2), 26,1 мин (димер).
g) Очистка и выделение
Неочищенный продукт c[Gly-Tyr-Ile-Gln-Asn-Glu]-Gly-Leu-Gly-NH2 растворяли в смеси воды и MeCN (10:1) и фильтровали. Фильтрат разбавляли, используя тот же объем воды. Раствор очищали препаративной ВЭЖХ на колонке Kromasil-C18-100 (250×80 мм, размер частиц 10 мкм, А: 0,1% TFA-вода, В: MeCN; скорость потока: 300 мл/мин; изократический режим 95/5 (А/В) в течение 2 мин, градиент от 95/5 (А/В) до 80/20 (А/В) в пределах 1 мин, градиент от 80/20 (А/В) до 77/23 (А/В) в пределах 17 мин, градиент от 77/23 (А/В) до 10/90 (А/В) в пределах 1 мин, изократический режим 10/90 (А/В) в течение 7 мин, градиент от 10/90 (А/В) до 95/5 (А/В) в пределах 1 мин, изократический режим 95/5 (А/В) в течение 6 мин. Фракции собирали и лиофилизировали, получая чистый продукт c[Gly-Tyr-lle-Gln-Asn-Glu]-Gly-Leu-Gly-NH2 1 (0,708 г) в виде белого порошка с чистотой 99,2% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1). В чистом продукте 1 никакого димера не обнаруживали. MS (m/z): 931,0 (М+Н)+.
Пример 2 (циклизация в жидкой Фазе)

Схема 4:
a) Отщепление Fmoc
В реактор для SPPS (100 мл) загружали смолу PL-Rink (загрузка 0,55 ммоль/г; 5,00 г; 2,75 ммоль) и 20%-ный пиперидин в ДМФА (50,0 мл). Затем эту смесь перемешивали при 25°С в течение 10 мин. После слива растворителя добавляли другую порцию 20%-ного пиперидина в ДМФА (50,0 мл) и смесь перемешивали при 25°С в течение 30 мин. После слива растворителя полученную смолу промывали ДМФА (8×50,0 мл), получая PL-Rink смолу без Fmoc группы.
b) Связывание Fmoc-производных аминокислот
К PL-Rink смоле без Fmoc группы добавляли раствор Fmoc-Gly-OH в смеси 0,35 М HOBt/ДМФА (32,0 мл; 11,2 ммоль), 0,92 М DIC в ДМФА (16,0 мл; 14,7 ммоль) и 10%-ный пиридин в ДМФА (16,0 мл; 19,8 ммоль) и перемешивали при 25°С в течение 3 ч. После слива растворителя полученную смолу промывали ДМФА (4×50,0 мл), получая Fmoc-Gly-смолу.
Стадии отщепления Fmoc и связывания с Fmoc-производными аминокислот повторяли 8 раз, используя вместо Fmoc-Gly-OH следующие Fmoc-производные аминокислот: Fmoc-Leu-OH, Fmoc-Gly-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH, с получением продукта X (Fmoc-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(tBu)-Pro-Leu-Gly-смолы). Образец отщепляли от смолы (см. ниже) для подтверждения правильности массы. MS (m/z): 1171,8 (М+Н)+.
c) Отщепление Fmoc
Отщепление Fmoc от концевого остатка Gly проводили так, как описано выше. После слива растворителя полученную смолу промывали ДМФА (8×50,0 мл), CH2Cl2 (3×50,0 мл) и МеОН (3×50,0 мл). Смолу сушили при давлении 10 мбар (1 кПа) при 25°С в течение 1 суток, получая H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(tBu)-Gly-Leu-Gly-cмoлy (10,8 г).
d) Удаление всех защитных групп и отщепление от смолы
К предварительно охлажденному (10-15°С) раствору триизопропилсилана (2,50 мл) в TFA (40,0 мл) и воде (10,0 мл) добавляли H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(tBu)-Gly-Leu-Gly-cмoлy (10,8 г) и перемешивали при 25°С в течение 3 ч. Смолу отфильтровывали и фильтрат концентрировали в вакууме. Остаток добавляли к MeTHF (100 мл) и смесь перемешивали при 25°С в течение 15 ч. Смесь фильтровали и осадок на фильтре промывали MeTHF (50,0 мл), затем сушили, получая LP1 (H-Gly-Tyr-Ile-Gln-Asn-Glu-Gly-Leu-Gly-NH2) (3,60 г) в виде белого твердого вещества с чистотой 67,4% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1). MS (m/z): 949,7 (М+Н)+.
e) Циклизация в растворе
К смеси LP1 (H-Gly-Tyr-Ile-Gln-Asn-Glu-Gly-Leu-Gly-NH2) (3,50 г) в NEP (60,0 мл) и DIPEA (3,13 мл; 18,4 ммоль) добавляли PyBOP (1,92 г; 3,69 ммоль) и перемешивали при 25°С в течение 1 ч. Для полного превращения добавляли другую порцию PyBOP (0,960 г; 1,84 ммоль) и перемешивали при той же температуре в течение 1 ч. Полученную смесь добавляли к раствору MTBE/MeTHF (400 мл/100 мл) и перемешивали при 25°С в течение 15 ч. Смесь фильтровали и осадок на фильтре промывали МТВЕ (50,0 мл), затем сушили, получая неочищенный продукт c[Gly-Tyr-Leu-Gln-Asn-Glu]-Gly-Leu-Gly-NH2 1 (4,30 г; результаты анализа: 18,0 масс. %, общий выход 31%) в виде белого твердого вещества с чистотой 56,6% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1). Соотношение продукт 1/димер составляло 15,1.
f) Очистка и выделение
Неочищенный продукт c[Gly-Tyr-Leu-Gln-Asn-Glu]-Gly-Leu-Gly-NH2 растворяли в смеси воды и MeCN (10:1) и отфильтровывали нерастворившийся материал. Фильтрат разбавляли, используя тот же объем воды. Раствор очищали препаративной ВЭЖХ на колонке Kromasil-C18-100 (250×80 мм, размер частиц 10 мкм, А: 0,1% TFA-вода, В: MeCN; скорость потока: 300 мл/мин; изократический режим 95/5 (А/В) в течение 2 мин, градиент от 95/5 (А/В) до 80/20 (А/В) в пределах 1 мин, градиент от 80/20 (А/В) до 77/23 (А/В) в пределах 17 мин, градиент от 77/23 (А/В) до 10/90 (А/В) в пределах 1 мин, изократический режим 10/90 (А/В) в течение 7 мин, градиент от 10/90 (А/В) до 95/5 (А/В) в пределах 1 мин, изократический режим 95/5 (А/В) в течение 6 мин. Фракции собирали и лиофилизировали, получая чистый продукт c[Gly-Tyr-Leu-Gln-Asn-Glu]-Gly-Leu-Gly-NH2 1 (444 мг) в виде белого порошка с чистотой 99,7% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1). В чистом продукте 1 присутствия димера не обнаружено. MS (m/z): 931,0.
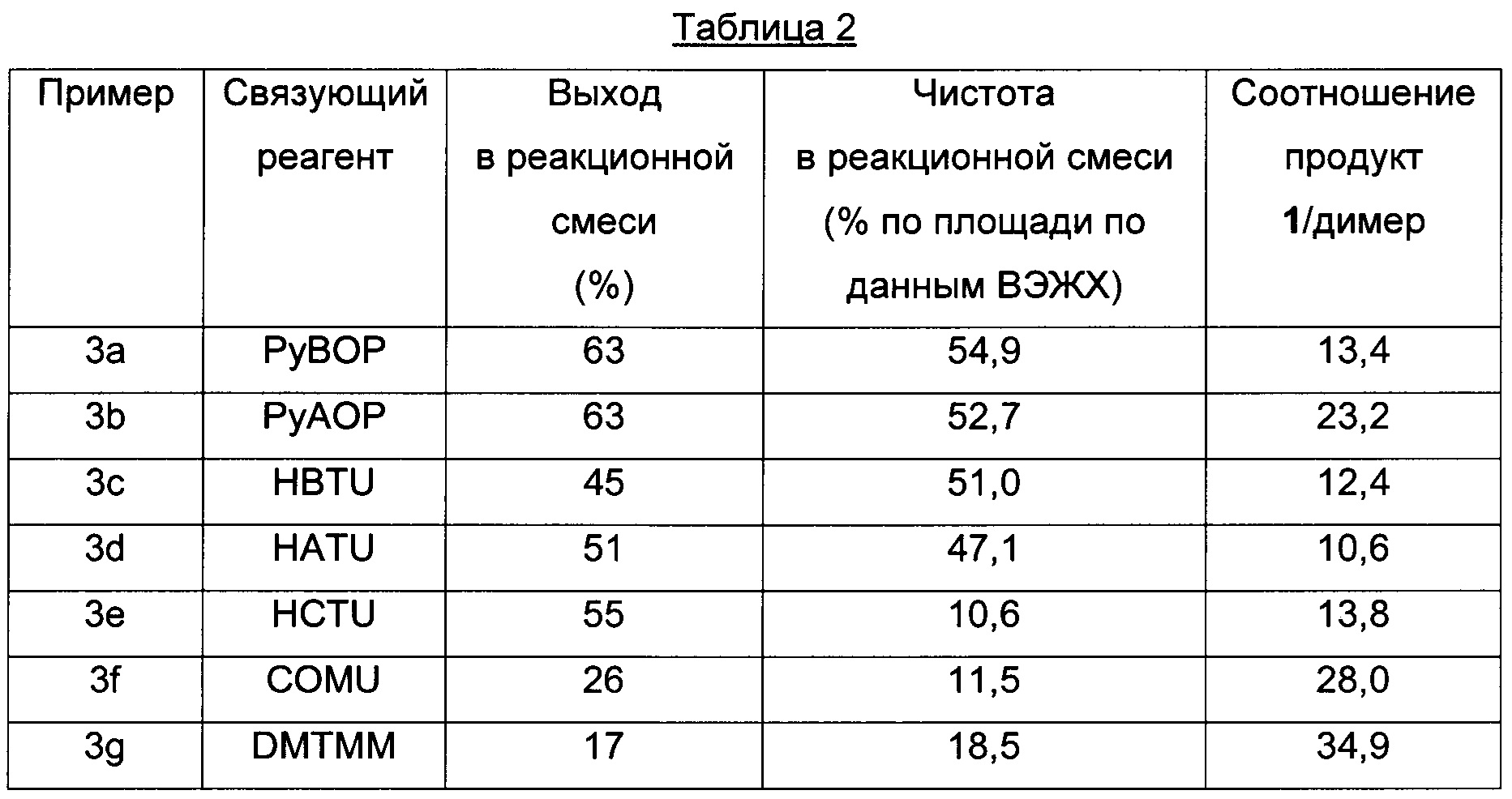
Пример 3 a-g (оптимизация связующих реагентов)

Способом, аналогичным таковому в примере 2, стадии циклизации осуществляли, используя связующие реагенты, приведенные в Таблице 2.
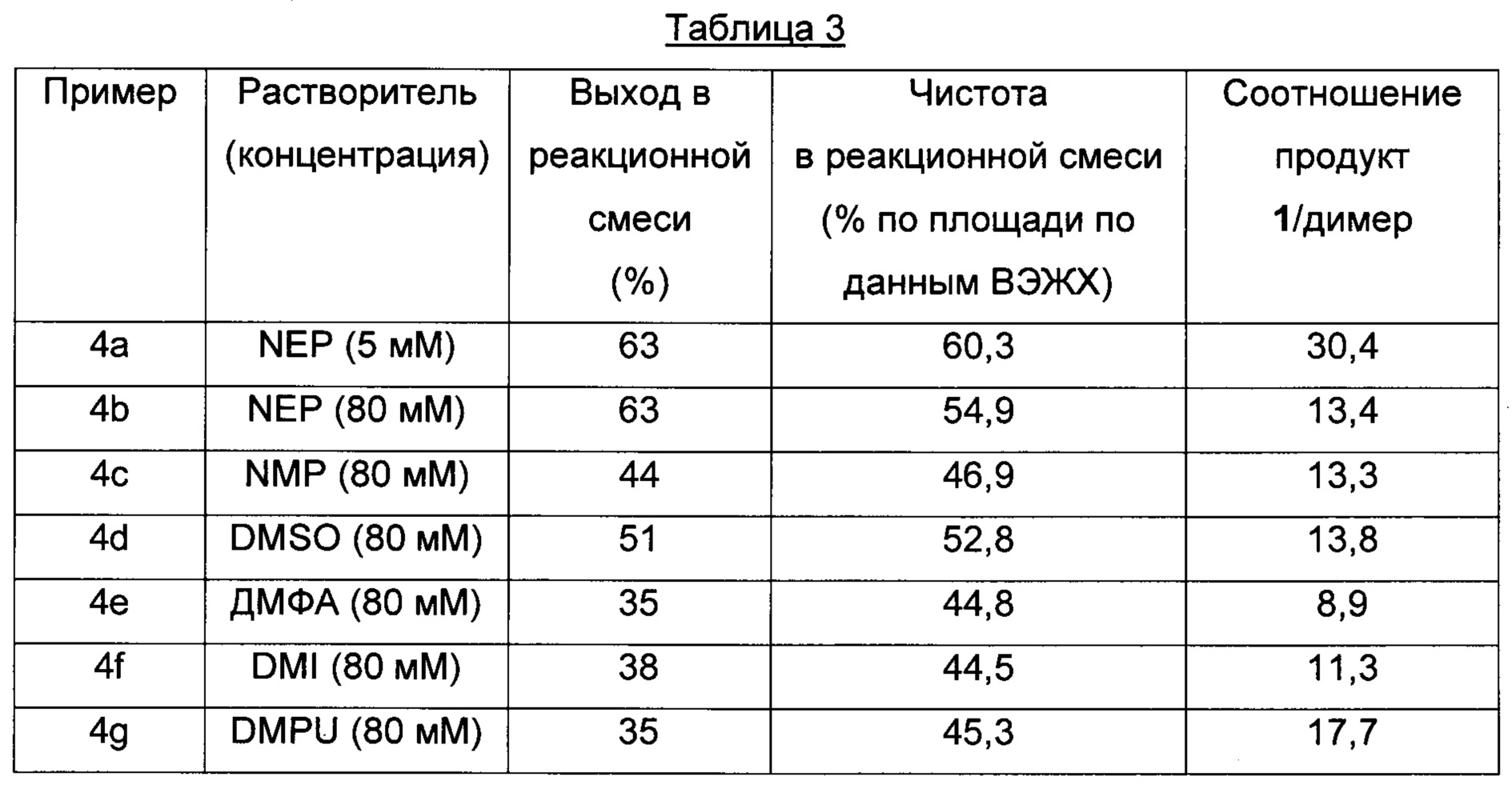
Пример 4 a-g (оптимизация растворителей)

Способом, аналогичным таковому в примере 2, стадии циклизации осуществляли, используя растворители, приведенные в Таблице 3.
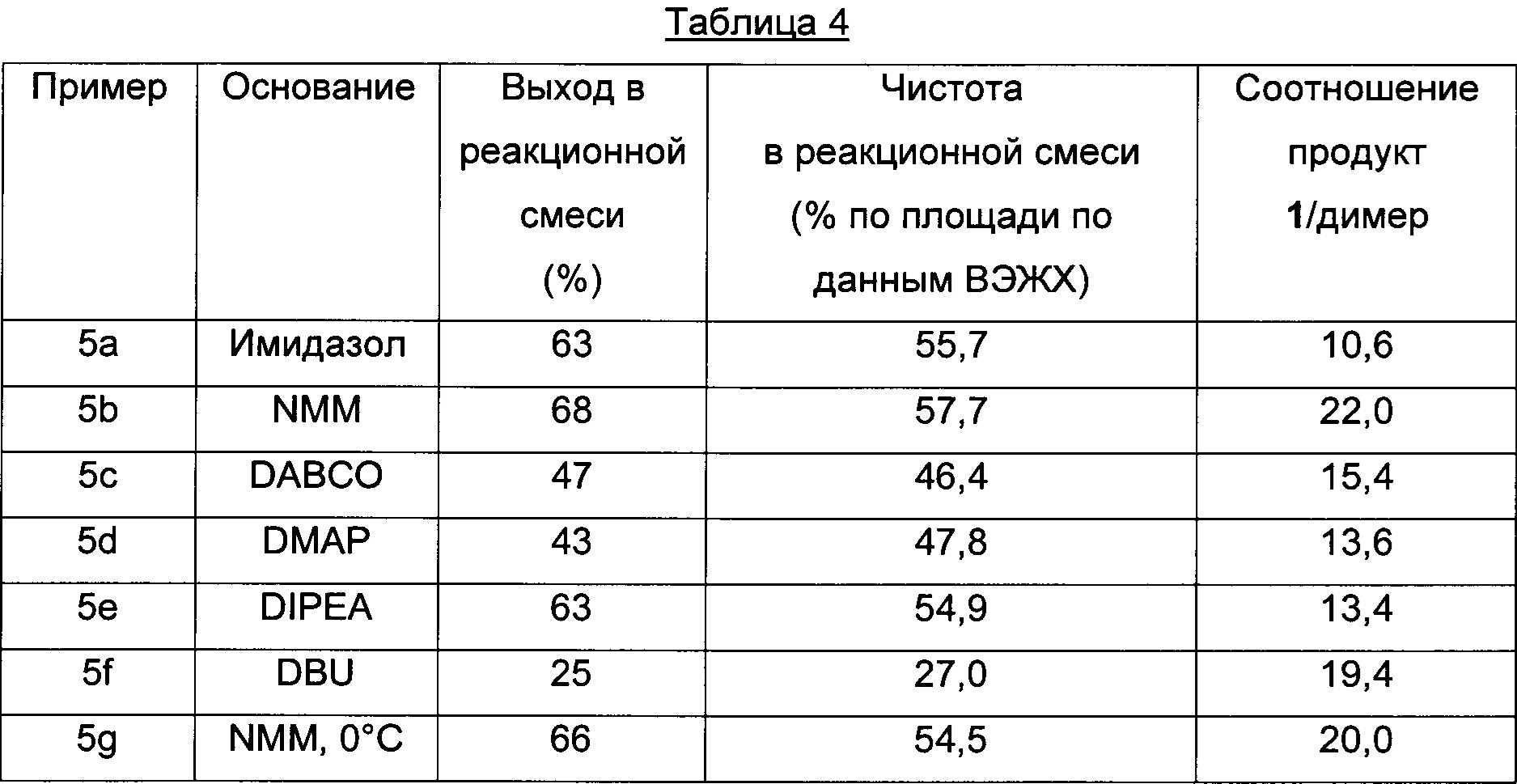
Пример 5 a-g (оптимизация оснований)
Способом, аналогичным таковому в примере 2, стадии циклизации осуществляли, используя основания, приведенные в Таблице 4.
Пример 6 a-d (сравнение нагрузки смолы/ эквивалентов аминокислоты)

Схема 5:
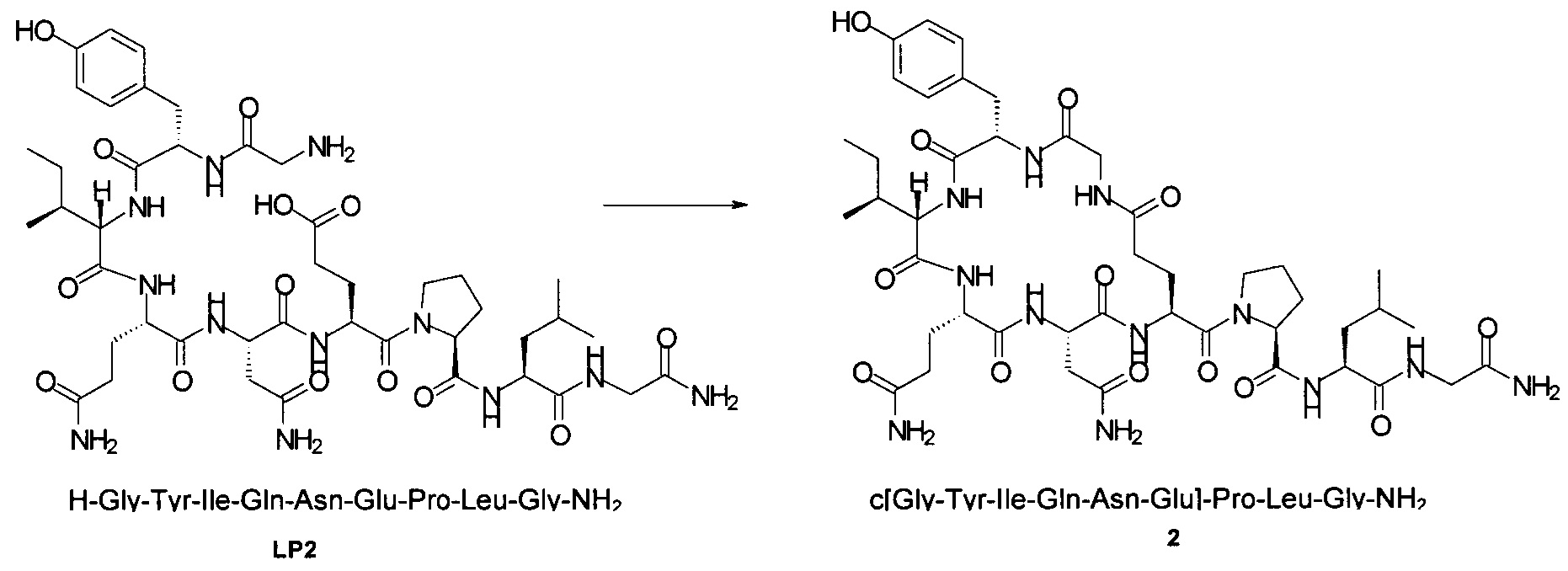
Способом, аналогичным таковому в примере 2, синтезировали чистый циклический пептид 2, используя следующие Fmoc-производные аминокислот: Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-Pro-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH.
Масштаб синтеза: 9,60 ммоль (нагрузка: см. пример 6а-d; смола 30,0 г).
Выход: 40% (после очистки).
Чистота: 98,2% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1).
Время удерживания: 29,8 мин (метод ВЭЖХ см. в примере 1).
MS (m/z): 971,5 (М+Н)+.
Чистоту и выход промежуточного линейного пептида LP2 (H-Gly-Tyr-Ile-Gln-Asn-Glu-Pro-Leu-Gly-NH2) определяли, используя нагрузку смолы/эквиваленты аминокислоты, которые приведены в Таблице 5.
Пример 7
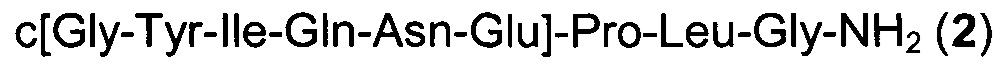
Получение продукта примера 7 осуществляли способом, аналогичным таковому в примере 2, за исключением того, что стадии циклизации осуществляли, используя в качестве основания N-метилморфолин.
a) Отщепление Fmoc
В реактор для SPPS (250 мл; пептидный синтезатор CS536XT от CSBio) загружали смолу PL-Rink (загрузка 0,55 ммоль/г; 10,0 г; 5,5 ммоль) и 20%-ный пиперидин в ДМФА (100,0 мл). Затем эту смесь перемешивали при 25°С в течение 10 мин. После слива растворителя добавляли другую порцию 20%-ного пиперидина в ДМФА (100,0 мл) и смесь перемешивали при 25°С в течение 30 мин. После слива растворителя полученную смолу промывали ДМФА (8×100,0 мл), получая PL-Rink смолу без Fmoc группы.
b) Связывание Fmoc-производных аминокислот
К PL-Rink смоле без Fmoc группы добавляли раствор Fmoc-Gly-OH в смеси 0,35 М HOBt/ДМФА (64,0 мл; 22,4 ммоль), 0,92 М DIC в ДМФА (32,0 мл; 29,4 ммоль) и 10%-ный пиридин в ДМФА (32,0 мл; 39,6 ммоль) и перемешивали при 25°С в течение 3 ч. После слива растворителя полученную смолу промывали ДМФА (4×100,0 мл), получая Fmoc-Gly-смолу.
Стадии отщепления Fmoc и связывания с Fmoc-производными аминокислот повторяли 8 раз, используя вместо Fmoc-Gly-OH следующие Fmoc-производные аминокислот: Fmoc-Leu-OH, Fmoc-Pro-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH, с получением Fmoc-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(tBu)-Pro-Leu-Gly-cмoлы. Образец отщепляли от смолы (разбавитель см. ниже) для подтверждения правильности массы. MS (m/z): 1211,1 (М+Н)+.
c) Отщепление Fmoc
Отщепление Fmoc от концевого остатка Gly проводили так, как описано выше. После слива растворителя полученную смолу промывали ДМФА (8×100 мл), CH2Cl2 (3×100 мл) и МеОН (3×100 мл). Смолу сушили при давлении 10 мбар (1 кПа) при 25°С в течение 1 суток, получая H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(tBu)-Pro-Leu-Gly-смолу (18,6 г). Образец отщепляли от смолы (разбавитель см. ниже) для подтверждения правильности массы. MS (m/z): 989,7 (М+Н)+.
d) Удаление всех защитных групп и отщепление от смолы
К предварительно охлажденному (10-15°С) раствору триизопропилсилана (3,00 мл) в TFA (48,0 мл) и воде (12,0 мл) добавляли H-Gly-Tyr(tBu)-Ile-Gln(Trt)-Asn(Trt)-Glu(tBu)-Pro-Leu-Gly-смолу (6,00 г) и перемешивали при 25°С в течение 3 ч. Смолу отфильтровывали и фильтрат концентрировали в вакууме. Остаток добавляли к MeTHF (120 мл) и смесь перемешивали при 25°С в течение 15 ч. Смесь фильтровали и осадок на фильтре промывали MeTHF (60,0 мл), затем сушили, получая H-Gly-Tyr-Ile-Gln-Asn-Glu-Pro-Leu-Gly-NH2 LP2 (1,84 г) в виде белого твердого вещества с чистотой 87,3% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1). Время удерживания: 23,9 мин (метод ВЭЖХ см. в примере 1); MS (m/z): 989,7 (М+Н)+.
е) Циклизация в растворе
К смеси H-Gly-Tyr-Ile-Gln-Asn-Glu-Pro-Leu-Gly-NH2 LP2 (300 мг) в N-этилпирролидоне (3,60 мл) и NMM (0,167 мл; 1,52 ммоль) добавляли PyBOP (237 мг; 0,455 ммоль) и перемешивали при 25°С в течение 1 ч. Для полного превращения добавляли другую порцию PyBOP (47,4 мг; 0,0910 ммоль) и перемешивали при той же температуре в течение 1 ч. Полученную смесь добавляли к раствору МТВЕ (24,0 мл) и MeTHF (6,00 мл) и затем перемешивали при 25°С в течение 15 ч. Смесь фильтровали и осадок на фильтре промывали МТВЕ (15,0 мл). Осадок на фильтре растворяли в воде/MeCN (10/1, 3,3 мл) и отфильтровывали нерастворившиеся материалы. Фильтрат лиофилизировали, получая неочищенный продукт c[Gly-Tyr-Leu-Gln-Asn-Glu]-Pro-Leu-Gly-NH2 2 (313 мг; результаты анализа: 54,0 масс. %, общий выход 60%) в виде белого твердого вещества с чистотой 71,4% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1). MS (m/z): 971,5 (М+Н)+.
Пример 8

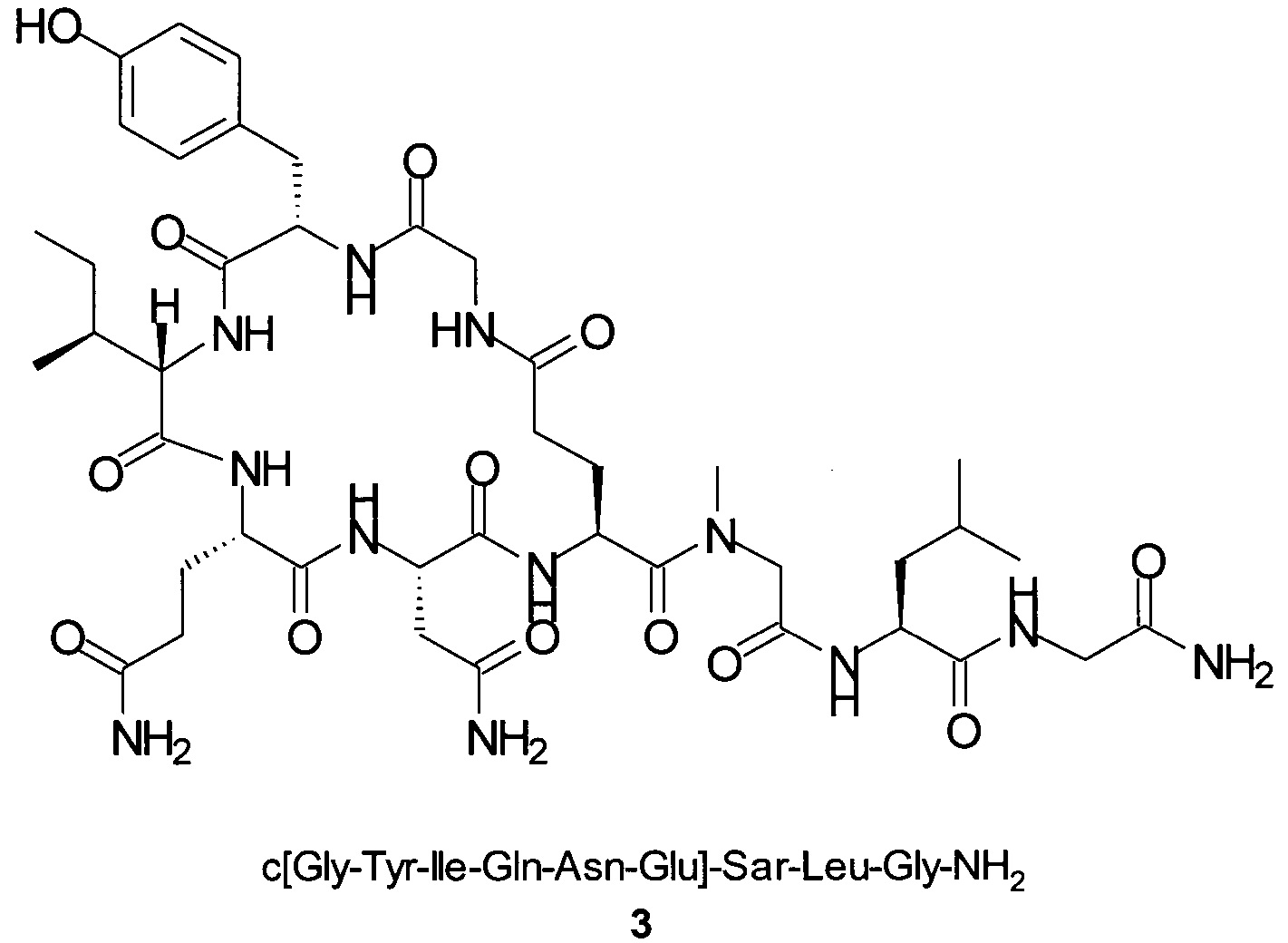
Способом, аналогичным таковому в примере 2, синтезировали чистый циклический пептид 3, используя следующие Fmoc-производные аминокислот: Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-Sar-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH.
Масштаб синтеза: 9,60 ммоль (загрузка: 0,32 ммоль/г, смола 30,0 г).
Выход: 41% (после очистки).
Чистота: 98,9% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1).
Время удерживания: 27,6 мин (метод ВЭЖХ см. в примере 1).
MS (m/z): 945,5 (М+Н)+.
Пример 9

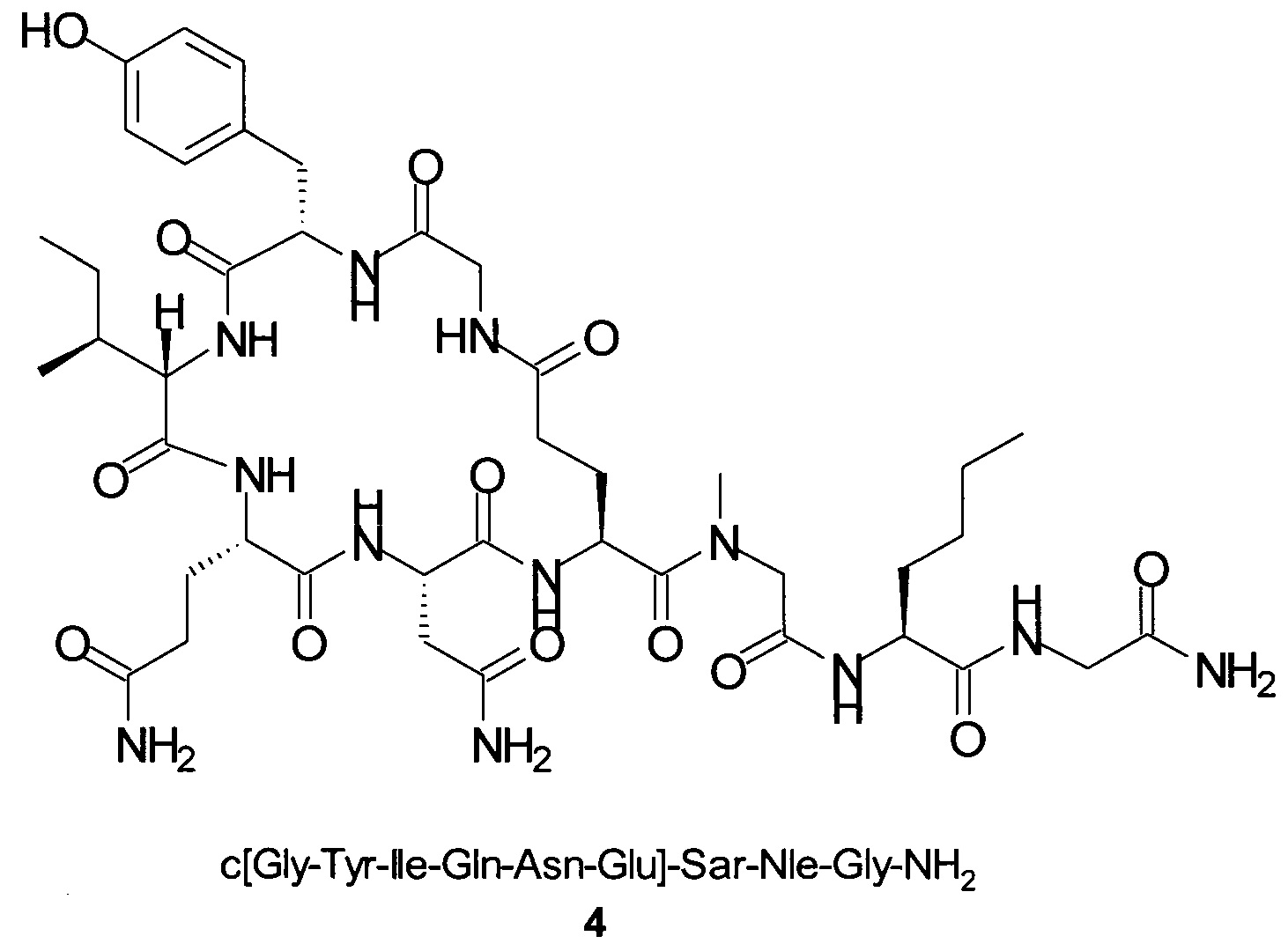
Способом, аналогичным таковому в примере 2, синтезировали чистый циклический пептид 4, используя следующие Fmoc-производные аминокислот: Fmoc-Gly-OH, Fmoc-Nle-OH, Fmoc-Sar-OH, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH.
Масштаб синтеза: 9,60 ммоль (загрузка 0,32 ммоль/г, смола 30,0 г).
Выход: 41% (после очистки).
Чистота: 99,2% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1).
Время удерживания: 25,9 мин (метод ВЭЖХ см. в примере 1).
MS (m/z): 945,5 (М+Н)+.
Пример 10
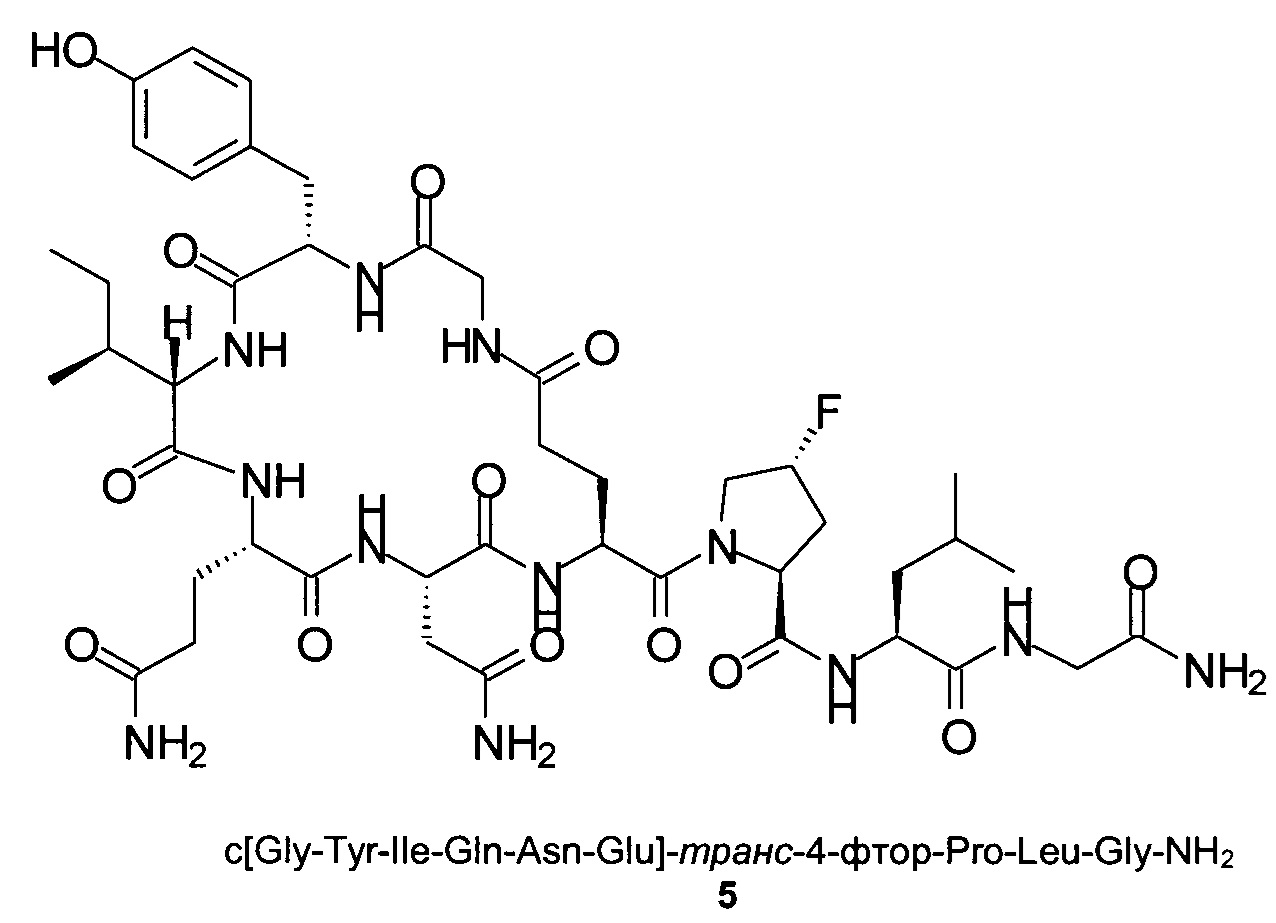
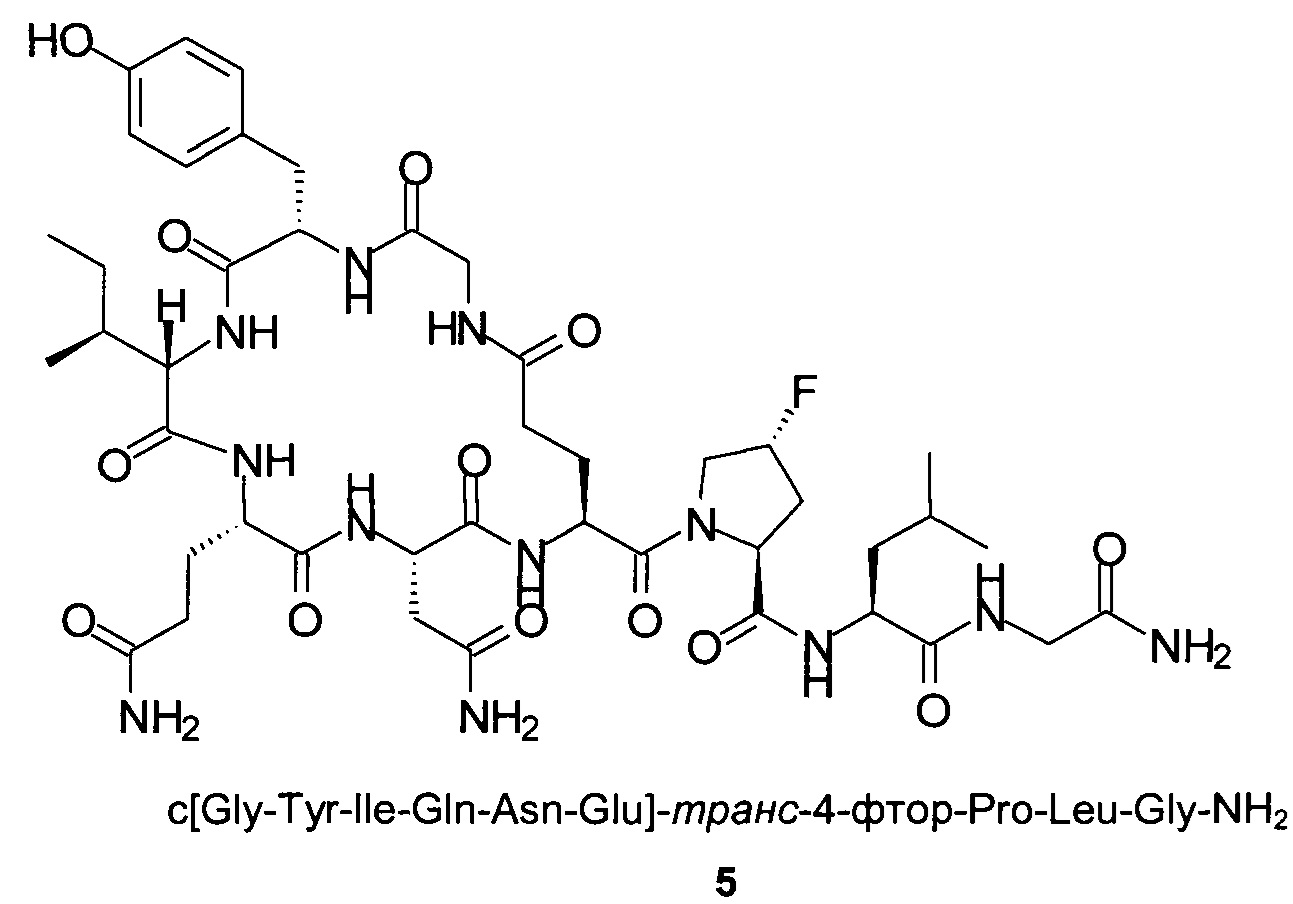
Способом, аналогичным таковому в примере 2, синтезировали чистый циклический пептид 5, используя следующие Fmoc-производные аминокислот: Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-транс-4-фтор-Pro-ОН, Fmoc-Glu(tBu)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH.
Масштаб синтеза: 9,60 ммоль (загрузка 0,32 ммоль/г, смола 30,0 г).
Выход: 39% (после очистки).
Чистота: 98,8% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1).
Время удерживания: 25,7 мин (метод ВЭЖХ см. в примере 1).
MS (m/z): 988,5 (М+Н)+.
Пример 11
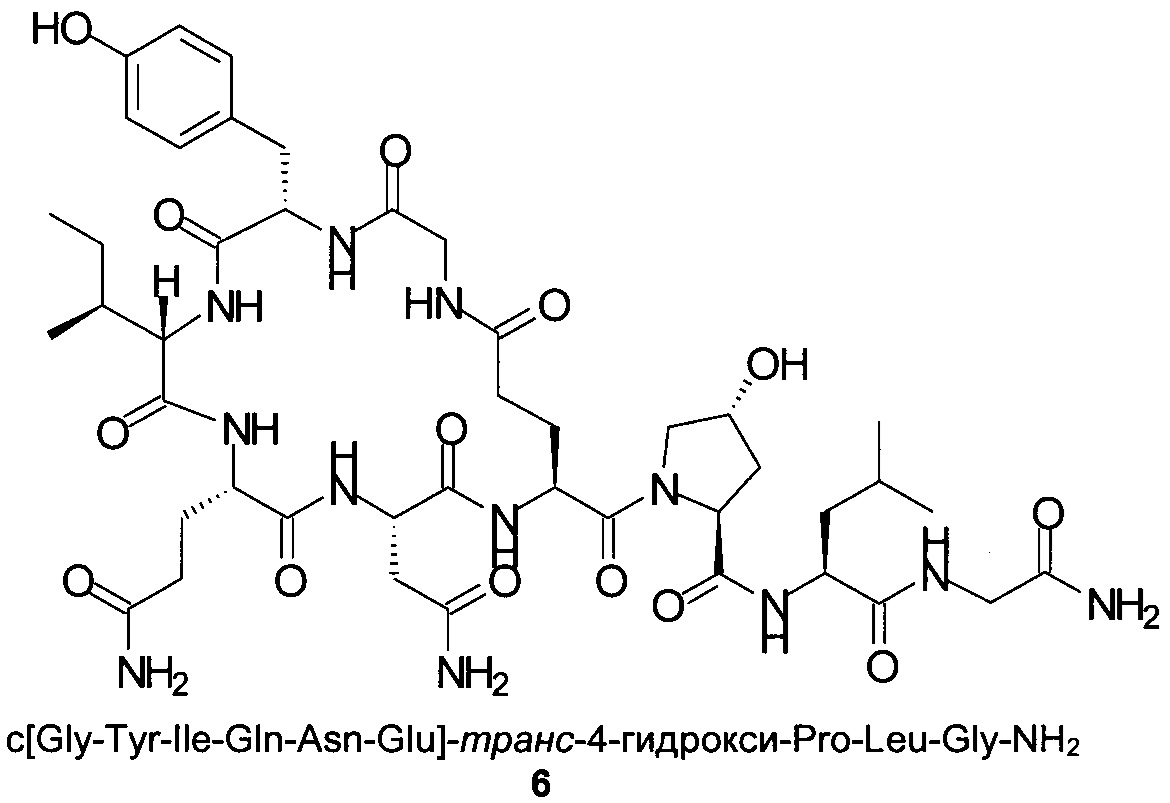
Способом, аналогичным таковому в примере 2, синтезировали чистый циклический пептид 6, используя следующие Fmoc-производные аминокислот: Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-транс-4-трет-бутокси-Pro-ОН, Fmoc-Glu(tBu)-ОН, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH.
Масштаб синтеза: 9,60 ммоль (загрузка 0,32 ммоль/г, смола 30,0 г). Выход: 22% (после очистки).
Чистота: 98,7% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1).
Время удерживания: 23,3 мин (метод ВЭЖХ см. в примере 1). MS (m/z): 987,5 (М+Н)+.
Пример 12 (циклизация на твердой Фазе)
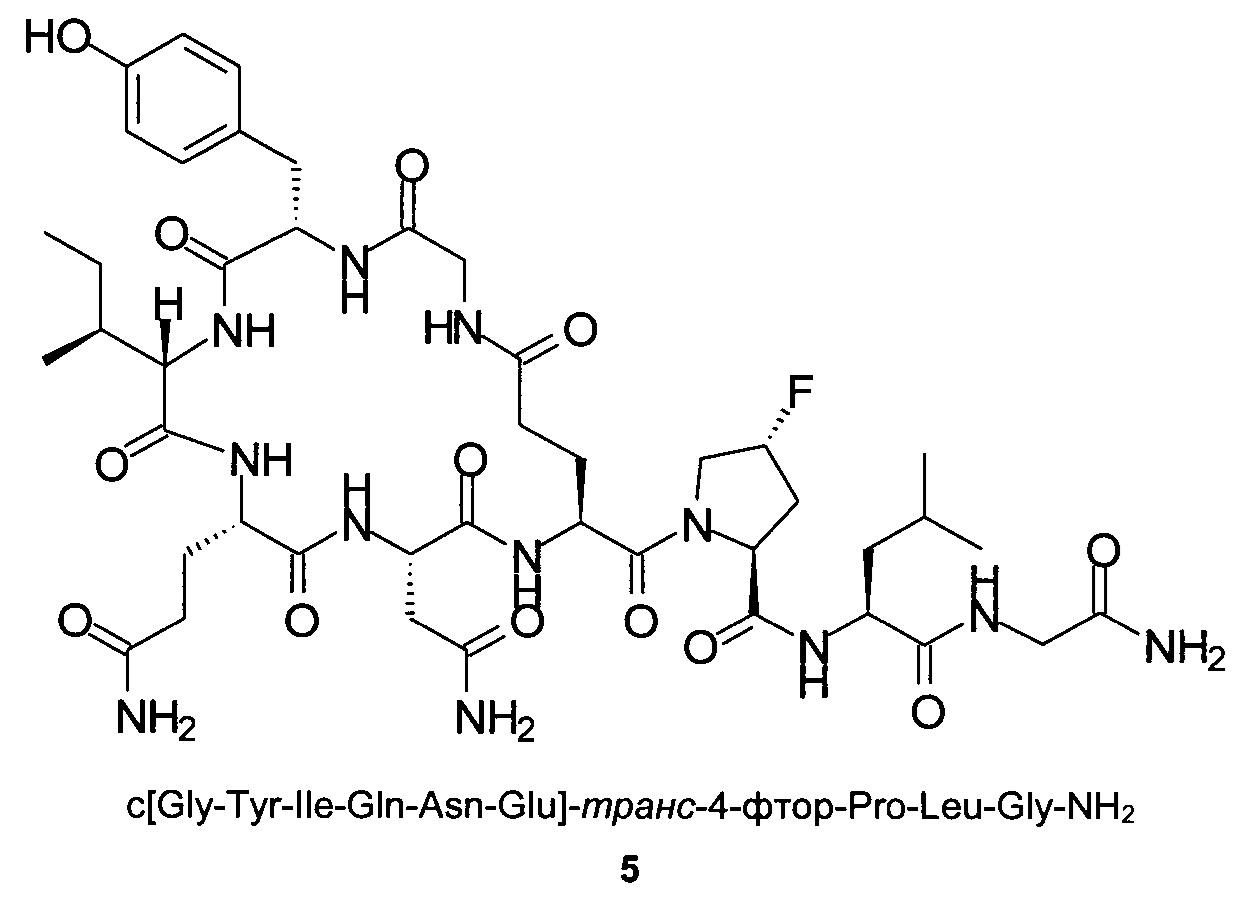
Способом, аналогичным таковому в примере 1 с использованием пептидного синтезатора CS536XT от CSBio, синтезировали чистый циклический пептид 5, используя следующие Fmoc-производные аминокислот: Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-транс-4-фтор-Pro-ОН, Fmoc-Glu(OAll)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH, Fmoc-Gly-OH. На протяжении всего синтеза, для отщепления Fmoc использовали 10%-ный раствор 4-метил-пиперидина в ДМФА вместо 20%-ного пиперидина в ДМФА и все реакции связывания аминокислот с образованием линейной последовательности проводили, используя HOPy вместо HOBt. На конечной PyBOP-стимулируемой стадии циклизации на смоле в качестве основания использовали 4-метилморфолин вместо DIPEA и циклизацию проводили, используя в качестве растворителя ДМФА вместо NEP. Очистку неочищенного продукта c[Gly-Tyr-Ile-Gln-Asn-Glu]-транс-4-фтор-Pro-Leu-Gly-NH2 препаративной ВЭЖХ проводили на колонке Kromasil-C18-100 (250×4,6 мм, размер частиц 10 мкм, А: 20 мМ NH4OAc, рН 5, В: MeCN; скорость потока: 1 мл/мин; изократический режим 90/10 (А/В) в течение 1 мин, градиент от 90/10 (А/В) до 80/20 (А/В) в пределах 1 мин, градиент от 80/20 (А/В) до 75/25 (А/В) в пределах 10 мин, градиент от 75/25 (А/В) до 10/90 (А/В) в пределах 1 мин, градиент от 10/90 (А/В) до в течение 5 мин, градиент от 10/90 (А/В) до 90/10 (А/В) в пределах 0,1 мин, изократический режим 90/10 (А/В) в течение 6,9 мин. Собранные фракции разбавляли водой (1:1) и концентрировали/обессоливали, нанося на предварительно обработанную (смесью вода/ACN (ацетонитрил), 90/10) колонку Kromasil С18-100-10 (250×4,6 мм), и после этого элюировали смесью вода/ACN (1:1). Собранные фракции (УФ 280 нм, порог 1000 миллиединиц оптической плотности (mAU; от milli Absorbance Unit) упаривали на роторном испарителе для удаления ACN и после этого лиофилизировали, получая чистый пептид в виде белого лиофилизированного продукта.
Масштаб синтеза: 5,50 ммоль (загрузка 0,55 ммоль/г, смола 10,0 г).
Выход: 34% (после очистки).
Чистота: 98,8% (% по площади по данным ВЭЖХ; метод ВЭЖХ см. в примере 1).
Время удерживания: 25,3 мин (метод ВЭЖХ см. в примере 1).
MS (m/z): 989,5 (М+Н)+.
Пример 13 (циклизация на твердой фазе)
Способом, аналогичным таковому в примере 13, синтезировали чистый циклический пептид 5, используя HOBt вместо HOPy на протяжении всего синтеза линейного пептида на смоле.
Масштаб синтеза: 5,50 ммоль (загрузка 0,55 ммоль/г, смола 10,0 г).
Выход: 25% (после очистки).
Препарат антитела
Производные гетероарилпирролидинил- и пиперидинилкетона
Производные изоксазоло-пиридина
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Ингибиторы jnk
Бензопирановые и бензоксепиновые ингибиторы рi3k и их применение
Арилциклогексилэфиры дигидротетраазабензоазуленов для применения в качестве антагонистов рецептора вазопрессина v1a
Пуриновые соединения, ингибирующие рi3к, и способы применения
Алкилциклогексиловые эфиры дигидротетраазабензоазуленов
Способ синтеза производных амино-метилтетралина
Производные имидазопиридина или имидазопиримидина в качестве ингибиторов фосфодиэстеразы 10а
Азотосодержащие производные гетероарилов
Производные n-(имидазопиримидин-7-ил)-гетероариламидов и их применение в качестве ингибиторов pde10a
Циклопентил- и циклогептилпиразолы в качестве модуляторов fxr
Способ получения производных 2-фенил[1,2,4]триазоло[1,5-а]пиридина
Производные дипептида лизин-глутаминовая кислота
Катализируемое палладием сочетание пиразоламидов

