Результат интеллектуальной деятельности: Способ получения комплекса физиологически активного полипептида
Вид РИД
Изобретение
Область изобретения
Настоящее изобретение относится к способу получения конъюгата физиологически активного полипептида и непептидного полимера путем связывания физиологически активного полипептида с непептидным полимером ковалентной связью с использованием органического растворителя и получения комплекса физиологически активного полипептида путем связывания конъюгата с носителем для повышения продолжительности действия in vivo и стабильности физиологически активного полипептида.
Предшествующий уровень техники
Обычно физиологически активные пептиды легко подвержены денатурации из-за их низкой стабильности и разрушаются гидролазами белков для их быстрого выведения через почки или печень. Поэтому для поддержания концентрации в крови и активности белковых лекарственных средств, содержащих физиологически активный полипептид в качестве фармакологического активного ингредиента, необходимо частое введение белкового лекарственного средства пациентам. Однако, в случае белковых лекарственных средств, вводимых пациентам преимущественно в форме композиций для инъекций, частые инъекции для поддержания концентрации активных полипептидов в крови могут приводить к излишним страданиям для пациентов. Для решения таких проблем постоянно предпринимаются попытки максимизировать фармакологическую эффективность путем повышения стабильности белковых лекарственных средств в крови и поддержания концентрации лекарственных средств в крови на протяжении более длительного времени. Такая постоянная разработка композиций необходима для повышения стабильности белковых лекарственных средств и, в то же время, поддержания активности самих лекарственных средств на достаточно высоком уровне, а также для предотвращения иммунных реакций у пациентов.
В предшествующем уровне техники для стабилизации белков и подавления контакта с гидролазой белков и выведения через почки использовали способ химического присоединения полимеров с высокой растворимостью, таких как полиэтиленгликоль (называемый далее PEG), к поверхности белковых лекарственных средств. Известно, что PEG эффективно стабилизируют белки и предотвращают гидролиз белков посредством неспецифического связывания PEG с определенным сайтом или различными сайтами белка-мишени, повышая растворимость белка и не приводя ни к каким нежелательным побочным эффектам (Sada et al., J. Fermentation Bioengineering 71: 137-139, 1991). Тем не менее, несмотря на то, что такое связывание с РЕО может повышать стабильность белков, оно приводит к значительному снижению активности физиологически активного полипептида, и реакционная способность РЕО в отношении белков снижается по мере увеличения молекулярной массы РЕО, снижая выход связывания. Поэтому авторами настоящего изобретения предложен конъюгат, полученный путем связывания физиологически активного полипептида и носителя с непептидным полимером. Тем не менее, потребность в способе получения конъюгата с высоким выходом и чистотой выросла, а недостатком предшествующего способа, где сначала проводят конъюгацию носителя с непептидным полимером, является его высокая стоимость.
Инсулин, тем временем, является полипептидом, секретируемым бета-клетками человеческой поджелудочной железы в качестве вещества, играющего очень важную роль в контролировании уровня глюкозы в крови в организме. В случаях, когда секреция инсулина не происходит должным образом, или когда секретированный инсулин не оказывает должного воздействия в организме, уровень глюкозы в крови не может контролироваться в организме и повышается, тем самым приводя к состоянию, называемому диабетом. Случай, описанный выше, называют сахарным диабетом 2 типа, а случай повышения уровня глюкозы в крови из-за отсутствия секреции инсулина из поджелудочной железы называют сахарным диабетом 1 типа.
Сахарный диабет 2 типа лечат пероральным гипогликемическим агентом, содержащим химическое вещество в качестве основного компонента, а некоторых пациентов также лечат инсулином. С другой стороны, для лечения диабета 1 типа необходимо введение инсулина.
Терапия инсулином, широко используемая в настоящее время, представляет собой способ введения инсулина посредством инъекции до и после приема пищи. Однако для такой терапии инсулином его необходимо вводить постоянно три раза в сутки, что приводит к значительным страданиям и неудобству. Было предпринято множество попыток преодоления таких проблем. Одна из них заключалась в попытке доставлять пептидные лекарственные средства в организм ингаляционным путем через полости рта или носа посредством повышения проницаемости биологических мембран для пептидных лекарственных средств. Однако, эффективность доставки в организм таким способом значительно ниже по сравнению с инъекцией, поэтому до сих пор имеется много сложностей в поддержании активности пептидных лекарственных средств in vivo в требуемых условиях.
Также была предпринята попытка разработки способа задержки всасывания после подкожного введения избытка лекарственного средства. В соответствии с этим был предложен способ поддержания концентрации лекарственного средства в крови посредством всего лишь одного введения в сутки. Некоторые такие продукты одобрены в качестве лекарственных средств (например, Lantus, Sanofi-aventis) и применяются для пациентов в настоящее время. Было проведено исследование с модификацией инсулина жирными кислотами для усиления связывания инсулинового полимера и для увеличения продолжительности действия путем связывания с альбумином, присутствующим в месте введения и в крови, и лекарственные средства, изготовленные с использованием такого способа, одобрены в качестве лекарственных средств (Levemir, NovoNordisk). Однако, побочным эффектом таких способов является боль в месте введения, и, кроме того, проведение инъекций один раз в сутки все еще приводит к значительным неудобствам для пациентов.
Также постоянно предпринимаются попытки максимизировать эффект пептидных лекарственных средств путем повышения стабильности в крови и поддержания концентрации лекарственного средства в крови на высоком уровне в течение длительных периодов времени после всасывания пептидных лекарственных средств в организм. Такая постоянная разработка пептидных композиций необходима для повышения стабильности пептидных лекарственных средств и, в то же время, поддержания активности самих лекарственных средств на достаточно высоком уровне, а также для предотвращения иммунных реакций у пациентов. Такие композиции пептидных лекарственных средств были получены способом химического присоединения полимерного вещества с высокой растворимостью, такого как полиэтилен гликоль (PEG), к поверхности пептидов.
PEG являются эффективными в ингибировании выведения пептида через почки и предотвращения гидролиза посредством неспецифического связывания с определенным сайтом или различными сайтами пептида-мишени, увеличивая молекулярную массу пептида, и, кроме того, не приводят ни к каким нежелательным побочным эффектам. Например, в WO 2006/076471 описано конъюгирование натрийуретических пептидов В-типа (BNP), которые снижают артериальное давление путем активации продуцирования cGMP (циклического гуанозинмонофосфата) посредством связывания с NPR-A и поэтому используются в качестве агента для лечения застойной сердечной недостаточности, с PEG для поддержания биологической активности таких пептидов. В US 6924264 раскрыт способ увеличения продолжительности действия in vivo путем конъюгирования лизинового остатка эксендина-4 с PEG. Однако, такие способы могут увеличивать продолжительность действия пептидных лекарственных средств in vivo при увеличении молекулярной массы PEG, но им свойственна проблема значительного снижения активности пептидных лекарственных средств и снижения реакционной способности PEG в отношении пептидов, приводящая таким образом к снижению выхода по мере увеличения молекулярной массы.
В WO 02/46227 раскрыт слитый белок GLP-1 и эксендина-4 или их аналогов с человеческим сывороточным альбумином или фрагментом иммуноглобулина (Fc) посредством рекомбинантной генетической методики, и в US 6756480 раскрыт слитый белок паратиреоидного гормона (PTH) и его аналога с Fc. Раскрытые в указанных источниках способы позволяют преодолеть проблемы низкого выхода и неспецифичности пегилирования, но, вопреки ожиданиям, приводят к проблемам недостаточно большого увеличения периода полувыведения из крови и, в некоторых случаях, к низкому титру. С целью максимизации эффекта увеличения периода полувыведения из крови можно также использовать различные типы пептидных линкеров, но они могут индуцировать иммунологическую реакцию. Кроме того, существуют проблемы, заключающиеся в том, что в случаях использования пептидов, имеющих дисульфидные связи, таких как BNP, их применение затруднительно из-за высокой вероятности неправильного сворачивания, а в случаях присутствия искусственных аминокислотных остатков получение в генетически рекомбинантной форме невозможно.
Описание изобретения
Техническая задача
Авторы настоящего изобретения предприняли попытку разработать способ одновременной максимизации увеличения периода полувыведения из крови и поддержания in vivo активности физиологически активных полипептидов, содержащих инсулин. В результате, они обнаружили, что в случаях, когда физиологически активные полипептиды сначала связывают с непептидными полимерами в реакционном растворе, содержащем органический растворитель, можно снизить стоимость получения и получить конъюгат физиологически активных полипептидов и непептидных полимеров с высоким выходом и чистотой, а также установили тот факт, что при получении комплексов физиологически активных полипептидов с использованием такого конъюгата активность конъюгата in vivo поддерживается на высоком уровне, и эффективность увеличения периода полувыведения из крови улучшается в превосходящей степени по сравнению с известным способом слияния с сохранением рамки считывания, создав тем самым настоящее изобретение.
Техническое решение
Задачей настоящего изобретения является предложение способа получения конъюгата физиологически активного полипептида и непептидного полимера для увеличения периода полувыведения из крови при поддержании in vivo активности физиологически активного полипептида.
Также задачей настоящего изобретения является предложение способа получения комплекса физиологически активного полипептида путем ковалентного связывания носителя с конъюгатом физиологически активного полипептида и непептидного полимера.
Полезные эффекты изобретения
Способ по настоящему изобретению позволяет получить конъюгат физиологически активного полипептида и непептидного полимера с высокой чистотой и выходом, и комплекс физиологически активного полипептида, полученный этим способом, может обеспечить последующее снижение стоимости получения, поддержание активности in vivo при относительно высоком уровне и значительное увеличение периода полувыведения из крови, и, таким образом, может быть эффективно использован для разработки композиций физиологически активного полипептида с длительным высвобождением, которые могут расширить возможности пациента по введению лекарственного средства.
Описание графических материалов
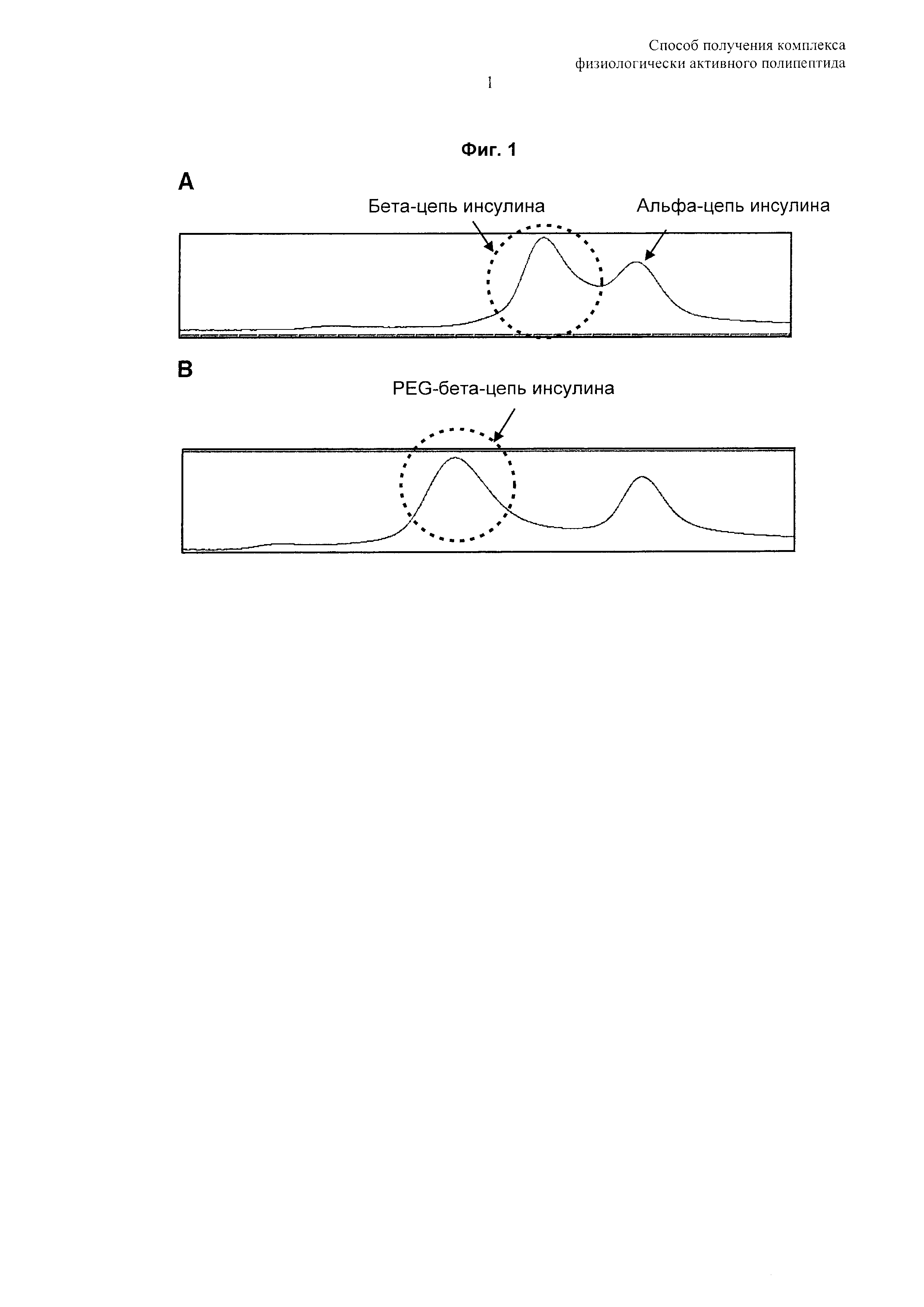
На Фиг. 1 показан результат анализа, подтверждающий модификацию бета-цепи конъюгата инсулин-PEG посредством эксклюзионной высокоэффективной хроматографии (SEC HPLC). А: восстанавливающие условия для инсулина, В: восстанавливающие условия для PEG-инсулина.
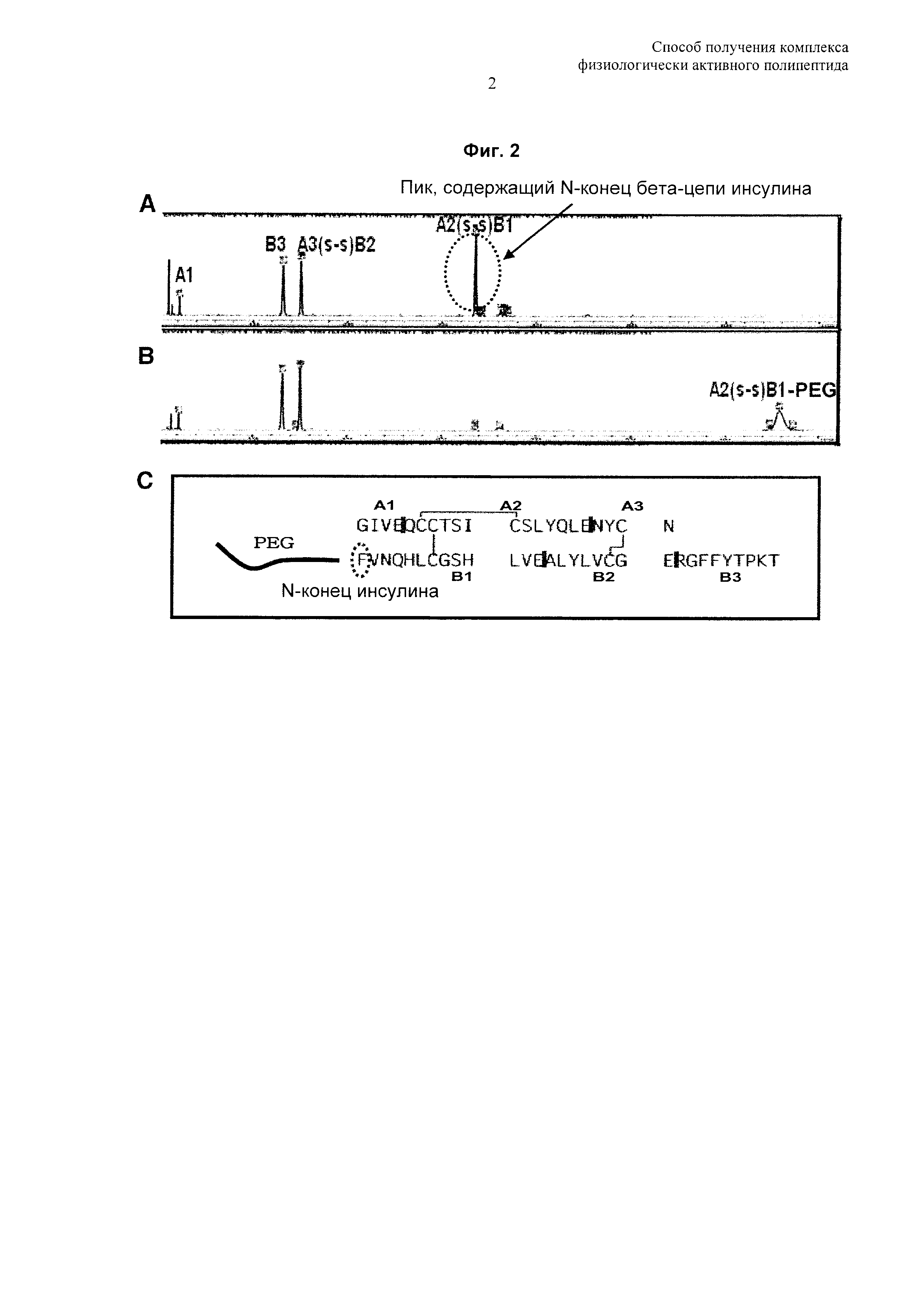
На Фиг. 2 показан результат анализа, подтверждающий модификацию фенилаланина №1 бета-цепи (B1F) конъюгата инсулин-PEG на 95% или более посредством пептидного картирования. А: результат пептидного картирования инсулина, В: результат пептидного картирования PEG-инсулина, С: последовательность PEG-инсулина и предполагаемые сайты расщепления при пептидном картировании (красная линия).
На Фиг. 3 показаны результаты ВЭЖХ с обращенной фазой (RP-HPLC), SE-HPLC-анализа конъюгата инсулин-PEG в соответствующих условиях буферных растворов. Сплошной стрелкой показаны пики монопегилированного инсулина, а пунктирной стрелкой показаны пики примесей, пегилированных в форме "мостика". Соответствующие условия буферных растворов являются следующими:
А: (а) буферный раствор 100 мМ фосфата калия (pH 6,0); (b) буферный раствор 100 мМ фосфата калия (pH 6,0), 30% изопропанола; (с) буферный раствор 100 мМ фосфата калия (pH 6,0), 45% изопропанола; (d) буферный раствор 100 мМ фосфата калия (pH 6,0), 55% изопропанола;
В: (а) буферный раствор 50 мМ цитрата натрия (pH 6,0), 45% изопропанола; (b) буферный раствор 50 мМ цитрата натрия (pH 6,0), 55% изопропанола;
С: (а) буферный раствор 50 мМ ацетата натрия (pH 4,0); (b) буферный раствор 50 мМ ацетата натрия (pH 4,0), 10% изопропанола; (с) буферный раствор 50 мМ ацетата натрия (pH 4,0), 20% изопропанола; (d) буферный раствор 50 мМ ацетата натрия (pH 4,0), 30% изопропанола; (е) буферный раствор 50 мМ ацетата натрия (pH 4,0), 40% изопропанола; (f) буферный раствор 50 мМ ацетата натрия (pH 4,0), 45% изопропанола; (g) буферный раствор 50 мМ ацетата натрия (pH 4,0), 50% изопропанола.
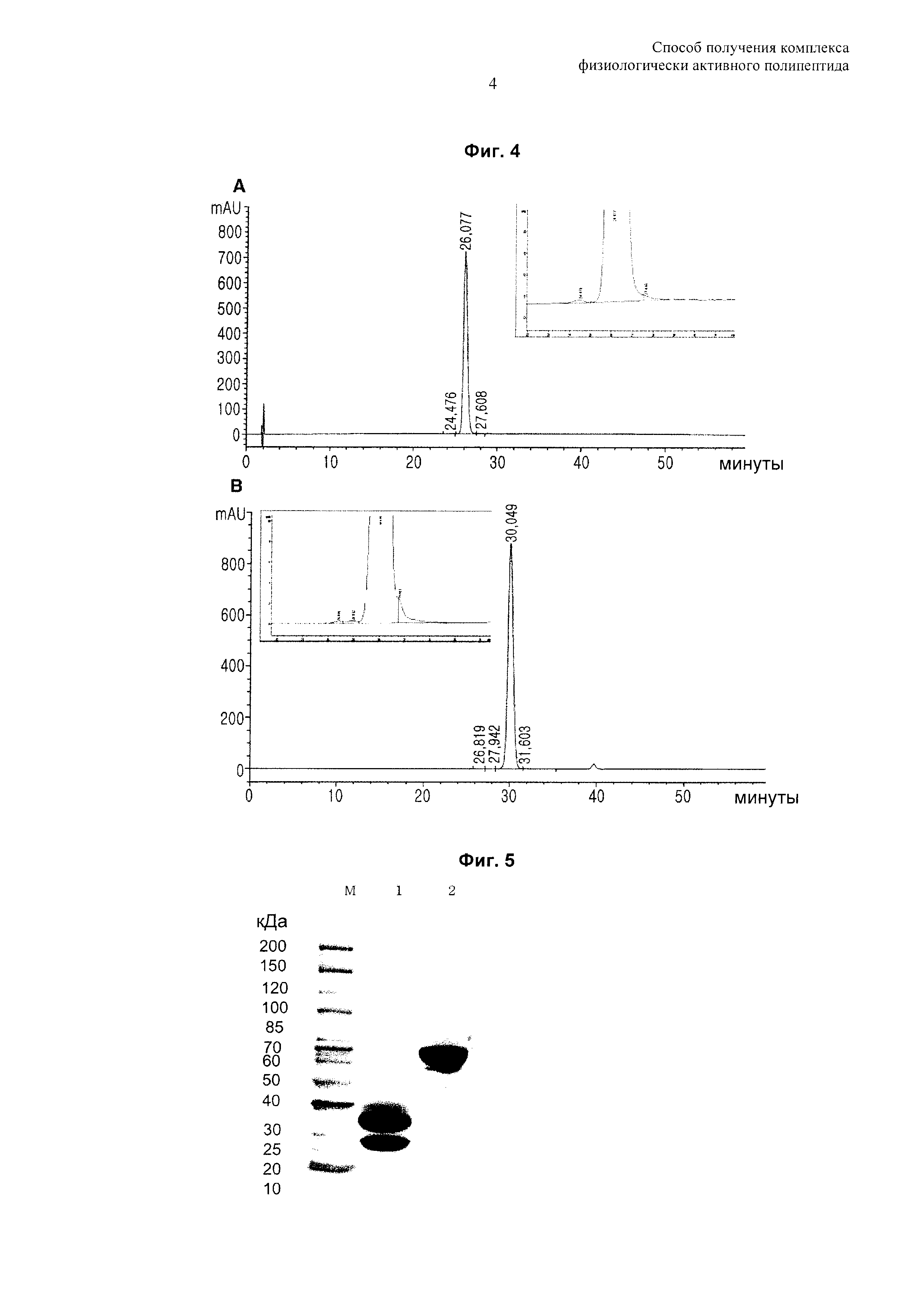
На Фиг. 4 показаны результаты RP HPLC-анализа (А), SE HPLC-анализа (В) комплекса инсулин-PEG-Fc иммуноглобулина.
На Фиг. 5 показаны результаты анализа методом электрофореза в полиакриламидном геле с додецилсульфатом натрия (SDS PAGE-анализ) комплекса инсулин-PEG-Fc иммуноглобулина. М: маркер молекулярной массы, 1: восстанавливающие условия для комплекса инсулин-PEG-Fc иммуноглобулина, 2: невосстанавливающие условия для комплекса инсулин-PEG-Fc иммуноглобулина.
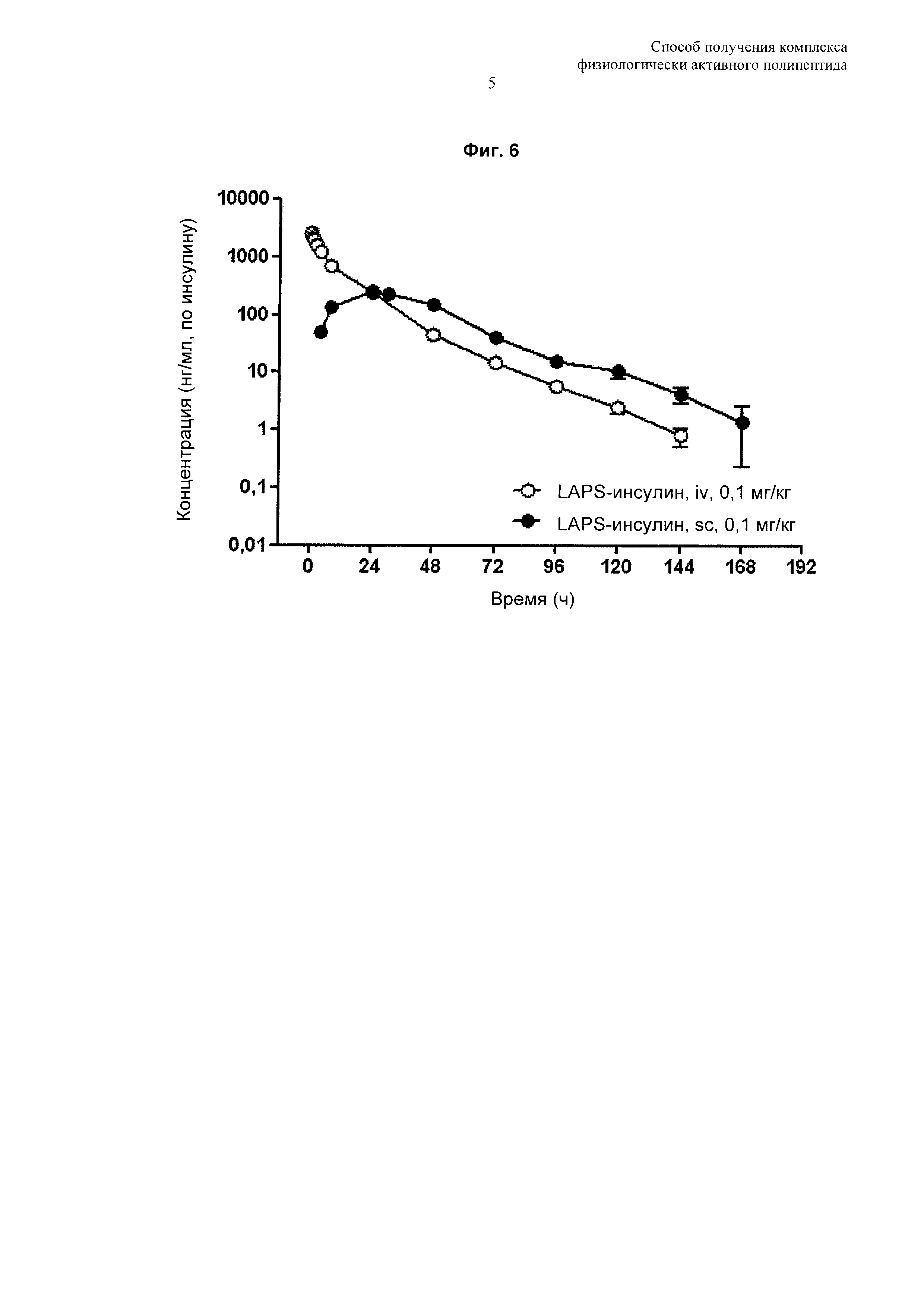
На Фиг. 6 показаны результаты фармакокинетического анализа с определением продолжительности действия in vivo комплекса инсулин-PEG-Fc иммуноглобулина. iv: внутривенное введение, sc: подкожное введение.
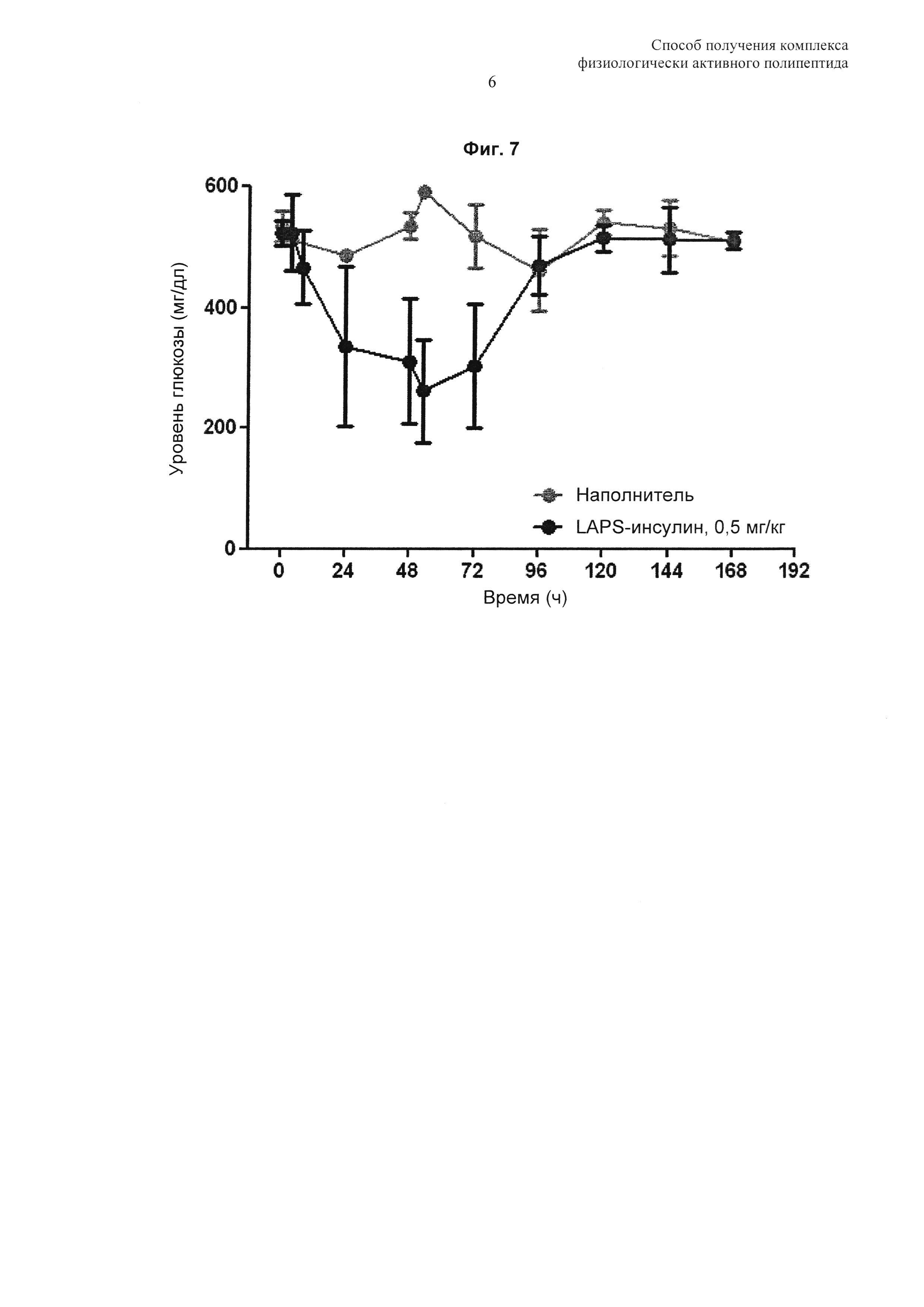
На Фиг. 7 показаны результаты анализа активности in vivo комплекса инсулин-PEG-Fc иммуноглобулина.
Наилучший вариант осуществления изобретения
В одном аспекте для выполнения указанной выше задачи согласно настоящему изобретению предложен способ получения конъюгата физиологически активного полипептида и непептидного полимера, включающий: (1) взаимодействие физиологически активного полипептида с непептидным полимером в реакционном растворе, содержащем органический растворитель, для связывания физиологически активного полипептида с непептидным полимером; и (2) выделение и очистку конъюгата физиологически активных полипептидов, ковалентно связанных с непептидными полимерами, из реакционной смеси со стадии (1).
При использовании в настоящем изобретении органический растворитель означает жидкое органическое соединение, в котором возможно растворение твердого вещества, газа и жидкости, и которое может быть включено в реакционный раствор для ковалентного связывания непептидных полимеров с N-концом физиологически активных полипептидов с высоким выходом и чистотой для задач настоящего изобретения.
В настоящем изобретении могут быть использованы любые органические растворители, обычно используемые в данной области техники. Органический растворитель, используемый в настоящем изобретении, предпочтительно может содержать, без ограничения, первичные, вторичные и третичные спирты. Могут быть использованы спирты, имеющие от 1 до 10 атомов углерода. Более предпочтительно, спирт может представлять собой изопропанол, этанол или метанол. В соответствии с типом физиологически активного полипептида может быть свободно выбран любой подходящий органический растворитель.
Органические растворители включают в реакционный раствор для связывания физиологически активного полипептида и непептидного полимера, и они могут быть включены, без ограничения, в количестве от 10 до 60%, предпочтительно в количестве от 30 до 55% и более предпочтительно в количестве от 45 до 55% по объему от общего объема реакционного раствора. Кроме того, pH реакционного раствора может предпочтительно составлять, без ограничения, от 4,5 до 7,0 и более предпочтительно от 5,5 до 6,5. В данном случае, несмотря на то, что физиологически активные полипептиды обычно демонстрируют тенденцию к снижению растворимости в реакционном растворе, имеющем слабокислый pH, применение органического растворителя по настоящему изобретению обладает тем преимуществом, что указанная проблема с растворимостью может быть решена при постепенном проведении реакции.
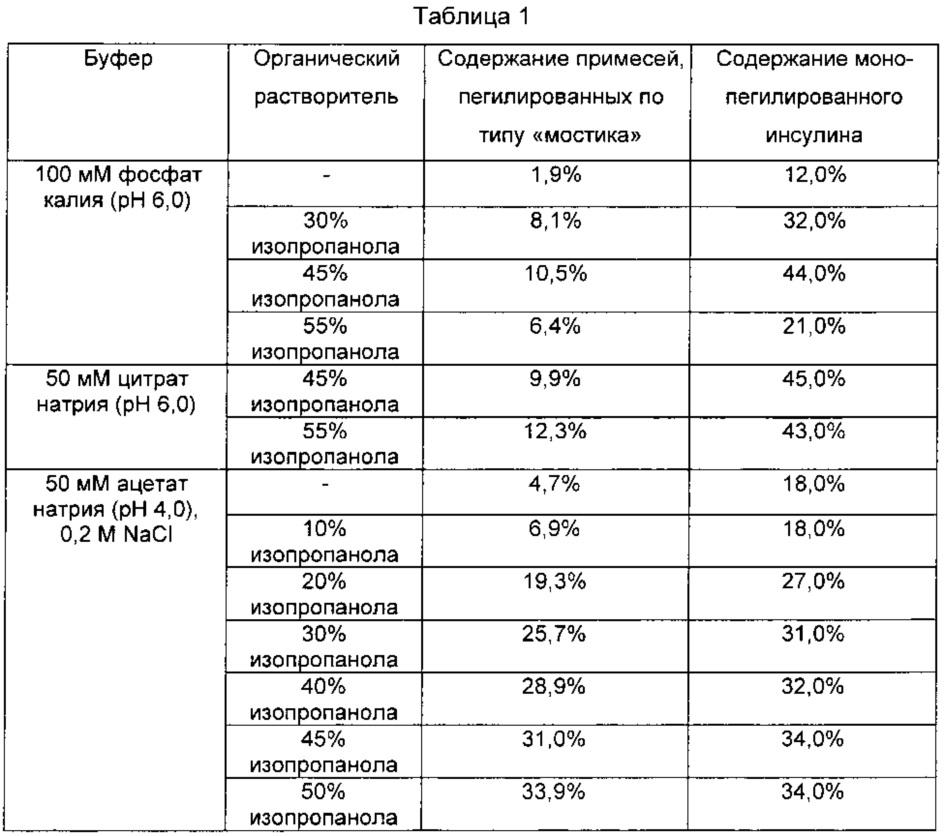
В одном воплощении настоящего изобретения для избирательной конъюгации PEG с N-концом бета-цепи инсулина изопропанол включали в реакционную смесь в качестве органического растворителя для проведения пегилирования инсулина при различных уровнях pH. Затем было обнаружено, что в случаях включения органического растворителя в реакционный раствор можно эффективно уменьшить количество различных типов примесей и получить конъюгат физиологически активного полипептида и непептидного полимера с высокой чистотой и выходом (Таблица 1), и принимая во внимание преимущества такого обнаружения, можно, в итоге, получить однородный и высокоочищенный комплекс физиологически активного полипептида. В одном воплощении настоящего изобретения содержание примесей и соотношение монопегилированного инсулина можно варьировать, изменяя содержание изопропанола и pH реакционного раствора. В частности, было установлено, что при содержании изопропанола на уровне от 45 до 55% от общего количества реакционного раствора при pH 6,0 содержание монопегилированного инсулина достигает максимально возможного уровня (Таблица 1).
При использовании в настоящем изобретении термин «физиологически активный полипептид» является общим понятием полипептидов, обладающих какой-либо физиологической функцией в живых организмах, и имеет общие признаки структуры полипептидов, а также разные физиологические активности. Физиологические активности играют роль в коррекции аномальных патологий, обусловленных недостаточной или избыточной секрецией веществ, участвующих в регуляции физиологических функций в живом организме путем контролирования генетического представления и физиологических функций. Физиологически активный полипептид может включать обычные белковые лекарственные средства.
При использовании в настоящем изобретении физиологически активные полипептиды включают все вещества, имеющие физиологические активности в живом организме, без какого-либо ограничения, и могут включать, например, инсулин, лютеотропный гормон (LTH), фолликулостимулирующий гормон (FSH), фактор свертывания крови VIII, фактор свертывания крови VII, адипонектин, антитело, фрагменты антител scFv, Fab, Fsb', F(ab')2, релаксин и так далее, и предпочтительно представляют собой инсулин. Физиологически активные полипептиды могут иметь два или более N-конца (аминоконца).
При использовании в настоящем изобретении инсулин представляет собой пептид, обладающий функцией контроля уровня сахара в крови в соответствии с механизмом, согласно которому инсулин секретируется из поджелудочной железы при высоком уровне сахара в крови в организме, приводит к поглощению сахара в печени, мышечной и жировой тканях для хранения в форме гликогена и ингибирует распад жиров и их использование в качестве источника энергии. Такие пептиды включают агонисты, предшественники, производные, фрагменты, варианты инсулина и так далее, и предпочтительно представляют собой нативный инсулин, инсулин немедленного высвобождения, инсулин длительного высвобождения.
Нативный инсулин представляет собой гормон, секретируемый из поджелудочной железы и обычно играющий роль в контроле уровня сахара в крови в организме, стимулируя поглощение глюкозы клетками и ингибируя распад жиров. Образование инсулина проходит посредством ряда процессов от проинсулиновых предшественников, не имеющих функции контроля уровня сахара в крови, до инсулина, имеющего функцию контроля уровня сахара в крови. Аминокислотная последовательность инсулина является следующей.
Альфа-цепь:
Gly-lle-Val-Glu-Gln-Cys-Cys-Thr-Ser-lle-Cys-Ser-Leu-Tyr-Gln-Leu-Glu-Asn-Tyr-Cys-Asn (SEQ ID NO: 1).
Бета-цепь:
Phe-Val-Asn-Gln-His-Leu-Cys-Gly-Ser-His-Leu-Val-Glu-Ala-Leu-Tyr-Leu-Val-Cys-Gly-Glu-Arg-Gly-Phe-Phe-Tyr-Thr-Pro-Lys-Thr (SEQ ID NO: 2).
Агонист инсулина означает вещество, связывающееся с рецептором инсулина in vivo, демонстрирующее те же биологические активности, что и инсулин, независимо от структуры инсулина.
Производное инсулина означает пептид, демонстрирующий по меньшей мере 80%-ую гомологию аминокислотной последовательности, по сравнению с нативным инсулином, имеющий некоторые группы аминокислотных остатков, измененные в форме химического замещения (например альфа-метилирования, альфа-гидроксилирования), удаления (например дезаминирования) или модификации (например N-метилирования, гликозилирования, жирной кислоты), и имеющий функцию контроля уровня сахара в крови в организме.
Фрагмент инсулина означает тип инсулина, где одна или более аминокислот добавлены или удалены с N- или C-конца инсулина, при этом добавленные аминокислоты могут также представлять собой искусственные аминокислоты (например, аминокислоты D-типа). Такие фрагменты инсулина сохраняют функцию контроля уровня сахара в крови в организме.
Вариант инсулина относится к пептиду, имеющему одно или более отличий от инсулина в аминокислотной последовательности, сохраняющий функцию контроля уровня сахара в крови в организме.
Соответствующие способы получения агонистов, производных, фрагментов и вариантов инсулина могут быть использованы независимо или в комбинации. Например, настоящее изобретение включает пептиды, имеющие одно или более отличий от инсулина в аминокислотной последовательности, имеющие дезаминирование N-концевого аминокислотного остатка и также имеющие функцию контроля уровня сахара в крови в организме.
В одном воплощении инсулин, используемый в настоящем изобретении, может быть получен рекомбинантным способом и может также быть получен синтетическим способом, включая твердофазный синтез.
Кроме того, инсулин, используемый в настоящем изобретении, может характеризоваться тем, что с N-концами (аминоконцами) бета-цепи конъюгирован непептидный полимер. В случае инсулина, поскольку модификация альфа-цепи приводит к снижению активности и стабильности, согласно настоящему изобретению непептидный полимер может быть связан с N-концом бета-цепи инсулина для сохранения активности инсулина при одновременном улучшении стабильности инсулина.
При использовании в настоящем изобретении термин «фактор свертывания крови» означает белок, вовлеченный в свертывание крови, действующий, защищая организм посредством свертывания крови в случаях кровотечения при повреждениях. Свертывание крови представляет собой серию реакций, в которую вовлечены 12 факторов. Среди них фактор свертывания крови VIII, также называемый антигемофильным фактором, при дефиците которого возникает гемофилия, и фактор свертывания крови VII, также называемый проконвертином, который может быть использован в качестве средства для активации свертывания крови.
При использовании в настоящем изобретении термин «адипонектин» означает белок, секретируемый жировыми клетками, представляющий собой вещество, вовлеченное в метаболизм жиров и сахара. Известно, что адипонектин является белком, снижающим риск заболеваний сердца и сахарного диабета во взрослом периоде и подавляющим аппетит, позволяя мышцам преобразовывать жир в энергию. Кроме того, термин «релаксин» означает гормон, секретируемый желтым телом яичника, расслабляющий лобковый симфиз и стимулирующий роды. Релаксин представляет собой гормон, играющий роль в расслаблении костей и суставов по всему организму.
При использовании в настоящем изобретении термин «непептидный полимер» означает биологически совместимый полимер, полученный сочетанием двух или более повторяющихся единиц, связанных друг с другом возможной ковалентной связью, а не пептидной связью.
Непептидные полимеры, которые могут быть использованы в настоящем изобретении, могут быть выбраны из группы, состоящей из полиэтиленгликоля, полипропиленгликоля, сополимера этиленгликоля и пропиленгликоля, полиоксиэтилированного полиола, поливинилового спирта, полисахарида, декстрана, поливинилэтилового эфира, биоразлагаемых полимеров, таких как полимолочная кислота (PLA) и полимолочная-гликолевая кислота (PLGA), липидных полимеров, хитинов, гиалуроновой кислоты и их комбинаций, и предпочтительно представляют собой полиэтиленгликоль. В объем настоящего изобретения включены все их производные, уже известные в данной области техники, и производные, которые могут быть легко получены на основании технических знаний в данной области техники.
Пептидные линкеры, используемые в слитых белках, полученных способом слияния с сохранением рамки считывания предшествующего уровня техники, имеют недостаток в том, что они легко расщепляются протеолитическими ферментами in vivo, и поэтому какое-либо увеличение периода полувыведения активных лекарственных средств из крови благодаря использованию соответствующего носителя не достигает ожидаемого. Тем не менее, в настоящем изобретении период полувыведения пептида из крови, как обнаружено, сходен с периодом полувыведения носителя из крови, благодаря использованию полимеров, устойчивых к протеолитическим ферментам. Таким образом, в настоящем изобретении любой полимер, имеющий указанную функцию, то есть, устойчивый к протеолитическим ферментам in vivo, может быть использован в качестве непептидного полимера, без каких-либо ограничений. Непептидные полимеры предпочтительно имеют молекулярную массу в диапазоне от 1 до 100 кДа и предпочтительно в диапазоне от 1 до 20 кДа. Кроме того, несмотря на то, что непептидный полимер, конъюгируемый с физиологически активным полипептидом, может представлять собой один тип полимера, в настоящем изобретении может также быть использована комбинация различных типов полимеров.
При использовании в настоящем изобретении непептидные полимеры могут иметь реакционноспособные группы на обоих концах или на трех концах, которые могут быть конъюгированы с физиологически активными полипептидами и носителями.
Реакционноспособные группы непептидных полимеров могут предпочтительно представлять собой, без ограничения, реакционноспособную альдегидную группу, пропиональдегидную группу, бутиральдегидную группу, малеимидную группу, орто-пиридилдисульфид и тиоловые или сукцинимидные производные. Среди указанных выше сукцинимидных производных могут также быть использованы сукцинимидилпропионат, гидроксисукцинимидил, сукцинимидилкарбоксиметил или сукцинимидилкарбонат. В частности, когда непептидные полимеры имеют реакционноспособные альдегидные группы в качестве реакционноспособных групп на их концах, они эффективны для минимизации неспецифического взаимодействия и для конъюгации непептидных полимеров с физиологически активными полипептидами и носителями на их соответствующих концах. Конечные продукты, полученные восстановительным алкилированием с альдегидными связями, значительно стабильнее продуктов, полученных связыванием амидными связями. Альдегидные реакционноспособные группы могут селективно взаимодействовать с N-концами при низком уровне pH и могут образовывать ковалентные связи с лизиновыми остатками при высоком уровне pH, например при pH 9,0.
Реакционноспособные группы, присутствующие на обоих концах или трех концах непептидных полимеров, могут быть идентичны друг другу или отличаться друг от друга. Например, малеимидная группа может присутствовать на одном конце, а альдегидная группа, пропиональдегидная группа или бутиральдегидная группа может присутствовать на другом конце.
При использовании полиэтиленгликоля, имеющего реакционноспособные гидроксигруппы, в качестве непептидного полимера белковый конъюгат по настоящему изобретению может быть получен активацией гидроксигрупп с получением различных реакционноспособных групп известными химическими реакциями или с использованием имеющихся в продаже полиэтиленгликолей, имеющих модифицированные реакционноспособные группы.
В то же время, в способе разделения и очистки конъюгата, содержащего физиологически активный полипептид, N-концы которого ковалентно связаны с непептидным полимером, на стадии (2) по настоящему изобретению может быть использован любой способ, известный в данной области техники, без каких-либо ограничений, и, предпочтительно, может быть использована ионообменная хроматография.
В другом аспекте согласно настоящему изобретению предложен способ получения комплекса физиологически активного полипептида, включающий (1) получение конъюгата физиологически активного полипептида и непептидного полимера указанным способом; и (2) ковалентное связывание носителя, выбранного из группы, состоящей из Fc-области иммуноглобулина, антитела, альбумина и трансферрина с непептидным полимером конъюгата с получением комплекса пептида, в котором концы непептидного полимера связаны соответственно с физиологически активным полипептидом и носителем.
В настоящем изобретении под «комплексом» подразумевают структуру, состоящую из по меньшей мере одного физиологически активного полипептида, по меньшей мере одного непептидильного полимера и по меньшей мере одного носителя, связанных друг с другом ковалентными связями. Для его разграничения с «комплексом», термин «конъюгат» использован здесь для обозначения структуры, в которой только пары физиологически активного полипептида и непептидного полимера связаны друг с другом ковалентной связью.
В настоящем изобретении «носитель» означает вещество, связываемое с лекарственным средством, и, как правило, вещество, связанное с лекарственным средством для повышения, снижения или устранения биологической активности лекарственного средства. Для задач настоящего изобретения лекарственное средство, связываемое с носителем, предпочтительно представляет собой физиологически активные полипептиды, которые могут также быть связаны с непептидными полимерами. Носитель представляет собой вещество для минимизации снижения активности связываемого физиологически активного полипептида in vivo и в то же время для повышения стабильности лекарственного средства in vivo. Носитель может предпочтительно содержать, без ограничения, Fc-область иммуноглобулина, антитело, альбумин и трансферрин.
Fc-область иммуноглобулина представляет собой биоразлагаемый полипептид, метаболизируемый в живом организме и поэтому безопасный при использовании в качестве носителя для лекарственного средства. Кроме того, поскольку молекулярная масса Fc-области иммуноглобулина меньше, чем у полноразмерной молекулы иммуноглобулина, она предпочтительна с точки зрения получения, очистки и выхода конъюгата. Кроме того, поскольку аминокислотные последовательности антител отличаются друг от друга, можно ожидать, что удаление Fab-областей, демонстрирующих высокую гетерогенность, приведет к значительному повышению однородности веществ и снизит вероятность индукции антигенности в крови.
В настоящем изобретении «Fc-область иммуноглобулина» означает фрагменты иммуноглобулина, содержащие константную область тяжелой цепи 2 (CH2) и константную область тяжелой цепи 3 (CH3) без вариабельных областей тяжелой и легкой цепи, константной области тяжелой цепи 1 (CH1) и константной области легкой цепи 1 (CL1) иммуноглобулина, и может также содержать шарнирную область в константной области тяжелой цепи. Кроме того, если Fc-область иммуноглобулина по настоящему изобретению имеет по существу эквивалентный или улучшенный эффект по сравнению с нативной формой, она может представлять собой расширенную Fc-область иммуноглобулина, содержащую полноразмерную константную область тяжелой цепи 1 (CH1) и/или константную область легкой цепи 1 (CL1) или их фрагмент, без вариабельных областей тяжелой цепи и легкой цепи иммуноглобулина. Кроме того, она может также представлять собой область, где удалены аминокислотные последовательности значительной длины, соответствующие CH2 и/или CH3. То есть, Fc-область иммуноглобулина по настоящему изобретению может представлять собой (1) домен CH1, домен CH2, домен CH3 и домен CH4, (2) домен CH1 и домен CH2, (3) домен CH1 и домен CH3, (4) домен CH2 и домен CH3, (5) комбинацию одного, двух или более доменов и шарнирной области иммуноглобулина (или фрагменты шарнирной области) и (6) димер соответствующих доменов константных областей тяжелой цепи и константных областей легкой цепи.
Кроме того, Fc-область иммуноглобулина по настоящему изобретению включает нативные аминокислотные последовательности, а также последовательности их мутантов. Мутанты аминокислотной последовательности означают последовательности, отличающиеся делецией, вставкой, неконсервативной или консервативной заменой одного или более аминокислотных остатков нативной аминокислотной последовательности и их комбинациями. Например, в случае Fc IgG, в качестве сайтов, подходящих для модификации, могут быть использованы аминокислотные остатки, присутствующие в положениях 214-238, 297-299, 318-322 или 327-331. Кроме того, доступны различные типы мутантов, в которых сайты, способные образовывать дисульфидные связи, удалены, некоторые аминокислоты удалены с N-конца нативного Fc, либо метиониновые остатки также могут быть добавлены на N-конце нативного Fc. Кроме того, для устранения эффекторной функции могут быть удалены сайты связывания с комплементом, например, сайты связывания с C1q, и также могут быть удалены ADCC-сайты. Методики получения таких мутантов последовательностей Fc-области иммуноглобулина раскрыты, среди прочего, в публикации международной заявки на патент №97/34631 и публикации международной заявки на патент №96/32478.
Некоторые изменения аминокислот белков и пептидов, не приводящие к полному изменению активности молекул, известны в данной области техники (H. Neurath, R.L. Hill, The Proteins, Academic Press, New York, 1979). Наиболее частыми заменами являются замены между следующими аминокислотными остатками: Ala/Ser, Val/IIe, Asp/Glu, Thr/Ser, Ala/Gly, Ala/Thr, Ser/Asn, Ala/Val, Ser/Gly, Thr/Phe, Ala/Pro, Lys/Arg, Asp/Asn, Leu/IIe, Leu/Val, Ala/Glu или Asp/Gly.
Если целесообразно, они могут быть модифицированы фосфорилированием, сульфатированием, акрилированием, гликозилированием, метилированием, фарнезилированием, ацетилированием, амидированием и так далее.
Мутанты Fc, как описано выше, являются мутантами, демонстрирующими ту же биологическую активность, что и Fc-область по настоящему изобретению, и, кроме того, имеющими повышенную структурную стабильность Fc-области к нагреванию, pH и так далее.
Кроме того, такая Fc-область может быть получена из нативной формы, выделенной из живого организма человека и животных, включая крупный рогатый скот, коз, свиней, мышей, кроликов, хомяков, крыс, морских свинок и так далее, и может также быть рекомбинантной, полученной от трансформированных животных клеток или микроорганизмов или их производных. В данном описании, Fc-области, получаемые из нативной формы, могут быть получены выделением полноразмерного иммуноглобулина из живого организма человека или животных и обработкой полноразмерного иммуноглобулина протеолитическими ферментами. При обработке иммуноглобулина папаином происходит его расщепление на Fab и Fc, и в случае обработки пепсином иммуноглобулин может быть расщеплен на pF'c и F(ab)2. Затем может быть проведена гель-хроматография реакционной смеси с выделением Fc или pF'c.
Предпочтительна рекомбинантная Fc-область иммуноглобулина, полученная из Fc-области, имеющей происхождение из человека, с использованием микроорганизмов.
Кроме того, Fc-область иммуноглобулина может быть представлена в формах с нативным гликозилированием цепи, с повышенным гликозилированием цепи по сравнению с нативной формой, со сниженным гликозилированием цепи по сравнению с нативной формой или в дегликозилированных формах. Повышение, снижение или устранение такого гликозилирования цепи Fc иммуноглобулина может быть проведено обычными способами, включая химические способы, энзимологические способы и генно-инженерные способы с использованием микроорганизмов. В данном случае, поскольку Fc-области иммуноглобулинов без гликозилирования цепи демонстрируют значительное уменьшение силы связывания с комплементом (c1q) и обеспечивают уменьшение или устранение антителозависимой цитотоксичности или комплемент-зависимой цитотоксичности, они не индуцируют никаких обязательных иммунных реакций в живом организме.
Поэтому формой, более подходящей для основной задачи настоящего изобретения в качестве носителя для лекарственных средств, может быть дегликозилированная или агликозилированная Fc-область иммуноглобулина.
В настоящем изобретении «дегликозилирование» относится к Fc-области, из которой гликозильная группа удалена ферментами, и «агликозилирование» означает Fc-область, полученную из прокариотических организмов, предпочтительно из E. coli, и не являющуюся гликозилированной.
Кроме того, Fc-область иммуноглобулина может представлять собой Fc-область, имеющую происхождение из IgG, IgA, IgD, IgE или IgM, или Fc-область, имеющую происхождение из их комбинации или гибрида. Предпочтительно, Fc-область иммуноглобулина может иметь происхождение из IgG или IgM, наиболее распространенных в крови человека, и наиболее предпочтительно имеет происхождение из IgG, о котором известно, что он улучшает период полувыведения белков, конъюгированных с лигандом.
В то же время, «комбинация» в настоящем изобретении означает, что связывание полипептидов, кодирующих одноцепочечную Fc-область иммуноглобулина, имеющих происхождение из одного и того же источника, с одноцепочечными полипептидами, имеющими происхождение из другого источника, происходит в форме димеров и мультимеров. То есть, возможно получение димеров или мультимеров из двух или более фрагментов, выбранных из группы, состоящей из Fc-фрагментов IgG, Fc-фрагментов IgA, Fc-фрагментов IgM, Fc-фрагментов IgD и Fc-фрагментов IgE.
В настоящем изобретении термин «гибрид» означает, что последовательности, соответствующие двум или более Fc-фрагментам иммуноглобулинов, имеющим происхождение из разных источников, присутствуют в одноцепочечной Fc-области иммуноглобулина. В определенном воплощении настоящего изобретения доступны различные типы гибридов. То есть, доступны гибриды, содержащие от одного до четырех доменов, выбранных из группы, состоящей из CH1, CH2, CH3 и CH4 Fc IgG, Fc IgM, Fc IgA, Fc IgE и Fc IgD, которые могут дополнительно содержать шарнирную область.
Кроме того, 1дС могут также быть разделены на подклассы IgG1, IgG2, IgG3 и IgG4, и в настоящем изобретении может также быть использована их комбинация или гибрид. Предпочтительны подклассы IgG2 и IgG4, и наиболее предпочтительна Fc-область IgG4, практически не имеющая эффекторной функции, такой как комплемент-зависимая цитотоксичность (CDC).
То есть, в настоящем изобретении наиболее предпочтительной Fc-областью иммуноглобулина для использования в качестве носителя для лекарственного средства является агликозилированный Fc-домен, имеющий происхождение из человеческого IgG4. Fc-области человеческого происхождения действуют в организме человека как антиген и поэтому предпочтительнее Fc-областей, имеющих происхождение из источника, не являющегося человеком, которые могут вызывать нежелательные иммунные реакции, такие как образование нового антитела.
Согласно способу по настоящему изобретению комплекс физиологически активного полипептида по настоящему изобретению может быть получен с высокой чистотой и выходом и может демонстрировать повышенную продолжительность действия и стабильность in vivo, что может привести к значительному улучшению адаптируемости приема лекарственного средства при лечении физиологически активным полипептидом. В одном примере настоящего изобретения комплексы инсулин-PEG-Fc иммуноглобулина, полученные способом по настоящему изобретению, были использованы для подтверждения их периода полувыведения in vivo. В результате, авторами изобретения выявлено, что они продемонстрировали продолжительность действия 15,73 часа в случае внутривенного введения и 16,98 часа в случае подкожного введения, что в 30 раз превосходит продолжительность действия в случае нативного инсулина, составляющую приблизительно 0,5 часа (Фиг. 6). Кроме того, по результатам анализа активности in vivo, авторами изобретения обнаружено, что их активность по снижению уровня сахара в крови сохраняется приблизительно 4 суток (Фиг. 7).
Согласно настоящему изобретению также предложена композиция физиологически активных полипептидов с длительным высвобождением, содержащая комплексы физиологически активных полипептидов.
В настоящем изобретении термин «введение» означает, что рассматриваемое вещество вводят пациенту любым подходящим способом. Комплексы можно вводить любым из обычных способов введения, при условии, что этот способ введения позволяет лекарственному средству достигнуть ткани-мишени. Введение комплекса может включать, без ограничения, внутрибрюшинное, внутривенное, внутримышечное, подкожное, внутрикожное, пероральное, местное, интраназальное, внутрилегочное, интраректальное введение и тому подобное. Тем не менее, вследствие расщепления пептидных лекарственных средств в случае их перорального введения, предпочтительно изготовление композиций для перорального приема путем нанесения покрытия на активное лекарственное средство для его защиты от расщепления в желудке. Предпочтительно, комплексы по настоящему изобретению можно вводить в форме инъекции. Кроме того, композиция с длительным высвобождением может быть введена с использованием любого возможного устройства, позволяющего переносить активное вещество к клеткам-мишеням.
Композиция с длительным высвобождением, содержащая комплекс по настоящему изобретению, может содержать фармацевтически приемлемый носитель. Фармацевтически приемлемые носители включают связывающие агенты, смазывающие агенты, разрыхлители, эксципиенты, солюбилизаторы, диспергирующие агенты, стабилизаторы, суспендирующие агенты, красители, отдушки и так далее, которые могут быть использованы для перорального введения; буферы, консерванты, обезболивающие агенты, солюбилизаторы, изотонические агенты, стабилизаторы и другие могут быть использованы в их комбинации для инъекций; и основы, эксципиенты, смазывающие агенты, консерванты и другие могут быть использованы для местного введения. Композиция с длительным высвобождением по настоящему изобретению может быть изготовлена по-разному путем смешивания с фармацевтически приемлемыми носителями, как описано выше. Например, композиция может быть изготовлена в форме таблеток, пастилок, капсул, эликсиров, суспензий, сиропов, пластинок и так далее для перорального введения, и в форме ампул для однократного или многократного введения для инъекций. Комплексы по настоящему изобретению могут также быть изготовлены в других формах, включая растворы, суспензии, таблетки, пилюли, капсулы, композиции с длительным высвобождением и так далее.
В то же время, в качестве примеров носителей, эксципиентов и разбавителей, подходящих для композиции, могут быть использованы лактоза, декстроза, сахароза, сорбит, маннит, ксилит, эритрит, мальтит, крахмал, аравийская камедь, альгинат, желатин, фосфат кальция, силикат кальция, целлюлоза, метилцеллюлоза, микрокристаллическая целлюлоза, поливинилпирролидон, вода, метилгидроксибензоат, пропилгидроксибензоат, тальк, стеарат магния, минеральные масла и так далее. Кроме того, дополнительно могут быть использованы наполнители, антикоагулянты, смазывающие агенты, смачивающие агенты, отдушки, консерванты и так далее.
Введение композиции с длительным высвобождением по настоящему изобретению может быть определено в зависимости от типа лекарственных средств, используемых в качестве активного ингредиента, наряду с различными факторами, связанными с введением, включающими заболевания, по поводу которых проводят лечение, способы введения, возраст, пол и массу тела пациентов, тяжесть заболеваний и так далее. Композиции с длительным высвобождением по настоящему изобретению имеют хорошую продолжительность действия и эффективность in vivo и поэтому могут привести к значительному снижению частоты введения фармацевтической композиции по настоящему изобретению. Кроме того, композиция с длительным высвобождением может поддерживать продолжительность действия и стабильность физиологически активных полипептидов in vivo и, таким образом, может быть эффективно использована для лечения заболеваний.
Вариант осуществления изобретения
Далее изобретение описано более подробно в примерах. Тем не менее, эти Примеры приведены лишь для более подробной иллюстрации настоящего изобретения и никак не ограничивают объем настоящего изобретения.
Пример 1: Реакция пегилирования инсулина и очистка монопегилированного инсулина
Порошок инулина растворяли в 10 мМ HCl и затем подвергали взаимодействию с 3.4К пропион-ALD2 PEG (PEG, имеющим две пропиональдегидные группы, IDB, Korea) при 4~8°C в течение приблизительно 2 часов в условиях, включающих молярное отношение инсулин:PEG 1:2-4 и концентрацию инсулина 3-5 мг/мл, для пегилирования N-конца бета-цепи инсулина. Эту реакцию проводили при 50 мМ цитрата натрия, pH 6,0, 45% изопропанола с добавлением 4-20 мМ NaCNBH3 в качестве восстановителя.
Реакционный раствор очищали на колонке SP-HP (GE Healthcare) с использованием буфера, содержащего цитрат натрия (pH 3,0) и 45% EtOH, и градиента концентрации KCl.
Посредством SE-HPLC и анализа пептидного картирования было установлено, что полученный таким образом монопегилированный инсулин был на 98% или более пегилирован по фенилаланину №1 бета-цепи (B1F) (Фиг. 1 и 2).
Пример 2: Изменение выхода и степени чистоты монопегилированного инсулина в зависимости от pH реакционного раствора и концентрации органического растворителя
Для сравнения выхода реакции получения монопегилированного инсулина и образования примесей во время получения при включении в реакционный раствор для взаимодействия инсулина с PEG органических растворителей, таких как изопропанол, инсулин и 3.4К пропион-ALD2 PEG подвергали взаимодействию в молярном отношении 1:2 при концентрации инсулина 3 мг/мл и 4°C в течение 4 часов. В данном примере в качестве реакционного раствора использовали буферный раствор 50 мМ цитрата натрия, pH 6,0, 45% изопропанола; буферный раствор 50 мМ цитрата натрия, pH 6,0, 55% изопропанола; буферный раствор 100 мМ фосфата калия, pH 6,0; буферный раствор 100 мМ фосфата калия, pH 6,0, 30% изопропанола; буферный раствор 100 мМ фосфата калия, pH 6,0, 45% изопропанола; буферный раствор 100 мМ фосфата калия pH 6,0, 55% изопропанола; буферный раствор 50 мМ ацетата натрия, pH 4,0; буферный раствор 50 мМ ацетата натрия, pH 4,0, 10% изопропанола; буферный раствор 50 мМ ацетата натрия, pH 4,0, 20% изопропанола; буферный раствор 50 мМ ацетата натрия, pH 4,0, 30% изопропанола; буферный раствор 50 мМ ацетата натрия, pH 4,0, 40% изопропанола; буферный раствор 50 мМ ацетата натрия, pH 4,0, 45% изопропанола; буферный раствор 50 мМ ацетата натрия, pH 4,0, 50% изопропанола, соответственно, к которым добавляли 20 мМ NaCNBH3 в качестве восстановителя. Соответствующие реакционные растворы очищали тем же способом, что был использован в Примере 1, и профили их очистки показаны на Фиг. 3. При одновременном взаимодействии обоих N-концов альфа- и бета-цепей инсулина с PEG в форме «мостика» реакция сочетания монопегилированного инсулина с Fc иммуноглобулина невозможна. Так, относительное содержание таких примесей показано в Таблице 1. Как видно из Таблицы 1, было установлено, что доля монопегилированного инсулина варьирует в зависимости от содержания изопропанола в реакционном растворе и pH, и содержание монопегилированного инсулина достигает максимума при pH 6,0, когда содержание изопропанола составляет приблизительно 45-55%. Низкое значение pH приводило к повышению содержания примесей, пегилированных по типу «мостика», по мере повышения содержания изопропанола, снижая таким образом выход монопегилированного инсулина.
Пример 3: Получение комплекса монопегилированного инсулина и Fc иммуноглобулина
Для получения комплекса инсулин-PEG-Fc иммуноглобулина монопегилированный инсулин, полученный способом по Примеру 1, и Fc иммуноглобулина подвергали взаимодействию в молярном отношении 1:1 при общем уровне белка 20-50 мг/мл при 25°C в течение 15-17 часов. В данной реакции реакционный раствор содержал 100 мМ HEPES, 22 мМ фосфата калия, 10% этанола, pH 8,2, и дополнительно содержал 20 мМ NaCNBH3 в качестве восстановителя.
По завершении реакции реакционный раствор сначала очищали через колонку Source 15Q (GE Healthcare) для удаления любых остаточных Fc иммуноглобулина и монопегилированного инсулина. В данном случае, элюирование проводили с использованием буфера трис-HCl (pH 7,5) и градиента концентрации NaCl.
Затем Source 15ISO (GE Healthcare) использовали в качестве второй колонки для удаления любого остаточного комплекса Fc иммуноглобулина и мультипегилированного инсулина, получая таким образом комплекс инсулин-PEG-Fc иммуноглобулина. В данном случае, элюирование проводили с использованием градиента концентрации сульфата аммония, содержащего трис-HCl (pH 7,5).
Элюированный комплекс инсулин-PEG-Fc иммуноглобулина анализировали посредством RP-HPLC, SE-HPLC и SDS PAGE, и было подтверждено, что он получен с высокой чистотой, составляющей по меньшей мере 98% (Фиг. 4 и 5).
Пример 4: Измерение периода полувыведения комплекса инсулина с длительным высвобождением in vivo
Для подтверждения продолжительности действия комплекса инсулина с длительным высвобождением in vivo в форме комплекса инсулин-PEG-Fc иммуноглобулина, полученного в Примере 3, нормального самца крысы (крысы Normal SD) использовали для определения фармакокинетического профиля лекарственного средства, вводимого внутривенно и подкожно. 0,1 мг/кг (по инсулину) комплекса инсулина с длительным высвобождением вводили внутривенно и подкожно однократно нормальному самцу крысы, после чего измеряли изменение концентрации в крови с течением времени с помощью набора для ELISA инсулина. По измеренным значениям вычисляли фармакокинетические параметры с использованием WinNonlin 5.2. В результате было установлено, что период полувыведения комплекса инсулина с длительным высвобождением in vivo составлял 15,73 часа в случае внутривенного введения и 16,98 часа в случае подкожного введения, что в 30 раз превосходит период полувыведения нативного инсулина, составляющего приблизительно 0,5 часа. Кроме того, было также установлено, что в случае подкожного введения биодоступность составила приблизительно 54% (Фиг. 6).
Пример 5: Анализ эффективности комплекса инсулина in vivo
Для сравнения активности комплексов инсулина IN vivo проводили сравнительный анализ по эффективности снижения уровня сахара в крови у крыс с индуцированным стрептозоцином сахарным диабетом. Сахарный диабет индуцировали внутрибрюшинным введением стрептозоцина, растворенного в буферном растворе 10 мМ лимонной кислоты (pH 4,5) при концентрации 60 мг/кг, нормальным крысам, лишенным еды в течение 16 часов. Затем проводили однократное подкожное введение комплекса инсулина в дозе 0,5 мг/кг крысам, у которых уровень сахара в крови был повышен до 500 мг/дл или более, и затем сравнивали эффективность снижения уровня сахара в крови. В результате, наблюдали сохранение эффекта снижения уровня сахара в крови для комплекса инсулина в течение приблизительно 4 суток, и уровень сахара в крови повышался до уровня, эквивалентного уровню в группе наполнителя, на 5-е сутки (Фиг. 7).


Способ получения солей тетразолметансульфоновой кислоты и новое соединение, используемое в нем
Жидкие препаративные формы для длительно действующего конъюгата g-csf
Конъюгат инсулина с применением фрагмента иммуноглобулина
Производные тиено[3,2-d]пиримидина, обладающие ингибирующей активностью в отношении протеинкиназы
Способ получения гидрохлорида 1-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она и используемых в нем промежуточных соединений
Фармацевтическая композиция, включающая производное амида или его фармацевтически приемлемую соль
Новые конденсированные пиримидиновые производные для ингибирования тирозинкиназной активности
Способ очистки человеческого фактора, стимулирующего колонии гранулоцитов, из рекомбинантных е. coli
Новые конденсированные пиримидиновые производные для ингибирования тирозинкиназной активности
Мультимер непептидильный полимер-инсулин и способ его получения
Способ получения солей тетразолметансульфоновой кислоты и новое соединение, используемое в нем
Жидкие препаративные формы для длительно действующего конъюгата g-csf
Конъюгат инсулина с применением фрагмента иммуноглобулина
Производные тиено[3,2-d]пиримидина, обладающие ингибирующей активностью в отношении протеинкиназы
Способ получения гидрохлорида 1-(3,4-дихлор-2-фторфениламино)-7-метоксихиназолин-6-илокси)пиперидин-1-ил)проп-2-ен-1-она и используемых в нем промежуточных соединений
Фармацевтическая композиция, включающая производное амида или его фармацевтически приемлемую соль
Новые конденсированные пиримидиновые производные для ингибирования тирозинкиназной активности
Способ очистки человеческого фактора, стимулирующего колонии гранулоцитов, из рекомбинантных е. coli
Новые конденсированные пиримидиновые производные для ингибирования тирозинкиназной активности
Мультимер непептидильный полимер-инсулин и способ его получения

