Результат интеллектуальной деятельности: СПОСОБ ОДНОВРЕМЕННОГО ОПРЕДЕЛЕНИЯ ПРИМЕСЕЙ ЭТИЛЕНДИАМИНТЕТРАУКСУСНОЙ КИСЛОТЫ, ДИМЕТИЛСУЛЬФОКСИДА И N-ЭТИЛМАЛЕИМИДА В ФАРМАЦЕВТИЧЕСКИХ СУБСТАНЦИЯХ МЕТОДОМ ОБРАЩЕННО-ФАЗОВОЙ ВЫСОКОЭФФЕКТИВНОЙ ЖИДКОСТНОЙ ХРОМАТОГРАФИИ
Вид РИД
Изобретение
Изобретение относится к исследованию или анализу материалов с использованием хроматографии и может быть использовано в биотехнологии для качественного и количественного определения этилендиаминтетрауксусной кислоты (ЭДТА), диметилсульфоксида (ДМСО) и N-этилмалеимида (ЭТМ) при производстве и контроле фармацевтических субстанций.
В настоящее время для повышения качества фармацевтических субстанций в процессе производства и для контроля конечного продукта, в соответствии с отечественными и международными нормативными документами (ГФ, ICH, Евр. фарм., фармакопея США), необходимо подтверждение отсутствия остаточных органических растворителей и примесей, которые могут быть использованы на определенных стадиях производства, или установлены нормы их содержания. Все остаточные органические растворители и примеси в максимальной степени должны быть удалены из фармацевтических субстанций, чтобы удовлетворять требованиям фармакопейных статей, GMP или иным требованиям к качеству.
Поэтому является актуальным разработка новых высокочувствительных аналитических методик для выявления посторонних примесей и остаточных органических растворителей в фармацевтических субстанциях. При этом аналитические методики должны выявлять примеси, присутствующие в лекарственной субстанции, на уровне выше, чем идентификационный порог. Наибольшей трудностью выявления примесей является то, что зачастую они имеют различную химическую природу, что приводит к использованию целого ряда разнородных аналитических методов, значительно увеличивающих время анализа и парк используемой аналитической техники. Другим подходом определения разнородных примесей является использование универсальных, высокотехнологичных методов исследований, таких, например, как масс-спектроскопия, спектроскопия ядерного магнитного резонанса (ЯМР) и т.д. Указанный подход позволяет анализировать одновременно несколько примесей, но зачастую требует специальной пробоподготовки при низкой сходимости результатов измерений и высокой стоимости анализа. Также возможны трудности с интерпретацией результатов.
Известен способ определения ЭДТА в майонезе методом ион-парной обращенно-фазовой высокоэффективной жидкостной хроматографии (ОФ ВЭЖХ) в форме комплекса с Fe (III) (Захарова A.M., Гринштейн И.Л., Карцова Л.А., Потолицина В.Е. Определение этилендиаминтетрауксусной кислоты в майонезе методом ион-парной обращенно-фазовой высокоэффективной жидкостной хроматографии. Сорбционные и хроматографические процессы, 2014, т. 14). Подготовка стандартного образца включает приготовление водного раствора ЭДТА требуемой концентрации, добавление раствора хлорида железа (III), выдерживание при комнатной температуре. Подготовка испытуемого образца майонеза включает извлечение целевого компонента из образца майонеза методом жидкостной экстракции фосфатным буферным раствором в ультразвуковой ванне в присутствии хлороформа с последующим получением комплексного соединения ЭДТА с хлоридом железа (III) и фильтрацию готового образца через нейлоновый мембранный фильтр с диаметром пор 0,45 мкм. Хроматографическое разделение осуществляют на колонке Marcherey-Nagel С18 Gravity размером 150×3,0 мм, заполненной сорбентом с зернением 3 мкм, при температуре 40°С, в качестве подвижной фазы используют смесь 20 мМ раствора калия дигидрофосфата (рН=7,5): ацетонитрил: 40% водный раствор гидроксида тетрабутиламмония в соотношении 90:10:0,2. Определение проводят в изократическом режиме со скоростью потока подвижной фазы 0,5 мл/мин и детекцией при 255 нм. Объем инжекции не указан. Предел детектирования составляет 0,005 мг/кг.
Недостатками данного способа являются: продолжительность анализа более 20 минут, низкий предел детектирования и сложный процесс пробоподготовки, а также использование дорогостоящих ион-парных агентов, которые значительно сокращают срок службы колонки.
Известен способ определения натриевой соли ЭДТА в субстанции Меропенем с УФ-детектированием и использованием техники предколоночной дериватизации в форме комплекса с Fe (III) методом ион-парной ОФ ВЭЖХ (Bhavil N. et al. A Validated Reverse Phase HPLC Method for the Determination of Disodium EDTA in Meropenem Drug Substance with UV-Detection using Precolumn Derivatization Technique // Anal Chem Insights. 2011. V. 6. P. 7-14). Подготовка раствора стандартного образца включает растворение навески натриевой соли ЭДТА в растворителе и проведение процесса предколоночной дериватизации, заключающегося в выдерживании стандартного образца в растворе хлорида железа (III) при температуре 70°С на водяной бане в течении 20 минут, в результате чего образуется комплекс ЭДТА с хлоридом железа (III). Подготовка испытуемого образца состоит в растворении навески препарата в растворителе (вода) и проведении процесса дериватизации. Хроматографическое разделение осуществляют на колонке Phenomenex Luna C18 размером 250×4,6 мм, заполненной сорбентом с зернением 5 мкм. Подвижная фаза состоит из раствора, содержащего 5% метанола и 95% раствора бромида тетрабутиламмония (0,7 г/л) с ацетатом натрия (4,6 г/л) в воде, рН=4,0. Определение проводят в изократическом режиме со скоростью потока подвижной фазы 1 мл/мин и детекцией при 254 нм, температура колонки 30°С, объем инжекции 100 мкл. Продолжительность анализа для стандартного раствора ЭДТА составляет 10 минут, для испытуемого раствора Меропенем - 60 минут. Предел детектирования составляет 0,023 мкг/мл, предел количественного определения - 0,034 мкг/мл.
Недостатками данного способа являются: продолжительность анализа испытуемого раствора субстанции (60 минут), низкий предел детектирования и сложный процесс пробоподготовки, использование дорогостоящих ион-парных агентов, которые значительно сокращают срок службы колонки и использование достаточно длинной колонки, что приводит к увеличению продолжительности анализа и увеличению расхода элюентов.
Известен способ определения ЭДТА в форме комплекса с Fe (III) в сточной воде и поверхностных водах методом обращенно-фазовой ион-парной жидкостной хроматографии (Xie С.Z. Determination of EDTA in dairy wastewater and adjacent surface water. International journal of civil and environmental engineering, 2010, 44-48). Подготовка образцов сточных вод включает добавление к образцам раствора железа (III), выдерживание в течение ночи без доступа света и фильтрование через фильтр из нитрата целлюлозы с диаметром пор 0,45 мкм. Подготовка образцов поверхностных вод включает высушивание образца при нагревании до 90°С, добавлении раствора подвижной фазы и раствора железа (III), а также выдерживание в течении ночи без доступа света и фильтрование, как описано выше. Хроматографическое разделение осуществляют с использованием колонки Phenomenex Hypersil C18 размером 200×4,6 мм, заполненной сорбентом с зернением 5 мкм и предколонки Phenomenex. Подвижной фазой является 2% раствор метанола, ион-парным агентом - 15 мМ раствор бромида тетрабутиламмония в формиатном буферном растворе (рН=3,3). Определение проводят в изократическом режиме со скоростью потока подвижной фазы 0,9 мл/мин и детекцией при 265 нм, температура колонки 30°С, объем инжекции 50 мкл. Предел детектирования составляет 5 мкг/л. Продолжительность анализа около 18 минут.
Недостатками данного способа являются продолжительность процесса пробоподготовки, использование дорогостоящих ион-парных агентов, которые значительно сокращают срок службы колонки и использование достаточно длинной колонки, что приводит к увеличению продолжительности анализа и увеличению расхода элюентов.
Известен способ определения ЭДТА в безалкогольных напитках методом обращенно-фазовой ион-парной жидкостной хроматографии (Cagnasso С.Е. et al. Development and validation of a method for the determination of EDTA in non-alcoholic drinks by HPLC. Journal of food composition and analysis, 2007, vol. 20, 248-251). Подготовка стандартного образца включает растворение навески Na2CaEDTA в воде, разведение ацетонитрилом, перемешивание и гомогенизацию с раствором хлорида железа (III). Подготовка испытуемых образцов безалкогольных напитков включает добавление ацетонитрила, центрифугирование, добавление раствора хлорида железа (III) к супернатанту, фильтрование через фильтр с диаметром пор 0,45 мкм. Предварительно некоторые образцы напитков могут быть дегазированы или приготовлены в соответствии с инструкцией. Хроматографическое разделение осуществляют с использованием колонки Marcherey-Nagel С18 (ЕТ 250/8/4 Nucleosil® 10 С18) размером 200 мм. Подвижной фазой является смесь 0,01 М раствора аммония фосфата одноосновного: ацетонитрил: 40%-ый водный раствор гидроксида тетрабутиламмония в соотношении 90:10:0,2 (рН=2,42). Определение проводят в изократическом режиме со скоростью потока подвижной фазы 0,9 мл/мин и детекцией при 257 нм, объем инжекции 50 мкл. Предел детектирования составляет 0,6 мг/л, предел количественного определения - 2,0 мг/л.
Недостатками данного способа являются низкий предел количественного определения (2,0 мкг/мл), использование дорогостоящих ион-парных агентов, которые значительно сокращают срок службы колонки и использование достаточно длинной колонки, что приводит к увеличению продолжительности анализа и увеличению расхода элюентов.
Известен способ определения следовых количеств ДМСО в лекарственной субстанции (Bisnafide) методом ОФ ВЭЖХ (Walker J.T. Quantitation of residual dimethylsulfoxide in a drug substance (Bisnafide) by reversed-phase high-performance liquid chromatography. Journal of chromatographic science, 1996, vol. 34, 513-516). Подготовка стандартного образца ДМСО включает приготовление растворов известных концентраций. Приготовление испытуемого образца включает растворение навески субстанции в воде. Хроматографическое разделение осуществляют на колонке Zorbax Rx-C8. В качестве подвижной фазы используют 0,01 М раствор натрия хлорида в 0,1% растворе фосфорной кислоты (фаза А) и смесь 90% ацетонитрила с 10% фазы А (Фаза Б). Определение проводят в режиме градиента: 0-10 мин фаза А, 10-15 мин фаза Б, 15-30 мин фаза А. Скорость потока подвижной фазы - 0,8 мл/мин, температура колонки 45±2°С, объем инжекции 200 мкл, детекция при 215 нм. Предел детектирования метода составляет 0,051 мкг/мл, предел количественного определения метода - 0,219 мкг/мл. Предел детектирования и предел количественного определения в испытуемом образце субстанции составляют 25,5 ppm ДМСО и 109,5 ppm ДМСО соответственно.
Недостатком данного способа является большой объем инжекции исследуемого образца.
Известен способ определения ДМСО в воде и сточных водах методом ВЭЖХ (Determination and quantification of dimethyl sulfoxide by HPLC, Chromatographia, vol. 32, 1991). Пробоподготовка включает фильтрование образца через нитроцеллюлозную мембрану с диаметром пор 0,2 мкм. Хроматографическое разделение осуществляют на колонке Hypersil ODS5 размером 250×4,6 мм, заполненной сорбентом с зернением 5 мкм. Также используют предколонку. В качестве подвижной фазы используют воду очищенную (фаза А) и ацетонитрил (фаза Б). Определение проводят в режиме градиента: 0-10 мин фаза А, 10-15 мин до 50% фаза Б, 15-20 мин до 100% фаза А. Скорость потока подвижной фазы - 0,8 мл/мин, объем инжекции 200 мкл, детекция при 195 нм. Предел количественного определения 0,5 ppm.
Недостатками данного способа являются большой объем инжекции исследуемого образца и использование достаточно длинной колонки, что приводит к увеличению продолжительности анализа и увеличению расхода элюентов.
В качестве ближайшего аналога принят способ определения органических кислот в белых винах методом ОФ-ВЭЖХ (Kordis-Krapez М., Abram V., Kас М., Ferjancic S. Determination of organic acids in white wines by RP-HPLC. Food technol. Biotechnol 2001; 39: 93-99). Подготовка стандартных растворов включает разведение растворителем до определенных концентраций. Подготовка испытуемых растворов включает разведение пробы вина растворителем в соотношении 1/1 и стерилизующую фильтрацию с помощью шприцевых фильтров. Растворителем для приготовления стандартных и испытуемых растворов служит раствор 96% этанола в бидистиллированной воде в соотношении 10/90. Непосредственно перед применением для удаления пузырьков воздуха растворитель обрабатывают ультразвуком на ультразвуковой бане в течение 5 минут. Хроматографическое разделение осуществляют с использованием хроматографической колонки LiChrosorb RP-18 размером 250×4,0 мм, заполненной сорбентом с зернением 10 мкм. Подвижной фазой является раствор кислоты ортофосфорной 6⋅10-3 моль/л (рН=2,1). Определение проводят в изократическом режиме со скоростью потока подвижной фазы 1,0 мл/мин и детекцией при 210 нм, объем инжекции 50 мкл. Продолжительность анализа составляет 9 мин.
Недостатками данного способа являются: ограничение ряда идентифицируемых веществ органическими кислотами и использование достаточно длинной колонки, что приводит к увеличению продолжительности анализа и увеличению расхода элюентов.
Признаки прототипа, совпадающие с признаками изобретения, состоят в разделении на хроматографической колонке, заполненной обращенно-фазовым носителем, содержащим привитую фазу С18, использовании раствора кислоты ортофосфорной в качестве подвижной фазы, детектировании с использованием спектрофотометрического детектора при длине волны 210 нм.
Причины, препятствующие получению технического результата в прототипе, который обеспечивается изобретением, заключаются в том, что длина колонки, средний размер частиц сорбента и объем инжекции приводят к снижению эффективности разделения примесей. Изократический режим элюирования приводит к увеличению продолжительности анализа.
Задачей, решаемой изобретением, является повышение эффективности способа и расширение спектра применения.
Технический результат, обеспечивающий решение поставленной задачи, заключается в увеличении количества определяемых во время одного анализа веществ и повышении точности количественного определения веществ с различной физико-химической природой, таких как ЭДТА, ДМСО и ЭТМ в фармацевтических субстанциях.
Технический результат достигается тем, что в способе определения примесей этилендиаминтетрауксусной кислоты, диметилсульфоксида и N-этилмалеимида методом обращенно-фазовой высокоэффективной жидкостной хроматографии в фармацевтических субстанциях, включающем разделение на хроматографической колонке, заполненной обращенно-фазовым носителем, содержащим привитую фазу С18, использование раствора кислоты ортофосфорной в качестве подвижной фазы, детектирование с использованием спектрофотометрического детектора при длине волны 210 нм, согласно изобретению определение ЭДТА, ДМСО и ЭТМ в фармацевтической субстанции проводится во время одного анализа с использованием хроматографической колонки длиной не более 150 мм, заполненной носителем с зернением не более 5 мкм, используя раствор кислоты ортофосфорной 10-30 мМ (рН 1,9-2,26) с градиентом органического растворителя от 0 до 100%, при температуре колонки 25-45°С, достигается предел детектирования для ЭДТА от 4,14 до 8,0 нг, ДМСО - от 0,8 до 3,0 нг, ЭТМ - 0,04 до 1 нг и предел количественного определения ЭДТА - от 12,9 до 30 нг, ДМСО - от 2,66 до 10 нг, ЭТМ - от 0,13 до 3 нг.
Длина колонки не более 150 мм, размер частиц носителя не более 5 мкм, раствор кислоты ортофосфорной 10-30 мМ (рН 1,9-2,26) в качестве подвижной фазы позволяют проводить разделение ЭДТА, ДМСО и ЭТМ в исследуемой фармацевтической субстанции с высокой точностью и селективностью, поддержание температуры колонки 25-45°С позволяет снизить давление на колонку и проводить анализ при более высокой скорости потока подвижной фазы, сокращая тем самым продолжительность анализа и расход элюентов, градиент ацетонитрила от 0 до 100% позволяет сократить время анализа при совместном определении ЭДТА, ДМСО и ЭТМ.
Новые признаки изобретения заключаются в том, что определение ЭДТА, ДМСО и ЭТМ в фармацевтической субстанции проводится во время одного анализа с использованием хроматографической колонки длиной не более 150 мм, заполненной носителем с зернением не более 5 мкм, используя раствор кислоты ортофосфорной 10-30 мМ (рН 1,9-2,26) с градиентом органического растворителя от 0 до 100%, при температуре колонки 25-45°С, достигаются пределы детектирования для ЭДТА - от 4,14 до 8,0 нг, ДМСО - от 0,8 до 3,0 нг, ЭТМ - 0,04 до 1 нг и предел количественного определения ЭДТА - от 12,9 до 30 нг, ДМСО - от 2,66 до 10 нг, ЭТМ - от 0,13 до 3 нг.
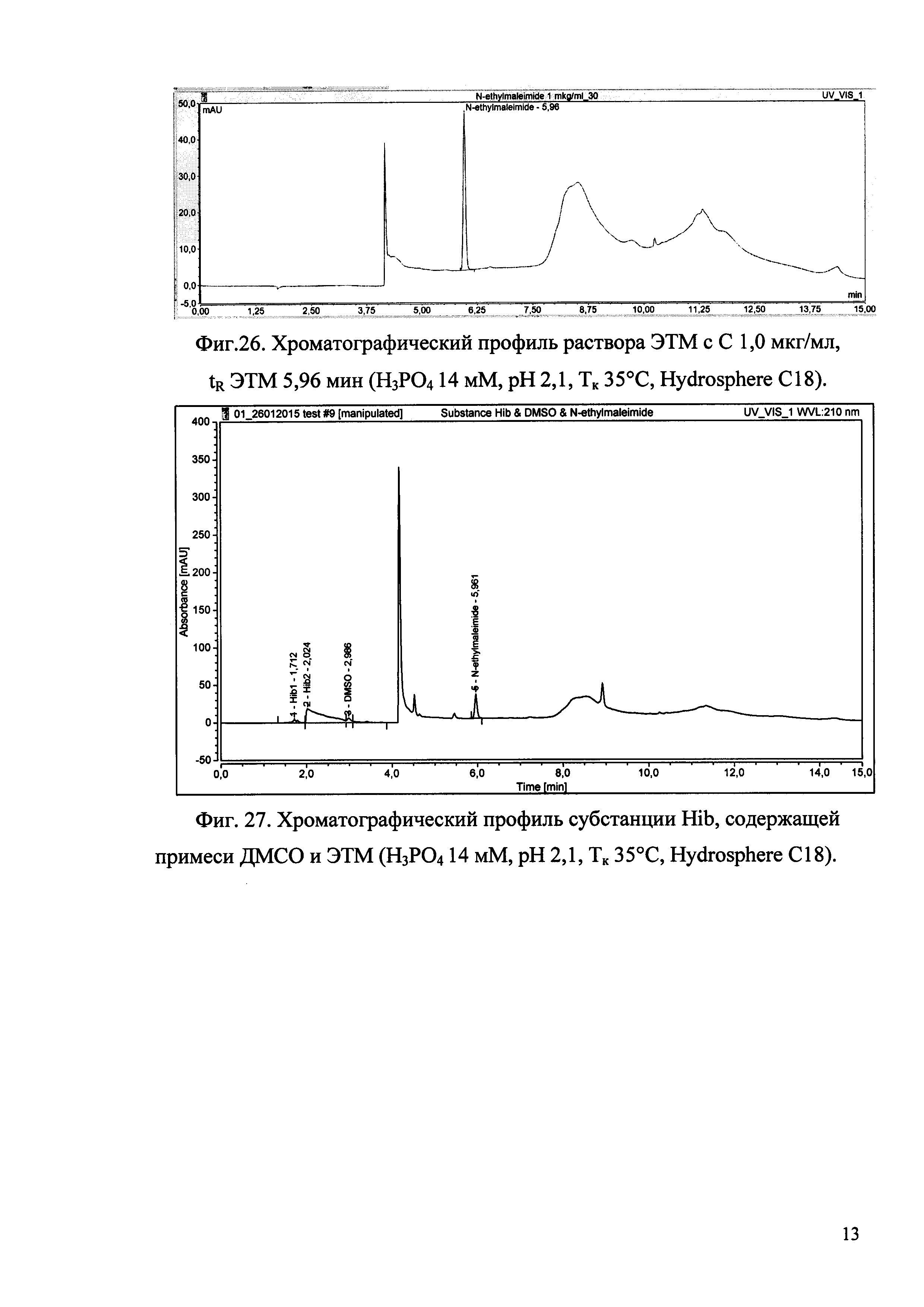
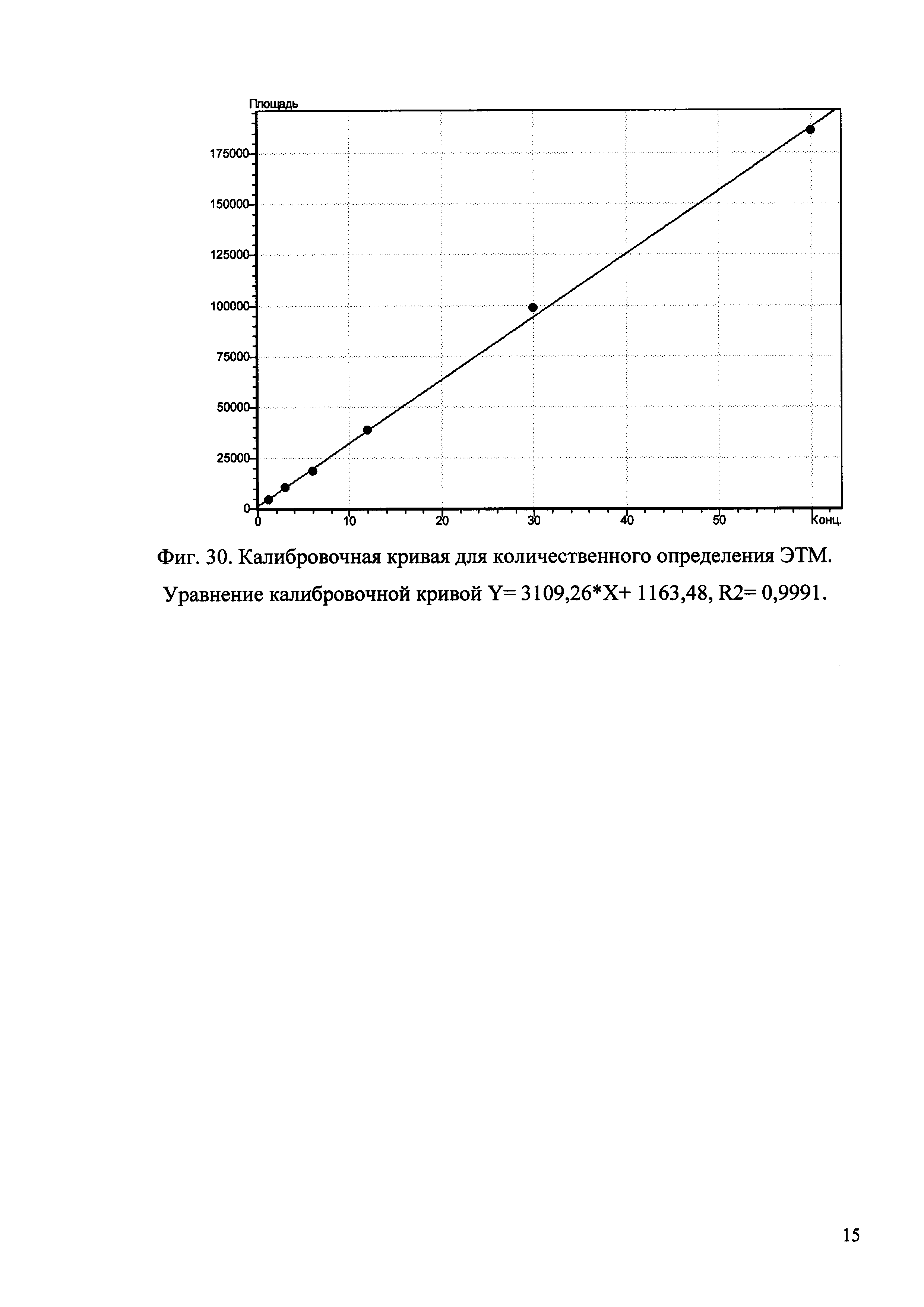
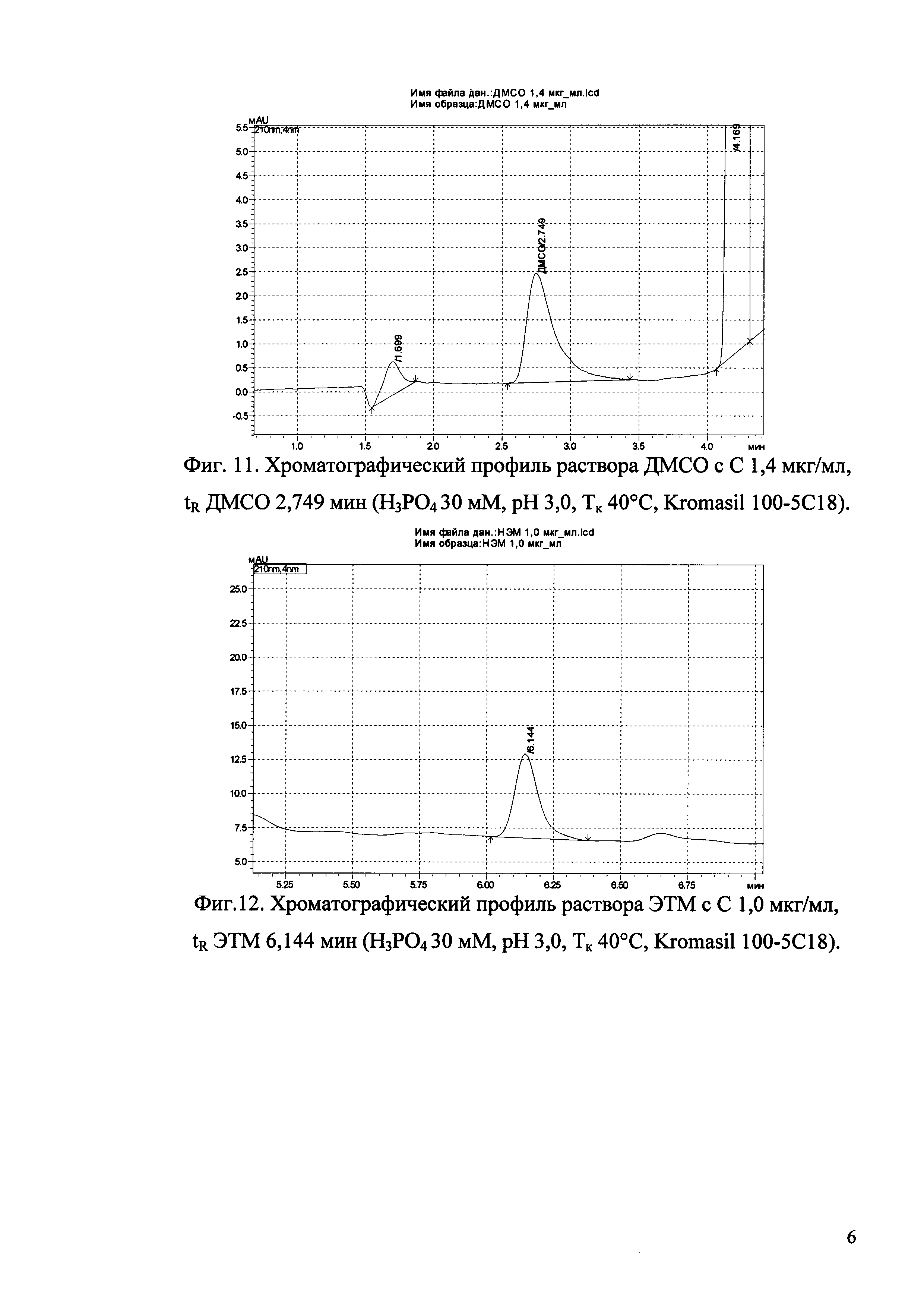
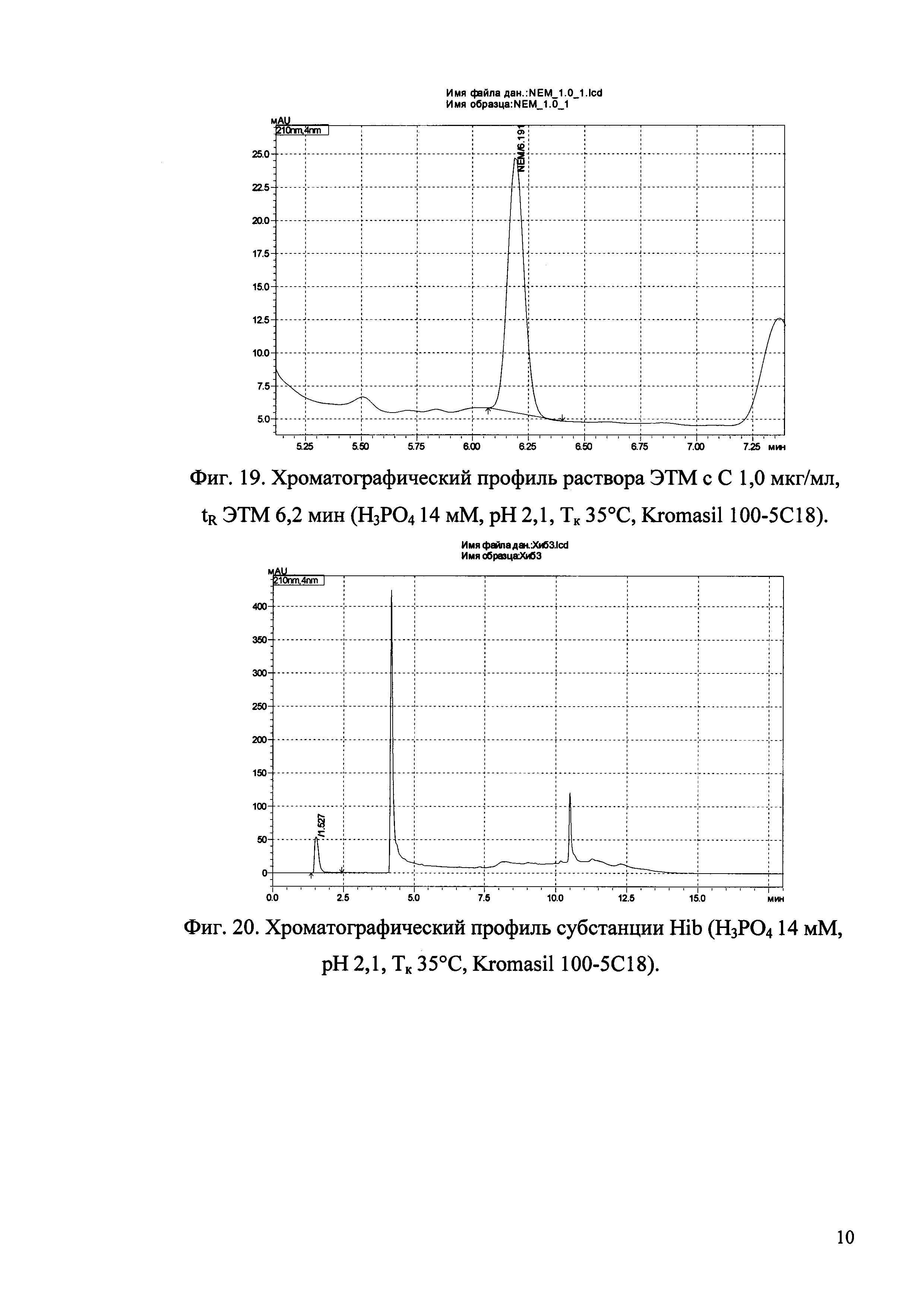
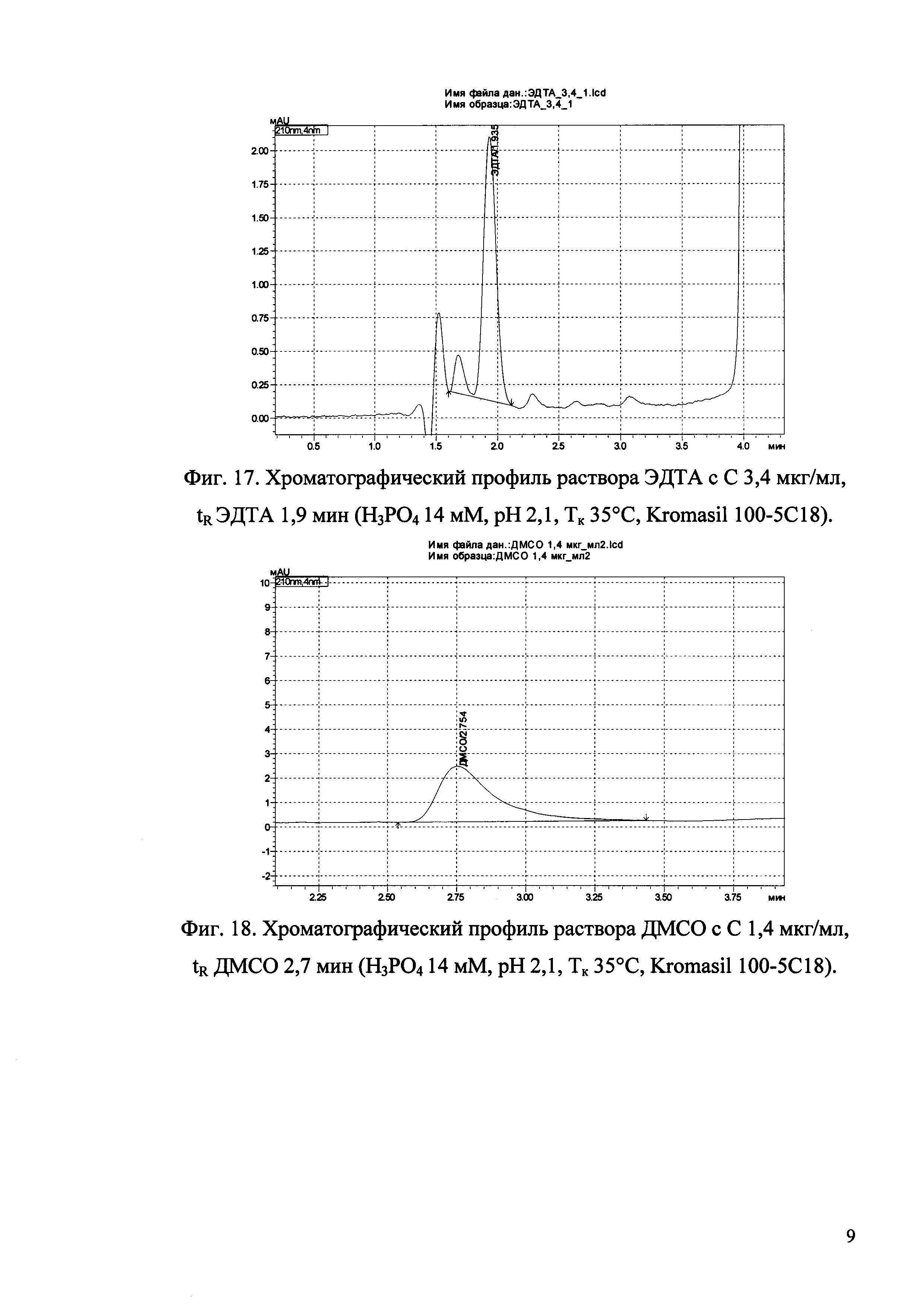
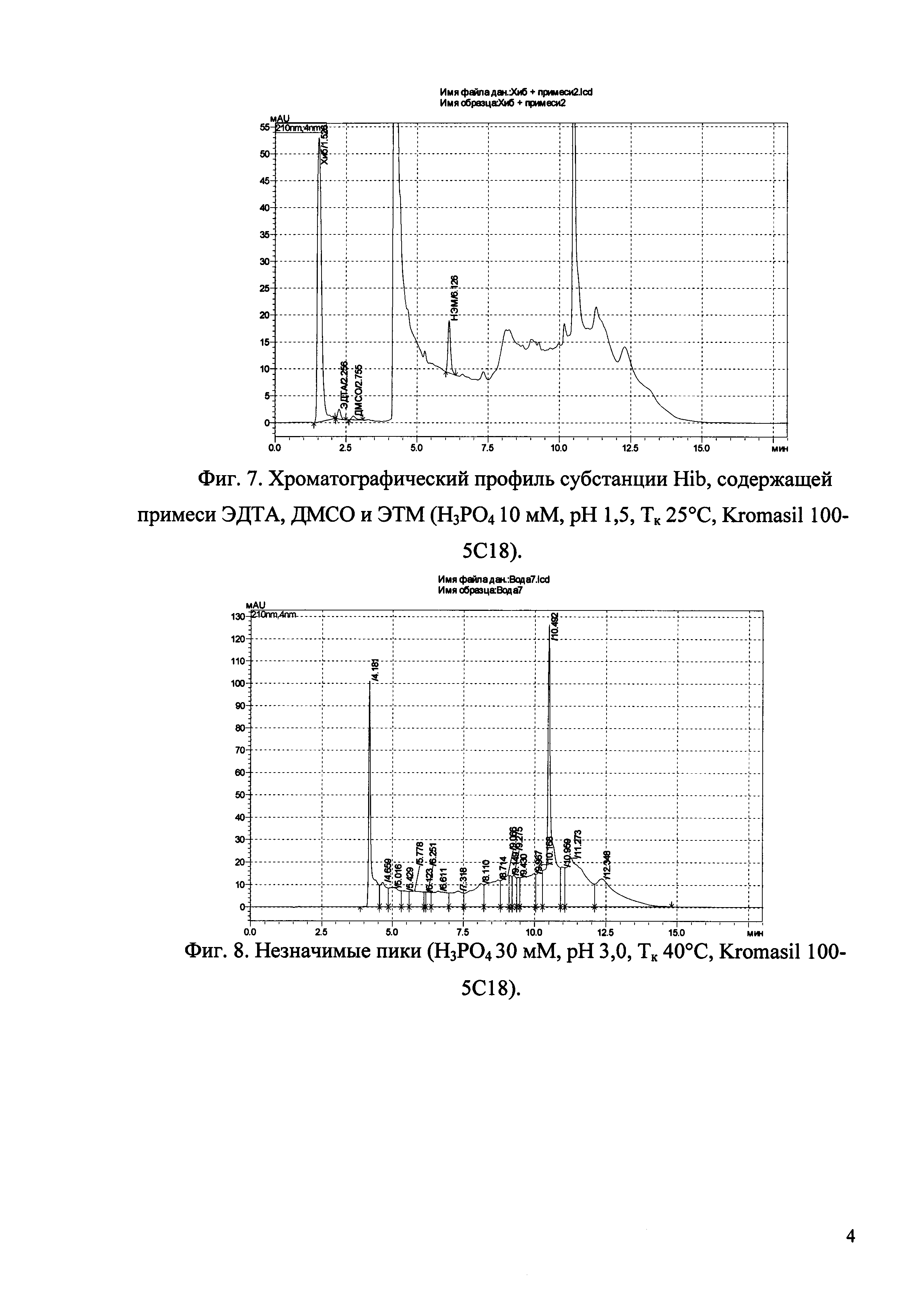
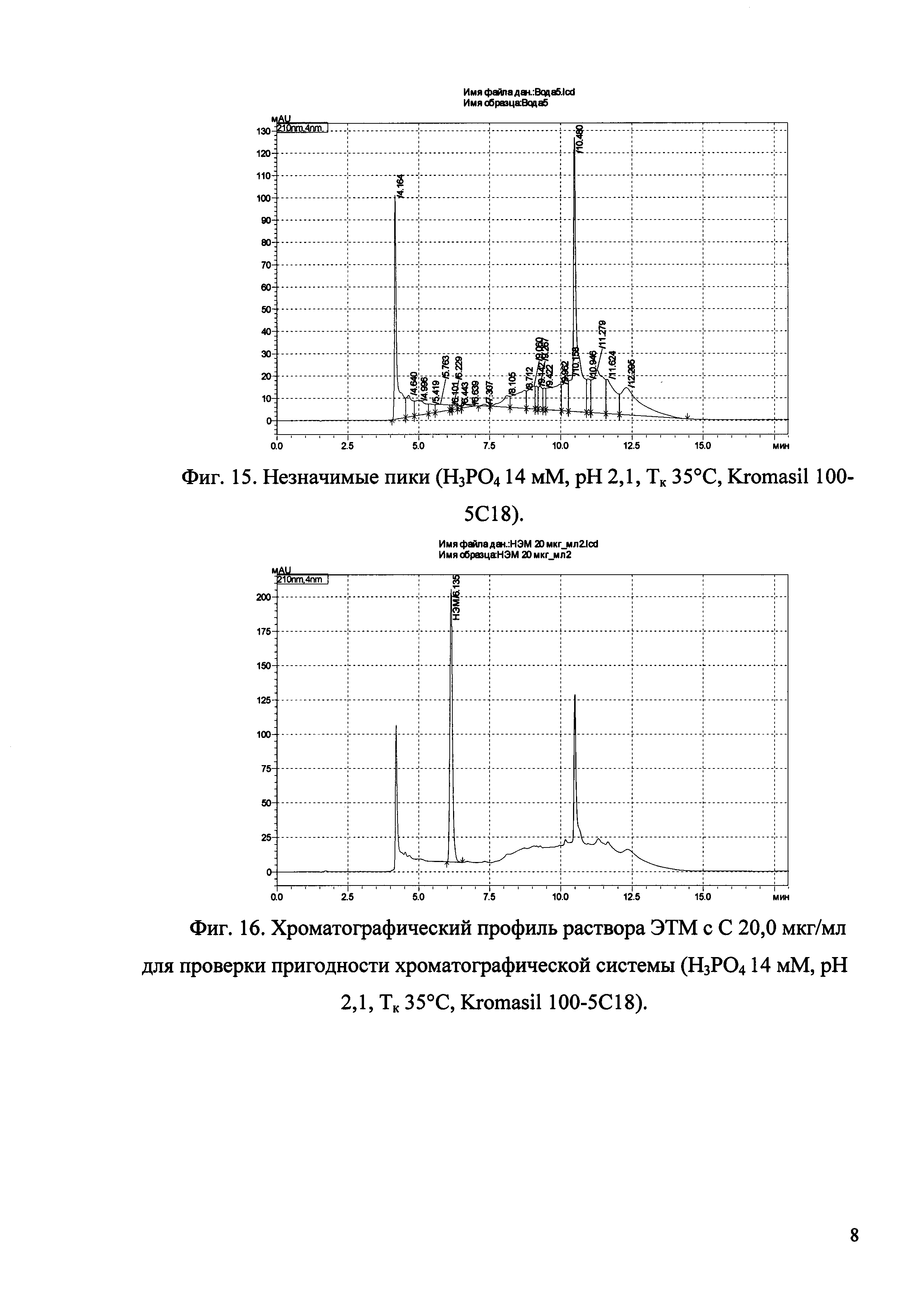
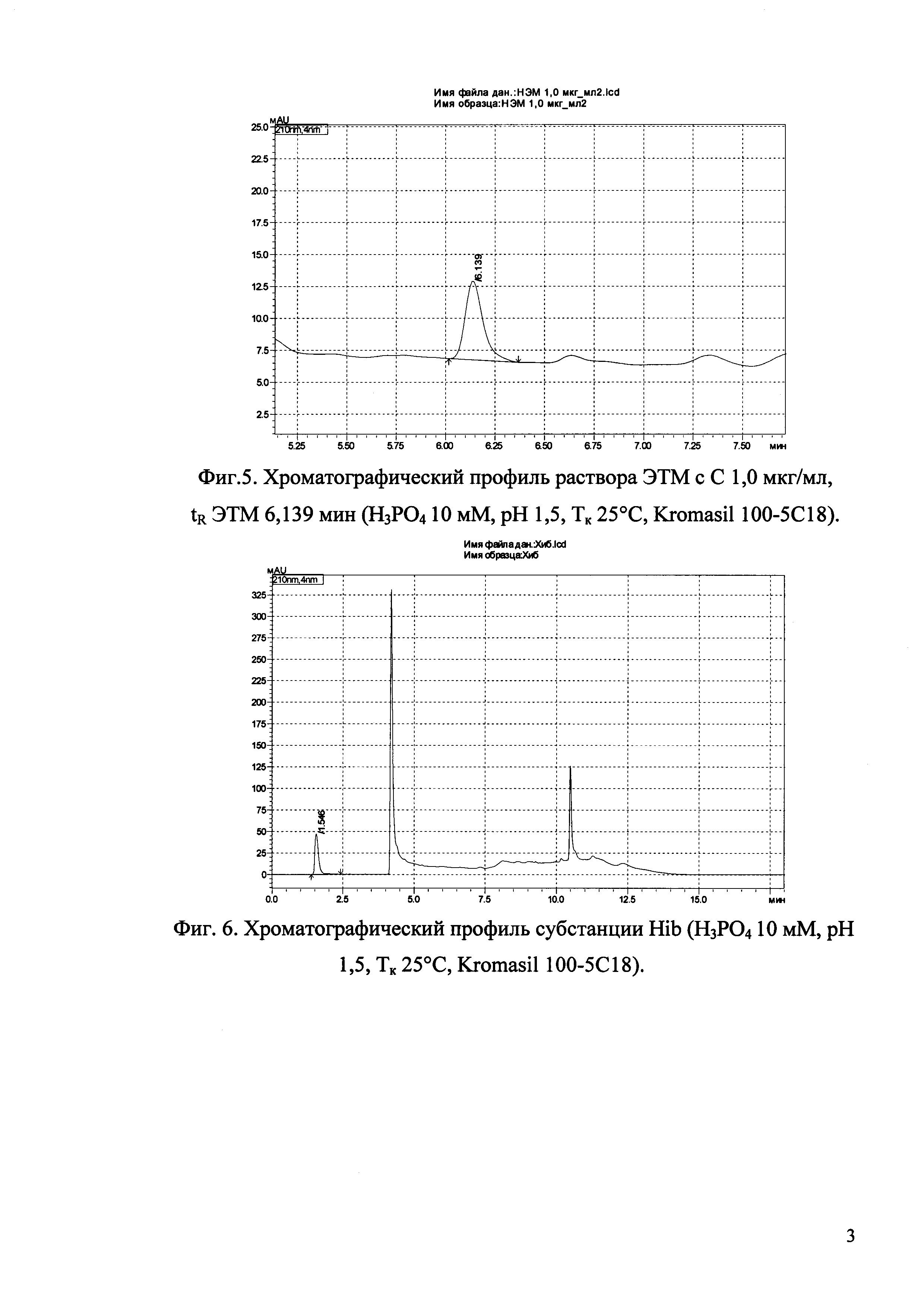
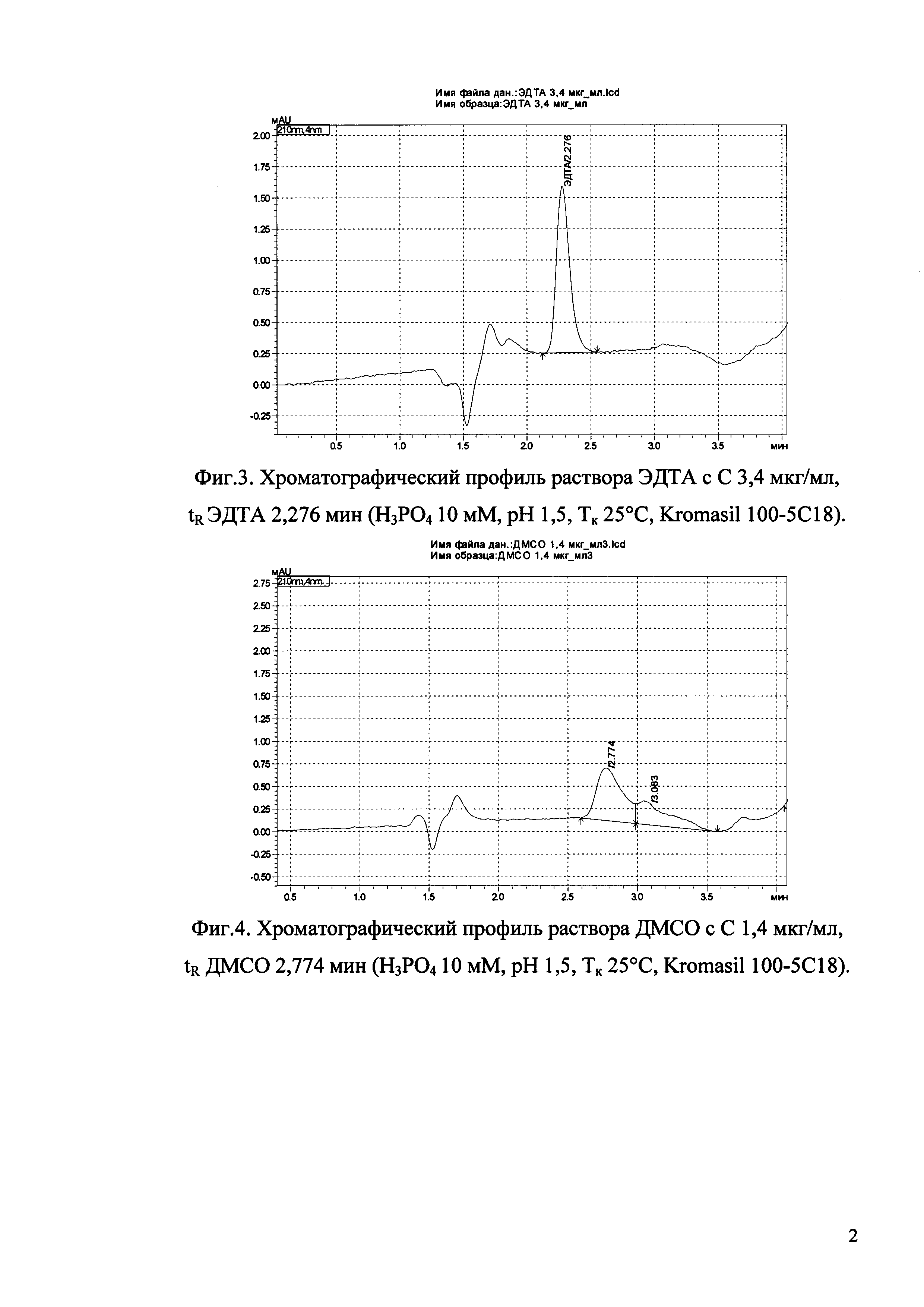
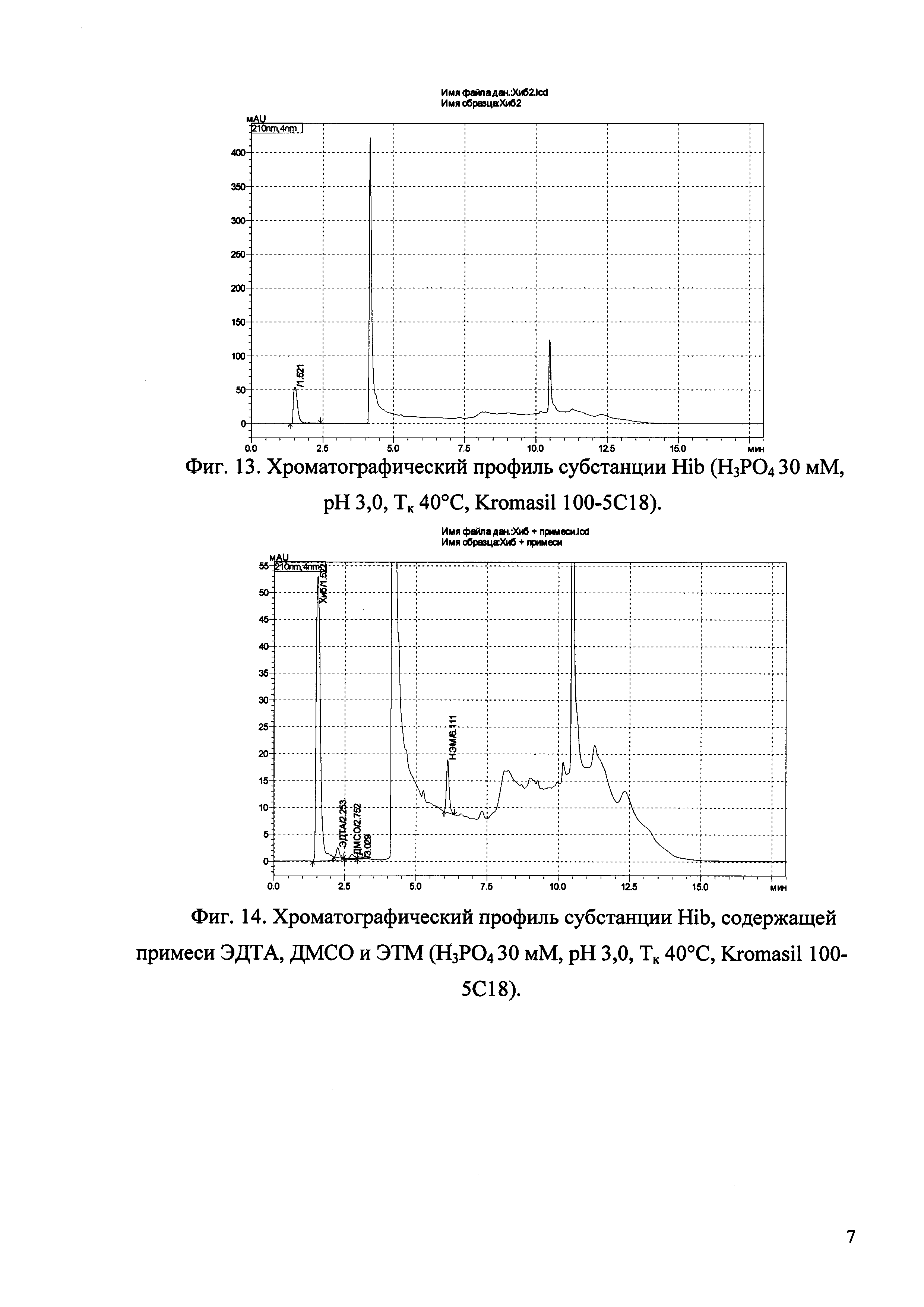
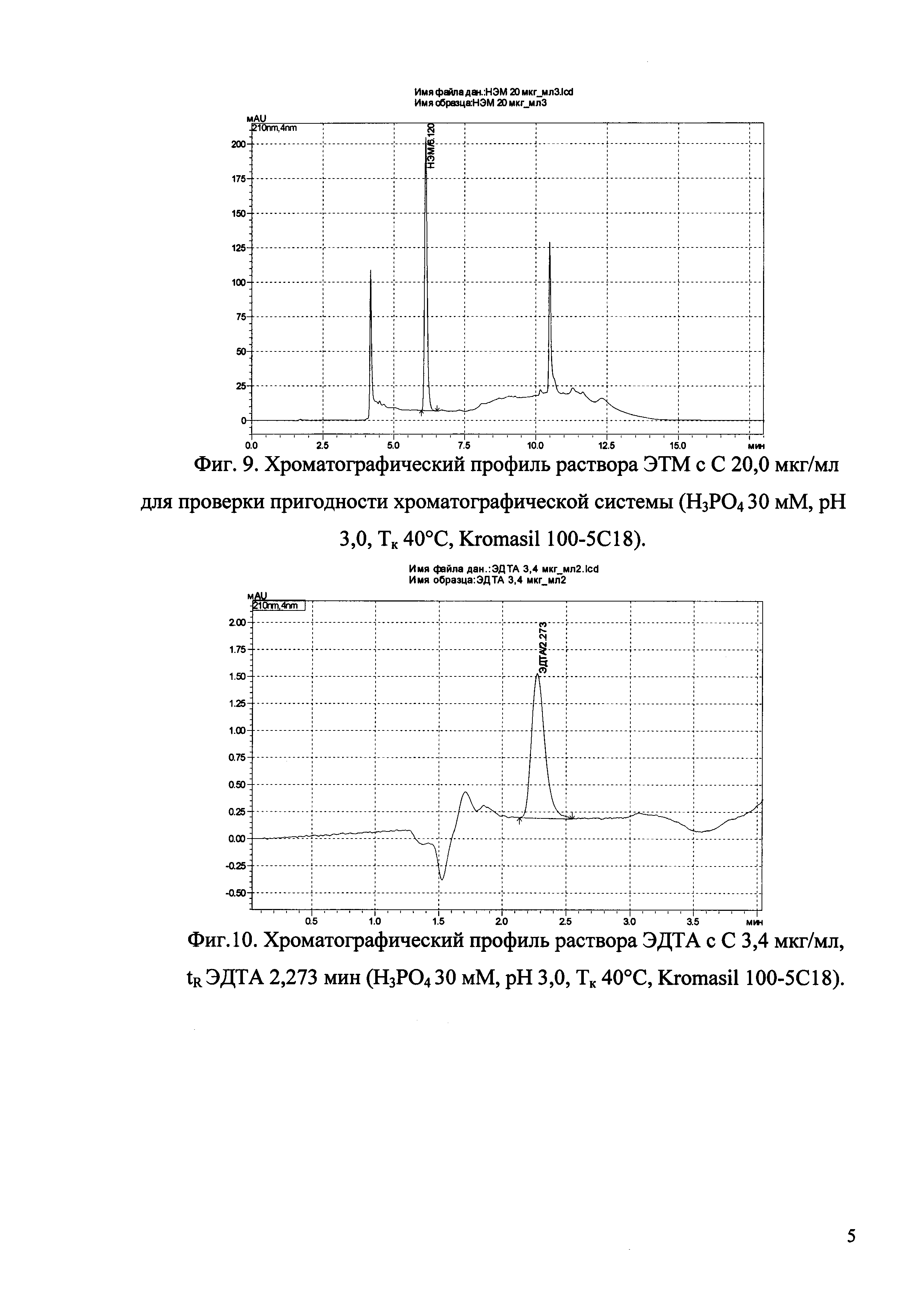
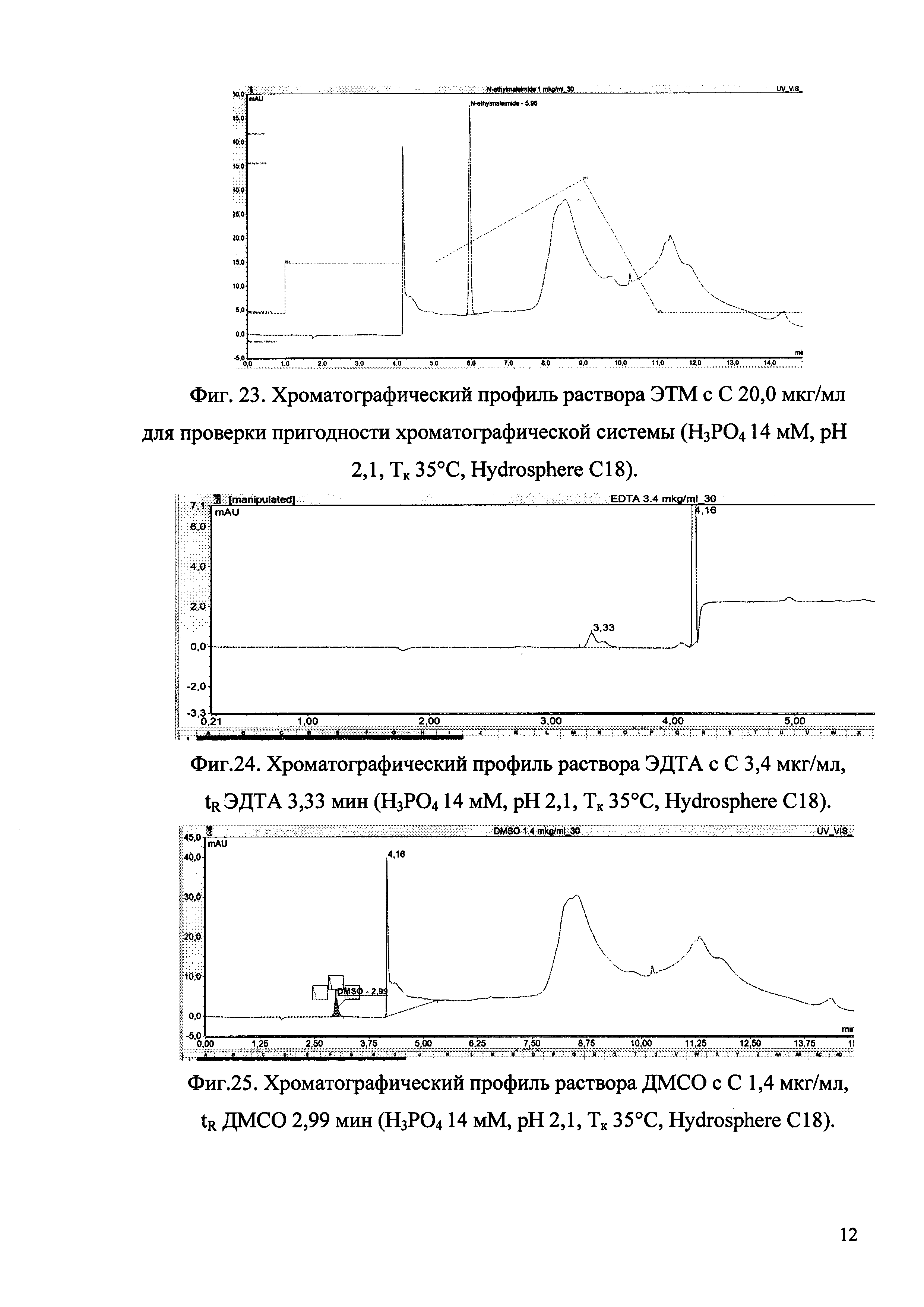
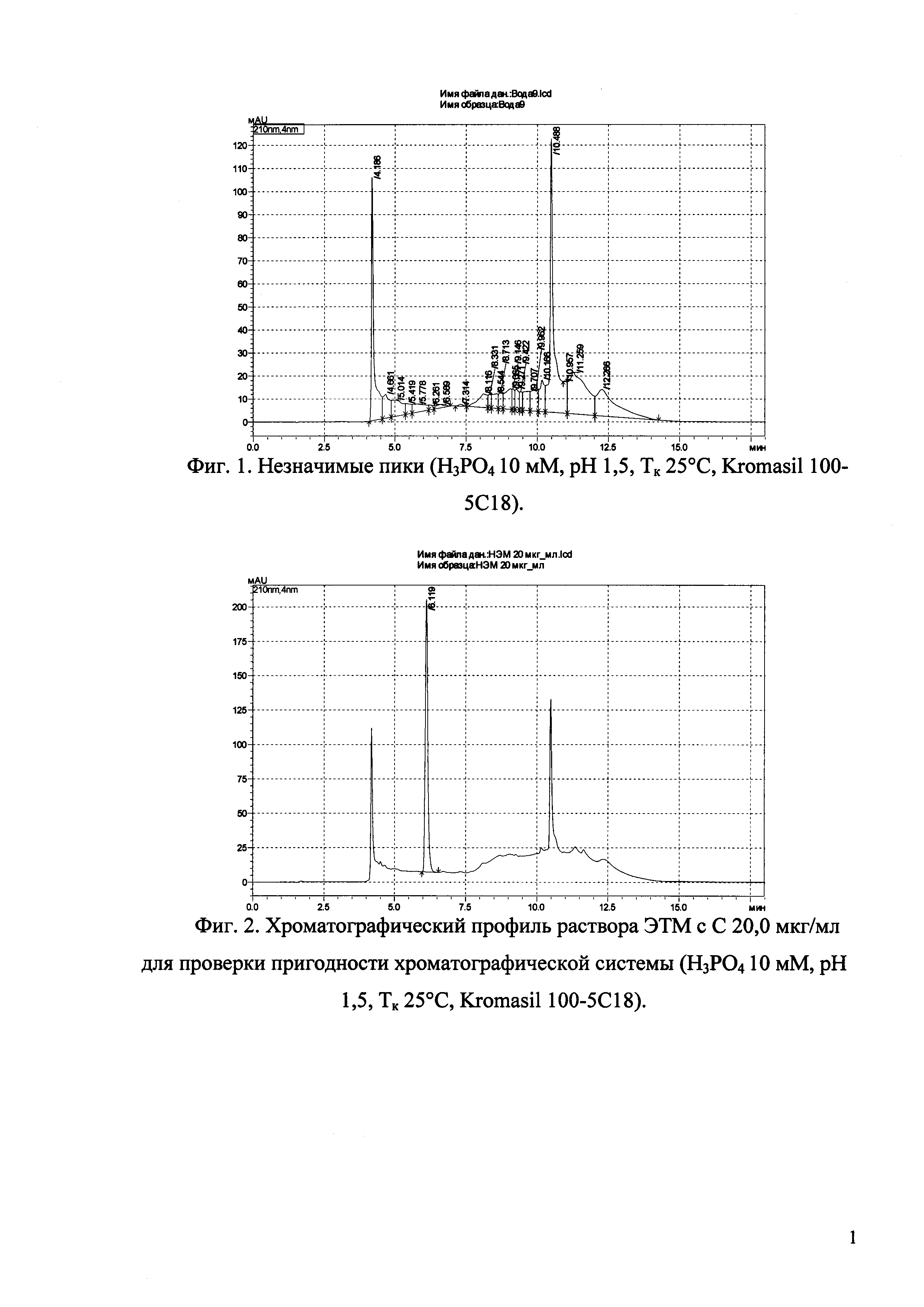
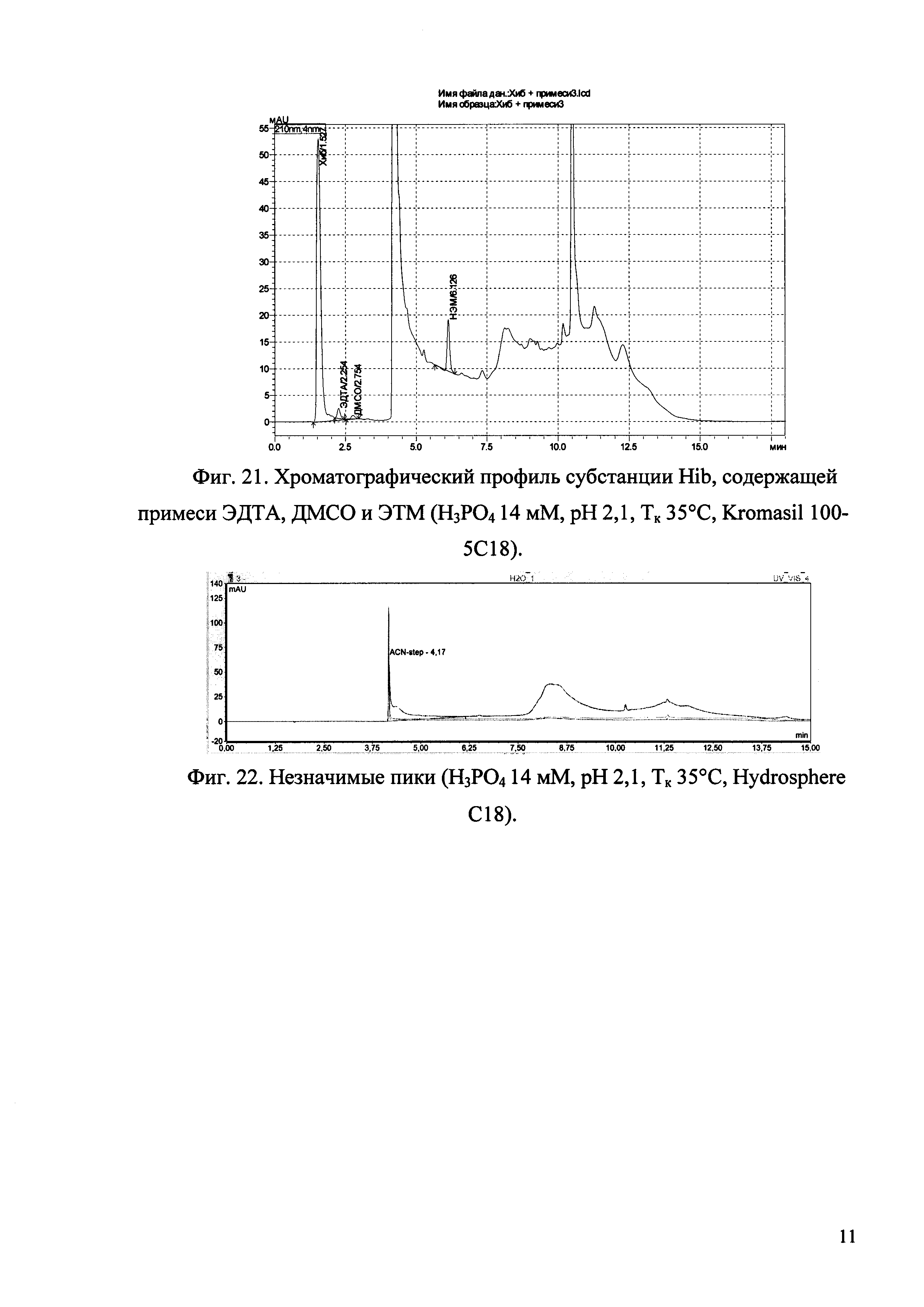
Изобретение иллюстрируется рисунками (хроматограммами), где на фиг. 1 представлена хроматограмма, отражающая незначимые пики (Н3РO4 10 мМ, рН 2,26, Тк 25°С, Kromasil 100-5С18); на фиг. 2 представлен хроматографический профиль раствора ЭТМ с С 20,0 мкг/мл для проверки пригодности хроматографической системы (Н3РO4 10 мМ, рН 2,26, Тк 25°С, Kromasil 100-5С18); на фиг. 3 представлен хроматографический профиль раствора ЭДТА с С 3,4 мкг/мл, tR ЭДТА 2,276 мин (Н3РO4 10 мМ, рН 2,26, Тк 25°С, Kromasil 100-5С18); на фиг. 4. представлен хроматографический профиль раствора ДМСО с С 1,4 мкг/мл, tR ДМСО 2,774 мин (Н3РO4 10 мМ, рН 2,26, Тк 25°С, Kromasil 100-5С18); на фиг. 5 представлен хроматографический профиль раствора ЭТМ с С 1,0 мкг/мл, tR ЭТМ 6,139 мин (Н3РO4 10 мМ, рН 2,26, Тк 25°С, Kromasil 100-5С18); на фиг. 6 представлен хроматографический профиль субстанции Hib (Н3РO4 10 мМ, рН 2,26, Тк 25°С, Kromasil 100-5С18); на фиг. 7 представлен хроматографический профиль субстанции Hib, содержащей примеси ЭДТА, ДМСО и ЭТМ (Н3РO4 10 мМ, рН 2,26, Тк 25°С, Kromasil 100-5С18); на фиг. 8 представлена хроматограмма, отражающая незначимые пики (Н3РO4 30 мМ, рН 1,9, Тк 45°С, Kromasil 100-5С18); на фиг. 9 представлен хроматографический профиль раствора ЭТМ с С 20,0 мкг/мл для проверки пригодности хроматографической системы (Н3РO4 30 мМ, рН 1,9, Тк 45°С, Kromasil 100-5С18); на фиг. 10 представлен хроматографический профиль раствора ЭДТА с С 3,4 мкг/мл, tR ЭДТА 2,273 мин (Н3РO4 30 мМ, рН 1,9, Тк 45°С, Kromasil 100-5С18); на фиг. 11 представлен хроматографический профиль раствора ДМСО с С 1,4 мкг/мл, tR ДМСО 2,749 мин (Н3РO4 30 мМ, рН 1,9, Тк 45°С, Kromasil 100-5С18); на фиг. 12 представлен хроматографический профиль раствора ЭТМ с С 1,0 мкг/мл, tR ЭТМ 6,144 мин (Н3РO4 30 мМ, рН 1,9, Тк 45°С, Kromasil 100-5С18); на фиг. 13 представлен хроматографический профиль субстанции Hib (Н3РО4 30 мМ, рН 1,9, Тк 45°С, Kromasil 100-5С18); на фиг. 14 представлен хроматографический профиль субстанции Hib, содержащей примеси ЭДТА, ДМСО и ЭТМ (Н3РO4 30 мМ, рН 1,9, Тк 45°С, Kromasil 100-5С18); на фиг. 15 представлена хроматограмма, отражающая незначимые пики (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Kromasil 100-5С18); на фиг. 16 представлен хроматографический профиль раствора ЭТМ с С 20,0 мкг/мл для проверки пригодности хроматографической системы (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Kromasil 100-5С18); на фиг. 17 представлен хроматографический профиль раствора ЭДТА с С 3,4 мкг/мл, tR ЭДТА 1,9 мин (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Kromasil 100-5С18); на фиг. 18 представлен хроматографический профиль раствора ДМСО с С 1,4 мкг/мл, tR ДМСО 2,7 мин (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Kromasil 100-5С18); на фиг. 19 представлен хроматографический профиль раствора ЭТМ с С 1,0 мкг/мл, tR ЭТМ 6,2 мин (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Kromasil 100-5С18); на фиг. 20 представлен хроматографический профиль субстанции Hib (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Kromasil 100-5С18); на фиг. 21 представлен хроматографический профиль субстанции Hib, содержащей примеси ЭДТА, ДМСО и ЭТМ (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Kromasil 100-5С18); на фиг. 22 представлена хроматограмма, отражающая незначимые пики (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Hydrosphere С18); на фиг. 23 представлен хроматографический профиль раствора ЭТМ с С 20,0 мкг/мл для проверки пригодности хроматографической системы (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Hydrosphere С18); на фиг. 24 представлен хроматографический профиль раствора ЭДТА с С 3,4 мкг/мл, tR ЭДТА 3,33 мин (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Hydrosphere С18); на фиг. 25 представлен хроматографический профиль раствора ДМСО с С 1,4 мкг/мл, tR ДМСО 2,99 мин (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Hydrosphere С18); на фиг. 26 представлен хроматографический профиль раствора ЭТМ с С 1,0 мкг/мл, tR ЭТМ 5,96 мин (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Hydrosphere С18); на фиг. 27 представлен хроматографический профиль субстанции Hib, содержащей примеси ДМСО и ЭТМ (Н3РO4 14 мМ, рН 2,1, Тк 35°С, Hydrosphere С18); на фиг. 28 представлена калибровочная кривая для количественного определения ЭДТА, уравнение калибровочной кривой Y=145,62*X+692,87, R2=0,9963; на фиг. 29 представлена калибровочная кривая для количественного определения ДМСО, уравнение калибровочной кривой Y=810,18⋅X-1248,88, R2=0,9998; на фиг. 30 представлена калибровочная кривая для количественного определения ЭТМ, уравнение калибровочной кривой Y=3109,26⋅Х+1163,48, R2=0,9991.
Изобретение осуществляют следующим образом.
Пример 1. Определение примесей ЭДТА, ДМСО и ЭТМ методом обращенно-фазовой ВЭЖХ в субстанции вакцины для профилактики инфекций, вызываемых Haemophilus influenzae типа b, конъюгированной синтетической (субстанции Hib).
Первоначально готовят водные растворы примесей ЭДТА, ДМСО и ЭТМ в концентрациях, соответствующих их предельно допустимому содержанию в фармацевтической субстанции Hib.
Для приготовления раствора ЭДТА с С 3,4 мкг/мл 25 мг (точная навеска) натрия эдетеата помещают в мерную колбу вместимостью 25 мл, растворяют в 10 мл воды, доводят объем раствора водой до метки и перемешивают. 340 мкл полученного раствора переносят в мерную колбу вместимостью 100 мл, растворяют в 50 мл воды и доводят объем раствора до метки. Раствор тщательно перемешивают и фильтруют через мембранный фильтр.
Для приготовления раствора ДМСО с С 1,4 мкг/мл 45 мкл диметилсульфоксида помещают в мерную колбу вместимостью 50 мл, растворяют в 20 мл воды, доводят объем раствора до метки и перемешивают. 140 мкл полученного раствора переносят в мерную колбу вместимостью 100 мл, растворяют в 50 мл воды и доводят объем раствора до метки. Раствор тщательно перемешивают и фильтруют через мембранный фильтр.
Для приготовления раствора ЭТМ с С 1,0 мкг/мл 50 мг N-этилмалеимида помещают в мерную колбу вместимостью 50 мл, растворяют в 20 мл воды, доводят объем раствора до метки и перемешивают. 100 мкл полученного раствора переносят в мерную колбу вместимостью 100 мл, растворяют в 50 мл воды и доводят объем раствора до метки. Раствор тщательно перемешивают и фильтруют через мембранный фильтр.
В качестве подвижной фазы для проведения анализа используют 10-30 мМ раствор кислоты ортофосфорной (рН 1,5-3,0) с градиентом ацетонитрила 0-100%.
10-30 мМ раствор кислоты ортофосфорной (Н3РO4) готовят непосредственно перед применением из 140 мМ раствора кислоты ортофосфорной.
Для приготовления раствора ортофосфорной кислоты 140 мМ 8,2 мл ортофосфорной кислоты концентрированной помещают в мерную колбу вместимостью 500 мл, растворяют в 200 мл воды, доводят объем раствора до метки и перемешивают. Полученный раствор фильтруют через мембранный фильтр.
Для приготовления раствора ортофосфорной кислоты 10-30 мМ (рН 1,5-3,0) 71,4-215 мл раствора Н3РO4 140 мМ переносят в мерную колбу вместимостью 1000 мл, растворяют в 500 мл воды и доводят объем раствора до метки, перемешивают и измеряют рН. Полученный раствор фильтруют через мембранный фильтр, используют свежеприготовленным.
Хроматографический анализ проводят с использованием хроматографической системы, снабженной спектрофотометрическим детектором, насосом для подачи подвижной фазы, дегазатором, автоматическим или ручным инжектором с возможностью поддержания температуры образцов +4°С, термостатируемым колоночным отделением и компьютером с установленной программой сбора и обработки хроматографических данных в следующих условиях:
- используют хроматографическую колонку длиной не более 150 мм, заполненную обращенно-фазовым носителем с зернением не более 5 мкм;
- скорость потока подвижной фазы 1 мл/мин;
- объем вводимой пробы 30 мкл;
- температура колонки (Тк) 25-45°С;
- длина волны детектирования 210 нм;
- продолжительность анализа 17 мин.
Устанавливают рабочую скорость потока и уравновешивают колонку, пропуская через нее раствор подвижной фазы до установления базовой линии.
Для установления незначимых пиков, пиков, относящихся к градиенту ацетонитрила, проводят несколько последовательных инжекций воды до получения идентичных результатов. Фиксируют пики, относящиеся к полному объему колонки, градиенту ацетонитрила и другие системные пики.
Проводят тест проверки пригодности хроматографической системы.
Для этого хроматографируют раствор ЭТМ с С 20,0 мкг/мл, получают не менее трех хроматограмм. Устанавливают время удерживания пика ЭТМ, исключая все незначимые пики.
Посредством программного обеспечения получают значения эффективности хроматографической колонки, фактора асимметрии пика и площади пика для каждого из анализов.
Хроматографическую систему считают пригодной, если
- эффективность хроматографической колонки не менее 15000 теоретических тарелок;
- фактор асимметрии пика (Т) не менее 1,0±0,15;
- относительное стандартное отклонение площади пика не более 5%.
В описанных выше условиях хроматографируют последовательно растворы примесей, получая не менее трех хроматограмм для каждого раствора. Устанавливают время удерживания для каждой примеси.
Рассчитывают относительное время удерживания (RRT) примесей относительно пика ЭТМ по следующей формуле:

где tR1 - время удерживания ЭТМ, мин;
tR2 - время удерживания ЭДТА и ДМСО, мин.
В описанных выше условиях хроматографируют раствор субстанции Hib, получая не менее трех хроматограмм. Устанавливают присутствие примесей в фармацевтической субстанции по наличию пика в установленных интервалах времени для каждой примеси.
Пример 2. Определение примесей ЭДТА, ДМСО и ЭТМ методом ОФ-ВЭЖХ в субстанции Hib (раствор кислоты ортофосфорной 10 мМ, рН 2,26, температура колонки 25°С).
Готовят водные растворы примесей ЭДТА, ДМСО и ЭТМ как описано в примере 1.
В качестве подвижной фазы для проведения анализа используют 10 мМ раствор кислоты ортофосфорной (рН 2,26) с градиентом ацетонитрила 30-80%.
10 мМ раствор кислоты ортофосфорной готовят непосредственно перед применением из 140 мМ раствора кислоты ортофосфорной.
Для приготовления раствора ортофосфорной кислоты 140 мМ 8,2 мл ортофосфорной кислоты концентрированной помещают в мерную колбу вместимостью 500 мл, растворяют в 200 мл воды, доводят объем раствора до метки и перемешивают. Полученный раствор фильтруют через мембранный фильтр.
Для приготовления раствора ортофосфорной кислоты 10 мМ (рН 2,26) 71,4 мл раствора Н3РО4 140 мМ переносят в мерную колбу вместимостью 1000 мл, растворяют в 500 мл воды и доводят объем раствора до метки, перемешивают и измеряют рН. Полученный раствор фильтруют через мембранный фильтр, используют свежеприготовленным.
Хроматографический анализ проводят с использованием хроматографической системы, снабженной спектрофотометрическим детектором, насосом для подачи подвижной фазы, дегазатором, автоматическим или ручным инжектором с возможностью поддержания температуры образцов +4°С, термостатируемым колоночным отделением и компьютером с установленной программой сбора и обработки хроматографических данных в следующих условиях:
- колонка Kromasil 100-5С18 (5 мкм, 150×4,8 мм) производства «AkzoNobel», Голландия, кат. №M05CLA15;
- скорость потока подвижной фазы 1 мл/мин;
- объем вводимой пробы 30 мкл;
- температура колонки 25°С;
- длина волны детектирования 210 нм;
- продолжительность анализа 17 мин.

Устанавливают рабочую скорость потока и уравновешивают колонку, пропуская через нее раствор подвижной фазы до установления базовой линии.
Для установления незначимых пиков, пиков, относящихся к градиенту ацетонитрила, проводят несколько последовательных инжекций воды до получения идентичных результатов. Фиксируют пики, относящиеся к полному объему колонки, градиенту ацетонитрила и другие системные пики.
Проводят тест проверки пригодности хроматографической системы.
Для этого хроматографируют раствор ЭТМ с С 20,0 мкг/мл, получают не менее трех хроматограмм. Устанавливают время удерживания пика ЭТМ, исключая все незначимые пики.
Посредством программного обеспечения получают значения эффективности хроматографической колонки, фактора асимметрии пика и площади пика для каждого из анализов.
Хроматографическую систему считают пригодной, если
- эффективность хроматографической колонки не менее 15000 теоретических тарелок;
- фактор асимметрии пика (Т) не менее 1,0±0,15;
- относительное стандартное отклонение площади пика не более 5%.

В описанных выше условиях хроматографируют последовательно растворы примесей, получая не менее трех хроматограмм для каждого раствора. Устанавливают время удерживания для каждой примеси.
Рассчитывают относительное время удерживания (RRT) примесей относительно пика ЭТМ по следующей формуле:

где tR1 - время удерживания ЭТМ, мин;
tR2 - время удерживания ЭДТА и ДМСО, мин.
Относительное время удерживания ЭДТА - 0,37±0,01, ДМСО - 0,45±0,01, ЭТМ - 1,0±0,01.
В описанных выше условиях хроматографируют раствор субстанции Hib, получая не менее трех хроматограмм. Устанавливают присутствие примесей в фармацевтической субстанции по наличию пика в установленных интервалах времени для каждой примеси.

Пример 3. Определение примесей ЭДТА, ДМСО и ЭТМ методом ОФ-ВЭЖХ в субстанции Hib (раствор кислоты ортофосфорной 30 мМ, рН 1,9, температура колонки 40°С).
Готовят водные растворы примесей ЭДТА, ДМСО и ЭТМ как описано в примере 1.
В качестве подвижной фазы для проведения анализа используют 30 мМ раствор кислоты ортофосфорной (рН 1,9) с градиентом ацетонитрила 30-80%.
30 мМ раствор кислоты ортофосфорной готовят непосредственно перед применением из 140 мМ раствора кислоты ортофосфорной.
Для приготовления раствора ортофосфорной кислоты 140 мМ 8,2 мл ортофосфорной кислоты концентрированной помещают в мерную колбу вместимостью 500 мл, растворяют в 200 мл воды, доводят объем раствора до метки и перемешивают. Полученный раствор фильтруют через мембранный фильтр.
Для приготовления раствора ортофосфорной кислоты 30 мМ (рН 1,9) 215 мл раствора Н3РО4 140 мМ переносят в мерную колбу вместимостью 1000 мл, растворяют в 500 мл воды и доводят объем раствора до метки, перемешивают и измеряют рН. Полученный раствор фильтруют через мембранный фильтр, используют свежеприготовленным.
Хроматографический анализ проводят с использованием хроматографической системы, снабженной спектрофотометрическим детектором, насосом для подачи подвижной фазы, дегазатором, автоматическим или ручным инжектором с возможностью поддержания температуры образцов +4°С, термостатируемым колоночным отделением и компьютером с установленной программой сбора и обработки хроматографических данных в следующих условиях:
- колонка Kromasil 100-5С18 (5 мкм, 150×4,8 мм) производства «AkzoNobel», Голландия, кат. №M05CLA15;
- скорость потока подвижной фазы 1 мл/мин;
- объем вводимой пробы 30 мкл;
- температура колонки 40°С;
- длина волны детектирования 210 нм;
- продолжительность анализа 17 мин.

Устанавливают рабочую скорость потока и уравновешивают колонку, пропуская через нее раствор подвижной фазы до установления базовой линии.
Для установления незначимых пиков, пиков, относящихся к градиенту ацетонитрила, проводят несколько последовательных инжекций воды до получения идентичных результатов. Фиксируют пики, относящиеся к полному объему колонки, градиенту ацетонитрила и другие системные пики.
Проводят тест проверки пригодности хроматографической системы.
Для этого хроматографируют раствор ЭТМ с С 20,0 мкг/мл, получают не менее трех хроматограмм. Устанавливают время удерживания пика ЭТМ, исключая все незначимые пики.
Посредством программного обеспечения получают значения эффективности хроматографической колонки, фактора асимметрии пика и площади пика для каждого из анализов.
Хроматографическую систему считают пригодной, если
- эффективность хроматографической колонки не менее 15000 теоретических тарелок;
- фактор асимметрии пика (Т) не менее 1,0±0,15;
- относительное стандартное отклонение площади пика не более 5%.

В описанных выше условиях хроматографируют последовательно растворы примесей, получая не менее трех хроматограмм для каждого раствора. Устанавливают время удерживания для каждой примеси.
Рассчитывают относительное время удерживания (RRT) примесей относительно пика ЭТМ по следующей формуле:

где tR1 - время удерживания ЭТМ, мин;
tR2 - время удерживания ЭДТА и ДМСО, мин.
Относительное время удерживания ЭДТА - 0,37±0,01, ДМСО - 0,45±0,01, ЭТМ - 1,0±0,01.
В описанных выше условиях хроматографируют раствор субстанции Hib, получая не менее трех хроматограмм. Устанавливают присутствие примесей в фармацевтической субстанции по наличию пика в установленных интервалах времени для каждой примеси.

Пример 4. Определение примесей ЭДТА, ДМСО и ЭТМ методом ОФ-ВЭЖХ в субстанции Hib (раствор кислоты ортофосфорной 14 мМ, рН 2,1, температура колонки 35°С).
Готовят водные растворы примесей ЭДТА, ДМСО и ЭТМ как описано в примере 1.
В качестве подвижной фазы для проведения анализа используют 14 мМ раствор кислоты ортофосфорной (рН 2,1) с градиентом ацетонитрила 30-80%.
14 мМ раствор кислоты ортофосфорной готовят непосредственно перед применением из 140 мМ раствора кислоты ортофосфорной.
Для приготовления раствора ортофосфорной кислоты 140 мМ 8,2 мл ортофосфорной кислоты концентрированной помещают в мерную колбу вместимостью 500 мл, растворяют в 200 мл воды, доводят объем раствора до метки и перемешивают. Полученный раствор фильтруют через мембранный фильтр.
Для приготовления раствора ортофосфорной кислоты 14 мМ (рН 2,1) 100 мл раствора Н3РО4 140 мМ переносят в мерную колбу вместимостью 1000 мл, растворяют в 500 мл воды и доводят объем раствора до метки, перемешивают и измеряют рН. Полученный раствор фильтруют через мембранный фильтр, используют свежеприготовленным.
Хроматографический анализ проводят с использованием хроматографической системы, снабженной спектрофотометрическим детектором, насосом для подачи подвижной фазы, дегазатором, автоматическим или ручным инжектором с возможностью поддержания температуры образцов +4°С, термостатируемым колоночным отделением и компьютером с установленной программой сбора и обработки хроматографических данных в следующих условиях:
- колонка Kromasil 100-5С18 (5 мкм, 150×4,8 мм) производства «AkzoNobel», Голландия, кат. №M05CLA15;
- скорость потока подвижной фазы 1 мл/мин;
- объем вводимой пробы 30 мкл;
- температура колонки 35°С;
- длина волны детектирования 210 нм;
- продолжительность анализа 17 мин.

Устанавливают рабочую скорость потока и уравновешивают колонку, пропуская через нее раствор подвижной фазы до установления базовой линии.
Для установления незначимых пиков, пиков, относящихся к градиенту ацетонитрила, проводят несколько последовательных инжекций воды до получения идентичных результатов. Фиксируют пики, относящиеся к полному объему колонки, градиенту ацетонитрила и другие системные пики.
Проводят тест проверки пригодности хроматографической системы.
Для этого хроматографируют раствор ЭТМ с С 20,0 мкг/мл, получают не менее трех хроматограмм. Устанавливают время удерживания пика ЭТМ, исключая все незначимые пики.
Посредством программного обеспечения получают значения эффективности хроматографической колонки, фактора асимметрии пика и площади пика для каждого из анализов.
Хроматографическую систему считают пригодной, если
- эффективность хроматографической колонки не менее 15000 теоретических тарелок;
- фактор асимметрии пика (Т) не менее 1,0±0,15;
- относительное стандартное отклонение площади пика не более 5%.

В описанных выше условиях хроматографируют последовательно растворы примесей, получая не менее трех хроматограмм для каждого раствора. Устанавливают время удерживания для каждой примеси.
Рассчитывают относительное время удерживания (RRT) примесей относительно пика ЭТМ по следующей формуле:

где tR1 - время удерживания ЭТМ, мин;
tR2 - время удерживания ЭДТА и ДМСО, мин.
Относительное время удерживания ЭДТА - 0,31±0,01, ДМСО - 0,44±0,01, ЭТМ - 1,0±0,01.
В описанных выше условиях хроматографируют раствор субстанции Hib, получая не менее трех хроматограмм. Устанавливают присутствие примесей в фармацевтической субстанции по наличию пика в установленных интервалах времени для каждой примеси. 
Пример 5. Определение примесей ЭДТА, ДМСО и ЭТМ методом ОФ-ВЭЖХ в субстанции Hib в оптимальных условиях с использованием альтернативной колонки.
Готовят водные растворы примесей ЭДТА, ДМСО и ЭТМ, раствор 14 мМ кислоты ортофосфорной как описано в примере 1.
Хроматографический анализ проводят с использованием хроматографической системы, снабженной спектрофотометрическим детектором, насосом для подачи подвижной фазы, дегазатором, автоматическим или ручным инжектором с возможностью поддержания температуры образцов +4°С, термостатируемым колоночным отделением и компьютером с установленной программой сбора и обработки хроматографических данных в следующих условиях:
- колонка Hydrosphere С18 150×4,6 мм ID, 3 мкм, 120 Å (кат №HS12S03-1546WT) производства компании YMC (Германия);
- скорость потока подвижной фазы 1 мл/мин;
- объем вводимой пробы 30 мкл;
- температура колонки 35°С;
- длина волны детектирования 210 нм;
- продолжительность анализа 15 мин.

Устанавливают рабочую скорость потока и уравновешивают колонку, пропуская через нее раствор подвижной фазы до установления базовой линии.
Для установления незначимых пиков, пиков, относящихся к градиенту ацетонитрила, проводят несколько последовательных инжекций воды до получения идентичных результатов. Фиксируют пики, относящиеся к полному объему колонки, градиенту ацетонитрила и другие системные пики.
Проводят тест проверки пригодности хроматографической системы как описано в примере 1.

В описанных выше условиях хроматографируют последовательно растворы примесей, получая не менее трех хроматограмм для каждого раствора. Устанавливают время удерживания для каждой примеси.
Рассчитывают относительное время удерживания (RRT) примесей относительно пика ЭТМ как описано в примере 1.
Относительное время удерживания ЭДТА - 0,56±0,01, ДМСО - 0,5±0,01, ЭТМ - 1,0±0,01.
В описанных выше условиях хроматографируют раствор субстанции Hib, получая не менее трех хроматограмм. Устанавливают присутствие примесей в фармацевтической субстанции по наличию пика в установленных интервалах времени для каждой примеси.

Пример 6. Количественное определение ЭДТА, ДМСО и ЭТМ методом ОФ-ВЭЖХ в субстанции Hib.
При детектировании в субстанции Hib примеси ЭДТА, ДМСО или ЭТМ необходимо установить их количественное содержание. Для этого проводят калибровку системы по каждому веществу.
1. Калибровка системы по ЭДТА.
1.1. Готовят раствор ЭДТА с С 1 мг/мл. Для этого 50 мг натрия эдетеата помещают в мерную колбу вместимостью 50 мл, растворяют в 20 мл воды, доводят объем раствора до метки и перемешивают. К 2 мл полученного раствора добавляют 8 мл воды и получают раствор с С 200 мкг/мл ЭДТА, из которого готовят калибровочные растворы в двукратном разведении в диапазоне концентраций 200-0,05 мкг/мл.
1.2. В условиях примера 1 последовательно хроматографируют растворы ЭДТА, начиная с наименьшей концентрации. Для каждого калибровочного раствора определяют значение шума (N) в интервале от 0,0 до 1,0 мин, а также значение высоты и площади пика ЭДТА, данные заносят в таблицу, рассчитывают соотношение сигнал/шум.

1.3. Определяют предел детектирования (LOD) и предел количественного определения (LOQ) ЭДТА.
Пределом детектирования метода считают количество ЭДТА, при котором значение соотношения сигнал/шум составляет 3.
Пределом количественного определения метода считают количество ЭДТА, при котором значение соотношения сигнал/шум составляет 10.
|
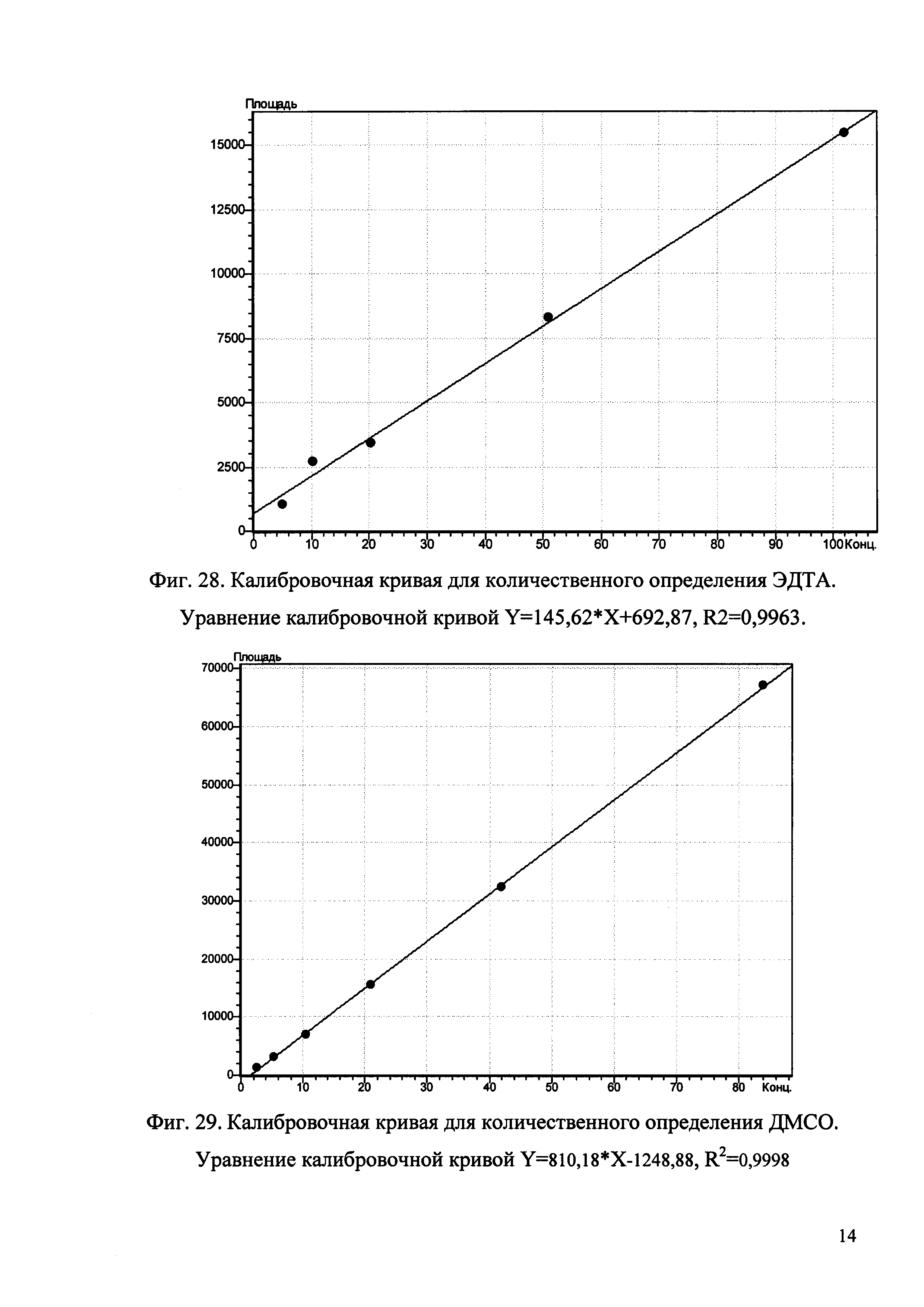
1.4. По полученным данным строят график зависимости площади пика, mAu (ось ординат) от количества ЭДТА, нг (ось абсцисс).
1.5. По уравнению рассчитывают содержание ЭДТА в субстанции Hib, подставляя вместо Y значение площади пика ЭДТА.
Количество ЭДТА в субстанции составляет:
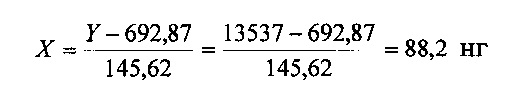
2. Калибровка системы по ДМСО.
2.1. Готовят раствор ДМСО с С 1 мг/мл. Для этого 25 мкл ДМСО помещают в мерную колбу вместимостью 25 мл, растворяют в 15 мл воды, доводят объем раствора до метки и перемешивают. К 2 мл полученного раствора добавляют 8 мл воды и получают раствор с С 200 мкг/мл ДМСО, из которого готовят калибровочные растворы в двукратном разведении в диапазоне концентраций 200-0,05 мкг/мл.
2.2. В условиях примера 1 последовательно хроматографируют растворы ДМСО начиная с наименьшей концентрации. Для каждого калибровочного раствора определяют значение шума (N) в интервале от 0,0 до 1,0 мин, а также значение высоты и площади пика ДМСО, данные заносят в таблицу, рассчитывают соотношение сигнал/шум.

2.3. Определяют предел детектирования (LOD) и предел количественного определения (LOQ) ДМСО.
Пределом детектирования метода считают количество ДМСО, при котором значение соотношения сигнал/шум составляет 3.
Пределом количественного определения метода считают количество ДМСО, при котором значение соотношения сигнал/шум составляет 10.
|
2.4. По полученным данным строят график зависимости площади пика, mAu (ось ординат) от количества ДМСО, нг (ось абсцисс).
2.5. По уравнению рассчитывают содержание ДМСО в субстанции Hib, подставляя вместо Y значение площади пика ДМСО.
Количество ДМСО в субстанции составляет:

3. Калибровка системы по ЭТМ.
3.1. Готовят раствор ЭТМ с С 1 мг/мл. Для этого 50 мг ЭТМ помещают в мерную колбу вместимостью 50 мл, растворяют в 20 мл воды, доводят объем раствора до метки и перемешивают. К 2 мл полученного раствора добавляют 8 мл воды и получают раствор с С 200 мкг/мл ЭТМ, из которого готовят калибровочные растворы в двукратном разведении в диапазоне концентраций 200-0,05 мкг/мл.
3.2. В условиях примера 1 последовательно хроматографируют растворы ЭТМ начиная с наименьшей концентрации. Для каждого калибровочного раствора определяют значение шума (N) в интервале от 0,0 до 1,0 мин, а также значение высоты и площади пика ЭТМ, данные заносят в таблицу, рассчитывают соотношение сигнал/шум.

3.3. Определяют предел детектирования (LOD) и предел количественного определения (LOQ) ЭТМ.
Пределом детектирования метода считают количество ЭТМ, при котором значение соотношения сигнал/шум составляет 3.
Пределом количественного определения метода считают количество ЭТМ, при котором значение соотношения сигнал/шум составляет 10.
|
3.4. По полученным данным строят график зависимости площади пика (ось ординат) от количества ЭТМ, нг (ось абсцисс).
3.5. По уравнению рассчитывают содержание ЭТМ в субстанции Hib, подставляя вместо Y значение площади пика ЭТМ.
Количество ЭТМ в субстанции составляет:

Суппозитории с интерфероном
Тест-система для количественного определения анти-hbs в биологическом образце
Способ получения миорелаксантного лекарственного средства для лечения мышечных дистоний
Устройство для дозированного разлива и укупорки жидкостей
Способ получения антигенов для вакцины против вирусов гриппа
Способ получения гидролизата сои
Способ получения и контроля пробиотических препаратов
Способ получения бактерийного препарата
Фармацевтическая композиция на основе секстафага (пиобактериофага поливалентного) или бактериофага сальмонеллезного и способ ее получения
Способ получения бетулина для использования в качестве адъюванта в вакцине против коронавируса sars-cov-2

