Результат интеллектуальной деятельности: АНТИСМЫСЛОВЫЕ КОМПОЗИЦИИ И СПОСОБЫ ИХ ПОЛУЧЕНИЯ И ПРИМЕНЕНИЯ
Вид РИД
Изобретение
РОДСТВЕННЫЕ ЗАЯВКИ
Для данной заявки испрашивается приоритет на основании заявок EP 08425727.8, поданной 12 ноября 2008 г. и U.S.S.N 61/152297, поданной 1 февраля 2009 г., обе из которых включены во всей полноте посредством ссылки на них.
ПРЕДПОСЫЛКИ СОЗДАНИЯ ИЗОБРЕТЕНИЯ
Язвенный колит и болезнь Крона являются главными формами хронических воспалительных заболеваний кишечника (ВЗК) у людей. Кишечное заболевание пищеварительного тракта является несоответствующей иммунной реакцией, которая происходит у генетически чувствительных лиц в результате сложного взаимодействия между факторами окружающей среды, микробными факторами и иммунной системой кишечника. Было показано, что чрезмерная иммунная реакция на антигены слизистой, неадекватно регулируемая нормальными антагонистичными механизмами, приводит к хроническому воспалению кишечника.
Болезнь Крона является хроническим, рецидивирующим заболеванием желудочно-кишечного тракта, характеризующимся сегментарным трансмуральным воспалением и грануломатозными изменениями. Типичные представления включают перемежающееся участие разных частей желудочно-кишечного тракта и развитие осложнений, включающих стриктуры, абсцессы или фистулы. Так как его причина неизвестна, медикаментозное лечение болезни Крона является большей частью эмпирическим и предназначено для снижения воспаления. Медикаментозная терапия включает кортикостероиды, антибиотики, иммуносупрессоры и средства против
TNFα. Из-за терапевтических неудач и серьезных побочных эффектов данных видов терапии необходимы альтернативы.
Важную роль в патогенезе ВЗК играет TGF-β1, многофункциональный цитокин, способный к регуляции роста, дифференцировки и функции иммунных и неиммунных клеток. Сниженная способность обеспечивать эффективную антагонистическую TGF-β1 реакцию на воспалительные стимулы, как полагают, значима для патогенеза такого заболевания ВЗК. TGF-β1 действует как сильный негативный регулятор воспаления слизистой, и это подавление его активности приводит к развитию колита, у которого показано иммуноморфологическое сходство с болезнью Крона или язвенным колитом.
В воспаленном кишечнике пациентов с ВЗК существует заметная сверхэкспрессия Smad7 (белок, который служит в качестве субстратов для рецепторов TGF-β1), а снижение фосфорилирования Smad 3, решающей стадии в опосредуемой TGF-β1 трансдукции сигнала. Таким образом, при ВЗК высокие уровни Smad7 могут приводить к дефектной трансдукции сигнала, приводящей к сверхэкспрессии генов провоспалительных молекул, и TGF-β1 не проявляет своей противовоспалительной роли.
Антисмысловые олигодезоксинуклеотидные лекарственные средства представляют собой короткие цепи нуклеотидов ДНК, которые подавляют трансляцию белка путем специфического связывания с небольшим сегментом информационной РНК (иРНК), ответственной за стимуляцию продукции вызывающих заболевание белков. Последовательность антисмыслового лекарственного средства создается так, чтобы быть комплементарной ее целевой иРНК, так что при гибридизации получаемый двухнитевой сегмент распознается клеткой как аномальный и разрушается, с предотвращением тем самым трансляции информации в белковый продукт.
Антисмысловые терапевтические средства, однако, обычно вводят парентерально, что может приводить к побочным реакциям из-за системных воздействий. При таком введении также невозможно локализовать препарат в месте необходимого лечения. Поэтому существует необходимость в применении антисмыслового лечения подобно местному для лечения ВЗК и родственных заболеваний с использованием таблетированных составов.
КРАТКОЕ ИЗЛОЖЕНИЕ СУЩНОСТИ ИЗОБРЕТЕНИЯ
Данное описание относится, по меньшей мере частично, к фармацевтическим составам для перорального введения антисмысловых олигонуклеотидов, таких как антисмысловые олигонуклеотиды к SMAD7.
В одном из воплощений представлен фармацевтический таблетированный состав для перорального введения антисмыслового олигонуклеотида, который включает внутригранулярную фазу, причем внутригранулярная фаза включает антисмысловой олигонуклеотид, такой как представляемый SEQ ID NO 1, или его фармацевтически приемлемую соль (такую, как натриевая соль) и фармацевтически приемлемый наполнитель, и который может также включать внегранулярную фазу, которая может включать фармацевтически приемлемое вспомогательное вещество, такое как разрыхлитель. Рассматриваемые олигонуклеотиды включают представляемые SEQ ID NO 1, причем, по меньшей мере, одна, или в некоторых воплощениях все межнуклеотидные связи представлены О,О-связанными фосфоротиоатами.
Данное описание раскрывает таблетку, которая включает описанный антисмысловой олигонуклеотид и содержит энтеросолюбильное покрытие. Такая таблетка может, например, включать наполнитель, разрыхлитель и/или смазывающее вещество. Например, здесь представлена пероральная дозированная форма, такая как таблетка, которая содержит от примерно 35 мг до примерно 500 мг антисмыслового олигонуклеотида, например, 40 мг олигонуклеотида, представляемого SEQ ID NO 1 или его фармацевтически приемлемую соль.
В одном из воплощений здесь представлена таблетка для перорального применения, содержащая: от примерно 0,5 до примерно 10% по массе антисмыслового олигонуклеотида, представляемого SEQ ID NO 1, или его фармацевтически приемлемой соли; от примерно 30% до примерно 50% по массе маннита и от примерно 10% до примерно 30% по массе микрокристаллической целлюлозы.
Например, данное описание представляет фармацевтически приемлемую таблетку для перорального применения, содержащую внутригранулярную фазу и внегранулярную фазу, причем, например, внутригранулярная фаза содержит от примерно 5% до примерно 10% по массе (например, от примерно 8% по массе) антисмыслового олигонуклеотида, представляемого SEQ ID NO 1 или его фармацевтически приемлемой соли, примерно 40% по массе маннита, примерно 8% по массе микрокристаллической целлюлозы, примерно 5% по массе гидропропилметилцеллюлозы и примерно 2% по массе
натрийкрахмалгликолята и, например, внегранулярная фаза содержит примерно 17% по массе микрокристаллической целлюлозы, примерно 2% по массе натрийкрахмалгликолята и примерно 0,4% по массе стеарата магния, где таблетка может дополнительно включать энтеросолюбильное покрытие.
Также здесь представлены способы лечения болезни Крона, язвенного колита и хронического воспалительного заболевания кишечника, включающие прием пациентом, нуждающимся в этом, таблетки, пероральной дозы или фармацевтического состава, описанного здесь. Например, при пероральном введении фармацевтического состава, таблетки или пероральной дозированной формы пациенту данные фармацевтический состав, таблетка или пероральная дозированная форма могут, главным образом, доставляться в терминальный отдел подвздошной кишки и/или в восходящий отдел ободочной кишки пациента.
КРАТКОЕ ОПИСАНИЕ ЧЕРТЕЖЕЙ
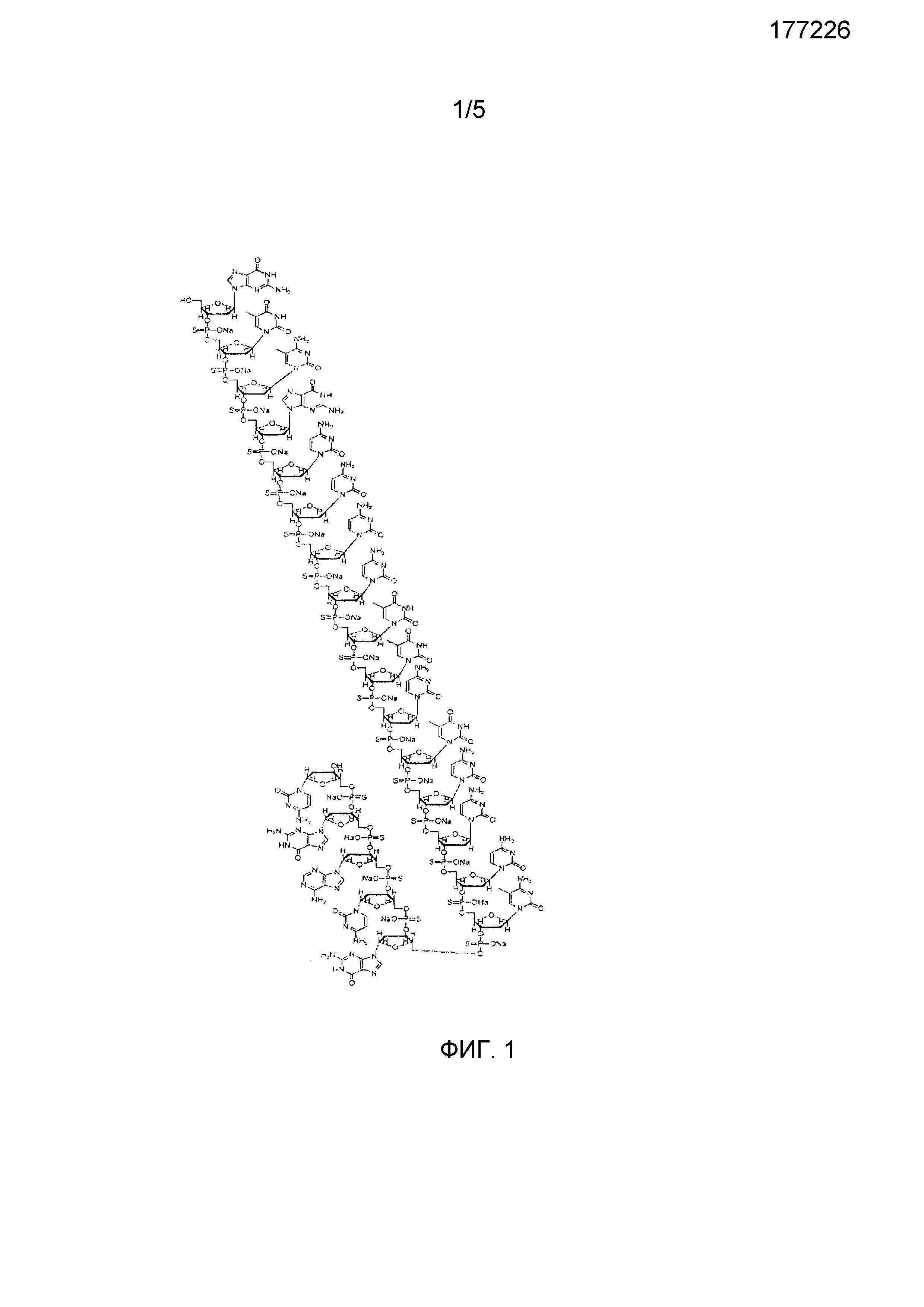
Фиг.1 представляет молекулярную структуру антисмыслового соединения, описанного здесь как AS1.
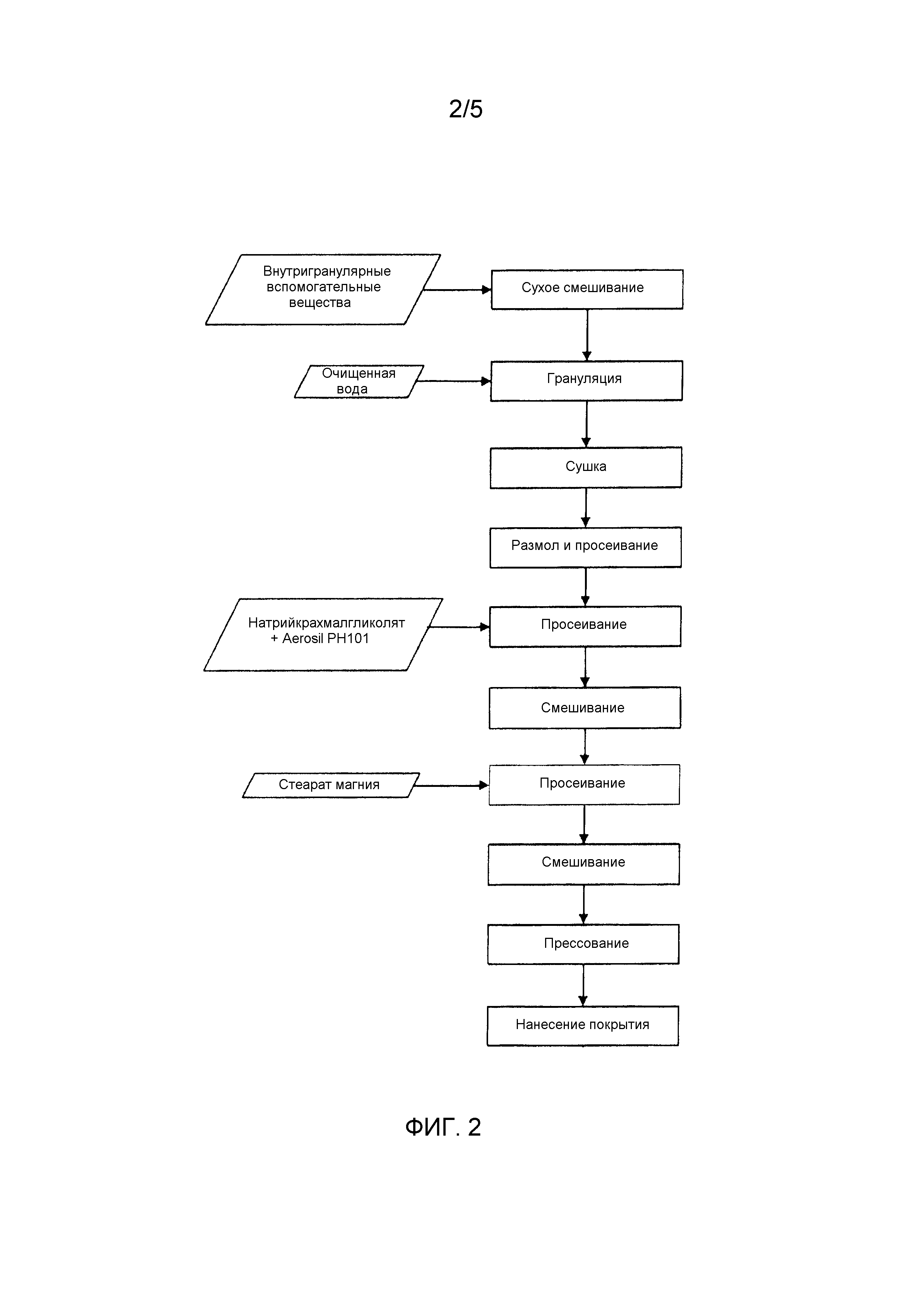
Фиг.2 является схематическим изображением процесса производства AS1.
Фиг.3 является столбчатой диаграммой, представляющей распределение частиц по размеру для дозы в 3,5 мг AS1.
Фиг.4 является столбчатой диаграммой, представляющей распределение частиц по размеру для дозы в 35 мг AS1.
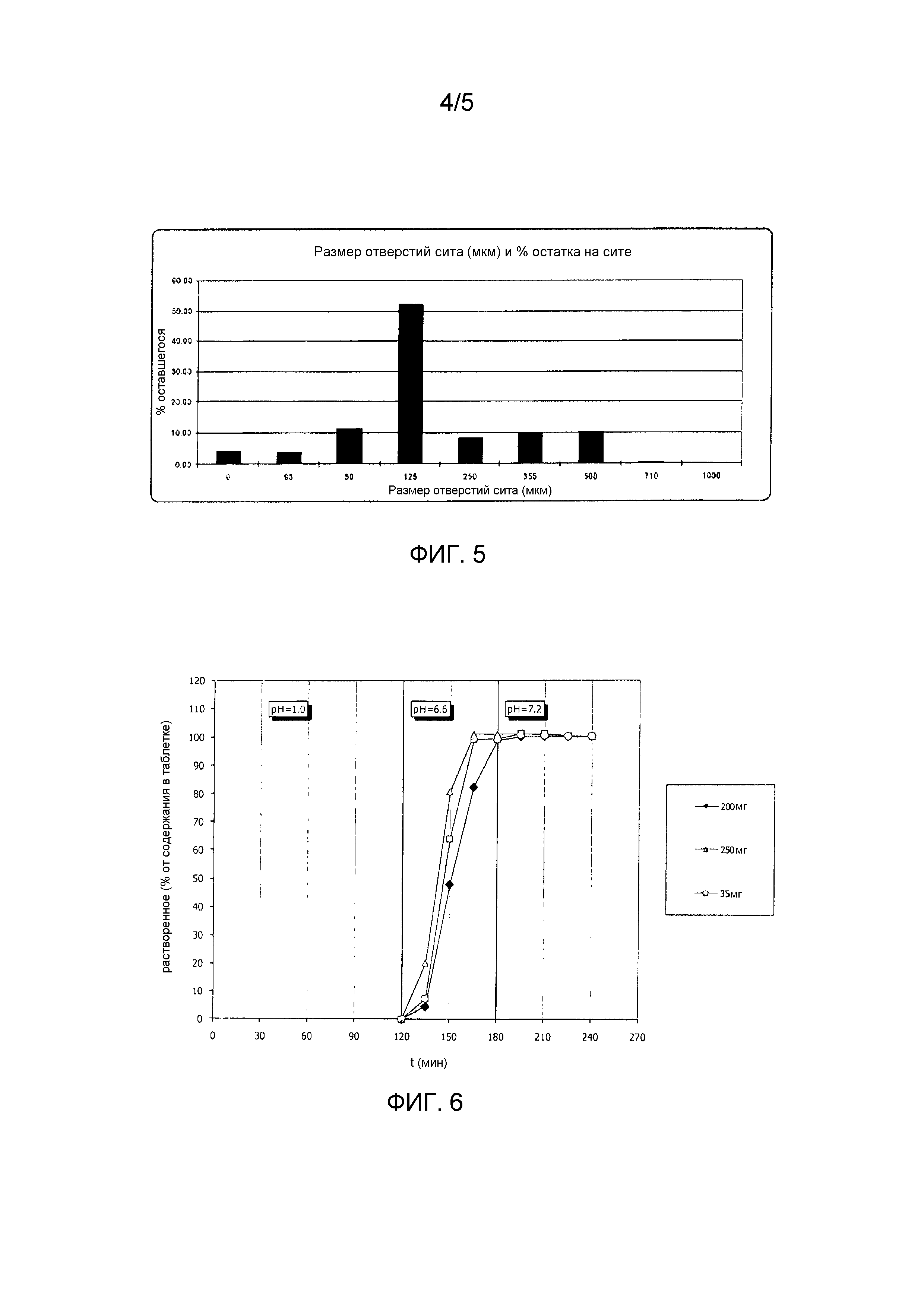
Фиг.5 является столбчатой диаграммой, представляющей распределение частиц по размеру для дозы в 250 мг AS1.
Фиг.6 является линейным графиком, представляющим характер
растворения (как процент растворенной таблетки) для трех таблеток с разными уровнями дозировки AS1 в трех разных средах (рН 1,0, рН 6,6 и рН 7,2).
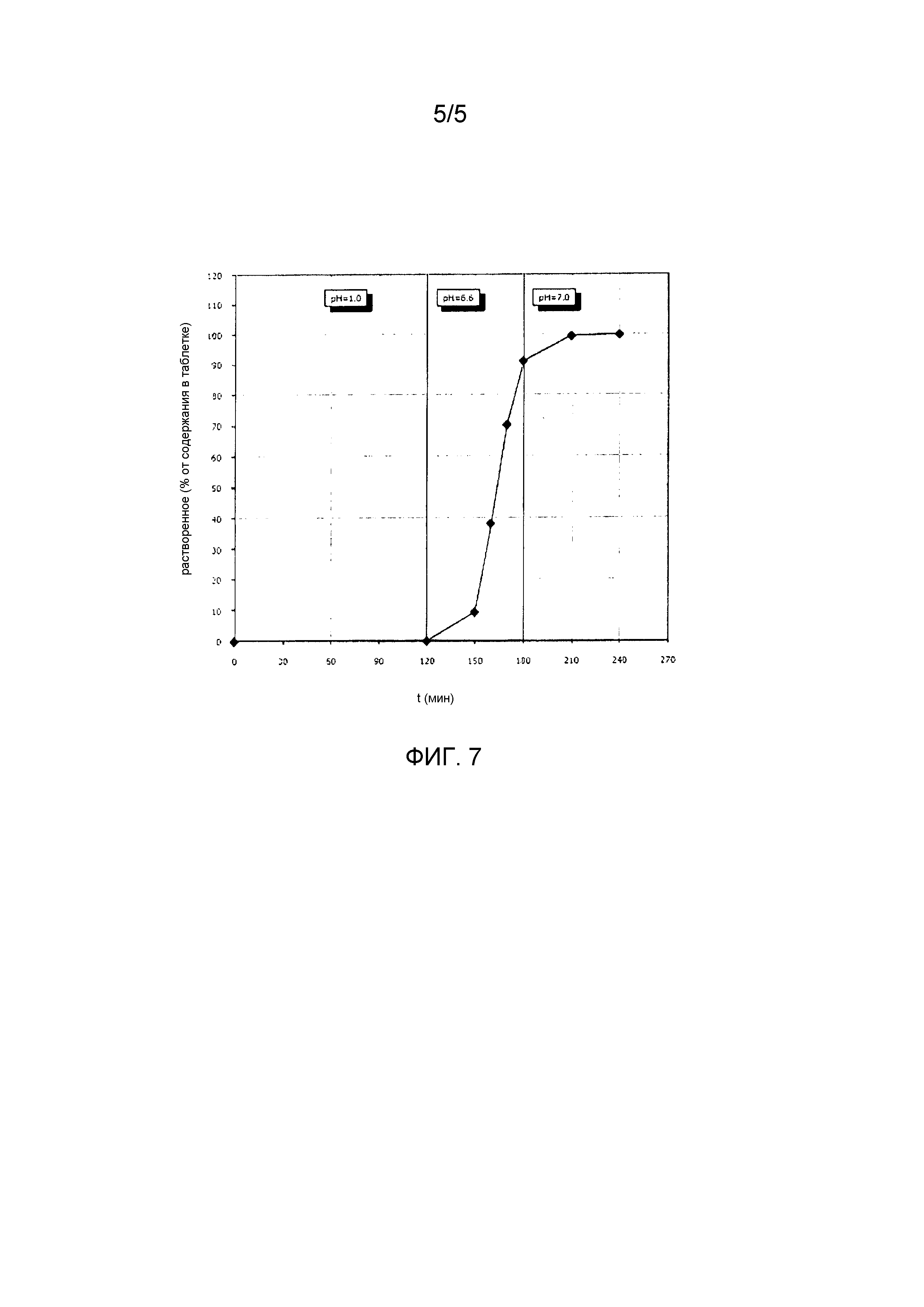
Фиг.7 представляет характер растворения партии, описанной здесь.
ПОДРОБНОЕ ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Данное описание в основном относится к фармацевтическим композициям, которые включают антисмысловой олигонуклеотид, такой как представленный на Фиг.1. Рассматриваемые композиции включают олигонуклеотиды, которые действуют против Smad7 и могут применяться перорально. Описанные композиции при пероральном введении могут доставлять эффективное количество антисмыслового олигонуклеотида в кишечную систему пациента, например, доставлять эффективное количество антисмыслового олигонуклеотида в терминальном отделе подвздошной кишки и/или в восходящем отделе ободочной кишки пациента.
Рассматриваемые антисмысловые олигонуклеотиды включают те, которые содержат SEQ ID NO 1 GTC* GCC CCT TCT CCC C*GC AGC, где С* представляет 5-метил-2'-дезоксицитидин. В некоторых воплощениях, по меньшей мере, одной из межнуклеотидных линкеров рассматриваемого антисмыслового олигонуклеотида является О,О-связанными фосфоротиоатами, например, каждая из 20 межнуклеотидных связей SEQ ID NO 1 может быть О,О-связанным фосфоротиоатом. В некоторых воплощениях рассматриваемые композиции, описанные здесь, могут включать фармацевтически приемлемую соль, например натриевую соль антисмыслового олигонуклеотида SEQ ID NO 1, которая, необязательно, может включать от 1 до 20 О,О-опосредованных фосфоротиоатных межнуклеотидных линкеров. Рассматриваемые соли олигонуклеотидов включают соли, которые полностью нейтрализованы, например, каждый фосфоротиоатный линкер соединен с ионом, таким как Na+. Олигонуклеотиды могут включать существующие в природе нуклеооснования, сахара и ковалентные межнуклеозидные (основной цепи) связи, а также не встречающиеся в природе части. Пример антисмыслового олигонуклеотида, приведенный здесь как AS1, представлен на Фиг.1.
В некоторых воплощениях, рассмотренных здесь, представлены композиции, пригодные для доставки антисмыслового олигонуклеотида при пероральном приеме, например таблетки, которые включают энтеросолюбильное покрытие, например, устойчивое в желудке покрытие, так что данные композиции могут доставлять антисмысловое соединение, например, в терминальный отдел подвздошной кишки и восходящий отдел ободочной кишки пациента. Например, способ введения может обеспечивать в результате местное действие, по существу местное применение антисмыслового соединения в пораженной части кишечника пациента. Такое применение в некоторых воплощениях может по существу избежать нежелательного системного всасывания антисмыслового соединения.
Например, представлена таблетка для перорального введения, которая содержит гранулы (например, является, по меньшей мере, частично образованной гранулами), которые включают антисмысловое соединение, например AS1, и фармацевтически приемлемые вспомогательные вещества. Такая таблетка может быть покрыта энтеросолюбильным покрытием. Рассмотренные таблетки могут включать фармацевтически приемлемые вспомогательные вещества, такие как наполнители, связующие вещества, разрыхлители и/или смазывающие вещества, а также красители, улучшающие высвобождение вещества, вещества для покрытия, подсластители, вкусовые вещества, такие как грушанковая, апельсиновая, ксилит, сорбит, фруктоза и мальтодекстрин и ароматизаторы, консерванты и/или антиоксиданты.
В некоторых воплощениях рассматриваемые фармацевтические составы включают внутригранулярную фазу, которая включает рассматриваемое антисмысловое соединение, например то, которое представлено в SEQ ID NO 1 или его фармацевтическую соль, например AS1, и фармацевтически приемлемый наполнитель. Например, AS1 и наполнитель могут быть смешаны между собой, с, необязательно, другими вспомогательными веществами и сформированы в гранулы. В некоторых воплощениях внутригранулярная фаза может быть сформирована с применением влажной грануляции, например жидкость (например, воду) добавляют к смешиваемым антисмысловому соединению и наполнителю, а затем комбинацию сушат, размалывают и/или просеивают с получением гранул. Специалист в данной области должен понять, что другие процессы могут быть применены для получения внутригранулярной фазы.
В некоторых воплощениях рассматриваемые составы включают внегранулярную фазу, которая может включать одно или более из фармацевтически приемлемых вспомогательных веществ и которые могут быть смешаны с внутригранулярной фазой с образованием описанного состава.
Описанный состав может включать внутригранулярную фазу, которая включает наполнитель. Примеры наполнителей включают, но не ограничиваются этим, целлюлозу, желатин, фосфат кальция, лактозу, сахарозу, глюкозу, манит, сорбит, микрокристаллическую целлюлозу, пектин, полиакрилаты, дектрозу, ацетатцеллюлозу, гидроксипропилметилцеллюлозу, частично пептизированный крахмал, карбонат кальция и другие включающие его комбинации.
В некоторых воплощениях описанный состав может включать внутригранулярную фазу и/или внегранулярную фазу, которая включает связующее вещество, которое может обычно функционировать с удержанием ингредиентов фармацевтического состава вместе. Примерами связующих веществ, включенных в изобретение, могут быть, но не ограничиваются этим, следующие: разные виды крахмала, сахара, целлюлозу или модифицированная целлюлоза, такая как гидроксипропилцеллюлоза, лактоза, пептизированный кукурузный крахмал, поливинилпирролидон, гидроксипропилцеллюлоза, гидроксипропилметилцеллюлоза, низкозамещенная гидроксипропилцеллюлоза, натрийкарбоксиметилцеллюлоза, метилцеллюлоза, этилцеллюлоза, сахара-спирты и другие включенные в нее комбинации.
Рассматриваемые составы, например те, которые включают внутригранулярную фазу и/или внегранулярную фазу, могут включать разрыхлитель, такой как, но без ограничения этим, крахмал, целлюлозу, структурированный поливинилпирролидон, натрийкрахмалгликолят, натрийкарбоксиметилцеллюлозу, альгинаты, зерновой крахмал, натрийкроскармеллозу, структурированную карбоксиметилцеллюлозу, низкозамещенную гидроксипропилцеллюлозу, камедь акации и другие включающие их комбинации. Например, внутригранулярная фаза и/или внегранулярная фаза могут включать разрыхлитель.
При некоторых воплощениях рассматриваемый состав включает внутригранулярную фазу, содержащую описанное антисмысловое соединение и вспомогательные вещества, выбранные из: маннита, микрокристаллической целлюлозы, гидроксипропилметилцеллюлозы и натрийкрахмалгликолята или их комбинаций, и внегранулярную фазу, содержащую одно или более из: микрокристаллической целлюлозы, натрийкрахмалгликолята и стеарата магния и их смесей.
В некоторых воплощениях рассмотренный состав может включать смазывающее вещество, например внегранулярная фаза может содержать смазывающее вещество. Смазывающие вещества включают, но не ограничиваются этим, тальк, кремнезем, жиры, стеарин, стеарат магния, фосфат кальция, диоксид кремния, силикат кальция, фосфат кальция, коллоидный диоксид кремния, стеараты металлов, гидрированное растительное масло, зерновой крахмал, бензоат натрия, полиэтиленгликоли, ацетат натрия, стеарат кальция, натрийлаурилсульфат, хлорид натрия, лаурилсульфат магния и стеариновую кислоту.
В некоторых воплощениях фармацевтический состав включает энтеросолюбильное покрытие. Обычно энтеросолюбильные покрытия создают барьер для пероральных лекарственных составов, который регулирует локализацию, в которой лекарственное вещество всасывается в пищеварительном тракте. Энтеросолюбильные покрытия могут включать полимер, который распадается в разной степени в соответствии с рН. Энтеросолюбильные покрытия могут включать, например, ацетофталатцеллюлозу, сополимеры метилакрилата-метакриловой кислоты, ацетосукцинатцеллюлозу, фталат гидроксипропилметилцеллюлозы, сополимеры метилметакрилата-метакриловой кислоты, сополимеры этилакрилата-метакриловой кислоты, сополимер метакриловой кислоты типа С, поливинилацетофталат и ацетофталатцеллюлозу.
Примеры энтеросолюбильных покрытий включают марки Opadry® AMB, Acryl-EZE®, Eudragit®. В некоторых воплощениях энтеросолюбильное покрытие может составлять от примерно 5% до примерно 10%, от примерно 5% до примерно 20%, от 8% до примерно 15%, от примерно 8% до примерно 18%, от примерно 10% до примерно 12%, от примерно 12% до примерно 16% рассматриваемой таблетки по массе. Например, энтеросолюбильные покрытия могут включать сополимер этилакрилата-метакриловой кислоты.
Например, представлена таблетка, которая включает или содержит по существу от примерно 0,5% до примерно 70%, например, от примерно 0,5% до примерно 10%, или от примерно 1% до примерно 20% по массе антисмыслового олигонуклеотида или его фармацевтически приемлемой соли (например, AS1). Такая таблетка может включать, например, от примерно 0,5% до примерно 60% по массе маннита, например, от примерно 30% до примерно 50% по массе маннита, например, примерно 40% по массе маннита; и/или от примерно 20% до примерно 40% по массе микрокристаллической целлюлозы или от примерно 10% до примерно 30% по массе микрокристаллической целлюлозы. Например, описанная таблетка может содержать внутригранулярную фазу, которая включает от примерно 30% до примерно 60%, например, от примерно 45% до примерно 65% по массе, или альтернативно, от примерно 5% до примерно 10% по массе AS1, от примерно 30% до примерно 50%, или альтернативно от примерно 5% до примерно 15% по массе маннита, от примерно 5% до примерно 15% микрокристаллической целлюлозы, от примерно 0% до примерно 4%, или от примерно 1% до примерно 7% гидроксипропилметилцеллюлозы и от примерно 0% до примерно 4%, например, от примерно 2% до примерно 4% натрийкрахмалгликолята по массе.
Примеры составов включают дозированные формы, которые включают или содержат от примерно 35 до примерно 500 AS1, например, здесь рассматриваются таблетки, которые включают примерно 35 мг, 40 мг, 50 мг, 60 мг, 70 мг, 80 мг, 90 мг, 100 мг, 150 мг, 200 мг или 250 мг AS1.
В одном из примеров воплощений представлена фармацевтически приемлемая таблетка для перорального применения, которая включает внутригранулярную фазу, которая может содержать примерно 50% по массе AS1 (или его соль), примерно 11,5% по массе маннита, примерно 10% по массе микрокристаллической целлюлозы, примерно 3% по массе гидропропилметилцеллюлозы и примерно 2,5% по массе натрийкрахмалгликолята и внегранулярную фазу, которая может содержать примерно 20% по массе микрокристаллической целлюлозы, примерно 2,5% по массе натрийкрахмалгликолята и примерно 0,5% по массе стеарата магния. Таблетка может также включать энтеросолюбильное покрытие.
В другом примере воплощения представлена фармацевтически приемлемая таблетка для перорального применения, которая включает указанные компоненты или состоит по существу из: внутригранулярной фазы, которая может содержать или по существу состоять из от примерно 5% до примерно 10%, например, от примерно 8% по массе AS1 (например, в котором межнуклеотидные связующие элементы представляют собой, каждый О,О-связанные фосфоротиоаты и/или его соль, например, натриевая соль), примерно 40% по массе маннита, примерно 8% по массе микрокристаллической целлюлозы, примерно 5% по массе гидропропилметилцеллюлозы и примерно 2% по массе натрийкрахмалгликолята; и внегранулярную фазу, которая может содержать примерно 17% по массе микрокристаллической целлюлозы, примерно 2% по массе натрийкрахмалгликолята и примерно 0,4% по массе стеарата магния.
Описываемые таблетки могут также включать энтеросолюбильное покрытие, например, описываемая таблетка может включать примерно 13%, примерно 15%, 16%, 17% по массе энтеросолюбильного покрытия, например, AcyrlEZE®.
Показатель, в момент которого покрытие растворяется и активный ингредиент высвобождается, является показателем его растворения. При одном из воплощений рассматриваемая таблетка может иметь показатели растворения, например, при испытании в аппарате (лопастная мешалка) типа 2 по USP/EP при 100 об/мин и 37°C в фосфатном буфере с рН 7,2 в от 50% до примерно 100% высвобождения олигонуклеотида через от примерно 120 минут до примерно 240 минут, например, через 180 минут. В другом воплощении рассматриваемая таблетка может иметь показатели растворения, например, при испытании в аппарате (лопастная мешалка) типа 2 по USP/EP при 100 об/мин и 37°C в разбавленной HCl с рН 1,0, когда олигонуклеотид по существу не высвобождается через 120 минут. В еще одном воплощении рассматриваемая таблетка может иметь показатели растворения, например, при испытании в аппарате (лопастная мешалка) типа 2 по USP/EP при 100 об/мин и 37°C в фосфатном буфере с рН 6,6 в от примерно 10% до примерно 30% или не более примерно 50% высвобождения олигонуклеотида через примерно 30 минут.
Описываемые составы, например, таблетки, в некоторых воплощениях при пероральном приеме пациентом могут обеспечивать в результате минимальные концентрации олигонуклеотида в плазме у пациента. В другом воплощении описываемые составы при пероральном приеме пациентом местная доставка в терминальный отдел подвздошной кишки и/или в восходящий отдел ободочной кишки пациента, например, в пораженном или больном месте кишечника пациента.
Здесь представлены также способы лечения болезни Крона, язвенного колита и/или хронического воспалительного заболевания кишечника у пациента, нуждающегося в этом применением раскрываемого состава.
ПРИМЕРЫ
Примеры, которые следуют далее, не предназначены никоим образом для ограничения объема этого изобретения, но представлены для иллюстрации способов данного изобретения. Многие другие воплощения этого изобретения будут очевидны специалисту в данной области.
Пример 1 - Таблетки
Влажные гранулы получали путем дозирования каждого внутригранулярного компонента в соответствующий контейнер. Все внутригранулярные вещества просеивали через 710 мкм сито и смешивали в чаше кухонного комбайна в течение примерно 5 минут. Медленно добавляли водную гранулирующую жидкость, используя шприц. Влажную массу пропускали через 2,00 мм ручное сито и сушили в термостате при 40°С в течение срока до 90 минут. После сушки гранулы просеивали через 1,00 мм сито. Для снижения размера крупных гранул использовали ступку и пестик. Гранулы смешивали с внутригранулярными вспомогательными веществами, за исключением стеарата магния, используя блендер Turbula в течение 10 минут при 42 об/мин. К смеси добавляли стеарат магния и дополнительно перемешивали в течение 2 минут при 42 об/мин. Составы прессовали на однопуансонной прессующей машине Manesty F3. Общая схема технологического маршрута производственного процесса для таблеток AS1 можно увидеть на Фиг.2.
Состав с уровнем дозировки в 250 мг на основе маннита, изготовленный в партии размером 50 г, имел содержание влаги в сухой смеси 5,21% и содержание влаги в высушенных гранулах в 6,42%. Состав с уровнем дозировки в 250 мг на основе маннита имел 5,0 г добавленной воды, время грануляции составляло 4 минуты и время сушки 60 минут.
Состав с уровнем дозировки в 35 мг на основе маннита, изготовленный в партии размером 100 г, имел содержание влаги 2,35% для высушенных гранул, было добавлено 28 г воды, время грануляции составило 6 минут, а время сушки составило 65 минут.
Прессование IPC обеспечивает в результате составы с уровнем дозировки 250 мг на основе маннита, которые представляют собой следующее: средний вес 453,0 мг, твердость 18,0 кп, толщина 3,82 мм, ломкость 0,23% и распадаемость 17 минут.
В Таблицу 1A включены композиции для трех уровней доз: 3,5 мг, 35 мг и 250 мг. Вес таблетки составлял 500 мг для всех составов.
|
Таблица 1В представляет состав партий ядер с этими тремя уровнями дозировки:
|
|
Пример 2 - Характеристики порошка
Составы оценивали на распределение частиц по размеру, плотность, показатель Кара и угол естественного откоса. Анализ распределения частиц по размеру показал, что частицы больше по размеру (335 мкм) для состава с уровнем дозировки 3,5 мг, тогда как составы с уровнем дозировки 35 мг и 250 мг проявляли обычное распределение частиц по размеру. На Фиг.3 представлено распределение частиц по размеру для дозировки в 3,5 мг при составе, указанном в Таблице 1. На Фиг.4 представлено распределение частиц по размеру для дозировки в 35 мг при составе, указанном в Таблице 1. И, наконец, на Фиг.5 представлено распределение частиц по размеру для дозировки в 250 мг при составе, указанном в Таблице 1. В Таблице 2 представлены результаты определения характеристик порошка для трех уровней дозировки.
|
Пример 3 - Энтеросолюбильное покрытие
Из покрытий Acryl-EZE® готовили 20% покрывающий раствор. Необходимые количества воды и Acryl-EZE® отмеряли в соответствующие контейнеры. При перемешивании Acryl-EZE® медленно добавляли в водоворот. Дисперсию перемешивали в течение 45 мин и пропускали через 500 мкм сито. Напыление продолжали до тех пор, когда получался прирост веса 10% или 16%.
Пример 4 - 40 мг таблетки
Состав партии таблеток по 40 мг AS1 представлен ниже.
|
Пример 5 - Состав 200 мг таблеток
Таблетки AS1 с уровнем дозировки 200 мг производили в основном по процедуре из Примера 1, как представлено в Таблице 4. Вес таблетки для всех составов был равен 500 мг.
|
Пример 6 - Анализ растворения по ВЭЖХ
Методика аналитического испытания описывает анализ растворения таблеток AS1 с энтеросолюбильным покрытием с помощью ВЭЖХ. Растворение выполняют по методике Европейской Фармакопеи для твердых дозированных форм с задержанным высвобождением с использованием метода А. Используемым прибором был прибор 2 (лопаточная мешалка) по Евр. Фармакопее/Фармакопее США.
Условия растворения для ВЭЖХ являются следующими: параметры среды состоят в: рН 1,0 HCl (120 мин), рН 6,6 (30 мин), рН 7,2 (60 мин); температура 37°С; скорость 100 об/мин; образец для рециркуляции 7,5 мл, размер образца 0,8 мл; время обработки образца 120 мин в HCl при рН 1,0; 15, 30 мин при рН 6,6; 15, 30, 45, 60 мин при рН 7,2; и 45 мкм линейный фильтр Disteck.
Среду в каждом сосуде доводили во время растворения при каждом состоянии следующим образом: добавляли первоначальный объем 750 мл HCl рН 1,0 при 120 мин; 200 мл 0,2 М Na3PO4 и 30 мл 1,0 М Na2HPO4 рН 6,7 с последующим доведением рН 6,60±0,05 с помощью 2,0 М NaOH; при 150 мин с последующим доведением рН до 7,20±0,05 с помощью 2,0 М NaOH.
Условия хроматографии являются следующими: аналитическая колонка для ВЭЖХ Dionex, DNAPPac-100, 4×250 мм; скорость потока 2,0 мл/мин; температура колонки 80°С; детекция по УФ при 260 нм; объем инъекции 100 мкл; промывание иглы водой; подвижная фаза А) 10% (объемн.) ацетонитрил в 100 мМ Трис (рН 8,0) и В) 10% (объемн.) ацетонитрил в 100 мМ Трис и 2М LiCl (pH 8,0); и время цикла ВЭЖХ 15 мин; и период элюирования AS1 в примерно 6 минут. Градиент представлен в Таблице 6.
|
Изготовление стандартных рабочих растворов таблеток 35 мг: 12,5 мг эталонного стандарта AS1 помещали в колбу объемом 50 мл и растворяли в воде с получением 50 мл раствора. 7 мл раствора разбавляли водой для изготовления 50 мл раствора с получением конечной концентрации AS1 0,035 мг/мл.
Изготовление стандартных рабочих растворов таблеток 250 мг: 12,5 мг эталонного стандарта AS1 помещали в колбу объемом 50 мл и растворяли в воде объемом 50 мл, получая конечную концентрацию AS1 0,25 мг/мл.
В Таблице 5 представлены результаты растворения в отношении партий с дозировками 35 мг, 250 мг и 200 мг и приростом веса в 12% при покрытии Acryl-EZE®. Фиг.6 представляет собой график по характеристикам растворения таблеток составов AS1 с тремя уровнями дозировки в трех разных средах.
|
Таблетки с 16% покрытием Acryl-EZE® изготавливают, используя таблетки, которые описаны выше для партии 1, и окончательное покрытие проводят до тех пор, когда прирост веса таблеток в чане для нанесения покрытия составит 16% от исходного значения. На Фиг.7 представлен график растворения.
Пример 7 - Дозировка AS1 in vivo при пероральном введении
Целями данного исследования были идентификация потенциального действия AS1 на сердечно-сосудистую, дыхательную и центральную нервную системы находящихся в сознании, содержащихся в свободных условиях макак cynomolgus, при применении единственной дозы путем перорального введения или внутривенного введения. Внутривенное введение при этом исследовании было включено для исследования потенциальных эффектов, связанных с системным воздействием. Внутривенный дозируемый состав применяли в виде нерасфасованного порошка, а для перорального введения в виде таблеток с энтеросолюбильным покрытием, помещенных в желатиновые капсулы. Четырех не первый раз используемых в эксперименте самцов макак cynomolgus в возрасте 3,6-3,8 лет и весящих 3,4-3,9 кг определяли в одну группу.
|
Все уровни дозирования представляют количество AS1, скорректированное на чистоту. Контрольными дозами для перорального введения были две капсулы, содержащие таблетки плацебо. Приближенный уровень дозировки, получали путем введения двух капсул, каждая из которых содержит таблетку с 26,1 мг AS1 на таблетку (скорректированных по чистоте) и на основе веса тела, который находился в пределах 3,5-3,9 кг на 4 день. Приближенный уровень дозировки, получали путем введения двух капсул, каждая из которых содержит таблетку с 169,9 мг AS1 на таблетку (скорректированных по чистоте) и на основе веса тела, который находился в пределах от 3,5 до 3,9 кг на 4 день.
Образцы крови для токсикокинетического анализа отбирали перед исследованием и через 1 и 6 часов после перорального введения препарата и через 5 минут, 1 час и 6 часов после внутривенного введения препарата. Образцы обрабатывали с получением плазмы в условиях охлаждения, и плазму хранили при -70°С до анализа. Образцы анализировали, используя анализ на специфическую гибридизацию с AS1.
В 1-й день все животные получали перорально названную контрольную дозу с последующим введением доз AS1 в дни 4 и 7 через введенный через рот желудочный зонд. Что касается доз для перорального введения, содержащие плацебо и AS1 таблетки были заключены в желатиновые капсулы соответствующего размера, чтобы облегчить введение пероральным путем. На 12-й день все животные получали внутривенно контрольную дозу (фосфатно-буферный физиологический раствор) путем медленной инъекции через шприц с последующим внутривенным введением доз AS1 на 14 и 19 дни.
Данные по сердечно-сосудистой системе и температуре тела регистрировали по телеметрии часто, через небольшие интервалы перед введением и в течение 24 часов после введения каждой дозы. Дыхательную функцию оценивали путем измерения показателей содержания газов в крови в пробах артериальной крови, взятых перед введением и через 1, 6 и 24 часа после введения каждой дозы, а также путем определения интенсивности дыхания (визуально) в те же самые моменты времени. Неврологические функции оценивали путем выполнения всестороннего неврологического обследования животных перед исследованием и в пределах примерно 24 часов после окончания периода регистрации телеметрии после последнего перорального введения дозы испытуемого препарата, и опять в пределах примерно 24 часов после окончания периода регистрации телеметрии после последнего внутривенного введения дозы. За животными наблюдали также в отношении клинических проявлений (смертность/заболеваемость, камерные наблюдения побочного действия, потребление пищи и вес тела).
Несмотря на очень низкий LLOQ для анализа (0,5 нг/мл), AS1 не был обнаружен в каких-либо образцах плазмы после перорального введения доз до примерно 100 мг/кг. В противоположность этому, средние максимальные концентрации в плазме после в/в инъекций составляли 28,309 и 180,352 нг/мл при дозах 3 и 10 мг/кг, соответственно. Выведение AS1 из плазмы после IC введения дозы проявлялась кинетика, подобная той, о которой сообщалось для структурно-родственных олигонуклеотидов (т.е. период полувыведения примерно 0,5 часа).
Пример 8 - 28-дневное исследование in vivo при пероральном введении AS1
Целью этого исследования была оценка потенциальной токсичности и токсикокинетики AS1 при пероральном введении один раз в сутки мышам в течение 28 дней с последующим 28-дневным периодом восстановления. Состав AS1 готовили как состав, содержащий защищающее от среды желудка покрытие в форме небольших покрытых гранул. AS1 наслаивали на инертные гранулы, которые затем покрывали Eudragit®S 100 с имитацией пероральных составов, подходящих для применения у человека. Использовали три группы, которым вводили дозы разных уровней (30 мг/кг/сутки (низкий); 100 мг/кг/сутки; 300 мг/кг/сутки).
Не существовало различий уровней препарата в плазме и тканях, связанных с полом. Несмотря на очень низкий НПКА для данного анализа, AS1 обнаруживался только в двух образцах плазмы от животных при очень низкой дозе (30 мг/кг/сутки), взятых в 1 день, а не в каких-либо образцах, взятых на 28 день. При более высоких уровнях доз в 100 и 300 мг/кг/сутки AS1 можно было обнаружить в большинстве образцов плазмы, и уровни в плазме в основном зависели от дозы. Однако даже при самом высоком уровне дозы уровни в плазме не превышали 21 нг/мл у любого животного (в образцах, собранных в разные сроки между 0,5 и 24 часами после введения). Концентрации AS1 были очень высокими в тканях желудочно-кишечного тракта со средними максимальными концентрациями при самом высоком уровне дозировки (после первого введения) равными примерно 536, 857, 825, 538, 137 и 127 мкг/грамм ткани для толстого кишечника, тонкого кишечника, кардиального отдела желудка, железистой части желудка, пищевода и прямой кишки, соответственно.
Интенсивное выведение из тканей ЖК тракта было очевидным через 24 часа после приема первой дозы. Не было явного накопления AS1 при ежедневном введении.
Максимальные средние концентрации в двух главных органах при системном введении, почки и печень, после первого введения дозы в 300 мг/кг были равны только 4,0 и 2,3 мкг/грамм, что в 100 раз ниже, чем порядок концентраций, определенных в тканях ЖКТ.
После введения самой низкой дозы в 30 мг/кг наивысшие средние концентрации в почках и печени составляли только 0,4 и 0,3 мкг/грамм. Не было данных в отношении накопления AS1 во внутренних органах в течение 28-дневного периода введения.
Системное воздействие AS1 у мышей было очень низким после перорального введения высоких доз (до 300 мг/кг/сутки) AS1, доставляемого в составе, защищенном от воздействия среды желудка, и не существовало накопления в тканях ЖКТ при тканях внутренних органов при ежедневном введении в течение 28 последовательных дней.
Пример 9 - Терапевтическое действие AS1 при колите
Чтобы оценить терапевтическое действие AS1 на течение воспаления кишечника на модели вызванного TNBS колита на мышах мышам вводили единственную дозу AS1 или смыслового олигонуклеотида Smad7 через один день после введения через прямую кишку TNBS. Однократные дозы в 125 или 250 мкг/мышь снижали потерю веса и заметно снижали тяжесть гистологических проявлений колита.
AS1 (при условии единственного введения дозы в 125 мкг один раз в сутки после индукции колита TNBS) значительно снижал продукцию в толстом кишечнике мономерной субъединицы р40, компонента цитокинов IL-12 и IL-23, что показывает подавление продукции в толстом кишечнике этих двух провоспалительных цитокинов.
Все публикации и патенты, упомянутые здесь, включая публикации, перечисленные ниже, включены тем самым в виде ссылки во всей их полноте, как если бы каждая отдельная публикация или патент был конкретно и отдельно включен сюда путем ссылки. В случае конфликта данная заявка будет проконтролирована, включая любые определения, представленные здесь.
ЭКВИВАЛЕНТЫ
Хотя обсуждены конкретные воплощения рассматриваемого изобретения, представленное выше описание является иллюстративным, а не ограничивающим. Многие варианты данного изобретения станут очевидны специалистам в данной области при рассмотрении этого описания. Полный объем данного изобретения должен быть определен путем обращения к формуле изобретения вместе с полным объемом его эквивалентов и описанию вместе с подобными вариантами.


Способы лечения диабета и/или улучшения выживания панкреатических островков после трансплантации
Способы лечения колоректального рака
Способы контроля восприимчивости анти-smad7 терапии
Способы контроля восприимчивости анти-smad7 терапии

