Результат интеллектуальной деятельности: ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ
Вид РИД
Изобретение
Настоящее изобретение относится к технической области, относящейся к крупномасштабному изготовлению фармацевтических соединений.
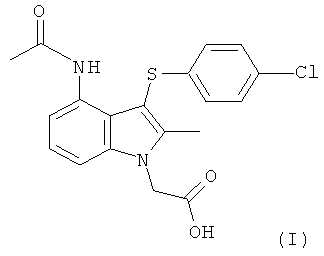
Международная патентная заявка PCT/SE2004/000808 (WO 2004/106302) относится к замещенным индолам, пригодным в качестве фармацевтических соединений для лечения респираторных расстройств, к фармацевтическим средствам, содержащим их, и к способам их получения. Более конкретно, на странице 25 в WO 2004/106302 раскрыта 4-(ацетиламино)-3-((4-хлорфенил)тио]-2-метил-1Н-индол-1-уксусная кислота (ниже упоминаемая как соединение формулы (I)) как Пример 1.
Способ получения соединения формулы (I), раскрытый на страницах 25 и 26 WO 2004/106302, показан на Схеме (I) ниже.
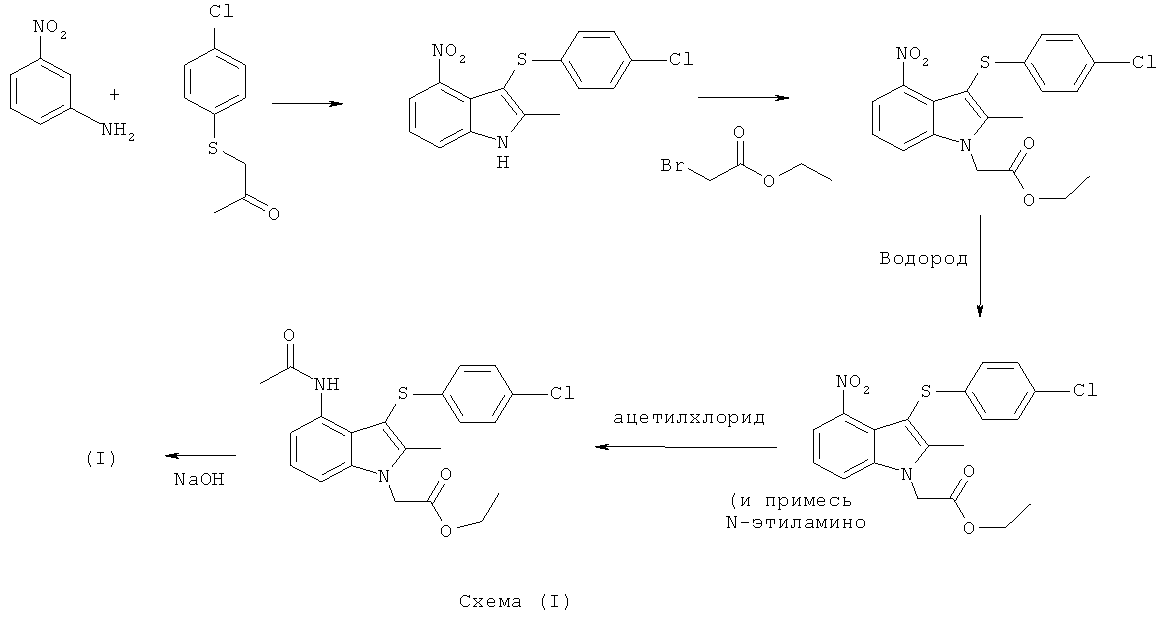
Синтез соединения формулы (I), раскрытый в WO 2004/106302, имеет ряд потенциальных недостатков.
Например, способ из уровня техники включает использование очень низких температур (-78°C). Такие низкие температуры может быть трудно достичь в крупном масштабе, и это требует специального фабричного оборудования и большого количества энергии.
Способ из уровня техники включает применение трет-бутил-гипохлорита, который рассматривается как нежелательный для крупномасштабного способа изготовления.
Способ из уровня техники включает использование дихлорметана, являющегося нежелательным растворителем для крупномасштабного использования из-за его сильного воздействия на окружающую среду.
Авторы данного изобретения установили, что одна или более реакций из уровня техники протекают относительно быстро. Быстрые экзотермические реакции могут быть неблагоприятными для более крупномасштабного изготовления в серийном производстве из-за необходимости контролировать и адекватно удалять выделяющееся тепло.
Международная патентная заявка PCT/GB2006/000060 (WO 2006/075139) относится к новому способу получения замещенных индолов, которые полезны в качестве терапевтических агентов. Более конкретно, в WO 2006/075139 раскрыты способы получения соединения формулы (I), где каждый способ начинается с 2-метил-4-нитроиндола.
Синтез соединения формулы (I), раскрытый в WO 2006/075139, имеет ряд недостатков.
Имеется основание предполагать, что 2-метил-4-нитроиндол не является хорошим исходным веществом для применения в крупномасштабном способе изготовления. Один из способов, известных для синтеза 2-метил-4-нитроиндола, как оказалось, обеспечивают низкий выход целевого продукта (Tetrahedron, 1990, 46 (17), 6085; и Tetrahedron Letters, 1983, 24 (34), 3665-8). Другой известный способ синтеза 2-метил-4-нитроиндола требует условий, которые нежелательны для крупномасштабного синтеза, потому что он включает использование воздушной атмосферы в присутствии органических растворителей, что может представлять трудности с точки зрения контроля и безопасности (Tetrahedron Letters, 1999, 40, 5395; и Tetrahedron, 2004, 60, 347). Действительно, сам 2-метил-4-нитроиндол, как было обнаружено, является высокоэнергетической молекулой (положительный результат в пробирочном тесте Кунена (Koenеn) при 2 мм), что значительно препятствует его применению в крупном масштабе. Кроме того, 2-метил-4-нитроиндол, как было обнаружено, является относительно дорогим в качестве исходного вещества, частично из-за требующихся больших объемов растворителя и необходимости в многократных кристаллизациях для удаления побочных продуктов. Следовательно "стоимость товара" для известных из уровня техники синтезов соединения формулы (I) из 2-метил-4-нитроиндола является потенциально неблагоприятно высокой.
Соединение формулы (I) разрабатывается как активное фармацевтическое соединение. Следовательно, желательны подходящие способы для безопасного, рентабельного, эффективного и учитывающего экологические факторы изготовления соединения формулы (I).
В настоящем изобретении предложено решение для устранения одного или более из вышеупомянутых недостатков. Дополнительные технические преимущества других аспектов изобретения описаны в данном описании изобретения ниже.
В первом аспекте изобретения предложено соединение формулы (X):

где:
X представляет собой =O, =N-OH или =N-OC(O)Me;
Y представляет собой водород или  ; и
; и
Q представляет собой водород или хлоро;
и Z представляет собой водород или -CH2COOR1, где R1 выбран из водорода, возможно замещенного гидрокарбила и возможно замещенного гетероциклила;
или его соль.
В еще одном аспекте предложено соединение формулы (X), как оно определено в описании изобретения.
В еще одном дополнительном аспекте изобретения предложено соединение формулы (II):
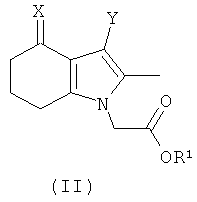
где:
R1 выбран из водорода, возможно замещенного гидрокарбила и возможно замещенного гетероциклила;
X представляет собой =O, =N-OH или =N-OC(O)Me;
Y представляет собой водород или ; и
Q представляет собой водород или хлоро;
или его соль.
Неожиданно авторы изобретения обнаружили, что фармацевтическое соединение формулы (I) может быть эффективно получено в крупном масштабе из соединения формулы (X), даже несмотря на то, что соединение формулы (X) не содержит бензольное кольцо в качестве ядра, которое в итоге присутствует в соединении формулы (I).
В еще одном аспекте предложено применение соединения формулы (II) или его соли, как они определены в данном описании изобретения, в качестве фармацевтического промежуточного соединения.
В одном воплощении предложено применение соединения формулы (II) или его соли, как они определены в данном описании изобретения, в качестве промежуточного соединения для изготовления соединения формулы (I) или его соли.
В еще одном аспекте предложено соединение формулы (II), как оно определено в данном описании изобретения.
Специалисту очевидно, что широкий диапазон групп R1 можно использовать для осуществления настоящего изобретения, так как группа R1 представляет собой только временный признак, который не предназначен играть ключевую роль в способе по настоящему изобретению. Следует понимать, что группа R1 может быть удалена с использованием щелочных условий, с высвобождением R1OH и соединения формулы (I) или его соли. В некоторых случаях группа R1 может быть отщеплена в кислых условиях или с использованием газообразного водорода с катализатором гидрирования или с использованием других установленных условий для отщепления данного типа сложноэфирной группы. Специалисту понятно, какие из вышеупомянутых условий могут быть предпочтительными в зависимости от природы группы R1.
Альтернативные воплощения и значения переменных групп описаны ниже, и следует понимать, что такие переменные группы в каком-либо аспекте или воплощении можно комбинировать с любыми другими переменными группами, или аспектом, или воплощением, описанными в данном описании изобретения выше или ниже. Воплощения и аспекты изобретения включают все такие комбинации переменных групп.
В одном воплощении предложено соединение формулы (II) с формулой (IIa):
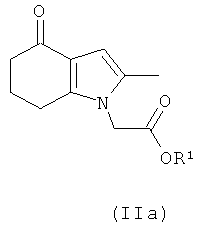
где R1 выбран из водорода, возможно замещенного гидрокарбила и возможно замещенного гетероциклила; или его соль.
В одном воплощении предложено применение соединения формулы (II) с формулой (IIa) или его соли, как они определены в данном описании изобретения, в качестве фармацевтического промежуточного соединения.
В одном воплощении предложено применение соединения формулы (II) с формулой (IIa) или его соли, как они определены в данном описании изобретения, в качестве промежуточного соединения для изготовления соединения формулы (I) или его соли.
Соединение формулы (IIа) может быть получено посредством реакции соединения формулы (III):
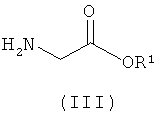
с 2-(2-оксопропил)циклогексан-1,3-дионом; где значения R1 такие, как они определены в данном описании изобретения.
2-(2-оксопропил)циклогексан-1,3-дион является известным соединением и может быть получен с использованием циклогексан-1,3-диона и хлорацетона в щелочных условиях, как описано в данном описании изобретения ниже.
Соединение формулы (III) либо имеется в продаже, либо может быть получено с использованием хорошо известных химических реакций из имеющихся в продаже исходных веществ. Например, группу NH2 соединения формулы (III) можно вводить путем замещения группы галогено из 2-галогеноацетатного эфира, например этил-2-хлорацетата. Специалисту понятно и что применение защитной группы для азота может быть полезно для такого превращения.
Альтернативно, N-защищенный глицин может быть подвергнут реакции сочетания со спиртом формулы R1OH с использованием агента сочетания, такого как EDCI (N-(3-диметиламинопропил)-N'-этилкарбодиимида гидрохлорид), или с использованием других условий эстерификации, которые хорошо известны в данной области техники.
Соединение формулы (IIa) может быть превращено в соединение формулы (I) путем выполнения следующих превращений:
когда R1 представляет собой водород:
(1) эстерификация - введение группы R1; и затем во всех случаях:
(2) тиоарилирование - введение тиоарильной группы;
(3) ацетилирование и ароматизация - создание бензольного кольца в качестве ядра и введение ацетильной группы;
и затем:
(4) снятие защиты (деэстерификация) - удаление группы R1;
где стадии (2) и (3) можно осуществлять в любом порядке.
В одном воплощении предложено соединение формулы (II) с формулой
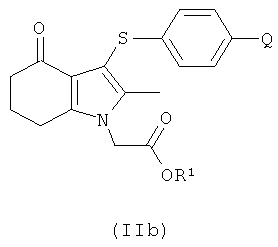
где R1 выбран из водорода, возможно замещенного гидрокарбила и возможно замещенного гетероциклила; или его соль.
В одном воплощении предложено применение соединения формулы (II) с формулой (IIb) или его соль, как они определены в данном описании изобретения, в качестве фармацевтического промежуточного соединения.
В одном воплощении предложено применение соединения формулы (II) с формулой (IIb) или его соли, как они определены в данном описании изобретения, в качестве промежуточного соединения для изготовления соединения формулы (I) или его соли.
Соединение формулы (IIb) может быть получено посредством реакции соединения формулы (IIa), как оно определено в данном описании изобретения, с соединением формулы (IVa) или (IVb):
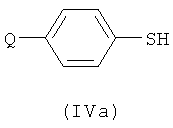
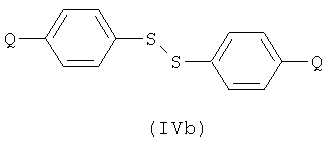
с использованием галогенирующего агента для активирования реагента или для превращения в сульфенилгалогенид. Например, можно использовать хлорирующий агент. Примеры подходящих хлорирующих агентов включают трихлоризоциануроновую кислоту (ТССА), сульфурилхлорид и хлор.
Соединение формулы (IIa) может быть превращено в соединение формулы (I) путем выполнения следующих превращений:
когда R1 представляет собой водород:
(1) эстерификация - введение группы R1;
и затем во всех случаях:
(2) ацетилирование и ароматизация - создание бензольного кольца в качестве ядра и введение ацетильной группы;
и затем:
(3) снятие защиты (т.е. деэстерификация) - удаление группы R1.
В одном воплощении предложено соединение формулы (II) с формулой
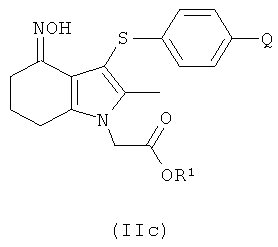
где R1 выбран из водорода, возможно замещенного гидрокарбила и возможно замещенного гетероциклила;
или его соль.
В одном воплощении предложено применение соединения формулы (II) с формулой (IIc) или его соли, как они определены в данном описании изобретения, в качестве фармацевтического промежуточного соединения.
В одном воплощении предложено применение соединения формулы (II) с формулой (IIc) или его соли, как они определены в данном описании изобретения, в качестве промежуточного соединения для изготовления соединения формулы (I) или его соли.
Соединение формулы (IIc) может быть получено посредством взаимодействия соединения формулы (IIb) с гидроксиламином или его солью, например с гидроксиламина гидрохлоридом.
Соединение формулы (IIc) может быть превращено в соединение формулы (I) путем выполнения следующих превращений:
когда R1 представляет собой водород:
(1) эстерификация - введение группы R1;
и затем во всех случаях:
(2) ацетилирование и ароматизация - создание бензольного кольца в качестве ядра и введение ацетильной группы;
и затем:
(4) снятие защиты (деэстерификация) - удаление группы R1.
В одном воплощении предложено соединение формулы (II) с формулой
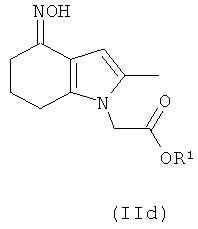
где R1 выбран из водорода, возможно замещенного гидрокарбила и возможно замещенного гетероциклила; или его соль.
В одном воплощении предложено применение соединения формулы (II) с формулой (IId) или его соли, как они определены в данном описании изобретения, в качестве фармацевтического промежуточного соединения.
В одном воплощении предложено применение соединения формулы (II) с формулой (IId) или его соли, как они определены в данном описании изобретения, в качестве промежуточного соединения для изготовления соединения формулы (I) или его соли.
Соединение формулы (IId) может быть получено посредством взаимодействия соединения формулы (IIa) с гидроксиламином или его солью, например гидроксиламина гидрохлоридом.
Соединение формулы (IId) может быть превращено в соединение формулы (I) путем выполнения следующих превращений:
когда R1 представляет собой водород:
(1) эстерификация - введение группы R1;
и затем во всех случаях.
(2) тиоарилирование - введение тиоарильной группы;
(3) ацетилирование и ароматизация - создание бензольного кольца в качестве ядра и введение ацетильной группы;
и затем:
(4) снятие защиты (деэстерификация) - удаление группы R1; где стадии (2) и (3) могут быть осуществлены в любом порядке. В другом аспекте предложено соединение формулы (XI):
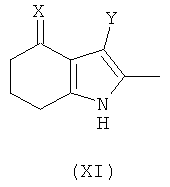
где:
X представляет собой =O, =N-OH или =N-OC(O)Me;
Y представляет собой ; и
Q представляет собой водород или хлоро; или его соль.
Соединение формулы (XI) можно использовать с получением соединения формулы (I), используя, например, способ, описанный на Схеме VI ниже. В одном воплощении предложено соединение формулы (XIa):
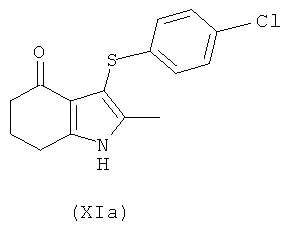
Соединение формулы (XIa) может быть получено из соединения формулы (XII):
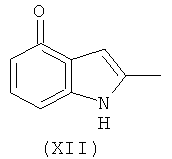
используя способы, аналогичные описанным в получении соединения формулы (IIb) из соединения формулы (IIa) и (IVa) или (IVb).
В другом воплощении предложено соединение формулы (XIb):
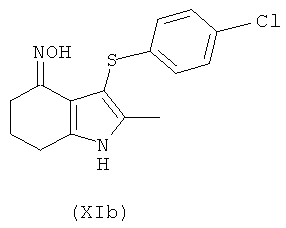
Соединение формулы (XIb) может быть получено из соединения формулы (XIa), используя способы, аналогичные описанным для получения соединения формулы (IIc) из (IIb).
В одном воплощении "гидрокарбил" представляет собой радикал, состоящий из атомов водорода и 1-15 атомов углерода, где гидрокарбил может быть насыщенным, частично насыщенным или полностью ненасыщенным и может содержать линейные, разветвленные или циклические элементы.
В одном воплощении "гетероциклил" представляет собой 4-12-членную моноциклическую или бициклическую кольцевую систему, где гетероциклил содержит 1-4 гетероатома, каждый из которых выбран из N, S и О, где гетероциклил может быть полностью насыщенным, частично насыщенным или полностью ненасыщенным.
Специалистам в данной области очевидно, что некоторые соединения по изобретению могут существовать в виде изомеров, например соединения формулы (II). Изобретение охватывает все изомерные формы соединений, изображенные в данном описании изобретения и их смеси, если не указано иное.
В данной заявке термин "алкил" включает алкильные группы как с прямой, так и с разветвленной цепью.
Упоминание индивидуальных алкильных групп, таких как "пропил" конкретно относится только к варианту с прямой цепью, а упоминание индивидуальных алкильных групп с разветвленной цепью, таких как "изопропил", конкретно относится только к варианту с разветвленной цепью. Данное обозначение применимо к другим радикалам, описанным в рамках данной заявки, таким как алкенильные радикалы, алкинильные радикалы, алкокси-радикалы и алканоильные радикалы.
Например "C1-6алкил" включает C1-4алкил, C1-3алкил, метил, этил, пропил, изопропил и трет-бутил.
В данной заявке "С2-6алкенил" включает С2-3алкенил, бутенил, изобутенил, 1,5-гексадиен-3-ил.
Примеры термина "С2-6алкинил" включают С2-3алкинил, бутинил, пропинил и этинил.
Примеры термина "C1-6алкокси" включают C1-6алкокси, трет-бутилокси, изопропокси, бутокси, этокси и метокси.
Примеры термина "(С1-6алкил)-5(S)а-, где a равен 0-2" включают "(С1-6алкил)-S-", "(С1-3алкил)-S(O)а-, где a равен 0-2", "(С1-3алкил)-S(O)2-", изопропилсульфанил, пропилсульфонил, мезил и этилсульфанил, бутансульфинил и изопентилсульфинил.
Примеры термина "C1-6алкоксикарбонил" включают C1-3алкоксикарбонил, метоксикарбонил, этоксикарбонил, изопропоксикарбонил и изопентоксикарбонил.
Примеры термина "C1-6алкилсульфонил" включают C1-3алкилсульфонил, мезил, этилсульфонил, изопропилсульфонил и изобутилсульфонил.
Примеры термина "C1-6алканоил" включают C1-3алканоил, формил, ацетил и пропионил.
Примеры термина "N-(С1-6алкил)амино" включают N-(C1-3алкил)амино, метиламино, изопропиламино и изогексиламино.
Примеры термина "N,N-(C1-6алкил)2амино" включают N,N-(C1-3алкил)2амино, N,N-диметиламино, N-изопропил-N-метиламино и N-пентил-N-этиламино.
Примеры термина "N-(С1-6алканоил)-N-(C1-6алкил)амино" включают N-(C1-3алканоил)-N-(C1-6алкил)амино, N-пропионоил-N-(C1-6алкил)амино, N-пропионоиламино, N-ацетил-N-метиламино и N'-ацетил-N-циклопропиламино.
Примеры "N-(C1-6алкил)карбамоила" включают N-(C1-3алкил)карбамоил, N-изопентиламинокарбонил, N-метиламинокарбонил и N-этиламинокарбонил.
Примеры "N,N(C1-6алкил)2карбамоила" включают
N,N(C1-3лкил)2карбамоил, N-изопентил-N-этиламинокарбонил,
N,N-диметиламинокарбонил и N-метил-N-этиламинокарбонил.
Примеры "N-(C1-6алкил)сульфамоила" включают
N-(C1-3алкил)сульфамоил, N-изопентилсульфамоил, N-метилсульфамоил и N-этилсульфамоил.
Примеры "N,N-(C1-6алкил)2сульфамоила" включают
N,N-(C1-3алкил)2сульфамоил, N-изопентил-N-этилсульфамоил,
N,N-диметилсульфамоил и N-метил-N-этилсульфамоил.
Соль, образованная соединением по изобретению, где R1 представляет собой водород, может быть солью щелочного металла, например солью натрия или калия, солью щелочноземельного металла, например солью кальция или магния, солью аммония или солью органического основания, например солью метиламина, диметиламина, триметиламина, пиперидина, морфолина или триэтаноламина.
Соль соединения по изобретению, где R1 содержит основную группу, представляет собой, например, соль присоединения кислоты, например неорганической или органической кислоты, например соляной, бромистоводородной, серной, фосфорной, трифторуксусной, лимонной или малеиновой кислоты.
Соль соединения по изобретению, где R1 содержит кислую группу, представляет собой, например, соль щелочного металла, например соль натрия или калия, соль щелочноземельного металла, например соль кальция или магния, соль аммония или соль с органическим основанием, например соль с метиламином, диметиламином, триметиламином, пиперидином, морфолином или триэтаноламином.
В одном воплощении R1 представляет собой водород или возможно замещенный гидрокарбил.
В одном воплощении возможные заместители на "гидрокарбиле" и "гетероциклиле" выбраны из галогено, нитро, циано, гидрокси, C1-6алкила, C1-6алкокси, C1-6алканоила, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, N-(C1-6алканоил)амино, C1-6алкоксикарбонила, N-(C1-6алканоил)-N-(C1-6алкил)амино, карбамоила, сульфамоила, N-(C1-6алкил)карбамоила, N,N-(C1-6алкил)2карбамоила, N-(C1-6алкил)сульфамоила, N,N-(C1-6алкил)2сульфамоила и (C1-6алкил)-S(O)а-, где а равен 0-2.
В еще одном воплощении R1 представляет собой водород или незамещенный гидрокарбил.
В еще одном воплощении R1 представляет собой водород.
В еще одном воплощении R1 представляет собой возможно замещенный гидрокарбил.
В еще одном воплощении R1 представляет собой незамещенный гидрокарбил.
В еще одном воплощении R1 выбран из водорода, C1-6алкила, С2-6алкенила, С2-6алкинила, С3-6циклоалкила, возможно замещенного фенила и возможно замещенного бензила.
В еще одном воплощении R1 выбран из водорода, C1-6алкила, С2-6алкенила, С2-6алкинила, С3-6циклоалкила, фенила и бензила; где фенил и бензил возможно замещены одним или более галогено, нитро, циано, гидрокси, C1-6алкилом, C1-6алкокси, C1-6алканоилом, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, N-(C1-6алканоил)амино, N-(C1-6алканоил)-N-(C1-6алкил)амино, карбамоилом, сульфамоилом, N-(C1-6алкил)карбамоилом, N,N-(C1-6алкил)2карбамоилом, N-(C1-6алкил)сульфамоилом, N,N-(C1-6алкил)2сульфамоилом или (С1-6алкил)-S(O)а-, где а равен 0-2.
В еще одном воплощении R1 выбран из C1-6алкила, С2-6алкенила, C2-6элкинила, С3-6циклоалкила, фенила и бензила; где фенил и бензил возможно замещены одним или более галогено, нитро, циано, гидрокси, C1-6алкилом, C1-6алкокси, C1-6алканоилом, N-(C1-6алкил)амино, N,N-(C1-6-алкил)2амино, N-(C1-6алканоил)амино, N-(C1-6алканоил)-N-(C1-6алкил)амино, карбамоилом, сульфамоилом, N-(C1-6алкил)карбамоилом, N,N-(C1-6алкил)2карбамоилом N-(C1-6алкил)сульфамоилом, N,N-(C1-6алкил)2сульфамоилом или (C1-6алкил)-S(O)а-, где а равен 0-2.
В еще одном воплощении R1 выбран из водорода, C1-6алкила, C2-6алкенила, C2-6алкинила, С3-6циклоалкила, фенила и бензила.
В еще одном воплощении R1 выбран из C1-6алкила, C2-6алкенила, C2-6алкинила, C3-6циклоалкила, фенила и бензила.
В еще одном воплощении R1 выбран из водорода и C1-6алкила.
В еще одном воплощении R1 выбран из водорода и этила.
В еще одном воплощении R1 представляет собой C1-6алкил.
В еще одном воплощении R1 представляет собой этил.
В еще одном воплощении Q представляет собой хлоро.
В еще одном воплощении X представляет собой =O или =N-OH.
В еще одном воплощении X представляет собой =N-OC(O)Me.
В еще одном воплощении X представляет собой =O.
В еще одном воплощении X представляет собой =N-OH.
Следовательно, в одном воплощении предложено соединение формулы (II), как оно изображено в данном описании изобретения выше, где:
R1 выбран из водорода, C1-6алкила, С2-6алкенила, С2-6алкинила, С3-6-циклоалкила, фенила и бензила; где фенил и бензил возможно замещены одним или более галогено, нитро, циано, гидрокси, C1-6алкилом, C1-6алкокси, C1-6алканоилом, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, N-(C1-6алканоил)амино, N-(C1-6алканоил)-N-(C1-6алкил)амино, карбамоилом, сульфамоилом, N-(С1-6алкил)карбамоилом N,N-(С1-6алкил)2карбамоилом, N-(С1-6алкил)сульфамоилом, N,N-(С1-6алкил)2сульфамоилом или (C1-6алкил)-S(O)а-, где а равен 0-2;
X представляет собой =O или =N-OH;
Y представляет собой водород или ; и
Q представляет собой водород или хлоро;
или его соль.
В еще одном воплощении предложено соединение формулы (II), как изображено в данном описании изобретения выше, где:
R1 выбран из C1-6алкила, С2-6алкенила, C2-6алкинила, С3-6циклоалкила, фенила и бензила; где фенил и бензил возможно замещены одним или более галогено, нитро, циано, гидрокси, алкилом, C1-6алкокси, C1-6алканоилом, N-(C1-6алкил)амино, N,N-(C1-6-алкил)2амино, N-(C1-6алканоил)амино, N-(C1-6алканоил)-N-(C1-6алкил)амино, карбамоилом, сульфамоилом, N-(C1-6алкил)карбамоилом, N,N-(C1-6алкил)2карбамоилом, N-(C1-6алкил)сульфамоилом, N,N-(C1-6алкил)2сульфамоилом или (C1-6алкил)-S(O)а-, где a равен 0-2;
X представляет собой =O или =N-OH;
Y представляет собой водород или ;
и Q представляет собой водород или хлоро;
или его соль.
В еще одном воплощении предложено применение соединения формулы (II), как оно изображено в данном списании изобретения выше, где:
R1 выбран из C1-6алкила, С2-6алкенила, С2-6алкинила, С3-6циклоалкила, фенила и бензила; где фенил и бензил возможно замещены одним или более галогено, нитро, циано, гидрокси, C1-6-алкилом, C1-6алкокси, C1-6алканоилом, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, N-(C1-6алканоил)амино, N-(C1-6алканоил)-N-(C1-6алкил)амино, карбамоилом, сульфамоилом, N-(C1-6алкил)карбамоилом, N,N-(C1-6алкил)2карбамоилом, N-(C1-6алкил)сульфамоилом, N,N-(C1-6алкил)2сульфамоилом или (C1-6алкил)-S(O)а-, где a равен 0-2;
X представляет собой =O или =N-OH;
Y представляет собой водород или ; и
Q представляет собой водород или хлоро; или его соль;
в качестве фармацевтического промежуточного соединения.
В еще одном воплощении предложено применение соединения формулы (II), как оно изображено в данном описании изобретения выше, где:
R1 выбран из C1-6алкила, С2-6алкенила, С2-6алкинила, C3-6циклоалкила, фенила и бензила; где фенил и бензил возможно замещены одним или более галогено, нитро, циано, гидрокси, C1-6алкилом, C1-6алкокси, C1-6алканоилом, N-(C1-6алкил)амино, N,N-(C1-6алкил)2амино, N-(C1-6алканоил)амино, N-(C1-6алканоил)-N-(C1-6алкил)амино, карбамоилом, сульфамоилом, N-(C1-6алкил)карбамоилом, N,N-(C1-6алкил)2карбамоилом, N-(C1-6алкил)сульфамоилом, N,N-(C1-6алкил)2сульфамоилом или (C1-6алкил)-S(O)а-, где a равен 0-2;
X представляет собой =O или =N-OH;
Y представляет собой водород или ; и
Q представляет собой водород или хлоро;
или его соль;
в качестве промежуточного соединения для получения соединения формулы (I) или его соли.
В еще одном воплощении предложено соединение формулы (II), как изображено в данном описании изобретения выше, где:
R1 выбран из C1-6алкила, С2-6алкенила, C2-6алкинила, C3-6циклоалкила, фенила и бензила;
X представляет собой =O или =N-OH;
Y представляет собой водород или 4-хлорфенилсульфанил; или его соль.
В еще одном воплощении предложено применение соединения формулы (II), как изображено в данном описании изобретения выше, где:
R1 выбран из C1-6алкила, С2-6алкенила, С2-6алкинила, С3-6циклоалкила, фенила и бензила;
X представляет собой =O или =N-OH;
Y представляет собой водород или 4-хлорфенилсульфанил;
или его соль;
в качестве фармацевтического промежуточного соединения.
В еще одном воплощении предложено применение соединения формулы (II), как изображено в данном описании изобретения выше, где:
R1 выбран из C1-6алкила, C2-6алкенила, C2-6алкинила, C3-6циклоалкила, фенила и бензила;
X представляет собой =O или =N-OH;
Y представляет собой водород или 4-хлорфенилсульфанил; или его соль;
в качестве промежуточного соединения для изготовления соединения формулы (I) или его соли.
В еще одном воплощении предложено соединение формулы (II), как изображено в данном описании изобретения выше, где:
R1 выбран из C1-6алкила;
X представляет собой =O или=N-OH;
Y представляет собой водород или 4-хлорфенилсульфанил; или его соль.
В еще одном воплощении предложено применение соединения формулы (II), как изображено в данном описании изобретения выше,
где: R1 выбран из C1-6алкила;
X представляет собой =O или =N-OH;
Y представляет собой водород или 4-хлорфенилсульфанил;
или его соль;
в качестве фармацевтического промежуточного соединения.
В еще одном воплощении предложено применение соединения формулы (II), как изображено в данном описании изобретения выше,
где: R1 выбран из C1-6алкила; X представляет собой =O или=N-OH;
Y представляет собой водород или 4-хлорфенилсульфанил;
или его соль;
в качестве промежуточного соединения для изготовления соединения формулы (I) или его соли.
В еще одном аспекте изобретения предложен способ получения фармацевтического соединения формулы (I) или его соли или сложного эфира, включающий взаимодействие соединения формулы (IIc), как изображено в данном описании изобретения выше, с ацетилирующим агентом;
где значения R1 такие, как они определены в данном описании изобретения выше, и где Q представляет собой хлоро;
и затем возможно взаимодействие с кислотой или основанием.
При взаимодействии с кислотой или основанием происходит гидролиз сложноэфирной группы (когда R1 не является водородом) с получением соединения формулы (I) или его соли.
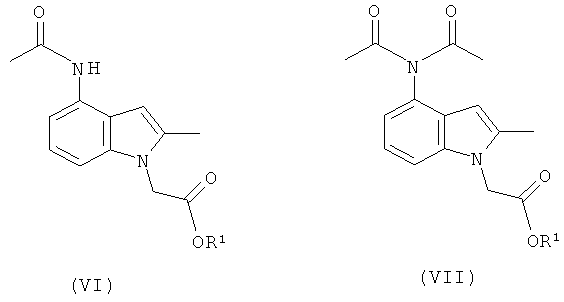
Смесь промежуточных продуктов может содержать главным образом целевой амид формулы (V) или может содержать смесь амида формулы (V) и имида формулы (VI), как показано на Схеме (II):
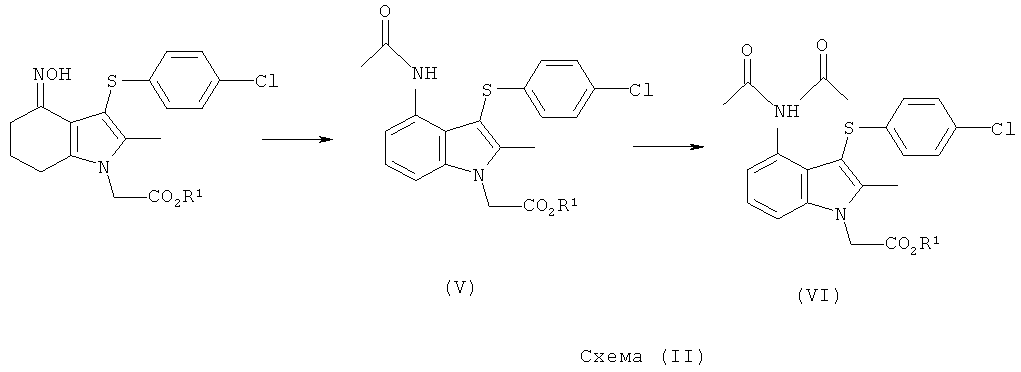
Смесь амида (V) и имида (VI) может быть превращена преимущественно в амид (V) посредством обработки водной кислотой в присутствии органического сорастворителя, например этанола. Альтернативно смесь может быть подвергнута воздействию деэстерифицирующих (гидролизующих) условий, в которых имидная группа превращается в амид, и выделенный продукт представляет собой соединение формулы (I) или его соль. Амид (V) возможно может быть перекристаллизован из растворителя, такого как этанол, для увеличения чистоты перед деэстерификацией.
В одном воплощении предложен способ, включающий взаимодействие соединения формулы (II) с формулой (IIc), как изображено в данном описании изобретения выше, с ацетилирующим агентом;
где значения R1 такие, как они определены в пп.1-8, и где Q представляет собой хлоро.
Следовательно, в одном воплощении предложен способ получения фармацевтического соединения формулы (I) или его соли, включающий взаимодействие соединения формулы (IIc), как изображено в данном описании изобретения выше, с ацетилирующим агентом;
где значения R1 такие, как они определены в данном описании изобретения выше, и где Q представляет собой хлоро;
и затем деэстерификацию с получением соединения формулы (I) или его соли.
Процесс деэстерификации может включать взаимодействие с основанием, или кислотой, или с водородом в присутствии катализатора.
В одном воплощении способ деэстерификации может включать взаимодействие с основанием.
Неожиданно было обнаружено, что некоторые условия взаимодействия преимущественно не способствуют образованию имида. Например, путем уменьшения количества уксусного ангидрида до 4 молярных экв. и используя натрия йодид при 85°C для поддержания времени взаимодействия примерно 4,5 ч.
Примерами подходящих оснований являются неорганические основания, например гидроксиды металлов, например LiOH, NaOH или КОН.
Как обсуждалось в данном описании изобретения выше, было обнаружено, что соединение формулы (IId) неожиданно полезно в качестве промежуточного соединения для получения фармацевтического соединения (I) или его соли. В еще одном аспекте изобретения предложен способ, включающий взаимодействие соединения формулы (IId), как оно изображено в данном описании изобретения выше, с ацетилирующим агентом, где R1 такой, как определено в данном описании изобретения.
Продуктами данного взаимодействия являются амид (VI) и имид (VII):
Эти продукты могут быть перенесены в виде смеси на следующую стадию и гидролизующие условия, используемые позже в синтезе, приводят к превращению имида в целевой амид с получением соединения формулы (I) или его соли.
Способы ацетилирования с участием соединения формулы (IIc) и (IId) можно проводить в растворителе, например в ароматическом углеводородном растворителе, например толуоле, ксилоле или мезитилене, или в кетонном растворителе, например метилизобутилкетоне (MIBK), метилэтилкетоне (МЕК), карбоновокислотных растворителях, например уксусной кислоте, или эфирных растворителях, например 2-метилтетрагидрофуране.
Способ ацетилирования с участием соединения формулы (IIc) и (IId), работает лучше всего при повышенной температуре, например вплоть до примерно 140°C.
В еще одном аспекте настоящего изобретения предложен улучшенный способ, где взаимодействие соединения формулы (IIc) или (IId) с ацетилирующим агентом осуществляют в присутствии йодидной соли.
Неожиданно присутствие йодидной соли в реакционной смеси, как было обнаружено, позволяет уменьшить температуру, необходимую для эффективного протекания реакции. Примерами йодидной соли являются йодиды металлов, например Kl, Nal, Lil и соли йодида аммония, например (C1-6алкил)4Nl, например тетра-N-бутиламмония йодид. Температура реакционной смеси, необходимая для этих способов, может быть снижена до 80-100°C по сравнению с обычными намного более высокими температурами за счет включения йодидной соли в реакционную смесь. Субстехиометрическое количество йодидной соли достаточно для обеспечения полезного эффекта.
Были изобретены другие условия, которые обеспечивают возможность эффективного взаимодействия при предпочтительно пониженной температуре без присутствия йодидной соли. Неожиданно применение карбоновой кислоты в качестве сорастворителя в реакционной среде приводит к эффективному взаимодействию при предпочтительно пониженной температуре.
Следовательно, в еще одном аспекте изобретения предложен улучшенный способ, в котором взаимодействие соединения формулы (IIc) или (IId) с ацетилирующим агентом проводят в присутствии карбоновокислотного сорастворителя.
Подходящими карбоновокислотными сорастворителями могут быть карбоновые кислоты, содержащие от 1 до 7 атомов углерода, например уксусная кислота.
Например, использование смеси 50:50 ксилола или мезитилена и уксусной кислоты обеспечивает возможность протекания реакции в отсутствии йодидного катализатора при температуре 105-110°C, в то время как в той же смеси растворителей в присутствии 5 мол.% либо йодида натрия, либо тетрабутиламмония йодида взаимодействие протекает при температура ниже 95-100°C.
В еще одном аспекте настоящего изобретения предложен улучшенный способ, где способ ацетилирования с участием соединения (IIc) или (IId) проводят в присутствии кислоты Льюиса. Одним примером кислоты Льюиса является FeCl3. Неожиданно было обнаружено, что присутствие кислоты Льюиса обеспечивает возможность эффективного протекания реакции ацетилирования при намного более низкой температуре, чем необходимо в иных условиях. Присутствие FeCl3 обеспечивает возможность протекания реакции при температуре вплоть до 70°C.
Ацетилирующие агенты хорошо известны специалисту. Ацетилирующие агенты, которые можно использовать для процесса ацетилирования с участием соединения формулы (IIc) и (IId) включают уксусный ангидрид, ацетилгалогениды, такие как ацетилхлорид, и сложные тиоэфиры, такие как фенилтиоацетат.Фенилтиоацетат, как было обнаружено, обеспечивает лучшее превращение, чем уксусный ангидрид сам по себе в сопоставимых условиях. Альтернативно в способе можно использовать другие ацилирующие агенты, например ангидрид бензойной кислоты или ангидрид пиваловой кислоты. Амидные группы в продуктах таких реакций затем необходимо гидролизовать, а полученные амины затем ацетилируют, используя агент ацетилирования.
В еще одном воплощении изобретения предложен способ получения соединения формулы (I):
включающий взаимодействие соединения формулы (IIAA):
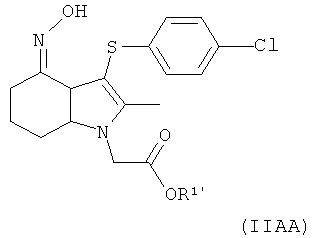
где R1′ представляет собой водород или C1-6алкил, с ацилирующим агентом с последующей деэстерификацией.
В одном воплощении R1 представляет собой водород или этил.
В одном воплощении ацилирующий агент представляет собой AC2O.
В одном воплощении способ проводят в присутствии ксилола и йодида натрия.
В еще одном воплощении изобретения предложено соединение формулы (I), полученное согласно способу, как он определен в формуле изобретения.
Различные аспекты изобретения проиллюстрированы следующими Примерами. Три способа показаны на Схемах (III), (IV), (V) и (VI) ниже.
Сокращения и общие методики
Используемые аналитические методики включают газовую хроматографию (GC), высокоэффективную жидкостную хроматографию (HPLC), жидкостную хроматографию-масс-спектрометрию (LC-MS) и ультраэффективную жидкостную хроматографию-масс-спектроскопию (UHPLC-MS). Масс-спектрометрические данные (m/z) представлены вместе с наблюдаемым распределением пика(ов). Данные ядерного магнитного резонанса (ЯМР) получали при 300 МГц, 400 МГц или 500 МГц в d6-диметилсульфоксиде, если не указано иное. Используются стандартные сокращения (s=синглет, d=дублет, m=мультиплет, dd=дублет дублетов, t=триплет, q=квартет, br=уширенный). Используемые растворители включают этанол (ETOH), метанол (МеОН), тетрагидрофуран (THF), этилацетат (EtOH), метилизобутилкетон (MIBK), метил-трет-бутиловый эфир (MTBE) и уксусную кислоту (AcOH). В общем случае реакции проводили в атмосфере азота. Если не указано иное, процедуры проводили при температуре окружающей среды (комнатная температура, к.т.), при перемешивании, в течение нескольких часов (ч) или минут (мин). Моль-экв. представляет собой молярные эквиваленты реагента относительно указанного ограничивающего реагента. Отн. об. (относительный объем) представляет собой количество растворителя относительно единицы массы указанного ограничивающего реагента, например л/кг. Отн. масс. (относительная масса) представляет собой количество вещества по массе относительно единицы массы указанного ограничивающего реагента, например кг/кг. Анализы посредством HPLC, GC и ЯМР осуществляли относительно полностью охарактеризованных стандартных образцов, используя стандартные методики, которые хорошо известны в данной области техники. ТССА=трихлоризоциануроновая кислота. GC-анализ может быть осуществлен на колонке DB-1 (30 м×0,25 мм вн. диам., 0,25 мкм) с использованием азота в качестве газа-носителя, используя подходящий градиент температуры и пламенно-ионизационное детектирование. HPLC-анализ может быть осуществлен либо на колонке Waters Symmetry С18 (150 мм×3,0 мм, 3,5 мкм), или колонке Zorbax SB-C8 (150 мм×3,0 мм, 3,5 мкм), или колонке Hichrom Асе Phenyl (50 мм×3,0 мм, 3 мкм), элюируя соответствующим градиентом водного ацетонитрила, забуференного TFA (трифторуксусной кислотой), и с УФ-детектированием (230 или 250 нм). UHPLC осуществляли либо на колонке ВЕН С18 (100 мм×2,1 мм, 1,7 мкм) или ВЕН Phenyl (100 мм×2,1 мм, 1,7 мкм), элюируя соответствующим градиентом водного ацетонитрила, забуференного TFA или ацетатом аммония соответственно, с УФ-детектированием (250 нм). UHPLC-MS может быть проведена, используя +ve или -ve ионизацию электрораспылением с капиллярным напряжением 3,5 кВ и конусным напряжением, возрастающим от 10 до 60 В. LC-MS может быть осуществлена на колонке Hichrom Асе Phenyl (50 мм×3,0 мм, 3 мкм), элюируя соответствующим градиентом водного ацетонитрила, забуференного TFA, и с УФ-детектированием (230 нм), используя комбинированную АРCl (химическая ионизация при атмосферном давлении)/+ve ионизацию электрораспылением.
Способ 1
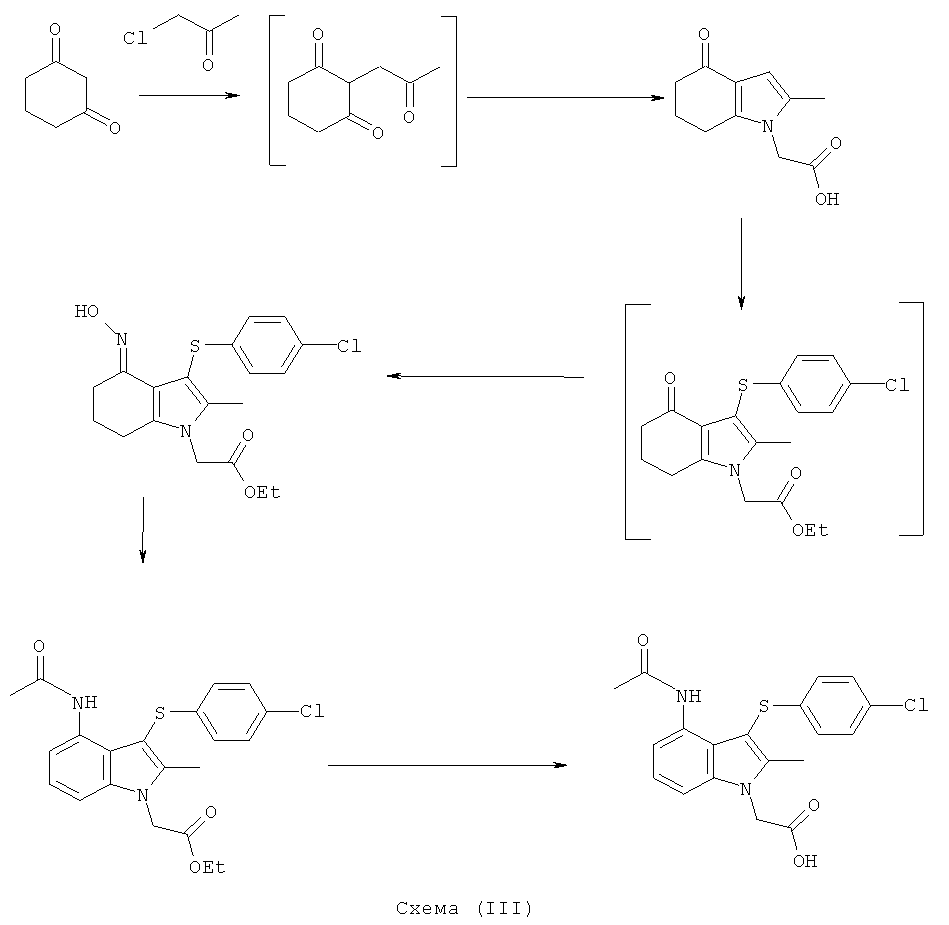
Этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)ацетат
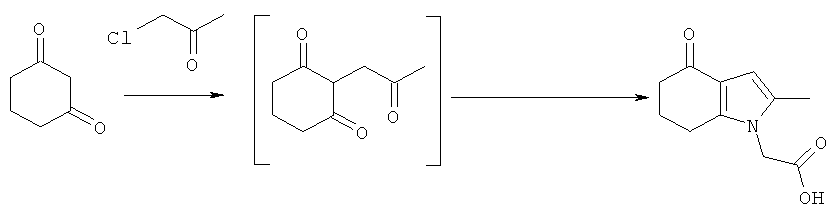
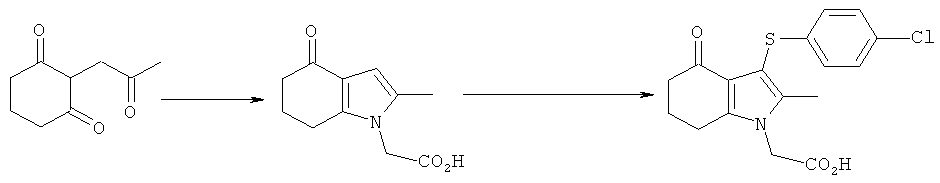
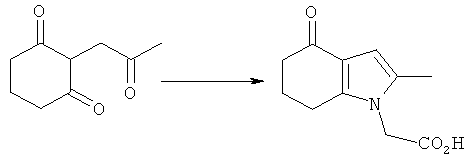
Раствор KOH (2,54 кг; 2,26 кг, скорректированные с учетом количественного анализа, 40,2 моль) в воде (4,0 л) добавляли в течение 30 мин к раствору циклогексан-1,3-диона (4,53 кг; 4,51 кг, скорректированные с учетом количественного анализа, 40,2 моль) в EtOH (16,2 л), поддерживая температуру ниже 30°C. После перемешивания в течение 15 мин при к.т., медленно добавляли к смеси свежеперегнанный хлорацетон (4,39 кг; 3,83 кг, скорректированные с учетом количественного анализа, 41,4 моль), поддерживая температуру 25-28°C. Полученную суспензию перемешивали при к.т. в течение 19 ч. Неорганические побочные продукты затем удаляли фильтрацией и промывали на фильтре EtOH (4,5 л). Фильтраты объединяли с получением 2-(2-оксопропил)циклогексан-1,3-диона в виде раствора в EtOH. Анализ посредством GC - 26,5% масс./масс.; чистота посредством GC: 82,3% по площади). Данный раствор переносили в реактор и добавляли при к.т. при перемешивании гидрохлоридную соль этилового эфира глицина (6,41 кг; 6,17 кг, скорректированные с учетом количественного анализа, 44,2 моль), затем ацетата натрия тригидрат (6,04 кг; 6,02 кг, скорректированные с учетом количественного анализа, 44,3 моль). Смесь разбавляли этанолом (28,9 л), затем нагревали с обратным холодильником в течение 2 ч. EtOH удаляли в вакууме (от -0,850 до -0,900 бар (от -0,850 до -0,900×105 Па)) при 30°C, пока не собрали 40 л дистиллята. После охлаждения остатка до 0°C добавляли воду (15 л), поддерживая температуру 0-5°C. После перемешивания в течение еще 1 ч при данной температуре отделившееся твердое вещество собирали фильтрацией и затем сушили в вакууме при 35°C, продувая азотом, с получением указанного в заголовке соединения в виде твердого вещества; 6,7 кг; анализ посредством 1Н-ЯМР: 86,6% масс./масс.; масса, скорректированная для анализа: 5,80 кг (61% за 2 стадии); чистота посредством GC: 96,0% по площади; m/z: 235 (МН+); 1Н-ЯМР: (CDCl3) 1.27-1.32 (3H, t), 2.10-2.17 (2Н, m), 2.19 (3H, s), 2.42-2.46 (2Н, t), 2.65-2.69 (2Н, t), 4.21-4.28 (2Н, m), 4.51 (2Н, s) и 6.27 (1Н, s).
Альтернативные методики синтеза этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил)ацетата
Альтернативная методика 1
Раствор гидроксида калия (23,58 г, 20,0 г, скорректированные с учетом количественного анализа, 357 ммоль) в воде (36 мл) при к.т. добавляли к раствору 1,3-циклогександиона (39,97 г, 356 ммоль) в этаноле (144 мл) при перемешивании (экзотермическое добавление). После перемешивания в течение 15 мин при 20°C добавляли одной порцией бидистиллированный хлорацетон (37,62 г, 33,9 г, скорректированные с учетом количественного анализа, 366 ммоль), и реакционную смесь перемешивали в течение 20 ч при 20°C. Неорганические побочные продукты удаляли фильтрацией, промывая остаток на фильтре этанолом (40 мл), и фильтраты объединяли с получением 2-(2-оксопропил)циклогексан-1,3-диона в виде раствора в водном этаноле. К данному раствору добавляли гидрохлорид этилового эфира глицина (54,73 г, 392 ммоль) и безводный ацетат натрия (32,19 г, 392 ммоль). Реакционную смесь нагревали до кипения с обратным холодильником (внутренняя температура 75°C) в течение 1 ч, затем охлаждали до 20°C и перемешивали в течение 19 ч. Смесь нагревали до кипения с обратным холодильником (внутренняя температура 75°C) и добавляли воду (116 мл), затем охлаждали до 20°C в течение 30 мин. Небольшое количество этилового эфира 2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил)уксусной кислоты добавляли в качестве затравки (10 мг) и через несколько минут наблюдалась кристаллизация продукта. Суспензию перемешивали в течение 30 мин, затем охлаждали до 5°C и перемешивали в течение 18 ч. Твердое вещество собирали фильтрацией, промывали водой (2×80 мл), затем трет-бутил-метиловым эфиром (2×80 мл) и затем сушили в вакууме при 40°C в течение 20 ч с получением этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)ацетата в виде бледно-желтого кристаллического твердого вещества, 41,77 г (выход 49,8%). Т.пл. (Точка плавления) от 105,1 до 105,4°C. Анализ посредством 1Н-ЯМР 99,3% масс./масс. Чистота посредством GC - 100% по площади.
Альтернативная методика 2
В реакционном сосуде 1 гидроксид калия (11,78 г, 10,0 г, скорректированные с учетом количественного анализа, 178 ммоль) растворяли в воде (72 мл) при перемешивании (сильно экзотермическое), затем раствор охлаждали опять до 20°C. Добавляли 1,3-циклогександион (20,0 г, 178 ммоль) (экзотермическое добавление), и полученный темно-красный раствор перемешивали при 20°C в течение 5 мин. Одной порцией добавляли бидистиллированный хлорацетон (18,9 г, 17,0 г, скорректированные с учетом количественного анализа, 184 ммоль), промывали этанолом (18 мл), и реакционную смесь перемешивали в течение ночи при 20°C. Полученный раствор 2-(2-оксопропил)циклогексан-1,3-диона, объем 124 мл, делили на 4 равные части. Гидрохлорид этилового эфира глицина (6,85 г, 49,0 ммоль), безводный ацетат натрия (4,02 г, 49,0 ммоль), воду (26,5 мл) и этанол (5,0 мл) загружали в реакционный сосуд 2 и начинали перемешивание. Одну порцию раствора 2-(2-оксопропил)циклогексан-1,3-диона, полученного выше (31 мл), затем добавляли к содержимому реакционного сосуда 2 при перемешивании, и полученную смесь нагревали до 75°C, выдерживали при данной температуре в течение 2 ч, затем охлаждали до 20°C в течение 55 мин. После перемешивания в течение ночи при 20°C твердое вещество собирали фильтрацией, промывали водой (10 мл), трет-бутил-метиловым эфиром (2×10 мл) и затем сушили в вакууме при 40°C в течение 20 ч с получением (2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)уксусной кислоты этилового эфира в виде бежевого твердого вещества, 5,88 г (56,0% выход). Т.пл. от 101,2 до 103,5°C. Анализ посредством 1Н-ЯМР 95,3% масс./масс. Чистота посредством GC - 99,25% по площади.
Альтернативная методика 3
Раствор гидроксида калия (1,0 моль-экв.) в воде (0,9 отн. об.) добавляли к суспензии 1,3-циклогександиона (1,0 моль-экв., ограничивающий реагент) в воде (1,1 отн. об.) при перемешивании в течение приблизительно 1 часа, поддерживая температуру ниже 30°C. Еще через 15 мин добавляли хлорацетон (1,03 моль-экв.) в течение приблизительно 4 часов, затем реакционную смесь перемешивали в течение ночи при 20°C. Добавляли гидрохлорид этилового эфира глицина (1,1 моль-экв.) и этилацетат (2 отн. об.), затем ацетат натрия (1,1 моль-экв.) и воду (2 отн. об.). Реакционную смесь нагревали при 60°C в течение 2 ч, затем охлаждали до 50°C, после чего водную фазу отделяли и отбрасывали. MTBE (4 отн. об.) добавляли к органической фазе, раствор нагревали опять вплоть до 50°C и затем охлаждали до 35°C в течение 20 мин. Добавляли затравку этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил)ацетата (0,0001 моль-экв.) и смесь охлаждали до 5°C в течение 60 мин и состаривали в течение ночи. Твердый продукт собирали фильтрацией, промывали водой (2 отн. об.), затем MTBE (2×2 отн. об.) и затем сушили при 40°C под вакуумом с получением указанного в заголовке соединения с 62% выходом; чистота 98% масс./масс.
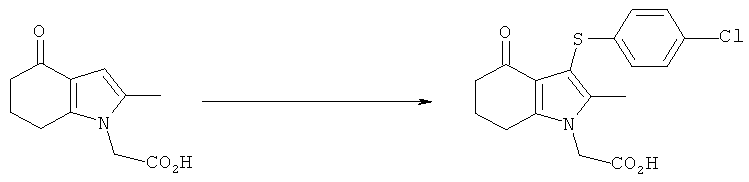
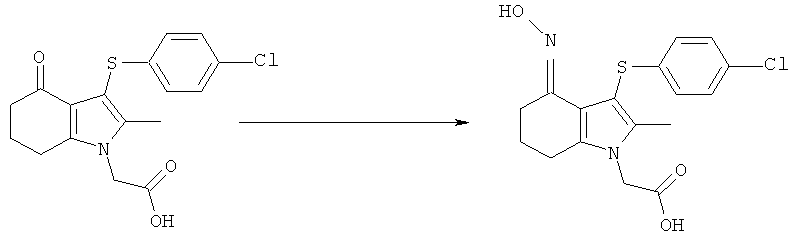
Этил-[3-(4-хлор-фенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]ацетат
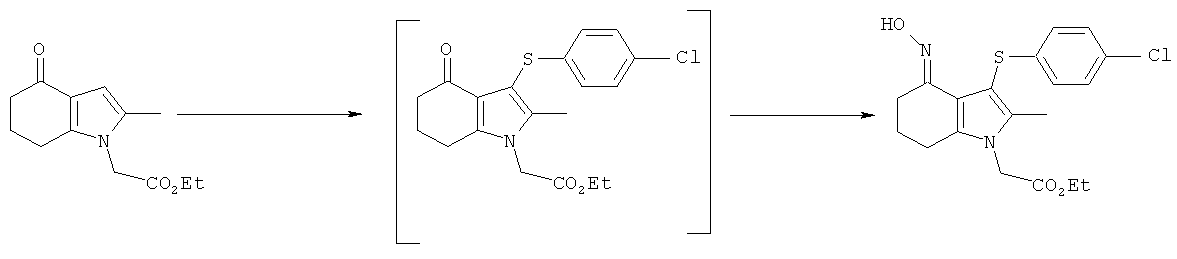
TCCA (1,16 кг, 5,01 Моль) добавляли к раствору бис-(4-хлорфенил)дисульфида (4,83 кг; 4,80 кг, скорректированные с учетом количественного анализа, 16,7 моль) в EtOAc (56 л). Смесь охлаждали до 0°C и выдерживали при данной температуре в течение 45 мин. Затем добавляли тремя порциями этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил)ацетат (6,48 кг; 5,61 кг, скорректированные с учетом количественного анализа, 23,9 моль) (умеренно экзотермическое). После перемешивания в течение 30 мин образец переносили для HPLC-анализа, затем добавляли раствор NaHCO3 (1,12 кг, 13,3 моль) в воде (22,5 л). Смесь нагревали до к.т. в течение 30 мин и после перемешивания в течение еще 15 мин твердый побочный продукт удаляли фильтрацией и промывали EtOAc (6,5 л). Фильтраты объединяли и фазы оставляли разделяться. Водный слой отделяли и экстрагировали EtOAc (11,2 л). Органические слои объединяли и анализировали, при этом анализ посредством HPLC показал 9,2% масс./масс., и чистота посредством HPLC составляла 77,7% по площади. Органический раствор затем концентрировали в вакууме, пока не оставался объем -10-12 л (объем дистиллята ~60 л). ETOH (38,9 л) добавляли к остатку и перегонку продолжали, пока не было собрано ~35 л дистиллята, с получением промежуточного соединения этил-[3-(4-хлорфенилсульфанил)-2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил]ацетата в виде раствора в EtOH. К данному раствору дополнительно добавляли EtOH (6,5 л), затем гидроксиламина гидрохлорид (2,15 кг, 30,9 моль) и безводный ацетат натрия (2,60 кг; 2,55 кг, скорректированные с учетом количественного анализа, 31,1 Моль) и смесь нагревали с обратным холодильником в течение 4 ч. После охлаждения до к.т. в течение 1 ч твердый продукт собирали фильтрацией. Реактор промывали смесью 50% об./об. этанол:вода (13 л), которую переносили на фильтр. После непродолжительной сушки на фильтре влажный продукт затем переносили в реактор и добавляли воду (42 л). Смесь нагревали до 50°C и перемешивали в течение 30 мин, затем охлаждали обратно до к.т. Твердый продукт собирали фильтрацией, затем загружали обратно в реактор влажным и суспендировали в этаноле (19,4 л) при к.т. в течение 30 мин. Твердое вещество собирали фильтрацией, промывали этанолом (9,7 л), затем свежим этанолом (6,5 л) и затем сушили в вакууме при 40°C с получением указанного в заголовке соединения в виде твердого вещества; 7,00 кг; анализ посредством 1Н-ЯМР 87,5% масс./масс.; 6,1 кг, скорректированные с учетом количественного анализа (65% за 2 стадии); чистота посредством HPLC: 88,4% по площади; m/z: 393 (МН+); 1Н-ЯМР: 1.20-1.25 (3H, t), 1.79-1.86 (2Н, m), 2.14 (3H, s), 2.50-2.51 (2Н, t), 2.54-2.59 (2Н, t), 4.15-4.22 (2Н, m), 4.83 (2Н, s), 6.92-6.95 (2Н, d), 7.21-7.24 (2Н: d) и 10,22 (1Н, s).
Данные для выделенного образца промежуточного этил-[3-(4-хлорфенилсульфанил)-2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил]ацетата: 1Н-ЯМР: (CDCl3) 1.33-1.37 (3H, t), 2.14-2.23 (2Н, m), 2.27 (3H, s), 2.45-2.49 (2Н, t), 2.74-2.78 (2Н, t), 4.27-4.35 (2Н, m), 4.64 (2Н, s), 7.03-7.08 (2.Н, d) и 7.13-717 (2Н, d).
Альтернативная методика синтеза промежуточного этил-[3-(4-хлорфенилсульфанил)-2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил]ацетата
Бис-(4-хлорфенил)дисульфид (7,39 г, 25,7 ммоль) растворяли в EtOAc (86,5 мл) в реакционном сосуде 1, охлаждая смесь до 5°C при перемешивании, с получением бледно-желтого раствора. Затем добавляли одной порцией сульфурилхлорид (2,1 мл, 25,7 ммоль) (слегка экзотермический) с получением через 15 мин раствора оранжевого цвета. Во втором реакционном сосуде этил-2-(2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил)ацетат (8,65 г, 36,8 ммоль) суспендировали в EtOAc (34,6 мл) при перемешивании с охлаждением смеси до 5°C. Предварительно охлажденный раствор в реакционном сосуде 1, полученный выше, добавляли во второй сосуд 4 равными частями в течение 10 мин, поддерживая температуру в диапазоне 5-10°C, что приводило к образованию темно-коричневого раствора, содержащего небольшое количество нерастворимого вещества. Смесь затем оставляли нагреваться до к.т. в течение 90 мин при перемешивании. Добавляли раствор гидрокарбоната натрия (1,73 г, 20,6 ммоль) в воде (34,6 мл), и полученную двухфазную смесь перемешивали в течение 15 мин. Слои оставляли разделяться, и водную фазу отбрасывали. Верхнюю органическую фазу сушили над сульфатом магния, который затем удаляли фильтрацией с получением раствора этил-2-(2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)ацетата в этилацетате, масса 132,6 г. Анализ посредством HPLC 8,4% масс./масс., следовательно присутствует 11,1 г этил-2-(2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)ацетата, (80%) выход.
Альтернативные методики синтеза этил-[3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]ацетата
Альтернативная методика 1
В сосуде 1 бис-(4-хлорбензол)дисульфид (0,53 моль-экв.) суспендировали в этилацетате (3,5 отн. об.) при перемешивании и смесь охлаждали до 0°C. Добавляли одной порцией сульфурилхлорид (0,53 моль-экв.), остатки промывали этилацетатом (0,5 отн. об.) и смесь перемешивали при 0°C в течение приблизительно 1 ч. Этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)ацетат (1,0 моль-экв., ограничивающий реагент) и этилацетат (5 отн. об.) загружали в сосуд 2 и смесь перемешивали при 20°C. Содержимое сосуда 1 добавляли к смеси в сосуде 2 в течение приблизительно 30 мин, промывая остатки этилацетатом (0,5 отн. об.). Медленно добавляли водный карбонат натрия (1M, 1,45 моль-экв.) (выделялся газ), смесь перемешивали, затем слои оставляли разделяться, отбрасывая нижнюю водную фазу. Раствор хлорида натрия (1,45 моль-экв.) в воде (5 отн. об.) добавляли к органической фазе, смесь перемешивали, затем слои оставляли разделяться, отбрасывая нижнюю водную фазу. Органический слой концентрировали посредством перегонки при атмосферном давлении до примерно 4 отн. об. Гидроксиламина гидрохлорид (1,0 моль-экв.) загружали к концентрату при 20°C, затем трибутиламин (1,0 моль-экв.) и этанол (2 отн. об.), и полученную смесь нагревали при 60°C в течение 4 ч. Смесь охлаждали до 20°C, твердое вещество собирали фильтрацией, промывали этилацетатом (2×2 отн. об.), затем сушили при 40°C под вакуумом с получением указанного в заголовке соединения с выходом 87,3%; чистота 99% масс./масс.
Альтернативная методика 2
В сосуде 1 бис-(4-хлорбензол)дисульфид (0,53 моль-экв.) суспендировали в этилацетате (4 отн. об.) при перемешивании и смесь охлаждали до 5°C. Сульфурилхлорид (0,53 моль-экв.) добавляли одной порцией и смесь перемешивали при 5°C в течение приблизительно 1 ч. Этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)ацетат (1,0 экв., ограничивающий реагент) и этилацетат (5 отн. об.) загружали в сосуд 2 и смесь перемешивали при 20°C. Содержимое сосуда 1 добавляли к смеси в сосуде 2 в течение приблизительно 30 мин, промывали остатки этилацетатом (0,5 отн. об.). Добавляли триэтиламин (1,0 моль-экв.), и смесь перемешивали в течение ночи. Твердый побочный продукт удаляли фильтрацией, остаток на фильтре промывали этилацетатом (1 отн. об.) и объединенные фильтраты упаривали в вакууме. Остаток растворяли в смеси этилацетата (4 отн. об.) и этанола (2 отн. об.), затем добавляли гидроксиламина гидрохлорид (1,0 моль-экв.) и трибутиламин (1,0 моль-экв.). Смесь нагревали при 60°C в течение 4 ч, затем охлаждали до 20°C. Продукт собирали фильтрацией, промывали этилацетатом (2×2 отн. об.), затем сушили при 40°C под вакуумом с получением указанного в заголовке соединения с выходом 83%; чистота посредством HPLC 95,95% по площади.
Альтернативная методика 3
В сосуде 1 бис-(4-хлорбензол)дисульфид (0,53 моль-экв.) суспендировали в этилацетате (4 отн. об.) и охлаждали до 5°C. Смесь обрабатывали хлором при 5°C в течение 15 минут, затем продували азотом и дегазировали под вакуумом. Этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)ацетат (1,0 экв., ограничивающий реагент) и этилацетат (4,5 отн. об.) загружали в сосуд 2 и смесь перемешивали при 20°C. Содержимое сосуда 1 добавляли к смеси в сосуде 2 в течение приблизительно 30 мин, промывая остатки этилацетатом (0,5 отн. об.). Добавляли триэтиламин (1,5 моль-экв.) и смесь перемешивали в течение ночи. Твердый побочный продукт удаляли фильтрацией и промывали этилацетатом (1 отн. об.). В другой сосуд загружали гидроксиламина гидрохлорид (1,0 моль-экв.), затем объединенные фильтраты. Добавляли трибутиламин (1,0 моль-экв.) и этанол (2 л/кг), и полученную смесь нагревали при 60°C в течение 4 ч. Дополнительно добавляли гидроксиламина гидрохлорид (0,5 моль-экв.) и трибутиламин (0,5 моль-экв.), затем смесь нагревали при 60°C в течение 4 ч. Смесь охлаждали до 20°C, твердый продукт собирали фильтрацией, промывали этилацетатом (2×2 отн. об.), затем сушили при 40°C под вакуумом с получением указанного в заголовке соединения с выходом 80,1%; чистота посредством HPLC 96,5% по площади.
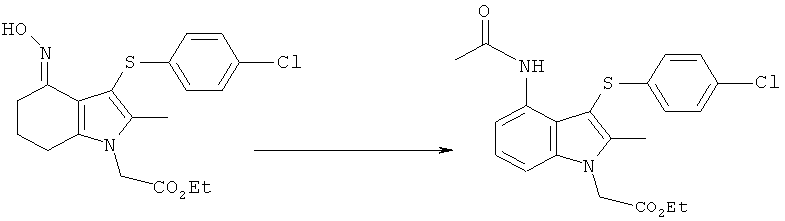
Этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-1-ил]ацетат
Способ 1
Перемешиваемую суспензию этил-[3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]ацетата (1,15 кг; 1,00 кг, скорректированные с учетом количественного анализа, 2,56 моль) и порошкообразного Nal (191,5 г, 1,28 моль) в ксилоле (7,0 л) нагревали до 85°C. Уксусный ангидрид (1,06 кг; 1,05 кг, скорректированные с учетом количественного анализа, 10,2 моль) затем добавляли в течение 1 ч при 83-85°C. Ацетилированный оксим, как ожидается, является промежуточным соединением, образующимся в данной реакции. После выдерживания при данной температуре в течение 4,5 ч смесь охлаждали до 45-50°C и растворитель удаляли в вакууме (от -830 до -850 мбар) (от -0,83 до -0,85×105 Па). К остатку добавляли ксилол (7,0 л), затем воду (2,0 л) и смесь нагревали до 60°C с получением 2 прозрачных фаз. Водный слой отделяли и органическую фазу концентрировали в вакууме при 45-50°C, пока не собирали -10 л дистиллята, а в остатке наблюдалась кристаллизация. EtOH (2,0 л) добавляли к остатку, который затем концентрировали в вакууме при 45-50°C. Дополнительный EtOH (2,0 л) добавляли к остатку, который затем охлаждали до 10°C в течение 30 мин и выдерживали при данной температуре в течение 1 ч. Твердый продукт собирали фильтрацией, промывали EtOH (1,0 л), затем сушили в вакууме при 40°C в течение 12 ч с получением неочищенного этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]ацетата в виде твердого вещества; 0,70 кг; анализ посредством 1Н-ЯМР: 96,8% масс./масс.; 0,68 кг, скорректированные с учетом количественного анализа (64%); чистота посредством HPLC: 97,9% по площади. Неочищенный этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-1-ил]ацетат (0,50 кг) объединяли с влажными продуктами из двух аналогичных получений, проведенных в масштабе 2,00 кг (скорректированный ввод) (суммарная масса 4,88 кг) в EtOH (42,8 л) и смесь нагревали до 75°C. После выдерживания в течение 15 мин при данной температуре полученный раствор затем охлаждали до 15°C в течение 2,5 ч, что приводило к кристаллизации. Твердый продукт собирали фильтрацией, промывали на фильтре EtOH (4,76 л), затем сушили в вакууме при 40°C с получением указанного в заголовке соединения в виде твердого вещества; 3,25 кг; анализ посредством 1Н-ЯМР 96,8% масс./масс.; выход, скорректированный для анализа: 3,15 кг (63%); чистота посредством HPLC: 98,6% по площади; m/z: 417 (МН+); 1Н-ЯМР: 1.12-1.24 (3H, t), 1.87 (3H, s), 2.40 (3H, s), 4.15-4.22 (2Н, q), 5.23 (2Н, s), 6.97-7.00 (2Н, d), 7.09-7.12 (1Н, m), 7.28-7.38 (3H, m), 7.48-7.51 (1Н, d), 9.50 (1Н, brs).
Способ 2
Перемешиваемую суспензию этил-[3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1Н-индол-1-ил]ацетата (571 мг, 500 мг, скорректированные с учетом количественного анализа, 1,11 ммоль) в ксилоле (2,5 мл) и уксусную кислоту (2,5 мл) нагревали до 107°C. Затем добавляли уксусный ангидрид (481 мкл, 4,45 ммоль). После нагревания до 95-100°C в течение 2 ч анализ посредством HPLC указывал на присутствие в реакционной смеси 79% по площади этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]ацетата (идентифицирован посредством сравнения времени удерживания со стандартным веществом).
Способ 3
В колбе 1 смесь йодида натрия (171,6 мг, 1,14 ммоль) в ксилоле (12,5 мл), уксусную кислоту (12,5 мл) и уксусный ангидрид (4,2 мл, 44,54 ммоль) нагревали до 97°C. В колбе 2 уксусный ангидрид (4,2 мл, 44,5 ммоль) добавляли к перемешиваемой суспензии этил-[3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1Н-индол-1-ил]ацетата (10 г, 22,3 ммоль, скорректированные с учетом количественного анализа) в ксилоле (12,5 мл) и уксусной кислоты (12,5 мл) при температуре окружающей среды. Смесь в колбе 2 добавляли к смеси в колбе 1 в течение 2-3 часов, поддерживая температуру 97°С. Реакционную смесь выдерживали при данной температуре в течение еще 2 часов после завершения добавления. Реакционную смесь охлаждали до 60°C и делили на 2 равные части. Одну из этих частей охлаждали до к.т. и добавляли пропан-1-ол (25 мл), затем добавляли в течение 15 минут воду (25 мл), вызывая осаждение. После перемешивания в течение 1 ч, твердый продукт собирали фильтрацией, промывали пропан-1-олом (2×10 мл) и сушили в вакууме при 40°C с получением неочищенного этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]ацетата в виде твердого вещества; 4,32 г (91% при обработке половины реакционной смеси). Чистота посредством HPLC 97,15% по площади.
Способ 4
Смесь йодида натрия (0,05 моль-экв.) в ксилоле (1,0 отн. об.), уксусной кислоте (1.0 отн. об.) и уксусном ангидриде (1,2 моль-экв.) нагревали до 97-103°C в сосуде 1 при перемешивании. В сосуде 2 уксусный ангидрид (1,3 моль-экв.) добавляли к перемешиваемой суспензия этил-[3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1Н-индол-1-ил]ацетата (1 моль-экв., ограничивающий реагент) в ксилоле (1,0 отн. об.) и уксусной кислоте (1,0 отн. об.) при 19-25°C. После перемешивания при данной температуре в течение 2 ч данную смесь добавляли к раствору в реакционном сосуде 1 в течение 2,5 ч, поддерживая температуру между 98 и 102°C. Сосуд 2 промывали смесью ксилола (0,25 отн. об.) и уксусной кислоты (0,25 отн. об.), которую добавляли в сосуд 1. Реакционную смесь выдерживали при 98-102°C в течение еще 1,5 ч, затем охлаждали до 60°C. Добавляли ксилол (0,9 отн. об.), затем теплый (60°C) раствор хлорида натрия (0,19 отн. масс.) в воде (1,5 отн. об.) и температуру доводили до 60°C. Водный слой отделяли и отбрасывали. Добавляли теплый (60°C) раствор тиосульфата натрия (0,1 моль-экв.) в воде (0,5 отн. об.) и после смешивания водный слой отделяли и отбрасывали. Продукт осаждали посредством добавления гептанов (3 отн. об.) к органическому слою, поддерживая температуру между 57 и 63°C. Полученную суспензию охлаждали до 20°C в течение 1 ч, затем твердый продукт собирали посредством центрифугирования, промывали этанолом (3,0 отн. об.) и сушили с получением неочищенного этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-1-ил]ацетата в виде твердого вещества с выходом 77%; чистота посредством UHPLC 97,4% по площади.
Неочищенный этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]ацетат (ограничивающий реагент) растворяли в смеси ацетонитрила (9,1 отн. об.) и воды (4,5 отн. об.) посредством нагревания до 80°C при перемешивании. Раствор охлаждали до 15°C, после чего полученный твердый продукт собирали центрифугированием, промывали этанолом (1,7 отн. об.), затем сушили с получением этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-1-ил]ацетата в виде твердого вещества с 92% выходом; чистота посредством UHPLC 99,4% по площади.
Способ 5
Смесь йодида натрия (0,0625 моль-экв.) в ксилоле (1,99 отн. об.), уксусной кислоте (0,27 отн. об.) и уксусном ангидриде (1,27 моль-экв.) нагревали до 102,5°C в сосуде 1 при перемешивании. В сосуде 2 уксусный ангидрид (1,27 моль-экв.) добавляли к перемешиваемой суспензии этил-[3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1Н-индол-1-ил]ацетата (1 моль-экв., ограничивающий реагент) в ксилоле (2,24 отн. об.) и уксусной кислоте (0,27 отн. об.) при 22°C. После перемешивания при данной температуре в течение 30 мин, эту смесь добавляли к содержимому реакционного сосуда 1 в течение 50 мин, поддерживая температуру 102,5°C. Реакционную смесь выдерживали при данной температуре в течение еще 2,5 ч. Реакционную смесь охлаждали до 60°C и добавляли тиосульфат натрия (0,05 моль-экв.) и воду (0,5 отн. об.). После смешивания и обеспечения возможности разделения слоев, нижний водный слой отбрасывали, затем органический слой дистиллировали под вакуумом, удаляя 1,8 отн. об. дистиллята. Температуру доводили до 95°C и продукт осаждали посредством добавления гептанов (3 отн. об.). Суспензию охлаждали до 20°C в течение 1 ч, затем твердый продукт собирали фильтрацией, промывали этанолом (2 отн. об.) и сушили в вакууме при 40°C с получением неочищенного этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-1-ил]ацетата в виде твердого вещества; 81% выход; чистота посредством UHPLC 98,3% по площади.
Получение и выделение образца промежуточного [3-[(4-хлорфенил)сульфанил]-(2-этокси-2-оксоэтил)-2-метил-4,5,6,7-тетрагидро-1H-индол-4-илиден]аминоацетата
Уксусный ангидрид (4,33 мл, 45,8 ммоль) добавляли к суспензии этил-[3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1Н-индол-1-ил]ацетата (15 г, 38,2 ммоль) в ксилоле (33,75 мл) и уксусной кислоте (3,75 мл) и смесь перемешивали при к.т. в течение примерно 20 мин. Образец указанного в заголовке соединения выделяли фильтрацией и сушили при 40°C. m/z 435/437 (МН+); 1Н-ЯМР: 7.27-7.23 (2Н, m), 7.00-6.96 (2Н, m), 4.92 (2Н, s), 4.19 (2Н, q, J=7,1 Гц), 2.68-2.59 (4H, m), 2.19 (3H, s), 1.90 (3H, s), 1.89-1.81 (2Н, m), 1.22 (3H, t, J=7,1 Гц).
[4-Ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]уксусная кислота
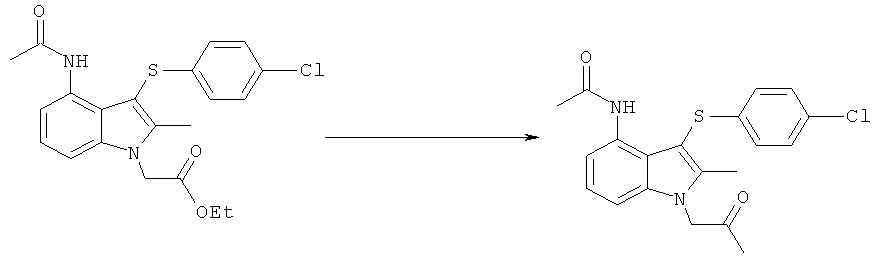
К перемешиваемой суспензии этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]ацетата (3,16 кг; 3,05 кг, скорректированные с учетом количественного анализа, 7,3 моль) в 1-пропаноле (15,3 л) добавляли водный NaOH (1M, 15,3 л). Смесь затем нагревали при 70°C в течение 2 ч, охлаждали до 40°C, затем добавляли MIBK (30,5 л) и смесь снова нагревали до 80°C. Приблизительно 20% полученной двухфазной смеси удаляли из реакционного сосуда для отдельной обработки. К остальной части, водную соляную кислоту (1M, 13,4 л) добавляли к раствору в течение 45 мин, затем полученную суспензию охлаждали до 15°C в течение 1 ч и перемешивание продолжали при данной температуре в течение еще 30 мин. Твердый продукт собирали фильтрацией, промывали водой (2×9,8 л), затем EtOAc (7,3 л), потом сушили на фильтре в течение 10 мин, затем в вакууме при 45°C с получением указанного в заголовке соединения в виде твердого вещества; 2,15 кг; анализ посредством 1Н-ЯМР: 99,4% масс./масс.; 2,14 кг, скорректированные с учетом количественного анализа (94%); чистота: 99,5% по площади посредством HPLC; m/z: 389 (МН+); 1Н-ЯМР: 1.86 (3H, s), 2.34 (3H, s), 5.11 (2Н, s), 6.97-7.00 (2Н, d), 7.08-7.11 (1Н, m), 7.27-7.30 (3H, m), 7.47-7.50 (1Н, d), 9.50 (1Н, brs).
Альтернативная методика 1
Смесь этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]ацетата (30,0 кг, 72,0 моль), 1-пропанола (120,6 кг) и водного NaOH (1M, 150,1 кг) нагревали до 68-72°C и выдерживали при данной температуре в течение 16 мин. Полученный раствор охлаждали до 18-22°C, затем фильтровали для удаления твердых веществ и фильтр промывали водой (15,0 кг). К объединенным фильтратам добавляли MIBK (240,3 кг) и двухфазную смесь нагревали до 83-87°C. Водную соляную кислоту (1M, 60,0 кг) добавляли к горячему раствору в течение периода примерно 15 мин, поддерживая температуру реакционной среды между 83 и 87°C, затем еще две части того же (52,6 кг в течение приблизительно 20 мин и 52,6 кг в течение приблизительно 20 мин). Полученную суспензию охлаждали до 13-17°C в течение 2 ч и перемешивание продолжали при данной температуре в течение еще 15 мин. Твердый продукт собирали фильтрацией, промывали водой (2×60 кг), затем EtOAc (81,1 кг), затем сушили на фильтре, используя азот, при 40°C с получением указанного в заголовке соединения в виде белого твердого вещества; 25,2 кг (80%).
Альтернативная методика 2
Смесь этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]ацетата (99,9 кг, 240 моль), этанола (393 кг), воды (450 кг) и водного NaOH (10 М, 72,3 кг) нагревали до 59-65°C и выдерживали при данной температуре в течение 30 мин. Полученный раствор охлаждали до 17-23°C, затем фильтровали для удаления твердых веществ, и фильтр промывали водой (50,9 кг). MIBK (403 кг) добавляли к объединенным фильтратам и смесь нагревали до 55-65°C. Смесь водной соляной кислоты (10M, 65,0 кг) и воды (496 кг) добавляли к горячему раствору в течение периода примерно 45 мин, поддерживая температуру реакционной смеси в рамках указанного диапазона. Полученную суспензию охлаждали до 12-18°C в течение приблизительно 60 мин и выдерживали при данной температуре в течение ночи. Твердый продукт собирали посредством центрифугирования, промывали водой (396 кг), затем этанолом (249 кг), затем сушили под вакуумом при максимальной температуре в рубашке 60°C, с получением указанного в заголовке соединения в виде белого твердого вещества; 79,1 кг (94%); чистота: 99,5% по площади посредством HPLC.
Способ 2
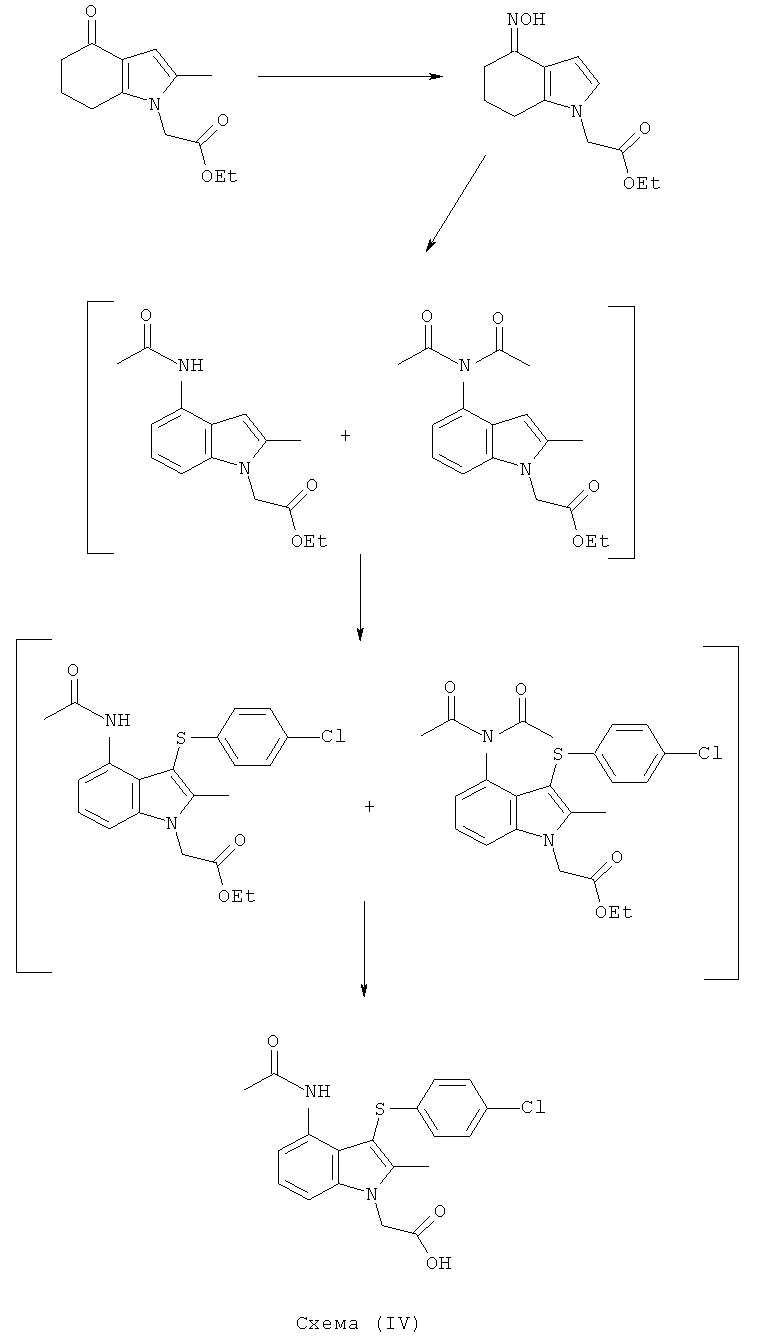
Этил[4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1Н-индол-1-ил]ацетат
Перемешиваемую смесь этил-(2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)ацетата (59,55 г, 54,06 г, скорректированные по чистоте, 0,23 моль), гидроксиламина гидрохлорида (24,44 г, 23,95 г, скорректированные по чистоте, 0,345 моль) и тригидрата ацетата натрия (47,14 г, 46,90 г, скорректированные по чистоте, 0,345 моль) в воде (108 мл) и EtOH (540 мл) нагревали с обратным холодильником в течение 2,5 ч. Смесь затем охлаждали до 10°C, твердый продукт собирали фильтрацией, затем сушили в вакууме при 40°C с получением указанного в заголовке соединения в виде смеси Е- и Z-изомеров; 55,4 г; анализ посредством HPLC: 99,0% масс./масс.; выход, скорректированный для анализа: 54,8 г (95,3%); чистота посредством HPLC: 98,9% по площади как сумма пиков 2 продуктов; LC-MS m/z: 250 для каждого пика продукта.
[4-Ацетиламино-3-(4-хлорфенилсульфанил)2-метил-1H-индол-1-ил]уксусная кислота
Перемешиваемую смесь этил-[4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]ацетата (2,068 г, 2,00 г, скорректированные по чистоте, 8,0 ммоль), уксусный ангидрид (12,37 г, 12,24 г, скорректированные по чистоте, 0,120 моль) и тетрабутиламмония йодида (2,997 г, 2,953 г, скорректированные по чистоте, 8,0 ммоль) в ксилоле (16 мл) нагревали с обратным холодильником в течение 2 ч. Ацетилированный оксим, как ожидается, является промежуточным соединением, образовавшимся в данной реакции. После охлаждения до к.т. уксусный ангидрид и ксилол удаляли в вакууме при 50°C. Воду (10 мл) добавляли к вязкому остатку и смесь упаривали досуха в вакууме при 50°C. После охлаждения до к.т. CH2CI2 (20 мл) добавляли к остатку и перемешивание продолжали в течение 10 мин. Нерастворимое твердое вещество удаляли фильтрацией и фильтрат концентрировали в вакууме. Ксилол (20 мл) добавляли к остатку, перемешивание продолжали в течение 10 мин, затем некоторое количество дополнительного твердого вещества удаляли фильтрацией. Фильтрат концентрировали в вакууме при 50°C и EtOAc (20 мл) добавляли к остатку с получением смеси продуктов в виде раствора в EtOAc; чистота согласно HPLC: 9,67% по площади этил-(4-ацетиламино-2-метил-1H-индол-1-ил)ацетата, и 74,79% по площади этил-(4-диацетиламино-2-метил-1Н-индол-1-ил)ацетата; LC-MS показал m/z: 275 и 317, оба согласуются с МН+.
К раствору ТССА (0,326 г, 0,316 г, скорректированные по чистоте, 1,36 ммоль) в EtOAc (10 мл) добавляли бис(4-хлорфенил)дисульфид (1,18 г, 1,14 г, скорректированные по чистоте, 4,0 ммоль). Раствор этил-(4-ацетиламино-2-метил-1H-индол-1-ил)ацетата и этил-(4-диацетиламино-2-метил-1H-индол-1-ил)ацетата, полученный выше, добавляли к смеси, по каплям, в течение 10 мин. После перемешивания в течение 1 ч нерастворимое твердое вещество удаляли фильтрацией. Фильтрат концентрировали в вакууме при 40°C и EtOH (10 мл) добавляли к остатку с получением смеси продуктов в виде раствора в EtOH; чистота посредством HPLC: 87,47% по площади в виде смеси этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-1-ил]ацетата и этил-[3-(4-хлорфенилсульфанил)-4-диацетиламино-2-метил-1Н-индол-1-ил]ацетата; m/z: 417 и 459, оба согласуются с МН+. Раствор NaOH (0,326 г, 0,319 г, скорректированные по чистоте, 8,0 ммоль) в воде (10 мл) добавляли к смеси этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]ацетата и этил-[3-(4-хлорфенилсульфанил)-4-диацетиламино-2-метил-1H-индол-1-ил]ацетата в EtOH, полученной выше. Через 2 ч EtOH удаляли в вакууме при 35°C. Оставшийся водный слой промывали EtOAc (10 мл), разбавляли водой (10 мл), затем подкисляли до pH 4 водной соляной кислотой. Полученный продукт собирали фильтрацией, затем суспендировали в EtOH (10 мл) при 50°C в течение 15 мин. После охлаждения обратно до к.т. твердое вещество собирали фильтрацией, промывали EtOH (4,0 мл), затем сушили в вакууме при 40°C с получением указанного в заголовке соединения в виде твердого вещества; 1,1 г (35,4% за 3 стадии); чистота посредством HPLC: 98,24% по площади.
Способ 3
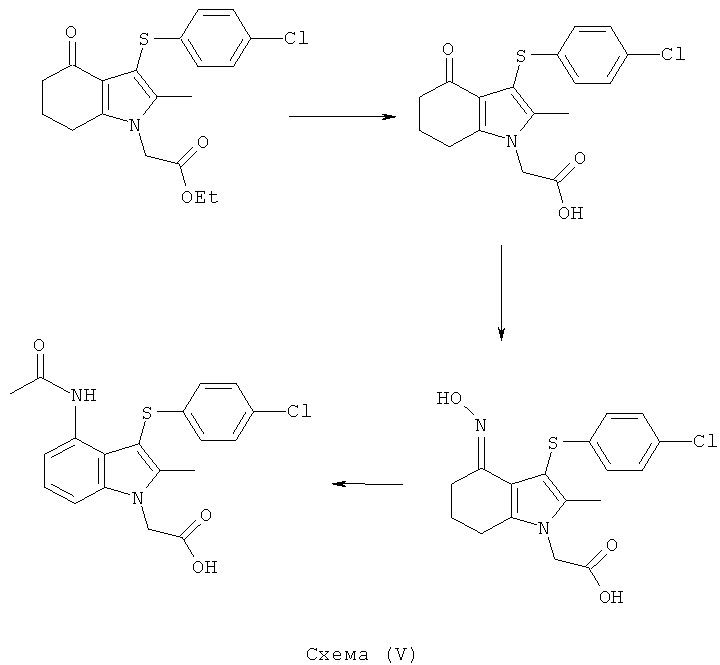
[3-(4-Хлорфенилсульфанил)-2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил]уксусная кислота
Раствор LiOH (1,79 г, 1,77 г, скорректированные с учетом количественного анализа, 42,2 ммоль) добавляли перемешиваемому раствору этил-[3-(4-хлорфенилсульфанил)-2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил]ацетата (7,5 г, 5,33 г, скорректированные с учетом количественного анализа, 14,1 моль) в смеси THF (27 мл) и МеОН (27 мл), Смесь перемешивали в течение 1 ч. Растворители удаляли в вакууме (45 мбар) (45×103 Па) при 45°C, пока объем смеси не составил примерно 20 мл. Добавляли воду (27 мл) и CH2Cl2 (27 мл) и смесь перемешивали в течение 5 мин. Слои оставляли разделяться, к водному слою добавляли концентрированную водную соляную кислоту (5 мл), и полученный твердый продукт собирали фильтрацией и промывали водой (5 мл). Этот неочищенный влажный продукт суспендировали в смеси EtOAc (20 мл), CH2Cl2 (20 мл) и гептана (20 мл) при к.т. Продукт непродолжительно сушили на фильтре с получением указанного в заголовке соединения; 4,64 г; анализ посредством 1Н-ЯМР: 89,3% масс./масс.; выход, скорректированный в соответствии с количественным анализом: 4,14 г (84%); чистота посредством LC 99,1% по площади; m/z: 350 (МН+); 1Н-ЯМР: 1.99-2.05 (2Н, m), 2.16 (3H, s), 2.26-2.31 (2Н, t), 2.73-2.77 (2Н, t), 4.83 (2Н, s), 6.95-6.99 (2Н, d), 7.23-7.27 (2Н, d).
Способ 3A
Альтернативный синтез [3-(4-хлорфенилсульфанил)-2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил]уксусной кислоты
2-(2-Метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил)уксусная кислота
Перемешиваемую смесь 2-(2-оксопропил)циклогексан-1,3-диона (полученную посредством выпаривания досуха этанольного раствора под вакуумом) (20,0 г, 119 ммоль) и глицина (17,9 г, 238 ммоль) в уксусной кислоте (100 мл) нагревали с обратным холодильником в течение 1,5 ч, затем оставляли охлаждаться до к.т. Затем добавляли воду (100 мл) и смесь упаривали до густого масла в вакууме. Ацетон (200 мл) и воду (40 мл) добавляли к остатку и смесь перемешивали в течение 30 мин при к.т., после чего твердое вещество собирали фильтрацией и фильтраты сохраняли. Твердое вещество суспендировали в дополнительном ацетоне (100 мл) и воде (20 мл) при к.т., затем удаляли фильтрацией. Объединенные фильтраты концентрировали в вакууме, затем растворяли в водном гидроксиде натрия (1M, 200 мл), добавляли небольшое количество 10М гидроксида натрия, чтобы довести pH до 14. После промывки этилацетатом (2×100 мл) смесь подкисляли посредством добавления водной соляной кислоты (5М, 60 мл), затем добавляли натрия хлорид (50 г). После перемешивания в течение 4 ч твердый продукт собирали фильтрацией, промывали ацетоном (2×25 мл), затем сушили под вакуумом при 40°C с получением указанного в заголовке соединения в виде оранжево-коричневого твердого вещества; 7,45 г (30%); чистота 99,1% по площади посредством HPLC; m/z: 208 (МН+); 1Н-ЯМР: 13.2 (1Н, brs), 6.02 (1Н, s), 4.67 (2Н, s), 2.65 (2Н, t, J=6.2 Гц), 2.29-2.24 (2Н, m), 2.10 (3H, s), 2.02-1.95 (2Н, m).
[3-(4-Хлорфенилсульфанил)-2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил]уксусная кислота
Сульфурилхлорид (0,2 мл, 2,5 ммоль) медленно добавляли при перемешивании к раствору бис-(4-хлорбензол)дисульфида (0,72 г, 2,5 ммоль) в этилацетате (7,5 мл) в реакционной колбе 1 при к.т. По окончании добавления смесь оставляли перемешиваться в течение еще 60 мин. Содержимое реакционной колбы 1 затем добавляли в течение 5 мин к перемешиваемой суспензии 2-(2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил)уксусной кислоты (0,9 г, 4,3 ммоль) в этилацетате (7,5 мл) в реакционной колбе 2 при к.т. Реакционную колбу 1 промывали этилацетатом (2 мл), который переносили в реакционную колбу 2. Перемешивание продолжали в течение 1 ч, затем реакцию гасили посредством добавления воды (15 мл). Оставляли на ночь, затем к двухфазной смеси при перемешивании добавляли водный гидроксид натрия (1M, 25 мл), затем небольшое количество 10М гидроксида натрия, чтобы довести pH до 14. Слои разделяли, водную фазу промывали этилацетатом (25 мл), затем подкисляли, используя водную соляную кислоту (5М, 7 мл). После перемешивания при к.т. масло, которое сначала разделилось, отвердевало, и его собирали фильтрацией, затем промывали водой (2×10 мл) и сушили под вакуумом при 40°C с получением указанного в заголовке соединения в виде бледно-коричневого твердого вещества; 1,0 г (66%); чистота: 79,5% по площади посредством HPLC; m/z: 350/352 (МН+); 1Н-ЯМР: 13.3 (1Н, brs), 7.25 (2Н, d, J=8,3 Гц), 6.96 (2Н, d, J=8,3 Гц), 4.84 (2Н, s), 2.75 (2Н, t, J=5,9 Гц), 2.28 (2Н, t, J=6,1 Гц), 2.16 (3H, s), 2.03-1.97 (2Н, m).
[3-(4-Хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]уксусная кислота
Смесь [3-(4-хлорфенилсульфанил)-2-метил-4-оксо-4,5,6,7-тетрагидро-1Н-индол-1-ил]уксусной кислоты (4,0 г, 3,57 г, скорректированные с учетом количественного анализа, 10,2 ммоль), гидроксиламина гидрохлорида (1,05 г, 1,04 г, скорректированные с учетом количественного анализа, 15,0 ммоль) и безводного ацетата натрия (1,24 г, 1,23 г, скорректированные с учетом количественного анализа, 15,0 ммоль) в EtOH (40 мл) нагревали с обратным холодильником в течение 6 ч. После охлаждения до 0°C твердый продукт собирали фильтрацией, затем суспендировали в смеси EtOH (14 мл) и воды (14 мл) в течение 15 мин при к.т. Твердое вещество собирали фильтрацией, промывали ацетоном (14 мл), затем сушили на фильтре с получением указанного в заголовке соединения; 3,42 г; анализ посредством ЯМР: 95,3% масс./масс.; выход, скорректированный для анализа: 3,26 г (87%); m/z: 365 (МН+); 1.79-1.83 (2Н, m), 2.13 (3H, s), 2.54-2.59 (4Н, t), 4.73 (2Н, s), 6.91-6.98 (2Н, d), 7.20-7.25 (2Н, d), 10,23 (1Н, brs).
Альтернативный синтез [3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1Н-индол-1-ил]уксусной кислоты
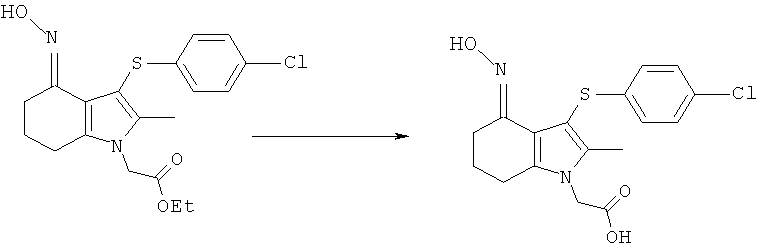
Водный гидроксид натрия (1M, 10 мл, 10 ммоль) и воду (20 мл) добавляли к перемешиваемой суспензии этил-[3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]ацетата (4,0 г, 10,2 ммоль) и этанола (40 мл) и смесь нагревали вплоть до 40°C. Через 1,5 ч при данной температуре реакционную смесь оставляли охлаждаться до к.т. и выдерживали в течение ночи. Суспензию снова нагревали до 33°C и к полученному раствору добавляли уксусную кислоту (1,7 мл, 29,7 ммоль). Смесь оставляли охлаждаться до к.т., твердый продукт собирали фильтрацией, промывали водой (2×20 мл), затем сушили под вакуумом при 30°C с получением указанного в заголовке соединения в виде твердого вещества; 2,94 г (79%); чистота посредством HPLC 98,9% по площади
[4-Ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-1-ил]уксусная кислота
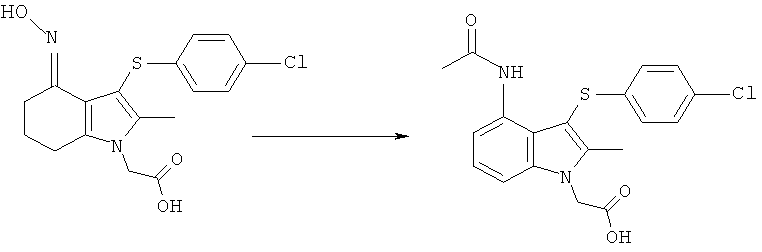
Смесь [3-(4-хлорфенилсульфанил)-4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]уксусной кислоты (1,0 г, 0,9 г, скорректированные с учетом количественного анализа, 2,6 ммоль), Nal (0,20 г, 1,3 ммоль) и уксусного ангидрида (1,0 мл, 10,6 ммоль) в ксилоле (6,7 мл) нагревали при 85°C в течение 5 ч. Ацетилированный оксим, как ожидается, является промежуточным соединением, образовавшимся в данной реакции. После охлаждения до к.т. образец анализировали посредством UPLC-MS, которая указывала на 39% по площади [4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1H-индол-1-ил]уксусной кислоты, со временем удерживания, соответствующим подлинному образцу.
Синтез 2-(4-ацетамидо-2-метил-1H-индол-1-ил)уксусной кислоты
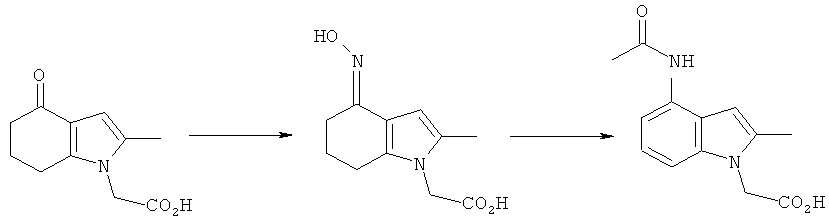
2-[4-(Гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]уксусная кислота
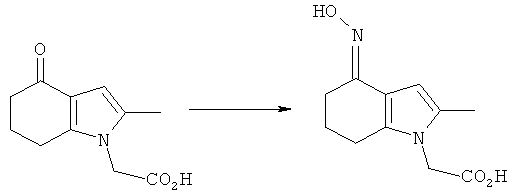
Смесь 2-(2-метил-4-оксо-4,5,6,7-тетрагидро-1H-индол-1-ил)уксусной кислоты (4,0 г, 19 ммоль), гидроксиламина гидрохлорида (2,0 г, 29 ммоль) и безводного ацетата натрия (2,4 г, 29 ммоль) в этаноле (40 мл) нагревали с обратным холодильником в течение 6 ч при перемешивании. После охлаждения до к.т. и выдерживания в течение ночи смесь дополнительно охлаждали до 4°C. Твердый продукт собирали фильтрацией, промывали водой (15 мл), затем этанолом (15 мл), затем сушили под вакуумом при 40°C с получением указанного в заголовке соединения в виде бледно-коричневого твердого вещества; 3,68 г (86%); чистота: 96,7% по площади посредством HPLC; m/z: 223 (МН+); 1Н-ЯМР: 9.82 (1Н, brs), 6.48 (1Н, d, J=0,90 Гц), 4.58 (2Н, s), 2.56-2.51 (2Н, m), 2.29-2.24 (2Н, m), 2.09 (3H, s), 1.86-1.79 (2Н, m).
2-(4-Ацетамидо-2-метил-1H-индол-1-ил)уксусная кислота
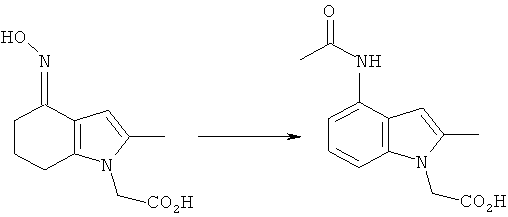
Уксусный ангидрид (1,0 мл, 10,6 ммоль) добавляли к перемешиваемой суспензии 2-[4-(гидроксиимино)-2-метил-4,5,6,7-тетрагидро-1H-индол-1-ил]уксусной кислоты (2,0 г, 9,0 ммоль) в смеси уксусной кислоты (10 мл) и ксилола (10 мл) при к.т. в реакционной колбе 1 (умеренно экзотермическое добавление), затем смесь выдерживали при перемешивании в течение 75 мин. В это время в реакционную колбу 2 загружали уксусную кислоту (5 мл), ксилол (5 мл), уксусный ангидрид (2,6 мл, 27,5 ммоль) и йодид натрия (270 мг, 1,80 ммоль) и смесь нагревали до 90-100°C при перемешивании. Содержимое реакционной колбы 1 переносили в реакционную колбу 2 аликвотами в течение 1,5 ч, поддерживая температуру реакционной смеси 90-100°C. Реакционную колбу 1 промывали смесью ксилола (2 мл) и уксусной кислоты (2 мл), которую переносили в реакционную колбу 2. Затем реакционную смесь поддерживали при 100°C в течение еще 2,5 ч и затем оставляли охлаждаться до к.т. Жидкую фазу декантировали с густого остатка в реакционной колбе и остаток промывали уксусной кислотой (5 мл), затем ксилолом (5 мл). Декантированную жидкость и промывки объединяли и упаривали досуха в вакууме с получением темного коричневого остатка. Анализ посредством HPLC и LC-MS показал пик 29,2% по площади с m/z 247 (МН+).
Способ 4
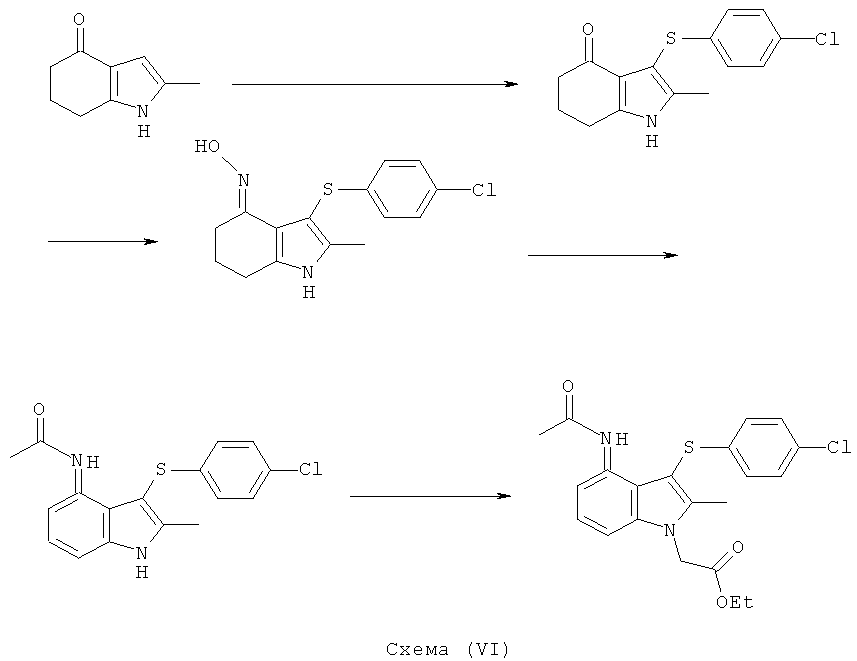
3-(4-Хлорфенилсульфанил)-2-метил-4,5,6,7-тетрагидро-1Н-индол-4-он
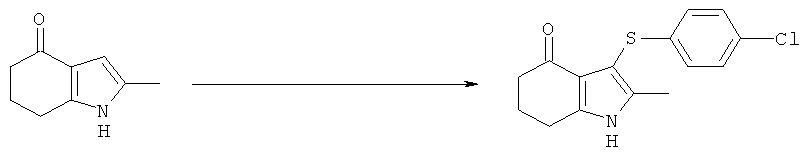
Сульфурилхлорид (1,7 мл, 21 ммоль) медленно добавляли при перемешивании к раствору бис-(4-хлорбензол)дисульфида (6,0 г, 21 ммоль) в этилацетате (45 мл) в реакционной колбе 1, которую охлаждали в бане лед-вода. По окончании добавления смесь оставляли перемешиваться при к.т. в течение 30 мин. Содержимое реакционной колбы 1 затем добавляли в течение 1 ч к перемешиваемой суспензии 2-метил-4,5,6,7-тетрагидро-1H-индол-4-она (5,0 г, 33,5 ммоль) в этилацетате (22,5 мл) в реакционной колбе 2, охлаждали на бане лед-вода, добавляя дополнительно этилацетат в процессе добавления, чтобы поддерживать гетерогенную реакционную смесь подвижной. Реакционную колбу 1 промывали этилацетатом (10 мл), который переносили в реакционную колбу 2. Перемешивание продолжали в течение ночи, в это время густой, гетерогенной смеси позволяли нагреваться до к.т. Твердый продукт собирали фильтрацией, промывали этилацетатом (2×15 мл), затем сушили под вакуумом при 40°C с получением указанного в заголовке соединения в виде бледно-желтого твердого вещества; 8,62 г (88%); чистота: 94,3% по площади посредством HPLC; m/z: 292/294 (МН+); 1Н-ЯМР: 11.77 (1Н, s), 7.24 (2Н, d, J=8,1 Гц), 6.95 (2Н, d, J=8,1 Гц), 2.80-2.74 (2Н, m), 2.29-2.21 (2Н, m), 2.16 (3H, s), 2.05-1.95 (2Н, m).
N-[-3-(4-хлорфенилсульфанил)-2-метил-4,5,6,7-тетрагидро-1Н-индол-4-илиден]гидроксиламин
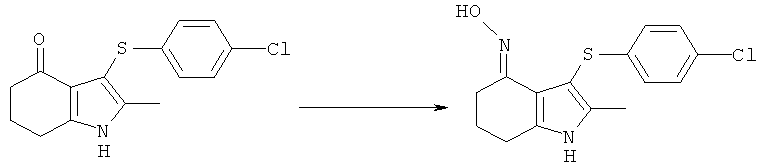
Трибутиламин (6,6 мл, 28 ммоль) добавляли к перемешиваемой суспензии 3-(4-хлорфенилсульфанил)-2-метил-4,5,6,7-тетрагидро-1Н-индол-4-она (8,0 г, 27 ммоль) и гидроксиламина гидрохлорида (1,9 г, 27 ммоль) в этаноле (40 мл). Смесь нагревали до 65°C и поддерживали при данной температуре в течение 4 ч, затем оставляли охлаждаться до к.т. и оставляли в течение ночи. Дополнительно добавляли гидроксиламина гидрохлорид (0,15 г, 2,2 ммоль) и трибутиламин (1,0 мл, 4,2 ммоль) и смесь снова нагревали до 65°C в течение еще 2 ч. После охлаждения до к.т. Твердый продукт собирали фильтрацией, промывали этанолом (2×10 мл), затем сушили в вакуумной печи при 40°C с получением указанного в заголовке соединения в виде не совсем белого твердого вещества; 5,71 г (68%); чистота: 96,3% по площади посредством HPLC; m/z: 307/309 (МН+); 1Н-ЯМР: 11.26 (1Н, s), 10,20 (1Н, s), 7.23 (2Н, m), 6.92 (2Н, m), 2.62-2.58 (2Н, m), 2.56-2.52 (2Н, m), 2.12 (3H, s), 2.83-1.76 (2Н, m).
N-[3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-4-ил]ацетамид
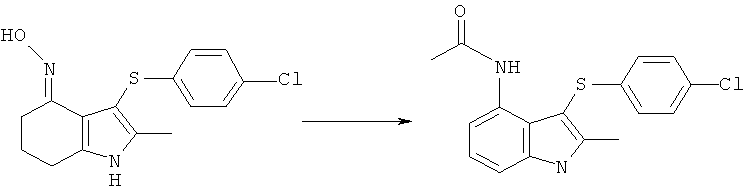
Уксусный ангидрид (1,1 мл, 11,6 ммоль) добавляли к перемешиваемой суспензии N-[-3-(4-хлорфенилсульфанил)-2-метил-4,5,6,7-тетрагидро-1Н-индол-4-илиден]гидроксиламина (3,0 г, 9,8 ммоль) в смеси уксусной кислоты (15 мл) и ксилола (15 мл) при к.т. в реакционной колбе 1 (умеренно экзотермическое добавление), затем смесь выдерживали при перемешивании в течение 45 мин. В это время в реакционную колбу 2 загружали уксусную кислоту (7,5 мл), ксилол (7,5 мл), уксусный ангидрид (1,1 мл, 11,6 ммоль) и йодид натрия (70 мг, 0,47 ммоль) и смесь нагревали до 110°C при перемешивании. Содержимое реакционной колбы 1 переносили в реакционную колбу 2 аликвотами в течение 2 ч, поддерживая температуру реакционной смеси при 110°C. Реакционную колбу 1 промывали смесью ксилола (2 мл) и уксусной кислоты (2 мл), которую переносили в реакционную колбу 2. Затем реакционную смесь поддерживали при 110°C в течение еще 3 ч, затем оставляли охлаждаться до к.т. и выдерживали в течение ночи. Реакционную смесь упаривали досуха в вакууме с получением темно-коричневого твердого остатка. Его очищали посредством хроматографии на сидикагеле, элюируя смесью дихлорметан/этилацетат, с получением указанного в заголовке соединения в виде не совсем белого твердого вещества; 2,74 г (85%); чистота 95,6% по площади посредством HPLC; m/z: 331/333 (МН+); 1Н-ЯМР; 11.84 (1Н, s), 9.45 (1Н, s), 7.45 (1Н, d, J=7,7 Гц), 7.29 (2Н, d, J=8,3 Гц), 7.17 (1Н, d, J=8,0 Гц), 7.04 (1Н, t, J=7,9 Гц), 6.98 (2Н, d, J=8,2 Гц), 2.40 (3H, s), 1.84 (3H, s).
Этил-[4-ацетиламино-3-(4-хлорфенилсульфанил)-2-метил-1Н-индол-1-ил]ацетат
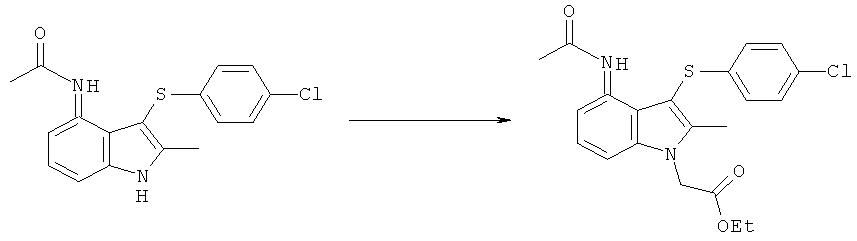
Смесь N-[3-(4-хлорфенилсульфанил)-2-метил-1H-индол-4-ил]ацетамида (1,0 г, 3,0 ммоль), безводного карбоната калия (0,63 г, 4,6 ммоль), ацетона (10 мл) и этил-бромацетата (0,50 мл, 4,5 ммоль) нагревали с обратным холодильником в течение 1,5 ч, затем охлаждали до к.т. и оставляли при перемешивании при данной температуре в течение ночи. После нагревания с обратным холодильником в течение еще 1 ч смесь охлаждали до к.т. Добавляли воду (2×10 мл), твердый продукт собирали фильтрацией, промывали водой (10 мл), затем сушили в вакуумной печи при 40°C с получением указанного в заголовке соединения в виде не совсем белого твердого вещества; 1,16 г (92%); чистота 95,4% по площади посредством HPLC; m/z: 417/419 (МН+); 1Н-ЯМР: 9.52 (1Н, s), 7.45 (1Н, d, J=7,9 Гц), 7.33 (1Н, d, J=8,2 Гц), 7.29 (2Н, d, J=8,6 Гц), 7.11 (1Н, t, J=8,0 Гц), 6.97 (1Н, d, J=8,6 Гц), 5.24 (2Н, s), 4.18 (2Н, q, J=7,1 Гц), 2.39 (3H, s), 1.86 (3H, s), 1.22 (3H, t, J=7,1 Гц).
![ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/4a/ac/ab/d14482f2600246344e3748ac149c9720.png)
![ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/4a/ac/ab/cc98ffaaf582b906ff333cc5e12b994a.png)
![ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/4a/ac/ab/8e449014f64f1afa034a8b231363e5bd.png)
![ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/4a/ac/ab/49cebac83ef508a7fc394712301d65b6.png)
![ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/4a/ac/ab/2d1e891d02a63e4ce09213745915b08b.png)
![ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/4a/ac/ab/119bde9e0e129890f0f1c2afc28a0f86.png)
![ПРОМЕЖУТОЧНЫЕ СОЕДИНЕНИЯ И СПОСОБЫ ПОЛУЧЕНИЯ 4-(АЦЕТИЛАМИНО)-3-[(4-ХЛОРФЕНИЛ)ТИО]-2-МЕТИЛ-1Н-ИНДОЛ-1-УКСУСНОЙ КИСЛОТЫ](https://fips.edrid.ru/images/rid/4a/ac/ab/7eb542b6b02bc60083b01b14e3fd6d72.png)
Новые соединения 707 и их применение
Новые соединения 951: бифенилоксипропановая кислота в качестве модулятора crth2-рецептора и промежуточные соединения
Новые 2-гетероарил-замещенные бензотиофены и бензофураны 709
Фармацевтическая композиция с модифицированным высвобождением и ее применение
Композиции, пригодные для перорального введения, содержащие производное триазоло[4,5-d]пиримидина
Способ получения (3ar,4s,6r,6as)-6-амино-2,2-диметилтетрагидро-3aн-циклопента[d][1,3]диоксол-4-ол-дибензоил-l-тартрата и продукты указанного способа
Производные пиразинона и их применение для лечения легочных заболеваний
Химические соединения 637: пиридопиримидиндионы в качестве ингибиторов pde4
Упаковка для раздаточного устройства, содержащая пакет
Устройство и способ дезагрегации порошкообразного средства 854
Новые соединения 707 и их применение
Аминные производные и их применение в бета-2-адренорецептор-опосредованных заболеваниях
Новые соединения 951: бифенилоксипропановая кислота в качестве модулятора crth2-рецептора и промежуточные соединения
Новые 2-гетероарил-замещенные бензотиофены и бензофураны 709
Фармацевтическая композиция с модифицированным высвобождением и ее применение
Имидазохинолины с иммуномодулирующими свойствами
Композиции, пригодные для перорального введения, содержащие производное триазоло[4,5-d]пиримидина
Способ получения (3ar,4s,6r,6as)-6-амино-2,2-диметилтетрагидро-3aн-циклопента[d][1,3]диоксол-4-ол-дибензоил-l-тартрата и продукты указанного способа
Производные пиразинона и их применение для лечения легочных заболеваний
Химические соединения 637: пиридопиримидиндионы в качестве ингибиторов pde4

