Результат интеллектуальной деятельности: АГОНИСТЫ ОКСИТОЦИНОВЫХ РЕЦЕПТОРОВ
Вид РИД
Изобретение
Область изобретения
Настоящее изобретение относится к соединениям - агонистам окситоциновых рецепторов, к фармацевтическим композициям, содержащим их, к применению таких соединений для изготовления лекарственного средства для лечения, среди прочего, боли в животе, синдрома раздраженного кишечника (IBS), аутизма, эректильной дисфункции, женской сексуальной дисфункции, стимуляции и поддержания родов, стимуляции и поддержания лактации, послеродового кровотечения, посттравматического стрессового расстройства (PTSD), боли, тревоги и других состояний, а также к способам лечения таких состояний, в которых вводят такие соединения.
Предшествующий уровень техники
Пептидные агонисты окситоциновых рецепторов включают природный гормон окситоцин и карбетоцин.

Окситоцин представляет собой сильнодействующий агент, стимулирующий сокращение матки, клинически используемый, чтобы вызывать роды и, как было показано, чтобы улучшить начало и сохранение лактации (Gimpl et al., Physiol. Rev., 81., (2001), 629-683; Ruis et al., BMJ, 283. (1981), 340-342). Карбетоцин (1-деамино-1-карба-2-тирозин(O-метил)-окситоцин) также представляет собой сильнодействующий агент, стимулирующий сокращение матки, клинически используемый для контроля за атонией матки и чрезмерным кровотечением. Дополнительное исследование показывает, что агонисты окситоцина полезны для лечения воспаления и боли, включая боли в животе и спине (Yang, Spine, 19, (1994), 867-71); сексуальной дисфункции как мужской (Lidberg et al., Pharrnakopsychiat., 10, (1977), 21-25), так и женской (Anderson-Hunt, et al., BMJ, 309 (1994), 929); синдрома раздраженного кишечника (IBS; Louvel et al., Gut, 39. (1996), 741-47), запора и желудочно-кишечной непроходимости (Ohlsson et al., Neurogastroenterol. Motil., 17, (2005), 697-704); аутизма (Hollander et al., Neuropsychopharm., 28, (2008), 193-98), стресса (включая посттравматическое стрессовое расстройство, PTSD; Pitman et al., Psychiatry Research, 48. 107-117), тревоги (включая тревожное расстройство) и депрессии (Kirsch et al., J. Neurosci., 25(49), 11489-93; Waldherr et al., PNAS, 104, (2007), 16681-84), потери крови при хирургической операции, контроля послеродового кровотечения (Fujimoto et al., Acta Obstet. Gynecol., 85, (2006), 1310-14), стимуляции и поддержания родов (Flamm et al., Obstet. Gynecol., 70, (1987) 709-12), заживления ран и инфекции; мастита и рождения плаценты; и остеопороза. Кроме того, агонисты окситоцина могут быть полезны для диагностики как рака, так и плацентарной недостаточности.
Продолжаются попытки идентифицировать и разработать соединения с достаточной активностью на окситоциновом рецепторе человека. Были синтезированы аналоги окситоцина. Такие аналоги описаны в Grzonka et al., J. Med. Chem., 26, (1983), 555-559 и J. Med. Chem., 26, (1983), 1786-1787 и в Engstrom et al., E. J. Pharmacol., 355, (1998), 203-210. Кроме того, аналоги окситоцина с антагонистической активностью к окситоциновым рецепторам были описаны в Fragiadaki et al., E. J. Med. Chem., (2007), 799-806.
Настоящее изобретение может предложить сильнодействующие и обладающие большой продолжительностью действия соединения, обеспечивая реальные альтернативы и/или улучшения в лечении, например, боли в животе, синдрома раздраженного кишечника (IBS), аутизма, эректильной дисфункции, женской сексуальной дисфункции, стимуляции и поддержания родов, стимуляции и поддержания лактации, послеродового кровотечения, посттравматического стрессового расстройства (PTSD), боли, тревоги, потери крови при хирургической операции, диагностики раковых заболеваний, запора, депрессии, бессонницы, мастита, OВ (акушерской) диагностики (в отношении плацентарной недостаточности), остеопороза, рождения плаценты и заживления ран/воспаления.
Подробное описание изобретения
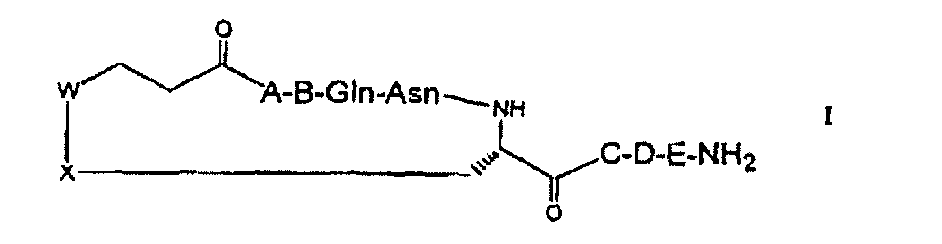
Настоящее изобретение относится к соединениям, представленным структурной Формулой (I):

или их фармацевтически приемлемым солям,
где:
W и Х независимо выбраны из Chb и S, но не могут оба представлять собой СН2;
А представляет собой аминокислоту, выбранную из: аланина, замещенного по боковой цепи 5- или 6-членным гетероароматическим кольцом; тирозина; и фенилаланина, замещенного по фенильному кольцу, например по положению 4 фенильного кольца, галогеном, С1-4алкокси, С1-4алкилгидрокси, С1-4алкилом или амино;
В представляет собой аминокислоту, выбранную из: изолейцина; и глицина, замещенного по α-углероду С4-6циклоалкилом;
С представляет собой аминокислоту, выбранную из: пролина, где пролин возможно замещен, например по положению 4, на боковой цепи гидроксилом, С1-4алкокси, галогеном или азидом; и пролина, где боковая цепь пролина возможно прервана гетероатомом и где возможно прерванная боковая цепь возможно замещена С1-4алкилом;
D представляет собой аминокислоту, выбранную из: лейцина; гомолейцина; изолейцина; и глицина, замещенного по α-углероду С4-6циклоалкилом; и
Е представляет собой аминокислоту, выбранную из: глицина и азаглицина,
при условии что, если С представляет собой 4-гидроксипролин, то А должен представлять собой либо фенилаланин, замещенный по фенильному кольцу галогеном, либо С1-4алкилгидрокси, и если С представляет собой 4-гидроксипролин и А представляет собой фенилаланин, замещенный по фенильному кольцу галогеном, то или В или D должен представлять собой глицин, замещенный по α-углероду С4-6циклоалкилом, или D должен представлять собой изолейцин,
при дополнительном условии что, если А представляет собой фенилаланин, замещенный по фенильному кольцу С1-4алкилом или галогеном, то С должен представлять собой пролин или пролин, замещенный по боковой цепи галогеном,
при дополнительном условии что, если А представляет собой фенилаланин, замещенный по фенильному кольцу галогеном, то или В, или D должен представлять собой глицин, замещенный по α-углероду С4-6циклоалкилом, или О должен представлять собой изолейцин.
В данном описании изобретения также предлагаются соединения, представленные Формулой I выше, где А представляет собой 4-галогенофенилаланин, например Сра (4-хлорфенилаланин); 4-бромфенилаланин, или где А представляет собой аланин, замещенный по боковой цепи 5- или 6-членным гетероароматическим кольцом, например Аlа(2-Fur) (2-фурилаланин), Ala(3-Fur) (3-фурилаланин); 2-Thi (2-тиенилаланин); 3-Thi (3-тиенилаланин); 2- или 3-пирролилаланин; 2-, 3- или 4-пиридилаланин; 2-, 4- или 5-имидазолилаланин; 2-, 4- или 5-тиазолилаланин; 2- или 5-тиадиазолил; 5-тетразолил; и тому подобное. Также в данном описании изобретения предлагаются новые соединения, представленные Формулой I выше, где А представляет собой тирозин, или где А представляет собой фенилаланин, замещенный по положению 4 фенильного кольца С1-4алкоксигруппами или аминогруппой, например Tyr(Ме) (4-метоксифенилаланин); 4-этоксифенилаланин; Aph (4-аминофенилаланин); 4-N,N-диметиламинофенилаланин; и тому подобное. Также в данном описании изобретения предлагаются новые соединения, представленные Формулой 1 выше, где А представляет собой фенилаланин, замещенный по положению 4 фенильного кольца С1-4алкилгидроксилом, С1-4алкилом или галогено, например Phe(4-Et) (4-этилфенилаланин); 4-метилфенилаланин; Phe(4-СН2OН) (4-гидроксиметилфенилаланин); 4-гидроксиэтилфенилаланин; Phe(Br) (4-бромфенилаланин); 4-хлорфенилаланин; 4-фторфенилаланин; и тому подобное.
В данном описании изобретения также предлагаются соединения, представленные Формулой I выше, где В представляет собой изолейцин, или глицин, замещенный С4-6циклоалкилом, такой как Gly(cPe) (циклопентилглицин), Gly(cBu) (циклобутилглицин), циклогексилглицин; и тому подобное.
В данном описании изобретения также предлагаются соединения, представленные Формулой I выше, где С представляет собой пролин, возможно замещенный по положению 4 пролинового кольца группами гидрокси, С1-4алкокси, галогено или азидо, например Hyp (4-гидроксипролин); Нyp(Ме) (4-метоксипролин); Pro(F) (4-фторпролин); Pro(N3) (4-азидопролин), и подобные. Также в данном описании изобретения предлагаются новые соединения, представленные Формулой I выше, где В представляет собой пролин, прерванный в пролиновом кольце гетероатомом и возможно замещенный по пролиновому кольцу С1-4алкилом, например Thz (4-тиапролин) или Dmt (5,5-диметилтиапролин), и подобные.
В данном описании изобретения также предлагаются соединения, представленные Формулой I выше, где D представляет собой лейцин, Hol (гомолейцин), изолейцин и глицин, замещенный С4-6циклоалкилом, такой как Gly(cPe) (циклопентилглицин) Gly(cBu) (циклобутилглицин), циклогексилглицин; и тому подобное.
В данном описании изобретения также предлагаются соединения, представленные Формулой I выше, где Е представляет собой глицин или AzGly (азаглицин).
Также в данном описании изобретения предлагаются соединения, которые описаны выше общей структурной Формулой I, и их конкретные структурные варианты для применения в качестве фармацевтических средств. Также предлагаются фармацевтические композиции, содержащие терапевтически эффективное количество по меньшей мере одного соединений, как оно определено выше, в качестве активного ингредиента вместе по меньшей мере с одним фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Для целей настоящего изобретения используют следующую терминологию.
С1-4алкил означает заместитель, имеющий от одного до четырех атомов углерода, включая изо-, втор- и трет-конфигурации, в то время как это выражение не относится к участку связывания рассматриваемой алкильной цепи или ее структурных изомеров.
С4-6циклоалкил означает карбоциклическую кольцевую систему, содержащую от четырех до шести атомов углерода. Кольцевая система может содержать ненасыщенные связи между атомами углерода, включать, например, циклогексенил, циклопентенил, циклогексадиенил, и тому подобное.
Пятичленная гетероароматическая кольцевая система представляет собой моноциклическую ароматическую кольцевую систему, имеющую пять кольцевых атомов, где 1, 2, 3 или 4 кольцевых атома независимо выбраны из N, О и S. Такие кольцевые системы могут представлять собой, например, тиенил, фурил, имидазолил, пирролил, пиразолил, тиазолил, тиадиазолил, тетразолил, и тому подобное.
Шестичленная гетероароматическая кольцевая система представляет собой моноциклическую ароматическую кольцевую систему, имеющую шесть кольцевых атомов, где 1, 2, 3 или 4 кольцевых атома независимо выбраны из N, О и S. Такие кольцевые системы могут представлять собой, например, пиридил, пиразинил, пиримидинил, пиридазинил, и тому подобное.
Группировки-заместители могут представлять собой, например, атомы галогена (фтор, хлор, бром) и алкил, циклоалкил, гидрокси (-ОН), алкокси (-O-алкил), алкилтио (-S-алкил), алкилгидрокси (-алкил-ОН), азид (N3), амино (-NR1R2, где R1 и R2 могут независимо представлять собой водород или С1-4алкил) или 5- или 6-членные гетероароматические группы.
Примеры фармацевтически приемлемых солей содержат соли присоединения кислоты, например соль, образованную посредством взаимодействия с галогеноводородными кислотами, такими как соляная кислота, и неорганическими кислотами, такими как серная кислота, фосфорная кислота и азотная кислота, а также с алифатическими, алициклическими, ароматическими или гетероциклическими? сульфоновыми или карбоновыми кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, яблочная кислота, винная кислота, лимонная кислота, бензойная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, пара-гидроксибензойная кислота, эмбоновая кислота, метансульфоновая кислота, этансульфоновая кислота, гидроксиэтансульфоновая кислота, галогенобензолсульфоновая кислота, трифторуксусная кислота, трифторметансульфоновая кислота, толуолсульфоновая кислота и нафталинсульфоновая кислота.
В частности, описание относится, но без ограничения ими, к конкретным иллюстративным соединениям, таким как соединения, описанные ниже,
или их фармацевтически приемлемые соли.
|








|






|








|


Кроме того, настоящее изобретение относится к соединению, как указано выше, для применения в качестве фармацевтического средства.
Таким образом, настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, как указано выше, в качестве активного ингредиента вместе с фармацевтически приемлемым вспомогательным веществом, разбавителем или носителем.
Фармацевтическая композиция может быть адаптирована для перорального, внутривенного, внутримышечного, местного, внутрибрюшинного, назального, буккального, внутриглазного, внутриушного, сублингвального или подкожного введения или для введения через дыхательные пути, например в форме аэрозоля или взвешенного в воздухе мелкодисперсного порошка. Таким образом, композиция может, например, находиться в форме таблеток, капсул, порошков, микрочастиц, гранул, сиропов, суспензий, растворов, чрескожных пластырей или суппозиториев.
Следует отметить, что композиция по настоящему изобретению возможно может включать два или более из указанных выше соединений.
Фармацевтическая композиция по настоящему изобретению возможно может содержать, например, по меньшей мере одну дополнительную добавку, выбранную из разрыхлителя, связующего вещества, смазывающего вещества, ароматизатора, консерванта, красителя и любую их смесь. Примеры таких и других добавок находятся в Handbook of Pharmaceuticals Excipients; Ed. A.M.Kibbe, 3rd Ed., American Pharmaceutical Association, USA and Pharmaceutical Press UK, 2000.
Фармацевтическая композиция по настоящему изобретению может быть адаптирована, например, для подкожного, внутривенного или внутримышечного введения. Она может содержать стерильный водный препарат соединений по изобретению, предпочтительно изотонический с кровью реципиента. Такую водную композицию можно приготовить в виде препарата согласно известным способам, используя подходящие диспергирующие или увлажняющие агенты и суспендирующие агенты. Внутривенная композиция DURATOCIN® (карбетоцин) является примером подходящей фармацевтической композиции, также применимой для соединений по изобретению, описанных в данном описании изобретения. Вода, раствор Рингера и изотонический раствор хлорида натрия являются примерами приемлемых разбавителей. Композиция также может включать такие эксципиенты, как фосфат натрия, лимонная кислота, хлорид натрия, глицерин, раствор сорбитола, метилпарабен, пропилпарабен и хлорбутанол.
Кроме того, настоящее изобретения относится к применению соединения, как оно описано выше, для лечения или для изготовления лекарственного средства для лечения одного или более медицинских состояний, таких как боли в животе, синдром раздраженного кишечника (IBS), аутизм, эректильная дисфункция, женская сексуальная дисфункция, стимуляция и поддержание родов, стимуляция и поддержание лактации, послеродовое кровотечение, посттравматическое стрессовое расстройство (PTSD), боль, тревога, потеря крови при хирургической операции, диагностика раковых заболеваний, запор, депрессия, бессонница, мастит, OВ (акушерская) диагностика (в отношении плацентарной недостаточности), остеопороз, рождение плаценты и заживления ран/воспаления. В данном описании изобретения термин тревога включает тревожное расстройство. Тревожное расстройство включает подпоказания - генерализованное тревожное расстройство, паническое расстройство, агорафобия, фобии, социальное тревожное расстройство, обсессивно-компульсивное расстройство, посттравматическое стрессовое расстройство и сепарационную тревогу.
В другом воплощении изобретение относится к способу лечения боли в животе, синдрома раздраженного кишечника (IBS), аутизма, эректильной дисфункции, женской сексуальной дисфункции, стимуляции и поддержания родов, стимуляции и поддержания лактации, послеродового кровотечения, посттравматического стрессового расстройства (PTSD), боли, тревоги, потери крови при хирургической операции, диагностики раковых заболеваний, запора, депрессии, бессонницы, мастита, OВ диагностики (в отношении плацентарной недостаточности), остеопороза, рождения плаценты и заживления ран/воспаления.
Обычная дозировка соединений по настоящему изобретению варьируется в широком диапазоне и зависит от различных факторов, таких как индивидуальные потребности каждого пациента и путь введения. Врач обычной в данной области техники квалификации сможет оптимизировать дозировку в имеющейся ситуации.
Например, если композиция по изобретению предназначена для послеродового кровотечения (например, для внутривенного или внутримышечного введения), обычная доза может находиться в интервале от 0,5 до 200 мкг/кг массы тела. Специалист или врач может рассмотреть релевантные варианты данного диапазона дозировок и практические варианты реализации для приспособления к имеющейся ситуации.
В еще одном примере композицию по изобретению можно вводить в виде интраназальной лекарственной формы, например, для лечения синдрома раздраженного кишечника, стимулирования и поддержания лактации или сексуальной дисфункции. В данном примере ее можно вводить в разделенных дозах, например, в 1, 2 или 3 субдозах (например, впрыскиваниях), например, доставляемых в одну или обе ноздри. Примерная дозировка для введения интраназальным путем может составлять 0,05-15,0 мкг/кг массы тела.
В еще одном примере композиция по изобретению может быть предназначена для подкожного (пк), интраназального или буккального введения, например для лечения тревожного расстройства или депрессии. Примерная дозировка для подкожного (пк) или буккального введения составляет 0,5-1000 мкг/кг массы тела. Дозировка может быть предназначена, например, для введения столько раз в сутки, сколько потребуется, например один раз или два раза в сутки.
Используемые сокращения:
|
Если не указано иное, использовали L-аминокислоты и придерживались обычной терминологии для аминокислот.
Экспериментальная часть (синтез)
Аминокислотные производные и смолы приобретали у коммерческих поставщиков (Bachem, BioQuadrant, Chemlmpex, Novabiochem, Peptides International, RSP Amino Acids and Synthetech). Fmoc-Cys (трет-бутоксикарбонилпропил)-ОН и Fmoc-Hcy (трет-бутоксикарбонилэтил)-ОН синтезировали согласно литературе [Prochazka et al., Collect. Czech. Chem. Commun., 57, (1992), 1335 и Wisniewski et al. в WO 03/072597]. Другие химические вещества и растворители получали от Sigma-Aldrich, Fluka and Acros Organics.
Соединения в данном изобретении синтезировали стандартными способами пептидной химии на твердой фазе, используя методику Fmoc. Все сочетания Fmoc-защищенных аминокислот были опосредованы DIC/HOBt/DMF. Удаление Fmoc-группы выполняли при помощи 20% пиперидина в DMF.
Если не предусмотрено иное, все реакции осуществляли при комнатной температуре. В дополнение к ссылкам, указанным выше, следующая стандартная справочная литература предлагает дополнительные указания в отношении общей постановки эксперимента, а также в отношении возможности использования необходимого исходного вещества и реагентов:
Kates and Albericio, Eds., "Solid Phase Synthesis: A Practical Guide," Marcel Dekker, New York, Basel, 2000;
Stewart and Young, "Solid Phase Synthesis," Pierce Chemical Company, 1984;
Bisello, et at., J. Вiоl. Chem., (1998), 273, 22498-22505; и
Merrifield, J. Am. Chem. Soc. (1963), 85, 2149-2154.
Чистоту синтезированного пептида можно определять посредством аналитической обращенно-фазовой ВЭЖХ. Структурную целостность пептидов можно подтверждать, используя аминокислотный анализ и масс-спектрометрию с электрораспылением.
Все аминокислотные сочетания следовали Fmoc-методике, если не указано иное.
Аминокислотное производное, введенное в положении 6, представляло собой одно из следующих: Fmoc-Cys(Trt)-OH; Fmoc-Hcy (трет-бутоксикарбонилэтил)-ОН или Fmoc-Cys (трет-бутоксикарбонилпропил)-ОН. Пептидные аналоги, где положение 6 представляло собой Fmoc-Cys (Trt)-OH, требовали сочетания Mpa(Trt)-OH с N-концом связанного со смолой остатка октапептида.
Пептиды, синтезированные с использованием подложки из амидной смолы Ринка, отщепляли от смолы вместе с любыми кислотолабильными защитными группами, такими как Вое, тритил и трет-бутил, раствором TFA/TIS/H2O 95/2,5/2,5 (об./об./об.). Указанные пептиды циклизовали после отщепления пептида от смолы.
Циклизации линейного нонапептида посредством образования дисульфида (кольцо) достигали путем окисления йодом линейных пептидов, растворенных в 10% TFA (водн.). Циклизации линейного нонапептида посредством образования амидной связи достигали при помощи HBTU/DI PEA/DM F или PyBOP/DIPEA/DMF с сильным разведением.
Пептиды очищали при помощи препаративной ВЭЖХ в триэтиламмониевых фосфатных буферах (водн.) и обессоливали буферной системой уксусная кислота (водн.)/ацетонитрил. Фракции с чистотой, превышающей 97%, объединяли и лиофилизировали.
Во всех синтезах аналитическую ВЭЖХ выполняли на жидкостном хроматографе Waters 600, используя колонку Vydac C18, 5 мкм, 4,6×250 мм при скорости потока 2 мл/мин. Препаративную ВЭЖХ выполняли на жидкостном хроматографе Waters 2000, используя картридж PrePak 47×300 мм при скорости потока 100 мл/мин. Окончательный анализ соединения выполняли на жидкостном хроматографе 1100 Agilent, используя колонку Vydac C18, 5 мкм, 2,1×250 мм при скорости потока 0,3 мл/мин. Масс-спектры регистрировали на спектрометре Finnigan MAT.
Следующие подробные примеры представлены, чтобы дополнительно проиллюстрировать синтез.
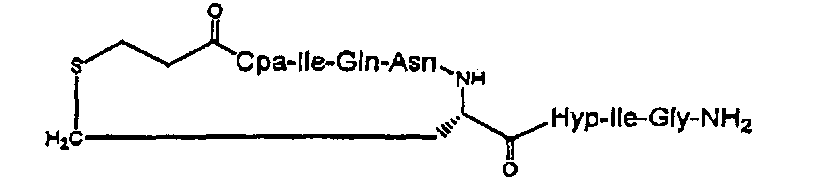
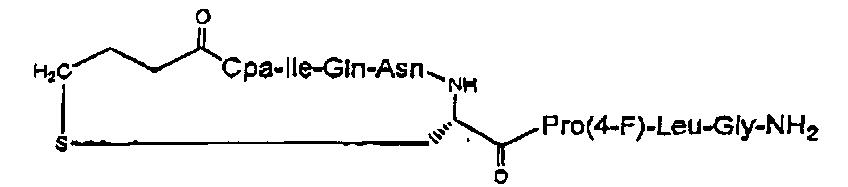
Получение Соединения 7: карба-1-[Cpa2, Pro(4-F)7]dOT.
Используемые аминокислотные производные представляли собой Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-Pro(4-F)-OH (BioQuadrant), Fmoc-Cys(трет-бутоксикарбонилпропил)-ОН, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH и Вос-Сра-ОН (Synthetech). Производные, для которых изготовитель не указан, получали из Peptide International. Fmoc-Cys(трет-бутоксикарбонил-пропил)-OH синтезировали, как указано выше.
Смолу с полностью защищенным пептидом синтезировали вручную, начиная с 0,5 г (0,125 ммоль) амидной смолы Ринка Tentagel (Peptide International). Выполняли опосредованные DIC/HOBt/DMF одинарные связывания с 3-кратным избытком аминокислотных производных. Fmoc-группы удаляли 20%-ным пиперидином в DMF. После завершения твердофазного синтеза смолу обрабатывали раствором (30 мл) TFA/TIS/H2O 96/2,5/1,5 (об./об./об.) в течение 1,5 ч и отфильтровывали. Фильтрат концентрировали в вакууме и неочищенный линейный пептид осаждали диэтиловым эфиром. Осадок растворяли в DMF (150 мл) и DIPEA (0,174 мл) и к интенсивно перемешиваемому раствору добавляли HBTU (50 мг). За взаимодействием наблюдали с помощью аналитической ВЭЖХ. Реакционный раствор концентрировали в вакууме и остаток растворяли в АсОН/СН3CN/Н2О. Смесь загружали на колонку для ВЭЖХ и очищали, используя триэтиламмоний-фосфатный буфер с рН 5,2. Соединение элюировали градиентом ацетонитрила. Фракции с чистотой, превышающей 97%, объединяли, разбавляли водой (2 объема) и загружали в колонку, предварительно уравновешенную 2% АсОН (водн.). Целевое соединение элюировали быстрым (3%/мин) градиентом СН3CN. Фракции, содержащие целевой продукт, объединяли и лиофилизировали. Получали 57,9 мг (выход ~40%, на основе загрузки исходной смолы и предполагая содержание пептида 85%) белого аморфного порошка. ВЭЖХ: Rt=12,4 мин, градиент: 5% В в течение 0,5 мин, 5→40% В в течение 0,5 мин, 40→60% В в течение 20 мин и 100% В в течение 5 мин, t=40°С, растворитель А 0,01% TFA (водн.), растворитель В 70% CH3CN, 0,01% TFA (водн.); чистота: 99,6%; MS (M+H+): ожидаемая 1010,4, наблюдаемая 1010,5.
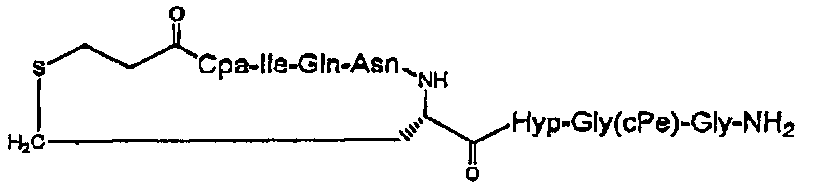
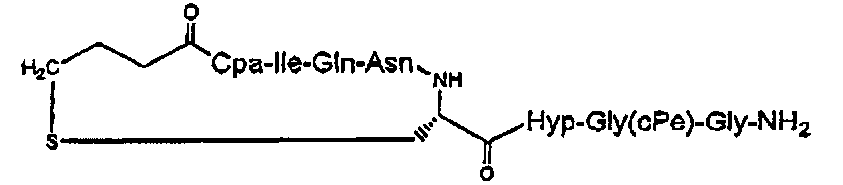
Получение Соединения 12: карба-6-[Сра2, Hyp7, Gly(cPe)8]dOT
Используемые аминокислотные производные представляли собой Fmoc-Gly-OH, Fmoc-Gly(cPe)-OH, Fmoc-Hyp (tBu)-OH (Novabiochem), Fmoc-Hcy (трет-бутоксикарбонилэтил)-ОН, Fmoc-Asn (Trt)-OH, Fmoc-Gln (Trt)-OH, Fmoc-Ile-OH и Boc-Cpa-OH (Synthetech). Производные, для которых изготовитель не указан, получали от Peptide International. Fmoc-Hcy (трет-бутоксикарбонил-этил)-ОН синтезировали, как указано выше.
Смолу с полностью защищенным пептидом синтезировали вручную, начиная с 0,2 г смолы Ринка Tentagel (0,05 ммоль, Peptide International). Выполняли опосредованные DIC/HOBt/DMF одинарные связывания с 3-кратным избытком аминокислотных производных. Fmoc-группы удаляли 20% пиперидином в DMF. После окончания твердофазного синтеза смолу обрабатывали раствором TFA/TIS/H2O 96/2,5/1,5 (об./об./об.) (20 мл) в течение 1,5 ч и отфильтровывали. Фильтрат концентрировали в вакууме, и неочищенный линейный пептид осаждали диэтиловым эфиром. Осадок растворяли в DMF (100 мл) и DIPEA (0,07 мл) и к интенсивно перемешиваемому раствору добавляли HBTU (20 мг). За реакцией наблюдали с помощью аналитической ВЭЖХ. Раствор реакционной смеси концентрировали в вакууме и остаток растворяли в АсОН/СН3CN/H2О. Смесь загружали на колонку для ВЭЖХ и очищали, используя триэтиламмоний-фосфатный буфер с рН 5,2. Соединение элюировали градиентом ацетонитрила. Фракции с чистотой, превышающей 97%, объединяли, разбавляли водой (2 объема) и загружали на колонку, предварительно уравновешенную 2% АсОН (водн.). Целевое соединение элюировали быстрым (3%/мин) градиентом. Фракции, содержащие целевой продукт, объединяли и лиофилизировали.
Получали 40,0 мг (выход ~66%, на основе загрузки исходной смолы и предполагая содержание пептида 85%) белого аморфного порошка. ВЭЖХ: Rt=10,8 мин, градиент: 5% В в течение 0,5 мин, 5-40% В в течение 0,5 мин, 40→60% В в течение 20 мин и 100% В в течение 5 мин, t=40°C, растворитель А 0,01% TFA (водн.), растворитель В 70% CH3CN, 0,01% TFA (водн.); чистота: 100,0%; MS (M+H+): ожидаемая 1020,5, наблюдаемая 1020,1.
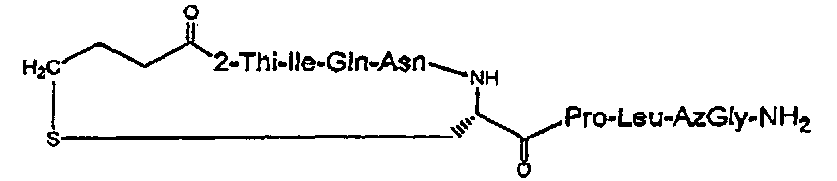
Получение Соединения 20: [AzGly9]dOT
Соединение получали посредством конденсации полностью защищенной N-концевой гептапептидной карбоновой кислоты и С-концевого дипептида H-Leu-AzGly-NH2. Используемые аминокислотные производные представляли собой Fmoc-Cys(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-Ile-OH, Fmoc-Tyr(tBu)-OH и Mpa(Trt)-OH (Peptide International) для гептапептида и Вос-Leu-OH (Bachem) для С-концевого дипептида. Производные, для которых изготовитель не указан, получали от Peptide International.
Полностью защищенную гептапептидную смолу синтезировали вручную, начиная из 1,5 г (0,9 ммоль) смолы H-Pro-2-CITrt (Novabiochem). Выполняли опосредованные DIC/HOBt/DMF одинарные связывания с 3-кратным избытком кислоты. Fmoc-группы удаляли 20%-ным пиперидином в DMF. Защищенный (1-7) пептид отщепляли от смолы с помощью 30% HFIP/DCM (60 мл) в течение 1,5 ч. Растворители выпаривали и продукт осаждали диэтиловым эфиром и использовали в последующей конденсации фрагментов без дополнительной очистки. С-концевой дипептид синтезировали в виде Boc-Leu-AzGly-NH2 посредством сочетания Boc-Leu-OH с полукарбазидом (H2N-NH-CO-NH2-HCl, Aldrich), опосредованным DCC/DCM/DIPEA. Дипептид (0,46 г) обрабатывали TFA/DCM (40 мл) в течение 1 ч. Растворители выпаривали и остаток растворяли в DMF (5 мл). Затем добавляли раствор гептапептида (1,67 г) в DMF (10 мл) с последующим добавлением DIPEA (3 мл) и РуВОР (0,546 г). Через 1 ч растворитель выпаривали и остаток обрабатывали смесью (50 мл) TIS/TFA 98/2 (об./об.) в течение 1 ч. Реакционную смесь концентрировали в вакууме и неочищенный линейный пептид осаждали диэтиловым эфиром. Осадок растворяли в неразбавленной TFA (50 мл), приливали к перемешиваемому магнитной мешалкой 5% водному раствору ацетонитрила (500 мл) и пептид окисляли добавлением 0,1 Мl2 в метаноле до устойчивого желтого цвета. Избыток йода снижали твердой аскорбиновой кислотой (Sigma-Aldrich) и рН раствора регулировали до примерно 4 путем добавления концентрированного аммония (водн.). Смесь загружали на колонку для ВЭЖХ и очищали, используя триэтиламмоний-фосфатный буфер с рН 5,2. Соединение элюировали градиентом ацетонитрила. Фракции с чистотой, превышающей 97%, объединяли, разбавляли водой (2 объема) и загружали на колонку, предварительно уравновешенную 2% АсОН (водн.). Целевое соединение элюировали быстрым (3%/мин) градиентом ацетонитрила. Фракции, содержащие целевой продукт, объединяли и лиофилизировали. Получали 411,9 мг (выход ~39%, на основании количества используемого N-концевого гептапептида и предполагая содержание пептида 85%) белого аморфного порошка. ВЭЖХ: Rt=17,3 мин, градиент: 0→20% В в течение 1 мин, 20→40% В в течение 20 мин и 100% В в течение 5 мин, t=40°C, растворитель А 0,01% TFA (водн.), растворитель В 70% СН3CN, 0,01% TFA (водн.); чистота: 100,0%; MS (М+Н+): ожидаемая 993,4, наблюдаемая 993,2.
Другие соединения получали посредством аналогичной вариации данных способов синтеза.
|
|
Экспериментальная часть (биологическое тестирование)
Анализы рецепторов in vitro.
Агонистические активности иллюстративных соединений в отношении hOT-рецепторов определяли в транскрипционном анализе гена-репортера либо посредством временной трансфекции ДНК экспрессии hOT-рецептора в клеточную линию яичников китайского хомяка (СНО) вместе с репортерной ДНК, содержащей внутриклеточные промоторные элементы кальциевого ответа, регулирующие экспрессию люциферазы светлячка, либо посредством трансфекции такой же конструкции репортерной ДНК в клеточную линию СНО, стабильно экспрессирующую hOT-рецепторы. Смотри, например, Boss et al., J. Biol. Chem., (1996), 271 (18), 10429-10432 относительно дополнительных указаний по данному анализу. На клетки воздействовали серийными разведениями соединений, разбавленных 10-кратно на дозу в течение 5 часов, с последующим лизисом клеток, определением люциферазной активности и определением эффективностей соединений и значений ЕС50 посредством нелинейной регрессии. В качестве внутреннего контроля в каждом эксперименте использовали карбетоцин. Данные показали нормальную вариацию в отдельных выполненных анализах.
Результаты анализа in vitro (величина EC50 для активности hOT в виде среднего геометрического, выраженного в наномоль/л (нМ)) для соединений, конкретно описанных в данном описании изобретения, находились в интервале от примерно 0,01 нМ до примерно 3,90 нМ, например от примерно 0,01 нМ до примерно 0,75 нМ, или, например, от примерно 0,01 нМ до примерно 0,50 нМ, или от примерно 0,01 нМ до примерно 0,25 нМ, или от примерно 0,01 нМ до примерно 0,10 нМ. В этих анализах каждое протестированное соединение было более активным в отношении hOT, чем карбетоцин.
Приведенные выше результаты показывают, что соединения, раскрытые в данном описании изобретения, входят в объем изобретения и могут быть полезными, например, в безопасном и эффективном лечении людей, находящихся в состояниях, включающих боль в животе, синдром раздраженного кишечника (IBS), аутизм, эректильную дисфункцию, женскую сексуальную дисфункцию, стимуляцию и поддержание родов, стимуляцию и поддержание лактации, послеродовое кровотечение, посттравматическое стрессовое расстройство (PTSD), боль, тревогу, потерю крови при хирургической операции, диагностику раковых заболеваний, запор, депрессию, бессонницу, мастит, OВ диагностику (в отношении плацентарной недостаточности), остеопороз, рождение плаценты и заживление/воспаление ран.
Объем настоящего изобретения далее определяется прилагаемой формулой изобретения.
Результаты анализа in vitro (величина ЕС50 для активности hOT в виде среднего геометрического, выраженного в наномоль/л (нМ)) для соединений, конкретно описанных в данном описании изобретения, находились в интервале от примерно 0,01 нМ до примерно 3,90 нМ, например от примерно 0,01 нМ до примерно 0,75 нМ, или, например, от примерно 0,01 нМ до примерно 0,50 нМ, или от примерно 0,01 нМ до примерно 0,25 нМ, или от примерно 0,01 нМ до примерно 0,10 нМ. В этих анализах каждое протестированное соединение было более активным в отношении hOT, чем карбетоцин.

Приведенные выше результаты показывают, что соединения, раскрытые в данном описании изобретения, входят в объем изобретения и могут быть полезными, например, в безопасном и эффективном лечении людей, находящихся в состояниях, включающих боль в животе, синдром раздраженного кишечника (IBS), аутизм, эректильную дисфункцию, женскую сексуальную дисфункцию, стимуляцию и поддержание родов, стимуляцию и поддержание лактации, послеродовое кровотечение, посттравматическое стрессовое расстройство (PTSD), боль, тревогу, потерю крови при хирургической операции, диагностику раковых заболеваний, запор, депрессию, бессонницу, мастит, ОВ диагностику (в отношении плацентарной недостаточности), остеопороз, рождение плаценты и заживление/воспаление ран.
Объем настоящего изобретения далее определяется прилагаемой формулой изобретения.
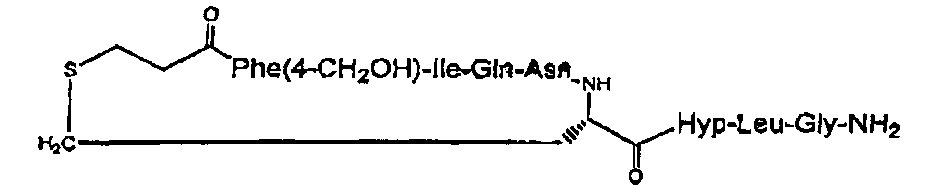
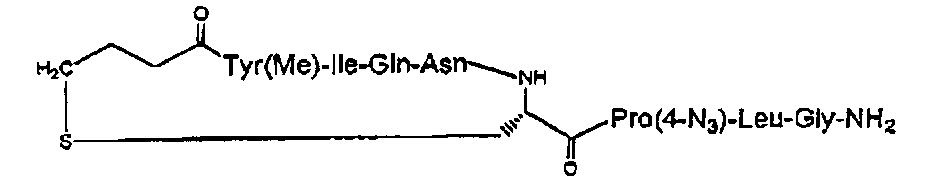
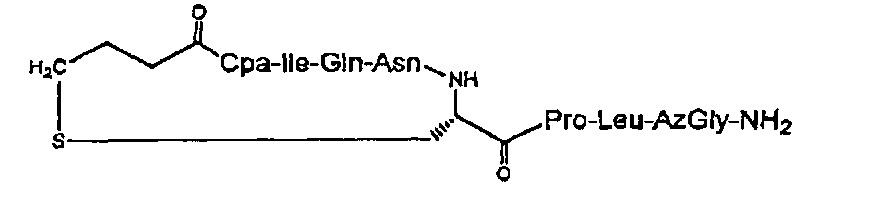
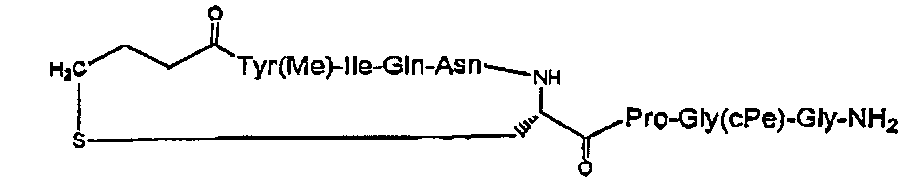
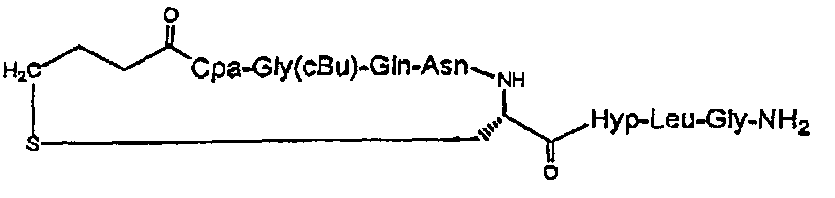
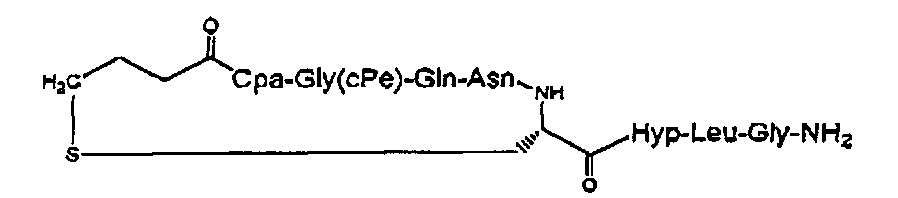
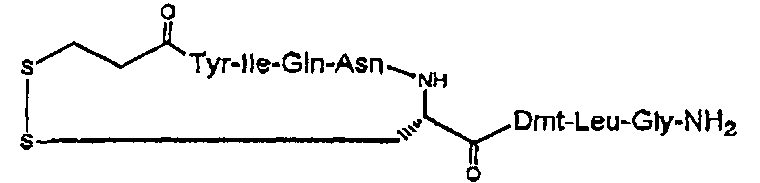
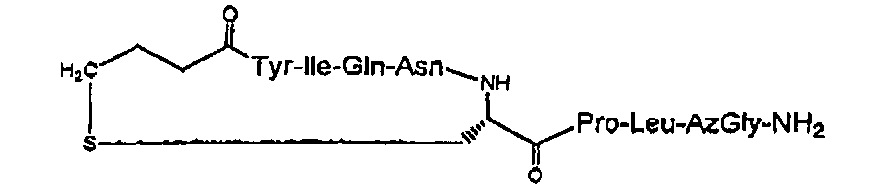
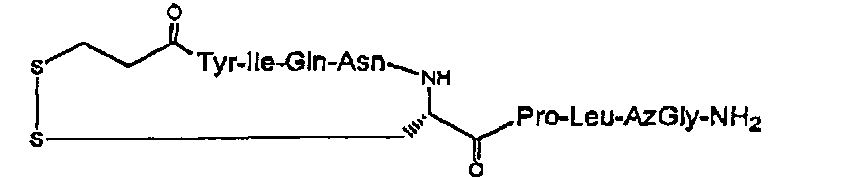
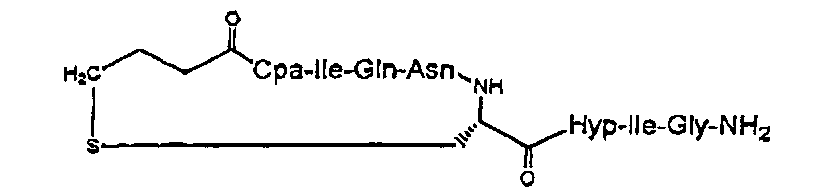
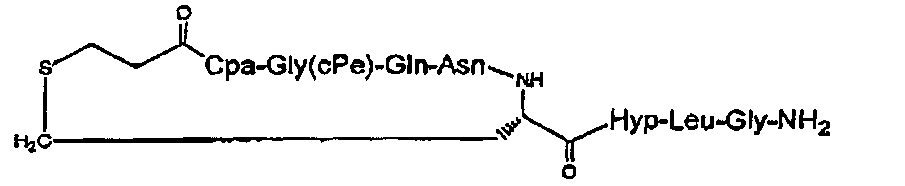
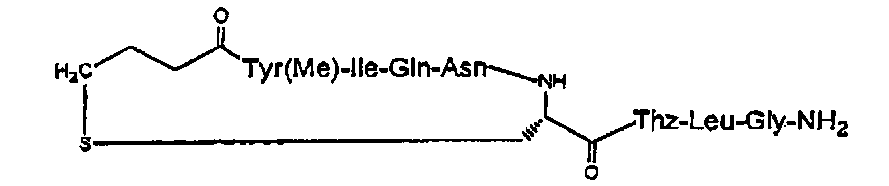
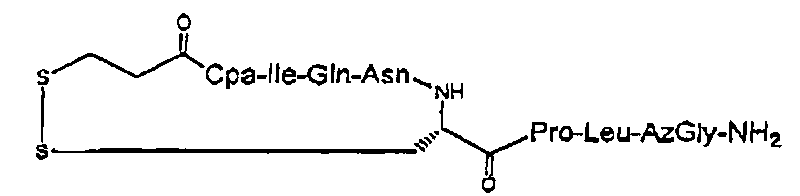
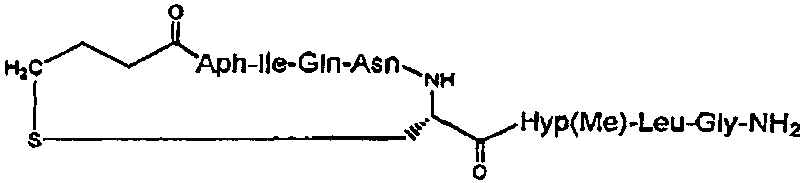
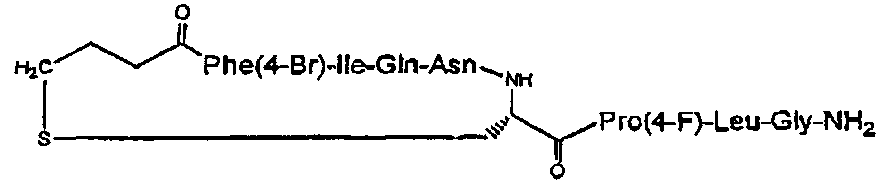
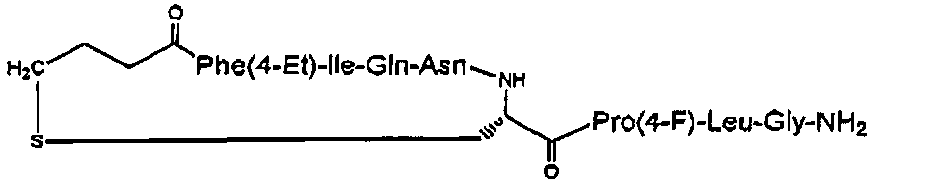
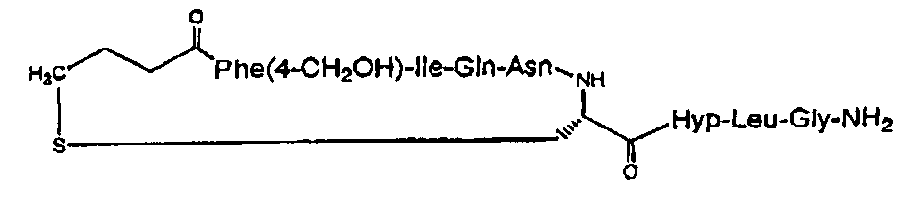
Аналоги окситоцина
Композиция для лечения рака предстательной железы
Саше месалазина с высокой нагрузкой лекарства
Пептидные агонисты glp-2
Быстрорастворимая фармацевтическая композиция
Быстрорастворимая фармацевтическая композиция
Стабилизация fsh
Гранулы для фармацевтических препаратов, способы и аппарат для их получения
Интравагинальное введение мизопростола
Фармацевтический препарат, содержащий рекомбинантный хорионический гонадотропный гормон человека
Аналоги окситоцина
Композиция для лечения рака предстательной железы
Саше месалазина с высокой нагрузкой лекарства
Пептидные агонисты glp-2
Быстрорастворимая фармацевтическая композиция
Быстрорастворимая фармацевтическая композиция
Стабилизация fsh
Гранулы для фармацевтических препаратов, способы и аппарат для их получения
Интравагинальное введение мизопростола
Фармацевтический препарат, содержащий рекомбинантный хорионический гонадотропный гормон человека

