Результат интеллектуальной деятельности: АНАЛОГИ ОКСИТОЦИНА
Вид РИД
Изобретение
Область изобретения
Настоящее изобретение относится к новым соединениям, содержащим их фармацевтическим композициям, применению указанных соединений для изготовления лекарственного средства для лечения, в том числе, нарушений лактации, а также к способу лечения указанных состояний, при котором вводят указанные соединения.
Предшествующий уровень техники
Пептидные агонисты рецептора окситоцина включают природный гормон окситоцин и карбетоцин.
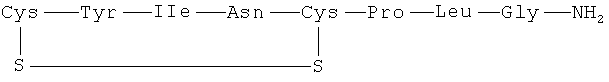 (окситоцин)
(окситоцин)
Окситоцин представляет собой мощный агент, стимулирующий сокращение матки, который клинически используют для того, чтобы вызвать родовую деятельность, и показано, что он усиливает наступление и поддержание лактации, Gimpl, G. et al., Physiol. Rev. 81 (2001) 629-683 и Ruis H. et al., British Medical Journal 283 (1981) 340-342. Карбетоцин (1-деамино-1-карбо-2-тирозин(O-метил)окситоцин) также представляет собой мощный стимулирующий сокращение матки агент, который клинически используют для контроля атонии матки и чрезмерного кровотечения. Дополнительные исследования свидетельствуют о том, что агонисты окситоцина полезны для лечения воспаления и боли, включая боль в животе и спине, сексуальной дисфункции у мужчин и женщин, синдрома раздраженного кишечника (IBS), запора и желудочно-кишечной непроходимости, аутизма, стресса, тревоги (включая тревожное расстройство) и депрессии (Pitman R. et al., Psychiatry Research, 48:107-117; Kirsch P et al., The Journal of Neuroscience, 25 (49): 11489-11493); потери крови при операции, для контроля послеродового кровотечения, заживления ран и инфекции, мастита и отхождения плаценты, а также остеопороза. Дополнительно агонисты окситоцина можно применять для диагностики рака и плацентарной недостаточности.
Недостатком как окситоцина, так и карбетоцина является отсутствие у них селективности в отношении рецепторов вазопрессина, особенно в отношении рецептора V2. Во время введения окситоцина этот недостаток наблюдают в виде таких побочных эффектов как прекращение мочеотделения и гипонатриемия.
Для улучшения фармакологических свойств окситоцина синтезированы аналоги окситоцина. Такие аналоги описаны Grozonka Z. et al. в J. Med. Chem. 26 (1983) 555-559 и J. Med. Chem. 26 (1983) 1786-1787, а также Engstroem T. et al. в E. J. Pharmacol. 355 (1998) 203-210. Дополнительно аналоги окситоцина, обладающие антагонистической активностью в отношении рецептора окситоцина, описаны Fragiadaki М. et al. в Е. J. Med. Chem. (2007) 799-806.
В настоящем изобретении предложены селективные эффективные соединения, обеспечивающие реальные альтернативы и/или улучшения, например, в лечении нарушений лактации.
Описание изобретения
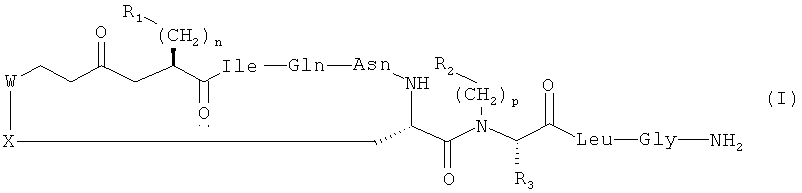
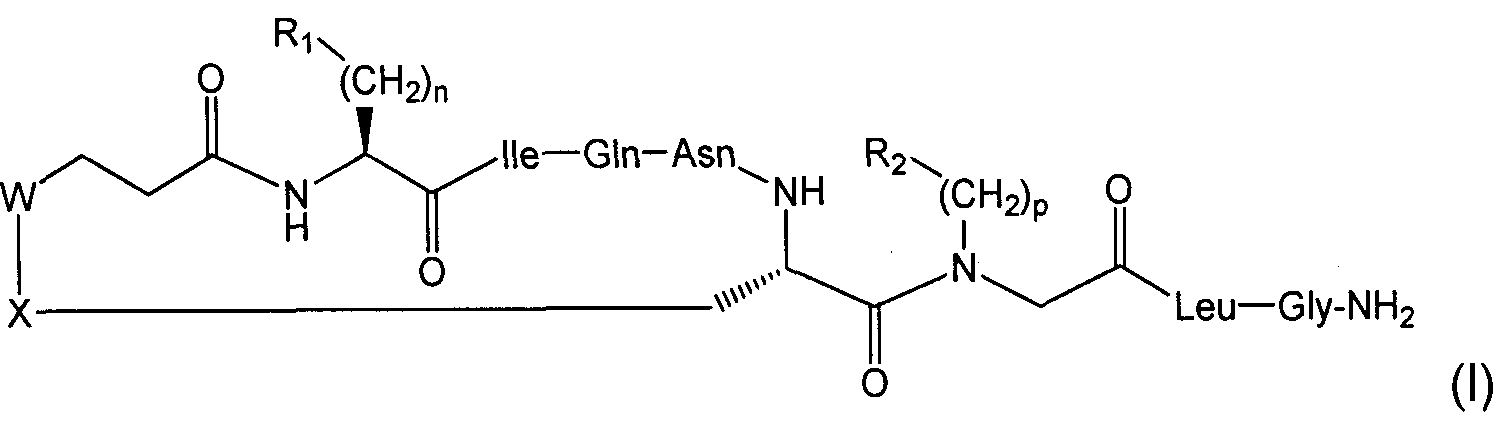
Настоящее изобретение относится к соединениям, представленным общей формулой (I):
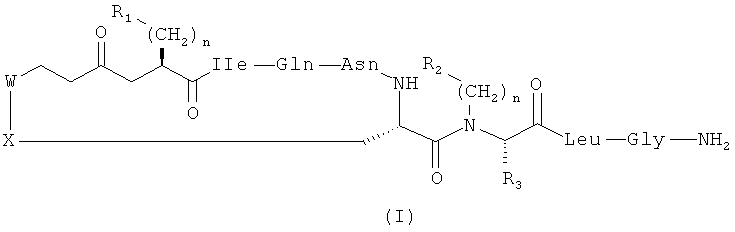 ,
,
где:
n выбран из 0, 1 и 2;
p выбран из 0, 1, 2, 3, 4, 5 и 6;
R1 выбран из арила, возможно замещенного по меньшей мере одним OH, F, Cl, Br, алкильным или O-алкильным заместителем;
R2 выбран из R4, H, алкила, циклоалкила, арила и 5- и 6-членных гетероароматических кольцевых систем;
R3 выбран из H и ковалентной связи с R2, когда R2 представляет собой R4, с образованием кольцевой структуры;
R4 представляет собой C1-6алкиленовую группировку, замещенную по меньшей мере одним O-алкилом, S-алкилом или заместителем OH;
каждый из W и X независимо выбран из CH2 и S, но оба они не могут представлять собой CH2;
алкил выбран из C1-6прямоцепочечного и C4-8вразветвленного алкила, возможно имеющего по меньшей мере один гидроксильный заместитель;
арил выбран из фенила и моно- или полизамещенного фенила;
при условии, что когда R2 представляет собой H, p равен 1, R3 представляет собой H, n равен 1, а оба из W и X представляют собой S, R1 не представляет собой 4-гидроксифенил;
циклоалкил выбран из C3-5циклоалкила и возможно имеет по меньшей мере один гидроксильный заместитель;
и их сольватам и фармацевтически приемлемым солям.
Настоящее изобретение далее относится к соединениям, представленным формулой (I) выше, при дополнительном условии, что когда R2 представляет собой H, p равен 0, R3 представляет собой H, n равен 1, а оба из W и X представляют собой S, R1 не представляет собой 4-гидроксифенил. Таким образом, настоящее изобретение может относиться к соединениям формулы (I), приведенной выше, при условии, что соединение не представляет собой [1-β-Mpa,7-Sar]OT и/или {деамино[7-глицин]окситоцин}.
Для задач настоящего изобретения используют следующую терминологию.
C1-6прямоцепочечный алкил обозначает цепь, имеющую от одного до шести атомов углерода, включая любое число между этими значениями.
C4-8разветвленный алкил обозначает все разветвленные алкильные группы, содержащие от четырех до восьми атомов углерода, включая изо-, втор- и трет-конфигурации, поскольку указанное выражение не относится к сайту связывания интересуемой алкильной цепи.
C3-6циклоалкил обозначает карбоциклическую кольцевую систему, содержащую от трех до шести атомов углерода, включая любое число между этими значениями. Кольцевая система может содержать ненасыщенные связи между атомами углерода.
Пятичленная гетероароматическая кольцевая система представляет собой моноциклическую ароматическую кольцевую систему, имеющую пять кольцевых атомов, где 1, 2, 3 или 4 кольцевых атома независимо выбраны из N, O и S. Предпочтительные кольцевые системы выбраны из группы, состоящей из тиенила, фурила, имидазолила, тиазолила, тиадиазолила и тетразолила.
Шестичленная гетероароматическая кольцевая система представляет собой моноциклическую ароматическую кольцевую систему, имеющую шесть кольцевых атомов, где 1, 2, 3 или 4 кольцевых атома независимо выбраны из N, O и S. Предпочтительные кольцевые системы выбраны из группы, состоящей из пиридила.
Арил обозначает ароматическую группу, выбранную из фенила и моно- или полизамещенного фенила.
Замещенные группировки могут быть выбраны из атомов фтора (F), хлора (Cl) и брома (Br), а также алкила, гидрокси (-OH), аклкокси (-O-алкил) и алкилтио (-S-алкил).
Примеры фармацевтически приемлемых солей включают соли присоединения кислоты, например, соли, образованные путем взаимодействия с галогеноводородными кислотами, такими как соляная кислота, и неорганическими кислотами, такими как серная кислота, ортофосфорная кислота и азотная кислота, а также алифатическими, алициклическими, ароматическими или гетероциклическими сульфокислотами или карбоновыми кислотами, такими как муравьиная кислота, уксусная кислота, пропионовая кислота, янтарная кислота, гликолевая кислота, молочная кислота, малеиновая кислота, винная кислота, лимонная кислота, бензойная кислота, аскорбиновая кислота, малеиновая кислота, гидроксималеиновая кислота, пировиноградная кислота, лара-гидроксибензойная кислота, эмбоновая кислота, метансульфоновая кислота, этансульфоновая кислота,
гидроксиэтансульфоновая кислота, галогенобензолсульфоновая кислота, трифторуксусная кислота, трифторметансульфоновая кислота, толуолсульфоновая кислота и нафталинсульфоновая кислота.
В предпочтительных воплощениях n равен 1.
В предпочтительных воплощениях p выбран из 1, 2, 3, 4 и 5.
В предпочтительных воплощениях R1 выбран из фенила, 4-гидроксифенила, 4-метоксифенила и 4-этилфенила.
В предпочтительных воплощениях R2 выбран из этила, н-пропила, н-бутила, циклопропила, 2-гидроксиэтила, 2-метоксиэтила, 2-фенилэтила, фенила, бензила, 2-метилфенила, 3-метилфенила, 4-метилфенила, 4-метоксифенила, 4-фторфенила, 3,4-дифторфенила, 2-тиенила, 2-тетрагидрофурила, 2-фурила, 2-пиридила и 4-пиридила.
В предпочтительных воплощениях R3 представляет собой H.
В предпочтительных воплощениях указанная кольцевая структура выбрана из (R)-4-метоксипирролидинила, (R)-4-метилтиопирролидинила и (S)-4-гидроксипирролидинила.
В предпочтительных воплощениях W представляет собой CH2, и X представляет собой S.
В предпочтительных воплощениях W представляет собой S, и X представляет собой CH2.
В предпочтительных воплощениях оба W и X представляют собой S.
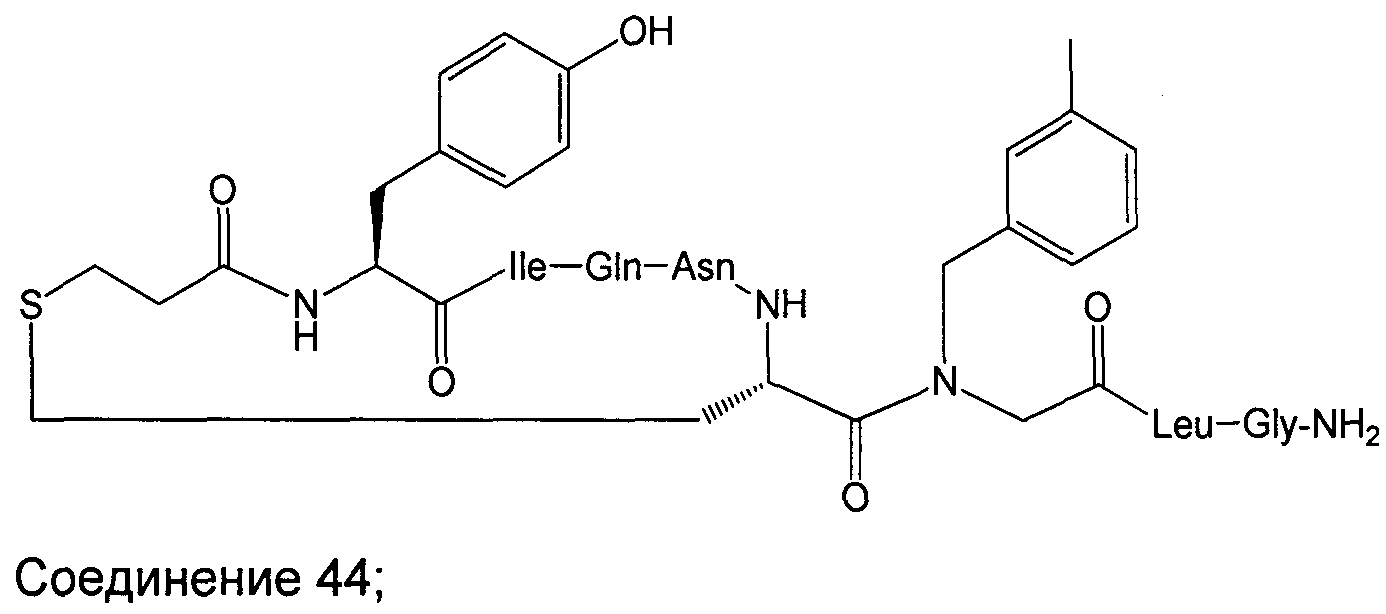
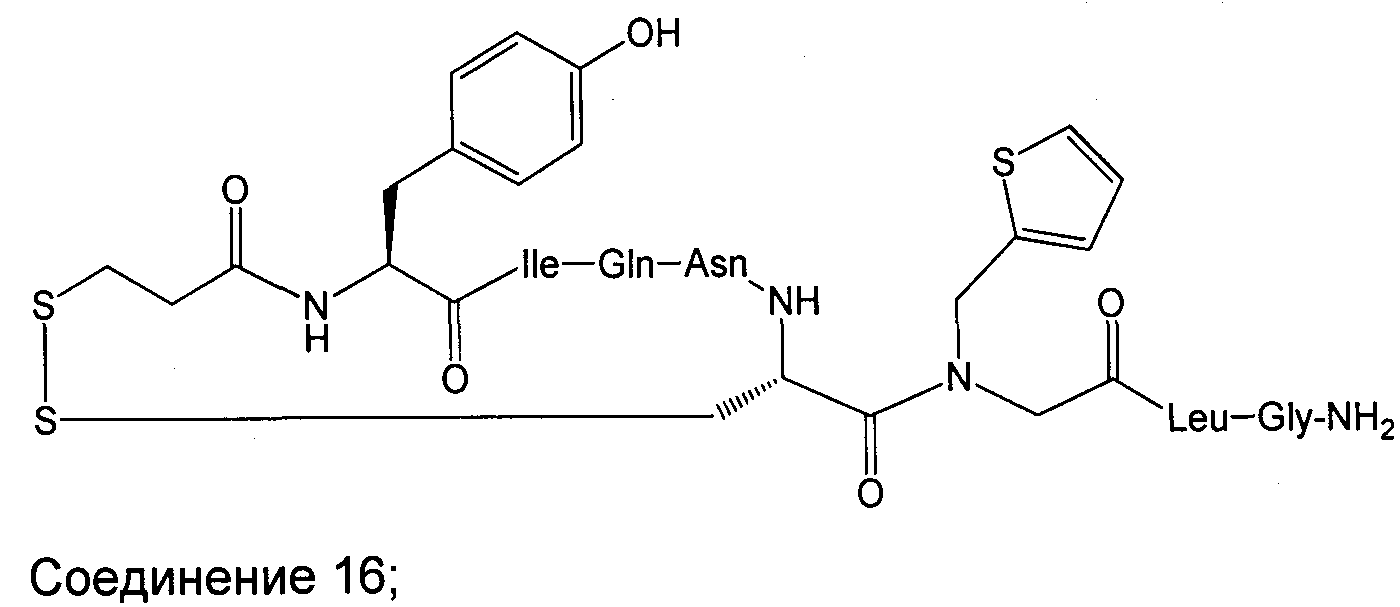
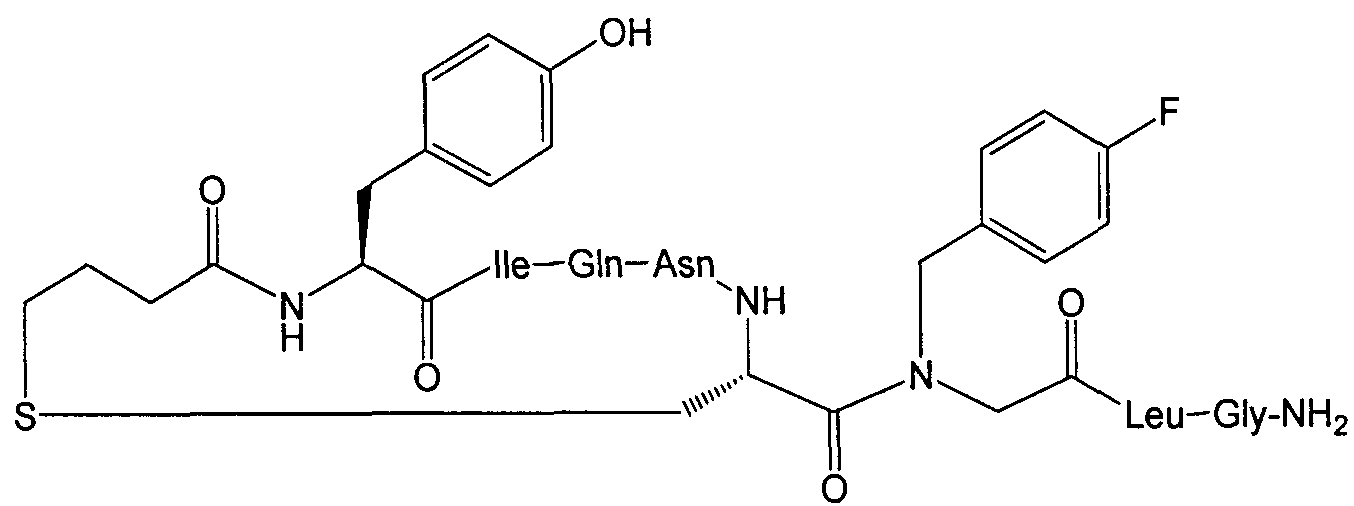
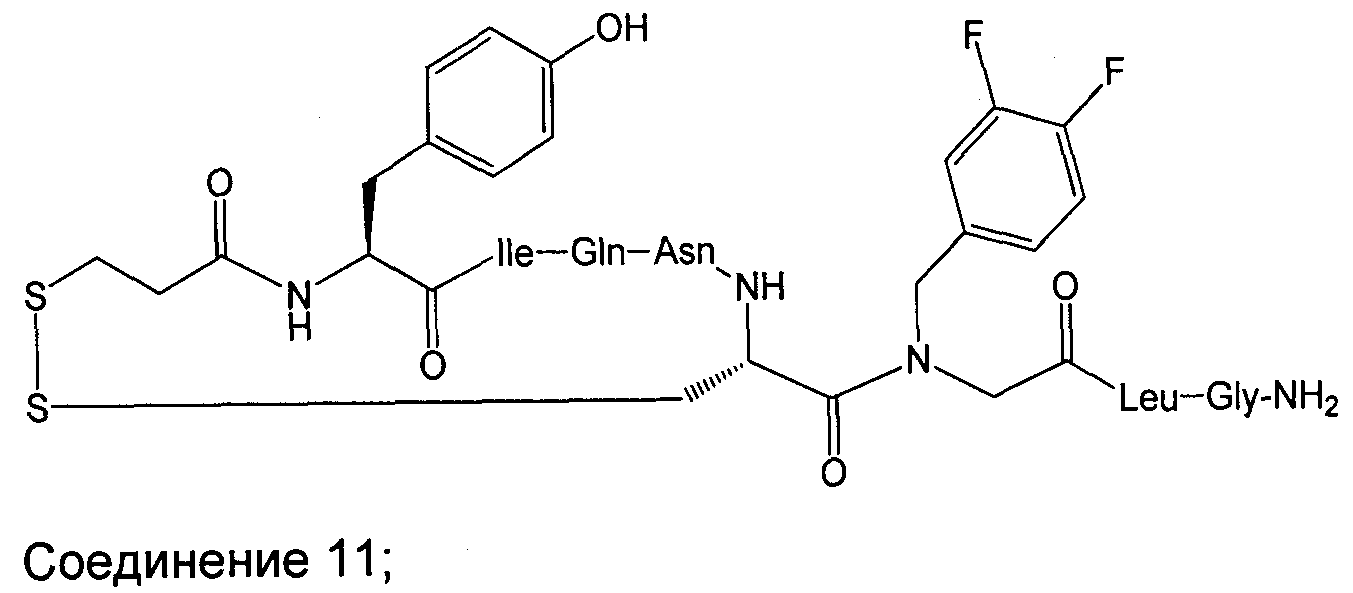
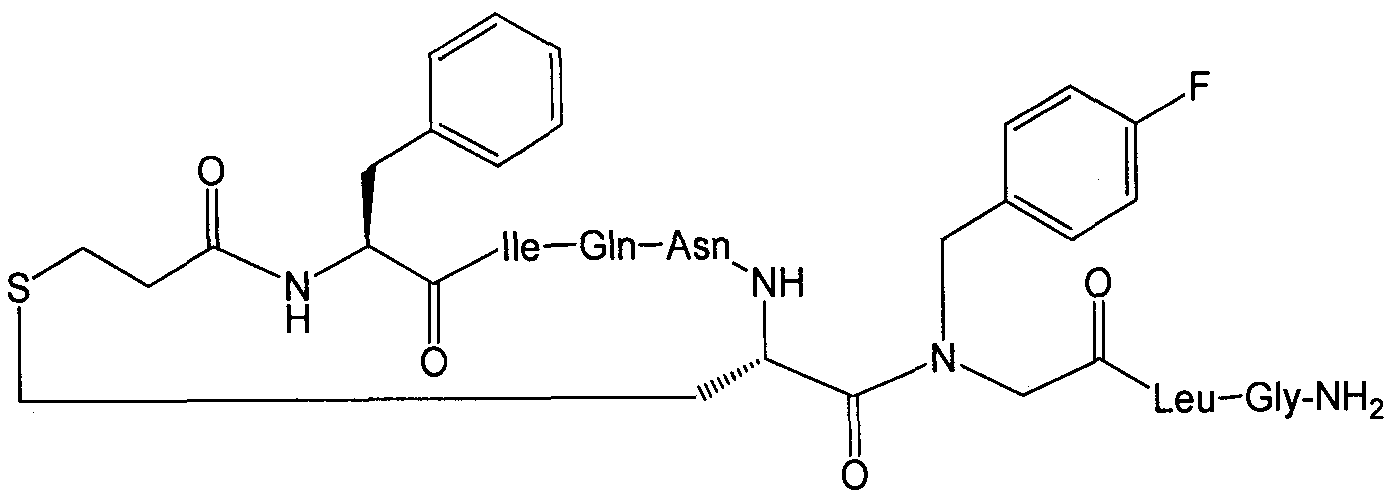
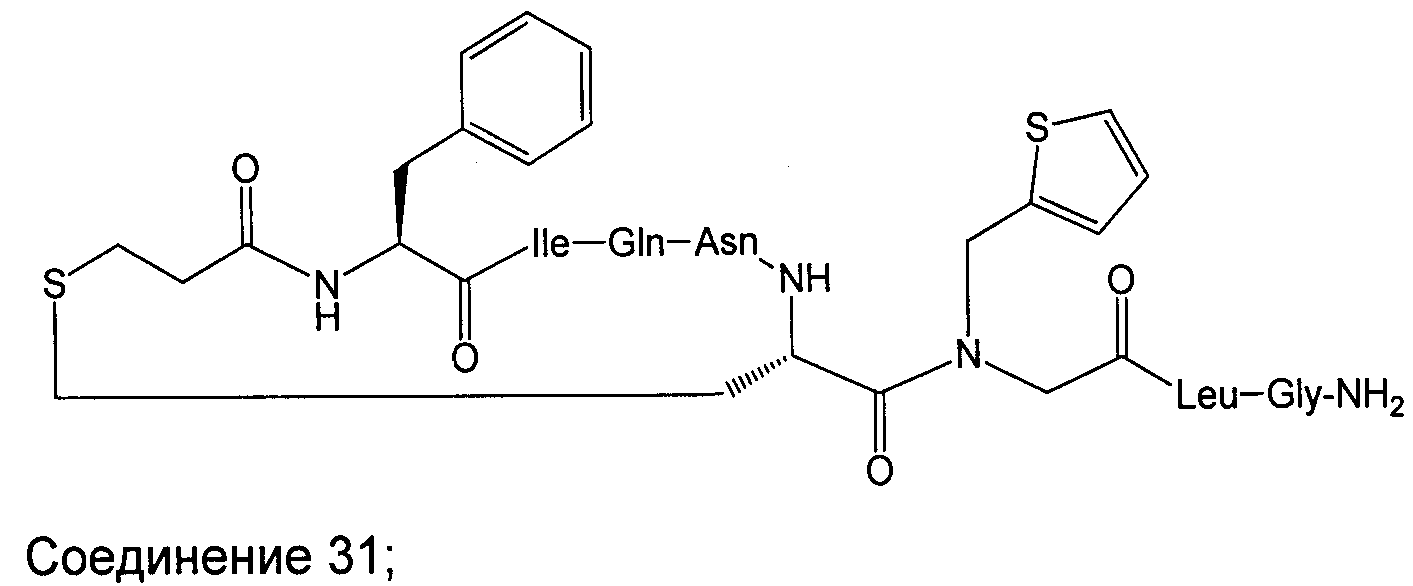
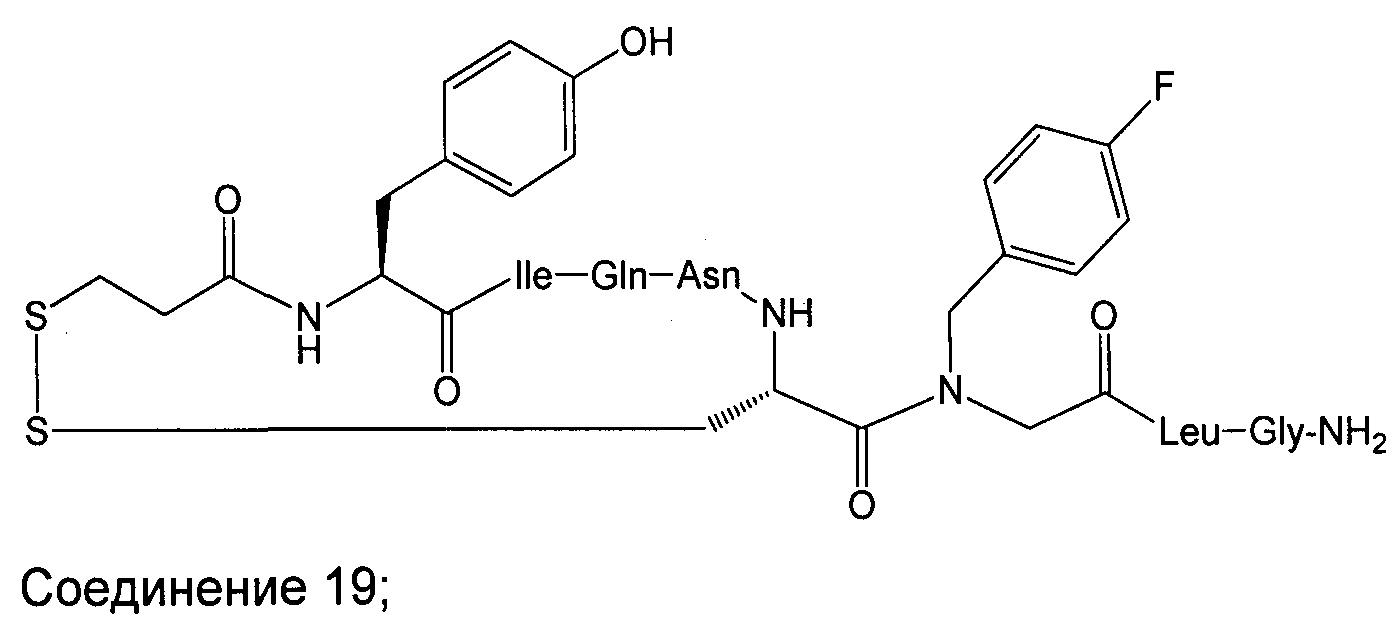
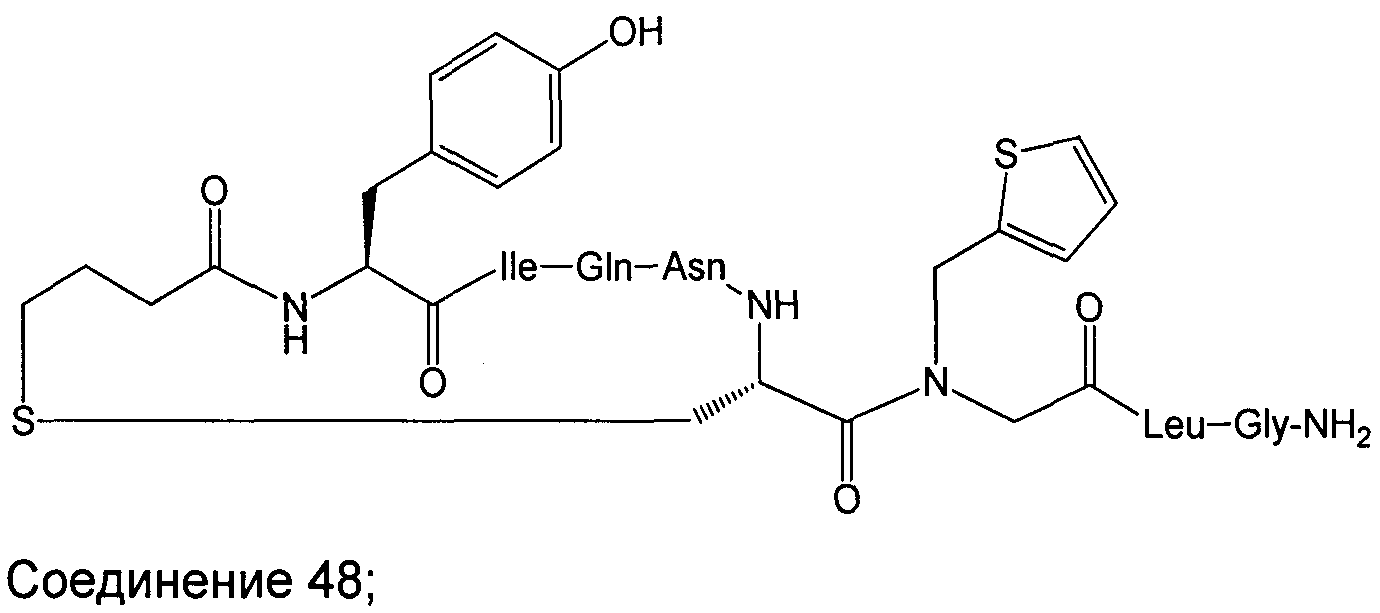
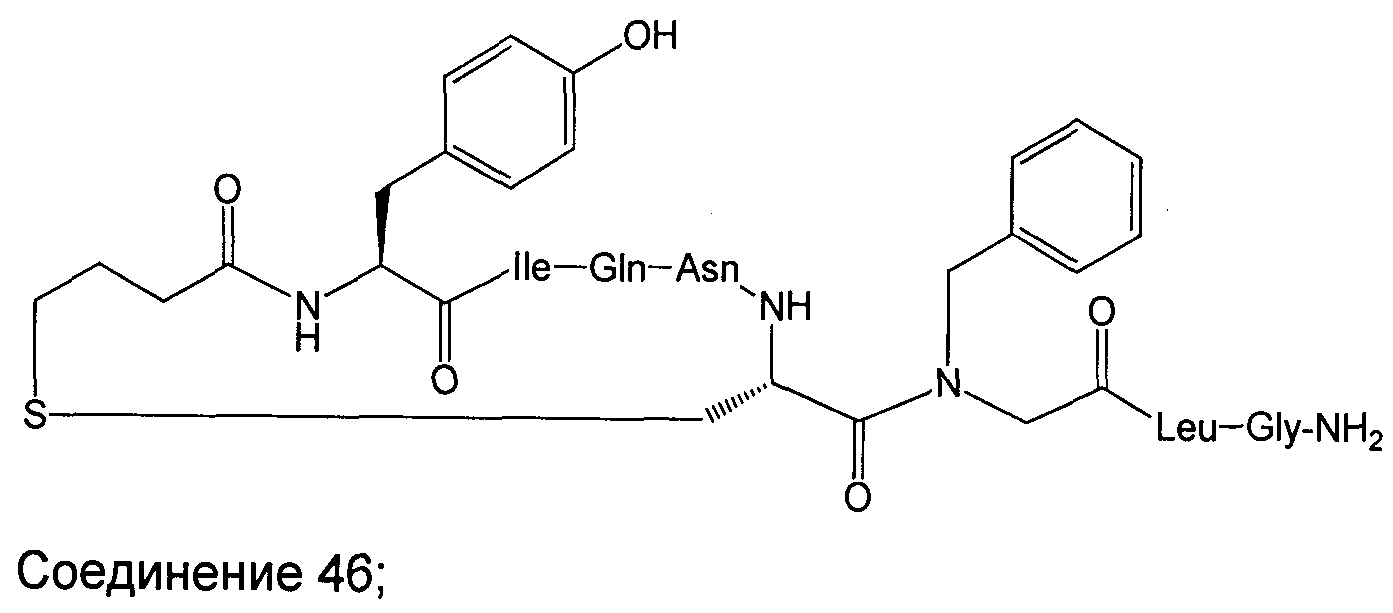
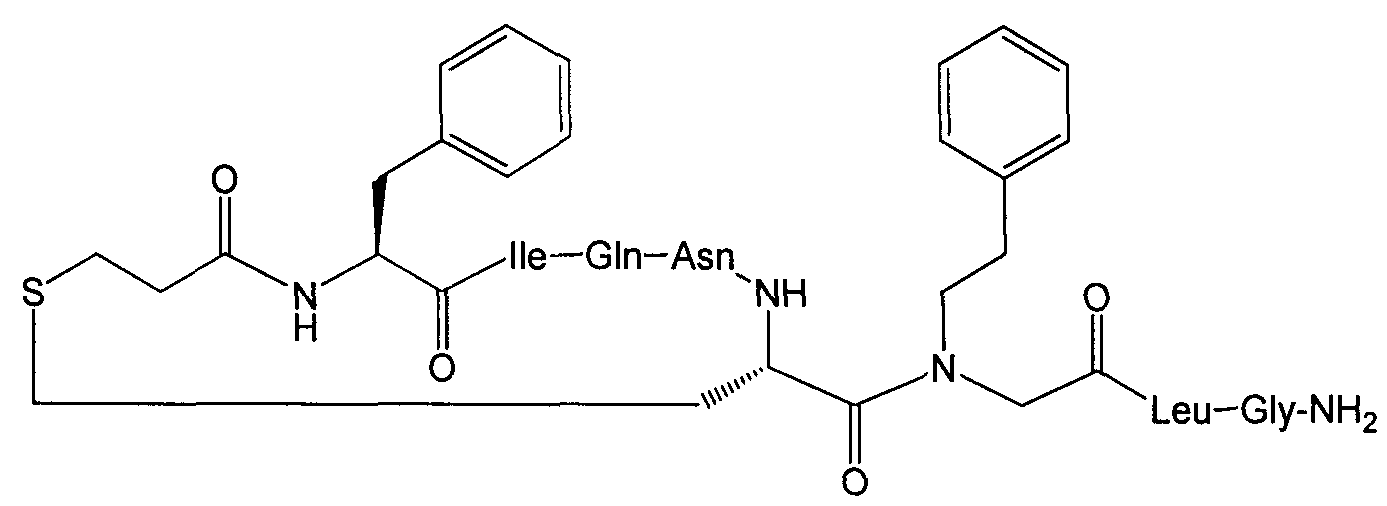
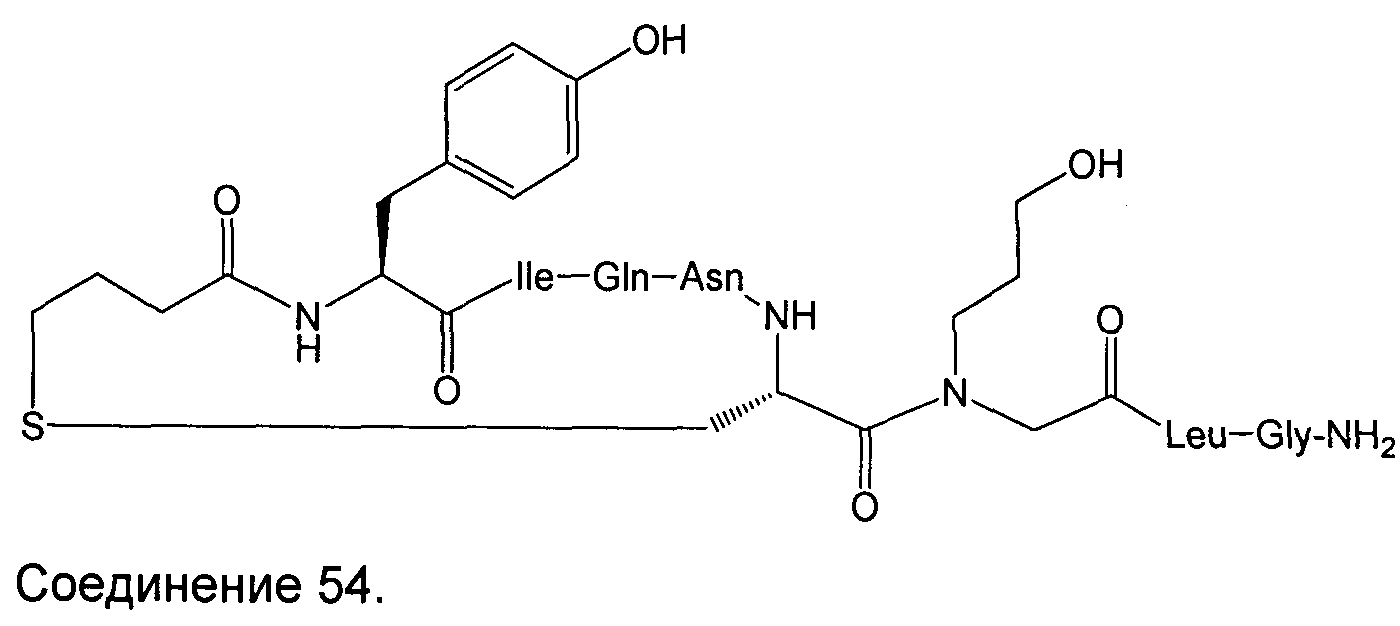
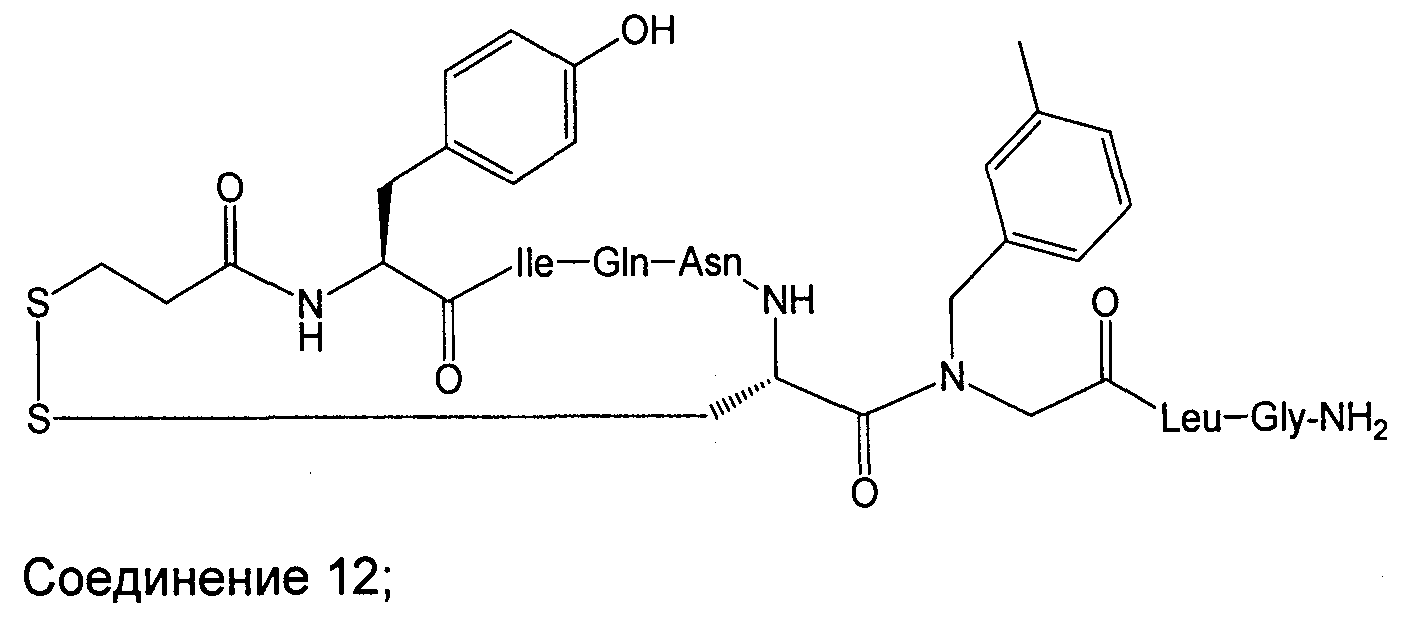
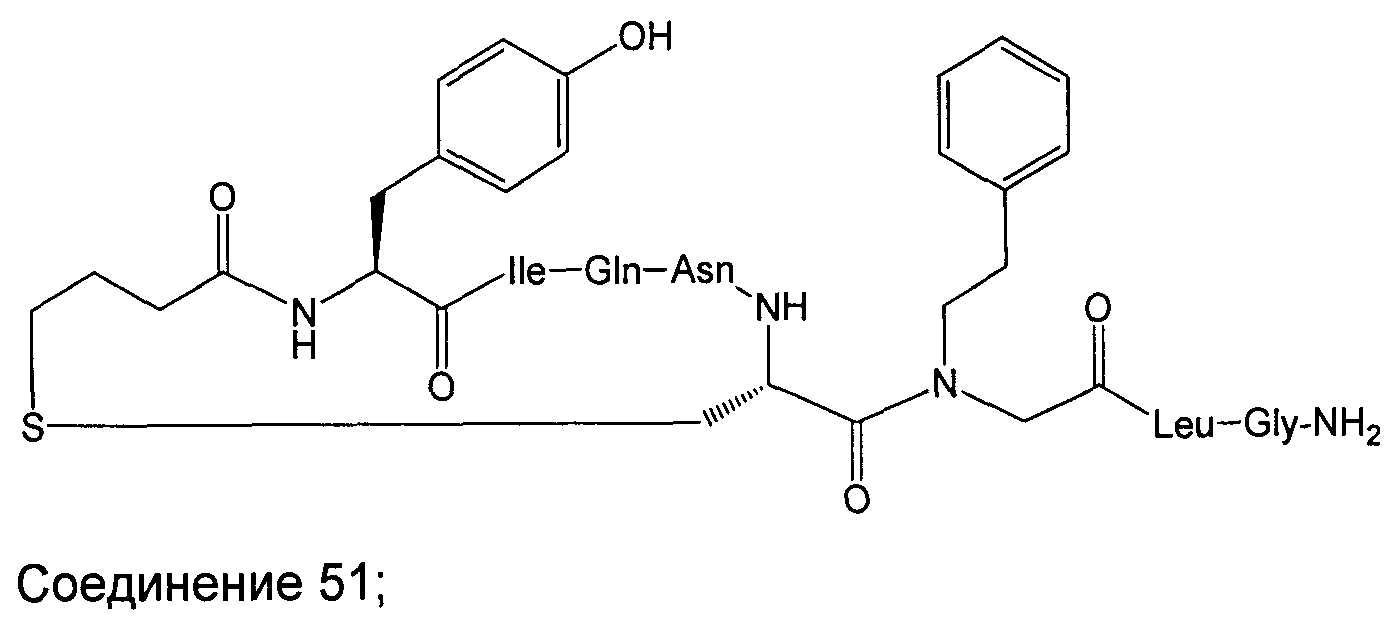
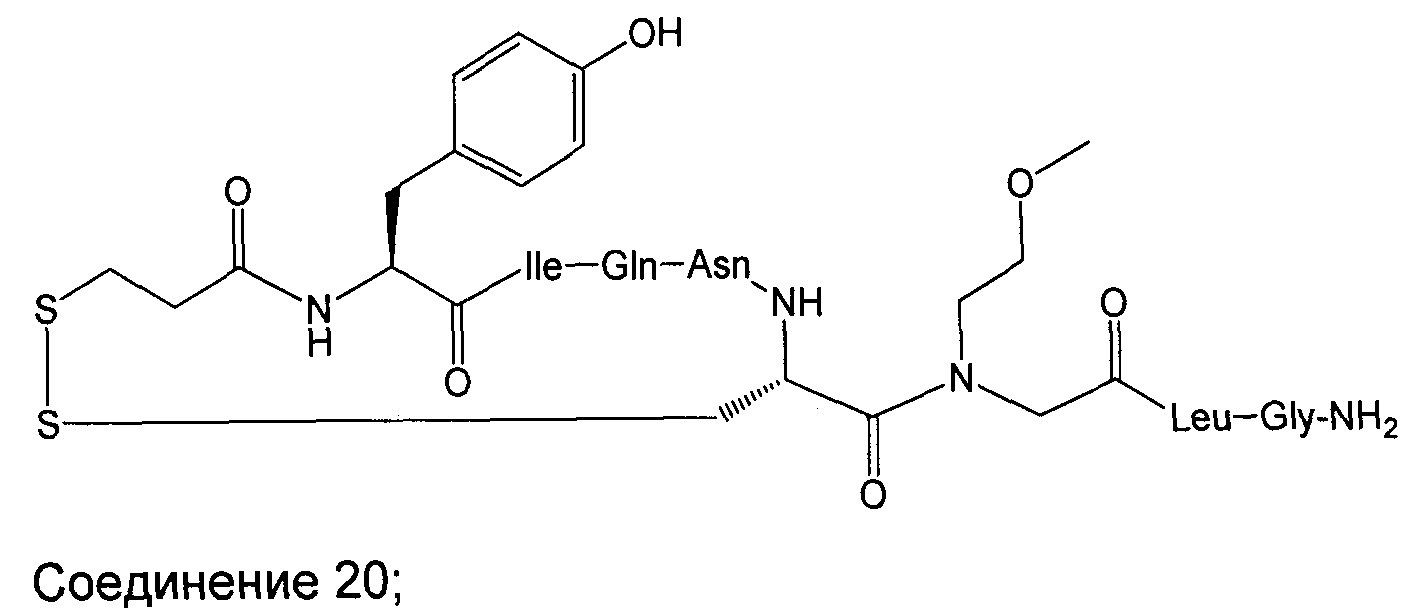
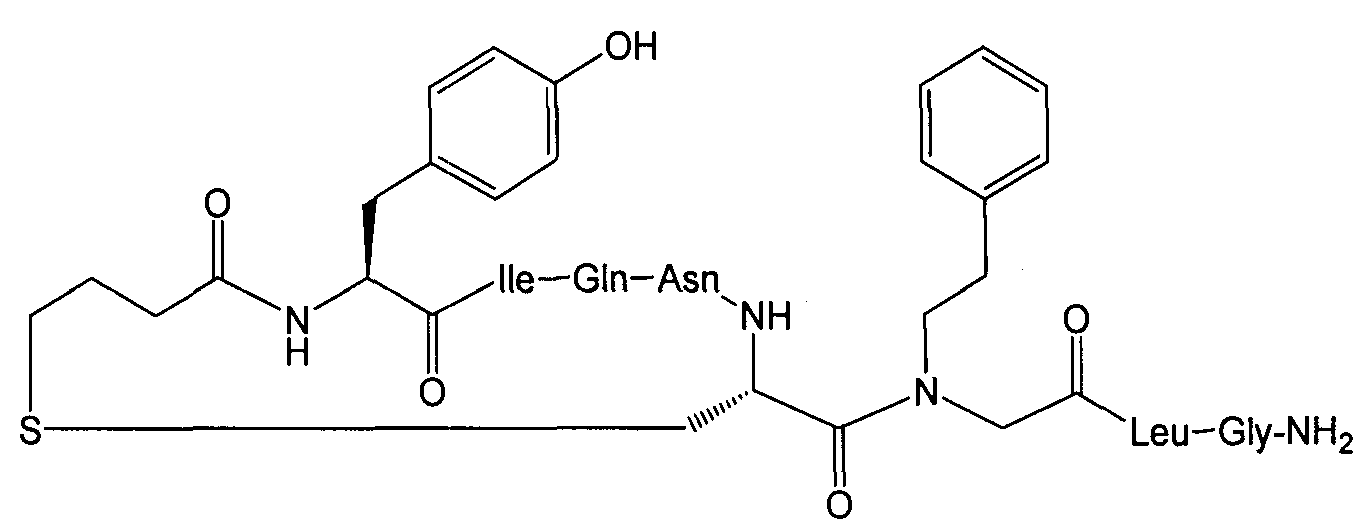
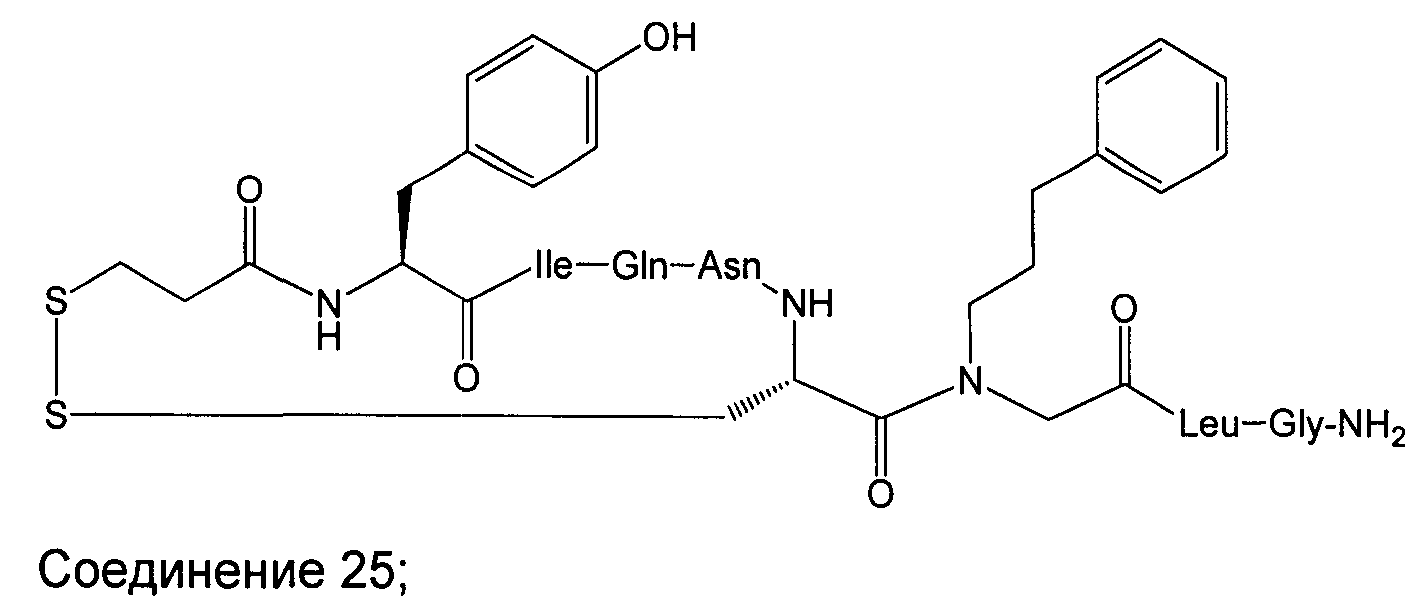
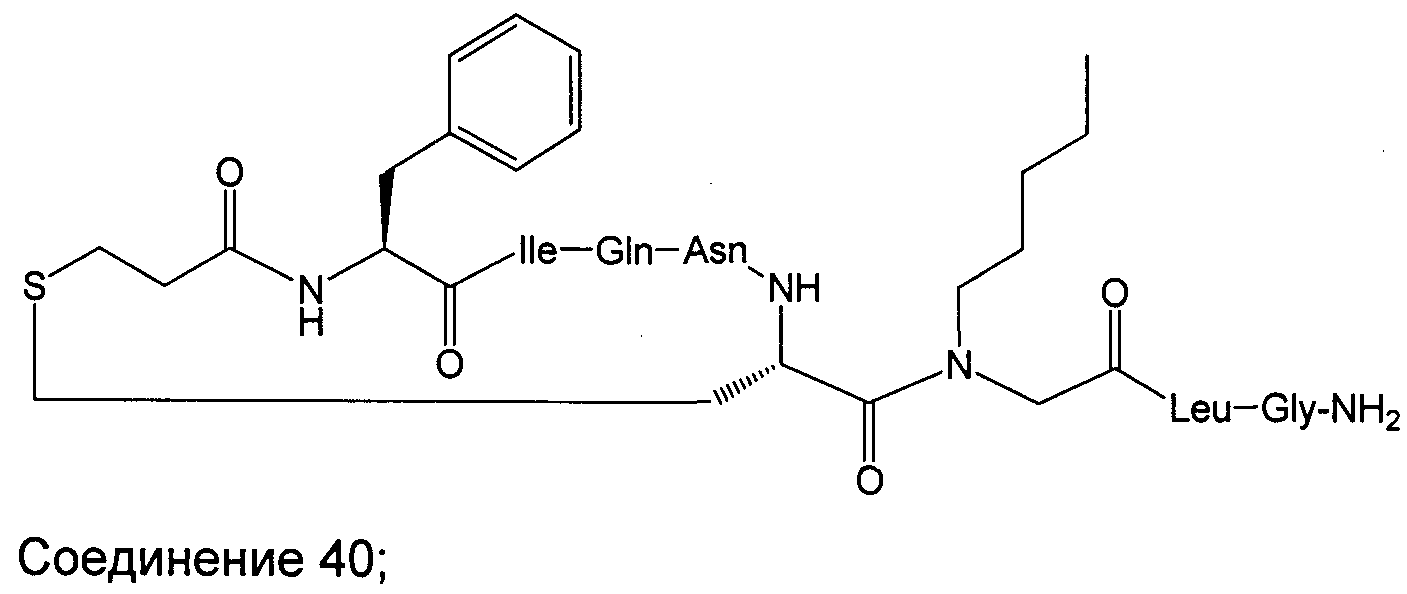
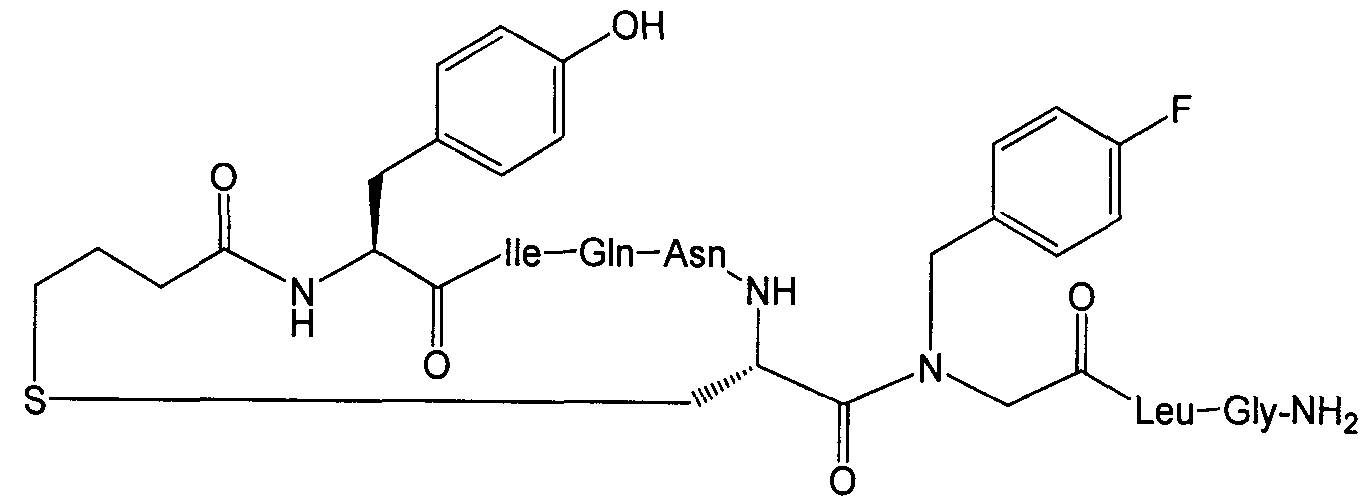
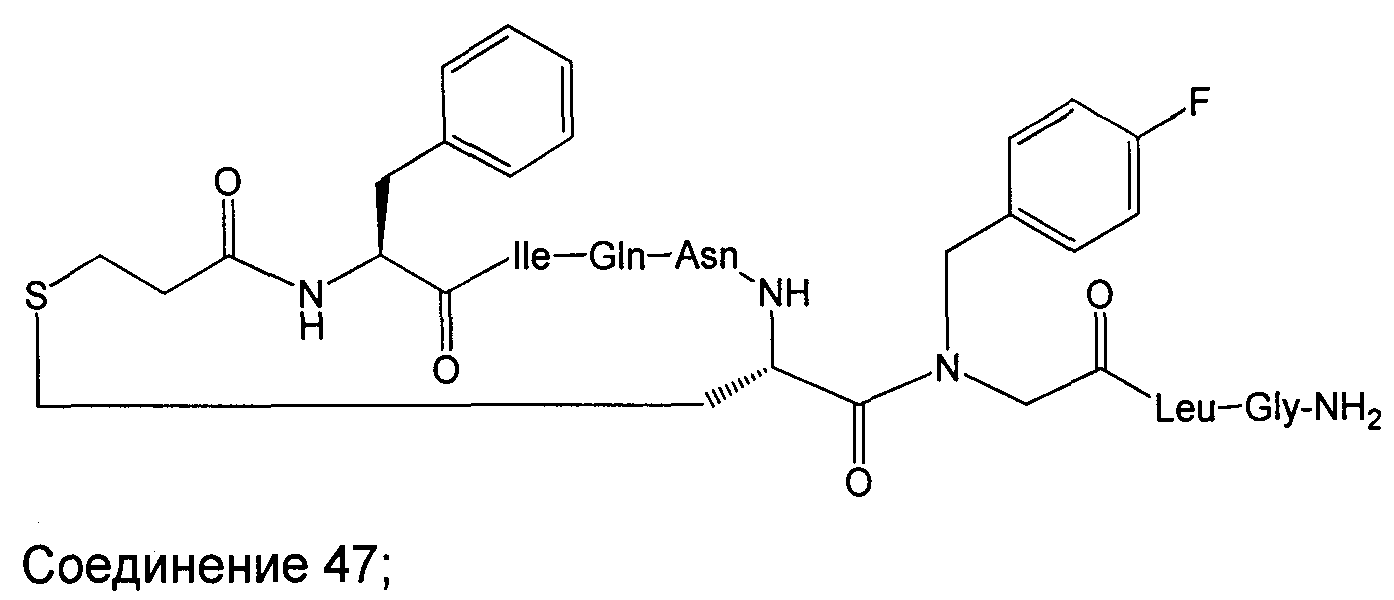
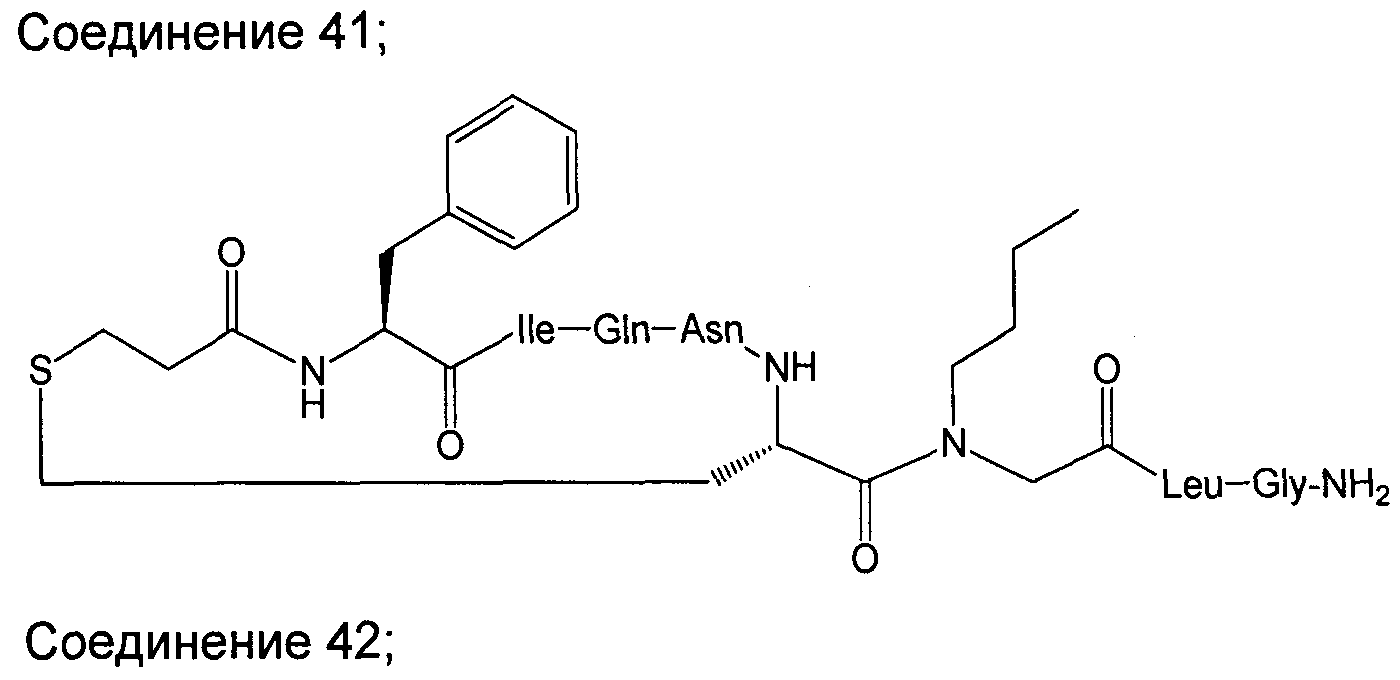
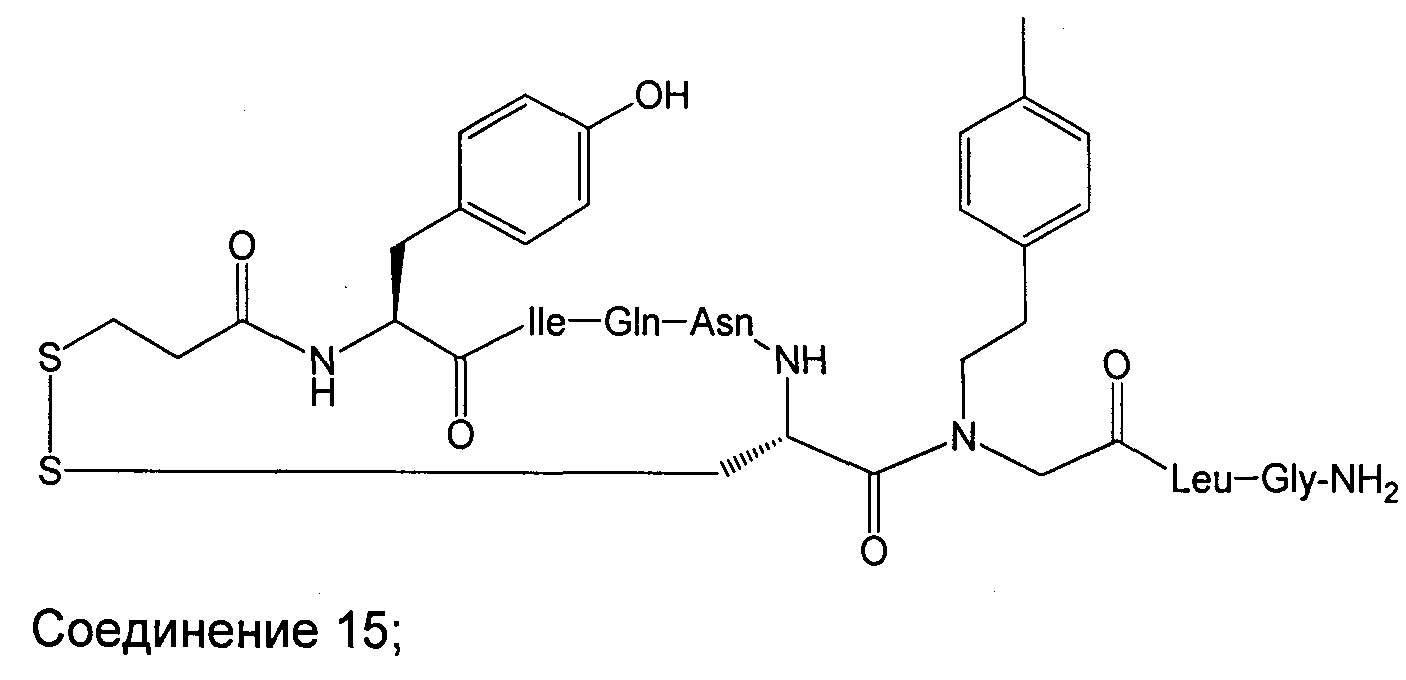
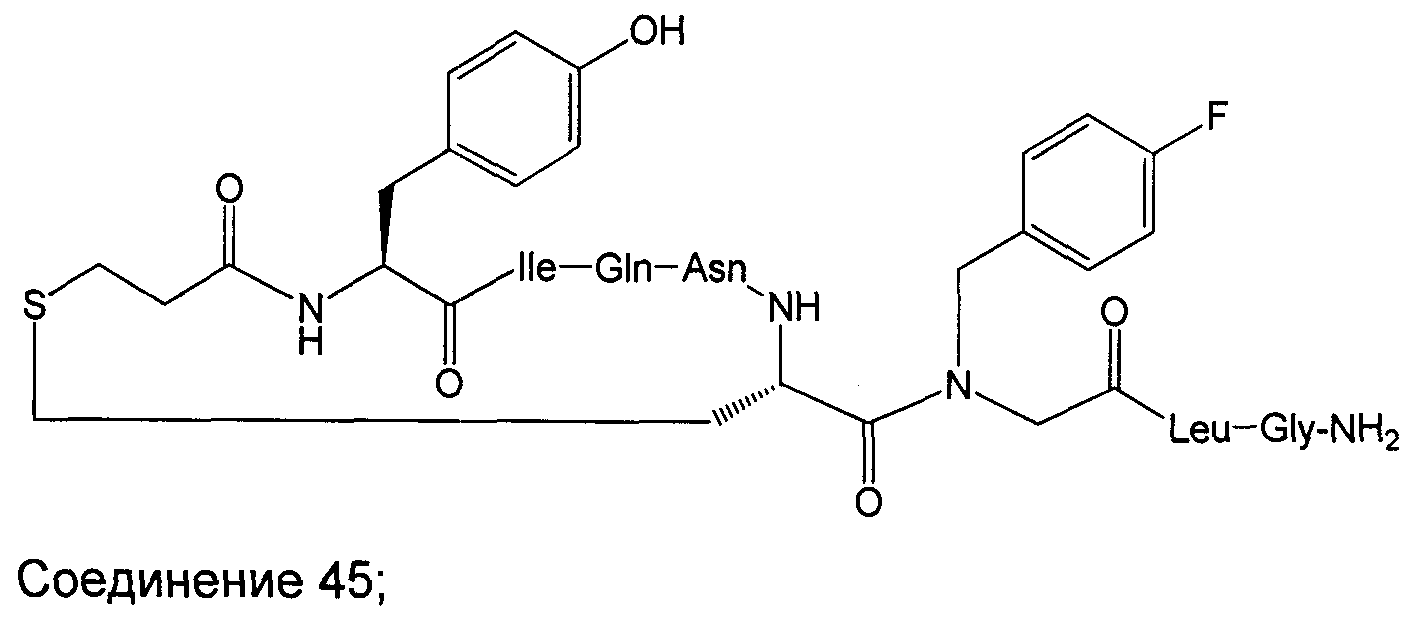
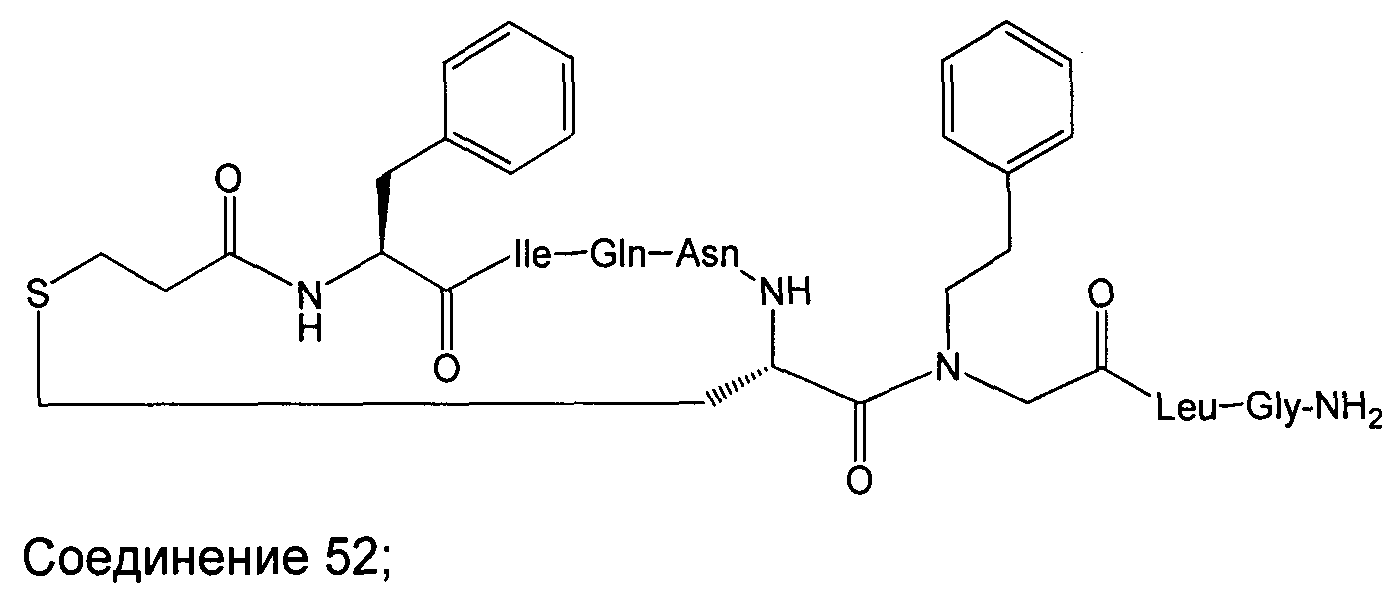
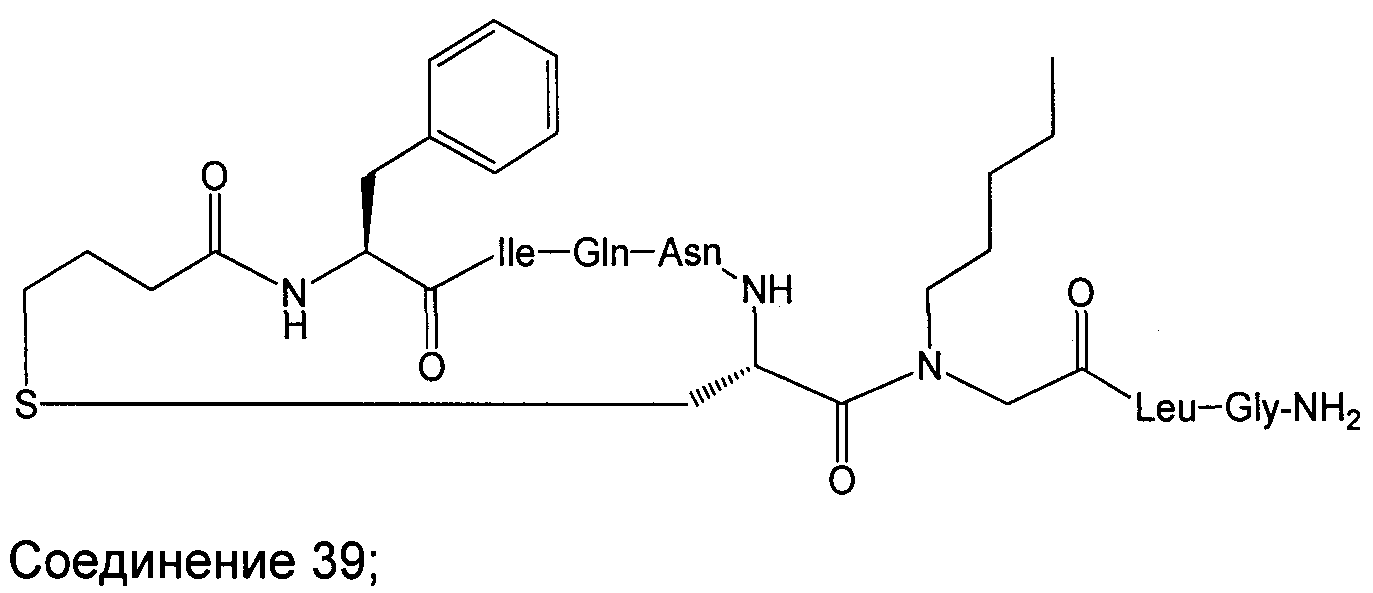
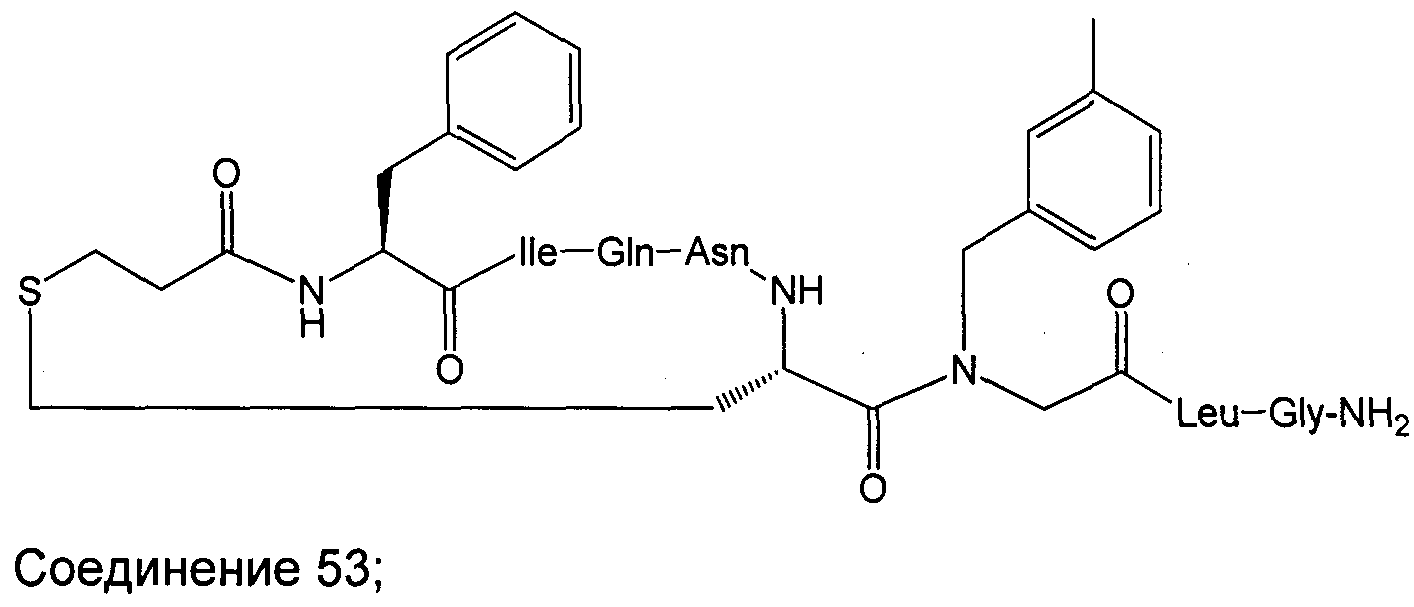
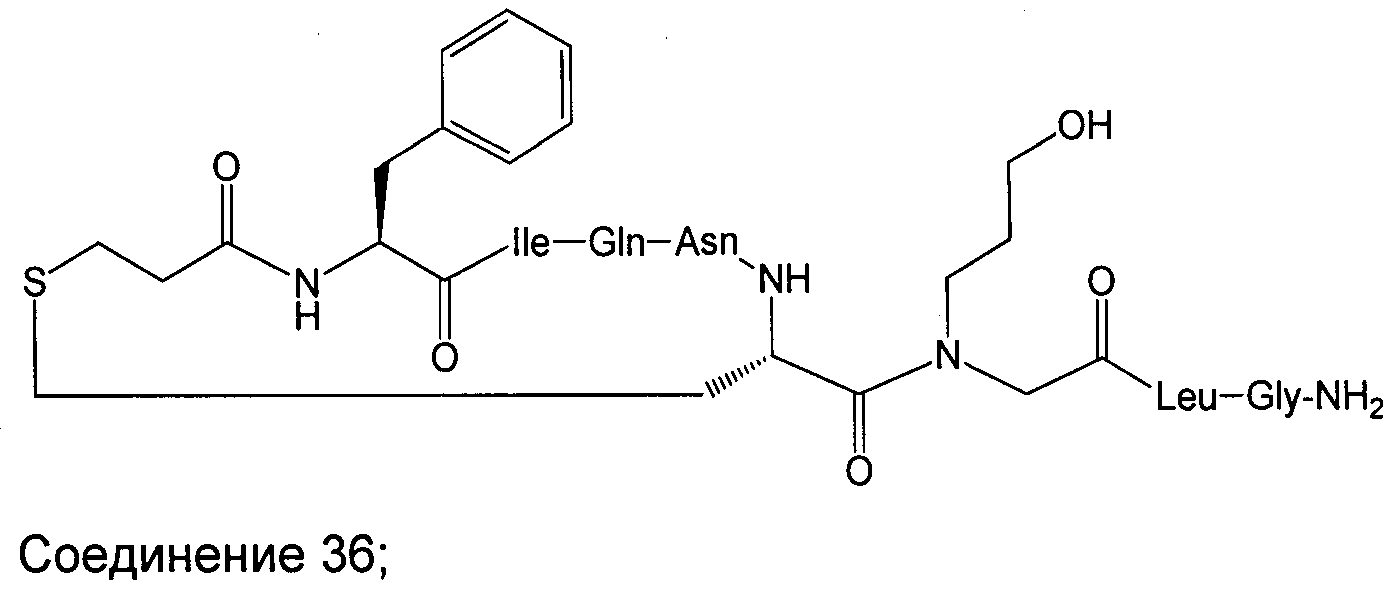
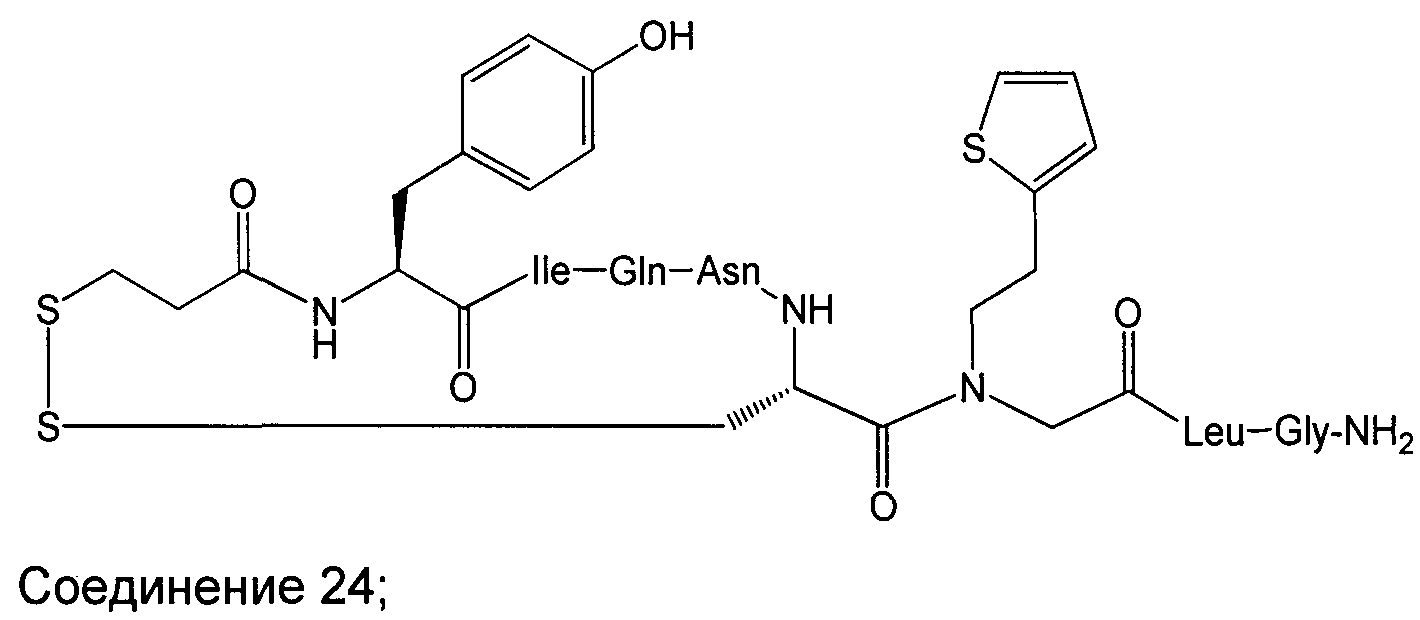
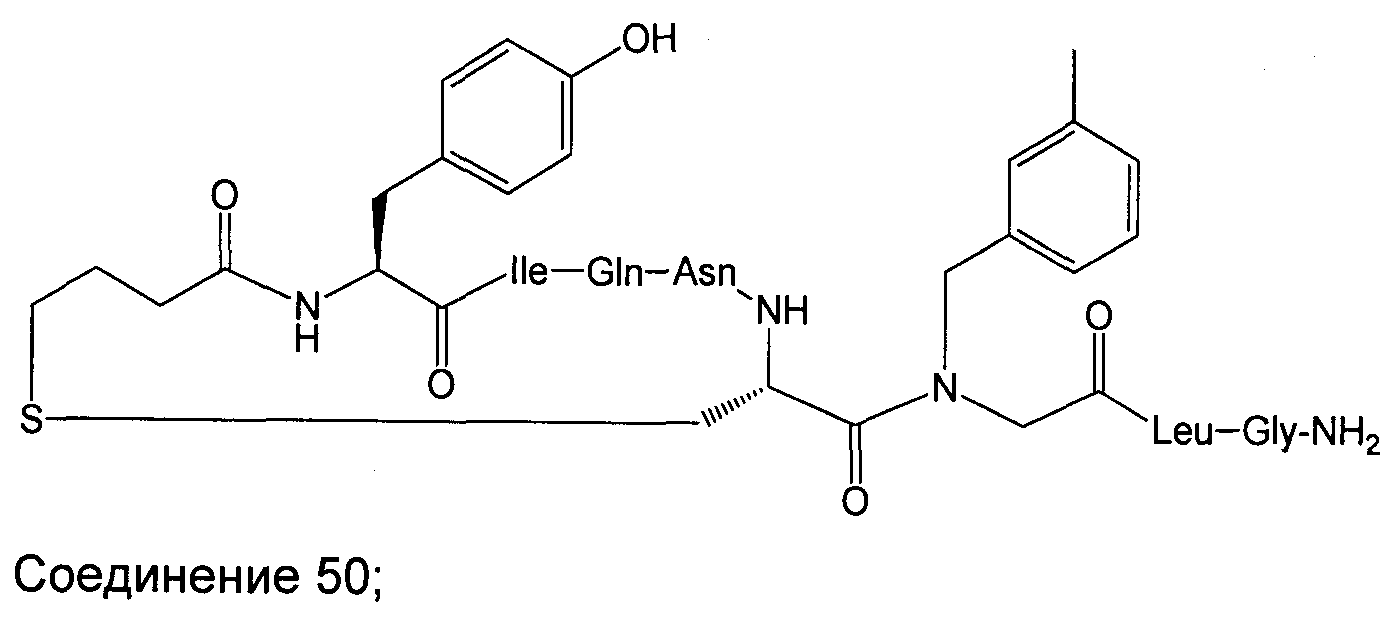
В наиболее предпочтительном воплощении изобретение представляет собой соединение, выбранное из группы, состоящей из:



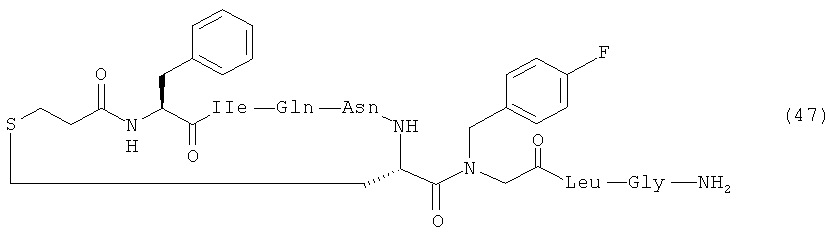
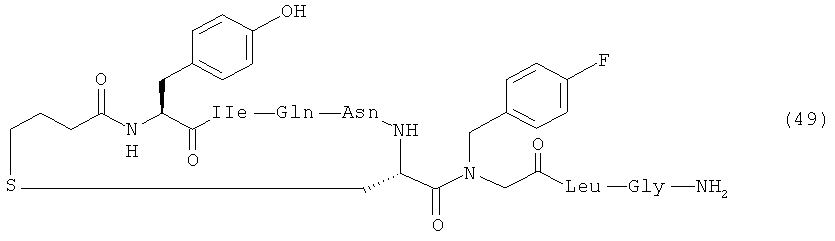
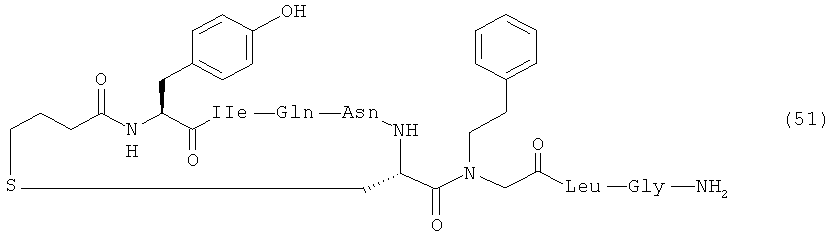
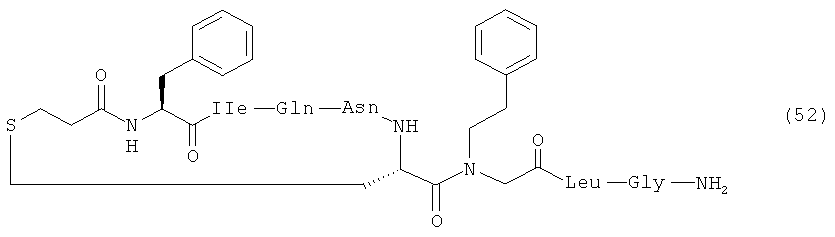
Кроме того, настоящее изобретение относится к соединению, как указано выше, для применения в качестве фармацевтического средства.
Соответственно, настоящее изобретение также относится к фармацевтической композиции, содержащей соединение, как указано выше, в качестве активного ингредиента вместе с фармацевтически приемлемым адъювантом, разбавителем или носителем.
Фармацевтическая композиция может быть адаптирована для перорального, внутривенного, местного, внутрибрюшинного, назального, трансбуккального, внутриглазного, внутриушного, подъязычного или подкожного введения или для введения через дыхательные пути, например, в форме аэрозоля или суспендированного в воздухе тонкоизмельченного порошка. Таким образом, композиция, например, может находиться в форме таблеток, капсул, порошков, микрочастиц, гранул, сиропов, суспензий, растворов, трансдермальных пластырей или суппозиториев.
Следует отметить, что композиция по настоящему изобретению может возможно включать два или более чем два вышеуказанных соединения.
Фармацевтическая композиция по настоящему изобретению может возможно содержать, например, по меньшей мере одну из дополнительных добавок, выбранных из разрыхлителя, связывающего вещества, смазывающего вещества, корригента, консерванта, красителя, а также любой их смеси. Примеры таких и других добавок можно найти в Handbook of Pharmaceutical Excipients'; Ed. A.H.Kibbe, 3rd Ed., American Pharmaceutical Association, USA and Pharmaceutical Press UK, 2000.
Фармацевтическая композиция по настоящему изобретению может быть адаптирована для назального введения. Она может содержать стерильный водный препарат соединений по изобретению, предпочтительно изотонический по отношению к крови реципиента. Этот водный препарат может быть изготовлен в соответствии с известными способами с использованием подходящих диспергирующих или увлажняющих агентов и суспендирующих агентов. Назальный препарат в виде спрея SYNTOCINON® (окситоцин) представляет собой пример подходящего фармацевтического препарата, применимого также и для раскрытых здесь заявленных соединений. Вода, раствор Рингера и изотонический раствор хлорида натрия представляют собой примеры подходящих растворителей. Препарат может также включать эксципиенты, такие как фосфат натрия, лимонную кислоту, хлорид натрия, глицерин, раствор сорбита, метилпарабен, пропилпарабен и хлорбутанол.
Дополнительно настоящее изобретение относится к применению соединения, как указано выше, для лечения или для изготовления лекарственного средства для лечения одного или более чем одного медицинского состояния, такого как нарушения лактации, нарушение индукции родовой деятельности; состояния атонии матки, чрезмерное кровотечение, воспаление и боль, включая внутрибрюшинную и боль в спине; сексуальная дисфункция у мужчин и женщин, синдром раздраженного кишечника (IBS), запор и желудочно-кишечная непроходимость, аутизм, стресс, тревога (включая тревожное расстройство) и депрессии; потеря крови при операции, для контроля послеродового кровотечения, заживления ран и инфекции, мастит и отхождение плаценты, а также остеопороз; а также для диагностики рака и плацентарной недостаточности. Здесь термин тревога включает тревожное расстройство. Тревожное расстройство включает вторичные показания генерализованного тревожного расстройства, панического расстройства, агорафобии, фобий, социального тревожного расстройства, обсессивно-компульсивного расстройства, посттравматического стрессового расстройства и тревожного расстройства, вызванного разлукой.
В еще одном воплощении изобретение относится к способу лечения нарушений лактации, нарушения индукции родовой деятельности; состояний атонии матки, чрезмерного кровотечения, воспаления и боли, включая внутрибрюшинную и боль в спине; сексуальной дисфункции у мужчин и женщин, синдрома раздраженного кишечника (IBS), запора и желудочно-кишечной непроходимости, аутизма, стресса, тревоги (включая тревожное расстройство) и депрессии; потери крови при операции, для контроля послеродового кровотечения, заживления ран и инфекции, мастита и отхождения плаценты, а также остеопороза; а также для диагностики рака и плацентарной недостаточности.
Типичные дозы соединений в соответствии с настоящим изобретением варьируют в широком диапазоне и будут зависеть от различных факторов, таких как индивидуальные потребности каждого пациента и способ введения. Врач или специалист в данной области техники способен оптимизировать дозу под конкретную ситуацию.
Например, если композиция по изобретению служит для усиления начала и поддержания лактации (например для интраназального введения), то обычная доза может находиться в диапазоне от 0,05 до 1,0 мкг/кг массы организма для каждой молокоотсасывающей сессии (breast pumping session). Интраназальная доза может быть разделена на, например, 1, 2 или 3 субдозы (например, впрыск), например, доставляемые в одну или обе ноздри при необходимости. Специалист в данной области техники или врач может учитывать значимые изменения для этого диапазона доз и практические обстоятельства для того, чтобы адаптироваться к конкретной ситуации.
В еще одном примере композицию по изобретению можно вводить в виде внутривенной (iv) инфузии, например, для лечения послеродового кровотечения или кровопотери при операции. В этом примере композицию можно вводить в течение более долгого периода. Например, доза для введения путем внутривенной инфузии составляет 0,5-200 мкг/кг массы организма в час.
В еще одном примере композиция по изобретению может быть предназначена для подкожного (sc), интраназального или трансбуккального введения, например для лечения тревожного расстройства или депрессии. Пример дозы для подкожного (sc), интраназального или трансбуккального введения составляет 0,5-1000 мкг/кг массы организма. Дозу можно, например, вводить столько раз в сутки, сколько необходимо, например один или два раза в сутки.
Используемые сокращения:
|
Если не указано иное, то использовали указанные L-аминокислоты, и придерживались общепринятой терминологии по аминокислотам.
Экспериментальная часть (синтез)
Производные аминокислот и смолы приобретены у коммерческих поставщиков (Bachem, Novabiochem and Peptides International). N-Fmoc-N-(R2(CH2)p)глицин, Fmoc-Cys(трет-бутоксикарбонилпропил)-OH и Fmoc-Hcy(трет-бутоксикарбонилэтил)-OH синтезировали в соответствии с литературой [Weber et al., J. Med. Chem., 46 1918 (2003), Prochazka et al. Collect. Czech. Chem. Commun., 51, 1335 (1992) и Wisniewski et al. в WO 03/072597]. Другие реактивы и растворители получены из Sigma-Aldrich, Fluka и Acros Organics.
Соединения синтезировали в соответствии со стандартными для твердофазной пептидной химии способами с использованием и Fmoc и Boc методологии. Все сочетания Fmoc-защищенных аминокислот опосредованы DIC/HOBt/DMF, а все сочетания Boc-защищенных аминокислот опосредованы DIC или DCC в DCM. Удаление Fmoc группы осуществляли с 20% пиперидином в DMF, а удаление Вос группы осуществляли в 50% TFA/DCM с 1% мета-крезолом в течение 5 и 25 минут. Необходимые промывки смол проводили DCM, IPA, DMF и MeOH. Нейтрализацию, при необходимости, осуществляли с 2 промывками смол 10% TEA/DCM в течение 5 минут.
Если не указано иное, то все взаимодействия осуществляли при комнатной температуре. В дополнение к цитируемым выше источникам, следующая литература, на которую обычно ссылаются, предоставляет дополнительное руководство по общей постановке эксперимента, а также по доступности требуемых исходных веществ и реагентов.
Kates, S.A., Albericio, F., Eds., Solid Phase Synthesis: A Practical Guide, Marcel Dekker, New York, Basel, 2000;
Stewart, J.M., Young, J.D., Solid Phase Synthesis, Pierce Chemical Company, 1984;
Bisello, et al., J. Biol. Chem. 1998, 273, 22498-22505 и
Merrifield, J. Am. Chem. Soc. 1963, 85, 2149-2154.
Чистоту синтезированного пептида определяли с помощью аналитической обращенно-фазовой HPLC. Структурную целостность пептидов подтверждали с использованием аминокислотного анализа и масс-спектрометрии с распылением электронов.
Fmoc и Boc способы использовали для синтеза дипептида, связанного со смолой в 8 позиции (Leu) и 9 позиции (Gly).
Аминокислотное производное в 7 позиции аминокислотного остатка вводили при помощи одного из двух путей: бромуксусную кислоту сочетали с дипептидом, связанным со смолой в условиях DIC/HOBt/DMF, и атом брома замещали на (R2(CH2)p)NH2 с получением связанного со смолой N-(R2(CH2)p)глицина, или N-Fmoc-N-(R2(CH2)p)глицин, или производное Fmoc-pro-OH сочетали со связанным со смолой дипептидом в соответствии с Fmoc способом. Если не указано иное, то все последующие сочетания аминокислот проводили в соответствии с Fmoc способом.
Аминокислотное производное, введенное в 6 позицию, представляло собой одно из: Fmoc-Cys(Trt)-OH, Fmoc-Hcy(трет-бутоксикарбонилэтил)-OH или Fmos-Cys(трет-бутоксикарбонилпропил)-OH. Пептидные аналоги, где позиция 6 представляла собой Fmoc-Cys(Trt)-OH, требовали сочетания Mpa(Trt)-OH с N-концом остатка, состоящего из девяти аминокислот, связанного со смолой.
Пептиды, синтезированные с использованием носителя со смолой Rink Amide, отщепляли от смолы вместе с любой из кислотных лабильных защитных групп, таких как Boc, тритил и трет-бутил, с использованием раствора TFA/TIS/H2O 95/2,5/2,5 (об./об./об.). Указанные пептиды подвергали циклизации после отщепления пептида от смолы. Пептиды, синтезированные с использованием носителя на основе смолы МВНА, отщепляли со смолы при помощи раствора HF/анизол 14/1 (об./об.). Указанные пептиды подвергали циклизации перед отщеплением пептида со смолы.
Циклизацию линейного нонапептида путем образования дисульфидной (кольцевой) связи осуществляли путем окисления линейных пептидов, растворенных в 10% TFA (водн.), йодом. Циклизацию линейного нонапептида путем образования амидной связи осуществляли при помощи HBTU/DIPEA/DMF или PyBOP/DIPEA/DMF с большим разбавлением.
Пептиды очищали при помощи препаративной HPLC в триэтиламмониевых фосфатных буферах (водн.) и обессаливали при помощи буферной системы уксусная кислота (водн.)/ацетонитрил. Фракции, имеющие чистоту больше 97%, объединяли и лиофилизировали.
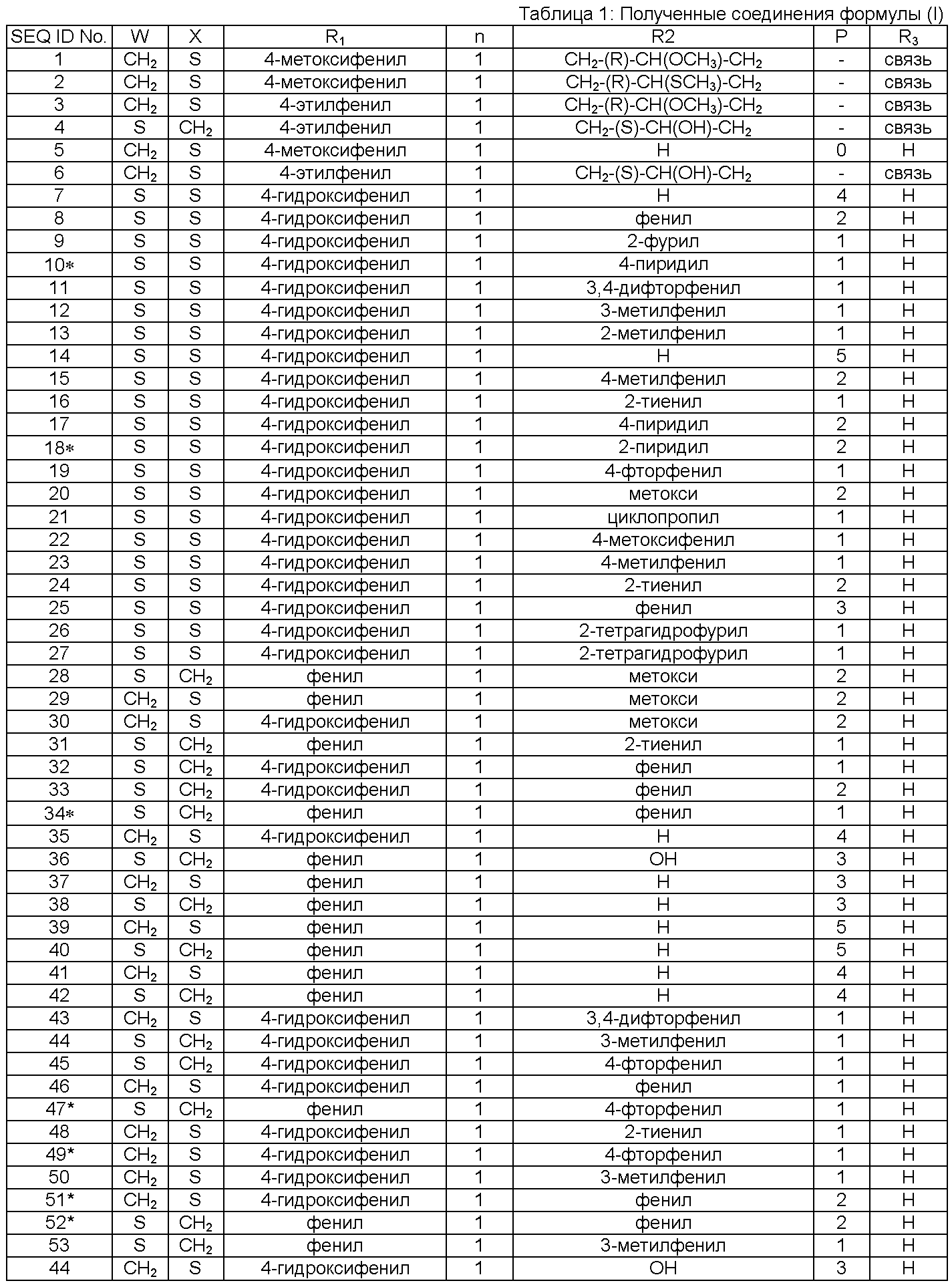
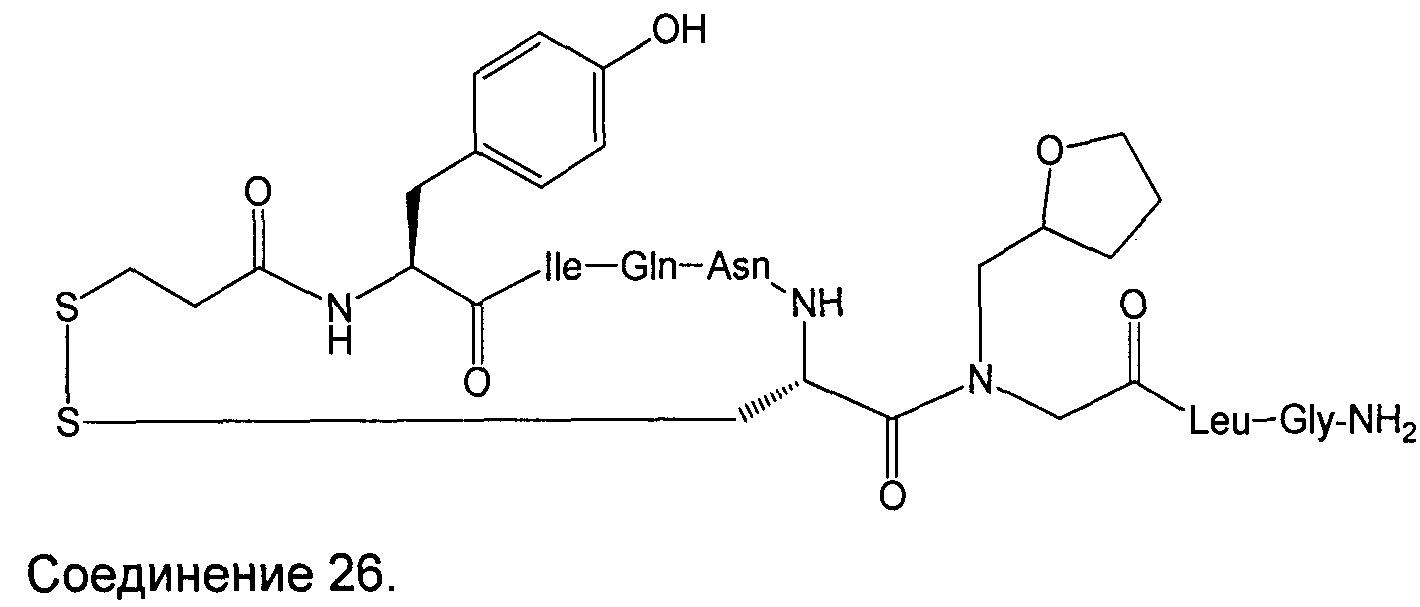
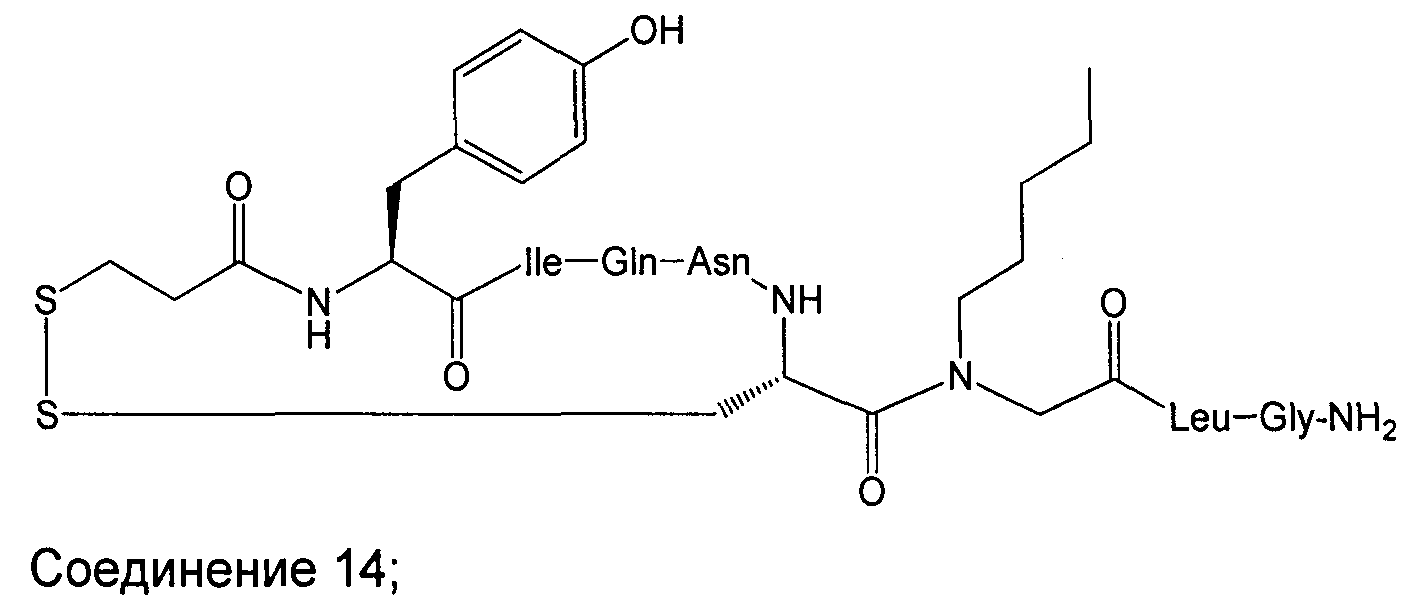
В Таблице 1 приведены соединения, полученные при помощи вышеприведенного способа. Звездочка '∗' обозначает наиболее предпочтительные воплощения.
Следующие подробные примеры приведены для того, чтобы дополнительно проиллюстрировать синтез:
Во всех синтезах аналитическую HPLC осуществляли на жидкостном хроматографе waters 600 с использованием колонки Vydac C18, 5 мкм, 4,6×250 мм при скорости потока 2 мл/мин. Препаративную HPLC осуществляли на жидкостном хроматографе Waters 2000 с использованием картриджа PrePak 47×300 мм при скорости потока 100 мл/мин. Окончательный анализ соединения осуществляли на жидкостном хроматографе 1100 Agilent с использованием колонки Vydac С18, 5 мкм, 2,1×250 мм при скорости потока 0,3 мл/мин. Масс-спектры регистрировали на спектрометре Finnigan МАТ.
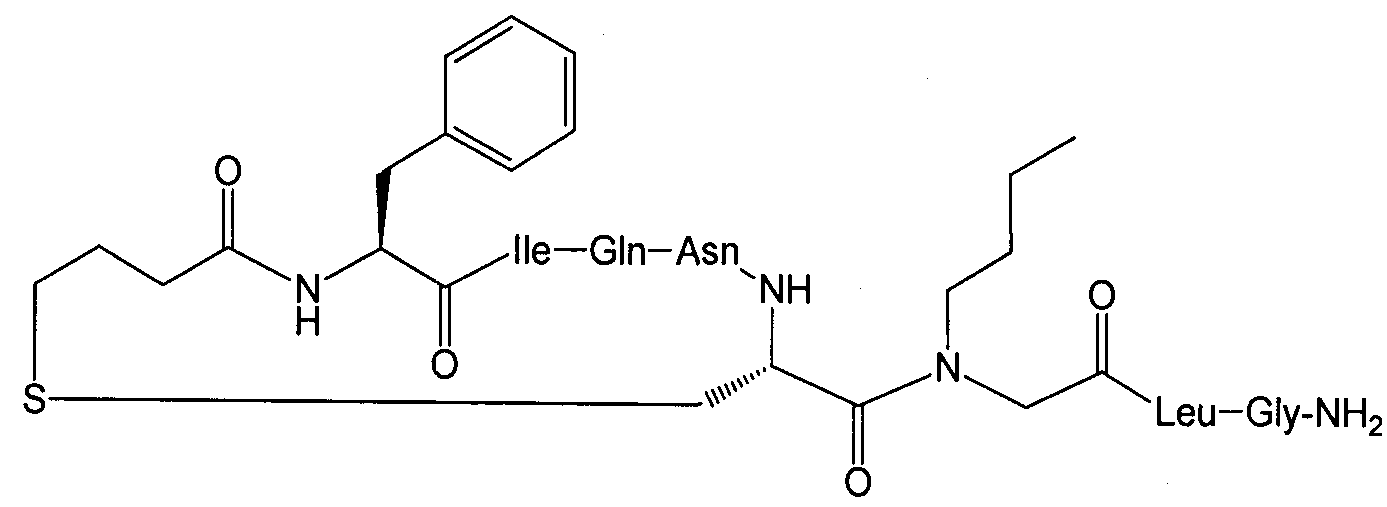
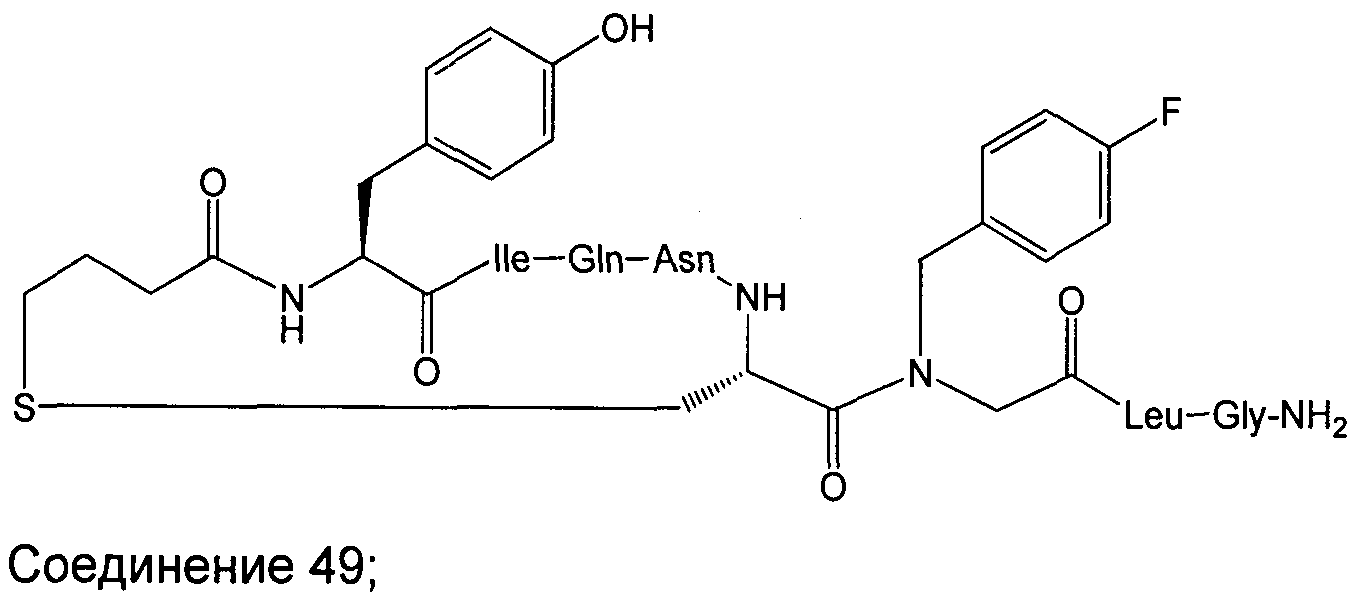
Соединение 49; Kapбo-1-[4-FBzlGly7]dOT:
Используемые аминокислотные производные представляли собой Fmoc-Gly-OH, Fmoc-Leu-OH, Fmos-Cys(трет-бутоксикарбонилпропил)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-IIe-OH и Boc-Tyr(fBu)-OH (Peptides International). Fmos-Cys(трет-бутоксикарбонилпропил)-OH синтезировали, как указано выше.
Полностью защищенную пептидную смолу синтезировали вручную, начиная с 1,45 г (0,87 ммоль) смолы Rink Amide AM (200-400 меш, Novabiochem). Осуществляли опосредованные DIC/HOBt/DMF единичные сочетания с 3-кратным избытком производных аминокислот Gly и Leu. Остаток N-(4-фторбензил)глицина вводили с 4-кратным избытком BrCH2CO2H/DIC/HOBt в DMF и затем замещали бром 10-кратным избытком 4-фторбензиламина в DMF. Осуществляли опосредованное DIC/DCM сочетание с 4-кратным избытком Fmos-Cys(трет-бутоксикарбонилпропил)-OH. Осуществляли последующие опосредованные DIC/HOBt/DMF единичные сочетания с 3-кратным избытком производных аминокислот Asn, Gln, IIe и Tyr. Группы Fmoc удаляли с 20% пиперидином в DMF. После завершения твердофазного синтеза смолу обрабатывали раствором TFA/TIS/H2O 96/2,5/1,5 (об./об./об.) (50 мл) в течение 1,5 ч и отфильтровывали. Фильтрат концентрировали в вакууме, и неочищенный линейный пептид осаждали диэтиловым эфиром. Осадок в DMF (300 мл) добавляли 3 порциями (3×100 мл) к интенсивно перемешиваемому раствору DIPEA (1 мл) в DMF (100 мл). HBTU (150 мг) в DMF (5 мл) добавляли к реакционной смеси после добавления каждой 100 мл порции пептидного раствора; pH реакционного раствора поддерживали на уровне pH 9 путем добавления, при необходимости, чистого DIPEA. Реакцию контролировали при помощи аналитической HPLC. Реакционный раствор концентрировали в вакууме, и остаток растворяли в AcOH/CH3CN/H2O. Смесь наносили на колонку HPLC и очищали с использованием триэтиламмонийфосфатного буфера с pH 5,2. Соединение элюировали градиентом ацетонитрила. Фракции, чистота которых превышала 97%, объединяли, разбавляли водой (2 объема) и наносили на колонку, предварительно уравновешенную с 2% AcOH (водн.). Желаемое соединение элюировали быстрым (3%/мин) градиентом CH3CN. Фракции, содержащие желаемый продукт, объединяли и лиофилизировали. Получали 434 мг (выход приблизительно 40% на основе загрузки исходной смолы и допуская 85% содержание пептида) белого аморфного порошка. HPLC: Rt=19,4 мин, градиент: 5% В в течение 0,5 мин, 5→30% В в течение 0,5 мин, 30→50% B в течение 20 мин и 100% B в течение 5 мин, t=40°C, растворитель A 0,01% TFA (водн.), растворитель B 70% CH3CN, 0,01% TFA (водн.); Чистота: 99,3%; MS (M+H+): ожидали 1042,4, найдено 1042,5.
Далее приведен пример крупномасштабного (т.е. в увеличенном масштабе) синтеза Соединения 49; карбо-1-[4-FBzlGly7] dOT:
Используемые аминокислотные производные представляли собой Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-4-FBzlGly-OH, Fmoc-Cys(трет-бутоксикарбонилпропил)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-lle-OH и Boc-Tyr(tBu)-OH (Peptides International). Fmoc-4-FBzlGly-OH и Fmoc-Cys(трет-бутоксикарбонилпропил)-OH синтезировали, как указано выше. Пептид синтезировали при помощи опосредованных DIC/HOBt/DMF единичных сочетаний с 3-кратным избытком аминокислотного производного. Оставшийся синтез и определение характеристик соединения 49 осуществляли в соответствии с определенным выше. Получали 434 мг (приблизительно 40% выход, на основе загрузки исходной смолы и допуская содержание пептида 85%) белого аморфного порошка.
Соединение 10; [4-PicGly7]dOT:
Используемые аминокислотные производные представляли собой Fmoc-Gly-OH, Fmoc-Leu-OH, Fmoc-Cys(Trt)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-IIe-OH, Fmoc-Tyr(fBu)-OH и Mpa(Trt)-OH (Peptides International). Полностью защищенную пептидную смолу синтезировали вручную, начиная с 1,33 г (0,65 ммоль) смолы Rink AM (200-400 меш, Novabiochem). Осуществляли опосредованные DIC/HOBt/DMF единичные сочетания с 3-кратным избытком производных аминокислот Gly и Leu. Остаток N-(4-пиколил)глицина вводили с 4-кратным избытком BrCH2CO2H/DIC/HOBt в DMF и затем бром замещали 10-кратным избытком 4-пиколиламина в DMF. Осуществляли опосредованное DIC/DCM сочетание с 4-кратным избытком Fmoc-Cys(Trt)-OH и опосредованные DIC/HOBt/DMF единичные сочетания с 3-кратным избытком производных аминокислот Asn, Gin, IIe, Tyr и Mpa. Группы Fmoc удаляли с 20% пиперидином в DMF. После завершения твердофазного синтеза смолу обрабатывали раствором TFA/TIS/H2O 96/2/2 (об./об./об.) (50 мл) в течение 1,5 ч и отфильтровывали. Фильтрат концентрировали в вакууме, и неочищенный линейный пептид осаждали диэтиловым эфиром. Осадок в DMF (300 мл) добавляли 3 порциями (3×100 мл) к интенсивно перемешиваемому раствору DIPEA (1 мл) в DMF (100 мл). Осадок растворяли в чистом TFA (50 мл), выливали в перемешиваемый на магнитной мешалке 5% водный раствор ацетонитрила (600 мл), и пептид окисляли путем добавления 0,1 М I2 в метаноле до появления устойчивого желтого окрашивания. Избыток йода восстанавливали при помощи твердой аскорбиновой кислоты (Sigma-Aldrich), и pH раствора доводили до приблизительно 4 путем добавления концентрированного аммиака (водн.). Смесь наносили на колонку HPLC и очищали с использованием триэтиламмонийфосфатного буфера с pH 5,2. Соединение элюировали градиентом ацетонитрила. Фракции, чистота которых превышала 97%, объединяли, разбавляли водой (2 объема) и наносили на колонку, предварительно уравновешенную с 2% AcOH (водн.). Желаемое соединение элюировали быстрым (3%/мин) градиентом ацетонитрила. Фракции, содержащие желаемый продукт, объединяли и лиофилизировали. Получали 348,7 мг (выход приблизительно 44% на основе загрузки исходной смолы и допуская 85% содержание пептида) белого аморфного порошка. HPLC: Rt=21,7 мин, градиент: 5% В в течение 0,5 мин, 5→10% В в течение 0,5 мин, 10→30% B в течение 20 мин и 100% B в течение 5 мин, t=40°C, растворитель A 0,01% TFA (водн.), растворитель В 70% CH3CN, 0,01% TFA (водн.); Чистота: 99,9%; MS (M+H+): ожидали 1043,4, найдено 1043,5.
Соединение 29; карбо-6-[Phe2,MeOEtGly7] dOT:
Используемые аминокислотные производные представляли собой Boc-Gly-OH и Boc-Leu-OH (Bachem), Fmos-Hcy(трет-бутоксикарбонилэтил)-OH, Fmoc-Asn(Trt)-OH, Fmoc-Gln(Trt)-OH, Fmoc-IIe-OH и Boc-Phe-OH (Peptides International). Fmos-Hcy(трет-бутоксикарбонилэтил)-OH синтезировали, как указано выше.
Полностью защищенную пептидную смолу синтезировали вручную, начиная с 1,33 г смолы МВНА (0,94 ммоль, Novabiochem). Смолу нейтрализовали 10% TEA в DCM. Осуществляли опосредованные DIC/DCM единичные сочетания с 1,7-кратным избытком аминокислот Boc-Gly-OH и Boc-Leu-OH. Остаток N-(2-метоксиэтил)глицина вводили с 3,6-кратным избытком BrCH2CO2H/DIC/HOBt в DMF и затем бром замещали 7-кратным избытком 2-метоксиэтиламина и 4-кратным избытком DIPEA в DMF (10 мл). Реакционную смесь перемешивали в течение 5 ч. Осуществляли опосредованное DIC/DCM единичное сочетание с 4-кратным избытком Fmoc-Hcy(трет-бутоксикарбонилэтил)-OH и опосредованные DIC/HOBt/DMF единичные сочетания с 3-кратным избытком производных аминокислот Asn и Gin. Два завершающих единичных сочетания с Fmoc-lle-OH и Boc-Phe-OH осуществляли с DIC/DCM с получением желаемого защищенного связанного со смолой линейного пептида. Группы Fmoc с 20% пиперидином в DMF. Смолу обрабатывали TFA/H2O/TIS 95/3/2 (об./об./об.) в течение 2 ч для удаления тритильной, Boc, и трет-бутильной групп. BOP (4 экв.) и DIPEA (10 экв.) добавляли к перемешиваемой суспензии смолы в DMF (10 мл); через 2 ч PyBOP (2 экв.) и DIPEA (5 экв.) удаляли. Пептид отщепляли со смолы с использованием 70 мл безводного HF, содержащего 5 мл анизола, при 0°C в течение 90 мин. HF удаляли в вакууме, и неочищенный линейный пептид промывали диэтиловым эфиром (300 мл). Пептид растворяли в AcOH/CH3CN/H2O 1/2/7 (об./об./об.) (400 мл). Получающуюся в результате смесь наносили на колонку HPLC и очищали с использованием триэтиламмонийфосфатного буфера с pH 2,3. Соединение элюировали градиентом ацетонитрила. Фракции, чистота которых превышала 97%, объединяли, разбавляли водой (2 объема) и наносили на колонку, предварительно уравновешенную с 2% уксусной кислоты (водн.). Желаемое соединение элюировали 1% градиентом AcOH/CH3CN. Фракции, содержащие желаемый продукт, объединяли и лиофилизировали.
Получали 292,7 мг (выход приблизительно 27% на основе загрузки исходной смолы и допуская 85% содержание пептида) белого аморфного порошка. HPLC: Rt=16,7 мин, градиент: 5% В в течение 0,5 мин, 5→30% B в течение 0,5 мин, 30→50% В в течение 20 мин и 100% В в течение 5 мин, t=40°C, растворитель A 0,01% TFA (водн.), растворитель В 70% CH3CN, 0,01% TFA (водн.); Чистота: 100,0%; MS (M+H+): ожидали 976,5, найдено 976,5.
Другие соединения получали путем аналогичных изменений этих процедур синтеза.
Экспериментальный пример (биологическое тестирование)
Анализы рецептора in vitro:
Агонистическую активность соединений в отношении рецептора hOT определяли в анализе транскрипции гена-репортера путем временной трансфекции ДНК, экспрессирующей рецептор hOT, в линию клеток яичника китайского хомячка (CHO) вместе с ДНК-репортером, содержащей внутриклеточные чувствительные к кальцию промотерные элементы, регулирующие экспрессию люциферазы светлячка. Смотри Boss, V., Talpade, D.J., Murphy, T.J. J. Biol. Chem. 1996, May 3; 271(18), 10429-10432 в отношении дополнительного руководства по этому анализу. Клетки подвергали серийным разведениям соединений, разведенным в 10 раз на дозу, в течение 5 ч, а затем клетки лизировали, определяли активность люциферазы и эффективности соединения и значения ЕС50 (средней эффективной концентрации) при помощи нелинейной регрессии.
Окситоцин (ОТ) использовали в качестве внутреннего контроля в каждом эксперименте, и соединения тестировали по меньшей мере в трех независимых экспериментах. Для определения селективности соединения дополнительно тестировали в основанных на люциферазе анализах транскрипции гена-репортера, экспрессирующего человеческий рецептор вазопрессина (hV2).
Для дополнительного сравнения карбетоцин также использовали в качестве референсного соединения.
Результаты анализов in vivo представлены в Таблице 2 ниже. Приведенное значение ЕС50 представляет собой среднее геометрическое, выраженное в наномоль/л (нМ). Значения селективности приведены как отношения ЕС50.
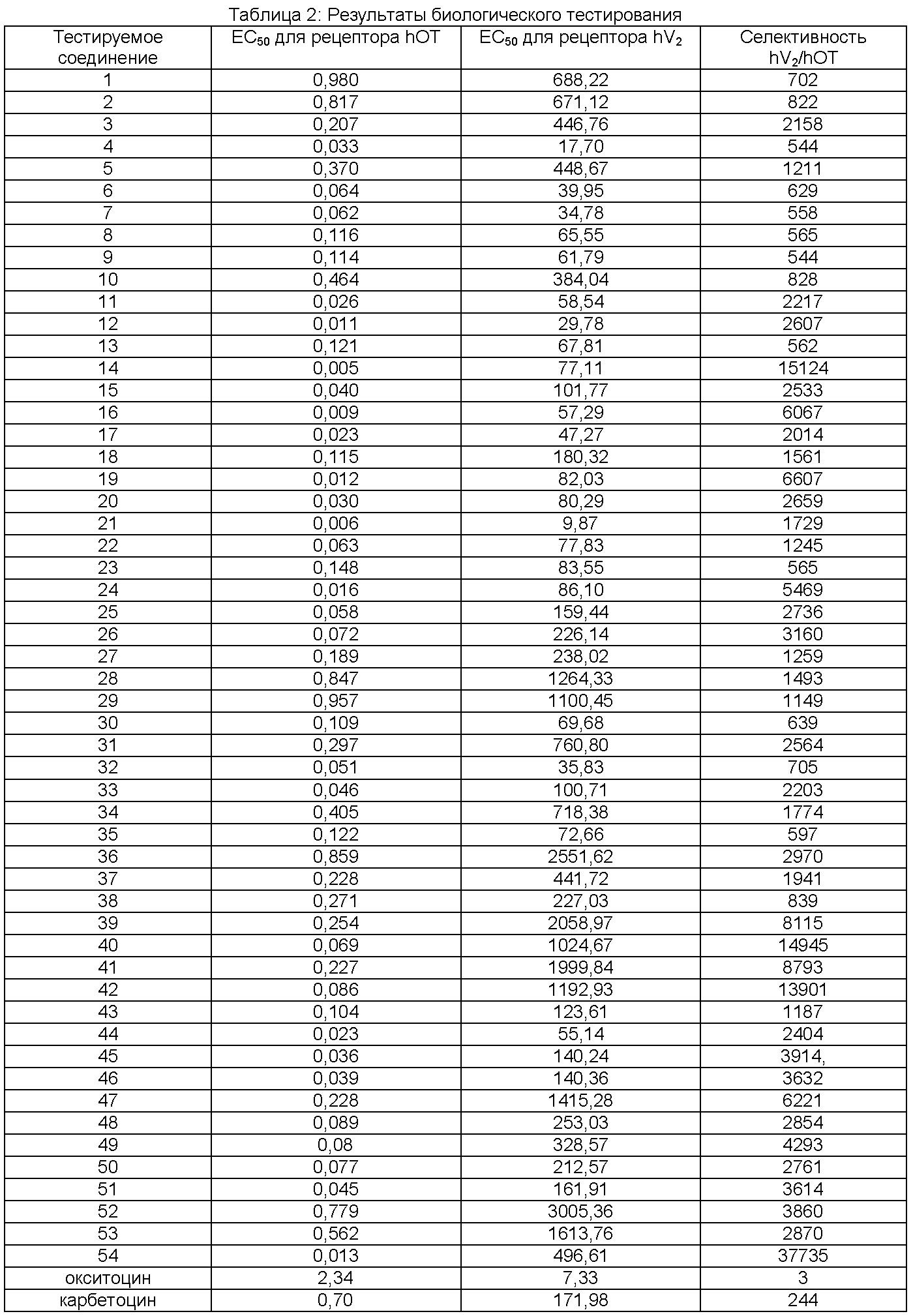
Вышеприведенные результаты свидетельствуют о том, что соединения Примеров находятся в объеме изобретения и могут, например, быть полезны для безопасного и эффективного лечения людей для того, чтобы вызвать родовую активность, контролировать маточную атонию, стимулировать и поддерживать лактацию и т.д.
Объем настоящего изобретения дополнительно определен в следующей формуле изобретения.
Композиция для лечения рака предстательной железы
Агонисты окситоциновых рецепторов
Саше месалазина с высокой нагрузкой лекарства
Пептидные агонисты glp-2
Быстрорастворимая фармацевтическая композиция
Быстрорастворимая фармацевтическая композиция
Стабилизация fsh
Гранулы для фармацевтических препаратов, способы и аппарат для их получения
Интравагинальное введение мизопростола
Фармацевтический препарат, содержащий рекомбинантный хорионический гонадотропный гормон человека
Композиция для лечения рака предстательной железы
Агонисты окситоциновых рецепторов
Саше месалазина с высокой нагрузкой лекарства
Пептидные агонисты glp-2
Быстрорастворимая фармацевтическая композиция
Быстрорастворимая фармацевтическая композиция
Стабилизация fsh
Гранулы для фармацевтических препаратов, способы и аппарат для их получения
Интравагинальное введение мизопростола
Фармацевтический препарат, содержащий рекомбинантный хорионический гонадотропный гормон человека

