Результат интеллектуальной деятельности: СПОСОБ ПОЛУЧЕНИЯ СОЛЕЙ ТЕТРАЗОЛМЕТАНСУЛЬФОНОВОЙ КИСЛОТЫ И НОВОЕ СОЕДИНЕНИЕ, ИСПОЛЬЗУЕМОЕ В НЕМ
Вид РИД
Изобретение
Область, к которой относится изобретение
Настоящее изобретение относится к способу получения солей тетразолметансульфоновой кислоты и к новому соединению, используемому в нем.
Предпосылки изобретения
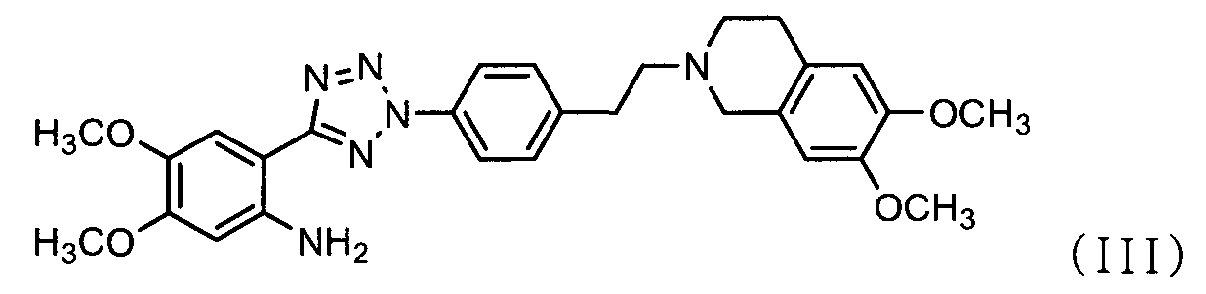
Фармацевтически приемлемые соли 4-оксо-4Н-хромен-2-карбоновой кислоты [2-(2-{4-[2-(6,7-диметокси-3,4-дигидро-1Н-изохинолин-2-ил)этил]фенил}-2Н-тетразол-5-ил)-4,5-диметоксифенил]амина в качестве ингибитора P-гликопротеина могут применяться в качестве ингибитора при устойчивости к множеству лекарственных средств. В публикации PCT № WO 2005/033097 описан способ их получения.
В соответствии с указанной публикацией, как показано в схемах реакции 1 и 2, нитросоединения (1 и 3) подвергаются гидрогенизации в растворителе, таком как метанол, этанол, хлороформ, дихлорметан, тетрагидрофуран, простой этиловый эфир и гексан толуол, в присутствии металлического катализатора, такого как палладий, платина и цинк, с получением амино-соединений (2 и 4). Затем полученное соединение подвергают ацилированию с использованием конденсирующего агента, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимид гидрохлорид, Ν,Ν-дициклогексилдиимид, Ν,Ν-диизопрокарбодиимид и метил-п-толуолсульфонат 1-циклогексил-3-(2-морфолинэтил)карбодиимида, в присутствии катализатора, такого как 4-(диметиламино)пиридин, в растворителе, таком как дихлорметан, хлороформ, Ν,Ν-диметилформамид, тетрагидрофуран и 1,4-диоксан, с получением соединения тетразола (5) в виде целевого продукта.
Схема реакции 1
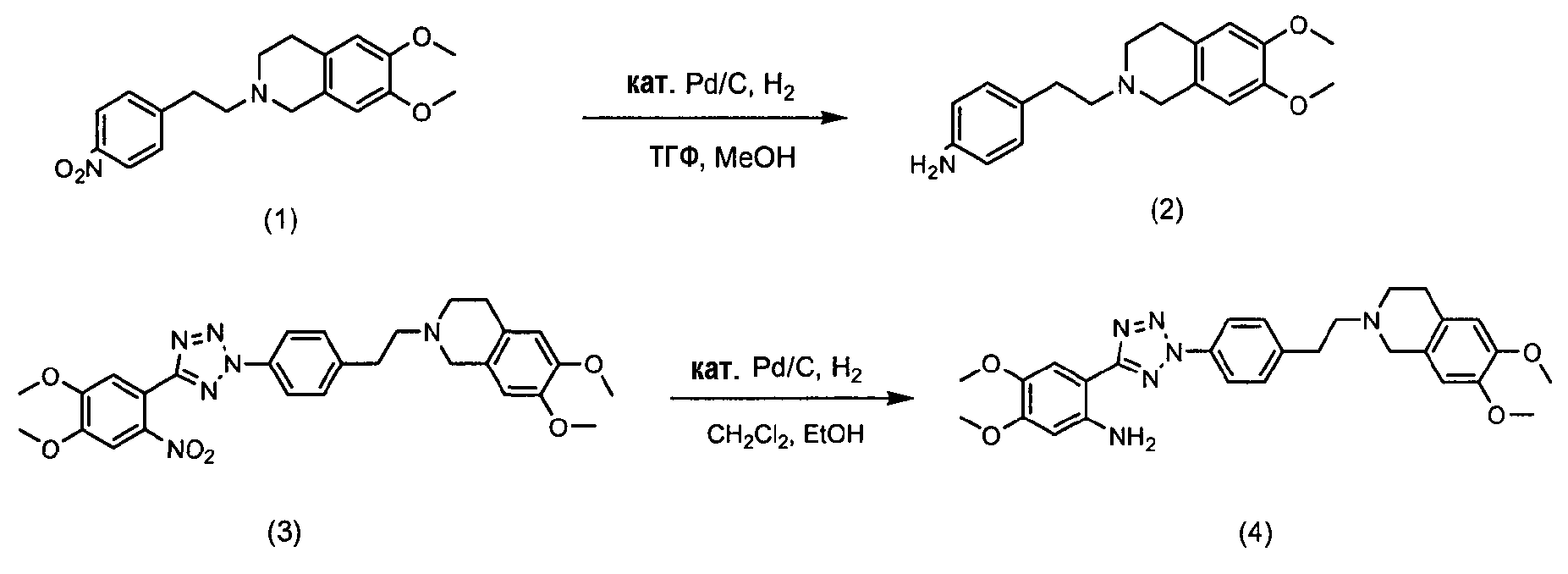
Схема реакции 2
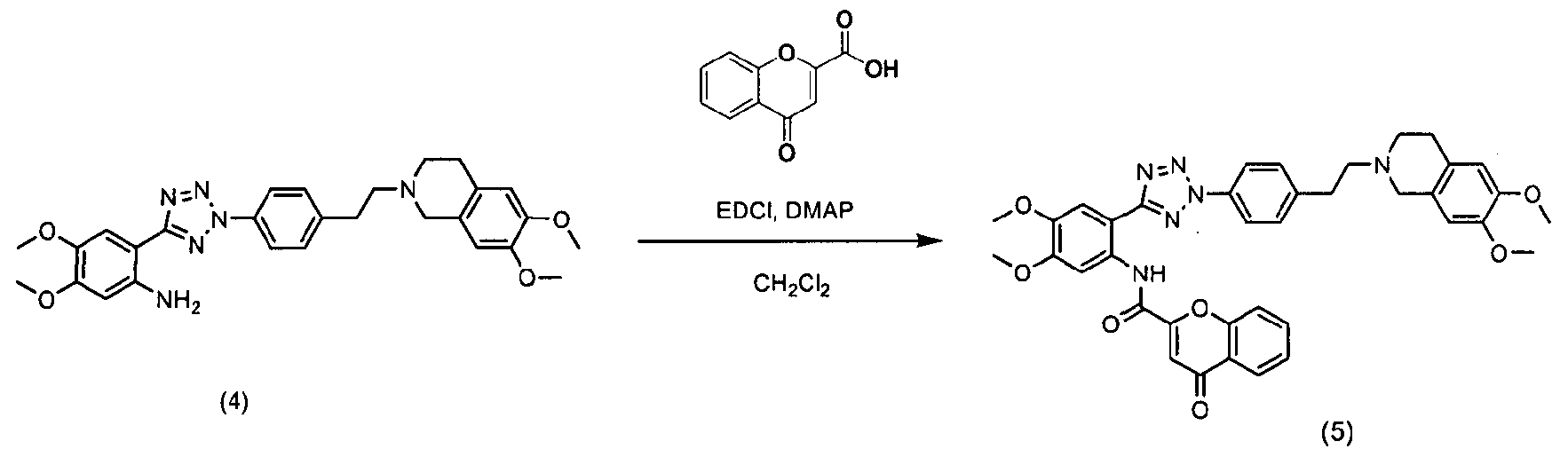
Однако обычный способ может вызвать риск таких опасностей, как взрыв и пожар, в связи с использованием водорода и металлического катализатора при крупномасштабном производстве. Он также требует процесса очистки с использованием колоночной хроматографии на силикагеле для отделения чистого тетразольного соединения, что нереально для крупномасштабного производства, поскольку имеются ограничения в отношении размера колонки и количества загружаемого материала при колоночной хроматографии. Кроме того, процесс хроматографии требует высоких рабочих затрат ввиду дорогостоящего материала набивки колонки, силикагеля и большого количества элюента, используемых для процесса.
Краткое изложение сущности изобретения
Соответственно, целью настоящего изобретения является способ получения солей тетразолметансульфоновой кислоты.
Другой целью настоящего изобретения является получение нового соединения, которое может использоваться для получения соли тетразолметансульфоновой кислоты, и способа ее получения.
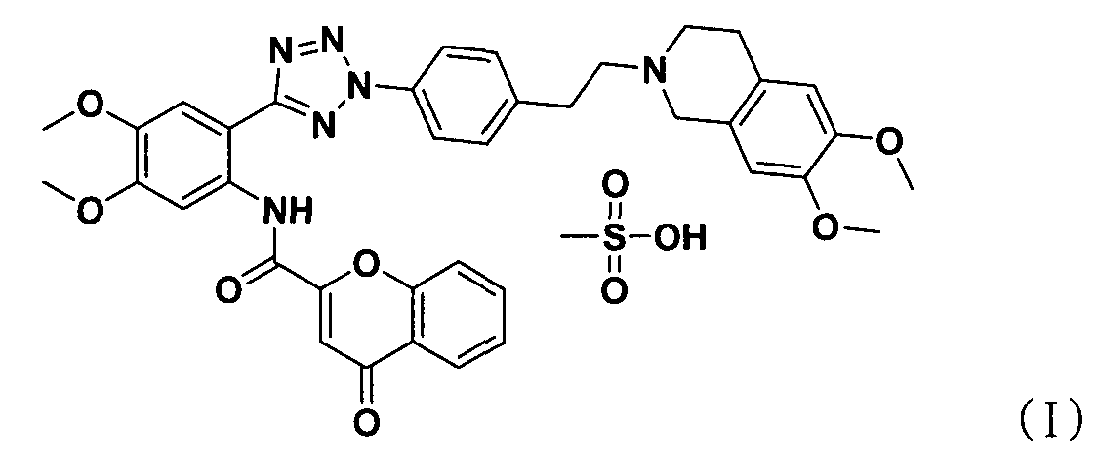
В соответствии с одним аспектом настоящего изобретения изобретение относится к способу получения соли тетразолметансульфоновой кислоты формулы (I), который включает стадии:
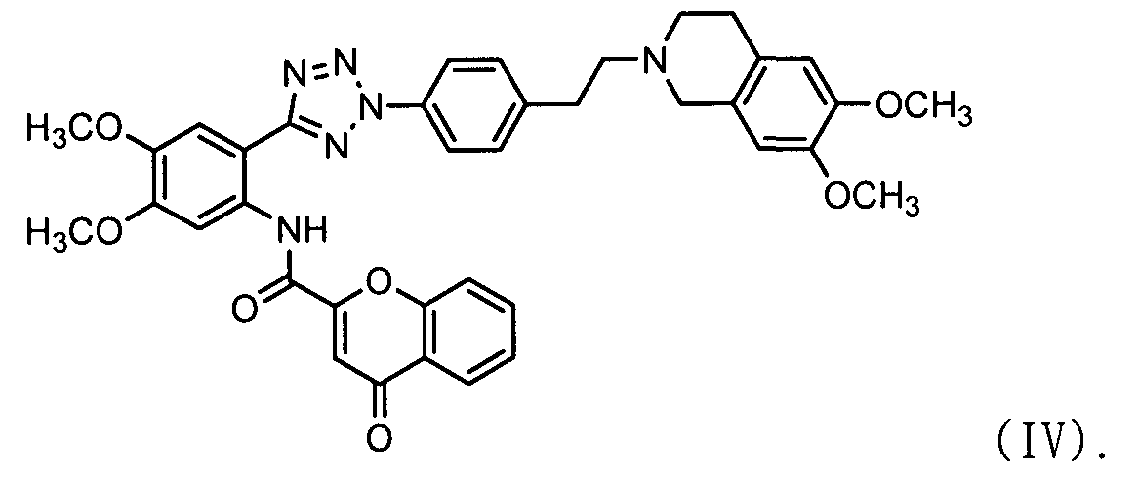
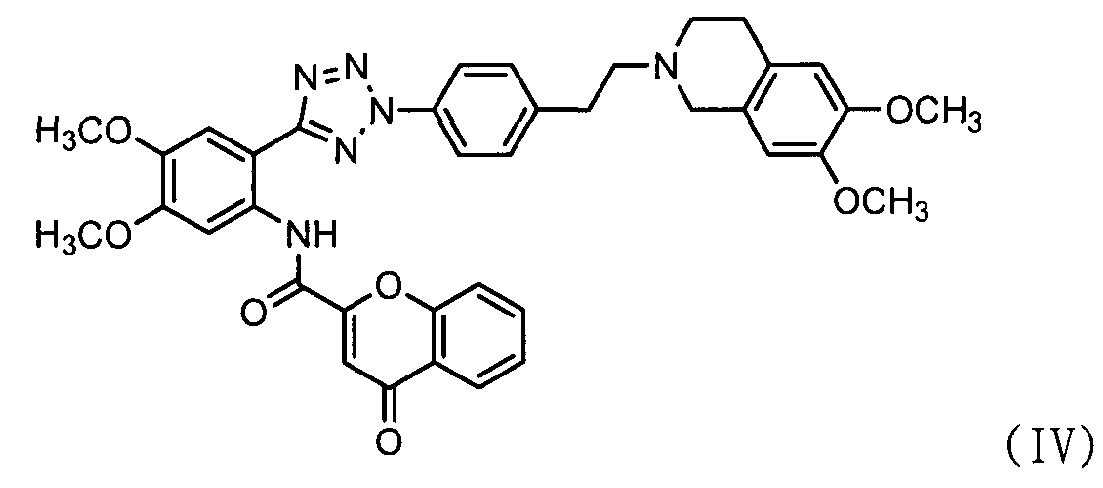
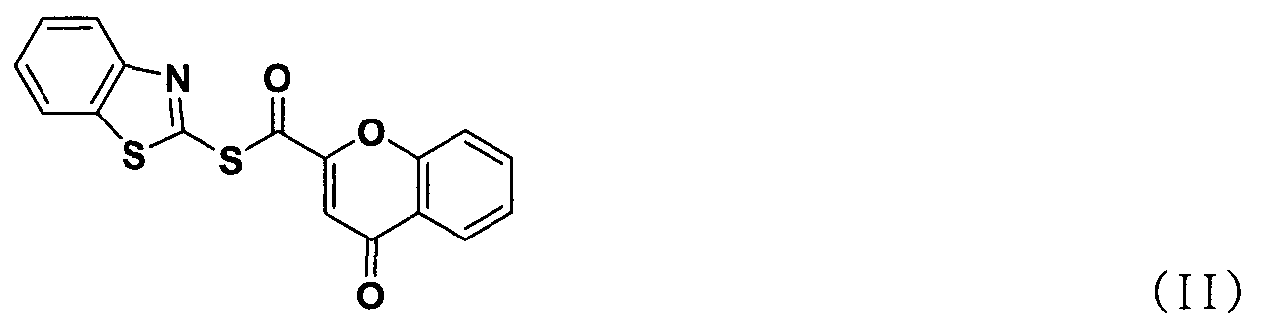
ацилирования соединения формулы (II) соединением формулы (III) с получением соединения формулы (IV); и
добавления метансульфоновой кислоты к соединению формулы (IV).
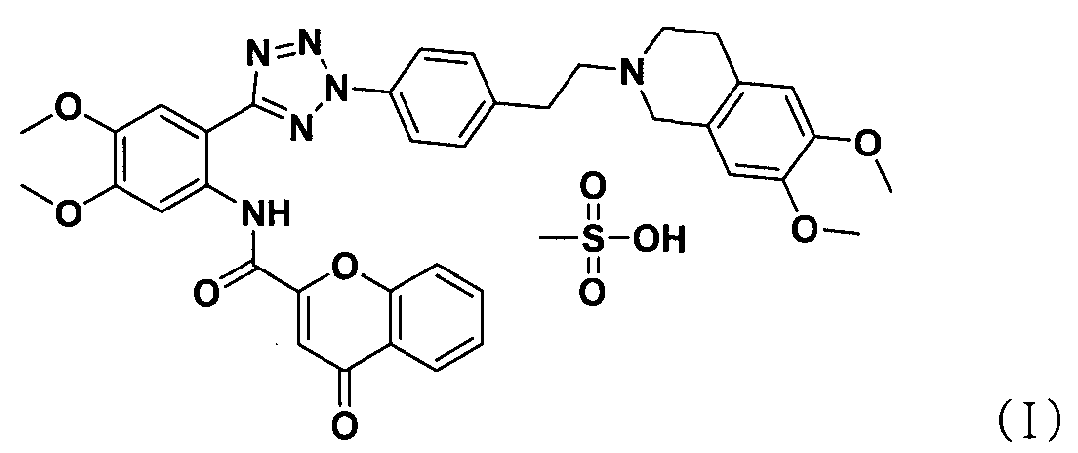
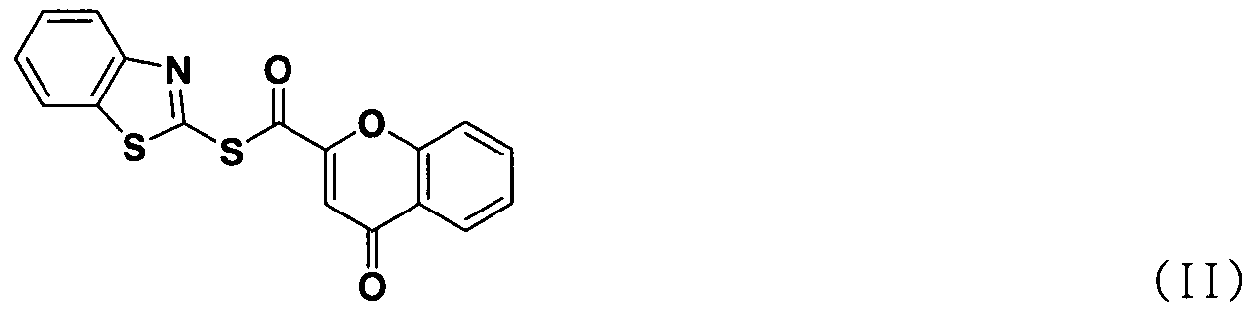
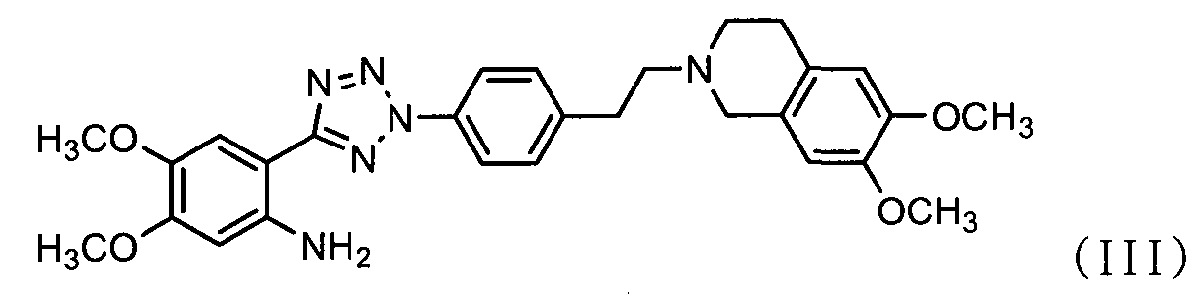
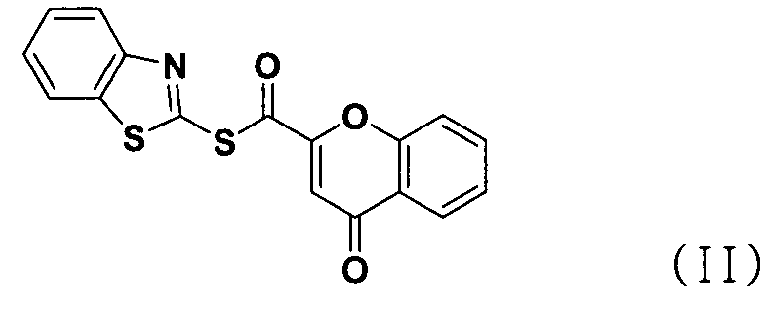
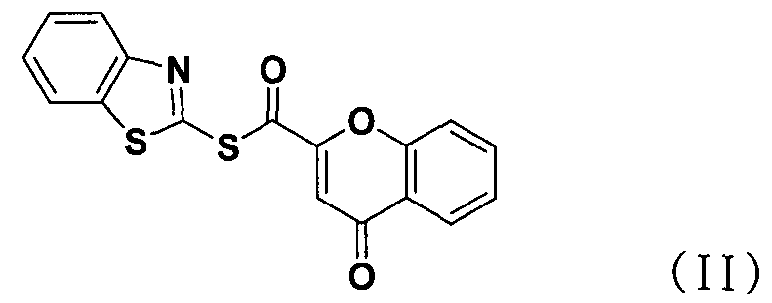
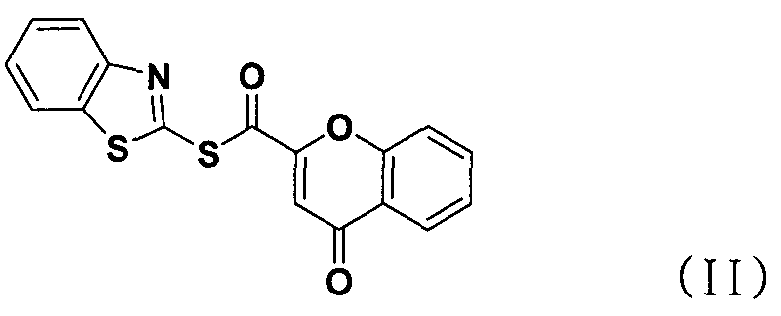
В соответствии с другим аспектом настоящее изобретение относится к соединению формулы (II), которое может использоваться для получения соли тетразолметансульфоновой кислоты.
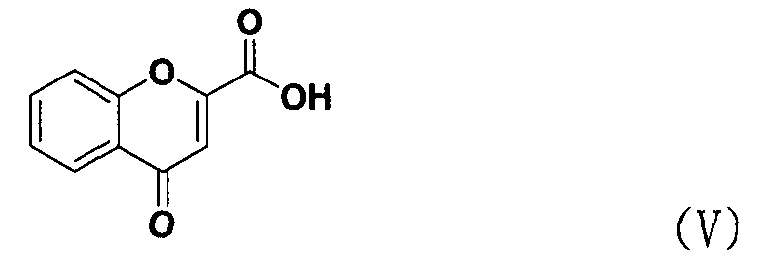
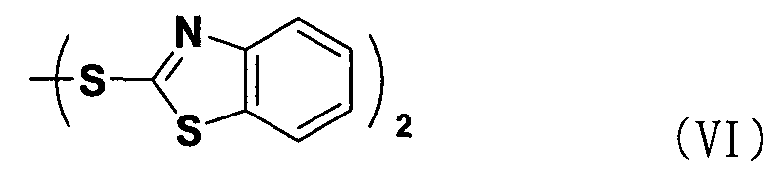
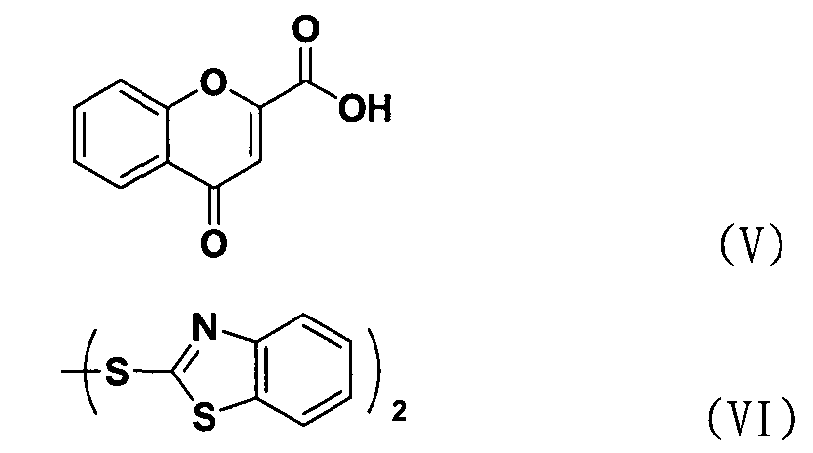
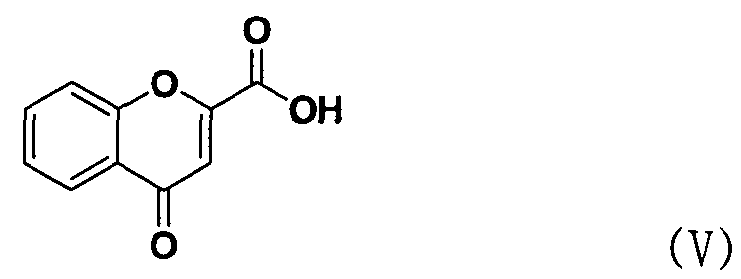
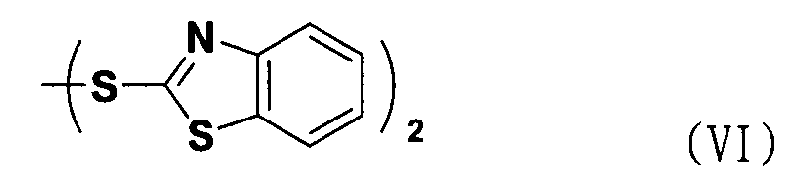
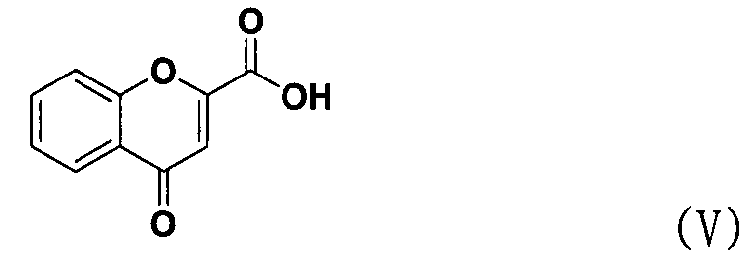
В соответствии с еще одним аспектом настоящее изобретение относится к способу получения соединения формулы формуле (II), включающему стадию взаимодействия соединения формулы (V) с соединением формулы (VI) в присутствии трифенилфосфина и основания.
Подробное описание изобретения
Далее настоящее изобретение будет описано более подробно.
В способе получения в соответствии с настоящим изобретением используется процесс ацилирования с использованием нового соединения, сложного S-бензотиазол-2-илового эфира 4-оксо-4Н-хромен-2-карботионовой кислоты вместо использования конденсирующего агента, как в обычных способах, для получения солей тетразолметансульфоновой кислоты с высокой чистотой при высоком выходе, без дополнительного процесса очистки, такого как колоночная хроматография.
Как показано ниже в схеме реакции 3, способ получения солей тетразолметансульфоновой кислоты в соответствии с настоящим изобретением включает: (Стадию 1) ацилирование соединения формулы (II) соединением формулы (III) с получением соединения формулы (IV); и (Стадию 2) добавление метансульфоновой кислоты к соединению формулы (IV), полученному на предыдущей стадии.
Схема реакции 3
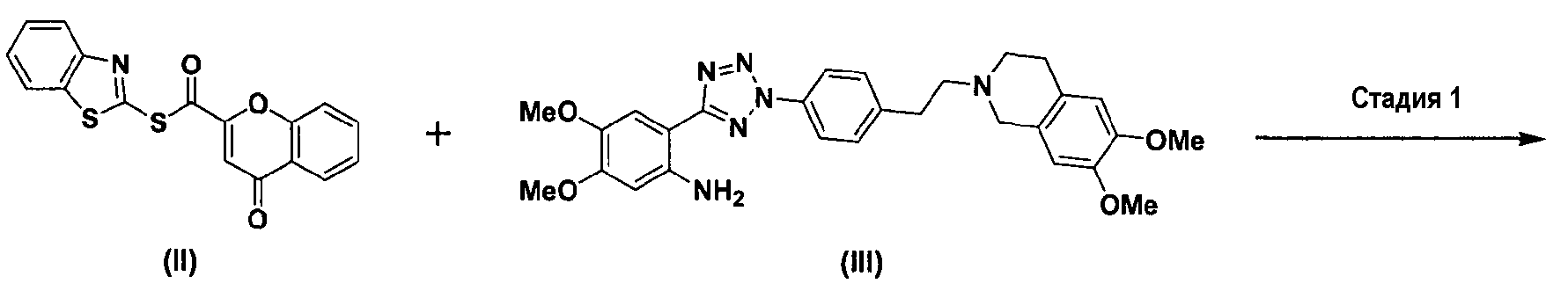
где Me представляет метильную группу.
Во-первых, соединения формул (II) и (III) подвергаются ацилированию в полярном апротонном растворителе с получением соединения формулы (IV).
В частности, сложноэфирное соединение формулы (II) и соединение формулы (III) подвергаются ацилированию в полярном апротонном растворителе, выбранном из группы, состоящей из дихлорметана, тетрагидрофурана, сложного этилового эфира, ацетона, N,N-диметилформамида, ацетонитрила, диметилсульфоксида и их смеси, предпочтительно дихлорметана. После завершения ацилирования, к нему добавляется метанол для деактивации остающегося соединения формулы (II). Затем к нему добавляют ацетон для очистки с тем, чтобы можно было естественно и эффективно обеспечить выход соединения формулы (IV) с чистотой 98% или более.
При ацилировании соединение формулы (II) предпочтительно используется в количестве от 1 до 5 эквивалентов на 1 эквивалент соединения формулы (III).
Для деактивации остающегося соединения формулы (II) после ацилирования метанол предпочтительно используется в отношении объема к массе (об./масс.) от 1 до 2, в объеме в диапазоне от 1 до 2 мл на 1 г соединения формулы (II).
Для очистки при ацилировании может также использоваться ацетон, где предпочтительной формой ацетона является водный раствор, предпочтительнее 95% водный ацетон. Ацетон предпочтительно используется в объеме в диапазоне от 35 до 45 мл на 1 г соединения формулы (III).
На следующей стадии метансульфоновую кислоту добавляют к соединению формулы (IV), полученному на предыдущей стадии, для выхода соли тетразолметансульфоновой кислоты формулы (I).
В частности, тетразольное соединение формулы (IV), полученное на предыдущей стадии, растворяют в органическом растворителе, таком как хлороформ и метанол, с последующим добавлением к нему метансульфоновой кислоты. Затем к нему последовательно добавляют этилацетат и ацетон для очистки с тем, чтобы безопасным и эффективным путем могла быть получена соль тетразолметансульфоновой кислоты формулы (I).
В описанном выше способе метансульфоновая кислота, которая действует в качестве сопряженной с основанием кислоты формулы (IV), предпочтительно используется в количестве от 1 до 1,5 эквивалентов на 1 эквивалент соединения формулы (IV).
Далее, для очистки может использоваться этилацетат и ацетон. Этилацетат предпочтительно используется в объеме в диапазоне от 1 до 5 мл на 1 г соединения формулы (IV). Ацетон может использоваться в водном растворе, предпочтительно 95% водном ацетоне, в объеме в диапазоне от 15 до 25 мл на 1 г соединения формулы (IV).
Как указано выше, в способе по изобретению с использованием соединения формулы (II) для получения соли тетразолметансульфоновой кислоты может использоваться только метанол и ацетон без использования колоночной хроматографии для получения соединения формулы (IV) с высокой чистотой и высоким выходом. Поэтому, по сравнению с обычными способами, включающими ацилирование с использованием конденсирующего агента, такого как 1-(3-диметиламинопропил)-3-этилкарбодиимид, Ν,Ν-дициклогексилдиимид, Ν,Ν-диизопрокарбодиимид и метил-п-толуолсульфонат 1-циклогексил-3-(2-(морфолинэтил)карбодиимида, способом по изобретению может быть получен продукт высокой чистоты посредством простой фильтрации, и, таким образом, он обеспечивает очень экономически рентабельный и удобный способ, подходящий для крупномасштабного производства.
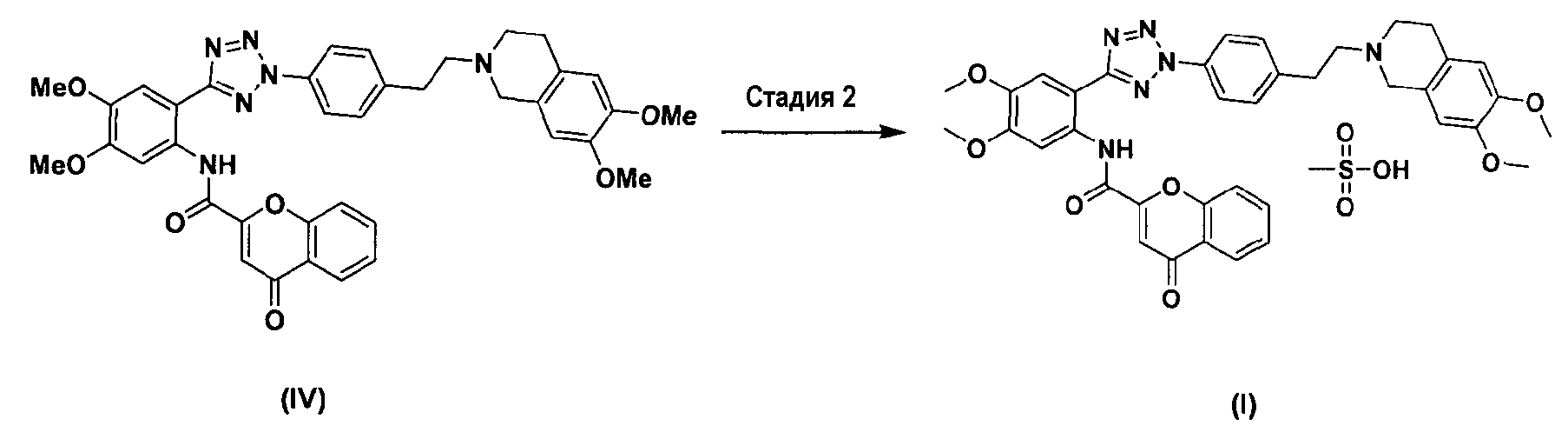
Между тем соединение формулы (II), используемое при указанном выше ацилировании (стадия 1), может быть получено взаимодействием соединения формулы (V) с соединением формулы (VI) в присутствии трифенилфосфина и основания, как показано ниже в схеме реакции 4.
Схема реакции 4
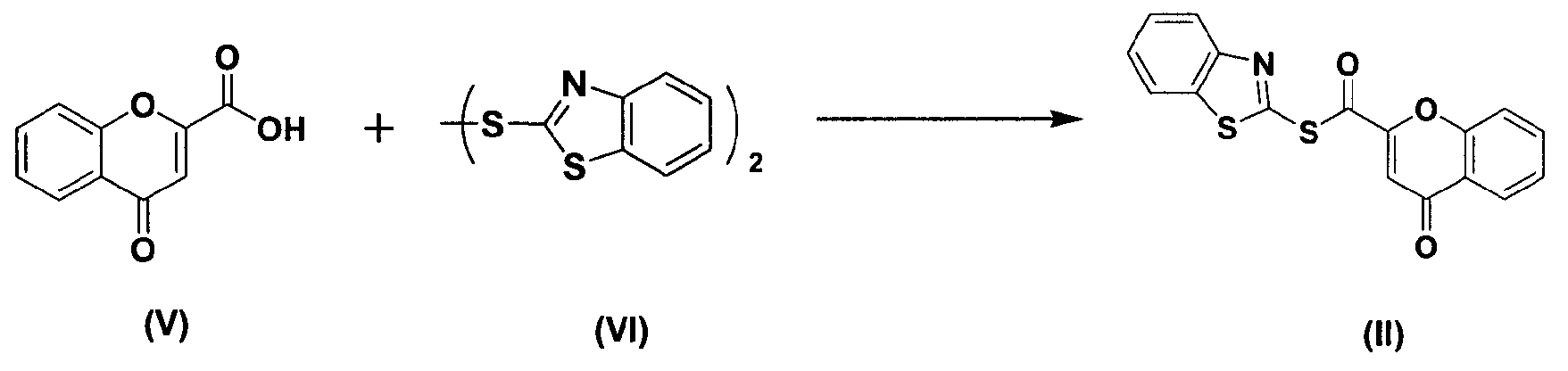
В частности, сложный S-бензотиазол-2-иловый эфир 4-оксо-4Н-хромен-2-карбоновой кислоты формулы (II) может быть получен взаимодействием хромоновой кислоты формулы (V) с 2,2'-дитиобисбензотиазолом формулы (VI) в органическом растворителе в присутствии основания и трифенилфосфина (PPH3) при температуре в диапазоне от 20 до 25°C в течение 1-3 часов, где органический растворитель выбран из группы, состоящей из дихлорметана (CH2Cl2), простого диэтилового эфира, сложного этилового эфира, тетрагидрофурана и их смеси, предпочтительно дихлорметана, и основания, выбранного из группы, состоящей из триэтиламина (NEt3), пиридина, имидазола, диизопропилэтиламина (DIPEA), 4-диметиламинопиридина (DMAP) и их смеси, предпочтительно триэтиламина. Если время реакции превышает 3 часа, то могут образовываться примеси в виде побочных продуктов.
При реакции используемое количество соединения формулы (VI) составляет предпочтительно от 1 до 2 эквивалентов на 1 эквивалент соединения формулы (V).
Также трифенилфосфин предпочтительно используется в количестве от 1 до 2 эквивалентов на 1 эквивалент соединения формулы (V).
Основание предпочтительно используется в количестве от 1 до 2 эквивалентов на 1 эквивалент хромоновой кислоты формулы (V).
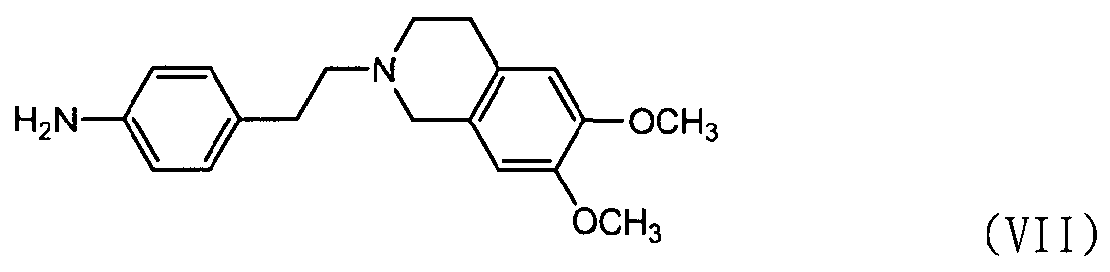
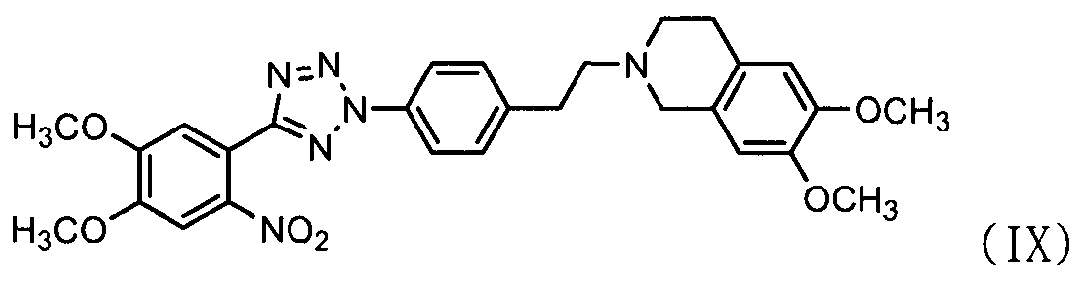
Кроме того, соединение формулы (III), которое используется в качестве исходного материала в способе по изобретению для получения соли тетразолметансульфоновой кислоты, может быть получено следующим способом, как показано в схеме реакции 5, включающим: (Стадию 1) циклизацию соединения формулы (VII) с соединением формулы (VIII) с получением соединения формулы (IX); и (Стадию 2) восстановление соединения формулы (IX), полученного на предыдущей стадии, путем использовании металла и кислоты.
Схема реакции 5
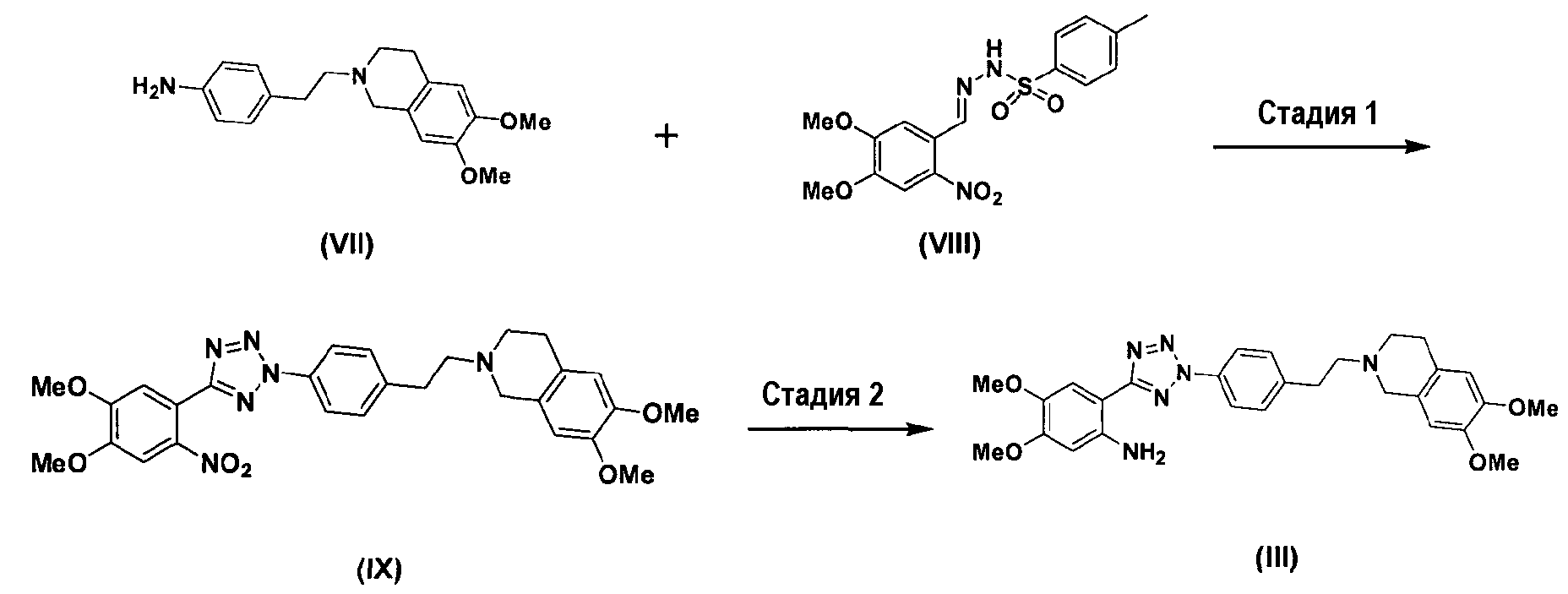
где Me представляет метильную группу.
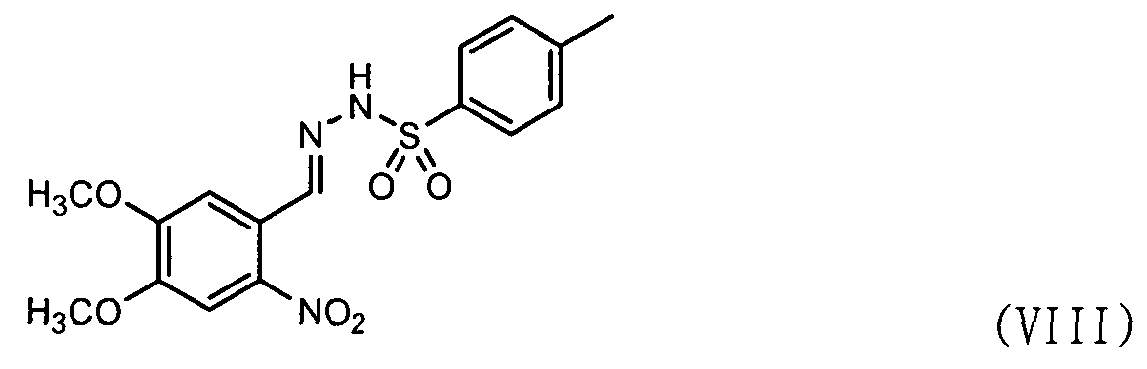
Во-первых, соединения формул (VII) и (VIII) подвергаются циклизации с получением соединения нитрофенилтетразола формулы (IX).
В частности, соединению формулы (VII) предоставляется возможность вступить в реакцию в присутствии нитрита натрия и хлористоводородной кислоты в водном растворе, например 50% растворе этанола, с получением соли диазония. К нему добавляют соединение формулы (VIII) и пиридин и перемешивают при поддержании реакционной температуры на уровне 10°С или ниже. Реакционную смесь нагревают до комнатной температуры и затем перемешивают, промывают 2,5н. раствором хлористоводородной кислоты, бикарбонатом натрия и водой и затем экстрагируют дихлорметаном. После удаления органического слоя добавляют метанол для кристаллизации с получением соединения формулы (IX) в виде целевого продукта [см. Suketaka Ito et al., Bulletin of Chemical Society of Japan, Vol.49(7), 1920-1923 (1976)].
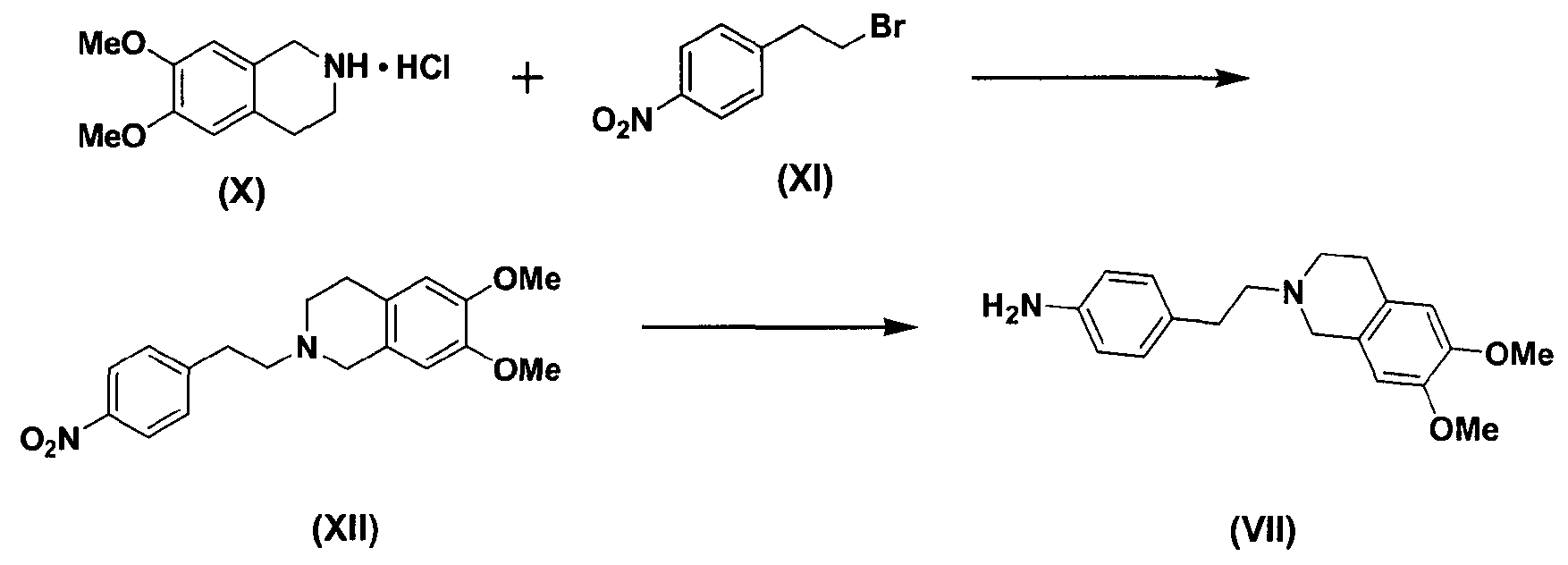
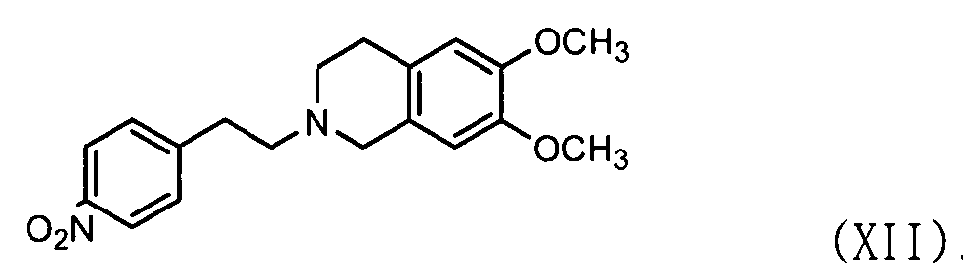
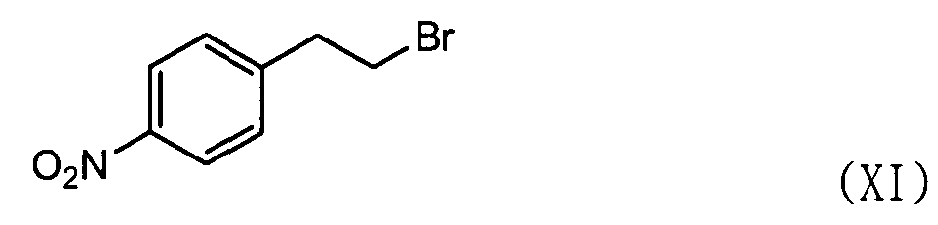
В этом случае соединение формулы (VII) может быть получено, как показано ниже в схеме реакции 6, путем взаимодействия соединения формулы (X) с соединением формулы (XI) с получением соединения формулы (XII) с последующим восстановлением соединения формулы (XII), используя металл и кислоту.
Схема реакции 6
где Me представляет метильную группу.
В частности, соединение формулы (X) подвергают взаимодействию с соединением формулы (XI) в растворителе, таком как Ν,Ν-диметилформальдегид, в присутствии безводного бикарбоната калия (K2CO3) и йодида натрия (NaI) при температуре в диапазоне от 70 до 100°C при перемешивании с получением соединения нитробензолизохинолина формулы (XII). Затем высушенное соединение формулы (XII) подвергают восстановлению с использованием металла и кислоты в водном растворе, например 50% растворе этанола, с получением соединения аминобензолизохинолина формулы (VII), где металл выбран из группы, состоящей из железа, олова, цинка и никеля, предпочтительно железа, а кислота выбрана из группы, состоящей из хлористоводородной кислоты, азотной кислоты, серной кислоты, уксусной кислоты и их смеси.
Восстановление может проводиться в течение 3 часов при 80°С. После восстановления к реакционной смеси может добавляться 10% водный раствор хлорида натрия для нейтрализации и фильтроваться через прокладку из целлита. После удаления органического слоя остаток отверждают простым этиловым эфиром с получением соединения формулы (VII). При восстановлении металл может использоваться в количестве от 2 до 10 эквивалентов на 1 эквивалент соединения формулы (XII), а кислота может использоваться в количестве от 0,1 до 0,5 эквивалентов на 1 эквивалент соединения формулы (XII).
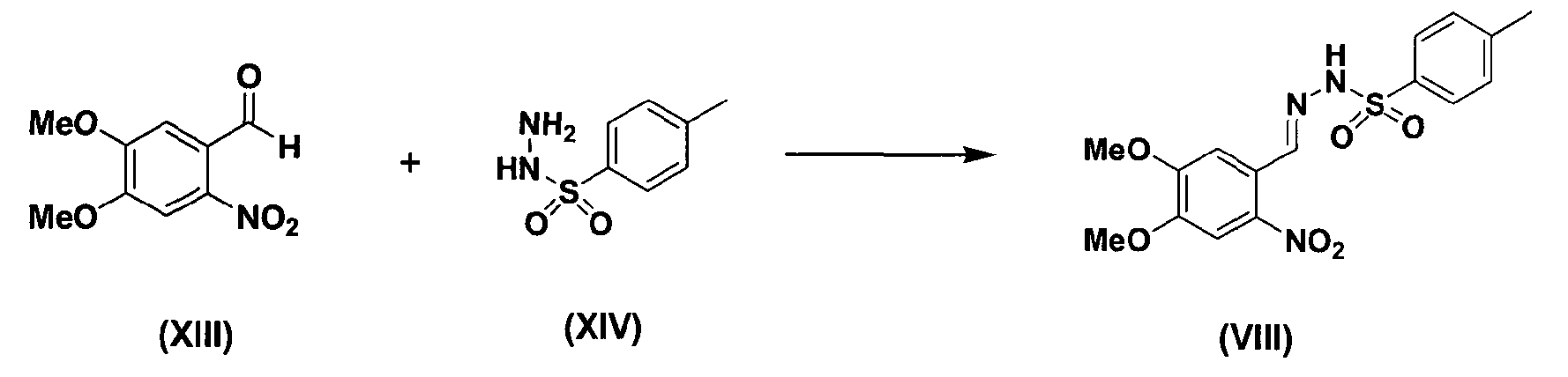
Кроме того, соединение формулы (VIII) может быть получено, как показано в схеме реакции 7, путем взаимодействия соединения формулы (XIII) с соединением формулы (XIV).
Схема реакции 7
где Me представляет метильную группу.
В частности, соединение формулы (XIII) подвергают взаимодействию с соединением формулы (XIV) в этаноле при температуре в диапазоне от 70 до 80°C с последующей фильтрацией и сушкой с получением соединения формулы (VIII) [см. Suketaka Ito et al., Bulletin of Chemical Society of Japan, Vol.49(7), 1920-1923 (1976)].
В реакционной схеме 5 соединение формулы (IX), полученное на стадии 1, затем подвергают восстановлению, используя металл и кислоту, с получением соединения формулы (III) (стадия 2).
В частности, соединение формулы (IX) подвергают восстановлению с использованием металла в кислоте для получения соединения формулы (III), где металл выбран из группы, состоящей из железа, олова, цинка и никеля, металла, предпочтительно железа, а кислота выбрана из группы, состоящей из хлористоводородной кислоты, азотной кислоты, серной кислоты, уксусной кислоты и их смеси, предпочтительно уксусной кислоты, например 50% водного раствора уксусной кислоты.
Восстановление предпочтительно проводят при 80°C в течение 3 часов. После восстановления полученную смесь фильтровали через прокладку из целлита и затем нейтрализовали. После удаления органического растворителя остаток перемешивают в метаноле с получением соединения формулы (III).
При восстановлении металл может использоваться в количестве от 5 до 15 эквивалентов на 1 эквивалент соединения формулы (IX), и кислота может использоваться в объеме в диапазоне от 2 до 5 мл на 1 г соединения формулы (IX).
Как указано выше, в настоящем изобретении в процессе восстановления при получении соединений формулы (ΙΙΙ) и (VII) используется металл и кислота, вместо палладия и газообразного водорода, используемых в традиционных способах, с тем, чтобы он мог сократить время реакции, а также риск взрыва вследствие повышенной безопасности восстановления.
В соответствии с другим аспектом настоящее изобретение относится к соединению формулы (II), которое может использоваться при получении соли тетразолметансульфоновой кислоты формулы (I):
Кроме того, настоящее изобретение относится к способу получения соединения формулы (II), включающему стадию взаимодействия соединения формулы (V) с соединением формулы (VI) в присутствии основания:
Далее настоящее изобретение будет описано более конкретно следующими примерами, но они представлены только в целях иллюстрации, и настоящее изобретение не ограничивается ими.
Пример: Получение соли N-(2-(2-(4-(2-(6,7-диметокси-3,4-дигидроизохинолин-2(1H)ил)этил)фенил)-2Н-тетразол-5-ил)-4,5-диметоксифенил)-4-оксо-4Н-хромен-2-карбоксамид метансульфоновой кислоты
Стадия 1-1) 6,7-диметокси-2-(4-нитрофенэтил)-1,2,3,4-тетрагидроизохинолин
5 л Ν,Ν-диметилформамида, 2-(4-нитрофенил)этилбромида (1,0 кг, 4,35 моль) и 6,7-диметокси-1,2,3,4-тетрагидроизохинолина гидрохлорида (DTIH, 1,0 кг, 4,35 моль) добавляли в реактор и перемешивали. К реакционной смеси добавляли карбонат калия (1,80 кг, 13 моль) и йодид натрия (780 г, 5,20 моль), реактор нагревали до 100°C и далее перемешивали в течение 12 часов. После проверки завершения реакции тонкослойной хроматографией (элюент: хлороформ/метанол = 15/1) реакционную смесь охлаждали до комнатной температуры.
В отдельный реактор добавляли 20 л холодной воды и затем реакционную смесь, полученную на предыдущей стадии, медленно добавляли к ней с последующим перемешиванием в течение 4 часов. Полученную смесь фильтровали, промывали 3 л воды и затем сушили горячим воздухом при 40°С в печи с получением указанного в заголовке соединения (1,34 кг, 90%).
1Н-ЯМР (CDCl3) d: 8,17 (д, 2H), 7,43 (д, 2H), 6,62 (c, 1H), 6,54 (c, 1H), 3,87 (c, 3H), 3,85 (c, 3H), 3,66 (c, 2H), 3,03 (т, 2H), 2,82-2,78 (м, 6H).
Стадия 1-2) 4-(2-(6,7-диметокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензоламин
Железо (1,27 кг, 23,48 моль), хлористоводородную кислоту (127 мл, 1,54 моль) и 6,7 л 50% водного этанола добавляли в реактор и перемешивали в течение 1 часа при 80°C. К указанной смеси медленно в течение 1 часа добавляли полученный на стадии 1-1 6,7-диметокси-2-(4-нитрофенэтил)-1,2,3,4-тетрагидроизохинолин (1,3 кг, 3,91 моль) с последующим перемешиванием в течение 3 часов при 80°C. После проверки завершения реакции тонкослойной хроматографией (элюент: хлороформ/метанол = 15/1) реакционную смесь охлаждали до комнатной температуры. К ней добавляли 5,3 л дихлорметана и 5,3 л воды, реакционную смесь нейтрализовали добавлением 804 мл 10% водного раствора гидроксида натрия. Полученную смесь фильтровали через прокладку целлита и промывали 3 л метиленхлорида. Органический слой собирали, сушили над сульфатом магния и затем фильтровали. Растворитель удаляли при пониженном давлении и сушили горячим воздухом при 40°С в сушильном шкафу с получением указанного в заголовке соединения (1,05 кг, 90%).
1Н-ЯМР (CDCl3) d: 7,02 (д, 2Н), 6,65-6,53 (м,4Н), 3,84 (с, 3Н), 3,83 (с, 3Н), 3,63 (с, 2Н), 3,57 (с, 2Н), 2,84-2,86 (м, 8Н).
Стадия 2) 4,5-диметокси-2-нитро-п-толуолсульфонилгидразон
12,5 л этанола, п-толуолсульфонилгидразид (1 кг, 5,37 моль) и 6-нитровератральдегида (1,2 кг, 5,907 моль) добавляли в реактор и затем нагревали до 80°C с последующим перемешивании в течение 6 часов. После проверки завершения реакции тонкослойной хроматографией (элюент: хлороформ/метанол = 15/1) реакционную смесь охлаждали до комнатной температуры. Полученную смесь фильтровали, промывали 12,5 л этанола и сушили горячим воздухом при 40°С в сушильном шкафу с получением указанного в заголовке соединения (1,47 кг, 99,6%).
1Н-ЯМР (CDCl3) d: 8,48 (c, 1H), 8,08 (c, 1H), 7,89 (д, 2H), 7,59 (c, 1H), 7,42 (c, 1H), 7,33 (д, 2H), 4,02 (c, 3H), 3,98 (c, 3H), 2,44 (c, 3H).
Стадия 3) Сложный S-бензотиазол-2-иловый эфир 4-оксо-4H-хромен-2-карботионовой кислоты
Хромон-2-карбоновую кислоту (700 г, 3,68 моль), 2,2'-битиобис-бензотиазол (1,47 кг, 4,42 моль), трифенилфосфин (1,16 кг, 4,42 моль) и 14,7 л дихлорметана добавляли в реактор и перемешивали. К реакционной смеси медленно добавляли триэтиламин (616 мл, 4,42 моль) в 2 л дихлорметана и перемешивали в течение 6 часов. После завершения реакции полученную смесь фильтровали, промывали 4 л ацетона и затем сушили горячим воздухом при 40°С в сушильном шкафу с получением указанного в заголовке соединения (0,99 кг, 80%).
1Н-ЯМР (CDCl3) d: 8,30 (д, 1H), 8,16 (д, 1H), 8,01 (д, 1H), 7,88 (т, 1H), 7,70 (д, 1H), 7,61-7,31 (м, 3H), 7,15 (c, 1H).
Стадия 4) 2-(4-(5-(4,5-диметокси-2-нитрофенил)-2Н-тетразол-2-ил)фенэтил)-6,7-диметокси-1,2,3,4-тетрагидроизохинолин
4-(2-(6,7-диметокси-3,4-дигидроизохинолин-2(1H)-ил)этил)бензоламин (1,0 кг, 3,2 моль), полученный на стадии 1-2, 3 л 50% водного этанола и хлористоводородную кислоту (850 мл, 12,5 моль) добавляли в реактор A и перемешивали при 0°C. К реакционной смеси медленно добавляли нитрит натрия (227 г, 3,3 моль) в 330 мл воды и перемешивали в течение 3 часов.
В 20-литровый реактор B добавляли 4,5-диметокси-2-нитро-п-толуолсульфонилгидрозон (1,2 кг, 3,2 моль), полученный на стадии 2, и 12 л пиридина и охлаждали до 0°С. В него медленно добавляли смесь из реактора A с последующим перемешиванием в течение 6 часов при комнатной температуре. После проверки завершения реакции тонкослойной хроматографией (элюент: хлороформ/метанол = 15/1) реакционную смесь подвергали экстракции 12 л дихлорметана и 12 л воды. Органический слой собирали, промывали три раза 18 л 2,5н. хлористоводородной кислоты и промывали раствором бикарбоната натрия. Органический слой сушили и дистиллировали при пониженном давлении. Остаток смешивали с 10 л метанола и перемешивали в течение 4 часов. Полученную смесь фильтровали и сушили горячим воздухом при 40°С в сушильном шкафу с получением указанного в заголовке соединения (1,08 кг, 62%).
1Н-ЯМР (CDCl3) d: 8,08 (д, 2H), 7,66 (c, 1H), 7,45 (д, 2H), 7,32 (c, 1H), 6,59 (д, 2H), 4,03 (c, 6H), 3,85 (c, 6H), 3,68 (c, 2H), 3,01 (м, 2H), 2,84 (м, 6H).
Стадия 5) 2-(2-(4-(2-(6,7-диметокси-3,4-дигидроизохинолин-2 (1H)-ил)этил)фенил)-2Н-тетразол-5-ил)-4,5-диметоксибензамин
Железо (433 г, 7,76 моль) и 5,4 л 50% водной уксусной кислоты добавляли в реактор и перемешивали в течение 1 часа при 80°C. В него медленно в течение двух часов добавляли 2-(4-(5-(4,5-диметокси-2-нитрофенил)-2Н-тетразол-2-ил)фенэтил)-6,7-диметокси-1,2,3,4-тетрагидроизохинолин (1,06 кг, 1,94 моль), полученный на стадии 4, с последующим перемешиванием в течение 1 часа. После проверки завершения реакции тонкослойной хроматографией (элюент: хлороформ/метанол = 15/1) реактор охлаждали до комнатной температуры медленно. В него добавляли 5,3 л хлороформа и 2,4 л воды, и полученную смесь фильтровали через прокладку целлита. Органический слой собирали, к нему медленно добавляли 6,6 л насыщенного раствора бикарбоната натрия при перемешивании. Органический слой собирали и водный слой далее экстрагировали с использованием 1,25 л хлороформа. Полученный органический слой сушили над сульфатом магния и затем дистиллировали при пониженном давлении для удаления растворителя. Остаток смешивали с 10,6 л метанола с последующим перемешиванием. Полученную смесь фильтровали и сушили горячим воздухом при 40°С в сушильном шкафу с получением указанного в заголовке соединения (0,87 кг, 87%).
1Н-ЯМР (CDCl3) d: 8,14 (д, 2H), 7,75 (c, 1H), 7,49 (д, 2H), 6,63 (д, 2H), 6,40 (c, 1H), 5,34 (д, 2H), 3,97 (д, 6H), 3,89 (c, 6H), 3,72 (c, 2H), 3,06 (т, 2H), 2,91-2,84 (м, 6H).
Стадия 6) N-(2-(2-(4-(2-(6,7-диметокси-3,4-дигидроизохинолин-2(1H)-ил)этил)фенил)-2H-тетразол-5-ил)-4,5-диметоксифенил)-4-оксо-4Н-хромен-2-карбоксамид
2-(2-(4-(2-(6,7-диметокси-3,4-дигидроизохинолин-2(1H)-ил)этил)фенил)-2Н-тетразол-5-ил)-4,5-диметоксибензамин (850 г, 1,6 моль), полученный на стадии 5, сложный S-бензотиазол-2-иловый эфир 4-оксо-4Н-хромен-2-карботионовой кислоты (723 г, 2,1 моль), полученный на стадии 3, и 17 л дихлорметана добавляли в реактор и перемешивали в течение 6 часов при комнатной температуре. После проверки завершения реакции тонкослойной хроматографией (элюент: хлороформ/метанол = 15/1) к смеси последовательно добавляли 1,1 л метанола и 35,7 л 95% водного ацетона с последующим перемешиванием в течение 16 часов при комнатной температуре. Полученную смесь фильтровали, промывали 4,3 л ацетона, сушили горячим воздухом при 40°С в сушильном шкафу с получением указанного в заголовке соединения (1,10 кг, 97%).
1Н-ЯМР (CDCl3) d: 12,53 (c,1H), 8,60 (c, 1H), 8,23 (д, 1H), 8,14 (д, 2H), 7,77 (д, 2H), 7,74 (c, 1H), 7,50-7,44 (м, 3H), 7,26 (д, 2H), 6,60 (д, 2H), 4,01 (c, 6H), 3,87 (c, 6H), 3,70 (c, 2H), 3,08 (т, 2H), 3,02-2,83 (м, 6H).
Стадия 7) соль N-(2-(2-(4-(2-(6,7-диметокси-3,4-дигидроизохинолин-2(1H)-ил)этил)фенил)-2H-тетразол-5-ил)-4,5-диметоксифенил)-4-оксо-4Н-хромен-2-карбоксамид метансульфоновой кислоты
N-(2-(2-(4-(2-(6,7-диметокси-3,4-дигидроизохинолин-2(1H)-ил)этил)фенил)-2H-тетразол-5-ил)-4,5-диметоксифенил)-4-оксо-4Н-хромен-2-карбоксамид (1,06 кг, 1,54 моль), полученный на стадии 6, растворяли в смешанном растворе 18,1 л хлороформа и 1,06 л метанола и полученную смесь фильтровали. К ней медленно в течение 30 минут добавляли метансульфоновую кислоту (102 мл, 1,57 моль) в 300 мл этилацетата. К нему в течение 1 часа медленно добавляли 3,95 л этилацетата с последующим перемешиванием в течение 16 часов при комнатной температуре. Полученную смесь фильтровали, промывали 1 л этилацетата и сушили сухим воздухом при 40°С в сушильном шкафу.
Продукт подвергали первой перекристаллизации, используя хлороформ, и затем фильтровали, промывали этилацетатом и сушили сухим воздухом при 40°С в сушильном шкафу. Высушенное твердое вещество повергали второй перекристаллизации, используя смешанный растворитель (дихлорметан/метанол/этилацетат = 17/1/4) с получением кристаллического твердого вещества. Для удаления остаточного растворителя, т.е. дихлорметана, продукт смешивали с 95% водным ацетоном с последующим перемешиванием в течение 16 часов. Полученную смесь фильтровали и сушили сухим воздухом при 40°С в сушильном шкафу с получением указанного в заголовке соединения (0,84 кг, 70%).
Масса (ESI), рассчитанная для C38H36N6O7;
m/z 688,26 (М+)+; найдено, m/z 689,32 (М+Н)+
1Н-ЯМР (CDCl3) d: 12,43 (c, 1H), 11,66 (c, 1H), 8,49 (c, 1H), 8,17 (д, 1H), 8,06 (д, 2H), 7,79-7,67 (м, 2H), 7,54 (д, 3H), 7,46 (т, 1H), 7,14 (c, 1H), 6,67 (д, 2H), 4,78 (д, 1H), 4,19-4,12 (м, 1H), 3,96-3,87 (м, 12H), 3,56-3,36 (м, 6H), 3,04 (д, 1H), 2,78 (c, 3H).
Сравнительный пример
Тетразолметансульфоновую кислоту получали в соответствии с процедурой, раскрытой в международной патентной публикации № WO 2005/033097, которая включает восстановление с использованием палладия и газообразного водорода и ацилирование с использованием и 1-этил-3-(3-диметиламинопропил)карбодиимида гидрохлорида (EDCI) и конденсирующего агента 4-диметиламинопиридина (DMAP)
Экспериментальный пример
Для сравнения способа по изобретению, включающего восстановление с использованием металла и кислоты, с обычным способом с использованием палладия и водорода, оценивали чистоту продукта, безопасность и экономическую рентабельность процессов восстановления в примере и сравнительном примере, и результаты сведены в таблице 1, где результат сравнительного примера сверяли с каталогом компании (Aldrich) 2009-2010 гг.
Как показано в таблице 1, оба процесса восстановления давали целевые продукты с одинаковой частотой, однако процесс восстановления в соответствии с настоящим изобретением мог обеспечить получение целевого продукта более безопасным, без рисков взрыва и экономически рентабельным путем, по сравнению с обычным способом.
|
Также для сравнения эффективности способа по изобретению, в котором используется соединение формулы II, с обычным способом, при котором используется 1-этил-3-(3-диметиламинопропил)карбодиимид гидрохлорид (EDCI) и 4-диметиламинопиридин (DMAP) в качестве конденсирующего агента, выход в процентах, чистота и способ очистки соединений формулы (IV) в примере и сравнительном примере сведены ниже в таблице 2.
Как показано в таблице 2, в способе по изобретению может быть также получено соединение формулы (IV) при более высоком процентном выходе и чистоте, по сравнению с обычным способом. Может быть также показано, что способом по изобретению можно получить чистый химический продукт только посредством перекристаллизации без дополнительного процесса колоночной хроматографии.
|
Хотя изобретение было описано в отношении описанных выше определенных вариантов осуществления, следует понимать, что специалисты в данной области техники могут внести в данное изобретение различные модификации и изменения, которые также входят в объем изобретения, определенный прилагаемой формулой изобретения.
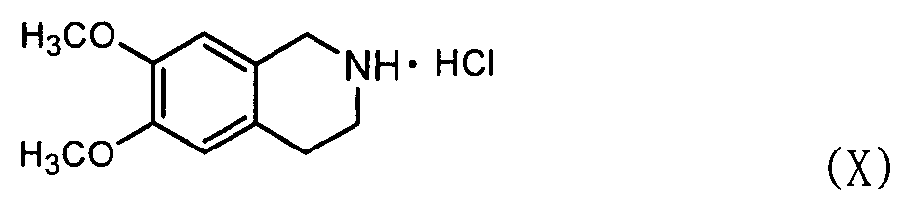

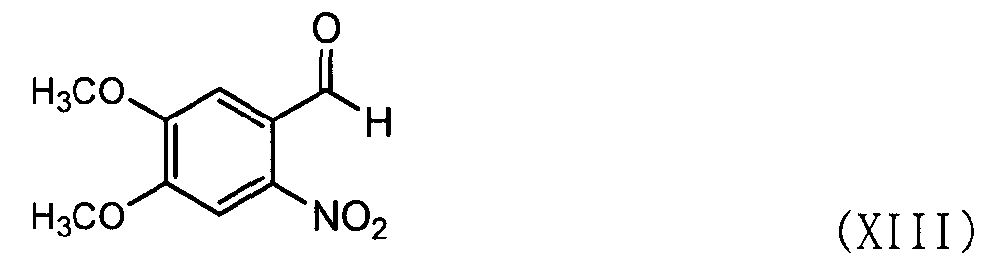
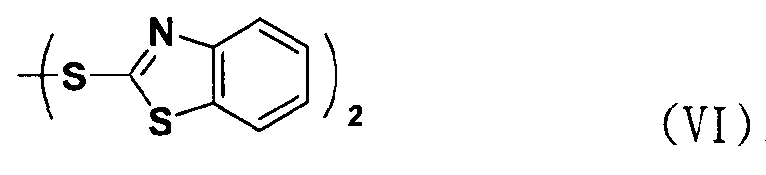
Конъюгат, содержащий оксинтомодулин и фрагмент иммуноглобулина, и его применение
Новые производные оксинтомодулина и содержащая их фармацевтическая композиция для лечения ожирения
Жидкие композиции длительно действующего конъюгата интерферона альфа
Производные факторов свертывания крови vii и viia, конъюгаты и комплексы, содержащие их, и их применение
Жидкий состав конъюгата гормона роста человека пролонгированного действия
Способ получения комплекса физиологически активного полипептида
Производные тиено[3,2-d]пиримидина, обладающие ингибирующей активностью в отношении протеинкиназ
Улучшенный способ получения конъюгата физиологически активного полипептида
Фармацевтическая композиция для предупреждения или лечения неалкогольной жировой болезни печени
Жидкие составы для конъюгата эритропоэтина длительного действия
Производные тиено[3,2-d]пиримидина, обладающие ингибирующей активностью в отношении протеинкиназ

