Результат интеллектуальной деятельности: ТВЕРДЫЕ ФОРМЫ ОРТАТАКСЕЛА
Вид РИД
Изобретение
ОБЛАСТЬ ИЗОБРЕТЕНИЯ
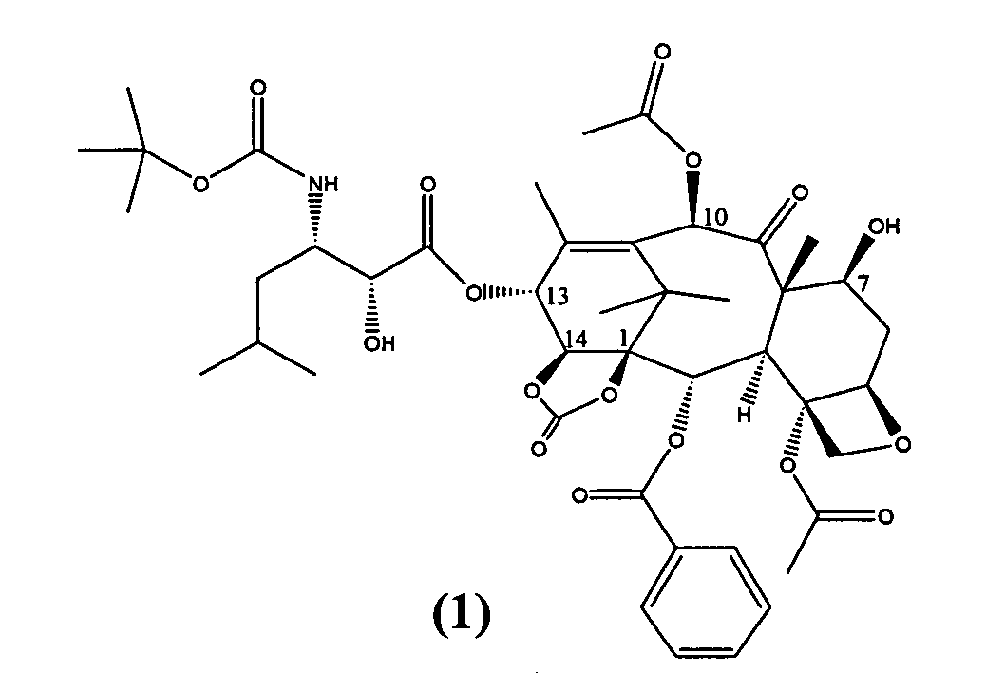
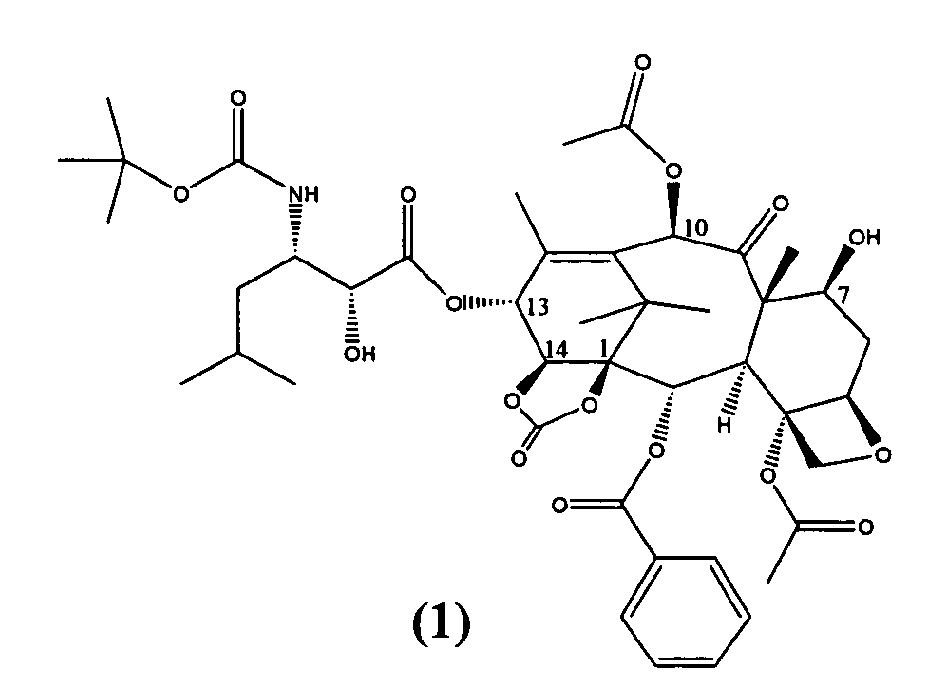
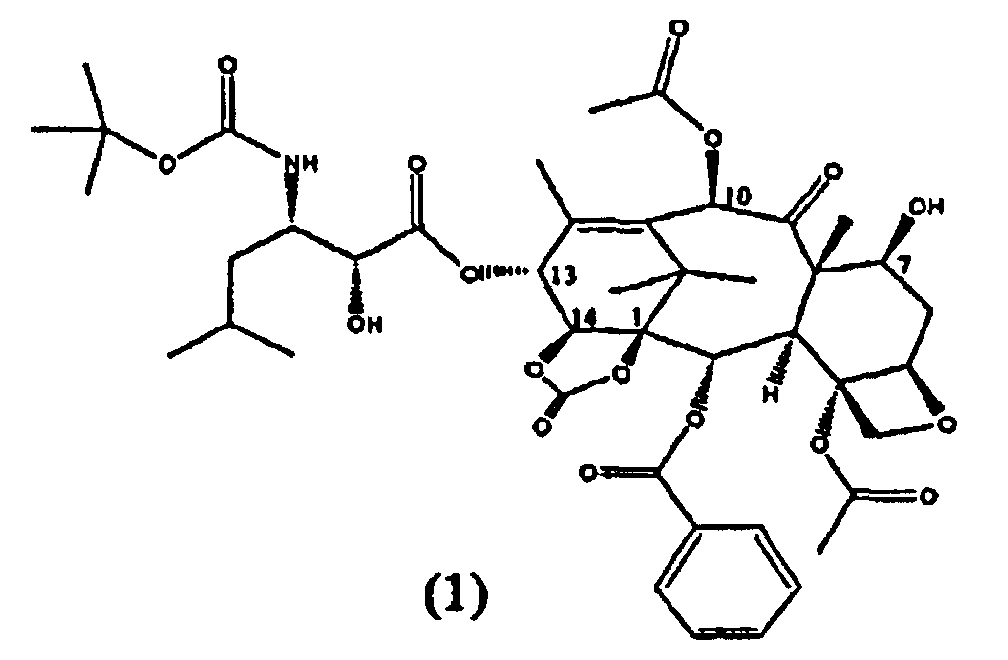
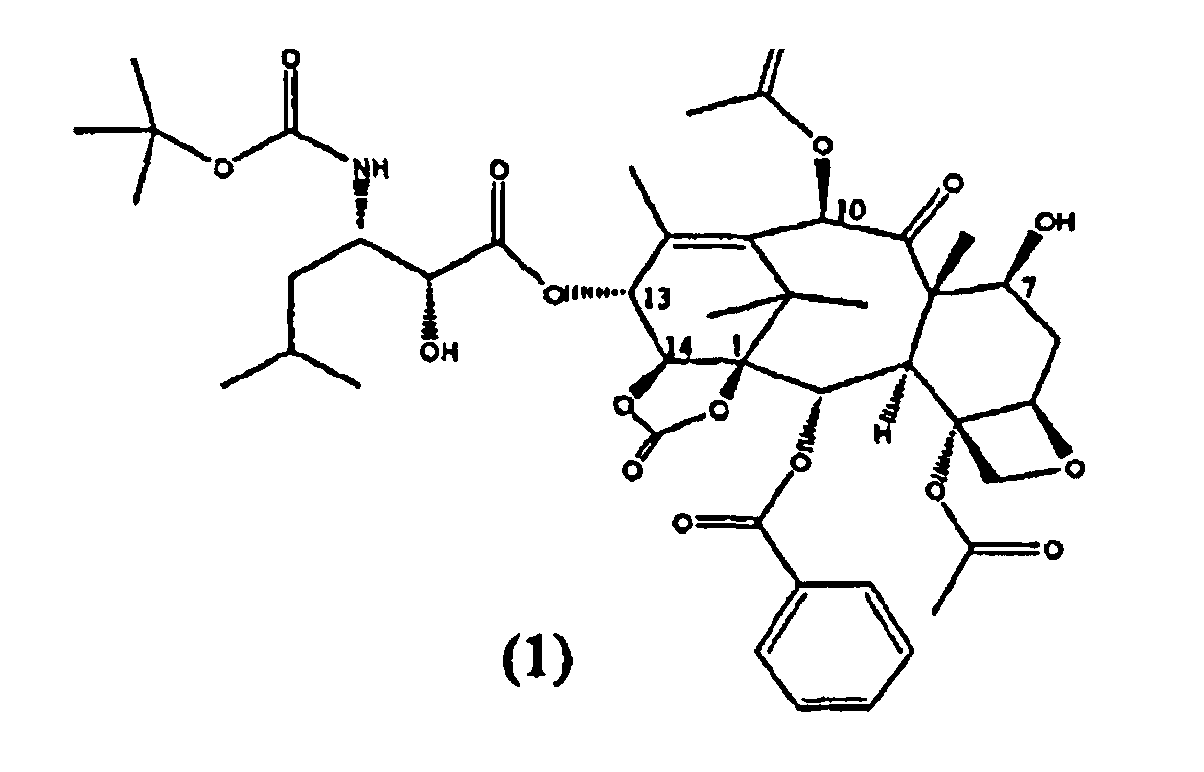
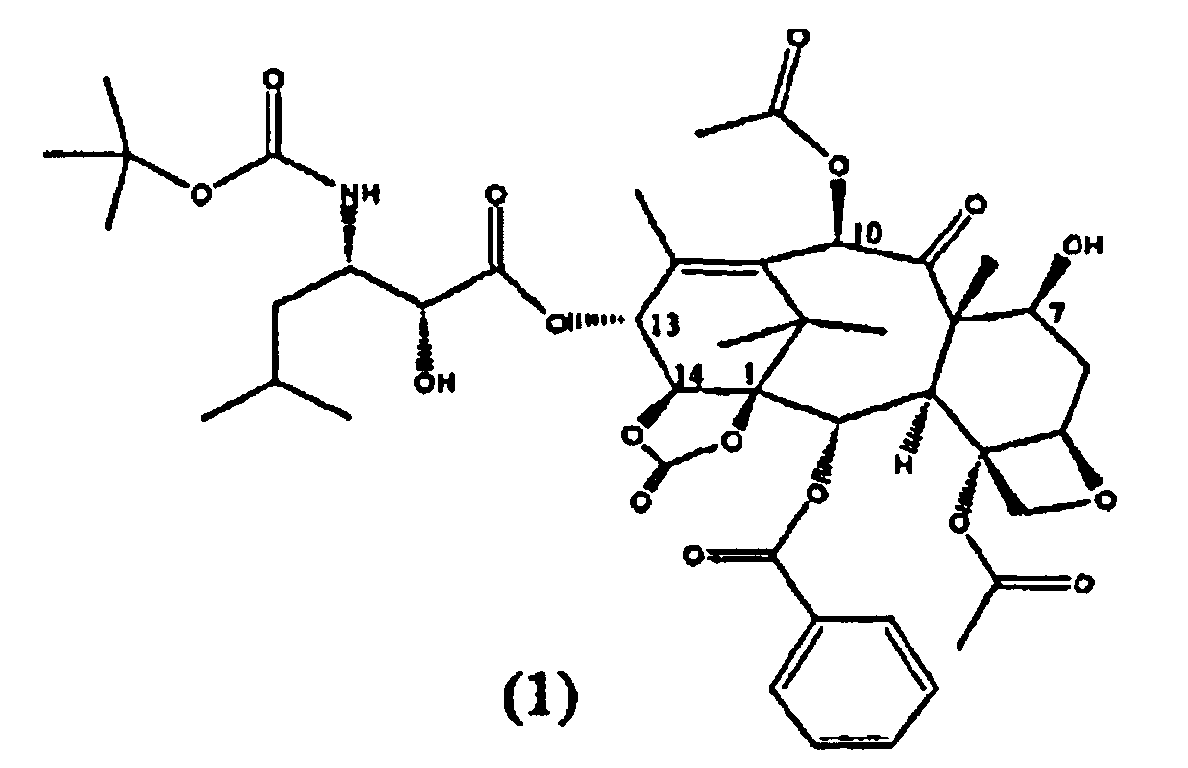
Настоящее изобретение относится к твердым формам Ортатаксела (1,14-карбонат 13-N-Boc-β-изобутилсеринил)-14-β-гидроксибаккатина III) (1), их смесям и способам их получения.
ПРЕДПОСЫЛКИ ИЗОБРЕТЕНИЯ
Ортатаксел (1) является противоопухолевым соединением, в частности, действующим на опухоли молочной железы, легкого, яичника, толстой кишки, предстательной железы, почки и поджелудочной железы даже в случае резистентности к известным противоопухолевым средствам, таким как адриамицин, винбластин и некоторые производные платины.
Ортатаксел может быть получен в соответствии с методами, описанными в патентах США №№7232916, 6737534 и 6906101. Эти патенты рассматривают в примерах конечную стадию очистки, состоящий из кристаллизации из смеси ацетона и гексана, приводящей к образованию Ортатаксела в форме сольвата с содержанием ацетона в диапазоне от 4,5 до 6,5%.
Рентгеновская порошковая дифрактометрия (XRPD) ацетонового сольвата показывает характерные пики приблизительно при 7,9, 9,8, 10,6, 10,9, 14,6, 16,9, 19,7, 21,3 град 2-тета. Кривая дифференциальной сканирующей калориметрии (DSC) показывает эндотермический пик, появляющийся приблизительно при 164°С в результате плавления и высвобождения растворителя кристаллизации [подтверждено потерей веса приблизительно на 5,0% в термогравиметрическом/дифференциальном термическом анализе TG/DTA)], и слабый экзотермический пик с максимумом приблизительно при 212°С с последующим интенсивным эндотермическим пиком с максимумом приблизительно при 247°С в результате плавления и начинающегося разложения. IR показывает характеристические частоты поглощения при 3521, 3321, 2971, 2953, 1826, 1762, 1706, 1526, 1366, 1238, 1165, 1072, 723 см-1.
Хорошо известно, что летучие примеси в активных фармацевтических ингредиентах должны отвечать требованиям руководства (Q3C) ICH (Международной Конференции по Гармонизации); в данном конкретном случае содержание ацетона в диапазоне от 4,5 до 6,5% не будет разрешено. Таким образом, было бы желательно найти стабильную кристаллическую форму Ортатаксела, которая не содержит остаточных примесей растворителей в количествах, недопустимых с нормативной точки зрения. Такая кристаллическая форма должна также быть химически и термодинамически стабильной, т.е. она должна сохранять неизменным качество за время хранения и должна быть доступна с помощью воспроизводимого способа.
ОПИСАНИЕ ИЗОБРЕТЕНИЯ
Найдено, что Ортатаксел существует в двух несольватированных физических Формах, далее в тексте как Формы А и В, которые также могут быть получены в виде смесей.
Форма А является аморфным веществом, поскольку она не демонстрирует ярко выраженных пиков на порошковой рентгенограмме. Она может быть легко получена из Ортатаксела, например, ацетонового сольвата Ортатаксела, полученного в соответствии с методами синтеза, описанными в вышеупомянутых патентах, путем растворения в подходящем смешивающемся с водой растворителе с последующим быстрым добавлением воды, содержащей следовые количества (обычно 0,001-0,003%, вес/объем) органической кислоты, такой как уксусная или аскорбиновая кислота, предпочтительно лимонная кислота. «Подходящий водорастворимый растворитель» означает кетоновый или апротонный биполярный растворитель или их смесь; предпочтительными растворителями являются ацетон, диметилсульфоксид или их смеси. Способ, как правило, выполняется при температуре в диапазоне от 20 до 30°С; и предпочтительной органической кислотой является лимонная кислота. Органическая кислота предотвращает нежелательное образование 7-эпимера и придает Форме А физическую и химическую стабильность в течение, как минимум, 36 месяцев. В соответствии с предпочтительным вариантом осуществления изобретения получение Формы А осуществляется путем растворения Ортатаксела в ацетоне (8 мл/гортатаксел) и его осаждения водой (40 мл/гортатаксел), содержащей 0,001-0,003% лимонной кислоты (вес/объем) при комнатной температуре.
Форма В является кристаллическим полиморфом, плавящимся при 159°С; в отношении псевдополиморфного ацетонового сольвата Форма В характеризуется низким содержанием растворителя, легким выделением путем фильтрования или центрифугирования и химической и физической стабильностью в течение, как минимум, 36 месяцев. Форма В может быть получена путем растворения Ортатаксела, например, ацетонового сольвата или вышеупомянутой Формы А, в протонном органическом растворителе, таком как метанол, этанол или изопропанол, предпочтительно этанол, содержащем следовые количества органической кислоты (0,01-0,03%, вес/объем) как, например, уксусной, аскорбиновой, но предпочтительно лимонной кислоты, с последующим добавлением воды до осаждения и перемешивания образующейся смеси при температуре в диапазоне от 0 до 60°С, предпочтительно при 40°С, в течение времени в диапазоне от 4 до 8 часов. В соответствии с предпочтительным вариантом осуществления изобретения, получение Формы В осуществляется растворением Ортатаксела в этаноле (8-12 мл/гортатаксел), содержащем 0,01-0,03% лимонной кислоты (вес/объем), с последующим добавлением воды (13-20 мл/гортатаксел) таким образом, чтобы отношение этанол/вода находилось в диапазоне 0,5-0,7, и перемешиванием в течение 6 часов. Если перемешивание выполняется менее чем в течение 4 часов, Ортатаксел получают в виде смеси Формы А и Формы В.
Формы А и В Ортатаксела и их смеси могут предпочтительно быть использованы для получения фармацевтических композиций для лечения рака. В частности, смеси формы А и В, которые имеют разные биодоступности, пригодны для получения твердых Форм с контролируемым высвобождением. В связи с этим, дополнительным объектом настоящего изобретения являются фармацевтические композиции, содержащие кристаллические Формы А или В Ортатаксела или их смеси со смесью фармацевтически приемлемых носителей и/или ингредиентов, например, тех, что рассмотрены в «Remington's Pharmaceutical Sciences», Mack Publishing Co., N.Y., USA.
Изобретение иллюстрируется теперь более подробно в следующем экспериментальном разделе.
ЭКСПЕРИМЕНТАЛЬНЫЙ РАЗДЕЛ
Описание чертежей
Рентгеновская порошковая дифрактометрия (XRPD), дифференциальная сканирующая калориметрия (DSC), термогравиметрический/дифференциальный термический анализ (TG/DTA), инфракрасная (IR) и оптическая микроскопия были использованы для характеристики новых твердых Форм, которые были сравнены с аналитическими данными ацетонового сольвата.
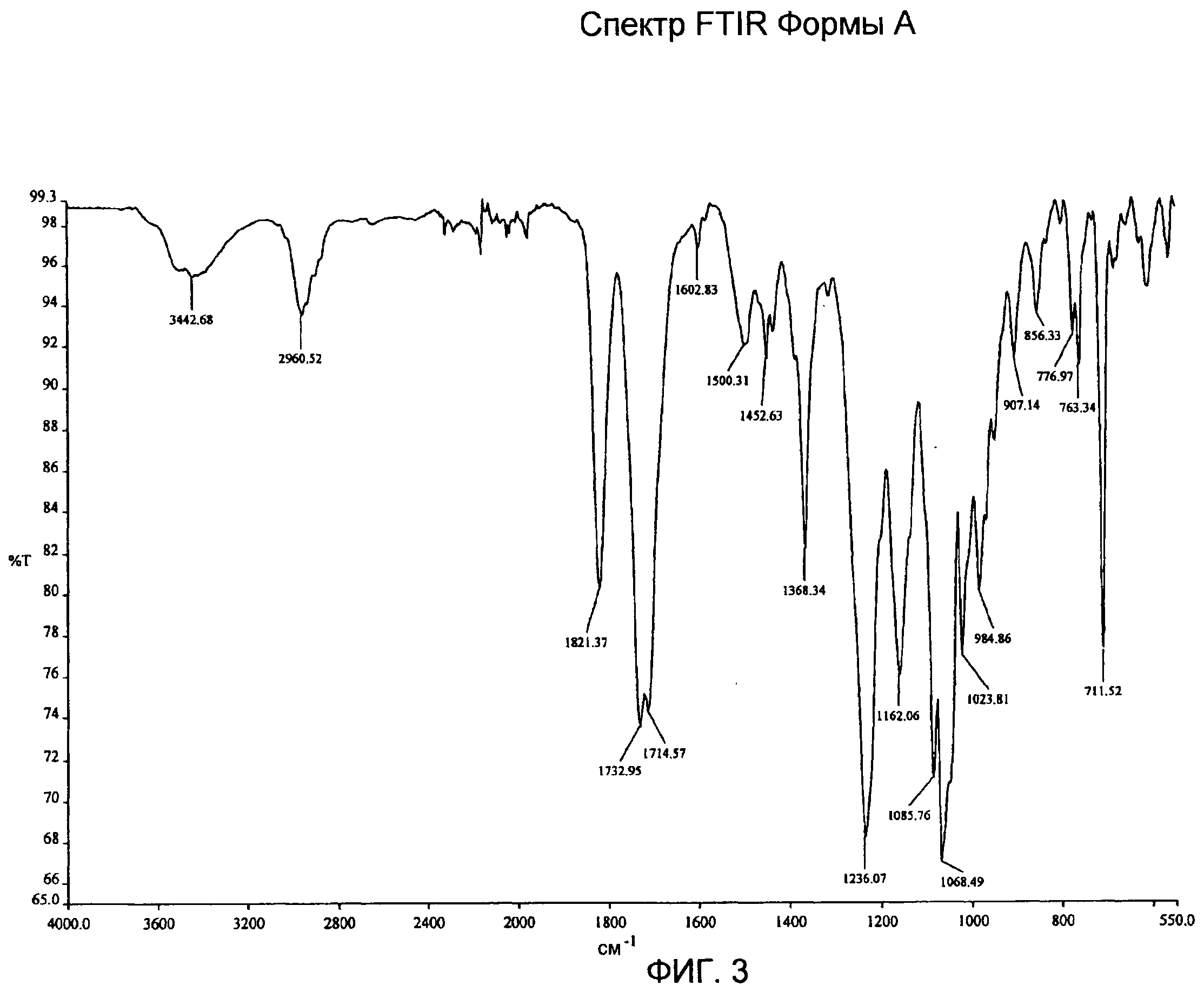
Фиг.1-4: спектры XRPD, DSC, TG/DTA и IR Формы А;
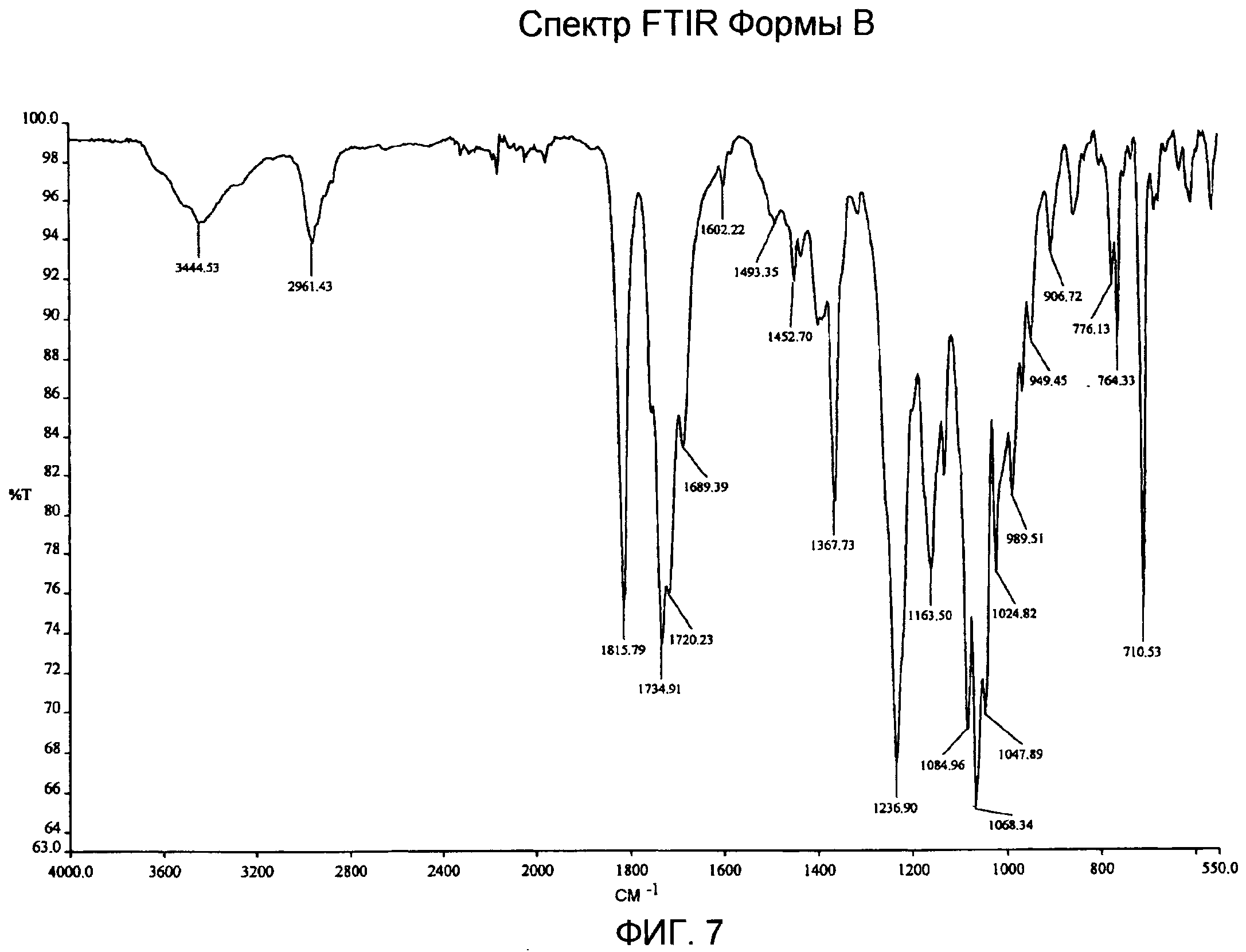
Фиг.5-8: спектры XRPD, DSC, TG/DTA и IR Формы В;
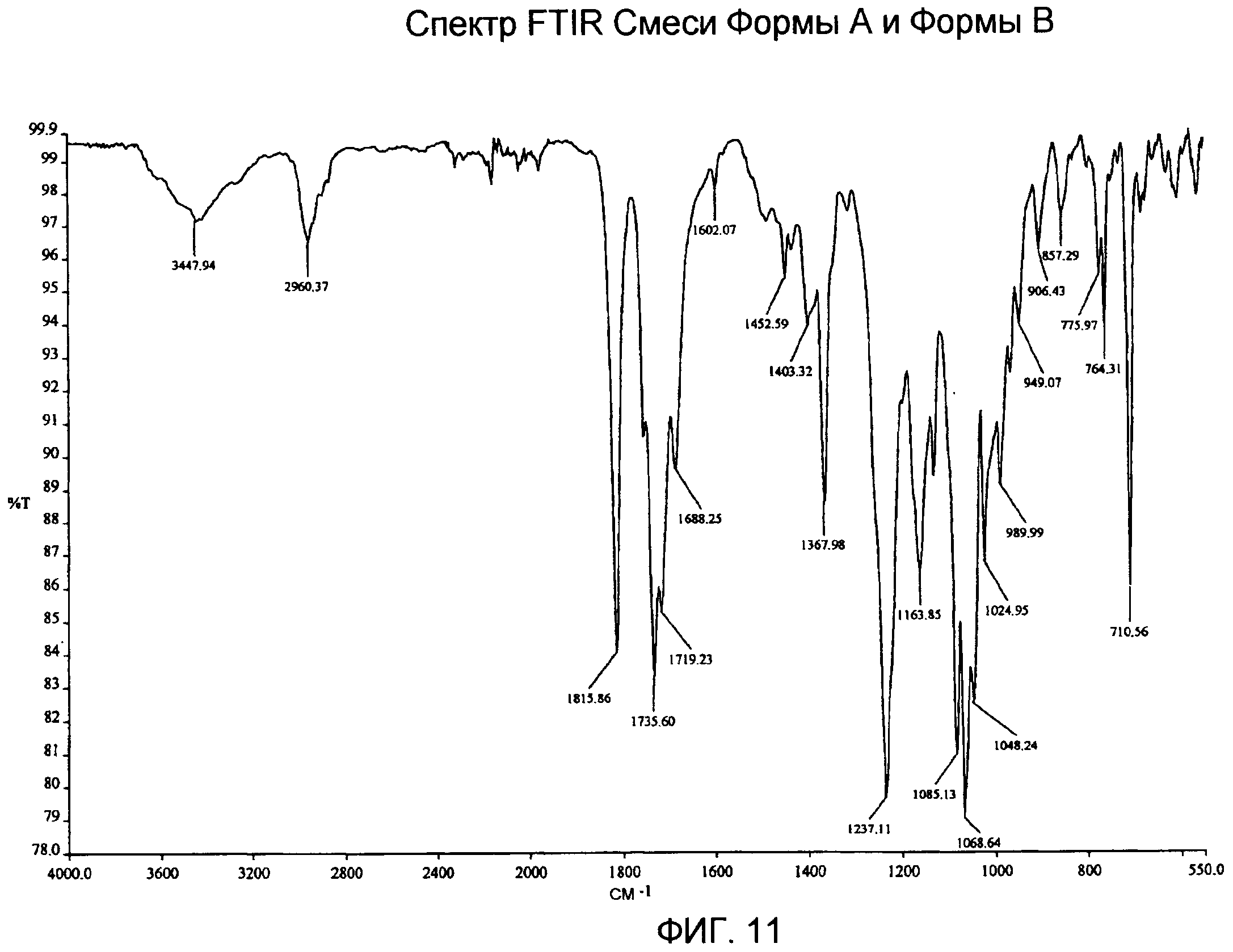
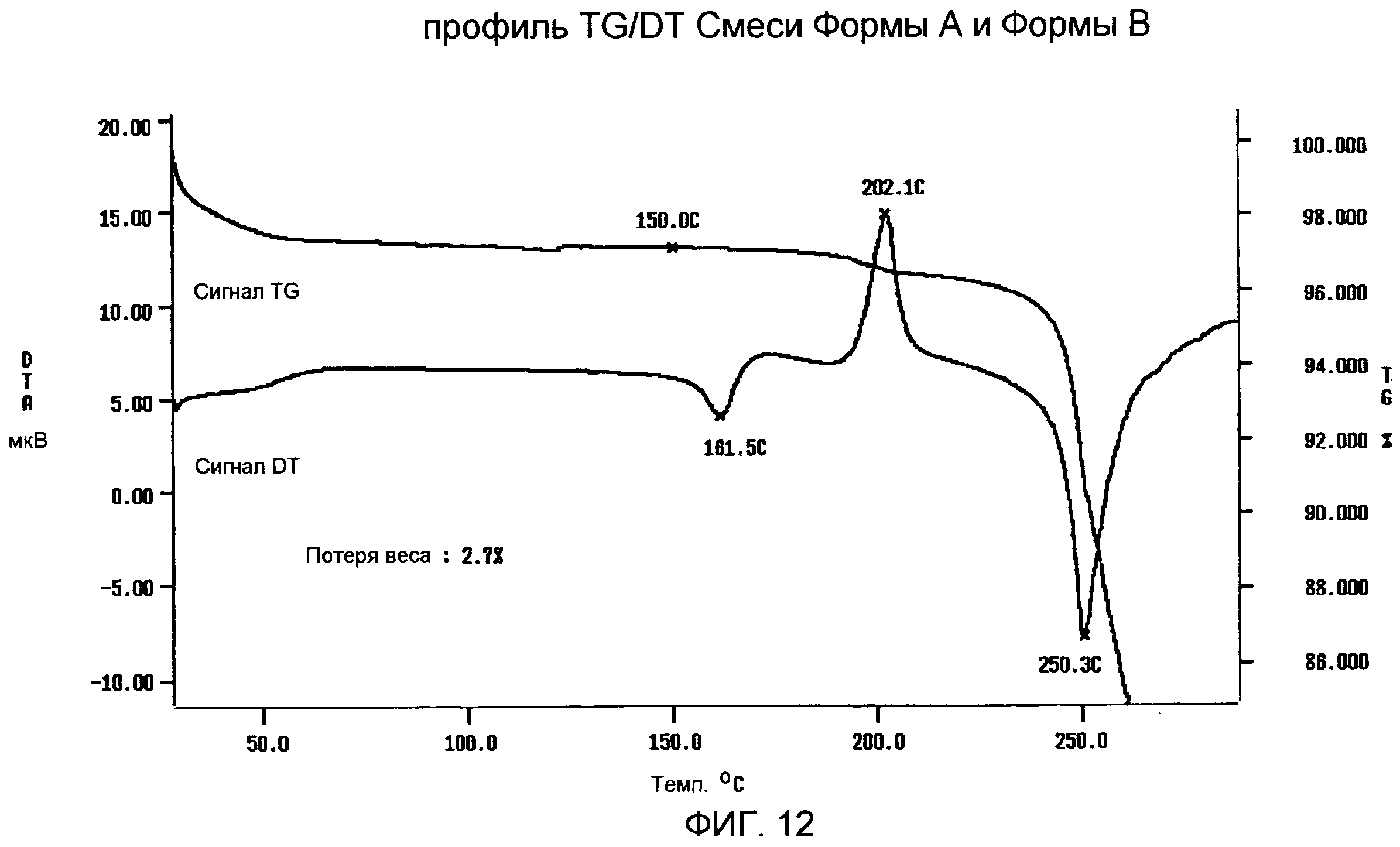
Фиг.9-12: спектры XRPD, DSC, TG/DTA и IR смеси Формы А и Формы В, содержащей приблизительно 75% Формы В;
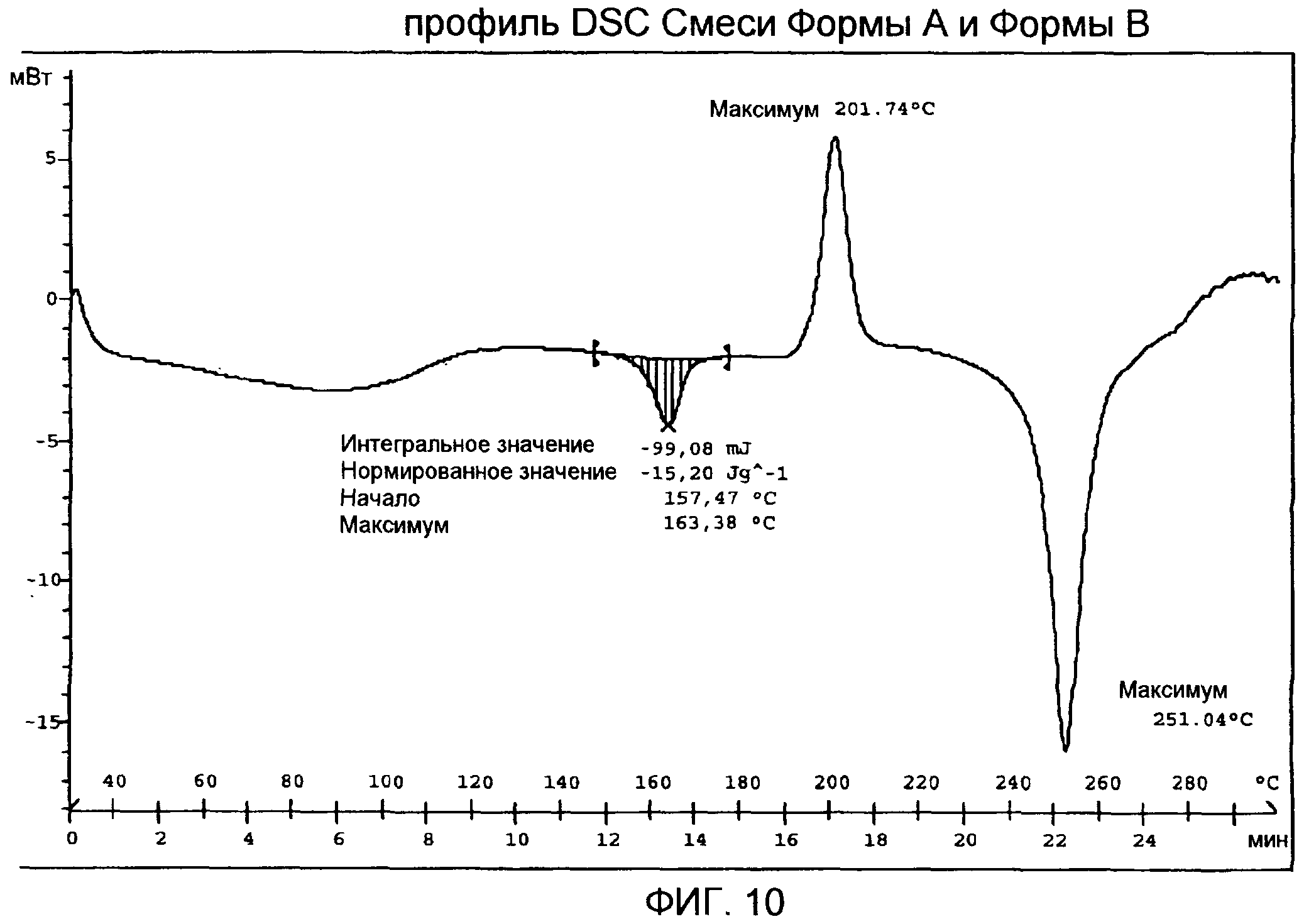
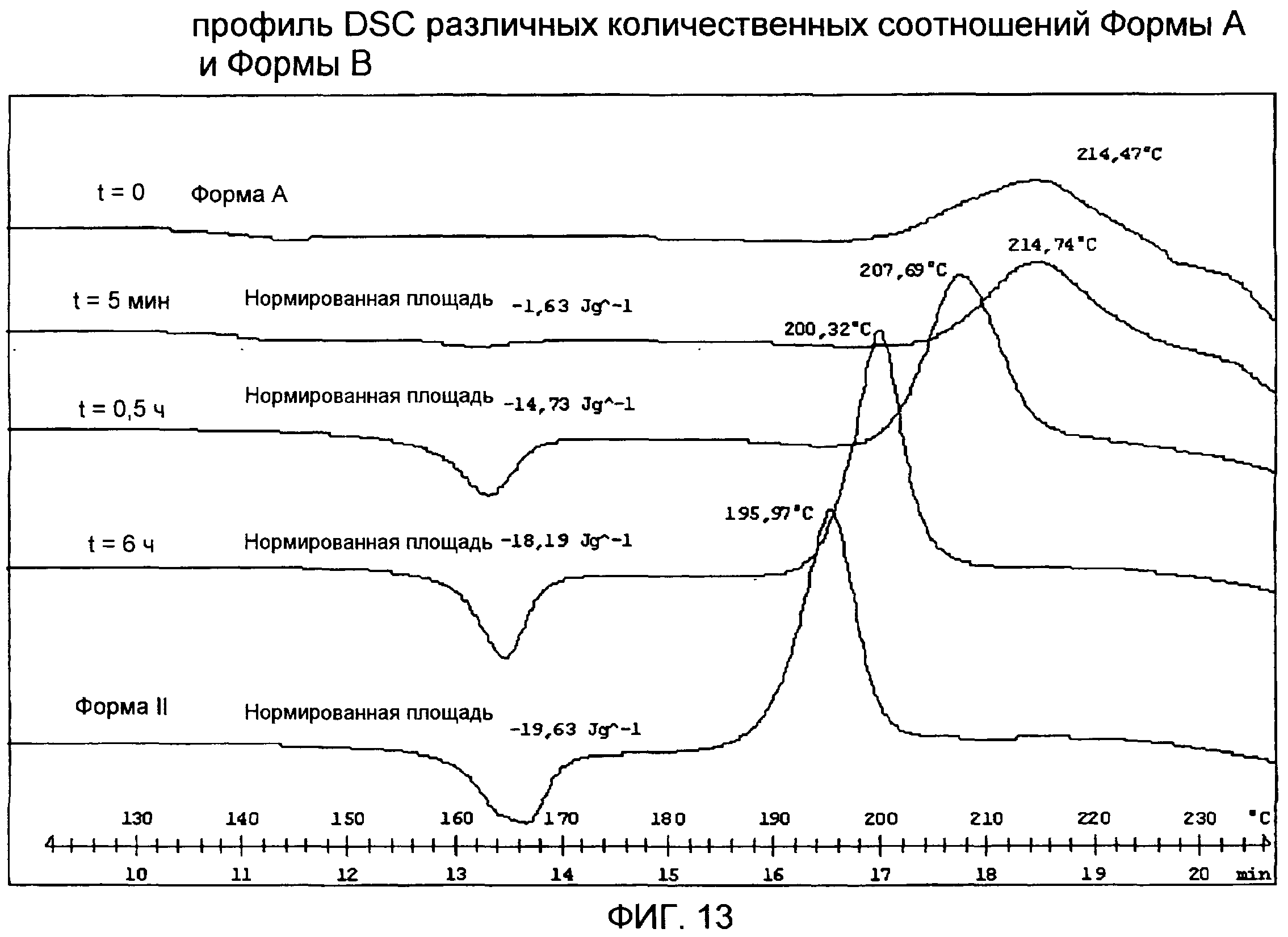
Фиг.13: профиль DSC разных количественных соотношений Формы А и Формы В.
Форма А
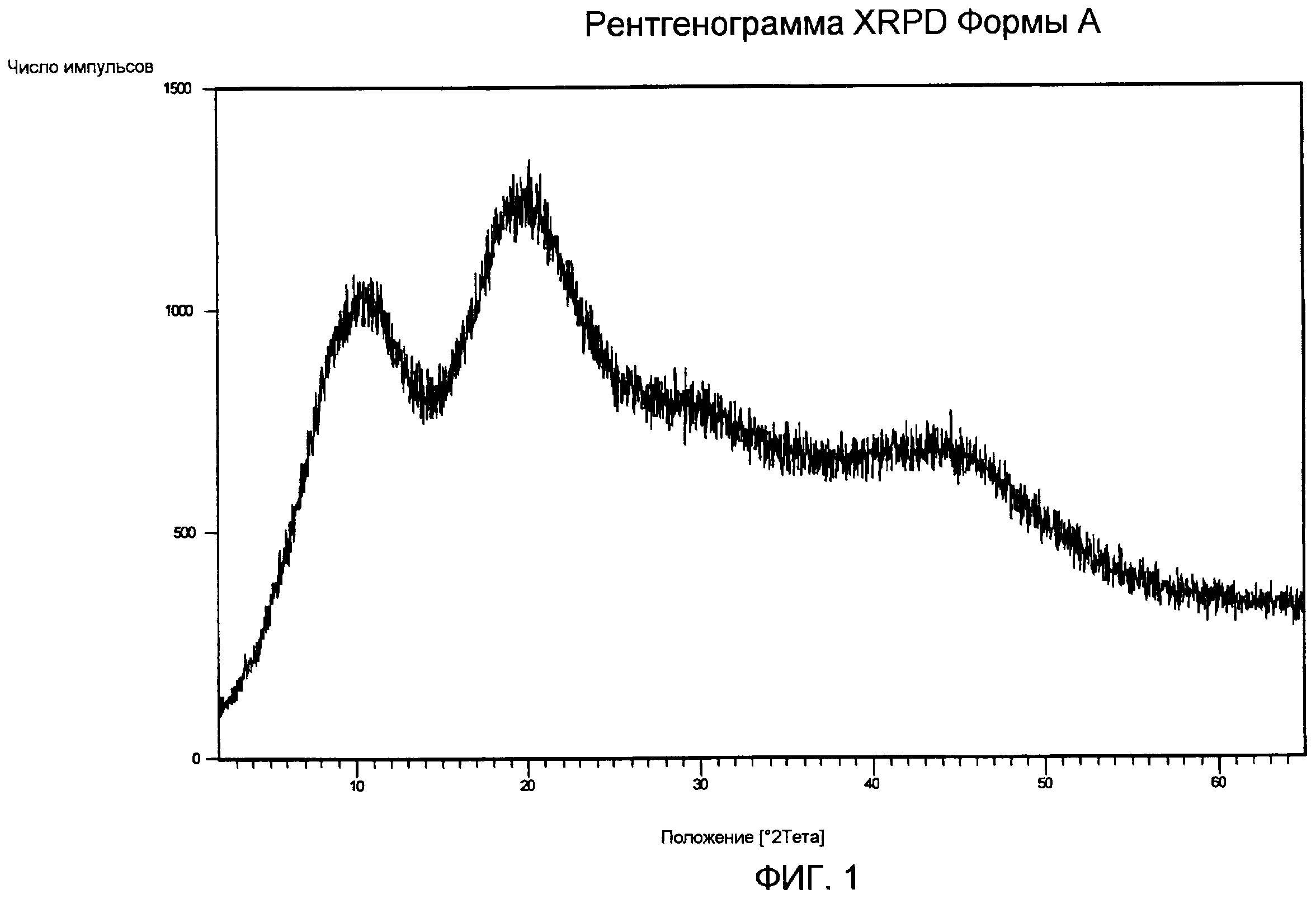
Порошковая рентгенограмма Формы А (фиг.1) типична для аморфных продуктов с полным отсутствием дифракционных пиков.
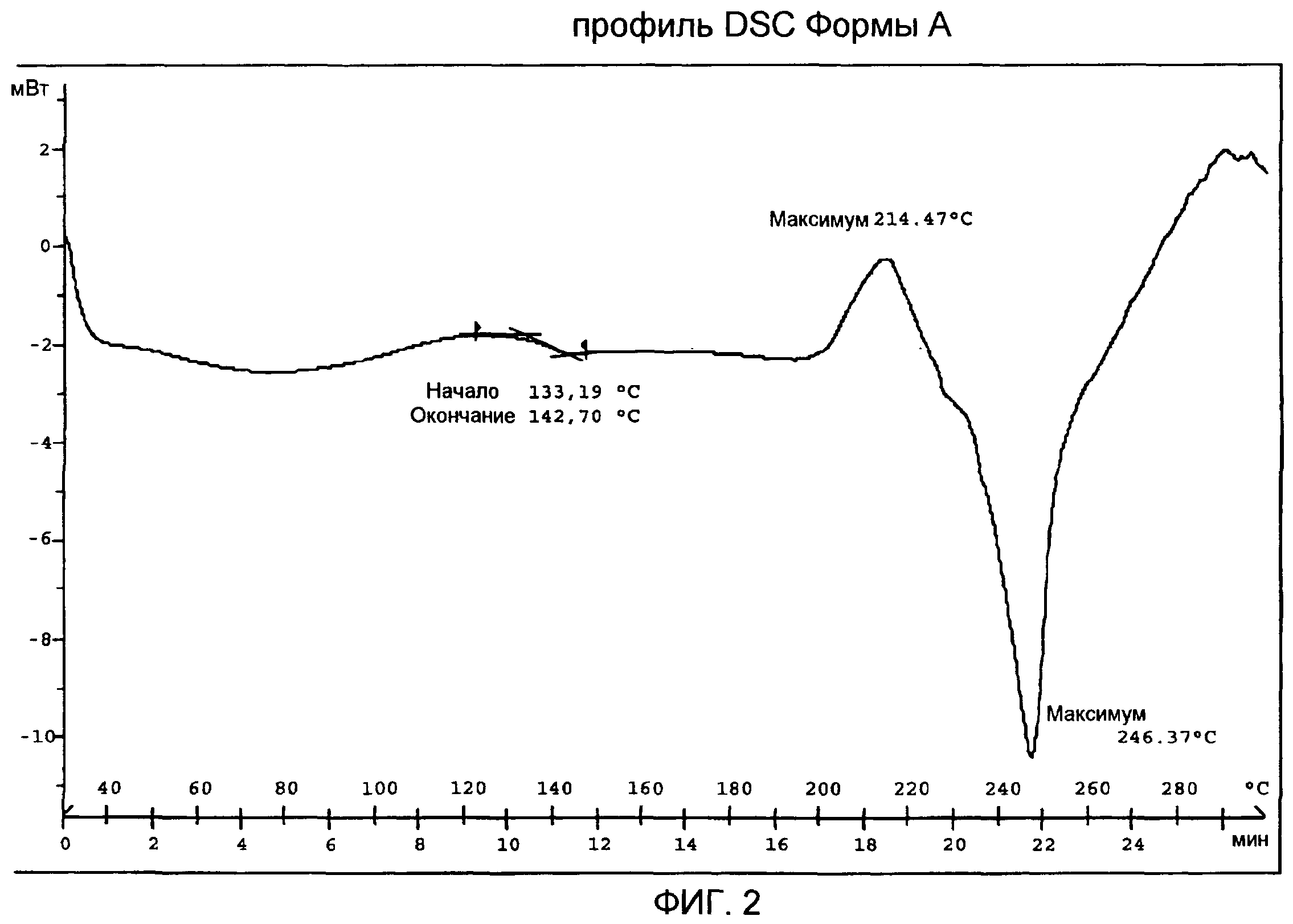
Кривая DSC Формы А (фиг.2) показывает слабый и широкий эндотермический сигнал с максимумом приблизительно при 80°С, смещением базовой линии в связи с Tg между 133°С и 143°С, экзотермический пик с максимумом приблизительно при 214°С в связи с рекристаллизацией расплавленного продукта и последующий пик плавления с максимумом приблизительно при 246°С, с последующим разложением.
Спектр IR Формы А (фиг.3) показывает характеристические частоты поглощения при 3442, 2960, 1821, 1732, 1714, 1368, 1236, 1162, 1085, 1068, 984, 907, 776, 763, 711 см-1.
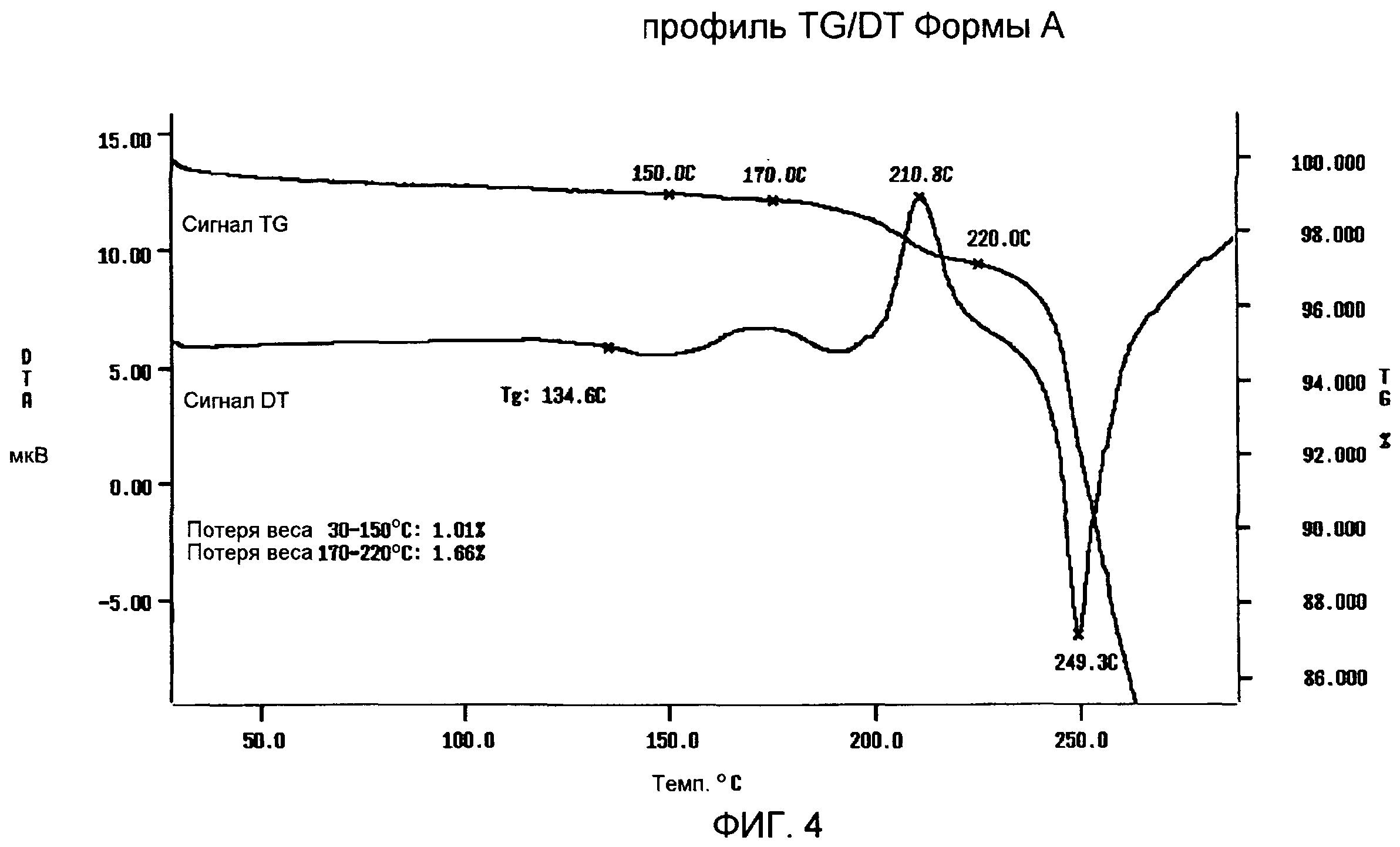
TG/DT анализ Формы А (фиг.4) подтверждает DSC анализ, демонстрирующий профиль DT, характеризуемый смещением базовой линии в связи с Tg между 130°С и 143°С, экзотермическим пиком с максимумом приблизительно при 211°С в связи с рекристаллизацией расплавленного продукта и последующим пиком плавления с максимумом приблизительно при 249°С, с последующим разложением. Профиль TG показывает потерю веса приблизительно на 1,0% от 30 до 150°С вследствие испарения остаточной влаги и потерю веса приблизительно на 1,6%, которая происходит при рекристаллизации с последующей огромной потерей веса в результате реакции разложения.
Оптическая микроскопия показывает, что твердая Форма А образована хрупкими, неровными частицами с большим разнообразием размеров и отсутствием четко обозначенных кристаллических Форм.
Форма В
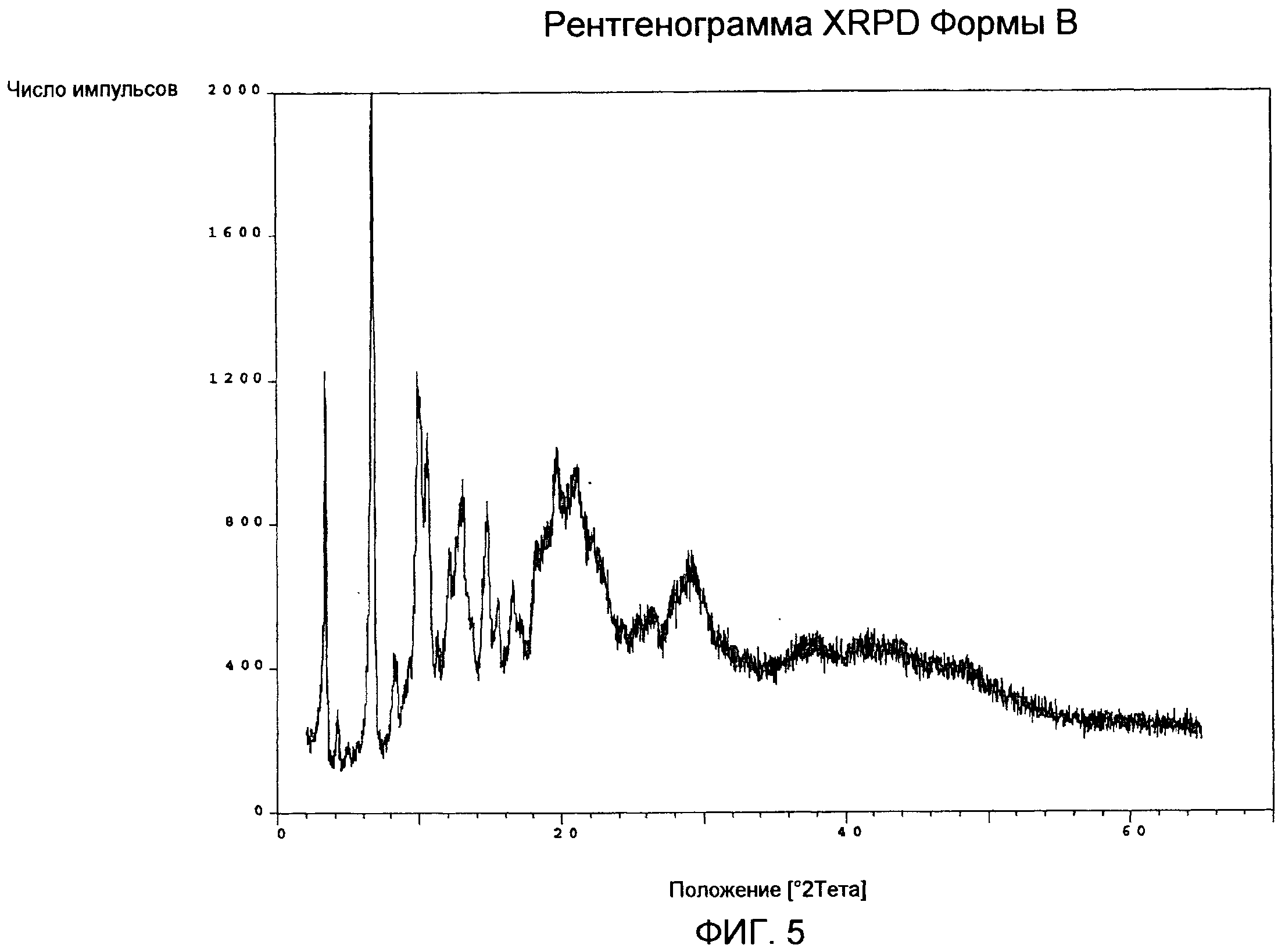
Порошковая рентгенограмма Формы В (фиг.5) демонстрирует кристаллическую структуру с соответствующими характерными пиками приблизительно при 3,5, 6,8, 9,9, 10,1, 10,7, 12,1, 13,1, 14,8, 18,2, 19,7, 21,3, 29,3 град 2-тета.
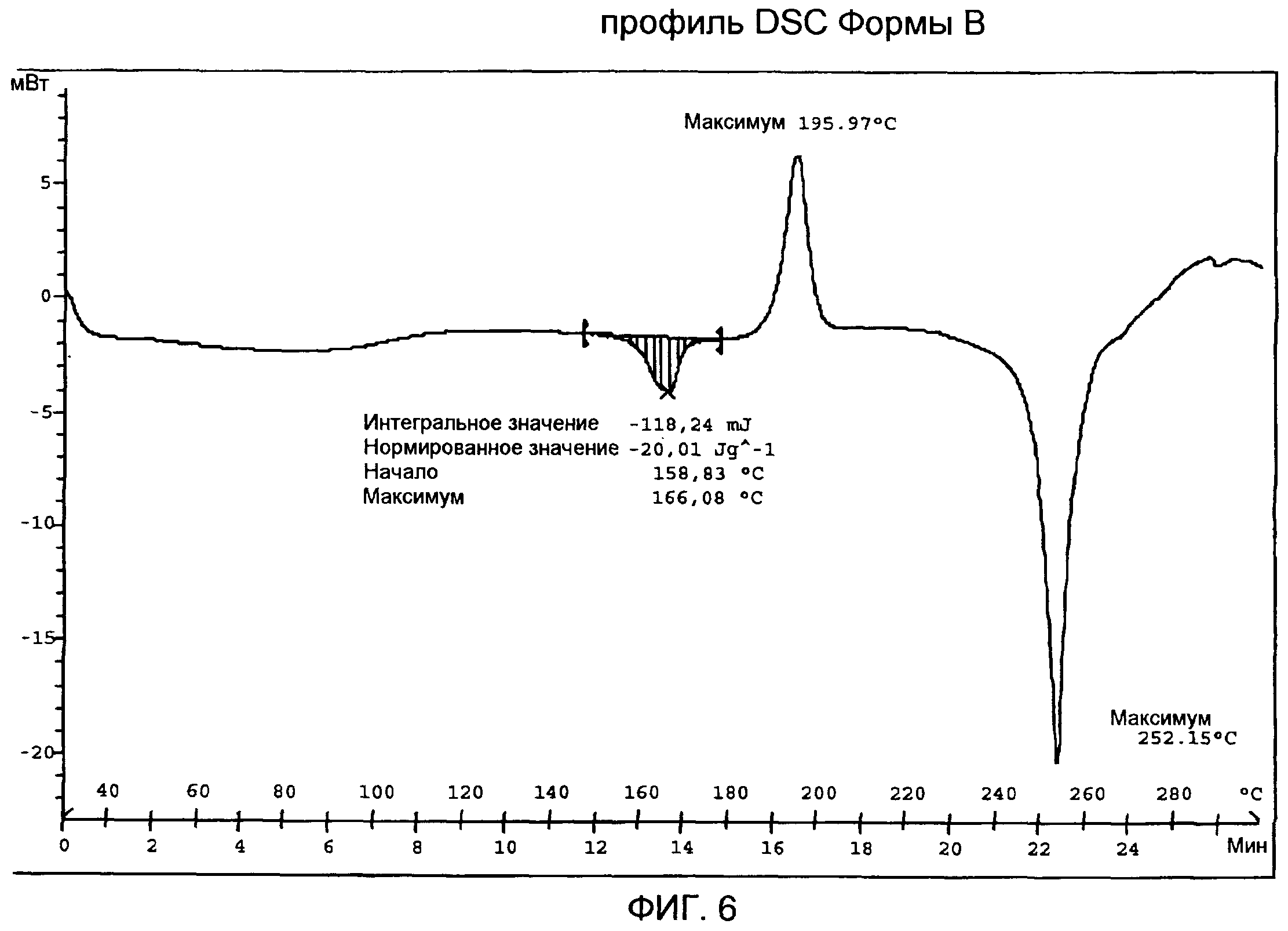
Кривая DSC Формы В (фиг.6) показывает слабый и широкий эндотермический сигнал с максимумом ниже 100°С, первый пик плавления с максимумом приблизительно при 166°С и ∆Hfus величиной приблизительно -20 Дж/г, экзотермический пик с максимумом приблизительно при 196°С в связи с рекристаллизацией расплавленного продукта и второй пик плавления с максимумом приблизительно при 252°С, с последующим разложением.
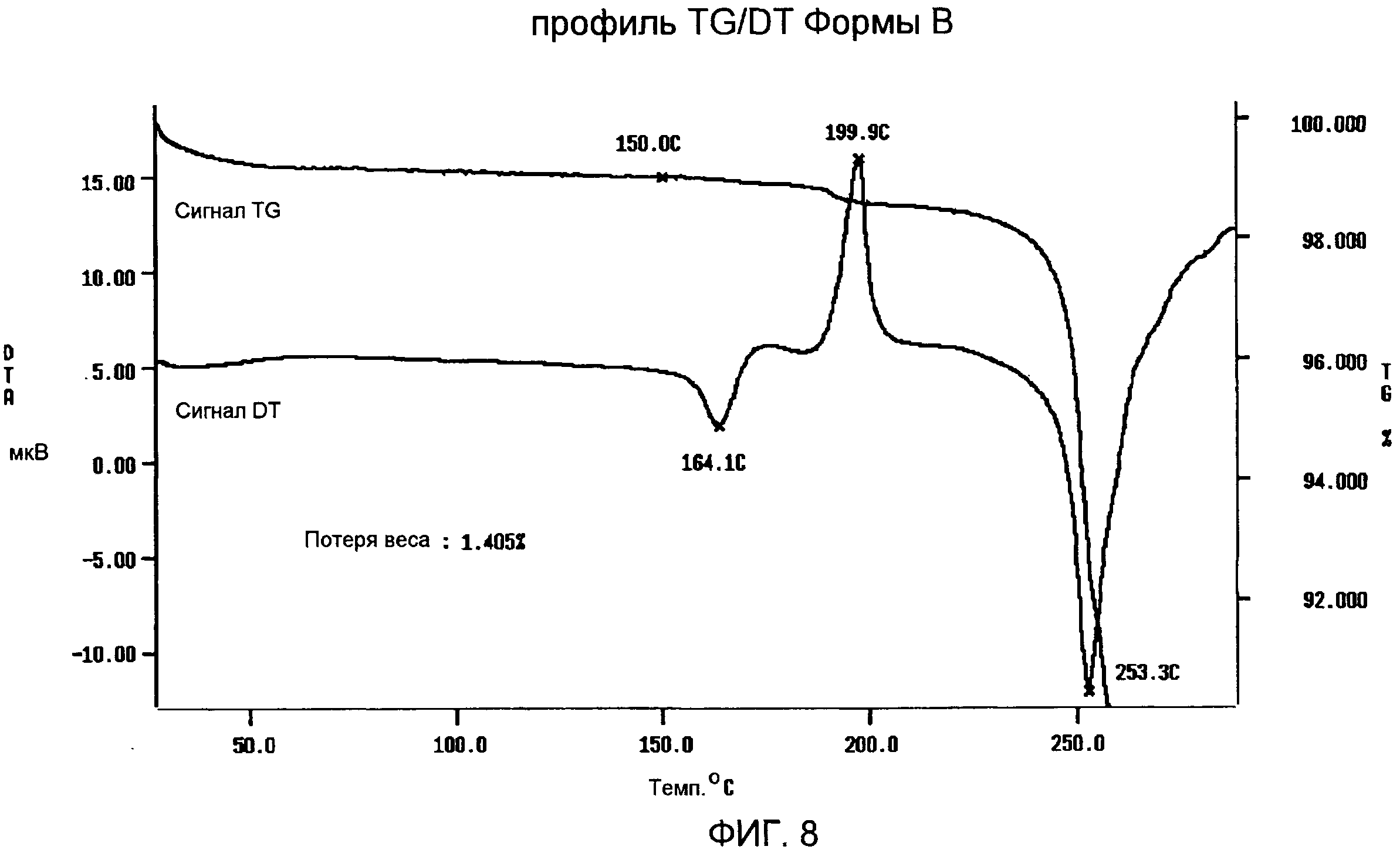
Спектр IR Формы В (фиг.7) показывает характеристические частоты поглощения при 3444, 2961, 1816, 1735, 1720, 1689, 1368, 1237, 1163, 1085, 1068, 1047, 989, 949, 907, 776, 764, 710 см-1. TG/DT анализ Формы В (фиг.8) подтверждает DSC анализ, выявляющий слабый и широкий эндотермический сигнал с максимумом ниже 100°С вследствие потери остаточной влаги, первый пик плавления с максимумом приблизительно при 164°С, экзотермический пик с максимумом приблизительно при 200°С в результате рекристаллизации расплавленного продукта и второй пик плавления с максимумом приблизительно при 253°С, с последующим разложением. В профиле TG за потерей веса приблизительно на 1,4% от 30 до 150°С вследствие испарения остаточной влаги следует огромная потеря веса, которая происходит выше 240°С в связи с реакцией разложения.
Оптическая микроскопия показывает, что твердая Форма В образована остроконечными (игольчатыми) кристаллами.
Смесь Формы А и Формы В
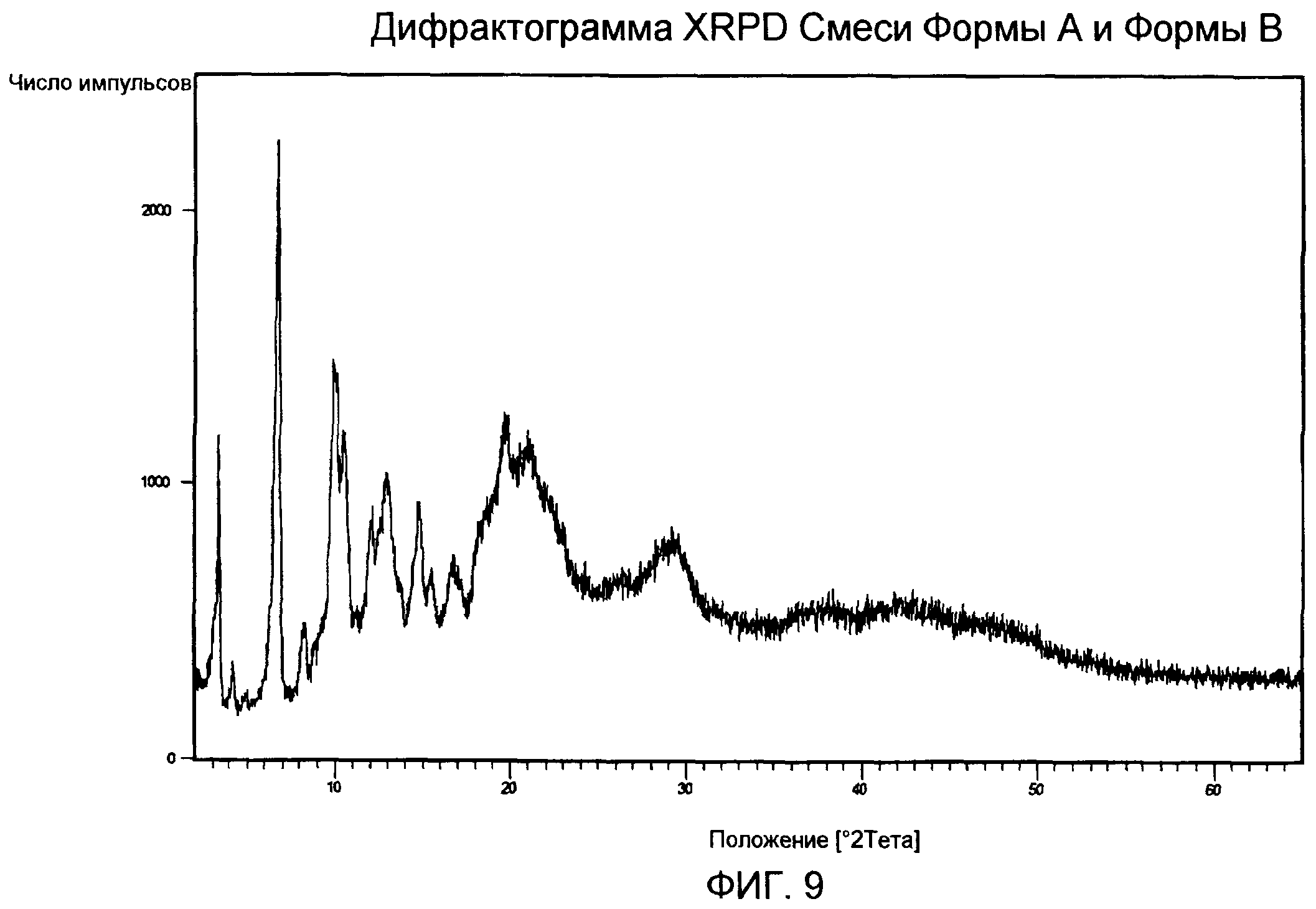
Порошковая рентгенограмма смеси Формы А и Формы В (фиг.9) демонстрирует кристаллическую структуру с характерными пиками приблизительно при 3,4, 6,8, 9,9, 10,6, 12,1, 13,1, 14,8, 18,1, 19,7, 21,2 град 2-тета за счет фракции Формы В в смеси.
Кривая DSC (фиг.10) показывает слабый и широкий эндотермический сигнал с максимумом ниже 100°С, первый пик плавления с максимумом приблизительно при 163°С и ∆Hfus величиной приблизительно -15 Дж/г, экзотермический пик с максимумом приблизительно при 202°С в результате рекристаллизации расплавленного продукта и второй пик плавления с максимумом приблизительно при 251°С, с последующим разложением.
Спектр IR (фиг.11) демонстрирует характеристические частоты поглощения при 3448, 2960, 1816, 1735, 1719, 1688, 1368, 1237, 1164, 1085, 1068, 1048, 989, 949, 906, 776, 764, 710 см-1.
TG/DT анализ (фиг.12) подтверждает DSC анализ, выявляя слабый и широкий эндотермический сигнал с максимумом ниже 100°С вследствие потери остаточной влаги, первый пик плавления с максимумом приблизительно при 162°С, экзотермический пик с максимумом приблизительно при 202°С в результате рекристаллизации расплавленного продукта и второй пик плавления с максимумом приблизительно при 250°С, с последующим разложением. В профиле TG за потерей веса приблизительно на 2,7% от 30 до 150°С вследствие испарения остаточной влаги следует огромная потеря веса, которая происходит при 240°С в результате реакции разложения.
Оптическая микроскопия показывает, что Смесь Формы А и Формы В образована призматическими кристаллами.
Эти данные отчетливо указывают на то, что полиморфные Формы А и В Ортатаксела легко отличимы от псевдополиморфного ацетонового сольвата с помощью XRPD, DSC, IR и анализов содержания растворителя (такими как термогравиметрия или газовая хроматография).
МАТЕРИАЛЫ И МЕТОДЫ
Порошковая рентгенограмма (XRPD)
Порошковые рентгенограммы были получены на дифрактометре Philips PW1800. Генератор рентгеновского излучения работал при 45 кВ и 40 мА с использованием трубки Cu Kα в качестве источника радиоактивного излучения. Образец располагали в соответствующей щели, и облучаемая длина составляла 10 мм. Данные были собраны в интервале углов от 2 до 65 град 2-тета с размером шага, равным 0,02 град 2-тета.
Дифференциальная Сканирующая Калориметрия (DSC)
Измерения дифференциальной сканирующей калориметрии были выполнены с использованием Системы Mettler TC15, оборудованной измерительной кюветой DSC20, используя закрытые алюминиевые тигли (объемом 40 мкл) с небольшим отверстием. Тепловой поток регистрировали от 30 до 300°С с линейной скоростью нагрева, равной 10°С/мин, при потоке азота 50 мл/мин. Приблизительно 5 мг порошка было использовано для каждого измерения.
Термогравиметрия и дифференциальный термический анализ
Исследования были выполнены, применяя одновременную систему Seiko TG/DTA6200 с использованием открытых алюминиевых кювет (объемом 40 мкл). Сигналы TG/DT регистрировались от 30 до 300°С с линейной скоростью нагрева (10°С/мин) при потоке азота 200 мл/мин. Приблизительно 10 мг порошка было использовано для каждого измерения.
Инфракрасная спектроскопия с Фурье-преобразованием (FTIR)
Инфракрасные спектры были записаны методом ATR, используя спектрометр с Фурье-преобразованием Perkin Elmer Spectrum One. Спектры были результатом получения и преобразования 16 наслоенных сканограмм в спектральной области 4000-550 см-1 с разрешением, равным 4 см-1.
Оптическая микроскопия
Исследования были выполнены с использованием микроскопа проходящего света Zeiss Axioskop. Для каждого исследования небольшое количество образца было диспергировано в силиконовом масле, нанесено на предметное стекло и покрыто покровным стеклом. Наблюдения были проведены в подходящих условиях освещения, контраста и увеличения.
ПРИМЕР 1 - Получение Формы А
Ортатаксел (13 г) был растворен в ацетоне (112,5 мл). Очищенная вода (555 мл), содержащая лимонную кислоту (12 мг), была мгновенно добавлена при перемешивании, вызывая осаждение аморфного твердого вещества, которое было профильтровано и промыто водой (65 мл), содержащей лимонную кислоту (18 мг). Образец был высушен при 40°С за 48 часов, обеспечивая получение 12 г белого твердого вещества, имеющего характеристики XRPD, DSC, IR и TG/DTA, описанные на фиг.1-4, соответственно.
ПРИМЕР 2 - Получение Формы В
Ортатаксел (14 г) был растворен в 95% этаноле (168 мл), содержащем лимонную кислоту (28 мг), при 50°С. Холодная деминерализованная вода (280 мл) была добавлена в полученный раствор в течение 15 минут. Суспензия была перемешана при 40°С в течение 6 часов. Смесь была охлаждена до 20°С, и белое твердое вещество было отфильтровано. Твердое вещество было промыто раствором этанола (168 мл) и водой (280 мл). Твердое вещество было высушено под вакуумом при 50°С за 40 часов, обеспечивая получение 13,4 г белого твердого вещества имеющего характеристики XRPD, DSC, IR и TG/DTA, описанные на фиг.5-8, соответственно.
ПРИМЕР 3 - Получение смеси приблизительно из 25% Формы А и 75% Формы В
Ортатаксел (14 г) был растворен в 95% этаноле (168 мл), содержащем лимонную кислоту (28 мг), при 50°С. Холодная деминерализованная вода (280 мл) была добавлена в конечный раствор в течение 15 минут. Смесь была сразу же охлаждена до 20°С, и белое твердое вещество было отфильтровано. Твердое вещество было промыто раствором этанола (168 мл) и водой (280 мл), содержащей лимонную кислоту (25 мг). Твердое вещество было высушено под вакуумом при 50°С за 40 часов, обеспечивая получение 13,4 г белого вещества, имеющего характеристики XRPD, DSC, IR и TG/DTA, описанные на фиг.9-12, соответственно.
ПРИМЕР 4 - Получение смесей Формы А и Формы В в различных количественных соотношениях
Форма А (1 г) была суспендирована в смеси из 95% этанола (12 мл) и воды (20 мл), содержащей лимонную кислоту (2 мг), при 40°С. Образцы были отобраны в разное время (t=0, t=5 мин, t=30 мин, t=6 ч) для демонстрации того, что разные количественные соотношения Формы А и Формы В могут быть получены. Фиг.13 иллюстрирует DSC анализ образцов при сравнении с кривой чистой Формы В.
Комбинации вазоактивных веществ с эстрогенами и их применение для лечения сексуальных дисфункций у женщин
Применение производных изотиоцианата в качестве противомиеломных средств
Комбинации вазоактивных веществ и их применение в лечении сексуальных дисфункций
Комбинации вазоактивных агентов, их применение в фармацевтической и косметической отраслях и содержащие их композиции
Составы с сангвинарином, хелеритрином или хелидонином для лечения бородавок, папиллом и псориатических бляшек
Способ гликозидирования колхицина и тиоколхицина
Комбинации вазопротекторных агентов и препаративные формы, содержащие такие комбинации
Противоопухолевые средства с бензофенантридиновой структурой и содержащие их препараты
Композиции для лечения и предупреждения инфекций полости рта
Композиции для лечения вагинальных инфекций с хроническим воспалением
Комбинации вазоактивных веществ с эстрогенами и их применение для лечения сексуальных дисфункций у женщин
Применение производных изотиоцианата в качестве противомиеломных средств
Комбинации вазоактивных веществ и их применение в лечении сексуальных дисфункций
Комбинации вазоактивных агентов, их применение в фармацевтической и косметической отраслях и содержащие их композиции
Составы с сангвинарином, хелеритрином или хелидонином для лечения бородавок, папиллом и псориатических бляшек
Способ гликозидирования колхицина и тиоколхицина
Комбинации вазопротекторных агентов и препаративные формы, содержащие такие комбинации
Противоопухолевые средства с бензофенантридиновой структурой и содержащие их препараты
Композиции для лечения и предупреждения инфекций полости рта
Композиции для лечения вагинальных инфекций с хроническим воспалением

