Результат интеллектуальной деятельности: Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида
Вид РИД
Изобретение
Область техники
Настоящее изобретение относится к кристаллу (6S,9aS)-N-бензил-6-[(4-гидроксифенил)метил]-4,7-диоксо-8-({6-[3-(пиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-2-(проп-2-ен-1-ил)-октагидро-1H-пиразино[2,1-c][1,2,4]триазин-1-карбоксамидного соединения.
Предпосылки изобретения
Сигнальный путь Wnt сохраняется независимо от отличия в видах организмов и известен как важный путь, участвующий в развитии, дифференциации и поддержании жизнедеятельности живых организмов. Однако в последние годы сообщалось о том, что конститутивная активация данного пути вовлечена в развитие злокачественной трансформации фиброза и рака. В частности, при колоректальном раке, меланоме, эндометриальном раке, раке печени и раке предстательной железы известно, что сигнальный путь Wnt может быть активирован конститутивно посредством супрессируемой мутации гена аденоматозного полипоза толстой кишки (APC) или активирующей мутации β-катенина или т.п. Также известно, что при раке поджелудочной железы, гемобластозах, раке печени и им подобных сигнальный путь Wnt может быть активирован после обработки известным противоопухолевым средством.
В непатентных литературных источниках 1 и 2 описывается, что превосходная противоопухолевая активность может достигаться посредством ингибирования сигнального пути Wnt. В непатентных литературных источниках 12, 13 и 14 описывается, что превосходный эффект в отношении фиброза может достигаться посредством ингибирования сигнального пути Wnt. Таким образом, сигнальный путь Wnt привлек к себе внимание в качестве новой мишени для лечения опухолей или лечения фиброза.
В непатентных литературных источниках 3, 4, 5, 6, 7, 8, 9, 10 и 11 раскрываются соединения или антитела, способные к ингибированию сигнальных путей Wnt, а также сообщается, что данные соединения или антитела могут воздействовать на танкиразу, Traf2- и Nck-взаимодействующую киназу (TNIK), Porcupine, рецептор Frizzled и им подобные.
Кроме того, каждое из соединений, содержащих октагидро-1H-пиразино[2,1-c][1,2,4]триазиновую главную цепь, известно в качестве модулятора сигнального пути Wnt, при этом отмечена взаимосвязь между соединениями и заболеваниями, такими как рак и фиброз (патентный литературный источник 1-3 литературы).
Список использованной литературы
Патентная литература
[Патентный литературный источник 1] WO 2009/051397 A
[Патентный литературный источник 2] US 2010/0286094 A
[Патентный литературный источник 3] WO 2009/148192 A
Непатентная литература
[Непатентный литературный источник 1] Nick Barker et al., "Mining the Wnt pathway for cancer therapeutics", Nature reviews Drug discovery 2006 Dec; 5(12):997-1014.
[Непатентный литературный источник 2] Katayoon H. Emami et al., "A small molecule inhibitor of beta-catenin/CREB-binding protein transcription", Proc. Natl. Acad. Sci. USA., 2004, 101(34), p.12682-12687.
[Непатентный литературный источник 3] Baozhi Chen et al., "Small molecule-mediated disruption of Wnt-dependent signaling in tissue regeneration and cancer", Nat Chem Biol., 2009, 5(2), p.100-107.
[Непатентный литературный источник 4] Shih-Min A. Huang et al., "Tankyrase inhibition stabilizes axin and antagonizes Wnt signalling", Nature, 2009, 461, p.614-620.
[Непатентный литературный источник 5] Lari Lehtio et al., "Tankyrases as drug targets", The FEBS Journal, 2013, 280, 3576-3593.
[Непатентный литературный источник 6] Miki Shitashige et al., "Traf2- and Nck-Interacting Kinase Is Essential for Wnt Signaling and Colorectal Cancer Growth", Cancer Res., 2010, 70(12), 5024-5033.
[Непатентный литературный источник 7] Austin Gurney et al., "Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumors", Proc. Natl. Acad. Sci. USA., 2012, 109(29), 11717-11722.
[Непатентный литературный источник 8] Xiaomo Jiang et al., "Inactivating mutations of RNF43 confer Wnt dependency in pancreatic ductal adenocarcinoma", Proc. Natl. Acad. Sci. USA., 2013, 110(31), 12649-12654.
[Непатентный литературный источник 9] Jo Waaler et al., "Novel Synthetic Antagonists of Canonical Wnt Signaling Inhibit Colorectal Cancer Cell Growth", Cancer Res, 2011, 71(1), 197-205.
[Непатентный литературный источник 10] H Yao et al., "AV-65, a novel Wnt/β-catenin signal inhibitor, successfully suppresses progression of multiple myeloma in a mouse model", Blood Cancer Journal, 2011, 1, e43.
[Непатентный литературный источник 11] De Robertis A et al., "Identification and characterization of a small-molecule inhibitor of Wnt signaling in glioblastoma cells", Mol Cancer Ther., 2013, 12(7), 1180-1189.
[Непатентный литературный источник 12] Anna P. Lam et al., "β-catenin signaling: a novel mediator of fibrosis and potential therapeutic target", Curr Opin Rheumatol. 2011 November; 23(6): 562-567.
[Непатентный источник 13] Sha Hao et al., ʺTargeted Inhibition of β-Catenin/CBP Signaling Ameliorates Renal Interstitial Fibrosisʺ, J. Am. Soc. Nephrol. 22: 1642-1653, 2011.
[Непатентный литературный источник 14] William R. Henderson, Jr. et al., "Inhibition of Wnt/β-catenin/CREB binding protein(CBP) signaling reverses pulmonary fibrosis", Proc. Natl. Acad. Sci. USA., 2010, 107(32), 14309-14314.
Сущность изобретения
Техническая задача
(6S,9aS)-N-бензил-8-({6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамид (в данном документе далее называемый как "соединение 1"), который является соединением, представленным следующей формулой, характеризуется активностью в отношении модуляции пути Wnt.
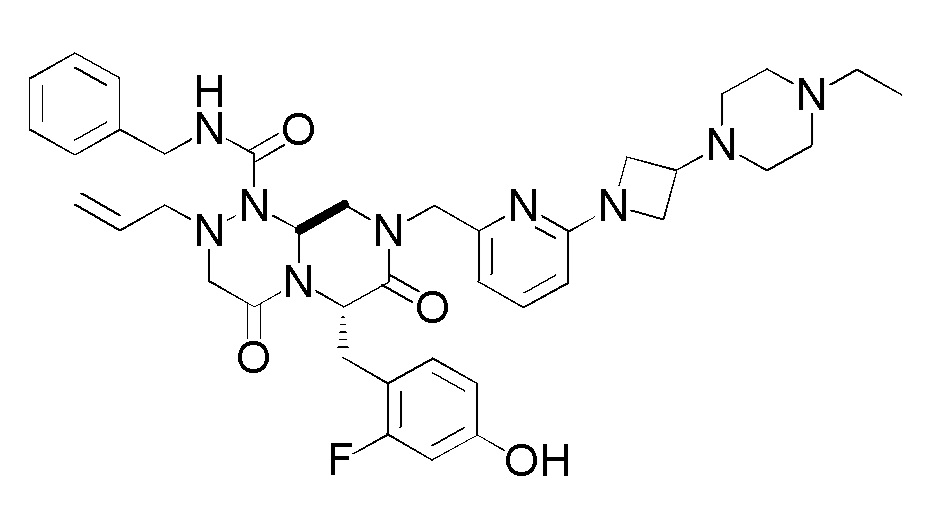
В целом физические свойства соединений и их солей и их кристаллов, используемых в качестве фармацевтических продуктов, оказывают большое влияние на биодоступность лекарственных средств, чистоту лекарственных веществ, фармацевтические составы и им подобные. Таким образом, задача, решаемая в настоящем изобретении, заключается в предоставлении кристалла соединения 1, которое характеризуется потенциально возможным применением в качестве лекарственного вещества для фармацевтических продуктов.
Решение задачи
Авторы настоящего изобретения завершили настоящее изобретение в результате целенаправленных усилий.
Таким образом, настоящее изобретение предусматривает следующие [1]-[12]:
[1] Кристалл (6S,9aS)-N-бензил-8-({6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида.
[2] Кристалл согласно [1], где кристалл характеризуется дифракционным пиком при угле дифракции (2θ±0,2°) 5,8° согласно измерению с помощью порошковой рентгеновской дифракции.
[3] Кристалл согласно [1], где кристалл характеризуется дифракционными пиками при углах дифракции (2θ±0,2°) 5,8°, 6,4° и 10,1° согласно измерению с помощью порошковой рентгеновской дифракции.
[4] Кристалл согласно [3], где кристалл также характеризуется дифракционными пиками при углах дифракции (2θ±0,2°) 8,0° и 12,8° согласно измерению с помощью порошковой рентгеновской дифракции.
[5] Кристалл согласно [4], где кристалл дополнительно характеризуется дифракционными пиками при углах дифракции (2θ±0,2°) 14,2°, 16,0°, 18,9°, 19,7° и 23,1° согласно измерению с помощью порошковой рентгеновcкой дифракции.
[6] Кристалл (6S,9aS)-N-бензил-8-({6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида, где кристалл характеризуется пиком при химическом сдвиге (δ±0,5 ppm) 154,7 ppm в спектре твердофазного 13C-ЯМР.
[7] Кристалл согласно [6], где кристалл дополнительно характеризуется пиками при химических сдвигах (δ±0,5 ppm) 141,1 ppm и 158,1 ppm в спектре твердофазного 13C-ЯМР.
[8] Кристалл согласно [7], где кристалл дополнительно характеризуется пиками при химических сдвигах (δ±0,5 ppm) 134,0 ppm и 165,1 ppm в спектре твердофазного 13C-ЯМР.
[9] Кристалл согласно [8], где кристалл дополнительно характеризуется пиками при химических сдвигах (δ±0,5 ppm) 12,6 ppm, 55,5 ppm и 118,5 ppm в спектре твердофазного 13C-ЯМР.
[10] Кристалл (6S,9aS)-N-бензил-8-({6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида, где кристалл характеризуется по существу такой же порошковой рентгеновской дифрактограммой, как порошковая рентгеновская дифрактограмма, показанная на фиг. 4.
[11] Кристалл (6S,9aS)-N-бензил-8-({6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида, где кристалл характеризуется по существу таким же спектром твердофазного 13C-ЯМР, как спектр твердофазного 13C-ЯМР, показанный на фиг. 6.
[12] Фармацевтическая композиция, содержащая кристалл согласно любому из [1] - [11].
Полезные эффекты изобретения
Кристалл соединения 1 согласно настоящему изобретению характеризуется такими свойствами, которые показаны в примерах, и характеризуется потенциально возможным применением в качестве лекарственного вещества для фармацевтических продуктов.
Краткое описание графических материалов
На фиг. 1 показаны результаты испытательного примера 2. На оси ординат показано количество полипов на мышь.
На фиг. 2 показаны результаты испытательного примера 3. На оси абсцисс показано количество прошедших дней, а на оси ординат показан относительный объем опухоли (RTV) по отношению к объему опухоли в день 0.
На фиг. 3 показаны результаты испытательного примера 3. На оси абсцисс показано количество прошедших дней, а на оси ординат показана относительная масса тела (RBW) по отношению к массе тела в день 0.
На фиг. 4 представлена порошковая рентгеновская дифрактограмма для кристалла соединения 1, полученная в испытательном примере 4. На оси абсцисс показан угол дифракции (2θ), а на оси ординат показана интенсивность пика.
На фиг. 5 показаны результаты испытательного примера 5. На оси абсцисс показана температура, на левой оси ординат показано изменение массы согласно термогравиметрии (TG), а на правой оси ординат показана теплопередача согласно дифференциальному термическому анализу (DTA).
На фиг. 6 представлен спектр твердофазного 13C-ЯМР для кристалла соединения 1 в испытательном примере 6. На оси абсцисс показан химический сдвиг (δ), а на оси ординат показана интенсивность пика.
Описание вариантов осуществления
В настоящей заявке примеры предпочтительных кристаллов включают:
кристалл соединения 1, характеризующийся дифракционным пиком при угле дифракции (2θ±0,2°) 5,8° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл соединения 1, характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 5,8°, 6,4° и 10,1° согласно измерению с помощью порошковой рентгеновcкой дифракции;
кристалл соединения 1, характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 5,8°, 6,4°, 8,0°, 10,1° и 12,8° согласно измерению с помощью порошковой рентгеновcкой дифракции; и
кристалл соединения 1, характеризующийся дифракционными пиками при углах дифракции (2θ±0,2°) 5,8°, 6,4°, 8,0°, 10,1°, 12,8°, 14,2°, 16,0°, 18,9°, 19,7° и 23,1° согласно измерению с помощью порошковой рентгеновcкой дифракции.
Каждый из вышеуказанных дифракционных пиков в порошковой рентгеновской дифракции является характерным для кристаллов соединения 1 и свойственным им.
В целом, поскольку угол дифракции (2θ) в порошковой рентгеновcкой дифракции может иметь погрешность в пределах±0,2°, следует понимать, что вышеуказанные значения углов дифракции также включают значения в пределах приблизительно±0,2°. Таким образом, не только кристаллы с полностью соответствующими углами дифракции пиков при измерении с помощью порошковой рентгеновcкой дифракции, но и также кристаллы с углами дифракции пиков, находящиеся в пределах погрешности, составляющей приблизительно±0,2°, включены в настоящее изобретение.
В настоящем описании, например, "характеризуется дифракционным пиком при угле дифракции (2θ±0,2°) 5,8°" означает "характеризуется дифракционным пиком при угле дифракции (2θ) от 5,6° до 6,0°", причем это применимо и к другим углам дифракции.
В целом, если даже формы кристаллов идентичны друг другу, интенсивность пика или ширина на половине высоты при угле дифракции (2θ) в порошковой рентгеновской дифракции изменяется в зависимости от измерения, из-за различия в условиях измерения и разницы в размере и форме частиц кристаллов порошка, используемых в качестве образцов для измерения, и постоянные интенсивности пиков или значения ширины на половине высоты не обязательно показаны. По этой причине при сравнении порошковых дифракционных рентгенограмм, если углы дифракции (2θ) идентичны друг другу, но их интенсивности пиков или значения ширины на половине высоты отличаются, разница не означает, что формы измеренных кристаллов отличаются друг от друга. Таким образом, это означает, что кристалл соединения, характеризующийся порошковой дифракционной рентгенограммой с таким отличием от дифракционного пика конкретного кристалла по настоящему изобретению, имеет такую же форму кристалла, как и кристалл соединения по настоящему изобретению. В настоящем описании "характеризуется по существу такой же порошковой рентгеновской дифрактограммой, как и порошковая рентгеновская дифрактограмма, показанная на фиг. 4" означает, что порошковая рентгеновская дифрактограмма с характерным дифракционным пиком является такой же, как и порошковая рентгеновская дифрактограмма, показанная на фиг. 4, не только тогда, когда она полностью соответствует порошковой рентгеновской дифрактограмме, показанной на фиг. 4, но и когда пик ее интенсивности или ширина на половине высоты отличается или находится в пределах диапазона погрешности, составляющего приблизительно±0,2°, порошковой рентгеновской дифрактограммы, показанной на фиг. 4. Таким образом, это означает, что все кристаллы с такими рентгеновскими порошковыми дифрактограммами являются такими же кристаллами, как и кристалл по настоящему изобретению.
В настоящей заявке примеры предпочтительных кристаллов включают:
кристалл соединения 1, характеризующийся пиком при химическом сдвиге (δ±0,5 ppm) 154,7 ppm в спектре твердофазного 13C-ЯМР;
кристалл соединения 1, характеризующийся пиками при химических сдвигах (δ±0,5 ppm) 141,1 ppm, 154,7 ppm и 158,1 ppm в спектре твердофазного 13C-ЯМР;
кристалл соединения 1, характеризующийся пиками при химических сдвигах (δ±0,5 ppm) 134,0 ppm, 141,1 ppm, 154,7 ppm, 158,1 ppm и 165,1 ppm в спектре твердофазного 13C-ЯМР; и
кристалл соединения 1, характеризующийся пиками при химических сдвигах (δ±0,5 ppm) 12,6 ppm, 55,5 ppm, 118,5 ppm, 134,0 ppm, 141,1 ppm, 154,7 ppm, 158,1 ppm и 165,1 ppm в спектре твердофазного 13C-ЯМР.
Каждый из вышеуказанных пиков в спектре твердофазного 13C-ЯМР является характерным для кристаллов соединения 1 и свойственным им.
В настоящем описании "характеризующийся пиками при химических сдвигах (δ±0,5 ppm) 12,6 ppm, 55,5 ppm, 118,5 ppm, 134,0 ppm, 141,1 ppm, 154,7 ppm, 158,1 ppm и 165,1 ppm" означает "характеризующийся пиками, по существу эквивалентными пикам при химических сдвигах (δ±0,5 ppm) 12,6 ppm, 55,5 ppm, 118,5 ppm, 134,0 ppm, 141,1 ppm, 154,7 ppm, 158,1 ppm и 165,1 ppm соответственно, когда твердофазную 13C-ЯМР спектроскопию проводят в нормальных условиях или по существу в таких же условиях, которые описаны в настоящем описании".
В определении "характеризуются ли по существу эквивалентными пиками или нет" в целом, учитывая, что химический сдвиг (δ) в спектре твердофазного 13C-ЯМР может иметь погрешность в пределах ±0,5 ppm, следует понимать, что вышеуказанные значения химических сдвигов также включают значения в пределах приблизительно ±0,5 ppm. Таким образом, не только кристаллы с полностью соответствующими химическими сдвигами в спектре твердофазного 13C-ЯМР, но и также кристаллы с химическими сдвигами, находящимися в пределах погрешности, составляющей приблизительно ±0,5 ppm, включены в настоящее изобретение. По этой причине в настоящем описании, например, "характеризуется пиком при химическом сдвиге (δ±0,5 ppm) 12,6 ppm" означает "характеризуется пиком при химическом сдвиге (δ) от 12,1 ppm до 13,1 ppm", причем это применимо и к другим химическим сдвигам в спектре твердофазного 13C-ЯМР. Кроме того, "кристалл, характеризующийся по существу таким же спектром твердофазного 13C-ЯМР, как и показанный на фиг. 6" означает, что не только тогда, когда спектр твердофазного 13C-ЯМР, характеризующийся пиком при определенном химическом сдвиге, полностью соответствует спектру твердофазного 13C-ЯМР, показанному на фиг. 6, но и также тогда, когда интенсивность его пика отличается или его характерный пик находится в пределах диапазона погрешности, составляющей приблизительно±0,5 ppm, кристалл представляет собой кристалл, характеризующийся таким же спектром твердофазного 13C-ЯМР, как и показанный на фиг. 6. Таким образом, под этим подразумевается, что все кристаллы, характеризующиеся таким спектром твердофазного 13C-ЯМР, являются такими же кристаллами, как и кристалл по настоящему изобретению.
Далее в данном документе описывается способ получения кристалла соединения 1 и ему подобных, что представляет собой вариант осуществления настоящего изобретения.
Получение соединения 1
Соединение 1 можно получать посредством способа, описанного в примерах и примерах получения, указанных ниже.
Способ получения кристалла соединения 1
Кристалл соединения 1 можно получать посредством вышеуказанного способа получения соединения 1 или можно получать посредством нагревания и растворения соединения 1 в растворителе и охлаждения его при перемешивании с целью его кристаллизации.
Соединение 1, используемое для кристаллизации, может быть в любой форме, может быть в форме сольвата, или гидрата, или ангидрата, может быть аморфным или кристаллическим (включая состоящие из множества полиморфов), и может быть любой их смесью, но предпочтительно представляет собой ангидрат.
Примеры растворителя для кристаллизации включают, например, спиртовые растворители, такие как метанол, этанол, 1-пропанол и 2-пропанол; ацетонитрил; амидные растворители, такие как N,N-диметилформамид; сложноэфирные растворители, такие как этилацетат и изопропилацетат; растворители на основе насыщенных углеводородов, такие как гексан и гептан; растворители на основе кетонов, такие как ацетон и 2-бутанон; растворители на основе простых эфиров, такие как простой трет-бутилметиловый эфир; или воду. Эти растворители можно использовать по отдельности или в смеси из двух или более из них. При осуществлении кристаллизации в смеси из двух или более растворителей, предпочтительно использовать, например, комбинацию гептана и 1-пропанола.
Количество растворителя, который будет использоваться, можно соответствующим образом подобрать, причем нижний предел представляет собой количество, в котором или выше которого соединение 1 растворяется при нагревании, или количество, в котором или выше которого можно получить его суспензию, а верхний предел представляет собой количество, в котором или ниже которого выход кристалла существенно не снижается.
Кристалл, полученный посредством вышеуказанного способа, одну кристаллическую форму. Эта форма кристалла стабильна, не переходит легко в другие кристаллические формы или в аморфную форму, характеризуется надлежащими физическими свойствами, а также является подходящей для составления.
В ходе кристаллизации затравочный кристалл (такой как требуемый кристалл соединения 1) можно добавлять или можно не добавлять. Температура добавления затравочных кристаллов конкретно не ограничена, но предпочтительно составляет от 0 до 100°C.
В отношении температуры, до которой соединение 1 нагревают для растворения, температуру, при которой соединение 1 растворяется, можно соответствующим образом выбирать в зависимости от растворителя, но предпочтительно в диапазоне от 50°C до температуры, при которой начинается рециркулирование растворителя для перекристаллизации, и более предпочтительно от 60 до 100°C.
Поскольку кристаллы, предусматривающие различные аспекты кристаллов (полиморфов), получают при гашении, охлаждение во время кристаллизации предпочтительно осуществляют путем соответствующего регулирования скорости охлаждения с учетом влияния на качество и размер частиц кристалла, и предпочтительно, например, при скорости охлаждения от 5 до 40°C/час. Это более предпочтительно при скорости охлаждения от 5 до 25°C/час.
Конечную температуру кристаллизации можно соответствующим образом подобрать в зависимости от выхода и качества кристалла и им подобных, и она предпочтительно составляет от -25 до 30°C.
Кристаллизованный кристалл отделяют посредством стандартной технологической операции фильтрации, и кристалл, отделенный посредством фильтрации, можно промывать растворителем, если в этом существует необходимость, а затем высушить с получением требуемого кристалла. В качестве растворителя, используемого для промывания кристалла, можно использовать тот же растворитель, что и при кристаллизации. Примеры такого растворителя предпочтительно включают, например, этанол, ацетон, 2-бутанон, этилацетат, простой диэтиловый эфир, простой трет-бутилметиловый эфир, гексан и гептан. Эти растворители можно использовать по отдельности или в смеси из двух или более из них.
Кристалл, выделенный посредством технологической операции фильтрации, можно сушить, оставляя его на воздухе или в атмосфере азота, если в этом существует необходимость, или посредством нагревания.
На протяжении периода сушки время, за которое количество остаточного растворителя опускается ниже заранее определенного количества, можно соответствующим образом подобрать в зависимости от производимого количества, аппарата для сушки, температуры сушки и им подобного. Высушивание можно проводить в условиях принудительной подачи воздуха или пониженного давления. Степень вакуума можно подходящим образом выбирать в зависимости от производимого количества, аппарата для сушки, температуры сушки и им подобных. После сушки полученный кристалл можно также оставлять на воздухе, если в этом существует необходимость.
Фармацевтические композиции можно получать путем добавления фармацевтически приемлемых добавок к кристаллу соединения 1, если в этом существует необходимость. Примеры лекарственной формы фармацевтической композиции включают препараты для перорального применения (таблетки, гранулы, порошки, капсулы, сиропы и т.д.), инъекционные препараты (для внутривенного введения, внутримышечного введения, подкожного введения, интраперитонеального введения и т.д.) и препараты для наружного применения (препараты для трансдермального всасывания (мази, пластыри и т.д.), глазные капли, назальные капли, суппозитории и т.д.).
Эти твердые препараты, такие как таблетки, капсулы, гранулы и порошки, могут содержать кристалл соединения 1 обычно в количестве от 0,001 до 99,5 масс. %, предпочтительно от 0,001 до 90 масс. %.
В случае получения твердого препарата для перорального применения наполнитель, связующее, дезинтегратор, смазывающее средство, краситель и им подобные добавляют к кристаллу соединения 1, если в этом существует необходимость, для получения таблеток, гранул, порошков или капсул посредством стандартного способа. На таблетки, гранулы, порошки, капсулы и им подобные можно также наносить покрытие, если в этом существует необходимость.
Доза лекарственного препарата в соответствии с настоящим изобретением обычно варьируется в зависимости от состояния организма, возраста, пола, веса тела и им подобных, при этом ее количество может быть достаточным для проявления необходимого эффекта. Например, в случае взрослого человека ежесуточно вводят от приблизительно 0,1 до 5000 мг (предпочтительно от 0,5 до 1000 мг) один раз в сутки или каждые несколько дней, или от 2 до 6 дробных доз в сутки.
Примеры
Кристалл соединения 1 в соответствии с настоящим изобретением можно получать посредством способов, описанных в примерах получения и примерах, приведенных ниже. Однако такие примеры приведены только для иллюстративных целей, и при этом кристалл соединения в соответствии с настоящим изобретением не ограничен каким-либо образом конкретными примерами, приведенными ниже.
В примерах получения и примерах, если конкретно не указано иное, силикагель для очистки с применением колоночной хроматографии на силикагеле представлял собой YMC GEL SILICA (YMC Co., Ltd, код по каталогу: SL06I52W), силикагель для очистки с применением колоночной хроматографии на силикагеле NH представлял собой силикагель NH (Fuji Silysia Chemical LTD., каталожный код: DM2035), силикагель для очистки с применением колоночной хроматографии на силикагеле ODS представлял собой YAMAZEN GEL ODS-SM (YAMAZEN Corporation, каталожные коды: W113, W116, и т.д.), пластина для TLC, предназначенная для очистки с применением тонкослойной хроматографии на силикагеле, представляла собой пластину для TLC на основе силикагеля 60F254 (Merck KGaA, код по каталогу: 1.05715.0001), пластина для TLC, предназначенная для очистки с применением тонкослойной хроматографии на силикагеле NH, представляла собой пластину для TLC на основе силикагеля NH (Fuji Silysia Chemical LTD., код по каталогу: T050908).
Аббревиатуры, используемые в данном документе, представляют собой следующее:
NMP: N-метилпирролидинон
THF: тетрагидрофуран
HATU: O-(7-азабензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат
HBTU: O-(бензотриазол-1-ил)-N,N,N',N'-тетраметилурония гексафторфосфат
TFA: трифторуксусная кислота
DMF: N,N-диметилформамид
DMSO: диметилсульфоксид
Химические сдвиги спектров 1H-ЯМР (протонного ядерного магнитного резонанса) регистрируются в δ единицах измерения (ppm) относительно тетраметилсилана, а константы взаимодействия регистрируются в герцах (Гц). Сокращения паттернов расщепления являются следующими: s: синглет; d: дуплет; t: триплет; q: квартет; m: мультиплет и br: уширенный.
[Пример 1]
(6S,9aS)-N-бензил-8-({6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамид
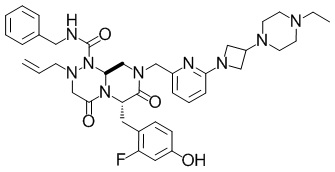
В раствор смеси из (6S,9aS)-N-бензил-6-((2-фтор-4-гидроксифенил)метил)-8-((6-фторпиридин-2-ил)метил)-4,7-диоксо-2-(проп-2-ен-1-ил)-октагидро-1H-пиразино[2,1-c][1,2,4]триазин-1-карбоксамида (397 мг, 0,689 ммоля), описанного в примере получения 1-1-6, и NMP (10 мл) добавляли смесь (1,16 г) 1-(азетидин-3-ил)-4-этилпиперазина и бензилбензола, описанную в примере получения 1-3-2, при комнатной температуре. Полученную в результате смесь подвергали воздействию микроволнового излучения при 140°C в течение 12 часов. Реакционную смесь охлаждали до комнатной температуры, затем в нее добавляли воду, затем полученный в результате раствор экстрагировали этилацетатом и органический слой промывали насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (метанол), а затем дополнительно очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол=20: 1) с получением искомого соединения (402 мг, выход: 80%).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(частей на миллион): 1,00(3H, t, J=6,8 Гц), 2,10-2,65(10H, m), 3,10-3,21(2H, m), 3,41-3,74(8H, m), 3,84-3,89(1H, m), 3,95-4,05(2H, m), 4,17-4,23(2H, m), 4,51(1H, dd, J=6,8 Гц, 15,6 Гц), 4,95(1H, d, J=13,6 Гц), 5,20-5,30(3H, m), 5,50-5,60(1H, m), 5,70-5,80(1H, m), 5,82-5,87(1H, m), 6,24(1H, d, J=8,0 Гц), 6,41(1H, dd, J=2,0 Гц, 11,2 Гц), 6,47(1H, dd, J=8,8 Гц, 8,8 Гц), 6,69(1H, d, J=7,2 Гц), 6,80-6,86(1H, m), 7,20-7,31(3H, m), 7,35-7,46(3H, m).
ESI-MS (масса/заряд): 726,57 [M+H]+.
[Пример получения 1-1-1]
(2,2-Диэтоксиэтил)((6-фторпиридин-2-ил)метил)амин
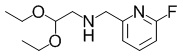
В раствор смеси из коммерчески доступного продукта 2,2-диэтоксиэтан-1-амина (926 мкл, 6,39 ммоль), THF (10,0 мл) и уксусной кислоты (1,00 мл) добавляли коммерчески доступный продукт 6-фторпиридин-2-карбальдегид (800 мг, 6,39 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 25 минут. Далее добавляли триацетоксиборогидрид натрия (2,71 г, 12,8 ммоль) в реакционную смесь при комнатной температуре, а затем перемешивали в течение 1 часа и 10 минут. В реакционную смесь добавляли гидрокарбонат натрия и воду для завершения реакции. Полученный в результате раствор экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле NH (гептан:этилацетат=1: 1) и затем дополнительно очищали с помощью колоночной хроматографии на силикагеле (этилацетат:метанол=20: 1) с получением искомого соединения (1,14 г, выход: 74%).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 1,22(6H, t, J=7,2 Гц), 2,76(2H, d, J=5,5 Гц), 3,50-3,61(2H, m), 3,65-3,76(2H, m), 3,89(2H, s), 4,64(1H, t, J=5,5 Гц), 6,80(1H, dd, J=2,8 Гц, 8,2 Гц), 7,22(1H, dd, J=2,4 Гц, 7,3 Гц), 7,74(1H, q, J=7,9 Гц).
[Пример получения 1-1-2]
9H-флуорен-9-илметил-N-((1S)-2-(4-(бензилокси)-2-фторфенил)-1-(2,2-диэтоксиэтил)((6-фторпиридин-2-ил)метил)карбамоил)этил)карбамат

В раствор смеси из (2,2-диэтоксиэтил)((6-фторпиридин-2-ил)метил)амина (3,50 г, 14,4 ммоль), описанного в примере получения 1-1-1, и дихлорметана (25 мл) добавляли (2S)-3-(4-(бензилокси)-2-фторфенил)-2-(((9H-флуорен-9-илметокси)карбонил)амино)пропановую кислоту (7,76 г, 15,1 ммоль), описанную в примере получения 1-2-7, N-метилморфолин (2,06 мл, 18,7 ммоль) и HATU (6,04 г, 15,8 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 13 часов. Добавляли гидрокарбонат натрия и воду в реакционную смесь и полученный в результате раствор экстрагировали дихлорметаном. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении с получением неочищенного продукта (14,4 г) на основе искомого соединения. Продукт использовали в последующей реакции без дополнительной очистки.
ESI-MS (масса/заряд): 758,50 [M+Na]+.
[Пример получения 1-1-3]
(2S)-2-амино-3-(4-(бензилокси)-2-фторфенил)-N-(2,2-диэтоксиэтил)-N-((6-фторпиридин-2-ил)метил)пропанамид
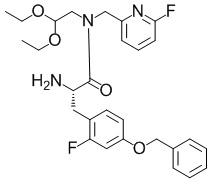
В раствор смеси из 9H-флуорен-9-илметил N-((1S)-2-(4-(бензилокси)-2-фторфенил)-1-((2,2-диэтоксиэтил)((6-фторпиридин-2-ил)метил)карбамоил)этил)карбамата (14,4 г), описанного в примере получения 1-1-2, и THF (30 мл) добавляли диэтиламин (5,27 мл, 50,4 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 2 часов. Реакционную смесь концентрировали при пониженном давлении, к остатку добавляли метанол, воду и гептан и разделяли полученную в результате смесь. Водный слой промывали гептаном и затем концентрировали при пониженном давлении. К остатку добавляли воду и полученный в результате раствор экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле NH (гептан:этилацетат=1: 1, а затем этилацетат) с получением искомого соединения (6,87 г, выход: 93%).
ESI-MS (масса/заряд): 514,32 [M+H]+.
[Пример получения 1-1-4]
(2S)-2-(2-(((бензилкарбамоил)амино)(проп-2-ен-1-ил)амино)ацетамидо)-3-(4-(бензилокси)-2-фторфенил)-N-(2,2-диэтоксиэтил)-N-((6-фторпиридин-2-ил)метил)пропанамид

В раствор смеси из (2S)-2-амино-3-(4-(бензилокси)-2-фторфенил)-N-(2,2-диэтоксиэтил)-N-((6-фторпиридин-2-ил)метил)пропанамида (4,87 г, 9,48 ммоль), описанного в примере получения 1-1-3, и дихлорметана (100 мл) добавляли известное вещество (WO2009/148192) 2-(((бензилкарбамоил)амино)(проп-2-ен-1-ил)амино)уксусную кислоту (2,62 г, 9,95 ммоль), триэтиламин (2,64 мл, 19,0 ммоль) и HBTU(3,96 г, 10,4 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 1 часа. Реакционную смесь концентрировали при пониженном давлении и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат и затем этилацетат:метанол=30: 1) с получением искомого соединения (7,28 г, выход: количественный).
ESI-MS (масса/заряд): 759,43 [M+H]+.
[Пример получения 1-1-5]
(6S,9aS)-N-бензил-6-((4-(бензилокси)-2-фторфенил)метил)-8-((6-фторпиридин-2-ил)метил)-4,7-диоксо-2-(проп-2-ен-1-ил)-октагидро-1H-пиразино[2,1-c][1,2,4]триазин-1-карбоксамид
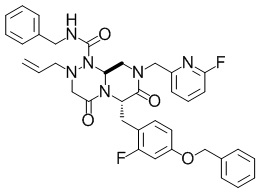
Раствор смеси из (2S)-2-(2-(((бензилкарбамоил)амино)(проп-2-ен-1-ил)амино)ацетамидо)-3-(4-(бензилокси)-2-фторфенил)-N-(2,2-диэтоксиэтил)-N-((6-фторпиридин-2-ил)метил)пропанамида (7,28 г, 9,48 ммоль), описанного в примере получения 1-1-4, и муравьиной кислоты (50 мл) перемешивали при комнатной температуре в течение 15 часов и 15 минут. Реакционную смесь концентрировали при пониженном давлении, к остатку добавляли водный раствор аммиака и полученный в результате раствор экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле NH (гептан:этилацетат=1: 1) с получением искомого соединения (5,04 г, выход: 80%).
ESI-MS (масса/заряд): 667,39 [M+H]+.
[Пример получения 1-1-6]
(6S,9aS)-N-бензил-6-((2-фтор-4-гидроксифенил)метил)-8-((6-фторпиридин-2-ил)метил)-4,7-диоксо-2-(проп-2-ен-1-ил)-октагидро-1H-пиразино[2,1-c][1,2,4]триазин-1-карбоксамид
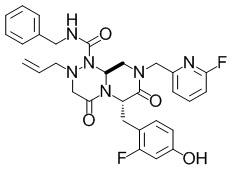
В раствор смеси из (6S,9aS)-N-бензил-6-((4-(бензилокси)-2-фторфенил)метил)-8-((6-фторпиридин-2-ил)метил)-4,7-диоксо-2-(проп-2-ен-1-ил)-октагидро-1H-пиразино[2,1-c][1,2,4]триазин-1-карбоксамида (5,04 г, 7,56 ммоль), описанного в примере получения 1-1-5, и TFA (20 мл) добавляли тиоанизол (3,55 мл, 30,2 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 13 часов и 50 минут. Реакционную смесь концентрировали при пониженном давлении, к остатку добавляли гидрокарбонат натрия и воду и полученный в результате раствор экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле NH (этилацетат:метанол=20: 1) с получением искомого соединения (4,34 г, выход: количественный).
ESI-MS (масса/заряд): 577,31 [M+H]+.
[Пример получения 1-2-1]
Метил-(2S)-2-(((9H-флуорен-9-илметокси)карбонил)амино)-3-гидроксипропаноат
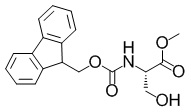
В раствор смеси из коммерчески доступного продукта гидрохлорида сложного метилового эфира L-серина (10,0 г, 64,3 ммоль), 1,4-диоксана (15 мл) и воды (90 мл) добавляли гидрокарбонат натрия (10,8 г, 129 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 15 минут. Далее раствор 2,5-диоксопирролидин-1-ил 9H-флуорен-9-илметилкарбоната (21,7 г, 64,3 ммоль) в 1,4-диоксане (60 мл) добавляли в полученный в результате раствор при комнатной температуре и полученную в результате смесь перемешивали при комнатной температуре в течение 14 часов. В реакционную смесь добавляли воду, полученный в результате раствор экстрагировали этилацетатом три раза и объединенный органический слой промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, в полученный в результате остаток добавляли простой диэтиловый эфир и гептан и собирали осадок с помощью фильтрации с получением искомого соединения (22,3 г, выход: количественный).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 2,00-2,15(1H, m), 3,81(3H, s), 3,89-4,07(2H, m), 4,20-4,28(1H, m), 4,39-4,53(3H, m), 5,63-5,74(1H, m), 7,29-7,37(2H, m), 7,38-7,46(2H, m), 7,55-7,65(2H, m), 7,74-7,82(2H, m).
[Пример получения 1-2-2]
Метил-(2S)-2-(((9H-флуорен-9-илметокси)карбонил)амино)-3-(((4-метилбензол)сульфонил)окси)пропаноат
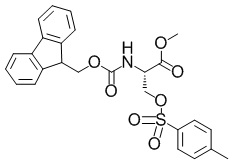
В раствор смеси из метил-(2S)-2-(((9H-флуорен-9-илметокси)карбонил)амино)-3-гидроксипропаноата (5,00 г, 14,6 ммоль), описанного в примере получения 1-2-1, и пиридина (25 мл) добавляли 4-диметиламинопиридин (18,0 мг, 0,146 ммоль) и п-толуолсульфонил хлорид (5,58 г, 29,3 ммоль) при 0°C и полученную в результате смесь перемешивали при 0°C в течение 7 часов. В реакционную смесь добавляли воду и полученный в результате раствор экстрагировали этилацетатом два раза. Объединенный органический слой промывали 1 н. хлористоводородной кислотой три раза, затем насыщенным водным раствором гидрокарбоната натрия и затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, затем в полученный в результате остаток добавляли этилацетат, простой диэтиловый эфир и гептан и затем собирали осадок с помощью фильтрации с получением искомого соединения (6,20 г, выход: 85%).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 2,37(3H, s), 3,74(3H, s), 4,16-4,23(1H, m), 4,23-4,31(1H, m), 4,32-4,40(2H, m), 4,41-4,48(1H, m), 4,54-4,62(1H, m), 5,63-5,66(1H, m), 7,26-7,37(4H, m), 7,38-7,45(2H, m), 7,56-7,64(2H, m), 7,72-7,81(4H, m).
[Пример получения 1-2-3]
Метил-(2R)-2-(((9H-флуорен-9-илметокси)карбонил)амино)-3-йодопропаноат
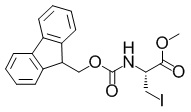
В раствор смеси из метил-(2S)-2-(((9H-флуорен-9-илметокси)карбонил)амино)-3-(((4-метилбензол)сульфонил)окси)пропаноата (6,20 г, 12,5 ммоль), описанного в примере получения 1-2-2, и ацетона (50 мл) добавляли йодид натрия (9,38 г, 62,6 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 90 часов и 50 минут. Фильтровали реакционную смесь и концентрировали фильтрат при пониженном давлении. К остатку добавляли воду и полученный в результате раствор экстрагировали этилацетатом. Органический слой промывали водой, затем насыщенным водным раствором тиосульфата натрия и затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, затем в полученный в результате остаток добавляли простой диэтиловый эфир и гептан и собирали осадок с помощью фильтрации с получением искомого соединения (3,82 г, выход: 68%).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 3,55-3,66(2H, m), 3,84(3H, s), 4,20-4,30(1H, m), 4,35-4,48(2H, m), 4,56-4,62(1H, m), 5,63-5,72(1H, m), 7,30-7,37(2H, m), 7,38-7,45(2H, m), 7,62(2H, d, J=7,2 Гц), 7,78(2H, d, J=7,5 Гц).
[Пример получения 1-2-4]
4-(Бензилокси)-1-бром-2-фторбензол
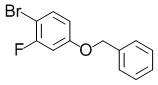
В раствор смеси из коммерчески доступного продукта 4-бром-3-фторфенола (15,0 г, 78,5 ммоль) и DMF (30 мл) добавляли карбонат калия (21,7 г, 157 ммоль) и бензилбромид (10,2 мл, 86,4 ммоль) при комнатной температуре и полученную в результате смесь перемешивали при комнатной температуре в течение 20 минут и затем при 70°C в течение 40 минут. Реакционную смесь охлаждали до комнатной температуры, затем в реакционную смесь добавляли воду и полученный в результате раствор экстрагировали этилацетатом. Органический слой промывали водой и затем насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, и полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (гептан:этилацетат=5: 1) с получением искомого соединения (22,7 г, выход: количественный).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 5,04(2H, s), 6,65-6,72(1H, m), 6,75-6,80(1H, m), 7,30-7,45(6H, m).
[Пример получения 1-2-5]
4-(Бензилокси)-2-фтор-1-йодбензол
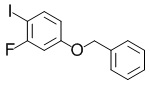
В раствор смеси из 4-(бензилокси)-1-бром-2-фторбензола (187 г, 665 ммоль), описанного в примере получения 1-2-4, и 1,4-диоксана (300 мл) добавляли йодид меди(I) (12,6 г, 66,1 ммоль), йодид натрия (200 г, 1,33 моль) и N,N'-диметилэтилендиамин (14,0 мл, 132 ммоль) при комнатной температуре и полученную в результате смесь перемешивали в атмосфере азота при 110-115°C в течение 19 часов. Реакционную смесь охлаждали до комнатной температуры, затем в реакционную смесь добавляли воду и этилацетат, полученную в результате смесь фильтровали с применением целита и разделяли фильтрат между водным слоем и органическим слоем. Водный слой экстрагировали этилацетатом. Объединенные органические слои фильтровали с применением стеклянного фильтра, содержащего силикагель, расположенный на нем. Силикагель промывали этилацетатом, объединяли полученные органические слои и выпаривали растворитель при пониженном давлении. Полученный в результате остаток очищали с помощью колоночной хроматографии на силикагеле (гептан:этилацетат=7: 1 и затем 4: 1) с получением искомого соединения (195 г, выход: 89%).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 5,04(2H, s), 6,57-6,62(1H, m), 6,73(1H, dd, J=2,8 Гц, 10,0 Гц), 7,31-7,43(5H, m), 7,55-7,60(1H, m).
[Пример получения 1-2-6]
Метил-(2S)-3-(4-(бензилокси)-2-фторфенил)-2-(((9H-флуорен-9-илметокси)карбонил)амино)пропаноат
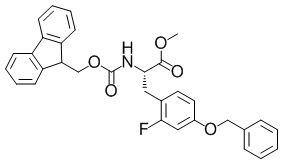
Добавляли цинковую пыль (51,6 г, 789 ммоль) в 1 н. хлористоводородную кислоту (100 мл), полученную в результате смесь обрабатывали ультразвуком, а затем оставляли отстояться, после чего удаляли из нее надосадочную жидкость. Данную процедуру повторяли два раза. К полученному в результате остатку цинка добавляли воду (300 мл), полученный в результате раствор перемешивали, и затем оставляли отстояться, и затем удаляли оттуда надосадочную жидкость. Данную процедуру повторяли три раза. Добавляли ацетон (300 мл) в полученный в результате продукт, смесь перемешивали и затем оставляли отстояться, удаляли из нее надосадочную жидкость, затем добавляли в раствор простой диэтиловый эфир (300 мл), полученный в результате раствор перемешивали и затем оставляли отстояться, удаляли оттуда надосадочную жидкость и затем сушили остаток при пониженном давлении с получением активированного цинка. В активированный цинк добавляли DMF (120 мл) и йод (3,36 г, 13,2 ммоль) в атмосфере азота при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 45 минут. В реакционную смесь добавляли раствор метил-(2R)-2-(((9H-флуорен-9-илметокси)карбонил)амино)-3-йодопропаноата (120 г, 266 ммоль), описанного в примере получения 1-2-3, в DMF (500 мл) в течение 30 минут в атмосфере азота при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 40 минут. В реакционную смесь добавляли 4-(бензилокси)-2-фтор-1-йодбензол (104 г, 318 ммоль), описанный в примере получения 1-2-5, трис(дибензилиденацетон)палладий(0) (6,00 г, 6,55 ммоль) и 2-дициклогексилфосфино-2',6'-диметоксибифенил (5,40 г, 13,2 ммоль) в атмосфере азота при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 20 часов и 40 минут. В реакционную смесь добавляли воду и этилацетат и полученный в результате раствор фильтровали с использованием целита. Фильтрат разделяли и водный слой дополнительно экстрагировали этилацетатом три раза. Объединенный органический слой промывали водой и насыщенным солевым раствором. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, затем в полученный в результате остаток добавляли простой диэтиловый эфир (1,00 л) и гептан (1,00 л) и затем собирали осадок с помощью фильтрации. Простой диэтиловый эфир (500 мл) и гептан (500 мл) добавляли к отфильтрованному твердому веществу и собирали осадок посредством фильтрации с получением искомого соединения (107 г, выход: 77%).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 3,03-3,20(2H, m), 3,75(3H, s), 4,20(1H, t, J=6,6 Гц), 4,25-4,38(1H, m), 4,43(1H, dd, J=7,1 Гц, 10,4 Гц), 4,58-4,70(1H, m), 4,99(2H, s), 5,33(1H, d, J=8,4 Гц), 6,63-6,72(2H, m), 6,94-7,03(1H, m), 7,26-7,48(9H, m), 7,52-7,62(2H, m), 7,77(2H, d, J=7,7 Гц).
[Пример получения 1-2-7]
(2S)-3-(4-(бензилокси)-2-фторфенил)-2-(((9H-флуорен-9-илметокси)карбонил)амино)пропановая кислота
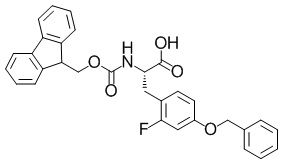
В раствор смеси из метил (2S)-3-(4-(бензилокси)-2-фторфенил)-2-(((9H-флуорен-9-илметокси)карбонил)амино)пропаноата (60,0 г, 114 ммоль), описанного в примере получения 1-2-6, и этилацетата (1331 мл) добавляли йодид лития (92,0 г, 685 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали с обратным холодильником в течение 23 часов и 45 минут. Реакционную смесь охлаждали до 0°C и собирали осадок посредством фильтрации. К полученному в результате твердому веществу добавляли 1 н. хлористоводородную кислоту (228 мл). Полученный в результате раствор экстрагировали этилацетатом. Органический слой сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении с получением искомого соединения (42,2 г, выход: 72%).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 3,05-3,15(1H, m), 3,18-3,30(1H, m), 4,15-4,23(1H, m), 4,25-4,50(2H, m), 4,60-4,70(1H, m), 4,99(2H, m), 5,29(1H, d, J=7,6 Гц), 6,64-6,73(2H, m), 7,06(1H, dd, J=8,0 Гц, 9,6 Гц), 7,24-7,44(9H, m), 7,55(2H, dd, J=6,4 Гц, 6,4 Гц), 7,76(2H, d, J=7,6 Гц).
ESI-MS (масса/заряд): 512,30 [M+H]+.
[Пример получения 1-3-1]
1-(1-(Дифенилметил)азетидин-3-ил)-4-этилпиперазин
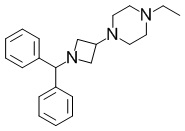
В раствор смеси из коммерчески доступного продукта 1-(дифенилметил)азетидин-3-она (10,1 г, 42,6 ммоль), THF (100 мл) и уксусной кислоты (5,00 мл) добавляли этилпиперазин (6,48 мл, 51,1 ммоль) при комнатной температуре. Полученную в результате смесь перемешивали при комнатной температуре в течение 45 минут. Триацетоксиборогидрид натрия (18,1 г, 85,1 ммоль) добавляли в реакционную смесь при комнатной температуре и затем перемешивали при комнатной температуре в течение 15 часов. В реакционную смесь добавляли гидрокарбонат натрия и воду и затем экстрагировали полученный в результате раствор этилацетатом. Органический слой промывали насыщенным солевым раствором, затем сушили над безводным сульфатом магния и затем фильтровали. Выпаривали растворитель при пониженном давлении, и очищали полученный в результате остаток с помощью колоночной хроматографии на силикагеле (этилацетат-метанол), и затем дополнительно очищали с помощью колоночной хроматографии на силикагеле NH (гептан:этилацетат=2: 1 и 1: 1) с получением искомого соединения (12,7 г, выход: 89%).
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 1,07(3H, t, J=7,6 Гц), 2,20-2,65(10H, m), 2,85-2,93(2H, m), 2,95-3,05(1H, m), 3,35-3,45(2H, m), 4,41(1H, s), 7,15-7,20(2H, m), 7,23-7,29(4H, m), 7,37-7,42(4H, m).
[Пример получения 1-3-2]
1-(Азетидин-3-ил)-4-этилпиперазин
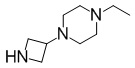
В раствор смеси из 1-(1-(дифенилметил)азетидин-3-ил)-4-этилпиперазина (12,7 г, 37,9 ммоль), описанного в примере получения 1-3-1, и метанола (50 мл) добавляли гидроксид палладия на угле (5,00 г) при комнатной температуре. Полученную в результате смесь перемешивали в атмосфере водорода при комнатной температуре и при от 0,35 МПа до 0,40 МПа в течение 10 часов. Реакционную смесь продували с использованием атмосферы азота и затем фильтровали с использованием целита. Концентрировали фильтрат при пониженном давлении с получением искомого соединения в форме смеси (12,4 г) с бензилбензолом. Продукт использовали в последующей реакции без дополнительной очистки.
Спектр 1H-ЯМР (400 МГц, CDCl3) δ(ppm): 1,09(3H, t, J=7,2 Гц), 2,10-2,80(10H, m), 3,20-3,30(1H, m), 3,53-3,60(2H, m), 3,60-3,70(2H, m).
[Пример 2]
Кристалл (6S,9aS)-N-бензил-8-({6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида
Изопропилацетат (1,5 мл) добавляли в (6S,9aS)-N-бензил-8-({6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил}метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамид (150 мг), описанный в примере 1, и перемешивали при 85°C в течение одного часа, а затем в течение ночи при комнатной температуре. Полученную суспензию собирали фильтрованием, промывали изопропилацетатом, охлажденным до 4°C, а затем сушили при пониженном давлением при 45°C с получением 141,2 мг белого твердого вещества. В 100 мг полученного твердого вещества добавляли 1-пропанол (0,4 мл) и смесь нагревали и растворяли при 95°C. Добавляли 1-пропанол (0,1 мл) и смесь перемешивали при комнатной температуре. Гептан (1,5 мл) добавляли в полученную смесь при комнатной температуре. Суспензию собирали фильтрованием, промывали гептаном, охлажденным до 4°C, а затем сушили при пониженном давлении при 45°C с получением 92,1 мг искомого соединения.
1H-ЯМР (600 MГц, CD3OD) δ (ppm): 1,15 (t, J=7 Гц, 3 H), 2,0-3,1 (br, 8 H), 3,13 (dd, J=14, 7 Гц, 1 H), 3,32 (m, 1 H), 3,33 (m, 1 H), 3,44 (dd, J=14, 5 Гц, 1 H), 3,46 (d, J=17 Гц, 1 H), 3,54 (dd, J=12, 4 Гц, 1 H), 3,62 (dd, J=13, 6 Гц, 1 H), 3,69 (dd, J=13, 7 Гц, 1 H), 3,79 (dd, J=8, 5 Гц, 1 H), 3,86 (dd, J=8, 6 Гц, 1 H), 3,89 (t, J=11 Гц, 1 H), 4,03 (t, J=8 Гц, 1 H), 4,06 (t, J=8 Гц, 1 H), 4,25 (d, J=15 Гц, 1 H), 4,33 (d, J=16 Гц, 1 H), 4,41 (d, J=16 Гц, 1 H), 4,77 (d, J=16 Гц, 1 H), 5,12-5,18 (m, 2H), 5,27 (dd, J=7, 5 Гц, 1 H), 5,84 (m, 2 H), 6,30 (d, J=8 Гц, 1 H), 6,38 (m, 1 H), 6,40 (m,1 H), 6,51 (d, J=8 Гц, 1 H), 6,88 (dd, J=9, 8 Гц, 1 H), 7,25 (dd, J=8, 7 Гц, 1 H), 7,28 (d, J=8 Гц, 2 H), 7,34 (dd, J=8, 7 Гц, 2 H), 7,47 (dd, J=8, 8 Гц, 1 H)
Испытательный пример 1. Выявление сигнала Wnt
pcDNA3.1(+) (invitrogen) расщепляли рестрикционными ферментами BglII и NotI и встраивали в нее адаптер BEHKS, имеющий последовательность, показанную ниже (содержащую сайты для рестрикционных ферментов BglII, EcoRI, HindIII, KpnI, SacI и NotI), посредством чего получали плазмиду pNeo-HKS.
BEHKS-F 5'-gatctgaattcaagcttctcgagggtacctctagagagctcgc-3' (SEQ ID NO: 1)
BEHKS-R 5'-ggccgcgagctctctagaggtaccctcgagaagcttgaattca-3' (SEQ ID NO: 2)
Далее фрагмент с длиной приблизительно 2700 п.о. (содержащий Wnt-чувствительную последовательность и ген люциферазы), который получали посредством расщепления из плазмиды TOPglow, содержащейся в наборе TOPglow/FOPglow TCF Reporter Kit (№ по каталогу 17-366 для северной части штата), с помощью рестрикционных ферментов HindIII и KpnI, встраивали между HindIII и KpnI в pNeo-HKS, посредством чего получали плазмиду pNeo-TOP. Плазмиду pNeo-TOP вводили в линию клеток почечного эпителия HEK293 человека, полученную из плода, затем проводили отбор клеток с использованием G418, а затем создавали линию клеточного клона посредством методики серийных разведений. Линию клеточного клона подвергали испытанию с выявлением сигнала Wnt.
Линию клеточного клона пересевали в обогащенную глюкозой культуральную среду D-MEM (Wako Pure Chemical Industries, Ltd.), содержащую 10% FBS, и в испытании использовали клетки в фазе роста. Собирали клетки с применением трипсин-EDTA, подсчитывали число клеток, а затем клетки суспендировали в обогащенной глюкозой культуральной среде D-MEM, содержащей 10% FBS, таким образом, что число клеток было равным 2 × 105 клеток/мл. Клеточную суспензию вносили в 96-луночный планшет для культивирования (Greiner Bio-One Co., Ltd., номер изделия: 655083) в количестве 0,1 мл/лунка и затем культивировали в течение ночи в термостате с 5% CO2 (37°C). После культивирования вещество, подлежащее испытанию, которое растворяли в DMSO, разбавляли обогащенной глюкозой культуральной средой D-MEM, содержащей 10% FBS и 80 мМ LiCl, с получением раствора образца. Вносили раствор образца (0,1 мл) в каждую лунку и затем культивировали в термостате с 5% CO2 (37°C) в течение ночи. Через шесть часов после добавления раствора образца надосадочную жидкость удаляли из каждой лунки, а затем в нее добавляли 50 мкл субстрата люциферазы Bright-Glo™ (Promega, номер изделия: E2620). Планшет помещали в перемешивающее устройство для планшетов на несколько секунд и затем измеряли испускание света из каждой лунки с использованием считывающего устройства для планшетов EnVision™ Multilabel (PerkinElmer Co., Ltd.). Определяли показатель активации сигнала Wnt (%) в каждой лунке и рассчитывали концентрацию (IC50), которая необходима для ингибирования активности сигнала Wnt веществом, представляющим интерес, на 50%, где количество света из лунки, в которую не добавляли раствор образца и добавляли LiCl, определяли как 100% активность сигнала Wnt, и количество света из лунки, в которую не добавляли либо раствор образца, либо LiCl, определяли как 0% активности сигнала Wnt. IC50 для соединения примера 1 составляла 0,06 мкM.
Испытательный пример 2. Эффект в отношении регрессирования небольших полипов в кишечнике у мыши APCMin/+
Ген APC (ген Adenomatous polyposis coli), представляющий собой фактор регуляции деградации сигнала Wnt, называется "геном-супрессором колоректального рака" и представляет собой причинный ген семейного аденоматозного полипоза. Если мутация происходит в гене APC, клетки слизистой оболочки ободочной и прямой кишки начинают бесконтрольно пролиферировать с образованием полипов, которые можно назвать предраковым очагом. Таким образом, известно, что ген играет важную роль в начальной стадии процесса возникновения колоректального рака.
У мыши с мутацией гена АРС (мышь APCMin/+), многие полипы развиваются в желудочно-кишечном тракте аналогично тому, как у пациента с семейным аденоматозным полипозом. Поэтому данная мышь применима для выяснения механизма возникновения или инвазии рака на основе сигнала WNT и представляет собой стандартную модель, которую использовали для исследований в отношении предупреждения, диагностики и лечения колоректального рака.
Мышей APCMin/+ (C57BL/6J-APC<Min>/J Hetero, самки, Sunplanet Co., Ltd.) разделяли на группы таким образом, чтобы средние значения веса тела мышей в группе практически соответствовали друг другу в первый день введения. Анализируемый образец получали посредством растворения испытуемого вещества (соединение из примера) в 0,1 н. HCl таким образом, чтобы концентрация соответствовала необходимой концентрации для введения, а затем хранили в холодильнике при 4°C. В контрольной группе (носитель) вводимый растворитель вводили перорально в тех же условиях, что и для испытуемого материала. Анализируемый образец непрерывно вводили перорально в дозах 50 мг/кг и 75 мг/кг два раза в сутки в течение 4 дней, а следующие три дня предусматривали "отдых от лекарств". Данную процедуру определяли как один цикл и введение проводили в общей сложности в течение 16 дней (то есть 4 дня × 4 цикла). Эксперимент проводили с 6-7 мышами в группе. Что касается каждой контрольной группы и группы с введением испытуемого вещества, то рассчитывали значение отношения веса тела в последний день к весу тела в первый день (то есть относительный вес тела: RBW). Группа с введением испытуемого вещества, в которой (RBW группы с введением испытуемого вещества)/(RBW контрольной группы) составлял 0,8 или более, определяли как группу, в которой возможно безопасное введение. Что касается группы с введением испытуемого вещества, то фактическое количество полипов после введения испытуемого вещества и стандартная погрешность фактического количества по сравнению с числом полипов в контроле в последний день (то есть в день 25, считая с первого дня введения) показаны на фигуре 1. В данном испытании подсчитывали полипы, образовавшиеся в тонкой кишке и толстой кишке. Проводили статистический анализ (тест Даннета) группы с введением испытуемого вещества относительно контрольной группы и приводили р-значение.
Испытательный пример 3. Противоопухолевый эффект в отношении модели с подкожной трансплантацией линии клеток K562 человека
Препарат на основе линии клеток K562 хронической миелогенной лейкемии человека (который культивировали в жидкой культуральной среде RPMI-1640, дополненной 10% FBS и пенициллином/стрептомицином), который получали таким образом с помощью PBS (Wako Pure Chemical Industries, Ltd.; № по каталогу 045-29795), чтобы плотность составила 2 × 108 клеток/мл, смешивали с MATRIGEL (BD Bioscience, № по каталогу: 354234) при соотношении компонентов смеси 1: 1, за счет чего получали суспензию клеток с плотностью 1 × 108 клеток/мл. Полученную в результате суспензию клеток трансплантировали подкожно в правый бок каждой из шестинедельных безтимусных мышей (CAnN.Cg-Foxn1nu/CrlCrlj, самки, Charles River Laboratories, Япония) в дозе 100 мкл. Через семь дней после трансплантации измеряли наименьший диаметр и наибольший диаметр опухоли с использованием электронного цифрового штангенциркуля (Digimatic TM Caliper, Mitutoyo Corporation) для расчета объема опухоли в соответствии со следующим уравнением.
Объем опухоли (мм3)=(наибольший диаметр (мм)) × (наименьший диаметр (мм)) × (наименьший диаметр (мм))/2
Мышей разделяли на группы по среднему значению объемов опухолей у мышей в группе, которые определяли на основе объема опухоли в первый день введения. Анализируемый образец получали посредством растворения соединения из примера 1 в 0,1 н. HCl таким образом, что количество дозы было равным 10 мл/кг. Раствор дазатиниба для введения получали посредством растворения свободного основания дазатиниба (LC Laboratories, №: по каталогу D-3307) в растворе 1: 1 дистиллированной воды Otsuka (Otsuka Pharmaceutical Co., Ltd., № по каталогу: 1324) и пропиленгликоля (Wako Pure Chemical Industries, Ltd., № по каталогу: 164-04996) таким образом, что количество было равным 10 мл/кг. Анализируемый образец вводили перорально непрерывно в течение 5 дней, начиная с первого дня введения в двух дробных дозах ежесуточно (bid). Раствор дазатиниба вводили перорально один раз в сутки (qd) непрерывно в течение 5 дней, а затем назначали двухдневный "отдых от лекарств". Данную процедуру определяли как один цикл и введение проводили в общей сложности в двух циклах. Контрольная группа представляла собой группу, в которой не вводили какое-либо соединение из примера. В эксперименте одна группа включала 9-10 мышей. Что касается контрольной группы, то есть группы, в которой вводили только соединение из примера, группы, в которой вводили только дазатиниб, и группы, в которой вводили как соединение из примера, так и дазатиниб (называемой в данном документе 'группа комбинированного введения'), то объемы опухоли и значения веса тела измеряли в динамике за период от первого дня до дня 28. Что касается контрольной группы и группы, в которой вводили только соединение из примера, то измерение проводили в течение периода от первого дня до дня 11. В каждом измерении рассчитывали объем опухоли (относительный объем опухоли: RTV) и вес тела (относительный вес тела: RBW) относительно значений для первого дня, при этом графики, определенные для периода с первого дня введения до дня 28, показаны на фигурах 2 и 3. Дополнительный статистический анализ (тест Даннета) проводили в группе, в которой вводили и соединение из примера, и дазатиниб, по сравнению с группой, в которой вводили только дазатиниб, с использованием значения RTV в день 28, при этом группу, в которой значение P составляло 0,05 или меньше, обозначали звездочкой (*). Кроме того, число особей, у которых опухоль не наблюдали при визуальной оценке и она была неощутима (т.е. с непальпируемой опухолью) в день 28, также показано в таблице 6. В это же время проводили статистический анализ (тест Даннета) в группе, в которой вводили и соединение из примера, и дазатиниб, по сравнению с группой, в которой вводили только дазатиниб, и группу, в которой значение P составляло 0,05 или меньше, обозначали звездочкой (*), а группу, в которой значение P составляло 0,01 или меньше, обозначали звездочками (***).
[Таблица 1]
|
Испытательный пример 4. Порошковая рентгеновская дифракция
Кристалл соединения 1, полученный в примере 2, помещали на пластину для образца устройства для порошковой рентгеновской дифракции и анализировали при следующих условиях. Результаты приведены на фигуре 4.
(Условия измерений)
Анод: медный
Детектор: сцинтилляционный счетчик
Напряжение в трубке: 50 кВ
Сила тока в трубке: 300 мА
Щель: щель расходимости: 0,5 мм; щель рассеивания: открыта; светоприемная щель: открыта
Скорость сканирования: 5°/минуту
Шаг сканирования: 0,02°
Диапазон сканирования: 5°-35°
Держатель образца: алюминиевый держатель
Испытательный пример 5. Термический анализ
Кристалл, полученный в примере 2, аккуратно взвешивали в алюминиевой кювете для образцов и подвергали термогравиметрии (TG) и дифференциальному термическому анализу (DTA) при следующих условиях.
Результаты приведены на фигуре 5.
(Условия измерений)
Атмосфера: в потоке газообразного азота при 40 мл/минута
Стандарт: пустая алюминиевая кювета для образцов
Скорость нагревания: 10°C/минута
Шаг сканирования: 1 секунда
Диапазон температуры измерения: 40-300°C
Испытательный пример 6. Спектр твердофазного 13C-ЯМР
Спектр твердофазного 13C-ЯМР измеряли при следующих условиях. Результаты приведены на фигуре 6.
(Условия измерений)
Используемый аппарат: Avance 400MГц (изготовленный компанией BRUKER) 7 мм-CPMAS датчик (изготовленный компанией BRUKER)
Измеряемое ядро: 13C (частота резонанса 100,6248425 MГц)
Температура при измерениях: комнатная температура (22°C)
Импульсный режим: Измерение CPTOSS
Частота вращения: 5000 Гц
Частота повторения импульсов: 4 секунды
Время контакта: 1 миллисекунда
Число сканирований: 8000
Эталонный материал: глицин (внешний стандарт: 176,03 ppm)
Спектр твердофазного 13C-ЯМР получали посредством способа CPTOSS (способа для исключения боковых полос вращения) с ядром углерода (частота резонанса 100,6 MГц) посредством устройства для ЯМР, BRUKER Avance 400 MГц, оснащенного 7 мм датчиком CPMAS (изготовленным компанией BRUKER). Пробирку с образцом, содержащую приблизительно 300 мг твердого образца, вращали с частотой 5 кГц и измеряли при времени контакта 1 миллисекунда, времени запаздывания импульса 4 секунды и числе сканирований 8000 при комнатной температуре (22°C). Химические сдвиги корригировали по резонансу карбонильного углерода глицина при 176,03 ppm, используемого в качестве внешнего стандарта.
![Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида](https://fips.edrid.ru/images/rid/5a/84/6c/6eb9a117eb63fddfc829f95a748d9c16.jpg)
![Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида](https://fips.edrid.ru/images/rid/5a/84/6c/4e4eb424133db8c822198ea6da751a76.jpg)
![Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида](https://fips.edrid.ru/images/rid/5a/84/6c/5c2aded0927a6d5d961b960ea6d0b2be.jpg)
![Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида](https://fips.edrid.ru/images/rid/5a/84/6c/4baa4e05ee8d4409a213e41a297beb9c.jpg)
![Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида](https://fips.edrid.ru/images/rid/5a/84/6c/ba11eaaa9cfbe77348811e2632efd995.jpg)
![Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида](https://fips.edrid.ru/images/rid/5a/84/6c/f36690f6d41bf21c8313b491adafabc3.jpg)
![Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида](https://fips.edrid.ru/images/rid/5a/84/6c/3daf74a9d8ebd91185007a70226050c8.jpg)
![Кристалл (6S,9aS)-N-бензил-8-({ 6-[3-(4-этилпиперазин-1-ил)азетидин-1-ил]пиридин-2-ил} метил)-6-(2-фтор-4-гидроксибензил)-4,7-диоксо-2-(проп-2-ен-1-ил)гексагидро-2H-пиразино[2,1-c][1,2,4]триазин-1(6H)-карбоксамида](https://fips.edrid.ru/images/rid/5a/84/6c/2cc0c77b9f684ba25e5614b9cd3f1427.jpg)
Липосомальная композиция
Конденсированное производное аминодигидротиазина
Производные пиридина, замещенные гетероциклическим кольцом и фосфоноксиметильной группой и содержащие их противогрибковые средства
Промежуточные соединения и способы синтеза аналогов галихондрина в
Реагенты и способы для бета-кетоамидного синтеза синтетического предшественника иммунологического адъюванта е6020
Новое конденсированное производное аминодигидротиазина
Азотсодержащие конденсированные гетероциклические соединения и их применение в качестве ингибиторов продукции бета-амилоида
Аналоги галихондрина в
Противоопухолевое средство, задействующее соединения с ингибирующим эффектом к киназам в комбинации
Биомаркер для болезни альцгеймера или умеренного когнитивного расстройства
Соль производного пирролидин-3-ил-уксусной кислоты и ее кристаллы

